Procédé pour la préparation de dérivés fonctionnalisés de β-(1.3)-glucanes.
La présente invention concerne d'une façon générale un nouveau procédé de préparation par voie chimique de dérivés fonctionnalisés de β-(l,3)-glucanes, permettant d'accéder à des oligosaccharides libres ou comportant des groupements spécifiques en des positions préalablement définies.
La présente invention trouve notamment application pour la préparation de composés biologiquement actifs utilisables dans le domaine agricole, cosmétique ou pharmaceutique.
On sait que de nombreux oligosaccharides présentent une activité biologique généralement liée à la présence de groupements spécifiques (c'est-à-dire non hydroxyles), tels que par exemple des groupements sulfate, phosphate, méthyle... sur des positions bien définies.
L'activité biologique est également liée, dans certains cas, à la longueur de l'oligosaccharide. Les oligosaccharides libres biologiquement actifs sont habituellement obtenus par hydrolyse ou acétolyse de polysaccharides naturels d'origine végétale et se présentent généralement sous la forme de mélanges complexes extrêmement difficiles à purifier.
On a déjà proposé, notamment dans le document FR 98.04610 un procédé de préparation par voie chimique du disaccharide libre communément appelé Laminaribiose, connu pour ses diverses activités biologiques.
Ce procédé qui permet d'obtenir un rendement global élevé en
Laminaribiose, et qui comprend généralement la réaction entre un donneur de glycosyle et un accepteur de glycosyle, est essentiellement caractérisé par un choix judicieux de ces composés, ainsi que du promoteur utilisé lors de la réaction de couplage.
Cependant, ce procédé est spécifique du seul Laminaribiose.
Les oligosaccharides biologiquement actifs comportant des groupements spécifiques sont généralement obtenus à partir d'oligosaccharides libres qui conduisent inévitablement à des mélanges complexes difficiles à purifier, y compris dans le cas où l'oligosaccharide de départ est de structure parfaitement définie. D'une façon générale, plus l'oligosaccharide est long et plus le mélange obtenu est complexe et difficile à purifier.
La préparation par voie chimique de dérivés de β-(l,3)-glucanes sélectivement fonctionnalisés permettant d'accéder à des oligosaccharides libres ou comportant des groupements spécifiques en des positions préalablement
définies se heurte notamment à la difficulté de différencier les fonctions hydroxyles tant au niveau de chaque unité monosaccharidique, et plus particulièrement au niveau des positions 2 et 3, qu'au niveau du polysaccharide.
Il existe donc un besoin non satisfait à ce jour, de disposer d'un procédé général permettant la préparation de dérivés fonctionnalisés de β-(l,3)-glucanes variés, présentant un nombre limité d'étapes, compatible avec un vaste choix de groupes protecteurs de différents types, d'une mise en oeuvre aisée et permettant d'obtenir les produits recherchés sous forme pure.
L'expression "dérivés sélectivement fonctionnalisés" utilisée dans le cadre de la présente description vise donc à couvrir des composés dont chaque position peut être sélectivement transformée par voie chimique en un groupement spécifique tel que par exemple un groupement sulfate, phosphate ou méthyle.
Il a été découvert, et ceci constitue le fondement de la présente invention, qu'il était possible de répondre à ce besoin par l'utilisation de donneurs de glycosyle et d'accepteurs de glycosyle originaux susceptibles en outre d'être préparés par une seule et même voie de synthèse à partir du glucose.
Ainsi, selon un premier aspect, la présente invention a pour objet un procédé pour la préparation de dérivés fonctionalisés de β-(l,3)-glucanes comprenant une réaction entre un donneur de glycosyle et un accepteur de glycosyle, caractérisé en ce que le donneur de glycosyle est choisi dans le groupe constitué des composés de formules générales la et Ib:
la
Ib
dans lesquelles :
X représente un groupe partant choisi parmi :
- un groupement de formule S(O)pRa, dans laquelle Ra représente un radical alkyle ayant 1 à 18 atomes de carbone, un radical 1,1-dicyclohexylméthyle, un radical aryle non substitué ou substitué par un groupement alkyle ou alcoxy ayant de 1 à 6 atomes de carbone, un groupement nitro ou acetamide et p est un nombre entier égal à 0 ou 1 ;
R représente : un radical alkyle, halogénoalkyle ou cétoalkyle ayant de 1 à 6 atomes de carbone ; un radical aryle non substitué ou substitué par un ou plusieurs groupements choisis parmi un atome d'halogène, un radical alcoxy ayant de 1 à 6 atomes de carbone ou un groupement nitro ;
R3 et R4, différents de -CO-R1 et de R2, représentent indépendamment un radical benzyle, chlorobenzyle, méthoxybenzyle, nitrobenzyle, allyle, méthylnaphtyle, chloroacétyle, trialkylsilyle ou triarylméthyle; ou forment ensemble un radical éthylidyle. isopropylidyle, hexafluoroisopropylidyle, cyclopentylidyle, cyclohexylidyle, cycloheptylidyle, butylidyle, 1-tertiobutyléthylidyle, benzylidyle, méthoxybenzylidyle, 1- phénylbenzylidyle .
R" représente :
- un groupement différent de -COR1 et choisi parmi un radical méthyle, allyle, méthylnaphthyle, benzyle, paraméthoxybenzyle ;
n est un nombre entier compris entre 1 et 4 ; étant précisé que dans le cas où n est supérieur à 1, -COR1, R3 et R4 peuvent être différents d'une unité glucosyle à l'autre ;
et en ce que l'accepteur de glycosyle est choisi dans le groupe constitué des composés de formule générale II:
dans laquelle :
Y représente un groupe choisi parmi :
- un groupement de formule -O-R5 dans laquelle Rt> représente un radical alkyle ayant de 1 à 24 atomes de carbone, alcényle ayant de 2 à 24 atomes de carbone ou un radical arylalkylaryle ou arylalkyle ayant de 6 à 18 atomes de carbone ;
- un résidu de serine ou de thréonine ;
- un résidu de stérol ;
- un résidu de glycérolipide ;
- un groupement de formule -S- Ra dans laquelle Ra est tel que défini précédemment ;
R i l , n R3J e _tt r R> 4 sont tels que définis précédemment et ;
m est un nombre entier compris entre 1 et 8, étant précisé que dans le cas où m est supérieur à 1, -COR1, R3 et R4 peuvent être différents d'une unité glucosyle à l'autre. Dans la description et les revendications, on entend :
- par radical alkyle, toute chaîne hydrocarbonée, linéaire ou ramifiée, un radical ayant de 1 à 6 atomes de carbone étant par exemple un radical méthyle, éthyle, propyle, isopropyle, butyle, isobutyle, tertiobutyle, pentyle. isopentyle. hexyle, isohexyle ; - par radical alcényle, toute chaîne hydrocarbonée, linéaire ou ramifiée, comportant une double liaison ;
- par radical alcoxy, tout radical de formule -O-R dans laquelle R est un radical alkyle tel que défini ci-dessus ;
- par radical halogénoalkyle ayant de 1 à 6 atomes de carbone, tout radical alkyle dont 1 à 7 atomes d'hydrogène ont été substitués par 1 à 7 atomes d'halogène, comme par exemple un radical chlorométhyle. un radical bromométhyle, un radical trifluorométhyle, un radical trifluoro-2,2,2-éthyle, un radical pentafluoroéthyle, un radical heptafluoropropyle ;
- par radical aryle. un cycle aromatique ayant 5 ou 6 atomes de carbone ou hétéroatomes, comme par exemple un radical phényle, pyridyle, thiényle, furannyle, pyrimidyle.
Le procédé selon l'invention est particulièrement intéressant puisqu'il permet d'accéder aisément à une multitude de composés sélectivement fonctionnalisés dans des positions prédéterminées. Les groupements spécifiques susceptibles d'être introduits dans ces positions prédéterminées selon le procédé de l'invention peuvent être de nature variée et seront généralement des groupements sulfate, phosphate, méthyle, notamment dans les positions 2, 3, 4 et 6. Ce procédé permet également de fixer des résidus originaux particulièrement intéressants d'un point de vue biologique sur la position anomérique de l'unité réductrice de l'oligosaccharide notamment dans le cas où Y représente un résidu de serine ou de thréonine, de stérol, de glycérolipide.
Selon une caractéristique particulière du procédé selon l'invention, le donneur de glycosyle précité est choisi dans le groupe constitué des composés de formules générales (la) ou (Ib) précitées dans lesquelles : X représente un groupe partant choisi parmi :
- un groupement de formule S(O)pRa, dans laquelle Ra représente un radical alkyle ayant 1 à 5 atomes de carbone, un radical aryle non substitué, de préférence un radical phényle ou un radical aryle substitué par un groupement alkyle ayant de 1 à 6 atomes de carbone, de préférence un radical toluyle, et p est un nombre entier égal à 0 ou 1 ;
R1 représente : un radical alkyle ayant de 1 à 6 atomes de carbone, de préférence méthyle, ou un groupement lévulinyle ; un radical aryle non substitué, de préférence un radical phényle ; R3 et R4, différents de -CO-R1 et de R2, représentent indépendamment un radical benzyle, chlorobenzyle, méthoxybenzyle, nitrobenzyle, allyle, méthylnaphtyle, chloroacétyle, trialkylsilyle ou triarylméthyle; ou forment ensemble un radical éthylidyle, isopropylidyle, benzylidyle ; R2 représente : - un groupement différent de -COR1 et choisi parmi un radical méthyle, allyle, méthylnaphthyle, benzyle, paraméthoxybenzyle ; n est un nombre entier égal à 1, 2 ou 3; étant précisé que dans le cas où n est égal à 2 ou 3, -COR1, R3 et R4 peuvent être différents d'une unité glucosyle à l'autre. Avantageusement, le donneur de glycosyle précité est choisi dans le groupe constitué des composés de formules générales (la) ou (Ib) précitées dans lesquelles :
X représente un groupe partant choisi parmi :
- un groupement de formule SRa dans laquelle Ra représente un radical éthyle, propyle, butyle, phényle ou toluyle, de préférence un radical éthyle ou phényle ;
R1 représente un radical méthyle ou phényle ;
R3 et R4 différents de -CO-R1 et de R2 représentent indépendamment un radical benzyle, méthoxybenzyle ou forment ensemble un radical benzylidyle ;
R2 représente un groupement différent de -COR1 et choisi parmi un radical allyle, méthylnaphtyle, de préférence un radical allyle ou méthlylnaphtyle ; n est un nombre entier égal à 1 ou 2; de préférence égal à 1 ; étant précisé que dans le cas où n est égal à 2, -COR1, R3 et R4 peuvent être différents d'une unité glucosyle à l'autre.
Selon une autre caractéristique particulière du procédé selon l'invention, l'accepteur de glycosyle précité est choisi dans le groupe constitué des composés de formule générale (II) précitée dans laquelle : Y représente un groupe choisi parmi : - un groupement de formule -O-Rb dans laquelle R^ représente un radical alkyle ayant de 1 à 24 atomes de carbone, alcényle ayant de 2 à 24 atomes de carbone ou un radical arylalkylaryle ou arylalkyle ayant de 6 à 18 atomes de carbone ;
- un résidu de serine ou de thréonine ; - un résidu de stérol ;
- un résidu de glycérolipide ;
- un groupement de formule -S- Ra dans laquelle Ra représente un radical alkyle ayant 1 à 5 atomes de carbone, un radical aryle non substitué, de préférence un radical phényle ou un radical aryle substitué par un groupement alkyle ayant de 1 à 6 atomes de carbone ; R1 représente : un radical alkyle ayant de 1 à 6 atomes de carbone, de préférence méthyle ; un radical aryle non substitué, de préférence un radical phényle ; R" et R4, différents de -CO-R1 et de R2, représentent indépendamment un radical benzyle, chlorobenzyle, méthoxybenzyle, nitrobenzyle, allyle, méthylnaphtyle, chloroacétyle, trialkylsilyle ou triarylméthyle; ou forment ensemble un radical éthylidyle, isopropylidyle, benzylidyle ; m est un nombre entier compris entre 1 et 8, étant précisé que dans le cas où m est supérieur à 1 , -COR , R et R peuvent être différents d'une unité glucosyle à l'autre.
Avantageusement, l'accepteur de glycosyle précité est choisi dans le groupe constitué des composés de formule générale (II) précitée dans laquelle l'une au moins des conditions suivantes est réalisée : Y représente un groupe choisi parmi :
- un groupement de formule -O-R^ dans laquelle Rj, représente un radical alkyle ayant de 1 à 24 atomes de carbone, alcényle ayant de 2 à 24 atomes de carbone ou un radical benzyle ; R1 représente un radical méthyle ou phényle ;
RJ et R4 différents de -CO-R1 représentent indépendamment un radical benzyle, méthoxybenzyle ou forment ensemble un radical benzylidyle ; m est un nombre entier compris entre 1 et 8, étant précisé que dans le cas où m est supérieur à 1 , -COR1, RJ et R4 peuvent être différents d'une unité glucosyle à l'autre.
Comme on le comprend, le procédé selon l'invention comporte une ou plusieurs étapes de couplage, selon le nombre d'unités de l'oligosaccharide.
D'une façon générale, chaque étape de couplage entre un donneur de glycosyle et un accepteur de glycosyle sera réalisée en solution dans un solvant organique anhydre à une température comprise entre -80°C et 40°C, pendant une durée de 1 minute à 8 heures en présence d'un promoteur approprié choisi parmi :
- une source d'ions halonium, associée ou non à un acide de Lewis ou un sel d'acide fort dans le cas des composés de formule générale (la) dans laquelle X représente un groupement S(O)pRa tel que défini ci-dessus et dans lequel p est égal à 0 ;
- un acide de Lewis associé à une aminé, dans le cas des composés de formule générale (la) dans laquelle X représente un groupement S(O)pRa tel que défini ci-dessus et dans lequel p est égal à 1 ; - un acide de Brδnstedt ou un acide de Lewis, dans le cas des composés de formule (Ib).
La nature chimique du promoteur, les quantités respectives de donneur de glycosyle, d'accepteur de glycosyle et de promoteur ainsi que les conditions réactionnelles de chaque étape de couplage pourront être facilement déterminées par l'homme de métier qui pourra se reporter notamment à la description du document FR 98.04610.
Les composés de formule (la), (Ib) et (II) pourront être préparés par diverses voies de synthèse connues dans la chimie des sucres.
Avantageusement, et ceci constitue une caractéristique originale de l'invention, tous ces composés pourront être préparés par une voie de synthèse unique à partir du glucose, ce qui constitue un avantage particulièrement intéressant du point de vue industriel.
Cette voie de synthèse, illustrée ci-dessous par le schéma réactionnel (I) dans le cas des monosaccharides, comprend dans l'ordre suivant :
- la préparation [ j d'un dérivé du glucose sous forme furanosique (X) dont les positions 1,2 et 5,6 sont protégées par exemple par des groupements acétals ;
- la fonctionnalisation sélective [Y] du composé ainsi obtenu en position 3 par un groupement correspondant au groupement R" défini précédemment pour former un composé (IX);
- le clivage [3] des groupements protecteurs des positions 1,2 et 5,6 dudit composé (IX) pour former un composé (VIII);
- l'introduction [4j dans ledit composé (Nffl) de groupes esters, de préférence acétyle ou benzoyle en position 1,2 (introduction du groupement COR1 en position 2), 4 et 6 pour former un composé (VII);
- l'introduction 5J en position anomérique d'un groupement thio de formule
S-Ra, de préférence un groupement thioéthyle ou thiophényle, pour former un composé de formule (VI) ;
- la déestérification [ό] des positions 2, 4 et 6 ou &_ des positions 4 et 6 du composé (NI) pour former ainsi les composés (IN) ou (V) ;
- l'introduction [7] , \ Ψj et 7^ dans lesdits composés (IN) ou (V) des groupements RJ et R4 tels que définis précédemment pour conduire au composé (III) ou au monosaccharide (la) recherché ;
- le cas échéant, l'estérification [ ] en position 2 dudit composé (III) pour conduire au monosaccharide voulu (la).
On obtient ainsi un composé sélectivement fonctionnalisé par l'introduction successive des groupements en position 3 puis 4 et 6 puis 2, ou en position 3 puis 2 puis 4 et 6 qui permet de conduire au composé recherché de formule (la), lequel permet de conduire subséquemment en une ou deux étapes de glycosylation et/ou de déprotection et/ou d'activation en position anomérique à un donneur de glycosyle ou un accepteur de glycosyle de formules (Ib) et (II) dans lesquelles n=l et m=l .
Ib (n=l) , II (m≈l)
SCHEMA REACTIONNEL I
Si le schéma réactionnel I précité est original dans certaines de ses séquences réactionnelles, l'homme de métier n'aura toutefois aucune difficulté pour déterminer les conditions réactionnelles les plus appropriées pour mettre en œuvre chacune des étapes mentionnées de ce schéma.
Les monosaccharides ainsi obtenus permettent d'accéder aisément aux autres oligosaccharides (di-, tri-, etc..) de formule (la), (Ib) et (II).
De préférence, un tel oligosaccharide sera préparé par une réaction de couplage entre un donneur de glycosyle monosaccharidique tel qu'obtenu selon le schéma réactionnel (I) ou disaccharidique et un accepteur de glycosyle monosaccharidique tel qu'obtenu selon le schéma réactionnel (I), ou le cas échéant polysaccharidique obtenu à partir de monosaccharides par une ou plusieurs opérations antérieures de couplage, et déprotection sélective du groupe R2.
Selon un second aspect, la présente demande vise à couvrir, en tant que produit nouveau, les synthons donneurs ou accepteurs de glycosyle de formule (la), (Ib) ou (II) définis précédemment.
L'invention sera maintenant illustrée par les exemples non limitatifs suivants préparés en suivant le schéma réactionnel I donné précédemment :
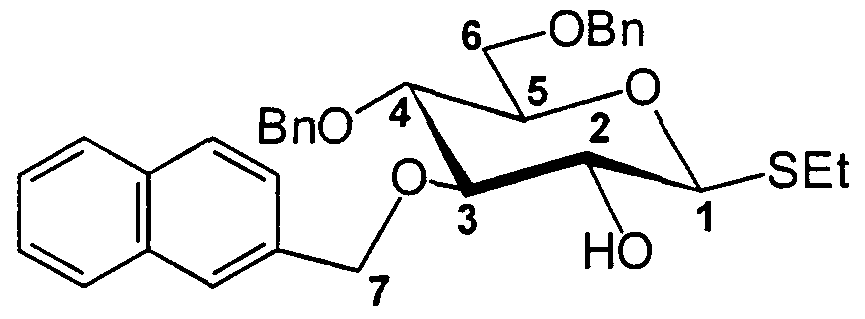
EXEMPLE 1 Préparation du 2,4,6-tri-O-acétyl-3-O-(2-méthylnaphtvI)-l-thio-β-D-gluco- pyranoside d'éthyle.
Etape g: préparation du 1,2:5, 6-di-O-isopropylidene-3-O-(2-méthylnaphtyl)-α-D- slucofuranose.
Dans un réacteur de 2 L sont introduits 117,7 g (1 éq.) de 1,2:5, 6-di-O- isopropylidene-α-D-glucofuranose commercial (M = 260,28) et 100 g de bromure de 2 -méthylnaphtyle (1 éq.) à une température de 2°C. 400 mL de diméthylformamide sont alors ajoutés (dilution 1 dans 2) et le milieu réactionnel est additionné progressivement de 21,7 g (1,2 éq.) d'hydrure de sodium à 60%
pendant 10 minutes. On laisse ensuite remonter doucement la température. Au bout de 2h20', l'excès de NaH est neutralisé par du méthanol et le produit est précipité par addition de 2 L d'eau glacé sous vive agitation. 2 heures plus tard, le surnageant est éliminé et le précipité repris au dichlorométhane (1 L). Après décantation, la phase organique est séchée et concentrée. Le produit brut (M = 400,5) est directement utilisé pour l'étape suivante. CCM: Rf = 0,5 [toluène/acétate d'éthyle (9/1; v/v)]. Solide blanc. RMN 13C (CDC13, 100 MHz): 135,12; 133,29; 133,11 (C quat. arom.); 128,29, 127,95, 127,78, 126,54, 126,25, 126,07, 125,71 (C arom.); 111,89, 109,011 (C quat. acetal); 105,39 (Cl); 82,73 (C2); 81,65 (C3); 81,41 (C4); 72,59 (C5); 72,49 (C7); 67,50 (C6); 26,90, 26,32, 25,54 (CH3).
RMN 'H (CDCI3, 400 MHz): 7,84-7,80 (m, 4H, H arom.); 7,49-7,45 (m, 3H, H arom.); 5,93 (d, 1H, Hl, JHι-H2 = 3,7 Hz); 4,85 (d, 1H, H7a, JH7a-H7b = 12,0 Hz); 4,79 (d, 1H, H7b, JH7b-H7a = 12,0 Hz); 4,63 (d, 1H, H2, JH2-HI = 3,7 Hz); 4,42 (td, 1H, H5, JH5-H4 = 7,8 Hz, JH5-H6a = T H5-H6b = 6,0 Hz); 4,16 (dd, 1H, H4, JH4-H3 = 3,0 Hz, JH4-H5 = 7,7 Hz); 4,14 (dd, 1H, H6a, JH6a-H5 = 6,1 Hz, JH6a-H6b = 8,6 Hz); 4,08 (d, 1H, H3, JH3-H4 = 3,0 Hz); 4,03 (dd, 1H, H6b, JH6b-H5 = 5,8 Hz; JH6 -H6a = 8,6 Hz); 1,49, 1,43, 1,40, 1,31 (4s, 12H, CH3).
Etape [3]: préparation du 3-O-(2-méthylnaphtyl)-D-glucopyranose.
Le produit obtenu précédemment (M = 400,5; 135 g théoriques) est dissous dans 270 mL d'acétone et introduit dans un réacteur de 2 L. 270 mL d'eau (dilution 1 dans 3 au total) puis 420 g de résine IR 120 (H+) sont alors ajoutés. Le milieu réactionnel est chauffé à 60°C pendant 1 à 2 jours (au départ le mélange est hétérogène puis devient homogène au cours de la réaction). La réaction terminée, la résine est filtrée, rincée avec du méthanol et le filtrat est neutralisé par addition de quelques mL de solution aqueuse de bicarbonate de sodium à 5%. On évapore à sec et on lave 2 fois le produit en réalisant une suspension de ce dernier dans 1 L de toluène (agitation pendant 15 min à 40 °C puis filtration après retour à
température ambiante). Finalement, 116 g de 3-O-(2-méthylnaphtyl)-D- glucopyranose recherché (M = 320,3) sont recueillis.
CCM: Rf = 0,2 [dichlorométhane/méthanol (9/1; v/v)].
Solide blanc.
Rendement (%) = 80 pour les étapes g et j.
RMN 13C (CD3OD, 100 MHz): 137,95, 134,77, 134,44 (C quat. arom.); 128,91,
128,76, 128,62, 127,49, 127,37, 127,32, 126,94, 126,75 (C arom.); anomère β:
98,29 (Cl); 86,40 (C3); 78,01, 76,50 (C2, C5); 73,03 (C7); 71,57 (C4); 62,80
(C6): anomère α: 94,16 (Cl); 83,64 (C3); 76,25, 74,02, 73,10, 71,66 (C2, C4, C5,
C7); 62,66 (C6).
RMN Η (CD3OD, 400 MHz): 7,78-7,31 (3 m, H arom.); 5,02 (d, IH, Hla,
H2α = 3,6 Hz); 4,97 (d, IH, H7aβ, JH7aβ-H7bβ = 11,3 Hz); 4,93 (d, IH, JH7bβ-H7aβ =
1 1,3 Hz); 4,42 (d, IH, Hlb, JH 1β-H2β = 7,8 Hz); 3,77 (dd, IH, H6aβ, JH6aβ-H5β = 2,4
Hz, JH6aβ-H6bβ = 11,9 Hz); 3,57 (dd, IH, H6bβ, JH6bβ-H5β = 5,9 Hz, JH6bβ-H6aβ = 11,8
Hz); 3,38 (t, IH, H4b, JH4β-H3β = H3β-H5β = 9,1 Hz): 3,32 (t, IH, JH3β-H2β = Jti3β-H4β
= 8,8 Hz); 3,24-3,19 (m, 2H, H2β, H5β).
Etape |4): préparation du 1 ,2,4,6-tétra-O-acétyl-3-O-(2-méthylnaρhtyl)-D- glucopyranose.
Dans un ballon de 1 L sont introduits successivement 116 g (1 éq.) de 3-O- (2-méthylnaphtyl)-D-glucopyranose (M = 320,3), 59,4 g (2 éq.) d'acétate de sodium et 680 mL (20 éq.) d'anhydride acétique. Le mélange est porté au reflux dans un bain d'eau bouillante pendant 2 heures puis coulé sur 5 L d'eau à température ambiante. Après une nuit d'agitation, le précipité obtenu est filtré puis lavé avec 6 L d'une solution aqueuse de bicarbonate de sodium à 5% pendant 10 min, filtré de nouveau et rincé à l'eau jusqu'à neutralité et séché. On obtient ainsi très majoritairement le produit de configuration β recherché (M = 488,5) sous la forme d'un mélange d'anomères β/α = 86/14. CCM: Rf = 0,3 [éther de pétrole/acétate d'éthyle (7/3; v/v)]. Solide blanc.
RMN 13C (CDCI3, 100 MHz): anomère : 170,88, 169,70, 169,37, 168,88 (C=O); 135,43, 133,26, 133,01 (C quat. arom.); (128,28, 127,91, 127,76, 126,37, 126,26, 126,15, 125,72, 125,54 (C arom.); 89,57 5 (Cl); 77,03 (C3); 74,89 (C7); 71,62 (C2); 70,33 (C5); 69,08 (C4); 61,81 (C6); 21,01, 20,83, 20,82, 20,71 (CH3). anomère β: 170,82, 169,38, 169,33, 169,18 (C=O); 135,01, 133,21, 133,03 (C quat. arom.); 128,33, 127,94, 127,72, 126,62, 126,35, 126,20, 125,70 (C arom.); 92,01 (Cl); 79,86 (C3); 74,34 (C7); 73,02 (C5); 71,61 (C2); 69,06 (C4); 61,79 (C6); 20,94, 20,82, 20,80, 20,77 (CH3). RMN Η (CDCI3, 400 MHz): anomère α: 7,83-7,81 (m, 3H, H arom.); 7,70 (s, IH, H arom.); 7,51-7,41 (m, 2H, H arom.); 7,37-7,35 (m, IH, H arom.); 6,32 (d, IH, Hl, JHι-H2 = 3,7 Hz); 5,20 (t, IH, H4, JH4.H3 = TH4-H5 = 9,8 Hz); 5,11 (dd, IH, H2, JH2-Hi = 3,7 Hz, JH2-H3 = 10,0 Hz); 4,87 (d, IH, H7a, JH7a-H7b = 12,1 Hz); 4,78 (d, IH, H7b, JH7b-H7a = 12,1 Hz); 4,21 (dd, IH, H6a, JH6a-H5 = 4,3 Hz, JH6a-H6b = 12,5 Hz); 4,07 (dd, IH, H6b, JH6b-H5 = 2,4 Hz, JH6b-H6a = 12,5 Hz); 4,02 (t, IH, H3, JH3- H2 = H3-H4 = 9,7 Hz); 4,01 (ddd, IH, H5, JH5-H4 = 10,2 Hz, JH5-H6a = 4,2 Hz, JH5-H6b = 2,4 Hz); 2,16, 2,09, 1,98, 1,92 (4s, 12H, CH3). anomère β: 7,83-7,80 (m, 3H, H arom.); 7,69 (s, IH, H arom.); 7,51-7,46 (m, 2H, H arom.); 7,36-7,33 (m, IH, H arom.); 5,66 (d, IH, Hl, JHι-H2 = 8,2 Hz); 5,21 (dd, IH, H2, JH -H. = 8,2 Hz, JH2-H3 = 9,5 Hz); 5,20 (t, IH, H4, JH4.H3 = JH4-H5 = 9,5 Hz); 4,78 (s, 2H, H7a, H7b); 4,23 (dd, IH, H6a, JH6a-H5 = 4,8 Hz, JH6a-H6b = 12,5 Hz); 4,09 (dd, IH, H6b, JH6b-H5 = 2,4 Hz, JH6b-H6a = 12,2 Hz); 3,81 (t, IH, H3, JH3-H2 = H3-H4 = 9,3 Hz); 3,74 (ddd, IH, H5, JH5-H4 = 10,0 Hz, JH5-H6a = 4,8 Hz, JH5-H6b = 2,3 Hz); 2,10, 2,08, 1,94, 1,93 (4s, 12H, CH3).
Etape |5j: préparation du 2,4,6-tri-O-acétyl-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle.
Dans un réacteur de 2 L sont dissous 177 g (1 éq.) théoriques de 1,2,4,6- tétra-O-acétyl-3-O-(2-méthylnaphtyl)-D-glucopyranose obtenu précédemment (M = 488,5) puis 885 mL (dilution 1 dans 5) de dichlorométhane. Le milieu est refroidi à 0°C puis on ajoute 20,5 mL (1,1 éq.) d'éthanethiol et doucement goutte à goutte 50,1 mL (1,1 éq.) d'éthérate de trifluorure de bore (addition pendant
20 min). Après 2 heures à 0°C, on lave 2 fois avec 1 L d'une solution aqueuse de bicarbonate de sodium à 5% (la solution de couleur rouge devient jaune pâle), une fois avec 500 mL d'eau puis la phase organique est séchée (MgSO4) et évaporée. Le produit brut isolé (M = 490,6) est utilisé comme tel pour la suite. CCM: Rf = 0,4 [éther de pétrole/acétate d'éthyle (7/3; v/v)]. Solide blanc.
RMN 13C (CDC13, 100 MHz): 170,89, 149,46, 169,43 (C=O); 135,27, 133,25, 133,02 (3 C quat. arom.); 128,29, 127,95, 127,74, 126,52, 126,32, 126,14, 125,73 (7 C quat. arom.); 83,75 (Cl); 81,51 (C3); 76,25, 74,32, 71,31, 69,70 (C2, C4, C5, C7); 62,54 (C6); 24,03 (CH2[SEt]); 21,03, 20,87 (CH3[Ac]); 14,85 (CH3[SEt]). RMN Η (CDC13, 400 MHz): 7,83-7,80 (m, 3H, H arom.); 7,68 (s, IH, H arom.); 7,50-7,46 (m, 2H, H arom.); 7,35-7,33 (m, IH, H arom.); 5,14 (t, IH, H2 ou H4, J = 9,9 Hz); 5,12 (t, IH, H2 ou H4, J = 9,7 Hz); 4,80 (d, IH, H7a, JH7a-H7b = 12,0 Hz); 4,75 (d, IH, H7b, JH7b-H7a = 12,1 Hz); 4,40 (d, IH, Hl, JHι-H2 = 10,0 Hz); 4,19 (dd, IH, H6a, JH6a-H5 = 5,1 Hz, JH6a-H6b = 12,3 Hz); 4,11 (dd, IH, H6b, JH6b-H5 = 2,4 Hz, JH6b-H6a = 12,2 Hz); 3,76 (t, IH, H3, JH3-H2 - JH3-H4 = 9,2 Hz); 3,60 (ddd, IH, H5, JH5-H4 = 10,0 Hz, JH5-H6a = 5,1 Hz, JH5.H6b = 2,5 Hz); 2,77-2,63 (m, 2H, CH2[SEt]); 2,07, 1,98, 1,92 (3s, 9H, CH3); 1,24 (t, 3H, CH3[SEt], J = 7,4 Hz).
EXEMPLE 2
Préparation d'un donneur de glvcosyle de formule générale (la) selon l'invention, produit de l'exemple 1 puis des étapes |6'| et |7"|
Etape |6]: préparation du 2-O-acétyl-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle.
178 g (1 éq.) de 2,4,6-tri-O-acétyl-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle (M = 490,6) théoriques sont dissous à chaud (40°C) dans 180 mL de toluène et coulés sur 900 mL (dilution 1 dans 5) de méthanol. La solution est limpide. Le retour à T. A. s'accompagne de la précipitation progressive du produit jusqu'à obtention d'une suspension. On ajoute alors doucement 0,05 éq. de sodium sous forme d'une solution de méthylate de sodium obtenue en
dissolvant 417 mg de sodium dans 50 mL de méthanol. Au bout d'une demi-heure, la solution redevient parfaitement limpide. Après 3h30' de réaction, le milieu est neutralisé par de la résine IR 120 (H+), filtré et concentré. Le résidu huileux obtenu est coulé sur 1,5 L d'heptane: le produit précipite. Après filtration, le résidu est repris par 1 L de dichlorométhane et les coproduits de réaction sont éliminés par 2 lavages à chaud (35-40°C) avec 1 L d'eau. La phase organique est récupérée, séchée et évaporée pour conduire à 95 g de 2-O-acétyl-3-O-(2-méthylnaphtyl)-l- thio-β-D-glucopyranoside d'éthyle recherché (M = 406,5). CCM: Rf = 0,5 [dichlorométhane/méthanol (9/1; v/v)]. Solide blanc.
Rendement (%) = 65 pour les étapes j4j, g] et |6J.
RMN 13C (CDC13, 100 MHz): 169,81 (C=O); 135,48, 133,27, 133,07 (3 C quat. arom.); 128,54, 127,99, 127,78, 126,70, 126,37, 126,18, 125,69 (C arom.); 83,86 (C3); 83,77 (Cl); 79,52 (C5); 74,85 (C7); 71,63 (C2); 70,46 (C4); 62,46 (C6); 24,17 (CH2[SEt]); 21,17 (CH3[Ac]); 14,90 (CH3[SEt]).
RMN Η (CDCI3, 400 MHz): 7,85-7,81 (m, 3H, H arom.); 7,74 (s, IH, H arom.); 7,50-7,45 (m, 3H, H arom.); 5,01 (dd, IH, H2, JH2-HI = 9,9 Hz, JH2-H3 = 9,2 Hz); 4,97 (d, IH, H7a, JH7a-H7b = 11,9 Hz); 4,85 (d, IH, H7b, JH7b-H7a = 11,8 Hz); 4,40 (d, IH, Hl, JH.-H2 = 10,0 Hz); 3,88 (dd, IH, H6a, JH6a.H5 = 3,2 Hz, JH6a.H6b = 12,0 Hz); 3,78 (dd, IH, H6b, JH6b-H5 = 4,7 Hz, JH6b-H6a = 12,0 Hz); 3,72 (t, IH, H4, JH4-H3 ≈ JH4-H5 = 9,3 Hz); 3,59 (t, IH, H3, JH3-H2 = JH3-H4 = 9,1 Hz); 3,37 (ddd, IH, H5, JH5-H4 = 9,5 Hz, JH5.H6a = 3,4 Hz, JH5-H6b = 4,6 Hz); 2,99 (si, IH, OH); 2,68 (qd, 2H, CH2[SEt], J = ); 2,41 (si, IH, OH); 2,00 (s, 3H, CH3[Ac]); 1,24 (t, 3H, CH3[SEt], J = 7,4 Hz).
Etape [7]: préparation du 2-O-acétyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-l- thio-β-D-glucopyranoside d'éthyle.
Dans un réacteur de 2 L, 56 g (1 éq.) de 2-O-acétyl-3-O-(2-méthylnaphtyl)- 1-thio-β-D-glucopyranoside d'éthyle (M = 406,5) sont dissous dans 300 mL (dilution 1 dans 5) d'acétonitrile puis additionnés de 31,0 mL (1,5 éq.) de benzaldéhyde diméthylacétal et 6,4 g (0,2 éq.) d'acide camphosulfonique anhydre
(le milieu rosit). Le mélange est chauffé à 55°C pendant 2 heures puis refroidi à T.A. et neutralisé avec 3,8 mL de triéthylamine (décoloration: le milieu devient jaune pâle). Après concentration, le résidu est dissous dans le minimum de dichlorométhane et coulé sur 1 L de méthanol: le produit précipite. Après une nuit au congélateur, le produit est filtré, rincé avec du méthanol glacé puis séché. 53 g de 2-O-acétyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-l-thio-β-D-gluco- pyranoside d'éthyle attendu (M = 494,6) sont ainsi isolés. CCM: Rf = 0,6 [toluène/acétate d'éthyle (17/3; v/v)]. Solide blanc. Rendement (%) = 78.
RMN 13C (CDC13, 100 MHz): 169,59 (C=O); 137,20, 135,61, 133,24, 133,03 (4 C quat. arom.); 129,15, 128,39, 128,13, 127,97, 127,72, 126,79, 126,17, 126,10, 126,02, 125,99 (C arom.); 101,36 (C8); 84,25 (Cl); 81,64, 79,48 (C3, C4); 74,37 (C7); 71,28 (C2); 70,70 (C5); 68,67 (C6); 23,98 (CH2[SEt]); 21,04 (CH3[Ac]); 14,86 (CH3[SEt]).
RMN Η (CDCI3, 400 MHz): 7,83-7,73 (m, 4H, H arom.); 7,53-7,45 (m, 4H, H arom.); 7,42-7,39 (m, 4H, H arom.); 5,61 (s, IH, H8); 5,12-5,07 (m, IH, H2); 5,03 (d, IH, H7a, JH7a-H7b = 12,2 Hz); 4,87 (d, IH, H7b, JH7b-H7a = 12,3 Hz); 4,45 (d, IH, Hl, JHι-H2 = 10,0 Hz); 4,39 (dd, IH, H6a, JH6a-H5 = 5,0 Hz, JH6a.H6b = 10,5 Hz); 3,83-3,78 (m, 3H, H3, H4, H6b); 3,50 (ddd, IH, H5, JH5-H4 = 9,5 Hz, JH5-H6a = 4,8 Hz, JH5-H6b = 9,4 Hz); 2,77-2,64 (m, 2H, CH2[SEt]); 2,00 (s, 3H, CH3[Ac]); 1,25 (t, 3H, CH3[SEt], J = 7,4 Hz).
EXEMPLE 3 Préparation d'un donneur de glvcosyle de formule générale (la) selon l'invention.
Etape 0: préparation du 4,6-di-O-benzyl-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle.
20 g (1 éq.) de 2-O-acétyl-3-O-(2-méthylnaphtyl)-l-thio-β-D-gluco- pyranoside d'éthyle préparé à l'étape |6] de l'exemple 2 (M = 406,5) sont dissous
dans 200 mL (dilution 1 dans 20) de diméthylformamide à 0°C et additionnés de 13 mL (2,2 éq.) de bromure de benzyle. On ajoute alors très progressivement 4,4 g (2,2 éq.) d'hydrure de sodium à 60% à 0°C. Après 2 heures de réaction à température ambiante, l'excès de NaH est détruit par addition de méthanol et le milieu est dilué avec 250 mL d'éther éthylique puis lavé 2 fois avec 30 mL d'eau, séché et concentré. La purification sur gel de silice [flash; éluant: éther de pétrole/acétate d'éthyle (9/1 ; v/v)] permet de recueillir 21,4 g de 4,6-di-O-benzyl- 3-O-(2-méthylnaphtyl)-l-thio-β-D-glucopyranoside d'éthyle recherché (M =544,7). CCM: Rf = 0,3 [éther de pétrole/acétate d'éthyle (8/2; v/v)]. Solide blanc. Rendement (%) = 80.
RMN 13C (CDC13, 100 MHz): 138,19, 138,10, 136,08, 133,36, 133,03 (5 C quat. arom.); 128,43, 128,38, 128,29, 127,98, 127,79, 127,72, 127,64, 126,69, 126,09, 126,04, 125,90 (C arom.); 86,16 (Cl); 85,98 (C3); 79,46 (C5); 77,46 (C4); 75,28 (CH2[Bn]); 75,12 (C7); 73,42 (CH2[Bn]); 73,34 (C2); 69,05 (C6); 24,32 (CH2); 15,46 (CH3).
RMN Η (CDCI3, 400 MHz): 7,73-7,67 (m, 4H, H arom.); 7,46-7,35 (m, 3H, H arom.); 7,26-7,17 (m, 8H, H arom.); 7,09-7,07 (m, 2H, H arom.); 5,02 (d, IH, CH2[Bn], J = 1 1,5 Hz); 4,92 (d, IH, CH2[Bn], J = 11,6 Hz); 4,78 (d, IH, H7a, JH7a-H7b = 10,9 Hz); 4,52 (d, IH, CH2[Bn], J = 11,9 Hz); 4,49 (d, IH, H7b, JH7b-H7a = 9,7 Hz); 4,45 (d, IH, CH2[Bn], J = 12,2 Hz); 4,23 (d, IH, Hl, JHι-H2 = 10,2 Hz); 3,67 (dd, IH, H6a, JH6a.H5 = 1,9 Hz, JH6a-H6b = 10,9 Hz); 3,62 (dd, IH, H6b, JH6b-H5 = 4,5 Hz, JH6b-H6a = 10,9 Hz); 3,59-3,47 (m, 3H, H2, H3, H4); 3,43 (m, IH, H5); 2,72-2,58 (m, 2H, CH2); 2,40 (s, IH, ΟH); 1,23 (t, 3H, CH3, J = 7,4 Hz).
Etape §: préparation du 4,6-di-O-benzyl-3-O-méthylnaphtyl- 1 -thio-β-D- glucopyranoside d'éthyle.
21 g (1 éq.) de 4,6-di-O-benzyl-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle recherché (M = 544,7) sont dissous dans 100 mL de pyridine et additionnés de 10,9 mL (3 éq.) d'anhydride acétique. La réaction terminée, le milieu est dilué à l'eau: le produit recherché précipite. Après filtration,
ce dernier est repris au dichlorométhane, lavé avec une solution d'acide chlorhydrique à 10%, une solution de bicarbonate de sodium à 5%, puis à l'eau.
21 g de 4,6-di-O-benzyl-3-O-méthylnaphtyl-l-thio-β-D-glucopyranoside d'éthyle
(M = 586,8) sont ainsi recueillis. CCM: Rf = 0,4 [éther de pétrole/acétate d'éthyle (8/2; v/v)].
Solide blanc.
Rendement (%) = 95.
RMN 13C (CDC13, 100 MHz): 169,76 (C=Ο); 138,20, 137,95, 135,71, 133,31,
133,02 (5 C quat. arom.); 128,50, 128,43, 128,25, 128,09, 127,99, 127,92, 127,77, 127,73, 127,67, 126,53, 126,20, 126,01, 125,89 (C arom.); 84,40 (C3 ou C4);
83,46 (Cl); 79,55 (C5); 78,00 (C3 ou C4); 75,31 (CH2[Bn]); 75,19 (C7); 73,52
(CH2[Bn]); 71,82 (C2); 68,91 (C6); 23,83 (CH2[SEt]); 21,07 (CH3[Ac]); 14,97
(CH3[SEt]).
RMN Η (CDCI3, 400 MHz): 7,75-7,69 (m, 3H, H arom.); 7,63 (s, IH, H arom.); 7,41-7,38 (m, 2H, H arom.); 7,32-7,30 (s, IH, H arom.); 7,26-7,19 (m, 10H, H arom.); 7, 1 1-7,08 (m, 2H, H arom.); 5,02-4,97 (m, IH, H2); 4,88 (d, IH, CH2[Bn],
J = 1 1,7 Hz); 4,77 (d, IH, CH2[Bn], J = 1 1,7 Hz); 4,73 (d, IH, H7a, JH7a-H7b =
10,8 Hz); 4,54 (d, IH, CH2[Bn], J = 12,0 Hz); 4,51 (d, IH, H7b, JH7b-H7a =
9,4 Hz); 4,48 (d, IH, CH2[Bn], J = 12,2 Hz); 4,28 (d, IH, Hl, JHι-H2 = 10,0 Hz); 3,69 (dd, IH, H6a, JH6a-H5 = 2,0 Hz, JH6a-H6b = 10,9 Hz); 3,68-3,63 (m, 3H, H3,
H4, H6b); 3,44 (ddd, IH, H5, JH5-H4 = 9,2 Hz, JH5-H6a = 2,1 Hz, JH5.H6b = 4,1 Hz);
2,71-2,56 (m, 2H, CH2[SEt]); 1,84 (s, 3H, CH3[Ac]); 1,18 (t, 3H, CH3[SEt], J =
7,4 Hz).
EXEMPLE 4
Préparation d'un donneur de glvcosyle de formule générale (la) selon l'invention, produit de l'exemple 1 puis des étapes 6, 7| et 8
Etape |6]: préparation du 3-O-(2-méthylnaphtyl)-l-thio-β-D-glucopyranoside d'éthyle.
A 100 g de 2,4,6-tri-O-acétyl-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle préparé à l'exemple 1 (M = 490,6) théoriques dissous dans 100 mL de toluène, sont progressivement ajoutés 2 L de méthanol contenant 7,03 g (1,5 éq.). Après 4 heures de réaction à température ambiante, le milieu est neutralisé par de la résine IR 120 (H
+), filtré et concentré. Le résidu huileux obtenu est coulé sur 1 L d'heptane: le produit précipite. Après filtration, le résidu est repris par 0,6 L de dichlorométhane et les coproduits de réaction sont éliminés par 2 lavages à chaud (35-40°C) avec 0,6 L d'eau. La phase organique est récupérée, séchée et évaporée pour conduire à 49,8 g de 3-O-(2-méthylnaphtyl)-l- thio-β-D-glucopyranoside d'éthyle (M = 364,5).
CCM: Rf = 0,5 [dichlorométhane/méthanol (9/1; v/v)].
Solide blanc.
Rendement (%) = 67 pour les étapes @, g| et jό).
RMN 13C (CDC13, 100 MHz): 135,83, 133,36, 133,12 (3 C quat. arom.); 128,62, 128,01, 127,83, 126,99, 126,34, 126,15, 125,91 (C arom.); 86,70 (Cl); 85,00 (C3); 79,48 (C5); 74,91 (C7); 73,23 (C2); 70,13 (C4); 62,74 (C6); 24,74 (CH2[SEt]); 15,49 (CH3[SEt]).
RMN Η (CDCI3, 400 MHz): 7,86-7,82 (m, 4H, H arom.); 7,51-7,47 (m, 3H, H arom.); 5,17 (d, IH, H7a, JH7a-H7b = 11,8 Hz); 4,94 (d, IH, H7b, JH7b-H7a = 11,8 Hz); 4,36 (d, IH, Hl, JHι-H2 = 9,6 Hz); 3,87 (dd, IH, H6a, HS = 3,3 Hz, JH6a-H6b = 12,0 Hz); 3,75 (dd, IH, H6b, JH6b-H5 = 4,9 Hz, JH6b-H6a = 12,0 Hz); 3,62 (t, IH, H4, JH4-H3 = Jtu-HS = 9,2 Hz); 3,53 (t, IH, H2, JH2-HI = JH2-H3 = 9,0 Hz); 3,46 (t, IH, H3, JH3-H2 = JH3-H4 - 8,6 Hz); 3,37 (ddd, IH, H5, JH5-H4 = 9,2 Hz, JH5-H6» ≈ 3,5 Hz, JH5-H6b = 4,8 Hz); 2,73 (qd, 2H, CH2[SEt]); 2,68 (si, IH, OH); 2,56 (s, IH, OH); 2,22 (si, IH, OH); 1,33 (t, 3H, CH3[SEt], J = 7,4 Hz).
Etape J7j: préparation du 4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle.
Dans un réacteur de 2 L, 50 g (1 éq.) de 3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle (M = 364,5) sont dissous dans 300 mL (dilution 1 dans 5) d'acétonitrile puis additionnés de 31,0 mL (1,5 éq.) de benzaldéhyde
méthylnaphtyl)-l-thio-β-D-glucopyranoside d'éthyle recherché (M = 556,7) sont ainsi recueillis.
CCM: Rf = 0,6 [toluène/acétate d'éthyle (9/1; v/v)].
Solide blanc.
Rendement (%) = 84.
RMN 13C (CDC13, 100 MHz): 165,25 (C=O); 137,27, 135,28, 133,25, 133,09,
132,95 (5 C quat. arom.); 129,95, 129,18, 128,42, 128,11, 127,92, 127,67, 127,06,
126,25, 126,14, 125,95, 125,83 (C arom.); 101,42 (C8); 84,38 (Cl); 81,81 (C4),
79,00 (C3); 74,29 (C7); 71,86 (C2); 70,76 (C5); 68,73 (C6); 24,12 (CH2[SEt]);
14,88 (CH3[SEfJ).
RMN Η (CDCI3, 400 MHz): 7,95-7,92 (m, 2H, H arom.); 7,69-7,36 (3m, 12H, H arom.); 7,22 (dd, IH, H arom.); 5,64 (s, IH, H8); 5,36 (dd, IH, H2, JH2-H I = 10,0
Hz, JH2-H3 = 8,5 Hz); 4,98 (d, IH, H7a, JH7a-H7b = 12,2 Hz); 4,86 (d, IH, H7b,
JH7b-H7a = 12,2 Hz); 4,59 (d, IH, Hl, JHι-H2 = 10,0 Hz); 4,41 (dd, IH, H6a, HS
= 5,0 Hz, JH6a-H6b ≈ 10,5 Hz); 3,93 (t, IH, H3, JH3-H2 = JH3-H4 = 9,2 Hz); 3,88 (dd,
IH, H4, JH4-H3 = 8,8 Hz, JH .H5 ≈ 9,2 Hz); 3,84 (t, IH, H6b, JH6b-H5 = JH6b-H6a =
10,3 Hz); 3,56 (ddd, IH, H5, JH5.H4 = 9,1 Hz, JH5-H6a = 5,0 Hz, JH5-H6b = 9,9 Hz);
2,77-2,64 (m, 2H, CH2[SEt]); 1,21 (t, 3H, CH3[SEt], J = 7,4 Hz).
EXEMPLE 5
Synthèse d'un svnthon accepteur de glvcosyle de formule générale (II) selon l'invention.
Etape 5A: préparation du précurseur 2-O-benzoyl-4,6-O-benzylidène-3-O-(2- méthylnaphtyl)-β-D-glucopyranoside de benzyle.
Dans un ballon, à 0°C, sont introduits successivement 31,2 g (1 éq) de 2- O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-l-thio-β-D-glucopyranoside d'éthyle (M = 556,7), 13,9 g (1,1 éq.) de N-iodosuccinimide, 10 g de tamis moléculaire 4Â, 200 mL (dilution 1 dans 5) de dichlorométhane anhydre et enfin 6,96 mL (1,2 éq.) d'alcool benzylique. On ajoute alors 1,27 mL (0,1 éq.) de triflate de triéthylsilyle. Après 1 heure de réaction à 0°C, le milieu est neutralisé par
diméthylacétal et 6,4 g (0,2 éq.) d'acide camphosulfonique anhydre (le milieu rosit). Le mélange est chauffé à 55°C pendant 2 heures puis refroidi à T.A. et neutralisé avec 3,8 mL de triéthylamine (décoloration: le milieu devient jaune pâle). Après concentration, le résidu est dissous dans le minimum de dichlorométhane et coulé sur 1 L de méthanol: le produit précipite. Après une nuit au congélateur, le produit est filtré, rincé avec du méthanol glacé puis séché. 48,5 g de 4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-l-thio-β-D-glucopyranoside d'éthyle attendu (M = 452,6) sont ainsi isolés. CCM: Rf = 0,4 [toluène/acétate d'éthyle (17/3; v/v)]. Solide blanc.
Rendement (%) = 78.
RMN 13C (CDC1 , 100 MHz): 137,30, 135,80, 133,33, 133,10 (4 C quat. arom.); 129,10, 128,35, 128,30, 128,01, 127,74, 126,90, 126,14, 126,11, 126,05, 125,95 (C arom.); 101,41 (C8); 86,71 (Cl); 81,47, 81,25 (C3, C4); 74,74 (C7); 73,19 (Cl); 70,83 (C5); 68,72 (C6); 24,64 (CH2); 15,31 (CH3).
RMN Η (CDCI3, 400 MHz): 7,82-7,74 (m, 4H, H arom.); 7,53-7,39 (m, 8 H, H arom.); 5,60 (s, IH, H8); 5,14 (d, IH, H7a, - ub = 12,0 Hz); 5,01 (d, IH, H7b, JH7b-H7a = 12,0 Hz); 4,46 (d, IH, Hl, JH1-H2 = 9,7 Hz); 4,37 (dd, IH, H6a, JH6a-H5 = 5,0 Hz, JH6a-H6b = 10,4 Hz); 3,79 (t, IH, H6b, JH6b-H5 = JH6b-H6a = 10,3 Hz); 3,76- 3,71 (m, 2H, H3, H4); 3,63 (dd, IH, H2, JH2-H I = 9,0 Hz, JH2-H3 = 8,4 Hz); 3,50 (ddd, IH, H5, JH5-H4 = JH5-H6b = 9,5 Hz, JH5-H6a = 5,1 Hz); 2,82-2,69 (m, 2H, CH2); 2,61 (s, IH, ΟH); 1,32 (t, 3H, CH3, J = 7,4 Hz).
Etape |: préparation du 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-l- thio-β-D-glucopyranoside d'éthyle.
A 48,5 g de 4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-l-thio-β-D- glucopyranoside d'éthyle (1 éq.) (M = 452,6) dans 300 mL de pyridine (dilution 1 dans 6) sont additionnés doucement en une seule fois 37 ml (3 éq.) de chlorure de benzoyle et la réaction est laissée une nuit à TA. La solution est ensuite coulée sur 1 1 de méthanol sous vive agitation. Le produit précipite doucement puis est filtré et rincé avec du méthanol glacé. 50 g de 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-
quelques gouttes de triéthylamine, filtré sur verre fritte et concentré. La purification sur gel de silice [flash; éluant: toluène/acétate d'éthyle (97/3; v/v)] permet de recueillir 30 g de 2-O-benzoyl-4,6-O-benzylidène-3-O-(2- méthylnaphtyl)-β-D-glucopyranoside de benzyle attendu (M = 602,7). CCM: Rf = 0,6 [toluène/acétate d'éthyle (9/1; v/v)]. Solide blanc. Rendement (%) = 89.
RMN 13C (CDCb, 100 MHz): 165,14 (C=Ο); 137,28, 136,75, 135,32, 133,13, 133,04, 132,89 (6 C quat. arom.); 129,93, 129,77, 129,13, 128,37, 128,32, 128,29, 128,05, 127,87, 127,77, 127,67, 127,61, 126,97, 126,20, 126,13, 125,89, 125,76 (C arom.); 101,38 (C8); 99,96 (Cl); 81,78 (C4); 77,57 (C3); 73,96 (C7); 73,31 (C2); 70.46 (C9); 68,76 (C6); 66,28 (C5).
RMN 'H (CDCI3, 400 MHz): 7,89-7,86 (m, 2H, H arom.); 7,67-7,07 (m, 17H, H arom.); 5,62 (s, IH, H8); 5,40 (t, IH, H2, JH -HI = JH2-H3 = 8,3 Hz); 4,95 (d, IH, H7a, JH7a-H7b = 12,4 Hz); 4,84 (d, 2H, H7b, H9a, JH7b-H7a = HÇ = 12,6 Hz); 4,58 (d, IH, H9b, JH9b-H9a = 13,6 Hz); 4,58 (d, IH, Hl, JHι-H2 = 7,8 Hz); 4,40 (dd, IH, H6a, JH6a-H5 = 5,0 Hz, JH6a-H6b = 10,5 Hz); 3,90 (t, IH, H4, JH4-H3 = JH4-H5 = 9,1 Hz); 3,86 (t, IH, H6b, JH6b-H5 = JH6b-H6a = 10,2 Hz); 3,84 (dd, IH, H3, JH3-H2 = 8,5 Hz, JH3-H4 = 9,1 Hz); 3,45 (ddd, IH, H5, JH5-H4 = 9,2 Hz, JH5-H6a = 5,0 Hz, JH5- Hόb = 9,8 Hz).
Etape 5B: préparation du synthon accepteur 2-O-benzoyl-4,6-O-benzylidène-β-D- glucopyranoside de benzyle (OH en position 3).
Dans 850 mL d'un mélange dichlorométhane/méthanol (4/1; v/v) sont introduits 42,0 g (1 éq.) de 2-O-benzoyl-4,6-benzylidène-3-O-(2-méthylnaphtyl)- β-D-glucopyranoside de benzyle (M = 602,7) et 47,5 g (3 éq.) de 2,3-dichloro-5,6- dicyano-l,4-benzoquinone (DDQ) à température ambiante. Au bout de 5h30', le milieu est dilué avec 1 L de dichlorométhane et lavé 2 fois avec une solution aqueuse de bicarbonate de sodium à 5%. Après séchage et evaporation de la phase organique, le résidu est purifié sur gel de silice [flash; éluant: toluène/acétate
d'éthyle (9/1; v/v)]. On obtient ainsi 27,4 g de 2-O-benzoyl-4,6-O-benzylidène-β- D-glucopyranoside de benzyle attendu (M = 462,5). CCM: Rf = 0,3 [toluène/acétate d'éthyle (9/1; v/v)]. Solide blanc. Rendement (%) = 85.
RMN 13C (CDC13, 100 MHz): 165,97 (C=O); 136,97, 136,73, 133,37 (3 C quat. arom.); 130,05, 129,59, 129,37, 128,41, 127,93, 127,84, 126,37 (C arom.); 101,96 (C7); 99,75 (Cl); 80,94 (C4); 74,82 (C2); 72,41 (C3); 70,63(C8); 68,66 (C6); 66,26 (C5).
RMN Η (CDCI3, 400 MHz): 8,03-8,00 (m, 2H, H arom.); 7,62-7,58 (m, IH, H arom.); 7,52-7,43 (m, 4H, H arom.); 7,41-7,36 (m, 3H, H arom.); 7,23-7,19 (m, 5H, H arom.); 5,57 (s, IH, H7); 5,26 (dd, IH, H2, JH2-H I = 7,9 Hz, JH2-H3 = 9,1 Hz); 4,89 (dd, IH, H8a, JH8a-H8b = 12,5 Hz); 4,70 (d, IH, Hl, JHι-H2 = 7,9 Hz); 4,66 (d, IH, H8b, JH8b-H8a = 12,6 Hz); 4,42 (dd, IH, H6a, JH6a.H5 = 5,0 Hz, JH6a-H6b = 10,5 Hz); 3,99 (t, IH, H3, JH3-H2 = JH3-H4 = 9,2 Hz); 3,85 (t, IH, H6b, JH6b-H5 = JH6b-H6a = 10,3 Hz); 3,68 (t, IH, H4, JH4-H3 = TH4-H5 = 9,4 Hz); 3,49 (ddd, IH, H5, JH5-H4 = JH5-H6b = 9,7 Hz, JH5-H6a = 5,1 Hz,); 2,84 (s, IH, OH).
EXEMPLE 6 Synthèse d'un disaccharide selon l'invention.
Cet exemple illustre l'intérêt du procédé conforme à l'invention pour la préparation d'un disaccharide par réaction de couplage entre deux monosaccharides, le disaccharide obtenu pouvant lui-même servir de précurseur d'un disaccharide accepteur de glycosyle (voir exemple 7) pour la synthèse d'un trisaccharide (voir exemple 8).
Synthèse du 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-β-D- glucopyranosyl-( 1 -- 3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle.

8,02 g (1,1 éq.) de 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)- 1 -thio-β-D-glucopyranoside d'éthyle préparé à l'exemple 4 (M = 556,7) et 6,06 g (1 éq.) de 2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle préparé à l'exemple 5 (M = 462,5) sont dissous dans 70 mL de dichlorométhane anhydre en présence de 2 g de tamis moléculaire 4Â à 0° C puis additionnés de 3,24 g (1 ,1 à 1 ,2 éq.) de N-iodosuccinimide (ΝIS) et 296 μL (0, 1 éq.) de trifluorométhanesulfonate de triéthylsilyle (TESOTf). Après 50 min de réaction, le milieu est neutralisé par de la triéthylamine, filtré puis lavé par un solution de thiosulfate de sodium à 10% puis à l'eau. Après séchage et evaporation de la phase organique, le produit attendu est purifié par chromatographie sur gel de silice [flash; éluant: toluène/acétate d'éthyle (95/5; v/v)]. 10,36 g de 2-O-benzoyl-4,6-O- benzylidène-3-O-(2-méthylnaphtyl)-β-D-glucopyranosyl-(l-→3)-2-O-benzoyl-4,6- O-benzylidène-β-D-glucopyranoside de benzyle (M =957,1) sont ainsi obtenus. CCM: Rf = 0,6 [toluène/acétate d'éthyle (8/2; v/v)]. Solide blanc.
Rendement (%) = 83%
RMΝ 13C (CDC13, 100 MHz): 164,74, 164,58 (C=O); 137,32, 137,19, 136,65, 135,33, 133,04, 132,96, 132,85, 132,72 (8 C quat. arom.); 129,82-125,67 (C arom.); 101 ,50, 101,08 (C7A, C7B); 100,58 (C1B); 99,62 (CIA); 80,89 (C4B); 79,30 (C4A); 78,22 (C3B); 77,79 (C3A); 73,69 (C2B); 73,52, 73,49 (C2A, C8B); 70,28 (C8A); 68,77, 68,73 (C6A, C6B); 66,48 (C5A); 66,00 (C5B). RMΝ Η (CDCI3, 400 MHz): 7,74-7,02 (m, 32H, H arom.); 5,53 (s, IH, H7A ou H7B); 5,34 (s, IH, H7A ou H7B); 5,31 (dd, IH, H2A, JH2A-H. A = 7,7 Hz, JH2A-H3A = 9,2 Hz); 5,28 (t, IH, H2B, JH2B-H IB = HSB = 7,4 Hz); 4,85 (d, IH, H1B, JHI B-H2B = 7,0 Hz); 4,84 (d, IH, H8aB, JH8aB-H8bB = 12,4 Hz); 4,74 (d, IH, H8bB, JH8bB-H8aB = 12,2 Hz); 4,74 (d, IH, H8aA, JH8aA-H8bA = 12,2 Hz); 4,56 (d, IH, H1A, JH IA-H2A = 7,6 Hz); 4,50 (d, IH, H8bA, JH8bA-H8aA = 12,6 Hz); 4,36 (dd, IH, H6aA,
JH6aA-H5A = 4,9 Hz, JH6aA-H6bA = 10,5 Hz); 4,16 (dd, IH, H6aB, JH6aB-H5B = 4,9 Hz,
JH6aB-H6bB = 10,4 Hz); 4,1 1 (t, IH, H3A, JH3A-H2A = JH3A-H4A - 8,8 Hz); 3,90 (t, IH, H4B, JH4B-H3B = JH4B-H5B = 9,4 Hz); 3,84 (t, IH, H4A, JH4A-H3A = JH4A-H A =
9,3 Hz); 3,80 (dd, IH, H6bA, JH6bA-H5A = JH6bA-H6aA = 10,3 Hz); 3,74 (dd, IH,
H3B, JH3B-H2B = 7,9 Hz, JH3B-H4B = 8,9 Hz); 3,68 (t, IH, H6bB, JH6bB-H5B =
JH6bB-H6aB = 10,3 Hz); 3,49 (ddd, IH, H5A, JH5A-H4A = JH5A-H6bA = 9,7 Hz, JH5A-H6aA
= 4,9 Hz,); 3,37 (ddd, IH, H5B, JH5B-H4B ≈ tiόbB = 9,7 Hz, JH5B-H6aB = 4,9 Hz).
EXEMPLE 7
Synthèse d'un disaccharide accepteur de glvcosyle selon l'invention.
Synthèse du 2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l→3)-2-O- benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle.
Après dissolution de 2,17 g (léq.) de 2-O-benzoyl-4,6-O-benzylidène-3-O- (2-méthylnaphtyl)-β-D-glucopyranosyl-(l-»3)-2-O-benzoyl-4,6-O-benzylidène-β- D-glucopyranoside de benzyle (M = 957,1) dans 43 mL d'un mélange dichlorométhane/méthanol (4/1; v/v), nous ajoutons 1,54 g (3 éq.) de 2,3-dichloro- 5,6-dicyano-l,4-benzoquinone (DDQ) et on laisse évoluer 4h30 à température ambiante. Le milieu est ensuite dilué au dichlorométhane, lavé par un solution d'hydrogénocarbonate de sodium à 5% puis à l'eau. La phase organique est séchée, évaporée et le produit recherché est obtenu après purification sur gel de silice [flash; éluant: toluène/acétate d'éthyle (9/1; v/v)]. 1,46 g de 2-O-benzoyl-4,6-O- benzylidène-β-D-glucopyranosyl-(l— >3)-2-O-benzoyl-4,6-O-benzylidène-β-D- glucopyranoside de benzyle (M = 815,9) sont ainsi recueillis. CCM: Rf = 0,4 [toluène/acétate d'éthyle (8/2; v/v)]. Solide blanc. Rendement (%) = 79%
RMN 13C (CDC13, 100 MHz): 165,64, 164,67 (C=O); 137,17, 136,97, 136,64, 133,10, 133,00 (5 C quat. arom.); 129,81-126,15 (C arom.); 101,69, 101,49 (C7A, C7B); 100,38 (C1B); 99,58 (CIA); 80,54 (C4B); 79,31 (C4A); 78,00 (C3A); 75,27 (C2B); 73,51 (C2A); 72,60 (C3B); 70,33 (C8A); 68,77 (C6A); 68,65 (C6B); 66,46 (C5A); 66,05 (C5B).
RMN Η (CDC13, 400 MHz): 7,70-6,96 (m, 25H, H arom.); 5,47 (s, IH, H7A ou H7B); 5,27 (s, IH, H7A ou H7B); 5,25 (t, IH, H2A, JH2A-H IA , = JH2A-H3A = 7,6 Hz); 5,03 (dd, IH, H2B, HI B = 7,4 Hz, H3B = 8,0 Hz); 4,83 (d, IH, H1B, JH I B-H2B = 7,2 Hz); 4,69 (d, IH, H8aA, JH8aA-H8bA = 12,5 Hz); 4,52 (d, IH, H1A, JH I A-H2A = 7,5 Hz); 4,44 (d, IH, H8bA, . -HSaA = 12,6 Hz); 4,30 (dd, IH, H6aA, JH6aA-H5A = 4,8 Hz, JHόâA-HόbA = 10,4 Hz); 4,10 (dd, IH, H6aB, JH6aB-H5B =
4,8 Hz, JH6aB-H6bB - 10,5 Hz); 4,08 (t, IH, H3A, JH3A-H2A = JH3A-H4A = 8,7 Hz); 3,77 (t, IH, H4A, JH4A-H3A = HSA = 9,3 Hz); 3,76 (t, IH, H3B, H2B = H4B = 10,2 Hz); 3,73 (t, IH, H6bA, . -HSA = JH6bA-H6aA = 10,3 Hz); 3,60 (t, IH,
H6bB, JH6bB-H5B = JH6bB-H6aB = 10,3 Hz); 3,57 (t, IH, H4B, JH4B-H3B = JH4B-H5B = 9,4 Hz); 3,44 (ddd, IH, H5A, JH5A-H4A = tiόbA = 9,7 Hz, JH5A-H6aA = 4,8 Hz,); 3,28 (ddd, IH, H5B, JH5B-H4B = H5B-H6bB = 9,7 Hz, JHSB-HOUB = 4,9 Hz,); 2,55 (s, 1H, 0H).
EXEMPLE 8 Synthèse d'un trisaccharide selon l'invention.
Cet exemple illustre l'intérêt du procédé conforme à la présente invention pour la préparation d'un trisaccharide par réaction de couplage entre un monosaccharide et un disaccharide, le trisaccharide obtenu pouvant lui-même servir de précurseur d'accepteur de glycosyle (voir exemple 9) pour la synthèse d'un tétrasaccharide (voir exemple 10).
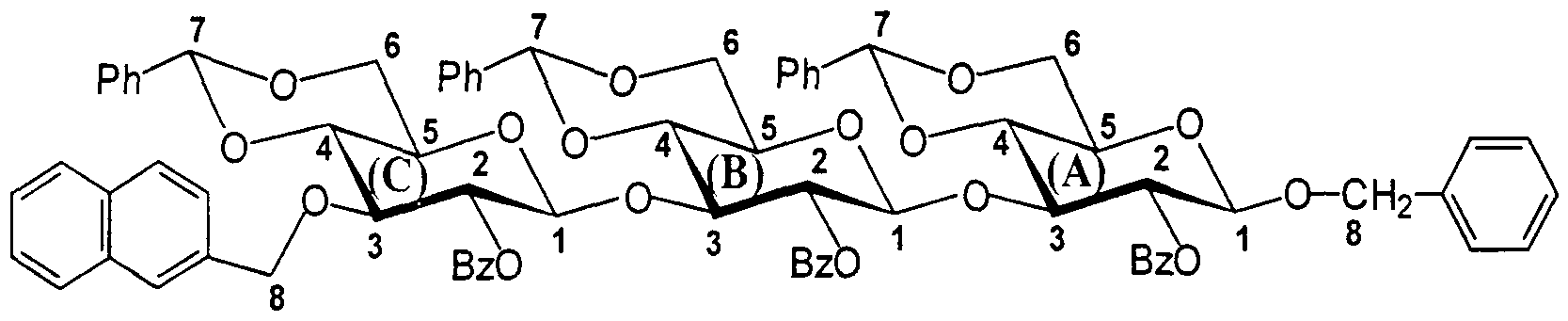
Synthèse du 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-β-D- glucopyranosyl-( 1 — »3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl- (1— >3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle.
5,03 g (1,1 éq.) de 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)- 1-thio-β-D-glucopyranoside d'éthyle préparé à l'exemple 4 (M = 556,7) et 6,70 g (1 éq.) de 2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l-»3)-2-O- benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle préparé à l'exemple 7 (M = 815,9) sont dissous dans 100 mL de dichlorométhane anhydre en présence de 10 g de tamis tamis moléculaire 4Â à 0°C puis additionnés de 2,03 g de NIS (1,1 à 1,2 éq.) et 190 μL de TESOTf (0,1 éq.). Après 50 min de réaction, le milieu est neutralisé par de la triéthylamine, filtré puis lavé par un solution de thiosulfate de sodium à 10% puis à l'eau. Après séchage et evaporation de la phase organique, le produit attendu est purifié par chromatographie sur gel de silice [flash; éluant:
[flash; éluant: toluène/acétate d'éthyle (95/5 puis 9/1 ; v/v). 9,28 g de 2-O-benzoyl- 4,6-O-benzylidène-3-O-méthylnaphtyl-β-D-glucopyranosyl-(l— >3)-2-O-benzoyl- 4,6-O-benzylidène-β-D-glucopyranosyl-(l- 3)-2-O-benzoyl-4,6-O-benzylidène-β- D-glucopyranoside de benzyle (M = 1310,5) sont ainsi isolés. CCM: Rf = 0,5 [toluène/acétate d'éthyle (17/3; v/v)]. Solide blanc. Rendement (%) = 86%
RMN 13C (CDC13, 100 MHz): 165,12, 164,77, 164,53 (3 C=O); 137,40, 137,36, 137,20, 136,78, 135,42, 133,43, 133,16 (2 C), 133,09, 132,90 (10 C quat. arom.); 129,93-125,37 (C arom.); 101,94, 101,22, 100,56 (C7A,B,C); 99,49 (CIA); 98,32 (C1C); 97,90 (C 1B); 81,38 (C4C); 78,67 (C4A); 78,19 (C3C); 77,61 (C4B); 76,22 (C3B); 74,31 (C2A); 74,1 1 (C3A); 73,87 (C8C); 73,28 (C2C); 72,58 (C2B); 70,22 (C8A); 68,81 (C6B); 68,77 (C6C), 68,67 (C6A); 66,49 (C5A); 66,14 (C5C); 65,37 (C5B). RMN Η (CDCI3, 400 MHz): 7,90-7,05 (m, 42H, H arom.); 5,47 (s, IH, H7); 5,46 (s, IH, H7); 5,34 (t, IH, H2C, JH2C-HIC = TH2C-H3C = 7,8 Hz); 5,08 (t, IH, H2B, JH2B-H . B = JH2B-H3B = 4,2 Hz); 5,06 (d, IH, HIC, JHIC-H2C = 7,4 Hz); 4,91 (d, IH, H8aC, JH8ac-H8bc = 12,3 Hz); 4,86 (d, IH, H1B, JH, B-H2B = 4,4 Hz); 4,86 (dd, IH, H2A, JH2A-H IA = 8,0 Hz, JH2A-H3A = 8,6 Hz); 4,80 (d, IH, H8bC, JH8bc-H8ac = 12,4 Hz); 4,75 (d, IH, H8aA, JH8aA-H8bA = 12,5 Hz); 4,57 (s, IH, H7); 4,49 (d, IH, H8bA, JH8bA-H8aA = 12,6 Hz); 4,45 (d, IH, H1A, JH,A-H2A = 7,7 Hz); 4,31 (dd, IH, H6aA, JH63A-H5A = 4,8 Hz, JH6aA-H6bA = 10,4 Hz); 4,22 (dd, IH, H6aC, JH6ac-H5c = 4,9 Hz, JH6ac-H6bc ≈ 10,4 Hz); 4,1 1 (dd, IH, H6aB, JH6aB-H5B = 3,5 Hz, JH6aB-H6bB = 8,9 Hz); 4,07 (t, IH, H3A, JH3A-H2A = JH3A-H4A = 9, 1 Hz); 4,07 (dd, IH, H4B, JH4B-H3B = 8,1 Hz, JH4B-H5B = 9,6 Hz); 3,97 (dd, IH, H3B, JH3B-H2B = 3,9 Hz, JH3B-H4B = 8,1 Hz); 3,91 (t, IH, H4C, JH4C-H3C = JMC-HSC = 9,1 Hz); 3,85 (dd, IH, H3C, JH3C-H2C = 8,1 Hz, JH3C-H4C = 9,0 Hz); 3,72 (dd, IH, H6bC, JH6bC-H5C = JH6bc-H6ac = 10,3 Hz); 3,64 (t, IH, H6bA, JH6bA-H5A = JH6bA-H6aA = 10,2 Hz); 3,55 (ddd, IH, H5B, JH5B-H4B = JH B-H6bB = 10,0 Hz, JHDB-HOUB = 4,3 Hz,); 3,52 (t, IH, H6bB, JH6bB-H5B = Jtl6bB-H6aB = 9,3 Hz); 3,50 (ddd, IH, H5C, JH C-H4C = JH5C-H6bC =
9,8 Hz, JH5CH6ac ≈ 3,8 Hz,); 3,37 (ddd, IH, H5A, JH5A-H4A = JH5A-H6bA = 9,8 Hz, JH5A-H6aA = 4,9 Hz,); 3,18 (t, IH, H4A, JH4A-H3A = JH4A-HSA = 9,4 Hz).
EXEMPLE 9
Synthèse d'un trisaccharide accepteur de glvcosyle selon l'invention.
Synthèse du 2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-( 1 —»3)-2-O- benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l- 3)-2-O-benzoyl-4,6-O- benzylidène-β-D-glucopyranoside de benzyle.
Après dissolution de 6,21 g (léq.) de 2-O-benzoyl-4,6-O-benzylidène-3-O- (2-méthylnaphtyl)-β-D-glucopyranosyl-(l->3)-2-O-benzoyl-4,6-O-benzylidène-β- D-glucopyranosyl-( 1 -»3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle (M = 1310,5) dans 125 mL d'un mélange dichlorométhane/méthanol (4/1; v/v), nous ajoutons 3,23 g (3 éq.) de DDQ et on laisse évoluer 7 h à température ambiante. Le milieu est ensuite dilué au dichlorométhane, lavé par un solution d'hydrogénocarbonate de sodium à 5% puis à l'eau. La phase organique est séchée, évaporée et le produit recherché est obtenu après purification sur gel de silice [flash; éluant: toluène/acétate d'éthyle (9/1 puis 17/3; v/v)]. 8,89 g de 2-O- benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l-»3)-2-O-benzoyl-4,6-O- benzylidène-β-D-glucopyranosyl-(l- 3)-2-O-benzoyl-4,6-O-benzylidène-β-D- glucopyranoside de benzyle (M = 1170,3) sont ainsi obtenus. CCM : Rf = 0,3 [toluène/acétate d'éthyle (17/3; v/v)] . Solide blanc. Rendement (%) = 78%
RMN 13C (CDC13, 100 MHz): 165,87, 164,81, 164,58 (3 C=O); 137,32, 137,18, 137,03, 136,75, 133,64, 133,23, 133,20 (7 C quat. arom.); 130,06-125,36 (C arom.); 101,96, 101,84, 100,57 (C7A,B,C); 99,48 (CIA); 98,18 (CIC); 98,03 (C1B); 80,85 (C4C); 78,72 (C4A); 77,64 (C4B); 76,31 (C3B); 74,73 (C2C); 74,40 (C3A); 74,28 (C2A); 72,66 (C2B); 72,54 (C3C); 70,26 (C8A); 68,78 (C6B); 68,67 (C6A, C6C), 66,48 (C5A); 66,09 (C5C); 65,40 (C5B). RMN Η (CDCI3, 400 MHz): 8,02-7,06 (m, 35H, H arom.); 5,47 (s, IH, H7); 5,42 (s, IH, H7); 5,20 (dd, IH, H2C, JH2C-HIC = 7,6 Hz, JH2C-H3C = 8,4 Hz); 5,13 (t, IH, H2B, JH2B-HIB = JH2B-H3B = 4,2 Hz); 5,11 (d, IH, HIC, JHιc-H2c = 7,5 Hz); 4,95
(dd, IH, H2A, JH2A-HiA = 8,0 Hz, JH2A-H3A = 8,6 Hz); 4,90 (d, IH, H1B, JH 1B-H2B = 4,4 Hz); 4,76 (d, IH, H8aA, JH8aA-H8bA = 12,6 Hz); 4,62 (s, IH, H7); 4,32 (d, IH, H8bA, JHSbA-HδaA = 12,6 Hz); 4,47 (d, IH, H1A, JHIA-H2A = 7,8 Hz); 4,31 (dd, IH, H6aA, JH63A-H5A = 4,8 Hz, JH6aA-H6bA = 10,4 Hz); 4,22 (dd, IH, H6aC, JH6ac-H5c = 4,8 Hz, JH6ac-H6bC = 10,4 Hz); 4,12 (dd, IH, H6aB, JH6aB-H5B = 3,3 Hz, JH6aB-H6bB =
8.8 Hz); 4,09 (t, IH, H3A, J
H3A-H2A = J
H3A-H4A = 9,0 Hz); 4,07 (dd, IH, H4B, JH4B-H3B = JH4B-H5B = 8,9 Hz); 4,01 (dd, IH, H3B, JH3B-H2B
= 3,8 Hz, JH3B-H4B
= 8,1 Hz); 3,96 (ddd, IH, H3C, J
H3C-H2C = JH3C-H4C = 8,7 Hz, J
H3C-OH = 3,6 Hz); 3,69 (dd, IH, H6bC, J
H6bc-
H5c = J
H6bc-H6
ac = 10,2 Hz); 3,67 (t, IH, H4C, J
H4C-H
3C = JH4C-H C = 9,4 Hz); 3,65 (t, IH, H6bA, J
H6bA-H5A =
= 10,6 Hz); 3,56 (ddd, IH, H5B, J
H5B-H4B =
τH5B-H6bB = 9,7 Hz, J
H5B-H
6aB = 4,1 Hz,); 3,55-3,47 (m, 2H, H6bB, H5C); 3,38 (ddd, IH, H5A, J
H5A-H4A = J
H5A-H6bA = 10,0 Hz, J
H5A-
H6aA =
4.9 Hz,); 3,21 (t, IH, H4A, JH4A-H3A = HSA = 9,4 Hz); 2,64 (d, IH, OH, J0H-H3C = 3,7 Hz).
EXEMPLE 10
Synthèse d'un tétrasaccharide selon l'invention.
Cet exemple illustre l'intérêt du procédé conforme à la présente invention pour la préparation d'un tétrasaccharide par réaction de couplage entre un monosaccharide et un trisaccharide, le tétrasaccharide obtenu pouvant lui-même servir de précurseur d'accepteur de glycosyle tétrasaccharidique (voir exemple 1 1) pour la synthèse d'un pentasaccharide, et ainsi de suite.
Synthèse du 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-β-D- glucopyranosyl-(l- 3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl- (l→3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l→3)-2-O-benzoyl- 4,6-O-benzylidène-β-D-glucopyranoside de benzyle.
3,23 g ( 1 , 1 éq.) de 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-
1-thio-β-D-glucopyranoside d'éthyle préparé à l'exemple 4 (M = 556,7) et 6,17 g (1 éq.) de 2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l-*3)-2-O-
benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l→3)-2-O-benzoyl-4,6-O- benzylidène-β-D-glucopyranoside de benzyle préparé à l'exemple 9 (M = 1 170,3) sont dissous dans 100 mL de dichlorométhane anhydre en présence de 10 g de tamis moléculaire 4Â à 0° C puis additionnés de 1,42 g (1,2 éq.) de NIS et 100 μL (0,1 éq.) de trifluorométhanesulfonate de triméthylsilyle (TMSOTf). Après 1 h de réaction, le milieu est neutralisé par de la triéthylamine, filtré puis lavé par un solution de thiosulfate de sodium à 10% puis à l'eau. Après séchage et evaporation de la phase organique, le produit attendu est purifié par chromatographie sur gel de silice [flash; éluant: toluène/acétate d'éthyle (95/5 puis 925/75 puis 9/1; v/v). 7,70 g de 2-O-benzoyl-4,6-O-benzylidène-3-O-(2-méthylnaphtyl)-β-D- glucopyranosyl-(l-→3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl- (l-→3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l-→3)-2-O-benzoyl- 4,6-O-benzylidène-β-D-glucopyranoside de benzyle (M = 1664,9) sont ainsi recueillis. CCM: Rf = 0,5 [toluène/acétate d'éthyle (8/2; v/v)]. Solide blanc. Rendement (%) = 88%
RMN 13C (CDC13, 100 MHz): 165,05, 164,68, 164,58, 164,55 (4 C=Ο); 137,36, 137,32, 137,24, 137,17, 136,69, 135,41, 133,41, 133,36, 133,09, 133,05 (2 C), 132,85 (12 C quat. arom.); 129,80-125,33 (C arom.); 101,82, 101,16, 101,08, 100,77 (C7A,B,C,D); 99,49 (CIA); 99,04 (C1D); 98,42 (C1B); 96,94 (CIC); 81,28 (C4D); 78,82 (C4A); 78,28, 78,16 (C3D, C4C); 77,41 (C4B); 76,90 (C3C); 75,07 (C3A); 74,16 (C3B); 73,96 (C2A); 73,77 (C8D); 73,47 (C2B); 73,39 (C2D); 72,54 (C2C); 70,24 (C8A); 68,71, 68,67 (C6A,B,C,D); 66,44, 66,04, 65,52 (C5A,B,C,D).
RMN Η (CDCI3, 400 MHz): 7,78-7,04 (m, 52H, H arom.); 5,53 (s, IH, H7); 5,43 (s, IH, H7); 5,33 (t, IH, H2D, JH2D-HID = H3D = 7,8 Hz); 5,12 (t, IH, H2C, JH2C-H I C = JH2C-H3C = 5,3 Hz); 4,98 (d, IH, H1D, JHID-H2D = 7,4 Hz); 4,98 (dd, IH, H2A, JH2A-HIA = 8,0 Hz, JH2A-H3A = 8,5 Hz); ); 4,95 (d, IH, HIC, JHιc-H2c = 5,4 Hz); 4,89 (d, IH, H8aD, JH8aD-H8bD = 12,5 Hz); 4,82 (s, IH, H7); 4,79 (d, IH, H8bD, JH8bD-H8aD = H,8 Hz); 4,76 (d, IH, H8aA, JH8aA-H8bA = 11,4 Hz); 4,75 (d, IH, H1B, JH I B-H2B = 3,1 Hz); 4,75 (m, IH, H2B); 4,74 (s, IH, H7); 4,50 (d, IH, H8bA, JH8bA-H8aA = 12,6 Hz); 4,47 (d, IH, H1A, JH 1 A-H2A = 7,8 Hz); 4,34 (dd, IH, H6a, JH6a-H5 = 4,6 Hz, JH6a-H6b = 10,3 Hz); 4,19 (dd, IH, H6a, JH6a-H5 = 4,9 Hz, JH6a-H6b = 10,4 Hz); 4,12-4,09 (m, 2H, H6); 4,04 (t, IH, H3A, JH3A-H2A = JH3A-H4A = 8,7 Hz); 4,03-4,00 (m, IH, H3C); 3,95 (t, IH, H4C, JH4c-H3c = JH4C-H5C = 8,8 Hz);
3,92-3,89 (m, IH, H3B); 3,87 (dd, IH, H4D, JH4D-H3D = 9,3 Hz, HSD = 8,7 Hz); 3,82 (dd, IH, H3D, JH3D-H2D = 8,0 Hz, JH3D-H4D = 9,1 Hz);3,75-3,68 (m, 2H, H6); 3,59-3,37 (m, 5H, H4B, 4 H5, 2 H6); 3,35 (t, IH, H4A, JH4A-H3A = JH4A-H5A = 9,2 Hz).
EXEMPLE 11
Synthèse d'un tétrasaccharide accepteur selon l'invention.
Synthèse du 2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l→3)-2-O- benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-(l-»3)-2-O-benzoyl-4,6-O- benzylidène-β-D-glucopyranosyl-(l— »3)-2-O-benzoyl-4,6-O-benzylidène-β-D- glucopyranoside de benzyle
Après dissolution de 3,79 g (léq.) de 2-O-benzoyl-4,6-O-benzylidène-3-O- méthylnaphtyl-β-D-glucopyranosyl-(l->3)-2-O-benzoyl-4,6-O-benzylidène-β-D- glucopyranosyl-(l→3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl- (l-»3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle (M = 1664,9) dans 55 mL d'un mélange dichlorométhane/méthanol (4/1; v/v), nous ajoutons 1,55 g (3 éq.) de DDQ et on laisse évoluer 7h30 à température ambiante. Le milieu est ensuite dilué au dichlorométhane, lavé par un solution d'hydrogénocarbonate de sodium à 5% puis à l'eau. La phase organique est séchée, évaporée et le produit recherché est obtenu après purification sur gel de silice [flash; éluant: toluène/acétate d'éthyle (17/3; v/v)]. 2,78 g de 2-O-benzoyl-4,6-O- benzylidène-β-D-glucopyranosyl-(l-»3)-2-O-benzoyl-4,6-O-benzylidène-β-D- glucopyranosyl-( 1 — >3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranosyl-
(l-»3)-2-O-benzoyl-4,6-O-benzylidène-β-D-glucopyranoside de benzyle (M =
1523,7) sont ainsi isolés.
CCM: Rf = 0,3 [toluène/acétate d'éthyle (8/2; v/v)].
Solide blanc. Rendement (%) = 80%
RMN I3C (CDC13, 100 MHz): 165,80, 164,66, 164,61, 164,59 (4 C=Ο); 137,28, 137,20, 137,13, 137,00, 136,65, 133,62, 133,42, 133,10, 133,06 (9 C quat. arom.);
129,87-125,31 (C arom.); 101,80, 101,71, 101,10, 100,71 (C7A,B,C,D); 99,46 (CIA); 98,76 (C1D); 98,45 (C1B); 96,98 (CIC); 80,73 (C4D); 78,80 (C4A); 78,20 (C4C); 77,42 (C4B); 76,84 (C3C); 75,14 (C3A); 74,75 (C2D); 74,30 (C3B); 73,93 (C2A); 73,47 (C2B); 72,53 (C2C); 72,38 (C3D); 70,22 (C8A); 68,62 (C6A,B,C,D); 66,41 , 66,00, 65,50 (C5 A,B,C,D).
RMN Η (CDC13, 400 MHz): 7,91-7,04 (m, 45H, H arom.); 5,51 (s, IH, H7); 5,39 (s, IH, H7); 5,16 (dd, IH, H2D, JH2D-HID = 7,6 Hz, HSD = 8,4 Hz); 5,15 (t, IH, H2C, JH2C-HIC = JH2C-H3C = 5,3 Hz); 5,02 (d, IH, H1D, JHID-H2D = 7,6 Hz); 4,99 (dd, IH, H2A, JH2A-HIA = 8,0 Hz, JH2A-H3A = 9,6 Hz); 4,97 (d, IH, HIC, JHιc-H2C = 5,3 Hz); 4,83 (s, IH, H7); 4,82 (t, IH, H2B, JH2B-HIB = JH2B-H3B = 5,2 Hz); 4,76 (d, IH, H1B, JHIB-H2B = 5,6 Hz); 4,75 (d, IH, H8aA, JH8aA-H8bA = 10,8 Hz); 4,74 (s, IH, H7); 4,50 (d, IH, H8bA, JH8bA-H8aA = 13,4 Hz); 4,47 (d, IH, H1A, JHIA-H2A = 7,9 Hz); 4,34 (dd, IH, H6a, JH6a.H5 = 4,6 Hz, JH6a-H6b = 10,4 Hz); 4,18 (dd, IH, H6a, JH6a-H5 = 4,9 Hz, JH6a-H6b = 10,4 Hz); 4,12-4,09 (m, 2H, H6a); 4,05 (dd, IH, H3C, JH3C-H2C = 4,9 Hz, JH3c-H4c = 8,3 Hz); 4,04 (t, 1 H, H3 A, JH3A-H2A = JH3A-H4A = 8,8 Hz); 3,95 (dd, IH, H4C, JH4C-H3C 8,7 Hz, JH4C-H5C = 9,1 Hz); ); 3,91 (dd, IH, H3B, JH3B-H2B = 5,6 Hz, JH3B-H4B = 8,5 Hz); 3,89 (ddd, IH, H3D, H2D ≈ JH3D-H4D = 8,9 Hz, OH = 4,0 Hz); 3,72 (t, IH, H6b, JH6b-H5 = JH6b-H6a = 9,9 Hz); 3,67 (t, IH, H6b, JH6b-H5 = JH6b-H6a = 10,1 Hz); 3,60 (t, IH, H4D, JH4D-H3D = JH4D-H5D = 9,4 Hz); 3,58-3,54 (m, IH, H5C); 3,51 (t, IH, H6b, JH6b-H5 = JH6b-H6a = 10,0 Hz); 3,47-3,37 (m, 5H, H4B, H5A,B,D, H6b); 3,35 (t, IH, H4A, JH4A-H3A = JH4A-H5A = 9,2 Hz); 2,73 (d, IH, OH, JOH-H3C = 3,8 Hz).
EXEMPLE 12 Autre exemple de synthèse d'un svnthon donneur de glycosyle de formule générale (la) selon l'invention.
Préparation du 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-l-thio-β-D-glucopyranoside d'éthyle selon les étapes [l] à |5J, |6| à |9| puis |7 dont l'homme de métier pourra détemiiner aisément les conditions réactionnelles les plus appropriées, notamment en suivant l'enseignement des exemples décrits précédemment.
M = 486 g/mol.
CCM: Rf = 0,6 [éther de pétrole/acétate d'éthyle (8/2; v/v)]. Huile incolore. RMN 13C (CDC13, 100 MHz) δ (ppm): 169,68 (C=O); 138,21, 137,97 (C quat. arom.); 134,74 (C8); 128,52, 128,44, 128,17, 127,96, 127,78, 127,68 (C arom.); 117,02 (C9) 84,17 (C3); 83,39 (Cl); 79,49 (C5); 77,66 (C4); 75,17 (ÇH2-Ar); 74,12 (C7); 73,51 (ÇH2-Ar); 71,83 (C2); 68,92 (C6); 23,85 (S-ÇH2); 21,18 (ÇH3- CO); 14,97 (S-CH2-ÇH3). RMN Η (CDCI3, 400 MHz) δ (ppm): 7,34-7,21 (m, 10H, H arom.); 5,87 (ddt, IH, H8, JH8-H7a = H8-H7b = 5,7 Hz, JH8-H9a = 17,2 Hz, JH8-H9b = 10,4 Hz); 5,24 (ddt, IH, H9a, JH9a-H7 = 1,6 Hz, JH9a-H8 = 17,2 Hz, JH9a-H9b = 1,7 Hz); 5,15 (ddt, IH, H9b, JH9b-H7 = 1,2 Hz, JH9b-H8 = 10,4 Hz, JH9b-H9a = 1,5 Hz); 4,97 (dd, IH, H2, JH2-H I = 9,9 Hz, JH2-H3 = 9,2 Hz); 4,80 (d, IH, CH2-Ar, 2J = 10,7 Hz); 4,60 (d, IH, CH -Ar, 2J = 12,2 Hz); 4,56 (d, IH, C/fc-Ar, 2J = 10,8 Hz); 4,54 (d, IH, CH2-Ar, 2J = 12,1 Hz); 4,35 (d, IH, Hl, JHI-H2 = 10,0 Hz); 4,27 (ddt, IH, H7a, JH7a-H7b = 12,6 Hz, JH7a-H8 = 5,6 Hz, JH7a-H9 = 1,4 Hz); 4,16 (ddt, IH, H7b, JH7b-H7a = 12,6 Hz, JH7b-H8 = 5,9 Hz, JH7b-H9 = 1,4 Hz); 3,74 (dd, IH, H6a, JH6a-H5 = 2,0 Hz, JH6a-H6b = 1 1,1 Hz); 3,69 (dd, IH, H6b, JH6b-H5 = 4,5 Hz, JH6b-H6a = 11,0 Hz); 3,63 (t, IH, H4, JH4-H3 = JH4-H5 = 9,4 Hz); 3,54 (t, IH, H3, JH3-H2 = JH3-H4 = 9,0 Hz); 3,47 (ddd, IH, H5, JH5-H4 = 9,7 Hz, JH5-H6a = 1,9 Hz, JH5.H6b = 4,4 Hz); 2,69 (qd, 2H, S-CH2-CH3, J = 7,4 Hz, J = 9,6 Hz); 2,11 (s, 3H, CH3-CO); 1,26 (t, 3H, S-CH2-CH3, J = 7,4 Hz).
EXEMPLE 13
Synthèse d'un svnthon selon l'invention.
Cet exemple illustre la préparation d'un synthon selon l'invention qui peut être donneur de glycosyle, ou accepteur de glycosyle par réaction avec un donneur plus actif comme par exemple un trichloroacétimidate.
Synthèse du 2-O-acétyl-4,6-di-O-benzyl-l-thio-β-D-glucopyranoside d'éthyle.
A 1,58 g de 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-l-thio-β-D- glucopyranoside d'éthyle (M = 486) dans 56 mL d'un mélange éthanol/toluène/eau (8/3/1; v/v/v) sont ajoutés 2,74 g (7,5 éq.) de Dabco et 450 mg (0,15 éq.) de catalyseur de Wilkinson. Le milieu est porté à reflux pendant 2 heures puis concentré. Le mélange est repris au dichlorométhane, lavé à l'eau glacée, avec HC1 5% glacé, NaHCÛ3 5% glacé, à l'eau glacée, séché (MgSO ) et mis à sec.
Le résidu est dissous dans 30 mL d'un mélange acétone/HCl aq. 10% (19/1; v/v) qui est porté à reflux pendant 8 min, puis refroidi à T. A., neutralisé avec NaHCÛ3 5% et concentré. Après reprise au dichlorométhane et lavages à l'eau, le produit est purifié sur gel de silice [flash; éluant: toluène/acétate d'éthyle (9/1 ; v/v)] et on isole 1 g de 2-O-acétyl-4,6-di-O-benzyl-l-thio-β-D- glucopyranoside d'éthyle (M = 446). CCM: Rf = 0,2 [toluène/acétate d'éthyle (9/1; v/v)]. Huile incolore. Rendement (%) = 69.
RMN 1 C (CDC13, 100 MHz) δ (ppm): 170,62 (C=O); 138,11, 138,04 (C quat. arom.); 128,65, 128,44, 128,14, 128,10, 127,84, 127,73 (C arom.); 83,12 (Cl); 79,20 (C5); 78,04 (C4); 76,92 (C3); 74,90 (ÇH2-Ar); 73,56 (Ç_H2-Ar); 72,54 (C2); 68,90 (C6); 23,97 (S-ÇH2); 21,10 (ÇH3-CO); 15,00 (S-CH2-ÇH3). RMN Η (CDCI3, 400 MHz) δ (ppm): 7,28-7,09 (m, 10H, H arom.); 4,80 (dd, IH, H2, JH2-Hi = 9,9 Hz, JH2-H3 = 9,2 Hz); 4,68 (d, IH, CH2-Ar, 2J = 11,2 Hz); 4,56 (d, IH, CH2-Ar, 2J = 11,2 Hz); 4,56 (d, IH, CH2-Ar, 2J = 12,0 Hz); 4,46 (d, IH, CH2- Ar, 2J = 12,0 Hz); 4,31 (d, IH, Hl, JHι-H2 = 10,0 Hz); 3,72-3,64 (m, 3H, H3, H6a, H6b); 3,51 (dd, IH, H4, JH4.H3 = JH4-HS = 9,3 Hz); 3,40 (m, IH, H5); 2,64 (qd, 2H, S-CH2, J = 7,4 Hz, J = 9,6 Hz); 2,34 (d, IH, OH, J = 3,9 Hz); 2,06 (s, 3H, CH3- CO); 1,20 (t, IH, CH -CH2-S, J = 7,4 Hz).
EXEMPLE 14
Synthèse d'un svnthon donneur de glvcosyle de formule générale (Ib) selon l'invention.
Synthèse du 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-α-D-glucopyranosyl trichloroacétimidate.
Etape 14A
A 4,68 g (1 éq.) de 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-l-thio-β-D- glucopyranoside d'éthyle (M = 486) dans 100 mL d'un mélange acétone/eau (8/2; v/v) sont additionnés de 3,41 g (2 éq.) de /V-bromosuccinimide. La réaction terminée, le milieu est dilué au dichlorométhane, lavé avec une solution d'hydrogénocarbonate de sodium à 5% puis à l'eau, séché (MgSO4) et concentré.
CCM: Rf = 0,3 [éther de pétrole/acétate d'éthyle (7/3; v/v)].
Etape 14B
Le résidu précédemment obtenu (M = 442) est alors dissous dans 40 mL de dichlorométhane anhydre et sont introduits 4,83 mL (5 éq.) de trichloroacétonitrile et 290 μL (0,2 éq.) goutte à goutte de DBU. Après 1 h de réaction à température ambiante, le milieu est concentré et la purification sur gel de silice [flash; éluant: éther de pétrole/acétate d'éthyle/triéthylamine (85/15/1; v/v/v) permet d'isoler 4,38 g de 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-α-D-glucopyranosyl trichloroacétimidate (M = 586,5). CCM: Rf = 0,7 [éther de pétrole/acétate d'éthyle (8/2; v/v)]. Huile incolore.
Rendement (%) = 78.
RMN 13C (CDCI3, 100 MHz) δ (ppm): 170,1 1 (C=Ο); 161,10 (C=NH); 137,92, 137,91 (C quat. arom.); 134,84 (C8); 128,55, 128,49, 128,30, 128,23, 128,17, 128,05, 128,00, 127,83 (C arom.); 1 16,92 (C9); 94,09 (Cl); 79,30 (C3); 76,86
(C5); 75,52 (Ç_H2-Ar); 74,37 (C7); 73,58 (ÇH2-Ar); 73,45 (C4); 72,49 (C2); 67,94 (C6); 20,82 (CH3).
RMN Η (CDC13, 400 MHz) δ (ppm): 8,57 (s, IH, NH); 7,34-7,20 (m, 10H, H arom.); 6,52 (d, IH, Hl, JH 1.H2 = 3,6 Hz); 5,91 (ddt, IH, H8, JH8-H7a = TH8-H7b = 5,7 Hz, JH8-H9a = 17,2 Hz, JH8-H9b = 10,4 Hz); 5,27 (ddt, IH, H9a, JH9a-H7 = 1,6 Hz, JH93-HS = 1 ,2 Hz, JH9a-H9b = 1,7 Hz); 5,16 (ddt, IH, H9b, JH9b-H7 = 1,2 Hz, JH9b-H8 = 10,4 Hz, JH9b-H9a = 1,5 Hz); 5,02 (dd, IH, H2, JH2-HI = 3,6 Hz, JH2-H3 = 9,9 Hz); 4,84 (d, IH, CH2-A1, 2J = 10,6 Hz); 4,63 (d, IH, CH2-Ar, 2J = 12,0 Hz); 4,56 (d, IH, CH2-Ar, 2J = 10,5 Hz); 4,50 (d, IH, CH2-Ar, 2J = 12,1 Hz); 4,33-4,32 (m, 2H, H7a, H7b); 3,97 (t, 2H, H3, H4, JH3-H2 = JH3-H4 = JH4-H3 = JH4-H5 = 9,5 Hz); 3,84- 3,78 (m, 2H, H5, H6a); 3,68 (dd, IH, H6b, JH6b-HS = 1,6 Hz, JH6b-H6a = 11,1 Hz); 2,04 (s, 3H, CH3).
EXEMPLE 15
Synthèse d'un svnthon accepteur de glvcosyle de formule générale (II) selon l'invention.
Etape 15A Préparation du précurseur 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-β-D-gluco- pyranoside de benzyle.
A 3,07 g (1 éq.) de 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-α-D-gluco- pyranosyl trichloroacétimidate (M = 586,5) dans 25 mL de dichlorométhane à 0°C en présence de 2,5 g de tamis moléculaire 4Â, sont additionnés 1,62 mL (3éq.) d'alcool benzylique et 130 μL (0,1 éq.) de TESOTf. Après 1 h de réaction à 0°C, le milieu est neutralisé avec de la triéthylamine, filtré et concentré. La purification sur gel de silice [flash; éluant: toluène/acétate d'éthyle (98/2; v/v)] permet d'isoler 2,52 g de 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-β-D-glucopyranoside de benzyle (M = 532).
CCM: Rf = 0,5 [toluène/acétate d'éthyle (9/1; v/v)]. Huile incolore. Rendement (%) = 91.
RMN 13C (CDC13, 100 MHz) δ (ppm): 169,55 (C=O); 138,20, 137,99, 137,47 (C quat. arom.); 134,75 (C8); 128,51, 128,46, 128,41, 128,13, 128,01, 127,94, 127,83, 127,74, 127,71, 127,67, 127,64 (C arom.); 117,04 (C9); 99,72 (Cl); 82,73 (C3); 77,78 (C4); 75,22 (C5); 75,09 (ÇH2-Ar); 73,94 (C7); 73,58 (ÇH2-Ar); 73,08 (C2); 70,36 (ÇH2-Ar sur Cl); 68,81 (C6); 21,13 (CH3). RMN Η (CDCI3, 400 MHz) δ (ppm): 7,36-7,20 (m, 15H, H arom.); 5,85 (ddt, IH, H8, JH8-H7a = JH8-H7b = 5,7 Hz, JH8-H9a = 17,2 Hz, JH8-H9b = 10,4 Hz); 5,22 (ddt, IH, H9a, JH9a-H7 = 1,6 Hz, JH9a-H8 = 17,2 Hz, JH9a-H9b = 1,7 Hz); 5,13 (ddt, IH, H9b, JH9b-H7 = 1,2 Hz, JH9b-H8 = 10,4 Hz, JH9b-H9a = 1,5 Hz); 5,02 (dd, IH, H2, JH2-HI = 8,0 Hz, JH2-H3 = 9,4 Hz); 4,89 (d, IH, CH2-Ar, 2J = 12,4 Hz); 4,80 (d, IH, CH2-Ar, 2J = 10,7 Hz); 4,62 (d, IH, CH2-Ar, J = 12,0 Hz); 4,61 (d, IH, CH2-Ar sur Cl, 2J = 1 1,3 Hz); 4,56 (d, IH, CH2-Ar, 2J = 12,2 Hz); 4,55 (d, IH, CH2-Ar, 2J = 10,7 Hz); 4,40 (d, IH, Hl. JHι-H2 = 8,0 Hz); 4,24 (ddt, IH, H7a, JH7a-H7b = 12,6 Hz, JH7a-H8 = 5,6 Hz, JH7a-H9 = 1 ,4 Hz); 4,12 (ddt, IH, H7b, JH7b-H7a = 12,6 Hz, JH7b-H8 = 5,9 Hz, JH7b-H9 = 1,4 Hz); 3,75 (dd, IH, H6a, JH6a-H5 = 2,0 Hz, JH6a-H6b = 11,0 Hz); 3,70 (dd, IH, H6b, JH6b.H5 = 4,7 Hz, JH6b-H6a = 10,9 Hz); 3,64 (t, IH, H4, JH4-H3 = JH -H5 = 9,4 Hz); 3,51 (t, IH, H3, JH3-H2 = JH3-H4 = 9,3 Hz); 3,45 (ddd, IH, H5, JH5-H4 = 9,6 Hz, JH5-H6a = 2,0 Hz, JH5-H6b = 4,6 Hz); 2,06 (s, 3H, CH3).
Etape 15B
Préparation du synthon accepteur 2-O-acétyl-4,6-di-O-benzyl-β-D-gluco- pyranoside de benzyle (OH en position 3).
2,44 g (1 éq.) de 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-β-D-gluco- pyranoside de benzyle (M = 532) sont dissous dans 85 mL d'un mélange éthanol/toluène/eau (8/3/1; v/v/v) puis additionnés de 3,86 g (7,5 éq.) de Dabco et 640 mg (0,15 éq.) de catalyseur de Wilkinson. Le milieu est porté à reflux pendant
2 heures puis concentré. Le mélange est repris au dichlorométhane, lavé à l'eau
glacée, avec HC1 5% glacé, NaHCÛ3 5% glacé, à l'eau glacée, séché (MgSO4) et concentré.
Le résidu est alors dissous dans 48 mL d'un mélange acétone/HCl aq. 10% (19/1; v/v) et la solution est porté à reflux pendant 8 min, puis refroidie à T. A., neutralisée avec NaHCÛ3 5% et concentrée. Après reprise au dichlorométhane et lavages à l'eau, la purification sur gel de silice [flash; éluant: toluène/acétate d'éthyle (85/15; v/v)] conduit à l'obtention de 1,32 g de 2-O-acétyl-4,6-di-O- benzyl-β-D-glucopyranoside de benzyle (M = 491). CCM: Rf = 0,3 [toluène/acétate d'éthyle (8/2; v/v)]. Solide Blanc.
Rendement (%) = 59.
RMN 13C (CDC13, 100 MHz) δ (ppm): 170,96 (C=O); 138,12, 138,09, 137,37 (C quat. arom.); 128,63, 128,49, 128,44, 128,13, 128,07, 127,90, 127,82, 127,78, 127,69 (C arom.); 99,47 (Cl); 78,34 (C3); 75,98 (C4); 74,98 (C5); 74,87 (ÇH2- Ar);74,40 (C2); 73,64 (ÇH2-Ar); 70,53 (ÇH2-Ar sur Cl); 68,77 (C6); 21,03 (CH3).
RMN Η (CDCI3, 400 MHz) δ (ppm): 7,38-7,23 (m, 15H, H arom.); 4,91 (d, IH, CH2-Ar, 2J = 12,4 Hz); 4,88 (dd, IH, H2, JH2-H I = 7,9 Hz, JH2-H3 = 9,4 Hz); 4,78 (d, IH, CH -Ar, 2J = 11,2 Hz); 4,66 (d, IH, CH2-Ar, 2J = 12,2 Hz); 4,62 (d, IH, CH-Ar, 2J = 12,2 Hz); 4,62 (d, IH, CH2-Ar, 2J = 1 1,4 Hz); 4,58 (d, IH, CH2-Ar, 2J = 12,1 Hz); 4,46 (d, IH, Hl, JH1-H2 = 7,9 Hz); 3,79 (dd, IH, H6a, JH6a-H5 = 2,2 Hz, JH6a-H6b = 10,9 Hz); 3,74 (dd, IH, H6b, JH6b-H5 = 4,4 Hz, JH6b-H6a = 10,9 Hz); 3,72 (t, IH, H3, JH3-H2 = JH3-H4 = 9,1 Hz); 3,60 (dd, IH, H4, JH4-H3 = 8,8 Hz, JH4-H5 = 9,6 Hz); 3,45 (ddd, IH, H5, JH5-H4 = 9,6 Hz, JH5-H6a = 2,2 Hz, JH5-H6b = 4,4 Hz); 2,39 (d, IH, OH, J = 4,1 Hz), 2,09 (s, 3H, CH3).
EXEMPLE 16
Synthèse d'un disaccharide selon l'invention.
Cet exemple illustre l'intérêt du procédé conforme à l'invention pour la préparation d'un disaccharide par réaction de couplage entre deux monosaccharides, le disaccharide obtenu pouvant lui-même servir de précurseur d'un disaccharide accepteur de glycosyle (voir exemple 17) pour la synthèse d'un trisaccharide (voir exemple 18).
Synthèse du 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-β-D-glucopyranosyl-(l— »3)-2- Ο-acétyl-4,6-di-O-benzyl-β-D-glucopyranoside de benzyle.
Dans 30 mL de dichlorométhane anhydre à 0°C en présence de tamis moléculaire 4Â (3 g) sont introduits 1,73 g (1,1 éq.) de 2-O-acétyl-3-O-allyl-4,6- di-O-benzyl-α-D-glucopyranosyl trichloroacétimidate préparé à l'exemple 14 (M = 586,5) et 1,32 g (1 éq.) de 2-O-acétyl-4,6-di-O-benzyl-β-D-glucopyranoside de benzyle préparé à l'exemple 15 (M = 491) puis 150 μL (0,25 éq.) de TESOTf. Après 1 h de réaction à 0°C, le milieu est neutralisé avec de la triéthylamine, filtré et concentré. Le produit est purifié sur gel de silice [flash; éluant: toluène/acétate d'éthyle (95/5; v/v)] et on recueille ainsi 1,87 g de 2-O-acétyl-3-O-allyl-4,6-di-O- benzyl-β-D-glucopyranosyl-(l→3)-2-O-acétyl-4,6-di-O-benzyl-β-D- glucopyranoside de benzyle (M = 915). CCM: Rf = 0,3 [toluène/acétate d'éthyle (9/1; v/v)]. Huile incolore.
Rendement (%) = 76.
RMN ,3C (CDC13, 100 MHz) δ (ppm): 170,04, 169,12 (C=O); 138,46, 138,28, 138,22, 137,84, 137,38 (C quat. arom.); 134,64 (C8B); 128,54, 128,45, 128,44, 128,40, 128,36, 128,22, 128,17, 128,00, 127,83, 127,77, 127,69, 127,63, 127,56 (C arom.); 1 17,10 (C9B); 100,86 (CIB); 99,22 (CIA); 82,95 (C3B); 80,44 (C3A); 77,93 (C4B); 75,88 (C4A); 75,50 (C5B); 75,15 (C5A); 75,10 (ÇH2-Ar); 74,98 (ÇH2-Ar); 73,99 (C7B); 73,63 (C2A); 73,53 (ÇH2-Ar); 73,48 (ÇH2-Ar); 73,05 (C2B); 70,28 (ÇH2-Ar sur CIA); 69,14, 69,04 (C6A, C6B); 21,07, 20,96 (CH3). RMN Η (CDCI3, 400 MHz) δ (ppm): 7,35-7,18 (m, 25H, H arom.); 5,85 (ddt, IH, H8B,JH8B-H73B = τH8B-H7bB = 5,7 Hz, τH8B-H9aB = 17,2 Hz, JH8B-H9bB = 10,4 Hz); 5,23 (ddt, IH, H9aB, JH9aB-H7B = 1,6 Hz, J^B-HSB = 17,2 Hz, JH9aB-H9bB = 1,7 Hz); 5,14 (ddt, IH, H9bB, JH9bB-H7B = 1,2 Hz, JH9bB-H8B = 10,4 Hz, JH9bB-H9aB = 1,5 Hz); 5,03 (d, IH, CH2-Ar, 2J = 1 1,0 Hz); 5,00 (dd, IH, H2A, H.A = 8,0 Hz, JH2A-H3A = 9,6 Hz); 4,95 (dd, IH, H2B, HIB = 8,2 Hz, JH2B-H3B = 9,6 Hz); 4,88 (d, IH, CH-Ar sur CIA, 2J = 12,4 Hz); 4,78 (d, IH, CH2-Ar, 2J = 10,8 Hz); 4,58 (d, IH,
CH-Ar, -J = 12,1 Hz); 4,57 (d, IH, H1B, JH I B-H2B = 8,0 Hz); 4,57 (d, IH, CH2-Ar sur CIA, 2J = 13,4 Hz); 4,54 (d, IH, CH2-Ar, 2J = 12,2 Hz); 4,52 (d, IH, CH2-Ar, 2J = 10,7 Hz); 4,47 (d, IH, CH2-Ax, 2J = 10,9 Hz); 4,41 (d, IH, CH-Ar, 2J = 12,1 Hz); 4,36 (d, IH, CH2-Ar, 2J = 14,0 Hz); 4,33 (d, IH, H1A, JH I A-H2A = 8,0 Hz); 4,25 (ddt, IH, H7aB, JH7aB-H7bB = 12,6 Hz, JH7aB-H8B = 5,6 Hz, JH7aB-H9B = 1,4 Hz); 4,11 (ddt, IH, H7bB, JH7bB-H7aB = 12,6 Hz, JH7bB-H8B = 5,9 Hz, JH7bB-H9B = 1,4 Hz); 3,94 (dd, IH, H3A, JH3A-H2A = 9,3 Hz, JH3A-H4A = 8,8 Hz); 3,74 (dd, IH, H6aA ou H6aB, JH6a-H5 = 2,0 Hz, JH6a-H6b = 10,8 Hz); 3,73 (dd, IH, H6aA ou H6aB, JH6a-H5 = 1,6 Hz, JH6a-H6b = 10,7 Hz); 3,64 (dd, IH, H6bA ou H6bB, JH6b-H5 = 5,1 Hz, JH6b-H6a = 10,8 Hz); 3,61 (t, IH, H4B, H3B = HSB = 9,2 Hz); 3,56 (dd, IH, H4A, JH4A-H3A =8,9 Hz, JH A-H5A = 9,4 Hz); 3,53 (dd, IH, H6bA ou H6bB,
JH6b-H5 = 5,2 Hz, JH6b-H6a = 10,9 Hz); 3,47 (t, IH, H3B, JH3B-H2B = JH3B-H4B =
9,2 Hz); 3,47-3,41 (m, 2H, H5A, H5B), 2,10 (s, 3H, CH3); 2,05 (s, 3H, CH3).
EXEMPLE 17
Synthèse d'un disaccharide accepteur de glvcosyle selon l'invention.
Synthèse du 2-O-acétyl-4,6-di-O-benzyl-β-D-glucopyranosyl-(l- 3)-2-O-acétyl- 4,6-di-O-benzyl-β-D-glucopyranoside de benzyle.
1,28 g (1 éq.) de 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-β-D-gluco- pyranosyl-( 1 — >3)-2-O-acétyl-4,6-di-O-benzyl-β-D-glucopyranoside de benzyle (M = 915) et 1,18 g de Dabco sont dissous dans 50 ml d'un mélange éthanol/toluène/eau (8/3/1; v/v/v) puis additionnés de 390 mg (0,3 éq.) de catalyseur de Wilkinson. Le milieu réactionnel est chauffé à reflux pendant lh30 puis mis à sec. Le mélange est alors repris au dichlorométhane et extrait à l'eau, avec HC1 1/2N glacé, avec NaHCO3 5% et encore à l'eau, séché et concentré. Le résidu est ensuite redissous dans 30 ml d'un mélange acétone/HCl aq. 10% (19/1; v/v) que l'on porte à reflux pendant 8 min. Après refroidissement rapide à T.A., la solution est neutralisée par quelques gouttes de NaHCθ3 5% et concentrée. On dilue de nouveau au dichlorométhane et on extrait à l'eau. Le produit est finalement purifié sur gel de silice [flash; éluant: toluène/acétate d'éthyle (85/15;
v/v)] ce qui permet d'isoler ainsi 600 mg de 2-O-acétyl-4,6-di-O-benzyl-β-D- glucopyranosyl-(l— >3)-2-O-acétyl-4,6-di-O-benzyl-β-D-glucopyranoside de benzyle (M = 875).
CCM: Rf = 0,3 [toluène/acétate d'éthyle (8/2; v/v)]. Huile incolore.
Rendement (%) = 49.
RMN 13C (CDC13, 100 MHz) δ (ppm): 171,65, 169,09 (C=Ο); 138,49, 138,24, 137,99, 137,37 (C quat. arom.); 128,61, 128,43, 128,39, 128,29, 128,23, 128,14, 128,07, 127,78, 127,76, 127,66, 127,61, 127,59, 127,57 (C arom.); 100,64 (CIB); 99,22 (CIA); 80,70 (C3A); 78,50 (C4B); 76,39 (C3B); 75,96 (C4A); 75,31, 75,17 (C5A, C5B); 74,97, 74,89 (Ç_H2-Ar); 74,31 (C2B); 73,53, 73,51, (C2A, 2 ÇH2- Ar); 70,25 (ÇH2-Ar sur CIA); 69,16, 69,01 (C6A, C6B); 21,07, 20,85 (CH3). RMN Η (CDCI3, 400 MHz) δ (ppm): 7,34-7,19 (m, 25H, H arom.); 5,04 (d, IH, CH,-Ar, 2J = 10,1 Hz); 5,03 (dd, IH, H2A, H IA = 8,1 Hz, JH2A-H3A = 9,8 Hz); 4,88 (d, IH, CH-Ar sur CIA, 2J = 12,4 Hz); 4,80 (dd, IH, H2B, HI B = 8,1 Hz, JH2B-H3B = 9,4 Hz); 4,76 (d, IH, CH2-Ar, 2J = 1 1,1 Hz); 4,62 (d, IH, H1B, JH I B-H2B = 8,5 Hz); 4,60 (d, IH, CH2-Ar, 2J = 11,5 Hz); 4,58 (d, IH, CH2-Ar, 2J = 12,2 Hz); 4,57 (d, IH, CH2-Ar sur CIA, 2J = 12,3 Hz); 4,53 (d, IH, CH-Ar, 2J = 12,2 Hz); 4,47 (d, IH, CH2-Ar, 2J ≈ 1 1,0 Hz); 4,40 (s, 2H, CH2-Ar); 4,34 (d, 1Η, Η1A, JΗIA-Η2A = 8,0 Hz); 3,95 (t, IH, H3A, JH3A-H2A = JH3A-H4A = 9,1 Hz); 3,76 (dd, IH, H6aA ou H6aB, JH6a-H = 1,6 Hz, JH6a-H6b = 11,6 Hz); 3,74 (dd, IH, H6aA ou H6aB, JH6a-H5 = 1,7 Hz, JH6a-H6b = 11,8 Hz); 3,69 (m, IH, H3B); 3,65 (dd, IH, H6bA ou H6bB, JH6b-H5 = 5,0 Hz, JH6b-H6a = 10,8 Hz); 3,57 (t, IH, H4A, JH A-H3A = JH4A-HSA = 9,3 Hz); 3,57 (dd, IH, H6bA ou H6bB, JH6b-H5 = 5,4 Hz, JH6b-H6a = 1 1,3 Hz); 3,55 (t, IH, H4B, JH4B-H3B = H5B = 8,6 Hz); 3,43 (m, 2H, H5A, H5B); 2,49 (d, IH, ΟH, J = 4,1 Hz); 2,13 (s, 3H, CH3); 2,04 (s, 3H, CH3).
EXEMPLE 18
Synthèse d'un trisaccharide selon l'invention.
Cet exemple illustre l'intérêt du procédé conforme à la présente invention pour la préparation d'un trisaccharide par réaction de couplage entre un monosaccharide et un disaccharide, le trisaccharide obtenu pouvant lui-même servir de précurseur d'accepteur de glycosyle pour la synthèse d'un tétrasaccharide, et ainsi de suite.
Synthèse du 2-O-acétyl-3-O-allyl-4,6-di-O-benzyl-β-D-glucopyranosyl-(l- 3)-2- O-acétyl-4,6-di-O-benzyl-β-D-glucopyranosyl-(l-→3)-2-O-acétyl-4,6-di-O-benzyl- β-D-glucopyranoside de benzyle.
Dans 10 mL de dichlorométhane anhydre en présence de 1 g de tamis moléculaire 4Â à 0°C sont introduits 600 mg (1,1 éq.) de 2-O-acétyl-3-O-allyl- 4,6-di-O-benzyl- -D-glucopyranosyl trichloroacétimidate préparé à l'exemple 14 (M = 586,5) et 540 mg (1 éq.) de 2-O-acétyl-4,6-di-O-benzyl-β-D-glucopyranosyl- (l—»3)-2-O-acétyl-4,6-di-O-benzyl-β-D-glucopyranoside de benzyle préparé à l'exemple 17 (M = 875) et additionnés ensuite de 35 μL (0,25 éq.) de TESOTf. Après 50 min de réaction à 0°C, le milieu est neutralisé avec de la triéthylamine, filtré et concentré. La purification sur gel de silice [flash; éluant: toluène/acétate d'éthyle (9/1; v/v)] conduit à l'obtention de 500 mg de 2-O-acétyl-3-O-allyl-4,6-di- O-benzyl-β-D-glucopyranosyl-(l—»3)-2-O-acétyl-4,6-di-O-benzyl-β-D- glucopyranosyl-( 1 -»3)-2-O-acétyl-4,6-di-O-benzyl-β-D-glucopyranoside de benzyle (M = 1299).
CCM: Rf = 0,4 [toluène/acétate d'éthyle (9/1; v/v)]. Huile incolore. Rendement (%) = 63. RMN 13C (CDC13, 100 MHz) δ (ppm): 170,09, 169,44, 169,29 (C=Ο); 138,46, 138,43, 138,35, 138,20, 138,17, 137,85, 137,36 (C quat. arom.); 134,58 (C8C); 128,47-127,37 (C arom.); 1 17,13 (C9C); 101,13 (CIC); 100,43 (CIB); 99,21 (CIA); 82,92 (C3C); 80,96 (C3B); 80,23 (C3A); 77,88 (C4C); 76,13 (C4B); 75,89 (C4A); 75,45, 75,08, 75,02, 74,99, 74,96 (2 ÇH2-Ar, C5A, C5B, C5C); 74,03 (C7C); 73,69, 73,52, 73,51, 73,46, 73,42, 72,95 (3 ÇH2-Ar, C2A, C2B, C2C); 70,30 (ÇH2-Ar sur CIA); 69,40, 69,18, 69,00 (C6A, C6B, C6C); 21,19, 21,02, 20,99 (CH3).
RMN Η (CDCI3, 400 MHz) δ (ppm): 7,34-7,19 (m, 35H, H arom.); 5,85 (ddt, IH, H8C, JH8C-H7UC = JH8C-H7bc = 5,7 Hz, JH8C-H9aC = 17,2 Hz, JH8C-H9bc = 10,4 Hz); 5,23 (ddt, IH, H9aC, JH9aC-H7C = 1,6 Hz, JH9ac-H8c = 17,2 Hz, JH9ac-H9bc = 1,7 Hz);
5,15 (ddt, IH, H9bC, JH9bc-H7c = 1,2 Hz, - -HSC = 10,4 Hz, JH9bc-H9ac = 1,5 Hz);
5.03 (d, IH, CH-Ar, 2J = 10,7 Hz); 5,01 (d, IH, CH2-Ar, 2J = 10,1 Hz); 4,97 (dd, IH, H2A, JH2A-HIA = 8,0 Hz, JH2A-H3A = 9,2 Hz); 4,96 (dd, IH, H2C, JH2C-H I C = 8,1 Hz, JH2C-H3C = 9,4 Hz); 4,92 (dd, IH, H2B, JH2B-HIB = 8,2 Hz, mB = 9,5 Hz); 4,87 (d, IH, CHJ-AI sur CIA, 2J = 12,4 Hz); 4,78 (d, IH, CH-Ar, 2J = 10,6 Hz); 4,58-4,28 (m, 3H, CH2-Ar); 4,56 (d, 1Η, Η1C, JΗιc-Η2c = 8,1 Hz); 4,56 (d, IH, CH-Ar sur CIA, 2J = 12,1 Hz); 4,52 (d, IH, CH2-Ar, J = 10,8 Hz); 4,49 (d, IH, H1B, JH I B-H2B = 8,0 Hz); 4,47 (d, IH, CH -Ar, J = 10,8 Hz); 4,46 (d, IH, CH-Ar, 2J = 1 1,2 Hz); 4,37 (d, IH, CH2-Ar, 2J ≈ 12,3 Hz); 4,33 (d, IH, C/fc-Ar, 2J = 1 1,9 Hz); 4,32 (d, IH, H1A, JH IA-H2A = 7,9 Hz); 4,30 (d, IH, CH2-Ar, 2J = 12,3 Hz); 4,24 (m, IH, H7aC); 4,12 (ddt, IH, H7bC, JH7bC-H7ac = 12,6 Hz,
JH7bC-H8C = 5,9 Hz, JH7bC-H9C = 1 ,4 Hz); 3,92 (t, IH, H3A, JH3A-H2A = JH3A-H4A =
9,0 Hz); 3,89 (dd, IH, H3B, JH3B-H2B = 9,4 Hz, JH3B-H4B = 8,6 Hz); 3,75-3,40 (m,
3H, H5A, H5B, H5C); 3,74-3,70 (m, 2H, H6); 3,64-3,59 (m, 2H, H6); 3,62 (t, IH, H4C, JH4c-H3c = JH C-H5C = 9,3 Hz); 3,53-3,40 (m, 2H, H6); 3,53 (t, IH, H4A,
JH4A-H3A = JH4A-H A = 9,1 Hz); 3,51 (dd, IH, H4B, JH4B-H3B = 8,7 Hz, JH4B-H5B =
9.4 Hz); 3,48 (t, IH, H3C, JH3C-H2C = JH3CH4C = 9,1 Hz); 2,12 (s, 3H, CH3); 2,10 (s, 3H, CH3); 2,02 (s, 3H, CH3).