WO2001083444A1 - Verfahren zur herstellung cyclischer lactame - Google Patents
Verfahren zur herstellung cyclischer lactame Download PDFInfo
- Publication number
- WO2001083444A1 WO2001083444A1 PCT/EP2001/004841 EP0104841W WO0183444A1 WO 2001083444 A1 WO2001083444 A1 WO 2001083444A1 EP 0104841 W EP0104841 W EP 0104841W WO 0183444 A1 WO0183444 A1 WO 0183444A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phase
- lactam
- diluent
- iii
- separated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D201/00—Preparation, separation, purification or stabilisation of unsubstituted lactams
- C07D201/02—Preparation of lactams
- C07D201/08—Preparation of lactams from carboxylic acids or derivatives thereof, e.g. hydroxy carboxylic acids, lactones or nitriles
Definitions
- the present invention relates to a process for the preparation of cyclic lactams of the formula (II)
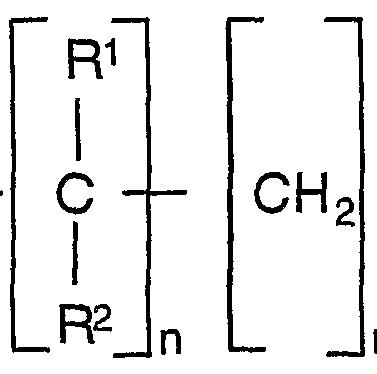
- n and m can each have the values 0, 1, 2, 3, 4, 5, 6, 7, 8 and 9 and the sum of n + is at least 3, preferably at least 4, R 1 and R 2 Ci C ⁇ -alkyl, C 5 -C cycloalkyl or C 6 -C ⁇ aryl groups mean
- R 1 , R 2 , m and n have the meaning given above and R is nitrile, carboxamide and carboxylic acid groups
- phase (V) is separated from phase (VI) and,
- phase (V) the diluent (III) and any by-products selected from the group consisting of low boilers, high boilers and unreacted compound (I) are separated off to give a lactam (II).
- WO 98/05636 describes the processing of reaction products as they occur in the gas phase cyclization of 6-aminocapronitrile with water vapor in the presence of solid catalysts. To do this
- this crude caprolacta is either subjected to a liquid / liquid extraction with the addition of a solvent containing acidic groups and / or treated with a cation exchanger. Measure b) extracts all amines or binds them to the cation exchanger.
- the main disadvantage of this workup is that unreacted aminocapronitrile is taken up by the ion exchangers in the cyclization and is obtained when the ion exchanger is regenerated as an ammonium salt in a mixture with ammonium salts of amine by-products. So it cannot be recycled.
- ammonium salts are also formed, which pose the same problems.
- the object of the present application was therefore to provide a process which enables the preparation of cyclic lacta en (II) from compounds (I) in a technically simple and economical manner and leads to high lactam yields with high conversions of compound (I) and also minimizes losses in yield during processing.
- compound (I) come aminocarboxylic acids and their derivatives, preferably those of the general formula I.
- R is carboxylic acid, nitrile and / or carboxamide groups and n and m can each have the values 0, 1, 2, 3, 4, 5, 6, 7, 8 and 9 and the sum of n + m is at least 3, preferably at least 4.
- R 1 and R 2 can in principle be substituents of any kind, it should only be ensured that the desired cyclization reaction is not influenced by the substituents.
- R and R 2 are independently Ci-Cg-alkyl or Cs-C-cycloalkyl groups or C 6 -C ⁇ aryl groups.
- Particularly preferred starting compounds are aminocarboxylic acid nitriles, preferably of the general formula
- a single compound (I) or a mixture of different compounds (I) can be used as the compound (I).
- a single compound is preferably used as compound (I).
- Omega-aminocarbonitriles are, for example, by partial hydrogenation of alpha, omega-dinitriles in the gas or liquid phase, for. B. according to WO 96/20166, WO 96/20916 or WO 96/20165, accessible.
- Omega-aminocarboxylic acids can be obtained, for example, by aminating hydrogenation of o-formylcarboxylic acids or by hydrolysis of omega-aminocarboxylic acid esters or omega-aminocarboxylic acid nitriles.
- Omega-aminocarboxamides can be obtained, for example, by reacting omega-aminocarboxylic acids and their esters with ammonia or omega-aminocarbonitriles with water.
- n, m, R 1 and R 2 have the meaning given above.
- the compound (I) described above is set in step a) with water vapor in the gas phase, advantageously in the presence of a heterogeneous catalyst, if appropriate in the presence of an organic dilution agent (III) to a mixture (IV) containing a lactam (II).
- Heterogeneous catalysts are all catalysts described for the gas phase cyclization of compound (I) to lactam (II), such as titanium dioxide, amorphous, as anatase or rutile, aluminum oxide, lanthanum phosphates, hafnium oxide, zirconium dioxide or mixtures thereof, preferably titanium dioxide, aluminum oxide, lanthanum phosphates , Zirconium dioxide or mixtures thereof.
- Diluent (III) can be added before the cyclization.
- diluent (III) is present in the gaseous reaction mixture during the cyclization of compound (I), preferably in amounts of 0.1 to 20 g of diluent (III) per gram of compound (I).
- diluent (III) added to the cyclization discharge is preferably suitable, for example by quenching the cyclization discharge.
- the diluent (III) can be added after the ammonia has been separated off from the mixture (IV).
- Such diluents (III) can be organic compounds which have a miscibility gap with water under certain quantity, pressure and temperature conditions, in particular below the reaction temperature during the cyclization.
- Suitable diluents (III) are C 4 -C 6 -alkanols, such as n-butanol, i-butanol, n-pentanol, preferably aliphatic hydrocarbons, such as n-hexane, cycloaliphatic hydrocarbons, such as cyclopentane or cyclohexane, in particular aromatic hydrocarbons, such as benzene, toluene, o-xylene, m-xylene, p-xylene, ethylbenzene, i-propylbenzene, di-i-propylbenzene and mixtures of such compounds, for example petroleum ether, into consideration.
- the hydrocarbons can carry functional groups, such as halogens, for example chlorine, as in chlorobenzene.
- Ammonia can also be present in the reaction according to step a).
- the reaction can advantageously be carried out in the gas phase at temperatures of generally 200 to 450 ° C., preferably 250 to 400 ° C.
- the pressure should generally be in the range from 0.01 to 20 bar, preferably from 0.1 to 5 bar.
- the reaction mixture should predominantly be gaseous. lie.
- the residence times are generally in the range from 0.1 to 100, preferably 1 to 50 seconds.
- the reaction can be carried out in the presence of an inert gas, such as nitrogen or noble gas or mixtures of such gases.
- an inert gas such as nitrogen or noble gas or mixtures of such gases.
- ammonia can be separated off in step b) after, preferably before phase separation from the mixture (IV), preferably by distillation, to give a mixture (IX) which is free or low in ammonia.
- the ammonia can also be separated off after the phase separation, preferably by distillation, from phase (V) and / or phase (VI).
- Mixture (IV) can contain ammonia, for example, if ammonia is formed in the reaction in step a) and / or if ammonia has been added to the reaction mixture in the reaction in step a).
- ammonia can be formed if R is a nitrile or carboxamide group.
- the separation can advantageously be carried out by distillation, in particular at bottom temperatures of 60 to 220 ° C. and pressures of 1 to 30 bar.
- mixture (IV) does not contain ammonia, which is understood to mean even small traces of ammonia which do not impair the subsequent process steps, mixture (IV) and mixture (IX) are identical.
- mixture (IX) according to the invention is transferred into quantity, pressure and temperature conditions under which diluent (III) and water are in liquid form and have a mixture gap, with the result that a two-phase system consisting of a phase (V) and a phase (VI ).
- diluent (III) and water are in liquid form and have a mixture gap
- a two-phase system consisting of a phase (V) and a phase (VI ).
- step a) was carried out in a homogeneous liquid phase
- the mixture (VII) can generally be separated into the two phases (V) and (VI) by selecting a suitable temperature.
- suitable quantitative ratios such as the addition of diluent (III), preferably water.
- phase (V) and phase (VI) according to step c) are separated according to the invention.
- phase separation can be carried out in a manner known per se in apparatuses described for such purposes, as described, for example, in: Ullmann's Encyclopedia of Industrial Chemistry, Vol. B3, 5th Ed., VCH Verlagsgesellschaft, Weinheim, 1988, pages 6-14 to 6- 22 are known.
- the optimal equipment and process conditions for phase separation can easily be determined by a few simple preliminary tests.
- the diluent (III) and optionally ammonia and optionally by-products are selected from the group consisting of low boilers (VIII), high boilers (VII) and unreacted compound (I) from phase (V) to give a lactam ( II).
- low boilers are understood to mean compounds with a boiling point below that of lactam (II), and high boilers (VII) are those compounds with a boiling point above that of lactam (II).
- This workup can advantageously be carried out by fractional distillation in one or more, such as 2 or 3, distillation apparatuses.
- the diluent (III) obtained in step d) can be partially or completely recycled in step a).
- any high boilers (VIII) and / or low boilers (VII) obtained in step d) can be partially or completely recycled in step a).
- any unreacted compound (I) obtained in step d) can be partially or completely recycled in step a).
- phase (VI) obtained in step c) can advantageously be recycled in step a).
- the lactam (II) can preferably be partially or completely separated from phase (VI) to obtain a mixture (X) and, if appropriate, low boilers (VIII) and / or high boilers (VII) can be separated off from the lactam (II) thus obtained.
- This lactam (II) can advantageously be worked up by fractional distillation in one or more, such as 2 or 3, distillation apparatuses.
- the high boilers (VII) and / or low boilers (VIII) can be separated off individually or together from the lactam (II).
- the high boilers (VII) and / or low boilers (VIII) can advantageously be partially or completely recycled in step a). Particularly preferred here is a cleavage of the high boilers (VII) before recycling in step a), as described, for example, in EP-A-793650, EP-A-793651, EP-A-794643 or EP-A-912508 ,
- lactam (II) obtained from phase (VI) can also be combined with the crude lactam (II) obtained from step d) and worked up together.
- Phase (X) can advantageously be returned in step a).
- Lactam (II) can be partially or completely separated from phase (VI) by extraction with a liquid extractant (XI) to give a mixture (XII) which contains an extractant (XI) and a lactam (II).
- the extracting agents (XI) are C 4 -C 9 -alkanols, such as n-butanol, i-butanol, n-pentanol, preferably aliphatic hydrocarbons, such as n-hexane, cycloaliphatic hydrocarbons, such as cyclopentane or cyclohexane, in particular aromatic hydrocarbons , such as benzene, toluene, o-xylene, m-xylene, p-xylene, ethylbenzene, i-propylbenzene, di-i-propylbenzene and mixtures of such compounds, for example petroleum ether, into consideration.
- the hydrocarbons can carry functional groups, such as halogens, for example chlorine, as in chlorobenzene.
- extractant (XI) and diluent (III) have the same or a similar composition.
- a diluent (III) which has been separated off from step d) can advantageously be used as the extractant (XI).
- the aqueous phase (X) remaining during the extraction can advantageously be returned in step a).
- the extractant (XI) and optionally low boilers (VIII), high boilers (VII) and / or unreacted compound (I) can advantageously be separated from mixture (XII) to give a lactam (II).
- This workup can advantageously be carried out by fractional distillation in one or more, such as 2 or 3, distillation apparatuses.
- the separation of extractant (XI) from mixture (XII) is preferably considered first.
- the high boilers, low boilers and, if appropriate, unreacted compound (I) can then be separated off individually or together from the lactam (II).
- the extractant (XI) obtained during processing can advantageously be partially or completely recycled in step a).
- any high boilers (VII) and / or low boilers (VIII) obtained in the workup can be partially or completely recycled in step a).
- any unreacted compound (I) obtained in the workup can be partially or completely recycled in step a).
- Mixture (XII) and phase (V) can advantageously be used together in step d) of the process according to the invention. Mixture (XII) and phase (V) can be combined before or during step d).
- lactams obtained in the process according to the invention can be used in a manner known per se in the production of technically important polymers, such as polyamides.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Hydrogenated Pyridines (AREA)
- Pyrrole Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description




Claims


Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001580873A JP2005508828A (ja) | 2000-05-03 | 2001-04-30 | 環式ラクタムの製造方法 |
| AU2001254821A AU2001254821A1 (en) | 2000-05-03 | 2001-04-30 | Method for producing cyclic lactams |
| MXPA02009712A MXPA02009712A (es) | 2000-05-03 | 2001-04-30 | Procedimiento para la obtencion de lactamas ciclicas. |
| AT01927931T ATE435851T1 (de) | 2000-05-03 | 2001-04-30 | Verfahren zur herstellung cyclischer lactame |
| US10/258,943 US6686465B2 (en) | 2000-05-03 | 2001-04-30 | Preparation of cyclic lactams |
| DE50114970T DE50114970D1 (de) | 2000-05-03 | 2001-04-30 | Verfahren zur herstellung cyclischer lactame |
| BR0110453-5A BR0110453A (pt) | 2000-05-03 | 2001-04-30 | Método para a preparação de lactamas cìclicas |
| EP01927931A EP1278726B1 (de) | 2000-05-03 | 2001-04-30 | Verfahren zur herstellung cyclischer lactame |
| CA002407813A CA2407813A1 (en) | 2000-05-03 | 2001-04-30 | Method for producing cyclic lactams |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10021193.3 | 2000-05-03 | ||
| DE10021193A DE10021193A1 (de) | 2000-05-03 | 2000-05-03 | Verfahren zur Herstellung cyclischer Lactame |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001083444A1 true WO2001083444A1 (de) | 2001-11-08 |
Family
ID=7640432
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2001/004841 Ceased WO2001083444A1 (de) | 2000-05-03 | 2001-04-30 | Verfahren zur herstellung cyclischer lactame |
Country Status (12)
| Country | Link |
|---|---|
| EP (1) | EP1278726B1 (de) |
| JP (1) | JP2005508828A (de) |
| KR (1) | KR20030013400A (de) |
| CN (1) | CN1199945C (de) |
| AR (1) | AR028549A1 (de) |
| AT (1) | ATE435851T1 (de) |
| AU (1) | AU2001254821A1 (de) |
| BR (1) | BR0110453A (de) |
| CA (1) | CA2407813A1 (de) |
| DE (2) | DE10021193A1 (de) |
| MX (1) | MXPA02009712A (de) |
| WO (1) | WO2001083444A1 (de) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109851543B (zh) * | 2019-01-24 | 2023-07-07 | 浙江华海药业股份有限公司 | 一种制备s-普瑞巴林内酰胺的方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0659741A1 (de) * | 1993-12-23 | 1995-06-28 | RHONE-POULENC FIBER & RESIN INTERMEDIATES | Verfahren zur Herstellung von Laktamen |
| US5495016A (en) * | 1994-11-25 | 1996-02-27 | Basf Aktiengesellschaft | Preparation of caprolactam |
| WO1996022974A1 (fr) * | 1995-01-27 | 1996-08-01 | Rhone-Poulenc Fiber And Resin Intermediates | Procede de preparation de lactame |
| US5693793A (en) * | 1996-07-17 | 1997-12-02 | Basf Aktiengesellschaft | Preparation of caprolactam from 6-aminocapronitrile |
| WO1998005636A1 (fr) * | 1996-08-02 | 1998-02-12 | Rhodia Fiber And Resin Intermediates | Procede de purification de lactames |
| WO1998037063A1 (en) * | 1997-02-19 | 1998-08-27 | Dsm N.V. | Process for the preparation of caprolactam in the absence of catalysts by contacting 6-aminocaproic acid derivatives with superheated steam |
| WO1999028296A1 (de) * | 1997-12-01 | 1999-06-10 | Basf Aktiengesellschaft | Verfahren zur herstellung von lactamen |
| WO1999047500A1 (de) * | 1998-03-18 | 1999-09-23 | Basf Aktiengesellschaft | Verfahren zur herstellung von lactamen mit hilfe von oligophosphat-katalysatoren |
-
2000
- 2000-05-03 DE DE10021193A patent/DE10021193A1/de not_active Withdrawn
-
2001
- 2001-04-27 AR ARP010101994A patent/AR028549A1/es not_active Application Discontinuation
- 2001-04-30 CA CA002407813A patent/CA2407813A1/en not_active Abandoned
- 2001-04-30 JP JP2001580873A patent/JP2005508828A/ja not_active Withdrawn
- 2001-04-30 EP EP01927931A patent/EP1278726B1/de not_active Expired - Lifetime
- 2001-04-30 WO PCT/EP2001/004841 patent/WO2001083444A1/de not_active Ceased
- 2001-04-30 AU AU2001254821A patent/AU2001254821A1/en not_active Abandoned
- 2001-04-30 BR BR0110453-5A patent/BR0110453A/pt not_active IP Right Cessation
- 2001-04-30 CN CNB018089534A patent/CN1199945C/zh not_active Expired - Fee Related
- 2001-04-30 AT AT01927931T patent/ATE435851T1/de not_active IP Right Cessation
- 2001-04-30 DE DE50114970T patent/DE50114970D1/de not_active Expired - Fee Related
- 2001-04-30 MX MXPA02009712A patent/MXPA02009712A/es unknown
- 2001-04-30 KR KR1020027014747A patent/KR20030013400A/ko not_active Ceased
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0659741A1 (de) * | 1993-12-23 | 1995-06-28 | RHONE-POULENC FIBER & RESIN INTERMEDIATES | Verfahren zur Herstellung von Laktamen |
| US5495016A (en) * | 1994-11-25 | 1996-02-27 | Basf Aktiengesellschaft | Preparation of caprolactam |
| EP0793650A1 (de) * | 1994-11-25 | 1997-09-10 | Basf Aktiengesellschaft | Verfahren zur herstellung von caprolactam |
| WO1996022974A1 (fr) * | 1995-01-27 | 1996-08-01 | Rhone-Poulenc Fiber And Resin Intermediates | Procede de preparation de lactame |
| US5693793A (en) * | 1996-07-17 | 1997-12-02 | Basf Aktiengesellschaft | Preparation of caprolactam from 6-aminocapronitrile |
| EP0912508A1 (de) * | 1996-07-17 | 1999-05-06 | Basf Aktiengesellschaft | Verfahren zur herstellung von caprolactam aus 6-aminocapronitril |
| WO1998005636A1 (fr) * | 1996-08-02 | 1998-02-12 | Rhodia Fiber And Resin Intermediates | Procede de purification de lactames |
| WO1998037063A1 (en) * | 1997-02-19 | 1998-08-27 | Dsm N.V. | Process for the preparation of caprolactam in the absence of catalysts by contacting 6-aminocaproic acid derivatives with superheated steam |
| WO1999028296A1 (de) * | 1997-12-01 | 1999-06-10 | Basf Aktiengesellschaft | Verfahren zur herstellung von lactamen |
| WO1999047500A1 (de) * | 1998-03-18 | 1999-09-23 | Basf Aktiengesellschaft | Verfahren zur herstellung von lactamen mit hilfe von oligophosphat-katalysatoren |
Also Published As
| Publication number | Publication date |
|---|---|
| DE10021193A1 (de) | 2001-11-08 |
| EP1278726A1 (de) | 2003-01-29 |
| DE50114970D1 (de) | 2009-08-20 |
| JP2005508828A (ja) | 2005-04-07 |
| KR20030013400A (ko) | 2003-02-14 |
| ATE435851T1 (de) | 2009-07-15 |
| AR028549A1 (es) | 2003-05-14 |
| BR0110453A (pt) | 2003-03-11 |
| CN1199945C (zh) | 2005-05-04 |
| CA2407813A1 (en) | 2002-10-30 |
| EP1278726B1 (de) | 2009-07-08 |
| CN1427821A (zh) | 2003-07-02 |
| MXPA02009712A (es) | 2003-04-22 |
| AU2001254821A1 (en) | 2001-11-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0912508B1 (de) | Verfahren zur herstellung von caprolactam aus 6-aminocapronitril | |
| EP2980069A1 (de) | Verfahren zur Herstellung von Cyclododecanon | |
| EP1261580B1 (de) | Verfahren zur abtrennung eines azepin-derivats aus einer mischung enthaltend ein amin und ein azepin-derivat | |
| EP0961767B1 (de) | Verfahren zur gewinnung von hexamethylendiamin aus hexamethylendiamin enthaltenden mischungen | |
| EP1125923B1 (de) | Verfahren zur Herstellung von N-Methyl-2-Pyrrolidon (NMP) | |
| DE102012215900A1 (de) | Verfahren zur Herstellung und Aufarbeitung eines Triacetonamin-haltigen Reaktionsgemisches | |
| EP0719757B1 (de) | Verfahren zur Herstellung von 3-Chlorphthalsäureanhydrid | |
| DE69709625T2 (de) | Verfahren zur herstellung von epsilon-caprolactam von 6-aminocaprosäure | |
| DE69717846T2 (de) | Behandlungsverfahren von laktamen | |
| DE2551055A1 (de) | Verfahren zur herstellung von 1,3- oder 1,4-bis-(aminomethyl)-cyclohexan | |
| EP0271815B1 (de) | Verfahren zur Herstellung von Caprolactam | |
| EP1278727B1 (de) | Verfahren zur herstellung cyclischer lactame | |
| EP1278726B1 (de) | Verfahren zur herstellung cyclischer lactame | |
| DE69717976T3 (de) | Gewinnung von epsilon-caprolactam aus wässrigen gemischen | |
| DE2611069A1 (de) | Verfahren zur gewinnung von im wesentlichen reinem triaethylendiamin | |
| EP1292569B1 (de) | Verfahren zur abtrennung von 6-aminocapronitril aus gemischen, die 6-aminocapronitril, adipodinitril und hexamethylendiamin enthalten | |
| EP1511720B1 (de) | Verfahren zur reduzierung des gehalts an einem ungesättigten amin in einer mischung enthaltend ein aminonitril, ein diamin, ein dinitril oder deren gemische | |
| US6686465B2 (en) | Preparation of cyclic lactams | |
| WO2001096294A1 (de) | Verfahren und herstellung von caprolactam | |
| EP1299347B1 (de) | Verfahren zur abtrennung von ammoniak | |
| EP0375612A2 (de) | Verfahren zur Herstellung von 4-Hydroxy-1,2,2,6,6-pentamethylpiperidin | |
| EP1339672A1 (de) | Verfahren zur reduzierung des gehalts an einem ungesättigten amin in einer mischung enthaltend ein amin und ein nitril | |
| CH361808A (de) | Verfahren zur Herstellung von Lactamen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2001927931 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/009712 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2001 580873 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10258943 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2407813 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018089534 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027014747 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IN/PCT/2002/1983/CHE Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001927931 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027014747 Country of ref document: KR |