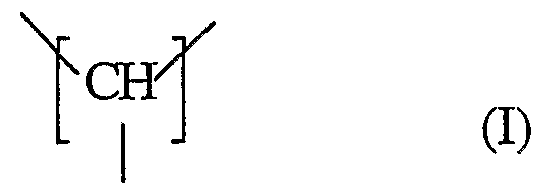Polymethylenamin, Verfahren zu seiner Herstellung und seine Verwendung
Die vorliegende Erfindung bezieht sich auf Polymethylenamin, ein Verfahren zu dessen Herstellung sowie dessen Verwendung.
Kationische Polymere mit hohen Ladungsdichten werden vor allem in der Papierindustrie verwendet. Sie dienen häufig als Fixier-, Entwässerungs-, Flockungs- und/oder Retentionsmittel und werden der Papiermasse vor bzw. bei dessen Herstellung zugegeben. Als Retentionsmittel bewirken sie eine Verminderung des Mitreißens der die Papiermasse formenden Faser- und Füllstoffe (Retention) beim Trennen der Feststoffe vom Wasser. Es wird also durch diese Retentionsmittel die Ausbeute der fest/flüssig-Trennung verbessert. Zur Herstellung von Papier werden grundsätzlich als Rohstoff omponenten Holzschliff, thermomechanischer Stoff, chemo- thermomechanischer Stoff, Druckschliff sowie Sulfit- und Sulfat- Zellstoff in kurz- und langfasriger Form, gegebenenfalls auch
Zellstoffe und Holzstoff sowie immer häufiger auch Altpapier eingesetzt. Als Füllstoffe eignen sich beispielsweise Clay, Kaolin, gefällte und natürliche, gemahlene Kreide, Talkum, Titandioxid, Calciu sulfat, Bariumsulfat, Aluminiumoxid und Satinweiß sowie Mischungen der genannten Füllstoffe.
Übliche kationische Polymere, die als Retentions- bzw. Fixiermittel eingesetzt werden, sind beispielsweise kationisches Poly- acrylamid (cPAM) Polyethylenimin (PEI) , Poly-Diallyldimethylammo- niumchlorid (Poly-DADMAC) und Polyvinylamin (PVAm) . Die theoretische Ladungsdichte von PEI und PVAm (vollständige Protonierung) liegt bei 23,2 meq/g.
Vinylamineinheiten enthaltende Polymere sind bekannt, vgl. US-A-4,421,602, ÜS-A-5 , 334 , 287 , EP-A-0 216 387, US-A-5 , 981 , 689 , WO-A-00/63295 und US-A-6 , 121, 409. Sie werden durch Hydrolyse von offenkettigen N-Vinylcarbonsäureamideinheiten enthaltenden Polymeren hergestellt. Diese Polymere sind z. B. erhältlich durch Po- lymerisieren von N-Vinylformamid, N-Vinyl-N-methylformamid, N-Vi- nylacetamid, N-Vinyl-N-methylacetamid, N-Vinyl-N-ethylacetamid und N-Vinylpropionamid. Die genannten Monomeren können entweder allein oder zusammen mit anderen Monomeren polymerisiert werden.
Als monoethylenisch ungesättigte Monomere, die mit den N-Vinyl- carbonsäureamiden copolymerisiert werden, kommen Vinylester von gesättigten Carbonsäuren von 1 bis 6 Kohlenstoffatomen, Vinyl-
ether wie Cι~ bis C6-Alkylvinylether und Ester, Amide und Nitrile von ethylenisch ungesättigten C3- bis Cß-Carbonsäuren in Betracht.
Weitere geeignete Carbonsäureester ungesättigter Carbonsäuren leiten sich von Glykolen oder Polyalkylenglykolen ab, bei denen jeweils nur eine OH-Gruppe verestert ist, weiterhin geeignet sind Acrylsäuremonoester von Polyalkylenglykolen einer Molmasse von 500 bis 10.000. Weitere geeignete Comonomere sind Ester von ethylenisch ungesättigten Carbonsäuren mit Aminoalkoholen. Die basischen Acrylate können in Form der freien Basen, der Salze mit Mineralsäuren wie Salzsäure, Schwefelsäure oder Salpetersäure, der Salze mit organischen Säuren wie Ameisensäure, Essigsäure, Propionsäure oder der Sulfonsäuren oder in quaternierter Form eingesetzt werden. Weitere geeignete Comonomere sind Amide ethylenisch ungesättigter Carbonsäuren, wie Acrylamid, Methacryl- amid sowie N-Alkylmono- und Diamide von monoethylenisch ungesättigten Carbonsäuren mit Alkylresten von 1 bis 6 C-Atomen sowie basische (Meth) acrylamide .
Weiterhin sind als Comonomere geeignet Dimethylaminoal- kyl (meth) acrylat CH3CI, N-Vinylpyrrolidon, N-Vinylcaprolactam, Acrylnitril, Methacrylnitiril, N-Vinylimidazol sowie substituierte N-Vinylimidazole.
Weiter geeignet sind Polyethylenimine, die beispielsweise durch Polymerisation von Ethylenimin in wässriger Lösung in Gegenwart von säureabspaltenden Verbindungen, Säuren oder Lewis-Säuren als Katalysator herstellbar sind. Polyethylenimine haben Molmassen bis zu 50.000, nach weiteren Vernetzungsmassnahmen können Mol- assen bis zu 2 Millionen, vorzugsweise von 200 bis 1.000.000, erreicht werden. Besonders bevorzugt werden Polyethylenimine mit Molmassen von > 1.000.000, die durch Ultrafiltration erhältlich sind, eingesetzt. Die Polyethylenimine können gegebenenfalls modifiziert werden, z. B. alkoxyliert, alkyliert oder amidiert wer- den. Sie können außerdem einer Michaeladdition oder einer Strek- kersynthese unterworfen werden.
Außerdem kommen mit Ethylenimin gepfropfte Polyamidoamine in Betracht, die beispielsweise durch Kondensieren von Dicarbonsäuren mit Polyaminen und anschließendem Aufpfropfen von Ethylenimin erhältlich sind. Höhere Molmassen erzielt man durch weitere Vernetzungen. Geeignete Polyamidoamine erhält man beispielsweise dadurch, dass man Dicarbonsäuren mit 4 bis 10 Kohlenstoffatomen mit Polyalkylenpolyaminen umsetzt, die 3 bis 10 basische Stickstoff- atome im Molekül enthalten.
Bei der Herstellung der Polyamidoamine kann man auch Mischungen von Dicarbonsäuren einsetzen, ebenso Mischungen aus mehreren Polyalkylenpolyaminen. Bei der Kondensation kann man gegebenenfalls auch Lactone oder Lacta e von Carbonεäuren mit 4 bis 8 C- Atomen einsetzen. Pro Mol einer Dicarbonsäure verwendet man beispielsweise 0,8 bis 1,4 Mol eines Polyalkylenpolyamins . Diese Polyamidoamine werden mit Ethylenimin gepfropft.
Außerdem eignen sich wasserlösliche vernetzte Polyethylenimine, die durch Reaktion von Polyethyleniminen mit Vernetzern wie
Epichlorhydrin oder Bischlorhydrinethern von Polyalkylenglykolen mit 2 bis 100 Ethylenoxid- und/oder Propylenoxid-Einheiten erhältlich sind und noch über freie primäre und/oder sekundäre Ami- nogruppen verfügen. Auch amidische Polyethylenimine sind geei- gnet, die beispielsweise durch Amidierung von Polyethyleniminen mit Cι~ bis C2 -Monocarbonsäuren erhältlich sind. Weitere geeignete kationische Polymere sind alkylierte Polyethylenimine und alkoxylierte Polyethylenimine. Bei der Alkoxylierung verwendet man z. B. pro NH-Einheit im Polyethylenimin 1 bis 5 Ethylenoxid- bzw. Propylenoxideinheiten.
Mit den genannten Polymeren lässt sich bei der Papierherstellung eine hohe Retention der Feststoffe erzielen. Es ist jedoch einsichtig, dass es im Prinzip erwünscht ist, das Retentionsverhal- ten so weit zu verbessern, dass von der Papiermasse nur noch minimale Mengen oder - im Idealfall - nichts mehr durch das ablaufende Wasser mitgerissen werden. Aus diesem Grund wird laufend versucht, die bekannten, als Retentionsmittel eingesetzten kationischen Polymere zu verbessern.
Es ist bekannt, dass die Ladungsdichte eines bestimmten Polymers dessen Retentionseigenschaften beeinflusst. Allgemein gilt, dass das Retentionsverhalten eines Polymers für bestimmte Papierstoffe optimiert werden kann. Insbesondere sind Fixiermittel mit höch- ster Ladungsdichte bevorzugt. Aus diesem Grund zielt die Entwicklung darauf ab, kationische Polymere mit hoher Ladungsdichte zu entwickeln.
Aus theoretischen Überlegungen ist bekannt, dass Polymethylenamin (PMAm) eine Ladungsdichte aufweisen sollte, die über denen der bisher erhältlichen kationischen Polymere liegt. PMAm, das aus wiederkehrenden Einheiten der Formel I
*2
besteht, ist bis jetzt in der Literatur nicht beschrieben.
Die Aufgabe der vorliegenden Erfindung besteht darin, Polymethy- lenamin bzw. ein Verfahren zu dessen Synthese bereitzustellen.
Diese Aufgabe wird gelöst durch wiederkehrende Einheiten der allgemeinen Formel I
NH,
enthaltendes Polymethylenamin.
Die vorliegende Erfindung bezieht sich weiterhin auf Verfahren zur Herstellung von PMAm. In allen diesen Verfahren wird als Edukt für die Synthese von PMAm von einer Verbindung der allgemeinen Formel (II) ausgegangen.
In der allgemeinen Formel (II) bedeutet die gestrichelte Linie eine Doppelbindung, die optionsweise vorhanden sein kann, R1 und R2 sind Substituenten, die optionsweise vorhanden sein können und unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus: Wasserstoff, gesättigten und ungesättigten, linearen und verzweigten, unsubstituierten und gegebenenfalls arylsubsti- tuierten Cι-C4o-Alkylgruppen; Formylgruppen und gesättigten und ungesättigten, linearen und verzweigten, unsubstituierten und gegebenenfalls arylsubstituierten aliphatischen Cx-C40-Acylgruppen; Gruppen des Typs R0C(0)-, in der R5 eine gesättigte oder ungesättigte, lineare oder verzweigte, unsubstituierte oder gegebenenfalls arylsubstituierte Cι-C4o-Alkylgruppe ist; Gruppen des
Typs -C(0)NR6R7, in der R6 und R7 unabhängig voneinander ausgewählt sind aus Wasserstoff, gesättigten und ungesättigten, linearen und verzweigten, unsubstituierten und gegebenenfalls aryl- substituierten Cι-C4o-Alkylgruppen; und gegebenenfalls substi- tuierten aromatischen C6-Acylgruppen;
R3 und R4 sind Substituenten, die optionsweiεe vorhanden sind und unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, gesättigten und ungesättigten, linearen und ver- zweigten, substituierten und unsubstituierten Cι-C4o-Alkylgruppen und Hydroxylgruppen; oder
R3, R4 sind zusammengenommen =0 oder =S.
In einer bevorzugten Ausführungsform der vorliegenden Erfindung sind R1 und R2 unabhängig voneinander ausgewählt aus der Gruppe bestehend aus: Wasserstoff, gesättigten linearen und verzweigten, unsubstituierten und gegebenenfalls arylsubstituierten Cι-Cι2-Alkylgruppen; Formylgruppen und gesättigten linearen und verzweigten, unsubstituierten und gegebenenfalls arylsubstituierten aliphatischen Cι-Cι-Acylgruppen; Gruppen des Typs R50C(0)-, in der R5 eine gesättigte lineare oder verzweigte, unsubstituierte oder gegebenenfalls arylsubstituierte Cι-C30-Alkylgruppe ist; Gruppen des Typs -C(0)NR6R7, in der R6 und R7 unabhängig voneinander ausgewählt sind aus Wasserstoff, gesättigten linearen und verzweigten, unsubstituierten und gegebenenfalls arylsubstituierten Cι-Cι-Alkylgruppen; und gegebenenfalls substituierten aromatischen Cδ-Acylgruppen;
R3 und R4 sind Substituenten, die optionsweise vorhanden sind und unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, gesättigten und ungesättigten, linearen und verzweigten, substituierten und unsubstituierten Cι-Cι2-Alkylgruppen und Hydroxylgruppen; oder
R3, R4 sind zusammengenommen =0.
In einer weiter bevorzugten Ausführungsform der vorliegenden Erfindung sind R1 und R2 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus: Wasserstoff, gesättigten linearen und verzweigten, unsubstituierten und gegebenenfalls arylsubstituierten Ci-Cs-Alkylgruppen; Formylgruppen und gesättigten linearen und verzweigten, unsubstituierten" und gegebenenfalls arylsubstituierten aliphatischen C-Cs-Acylgruppen; Gruppen des Typs R5OC(0)-, in der R5 eine gesättigte lineare oder verzweigte, unsubstituierte oder gegebenenfalls arylsubstituierte
Cι-Cι2-Alkylgruppe ist; und gegebenenfalls substituierten aromatischen Cξ-Acylgruppen;
R3 und R4 sind Substituenten, die optionsweise vorhanden sind und unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, gesättigten und ungesättigten, linearen und verzweigten, substituierten und unsubstituierten Cι-Cχ2-Alkylgruppen und Hydroxylgruppen; oder
R3 , R4 sind zusammengenommen =0.
In der einfachsten Ausführungsform der vorliegenden Erfindung ist das Molekül der Formel (II) Imidazol.
In einer Ausführungsform der vorliegenden Erfindung wird als Eduktmonomer ein 1, 3-Diacyl-4-imidazolin-2-on oder ein 1, 3-Diacyl-4-imidazol-2-thion eingesetzt. Man erhält so durch Polymerisation Poly(1, 3-diacyl-4-imidazolin-2- (thi) on) (II), das wiederkehrende Einheiten der allgemeinen Formel III
enthält.
In der Formel III bedeuten:
R ist Wasserstoff, eine lineare oder verzweigte Cι-C4o-Alkyl- gruppe, eine lineare oder verzweigte Cι-C4o-Alkoxygruppe, oder Phenyl, vorzugsweise Wasserstoff oder eine lineare oder verzweigte Ci-Cs-Alkylgruppe, insbesondere eine Methylgruppe;
X ist 0 oder S, vorzugsweise 0.
Anschließend wird zur Darstellung von PMAm Poly (1, 3-dia- cyl-4-imidazolin-2- (thi) on) unter Abspaltung der Acylgruppen und Öffnung des N-Heterocyclus zu PMAm verseift. Die Reaktion ist in der nachfolgenden Gleichung (1) wiedergegeben.
+ n RC(0)0-B+ + n/2 C(0)Cr2 2B+ Gleichung 1
In der Gleichung (1) beträgt der Polymeriεationsgrad n des PMAm Ia > 2, vorzugsweise > 10. Es ist mehr bevorzugt, wenn der Polymerisationsgrad n bei Werten von 1000 bis 1.000.000 liegt, ins- besondere bei Werten von 10.000 bis 100.000. Das Molgewicht des PMAm liegt bei Werten von > 100.000 D, vorzugsweise > 300.000 D, mehr bevorzugt 300.000 D bis 10.000.000 D, insbesondere 300.000 D bis ca. 2.000.000 D.
Das PMAm nach der vorliegenden Erfindung kann dabei ausschließlich Einheiten der Formel (I) enthalten, also als Homopolymer vorliegen. Es können aber auch ein oder mehrere weitere, von (I) verschiedene Monomereinheiten eingebaut sein. Es liegen dann Copolymere vor, beispielsweise Bi- oder Terpolymere . Diese können in Form von Block- oder statistischen Polymeren vorliegen.
In einem entsprechenden Copolymer nach der vorliegenden Erfindung können als weitere Einheiten neben den Methylenamineinheiten sämtliche damit kompatiblen Monomereinheiten vorliegen. Vorzugs- weise werden bei der Copolymerisation Comonomere eingesetzt, die mit den Monomeren nach Formel (II) oder (IV) copolymerisiert werden können. Beispiele hierfür sind Vinylester von gesättigten Carbonsäuren mit 1 bis 6 Kohlenstoffatomen wie Vinylformiat, Vinylacetat, Vinylpropionat und Vinylbutyrat und Vinylether wie Ci- bis C6-Alkylvinylether, z.B. Methyl- oder Ethyl inylether. Weitere geeignete Comonomere sind Ester, Amide und Nitrile von ethylenisch ungesättigten C3- bis C6-Carbonsäuren, beispielsweise Methylacrylat, Methylmethacrylat, Ethylacrylat und Ethylmeth- acrylat, Acrylamid und Methacrylamid sowie Acrylnitril und Meth- acrylnitril. Schließlich sind auch ethylenisch ungesättigte
C3-C6-Carbonsäuren geeignet, beispielsweise Acrylsäure und Meth- acrylsäure.
Weitere geeignete Carbonsäureester leiten sich von Glykolen oder bzw. Polyalkylenglykolen ab, wobei jeweils nur eine OH-Gruppe vereεtert ist, z.B. Hydroxyethylacrylat, Hydroxyethylmethacrylat, Hydroxypropylacrylat, Hydroxy-butylacrylat, Hydroxypropylmeth-
acrylat, Hydroxybutylmethacrylat sowie Acrylsäuremonoester von Polyalkylenglykolen einer Molmasse von 500 bis 10.000. Weitere geeignete Comonomere sind Ester von ethylenisch ungesättigten Carbonsäuren mit Aminoalkoholen wie beispielsweise Dimethylamino- ethylacrylat, Dimethylamino-methylmethacrylat, Diethylaminoethyl- acrylat, Diethylaminoethylmethacrylat, Dimethyl-aminopropylacry- lat, Dimethylaminopropylmethacrylat, Diethylaminopropylacrylat, Dimethylaminobutylacrylat und Diethylaminobutylacrylat . Die basischen Acrylate können in Form der freien Basen, der Salze mit Mi- neralsäuren wie Salzsäure, Schwefelsäure oder Salpetersäure, der Salze mit organischen Säuren wie Ameisensäure, Esεigεäure, Propionεäure oder der Sulfonsäuren oder in quaternierter Form eingesetzt werden. Geeignete Quaternierungsmittel εind beispielsweise Dimethylsulfat, Diethylεulfat, Methylchlorid, Ethylchlorid oder Benzylchlorid.
Weitere geeignete Comonomere sind Amide ethylenisch ungesättigter Carbonsäuren wie Acrylamid, Methacrylamid sowie N-Alkylmono- und Diamide von monoethylenisch ungesättigten Carbonsäuren mit Alkyl- resten von 1 bis 6 C-Atomen, z.B. N-Methylacrylamid, N,N-Dimethy- lacrylamid, N-Methylmethacrylamid, N-Ethylacrylamid, N-Propyla- crylamid und tert . Butylacrylamid sowie basische (Meth)acryl- amide, wie z.B. Dimethylaminoethylacrylamid, Dimethylaminoethyl- ethacrylamid, Diethylaminoethyl-acrylamid, Diethylaminoethyl- methacrylamid, Dimethylaminopropylacrylamid, Diethylaminopropyl- methacrylamid, Dimethylaminopropylmethacrylamid und Diethyl-ami- nopropylmethacrylamid.
Weiterhin sind als Comonomere geeignete N-Vinylpyrrolidon, N-Vi- nylcaprolactam, Acrylnitril, Methacrylnitril, N-Vinylimidazol sowie substituierte N-Vinylimidazole wie z.B. N-Vinyl-2-methyl-imi- dazol, N-Vinyl-4-methylimidazol, N-Vinyl-5-methylimidazol, N-Vi- nyl-2-ethylimidazol und N-Vinylimidazoline wie N-Vinylimidazolin, N-Vinyl-2-methylimidazolin und N-Vinyl-2-ethylimidazolin. N-Viny- limidazole und N-Vinylimidazoline werden außer in Form der freien Basen auch in mit Mineralεäuren oder organischen Säuren neutralisierter oder in quaternisierter Form eingesetzt, wobei die Quaternisierung vorzugεweise mit Dirnethylsulfat, Diethylsulfat, Methylchlorid oder Benzylchlorid vorgenommen wird. In Frage ko - en auch Diallyldialkyl-ammoniumhalogenide wie z.B. Diallyldi- methylammoniumchlorid.
Schließlich sind auch Vinylformamid, Styrol, Butadien, Malein- säureanhydrid (MSA) , Fumarsäure und Maleinsäure geeignet.
Die Hydrolyεe kann in Löεung oder in Substanz durchgeführt werden, wobei dem Fachmann bekannte Bedingungen und Reagenzien eingesetzt werden. Vorzugsweise wird diese Hydrolyse mit starken Basen durchgeführt. Geeignet εind die Alkoholate der bekannten nieder und höheren Alkohole, beispielsweise Methanolate oder Ethanolate. Besonders geeignete Basen umfasεen NaOH, KOH, LiOH, CaOH und Mischungen davon. Insbesondere ist als Base ein eutekti- sches Gemisch oder ein nahe dem Eutektikum liegendes Gemisch von NaOH und KOH geeignet.
Die Copolymerisate enthalten beispielsweise
99 bis 1 mol-%, vorzugsweise 90 bis 10 mol-% Methylenamin- Einheiten - 1 bis 99 mol-%, vorzugsweise 10 bis 90 mol-% andere, einpoly- meriεierte Einheiten
in einpolymerisierter Form.
Poly(1, 3-diacyl-4-imidazolin-2- (thi) on) wird durch radikalische oder ionische, vorzugsweise radikalische Polymerisation aus 1, 3-Diacyl-4-imidazolin-2-on bzw. -thion hergestellt. Die Polymerisation kann dabei in Substanz, in Lösung, Suspension, Emulsion oder Dispersion durchgeführt werden. Vorzugsweise wird sie in Substanz durchgeführt. Es werden die üblichen, dem Fachmann bekannten Radikalstarter eingesetzt.
Als Initiatoren kommen grundsätzlich alle die Verbindungen in Betracht, durch die eine radikalische Polymerisation ethylenisch ungesättigter Monomere erreicht werden kann. Beispiele hierfür sind Azoverbindungen, organische oder anorganische Peroxide, Salze der Peroxodischwefelsäure und Redox-Initiatorsysteme. In der Regel setzt man im erfindungsgemäßen Verfahren den Initiator in einer Menge von 0,1 bis 5 Gew.-%, bezogen auf die Menge der zu polymerisierenden Monomere ein. Die Menge an eingesetztem Initiator richtet sich dabei in bekannter Weise nach der Wirksamkeit, mit der die radikalische Polymerisation der Monomere ausgelöst wird. Bei der Polymerisation in einer Wasser-in-Öl-Emulsion wird man häufig geringe Initiatormengen einsetzen, insbesondere wenn man hydrophobe Initiatoren verwendet. Selbstverständlich kann die Polymerisation auch durch aktinische Strahlung, Mikrowellenstrahlung oder α-, ß- oder γ-Strahlung ausgelöst werden.
Beispiele für Azo-Initiatoren umfasεen 2 , 2 '-Azobisisobutyro- nitril, 2 , 2 '-Azobis (2 , 4-dimethylvaleronitril) , 2,2'-Azo- bis (2-amidinopropan) dihydrochlorid (V50) und 2,2'-Azo- bis (2- (2-rmidazolin-2-yl)propan) dihydrochlorid. Beispiele für an-
organische Peroxide sind Wasserstoffperoxid und Percarbonate, Beispiele für organische Peroxide umfassen Alkylhydroperoxide wie tert .-Butylhydroperoxid, Cumolhydroperoxid, weiterhin Peroxycar- bonsäureester wie tert . -Butylperoctoat und Diacylperoxide wie Dibenzoylperoxid. Geeignete Salze der Peroxodischwefelsaure sind inεbesondere die Natrium-, Kalium- und Ammoniumsalze. Beispiele für RedoxinitiatorSysteme umfassen Systeme, die eines der obengenannten organischen oder anorganischen Peroxide, insbesondere Wasεerstoffperoxid, als Oxidationskomponente und wenigstens eine weitere Reduktionskomponente wie Ascorbinsäure, Hydroxylamin oder Addukte der schwefligen Säure an Aldehyde, z.B. das Biεulfitad- dukt an Aceton oder Hydroxymethanεulfinεäure-Natriumεalz als Reduktionsmittel enthalten. Dabei können sowohl die vorgenannten Peroxide alε auch die Redoxinitiatorsysteme in Gegenwart redoxak- tiver Übergangsmetalle wie Eisen, Vanadium oder Kupfer, vorzugsweise in Form wasεerlöslicher Salze, eingesetzt werden.
Beispiele für weitere Initiatorsyεteme umfaεsen Wasserstoffperoxid, tert . -Butylperoxid und salzartige Azoverbindungen, z. B. die vorgenannten Hydrochloride. Beispiele sind Bisazo (alkylni- trile) wie 2 , 2 '-Azobis (valeronitril) , 2 , 2-Azobis (2 , 4-dimethyl- valeronitril) , 2, 2 '-Azobis (4-methoxy-2-4-dimethylvaleronitril) , 2, 2 '-Azobis (isobutyronitril) ; Alkyl- und Cycloalkylpercarbonate wie Dicyclohexylperoxodicarbonat, Di-2-Ethylhexylperoxidcarbonat, tert . -Butylperoxoisopropylcarbonat; Diacylperoxide wie Acetylpe- roxid, Dioctanoylperoxid, Dilauroylperoxid, Dibenzoylperoxid; Peroxyester, insbesondere tert .-Butylperoxycarbonsäureester wie tert . -Butylperpivalat, -per-2-ethylhexanoat, -perneodecanoat; Hydroperoxide wie Cumolhydroperoxid, p-Menthanhydroperoxid, Pinanhydroperoxid und tert . -Butylhydroperoxid und Peroxide wie Dicumolperoxid, Di-tert .-butylperoxid und Di-tert. -amylperoxid.
Bei der radikalischen Polymerisation werden in der Regel Kettenübertragungsmittel (Regler) eingesetzt. Die eingesetzte Menge an Kettenübertragungsmittel hängt naturgemäß von der Wirksamkeit des Kettenübertragungsmittels ab und liegt in der Regel im Bereich von 0,0001 bis 5 Gew.-%, bezogen auf die Gesamtmonomerenmenge . Beiεpiele für Kettenübertragungsmittel sind: Aliphatische Mercaptane wie C6-Cι-Alkylmercaptane, wie n-Hexylmercaptan; n-Oc- tylmercaptan, tert .-Octylmercaptan, 2-Ethylhexylmercaptan, n-De- cylmercaptan, 2-Propylheptylmercaptan, n-Dodecylmercaptan und tert .-Dodecylmercaptan; wasserlösliche Mercaptane wie 2-Mercapto- ethanol, Mercaptopropanol , Mercaptobutanol , Mercaptopropionεäure und Mercaptoeεεigsäure, weiterhin Ameisensäure, Isopropanol, Allylalkohole, Aldehyde wie Butyraldehyd, Halogenkohlenwasεer-
εtoffe wie Chloroform, Bromoform, Tetrachlorkohlenstoff und dergleichen.
Die Polymerisationstemperatur richtet sich in der Regel nach dem eingesetzten Initiatorsyεtem und liegt häufig im Bereich von 20 bis 160°C und insbesondere im Bereich von 130 bis 150 °C.
Die Polymerisationsdauer liegt in der Regel im Bereich von 1 min bis 24 h.
Der pH-Wert des Reaktionsmediums liegt vorzugsweise im Bereich von 3 bis 10.
Die Umsetzung kann als Batch-Verfahren erfolgen. Dabei werden die zu polymerisierenden Monomere in dem wässrigen Reaktionsmediu vorgelegt und der Polymerisationsinitiator unter Polymerisationsbedingungen, vorzugsweise abhängig von dessen Verbrauch, zugeführt oder im Polymerisationsgefäß zusammen mit den Monomeren vorgelegt und anschließend auf Polymerisationstemperatur erwärmt.
Vorzugsweise wird tert-Butylhydroperoxid als Radikalstarter eingesetzt.
Poly (1, 3-diacyl-4-imidazolin-2- (thi) on) -Polymere sind ein weite- rer Gegenstand der vorliegenden Erfindung.
Poly (1, 3-diacetyl-4-imidazolin-2-on) mit einem Polymerisationsgrad n von 3 bis 4 wird beschrieben von G. 0. Schenck in Chem. Ber. 100, Seiten 369 bis 3978 (1967) .
1, 3-Diacyl-4-imidazolin-2-one und -thione sind durch konventionelle, dem Fachmann bekannte Acylierung von 4-Imidazolin-2-on bzw. -2-thion erhältlich.
Poly (4-Imidazolin-2-on) , das durch Teilhydrolyse von
Poly (1, 3-diacyl-4-imidazolin-2-on) erhältlich ist, ist ebenfalls ein Gegenstand der vorliegenden Erfindung.
Ein weiterer Gegenstand der vorliegenden Erfindung ist die Dar- Stellung von PMAm durch Polymerisation von 1, 2-Diaminoethen-Deri- vaten der nachfolgenden Formel IV, die aus Imidazol erhältlich sind.
In der Formel IV sind R1 und R2 unabhängig voneinander ein Rest der Formel R5, OR6 oder NR7R8, wobei R5, R6, R7 und R8 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, linearen und verzweigten Cχ-C4o-Alkylgruppen, die gegebenenfalls einen oder mehrere Substituenten aus der Gruppe der gegebenenfalls alkylsubstituierten Cε-Aromaten aufweisen, und Cg-Aromaten, die gegebenenfalls einen oder mehrere Substituenten aus der Gruppe bestehend aus Cι-C4o-Alkylgruppen aufweisen; R3 und R4 sind unabhängig voneinander ausgewählt aus der Gruppe bestehend aus Wasserstoff, Cι-C4o-Alkylgruppen und Aromaten.
Die Diaminoethen-Derivate der Formel (IV) können in der eis- oder trans-Form vorliegen bzw. in die Polymerisationsreaktion eingesetzt werden.
Durch Polymerisation sind aus den Monomeren der Formel IV substituierte Polymethylenamine erhältlich, bei denen ein Wasserstoff der Aminogruppe durch eine -C (0) R-Gruppe, wie in Formel IV dargestellt, ersetzt ist. Die Polymerisation wird kationisch oder radikalisch durchgeführt. Vorzugsweise wird die Polymerisation kationisch durchgeführt, wobei die üblichen Lewis-Säuren benutzt werden können.
Selbstverständlich kann die Polymerisation auch durch aktinische Strahlung, Mikrowellenstrahlung oder α-, ß- oder γ-Strahlung ausgelöst werden.
Die Polymerisationstemperatur richtet sich in der Regel nach dem eingesetzten Initiatorsystem und liegt häufig im Bereich von -10 bis 110°C und insbesondere im Bereich von 0 bis 40°C.
Die Polymerisationsdauer liegt in der Regel im Bereich von 1 bis 10 h, können jedoch in einzelnen Fällen auch über oder unter diesen Werten liegen.
Der pH-Wert des Reaktionsmediums liegt vorzugsweise im Bereich von 3 bis 10.
Die Umsetzung kann als Batch-Verfahren erfolgen. Dabei werden die zu polymerisierenden Monomere in dem Reaktionsmedium vorgelegt und der Polymerisationsinitiator unter Polymerisationsbedingungen, vorzugsweise abhängig von dessen Verbrauch, zugeführt oder im Polymerisationsgefäß zusammen mit den Monomeren vorgelegt und anschließend auf Polymerisatiostemperatur erwärmt.
Zur Polymerisation werden vorzugsweise Lewis-Säuren, beispielsweise BF3-Diethylether, SnCl4, TiCl3, oder C0 , eingesetzt. Vorzugsweise wird die Polymerisation kationisch durchgeführt, wobei in diesem Fall vorteilhafte Resultate mit BF3/Diethylether als Lewis-Säure erzielt wurden.
Die besten Resultate wurden bei Einsatz von Monomeren erhalten, bei denen R1 und R2 gleich OR6 mit R6 gleich Ci-Cε-Alkyl, Benzyl oder Phenyl sind. R3 und R4 sind vorzugsweise Wasserstoff.
Die kationische Polymerisation der Verbindungen der Formel (IV) wird vorzugsweise in homogener Lösung in einem organischen Löse- mittel durchgeführt. Es kommen diejenigen Lösemittel in Betracht, die die Verbindungen der Formel (IV) lösen und dem Fachmann bekannt sind, vorzugsweise Dimethylsulfoxid (DMSO) , N-Methylpyrro- lidon (NMP) , Dimethylformamid (DMF) , Formamid, oder Hexamethyl- phosphortriamid (HMPTA) . Besonders geeignet ist DMSO. Auf diese Weise sind Oligomere und Polymere zugänglich.
Die entstandenen Polymere, die aus den Moleküleinheiten mit der Formel IV aufgebaut sind, werden zur Herstellung von PMAm hydrolysiert, auf dem Fachmann bekannte Weise.
Ein Teil der substituierten 1, 2-Diaminoethene der allgemeinen Formel IV ist erhältlich durch eine sogenannte Bamberger-Spaltung (E. Bamberger, Ann. 273, 267 (1893)) von Imidazol. Die Reaktion ist in der nachfolgenden Gleichung (2) wiedergegeben, wobei X für Cl, Br oder I, vorzugsweise Cl oder Br, steht.
(V) (VI) Gleichung 2
Die erhaltenen Verbindungen können in einer dem Fachmann bekannten Reaktion umgesetzt und N-alkyliert oder -aryliert werden, um zu den Verbindungen der Formel (IV) zu gelangen. Der Rest R* in der CarbonylVerbindung (VI) entspricht dabei den Resten R1 und R2 in der Formel (IV) . Die Reaktion verläuft über das mono- und diacylierte Imidazol, das unter Hydrolyse und Ringöffnung zu den Dia inoethen-Derivaten der Formel (IVa) reagiert. Bei der Umsetzung werden die üblichen, dem Fachmann bekannten Bedingungen eingehalten. Vorzugsweise wird die Reaktion in einem neutralen Reak- tionsmedium durchgeführt, da hier die besten Resultate erhalten wurden.
Selbstverständlich können auch 1, 2-Diaminoethene eingesetzt werden, die nicht aus Imidazol erhalten werden.
Die erfindungsgemäßen Polymere sind insbesondere als Ent- wässerungs-, Flockungs- und Retentionsmittel sowie als Fixiermittel bei der Herstellung von Papier geeignet. Zu diesem Zweck werden sie dem jeweiligen Papierstoff in einer Menge von 0,01 bis 2 und vorzugsweise in einer Menge von 0,01 bis 0,5 Gew.-%, z.B.
0,01 bis 0,1 Gew.-% zugesetzt, jeweils bezogen auf die im Papierstoff enthaltenen Feststoffe.
Die erfindungsgemäßen Polymere führen insbesondere zu einer ver- besserten Retention von Feinstoffen gegenüber den Polymeren des Standes der Technik. Besonders vorteilhafte Effekte weisen sie zur Retention von Füllstoffen wie Calciumcarbonat auf. Die Polymere zeichnen sich insbesondere durch gute Fixiereigenschaften aus. Insbesondere zur Fixierung von Störstoffen, gestrichenem Ausschuss und Stickies sind die erfindungsgemässen Polymere geeignet.
Die erfindungsgemäßen Polymere können zur Herstellung sämtlicher Papier-, Pappe- und Kartonqualitäten eingesetzt werden, beispielsweise für die Herstellung von Papieren für den Zeitungsdruck (Hochdruck/Offset-Druck) , sogenannte mittelfeine Schreibund Druckpapiere, Naturtiefdruckpapiere und auch leichtgewichtige Streichrohpapiere. Zur Herstellung der genannten Papiere verwendet man als Rohstoffko ponente Holzschliff, thermomechani- sehen Stoff (TMP) , chemo-thermomechanischen Stoff (CTMP) , Druckschliff (PGW) , sowie Sulfit- und Sulfatzellstoff. Die genannten Stoffe können sowohl kurz- als auch langfasrig sein. Als Rohstoff für die Herstellung des Papierstoffs kommen auch Altpapier, Zellstoffe und Holzschliff in Betracht, die in den sogenannten inte- grierten Fabriken in mehr oder weniger feuchter Form direkt ohne vorherige Eindickung oder Trocknung weiter zu Papier verarbeitet werden. In diesen Stoffen sind zum Teil noch Reste von Verunrei-
nigungen enthalten, die beim Aufschluss entstehen und die den üblichen Papierherstellprozess stark stören. Diese Verunreinigungen werden durch die erfindungsgemäßen Polymere auf dem Papier in besonders guter Weise fixiert.
Mit Hilfe der erfindungsgemäßen Polymere können sowohl Füllstofffreie als auch Füllstoff-haltige Papiere hergestellt werden. Der Füllstoffgehalt Füllstoff-haltiger Papiere kann bis maximal 40 Gew.-% betragen und liegt vorzugsweise im Bereich von 5 bis 30 Gew.-%. Geeignete Füllstoffe sind beispielsweise Clay, Kaolin, Kreide, Talkum, Titandioxid, Calciumsulfat, Bariumsulfat, Aluminiumoxid, Satinweiß oder Mischungen der vorgenannten Füllstoffe.
Insbesondere kommt die gute Fixierwirkung bei der Herstellung von Pappe, Karton, Zeitungspapier aus Altpapier und solchen StoffSystemen in Betracht, die Störstoffe enthalten und die sich in den zumindest teilweise geschlossenen Wasserkreisläufen von Papiermaschinen anreichern.
Beispiele für den Einsatz der erfindungsgemässen Polymethylena- mine bzw der Copoly eren sind die Verwendung als Retentionsmittel bei der Herstellung von Papier, Pappe und Karton, als Fixiermittel zum Unschädlichmachen von Störstoffen bei der Papierherstel- lung, als Fixiermittel für anionische stabilisierte Leimungsmittel, als Fixiermittel für die Fixierung von anionischen Farbstoffen, als Primär für die Haftung auf anionischen Oberflächen, als Hilfsmittel bei der Ölförderung, als Textil- und Lederhilfsmittel, als Bestandteil in Waschmitteln, als Emulgator für Emul- sionen/Dispersionen, als Vernetzer bei der PUR-Herstellung, als Fänger von Formaldehyd und höheren Aldehyden, als Bindemittel für Faserplatten, als Hilfsmittel für die Schlammentwässerung, sowie als Vektor in der Krebstherapie.
Die Herstellung der erfindungsgemässen Polymere wird nun in den nachfolgenden Beispielen erläutert.
Beispiele
A Synthese von Polymethylenamin ausgehend von Poly ( 1 , 3-Diacetyl-4-Imidazolin-2-on)
1. Synthese von 4-Imidazolin-2-on
In einem 2 1 Vierhalsrundkolben mit Destillationsaufsatz, Tropf- trichter und Thermometer werden 285 g ( 1,63 ol ) alpha-Ureidoa- cetaldehyddiethylacetal bei Raumtemperatur (RT) in 1245 ml voll-
entsalztem Wasser aufgeschlämmt vorgelegt. Mit 14 g Ameisensäure 98-100% stellt sich der pH-Wert auf 2,45 ein. Die Reaktionsmischung wird auf 55°C aufgeheizt. Es wird Wasserstrahlvakuum von 350 bis 400 mbar angelegt, um das entstandene Ethanol abzu- destillieren. Nach 1 Stunde bei 55°C wird das Vakuum abgeschaltet und die Reaktionsmischung auf 0 bis 5°C abgekühlt. Die ausgefallenen Kristalle werden abgesaugt und im Vakuumtrockenschrank bei 40 bis 50°C und 200 mbar getrocknet (62,2 g sauberes Produkt) . Aus der Mutterlauge konnten noch 42,4 g Produkt isoliert werden.
Ausbeute gesamt: 104.6 g = 76.5% d. Theorie
FP: 242 bis 244°C (Zers. rötlich braune Färbung)
C,H,N-Analyse: Theorie gefunden
C 42 , 8 42 , 3
H 4 , 76 4 , 9
N 33 , 33 33 , 0
iH-NMR (DMSO-D6 , TMS , 400 MHz ) : 6 , 25 ( s , 2 H ; HC = CH) ; 9 ,
( s , 2 H; HN-CO-NH) .
IR-Spektrum: 1652 cm"1 (s) ; 1399 (m) ; 1073 (m) ; 697 (s)
2. Synthese von 1, 3-Diacetyl-4-imidazolin-2-on
In einem 1000 ml Vierhalskolben mit Kühler und Thermometer werden 31,7 g (127 Mol) 4-Imidazolin-2-on, 307,3 g (vierfacher molarer Uberschuss) Acetanhydrid und 1,5 g 4-Dimethylaminopyridin als Katalysator zugegeben. Die Lösung wird vorsichtig zum Rückfluss bei zunächst 106, dann 116 und schließlich 124°C gebracht. Die Reaktion ist nach 5 Stunden beendet. Beim Abkühlen auf RT fallen farblose Kristalle aus. Fp. 102°C (aus Ethylacetat bzw. Cyclohexan) . Gesamtausbeute 51 g entspricht ca. 80% d. Theorie.
H-NMR (CDC13, TMS, 400 MHz): 2,63 (s, 6H, C0-CH3) ; 7,05 (s, 2H, - HC=CH -) .
"C-NMR (CDCI3, TMS, 100 MHz) : 24,1 (-CH3) ; 109,5 (=CH-) ; 149,5 (-HN-CO-NH-) ; 167,7 (-CO-CH3)
3. Substanz-Polymerisation von 1, 3-Diacetyl-4-imidazolin-2-on
In einem 50 ml Rundkolben werden unter N2-Spülung 10 g 1,3
Diacetyl-4-imidazolin-2-on vorgelegt und dann die Innentemperatur auf 140°C gebracht. Hierbei entsteht eine klare Schmelze des
1, 3-Diacetyl-4-imidazolin-2-on. Man gibt zunächst 10 Tropfen einer 70%igen wässrigen Lösung von tert . -Butyl-Hydroperoxid und dann im Abstand von 5 Minuten jeweils 10 weitere Tropfen zu bis zu einer Gesamtmenge von 40 Tropfen. Drei Minuten nach der Zugabe der ersten 10 Tropfen reagiert das Monomer zu einem festen Polymer-Block. Man hält die Temperatur noch bei 140°C während 1 Stunde. Nach dem Abkühlen auf Raumtemperatur isoliert man 9,5 g eines farblosen spröden Polymers, welches sich leicht pulverisieren lässt. Das Polymer löst sich in THF und Chloroform, es ist unlöslich in Wasser.
Die C,H,N-Analyse ergab:
iH- MR (CDC13, TMS, 400 MHz): 2,43 (br. S, 6H, (-CO-CH3) ) ; 3,85 • 4,65 (br. , 2H, -CH-CH-)
13C-NMR (CDCI3, TMS, 100 MHz): 23,6 (br. q, CH3) ; 54,8 (br. d, -CH-) ; 149,9 (br. s, N-CO-N) ; 171,2 (br. s, -CO-CH3)
Nach der Maldi-Analyse weist das Polymer Wiederholungseinheiten aus 1, 3-Diacetyl-4-imidazolin-2-on auf.
4. Hydrolyse des Polymers in einer NaOH/KOH Schmelze
In 13 g einer eutektischen Mischung von 5,4 g NaOH und 7,-6 g KOH werden bei 200°C 1 g des Polymers eingetragen und 1 1/2 Stunden bei dieser Temperatur gehalten. Die Schmelze verfärbt sich braun und wird nach kurzer Zeit dünnflüssiger. Anschließend gibt man die Schmelze auf 110 g Wasser, wobei sich ein pH von > 14 einstellt. Mit 3,22 g HCl konz. wird ein pH von 7,6 eingestellt. Die Probe wird filtriert. Mit Hilfe einer enzymatischen Bestimmungs- methode wurde der Essigsäuregehalt der Lösung auf 16,5 mMol/lOOg bestimmt, was einer Ausbeute von 48% entspricht.
Der Erweichungspunkt des Polymers war > 260°C, die Ladungsdichte des Polymers wurde zu 1,94 mMol/g bestimmt. Mit statischer Lichtstreuung wurde eine Molmasse von 300.000 D bestimmt.
B Synthese von Polymethylenamin ausgehend von 1, 2-Diaminoethe- nen
B.1 Synthesen von Diaminoethenen
1) N,N'-Dibenzoyl-l, 2-diaminoethen
In einem 0,25 1 Vierhaiskolben mit Intensivkühler, Thermometer , Tropftrichter, Rührer, Mehrfachaufsatz und pH-Elektrode werden 216 g Wasser vorgelegt und 6.8 g Imidazol (0,1 Mol) darin gelöst. Bei einer Temperatur von 20-25°C stellt sich ein pH-Wert von 9.6 ein. Unter leichter Kühlung werden dann bei 20-25°C 28,1 g (0,2 Mol) Benzoylchlorid zugetropft (leicht exotherme Reaktion). Die Reaktionslösung verfärbt sich gelblich und es fällt ein weißer Feststoff aus. Der pH-Wert sinkt langsam unter 8 ab. Innerhalb von 60 Minuten wurden weitere 7.0 g Benzoylchlorid (0.05 mol) unter Einhaltung eines pH-Wertes von 7-8 zugetropft. Hierzu wurden 25.2 g NaOH 50 %ig benötigt. Nach weiteren 4 Stunden Reaktionszeit bei 20-25 °C, wobei der pH-Wert bei 7.5 1 Stunde konstant blieb, wurde die Temperatur auf 40-45°C erhöht, wobei dieser von 7 auf 7.25 hochgestellt wurde und dort dann 1 Stunde stehen blieb.
Zur pH-Einstellung wurden weitere 7.2 g NaOH 50 %ig benötigt.
Die Reaktionsmischung wurde dann über eine Glasfilternutsche G 3 abgesaugt. Der Rückstand bei 50-60°C /13 mbar am Rotationsverdamp- fer 3 Stunden getrocknet. Ausbeute: 10 g.
Das Filtrat, welches laut HPLC noch nicht vollständig umgesetztes Monoaddukt enthielt, wurde bei 20-25°C langsam unter Einhaltung eines pH-Wertes von 7-8 in 2.75 Stunden mit weiteren 7,0 g Benzoylchlorid (0,05 Mol) umgesetzt. Nachreaktionszeit 1 Stunde bei 20-25°C. Zur pH-Einstellung wurden 3.4 g NaOH 50 %ig benötigt. Dann wurde die Reaktionsmischung über' eine Glasfilternutsche G 3 abgesaugt. Der Rückstand bei 50-60°C/17 mbar am Rotationsverdampfer 3 Stunden getrocknet. Ausbeute: 3.4 g (laut HPLC enthielt der Feststoff noch 35 % Wertprodukt) = 1.2 g. Das Filtrat wurde nochmals langsam bei 20-25°C unter Einhaltung des pH-Wertes 7-8 in 2 Stunden mit 3,5 g Benzoylchlorid (0,025 Mol) umgesetzt. Nachreaktionszeit 1 Stunde bei 20-25°C. Zur pH-Einstellung wurden 3.7 g NaOH 50 % ig benötigt. Der Rückstand wurde über eine Glas- filternutsche G 3 abgesaugt und bei 40-50°C/15 mbar am Rotationsverdampfer 3 Stunden getrocknet. Ausbeute: l g 66.4 % ig = 0.7 g.
Ausbeute aus drei Fraktionen: 11.5 g entsprechen 43.2 % der Theo- rie
Die C,H,N-Analyse einer zweimal aus Ethanol umkristallisierten
Probe (Fp. 206-208°C) lieferte:
Theoretisch Gefunden
5
C 72 . 18 % 72 . 2 % H 5 . 26 % 5 . 2 % N 10 . 52 % 10 . 5 %
10 l3C-NMR (100 MHz, DMS0-d6) : 106, 7/d; 127, 9/d; 128,45/d; 131, 7/d; 133, 8/s; 163,87/s ; vermutlich nur geringe Mengen an trans-Iso- mer .
2) N,N%-Ethoxycarbonyl-l, 2-diaminoethen
15
In einem 21 Vierhalskolben mit Intensivkühler, Thermometer , Tropftrichter, Rührer, Mehrfachaufsatz und pH-Elektrode wurden 1306.8 g Wasser vorgelegt und 41.14 g (0,605 Mol) Imidazol darin gelöst. Bei einer Temperatur von 20-25°C stellt sich ein pH-Wert
20 von 9.8 ein. Unter leichter Kühlung werden dann bei 20-25°C 131.3 g (1,21 Mol) Ethylchlorformiat zugetropft (leicht exotherme Reaktion) . Der pH-Wert fällt langsam und wird durch Zutropfen von 147,2 g NaOH 50%ig bei 8-9 gehalten. Die Reaktionslösung wird trübe und es fällt ein weißer Feststoff aus . Nach 1 Stunde und
25 10 Minuten war das Ethylchlorformiat zugetropft. Nach weiteren 30 Minuten wurde der Versuch abgestellt. Das Produkt wurde über eine Glasfilternutsche G 3 abgesaugt und drei mal mit kaltem Wasser gewaschen, trockengesaugt, und dann am Rotationsverdampfer 2 Stunden bei 40-45°C/15 mbar getrocknet. Der Feststoff wurde aus
30 Ethanol umkristallisiert und nochmals 2 Stunden bei 40-45°C/<15 mbar getrocknet .
Ausbeute: 74.2 g entsprechen 60.6 % der Theorie.
35 Die C,H,N-Analyse einer aus Ethanol umkristallisierten Probe (Fp: 136-138°C ) lieferte:
.eo. retisch Gefunde sn
40 C 47 , . 50 % 47 . 7 %
H 6 , . 93 % 7 . 0 % N 13 , . 86 % 13 . 8 % 0 31 , . 68 % 31 . 9 %
45
13C-NMR (100 MHz, DMSO-d6) : 14,38/t; 60,46/d (Hauptkomponente);
60,85/d (Nebenkomponente); 104,68/d (Nebenkomponente); 105,31/d
(Hauptkomponente); 152,54 (Nebenkomponente);
5 Es können nur Spuren des trans-Isomeren nachgewiesen werden.
3 ) N,N* -Methoxycarbonyl-1 , 2-diaminoethen
In einem 21 Vierhalskolben mit Intensivkühler, Thermometer, 10 Tropftrichter, Rührer, Mehrfachaufsatz und pH-Elektrode wurden 1306.8 g Wasser vorgelegt und 41.14 g (0,605 Mol) Imidazol darin gelöst. Bei einer Temperatur von 20-25°C stellt sich ein pH-Wert von 10,3 ein. Unter Eis-Kühlung werden dann bei 20-25°C 114,4 g (1,21 Mol) Methylchlorformiat zugetropft (leicht exotherme Reak- 15 tion) . Der pH-Wert fällt langsam und wird durch Zutropfen von 147,2 g NaOH 50%ig bei 8-9 gehalten. Die Reaktionslösung wird trübe und es fällt ein weißer Feststoff aus . Nach 1 Stunde war das Methylchlorformiat zugetropft. Nach weiteren 3 Stunden wurde die Temperatur auf 65°C angehoben und 4 Stunden gehalten. Das 20 Produkt wurde über eine Glasfilternutsche G 3 abgesaugt und dreimal mit jeweils 1 1 kaltem Wasser gewaschen. Die Rohausbeute betrug 55,1 g feucht. Die kristalline Masse wurde dann 2 Stunden bei 40-45°C/15 mbar getrocknet. Ausbeute: 30,1g entsprechen 28.6 % der Theorie.
25
Die C,H,N-Analyse einer aus Ethanol umkristallisierten Probe (Fp: 158-160°C) lieferte:
Theoretisch Gefunden
30 c 47 . 3 % 47 . 1 %
H 5 , 8 % 6 . 0 % N 16 , 1 % 16 . 1 %
35
13C-NMR (100 MHz, DMS0-d6) : 51,97/t (Haupt); 52,29/t (Neben); 104,87/d; 105,41/d (Haupt); 152,95/s (Neben); 153,72/s (Haupt)
Die Probe liegt als Gemisch der eis- und trans-Isomeren vor.
40
4) N,N -Benzyloxycarbonyl-l, 2-diaminoethen
In einem 0,5 1 Vierhalskolben mit Intensivkühler, Thermometer , Tropftrichter, Rührer, Mehrfachaufsatz und pH-Elektrode wurden 45 240 g einer 5 %igen NaOH vorgelegt und darin 6,8 g (0,1 Mol)
Imidazol gelöst. Bei einer Temperatur von 20-25°C stellt sich ein pH-Wert von 13.8 ein. Unter leichter Kühlung wird dann bei 20-25°C
34,1 g (0,2 Mol) Benzyloxychlorformiat zugetropft (leicht exotherme Reaktion) . Bei einem pH-Wert von 13.2 trübt sich die Reaktionslösung ein und ein Feststoff flockt aus. Nach ca. 1.5 Stunden bei 25-30°C ist das Chlorformiat zugetropft. Der pH-Wert ist auf 10.5 gefallen. Die Temperatur wird auf 30-35°C erhöht. Nach 45 Minuten bei dieser Temperatur steigert man die Temperatur nochmals auf 60-65°C. Nach einer weiteren Stunde bei 60-65°C fällt der pH-Wert auf 9.5 ab. Nach weiteren 20 Minuten wurde der Versuch abgestellt.
10
Der Ansatz wurde über eine Glasfilternutsche G 3 abgesaugt, die wässrige Phase wurde verworfen.
Die feste, knetartige Masse wird mit kaltem Wasser gewaschen und 15 anschließend in Ethanol gelöst mit Na S04 wasserfrei getrocknet und abfiltriert.
Am Rotationsverdampfer wird die Lösung bei 50-55°C/ 15 mbar einrotiert.
20
Ausbeute: 25.8 g entsprechen 79.1 % der Theorie.
Die C,H,N-Analyεe einer aus Cyclohexan umkristallisierten Probe 25 (Fp: 89-90°C) lieferte:
Theoretisch Gefunden
C 66.20 % 66.2 % 30 H 5.52 % 5.4 %
N 8.58 % 8.7 %
13C-NMR (100 MHz, DMS0-d6) : 66,01/t; 105, 5/d; 127, 8/d; 128, 0/d; 128, 3 /d; 136, 3 /s; 153, 02 /s geringste Mengen an trans-Isomer
35
5) N,Nλ-2-Ethylhexyloxycarbonyl-l, 2-diaminoethen
In einem 0,25 1 Vierhalskolben mit Intensivkühler, Thermometer , Tropftrichter, Rührer, Mehrfachaufsatz und pH-Elektrode wurden
40 158 ml m-Xylol, 3,4 g (0,05 Mol) Imidazol und 1,1 g Tetrabutyl- ammoniumhydroxid 40 % ig vorgelegt. Bei einer Temperatur von 20-25 °C stellt sich ein pH-Wert von > 13 ein. Unter leichter Kühlung werden dann bei 20-25°C 19,2 g '(0,1 Mol) 2-Ethylhexylchlor- formiat zugetropft (leicht exotherme Reaktion) . Der pH-Wert fällt
45 langsam, die Reaktionslösung trübt sich ein. Hat der pH-Wert 7-8 erreicht, wird dieser mit 50 % iger Natronlauge gehalten. Nach ca. 3.5 Stunden bei 20-25°C ist das Chlorformiat zugetropft. Die
Temperatur wird auf 30-35°C erhöht. Nach 3 Stunden bei dieser Temperatur steigert man diese nochmals auf 60-65°C. Nach weiteren 6,5 Stunden bei 60-65°C ist der pH-Wert bei 7,55 konstant. Zur pH- Einstellung wurden 10,8 g (0,14 Mol) Natronlauge 50 % ig benö- 5 tigt. Der Ansatz wurde über eine Glasfilternutsche G 4 abgesaugt. Der weiße Niederschlag wurde mit wenig m-Xylol gewaschen. Rückstand 3,1 g wasserlöslich. Im Scheidetrichter wurde die wässrige Phase abgetrennt. Die organische Phase wird mit 100 ml THF verdünnt und mit Natriumsulfat wasserfrei getrocknet, dann ab- 10 filtriert. Am Rotationsverdampfer wurde bei 50-55°C/ll mbar m-Xy- lol und THF abrotiert. Man erhielt 21,9 g eines gelblichen Öls enthaltend ca. 90 % Produkt.
13C-NMR (100 MHz, DMSO-d6) : 66,01/t; 105, 5/d; 127, 8/d; 128,0/d; 15 128, 3/d; 136, 3/s; 153,02/s; geringste Mengen an trans-Isomer
Ausbeute: 18,17 g entsprechen ca. 98 % der Theorie
6) N,Nx-MyriΞtyloxycarbonyl-l, 2-diaminoethen
20
In einem 0,25 1 Vierhalskolben mit Intensivkühler, Thermometer, Tropftrichter, Rührer, Mehrfachaufsatz und pH-Elektrode wurden 108 ml m-Xylol 3,4 g (0,05 Mol) Imidazol und 1,1 g Tetrabutyl- ammoniumhydroxid 40 % ig vorgelegt. Bei einer Temperatur von
25 20-25°C stellt sich ein pH-Wert von > 13 ein. Unter leichter Kühlung wird dann bei 20-25°C 27,6 g (0,1 Mol) Myristylchlorformiat zugetropft (leicht exotherme Reaktion) . Der pH-Wert fällt langsam, die Reaktionslösung trübt sich ein. Hat der pH-Wert 7-8 erreicht, wird dieser mit 50 % iger Natronlauge gehalten. Nach ca.
30 3 Stunden bei 20-25°C ist das Chlorformiat zugetropft. Die Temperatur wird 3 Stunden bei 20-25°C gehalten. Dann wird die Temperatur nochmals auf 40-45°C gesteigert. Nach einer weiteren Stunde bei 40-45°C ist der pH-Wert konstant bei 8,2. Zur pH-Einstellung wurden 11 g (0,14 Mol) Natronlauge 50 % ig benötigt. Zur
35 Entfernung des Natriumchlorids wurde der Ansatz über eine Glasfilternutsche G 4 abgesaugt. Dabei erhielt man eine wässrige, klare und eine organische, trübe Phase. Die wässrige Phase wurde in THF aufgenommen mit Natriumsulfat wasserfrei getrocknet, abfiltriert und am Rotationsverdampfer bei 30-35°C einrotiert. Es
40 ergab sich ein Rückstand von 10 g. Dieser wurde aus Ethanol und Cyclohexan umkristallisiert. Ausbeute 6,8 g.
Die organische Phase ist nicht ganz in THF löslich. Der nicht lösliche Teil wurde über eine Glasfilternutsche G 3 abgesaugt und 45 betrug 4,4 g (NaCl) . Die THF-Phase wurde mit Natriumsulfat wasserfrei getrocknet und am Rotationsverdampfer bei 35-40°C/ll
mbar einrotiert und bei 65-70°C/9 mbar am Rotationsverdampfer getrocknet. Die Ausbeute betrug 15 g.
Ausbeute: 21,8 g entsprechen 81 % der Theorie (MG 538) 5
Die C,H,N-Analyse einer aus Cyclohexan umkristallisierten Probe (Fp: 88-89 °C) lieferte:
Theoretisch Gefunden
10 c 71 , 30 % 71 , 5 %
H 11 , 50 % 11 , 3 % N 5 , 20 % 5 , 1 %
15 13C-NMR (100 MHz, CDC13) : 14,11/q; 22,7; 25,8; 28,9; 29,36; 29,39; 29,6; 29,65; 29,67; 31,96 (alle ts) ; 65,6 und 66,78 (beide ts) ; 104,9 und 107,2 (beide ds) ; 154,18 und 155,35 (beide Singlets) ; bei der Substanz handelt es sich um ein Gemisch aus eis- und trans-Isomer .
20
7) N,N -Cetyloxycarbonyl-l, 2-diaminoethen
In einem 0,25 1 Vierhalskolben mit Intensivkühler, Thermometer, Tropftrichter, Rührer, Mehrfachaufsatz und pH-Elektrode wurden
25 108 ml m-Xylol, 3,4 g (0,05 Mol) Imidazol und 1,1 g Tetrabutyl- ammoniumhydroxid 40 % ig vorgelegt. Bei einer Temperatur von 20-25°C stellt sich ein pH-Wert von >13 ein. Unter leichter Kühlung wird dann bei 20-25°C 30,4 g (0,1 Mol) Cetylchlorformiat zugetropft (leicht exotherme Reaktion) . Der pH-Wert fällt langsam
30 und die Reaktionslösung trübt sich ein. Hat der pH-Wert 7-8 erreicht, wird dieser mit 50 % iger Natronlauge gehalten. Nach ca. 3,5 Stunden bei 20-25°C ist das Chlorformiat zugetropft. Es wurden nochmals 50 ml m-Xylol zugetropft. Die Temperatur wurde auf 40-45°C erhöht. Nach weiteren 3 Stunden Rühren steigert man die
35 Temperatur nochmals auf 60-65°C. Nach 4 Stunden bei 60-65°C ist der pH-Wert bei 7,45 konstant. Zur pH-Einstellung wurden 10,85g (0,14Mol) Natronlauge benötigt.
Zur Entfernung des Natriumchlorids wurde der gesamte Ansatz in 40 ca. 600 ml THF aufgenommen, mit Natriumsulfat wasserfrei getrocknet und dann über eine Glasfilternutsche G 4 abgesaugt. Am Rota- tionsverdampfer wird die Lösung bei 60-65°C/ 13 mbar einrotiert. Die rohe Ausbeute betrug 31,5 g.
45
Es wurde aus n-Hexan umkristallisiert und am Rotationsverdampfer bei 50-55
°C/< 10 mbar getrocknet.
5 Ausbeute: 24,8 g entsprechen 83,5 % der Theorie
Die C,H,N-Analyse einer aus n-Hexan umkristallisierten Probe (Fp: 66-67°C) lieferte:
10 Theoretisch Gefunden
C 72,7 % 71,5 %
H 11,8 % 11,4 %
N 4,7 % 4,5 %
15 13C-NMR (100 MHz, DMS0-d6) : 66,01/t; 100, 5/d; 153, 2/s; die Substanz war laut 13C-NMR gering mit Cetylchlorformiat verunreinigt; geringste Mengen an trans-Isomer .
B.2 Radikalische Polymerisation mit Diaminoethenen 20
1. Radikalische Copolymerisation von N,Nv-Ethoxicarbo- nyl-1, 2-diaminoethen mit MSA
0,828 g N,Nx-Ethoxycarbonyl-l,2-diaminoethen und 0,172 g Malein- 25 säureanhydrid werden in 2,5 g NMP eingewogen, mit N begast und dann wird die Temperatur auf 140°C angehoben und 0,2 g tertiär- Butylhydroperoxid zugegeben. Die Reaktionsmischung wird darauf 30 min bei 140°C gehalten und dann auf RT abgekühlt.
30 Die Analyse des Reaktionsgemisches erfolgte mittels GPC (Laufmittel THF) und MALDI-TOF bis ca. 800 D.
2. Radikalische Polymerisation von N,Nλ-Ethoxicarbonyl-l, 2-dia- minoethen in Substanz
35
1 g N,Nx-Ethoxicarbonyl-l,2-diaminoethen wird bei 140°C geschmolzen und die Schmelze mit N begast und anschließend wird 0,2 g tertiär-Butyloxiacetat zugegeben. Die Reaktionsmischung wird 1 h bei dieser Temperatur gehalten und anschließend auf RT abge-
40 kühlt. Die Analyse des Reaktionsgemisches erfolgte mittels GPC (Laufmittel THF) .
Die Molmasse liegt bei ca. 1.000 D.
45 B.3. Kationische Polymerisation mit 1,2-Diaminoethenen
1. Kationische Oligomerisation von N,N'-Ethoxicarbonyl-l, 2-dia- minoethen
1 g einer 10%igen DMSO-Lösung von N,N -Ethoxicarbonyl-l, 2-diami- noethen wird unter Eiskühlung mit 0,3 ml BF3/EtOEt versetzt. Diese Mischung wird dann auf RT aufgewärmt und bei dieser Temperatur, maximal bis 50°C gehalten. Die Reaktionadauer beträgt mehrere Tage. Die Analyse des Reaktionsge isches erfolgt mittels GPC (Laufmittel=THF) und MALDI-TOF Spektroskopie. Die maximale Mol- asse der erhaltenen Oligomere beträgt ca. 1000 D.