WO2004085373A1 - 新規ショウガオール系化合物および当該化合物によるチロシナーゼ活性阻害剤 - Google Patents
新規ショウガオール系化合物および当該化合物によるチロシナーゼ活性阻害剤 Download PDFInfo
- Publication number
- WO2004085373A1 WO2004085373A1 PCT/JP2004/004316 JP2004004316W WO2004085373A1 WO 2004085373 A1 WO2004085373 A1 WO 2004085373A1 JP 2004004316 W JP2004004316 W JP 2004004316W WO 2004085373 A1 WO2004085373 A1 WO 2004085373A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- group
- choh
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/76—Unsaturated compounds containing keto groups
- C07C59/90—Unsaturated compounds containing keto groups containing singly bound oxygen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/14—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D317/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/16—Emollients or protectives, e.g. against radiation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/20—Unsaturated compounds containing keto groups bound to acyclic carbon atoms
- C07C49/255—Unsaturated compounds containing keto groups bound to acyclic carbon atoms containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/76—Unsaturated compounds containing keto groups
- C07C59/84—Unsaturated compounds containing keto groups containing six membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/38—Compounds containing oxirane rings with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D303/40—Compounds containing oxirane rings with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals by ester radicals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to a novel compound having a chemical structure similar to shogaol and gingerol, which is useful in the fields of foods, pharmaceuticals, quasi-drugs, cosmetics, and the like, and an intermediate for producing the same. Further, the present invention relates to a tyrosinase activity inhibitor, a hyaluronic acid degrading enzyme inhibitor or an antioxidant containing the novel compound as an active ingredient.
- Pigmentation due to spots, freckles, etc. occurs when melanin pigments are formed in epidermal pigment cells due to exposure to ultraviolet rays and abnormally deposit on the epidermis.
- the melanin pigment is synthesized by the metabolism of L-tyrosine, a kind of amino acid, to L-dopa and L-dopaquinone by the action of oxidase, tyrosinase, and through various processes. (For example, Fine Chemicals Magazine, March 15, 1990, “Special Feature Development and Product Development of Whitening Agents” and Frederance Journal, September 1997, September, “Special Features Recent Whitening Research and Development Trends in Drugs ”). Therefore, in order to prevent pigmentation due to spots, freckles, etc., it is important to inhibit the activity of tyrosinase, which plays an important role in the synthesis of melanin pigment.
- drugs such as placenta extract, vitamin C, a vitamin C derivative, kojic acid, and arbutin have been used to improve the prevention of spots and freckles.
- drugs such as placenta extract, vitamin C, a vitamin C derivative, kojic acid, and arbutin have been used to improve the prevention of spots and freckles.
- these have not achieved sufficient effects.
- hydroquinone is used for depigmentation of pigment spots, but its use is restricted due to safety issues.
- kojic acid may be carcinogenic, which has become a problem.
- shogaol and gingerol are components of the ginger extract, and include, for example, a blood circulation promoting action (Japanese Patent Application Laid-Open No. 6-189359) and a body odor suppressing effect (US Pat. 928), antioxidant effect (H.Kikuzaki, N.Nakatani, "Antioxidant Effects of Some Ginger Constituents", J. FoodScL, Vol58, No6, 1407-1410 (1993)), moisturizing effect (supervised by Masato Suzuki, It is known to have "New Cosmetic Functional Materials 300, Volume 1", pages 311-312, CMC Publishing, 2002).
- the present inventors studied a method capable of mass-producing shogaol, created an industrial production method for a specific shogaol, and filed a patent application for the specific shogaol and the production method (Japanese Patent Application No. 20-203). 0 3—3 2 7 5 7 4) and found that ginger has the property of inhibiting tyrosinase activity.
- shogaol obtained by this production method may be difficult to apply to humans because of insufficient water solubility, and a more water-soluble one is required.
- the present invention is a compound similar to shogaol and gingerol which is useful in the fields of foods, pharmaceuticals, quasi-drugs, cosmetics, etc., and has higher activity than conventional shogaol and gingerol in tyrosinase activity inhibition and the like. It is an object to provide a compound.
- the present inventors have conducted intensive studies to solve the above problems, and as a result, have found a novel compound having the same basic structure as that of gingerol and zingediol and having the same properties as known gingerol and gingerol. Furthermore, they have found that the compound works well as a tyrosinase activity inhibitor and the like, and completed the present invention.
- the present invention is a compound represented by the following general formula (1).
- R 1 is a protecting group for a hydrogen atom, a lower alkyl group or a phenolic hydroxyl group
- R 2 is a protecting group for a hydrogen atom or a phenolic hydroxyl group
- A is an alkyl group having 1 to 4 carbon atoms.
- B is an alkylene group having 1 to 12 carbon atoms
- the lower alkyl group in the formula (1) has 1 to 4 carbon atoms, and is preferably a methyl group.
- one CHOH-1,2-epoxy is represented by the following formula (3).
- the ketal derivative in the formula (1) is an acyclic ketal or a cyclic ketal, and is preferably a cyclic ketal.
- this cyclic ketal include those obtained from ethylene glycol, 1,3-propanediol or 2,2-dimethyl-1,3-propanediol with respect to the carbonyl group.
- the present invention relates to a tyrosinase inhibitor comprising a compound represented by the above formula (1), and a hyaluronic acid degrading enzyme inhibitor comprising a compound represented by the formula (1). And an antioxidant characterized by having a compound represented by the formula (1).
- the present invention also provides a tyrosinase activity inhibitor, a hyaluronan degrading enzyme inhibitor and Z or an antioxidant, which comprises a compound represented by the following formula (2).
- R 6 in the formula (2) is a hydrogen atom, a lower alkyl group or a protecting group for a phenolic hydroxyl group
- A is an alkylene group having 1 to 4 carbon atoms
- B is an alkylene group having 1 to 12 carbon atoms.
- R 7 is one COOR 8 (where R 8 is a protecting group for the lipoxyl group), lipoxyl group or one CH 2 ⁇ H
- One CHOH-1, 2-epoxy one, single CO- CH 2 CH 2 -, one CH_ ⁇ _H-CH 2 CH 2 _, one CO- CH 2 CHOH- one CHOH- CH 2 CH_ ⁇ _H- one C ⁇ One CH 2 CHOR 9 —, one CHOH—CH 2 CHOR 9 —, _ CO—CH CH— ketal derivative, or — CO—CH 2 CH 2 — ketal derivative, and R 9 is a lower alkyl group is there.
- R 1 is a protecting group for a hydrogen atom, a lower alkyl group or a phenolic hydroxyl group.
- R 2 is a hydrogen atom or a protecting group for a phenolic hydroxyl group.
- the protecting group for the phenolic hydroxyl group of R 1 and R 2 it is preferable that the protecting group is easily introduced and removed, and a silyl protecting group, an acyl protecting group, a benzyl protecting group, and an ether protecting group Etc. are shown.
- a t-butyldimethylsilyl group, a propionyl group, a petyloyl group, an isoptyloyl group, a piperoyl group, a benzoyl group, a toluoyl group, a benzyl group, a tetrahydrobiranyl group, and a methoxymethyl group are preferred.
- A is an alkylene group having 1 to 4 carbon atoms, preferably an ethylene group or a butylene group, and more preferably an ethylene group.
- B is an alkylene group having 1 to 12 carbon atoms, preferably It is an alkylene group having 1 to 9 and more preferably an alkylene group having 2 to 6 carbon atoms.
- R 3 is one C ⁇ R 4 group (where R 4 is a protecting group for a carbonyl group), a carboxyl group or one CH 2 ⁇ H.
- R 4 is a protecting group for a carbonyl group
- a protective group for a propyloxyl group a methyl group, an ethyl group, a propyl group, a butyl group and a benzyl group are preferred. More preferably, it is a methyl group or an ethyl group.
- R 3 is a carbonyl group
- one CHOH_CH CH—, -CHOH-1, 2, 2-epoxy one, one CHOH— CH 2 CH 2 -, - CHOH- CH 2 CHOH- , _ CO- CH 2 CHOR 5 -,
- R 5 is a lower alkyl group.
- R 6 is a protecting group for a hydrogen atom, a lower alkyl group or a phenolic hydroxyl group.
- the phenolic hydroxyl-protecting group for R 6 it is preferable that the protecting group is easily introduced and removed, and examples thereof include a silyl-type protecting group, an acyl-type protecting group, a benzyl-type protecting group, and an ether-type protecting group. Is done.
- a t-butyldimethylsilyl group, a propionyl group, a butyroyl group, an isoptyloyl group, a piperoyl group, a benzoyl group, a toluoyl group, a benzyl group, a tetrahydrovinylil group, and a methoxymethyl group are preferred.
- A is an alkylene group having 1 to 4 carbon atoms, preferably an ethylene group or a butylene group, and more preferably an ethylene group.
- B is an alkylene group having 1 to 12 carbon atoms, preferably an alkylene group having 1 to 9 carbon atoms, and more preferably an alkylene group having 2 to 6 carbon atoms.
- R i—COOR 8 (where R 8 is a protective group for a lipoxyl group), a lipoxyl group or 1 CH 2 ⁇ H.
- a methyl group, an ethyl group, a propyl group, a butyl group and a benzyl group are preferred. More preferably, it is a methyl group or an ethyl group.
- the compound of the formula (4) can be synthesized from a compound represented by the formula (7) (hereinafter, referred to as a compound of the formula (7).
- the following compounds represented by other formulas are similarly abbreviated).
- the compound of formula (7) is obtained by reacting a compound of formula (5) described below with a compound of formula (6).
- the compound of formula (5) can be synthesized from the compound of formula (10).
- the compound of formula (10) is obtained by reacting a compound of formula (8) described below with a compound of formula (9).
- the compound of the formula (4) is prepared by preparing the compound of the following formula (7) using the compound of the following formula (5) and the compound of the following formula (6) as raw materials, and further removing HX from the compound of the following formula (7) it can.
- R 4 represents a protecting group for a carbonyl group
- B represents an alkylene group having 1 to 12 carbon atoms
- X represents a benzenesulfonyl group or a toluenesulfonyl group.
- R 1 is a protecting group for a hydrogen atom, a lower alkyl group or a phenolic hydroxyl group
- R 2 is a protecting group for a hydrogen atom or a phenolic hydroxyl group
- A is an alkyl group having 1 to 4 carbon atoms. Shows a kylene group.
- I 1 , R 2 , R 4 , A, and B are as defined in Formula (1), and X is as defined in Formula (5).
- the compound of the formula (5) can be produced by performing a rearrangement reaction of the X group of the compound of the formula (10).
- the compound of the formula (10) can be produced by reacting a compound of the following formula (8) with an alkyl metal compound and then reacting a compound of the following formula (9).
- X in equation (8) is as defined in equation (5).
- R 4 and B in the formula (9) are as defined in the formula (1), and I is an iodine atom.
- R 4 and B in equation (10) are as defined in equation (1), and X is as defined in equation (5).
- the ratio of the kill metal compound is preferably from 0.7 to 1.3 chemical equivalents, more preferably from 0.9 to 1.1 chemical equivalents.
- the temperature of the above reaction is preferably from 100 ° C to 0 ° C, more preferably from 180 ° C to 120 ° C. If the reaction temperature is too low, cost is required to maintain the temperature, and if the reaction temperature is too high, side reactions may proceed.
- the reaction between the compound of the formula (8) and the alkyl metal compound is preferably carried out in an aprotic solvent, such as tetrahydrofuran, 1,4-dioxane, geethylether, 1,2-dimethoxetane, and hexamethylphosphoric acid.
- an aprotic solvent such as tetrahydrofuran, 1,4-dioxane, geethylether, 1,2-dimethoxetane, and hexamethylphosphoric acid.
- Triamide, N, N-dimethylpropyleneurea, a mixed solvent thereof and the like can be suitably used.
- the reaction time varies depending on conditions, but is usually several minutes to several ten minutes.
- alkyl metal compound examples include alkyl lithium compounds such as n-butyllithium, s-butyllithium, t-butyllithium, and phenyllithium; n-butylmagnesium chloride, s-butylmagnesium chloride, t-butylmagnesium chloride, and n-butylmagnesium chloride.
- alkyl lithium compounds such as n-butyllithium, s-butyllithium, t-butyllithium, and phenyllithium
- n-butylmagnesium chloride examples include Grignard compounds such as —butyl magnesium bromide, s-butyl magnesium bromide, t-butyl magnesium bromide, and n-butyl lithium, n-butyl magnesium chloride, and n-butyl magnesium bromide are preferred.
- an alkali metal such as lithium metal or sodium metal may be used instead of the alkyl metal compound.
- the compound of formula (10) is obtained by reacting the compound of formula (8) with the alkyl metal compound as described above and subsequently reacting the compound of formula (9).
- the temperature of the reaction system at the time of adding the compound of the formula (9) to the reaction product is preferably ⁇ 100 ° C. and preferably 0 ° C., and more preferably —8 O X to 120 ° C. If the reaction temperature is too low, it will be costly to maintain the temperature. If the reaction temperature is too high, side reactions may proceed.
- the reaction time varies depending on the conditions, but is usually several minutes to several ten minutes.
- the compound of the formula (10) can be isolated and purified by a known purification method such as solvent extraction or column chromatography.
- the compound of formula (5) can be obtained by rearranging the X group in the compound of formula (10).
- a palladium catalyst can be exemplified as a preferable catalyst for the rearrangement reaction of the compound of the formula (10).
- the palladium catalyst examples include tetrakistriphenylphosphine palladium (0), tris (dibenzylideneacetone) dipalladium (0), form-adduct, palladium chloride (II) / triphenylphosphine mixture, palladium (II) acetate A z-triphenylphosphine mixture or a palladium acetate (II) Z-tributylphosphine mixture is preferably used.
- the amount of the palladium catalyst to be used is preferably from 0.001 to 1mo1, more preferably from 0.001 to 0.1mo1, based on the compound (lmo1) of the formula (10). If the amount of the catalyst used is too small, the progress of the reaction may be slow, and if the amount used is too large, labor may be required to remove the catalyst.
- This rearrangement reaction is preferably carried out in the presence of a solvent.
- a mixed solvent of tetrahydrofuran and methanol can be used. Is preferred.
- the reaction temperature of the rearrangement reaction is preferably from 0 ° C to 1201 and more preferably from 20 to 100 ° C.
- the reaction time varies depending on conditions, but is usually several hours to several tens of hours.
- the compound of the formula (5) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound of the formula (6) can be synthesized by a method described in a literature such as G.Solidadie, eta1, J. Org. Chem., —58.2. 2181 (1993).
- R 1 represents a hydrogen atom, a lower alkyl group or a protecting group for a phenolic hydroxyl group
- R 2 represents a hydrogen atom or a protecting group for a phenolic hydroxyl group.
- the lower alkyl group for R 1 in the formula (6) preferably has 1 to 3 carbon atoms, and more preferably is a methyl group.
- the protecting group for the phenolic hydroxyl group of R 1 and R 2 in the formula (6) it is preferable that the protecting group is easily introduced and removed, and a silyl-type protecting group, an acyl-type protecting group, a benzyl-type protecting group, and Examples include ether-type protecting groups.
- a silyl-type protecting group, an acyl-type protecting group, a benzyl-type protecting group, and Examples include ether-type protecting groups.
- t-butyldimethylsilyl group Preferred are a pionyl group, a butyroyl group, an isobutyroyl group, a bivaloyl group, a benzoyl group, a toluene group, a benzyl group, a tetrahydroviranyl group, and a methoxymethyl group.
- a in the formula (6) is an alkylene group having 1 to 4 carbon atoms, preferably an ethylene group or a butylene group, and more preferably an ethylene group.
- the compound of the formula (7) which is a compound for obtaining the compound of the formula (4), is obtained by reacting the compound of the formula (5), which can be produced by the above method, with an alkyl metal compound, and subsequently reacting the compound of the formula (6). It can be manufactured by doing.
- the alkyl metal compound a compound which does not adversely affect the protecting group of the hydroxyl group in the compound of the formula (5) is preferable.
- An amide can be preferably used.
- the amount of the alkyl metal compound used is basically preferably from 0.7 to 1.3 chemical equivalents, more preferably from 0.9 to 1.1 chemical equivalents, based on the compound of the formula (5).
- the above reaction is preferably performed in an aprotic solvent, such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, hexamethylphosphoric triamide, ⁇ , ⁇ -dimethylpropylene ⁇ Rare and mixed solvents thereof can be suitably used.
- an aprotic solvent such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, hexamethylphosphoric triamide, ⁇ , ⁇ -dimethylpropylene ⁇ Rare and mixed solvents thereof can be suitably used.
- the reaction temperature of the compound of formula (5) with the alkyl metal compound is preferably ⁇ 80 ° C. to 25, more preferably 150 to 0.
- the reaction time is usually several minutes to several hours.
- the compound of the formula (7) can be produced by reacting the compound of the formula (5) with the alkyl metal compound and subsequently reacting the compound of the formula (6).
- the temperature of the reaction system at the time of adding the compound of the formula (6) to the reaction product is preferably from 10 to 25 ° C, more preferably from 180 to 0 ° C.
- the reaction time is usually several minutes to several hours.
- the compound of the formula (7) can be isolated and purified by a known purification method such as solvent extraction or column chromatography.
- the compound of the formula (4) in the present invention is prepared by reacting the compound of the formula (7) produced by the above-mentioned method and the like with a basic compound in the presence of a metal catalyst which forms a ⁇ -aryl complex. Combination Can be achieved.
- a palladium complex can be preferably used as the metal catalyst for forming the ⁇ -aryl complex.
- tetrakistriphenylphosphine palladium (0), tris (dibenzylideneacetone) dipalladium (0) examples thereof include a form adduct, a palladium (II) chloride "triphenylphosphine mixture, a palladium (II) acetate / triphenylphosphine mixture, and a palladium (II) tributylphosphine mixture.
- the amount is preferably from 0.000 l to lmo 1, more preferably from 0.001 to 0.1 lmo 1 based on the compound lmo 1 of the formula (7).
- Preferred examples of the basic compound include triethylamine, diisopropylethylamine, and tertiary amines such as N-methylimidazole and pyridine.
- the amount of the basic compound used is 0.9 mol or more with respect to the compound lmo 1 of the formula (7), and the range of 1. Omol to 1 Omo 1 is suitable. In addition, you may use such a basic compound as a solvent.
- the above reaction is preferably carried out in the presence of a solvent, for example, tetrahydrofuran, 1,4-dioxane, geethylether, 1,2-dimethoxyethane, acetonitrile, chloroform, dichloromethane, 1,2-dichloroethane, Hexamethylphosphoric triamide, N, N-dimethylpropylene perylene, methanol, ethanol, isopropyl alcohol, ethylene glycol, glycerin, and a mixed solvent thereof can be used.
- 1,2-dichloroethane A mixed solvent of alcohol and alcohol is preferred.
- the reaction temperature is preferably from room temperature to 150 ° C, more preferably from 50 ° C to 120.
- a suitable reaction time is several hours to several tens of hours.
- the compound of formula (4) can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 2 in the compound of the formula (4) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the method for removing the protecting group of the phenolic hydroxyl group is generally used in the field of synthetic organic chemistry.
- the method can be carried out according to a method known in the art, for example, the method described in TW Greene., “Protecti ve Grosin Sin Organic Synthes sis J, John Wi i ey & Sons.
- R 3 is —COOR 4 and Z is
- R 3 is -COOR 4 and Z is
- a compound that is -CHOH-1,2-epoxy (hereinafter referred to as a compound of formula (12)) can be produced by epoxidizing a compound of formula (11).
- R 1 , R 2 , R 4 , A and B in the equations (11) and (12) are as defined in the equation (1).
- the metal hydride complex when the ketone of the compound of the formula (4) is reduced to a hydroxyl group lithium borohydride, sodium borohydride and the like can be preferably used.
- the reaction can be carried out under the condition of using sodium borohydride and cerium (III) chloride in combination. In this reaction, it is preferable to use sodium borohydride and cerium (III) salt in combination.
- the ratio of the metal-hydrogen complex compound to the compound of the formula (4) is preferably 0.5 to 10 chemical equivalents, more preferably 1 to 2 chemical equivalents.
- the amount of cerium (III) chloride used is 0.01 to 5 chemical equivalents relative to the compound of formula (4). And more preferably Preferably 0.1 to 3 chemical equivalents.
- the above reaction is preferably carried out in the presence of a solvent, such as tetrahydrofuran, 1,4-dioxane, dimethyl ether, 1,2-dimethoxyethane, acetonitrile, methanol, ethanol, isopropyl alcohol, and a mixed solvent thereof.
- a solvent such as tetrahydrofuran, 1,4-dioxane, dimethyl ether, 1,2-dimethoxyethane, acetonitrile, methanol, ethanol, isopropyl alcohol, and a mixed solvent thereof.
- the reaction temperature for producing the compound of the formula (11) is preferably from 100 to 100, more preferably from 178 ° C to room temperature. A suitable reaction time is several minutes to several hours.
- the compound of the formula (11) can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 2 is a hydrogen atom
- R 7 is —CQOR 8
- R 2 in the compound of the formula (11) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- Examples of the epoxidizing agent for producing the compound of formula (12) from the compound of formula (11) include peracids such as peracetic acid, trifluoroperacetic acid, perbenzoic acid and m-mouth perbenzoic acid, and hydrogen peroxide and t.
- a peroxide such as butyl hydroperoxide can be suitably used.
- the amount of the peroxide or peroxide used is preferably 1 to 10 chemical equivalents, more preferably 1.1 to 2 chemical equivalents.
- the above reaction is preferably carried out in the presence of a solvent, such as dichloromethane, chloroform, 1,2-dichloroethane, tetrahydrofuran, 1,4-dioxane, dimethyl ether, 1,2-dimethoxyethane, acetonitrile, toluene, Water, a mixed solvent thereof and the like can be suitably used.
- a solvent such as dichloromethane, chloroform, 1,2-dichloroethane, tetrahydrofuran, 1,4-dioxane, dimethyl ether, 1,2-dimethoxyethane, acetonitrile, toluene, Water, a mixed solvent thereof and the like can be suitably used.
- the reaction temperature at the time of producing the compound of the formula (12) is preferably from 100 to 100 ° C, more preferably from 178 to 50 ° C.
- the reaction time is suitably from tens of minutes to several hours.
- the compound of formula (12) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound wherein R 2 is a hydrogen atom is a compound represented by the general formula (2) of the present invention wherein R 7 is —COR 8 and Z is —CHOH—1, It represents the same compound as 2-epoxy.
- R 2 of the compound of formula (12) is a phenolic hydroxyl group
- the compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group of the phenolic hydroxyl group.
- a compound in which R 3 is —C ⁇ R 4 and Z is —CO—CH 2 CH 2 — (hereinafter, referred to as a compound of the formula (13)) Can be produced by catalytic hydrogenation of the compound of formula (4).
- a compound in which R 3 is —C ⁇ R 4 and Z is —CHO H—CH 2 CH 2 — (hereinafter, referred to as a compound of the formula (14)) Can be produced by reducing the compound of the formula (13) with a metal hydride complex compound.
- RR 2 , R 4 , A and B in Equations (13) and (14) are as defined in Equation (1).
- metal such as palladium monocarbon, platinum (IV) oxide, Raney nickel, platinum black, rhodium aluminum monoxide, triphenylphosphine rhodium monochloride, and palladium monosulfate It is preferable to use a catalyst and make the inside of the reaction vessel under a hydrogen atmosphere. In this reaction, the ratio of the metal catalyst to the compound of the formula (4) is preferably from 0.0 :! to 100% by weight, more preferably from 0.1 to 10% by weight.
- the production of the compound of the formula (13) is preferably carried out in the presence of a solvent, such as methanol, ethanol, n-propyl alcohol, isopropyl alcohol, n-butyl alcohol, t-butyl alcohol, and n-pentane.
- a solvent such as methanol, ethanol, n-propyl alcohol, isopropyl alcohol, n-butyl alcohol, t-butyl alcohol, and n-pentane.
- N-hexane, toluene, ethyl acetate, Acetic acid, tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxetane, acetonitrile, N, N-dimethylformamide, N, N-dimethylpropionerea, dimethylsulfoxide, water, and a mixed solvent thereof Can be used.
- the reaction temperature for producing the compound of the formula (13) is preferably from 0 ° C to 100 ° C, more preferably from room temperature to 50 ° C.
- the reaction time is suitably from tens of minutes to tens of hours.
- the compound of the formula (13) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is —COOR 8 and Z is —C ⁇ —CH 2 Represents the same compound as CH 2 —.
- R 2 in the compound of the formula (13) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the metal hydride complex compound used in producing the compound of the formula (14) those which do not adversely affect the protecting group for the carboxyl group are preferable, and lithium borohydride, sodium borohydride and the like can be preferably used. More preferred is sodium borohydride.
- the ratio of the metal hydride complex compound to the compound of the formula (13) is preferably 0.5 to 10 chemical equivalents, more preferably 1 to 2 chemical equivalents.
- the production of the compound of the formula (14) is preferably carried out in the presence of a solvent, for example, tetrahydrofuran, 1,4-dioxane, geethylether, 1,2-dimethoxyethane, acetonitrile, methanol, ethanol , Isopropyl alcohol and a mixed solvent thereof can be used.
- a solvent for example, tetrahydrofuran, 1,4-dioxane, geethylether, 1,2-dimethoxyethane, acetonitrile, methanol, ethanol , Isopropyl alcohol and a mixed solvent thereof can be used.
- the reaction temperature for producing the compound of the formula (14) is preferably from 20 ° C to 100 ° C, more preferably from 0 ° C to room temperature.
- a suitable reaction time is several minutes to several hours.
- the compound of the formula (14) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is —COOR 8 and Z is —CHOH—CH 2 It represents the same compound as CH 2 —.
- R 2 of the compound of the formula (14) is a protecting group for a phenolic hydroxyl group
- R 2 is a hydrogen atom by removing the protecting group for the phenolic hydroxyl group.
- a compound in which R 3 is _C ⁇ R 4 and Z is —CHO H—CH 2 CHOH— (hereinafter, referred to as a compound of the formula (16)) is It can be produced by reducing the compound of the formula (15) with a metal hydride complex compound.
- RR 2 , R 4 , A and B in equations (15) and (16) are as defined in equation (1).
- a metal hydroxide such as sodium hydroxide or potassium hydroxide
- the amount of the metal 7_K oxide to be used is preferably 1 to 10 chemical equivalents, more preferably 2 to 5 chemical equivalents, relative to the compound of the formula (4).
- a phase transfer catalyst may be used in combination.
- phase transfer catalyst quaternary ammonium salts such as tetrabutylammonium bromide, benzyltriptylammonium bromide, and trioctylmethylammonium bromide are preferably used. It can.
- the amount of the phase transfer catalyst to be used is preferably 0.001 to 1 chemical equivalent, more preferably 0.01 to 0.2 chemical equivalent to the compound of the formula (4).
- the above reaction is preferably carried out in the presence of a solvent, such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, acetonitrile, toluene,
- a solvent such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, acetonitrile, toluene
- a solvent such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, acetonitrile, toluene
- a solvent such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, acetonitrile, toluene
- chloroform dichloromethane, 1,2-dichloroethane, t-buty
- the reaction temperature at the time of producing the compound of the formula (15) is 0 to 150 ° C., preferably room temperature to 100 ° C.
- a suitable reaction time is several hours to several tens of hours.
- the compound of formula (15) can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 4 is an alkyl group such as a methyl group or an ethyl group during the above addition reaction of water molecules
- hydrolysis may occur and R 4 may be removed.
- esterification may be performed after isolation and purification to synthesize the compound of formula (15).
- the conditions for this esterification are generally known in the field of synthetic organic chemistry, for example, described in TW Greene., "Protective Group Organic Synthesis", John Wi 1 e y & Sons. Can be performed according to the method described in
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is —COOR 8 and Z is —CO—CH 2 It represents the same compound as the compound that is CHOH_.
- R 2 of the compound of the formula (15) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the reducing agent used in producing the compound of the formula (16) is the same as the reducing agent used in producing the compound of the formula (14) from the compound of the formula (13), and is preferably sodium borohydride.
- the solvent used in producing the compound of formula (16) is the same as the solvent used in producing the compound of formula (14) from the compound of formula (13).
- the reaction temperature for producing the compound of the formula (16) is preferably from 20 to 100 ° C, more preferably from 0 ° C to room temperature. A suitable reaction time is several minutes to several hours. After completion of the reaction, the compound of formula (16) can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 2 is a hydrogen atom
- R 7 is —C ⁇ R 8 and Z is —CHOH — CH 2 CH ⁇ H— And the same compound.
- R 2 of the compound of the formula (16) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a compound in which R 3 is —COOR 4 and Z is one CO—CH 2 CHOR 5 — (hereinafter, referred to as a compound of the formula (17)) is The compound can be produced by adding a lower alcohol to the compound of the formula (4) in the presence of a basic compound.
- R 1 , R 2 , R 4 , A and B in the formulas (17) and (18) are as defined in the formula (1), and R 5 represents a lower alkyl group.
- an alkali metal hydroxide such as sodium hydroxide or potassium hydroxide.
- the amount of the alkali metal hydroxide to be used is preferably 1 to 10 chemical equivalents, more preferably 2 to 5 chemical equivalents to the formula (4).
- the lower alcohol used for producing the compound of the formula (17) from the formula (4) include methanol, ethanol, n-propyl alcohol, isopropyl alcohol and n-butyl alcohol.
- the reaction temperature for producing the compound of the formula (17) from the formula (4) is preferably 0 to 150 ° C, more preferably room temperature to 100 ° C. An appropriate reaction time is several hours to several tens of hours. After completion of the reaction, the compound of formula (17) can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 4 is an alkyl group such as a methyl group or an ethyl group during the above addition reaction of a lower alcohol
- hydrolysis may occur and R 4 may be removed.
- esterification may be performed after isolation and purification to synthesize the compound of formula (17).
- Conditions for this esterification include generally well-known methods in the field of synthetic organic chemistry, for example, TW Greene., "Protecti ve Grosin sin Organic Synthesis", John Wiiey ey & S. The method can be performed according to the method described in ons.
- the compound in which R 2 is a hydrogen atom is a compound represented by the general formula (2) of the present invention, wherein R 7 is —COOR 8 and Z is one CO—CH 2 This is the same compound as the compound in which CH ⁇ R 5 _.
- R 2 is a phenolic hydroxyl-protecting group in the compound of the formula (17)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the phenolic hydroxyl-protecting group.
- the reducing agent used in producing the compound of formula (18) from the compound of formula (17) is the same as the reducing agent used in producing compound of formula (14) from compound of formula (13), Preferred is sodium borohydride.
- the solvent used in producing the compound of formula (18) from the compound of formula (17) is the same as the solvent used in producing the compound of formula (14) from the compound of formula (13).
- the reaction temperature for producing the compound of the formula (18) from the compound of the formula (17) is preferably from -20 ° C to 100 ° C, more preferably from 0 ° C to room temperature.
- a suitable reaction time is several minutes to several hours.
- the compound of formula (18) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound wherein R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention wherein R 7 is —COOR 8 and Z is —CHOH—CH 2 It is the same compound as CH ⁇ R 5 —.
- R 2 is a protecting group for a phenolic hydroxyl group in the compound of the formula (18)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- Equation (4) The conversion from Equation (4) to Equation (19) and Equation (20) is shown below.
- RR 2 , R 4 , A and B in the formulas (19) and (20) are as defined in the formula (1), and Y represents an acyclic ketal or a cyclic ketal.
- acyclic ketones of the formulas (19) and (20) include dimethyl ketal and diacetyl ketal.
- the cyclic ketals of the formulas (19) and (20) include those obtained from ethylene glycol, 1,3-propanediol or 2,2-dimethyl-1,1,3-propanediol with respect to the carbonyl group. .
- the ketalization reaction of the formula (4) is generally performed by a method well known in the field of synthetic organic chemistry, for example, TW Greene., Protecti ve Grown Up Organic Synt he si sj, J on Wi 1ey & S. This can be performed according to the method described in ons.
- the compound in which R 2 is a hydrogen atom is a compound represented by the general formula (2) of the present invention, wherein R 7 is —C ⁇ R 8 and Z is —C ⁇ —
- R 7 is —C ⁇ R 8 and Z is —C ⁇ —
- R 7 is —C ⁇ R 8 and Z is —C ⁇ —
- R 7 is —COOR 8 and Z is a ketal derivative of —CO—CH—CH—.
- R 2 in the compound of the formula (19) or the compound of the formula (20) is a protecting group for the phenolic hydroxyl group
- the compound in which R 2 is a hydrogen atom is obtained by removing the protecting group for the phenolic hydroxyl group.
- RR 2 , R 4 , A, and B in the formula (21) are as defined in the formula (1).
- R 1 R 2 , R 4 , A and B in the formula (22) are as defined in the formula (1).
- compounds in which R 3 is a carbonyl group and Z is —CHOH—1,2_epoxy (hereinafter, a compound of the formula (23) ) Can be synthesized from the compound of the above formula (12). See the formula below.
- R 1 in the formula (23), R 2, R 4, A, B are as defined in formula (1) £
- R 3 in the formula (1) of the present invention compounds in which R 3 is a carboxyl group and Z is —CHOH—CH 2 CH 2 — (hereinafter, referred to as a compound of the formula (24) ) Can be synthesized from the compound of the above formula (14). See the formula below.
- R 1 R 2 in the formula (24), R 4, A, B in the same manner c are as defined in formula (1), among the compounds represented by the general formula (1) of the present invention, R 3 Is a carbonyl group and Z is —CH ⁇ H—CH 2 CHOH— (hereinafter referred to as the compound of formula (25)) can be synthesized from the compound of formula (16) described above. See the formula below.
- RR 2 , R 4 , A, and B in equation (25) are as defined in equation (1).
- a compound in which R 3 is a carbonyl group and Z is —CO—CH 2 CH ⁇ R 5 _ (hereinafter, a compound represented by the formula (26) Can be synthesized from the compound of the above formula (17). See the formula below.
- R 1 R 2 , R 4 , R 5 , A and B in the formula (26) are as defined in the formula (1).
- compounds in which R 3 is a carboxyl group and Z is —CH0H—CH 2 CH ⁇ R 5 — (hereinafter, a compound of the formula (27) ) Can be synthesized from the compound of the above formula (18). See the formula below.
- R 1 R 2 , R 4 , R 5 , A and B in the formula (27) are as defined in the formula (1).
- R 1 , R 2 , R 4 , A, B, and Y in the formula (28) are as defined in the formula (1).
- compounds in which R 3 is a carbonyl group and Z is a ketal derivative of —CO—CH 2 CH 2 — ) Compound can be synthesized from the compound of the above formula (20). See the formula below.
- R 1 R 2 , R 4 , A, B, and Y in the formula (29) are as defined in the formula (1).
- C The reaction for removing a protecting group from a compound (protected compound) in which the carbonyl group is protected.
- the conditions vary depending on the type of protecting group used. For example, when the protecting group of the lipoxyl group is an ethyl group, it can be removed in the presence of an alkali catalyst such as sodium hydroxide or potassium hydroxide.
- the amount of the alkali catalyst to be used varies depending on the structures of R 1 , R 2 , and R 4 of the protective compound and the type of the catalyst, but is preferably 1 to 10 chemical equivalents relative to the protective compound, and more preferably, 2 to 5 chemical equivalents. If the amount of the alkali catalyst used is too small, the progress of the reaction may be slow.If the amount is too large, a large amount of neutralizing agent is required for post-reaction treatment. There are cases.
- a solvent may be used for the above reaction. Tetrahydrofuran, 1,4-dioxane, methyl ether, 1,2-dimethoxyethane, N, N-dimethylformamide, N, N-dimethylpropylene urea, water, methanol , Ethanol, isopropyl alcohol, t-butyl alcohol, and a mixed solvent thereof can be suitably used.
- the reaction temperature of the above reaction is preferably from 20 to 80 ° C, more preferably from 0 ° C to 50.
- the reaction time varies depending on conditions, but is usually several tens minutes to several hours.
- the desired compound can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 2 of the compound of the formula (21) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- R 2 is a phenolic hydroxyl-protecting group in the compound of the formula (22)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the phenolic hydroxyl-protecting group.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is a carbonyl group, and Z is —CHOH— 1, It is the same compound as 2-epoxy.
- R 2 is a protecting group for a phenolic hydroxyl group in the compound of the formula (23)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the compound wherein R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is a carbonyl group and Z is —CHOH—CH— This is the same compound as CH—.
- R 2 is a phenolic hydroxyl group protecting compound. When it is a group, the compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group of the phenolic hydroxyl group.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, in which R 7 is a carbonyl group and Z is —CH ⁇ H— It is the same compound as CH 2 CH ⁇ H—.
- R 2 is a protecting group for a phenolic hydroxyl group in the compound of the formula (25)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the compound wherein R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is a carbonyl group and Z is —CO—CH 2 It is the same compound as CH ⁇ R 5 —.
- R 2 is a phenolic hydroxyl-protecting group in the compound of the formula (26)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the phenolic hydroxyl-protecting group.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, in which R 7 is a carbonyl group and Z is —CHOH—CH 2 This is the same compound as CHOR 5 _.
- R 2 is a protecting group for a phenolic hydroxyl group in the compound of the formula (27)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- R 2 of the formula (28) compound is a protective group of phenolic hydroxyl group, by removal of the protecting group of the phenolic hydroxyl group, to give compound R 2 is water atom.
- the compound wherein R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is a carbonyl group and Z is —CO—CH 2 This is the same compound as the compound that is a ketal derivative of CH 2 —.
- R 2 of the formula (29) compound is a protective group of phenolic hydroxyl group, by removal of the protecting group of Fueno Ichiru hydroxyl group, to give compound R 2 is water atom.
- R 3 is —CH 2 ⁇ H
- Equation (4) The conversion from Equation (4) to Equation (30) and Equation (31) is shown below.
- RR 2 , R 4 , A and B in equations (30) and (31) are as defined in equation (1).
- the conditions for the reduction reaction for producing the compound of the formula (30) from the formula (4) differ depending on the structures of RR 2 and R 4 in the formula (4).
- the protecting group of the carbonyl group is an ethyl group.
- the compound of the formula (30) can be obtained by using a metal hydride complex compound or a metal hydride.
- the metal hydride complex include lithium aluminum hydride, aluminum tributoxy hydride, sodium aluminum bis (2-methoxyethoxy) hydride, and the like.
- the metal hydride include diisobutylaluminum hydride.
- Particularly preferred reducing agents are lithium aluminum hydride and aluminum disodium hydride.
- the amount of the reducing agent used varies depending on the structure of R 1 , R 2 and R 4 in the formula (4) and the type of the reducing agent, but is 1 to 10 chemical equivalents relative to the compound represented by the formula (4). And more preferably 2 to 5 chemical equivalents.
- the above reduction reaction is preferably carried out in the presence of a solvent, such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, diglyme, toluene, Dichloromethane and a mixed solvent thereof can be used.
- a solvent such as tetrahydrofuran, 1,4-dioxane, getyl ether, 1,2-dimethoxyethane, diglyme, toluene, Dichloromethane and a mixed solvent thereof can be used.
- the reaction temperature of the above-mentioned reduction reaction is preferably in the range of 100 ° C to 80 ° C, and more preferably in the range of -78 ° C to room temperature.
- a suitable reaction time is several minutes to several hours.
- the compound of formula (30) can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 2 in the compound of the formula (30) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the epoxidizing agent used in producing the compound of the formula (31) is the same as the epoxylating agent used in producing the compound of the formula (12) from the compound of the formula (11).
- the solvent used in producing the compound of the formula (31) is the same as the solvent used in producing the compound of the formula (12) from the compound of the formula (11).
- the reaction temperature at the time of producing the compound of the formula (31) is preferably from 100 ° C to 80 ° C, more preferably from ⁇ 78 ° C to room temperature.
- a suitable reaction time is several tens minutes to several hours.
- the compound of formula (31) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is —CH 2 ⁇ H, and Z is —CHOH— This is equivalent to a compound that is 1,2-epoxy.
- R 2 of the compound of the formula (31) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a compound in which R 3 is —CH 2 ⁇ H and Z is one CHOH—CH 2 CHOH— (hereinafter, referred to as a compound of the formula (32)) is The compound can be synthesized by reducing the compound of the above formula (12).
- Equation (12) Force The conversion to equation (32) is shown below.
- R 1 R 2 R 4 A and B in the formula (32) are as defined in the formula (1).
- the reducing agent used in producing the compound of the formula (32) from the compound of the formula (12) is the same as the reducing agent used in producing the compound of the formula (30) from the compound of the formula (4), and is preferably used.
- the solvent used in producing the compound of the formula (32) is the same as the solvent used in producing the compound of the formula (30) from the compound of the formula (4).
- the above reaction temperature is preferably ⁇ 100 ° C. and 80 ⁇ , more preferably in the range of ⁇ 78 ° C. to room temperature.
- a suitable reaction time is several minutes to several hours.
- the compound of the formula (32) can be obtained by a known method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is —CH 2 ⁇ H and Z is —CH ⁇ This is the same compound as H—CH 2 CH ⁇ H—.
- R 2 is a protecting group for a phenolic hydroxyl group in the compound of the formula (32)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a compound in which R 3 is a —CH 2 ⁇ H group and Z is —CO—CH 2 CH ⁇ R 5 — (hereinafter, a compound of the formula (34) )
- a compound of the formula (34) Can be synthesized by adding a lower alcohol to a compound represented by the formula (33) described below (hereinafter, referred to as a compound of the formula (33)) in the presence of a basic compound. .
- R 1 , R 2 R 5 A and B in the equation (34) are as defined in the equation (1).
- the basic compound used in producing the compound of formula (34) from the compound of formula (33) is the same as the basic compound used in producing the compound of formula (17) from the compound of formula (4). is there. Also the formula
- the lower alcohol used in producing the compound is the same as the lower alcohol used in producing the compound of formula (17) from the compound of formula (4).
- the reaction temperature at the time of producing the compound of the formula (34) is preferably from 0 ° C to 150 ° C, more preferably from room temperature to 10 ox.
- a suitable reaction time is several hours to several tens of hours.
- the compound of the formula (34) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is a compound represented by the general formula (2) of the present invention, wherein R 7 is —CH 2 OH and Z is —CO—CH This is the same compound as 2 CHOR 5 —.
- R 2 of the compound of the formula (34) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a compound in which R 3 is _CH 2 OH and Z is —CHOH—CH 2 CH ⁇ R 5 — (hereinafter, referred to as a compound of the formula (35)) Can be synthesized by reducing the ester and ketone of the compound of the above formula (17) to alcohol.
- Equation (17) The conversion to force equation (35) is shown below.
- RR 2 , R 4 , R 5 , A, and B in the formula (35) are as defined in the formula (1).
- the agent is the same as the reducing agent used in producing the compound of the formula (30) from the compound of the formula (4), and is preferably lithium aluminum hydride and hydridiisobutylaluminum hydrogen.
- the solvent used in producing the compound of the formula (35) is the same as the solvent used in producing the compound of the formula (30) from the compound of the formula (4).
- the reaction temperature for producing the compound of the formula (35) is preferably from -100 ° C to 80 ° C, more preferably from -78 to room temperature. A suitable reaction time is several minutes to several hours. After completion of the reaction, the compound of formula (35) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is —CH 2 ⁇ H, and Z is —CHOH— The same compound as CH 2 CHOR 5 —.
- R 2 is a protecting group for a phenolic hydroxyl group in the compound of the formula (35)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- compounds in which R 3 is —CH 2 OH and Z is a ketal derivative of —CO—CH CHCH— (hereinafter, referred to as a compound of formula (36) ) can be synthesized by reducing the ester of the compound of the formula (19) to an alcohol. Further, by removing the ketal of the compound of the formula (36), the compound of the above formula (33) can be synthesized.
- R: R 2 , A, B and Y in the equations (33) and (36) are as defined in the equation (1).
- the reducing agent used in producing the compound of the formula (36) from the compound of the formula (19) is the same as the reducing agent used in producing the compound of the formula (30) from the compound of the formula (4).
- the solvent used for producing the compound of the formula (36) is obtained by converting the compound of the formula (4) into the compound of the formula (30). It is the same as the solvent used when producing the product.
- the reaction temperature at the time of producing the compound of the formula (36) is preferably from 100 ° C to 80 ° C, more preferably from 178 ° C to room temperature. A suitable reaction time is several minutes to several hours.
- the compound of formula (36) can be obtained by a known purification method such as solvent extraction or column chromatography.
- R 2 is a phenolic hydroxyl-protecting group in the compound of the formula (36)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the phenolic hydroxyl-protecting group.
- the reaction for removing the ketal from the compound of the formula (36) is generally carried out by a method well known in the field of synthetic organic chemistry, for example, TW Greene., "Protecti ve Group Sin Organic Synthesis, John”. This can be performed according to the method described in Wi 1 ey & Sons, and the compound of formula (33) can be obtained.
- R 2 of the compound of the formula (33) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a compound of the formula (37) can be synthesized by reducing the ester of the compound of formula (20) to an alcohol. Further, by removing the ketal of the compound of the formula (37), a compound represented by the formula (38) (hereinafter, referred to as a compound of the formula (38)) can be synthesized.
- R 1 , R 2 , A, B and Y in equations (37) and (38) are as defined in equation (1).
- the reducing agent used in producing the compound of the formula (37) from the compound of the formula (20) is the same as the reducing agent used in producing the compound of the formula (30) from the compound of the formula (4).
- the solvent used in producing the compound of the formula (37) is the same as the solvent used in producing the compound of the formula (30) from the compound of the formula (4).
- the reaction temperature for producing the compound of the formula (37) is preferably from -100 ° C to 80, and more preferably from 78 ° C to room temperature.
- a suitable reaction time is several minutes to several hours.
- the compound of the formula (37) can be obtained by a known purification method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is one CH 2 ⁇ H and Z is —CO— CH— This is the same compound as the compound that is the ketal derivative of CH—.
- R 2 in the compound of the formula (37) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the reaction for removing the ketal from the compound of the formula (37) is generally carried out by a method well known in the field of organic synthesis, for example, TW Greene., "Protective Growup sin Organic Synthesis", J
- the method can be carried out according to the method described in ohn Wieey & Sons, and the compound of formula (38) can be obtained.
- the compound wherein R 2 is a hydrogen atom is a compound represented by the general formula (2) of the present invention, wherein R 7 is —CH 2 ⁇ H and Z is —CO— Compounds that are CH 2 CH 2 — Is the same compound as
- R 2 is a protecting group for a phenolic hydroxyl group in the compound of the formula (38)
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a compound represented by the general formula (38) (hereinafter referred to as the compound of the formula (39)) can be synthesized.
- a compound represented by the general formula (40) (hereinafter, referred to as a compound of the formula (40)) can be synthesized from the compound of the formula (15) of the present invention. Wear.
- the reaction conditions for producing the compound of formula (40) from the compound of formula (13) from the compound of formula (13) and the compound of formula (40) from the compound of formula (15) are the reaction conditions for producing the compound of formula (21) from the compound of formula (4). Same as the case.
- the solvent used in producing the compound of the formula (39) and the compound of the formula (40) is the same as the solvent used in producing the compound of the formula (21) from the compound of the formula (4).
- the reaction temperature for producing the compound of the formula (39) and the compound of the formula (40) is preferably from 20 to 80 ° C, more preferably from 0 to 50 ° C. If the reaction temperature is too low, the progress of the reaction may be slow, and if the reaction temperature is too high, a side reaction may proceed.
- the reaction time varies depending on the conditions, but is usually several tens of minutes to several hours. After the reaction is complete, extract the solvent, The desired compounds can be obtained by known methods such as-.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, in which R 7 is a carboxyl group and Z is —CO—CH 2 CH It is the same compound as the compound that is 2— .
- R 2 in the compound of the formula (39) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, in which R 7 is a carboxyl group and Z is —CO—CH 2 CH It is the same compound as the compound having ⁇ H—.
- R 2 of the compound of the formula (40) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a compound represented by formula (41) hereinafter, referred to as compound of formula (41)
- R 1 , R 2 , A and B in the equation (41) are as defined in the equation (1).
- the reducing agent used in producing the compound of the formula (41) from the compound of the formula (13) is the same as the reducing agent used in producing the compound of the formula (30) from the compound of the formula (4), and is preferably used.
- the solvent used in producing the compound of the formula (41) is the same as the solvent used in producing the compound of the formula (30) from the compound of the formula (4).
- the reaction temperature for producing the compound of the formula (41) is preferably from 1 to 80, more preferably from 1 to 78 and room temperature. A suitable reaction time is several minutes to several hours. After completion of the reaction, the compound of the formula (41) can be obtained by a known method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is a compound represented by the general formula (2) of the present invention, wherein R 7 is —CH 2 OH group, and Z is —CHOH— This is the same compound as CH 2 CH 2 —.
- R 2 in the compound of the formula (41) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- a water molecule is added to the compound of the formula (33) of the present invention, a compound represented by the general formula (42) (hereinafter, referred to as a compound of the formula (42)) can be obtained.
- RR 2 , A, and B in equation (42) are as defined in equation (1).
- the reaction conditions for producing the compound of the formula (42) from the compound of the formula (33) are the same as the reaction conditions for producing the compound of the formula (15) from the compound of the formula (4).
- the solvent used in producing the compound of the formula (42) is the same as the solvent used in producing the compound of the formula (15) from the compound of the formula (4).
- the reaction temperature for producing the compound of the formula (42) is preferably from 0 ° C to 150 ° C, more preferably from room temperature to 100 ° C.
- a suitable reaction time is several hours to several tens of hours.
- the compound of the formula (42) can be obtained by a known method such as solvent extraction or column chromatography.
- the compound in which R 2 is a hydrogen atom is the compound represented by the general formula (2) of the present invention, wherein R 7 is —CH 2 OH and Z is —CO—CH This is the same compound as the compound that is 2 CH ⁇ H—.
- R 2 of the compound of the formula (42) is a protecting group for a phenolic hydroxyl group
- a compound in which R 2 is a hydrogen atom can be obtained by removing the protecting group for the phenolic hydroxyl group.
- the compounds represented by the general formulas (1) and (2) of the present invention there are compounds having one or two asymmetric carbon atoms. These are usually obtained as a racemic mixture, Separate and use only one optically active compound by asymmetric synthesis, if necessary, or by a method such as high-performance liquid chromatography using an optically active column. Is also possible.
- the compound represented by the general formula (1) and the compound represented by the general formula (2) of the present invention can inhibit the activity of tyrosinase, an oxidase of L-tyrosine. Further, it is considered that the compound represented by the general formula (1) and the compound represented by the general formula (2) also have a hydroxyradical elimination action and a lipid peroxide production inhibitory action.
- R 1 of the compound represented by the formula (1) is a lower alkyl group or a protecting group for a phenolic hydroxyl group
- R 2 is a protecting group for a phenolic hydroxyl group
- melanin pigment and suppress the pigment deposition on the skin and the like.
- All of them can be used in the formulation of foods, pharmaceuticals, quasi-drugs, cosmetics, etc., and when used as an external preparation for the purpose of preventing or improving spots and freckles, It is preferably incorporated into lotions, serums, emulsions, creams, packs and the like.
- the compound represented by the general formula (2) has an inhibitory effect on hyaluronan degrading enzyme, a free radical scavenging effect, and an inhibitory effect on lipid peroxide production, it can be used for prescription of foods, pharmaceuticals, quasi-drugs, and cosmetics. It can be suitably blended and used.
- Compounds 6, 12, 17, 22, 26, 27, 28, 29, 30, 31, 32 and Z or 33 are tyrosinase activity inhibitors, hyaluronan degrading enzyme inhibitors, antioxidants, and hydroxyl radical scavengers And Z or can be used as a lipid peroxide production inhibitor.
- Compounds 7, 8, 13, 18, 19, 23, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47 and / or 48 inhibit tyrosinase activity Agents, hyaluronan degrading enzyme inhibitors, antioxidants, hydroxy radical scavengers and / or lipid peroxide production inhibitors.
- T s represents a p-toluenesulfonyl group.
- compound 5 was prepared via compound 1 and compound 3 using compound 1 as a starting material.
- the compound 1 is a compound of the formula (8)
- the compound 2 is a compound of the formula (10)
- the compound 3 is a compound of the formula (5)
- the compound 4 is a compound of the formula (6)
- Compound 5 corresponds to the compound of formula (7).
- the reaction was stopped by adding 5 Om 1 of a 5% aqueous solution of citrate.
- the reaction mixture was partitioned by adding 10 Om 1 of a 10% aqueous sodium thiosulfate solution, 150 ml of saturated saline and 50 ml of ethyl acetate.
- the organic layer was separated, and the aqueous layer was extracted with 5 Oml of ethyl acetate.
- the combined organic layers were dried over anhydrous magnesium sulfate. After drying, the solvent was distilled off, and the residue was purified by silica gel column chromatography to obtain 49.5 g (yield: 94%) of a pale yellow, low-viscosity liquid compound.
- the wave number (cm- 1 ) of the absorption in the infrared absorption spectrum was 2930, 2860, 1730, 1600, 1300, 1290, 1180, 1140, 940, 670.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 2930, 2860, 1730, 1600, 1320, 1150, 1090, 1030, 820, and 740.
- the wave number (cm- 1 ) of the absorption in the infrared absorption spectrum was 3510, 2940, 2860, 1740, 1600, 1510, 1290, 1270, 1200, 1140, 1061, 1030, 710. there were.
- Compound 6 of the present invention was prepared using compound 5 obtained in Synthesis Example 1 described above as a raw material.
- the wave number (cm- 1 ) of the absorption in the infrared absorption spectrum was 2930, 2860, 1730, 1700, 1670, 1630, 1600, 1510, 1270, 1200, 1150, 1060, 1030, It was 710.
- Compound 7 of the present invention was prepared using compound 6 obtained in Example 1 as a starting material.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 3530, 2930, 2850, 1700, 1660, 1640, 1520, 1280, 1230, and 1030.
- compound 11 was prepared via compound 9 and compound 10, using compound 1 as a starting material in the same manner as in Synthesis Example 1.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 2980, 2930, 1730, 1600, 1300, 1290, 1140, 1090, 820, and 670.
- this product contains 81% of compound 10 by mole fraction, and the remaining 19% is carbon in compound 10. -It was confirmed that the carbon double bond was a geometric isomer arranged in cis form.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 2980, 2930, 1730, 1600, 1320, 1300, 1150, 1090, 820, and 740.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 2980, 2930, 1730, 1600, 1320, 1300, 1150, 1090, 820, and 740.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 3510, 2940, 1730, 1600, 1510, 1290, 1270, 1150, 1060, and 710.
- Compound 12 of the present invention was prepared using compound 11 obtained in Synthesis Example 2 as a raw material.
- Compound 13 of the present invention was prepared using compound 12 obtained in Example 4 as a raw material.
- the wave number (cm—) of the absorption in the infrared absorption spectrum is 3400, 2940, 1710, 1660, 1630, 1520, 1270, 1240, 1150, 1030, 820, 720.
- compound 16 was prepared via compound 14 and compound 15 using compound 1 as a starting material in the same manner as in Synthesis Example 1.
- the chemical shift values of the 1 H-NMR spectrum of this product measured in heavy-duty mouth form are 1.23 (3H, t), 1.80 -2.00 (1H, m), 2.30-2. 49 (6H, m), 3.64 (1H, dt), 4.10 (2H, d), 5.10 (1H, d), 5.33 (1H, d), 5.56-5. 68 (1 H, m), 7.33 (2H, d) and 7.71 (2H, d).
- the wave numbers (cm—) of absorption in the infrared absorption spectrum were 2980, 1730, 1600, 1380, 1290, 1150, 1090, 940, 820, and 670.
- the wave number (cm— 1 ) of the absorption in the infrared absorption spectrum is 2980, 2930, 1730, 1600, 1320, 1300, 1180, 1150, 1090, 820, 740.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 3450, 2940, 1740, 1600, 1510, 1270, 1200, 1150, 1060, 1030, and 710.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 2940, 1740, 1670, 1650, 1510, 1270, 1200, 1150, 1060, 1030, and 710.
- the reaction mixture was partitioned by adding 6 Oml of saturated saline. The organic layer was separated, and the aqueous layer was extracted with 5 Oml of ethyl acetate. The combined organic layers were dried over anhydrous magnesium sulfate. The solvent was distilled off, and the residue was purified by silica gel column chromatography to obtain 6.03 g (yield 29%) of a pale yellow compound having a medium viscosity.
- the wave number (cm- 1 ) of the absorption in the infrared absorption spectrum was 3510, 2970, 2940, 2860, 1750, 1730, 1600, 1510, 1280, 1200, 1120, 1030, and 670. there were. In addition, elemental analysis showed 65.51% carbon and 7.45% hydrogen.
- Compound 22 of the present invention was prepared using compound 21 obtained in Synthesis Example 4 as a raw material.
- Compound 23 of the present invention was prepared using compound 22 obtained in Example 8 as a starting material.
- Compound 25 was prepared from Compound 3 and Compound 24 as starting materials for obtaining the compound of Formula (4).
- the wave number (cm- 1 ) of the absorption in the infrared absorption spectrum was 3510, 2970, 2940, 2860, 1750, 1730, 1600, 1510,
- Compound 26 of the present invention was prepared using compound 25 obtained in Synthesis Example 5 as a starting material.
- the wave numbers (cm— 1 ) of absorption in the infrared absorption spectrum were 2979, 1760, 1732, 1668, 1603, 1512, 1468, 1369, 1267, 1150, 1127, and 1038. .
- the wave numbers (cm— 1 ) that were absorbed in the infrared absorption spectrum were 3504, 2974, 1761, 1736, 1606, 1511, 1467, 1418, 1280, 1182, 1127, 1098, and 1036. there were.
- Example 11 The compound 27 obtained in Example 11 was epoxidized to obtain a compound 28 of the present invention.
- Example 8 The compound 22 obtained in Example 8 was catalytically hydrogenated to obtain a compound 29 of the present invention.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 2,930, 1760, 1730, 1639, 1511, 1452, 1419, 1369, 1267, 1183, and 1126.
- Example 10 The compound 26 obtained in Example 10 was catalytically hydrogenated to obtain a compound 30 of the present invention.
- the wave number (cm—) of the absorption in the infrared absorption spectrum was 2930, 1760, 1730, 1639, 1511, 1452, 1419, 1369, 1267, 1183, 1126, 1098, 1029, and 1015. there were.
- Example 10 The compound 26 obtained in Example 10 was ketalized to give a compound 32 of the present invention.
- the wave number (cm— 1 ) at which the absorption was observed in the infrared absorption spectrum was 2924 1758 1737 1608 1510 1466 1420 1127 1042.
- elemental analysis showed that carbon was 68.04% and hydrogen was 8.46%.
- Example 13 The compound 29 obtained in Example 13 was ketalized to obtain a compound 33 of the present invention.
- a water molecule was added to the compound 26 obtained in Example 10, and a protecting group for a phenolic hydroxyl group and a protecting group for a hydroxyl group were removed to obtain a compound 3 of the present invention.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 3336, 2943, 1723, 1710, 1603, 1514, 1426, 1275, and 1212.
- the wave numbers (cm— 1 ) at which absorption was observed in the infrared absorption spectrum were 3448, 2924, 1701, 1621, 1560, 1524, 1410, 1212, 1127, 1098, and 1028.
- Example 10 The compound 26 obtained in Example 10 was subjected to addition of methanol and removal of a protecting group for a phenolic hydroxyl group, to obtain a compound 36 of the present invention.
- the ethyl group which was a protecting group for the carbonyl group, was substituted with a methyl group.
- the wave number (cm—) of the absorption in the infrared absorption spectrum was 3413, 2931, 1733, 1604, 1515, 1436, 1364, 1267, 1238, 1196, 1155, 1127, 1084, 1072,
- the result of elemental analysis was 66.29% for carbon and 8.48% for hydrogen.
- the wave numbers (cm— 1 ) at which absorption was observed in the infrared absorption spectrum were 3403, 2926, 2858, 2368, 1700, 1515, 1417, 1274, and 1034.
- the wave numbers (cm—) at which absorption was observed in the infrared absorption spectrum were 3414, 2935, 1703, 1611, 1520, 1466, 1409, 1279, 1228, 1122, and 1028.
- the wave numbers (cm- 1 ) at which absorption was observed in the infrared absorption spectrum were 3413, 2931, 1705, 1612, 1515, 1435, 1367, 1264, 1154, and 1038.
- the wave numbers (cm—) of absorption in the infrared absorption spectrum were 3349, 2930, 1666, 1602, 1516, 1464, 1453, 1431, 1368, 1273, 1236, 1154, and 1036. .
- the wave numbers (cm— 1 ) at which absorption was observed in the infrared absorption spectrum were 3394, 2931, 1723, 1603, 1517, 1453, 1430, 1368, 1270, and 1035.
- the wave numbers (cm— 1 ) that were absorbed in the infrared absorption spectrum were 3426, 2930, 1607, 1515, 1481, 1468, 1437, 1365, 1268, 1239, 1155, 1124, and 1039. there were.
- Example 16 The compound 32 obtained in Example 16 was reduced with lithium aluminum hydride to obtain a compound 43 of the present invention.
- the wave numbers (cm— 1 ) that were absorbed in the infrared absorption spectrum were 3415, 2920, 1705, 1611, 1520, 1463, 1455, 1365, 1278, 1236, 1151, 1122, 1064, 1028.
- wave numbers had absorbed in the infrared absorption spectrum (KB r pellet method) (cm one 1), 3416, 2930, 1653, 1609, 1516, 1453, 1429, 1368, 1274, 1239, 1154, was 1030 .
- the wave numbers (cm— 1 ) at which absorption was observed in the infrared absorption spectrum were 3389, 2932, 2857, 1602, 1516, 1453, 1428, 1371, 1272, 1238, 1084, and 1036. .
- XI 00 T 1 Difference in absorbance of the sample-free solution after 2 minutes 45 seconds and 1 minute 45 seconds
- ⁇ 2 Difference in absorbance between 2 min 45 sec and 1 min 45 sec of the solution to which the sample was added
- the compound of the present invention and arbutin were added to the enzyme reaction mixture at 0.5 mgZml and 1 mgZm1, respectively. The results are shown in Table 1.
- Example 24 Using compound 40 obtained in Example 24 and arbutin (manufactured by Tokyo Kasei Kogyo Co., Ltd.) as a comparative control, a tyrosinase activity inhibition test using L-dopa as a substrate was performed.
- the test method was in accordance with Test Example 1.
- Table 2 shows the results when the compound of the present invention and arbutin were used in the enzyme reaction solution at 0.125 mg / m 1, 0.25 Omg / m 1, and 0.5 Omg / m 1, respectively.
- the absorbance at 490 nm after 2 s and 2 min 45 s was measured with a microplate reader.
- Tyrosinase activity inhibition rate (%) [(T 1 — T 2 ) / T 1 ] XI 00
- T 1 Difference in absorbance of the sample-free solution after 2 minutes 45 seconds and 1 minute 45 seconds
- T 2 Difference in absorbance between 2 minutes 45 seconds and 1 minute 45 seconds of the solution to which the sample was added
- the compound of the present invention and arbutin were added to the enzyme reaction mixture at 0.50 mg / ml and 1.0 OmgZml, respectively.
- Table 3 shows the results of the use.
- Table 4 shows the results when 0.02 mgZm 1, ⁇ and 0.125 mgZm1 were used.
- Table 5 shows the results obtained when the compound of the present invention and arbutin were used in the enzyme reaction mixture at 1.0 mg / ml.
- Table 6 shows the results when the compound of the present invention and arbutin were used in the enzyme reaction solution at 0.25 mgZml and 0.5 Omg / ml, respectively.
- a 0.1 M acetate buffer was prepared by dissolving 0.60 g of acetic acid in 100 ml of distilled water.
- a hyaluronidase solution was prepared by dissolving 7.2 mg of hyaluronan degrading enzyme (derived from bovine testes, type IV-S, manufactured by Sigma) in 3. Oml of acetate buffer (prepared in A above). .
- An activator solution was prepared by dissolving Omg of compound48Z80 (a hyaluronidase activator manufactured by Sigma) in 10. Oml of acetate buffer (prepared in A).
- a hyaluronic acid solution was prepared by dissolving 12.0 mg of potassium hyaluronate (Wako Pure Chemical Industries, Ltd.) in 15.Oml of acetate buffer (prepared in A).
- Table 7 shows the results when the compound of the present invention and magnesium ascorbate were used in the enzyme reaction mixture at 0.1 mg / m 1, 0.2 mg / ml, and 0.4 mg Zm1, respectively. (Compound 7 was not tested because it did not dissolve at a concentration of 0.4 mg Zml.)
- the compound of the present invention was found to have a radical scavenging activity higher than that of d 1 -hytocopherol. Therefore, it is considered that the compounds of the present invention can be used as scavengers for active oxygen, especially hydroxy radicals.
- Linoleic acid 0.2 g, manufactured by Tokyo Kasei Kogyo Co., Ltd.
- compound 7 (0.01 g)
- surfactant Nissan OT-221 0.4 g, Japan
- distilled water 17.39 g
- HP LC analysis conditions were: detection wavelength: 210 nm, mobile phase: mixed solution of McI 1 Vaine buffer adjusted to pH 2.6 and methanol (10:90), flow rate: 1 ml 1 Zmin, power column : ODS-80Ts (4.6 ⁇ 150 mm), column temperature 40 ° C.
- the product of the present invention can be industrially produced, and has excellent tyrosinase activity inhibitory activity, hyaluronan degrading enzyme, hydroxyl radical scavenging activity, and inhibitory activity on lipid peroxide production. is there. From this, the compound of the present invention can be applied to fields such as foods, flavorings, pharmaceuticals, quasi-drugs, and cosmetics. As a specific example, it can be used to prevent freckles, freckles, skin aging and allergies.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Dermatology (AREA)
- Pulmonology (AREA)
- Immunology (AREA)
- Toxicology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Anti-Oxidant Or Stabilizer Compositions (AREA)
- Epoxy Compounds (AREA)
- Cosmetics (AREA)
Description





























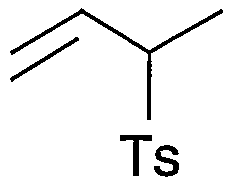
















































Claims


Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005504124A JP4329759B2 (ja) | 2003-03-27 | 2004-03-26 | 新規ショウガオール系化合物および当該化合物によるチロシナーゼ活性阻害剤 |
| US10/549,117 US7355063B2 (en) | 2003-03-27 | 2004-03-26 | Shogaol compound and tyrosinase activity inhibitor comprising the compound |
| EP04723805A EP1612201A4 (en) | 2003-03-27 | 2004-03-26 | NEW SHOGAOL COMPOUND AND AN INHIBITOR OF TYROSINASE ACTIVITY CONTAINING THE COMPOUND |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003-86818 | 2003-03-27 | ||
| JP2003086818 | 2003-03-27 | ||
| JP2003417389 | 2003-12-16 | ||
| JP2003-417389 | 2003-12-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004085373A1 true WO2004085373A1 (ja) | 2004-10-07 |
Family
ID=33100396
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2004/004316 Ceased WO2004085373A1 (ja) | 2003-03-27 | 2004-03-26 | 新規ショウガオール系化合物および当該化合物によるチロシナーゼ活性阻害剤 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US7355063B2 (ja) |
| EP (1) | EP1612201A4 (ja) |
| JP (1) | JP4329759B2 (ja) |
| KR (1) | KR100778191B1 (ja) |
| WO (1) | WO2004085373A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006073042A1 (ja) * | 2005-01-05 | 2006-07-13 | Toagosei Co., Ltd. | Nrf2依存遺伝子の活性化剤 |
| JP2010000054A (ja) * | 2008-06-23 | 2010-01-07 | Kao Corp | ジンジャーオレオレジン精製物の製造方法 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100158842A1 (en) * | 2008-10-09 | 2010-06-24 | Spectrum Dynamics Llc | Natural skin whitener: 4-hydroxy-oxindole-3-acetic acid |
| US8198337B2 (en) * | 2010-01-27 | 2012-06-12 | Momentive Performance Materials Inc. | Demulsifier compositions and methods for separating emulsions using the same |
| US11534382B2 (en) | 2017-06-19 | 2022-12-27 | Novan, Inc. | Topical compositions and methods of using the same |
| CN114520441B (zh) | 2020-11-20 | 2024-06-25 | 财团法人工业技术研究院 | 导电元件、电连接器的端子元件装置以及电连接器装置 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001131033A (ja) * | 1999-11-09 | 2001-05-15 | Shiseido Co Ltd | 頭皮頭髪用化粧料 |
-
2004
- 2004-03-26 EP EP04723805A patent/EP1612201A4/en not_active Withdrawn
- 2004-03-26 JP JP2005504124A patent/JP4329759B2/ja not_active Expired - Fee Related
- 2004-03-26 KR KR1020057018087A patent/KR100778191B1/ko not_active Expired - Fee Related
- 2004-03-26 US US10/549,117 patent/US7355063B2/en not_active Expired - Fee Related
- 2004-03-26 WO PCT/JP2004/004316 patent/WO2004085373A1/ja not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001131033A (ja) * | 1999-11-09 | 2001-05-15 | Shiseido Co Ltd | 頭皮頭髪用化粧料 |
Non-Patent Citations (2)
| Title |
|---|
| NAKAZAWA T, ET AL: "Metabolism of [6]-gingerol in rats", LIFE SCIENCES, vol. 70, no. 18, 2002, pages 2165 - 2175, XP002980564 * |
| See also references of EP1612201A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006073042A1 (ja) * | 2005-01-05 | 2006-07-13 | Toagosei Co., Ltd. | Nrf2依存遺伝子の活性化剤 |
| JP2010000054A (ja) * | 2008-06-23 | 2010-01-07 | Kao Corp | ジンジャーオレオレジン精製物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20050115314A (ko) | 2005-12-07 |
| US20060173072A1 (en) | 2006-08-03 |
| US7355063B2 (en) | 2008-04-08 |
| EP1612201A1 (en) | 2006-01-04 |
| JP4329759B2 (ja) | 2009-09-09 |
| KR100778191B1 (ko) | 2007-11-28 |
| EP1612201A4 (en) | 2006-07-05 |
| JPWO2004085373A1 (ja) | 2006-06-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004085373A1 (ja) | 新規ショウガオール系化合物および当該化合物によるチロシナーゼ活性阻害剤 | |
| KR102068635B1 (ko) | 3,4,5-트리메톡시 신남산 에스테르 유도체와 그 제조방법 및 이를 포함하는 피부 미백용 조성물 | |
| JP2006052152A (ja) | 抗酸化性化合物 | |
| EP1506958B1 (en) | Process for producing shogaol and intermediates for the synthesis thereof | |
| EP1510511A1 (en) | Gingerol analogues and use thereof | |
| CA1136638A (en) | N-hydroxylpropylamides of all-e retinic acid and 13-z-retinic acid, their preparation, and pharmaceutical formulations containing these compounds | |
| JP4986110B2 (ja) | レゾルシノール誘導体を含有するメラニン生成抑制剤 | |
| CN100372829C (zh) | 新型的姜烯酚化合物及包括该化合物的酪氨酸酶活性抑制剂 | |
| JP4210573B2 (ja) | 置換フェノキシプロパノールアミン類 | |
| JP4345311B2 (ja) | 抗酸化剤 | |
| WO2006049184A1 (ja) | 4-アルキルレソルシノール誘導体及びこれを有効成分とする美白剤 | |
| JPH07188208A (ja) | 皮膚外用剤 | |
| JP5886121B2 (ja) | メラニン生成抑制剤 | |
| JP4622310B2 (ja) | 低比重リポタンパクの酸化変性防止剤および抗アテローム性動脈硬化剤 | |
| JP2003201264A (ja) | ヒノキチオール誘導体、これを含有する皮膚外用剤及び毛髪用化粧料 | |
| JPH09241128A (ja) | チロシナーゼ活性阻害剤 | |
| JP3989103B2 (ja) | 新規フラバン誘導体、その製造方法および該誘導体を有効成分として配合した化粧料 | |
| JP2003342224A (ja) | チロシナーゼ活性阻害剤 | |
| JPH08231367A (ja) | チロシナーゼ活性阻害剤 | |
| JP2006052150A (ja) | 抗酸化剤 | |
| JP2006052186A (ja) | 新規ジヒドロフェルラ酸縮合物 | |
| JP2013209321A (ja) | メラニン生成抑制剤 | |
| WO2011126098A1 (ja) | 皮膚外用剤 | |
| JPS6396149A (ja) | 新規テルペン化合物およびこれを含有する抗炎症剤 | |
| JP2000007631A (ja) | 皮膚外用剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004723805 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005504124 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2006173072 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10549117 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020057018087 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048083189 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057018087 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004723805 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10549117 Country of ref document: US |