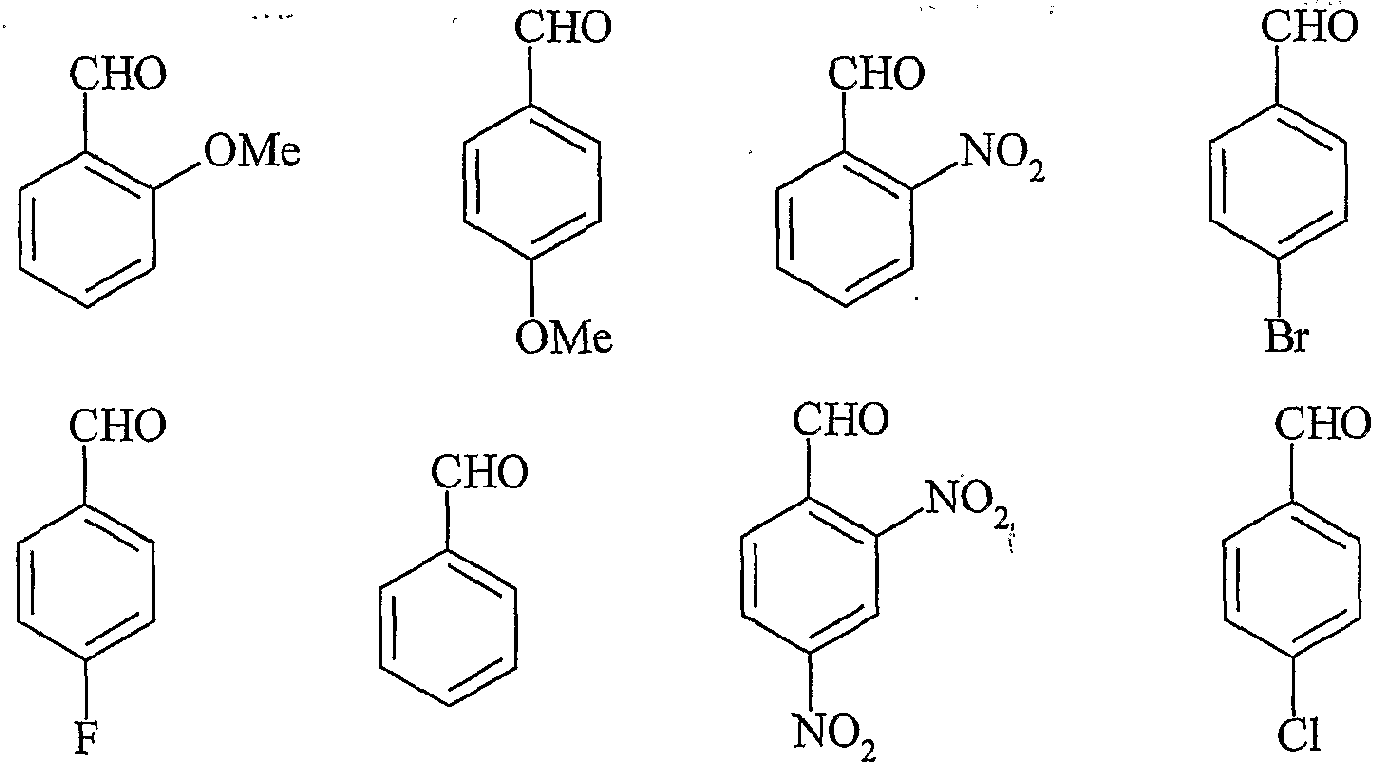
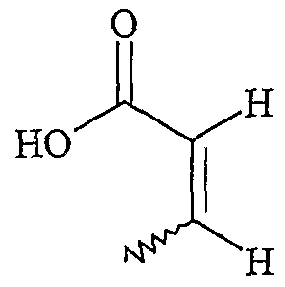
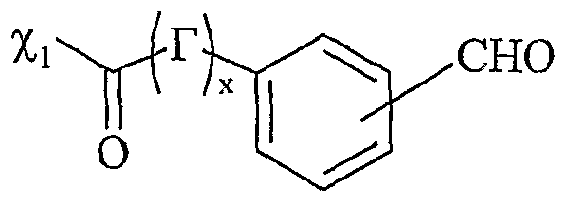
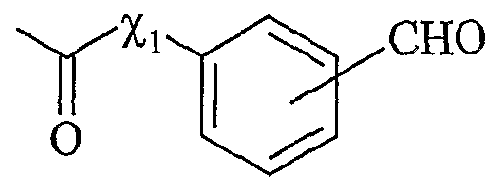
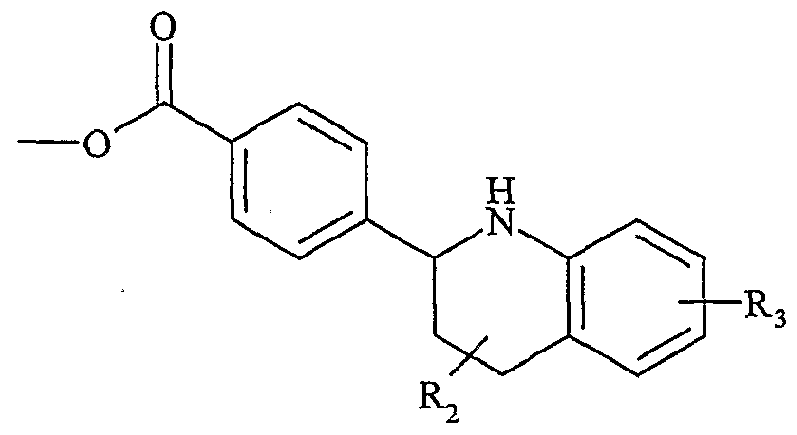
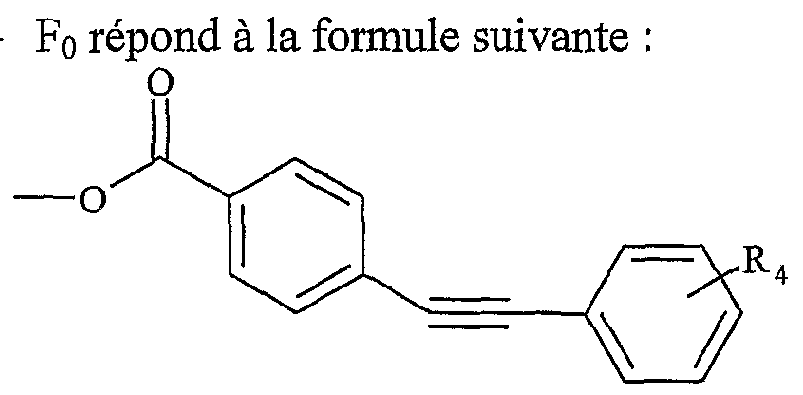
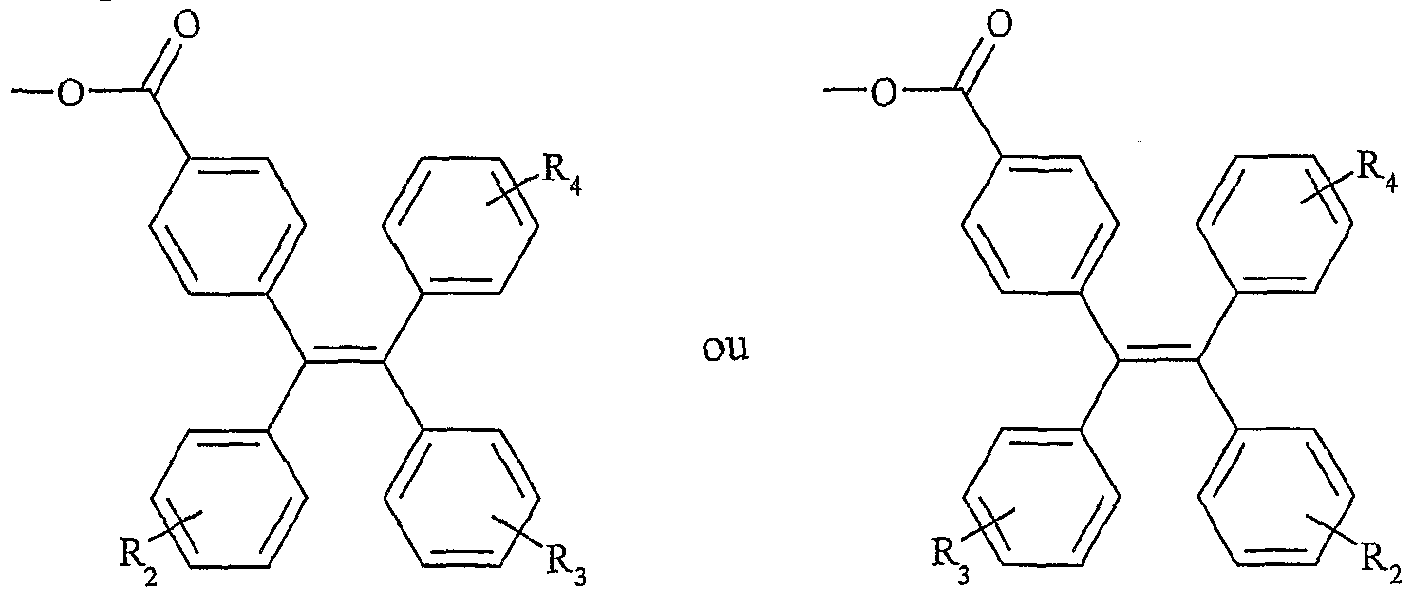
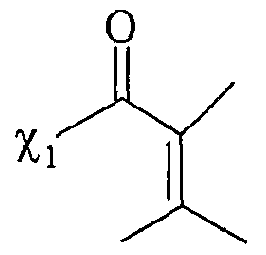
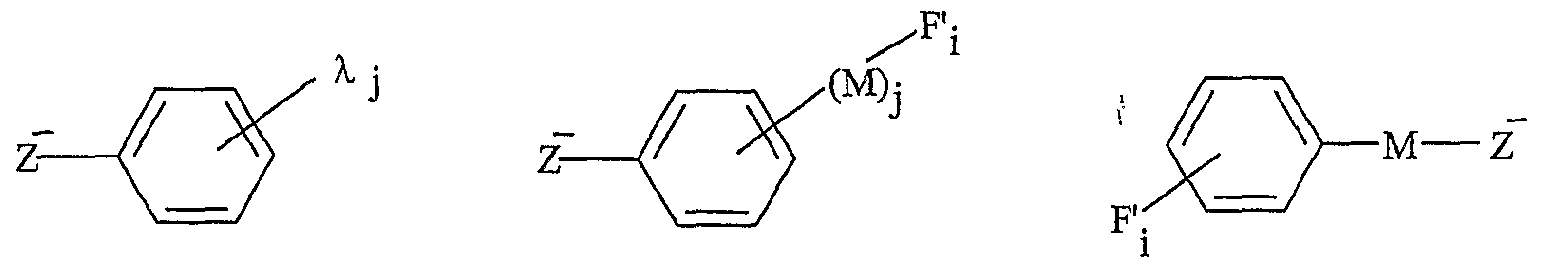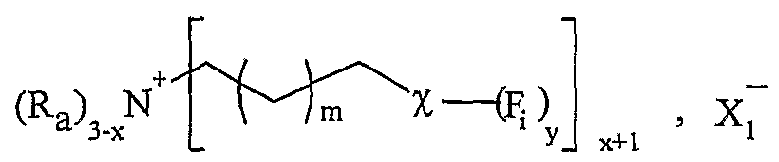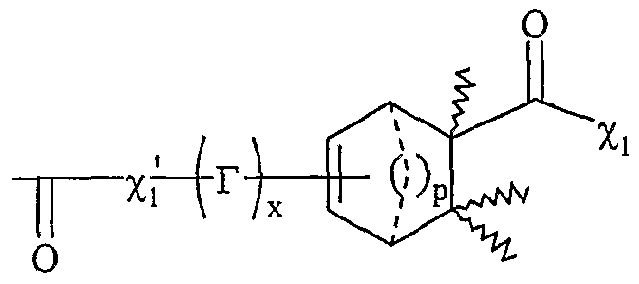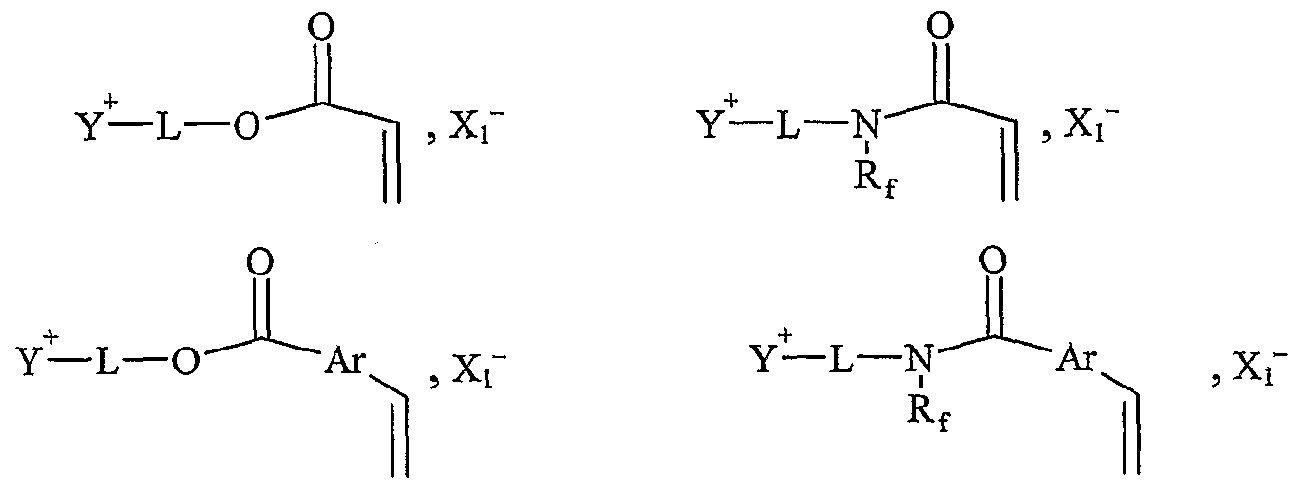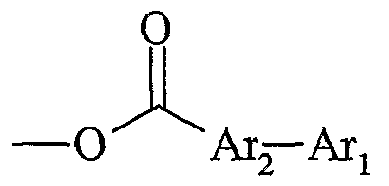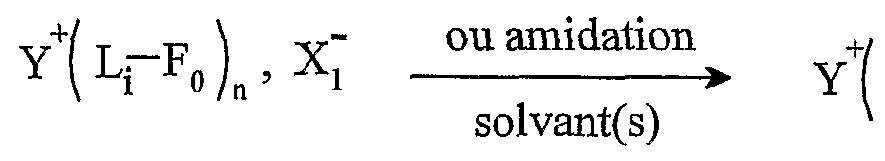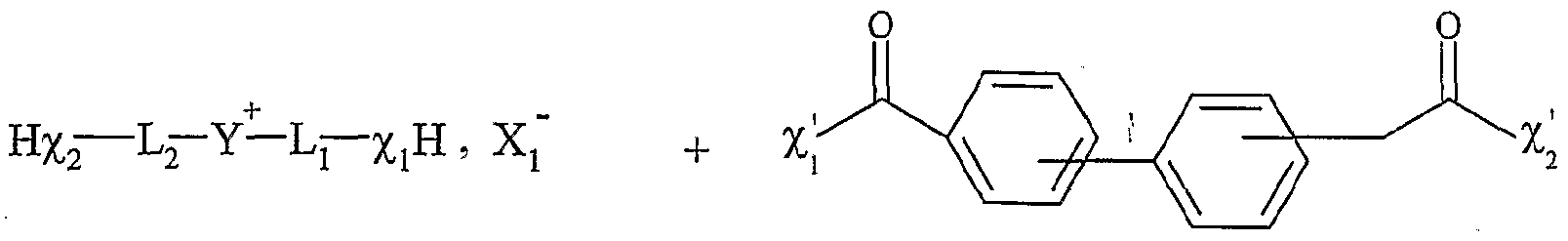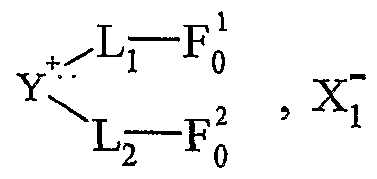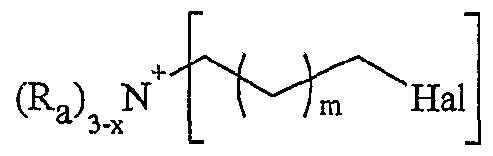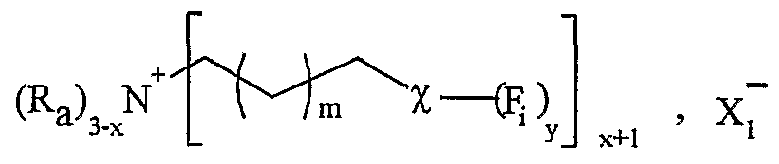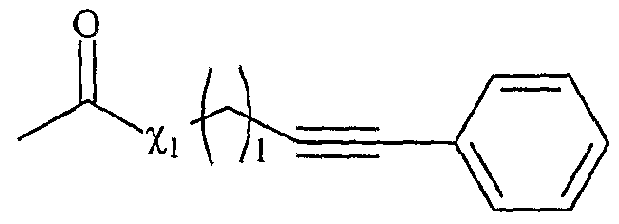UTILISATION DE SELS D'ONIUM FONCTIONNALISÉS EN TANT QUE SUPPORT SOLUBLE POUR LA SYNTHÈSE ORGANIQUE La présente invention a pour objet l'utilisation de sels d'onium fonctionnalisés en tant que support soluble pour la synthèse organique. Depuis l'introduction de la méthode de Merrifield pour la synthèse de peptides (Merrifield, 1963), les supports insolubles de type résines ont été introduits dans beaucoup de synthèses pour faciliter la purification des produits et plus particulièrement dans le domaine de la chimie combinatoire au cours de ces 10 dernières années
(Thompson et al., 1996 ; Toy et al., 2000 ; P.; Seeberger et al., 2000 ; V.,Krchnja'k et M., W. Holladay, 2002 ; Mϋtter et al., 1979 ; Han et al, 1995 ; Harris et al., 1992 ; Nicolaou et al., 2002 ; Kates et al, 2000). Quoique très efficace, la synthèse sur support solide souffre toujours d'un certain nombre de problèmes liés à la nature hétérogène des conditions de réactions. En effet, les comportements cinétiques non linéaires, les distributions inégales, les sites non accessibles aux réactifs, les problèmes de solvatation, de synthèse pure posés par la phase solide ainsi que l'identification complexe des résines greffées restent des handicaps majeurs pour cette méthodologie. Les inconvénients des supports solides ont conduit à l'exploration d'alternatives pour retrouver des conditions de réactions homogènes. De fait, l'utilisation des polymères solubles contourne les difficultés de la synthèse sur supports solides tout en conservant une grande partie de ses aspects positifs. La dénomination "Synthèse sur polymère soluble ou SPOS" est utilisée pour les réactions en phase liquide homogène réalisée sur un polymère fonctionnel soluble qui sert de groupement protecteur et dont les propriétés macromoléculaires facilite la purification des produits (Haag, 2001 ; Haag et al, 2002 ; Kim et al., 2000 ; Kim et al., 1996 ; Hovestad et al, 2000 ; Hodge, 1997 ; Frank et al, 1975 ; Han et al, 1997). Ces méthodologies en phase-liquide évitent les difficultés de la synthèse en phase solide, par exemple le comportement cinétique non linéaire, la distribution (l'accès inégal aux sites d'interaction), les problèmes de solvatation liés à la nature du support, et les conditions opératoires qui ne sont pas transposables de manière triviale entre les réactions organiques standards en solution et la phase solide. Cependant, en remplaçant les résines réticulées insolubles par des supports polymères solubles, les avantages du support solide sont préservés : conditions de réactions classiques en milieu homogène
mais aussi purification aisée des produits. De plus, les supports solubles présentent la possibilité d'analyse par les moyens classiques utilisés en chimie organique tels que les spectroscopies UN-visible, JR.TF, et RMΝ aussi bien que la spectrométrie de masse à haute résolution. En outre, la chromato graphie sur couches minces peut être employée poux le suivi des réactions sans exiger le clivage préhminaire du support (Mϋtter et al.,
1979 ; Han et al., 1995 ; Harris, 1992), ce qui est un avantage de cette technologie. Une caractérisation rapide et efficace du support est un outil important, particulièrement pour les synthèses parallèle, combinatoire ou multiétape. De la même façon qu'en phase solide, les supports solubles peuvent être séparés des molécules de faible poids moléculaire après chaque étape par ultrafiltration, dialyse, chromatographie préparative par exclusion (SEC), ou précipitation. Même si l'automatisation de ces techniques n'est pas aussi avancée qu'avec les résines, des progrès importants ont été faits ces dernières années. Les polymères utilisables comme supports solubles doivent être commercialement accessibles ou facilement préparés, être chimiquement stables, être correctement fonctionnalisés pour pouvoir accrocher une partie organique et être très solubles dans les solvants usuels. En général les polymères sont un mélange de molécules de différentes tailles qui ont des propriétés différentes. Les supports solubles devront avoir une polydispersité aussi proche de 1 que possible et une masse moléculaire assez élevée pour être cristallisés à la température ambiante. La plupart des polymères solubles utilisés ont des squelettes hydrocarbonés (polymères de
Janda) ou alkyle polyéthers et plus particulièrement les pol éthylènes glycols (PEGs). Jusqu'à maintenant, le polymère le plus utilisé comme support soluble en synthèse organique est le polyéthylène glycol monométhylé (MPEG 5000) ne contenant qu'une fonction OH ou diol et de ce fait a une faible charge spécifique (0,2 m ol OH/g)(Mutter et al., 1974 ; Mutter et al., 1975 ; Gravert et al., 1997 ; Toy et al, 2000).
Plus récemment, des PEGs de plus grande charge spécifique ont été préparés, pouvant être purifiés par précipitation. Ces PEGs en forme d'étoile (Chang et al., 1999 ; Knischka et al., 2000 ; Reed et al., 2000) ou ramifiés (Benaglia et al., 1998 ; Annunziata et al., 2000) sur les positions terminales peuvent atteindre des charges spécifiques de 1 mol de OH/g de polymère mais sont longs à préparer. Les polymères linéaires portant des groupes fonctionnels sur chaque unité monomère, comme l'alcool polyvinylique (EUas, 1997), les polyacrylamides (Wellings et al., 1987 ; Ranucci et al., 1994) et les polymères préparés par ROMP ("ring opening metathesis polymerisation")(Barrett et al., 1999 ; Barrett et al., 2000 ; Barrett et al,
2000) ont aussi été utilisés en SPOS. Ces polymères à forte charge spécifique sont d'utilisation problématique dans certains cas à cause de leur solubilité et stabilité limitées (Meier et al. , 2001 ) . Aussi, des dendrimères parfaits (polyamidoamine, polysilane, polyester) ont-ils été utilisés comme supports en synthèse combinatoire. Ces supports solubles polymériques de grande charge spécifique théorique sont fragiles, de poids moléculaires relativement faibles et sont préparés par voie multiétape, ce qui limite leur utilisation en chimie combinatoire (Burgath et al., 2000). Les polyéthers dendritiques aliphatiques (analogues ramifiés des PEGs) sont par contre chimiquement stables dans beaucoup de conditions de réaction et donc devraient être utiles comme supports polymères en synthèse organique. De plus, les propriétés chimiques de ces matériaux sont idéales pour la synthèse supportée en solution. Par ailleurs, la forme globulaire de ces polymères dendritiques peut favoriser leur purification par les techniques de membranes (dialyse et ultrafiltration) Les polyéthers aliphatiques dendrimériques contenant des unités 1-3 diol et 1-2 diol ont été préparés récemment en 6-7 étapes. Ils ont des charges spécifiques de 6-7 mmol de OH/g mais ils sont longs à préparer et de poids moléculaires relativement faibles (Jayaraman et al., 1998 ; Grayson et al., 1999 ; Haag et al., 2000). Haag a rapporté récemment la synthèse contrôlée de polyglycérols dendritiques. Ces polyéthers polyols aliphatiques ont un squelette stable et sont facilement préparés en une étape à l'échelle de 1 kg (Haag et al., 2002). Ils ont des poids moléculaires pouvant atteindre 30 000 g/mole avec une polydispersité Mw Mn~l,5. La structure dendrimérique contient statistiquement des unités glycérol incorporées linéairement (OH primaires et secondaires) et des 1,2 diols terminaux. La densité totale de groupes fonctionnels atteint 13,5 mmol de OH/g de polymère dont environ 30% (4,1 mmol/g) sont des 1,2 diols terminaux facilement accessibles et qui peuvent être utilisés directement pour greffage d'aldéhydes ou de cétones sur ces polymères sous forme d'acétals. Ces polyglycérols dendritiques ont été utilisés en SPOS pour l'aménagement fonctionnel de cétones ω-halogénées et pour le couplage de Suzuki. Un autre problème vient de leur structure : les fonctions OH à la périphérie utilisables pour la synthèse ne sont pas identiques, dans la mesure où il y a un mélange de OH secondaires et primaires qui n'auront pas la même réactivité et entraîneront donc des problèmes de sélectivité et probablement de réactivité secondaire.
Malgré les différents avantages présentés par les supports polymères solubles actuellement utilisés, les inconvénients liés à leur poids moléculaire élevé et à leur charge spécifique utilisable limitée sont de sévères handicaps à une utilisation généralisée. En effet, les PEGs les plus souvent utilisés pour la synthèse organique et la chimie combinatoire sur supports solubles ont des poids moléculaires compris entre
2000 et 10000 daltons et ne présentent qu'une charge spécifique comprise entre 0,1 et 1 mmol/g de polymère. L'analyse par spectroscopies RMN 1H et 13C de ces polymères en solution est réalisable mais présente des difficultés car les signaux correspondant aux protons et aux carbones des PEGs ont une forte intensité par rapport aux signaux relatifs aux protons et carbones du substrat supporté. Des problèmes de viscosité des solutions sont aussi rencontrés à forte concentration. Les problèmes de purification de ces polymères sont aussi une sévère limitation à leur utilisation et à leur recyclage, en particulier dans une optique d'automatisation des synthèses. Depuis quelques années, certains sels d'onium sous l'appellation "liquides ioniques" (Welton et al., 1999 ; Wasserscheid et al., 2000 ; Wasserscheid et Welton,
2003), moyennant un choix approprié des anions, sont de plus en plus utilisés en synthèse organique et en catalyse car ils présentent un certain nombre de propriétés • physico-chimiques intéressantes et importantes telles que leur grande stabilité thermique, leur faible volatilité et leur tension de vapeur très faible, leur faible inflainmabilité, leur fort pouvoir de solubilisation aussi bien des sels que des molécules organiques neutres et des polymères et enfin la possibilité d'un recyclage aisé. Cependant, les sels d'onium décrits dans la littérature sont généralement synthétisés et utilisés en tant que tels pour leurs propriétés biologiques ou physiques (tensioactifs par exemple). La présente invention a pour but de fournir une nouvelle utilisation des sels d'onium en tant que supports solubles pour la synthèse organique en phase homogène en présence d'au moins un solvant organique. La présente invention a pour but de fournir de nouveaux supports solubles pour la synthèse organique en présence d'au moins un solvant, en remplacement des supports solubles de l'art antérieur tels que les PEG, lesdits nouveaux supports solubles étant plus faciles à préparer, à utiliser et à purifier, parfaitement définis et identifiés, moins coûteux et de fonctionnalisation aisée. La présente invention a pour but de fournir de nouveaux supports solubles présentant une charge spécifique élevée et dont le recyclage est aisé.
La présente invention concerne l'utilisation d'un sel d'onium fonctionnalisé par au moins une fonction organique, en tant que support soluble, en présence d'au moins un solvant organique, pour la synthèse organique d'une molécule, en phase homogène, par au moins une transformation de ladite fonction organique, ledit sel d'onium permettant la libération de la molécule synthétisée, ledit sel d'onium se présentant sous forme liquide ou solide à température ambiante, et répondant à la formule Aι+, Xf , dans laquelle : - A\ représente un cation, - Xi- représente un anion, Aι+ étant un cation fonctionnel ou polyfonctionnel, et/ou Xi- étant un anion fonctionnel ou polyfonctionnel, le sel d'onium étant tel que sous sa forme initiale, c'est-à-dire avant la première transformation de ladite fonction organique, Aι+ et XX ne sont pas liés entre eux par une liaison covalente, et lorsque F anion et le cation portent respectivement une fonction organique, celles-ci ne peuvent pas réagir entre elles avant la première transformation de ladite fonction organique. L'utilisation en tant que support soluble, donc en solution dans un solvant ou un mélange de solvants, de sels d'onium est démontrée dans le cadre de l'invention. Ceci n'était pas a priori évident compte tenu des connaissances sur ces sels. Sous forme d'halogénures, ils sont généralement peu solubles dans les solvants usuels utilisés en chimie organique. Par ailleurs, leur fonctionnalisation peut poser, a priori, des problèmes de chimiosélectivité à cause de la présence d'un groupement onium chargé positivement qui est sujet à la β-élimination ou à la déprotonation en α en milieu basique. L'expression "sel d'onium fonctionnalisé" désigne les sels d'ammonium, de phosphonium, de sulfonium, ainsi que tous les sels résultant de la quaternarisation d'une aminé, d'une phosphine, d'une arsine, d'un thioéther ou d'un hétérocycle contenant l'un ou plusieurs de ces hétéroatomes, et portant au moins une fonction organique F; ou F',. Cette expression désigne aussi un sel d'onium dont le cation tel que défini ci-dessus n'est pas fonctionnalisé mais dont l'a-nion porte une fonction F',. Cette expression peut
également désigner un sel dont Fanion et le cation portent au moins une fonction organique. L'expression "support soluble" désigne une molécule fonctionnelle polymère ou un sel servant d'"ancre" pour effectuer, en solution, des transformations successives d'une molécule accrochée par la fonction. Cette ancre confère des propriétés à la molécule accrochée (donc finalement à l'ensemble formé par l'ancre et la molécule accrochée) qui permettent de purifier aisément par lavage, évaporation ou tout autre technique. Ceci ne pourrait être fait facilement avec des molécules volatiles et/ou solubles dans les solvants usuels par exemple. En utilisant cette technique, on peut utiliser des excès de réactifs, par exemple, comme dans le cas des résines de Merrifield insolubles. Un support soluble doit par définition être soluble dans un solvant. Ceci confère l'avantage d'effectuer les réactions en solution et de pouvoir en suivre l'avancement à l'aide de techniques d'analyse classiquement utilisées dans le domaine de la chimie organique. Un support soluble doit également être récupérable à la fin des transformations. En d'autres termes, les molécules synthétisées sur ce support doivent pouvoir être facilement décrochées. Par ailleurs, le squelette du support soluble ne doit pas réagir avec les réactifs utilisés, les réactions ayant lieu sélectivement sur .les fonctions accrochées sur le squelette de base. L'expression "synthèse organique d'une molécule en phase homogène" désigne la ou les transformations) de la ou des fonctions chimiques portée(s) par ledit sel d'onium, suivie d'une réaction de clivage libérant la molécule recherchée en solution dans un solvant donné ou dans un mélange de solvants et le sel de départ ou un sel recyclable en le support de départ. L'expression "cation fonctionnel" désigne un groupe moléculaire qui possède au moins une fonction chimique, ainsi qu'une tête portant une charge positive. L'expression "anion fonctionnel" désigne un groupe moléculaire qui possède au moins une fonction chimique, ainsi qu'une tête portant une charge négative. Les sels d'onium susmentionnés sont solubilisés dans un solvant organique ou dans un mélange de solvants organiques puis mis en présence d'un excès ou non de réactif. Ils sont alors utilisés comme supports solubles. Une autre propriété de ces sels est qu'ils ne sont pas solubles dans certains solvants usuels tels que Féther, les alcanes ou les hydrocarbures par exemple. De plus, ils ont des tensions de vapeur extrêmement faibles et peuvent donc être placés sous un vide poussé sans pertes. Ces deux propriétés permettent l'utilisation d'un excès de réactif qui sera ensuite facilement éliminé par
lavage, par extraction ou sous vide poussé comme dans le cas des réactions sur résines ou sur PEG. De très nombreuses transformations chimiques des sels d'onium fonctionnels sont possibles. On peut utiliser ces sels comme des résines ou des polymères solubles. La charge spécifique d'un support est définie par la quantité de réactif qui peut être supportée par gramme de support et s'exprime en mmol/g. Cela correspond en fait à ce que l'on pourrait appeler la fonctionnalité spécifique d'un support notée /que l'on pourra exprimer en millifonction par gramme (mf/g). Si le sel est monofonctionnel, la fonctionnalité spécifique (f exprimée en mf/g) sera égale à la charge spécifique exprimée en mmol/g. Si le sel porte n fois la même fonction, la fonctionnalité spécifique
/ sera égale à n fois la charge spécifique x du sel. Lorsque les sels d'onium sont en solution, ce qui est pratiquement toujours le cas, il faut adapter les notions définies ci- dessus. La molarité d'une solution sera exprimée en mol/1 ou en mmol/ml. Connaissant la densité des solutions, il est alors facile de convertir en mmol/g de solution donnant ainsi des éléments précis sur la charge spécifique des solutions pour comparaison avec les résines de Merrifield ou les solutions de supports solubles de type polymères (PEG ou autres). Si le sel porte n fois la même fonction, une solution contenant par exemple une millimole de ce sel par gramme aura une fonctionnalité spécifique/de n mf/g. Les sels d'onium monofonctionnalisés tels qu'utilisés dans le cadre de la présente invention présentent une charge spécifique supérieure à 1 mmol.g"1 et pouvant atteindre jusqu'à 7 mmol.g"1, à comparer avec celle du PEG 5000 qui est de 0,2 mmol.g"1. L'expression "transformation de la fonction organique" désigne la modification d'une fonction F; par un ou plusieurs réactifs et/ou catalyseurs et/ou par activation physique (chauffage, micro-ondes, ultrasons, radiations hv, pression, électrochimie...). L'expression "sel d'onium sous sa forme initiale" désigne le sel dans lequel la fonction organique initiale n'a pas encore subi de transformation, c'est-à-dire n'a pas encore été impliquée dans une réaction, cette fonction étant dans ce qui suit désignée par F0. L'expression "première transformation de la fonction organique" désigne la modification de la fonction organique initiale portée par le sel d'onium sous sa forme initiale et qui sera symbolisée dans ce qui suit par la modification de Fo en V\. Un sel dans lequel le cation et Fanion portent respectivement une fonction organique initiale nommée FQ et F'o, et dans lequel Fanion et le cation ne réagissent pas
8 I U I II I\ i. W W "»' » M « »
ensemble avant la première transformation desdites fonctions organiques, est un sel dans lequel les fonctions Fo et F'o sont chimiquement compatibles ou encore chimiquement inertes l'une par rapport à l'autre. Ceci signifie donc que le sel en question est stable. Fo et F'o pourront par contre réagir l'une sur l'autre sous l'effet d'une activation quelconque qui pourra être physique (radiations hv, micro-ondes, pression, chauffage...) ou chimique (catalyseur, autre réactif...) Cette nouvelle façon de faire de la synthèse supportée est également nommée
SOSSO pour Synthèse Organique Supportée sur Sel d'Onium (OSSOS en anglais pour
Onium Sait Supported Organic Synthesis). Une utilisation avantageuse selon l'invention est caractérisée en ce que le sel d'onium est purifié et/ou recyclé sous sa forme initiale après la libération de la molécule synthétisée. Les procédés préférés de purification et/ou de recyclage utilisés sont notamment un procédé simple de lavage ou de recristallisation dans un solvant approprié. Une utilisation avantageuse de la présente invention est caractérisée en ce que les cations et anions fonctionnels correspondent à une entité ionique, respectivement cationique Y"1"- et anionique Z~-, liée, éventuellement par l'intermédiaire d'un -bras, respectivement L et M, notamment un groupe alkyle ou aralkyle ou aïkaryle comprenant de 1 à 30 atomes de carbone, à au moins respectivement une fonction F; et F'j, Fi variant de Fo à Fn, F'j variant de F'o à F'n, n étant un nombre entier variant de 1 à 20, le cation fonctionnel A + pouvant être représenté sous la forme Y+-L-Fi, et Fanion fonctionnel X ~ sous la forme Z~-(M-)ir-F'i, k étant égal à 0 ou 1. L'expression "entité ionique" désigne la partie du cation ou de Fanion, qui porte la charge, respectivement positive ou négative. Les fonctions Fi et F' i sont notamment choisies parmi les fonctions suivantes : hydroxyle, acide carboxylique, amide, sulfone, aminé primaire, aminé secondaire, aldéhyde, cétone, éthényle, éthynyle, diényle, éther, époxyde, phosphine (primaire, secondaire ou tertiaire), azoture, i ine, cétène, cumulène, hétérocu ulène, thiol, thioéther, sulfoxyde, groupements phosphores, hétérocycles, acide sulfonique, silane, stannane ou aryle fonctionnel, et toute fonction résultant d'une transformation des fonctions précédentes par voie c-himique ou induite par activation thermique, électrochimique, photochimique ou par toute autre technique physique telle que les irradiations micro-ondes, les ultrasons ou par pression.
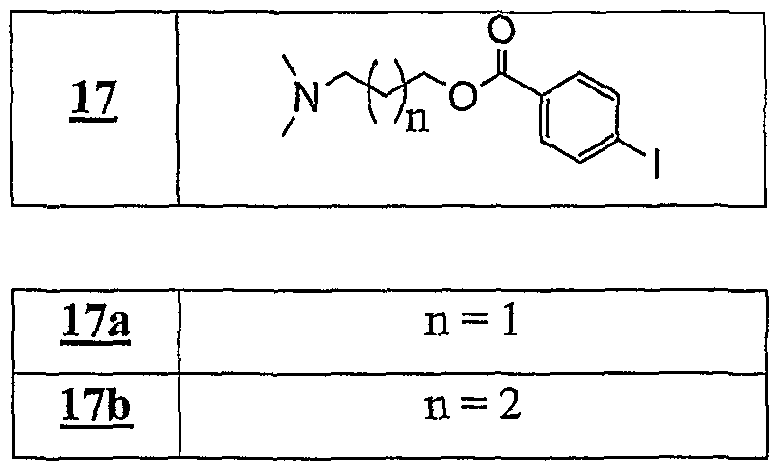
Dans le terme "Y*-", le tiret "-" représente la liaison éventuelle entre l'entité cationique et le bras L. Dans le terme "Z"-", le tiret "-" représente la liaison éventuelle entre l'entité anionique et le bras L. Le terme "bras L" désigne une chaîne alkyle ou aralkyle ou aïkaryle pouvant contenir un ou plusieurs hétéroatomes tels que l'azote, le phosphore, le soufre, l'oxygène, le silicium, étain, contenant entre 1 et 30 atomes de carbone, et ledit bras est notamment choisi parmi une chaîne alkyle contenant de 2 à 20 atomes de carbone et de 1 à 6 atomes d'oxygène ou d'azote. Selon un mode de réalisation avantageux, le sel d'onium fonctionnalisé A_+, X ~ utilisé dans le cadre de la présente invention est soluble dans un solvant organique. Selon un mode de réalisation avantageux, le sel d'onium fonctionnalisé Aι+, Xi- utilisé dans le cadre de la présente invention est liquide à température ambiante. Selon un mode de réalisation avantageux, le sel d'onium Aι+, Xi- utilisé dans le cadre de la présente invention est solide à température ambiante et est liquéfiable dans une gamme de températures allant d'environ 25°C à environ 450°C, notamment d'environ 30°C à environ 15Ô°C. Une utilisation avantageuse de l'invention est caractérisée en ce que les fonctions organiques Fi et F'i sont choisies parmi les fonctions classiques de la chimie organique, telles que les fonctions hydroxyle, acide carboxylique, a ide, sulfone, a iné primaire, aminé secondaire, aldéhyde, cétone, éthényle, éthynyle, diényle, éther. époxyde, phosphine (primaire, secondaire ou tertiaire), azoture, imine, cétène, cumulène, hétérocumulène, thiol, thioéther, sulfoxyde, groupements phosphores, hétérocycles, acide sulfonique, silane, stannane ou aryle fonctionnel. Une utilisation avantageuse de la présente invention est caractérisée en ce que le poids moléculaire du sel d'onium fonctionnalisé est inférieur à 1500 g.mol"1, notamment inférieur à 750 g.mol"1, et est de préférence compris de 130 à 500 g.mol"1. Pour assurer une bonne productivité du support, il est nécessaire que la masse moléculaire du sel soit la plus faible possible (en raison de la charge spécifique telle que définie précédemment). Ainsi, on utilise de préférence des anions de masse la plus faible possible comme les chlorures afin de pouvoir utiliser éventuellement des cations de masse plus élevée (voir tableau 17 plus loin représentant les variations de charge spécifique en fonction de la masse).
10 rbli K fC W l y . υ u * %* ~e
Une utilisation avantageuse de la présente invention est caractérisée en ce que Aι+ est un cation fonctionnel et en ce que Xj~ est un anion non fonctionnel. L'expression "anion non fonctionnel" désigne un groupe moléculaire qui ne possède pas de fonction chimique, une partie de ce groupe portant une charge négative. Ce mode de réalisation permet d'effectuer des réactions spécifiques sur la partie cationique du sel d'onium. On peut donc maîtriser la sélectivité et la réactivité qui pourrait être différente sur anion et le cation. La présente invention concerne également une utilisation telle que définie ci- dessus, dans laquelle le sel d'onium A_+, Xf" a comme forme initiale Y+-L-Fo, Xf, pour l'obtention d'une molécule G, par transformation de ladite fonction initiale Fo selon le schéma
Y-L-F0 » X " *- Y-L-F, . Xj ~ — *- - — *- Y-L-Fn » — *- G + YtL-F0 > X~
L étant tel que défini ci-dessus, ladite molécule G étant obtenue par clivage au niveau de la fonction Fn, et le sel d'onium fonctionnalisé pouvant être récupéré ou recyclé sous sa forme initiale Y -L-Fo, Xf", après la libération de G. Les réactions utilisées pour la libération de G par clivage sont notamment les suivantes : τransestérification, transamidation, réduction, lactonisation, lactamisation, cyclisation décrochante et couplage décrochant. Une utilisation avantageuse de l'invention est caractérisée en ce que le cation fonctionnel Aι+ est choisi parmi les cations pyridinium, imidazolium, ammonium, phosphonium ou sulfonium, cycliques ou non, substitués ou non, et de préférence ammonium ou phosphonium. Une utilisation avantageuse selon l'invention est caractérisée en ce que le cation fonctionnel Aj+ est choisi parmi les cations ammonium quaternaire, cycliques ou non. Une utilisation avantageuse selon l'invention est caractérisée en ce que X est un anion fonctionnel et A_+ est un cation non fonctionnel. L'expression "cation non fonctionnel" désigne un groupe moléculaire qui ne possède pas de fonction chimique, une partie de ce groupe portant une charge positive. Ce mode de réalisation permet d'effectuer des modifications fonctionnelles uniquement sur la partie anionique.
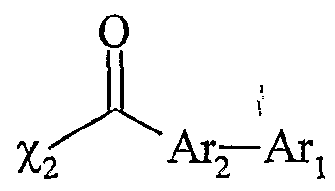
La présente invention concerne également une utilisation telle que définie ci- dessus, dans laquelle le sel d'onium Aχ+, Xf a comme forme initiale A_+, Z~-(M)k-FO, pour l'obtention d'une molécule G, par transformation de ladite fonction initiale F'o selon le schéma Z-(M)k-F; *- A* , Z-(M)k-Fj — A^ , Z-(M) Fi -→- G + A* , z"-(M) F; k et M étant tels que définis ci-dessus, ladite molécule G étant obtenue par clivage de la fonction F'n, et le sel d'onium fonctionnalisé pouvant être récupéré ou recyclé sous sa forme initiale A_ , Z~-(M)j--F'o, après la libération de G. Les réactions utilisées pour la libération de G par clivage sont notamment les suivantes : transestérification, transamidation, féduction, lactonisation, lactamisation, cyclisation décrochante et couplage décrochant. Une utilisation avantageuse de l'invention est caractérisée en ce que Xf est choisi parmi : - la famille des phosphates : RιPO4 2", RIR2P0 ", - la famille des sulfates : RtSO^, - la famille des sulfonates : RtSOf, - la famille des carboxylates : RιC02 ", ou parmi les anions suivants : λ R1NSO2R2 R2SO2NSO2R2 Rj-Cf Y
77, M et F'; étant tels que définis ci-dessus, 77 représentant notamment O-, S0
3 ~, CO2
" RιPO
3 " ou RιPθ2
", j représentant un nombre entier compris de 1 à 5, Ri et R
2 pouvant représenter indépendamment l'un de l'autre un groupe alkyle fonctionnel, un groupe vinyle ou alcynyle, éventuellement fonctionnel, comprenant de 1 à 20 atomes de carbone, ou pouvant représenter un groupement aryle fonctionnel comprenant de 6 à 30 atomes de carbone,
γ et λ représentant un groupe électroattracteur, notamment choisi parmi les groupes : CO
2R'ι, SO^, CN, NO
2, P(O)(OR'
2, C(O)R'ι et SO
3R'
l5 R' i représentant un groupe alkyle, éventuellement fonctionnel, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle, éventuellement fonctionnel, comprenant de 6 à 30 atomes de carbone. Une utilisation avantageuse selon l'invention est caractérisée en ce que Aι
+ est un cation fonctionnel et Xf est un anion fonctionnel. Ce mode de réalisation permet d'effectuer des transformations en parallèle ainsi que la réaction intra- ou intermoléculaire d'une fonction de Af
" avec une fonction de Xf. La présente invention concerne également une utilisation telle que définie ci- dessus, dans laquelle le sel d'onium Aι
+, Xf a comme forme initiale
Y^-L-Fo, Z"""-(M)k-F'o, pour l'obtention d'une molécule G, par transformation desdites fonctions initiales F0 et F'o selon le schéma Y-L-F0 : Z"-(M)k-F; »- Y^L-F. . Z-(M)k-F; -→- - — * Y-L-Fn . Z~-(M) Εn <
L, k et M étant tels que définis ci-dessus, et par réaction de Fn sur F'n dans le sel d'onium fonctionnalisé Y-L-Fn . Z-(M)— Fn conduisant à la formation d'un sel interne de formule : Y+-L-F— F;— (M) z- ladite molécule G étant obtenue par clivage du sel interne susmentionné et correspondant à la formule Fn+2-F'n+2, et le sel d'onium fonctionnalisé pouvant être récupéré ou recyclé sous sa forme initiale Y+-L-F0, Z~-(M)k-F'o, après la libération de G. Ce mode de réalisation permet d'effectuer des réactions intramoléculaires. L'expression "sel interne" désigne une entité portant simultanément au moins un groupe chargé positivement et un groupe chargé négativement, distants d'au moins 2 atomes reliés par des liaisons covalentes. Une utilisation selon la présente invention est caractérisée en ce que le sel d'onium est choisi parmi les sels suivants :
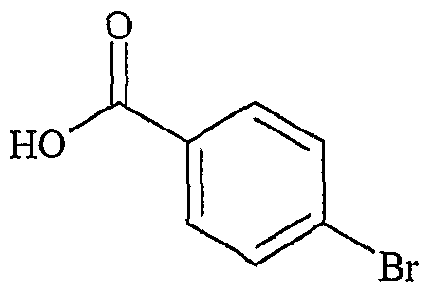
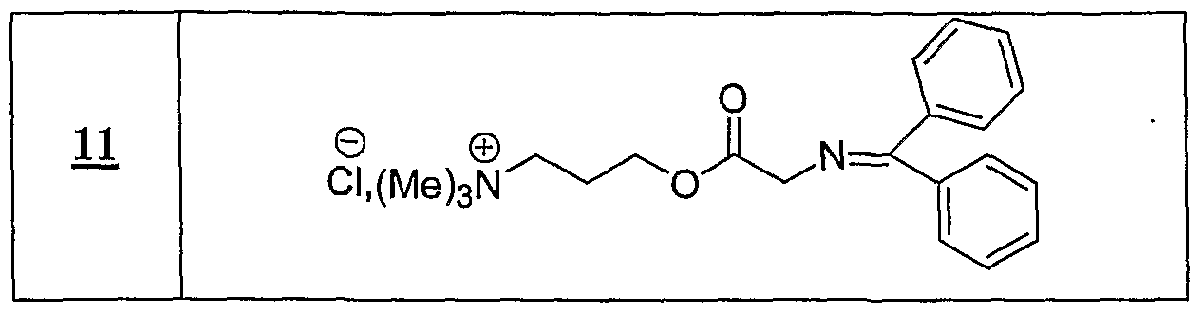
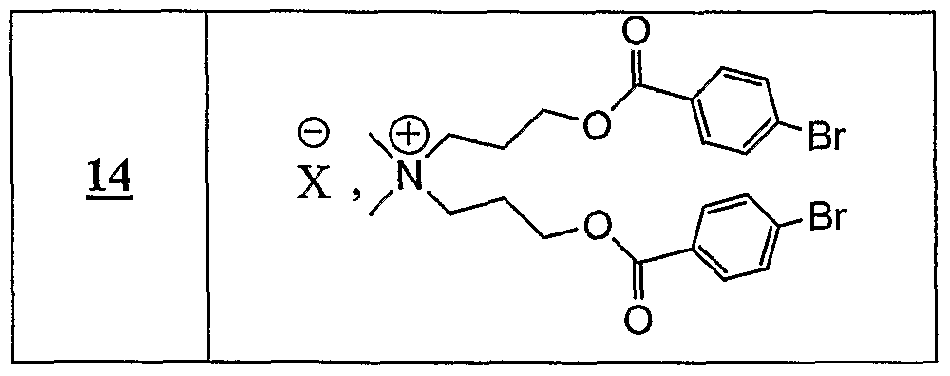
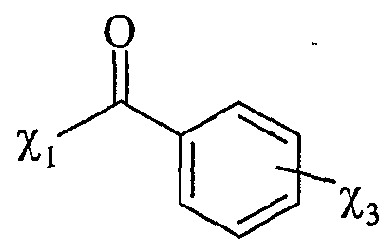
(Ru), P NHMe , X (Ra) '3,-x P COOH X, x+l
R représentant un atome d'hydrogène, un groupe alkyle, aïkaryle ou aralkyle, fonctionnel ou non, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle, fonctionnel ou non, comprenant de 6 à 30 atomes de carbone, x représentant un nombre entier compris de 0 à 3, y représentant un nombre entier compris de 1 à 5, -Ar représentant un noyau aromatique fonctionnel ou polyfonctionnel, F,- étant tel que défini précédemment, Hal représentant un atome d'halogène, notamment choisi parmi le chlore, le brome et l'iode, χ représentant un carbocycle ou un hétérocycle fonctionnel, Xf étant choisi parmi : NTff, PF
5 "", BF , CF, Br
", T, CF
3SO
3 ", MeSO , EtSO , MeSO
3 ", C
fiH
5SOf , pMeC
6H
4SOf, m étant un nombre entier compris de 0 à 20, Rp représentant un groupe diényle, vinyle, substitué ou non, alkyle fonctionnel comprenant de 1 à 20 atomes de carbone, ou aryle fonctionnel comprenant de 6 à 30 atomes de carbone, alcynyle substitué ou non, et étant notamment un groupe alkylvinyle, alkylalcynyle, alkylaryle, alkyldiényle, alkylmalonyle, acyle, et R
a représentant un groupe alkyle ramifié ou non comprenant de 1 à 20 atomes de carbone, notamment un groupe éthyle, propyle, butyle, pentyle, hexyle, heptyle ou octyle. La présente invention concerne également une utilisation telle que définie ci- dessus, caractérisée en ce que le ou les solvant(s) utilisé(s) est un solvant aprotique, choisi parmi : - les solvants dont la constante diélectrique ε est inférieure ou égale à 2, tels que les alcanes, les carbures aromatiques comme le benzène, le toluène ou le xylène, - les solvants dont la constante diélectrique ε est comprise entre environ 2 et 15, tels que les éthers, les halogénobenzènes ou le dichlorométhane, et - les solvants dont la constante diélectrique ε est supérieure à 15, tels que acétonitrile, le nitrométhane, le DMF ou la diméthylacétamide. On utilise souvent la constante diélectrique ε et/ou le moment dipolaire pour caractériser la polarité d'un solvant. Plus récemment, le paramètre de Dimroth-
Reichardt ET a été proposé pour mieux décrire la polarité des solvants (Reichardt, 1988). Les solvants avantageusement utilisés dans l'invention sont le toluène, le dichlorométhane, le THF, l'acétonitrile et le DMF. La présente invention concerne également une utilisation telle que définie ci- dessus, pour la synthèse organique, en continu, en discontinu, combinatoire, ou parallèle, et/ou pour la préparation de banques de produits. Les avantages de l'utilisation des sels d'onium susmentionnés sont les suivants : - de très nombreux sels d'onium fonctionnels sont connus, facilement accessibles et certains sont commerciaux ; - les modifications fonctionnelles des sels d'onium sont généralement simples et facilement réalisables selon des méthodes décrites dans la littérature ; si elles n'existent pas, elles pourront être mises au point à partir des connaissances en chimie organique ; - les réactions ont lieu en phase homogène, ce qui implique que toutes les connaissances de la réactivité en chimie organique, organométallique et catalytique sont applicables ; de plus, toutes les techniques d'analyse, incluant les RMN 1H,
13C,
19F,
31P,
πB,
15N etc ., la CLHP, FIRTF, FUV-visible, la fluorescence, les techniques électrochimiques, Félectrophorèse, la spectrométrie de masse etc ., peuvent être utilisées dans les conditions normales sans complications particulières ; - les réactions sont réalisées aux concentrations habituelles de 0,5 à 1 mole par litre (voir beaucoup plus) ce qui représente un énorme avantage en terme de charge spécifique ; - la purification des intermédiaires est généralement facile ; - le recyclage de ces supports est aisé ; - les solutions de sels dans les solvants habituels sont facilement transférables à l'aide des techniques de seringue et ou de pompage ; - les solutions de sels d'onium dans les solvants organiques se prêtent aisément aux techniques de partition et donc aux techniques de synthèse en parallèle ou combinatoire ; des bibliothèques de produits peuvent donc être facilement synthétisées ; - les réactivités et les sélectivités dépendent de la nature de Fanion ou du cation ; - la montée en échelle ("scale up") ne pose pas de problèmes ce qui présente un avantage majeur par rapport aux résines et aux polymères solubles ; - une analogie est facilement établie entre cette nouvelle technologie et les techniques de synthèse sur résines de type Merrifield ou sur polymères solubles de type
PEG, PG ou JANDA ; ces sels d'onium peuvent être fonctionnalisés comme les résines de Wang, Rink, silylalkyle, carbonate, carboxylique, formyle, hydroxyle, amino, oxime etc. ou les polymères fonctionnalisés mais sont d'une utilisation beaucoup plus aisée et avantageuse ; - ils sont beaucoup moins coûteux ; cet avantage économique est très important car de nature à ouvrir un marché de substitution de grande ampleur. La présente invention concerne également une utilisation telle que définie ci- dessus, pour la mise en œuvre de réactions de cycloaddition, de préférence pour la mise en œuvre de la réaction de Diels-Alder, selon l'un des schémas réactionnels suivants :
a)
solvant(s) cycloaddition 4+2 de Diels-Alder
clivage par transestérification v+ T _τ? v r ou transamidation X .τ _p Y _ Y— L— - 0 , λ.- + G < L r 2 ' ι solvant(s) p étant un nombre entier variant de 0 à 2, Y"1"- représentant un cation onium tel que défini précédemment, et étant de préférence un cation triméthylalkylammonium, triéthylalkylammonium, tributylalkylphosphonium, N-méthylimidazolium ou pyridinium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle éventuellement fonctionnel, comprenant de 6 à 30 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à 20, et de préférence de 2 à 10,
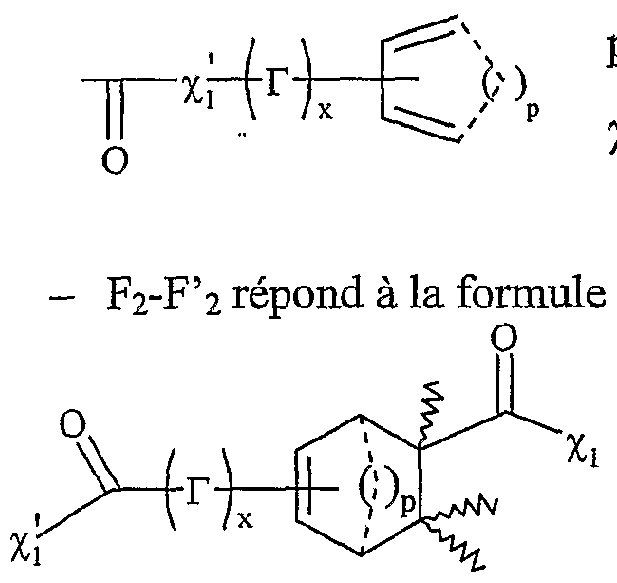
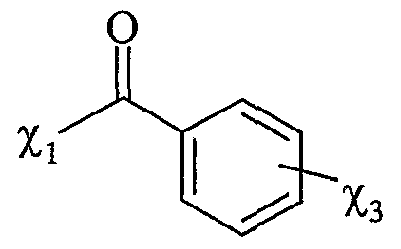
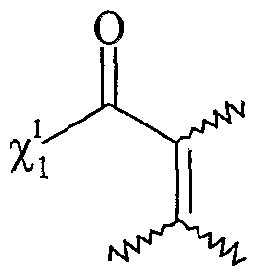
Xi- étant tel que défini précédemment, et étant notamment CL, Bf, -T, CF3CO2 "", CH3CO2 "", BFf, PF6 ", CF3SO3-, Tvf(SO2CF3)2, SO4 2"", R^O , SbF<f, R1SO3-, FSO3 ~ PO4 3~, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le téti-ihydrofuranne, le dioxane, Facétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions F0, Fi et F2 étant telles que définies ci-dessous : - Fo correspond à un groupe -χ_Η., dans lequel %χ représente un atome d'oxygène ou un groupe -NRf, R correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi répond à la formule suivante :
%ι étant tel que défini ci-dessus,
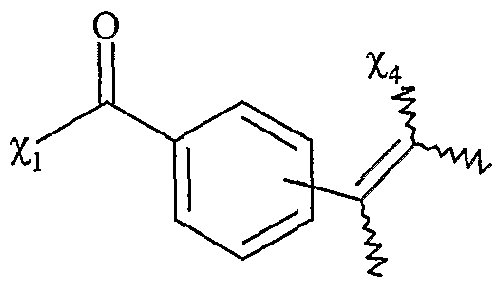
F
2 répond à la formule suivante %ι étant tel que défini ci-dessus,
G répondant à la formule suivante
dans laquelle χ
2 représente soit un groupe OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NR
hR
u, R-
h et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, la réaction d'estérification ou d'amidation dans ce schéma réactionnel étant effectuée par addition de l'acide carboxylique de formule suivante :
estérification -j, -.χ ou amidation + , _
v || Y-L-F-, , X, Y-L— F
j , X + || b) solvant(s) r~χ Xι,
solvant(s) cycloaddition 4+2 de Diels-Alder
clivage par transestérification Y-L-Fn , X, ou + G < transamidation Y-L-F. , X, solvant(s)
Y
"1"-, L et Xf étant tels que définis précédemment, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, ou un mélange de ces solvants, les fonctions F
0, F
1 et F
2 étant telles que définies ci-dessous : - Fo représente toute fonction permettant d'agrafer un diène-1,3, et est notamment choisie parmi les fonctions carbonyles, aminés, alcoxy, silanes, stannanes et boranes, comprenant de 1 à 20 atomes de carbone, - F] répond à la formule suivante : p étant un nombre entier variant de 0 à 2,
F répond à la formule suivante : χ
3 représentant un groupement électroattracteur, notamment choisi parmi les groupes cyano, alkoxycarbonyle, comprenant de 1 à 20 atomes de carbone, acyle comprenant de 2 à 20 atomes de carbone, benzoyle,
sulfonyle, dialkoxyphosphonyle comprenant de 1 à 10 atomes de carbone,
G répondant à la formule suivante : χ
3 étant tel que défini ci-dessus.
Le passage de Fo à Fi s'effectue de la façon suivante : et le composé de chlorure d'acide
t le composé de chlorure d'acide
Le produit de formule
> Xf s'obtient par estérification
des alcools avec Y— — COOH , Xf ou Y— L— coci, Xf
Le produit de
aminés de formule
n, R
f, p, X
\ , Y et L étant tels que définis précédemment,
C) Y~L-F
0 , Xj Y-L-F
0 , X
Γ Y-L-F
0 , X, Y-L-F"
0, X- estérification estérification solvant(s) ou amidation solvant(s) solvant(s) solvant(s) ou amidation
Y-L-FJ , x; Y-L-F , X] Y-L-F, , x; Y-L-FY Xj
OU solvant(s) réaction de solvant(s) réaction de Diels-Alder Diels-Alder
Y-L-F— F'rL-Y , 2 X Y-L-F— F" L-Y+, 2 X clivage clivage
Y-L-F0 , X! + Y-L-F. , X] + G Y-L-Fn , X, + Y-L- -F'V ! + G"
Y "-, L et Xf étant tels que définis précédemment, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofurane, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions F0, F'o, F"0, Fi, F'.-, F"ι, F2, F'2 et F"2 étant telles que définies ci- dessous : - Fo et F'o correspondent respectivement à un groupe -χiH et -χ'tH, dans lequel Xi et χ'î, identiques ou différents, représentent un atome d'oxygène ou un groupe -NRf,
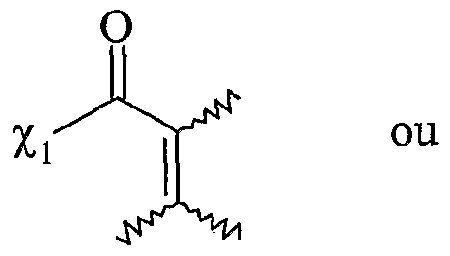
Rf correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - F'O correspond à une fonction -COOH ; - Fi répond à la formule suivante :
Xi étant tel que défini ci-dessus,
V répond à la formule suivante : p étant un nombre entier variant de 0 à 2, O, χ'i étant tel que défini ci-dessus, V (.-) x étant égal à 0 ou 1 , r représentant une chaîne alkyle comprenant de 1 à 30 atomes de carbone, aïkaryle, aralkyle, aryle comprenant de 6 à 30 atomes de carbone, - F"ι répond à la formule suivante : , x et r étant tels que définis ci-dessus, χ'i étant tel que défini ci-dessus, suivante p, χi, χ'i, x et T étant tels que définis ci-
dessus,
- F
2-F"
2 répond à la formule suivante : P
» Xi. X'i. x et T étant tels que définis ci- dessus,
G répond à la formule suivante :
G' ' répond à la formule suivante
χ
2 et χ'
2, identiques ou différents, représentent soit un groupe OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NR Ru, R-
h et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone. Dans le dernier schéma réactionnel (cas c) : - le passage de Fo à Fi s'effectue par estérification ou amidation de l'acide carboxylique de formule
- le passage de F'o à F'i s'effectue par estérification ou amidation de l'acide carboxylique de formule
T, x. et p étant tels que définis ci-dessus,
- le passage de F'O à F" ι s'effectue par l'addition du composé de formule
T, x, p et χ'i étant tels que définis ci-dessus.
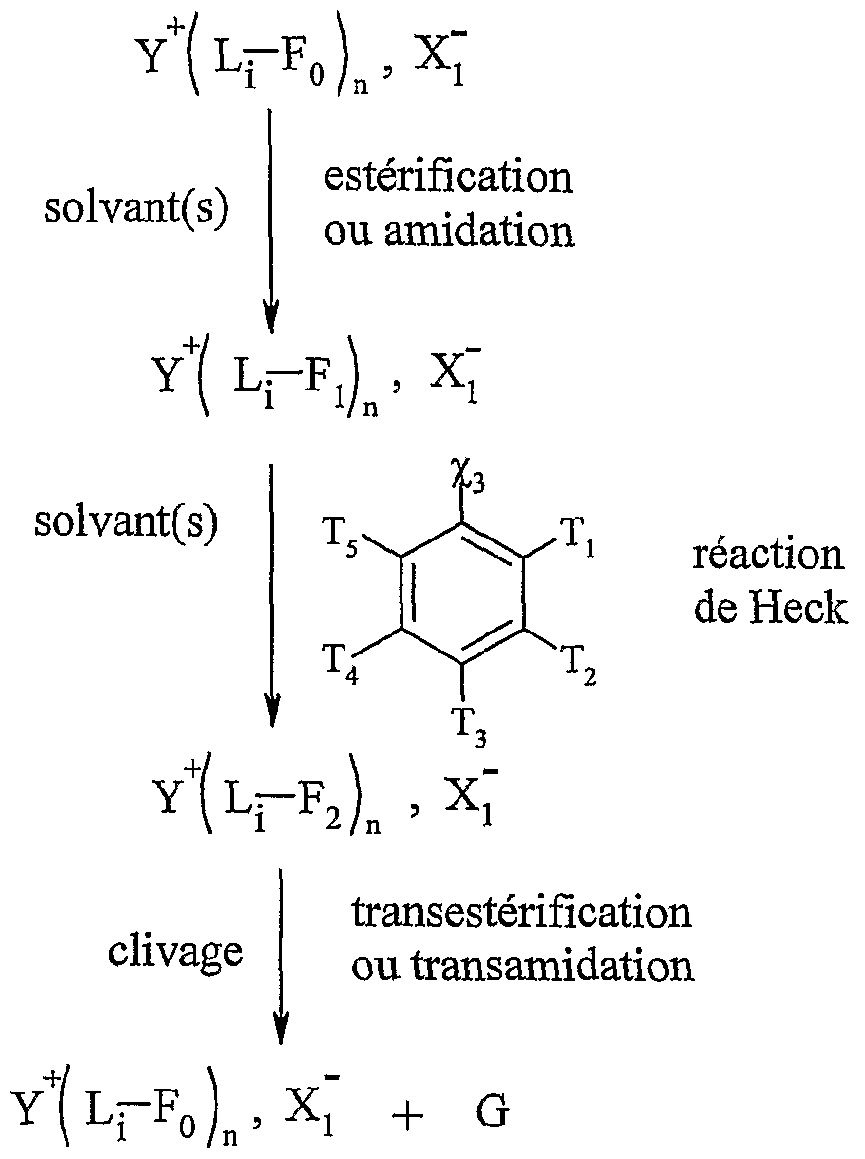
La présente invention concerne l'utilisation telle que définie ci-dessus, pour la mise en œuvre de réactions de couplage comme les réactions de Heck, de Suzuki, de Sonogashira ou d'Ullmann.
La présente invention concerne également l'utilisation telle que définie ci-dessus pour la mise en œuvre de la réaction de Heck, selon l'un des schémas réactionnels suivants :
Y-L-Fn , X, estérification ou amidation solvant(s)
Y-L-F! , Xï Y-L-F. , Xj
Y-L-F
0 ,
"ι + G Y-L-F
0 , X,- + G
1
Y "- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation triméthylalkylammonium, triéthylalkylammonium, tributylalkylphosphonium, tricyclohexylalkylphosphonium, N-méthyl-N' -alkylimidazolitrm, N-alkylpyridinium, diméthylalkylsulfonium, diéthyl-alkylsulfonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à 20, et de préférence de 2 à 10, Xf étant tel que défini ci-dessus, et étant notamment CL, Bf, T, CF3CO2 _, CH3CO2 ", BF4 ", PF6- CF3SO3-, "Η(S02CF3)2, S04 2-, R^O , SbFg", R^Of, FSOf, P0 3_, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone,
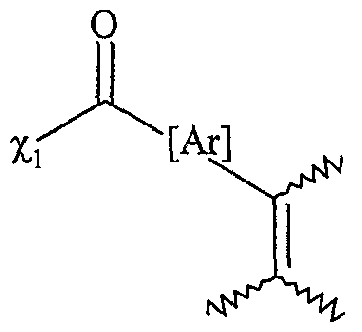
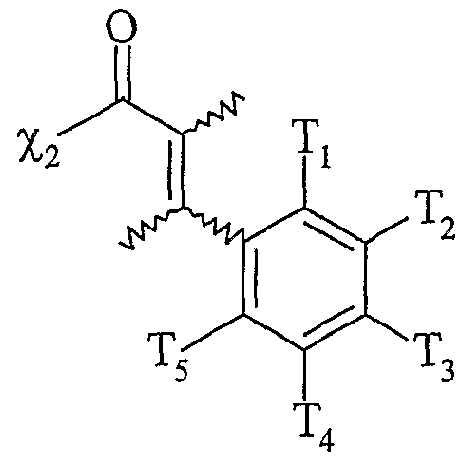
le ou les solvants étant choisis parmi : le dichlorométhane, le tétiahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions Fo, Fb F'i, F2 etF'2 étant telles que définies ci-dessous : - Fo correspond à un groupe -χiH, dans lequel χi représente un atome d'oxygène ou un groupe -NRf, R correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi répond à l'une des formules suivantes :
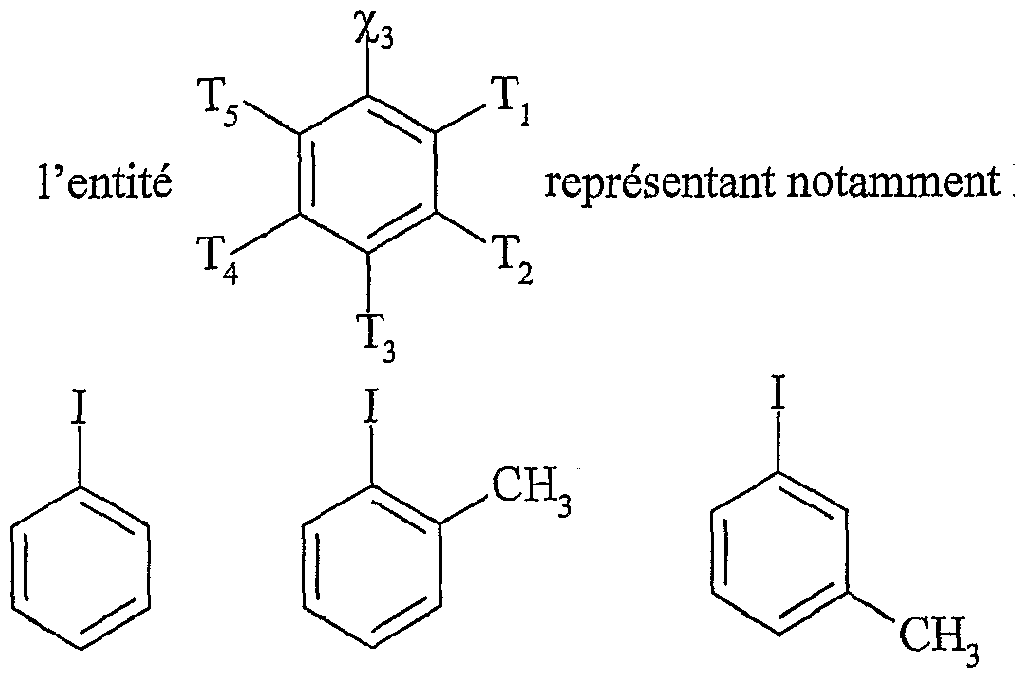
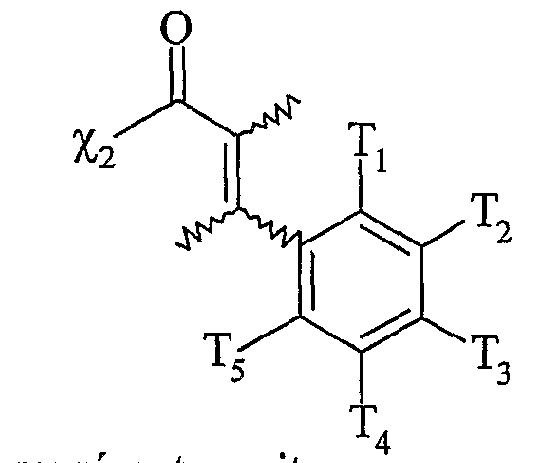
χi étant tel que défini ci-dessus,
[Ar] représentant un noyau aromatique, éventuellement substitué par un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, ou un groupe fonctionnel notamment choisi parmi N0
2, CN, COOR, OR, COR, NHCOR, NRR', S0
2R, I, Br, R et R' représentant indépendamment l'un de l'autre un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, [Ar] répondant de préférence à la formule suivante : dans laquelle T'i, T'
2; T'
4 et T'
5 représentent indépendamment les uns des autres un atome d'hydrogène, un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, ou un groupe fonctionnel
notamment choisi parmi N0
2, CN, COOR, OR, COR, NHCOR, NRR', S0
2R, I, Br, R et R' représentant indépendamment l'un de l'autre un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone,
- F
2 répond à l'une des formules suivantes
χi et r étant te s que éfinis ci-dessus, Ti, T
2, T
3, T et T
5 répondant à la définition donnée ci-dessus pour T , T'
2, T'
4 et T'
5
G ré ondant à l'une des formules suivantes
dans laquelle χ
2 représente soit un groupe -OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NRhR
u, R-
h et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, χ
3 représentant un groupement partant, notamment choisi parmi les halogénures I, Cl et Br, les groupes mésylate, tosylate, triflate, sulfonate, sulfate ou phosphate,
les groupes suivants :
F'
1 répond à la formule suivante : χ! et χ
3 étant tels que définis ci-dessus,
- F'
2 répond à la formule suivante : χι étant tel que défini ci-dessus, χ
4 représentant un groupe fonctionnel de type ester, amide, sulfone, phosphonate, silane, borane, ou un groupe alkyle, fonctionnel ou non, comprenant de 1
à 20 atomes de carbone, ou un groupe aryle, fonctionnel ou non, comprenant de 6 à 30 atomes de carbone,
G' répondant à la formule suivante : χ
2 et χ
4 étant tels que définis ci-dessus.
La partie gauche du schéma réactionnel ci-dessus correspond à la fixation du reste acrylique sur le support et la partie droite du schéma ci-dessus correspond à la fixation du reste arylique sur le support. Le passage de F
0 à Fi s'effectue par Festérification de l'acide carboxylique de formule
Le passage de F'o à F'i s'effectue par Festérification de l'acide carboxylique de formule χ
3 étant tel que défini ci-dessus.
La réaction de Heck peut être effectuée de trois façons différentes — en supportant la partie acrylique de la façon suivante :
Y4"-, Xi , L, Ar et Rf étant tels que définis ci-dessus, en supportant la partie arylique de la façon suivante :
Y
1"-, Xi , L, R
f et χ
3 étant tels que définis ci-dessus,
en supportant les parties acrylique et arylique notamment de la façon suivante
Y
4"-, X , L et R
g étant tels que définis ci-dessus.
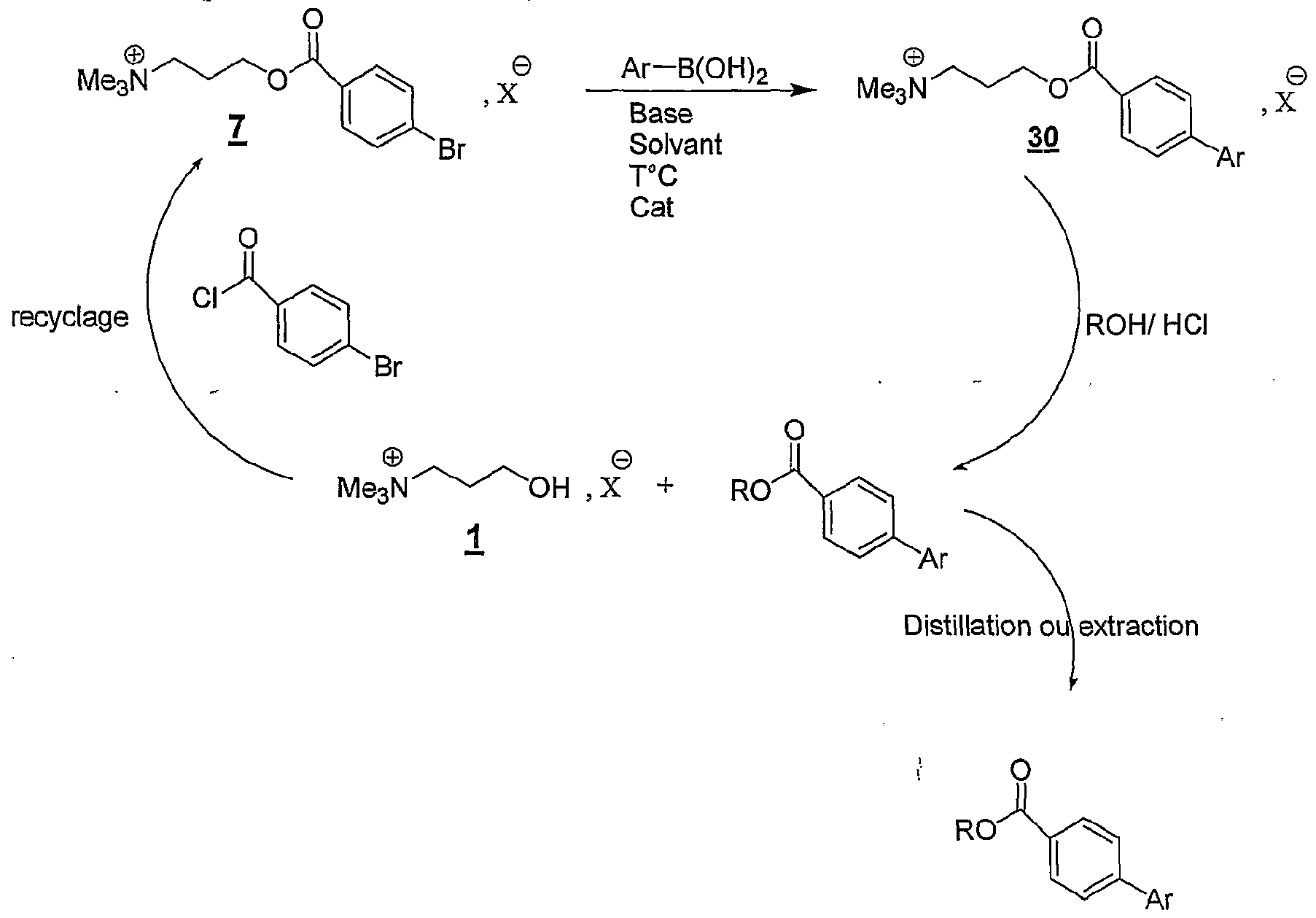
La présente invention concerne également l'utilisation telle que définie ci-dessus pour la mise en œuvre du couplage de Suzuki, selon l'un des schémas réactionnels suivants : estérification a) Y-L-F0 , X 0U amidati0n> Y-L-F. , Xr solvant(s) réaction de Suzuki solvant(s) avec R3B(OR7)2 clivage par transestérification ou transamidation Y-L-F0 , X + G - Y-L-F, X, solvant(s)
R3 étant choisi parmi les groupes aryle, hétéroaryle, éthényle, diényle, allyle, éthynyle, substitués ou non, comprenant de 2 à 30 atomes de carbone, R représentant un atome d'hydrogène ou un groupe alkyle, ramifié ou linéaire ou un groupe cycloalkyle comprenant de 1 à 12 atomes de carbone,
Y+- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation tiimé ylal-kylammomum, triéthylalkylammonium, tributylalkylphosphonium, N-méthylimidazolium ou pyridinium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle éventuellement fonctionnel comprenant de 6 à 30 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à
20, et de préférence de 1 à 10, Xf" étant tel que défini ci-dessus, et étant notamment CF, Br"", T, CF3C02 ~, CH3CO2", BF , PF<f, CF3S03 _, "K(S02CF3)2, SO4 2~, RiSOf, SbF<f, RiSOf , FSO ,
PO4 3", Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétiahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions F0, Fi et F2 étant telles que définies ci-dessous - Fo est de la forme -χiH, χ_ représentant un atome d'oxygène ou un groupe -NRf, Rf correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi est de la forme -Re-χ, Rβ représentant un groupe aromatique ou hétéroaromatique comprenant de 6 à 30 atomes de carbone, x représentant un groupe partant choisi de préférence parmi Cl, Br, I, OTf, 0-C02R5 ou OS03-R5, R5 représentant un groupe alkyle comprenant de 1 à 10 atomes de carbone ou un groupe aralkyle comprenant de 6 ndant de préférence à la formule suivante :
- F
2 est de la forme -R-.-R
2, R
e étant te que n c -dessus et R
2 étant choisi parmi les groupes aryle, hétéroaryle, éthényle, diényle, allyle, éthynyle, substitués ou non, comprenant de 2 à 30 atomes de carbone, F
2 répondant de préférence à la formule suivante :
Aïi représentant un groupe aromatique choisi de préférence parmi :
la molécule G étant de la forme R -R
3, R
2 et R
3 étant tels que définis ci-dessus, et répond notamment à la formule suivante :
dans laquelle χ
2 représente soit un groupe -OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NRhRu, Rh et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, Ari est tel que défini ci-dessus. Lorsque F
0 représente un groupe -OH, on obtient la fonction Fi par estérification notamment avec l'acide carboxylique de formule
Lorsque Fo représente un groupe -NR
fH, on obtient la fonction Fi par amidation notamment avec l'acide carboxylique de formule
estérification b) +
τ -_,
v ou amidation , +
τ --,
v Y— L— F
0 , X
j-
> Y-L— F
j , solvant(s) réaction de Suzuki solvant(s) avec R
2χ clivage par transestérification +
τ --- m-m ou transamidation , + , „
v Y-L— F
0 , X
χ- A- G -* Y— L— F
2 , X - solvant(s) Y
"^- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation
triéthylalkylammonium, tributylalkylphosphonium, N-méthylimidazolium ou pyridinium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle éventuellement fonctionnel comprenant de 6 à 30 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)
r, r variant de 1 à
20, et de préférence de 1 à 10, Xι~ étant tel que défini ci-dessus, et étant notamment Cl-, Br~, F, CF3C02 _, CH3CO , BFf, PF<f, CF3SO3-, "Η(SO2CF3)2, S04 2~, RiSOf, SbF<f, RiSO , FSOf ,
P0
4 3_, Rj représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétiahydrofura-nne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R
2 étant choisi parmi les groupes aryle, hétéroaryle, éthényle, diényle, allyle, éthynyle, substitués ou non, comprenant de 2 à 30 atomes de carbone, les fonctions Fo, Fj et F
2 étant telles que définies ci-dessous : - Fo est de la forme -χiH, χi étant tel que défini ci-dessus, - Fi est de la forme -Rq-B(0R )
2, R étant tel que défini ci-dessus, et Rq correspondant à un groupe aryle comprenant de 6 à 30 atomes de carbone, hétéroaryle comprenant de 4 à 20 atomes de carbone, éthényle comprenant de 2 à 20 atomes de
carbone, diényle comprenant de 3 à 20 atomes de carbone, allyle comprenant de 3 à 20 atomes de carbone, éthynyle comprenant de 2 à 20 ou non, Fi répondant de préférence à la formule suivante
Ar
2 correspondant à un groupe aryle substitué ou non comprenant de 6 à 30 atomes de carbone, - F
2 est de la forme -R
q-R
e, Rq et R
e étant tels que définis ci-dessus, F
2 répondant de préférence à la formule suivante :
Ar
t représentant un groupe aromatique choisi de préférence parmi
la molécule G étant de la forme R
2-R
3, R
2 et R
3 étant tels que définis ci-dessus, et répondant notamment à la formule suivante :
dans laquelle χ
2, An et Ar
2 sont tels que définis ci-dessus,
Y-L , X
réaction de Suzuki avec R^χ solvant(s) avec clivage par transestérification ou transamidation
Y-L-N(CH2CH2OH)2 , Xr + R— R3
Y"^-, L, Xι~, R2 et R3 étant tels que définis ci-dessus, 3 étant de préférence un groupe phényle, le ou les solvants étant choisis parmi : le dichlorométhane, le tétiahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants. Le mode de réalisation a) correspond au cas où Fhalogénure d' aryle est supporté et où l'acide boronique est libre. Le mode de réalisation b) correspond au cas où l'acide boronique est supporté et où Fhalogénure d'aryle est libre. Les modes de réalisation a), b) et c) permettent de purifier facilement les produits de couplage sous forme de sels. En particulier, il est facile d'éliminer les produits d'homocouplage qui ne sont pas des sels par simple lavage avant transestérification. Les avantages de ces différents modes de réalisation sont les suivants : - les réactions sur support soluble se prêtent aux techniques de la chimie combinatoire et de la synthèse parallèle ; - ces supports sont faciles à purifier car ce sont des sels insolubles dans un certain nombre de solvants ; ils peuvent donc être lavés et/ou recristallisés ; en particulier, dans le cas de la réaction de Suzuki, le produit d'homocouplage peut être facilement éliminé par simple lavage du sel avant la transestérification.
La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre du couplage de Sonogashira, selon l'un des schémas réactionnels suivants : estérification a. v + T . v o mu aammiiddaattiioi aJ Y-L— F0 , Xr on . Y +-L τ — - F,j . γf solvant(s) solvant(s) réaction de Sonogashira avec R8C--≡CH clivage par transestérification _ + -- ,--, _r ou transamidation + -. --, v Y~L-F0 , Xr + G Y-L-F2 , Xr solvant(s)
Y4- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation tr-iméthylalkylammonium, triéthylalkylammonix-un, tributylalkylphosphonium, N-méthylimidazolium ou pyridinium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à 20, et de préférence de 1 à 10, Xi- étant tel que défini ci-dessus, et étant notamment CL, Br~, F, CF3CO2 "*, CH3CO2 ", BF4 ~, PF6 ~ CF3SOf, -N(S02CF3)2, S04 2~, RjSO , SbF6 ", RιS03 ~, FSO , P04 3-, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R8 représentant un groupe ORh, NRhRu, CORh, CN, S02Rh, SRh, un groupe alcényle, éthynyle, diényle, R et Ru représentant, indépendamment l'un de l'autre, un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone,
ou R8 représentant un groupe alkyle, ramifié ou linéaire, éventuellement fonctionnel, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle, ou un groupe aïkaryle ou aralkyle, comprenant de 6 à 30 atomes de carbone, substitué ou non, lesdits groupes alkyle ou aryle pouvant être substitués par l'un des groupes fonctionnels suivants : un atome d'halogène, notamment Cl, un groupe ORh, NR-hRu, CORh, CN, SO2R11-- SRh, un groupe alcényle, éthynyle, diényle, vinyle, alcynyle, Rh et Ru étant tels que définis précédemment, Rg étant notamment l'un des groupes suivants : -(CH2)S-CH3, -(CH2)s-CH2OH, -(CH2)s-CH2OMe, s représentant un nombre entier compris entre 0 et 10,
les fonctions Fo, Fi et F
2 étant telles que définies ci-dessous : - Fo correspond à un groupe -χiH, dans lequel Xi représente un atome d'oxygène ou un groupe -NR
f, R
f correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi répond à la formule suivante : χi étant tel que défini ci-dessus, et Hal représentant un halogène, et étant de préférence l'iode,
- F
2 répond à la formule suivante : χi et R
8 étant tels que définis ci-dessus,
G répondant à la formule suivante
dans laquelle χ2 représente soit un groupe -OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NRhRu, Rh et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, χ
2 représentant notamment un groupe OMe, OEt, OPr ou OBu. La transformation de la fonction Fo en Fi s'effectue par une réaction d' estérification ou d' amidation avec l'acide carboxylique de formule :
estérification >u amidatioi Y-L-F
0 , X
r °
U amidati0n> Y-L-F, , j- solvant(s) réaction de Sonogashira solvant(s) clivage par transestérification + ., ,-, „ ou transamidation +
τ --,
v Y-L-F
0 , X
r + G < Y-L-F
2 , X
r solvant(s)
Y - représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation triméthylalkyl-immo-nium, triéthylalkylammonium, tributylalkylphosphonium, N-méthylimidazolium, alkylpyridinium, diméthylalkylsulfonium ou diéthylalkyl- sulfonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)r, r variant de 1 à 20, et de préférence de 1 à 10,
Xi- étant tel que défini ci-dessus, et étant notamment CF, Br-, F, CF
3C0
2-, CH
3CO
2-, BF
4-, PF
6 ", CF3SO3-,
"Η(S0
2CF
3)
2, S0
4 2", RiSOf, SbF
6-, RiS0
3-, FSO
3 ", P0
4 3_, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, GP représentant un groupe partant, et étant notamment Cl, Br, I ou OTf, les fonctions F
o, Fi et F
2 étant telles que définies ci-dessous : - Fo correspond à un groupe -COOH, - Fi répond à la formule suivante :
dans laquelle 1 représente un nombre entier variant de 1 à 20, et χi représente xm atome d'oxygène ou un groupe -NR
f, R
f correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à
30 atomes de carbone, - F2 répond à la formule suivante : O -^^•χj A ]X \ Xi et 1 étant tels que définis ci-dessus,
G répondant à la formule suivante :
dans laquelle χi et 1 sont tels que définis ci-dessus. Le mode de réalisation a) correspond au cas où l'aromatique est supporté et où l'acétylénique est libre. Le mode de réalisation b) correspond au cas où l'acétylénique est supporté et où l'aromatique est libre.
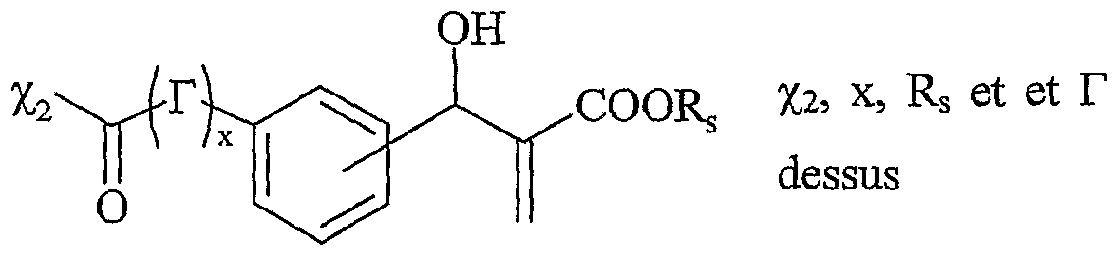
La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre de la réaction de Baylis-Hilman, selon l'un des schémas réactionnels suivants : estérification a) Y-L-F
0 , X
r °
U amidati°
n> Y-L-F, , X
r solvant(s) solvant(s) réaction de Baylis-Hilman avec ArCHO clivage par transestérification , ,+
τ ..-, , ou transamidation +
v Y-L-F
0 , X
r + G Y-L-F
2 , X
r solvant(s)
Y4- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation friméthyla-Jcylammonium, tiiéthyla-U- ylammonium, tributylal-kylphosphonium, N-méthylimidazolium ou pyridinium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à 20, et de préférence de 1 à 10, Xi- étant tel que défini ci-dessus, et étant notamment CF, Br", T, CF3CO2", CH3C02-, BF4 ", PFtf, CF3SO3-, T^(SO2CF3)2, S04 2-, RιS04 ~, SbF<f, RιS03 ~, FSOf, PO4 3"", Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions Fo, Fi et F2 étant telles que définies ci-dessous : - Fo représente un groupe -OH,
- Fi répond à la formule suivante :
F
2 répond à la formule suivante :
G répondant à la formule suivante : Xi représentant un groupe -OH, ou un groupe -OR
g, R
g représentant un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone,
Ar représentant un groupe aromatique ou hétéroaromatique, substitué ou non, ArCHO étant notamment choisi parmi :
La transformation de la fonction Fo en Fi s'effectue par une réactionérification ou d' amidation avec l'acide carboxylique de formule :
b) estérification Y
+-L-F
0 , X
r 0U amidati0n> Y-L-F, , Xi- solvant(s) réaction de Baylis-Hilman solvant(s) avec R
jOOC clivage par transestérification , + _ --, „ ou transamidation +
τ _
v Y-L-F
0 , X
r + G Y-L
_F 2 •
Xf solvant(s) Y
+- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation tiiméthylalkylammonium, triéthylalkylammonium, tributylalkylphosphonium, N-méthylimidazolium, alkylpyridinium, diméthylalkylsulfonium ou diéthylalkyl- sulfonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de - préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)
r, r variant de 1 à 20, et de préférence de 1 à 10, Xf étant tel que défini ci-dessus, et étant notamment CF, Br-, F, CF
3C0
2 ~, CH
3CO
2 ", BF
4-, PF
6-, CF
3SO , -N(S0
2CF
3)
2, S0
4 2-, RiSOf, SbF
6-, RiSOf, FS0
3 ~, P0
3~, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le téti- ydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R
s représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone ou aralkyle ou aïkaryle comprenant de 7 à 30 atomes de carbone, les fonctions Fo, Fi et F
2 étant telles que définies ci-dessous : - Fo correspond à un groupe -χιH, dans lequel χι représente un atome d'oxygène ou un groupe -NR
f, R
f correspondant à un groupe alkyle, linéaire ou ramifié,
comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fj répond à la formule suivante : χi étant tel que défini ci-dessus, x étant égal à O ou 1, T représentant une chaîne alkyle
comprenant de 1 à 20 atomes de carbone, aïkaryle, aralkyle comprenant de 6 à 30 atomes de carbone, - F
2 répond à la formule suivante ι, x, R
s et T étant tels que définis ci-dessus
G répondant à la formule suivante : χ
2, x, R
s et et T étant tels que définis ci- dessus
dans laquelle χ
2 représente soit un groupe -OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NR
hR
u, R
h et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, χ
2 représentant notamment un groupe OMe, OEt OPr ou OBu.
estérification c) +
τ „
v ou amidation + _ Y-L— F
0 , X
> Y-L— F, , X
r solvant(s) réaction de Baylis-Hilman solvant(s)
clivage par transestérification +
τ _ .,- ou transamidation _ -.+
τ _
v Y-L-F
0 , x
r + G < Y-L-
F2
' Xf solvant(s)
Y
"^- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation triméthylalkylammonium, triémylalkylammonium, tributylalkylphosphonium, N-méthylimidazolium ou pyridinium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)
r, r variant de 1 à 20, et de préférence de 1 à 10, Xf étant tel que défini ci-dessus, et étant notamment CF, Br
", T, CF3CO2
", CH
3C0
2- BFf, PF<f , CF3SO3
",
"Η(S0
2CF
3)
2, S0
4 2- Rj-SOf, SbF
6-, RiSOf, FSO , P0
3~, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le téfrahydrofi-u anne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R
s représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone ou aralkyle ou aïkaryle comprenant de 7 à 30 atomes de carbone, les fonctions Fo, Fi et F
2 étant telles que définies ci-dessous : - Fo correspond à un groupe -COχiH, dans lequel %
\ représente un atome d'oxygène ou un groupe -NRf, R correspondant à un groupe alkyle, linéaire ou ramifié,
comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi répond à la formule suivante :
F
2 répond à la formule suivante :
G répondant à la formule suivante :
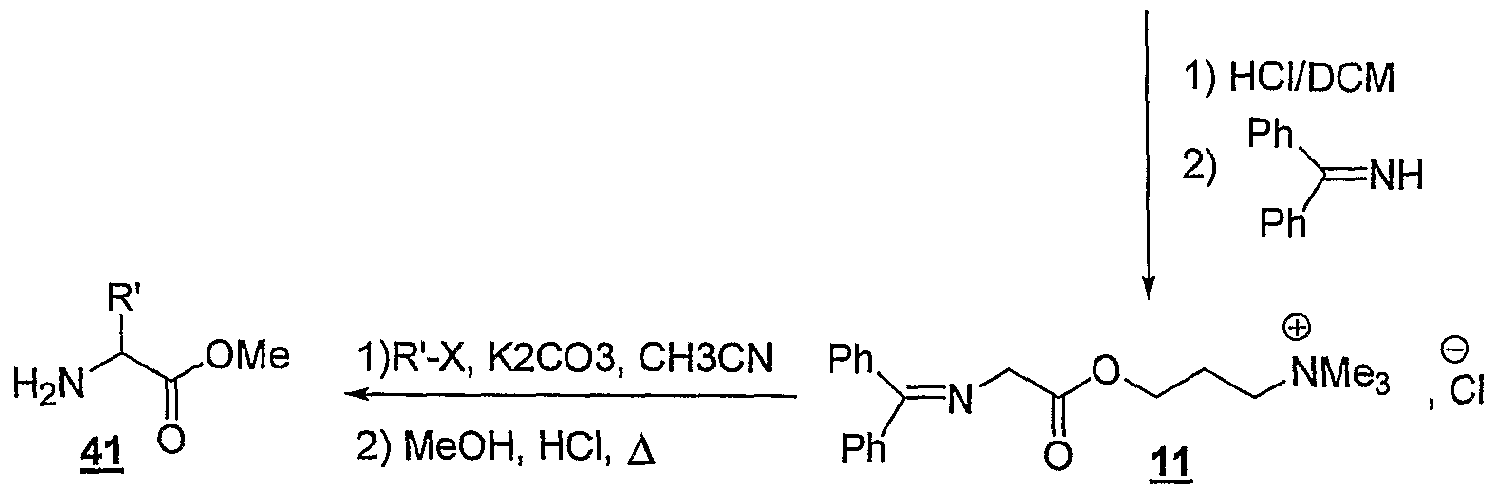
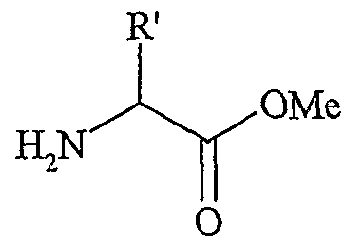
La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la synthèse, éventuellement asymétrique, d'α-aminoacidës, selon le schéma réactionnel suivant :
Y-L- X,
1) déprotection solvant(s)
+ 1) RX, K,CO,, CH
3CN, S
* + -- _
v Y-L~F
0 , X,- +
G — -^ Y
~L-F
2 , X
r 2) MeOH, HC1 Y
4"- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation triméthylalkylammonium, triéthylalkylatnmonium ou tributylalkylphosphonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle éventuellement
fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)
r, r variant de 1 à 20, et de préférence de 3 à 6, Xf étant tel que défini ci-dessus, et étant notamment CF, Br-, F, TS (S0
2CF
3)
2, BF
4 ", PF
6 ~, le ou les solvants étant choisis parmi : l'acétonitrile, le dichlorométhane, le tétrahydrofuranne, le dioxane, le toluène, le chlorobenzène ou un mélange de ces solvants, R' représentant un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 30 atomes de carbone, éventuellement fonctionnel, S représentant un agent chiral de transfert de phase tel que le bromure de 0(9)- allyl-N-9-anthracényl-méthylcinchonidinium (voir Corey et al., 1998), les fonctions Fo,
et F
2 étant telles que définies ci-dessous : - Fo correspond à -OH, - Fi répond à la formule suivante :
F
2 répond à la formule suivante
G répondant à la formule suivante
La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre de réactions multi-composants. Les réactions multi-composants (RMC) mettent en présence simultanément au moins trois partenaires dans des conditions expérimentales qui ne varient pas au cours du temps et permettent la création de plusieurs liaisons covalentes en cascade dans un seul réacteur, à la différence des réactions classiques ou deux réactifs conduisent à un produit par création d'une nouvelle liaison. Ainsi il est possible d'accéder en une seule étape à une molécule hautement fonctionnalisée à partir d'entités relativement simples. De plus les RMC allient convergence et économie d'atomes, deux principes essentiels
en synthèse organique mais aussi en chimie combinatoire. Enfin, ces réactions ont généralement lieu avec un rendement élevé, puisqu'elles évitent la succession d'étapes des synthèses linéaires ou multiétapes qui, font, à chaque pas, chuter le rendement. Les RMC les plus connues et les plus développées sont celles de Passérini et de Ugi (Ugi et al., 1999 ; Ugi et al., 2001 ; Domling et al., 2000 ; Ugi, 2001 ; Bienayme et al, 2000 ; Vanden Eynde et al, 2000 ; Domling, 2002). La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre de réactions multi-composants de type UGI, notamment pour la réaction de type Grieco selon l'un des schémas réactionnels suivants : a) estérification ou amidation Y-L-F„ , X, Y-L- *τ solvant(s) réaction de type Grieco avec solvant(s)
clivage par transestérification ou transamidation Y-L-F
π , X,-
+ G ^ Y-L-F
2 , X
r solvant(s)
Y+- représentant un cation onium tel que défini ci-dessύs, et étant de préférence un cation Ijiméthylalkylammonium, triéthylalkylammonium, tributylalkylphosphonium, N-méthyl-N' -alkylhnidazolium, N-alkylpyridinium, diméthylalkylsulfonium ou diéthylalkylsulfonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à 20, et de préférence de 1 à 10,
Xi étant tel que défini ci-dessus, et étant notamment Cl , Br , T, CF3C02 ~ CH3Cθ2~, BF4 ", PF6-, CF3S03-, "N(S02CF3)2, S04 2~, RiSOf, SbF6-, RiS03-, FS03- P0 , Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétiahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R représentant un atome d'hydrogène, un groupe nitro, de préférence en position para, un atome de chlore, de préférence en position para ou un groupe méthoxy, de préférence en position para, les fonctions Fo, F] et F2 étant telles que définies ci-dessous : - Fo représente un groupe -OH, - Fi répond à la formule suivante :
F
2 répond à la formule suivante :
G répondant à la formule suivante Xi représentant un groupe -OH, ou un groupe -OR
g, R
g représentant un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone,
b) estérification Y-L-F
0 , X,- «nidation^
γ +_
L_
Fι , χ
f solvant(s) réaction de type Grieco avec solvant(s) H
2N
clivage par transestérification ou transamidation Y-L-F
0 , X,- + G , Y-L-F
2 , X,- solvant(s)
Y+- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation teiméthyla-Ucylammonium, triéthylalkylammonium, tributylalkylphosphonium, N-méthyl-N' -alkylimidazolium, N-alkylpyridinium, diméthylalkylsulfonium ou diéthylalkylsulfonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à 20, et de préférence de 1 à 10, Xf étant tel que défini ci-dessus, et étant notamment CF, Br-, T, CF3C02~, CH3CO , BF4-, PF6-, CF3SO3-, -N(S02CF3)2, S04 2-, RiSOf SbF6-, R1SO3", FSOf , P04 3-, i représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R2 représentant un groupe alkyle, fonctionnel ou non, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle, fonctionnel ou non, comprenant de 6 à 30
atomes de carbone, ou un groupe aralkyle ou aïkaryle, fonctionnel ou non, comprenant de 7 à 50 atomes de carbone, R3 représentant un atome d'hydrogène, un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, ou un groupe aralkyle ou aïkaryle, fonctionnel ou non, comprenant de 7 à 50 atomes de carbone, ou un groupe fonctionnel notamment choisi parmi NO2, CN, COOR, OR, COR, NHCOR, NRR', S02R, I, Br, R et R' représentant indépendamment l'un de l'autre un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, les fonctions Fo, Fi et F2 étant telles que définies ci-dessous : - Fo représente un groupe -OH, - Fi répond à la formule suivante :
- F
2 répond à la formule suivante :
G répondant à la formule suivante : Xi représentant un groupe -OH, ou un groupe -OR
g, R
g représentant un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone.
estérification c) + ou amidation +
v Y-L-F
0 , X,-
> Y-L-F, , Xj- solvant(s) réaction de type Grieco avec solvant(s)
clivage par transestérification ou transamidation Y-L-F. , X, + Y +—
τ L— _ 1,
2 ,
v Λ.J- solvant(s)
1"- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation tiimémylalkylammonium, triéthylalkylammonium, tributylalkylphosphonium, N-méthyl-N' -alkylimidazolium, N-alkylpyridinium, diméthylalkylsulfonium ou diéthylalkylsulfonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)
r, r variant de 1 à 20, et de préférence de 1 à 10, Xf étant tel que défini ci-dessus, et étant notamment CF, Br-, F, CF
3C0
2-, CH
3C0
2-, BF , PF
6-, CF3SO3
",
" (S0
2CF
3)
2, S0
4 2-, RiSO l SbF
6-, RiS0
3 ", FS0
3-, P0
3-, R
1 représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R représentant un atome d'hydrogène ou un groupe fonctionnel tel qu'un groupe nitro en position para, un atome de chlore en para ou un groupe méthoxy en position ortho, ou un groupe alkyle, fonctionnel ou non, comprenant de 1 à 20 atomes de
carbone, ou un groupe aryle, fonctionnel ou non, comprenant de 6 à 30 atomes de carbone, ou un groupe aralkyle ou aïkaryle, fonctionnel ou non, comprenant de 7 à 50 atomes de carbone, R
3 représentant un atome d'hydrogène, un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, ou un groupe aralkyle ou aïkaryle, fonctionnel ou non, comprenant de 7 à 50 atomes de carbone, ou xm groupe fonctionnel notamment choisi parmi N0
2, CN, COOR, OR, COR, NHCOR, NRR', S0
2R, I, Br, R et R' représentant indépendamment l'un de l'autre un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, les fonctions Fo, Fi et F
2 étant telles que définies ci-dessous : - Fo représente toute fonction permettant d'accrocher et de décrocher un reste portant une oléfine, de préférence un ester, ou un amide. - Fi répond à l'une des formules générales suivantes : n représentant un nombre entier variant de 1 à 10
F
2 répond à l'une des formules générales suivantes :
n, R et R3 étant tels que définis ci-dessus, et Xi représentant un groupe -OH, ou un groupe -OR
g, R
g représentant un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone. La transformation de la fonction Fo en F
ls dans le premier cas de figure, s'effectue par une réaction d' estérification avec l'acide
La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre de réactions multi-composants, notamment pour la synthèse d'oléfines tétiasubstituées selon R.C. Larock et al. (2003), selon le schéma réactionnel stuvant :
Y-L-F
0 , X,- Y-L-F, , X,- solvant(s) clivage par transestérification ou transamidation
Y-L-F0 , Xf + G Y"1"- représentant un cation onium tel que défini ci-dessus, et étant de préférence un cation triméthylalkyl-immonium, triéthylalkylammonium, tributylalkylphosphonium, N-méthyl-N' -alkylimidazolium, N-alkylpyridinium, diméthylalkylsulfonium ou diéthylalkylsulfonium, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle,
éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH2)r, r variant de 1 à 20, et de préférence de 1 à 10, Xi- étant tel que défini ci-dessus, et étant notamment CF, Br-, T, CF3C02 ", CH3C02-, BF , PF«-, CF3SO3-, - S02CF3 2, S04 2", RiSOf, SbF , RiS03-, FSOf PO4 3-, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, R2 et R3, de préférence en para, représentant un atome d'hydrogène, un groupe alkyle, linéaire ou ramifié, éventuellement fonctionnel, comprenant de 1 à 30 atomes de carbone, un groupe aryle, éventuellement substitué et/ou fonctionnel, comprenant de 6 à 30 atomes de carbone, un groupe fonctionnel, de préférence un groupe méthoxy, mono- alkylamino, dialkylamino, aryla ino, cyano, ester, nitro, cétone, sulfonyle, alkylthio, sulfoxyde, les fonctions Fo et Fi étant telles que définies ci-dessous :
R
t représentant un groupe tel que défini pour R
2 et R
3 ci-dessus,
Fi répond à l'une des formules suivantes :
R2, R3 et R4 étant tels que définis ci-dessus,
G répond à l'une des formules suivantes :
χι représentant un groupe -OH, ou xm groupe -OR
g, R
g représentant un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone. La transformation de F
0 en ¥_ s'effectue par une cis-addition du groupement aryle provenant de Fhalogénure d' aryle du côté le moins encombré ou à l'extrémité la plus riche en électrons de l'alcyne de départ, tandis que le groupement acyle provenant de l'acide arylboronique s'additionne à l'autre extrémité. Dans le cadre de la présente invention, il est également possible d'utiliser des cations polyfonctionnels, que l'on définit comme étant des cations portant plusieurs fonctions, lesdites fonctions étant identiques ou différentes. La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre de réactions de cycloaddition, de préférence pour la mise en œuvre de la réaction de Diels-Alder, selon le schéma réactionnel suivant :
solvant(s) cycloaddition 4+2 de Diels-Alder
clivage par transestérification ou transamidation Y i L F0 X, + G « γ i LrF2/n • Xi solvant(s)
n étant un nombre entier variant de 2 à 4, tel que défini ci-dessous, i étant un nombre entier variant de 1 à n, p étant xm nombre entier variant de 0 à 2, Y+ représentant un cation onium tel que défini ci-dessus, de formule (Rb- )κ-nΛ+ dans laquelle x représente un nombre entier égal à 3 ou 4, n étant égal à 2, 3 ou 4 lorsque x est égal à 4 et n étant égal à 2 ou 3 lorsque x est égal à 3, Rb- représente un groupe alkyle comprenant de 1 à 20 atomes de carbone, un groupe aryle comprenant de
6 à 30 atomes de carbone ou un groupe aralkyle ou aïkaryle comprenant de 6 à 30 atomes de carbone, lesdits groupes alkyle, aryle, aralkyle ou aïkaryle susmentionnés étant non fonctionnels, et dans laquelle Λ+ représente un cation ammonium, imidazolium, phosphonium ou sulfonium, Y* représentant notamment un cation alkylammonium, alkylphosphonium ou alkylsulfonium, et étant de préférence un cation tétraalkylammonium, tètraalkylphosphonium, di--dkylimidazolium, trialkylsulfonixim, L représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle éventuellement fonctionnel, comprenant de 6 à 30 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type
(CH2)r, r variant de 1 à 20, et de préférence de 2 à 10, les bras L pouvant être identiques ou différents, Xi étant tel que défini ci-dessus, et étant notamment Cl , Br , F, CF3CO2-,
CH3CO2- BF4-, PF6 ", CF3SO3-, T^(S02CF3)2, S04 2-, RιSO4-, SbF6-, RjS03-, FS03-, P0 3-, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le téteahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions Fo, Fi et F2 étant telles que définies ci-dessous : - Fo correspond à un groupe -χiH, dans lequel χι représente un atome d'oxygène ou un groupe -NRf, Rf correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone,
Fi répond à la formule suivante :
%ι étant tel que défini ci-dessus,
F
2 répond à la formule suivante : χι étant tel que défini ci-dessus,
G répondant à la formule suivante
dans laquelle χ2 représente soit xm groupe OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NR
hRu, R
h et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone. La notation Y
+(Li-F
0)
n signifie que l'entité Y est substituée par n entités Lj-Fo reliées par liaison covalente. L'utilisation d'un sel plurifonctionnel dans le cadre de la réaction de Diels-Alder permet d'augmenter la productivité des solutions utilisées. Par ailleurs, les bras peuvent être fonctionnalisés différemment et permettre soit des réactions intramoléculaires, soit des réactions en cascades, soit des réactions multicomposants.
La présente invention concerne l'utilisation telle que définie ci-dessus, pour la mise en œuvre de la réaction de Heck, selon le schéma réactionnel suivant :
" n étant un nombre entier variant de 2 à 4, tel que défini ci-dessous, i étant un nombre entier variant de 1 à n, Y
"^ représentant un cation onium tel que défini ci-dessus, de formule (R
b)
x.
nΛ
+ dans laquelle x représente un nombre entier égal à 3 ou 4, n étant égal à 2, 3 ou 4 lorsque x est égal à 4 et n étant égal à 2 ou 3 lorsque x est égal à 3, R
b représente un groupe alkyle comprenant de 1 à 20 atomes de carbone, un groupe aryle comprenant de 6 à 30 atomes de carbone ou un groupe aralkyle ou alkarylè comprenant de 6 à 30 atomes de carbone, lesdits groupes alkyle, aryle, aralkyle ou aïkaryle susmentionnés étant non fonctionnels, et dans laquelle Λ
+ représente un cation ammonium, imidazolium, phosphonium ou sulfonium, Y
1" représentant notamment un cation alkylammonium, alkylphosphonium ou alkylsulfonium, et étant de préférence un cation tétraalkylammonium, tétraalkylphosphonium, dialkylimidazolium, trialkylsulfonium, L; représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type
(CH2)
r, r variant de 1 à 20, et de préférence de 2 à 10, les bras L pouvant être identiques ou différents, Xi- étant tel que défini ci-dessus, et étant notamment CF, Br
~, T, CF
3C0
2-, CH
3C0
2-, BF
4-, PF
6-, CF
3S0
3-, -N(S0
2CF
3)
2, S0
4 2-, RiSOf, SbF
6-, RjSOa-, FS0
3 ~, PO
4 3~, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions Fo, Fi et F2 étant telles que définies ci-dessous : - Fo correspond à un groupe -χiH, dans lequel χi représente un atome d'oxygène ou un groupe -NR
f, R
f correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi répond à la formule suivante : Xi étant tel que défini ci-dessus,
- F
2 répond à la formule suivante :
Xi étant tel que défini ci-dessus,
G répondant à la formule suivante :
dans laquelle χ
2 représente soit un groupe -OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NRh u, h et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, χ
3 représentant un groupement partant, notamment choisi parmi les halogénures I, Cl et Br, les groupes mésylate, tosylate, triflate, sulfonate, sulfate ou phosphate, Ti, T
2, T
3, T
4 et T
5 représentant indépendamment les uns des autres un atome d'hydrogène, un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone, ou un groupe fonctionnel notamment choisi parmi N0
2, CN, COOR, OR, COR, NHCOR, NRR", S0
2R, I, Br, R et R" représentant indépendamment l'un de l'autre un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone,
l'entité représentant notamment les groupes suivants
L'utilisation d'un sel plurifonctionnel dans le cadre de la réaction de Heck permet d'augmenter la productivité des solutions utilisées. Par ailleurs, les bras peuvent être fonctionnalisés différemment et permettre soit des réactions intramoléculaires, soit des réactions en cascades, soit des réactions multicomposants. La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre du couplage de Suzuki, selon le schéma réactionnel suivant : estérification
Lr réaction de Suzuki solvant(s) avec R
3B(OR
7)
2 clivage par transestérification
R3 étant choisi parmi les groupes aryle, hétéroaryle, éthényle, diényle, allyle, éthynyle, substitués ou non, comprenant de 2 à 30 atomes de carbone, R représentant un atome d'hydrogène ou un groupe alkyle, ramifié ou linéaire ou un groupe cycloalkyle comprenant de 1 à 12 atomes de carbone, n étant un nombre entier variant de 2 à 4, tel que défini ci-dessous,
i étant x nombre entier variant de 1 à n, Y*" représentant un cation onium tel que défini ci-dessus, de formule (Rb)x-nΛ+ dans laquelle x représente un nombre entier égal à 3 ou 4, n étant égal à 2, 3 ou 4 lorsque x est égal à 4 et n étant égal à 2 ou 3 lorsque x est égal à 3, Rb représente un groupe alkyle comprenant de 1 à 20 atomes de carbone, un groupe aryle comprenant de
6 à 30 atomes de carbone ou un groupe aralkyle ou aïkaryle comprenant de 6 à 30 atomes de carbone, lesdits groupes alkyle, aryle, aralkyle ou aïkaryle susmentionnés étant non fonctionnels, et dans laquelle Λ+ représente un cation ammonium, imidazolium, phosphonium ou sulfonium, Y1" représentant notamment un cation alkylammonium, alkylphosphonium ou alkylsulfonium, et étant de préférence un cation tétiaalkylammonium, tétraalkylphosphonium, dialkylimidazolium, trialkylsulfonium, Li représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type
(CH2)r, r variant de 1 à 20, et de préférence de 2 à 10, les bras L pouvant être identiques ou différents, Xf étant tel que défini ci-dessus, et étant notamment CF, Br-, T, CF3C02 _, CH3C02-, BF-f, PF6-, CF3SO3-, "N(S02CF3)2, S04 2-, RiSOf SbF<f, I^SOf, FSOf , P0 3~, i représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le téti-mydrofuranne, le dioxane, Facétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, les fonctions Fo, Fi et F2 étant telles que définies ci-dessous : - Fo est de la forme -χiH, χi représentant un atome d'oxygène ou un groupe -NRf, Rf correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi est de la forme --Re-χ, Re représentant un groupe aromatique ou hétéroaromatique comprenant de 6 à 30 atomes de carbone, χ représentant un groupe partant choisi de préférence parmi Cl, Br, I, OTf, 0-C02R5 ou OSO3-R5, R5 représentant un groupe alkyle comprenant de 1 à 10 atomes de carbone ou un groupe
aralkyle comprenant de 6 à 30 atomes de carbone, Fi répondant de préférence à la formule suivante :
- F
2 est de la forme -R
e-R
2, R
e étant tel que défini ci-dessus et R
2 étant choisi parmi les groupes aryle, hétéroaryle, éthényle, diényle, allyle, éthynyle, substitués ou non, comprenant de 2 à 30 atomes de carbone, F
2 répondant de préférence à la formule suivante :
Ari représentant un groupe aromatique choisi de préférence parmi
la molécule G étant de la forme R2-R3, R2 et R
3 étant tels que définis ci-dessus, et répond notamment à la formule suivante :
dans laquelle χ
2 représente soit un groupe -OR
g, R
g représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NR
hR
u, R
h et R
u représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone,
Ari est tel que défini ci-dessus. La présente invention concerne également l'utilisation telle que définie ci-dessus, pour la mise en œuvre du couplage de Suzuki, selon le schéma réactionnel suivant :
Hχ2— Lj-Y-Lr 1 -x Λ-.H , X estérification solvant(s) ou amidation
solvant(s) couplage de Suzuki
m représentant un nombre entier compris entre 1 et 50, Y
4" représentant un cation onium tel que défini ci-dessus, de formule (R
b)
x.
2Λ
+ dans laquelle x représente un nombre entier égal à 3 ou 4 selon la nature de A
+, à savoir un cation ammonium, phosphonium ou sulfonium respectivement, R
b représente un groupe alkyle comprenant de 1 à 20 atomes de carbone, un groupe aryle comprenant de 6 à 30 atomes de carbone ou un groupe aralkyle ou aïkaryle comprenant de 6 à 30 atomes de carbone, lesdits groupes alkyle, aryle, aralkyle ou aïkaryle susmentionnés étant non fonctionnels, et dans laquelle Λ
+ représente un cation ammonium,
imidazolium, phosphonium ou sulfonium, Y*
" représentant notamment un cation alkylammonium, alkylphosphonium ou alkylsulfonium, et étant de préférence un cation tétraalkylammonium, tétraalkylphosphonium, dialkylimida-zolium, trialkylsulfonium, Li et L
2 représentant un bras, identiques ou différents, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle éventuellement fonctionnel comprenant de 6 à 30 atomes de carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)
r, r variant de 1 à 20, et de préférence de 1 à 10, Xi- étant tel que défini ci-dessus, et étant notamment CF, Br-, T, CF
3CO
2-, CH
3CO
2-, BF
4-, PF<f, CF
3SO
3 - "Η(S0
2CF
3)
2, S0
4 2-, Ri-SO , SbF
6-, RiSOf, FSOf
P04 3-, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétiahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, %x et χ2, identiques ou différents, représentant un atome d'oxygène ou un groupe - NRf, f correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, χ représentant un groupe partant choisi de préférence parmi Cl, Br, I, OTf, O-
C02R5 ou OSO3-R5, R5 représentant un groupe alkyle comprenant de 1 à 10 atomes de carbone ou un groupe aralkyle comprenant de 6 à 30 atomes de carbone, R7 représentant un atome d'hydrogène, un groupe alkyle, ramifié ou non, ou cycloalkyle, comprenant de 1 à 12 atomes de carbone, ou un groupe aryle, comprenant de 6 à 30 atomes de carbone, χ'i et χ'2, identiques ou différents, représentant soit un groupe -ORg, Rg représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NRhRu, Rh et Ru représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone. L'utilisation d'un sel plurifonctionnel dans le cadre de la réaction de Suzuki permet d'augmenter la productivité des solutions utilisées. Par ailleurs, les bras peuvent
être fonctionnalisés différemment et permettre soit des réactions intramoléculaires, soit des réactions en cascades, soit des réactions multicomposants. La présente invention concerne l'utilisation telle que définie ci-dessus, pour la mise en œuvre de la réaction de Heck, selon le schéma réactionnel suivant :
solvant(s) estérification ou amidation
réaction solvant(s) de Heck transestérification clivage ou transamidation
Y
"1" représentant un cation onium tel que défini ci-dessus, de formule (R
b)
x-
2Λ
+ dans laquelle x représente un nombre entier égal à 3 ou 4, R
b représente un groupe alkyle comprenant de 1 à 20 atomes de carbone, un groupe aryle comprenant de 6 à 30 atomes de carbone ou un groupe aralkyle ou aïkaryle comprenant de 6 à 30 atomes de carbone, lesdits groupes alkyle, aryle, aralkyle ou aïkaryle Susmentionnés étant non fonctionnels, et dans laquelle Λ
+ représente un cation ammonium, imidazolium, phosphonium ou sulfonium, Y
+ représentant notamment un cation alkylammonium, alkylphosphonium ou alkylsulfonium, et étant de préférence un cation tétraalkylammonium, tétraalkylphosphonium, dialkylimidazolium, trialkylsulfonium, A
+ représentant un cation ammonium ou phosphonium lorsque x = 4 et un cation sulfonium lorsque x = 3, Li et L
2, identiques ou différents, représentant un bras, notamment un groupe alkyle linéaire ou ramifié comprenant de 1 à 20 atomes de carbone, ou un groupe aralkyle ou aïkaryle, éventuellement fonctionnel comprenant de 1 à 20 atomes de
carbone, et étant de préférence un groupe alkyle linéaire de préférence un groupe alkyle linéaire de type (CH
2)
r, r variant de 1 à 20, et de préférence de 2 à 10, Xi- étant tel que défini ci-dessus, et étant notamment CF, Br-, F, CF
3C0
2 _, CH
3C0
2 ~, BF
4-, PF
6 ", CF
3S0
3 ", -N(S0
2CF
3)
2, S0
4 2-, RiSOf, SbF
6 ", RiS0
3-, FS0
3-, PO
4 3-, Ri représentant un groupe alkyle comprenant de 1 à 20 atomes de carbone, le ou les solvants étant choisis parmi : le dichlorométhane, le tétrahydrofuranne, le dioxane, l'acétonitrile, le diméthylformamide, le diméthylacétamide, la N- méthylpyrrolidinone, le propionitrile, l'acétone, le toluène, le chlorobenzène, le nitrobenzène, le dichlorobenzène, le nitrométhane, le nitroéthane, ou un mélange de ces solvants, 1 1 0 les fonctions F
0 , Fi , F
0 et Fi étant telles que définies ci-dessous : - Fo
1 correspond à un groupe -χ H, dans lequel γ
\ représente un atome d'oxygène ou un groupe -NR
f, R
f correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fo
2 correspond à un groupe -χ
2iH, dans lequel χ
2i représente un atome d'oxygène ou un groupe -NR
f, R
f correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou un groupe aryle comprenant de 6 à 30 atomes de carbone, - Fi
1 répond à la formule suivante : χ
1 ! étant tel que défini ci-dessus,
- Fi
2 répond à la formule suivante : χ
2ι représentant un atome d'oxygène ou un groupe - NR
f, R correspondant à un groupe alkyle, linéaire ou ramifié, comprenant de 1 à 20 atomes de carbone, ou
un groupe aryle comprenant de 6 à 30 atomes de carbone, et χ
3 représentant un groupement partant, notamment choisi parmi les halogénures I, Cl et Br, les groupes
mésylate, tosylate, triflate, sulfonate, sulfate ou phosphate, G répondant à la formule suivante :
dans laquelle χ
2 et χ
2 , identiques ou différents, représentent soit un groupe -OR
g,
Rg représentant un atome d'hydrogène ou un groupe alkyle comprenant de 1 à 20 atomes de carbone, soit un groupe -NRhRu, Rh et Ru représentant indépendamment l'un de l'autre un atome d'hydrogène, un groupe alkyle comprenant de 1 à 20 atomes de carbone ou un groupe aryle comprenant de 6 à 30 atomes de carbone. L'utilisation d'un sel plurifonctionnel dans le cadre de la réaction de Heck permet d'augmenter la productivité des solutions utilisées. Par ailleurs, les bras peuvent être fonctionnalisés différemment et permettre soit des réactions intramoléculaires, soit des réactions en cascades, soit des réactions multi-composants.
DESCRIPTION DES FIGURES
La Figure 1 représente des spectres de RMN du proton enregistrés à 200 MHz dans l'acétone D6, correspondant au suivi de la réaction de couplage de Heck entre le sel 5 (Y = (Me)3N, n = 0) et l'iodobenzène. Le spectre du bas correspond au produit de départ 5d dans l'acétone D6 et le spectre du haut au produit brut d'arrivée 24d dans l'acétone D6.
La Figure 2 représente un chromatogramme correspondant au mélange des trois esters méthyliques 27a à 27ç dont les spectres de masse sont décrits dans le tableau 7.
La Figure 3 représente un chromatogramme correspondant au mélange des sept esters méthyliques biaryliques 29a à 29g dont les spectres de masse sont décrits dans le tableau 8.
La Figure 4 représente un chromatogramme correspondant au mélange des neuf esters méthyliques biaryliques 31a à 31i dont les spectres de masse sont décrits dans le tableau 10. La Figure 5 représente un chromatogramme correspondant au mélange des neuf esters ethyliques biaryliques 32a à 32i dont les spectres de masse sont décrits dans le tableau 11.
La Figure 6 représente un chromatogramme correspondant au mélange des neuf esters propyliques biaryliques 33 a à 33i dont les spectres de masse sont décrits dans le tableau 12.
La Figure 7 représente des spectres de R-V-QST du proton enregistrés à 200 MHz dans l'acétone D6, correspondant au suivi de la réaction de Sonogashira à partir du sel 6b. Le spectre du bas correspond au produit de couplage de 6b avec l'heptynol dans l'acétone D6 et le spectre du haut au produit de départ 6b dans l'acétone D6.
La Figure 8 représente un chromatogramme correspondant au mélange des cinq esters méthyliques acétyléniques 35a à 35e dont les spectres de masse sont décrits dans le tableau 17. La Figure 9 représente un chromatogramme correspondant au mélange des cinq esters ethyliques acétyléniques 36a à 36e dont les spectres de masse sont décrits dans le tableau 18.
La Figure 10 représente un chromatogramme correspondant au mélange des cinq esters propyliques acétyléniques 37a à 37e dont les spectres de masse sont décrits dans le tableau 19.
La Figure 11 représente un chromatogramme correspondant au mélange des cinq esters butyliques acétyléniques 38a à 38e dont les spectres de masse sont décrits dans le tableau 20.
La Figure 12 représente un chromatogramme correspondant au mélange des six alcools 40a à 40f dont les spectres de masse sont décrits dans le tableau 22. La Figure 13 représente des spectres de RMN du proton enregistrés à 200 MHz dans l'acétone D6, correspondant au suivi de la réaction de Grieco réalisée sur un sel d'onium. Le spectre du haut correspond au produit de départ 7g et le spectre du bas correspond au produit d'arrivée 42. La Figure 14 représente le chromatogramme HPLC des produits 47a à 47e (produits de couplage de Sonogashira).
La Figure 15 représente le chromatogramme GC MS du mélange d'oléfines tétrasubstituées 49a à 49e (tableau 26).
La Figure 16 représente un chromatogramme du produit de couplage de Suzuki de 14a (X = Cl) avec l'acide phénylboronique après transestérification par du méthanol.
La Figure 17 représente des spectres de RMN du proton enregistrés à 200 MHz dans l'acétone D6, correspondant au suivi de la réaction de cycloaddition entre le bis- ester d'acryloyle 13 et le cyclopentadiène. Le spectre du haut correspond au produit de départ 13 et le spectre du bas correspond au produit de cycloaddition obtenu après réaction avec le cyclopentadiène.
PARTIE EXPERIMENTALE - PRÉPARATION DES COMPOSÉS
I) SYNTHÈSE DES SELS FONCTIONNALISÉS 1/ Sels d'ammonium 1 :
* -lâ : Une solution de 2 g (0,01 mmol) du t-riméthyl-2-hydroxyéthyl--mmonium dans lml d'eau est ajoutée à une solution de 1 g (0,018 mmol) de bis- trifluorométhanesulfonamidure de lithium. Le mélange est agité pendant 2 heures. Les deux phases obtenues sont séparées, et la phase aqueuse est extraite 2 fois par 15 ml de chlorure de méthylène. Le solvant est ensuite évaporé et le produit est séché sous vide. Huile visqueuse incolore Rdt = 90 % RMN1 H (200 MHz, Acétone D6) : 3,35 (s, 9H) ; 4,14-4,40 (m, 2H) ; 4,05-4,63(m, 2H). RMN 13C (50 MHz, Acétone D6) : 55,02 (t ; JC-N=4,03 Hz ) ; 57,23 ; 68,91 ; 121,05 (q, J=321,2 Hz). * 1b : 25 g (0,1 mol) de 3-chloropropanol, 30 ml d'une solution de triméthylamine à 45% dans l'eau (0,2 mol) et 100 ml d'acétonitrile sont portés à reflux pendant 36
heures. Le solvant est ensuite évaporé sous vide et le solide blanc obtenu est lavé par 2 fois 30 ml d'éther. Solide blanc Rdt = 82% Pf = 158-160°C RMN ^ (200 MHz, D20) : 1,80-2,05 (m, 2H) ; 3,00 (s, 9H) ; 3,20-3,41 (m, 2H) ; 3,60 (t, 2H, -T = 7,l Hz) RMN13C (50 MHz, D20) : 25,68 ; 53,31 (t, Jc-N= 4,1 Hz) ; 58,52 ; 64,52. Spectrométrie de masse (FAB) pour C12H32N2O2CI Masse Théorique calculée pour (2C+, CF)+ 271,2152 Masse Trouvée 271,2149
* le : Un mélange d'une solution de 10 g (65,3 mol) de chlorure de N,N',N"- tr-iméthyle-3-hydroxypropylammonium (Ib) dans 15ml d'eau et 13,23 ml (0,15 mol) de l'acide hexafluorophosphorique en solution à 60% dans l'eau est agité à température ambiante pendant 2 heures. Le milieu devient hétérogène immédiatement et le précipité formé est filtré et lavé à l'éther. Le solide blanc obtenu est séché sous vide. Solide blanc .. Rdt = 67% Pf = 124-126°C RMN H (200 MHz, CD3CN) : 1,70 (m, 2H) ; 2,82 (s, 9H) ; 3,15 (m, 2H) ; 3,40 (t, 2H, J = 6,l Hz). RMNI3C (50 MHz, CD3CN) : 25,44 ; 52,59 (t ; J = 4,2 Hz) ; 57,67 ; 64,26 (t, J = 3,8 Hz). Spectrométrie de masse (FAB) pour C12H32N2O2F6P Masse Théorique calculée pour (2C+, PF6 ")+ 381 ,2106 Masse Trouvée 381 ,2θ * ld : Un mélange d'une solution de 10 g (65 mmol) de chlorure de N,N',N"- triméthyie-3-hydroxypropylammoinum (lb) dans 15 ml d'eau et 9,1 ml (0,15 mol) de l'acide tétrafluoroborique à 50% dans eau est agité à température ambiante. Le milieu reste homogène. Après 12 heures, l'eau est évaporée à sec, le solide blanc obtenu est lavé par 2 fois 15 ml d'éther anhydre. Solide blanc Rdt = 82% Pf = 110- 112°C RMN J (200 MHz, Acétone D^ : 2,10-2,241 (m, 2H) ; 3,05 (s, 9H) ; 3,24-3,45 (m,H) ; 3,61 (t, J = 7,l Hz, 2H).
RMN I3C (50 MHz, Acétone D6) : 27,52 ; 53,35 (t ; JC-N = 4,1 Hz) ; 58,25 ; 64,58.
* le : 10 g du sel d'ammonium (lb) (65,3 mmol) sont dissous dans 10 ml d'eau. Cette solution est ajoutée à 20 g de bis-trifluorométhanesulfonamidure de lithium (71,9 mmol) dans 10 ml d'eau. Le mélange est agité pendant 2 heures à température ambiante. Les deux phases obtenues sont séparées, et la phase aqueuse est extraite 2 fois par 15 ml de chlorure de méthylène. Enfin le solvant est évaporé et le produit est séché sous vide. Huile visqueuse incolore Rdt = 86% RMN XH (200 MHz, Acétone D^ : 2,00-2,21 (m, 2H) ; 3,25 (s, 9H) ; 3,50-3,80 (m, 4H). RMN 13C (50 MHz, Acétone D6) : 29,14 ; 54,27 (t ; Jc-N = 4,1 Hz ) ; 60,05 ;
66,09 ; 121,05 (q, J = 321,2 Hz).
* lf Dans un ballon de 250 ml on met 2 ml (23,90 mmol) de 3-chloropropanol, 4 ml d'une solution de triméthylamine à 45% dans l'eau (40 mmol). Le mélange est ensuite porté à reflux pendant 24 heures. Les solvants sont ensuite évaporés sous vide. Le solide blanc obtenu est lavé par 2 fois 30 ml d'éther. Solide blanc Rdt = 94% Pf = 118-120°C RMN XH (200 MHz, D20) : 1,6-1,78 (m, 2H) ; 1,85-2,05 (m, 2H) ; 3,25 (s, 9H) ; 3,4-3,5 (m, 2H) ; 3,60 (t, 2H, J = 7,2 Hz) RMN13C (50 MHz, D20) : 19,50 ; 28,43 ; 53,18 (t, JC-N = 4,1 Hz) ; 61,11 ; 66,66.
* lg : Dans un bêcher, on prépare une solution de 1 g (0,01mole) du sel d'ammonium (lf) dans 1ml d'eau. Dans un autre bêcher, on dissout de la même manière 2 g (0,018 mmol) de bis-trifluorométhanesulfonamidure de lithium. On mélange les deux solutions et on agite pendant 2 heures à température ambiante pour que l'échange soit total. Les deux phases obtenues sont séparées dans une ampoule à décanter, et la phase aqueuse est extraite 2 fois par 15 ml de chlorure de méthylène. Enfin le solvant est évaporé et le produit est séché sous vide.
Huile visqueuse incolore Rdt = 86% RMN JH (200 MHz, Acétone D6) : 1,5-1,65 (m, 2H) ; 1,9-2,2 (m, 2H) ; 3,3 (s, 9H) ; 3,50-3,65 (m, 5H). RMN 13C (50 MHz, Acétone D6) : 20,91 ; 30,47 ; 53,99 (t ; Jc-N = 4,03 Hz) ; 61,88 ; 67,90 ; 121,05 (q, J = 321,2 Hz)
* lh : A une solution de 4 g (34 mmol) de N,N'-diméthylamino-l-butanol dans 10 ml d'acétonitrile, on ajoute 4,71 g (37,4 mmol) de méthyltriflate à 0°C. On laisse remonter à température ambiante sous agitation et on agite ensuite pendant 2 heures. Après quoi le solvant est évaporé à sec et l'huile incolore obtenue est lavée par 3 x 10 ml d'éther et séchée sous vide. Huile incolore Rdt = 97% RMN 2H (200 MHz, Acétone D6) : 1,65-1,80 (m, 2H) ; 1,90-2,00 (m, 2H) ; 3,12 (s, 9H) ; 3,3-3,5 (m, 2H) ; 3,68 (t, 2H, J = 6,13 Hz) RMN I3C (50 MHz, Acétone Dç) : 28,36 ; 28.99 ; 51,93 (t ; Jc-N ≈ 4,03 Hz) ; 60,47 ; 68,75 (t ; Jc-N = 4,03 Hz) ; 121,05 (q, J = 321,2 Hz) -
* li : Dans un ballon de 250 ml on met 5 g (36 mmol) de 6-chlorohexanol, 10 ml d'une solution de triméthylamine à 45% dans l'eau (0,1 mol) et 100 ml d'acétonitrile pour homogénéiser le milieu. On porte ensuite le mélange à reflux pendant 36 heures. Le mélange eau/acétonitrile est évaporé sous vide et le solide blanc obtenu est lavé par 2 fois 30 ml d'éther. Solide blanc Rdt = 62% Pf = 178-180°C RMN H (200 MHz, MeOH) : 1,30-1,65 (m, 6H) ; 1,80-1,95 (m, 2H) ; 3,18 (s, 9H) ; 3,4-3,6 (m, 2H) ; 3,55 (t, 2H, J ≈ 6,1 Hz). RMN 13C (50 MHz, MeOH) : 22,93 ; 25,48 ; 26,15 ; 32,35 ; 52,60 (t ; J = 4,1 Hz) ; 61,67 ; 66,76.
* 1] : Un mélange d'une solution de 10 g (51,2 mmol) de chlorure de N,N',N"- triméthyle-6-hydroxybutylammonium (li)dans 15ml d'eau et 18,7g (6,66 mmol) de bis-
trifluorométhanesulfonamidure de lithium est agité à température ambiante. Le milieu devient hétérogène immédiatement, et les deux phases sont séparées dans une ampoule à décanter. L'huile incolore obtenue est ensuite lavée deux fois par 3 ml d'eau et séchée à 50°C sous vide poussé. Huile incolore Rdt = 93% RMN *H (200 MHz, Acétone, D6) : 1,41-1,60 (m, 6H) ; 1,88-2,01 (m, 2H) ; 3,30 (s, 9H) ; 3,50-3,65 (m, 4H) ; 3,55(t, 2H, J = 6,1 Hz). RMN 13C (50 MHz, Acétone, D6) : 23,02 ; 25,60 ; 26,22 ; 53,01 (t ; J = 4,1 Hz) ; 61,73 ; 66,99 ; 121,05 (q, J = 324,2 Hz).
2/ Sels de phosphonium 2 :
* 2a : 2 ml (23,90 mmol) de 3-chloropropanol et 7,2 ml (28,68 mmol) de tributylphosphine sont portés à reflux pendant 14 h. L'excès de la 1-ributylpho-φhine est ensuite éliminé par lavage à l'éther (3 x 10 ml). Huile incolore Rdt ≈ 72% RMN *H (200 MHz, D20) : 0,90 (t; 9H ; J = 5,57 Hz) ; 1,30-1,60 (m, 12H ) ; 1,65-1,85 (m, 2H) ; 2,05-2,30 (m, 8H) ; 3,55-3,70(m, 2H). RMN 13C (50 MHz, D20) : 13,15 ; 14,91 ; 15,90 ; 17,57 ; 18,53 ; 23,10, 23,59 ; 23,89 ; 34,62 ; 42,52 ; 58,77 ; 61,32 ; 61,64.
* 2b : 2 g (6,97 mmol) de bis-trifluorométhanesulfonamidure de lithium et 1 g (3,36 mmol) de chlorure de tributyl-3-hydroxypropyl phosphonium (2a) sont dissous dans 3 ml d'eau. Les deux solutions sont ensuite mélangées et agitées pendant 2 h à température ambiante.
La phase aqueuse est extraite par 2 fois 15 ml de chlorure de méthylène et les phases organiques sont rassemblées et séchées sur MgS04. Le solvant est ensuite évaporé à sec, puis le produit obtenu est séché sous vide. huile incolore Rdt = 90% RMN1 H (200 MHz, Acétone D<) : 0,75 (t ; 9H ; J = 7,13) ; 1,22-1,60 (m, 12H) ; 1,61-1,82 (m, 2H) ; 2,12-2,34 (m, 8H) ; 3,45-3,60 (m, 2H) ; 3,8 (t, 1H, J = 5 Hz) RMN 13C (50 MHz, Acétone DΛ ; 13,99 ; 16,06 ; 17,05 ; 18,90 ; 19,86 ; 24,29 ; 24,38 ; 24,81 ; 25,12 ; 25,61 ; 25,70 ; 62,05 ; 62,35 ; 121,05 (q, JCF = 374,2 Hz )
3/ Sels de pyridinium 3
* 3a : Dans un ballon de 100 ml on introduit 2 ml (23,9 mmol) de 3-chloropropanol et 6 ml de pyridine. Le mélange réactionnel est chauffé à 80°C durant une nuit. L'excès de pyridine est éliminé à l'éther par lavage par 3 x 10 ml. Enfin, on cristallise le produit dans l'acétone. Solide blanc Rdt = 73 % Pf = 68-70°C RMN1H(200MHz, D20) : 2,1-2,3 (m, 2H) ; 3,6 (t, J = 5,99 Hz, 2H) ; 3,75 (t, J = 7,19 Hz, 2H) ; 8,01 (t, J = 7,04 Hz, 2H) ; 8,55 (t, J = 9,97 Hz, 1H) ; 8,83 (d, J = 6,04Hz, 2H) RMN13C (50 MHz, D20) : 33,14 ; 58,22 ; 59,51 ; 128,72 ; 144,89 ; 146,12
* 3b : On dissout dans un bêcher 2 g (6,97 mmol) de bis-trifluorométhane- sulfonamidure de lithium dans 3 ml d'eau. De la même manière, on dissout dans un autre bêcher environ 1 g (5,76 mmol) de chlorure de 3-hydroxypropylpyridinium (3a) dans Feau puis on mélange les deux solutions et on laisse agiter durant deux heures. Le contenu du bêcher est transvasé dans une ampoule à décanter. La phase aqueuse est extraite avec 2 fois 15 ml de chlorure de méthylène. Les phases organiques
sont rassemblées et séchées sur MgS0 . Le solvant est ensuite évaporé à sec puis le produit est séché sous vide. huile incolore Rdt = 90% RMN 2H (200 MHz, Acétone) : 2,03-2,21 (m, 2H) ; 3,5 (t, J = 6,1Hz, 2H) ; 3,8 (t, J = 6,9 Hz, 1H) ; 4,73 (t, J = 7Hz, 2H) ; 8,1 (t, J ≈ 7 Hz, 2H) ; 8,52 (t, J = 9,8 Hz, 1H) ; 8,98(d, J = 6,l Hz, 2H) RMN 13C (50 MHz, Acétone) : 34,57 ; 58,95 ; 60,95 ; 121,37 (q, JC-F = 320,9 Hz) ; 129,67 ; 146,46 ; 147,11
4/ Sels d'imidazolium 4
* 4a : Dans un ballon de 100 ml on introduit 2 ml (23,90 mmol) de 3-chloropropanol et 2,87 g (35 mmol) de la N-méthylimidazole. Le mélange réactionnel est chauffé à 80°C durant une nuit. L'excès de la N-méthylimidazole est éliminé par lavage à l'éther (3 10 ml) et le solide blanc obtenu est séché sous vide. Solide blanc Rdt = 80 % RMN
2H(200MHz, D
20) : 1,96-2,25 (m, 2H) ; 3,55 (t, 2H, J = 6,11 Hz) ; 3,85 (s, 3H) ; 4,15 (t, 2H, J = 7,12 Hz) ; 7,43 (t apparent, 1H, J = 1,73 Hz) ; 7,48 (t apparent, 1H, J = 1,74 Hz) ; 8,75 (s, 1H) RMN
13C (50MHz, D
20) : 32,87 ; 35,40 ; 47,23 ; 58,29 ; 123,03 ; 124,26 ; 137,06
* 4b : On dissout dans un bêcher 2 g (6,97 mmol) de LiNTf2 dans 3 ml d'eau. De la même manière, on dissout dans un autre bêcher environ 1 g de chlorure d'imidazolium 4a dans l'eau. Les deux solutions sont mélangées et laissées agiter durant deux heures. Le contenu du bêcher est transvasé dans une ampoule à décanter. La phase aqueuse est extraite avec 2 fois 15 ml de chlorure de méthylène. Les phases organiques sont rassemblées et séchées sur MgS04. Le solvant est ensuite évaporé à sec puis le produit est séché sous vide.
huile incolore Rdt = 90% RMN 2H (200 MHz, Acétone) : 2,15-2,21 (m, 2H ) ; 3,65 (t, 2H, J = 6,11 Hz) ; 4,05 (s, 3H) ; 4,15 (t, 2H, J ≈ 7,12 Hz) ; 7,68 (t apparent, IH, J = 1,73 Hz) ; 7,73 (t apparent, IH, J = 1,74 Hz) ; 8,95 (s, IH) ItMN 13C (50 MHz, Acétone) : 32,32 ; 35,92 ; 47,01 ; 58,35 ; 121,37 (q, JC-F = 320,9 Hz) ; 123,25 ; 124,01 ; 136,50
II) FONCTIONNALISATION DES SELS PRÉCÉDENTS : 1/ Ester acrylique 5 :
Procédure générale de V estérification par l'acide acryliqu ,
Une solution du sel d'onium et de 3 équivalents de chlorure d'acryloyle dans l'acétonitrile est chauffée à 80°C en présence de 5 équivalents de K2C03 solide pendant 2 heures. Le mélange réactionnel est ensuite placé sous vide à 40 °C pour éliminer le solvant et l'excès du réactif. L'acrylate d'onium est ensuite extrait par le chlorure de méthylène.
* 5a : huile rose Rdt = 90% RMN1 H (200 MHz, Acétone D6) : 3,5 (s, 9H) ; 4,03-4,40 (m, 2H) ; 4,73-4,85 (m, 2H) ; 6,05 (dd, IH, L = 1,92 Hz, J2 = 10,5 Hz) ; 6,25 (dd, IH, Ji ≈ 10,5 Hz, J2 = 17,3 Hz) ; 6,45 (dd, IH, Ji = 1,9 Hz, J2 = 17,3 Hz) RMN13C (50MHz, Acétone D6) : 54,01 (t, J = 4,2 Hz) ; 58,25 ; 65,23 ; 121,05 (q, JCF = 374,2 Hz ) ; 128,80 ; 132,24 ; 165,46
* 5b : Solide blanc Rdt = 100% Pf = 175-177°C RMN H (200 MHz, Acétone Z : 2,15-2,20 (m, 2H) ; 3,15 (s, 9H) ; 3,48-3,52 (m, 2H) ; 4,18 (t, 2H, J = 6,0 Hz) ; 5,75 (dd, IH, Ji = 1,92 Hz, J2 = 10,5 Hz) ; 6,15 (dd, IH, Ji = 10,5 Hz, J2 ≈ 17,3 Hz) ; 6,15 (dd, IH, Ji = 1,9 Hz, J2 = 17,3 Hz) -R N 13C (50 MHz, Acétone D6) : 21,74 ; 52,23 (t, J = 4,2 Hz) ; 60,44 (t, J = 3,02) ; 62,6 ; 127,41 ; 130,65 ; 165,04
* 5ç : Huile incolore Rdt = 93% RMN IH (200 MHz, Acétone D6) : 2,28-3,31 (m, 2H ) ; 3,32 (s, 9H) ; 3,06-3,15 (m, 2H) ; 4,52 (t, 2H, J = 6,6 Hz ) ; 5,80 (dd, IH, Ji = 1,9 Hz, J2 = 10,0 Hz) ; 6,05 (dd,
IH, = 18,3 Hz, J2 = 10,0 Hz) ; 6,15 (dd, IH, Ji ≈ 1,9 Hz, J2 = 18,3 Hz) RMN13 C (5 MHz, Acétone D6) : 22,81 ; 53,28 ; 61,46 ; 63,83 ; 128,51 ; 131,72 ; 167,31 * 5d : Huile incolore Rdt = 100%. RMN 2H (200 MHz, Acétone D6) : 2,22-2,25 (m, 2H) ; 3,25 (s, 9H) ; 3,60-3,75 (m, 2H) ; 4,15 (t, 2H, J = 6,0 Hz) ; 5,80 (dd, IH, Jj = 1,92 Hz ; J2 = 10,68 Hz) ; 6,05 (dd, IH, Jj = 17,2 ; J2 = 10,7) ; 6,15 (dd, IH, = 1,9 Hz ; J2 ≈ 17,2 Hz) RMN 13C (50 MHz, Acétone D6) : 29,17 ; 54,16 (t, J = 4,0) ; 65,16 ; 65,23 ; 121,05 (q, JCF = 374,2 Hz) ; 129,40 ; 132,15 ; 165,61 RMN 19F (282 MHz, Acétone D^ : - 79,8
Spectrométrie de masse (FAB) pour C9Hi8NO2 Masse Théorique calculée pour (C ) 172,1338 Masse Trouvée 172, 1346
* 5e : Huile incolore Rdt = 77% RMN *H (200 MHz, Acétone Dt) : 1,7-1,9 (m, 2H) ; 2,00-2,20 (m, 2H) ; 3,40 (s, 9H) ; 3,58-3,72 (m, 2H) ; 4,25 (t, 2H, J = 6,0 Hz) ; 5,80 (dd, IH, lx = 1,92 Hz ; J2 =
10,68 Hz) ; 6,05 (dd, IH, = 17,2 ; J2 = 10,7) ; 6,15 (dd, IH, Ji ≈ 1,9 Hz ; J2 ≈ 17,2 Hz) RMN 13C (50 MHz, Acétone J>6) : 20,86 ; 26,44 ; 53,99 ; 64,52 ; 67,37 ; 122,19 (q, JCF ≈ 374,2 Hz) ; 129,69 ; 131,83 ; 166,99 * 5f : Huile incolore Rdt = 77% RMN JH (200 MHz, Acétone D6) : 1 (t, 9H, J = 7,2 Hz) ; 1,45-1,85 (m, 12H) ; 2,10-2,28 (m, 2H) ; 2,48-2,7 (m, 8H) ; 4,3 (t, 2H, J = 6,27 Hz) ; 5,58 (dd, IH, Ji = 0,3 Hz ; J2 = 1,96 Hz) ; 6,18 (dd, IH, = 17,16 Hz ; J2 = 0,3 Hz) ; 6,4 (dd, IH, i = 17,16 Hz ; J2 = l,97 Hz) RMN 13C (50 MHz, Acétone Ω6) : 13,09 ; 15,10 ; 16,09 ; 17,84 ; 18,80 ; 21,15 ; 21,22 ; 23,38 ; 23,47 ; 23,86 ; 24,18 ; 63,77 ; 64,11 ; 121,05 (q, JCF = 374,2 Hz) ; 128,63 ; 131,12 ; 165,74 * 5g : Huile incolore Rdt = 80% RMN *H (200 MHz, Acétone D< : 2,15-2,27 (m, 2H) ; 3,8 (t, J = 6,1 Hz, 2H) ; 4,96 (t, J= 6,8 Hz, 2H) ; 5,54 (dd, IH, Ji = 0,5 Hz ; J2 = 2 Hz) ; 6,18 (dd, IH, Ji ≈ 17,2 Hz ; J2 = 0,3 Hz) ; 6,4 (dd, IH, Ji = 17,2 Hz ; J2 = 2 Hz) ; 8,1 (t, J = 7 Hz, 2H) ; 8,52 (t, J ≈ 9,8 Hz, IH) ; 8,98 (d, J = 6,2 Hz, 2H) RMN 13C (50 MHz, Acétone Dg) : 30,39 ; 59,98 ; 61,42 ; 121,05 (q, JCF ≈ 374,2 Hz) ; 128,75 ; 128,98 ; 131,38 ; 145,48 ; 146,50 ; 165,78
* 5h : Huile incolore Rdt = 85 % RMN λH (200 MHz, Acétone D6) : 2,3-2,5 (m, 2H ) ; 4,15 (s, 3H) ; 4,28 (t, 2H, J = 6,03 Hz) ; 4,63 (t, 2H, J = 6,99 Hz) ; 5,95 (dd, IH, Ji = 1,92 ; J2 = 10,68 ) ; 6,18 (dd, IH, Ji = 17,2 Hz ; J2 = 10,7 Hz) ; 6,4 (dd, IH, Ji = 1,9 Hz ; J2 = 17,2 Hz) ; 7,8 (t apparent, IH, J ≈ 1,70 Hz) ; 7,95 (t apparent, IH, J = 1,82 Hz) ; 9,2 (s, IH) RMN 13C (50 MHz, Acétone D^ : 29,32 ; 36,17 ; 47,38 ; 61,36 ; 121,05 (q, JCF ≈ 374,2 Hz) ; 122,97 ; 124,30 ; 128,44 ; 131,31 ; 136,948 ; 166,07
2/ Esters 4-iodobenzoïques 6
* 6a : A une solution de 4g (10,96 mmole) de para-iodobenzoate de 3-diméthyl- aminopropyle dans 10 ml d'acétonitrile, on ajoute goutte à goutte à 0°C 1,1 ml (13,15 mmole) d'iodure de méthyle. Le solide blanc ainsi formé est filtré puis lavé par 3 10 ml d'éther. Solide blanc Rdt ≈ 99% Pf = 248-250°C
RMN2H (200MHz, Acétone D6) : 1,9-2,05 (m, 4H) ; 3,20 (s, 9H) ; 3,42-3,55 (m, 2H) ; 4,45 (t, 2H, J = 6,05 Hz) ; 7,8 (d, 2H, J ≈ 8,73 Hz) ; 7,95 (d, 2H, Ji = 8,76 Hz) RMN13C (50MHz, Acétone Dà : 19,89 ; 25,45 ; 53,37 (t, JC-N = 4,1 Hz) ; 65,13 ; 66,42 ; 101,57 ; 131,19 ; 131,45 ; 138,38 ; 166,78 Spectrométrie de masse (FAB) pour C28H 2N2O I3 Masse Théorique calculée pour (2C+, T)+ 851 ,03 Masse Trouvée 851,10
* 6b : A une solution de 1 g (2,04 mmol)de 6a dans 30 ml du mélange eau/acétone, on ajoute 2,65 mmol de LiNTf2 en solution dans l'eau. Après 2 heures d'agitation à température ambiante, on évapore le solvant et on extrait le sel par 3 x 10 ml de chlorure de méthylène. Après évaporation de ce dernier, on obtient un solide blanc. Solide blanc Rdt = 90% Tf = 48-50°C
RMN 2H (200 MHz, Acétone D6) : 1,82-2 (m, 2H) ; 2,1-2,22 (m, 2H) ; 3,38 (s, 9H) ; 3,6-3,72 (m, 2H) ; 4,4 (t, 2H, J ≈ 6,24 Hz) ; 7,8 (d, 2H, J = 8,73 Hz) ; 7,95 (d, 2H, Ji ≈ 8,36 Hz) RMN13C(50MHz, Acétone Dt) : 26,66 ; 29,12 ; 54,12 (t, JC.N ≈ 4,1 Hz) ; 65,28 ; 67,40 ; 101,59 ; 121,17 (q, Jc-F = 320,9 Hz) ; 131,17 ; 132,28 ; 139,23 ; 166,74 Spectrométrie de masse (FAB) pour C3oH 2N3O8F6I2S2 Masse Théorique calculée pour (2C+, NTf2 ")+ 1004,0407 Masse Trouvée 1004,0427
* 6ç : A xine solution de 1 g (2,04 mmol) de l'ammonium 6a dans 30 ml du mélange eau/acétone on ajoute 2,65 mmol de HPf6 en solution (60%) dans l'eau. Après 2h d'agitation à température ambiante le solvant est évaporé et le solide obtenu est lavé à l'éther (3 10ml). Solide blanc Rdt = 96% Tf = 204-206°C RMN XH (200 MHz, Acétone D6) : 2,05-2,2 (m, 2H) ; 2,28-2,47 (m, 2H) ; 3,6 (s,
9H) ; 3,8-3,97 (m, 2H) ; 4,6 (t, 2H, J = 6,3 Hz) ; 8 (d, 2H, J = 8,58 Hz) ; 8,15 (d, 2H, Ji = 8,58 Hz )
RMN 13C (50 MHz, Acétone D6) : 20,97 ; 26,65 ; 54,06 (t ; J = 4,03 Hz) ; 65,31 ; 67,4 (t ; J = 3,17 Hz) ; 101,57 ; 131,19 ; 132,32 ; 139,23 ; 166,75 Spectrométrie de masse (FAB) pour C28H42N2O F-5l2P Masse Théorique calculée pour (2C+, PF6 ")+ 869,0876 Masse Trouvée 869,0879
* 6d : A une solution de 1 g (2,04 mmol) de 6a dans 3 ml d'eau on ajoute 2,65 mmol de
HBF en solution dans l'eau à 40 %. Après l'ajout de ce dernier, on observe la formation d'un solide blanc. Le mélange réactionnel est laissé agité deux heures à température ambiante. Le solide blanc obtenu après filtration est lavé par l'eau (pour éliminer l'excès de HBF ) puis deux fois par 30 ml d'éther et enfin séché sous vide. Solide blanc Rdt = 86% Tf = 184-186°C Ε2MN1H (200 MHz, Acétone Dβ) : 1,90-2,00 (m, 2H) ; 2,15-2,28 (m, 2H) ; 3,4 (s, 9H) ; 3,65-3,80 (m, 2H) ; 4,42 (t, 2H, J = 6,3 Hz) ; 7,85 (d, 2H, J = 8,58 Hz) ; 8,15 (d, 2H, J! = 8,58 Hz) RMN 13C (50 MHz, Acétone D6) : 20,96 ; 26,62 ; 53,97 (t ; J = 4,03 Hz) ; 65,34 ; 101,53 ; 131,22 ; 132,34 ; 139,23 ; 166,73 Spectrométrie de masse (FAB) pour C28H 2N2O F4I2B Masse Théorique calculée pour (2C+, BF4 ")+ 811,1263 Masse Trouvée 811,1259
A une solution de 1 g (10,96 mmol) de para-iodobenzoàte de 3-diméthylamino- propyle dans 10 ml d'acétonitrile, on ajoute goutte à goutte à 0°C 1,1ml (13,15 mmol) de triflate de méthyle. Le solide blanc ainsi formé est filtré puis lavé par 3 10 ml d'éther. Solide blanc Rd<= 96% Pf= 176-178°C RMN 2H (200 MHz, CD3CN) : 1,8-2,1 (m, 4H) ; 3,06 (s, 9H) ; 3,35-3,45 (m, 2H) ; 4,4 (t, 2H, J = 6,05 Hz) ; 7,85 (d, 2H, J = 8,58 Hz) ; 8 (d, 2H, J, = 8,58 Hz) RMN 13C (50 MHz, CD3CN) : 19,16 ; 24,66 ; 52,50 (t ; J = 4,03 Hz) ; 63,66 ; 65,62 (t ; J = 3,17 Hz) ; 99,89 ; 121,17 (q, JC.F = 320,9 Hz) ; 129,58 ; 130,59 ; 137,57 ; 165,39
Spectrométrie de masse (FAB) pour C29H 2N2O7F3I2S Masse Théorique calculée pour (2C+, OTf)+ 873, 1 Masse Trouvée 873,1
* : A un mélange du chlorure de (2-hydroxy-éthyl)-1rimémylammonium commercial dans 100 ml de CH3CN on ajoute : 1,5 éq de DCC, 1,5 éq de l'acide 4-iodobenzoïque et 0,2 éq de DMAP. Le mélange réactionnel est agité 4 heures à température ambiante puis évaporé à sec. L'ester formé est extrait par 3 x 30 ml d'eau. A cette solution on ajoute 1,5 eq de HBF4 en solution à 50% dans l'eau. Le mélange réactionnel est agité deux heures à température ambiante. Le solide blanc obtenu après filtration est lavé par l'eau (pour éliminer l'excès de HBF4) puis deux fois par 30 ml d'éther et enfin séché sous vide. Aspect du produit : Solide blanc Rdt = 90% Pf = 200-202°C RMN 2H (CD3CN, 300Mhz) : 3,18 (s, 9H) ; 3,68-3,80 (m, 2H) ; 4,50-4,73 (m, 2H) ; 7,78 (dd, Ji=l,9 Hz, J2=6,7 Hz ; 2H) ; 7,90 (dd, Jι=l,8Hz, J2=6,6 Hz, 2H). RMN 13C (CD3CN, 75Mhz) : 53.40 (t, J=3,8Hz) ; 58,27 ; 64,38 (t, J=3,8Hz) ; 100,57 ; 128,65 ; 130,68 ; 137,76 ; 164,73.
Spectrométrie de masse (APCI) pour [Cι2 Hι7INO2][BF4] : Masse Théorique calculée pour (C4) 334.2 Masse Trouvée 334.0
* 6g : A une solution de 12,6 m oles de l'alcool le dans 60 ml de CH CN on ajoute ; 1.5 eq de DCC, 1.1 eq de l'acide 4-iodobenzoique et 0.2eq de DMAP. Le mélange réactionnel est agité 4 heures à température ambiante ; filtré et ensuite évaporé à sec. Le solide blanc obtenu est lavé plusieurs fois à l'éther et enfin séché sous vide. Aspect du produit : solide blanc Rdt = 95 % Pf = 48-50°C RMN JH (300 MHz, Acétone De) : 2,40-2,60 (m, 2H) ; 3,47 (S, 9H) ; 3,80-4,00 (m, 2H) ; 4,53 (t, 2H, J=5,9 Hz) ; 7,83 (d, 2H, J=8,3Hz) ; 7,97 (d, 2H, J=8,3Hz). RMN 13C (50 MHz, Acétone D6) : 23,07 ; 53,37 (t, J=3,8Hz) ; 62,19 ; 64,46 ; 100,91 ; 120,50 (q, JCF=321,2 Hz); 129,97 ; 131,51 ; 138,33 ; 165,75.
Spectrométrie de masse (APCI) pour [C13 Hι9INO2][C2NS2O4F6] Masse Théorique calculée pour (C4) 348.2 Masse Trouvée 348.3
3/ Esters benzoïques 4-substitués 7 :
* 7a : Dans un ballon de 250 ml on introduit 2 g (13,1 mmol) de chlorure de N,N',N"- triméthyl-3-hydroxypropylammonium, 25 ml d'acétonitrile, 20 g de K2C03 en poudre et 4 g (17,5 mmol)du chlorure d'acide du 4-bromobenzoïque. Après une nuit d'agitation à température ambiante, on filtre et on lave K-2C03 par 3 fois 15 ml de chlorure de méthylène et enfin on évapore à sec. On reprend à Feau et on élimine l'excès de l'acide 4-bromobenzoïque qui cristallise par filtration. Le produit est ensuite cristallisé dans l'acétone après évaporation d'eau. Solide blanc Rdt ≈ 60% Pf = 164-166°C RMN1H(200MHz, D20) : 2,21-2,34 (m, 2H) ; 3,12 (s, 9H) ; 3,30-3,58 (m, 2H) ; 4,35 (t, 2H, J = 6,8 Hz) ; 7,57 (d, 2H, J = 7,4 Hz) ; 7,80 (d, 2H, = 7,4 Hz)
RMN 13C (50 MHz, D20) : 22,55 ; 30,61 ; 53,34 (t, JC-N = 4,2 Hz) ; 62,69 ; 64,41 (t, JC-N = 4,09 Hz) ; 128,34 ; 128,66 ; 131,37 ; 132,20 ; 167,84 Spectrométrie de masse (FAB) pour Cι3Hι9NO2Br Masse Théorique calculée pour (C ) 300,0599 Masse Trouvée 302,0607
* 7b : Dans ce cas, la synthèse du substrat a été envisagée selon deux approches : par estérification directe du bis-trifluorométhanesulfonamidure de N,N,N-trimethyl-3- hydroxypropylammonium ou par métathèse à partir du chlorure correspondant. Estérification : Dans un ballon de 250 ml, on introduit 4 g (10,5 mmol) de l'alcool, 20 ml d'acétonitrile, 2 ml d'une solution saturée de Na2C03 dans Feau et 4 g (17,5 mmol) de chlorure de l'acide 4-bromobenzoïque. Le mélange réactionnel est chauffé à 60°C durant une nuit. On évapore ensuite à sec, et on solubilise le résidu obtenu dans le chlorure de méthylène. On lave cette solution successivement par 2 20ml d'eau, 2 20 ml d'une solution de soude (1 N) et enfin par 2 x 20ml d'eau. On sèche la solution sur sulfate de magnésium et on évapore à sec le solvant. On reprend dans l'acétone et élimine les traces d'acides par précipitation à 4°C. Après séchage sous vide, on obtient un solide blanc pur. Rdt = 90 %. Métathèse Dans un ballon de 100 ml on solubilise 1 g (2,98 mmol) de lb dans 5 ml d'eau. A cette solution on ajoute 1,1 g (3,19 mmol) de bis-trifluorométhanesulfonamidure de lithixim (LiNTf2) en solution dans 3 ml d'eau. Le mélange réactionnel est agité 2 heures à température ambiante avant d'extraire notre produit par 20 ml de chlorure de méthylène. Après évaporation de ce dernier, on obtient un solide blanc qui est séché sous vide. Solide blanc Rdt = 90% Tf= 86-88°C RMN !H (200 MHz, Acétone D<2> : 2,64-2,83 (m, 2H) ; 3,59 (s, 9H) ; 3,96-4,06 (m, 2H) ; 4,71 (t, 2H, J = 6,76 Hz) ; 7,90 (d, 2H, J = 8,9 Hz) ; 8,19 (d, 2H, J ≈ 8,9 Hz)
RMN 13C (50 MHz, Acétone D6) : 23,96 ; 54,24 (t, Jc-N = 4,2 Hz) ; 63,09 ; 65,35 (t, Jc-N = 4,0 Hz) ; 121,46 (q, Jc-F = 322,0 Hz) ; 128,95 ; 130,42 ; 132,58 ; 133,12 ; 166,34 Spectrométrie de masse (FAB) pour C28H38F-3N3O8S2Br2 Masse Théorique calculée pour (2C+, -OTO 880,0371 Masse Trouvée 880,0375 * 7ç : A une solution de 1 g (2,98 mmol) de lb dans 3 ml d'eau on ajoute 0,5 ml (5,7 mmol) de HPFδ à 60% dans l'eau. Le mélange réactionnel est agité deux heures à température ambiante. Le solide blanc obtenu après filtration est lavé à Feau puis deux fois par 30 ml d'éther et enfin séché sous vide. Solide blanc Rdt = 96% Pf = 154-156°C RMN JH (200 MHz, Acétone D6) : 2,45-2,59 (m, 2H) ; 3,40 (s, 9H) ; 3,79-3,85 (m, 2H) ; 4,50 (t, 2H, j=6.0 Hz) ; 7,55 (dd, 2H, Jι=l,9 Hz ; J2= 7,7 Hz ) ; 8,00 (dd, 2H, Jι=l,9 Hz ; J2= 7,7 Hz) RMN 13C (50 MHz, Acétone D6) : 22,96 ; 53,23 (t, J = 4,0 Hz) ; 62,31 ; 64,34 ; 128,06 ; 129,48 ; 131,79 ; 132,25 ; 165,63 RMN 19F (282 MHz, Acétone Dg) : -71,6 (d ; J = 707,3Hz ; P-F) RMN31P (Acétone, 129,5 MHz) δ : -142 (m, J = 0,7 Hz, P-F6) Spectrométrie de masse (FAB) pour C26H38F6N2Br2O4P Masse Théorique calculée pour (2C+, PF6 ~) 745,0840 Masse Trouvée 745,0824
* 7d : A une solution de 1 g (2,98 mmol) de lb dans 3 ml d'eau on ajoute 1 ml de HBF4 en solution dans Feau à 40%. Après l'ajout de ce dernier, on observe la formation d'un solide blanc. Le mélange réactionnel est agité deux heures à température ambiante. Le solide blanc obtenu après filtration est lavé par Feau (pour éliminer l'excès de HBF ) puis deux fois par 30 ml d'éther et enfin séché sous vide. Solide blanc Rdt = 98% Tf = 154-156°C
RMN TH (200 MHz, Acétone De) : 2,39-2,57 (m, 2H) ; 3,35 (s, 9H) ; 3,70-3,87 (m, 2H) ; 4,50 (t, 2H,J = 5,91 Hz), 7,73 (dd, 2H, Ji = 1,97 Hz ; J2 = 6,77 Hz) ; 8,02 (dd, 2H, Ji = l,77 Hz ; J2 = 6,47 Hz) RMN 13C (50 MHz, Acétone De) : 22,96 ; 53,14 (4,1 Hz) ; 62,35 ; 64,29 ; 128,01 ; 129,52 ; 131,85 ; 132,24 ; 165,61 RMN19F (282 MHz, Acétone De) : -150.16(s, B-F) Spectrométrie de masse (FAB) pour C26H38F4N2O Br2B Masse Théorique calculée pour (2C+, BF4") 689,1228 Masse Trouvée 689,1234
* 7e : A un mélange de l'alcool lb dans 100 ml de CH3CN on ajoute : 1,5 eq de DCC 1,5 eq de l'acide 4-vinylbenzoïque en présence de 0,02 eq de DMAP. Le mélange réactionnel est agité pendant 4 heures à température ambiante puis évaporé à sec. L'ester formé est extrait par 3 x 30 ml d'eau. A cette solution on ajoute 1,5 eq de NaBF en solution dans Feau. Le mélange réactionnel est agité deux heures à température ambiante. Le solide blanc obtenu après filtration est lavé à Feau (pour éliminer l'excès de NaBF4) puis deux fois par 30 ml d'éther et enfin séché sous vide. Aspect du produit : solide blanc Rdt = 75 % Pf = 138-140°C RMN XH (300 MHz, Acétone D6): 2.30-2.48 (m, 2H) ; 3.38 (S, 9H) ; 3.69-3.82 (m, 2H) ; 4.36 (t, 2H, J=6.0 Hz), 6.58 (d, IH, J=l 6.1Hz) ; 7.40-7.50 (m, 3H) ; 7.63-7.78 (m, 3H). RMN 13C (75MHz, Acétone D6) : 22.68 ; 52.79 (t, J≈ 3.8 Hz) ; 60.88 ; 63.86 (t, J=
3.32Hz) ; 117.76 ; 128.20 ; 128.95 ; 130.43 ; 134.42 ; 144.86 ; 165.99. Spectrométrie de masse (APCI) pour [ s H22NO2][BF4] Masse Théorique calculée pour (C4) 248.3 Masse Trouvée 248.2
* If : Méthode 1 A une solution de 5 g d'acide 4-aminobenzoïque (36,46 mmol) dans 30 ml de 3- chloropropanol, on ajoute, goutte à goutte et à 0°C, 5 ml de chlorure de thionyle (68,54 mmol). Le mélange réactionnel est ensuite porté à 100°C pendant 3 heures. 50 ml
d'éther sont ensuite ajoutés et le solide blanc ainsi formé est filtré puis lavé par 3 10 ml d'éther. Le solide est ensuite dissous dans 30 ml d'eau et l'ester est ensuite extrait par 3 20 ml d'acétate d'éthyle. La phase organique est enfin lavée par 20 ml d'une solution saturé de K-2C03 et évaporée. Le mélange constitué du solide blanc obtenu, de 5 ml d'une solution de triméthylamine à 45% dans l'eau et de 0.5g de Nal sont dissout dans 20 ml d'acétonitrile. Puis porté au reflux pendant 16 heures. Après quoi le solvant est évaporé et le produit est cristallisé dans l'acétone. Solide blanc Rdt (en deux étapes)= 58% Pf= 120-122°C Méthode 2 A xm mélange de l'alcool lb dans 100 ml de CH3CN on ajoute : 1,5 eq de DCC 1,5 eq de l'acide 4-nitrobenzoïque en présence de 0,02 eq de DMAP. Le mélange réactionnel est laissé sous agitation 4 heures à température ambiante puis évaporé à sec et l'ester formé est extrait par 3 x 30 ml d'eau. La réduction du groupement nitro est réalisée en ajoutant à cette solution 0,01 eq de Pd/C 5% et sous agitation à température ambiante sous une pression de 5 bars d'hydrogène pendant 8 heures. Après filtration et évaporation de Feau on recristallise le produit dans l'acétone. Rdt= 90% RMN1H(20 MHz, D20 ) : 2,1-2,25 (m, 2H ) ; 3,09 (s, 9H) ; 3,32-3,45 (m, 2H) ; 4,48 (t, 2H, J = 6,15 Hz) ; 6,75 (d, 2H, J = 7,78 Hz) ; 7,72 (d, 2H, J = 7,78 Hz). RMN13 C (50 MHz, D20) : 22,61 ; 30,63 ; 53,37 (t, JC-N = 4,1 Hz) ; 61,88 ; 64,23 ; 114,91 ; 118,20 ; 132,02 ; 153,03 ; 168,80
* 7g : A une solution de 1 g (3,67 mmol) de 7f dans 10 ml d'eau on ajoute 4,77 mmol de
LiNTf2 en solution dans l'eau. Après 2 heures d'agitation à température ambiante, le solide blanc obtenu est filtré puis séché. Solide blanc Rdt = 93% Pf= 109-110°C RMN } (200 MHz, Acétone D6) : 2,35-2,51 (m, 2H) ; 3,41 (s, 9H) ; 3,78-3,92 (m, 2H) ; 4,5 (t, 2H, J = 6,1 Hz) ; 5,51 (signal large, 2H) ; 6,58 (d, 2H, J = 7,9 Hz) ; 7,84 (d, 2H, Ji = 1,1 Hz) RMN13 C (50 MHz, Acétone D6) : 24,17 ; 29.12 ; 54,12 (t, JC-N = 4,1 Hz) ; 61,72 ; 65,60 ; 114,25 ; 118,33 ; 121,17 (q, JC.F = 320,9 Hz) ; 132,72 ; 154,74 ; 167,08
* 7h : A un mélange de l'alcool lb dans 100 ml de CH3CN on ajoute : 1,5 eq de DCC, 1,5 eq de 4-carboxybenzaldéhyde et 0,2 eq de DMAP. Le mélange réactionnel est agité 4 heures à température ambiante puis évaporé à sec. L'ester formé est extrait par 3 x 30 ml d'eau. A cette solution de l'ester dans Feau on ajoute 1,5 eq de HBF en solution à 50% dans Feau. Le mélange réactionnel est agité deux heures à température ambiante. Le solide jaune obtenu après filtration est lavé par l'eau (pour éliminer l'excès de HBF4) puis deux fois par 30 ml d'éther et enfin séché sous vide. Aspect du produit : solide jaune Rdt = 90% Pf = 146-148°C RMN XH (200 MHz, CD3CN) : 2,10-2,33 (m, 2H) ; 3,05 (s, 9H) ; 3,36-3,55 (m, 2H) ; 4,45 (t, 2H, J=5,8 Hz), 8,00 (dd, 2H, L=l,6Hz, J2=6,6Hz) ; 8,13 (dd, 2H, Jι=l,4Hz, J2=8,3 Hz) ; 10,10 (s, IH). RMN 13C (SOMHz, CD3CN) : 22,08 ; 52,66 (t, J= 3,8Hz) ; 61,53 ; 63,52 (t, J= 3,02Hz) ; 129,10 ; 129,80 ; 134,39 ; 139,27 ; 164,89 ; 192,04.
4/ Alcyne supporté
A une solution de 25 mmol d'alcool propargylique dans 25 ml de chlorure de méthylène anhydre et 5eq de Et
3N sont ajoutés 0,9eq du chlorure de 4-chlorobutyryle à 0°C au goutte à goutte. Le mélange réactionnel est agité 3 heures à température, évaporé à sec et l'ester formé est isolé après extraction par 3 x 10 ml d'éther suivie d'une distillation au four à boules (60°C, 0.2mmHg). Le sel obtenu après réaction de quaternarisation à 80°C en présence de la triéthyl-irnine, de l'acétonitrile et de Nal est engagée dans la réaction de métathèse dans eau en présence de LiNTf
2 pour donner le produit 8. Aspect du produit : huile orange Rdt = 85 % RMN
2H (200 MHz, Acétone D
6) : 1,36-1,55 (m, 9H) ; 2,05-2,30 (m, 2H) ; 2,65 (t, J=6,7 Hz, 2H) ; 3,05-3,15 (m, IH) ; 3,39-3,68 (m, 8H) ; 4,76 (d, J=2,5Hz, 2H).
RMN
13C (50 MHz, Acétone D
6) : 7,13 ; 17,20 ; 52,19 ; 53,19 (t, J≈2,8Hz) ; 56,07 ; 120,46 (q, J
CF=321,4 Hz) ; 171,68. Spectrométrie de masse (APCI) pour [Cι
3 H
24NO2] [C
2NS
2O
4F
6] Masse Théorique calculée pour (C ) 226.3 Masse Trouvée 226.4
III) SELS D'ONIUM A ANION FONCTIONNALISE
Dans un b
1 mmol) de fluorure de tétraméthylammonium anhydre. Puis on ajoute 1 mL de THF anhydre et on homogénéise la solution en chauffant si nécessaire. Enfin, on introduit 0,13 g d'acide phénylboronique (2,1 mmol). On laisse agiter durant environ 2 heures à température ambiante. Après 2 heures d'agitation, on rajoute de l'éther anhydre puis on filtre le solide sur verre fritte. Le solide est lavé 2 à 3 fois par 20 ml d'éther et pour finir, il est placé sous vide pour le sécher. Solide blanc Rdt= 82% Pf = 162-164°C RMN
!H (200 MHz, Acétone De) : 3,15 (s, 12H) ; 6,8-7,4 (m, 3H) ; 7,50-7,70 (m, 2H) RMN
13C (50 MHz, Acétone. D
6) : 56,19 (t, J
C.
N = 3,97 Hz) ; 127,47 ; 128,36 ; 130,91 ; 132,98 ; 135,96 Spectre RMN
19 F (300 MHz, Acétone. D
6) : -136,40 (multiplet) Spectre RMN
UB (300 MHz, Acétone. D
6) : 4,66 (D, J
B-
F = 27,4 Hz) (56%) ; 28,5 (44%)
IV) ESTERS DÉRIVES DE LA GLYCINE
2,0 g (11,3 m o de - oc-g yc ne et , g e b 1, mmo sont dissous dans 15 ml de chlorure de méthylène, on ajoute 11,2 mmol de la DCC et 5% molaire de DMAP. Le mélange est agité à température ambiante pendant 18 heures. Après
filtration du précipité formé au cours de la réaction, le filtrat est évaporé à sec et le produit obtenu est lavé par 5 20 ml d'éther puis séché sous vide. Huile incolore Rdt = 92 % RMN
2H (200 MHz, D
20) : 1,39 (s, 9H) ; 2,4-2,5 (m, 2H) ; 3,08 (s, 9H) ; 3,35- 3,45 (m, 2H) ; 3,75 (s, 2H) ; 3,32 (t, 2H, J ≈ 6,9Hz) RMN
13C(50MHz, D
20) : 23,82 ; 39,07 ; 47,31 ; 53,69 (t, J
C.
N = 4.0 Hz) ; 61,62 ; 65,33 ; 166,92 ; 178,06
On fait barboter de l'acide chlorhydrique dans une solution de l'ester 10 dans 15 ml de chlorure de méthylène pendant 6 heures. Le précipité blanc qui se forme après 24 heures d'agitation à température ambiante est lavé deux fois à l'éther. Le produit ainsi obtenu est ajouté à une solution de diphénylméthylène imine (2,0 g ; 11 mmol) dans 15 ml de chlorure de méthylène. Le mélange réactionnel est ensuite agité à température ambiante pendant 24 heures. On filtre le précipité qui se forme,' on évapore le solvant puis on lave par 3 10 ml d'éther. Rdt en 2 étapes : 62% RMN 1H(200 MHz,D2O) : 2,49-2,52 (m, 2H) ; 3,18 (s, 9H) ; 3,43-3,49 (m, 2H) ; 4,19 (s, 2H) ; 4,31 (t, 2H, J = 6,9 Hz) ; 7,18-7,67 (m, 8H) ; 7,82 (m, 2H) RMN13C(50MHz, D20) : 23,89 ; 54,03 ; 55,14 (t, Jc-N = 4,0 Hz) ; 61,98 ; 65,74 ; 127,98 ; 128,33 ; 128,94 ; 129,19 ; 130,67 ; 130,93 ; 140,03 ; 167,68 ; 177,38
V) SELS D'ONIUM BIFONCTIONNELS 1/ Synthèse des sels 12 :
* 12a : A une solution de 4,5g de diméthylamine (0,1 mole) à 45 % dans Feau est mélangée à 30 ml d'acétonitrile. On ajoute 17 g de K C0
3, 150 mg de Nal et 25 ml (0,3 mol) de chloropropanol. Le mélange réactionnel est chauffé à 70°C durant une nuit. Après évaporation de l'acétonitrile on extrait le sel par 3 10ml de méthanol et il est ensuite cristallisé dans l'acétone. Solide blanc Rdt = 87% Tf=112-114°C RMN
1H(200MHz, D
20) : 1,7-1,85 (m, 4H) ; 2,85 (s, 6H) ; 3,05-3,2 (m, 4H) ; 3,4 (t, 4H, J = 6,l Hz) RMN
13C(50MHz, D
20) : 25,25 ; 51,10 (t, J
C-
N = 4,1 Hz) ; 58,94 ; 62,01 (t, J
C.
N = 4,0 Hz)
Spectrométrie de masse (FAB) pour Ci6H oN2O Cl Masse Théorique calculée pour (2C+! Cl") 359,2677 Masse Trouvée 359,2675
* 12b : Dans un ballon de 100ml on introduit une solution de 5,0 g (25,30 mmol) de 12a dans 10 ml d'eau distillée. A cette solution on ajoute une solution de 10 g (32,89 mmol) de LiNTf2 dans Feau. Après 2 heures d'agitation à température ambiante on évapore Feau et on extrait le produit par 3 10 ml d'acétone. Huile incolore Rdt = 90% RMN XH (200 MHz, D20) : 1,82-2,05 (m, 4H) ; 3 (s, 6H) ; 3,25-3,40 (m, 4H) ; 3,46(t, 4H, J = 6,9 Hz) RMN13C (50 MHz, DzO) : 25,34 ; 51,09 ; 58,53 ; 62,13 ; 121,05 (q, J = 321,2 Hz)
2/ Synthèse des diesters 13
* X = NTf
2 Une solution de 2,7 g (6,02 mmol) de 12b et de 6 équivalents de chlorure d'acryloyle dans l'acétonitrile est chauffée à 80°C en présence de 10 équivalents de K
2C0
3 solide pendant 2 heures. Le mélange est ensuite placé sous vide à 40°C pour éliminer le solvant et l'excès du réactif. L'acrylate d'ammonium ainsi obtenu par extraction par le dichlorométhane est stable à 4°C et peut être stocké pendant plusieurs mois. Huile j aune pâle Rdt = 87 % RMN
JH (200 MHz, Acétone De) : 2,3-2,5 (m, 4H) ; 3,42 (s, 6H) ; 3,72-3,82 (m, 4H) ; 4,32 (t, 2H, J= 5,8 Hz) ; 5,95 (dd, 2H, Ji = 1,8 ; J
2 = 10,1) ; 6,18 (dd, 2H, J_ = 10,1 Hz ; J
2 = 17,1 Hz) ; 6,4 (dd, 2H, J
! = 2,1 Hz ; J
2 = 17,1 Hz) RMN
13C (50 MHz, Acétone De) : 22.52 ; 51,14 ; 61,89 ; 61,95 ; 121,05 (q, J=321,2Hz) ; 128,78 ; 131,02 ; 165,81 Spectrométrie de masse (FAB) pour Cι H
2 O N Masse Théorique calculée pour (C
4) 270, 1750 Masse Trouvée 270, 1703
2/ Synthèse des diesters 14 :
* 14a : Dans un ballon de 100 ml on introduit 1 g (5,06 mmol) de chlorure de 12a, 10 ml d'acétonitrile, 4,3 g de K2C03 en poudre et 4,2 g (19,14 mmol) du chlorure d'acide du 4-bromobenzoïque. Après une nuit d'agitation à température ambiante, on filtre et on lave K2CO3 par 3 fois 15 ml de chlorure de méthylène et enfin on évapore à sec. On reprend à Feau et on élimine l'excès de l'acide 4-bromobenzoïque qui cristallise par filtration. Le produit est ensuite cristallisé dans l'acétone après évaporation d'eau.
Solide blanc Rdt = 60% Pf= 112-114°C RMN1H (200 MHz, MeOH-d4) : 2,05-2,25 (m, 4H) ; 3,1 (s, 6H) ; 3,35-3,53 (m,
4H) ; 4,21 (t, 4H, J = 5,8 Hz) ; 7,1 (d, 4H, J = 8,6 Hz) ; 7,35 (d, 4H, = 8,6 Hz ). RMN 13C (50 MHz, MeOH-d4) : 23,87 ; 52,50 ; 62,50 ; 63,64 ; 130,08 ; 130,16 ;
132,78 ; 133,63 ; 168,22
Spectrométrie de masse (FAB) pour C22H26Nθ4Br2 Masse Théorique calculée pour (C4) 526,0229 Masse Trouvée 526,0221
* 14b : A une solution de 100 mg (0,18 mmol) de 14a dans 5ml d'eau on ajoute 0,2 ml de HBF en solution dans l'eau à 40 %. Après l'ajout de ce dernier, on observe la formation d'un solide blanc. Le mélange réactionnel est agité deux heures à température ambiante. Le solide blanc obtenu après filtration est lavé à l'eau (pour éliminer l'excès de HBF ) puis deux fois par 30 ml d'éther et enfin séché sous vide. Solide blanc Rdt = 80% Pf= 172-174°C RMN 2H (200 MHz, Acétone D6) : 2,4-2,57 (m, 4H) ; 3,4 (s, 6H) ; 3,8-3,94 (m, 4H) ; 4,45 (t, 4H, J = 5,8 Hz) ; 7,67 (d, 4H, J = 8,6 Hz) ; 7,95 (d, 4H, Ji = 8,6 Hz) RMN 13C (50 MHz, Acétone De) : 23,55 ; 51,99 (t, J = 4,1 Hz) ; 62,81 ; 63,12 ; 128,92 ; 130,41 ; 132,65 ; 133,14 ; 166,36 Spectrométrie de masse (FAB) pour C22H26NO4Br2 Masse Théorique calculée pour (C1) 526,0229 Masse Trouvée 526,0216
VI) SELS D'ONIUM TRI- ET TETRAFONCTIONNELS PORTANT DES FONCTIONS IDENTIQUES : 1/ Sels trifonctionnels 15 :
* 15a : Dans un tube de Schlenk, on introduit 3 ml (87,15 mmol) de méthylamine, 20 ml d'acétonitrile, 12 g de K
2C0
3, 53 mg de Nal et 12 ml de 3-chloropropanol. Le mélange réactionnel est chauffé à 80°C durant 48 heures. Après évaporation du solvant on extrait le sel par 3 x 10ml du méthanol. Il est ensuite cristallisé dans l'acétone après évaporation du solvant. Solide blanc Rdt = 75% Tf> 260°C RMN
1H(200 MHz, D
20) : 1,8-2,03 (m, 6H) ; 3,1 (s, 3H) ; 3,25-3,4 (m, 6H) ; 3,4 (t, 6H, J = 5,8 Hz) RMN
13C(50MHz, D
20) : 24,89 ; 48,6 ; 58,47 ; 59,60
* 15b : Dans xm ballon de 100 ml on introduit une solution de 0,4 g (1,65 mmol) de 15a dans 1 ml d'eau distillée. A cette solution on ajoute une solution de 0,7 g (2,14 mmol) de LiNTf2 dans Feau. Après 2 heures d'agitation à température ambiante, on évapore l'eau et on extrait 14b par 3 x 10 ml d'acétone. Après évaporation du solvant et séchage sous vide, on obtient 0,64 g d'une huile incolore soit xm rendement de 80%>. RMN1H(200 MHz, MeOH-d4) : 1,92-2,01 (m, 6H) ; 3,1 (s, 3H) ; 3,38-3,52 (m, 6H) ; 3,7 (t, 6H, J = 5,5 Hz) RMN 13C (50 MHz, MeOH-d4) : 25,25 ; 29,92 ; 58,35 ; 59,80 ; 121,05 (q, J = 321,2 Hz)
2/ Sels tétrafonctionnels 16
* 16a : Dans un ballon de 100 ml on introduit 1 g (5,23 mmol) de propanolamine et 2 ml (23,90 mmol) de 3-chloro-l-propanol. Après une nuit de chauffage du mélange
réactionnel à 150°C, on élimine l'excès de chloropropanol par lavage à l'éther (3 x 10ml). On isole ainsi le produit par filtration après cristallisation dans l'acétone. Solide blanc Rdt = 58% RMN !H (200 MHz, D20) : 2-2,2 (m, 8H) ; 3,4-3,6 (m, 8H) ; 3,8(t, 8H, J = 5,1 Hz) RMN13C (50MHz, D20) : 24,49 ; 56,56 ; 58,40
* 16b : Dans un ballon de 100ml on introduit une solution de 1 g (3,5 mmol) de 16a dans
5 ml d'eau distillée. A cette solution on ajoute une solution de 2 g (4,55 mmol) de LiNTf2 dans l'eau. Après 2 heures d'agitation à température ambiante on évapore Feau et on extrait 15b par 3 5 ml de méthanol. Après évaporation du solvant et séchage sous vide, on obtient une huile incolore. Huile incolore Rdt= 85%, RMN1H(200MHz, D20) : 1,7-1,93 (m, 8H) ; 3,15-3,30 (m, 8H) ; 3,58 (t, 8H, J = 5,8 Hz) RMN13C(50MHz, D20) : 24,56 ; 56,67 ; 58,38 ; 121,05 (q, J = 321,2 Hz)
VII) SYNTHÈSE DES SELS D'ONIUM PLURIFONCTIONNELS PORTANT DES FONCTIONS DIFFÉRENTES :
1/ Préparation des aminés tertiaires précurseurs 17
* 17a : Dans un ballon de 100 ml, on introduit 1 g (9,1 mmol) de l-diméthylamino-3- propanol, 10 ml de chlorure de méthylène anhydre, 3 g de K2C03 et 2,84 (10,66 mmol) de chlorure dérivé de l'acide 4-iodobenzoïque. Après 3 heures d'agitation à température
ambiante la réaction est totale. Le mélange réactionnel est ensuite filtré et évaporé à sec. On obtient un solide blanc avec un rendement de 90% (Pf = 48-50°C). RMN !H (200 MHz, CDCl3) : 1,87-2,03 (m, 2H) ; 2,3(s, 6H) ; 2,45 (t, 2H, J = 7,0 Hz) ; 4 (t, 2H, J = 6,5 Hz) ; 7,3-7,5 (m, 4H) RMN 13C (50 MHz CDCl3) : 27,36 ; 45,85 ; 56,58 ; 63,96 ; 101,15 ; 130,17 ; 131,37 ; 138,04 ; 166,34
* 17b : Dans un ballon de 100 ml, on introduit 2,8 g (23,93 mmol) de 1-N,N'- diméthylamino-4-butanol, 10 ml de chlorure de méthylène anhydre, 6,6g de K2C03 et 7,0 g (26,27 mmol) de chlorure d'acide. La réaction est exothermique. Après 3 heures d'agitation à température ambiante la réaction est totale. Le mélange réactionnel est ensuite filtré et évaporé à sec pour donner un solide blanc, avec un rendement de 88%. RMN }H (200 MHz, CDCl3) : 1,78-1,9 (m, 4H) ; 2,5(s, 6H) ; 2,65 (t, 2H, J = 5,3 Hz) ; 4,35 (t, 2H, J = 6,0 Hz) ; 7,7-7,85 (m, 4H) RMN13 C (50 MHz CDCl3) : 20,25 ; 24,86 ; 43,51 ; 57,34 ; 63,47 ; 98,97 ; 128,39 ; 129,39 ; 136,16 ; 164,26
2/ Synthèse des sels 18
* 18a : Dans xm ballon de 50 ml on introduit 1,0g (3 mmol) de 17a et 0,5 ml de 3-chloro- 1-propanol. Après 30 minutes de chauffage du mélange réactionnel à 110°C, on observe la formation d'un solide blanc. Ce dernier est isolé par filtration après cristallisation dans l'acétone et lavage par 3 x 10ml d'éther. Solide blanc Rdt = 94% Tf = 180-182°C.
RMN JH (200 MHz, D20) : 1,8-2 (m, 2H) ; 2,10-2,30(m, 2H) ; 3,05 (s, 6H) ; 3,25-3,45(m, 4H) ; 3,6 (t, IH, J = 0,7 Hz) ; 4,35(t, 2H, J = 0,7 Hz) ; 7,85 (d, 2H, J = 8,1 Hz) ; 8 (d, 2H, J= 8,1 Hz) RMN 13C (50 MHz, D20) : 22,18 ; 25,29 ; 51,27 (t, J = 4,3 Hz) ; 58,42 ; 62,00 ; 101,85 ; 129,94 ; 131,13 ; 138,26 ; 165,49 Spectrométrie de masse (FAB) pour CιSH23NO3I Masse Théorique calculée pour (C ) 392,0723 Masse Trouvée 392,0720
* 18b : Dans un ballon de 100ml on introduit une solution de 1,0 g (2,34 mmol) de 18a dans 5 ml d'eau distillée. A cette solution on ajoute une solution de 3,04 mmol de
LiNTf2 dans Feau. Après 2 heures d'agitation à température ambiante on extrait 18b par
3 x 5 ml de chlorure de méthylène qui est ensuite chassé sous vide pour donner un solide blanc. Solide blanc Rdt ≈ 91% Tf = 90-92°C RMN 2H (200 MHz, Acétone De) : 2,12-2,63 (m, 2H) ;2,45-2,63(m, 2H) 3,4 (s,
6H) ; 3,7-3,93 (m, 6H) ; 4,1 (t, IH, J = 0,3 Hz), 4,55 (t, 2H,J = 0,7 Hz) ; 7,85 (d, 2H, J =
8,l Hz) ; 8 (d, 2H, J = 8,l Hz) RMN 13C (50 MHz, Acétone D6) : 22,62 ; 26,03 ; 51,16 (t, J = 4,3 Hz) ; 58,56 ;
62,21 ; 100,96 ; 121,01 (q ; JCF3 = 321,0 Hz ) ; 129,94 ; 131,51 ; 138,33 ; 165,79. Spectrométrie de masse (FAB) pour C32H46N3O1oF6l2S2 Masse Théorique calculée pour (2C+, NTf2") 1064,0618 Masse Trouvée 1064,0607
3/ Synthèse des sels à fonctionnalités différentes 19 :
Une solution de 1 mmol de 18a ou 18b et de 3 équivalents de chlorure d'acryloyle dans l'acétonitrile est chauffée à 80°C en présence de 5 équivalents de
2C0
3 solide pendant 2 heures. Le mélange est ensuite filtré puis évaporé sous vide à 40°C. L'ester d'ammonium est ensuite extrait par le chlorure de méthylène et stocké après évaporation du solvant à 4°C. 19a : Solide blanc Rdt =75 %. RMN
1H (200 MHz,D
2O) : 2,05- 2,32 (m, 4H) ; 3,1 (s, 6H) ; 3,35-3,55 (m, 2H) 4,18 (t, 2H, J ≈ 5,5 Hz) ; 4,35 (t, 2H, J = 5,8 Hz) 5,90 (dd, IH, Ji = 1,9 Hz ; J
2 = 10,7) 6,05 (dd, IH, Ji = 17,2 Hz ; J
2 = 10,7 Hz) ; 6,3 (dd, IH, Jj = 1,9 Hz ; J
2 = 17,2 Hz) 7,62 (d, 2H, J = 8,6 Hz ) ; 7,82 (d, 2H, J = 7,8 Hz) RMN
13C (50 MHz, D
20) : 22,09 ; 22,86 ; 51,39 (t, J = 4,2 Hz) ; 62,13 ; 62,65 ; 63,03 ; 63,35 ; 101,78 ; 127,53 ; 128,92 ; 131,13 ; 133,05 ; 139,63 ; 168,07 ; 168,45 Spectrométrie de masse (FAB) pour Cι
8H
2sNO I Masse Théorique calculée pour (C
4) 426,0828 Masse Trouvée 446,0821
19b : Dans un ballon de 100ml on introduit une solution de 1,0g (1,80 mmol) de 19a dans 5 ml d'eau distillée. A cette solution on ajoute une solution de 2 mmol de LiNTf2 en solution dans Feau. Après 2 heures d'agitation à température ambiante on extrait 19b par 3 5 ml de chlorure de méthylène. Après élimination du solvant sous vide, on obtient une huile incolore. Rdt= 78% RMN^ (200 MHz, Acétone D ) : 2,31-2,65 (m, 4H) ; 3^5 (s, 6H) ; 3,75-3,93 (m,
4H) ; 4,32 (t, 2H, J = 6,0 Hz), 4,55 (t, 2H, J = 6,1 Hz) ; 5,95 (dd, IH, Ji = 1,9 Hz, J2 = 10,3 Hz) ; 6,2 (dd ; IH ; Jt = 17,2 Hz ; J2 =10,3 Hz) ; 6,43 (dd ; IH ; Ji =1,9 Hz ; J2 = 17,2 Hz) ; 7,82 (d, 2H, J = 8,1 Hz) ; 7,78(d, 2H, J = 8,1 Hz) RMN 13C (50 MHz, Acétone D6) : 23,49 ; 23,54 ; 52,12 (t, J = 4,31 Hz) ; 62,16 ; 62,89 ; 100,84 ; 121,09 (q, JCF3 = 321 Hz ) ; 124,56 ; 129,38 ; 130,82; 132,22 ; 132,38 ; 139,23 ; 166,66. 34i : A une solution de lg (1,55 mmol ) de 6b dans 10 ml du mélange CH CN/NEt3 (2/1), on additionne 4 eq du phénylacétylène et 5% molaire de Cul. Le mélange réactionnel est laissé agité 5mn à température ambiante avant d'ajouter 2,5% de PdCl2 (PPh3)2.
Après 15 minutes d'agitation à température ambiante, le mélange réactionnel est évaporé à sec et lavé à l'éther pour éliminer l'excès des réactifs. L'huile ainsi obtenue est solubilisée dans le chlorure de méthylène et ensuite lavée p r un solution diluée de 2C03 dans l'eau pour libérer Et3N de son chlorhydrate formé au cours de la réaction. Après traitement par Na2S0 et évaporation du chlorure de méthylène, le produit est isolé par filtration après cristallisation dans l'éther et séchage sous vide. Aspect du produit : solide blanc Rdt = 87% Pf = 228-230°C --RΛfiV ;H (CD3CN, 300Mhz) : 1,72-1,93 (m, 4H) ; 3,05 (s, 9H) ; 3,23-3,40 (m, 2H) ; 4,38 (t, J=6,l Hz, 2H) ; 7,38-7,45 (m, 3H) ; 7,55-7,64 (m, 2H) ; 7,70 (dd, J=8,5 Hz, 2H) ; 8, 10 (dd, J=8,5 Hz, 2H). RMN 13C (CD3CN, 75Mhz) : 19,17 ; 24,70 ; 52,61 (t, J=3,8 Hz) ; 63,86 ; 65,65 (t, J=3,0 Hz) ; 87,93 ; 91,93 ; 124,63 (q, JCF=412 Hz) ; 128,42 ; 128,85 ; 129,24 ; 129,44 ; 131,20 ; 131,24 ; 165,63. Spectrométrie de masse (APCI) pour [C22 H26NO2][C2NS2O4F6] : Masse Théorique calculée pour (C4) 336,4 Masse Trouvée 336,0 Applications à la synthèse - Protocole général des différentes réactions :
1/ réaction de Diels-Alder : a- Pour le cyclopentadiène : Une solution de l'ester acrylique supporté (du mono au tiétra-fonctionnalisé) et de 10 équivalents de cyclopentadiène dans 2 ml de chlorure de méthylène est agitée pendant deux heures à température ambiante. L'excès du réactif et le solvant sont ensuite éliminés sous vide.Le produit de réaction ainsi obtenu est mis en solution dans du méthanol en présence de trois gouttes d'acide chlorhydrique 12 N. Après douze heures au reflux, la transestérification est totale et le produit est alors extrait avec du pentane après évaporation de l'alcool sous vide. Le pentane est ensuite éliminé sous vide pour donner les esters méthyliques purs. b- Pour les divers autres diènes : A une solution de 5a dans l'acétonitrile, on ajoute 0,01eq d'hydroquinone et 5eq de diènes. Le mélange réactionnel est chauffé à 120°C dans des tubes scellés, évaporé à
sec puis lavé à l'éther. Le produit de réaction ainsi obtenu est mis en solution dans du méthanol en présence d'une quantité catalytique d'acide chlorhydrique 12 N. Après douze hexires au reflux, le produit est alors extrait avec de l'éther après évaporation de l'alcool sous vide. L'éther est ensuite éliminé sous vide pour donner les esters méthyliques purs.
2/ réaction de Heck : a- Avec l'ester acrylique supporté 5_: 1,5 mmol de substrat supporté sont dissous dans 2 ml de solvant. A cette solution, on ajoute 5 équivalents d'halogenure d' aryle, 1 équivalent de K2C03 comme base et
1 % molaire d'acétate de palladium. A la fin de la réaction on élimine le solvant et l'excès du réactif par lavage à l'éther puis on ajoute le méthanol (2ml), l'acide chlorhydrique 12 N (3 gouttes) et on porte à reflux. Après 12 heures, le produit de couplage est extrait à l'éther après évaporation de l'alcool. La solution éthérée est ensuite évaporée à sec conduisant au produit attendu. b- Avec l'ester iodoaryle supporté 6g : 0,5 mmol de substrat supporté 6g est dissout dans 1 ml de DMF. A cette solution, on ajoute 5 éq d'oléfine (acrylate de tertiobutyle, diméthylacrylamide ou styrène), 1.5éq de K2CO3 comme base et 1 à 5% molaire d'acétate de palladium, A la fin de la réaction, on élimine le solvant et l'excès du réactif par lavage à l'éther puis on ajoute le méthanol
(2 ml), l'acide chlorhydrique 12N (3 gouttes) et on porte à reflux. Après 12 heures, le produit de couplage est extrait à l'éther après évaporation de Falcool et neutralisation du milieu par l'ajout d'une solution diluée de K2CO3 dans Feau. La solution éthérée est ensuite évaporée à sec. c- Avec le styrène supporté 7e : A une solution de 800 mg (2.38mmole) de 7e dans 3 ml de DMF, nous ajoutons 1 ml de NEt3 (3eq), 27mg de Pd(OAc)2 (0,05eq) et 0,125eq de chaque iodure utilisé. Après deux heures de chauffage à 110°C, l'huile ainsi obtenue après l'ajout de l'éther est transestérifiée en présence du méthanol et de quelques gouttes de l'acide chlorhydrique.
3/ Réaction de couplage de Suzuki : Procédure générale pour les sels mono et bifonctionnalisés : A une solution de 100 mg de Fhalogénure d' aryle supporté dans 1ml de DMF on additionne 0,95 équivalents de l'acide boronique (par fonction), 2 équivalents de K2CO3 et 1% molaire d'acétate de palladium. Le mélange réactionnel est chauffé 5 heures à 80°C. Après quoi, on ajoute un alcool et on porte le mélange à reflux pendant 12 heures, en présence de 0,1 ml d'acide chlorhydrique concentré (12 N). Après transestérification et évaporation de l'alcool, l'ester formé est extrait du milieu par lavage à l'éther (3 10 ml).
4/ Réaction de couplage de Sonogashira : a- Pour l'ester iodoaryle supporté : A une solution de 100 mg de Fhalogénure d' aryle supporté dans 1 ml de solvant on additionne 4 équivalents de l'alcyne, 1 équivalent de K2C03 et un mélange (1/2 catalyseur/Cul). Le mélange réactionnel est chauffé 1 heure à 40°C. Après quoi, on ajoute un alcool et on porte le mélange à reflux pendant 12 heures, en présence de 0,1 ml d'acide chlorhydrique concentré (12 N). Après transestérification et évaporation de l'alcool, l'ester formé est extrait du milieu par lavage à l'éther (3 x 10 ml) et isolé après évaporation de l'éther. b- Pour l'alcyne supporté : A une solution de 100 mg de l'alcyne supporté 8 dans 1 ml du mélange CH3CN/NEt3 (2/1) on additionne 4 eq de l'iodure d'aryle et 0,2 eq de Cul. Le mélange réactionnel est agité 5 mn avant d'ajouter 0,leq de PdCl2(PPh3)2. Après réaction, les alcools formés sont extraits à l'éther après évaporation du solvant et élimination de l'excès des réactifs suivie d'une réaction de saponification en présence de 5ml de
NaOH(2N).
5/Alkylation de la Base de Schiff supportée : A un mélange de la base de Schiff supportée 11 (lg ; 2,6mmol), et de 2 équivalents de K2CO3 dans l'acétonitrile (2 ml) on ajoute le dérivé halogène RX (4 mmol) à température ambiante. Le mélange est ensuite porté à reflux sous une agitation vigoureuse. Après 12 heures, le milieu réactionnel est filtré puis évaporé à sec. La transestérification et l'hydrolyse de l'imine sont réalisées au reflux du méthanol en présence d'acide chlorhydrique concentré pendant 12 heures. Après
évaporation du solvant le mélange est dissous dans 1 ml d'eau. L'aminoacide libre est extrait par le dichlorométhane après neutralisation du milieu.
6/ Réaction multicomposant de type Grieco : Sous courant d'argon on mélange 100 mg de Famine 34 supporté, 500 μl d'une solution 1 M de l'aldéhyde dans l'acétonitrile, 500 μl d'une solution 1 M de cyclopentadiène dans l'acétonitrile et 50 μl d'une solution de 1% de TFA dans l'acétonitrile. Le mélange réactionnel est agité une nuit à température ambiante. Après évaporation à sec et lavage à l'éther, on obtient un solide.
7/ Synthèse des oléfines tétrasubstituées : A une solution de 100 mg (0,16 mmole) de 34i dans 0,5 ml du mélange DMF/H20, on ajoute 2eq de Fiodure, 3eq de l'acide boronique et 1,2 mg de PdCl2(PPh3)2 (0,01eq). Après trois heures de chauffage à 100°C, l'huile orange obtenue après ajout de l'éther est solubilisée dans 10ml de chlorure de méthylène et lavée par2 x 3ml d'H20. Après traitement de la phase aqueuse par MgS0 et évaporation à sec, les
" oléfines tétrasubstituées sont isolées par filtration après cristallisation dans l'éther.
EXEMPLES
Pour montrer l'intérêt des sels d'ammonium comme nouveaux supports solubles, les réactifs 5, 6, 7 et 11 permettant de mettre en oeuvre plusieurs types de réactions fondamentales en chimie organique ont été choisis: ; - les esters acryliques 5 ont été engagés dans des réactions de cycloadditions et dé couplages
- un ester arylique substitué par un R (Br ,1, ou CH2=CH) sur le noyau aromatique qui a été testé dans trois exemples de réactions de couplage.
- x ester arylique substitué par un R = NH
2 sur le noyau aromatique qui a été testé dans la réaction de Grieco.
la base de Schiff dérivée de la glycine qui, après alkylation, conduira à des aminoacides supérieurs /f
un ester arylique substitué par un R = PhC≡≡CH sur le noyau aromatique qui a été testé dans la synthèse des oléfines tétrasubstituées.
Afin d'augmenter la charge spécifique des sels d'onium, les présents Inventeurs ont également développé de nouveaux sels d'onium portant plus d'un bras fonctionnalisé. La synthèse de supports et de réactifs supportés ainsi que leurs applications dans quelques exemples est décrite en détail dans la partie expérimentale qui suit.
A - Exemple 1 : Réaction de Diels-Alder : 1) Réaction avec le cyclopentadiène. La richesse et le potentiel synthétique de la réaction de Diels-Alder ont incité les chimistes à rechercher des méthodes permettant d'en augmenter d'une part la vitesse et le rendement, d'autre part la régio et la stéréosélectivité. Cette réaction est le premier exemple choisi pour montrer l'efficacité de la synthèse supportée sur sels d'onium. La réaction de Diels-Alder entre un diénophile supporté sur un sel d'onium 5 et le cyclopentadiène dans le chlorure de méthylène comme solvant a donc été étudiée selon le schéma suivant :
Dans cette partie du travail les Inventeurs ont étudié de façon précise l'influence de : 1) la longueur de chaîne carbonée qui sépare la fonction acryloyle de la fonction ammonium 2) la nature du cation et de Fanion sur la réactivité 3) le recyclage du support
Procédure pour la réaction de Diels-Alder : L'acrylate supporté 5 et 10 équivalents du cyclopentadiène sont dissous dans 2 ml de chlorure de méthylène. La solution est ensuite agitée pendant deux heures à température ambiante. Le solvant et l'excès de réactif sont ensuite éliminés sous vide puis le produit de réaction est solubilisé dans du méthanol en présence de quelques gouttes d'acide chlorhydrique 12 N. Après douze heures à reflux, la transestérification est totale et le produit 21 est alors extrait avec du pentane.
Les résultats obtenus sont regroupés dans le tableau 1 : nature du cation oni
Les résultats du tableau montrent que la longueur de la chaîne alkyle du bras de greffage influence la réactivité de l'ester acrylique. En effet, l'augmentation de la longueur de la chaîne alkyle séparant les deux fonctions ammonium et ester réduit la vitesse de réaction. Ceci suggère qu'une activation de l'acryloyle due à l'effet électroattracteur de la fonction ttiméthylammonium appauvrit en électrons la double liaison acrylique et la rend plus réactive (comparer les essais 1, 2 et 3). De même, la nature du cation influence la réactivité (comparer les essais 2, 4 et 5). Il faut en outre noter que la sélectivité endo/exo est la même quelque soit la nature et la composition du support. Enfin, la possibilité de recyclage du support a été testée dans le cas des sels d'ammonium. Les résultats obtenus sont regroupés dans le tableau 2.
Tableau 2 : Recyclage du support 5
Au cours des opérations de recyclage, une stabilité au niveau de la réactivité, de la sélectivité et du rendement des réactions a été constatée.
2) Réaction de 5a avec divers autres diènes : On a utilisé l'acide acrylique 5a comme diénophile dans la réaction de Diels- Alder avec différents diènes. Pour ceci, une solution de 0,85 mol/1 de 5a dans l'acétonitrile est chauffée à 120°C dans des tubes scellés en présence de 0,01% d'hydroquinone selon le schéma ci-dessus. Après réaction, le mélange réactionnel est évaporé à sec puis lavé à l'éther. Ceci nous a permis d'isoler les cycloadduits 22aa-ad avec des bons rendements (Tableau 3).
θ ® 22aa-ad R = NTN Me
3 -^ . 23aa-ad R = Me
Tableau 3 : Rendement en cycloadduits 22aa-ad. Essai Diènes Temps (h) Rdt (%) ) { 4 90 2 Q 6 85 3 \JT 4 80
Les esters méthyliques 23aa-ad sont obtenus par réaction de transestérification des cycloadduits 22aa-ad après 12 heures au reflux du méthanol en présence d'une quantité catalytique d'acide chlorhydrique. Le tableau 4 regroupe les rendements en 23aa-ad isolés. Tableau 4 : Rendement en cycloadduits 23aa-ad. 23a Diènes Rdt (%) 23aa 85 23ab Q 83 23ac J
~ 73

B - Exemple 2 : Réactions de Couplage La formation de liaisons carbone-carbone est une opération fondamentale en chimie organique. Parmi le grand nombre de réactions possibles, les méthodes catalytiques utilisant des catalyseurs organométalliques sont extrêmement importantes. Pour notre part, nous avons testé ces nouveaux supports dans les réactions de couplage qui ont déjà fait l'objet de nombreux travaux sur résines et polymères solubles (Franzen, 2000 ; Bertineina et al.,1998 ; Wendeborn et al.,1998). Les différentes réactions de couplage étudiées sont les suivantes : 1) le couplage de Heck 2) le couplage de Suzuki 3) le couplage de Sonogashira Dans les différents exemples qui suivent, nous avons supporté des esters acryliques, ou des esters aryliques ou encore les deux réactifs : 1) Couplage de Heck : -> Avec un ester acrylique supporté Dans cet exemple un ester acrylique supporté est engagé dans la réaction de Heck en présence d'acétate de palladium comme catalyseur, de bicarbonate de potassium comme base et d'un iodure d'aryle en large excès comme réactif selon le schéma qui
Toutes les réactions de couplage ont été réalisées à 100°C. Le suivi des réactions a été effectué par RMN du proton (Figure l)(essai 3 du tableau 5), La Figure 1 illustre la possibilité du suivi par RMN Η des réactions et sa simplicité. Il est facile de constater la disparition complète des signaux entre 5,9 et 6,5 ppm correspondant aux trois protons de la double liaison du substrat 5d, et l'apparition des signaux de la double liaison du produit de couplage 24.
Les différents paramètres qui influencent cette réaction ont ensuite été étudiés pour mettre au point les conditions optimales. Les résultats obtenus sont regroupés dans le tableau 5 : Tableau 5 : Influence de la nature du cation, du support et du bras espaceur sur la réaction de couplage de Heck
i : % de conversion de 5 en 24 déterminé par RMN. ii : déterminé par RMN et confirmé par GC après transestérification.
Les essais 1 et 2 montrent que la concentration du milieu réactionnel, outre qu'elle réduit la quantité du solvant, influence aussi bien la réactivité que la sélectivité de la réaction. Ainsi en doublant la concentration, l'isomère trans est obtenu exclusivement. On a remarqué une relation directe entre réactivité et nature du cation du substrat supporté. En effet, en présence du cation pyridinium, la vitesse de réaction est réduite et 87% seulement de produit de couplage sont formés dans les conditions standards. En revanche la longueur de la chaîne alkyle séparant l'ester acrylique et la fonction ammonium n'a aucune influence ni sur la réactivité, ni sur la sélectivité (comparer les essais 2, 3 et 4).
--> Avec l'ester d'iodoaryle supporté 6g L'ester d'iodoaryle supporté sur sel d'ammonium 6g a été utilisé dans la réaction de Heck. Une solution (0.85 mol/1) du sel 6g dans le DMF est chauffée à 100 °C, en présence d'acétate de palladium comme catalyseur, de bicarbonate de potassium comme base et d'un large excès de l'oléfine. Les rendements en produits isolés sont regroupés dans le tableau 6 :
Tableau 6 : Rendements des produits de couplage 26 N° R Quantité de Pd(OAc)
2(%) Rdt(%)(Temps h) 26a C0
2tBu ï 84(1) 26b C0NMe
2 5 80(3) 26c Ph 5 80(3) Le clivage des produits de couplage est réalisé par réaction de transestérification d'un mélange de ces trois sels en présence du méthanol et d'une quantité catalytique d'acide chlorhydrique. La réaction est quantitative et les esters méthyliques obtenus sont extraits à l'éther après neutralisation par une solution de K
2C0
3 et ensuite injectés en GC MS. Le chromatogramme suivant montre les temps de retentions des différent esters (Figure 2 et tableau 7).
Tableau 7 : Caractérisation par GC/MS de la librairie des esters 27 Temps de rétention N° R Masse moléculaire en minutes 27a C02tBu 14.87 220 27b CONMe2 20.92 233 27c Ph 21.24 238
- Avec le styrène supporté 7e Dans cet exemple, le styrène 7e est mis en réaction avec un mélange équimoléculaire de 7 iodures d'aryle différents conduisant ainsi, après réaction, à un petite bibliothèque des 7 produits 28 ou 29. Ainsi, dans un même puits, on introduit une solution 0,85 mol/1 de 7e dans le DMF à laquelle on ajoute 3 équivalents de triéthylamine, 5% d'acétate de palladium et un mélange des sept iodures d'aryle en quantité stoechiométrique (Schéma ci-dessous).
L'avancement de la réaction est suivi par HPLC en suivant la disparition des iodures d'aryle. Le mélange des sels 28a-g est ensuite transestérifié par ajout de méthanol et d'une quantité catalytique d'acide chlorhydrique. Après évaporation du solvant, les esters formés 29a-g sont extraits à l'éther et injecté en GC/MS. (Figure 3) Le tableau 8 regroupe l'attribution des esters 29 obtenus.
Tableau 8 : Caractérisation par GC/MS de la librairie des esters. 29 R Temps de rétention Masse moléculaire 29a F 15.46 256 29b H 15.59 238 29e CH3 16.89 et 17.15 252 17.33 29d et 29e 2 et 4-MeO 268 20.15 29f Br 20.80 317 29g Napht 25.36 et 26.00 290
2) Couplage de Suzuki : Le second exemple de réaction de couplage où les supports onium ont été utilisés est la réaction de Suzuki qui consiste en un couplage d'un halogénxire d'aryle avec un acide aryl boronique. Cette étude a été réalisée selon deux approches différentes :
1) en supportant un halogénxire d'aryle 2) en supportant simultanément un halogénure d'aryle et un acide boronique.
* Halogénure d'aryle supporté. Dans cette étude on a utilisé l'acide 3-iodobenzoïque et l'acide 4-bromobenzoïque supporté sur un sel d'onium, et on a choisi d'utiliser le DMF et le dioxane comme solvants très utilisés dans ce type de réaction sur résine ou polymère soluble. Nous avons donc étudié l'effet de la température et de Fanion du support. Ces différentes études ont été réalisées en utilisant l'acide phénylboronique et l'acétate de palladium comme catalyseur selon le schéma réactionnel suivant :
31, 32 6133 Avec Ar≈cycle aromatique et R≈Me, Et ou Pr ou Ar'-Ar
Tableau 9: Influence de la température et du solvant sur la réaction de couplage de Suzuki en présence de K
2CO
3 (2 équivalents) et de Pd(OAc)
2 (1% molaire).
L'examen du tableau 9 montre que dans le DMF après cinq heures à 80°C, la réaction est totale avec une excellente sélectivité. Par contre, dans le cas du dioxane, dans les mêmes conditions, 70% de conversion ont été observés avec formation de 30% de produit d'homocouplage (Ar-Ar). Il faut noter que le produit désiré est isolé après transestérification avec un rendement de 95% et une pureté de 99,9% (essai 2). Au vu des résultats obtenus lors de cette étude non exhaustive, il a été choisi de travailler dans les conditions suivantes pour la préparation d'une librairie d'esters biaryliques : Solvant : DMF Température : 80°C Base : K
2C0
3 solide pour la simplicité du traitement de réaction Pré-catalyseur : Pd(OAc)
2 Pour la préparation de cette librairie d'esters biaryliques, on a dans un premier temps réalisé une série de réactions de couplage en parallèle avec 9 acides arylboroniques et l'acide 4-bromobenzoique supporté. Ensuite, les 9 esters biaryliques supportés sont mélangés pour former une solution homogène, qui est alors divisée en trois portions égales. Après quoi, chacune des solutions est solubilisée dans un alcool différent après évaporation du DMF sous vide. Quelques gouttes d'acide chlorhydrique concentré (12 N) sont ensuite ajoutées puis le mélange est porté 18 heures à reflux. Après évaporation à sec, le mélange des biarylesters est extrait par l'éther. 3 séries de 9 esters sont donc obtenues et sont analysées en GC/MS. Les différents biarylesters attendus sont tous obtenus quantitativement (aucune trace d'esters aryliques correspondant au produit de départ n'a été détectée par GC/MS) et identifiés sans ambiguïté.
Tous les résultats sont rassemblés ci-dessous dans les tableaux 9 à 11 et les chromatogrammes correspondant aux mélanges des esters biaryliques représentés dans les figures 4 à 6. a/ Esters méthyliques biaryliques 31a-i : Le tableau 10 ci-après correspond au cl-tromatogramme de la figure 4.
Tableau 10 : Caractéristiques GC MS de la librairie des esters méthyliques 31a-i
b/ Esters ethyliques biaryliques 32a-i : Le tableau 11 ci-après correspond au chromatogramme de la figure 5.
Tableau 11 : Caractéristiques GC/MS de la librairie des esters ethyliques 32a-i
cl Esters biaryliques propyliques 33a-i Le tableau 12 ci-après correspond au chromatogramme de la figure 6.
Tableau 12 : Caractéristiques GC/MS de la librairie des esters propyliques 33 -i
* Halogénure d'aryle et acide boronique supportés simultanément : On a greffé un acide arylboronique sur un anion afin de l'engager ensuite dans une réaction de couplage de Suzuki décrochant. L'acide phénylboronique est greffé sur un support onium via Fanion. En effet, si Fanion X
" du sel support est assez nucléophile, il va réagir avec l'acide phénylboronique en quatemarisant l'atome de bore pour donner un borate. Le nucléophile de choix est le fluorure.
Cette réaction de quaternarisation a été réalisée en solubilisant, à température ambiante, le fluorure de tétiaméthylammonium dans le THF (anhydre) puis ajout de l'acide phénylboronique. Après 18 heures d'agitation à température ambiante, le précipité qui se forme est filtré puis lavé à l'éther. Le rendement en produit isolé est de l'ordre de 80%>. Le suivi de cette réaction a été réalisé à l'aide de la RMN du bore et du fluoré. Le couplage de Suzuki est réalisé dans les mêmes conditions que celles décrites dans la première partie, entre l'acide 4-bromobenzoique supporté et l'acide phénylboronique supporté selon le schéma réactionnel suivant :
La réaction conduit au produit de couplage avec un excellent rendement (98% en produit isolé pur après transestérification) et une réactivité supérieure à celle observée dans le cas de l'acide phénylboronique. Le support ld est récupéré quantitativement et peut être réutilisé.
3) Couplage de Sonogashira : Un autre exemple de couplage où cette famille de supports solubles a été testée est celui de Sonogashira qui consiste en un couplage d'un halogénure d'aryle et d'un alcyne vrai. Cette étude a été effectuée en supportant Fhalogénure d'aryle ou l'alcyne sur un sel d'onium. • Halogénure d'aryle supporté : Dans un premier temps et afin de mettre au point les conditions optimales, on a étudié l'influence des différents paramètres sur la réaction de couplage. On a donc examiné l'effet de la température, de la nature du solvant, du catalyseur, de la base et du contre-ion du support ionique.
Ces différentes études ont été réalisées en utilisant l'acide 4-iodobenzoique selon le schéma réactionnel suivant
^(R'≈WIeh R'≈Et), 37j[R'=Pr) et 38iR'=Bu) (R = Me, Et, Pr ou Bu)
1 - Effet de la nature du solvant et de la base : Cette étude a été effectuée en opérant avec 2,5 % de catalyseur, 5 % de Cul et une température de 40°C afin de déterminer l'influence de la naturç de la base et du solvant sur la réaction de couplage. On a donc fait varier la nature de la base ou le solvant :
Les différents essais réalisés à 40°C après 1 heure de réaction sont regroupés dans le tableau 13 :
Tableau 13 : couplage de Sonogashira à 40°C pendant 1 heure (influence de la base et du solvant)
i : X = PF
6 ; R = butyle ii: X = NTf
2; R = pentyle
L'examen du tableau 13 montre que l'emploi de la triéthylamine permet d'observer une meilleure réactivité comparée à l'utilisation du bicarbonate de potassium solide comme base (comparer les essais 1 et 2). Les essais 2 à 8 montrent que différents solvants organiques usuels peuvent être utilisés. En revanche, une diminution de réactivité a été observée dans les cas du toluène et du tétrahydrofurane où le milieu réactionnel est hétérogène.
2 - Effet de la nature de l' anion du sel d'onium : L'étude comparative a été effectuée sur la réaction de couplage du 1-heptyne sur le 4-iodobenzoate de N,N',N''-tiimémylbutylammonium, en utilisant l'acétonitrile comme solvant, la triéthylamine comme base et le PdCl2(PPh3)2 comme catalyseur, selon le schéma réactionnel suivant :
Les résultats obtenus après 15 min d'agitation à 40°C sont regroupés dans le tableau 14 suivant : Tableau 14 : Couplage de Sonogashira (15' à 40°C) : influence de P anion.
L'examen du tableau 14 montre que la réaction de couplage a lieu quelle que soit la nature de Fanion. En revanche, une meilleure réactivité est observée dans le cas du triflate, du méthylsulfate, de Fhexafluorophosphate et du bis-trifluorométhane sulfonamidure (triflimide).
3 - Effet de la nature de l'alcyne : Afin de généraliser cette méthodologie on a réalisé le couplage de Fhalogénure d'aryle supporté 6b avec divers alcynes fonctionnalisés ou non.
Ces différentes réactions ont été suivies par RMN du proton. La figure 7 montre la simplicité et la facilité de l'interprétation de tels spectres.
Les résultats obtenus en faisant varier les alcynes sont regroupés dans le tableau 15.
4 - Effet de la nature et de la quantité du catalyseur: La réaction de couplage du 4-iodobenzoate de N,N,N-tiiméthylbutylammonium et de Fhexyne a été effectuée en présence de triéthylamine comme base et de l'acétonitrile comme solvant selon le schéma réactionnel suivant :
Les résultats obtenus sont regroupés dans le tableau 16 suivant :
Tableau 16 : conditions du couplage de Sonogashira dans l'acétonitrile de l'iodoaryle 6ç avec le 1-hexyne.
i : rendement déterminé par RMN. ii : rendement déterminé par GC/MS après transestérification.
On constate les points suivants : - la réaction a lieu à température ambiante avec des temps records même en présence de 2,5% en catalyseur. Ces résultats sont d'un intérêt majeur sachant que, dans le cas du support solide la même réaction n'est totale qu'après 24h en présence de 10% de catalyseur ; - la présence de ligand phosphore accélère nettement la réaction, et le PdCl2(PPh3)2 reste, de loin, le catalyseur de choix pour cette réaction (comparer les essais 7, 8 et 9) ; ! - la réactivité reste identique en diminuant le pourcentage de catalyseur de 10 à 2,5%. En revanche, la réduction à 1% provoque une chute de la réactivité (essai 5). De même, Fessai 6 montre que la présence de Cul n'est pas indispensable mais elle accélère la réaction.
5 - Application en chimie combinatoire : Après avoir analysé les résultats obtenus lors de cette étude, il a été décidé de travailler dans les conditions suivantes pour la préparation d'une librairie d' alcynes aromatiques : Solvant : CH3CN Température : 40°C Base : Triéthylamine Pour la préparation de cette librairie d'esters on a opéré comme suit : Dans un premier temps, une série de réactions de couplage en parallèle avec 5 alcynes et l'acide 4-iodobenzoique supporté a été réalisée. Ensuite, les 5 miheux réactionnels ont été mélangés pour former une solution homogène. Après évaporation de Pacétonitrile et lavage à l'éther, le résidu est alors divisé en quatre portions égales. Chaque partie est solubilisée dans un alcool en présence de 3 gouttes d'acide chlorhydrique concentré (12 N) puis le mélange est porté à reflux pendant 18 heures. Après évaporation à sec de l'alcool, le mélange des produits est extrait à l'éther. Les 4 séries de 5 alcynes obtenues sont alors analysées en GC/MS. Les différents alcynes attendus sont tous identifiés sans ambiguïté. " Les résultats obtenus sont répertoriés ci-dessous sous forme de tableaux. Les chromatogrammes des mélanges d' alcynes sont également reproduits (Figures 8 à 11). a/ Esters méthyliques acétyléniques 35a-e : Le tableau 17 ci-après correspond au chromatogramme de la Figure 8.
Tableau 17 : Caractérisation par GC/MS de la librairie des esters méthyliques
b/ Esters ethyliques 36a-e : Le tableau 18 ci-après correspond au chromatogramme de la Figure 9.
Tableau 18 : Caracterisation par GC/MS de la librairie des esters ethyliques
c/ Esters propyliques 37a-e : Le tableau 19 ci-après correspond au chromatogramme de la Figure 10.
Tableau 19 : Caracterisation par GC/MS de la librairie des esters propyliques
d/ Esters butyliques 38a-e : Le tableau 20 ci-après correspond au chromatogramme de la Figure 11.
Tableau 20 : Caracterisation par GC/MS de la librairie des esters butyliques
• Alcyne supporté 8 : Nous avons engagé l'alcyne supporté 8 dans la réaction de Sonogashira avec différents iodures. Ceci en présence de PdCl2(PPh3)2 comme catalyseur et Cul comme co-catalyseur. Après 20mn d'agitation à température ambiante, la réaction est totale conduisant à 39a-f.
Après réaction, les mélanges réactionnels sont évaporés à sec et lavés à l'éther pour éliminer l'excès des réactifs. Les huiles ainsi obtenues sont solubilisées dans le chlorure de méthylène puis les solutions sont lavées par une solution aqueuse de K
2C0 pour libérer Et
3N de son chlorhydrate formé au cours de la réaction. Après traitement par Na
2S0
4 et évaporation du chlorure de méthylène, les sels sont isolés avec un bon rendement. Ceci est illustré dans le tableau 21.
Tableau 21 : Rendement en produits 39a-f Entrée R Rdt(%) 39a H 77 39b 4-CH
3 89 39c 2-N0
2 81 39d 4-OCH
3 92 39e 4-Br 80 39f 1-Naρht 86
Les alcools formés sont extraits à l'éther après saponification en présence d'une solution aqueuse de NaOH à 5% dans Feau des mélanges des sels isolés précédemment et ensuite injectés en GC-MS(Figure 12 et tableau 22).
Tableau 22 ; : Caracterisation par GC MS de la librairie des alcools 40a-f Masse 40 R Temps de rétention moléculaire 40a H 11,47 132 40b 4-CH
3 12,40 146
40d 4-OCH3 13,59 162 40e 4-Br 13,71 211 40f 1-Napht 16,15 182
C - Exemple 3 : Synthèse d' -aminoacides : On a choisi l'alkylation des imines dérivées de la glycine et de la benzophenone comme réaction modèle afin d'explorer le potentiel des sels d'onium comme supports dans ce type de réaction. L'intérêt vis-à-vis de ces substrats réside notamment dans la possibilité d'accéder à des α-aminoacides substitués et éventuellement de réaliser leur synthèse asymétrique. La séquence utilisée met en œuvre l'al- ylation d'un iminoester dérivé de la glycine et de la benzophenone dans les conditions de transfert de phase selon la méthode décrite par O'Donnel et al. (1989) en solution et permet de synthétiser des ---u-nino acides supérieurs selon le schéma suivant :
Hormis les vingt et un (L)-α-aminoacides naturels connus et isolés à partir d'hydrolysats de protéines, d'autres non naturels possèdent des propriétés biologiques intéressantes. Par exemple, l'incorporation au niveau d'un peptide d'un aminoacide ayant des substituants bien choisis peut provoquer des contraintes conformationnelles et augmenter la sélectivité vis-à-vis d'un récepteur. On peut citer par exemple l'inhibition des décarboxylases, hydrolases ou transférases par un α-alkyl aminoacide qui joue le rôle de substrat suicide (Williams, R.M. "Synthesis of Optically Active α-Amino
Acids" Pergamon Press, Oxford, 1989). La (D)-phénylglycine (A) efla (D)-p-hydroxyphénylglycine (B), commercialisées par la compagnie hollandaise DSM, constituent respectivement les matières premières pour la production des antibiotiques ampicilline (A) et amoxicilline (B). Les ventes de ceux-ci atteignent 1,5-1,7 milliards de dollars par an. Malgré la demande accrue en α-aminoacides, la fermentation reste la principale méthode pour leur préparation. Depuis la fin des années 80, des méthodes de synthèse ont vu leur application se concrétiser dans le domaine industriel Les α-a-minoacides non naturels sont obtenus par deux méthodes : • la synthèse asymétrique. • Le dédoublement qui reste la méthode de choix. Par ailleurs, la synthèse sur support solide ou soluble de type PEG reste parmi les méthodes les plus efficaces et simples à mettre en œuvre pour accéder à ces molécules (Lindstrôm et al, 2002). On a donc testé les sels d'onium comme supports solubles pour la préparation des α-aminoacides supérieurs selon le schéma réactionnel suivant :
BocHN OH I
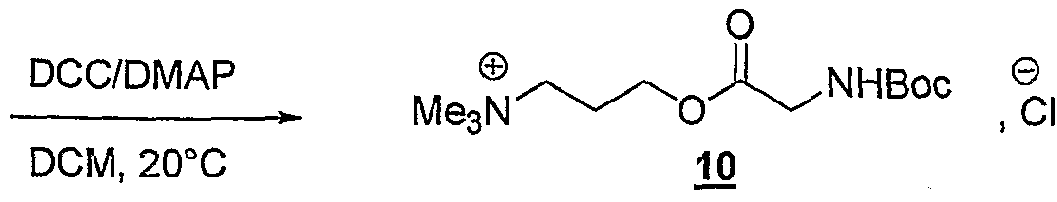
R" = Bu: Rdt= 73% R' = Allyle: Rdt= 82%
La réaction d'estérification est effectuée directement sur la N-Boc-glycine en présence d'un équivalent de DCC et de 5% de DMAP à température ambiante dans le dichlorométhane (DCM). Le groupement protecteur Boc est ensuite éliminé en présence d'acide chlorhydrique dans le DCM avec un bon rendement. De même, la condensation de Faminoester avec la diphénylméthylène imine conduit à Fiminoester 11 avec un excellent rendement et peut-être conservé plusieurs jours' à température ambiante.' Il faut noter que le support qui est un sel d'ammonium joue aussi le rôle du catalyseur de transfert de phase dans l'étape d'alkylation de la base de Schiff 11, et nous avons constaté une réactivité plus importante que celle décrite dans la littérature pour l'alkylation du même substrat supporté sur PEG ou support solide.
D - Exemple 4 : les Réactions Multi-Composants RMÇ Les réactions multi-composants mettent en présence simultanément au moins trois partenaires dans des conditions expérimentales qui ne varient pas au cours du temps et permettent la création de plusieurs liaisons covalentes en cascade dans un seul réacteur, à la différence des réactions classiques ou deux réactifs conduisent à un produit par création d'une nouvelle liaison. Ainsi il est possible d'accéder en une seule étape à une molécule hautement fonctionnalisée à partir d'entités relativement simples. De plus les RCM allient convergence et économie d'atomes, deux principes essentiels en synthèse organique mais aussi en chimie combinatoire. Signalons enfin que ces réactions ont généralement lieu avec un rendement élevé, puisqu'elles évitent la succession d'étapes des synthèses linéaires ou multiétapes qui, font, à chaque pas, chuter le rendement.
Les RMC les plus connues et les plus développées sont celles de Passérini et de Ugi. Un des réactifs clef de ces réactions est un isonitrile de formule générale RN=C, dont la structure électronique du carbone terminal compte un doublet et une lacune électronique (structure de type carbonique) et permet le passage d'un atome de carbone formellement divalent à un atome de carbone tétravalent par addition d'un électrophile et d'un nucléophile. Le schéma qui suit représente un exemple de réaction de Passérini (réaction 3CC pour 3 component condensation).
α-acyloxyamide Bien sûr les RMC ont été transposées sur support solide. Par exemple une résine à terminaison aminé a été engagée dans une réaction de type Ugi pour conduire après clivage à une série d'adduits d'une grande pureté avec des rendements s'échelonnant de moyens à excellents :
Bien que les réactions de Ugi et Passérini soient les plus connues et les plus développées, il existe d'autres RMC, qui répondent au critère essentiel que tous les réactifs sont présents dès le début de la réaction et les conditions ne varient pas au cours de celles-ci. A la différence des réactions de Ugi et Passérini, ces autres réactions ne reposent pas sur l'utilisation d'un isonitrile comme un des acteurs centraux de la création de nouvelles liaisons covalentes. Ces différents types de réactions permettent d'accéder à des structures hautement fonctionnalisées variées en une seule étape. • Synthèse de quinoléines substituées selon la réaction de Grieco: Les quinoléines substituées sont des pharmacophores intéressants. Leur synthèse sur support solide a été réalisée par une RCM dite de Doebner, mettant en jeu une aniline, un aldéhyde et un composé -dicarbonylé. Les quinoléines sont obtenues avec une grande pureté et de très bons rendements.
R< = H, OCH
3 R2 ≈ H, 4-N0
2, 4-CN, 4-CI Il a été décidé ici de profiter des nombreux avantages offerts par les supports onium, comme cela a été mis en évidence dans les différents exemples décrits précédemment. Ainsi, on a choisi de les tester dans les RMC de type Grieco (Grieco et al., 1988). Cet exemple a fait l'objet de plusieurs travaux décrits par W. Armstrong et al. (1997 et 1998) sur support solide et il a permis la préparation d'une librairie de 80 produits avec des rendements allant de 50 à 93%. a- Utilisation de l'aniline supporté 7g dans la réaction de Grieco : Pour ce faire, on a supporté l'aniline 7g qui a été mise en présence d'un aldéhyde et du cyclopentadiène pour conduire à des tétiahydroqxiinoléines. Cet exemple à trois composants, consiste en une première condensation de l'aldéhyde et de l'aniline pour conduire à Fimine. Celle-ci réagit ensuite dans ce qui est formellement une réaction de Diels-Alder avec le cyclopentadiène en présence d'une quantité catalytique d'acide trifl-uoroacétique.
R= 4-N0
2, H, 4-CI, 2-MeO
Le suivi des différentes réactions a été effectué par RMN 1H (Figure 13) et on a observé une conversion allant de 80% à 100% selon la nature de l'aldéhyde. En effet, en présence d'aldéhyde riche en élections la réaction est ralentie. Ainsi en présence du 4- nitrobenzaldéhyde la réaction est totale au bout de 12 heures alors que l'avancement n'est que de 70% dans le cas du benzaldéhyde. Le schéma réactionnel ci-dessus illustre le cas du 4-nitrobenzaldéhyde, après évaporation du solvant (CH CN) et lavage à l'éther pour éliminer l'excès des deux réactifs et de l'acide trifluoroacétique. Ce schéma montre aussi que le suivi d'une réaction qui conduit à des composés complexes est possible et d'une clarté remarquable. La transestérification par le méthanol conduit à des produits très propres qui sont extraits à l'éther et purifiés par filtration sur silice. Les différents exemples que nous avons réalisés et les résultats obtenus sont regroupés dans le tableau 23 ci-dessous. Tableau 23 : Réaction de Grieco réalisée sur sels d'ammonium utilisés comme support soluble : R Temps eu (h) Rdt de 42 en (%) Rdt de 43 en (%) 4-ΝO
2 12 98 82 H 12 70 68 4-OMe 12 60 54 4-Cl 14 96 83
Exemple : synthèse de la tétrahydroquinoléine 43 dérivée du benzaldéhyde : Sous courant d'argon on mélange 100 mg de Famine 7g supporté, 500 μl d'une solution 1M du ara-nitrobenzaldéhyde dans l'acétonitrile, 500, μl d'une solution 1M de cyclopentadiène dans l'acétonitrile et 50 μl d'une solution à 1% de TFA dans l'acétonitrile. Le mélange réactionnel est agité une nuit à température ambiante. Après évaporation à sec et lavage à l'éther, on obtient un solide jaune. Rdt = 71% R N !H (200 MHz, Acétone De) : 1,55-1,8 (m, IH) ; 2,35-2,58 (m, 3H) ; 3,01- 3,2 (m, IH) ; 3,4 (s, 9H) ; 3,7-3,9 (m, 2H) ; 4,1-4,22 (m, IH) ; 4,42 (t, 2H, J - 5,81 Hz) ; 4,9-5,0 (m, IH) ; 5,58-5,75 (m, IH) ; 5,7-5,8 (m, IH) ; 5,93-6,05 (m, IH) ; 6,9 (d, IH, J = 8,4 Hz) ; 7,6- 7,95 (m, 4H) ; 8,28 (d, 2H, J ≈ 8,6 Hz). RMN 13C (50 MHz, Acétone De) : 22,92 ; 31,25 ; 45,35 ; 45,40 ; 52,96 (t, JC.Ν = 4,07 Hz) ; 56,46 ; 60,67 ; 64,28 ; 115,40 ; 119,45 ; 120,13 (q, JC.F = 321,194 Hz),
123,40 ; 124,65 ; 127,68 ; 128,17 ; 129,70 ; 130,98, 134,26 ; 147,20 ; 150,12 ; 150,50 ; 165,77 b- Utilisation de l'aldéhyde supporté 7h dans la réaction de Grieco: Dans ce travail, l'aldéhyde supporté 7h a été engagé dans la réaction de Grieco avec différentes aminés et oléfines, dans l'acétonitrile comme solvant et en présence de TFA comme catalyseur comme le montre le schéma suivant :
Après deux heures d'agitation à température ambiante d'une solution de 0,85 mol/1 de 7h dans l'acétonitile en présence de 1,2 équivalents de TFA, 1 équivalent de Famine et 2 équivalents de l'oléfine. Le suivi de la réaction réalisé par HPLC montre qu'elle est totale après 2 heures. Après l'ajout de l'éther les sels formés 44a-d sont isolés par filtration suivie d'une étape de lavage à l'éther. Les produits 45a-d sont isolés par extraction à l'éther après réaction de transestérification au reflux du méthanol en présence d'une quantité catalytique de l'acide chlorhydrique. Le milieu est ensuite neutralisé par ajout d'une solution diluée de de K
2CO
3 après évaporation du solvant. Le tableau 24 regroupe les rendements en 44 et 45 isolés.
Tableau 24 : Rendements en produits 44 et 45 isolés. Produits Ri R2 Rdt de 44 en % Rdt de 45 en % 44a Cyclopentadiène 3-NO2 90 85 44b Cyclopentadiène 4-Br 88 77 44c Cyclopentadiène H 77 70 44d Indène 4-Br 92 67
Exemple : Synthèse de 44ç : A une solution de 100 mg (0,3 mmole) de 7h dans 0,4 ml d'acétonitrile, on a ajouté 1 eq de Pa-niline, 2 eq de cyclopentadiène et 1,2 eq de TFA. Le mélange réactiormel est agité deux heures à température ambiante, évaporé à sec et ensuite lavé à l'éther. Le produit de la réaction est isolé par filtration après cristallisation dans l'éther. RMN*H (300 MHz, Acétone D6) : 1,58-1,75 (m, IH) ; 2,40-2,65 (m, 3H) ; 2,97- 2,98 (m, IH) ; 3,00-3,15(m, IH) ; 3,40 (s, 9H) ; 3,77-3,92 (m, 2H) ; 4,03-4,15 (m, IH) ; 4,46 (t, 2H, J = 5,94 Hz) ; 4,68-4,76 (m, IH) ; 5,55-5,62 (m, IH) ; 6,58-7,13 (m, 4H) ; 7,60 (d, 2H, J = 8,3 Hz) ; 8,05 (d, 2H, J ≈ 8,4 Hz). RMN13C (75 MHz, Acétone D6) : 23,58 ; 32,19 ; 46,69 ; 47,06 ; 53,67 (t, J = 3,8 Hz) ; 58,16 ; 62,42 ; 64,81 ; 117,00 ; 119,49 ; 126,47 ; 126,88 ; 127,59 ; 129,52 ; 129,63 ; 130,18 ; 130,43 ; 135,44 ; 146,90 ; 149,82 ; 166,57.
Spectrométrie de masse (APCI) pour [C
25 H
31N
2O
2HBF
4] : Masse Théorique calculée pour (C ) 391,5 Masse Trouvée 391,4 • Synthèse des oléfines tétrasubstituées : a- Synthèse individuelle d'oléfines tétrasubstituées : Les oléfines tétrasubstituées peuvent être obtenues par réaction de Me Murry ou par réaction d'oléfination de Wittig. Cependant, les régio- et stéréo- sélectivités sont les problèmes majeurs associés à ces deux procédés. D'autres approches, mettent en œuvre la carbolithiation des alcynes, des réactions des oxiranes portant un groupement CF
3, les organosilanes, Félectrotelluration. Cependant, ces approches utilisent, généralement, des réactifs non facilement disponibles et en plus s'accompagnent parfois d'un défaut de la régio- et la stéreosélectivité. Les oléfines tétrasubstituées peuvent être préparées par réaction d'addition intermoléculaire d'un intermédiaire arylpalladium à un alcyne interne, suivie d'une réaction de couplage avec les organométalliques dérivés du bore, de l'étain ou du zinc. En 2003, Zhou et al ont développé un procédé de synthèse d'oléfines tétrasubstituées utilisant le palladium comme catalyseur. Ce procédé met en œuvre le couplage intermoléculaire d'un iodoaryle, d'un alcyne interne et d'un acide arylboronique
7. PdCI
2(PhCN)
2 R. F-, „,-_,
+
KHCθ3,
100.
c R <
R3
Ces oléfines ont été synthétisées sur support sel d'onium. Pour cela, nous avons utilisé comme alcyne interne le sel 341 obtenu par réaction de Sonogashira entre Fiodure supporté 6b et le phénylacétylène. La réaction est réalisée à 100°C dans un mélange DMF/H
20 (80/20) comme solvant, HCO
3 comme base et PdCl
2 (PPh
3)
2 comme catalyseur.
i = 1 ou 2 j = 2 ou 1 respectivement
Le tableau 25 regroupe les rendements en sels isolés 46 après 3 heures de chauffage à 100°C.
Tableau 25 : Synthèse des oléfines 46. Entrée Ri R2 Rdt(%) 1 H H 67 2 CH3 H 60 3 CH3O H 70 4 H 1-Napht 68 5 CH3O 1-Napht 86 6 CH3 1-Napht 83 Mélange de 2 oléfines régioisomères b- Synthèse d'oléfines tétrasubstituées 47 en mélange : L'iodure supporté 6f est engagé dans le même puit et dans les mêmes conditions que précédemment dans la réaction de Sonogashira en l'absence de Cul avec cinq alcynes différents.
Le suivi de la réaction par HPLC montre l'apparition des produits de couplage de Sonogashira 47 dont les temps de rétention sont 1.92, 3.09, 5.33, 6.57 et 9.35 après une nuit d'agitation à température ambiante (Figure 14). Après évaporation à sec du solvant et lavage à l'éther, le mélange obtenu est divisé en trois parties. Chaque partie est engagée pour former des oléfines tétrasubstituées sans l'ajout de catalyseur. Ceci, en présence de l'acide phénylboronique et trois iodures d'aryle. Après trois heures de chauffage à 100°C les mélanges réactionnels sont transestérifiés séparément en présence du méthanol et d'acide chlorhydrique.
Les oléfines tétrasubstituées sont extraites à l'éther après évaporation du méthanol et injectées en GC/MS. Le chromatogramme présenté en figure 15 illustre le cas du 4-iodotoluène (49a-e).
Tableau 26 : Caracterisation par GC/MS de la librairie d'oléfines tétrasubstituées 49a-e. 49 R Temps de rétention Masse moléculaire 49a CH3(CH2)3 31.37 et 31.59 384 49b CH3(CH2)4 33.12 et 33.42 398 49c et49d HO(CH2)3 et Ph 37.50-38.50 , 386 et 404 49e CH3(CH2)5 38.80 426
E - AUGMENTATION DE LA PRODUCTIVITE DES SOLUTIONS DE SELS D'ONIUM DANS LES SOLVANTS POLAIRES CONVENTIONNELS. On rappelle que la charge spécifique d'un support est définie par la quantité de réactif qui peut être supportée par gramme de support et s'exprime en mmol/g. Cela correspond en fait à ce que l'on pourrait appeler une fonctionnalité spécifique d'un support notée /que l'on pourra exprimer en millifonction par gramme (mf7g.) Dans le cas des solutions de sels d'onium dans les solvants polaires, la molarité sera exprimée en mol/1 ou en mmol/ml. Connaissant la densité des solutions, il est alors facile de convertir en mmol/g pour obtenir des éléments de comparaison avec les résines de
Merrifield ou les solutions de polymères solubles de type PEGs ou autre. Si le sel est monofonctionnel, la fonctionnalité spécifique (f exprimée en mf7g) sera égale à la charge spécifique exprimée en mmol g. Si le sel porte n fois la même fonction, une solution contenant par exemple une millimole de ce sel par gramme aura une fonctionnalité spécifique/de n-mf/g. Dans le cas des sels d'onium testés dans la partie précédente des exemples (cas des sels d'onium mono fonctionnalisés), la charge spécifique est supérieure à 1 mmol. g"1 et peut atteindre jusqu'à 7 mmol.g"1 (voir tableau 27 ci-dessous).
Tableau 27
A titre de comparaison, la charge spécifique des PEG, qui sont les supports solubles les plus utilisés, est habituellement comprise entre 0,1 et 1 mmol.g"1. Des valeurs supérieures (de l'ordre de 10 mmol g"1) sont atteintes dans le cas bien particulier d'un PEG possédant une structure de type dendrimère telle que celle décrite par Haag (Haag et al., 2002). On a synthétisé des sels d'ammonium portant deux bras fonctionnels (ou plus). Les 4 substituants de l'atome d'azote peuvent être simultanément fonctionnalisés multipliant d'autant la densité de fonction de ces sels. On peut aussi envisager de synthétiser des sels d'onium à structure dendrimérique, dont la charge spécifique sera ainsi naturellement multipliée.
Il faut également signaler que les solutions de sels d'onium dans les solvants couramment utilisés (CH2C12, CH3CN, DMF, H2O...) restent peu visqueuses même en opérant à des concentrations supérieures à 4 moles par litre. Ceci représente un avantage majeur par rapport aux solutions de PEG dont la concentration est généralement inférieure à 1 mol.l" 1 - Sels d'ammonium bifonctionnels : La condensation de 3-chloropropanol et de la diméthylamine est réalisée à reflux dans Feau pour conduire après 24 heures au sel bifonctionnalisé avec un bon rendement s
Rdt = 87 % Ces nouveaux supports ont ensuite été testés dans les mêmes réactions que leurs analogues monofonctionnalisés. Pour cela on a supporté des esters d'acryloyle et aryliques halogènes, qui sont ensuite engagés dans des réactions de cycloaddition de
Diels-Alder et de couplages. . Les séquences réactionnelles et les résultats obtenus sont présentés sur les schémas ci-dessous.
a- Réaction de cycloaddition
La transposition des réactions réalisées sur les supports monofonctionnalisés aux analogues bifonctionnahsés n'entraîne aucune différence de réactivité ou de sélectivité. De même, les rendements en produits isolés et leur pureté restent excellents.
Les métathèses d'anions ont été réalisées soit avant le greffage des substrats (cas de Fanion bis-trifluorométhanesulfonamidure) ou après (cas de Fanion tétr afluorobor ate) . -Le chromatogramme correspondant aux produits de couplage de Suzuki réalisé sur le chlorure d'ammonium bifonctionnel (14, X=C1) est présenté dans la Figure 16. Ce chromatogramme met en évidence la pureté du produit brut isolé après transestérification par du méthanol et extraction à l'éther diéthylique avec un rendement de 92%. Le suivi de ces différentes réactions a été réalisé par RMN du proton comme dans le cas des supports monofonctionnalisés (voir Figure 17).
Dans un deuxième volet, nous nous sommes intéressés à augmenter davantage la densité de fonction des sels d'ammonium par tri- et tétrafonctionnalisation selon les séquences réactionnelles suivantes :
H2N— OH + C,— -OH
Les sels tri- et tétra-fonctionalisés ont été préparés à partir de la tripropanolamine par quatemarisation au moyen d'iodure de méthyle ou du 3-chloropropanol conduisant respectivement à 15a ou 16a. On a ensuite utilisé les sels de bis-trifluorométhanesulfonamidure d'ammonium dérivés de 15a et 16a obtenus par métathèse dans Feau pour supporter l'ester acrylique. Ces derniers sont ensuite engagés dans les réactions de cycloaddition comme diénophiles en présence du cyclopentadiène et dans la réaction de couplage de Heck. La réaction de cycloaddition est totale après 2 heures et permet d'isoler le produit avec des rendements supérieurs à 75% et une pureté de 97%. De même le couplage de Heck a été réalisé avec les mêmes rendements que ceux obtenus avec les sels mono et bi- fonctionnalisés (> 95%»).
a- Réactions de cycloaddition :
b- Couplages de Heck :
3 - Sels d'onium supportant des fonctions différentes : Au cours de cette étude, on a montré que les sels d'onium présentent des potentialités et des propriétés très intéressantes comme supports solubles en synthèse organique. En effet, en plus des avantages que nous avons déjà exposés dans les premières parties de ce manuscrit, une des possibilités offertes par ces nouveaux supports solubles est de porter deux fonctions (ou plus) de nature différente sur un même cation à condition qu'elles n'interagissent pas entre elles.
A titre d'illustration, nous nous limiterons à la description de sels d'ammonium portant simultanément un ester acrylique et un halogénure d'aryle de formule suivante :
Le sel d'ammonium préparé selon le schéma réactionnel ci-dessous a été engagé dans la réaction de couplage de Heck. Après une heure à 100°C en présence d'acétate de palladium des produits ont été isolés, qui après transestérification par le méthanol en présence de quelques gouttes d'acide chlorhydrique concentré conduisent au produit de couplage de Heck avec un rendement de 66% et une pureté supérieure à 98%. Il faut noter que seul l'isomère trans a été observé dans les deux cas.
L'insolubilité du produit de réaction avant la transestérification dans la dernière étape dans différents solvants laisse penser qu'une structure de type polymère se forme :
Si le nombre d'atomes est choisi de façon judicieuse pour conduire au produit de cyclisation (couplage intramoléculaire) la forme cis sera obtenue exclusivement. En effet, une telle cyclisation a été déjà observée dans des conditions similaires.
Les résultats préliminaires démontrent l'importance de cette famille de nouveaux supports. Les applications potentielles peuvent en outre encore être enrichies en supportant sur un des bras un catalyseur ou ligand, et sur ceux restants un ou plusieurs réactifs identiques ou non. Cette nouvelle technologie offre un choix énorme et illimité d'applications.
REFERENCES
- Alexander, S. . et Armstrong, R.W. (1997) Tetrahedron Lett., 38, 6163, - Alexander, S.K.; Leonsmith, AN.; Armstrong, R. . (1998) Tetrahedron Lett, 54, 7987,
- Annunziata, R.; Benaglia, M.; Cinquini, M.; Cozzi, F. (2000) Chem.-Eur. J., 6, 133-138,
- Barrett, A. G. M.; Cramp, S. M.; Roberts, R. S.; Zecri, F. (1999) J. Org. Lett., 1, 579-582,
- Barrett, A. G. M.; Cramp, S. M.; Roberts, R. S.; Zécri, F. (2000) J. Org. Lett, 2, 261-264,
- Barrett, A. G. M.; Cramp, S. M.; Roberts, R. S.; Zécri, F. (2000) J. Comb. Chem. High Throughput Screening, 3, 131-133, - Benaglia, M.; Annunziata, R.; Cinquini, M.; Cozzi, F.; Ressel, S. (1998) J. Org. Chem., 63, 8628-8629,
- Berteina, S.; Wendeborn, S.; Brill, K.-D.; De Mesmaeker, A. (1998) Synlett, 6, 676,
- Bienayme, H.; Schmitt, P. (2000) Actualité Chimique, 9, 29-34, - Bleicher, K.H.; Wareing, J.R. (1998) Tetrahedron Letters, 39 (26), 4587,
- Burgath, A.; Sunder, A.; Frey, H. (2000) Macromol. Chem. Phys., 201, 782- 791,
- Chang, J.; Oyelaran, O.; Esser, C. K.; Kath, G. S.; Ki g, G. W.; Uhrig, B. G.; et al. (1999) Tetrahedron Lett, 40, 4477-4480, - Corey et al. (1998) Tetraedron Lett., 39, 5347-5350,
- Domling, A.; Ugi, I. (2000) Angew. Chem., Int. Ed., 39(18), 3168-3210,
- Domling, A. (2002) Current Opinion in Chemical Biology, 6(3), 306-313,
- Elias, H. G. (1997) An Introduction to Polymer Science; VCH: Weinheim; p 163, - Frank, H.; Hagenmaier, H. (1975) Experientia, 31, 131-133,
- Franzen, R. (2000) Canadian Journal ofChemisttγ, 78(7), 957-962,
- Gravert, D. J.; Janda, K. D. (1997) Chem. Rev., 97, 489-509, - Grayson, S. M.; Jayaraman, M.; Fréchet, J. M. (1999) J. Chem. Commun., 1329-1330,
- Grieco, P.; Bahsas, A. (1988) Tetrahedron Lett, 29, 5855,
- Haag, R. (2001) Chem.-Eur. J, 7, 327-335,
- Haag, R.; Hebel, A.; Stumbé, J. F. (2002) In Handbook of Combinatorial Chemistry; Nicolaou, K. C, Hanko, R., Hartwig, W., Eds.; Wiley-VCH; einheim; pp 24-58,
- Haag, R.; Sunder, A.; Hebel, A.; et Roller, S. (2002) J. Comb. Chem., 4, 112- 119,
- Haag, R.; Sunder, A.; Stumbé, J.-F. (2000) J. Am. Chem. Soc, 122, 2954-2955,
- Han, H.; Janda, K. D. (1997) Angew. Chem., Int. Ed. Engl, 36, 1731-1733, - Han, H.; Wolfe, M. M.; Brenner, S.; Janda, K. D. (1995) Proc. Natl. Acad. Sci. U.S.A., 92, 6419,
- Harris, J. M. (1992) In Poly(Ethylene Glycol) Chemistry: Bioîechnical and Biomédical Applications; Harris, J. M., Ed.; Plénum Press: New York; p 2,
- Hodge, P. (1997) Chem. Soc. Rev., 26, 417-424, - Hovestad, N. J.; Ford, A.; Jastrzebski, J. T. B. H.; van Koten, G. (2000) J. Org. Chem., 65, 6338-6344,
- Jayaraman, M.; Fréchet, J. M. J. (1998) J. Am. Chem. Soc, 120, 12996-12997,
- Kates, Steven A.; Albericio, Fernando (2000) Solid-Phase Synthesis,
- Kim, R. M.; Chang, J. (2000) hi Combinatorial Chemistry; Fenniri, H., Ed.; Oxford University Press: Oxford; pp 353-371, - Kim, R. M.; Manna, M.; Hutchins, S. M.; Griffin, P. R.; Yates, N. A; Bernick, A. M.; Chapman, K. T. (1996) Proc. Natl. Acad. Sci. U.S.A., 93, 10012-10017, - Knischka, R.; Lutz, P.; Sunder, A.; Mxilhaupt, , R.; Frey, H. (2000) Macromolecules, 33, 315-320, - Lindstrôm Ulf M. (2002) Chem. Rev. 102 (8), 2751-2772, - Meier, S.; Reisinger, H.; Haag, R.; Mecking, S.; Mûlhaupt, R.; Stelzer, F. (2001) Chem. Commun., 855-856, - Merrifield, R. B. (1963) J. Am. Chem. Soc, 85, 2149, - Mutter, M.; Baeyer, E. (1914) Angew. Chem., Int. Ed. Engl, 13, 88-89, - Mutter, M.; Bayer, E. (1979) In The Peptides; Académie Press: New York, Vol. 2, p 285, - Mutter, M.; Uhmann, R.; Baeyer, E. (1975) Liebigs Ann. Chem., 901-915, - Nicolaou, K.C.; Hanko, R.; Hartwig, W. (2002) Handbook of Combinatorial Chemistry, Volume 2: Drugs, Catalysts, Materials,
- Nicolaou, K.C.; Hanko, R.; Hartwig, W. (2002) Handbook of Combinatorial Chemistry, Volume 1 : Drugs, Catalysts, Materials,
- O'Donnel, M.L; Wu, S.; Bennett, W.D. (1989) J. Am. Chem. Soc, 111, 2353,
- Ranucci, E.; Spagnoli, G.; Sartore, L.; Ferruti, P.; Caliceti, P.; Schiavon, O.; Veronese, F. (1994) Macromol. Chem. Phys., 195, 3469-3479,
- Haag, R.; Sunder, A.; Hebel, A. et Roller, S. (2002) J. Comb. Chem., 4, 112- 119,
- Reed, N. N.; Janda, K. D. (2000) Org. Lett, 2, 1311-1313,
- Reichardt, Christian (1988) Solvents and solvent effects in organic chemistry, Weinheim, Bâle, Suisse ; Cambridge ; New York, NY : VCH,
- Seeberger, P. H.; Haase, .-C. (2000) Chem. Rev., 100, 4349,
- Thompson, L. A.; Ellman, J. A. (1996) Chem. Rev., 96, 555,
- Toy, P.; Janda, K. D. (2000) Ace Chem. Res., 33, 546-554,
- Ugi, L; Domling, A.; Ebert, B. (1999) Combinatorial Chemistry, 125-165, - Ugi, I. (2001) Pure and Applied Chemistry, 73(1), 187-191,
- Ugi, L; Heck, S. (2001) J. Comb. Chem. High Throughput Screening, 4(1), 1- •34,
- V.,Krc-hnja'k et M., W. Holladay (2002) Chem. Rev., 102, 61-91,
- Vanden Eynde, J.J.; Mayence, A. (2000) International Journal of Medicine, Biology and the Environment, 28(1), 89-95,
- Wasserscheid, P.; Keim, W. (2000) Ang. Chem. Int. Ed., 39, 3772-3789, - Wellings, D. A.; Williams, A. (1987) React. Polym., 6, 143-157, - Welton, TN1999) Chem. Rev., 99, 2071-2083, - Wendeborn, S.; Berteina, S.; Brill, K.-D.; De Mesmaeker, A. (1998) Synlett, 6, 671, - Zhou, C; Emrich, D.E.; Larock, R.C. (2003) Org. Lett, 5, 1579-1582.