WO2005073229A1 - 7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel - Google Patents
7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel Download PDFInfo
- Publication number
- WO2005073229A1 WO2005073229A1 PCT/EP2005/000363 EP2005000363W WO2005073229A1 WO 2005073229 A1 WO2005073229 A1 WO 2005073229A1 EP 2005000363 W EP2005000363 W EP 2005000363W WO 2005073229 A1 WO2005073229 A1 WO 2005073229A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- phenyl
- substituted
- agonists
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1cc(N)nc(*)c1* Chemical compound *c1cc(N)nc(*)c1* 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/60—Quinoline or hydrogenated quinoline ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/36—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/54—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D215/54—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3
- C07D215/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 3 with oxygen atoms in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the invention relates to 7-phenylamino-4-quinolone-3-carboxylic acid derivatives and their physiologically acceptable salts and physiologically functional derivatives.
- the invention had the object of providing compounds that develop a therapeutically useful blood sugar lowering effect.
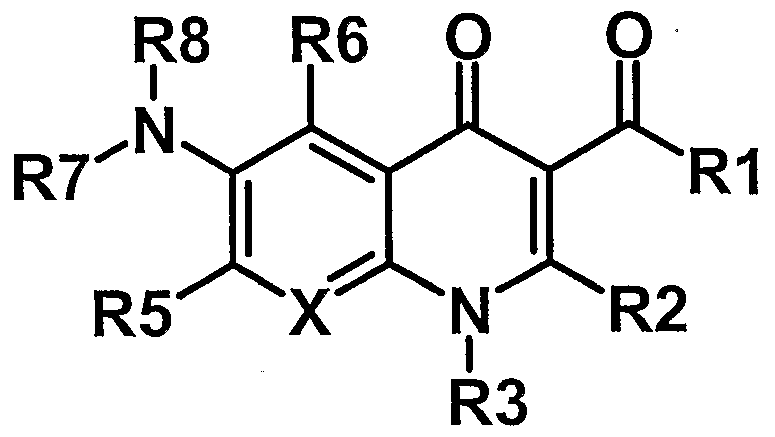
- the invention therefore relates to compounds of the formula I,
- R1 OH, 0- (C 1 -C 6) alkyl or 0- (C 1 -C 6) alkyl-OCO- (C 1 -C 6) alkyl;
- R2 H (CC 6) alkyl or phenyl
- R 3 is H, (C 1 -C 8 ) -alkyl, (C 3 -C 7 ) -cycloalkyl, pyridyl or phenyl, where alkyl may be substituted by R 9 and wherein phenyl or pyridyl may be substituted by R 10;
- R 9 is NH 2 , NH- (C 1 -C 6 ) -alkyl, N - ((C 1 -C 6 ) -alkyl) 2 , COOH, COO- (CC 6 ) -alkyl, (C 3 -C 7 ) - Cycloalkyl, heteroalkyl, heteroaryl, O-phenyl or phenyl, wherein phenyl and heteroaryl may be substituted by R11;
- R 10 is F, Cl, Br, (C 1 -C 6 -alkyl), 0- (C 1 -C 6 ) -alkyl, COOH, COO- (C 1 -C 6 ) -alkyl, NH 2 , NH- (CC 6 ) -alkyl or N - (( C 1 -C 6 ) alkyl) 2 ;
- R 11 is F, Cl, (CC 6 -alkyl), 0- (dC 6 ) -alkyl, NH 2 , NH- (CC 6 ) -alkyl, N - ((CC 6 ) -alkyl) 2 , COOH or COO- ( dC 4 ) -alkyl;
- X is C-R4 or N
- R 4 is H, F, Cl, Br, OH, NO 2 , CN, (C 1 -C 6 ) -alkyl or O- (C 1 -C 6 ) -alkyl, where alkyl may be substituted several times by F, Cl or Br;
- R 5 is H, F, Cl, Br, OH, NO 2 , CN, (C 1 -C 6 ) -alkyl or O- (CC 6 ) -alkyl, where alkyl may be substituted several times by F, Cl or Br;
- R 6 is H, F, Cl, Br, NO 2 , CN or (C 1 -C 6 ) -alkyl, where alkyl may be substituted several times by F, Cl or Br;
- R7 is H or (CC 6) alkyl
- R 8 is phenyl, where phenyl may be substituted up to five times by F, Cl, Br, CN, NO 2 , (C 1 -C 8 ) -alkyl, O- (C 1 -C 8 ) -alkyl, S- (CC 8 ) -alkyl, ( C 2 -C 8 ) - alkenyl, (C 3 -C 7 ) -cycloalkyl, CO- (C 1 -C 4 ) -alkyl, phenyl, benzyl, benzoyl, NH 2 , NH- (CC 6 ) -alkyl, N (C 1 -C 6 ) -alkyl) 2 , P (O) - (O- (CC 4 ) -alkyl) 2 or heteroalkyl, where alkyl and alkenyl are polysubstituted by F, Cl, Br, COOH, or COO- ( C 1 -C 4) alkyl may be substitute
- Heteroalkyl heterocyclic, saturated or unsaturated 4- to 7-membered ring which may contain up to 3 heteroatoms N, O or S as ring members, wherein the ring may be substituted by F, Cl, Br, CN, NO 2 , (dd) alkyl, OH, COOH, COO- (dC 4 ) alkyl;
- X is N, R 1 is OH, R 2, R 3, R 4, R 5 and R 7 are H and R 8 is unsubstituted phenyl;
- R1 OH, 0- (dC 6) -alkyl or 0- (CC 6) alkyl-OCO- (C 1 -C 6) alkyl;
- R 2 is H, (C 1 -C 6 ) -alkyl or phenyl
- R 3 is (C 1 -C 8 ) -alkyl, (C 3 -C 7 ) -cycloalkyl, pyridyl or phenyl, where alkyl is represented by R 9 and where phenyl . or pyridyl may be substituted by R10;
- R 9 is NH 2 , NH- (C 1 -C 6 ) -alkyl, N - ((C 1 -C 6 ) -alkyl) 2 , COOH, COO- (C 1 -C 6 ) -alkyl, (C 3 -C 7 ) -cycloalkyl, heteroalkyl, heteroaryl, O-phenyl or phenyl, wherein phenyl and heteroaryl may be substituted by R11;
- R 10 is F, Cl, Br, (CC 6 -alkyl), 0- (dC 6 ) -alkyl, COOH, COO- (dC 6 ) -alkyl, NH 2 , NH- (dC 6 ) -alkyl or N - (( dC 6 ) -alkyl) 2 ;
- R 11 is F, Cl, (C 1 -C 6 -alkyl), 0- (C 1 -C 6 ) -alkyl, NH 2 , NH- (C 1 -C 6 ) -alkyl, N - ((CC 6 ) -alkyl) 2 , COOH or COO- (C 1 -C 4 ) -alkyl;
- X is C-R4 or N;
- R 4 is H, F, Cl, Br, OH, NO 2 , CN, (C 1 -C 6 ) -alkyl or O- (C 1 -C 6 ) -alkyl, where alkyl may be substituted several times by F, Cl or Br;
- R 6 is H, F, Cl, Br, NO 2 , CN or (C 1 -C 6 ) -alkyl, where alkyl may be substituted several times by F, Cl or Br;
- R7 is H or (CC 6) alkyl
- R 8 is phenyl, where phenyl may be substituted up to five times by F, Cl, Br, CN, NO 2 , (C 1 -C 8 ) -alkyl, O- (C 1 -C 8 ) -alkyl, S- (CC 8 ) -alkyl, ( C 2 -C 8 ) -alkenyl, (C 3 -C 7 ) -cycloalkyl, CO- (C 1 -C 4 ) -alkyl, phenyl, benzyl, benzoyl, NH 2 , NH- (C 1 -C 6 ) -alkyl, N- ((CC 6 ) -alkyl) 2 , P (O) - (O- (C 1 -C 4 ) -alkyl) 2 or heteroalkyl, where alkyl and alkenyl are polysubstituted by F, Cl, Br, COOH, or COO- (dC 4 ) Alky
- R 1 is OH, O- (C 1 -C 6 ) -alkyl or O- (C 1 -C 6 ) -alkyl-OCO- (C 1 -C 6 ) -alkyl; R2H;
- R3 is phenyl, wherein phenyl may be substituted by R10;
- R 10 is F, Cl, Br, (CC 6 -alkyl), 0- (dC 6 ) -alkyl, COOH, COO- (dC 6 ) -alkyl, NH 2 , NH- (CC 6 ) -alkyl or N - (( CC 6 ) -alkyl) 2 ;
- R 4 is H, (C 1 -C 6 ) -alkyl
- R8 is phenyl, wherein phenyl may be substituted up to five times by F ,, Cl;
- R 1 is OH, O- (C 1 -C 6 ) -alkyl
- R3 is phenyl wherein phenyl is substituted by R10;
- R 10 is COOH, COO- (CC 6 ) -alkyl; X C-R4;
- R 4 is H, (CC 6 ) -alkyl
- R8 is phenyl wherein phenyl is substituted one to five times by F, Cl;
- F or Cl is substituted in ortho, ortho and para position.
- the invention relates to compounds of the formula I, in the form of their racemates, racemic mixtures and pure enantiomers, and to their diastereomers and mixtures thereof.
- alkyl radicals in the substituents R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11 and heteroalkyl can be both straight-chain and branched.
- radicals or substituents can occur several times in the compounds of the formula I, they may all independently of one another have the meanings indicated and be identical or different.
- Pharmaceutically acceptable salts are particularly suitable for medical applications because of their higher water solubility compared to the starting or basic compounds. These salts must have a pharmaceutically acceptable anion or cation. Suitable pharmaceutically acceptable
- Acid addition salts of the compounds according to the invention are salts of inorganic acids, such as hydrochloric acid, hydrobromic acid, phosphoric acid, metaphosphoric acid, nitric acid and sulfuric acid, as well as organic acids, e.g.
- Suitable pharmaceutically acceptable basic salts are ammonium salts, alkali metal salts (such as sodium and potassium salts), alkaline earth salts (such as magnesium and calcium salts), trometamol (2-amino-2-hydroxymethyl-1, 3-propanediol), diethanolamine, lysine, or ethylenediamine.
- Salts with a non-pharmaceutically acceptable anion are also within the scope of the invention as useful intermediates for the preparation or purification of pharmaceutically acceptable salts and / or for use in non-therapeutic, for example, in vitro applications.
- physiologically functional derivative refers to any physiologically acceptable derivative of a compound of formula I, e.g. an ester which, when administered to a mammal, e.g. humans, is able to form (directly or indirectly) a compound of formula I or an active metabolite thereof.
- the physiologically functional derivatives also include prodrugs of the compounds according to the invention.
- prodrugs can be metabolized in vivo to a compound of the invention. These prodrugs may or may not be effective.
- the compounds according to the invention can also be present in various polymorphic forms, for example as amorphous and crystalline polymorphic forms. All polymorphic forms of the compounds of the invention are within the scope of the invention and are a further aspect of the invention.
- heteroaryl radical is meant a pyridinyl, pyrrolyl, pyrimidinyl, pyrazinyl, indolyl, benzimidazolyl, imidazolyl, pyrazolyl, thiazolyl, thiophenyl or furanyl radical.
- the compound (s) of formula (I) may also be administered in combination with other active ingredient.
- the amount of a compound of Formula I required to achieve the desired biological effect is dependent upon a number of factors, e.g. the chosen specific compound, the intended
- the daily dose is in the range of 0.3 mg to 100 mg (typically 3 mg and 50 mg) per day per kilogram of body weight, eg 3-10 mg / kg / day.
- an intravenous dose may range from 0.3 mg to 1.0 mg / kg, which may suitably be administered as an infusion of 10 ng to 100 ng per kilogram per minute.
- Suitable infusion solutions for these purposes may contain, for example, from 0.1 ng to 10 mg, typically from 1 ng to 10 mg per milliliter.
- Single doses may contain, for example, from 1 mg to 10 g of the active ingredient.
- vials for injections, and orally administrable unit dose formulations may contain, for example, from 1.0 to 1000 mg, typically from 10 to 600 mg.
- the compounds according to formula I can themselves are used as the compound, but preferably they are with a compatible carrier in the form of a pharmaceutical composition.
- the carrier must of course be compatible in the sense that it is compatible with the other ingredients of the composition and is not harmful to the patient.
- the carrier may be a solid or a liquid, or both, and is preferably formulated with the compound as a single dose, for example, as a tablet, which may contain from 0.05% to 95% by weight of the active ingredient.
- compositions according to the invention can be prepared by one of the known pharmaceutical methods, which essentially consist in mixing the ingredients with pharmacologically acceptable carriers and / or excipients ,
- compositions according to the invention are those which are suitable for oral, rectal, topical, peroral (eg sublingual) and parenteral (eg subcutaneous, intramuscular, intradermal or intravenous) administration, although the most suitable mode of administration in each individual case is the nature and severity of the treatment State and on the nature of the particular compound used in accordance with formula I is dependent.
- coated formulations and coated slow release formulations are within the scope of the invention. Preference is given to acid and enteric formulations. Suitable enteric coatings include cellulose acetate phthalate, polyvinyl acetate phthalate, hydroxypropylmethylcellulose phthalate and anionic polymers of methacrylic acid and methyl methacrylate.
- Suitable pharmaceutical compounds for oral administration may be in separate units, such as capsules, cachets, lozenges or tablets, each containing a specified amount of the compound of formula I as a powder or granules, as a solution or suspension in an aqueous or aqueous solution non-aqueous liquid, or as an oil-in-water or water-in-oil emulsion.
- these compositions may be prepared by any suitable pharmaceutical method comprising a step of contacting the active ingredient and the carrier (which may consist of one or more additional ingredients).
- the compositions are prepared by uniformly and homogeneously mixing the active ingredient with a liquid and / or finely divided solid carrier, after which the product is molded, if necessary.
- a tablet can be made by compressing or molding a powder or granules of the compound, optionally with one or more additional ingredients.
- Compressed tablets can be prepared by tableting the compound in free-flowing form, such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent and / or surfactant / dispersant in a suitable machine.
- Molded tablets may be prepared by shaping the powdered compound moistened with an inert liquid diluent in a suitable machine.
- compositions suitable for peroral (sublingual) administration include lozenges containing a compound of formula I with a flavor, usually sucrose and gum arabic or tragacanth, and lozenges containing the compound in an inert base such as gelatin and glycerol or sucrose and gum arabic.
- Suitable pharmaceutical compositions for parenteral administration preferably comprise sterile aqueous preparations of a compound according to formula I which are preferably isotonic with the blood of the intended recipient. These preparations are preferably administered intravenously, although the administration may also be subcutaneous, intramuscular or intradermal as an injection. These preparations may preferably be prepared by mixing the compound with water and rendering the resulting solution sterile and isotonic with the blood. Injectable compositions of the invention generally contain from 0.1% to 5% by weight of the active compound. Suitable pharmaceutical compositions for rectal administration are preferably as single dose suppositories. These can be prepared by mixing a compound according to formula I with one or more conventional solid carriers, for example cocoa butter, and shaping the resulting mixture.
- Suitable pharmaceutical compositions for topical application to the skin are preferably as an ointment, cream, lotion, paste, spray, aerosol or oil.
- Vaseline, lanolin, polyethylene glycols, alcohols and combinations of two or more of these substances can be used as the carrier.
- the active ingredient is generally present at a level of from 0.1% to 15% by weight of the composition, for example from 0.5% to 2%.
- Suitable pharmaceutical compositions for transdermal applications may exist as single patches suitable for long-term close contact with the epidermis of the patient. Such patches suitably contain the active ingredient in an optionally buffered aqueous solution, dissolved and / or dispersed in an adhesive or dispersed in a polymer.
- a suitable active ingredient concentration is about 1% to 35%, preferably about 3% to 15%.
- the active ingredient can be released by electrotransport or iontophoresis as described, for example, in Pharmaceutical Research, 2 (6): 318 (1986).
- active ingredients for the combination preparations are suitable: All Antidiabetika, which are mentioned in the red list 2003, chapter 12. They can be combined with the compounds of the formula I according to the invention in particular for the synergistic effect improvement.
- the administration of the active ingredient combination can be carried out either by separate administration of the active ingredients to the patient or in the form of combination preparations in which several active ingredients are present in a pharmaceutical preparation.
- Most of the drugs listed below are disclosed in USP Dictionary of USAN and International Drug Names, US Pharmacopeia, Rockville 2001.
- Antidiabetics include insulin and insulin derivatives, such as Lantus ® (see www.lantus.com) or HMR 1964 fast-acting insulins (see US 6,221, 633), GLP-1 derivatives such as those described in WO 98/08871 of Novo Nordisk A / S, as well as orally active hypoglycemic agents.
- the orally active hypoglycemic agents preferably comprise
- Sulfonylfhureas biguanidines, meglitinides, oxadiazolidinediones, thiazolidinediones, glucosidase inhibitors, glucagon antagonists, GLP-1 agonists, potassium channel openers, e.g.
- the compounds of formula I are administered in combination with an HMGCoA reductase inhibitor such as simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin.
- an HMGCoA reductase inhibitor such as simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin.
- the compounds of formula I are administered in combination with a cholesterol resorption inhibitor, e.g. Ezetimibe, Tiqueside, Pamaqueside.
- a cholesterol resorption inhibitor e.g. Ezetimibe, Tiqueside, Pamaqueside.
- PPAR gamma agonist e.g. Rosiglitazone, pioglitazone, JTT-501, Gl 262570.
- the compounds of the formula I are administered in combination with PPAR alpha agonist, such as, for example, GW 9578, GW 7647.
- PPAR alpha agonist such as, for example, GW 9578, GW 7647.
- the compounds of formula I are used in combination with a mixed PPAR alpha / gamma agonist such as GW 1536, AVE 8042, AVE 8134, AVE 0847 or as in PCT / US 11833, PCT / US 11490, DE10142734 .4 administered described.
- the compounds of formula I are used in combination with a fibrate, e.g. Fenofibrate, clofibrate, bezafibrate.
- a fibrate e.g. Fenofibrate, clofibrate, bezafibrate.
- the compounds of formula I are administered in combination with an MTP inhibitor, e.g. Implitapide, BMS-201038, R-103757.
- an MTP inhibitor e.g. Implitapide, BMS-201038, R-103757.
- the compounds of formula I are used in combination with bile acid resorption inhibitor (see, e.g., U.S. 6,245,744 or U.S. 6,221,897), e.g. HMR 1741 administered.
- the compounds of formula I are administered in combination with a CETP inhibitor, e.g. JTT-705.
- a CETP inhibitor e.g. JTT-705.
- the compounds of formula I are used in combination with a polymeric bile acid adsorber, e.g. Cholestyramine, colesevelam.
- a polymeric bile acid adsorber e.g. Cholestyramine, colesevelam.
- the compounds of formula I are used in combination with an LDL receptor inducer (see US 6,342,512), e.g. HMR1171, HMR1586.
- the compounds of the formula I are administered in combination with an ACAT inhibitor, such as avasimibe. In one embodiment of the invention, the compounds of formula I are administered in combination with an antioxidant, such as OPC-14117.
- the compounds of formula I are used in combination with a lipoprotein lipase inhibitor, e.g. NO-1886, administered.
- a lipoprotein lipase inhibitor e.g. NO-1886
- the compounds of formula I are used in combination with an ATP citrate lyase inhibitor, e.g. SB-204990 administered.
- the compounds of formula I are administered in combination with a squalene synthetase inhibitor, e.g. BMS-188494.
- a squalene synthetase inhibitor e.g. BMS-188494.
- the compounds of formula I in combination with a lipoprotein (a) antagonist, e.g. CI-1027 or nicotinic acid.
- a lipoprotein (a) antagonist e.g. CI-1027 or nicotinic acid.
- the compounds of formula I are used in combination with a lipase inhibitor, e.g. Orlistat, administered.
- a lipase inhibitor e.g. Orlistat
- the compounds of the formula I are administered in combination with insulin.
- the compounds of formula I are used in combination with a sulphonylurea, e.g. Tolbutamide, glibenclamide, glipizide or glimepiride.
- a sulphonylurea e.g. Tolbutamide, glibenclamide, glipizide or glimepiride.
- the compounds of formula I are used in combination with a biguanide, e.g. Metformin, administered.
- a biguanide e.g. Metformin
- the compounds of formula I are administered in combination with a meglitinide, such as repaglinide.
- the compounds of formula I are used in combination with a thiazolidinedione, such as troglitazone, ciglitazone, pioglitazone, rosiglitazone or those described in WO 97/41097 by Dr. med. Reddy's Research Foundation disclosed compounds, particularly 5 - [[4 - [(3,4-dihydro-3-methyl-4-oxo-2-quinazolinylmethoxy] phenylmethyl] -2,4-thiazolidinedione.
- the compounds of formula I are used in combination with a ⁇ -glucosidase inhibitor, e.g. Miglitol or acarbose, administered.
- a ⁇ -glucosidase inhibitor e.g. Miglitol or acarbose
- the compounds of formula I are administered in combination with an agent which acts on the ATP-dependent potassium channel of the beta cells, e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- an agent which acts on the ATP-dependent potassium channel of the beta cells e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- the compounds of formula I are used in combination with more than one of the aforementioned compounds, e.g. in combination with a sulphonylurea and metformin, a sulphonylurea and acarbose, repaglinide and metformin, insulin and a sulphonylurea, insulin and metformin, insulin and troglitazone, insulin and lovastatin, etc.
- the compounds of the formula I are used in combination with CART modulators (see “Cocaine-amphetamine-regulated transcript-influenced transient influenza energy metabolism, anxiety and gastric emptying in mice" Asakawa, A, et al., M .: Hormones and Metabolism Research (2001), 33 (9), 554-558), NPY antagonists eg naphthalene-1-sulfonic acid ⁇ 4 - [(4-amino-quinazolin-2-ylamino) -methyl] -cyclohexylmethyl ⁇ -amide hydrochloride ( CGP 71683A)), MC4 agonists (eg, 1-amino-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [2- (3a-benzyl-2-methyl-3-oxo-2,3,3a , 4,6,7-hexahydro-pyrazolo [4,3-c] pyridin-5-yl) -1- (4
- Leptin agonists as a potential approach to the treatment of obesity. Drugs of the Future (2001), 26 (9), 873-881), DA agonists (bromocriptine, doprexin), lipase / amylase inhibitors (eg WO 00/40569), PPAR modulators (eg WO 00/78312) , RXR modulators or TR-agonists.
- the further active ingredient is leptin; see, e.g. "Perspectives in the therapeutic use of leptin", Salvador, Javier; Gomez-Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001), 2 (10), 1615-1622.
- the further active ingredient is dexamphatamine or amphetamine.
- the other active ingredient is fenfluramine or dexfenfluramine.
- the other active ingredient is sibutramine.
- the other active ingredient is orlistat.
- the other active ingredient is mazindol or phentermine.
- the compounds of formula I in combination with bulking agents, preferably insoluble bulking agents see, for example, carob / Caromax ® (Zunft HJ; et al, Carob pulp preparation for treatment of hypercholesterolemia, ADVANCES IN THERAPY (2001 Sep-Oct). 18 (5), 230-6.)
- Caromax is a carob-containing product of the company Nutrinova, Nutrition Specialties & Food Ingredients GmbH, Industriepark availability, 65926 Frankfurt / Main)) administered.
- Combination with Caromax ® is possible in one preparation or by separate administration of compounds of the formula I and Caromax ®.
- Caromax ® can also be administered in the form of food, such as in baked goods or muesli bars.
- the compounds of the formula II can be prepared by various generally known rules, for example in WO2002 048113.
- compounds of the general formula II in which X represents a carbon atom can be synthesized from the corresponding anilines of the formula V via the Gould Jacobs route, as shown in Scheme 2.
- the alkylation on the nitrogen can be carried out at any point in the synthesis.
- Another object of this invention is a novel preparation process for the preparation of the quinolones of the formula I according to Scheme 2, characterized in that the radical Y represents an unsubstituted or substituted aniline radical.
- compounds of the formula II in which X represents a carbon atom can be prepared starting from carboxylic acids of the general formula VIII by conversion into the acid chloride and reaction with malonic acid ester and orthoformic acid ester, reaction with amines and subsequent cyclization (Scheme 3).
- Another object of this invention is a novel preparation process for the preparation of the quinolones of the formula I according to Scheme 3a, characterized in that the radical Y represents an unsubstituted or substituted aniline radical.
- 2-Amino-pyridines are converted by heating with EMME to the compounds of the structure type XI. These cyclize to the desired Naldixinklarederivaten XII at temperatures above 200 ° C in a suitable Solvents such as DOWTHERM A or diphenyl ether. The cyclization succeeds only in the form described above if the substituent R5 does not represent a hydrogen atom. (References: Edmont, Rocher, Plisson, Chenault, Bioorg. Med. Chem. Lett., 2000, 1831)
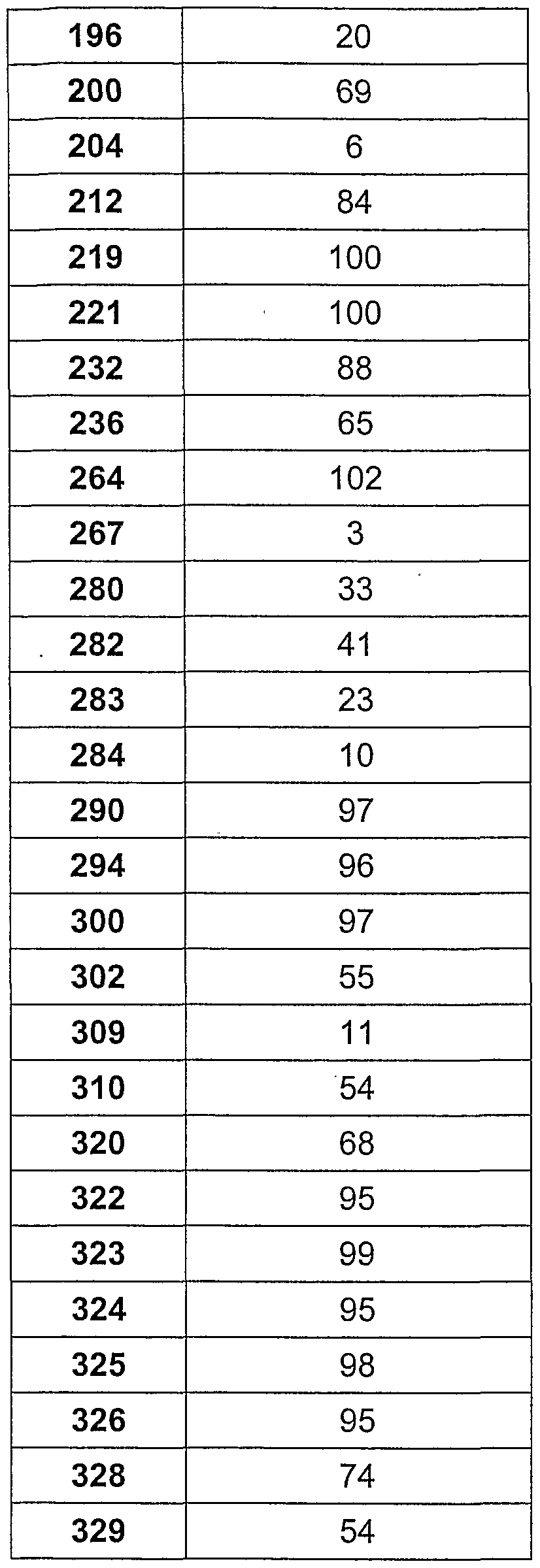
- the effect of compounds on the activity of the active form of glycogen phosphorylase (GPa) was measured in the reverse direction by following the glycogen synthesis from glucose-1-phosphate by determination of the release of inorganic phosphate. All reactions were performed as duplicates in 96-well microtiter plates (Costar # 3696), the change in absorbance due to the formation of the reaction product at the wavelength specified below in a Multiskan Ascent Elisa Reader (Lab Systems. Finland). In order to measure the GPa enzyme activity in the reverse direction, the conversion of glucose-1-phosphate to glycogen and inorganic phosphate was determined by the general method of Engers et al.
- glycerophosphate pH 7.0, 1 mM EDTA and 1 mM dithiothreitol
- buffer T 50 mM Hepes, pH 7.0, 100 mM KCl, 2.5 mM EDTA, 2.5 mM MgCl 2 6H 2 0
- Diluted 5 mg / ml glycogen to a concentration of 10 ⁇ g protein / ml Test Substances were prepared as 10 mM solution in DMSO and diluted to 50 ⁇ M with buffer solution T.
- the pure product was isolated from the reaction solution by chromatography on an HPLC. This was a Merck Purospher RP-18 column and a
- AcetonitriLWasser mixture used as eluent the initial content of acetonitrile was 15% and increased within 20 minutes to 95%.
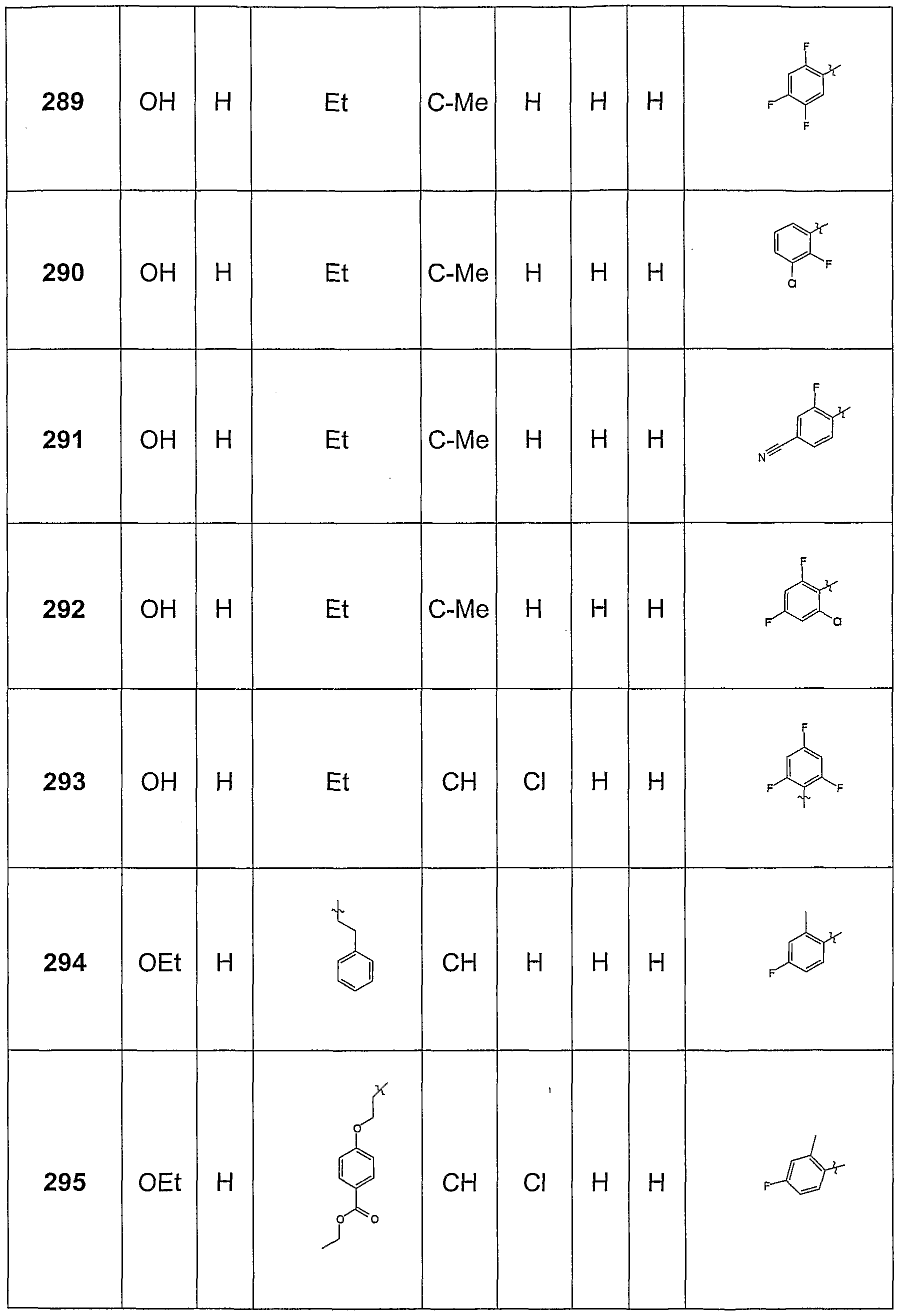
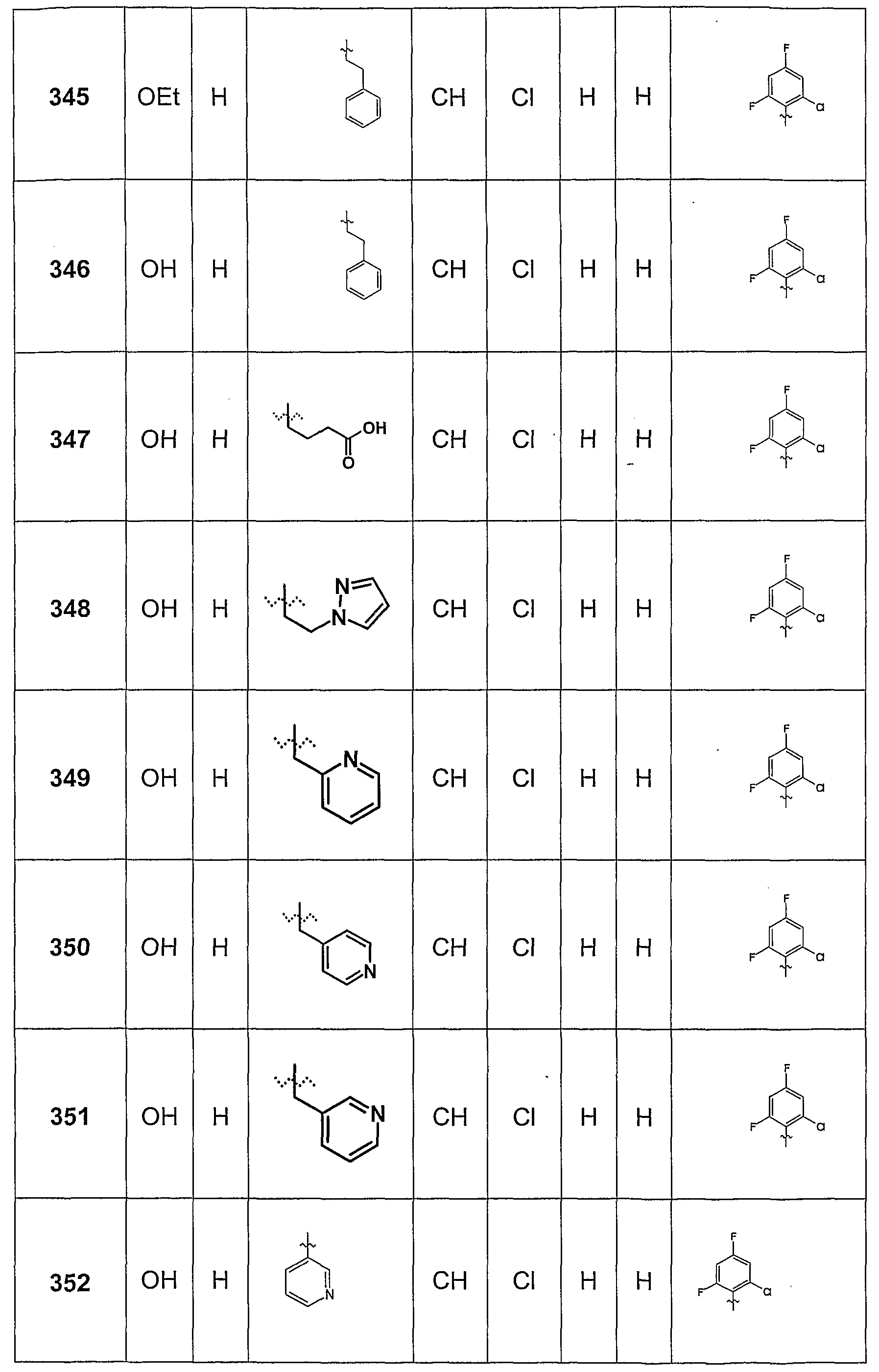
- Methyl 1-ethyl-6- (4-methoxy-2-methylphenylamino) -8-methyl-4-oxo-1,4-dihydro-quinolone-3-carboxylate (30 mg) was dissolved in 5 ml of dioxane, added with 2.5 equivalents of a 1 N NaOH solution and heated to 60 ° C for 4 h. After removal of the solvent in vacuo, the product was purified by chromatography on an HPLC. A Merck Purospher RP18 column and an acetonitrile / water mixture were used as eluent, the initial acetonitrile content was 15% and increased to 95% within 20 minutes. Yield: 75%
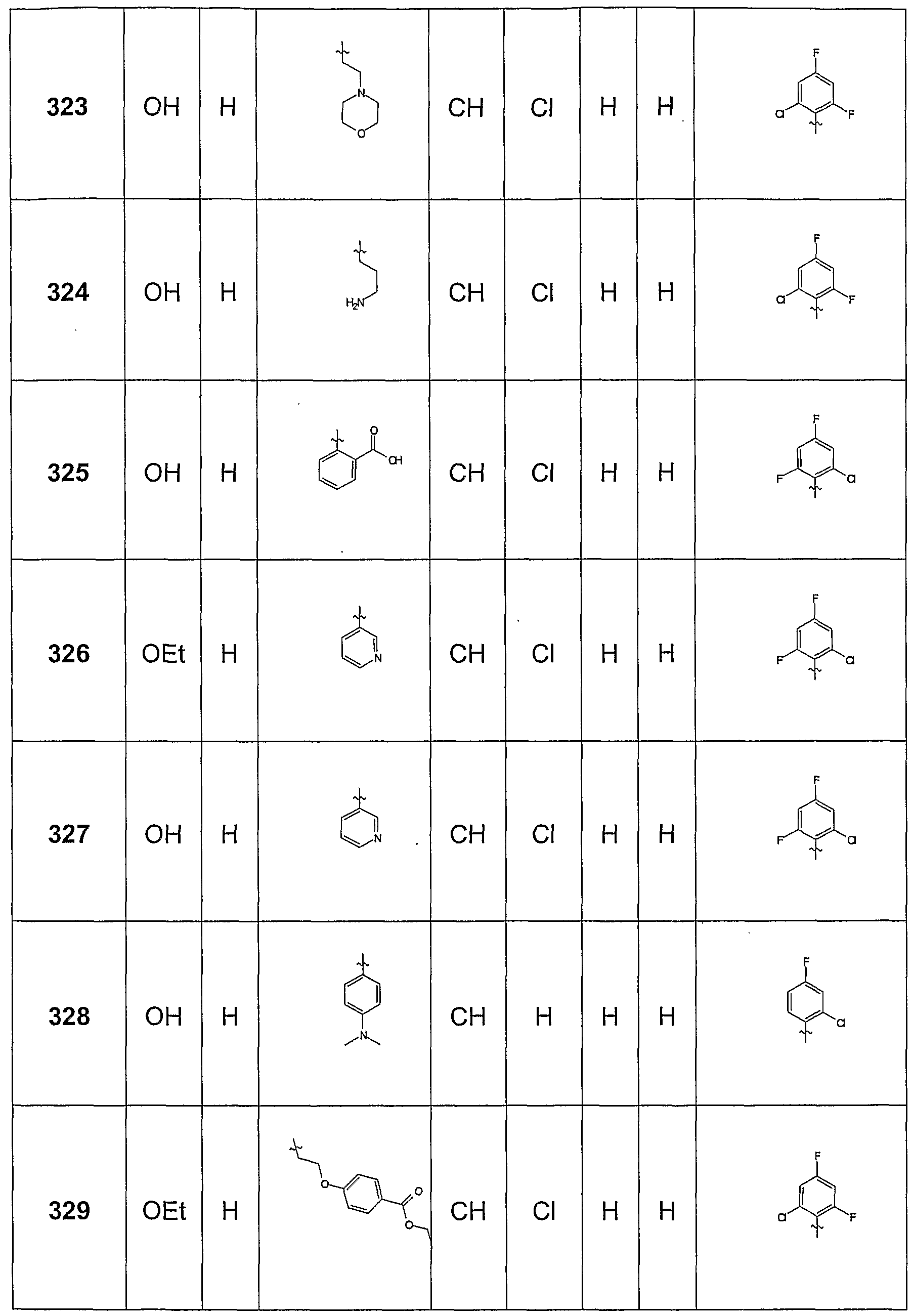
- This intermediate was used, for example, for the synthesis of Beipsiel 327.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Diabetes (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Quinoline Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

















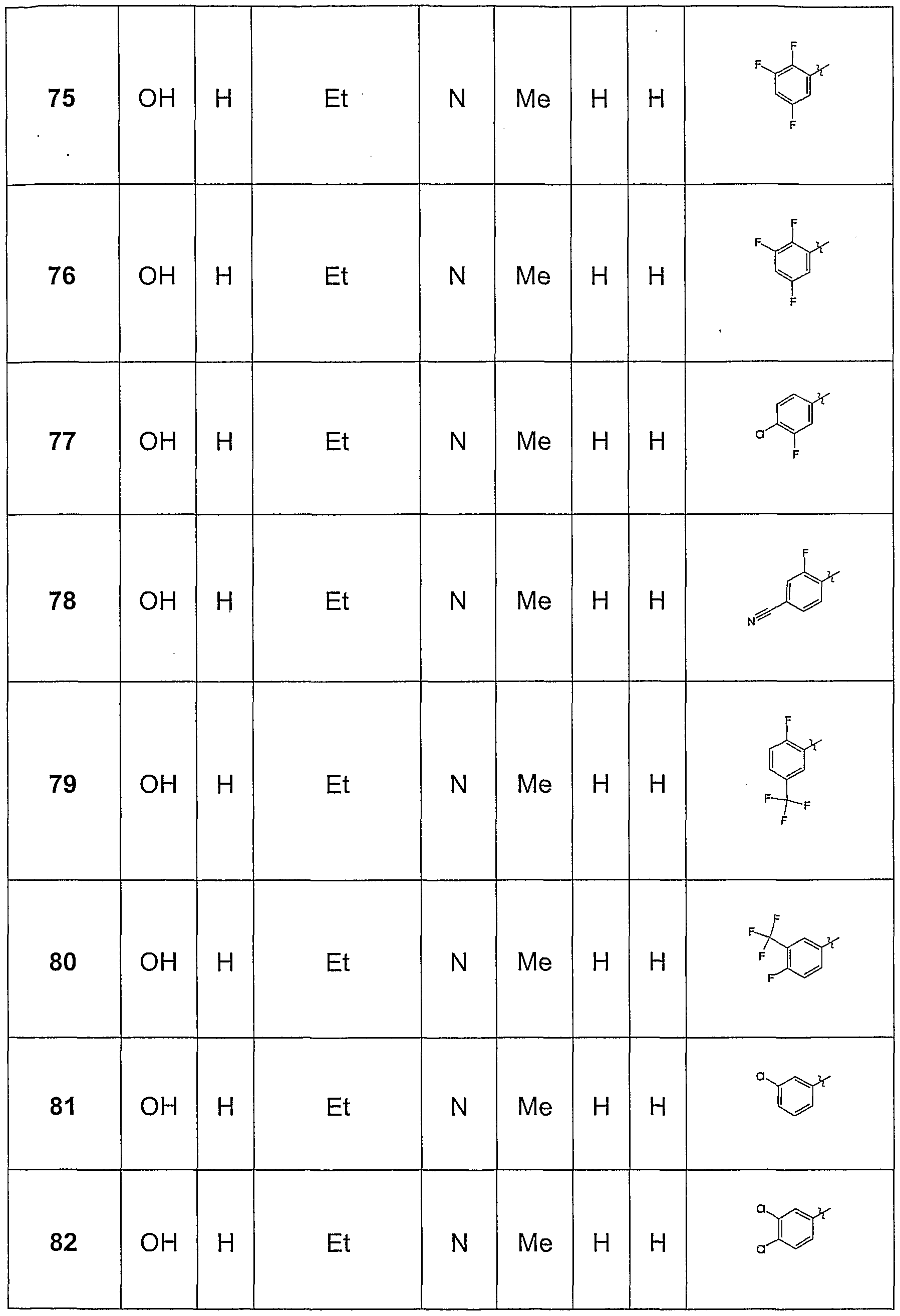

















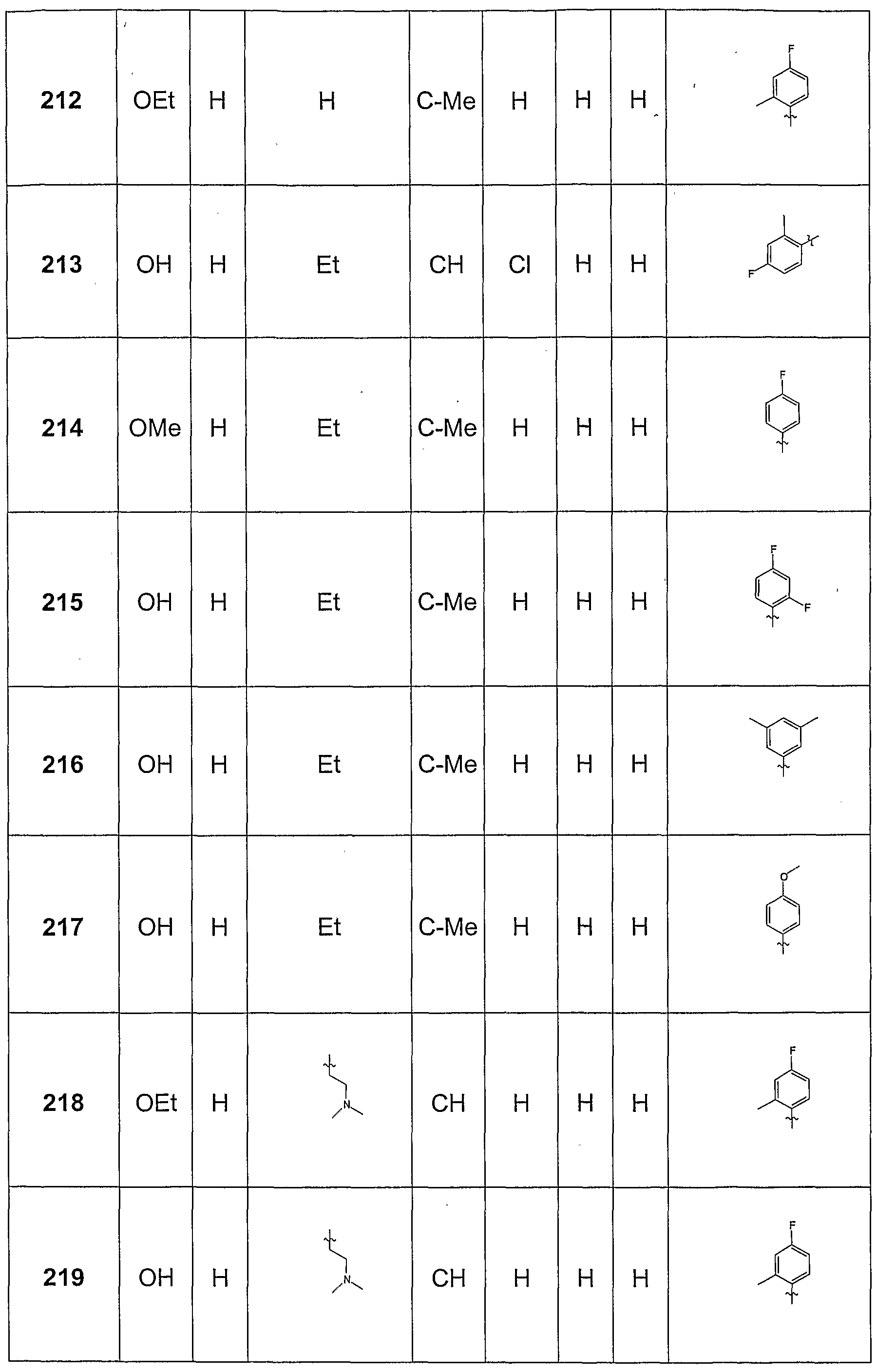







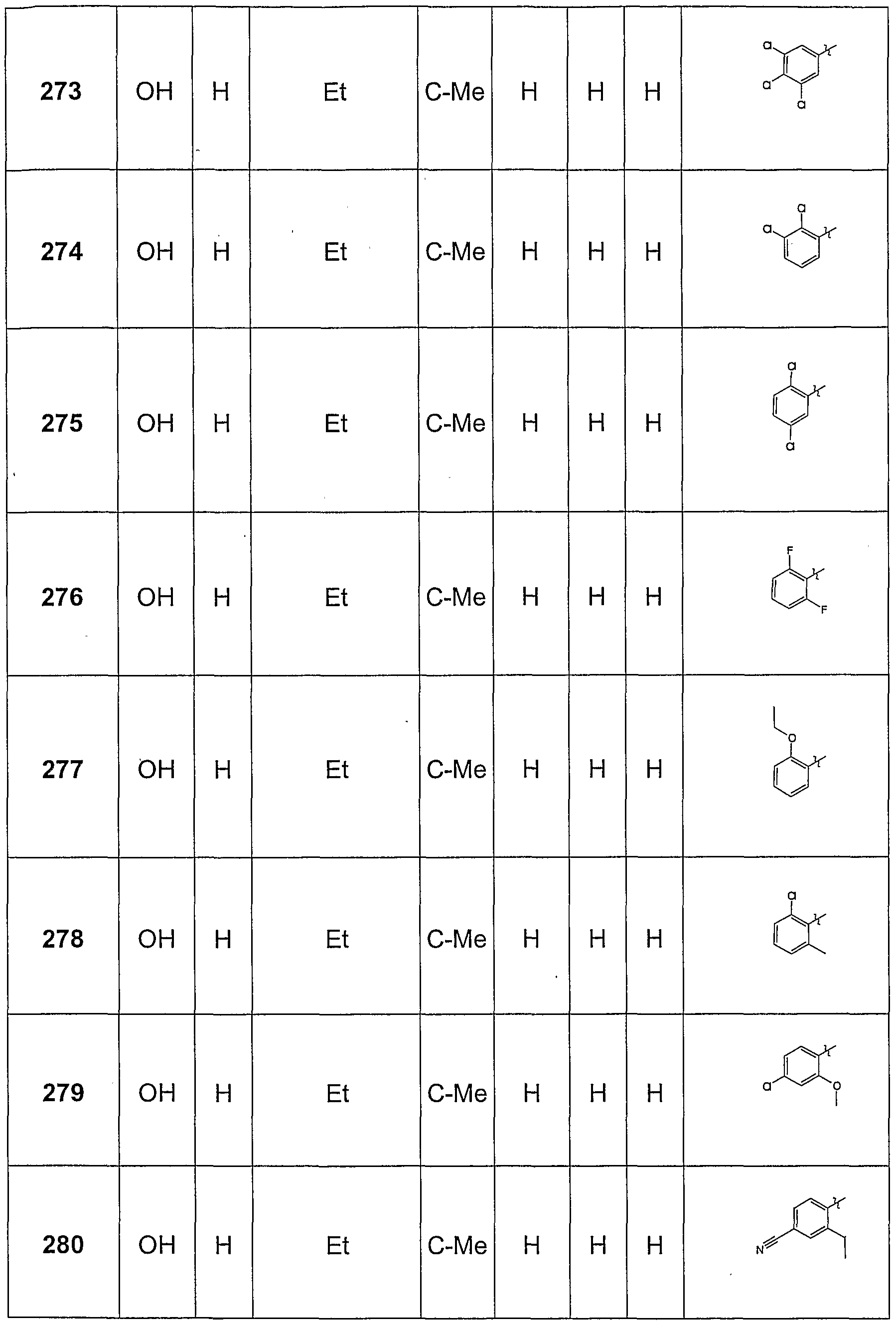











Claims

Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE502005007840T DE502005007840D1 (de) | 2004-01-31 | 2005-01-15 | 7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| AT05700953T ATE438645T1 (de) | 2004-01-31 | 2005-01-15 | 7-phenylamino-4-chinolon-3-carbonsäure-derivate verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| AU2005209365A AU2005209365A1 (en) | 2004-01-31 | 2005-01-15 | 7-phenylamino-4-quinolone-3-carboxylic acid derivatives, methods for production and use thereof as medicaments |
| CA002554522A CA2554522A1 (en) | 2004-01-31 | 2005-01-15 | 7-phenylamino-4-quinolone-3-carboxylic acid derivatives, methods for production and use thereof as medicaments |
| BRPI0507309-0A BRPI0507309A (pt) | 2004-01-31 | 2005-01-15 | derivados de ácido 7-fenilamino-4quinolon-3-carboxìlico, processos para produção e seu uso como medicamento |
| EP05700953A EP1713803B1 (de) | 2004-01-31 | 2005-01-15 | 7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| JP2006549988A JP2007519648A (ja) | 2004-01-31 | 2005-01-15 | 7−フェニルアミノ−4−キノロン−3−カルボン酸誘導体、その製造方法及び医薬としての使用 |
| IL176913A IL176913A0 (en) | 2004-01-31 | 2006-07-17 | 7-phenylamino-4-quinolone-3-carboxylic acid derivatives, methods for production and use thereof as medicaments |
| TNP2006000233A TNSN06233A1 (en) | 2004-01-31 | 2006-07-28 | 7-phenylamino-4-quinolone-3-carboxylic acid derivatives, methods for production and use thereof as medicaments |
| NO20063860A NO20063860L (no) | 2004-01-31 | 2006-08-29 | 7-fenylamino-4-kinolon-3-karboksylsyrederivater, fremgangsmate for fremstilling derav samt deres anvendelse som legemidler |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102004004973.4 | 2004-01-31 | ||
| DE102004004973A DE102004004973A1 (de) | 2004-01-31 | 2004-01-31 | 7-Phenylamino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| DE102004033405.6 | 2004-07-10 | ||
| DE102004033405A DE102004033405A1 (de) | 2004-07-10 | 2004-07-10 | 7-Phenylamino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005073229A1 true WO2005073229A1 (de) | 2005-08-11 |
Family
ID=34828328
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2005/000363 Ceased WO2005073229A1 (de) | 2004-01-31 | 2005-01-15 | 7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
Country Status (19)
| Country | Link |
|---|---|
| EP (1) | EP1713803B1 (de) |
| JP (1) | JP2007519648A (de) |
| KR (1) | KR20060132671A (de) |
| AR (1) | AR047516A1 (de) |
| AT (1) | ATE438645T1 (de) |
| AU (1) | AU2005209365A1 (de) |
| BR (1) | BRPI0507309A (de) |
| CA (1) | CA2554522A1 (de) |
| DE (1) | DE502005007840D1 (de) |
| EC (1) | ECSP066734A (de) |
| IL (1) | IL176913A0 (de) |
| MA (1) | MA28337A1 (de) |
| OA (1) | OA13364A (de) |
| PA (1) | PA8622401A1 (de) |
| PE (1) | PE20051094A1 (de) |
| RU (1) | RU2006131306A (de) |
| TW (1) | TW200533353A (de) |
| UY (1) | UY28732A1 (de) |
| WO (1) | WO2005073229A1 (de) |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| DE102007005045A1 (de) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | Phenothiazin Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010073078A3 (en) * | 2008-12-22 | 2010-11-04 | Orchid Research Laboratories Ltd. | Heterocyclic compounds as hdac inhibitors |
| EP1881974A4 (de) * | 2005-04-21 | 2011-03-16 | Targanta Therapeutics Inc | Phosphonierte fluorchinoline, antibakterielle analoga davon und verfahren zur vorbeugung und behandlung von knochen- und gelenkinfektionen |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120051A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit adamantan- oder noradamantan substituierte benzyl-oxathiazinderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120050A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120057A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120058A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit benzyl- oder heteromethylengruppen substituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| US8293737B2 (en) | 2006-10-16 | 2012-10-23 | Bionomics Limited | Anxiolytic compounds |
| EP2567959A1 (de) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridin-4-Carbonsäureamid-Derivate als Kinaseinhibitoren |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9023848B2 (en) | 2011-03-02 | 2015-05-05 | Bionomics Limited | Small-molecules as therapeutics |
| US9133188B2 (en) | 2011-05-12 | 2015-09-15 | Bionomics Limited | Methods for preparing naphthyridines |
| WO2020033919A1 (en) | 2018-08-10 | 2020-02-13 | Diapin Therapeutics, Llc | Tri-peptides and treatment of metabolic, cardiovascular and inflammatory disorders |
| US10954231B2 (en) | 2006-10-16 | 2021-03-23 | Bionomics Limited | Anxiolytic compounds |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003074532A1 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Heterocyclic amide derivatives as inhibitors of glycogen phosphorylase |
-
2005
- 2005-01-15 AU AU2005209365A patent/AU2005209365A1/en not_active Abandoned
- 2005-01-15 KR KR1020067015355A patent/KR20060132671A/ko not_active Withdrawn
- 2005-01-15 WO PCT/EP2005/000363 patent/WO2005073229A1/de not_active Ceased
- 2005-01-15 JP JP2006549988A patent/JP2007519648A/ja not_active Abandoned
- 2005-01-15 DE DE502005007840T patent/DE502005007840D1/de not_active Expired - Lifetime
- 2005-01-15 AT AT05700953T patent/ATE438645T1/de not_active IP Right Cessation
- 2005-01-15 CA CA002554522A patent/CA2554522A1/en not_active Abandoned
- 2005-01-15 OA OA1200600243A patent/OA13364A/en unknown
- 2005-01-15 RU RU2006131306/04A patent/RU2006131306A/ru unknown
- 2005-01-15 BR BRPI0507309-0A patent/BRPI0507309A/pt not_active Application Discontinuation
- 2005-01-15 EP EP05700953A patent/EP1713803B1/de not_active Expired - Lifetime
- 2005-01-27 TW TW094102386A patent/TW200533353A/zh unknown
- 2005-01-28 AR ARP050100321A patent/AR047516A1/es unknown
- 2005-01-28 PE PE2005000109A patent/PE20051094A1/es not_active Application Discontinuation
- 2005-01-31 PA PA20058622401A patent/PA8622401A1/es unknown
- 2005-01-31 UY UY28732A patent/UY28732A1/es unknown
-
2006
- 2006-07-17 IL IL176913A patent/IL176913A0/en unknown
- 2006-07-28 EC EC2006006734A patent/ECSP066734A/es unknown
- 2006-07-28 MA MA29216A patent/MA28337A1/fr unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003074532A1 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Heterocyclic amide derivatives as inhibitors of glycogen phosphorylase |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 1 January 2004 (2004-01-01), XP002331735, Database accession no. 2003:2814273 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 27 January 2004 (2004-01-27), XP002331734, Database accession no. 641992-79-2 * |
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1881974A4 (de) * | 2005-04-21 | 2011-03-16 | Targanta Therapeutics Inc | Phosphonierte fluorchinoline, antibakterielle analoga davon und verfahren zur vorbeugung und behandlung von knochen- und gelenkinfektionen |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| EP2351568A2 (de) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Verwendungen von dpp iv Inhibitoren |
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| US9573945B2 (en) | 2006-10-16 | 2017-02-21 | Bionomics Limited | Anxiolytic compounds |
| US8293737B2 (en) | 2006-10-16 | 2012-10-23 | Bionomics Limited | Anxiolytic compounds |
| EP2540722A1 (de) * | 2006-10-16 | 2013-01-02 | Bionomics Limited | Neuartige angstlösende Verbindungen |
| US8551990B2 (en) | 2006-10-16 | 2013-10-08 | Bionomics Limited | Anxiolytic compounds |
| US8614212B2 (en) | 2006-10-16 | 2013-12-24 | Bionomics Limited | Anxiolytic compounds |
| US8906912B2 (en) | 2006-10-16 | 2014-12-09 | Bionomics Limited | Anxiolytic compounds |
| US10954231B2 (en) | 2006-10-16 | 2021-03-23 | Bionomics Limited | Anxiolytic compounds |
| US10233181B2 (en) | 2006-10-16 | 2019-03-19 | Bionomics Limited | Anxiolytic compounds |
| US9975892B2 (en) | 2006-10-16 | 2018-05-22 | Bionomics Limited | Anxiolytic compounds |
| JP2017105818A (ja) * | 2006-10-16 | 2017-06-15 | バイオノミクス リミテッド | 新規な抗不安薬化合物 |
| DE102007005045A1 (de) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | Phenothiazin Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010073078A3 (en) * | 2008-12-22 | 2010-11-04 | Orchid Research Laboratories Ltd. | Heterocyclic compounds as hdac inhibitors |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US9023848B2 (en) | 2011-03-02 | 2015-05-05 | Bionomics Limited | Small-molecules as therapeutics |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120050A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120058A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit benzyl- oder heteromethylengruppen substituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120057A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120051A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit adamantan- oder noradamantan substituierte benzyl-oxathiazinderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| US9133188B2 (en) | 2011-05-12 | 2015-09-15 | Bionomics Limited | Methods for preparing naphthyridines |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP2567959A1 (de) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridin-4-Carbonsäureamid-Derivate als Kinaseinhibitoren |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2020033919A1 (en) | 2018-08-10 | 2020-02-13 | Diapin Therapeutics, Llc | Tri-peptides and treatment of metabolic, cardiovascular and inflammatory disorders |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1713803B1 (de) | 2009-08-05 |
| ATE438645T1 (de) | 2009-08-15 |
| DE502005007840D1 (de) | 2009-09-17 |
| MA28337A1 (fr) | 2006-12-01 |
| TW200533353A (en) | 2005-10-16 |
| CA2554522A1 (en) | 2005-08-11 |
| IL176913A0 (en) | 2006-12-10 |
| UY28732A1 (es) | 2005-08-31 |
| AU2005209365A1 (en) | 2005-08-11 |
| KR20060132671A (ko) | 2006-12-21 |
| OA13364A (en) | 2007-04-13 |
| PE20051094A1 (es) | 2006-02-07 |
| ECSP066734A (es) | 2006-10-31 |
| AR047516A1 (es) | 2006-01-25 |
| BRPI0507309A (pt) | 2007-06-26 |
| JP2007519648A (ja) | 2007-07-19 |
| PA8622401A1 (es) | 2005-08-10 |
| EP1713803A1 (de) | 2006-10-25 |
| RU2006131306A (ru) | 2008-03-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1713803B1 (de) | 7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP1497262B1 (de) | Acyl-4-carboxy-phenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung | |
| WO2004007455A1 (de) | Heterozyklisch substituierte benzoylharnstoffe, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| DE10225635C1 (de) | N-Benzoylureido-Zimtsäurederivate, Verfahren zu deren Herstellung und deren Verwendung | |
| EP1523471B1 (de) | Harnstoff- und urethan-substituierte acylharnstoffe, verfahren zu deren herstellung und deren verwendung als arzneimittel | |
| DE102004004972B3 (de) | Heterocyclisch substituierte 7-Amino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| EP1497263B1 (de) | Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung | |
| EP1713804B1 (de) | Cycloalkyl substituierte 7-amino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| DE10309929B4 (de) | Substituierte Benzoylureidopyridyl-piperidin- und -pyrrolidin-carbonsäurederivate, Verfahren zu deren Herstellung und deren Verwendung | |
| DE10306502B4 (de) | Substituierte 3-(Benzoylureido)-thiophenderivate und sie enthaltende Arzneimittel | |
| EP1590322B1 (de) | Carbonylamino-substituierte acyl-phenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung | |
| DE102004004973A1 (de) | 7-Phenylamino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| EP1556339B1 (de) | Carboxyalkoxy-substituierte acyl-carboxyphenyl-harnstoffderivate, verfahren zu ihre herstellung und ihre verwendung als arzneimittel | |
| DE102004033405A1 (de) | 7-Phenylamino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| DE10231627B4 (de) | Heterozyklisch substituierte Benzoylharnstoffe und ihre Verwendung als Arzneimittel | |
| DE10306503A1 (de) | Substituierte Benzoylureidophenyl-piperidin- und pyrrolidin-carbonsäurederivate, Verfahren zu deren Herstellung und deren Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 200605209 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2006-008489 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 176913 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005700953 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2554522 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/008403 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 06073780 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2791/CHENP/2006 Country of ref document: IN Ref document number: 200580003532.X Country of ref document: CN Ref document number: 548837 Country of ref document: NZ Ref document number: 1020067015355 Country of ref document: KR Ref document number: 2005209365 Country of ref document: AU Ref document number: 2006549988 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006501476 Country of ref document: PH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200601340 Country of ref document: VN |
|
| ENP | Entry into the national phase |
Ref document number: 2005209365 Country of ref document: AU Date of ref document: 20050115 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005209365 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006131306 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005700953 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067015355 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0507309 Country of ref document: BR |