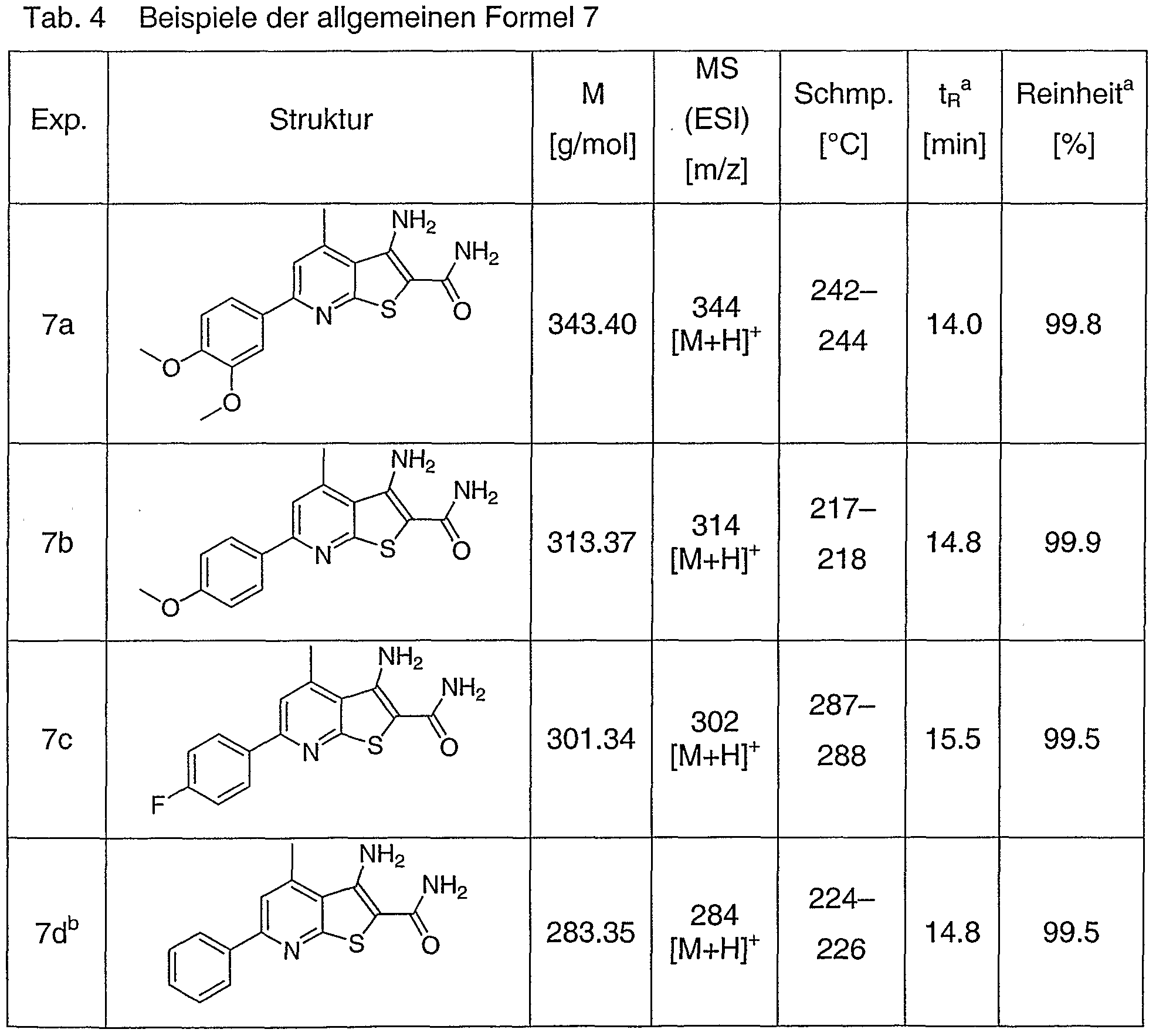
Substituierte Pyrido[3',2':4J5]thieno[3!2-d]pyrimidin-2,4(1 HJ3H)dione und - 4(3H)one, substituierte Thieno[2J3-d:4,5-d']dipyrimidin-254(1 H,3H)-dione und - 4(3H)-one, substituierte Pyrido[3',2':4,5]f uro[3,2-d]-pyrimidin-2,4(1 H,3H)-dione und -4(3H)one sowie substituierte Furo[2,3-d:4,5-d']dipyrimidin-2,4(1 H,3H)- dione und -4(3H)-one, diese enthaltende pharmazeutische Zusammensetzungen und die Verwendung ais Inhibitoren der TN Fa- Freisetzung sowie Verfahren zu deren Herstellung
Beschreibung
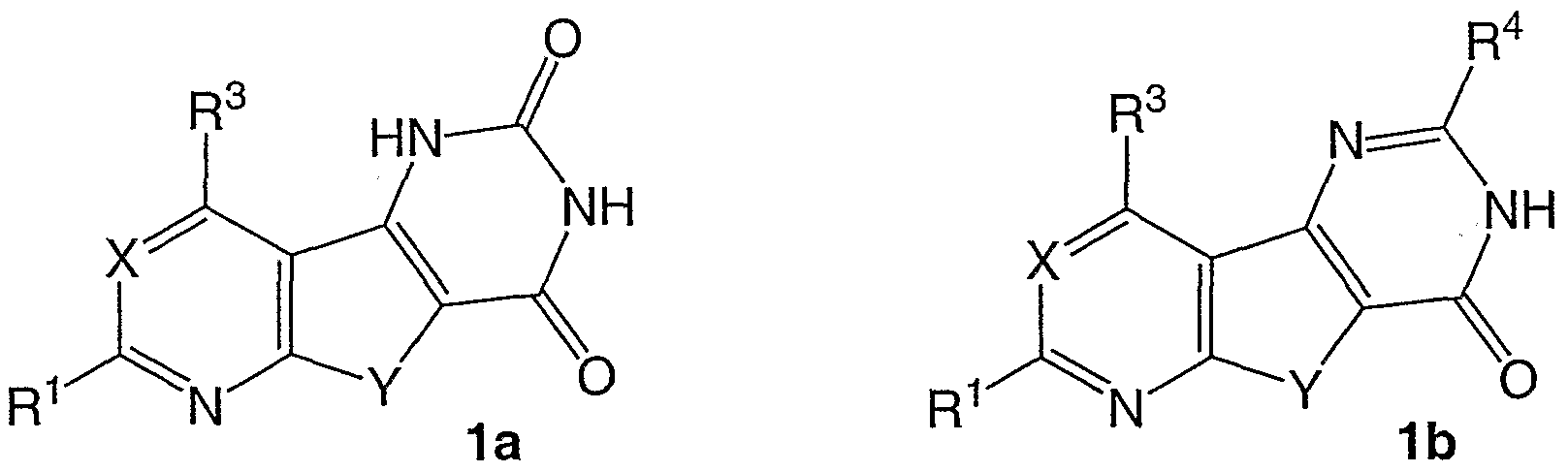
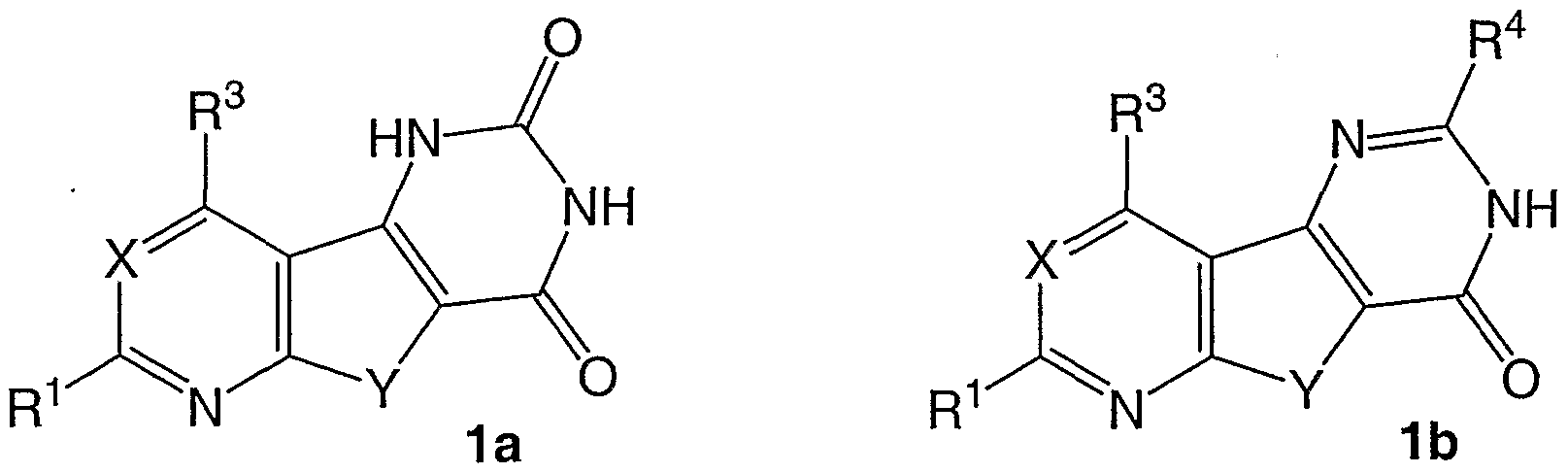
Die Erfindung betrifft Heterocyclen, die als Bislactame der allgemeinen Formel 1a bzw. Monolactame der allgemeinen Formel 1b des bisher wenig bekannten tricyclischen Pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-Systems sowie des Thieno[2,3- d:4,5-d']dipyrimidin-Systems anzusehen sind. Die Erfindung betrifft weiterhin die Eignung entsprechender Verbindungen als TNFα-Freisetzungshemmer.
Gegenstand der Erfindung sind demzufolge die Verbindungen selbst, Verfahren zu deren Herstellung, pharmazeutische Zubereitungen, die diese Verbindungen und/oder deren Tautomere, pharmakologisch übliche Prodrug-Formulierungen und daraus herstellbare physiologisch verträgliche Salze und/oder deren Solvate enthalten, sowie die pharmazeutische Verwendung dieser Verbindungen, deren Tautomere, Salze oder Solvate, einschließlich Prodrug-Formulierungen als Inhibitoren der TNFα-Freisetzung. Prodrug-Formulierungen umfassen hierbei alle jene Substanzen, die durch einfache Transformation einschließlich Hydrolyse, Oxidation, oder Reduktion entweder enzymatisch, metabolisch oder auf andere Art und Weise entstehen. Insbesondere, wenn ein solches Prodrug dieser
erfindungsgemäßen Verbindungen einem Patienten appliziert wurde, und dieses Prodrug in eine Substanz der allgemeinen Formeln Ia und Ib transformiert wird, wodurch der gewünschte pharmakologische Effekt erzielt wird.
Stand der Technik
Das Zytokin Tumour necrosis factor (TNFα) ist eines von heute 17 bekannten Mitgliedern einer strukturell sehr ähnlichen Proteinfamilie. Seinen Namen verdankt es der Fähigkeit, eine Nekrose von transplantierten Tumorzellen im Mausmodell zu triggem. Neben seiner Apoptose-induzierenden Wirkung wurde sehr schnell erkannt, dass TNFα auch ganz maßgeblich in die Regulation der Entzündungsantwort und der Immunantwort eingebunden ist. Eine Überproduktion von TNFα oder die Aktivierung der TNFα-vermittelten Signalkaskaden spielen in der Pathogenese einer Vielzahl von Erkrankungen, wie z.B. Sepsis, cerebrale Form der Malaria, neurodegenerativen Erkrankungen wie z.B. Mb. Alzheimer, Mb. Parkinson, bei Diabetes mellitus, COPD/Asthma, Tumorerkrankungen und hier insbesondere Tumoren des Blutbildenden Systems wie z.B. Leukämien und Lymphome, virale Erkrankungen und hier insbesondere retrovirale Erkrankungen wie z.B. das erworbene Immundefizienz Syndrom (AIDS), Guillain-Barre Syndrom, Rhinitis allergica, allergische Konjunktivitis, systemische Sklerodermie, Graft versus host disease (GvHD), Systemischer Lupus Erythematodes (SLE), Osteoporosis, Toxisches Schocksyndrom, Akute Glomerulonephritis, akute und chronische Schmerzen, Arteriosklerose, Herzinfarkt, Schlaganfall, bei Sarkoidose, Multiple Sklerose, Rheumatoid Arthritis (RA), Osteoarthritis, Colitis ulcerosa, Vasculitis, Uveitis, Mb. Crohn, Mb. Behcet, Myastenia Gravis und chronisch entzündlichen Hauterkrankungen wie Psoriasis, atopische Dermatitis, Ekzeme und Alopecie, eine zentrale Rolle (Chen G, Goeddel DV (2002 TNF-R1 signaling: a beautiful pathway. Science 296 1634-1635; Ware CF (2003) The TNF superfamily. Cytokine & Growth Factor reviews 1_4 181-184; Dempsey PW (2003) The signaling adaptors and pathways activated by TNF superfamily. Cytokine & Growth Factor reviews 14 193-209).
Bei TNFα handelt es sich um eines der wichtigsten pro-inflammatorischen Zytokine, der in die Pathogenese fast aller chronisch entzündlichen Erkrankungen maßgeblich
eingebunden ist. TNFα, welches auch als Chachektin, Makrophagen-Cytotoxin (MCT), E tumor necrosis factor-a und als macrophage cytotoxic factor (MCF) beschrieben wurde, wird von verschiedensten Zellen nach Stimulation mit Lipopolysaccharid (LPS), Interferronen (IFN's), IL-2, Bradykinin, GM-CSF, Antigen- Antikörper-Komplexen, Substanz P und zahlreichen weiteren biologisch aktiven Verbindungen synthetisiert und sezemiert. TNFα wird unter physiologischen Bedingungen hauptsächlich von aktivierten Makrophagen, T-Lymphozyten, Mikrogliazellen und NL-Zellen gebildet. Stimulierte und somit aktivierte Fibroblasten, glatte Muskelzellen, Astrozyten, Keratinozyten, Endothelzellen und Lungen- Epithelzellen sezernieren gleichfalls TNFα.
Humanes TNFα ist ein 17 kDa großes Protein, welches aus 157 Aminosäuren besteht und zu Dimeren und Trimeren assoziiert. Es existiert eine weitere höhermolekulare Variante dieses Moleküls mit einer Molmasse von 26 kDa, das als Transmembranprotein in der Zellmembran verankert ist. Man weiß heute, dass zunächst die höhermolekulare Transmembranform synthetisiert in die Zellmembran eingelagert wird und bei Bedarf deren extrazelluläre Domäne durch das TNFα Converting enzyme (TACE) abgespalten wird. Das lösliche TNFα zirkuliert als ein Homotrimer und bindet sich an seine spezifischen Rezeptoren an Zelloberflächen. Die Bindung von TNFα an seine Rezeptoren (TNFR1 , TNFR2) bewirkt bei diesen eine konformative Änderung und Dimerisierung bzw. Clusterung, welche über eine Signalkaskade den biologischen Effekt von TNFα vermitteln. In zahlreichen Untersuchungen konnte gezeigt werden, dass über die Bindung von TNFα an den TNFR1 die meisten biologischen Effekte realisiert werden. Dies beinhaltet die Induktion der Apoptose über eine Aktivierung der Caspase 8 und nachfolgender Aktivierung der Caspasen 3, 6 und 7, die dann zur Apoptose der Zelle führen.
Ein weiterer wichtiger Signalweg durch TNFα ist die Aktivierung von zwei wichtigen Transkriptionsfaktoren, dem nuclear factor-kappaB (NF-KB) und c-Jun. Diese beiden Transkriptionsfaktoren spielen eine außerordentlich wichtige Rolle in der Regulation der Genexpression bei der Zelldifferenzierung, dem Zellwachstum, bei der Immun- und Entzündungsantwort, bei Zellstressregulationsvorgängen und bei der Tumorgenese.
NF-KB reguliert unter anderem die Gene für IL-1α, IL-1ß, IL-2, IL-3, IL-6, IL-8, IL-12, TNFα, LT-α, IFN-α/ß, G-CSF, M-CSF, GM-CSF1 für den Zytokinrezeptor IL-2Rα, für die Adhäsionsmoleküle ICAM-1 , VCAM-1 , MAdCAM, E-Selektin, für die immunregulatorischen Moleküle leichte Kette des lgγ, MHC Class I und II, TCRα und ß, ß2 Mikroglobulin, TAP1 , iNOS und für die Akute Phase Proteine SAA, αrsaures Glycoprotein und TSG-14/PTX3.
Über die tatsächliche physiologische Bedeutung der Bindung von TNFα an den TNFR2 existieren heute noch erhebliche Widersprüche. Deshalb ist die Aufdeckung der genauen molekularen Signaltransduktionsabläufe noch Gegenstand der Grundlagenforschung. Mehrheitlich geht man heute davon aus, dass die Bindung von TNFα an den TNFR2 auch die Mitogen-aktivierten Proteinkinase Kinasen (MAPKK) aktiviert, im speziellen die MEKK1 und die ASK1 , die über eine Aktivierungskaskade zur Aktivierung der c-Jun Kinase (JNK) und damit zu einer Aktivierung des Transkriptionsfaktors c-Jun führt. In diesen Regulationsweg ist auch die Aktivierung der p38 Kinase eingebunden, die zur Aktivierung von p38 führt. Die Aktivierung von p38 ist essentiell für die Produktion der pro-inflammatorischen Zytokine IL-lß, TNFα und IL-6 und ist darüber hinaus auch verantwortlich für die Induktion und Expression der mit chronischen Entzündung vergesellschafteten Enzyme COX-2 und iNOS (Ono K, Han J (2000) The p38 Signal transduction pathway: activation and function. Cell Signal 12 1-13). Über weitere Aktivierungswege werden auch die wichtigen Transkriptionsfaktoren activating- transcrip-tion factor 2 (ATF2) und das Aktivatorprotein-1 (AP-1) induziert, welche unmittelbar stimulierenden Einfluss auf die Expression pro-inflammatorischer Moleküle wie E-Selectin, RANTES, IL-12, IL-6 und IL-8 ausüben (Guicciardi ME, Gores GJ (2003)J Clin Invest rπ 1813-1815).
Die biologische Bedeutung von TNF erkannten zuerst 1969 GRANGER et al. (Granger GA, Shacks SJ, Williams TW, KoIb WP (1969) Lymphocyte in vitro cytotoxicity: specific release of lymphotoxin-like materials from tuberculin-εensitive lymphoid cells. Nature 221 1155-1157) die zeigen konnten, dass ein von Lymphozyten und Makrophagen sezemiertes Protein (Lymphotoxin) zur Lyse von Zellen, insbesondere von Tumorzellen, führt. 1984 konnten GRAY et al. (Gray PW, Aggarwal BB, Benton CV, Briingman TS, Henzel WJ, Jarrett JA, Leung DW, Maffatt
B, Ng P, Svedersky LP et al. (1984) Cloning and expression of cDNA for human lymphotoxin, a lymphokine with tumor necrosis activity. Nature 312 721-724) und PENNICA et al. {Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Deyrynck R, Palladino MA, Kohr WJ, Aggarwal BB, Goeddel DV (1984) Human tumor necrosis factor: precursor structure, expression and homology to lymphotoxin. Natur 312 724- 729) die cDNA für TNFα klonieren und das Protein exprimieren.
Die biologische Aktivität von TNFα wird hauptsächlich über zwei spezifische Rezeptortypen (TNFR1 , TNFR2) vermittelt, die sich transmembran und mit einem extra- und intrazellulären Anteil auf einer Vielzahl Zellen des menschlichen Körpers befinden.
TNFα besitzt ein sehr breites Spektrum an biologischen Aktivitäten und reguliert fast alle Zellen. Er ist aus heutiger Sicht ein wesentlicher Mediator bei Entzündungs- und Immunreaktionen, aber auch bei der Apoptose, der Zelldifferenzierung, bei der Induktion von Fieber und zahlreichen weiteren pathophysiologischen Regulationsprozessen.
Eine zentrale Stellung nimmt TNFα bei der Endothelzellaktivierung während des Entzündungsprozesses ein. Hierbei stellt die Aktivierung der vaskulären Endothelzellen einen wesentlichen Schritt in der Initiationsphase der entzündlichen Reaktionen im Gewebe dar. So führen pro-inflammatorische Zytokine, mit TNFα an der Spitze, zur Expression endothelialer Adhäsionsmoleküle und chemotaktisch wirksamer Chemokine, die ihrerseits Makrophagen und T-Lymphozyten die Möglichkeit geben, am Endothel anzudocken und über eine aktive Wanderung ins entzündliche Gewebe (Extravasion) zu kommen. Man unterscheidet heute in diesem Zusammenhang eine lokale Wirkung von TNFα von einer systemischen. Die lokalen Effekte sind wie oben angeführt eine verstärkte Diapetese von Immun- und Entzündungszellen ins entzündliche Gewebe und eine starke Adhäsion von Thrombozyten an den Blutgefäßwänden. Der systemische Effekt von TNFα führt zu Ödemen, einer Verringerung des Blutvolumens, Hypoproteinämie, verbreitete intravaskuläre Blutgerinnung und in ihrer Maximalvariante zu multiplem Organversagen (septischer Schock).
TN Fa bewirkt also eine lokale Aktivierung des vaskulären Endothels, eine Freisetzung von Stickoxid (NO) mit nachfolgender Steigerung der vaskulären Permeabilität, eine erhöhte Expression von Adhäsionsmolekülen und eine erhöhte Expression von „class I! major histocompatibility molecules" (MHC II). Das Ergebnis ist ein Einwandern von Entzündungs- und Immunzellen, Antikörpern und Komplementfaktoren in das entzündliche Gewebe. TNFα verursacht gleichfalls in den lokalen Lymphknoten eine antigenspezifische Aktivierung der B- und T-Lymphozyten. Des Weiteren aktiviert TNFα Thrombozyten und verstärkt deren Adhäsion an den Gefäßwänden.
TNFα selbst induziert die Synthese anderer pro-inflammatorischer Zytokine wie 1L-1 , IL-6, IL-8 und GM-CSF und führt dadurch zu einem Circulus vitiosus des entzündlichen Prozesses. Zusätzlich ist TNFα noch maßgeblich in weitere pathophysiologische Prozesse, wie die Gelenkknorpelzerstörung bei rheumatischen Erkrankungen, Knochenresorptionsprozesse, Hemmung der Knochenbildung, Hemmung der Proteoglycansynthese und Induktion von Matrix Metalloproteinasen (MMP's) und Prostaglandin E2 (Mease P (2002) Psoriatic arthritis: The role of TNF Inhibition and the effect of its inhibition with etanercept. Clin Exp Rheumatol 20 (Suppl. 28) S116-S121) involviert.
Übersicht der abgesicherten biologischen Wirkung von TNFα auf humane Zellen/Organe
Tabelle 1
Entwicklung von TNFα Inhibitoren
In der Vergangenheit gab es zahlreiche therapeutische Strategien, um die biologische Aktivität von TNFα zu hemmen und damit den chronischen Entzündungsprozess zu unterbrechen.
• An erster Stelle standen Bemühungen, die Synthese von TNFα zu hemmen. Hierzu kamen anti-inflammatorische Zytokine, wie z.B. das IL-10, Pentoxifylline, Thalidomid bzw.-Analoga, Kortikosteroide, Ciclosporin A, PDE- 4 Inhibitoren und Antisense Oligonukleotide zum Einsatz.
Am erfolgreichsten werden seit Jahren Kortikosteroide in der akuten Phase eines schweren entzündlichen Prozesses eingesetzt. Ihre breitere und besonders längere Anwendung ist auf Grund der schweren unerwünschten Arzneimittelwirkungen stark limitiert. Diese Gründe treffen auch für das gleichfalls seit Jahren zugelassene Immunsuppresivum Cyclosporin A zu. Pentoxifylline und Thalidomidanaloga zeigten in den klinischen Studien nur eine unzureichende therapeutische Wirksamkeit.
• Der Einsatz von PDE-4 Inhibitoren zeigt über die intrazelluläre Steigerung der cAMP Konzentration einen inhibierenden Einfluss auf die TNFα Freisetzung. Momentan sind mit Cilomilast, AWD 12-81 (GSK) und Roflumilast (Altana) drei Entwicklungskandidaten in der fortgeschrittenen klinischen Prüfung bzw. stehen kurz vor der Zulassung. Die klinische Anwendung dieser Substanzen
geht jedoch auch mit unerwünschten Arzneimittelwirkungen, hauptsächlich emetischer Natur, einher.
• Die Antisense-Therapie befindet sich noch in einer sehr frühen Entwicklungsphase und hat zumindest in den ersten Tieruntersuchungen die erhoffte Wirksamkeit nachweisen können, aber auch hier sind noch umfängliche grundlagenorientierte Arbeiten notwendig.
• Ein weiterer Ansatz bestand in der Inhibierung des TNFα Prozessings durch Inhibitoren der Metalloproteinase TNF Converting enzyme (TACE).
Die Entwicklung von niedermolekularen TAGE Inhibitoren befindet sich noch in der Phase der angewandten Grundlagenforschung. So beschrieben TSUKIDA et al. 2004 Hydroxamsäurederivate, die TAGE in vitro hemmen (Tsukida T, Moriyama H, Inoue Y, Kondo H, Yoshino K, Nishimura S (2004) Synthesis and biological activity of selective azasugar-based TACE Inhibitors. Bioorg Med Chem Lett. 22 1569-1572). Einige Matrix Metalloproteinase Inhibitoren hemmen auch unspezifisch TACE, sind aber auf Grund ihrer MMP- inhibitorischen Aktivität für eine pharmazeutische Entwicklung nicht geeignet. WILLIAMS et al. konnten überraschend zeigen, dass der MMP Inhibitor BB- 2275 paradoxerweise sowohl TAGE, als auch das Shedding der TNFα Rezeptoren (TNFR1, TNFR2) hemmt und dadurch keinen TNFα inhibierenden Effekt hatte {Williams LM, Gibbons DL, Gearing A, et al. (1996) Paradoxical effects of a synthetic metalloproteinase inhibitor that blocks both p55 and p75 TNF receptor shedding and TNF alpha processing in RA synovial membrane cell cultures. J Clin Invest 97 2833-2841). Alle heute bekannten TACE Inhibitoren sind in ihrer hemmenden Wirkung unspezifisch, d.h. auch andere wichtige Metalloenzyme werden durch sie gehemmt, mit der Wahrscheinlichkeit unerwünschter (Neben)Wirkungen.
• Momentan sind zahlreiche nichtproteinogene (small molecules) TNFα Inhibitoren in der präklinischen und klinischen Entwicklung. Haupttarget dieser Wirkstoffe sind intrazelluläre Proteinkinasen, die über eine Phosphorylierung Transkriptionsfaktoren aktivieren und dadurch unmittelbar in die Genexpression eingreifen. Am besten untersucht ist in diesem
Zusammenhang die Regulation des Transkriptionsfaktors NFKB. NFKB befindet sich komplexiert mit IKB, der als Inhibitor für NFKB wirkt. Wird dieser Inhibitor durch die Proteinkinasen IKK-1 und IKK-2 phosphoryliert, kommt es zu einer partiellen Degradation von IKB, die die Freisetzung des NFKB aus dem Komplex verursacht. Jetzt kann der Transkriptionsfaktor NFKB vom Zytosol in den Zellkern wandern und dort direkt die Expression von z.B. TNFα erhöhen. Es ist hierbei offensichtlich, dass es bei einer Hemmung der Proteinkinase IKK zu keiner Phosphorylierung der IKB und damit zu keiner Aktivierung des NFKB kommt, mit dem Resultat, dass die Expression von z.B. TNFα, aber auch anderer NFκB-abhängiger Mediatoren, nicht stimuliert wird. Die Firma Boehringer Ingelheim beschrieb in zwei Patenten (US 2004/0180922 A1 und US 2005/0038104 A1) Aminoamide und Benzothiophene, die sehr effektiv die Aktivität der beiden Proteinkinasen IKK- 1 und IKK-2 hemmten und dadurch u.a. einen anti-entzündlichen Effekt erzielten. Ausgewählte Vertreter der in diesem Patent beschriebenen Verbindungen, hemmten jedoch nicht die IKK-1 bzw. IKK-2. Es kann deshalb zu Grunde gelegt werden, dass die in diesem Patent beschriebenen Substanzen über einen gänzlich anderen Regulationsweg die Freisetzung von TNFα hemmen.
In neuerer Zeit haben sich Strategien zur Blockierung des TNFα durch Antikörper gegen TNFα und lösliche TNF-Rezeptoren durchgesetzt.
Zum jetzigen Zeitpunkt sind mit Remicade® und Humira™ zwei monoklonale anti-TNF Antikörper von der FDA und auch von der EMEA als anti-inflammato¬ rische Therapeutika zugelassen worden.
Remicade (Essex/Centrocor) wurde durch die FDA 1998 für die Indikation Mb. Crohn und in 2000 für die Indikation Rheumatoide Arthritis zugelassen. Momentan laufen klinische Studien für die Anwendung bei Psoriasis vulgaris und Psoriasis arthropatica. Bei Remicade handelt es sich um einen chimaeren monoklonalen Antikörper gegen das humane TNFα. In den klinischen Studien zeigte das Präparat gute bis sehr gute Wirkeigenschaften beim Mb. Crohn. Jedoch wurde über teilweise erhebliche Nebenwirkungen, wie erhöhte
Infektionsgefahr, Magen-Darm Beschwerden, Kopfschmerz und allergische Reaktionen berichtet. Ein Teil der Nebenwirkungen wird auf den Maus-Anteil des monoklonalen Antikörpers zurückgeführt, der vom menschlichen Organismus als „fremd" erkannt wird, wodurch Antikörper dagegen gebildet werden. Remicade wird intravenös verabreicht und die jährlichen Medikamentenkosten belaufen sich auf über $ 12.000 pro Patient.
Humira (Abbott) ist für die Behandlung der Rheumatoiden Arthritis seit 2002 in USA und seit 2003 in Europa zugelassen. Klinische Studien zur Behandlung der Psoriasis vulgaris zeigten sehr gute Therapieerfolge. Als häufige Nebenwirkungen wurden Kopfschmerz, erhöhte Infektanfälligkeit, Magen- Darm Beschwerden und allergische Reaktionen beobachtet. Bei Humira handelt es sich um einen vollhumanisierten monoklonalen Antikörper gegen humanes TNFα. Das Präparat wird subcutan (s.c.) verabreicht. Die jährlichen Behandlungskosten belaufen sich auch bei diesem Präparat auf über $ 12.000 pro Patient.
Enbrel (Immunex/Wyeth) wurde erstmals durch die FDA 1998 für die Indikation Rheumatoide Arthritis zugelassen und seit 2000 ist das Präparat auch auf dem europäischen Markt. Die Zulassung für die Indikation Psoriasis vulgaris und Psoriasis arthropatica wird noch 2004 von der FDA erwartet. Bei Enbrel handelt es sich um ein rekombinantes (CHO-Zellen) dimeres Fusionsprotein, bei dem zwei extrazelluläre Bindungsdomänen des p75- Anteils des TNF Rezeptors an den Fc-Anteil des humanen IgGI -Moleküls angekoppelt sind und dadurch lösliches TNFα im Blut/Gewebe abbinden und somit neutralisieren kann. Laut Herstellerangaben besitzt dieses Fusionsprotein ein geringes immunogenes Potential, wohingegen aber auch Fälle beschrieben wurden, in denen eine Antikörperbildung gegen das Fusionsprotein beobachtet wurde. Als häufige Nebenwirkungen wurden allergische Reaktionen, Infektanfälligkeit und die Bildung von Auto-Antikörpern (ANA) beschrieben. Das Präparat wird subcutan verabreicht und die jährlichen Behandlungskosten belaufen sich gleichfalls auf über $ 10.000 pro Patient.
Zusammenfassend kann festgestellt werden, dass sich das therapeutische Konzept der Hemmung von TNFα als biologischer Endpunkt, als tragfähig in der Klinik erwiesen hat. Momentan stehen nur proteinogene Präparate (monoklonale Antikörper, Fusionsproteine) zur Verfügung, die eine Reihe von unerwünschten Arzneimittelnebenwirkungen aufweisen. Darüber hinaus ist deren intravenöse bzw. subkutane Applikationsform für Patienten sehr belastend und geht mit einer entsprechend schlechten Compliance einher. Die sehr hohen Herstellungs- und somit auch Behandlungskosten limitieren ebenfalls deren Einsatz.
Es besteht deshalb nach wie vor Bedarf an niedermolekularen, nichtproteinogenen Wirkstoffen, die oral verabreicht werden können und eine bessere Verträglichkeit aufweisen. Zurzeit existieren noch keine bekannten niedermolekularen Verbindungen, die selektiv die Synthese von TNFα hemmen, ohne mit anderen Stoffwechselwegen zu interagieren und die oben beschriebenen unerwünschten Arzneimittelwirkungen nicht aufweisen.
Es besteht deshalb die Aufgabe, neuartige und oral applizierbare niedermolekulare Wirkstoffe zu entwickeln, die zu einer signifikanten Inhibition der TNFα-Freisetzung aus relevanten humanen Zellpopulationen führen.
Diese Aufgabe wird von Verbindungen
der allgemeinen Formeln 1a und 1b gelöst.
In bisher nur wenigen Fällen ist über biologisch aktive Pyrido[3',2':4,5]thieno- [3,2-d]pyrimidin-Derivate, jedoch noch nicht über entsprechende -2,4(1 H,3H)-dione berichtet worden:
So wurden von J. M. Quintθla etai: Bioorg. Med. 6 (1998) 1911-1925, die Synthese von mehreren 2-Dimethylamino-(oder 2-H)-4-sek.amino-7-ethoxy-8-cyano-(oder 8- H)-9-phenyl- und auch das strukturanaloge 4-Ethoxy-pyrido[3',2':4,5]thieno[3,2- d]pyrimidin-Derivat beschrieben. Einige dieser Verbindungen zeigten eine die Histamin-Freisetzung aus Mastzellen von Ratten inhibierende Wirkung. Abdel- Rahmann et al.: Pharmazie 58 (2003) 372-377 berichteten über die Synthese von 8- Acetyl-3-amino-7-methyl-4-imino-9-substituierten Phenyl-pyrido[3',2':4,5]thieno[3,2- d]pyrimidinen mit antimikrobieller Wirksamkeit. Weiterhin wurden Derivate dieses Heterosystems mit unter anderem signifikanter cholesterolsenkender {C.J.Shishoo, M.B.Devani und V.S.Bhadti: Indian Patent 151.456 (1983); Chem. Abstr.: 100, 209858 (1984); und V.P.Arya, Drugs Future, 10, 123 (1985)), mit analgetischer (C.G.Dave et al.: J. Indian Chem.Soc. 66, 48 (1989)), mit antipyretischer (E.Bousquet et al.: Farmaco Ed.Sci. 40, 869 (1985); und E.Bousquet et al.: Farmaco Ed.Sci. 39, 110 (1984)), mit antianaphylaktischer (H.Vieweg, S.Leistner, G.Wagner et al.: East German Patent DD 257830 (1988); Chem. Abstr.: 110, 95262p (1989); und H. Vieweg, S.Leistner, G. Wagner et al.: East German Patent DD 258234 (1988)); mit antiinflammatorischer (E.F.EIslager, P.W.Jacob und M.Leslic: J.Heterocyclic Chem. 9, 775 (1972); und M.Chaykovskyet al.: J.Med.Chem. W, 188 (1973); und LA.Radinovskaya und A.Sharanin: Khim.Geterotsikl.Soedin. 805 (1988); und S.Leistner et al.: Pharmazie 41_, 54 (1986)); mit klinisch effektiver antiallergischer (G.D.Madding und M.D.Thompson: J.Heterocyclic Chem. 24, 581 (1987)); und mit potentiell antineoplastischer Wirksamkeit (C.C.Cheng:in Progress in Medicinal Chemistry25, 35 (1989) beschrieben.
Von M.A.A.EI-Neairy (Phosphorys, Sulfur and Silicon 1999, 189) wurden 8-Acetyl-7- methyl-θ-p-nitrophenyl-pyridoCS'^'^.δlthienofS^-dlpyrimidin^^CI H.SHJ-dion und dessen 9-p-Dimethylamino-Analogon und von V.A.Artemov et al. (Chemistry of Heterocyclic Compounds 30, 110 (1994) das entsprechende 7,9-Dimethyl-2,4-dion- Derivat beschrieben. Angaben über pharmakologische Eigenschaften dieser Pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-2,4(1 H,3H)-dione sind bisher nicht bekannt.
Als Inhibitoren der TNFα-Freisetzung sind Verbindungen der allgemeinen Formeln 1a und 1b (jeweils für X=C-R2) jedoch bisher völlig unbekannt.
Einige Vertreter des tricyclischen Thieno[2,3-d:4,5-d']dipyrimiclin-heterosystems sind als solche bereits bekannt.
Es handelt sich bei den Verbindungen der allgemeinen Formel 1a (für X=N) um R1- und R3-substituierte Bislactame und bei den Verbindungen der allgemeinen Formel 1 b (für X=N) um R1-, R3- und R4-substituierte Monolactame des Thieno[2,3-d:4,5- d']dipyrimidin-heterosystems.
Von den Verbindungen der allgemeinen Formel 1a (für X=N) ist eine Substanz (mit R1 = H und R3 = NHMe) beschrieben (Clark,J. und Hitiris,G. J.Chem.Soc, Perkin Trans. 1984, 2005). Ein Hinweis auf pharmakologische Eigenschaften findet sich nicht.
Von den Verbindungen der allgemeinen Formel 1b (für X=N) sind dagegen derzeit 24 Substanzen literaturbekannt.
Es handelt sich dabei vorwiegend um Verbindungen mit R1 = Ph und R3 = Me und verschiedenen R4-Substituenten wie Me, SH, SAIk, SCH2COR5, NHNH2 und daraus dargestellte Hydrazone sowie substituierte 3,5-Pyrazol-1-yl-Reste. Als R1 tritt auch H, Me, OH und 3,5-Dimethylpyrazol-1 -yl und als R3 auch Ph, NHMe (in diesem Fall R1 = H; R4 = NH2). In der Veröffentlichung von Wagner.G. Vieweg.H. und Leistner.S. Pharmazie 48, 588 (1993) ist ein Hinweis auf nicht sehr ausgeprägte Wirkung am PCA-Test der Ratte gegeben. Ein Hinweis auf andere pharmakologische Eigenschaften findet sich laut Recherche nicht.
Vor der vorliegenden Erfindung bekannte Struktur-Wirkungsbeziehungen erlaubten keine seriöse Voraussage, ob Verbindungen der allgemeinen Formel 1a (für X=N), aber auch 1b (für X=N) nützliche pharmakologische Eigenschaften aufweisen. Es ist daher überraschend, daß erstmals festgestellt werden konnte, daß Derivate der Titelverbindungen 1a und 1b die TNFα-Freisetzung inhibieren.
Beschreibung der Erfindung
Die Erfindung betrifft neue Pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-2,4(1 H,3H)-clione und -4(3H)-one (X=C-H1 Y=S), Thieno[2,3-d:4,5-d']dipyrimidin-2,4(1 H,3H)-dione und -4(3H)-one (X=N, Y=S) sowie Pyrido[3',2':4,5]furo[3,2-d]pyrimidin-2,4(1H,3H)-dione und -4(3H)-one (X=C-H, Y=O) und Furo[2,3-d:4,5-d']dipyrimidin-2l4(1 H,3H)-dione und -4(3H)-one (X=N, Y=O) der allgemeinen Formeln 1a und 1 b
worin bedeuten:
X C-R2 oder Stickstoff
Y Schwefel oder Sauerstoff und
R1 und R3, gleich oder ungleich unabhängig voneinander,
- Wasserstoff,
- Ci-i2Alkyl (ggf. mit R§ substituiert),
- C-2-12 Alkenyl und C-2-12 Alkinyl (jeweils ggf. mit R§ substituiert ),
- Benzyl (ggf. ein- bis fünffach unabhängig voneinander mit R§ substituiert)), Phenyl-(C2-β)alkyl , Phenyl (ggf. ein- bis fünffach unabhängig voneinander mit R§ substituiert),
- Monofluormethyl, Difluormethyl, Trifluormethyl,
- 1-Naphthyl, 2-Naphthyl (jeweils ggf. mit R§ substituiert),
- C3-i4Cycloalkyl, C3-i4Cycloalkenyl (jeweils ggf. mit R§-substituiert),
- mono- oder bicyclische gesättigte oder ein- oder mehrfach ungesättigte Heterocyclen (ggf. ein oder mehrfach mit R§ substituiert) mit 5 - 14 Ringatomen darunter 1 - 4 Heteroatome, die vorzugsweise N, O und S sind und ggf. am Heteroatom ein oder mehrfach oxidiert sind,
- Ci-i2Alkylacyl (ggf. mit R§ substituiert),
- Benzoyl, 1- und 2-Naphthoyl (jeweils ggf. mit R§ substituiert),
- Heterocyclylacyl [z.B. Nicotinoyl, Isonicotinoyl, 2-Picolinoyl, 2-Thienoyl, 2- Furoyi] (ggf. mit R§ substituiert)
- Hydroxy,
- Sulfhydryl,
- C1-10 Alkoxy,
- Alkylthio, Alkylsulfinyl und Alkylsulfonyl (jeweils Ci-6 )
- Formyl, Carboxyl, Ci-4Alkoxycarbonyl;
- CONH2, CONHAIk und CONAIk2 (mit „Alk" jeweils Ci-6),
- Cyano, Rhodano, Nitro, SO3H, SO2OAIk (mit „Alk": Ci-5),
- Chlor, Brom, lod, Fluor,
- Amino, Ci-6Alkylamino, Di(Ci-5)alkylamino (jeweils ggf. mit R§ am Alkylrest substituiert),
- Morpholino, Thiomorpholino, Thiomorpholino-S,S-Dioxid, Pyrrolidino,
- Piperidino, 1-Piperazino, 4-Methyl-1-piperazino, 4-Hydroxyethyl-1-piperazino, 4-Phenyl-1 -piperazino,
- Cycloalkylamino, C3-I4 Arylamino und Heteroarylamino [z.B. Phenyl-, 1τ und 2-Naphthyl-, 2-, 3- oder 4-Pyridyl-, Chinolinyl-, Isochinolinyl-, Acridinyl-, Phenothiazinyl-, 2-Thienyl- und 2-Furylamino] (ggf. jeweils an den carbo- bzw. heterocyclischen Ringen mit R§ substituiert);
R2
- Wasserstoff,
- Ci-i2Alkyl>
- C2-i2Alkenyl und C2-12 Alkinyl,
- Benzyl, Phenyl(C2-6)alkyl (jeweils ggf. mit R§ ein oder mehrmals, gleich oder ungleich am aromatischen und/oder aliphatischen Molekülteil substituiert);
- Phenacyl (ggf. mit R§ ein oder mehrmals, gleich oder ungleich am aromatischen Molekülteil substituiert);
- Carboxyl, C1-4 Alkoxycarbonyl, CONH2, CONHAIk und CONAIk2 (mit „Alk" jeweils C1-6),
- FTCO- (worin R* Wasserstoff, Cπ2Alkyl bedeuten sowie ggf. mit R§ substituiert),
- Cyano, Nitro, Amino, Ci-6Alkylamino, Di(Ci-6)alkylamino, -N=N-C6H5, -N=N- C6H4-R§,
- 1 ,3-DiphenyI-pyrazol-4-yl, Thiazolin-2-yl, lmidazolin-2-yl und 3,4,5,6- Tetrahydro-pyrimidinyl;
RA
- C2-i4A!kyl, C3-i4Cycloalkyl, C2-i4Alkenyl, Ce-nCycloalkenyl, C2-i4Alkinyl (jeweils ggf. am C-Skelett der vorgenannten aliphatischen oder cycloaliphatischen Reste mit R§ substituiert);
- Phenyl, 2-R§-Phenyl, 3-R§-Phenyl, 4-R§-Phenyl, 2-R§,5-R§-Phenyl,
3-R§,5-R§-Phenyl, 3-R§,4-R§-Phenyl, 3-R§,4-R§,5-R§-Phenyl, 2-R§,3-R§,4-R§-Phenyl,
- 1-Naphthyl, 2-Naphthyl (jeweils ggf. mit R§ substituiert);
- mono-, bi- oder tricyclische gesättigter oder ein- oder mehrfach ungesättigter heterocyclischer Rest mit insgesamt 4-14 Ring-Atomen, davon 1 -5 Heteroatomen, die vorzugsweise N, O, S und Se sind (jeweils ggf. mit R§ substituiert);
- CH2OAIk* (mit Alk*: C1-6Alkyl),
- CH2OCOR' (mit R': Ci-6Alkyl, C2-6Alkenyl, Phenyl sowie mit R§ substituiert),
- COOH, COOAIk** (mit Alk**: Ci-5Alkyl),
- NH2, NHAIk*, (mit Alk*: C1-6Alkyl )
- NHNH2, NHNHCOR' (mit R': Ci-6Alkyl, Ci-6Alkenyl, Phenyl sowie mit R§ substituiert),
- SO3H, S-Phenacyl, S-Alkyl, SO-Alkyl, SO2-Alkyl (jeweils C1-S )
- S-Alkenyl, SO-Alkenyl, SO2-Alkenyl (jeweils C2-8), S-Alkinyl, SO-Alkinyl, SO2-Alkinyl (jeweils C2-6) (ggf- jeweils am C-Skelett der oben genannten aliphatischen Reste mit -OH, -CN, -SCN, -NO2, Phenyl oder (C3-7)Cycloalkyl substituiert)
Der oben erwähnte Ausdruck „ggf. mit R§ substituiert" bedeutet, daß die genannten Reste einfach oder mehrfach, gleich oder ungleich unabhängig voneinander, substituiert sein können, wobei R§ folgende Bedeutung hat:
OH1 -SH, -O-C1-8Alkyl, -0-C6-I4 Aryl, -S-Ci-4 Alkyl, -S-C6-i4Aryl, -SO-C1-4AIkVl, -SO-C6-I4ArVl1 -SO2-C1-4Alkyl, -SO2-C6-I4AIyI1 -SO3H, -OSO2C1-8Alkyl, -OSO2C6-14Ary I, -COOH, -COOCi-8Alkyl, -(CO)Ci-8Alkyl, -COOH, -COOCi-8Alkyl, -CONH2, -CONHCi-6Alkyl, -CON(Ci-6Alkyl)2, -NH2, -NHC1-6Alkyl, -N(C1-6Alkyl)2, -NHC6-i4Aryl, -NH-Hetaryl, -N(C6-I4AIyI)2, -N(CiVMM)(C6-I4ArVl),
-CH3, -CHF2, -CH2F1 -CF3, -C2HO, -C(CHa)2, -(CH2)2CH3, -(CH2)3CH3, -CH2CH2OH, -CH2CH2SH, -CH2CH2SCHa, -CH2CF3, -CH2CCIs, -CH2CBr3 -CH2CHF2, -CH2CHCI2, -CH2CHBr21 -CH2CH2F, -CH2CH2CI, -CH2CH2Br, -Cylopropyl, -Cylopropylmethyl, -Cylobutyl, -Cyclobutylmethyl, -Cyclopentyl, - -Cyclopentylmethyl, -Cylohexyl, -Cyclohexylmethyl, -F, -Cl, -Br, -I, -CN1 -NO2, und -SCN
Ausgenommen vom Stoffschutz sind die folgenden Verbindungen der Formel 1a:
R1 X R3
1. Me C-H Me
2. Me C-Ac 4-NMe2Ph
3. Me C-Ac 4-NO2Ph
Ausgenommen vom Stoffschutz sind auch die folgenden Verbindungen der Formel 1 b mit folgenden Resten:
jeweils R1 = OEt, X = C-CN, R3 = Ph mit R4= Benzyl, Phenyl, z.T. mehrfach mit Me,
OMe, NO2 und Cl substituiert und 3,4-Methylendioxy-phenyl; jeweils R1 = Ph, X = C-H, R3 = Ph mit R4= Phenyl, p-OCH3-Ph, p-CI-Ph,
2,3,4-Tn-OCH3-Ph und 2-Thienyl; jeweils R1 = Me, X =C-H, R3 = Me mit R4 = CH2OMe, CH2OEt, CH2OCOMe,
CH2COOPh, SCH2COPh, NH2, NHNH2, COOH1 Ph; sowie:
R1 = Me, R3 = Ph1 R4 = COOEt; X = C-H;
R1 = Me, R3 = p-Br-Ph, R4 = COOEt; X = C-H; R1 = p-Br-Ph, R3 = Me, R4 = COOMe; X = C-H; R1 = Me, R3 = Me, R4 = COOMe; X = C-Benzyl R1 = R3 = Me, R4 = COOMe, X = C-Me R1 = Ph, R3 = Me, R4 = SEt, X = N.
In der Erfindungsbeschreibung bedeuten die Begriffe "Alkyl, Alkenyl, Alkinyl, Alkoxy, usw.", auch in Wortzusammensetzungen wie Alkylsulfonyl, Alkylamino oder Alkoxycarbonyl usw. sowohl die unverzweigten wie auch die verzweigten möglichen Verbindungen. Ebenso bedeuten „Alkenyl und Alkinyl" die entsprechend möglichen einfach oder mehrfach ungesättigten Verbindungen. Das gleiche gilt auch für die entsprechenden cyclischen Verbindungen.
Im Sinne der Erfindung gelten alle Reste als miteinander kombinierbar, soweit bei der Definition der Reste nichts anderes angegeben ist. Es sollen alle denkbaren Untergruppierungen als offenbart gelten.
Die Erfindung betrifft auch physiologisch verträgliche Salze der Verbindungen der allgemeinen Formeln 1a und 1b.
Die physiologisch verträglichen Salze werden auf übliche Weise durch Umsetzung basischer Verbindungen der allgemeinen Formeln 1a und 1b mit anorganischen oder organischen Säuren, ggf. auch bei Vorliegen von Verbindungen mit aciden Eigenschaften, wenn z.B. einer der Substituenten R1, R2, R3 oder R4 in diesen Verbindungen -COOH bzw. -SO3H bedeutet, durch Neutralisation mit anorganischen oder organischen Basen, erhalten.
Als anorganische Säuren kommen vorzugsweise Salzsäure, Schwefelsäure, Salpetersäure oder Bromwasserstoffsäure, als organische Säuren zum Beispiel Ameisensäure, Essigsäure, Propionsäure, Glykolsäure, Milchsäure, Mandelsäure, Weinsäure, Äpfelsäure, Zitronensäure, Malonsäure, Maleinsäure, Fumarsäure, Succinsäure, Alginsäure, Benzoesäure, 2-, 3- und 4-Alkyloxy- und Acyloxy- benzoesäuren, Ascorbinsäure, Ci-C3,Alkylsulfonsäuren, Benzolsulfonsäure, Nicotinsäure, Isonicotinsäure und Aminosäuren zur Anwendung.
Als anorganische Basen kommen zum Beispiel Ammoniak, Natron- und Kalilauge sowie als organische Basen Alkylamine, CrC3, Pyridin, Chinolin, Isochinolin, Piperazin und -Derivate, Picoline, Chinaldin oder Pyrimidin zur Anwendung.
Weiterhin können physiologisch verträgliche Salze der Verbindungen gemäß der allgemeinen Formeln 1a und 1 b dadurch gewonnen werden, dass jene Substanzen, die als Substituenten eine tertiäre Amino-Gruppe besitzen, in prinzipiell bekannter Weise mit alkylierenden Agentien - wie zum Beispiel Alkyl- oder Aralkylhalogeniden - in die entsprechenden quatemären Ammoniumsalze übergeführt werden können.
Die Erfindung betrifft auch Solvate der Verbindungen, einschließlich der pharmazeutisch akzeptablen Salze, Säuren, Basen und Ester sowie deren aktive Metabolite und gegebenenfalls deren Tautomere gemäß der allgemeinen Formeln 1 a und 1 b einschließlich Prodrug-Formulierungen. Prodrug-Formulierungen umfassen hierbei alle jene Substanzen, die durch einfache Transformation einschließlich Hydrolyse, Oxidation, oder Reduktion entweder enzymatisch, metabolisch oder auf andere Art und Weise entstehen. Ein geeignetes Prodrug enthält beispielsweise eine Substanz der allgemeinen Formeln 1 a und 1 b, die über einen enzymatisch spaltbaren Linker (z.B. Carbamat, Phosphat, N-Glycosid oder eine Disulfidgruppe) an eine lösungsverbessemde Substanz (z.B. Tetraethylenglykol, Saccharide, Aminosäuren oder Glucuronsäure, etc.) gebunden ist. Ein solches Prodrug einer erfindungsgemäßen Verbindung kann einem Patienten appliziert werden, und dieses Prodrug kann in eine Substanz der allgemeinen Formeln 1a und 1b transformiert werden, wodurch der gewünschte pharmakologische Effekt erzielt wird.
Die durch die erfindungsgemäßen Verbindungen behandelbaren Erkrankungen schließen alle ein, bei denen TNF-alpha eine Rolle spielt und die durch eine Hemmung oder Inhibierung desselben positiv zu beeinflussen sind, z.B. chronische Entzündungserkrankungen, Autoimmun-Erkrankungen, cardiovaskuläre
Erkrankungen, virale Erkrankungen und hier insbesondere retrovirale Erkrankungen wie z.B. das erworbene Immundefizienz Syndrom (AIDS) sowie Krebs, insbesondere Entartungen des blutbildenden Systems. Insbesondere sind dies Rheumatoide Arthritis, Osteoarthritis, Osteoporosis, Asthma bronchiale, chronische obstruktive pulmonäre Erkrankung (COPD), Multiple Sklerose, Sepsis, cerebrale Form der
Malaria, neurodegenerativen Erkrankungen wie z.B. Mb. Alzheimer, Mb. Parkinson, Guillain-Barre-Syndrom, Crohns Disease, Colitis ulcerosa, Psoriasis, Graft-versus- Host-Disease (GvHD), systemischer Lupus erythematodes (SLE), Vasculitis, Uveitis, insulin-abhängiger Diabetes mellitus, Respiratorisches Distress-Syndrom beim Erwachsenen (ARDS), multiples Organversagen nach Trauma, aktute Glomerulonephritis, akute und chronische Schmerzen, Arteriosklerose, Herzinfarkt, Schlaganfall, entzündliche Dermatosen, atopische Dermatitis, Psoriasis vulgaris, Alopecie, Rhinitis allergica, allergische Konjunktivitis, akute Meningitis, Myastenia Gravis, Sklerodermie und Sarkoidose.
Die erfindungsgemäßen Verbindungen können auf verschiedenen Wegen verabreicht werden, z.B. oral, parenteral, kutan, subkutan, intravenös, intramuskulär, rektal oder inhalativ. Bevorzugt ist die intravenöse oder inhalative Verabreichung. Die Verbindung wird einem Patienten, der eine Therapie einer unter das Indikationsspektrum der erfindungsgemäßen Verbindungen fallenden Krankheit bedarf, über einen vom Arzt zu bestimmenden Zeitraum verabreicht. Die Verbindung kann sowohl Menschen als auch anderen Säugern verabreicht werden.
Die Dosierung der erfindungsgemäßen Verbindungen wird vom Arzt anhand der patientenspezifischen Parameter wie z.B. Alter, Gewicht, Geschlecht, Schwere der Erkrankung, etc. bestimmt. Bevorzugt beträgt die Dosierung zwischen 0,001 mg/kg bis 1000 mg/kg Körpergewicht, bevorzugt 0,01 bis 500 mg/kg Körpergewicht und ganz bevorzugt 0,1 bis 100 mg/kg Körpergewicht.
Entsprechend der Art der Verabreichung wird das Medikament in geeigneter Weise formuliert, z.B. in Form von Lösungen bzw. Suspensionen, einfachen oder dragierten Tabletten, Hart- oder Weichgelatinekapseln, Pulver zur Rekonstitution vor Gebrauch, Aerosolen, Inhalationssprays, Wirkstoffpflastern, Granulaten, Suppositorien, Ovula, Injektionspräparaten, Cremes, Salben, Gels, Mikrospheren, Implantaten, die nach üblichen galenischen Verfahren hergestellt werden.
Die erfindungsgemäßen Verbindungen können gegebenenfalls zusammen mit weiteren Wirkstoffen und mit in pharmazeutischen Zusammensetzungen üblichen Exzipientien formuliert werden, z.B. je nach herzustellendem Präparat Talk, Gummi
arabicum, Lactose, Stärke, Magnesiumstearat, Kakaobutter, wäßrige und nichtwäßrige Träger, Fettkörper mit tierischem oder pflanzlichem Ursprung, Paraffinderivate, Glykole (insbesondere Polytethylenglykol), verschiedene Weichmacher, Dispergiermittel oder Emulgatoren, pharmazeutisch verträgliche Gase (z.B. Luft, Sauerstoff, Kohlendioxid usw.), Konservierungsstoffe.
Zur Herstellung flüssiger Präparate können Additive wie Natriumchloridlösung, Ethanol, Sorbit, Glycerin, Olivenöl, Mandelöl, Propylenglycol oder Ethylenglycol verwendet werden.
Bei der Verwendung von Infusions- oder Injektionslösungen sind diese sind bevorzugt wäßrige Lösungen oder Suspensionen, wobei es möglich ist, diese vor Gebrauch herzustellen, beispielsweise aus lyophilisierten Präparaten, die den Wirkstoff alleine oder zusammen mit einem Träger, wie Mannit, Lactose, Glucose, Albumin und dergleichen, enthalten. Die gebrauchsfertigen Lösungen werden sterilisiert und gegebenenfalls mit Hilfsmitteln vermischt, beispielsweise mit Konservierungsstoffen, Stabilisatoren, Emulgatoren, Lösungsvermittlern, Puffern und/oder Salzen zur Regulierung des osmotischen Drucks. Die Sterilisierung kann durch Sterilfiltration durch Filter mit einer kleinen Porengröße erzielt werden, wonach die Zusammensetzung gegebenenfalls lyophilisiert werden kann. Geringe Mengen an Antibiotika können auch zugesetzt werden, um die Beibehaltung der Sterilität zu gewährleisten.
Weiter bevorzugt werden Inhalationszusammensetzungen, z.B. in Form von Aerosolen, Sprays, oder als mikronisiertes Pulver hergestellt. Dazu werden die erfindungsgemäßen Verbindungen entweder als in pharmazeutisch üblichen Lösungsmitteln gelöst bzw. suspendiert und mittels Überdruck in einem bestimmten Volumen fein verteilt und inhaliert. Ein entsprechendes Vorgehen erfolgt bei den zu inhalierenden Festsubstanzen, die gleichfalls mittels Überdruck fein verteilt und inhaliert werden. Ebenfalls andere als mit Überdruck funktionierende Applikatoren sind hierbei eingeschlossen.
Die Erfindung betrifft auch pharmazeutische Zubereitungen, die eine therapeutisch wirksame Menge des aktiven Inhaltsstoffs (erfindungsgemäße Verbindung der
Formel (1 a) oder (1 b)) zusammen mit organischen oder anorganischen festen oder flüssigen pharmazeutisch verträglichen Trägern, die für die beabsichtigte Verabreichung geeignet sind, und die mit den aktiven Inhaltsstoffen nicht nachteilig wechselwirken, enthalten.
Die Erfindung betrifft auch Verfahren zur Herstellung pharmazeutischer Zubereitungen, die dadurch gekennzeichnet sind, dass die erfindungsgemäße Verbindung mit einem pharmazeutisch verträglichen Träger vermischt wird.
Die erfindungsgemäßen Verbindungen eignen sich auch im Rahmen von Kombinationstherapien mit schon bekannten Wirkstoffen zur Behandlung der oben genannten Erkrankungen. Dabei sollen überraschende Synergieeffekte zur Steigerung der therapeutischen Wirksamkeit der erfindungsgemäßen Substanzen genutzt werden. Die Kombination kann zum einen darin bestehen, eine einzige pharmazeutische Zusammensetzung anzubieten, die mindestens eine der erfindungsgemäßen Verbindungen in Kombination mit einem oder mehrere der nachfolgend genannten Wirkstoffen enthält oder dem Patienten werden gleichzeitig oder zeitlich versetzt mehrere Mittel, die einen oder mehreren der nachfolgenden Wirkstoffe enthalten, verabreicht.
Es ist bevorzugt eine oder mehrere der erfindungsgemäßen Verbindungen mit einem oder mehreren der folgenden Wirkstoffe zu kombinieren:
Corticosteroide
(monoklonale) Antikörpern gegen TNF-alpha oder andere Wirkstoffe, die die
Bildung bzw. Freisetzung von TNF-alpha oder die Aktivität von TNF-alpha hemmen (z.B. rekombinante TNFα-Rezeptorkonstrukte)
Zytokin-Antagonisten (z.B. IL-1 ß, IL-6, IL-12)
Chemokin-Antagonisten
Zytokin-Agonisten (z.B. IL-10) immunmodulatorische Wirkstoffe wie z.B. Cyclosporin A, Methodrexat,
Leflunomid, D-Penicillamin, Auranofine
Substanz P-Antagonisten
Bradykinin-Antagonisten
PAF-Antagonisten
Adenosin-Rezeptor-Antagonisten
Antibiotika/Virostatika α-Mimetika
Zytostatika ß2-Adrenoceptor Agonisten (z.B. Terbutalin, Salbutanol, Salmetanol,
Fenoterol, Formoterol)
Leukotrien-Antagonisten (entweder Enzym-Inhibitoren [wie 5-
Lipoxygenaseinhibitoren oder Arachidonsäure-Enzyminhibitoren] oder
Rezeptorantagonisten) , z.B. Pranlukast, Montelukast, Zafirlukast, Zileuton
Antihistaminika (bevorzugt solche mit Mastzellen-stabilisierenden
Eigenschaften oder Leukotrien-antagonisierenden Aspekten, wie z.B.
Loratadin, Astemizol, Mizolastin, , Olopatadin
Theophyllin
Muscarinrezeptor-Antagonisten, z.B. Spiriva
Die Kombination mit oben aufgeführten Arzneimitteln bzw. Wirkprinzipien dient besonders dazu, den akut zu behandelnden Krankheitszustand in einem möglichst frühen Stadium in seiner Manifestation zu beeinflussen und nicht chronisch werden zu lassen, da die erfindungsgemäßen Verbindungen in Kombination mit den anderen Wirkstoffen komplementäre/additive Aspekte ermöglichen. In der Kombination ergibt sich ein positiver Effekt u.a. daraus, dass eine geringere Substanzmenge pro Prinzip angewendet werden kann und damit zum einen eine Verbesserung des therapeutischen Effektes, geringere UAWs und zum anderen ein Spareffekt zu erreichen ist.
Abhängig von der Krankheitsausprägung und den zugrunde liegenden Symptomen können die erfindungsgemäßen Verbindungen zu den anderen Wirkstoffen in der Kombination im Verhältnis von 1 :10.000 bis 10.000:1 , bevorzugt 1 :1000 bis 1000:1 , ganz bevorzugt 1 :10 bis 10:1 , vorliegen.
Die Erfindung betrifft weiterhin Verfahren zur Herstellung der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verfahren zur Herstellung der Verbindungen der
allgemeinen Formeln 1a und 1 b mit den zuvor aufgeführten Bedeutungen von R1, R3, R4, X und Y sind gekennzeichnet durch folgende Verfahrensweisen:
A)
Für X=N: Darstellung der 2-Aminonitrile der allgemeinen Formel 2
durch Umsetzung von Acetonitril mit einem Nitril der allgemeinen Formel 3 (mit identischer Bedeutung von R3)
in Gegenwart eines Alkoxides, vorzugsweise Kalium-te/t-butoxid in einem geeigneten Lösungsmittel, vorzugsweise Toluol.
Umsetzung eines Säurehalogenids der allgemeinen Formel 4 (mit identischer Bedeutung von R1)
R1— COCI 4
mit einem Thiocyanat, vorzugsweise Ammoniumthiocyanat, in einem geeignetem Lösungsmittel, vorzugsweise Dioxan, zum Carbonsäure-isothiocyanat welches mit einem 2-Aminonitril der allgemeinen Formel 2 zum Pyrimidin-5-carbonitril der allgemeinen Formel 5 (mit X=N) umgesetzt wird.
- Umsetzung der dargestellten Pyridin- bzw Pyrimidin-3-carbonitrile der allgemeinen Formel 5,
mit identischer Bedeutung von R
1, R
3, X und Y wie oben, in an sich bekannter Weise mit Chloracetamid, CICH
2CONH
2, in methanolischer oder ethanolischer Lösung in Gegenwart eines Natriumalkoxides, vorzugsweise Natriummethoxid oder Natriumethoxid, zunächst zu den Verbindungen der allgemeinen Formel 7, mit identischer Bedeutung von X, R
1, R
3 und Y
- Die Verbindungen der allgemeinen Formel 7 (mit identischer Bedeutung von X, Y, R1, und R3 wie oben) sind aus den Verbindungen der allgemeinen Formel 5, auch dadurch darstellbar, dass diese Verbindungen mit Choracetamid zunächst in vorzugsweise ethanolischer Lösung in Gegenwart von vorzugsweise Triethylamin oder in wasserfreier acetonischer Lösung in Gegenwart von Natrium- oder Kaliumhydrogencarbonat zu den Verbindungen der allgemeinen Formel 6,
worin X, Y, R1 und R3 die oben genannten Bedeutungen aufweisen, umgesetzt werden und diese Verbindungen in einem weiteren Syntheseschritt in vorzugsweise wasserfreier ethanolischer Lösung mit einer katalytischen Menge Natrium¬ methoxid oder Natriumethoxid durch Erhitzen unter Rückfluss gleichfalls in die oben genannten Verbindungen der allgemeinen Formel 7 übergeführt werden;
- Umsetzung der Verbindungen der allgemeinen Formel 7 mit Phosgen oder Trichlormethylchlorformiat (Diphosgen) in einem vorzugsweise aprotischen, hochsiedenden Lösungsmittel wie Dioxan, Tetrahydrofuran oder Toluol bzw. deren Mischungen unter Erhitzen, ggf. in Anwesenheit katalytischer Mengen eines
Matriumalkoxides, wie zum Beispiel
Natriumethylat, zu Verbindungen der allgemeinen Formel 1a,
worin X, Y, R
1 und R
3 dasselbe wie oben bedeuten;
oder
B)
Umsetzung der bekannten bzw. nach prinzipiell bekannten Methoden dargestellten Pyridin- bzw. Pyrimidin-3-carbonitrile der allgemeinen Formel 5,
mit identischer Bedeutung von X, Y, R
1 und R
3 wie oben, mit N-Chloracetylurethan, CICH
2CONHCOOC
2H
5, in methanolischer oder ethanolischer Lösung oder in Butoxyethoxyethanol in Gegenwart einer organischen Base vorzugsweise Triethylamin, unter Erwärmen zu den Verbindungen der allgemeinen Formel 1a,
worin X, Y, R
1 und R
3 dasselbe wie oben bedeuten; oder
C)
- Umsetzung der Verbindungen der allgemeinen Formel 7, worin X, Y, R1 und R3 dasselbe wie oben bedeuten, mit Carbonsäurehalogeniden der allgemeinen Formel 8,
R4-CO— Z 8 worin R4
C2-i4Alkyl, Ca-uCycloalkyl, C2-i4Alkenyl, C3-i4Cycloalkenyl, C2-i4Alkinyl (jeweils ggf. am C-Skelett der vorgenannten aliphatischen oder cycloaliphatischen Reste mit R§ substituiert);
- 1-Naphthyl, 2-Naphthyl (jeweils ggf. mit R§ substituiert);
- mono-, bi- oder tricyclische gesättigter oder ein- oder mehrfach ungesättigter heterocyclischer Rest mit insgesamt 4-14 Ring-Atomen, davon 1-5 Heteroatomen, die vorzugsweise N, O, S und Se sind (jeweils ggf. mit R§ substituiert);
- CH2OAIk* (mit Alk*: C1-6AIkyl),
- CH2OCOR' (mit R': Ci-6AIkyl, C2-6Alkenyl, Phenyl sowie mit R§ substituiert),
- COOH, COOAIk** (mit Alk**: C1-5Alkyl),
bedeutet und
Z = Chlor oder Brom
oder
- Umsetzung der Verbindungen der allgemeinen Formel 7 (worin X, Y, R1 und R3 dasselbe wie oben bedeuten), mit Carbonsäureanhydriden der allgemeinen Formel 9,
(R4CO)2O 9
worin R4 dasselbe wie bei den Verbindungen der allgemeinen Formel 8 bedeutet, jeweils im Überschuss und in der Siedehitze, gegebenenfalls zusammen mit Pyridin oder einem aprotischen, hochsiedenden
Lösungsmittel, wie zum Beispiel Toluol oder XyIoI,
Behandlung des gebildeten Niederschlages mit wässerig-ethanolischer Natronlauge oder mittels konz. wässeriger Ammoniak-Lösung, jeweils bis zu einem pH-Wert von
8-10, unter Erwärmen, nachfolgendem Ansäuern mit verdünnter Salzsäure unter Bildung der Verbindungen der allgemeinen Formel 1b,
worin X, Y, R1 und R3 dasselbe wie oben bedeuten und R4 die bei den Verbindungen der allgemeinen Formel 8 angegebene Bedeutung aufweist
oder
D)
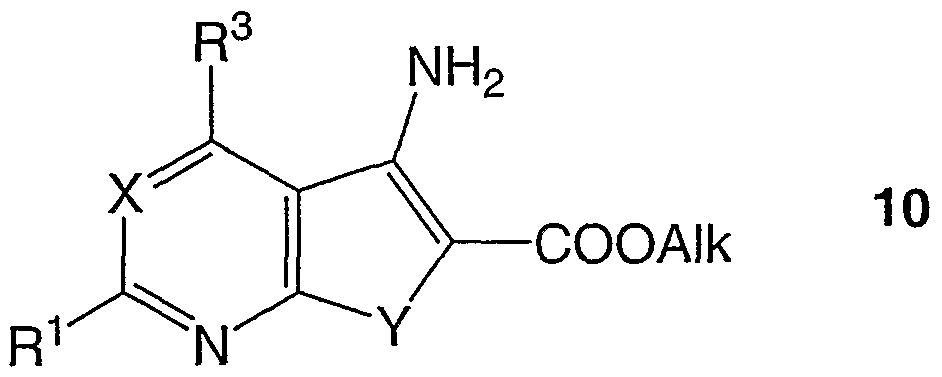
- Umsetzung der aus den Verbindungen der allgemeinen Formel 5 und Chloressigsäurealkylestem, C1-Cs, darstellbaren bekannten oder nach prinzipiell bekannten Methoden darstellbaren Verbindungen der allgemeinen Formel 10,
worin X, Y, R
1 und R
3 dasselbe wie oben und Alk Alkyl, CrC
3, bedeuten, mit Benzoylisothiocyanat, PhCONCS, in aprotischen, dipolaren Lösungsmitteln, vorzugsweise Aceton oder Dioxan unter Erwärmen zu den Verbindungen der allgemeinen Formel 11 ,
worin X, Y, R1 und R3 dasselbe wie oben und Alk Alkyl, CrC3, bedeuten,
- Umsetzung der Verbindungen der allgemeinen Formel 11 mit Natrium- oder
Kaliumhydroxid in protischen oder aprotischen polaren Lösungsmitteln oder
Lösungsmittelgemischen unter Erwärmen,
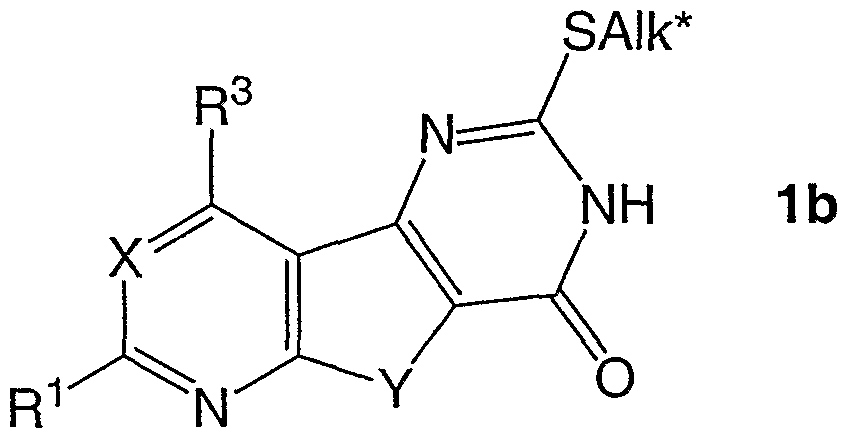
Zugabe von Brom- oder lodalkanen, -alkenen oder alkinen, C3-C7ZU der erkalteten alkalischen Reaktionslösung, mäßiges Erwärmen bis zur Beendigung der Umsetzung,
Ansäuern mit verdünnter Salzsäure unter Entstehung der Verbindungen der allgemeinen Formel 1 b,
worin X, Y1 R1 und R3 dasselbe wie oben und Alk* (Ci-8 )Alkyl, (C2-8,)Alkenyl, (C3-6) Alkinyl, jeweils unverzweigt oder gegebenenfalls verzweigt und ggf. mit einem Rest wie -CN, -SCN, -NO2, Phenyl und (C3-7)Cycloalkyl substituiert ist, bedeuten,
oder
E)
- Umsetzung von Verbindungen der allgemeinen Formel 1b bei denen R4 SAIk* und
Alk* (Ci-8)Alkyl, (C2-8,)Alkenyl, (C3-6) Alkinyl, jeweils unverzweigt oder verzweigt und ggf. mit einem Rest wie -CN, -SCN, -NO2, Phenyl und (C3-7)CycloaIkyl substituiert ist, bedeutet, mit annähernd äquimolarer Menge Dihydrogenperoxid in Essigsäure oder
Ameisensäure unter dc-Kontrolle bei Raumtemperatur oder mit KIO4 in protischen oder aprotischen, dipolaren Lösungsmitteln unter
Erwärmen zu jenen Verbindungen der allgemeinen Formel 1 b, worin X, Y, R1 und R3 dasselbe wie oben bedeuten und R4 SOAIk* und Alk* (d-8)Alkyl, (C2-8,)Alkenyl, (C3-6) Alkinyl, jeweils unverzweigt oder verzweigt und ggf. mit einem Rest wie -CN, -SCN, -NO2,
Phenyl und (Cs-7) Cycloalkyl substituiert ist, bedeutet
oder
F)
- Umsetzung jener Verbindungen der allgemeinen Formel 1b bei denen R4 SAIk* oder SOAIk* und Alk* (Ci-8 )Alkyl, (C2-8,)Alkenyl, (C3-6) Alkinyl, jeweils unverzweigt oder verzweigt und ggf. mit einem Rest wie -CN, -SCN, NO2, Phenyl und (C3-7) Cycloalkyl substituiert ist, bedeutet, mit überschüssigem Dihydrogenperoxid in Essigsäure oder Ameisensäure unter dc- Kontrolle bei Raumtemperatur, gegebenenfalls auch unter mäßigem Erwärmen oder mit KMnO4 in protischen oder aprotischen, dipolaren Lösungsmitteln unter Erwärmen zu den Verbindungen der allgemeinen Formel 1b, worin X, Y, R1 und R3 dasselbe wie oben bedeuten und R4SO2AIk* und Alk*(C1-8) Alkyl, (C2-8,)Alkenyl, (C3-6) Alkinyl, jeweils unverzweigt oder verzweigt und ggf. mit einem Rest wie -CN, -SCN, NO2, Phenyl und (C3-7)Cycloalkyl substituiert ist, bedeutet.
Entsprechend der vorliegenden Erfindung können die unter A) bis F) beschriebenen Verfahren innerhalb weiter Grenzen variiert werden.
Weitere Ausführungsformen der Erfindung können beispielsweise darin bestehen,
dass die Darstellung der tricyclischen Pyrimidin-2,4-dione der allgemeinen Formel 1a bzw. die der tricyclischen Pyrimidin-4-one der allgemeinen Formel 1 b unter Verwendung eines Mikrowellengeräts realisiert wird,
Methodenbeschreibung der Hemmung der TNFα Freisetzung nach LPS Stimulation von humanem Vollblut
Die Stimulierung isolierter Leukozyten für die Freisetzung von Zytokinen kann auf verschiedenen Wegen erfolgen. Lipopolysaccharide (LPS) stellen einen Stimulus für die Untersuchung der Freisetzung von TNFα dar. LPS ist Bestandteil bakterieller Zellwände und wird beim Abtöten der Bakterien (durch Antibiotika oder das natürliche Immunsystem) freigesetzt. LPS stimuliert insbesondere die Aktivität phagozytierender Leukozyten (Gewebsmakrophagen, Granulozyten, Monozyten) und verursacht die Infiltration von Leukozyten vom peripheren Blut in das betroffene Gewebe. Ein Zytokin von besonderer Bedeutung für diese Mechanismen ist TNFα, das in großen Mengen durch die betroffenen Zellen sezemiert wird. Hauptquelle dabei sind Monozyten und Makrophagen. TNFα initiiert und prolongiert den Entzündungsprozess im Zusammenspiel mit anderen Mediatoren.
Für die Untersuchung des Effektes auf die LPS-induzierte TNFα-Freisetzung wurde eine Methode verwendet, die von MARX et al. {Marx D, Tassabehji M, Heer S, Hüttenbrink KB, Szelenyi I (2002) Pulmonary Pharmacology & Therapeutics 15 7-15) beschrieben wurde. Die Methode in Kürze: Humanes Blut wird von verschiedenen Spendern entnommen, durch Zusatz von 10 mM Na-Citrat ungerinnbar gemacht und 1 :5 mit RPMI 1640 Zellkulturmedium verdünnt. Die Testsubstanzen werden den Blutproben in verschiedenen Konzentrationen zugefügt. 15 Minuten später werden die Leukozyten durch Zusatz von Lipopolysacchariden (LPS) aus Salmonella abortus equi in einer Endkonzentration von 1μg/ml stimuliert. Nach Inkubation der Testansätze für 24 Stunden bei 37°C. und unter 5% CO2 in wassergesättigter Luft, wird das Blut zentrifugiert und die Konzentration an TNFα im zellfreien Überstand unter Verwendung eines käuflichen ELISA (BD Biosciences) nach Angaben des Herstellers exakt vermessen.
ICδo-Werte im Bereich von 10"6"bis 10'10 M wurden für die in der Erfindung beschriebenen Substanzen bestimmt.
Die Verbindungen der allgemeinen Formeln 1 a und 1 b sind schwache Inhibitoren der Phosphodiesterase 4 und äußerst starke Inhibitoren der Freisetzung von TNFα.
Die nachfolgende Auflistung beinhaltet erfindungsgemäße Substanzen, die im TNFα- Hemmassay bei 1OnM die TNFα-Freisetzung >20% hemmen:
52 9-Methyl-7-naphthalen-2-yl-thieno[2,3-d:4
)5-d']dipyrimidin-2
)4-(1 H,3H)-dion
58 9-Methyl-7-biphenyl-2-ethyl-thieno[2,3-d:4,5-d']dipyrimidin-4(3H)-on
Die nachfolgende Auflistung beinhaltet erfindungsgemäße Substanzen, die im TNFα- Hemmassay einen IC50-Wert von <10 nM aufweisen:
Besonders bevorzugte Verbindungen der vorliegenden Erfindungen sind nachfolgend aufgelistet:
Formel 1 a mit Y = S, wobei die Substituenten Ri , R3, X die folgenden Bedeutungen haben:
Formel 1 a mit Y = O und R1 = Ph, X = C-H und R3 = Me.
Formel 1 b mit Y = S , wobei die Substituenten R1 , X , R3 ,R4 die folgenden Bedeutungen haben:
Ganz speziell seien die nachfolgenden Verbindungen als bevorzugt genannt:
7-(4-Methoxy-phenyl)-9-methyl-pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-2,4(1H,3H)- dion;
9-Methyl-7-phenyl-pyrido[3l J2':4J5]thieno[3l2-d]pyrimidin-2,4(1 H]3H)-dion;
7-(4-Methoxy-phenyl)-9-methyl-thieno[2,3-d:4J5-d']dipyrimidin-2,4-(1 H,3H)-dion;
9-Methyl-7-(4-methyl-phenyl)-thieno[2J3-d:4,5-d>]dipyrimidin-2,4-(1 H,3H)-dion;
7-(4-Cyano-phenyl)-9-methyI-thieno[2I3-d:4,5-d']dipyrimidin-2)4-(1 Hl3H)-clion;
7-(4-Brom-2-fluor-phenyl)-9-methyl-thieno[2,3-d:4l5-d']dipyrimidin-2,4-(1 H,3H)-dion;
9-Methyl-7-biphenyI-2-ethyl-thieno[2,3-d:4,5-d']dipyrimidin-4(3H)-on
2-Ethyl-9-methyl-7-phenyl-pyrido[3',2':4,5]thieno[3,2d]pyrimidin-4(3H)-on
7-(3)4-Dimethoxy)-9-methyl-pyndo[3',2':4,5]thieno[3,2-d]pyrinnidin-2,4(1 Hl3H)-dion;
7-Phenyl-2-n-propyl-9-(pyrid-3-yl)-pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-on;
7-(4-Fluor-phenyl)-9-methyl-pyrido[3',2':4,5]thieno[3>2-d]pyrimidin-2,4(1 H)3H)-dion;
7-(4-Methoxy-phenyl)-9-methyi-pyrido[3',2':4l5]thieno[3,2-d]pyrimidin-2,4(1 H,3H)- dion;
7-Phenyl-pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-on-2-carbonsäure;
9-Methyl-7-phenyl-pyrido[3l >2':4>5]thieno[3I2-d]pyrimidin-2,4(1 H,3H)-dion;
7-Phenyl-pyrido[3',2':4,5]thieno[3I2-d]pyrimidin-4(3H)-on-2-carbonsäure;
7,9-Dimethyl-pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-2,4(1 H,3H)-dion;
Die Erfindung wird durch die nachfolgenden Beispiele näher erläutert:
Beispiel 1
7-(3,4-DimethoxyphenyI)-9-methyI-pyrido[3'52':4,5]thieno[3,2-c(Ipyrimidin-2,4- dion
(allgemeine Formel 1a: R1=S^-(MeO)2-Ph, X=C-H, R3=Me, Y=S)
3.0 g (8.7 mmol) 3-Amino-6-(3,4-dimethoxyphenyl)-4-methyl-thieno[2,3-Jb]pyridin-2- carbonsäureamid (Beispiel 62; Experiment 7a; allgemeine Formel 7: Ri=3,4-(CH3O)2- Ph, X=C-H, R3=Me, Y=S) werden in 100 ml abs. Dioxan suspendiert. Anschließend werden 1.1 ml (8.7 mmol) Chlorameisensäuretrichlormethylester (Diphosgen) zugegeben und 3 Stunden unter Rückfluß erhitzt. Nach Abkühlen wird der Niederschlag abgesaugt und anschließend in 50 ml Wasser suspendiert. Die Suspension wird mit konz. Ammoniak-Lösung auf pH 8 eingestellt und der Niederschlag abgesaugt, mit wenig Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 °C getrocknet.
Ausbeute: 3.08 g (96 %); HPLC tR: 13.8 min, Reinheit: 99.9 % (s. a in Tab. 1); MS (ESl, m/e): 368 [M-H]"
Ausgehend von den nachfolgend beschriebenen Vorstufen werden durch Umsetzung der entsprechenden Verbindungen der allgemeinen Formel 7 mit Chlorameisen- säuretrichlormethylester (Diphosgen) analog Beispiel 1 die folgenden Beispiele 2-8, 11-14, 16-38, 42 und 45-52 erhalten:
Beispiel 2
7-(4-l\/lethoxyphenyl)-9-methyl-pyrido[3',2':4,5]thieno[3,2-c/lpyrJmidin-2,4-dion (allgemeine Formel 1a: R1=4-MeO-Ph, X=C-H, R3=Me, Y=S)
Analog Beispiel 1 aus 3-Amino-6-(4-methoxyphenyl)-4-methyl-thieno[2,3-ö]pyridin-2- carbonsäureamid (Beispiel 62; Experiment 7b; allgemeine Formel 7: R-ι=4-MeO-Ph, X=C-H, R3=Me1 Y=S) (s. Tab. 1)
Beispiel 3
7-(4-Fluorphenyl)-9-methyl-pyrido[3',2':4,5]thieno[3,2-c(lpyrimidin-2,4-dion (allgemeine Formel 1a: R1=4-F-Ph, X=C-H, R3=Me5 Y=S)
Analog Beispiel 1 aus 3-Amino-6-(4-fluorphenyl)-4-methyl-thieno[2,3-jb]pyridin-2- carbonsäureamid (Beispiel 62; Experiment 7c; allgemeine Formel 7: R-ι=4-F-Ph, X=C-H, R3=Me, Y=S) (s. Tab. 1)
Beispiel 4
9-Methyl-7-phenyl-pyrido[3^2':4,5]thieno[3,2-c(]pyrimidin-2,4-dion (aligemeine Formel 1a: R1=Ph, X=C-H, R3=Me, Y=S)
Analog Beispiel 1 aus 3-Amino-4-methyl-6-phenyl-thieno[2,3-/?]pyridin-2- carbonsäureamid (Beispiel 62; Experiment 7d; allgemeine Formel 7: Ri=Ph, X=C-H, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 5
7-Methyl-9-phenyl-pyrido[3',2':4,5]thieno[3,2-c(]pyrimidin-2,4-dion (Beispiel 5; allgemeine Formel 1a: Ri=Me5 X=C-H, R3=Ph, Y=S)
Analog Beispiel 1 aus 3-Amino-6-methyl-4-phenyl-thieno[2,3-£>]pyridin-2-carbon- säureamid (Beispiel 62; Experiment 7e; allgemeine Formel 7: Ri=Me, X=C-H, R3=Ph, Y=S) (S. Tab. 1)
Beispiel 6
7,9-Diphenyl-pyrido[3',2':455]thieno[3,2-d|pyrimidin-2,4-dion (allgemeine Formel 1a: R1=Ph, X=C-H, R3=Ph, Y=S)
Analog Beispiel 1 aus 3-Amino-4,6-diphenyl-thieno[2,3-ό]pyridin-2-carbonsäureamid (Beispiel 62; Experiment 7f; allgemeine Formel 7: Ri=Ph, X=C-H, R3=Ph, Y=S) (s. Tab. 1)
Beispiel 7
7,9-Dimethyl-pyrido[3',2':4,5]thieno[3,2-cdpyrimidin-2,4-dion (allgemeine Formel 1a: R1=Me, X=C-H, R3=Me, Y=S)
Analog Beispiel 1 aus 3-Amino-4,6-dinnethyl-thieno[2,3-ib]pyπdin-2-carbonsäureamici (Beispiel 62; Experiment 7g; allgemeine Formel 7: Ri=Me, X=C-H, R3=Me, Y=S) (s. Tab. 1)
Beispiel 8
7,9-Dimethyl-8-(4 -nitrobenzyl)-pyrido[3',2':4,5]thieno[3,2-cGpyrimidin-254-dion (allgemeine Formel 1a: Ri=Me, X=C-CH2-Ph-4'-NO2 , R3=Me5 Y=S)
Analog Beispiel 1 aus 3-Amino-4,6-dimethyl-5-(4'-nitrobenzyl)-thieno[2,3-jb]pyridin-2- carbonsäureamid (Beispiel 62; Experiment 7h; allgemeine Formel 7: Ri=Me, X=C- CH2-Ph-^-NO2, R3=Me, Y=S) (s. Tab. 1)
Beispiel 9
9-Methyl-7-phenyl-pyrido[3
',2
':4,5]furo[3
J2-c0pyrimidin-2,4-dion (allgemeine Formel 1a:
R
3=Me, Y=O)
Zu 1.0 g (3.4 mmol) 3-Amino-4-methyl-6-phenyl-furo[2,3-b]pyridin-2-carbonsäure- ethylester (allgemeine Formel 10: Ri=Ph, X=C-H, R3=Me, Y=O)* in 20 ml abs. Aceton werden 1.0 g (6.8 mmol) Benzoylisocyanat gegeben und 30 Minuten bei Raumtemperatur gerührt. Das Lösungsmittel wird entfernt und der nach Zugabe von Wasser ausgefallene Niederschlag abgesaugt und trocken gesaugt. Der Niederschlag wird in 6.8 ml (6.8 mmol) Natriumhydroxid-Lösung (1 M) und 20 ml Ethanol 15 Minuten unter Rückfluss erhitzt. Das Lösungsmittel wird entfernt und der Rückstand mit 100 ml abs. Dichlormethan aufgenommen. Nach Zugabe von 0.44 ml (5.1 mmol) Oxalylchlorid wird 1 Stunde bei Raumtemperatur gerührt. Das Lösungsmittel wird entfernt und der Rückstand mit Wasser suspendiert. Der entstandene Niederschlag wird abgesaugt, mit wenig Ethanol gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet.
Ausbeute: 0.85 g (85 %); HPLC tR: 15.0 min, Reinheit: 85.1 % (s. a in Tab. 1); MS (ESI, m/e): 294 [M+H]+ (* Lit.: Wagner G., Prantz J. Pharmazie (1990); 45, 213-214)
Beispiel 10
θ-Trifluormethyl-y^-methoxy-phenyO-pyridoja'^'^jδlthienotS^-dlpyrimidin-
2,4-dion*
(allgemeine Formel 1a: R1=^MeO-Ph, X=C-H, R3=CF3, Y=S)
Zu 1 ,55 g (5 mmol) 4-Trifluormethyl-2-mercapto-6-(4-methoxyphenyl)-pyridin-3- carbonitril (Beispiel 61 ; Experiment 5i; allgemeine Formel 5: R1=4-MeO-Ph, X=C-H, R3=CF3, Y=S) in 25 ml Butoxyethoxyethanol werden 1 ,00 g (5.5 mmol) N- chloracetylurethan sowie 0.96 ml (12.5 mmol) Triethylamin gegeben und der Ansatz 30 Minuten bei 80 °C gerührt. Anschließend wird der gerührte Ansatz für 30- 60 Minuten auf 180 °C erwärmt. Nach Abkühlen wird der Niederschlag abgesaugt, erst mit Ethanol, anschließend mit Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet.
Ausbeute: 1.62g (82%); HPLC tR: — min, Reinheit: 99.9 % (s. a in Tab. 1); MS (ESI, m/e): 394 [M+H]+
(* in Anlehnung an: Shestopalov, A. M.; Nikishin, K. G.; Gromova, A. V.; Rodinovskaya, L A. Russ.Chem. Bull. (2003), 52(10), 2203-2206)
Ausgehend von den nachfolgend beschriebenen Vorstufen werden durch Umsetzung der entsprechenden Verbindungen der allgemeinen Formel 5 mit N-chloracetyl- urethan analog Beispiel 10 die folgenden Beispiele 39-41 , 43 und 44 und erhalten:
Beispiel 11
9-Methyl-7-(thiophen-2-yl)-thieno[2,3-d:455-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=Thiophen-2-yl, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-(thiophen-2-yl)-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7j; allgemeine Formel 7: Ri=Thiophen-2-yl, X=N1 R3=Me1 Y=S) (S. Tab. 1)
Beispiel 12
θ-Methyl-T^-nitro-phenylHhieno^^-d^^-d'ldipyrimidin-a^iH^HJ-dion (allgemeine Formel 1a: Ri=4-NO2-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-(4-nitro-phenyl)-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7k; allgemeine Formel 7: R-ι=4-Nθ2-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 13
9-Methyl-7-naphthalen-1-yl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1 H,3H)-dion (allgemeine Formel 1a: Ri=Naphthalen-1-yl, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4~methyl-2-naphthalen-1-yl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7I; allgemeine Formel 7: Ri=Naphthalen-1-yl, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 14
7-(Chinolin-6-yl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R^Chinolin-6-yl, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(chinolin-6-yl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7m; allgemeine Formel 7: R-ι=Chinolin-6-yl, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 15
7-(4-Amino-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=4-NH2-Ph, X=N, R3=Me, Y=S)
5.5 g (15.5 mmol) 9-Methyl-7-(4-nitro-phenyl)-thieno[2,3-d:4,5-d']dipyπmidin-2,4- (1H,3H)-dion (Beispiel 12, allgemeine Formel 1a: R1=4-NO2-Ph, X=N1 R3=Me, Y=S) werrden mit 9.9 g (56.9 mmol) Natriumdithionit und 31.5 ml Glykolmonomethylester in 30 ml Wasser suspendiert und 3 Stunden unter Rückfluß erhitzt. Danach werden 20 ml Wasser und 20 ml konz. HCl zugetropft und es wird weitere 30 Minuten unter Rückfluss erhitzt. Nach Erkalten wird auf 100 ml Eiswasser gegossen und mit Natriumcarbonat alkalisiert. Der Niederschlag wird abgesaugt, mit Wasser gewaschen und getrocknet.
Ausbeute 4.8 g (95%); HPLC tR: 10.6 min, Reinheit: 93.0 % (s. a in Tab. 1); MS (ESI, m/e): 324 [M+H]~
Beispiel 16
g-Wlethyl-T^-phenyloxy-phenyO-thienota^-d^^-d'ldipyrimidin-a^-OH^H)- dion (allgemeine Formel 1a: Ri-=4-Ph-O-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-(4-phenyloxy-phenyl)-thieno[2,3- d]pyrimidin-6-carboxamid (Beispiel 62; Experiment 7o; allgemeine Formel 7: R-i=4- Ph-O-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 17
9-Methyl-7-(4-n-propyl-phenyl)-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R1=4-n-Pr-Ph, X=N, R3=Me5 Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-(4-n-propyl-phenyl)-thieno[2,3-d]pyrimidin- 6-carboxamid (Beispiel 62; Experiment 7p; allgemeine Formel 7: R-ι=4-n-Pr-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 18
y^-Trifluormethyl-phenyO-θ-methyl-thienofa^-d^^-d'ldipyrimidin^^-CI H^H)- dion (allgemeine Formel 1a: R1=^CF3-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-trifluormethyl-phenyl)-4-methyl-thieno[2,3- d]pyrimidin-6-carboxamid (Beispiel 62; Experiment 7q; allgemeine Formel 7: Ri=4- CF3-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 19
7-(4-Brom-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=4-Br-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-brom-phenyl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7r; allgemeine Formel 7: R-ι=4-Br-Ph, X=N, R3-=Me, Y=S) (s. Tab. 1)
Beispiel 20
4-(9-Methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion-7-yl)- benzoesäureethylester
(allgemeine Formel 1a: R1=EtOCO-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 4-(5-Amino-6-carbamoyl-4-methyl-thieno[2,3-d]pyrimidin-2-yl)- benzoesäureethylester (Beispiel 62; Experiment 7s; allgemeine Formel 7: R1=EtOCO-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 21
9-l\Λethyl-7-(4-iso-propoxy-phenyl)-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1 H,3H)- dion (allgemeine Formel 1a: R1=4-iso-PrO-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-iso-propoxy-phenyl)-4-methyl-thieno[2,3- d]pyrimidin-6-carboxamid (Beispiel 62; Experiment 7t; allgemeine Formel 7: R-ι=4- iso-PrO-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 22
7-(4-Iod-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R1=4-I-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-lod-phenyl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7u; allgemeine Formel 7: Ri=4-I-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 23
θ-Methyl^^-methyl-phenyO-thienota^-d^^-d'ldipyrimidin^^-OH^HJ-dion (allgemeine Formel 1a: Ri=4-Me-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-(4-methyl-phenyl)-thieno[2,3-d]pyrimidin- 6-carboxamid (Beispiel 62; Experiment 7v; allgemeine Formel 7: Ri=4-Me-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 24
7-(4-Chlor-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R1=4-CI-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-chlor-phenyl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7w; allgemeine Formel 7: Ri=4-Cl-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 25
T^-Cyano-phenyO-θ-methyl-thienota^-d^^-d'ldipyrimidin^j^iH^HHion (allgemeine Formel 1a: R1=4-CN-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-cyano-phenyl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7x; allgemeine Formel 7: R1=^CN-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 26
7-(3-Fluor-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H53H)-dion (allgemeine Formel 1a: R1=S-F-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(3-fluor-phenyl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7y; allgemeine Formel 7: Ri=3-F-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 27
7-(4-Methoxy-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R1=4-MeO-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-methoxy-phenyl)-4-methyl-thieno[2,3-d]- pyrimidin-6-carboxamid (Beispiel 62; Experiment 7z; allgemeine Formel 7: R-ι=4- MeO-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 28
7-(4-Brom-2-fluor-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)- dion (allgemeine Formel 1a:
X=N, R
3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-brom-2-fluor-phenyl)-4-methyl-thieno[2,3-d]- pyrimidin-6-carboxamid (Beispiel 62; Experiment 7aa; allgemeine Formel 7: Ri=4- Br,2-F-Ph, X=N, R
3=Me, Y=S) (s. Tab. 1)
Beispiel 29
θ-Methyl-T-phenyl-thienop^-d^^-d'Jdipyrimidin-a^-CIH^HJ-dion (allgemeine Formel 1a: R1=Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-phenyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ab; allgemeine Formel 7: Ri=Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 30
7,9-DimethyI-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=Me, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2,4-dimethyl-thieno[2,3-d]pyrimidin-6-carboxamid (Beispiel 62; Experiment 7ac; allgemeine Formel 7: Ri=Me, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 31
7-Benzyl-9-methyl-thieno[2,3-d:4,5-d'3dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a:
X=N, R
3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-benzyl-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ad; allgemeine Formel 7: R1=Bn, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 32
T-Cyclopropyl-θ-methyl-thieno^jS-di^δ-d'jdipyrimidin-a^-CI H^HJ-dion (allgemeine Formel 1a: Ri=CyPr, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-cyclopropyl-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ae; allgemeine Formel 7: Ri=CyPr, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 33
7-CyclohexyI-9-methyl-thieno[2,3-d:4,5-d']dipyπmidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=CyHeX, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-cyclphexyl-4-methyI-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7af; allgemeine Formel 7: R1=CyHeX, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 34
T-Benzhydryl-S-methyl-thienoI^S-dϊ^δ-d'Jdipyrimidin-a^-OH^HHion (allgemeine Formel 1a: Ri=Benzhydryl, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-benzhydryl-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ag; allgemeine Formel 7: Ri=Benzhydryl, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 35
9-Methyl-7-pyridin-4-yl-thieno[2,3-d:4,5-d'3dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R^Pyridin-4-yl, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-pyridin-4-yl-thieno[2,3-d]pyπmidin-6- carboxamid (Beispiel 62; Experiment 7ah; allgemeine Formel 7: R-ι=Pyridin-4-yl, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 36
θ-Methyl-T-styryl-thieno^^-d^^-d'ldipyrimidin^^-CIHjSHJ-dion (allgemeine Formel 1a: Ri=Styryl, X=N, R3=WIe, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-styryl-thieno[2,3-d]pyrimidin-6-carboxamid (Beispiel 62; Experiment 7ai; allgemeine Formel 7: R-ι=Styryl, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 37
7-(2-Fluor-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dlon (allgemeine Formel 1a: R1=2-F-Ph, X=N, R3=IVIe, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(2-fluor-phenyl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7aj; allgemeine Formel 7: R1=2-F-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 38
7-(2,6-Difluor-phenyl)-9-methyl-thieno[2,3-d:455-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=2,4-F2-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(2,4-difluor-phenyl)-4-methyl-thieno[2,3-d]pyrimidin- 6-carboxamid (Beispiel 62; Experiment 7ak; allgemeine Formel 7: R1=2,4-F2-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 39
y^-Brom-phenyO-θ-methyl-thieno^^-d^^-d'ldipyrimidin^^i H.SHJ-dion (allgemeine Formel 1a: Ri=2-Br-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 10 aus 2-(2-Brom-phenyl)-4-mercapto-6-methyl-pyrimidin-5- carbonitril (Beispiel 61 ; Experiment 5al; allgemeine Formel 5: Ri=2-Br-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 40
7-(3-Methoxy-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=3-MeO-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 10 aus 4-Mercapto-2-(3-methoxy-phenyI)-6-methyl-pyrimidin-5- carbonitril (Beispiel 61 ; Experiment 5am; allgemeine Formel 5: R1=S-MeO-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1 )
Beispiel 41
7-(2-Methoxy-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R1=2-MeO-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 10 aus 4-Mercapto-2-(2-methoxy-phenyl)-6-methyl-pyrimidin-5- carbonitril (Beispiel 61 ; Experiment 5an; allgemeine Formel 5: Ri=2-MeO-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 42
9-Methyl-7-(2-methyl-phenyl)-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R1=2-Me-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-4-methyl-2-(2-methyl-phenyl)-thieno[2,3-d]pyrimidin- 6-carboxamid (Beispiel 62; Experiment 7ao; allgemeine Formel 7: Ri=2-Me-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 43
θ-Methyl-T-pyridin-S-yl-thienop^-d^^-d'ldipyrimidin-aAOH.SHJ-dion (allgemeine Formel 1a: R^Pyridin-3-yI, X=N, R3=Me, Y=S)
Analog Beispiel 10 aus 4-Mercapto-6-methyl-2-pyridin-3-yl-pyrimidin-5-carbonitril (Beispiel 61 ; Experiment 5ap; allgemeine Formel 5: Ri=Pyridin-3-yl, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 44
7-(254-Dimethoxy-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)- dion
(allgemeine Formel 1a:
X=N, R
3=Me, Y=S)
Analog Beispiel 1 aus 4-Mercapto-2-(2,4-dimethoxy-phenyl)-6-methyl-pyrimidin~5- carbonitril (Beispiel 61 ; Experiment 5aq; allgemeine Formel 5:
X=N, R
3=Me, Y=S) (s. Tab. 1)
Beispiel 45
7-Furan-2-yl-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a:
Analog Beispiel 1 aus 5-Amino-2-furan-2-yl-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ar; allgemeine Formel 7: Ri=Furan-2-yl, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 46
7-(3,4-Dimethoxy-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)- dion
(allgemeine Formel 1a: Ri=3,4-(MeO)2-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(3,4-dimethoxy-phenyl)-4-methyl-thieno[2,3- d]pyπmidin-6-carboxamid (Beispiel 62; Experiment 7as; allgemeine Formel 7:
Beispiel 47
7-Methyl-9-phenyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: Ri=Me, X=N, R3=Ph, Y=S)
Analog Beispiel 1 aus 5-Amino-2-methyl-4-phenyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7at; allgemeine Formel 7: Ri=Me, X=N, R3=Ph, Y=S) (S. Tab. 1)
Beispiel 48
9-Methyl-7-(3-methyl-phenyl)-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1H,3H)-dion (allgemeine Formel 1a: R1=3-Me-Ph, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(3-methyl-phenyl)~4-methyl-thieno[2,3-d]pyrimidin~ 6-carboxamid (Beispiel 62; Experiment 7au; allgemeine Formel 7: Ri=3-Me-Ph, X=N, R3=Me1 Y=S) (S. Tab. 1)
Beispiel 49
9-Methyl-7-(4-(methylsulfonyl)-phenyI)-thieno[2,3-d:4,5-d']dipyrirnidin-2,4-
(1H,3H)-dion
(allgemeine Formel 1a: R1=4-WIe-SO2-Ph, X=N, R3=WIe, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-(methylsulfonyl)-phenyl)-4-methyl-thieno[2,3- d]pyrimidin-6-carboxamid (Beispiel 62; Experiment 7av; allgemeine Formel 7: Ri=4- Me-SO2-Ph, X=N1 R3=Me, Y=S) (s. Tab. 1)
Beispiel 50
7-Biphen-4-yI-9-methyl-thieno[2,3-d:455-d']diρyrimidin-2,4-(1H)3H)-dion (allgemeine Formel 1a: R^Biphen-4-yl, X=N, R3=Me, Y=S)
Analog Beispiel 1 aus 5-Amino-2-biphen-4-yl-4-methyl-thieno[2,3-d]pyrimidin-6~ carboxamid (Beispiel 62; Experiment 7aw; allgemeine Formel 7: Ri=Biphen-4-yl, X=N1 R3=Me1 Y=S) (S. Tab. 1)
Beispiel 51
7-(4-t-Butyl-phenyl)-9-methyl-thieno[2,3-d:4,5-d']dipyrimidin-2,4-(1HJ3H)-dion (allgemeine Formel 1a: R1=4-t-Bu-Ph, X=N, R3=WIe, Y=S)
Analog Beispiel 1 aus 5-Amino-2-(4-t-butyl-phenyl)-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ax; allgemeine Formel 7: R1=4-t-Bu-Ph, X=N, R3=Me, Y=S) (s. Tab. 1)
Beispiel 52
θ-Methyl^-naphthalen^-yl-thieno^-d^^-d'ldipyrimidin^AOH^HHion (allgemeine Formel 1a:
Analog Beispiel 1 aus 5-Amino-2-naphthalen-2-yl-4-methyl-thieno[2,3-cl3pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ay; allgemeine Formel 7: Ri=Naphthalen-2-yl, X=N, R
3=Me, Y=S) (s. Tab. 1)
Beispiel 53
2-EthyI-9-methyI-7-phenyl-pyrido[3',2':4,5]thieno[3,2-c/lpyrimiciin-4-on (allgemeine Formel 1b: Ri=Ph, X=C-H5 R3=Me, R4=Et, Y=S)
Es werden 0.86 g (3.0 mmol) 3-Amino-6-phenyI-4-methyl-thieno[2,3-6]pyridin-2~ carboxamid (Beispiel 62; Experiment 7d; allgemeine Formel 7: Ri=Ph, X=C-H, R3=Me, Y=S) und 5.0 ml (39.2 mmol) Propionsäureanhydrid in 20 ml Toluol 2 Stunden unter Rückfluß erhitzt. Nach Abkühlen wird der Niederschlag abgesaugt, mit ca. 5 m( Ethanol gewaschen und trocken gesaugt. Der Rückstand wird in 5 ml (15.0 mmol) Natriumhydroxid-Lösung (3N) 15 Minuten unter Rückfluß erhitzt. Nach Abkühlen werden 5 ml Wasser zugegeben und es wird mit Essigsäure (10 %) neutralisiert. Anschließend wird der Niederschlag abgesaugt, mit ca. 10 ml Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet. Ausbeute: 0.29 g (30 %); HPLC tR: 17.6 min, Reinheit: 96.2 % (s. Tab. 2); MS (ESI, m/e): 322 [M+H]+
Beispiel 54
9-Methyl-7-phenyl-2-propyl-pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4-on (allgemeine Formel 1b: Ri=Ph, X=C-H, R3=Me, R4=πPr, Y=S)
Analog Beispiel 53 aus 3-Amino-6-phenyl-4-methyl-thieno[2,3-ö]pyridin-2-carboxamid
(Beispiel 62; Experiment 7d; allgemeine Formel 7: Ri=Ph, X=C-H, R3=Me, Y=S) und
Buttersäureanhydrid.
Es wird bei 170 0C gerührt. Nach Abkühlen wird mit Natriumhydroxid alkalisiert und kurz erwärmt. Der Niederschlag wird abgesaugt, mit Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet.
Ausbeute: 0.55 g (91 %); HPLC tR: 20.3 min, Reinheit: 46.8 % (s. Tab. 2); MS (ESI, m/e): 336 [MH-H]+
Beispiel 55
7-(3,4-Dimethoxyphenyl)-9-methy[-2-propyl-pyrido[3',2':4,5]thieno[3,2- d3pyrimidin-4-on (allgemeine Formel 1b: Ri=3,4-(MeO)2-Ph, X=C-H, R3=Me, R4=nPr, Y=S)
Analog Beispiel 53 aus 3-Amino-6-(3,4-dimethoxyphenyl)-4-methyl-thieno[2,3~£>]- pyridin-2-carboxamid (Beispiel 62; Experiment 7a; allgemeine Formel 7: Ri=3,4-
(MeO)2-Ph, X=C-H, R3=Me, Y=S) und Buttersäureanhydrid.
Der Rückstand wird in 5 ml (15.0 mmol) Natriumhydroxid-Lösung (3N) und 5 ml
Ethanol 6 Stunden unter Rückfluß erhitzt. Nach Ansäuern mit Salzsäure (10 %) wird der Niederschlag mit Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 °C getrocknet.
Ausbeute: 0.59 g (47 %); HPLC tR: 17.4 min, Reinheit: 89.8 % (s. Tab. 2);
MS (ESI, m/e): 396 [M+H]+
Beispiel 56
2-Ethylthio-9-methyl-7-phenyl-pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-4-on (allgemeine Formel 1b: Ri=Ph, X=C-H, R3=WIe, R4=SEt, Y=S)
Zu 2.00 g (4.2 mmol) 3-(3-Benzoylthioureido)-4-methyl-6-phenyl-thieno[2,3-/?]pyridin- 2-carbonsäureethylester (Beispiel 64; Experiment 11 ; allgemeine Formel 11 : Ri=Ph, X=C-H, R3=Me, Y=S) in 25 ml Ethanol werden 10.5 ml (10.5 mmol) Natriumhydroxid- Lösung (1N) gegeben und 30 Minuten unter Rückfluß erhitzt. Nach Zugabe von 10 ml Λ/,Λ/-Dimethylformamid, 5 ml (5 mmol) Natriumhydroxid-Lösung (1 N) und 0,50 ml lodethan (6.2 mmol) wird 30 Minuten bei Raumtemperatur gerührt. Der nach Ansäuern mit verd. Salzsäure ausgefallene Niedeschlag wird abgesaugt, mit Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 °C getrocknet. Ausbeute: 1.23 g (83 %), HPLC tR: 18.5 min, Reinheit: 98.5 % (s. Tab. 2);
MS (ESI, m/e): 354 [M+H]+
Beispiel 57
9-Methyl-7-(4-iso-propoxy-phenyl)-2-propyl-thieno[2,3-d:4,5-d']dipyrimidin-
4(3H)-on
(allgemeine Formel 1b: Ri=4-isoPropO-Ph, X=N, R3=WIe, R4=Prop, Y=S)
100 mg (0.3 mmol) 5-Amino-4-methyl-2-(4-iso-propoxy-phenyl)-thieno[2,3-d]- pyrimidin-6-carboxamid (Beispiel 62; Experiment 7t; allgemeine Formel 7: R-ι=4- isoPropO-Ph, X=N, R3=Me1 Y=S) werden zusammen mit 3 ml (18 mmol) Buttersäureanhydrid 45 Minuten in einer Mikrowelle (300 W) auf 160 0C erhitzt. Der entstandene Niederschlag wird abgesaugt und in Ethanol resuspendiert. Man stellt mit 1 M LiOH-Lösung pH 11 ein und rührt 1 Stunde bei Raumtemperatur. Anschließend wird mit verd. HCl angesäuert, der Niederschlag abgesaugt, mit Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet. Ausbeute: 15 mg (13 %), HPLC tR: 6.7 min, Reinheit: 98.1 % (s. Tab. 2); MS (ESI, m/e): 393 [M-H]"
Beispiel 58
9-Methyl-7-biphenyl-2-ethyl-thieno[2,3-d:4,5-d']dipyrimidin-4(3H)-on (allgemeine Formel 1b: R1=4-Ph-Ph, X=N5 R3=Me, R4=Et, Y=S)
Analog Beispiel 57 aus 5-Amino-2-biphen-4-yl-4-methyl-thieno[2,3-/b]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7aw; allgemeine Formel 7: Ri=Biphen-4-yl, X=N, R3=Me, Y=S) und Propionsäureanhydrid (s. Tab. 2)
Beispiel 59
9-I\Λethyl-7-biphenyl-2-propyl-thieno[2,3-d:4,5-d']dipyrimidin-4(3H)-on
(allgemeine Formel 1b: Ri=4-Ph-Ph, X=N, R3=Me5 R4=Prop, Y=S)
Analog Beispiel 57 aus 5-Amino-2-biphen-4-yl-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7aw; allgemeine Formel 7: Ri=Biphen-4-yl, X=N, R3=Me, Y=S) und Buttersäureanhydrid (s. Tab. 2)
Beispiel 60
9-Methyl-7-naphthalen-2-yl-2-propyl-thieno[2,3-d:4,5-d']dipyrimidin-4(3H)-on (allgemeine Formel 1b: Ri=Naphthalen-2-yl, X=N, R3=Me, R4=Prop, Y=S)
Analog Beispiel 57 aus 5-Amino-2-naphthaIen-2-yl-4-methyl-thieno[2,3-d]pyrimidin-6- carboxamid (Beispiel 62; Experiment 7ay; allgemeine Formel 7: R^Naphthalen^-yl, X=N, R3=Me, Y=S) und Buttersäureanhydrid (s. Tab. 2)
a Retentionszeit (t
R) und Reinheit durch HPLC bestimmt:
Säule: RP-18 (Waters Nova-Pak Ci8, 60 Angström, 4μm, 3.9x150mm); Eluent A: 0.1% TFA, B: 0.09% TFA / 80% Acetonitril; Flussrate: 1 ml/min; Temp.: 25°C; Detektion: 254nm; Gradient (linear) B: 0-2min: 0%, 2- 20min: 0-100%, 20-25min: 100%, 25-35min: 100-0% b Lit.: Wagner G., Böhm N. Pharmazie (1993); 48, 95-99 c Beispiel für Verbindungen der allgemeinen Formel 1 a mit Y=O d Darstellung erfolgt in Anlehnung an Shestopalov, A. M.; Nikishin, K. G.; Gromova, A. V.; Rodinovskaya, L A. Russ.Chem. Bull. (2003), 52(10), 2203-2206
Säule: RP-18 (CROM Resolution Star Ci8, 100 Ä, 5 μm, 4.6x50 mm); Eluent A: 0.1 % TFA / Wasser, B: 0.09% TFA / 80% Acetonitril; Flussrate: 1 ml/min; Temp.: 25°C; Detektion: 254 nm; Gradient (linear) B: 0- 0.5 min: 20 %, 0.5-5 min: 20-100 %, 5-7 min: 100 %, 7-7.3 min: 100-20 %, 7.3-9 min 20 % Säule: RP-18 (CROM Resolution Star Ci8, 100 Ä, 5 μm, 4.6x50 mm); Eluent A: 0.1 % TFA / Wasser, B: 0.09 % TFA Acetonitril; Flussrate: 1 ml/min; Temp.: 25 0C; λ: 254 nm; Gradient (linear) B: 0-0.5 min: 50 %, 0.5-4 min: 50-100 %, 4-6 min: 100 %, 6-6.3 min: 100-50 %, 6-8 min: 50 % n.b. nicht bestimmt
ab. 2 Beispiele der allgemeinen Formel 1b (Y=S)
Retentionszeit (tR) und Reinheit durch HPLC bestimmt:
Säule: RP-18 (Waters Nova-Pak Ci8, 60 Angström, 4μm, 3.9x150mm); Eluent A: 0.1% TFA / Wasser, B:
0.09% TFA / 80% Acetonitril; Flussrate: 1 ml/min; Temp.: 25°C; Detektion: 254nm; Gradient (linear) B: 0-
2min: 0%, 2-20min: 0-100%, 20-25min: 100%, 25-35min: 100-0%
Säule: RP-18 (CROM Resolution Star Ci8, 100 Ä, 5 μm, 4.6x50 mm); Eluent A: 0.1 % TFA / Wasser, B:
0.09% TFA / 80% Acetonitril; Flussrate: 1 ml/min; Temp.: 25°C; Detektion: 254 nm; Gradient (linear) B: 0-
0.5 min: 20 %, 0.5-5 min: 20-100 %, 5-7 min: 100 %, 7-7.3 min: 100-20 %, 7.3-9 min 20 %
Säule: RP-18 (CROM Resolution Star Ci8, 100 A, 5 μm, 4.6x50 mm); Eluent A: 0.1 % TFA / Wasser, B:
0.09 % TFA Acetonitril; Flussrate: 1 ml/min; Temp.: 25 0C; λ: 254 nm; Gradient (linear) B: 0-0.5 min: 50 %,
0.5-4 min: 50-100 %, 4-6 min: 100 %, 6-6.3 min: 100-50 %, 6-8 min: 50 % n.b. nicht bestimmt
Beispiel 61
Beispiele zur Herstellung von Verbindungen der allgemeinen Formel 5
Methode A: Darstellung von 5-unsubstituierten Verbindungen der allgemeinen Formel 5 (X= C-H)
2-Mercapto-6-(3,4-dimethoxyphenyl)-4-methyl-pyridin-3-carbonitril (Experiment 5a; allgemeine Formel 5: Ri=3,4-(MeO)2-Ph, X=C-H, R3=Me, Y=S)
5.0 g (22.5 mmol) 1-(3,4-Dimethoxyphenyl)butan-1 ,3-dion*, 2.9 g (29.2 mmol) Cyanthioacetamid und 4.0 g (29.2 mmol) Kaliumcarbonat werden bei Raumtemperatur 21 Stunden in 170 ml Aceton gerührt. Anschließend wird das Lösungsmittel entfernt und der Rückstand in Ethanol/Wasser (1 :1) gerade gelöst. Nach Ansäuern mit Eisessig wird der erhaltene Niederschlag abgesaugt, mit wenig Wasser gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet.
Ausbeute: 6.34 g (98 %); HPLC tR: 12.9 min, Reinheit: 87.5 % (s. a in Tab. 3); MS (ESI, m/e): 287 [M+H]+
(* Nicht kommerziell verfügbare! ,3-Diketone werden nach bekannten Verfahren erhalten und sind in der Fachliteratur beschrieben.)
Analog Methode A werden die folgenden Experimente durchgeführt: 5a-5g, 5i (s. Tab.3)
Methode B: Darstellung von 5-Benzyl-substituierten Verbindungen der allgemeinen Formel 5 mit X=C-CH2-Ph-4-NO2
4,6-Dimethyl-2-mercapto-5-(4'-nitrobenzyl)-pyridin-3-carbonitril
(Experiment 5h; allgemeine Formel 5: R1=Me, X=C-CH2-Ph-4-NO2, R3=Me, Y=S)
Zu einer Lösung von 1.28 g (55.7 mmol) Natrium in 50 ml abs. Ethanol werden 5.70 ml (55.2 mmol) Acetylaceton und 0.75 g (5.0 mmol) Natriumiodid gegeben. Nach Eintragen einer Suspension von 13.20 g (61.1 mmol) 4'-Nitrobenzylbromid in 60 ml abs. Ethanol wird 3 Stunden unter Rückfluß erhitzt. Nach Abkühlen und Filtrieren werden 5.23 g (52.2 mmol) Cyanthioacetamid und 5 ml (36.1 mmol) Triethylamin zum Filtrat hinzugefügt. Anschließend wird 4 Stunden unter Rückfluß erhitzt. Der nach Kühlen erhaltene Niederschlag wird abgesaugt, mit ca. 50 ml Ethanol und ca. 10 ml Ether rasch gewaschen, trocken gesaugt und bei i. Vak. bei 50 0C getrocknet.
Ausbeute: 5.30 g (32 %); HPLC tR: 13.3 min, Reinheit: 95.9 % (s. Tab. 3); MS (ESI, m/e): 300 [M+H]+
Methode C: Darstellung von Verbindungen der allgemeinen Formel 5 mit X= N
2-Mercapto-6-(4-/so-propoxy-phenyl)-4-methyl-pyrimidin-3-carbonitril (Experiment 5t; allgemeine Formel 5: Ri=4-(isoPropO)-Ph, X=N, R3=Me, Y=S)
5.5 g (28 mmol) 4-/so-Propoxybenzoylchlorid und 2.5 g (33 mmol) Ammoniumthiocyanat werden in 100 ml Dioxan suspendiert und 15 min unter
Rückfluss erhitzt. Anschließend werden 3.0 g (36 mmol) (E/Z)-3-Amino- crotonsäurenitril zugegeben. Das Reaktionsgemisch wird 3 h unter Rückfluss erhitzt. Danach wird das Gemisch mit 500 ml Eiswasser gemischt und über Nacht stehen gelassen, anschließend filtriert und i. Vak. bei 50 0C getrocknet. Ausbeute: 7.3 g (92 %); MS (ESI, m/e): 286 [M+H]+ (s. Tab. 3);
Analog Methode C werden die folgenden Experimente durchgeführt: 5j-5ay (s. Tab.3)
Beispiel 62
Beispiele zur Herstellung von Verbindungen der allgemeinen Formel 7
3-Amino-6-(3,4-dimethoxyphenyl)-4-methyl-thieno[2,3-b]pyridin-2- carbonsäureamid
(Experiment 7a; allgemeine Formel 7: R1=S^-(MeO)2-Ph, X=C-H5 R3=Me, Y=S)
2.1 g (31.4 mmol) Natriumethylat werden in 40 ml abs. Ethanol gelöst. Es werden 3.0 g (10.5 mmol) 2-Mercapto-6-(3,4-dimethoxyphenyl)-4-methyl-pyridin-3-carbonitril (Beispiel 61 ; Experiment 5a; allgemeine Formel 5: Ri=3,4-(MeO)2~Ph, X=C-H, R3=Me, Y=S) und 1.2 g (12.6 mmol) 2-Chloracetamid zugegeben und 1 Stunde unter Rückfluß erhitzt. Nach dem Abkühlen werden 10 ml Wasser zugegeben und der Niederschlag abgesaugt, trocken gesaugt und i. Vak. bei 50 0C getrocknet. Ausbeute: 3.4 g (94 %); HPLC tR: 14.0 min, Reinheit: 99.8 % (s. a in Tab. 4); MS (ESI1 m/e): 344 [M+H]+
Analog dargestellte Beispiele der allgemeinen Formel 7 sind in Tab. 4 aufgeführt.
Beispiel 63
Beispiele zur Herstellung von Verbindungen der allgemeinen Formel 10
3-Amino-4-methyl-6-phenyl-thieno[2,3-b]pyridin-2-carbonsäureethylester (Experiment 10a; allgemeine Formel 10: Ri=Ph5 X=C-H, R3=Me, Y=S)
6.45 g (28.5 mmol) 2-Mercapto-6-phenyl-4-methyl-pyridin-3-carbonitril (Beispiel 61 ; Experiment 5d; allgemeine Formel 5: Ri=Ph, X=C-H, Re=Me, Y=S) werden in 100 ml abs. Ethanol vorgelegt und 5.3 ml (50.0 mmol) 2-Chloressigsäureethylester sowie 10 ml (72.1 mmol) Triethylamin hinzugefügt. Es wird 1 Stunde unter Rückfluß erhitzt und der nach Kühlen ausgefallene Niederschlag abgesaugt, mit wenig Wasser und Ethanol gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet. Anschließend wird der Niederschlag in einer Lösung aus 0.14 g (1.6 mmol) Natriumethylat in abs. Ethanol 1 Stunde unter Rückfluß erhitzt. Der nach Abkühlen ausgefallene Niederschlag wird abgesaugt, mit 30 ml Wasser und 15 ml Ethanol gewaschen, trocken gesaugt und i. Vak. bei 50 0C getrocknet. Ausbeute: 5.0 g (91 %); HPLC tR: 18.0 min, Reinheit: 98.4 % ; MS (ESI, m/e): 313 [M+H]+
Beispiel 64
Beispiele zur Herstellung von Verbindungen der allgemeinen Formel 11
3-(3-Benzoylthioureido)-4-methyl-6-phenyl-thieno[2,3-b]pyridin-2-carbonsäure- ethylester (Beispiel 11 ; allgemeine Formel 11 : R1=Ph, X=C-H, R3=Me, Y=S)
Zu 2.78 g (8.9 mmol) 3-Amino-4-methyl-6-phenyl-thieno[2,3-ö]pyridin-2-carbonsäure- ethylester (Beispiel 63; Experiment 10a; allgemeine Formel 10: Ri=Ph, X=C-H, R3=Me, Y=S) in 10 ml Aceton werden 5 ml (13.3 mmol) acetonischer Benzoylisothiocyanat-Lösung (2M) gegeben und 2 Stunden unter Rückfluß erhitzt. Der nach Abkühlen ausgefallene Niederschlag wird abgesaugt, mit wenig Wasser und Ethanol gewaschen, trocken gesaugt und i. Vak. Bei 50 0C getrocknet. Ausbeute: 3.93 g (93 %); HPLC tR: 19.3 min, Reinheit: 99.9 % (s. ain Tab. 4); MS (ESI, m/e): 476 [M+H]+
Tab. 3 Beispiele der allgemeinen Formel 5 (Y=S)
a Darstellung erfolgt nach Methode A: Erhitzen unter Rückfluß für 3-4 Stunden; nach Abkühlen wird der Ansatz ggf. filtriert und das Filtrat weiterverarbeitet b LJt.: Vieweg H., Leistner S., Wagner G. Pharmazie (1986); 41_, 827-830 c Lit.: Vieweg H., Krasselt U., Boehm N., Prantz J., Wagner G., Pharmazie (1990); 45, 731-733 d Lit.: Schmidt U., Kubitzek H., Chem Ber (1960); 93, 1559-1565 e Darstellung erfolgt nach Methode A: Erhitzen unter Rückfluß für 3-4 Stunden; nach Abkühlen wird der Niederschlag abgesaugt und analog dem Rückstand weiterverarbeitet. f Lit.: Krauze A., Bomika Z, Shestopalov A.M., Rodinovskaya LA., Pelcers J., Duhurs G., Sharanin Y.A., Promonenkov VK. KNm Geterotsikl Soedin (1981); 377-382 g Lit.: Guerrera F., Siracusa M.A., Tornetta B. Farmaco, Ediz Scientif (1976); 3±, 21-30 h Lit.: Wallenfels K, Schuly H„ Ann (1959); 621, 215-221 i Darstellung erfolgt nach Methode B, in Anlehnung an Wagner G., Vieweg H., Leistner S., Böhm N.,
Krasselt U., Hanfeld V., Prantz J., Grupe R. Pharmazie (1990); 45, 102-109 j Darstellung erfolgt nach Methode C
Retentionszeit (tR) und Reinheit durch HPLC bestimmt: ,
Säule: RP-18 (Waters Nova-Pak Ci8, 60 Angström, 4μm, 3.9x150mm); Eluent A: 0.1% TFA, B: 0.09% TFA
/ 80% Acetonitril; Flussrate: 1 ml/min; Temp.: 25°C; Detektion: 254nm; Gradient (linear) B: 0-2min: 0%,
2-20min: 0-100%, 20-25min: 100%, 25-35min: 100-0%
Säule: CROM Resolution Star Ci8, 100 A, 5 μm, 4.6x50 mm; Eluent A: 0.1 % TFA / Wasser, B: 0.09 % TFA
/
Acetonitril; Flussrate: 1 ml/min; Temp.: 25 0C; λ: 254 nm; Gradient (linear) B: 0-0.5 min: 20 %, 0.5-5 min: 20-100 %, 5-7 min: 100 %, 7-7.3 min: 100-20 %, 7.3-9 min 20 % Säule: CROM Resolution Star Ci8, 100 Ä, 5 μm, 4.6x50 mm; Eluent A: 0.1 % TFA / Wasser, B: 0.09 %
TFA/
Acetonitril; Flussrate: 1 ml/min; Temp.: 25 0C; λ: 254 nm; Gradient (linear) B: 0-0.5 min: 50 %, 0.5-4 min: 50-100 %, 4-6 min: 100 %, 6-6.3 min: 100-50 %, 6-8 min: 50 %
Lit.: Litvinov V.P., Sharanin Y.A., Rodinovskaya LA, ShestopalovA.M., Mortikov V.Yu., Promonenkov V.K. SerKhimich (1984); 12, 2760-2765
C Lit.: Sharanin Y.A., Shestopalov AM, Promonenkov V.K. Zhurnal Organ Khimii (1984); 20, 2012-2020 d Ut.: Shestopalov AM, Sharanin Y.A. Zhurnal Organ Khimii (1984); 20, 1991-2002 e Lit.: Tornetta B., Siracusa M.A., Ronsisvalle G., Guerrera F. Gazz. Chim. Ital. (1978); 108, 57-62 und Shvedov V.l., Sycheva T.P., Sakovlch T.V. Khimiya Geterot Soedin (1979); 1331-1335 n.b. nicht bestimmt