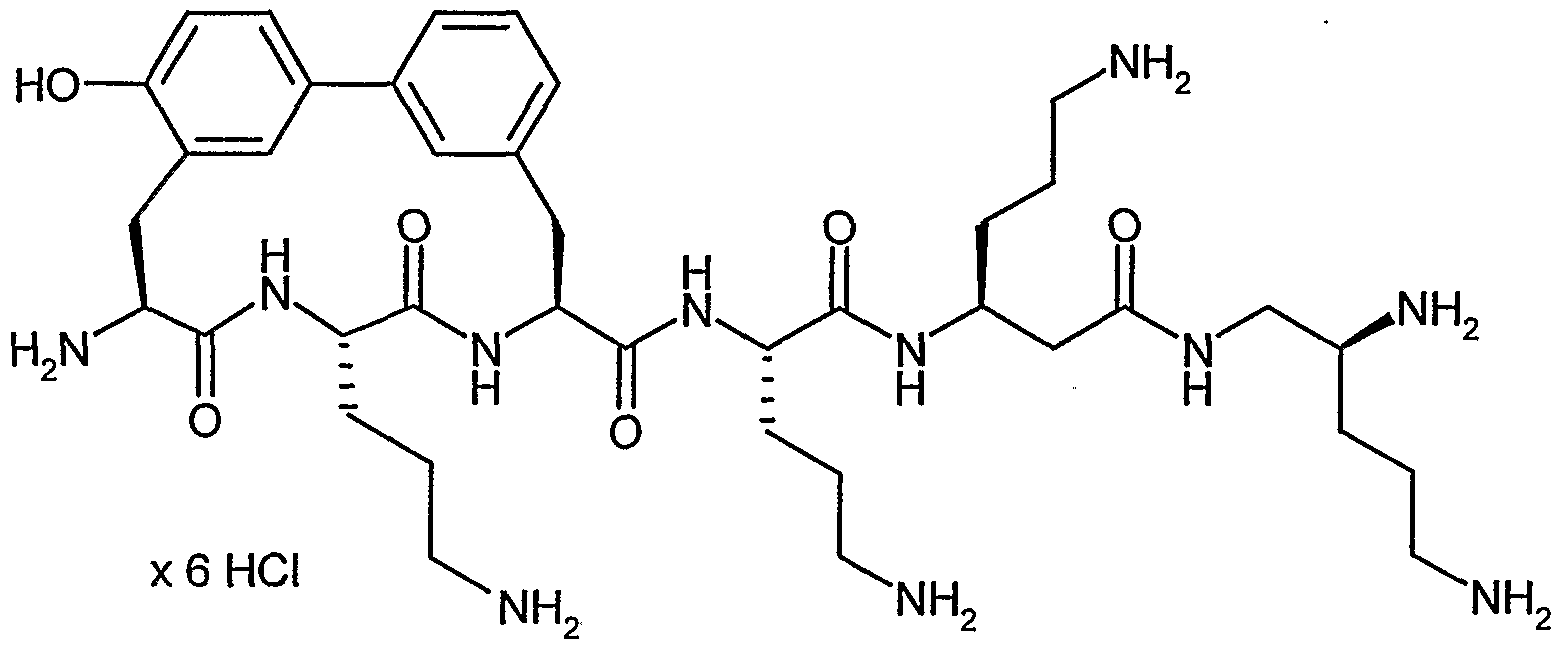
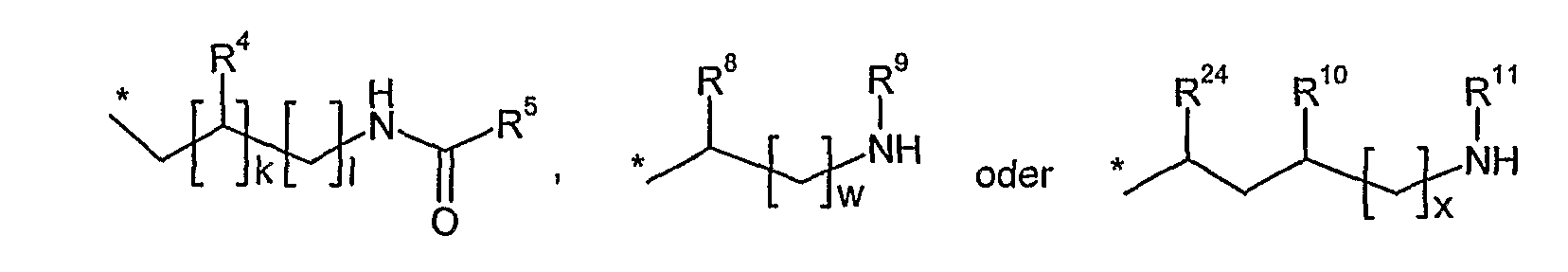
Antibakterielle Amid-Makrozyklen V
Die Erfindung betrifft antibakterielle Amid-Makrozyklen und Verfahren zu ihrer Herstellung, ihre Verwendung zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, insbeson- dere von bakteriellen Infektionen.
In WO 03/106480 und WO 04/012816 werden antibakteriell wirkende Makrozyklen vom Biphenomycin B Typ mit Amid- bzw. Estersubstituenten beschrieben.
In US 3,452,136, Dissertation R. U. Meyer, Universität Stuttgart, Deutschland 1991, Dissertation V. Leitenberger, Universität Stuttgart, Deutschland 1991, Synthesis (1992), (10), 1025-30, J. Chem. Soc, Perkin Trans. 1 (1992), (1), 123-30, J. Chem. Soc, Chem. Commun. (1991), (10), 744, Synthesis (1991), (5), 409-13, J. Chem. Soc, Chem. Commun. (1991), (5), 275-7, J. Antibiot. (1985), 38(11), 1462-8, J. Antibiot. (1985), 38(11), 1453-61, wird der Naturstoff Biphenomycin B als antibakteriell wirksam beschrieben. Teilschritte der Synthese von Biphenomycin B werden in Synlett (2003), 4, 522-526 beschrieben.
Chirality (1995), 7(4), 181-92, J. Antibiot. (1991), 44(6), 674-7, J. Am. Chem. Soc. (1989), 111(19), 7323-7, J. Am. Chem. Soc. (1989), 111(19), 7328-33, J. Org. Chem. (1987), 52(24), 5435-7, Anal. Biochem. (1987), 165(1), 108-13, J. Org. Chem. (1985), 50(8), 1341-2, J. Antibiot. (1993), 46(3), C-2, J. Antibiot. (1993), 46(1), 135-40, Synthesis (1992), (12), 1248-54, Appl. Environ. Microbiol. (1992), 58(12), 3879-8, J. Chem. Soc, Chem. Commun. (1992), (13), 951-3 beschreiben einen strukturell verwandten Naturstoff, Biphenomycin A, der am Makrozyklus eine weitere Substitution mit einer Hydroxygruppe aufweist.
Die Naturstoffe entsprechen hinsichtlich ihrer Eigenschaften nicht den Anforderungen, die an antibakterielle Arzneimittel gestellt werden. Auf dem Markt sind zwar strukturell andersartige antibakteriell wirkende Mittel vorhanden, es kann aber regelmäßig zu einer Resistenzentwicklung kommen. Neue Mittel für eine gute und wirksamere Therapie sind daher wünschenswert.
Eine Aufgabe der vorliegenden Erfindung ist es daher, neue und alternative Verbindungen mit gleicher oder verbesserter antibakterieller Wirkung zur Behandlung von bakteriellen Erkrankungen bei Menschen und Tieren zur Verfügung zu stellen.
Überraschenderweise wurde gefunden, dass bestimmte Derivate dieser Naturstoffe, worin die Carboxylgruppe des Naturstoffs gegen eine Amidgruppe ausgetauscht wird, die eine basische Gruppe enthält, gegen Biphenomycin resistente S. aureus Stämme (RN4220BiR und Tl 7) antibakteriell wirksam sind.
Weiterhin zeigen die Derivate gegen S. aureus Wildtyp-Stämme und Biphenomycin resistente S. aureus Stämme eine verbesserte Spontanresistenz-Frequenz.
Gegenstand der Erfindung sind Verbindungen der Formel
bei denen
R26 gleich Wasserstoff, Halogen, Amino oder Methyl ist,
R7 gleich eine Gruppe der Formel
*
ist,
wobei
R1 gleich Wasserstoff oder Hydroxy ist,
* die Anknüpfstelle an das Kohlenstoffatom ist,
R2 gleich Wasserstoff oder Methyl ist,
R
3 gleich eine Gruppe der Formel
ist,
wobei
* die Anknüpfstelle an das Stickstoffatom ist,
A gleich eine Bindung oder Phenyl ist,
R4 gleich Wasserstoff, Amino oder Hydroxy ist,
R5 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
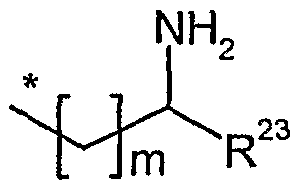
R23 Wasserstoff oder eine Gruppe der Formel *-(CH2)„-OH oder *-CCH2)0- NH2 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
n und o unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
m eine Zahl 0 oder 1 ist,
R8 und R12 unabhängig voneinander eine Gruppe der Formel *-CONHR14 oder *-CH2CONHR15 sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R14 und R15 unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4a gleich Wasserstoff, Amino oder Hydroxy ist,
R5a gleich Wasserstoff, Methyl oder Aminoethyl ist,
Rδa gleich Wasserstoff oder Aminoethyl ist,
oder
R5a und R6a bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8a und Rl2a unabhängig voneinander *-(CΗ2)zia-OH, *_(CH2)Z2a- NHR13a, *-CONHRUa oder *-CH2CONHR15a sind,
woπn
die Anknüpfstelle an das Kohlenstoffatom ist,
ZIa und Z2a unabhängig voneinander eine Zahl 1 , 2 oder 3 sind,
R1 Ja gleich Wasserstoff oder Methyl ist
und
R a und R a unabhängig voneinander eine Gruppe der Formel
sind,
worin
10 die Anknüpfstelle an das Stickstoffatom ist,
R c gleich Wasserstoff, Amino oder Hydroxy ist,
R c gleich Wasserstoff, Methyl oder Aminoethyl ist,
R ° gleich Wasserstoff oder Aminoethyl ist,
kc eine Zahl 0 oder 1 ist
15 und
Ic eine Zahl 1, 2, 3 oder 4 ist,
R9a und R1 la unabhängig voneinander Wasserstoff oder Methyl sind,
RIOa gleich Amino oder Hydroxy ist,
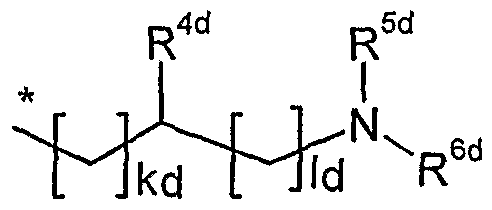
R16a eine Gruppe der Formel
20
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
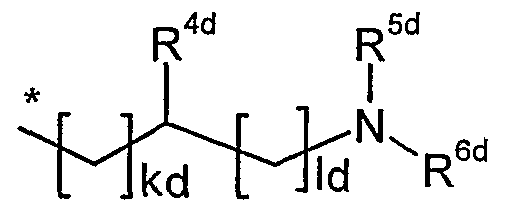
R4d gleich Wasserstoff, Amino oder Hydroxy ist,
5 R5d gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6d gleich Wasserstoff oder Aminoethyl ist,
kd eine Zahl 0 oder 1 ist
und
Id eine Zahl 1 , 2, 3 oder 4 ist,
10 R18a und RI9a unabhängig voneinander Wasserstoff oder eine Gruppe der
Formel
sind,
worin
15 * die Anknüpfstelle an das Stickstoffatom ist,
R4h gleich Wasserstoff, Amino oder Hydroxy ist,
R5h gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6h gleich Wasserstoff oder Aminoethyl ist,
oder
20 R5h und R6h bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
kh eine Zahl 0 oder 1 ist
und
Ih eine Zahl 1, 2, 3 oder 4 ist,
wobei R1Sa und R19a nicht gleichzeitig Wasserstoff sind,
ka eine Zahl 0 oder 1 ist,
ea eine Zahl 1, 2 oder 3 ist,
und
Ia, wa, xa und ya unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
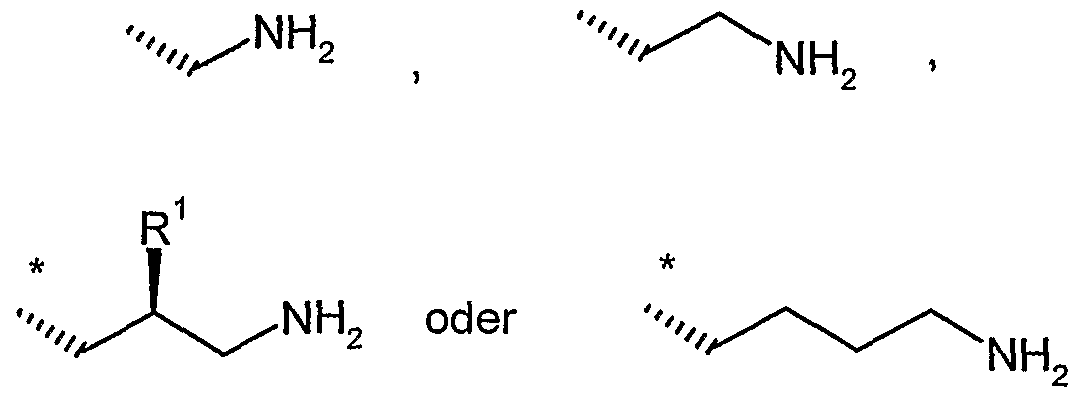
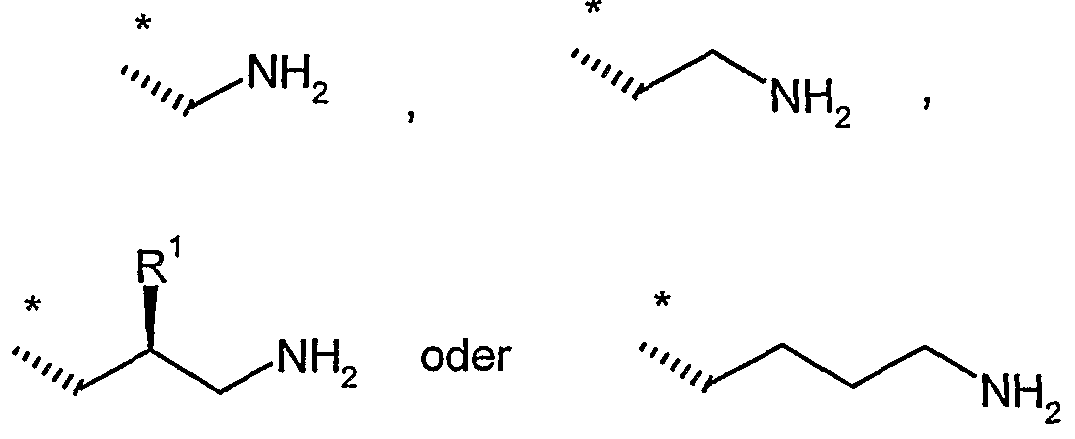
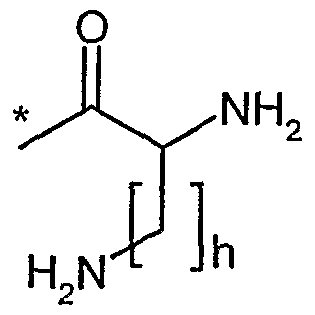
R9 und R11 unabhängig voneinander Wasserstoff, Methyl, *-C(NH2)=NH oder eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R20 gleich Wasserstoff oder *-{CH2)i-NHR22 ist,
worin
R22 gleich Wasserstoff oder Methyl ist
und
i eine Zahl 1, 2 oder 3 ist,
R21 gleich Wasserstoff oder Methyl ist,
f eine Zahl 0, 1, 2 oder 3 ist,
g eine Zahl 1, 2 oder 3 ist
und
h eine Zahl 1, 2, 3 oder 4 ist,
oder
R8 gleich *-(CH2)zi-OH ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
Zl eine Zahl 1 , 2 oder 3 ist,
und
R9 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
und
h eine Zahl 1 , 2, 3 oder 4 ist,
R10 gleich Amino oder Hydroxy ist,
R
16 und R
17 unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4b gleich Wasserstoff, Amino oder Hydroxy ist,
R5b gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6b gleich Wasserstoff oder Aminoethyl ist,
oder
R5b und R6b bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
RSb und R12b unabhängig voneinander *-(CH2)zib-OH, *-{CH2)Z2b-NHR13b, *-CONHRI4b oder *-CH2CONHR15b sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R13b gleich Wasserstoff oder Methyl ist
und
ZIb und Z2b unabhängig voneinander eine Zahl 1 , 2 oder 3 sind,
und
R
I4b und R
15b unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4ε gleich Wasserstoff, Amino oder Hydroxy ist,
R5ε gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6g gleich Wasserstoff oder Aminoethyl ist,
kg eine Zahl 0 oder 1 ist
und
Ig eine Zahl 1 , 2, 3 oder 4 ist,
R9b und R1 lb unabhängig voneinander Wasserstoff oder Methyl sind,
R1Ob gleich Amino oder Hydroxy ist,
kb eine Zahl 0 oder 1 ist,
Ib, wb, xb und yb unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
R19 unabhängig voneinander Wasserstoff oder eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4e gleich Wasserstoff, Amino oder Hydroxy ist,
R5e gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6e gleich Wasserstoff oder Aminoethyl ist,
oder
R5e und R6e bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8e und R12e unabhängig voneinander *-(CH2)z,e-OH oder *-(CH2)Z2e-NHR13e sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R13e gleich Wasserstoff oder Methyl ist
und
ZIe und Z2e unabhängig voneinander eine Zahl 1 , 2 oder 3 sind,
R9e und R1 Ie unabhängig voneinander Wasserstoff oder Methyl sind,
R1Oe gleich Amino oder Hydroxy ist,
ke eine Zahl 0 oder 1 ist
und
Ie, we, xe und ye unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
wobei RIS und R19 nicht gleichzeitig Wasserstoff sind,
eine Gruppe der Formel *-CONHR25 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R25 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4f gleich Wasserstoff, Amino oder Hydroxy ist,
R5f gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6f gleich Wasserstoff oder Aminoethyl ist,
oder
R5f und Rδf bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
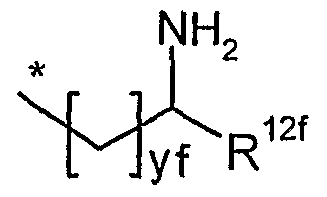
R8fund R12f unabhängig voneinander ^-(CH2)ZIrOH oder *-(CH2)Z2rNHR13f sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R13f gleich Wasserstoff oder Methyl ist
und
ZIf und Z2f unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R9f und R1 lf unabhängig voneinander Wasserstoff oder Methyl sind,
R1Of gleich Amino oder Hydroxy ist,
kf eine Zahl 0 oder 1 ist
und
If, wf, xf und yf unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
d und e unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
k eine Zahl 0 oder 1 ist,
1, w, x und y unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
unabhängig voneinander bei w, x oder y gleich 3 eine Hydroxy-
Gruppe tragen kann,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Erfϊndungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, sowie die von Formel (I) umfassten, nachfolgend als Ausführungs- beispiel(e) genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung betrifft deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich durch bekannte Verfahren wie Chromatographie an chiraler Phase oder Kristallisation mit chiralen Aminen oder chiralen Säuren die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Die Erfindung betrifft in Abhängigkeit von der Struktur der Verbindungen auch Tautomere der Verbindungen.
Als Salze sind im Rahmen der Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt.
Physiologisch unbedenkliche Salze der Verbindungen (T) umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasser-
stoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethansulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure, Trifluoressigsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der Verbindungen (I) umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze
(z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze., abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Di- ethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin,
Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methylmorpholin, Dihydro- abietylamin, Arginin, Lysin, Ethylendiamin und Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt.
Halogen steht für Fluor, Chlor, Brom und Jod.
Ein Symbol # an einem Kohlenstoffatom bedeutet, dass die Verbindung hinsichtlich der Konfiguration an diesem Kohlenstoffatom in enantiomerenreiner Form vorliegt, worunter im Rahmen der vorliegenden Erfindung ein Enantiomerenüberschuss (enantiomeric excess) von mehr als 90% verstanden wird (> 90% ee).
In den Formeln der Gruppen, für die R3 stehen kann, steht der Endpunkt der Linie, neben der jeweils ein * steht, nicht für ein Kohlenstoffatom beziehungsweise eine CH2-Grupρe sondern ist Bestandteil der Bindung zu dem Stickstoffatom, an das R3 gebunden ist.
In den Formeln der Gruppen, für die R7 stehen kann, steht der Endpunkt der Linie, neben der jeweils ein * steht, nicht für ein Kohlenstoffatom beziehungsweise eine CH2-Gruppe sondern ist Bestandteil der Bindung zu dem Kohlenstoffatom, an das R7 gebunden ist.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), bei denen
R26 gleich Wasserstoff, Halogen, Amino oder Methyl ist,
R
7 gleich eine Gruppe der Formel
ist,
wobei
R1 gleich Wasserstoff oder Hydroxy ist,
die Anknüpfstelle an das Kohlenstoffatom ist,
R gleich Wasserstoff oder Methyl ist,
R3 gleich eine Gruppe der Formel
ist,
wobei
* die Anknüpfstelle an das Stickstoffatom ist,
A gleich eine Bindung oder Phenyl ist,
R4 gleich Wasserstoff, Amino oder Hydroxy ist,
R5 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R23 Wasserstoff oder eine Gruppe der Formel *-(CH2)n-OH oder
*-(CH2)0-NH2 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
n und o unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
m eine Zahl 0 oder 1 ist,
R8 und R12 unabhängig voneinander eine Gruppe der Formel *-CONHR14 oder *-CH2CONHR15 sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R14 und RIS unabhängig voneinander eine Gruppe der Formel
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4a gleich Wasserstoff, Amino oder Hydroxy ist,
R5a gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6a gleich Wasserstoff oder Aminoethyl ist,
oder
R5a und R6a bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8a und R12a unabhängig voneinander *-(CH2)zia-OH,
*-(CH2)Z2a-NHR13a, *-CONHR14a oder *-CH2CONHR15a sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
ZIa und Z2a unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R13a gleich Wasserstoff oder Methyl ist
und
R14a und R15a unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4° gleich Wasserstoff, Amino oder Hydroxy ist,
R5c gleich Wasserstoff, Methyl oder Aminoethyl ist,
Rδc gleich Wasserstoff oder Aminoethyl ist,
kc eine Zahl 0 oder 1 ist
und
Ic eine Zahl 1, 2, 3 oder 4 ist,
R9a und R1 la unabhängig voneinander Wasserstoff oder Methyl sind,
R1Oa gleich Amino oder Hydroxy ist,
R16a eine Gruppe der Formel
10 sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4d gleich Wasserstoff, Amino oder Hydroxy ist,
R5d gleich Wasserstoff, Methyl oder Aminoethyl ist,
15 R6d gleich Wasserstoff oder Aminoethyl ist,
kd eine Zahl 0 oder 1 ist
und
Id eine Zahl 1, 2, 3 oder 4 ist,
ka eine Zahl 0 oder 1 ist
20 und
Ia, wa, xa und ya unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
R9 und R11 unabhängig voneinander Wasserstoff, Methyl, *-C(NH2)=NH oder eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R20 gleich Wasserstoff oder *-<CH2)i-NHR22 ist,
worin
R22 gleich Wasserstoff oder Methyl ist
und
i eine Zahl 1, 2 oder 3 ist,
R21 gleich Wasserstoff oder Methyl ist,
f eine Zahl 0, 1 , 2 oder 3 ist,
g eine Zahl 1, 2 oder 3 ist
und
h eine Zahl 1, 2, 3 oder 4 ist,
oder
R8 gleich *-(CH2)zl-OH ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
Zl eine Zahl 1 , 2 oder 3 ist,
und
R9 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
und
h eine Zahl 1 , 2, 3 oder 4 ist,
R10 gleich Amino oder Hydroxy ist,
R16 und R17 unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4b gleich Wasserstoff, Amino oder Hydroxy ist,
R5b gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6b gleich Wasserstoff oder Aminoethyl ist,
oder
R5b und R6b bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8b und R12b unabhängig voneinander *-(CH2)Zib-OH, *_(CH2)Z2b-NHRI3b, *-CONHR14b oder *-CH2CONHR15b sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R13b gleich Wasserstoff oder Methyl ist
und
ZIb und Z2b unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
und
R14b und R15b unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4g gleich Wasserstoff, Amino oder Hydroxy ist,
R5g gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6g gleich Wasserstoff oder Aminoethyl ist,
kg eine Zahl 0 oder 1 ist
und
Ig eine Zahl 1, 2, 3 oder 4 ist,
R9b und R1 Ib unabhängig voneinander Wasserstoff oder Methyl sind,
R1Ob gleich Amino oder Hydroxy ist,
kb eine Zahl 0 oder 1 ist,
Ib, wb, xb und yb unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
R19 unabhängig voneinander Wasserstoff oder eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4e gleich Wasserstoff, Amino oder Hydroxy ist,
R5e gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6e gleich Wasserstoff oder Aminoethyl ist,
oder
R5e und R6e bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
RSe und R12e unabhängig voneinander *-(CH2)Zie-OH oder ^CH2)Z26-NHR13e sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R13e gleich Wasserstoff oder Methyl ist
und
ZIe und Z2e unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R9e und R1 le unabhängig voneinander Wasserstoff oder Methyl sind,
RIOe gleich Amino oder Hydroxy ist,
ke eine Zahl 0 oder 1 ist
und
Ie, we, xe und ye unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
wobei R18 und R19 nicht gleichzeitig Wasserstoff sind,
eine Gruppe der Formel *-CONHR25 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R25 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4f gleich Wasserstoff, Amino oder Hydroxy ist,
R5f gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6f gleich Wasserstoff oder Aminoethyl ist,
oder
R5f und R6f bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R
8fund R
I2f unabhängig voneinander oder
*-(CH
2)
Z2rNHR
I3f sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R13f gleich Wasserstoff oder Methyl ist
und
ZIf und Z2f unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R9f und R1 If unabhängig voneinander Wasserstoff oder Methyl sind,
RIOf gleich Amino oder Hydroxy ist,
kf eine Zahl 0 oder 1 ist
und
If, wf, xf und yf unabhängig voneinander eine Zahl 1 , 2, 3 oder 4 sind,
d und e unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
k eine Zahl 0 oder 1 ist,
1, w, x und y unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
χ ocjer y unabhängig voneinander bei w, x oder y gleich 3 eine Hydroxy-
Gruppe tragen kann,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel
(Ia),
R26 gleich Wasserstoff, Halogen, Amino oder Methyl ist,
R1 gleich Wasserstoff oder Hydroxy ist,
R2 gleich Wasserstoff oder Methyl ist,
R3 wie oben definiert ist,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I) oder (Ia), bei denen
R26 gleich Wasserstoff, Chlor oder Methyl ist.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I) oder (Ia), bei denen
,26 gleich Wasserstoff ist.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I) oder (Ia), bei denen
R3 gleich eine Gruppe der Formel
wobei
* die Anknüpfstelle an das Stickstoffatom ist,
R4 gleich Wasserstoff, Amino oder Hydroxy ist,
Rs eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R23 Wasserstoff oder eine Gruppe der Formel *-{CH2)n-OH oder *-
(CHa)0-NH2 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
n und o unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
m eine Zahl 0 oder 1 ist,
R8 eine Gruppe der Formel *-CONHR14 oder *-CH2CONHR15 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R
14 und R
15 unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4a gleich Wasserstoff, Amino oder Hydroxy ist,
R5a gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6a gleich Wasserstoff oder Aminoethyl ist,
oder
R5a und R6a bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8a und R12a unabhängig voneinander MCH2WOH, *-(CH2)Z2a- NHRI3a, *-CONHR14a oder *-CH2CONHR15asind,
worin
* . die Anknüpfstelle an das Kohlenstoffatom ist,
Zla und Z2a unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R13a gleich Wasserstoff oder Methyl ist
und
R
14a und R
15a unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
5 R4c gleich Wasserstoff, Amino oder Hydroxy ist,
R5c gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6c gleich Wasserstoff oder Aminoethyl ist,
kc eine Zahl 0 oder 1 ist
und
10 Ic eine Zahl 1, 2, 3 oder 4 ist,
R9a und R1 la unabhängig voneinander Wasserstoff oder Methyl sind,
RIOa gleich Amino oder Hydroxy ist,
R16a eine Gruppe der Formel
15 sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4d gleich Wasserstoff, Amino oder Hydroxy ist,
R5d gleich Wasserstoff, Methyl oder Aminoethyl ist,
20 R6d gleich Wasserstoff oder Aminoethyl ist,
kd eine Zahl 0 oder 1 ist
und
Id eine Zahl 1, 2, 3 oder 4 ist,
ka eine Zahl 0 oder 1 ist
und
Ia, wa, xa und ya unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
R9 und R11 unabhängig voneinander Wasserstoff, Methyl, *-C(NH2)=NH oder eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R20 gleich Wasserstoff oder MCH2VNHR22 ist,
worin
R22 gleich Wasserstoff oder Methyl ist
und
i eine Zahl 1, 2 oder 3 ist,
R21 gleich Wasserstoff oder Methyl ist,
f eine Zahl 0, 1, 2 oder 3 ist,
g eine Zahl 1 , 2 oder 3 ist
und
h eine Zahl 1 , 2, 3 oder 4 ist,
oder
R8 gleich *-(CH2)Z1-OH ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
Zl eine Zahl 1, 2 oder 3 ist,
und
R9 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
und
h eine Zahl 1, 2, 3 oder 4 ist,
R10 gleich Amino oder Hydroxy ist,
R24 eine Gruppe der Formel *-CONHR25 ist,
worin
die Anknüpfstelle an das Kohlenstoffatom ist,
R -.25 eine Gruppe der Formel
ist,
worin
die Anknüpfstelle an das Stickstoffatom ist,
R ,4f gleich Wasserstoff, Amino oder Hydroxy ist,
R5f gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6f gleich Wasserstoff oder Aminoethyl ist,
oder
R5f und R bilden zusammen mit dem Stickstoffatom an das sie
10 gebunden sind einen Piperazin-Ring,
R und R unabhängig voneinander *-(CH2)zirOH oder
*-(CH2)Z2rNHR 113Jft s , ind,
worin
die Anknüpfstelle an das Kohlenstoffatom ist,
15 R , 13f gleich Wasserstoff oder Methyl ist
und
ZIf und Z2f unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R und R unabhängig voneinander Wasserstoff oder Methyl sind,
R gleich Amino oder Hydroxy ist,
20 kf eine Zahl 0 oder 1 ist
und
If, wf, xf und yf unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
k eine Zahl 0 oder 1 ist,
1, w und x unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
unabhängig voneinander bei w oder x gleich 3 eine Hydroxy-Gruppe
tragen kann,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I) oder (Ia), bei denen
R3 gleich eine Gruppe der Formel
ist,
wobei
* die Anknüpfstelle an das Stickstoffatom ist,
R4 gleich Wasserstoff, Amino oder Hydroxy ist,
R5 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R23 Wasserstoff oder eine Gruppe der Formel *-(CH2)n-OH oder *-(CH2)0-NH2 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
n und o unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
m eine Zahl 0 oder 1 ist,
k eine Zahl 0 oder 1 ist,
1 eine Zahl 1 , 2, 3 oder 4 ist,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I) oder (Ia), bei denen
R3 gleich eine Gruppe der Formel
( ist,
wobei
* die Anknüpfstelle an das Stickstoffatom ist,
R8 eine Gruppe der Formel *-CONHR14 oder *-CH2CONHR15 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R
14 und R
15 unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4a gleich Wasserstoff, Amino oder Hydroxy ist,
RSa gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6a gleich Wasserstoff oder Aminoethyl ist,
oder
R5a und R6a bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8a und R12a unabhängig voneinander *-(CH2)Zia-OH, *-(CH2)Z2a- NHR13a, *-CONHR14a oder *-CH2CONHR15asind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
ZIa und Z2a unabhängig voneinander eine Zahl 1 , 2 oder 3 sind,
R13a gleich Wasserstoff oder Methyl ist
und
R
I4a und R
15a unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
5 R4c gleich Wasserstoff, Amino oder Hydroxy ist,
R5c gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6c gleich Wasserstoff oder Aminoethyl ist,
kc eine Zahl 0 oder 1 ist
und
10 Ic eine Zahl 1, 2, 3 oder 4 ist,
R9a und R1 la unabhängig voneinander Wasserstoff oder Methyl sind,
R1Oa gleich Amino oder Hydroxy ist,
R16a eine Gruppe der Formel
15 sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4d gleich Wasserstoff, Amino oder Hydroxy ist,
R5d gleich Wasserstoff, Methyl oder Aminoethyl ist,
20 R6d gleich Wasserstoff oder Aminoethyl ist,
kd eine Zahl O oder 1 ist
und
Id eine Zahl 1 , 2, 3 oder 4 ist,
ka eine Zahl 0 oder 1 ist
und
Ia, wa, xa und ya unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
R9 und R11 unabhängig voneinander Wasserstoff, Methyl, *-C(NH2)=NH oder eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R20 gleich Wasserstoff oder MCH2VNHR22 ist,
worin
R22 gleich Wasserstoff oder Methyl ist
und
i eine Zahl 1, 2 oder 3 ist,
R21 gleich Wasserstoff oder Methyl ist,
f eine Zahl 0, 1 , 2 oder 3 ist,
g eine Zahl 1, 2 oder 3 ist
und
h eine Zahl 1, 2, 3 oder 4 ist,
oder
R8 gleich MCH2)Zi-OH ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
Zl eine Zahl 1, 2 oder 3 ist,
und
R9 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
und
h eine Zahl 1, 2, 3 oder 4 ist,
R10 gleich Amino oder Hydroxy ist,
R24 eine Gruppe der Formel *-CONHR25 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R
25 eine Gruppe der Formel
ist,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
5 R4f gleich Wasserstoff, Amino oder Hydroxy ist,
R5f gleich Wasserstoff, Methyl oder Aminoethyl ist,
Rδf gleich Wasserstoff oder Aminoethyl ist,
oder
R5f und R6f bilden zusammen mit dem Stickstoffatom an das sie ge- 10 bunden sind einen Piperazin-Ring,
R8f und R12f unabhängig voneinander *-(CH2)ZirOH oder *-{CH2)Z2r NHRI3f sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
15 RI3f gleich Wasserstoff oder Methyl ist
und
ZIf und Z2f unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R9f und R1 ' f unabhängig voneinander Wasserstoff oder Methyl sind,
R1Of gleich Amino oder Hydroxy ist,
20 kf eine Zahl 0 oder 1 ist
und
If, wf, xf und yf unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
w und x unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
der x
unabhängig voneinander bei w oder x gleich 3 eine Hydroxy-Gruppe
tragen kann,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I) oder (Ia), bei denen
R3 gleich eine Gruppe der Formel
ist,
wobei
* die Anknüpfstelle an das Stickstoffatom ist,
R12 eine Gruppe der Formel *-CONHR14 oder *-CH2CONHR15 ist,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
R
14 und R
15 unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatorn ist,
R4a gleich Wasserstoff, Amino oder Hydroxy ist,
R5a gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6a gleich Wasserstoff oder Aminoethyl ist,
oder
R5a und R6a bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8a und R12a unabhängig voneinander *-{CH2)zia-OH, MCH2)Z23- NHR13a, *-CONHR14a oder *-CH2CONΗRI5asind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
ZIa und Z2a unabhängig voneinander eine Zahl 1 , 2 oder 3 sind,
R13a gleich Wasserstoff oder Methyl ist
und
R
14a und R
I5a unabhängig voneinander eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
5 R4c gleich Wasserstoff, Amino oder Hydroxy ist,
R5c gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6c gleich Wasserstoff oder Aminoethyl ist,
kc eine Zahl 0 oder 1 ist
und
10 Ic eine Zahl 1, 2, 3 oder 4 ist,
R9a und RUa unabhängig voneinander Wasserstoff oder Methyl sind,
R1Oa gleich Amino oder Hydroxy ist,
R16a eine Gruppe der Formel
15 sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4d gleich Wasserstoff, Amino oder Hydroxy ist,
R5d gleich Wasserstoff, Methyl oder Aminoethyl ist,
20 R6d gleich Wasserstoff oder Aminoethyl ist,
kd eine Zahl 0 oder 1 ist
und
Id eine Zahl 1, 2, 3 oder 4 ist,
ka eine Zahl 0 oder 1 ist
und
Ia, wa, xa und ya unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
y eine Zahl 1, 2, 3 oder 4 ist,
bei y gleich 3 eine Hydroxy-Gruppe tragen kann,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I) oder (Ia), bei denen
R3 gleich eine Gruppe der Formel
ist,
wobei
* die Anknüpfstelle an das Stickstoffatom ist,
A gleich eine Bindung oder Phenyl ist,
R
16 und R
17 unabhängig voneinander eine Gruppe der Formel
sind,
worin
die Anknüpfstelle an das Stickstoffatom ist,
R ,4b gleich Wasserstoff, Amino oder Hydroxy ist,
R gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6b gleich Wasserstoff oder Aminoethyl ist,
oder
R5b und R6b bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R )8SbÖ . u,„ndj D R1U2bB unabhängig voneinander *-(CH2)zib-OH oder *-(CH2)Z2b-NHR 13b sind,
worin
die Anknüpfstelle an das Kohlenstoffatom ist,
R gleich Wasserstoff oder Methyl ist
und
ZIb und Z2b unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R und R unabhängig voneinander Wasserstoff oder Methyl sind,
R gleich Amino oder Hydroxy ist,
kb eine Zahl 0 oder 1 ist,
Ib, wb, xb und yb unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
eine Zahl 1, 2 oder 3 ist,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Besonders bevorzugt sind darunter Verbindungen, bei denen R3 eine Gruppe der Formel
insbesondere eine Gruppe der Formel
ist.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I) oder (Ia), bei denen
R3 gleich eine Gruppe der Formel
ist,
wobei
die Anknüpfstelle an das Stickstoffatom ist,
R unabhängig voneinander Wasserstoff oder eine Gruppe der Formel
sind,
worin
* die Anknüpfstelle an das Stickstoffatom ist,
R4e gleich Wasserstoff, Amino oder Hydroxy ist,
R5e gleich Wasserstoff, Methyl oder Aminoethyl ist,
R6e gleich Wasserstoff oder Aminoethyl ist,
oder
R5e und R6e bilden zusammen mit dem Stickstoffatom an das sie gebunden sind einen Piperazin-Ring,
R8β und R12e unabhängig voneinander *-(CH2)zie-OH oder MCH2K-NHR136 sind,
worin
* die Anknüpfstelle an das Kohlenstoffatom ist,
Rl3e gleich Wasserstoff oder Methyl ist
und
ZIe und Z2e unabhängig voneinander eine Zahl 1, 2 oder 3 sind,
R9e und Rllε unabhängig voneinander Wasserstoff oder Methyl sind,
R1Oe gleich Amino oder Hydroxy ist,
ke eine Zahl 0 oder 1 ist
und
Ie, we, xe und ye unabhängig voneinander eine Zahl 1, 2, 3 oder 4 sind,
wobei R18 und R19 nicht gleichzeitig Wasserstoff sind,
e eine Zahl 1, 2 oder 3 ist,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Gegenstand der Erfindung ist weiterhin ein Verfahren zur Herstellung der Verbindungen der Formel (I) oder ihrer Salze, ihrer Solvate oder der Solvate ihrer Salze, wobei nach Verfahren
[A] Verbindungen der Formel
(H),
worin R
2, R
7 und R
26 die oben angegebene Bedeutung haben und boc gleich te/t-Butoxycarbonyl ist,
in einem zweistufigen Verfahren zunächst in Gegenwart von einem oder mehreren De- hydratisierungsreagenzien mit Verbindungen der Formel
H2NR3 (III),
worin R3 die oben angegebene Bedeutung hat,
und anschließend mit einer Säure und/oder durch Hydrogenolyse umgesetzt werden,
oder
[B] Verbindungen der Formel
σv),
worin R2, R7 und R26 die oben angegebene Bedeutung haben und Z gleich Benzyloxycarbonyl ist,
in einem zweistufigen Verfahren zunächst in Gegenwart von einem oder mehreren De- hydratisierungsreagenzien mit Verbindungen der Formel
H2NR3 (HI),
worin R3 die oben angegebene Bedeutung hat,
und anschließend mit einer Säure oder durch Hydrogenolyse umgesetzt werden.
Die freie Base der Salze kann zum Beispiel durch Chromatographie an einer Reversed Phase Säule mit einem Acetonitril- Wasser-Gradienten unter Zusatz einer Base erhalten werden, insbesondere durch Verwendung einer RPl 8 Phenomenex Luna Cl 8(2) Säule und Diethylamin als Base.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der Verbindungen der Formel (I) oder ihrer Solvate nach Anspruch 1, bei dem Salze der Verbindungen oder Solvate der Salze der Verbindungen durch Chromatographie unter Zusatz einer Base in die Verbindungen überführt werden.
Die Hydroxygruppe an R1 ist gegebenenfalls während der Umsetzung mit Verbindungen der Formel (III) mit einer tert-Butyldimethylsilyl-Gruppe geschützt, die im zweiten Reaktionsschritt abgespalten wird.
Reaktive Funktionalitäten in dem Rest R3 von Verbindungen der Formel (IH) werden bereits geschützt mit in die Synthese eingebracht, bevorzugt sind säurelabile Schutzgruppen (z.B. boc). Nach erfolgter Umsetzung zu Verbindungen der Formel (I) können die Schutzgruppen durch Entschützungsreaktion abgespalten werden. Dies geschieht nach Standardverfahren der Schutzgruppenchemie. Bevorzugt sind Entschützungsreaktionen unter sauren Bedingungen oder durch Hydrogenolyse.
Die Umsetzung der ersten Stufe der Verfahren [A] und [B] erfolgt im Allgemeinen in inerten Lösungsmitteln, gegebenenfalls in Gegenwart einer Base, bevorzugt in einem Temperaturbereich von 0°C bis 4O°C bei Normaldruck.
Als Dehydratisierungsreagenzien eignen sich hierbei beispielsweise Carbodiimide wie z.B. N,N- Diethyl-, N,N'-Dipropyl-, N,N'-Diisopropyl-, N,N'-Dicyclohexylcarbodiimid, N-(3-Dimethylamino- isopropy I)-N -ethy Icarbodiimid-Hydrochlorid (EDC), N-Cyclohexylcarbodiimid-N '-propyloxy- methyl-Polystyrol (PS-Carbodiimid) oder Carbonylverbindungen wie Carbonyldiimidazol, oder 1,2-Oxazoliumverbindungen wie 2-Ethyl-5-phenyl-l,2-oxazolium-3-sulfat oder 2-tert.-Butyl-5- methyl-isoxazolium-perchlorat, oder Acylaminoverbindungen wie 2-Ethoxy-l-ethoxycarbony 1-1,2- dihydrochinolin, oder Propanphosphonsäureanhydrid, oder Isobutylchloroformat, oder Bis-(2-oxo- 3 -oxazolidiny l)-phosphorylchlorid oder Benzotπazolyloxy-tri(dimethylamino)phosphoniumhexa- fluorophosphat, oder O-(Benzotriazol- 1 -yϊ)-N,N,N', N'-tetra-methyluroniumhexafluorophosphat (HBTU), 2-(2-Oxo-l-(2H)-pyridyl)-l,l,3,3-tetramethyluroniumtetrafluoroborat (TPTU) oder 0-(7-Azabenzotriazol-l -yl)-N,N,N' N -tetramethyluroniumhexafluorophosphat (HATU), oder 1-Hydroxybenztriazol (HOBt), oder Benzotriazol-l-yloxytris(dimethylamino)-phosphoniumhexa- fluoro-phosphat (BOP), oder Mischungen aus diesen, oder Mischung aus diesen zusammen mit Basen.
Basen sind beispielsweise Alkalicarbonate, wie z.B. Natrium- oder Kaliumcarbonat, oder -hydrogencarbonat, oder organische Basen wie Trialkylamine z.B. Triethylamin, N-Methyl- morpholin, N-Methylpiperidin, 4-Dimethylaminopyridin oder Diisopropylethylamin.
Vorzugsweise wird die Kondensation mit HATU in Gegenwart einer Base, insbesondere Diisopropylethylamin, oder mit EDC und HOBt in Gegenwart einer Base, insbesondere Triethylamin, durchgeführt.
Inerte Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan oder Trichlormethan, Kohlenwasserstoff wie Benzol, oder Νitromethan, Dioxan, Dimethylformamid oder Acetonitril. Ebenso ist es möglich, Gemische der Lösemittel einzusetzen. Besonders bevorzugt ist Dimethylformamid.
Die Umsetzung mit einer Säure in der zweiten Stufe der Verfahren [A] und [B] erfolgt bevorzugt in einem Temperaturbereich von O°C bis 40°C bei Normaldruck.
Als Säuren eignen sich hierbei Chlorwasserstoff in Dioxan, Bromwasserstoff in Essigsäure oder Trifluoressigsäure in Methylenchlorid.
Die Hydrogenolyse in der zweiten Stufe des Verfahrens [B] erfolgt im Allgemeinen in einem Lösungsmittel in Gegenwart von Wasserstoff und Palladium auf Aktivkohle, bevorzugt in einem Temperaturbereich von O°C bis 40°C bei Normaldruck.
Lösungsmittel sind beispielsweise Alkohole wie Methanol, Ethanol, n-Propanol oder iso-Propanol, in einem Gemisch mit Wasser und Eisessig, bevorzugt ist ein Gemisch aus Ethanol, Wasser und Eisessig.
Die Verbindungen der Formel (EI) sind bekannt oder können analog bekannten Verfahren hergestellt werden.
Die Verbindungen der Formel (E) sind bekannt oder können hergestellt werden, indem Ver- bindungen der Formel
worin R2, R7 und R26 die oben angegebene Bedeutung haben,
mit Di-(tert-butyl)-dicarbonat in Gegenwart einer Base umgesetzt werden.
Die Umsetzung erfolgt im Allgemeinen in einem Lösungsmitteln, bevorzugt in einem Tem- peraturbereich von 0°C bis 4O°C bei Normaldruck.
Basen sind beispielsweise Alkalihydroxide wie Natrium- oder Kaliumhydroxid, oder Alkali- carbonate wie Cäsiumcarbonat, Natrium- oder Kaliumcarbonat, oder andere Basen wie DBU, Triethylamin oder Diisopropylethylamin, bevorzugt ist Natriumhydroxid oder Natriumcarbonat.
Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Methylenchlorid oder 1,2-Di- chlorethan, Alkohole wie Methanol, Ethanol oder iso-Propanol, oder Wasser.
Vorzugsweise wird die Umsetzung mit Natriumhydroxid in Wasser oder Natriumcarbonat in Methanol durchgeführt.
Die Verbindungen der Formel (V) sind bekannt oder können hergestellt werden, indem Verbindungen der Formel
(VI),
worin R
2, R
7 und R
26 die oben angegebene Bedeutung haben, und
R27 gleich Benzyl, Methyl oder Ethyl ist,
mit einer Säure oder durch Hydrogenolyse, wie für die zweite Stufe des Verfahrens [B] beschrieben, gegebenenfalls durch anschließende Umsetzung mit einer Base zur Verseifung des Methyl- oder Ethylesters, umgesetzt werden.
Die Verseifung kann zum Beispiel erfolgen, wie bei der Umsetzung von Verbindungen der Formel (VI) zu Verbindungen der Formel (IV) beschrieben.
Die Verbindungen der Formel (IV) sind bekannt oder können hergestellt werden, indem in Verbindungen der Formel (VI) der Benzyl-, Methyl- oder Ethylester verseift wird.
Die Umsetzung erfolgt im Allgemeinen in einem Lösungsmitteln, in Gegenwart einer Base, bevorzugt in einem Temperaturbereich von O°C bis 4O°C bei Normaldruck.
Basen sind beispielsweise Alkalihydroxide wie Lithium-, Natrium- oder Kaliumhydroxid, bevorzugt ist Lithiumhydroxid.
Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan oder Trichlor- methan, Ether wie Tetrahydrofuran oder Dioxan, oder Alkohole wie Methanol, Ethanol oder Iso- propanol, oder Dimethylformamid. Ebenso ist es möglich, Gemische der Lösungsmittel oder Gemische der Lösungsmittel mit Wasser einzusetzen. Besonders bevorzugt sind Tetrahydrofuran oder ein Gemisch aus Methanol und Wasser.
Die Verbindungen der Formel (VI) sind bekannt oder können hergestellt werden, indem Verbindungen der Formel
(vπ),
worin R ,2 , r R,7 , τ R,26 , und R >27 die oben angegebene Bedeutung haben,
in der ersten Stufe mit Säuren, wie für die zweite Stufe der Verfahren [A] und [B] beschrieben, und in der zweiten Stufe mit Basen umgesetzt werden.
In der zweiten Stufe erfolgt die Umsetzung mit Basen im Allgemeinen in einem Lösungsmitteln, bevorzugt in einem Temperaturbereich von O°C bis 40°C bei Normaldruck.
Basen sind beispielsweise Alkalihydroxide wie Natrium- oder Kaliumhydroxid, oder Alkalicarbo- nate wie Cäsiumcarbonat, Natrium- oder Kaliumcarbonat, oder andere Basen wie DBU, Triethyl- amin oder Diisopropylethylamin, bevorzugt ist Triethylamin.
Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Chloroform, Methylenchlorid oder 1,2-Dichlorethan, oder Tetrahydrofuran, oder Gemische der Lösungsmittel, bevorzugt ist Methylenchlorid oder Tetrahydrofuran.
Die Verbindungen der Formel (VII) sind bekannt oder können hergestellt werden, indem Verbindungen der Formel
(VTS),
worin R , R
7, R und R die oben angegebene Bedeutung haben,
mit Pentafluorphenol in Gegenwart von Dehydratisierungsreagenzien, wie für die erste Stufe der Verfahren [A] und [B] beschrieben, umgesetzt werden.
Die Umsetzung erfolgt bevorzugt mit DMAP und EDC in Dichlormethan in einem Temperaturbereich von -4O°C bis 40°C bei Normaldruck.
Die Verbindungen der Formel (VIII) sind bekannt oder können hergestellt werden, indem Verbindungen der Formel
(IX),
worin R
2, R
7, R
26 und R
27 die oben angegebene Bedeutung haben,
mit Fluorid, insbesondere mit Tetrabutylammoniumfluorid, umgesetzt werden.
Die Umsetzung erfolgt im Allgemeinen in einem Lösungsmitteln, bevorzugt in einem Temperaturbereich von -1O°C bis 3O°C bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, oder Kohlenwasserstoffe wie Benzol oder Toluol, oder Ether wie Tetrahydrofuran oder Dioxan, oder Dimethylformamid. Ebenso ist es möglich, Gemische der Lösemittel einzusetzen. Bevorzugte Lösungsmittel sind Tetrahydrofuran und Dimethylformamid.
Die Verbindungen der Formel (IX) sind bekannt oder können hergestellt werden, indem Verbindungen der Formel
(X),
worin R
2, R
26 und R
27 die oben angegebene Bedeutung haben,
mit Verbindungen der Formel
(XI),
worin R
7 die oben angegebene Bedeutung hat,
in Gegenwart von Dehydratisierungsreagenzien, wie für die erste Stufe der Verfahren [A] und [B] beschrieben, umgesetzt werden.
Die Verbindungen der Formel (X) sind bekannt oder können analog den im Beispielteil beschriebenen Verfahren hergestellt werden.
Die Verbindungen der Formel (XI) sind bekannt oder können analog bekannten Verfahren herge- stellt werden.
Die erfindungsgemäßen Verbindungen zeigen ein nicht vorhersehbares, wertvolles pharmakologisches und pharmakokinetisches Wirkspektrum.
Sie eignen sich daher zur Verwendung als Arzneimittel zur Behandlung und/oder Prophylaxe von Krankheiten bei Menschen und Tieren.
Die erfindungsgemäßen Verbindungen können aufgrund ihrer pharmakologischen Eigenschaften allein oder in Kombination mit anderen Wirkstoffen zur Behandlung und/oder Prophylaxe von Infektionskrankheiten, insbesondere von bakteriellen Infektionen, eingesetzt werden.
Beispielsweise können lokale und/oder systemische Erkrankungen behandelt und/oder verhindert werden, die durch die folgenden Erreger oder durch Mischungen der folgenden Erreger verursacht werden:
Gram-positive Kokken, z.B. Staphylokokken (Staph. aureus, Staph. epidermidis) und Streptokokken (Strept. agalactiae, Strept. faecalis, Strept. pneumoniae, Strept. pyogenes); gram-negative Kokken (neisseria gonorrhoeae) sowie gram-negative Stäbchen wie Enterobakteriaceen, z.B. Escherichia coli, Hämophilus influenzae, Citrobacter (Citrob. freundii, Citrob. divernis), Salmonella und Shigella; ferner Klebsiellen (Klebs. pneumoniae, Klebs. oxytocy), Enterobacter (Ent. aerogenes, Ent. agglomerans), Hafnia, Serratia (Serr. marcescens), Proteus (Pr. mirabilis, Pr. rettgeri, Pr. vulgaris), Providencia, Yersinia, sowie die Gattung Acinetobacter. Darüber hinaus
umfaßt das antibakterielle Spektrum die Gattung Pseudomonas (Ps. aeruginosa, Ps. maltophilia) sowie strikt anaerobe Bakterien wie z.B. Bacteroides fragilis, Vertreter der Gattung Peptococcus, Peptostreptococcus sowie die Gattung Clostridium; ferner Mykoplasmen (M. pneumoniae, M. hominis, M. urealyticum) sowie Mykobakterien, z.B. Mycobacterium tuberculosis.
Die obige Aufzählung von Erregern ist lediglich beispielhaft und keineswegs beschränkend aufzufassen. Als Krankheiten, die durch die genannten Erreger oder Mischinfektionen verursacht und durch die erfindungsgemäßen topisch anwendbaren Zubereitungen verhindert, gebessert oder geheilt werden können, seien beispielsweise genannt:
Infektionskrankheiten beim Menschen wie z. B. septische Infektionen, Knochen- und Gelenk- Infektionen, Hautinfektionen, postoperative Wundinfektionen, Abszesse, Phlegmone, Wundinfektionen, infizierte Verbrennungen, Brandwunden, Infektionen im Mundbereich, Infektionen nach Zahnoperationen, septische Arthritis, Mastitis, Tonsillitis, Genital-Infektionen und Augeninfektionen.
Außer beim Menschen können bakterielle Infektionen auch bei anderen Spezies behandelt werden. Beispielhaft seien genannt:
Schwein: Coli-diarrhoe, Enterotoxamie, Sepsis, Dysenterie, Salmonellose, Metritis-Mastitis-Aga- laktiae-Syndrom, Mastitis;
Wiederkäuer (Rind, Schaf, Ziege): Diarrhoe, Sepsis, Bronchopneumonie, Salmonellose, Pasteurellose, Mykoplasmose, Genitalinfektionen;
Pferd: Bronchopneumonien, Fohlenlähme, puerperale und postpuerperale Infektionen, Salmonellose;
Hund und Katze: Bronchopneumonie, Diarrhoe, Dermatitis, Otitis, Harnwegsinfekte, Prostatitis;
Geflügel (Huhn, Pute, Wachtel, Taube, Ziervögel und andere): Mycoplasmose, E. coli-Infektionen, chronische Luftwegserkrankungen, Salmonellose, Pasteurellose, Psittakose.
Ebenso können bakterielle Erkrankungen bei der Aufzucht und Haltung von Nutz- und Zierfischen behandelt werden, wobei sich das antibakterielle Spektrum über die vorher genannten Erreger hinaus auf weitere Erreger wie z.B. Pasteurella, Brucella, Campylobacter, Listeria, Erysipelothris, Corynebakterien, Borellia, Treponema, Nocardia, Rikettsie, Yersinia, erweitert.
Weiterer Gegenstand der vorliegenden Erfindung ist der Einsatz der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, vorzugsweise von bakteriellen Krankheiten, insbesondere von bakteriellen Infektionen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer antibakteriell wirksamen Menge der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende schnell und/oder modifiziert die erfindungsgemäßen Verbindungen abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/ oder amorphisierter und/oder gelöster
Form enthalten, wie z.B. Tabletten (nichtüberzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die
Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfal- lende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder
Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen, -sprays; lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augenpräparationen, Vaginalkapseln, wässrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (wie beispielsweise Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (bei- spielsweise mikrokristalline Cellulose, Laktose, Mannitol), Lösungsmittel (z.B. flüssige PoIy- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdode- cylsulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisen- oxide) und Geschmacks- und / oder Geruchskorrigentien.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfϊn- dungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 5 bis 250 mg/kg Körpergewicht je 24 h zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Menge etwa 5 bis 100 mg/kg Körpergewicht je 24 h.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Verwendete Abkürzungen:
abs. absolut aq. wässrig
Bn Benzyl boc tert-Butoxycarbonyl
Bsp. Beispiel
CDCl3 Chloroform
CH Cyclohexan d dublett (im 1H-NMR) dd dublett von dublett (im 1H-NMR)
DC Dünnschichtchromatographie
DCC Dicyclohexylcarbodiimid
DIC Diisopropylearbodiirnid
DIEA Diisopropylethylamin (Hünig-Base)
DMSO Dimethylsulfoxid
DMAP 4-N,N-Dimethylaminopyridin
DMF Dimethylformamid d. Th. der Theorie
EDC N'-(3-Dimethylaminopropyl)-N-ethylcarbodiimid x HCl
EE Ethylacetat (Essigsäureethylester)
ESI Elektrospray-Ionisation (bei MS)
Fraoc 9-Fluorenylmethoxycarbonyl ges. gesättigt
HATU O-(7-Azabenzotriazol-l-yl)-N,NN'N'-tetramethyluronium hexafluorophosphat
HBTU 0-(Benzotriazol-l-yl)-NN,N'N'-tetramethyluroniurn- hexafluorophosphat
HOBt 1-Hydroxy-lH-benzotriazol x H2O h Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
LC-MS Flüssigchromatographie-gekoppelte Massenspektroskopie m multiple« (im 1H-NMR) min Minute
MS Massenspektroskopie
NMR Kernresonanzspektroskopie
MTBE Methyl-tert-butylether
Pd/C Palladium/Kohle
PFP Pentafluorphenol proz. Prozent q quartett (im 1H-NMR)
Rf Retentionsindex (bei DC)
RP Reverse Phase (bei HPLC)
RT Raumtemperatur
Rt Retentionszeit (bei HPLC)
S singulett (im 1H-NMR) t triplett (im 1H-NMR)
TBS tert-Butyldimethylsilyl
TFA Trifluoressigsäure
THF Tetrahydrofuran
TMSE 2-(Trimethylsilyl)-ethyl
TPTU 2-(2-Oxo- 1 (2H)-pyridyl)- 1,1,3,3 -tetramethy luroniumtetrafluoroborat
Z Benzyloxycarbonyl
LC-MS- und HPLC-Methoden:
Methode 1 (LC-MS): Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A -» 2.5 min 30%A -> 3.0 min 5%A -> 4.5 min 5%A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 5O°C; UV-Detektion: 208- 400 nm.
Methode 2 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A -> 2.5 min 30%A -> 3.0 min 5%A -> 4.5 min 5%A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 5O°C; UV-Detektion: 210 nm.
Methode 3 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV
DAD; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient:
0.0 min 90%A -> 2.5 min 30%A -» 3.0 min 5%A -> 4.5 min 5%A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min. 2 ml/min; Ofen: 5O°C; UV-Detektion: 210 nm.
Methode 4 (LC-MS); Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Grom-SIL120 ODS-4 HE, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50%ige Ameisensäure; Gradient: 0.0 min 100%A -> 0.2 min 100%A -> 2.9 min 30%A -> 3.1 min 10%A -> 4.5 min 10%A; Ofen: 55°C; Fluss: 0.8 ml/min; UV-Detektion: 208-400 nm.
Methode 5 (LC-MS); Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith SpeedROD RP-ISe 50 mm x 4.6 mm; Eluent A: Wasser + 500 μl 50%ige Ameisensäure / 1; Eluent B: Acetonitril + 500 μl 50%ige Ameisensäure / 1; Gradient: 0.0 min 10%B -> 3.0 min 95%B -> 4.0 min 95%B; Ofen: 35°C; Fluss: 0.0 min 1.0 ml/min -> 3.0 min 3.0 ml/min -> 4.0 min 3.0 ml/min; UV-Detektion: 210 nm.
Methode 6 (LC-MS); Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Grom-Sil 120 ODS-4 HE 50 mm x 2 mm, 3.0 μm; Eluent A: Wasser + 500 μl 50%ige Ameisensäure / 1, Eluent B: Acetonitril + 500 μl 50%ige Ameisensäure / 1; Gradient: 0.0 min 0%B -> 2.9 min 70%B -> 3.1 min 90%B -» 4.5 min 90%B; Ofen: 50 °C; Fluss: 0.8 ml/min; UV- Detektion: 210 nm.
Methode 7 (LC-MS); Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Grom-Sil 120 ODS-4 HE 50 mm x 2 mm, 3.0 μm; Eluent A: Wasser + 500 μl 50%ige Ameisensäure; Eluent B: Acetonitril + 500 μl 50%ige Ameisensäure / 1; Gradient: 0.0 min 5%B -> 2.0 min 40%B -» 4.5 min 90%B -> 5.5 min 90%B; Ofen: 45°C; Fluss: 0.0 min 0.75 ml/min -> 4.5 min 0.75 ml/min 5.5 min -> 5.5 min 1.25 ml/min; UV-Detektion: 210 nm.
Methode 8 (LC-MS); Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Thermo HyPURITY Aquastar 3μ 50 mm x 2.1 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 100%A -> 0.2 min 100%A -> 2.9 min 30%A -> 3.1 min 10%A -> 5.5 min 10%A; Ofen: 5O°C; Fluss: 0.8 ml/min; UV-Detektion: 210 nm.
Methode 9 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Grom-Sil 120 ODS-4 HE 50 x 2 mm, 3.0 μm; Eluent B: Acetonitril + 0.05% Ameisensäure, Eluent A: Wasser + 0.05% Ameisensäure; Gradient: 0.0 min 70%B -> 4.5 min 90%B -> 5.5 min 90%B; Ofen: 45°C; Fluss: 0.0 min 0.75 ml/min -> 4.5 min 0.75 ml/min -» 5.5 min 1.25 ml/min; UV-Detektion: 210 nm.
Methode 10 (LCMS): Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Thermo Hypersil GOLD-3μ 20 x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 100%A -» 0.2 min 100%A ■» 2.9 min 30%A -» 3.1 min 10%A -» 5.5 min 10%A; Ofen: 5O°C; Fluss: 0.8 ml/min; UV-Detektion: 210 nm.
Methode 11 (HPLC): Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil RP-18, 60 mm x 2 mm, 3.5 μm; Eluent A: 5 ml HC1O4/1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2%B, 0.5 min 2%B, 4.5 min 90%B, 6.5 min 90%B; Fluss: 0.75 ml/min; Ofen: 30°C; UV-Detektion: 210 nm.
Methode 12 (HPLC): Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil RP-18, 60 mm x 2 mm, 3.5 μm; Eluent A: 5 ml HC1O4/1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2%B, 0.5 min 2%B, 4.5 min 90%B, 15 min 90%B; Fluss: 0.75 ml/min; Ofen: 30°C; UV-Detektion: 210 nm.
Ausgangsverbindungen
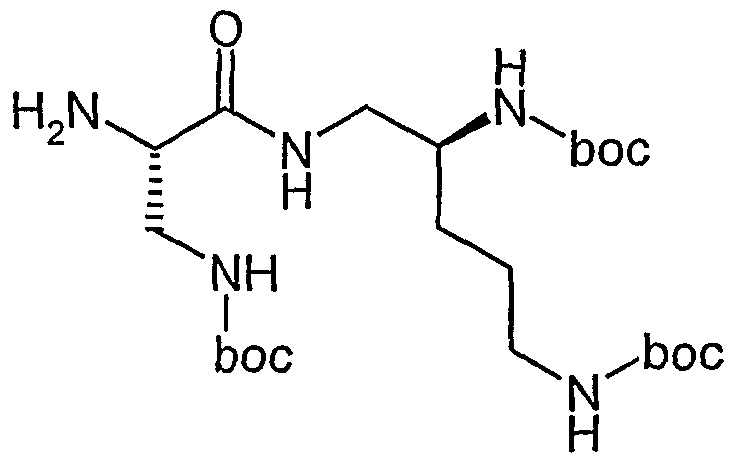
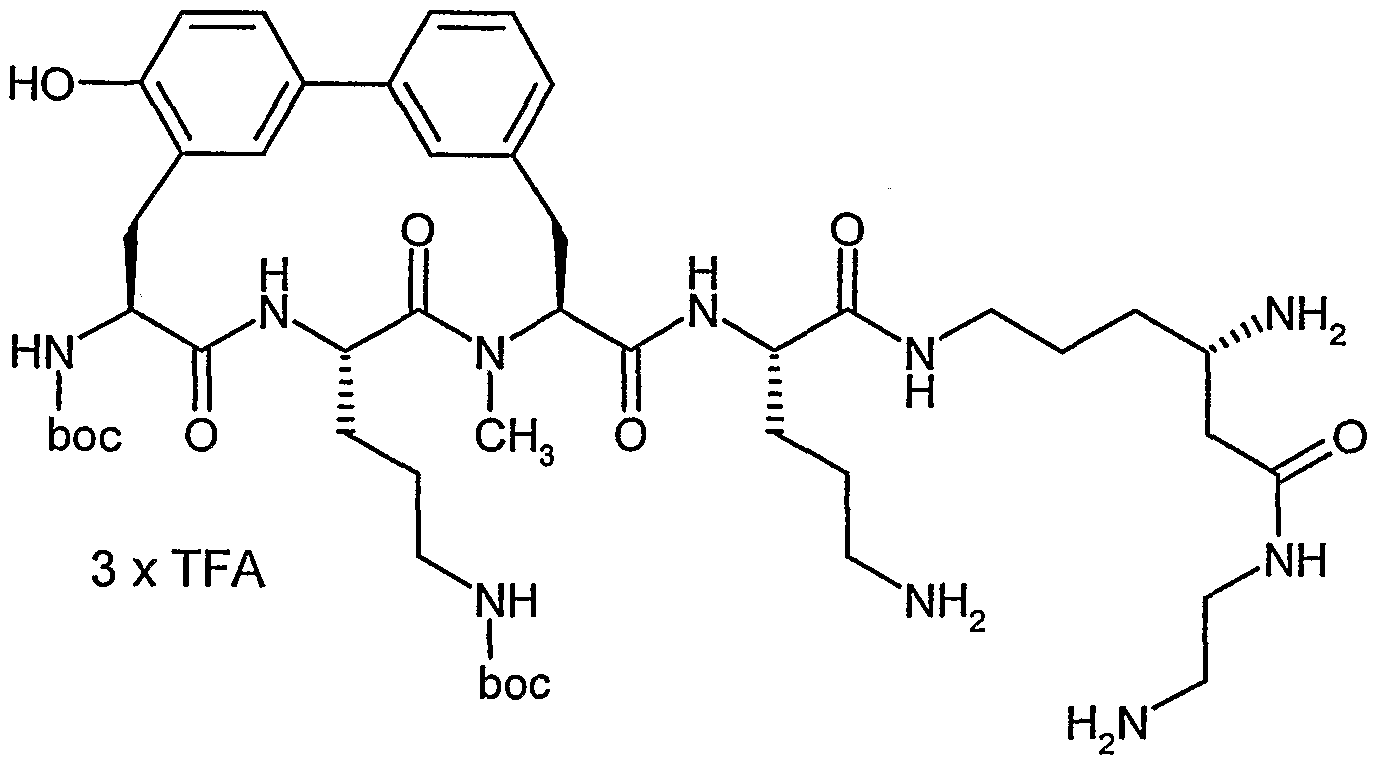
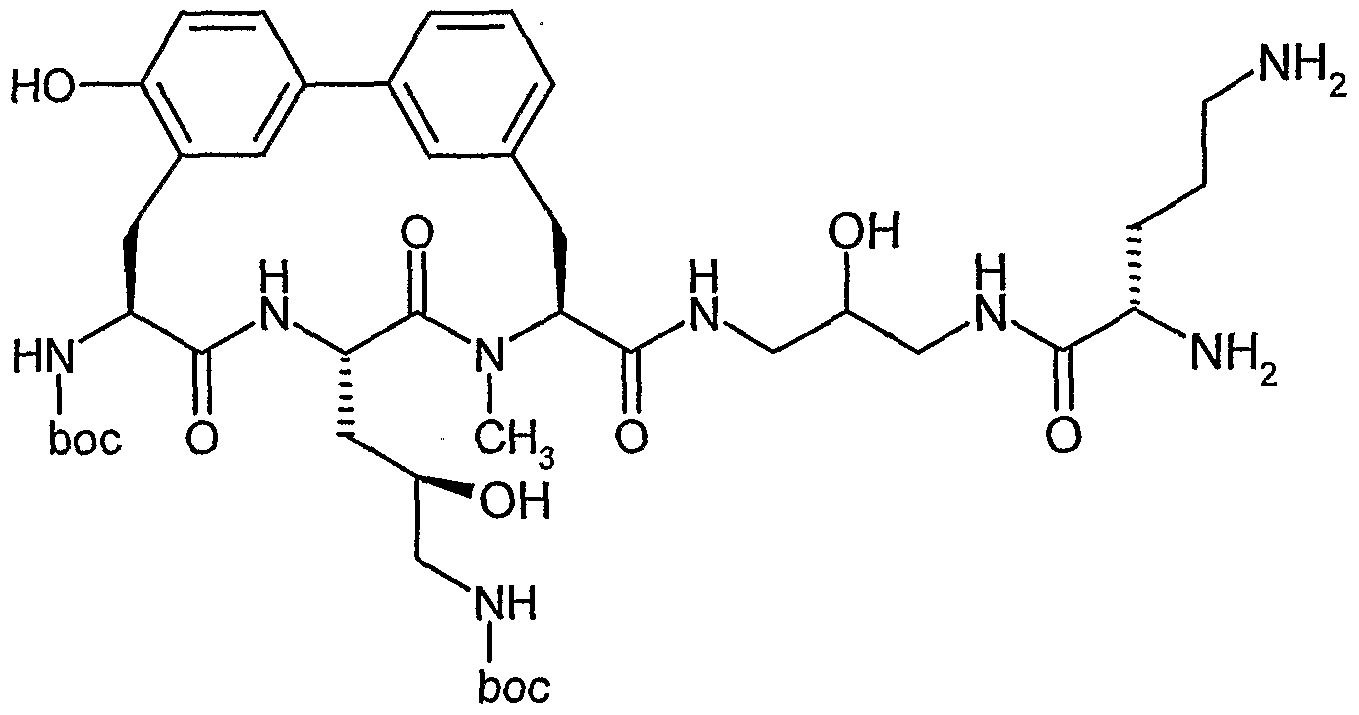
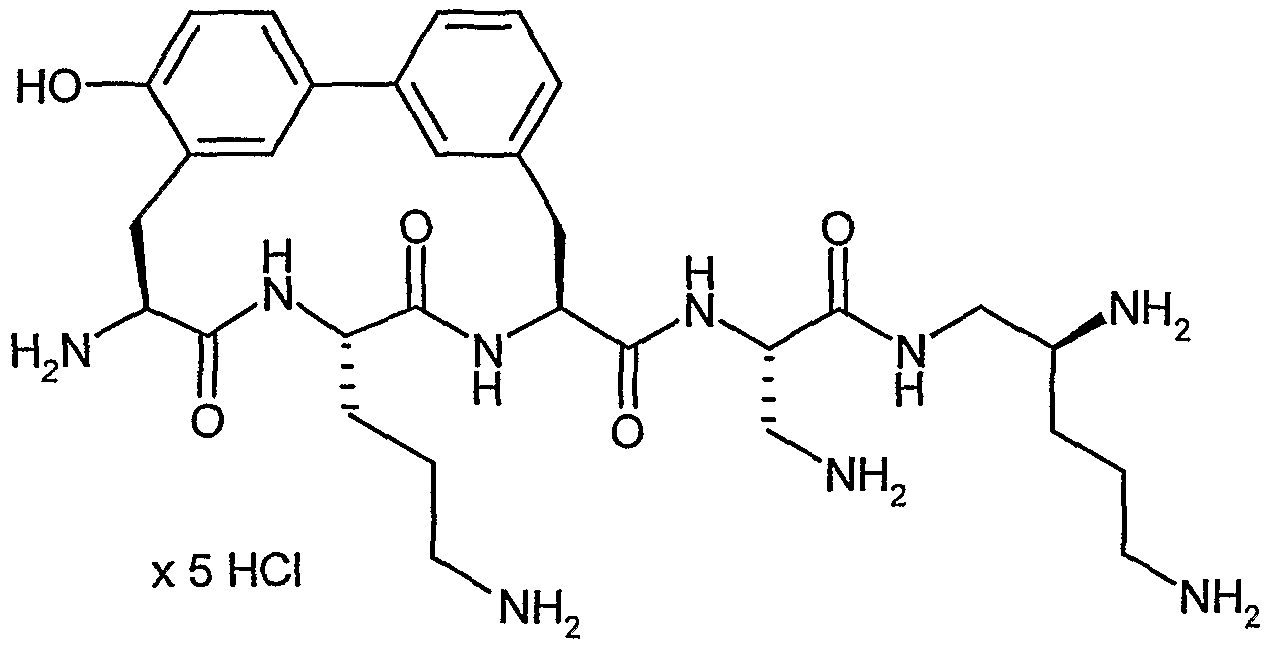
Beispiel IA
5-Brom-2-methylbenzaldehyd
77.7 g (583 mmol) Aluminiumtrichlorid werden in 200 ml Dichlormethan suspendiert und auf 0°C gekühlt. 40.0 g (333 mmol) 2-Methylbenzaldehyd werden innerhalb von 30 min zugetropft. Anschließend gibt man 53.2 g (333 mmol) Brom innerhalb von 6 h bei 0°C zu, lässt auf RT erwärmen und rührt 12 h nach. Die Reaktionslösung wird auf 500 ml Eiswasser gegeben. Die wässrige Phase wird mehrfach mit Dichlormethan extrahiert. Die vereinigten organischen Phasen werden nacheinander mit 2N Salzsäure, gesättigter wässriger Natriumhydrogencarbonat-Lösung und gesättigter wässriger Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet und im Vakuum eingeengt. Man reinigt per Kieselgelchromatographie und anschließend über Kristallisation aus Cyclohexan. Das ausgefallene Produkt wird abfiltriert.
Ausbeute: 3.2 g (5% d.Th.)
LC-MS (Methode 7): Rt = 3.26 min
MS (EI): m/z = 199 (M+H)+
Beispiel 2A
Methyl-(2Z)-3-(3-bromphenyl)-2-[(tert-butoxycarbonyl)amino]acrylat
Zu einer auf -7O°C gekühlten Lösung von 10 g (54.1 mmol) 3-Brombenzaldehyd und 17.7 g (59.5 mmol) Methyl-[(ter/-butoxycarbonyl)amino](dimethoxyphosphoryl)acetat in 200 ml wasserfreiem Tetrahydrofuran werden 7.48 ml (59.5 mmol) N,N,N,N-Tetramethylguanidin hinzugegeben. Nach 4 h Rühren bei -70°C wird das Reaktionsgemisch 15 h bei RT gerührt. Die Mischung wird mit 500 ml Wasser und 500 ml Essigsäureethylester versetzt. Die organische Phase wird mit Wasser gewaschen, über Natriumsulfat getrocknet und eingeengt. Das Rohprodukt wird säulen- chromatographisch an Silicagel (Laufmittel: Cyclohexan:Essigsäureethylester 4:1) gereinigt.
Ausbeute: quant.
LC-MS (Methode 3): Rj = 2.61 min.
MS (EI): m/z = 356 (M+H)+.
1H-NMR (300 MHz, DMSO-d6): δ = 1.40 (s, 9H), 3.73 (s, 3H), 7.15 (br.s, IH), 7.48 (m, IH), 7.56 (dd, IH), 7.63 (dd, IH), 7.86 (s, IH), 8.82 (br.s, IH).
Analog zu obiger Vorschrift wird Beispiel 3A aus den entsprechenden Edukten hergestellt:
Beispiel 4A
Methyl-3-brom-N-(^ert-butoxycarbonyl)-Z-phenylalaninat
10 g (28.1 mmol) Methyl-(2Z)-3-(3-bromphenyl)-2-[(tert-butoxycarbonyl)amino]aciylat (Beispiel 2A) werden in einer Mischung aus 150 ml Ethanol und 100 ml Dioxan gelöst. Unter Argonatmosphäre gibt man 100 mg (0.14 mmol) Hydrierkatalysator [(+)-l,2-Bis((2<S',55)-2,5- diethylphospholano)benzol-(cyclooctadien)rhodium(I)trifluormethansulfonat] hinzu und leitet 30 min Argon durch die Lösung. Anschließend wird für 5 Tage unter einem Wasserstoffdruck von 3 bar hydriert. Es wird über Kieselgel filtriert und sorgfaltig mit Ethanol nachgewaschen. Das Filtrat wird im Vakuum eingeengt und das Rohprodukt am Hochvakuum getrocknet.
Ausbeute: 9.2 g (89% d.Th.)
LC-MS (Methode 3): R1 = 2.63 min.
MS (EI): m/z = 358 (M+H)+
1H-NMR (400 MHz, DMSOd6): δ - 1.32 (s, 9H), 2.74 (mg, IH), 3.03 (m0, IH), 3.62 (s, 3H), 4.70 (mc, IH), 7.20-7.5 (m, 5H).
Analog zu obiger Vorschrift wird Beispiel 5A aus den entsprechenden Edukten hergestellt:
Beispiel 6A
Methyl-3-brom-N-(ter^butoxycarbonyl)-N-methyl-L-phenylalaninat
Zu einer Lösung von 16.5 g (43.86 mmol) Methyl-3-brom-N-(terM)utoxycarbonyl)-L-phenyl- alaninat (Beispiel 4A) in 220 ml wasserfreiem Tetrahydrofuran werden 49.8 g (350.86 mmol) Iodmethan und 2.28 g (57.01 mmol) Νatriumhydrid hinzugegeben. Die Reaktionsmischung wird bei RT über Nacht gerührt. Die Mischung wird mit 1000 ml Wasser und 1000 ml Essigsäureethylester versetzt. Die organische Phase wird nacheinander mit Wasser und gesättigter Natriumchlorid-lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Das Rohprodukt wird säulenchromatographisch an Silicagel (Laufmittel: Cyclohexan:Essigsäureethylester 3:1) gereinigt.
Ausbeute: quant.
HPLC (Methode 11): Rt = 5.1 min.
MS (DCI(NH3)): m/z = 390 (M+H)+.
1H-NMR (400 MHz, CDCl3): δ = 1.48 (d, 9H), 2.23 (d, 3H), 3.09 (dd, IH), 3.30 (dd, IH), 3.75 (s, 3H), 4.70 (ddd, IH), 6.92 (dd, IH), 7.30 (m, 2H).
Beispiel 7A
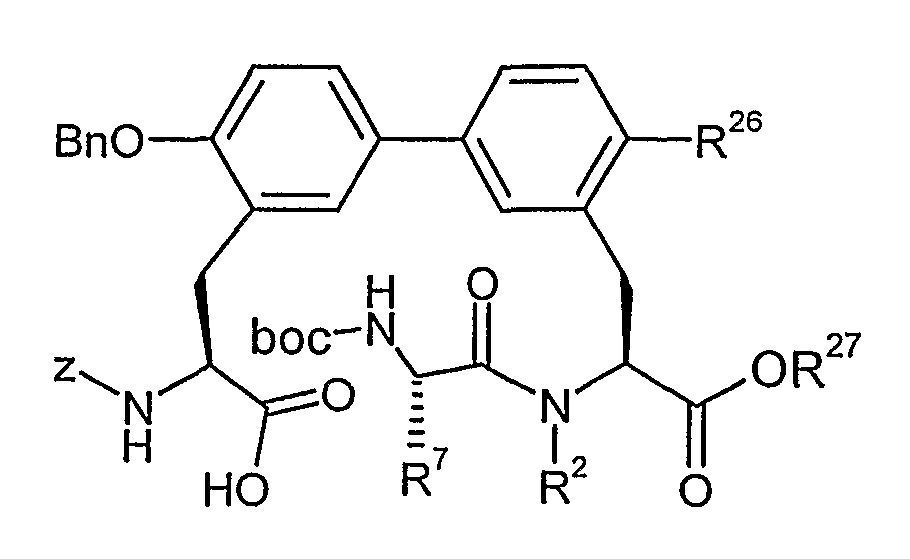
Methyl-(21S)-3-(4'-(benzyloxy)-3'-{(2S)-2-{[(benzyloxy)carbonyl]amino}-3-oxo-3-[2-(trimethyl- silyl)ethoxy]propyl}biphenyl-3-yl)-2-[(ter^butoxycarbonyl)amino]propanoat
Eine Lösung von 6.0 g (16.8 mmol) Methyl-3-brom-N-(fer/-butoxycarbonyl)-N-methyl--L-phenyl- alaninat (Beispiel 4A) und 11.7 g (18.4 mmol) 2-(Trimethylsilyl)ethyl-2-(benzyloxy)-N-[(benzyl- oxy)carbonyl]-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-Z-phenylalaninat (Beispiel 84A aus WO03/106480) in 80 ml l-Methyl-2-pyrrolidon und 4 ml Wasser wird inertisiert und mit Argon gesättigt. Anschließend gibt man 1.37 g (1.67 mmol) Bis(diphenylphosphino)ferrocen-palla- dium(II)chlorid (PdCl2(dppf)) und 11 g (34 mmol) Cäsiumcarbonat hinzu. Das Reaktionsgemisch wird mit Argon leicht überströmt und für 10 h bei 5O°C gerührt. Die Mischung wird abgekühlt, in Dichlormethan aufgenommen und mit Wasser gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet und das Lösungsmittel wird im Vakuum eingeengt. Der Rückstand wird säulenchromatographisch an Kieselgel gereinigt (Cyclohexan:Essigsäureethylester 15:1 -^ 7:1).
Ausbeute: 6.82 g (52% d. Th.).
LC-MS (Methode 1): R, = 3.41 min
MS (EI): m/z = 783 (M+H)+.
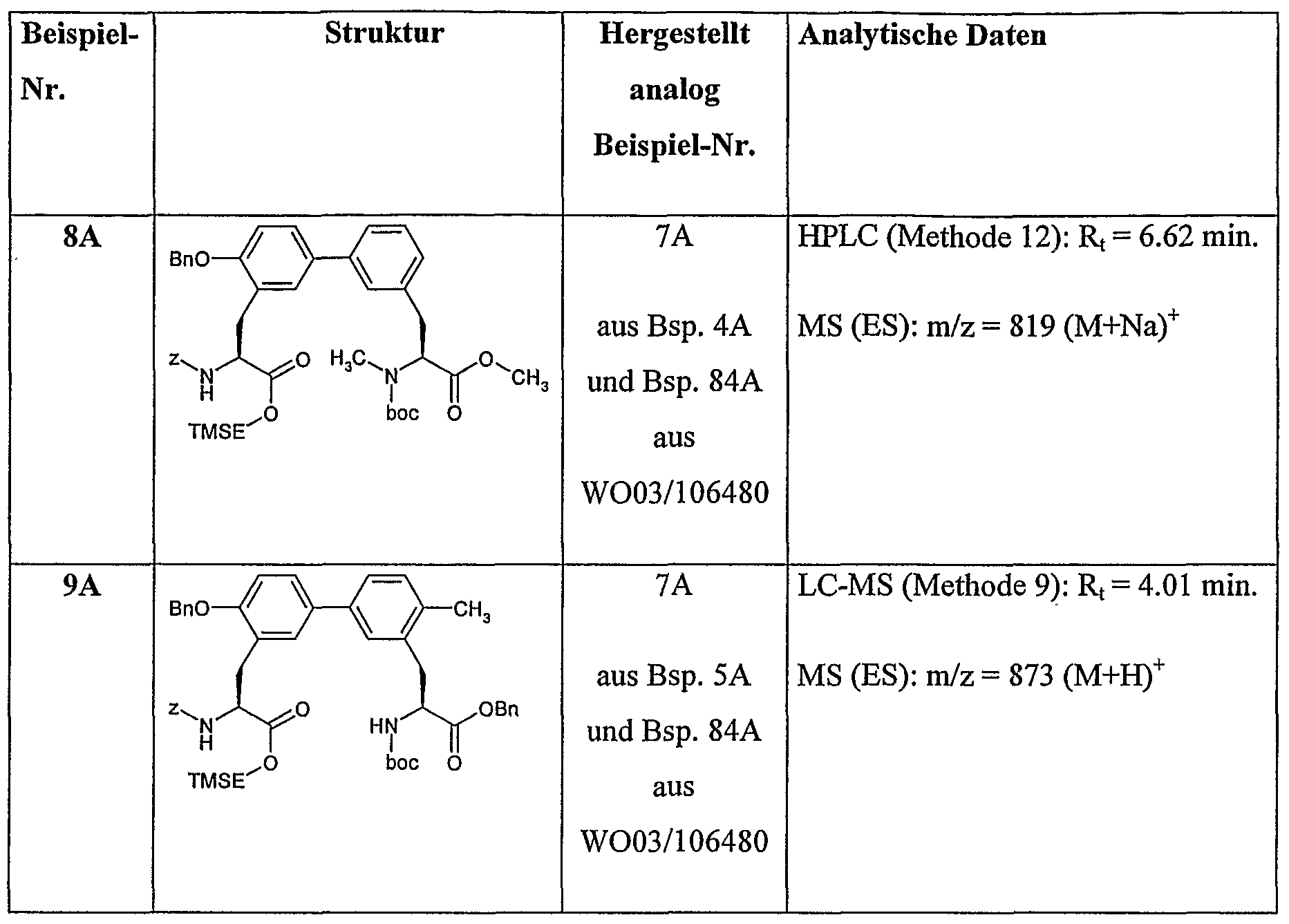
Analog zu obiger Vorschrift werden die in der folgenden Tabelle aufgeführten Beispiele 8A und 9A aus den entsprechenden Edukten hergestellt:
Beispiel IQA
Methyl-(25)-2-amino-3-(4'-(benzyloxy)-3'-{(2>S)-2-{[(benzyloxy)carbonyl]amino}-3-oxo-3-[2- (trimethylsilyl)ethoxy]propyl}biphenyl-3-yl)propanoat Hydrochlorid
Zu einer auf O°C gekühlten Lösung von 4.0 g (3.6 mmol) der Verbindung aus Beispiel 7A in 10 ml wasserfreiem Dioxan werden 54 ml einer 4M Chlorwasserstoff-Dioxan-Lösung hinzugegeben. Nach 3 h Rühren wird das Lösungsmittel im Vakuum eingedampft, mehrmals mit Dichlormethan coevaporiert und im Hochvakuum bis zur Gewichtskonstanz getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 2): R, = 2.24 min.
MS (EI): m/z = 683 (M-HCB-H)+.
Analog zu obiger Vorschrift werden die in der folgenden Tabelle aufgeführten Beispiele 1 IA und 12A aus den entsprechenden Edukten hergestellt:
Beispiel 13A
2-(Trimethylsilyl)ethyl-(21y)-3-(4-(benzyloxy)-3T-{(2lS)-2-[((2S,4R)-5-{[(benzyloxy)carbonyl]- amino}-2-[(tert-butoxycarbonyl)amino]-4-{[^ert-butyl(dimethyl)silyl]oxy}pentanoyl)amino]-3- methoxy-3 -oxopropy 1 } bipheny 1-3 -y l)-2- { [(benzy loxy)carbony 1] amino } propanoat
Zu einer Lösung von 1.91 g (2.66 mmol) der Verbindung aus Beispiel 1OA und 1.45 g (2.92 mmol) (25',4i?)-5-{[(Benzyloxy)carbonyl]amino}-2-[(tert-butoxycarbonyl)amino]-4-{[fe^-butyl-(di- methyl)silyl]oxy}pentansäure (Beispiel 14A aus WO03/106480) in 20 ml abs. DMF werden bei O°C (Badtemperatur) 1.26 g (3.32 mmol) HATU und 1.1 ml (6.2 mmol) Hünig-Base gegeben. Man rührt 30 min. bei dieser Temperatur, versetzt dann mit weiteren 0.55 ml (1.1 mmol) Hünig-Base und lässt die Temperatur auf RT ansteigen. Nach Reaktion über Nacht engt man alles im Vakuum zur Trockne ein und der Rückstand wird in Dichlormethan aufgenommen. Die organische Phase wird mit Wasser und gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Das Rohprodukt wird chromatographisch an Silicagel gereinigt (Laufmittel: Cyclohexan / Essigsäureethylester 5:1 -> 3:1).
Ausbeute: 1.89 g (61% d.Th.)
LC-MS (Methode 3): R, = 3.66 min.
MS (EI): m/z = 1161 (M+H)+
Beispiel 14A
2-(Trimethylsilyl)ethyl-(2S)-3-{4-(benzyloxy)-3'-[(2S)-2-({(2,S)-5-{[(benzyloxy)carbonyl]amino}-
2-[(te^butoxycarbonyl)amino]pentanoyl}amino)-3-methoxy-3-oxopropyl]biphenyl-3-yl}-2-
{[(benzyloxy)carbonyl]amino}propanoat
Zu einer Lösung von 1.55 g (2.16 mmol) der Verbindung aus Beispiel 1OA und 0.95 g (2.59 mmol)
N5-[(Benzyloxy)carbonyl]-N2-(tert-butoxycarbonyl)-I-ornithin in 28 ml abs. DMF werden bei O°C
(Badtemperatur) 1.03 g (2.7 mmol) HATU und 1.1 ml (6.1 mmol) Hünig-Base gegeben. Man rührt 30 min. bei dieser Temperatur, versetzt dann mit weiteren 0.3 ml (1.5 mmol) Hünig-Base und lässt die Temperatur auf RT ansteigen. Nach Reaktion über Nacht engt man alles im Vakuum zur
Trockne ein und der Rückstand wird in Dichlormethan aufgenommen. Die organische Phase wird mit Wasser und gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Das Rohprodukt wird chromatographisch an Silicagel gereinigt (Laufmittel: Dichlormethan / Essigsäureethylester 30:1 -> 5:1).
Ausbeute: 1.67 g (75% d.Th.)
LC-MS (Methode 1): R, = 3.40 min.
MS (EI): m/z = 1031 (M+H)+
Analog zu den angegebenen Vorschriften werden die in der folgenden Tabelle aufgeführten Beispiele 15A bis 17A aus den entsprechenden Edukten hergestellt:
Beispiel 18A
(2S)-3-{4-(Benzyloxy)-3'-[(2S)-2-({(2S',4R)-5-{[(benzyloxy)carbonyl]amino}-2-[(tert-butoxy- carbonyl)amino]-4-hydroxypentanoyl}amino)-3-methoxy-3-oxopropyl]biphenyl-3-yl}-2-{[(benzyl- oxy)carbonyl]amino}propansäure
Zu einer Lösung von 1.89 g (1.63 mmol) der Verbindung aus Beispiel 13A in 10 ml abs. DMF werden unter Rühren 4.88 ml (4.88 mmol) einer IN Tetra-n-butylammoniumfluorid-Lösung in THF gegeben. Nach 2 h bei RT wird auf O°C abgekühlt und mit Eiswasser und etwas 0.5 N Salzsäure versetzt. Es wird sofort mit Essigsäureethylester extrahiert. Die organische Phase wird über Magnesiumsulfat getrocknet, im Vakuum eingeengt und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 3): R, = 2.90 min.
MS (EI): m/z = 947 (M+H)+
Beispiel 19A
(2_S)-3 - {4-(Benzyloxy)-3'-[(2iS)-2-( {(2S)-5- { [(benzy loxy)carbonyl]amino } -2-[(tert-butoxy- carbonyl)amino]pentanoyl}amino)-3-methoxy-3-oxopropyl]biphenyl-3-yl}-2-{[(benzyloxy)- carbonyl]amino}propansäure
Zu einer Lösung von 2.38 g (1.79 mmol) der Verbindung aus Beispiel 14A in 35 ml absolutem DMF werden tropfenweise 3.58 ml IN Tetra-n-butylammoniumfluorid-Lösung in THF hinzugegeben. Nach 2 h bei RT wird auf O°C abgekühlt und mit Eiswasser und etwas 0.5 N Salzsäure versetzt. Es wird sofort mit Essigsäureethylester extrahiert. Die organische Phase wird über Magnesiumsulfat getrocknet, im Vakuum eingeengt und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 2): Rt = 2.88 min.
MS (EI): m/z = 931 (M+H)+.
Analog zu den angegebenen Vorschriften werden die in der folgenden Tabelle aufgeführten Beispiele 2OA bis 22A aus den entsprechenden Edukten hergestellt:
Beispiel 23A
PentafluorphenyI-(2S)-3-{4-(benzyloxy)-3'-[(2S)-2-({(2S,4R)-5-{[(benzyloxy)carbonyl]amino}-2- [(tert-butoxycarbonyl)amino]-4-hydroxypentanoyl}amino)-3-methoxy-3-oxopropyl]biphenyl-3- yl } -2- { [(benzy loxy)carbony I]amino } propanoat
Eine Lösung aus 1.54 g (1.63 mmol) der Verbindung aus Beispiel 18A in 50 ml abs. Dichlor- methan wird auf -20°C abgekühlt und unter Rühren mit 1.2 g (6.52 mmol) Pentafluorphenyl, 0.02 g (0.16 mmol) DMAP und 0.48 g (2.12 mmol) EDC versetzt. Man lässt die Temperatur langsam auf RT ansteigen und rührt über Nacht nach. Es wird im Vakuum eingeengt und das Rohprodukt im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 1.8 g (99% d.Th.)
LC-MS (Methode 2): R1 = 3.14 min.
MS (EI): m/z = 1113 (MH-H)+
Beispiel 24A
Pentafluorpheny l-(25)-3- {4-(benzyloxy)-3 '-[(2$)-2-({(2S)-5-{ [(benzy loxy)carbonyl]amino } -2- [(^er/-butoxycarbonyl)amino]pentanoyl}amino)-3-methoxy-3-oxopropyl]biphenyl-3-yl}-2- { [(benzy loxy)carbonyl] amino} propanoat
Eine Lösung aus 1.67 g (1.79 mmol) der Verbindung aus Beispiel 19A in 70 ml abs. Dichlor- methan wird auf -20°C abgekühlt und unter Rühren mit 1.65 g (8.95 mmol) Pentafluorphenyl, 0.025 g (0.18 mmol) DMAP und 0.53 g (2.33 mmol) EDC versetzt. Man lässt die Temperatur langsam auf RT ansteigen und rührt über Nacht nach. Es wird im Vakuum eingeengt und das Rohprodukt im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: quant.
LC-MS (Methode 3): R, = 3.47 min.
MS (EI): m/z = 1097 (M+H)+
Analog zu den angegebenen Vorschriften werden die in der folgenden Tabelle aufgeführten Beispiele 25A bis 27A aus den entsprechenden Edukten hergestellt:
Beispiel 28A
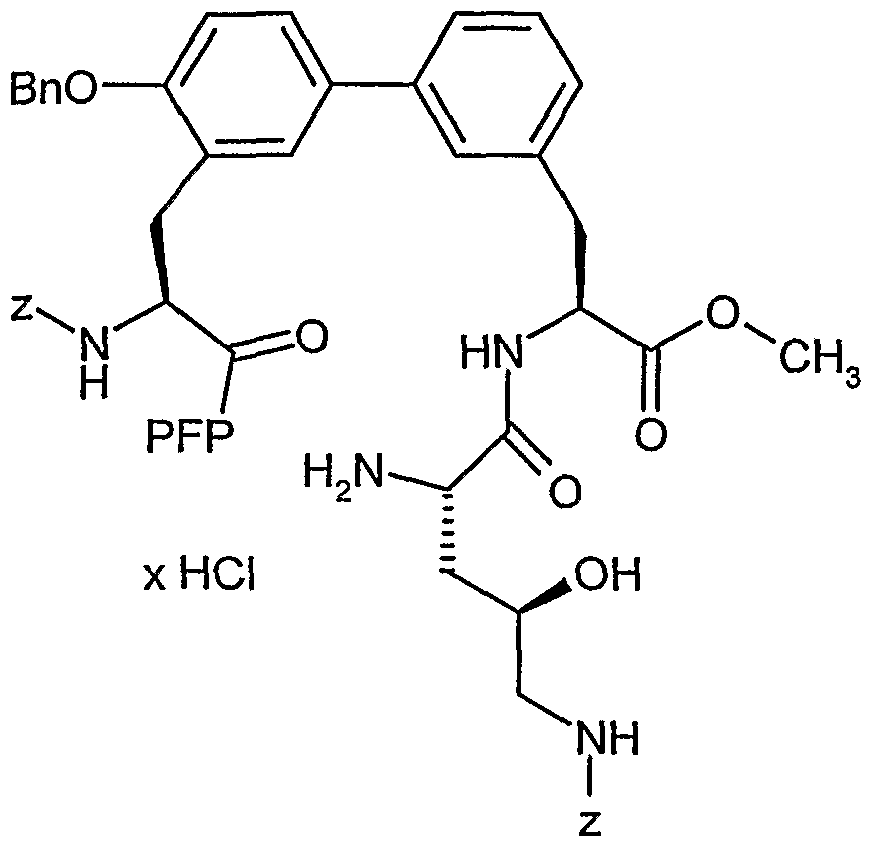
Methyl-(25)-2-[((2S,4R)-2-amino-5-{[(benzyloxy)carbonyl]amino}-4-hydroxypentanoyl)amino]-3- {4'-(benzyloxy)-3'-[(2
1S)-2-{[(benzyloxy)carbonyl]amino}-3-oxo-3-(pentafluorphenoxy)propyl]- biphenyI-3-yI}propanoat Hydrochlorid
Eine Lösung aus 1.81 g (1.63 mmol) der Verbindung aus Beispiel 23A in 10 ml Dioxan wird unter Rühren bei O°C mit 20 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Man rührt 30 min bei 0°C, lässt die Temperatur auf RT ansteigen, rührt eine weitere Stunde und dampft dann alles im Vakuum zur Trockne ein. Nach Trocknen im Hochvakuum bis zur Gewichtskonstanz erhält man das Produkt.
Ausbeute: quant.
LC-MS (Methode 3): Rt = 2.62 min.
MS (EI): m/z = 1013 (M-HCB-H)+
Beispiel 29A
Methyl-(25)-2-[((2iS)-2-amino-5-{[(benzyloxy)carbonyl]amino}pentanoyl)amino]-3-{4'-(benzyl- oxy)-3'-[(2ιS)-2-{[(benzyloxy)carbonyl]amino}-3-oxo-3-(pentafluorphenoxy)propyl]biphenyl-3- yl}propanoat Hydrochlorid
Eine Lösung aus 1.96 g (1.79 mmol) der Verbindung aus Beispiel 24A in 20 ml Dioxan wird unter Rühren bei O°C mit 60 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Man rührt 60 min bei O°C, lässt die Temperatur auf RT ansteigen, rührt eine weitere Stunde und dampft dann alles im Vakuum zur Trockne ein. Nach Trocknen im Hochvakuum bis zur Gewichtskonstanz erhält man das Produkt.
Ausbeute: quant.
LC-MS (Methode 1): R, = 2.73 min.
MS (EI): m/z = 997 (M-HC1+H)+
Analog zu den angegebenen Vorschriften werden die in der folgenden Tabelle aufgeführten Beispiele 3OA bis 32A aus den entsprechenden Edukten hergestellt:
Beispiel 33A
Methyl-(8S, 11 S, 14S)- 17-(benzy loxy)-l 4- { [(benzy loxy)carbonyl] amino}- 11 -((2R)-3- { [(benzy loxy)- carbonyl]amino}-2-hydroxypropyl)- 10, 13-dioxo-9, 12-diazatricyclo[ 14.3.1.1
2>6]henicosa- l(20),2(21),3,5,16,18-hexaen-8-carboxylat
Eine Lösung von 1.71 g (1.63 mmol) der Verbindung aus Beispiel 28A in 600 ml abs. Dichlormethan wird unter kräftigem Rühren tropfenweise in 20 min mit einer Lösung von 4.5 ml (32.6 mmol) Triethylamin in 150 ml Dichlormethan versetzt. Man lässt über Nacht weiterrühren und dampft alles im Vakuum ein (Badtemperatur ca. 4O°C). Der Rückstand wird mit Acetonitril verrührt und der zurückbleibende Feststoff wird abfiltriert und im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 0.611 g (45% d.Th.)
LC-MS (Methode 3): R4 = 2.92 min.
MS (EI): m/z = 829 (M+H)+
Beispiel 34A
Methyl-(8S, 11 S, 14S)- 17-(benzy loxy)- 14- { [(benzy loxy)carbony l]amino} - 11 -(3 - { [(benzy loxy)- carbony 1] amino } propy I)- 10, 13 -dioxo-9, 12-diazatricyclo [14.3.1.12>6]henicosa- 1 (20),2(21 ),3,5, 16, 18-hexaen-8-carboxylat
Eine Lösung von 1.85 g (1.79 mmol) der Verbindung aus Beispiel 29A in 600 ml abs. Chloroform wird unter kräftigem Rühren tropfenweise in 20 min mit einer Lösung von 5 ml (35.8 mmol) Triethylamin in 150 ml Chloroform versetzt. Man lässt über Nacht weiterrühren und dampft alles im Vakuum ein (Badtemperatur ca. 4O°C). Der Rückstand wird mit Acetonitril verrührt und der zurückbleibende Feststoff wird abfiltriert und im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 1.21 g (83% d.Th.)
LC-MS (Methode 1): R1 = 3.0 min.
MS (EI): m/z = 813 (M+H)+
Analog zu den angegebenen Vorschriften werden die in der folgenden Tabelle aufgeführten Beispiele 35A bis 37A aus den entsprechenden Edukten hergestellt:
Beispiel 38A
Methy 1-(8S, 11 S, 14S)-14-amino- 11 -[(2i?)-3 -amino-2-hydroxypropy I]- 17-hydroxy- 10,13 -dioxo-9, 12- diazatricyclo[ 14.3.1.12>6]henicosa- 1 (20),2(21 ),3 ,5, 16, 18-hexaen-8-carboxylat Dihydroacetat
Es werden 0.50 g (0.61 mmol) der Verbindung aus Beispiel 33 A in ein Gemisch aus 60 ml Essigsäure/Wasser/Ethanol (4:1:1) gegeben. Dazu gibt man 100 mg Palladium auf Aktivkohle (10% ig) und hydriert anschließend 36 h bei RT und Normaldruck. Das Reaktionsgemisch wird
über vprgewaschenem Kieselgur filtriert, mit Ethanol gewaschen und das Filtrat im Vakuum einrotiert. Der Rückstand wird im Hochvakuum bis zur Gesichtskonstanz getrocknet.
Ausbeute: quant.
LC-MS (Methode 2): R1 = 0.88 min.
MS (EI): m/z = 471 (M-2HOAc+H)+.
Beispiel 39A
Methyl-(85',115'314>S)-14-amino-l l-(3-ammopropyl)-17-hydroxy-10,13-dioxo-9,12-diazatri- cyclo[ 14.3.1.12>δ]henicosa- 1 (20),2(21 ),3,5, 16, 18-hexaen-8-carboxylat Dihydroacetat
Es werden 1.19 g (1.46 mmol) der Verbindung aus Beispiel 34A in ein Gemisch aus 440 ml Essigsäure/Wasser/Ethanol (4:1 :1) gegeben. Dazu gibt man 200 mg Palladium auf Aktivkohle (10% ig) und hydriert anschließend 36 h bei RT und Normaldruck. Das Reaktionsgemisch wird über vorgewaschenem Kieselgur filtriert, mit Ethanol gewaschen und das Filtrat im Vakuum einrotiert. Der Rückstand wird im Hochvakuum bis zur Gesichtskonstanz getrocknet.
Ausbeute: quant.
LC-MS (Methode 8): R, = 2.33 min.
MS (EI): m/z = 455 (M-2HOAc+H)+.
Analog zu den angegebenen Vorschriften werden die in der folgenden Tabelle aufgeführten Beispiele 4OA bis 42A aus den entsprechenden Edukten hergestellt:
Beispiel 43A
(8S,11S,14S)-14-[(tert-Butoxycarbonyl)amino]-l l-{(2R)-3-[(tert-butoxycarbonyl)amino]-2- hydroxypropy 1}- 17-hydroxy- 10, 13-dioxo-9, 12-diazatricyclo[ 14.3.1.1
2,6]henicosa- 1 (20),2(21 ),3,5, 16, 18-hexaen-8-carbonsäure
Eine Lösung von 150 mg (0.26 mmol) der Verbindung aus Beispiel 38A in 1 ml Wasser wird mit 1.3 ml IN Natronlauge versetzt. Unter Rühren wird eine Lösung von 170 mg (0.78 mmol) Oi-tert- butyldicarbonat in 0.5 ml Methanol bei RT hinzugegeben und für 4 h gerührt. Der Ansatz wird auf 15 ml Wasser gegeben, mit 0.1N Salzsäure stellt man pH 3 ein und schüttelt zweimal mit Essigsäureethylester aus. Die organischen Phasen werden vereinigt, mit Magenisiumsulfat getrocknet und im Vakuum zur Trockne eingedampft. Der verbleibende Feststoff wird chromatographisch (Sephadex LH20, Laufmittel: Methanol / Essigsäure (0.25%)) gereinigt.
Ausbeute: 137 mg (81% d. Th.)
LC-MS (Methode 1 ) : R, = 1.94 min.
MS (EI): m/z = 657 (M+H)+
Beispiel 44A
(8S, \ IS,14S)- 14-[(ter/-Butoxycarbonyl)amino]- 11 -{3-[(ter?-butoxycarbonyl)amino]propyl}- 17- hydroxy-10,13-dioxo-9,12-diazatricyclo[14.3.1.12'6]henicosa-l(20),2(21),3,5,16,18-hexaen-8- carbonsäure
Eine Lösung von 0.85 g (1.45 mmol) der Verbindung aus Beispiel 39A in 5 ml Wasser wird mit 7.3 ml IN Natronlauge versetzt. Unter Rühren wird eine Lösung von 0.95 g (4.36 mmol) Oi-tert- butyldicarbonat in 2 ml Methanol bei RT hinzugegeben und für 6 h gerührt. Der Ansatz wird auf 25 ml Wasser gegeben, mit 0.1N Salzsäure stellt man pH 3 ein und schüttelt zweimal mit Essigsäureethylester aus. Die organischen Phasen werden vereinigt, mit Magenisiumsulfat getrocknet und im Vakuum zur Trockne eingedampft. Der verbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz gereinigt.
Ausbeute: 0.75 g (81% d. Th.)
LC-MS (Methode 1): R1 = 2.20 min.
MS (EI): m/z = 641 (M+H)+
Analog zu den angegebenen Vorschriften werden die in der folgenden Tabelle aufgeführten Beispiele 45A bis 47A aus den entsprechenden Edukten hergestellt:
Beispiel 48A
Benzyl-{(1 S)-4-[(tert-butoxycarbonyl)amino]-l-[({2-[(tert-butoxycarbonyl)ammo]ethyl}amino)- carbonyl]butyl} carbamat
Unter Argon werden 300 mg (0.82 mmol) N2-[(Benzyloxy)carbonyl]-N5-(/er/-butoxycarbonyl)-Z- ornithin und 171 mg (1.06 mmol) te/t-Butyl-(2-aminoethyl)carbamat in 6 ml Dimethylformamid gelöst. Bei O°C (Eisbad) werden dann 204 mg (1.06 mmol) EDC und 33 mg (0.25 mmol) HOBt
zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Natriumhydrogencarbonat- und Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum getrocknet.
Ausbeute: 392 mg (94% d. Th.)
LC-MS (Methode 2): Rf = 2.36 min.
MS (ESI): m/z = 509 (M+H)+
Beispiel 49A
N5-(tert-Butoxycarbony I)-N- {2-[(ter£-butoxycarbonyl)amino]ethyl} -Z-ornithinamid
Eine Lösung von 390 mg (0.77 mmol) Benzyl-{(lS)-4-[(re^butoxycarbonyl)ammo]-l-[({2-[(tert- butoxycarbonyl)amino]ethyl}amino)carbonyl]butyl}carbamat (Beispiel 48A) in 50 ml Ethanol wird nach Zugabe von 40 mg Palladium auf Aktivkohle (10%ig) 4 h bei RT und Normaldruck hydriert. Es wird über Kieselgur filtriert und der Rückstand mit Ethanol gewaschen. Das Filtrat wird im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 263 mg (91% d. Th.)
MS (ESI): m/z = 375 (M+H)+; 397 (M+Na)+.
Beispiel 50A
terf-Butyl-[(15)-4-[(^rt-butoxycarbonyl)amino]-l-(hydroxymethyl)butyl]carbamat
Eine Lösung von 300 mg (0.90 mmol) N2,N5-Bis(tert-butoxycarbonyl)-.L-ornithin in 10 ml Tetrahydrofuran wird bei -1O°C mit 91 mg (0.90 mmol) 4-Methylmorpholin und 98 mg (0.90 mmol) Chlorameisensäureethylester versetzt und 30 min gerührt. Bei dieser Temperatur werden 1.81 ml (1.81 mmol) einer IM Lösung von Lithiumaluminiumhydrid in Tetrahydrofuran langsam zugetropft. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Unter Eiskühlung gibt man vorsichtig 0.1 ml Wasser und 0.15 ml 4.5%ige Natriumhydroxid-Lösung hinzu und rührt weitere 3 h bei RT. Der Ansatz wird filtriert und das Filtrat wird im Vakuum eingeengt. Der Rückstand wird in Essigsäureethylester gelöst, mit Wasser gewaschen, über Magnesiumsulfat getrocknet und erneut im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 239 mg (83% d. Th.)
MS (ESI): m/z = 319 (M+H)+; 341 (M+Na)+.
Beispiel 51A
(26)-2,5-Bis[(tert-butoxycarbonyl)amino]pentyl-methansulfonat
Eine Lösung von 240 mg (0.75 mmol) tert-Butyl-[(lιS)-4-[(tert-butoxycarbonyl)amino]-l- (hydroxymethyl)butyl]carbamat (Beispiel 50A) in 20 ml Dichlormethan wird mit 103 mg (0.90 mmol) Methansulfonsäurechlorid und 0.21 ml (1.5 mmol) Triethylamin versetzt und für 16 h bei RT gerührt. Es wird mit Dichlormethan verdünnt und zweimal mit 0.1N Salzsäure gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet und im Vakuum bis zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 218 mg (73% d. Th.)
MS (ESI): m/z = 419 (M+Na)+.
Beispiel 52A
tert-Butyl-{(4S)-5-azido-4-[( tert-butoxycarbonyl)amino]pentyl}carbamat
Eine Lösung von 218 mg (0.55 mmol) (2S)-2,5-Bis[(?ert-butoxycarbonyl)amino]pentyl-methan- sulfonat (Beispiel 51A) in 15 ml Dimethylformamid wird mit 36 mg (0.55 mmol) Natriumazid versetzt und 12 h bei 7O°C gerührt. Ein Großteil des Lösungsmittel wird im Vakuum abdestilliert und der Rückstand wird mit Essigsäureethylester verdünnt. Es wird mehrmals mit gesättigter Natriumhydrogencarbonat-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 188 mg (99% d. Th.)
MS (ESI): m/z = 344 (M+H)+.
Beispiet 53A
tert-Butyl-{(4S)-5-amino-4-[( tert-butoxycarbonyl)amino]pentyl}carbamat
Eine Lösung von 188 mg (0.55 mmol) tert-Butyl-{(45)-5-azido-4-[( tert-butoxycarbonyl)amino]- pentyl}carbamat (Beispiel 52A) in Ethanol wird nach Zugabe von 20 mg Palladium auf Aktivkohle (10%ig) 12 h bei RT und Normaldruck hydriert. Es wird über Kieselgur filtriert und der Rückstand mit Ethanol gewaschen. Das Filtrat wird im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 102 mg (59% d. Th.)
MS (ESI): m/z = 318 (M+H)+; 340 (M+Na)+.
Beispiel 54A
Benzyl-[2-({(25)-2,5-bis[(^7'/-butoxycarbonyl)amino]pentyl}amino)-2-oxoethyl]carbaraat
Die Herstellung erfolgt analog zu Beispiel 48A aus 92 mg (0.44 mmol) N-[(Benzyloxy)- carbonyljglycin und 181 mg (0.57 mmol) tert-Butyl-{(4S)-5-arnino-4-[(tert-butoxycarbonyl)- amino]pentyl}carbamat (Beispiel 53A) in 6 ml Dimethylformamid unter Zusatz von 110 mg (0.57 mmol) EDC und 18 mg (0.13 mmol) HOBt. Das Produkt wird mittels präparativer RP-HPLC gereinigt (Laufmittel Wasser / Acetonitril Gradient: 90:10 -> 5:95).
Ausbeute: 105 mg (47% d. Th.)
LC-MS (Methode 2): R, = 2.12 min.
MS (ESI): m/z = 509 (M+H)+
Beispiel 55A
tert-Butyl-{(4S)-5-[(aminoacetyl)amino]-4-[(tert-butoxycarbonyl)amino]pentyl}carbamat
Die Herstellung erfolgt analog Beispiel 49A aus 105 mg (0.21 mmol) Benzyl-[2-({(2S)-2,5- bis[(tert-butoxycarbonyl)amino]pentyl}amino)-2-oxoethyl]carbamat (Beispiel 54A) in 50 ml Ethanol unter Zusatz von 11 mg Palladium auf Aktivkohle (10%ig). Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 64 mg (83% d. Th.)
MS (ESI): m/z = 375 (M+H)+
Beispiel 56A
Benzyl-{(lιS)-l-[({(25)-2,5-bis[(tert-butoxycarbonyl)amino]pentyl}amino)carbonyl]-4-[(tert- butoxycarbonyl)amino]butyl}carbamat
Die Herstellung erfolgt analog zu Beispiel 48A aus 120 mg (0.33 mmol) N5-(tert-Butoxycarbonyl)- N2-[(benzyloxy)carbonyl]-I-ornithm und 136 mg (0.43 mmol) tert-Butyl-{(4S)-5-amino-4-[(tert- butoxycarbonyl)amino]pentyl}carbamat (Beispiel 53A) in 6 ml Dimethylformamid unter Zusatz von 82 mg (0.43 mmol) EDC und 13 mg (0.1 mmol) HOBt. Das Produkt wird mittels präparativer RP-HPLC gereinigt (Laufmittel Wasser / Acetonitril Gradient: 90:10 -> 5:95).
Ausbeute: 132 mg (61% d. Th.)
LC-MS (Methode 3): Rt = 2.68 min.
MS (ESI): m/z = 666 (M+H)+
Beispiel 57A
tert-Butyl-[(4ιS)-4-amino-5-({(2S)-2,5-bis[(tert-butoxycarbonyl)amino]pentyl}amino)-5- oxopentyl]carbamat
Die Herstellung erfolgt analog Beispiel 49A aus 132 mg (0.20 mmol) Benzyl-{(1S)-l-[({(2,S)-2,5- bis[(tert-butoxycarbonyl)amino]pentyl}amino)carbonyl]-4-[(tert-butoxycarbonyl)amino]butyl}- carbamat (Beispiel 56A) in 50 ml Ethanol unter Zusatz von 13 mg Palladium auf Aktivkohle (10%ig). Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 532 (M+H)+
Beispiel 58A
Benzyl-[( 1 S)- 1 -[(benzyloxy)methy 1] -2-( {(2£)-2,5-bis [(tert-butoxycarbony l)amino]penty 1 } amino)- 2-oxoethyl] carbamat
Die Herstellung erfolgt analog zu Beispiel 48 A aus 150 mg (0.46 mmol) O-Benzyl-N- [(benzyloxy)carbonyl]-L-serin und 188 mg (0.59 mmol) tert-Buty\-{(4S)-5-ammo-4-[(tert- butoxycarbonyl)amino]pentyl} carbamat (Beispiel 53A) in 6 ml Dimethylformamid unter Zusatz von 114 mg (0.57 mmol) EDC und 18 mg (0.13 mmol) HOBt. Das Produkt wird mittels präparativer RP-HPLC gereinigt (Laufmittel Wasser / Acetonitril Gradient: 90:10 -^ 5:95).
Ausbeute: 129 mg (45% d. Th.)
LC-MS (Methode 3): R1 = 2.81 min.
MS (ESI): m/z = 629 (M+H)+
Beispiel 59A
?e/"/-Butyl-{(41S)-5-{[(2»S}-2-amino-3-hydroxypropanoyl]amino}-4-[(^/"t-butoxycarbonyl)amino]- penty 1} carbamat
Eine Lösung von 128 mg (0.77 mmol) Benzyl-[(15)-l-[(benzyloxy)methyl]-2-({(25)-2,5-bis[(^^- butoxycarbonyl)amino]pentyl}amino)-2-oxoethyl]carbamat (Beispiel 58A) in 50 ml Ethanol wird nach Zugabe von 13 mg Palladium auf Aktivkohle (10%ig) 48 h bei RT und Normaldruck hydriert. Es wird über Kieselgur filtriert und der Rückstand mit Ethanol gewaschen. Das Filtrat wird im Vakuum zur Trockne eingeengt. Das Produkt wird mittels präparativer RP-HPLC gereinigt (Laufmittel Wasser / Acetonitril Gradient: 90:10 -> 5:95).
Ausbeute: 22 mg (27% d. Th.)
LC-MS (Methode 1): R1= 1.43 min.
MS (ESI): m/z = 405 (M+H)+
Beispiel 6OA
Benzyl-[2-({(3S)-3-{[(benzyloxy)carbonyl]amino}-6-[(^ert-butoxycarbonyl)amino]hexanoyl}- amino)ethyl]carbamat
Zu einer Lösung von 500 mg (1.31 mmol) (35)-3-{[(Benzyloxy)carbonyl]amino}-6-[(tert- butoxycarbonyl)amino]hexansäure in 25 ml wasserfreiem DMF werden 549.7 mg (1.446 mmol) HATU und 339.7 mg (2.629 mmol) N,N-Diisopropylethylamin hinzugegeben. Nach 15 min Rühren bei RT werden 333.5 mg (1.446 mmol) Benzyl-(2-aminoethyl)carbamat Hydrochlorid hinzugegeben. Das Reaktionsgemisch wird 15 h bei RT gerührt. Das Lösungsmittel wird dann eingedampft und der Rückstand in Dichlormethan aufgenommen. Die organische Phase wird mit Wasser gewaschen, über Magnesiumsulfat getrocknet und eingeengt. Das Rohprodukt wird durch präparative HPLC aufgereinigt.
Ausbeute 556.6 mg (44% d. Th.)
LC-MS (Methode 3): R1 = 2.41 min.
MS (ESI): m/z = 557 (M+H)+.
Beispiel 61A
Benzyl-((l1S)-4-amino-l-{2-[(2-{[(benzyloxy)carbonyl]amino}ethyl)amino]-2-oxoethyl}butyl)- carbamat Hydrochlorid
Zu einer Lösung von 320 mg (0.287 mmol) Benzyl-[2-({(3<S)-3-{[(benzyloxy)carbonyl]amino}-6- [(fe/"/-butoxycarbonyl)amino]hexanoyl}amino)ethyl]carbamat (Beispiel 60A) in 2 ml Dioxan
werden bei O°C 8 ml einer 4M Chlorwasserstoff-Dioxan-Lösung hinzugegeben. Nach 1 h bei RT wird die Reaktionslösung im Vakuum eingeengt, mehrmals mit Dichlormethan coevaporiert und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 2): R1 = 2.84 min.
MS (ESI): m/z = 457 (M-HCRH)+.
Beispiel 62A
Benzyl-{2-[((3S)-3-{[(benzyloxy)carbonyl]amino}-6-{[N5-[(benzyloxy)carbonyl]-N2-(tert- butoxycarbonyl)-Z,-omithyl]amino}hexanoyl)amino]ethyl}carbatnat
Zu einer Lösung von 78.4 mg (0.214 mmol) N5-[(Benzyloxy)carbonyl]-N2-(ter/-butoxycarbonyl)-Z,- ornithin in 5 ml wasserfreiem DMF werden 89.5 mg (0.235 mmol) HATU und 55.3 mg (0.428 mmol) N.N-Diisopropylethylamin hinzugegeben. Nach 15 min Rühren bei RT wird eine Lösung von 116 mg (0.235 mmol) Benzyl-((15}-4-amino-l-{2-[(2-{[(benzyloxy)carbonyl]arnino}- ethyl)amino]-2-oxoethyl}butyl)carbamat Hydrochlorid (Beispiel 61A) in 5 ml wasserfreiem DMF hinzugegeben. Das Reaktionsgemisch wird 15 h bei RT gerührt. Das Lösungsmittel wird dann eingedampft und der Rückstand in Dichlormethan aufgenommen. Die organische Phase wird mit Wasser gewaschen, über Magnesiumsulfat getrocknet und eingeengt. Das Rohprodukt wird durch präparative HPLC aufgereinigt.
Ausbeute 48 mg (28% d. Th.)
LC-MS (Methode 2): R, = 2.33 min.
MS (ESI): m/z = 805 (M+H)+.
Beispiel 63A
Benzy \-((4S, 1 OSΗ-amino- 10- { [(benzy loxy)carbonyl]amino } -5, 12, 17-trioxo- 19-phenyl- 18-oxa- 6,13,16-triazanonadec- 1 -y l)carbamat Hydrochlorid
Zu einer Lösung von 48 mg (0.060 mmol) Benzyl-{2-[((3«S)-3-{[(benzyloxy)carbonyl]amino}-6- { [N5-[(benzyloxy)carbonyl]-N2-(ter^butoxycarbonyl)-£-omithyl] amino} hexanoyl)amino]ethyl} - carbamat (Beispiel 62A) in 1 ml Dioxan werden bei RT 2.5 ml einer 4M Chlorwasserstoff-Dioxan- Lösung hinzugegeben. Nach 4 h bei RT wird die Reaktionslösung im Vakuum eingeengt, mehrmals mit Dichlormethan coevaporiert und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 2): Rt = 1.69 min.
MS (ESI): m/z = 705 (M-HC1+H)+.
Beispiel 64A
Benzyl[(5lS}-5-[(/er^-butoxycarbonyl)amino]-7-({2-[(tert-butoxycarbonyl)amino]ethyl}amino)-7- oxohepty 1] carbamat
Unter Argon werden 1 g (2.54 mmol) (3S)-7-{[(Benzyloxy)carbonyl]amino}-3-[(tert-butoxy- carbonyl)amino]heptancarbonsäure, 406 mg (2.54 mmol) terf-Butyl-(2-aminoethyl)carbamat und 0.96 ml Triethylamin (6.85 mmol) in 20 ml Dimethylformamid gelöst. Bei O°C (Eisbad) werden dann 826 mg (4.3 mmol) EDC und 113 mg (0.84 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand
wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Natriumhydrogencarbonat- und Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum getrocknet.
Ausbeute: quant.
LC-MS (Methode 2): R1 = 2.21 min.
MS (ESI): m/z = 537 (M+H)+
Beispiel 65A
^e^-Butyl-((liS)-5-amino-l-{2-[(2-{[(benzyloxy)carbonyl]amino}ethyl)amino]-2-oxoethyl}pentyl)- carbamat Hydroacetat
Es werden 1.3 g (2.42 mmol) Benzyl-[(5S)-5-[(te^butoxycarbonyl)amino]-7-({2-[(ferf-butoxy- carbonyl)amino]ethyl}arnino)-7-oxoheptyl]carbamat (Beispiel 64A) in 100 ml einer Mischung Eisessig/Wasser 4/lgelöst. Dazu gibt man 70 mg Palladium auf Aktivkohle (10%ig) und hydriert anschließend 15 h bei Normaldruck. Das Reaktionsgemisch wird über vorgewaschenem Kieselgur filtriert und das Filtrat im Vakuum einrotiert. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 1): R1 = 1.35 min.
MS (ESI): m/z = 403 (M-HOAc+H)+
Beispiel 66A
Benzyl-tert-butyl[(2ιS)-3-({(2S)-2,5-bis[(/err-butoxycarbonyl)amino]pentyl}amino)-3-oxoρropan- l,2-diyl]biscarbamat
Unter Argon werden 0.127 g (0.37 mmol) N-[(Benzyloxy)carbonyl]-3-[(tert-butoxycarbonyl)- amino]-L-alanin und 0.193 g (0.49 mmol) te^Butyl-{(4S)-5-amino-4-[(tert-butoxycarbonyl)- amino]pentyl}carbamat (Beispiel 53A) in 6 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 0.093 g (0.49 mmol) EDC und 0.015 g (0.11 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Natriumhydrogencarbonat- und Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird per präparativer HPLC (Kromasil, Laufmittel Acetonitril/ 0.25% wässrige Trifluoressigsäure 5:95 -^ 95:5) gereinigt.
Ausbeute: 0.126 g, (53% d. Th.)
LC-MS (Methode 1): R1 = 2.65 min.
MS (ESI): m/z = 638 (M+H)+
Beispiel 67 A
tert-Butyl-[(2S)-2-amino-3-({(2S)-2,5-bis[(tert-butoxycarbonyl)amino]pentyl}amino)-3- oxopropyl]carbamat
Zu einer Mischung aus 0.122 g (0.19 mmol) der Verbindung aus Beispiel 66A in 50 ml Ethanol gibt man 20 mg Palladium auf Aktivkohle (10%ig) und hydriert anschließend 4 h bei Normaldruck. Das Reaktionsgemisch wird über Kieselgur filtriert, das Filtrat im Vakuum eingeengt und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 504 (M+H)+
Beispiel 68A
Benzyl-{(l»S)-4-[(tert-butoxycarbonyl)amino]-l-[2-({2-[(^r^-butoxycarbonyl)amino]ethyl}amino)- 2-oxoethyl]butyl}carbamat
Zu einer Lösung von 760.9 mg (2 mmol) (35)-3-{[(Benzyloxy)carbonyl]amino}-6-[(tert- butoxycarbonyl)amino]hexansäure in 25 ml wasserfreiem DMF werden 836.5 mg (2.2 mmol) HATU und 517.0 mg (4 mmol) NN-Diisopropylethylamin hinzugegeben. Nach 15 min Rühren bei RT werden 352.5 mg (2.2 mmol) tert-Butyl-(2-aminoethyl)carbamat Hydrochlorid hinzugegeben. Das Reaktionsgemisch wird 15 h bei RT gerührt. Das Lösungsmittel wird dann eingedampft und der Rückstand in Dichlormethan aufgenommen. Die organische Phase wird mit Wasser gewaschen, über Magnesiumsulfat getrocknet und eingeengt. Das Rohprodukt wird durch präparative HPLC aufgereinigt.
Ausbeute 400 mg (38% d. Th.)
LC-MS (Methode 1): Rt = 2.33 min.
MS (ESI): m/z = 523 (M+H)+.
Beispiel 69A
tert-Butyl-[(4iS)-4-amino-6-({2-[(tert-butoxycarbonyl)amino]ethyl}amino)-6-oxohexyl]carbamat
Es werden 400 mg (0.765 mmol) Benzyl-{(15>4-[(ter^butoxycarbonyl)amino]-l-[2-({2-[(terf- butoxycarbonyl)amino]ethyl}amino)-2-oxoethyl]butyl}carbamat (Beispiel 68A) in 50 ml Ethanol gelöst. Dazu gibt man 80 mg Palladium auf Aktivkohle (10%ig) und hydriert anschließend 15 h bei Normaldruck. Das Reaktionsgemisch wird über vorgewaschenem Kieselgur filtriert und das Filtrat im Vakuum einrotiert. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 3): R4 = 1.42 min
MS (ESI): m/z = 389 (M+H)+.
Beispiel 7OA
Benzyl-((l1S',4S)-l,4-bis{3-[(fe^-butoxycarbonyl)amino]propyl}-13,13-dimethyl-2,6,l l-trioxo-12- oxa-3 ,7,10-triazatetradec-l -y l)carbamat
Unter Argon werden 72 mg (0.197 mmol) N2-[(Benzyloxy)carbonyl]-N5-(te/Y-butoxycarbonyl)-Z,- Ornithin und 100 mg (0.26 mmol) der Verbindung aus Beispiel 69A in 8 ml Dimethylformamid gelöst. Bei O°C (Eisbad) werden dann 49 mg (0.26 mmol) EDC und 8 mg (0.059 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im
Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Νatriumhydrogencarbonat- und Νatrium- chlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum getrocknet
Ausbeute 121 mg (83% d. Th.)
LC-MS (Methode 1): Rt = 2.24 min.
MS (ESI): m/z = 737 (M+H)+.
Beispiel 71A
tert-Butyl-[(45)-4-({(2jS)-2-amino-5-[(tert-butoxycarbonyl)amino]pentanoyl}amino)-6-({2-[(tert- butoxycarbonyl)amino]ethyl}amino)-6-oxohexyl]carbamat
Es werden 120 mg (0.16 mmol) der Verbindung aus Beispiel 7OA in 10 ml Ethanol gelöst. Dazu gibt man 15 mg Palladium auf Aktivkohle (10%ig) und hydriert anschließend 15 h bei Normaldruck. Das Reaktionsgemisch wird über vorgewaschenem Kieselgur filtriert und das Filtrat im Vakuum einrotiert. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 603 (M+H)+.
Beispiel 72A
Benzyl-[(4,S)-4-[(tert-butoxycarbonyl)amino]-6-( {2-[(tert-butoxycarbonyl)amino]ethyl} amino)-6- oxohexy 1] carbamat
Unter Argon werden 100 mg (0.26 mmol) (3,S)-6-{[(Benzyloxy)carbonyrjamino}-3-[(tert- butoxycarbonyl)amino]hexansäure und 55 mg (0.34 mmol) te^Butyl-(2-aminoethyl)carbamat in 6 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 66 mg (0.34 mmol) EDC und 11 mg (0.08 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Natriumhydrogencarbonat- und Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum getrocknet.
Ausbeute: 71 mg (51% d. Th.)
LC-MS (Methode 3): R4 = 2.43 min.
MS (ESI): m/z = 523 (M+H)+
Beispiet 73A
^r^-Butyl-{(liS)-4-amino-l-[2-({2-[(fe^-butoxycarbonyl)amino]ethyl}amino)-2-oxoethyl]butyl}- carbamat
Eine Lösung von 71 mg (0.135 mmol) der Verbindung aus Beispiel 72A in 10 ml Ethanol wird nach Zugabe von 15 mg Palladium auf Aktivkohle (10%ig) 12 h bei RT und Normaldruck hydriert. Es wird über Kieselgur filtriert und der Rückstand mit Ethanol gewaschen. Das Filtrat wird im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quänt.
MS (ESI): m/z = 389 (M+H)+.
Beispiel 74A
Benzyl-((15',7ιS)-7-[(te^-butoxycarbonyl)amino]-l-{3-[(te^-butoxycarbonyl)amino]propyl}-16,16- dimethy 1-2,9, 14-trioxo-l 5-oxa-3 ,10,13 -triazaheptadec- 1 -y l)carbamat
Unter Argon werden 40 mg (0.11 mmol) N2-[(Benzyloxy)carbonyl]-N5-(te/t-butoxycarbonyl)-Z,- ornithin und 55 mg (0.14 mmol) der Verbindung aus Beispiel 73A in 8 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 27 mg (0.14 mmol) EDC und 4.4 mg (0.033 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Νatriumhydrogencarbonat- und Νatrium- chlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum getrocknet
Ausbeute: 72 mg (89% d. Th.)
LC-MS (Methode 1): R1 = 2.2 min.
MS (ESI): m/z = 737 (M+H)+
Beispiel 75A
tert-Butyl- {(4S, 10S)-4-amino- 10-[(tert-butoxycarbonyl)amino]-l 9, 19-dimethyl-5, 12, 17-trioxo-l 8- oxa-6, 13,16-tr iazaicos- 1 -y 1 } carbamat
Eine Lösung von 72 mg (0.097 mmol) der Verbindung aus Beispiel 74A in 10 ml Ethanol wird nach Zugabe von 10 mg Palladium auf Aktivkohle (10%ig) 12 h bei RT und Normaldruck hydriert. Es wird über Kieselgur filtriert und der Rückstand mit Ethanol gewaschen. Das Filtrat wird im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 603 (M+H)+.
Beispiel 76A
Benzyl-{(45)-6-({(2iS)-2,5-bis[(?ert-butoxycarbonyl)amino]pen1yl}amino)-4-[(^erf-butoxy- carbony l)amino] -6-oxohexyl} carbamat
Unter Argon werden 0.1 g (0.263 mmol) (3ιS)-6-{[(Benzyloxy)carbonyl]amino}-3-[(tert-butoxy- carbonyl)amino]hexancarbonsäure (Bioorg. Med. Chem. Lett. 1998, 8, 1477-1482) und 0.108 g (0.342 mmol) ?er?-Butyl-{(45)-5-amino-4-[(tert-butoxycarbonyl)amino]pentyl}carbamat (Beispiel 53 A) in 6 ml Dimethylformamid gelöst. Bei O°C (Eisbad) werden dann 0.066 g (0.342 mmol) EDC und 0.011 g (0.079 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Natriumhydrogencarbonat- und Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum
eingedampft. Der verbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 0.127 g (71% d. Th.)
LC-MS (Methode 1): Rt = 2.36 min.
MS (ESI): m/z = 680 (M+H)+
Beispiel 77A
tert-Butyl-{(1 S)-4-amino-l-[2-({(2S)-2,5-bis[ tert-butoxycarbonyl)amino]pentyl}amino)-2- oxoethyl]butyl} carbamat
Zu einer Mischung aus 0.127 g (0.19 mmol) der Verbindung aus Beispiel 76A in 10 ml Ethanol gibt man 20 mg Palladium auf Aktivkohle (10%ig) und hydriert anschließend 12 h bei Normaldruck. Das Reaktionsgemisch wird über Kieselgur filtriert, das Filtrat im Vakuum eingeengt und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 546 (M+H)+
Beispiel 78A
Benzyl-(( 1 S,7S, 125)-7, 12-bis[(tert-butoxycarbonyl)amino]- 1 - {3-[(tert-butoxycarbonyl)amino]- propy 1 } - 19, 19-dimethy 1-2,9, 17-trioxo- 18-oxa-3 ,10,16-triazaicos- 1 -y l)carbamat
Unter Argon werden 44 mg (0.12 mmol) N2-[(Benzyloxy)carbonyl]-N5-(tert-butoxycarbonyl)-Z- ornithin und 85 mg (0.16 mmol) der Verbindung aus Beispiel 77A in 8 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 30 mg (0.16 mmol) EDC und 4.9 mg (0.036 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Νatriumhydrogencarbonat- und Νatrium- chlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum getrocknet
Ausbeute: 91 mg (85% d. Th.)
LC-MS (Methode 1): R1 = 2.35 min.
MS (ESI): m/z = 894 (M+H)+
Beispiel 79A
tert-Butyl-{(45',10S,15)S)-4-ammo-10,15-bis[(^rt-butoxycarbonyl)amino]-22,22-dimethyl-5,12,20- trioxo-21 -oxa-6, 13,19-triazatricos-l -yl} carbamat
Eine Lösung von 91 mg (0.10 mmol) der Verbindung aus Beispiel 78A in 10 ml Ethanol wird nach Zugabe von 10 mg Palladium auf Aktivkohle (10%ig) 12 h bei RT und Normaldruck hydriert. Es wird über Kieselgur filtriert und der Rückstand mit Ethanol gewaschen. Das Filtrat wird im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 760 (M+H)+.
Beispiel 8OA
Benzyl-{(16)-l-[2-({(25)-2,5-bis[(tert-butoxycarbonyl)amino]pentyl}amino)-2-oxoethyl]-4-[(tert- butoxycarbonyl)amino]butyl}carbamat
Unter Argon werden 0.1 g (0.26 mmol) (3iS)-3-{[(Benzyloxy)carbonyl]amino}-6-[(^e7Y-butoxy- carbonyl)amino]hexansäure (J. Med. Chem. 2002, 45, 4246-4253) und 0.11 g (0.34 mmol) tert- Butyl-{(4S)-5-amino-4-[(tert-butoxycarbonyl)amino]pentyl}carbamat (Beispiel 53A) in 6 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 0.065 g (0.34 mmol) EDC und 0.011 g (0.079 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester
aufgenommen. Die organische Phase wird nacheinander mit gesättigter Natriumhydrogencarbonat- und Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 0.146 g (82% d. Th.)
LC-MS (Methode 2): R4 = 2.5 min.
MS (ESI): m/z = 680 (M+H)+
Beispiel 81A
/e^-Butyl-[(45}-4-amino-6-({(2S)-2,5-bis[(^rt-butoxycarbonyl)amino]pentyl}amino)-6- oxohexy 1] carbamat
Zu einer Mischung aus 0.146 g (0.22 mmol) der Verbindung aus Beispiel 80A in 10 ml Ethanol gibt man 22 mg Palladium auf Aktivkohle (10% ig) und hydriert anschließend 12 h bei Normaldruck. Das Reaktionsgemisch wird über Kieselgur filtriert, das Filtrat im Vakuum eingeengt und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 546 (M+H)+
Beispiel 82A
Benzyl-((l,S',4S,9iS)-9-[(rerr-butoxycarbonyl)amino]-l,4-bis{3-[(^^-butoxycarbonyl)amino]- propyl}-l6,16-dimethyl-2,6,14-trioxo-15-oxa-3,7,13-triazaheptadec-l-yl)carbamat
Unter Argon werden 40 mg (0.11 mmol) N2-[(Benzyloxy)carbonyl]-N5-(/e^-butoxycarbonyl)-Z- ornithin und 77 mg (0.14 mmol) der Verbindung aus Beispiel 81 A in 8 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 27 mg (0.14 mmol) EDC und 4.4 mg (0.032 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Νatriumhydrogencarbonat- und Νatrium- chlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum getrocknet
Ausbeute: 78 mg (81 % d. Th.)
LC-MS (Methode 1): R, = 2.43 min.
MS (ESI): m/z = 894 (M+H)+
Beispiel 83A
tert-Butyl-((l
)S',65
r,9
ιS)-9-amino-l,6-bis{3-[(/ert-butoxycarbonyl)amino]propyl}-16,16-dimethyl- 4,8, 14-trioxo- 15-oxa-3,7, 13-triazaheptadec-l -yl)carbamat
Eine Lösung von 78 mg (0.088 mmol) der Verbindung aus Beispiel 82A in 10 ml Ethanol wird nach Zugabe von 10 mg Palladium auf Aktivkohle (10% ig) 12 h bei RT und Normaldruck hydriert. Es wird über Kieselgur filtriert und der Rückstand mit Ethanol gewaschen. Das Filtrat wird im Vakuum zur Trockne eingeengt. Das Produkt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
MS (ESI): m/z = 760 (M+H)+.
Beispiel 84A
N5-[N2-[(Benzyloxy)carbonyl]-N5-(tert-butoxycarbonyl)-Z)-ornithyl]-N2-(^r^-butoxycarbonyl)-N- {2-[(fert-butoxycarbonyl)amino]ethyl} -Z-ornithinamid
Unter Argon werden 286 mg (0.78 mmol) N2-[(Benzyloxy)carbonyl]-N5-(tert-butoxycarbonyl)-D- ornithin und 439 mg (1.17 mmol) der Verbindung aus Beispiel 104A in 16 ml Dimethylfbrmarnid gelöst. Bei O°C (Eisbad) werden dann 255 mg (1.33 mmol) EDC und 106 mg (0.78 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 48 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand mit Dichlormethan aufgenommen und mit gesättigter
Natriumhydrogencarbonat-Lösung, 0.1 N Salzsäure und Wasser gewaschen. Die vereinigten organischen Phasen werden im Vakuum eingeengt und der so erhaltene Feststoff ohne Reinigung weiter umgesetzt.
Ausbeute: 0.58 g (quant.)
LC-MS (Methode 3): Rt = 2.59 min.
MS (ESI): m/z = 723 (M+H)+
Beispiel 85A
N5-[N5-(tert-Butoxycarbonyl)-D-ornithyl]-N2-(tert-butoxycarbonyl)-N-{2-[(tert-butoxycarbonyl)- amino]ethyl} -L-ornithinamid
0.58 g (0.80 mmol) der Verbindung aus Beispiel 84A werden in 27 ml Ethanol gelöst und mit 0.06 g (0.06 mmol) Pd/C versetzt. Man hydriert 12 h bei Normaldruck, filtriert über Celite und engt das Filtrat im Vakuum ein. Der so erhaltene Feststoff wird ohne Reinigung weiter umgesetzt.
Ausbeute: 0.47 g (97% d. Th.)
LC-MS (Methode 1): R, = 1.61 min.
MS (ESI): m/z = 589 (M+H)+
Beispiel 86A
Benzyl-[(2S)-2-[(tert-butoxycarbonyl)amino]-3-({2-[(tert-butoxycarbonyl)amino]ethyl}amino)-3- oxopropyl]carbamat
Unter Argon werden 0.50 g (0.96 mmol) 3-{[(Benzyloxy)carbonyl]amino}-N-(tert-butoxy- carbonyl)-Z,-alanin - N-Cyclohexylcyclohexanamin (1:1) und 0.154 g (0.96 mmol) tert-Butyl-(2- aminoethyl)carbamat in 10 ml Dimethylformamid und 0.5 ml Triethylamin gelöst. Bei O°C (Eisbad) werden dann 0.314 g (1.64 mmol) EDC und 0.043 g (0.32 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Νatriumhydrogencarbonat- und Νatriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 0.41 g (88% d. Th.)
LC-MS (Methode 2): R. = 2.17 min.
MS (ESI): m/z = 481 (M+H)+
Beispiel 87A
3-Amino-N2-(tert-butoxycarbonyl)-N-{2-[(^er/-butoxycarbonyl)amino]ethyl}-X-alaninamid Hydroacetat
Zu einer Mischung aus 0.41 g (0.847 mmol) der Verbindung aus Beispiel 86A in 80 ml Essigsäure/Ethano l/Wasser (4:1:1) gibt man 50 mg Palladium auf Aktivkohle (10%ig) und hydriert anschließend 12 h bei Normaldruck. Das Reaktionsgemisch wird über Kieselgur filtriert, das Filtrat im Vakuum eingeengt und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: quant.
LC-MS (Methode 2): R, = 1.09 min.
MS (ESI): m/z = 347 (M-HOAc+H)+
Beispiet 88A
N5-{N-[(Benzyloxy)carbonyl]glycyl}-N2-(fe/*^-butoxycarbonyl)-N-{2-[(tert-butoxycarbonyl)- amino] ethy 1 } -Z,-ornithinamid
Unter Argon werden 300 mg (1.43 mmol) N-[(Benzyloxy)carbonyl]glycin und 830 mg (2.15 mmol) der Verbindung aus Beispiel 104A in 28 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 467 mg (2.44 mmol) EDC und 194 mg (1.43 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 48 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand mit Dichlormethan aufgenommen und mit gesättigter Νatriumhydrogencarbonat- Lösung, 0.1 Ν Salzsäure und Wasser gewaschen. Die vereinigten organischen Phasen werden im Vakuum eingeengt und der so erhaltene Feststoff ohne Reinigung weiter umgesetzt.
Ausbeute: quant.
LC-MS (Methode 2): R, = 1.98 min.
MS (ESI): m/z = 566 (M+H)+
Beispiel 89A
N5-Glycyl-N2-(^e^-butoxycarbonyl)-N-{2-[(te/"/-butoxycarbonyl)amino]ethyl}-Z-ornithinamid
1.03 g (1.82 mmol) der Verbindung aus Beispiel 88A werden in 60 ml Ethanol gelöst und mit 100 mg (0.09 mmol) Pd/C (10%ig) versetzt. Man hydriert über Nacht bei Normaldruck, filtriert über Celite und engt das Filtrat im Vakuum ein. Der so erhaltene Feststoff wird ohne Reinigung weiter umgesetzt.
Ausbeute: 693 mg (84% d. Th.)
LC-MS (Methode 3): R4 = 1.41 min.
MS (ESI): m/z = 432 (M+H)+
Beispiel 9OA
Ben2yl- tert-butyl-[5-({(25)-2,5-bis[( tert-butoxycarbonyl)amino]pentyl}amino)-5-oxopentan-l,3- diyl]biscarbamat
Unter Argon werden 0.146 g (0.40 mmol) 3-{[(Benzyloxy)carbonyl]amino}-5-[( tert-butoxy- carbonyl)amino]pentansäure (Bioorg. Med. Chem. 2003, 13, 241-246) und 0.164 g (0.52 mmol) tert-Butyl-{(4ιS)-5-amino-4-[( tert-butoxycarbonyl)amino]pentyl}carbamat (Beispiel 53A) in 8 ml Dimethylformamid gelöst. Bei O°C (Eisbad) werden dann 0.10 g (0.52 mmol) EDC und 0.009 g (0.12 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Essigsäureethylester aufgenommen. Die organische Phase wird nacheinander mit gesättigter Natriumhydrogencarbonat- und Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingedampft. Der verbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 0.232 g, (87% d. Th.)
LC-MS (Methode 3): Rt = 2.73 min.
MS (ESI): m/z = 666 (M+H)+
Beispiel 91A
tert-Butyl-[3-amino-5-({(2>S)-2,5-bis[( tert-butoxycarbonyl)amino]pentyl}amino)-5-oxopentyl]- carbamat
Zu einer Mischung aus 0.232 g (0.35 mmol) der Verbindung aus Beispiel 9OA in 10 ml Ethanol gibt man 35 mg Palladium auf Aktivkohle (10% ig) und hydriert anschließend 12 h bei Normaldruck. Das Reaktionsgemisch wird über Kieselgur filtriert, das Filtrat im Vakuum eingeengt und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 0.175 g (94% d. Th.)
LC-MS (Methode 3): R1 = 1.8 min.
MS (ESI): m/z = 532 (M+H)+
Analog zu der oben aufgeführten Vorschrift von Beispiel 5OA werden die in der folgenden Tabelle aufgeführten Beispiele 92A und 93A aus den entsprechenden Ausgangsverbindungen hergestellt:
Beispiel 94A
Benzyl-[(l<S}-2-amino-l-(hydroxymethyl)ethyl]carbamat Hydrochlorid
Eine Mischung von 269 mg (0.83 mmol) Benzyl-fe^butyl[(2,S)-3-hydroxypropan-l,2- diyljbiscarbamat (Beispiel 93A) und 5 ml einer 4M Chlorwasserstoff-Dioxan-Lösung wird 2 h bei RT gerührt. Die Reaktionslösung wird eingeengt, mehrmals mit Dichlormethan coevaporiert und im Hochvakuum getrocknet. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 212 mg (98% d. Th.)
LC-MS (Methode 2): R1 = 0.55 min.
MS (ESI): m/z = 225 (M-HCH-H)+.
Analog zu der oben aufgeführten Vorschrift von Beispiel 48A werden die in der folgenden Tabelle aufgeführten Beispiele 95A bis 102A aus den entsprechenden Edukten hergestellt:
Analog zu der oben aufgeführten Vorschrift von Beispiel 49A werden die in der folgenden Tabelle aufgeführten Beispiele 103 A bis 11 IA aus den entsprechenden Edukten hergestellt:
Beispiel 112 A
tert-Buty l-(2- { [(2S)-2-[(tert-butoxycarbonyl)amino]-5-( { [(8S,11S,14S)- 14-[(tert-butoxycarbonyl)- amino] - 1 1 - {3 -[(tert-butoxycarbony l)amino]propy 1} - 17-hydroxy-9-methy 1- 10,13 -dioxo-9, 12- diazatricyclo[14.3.1.12,6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-yl]carbonyl}amino)pentanoyl]- amino } ethy l)carbamat
Es werden 50 mg (0.05 mmol) (8S,11S,14S)-14-[(tert-Butoxycarbonyl)amino]-11-{3-[(tert- butoxy carbony l)amino] propy 1 } - 17-hy droxy-9-methy 1- 10, 13 -dioxo-9, 12-diazatr icy c Io [14.3.1.12-6]- henicosa-l(20),2(21),3,5,16,18-hexaen-8-carbonsäure (Beispiel 46A) und 34 mg (0.09 mmol) N2- (tert-Butoxycarbonyl)-N-{2-[(tert-butoxycarbonyl)amino]ethyl}-L-oraithinamid (Beispiel 104A) in 2.5 ml DMF gelöst und auf O°C gekühlt. Man versetzt mit 15 mg (0.08 mmol) EDC und 6 mg (0.05 mmol) HOBt und rührt 12 h bei Raumtemperatur. Das Reaktionsgemisch wird im Vakuum einrotiert. Das Rohprodukt wird ohne weitere Reinigung umgesetzt.
Ausbeute: 215 mg (88% d. Th.)
LC-MS (Methode 3): R, = 2.70 min.
MS (ESI): m/z = 1011 (M+H)+
Beispiel 113A
tert-Butyl[(45)-5-({(2IS)-2,5-bis[(tert-butoxycarbonyl)amino]pentyl}amino)-4-({[(8S,11S,14S)-14- [(tert-butoxycarbonyl)amino]-l l-{3-[(tert-butoxycarbonyl)amino]propyl}-17-hydroxy-10,13- dioxo-9,12-diazatricyclo[14.3.1.12,6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-yl]carbonyl}amino)- 5-oxopentyl]carbamat
Es werden 29 mg (0.05 mmol) (8S,11S,14S)-14-[tert-ButoxycarbonyOamino]-11-{3-[(tert-- butoxycarbonyl)amino]propyl}-17-hydroxy-10,13-dioxo-9,12-diazatricyclo[14.3.1.12'6]henicosa- l(20),2(21),3,5,16,18-hexaen-8-carbonsäure (Beispiel 44A) und 24 mg (0.05 mmol) tert-Butyl- [(4S)-4-amino-5-({(2S)-2,5-bis[(^ert-butoxycarbonyl)amino]pentyl}amino)-5-oxopentyl]carbamat (Beispiel 57A) in 2.0 ml DMF gelöst und auf O°C gekühlt. Man versetzt mit 15 mg (0.08 mmol) EDC und 6 mg (0.05 mmol) HOBt und rührt 12 h bei Raumtemperatur. Das Reaktionsgemisch wird im Vakuum einrotiert und chromatographisch über Sephadex-LH20 (Laufmittel: Methanol / Essigsäure 0.25%) gereinigt.
Ausbeute: 53 mg (54% d. Th.)
LC-MS (Methode 2): R4 = 2.68 min.
MS (ESI): m/z = 1154 (M+H)+
Beispiel 114A
tert-Butyl-(2-{[(31S)-3-[(fe^-butoxycarbonyl)amino]-7-({[(85',115',145)-14-[(tert-butoxycarbonyl)- amino]-l l-{3-[(tert-butoxycarbonyl)amino]propyl}-17-hydroxy-9-methyl-10,13-dioxo-9,12- diazatricyclo[14.3.1.12,6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-yl]carbonyl}amino)heptanoyl]- amino } ethy l)carbamat
Es werden 40 mg (0.06 mmol) ($S,\ lS,14S)-l4-[(tert-Butoxycarbony\)ammo]-l l-{3-[(tert- butoxycarbonyl)amino]propy 1} - 17-hydroxy-9-methy 1- 10, 13-dioxo-9, 12-diazatricyclo [14.3.1.12>6]- henicosa-l(20),2(21),3,5,16,18-hexaen-8-carbonsäure (Beispiel 46A) und 46 mg (0.08 mmol) tert- Butyl{(15)-5-amino-l-[2-({2-[(fert-butoxycarbonyl)amino]ethyl}amino)-2-oxoethyl]pentyl}- carbamat (Beispiel 65A) in 2.0 ml DMF gelöst und auf 0°C gekühlt. Man versetzt mit 15 mg (0.08 mmol) EDC, 3 mg (0.02 mmol) HOBt und O.Ol ml (0.08 mmol) Triethylamin und rührt 12 h bei Raumtemperatur. Das Reaktionsgemisch wird im Vakuum einrotiert und via präparativer HPLC gereinigt.
Ausbeute: 6 mg (9% d. Th.)
LC-MS (Methode 2): R. = 2.47 min.
MS (ESI): m/z = 1039 (M+H)+
Beispiel 115A
Benzyl-((lS)-4-{[(2S)-5-{[(benzyloxy)carbonyl]amino}-2-({[(8S,l l
1S',14
JS)-14-[(^^-butoxy- carbonyl)amino]- 11 - {3-[(fert-butoxycarbonyl)amino]propyl }-l 7-hydroxy-9-methyl- 10, 13 -dioxo- 9,12-diazatricyclo[14.3.1.1
2i6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-yl]carbonyl}amino)- pentanoyl]amino}-l-{2-[(2-{[(benzyloxy)carbonyl]amino}ethyl)amino]-2-oxoethyl}butyl)- carbamat
Es werden 65 mg (0.06 mmol) (ßS,l lS,\4S)-^-[(^rt-Butoxyceιτhonyϊ)ammo]-l l-{3-[(tert- butoxycarbonyl)amino]propyl}- 17-hydroxy-9-methyl- 10, 13-dioxo-9, 12-diazatricyclo[l 4.3.1.12>6]- henicosa-l(20),2(21),3,5,16,18-hexaen-8-carbonsäure (Beispiel 46A) und 120 mg (0.13 mmol) Benzyl-((5S,1 lS)-5-amino-l l-{[(benzyloxy)carbonyl]amino}-6,13,l 8-trioxo-20-phenyl-19-oxa- 7,14,17-triazaicos-l-yl)carbamat Hydrochlorid (Beispiel 63A) in 3.0 ml DMF gelöst und auf 0°C gekühlt. Man versetzt mit 25 mg (0.13 mmol) EDC, 4 mg (0.03 mmol) HOBt und 0.02 ml (0.13 mmol) Triethylamin und rührt 12 h bei Raumtemperatur. Das Reaktionsgemisch wird im Vakuum einrotiert und via präparativer HPLC gereinigt.
Ausbeute: 50 mg (25% d. Th.).
LC-MS (Methode 3): Rt = 2.92 min.
MS (ESI): m/z = 1341 (M+H)+
Beispiel 116A
tert-Butyl{3-[(8S,l l,S;i45)-8-[({(lS)-4-amino-l-[({(45)-4-amino-6-[(2-aminoethyl)amino]-6- oxohexyl}amino)carbonyl]butyl}amino)carbonyl]-14-[(?ert-butoxycarbonyl)amino]-17-hydroxy-9- methyl-10,13-dioxo-9,12-diazatricyclo[14.3.1.1
2'
6]henicosa-l(20),2(21),3,5,16,18-hexaen-l l- yl]propyl} carbamat Tris(hydrotrifluoracetat)
49 mg (0.04 mmol) Benzyl-((l,S}-4-{[(21y)-5-{[(benzyloxy)carbonyl]amino}-2-({[(85,l lS,14S)-14- [(tert-butoxycarbonyl)amino]-l l-{3-[(tert-butoxycarbonyl)amino]propyl}-17-hydroxy-9-methyl- 10, 13-dioxo-9, 12-diazatricyclo[ 14.3.1.12,6]henicosa- 1 (20),2(21 ),3,5, 16, 18-hexaen-8-yl]carbony 1} - amino)pentanoyl]amino } - 1 - {2-[(2- { [(benzyloxy)carbonyl]amino } ethy l)amino]-2-oxoethyl} butyl)- carbamat (Beispiel 115A) wird in 10 ml Eisessig/Wasser (4:1) gelöst, mit 5 mg Pd/C (10%) versetzt und 12 h bei Normaldruck und Wasserstoffatmosphäre hydriert. Man saugt ab, engt das Reaktionsgemisch im Vakuum ein und reinigt durch präparative HPLC (Kromasil 100 C 18, 5 μm 250 mm x 20 mm; Laufmittel Acetonitril / 0.2% wässrige Trifluoressigsäure 5:95 -> 95:5).
Ausbeute: 9 mg (19% d. Th.)
LC-MS (Methode 3): Rt = 1.45 min.
MS (ESI): m/z = 939 (M+H)+
Beispiel 117A
tert-Butyl-(2-{[(2
1S)-2-[tert-butoxycarbonyl)amino]-5-({[(8S,116',14S)-14-[tert-butoxycarbonyl)- amino]-l l-{(2i?)-3-[(tert-butoxycarbonyl)amino]-2-hydroxypropyl}-17-hydroxy-10,13-dioxo-9,12- diazatricyclo[14.3.1.1
2,6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-yl]carbonyl}amino)pentanoyl]- amino } ethy l)carbamat
Unter Argon werden 50 mg (0.076 mmol) der Verbindung aus Beispiel 43 A und 37 mg (0.1 mmol) N2-tert-Butoxy carbony I)-N- {2-[(tert-butoxycarbonyl)amino] ethyl} -L-ornithinamid (Beispiel
104A) in 2 ml Dimethylformamid gelöst. Bei 0°C (Eisbad) werden dann 19 mg (0.1 mmol) EDC und 3.1 mg (0.023 mmol) HOBt zugegeben. Es wird langsam auf RT erwärmt und für 12 h bei RT gerührt. Die Lösung wird im Vakuum eingeengt und der Rückstand wird mit Wasser verrührt. Der verbleibende Feststoff wird abgesaugt und über präparative HPLC gereinigt.
Ausbeute: 6 mg (7% d. Th.)
LC-MS (Methode 3): R, = 2.49 min.
MS (ESI): m/z = 1013 (M+H)+
Beispiel 118 A
Di-tert-butyI-(5-{[(3S)-6-[tert-butoxycarbonyl)amino]-3-({[(8S',115,145)-14-[tert-butoxy- carbonyl)amino]-l l-{(2 R)-3-[(tert-butoxycarbonyl)amino]-2-hydroxypropyl}-17-hydroxy-9- methy 1- 10,13 -dioxo-9, 12-diazatr icyclo[l 4.3.1.1
2,6]henicosa- 1 (20),2(21 ),3 ,5 , 16, 18-hexaen-8- yl]carbonyl}ammo)hexanoyl]amino}pentan-l,4-diyl)biscarbamat
Es werden 30.7 mg (0.046 mmol) (8S,ll,S',145)-14-[(rerr-Butoxycarbonyl)amino]-ll-{(2i?)-3- [(^r^-butoxycarbonyl)amino]-2-hydroxypropyl}-17-hydroxy-9-methyl-10,13-dioxo-9,12-diazatri- cyclo[14.3.1.12'6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-carbonsäure (Beispiel 45A) und 30 mg (0.055 mmol) der Verbindung aus Beispiel 81 A in 2.0 ml DMF gelöst und auf 0°C gekühlt. Man versetzt mit 11.4 mg (0.06 mmol) EDC und 2 mg (0.015 mmol) HOBt und rührt 12 h bei Raumtemperatur. Das Reaktionsgemisch wird im Vakuum einrotiert und chromatographisch über Sephadex-LH20 (Laufmittel: Methanol / Essigsäure 0.25%) gereinigt.
Ausbeute: 13 mg (24% d. Th.)
LC-MS (Methode 3): Rt = 2.84 min.
MS (ESI): m/z= 1198 (M+H)+
Analog zur Vorschrift des Beispiels 112A wird das in der folgenden Tabelle aufgeführte Beispiel 119A hergestellt.
Analog zur Vorschrift des Beispiels 117A werden die in der folgenden Tabelle aufgerührten Beispiele 120A bis 126A hergestellt.
Analog zur Vorschrift des Beispiels 113A werden die in der folgenden Tabelle aufgeführten Beispiele 127A bis 149A hergestellt.
Analog zur Vorschrift des Beispiels 48A werden die in der folgenden Tabelle aufgeführten Beispiele 150A bis 187A aus den entsprechenden Edukten hergestellt.
Analog zur Vorschrift des Beispiels 49A werden die in der folgenden Tabelle aufgeführten Beispiele 188A bis 224A aus den entsprechenden Edukten hergestellt.
Benzyl-((45)-5-[(3-amino-2-hydroxyρropyl)amino]-4-{[(benzyloxy)carbonyl]amino}-5-oxo- pentyl)carbamat Hydrochlorid
Eine Lösung von 0.263 g (0.46 mmol) der Verbindung aus Beispiel 187A in 1 ml Dioxan wird bei O°C mit 6.8 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 2 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 0.205 g (88% d. Th.)
LC-MS (Methode 2): Rt = 1.47 min.
MS (EI): m/z = 473 (M-HCH-H)+
Beispiel 226A
Benzyl-[(1 S)-4-{[(benzyloxy)carbonyl]amino}-l-({[3-({[(8S,l 1 S,14S)-14-[(tert-butoxycarbonyl)- amino]-l l-{(2R)-3-[(tert-butoxycarbonyl)amino]-2-hydroxypropyl}-l 7-hydroxy-9-methyl-l 0, 13- dioxo-9, 12-diazatricyclo[ 14.3.1.12>6]henicosa- 1 (20),2(21),3,5, 16, 18-hexaen-8-yl]carbonyl} amino)- 2-hydroxypropyl]amino}carbonyl)butyl]carbarnat
Es werden 25 mg (0.037 mmol) der Verbindung aus Beispiel 45A in 1.0 ml DMF gelöst und auf O°C gekühlt. Man versetzt mit 21 mg (0.O41 mmol) PyBOP und 15 mg (0.11 mmol) Diisopropylamin. Nach 30 min werden 24.7 mg (0.048 mmol) der Verbindung aus Beispiel 225A hinzugegeben und die Mischung wird 12 h bei Raumtemperatur gerührt. Das Reaktionsgemisch wird im Vakuum einrotiert und chromatographisch über Sephadex-LH20 (Laufmittel: Methanol / Essigsäure 0.25%) gereinigt.
Ausbeute: 12.7 mg (30% d. Th.)
LC-MS (Methode 3): R1 = 2.61 min.
MS (ESI): m/z = 1125 (M+H)+
Beispiel 227A
tert-Butyl-{(2R)-3-[(8S,l lS,14S)-14-[(tert-butoxycarbonyl)amino]-17-hydroxy-8-({[2-hydroxy-3- (L-omithylamino)propy l]amino} carbony l)-9-methyl-l 0, 13-dioxo-9, 12-diazatricyclo[ 14.3.1.12>6]- henicosa-l(20),2(21),3,5,16,18-hexaen-l l-yl]-2-hydroxypropyl}carbamat
12.7 mg (0.011 mmol) der Verbindung aus Beispiel 226A werden in 5 ml Ethanol gelöst, mit 5 mg Pd/C (10%ig) versetzt und 12 h bei Normaldruck und Wasserstoffatmosphäre hydriert. Man saugt ab, engt das Reaktionsgemisch im Vakuum ein und verwendet das Rohprodukt ohne weitere Reinigung im nächsten Schritt.
Ausbeute: 11 mg (95% d. Th.)
LC-MS (Methode 2): R1 = 1.26 min.
MS (ESI): m/z = 857 (M+H)+
Analog zur Vorschrift des Beispiels 112A werden die in der folgenden Tabelle aufgeführten Beispiele 228A und 229A hergestellt.
Analog zur Vorschrift des Beispiels 117A werden die in der folgenden Tabelle aufgeführten Beispiele 230A bis 254A hergestellt.
Analog zur Vorschrift des Beispiels 113A werden die in der folgenden Tabelle aufgeführten Beispiele 255A bis 28 IA hergestellt.
Ausführungsbcispiele
Beispiel 1
(8S,l lS,14S)-14-Amino-N-((lS)-4-amino-l-{[(2-:aminoethyl)atnino]carbonyl}butyl)-ll-[(2i?)-3- amino-2-hydroxypropyl] - 17-hydroxy- 10, 13 -dioxo-9, 12-diazatricyclo [14.3.1.12>6]henicosa- l(20),2(21),3,5,16518-hexaen-8-carboxamid Tetrahydrochlorid
Zu einer Lösung von 5.7 mg (0.006 mmol) der Verbindung aus Beispiel 120A in 1 ml Dioxan werden bei O°C 0.084 ml einer 4Ν Chlorwasserstoff-Dioxan-Lösung hinzugegeben. Nach 2 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 3.3 mg (77% d. Th.)
MS (ESI): m/z = 612 (M-4HC1+H)+.
Beispiel 2
(85,115,141S)-14-Amino-l l-[(2R)-3-amino-2-hydroxypropyl]-N-(2-{[(25)-2,5-diaminopentyl]- amino } -2-oxoethyl)- 17-hydroxy- 10, 13-dioxo-9, 12-diazatricyclo [14.3.1.12>6]henicosa- 1 (20),2(21),3,5, 16,18-hexaen-8-carboxamid Tetrahydrochlorid
Zu einer Lösung von 4.2 mg (0.004 mmol) der Verbindung aus Beispiel 121A in 1 ml Dioxan werden bei 0°C 0.062 ml einer 4N Chlorwasserstoff-Dioxan-Lösung hinzugegeben. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 2 mg (64% d. Th.)
MS (ESI): m/z = 613 (M-4HC1+H)+.
Beispiel 3
(85,115,145)-14-Amino-N-[(15)-4-amino-l-({[(25)-2,5-diaminopentyl]amino}carbonyl)butyl]-l 1- (3 -aminopropy I)- 17-hydroxy- 10,13 -dioxo-9, 12-diazatricyclo [14.3.1.12>6]henicosa- 1 (20),2(21 ),3,5, 16, 18-hexaen-8-carboxamid Pentahydrochlorid
Zu einer Lösung von 22.8 mg (0.02 mmol) der Verbindung aus Beispiel 113A in 1 ml Dioxan werden bei O°C 0.4 ml einer 4N Chlorwasserstoff-Dioxan-Lösung hinzugegeben. Nach 3 h bei RT
wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 15.3 mg (93% d. Th.)
+
MS (ESI): m/z = 654 (M-5HC1+H)
1H-NMR (400 MHz, D2O): δ = 1.55-1.95 (m, 12H), 2.8-3.2 (m, 9H), 3.3-3.7 (m, 4H), 4.29 (mc, IH), 4.47 (mc, IH), 4.7-4.9 (m, 2H, unter D2O)3 6.94 (d, IH), 6.99 (s, IH), 7.16 (d, IH), 7.31 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 4
(85r,l lS,145)-14-Amino-N-[(lS)-4-amino-l-({[(2S)-2,5-diaminopentyl]amino}carbonyl)butyl]-l l- (3 -aminopropyl)- 17-hydroxy- 10, 13 -dioxo-9, 12-diazatricyclo[ 14.3.1.12l6]henicosa- 1 (20),2(21 ),3 ,5, 16, 18-hexaen-8-carboxamid Penta(hydrotrifluoracetat)
Beispiel 3 als Tetrahydrochlorid-Salz wird durch präparative HPLC (Reprosil ODS-A, Laufmittel Acetonitril / 0.2% wässrige Trifluoressigsäure 5:95 -> 95:5) in das Tetra(hydrotrifluoracetat) überführt.
LC-MS (Methode 10): R, = 2.21 min.
MS (ESI): m/z = 654 (M-5TFA+H)+.
Beispiel 5
(85',l l^,145)-14-Amino-N-{(4S)-4-amino-5-[(2-aminoethyl)amino]-5-oxopentyl}-l l-[(2/?)-3- amino-2-hydroxypropyl]-l 7-hydroxy-l 0, 13-dioxo-9, 12-diazatricyclo[l 4.3.1.12>6]henicosa- l(20),2(21),3,5,16,18-hexaen-8-carboxamid Tetrahydrochlorid
Eine Lösung von 4.6 mg (0.005 mmol) der Verbindung aus Beispiel 117A in 1 ml Dioxan wird bei 0°C mit 0.27 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 3.4 mg (99% d. Th.)
MS (ESI): m/z = 613 (M-4HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.47-1.67 (m, 2H), 1.75-2.09 (m, 4H), 2.89 (m0, IH), 2.95-3.25 (m, 7H), 3.3 (mc, IH), 3.4 (mc, IH), 3.5-3.7 (m, 2H), 3.86 (m0, IH), 3.98 (m0, IH), 4.44 (mc, IH), 4.7- 4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.16 (d, IH), 7.31 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 6
(8S, 11 S, 14S)- 14-Amino-N-[( l>S>4-amino- 1 -({ [(5S)-5-amino-6-hydroxyhexyl]amino } carbonyl)- buty I]- 11 -(3 -aminopropyl)- 17-hydroxy- 10, 13 -dioxo-9, 12-diazatricyclo[ 14.3.1.12>6]henicosa- l(20),2(21),3,5,16,18-hexaen-8-carboxamid Tetrahydrochlorid
Eine Lösung von 62 mg (0.058 mmol) der Verbindung aus Beispiel 128A in 1 ml Dioxan wird bei O°C mit 0.87 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 46 mg (97% d. Th.)
LC-MS (Methode 10): R. = 1.84 min.
MS (ESI): m/z = 669 (M-4HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.25-1.95 (m, 14H), 2.9-3.3 (m, 10H), 3.5-3.8 (m, 3H)3 4.19 (ItI0, IH), 4.46 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.16 (d, IH), 7.31 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 7
(85, 115, 145)- 14-Amino-N-(( 15)- 1 -(aminomethyl)-2- { [(25)-2,5-diaminopentyl]amino } -2- oxoethyl)- 11 -(3 -aminopropy I)- 17-hydroxy- 10,13 -dioxo-9, 12-diazatricyclo [14.3.1.12>6]henicosa- l(20),2(21),3,5,16,l 8-hexaen-8-carboxamid Pentahydrochlorid
Eine Lösung von 70 mg (0.062 mmol) der Verbindung aus Beispiel 129A in 1 ml Dioxan wird bei O°C mit 0.94 ml einer 4Ν Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 50 mg (99% d. Th.)
MS (ESI): m/z = 626 (M-5HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.55-1.95 (m, 8H), 2.9-3.2 (m, 6H), 3.26 (m0, IH), 3.3-3.7 (m, 7H), 4.47 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.16 (d, IH), 7.31 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 8
(85, 115, 145)- 14-Amino-N-(( 15)-4-amino- 1 - { [(2-aminoethy l)amino]carbony 1} buty I)- 11 -(3 -amino- propyl)-17-hydroxy-10,13-dioxo-9,12-diazatricyclo[14.3.1.12-6]henicosa-l(20),2(21),3,5,16,18- hexaen-8-carboxamid Tetrahydrochlorid
Eine Lösung von 12 mg (0.012 mmol) der Verbindung aus Beispiel 130A in 1 ml Dioxan wird bei O°C mit 0.181 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 8.8 mg (99% d. Th.)
MS (ESI): m/z = 597 (M-4HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.55-1.95 (m, 8H)5 2.9-3.2 (m, 8H), 3.4-3.7 (m, 4H), 4.25 (mc, IH), 4.46 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.17 (d, IH), 7.32 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 9
(85r,l l.S',14)S)-14-Ammo-N-((15)-4-ammo-l-{[((15)-4-ammo-l-{2-[(2-aminoethyl)amino]-2- oxoethy 1} buty l)amino] carbony 1} buty I)- 11 -(3 -aminopropyl)- 17-hydroxy- 10,13 -dioxo-9, 12- diazatricyclo[14.3.1.12>6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-carboxamid Pentahydrochlorid
Eine Lösung von 24 mg (0.02 mmol) der Verbindung aus Beispiel 133A in 1 ml Dioxan wird bei 0°C mit 0.29 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 17.5 mg (99% d. Th.)
MS (ESI): m/z = 725 (M-5HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.45-2.0 (m, 12H), 2.36 (mc, IH), 2.9-3.2 (m, HH), 3.4-3.7 (m, 4H), 4.1-4.25 (m, 2H), 4.47 (mOJ IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.98 (s, IH), 7.17 (d, IH), 7.32 (s, IH), 7.35 (t, IH)5 7.4-7.5 (m, 2H).
Beispiet 10
(8S, 11 S, 14S)- 14-Amino-N-((l ,S)-4-amino- 1 - { [(( 1 <S)-4-amino- 1 - {2-[(2-aminoethyl)amino]-2- oxoethyl} butyl)amino] carbony 1} buty I)- 11 -[(2R)-3 -amino-2-hydroxypropyl]- 17-hydroxy- 10,13- dioxo-9,12-diazatricyclo[14.3.1.12>6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-carboxamid Pentahydrochlorid
Eine Lösung von 13 mg (0.01 mmol) der Verbindung aus Beispiel 134A in 1 ml Dioxan wird bei O°C mit 0.16 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 9.5 mg (99% d. Th.)
MS (ESI): m/z = 741 (M-5HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.4-2.05 (m, 10H), 2.37 (m-, IH), 2.53 (mc, IH), 2.8-3.2 (m, 10H), 3.3-3.7 (m, 3H), 3.86 (m0, IH), 4.1-4.21 (m, 2H), 4.44 (m0, IH), 4.7-4.9 (m, 2H, unter D2O), 6.95 (d, IH), 7.0 (s, IH), 7.18 (d, IH), 7.3-7.4 (m, 2H), 7.4-7.5 (m, 2H).
Beispiel 11
(85, 115, 145)- 14-Amino-N- {( 15)-4-amino-l -[({(45)-4-amino-6-[(2-aminoethyl)amino]-6- oxohexy 1 } amino)carbony 1] buty 1} - 11 -[(2R)-3 -amino-2-hydroxypropy 1] - 17-hydroxy- 10, 13-dioxo- 9, 12-diazatricyclo[ 14.3.1.12>6]henicosa-l (20),2(21 ),3 ,5, 16, 18-hexaen-8-carboxamid Pentahydrochlorid
Eine Lösung von 24 mg (0.02 mmol) der Verbindung aus Beispiel 135A in 1 ml Dioxan wird bei O°C mit 0.29 ml einer 4Ν Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 17.5 mg (99% d. Th.)
MS (ESI): m/z = 741 (M-5HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.45-2.05 (m, 10H), 2.55 (m0, IH), 2.68 (m0, IH), 2.8-3.2 (m, 10H), 3.3-3.7 (m, 4H), 3.86 (mc, IH), 4.21 (mc, 2H), 4.44 (m0, IH), 4.7-4.9 (m, 2H, unter D2O)3 6.94 (d, IH), 6.99 (s, IH), 7.17 (d, IH), 7.33 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 12
(85, 115, 145)- 14-Amino-N- {( 15)-4-amino- 1 -[({(45)-4-amino-6-[(2-aminoethyl)amino]-6- oxohexy 1 } amino)carbony l]buty 1} - 11 -(3 -aminopropy I)- 17-hydroxy- 10, 13 -dioxo-9, 12- diazatricyclo[ 14.3.1.12'6]henicosa- 1 (20),2(21 ),3 ,5, 16, 18-hexaen-8-carboxamid Pentahydrochlorid
Eine Lösung von 21 mg (0.017 mmol) der Verbindung aus Beispiel 136A in 1 ml Dioxan wird bei 0°C mit 0.26 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 15 mg (99% d. Th.)
MS (ESI): m/z = 716 (M-5HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.45-1.95 (m, 12H), 2.55 (mc, IH), 2.68 (mc, IH), 2.9-3.2 (m, 10H), 3.42 (mc, 2H), 3.5-3.7 (m, 3H), 4.2 (mc, IH), 4.46 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.98 (s, IH), 7.17 (d, IH), 7.32 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 13
(8S,11S,14S)-Amino-N--[(1S)-4-amino- ({(2S)-2,5--diaminopentyl]amino}carbonyl)butyl]-11- [(2R)-3-amino-2-hydroxypropyl]-17-hydroxy-10,13-dioxo-9,12-diazatricyclo[14.3.1.12,6]henicosa- 1 (20),2(21 ),3,5, 16, 18-hexaen-8-carboxamid Pentahydrochlorid
Eine Lösung von 20 mg (0.017 mmol) der Verbindung aus Beispiel 137A in 1 ml Dioxan wird bei O°C mit 0.256 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 13.5 mg (93% d. Th.)
MS (ESI): m/z = 670 (M-5HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.5-2.05 (m, 10H), 2.8-3.2 (m, 8H), 3.3-3.7 (m, 5H), 3.86 (mc, IH), 4.30 (mc, IH), 4.44 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.17 (d, IH), 7.33 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 14
(δ^l l.S'jH^-M-Amino-N-^l^^-amino-l-IC^^-amino-ό-l^^^^-diaminopenty^amino}- 6-oxohexyl)amino]carbonyl} butyl)- 11 -[(2R)-3-amino-2-hydroxypropyl]- 17-hydroxy- 10, 13-dioxo- 9, 12-diazatricyclo[ 14.3.1.12'6]henicosa-1 (20),2(21),3,5, 16, 18-hexaen-8-carboxamid Hexahydrochlorid
Eine Lösung von 29 mg (0.021 mmol) der Verbindung aus Beispiel 138A in 1 ml Dioxan wird bei 0°C mit 0.31 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 16.5 mg (78% d. Th.)
MS (ESI): m/z = 798 (M-6HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.45-2.05 (m, 14H), 2.50 (mc, IH), 2.72 (mc, IH), 2.8-3.7 (m, 15H), 3.89 (mc, IH), 4.23 (mc, IH), 4.46 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.17 (d, IH), 7.33 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiet 15
(8S,115,145)-14-Amino-N-((16)-4-amino-l-{[((4^-4-amino-6-{[(2S)-2,5-diaminopentyl]amino}- 6-oxohexy l)amino] carbonyl} buty I)- 11 -(3 -aminopropyl)- 17-hydroxy- 10, 13 -dioxo-9, 12- diazatricyclo[14.3.1.12>6]henicosa-l(20),2(21),3,5,16,18-hexaen-8-carboxamid Hexahydrochlorid
Eine Lösung von 29 mg (0.021 mmol) der Verbindung aus Beispiel 139A in 1 ml Dioxan wird bei O°C mit 0.31 ml einer 4Ν Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 16.5 mg (78% d. Th.)
MS (ESI): m/z = 782 (M-6HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.45-1.95 (m, 16H), 2.60 (mc, IH), 2.83 (m0, IH), 2.9-3.3 (m, 10H), 3.3-3.75 (m, 6H), 4.24 (mc, IH), 4.49 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.17 (d, IH), 7.33 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 16
(8S,l l.S',145)-14-Amino-N-[(15)-4-amino-l-({[(l
1S)-4-amino-l-(2-{[(2S)-2,5-diaminopentyl]- amino}-2-oxoethyl)butyl]amino}carbonyl)butyl]-l l-(3-aminopropyl)-17-hydroxy-10,13-dioxo- 9, 12-diazatricyclo[14.3.1.1
2'
6]henicosa-1 (20),2(21),3,5, 16, 18-hexaen-8-carboxamid Hexahydrochlorid
Eine Lösung von 28 mg (0.02 mmol) der Verbindung aus Beispiel 140A in 1 ml Dioxan wird bei 0°C mit 0.3 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 20 mg (99% d. Th.)
MS (ESI): m/z = 782 (M-6HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.4-1.9 (m, 16H), 2.4 (mc, IH), 2.54 (mc, IH), 2.85-3.2 (m, HH), 3.29 (mc, IH), 3.39 (mc, IH), 3.45-3.65 (m, 2H), 4.1-4.25 (m, 2H), 4.47 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.17 (d, IH), 7.33 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 17
(8S,11S,14S)-14-Amino-N-[(1S)-4 -amino-1-({[(1S)-4--amino-1-(2-{[(2S)-2,5-diaminopentyl]- amino } -2-oxoethy l)buty 1] amino } carbonyl)butyl]- 11 -[(2R)-3 -amino-2-hydroxypropyl] - 17-hydroxy- 10, 13-dioxo-9, 12-diazatricyclo[ 14.3.1.12,6]henicosa- 1 (20),2(21),3,5, 16, 18-hexaen-8-carboxamid Hexahydrochlorid
Eine Lösung von 36 mg (0.026 mmol) der Verbindung aus Beispiel 141A in 1 ml Dioxan wird bei O°C mit 0.39 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 26 mg (99% d. Th.)
MS (ESI): m/z = 798 (M-6HC1+H)+.
1H-NMR (400 MHz5 D2O): δ = 1.4-2.05 (m, 14H), 2.41 (m,, IH), 2.54 (mc, IH), 2.85-3.2 (m, HH), 3.29 (mc, IH), 3.39 (m05 IH), 3.45-3.65 (m, 2H), 3.85 (mc, IH), 4.1-4.25 (m, 2H), 4.45 (mc, IH), 4.7-4.9 (m, 2H, unter D2O)5 6.95 (d, IH), 7.0 (s, IH)5 7.17 (d, IH), 7.29-7.6 (m, 4H).
Beispiel 18
N5-(N2-{ [(8S, 11 S, 14S)-14-Amino- 11 -(3 -aminopropy I)- 17-hydroxy- 10, 13 -dioxo-9, 12- diazatricyclof 14.3.1.12>6]henicosa- 1 (20),2(21 ),3 ,5, 16, 18-hexaen-8-yl]carbonyl} -Z,-ornithyl)-N-(2- aminoethyl)-Z-omithinamid Pentahydrochlorid
Eine Lösung von 47 mg (0.039 mmol) der Verbindung aus Beispiel 142A in 1 ml Dioxan wird bei 0°C mit 0.58 ml einer 4Ν Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 34 mg (99% d. Th.)
MS (ESI): m/z = 711 (M-5HC1+H)+.
1H-NMR (400 MHz, D2O): δ = 1.45-1.95 (m, 12H), 2.9-3.25 (m, 10H), 3.38 (m0, IH), 3.5-3.7 (m, 2H), 3.96 (mc, IH), 4.26 (mc, IH), 4.47 (m0, IH), 4.7-4.9 (m, 2H3 unter D2O), 6.94 (d, IH), 6.99 (s, IH), 7.17 (d, IH), 7.33 (s, IH), 7.35 (t, IH), 7.4-7.5 (m, 2H).
Beispiel 19
(S^l l^H^-M-Amino-N-^l^^-amino-l^-I^^^S-diaminopentylJaminoJ^-oxoethyl)- buty I]- 11 -(3 -aminopropy I)- 17-hydroxy- 10, 13 -dioxo-9, 12-diazatricyclo [ 14.3.1.12|6]henicosa- l(20),2(21),3,5,16,18-hexaen-8-carboxamid Penta(hydrotrifluoracetat)
X 5 TFA
Zu einer Lösung von 15 mg (0.013 mmol) der Verbindung aus Beispiel 143 A in 1 ml Dioxan werden bei O°C 0.19 ml einer 4N Chlorwasserstoff-Dioxan-Lösung hinzugegeben. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet. Das Rohprodukt wird durch präparative HPLC (Reprosil ODS-A, Laufmittel Acetonitril / 0.2% wässrige Trifiuoressigsäure 5:95 -^ 95:5) in das Tetra(hydrotrifluoracetat) überfuhrt.
Ausbeute: 5.4 mg (34% d. Th.)
MS (ESI): m/z = 668 (M-5TFA+H)+.
1H-NMR (400 MHz, D2O): δ = 1.4-1.9 (m, 12H), 2.39 (m0, IH), 2.57 (mc, IH), 2.83-3.17 (m, 9H), 3.32 (m0, IH), 3.41 (m0, IH), 3.5-3.7 (m, 2H), 4.21 (mc, IH), 4.46 (mc, IH), 4.7-4.9 (m, 2H, unter D2O), 6.94 (d, IH), 6.98 (s, IH), 7.11 (d, IH), 7.32 (s, IH), 7.35 (t, IH), 7.44-7.55 (m, 2H).
Beispiel 20
(86',115',145)-14-Amino-N-(l-(2-aminoethyl)-3-{[(25)-2,5-diaminopentyl]aniino}-3-oxopropyl)-
H^S-aminopropyO-π-hydroxy-lO^S-dioxo-Pj^-diazatricyclofM.S.l.l^henicosa- l(20),2(21),3,5,16,18-hexaen-8-carboxamid Penta(hydrotrifluoracetat)
Zu einer Lösung von 14.8 mg (0.013 mmol) der Verbindung aus Beispiel 144A in 1 ml Dioxan werden bei O°C 0.19 ml einer 4N Chlorwasserstoff-Dioxan-Lösung hinzugegeben. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet. Das Rohprodukt wird durch präparative HPLC (Reprosil ODS-A, Laufmittel Acetonitril / 0.2% wässrige Trifluoressigsäure 5:95 -> 95:5) in das Tetra(hydrotrifluoracetat) überführt.
Ausbeute: 8.9 mg (57% d. Th.)
MS (ESI): m/z = 654 (M-5TFA+H)+.
1H-NMR (400 MHz, D2O): δ = 1.5-2.0 (m, 10H), 2.4-2.65 (m, 2H), 2.85-3.2 (m, 9H), 3.25-3.47 (m, 2H), 3.53-3.68 (m, 2H), 4.27 (mc, IH), 4.46 (mC5 IH), 4.7-4.9 (m, 2H, unter D2O), 6.9-7.0 (m, 2H), 7.05-7.15 (m, IH), 7.3-7.4 (m, 2H), 7.42-7.52 (m, 2H).
Beispiel 21
(86',l lS514S)-14-Amino-N-[(15)-4-amino-l-(2-{[(25)-2,5-diaminopentyl]amino}-2-oxoethyl)- butyl]-l l-[(2i?)-3-amino-2-hydroxypropyl]-17-hydroxy-9-methyl-10,13-dioxo-9,12-diazatri- cyclo[ 14.3.1.12>6]henicosa-l (20),2(21),3 ,5, 16, 18-hexaen-8-carboxamid Pentahydrochlorid
Eine Lösung von 12.9 mg (0.011 mmol) der Verbindung aus Beispiel 118A in 1 ml Dioxan wird bei 0°C mit 0.161 ml einer 4N Chlorwasserstoff-Dioxan-Lösung versetzt. Nach 3 h bei RT wird die Reaktionslösung im Vakuum eingeengt und mehrmals mit Dichlormethan coevaporiert. Der zurückbleibende Feststoff wird im Hochvakuum bis zur Gewichtskonstanz getrocknet.
Ausbeute: 9 mg (95% d. Th.)
MS (ESI): m/z = 698 (M-5HC1+H)+.
Analog zur Vorschrift des Beispiels 1 werden die in der folgenden Tabelle aufgeführten Beispiele hergestellt, entsprechend der jeweiligen Isolierungsmethode als Hydrochlorid- oder Hydro(trifluoracetat)-Salz.
Analog zur Vorschrift des Beispiels 1 werden die in der folgenden Tabelle aufgeführten Beispiele 39 bis 93 hergestellt, entsprechend der jeweiligen Isolierungsmethode als Hydrochlorid- oder Hydro(trifluoracetat)-Salz.
Bewertung der physiologischen Wirksamkeit
Verwendete Abkürzungen:
AMP Adenosinmonophosphat
ATP Adenosintriphosphat
BHI Medium Brain heart infϊision medium
CoA Coenzym A
DMSO Dimethylsulfoxid
DTT Dithiothreitol
EDTA Ethylendiamintetraessigsäurβ
KCl Kaliumchlorid
KH2PO4 Kaliumdihydrogenphosphat
MgSO4 Magnesiumsulfat
MHK Minimale Hemmkonzentration
MTP Mikrotiterplatte
NaCl Natriumchlorid
Na2HPθ4 Dinatriumhydrogenphosphat
NH4Cl Ammoniumchlorid
NTP Nukleotidtriphosphat
PBS Phosphat Buffered Saline
PCR Polymerase Chain Reaction
PEG Polyethylenglykol
PEP Phosphoenolpyruvat
Tris Tris[hydroxymethyl]aminomethan
Die in vitro-Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
In vitro Transkription-Translation mit E. coli Extrakten
Zur Herstellung eines S30-Extraktes werden logarithmisch wachsende Escherichia coli MRE 600 (M. Müller; University Freiburg) geerntet, gewaschen und wie beschrieben für den in vitro Transkriptions-Translations-Test eingesetzt (Müller, M. and Blobel, G. Proc Natl Acad Sei U S A (1984) 81, pp.7421-7425).
Dem Reaktionsmix des in vitro Transkriptions-Translations-Tests werden zusätzlich 1 μl cAMP (11.25 mg/ml) je 50 μl Reaktionsmix zugegeben. Der Testansatz beträgt 105 μl, wobei 5 μl der zu
testenden Substanz in 5%igem DMSO vorgelegt werden. Als Transkriptionsmatrize werden 1 μg/100μl Ansatz des Plasmides pBESTLuc (Promega, Deutschland) verwendet. Nach Inkubation für 60 min bei 3O°C werden 50 μl Luziferinlösung (20 mM Tricine, 2.67 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT pH 7.8, 270 μM CoA5 470 μM Luziferin, 530 μM ATP) zugegeben und die entstehende Biolumineszenz für 1 Minute in einem Luminometer gemessen. Als IC50 wird die Konzentration eines Inhibitors angegeben, die zu einer 50%igen Inhibition der Translation von Firefly Luziferase führt.
In vitro Transkription-Translation mit S. aureus Extrakten
Konstruktion eines S. aureus Luziferase Reporterplasmids
Zur Konstruktion eines Reporterplasmids, welches in einem in vitro Transkriptions-Translations- Assay aus S. aureus verwendet werden kann, wird das Plasmid pBESTluc (Promega Corporation, USA) verwendet. Der in diesem Plasmid vor der Firefly Luziferase vorhandene E. coli tac Promoter wird gegen den capAl Promoter mit entsprechender Shine-Dalgarno Sequence aus S. aureus ausgetauscht. Dazu werden die Primer CAPFor 5'-CGGCC- AAGCTTACTCGGATCCAGAGTTTGCAAAATATACAGGGGATTATATATAATGGAAAAC AAGAAAGGAAAATAGGAGGTTTATATGGAAGACGCCA-3' und CAPRev 5'- GTCATCGTCGGGAAGACCTG-3' verwendet. Der Primer CAPFor enthält den capAl Promotor, die Ribosomenbindestelle und die 5'-Region des Luziferase Gens. Nach PCR unter Verwendung von pBESTluc als Template kann ein PCR-Produkt isoliert werden, welches das Firefly Luziferase Gen mit dem fusionierten capÄl Promotor enthält. Dieses wird nach einer Restriktion mit CIaI und Hindin in den ebenfalls mit CIaI und Hindlll verdauten Vektor pBESTluc ligiert. Das entstandene Plasmid pla kann in E. coli repliziert werden und als Template im S. aureus in vitro Transkriptions-Translations-Test verwendet werden.
Herstellung von S30 Extrakten aus S. aureus
Sechs Liter BHI Medium werden mit einer 250 ml Übernachtkultur eines S. aureus Stammes inokuliert und bei 37°C bis zu einer OD600nm von 2-4 wachsen gelassen. Die Zellen werden durch Zentrifugation geerntet und in 500 ml kaltem Puffer A (10 mM Tris-acetat, pH 8.0, 14 mM Magnesiumacetat, 1 mM DTT, 1 M KCl) gewaschen. Nach erneutem Abzentrifugieren werden die Zellen in 250 ml kaltem Puffer A mit 50 mM KCl gewaschen und die erhaltenen Pellets bei -20°C für 60 min eingefroren. Die Pellets werden in 30 bis 60 min auf Eis aufgetaut und bis zu einem Gesamtvolumen von 99 ml in Puffer B (10 mM Tris-acetat, pH 8.0, 20 mM Magnesiumacetat, 1 mM DTT, 50 mM KCl) aufgenommen. Je 1.5 ml Lysostaphin (0.8 mg/ml) in Puffer B werden in 3 vorgekühlte Zentrifugenbecher vorgelegt und mit je 33 ml der Zellsuspension vermischt. Die
Proben werden für 45 bis 60 min bei 37°C unter gelegentlichem Schütteln inkubiert, bevor 150 μl einer 0.5 M DTT Lösung zugesetzt werden. Die lysierten Zellen werden bei 30.000 x g 30 min bei 4°C abzentrifugiert. Das Zellpellet wird nach Aufnahme in Puffer B unter den gleichen Bedingungen nochmals zentrifugiert und die gesammelten Überstände werden vereinigt. Die Überstände werden nochmals unter gleichen Bedingungen zentrifugiert und zu den oberen 2/3 des Überstandes werden 0.25 Volumen Puffer C (670 mM Tris-acetat, pH 8.0, 2O mM Magnesiumacetat, 7 mM Na3-Phosρhoenolpyruvat, 7 mM DTT, 5.5 mM ATP, 70 μM Aminosäuren (complete von Promega), 75 μg Pyruvatkinase (Sigma, Deutschland))/ml gegeben. Die Proben werden für 30 min bei 37°C inkubiert. Die Überstände werden über Nacht bei 4°C gegen 2 1 Dialysepuffer (10 mM Tris-acetat, pH 8.0, 14 mM Magnesiumacetat, 1 mM DTT5 60 mM Kaliumacetat) mit einem Pufferwechsel in einem Dialyseschlauch mit 3500 Da Ausschluss dialysiert. Das Dialysat wird auf eine Proteinkonzentration von etwa 10 mg/ml konzentriert, indem der Dialyseschlauch mit kaltem PEG 8000 Pulver (Sigma, Deutschland) bei 4°C bedeckt wird. Die S30 Extrakte können aliquotiert bei -7O°C gelagert werden.
Bestimmung der ICgn im S. aureus in vitro Transcriptions-Translations-Assay
Die Inhibition der Proteinbiosynthese der Verbindungen kann in einem in vitro Transkriptions- Translations-Assay gezeigt werden. Der Assay beruht auf der zellfreien Transkription und Translation von Firefly Luziferase unter Verwendung des Reporterplasmids pla als Template und aus S. aureus gewonnenen zellfreien S30 Extrakten. Die Aktivität der entstandenen Luziferase kann durch Lumineszenzmessung nachgewiesen werden.
Die Menge an einzusetzenden S30 Extrakt bzw. Plasmid pla muss für jede Präparation erneut ausgetestet werden, um eine optimale Konzentration im Test zu gewährleisten. 3 μl der zu testenden Substanz gelöst in 5% DMSO werden in eine MTP vorgelegt. Anschließend werden 10 μl einer geeignet konzentrierten Plasmidlösung pla zugegeben. Anschließend werden 46 μl eines Gemisches aus 23 μl Premix (500 mM Kaliumacetat, 87.5 mM Tris-acetat, pH 8.0, 67.5 mM Ammoniumacetat, 5 mM DTT, 50 μg Folsäure/ml, 87.5 mg PEG 8000/ml, 5 mM ATP5 1.25 mM je NTP, 20 μM je Aminosäure, 50 mM PEP (Na3-SaIz), 2.5 mM cAMP, 250 μg je E. coli tRNA/ml) und 23 μl einer geeigneten Menge S. aureus S30 Extrakt zugegeben und vermischt. Nach Inkubation für 60 min bei 3O°C werden 50 μl Luziferinlösung (20 mM Tricine, 2.67 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT pH 7.8, 270 μM CoA, 470 μM Luziferin, 530 μM ATP) und die entstehende Biolumineszenz für 1 min in einem Luminometer gemessen. Als IC50 wird die Konzentration eines Inhibitors angegeben, die zu einer 50%igen Inhibition der Translation von Firefly Luziferase führt.
Bestimmung der Minimalen Hemmkonzentration (CLSI-Standard)
Die minimale Hemmkonzentration (MHK) ist die minimale Konzentration eines Antibiotikums, mit der ein Testkeim in seinem Wachstum über 18-24 h inhibiert wird. Die Hemmstoffkonzentration kann dabei nach mikrobiologischen Standardverfahren bestimmt werden (siehe z.B. The National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-fifth edition. NCCLS document M7-A5 [ISBN 1-56238-394-9]. NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2000). Dabei werden die Testsubstanzen in 1:2- Verdünnungsreihen in 96-Loch-Rundboden-Mikrotiterplatten (Greiner) doppelt konzentriert in 50 μl Testmedium vorgelegt. Die aerob wachsenden Testkeime (z.B. Staphylokokken und Enterokokken), die über Nacht auf Columbia-Blutagarplatten (Becton-Dickinson) inkubiert werden, werden nach Resuspension in 0.9% NaCl auf eine Keimzahl von ca. 5x107 Keime/ml eingestellt und anschließend 1 :150 in Kationen-angepaßtem MH-Medium (Testmedium) verdünnt. Von dieser Suspension werden 50 μl auf die in Mikrotiterplatten vorgelegten Testpräparate pipettiert. Die Kulturen werden bei 37°C für 18-24 Stunden inkubiert. Für mikroaerophil wachsende Keime (z.B. Streptokokken) wird dem Medium 2% lysiertes Pferdeblut in der Endkonzentration zugesetzt und die Kulturen in Gegenwart von 5% CO2 inkubiert. Die jeweils niedrigste Substanzkonzentration, bei der kein sichtbares Bakterienwachstum mehr auftritt, wird als MHK definiert und wird in μg/ml angegeben.
Bestimmung der Minimalen Hemmkonzentration (MHK)
Die minimale Hemmkonzentration (MHK) ist die minimale Konzentration eines Antibiotikums, mit der ein Testkeim in seinem Wachstum über 18-24 h inhibiert wird. Die Hemmstoffkonzentration kann dabei nach mikrobiologischen Standardverfahren bestimmt werden (siehe z.B. The National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-fifth edition. NCCLS document M7-A5 [ISBN 1-56238-394-9]. NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2000). Die MHK der erfindungsgemäßen Verbindungen wird im Flüssigdilutionstest im 96er-Mikrotiter-Platten-Maßstab bestimmt. Die Bakterienkeime werden in einem Minimalmedium (18.5 mM Na2HPO4, 5.7 mM KH2PO4, 9.3 mM NH4Cl, 2.8 mM MgSO4, 17.1 mM NaCl, 0.033 μg/ml Thiaminhydrochlorid, 1.2 μg/ml Nicotinsäure, 0.003 μg/ml Biotin, 1% Glucose, 25 μg/ml von jeder proteinogenen Aminosäure mit Ausnahme von Phenylalanin; [H.- P. Kroll; unveröffentlicht]) unter Zusatz von 0.4% BH-Bouillon kultiviert (Testmedium). Im Fall von Enterococcus faecium L4001 wird dem Testmedium hitzeinaktiviertes fötales Kälberserum (FCS; GibcoBRL, Deutschland) in einer Endkonzentration von 10% zugesetzt. Übernachtkulturen
der Testkeime werden auf eine OD578 von 0.001 (im Falle der Enterokokken auf 0.01) in frisches Testmedium verdünnt und 1:1 mit Verdünnungen der Testsubstanzen (Verdünnungsstufen 1:2) in Testmedium inkubiert (200 μl Endvolumen). Die Kulturen werden bei 37°C für 18-24 Stunden inkubiert; Enterokokken in Gegenwart von 5% CO2.
Die jeweils niedrigste Substanzkonzentration, bei der kein sichtbares Bakterienwachstum mehr auftritt, wird als MHK definiert.
Alternative Bestimmungsmethode der Minimalen Hemmkonzentration (MHK)
Die minimale Hemmkonzentration (MHK) ist die minimale Konzentration eines Antibiotikums, mit der ein Testkeim in seinem Wachstum über 18-24 h inhibiert wird. Die Hemmstoff- konzentration kann dabei nach mikrobiologischen Standardverfahren mit modifiziertem Medium im Rahmen eines Agardilutionstests bestimmt werden (siehe z.B. The National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-fifth edition. NCCLS document M7-A5 [ISBN 1-56238- 394-9]. NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2000). Die Bakterienkeime werden auf 1.5%igen Agarplatten kultiviert, die 20% defibriniertes Pferdeblut enthalten. Die Testkeime, die über Nacht auf Columbia-Blutagarplatten (Becton- Dickinson) inkubiert werden, werden in PBS verdünnt, auf eine Keimzahl von ca. 5x105 Keime/ml eingestellt und auf Testplatten getropft (1-3 μl). Die Testsubstanzen enthalten unterschiedliche Verdünnungen der Testsubstanzen (Verdünnungsstufen 1:2). Die Kulturen werden bei 37°C für 18-24 Stunden in Gegenwart von 5% CO2 inkubiert.
Die jeweils niedrigste Substanzkonzentration, bei der kein sichtbares Bakterienwachstum mehr auftritt, wird als MHK definiert und in μg/ml angegeben.
Tabelle A (mit Vergleichsbeispiel Biphenomycin B)
Konzentrationsangaben: MHK in μg/ml; IC5O in μM.
Systemische Infektion mit S. aureus 133
Die Eignung der erfindungsgemäßen Verbindungen zur Behandlung von bakteriellen Infektionen kann in verschiedenen Tiermodellen gezeigt werden. Dazu werden die Tiere im allgemeinen mit einem geeigneten virulenten Keim infiziert und anschließend mit der zu testenden Verbindung, die in einer an das jeweilige Therapiemodell angepassten Formulierung vorliegt, behandelt. Speziell kann die Eignung der erfindungsgemäßen Verbindungen zur Behandlung von bakteriellen Infektionen in einem Sepsismodell an Mäusen nach Infektion mit S aureus demonstriert werden.
Dazu werden S aureus 133 Zellen über Nacht in BH-Bouillon (Oxoid, Deutschland) angezüchtet. Die Übernachtkultur wurde 1:100 in frische BH-Bouillon verdünnt und für 3 Stunden hochgedreht. Die in der logarithmischen Wachstumsphase befindlichen Bakterien werden abzentrifugiert und zweimal mit gepufferter, physiologischer Kochsalzlösung gewaschen. Danach wird am Photometer (Dr. Lange LP 2W) eine Zellsuspension in Kochsalzlösung mit einer Extinktion von 50 Einheiten eingestellt. Nach einem Verdünnungsschritt (1:15) wird diese Suspension 1 :1 mit einer 10%-igen Mucinsuspension gemischt. Von dieser Infektionslösung wird 0.2 ml/20 g Maus i.p. appliziert. Dies entspricht einer Zellzahl von etwa 1-2 x 106 Keimen/Maus. Die i.v. -Therapie erfolgt 30 Minuten nach der Infektion. Für den Infektions versuch werden weibliche CFWl -Mäuse verwendet. Das Überleben der Tiere wird über 6 Tage protokolliert. Das Tiermodell ist so eingestellt, daß unbehandelte Tiere innerhalb von 24 h nach der Infektion versterben. Für die Beispielverbindung 2 konnte in diesem Modell eine therapeutische Wirkung von EDioo = 1.25 mg/kg demonstriert werden.
Bestimmung der Spontanresistenzfrequenzen gegen S. aureus
Die Spontanresistenzraten der erfindungsgemäßen Verbindungen werden wie folgt bestimmt: die Bakterienkeime werden in 30 ml eines Minimalmediums (18.5 mM Na2HPC>4, 5.7 mM KH2PO4,
9.3 DiM NH4Cl, 2.8 mM MgSO4, 17.1 mM NaCl, 0.033 μg/ml Thiaminhydrochlorid, 1.2 μg/ml Nicotinsäure, 0.003 μg/ml Biotin, 1% Glucose, 25 μg/ml von jeder proteinogenen Aminosäure unter Zusatz von 0,4% BH Bouillon) bei 37°C über Nacht kultiviert, 10 min bei ό.OOOxg abzentrifugiert und in 2 ml phosphat-gepufferter physiologischer NaCl-Lösung resuspendiert (ca. 2x109 Keime/ml). 100 μl dieser Zellsuspension bzw. 1:10 und 1:100 Verdünnungen werden auf vorgetrockneten Agarplatten (1.5% Agar, 20% defϊbriniertes Pferdeblut bzw. 1.5% Agar, 20% Rinderserum in 1/10 Müller-Hinton-Medium verdünnt mit PBS), welche die zu testende erfindungsgemäße Verbindung in einer Konzentration entsprechend 5xMHK bzw. 1 OxMHK enthalten, ausplattiert und 48 h bei 37°C bebrütet. Die entstehenden Kolonien (cfu) werden ausgezählt.
Isolierung der Biphenomvcin-resistenten S. aureus Stämme RN4220BiR und T17
Der S. aureus Stamm RN4220BiR wird in vitro isoliert. Dazu werden jeweils 100 μl einer S. aureus RN4220 Zellsuspension (ca. 1.2x108 cfu/ml) auf einer antibiotikafreien Agarplatte (18.5 mM Na2HPO4, 5.7 mM KH2PO4, 9.3 mM NH4Cl, 2.8 mM MgSO4, 17.1 mM NaCl, 0.033 μg/ml Thiaminhydrochlorid, 1.2 μg/ml Nicotinsäure, 0.003 μg/ml Biotin, 1% Glucose, 25 μg/ml von jeder proteinogenen Aminosäure unter Zusatz von 0.4% BH-Bouillon und 1% Agarose) und einer Agarplatte, die 2 μg/ml Biphenomycin B (lOxMHK) enthält, ausplattiert und über Nacht bei 37°C bebrütet. Während auf der antibiotikafreien Platte ca. 1x107 Zellen wachsen, wachsen auf der antibiotikahaltigen Platte ca. 100 Kolonien, entsprechend einer Resistenzfrequenz von IxIO-5. Einige der auf der antibiotikahaltigen Platte gewachsenen Kolonien werden auf MHK gegen Biphenomycin B getestet. Eine Kolonie mit einer MHK > 50 μM wird zur weiteren Verwendung ausgewählt und der Stamm mit RN4220BiR bezeichnet.
Der S. aureus Stamm T17 wird in vivo isoliert. CFWl-Mäuse werden mit 4x10^ S. aureus 133 - Zellen pro Maus intraperitoneal infiziert. 0.5 Std. nach der Infektion werden die Tiere mit 50 mg/kg Biphenomycin B intravenös behandelt. Den überlebenden Tieren werden am Tag 3 nach der Infektion die Nieren entnommen. Nach dem Homogenisieren der Organe werden die Homogenate, wie bei RN4220BiR beschrieben, auf antibiotikafreien und antibiotikahaltigen Agarplatten, ausplattiert und über Nacht bei 37°C bebrütet. Etwa die Hälfte der aus der Niere isolierten Kolonien zeigen ein Wachstum auf den antibiotikahaltigen Platten (2.2x106 Kolonien), was die Anreicherung von Biphenomycin B resistenten S. aureus Zellen in der Niere der behandelten Tiere belegt. Ca. 20 dieser Kolonien werden auf MHK gegen Biphenomycin B getestet und eine Kolonie mit einer MHK > 50 μM wird zur Weiterkultivierung ausgewählt und der Stamm mit Tl 7 bezeichnet.
B. Ausfühiungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Intravenös applizierbare Lösung;
Zusammensetzung:
1 mg der Verbindung von Beispiel 1, 15 g Polyethylenglykol 400 und 250 g Wasser für Inj ektionszwecke.
Herstellung:
Die erfindungsgemäße Verbindung wird zusammen mit Polyethylenglykol 400 in dem Wasser unter Rühren gelöst. Die Lösung wird sterilfiltriert (Porendurchmesser 0.22 μm) und unter aseptischen Bedingungen in hitzesterilisierte Infusionsflaschen abgefüllt. Diese werden mit Infusionsstopfen und Bördelkappen verschlossen.