WO2007013469A1 - (アルキルフェニル)アルキルシクロヘキサン及び(アルキルフェニル)アルキルシクロヘキサン、アルキルビフェニルの製造方法 - Google Patents
(アルキルフェニル)アルキルシクロヘキサン及び(アルキルフェニル)アルキルシクロヘキサン、アルキルビフェニルの製造方法 Download PDFInfo
- Publication number
- WO2007013469A1 WO2007013469A1 PCT/JP2006/314693 JP2006314693W WO2007013469A1 WO 2007013469 A1 WO2007013469 A1 WO 2007013469A1 JP 2006314693 W JP2006314693 W JP 2006314693W WO 2007013469 A1 WO2007013469 A1 WO 2007013469A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- production method
- acid
- formula
- catalyst
- alkylcyclohexane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2/00—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms
- C07C2/86—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by condensation between a hydrocarbon and a non-hydrocarbon
- C07C2/862—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by condensation between a hydrocarbon and a non-hydrocarbon the non-hydrocarbon contains only oxygen as hetero-atoms
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/26—Chromium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/46—Ruthenium, rhodium, osmium or iridium
- B01J23/464—Rhodium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/18—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of the mordenite type
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/82—Phosphates
- B01J29/84—Aluminophosphates containing other elements, e.g. metals, boron
- B01J29/85—Silicoaluminophosphates [SAPO compounds]
Definitions
- the present invention relates to a process for producing an (alkylphenyl) alkylcyclohexane comprising a step of condensing an alkylbenzene and an alkylcyclohexene or an alkylcyclohexanol, and the (alkylphenyl) alkylcyclohexane. Further, the present invention relates to a method for producing an alkyl biphenyl and a related compound thereof, which further comprises a step of dehydrogenating.
- Alkylphenyl) alkylcyclohexane and alkylbiphenyl are important compounds in organic synthetic chemistry that are used as liquid crystal raw materials, pharmaceutical raw materials, and monomer raw materials. Above all, the following formula (Alkylphenyl) alkylcyclohexane and alkylbiphenyl are important compounds in organic synthetic chemistry that are used as liquid crystal raw materials, pharmaceutical raw materials, and monomer raw materials. Above all, the following formula (Alky
- Alkylbenzene and alkylcyclohexene or alkylcyclohexanol A method for producing an (alkylphenol) alkylcyclohexane as represented by the above (1) or (2) by condensation is not known. Instead of alkylcyclohexene, 1,4-dimethyl 1-chlorocyclohexane is condensed with toluene and Friedd-Crafts in the presence of aluminum chloride to produce 1 (4-methylphenol) -1,4-dimethylcyclohexane. The method is known (Non-Patent Document 1). However, it is not easy to obtain 1,4 dimethyl 1-chlorocyclohexane, and the salt and aluminum are expensive and require an equivalent amount to the halogen compound. In addition, it is difficult to say that this method is an industrially advantageous method because it is necessary to treat the waste salt and aluminum after the reaction.
- Non-Patent Document l Azerb. Khim. Zh. '67, 1, 69 (71
- Patent Document 1 Japanese Patent Laid-Open No. 4-5246
- alkylbenzene was found to be an alkylcyclohexene.
- it has been found that it is easily condensed with an alkylcyclohexanol in the presence of an acid catalyst to give an (alkylphenol) alkylcyclohexane.
- the desired alkylbiphenyl can be obtained easily and selectively by dehydrogenating and dealkylating the (alkylphenol) alkylcyclohexane thus obtained.
- the present invention is based on these findings.
- R 1 represents an alkyl group having 1 to 4 carbon atoms
- R 2 represents an alkyl group having 1 to 4 carbon atoms that is the same as or different from R 1
- m represents an integer of 0 to 2
- m represents 1
- R 2 is located in the ortho position or meta position of R 1 , and when m is 2, two R 2 are located in both ortho positions of R 1
- Equation (4) Equation (4):
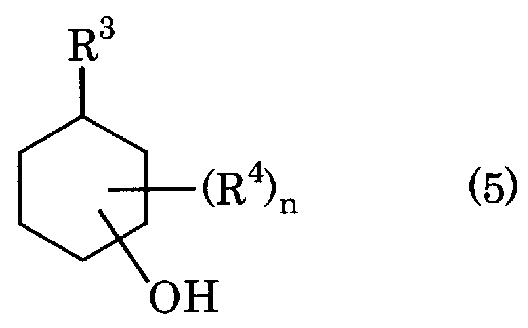
- R 3 represents an alkyl group having 1 to 4 carbon atoms
- R 4 represents an alkyl group having 1 to 4 carbon atoms that is the same as or different from R 3
- n represents an integer of 1 to 5
- n is 2 In the case of an integer of ⁇ 5, a plurality of R 4 may be the same or different, except for R 3 and R 4 , or when two R 4 are bonded to the same carbon).
- n are as defined above, if either R 2 absent R 2 is located in the O belt-position of R 1, cyclohexane ring is located in the para-position of R 1, R 2 is R 1 When located at the meta position, the cyclohexane ring is located at the meta position of R 1 )
- the present invention includes a step of simultaneously dehydrogenating and dealkylating the (alkylphenyl) alkylcyclohexane obtained by the above production method in the presence of a catalyst:
- R 2, R 4, m and n are as defined above, if does not exist or R 2 is located at the ortho position of R 1, the benzene ring having R 4 is located at the para-position of R 1, R When 2 is located in the meta position of R 1, the benzene ring having R 4 is located in the meta position of R 1 ).
- R 4 and m are the same as above, n represents an integer of 2 to 5, and when R 2 is absent or R 2 is located at the ortho position of R 1 , the cyclohexane ring is R 1 In the case where R 2 is located at the meta position of R 1 , the cyclohexane ring is located at the meta position of R 1 ).
- FIG. 2 is a 1 H-NMR ⁇ vector of isomer 2 of 1- (3,4-dimethylphenol) 1,3,4-trimethylcyclohexane obtained in Example 7.
- the process for producing an (alkylphenol) alkylcyclohexane of the present invention includes a step of condensing alkylbenzene with alkylcyclohexene or alkylcyclohexanol in the presence of an acid catalyst.
- Alkylbenzene has the following formula (3):
- R 1 represents an alkyl group having 1 to 4 carbon atoms
- R 2 represents an alkyl group having 1 to 4 carbon atoms that is the same as or different from R 1
- m represents an integer of 0 to 2
- m represents 1
- R 2 is located in the ortho or meta position of R 1 , and when m is 2, two R 2 are located in both ortho positions of R 1 ).
- the speed of reaction and the ability to obtain raw materials are preferable.
- Polymethylbenzene having an alkyl group is preferred, and toluene, orthoxylene, metaxylene, and 1,2,3 trimethylbenzene are particularly preferred.
- alkylcyclohexene is represented by the following formula (4):
- R 4 represents an alkyl group having 1 to 4 carbon atoms
- R 4 represents an alkyl group having 1 to 4 carbon atoms that is the same as or different from R 3
- n represents an integer of 1 to 5
- n is 2
- a plurality of R 4 may be the same or different, except for R 3 and R 4 , or when two R 4 are bonded to the same carbon). Is done.
- Alkylcyclohexene can be produced by a known method, for example, a Diels-Alder reaction of a gen compound and an aryl compound, dehydration of an alkylcyclohexanol, dehydrohalogenation of a halogenated cyclohexane, or the like.
- a Diels-Alder reaction of a gen compound and an aryl compound dehydration of an alkylcyclohexanol
- dehydrohalogenation of a halogenated cyclohexane or the like.
- R 5 represents an alkyl group having 1 to 4 carbon atoms
- p represents an integer of 2 to 6, and a plurality of may be the same or different
- catalytic partial hydrogenation of alkylbenzene It can be manufactured by
- the alkylbenzene of formula (9) includes ortho, meta, para xylene, 1, 2, 3-trimethylbenzene, 1, 2, 4-trimethylbenzene (pseudotamen), 1, 3, 5-trimethylbenzen. 1, 2, 3, 4—tetramethylbenzene, 1, 2, 4, 5—tetramethylbenzene, 1, 2, 3, 4, 5—pentamethylbenzene, 1, 2, 3, 4, 5, 6 —Tetramethylbenzene, Jetylbe Benzene, dinormalpropyl benzene, diisopropylbenzene, dinormal butylbenzene, di-t-butylbenzene, 1,2-methylethylbenzene, 1,3-methylethylbenzene, 1,4-methylethylbenzene, and the like.
- Xylene including isomers
- trimethylbenzene including isomers
- metaxylene, paraxylene and pseudodex are more preferable. Men are more preferred.
- the catalyst used for partial hydrogenation of the alkylbenzene of the formula (9) is not limited as long as it is a catalyst used for ordinary hydrogenation.
- Specific examples include ruthenium, rhodium, rhenium, platinum, palladium,
- Examples include metals such as nickel, cobalt, chromium, iridium, and copper. These metals can be used by supporting them on a carrier such as carbon, alumina, silica, zircoia, /, funi-ryo, tita, and magnesia, or can be used as metal fine particles as they are.
- the catalyst amount is preferably in a weight ratio with respect to the alkylbenzene of formula (9) ⁇ 0.0001 to 10, more preferably ⁇ 0.0001 to 1.
- a promoter such as zinc sulfate, cobalt sulfate, barium sulfate, zinc chloride, zinc oxide, zinc bromide, zinc iodide, or zinc hydroxide is used. Also good.
- the amount of the co-catalyst is preferably from 0.0001 to 100, more preferably from 0.0001 to 10 by weight ratio to the catalyst.
- the deteriorated catalyst can be activated by a conventional method, for example, a method of circulating air at a high temperature.
- the reaction mode of the partial hydrogenation of the alkylbenzene of formula (9) is preferably a tube-type fixed bed or a tank-type suspension bed.
- water can be added to form a two-phase system of oil and water, and additives such as alcohol can be added.
- the weight ratio of the water Z formula (9) alkylbenzene is preferably 0.001 to 100, more preferably 0.1 to 10. If the amount is too small, the water-time yield is too small. Go down.
- the reaction temperature is preferably in the range of 0 ° C to 300 ° C, more preferably in the range of 50 to 200 ° C.
- the hydrogen pressure is in the range of 0.1 to 30 MPa, preferably 1 to 15 MPa. If it is too high, a high pressure device is required, and if it is too low, the conversion rate does not increase.
- the reaction time is in the range of 10 minutes to 20 hours for batch systems.
- the alkylcyclohexene of the formula (4) obtained by the above hydrogenation has an alkyl group and a double It includes isomers depending on the positional relationship of bonds.
- para-xylene forms two isomers: 1,4-dimethyl-1-cyclohexene and 1,4 dimethyl-2-cyclohexene.
- 1, 2, 4 trimethylbenzene is derived from 1, 2, 4 trimethyl-1-cyclohexene, 1, 3, 6 trimethyl 1-cyclohexene, 2, 5, 6 trimethyl 1-cyclohexene. 1, 4, 5 Trimethyl 1-cyclohexene and 2, 3, 5 Trimethyl 1-cyclohexene are produced.
- These isomers can be used in the condensation reaction with the alkylbenzene of formula (3) after or without separation. Isomers can also be isomerized in the presence of an acid catalyst.
- alkylcyclohexane with all aromatic rings hydrogenated is by-produced.
- Alkylcyclohexene, alkylcyclohexane and unreacted alkylbenzene can be separated by conventional methods such as distillation, extractive distillation, liquid separation, extraction, filtration and crystallization, and unreacted alkylbenzene can be separated from alkylcyclohexene. It can be reused as a raw material for manufacturing.
- alkylcyclohexanol examples include 1,2 dimethylcyclohexanol, 2,3 dimethylcyclohexanol, 3,4-dimethylcyclohexanol, 4,5-dimethylcyclohexanol, 5,6-dimethyl Cyclohexanol, 1,3-dimethylcyclohexanol, 1,4-dimethylcyclohexanol, 1,5-dimethylcyclohexanol, 1,6-dimethylcyclohexanol, 2,3-dimethylcyclohexanol, 2,4-dimethylcyclohexanol, 2,5-dimethylcyclohexanol, 2,6-dimethylcyclohexanol, 3,4-dimethylsilane Clohexanol, 3, 5-dimethylcyclohexanol, 2,3 dimethylcyclohexanol, 3,4-dimethylcyclohexanol, 4,5-dimethylcyclohex
- Dimethylhexanol and trimethylhexanol are more preferred because polymethylcyclohexanol is preferred because of the speed of reaction and the availability of raw materials.
- the production method of the alkylcyclohexanol is not limited, but it can be obtained by, for example, nuclear hydrogenation of alkylphenol, hydration of alkylcyclohexene, or the like.
- the molar ratio of alkylbenzene to alkylcyclohexene is preferably from 0.001 to 1000, more preferably from 0.1 to 100. If the amount is too small, the reaction between the alkylcyclohexenes proceeds.
- the molar ratio of alkylbenzene to alkylcyclohexanol is preferably from 0.001 to 1000, more preferably from 0.1 to 100.
- the molar ratio of toluene / 1,4-dimethylcyclohexene is preferably 2 to 20, and more preferably 3 to 10.
- the addition reaction between 1,4-dimethylcyclohexenes is prevented, and the selectivity to 1- (4-methylphenol) 1,4-dimethylcyclohexane is high.
- the condensation of orthoxylene and 1, 2, 4 trimethylcyclohexene is preferably 1 to 100 forces S, more preferably 2 to 50.
- the condensation reaction may be carried out in a solvent.
- a solvent an inactive compound is used for the condensation reaction, and it is widely used for the Friedd-Crafts reaction.
- Aliphatic hydrocarbons such as can be used.
- Alkylcyclohexane by-produced in the above-described production of alkylcyclohexene may be used.
- Alkylbenzene which is all alkylated at the ortho and V positions of the alkyl group, can also be used as a solvent. If all para positions are alkylated, alkylbenzene is preferred as a solvent because it participates in the condensation reaction.
- Acid catalysts for the condensation reaction include sulfuric acid, hydrochloric acid, phosphoric acid, polyphosphoric acid, hydrogen fluoride, borohydrofluoric acid, boron trifluoride, aluminum trichloride, aluminum tribromide, and gallium trichloride.
- proton acids such as gallium tribromide, iron trichloride, pentachloride-antimony, tin tetrachloride, titanium tetrachloride, zinc chloride, and Lewis acid.
- cation exchange resins such as naphthions, and solid acids such as silica alumina and zeolite can also be used as catalysts.
- zeolite examples include A type, ferrierite type, ZSM 5 (MFI type), ZSM-12 (MTW type), mordenite type, beta type, X type, Y type, etc. Replacing phosphates, exchanging cations (H +, NH +, metal ions, etc.), silica alumina
- an undesired isomer may be produced as a by-product in a negligible amount.
- 1- (4 methylphenol) 1,4-dimethylcyclohexane plus 2- (4 methylphenol) of formula (10) 1 4-Dimethylcyclohexane, one of the formula (11) (3 methylphenol) -1,4-dimethylcyclohexane Hexane and isomers of 2- (3 methylphenol) -1,4-dimethylcyclohexane of formula (12) may be by-produced.
- 11 (4-methylphenol) 1,4-dimethylcyclohexane, which is the target product can be obtained in high yield by using sulfuric acid as a catalyst.
- H type zeolite or NH type zeolite especially when using liquid catalysts such as sulfuric acid and hydrogen fluoride, especially H type mordenite, NH type mordenite, H type
- MFI-type zeolite such as ZSM-5
- MTW-type zeolite such as ZSM-12
- by-product formation of isomers may be suppressed.
- These zeolites have a pore diameter of 0.5 to 0.8 nm, and the formation of relatively bulky isomers can be suppressed in the zeolites having such pore diameters, so the double bond of the raw material trimethylcyclohexene Regardless of the position, the target 1- (3,4-dimethylphenol) -1,3,4-trimethylcyclohexane is obtained as the main product.
- the amount of the acid catalyst is preferably in the range of 0.0001 to 1000, more preferably 0.001 to 1000 in terms of a weight ratio with respect to the alkylcyclohexene. It ’s right.
- the catalyst amount is often required as compared with alkylcyclohexene, and the weight ratio to alkylcyclohexanol is preferably from 0.0001 to 10000, more preferably in the range of 0.001 to 1000.
- a condensation reaction between toluene and 1,4 dimethylcyclohexene it is preferable to use 0.1 to 100 moles of protonic acid and Lewis acid catalyst for 1 mole of 1,4 dimethylcyclohexene. It is preferable to use 0.1 to 10 g of solid acidic catalyst such as cation exchange resin or zeolite, based on 1,4 dimethylcyclohexene lg.
- solid acidic catalyst such as cation exchange resin or zeolite
- the condensation reaction is carried out in a reaction type such as a batch type or a continuous flow type fixed bed, suspension bed, and uniform tank type.
- a fixed bed is preferable because the catalyst can be easily separated and recovered.
- alkylcyclohexene or alkylcyclohexanol is flown continuously in the reactor so that the weight hourly space velocity (WHSV) is in the range of 0.001 to 10 hr- 1. It is preferable to supply to.
- the reaction temperature varies depending on the type and amount of catalyst used, and the type and concentration of reaction substrate, but it is in the range of -50 to 250 ° C.
- reaction pressure can be arbitrarily selected from normal pressure, reduced pressure, and increased pressure.
- the boiling point of the raw material or the solvent used is higher than the reaction temperature, it is preferable to carry out the reaction under a pressure of 0.1 to: LOMPa. It is preferable to introduce an inert gas such as nitrogen or argon into the reaction system and pressurize it.
- the reaction time is preferably 10 minutes to 20 hours for batches, more preferably 10 minutes to 3 hours.
- the (alkylphenyl) alkylcyclohexane produced by the above condensation reaction may be obtained as a mixture of cis and trans isomers. Isomeric mixtures can be separated from raw materials and by-products by conventional methods such as distillation, crystallization and column separation. The isomer mixture of (alkylphenyl) alkylcyclohexane may be separated into each isomer or used as a raw material for the next step (simultaneous dehydrogenation Z dealkylation) as the mixture.
- R 2, R 4, m and n are as defined above, if does not exist or R 2 is located at the ortho position of R 1, the benzene ring having R 4 is located at the para-position of R 1, R When 2 is located at the meta position of R 1, the benzene ring having R 4 is located at the meta position of R 1 ).
- the simultaneous dehydrogenation Z dealkylation catalyst includes, for example, a metal such as platinum, noradium, rhodium, rhenium, ruthenium, iron, chromium, connort, nickel, molybdenum, copper, zinc, iridium or the like.
- a metal such as platinum, noradium, rhodium, rhenium, ruthenium, iron, chromium, connort, nickel, molybdenum, copper, zinc, iridium or the like.
- Specific examples of compounds containing chromium and zinc include chromium (III) oxide, zinc oxide, Cu—Cr ⁇ Cu—Zn, Zn— Cr.
- alkaline metals such as lithium, sodium and potassium, alkaline earth metals such as magnesium and calcium can be added to these compounds, and potassium is particularly preferred.
- These catalysts can also be used by supporting them on a support such as carbon, alumina, zircoure, silica alumina and the like.
- the catalyst can be pretreated by heating in a conventional manner, for example, in the presence of an acidic gas such as air or a reducing gas such as hydrogen. Even when the activity of the catalyst decreases, activation can be performed by the same treatment.
- the method for producing the catalyst is not limited.
- a metal salt such as nitric acid or acetic acid is dissolved in water, impregnated in a carrier such as ⁇ -alumina, dried, and then 300 ° C to 600 ° C in air. It can be produced by an impregnation method of baking with C.
- Simultaneous dehydrogenation Z dealkylation reaction mode is preferably fixed bed flow system.
- the reaction temperature is preferably 200 to 600 ° C, more preferably 350 to 550 ° C. If it is lower than this, the reaction does not proceed sufficiently, and if it is higher, the selectivity is lowered.
- the reaction pressure can be any of reduced pressure, normal pressure and increased pressure. During the reaction, nitrogen, hydrogen and water vapor can be circulated simultaneously, and it is preferable to circulate hydrogen since deterioration of the catalyst is suppressed.
- Alkylphenyl Alkylcyclohexane can be supplied to the reactor as it is, but a solvent inert to the reaction, for example, benzene, toluene and the like can also be used.
- the by-product of the alkylcyclohexene production process is substantially only alkylcyclohexane, and this by-product alkylcyclohexane is converted into alkylbenzene of formula (9) by the simultaneous dehydrogenation Z dealkylation process.
- As raw material for alkylcyclohexene production Can be reused. Therefore, when the alkylcyclohexene production process, the alkylcyclohexene condensation process, and the (alkylphenol) alkylcyclohexane simultaneous dehydrogenation Z dealkylation process are carried out in succession, the alkylcyclohexene is obtained.
- Alkyl biphenyl without loss of the alkylbenzene raw material of formula (9) used in the production can be produced by an economical process.
- the obtained alkyl biphenyl can be separated by a conventional method such as distillation, crystallization, column separation or the like.
- the corresponding biphenyldicarboxylic acid, biphenyltetracarboxylic acid, biphenyltetracarboxylic acid anhydride can be produced from alkylbiphenyl by a conventionally known method.
- a glass 3000 ml flask equipped with a thermometer and condenser was charged with 2700 g of toluene, 15 g of concentrated sulfuric acid, and 55 g of 1,4 dimethylcyclohexene, and the temperature of the reaction solution was set to -20 ° C. After reacting for 5 hours, the organic layer was separated, and the separated organic layer was washed twice with 500 ml of distilled water, once with a 3% aqueous sodium hydrogen carbonate solution and twice with distilled water. When the organic layer was analyzed by gas chromatography, the conversion of 1,4 dimethylcyclohexene was 96%, and the selectivity for 1- (4-methylphenol) 1,4-dimethylcyclohexane was 92%.
- a 500-m Lesteloy C autoclave was charged with 130 g of hot hydrogen and 100 g of toluene, and cooled to 27 ° C. Thereto, a mixed liquid of 10 g of 1,4 dimethylcyclohexene and 100 g of toluene was added over 1 hour. After completion of the addition, the mixture was further held for 20 minutes, and the liquid was drained to recover hydrogen fluoride. During the reaction, the pressure in the system was 0.2 MPa or less. The obtained organic layer was washed with 200 ml of distilled water three times, 5% aqueous sodium hydrogen carbonate solution once, and distilled water twice. When the organic layer is analyzed by gas chromatography, 1,4 dimethylcyclohex The conversion rate of cene was 100%, and the selectivity for 1- (4-methylphenol) -1,4 dimethylcyclohexane was 28.5%.
- a 500 ml SUS304 electromagnetic stirring autoclave equipped with a condenser and a thermometer was prepared. The inside was replaced with nitrogen gas. The system was filled with 3 MPa of nitrogen gas, and then heated and stirred at 180 ° C. During the reaction, the pressure in the system rose to 4 MPa. After the reaction for 3 hours, the reaction solution was taken out, the catalyst was filtered, and the organic layer was analyzed by gas chromatography. The conversion rate of 1,4-dimethylcyclohexene was 85.8%, 1 (4 methylphenol) — 1 The selectivity for dimethylcyclohexane was 59.5%.
- the reaction was carried out under the same conditions as in Example 3, except that the reaction temperature was 110 ° C., the reaction pressure was normal pressure, toluene was refluxed, and the reaction time was 50 hours. After completion of the reaction, the reaction solution was taken out, the catalyst was filtered, and the organic layer was analyzed by gas chromatography. The conversion of 1,4 dimethyl hexene to 70,1%, 1- (4-methylphenol) 1 The selectivity for dimethylcyclohexane was 63.2%.
- Type H SAPO-5 Zeolite (prepared by referring to the method described in H. Robson ⁇ “Verified Syntheses of Zeoritic Materials”, Elsevier Science BV p93) 0. Under the same conditions as in Example 5 except that lg was used. Reaction was performed. During the reaction, the pressure in the system is 3MP. It rose to a. After the reaction, the reaction solution was extracted, the catalyst was filtered, and the organic layer was analyzed by gas chromatography. The conversion of 1,4-dimethylcyclohexene was 42.6% and 1- (4-methylphenol) 1, The selectivity for 4-dimethylcyclohexane was 60.6%.
- a glass 3000 ml flask equipped with a thermometer and a condenser was charged with 2000 g of orthoxylene, 24 g of concentrated sulfuric acid and 50 g of 1,2,4 trimethylcyclohexene, and the temperature of the reaction solution was set to 20 ° C. After reacting for 1 hour, the organic layer was separated. The separated organic layer was washed twice with 50 ml of distilled water, once with a 3% aqueous sodium hydrogen carbonate solution, and twice with distilled water. The organic layer was analyzed by gas chromatography. The conversion of 1,2,4 trimethylcyclohexene was 87%, and the selectivity for 1— (3,4 dimethylphenol) —1,3,4 tetramethylcyclohexane 43%.
- a glass 300 ml flask equipped with a thermometer and a condenser was charged with 10 g of orthoxylene, 0.3 g of concentrated sulfuric acid, and 0.5 g of 1,4-dimethylcyclohexene, and the temperature of the reaction solution was set to 20 ° C. After reacting for 1 hour, the organic layer was separated. The separated organic layer was washed twice with 30 ml of distilled water, once with a 3% aqueous sodium hydrogen carbonate solution and twice with distilled water. The organic layer was analyzed by gas chromatography. The conversion of 1,4 dimethylcyclohexene was 94%, and the selectivity of 1- (3,4 dimethylphenol) 1,4 dimethylcyclohexane was 71%. .
- the reaction was performed in the same manner as in Example 13 except that meta-xylene was used instead of ortho-xylene.
- the organic layer was analyzed by gas chromatography.
- the conversion of 1,4 dimethylcyclohexene was 92%, 1— (3,5 dimethylphenol) 1,4 dimethylcyclohexane.
- the selectivity of N was 11%.
- the reaction was performed in the same manner as in Example 13 except that propylbenzene was used instead of orthoxylene.
- the organic layer was analyzed by gas chromatography.
- the conversion of 1,4-dimethylcyclohexene was 93%, and the selectivity of 1- (4-propylphenol) — 1,4-dimethylcyclohexane was 31%. there were.
- the reaction was performed in the same manner as in Example 13 except that isopropylbenzene was used instead of orthoxylene.
- the organic layer was analyzed by gas chromatography.
- the conversion of 1,4-dimethylcyclohexene was 85% and the selectivity of 1- (4-isopropylphenol) -1,4-dimethylcyclohexane was 28%. there were.
- the reaction was conducted in the same manner as in Example 13 except that 1,2,3-trimethylbenzene was used instead of orthoxylene.
- the organic layer was analyzed by gas chromatography.
- the conversion of 1,4-dimethylcyclohexene was 97% and 1- (3, 4, 5-trimethylphenol) — 1,4-dimethylcyclohexane was selected. The rate was 46%.
- a 100 ml glass flask equipped with a thermometer and condenser was charged with 10 g of metaxylene, 0.3 g of concentrated sulfuric acid and 0.5 g of 1,2,4-trimethylcyclohexene, and the temperature of the reaction solution was adjusted to 20 ° C. Set. After reacting for 1 hour, the organic layer was separated, and the separated organic layer was washed with 30 ml of distilled water twice, 3% aqueous sodium hydrogen carbonate solution once, and distilled water twice. The organic layer was analyzed by gas chromatography. The conversion of 1,2,4-trimethylcyclohexene was 98% and 1- (3,5-dimethylphenol) —1,3,4-trimethylcyclohexene. The selectivity for Sun was 17%.
- the reaction was conducted in the same manner as in Example 19 except that 1,2,3 trimethylbenzene was used instead of metaxylene.
- the organic layer was analyzed by gas chromatography.
- the conversion of 1,2,4-trimethylcyclohexene was 95% and 1,3,4-trimethyl-1- (3,4,5 trimethylphenol) cyclohexane.
- the selectivity was 53%.
- a glass 300 ml glass flask equipped with a thermometer and a condenser was charged with 65 g of toluene, 3.5 g of concentrated sulfuric acid, and 2.5 g of 2,5 dimethylcyclohexanol, and the temperature of the reaction solution was set to 20 ° C. After reacting for 1 hour, the organic layer was separated. The separated organic layer was washed twice with 20 ml of distilled water, once with a 3% aqueous sodium hydrogen carbonate solution and twice with distilled water. The organic layer was analyzed by gas chromatography. The conversion of 2,5 dimethylcyclohexanol was 88%, and the selectivity for 1- (4-methylphenol) -1,4 dimethylcyclohexane was 74%.
- a glass 100 ml glass flask equipped with a thermometer and a condenser was charged with 20 g of orthoxylene, 3 g of concentrated sulfuric acid, and 2 g of 3,4-dimethylcyclohexanol, and the temperature of the reaction solution was set to 20 ° C. After reacting for 1 hour, the organic layer was separated. The separated organic layer was washed twice with 20 ml of distilled water, once with a 3% aqueous sodium hydrogen carbonate solution and twice with distilled water. When the organic layer was analyzed by gas chromatography, the conversion of 3,4-dimethylcyclohexanol was 68% and the selectivity for 1- (3,4 dimethylphenol) 1,2 dimethylcyclohexane was 74%.
- a 500 ml magnetic stirring autoclave made of SUS316 equipped with a thermometer was charged with 100 g of water, 1 g of 5% Ru alumina catalyst, 0.06 g of zinc sulfate, and then 200 g of paraxylene.
- the system was pressurized to 5 MPa with hydrogen and reacted at 150 ° C for 4 hr. After cooling, the oil phase was separated from the reaction mixture and analyzed by gas chromatography.
- Paraxylene conversion was 80%, 1,4-dimethylcyclohexene selectivity was 39%, and 1,4-dimethylcyclohexane selectivity was 61. %Met. 1,4-Dimethylcyclohexene and 1,4-dimethylcyclohexane were separated from the resulting reaction solution by distillation.
- a glass 3000 ml flask equipped with a thermometer and condenser was charged with 2700 g of toluene, 15 g of concentrated sulfuric acid, and 55 g of 1,4 dimethylcyclohexene, and the temperature of the reaction solution was set to -20 ° C. After reacting for 5 hours, the organic layer was separated, and the separated organic layer was washed twice with 500 ml of distilled water, once with a 3% aqueous sodium hydrogen carbonate solution and twice with distilled water. When the organic layer was analyzed by gas chromatography, the conversion of 1,4 dimethylcyclohexene was 96%, and the selectivity for 1- (4-methylphenol) 1,4-dimethylcyclohexane was 92%. 1- (4-Methylphenol) 1,4 dimethylcyclohexane was also separated by distillation of the resulting reaction solution.
- the reaction was started by supplying 10% benzene solution of 11- (4-methylphenol) -1,4-dimethylcyclohexane in lOgZhr from the raw material supply line. The reaction solution was collected and analyzed for 2 to 5 hours after the start of the reaction. The conversion rate of 1- (4-methylphenol) -1,4 dimethylcyclohexane was 100%, and that of 4, 4, monodimethylbifurol. The yield was 78%.
- the reaction was conducted in the same manner as in Example 24 except that the catalyst in the third step was changed to acid zinc.
- the conversion rate of 1- (4-methylphenol) -1,4 dimethylcyclohexane was 70%, and the yield of 4,4, dimethylbiphenyl was 49%.
- the reaction was carried out in the same manner as in Example 24 except that the solvent in the third step was changed to benzene to obtain 1,4-dimethylcyclohexane by-produced in the first step.
- the conversion of 1- (4-methylphenol) 1,4-dimethylcyclohexane was 80%, and the yield of 4,4′-dimethylbiphenyl was 45%.
- the conversion of 1,4-dimethylcyclohexane was 14%, and the yield of paraxylene was 14%.
- the reaction solution was heated to reflux temperature (150 ° C) at normal pressure.
- the reaction mixture which was allowed to react for 1 hour and filtered the catalyst, was analyzed by gas chromatography. , 4 Trimethylcyclohexene conversion was 95%, and 1- (3,4 dimethylphenol) -1,3,4, trimethylcyclohexane selectivity was 72%. From the resulting reaction solution, 11 (3,4 dimethylphenol) 1,3,4 tetramethylcyclohexane was separated by distillation. 3rd process
- a 100 ml magnetic stirring autoclave made of SUS316 equipped with a thermometer was charged with 10 g of water, 0.2 g of 5% Ru alumina catalyst, and then 20 g of mesitylene.
- the system was pressurized to lOMPa with hydrogen and reacted at 150 ° C for 2 hr. After cooling, the oil phase was separated from the reaction liquid and analyzed by gas chromatography.
- the conversion of mesitylene was 49%, the selectivity for 1,3,5 trimethylcyclohexene was 21%, and 1,3,5 trimethylcyclohexane. The selectivity of was 79%.
- a 100 ml glass flask equipped with a thermometer and a condenser was charged with 10 g of orthoxylene, 0.3 g of sulfuric acid, and 0.5 g of 1,3,5 trimethylcyclohexene, and the temperature of the reaction solution was set to 20 ° C. After reacting for 1 hour, the organic layer was separated, and the separated organic layer was washed twice with 30 ml of distilled water, once with a 3% aqueous sodium hydrogen carbonate solution and twice with distilled water. The organic layer was analyzed by gas chromatography. The conversion of 1,3,5 trimethylcyclohexene was 92%, and the selectivity for 1,3,5-trimethylcyclohexane was 1,3,5. %Met.
- the desired (alkylphenol) alkylcyclohexane and alkylbiphenyl can be produced efficiently and with high selectivity from an inexpensive raw material in a step that does not require post-treatment. Therefore, it is very advantageous industrially.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
(アルキルフエニル)アルキルシクロへキサン及び(アルキルフエニル)アル キルシクロへキサン、アルキルビフエ二ルの製造方法
技術分野
[0001] 本発明は、アルキルベンゼンとアルキルシクロへキセンまたはアルキルシクロへキ サノールとを縮合する工程を含む(アルキルフエニル)アルキルシクロへキサンを製造 する方法、該(アルキルフエ-ル)アルキルシクロへキサンをさらに脱水素する工程を 含むアルキルビフヱ-ルおよびその関連化合物を製造する方法に関する。(アルキ ルフエ-ル)アルキルシクロへキサンおよびアルキルビフエ-ルは、液晶原料、医薬 品原料、モノマー原料となる有機合成化学上重要な化合物である。中でも、下記式(
1):
[化 1]
 で表される 1一(4 メチルフエ-ル)—1, 4ージメチルシクロへキサンを脱水素、脱ァ ルキルして得られる 4, 4' ジメチルビフヱ-ルをさらに酸化して製造される 4, 4' ビフエ-ルジカルボン酸はポリエステル原料として有用である。また、下記式(2): [化 2]
で表される 1一(4 メチルフエ-ル)—1, 4ージメチルシクロへキサンを脱水素、脱ァ ルキルして得られる 4, 4' ジメチルビフヱ-ルをさらに酸化して製造される 4, 4' ビフエ-ルジカルボン酸はポリエステル原料として有用である。また、下記式(2): [化 2]

で表される 1— (3, 4 ジメチルフエ-ル) 1, 3, 4 トリメチルシクロへキサンを脱 水素、脱アルキルして得られる 3, 4, 3' , 4'ーテトラメチルビフエ-ルをさらに酸化、 無水化して製造される 3, 4, 3' , 4' ビフヱニルテトラカルボン酸無水物は耐熱性の ポリイミドの原料として有用である。
背景技術
アルキルベンゼンとアルキルシクロへキセンまたはアルキルシクロへキサノールとを
縮合させ上記(1)、 (2)で表されるような(アルキルフエ-ル)アルキルシクロへキサン を製造する方法は知られていない。アルキルシクロへキセンの代わりに 1, 4 ジメチ ルー 1—クロロシクロへキサンを塩化アルミニウム存在下でトルエンと Friedd-Crafts縮 合させて 1一(4 メチルフエ-ル)—1, 4ージメチルシクロへキサンを製造する方法 が知られている(非特許文献 1)。しかし、 1, 4 ジメチル一 1—クロロシクロへキサン の入手が容易ではないこと、塩ィ匕アルミニウムは高価で、かつ、ハロゲン化合物に対 して当量以上必要であること、副生する塩ィ匕水素及び反応後の廃塩ィ匕アルミニウム の処理が必要であることから、この方法は工業的に有利な方法とは言い難い。
[0003] またアルキルビフヱ-ルの製造法は種々知られている。例えばブロモアルキルベン ゼンとグリニャール試薬との反応は選択的にアルキルビフエ-ルが製造できる力 原 料の調達の点から工業的には非常に困難である。ビフエ-ルのアルキルィ匕によりァ ルキルビフ -ルを得る方法ではアルキルビフヱ-ルの混合物が得られ目的とするァ ルキルビフエ-ルを効率よく得ることは難し 、(特許文献 1)。このように、 目的とするァ ルキルビフエニルを高 、選択率で工業的に製造する方法は従来知られて 、な 、。
[0004] 非特許文献 l :Azerb. khim. Zh. '67, 1, 69(71
特許文献 1:特開平 4— 5246
発明の開示
[0005] 力かる状況に鑑み、発明者らは(アルキルフエ-ル)アルキルシクロへキサンおよび アルキルビフエ-ルを工業的に有利に製造する方法にっ 、て検討したところ、アルキ ルベンゼンはアルキルシクロへキセンまたはアルキルシクロへキサノールと酸触媒存 在下で容易に縮合して(アルキルフエ-ル)アルキルシクロへキサンを与えることを見 出した。また、このようにして得られた(アルキルフエ-ル)アルキルシクロへキサンを 脱水素、脱アルキルすることにより目的とするアルキルビフエ二ルが容易、かつ、選択 的に得られることを見出した。本発明はこれらの知見に基づく。
[0006] 即ち本発明は、下記式(3):

(式中 R1は炭素数 1〜4のアルキル基を表し、 R2は R1と同一又は異なる炭素数 1〜4 のアルキル基を表し、 mは 0〜2の整数を表し、 mが 1である場合、 R2は R1のオルト位 またはメタ位に位置し、 mが 2である場合、 2個の R2は R1の両オルト位に位置する)で 表されるアルキルベンゼンと、下記式 (4):
[化 4]

(式中 R3は炭素数 1〜4のアルキル基を表し、 R4は R3と同一又は異なる炭素数 1〜4 のアルキル基を表し、 nは 1〜5の整数を表し、 nが 2〜5の整数である場合、複数個 の R4は同一でも異なっていてもよぐ R3と R4、または、 2個の R4が同一の炭素に結合 している場合は除く)で表されるアルキルシクロへキセン、または、下記式(5): [化 5]

(式中 R3、 R4および nは上記と同様)で表されるアルキルシクロへキサノールとを酸触 媒存在下で縮合させる工程を含む下記式 (6):
[化 6]

で表される(アルキルフエ-ル)アルキルシクロへキサンの製造方法に関する。
[0007] さらに本発明は、上記製造方法により得られた (アルキルフエニル)アルキルシクロ へキサンを触媒の存在下で同時に脱水素および脱アルキルする工程を含む下記式 (7) :
[化 7]

[0008] さらに本発明は、下記式 (8):
[化 8]

図面の簡単な説明
[0009] [図 1]実施例 7で得た 1— (3, 4 ジメチルフエニル) 1, 3, 4 トリメチルシクロへキ
サンの異性体 1の1 H— NMR ^ベクトルである。
[図 2]実施例 7で得た 1一(3, 4 ジメチルフエ-ル) 1, 3, 4ートリメチルシクロへキ サンの異性体 2の1 H— NMR ^ベクトルである。
発明を実施するための最良の形態
[0010] 本発明の(アルキルフエ-ル)アルキルシクロへキサンの製造方法は、酸触媒存在 下でアルキルベンゼンをアルキルシクロへキセンまたはアルキルシクロへキサノール と縮合させる工程を含む。
アルキルベンゼンは下記式(3):
[化 9]

(式中 R1は炭素数 1〜4のアルキル基を表し、 R2は R1と同一又は異なる炭素数 1〜4 のアルキル基を表し、 mは 0〜2の整数を表し、 mが 1である場合、 R2は R1のオルト位 またはメタ位に位置し、 mが 2である場合、 2個の R2は R1の両オルト位に位置する)で 表される。具体的には、トルエン、オルトキシレン、メタキシレン、 1, 2, 3 トリメチル ベンゼン、ェチノレベンゼン、 n プロピルベンゼン、イソプロピルベンゼン、 n—ブチ ルベンゼン、 t ブチルベンゼン、 1, 2 メチルェチルベンゼン、 1, 3—メチルェチ ルベンゼン、 1, 2—ジェチルベンゼン等が挙げられる。この中では反応の速さ、原料 の入手のしゃすさ力 好ましくはアルキル基カ チル基のポリメチルベンゼンであり、 トルエン、オルトキシレン、メタキシレン、 1, 2, 3 トリメチルベンゼンが特に好ましい
[0011] アルキルシクロへキセンは下記式(4):
[化 10]
R3
(R4)n (4)
(式中 R3は炭素数 1〜4のアルキル基を表し、 R4は R3と同一又は異なる炭素数 1〜4 のアルキル基を表し、 nは 1〜5の整数を表し、 nが 2〜5の整数である場合、複数個 の R4は同一でも異なっていてもよぐ R3と R4、または、 2個の R4が同一の炭素に結合 している場合は除く)で表される。具体的には、 1, 2—ジメチルシクロへキセン、 1, 3 ージメチノレシクロへキセン、 1, 4ージメチノレシクロへキセン、 1, 2, 3—トリメチノレシクロ へキセン、 1, 2, 4—トリメチルシクロへキセン、 1, 3, 5—トリメチルシクロへキセン、 1 , 2, 3, 4ーテトラメチノレシクロへキセン、 1, 2, 4, 5—テトラメチノレシクロへキセン、 1, 2, 3, 4, 5—ペンタメチノレシクロへキセン、 1, 2, 3, 4, 5, 6—テトラメチノレシクロへキ セン、ジェチノレシクロへキセン、ジノノレマノレシクロへキセン、ジイソプロビルシクロへキ セン、ジノルマルブチルシクロへキセン、ジー tーブチルシクロへキセン、 1, 2—メチ ルェチルシクロへキセン、 1, 3—メチルェチルシクロへキセン、 1, 4ーメチノレエチノレ シクロへキセンが挙げられ、 1, 2—ジメチルシクロへキセン、 1, 3—ジメチルシクロへ キセン、 1, 4—ジメチルシクロへキセン、 1, 2, 4—トリメチルシクロへキセン、 1, 3, 5 -トリメチルシクロへキセンが好まし!/、。
[0012] アルキルシクロへキセンは、公知の方法、例えばジェン化合物とァリル化合物のデ イールズアルダー反応、アルキルシクロへキサノールの脱水、ハロゲン化シクロへキ サンの脱ハロゲン化水素等により製造することができるが、下記式(9):
[化 11]

(式中、 R5は炭素数 1〜4のアルキル基を表し、 pは 2〜6の整数を表し、複数個の は同一でも異なっていてもよい)で表されるアルキルベンゼンの接触部分水素化によ り製造することちでさる。
[0013] 式(9)のアルキルベンゼンとしては、オルト、メタ、パラのキシレン、 1, 2, 3—トリメチ ルベンゼン、 1, 2, 4—トリメチルベンゼン(プソイドタメン)、 1, 3, 5—トリメチルベンゼ ン、 1, 2, 3, 4—テトラメチルベンゼン、 1, 2, 4, 5—テトラメチルベンゼン、 1, 2, 3, 4, 5—ペンタメチルベンゼン、 1, 2, 3, 4, 5, 6—テトラメチルベンゼン、ジェチルべ
ンゼン、ジノルマルプロピルベンゼン、ジイソプロピルベンゼン、ジノルマルブチルベ ンゼン、ジー t ブチルベンゼン、 1, 2 メチルェチルベンゼン、 1, 3—メチルェチ ルベンゼン、 1, 4 メチルェチルベンゼン等が挙げられる。反応の速さ、入手のしゃ すさから R5カ チル基であることが好ましぐキシレン (異性体を含む)およびトリメチル ベンゼン (異性体を含む)がより好ましぐメタキシレン、パラキシレンおよびプソィドク メンがさらに好ましい。
[0014] 式(9)のアルキルベンゼンの部分水素化に使用する触媒としては通常の水素化に 用いられる触媒であれば制限されないが、具体例としてはルテニウム、ロジウム、レニ ゥム、白金、パラジウム、ニッケル、コバルト、クロム、イリジウム、銅等の金属が挙げら れる。これらの金属は炭素、アルミナ、シリカ、ジルコユア、 /、フニ了、チタ-ァ、マグ ネシァ等の担体に担持させて使用することもできるし、金属の微粒子としてそのまま 使用することもできる。触媒量は式(9)のアルキルベンゼンに対する重量比で好まし <は 0. 000001〜10、より好まし <は 0. 00001〜1である。またアルキルシクロへキ センの選択率を上げる目的で、硫酸亜鉛、硫酸コバルト、硫酸バリウム、塩化亜鉛、 酸化亜鉛、臭化亜鉛、ヨウ化亜鉛、水酸ィ匕亜鉛等の助触媒を使用してもよい。助触 媒量は触媒に対する重量比で好ましくは 0. 00001〜100、より好ましくは 0. 0001 〜10である。また劣化した触媒は常法、例えば高温で空気を流通させる等の方法で 賦活することちできる。
[0015] 式(9)のアルキルベンゼンの部分水素化の反応形式は管型固定床、槽型懸濁床 方式が好ましい。槽型懸濁床方式の場合には水を加えて油、水の二相系とすること もでき、アルコール等の添加剤を添加することもできる。このときの水 Z式(9)のアル キルベンゼンの重量比は好ましくは 0. 001〜100、より好ましくは 0. 1〜10であり、 少なすぎると水の効果がなぐ多すぎると空時収率が下がる。反応温度は好ましくは 0 °C〜300°C、より好ましくは 50〜200°Cの範囲であり高すぎると選択率が低下し、低 すぎると転化率が上がらない。水素圧は 0. l〜30MPa、好ましくは l〜15MPaの範 囲であり高すぎると高耐圧の装置が必要であり、低すぎると転ィ匕率が上がらない。反 応時間は回分方式であれば 10分から 20時間の範囲である。
[0016] 上記水素化により得られる式 (4)のアルキルシクロへキセンは、アルキル基と二重
結合の位置関係による異性体を含む。例えば、パラキシレンからは、 1, 4ージメチル —1—シクロへキセン、 1, 4 ジメチルー 2 シクロへキセンの 2つの異性体が生成 する。また、 1, 2, 4 トリメチルベンゼンからは、 1, 2, 4 トリメチル—1—シクロへキ セン、 1, 3, 6 トリメチル 1—シクロへキセン、 2, 5, 6 トリメチル 1—シクロへキ セン、 1, 4, 5 トリメチル 1—シクロへキセン、 2, 3, 5 トリメチル 1—シクロへキ センの 5つの異性体が生成する。これらの異性体は分離した後、あるいは、分離する ことなく式(3)のアルキルベンゼンとの縮合反応に使用することができる。また異性体 は酸触媒の存在下で異性化することもできる。
[0017] 式(9)のアルキルベンゼンの水素化では芳香環が全て水素化されたアルキルシク 口へキサンが副生する。アルキルシクロへキセン、アルキルシクロへキサン及び未反 応のアルキルベンゼンは常法、例えば蒸留、抽出蒸留、分液、抽出、ろ過、晶析によ り分離可能であり、未反応アルキルベンゼンはアルキルシクロへキセン製造の原料と して再利用できる。
[0018] 本発明の(アルキルフエ-ル)アルキルシクロへキサンの製造方法では、アルキルシ クロへキセンにかえて下記式(5):
[化 12]

(式中 R3、 R4および nは上記と同様)で表されるアルキルシクロへキサノールを式(3) のアルキルベンゼンと酸触媒存在下で縮合させることもできる。アルキルシクロへキサ ノールの具体的としては、 1、 2 ジメチルシクロへキサノール、 2, 3 ジメチルシクロ へキサノール、 3,4-ジメチルシクロへキサノール、 4, 5-ジメチルシクロへキサノール、 5, 6-ジメチルシクロへキサノール、 1, 3-ジメチルシクロへキサノール、 1, 4-ジメチ ルシクロへキサノール、 1, 5-ジメチルシクロへキサノール、 1, 6-ジメチルシクロへキ サノール、 2, 3-ジメチルシクロへキサノール、 2, 4-ジメチルシクロへキサノール、 2, 5-ジメチルシクロへキサノール、 2, 6-ジメチルシクロへキサノール、 3, 4-ジメチルシ
クロへキサノール、 3, 5-ジメチルシクロへキサノール、 3, 6-ジメチルシクロへキサノ ール、 4, 5-ジメチルシクロへキサノール、 4, 6-ジメチルシクロへキサノール、 1, 2, 3 —トリメチルシクロへキサノール、 2, 3, 4 トリメチルシクロへキサノール、 3, 4, 5 ト リメチルシクロへキサノール、 4, 5, 6 トリメチルシクロへキサノール、 1, 3, 4 トリメ チルシクロへキサノール、 2, 4, 5 トリメチルシクロへキサノール、 3, 5, 6 トリメチ ルシクロへキサノール、 1, 3, 6 トリメチルシクロへキサノール、 2, 5, 6 トリメチル シクロへキサノール、 1, 4, 5 トリメチルシクロへキサノール、 3, 4, 6 トリメチルシク 口へキサノール、 2, 3, 5 トリメチルシクロへキサノール、 1, 3, 5 トリメチルシクロ へキサノーノレ、 2, 4, 6 トリメチノレシクロへキサノーノレ、 1, 2, 3, 4ーテトラメチノレシク 口へキサノーノレ、 1, 2, 3, 5—テトラメチノレシクロへキサノーノレ、 1, 2, 3, 6—テトラメ チノレシクロへキサノーノレ、 1, 2, 4, 5—テトラメチノレシクロへキサノーノレ、 1, 2, 3, 4, 5 ペンタメチルシクロへキサノール、 1, 2, 3, 4, 5, 6 へキサメチルシクロへキサ ノール、及び上記化合物のメチル基をェチル、ノルマルプロピル、イソプロピル、ノル マルブチル、 sec ブチル、 t ブチルに置換した化合物が挙げられる。反応の速さ 、原料の入手のしゃすさからポリメチルシクロへキサノールが好ましぐジメチルへキ サノール、トリメチルへキサノールがより好ましい。アルキルシクロへキサノールの製造 方法は限定されないが、例えばアルキルフエノールの核水素化、アルキルシクロへキ センの水和等により得られる。
アルキルベンゼンのアルキルシクロへキセンに対するモル比は好ましくは 0. 001〜 1000、より好ましくは 0. 1〜100である。少なすぎるとアルキルシクロへキセン同士の 反応が進みやすぐ多すぎると経済的でない。アルキルベンゼンのアルキルシクロへ キサノールに対するモル比は好ましくは 0. 001〜1000、より好ましくは 0. 1〜100 である。例えば、トルエンと 1, 4 ジメチルシクロへキセンの縮合反応の場合は、トル ェン /1, 4ージメチルシクロへキセンのモル比は 2〜20が好ましぐ 3〜10がより好 ましい。上記範囲内であると、 1, 4ージメチルシクロへキセン同士の付加反応が防止 され、 1一(4 メチルフエ-ル) 1, 4ージメチルシクロへキサンへの選択率が高い。 選択率をより高くするためには、トルエンに 1, 4ージメチルシクロへキセンを徐々に添 加するのが好ましい。また、オルトキシレンと 1, 2, 4 トリメチルシクロへキセンの縮
合反応の場合は、オルトキシレン Zl, 2, 4ートリメチルシクロへキセンのモル比は 1 〜 100力 S好ましく、 2〜50がより好ましい。上記範囲内であると、 1, 2, 4 トリメチル シクロへキセン同士の付加反応が防止され、 1ー(3, 4 ジメチルフエ-ル) 1, 3, 4—トリメチルシクロへキサンへの選択率が高い。選択率をより高くするためには、ォ ルトキシレンに 1, 2, 4 トリメチルシクロへキセンを徐々に添カ卩するのが好ましい。
[0020] 縮合反応は溶媒中で行ってもょ ヽ。溶媒としては縮合反応に不活性な化合物が使 用され、 Friedd— Crafts反応に汎用される-トロメタン、ニトロベンゼン、二硫化炭素、 ァセトニトリルの他に、へキサン、シクロへキサン、石油エーテル、オクタン、デカリン 等の脂肪族炭化水素を使用することができる。前記したアルキルシクロへキセン製造 で副生したアルキルシクロへキサンを使用してもよ 、。アルキル基のオルト位とならな V、パラ位が全てアルキル化されて 、るアルキルベンゼンも溶媒として用いることがで きる。パラ位が全てアルキル化されて 、な 、アルキルベンゼンは縮合反応に関与す るので溶媒として好ましくな 、。
[0021] 縮合反応の酸触媒としては、硫酸、塩酸、リン酸、ポリリン酸、フッ化水素、ホウフッ 化水素酸、三フッ化ホウ素、三塩ィ匕アルミニウム、三臭化アルミニウム、三塩化ガリゥ ム、三臭化ガリウム、三塩化鉄、五塩ィ匕アンチモン、四塩化スズ、四塩化チタン、塩ィ匕 亜鉛等のプロトン酸およびルイス酸が挙げられる。又、ナフイオン等の陽イオン交換 榭脂、シリカアルミナ、ゼォライト等の固体酸も触媒として使用する事が出来る。ゼォ ライトとしては例えば A型、フェリエライト型、 ZSM 5 (MFI型)、 ZSM—12 (MTW 型)、モルデナイト型、ベータ型、 X型、 Y型等が挙げられ、これらのケィ酸塩をリン酸 塩にかえたもの、カチオン (H+、 NH +、金属イオンなど)交換したもの、シリカアルミナ
4
比を変えたものも使用することができる。これらの酸触媒は単独、あるいは組み合わ せて使用する事が出来る。
[0022] 原料とするアルキルシクロへキセンによってはこれらの酸触媒を使用して反応した 場合、 目的外の異性体が無視できない量副生する場合がある。たとえば、トルエンと 1, 4ージメチルシクロへキセンとの縮合反応の場合、 1一(4 メチルフエ-ル) 1, 4 ジメチルシクロへキサンに加えて、式(10)の 2— (4 メチルフエ-ル) 1, 4— ジメチルシクロへキサン、式(11)の 1一(3 メチルフエ-ル)—1, 4ージメチルシクロ
へキサン、および式(12)の 2—(3 メチルフエ-ル)—1, 4ージメチルシクロへキサ ンの異性体が副生することがある。この場合には硫酸を触媒とすることにより目的物 である 1一(4 メチルフエ-ル) 1, 4ージメチルシクロへキサンが高収率で得られ る。
[化 13]

[化 14]

4 4
βーゼオライト、 ZSM— 5などの MFI型ゼオライト、 ZSM— 12などの MTW型ゼオラ イトを使用すると異性体の副生を抑制できることがある。これらのゼォライトは、細孔径 力 0. 5〜0. 8nmであり、このような細孔径を有するゼォライトでは比較的かさ高い 異性体の生成が抑えられるため、原料のトリメチルシクロへキセンの二重結合の位置 に関わらず、 目的とする 1— (3, 4 ジメチルフエ-ル)一 1, 3, 4 トリメチルシクロへ キサンが主生成物として得られる。
[0023] 酸触媒の量はアルキルシクロへキセンに対する重量比で好ましくは 0. 0001〜100 00、より好ましくは 0. 001〜1000の範囲であり、少なすぎると効果が少なぐ多すぎ ると経済的でな 、。アルキルシクロへキセンの代わりにアルキルシクロへキサノールを 使用する場合には触媒量はアルキルシクロへキセンに比べて多く必要となることが多 く、アルキルシクロへキサノールに対する重量比で好ましくは 0. 0001〜10000、より 好ましくは 0. 001〜1000の範囲である。たとえば、トルエンと 1, 4 ジメチルシクロ へキセンとの縮合反応の場合、プロトン酸およびルイス酸触媒は、 1, 4 ジメチルシ クロへキセン 1モルに対して 0. 1モルから 100モル使用するのが好ましぐ陽イオン交 換榭脂、ゼォライト等の固体酸性触媒は、 1, 4 ジメチルシクロへキセン lgに対して 0. l〜10g使用するのが好ましい。ゼォライト触媒の活性が低下した場合には常法、 例えば空気下焼成により賦活することもできる。
[0024] 縮合反応は回分式または流通連続式の固定床、懸濁床、均一槽型などの反応形 式で行われる。固定床は触媒の分離回収が容易であるので好ましい。固体酸性触媒 を使用した流通方式の場合は、重量時間空間速度 (WHSV)が 0. 001〜10hr— 1の 範囲になるようにアルキルシクロへキセンまたはアルキルシクロへキサノールを流通 反応器内に連続的に供給するのが好ましい。反応温度は触媒の種類と使用量、反 応基質の種類と濃度により異なるが— 50〜250°Cの範囲である。硫酸や塩酸の場合 は 0〜50°Cが好ましぐフッ化水素を使用する場合は 50〜50°Cが好ましい。ゼォ ライトやイオン交換榭脂を使用する場合は 50〜200°Cが好ましい。これらの温度より 高 、場合には異性ィ匕等の副反応が増大し、これらの温度より低 、場合には十分な反
応速度が得られない。反応圧力は常圧、減圧、加圧のいずれでも任意に選択できる 。原料や使用する溶媒の沸点が反応温度より高い場合には 0. 1〜: LOMPaの加圧下 で反応を行うことが好ましい。窒素、アルゴン等の不活性気体を反応系に導入してカロ 圧するのが好ましい。反応時間は回分の場合 10分から 20時間が好ましぐ 10分から 3時間がより好ましい。
[0025] 上記縮合反応で生成する(アルキルフエニル)アルキルシクロへキサンはシス、トラ ンス異性体の混合物として得られる場合もある。異性体混合物は蒸留、晶析、カラム 分離等の常法により原料や副生物と分離できる。(アルキルフ ニル)アルキルシクロ へキサンの異性体混合物は各異性体に分離しても良いし、混合物のまま次工程(同 時脱水素化 Z脱アルキル化)の原料として用いることもできる。
[0026] 上記のようにして得られた(アルキルフエ-ル)アルキルシクロへキサンのシクロへキ サン環を触媒の存在下で脱水素化すると同時に R3を脱アルキルすることにより下記 式 (7) :
[化 15]

[0027] 同時脱水素化 Z脱アルキルィ匕触媒としては、例えば、白金、ノラジウム、ロジウム、 レニウム、ルテニウム、鉄、クロム、コノルト、ニッケル、モリブデン、銅、亜鉛、イリジゥ ム等の金属または該金属を含む化合物、ゼォライト、アルミナ、シリカアルミナ及びこ れらの混合物が挙げられる。これらの中でもクロム、亜鉛を含む化合物が活性及び選 択性の点カゝら特に好ましい。クロム、亜鉛を含む化合物の具体例としては酸化クロム( III)、酸化亜鉛、 Cu—Crゝ Cu—Zn、 Zn—
Crが挙げられる。またこれらの化合物にリチウム、ナトリウム、カリウム等のアルカリ金 属ゃマグネシウム、カルシウム等のアルカリ土類金属を添加することもでき、カリウム が特に好ましい。これらの触媒は炭素、アルミナ、ジルコユア、シリカアルミナ等の担 体に担持させて使用することもできる。触媒は常法、例えば、空気等の酸ィ匕性ガスま たは水素等の還元性ガスの流通下、加熱による前処理を行うこともできる。また触媒 の活性が低下した場合にも同様の処理により賦活が可能である。触媒の製造方法は 限定されるものではないが、例えば硝酸や酢酸等の金属塩を水に溶解させて γ—ァ ルミナ等の担体に含浸させた後乾燥し、空気下 300°C〜600°Cで焼成する含浸法で 製造することができる。流通方式の場合の重量時間空間速度 (WHSV)は 0. 0001 〜100hr— 1の範囲が好ましぐ 0. 001〜10hr_1がより好ましい。
同時脱水素化 Z脱アルキルィ匕の反応形式は固定床流通方式が好まし 、。反応温 度は好ましくは 200〜600°C、より好ましくは 350〜550°Cである。これより低いと反 応が十分に進行せず、高いと選択性が低下する。反応圧力は減圧、常圧、加圧のい ずれでも行うことができる。反応中、窒素、水素や水蒸気を同時に流通させることもで き、触媒の劣化が抑制されるので水素を流通するのが好ましい。 (アルキルフエニル) アルキルシクロへキサンはそのまま反応器に供給することもできるが、反応に不活性 な溶媒、例えばベンゼン、トルエン等を使用することもできる。また上記した、式(9)の アルキルベンゼンの接触部分水素化による式 (4)のアルキルシクロへキセン製造で 副生した下記式(15) :
[化 16]

(式中、 R5および pは前記と同様)で表されるアルキルシクロへキサンを溶媒として用 いることちでさる。
前記アルキルシクロへキセン製造工程の副生物は実質的にアルキルシクロへキサ ンのみであり、この副生アルキルシクロへキサンは同時脱水素化 Z脱アルキル化工 程で式(9)のアルキルベンゼンに変換され、アルキルシクロへキセン製造の原料とし
て再利用することができる。そのため、アルキルシクロへキセンの製造工程、アルキル シクロへキセンの縮合工程、および(アルキルフエ-ル)アルキルシクロへキサンの同 時脱水素化 Z脱アルキル化工程を連続して行うと、アルキルシクロへキセン製造で 使用した式(9)の原料アルキルベンゼンのロスがなぐアルキルビフエ二ルを経済的 なプロセスで製造することができる。
[0030] 得られたアルキルビフヱ-ルは常法、例えば蒸留、晶析、カラム分離等により分離 可能である。アルキルビフヱニルから、従来公知の方法により、対応するビフエニルジ カルボン酸、ビフエ-ルテトラカルボン酸、ビフエ-ルテトラカルボン酸無水物を製造 することができる。
実施例
[0031] 以下実施例により本発明を具体的に説明するが、本発明は以下の実施例に限定さ れるものではない。転化率、選択率および収率は、内部標準法によるガスクロマトダラ フィ一から求めた。
[0032] 実施例 1
温度計、コンデンサーを備えたガラス製 3000mlの 3ッロフラスコに、トルエン 2700 g、濃硫酸 15g、 1, 4 ジメチルシクロへキセン 55gを仕込み、反応液の温度を— 20 °Cに設定した。 5時間反応させてから有機層を分離し、分離した有機層を、 500mlの 蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗浄した。 有機層をガスクロマトグラフィーで分析したところ、 1, 4 ジメチルシクロへキセンの転 ィ匕率 96%、 1一(4 メチルフエ-ル) 1, 4ージメチルシクロへキサンの選択率 92 %であった。
[0033] 実施例 2
500mレヽステロイ C製オートクレーブに、フツイ匕水素 130g、トルエン 100gを仕込み 、— 27°Cに冷却した。そこへ、 1, 4 ジメチルシクロへキセン 10g、トルエン 100gの 混合液を 1時間かけて添加した。添加終了後、更に 20分保持して力ゝら抜液し、フッ化 水素を回収した。反応中、系内の圧力は 0. 2MPa以下であった。得られた有機層は 、 200mlの蒸留水で 3回、 5%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順 で洗浄した。有機層をガスクロマトグラフィーで分析すると、 1, 4 ジメチルシクロへキ
センの転化率は 100%、 1— (4—メチルフエ-ル)— 1, 4 ジメチルシクロへキサン の選択率は 28. 5%であった。
[0034] 実施例 3
コンデンサー、温度計を備えた 500mlの SUS304製電磁攪拌式オートクレープに 、トルエン 250g、 H型モルデナイト(東ソ一製: HSZ— 640HODlC) 5g、 1, 4 ジメ チルシクロへキセン 25gをカ卩え、系内を窒素ガスで置換した。系内に窒素ガス 3MPa を充填してから、 180°Cに加熱、攪拌した。反応中、系内の圧力は 4MPaまで上昇し た。 3時間反応後、反応液を抜き出し触媒を濾過して、有機層をガスクロマトグラフィ 一で分析すると、 1, 4ージメチルシクロへキセンの転化率 85. 8%、 1一(4 メチル フエ-ル)— 1, 4 ジメチルシクロへキサンの選択率 59. 5%であった。
[0035] 実施例 4
反応温度を 110°C、反応圧力を常圧にしトルエン還流下、反応時間を 50時間とし た以外は、実施例 3と同様の条件で反応を行った。反応終了後、反応液を抜き出し 触媒を濾過して、有機層をガスクロマトグラフィーで分析すると、 1, 4 ジメチルシク 口へキセンの転化率 70. 1%、 1— (4—メチルフエ-ル)一 1, 4 ジメチルシクロへキ サンの選択率 63. 2%であった。
[0036] 実施例 5
50mlの SUS304製オートクレーブに、トルエン 5. 0g、 H型 j8ゼォライト(東ソ一製: HSZ— 930HOD1A) 0. lg、 1, 4 ジメチルシクロへキセン 0. 25gを加え、系内を 窒素ガスで置換した。系内に窒素ガス 2MPaを充填してから、 200°Cに加熱、攪拌し た。反応中、系内の圧力は 3MPaまで上昇した。 20時間反応後、反応液を抜き出し 触媒を濾過して、有機層をガスクロマトグラフィーで分析すると、 1, 4 ジメチルシク 口へキセンの転化率 77. 5%、 1— (4—メチルフエ-ル)一 1, 4 ジメチルシクロへキ サンの選択率 62. 5%であった。
[0037] 実施例 6
H型 SAPO— 5ゼォライト(H. Robsonゝ" Verified Syntheses of Zeoritic M aterials", Elsevier Science B. V. p93記載の方法を参考に調製) 0. lgを使 用した以外は、実施例 5と同様の条件で反応を行った。反応中、系内の圧力は 3MP
aまで上昇した。反応後、反応液を抜き出し触媒を濾過して、有機層をガスクロマトグ ラフィ一で分析すると、 1, 4ージメチルシクロへキセンの転化率 42. 6%、 1ー(4ーメ チルフエ-ル) 1, 4ージメチルシクロへキサンの選択率 60. 6%であった。
実施例 7
温度計、コンデンサーを備えたガラス製 3000mlの 3ッロフラスコに、オルトキシレン 2000g、濃硫酸 24g、 1, 2, 4 トリメチルシクロへキセン 50gを仕込み、反応液の温 度を 20°Cに設定した。 1時間反応させてから有機層を分離した。分離した有機層は、 50mlの蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗 浄した。有機層をガスクロマトグラフィーで分析すると、 1, 2, 4 トリメチルシクロへキ センの転化率 87%、 1— (3, 4 ジメチルフエ-ル)— 1, 3, 4 トリメチルシクロへキ サンの選択率 43%であった。
反応液力 低沸点留分を除去した後、理論段数 10段の蒸留塔にて留出圧力 4tor r、留出温度 140°C及び 142°Cの 2つの留分を得た。 GC— MS、 — NMR、 13C— NMRにより分析した結果、 2つの留分は下記に示す 1一(3, 4 ジメチルフエ-ル) —1, 3, 4 トリメチルシクロへキサンの異性体 (異性体 1および 2)であった。
異性体 1
GC— MS : M+ = 230
一 NMR(CDC1溶媒)、 δ (ppm) :
3
7. 0〜7. 2 (m、 3H、芳香環水素)
2. 2及び 2. 3 (s、 3H、芳香環結合メチル水素)
1. 2 (s、 3H、シクロへキサン環 4級炭素結合メチル水素)
0. 8〜0. 9 (m、 6H、シクロへキサン環結合メチル水素)
1. 7〜2. 0 (m、 2H、シクロへキサン環メチン水素)
1. 3〜1. 6 (m、 6H、シクロへキサン環メチレン水素)
13C— NMR(CDC1溶媒)、 δ (ppm) :
3
133、 135、 151 (芳香環四級炭素)
122、 126、 129 (芳香環メチン炭素)
41、 31、 29 (シクロへキサン環メチレン炭素)
37 (シクロへキサン環四級炭素)
32、 31 (シクロへキサン環メチン炭素)
19、 20 (芳香環結合メチル炭素)
11、 20 (シクロへキサン環メチン結合メチル炭素)
26 (シクロへキサン環四級炭素結合メチル炭素)
異性体 1の1 H— NMRスペクトルを図 1に示す。
異性体 2
GC-MS : M+ = 230
一 NMR(CDC1溶媒)、 δ (ppm) :
3
7. 0〜7. 2 (m、 3H、芳香環水素)
2. 2及び 2. 3 (s、 3H、芳香環結合メチル水素)
1. 2 (s、 3H、シクロへキサン環 4級炭素結合メチル水素)
0. 9〜1. 0 (m、 6H、シクロへキサン環結合メチル水素)
0. 9〜1. 5 (m、 2H、シクロへキサン環メチン水素)
I. 2〜1. 9 (m、 6H、シクロへキサン環メチレン水素)
13C— NMR(CDC1溶媒)、 δ (ppm) :
3
133、 136、 150 (芳香環四級炭素)
122、 126、 129 (芳香環メチン炭素)
47、 38、 32 (シクロへキサン環メチレン炭素)
37 (シクロへキサン環四級炭素)
39、 35 (シクロへキサン環メチン炭素)
19、 20 (芳香環結合メチル炭素)
I I、 20 (シクロへキサン環メチン結合メチル炭素)
25 (シクロへキサン環四級炭素結合メチル炭素)
異性体 2の1 H— NMRスペクトルを図 2に示す。
実施例 8
温度計、コンデンサーを備えたガラス製 200mlの 3ッロフラスコに、オルトキシレン 1 00g、 H型モルデナイト(シリカ/アルミナ比 = 200、東ソー製: HSZ— 690HOA) 2
5g、 1, 2, 4 トリメチルシクロへキセン 5gを仕込み、常圧で反応液を還流温度(150 °C)まで加熱した。 1時間反応させて力も触媒をろ別した反応液をガスクロマトグラフィ 一で分析すると、 1, 2, 4 トリメチルシクロへキセンの転化率 95%、 1— (3, 4 ジメ チルフエ-ル) 1, 3, 4ートリメチルシクロへキサンの選択率 72%であった。
[0040] 実施例 9〜12
H型モルデナイトに変えて表 1に示す各種ゼォライトを用いたほかは実施例 8と同 様にして反応を行った。結果を表 1にまとめて示す。
[表 1] 表 1
1 , 2 , 4—トリメチル 1 - ( 3 , 4—ジメチルフエ-ル) 1, 実施例 ゼォライ ト シク口へキセンの転 {匕率 3, 4—トリメチルシクロへキサンの選
(%) 択率 (%)
9 Z S M- 5 7 9 4 1
N H 4型
1 0 Z S M— 5 5 7 3 0
1 1 Z S M- 1 2 8 5 2 6
H型
1 2 H - i3 4 6 3
[0041] 実施例 13
温度計、コンデンサーを備えたガラス製 300ml3ッロフラスコに、オルトキシレン 10 g、濃硫酸 0. 3g、 1, 4ージメチルシクロへキセン 0. 5gを仕込み、反応液の温度を 20 °Cに設定した。 1時間反応させてから有機層を分離し、分離した有機層は、 30mlの 蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗浄した。 有機層をガスクロマトグラフィーで分析したところ、 1, 4 ジメチルシクロへキセンの 転化率 94%、 1— (3, 4 ジメチルフエ-ル) 1, 4 ジメチルシクロへキサンの選 択率 71%であった。
[0042] 実施例 14
オルトキシレンにかえてメタキシレンを使用した以外は実施例 13と同様にして反応 を行った。有機層をガスクロマトグラフィーで分析したところ、 1, 4 ジメチルシクロへ キセンの転化率 92%、 1— (3、 5 ジメチルフエ-ル) 1, 4 ジメチルシクロへキサ
ンの選択率 11 %であった。
[0043] 実施例 15
オルトキシレンにかえてェチルベンゼンを使用した以外は実施例 13と同様にして 反応を行った。有機層をガスクロマトグラフィーで分析したところ、 1, 4—ジメチルシク 口へキセンの転化率 89%、 1— (4—ェチルフエ-ル)— 1, 4—ジメチルシクロへキサ ンの選択率 60%であった。
[0044] 実施例 16
オルトキシレンにかえてプロピルベンゼンを使用した以外は実施例 13と同様にして 反応を行った。有機層をガスクロマトグラフィーで分析したところ、 1, 4—ジメチルシク 口へキセンの転化率 93%、 1— (4—プロピルフエ-ル)— 1, 4—ジメチルシクロへキ サンの選択率 31 %であった。
[0045] 実施例 17
オルトキシレンにかえてイソプロピルベンゼンを使用した以外は実施例 13と同様に して反応を行った。有機層をガスクロマトグラフィーで分析したところ、 1, 4—ジメチル シクロへキセンの転化率 85%、 1— (4—イソプロピルフエ-ル)一 1, 4—ジメチルシク 口へキサンの選択率 28%であった。
[0046] 実施例 18
オルトキシレンにかえて 1, 2, 3—トリメチルベンゼンを使用した以外は実施例 13と 同様にして反応を行った。有機層をガスクロマトグラフィーで分析したところ、 1, 4— ジメチルシクロへキセンの転化率 97%、 1— (3, 4, 5—トリメチルフエ-ル)— 1, 4— ジメチルシクロへキサンの選択率 46%であった。
[0047] 実施例 19
温度計、コンデンサーを備えたガラス製 100mlの 3ッロフラスコに、メタキシレン 10g 、濃硫酸 0. 3g、 1, 2, 4—トリメチルシクロへキセン 0. 5gを仕込み、反応液の温度を 20°Cに設定した。 1時間反応させてから有機層を分離し、分離した有機層を、 30ml の蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗浄し た。有機層をガスクロマトグラフィーで分析したところ、 1, 2, 4—トリメチルシクロへキ センの転化率 98%、 1— (3, 5—ジメチルフエ-ル)— 1, 3, 4—トリメチルシクロへキ
サンの選択率 17%であった。
[0048] 実施例 20
メタキシレンにかえて 1, 2, 3 トリメチルベンゼンを使用した以外は実施例 19と同 様にして反応を行った。有機層をガスクロマトグラフィーで分析したところ、 1, 2, 4— トリメチルシクロへキセンの転化率 95%、 1, 3, 4 トリメチル—1— (3, 4, 5 トリメチ ルフエ-ル)シクロへキサンの選択率 53%であった。
[0049] 実施例 21
温度計、コンデンサーを備えたガラス製 300mlの 3ッロフラスコに、トルエン 65g、 濃硫酸 3. 5g、 2, 5 ジメチルシクロへキサノール 2. 5gを仕込み、反応液の温度を 2 0°Cに設定した。 1時間反応させてから有機層を分離した。分離した有機層を、 20ml の蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗浄し た。有機層をガスクロマトグラフィーで分析すると 2, 5 ジメチルシクロへキサノール の転化率 88%、 1— (4—メチルフエ-ル)— 1, 4 ジメチルシクロへキサンの選択率 74%であった。
[0050] 実施例 22
温度計、コンデンサーを備えたガラス製 100mlの 3ッロフラスコに、オルトキシレン 2 0g、濃硫酸 3g、 3, 4ージメチルシクロへキサノール 2gを仕込み、反応液の温度を 20 °Cに設定した。 1時間反応させてから有機層を分離した。分離した有機層を、 20ml の蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗浄し た。有機層をガスクロマトグラフィーで分析すると 3, 4—ジメチルシクロへキサノール の転化率 68%、 1— (3, 4 ジメチルフエ-ル) 1, 2 ジメチルシクロへキサンの選 択率 74%であった。
[0051] 実施例 23
温度測定用のさや管を付したガラス製の反応管 (長さ 250mm X内径 12mm φ )の 上部に原料供給ライン及び水素ガス供給ライン、下部に冷却用のジムロート及び反 応液捕集用のフラスコと排気用のベントラインを設置した。反応管に 10%Cr O—1
2 3
%K/A1 O触媒 8gを仕込み、 50ml/minの速度で水素を流通しながら 450°Cにカロ
2 3
熱した。原料供給ラインより実施例 7で得た 1— (3, 4 ジメチルフエ-ル)— 1, 3, 4
ートリメチルシクロへキサン異性体混合物(異性体 1Z異性体 2 = 60Z40 (重量比)) の 10wt%ベンゼン溶液を lOgZhrで供給して反応を開始した。反応開始後 2〜5時 間の反応液を捕集し GC及び NMRにて分析したところ 3, 4, 3' , 4'—テトラメチルビ フエニルが 92%の収率で得られ、そのほかのテトラメチルビフエニル異性体は見られ なかった。
実施例 24
第 1工程
温度計を備えた SUS316製の 500ml電磁撹拌オートクレーブに水 100g、 5%Ru アルミナ触媒 lg、硫酸亜鉛 0. 06g、次いでパラキシレン 200gを仕込んだ。系内を水 素で 5MPaに加圧し 150°Cで 4hr反応させた。冷却後反応液から油相を分離しガス クロマトグラフィーで分析したところパラキシレンの転ィ匕率 80%、 1, 4 ジメチルシク 口へキセンの選択率 39%、 1, 4ージメチルシクロへキサンの選択率 61%であった。 得られた反応液から蒸留により 1, 4ージメチルシクロへキセン及び 1, 4ージメチルシ クロへキサンを分離した。
第 2工程
温度計、コンデンサーを備えたガラス製 3000mlの 3ッロフラスコに、トルエン 2700 g、濃硫酸 15g、 1, 4 ジメチルシクロへキセン 55gを仕込み、反応液の温度を— 20 °Cに設定した。 5時間反応させてから有機層を分離し、分離した有機層を、 500mlの 蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗浄した。 有機層をガスクロマトグラフィーで分析したところ、 1, 4 ジメチルシクロへキセンの転 ィ匕率 96%、 1一(4 メチルフエ-ル) 1, 4ージメチルシクロへキサンの選択率 92 %であった。得られた反応液力も蒸留により 1— (4—メチルフエ-ル)一 1, 4 ジメチ ルシクロへキサンを分離した。
第 3工程
温度測定用のさや管を付したガラス製の反応管 (長さ 250mm X内径 12mm φ )の 上部に原料供給ライン及び水素ガス供給ライン、下部に冷却用のジムロート及び反 応液捕集用のフラスコと排気用のベントラインを設置した。反応管に 10%Cr O—1
2 3
%K/A1 O触媒 8gを仕込み、 50ml/minの速度で水素を流通しながら 450°Cにカロ
熱した。原料供給ラインより上記 1一(4 メチルフエ-ル)—1, 4ージメチルシクロへ キサンの 10%ベンゼン溶液を lOgZhrで供給して反応を開始した。反応開始後 2〜 5時間の反応液を捕集し分析したところ 1— (4—メチルフエ-ル)一 1, 4 ジメチルシ クロへキサンの転化率は 100%、 4, 4, 一ジメチルビフ -ルの収率は 78%の収率 であった。
[0053] 実施例 25
第 3工程の触媒を酸ィ匕亜鉛に変えた以外は実施例 24と同様にして反応を行った。 1— (4—メチルフエ-ル)— 1, 4 ジメチルシクロへキサンの転化率は 70%、 4, 4, ジメチルビフエ-ルの収率は 49%であった。
[0054] 実施例 26
第 3工程の溶媒をベンゼンに変えて第一工程で副生した 1, 4ージメチルシクロへキ サンとした以外は実施例 24と同様にして反応を行った。 1— (4—メチルフエ-ル) 1, 4ージメチルシクロへキサンの転化率は 80%、 4, 4' ジメチルビフエ-ルの収率 は 45%であった。また 1, 4ージメチルシクロへキサンの転化率は 14%、パラキシレン の収率は 14%であった。
[0055] 実施例 27
第 1工程
実施例 24と同様の反応器に水 100g、 5%Ruアルミナ触媒 2g、硫酸亜鉛 0. 06g、 次いでプソイドクメン 200gを仕込んだ。系内を水素で lOMPaに加圧し 160°Cで 2. 5 hr反応させた。冷却後反応液力 油相を分離しガスクロマトグラフィーで分析したとこ ろプソィドクメンの転ィ匕率 65%、 1, 2, 4 トリメチルシクロへキセンの選択率 31%、 1 , 2, 4 トリメチルシクロへキサンの選択率 69%であった。得られた反応液力 蒸留 により 1, 2, 4 トリメチルシクロへキセン異性体混合物を分離した。
第 2工程
実施例 24と同様の反応器にオルトキシレン 100g、 H型モルデナイト(シリカ/アル ミナ比 = 200、東ソー製: HSZ— 690HOA) 25g、 1, 2, 4 卜リメチルシクロへキセ ン異性体混合物 5gを仕込み、常圧で反応液を還流温度(150°C)まで加熱した。 1時 間反応させて力も触媒をろ別した反応液をガスクロマトグラフィーで分析すると、 1, 2
, 4 トリメチルシクロへキセンの転化率 95%、 1— (3, 4 ジメチルフエ-ル)— 1, 3 , 4 トリメチルシクロへキサンの選択率 72%であった。得られた反応液から蒸留によ り 1一(3, 4 ジメチルフエ-ル) 1, 3, 4 トリメチルシクロへキサンを分離した。 第 3工程
原料として 1— (3, 4 ジメチルフエ-ル)— 1, 3, 4 トリメチルシクロへキサンを使 用した以外は実施例 24と同様にして反応を行った。 1— (3, 4 ジメチルフエニル) —1, 3, 4ートリメチルシクロへキサンの転化率は 94%、 3, 4, 3' , 4,ーテトラメチル ビフエ-ルの収率は 85%であった。
[0056] 実施例 28
温度計を備えた SUS316製の 100ml電磁撹拌オートクレーブに水 10g、 5%Ruァ ルミナ触媒 0. 2g、次いでメシチレン 20gを仕込んだ。系内を水素で lOMPaに加圧 し 150°Cで 2hr反応させた。冷却後反応液から油相を分離しガスクロマトグラフィーで 分析したところメシチレンの転ィ匕率 49%、 1, 3, 5 トリメチルシクロへキセンの選択 率 21%、 1, 3, 5 トリメチルシクロへキサンの選択率 79%であった。
温度計、コンデンサーを備えたガラス製 100mlの 3ッロフラスコにオルトキシレン 10 g、硫酸 0. 3g、 1, 3, 5 トリメチルシクロへキセン 0. 5gを仕込み反応液の温度を 20 °Cに設定した。 1時間反応させてから有機層を分離し、分離した有機層を、 30mlの 蒸留水で 2回、 3%炭酸水素ナトリウム水溶液で 1回、蒸留水で 2回の順で洗浄した。 有機層をガスクロマトグラフィーで分析したところ、 1, 3, 5 トリメチルシクロへキセン の転化率 92%、 1一(3、 4ージメチルフエ-ル)一 1, 3, 5 トリメチルシクロへキサン の選択率 15%であった。
産業上の利用可能性
[0057] 本発明の方法によれば、後処理を必要としない工程で、安価な原料から効率的か つ高選択率で目的の(アルキルフエ-ル)アルキルシクロへキサンおよびアルキルビ フエニルを製造することが出来るため、工業的に極めて有利である。
Claims
[化 1]

(式中 R1は炭素数 1〜4のアルキル基を表し、 R2は R1と同一又は異なる炭素数 1〜4 のアルキル基を表し、 mは 0〜2の整数を表し、 mが 1である場合、 R2は R1のオルト位 またはメタ位に位置し、 mが 2である場合、 2個の R2は R1の両オルト位に位置する)で 表されるアルキルベンゼンと、下記式 (4):
[化 2]

(式中 R3は炭素数 1〜4のアルキル基を表し、 R4は R3と同一又は異なる炭素数 1〜4 のアルキル基を表し、 nは 1〜5の整数を表し、 nが 2〜5の整数である場合、複数個 の R4は同一でも異なっていてもよぐ R3と R4、または、 2個の R4が同一の炭素に結合 している場合は除く)で表されるアルキルシクロへキセン、または、下記式(5): [化 3]
(式中 R3、 R4および nは上記と同様)で表されるアルキルシクロへキサノールとを酸触 媒存在下で縮合させる工程を含む下記式 (6):
[化 4]
 び nは上記と同様であり、 R2が存在しないか R2が R1のォ ルト位に位置する場合、シクロへキサン環は R1のパラ位に位置し、 R2が R1のメタ位に 位置する場合、シクロへキサン環は R1のメタ位に位置する)で表される(アルキルフエ -ル)アルキルシクロへキサンの製造方法。
び nは上記と同様であり、 R2が存在しないか R2が R1のォ ルト位に位置する場合、シクロへキサン環は R1のパラ位に位置し、 R2が R1のメタ位に 位置する場合、シクロへキサン環は R1のメタ位に位置する)で表される(アルキルフエ -ル)アルキルシクロへキサンの製造方法。
[2] 前記酸触媒が硫酸、塩酸、リン酸、ポリリン酸、フッ化水素、ホウフッ化水素酸、三フッ 化ホウ素、三塩ィ匕アルミニウム、三臭化アルミニウム、三塩ィ匕ガリウム、三臭化ガリウム 、三塩化鉄、五塩ィ匕アンチモン、四塩化スズ、四塩化チタン、および塩化亜鉛からな る群より選ばれた少なくとも一種の触媒である請求項 1記載の製造方法。
[3] 前記酸触媒が陽イオン交換榭脂、シリカアルミナ、およびゼォライトからなる群より選 ばれた少なくとも一種の触媒である請求項 1記載の製造方法。
[4] 前記式 (4)のアルキルシクロへキセンを下記式(9):
[化 5]

(式中、 R5は炭素数 1〜4のアルキル基を表し、 pは 2〜6の整数を表し、複数個の は同一でも異なっていてもよい)で表されるアルキルベンゼンの接触部分水素化によ り製造する請求項 1記載の製造方法。
[5] 前記式(3)〜(6)にお 、て、
 R2、 R3および R4カ チル基である請求項 1記載の製 造方法。
R2、 R3および R4カ チル基である請求項 1記載の製 造方法。
[6] 前記式(3)のアルキルベンゼンとしてトルエン、および前記式(4)のアルキルシクロへ キセンとして 1, 4 ジメチルシクロへキセンを使用して 1— (4—メチルフエ-ル)一 1, 4 ジメチルシクロへキサンを製造する請求項 1記載の製造方法。
[7] 前記酸触媒が硫酸、フッ化水素、 H型モルデナイト、および H型 βーゼオライトからな る群より選ばれた少なくとも一種の触媒である請求項 6記載の製造方法。
[8] 前記式(3)のアルキルベンゼンとしてオルトキシレン、および前記式(4)のアルキル シクロへキセンとして 1, 2, 4 トリメチルシクロへキセンを使用して 1— (3, 4 ジメチ ルフヱニル) 1, 3, 4ートリメチルシクロへキサンを製造する請求項 1記載の製造方 法。
[9] 前記酸触媒が硫酸、 H型モルデナイト、 NH型モルデナイト、 H型または NH型の M
4 4
FI型ゼオライト、 H型または NH型の MTW型ゼオライト、および H型 |8—ゼォライト
4
からなる群より選ばれた少なくとも一種の触媒である請求項 8記載の製造方法。
[10] 請求項 1に記載の製造方法で得られた式(6)の(アルキルフ -ル)アルキルシクロ へキサンを、触媒の存在下で同時に脱水素および脱アルキルする工程を含む下記 式 (7) :
[化 6]

[11] 前記触媒が、白金、パラジウム、ロジウム、レニウム、ルテニウム、鉄、クロム、コバルト 、ニッケル、モリブデン、銅、亜鉛、イリジウム等の金属、該金属を含む化合物、ゼオラ イト、アルミナ、およびシリカアルミナ力 なる群より選ばれた少なくとも一種の触媒で ある請求項 10に記載の製造方法。
[12] 請求項 4に記載のアルキルベンゼンの接触部分水素化にぉ 、て副生した下記式(1 5) :
[化 7]
(15)
(R
(式中、 R5および pは前記と同様)で表されるアルキルシクロへキサンを溶媒として用 いる請求項 10に記載の製造方法。
前記式(15)のアルキルシクロへキサンを前記同時に脱水素および脱アルキルする 工程で脱水素し、下記式(9):
[化 8]

(式中、 R5および pは前記と同様)で表されるアルキルベンゼンに変換する請求項 10 記載の製造方法。
[14] 請求項 13で得られた式(9)のアルキルベンゼンを請求項 4に記載のアルキルシクロ へキセンの製造工程の原料として使用する請求項 10記載の製造方法。
[15] 前記アルキルビフヱニルを酸化して対応するビフヱ二ルポリカルボン酸にする工程を さらに含む請求項 10に記載の製造方法。
[16] 前記ビフエ-ルポリカルボン酸が 4, 4'ービフエ-ルジカルボン酸または 3, 4, 3' , 4
, -ビフエ-ルテトラカルボン酸である請求項 15に記載の製造方法。
[17] 前記 3, 4, 3,, 4,一ビフエ-ルテトラカルボン酸を 3, 4, 3,, 4,一ビフエ-ルテトラ力 ルボン酸無水物に無水化する工程をさらに含む請求項 16に記載の製造方法。
[18] 下記式(8) :
[化 9]

(式中 R1は炭素数 1〜4のアルキル基を表し; R2は R1と同一又は異なる炭素数 1〜4 のアルキル基を表し; mは 0〜2の整数を表し、 mが 1である場合、 R2は R1のオルト位 またはメタ位に位置し、 mが 2である場合、 2個の R2は R1の両オルト位に位置し; R3は 炭素数 1〜4のアルキル基を表し; R4は R3と同一又は異なる炭素数 1〜4のアルキル 基を表し; R2が存在しないか R2が R1のオルト位に位置する場合、シクロへキサン環は
R1のパラ位に位置し、 R2が R1のメタ位に位置する場合、シクロへキサン環は R1のメタ 位に位置し; n'は 2〜5の整数を表し、複数個の R4は同一でも異なっていてもよぐ 2 個の R4が同一の炭素に結合している場合は除く)で表される(アルキルフエ-ル)ァ ルキルシクロへキサン。
下記式(2) :
[化 10]
 で表される 1— (3, 4 ジメチルフエ-ル) 1, 3, 4 トリメチルシクロへキサンである 請求項 18に記載の(アルキルフエ-ル)アルキルシクロへキサン。
で表される 1— (3, 4 ジメチルフエ-ル) 1, 3, 4 トリメチルシクロへキサンである 請求項 18に記載の(アルキルフエ-ル)アルキルシクロへキサン。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007528485A JPWO2007013469A1 (ja) | 2005-07-26 | 2006-07-25 | (アルキルフェニル)アルキルシクロヘキサン及び(アルキルフェニル)アルキルシクロヘキサン、アルキルビフェニルの製造方法 |
| US11/996,147 US20090299111A1 (en) | 2005-07-26 | 2006-07-25 | (alkylphenyl) alkylcyclohexane and method for producing (alkylphenyl) alkylcyclohexane or alkylbiphenyl |
| EP06781605A EP1908743A4 (en) | 2005-07-26 | 2006-07-25 | (ALKYLPHENYL) ALKYLCYCLOHEXAN AND METHOD FOR THE PRODUCTION OF (ALKYLPHENYL) ALKYLCYCLOHEXAN OR ALKYLBIOPHENYL |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005215382 | 2005-07-26 | ||
| JP2005-215382 | 2005-07-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007013469A1 true WO2007013469A1 (ja) | 2007-02-01 |
Family
ID=37683362
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/314693 Ceased WO2007013469A1 (ja) | 2005-07-26 | 2006-07-25 | (アルキルフェニル)アルキルシクロヘキサン及び(アルキルフェニル)アルキルシクロヘキサン、アルキルビフェニルの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20090299111A1 (ja) |
| EP (1) | EP1908743A4 (ja) |
| JP (1) | JPWO2007013469A1 (ja) |
| CN (1) | CN101228102A (ja) |
| WO (1) | WO2007013469A1 (ja) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012157749A1 (ja) * | 2011-05-18 | 2012-11-22 | 宇部興産株式会社 | 3,3',4,4'-テトラアルキルシクロヘキシルベンゼン及びその製造方法 |
| WO2014078652A1 (en) | 2012-11-16 | 2014-05-22 | Zs Genetics, Inc. | Heavy atom labeled nucleosides, nucleotides, and nucleic acid polymers, and uses thereof |
| US9085669B2 (en) | 2013-01-28 | 2015-07-21 | Exxonmobil Chemical Patents Inc. | Alkyl aromatic hydroalkylation for the production of plasticizers |
| US9328053B2 (en) | 2013-03-14 | 2016-05-03 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9534104B2 (en) | 2013-01-28 | 2017-01-03 | Exxonmobil Chemical Patents Inc. | Plasticizer blends and use thereof |
| US9556087B2 (en) | 2013-03-14 | 2017-01-31 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9580572B2 (en) | 2013-03-14 | 2017-02-28 | Exxonmobil Chemical Patents Inc. | (Methylcyclohexyl)toluene isomer mixtures,their production and their use in the manufacture of plasticizers |
| US9663417B2 (en) | 2013-03-14 | 2017-05-30 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9688602B2 (en) | 2013-03-14 | 2017-06-27 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9725377B2 (en) | 2013-03-14 | 2017-08-08 | Exxonmobil Chemical Patents Inc. | Hydroalkylation catalyst and process for use thereof |
| US9758447B2 (en) | 2014-10-24 | 2017-09-12 | Exxonmobil Chemical Patents Inc. | Activation of dehydrogenation catalysts |
| US9856186B2 (en) | 2014-12-19 | 2018-01-02 | Exxonmobil Chemical Patents Inc. | Production and use of dialkylbiphenyl isomer mixtures |
| US9896393B2 (en) | 2014-06-13 | 2018-02-20 | Exxonmobil Chemical Patents Inc. | Process for preparing dialkylbiphenyl isomer mixtures |
| WO2022102600A1 (ja) * | 2020-11-13 | 2022-05-19 | Eneos株式会社 | シクロヘキシルベンゼンの製造方法及びこれを用いたシクロヘキシルベンゼン組成物 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2976580B1 (fr) * | 2011-06-14 | 2013-05-31 | Coatex Sas | Epaississants non ioniques associatifs contenant des alkyls cyclohexylols, formulations les contenant et leurs utilisations. |
| FR2976579B1 (fr) * | 2011-06-14 | 2013-07-05 | Coatex Sas | Epaississants non ioniques associatifs contenant des alkyls cyclohexylols, formulations les contenant et leurs utilisations |
| US10550050B2 (en) * | 2017-02-01 | 2020-02-04 | Exxonmobil Research And Engineering Company | Processes for separating dimethyl biphenyl isomers using zeolite adsorbents |
| CN113376262A (zh) * | 2020-03-10 | 2021-09-10 | 中海油能源发展股份有限公司 | 一种气质联用分离检测全氟环烷烃的方法及其应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4911860A (ja) * | 1972-05-15 | 1974-02-01 | ||
| JPH0820548A (ja) * | 1994-07-07 | 1996-01-23 | Mitsubishi Gas Chem Co Inc | ビフェニル化合物の製造法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3681316A (en) * | 1968-08-07 | 1972-08-01 | Hercules Inc | T-butyl carbocyclic substituted cumene peroxides as curing agents |
| JPS5528904A (en) * | 1978-08-18 | 1980-02-29 | Mitsubishi Petrochem Co Ltd | Preparation of biphenyl derivative |
| US4879061A (en) * | 1986-09-29 | 1989-11-07 | Crystaloid Electronics Co. | Liquid crystalline materials and method of making same |
| DE4125261A1 (de) * | 1991-07-31 | 1993-02-04 | Huels Chemische Werke Ag | Verwendung von substituierten biphenylen als waermeuebertragungsfluessigkeiten |
| US5189234A (en) * | 1991-10-31 | 1993-02-23 | Amoco Corporation | Selective dehydrogenation processes and catalysts |
| NL9201932A (nl) * | 1992-11-05 | 1994-06-01 | Dsm Nv | Werkwijze voor de bereiding van een alkylbenzeen. |
-
2006
- 2006-07-25 WO PCT/JP2006/314693 patent/WO2007013469A1/ja not_active Ceased
- 2006-07-25 JP JP2007528485A patent/JPWO2007013469A1/ja active Pending
- 2006-07-25 CN CNA200680026983XA patent/CN101228102A/zh active Pending
- 2006-07-25 US US11/996,147 patent/US20090299111A1/en not_active Abandoned
- 2006-07-25 EP EP06781605A patent/EP1908743A4/en not_active Withdrawn
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4911860A (ja) * | 1972-05-15 | 1974-02-01 | ||
| JPH0820548A (ja) * | 1994-07-07 | 1996-01-23 | Mitsubishi Gas Chem Co Inc | ビフェニル化合物の製造法 |
Non-Patent Citations (11)
| Title |
|---|
| AZERB. KHIM. ZH., vol. 67, no. 1, pages 69 - 71 |
| AZERBAIDZHANSKII KHIMICHESKII ZHURNAL, no. 3, 1975, pages 37 - 40, XP003003646 * |
| DATABASE CAPLUS [online] LIPOVICH V.G. ET AL.: "Alkylation of benzene by 3-methylcyclohexanol in the presence of aluminum chloride and additives of chlorides of different metal salts", XP003003650, Database accession no. (85135) * |
| DATABASE CAPLUS [online] LIPOVICH V.G. ET AL.: "Alkylation of benzene with 4-methylcyclohexanol in the presence of sulfuric acid", XP003003649, Database accession no. (40020) * |
| DATABASE CAPLUS [online] OBUKHOVA T.A. ET AL.: "synthesis of p-(1-alkylcyclohexyl)benzoic acids", XP003003648, Database accession no. (32475) * |
| H. ROBSON: "Verified Syntheses of Zeoritic Materials", ELSEVIER SCIENCE, pages: 93 |
| IZVESTIYA ADAKEMII NAUK SSSR, SERIYA KHIMICHESKAYA, no. 1, 1990, pages 121 - 125 * |
| See also references of EP1908743A4 |
| SINGH P.K. ET AL., INDIAN J. CHEM., SECT. B: ORG. CHEM. INCL. MED. CHEM., vol. 24B, no. 6, 1985, pages 651 - 653, XP003003647 * |
| ZHURNAL OBSHCHEI KHIMII, vol. 51, no. 11, 1981, pages 2517 - 2520 * |
| ZHURNAL ORGANICHESKOI KHIMII, vol. 22, no. 2, 1986, pages 398 - 400 * |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140009563A (ko) * | 2011-05-18 | 2014-01-22 | 우베 고산 가부시키가이샤 | 3,3'',4,4''-테트라알킬 시클로헥실벤젠 및 그 제조 방법 |
| KR101581101B1 (ko) | 2011-05-18 | 2015-12-29 | 우베 고산 가부시키가이샤 | 3,3'',4,4''-테트라알킬 시클로헥실벤젠 및 그 제조 방법 |
| WO2012157749A1 (ja) * | 2011-05-18 | 2012-11-22 | 宇部興産株式会社 | 3,3',4,4'-テトラアルキルシクロヘキシルベンゼン及びその製造方法 |
| JP5979140B2 (ja) * | 2011-05-18 | 2016-08-24 | 宇部興産株式会社 | 3,3’,4,4’−テトラアルキルシクロヘキシルベンゼン及びその製造方法 |
| US9522857B2 (en) | 2011-05-18 | 2016-12-20 | Ube Industries, Ltd. | 3,3′,4,4′-tetraalkyl cyclohexylbenzene and method for producing same |
| WO2014078652A1 (en) | 2012-11-16 | 2014-05-22 | Zs Genetics, Inc. | Heavy atom labeled nucleosides, nucleotides, and nucleic acid polymers, and uses thereof |
| US9534104B2 (en) | 2013-01-28 | 2017-01-03 | Exxonmobil Chemical Patents Inc. | Plasticizer blends and use thereof |
| US9085669B2 (en) | 2013-01-28 | 2015-07-21 | Exxonmobil Chemical Patents Inc. | Alkyl aromatic hydroalkylation for the production of plasticizers |
| US9321898B2 (en) | 2013-01-28 | 2016-04-26 | Exxonmobil Chemical Patents Inc. | Alkyl aromatic hydroalkylation for the production of plasticizers |
| US9556087B2 (en) | 2013-03-14 | 2017-01-31 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9328053B2 (en) | 2013-03-14 | 2016-05-03 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9580572B2 (en) | 2013-03-14 | 2017-02-28 | Exxonmobil Chemical Patents Inc. | (Methylcyclohexyl)toluene isomer mixtures,their production and their use in the manufacture of plasticizers |
| US9663417B2 (en) | 2013-03-14 | 2017-05-30 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9688602B2 (en) | 2013-03-14 | 2017-06-27 | Exxonmobil Chemical Patents Inc. | Methyl-substituted biphenyl compounds, their production and their use in the manufacture of plasticizers |
| US9725377B2 (en) | 2013-03-14 | 2017-08-08 | Exxonmobil Chemical Patents Inc. | Hydroalkylation catalyst and process for use thereof |
| US9896393B2 (en) | 2014-06-13 | 2018-02-20 | Exxonmobil Chemical Patents Inc. | Process for preparing dialkylbiphenyl isomer mixtures |
| US9758447B2 (en) | 2014-10-24 | 2017-09-12 | Exxonmobil Chemical Patents Inc. | Activation of dehydrogenation catalysts |
| US9856186B2 (en) | 2014-12-19 | 2018-01-02 | Exxonmobil Chemical Patents Inc. | Production and use of dialkylbiphenyl isomer mixtures |
| WO2022102600A1 (ja) * | 2020-11-13 | 2022-05-19 | Eneos株式会社 | シクロヘキシルベンゼンの製造方法及びこれを用いたシクロヘキシルベンゼン組成物 |
| JPWO2022102600A1 (ja) * | 2020-11-13 | 2022-05-19 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1908743A1 (en) | 2008-04-09 |
| JPWO2007013469A1 (ja) | 2009-02-05 |
| EP1908743A4 (en) | 2010-09-29 |
| CN101228102A (zh) | 2008-07-23 |
| US20090299111A1 (en) | 2009-12-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007013469A1 (ja) | (アルキルフェニル)アルキルシクロヘキサン及び(アルキルフェニル)アルキルシクロヘキサン、アルキルビフェニルの製造方法 | |
| JP6003643B2 (ja) | アルキル化反応用触媒及び該触媒を用いたアルキル芳香族炭化水素化合物の製造方法 | |
| RU2525122C2 (ru) | Способ алкилирования бензола изопропиловым спиртом или смесью изопропилового спирта и пропилена | |
| US7193093B2 (en) | Process for producing alkylene oxide | |
| JP5979140B2 (ja) | 3,3’,4,4’−テトラアルキルシクロヘキシルベンゼン及びその製造方法 | |
| EP0202752B1 (en) | Alkylation process | |
| JPH0820548A (ja) | ビフェニル化合物の製造法 | |
| RU2340587C2 (ru) | Способ получения алкилбензола | |
| ITMI992171A1 (it) | Processo per preparare 2,6-dimetilnaftalene | |
| JP2756828B2 (ja) | p―イソブチルエチルベンゼンの製造方法 | |
| JP2018509461A (ja) | トランスアルキル化シクロヘキシルベンジル及びビフェニル化合物 | |
| JP2975702B2 (ja) | アルキル置換芳香族化合物の製造方法 | |
| US5550284A (en) | Production method for alkoxynaphthalenecarboxylic acid | |
| JP2805107B2 (ja) | 1,5―ジアルキルナフタレンの製造方法 | |
| EP0405508A2 (en) | Process for producing pyromellitic dianhydride | |
| JP4336045B2 (ja) | 改良された芳香族化合物の製造方法 | |
| JP4059531B2 (ja) | アルコキシナフタレンカルボン酸の製造方法 | |
| JP3110531B2 (ja) | アルキル置換芳香族炭化水素の製造方法 | |
| JP4452554B2 (ja) | 単環芳香族炭化水素−スチレン類付加体の製造方法 | |
| JP2980758B2 (ja) | アルケニルベンゼン及びその誘導体の製造方法 | |
| JPH0798769B2 (ja) | α―(4―イソブチルフェニル)プロピオンアルデヒドの製造方法 | |
| JP2980760B2 (ja) | アルケニルベンゼン及びその誘導体の製造方法 | |
| JPWO2000010947A1 (ja) | 改良された芳香族化合物の製造方法 | |
| JPH05140003A (ja) | アルキル置換芳香族炭化水素の製造方法 | |
| KR20100077501A (ko) | 고수율의 1,5-디메틸나프탈렌의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680026983.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007528485 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006781605 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11996147 Country of ref document: US |