WO2007057265A2 - Triazinverbindungen mit aminogruppen- und carboxygruppen-haltigen substituenten - Google Patents
Triazinverbindungen mit aminogruppen- und carboxygruppen-haltigen substituenten Download PDFInfo
- Publication number
- WO2007057265A2 WO2007057265A2 PCT/EP2006/067401 EP2006067401W WO2007057265A2 WO 2007057265 A2 WO2007057265 A2 WO 2007057265A2 EP 2006067401 W EP2006067401 W EP 2006067401W WO 2007057265 A2 WO2007057265 A2 WO 2007057265A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- hydrogen
- mol
- triazine compounds
- substituent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1nc(*)nc(I)n1 Chemical compound *c1nc(*)nc(I)n1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D251/00—Heterocyclic compounds containing 1,3,5-triazine rings
- C07D251/02—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings
- C07D251/12—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D251/26—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with only hetero atoms directly attached to ring carbon atoms
- C07D251/40—Nitrogen atoms
- C07D251/54—Three nitrogen atoms
- C07D251/70—Other substituted melamines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the invention relates to triazine compounds which have both amino and carboxy-containing substituents, and to a process for their preparation.
- EP 0 466 647 describes a process for the photochemical and thermal stabilization of polyamide fiber materials and water-soluble triazine derivatives according to the formula I.
- EP 0 702 011 also describes a process for the photochemical and thermal stabilization of polyamide fiber materials and dyeings.
- water-soluble piperidine triazine compounds of formula III are also described.
- reactive stabilizers are understood to mean that the stabilizers according to the invention, by virtue of their amino- and carboxy-containing substituents, are capable of being incorporated into the polymer and thus are a building block in the polymer chain.
- This has the advantage over stabilizers according to the prior art that the stabilizers according to the invention can already be added during the polymerization and incorporated into the polymer chain. Thus, the additional reaction step for the admixture of the stabilizer to the polymer may be omitted.
- these stabilizers according to the invention have the advantage that they can not be dissolved out of the polymer and thus a permanently effective stabilizer is available.
- the solution of the problem was all the more surprising, especially since it was found that this compound can be prepared according to an economical process.
- the invention relates to triazine compounds of the formula (1)
- R 5 hydrogen or alkyl group
- Aryl group which is respectively substituted or unsubstituted.
- R 5 hydrogen or alkyl group
- composition which is characterized in that the composition comprises at least two different triazine compounds according to the formula (1).
- subject of this invention is a solution which is characterized in that the solution comprises at least one triazine compound according to the formula (1).
- the triazine compounds according to the invention have a structure according to the formula (1)
- R4 hydrogen or alkyl group
- R 5 hydrogen or alkyl group
- Aryl group which is in each case substituted or unsubstituted, is on.
- the structural fragment A in the Ri-type substituent of the triazine compounds of the invention may be both -O- and -NR 4 -, wherein the structural fragment A in the Ri and R 3 substituents may be the same or different. This is preferred
- the substituent of the R 4 type may be both hydrogen and an alkyl group.
- the substituent of type R 4 is hydrogen or a
- This alkyl group of the structural fragment A may be branched or unbranched, but is preferably unbranched. Furthermore, this alkyl group is preferably unsubstituted.
- the substituent of the type R 4 is particularly preferably hydrogen.
- the substituent of type B is an amino group-containing substituent, wherein the amino group may be on an aliphatic skeleton or the amino group may be an aliphatic cyclic amine.
- the B-type substituent comprises an aliphatic cyclic amine.
- the triazine compounds according to the invention preferably have a substituent of the B type according to the formula (5)
- Rn hydrogen, alkyl, cycloalkyl or heterocycloalkyl group and R 1 and R n are identical or different,
- the substituents of the Rio and Rn type may be identical or different and are preferably hydrogen, an alkyl, a cycloalkyl or a heterocycloalkyl group, in particular each having a carbon number of from 1 to 20, preferably from 2 to 10 or a number of carbon atoms and heteroatoms of 1 to 20, preferably of 2 to 10.
- This alkyl group of the substituents of the type of Rio and Rn are preferably branched or unbranched, but are preferably unbranched. Further, they are preferably unsubstituted or substituted with an amino group, but are preferably unsubstituted.
- the cycloalkyl group of the substituents of the type R 1 and R n is preferably unsubstituted or substituted, in particular this cycloalkyl group is unsubstituted.
- the heterocycloalkyl group of the substituents of the type Ri 0 and Rn is preferably unsubstituted or substituted, this heterocycloalkyl group is preferably substituted by one or more methyl groups, preferably it is a heterocycloalkyl group having as heteroatom one or more nitrogen atoms, preferably it is a heterocycloalkyl group according to the formula (5).
- the substituent of the type Rg is preferably hydrogen or an alkyl group having a carbon number of 1 to 16, preferably 1 to 8 or an alkoxy group having a branched or unbranched alkyl group or a cycloalky group, more preferably the substituent of the type R 9 is hydrogen.
- the triazine compounds according to the invention preferably have a substituent of the B type according to the formula (6).
- the triazine compound according to the invention particularly preferably has a substituent of the B type according to the formulas (6a) or (6b):
- these have a substituent of the type B according to the formula (5).
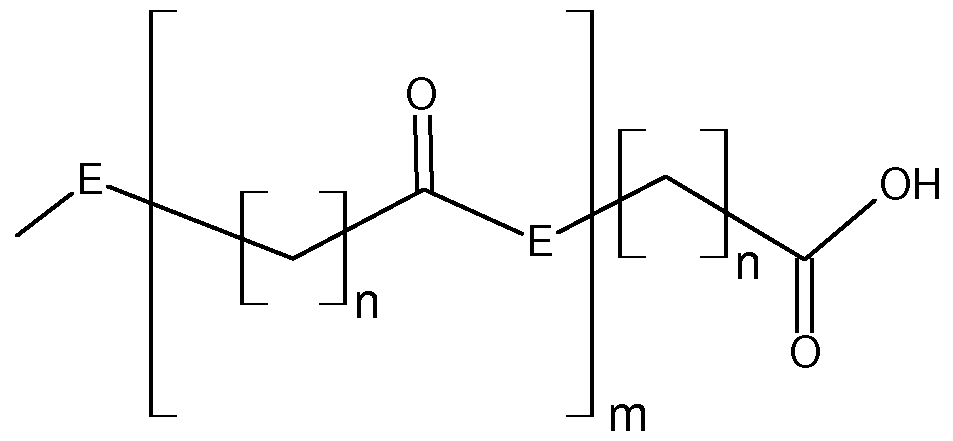
- the substituents of type R 2 in the triazine compounds of the invention preferably have a structural fragment E with -O- or -NR 5 -, wherein the substituent of type R 5 is preferably hydrogen or an alkyl group having a carbon number of 1 to 16, preferably from 1 to 4, preferably the substituent of type R 5 is hydrogen.
- the alkyl group of the substituent R 5 is branched or unbranched, but is preferably unbranched. Further, this alkyl group of the substituent R 5 is preferably unsubstituted.
- n is preferably from 3 to 15, preferably from 5 to 11, and more preferably 5, and m is preferably from 0 to 10, preferably from 0 to 4, and more preferably m is 0.
- the triazine compounds according to the invention preferably have, as substituents of the R 3 type, a substituent of the Ri type.
- the triazine compounds according to the invention have, as substituents of the R 3 type, a substituent of the R 2 type.
- the two substituents of the type Ri or R 2 can be identical or different, preferably the substituents of the same type are identical.
- the triazine compounds according to the invention have a substituent of the Ri type according to the following structures (7a), (7b) or (7c):
- the substituent of type R 3 is preferably a substituent of the Ri type, and more preferably these two substituents are identical.
- the substituent of type R 3 in this preferred embodiment may also be a substituent of the type R 2 , and here too these two substituents are preferably identical.
- R 6 , R 7 and R 8 is hydrogen, alkyl or aryl group, wherein the alkyl or aryl group unsubstituted or with one or more substituents of the formula R 6 , R 7 , R 8 , -SO 3 H or -SO 3 M where M is an alkali metal cation, preferably a lithium, sodium or potassium cation is substituted.
- the alkyl and / or aryl groups of the substituents of the R 6 , R 7 or R 8 type preferably have from 1 to 4, particularly preferably from 1 to 2 and very particularly preferably a substituent of the formula -SO 3 H or -SO 3 M ,
- R 6 , R 7 and R 9 substituents are hydrogen or an alkyl group having a carbon number of 1 to 18, preferably 2 to 16.
- This alkyl group of the substituent R 6 , R 7 and R 5 may be branched or unbranched, is preferred However, this is unbranched.
- the aryl group of the substituents R 6 , R 7 and Rs is preferably a phenyl group or a phenyl group monosubstituted with -SO 3 H or -SO 3 M, preferably in the para position.
- the substituents of the type R 6 , R 7 and R 8 may all be identical, all different or even in pairs identical.
- the substituents of the R 6 , R 7 and Rg type are preferably hydrogen, phenyl or a phenyl group which is monosubstituted with -SO 3 H or -SO 3 M in the para position.
- R 4 hydrogen or alkyl group
- R 5 hydrogen or alkyl group
- Aryl group which is in each case substituted or unsubstituted, is
- the substituent of type R 5 may be both hydrogen and an alkyl group.
- the substituent of type R 5 is hydrogen or an alkyl group having a
- This alkyl group of the substituent of type R 5 may be branched or unbranched, but is preferably unbranched.
- this alkyl group of the substituent R 5 is preferably unsubstituted.
- R 5 is preferably hydrogen.
- o is preferably from 0 to 12, preferably from 2 to 8, and particularly preferably 2.
- lactams or lactones preference is given to using lactams.
- Caprolactam is particularly preferably used in the process according to the invention.
- a compound according to the formula (4) is used, particularly preferably sodium aminocapronate is used.
- the starting material according to formula (4) is formed in situ, for example by the use of a compound of the formula
- the two starting materials for this purpose in a molar ratio of 5: 1 to 1: 1, preferably from 4: 1 to 1: 1, more preferably from 2: 1 to 1: 1 used.
- an additional starting material is preferably an amine according to formula (2), wherein the structural fragment A can be both -O- and -NR 4 -.
- the structural fragment A can be both -O- and -NR 4 -.
- the substituent of the R 4 type may be both hydrogen and an alkyl group.
- the R 4 substituent is hydrogen or an alkyl group having a carbon number of 1 to 10, preferably 2 to 5.
- This alkyl group of the structural fragment A may be branched or unbranched, but is preferably unbranched.
- this alkyl group is preferably unsubstituted.
- the substituent of the type R 4 is particularly preferably hydrogen.
- an amine according to the formula (2) which preferably has a substituent of type B as an amino group-containing substituent, where the amino group can be on an aliphatic skeleton or the amino group can be an aliphatic cyclic amine.
- this type B substituent comprises an aliphatic cyclic amine.
- the substituents of the type R 1 and R n may be identical or different and are preferably hydrogen, an alkyl, a cycloalkyl or a heterocycloalkyl group each having a carbon number of 1 to 20, preferably 2 to 10 or a number of carbon atoms and heteroatoms of 1 to 20, preferably from 2 to 10.
- This alkyl group of the substituents of the type Ri 0 and Rn are preferably branched or unbranched, but these are preferably unbranched. Further, they are preferable unsubstituted or substituted with an amino group, but it is preferably unsubstituted.
- the cycloalkyl group of the substituents of the type R 1 and R n is preferably unsubstituted or substituted, in particular this cycloalkyl group is unsubstituted.
- the heterocycloalkyl group of the substituents of the type R 1 and R n is preferably unsubstituted or substituted, this heterocycloalkyl group is preferably substituted by one or more methyl groups, preferably a heterocycloalkyl group which has one or more nitrogen atoms as the heteroatom, preferably a heterocycloalkyl group according to the formula ( 5).
- the substituent of the R 9 type is preferably hydrogen or an alkyl group having a carbon number of 1 to 16, preferably 1 to 8, or an alkoxy group having a branched or unbranched alkyl group or a cycloalkyl group, more preferably the substituent of the R 9 type is hydrogen.
- R 6 , R 7 and Rg is hydrogen, alkyl or aryl group, wherein the alkyl or aryl group unsubstituted or with one or more substituents of the formula R 6 , R 7 , Rg, -SO 3 H or -SO 3 M, wherein M is an alkali metal cation, preferably a lithium, sodium or potassium cation is substituted.
- the alkyl and / or aryl groups of the substituents of the R 6 , R 7 or R 8 type preferably have from 1 to 4, particularly preferably from 1 to 2 and very particularly preferably a substituent of the formula -SO 3 H or -SO 3 M ,
- the substituents R 6 , R 7 and R 8 are hydrogen or an alkyl group having a carbon number of 1 to 18, preferably 2 to 16.
- This alkyl group of the substituent R 6 , R 7 and R 8 may be branched or unbranched However, this is preferably unbranched.
- the aryl group of the substituents of the R 6 , R 7 and R 8 type is preferably a phenyl group or a phenyl group monosubstituted with -SO 3 H or -SO 3 M, preferably in the para position.
- the substituents of the type R 6 , R 7 and R 8 may all be identical, all different or even in pairs identical.
- the substituents of the R 6 , R 7 and R 8 type are preferably hydrogen, phenyl or a phenyl group which is monosubstituted with -SO 3 H or -SO 3 M in the para position.
- the process according to the invention may consist of two or three reaction steps for the actual reaction or reaction.
- cyanuric chloride in a first reaction step, is reacted with an amine according to formula (2) in the presence of a base in a solvent.
- the base used is preferably sodium hydroxide solution.
- a molar ratio of amine to base of 1: 1 is used here.
- a solvent selected from water, aromatic hydrocarbons, especially toluene, xylene, alkanes, ethers, ketones, such as acetone, or esters may be used, preferably water is used as the solvent.
- alcohols, primary or secondary amines as solvents.
- the reaction then takes place with a compound selected from compounds of the formula (3) or (4).
- cyanuric chloride in a first reaction step is reacted with a compound selected from the compounds of the formula (3) or (4) and subsequently in a further reaction step with an amine of the formula (2).
- the solvents and the molar ratio of amine to base can be selected analogously to the preferred embodiment.
- a base is preferably added with respect to the ring opening.
- the solvents used here are, in particular, water, toluene, xylene, alkanes, ethers, ketones, for example acetone, or esters, but preferably water.
- a lactam is used as the solvent, in particular a compound according to formula (3) is used here as solvent, particularly preferably the same compound is used as solvent and reactant.
- the cyanuric chloride is preferably from 0.5 to 3, preferably from 1 to 2 molar equivalents reacted to the amount of cyanuric chloride to reactant A, wherein as reactant A either an amine according to formula (2) or a compound selected from compounds of formula (3) or (4) can be used.
- the intermediate obtained is then reacted with from 0.5 to 5, preferably from 1 to 4, mole equivalents of reactant B relative to the amount of cyanuric chloride, using as reactant B a compound selected from compounds of the formula ( 3) or (4), is used when reactant A is an amine according to formula (2), or an amine according to formula (2), if as reactant A is a compound selected from compounds of formula (3) or (4).
- the first reaction step of the process according to the invention is preferably carried out at a temperature of -20 to 100 ° C., preferably at a temperature of -10 to 80 ° C., and more preferably from 0 to 60 ° C.
- the second reaction step is preferably carried out at a temperature of 0 to 200 0 C, preferably at 10 to 180 0 C and particularly preferably at a temperature of 20 to 170 0 C.
- the first reaction step of the method according to the invention is preferably in a Pressure of 0.5 to 1.5 bar, preferably at a pressure of 0.8 to 1.2 bar and more preferably carried out at atmospheric pressure.
- the second reaction step is preferably carried out at a pressure of 1 to 11 bar, preferably at a pressure of 1 to 9 bar and more preferably at a pressure of 1 to 8 bar.
- cyanuric chloride in a first reaction step, is reacted with a compound selected from compounds of the formula (3) or (4) in a solvent.
- the reaction then takes place with an amine according to the formula (2).
- the cyanuric chloride in the first reaction step, is reacted with from 1 to 3, preferably with 2 molar equivalents based on the amount of cyanuric chloride to reactant A and in a second reaction step, the resulting intermediate with from 0.5 to 5, preferably from 1 to 3 molar equivalents based on the amount of cyanuric chloride to reactant B reacted.
- the first reaction step is preferably carried out at a temperature of 0 to 100 0 C, preferably at a temperature of 10 to 80 0 C and particularly preferably from 20 to 60 0 C.
- the second reaction step is preferably carried out at a temperature of 80 to 200 0 C, preferably at 90 to 180 0 C and particularly preferably at a temperature of 100 to 170 0 C.
- the intermediate obtained is then reacted with from 0.5 to 2, preferably 1 molar equivalents based on the amount of cyanuric chloride to Reaktant B, wherein as reactant B, a compound selected from compounds according to formulas (3) , (4), (16) or (17) is used when a compound of the formula (2) is used as reactant A, or a compound selected from compounds of the formula (2), (16) or (17) is used, if as reactant A a compound, selected from compounds of the formula (3) or (4), or a compound selected from compounds of the formula (2), (3) or (4) is used, when as reactant A a compound selected from compounds according to Formula (16) or (17) is used.
- reactant B a compound selected from compounds according to formulas (3) , (4), (16) or (17) is used when a compound of the formula (2) is used as reactant A, or a compound selected from compounds of the formula (2), (16) or (17) is used.
- the resulting intermediate is then reacted with from 0.5 to 2, preferably 1 molar equivalents based on the amount of cyanuric chloride to Reactant C, wherein as reactant C is a compound selected from compounds according to formulas (16) or (17), when reactants A and B compounds of formula (2) and
- the first reaction step of this particular embodiment of the process according to the invention is preferably carried out at a temperature of -20 to 80 ° C., preferably at a temperature of -10 to 60 ° C. and more preferably from 0 to 40 ° C.
- the second reaction step is preferably carried out at a temperature of 0 to 100 0 C, preferably at 10 to 80 0 C and particularly preferably at a temperature of 20 to 60 0 C.
- the third reaction step is preferably carried out at a temperature of 80 to 200 0 C, preferably at 90 to 180 0 C and particularly preferably at a temperature of 100 to 170 0 C.
- the first and second reaction steps of this particular embodiment of the invention is preferably carried out at a pressure of 0.5 to 1.5 bar, preferably at a pressure of 0.8 to 1.2 bar and particularly preferably at atmospheric pressure.
- the third reaction step is preferably carried out at a pressure of 1 to 11 bar, preferably at a pressure of 1 to 9 bar and more preferably at a pressure of 1 to 8 bar.
- the individual reaction steps of the process according to the invention can be carried out in each case in one process step of the process according to the invention, wherein the respective resulting intermediates can be separated and isolated and thus used as starting material for the subsequent process step.
- the intermediates of the respective process stage are not separated and isolated - with the exception of the last stage of the process - but fed directly to the next process stage as starting material.
- the formation of the intermediates thus takes place "in situ”.
- all the reaction steps are carried out in a reactor, in particular in an autoclave.
- the reaction of the compounds of the formula (3) with a base can be carried out in a separate process stage or in a separate reaction vessel.
- the reaction of the compounds of the formula (3) with a base can also be carried out in the same reaction vessel, so that all the reaction steps of the process according to the invention are carried out in the same reaction vessel. In this way, all three substitutions on the triazine ring of the process according to the invention can be carried out in a reaction vessel.
- reaction of the compounds of the formula (3) with a base can be carried out in a separate process stage or in a separate reaction vessel.
- reaction of the compounds of the formula (3) with a base can also be carried out in the same reaction vessel in which the particular reaction step is also taking place.
- the workup of the reaction mixture is used mainly to sodium chloride as Separate reaction by-product.
- the sodium chloride is dissolved in water and removed by filtration of the aqueous suspensions and subsequent washing of the filter cake or by extraction of the target product with an organic solvent, preferably the organic solvent used during the reaction.
- the separation of the likewise water-soluble sodium chloride is preferably carried out by means of electrodialysis via membranes or by ion exchange chromatography, preferably the separation of the sodium chloride is carried out by means of ion exchange chromatography.
- reaction mixture worked up in this way as a solution in water or in an organic solvent can then be used directly, in a particular embodiment drying takes place before use as a stabilizer.
- the intermediates occurring after the individual reaction steps can be isolated and purified from the reaction mixture in a particular embodiment of the process according to the invention. This is preferably done by crystallization, filtration and optionally a wash from the reaction mixture.
- the isolation and purification of these intermediates can also be carried out by extraction with an organic solvent, preferably with the organic solvent that has already been used for the reaction.
- the thus isolated and purified intermediates, which are usually present as solids, can then be used in the next reaction step of the process according to the invention.
- compositions of the invention have at least two different triazine compounds according to the formula (1).
- composition of the invention 85 to 95% by weight of triazine compounds of the formula (18) according to the invention,
- this composition can be obtained if the starting materials cyanuric chloride, amine of formula (2) and a compound according to the formula (3) or (4) in a molar ratio of 1: 2: 1 are used in the process according to the invention.
- composition according to the invention has
- This composition can be obtained if the educts cyanuric chloride, amine of the formula (2), a compound according to the formula (3) or (4) and a compound according to the formula (16) or (17) in a molar ratio of 1: ( from 0.5 to 2): (from 0.5 to 2): (from 0.5 to 5), preferably in a molar amount Ratio of 1: 1: 1: (from 1 to 4) can be used in the inventive method.
- the solution according to the invention has at least one triazine compound according to the formula (1), preferably it has from 1 to 50% by weight, preferably from 20 to 45% by weight
- Hydrocarbons in particular toluene, xylene, alkanes, ethers, ketones, such as
- Acetone, or esters are used, or the compound according to the formula (3) used for the preparation of the triazine compounds according to the invention.
- the solution according to the invention preferably has water as solvent.
- the triazine compounds according to the invention can be used for the stabilization of polyamides.
- Suitable polyamides are primarily aliphatic homo- and copolycondensates, for example PA 46, PA 66, PA 68, PA 610, PA 612, PA 410, PA 810, PA 1010, PA 412, PA 1012, PA 1212, PA6, PA 7, PA 8, PA 9, PA 10, PA 11 and PA 12.
- the triazine compounds according to the invention are preferred for stabilizing PA 410, PA 810, PA 1010, PA 412, PA 1012, PA 1212, PA 6, PA 7, PA 8, PA 9, PA 10, PA 11 and PA 12 used.
- the triazine compounds of the invention can be used for the stabilization of polyester.
- they can be used to stabilize polyesters prepared by polycondensation of diols with dicarboxylic acid or its polyester-forming derivatives such as dimethyl esters.
- diols the formula HO-R-OH, where R is a divalent, branched or unbranched aliphatic and / or cycloaliphatic radical having 2 to 18, preferably 2 to 12, carbon atoms.
- Suitable dicarboxylic acids have the formula HOOC-R'-COOH, where R 'is a divalent aliphatic, cycloaliphatic or aromatic radical having 2 to 18, preferably 4 to 12, carbon atoms.
- the preparation of these polyesters belongs to the prior art (DE-OSS 24 07 155, 24 07 156, Ullmann's Encyclopedia of Industrial Chemistry, 4th ed., Vol. 19, pages 65 ff., Verlag Chemie, Weinheim, 1980).
- the triazine compounds according to the invention can be added already during the polycondensation, this has the advantage that the polyamides thus prepared contain the additives which improve the mechanical strength, the stability against oxidative and light-induced degradation and dyeability covalently and thus permanently bound.
- composition of the products was carried out by means of HPLC and a CLND detector (Chemiluminescence Nitrogen Detector). This type of detector allows equimolar detection of nitrogenous compounds.
- Example 1.2 a solution of sodium aminocaproate (1.0 mol) melted at 110 ° C. was prepared in ⁇ -caprolactam (4 mol). After addition of 536.3 g of the intermediate prepared according to Example 3.1 (1.0 mol) was heated at atmospheric pressure and 128 to 136 0 C under reflux for 3 hours. After crystallization, filtration, washing by means of ethyl acetate and drying analogously to Example 1.2, 670.8 g of crude product are obtained (91%, based on the amount of intermediate used from Example 3.1).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Treatments For Attaching Organic Compounds To Fibrous Goods (AREA)
Abstract
Description



































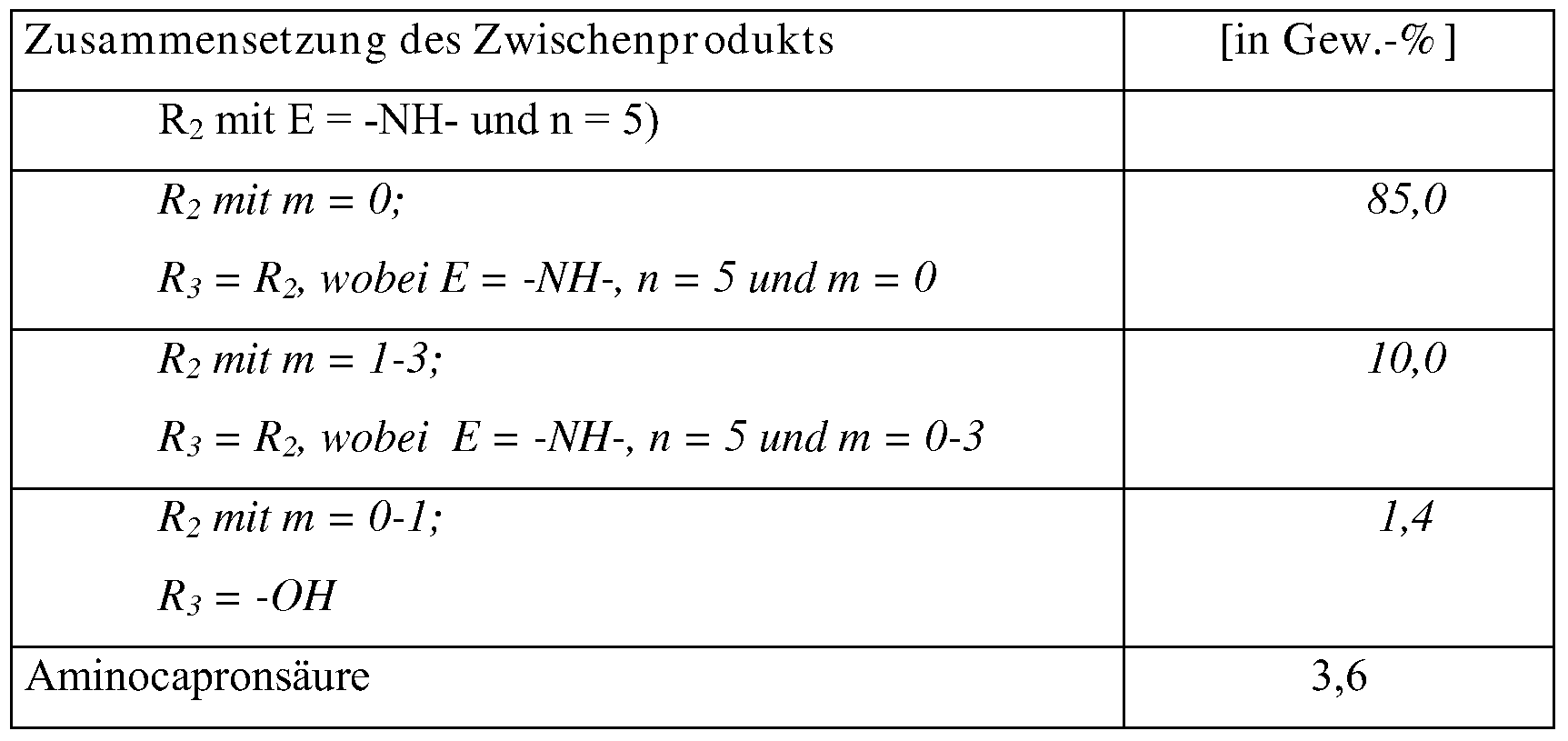


















Claims











Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/090,541 US20080251758A1 (en) | 2005-11-17 | 2006-10-13 | Triazine Compounds Comprising Substituents Containing Amino Groups and Carboxyl Groups |
| JP2008540551A JP2009515923A (ja) | 2005-11-17 | 2006-10-13 | アミノ基及びカルボキシ基含有置換基を有するトリアジン化合物 |
| EP06807263A EP1948640A2 (de) | 2005-11-17 | 2006-10-13 | Triazinverbindungen mit aminogruppen- und carboxygruppen-haltigen substituenten |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05110842A EP1787989A1 (de) | 2005-11-17 | 2005-11-17 | Triazinverbindungen mit Aminogruppen- und Carboxygruppen-haltigen Substituenten |
| EP05110842.1 | 2005-11-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007057265A2 true WO2007057265A2 (de) | 2007-05-24 |
| WO2007057265A3 WO2007057265A3 (de) | 2008-12-24 |
Family
ID=36087844
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/067401 Ceased WO2007057265A2 (de) | 2005-11-17 | 2006-10-13 | Triazinverbindungen mit aminogruppen- und carboxygruppen-haltigen substituenten |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20080251758A1 (de) |
| EP (2) | EP1787989A1 (de) |
| JP (1) | JP2009515923A (de) |
| KR (1) | KR20080058493A (de) |
| CN (1) | CN101466700A (de) |
| TW (1) | TW200730521A (de) |
| WO (1) | WO2007057265A2 (de) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012055965A1 (en) | 2010-10-28 | 2012-05-03 | 3V Sigma S.P.A. | New sterically hindered polymeric amines and their use as polymer stabilizers |
| WO2012153260A1 (en) | 2011-05-10 | 2012-11-15 | 3V Sigma S.P.A. | Mixture of sterically hindered amines for polymer stabilization |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102005036693A1 (de) * | 2005-08-04 | 2007-02-08 | Degussa Ag | Verfahren zur Herstellung von 2,4,6-Trimercapto-1,3,5-triazin |
| DE102008040045A1 (de) * | 2008-02-01 | 2009-08-06 | Evonik Degussa Gmbh | Verfahren zur Herstellung von 4-Amino-2,2,6,6-tetramethylpiperidin |
| MX2016000701A (es) | 2015-01-22 | 2016-11-24 | Evonik Degussa Gmbh | Proceso para preparar un compuesto de triacetonamina n-metil sustituida. |
| DE102016212378A1 (de) | 2016-07-07 | 2018-01-11 | Evonik Degussa Gmbh | Synthese von Triacetondiaminverbindungen durch reduktive Aminierung ausgehend von Triacetondiamin und dessen Derivaten |
| DE102016212379A1 (de) | 2016-07-07 | 2018-01-11 | Evonik Degussa Gmbh | Verfahren zur Herstellung einer N-methylsubstituierten Triacetonaminverbindung |
| CN110590689B (zh) * | 2019-10-11 | 2023-04-07 | 中北大学 | 一种具表面活性的缓蚀剂及其制备方法和应用 |
| CN114874107B (zh) * | 2022-07-12 | 2022-11-29 | 中山大学附属第七医院(深圳) | 一种氨基脂质及其制备方法和应用 |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1093318B (de) * | 1957-06-05 | 1960-11-24 | Bayer Ag | Aufhellungsmittel |
| US3700662A (en) * | 1968-08-28 | 1972-10-24 | Tatsuo Ishikawa | Triazine derivatives and compositions thereof |
| GB1595855A (en) * | 1977-05-09 | 1981-08-19 | Ciba Geigy Ag | Halogen-containing 1,3,5-triazine-dicarboxylic acid derivatives and their use as flameproofing agents in synthetic plastics |
| US4228281A (en) * | 1977-09-23 | 1980-10-14 | Ciba-Geigy Corporation | Dicarboxylic acids containing triazine rings |
| US4281103A (en) * | 1978-09-18 | 1981-07-28 | Ciba-Geigy Corporation | Dicarboxylic acids containing triazine rings, and polyesters formed from these dicarboxylic acids |
| US4883860A (en) * | 1988-12-15 | 1989-11-28 | Ici Americas Inc. | Triazine-based light stabilizers for plastics |
| US5019613A (en) * | 1989-03-21 | 1991-05-28 | Ciba-Geigy Corporation | N-hydrocarbyloxy derivatives of hindered amine-substituted S-triazines |
| EP0466647B1 (de) * | 1990-07-12 | 1995-11-29 | Ciba-Geigy Ag | Verfahren zur photochemischen und thermischen Stabilisierung von Polyamid-Fasermaterialien |
| US5216156A (en) * | 1992-05-05 | 1993-06-01 | Ciba-Geigy Corporation | Non-migrating 1-hydrocarbyloxy-2,2,6,6-tetramethylpiperidine 1,3,5-triazine derivatives |
| DE4219459A1 (de) * | 1992-06-13 | 1993-12-16 | Huels Chemische Werke Ag | Verfahren zur Herstellung von 2,2,6,6-Tetramethylpiperidin-N-oxyl und seinen in 4-Stellung substituierten Derivaten |
| JP3440544B2 (ja) * | 1994-04-11 | 2003-08-25 | 三菱化学株式会社 | 記録液 |
| DE19531995A1 (de) * | 1994-09-02 | 1996-03-07 | Ciba Geigy Ag | Verfahren zum photochemischen und thermischen Stabilisieren von Polyamid-Fasermaterialien |
| JP3486982B2 (ja) * | 1994-09-30 | 2004-01-13 | 三菱化学株式会社 | 記録液 |
| DE4442990A1 (de) * | 1994-12-02 | 1996-06-05 | Huels Chemische Werke Ag | Verfahren zur Herstellung von 4-Amino-2,2,6,6-tetramethylpiperidin |
| DE19532215B4 (de) * | 1995-09-01 | 2009-06-25 | Evonik Degussa Gmbh | Verfahren zur Herstellung von 4-Acylamino-2,2,6,6-tetramethylpiperidinen |
| DE19544599A1 (de) * | 1995-11-30 | 1997-06-05 | Huels Chemische Werke Ag | Kontinuierliches Verfahren zur Herstellung von 4-Amino-2,2,6,6-tetramethylpiperidin |
| BE1009976A3 (nl) * | 1996-01-19 | 1997-11-04 | Dsm Nv | Sterk vertakte polymeren. |
| GB9611614D0 (en) * | 1996-06-04 | 1996-08-07 | Ciba Geigy Ag | Process for inhibiting the effect of flourescent whitening agents |
| DE19704460A1 (de) * | 1997-02-06 | 1998-08-13 | Huels Chemische Werke Ag | Kontinuierliches Verfahren zur Herstellung von 4-Aminopiperidinen |
| SE512919C2 (sv) * | 1998-10-15 | 2000-06-05 | Borealis As | KOmbinerat ljus- och värmestabiliseringsmedel för polymerer |
| DE102004023640A1 (de) * | 2004-05-10 | 2005-12-08 | Degussa Ag | Verfahren zur Herstellung von 4-substituierten 2,2,6,6-Tetramethyl-piperidin-N-oxy- und 2,2,6,6-Tetramethyl-piperidin-N-hydroxy-verbindungen |
| US7030279B1 (en) * | 2004-12-23 | 2006-04-18 | Degussa Ag | Process for transition metal free catalytic aerobic oxidation of alcohols under mild conditions using stable free nitroxyl radicals |
| DE102006010347A1 (de) * | 2006-03-03 | 2007-09-06 | Degussa Gmbh | Polymerisationsinhibitor zur Stabilisierung von olefinisch ungesättigten Monomeren |
-
2005
- 2005-11-17 EP EP05110842A patent/EP1787989A1/de not_active Withdrawn
-
2006
- 2006-10-13 EP EP06807263A patent/EP1948640A2/de not_active Withdrawn
- 2006-10-13 WO PCT/EP2006/067401 patent/WO2007057265A2/de not_active Ceased
- 2006-10-13 US US12/090,541 patent/US20080251758A1/en not_active Abandoned
- 2006-10-13 CN CNA2006800009508A patent/CN101466700A/zh active Pending
- 2006-10-13 JP JP2008540551A patent/JP2009515923A/ja not_active Withdrawn
- 2006-10-13 KR KR1020087011760A patent/KR20080058493A/ko not_active Ceased
- 2006-11-13 TW TW095141924A patent/TW200730521A/zh unknown
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012055965A1 (en) | 2010-10-28 | 2012-05-03 | 3V Sigma S.P.A. | New sterically hindered polymeric amines and their use as polymer stabilizers |
| WO2012153260A1 (en) | 2011-05-10 | 2012-11-15 | 3V Sigma S.P.A. | Mixture of sterically hindered amines for polymer stabilization |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20080058493A (ko) | 2008-06-25 |
| CN101466700A (zh) | 2009-06-24 |
| TW200730521A (en) | 2007-08-16 |
| WO2007057265A3 (de) | 2008-12-24 |
| EP1787989A1 (de) | 2007-05-23 |
| US20080251758A1 (en) | 2008-10-16 |
| JP2009515923A (ja) | 2009-04-16 |
| EP1948640A2 (de) | 2008-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69233001T2 (de) | N-Halogenierte Säureamide | |
| DE3315115C2 (de) | ||
| EP0029522A1 (de) | s-Triazinderivate, ihre Herstellung aus Halogentriazinylaminen und Polyaminen, und ihre Verwendung als Stabilisatoren | |
| DE3236070A1 (de) | Piperidylderivate makrocyclischer triazinverbindungen mit stabilisierender wirkung auf polymere sowie verfahren zu deren herstellung | |
| DE3226889A1 (de) | Verfahren zur herstellung von bis(aminocyclohexyl) dialkylmethanen | |
| EP1948640A2 (de) | Triazinverbindungen mit aminogruppen- und carboxygruppen-haltigen substituenten | |
| EP0779280B1 (de) | Verfahren zur Herstellung von 2-(2,4-Dihydroxyphenyl)-4,6-bis-(2,4-dimethylphenyl)-s-triazin | |
| DE2453326A1 (de) | Halogensubstituierte benzimidazolonverbindungen und verfahren zu ihrer herstellung | |
| EP0072009A2 (de) | Polytriazinylalkohole, -ester und -urethane, ein Verfahren zu ihrer Herstellung und ihre Verwendung als Stabilisatoren | |
| DE1620236B2 (de) | N,n'-dilactamidsulfide | |
| DE1620638A1 (de) | Verfahren zur Herstellung von Dihydrotiazinen | |
| DE2166766C2 (de) | Verfahren zur Herstellung von polyfluorierten Alkylsulfonsäureamidaminen | |
| DE2505704C3 (de) | Verfahren zur Herstellung von gegebenenfalls substituierten 2-Alkylamino-4,6-dichlor-s-triazinen und 2,4-Bis alkylamino-6-chlor-s-triazinen | |
| DE19729358A1 (de) | Polyaminotriazine, ihre Herstellung und Verwendung | |
| CH621118A5 (de) | ||
| EP0648756A1 (de) | Verfahren zur Herstellung von Hydroxyoxaalkylmelaminen | |
| EP0304685B1 (de) | Verfahren zur Herstellung von N-alkylierten Caprolactamen | |
| EP0198795B1 (de) | Verfahren zur Herstellung von 1,4-Diaminoanthrachinonen | |
| DE2600202C2 (de) | Verfahren zur Herstellung von Benzodiazepinen | |
| DE69113090T2 (de) | Verfahren zur Herstellung von Meta-Aminophenolen aus Resorcin. | |
| DE1670042A1 (de) | Verfahren zur Herstellung von 2,4-Bis-alkylamino-6-chlor-s-triazinen | |
| DE2363851A1 (de) | Verfahren zur herstellung von unsymmetrisch substituierten alkanolamino-striazinen | |
| DE1245973B (de) | Verfahren zur Herstellung von aminosubstituierten l^-Dihydro-i-hydroxy-lJ.S-triazinen | |
| EP0722938B1 (de) | Verfahren zur Herstellung eines radikalisch polymerisierbaren UV-Absorbers auf Basis Benz-triazolylphenol | |
| DE1545761A1 (de) | Stickstoffhaltige organische Phosphorylderivate und Verfahren zu deren Herstellung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680000950.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006807263 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12090541 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087011760 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2008540551 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006807263 Country of ref document: EP |