WO2007063928A1 - 新規な非環状アミンカルボキシアミド誘導体及びその塩 - Google Patents
新規な非環状アミンカルボキシアミド誘導体及びその塩 Download PDFInfo
- Publication number
- WO2007063928A1 WO2007063928A1 PCT/JP2006/323889 JP2006323889W WO2007063928A1 WO 2007063928 A1 WO2007063928 A1 WO 2007063928A1 JP 2006323889 W JP2006323889 W JP 2006323889W WO 2007063928 A1 WO2007063928 A1 WO 2007063928A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- methyl
- added
- stirred
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/382—Heterocyclic compounds having sulfur as a ring hetero atom having six-membered rings, e.g. thioxanthenes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4468—Non condensed piperidines, e.g. piperocaine having a nitrogen directly attached in position 4, e.g. clebopride, fentanyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/28—Radicals substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D335/00—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom
- C07D335/02—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to a novel orally administrable or preventive agent for diabetes, and an acyclic amine carboxyamide derivative or a pharmaceutically acceptable salt thereof.
- Diabetes is a group of metabolic diseases mainly characterized by chronic hyperglycemia due to insufficient insulin action, and is often roughly classified into type 1 and type 2.
- Type 1 diabetes synthesizes and secretes' splenic splenic Langerhans islet ⁇ cell destruction / disappearance power is the main cause of S insulin deficiency
- type 2 diabetes is a predisposing factor for decreased insulin secretion and insulin resistance It develops with multiple genetic factors, including environmental factors such as overeating (especially high-fat diet), lack of exercise, obesity, and stress, and aging.
- Currently used therapeutic agents for diabetes include insulin preparations, tolptamide, acetohexamide, chlorpropamide, tolazamide, glycloviramide, daruribole, darifenclamide, daliclazide, glimepiride and other sulfonylureas (nateglinide, (Including fast-acting insulin secretagogues such as mitiglinide and repaglinide), metformin (biguanide), a -darcosidase inhibitors such as voglibose, carbolose and miglitol, and thiazolidinediones such as pioglitazone and rosiglitazone.
- Insulin preparations have a strong hypoglycemic effect, but often cause severe hypoglycemia. It can also cause weight gain. In addition, many insulin preparations require administration by injection, which places a heavy burden on patients. While sulfonylureas also have a powerful hypoglycemic effect, they often cause severe hypoglycemia. In addition, the effect is low when gaining weight or when administered for a long time. So-called secondary invalidation may occur. Metformin has a hypoglycemic effect, an insulin resistance improving effect, etc., but it often causes gastrointestinal disorders such as diarrhea and bloating. Infrequent but severe lactic acidosis may occur and is contraindicated in patients with renal or liver dysfunction. a-Dalcosidase inhibitor is less effective than sulfonylurea, but has a blood glucose lowering effect, but abdominal pain
- Non-patent Document 15 May cause gastrointestinal disorders such as diarrhea and bloating.
- Thiazolidinediones like metformin, have the effect of improving insulin resistance in addition to the hypoglycemic effect, but often cause body weight gain and edema, which may cause heart failure patients and those with a history of heart failure. Not suitable.
- GLP-1 glucagon-like peptide-1
- incretin one of the gastrointestinal hormones called incretin, which is useful for improving glucose metabolism in the body, such as insulin secretion promoting action and glucagon secretion inhibiting action, and protection of knee Langernos island ⁇ cells It is said to have an effect.
- GLP-1 is secreted by dietary intake, that is, it exhibits an insulin secretion promoting action when blood sugar level rises to some extent, so it is generally easy to occur before or during exercise or after exercise, and hypoglycemia is unlikely to occur. It is thought that.
- GLP-1 is also considered to be unlikely to cause weight gain, which is one of the risk factors for diabetes, due to its gastric motility-inhibiting and appetite-inhibiting effects.
- GLP-1 is hydrolyzed and inactivated by an enzyme called dipeptidyl peptidase IV (hereinafter abbreviated as DPP-IV), and its half-life is 2 minutes. Because of its very short duration, it was difficult to use GLP-1 itself as a therapeutic drug for diabetes (Non-patent Documents 6-8). Recently, clinical studies of GLP-1 analogues that are not susceptible to enzymatic hydrolysis have been conducted, but none of them can be administered orally (Non-patent Document 8).
- DPP-IV blocking Pesticides are also expected not to have side effects such as hypoglycemia and weight gain found in conventional oral diabetes drugs.
- DPP-IV inhibitors including sulfostin derivatives, xanthine derivatives, isoquinolone derivatives and their analogs, isoquinoline derivatives and their analogs, pyridine or pyrimidine derivatives, but most of them.
- Non-patent Documents 9-11 It is a cyclic amine carboxamide derivative (Non-patent Documents 9-11).
- DPP-IV inhibitors are cyclic amine carboxamide derivatives, but examples of acyclic amine carboxamide derivatives are also known (Patent Document 1). - Four).
- Patent Document 3 the DPP-IV inhibitory activity is specifically disclosed only in Example Compounds 1 and 10 of International Publication No. WO05Z40095 (Patent Document 3). Are 0.46 ⁇ and 0.45 ⁇ .
- Non-Patent Document 1 The Diabetes Society of Japan, Diabetes Treatment Guide 2004— 2005
- Non-patent document 2 Moller DE, “Nature”, 2001, 414, pp.821-827
- Non-patent document 3 Skyler JS, “Journal 'Ob' 'Medicine' Chemistry” (Journal of Medicinal Chemistry), 2004, 47th, p. 4113-4117
- Non-Patent Document 4 Ross S. A. et al., “Chemical Review”, 2004, 104, p. 1225-1282
- Non-Patent Document 5 Stumvoll M. et al., “Lancet”, 2005, 365, p. 1333-1346
- Non-Patent Document 6 Vilsboll T. et al., “Diabetologia”, 2004, 47th pp. 357-366
- Non-Patent Document 7 D'Alessio DA et al., "American Journal of Physiology-Endocrinology and Metabolism", 2004, 286th , P. E882- E890
- Non-Patent Document 8 Knudse L. B., "Journ of Medicinal Chemistry", 2004, 47th, p. 4128-4134
- Non-Patent Document 9 Augustyns K. et al., "Current 'Medicine'Chemistry” (Current Me dicinal Chemistry), 1999, VIII, p. 311-327
- Non-Patent Document 10 Augustyns K. et al., "Expert Opinion on Therapeutic Patens", 2003, 13 pp. 499-510
- Non-Patent Document 11 Weber AE Author, Journal of Medicinal Chemistry, 2004, 47th, p. 4135-4141
- Patent Document 1 International Publication No. 04Z37181 Pamphlet
- Patent Document 2 Pamphlet of International Publication No. 05Z25554
- Patent Document 3 International Publication No. 05Z40095 Pamphlet
- Patent Document 4 International Publication No. 05Z95343 Pamphlet
- An object of the present invention is to provide an orally administrable treatment or prevention agent for diabetes with few side effects and a novel compound useful as the treatment or prevention agent.
- Ar represents a phenyl optionally substituted with 1 to 5 R 3 ,
- R 3 represents halogen, hydroxy, alkyl having 1 to 6 carbon atoms, or alkyl having 1 to 6 carbon atoms, preferably 1 to 3 carbon atoms (wherein alkyl or alkyloxy is 1 to 5 carbon atoms). Substituted with halogen !, may! /,),
- R 1 represents hydrogen or alkyl having 1 to 6 carbon atoms (wherein alkyl may be substituted with any 1 to 5, preferably 1 to 3, halogen or hydroxy);
- R 5 represents alkyl having 1 to 6 carbon atoms, aryl or heteroaryl
- p 0, 1 or 2
- n each independently represent 1, 2 or 3
- R 2 represents aryl or heteroaryl optionally substituted with any 1 to 3 substituents for which R 6 or R 7 forces are also selected
- R 6 is alkyl having 1 to 6 carbon atoms, alkyloxy having 1 to 6 carbon atoms (wherein alkyl or alkyloxy may be substituted with 1 to 5 halogen atoms), cycloalkyl, aryloxy, heteroalkyl. Riloxy, hydroxy, halogen, with shear 0— (CH) q
- R 9 and R 9 ′ are each independently hydrogen, alkyl having 1 to 6 carbons (wherein alkyl is substituted with 1 alkyl having 1 to 3 carbons or 1 to 5 halogens).
- R 9 , R 9 ′ and nitrogen together may be pyrrolidine, piperidine (wherein pyrrolidine or piperidine may be substituted with 1 to 5 halogens), Piperazine, N-alkylpiperazine (wherein the alkyl has 1 to 6 carbon atoms), morpholine, thiomorpholine or thiomorpholine 4,4 dioxide,
- R 1Q and R 1Q ′ each independently represent hydrogen, alkyl having 1 to 6 carbon atoms (which may be substituted with one alkyl group having 1 to 3 carbon atoms or 1 to 5 halogen atoms). Or R 1Q , R 1Q ', combined with nitrogen, pyrrolidine, piperidine (where pyrrolidine or piperidine is 1 May be substituted by 5 halogens), piperidines Rajin, N Arukirupi Bae Rajin (number of carbon atoms in the alkyl here is 1 to 6), morpholine, thiomorpholine or Chiomoruhori Hmm 4, 4 Jiokishido Where pyrrolidine, piperidine, piperazine, N-alkylbiperazine, morpholine, thiomorpholine or thiomorpholine 4,4 dioxide, one methylene may be substituted with oxo,
- q 0, 1, 2 or 3
- R 7 represents aryl or heteroaryl (wherein aryl or heteroaryl may be optionally substituted with 1 to 3 R 11 ),
- R 11 represents alkyl having 1 to 3 carbon atoms, halogen, alkyloxy having 1 to 6 carbon atoms, trifluoromethyl, —SO—R 5 or one NR 9 R 3 ;
- X represents a valence bond or alkylene having 1 to 6 carbon atoms.
- the present invention also provides the use of the non-cyclic aminocarboxamide derivative represented by the above general formula (I) or a pharmaceutically acceptable salt thereof for the treatment or prevention of diabetes and the production of a hypoglycemic agent. .
- a method for treating or preventing diabetes and a method for lowering blood glucose comprising administering to a patient an effective amount of the acyclic amine carboxamide derivative represented by the general formula (I) or a pharmaceutically acceptable salt thereof.
- the compound of the general formula (I) has one or more asymmetric points. Therefore, the compound of the present invention can be used in which stereoisomers such as enantiomers, racemates or diastereomers may exist. Stereoisomers or mixtures of these isomers are also included.
- alkyl means a linear or branched saturated hydrocarbon group, specifically, methyl, ethyl, 1-propyl, 2-propyl, 1-butyl. , 2-butyl, 2-methyl-2-propyl, 1,1-dimethylethyl, 1 pentyl, 2-pentyl, 3 pentyl, 1 monohexyl, and the like.
- Alkyloxy represents a functional group in which the alkyl is bonded to an oxygen atom, and specifically includes methoxy, ethoxy, 1-propoxy, 2-propoxy, 1-butoxy and the like. The powers to be listed are not limited to these.
- Cycloalkyl means a saturated alicyclic hydrocarbon group having 3 to 8 carbon atoms, preferably 3 to 6 carbon atoms. Specifically, cyclopropyl, cyclobutyl, cyclohexane Examples include benzyl and cyclohexyl.
- Alkylene means a straight or branched saturated hydrocarbon chain, specifically,
- Forces including, but not limited to, methylene, ethylene, trimethylene, tetramethylene and the like.
- Aryl means an aromatic group having 6 to 10 carbon atoms, and specifically includes forces such as phenyl, naphthyl and the like.
- the "heteroaryl” includes 1 to 4 heteroatoms arbitrarily selected from S, N, and O.
- the total number of atoms is 5 to 17, preferably the total number of atoms is 5 to 16.
- R 8 may be substituted with the same definition as above.
- the nitrogen atom may be oxidized.
- chanel (2 chaels, 3 chaels), pyrrolyl (1 pyrrolyl, 2 pyrrolyl, 3 pyrrolyl), 1 methyl pyrrolyl (1 methyl pyrrole, 2 yl, 1-methyl pyrrole, 1 3 -Yl), 1-ethylpyrrolyl (1-ethyl pyrrole 2-yl, 1-ethynole pyrrole 3-yl), furyl (2-furyl, 3-furyl), thiazolyl (thiazole-2-yl, thiazole-4-yl) , Thiazole-5-yl), thiazole-3-oxide 2-yl (ie, thiazole-2-yl N-oxide), thiazole-3-oxide 4-yl, thiazole-3-oxide 5-yl, thiazolyl 3-oxide (ie , Thiazole
- alkyl substituted with aryl or “alkyl substituted with heteroaryl” is an alkyl having 1 to 3, preferably 1 to 2 carbon atoms substituted with 1 or 2 of the aryl or heteroaryl.
- benzyl diphenylmethyl, 1-phenylethyl, 2-phenylethyl, pyridine-2-ylmethyl, pyridine 1-oxide 2-ylmethyl, pyridine-3-ylmethyl, pyridine 1-oxide 3-ylmethyl, pyridine 4-methyl methyl, pyridine 1-xoxide 4-phenyl methyl, phenyl (pyridine-2-yl) methyl, phenyl (pyridine-1-oxyd-2-yl) methyl, phenyl (3-yl) pyridine, phenyl (1 pyridine 1 3 yl) methyl, phenol (4 1 pyridine) methyl, phenol (1 pyridine 1 4-methyl) methyl, furan-2-ylmethyl, furan-3-yl.
- Halogen means fluorine, chlorine, bromine or iodine.
- diabetes is a disease diagnosed as diabetes according to diagnostic criteria such as WHO (World Health Organization), Japan Diabetes Society, American Diabetes Society or European Diabetes Society. It means state, including type 1 diabetes, type 2 diabetes and gestational diabetes.
- WHO World Health Organization
- Therapeutic or prophylactic agent includes not only those used for either treatment or prevention, but also those used simultaneously for both treatment and prevention.
- “Prophylactic agent” is used for a condition in which diabetes is not diagnosed but shows either glucose tolerance abnormality or fasting blood glucose abnormality, or both glucose tolerance abnormality and fasting blood glucose abnormality Are also included.
- the “treatment or prevention method” includes not only a method of performing either treatment or prevention, but also a method of performing both treatment and prevention at the same time.
- diabetes is not diagnosed, but it indicates either glucose tolerance abnormality or fasting blood glucose abnormality, or a state of indicating glucose tolerance abnormality or fasting blood glucose abnormality!
- the method of carrying out is also included.
- the “hypoglycemic agent” means a drug having an effect of lowering blood glucose level, and includes effects such as an improvement effect of postprandial hyperglycemia in diabetes and an improvement effect of postprandial blood glucose transition in diabetes.
- the “blood glucose lowering method” means a method for lowering blood glucose level, and includes a method for improving postprandial hyperglycemia in diabetes, a method for improving postprandial blood glucose transition in diabetes, and the like.
- the acyclic amine carboxamide derivative of the present invention exhibited a blood glucose lowering action having good oral activity. Furthermore, as a result of intensive studies on the action mechanism of the hypoglycemic action of the acyclic amine carboxamide derivative of the present invention, it has become clear that DPP-IV inhibitory activity is one of the action mechanisms. Surprisingly, the compounds of the present invention showed potent DPP-IV inhibitory activity, as shown in the following examples, despite being acyclic amine carboxamide derivatives. From these facts, it can be said that the compound of the present invention is extremely useful as an agent for treating or preventing diabetes that can be administered orally and can be expected to have no side effects such as hypoglycemia and weight gain.
- R 3 that can be substituted with Ar is preferably fluorine or chlorine among halogen (fluorine, chlorine, bromine, iodine) or trifluoromethyl.
- the number of substitution is preferably 1 to 3.
- R 1 is preferably hydrogen, among hydrogen, methyl or ethyl.
- R 4 is hydrogen, alkyl having 1 to 6 carbon atoms (eg, methyl, ethyl, 1 propyl, 2-propyl, 1-butyl, 2-butyl, 2-methylpropyl, 1 pentyl, 1 monohexyl, etc.
- Heteroaryl eg, pyridine-2-yl, pyridine-3-yl, pyridine-4-yl, pyrimidine-2-yl, pyrimidine-4-yl, virazile, etc.
- 1 carbon substituted with aryl ⁇ 2 alkyls eg benzyl, phenyl (2-phenyl), etc.
- m and n are preferably the case where both m and n are 2, even though the sum of m and n is preferably 2, 3, or 4.
- R 2 is a monocyclic aryl or heteroaryl having 1 to 3, preferably 1 or 2, heteroaryl (where aryl or heteroaryl is selected from R 6 or R 7 forces)
- aryl or heteroaryl is selected from R 6 or R 7 forces
- Preferred is an aryl or heteroaryl having the following structure (which may be substituted with any one to three substituents), wherein aryl or heteroaryl is R 6 or R 7 force selection Substituted with any 1 to 3 substituents, which is more preferred).
- aryl or heteroaryl having the following structure (wherein aryl or heteroaryl may be substituted with any one to three substituents selected as R 6 or R 7 force) is most preferable. .
- R is preferably hydrogen, methyl, ethyl, 2-methoxyethyl, 2 ethoxyethyl or 2 morpholinoethyl.
- R 6 represents alkyl having 1 to 6 carbon atoms, preferably 1 to 3 carbon atoms (which may be substituted with 1 to 5 halogen atoms) (eg, methyl, ethyl, 1 propyl, 2-propyl).
- R 7 is Hue - Le, pyridine one 2-I le, pyridine one 3-I le, pyridine one 4-I le, pyridine Hmm 1 Okishido 2 I le, pyridine 1 Okishido 3 I le, pyridine 1 Okishi Dough 4-inole, pyrrole 1-yl, pyrrole 2-yl, 1-methylpyrrole 2-yl, pyrrole 3-il, 1 methyl pyrrole 3-yl, furan 2-il, furan 3 yl, thiophene 2 yl, thiophene 3 yl, imidazole 1 yl, imidazole 2 yl, 1 methyl imidazole 2 yl, imidazole 4 yl, 1-methyl imidazole 4 yl, 1 methyl imidazole -5 yl, pyrazole-1 yl, pyrazole-3 yl, 1-methyl pyrazole-3 yl, 2-methylpyrazole-3 yl, 2-methylpyrazo
- R 11 is alkyl having 1 to 6 carbon atoms, preferably 1 to 3 carbon atoms (eg, methyl, ethyl, 1 propyl, 2-propyl, etc.), halogen (fluorine, chlorine, bromine, iodine), carbon C 1 -C 3, preferably C 1 -C 3 alkyloxy (for example, methoxy, ethoxy, 1-propoxy, etc.), trifluoromethyl, —SO—R 5 (for example, methylsulfol, ethylsulfo
- X is preferably a valence bond, among which a valence bond, methylene or ethylene is preferred.
- an inorganic acid salt such as hydrochloride, sulfate, nitrate, hydrobromide, hydroiodide, phosphate, Acetate, trifluoroacetate, lactate, citrate, oxalate, dartrate, malate, tartrate, fumarate, mandelate, maleate, benzoate, phthalate, etc.
- Organic carboxylates methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, camphorsulfonate, and other organic sulfonates, aspartate, glutamate, and other acidic amino acids Salt, etc., among which hydrochloride, hydrobromide, phosphate, trifluoroacetate, citrate, fumaric acid, tartrate, methanesulfonate, etc. are preferred, but not limited thereto .
- an acidic substituent such as a phenolic hydroxyl group is present in the general formula (I)
- a salt with a base is also possible.
- Alkaline earth metal salts such as salts and magnesium salts, aluminum salts, ammonium salts, trimethylamine salts, triethylamine salts, pyridine salts, picoline salts, ethanolamine salts, diethanolamine salts, triethanolamine salts, dicyclohexane
- Organic base salts such as xyllamine salt, N, N-dibenzylethylenediamine salt, and basic amino acid salts such as arginine salt, lysine salt and orthotin salt, etc., among which sodium salt and potassium salt Ethanolamine salts and the like are preferable, but not limited thereto.
- Ar is 2-fluorophenyl
- R 1 is hydrogen
- Y is NR 4 —
- R 4 is benzyl
- m and n are Both are compounds wherein X is a valence bond
- R 2 is phenol
- R 7 is phenyl
- R 11 is methyl sulfone (free base of Example Compound 44)
- Ar is 2, 4, 5 trifluorophenyl
- R 1 is hydrogen
- Y is 0-0
- m and n are Both compounds are compounds in which X is a valence bond, R 2 is methyl at the 1-position, and benzimidazol-2-yl having a 2-methoxyethoxy group as the substituent at the 5-position (free of Example Compound 141) base):
- the compounds of the present invention include the following general formulas (II) and (III):
- Y is as defined above.
- R & R 6b is the same as the definition of hydrogen or R 6 above.
- R 3a , R 3b and R 3e are the same as defined above for hydrogen or R 3 .
- R 6 R 6b is the same as defined for hydrogen or R 6 above.
- the acyclic amine carboxamide derivative used as an active ingredient of an orally administrable agent for treating or preventing diabetes represented by the general formula (I) is specifically the following scheme.
- the force that can be produced by the method shown in FIG. The powerful methods not shown by the scheme below have been specifically disclosed in the examples.
- the compound represented by general formula (I) can be produced by condensing amino acid (IV) and amine (V) to give compound (VI), and removing protecting group P of compound (VI) ( Scheme 1).
- Amino acid (IV) can be obtained from commercial products, or Cole DC et al., “Tetrahedron”, 1994, 50 ⁇ , p 9517-9582 .; Cardillo G. et al., “Chemical 'Society'” Reviews (Chemical Society Reviews), 1996, 25 ⁇ , ⁇ 117-128 .; Juaristi
- the removal of the protecting group P can be accomplished by a general deprotection method (Green TW et al., “Protective Groups in Organic Synthesis”, 5th edition, 1999, John '
- the compound represented by the general formula (I) can be produced by using “Iriichi & And Sons”.
- Ar in the scheme, Y, m, n, X and R 2 are the same as defined above, and P represents an appropriate protecting group.
- Acidic agents include peracetic acid, pertrifluoroacetic acid, m-chloroperbenzoic acid (hereinafter abbreviated as mCPBA), t-butyl hydroperoxide, Use organic peroxides such as menhydroperoxide, bis (trimethylsilyl) peroxide, dimethyldioxysilane, and benzoyl peroxide, and inorganic peroxides such as hydrogen peroxide, potassium persulfate, and sodium periodate.
- mCPBA m-chloroperbenzoic acid
- Use organic peroxides such as menhydroperoxide, bis (trimethylsilyl) peroxide, dimethyldioxysilane, and benzoyl peroxide
- inorganic peroxides such as hydrogen peroxide, potassium persulfate, and sodium periodate.
- TPAP Perruthenium (VII) tetra-n-propylammonium
- TPAP Perruthenium tetra-n-propylammonium
- NMO N-methylmorpholine N-oxide
- it can be carried out at 0.001 mol% to 90 mol%, but 0.1 mol% to 50 mol% is good. Often gives fruit .
- the amount of co-oxidant can be 1 to 50 equivalents, but usually 1 to 10 equivalents gives good results.
- halogen solvents such as dichloromethane and chloroform
- -tolyl solvents such as acetonitrile and propio-tolyl, N, N-dimethylformamide (hereinafter abbreviated as DMF) and a mixture of these solvents
- DMF N, N-dimethylformamide
- the reaction temperature can be 0 ° C to 50 ° C, but usually good results are obtained at room temperature.
- the reaction is often completed in 1 hour to 3 days. If the reaction is slow, better results may be obtained by adding an oxidizing agent and / or a co-acid additive rather than lengthening the reaction time. Addition of molecular sieves (hereinafter abbreviated as MS) is also an effective means.
- Ar, P, in the scheme m, n, p, X and R 2 are the same as defined above.
- amino acid (IV) can be obtained from a commercial product, or can be obtained by synthesis by a method described in the literature. Racemic amino acid (IV) and optically active are listed below. A specific method for synthesizing amino acid (IV ′) is shown (Schemes 3 and 4).
- Amino acid (IV) is obtained by reacting compound (VII), which is commercially available, and ester bromoacetate (VIII) to give compound (IX), and reducing compound (IX). It can be produced by introducing a protecting group P into the amino group of the amino acid ester (X) and hydrolyzing the ester (Scheme 3).
- Compound (IX) can be prepared according to Enishimino described in Kishi Y. et al., “Journal of Organic Chemistry” (1983, 483-3, p 3833-3835). It can be carried out by the method described in the general synthesis method of esters, etc.
- the production of I) is a common j8-ketoester described in Kishi Y. et al., “Journal of Organic Chemistry” (1983, 48 ⁇ , p 3833-3835). It can be performed by the method described in the synthesis method and the like. Preparation of ⁇ compounds ( ⁇ ) by hydrolysis of esters is a common method (Green TW et al., “Protective Groups in Organic Synthesis”, 5th edition, 1 999, “John Wiley & Sons” etc.). The asymmetric reduction reaction can be carried out by the method described in Wang Z. et al., “Tetrahedron A symmetry”, 1999, 10 ⁇ , p 225-228.
- the amidei reaction is a general carboxylic acid amide synthesis method (5th edition, Experimental Chemistry Course 16 Synthesis of Organic Compounds IV, pp. 118-134; Comprehensive 'Organic' Synthesis) by Benz G.
- the ring-opening reaction of ratatam can be carried out with lithium salt, potassium salt, sodium salt, etc. of alcohol R 13 OH Usually, good results are obtained with sodium salt.
- Commercially available alcohol salts may be used, and good results are obtained in the case of sodium methoxide and sodium ethoxide as alcohol salts that may be prepared before use.
- the amount of salt of the alcohol is 1 to: a power that can be carried out with L00 equivalents, preferably 1 to 10 equivalents, more preferably 1 to 5 equivalents.
- alcohol solvents such as methanol, ethanol and isopropyl alcohol, ether solvents such as tetrahydrofuran (hereinafter abbreviated as THF) and 1,4 dioxane, halogen solvents such as dichloromethane and dichloroethane, DMF and the like can be used.
- THF tetrahydrofuran
- halogen solvents such as dichloromethane and dichloroethane, DMF and the like
- the reaction can be carried out at a temperature from 0 ° C to the reflux temperature of the solvent, but good results are often obtained at room temperature. The reaction is often completed within one day, and usually complete within one hour.
- Removal of the benzyloxy group can be carried out by a catalytic hydrogenation reaction using a noradium catalyst.
- the hydrogen pressure is a force that can be carried out at 1 to 10 atmospheres. Usually, good results are obtained at 1 to 3 atmospheres.
- the amount of palladium can be practiced from 0.001 mol% to 1000 mol%, but is usually from 0.1 mol% to 10 mol%. Good results are obtained at 0 mol%. Satisfactory results are often obtained when an alcohol solvent such as methanol or ethanol is used as the solvent.
- the reaction temperature can be 0 to 100 ° C, but it is usually carried out at room temperature. The reaction is often completed within one day, and is usually completed within one hour.
- molybdenum hexacarbox described in Naito T. et al., “Journal of Organic Chemistry”, 2000, 65 ⁇ , p 176-185.
- This method can also be implemented by using a method.
- Introduction of the protecting group P to the amino group and hydrolysis of the ester is a common method (Gren TW et al., “Protective Groups in Organic Synthesis”, 5th edition. 1999, John Wiley & Sons etc.).
- Ar, P and R 13 in the scheme are the same as defined above.
- Amine (XX) (compound with ammine (V)! /, Where R 1 is hydrogen) can be produced mainly by the methods ⁇ to ⁇ .
- Amine (XX) is an alcohol (XVIII) obtained by the reaction of a commercially available ketone (XVI) with an organometallic compound (XVII), Krimen LI et al., “Organic Reactions”. ), 1969, 17 ⁇ , p 213-325 .; Bishop R.
- the compound (XIX) was introduced into the compound (XIX) by the method described in “Comprehensive Organic Synthesis”, 1991, 6300, p.2 61-300, Pergamon Press (Pergamon Press).
- M represents a metal such as lithium, magnesium, aluminum, zinc, copper, titanium, cerium, etc., and usually good results are obtained with lithium and magnesium.
- ether solvents such as jetyl ether, THF, and 1,4 dioxane, and aromatic hydrocarbon solvents such as benzene and toluene can be used. Usually, good results are obtained with THF.
- the reaction can be carried out at a temperature of -100 ° C to 100 ° C, but usually good results are obtained at 78 ° C to 30 ° C.
- Organometallic compound (XVII) is a force that usually gives good results when used in an equivalent amount of 1 to 2 equivalents to ketone (XVI). It is also possible. The reaction is often completed within one day, and the reaction is usually completed within one hour.
- the reaction to remove the acyl group by the compound (XIX) force is a general method (Green TW et al., “Protective Groups in Organic Synthesis”, 5th edition, 1999, John Wiley & Sons. Etc.) or WO97 / 32880; Lee GT et al. “Synthetic Communications” (1998) 28 ⁇ , ⁇ 4009-4018. It can be implemented by the method described in 1.
- R 14 when R 14 is a chloromethyl group, it can be carried out by the method described in Jirgensons A. et al., “Synthesis”, 2000, p. 1709-1711.
- Y, m, n, X, and R 2 are the same as defined above, and R 14 may be substituted with one halogen and may represent alkyl having 1 to 3 carbon atoms.
- Amine (XX) is obtained by oximation of ketone (XVI) with benzyloxyamine 'hydrochloride. It can be produced by removing the benzyloxy group of compound ( ⁇ ) by leading to compound (XXII) by reaction of the obtained oxime (XXI) and organometallic compound (XVII).
- the oximation reaction can be carried out in the presence of a base with a commercially available ketone (XVI) and a commercially available benzyloxyamine 'hydrochloride.
- Examples of the base that can be used include organic bases such as pyridine and triethylamine (hereinafter abbreviated as TEA), organic sulfonates such as sodium acetate and potassium acetate, and the like.
- the amount of the base can be used up to 1 equivalent force solvent amount.
- the amount of benzyloxyamine hydrochloride can be 1 to 10 equivalents, but usually 1 to 3 equivalents gives good results.
- the solvent alcohol solvents such as methanol and ethanol are usually used, and water can be used as a cosolvent. It is also possible to use a base as a solvent.
- the reaction can be carried out at a temperature ranging from 0 ° C. to the reflux temperature of the solvent, preferably the reflux temperature of the solvent at room temperature.
- reaction is often completed within one day, and usually the reaction is completed within 5 hours.
- M represents a metal such as lithium, magnesium, aluminum, zinc, copper, titanium, cerium, and usually good results are obtained with lithium and magnesium.
- ether solvents such as jetyl ether, THF, and 1,4 dioxane, and aromatic hydrocarbon solvents such as benzene and toluene can be used. Usually, good results are obtained with jetyl ether. .
- the reaction temperature is a force that can be carried out at -100 ° C to 100 ° C.
- the organometallic compound (XVII) is usually used in an amount equivalent to 1 to 2 equivalents of the oxime (XXI). Use more than that amount, or add it sequentially until the reaction is complete. Is also possible.
- the reaction is often completed within one day, and the reaction is usually completed within one hour.
- Removal of the benzyloxy group can be carried out by a catalytic hydrogenation reaction using a noradium catalyst.
- the hydrogen pressure is a force that can be implemented at 1 to 10 atmospheres. Usually, good results are obtained at 1 to 3 atmospheres.
- the amount of palladium, 0.001 mol% to 1000 mol 0/0 can be carried 1S Usually, good results have been obtained at 0.1 mol% to 100 mol%.
- reaction temperature is from 0 to: force that can be carried out at L00 ° C Usually carried out at room temperature. The reaction is often completed within a day, and the reaction is usually completed within an hour.
- Catalytic hydrogenation reaction the molybdenum hexacarbonyl described in Naito T. et al., "Journal 'Ob' Organic 'Chemistry” (Journal of 0 rganic Chemistry), 2000, 65 ⁇ , p 176-185. It can also be implemented by a method. In the scheme, Y, m, n, X and R 2 are the same as defined above.
- Amin (XX) can be obtained as a commercial product by the method described in Ellman JA et al., “Accounts of Chemical Research”, 2002, 35 ⁇ , p 984-995. It can be produced from a new ketone (XVI) via compound ( ⁇ ) and compound (XXIV).
- XVI new ketone
- Y, m, n, M, X, and R 2 are the same as defined above.
- the organometallic compound (XVII) used in the above-mentioned methods A to C is a commonly used halogen metal exchange reaction or deprotonation reaction (Tomooka K. et al. “Main Group Metals in Organic Synthesis”, 2004, 1 ⁇ , p 1-34. Weilly.; Oshima K. et al. Group Metals in Organic Synthesis), 2004, 1 ⁇ , p 51-154. Weilly.; Katritzky AR et al. “Comprehensive Heterocyclic Chemistry” (1984) 5 ⁇ , p 39-11 0. Pergamon Press, etc.).
- the compound (XXV) used in the production of the organometallic compound (XVII) is commercially available or, if not commercially available, can be synthesized and obtained by the method shown in Scheme 8-: L1. .
- X and R 2 are the same as defined above, and Z represents hydrogen or halogen.
- XXV Compound (XXV ') (compound (XXV) in which R 2 is thiazole- 2- yl, Z is hydrogen, and X is a valence bond) is a commercially available promoketone ( XXVII), or a bromoketone (XXVII) obtained by bromination of a commercially available ketone (XXVI) is cyclized to thiazole (XXVIII) by thiourea or ethyl oxalate, and the thiazole (XXVIII) force is also R 15 (Scheme 8). Bromoketone (XXVII) is produced by Jaeques J.
- thiourea or thioxamic acid ethyl By using 0.1 to 10 equivalents, preferably 0.5 to 2 equivalents, of thiourea or thioxamic acid ethyl, good results are obtained.
- the solvent alcohol solvents such as methanol and ethanol, and aprotic polar solvents such as acetone and DMF are preferably used.
- the reaction can be carried out at a temperature from 0 ° C to the reflux temperature of the solvent, but usually good results are obtained from room temperature to the reflux temperature of the solvent. The reaction is often completed within a day, and usually the reaction is completed within 3 hours.
- R 15 is an ethoxycarbo group
- the ethoxycarbo group of thiazole (XXVI ⁇ ) can be synthesized by a general method (Green TW et al., “Protective 'Gnorapes'In'Onoreganic' Synthesis” (Protective Groups). in Organic Synthesis), fifth edition, 1999, John Wiley & Sons, etc.) and then decarboxylated using acids such as hydrochloric acid and hydrobromic acid. It is possible to implement it.
- an acid used for the reaction may be used as a solvent, or an ether solvent such as THF or 1,4 dioxane may be used as a cosolvent.
- the reaction temperature can be from room temperature to the reflux temperature of the solvent, but usually favorable results are obtained at the reflux temperature of the solvent.
- the reaction is often completed within one day, and the reaction is usually completed within one hour.
- R 7 is the same as defined above, and R 15 represents amino or ethoxycarbonyl.
- isothiocyanate (XXX) or thiourea (XXXI) is commercially available, it can also be produced from them), and can be produced by cyclization.
- the isothiocyanate (XXX) can be produced by the reaction of a-line (XXIX) with a thiocarbonylated reagent.
- a thiocarbonylated reagent Preferable examples of the thiocarbo-Ruyi reagent include thiophosgene, thiocarbodiimidazole (hereinafter abbreviated as TCDI), 1,1, -thiocarbonylji 2 (1H) -pyridinone (hereinafter abbreviated as TCDP), and the like.
- the amount of the thiocarbonylating reagent can be carried out at 1 to 10 equivalents. Usually, 1 to 3 equivalents gives preferable results.
- the solvent ether solvents such as THF and 1,4 dioxane, halogen solvents such as dichloromethane and chloroform, and aromatic hydrocarbon solvents such as benzene and toluene are usually used. Satisfactory results are obtained.
- the reaction can be carried out at a temperature of -100 ° C to 100 ° C, but satisfactory results are often obtained at -20 ° C to room temperature. The reaction is often completed within 1 day, and usually the reaction is completed within 3 hours.
- Thiourea (XXXI) can be produced by reacting isothiocyanate (XXX) with ammonia.
- Ammonia may be a gas or an ammonia solution such as ammonia water, methanol, ethanol, 2 propanol, or 1,4 dioxane, but the method using an ammonia solution is simple and has good results. This is the method obtained.
- the amount of ammonia is not particularly limited, but usually a satisfactory result is obtained with 1 to: LO equivalent.
- the solvent, THF, 1, 4 ether solvents such Jiokisan, dichloromethane emissions, black hole halogen solvents such Holm, benzene, force normally can be used aromatic hydrocarbon-based Solvent such as toluene, T HF Satisfactory results can be obtained with dichloromethane, etc. Satisfactory results are often obtained at a reaction temperature of ⁇ 100 ° C. to 100 ° C., a force that can be carried out at ⁇ 20 ° C. to room temperature. The reaction is often completed within one day, and usually within 3 hours.
- Thiourea (XXXI) can also be produced directly by the method described in Meckler H. et al., “Synthesis”, 2000, p 1569-1574. .
- the compound (XXV) having the substituent R 6 or R 7 can be produced using the compound having these substituents as a raw material as described above, but can also be introduced later.
- An example of a method to be introduced later is shown in Scheme 11.
- Other common Suzuki coupling conditions (Suzuk i A. et al. “Chemical Review”, 1995, 95 ⁇ , p 2457-2483; Suz uki A. Cross-Coupling Reactions (Meta ⁇ Catalyzed Cross-Coupling Reactions), 1998, p 49-97.
- Benzothiazole (XXV,,,) is a copy of Haddock E. et al. "Courtesy ⁇ ⁇ ⁇ Sea” (Journal of Chemical Society C), 1971, p 3994-3999 .; Cadog an JIG. Hemicai Society, Parkin Transaction 1), 1973, p 541-542.; Doyl e MP et al., “Journal of Organic Chemi stry”, 1977, 42 ⁇ , p 3494 -3498;. Chedekel MR et al., ".
- Compound (XXV '''') is a general phenolic hydroxyl group alkylation condition (Protective Groups in Organic Synthesis, Green TW et al., Protective Groups in Organic Synthesis), 5th edition, 1999, John Wiley & Sons etc.) or general Mitsunobu reaction conditions (Mitsunobu 0. “Synthesis”, 1981, p 1- 28 ; Benzothiazole (XXV,,, ') force can be produced by Hughes DL, “Organic Reactions”, 1992, 42 ⁇ , ⁇ 335-656.
- R 17 represents alkyl having 1 to 6 carbon atoms or (CH 3) q—R 8 (q and R 8 are as defined above).
- Amine (XX,) (a compound in which R 2 is benzimidazol-2-yl and X is a valence bond in Amine (XX)) is a commercially available ketone (XVI).
- Strecker Reaction, followed by hydrolysis of the cyano group (Steiger RE, “Organic Synthesis”, 1942, 22 ⁇ , p 13-15, etc.) leads to the amino acid (XXXIV) and amino acid It can be produced by introducing an Fmoc group into the (XXXIV) amino group, then subjecting it to a phenylenediamine (XXX VI), followed by an amide reaction followed by a cyclization reaction, and then removing the Fmoc group It is.
- Fmoc group Introduction of the Fmoc group can be carried out by the method described in McLaughlm M. et al., “Tetrahedron Letters”, 1997, 3813, p 4013-4016.
- the amidation reaction of amino acid (XXXIV) and phenylenediamine (XXXVI) is carried out using a general carboxylic acid amide synthesis method (5th edition, Laboratory Chemistry Course 16 Synthesis of Organic Compounds IV, pp. 118-134 .; Benz G. “Comprehensive Organic Synthesis”, 1991, 6 ⁇ , p.381-417, Pergamon Press .; Bailey PD et al.
- acids include organic carboxylic acids such as formic acid, acetic acid, propionic acid, and trifluoroacetic acid, inorganic acids such as hydrochloric acid and hydrobromic acid, methanesulfonic acid, and P-toluenesulfonic acid.
- organic carboxylic acids such as formic acid, acetic acid, propionic acid, and trifluoroacetic acid
- inorganic acids such as hydrochloric acid and hydrobromic acid, methanesulfonic acid, and P-toluenesulfonic acid.
- the ability to use sulfonic acids such as polyphosphoric acid etc.
- the acid is preferably used in a solvent amount.
- the reaction temperature can be carried out at room temperature or the reflux temperature of the solvent, and is preferably from 50 ° C to the reflux temperature of the solvent.
- the reaction is often completed within 3 days, and the reaction is usually completed within 1 day.
- Phenylenediamine may be available as a commercial product. If it is not available, it can be obtained by synthesis.
- An example of a process for producing phenylenediamine (XXXVI) is shown in Scheme 13.
- Phenol-Diamine (XXXVI) is commercially available-Toro-Rin (XXXVII) amino group Voskresens S. et al. "Synthetic Communications" (Synthetic Communications), 2000, 30 ⁇ , p .
- Amine (XX ,,) (a compound in which R 2 is thiazole-4-yl and X is a valence bond in Amine (XX)) is a commercially available amino acid ester (XXXIX) by Chen P. et al. Tetrahedron Letters, 1997, 38 ⁇ , ⁇ 3175-3178.
- the compound (XL) can be prepared by the method described in the above, and the compound (XL) and thioamide (XLI) can be reacted and cyclized, and then the Boc group can be removed.
- Thioamide (XLI) may be available as a commercial product, or if it is not available as a commercial product, Taylor EC et al.
- the anti-diabetic effect and the hypoglycemic effect of the compounds of the present invention are described in, for example, glucose tolerance using experimental animals described in Takeni Hironi et al. Although it can be confirmed by a test (Oral Glucose Tolerance Test, hereinafter abbreviated as OGTT test), it is not limited to this method.
- OGTT test Oral Glucose Tolerance Test
- experimental animals Normal animals such as mice, rats, dogs, monkeys, etc. or diabetic model animals (for example, the obesity diabetes model described in Winzell MS et al. Induced Obesity Model (hereinafter abbreviated as DIO model), etc.
- DIO model Induced Obesity Model
- the drug when the compound of the present invention is clinically used as a drug such as a therapeutic drug for diabetes, the drug may be a free form or a salt thereof itself, or an excipient, a stabilizer, a preservative, a buffer, a dissolution agent.
- Additives such as auxiliary agents, emulsifiers, diluents, tonicity agents and the like may be appropriately mixed.
- the dosage form includes tablets' capsules, granules, powders, syrups and other oral preparations, as well as parenterals such as injections, suppositories, etc., or ointments, creams, and patches. Topical administration is also possible. These preparations can be made by generally known manufacturing methods.
- the therapeutic or prophylactic agent for diabetes of the present invention preferably contains 0.00001 to 90% by weight, more preferably 0.0001 to 70% by weight of the active ingredient.
- the amount to be used is appropriately selected according to the symptom, age, body weight, administration method, etc. For adults, the amount of active ingredient is 1 ⁇ g to 10 g per day for oral preparations. Or it can be administered in several divided doses.
- the compound represented by the general formula (I) and a salt thereof of the present invention can be used as a therapeutic or preventive agent for diabetes and a hypoglycemic agent.
- the compound represented by the general formula (I) and the salt thereof of the present invention can be used in combination with other antidiabetic agents (hereinafter abbreviated as concomitant drugs).
- concomitant drugs antidiabetic agents
- the administration time of the compound represented by the general formula (I) of the present invention and the salt thereof and the concomitant drug is not limited, and these may be administered to the administration subject at the same time or with a time difference. It may be administered.
- the dose of the concomitant drug can be appropriately selected based on the clinically used dose.
- the compounding ratio of the compound represented by the general formula (I) of the present invention and a salt thereof and the concomitant drug can be appropriately selected depending on the administration subject, administration route, symptom, combination and the like.
- Concomitant drugs include insulin preparations (super fast-acting insulin preparations, fast-acting insulin preparations, mixed insulin preparations, intermediate-type insulin preparations, continuous-type insulin preparations, long-acting dissolved insulin preparations, transpulmonary insulin preparations, oral insulin preparations) ), Insulin sensitizers (Pioglitazone, Rosiglitazone, Netoglitazon, Farglitazar, Ribo) Gritazone (Rivoglitazone), Oxeglitazar, Naveg Litazar, Balaglitazone, ONO-5129, AVE-0847, LBM-642, CKD-501, AVE-5376, etc., oc— Darcosidase inhibitors (Acarbose, Voglibose, Mi
- Fructose 1, 6 bisphosphatase inhibitor (MB-6322, MB-07803, etc.), SGLT (somum-dependent renal glucose transporter) inhibitor: ⁇ (J — 033, KGA—2727, SAR—7226, etc., 11 ⁇ — HSD1 (11 ⁇ -hydroxysteroid dehydrogenase 1) inhibitors (BVT-3498, AMG-221, INCB-13739, etc.), PTP-1B (protein tyrosine phosphatase- IB) inhibitors (ISIS-113715, JTT-551, etc.), GSK3 ⁇ (glycogen synthase kinase 3 ⁇ ) inhibitors (SAR-502250, etc.), glucagon antagonists (BAY-27-9955, NN-2501, etc.), glycogen Phosphorylase inhibitors (such as Isofagomine, PSN-357), CPT1 (Cartin 0—palmitoyltransferase 1) inhibitors (such
- FIG. 1 is a graph showing the blood glucose elevation inhibitory effect of Example Compound 8.
- FIG. 2 is a graph showing the blood glucose increase inhibitory effect of Example Compound 60.
- FIG. 3 is a graph showing the blood glucose increase inhibitory effect of Example Compound 73.
- FIG. 4 is a graph showing the blood glucose increase inhibitory effect of Example Compound 75.
- FIG. 5 is a graph showing the blood glucose increase inhibitory effect of Example Compound 121.
- FIG. 6 is a graph showing the blood glucose increase inhibitory effect of Example Compound 122.
- FIG. 7 is a graph showing the blood glucose increase inhibitory effect of Example Compound 125.
- FIG. 8 is a graph showing the blood glucose elevation inhibitory effect of Comparative Compounds 1 and 2.
- FIG. 9 is a graph showing the blood glucose elevation inhibitory effect of Example Compound 60 in a diabetes model mouse.
- Step 1 Synthesis of benzyl 4 ferrobiperidine 4 ol (reference compound 1)
- Step 3 1 Synthesis of Benjirou 4 ferrobiperidine 4-amamine (Reference compound 3)
- N— (1-benzyl-4-phenolbiperidine-4-yl) acetamide (lOlmg, 0.33 mmol) was suspended in THF (0.2 mL) and titanium tetraisopropoxide (96 ⁇ L, 0.32 mmol) and diphenylsilane (0.15 mL, 0.81 mmol) were added, and the mixture was stirred at room temperature. After 12 hours, the mixture was diluted with ethyl acetate, and saturated aqueous sodium hydrogen carbonate solution was added and stirred vigorously. The resulting white solid was filtered off, and the filtrate was separated and extracted with ethyl acetate.
- Step 4 (3R) —N— (1—Benzyl—4-phenolbiperidine—4-yl) —4— (2 fluorophenol) -3— (2-methyl-2-propoxycarbolamamino) butanamide (Reference Compound 4) Synthesis
- EDCI 1-ethyl-3- (3 dimethylaminopropyl) carbodiimide
- EDCI 1-ethyl-3- (3 dimethylaminopropyl) carbodiimide
- Step 5 Synthesis of 3-amino-1-N- (l-benzyl-1-4-biruberidine-1-4-yl) 4- (2-fluorophenyl) butanamide dihydrochloride (compound 1)
- Step 1 Synthesis of methyl 4 ferrobiperidine 4 ol (Reference compound 5)
- ferrous lithium Z cyclohexane-jetyl ether solution (1.05M, 7.7 mL, 8. lmmol) was added to THF (5 mL).
- THF 5 mL
- a solution of peridone (0.5 mL, 4. 1 mmol) in THF (3 mL) was added dropwise. After completion of the dropwise addition, the ice bath was removed and the mixture was stirred at room temperature. 1.5 hours later, a saturated aqueous sodium hydrogen carbonate solution was added. The mixture was extracted with ethyl acetate, and the organic layer was washed with saturated brine, dried and concentrated.
- the obtained crude product was purified by silica gel column chromatography to obtain the title compound (877 mg, 97%).
- Step 3 (3R) — 4— (2 Fluorophenol) — N— (l-Methyl—4-Ferruberidine-4-yl) -3— (2-Methyl-2-propoxycarbolamamino) butanamide (Reference Compound 7) Synthesis
- N— (1-methyl-4-phenol-biperidine-4-yl) acetamide (58 mg, 0.25 mmol) was suspended in THF (0.1 mL) and titanium tetraisopropoxide (75 ⁇ L , 0.25 mmol) and diphenylsilane (0.12 mL, 0.65 mmol) were added and stirred at room temperature. After 12 hours, the mixture was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was vigorously stirred. The resulting white solid was filtered off, and the filtrate was separated and extracted with ethyl acetate. The organic layers were combined, washed with saturated brine, dried and concentrated.
- Step 4 (3R) — 3 Amino 4— (2 Funoleo mouth Hue-Nole) 1 N— (l—Methinore 1— Synthesis of phthalbiperidine 4-yl) butanamide dihydrochloride (compound 2) Under argon atmosphere, (3R) —4— (2 fluorophenyl) N— (1-methyl-4 ferrobiperidine 4-yl) 3- (2-Methyl-2-propoxycarbo-lumino) butanamide (49 mg, 0.1 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (3 mL), stirred at room temperature for 3 hours, concentrated and concentrated. 31 mg (67%) of product was obtained.
- N— (1-methyl-4-phenol-biperidine-4-yl) acetamide (Reference compound 6) (HOmg, 0.47 mmol) was suspended in THF (0.1 mL), and titanium tetrisopropoxide (0.15 mL, 0.51 mmol) and diphenylsilane (0.22 mL, 1.2 mmol) were added and stirred at room temperature. After 12 hours, the reaction mixture was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was vigorously stirred. The resulting white solid was separated by filtration, and the filtrate was separated and extracted with ethyl acetate.
- Step 2 (3R) —3 amino 4- (2, 4, 5 trifluorophenol) 1 N— (l-methyl 4-phenylbiperidine 4-yl) butanamide dihydrochloride (compound 3 )
- Propoxy ball Amino) butanamide (81 mg, 0.16 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (3 mL), stirred at room temperature for 3 hours and concentrated to give 59 mg (77%) of the title compound.
- Step 1 4 Synthesis of 2H-tetrahydropyran 4ol (Reference Compound 9)
- N— (4-Ferro 2H-tetrahydropyran-4-yl) acetamide 63 mg, 0.29 mmol
- THF 0. ImL
- titanium tetraisopropoxide 80 0
- diphenylsilane 0.13 mL, 0.70 mmol
- the resulting white solid was filtered off, and the filtrate was separated and extracted with ethyl acetate. The organic layers were combined, washed with saturated brine, dried and concentrated.
- Step 4 (3R) — 3 Amino 4— (2 Funoreux mouth-nore) 1 N— (4 Hue-Nole 1 H-tetrahydropyran 4 yl) butanamide hydrochloride (Compound 4)
- N— (4 Fethiane-4-yl) acetamide 60 mg, 0.25 mmol was suspended in THF (0.1 mL) and titanium tetraisopropoxide (76 L, 0.26 mmol).
- diphenylsilane (0.12 mL, 0.65 mmol) were added and stirred at room temperature. After 13 hours, the mixture was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was vigorously stirred. The resulting white solid was filtered off, and the filtrate was separated and extracted with ethyl acetate. The organic layers were combined, washed with saturated brine, dried and concentrated.
- Step 4 (3R) —3-amino 4- (2-fluorophenol) -one N— (4-phenol cyan 4-yl) butanamide hydrochloride (Compound 5)
- Step 2 (3R) — 3 Amino 4— (2 Funeo-Leo Fue-Nole) 1 N— (4 F-Nole 1, 1 Dioxothian 4 yl) butanamide hydrochloride (Compound 6)
- N— (4 ferrothane-4-yl) acetamide (Reference compound 13) (181 mg, 0.77 mmol) was suspended in THF (0.3 mL), and titanium tetraisopropoxide (0.23 mL, 0 77 mmol) and diphenylsilane (0.36 mL, 1.94 mmol) were added at room temperature. And stirred. After 20 hours, the mixture was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was vigorously stirred. The resulting white solid was separated by filtration, and the filtrate was separated and extracted with ethyl acetate.
- Step 2 (3R) — 3 amino 1- 4 — (2, 4, 5 trifluorophenol) 1 N— (4 phenyl 2- 1 4-yl) butanamide 'hydrochloride (compound 7 )
- Step 2 (3R) — 3 amino 1- 4 — (2, 4, 5 trifluorophenol) 1 N— (4 phenol 2 1, 1 dioxothian 4 yl) butanamide hydrochloride (compound 8)
- Step 1 Synthesis of 4- (3-fluorophenyl) thiane-4-1ol (Reference Compound 18) Under argon atmosphere, 1-bromo-3fluorobenzene (753 mg, 4.30 mmol) in T HF (15 mL) was cooled to -78 ° C, n-Butyllithium Z-hexane solution (1.39M, 3.4mL, 4.73mmol) was added. After 10 minutes, a solution of 4-oxothiane (500 mg, 4.30 mmol) in THF (10 mL) was added and stirred at 78 ° C. for 2.5 hours, and then the mixture was further warmed to room temperature and stirred.
- Step 3 Synthesis of 4— (4 fluorophenyl) thian-4-amamine (Reference compound 25)
- a solution of N- (4- (4 fluorophenyl) thian-4-yl) acetamide (530 mg, 2. O5 mmol) in THF (1 mL) was added to titanium tetraisopropoxide (0.53 mL, 1.93 mmol), Diphenylsilane (0.90 mL, 2.50 mmol) was added and stirred at room temperature. After 12 hours, ethyl acetate and saturated aqueous sodium hydrogen carbonate solution were added, and the resulting insoluble material was removed by Celite filtration. The filtrate was extracted with ethyl acetate, washed with saturated brine, dried and concentrated. The obtained crude product was purified by silica gel column chromatography to obtain 220 mg (51%) of the title compound.
- Step 4 (3R) —4— (2 Funoleo Mouth Hue-Nole) One N— (4 One (4-Fonoleo Mouth Hue-Nole)) ) Synthesis of butanamide (reference compound 26)
- Step 5 (3R) —4— (2 Funoleo Mouth Hue-Nole) N— (4 One (4 Funoleo Mouth Hue-Nole)
- Step 6 (3R) — 3 amino 4— (2 fluorophenol) 1 N— (4— (4 fluoro 1) 1-Dioxothian 4 4-yl) butanamide 'hydrochloride (Compound 10) Synthesis under argon atmosphere (3R) —4 1 (2 Fluoro-F) N— (4 1 (4 Fluorophenyl) 1, 1, 1-Dioxothian 1 4-yl) 3— (2-Methyl 2-propoxyl-amino) butanamide (33 mg, 0.06 mmol) in 10% hydrogen chloride Z methanol solution (3 mL The mixture was stirred at room temperature for 12 hours and concentrated to give 30 mg (quantitative) of the title compound.
- Step 3 Synthesis of 4— (2,3 difluorophenol) thian-4-amamine (Reference compound 30) Under argon atmosphere, titanium tetraisopropoxide (0.3 mL , 1. lOmmol) and diphenylsilane (0.51mL, 2.70mmol) were added and stirred at room temperature. After 17 hours, ethyl acetate and saturated aqueous sodium hydrogen carbonate solution were added, and the resulting insoluble material was removed by Celite filtration. The filtrate was extracted with ethyl acetate, washed with saturated brine, dried and concentrated. The obtained crude product was purified by silica gel column chromatography to obtain 100 mg (40%) of the title compound.
- Step 5 (3R) — 4— (2 Fluorophenol) 1 N— (4— (2, 3 Difluorophenol) —1, 1-Dioxothian 4 yl) -3— ( Synthesis of 2-methyl-2-propoxycarbo-lamino) butanamide (Reference compound 32)
- Step 6 (3R) —3 amino 1- 4— (2 fluorophenol) 1 N— (4— (2, 3 diph Fluorophylol) -1,1,1-Dioxothiane-4yl) butanamide 'hydrochloride (compound 11) synthesis
- Step 3 4 (2,4 Difluorophenol) thiane 4-amamine (Reference compound 35) Composition
- Step 5 (3R) — 4— (2 Fluorophenol) 1 N— (4— (2, 6 Difluorophenol) —1, 1-Dioxothian-4 yl) -3— ( Synthesis of 2-methyl-2-propoxycarbo-lamino) butanamide (Reference compound 47)
- Step 3 4 Synthesis of (3,5 difluorophenol) thian-4-amamine (Reference compound 50)
- Step 3 4 Synthesis of (m-Tolyl) thian-4-amamine (Reference Compound 55)
- Step 5 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force Rubonylamino) N— (4— (m—Tolyl) — 1, 1—Dioxothian— 4— B) Synthesis of butanamide (Reference compound 57)
- Step 6 (3R) — 3 amino 1 4 — (2 fluorophenol) 1 N— (4 — (m-tolyl) — 1, 1 dioxothian 4 yl) butanamide hydrochloride (compound 16) Under an argon atmosphere, (3R) —4— (2 fluorophenyl) —3— (2—methyl—2, double-carboxylamino) N— (4— (m—tolyl) — 1, 1—dioxanthian—4— Yl) butanamide (72 mg, 0.08 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (4 mL), stirred at room temperature for 12 hours and concentrated to obtain 66 mg (quantitative) of the title compound. .
- Step 3 Synthesis of 4— (p-Tolyl) thian-4-amamine (Reference Compound 60) Under argon atmosphere, titanium tetraisopropoxide (0.47mL, 1.57mmol) in THF (lmL) solution of N— (4- (p-tolyl) thian-4-yl) acetamide (391mg, 1.57mmol), Diphenylsilane (0.73 mL, 3.93 mmol) was added and stirred at room temperature. After 17 hours, ethyl acetate and saturated aqueous sodium hydrogen carbonate solution were added, and the resulting insoluble material was removed by Celite filtration. The filtrate was extracted with ethyl acetate, washed with saturated brine, dried and concentrated. The obtained crude product was purified by silica gel column chromatography to obtain 215 mg (94%) of the title compound.
- Step 4 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 propoxy force, Rolamino) N— (4— (p-tolyl) thian—4-yl) butanamide ( Synthesis of Reference Compound 61)
- Step 5 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) N— (4— (p Tolyl) — 1, 1—Dioxothian— 4— Yl) butanamide (Reference compound 62)
- Step 6 (3R) —3 amino 1 4 -— (2 Fluorophenol) 1 N— (4— (p-Tolyl) 1 1, 1 Dioxothian 4yl) butanamide hydrochloride (Compound 17) synthesis (3R) —4— (2 fluorophenol) —3— (2-methyl-2-polyoxyboramino) N under argon atmosphere — (4- (p-tolyl)-1,1-dioxothian-4-yl) butanamide (89 mg, 0.17 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (4 mL) and stirred at room temperature for 12 hours. After concentrating, 80 mg (quantitative) of the title compound was obtained.
- Step 2 Synthesis of N-methyl-4-phenol-4-amine (Reference compound 64)
- N-methyl-N- (4-phenol-4-yl) acetamide 530 mg, 2. 13 mmol
- Titanium tetraisopropoxide (0.63 mL, 2.13 mmol
- diphenylsilane (0.99 mL, 5.31 mmol) were added and stirred at room temperature.
- ethyl acetate and saturated aqueous sodium hydrogen carbonate solution were added, and the resulting insoluble material was removed by celite filtration.
- the filtrate was extracted with ethyl acetate, washed with saturated brine, dried and concentrated.
- the resulting crude product was purified by silica gel column chromatography to obtain 324 mg (73%) of the title compound.
- Step 3 (3R) — 4— (2 Fluorophenol) — N—Methyl—3— (2—Methyl—2—propoxycarbolamino) N— (4-Feltian-4-yl) butanamide ( Reference Synthesis of Compound 65) Under an argon atmosphere, add (3R) —4 ((2-fluorophenyl) 3- (2 methyl —2 propoxycarbo) to a DMF (2 mL) solution of N-methyl-4 phenthian—4 amine (59 mg, 0.28 mmol).
- Step 4 (3R) — 4— (2 Fluorophenol) — N—Methyl—3— (2—Methyl—2—propoxycarbolumino) N— (4-Fue-Lu 1, 1—Dioxothian— 4-yl) butanamide (reference compound 66)
- Step 5 (3R) — 3 Amino 4— (2 Funoleo Mouth Hue-Nole) 1 N—Methinore 1 N— (4—Fel—1, 1 Dioxothian— 4 -yl) butanamide hydrochloride ( Synthesis of Compound 18) Under an argon atmosphere, (3R) —4— (2 fluorophenyl) N-methyl 3— (2-methyl-2-propoxycarbolamino) N— (4 phenyl-1,1, 1-dioxothiane 4 Yl) butanamide (90 mg, 0.17 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (4 mL), stirred at room temperature for 12 hours and concentrated to obtain 70 mg (quantitative) of the title compound. .
- Step 1 Synthesis of 4- (2-fluorophenyl) thiane-4-1ol (reference compound 67) Under an argon atmosphere, a solution of 2 fluorob-mouthed mobenzene (3.91 mL, 36. Ommol) in THF (50 mL) was cooled to -78 ° C, and n-butyllithium Z-hexane solution (1.59 M, 21.6 mL, 34 4 mmol) was added dropwise. After 20 minutes, a solution of 4-oxothiane (4. OOg, 34.4 mmol) in THF (25 mL) was added dropwise.
- Step 3 Synthesis of (2 fluorophenyl) thian-4 amine 'hydrochloride (Reference Compound 69)
- N— (4- (2 fluorophenyl) thian-4-yl) acetamide (5.OOg, 19.7 mmol) was suspended in THF (3 mL), and titanium tetraisopropoxide (5.85 mL, 19.7 mmol) and diphenylsilane (5.50 mL, 29.6 mmol) were added and stirred for 15 hours. After diluting the reaction solution by adding ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added and the mixture was further stirred for 30 minutes. After filtration through Celite, distilled water was added to the filtrate, and the mixture was extracted with ethyl acetate.
- Step 4 (R) — 4— (3 Black Mole) 1 N— (4— (2 Fluorophenyl) Thiane-4-yl) 3-— (2-Methyl-2-propoxycarbolamamino) butanamide (reference Synthesis of compound 70)
- Step 5 (3R) —4 (3 black mouth-nore) N— (4— (2 funoreo mouth-nore) 1, 1-Dioxochian 4 ill) -3— (2— Synthesis of methyl-2-propoxycarbonylamino) butanamide (Reference compound 71)
- Step 6 (3R) — 3 Amino 4— (3 Black Mole) 1 N— (4— (2 Fluorophenol) 1, 1—Dioxothian 1 4-yl) Butanamide Hydrochloride ( Synthesis of compound 19) Under argon atmosphere, (3R) -4- (3 black mouth ring) N— (4- (2 fluorophenol) —1, 1-dioxothiane 4 yl) 3— (2 —Methyl-2-propoxycarbonylaminoamino) butanamide (148 mg, 0.28 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (3 mL) and black mouthform (3 mL), stirred at room temperature for 14 hours and concentrated to give the title compound 1 29 mg (98%) were obtained.
- Step 1 (3R) —4— (2 Black Mole) N— (4— (2 Fluorophenyl) Thiane-4-yl) 3 -— (2-Methyl-2-propoxycarbolamino) butanamide (Reference Compound 74) )
- Step 2 (3R) —4— (2 black mouth-nore) N— (4— (2 funoreo mouth-nore)-1, 1-Dioxochian 4 il) -3— ( Synthesis of 2-methyl-2-propoxycarbonylamino) butanamide (Reference Compound 75)
- Step 3 (3R) — 3 Amino 4— (2 Black Mole) 1 N— (4— (2 Fluorophenol) 1, 1—Dioxothian 1 4-yl) Butanamide Hydrochloride ( Synthesis of compound 21) Under argon atmosphere, (3R) -4- (2 black-mouthed) N— (4- (2 fluorophenol) -1, 1, 1-dioxothiane 4) 3— (2 —Methyl-2-propoxycarbonylaminoamino) butanamide (l lOmg, 0.20mmol) in 10% hydrogen chloride Z methanol solution (3m L), dissolved in black mouth form (3 mL), stirred at room temperature for 14 hours and then concentrated to obtain 9 lmg (94%) of the title compound.
- Step 2 (3R) -4- (4 Black-headed Hue-Nole) N— (4— (2 Funoleo-mouthed Hue-Nole) 1, 1-Dioxothiane 4 Fil) -3 (2 —Synthesis of methyl-2-propoxycarbonylamino) butanamide (Reference Compound 77)
- Step 2 (3R) — 4— (2, 4 Dichlorophenol-Nole) One N— (4— (2 Funoleo-Fue-Nole) —1, 1-Dioxothiane 4-yl) -3— Synthesis of (2-Methyl-2-propoxycarbolamamino) butanamide (Reference Compound 79)
- Step 2 (3R) — 4— (3, 4 Dichlorophenol-NO) 1 N— (4— (2 Funeo Leo-Fue-Nole) —1, 1-Dioxothiane 4) 3— Synthesis of (2-Methyl-2-propoxycarbolamamino) butanamide (Reference Compound 81)
- Step 1 Synthesis of 4- (3-chlorophenyl) thian-4-ol (reference compound 82) 1—Cro-neck 3 1-benzene (1 lmL, 8.7 mmol) in THF (20 mL) was cooled to —78 ° C under argon atmosphere. N-Butyllithium Z-hexane solution (1.59M, 5.4 mL, 8.6 mmol) was added dropwise. After 20 minutes, a solution of 4-oxothiane (1. Og, 8.6 mmol) in THF (10 mL) was added dropwise. 30 minutes after completion of the dropwise addition, the temperature was slowly raised to room temperature, and 30 minutes later, an aqueous salt ammonium solution was added.
- Step 3 4 Synthesis of (3 Chlorophenyl) thian-4-amamine (Reference Compound 84)
- N— (4- (3 chlorophenyl) thian-4-yl) acetamide (62 Omg, 2.30 mmol) was suspended in THF (2 mL), and titanium tetraisopropoxide (678 L, 2.30 mmol) was suspended.
- diphenylsilane (640 / z L, 3.54 mmol) were added, and the mixture was stirred at room temperature for 15 hours. After diluting with ethyl acetate, a saturated aqueous sodium hydrogen carbonate solution was added and the mixture was further stirred for 30 minutes. After filtration through celite, the filtrate was poured into distilled water and extracted with ethyl acetate. The organic layer was washed with distilled water and saturated brine, dried and concentrated. The resulting crude product was purified by silica gel column chromatography to obtain 320 mg (61%) of the title compound.
- Step 5 (313 ⁇ 4 —? ⁇ -(4- (3-chlorophenol) -1,1,1-dioxanthane-4-yl) 4 (2-fluorophenol) 3- (2-methyl-2-propoxy) Carbo-Lamino) Butanamide (Reference Compound 86)
- Step 6 (3R) — 3 Amino N— (4— (3 Closing Fell) 1 1, 1-Dioxothian 4 yl) 4 1 (2 Fluorophenyl) Butanamide Hydrochloride Synthesis of Compound 25) Under an argon atmosphere, ( 3 R) — N— (4— (3 black mouth file) — 1, 1-Dioxothian 4 yl) -4 one (2 Fluorophenol) -3— (2-Methyl-2-propoxycarbonylamino) butanamide (180 mg, 0.33 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (4 mL) and chloroform (2 mL), stirred at room temperature for 15 hours, concentrated. The title compound 1 52 mg (96%) was obtained.
- Step 1 4-—Synthesis of (4 trifluoromethylphenol) thian—4-ol (reference compound 87)
- N— (4 trifluoromethylphenol) thian-4-yl) acetamide (1.20 g, 3.96 mmol) was suspended in THF (3 mL), and titanium tetraisopropoxide ( 1. 17 mL, 3.96 mmol) and diphenol-noresilane (1.10 mL, 5.94 mmol) were added and stirred for 15 hours. After diluting with ethyl acetate, a saturated aqueous sodium hydrogen carbonate solution was added and the mixture was further stirred for 30 minutes.
- Step 4 (3R) —N— (4— (4 trifluoromethylphenol) thiane — 4—yl) —4 1 (2, 4, 5 trifluorotation) -3— ( Synthesis of 2-Methyl-2-propoxycarbolamino) butanamide (Reference Compound 90)
- Step 5 (3R) —N— (4— (4 trifluoromethyl filed) 1, 1, 1-dioxothione 4 yl) 4 1 ( 2 , 4) 5 trifluorophenol) Synthesis of 3- (2-Methyl-2-propoxycarbonylamino) butanamide (Reference Compound 91)
- Step 3 Synthesis of (3 Trifluoromethyl Phenol) thian — 4 Amine (Reference Compound 94)
- N— (4- (3-trifluoromethylphenol) thian-4-yl) acetamide (1.20 g, 3.96 mmol) was suspended in THF (3 mL), and titanium tetra Isopropoxide (1.17 mL, 3.96 mmol) and diphenol-noresisilane (1.10 mL, 5.94 mmol) were added and stirred for 15 hours. After diluting with ethyl acetate, a saturated aqueous sodium hydrogen carbonate solution was added and the mixture was further stirred for 30 minutes.
- Step 4 (3R) —N— (4— (3 trifluoromethylphenol) thiane — 4—yl) —4 1 (2, 4, 5 trifluorotation) -3— ( Synthesis of 2-Methyl-2-propoxycarbolamino) butanamide (Reference Compound 95)
- Step 2 4 Synthesis of 2- (2-Black 6-Funoleo-Fue-Nore) 1 3-— (2-Methinore 2 Propoxycarbonylamino) Butyrate Ethyl (Reference Compound 98)
- Step 3 4 Synthesis of 2- (2 Black Mouth 6 Funoleo Mouth Hue-Nole) 1 3-— (2-Methinore 1 Propoxycarbonylamino) Butyric Acid
- Step 4 (2 Black mouth 6 Funoleo mouth fuel) One N— (4— (2 Funore mouth mouth) Thiane 4 ill) -3— (2-Methyl-2-propoxycarbolamamino) ) Synthesis of butanamide (Reference compound 100)
- Step 5 (2 Black mouth 6 Funoleo mouth fue-nore) 1 N— (4— (2 Funoleo mouth feel) —1, 1-Dioxochian 4 il) -3— Synthesis of (2-Methyl-2-propoxycarbo-lamino) butanamide (Reference Compound 101)
- Step 2 4 (2 Black mouth— 4 Fluorophenol) —3— Synthesis of (2-Methyl-2 propoxycarbonylamino) butyrate (Reference compound 103)
- Step 3 4 Synthesis of 2- (2-chlorotetrafluoro) -3- (2-methyl-2-propoxycarbonylamino) butyric acid (Reference Compound 104)
- Step 4 (2 Black mouth 4 Funoleo mouth fuel) One N— (4— (2 Funore mouth mouth) Thiane 4 ill) -3— (2-Methyl-2-propoxycarbolamamino) ) Butanamide Synthesis of Reference Compound 105
- Step 5 (2 Black mouth 4 Funoleo mouth fuel) 1 N— (4— (2 Funore mouth mouth) —1, 1-Dioxochian 4 ill) -3— Synthesis of (2-Methyl-2-propoxycarbo-lamino) butanamide (Reference Compound 106)
- reaction solution was allowed to cool, THF (30 mL) and an aqueous potassium carbonate solution were added, and the mixture was further stirred for 20 minutes. After decanting the supernatant of the reaction solution in two layers, the aqueous layer was washed with THF.
- the crude product obtained by drying and concentrating the organic layer was roughly purified by silica gel column chromatography. Under an argon atmosphere, sodium borohydride (918 mg, 24.3 mmol) was slowly added to acetic acid (15 ml) at room temperature, and then the acetic acid (5 mL) solution of the crude product was added to the reaction mixture stirred for 30 minutes at room temperature. Stir for 7 hours.
- Step 2 4 Synthesis of 2- (2 Black Mouth 3, 6 Difunoleo Mouth Hue-Nole) 1 3-— (2-Methinore 2 Propoxycarbolamino) Ethyl Butyrate (Reference Compound 108)
- Step 3 4 Synthesis of 2- (2 Black Mouth 3, 6 Difunoleo Mouth Hue-Nole) 1 3-— (2-Methinore 2 Propoxycarbonylamino) Butyric Acid (Reference Compound 109)
- To the solution was added IN sodium hydroxide aqueous solution (6 mL) and stirred for 14 hours.
- 1N Hydrochloric acid was added to weakly acidify, and the mixture was further stirred for 3 hr.
- the precipitated white solid was collected by filtration and dried to obtain 1.17 g (90%) of the title compound.
- Step 4 (2 Black mouth 3,3 Diphnoreo mouth Fu-Nole) One N— (4— (2 Funoleolophane) Cyan-4 Fil) -3— (2-Methyl-2-propoxycarbo- Synthesis of Ruamino) butanamide (Reference Compound 110)
- Step 5 4 (2 Black-and-white 3, 6 Diphnoreo-mouthed Fu-Nore) One N— (4— (2 Funoleolophenyl) 1, 1-Dioxothian— 4-yl) —3— (2-Methyl —2 Synthesis of Propoxybonylamino) butanamide (Reference Compound 111)
- Step 6 3 Amino 4— (2 Black mouth 3, 6 Difluorophenol) 1 N— (4— (2 Fluorophenol) —1, 1-Dioxothian 4 yl) Butanamide hydrochloride Synthesis of compound 30) 4 (2 chloro-3, 6 difluorophenol) N— (4— (2—fluorophenol) —1, 1—dioxothian— 4—yl) —3— (2-methyl-2 pro Poxycarbolamino) butanamide (135 mg, 0.25 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (3 mL) and black mouth form (4 mL), stirred at room temperature for 15 hours and concentrated to give the title compound 116 mg ( 97%).
- Step 2 4 (3 Trifluoromethylphenol) — 3— (2-Methyl-2 propoxycarbonylamino) butyric acid (Reference compound 113)
- Step 3 4 (3 trifluoromethyl phenol) —N— (4— (2 Fluorophenyl) tian-4 yl) -3— (2-Methyl-2-propoxycarbolamamino) butanamide (see Synthesis of compound 114)
- Step 4 4 (3 trifluoromethyl phenol) —N— (4— (2 fluorophenol)
- Step 2 4 (3 Black mouth 6 Funoreo mouth Hue-nore) 1 3— (2—Metinore 1 Propoxy Synthesis of Cycarbonylamino) Ethyl Butyrate (Reference Compound 117)
- Step 3 4 (3 Black mouth 6 Funoleo mouth Hue-Nole) 1 3— (2-Methinore 2 Propoxycarbonylamino) Butyric acid (Reference compound 118) Synthesis
- Step 4 (3 Black mouth 6 Funoleo mouth Fuerole) One N— (4— (2 Funoleo mouth Fuer) Thiane 4 ill) -3 (2-Methyl-2-propoxycarbolamamino) ) Synthesis of butanamide (Reference compound 119)
- Step 5 (3 Black mouth 6 Funoleo mouth Hue-Nole) 1 N— (4— (2 Funoleo mouth Huel) —1, 1-Dioxochian 4 Fil) -3— Synthesis of (2-Methyl-2-propoxycarbo-lamino) butanamide (Reference Compound 120) 4— (3 Black mouth 6 Fluorophenol) -N- (4— (2 Fluorophenol) thian 4 yl) -3— (2-Methyl-2-propoxycarbolamamino) butanamide (115 mg, 0.22 mmol) was dissolved in dichloromethane (10 mL), and mCPBA (> 65%) (95 mg,> 0.36 mmol) was added under ice cooling.
- Step 6 Amino-4 (3 Chloro-6 Funoleo Oral Fenol) N— (4 1 (2 Orenol Orophol) —1, 1-Dioxothian-4-yl) butanamide Hydrochloride ( Synthesis of compound 32)
- reaction solution was poured into distilled water and extracted with ethyl acetate, and the organic layer was washed with dilute hydrochloric acid, distilled water and saturated brine. The organic layer was dried and concentrated, and the resulting crude product was purified by silica gel column chromatography to obtain 493 mg (20%) of the title compound.
- Step 2 4 (3 Black-headed 1, 6 Diphnoreo-mouthed Fu-Nore) One N— (4— (2 Funoleolophane) Thiane 4 ill) -3— (2-Methyl-2-propoxycarbo- Synthesis of Ruamino) butanamide (Reference Compound 122)
- Step 3 4 (3 Black-and-white 2, 6 Diphnoreo-mouth Fue-Nole) One N— (4— (2 Funoleolophenyl) 1, 1-Dioxothiane — 4-yl) —3— (2-Methyl —2 Synthesis of propoxybonylamino) butanamide (reference compound 123)
- Step 1 (3R) —4— (2 Fluorophenol) —3— (2—Methyl-2 propoxyl force Rupolumino) N— (4 Phenylbiperidine 4 yl) butanamide (Reference compound 12 4)
- Step 2 Synthesis of 3 amino 1- 4 — (2 fluorophenol) 1 N— (4 vinylbiperidine-4-yl) butanamide dihydrochloride (compound 34) Under an argon atmosphere, (3R) —4— (2 fluorophenyl) —3— (2-methyl-2-propylcarboxylamino) N— (4-phenylbiperidine-4-yl) butanamide (50 mg, (0.1 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (2 mL), stirred at room temperature for 3 hours and concentrated to obtain 42 mg (89%) of the title compound.
- Step 2 Synthesis of 3-amino 4- (2 Fluoro-Fe) -N- (l-methylsulfo di-ru 4 4-biruberidine 4 yl) butanamide 'hydrochloride (Compound 35) Under argon atmosphere, (3R) —4— (2 fluorophenyl) —3— (2—Methyl-2 propoxycarbolamino) N— (1-methylsulfolulu 4-phenolbiperidine—4 yl) butanamide (55 mg, 0.1 mmol) was dissolved in 10% hydrogen chloride Z methanol solution (3 mL), stirred at room temperature for 9 hours and concentrated to give 51 mg (quantitative) of the title compound.
- Step 2 4 (2-Methyl-2 propoxycarbolamino) — Synthesis of 4-phenolbiperidine (Reference compound 127)
- Step 3 4 (2-Methyl-2 propoxycarbolamino) — Synthesis of 4-ferro- 1- (pyrimidine 2-yl) piperidine (reference compound 128)
- Step 4 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) N— (1— (Pyrimidine 1—2) 4-Ferbiperidine 1 4-yl) butanamide (reference compound 129)
- Step 2 1 Synthesis of Acetyl-4 ferrobiperidine-4-amamine (Reference Compound 131)
- Step 3 (3R) — N— (1-acetyl- 4-phenolbiperidine — 4-yl) —4— (2 fluorophenol) -3— (2-methyl-2-propoxycarbolamamino) butanamide (Reference Compound 132) Synthesis
- Step 4 (3R) — N— (1—Acetyl— 4—Ferbiperidine — 4—yl) 3 Amino— 4— (2 Fluorophenyl) butanamide hydrochloride (Compound 37)
- Step 2 Synthesis of Phenacyl-4 Phenolbiperidine-4-amamine (Reference Compound 134)
- Step 4 (3R) — 3 amino 1 4 — (2 fluorophenol) 1 N— (l phenacyl 1 4 phenylbiperidine 4 yl) butanamide hydrochloride (compound 38)
- Step 3 (3R) — N— (1—Benzyl 1- 4- (2 Fluoro-Phenol) Piperidine 4-4-yl) -4 (2-Fluoro-Phenol) -3— (2-Methyl-2 Synthesis of propoxycarboamino) butanamide (Reference compound 138)
- N— (1—benzyl—4- (2 fluorophenyl) piperidine—4 yl) acetamide (880 mg, 2.7 mmol) was suspended in THF (0. 1 mL) and titanium tetrisoproboxide (800 / z L, 2.7 mmol) and diphenylsilane (750 / z L, 4. Ommol) were added and stirred at room temperature. After 18 hours, the reaction mixture was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was vigorously stirred. The produced white solid was separated by filtration, and the filtrate was separated and extracted with ethyl acetate.
- N— (1-benzyl-4- (3-phenol) piperidine-4-yl) acetamide 200 mg, 0.46 mmol was dissolved in 1,4 dioxane (3 mL).
- copper (I) iodide (4 mg, 0.023 mmol)
- trans-N, N, -dimethyl 1,2 cyclohexanediamine 13 mg, 0.092 mmol
- potassium phosphate 258 mg, 0.997 mmol
- Step 5 (3R) — N— (1—Benzyl 1- 4— (3— (Pyrrol 1-yl) -Fel) Piperidine 4-4-1) 2-4 (2-Fluoro-Fel) Synthesis of 3- (2-Methyl-2-propoxycarbonylamino) butanamide (Reference Compound 143)
- N— (1-benzyl-4- (3-phenol) piperidine-4-yl) acetamide (reference compound 140) 400 mg, 0.92 mmol was added to 1,4 dioxane (4 mL).
- Imidazolene (62mg, 0.92mmol) Yowi copper (I) (9mg, 0.047mmol), 1, 10 phenanthr phosphorus (166mg, 0.092mmol), cesium carbonate (600mg, 1.8) mmol) was added, and the mixture was stirred at 110 ° C for 15 hours. After allowing to cool, the solid was filtered, and the filtrate was concentrated. The resulting crude product was purified by silica gel column chromatography to obtain 217 mg (63%) of the title compound.
- Step 2 Synthesis of benzyl-4- (3- (imidazole-1-yl) phenol) piperidine-4-amine (reference compound 145)
- Step 3 (3R) —N— (1—Benjirou 4— (3— (Imidazol-1 Fil) Fel) piperidine—4—yl) —4— (2 Fluorophenol) —3— ( Synthesis of 2-Methyl-2-propoxycarbonylamino) butanamide (Reference Compound 146)
- N— (1—benzyl— 4— (3 pseudophenol) piperidine — 4— Yl) acetamide (reference compound 140) 200 mg, 0.46 mmol) dissolved in 1,4 dioxane (3 mL), pyrazonole (31 mg, 0.46 mmol), yowi copper (I) (4 mg, 0.023 mmol) ), Trans-N, N, monodimethyl 1,2 cyclohexanediamine (13 mg, 0.092 mmol) and potassium phosphate (258 mg, 0.97 mmol) were added and stirred at 110 ° C. for 15 hours. After allowing to cool, the solid was filtered, and the crude product obtained by concentrating the filtrate was purified by silica gel column chromatography to obtain 165 mg (96%) of the title compound.
- Step 2 1 Synthesis of benzyl-4- (3- (pyrazole-1-yl) phenol) piperidine-4-amine (reference compound 148)
- the resulting crude product was purified by silica gel column chromatography to obtain 1.9 mg (8%) of the title compound (free base).
- the title compound (free base) was made into a methanol solution, salted with 10% salt-hydrogen Z methanol solution, and concentrated to obtain the title compound (trihydrochloride).
- Step 2 (3R) — 4— (2, 4, 5 trifluorophenyl) — N— (4— (2 Fluorophenyl) 1, 1—Dioxothian— 4—yl) —3— (2 —Methyl—2 (propoxycarbonylamino) butanamide (Reference Compound 151)
- N— (1-benzyl-4- (3-phenol) piperidine-4-yl) acetamide (reference compound 140) 400 mg, 0.92 mmol was added to 1,4 dioxane (6 mL).
- 4- (methylthio) furboronic acid 155mg, 0.92mmol
- tetrakis (triphenylphosphine) palladium 106mg, 0.092mmol
- potassium phosphate 7.36mg, 2.8mmol
- Step 2 N (l Benzyl— 4— (3— (4-Methylsulfurylphenol) phenol) Synthesis of piperidine-4-yl) acetamide (reference compound 153)
- N— (1-Benzylru 4-1- (3- (4-methylsulfururfel) phenol) piperidine-4-yl) acetamide (242 mg, 0.52 mmol) was dissolved in THF ( The suspension was suspended in 0.05 mL), and titanium tetraisopropoxide (154 L, 0.52 mmol) and diphenylsilane (146 0.78 mmol) were added and stirred at room temperature. After 16 hours, the mixture was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was vigorously stirred. The resulting white solid was separated by filtration, and the filtrate was separated and extracted with ethyl acetate. The combined organic layers were dried and concentrated, and the resulting crude product was purified by silica gel column chromatography to obtain 146 mg (66%) of the title compound.
- Step 4 (3R) —N— (1—Benzylue 4— (3— (4-Methylsulfurylphenol) tiv) piperidine—4—yl) —4— (2 —Fluorophenol) —3— (2-Methyl-2-propoxycarbonylamino) butanamide (Reference Compound 155)
- Step 1 4 i (3 Yodofueniru) Chian 4 Under argon atmosphere blanket ol (Reference Compound 156), 1, 3 over the jaw de benzene (5. 7 g, 17 mmol) and THF (50 mL) dissolved solution was cooled to 78 ° C N-Butyllithium Z-hexane solution (2.55 M, 6.8 mL, 17 mmol) was added dropwise. After 10 minutes, a solution of 4-oxothiane (2 g, 17 mmol) in THF (30 mL) was added dropwise. After completion of the dropwise addition, the temperature was slowly raised to room temperature. After 2 hours, distilled water was added and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried and concentrated. The resulting crude product was purified by silica gel column chromatography to obtain 4.7 g (85%) of the title compound.
- the obtained crude product (5.8 g) was dissolved in ethanol (90 mL) and acetic acid (18 mL), and thiourea (1.3 g, 18 mmol) was added and heated to reflux. After 12 hours, the mixture was allowed to cool, an aqueous solution of potassium carbonate was added and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried and concentrated.
- the obtained crude product was purified by silica gel column chromatography, Compound 1.8g (38%) was obtained.
- Step 2 (313 ⁇ 4—4— (2-Fluorofer)-? ⁇ -(4— (3--Fodefer) —1, 1—Dioxothian-4 Fil) —3— (2 —Synthesis of methyl-2-propoxycarbonylamino) butanamide (Reference Compound 161)
- Step 4 (3R) — 3 amino 1 4— (2 Fluoro-Fel) 1 N— (4— (3— (Pyridine 4 Fil) file) —1, 1-Dioxothian 4yl) butanamide dihydrochloride (Compound 46)
- Step 1 Synthesis of 2 (Tributyl Star) thiazole (Reference Compound 163) 78.
- n-butyllithium Z-hexane solution (2.55M, 2.5mL, 6.5mmol)
- THF 24mL
- a THF solution (15 mL) of thiazole (500 mg, 5.9 mmol) was added dropwise over 20 minutes and stirred for 1 hour.
- a solution of tryptyl chloride (1.8 mL, 6.5 mmol) in THF (9 mL) was added dropwise over 15 minutes, stirred for 1 hour, saturated aqueous sodium hydrogen carbonate solution and jetyl ether were added, and the mixture was stirred at room temperature for 2 hours. did. After separation, the organic layer was dried and concentrated to give 2.4 g of the title compound as a crude product.
- Step 2 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) -N- (4— (3- (Thiazol-2-yl) phenol -L) — 1, 1—Dioxosothian 4 yl) butanamide (Reference Compound 164)
- Step 4 (3R) —3 Amino— 4— (2 Fluorophenol) — N— (4— (3— (Thiazo One-Loop 5-Fil)) — 1, 1—Dioxothian— 4 —Yil) Butanamide 'Synthesis of hydrochloride (I compound 48)
- Step 2 Synthesis of tributyl star 1 Tritylpyrazole (Reference Compound 169) Under an argon atmosphere, THF (30 mL) was added with n-butyllithium Z-hexane solution (2.55 M, 1. OmL, 2.5 mmol). Cooled to -78 ° C. 4 A THF solution (6 mL) of 1-tritylpyrazole (1. Og, about 2.3 mmol) was added dropwise over 20 minutes and stirred for 1 hour. A THF solution (6 mL) of tributyltin chloride (0.62 mL, 2.3 mmol) was added dropwise over 10 minutes, stirred for 30 minutes, warmed to room temperature, and stirred for 2 hours. An aqueous sodium chloride solution was added, and the mixture was extracted with jetyl ether. The organic layer was dried and concentrated. The resulting crude product was purified by silica gel column chromatography to obtain 1.2 g (84%) of the title compound.
- Step 3 (R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) N— (4— (3— (1—Tritylpyrazole—4-yl) ) Fuel) — 1, 1 Dioxothian 4 yl) Butanamide (Reference Compound 170)
- Step 4 (3R) — 3 Amino 1 4— (2 Fluoro-Fel) 1 N— (4— (3— (Virazo 1-Lu 4-Fil) Fail) — 1, 1—Dioxothian— 4 —Yl) butanamide 'hydrochloride (I compound 49) synthesis
- Step 2 (3R) — 4— (2 Fluorophenol) — N— (4— (3— (1—Methylimidazo One-Loop 5-Fil) Fuel) — 1, 1—Dioxothian— 4 —Yl) —3— (2-Methyl-2 propoxycarbonylamino) butanamide (Reference Compound 172)
- Step 2 (3R) — 4— (2, 4, 5 trifluorofeles) 1 N— (4— (3 pseudofeles) —1, 1-Dioxothian 4 ills) 3— (2—Methyl 2 Propoxycarbo-Luamino) butanamide (Reference Compound 174)
- Step 3 (3R) — 4— (2, 4, 5 trifluorophenol) —3— (2—Methyl-2 propoxycarbolamino) —N— (4— (3 (thiazole-5 yl) ) Fuel) -1,1,1-Dioxothian 4 yl) butanamide (Reference compound 175)
- Step 1 Synthesis of 4- (4-phenyl) thiane-4-1ol (Reference Compound 176) 1,4-Jodobenzene (5.7 g, 17 mmol) in THF (50 mL) was cooled to 78 ° C under an argon atmosphere. Then, n-butyllithium Z-hexane solution (2.55 M, 6.8 mL, 17 mmol) was added dropwise. After 30 minutes, a solution of 4-oxothiane (2. Og, 17 mmol) in THF (30 mL) was added dropwise.
- Step 2 Synthesis of (4 odophenol) thiane 4-amamine (reference compound 177)
- Step 4 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) N— (4— (4— (Thiazol-5-yl) phenol Lu) thian-4-yl) butanamide (reference compound 179) synthesis
- Step 5 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) N— (4— (4— (Thiazol-5-yl) Fe- ) — 1, 1—Dioxosothian — 4-yl) butanamide (Reference Compound 180)
- Step 6 (3R) — 3 Amino 1 4 — (2 Fluoro-Fel) 1 N— (4— (4— (Thiazo 1-Lu 5-F))) 1, 1, 1-Dioxothian 4 —Yl) butanamide 'hydrochloride (compound 52) synthesis
- Step 2 (3R) —4— (2,4 Diclonal Fuenole) —3— (2-Methanole 2 Propoxy carbolumino) N— (4— (3— (Thiazole 5-yl) Hue -L) Synthesis of 1, 1-dioxothiane 4-yl) butanamide (Reference compound 185)
- Step 1 5 Synthesis of tributylstar-2-methylthiazole (reference compound 186) Under an argon atmosphere, add n-butyllithium Z-hexane solution (2.55M, 4.4mL, llmmol) to THF (40mL), 78. Cooled to C. A THF solution (15 mL) of 2-methylthiazole (1. Og, 10 mmol) was added dropwise over 50 minutes and stirred for 1 hour. A THF solution (15 mL) of sodium chloride triptylutin (2.7 mL, lOmmol) was added dropwise over 20 minutes, and the mixture was stirred for 1 hour.
- Step 4 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) -N- (4- (3- (2-Methylthiazole-5 yl) ) Fuel) -1,1,1-Dioxothian-4 yl) butanamide (Reference compound 189)
- n-butyllithium Z-hexane solution (2.55 M, 0.81 mL, 2. 1 mmol) was added to THF (8 mL), and the mixture was cooled to 78 ° C. 2
- a THF solution (3 mL) of piperidinothiazole (316 mg, 1.9 mmol) was added dropwise over 20 minutes, and the mixture was stirred for 1 hour.
- a THF solution (3 mL) of tributyltin chloride (0.51 mL, 1.9 mmol) was added dropwise over 20 minutes and stirred for 1 hour.
- Step 4 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Lolamino) N— (4— (3— (2 Piperidinothiazole—5-—) L) Fer) thian — 4yl) butanamide (Reference Compound 193)
- Step 5 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, L-amino) —N— (4— (3— (2 Piperidinothiazole-5 L) Ferl) 1, 1 Dioxothian 4 yl) butanamide (Reference compound 194)
- Step 3 Synthesis of 4 (thiazole-2-yl) thian-4-amamine (Reference Compound 197) Under argon atmosphere, N— (4 (thiazol-2-yl) thian-4-yl) acetamide (70 mg, 0.29 mmol) Suspended in THF (0.2 mL), titanium tetraisopropoxide (86 / z L, 0.29 mmol) and diphenylsilane (0.15 mL, 0.76 mmol) were added and stirred at room temperature. After 15 hours, the reaction mixture was diluted with ethyl acetate, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was vigorously stirred.
- Step 4 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Lolamino) N— (4— (Thiazol-2-yl) thian — 4— B) Synthesis of butanamide (reference compound 198)
- Step 5 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 propoxy force rubonylamino) —N— (4— (thiazole 2-yl) 1, 1—Dioxothian 1 — Synthesis of butanamide (Reference Compound 199)
- Step 1 Synthesis of 4-oxothiane O benzyloxime (reference compound 200)
- 4-benzothiane 500 mg, 4.30 mmol
- 16% aqueous methanol 60 mL
- O-bendyrocyanine hydrochloride 1.37 g, 8. 60 mmol
- sodium acetate 1.76 g, 21.5 mmol
- the mixture was extracted with ethyl acetate, and the organic layer was washed with saturated brine, dried and concentrated.
- the resulting crude product was purified by silica gel column chromatography to obtain 1.95 g (quantitative) of the title compound.
- Step 3 Synthesis of 4- (Pyridine-2-yl) thian-4-amamine (Reference Compound 202) N-Benzyloxy 4- (Pyridine-2-yl) thian-4amine (300 mg, 1. OOmmol) under argon atmosphere ) was added to a solution of acetonitrile (17 mL) with distilled water (1 mL) and molybdenum hexacarbonyl (264 mg, 1.OOmmol) and heated to reflux. After 3 hours, the mixture was allowed to cool, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with dichloromethane. The organic layer was washed with saturated brine, dried and concentrated. The resulting crude product was purified by silica gel column chromatography to obtain 60 mg (31%) of the title compound.
- Step 5 (3R) — 4— (2 Fluorophenol) —3— (2—Methyl—2 Propoxy force, Rolamino) N— (4— (Pyridine 2-yl) 1, 1—Dioxothian Synthesis of 4-anil) butanamide (Reference compound 204)
- Step 3 (3R) —N— (4— (Benzothiazole-2-yl) thiane — 4-yl) —4 1 (2 fluorophenol) -3— (2-methyl-2-propoxycarbo- Synthesis of Ruamino) butanamide (Reference Compound 207)
- Step 2 (3R) — N— (4— (Benzothiazole 2-yl) 1, 1-Dioxothian —4—yl) —4— (2, 4, 5 Trifluorophenol) —3 — Synthesis of (2-Methyl-2 propoxycarbonylamino) butanamide (Reference Compound 210)
- Step 3 (3R) — 4— (2 Fluorophenol) 1 N— (4— (6—Methoxybenzothiazo 1 lou 2 yl) thian 4 yl) 3— (2—Methyl-2-propoxycarbo) -Luamino) butanamide (Reference Compound 213)
- Step 4 (3R) — 4— (2 Fluorophenyl) 1 N— (4— (6—Methoxybenzothiazo 1 Lu 2 yl) 1, 1—Dioxothian— 4—yl) —3— (2-Methyl-2-Provo Synthesis of (xycarbonylamino) butanamide (Reference compound 214)
- Step 5 (3R) — 3 Amino 4— (2 Fluorophenol) 1 N— (4— (6-Methoxybenzothiazol 2 yl) 1, 1-dioxothian 4 yl) butanamide 'hydrochloride (compound 61)
- the reaction mixture was diluted with chloroform, and saturated aqueous sodium hydrogen carbonate solution was slowly added under ice-cooling. After liquid separation, the mixture was extracted with black mouth form, dried and concentrated. The resulting crude product was purified by silica gel column chromatography to obtain 27 mg (71%) of the title compound (free base).
- the title compound (free base) was dissolved in methanol, and 10% salt-hydrogen Z methanol solution was added dropwise for chlorination. By concentrating this solution, 21 mg of the title compound (hydrochloride) was obtained.
- Step 2 (3R) — 4— (2, 4, 5 trifluorophenol) 1 N— (4— (6-Methoxybenzothiazole-2-yl) —1, 1-dioxothiane 4-yl) 3 Synthesis of — (2-Methyl-2-propoxycarbonylamino) butanamide (Reference Compound 216)
- Step 3 3-amino 4- (2, 4, 5 trifluorophenol) 1 N— (4- (6-methoxybenzothiazol-2-yl) 1, 1-dio Synthesis of xotian-4yl) butanamide 'hydrochloride (compound 63)
- Step 2 (3R) —N— (4— (6 Black Mouth Benzothiazole-2-yl) thiane-4-yl) — 4— (2 Fluorophenol) —3— (2-Methyl- 2 Synthesis of propoxycarboamino) butanamide (Reference compound 218) Under an argon atmosphere, a solution of 6-clobenzothiazole (95 mg, 0.56 mmol) in THF (5 mL) was cooled to ⁇ 78 ° C., and n-butyllithium Z-hexane solution (1.57 M, 0.31 mL, 0 49 mmol) was added dropwise.
- the obtained crude product was dissolved in methanol (2 mL) under an argon atmosphere, and 10% hydrochloric acid-hydrogen Z-methanol (4 mL) was added thereto, followed by stirring at room temperature for 19 hours.
- the crude product obtained by concentration in an argon atmosphere was dissolved in DMF (3 mL) and dissolved in (3R) —4— (2-Fluorophthalate) -3— (2-Methyl-2-propoxycarbo- Luamino) butyric acid (22 mg, 0.08 mmol), ⁇ (32 ⁇ ⁇ , 0.25 mmol), and HATU (31 mg, 0.08 mmol) were added, and the mixture was stirred at room temperature.
- Step 3 (3R) —N— (4— (6 Black mouth benzothiazole 2-yl) 1, 1-dioxothiane 4-yl) —4— (2 Fluorophenol) —3— (2 Synthesis of —methyl-2-propoxycarbonylamino) butanamide (Reference Compound 219)
- Step 2 (3R) —4 (2 Fluorophenyl) —N— (4— (4 Methylbenzothiazo 1 Ru 2 yl) 1, 1—Dioxothian— 4—yl) —3— (2 Synthesis of —Methyl-2-propoxycarbonylamino) butanamide (Reference Compound 221)
- Step 3 (3R) —4 (2 Fluorophenyl) —N— (4— (6 Methylbenzothiazo 1 Lu 2 yl) 1, 1—Dioxothian — 4—yl) —3— (2 Synthesis of —Methyl-2-propoxycarbonylamino) butanamide (Reference Compound 224)
- Step 4 (3R) —3 amino 1- 4— (2 fluorophenol) 1 N— (4 -— (6-methylbenzothiazole-2-yl) 1,1-dioxothiane 4-yl) butanamide 'hydrochloride (compound 67 )
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
明 細 書
新規な非環状アミンカルボキシアミド誘導体及びその塩
技術分野
[0001] 本発明は、新規な経口投与可能な糖尿病の治療又は予防剤、及び非環状アミンカ ルポキシアミド誘導体又はその薬学的に許容される塩に関する。
背景技術
[0002] 糖尿病とは、インスリン作用不足による慢性の高血糖状態を主徴とする代謝疾患群 であり、 1型と 2型に大別されることが多い。 1型糖尿病がインスリンを合成 '分泌する 脾ランゲルハンス島 β細胞の破壊 ·消失力 Sインスリン作用不足の主要な原因であるの に対して、 2型糖尿病は、インスリン分泌低下やインスリン抵抗性をきたす素因を含む 複数の遺伝因子に、過食 (特に高脂肪食)、運動不足、肥満、ストレスなどの環境因 子及び加齢が加わり発症する。また、糖尿病による長期にわたる高血糖状態の持続 は、糖尿病網膜症、糖尿病腎症、糖尿病神経障害のいわゆる糖尿病三大合併症の 他、失明、心血管障害等につながる危険性が高い。従って、糖尿病及び糖尿病の前 段階 (いわゆる、境界型)において認められる耐糖能異常、食後高血糖、空腹時高血 糖などを含めて、血糖の適正なコントロールは、 QOL (quality of life,生活の質)の観 点からも極めて重要である(非特許文献 1)。
[0003] 現在、用いられて ヽる糖尿病治療剤は、インスリン製剤、トルプタミド、ァセトへキサ ミド、クロルプロパミド、トラザミド、グリクロビラミド、ダリブゾール、ダリベンクラミド、ダリ クラジド、グリメピリド等のスルホニルゥレア剤(ナテグリニド、ミチグリニド、レパグリニド 等の速効型インスリン分泌促進薬を含む)、メトホルミン (ビグアナイド剤)、ボグリボー ス、ァカルボース、ミグリトール等の a—ダルコシダーゼ阻害剤、ピオグリタゾン、ロシ グリタゾン等のチアゾリジンジオン類に分類できる。インスリン製剤は強力な血糖低下 作用を有する反面、しばしば重篤な低血糖を引き起こす。また、体重増加も引き起こ すことがある。更には、多くのインスリン製剤は注射による投与が必要で、患者に対す る負担も大きい。スルホニルゥレア剤も強力な血糖低下作用を有する反面、しばしば 重篤な低血糖を引き起こす。また、体重増加や、長期間投与した場合には効果が低
くなるいわゆる 2次無効が起こることがある。メトホルミンは、血糖低下作用の他、イン スリン抵抗性改善作用等も有するが、しばしば下痢、膨満等の胃腸管障害を引き起 こすことがある。また、頻度は低いが重篤な乳酸アシドーシスを起こすことがあり、腎 障害、肝機能障害のある患者には禁忌である。 a—ダルコシダーゼ阻害剤は、スル ホニルゥレア剤等と比較するとその効力は弱いものの血糖低下作用を示すが、腹痛
、下痢、膨満等の胃腸障害を引き起こすことがある。チアゾリジンジオン類は、メトホ ルミンと同様、血糖低下作用に加えてインスリン抵抗性改善作用等も有するが、しば しば体重増カロ、浮腫を起こすことがあり、心不全患者や心不全の既往者には適さな い。また、類薬で重篤な肝障害を起こし死亡例が報告されていることより、定期的な 肝機能検査が必要である。いずれの薬剤も血糖低下作用を示すが、種々の欠点、副 作用を有しており、効果的で使い勝手のよい糖尿病治療薬が望まれている (非特許 文献 1 5)。
最近注目されて 、る糖尿病治療薬の新たな作用機序の 1つとして、グルカゴン様ぺ プチドー 1 (glucagon- like peptide- 1、以下 GLP- 1と略す)が挙げられる。 GLP- 1はイン クレチンと称される消化管ホルモンの 1つで、インスリン分泌促進作用、グルカゴン分 泌抑制作用等の生体の糖代謝改善に有用な作用や、膝ランゲルノヽンス島 β細胞の 保護作用があるとされている。また、 GLP-1は食事摂取により分泌される、すなわち、 血糖がある程度高くなつた時にインスリン分泌促進作用を示すため、一般に食前や 強 ヽ運動中及び運動後に起こりやす 、低血糖が起こりにく 、と考えられて 、る。更に は、 GLP-1は胃運動抑制作用や食欲抑制作用も有していることより、糖尿病の危険 因子の 1つである体重増加が起こりにくいとも考えられている。し力し、 GLP-1は主に ジぺプチジルぺプチダーゼ IV (dipeptidyl peptidase- IV.以下、 DPP- IVと略す)と 呼ばれる酵素により加水分解されて不活性ィ匕し、その半減期が 2分以下と非常に短 いため、 GLP-1自身を糖尿病治療薬として用いることは困難であった (非特許文献 6 — 8)。最近、酵素的加水分解を受けにくい GLP-1類縁体の臨床研究が実施されて いるが、いずれも経口投与は不可能である(非特許文献 8)。血中の GLP-1濃度を相 対的に上昇させる方法としては、先に述べた非加水分解性 GLP-1を投与する方法の 他に、加水分解酵素である DPP-IVを阻害する方法が考えられる。従って、 DPP-IV阻
害薬もまた、従来の経口糖尿病薬において認められる低血糖、体重増加などの副作 用は起こらないことが期待されている。今までに、 DPP-IV阻害薬は数多く報告されて おり、スルホスチン誘導体、キサンチン誘導体、イソキノロン誘導体及びその類縁体、 イソキノリン誘導体及びその類縁体、ピリジン又はピリミジン誘導体などがあるが、そ の大部分は環状アミンカルボキシアミド誘導体である(非特許文献 9— 11)。このよう に、今までに知られて!/、る DPP-IV阻害薬の大部分は環状アミンカルボキシアミド誘 導体であるが、非環状アミンカルボキシアミド誘導体の例も知られている(特許文献 1 — 4)。し力しながら、これらのなかで具体的に DPP-IV阻害活性が開示されているの は国際公開第 WO05Z40095号 (特許文献 3)の実施例化合物 1及び 10のみであ り、各々の IC 値は 0. 46 μ Μ及び 0. 45 μ Μである。更には、特許文献 3記載の実
50
施例化合物 1及び 10を含めて、糖尿病治療効果を示す実験結果を開示して!/、るも のは皆無である。
非特許文献 1 :日本糖尿病学会編、糖尿病治療ガイド 2004— 2005
非特許文献 2 : Moller D. E.著、「ネイチヤー」 (Nature)、 2001年、第 414卷、 p.821-827 非特許文献 3 : Skyler J. S.著、「ジャーナル'ォブ 'メデイシナル 'ケミストリー」(Journal of Medicinal Chemistry)、 2004年、第 47卷、 p. 4113-4117
非特許文献 4 : Ross S. A.他著、「ケミカル 'レビュー」(Chemical Review)、 2004年、第 104卷、 p. 1225-1282
非特許文献 5 : Stumvoll M.他著、「ランセット」(Lancet)、 2005年、第 365卷、 p. 1333- 1346
非特許文献 6 :Vilsboll T.他著、「ダイアベトロジァ」(Diabetologia)、 2004年、第 47卷 ゝ p. 357-366
非特許文献 7 : D'Alessio D. A.他著、「アメリカン 'ジャーナル'ォブ 'フィジオロジー —エンドクライノロジ一'アンド'メタボリズム」 (American Journal of Physiology - Endoc rinology and Metabolism)、 2004年、第 286卷、 p. E882- E890
非特許文献 8 : Knudse L. B.著、「ジャーナル'ォブ 'メデイシナル 'ケミストリー」(Journ al of Medicinal Chemistry)、 2004年、第 47卷、 p. 4128-4134
非特許文献 9 :Augustyns K.他著、「カレント'メディシナル 'ケミストリー」(Current Me
dicinal Chemistry) , 1999年、第 6卷、 p. 311-327
非特許文献 10 :Augustyns K.他著、「エキスパート'オピニオン'オン'セラピュティック 'パテンッ」 (Expert Opinion on Therapeutic Patens)、 2003年、 13卷、 p. 499-510 非特許文献 11 : Weber A. E.著、「ジャーナル'ォブ 'メデイシナル 'ケミストリー」(Jour nal of Medicinal Chemistry)、 2004年、第 47卷、 p. 4135-4141
特許文献 1:国際公開第 04Z37181号パンフレット
特許文献 2:国際公開第 05Z25554号パンフレット
特許文献 3:国際公開第 05Z40095号パンフレット
特許文献 4:国際公開第 05Z95343号パンフレット
発明の開示
発明が解決しょうとする課題
[0005] 本発明は、副作用が少な 、経口投与可能な糖尿病の治療又は予防剤、及び該治 療又は予防剤として有用な新規化合物を提供することを目的とする。
課題を解決するための手段
[0006] 本発明者らは、前記目的を達成するため鋭意検討した結果、新規な非環状アミン カルボキシアミド誘導体を見出し、本発明を完成した。
[0007] すなわち本発明は、一般式 (I)
[化 1]

[0008] [式中、 Arは、任意の 1〜5個の R3で置換されていてもよいフエ-ルを表し、
R3は、ハロゲン、ヒドロキシ、シァ入炭素数 1〜6のアルキル又は炭素数 1〜6、好まし くは炭素数 1〜3のアルキルォキシを表し(ここで、アルキル又はアルキルォキシは 1 〜5個のハロゲンで置換されて!、てもよ!/、)、
R1は、水素又は炭素数 1〜6のアルキルを表し(ここでアルキルは、任意の 1〜5個、 好ましくは 1〜3個のハロゲン又はヒドロキシで置換されていてもよい)、
Yは、 NR4—、 一 O 、 一 S(=0)p 又は。一フエ-レンを表し、
R4は、水素、炭素数 1〜6のアルキル、ァリール、ヘテロァリール、ァリールで置換され た炭素数 1〜3のアルキル、ヘテロァリールで置換された炭素数 1〜3のアルキル、 C(=0)— R5、 一 CH— C(=0)— R5又は一 SO— R5を表し、
2 2
R5は、炭素数 1〜6のアルキル、ァリール又はへテロアリールを表し、
pは、 0、 1又は 2を表し、
m及び nは、各々独立して、 1、 2又は 3を表し、
R2は、 R6又は R7力も選択される任意の 1〜3個の置換基で置換されていてもよいァリ ール又はへテロアリールを表し、
R6は、炭素数 1〜6のアルキル、炭素数 1〜6のアルキルォキシ(ここで、アルキル又 はアルキルォキシは 1〜5個のハロゲンで置換されていてもよい)、シクロアルキル、 ァリールォキシ、ヘテロァリールォキシ、ヒドロキシ、ハロゲン、シァ入 0— (CH )q
2
— R8、 —SO— R5又は— NR 9'を表す力、 2個の隣接する R6が一緒になつてメチレン
2
ジォキシ又は炭素数 3又は 4のアルキレンを表し、
R8は、炭素数 1〜3のアルキルォキシ(ここで、アルキルォキシは 1個の炭素数 1〜3 のアルキルォキシ又は 1〜5個のハロゲンで置換されていてもよい)、ァリールォキシ 、ヘテロァリールォキシ、シァ入シクロアルキル(ここで、シクロアルキルの 1個のメチ レンは、— 0—、— NR9 又は— S(=0)p のいずれかで置換されていてもよい)、 N — (C=0)— R5、— (C=0)— NR9R9'又は— S(=0)p— R5を表し、
R9及び R9'は、各々独立して、水素、炭素数 1〜6のアルキル(ここで、アルキルは 1個 の炭素数 1〜3のアルキルォキシ又は 1〜5個のハロゲンで置換されていてもよい)を 表すか、又は R9、 R9'及び窒素が一緒になつてピロリジン、ピぺリジン (ここで、ピロリジ ン又はピペリジンは 1〜5個のハロゲンで置換されていてもよい)、ピぺラジン、 N—ァ ルキルピペラジン(ここでアルキルの炭素数は 1〜6である)、モルホリン、チオモルホ リン又はチオモルホリン 4, 4 ジォキシドを表し、
R1Q及び R1Q'は、各々独立して、水素、炭素数 1〜6のアルキル(1個の炭素数 1〜3の アルキルォキシ又は 1〜5個のハロゲンで置換されていてもよい)を表すか、又は R1Q、 R1Q'、窒素が一緒になつてピロリジン、ピぺリジン(ここで、ピロリジン又はピぺリジンは 1
〜5個のハロゲンで置換されていてもよい)、ピぺラジン、 N アルキルピぺラジン(こ こでアルキルの炭素数は 1〜6である)、モルホリン、チオモルホリン又はチオモルホリ ンー 4, 4 ジォキシドを表し(ここで、ピロリジン、ピぺリジン、ピぺラジン、 N—アルキ ルビペラジン、モルホリン、チオモルホリン又はチオモルホリン 4, 4 ジォキシドの 1個のメチレンは、ォキソで置換されていてもよい)、
qは、 0、 1、 2又は 3を表し、
R7は、ァリール又はへテロアリールを表し(ここで、ァリール又はへテロアリールは、任 意に選択される 1〜3個の R11で置換されていてもよい)、
R11は、炭素数 1〜3のアルキル、ハロゲン、炭素数 1〜6のアルキルォキシ、トリフルォ ロメチル、 -SO— R5又は一 NR9R3を表し、
2
Xは、原子価結合又は炭素数 1〜6のアルキレンを表す。 ]
で示される非環状アミンカルボキシアミド誘導体又はその薬学的に許容される塩、並 びに該誘導体又は塩を有効成分として含有する医薬組成物、特に糖尿病の治療又 は予防剤、及び血糖低下剤を提供する。また、前記一般式 (I)で示される非環状アミ ンカルボキシアミド誘導体又はその薬学的に許容される塩の、糖尿病の治療又は予 防剤、及び血糖低下剤の製造のための使用を提供する。更に、前記一般式 (I)で示 される非環状アミンカルボキシアミド誘導体又はその薬学的に許容される塩の有効量 を患者に投与することを含む、糖尿病の治療又は予防方法及び血糖低下方法を提 供する。
[0009] 一般式 (I)の化合物は 1つ以上の不斉点を有しており、このため、ェナンチォマー、 ラセミ体又はジァステレオマー等の立体異性体が存在し得る力 本発明化合物は、 いずれの立体異性体又はこれら異性体の混合物も含むものである。
[0010] 本明細書にお!、て、「アルキル」とは、直鎖又は分岐の飽和炭化水素基を意味し、 具体的に例えれば、メチル、ェチル、 1 プロピル、 2—プロピル、 1ーブチル、 2—ブ チル、 2—メチルー 2—プロピル、 1, 1ージメチルェチル、 1 ペンチル、 2—ペンチ ル、 3 ペンチル、 1一へキシル等が挙げられる力 これらに限られるわけではない。
[0011] 「アルキルォキシ」とは、前記アルキルが酸素原子に結合した官能基を表し、具体 的に例えれば、メトキシ、エトキシ、 1 プロボキシ、 2—プロボキシ、 1 ブトキシ等が
挙げられる力 これらに限られるわけではない。
[0012] 「シクロアルキル」とは、炭素数 3〜8個、好ましくは炭素数 3〜6個からなる飽和脂環 式炭化水素基を意味し、具体的に例えれば、シクロプロピル、シクロブチル、シクロべ ンチル、シクロへキシルが挙げられる。
[0013] 「アルキレン」とは、直鎖又は分岐の飽和炭化水素鎖を意味し、具体的に例えれば
、メチレン、エチレン、トリメチレン、テトラメチレン等が挙げられる力 これらに限られる わけではない。
[0014] 「ァリール」とは、炭素数 6〜10個からなる芳香族基を意味し、具体的に例えれば、 フエニル、ナフチル等が挙げられる力 これらに限られるわけではない。
[0015] 「ヘテロァリール」とは、 S、 N、 Oから任意に選択されるへテロ原子 1〜4個を含む、 総原子数 5〜17個、好ましくは総原子数 5〜16個、更に好ましくは総原子数 5〜: LO 個からなる複素環式芳香族基を意味し、その複素環が NHを含む場合、その水素は 炭素数 1〜6、好ましくは炭素数 1〜3のアルキル又は (CH )q— R8 (q
2 、 R8は前記定 義に同じ)で置換されていてもよい。また、窒素原子は酸化されていてもよい。具体的 に例えれば、チェ-ル(2 チェ-ル、 3 チェ-ル)、ピロリル(1 ピロリル、 2 ピロ リル、 3 ピロリル)、 1 メチルピロリル(1 メチルピロ一ルー 2 ィル、 1ーメチルピ ロール一 3—ィル)、 1—ェチルピロリル(1—ェチルピロール一 2—ィル、 1—ェチノレ ピロ一ルー 3 ィル)、フリル(2 フリル、 3 フリル)、チアゾリル(チアゾールー 2—ィ ル、チアゾールー 4 ィル、チアゾールー 5 ィル)、チアゾールー 3 ォキシドー 2 ーィル(すなわち、チアゾールー 2 ィルの N—ォキシド)、チアゾールー 3—ォキシド 4 ィル、チアゾールー 3—ォキシドー 5—ィル、チアゾリルー 3—ォキシド(すなわ ち、チアゾリルの N—ォキシド)、イミダゾリル(イミダゾールー 1 ィル、イミダゾールー 2 ィル、イミダゾール 4 ィル)、 1 メチルイミダゾリル( 1 メチルイミダゾール 2 ィル、 1 メチルイミダゾールー 4 ィル、 1 メチルイミダゾールー 5 ィル)、ォ キサゾリル(ォキサゾールー 2 ィル、ォキサゾールー 4 ィル、ォキサゾールー 5— ィル)、ビラゾリル(ピラゾールー 1 ィル、ピラゾールー 3 ィル、ピラゾールー 4ーィ ル)、 1 メチルピラゾリル( 1 メチルピラゾール 3 ィル、 1 メチルピラゾール 4 ィル、 1ーメチルビラゾールー 5 ィル)、イソォキサゾリル (イソォキサゾールー 3
ィル、イソォキサゾールー 4 ィル、イソォキサゾールー 5—ィル)、 1H- 1, 2, 3 ートリアゾリノレ(1H—1, 2, 3 トリァゾーノレ 1ーィノレ、 1H- 1, 2, 3 トリァゾーノレ —4—ィル、 1H- 1, 2, 3 トリァゾール— 5—ィル)、 1H- 1, 2, 4 トリァゾリル(1 H- 1, 2, 4 トリァゾール— 1—ィル、 1H- 1, 2, 4 トリァゾール— 3—ィル)、 2H 1, 2, 3 卜ジァゾジノレ(2H— 1, 2, 3 卜ジァゾ一ノレ 2—ィノレ、 2H- 1, 2, 3 卜!; ァゾーノレ一 4—ィノレ)、 1—メチノレ一 1, 2, 3 トリアゾリノレ(1—メチノレ一 1, 2, 3 トリ ァゾールー 4 ィル、 1ーメチルー 1, 2, 3 トリァゾールー 5—ィル)、 1ーメチルー 1 , 2, 4 トリアゾリノレ(1—メチノレ一 1, 2, 4 トリァゾーノレ一 3—ィノレ、 1—メチノレ一 1, 2, 4 トリァゾーノレ一 5—ィノレ)、 2—メチノレ一 1, 2, 3 トリアゾリノレ(2—メチノレ一 1, 2, 3 トリァゾール— 4—ィル)、 1, 3, 4—ォキサジァゾール— 2—ィル、 1H—テトラ ゾリル(1H—テトラゾールー 1 ィル、 1H—テトラゾールー 5—ィル)、 2H—テトラゾリ ル(テトラゾールー 2 ィル)、 1ーメチルテトラゾリル(1ーメチルテトラゾールー 5—ィ ル)、 2—メチルテトラゾリル(2—メチルテトラゾールー 5 ィル)、ピリジル(ピリジン 2 ィル、ピリジン一 3—ィル、ピリジン一 4—ィル)、ピリジン一 1—ォキシド一 2—ィル (すなわち、ピリジンー2—ィノレの N—ォキシド)、ピリジンー1 ォキシドー 3—ィノレ、 ピリジン一 1—ォキシド一 4—ィル、ピリジル一 1—ォキシド(すなわち、ピリジルの N— ォキシド)、ピリミジ-ル(ピリミジン一 2—ィル、ピリミジン一 4—ィル、ピリミジン一 5—ィ ル)、ピリミジン一 1—ォキシドー 2—ィル(すなわち、ピリミジン一 2—ィルの N—ォキ シド)、ピリミジン一 1—ォキシド一 4—ィル、ピリミジン一 3—ォキシド一 4—ィル、ピリミ ジン— 1—ォキシド— 5—ィル、ピリミジン— 3—ォキシド— 5—ィル、ピリミジン— 1— ォキシドー 6 ィル、ピリミジンー3 ォキシドー 6 ィル、ピリミジニルー 1 ォキシド( すなわち、ピリミジ -ルの 1位窒素の N—ォキシド)、ピリミジ -ル— 3—ォキシド (すな わち、ピリミジ -ルの 3位窒素の N ォキシド)、ピラジ -ル、ピラジュルー 1ーォキシド (すなわち、ピラジュルの N—ォキシド)、ビラジニル 1—ォキシド (すなわち、ピラジ ニルの N—ォキシド)、インドリル(インドールー 1 ィル、インドールー 2—ィル、インド 一ルー 3—ィル、インドールー 4 ィル、インドールー 5—ィル、インドールー 6—ィル 、インドールー 7—ィル)、 1 メチルインドリル(1 メチルインドールー 2 ィル、 1 メチルインドールー 3 ィル、 1 メチルインドールー 4 ィル、 1 メチルインドール
5 ィル、 1 メチルインドールー 6 ィル、 1 メチルインドールー 7 ィル)、ベン ゾイミダゾリル(ベンゾイミダゾールー 1—ィル、ベンゾイミダゾールー 2—ィル、ベンゾ イミダゾールー 4 ィル、ベンゾイミダゾールー 5—ィル、ベンゾイミダゾールー 6—ィ ル、ベンゾイミダゾール 7 ィル)、 1 メチルベンゾイミダゾール 2 ィル、 1 メ チルベンゾイミダゾールー 4 ィル、 1 メチルベンゾイミダゾールー 5 ィル、 1ーメ チルベンゾイミダゾールー 6 ィル、 1 メチルベンゾイミダゾールー 7 ィル、 1ー(2 ーメトキシェチル)ベンゾイミダゾールー 2—ィル、 1一(2—メトキシェチル)ベンゾイミ ダゾールー 4—ィル、 1— (2—メトキシェチル)ベンゾイミダゾールー 5—ィル、 1— (2 ーメトキシェチル)ベンゾイミダゾールー 6 ィル、 1一(2—メトキシェチル)ベンゾイミ ダゾール - 7-ィル、ベンゾチアゾール - 2-ィル、ベンゾチアゾール 4 ィル、ベ ンゾチアゾール 5—ィル、ベンゾチアゾール 6—ィル、ベンゾチアゾール 7—ィ ル、ベンゾチアゾールー 3 ォキシドー 2—ィル(すなわち、ベンゾチアゾールー 2— ィノレの N—ォキシド)、ベンゾチアゾーノレ 3—ォキシドー 4ーィノレ、ベンゾチアゾー ルー 3—ォキシド 5—ィル、ベンゾチアゾール 3—ォキシド 6—ィル、ベンゾチ ァゾールー 3—ォキシドー 7—ィル、ベンゾイミダゾリルー 3—ォキシド(すなわち、ベ ンゾイミダゾリルの N—ォキシド)、キノリル(キノリン一 2—ィル、キノリン一 3—ィル、キ ノリンー 4 ィル、キノリン一 5—ィル、キノリン一 6—ィル、キノリン一 7—ィル、キノリン —8—ィル)、キノリン一 1—ォキシド一 2—ィル(すなわち、キノリン一 2—ィルの N— ォキシド)、キノリン 1 ォキシドー 3 ィル、キノリン 1 ォキシドー 4 ィル、キノ リン一 1—ォキシド一 5—ィル、キノリン一 1—ォキシド一 6—ィル、キノリン一 1—ォキ シド一 7—ィル、キノリン一 1—ォキシドー 8—ィル、キノリル一 1—ォキシド(すなわち、 キノリンの N—ォキシド)、イソキノリル (イソキノリン一 1—ィル、イソキノリン一 3—ィル、 イソキノリン一 4—ィル、イソキノリン一 5—ィル、イソキノリン一 6—ィル、イソキノリン一 7—ィル、イソキノリン一 8—ィル)、イソキノリン一 2—ォキシド一 1—ィル(すなわち、ィ ソキノリン 1 ィルの N—ォキシド)、イソキノリン 2 ォキシドー 3 ィル、イソキノ リンー2 ォキシドー 4ーィノレ、イソキノリンー2 ォキシドー 5 ィル、イソキノリンー2 —ォキシド一 6—ィル、イソキノリン一 2—ォキシド一 7—ィル、イソキノリン一 2—ォキ シド一 8 ィルイソキノリル 2 ォキシド(すなわち、イソキノリルの N -ォキシド)等
が挙げられるが、これらに限られるわけではない。
[0016] 「ァリールで置換されたアルキル」又は「ヘテロァリールで置換されたアルキル」とは 、前記ァリール又はへテロアリールが 1又は 2個置換した炭素数 1〜3、好ましくは炭 素数 1〜2のアルキルを意味し、具体的に例えれば、ベンジル、ジフエ-ルメチル、 1 フエニルェチル、 2—フエニルェチル、ピリジン 2—ィルメチル、ピリジン 1ーォ キシドー 2 ィルメチル、ピリジンー3 ィルメチル、ピリジン 1 ォキシドー 3—ィル メチル、ピリジンー4 ィルメチル、ピリジン 1 ォキシドー 4 ィルメチル、フエニル (ピリジン— 2—ィル)メチル、フエ-ル(ピリジン— 1—ォキシドー 2—ィル)メチル、フ ェ-ル(ピリジン一 3 ィル)メチル、フエニル(ピリジン一 1 ォキシド一 3 ィル)メチ ル、フエ-ル(ピリジン一 4—ィル)メチル、フエ-ル(ピリジン一 1—ォキシド一 4—ィル )メチル、フランー2 ィルメチル、フランー3 ィルメチル、ピロ一ルー 1ーィルメチル 、ピロ一ルー 2 ィルメチル、 1 メチルピロ一ルー 2 ィルメチル、ピロ一ルー 3—ィ ルメチル、 1 メチルピロ一ルー 3 ィルメチル、インドールー 1 ィルメチル、インド 一ルー 2 ィルメチル、インドールー 3 ィルメチル、インドールー 4 ィルメチル、ィ ンドール 5—ィルメチル、インドールー 6—ィルメチル、インドールー 7—ィルメチル 等が挙げられる力 これらに限られるわけではない。
[0017] 「ァリールォキシ」又は「ヘテロァリールォキシ」とは、前記ァリール又はへテロアリー ルが酸素原子に結合した官能基を表し、具体的に例えれば、フエノキシ、 1 ナフチ ルォキシ、 2—ナフチルォキシ、ピリジン 2—ィルォキシ、ピリジン 1 ォキシドー 2 ィルォキシ、ピリジン 3 ィルォキシ、ピリジン 1 ォキシドー 3 ィルォキシ、 ピリジン 4 ィルォキシ、ピリジン 1 ォキシドー 4 ィルォキシ、ピラジュルォキ シ、ピラジン 1 ォキシドー 2—ィルォキシ、ピリミジン 2—ィルォキシ、ピリミジン 1 ォキシドー 2 ィルォキシ、ピリミジン 4 ィルォキシ、ピリミジン 1 ォキシ ドー 4—ィルォキシ、ピリミジン一 3—ォキシドー 4—ィルォキシ等が挙げられる力 こ れらに限られるわけではない。
[0018] 「ハロゲン」とは、フッ素、塩素、臭素又はヨウ素を意味する。
[0019] また、本明細書にお!ヽて、「糖尿病」とは、 WHO (世界保健機構)、日本糖尿病学会 、米国糖尿病学会又は欧州糖尿病学会等の診断基準に従い糖尿病と診断された病
態を意味し、 1型糖尿病、 2型糖尿病、妊娠糖尿病等が含まれる。「治療又は予防剤 」には、治療及び予防のいずれか一方に用いられるもののみならず、治療及び予防 の両方に同時に用いられるものも包含される。「予防剤」には、糖尿病とは診断されな いが、耐糖能異常又は空腹時血糖異常のいずれかを示す、又は耐糖能異常及び空 腹時血糖異常のいずれをも示す状態に用いられるものも包含される。「治療又は予 防方法」には、治療及び予防のいずれか一方を実施する方法のみならず、治療及び 予防の両方を同時に実施する方法も包含される。「予防方法」には、糖尿病とは診断 されないが、耐糖能異常又は空腹時血糖異常のいずれかを示す、又は耐糖能異常 及び空腹時血糖異常の!/ヽずれをも示す状態にお!ヽて実施する方法も包含される。ま た、「血糖低下剤」とは、血糖値を低下させる効果を示す薬剤のことを意味し、糖尿病 の食後過血糖の改善作用や糖尿病における食後血糖推移の改善作用などの効果も 含まれる。「血糖低下方法」とは、血糖値を低下させる方法のことを意味し、糖尿病の 食後過血糖の改善方法や糖尿病における食後血糖推移の改善方法なども含まれる 発明の効果
[0020] 本発明の非環状アミンカルボキシアミド誘導体は、以下の実施例において示すとお り、良好な経口活性を有する血糖低下作用を示した。更に、本発明の非環状アミンカ ルポキシアミド誘導体による血糖低下作用の作用機序を鋭意検討した結果、作用機 序の 1つとして DPP-IV阻害活性があることが明ら力となった。驚くべきことに、本発明 化合物は、非環状アミンカルボキシアミド誘導体であるにもかかわらず、以下の実施 例において示すとおり、強力な DPP-IV阻害活性を示した。これらの事実より、本発明 化合物は経口投与可能で、かつ低血糖や体重増加などの副作用が起こらな 、ことが 期待できる糖尿病の治療又は予防剤として、極めて有用であるといえる。
発明を実施するための最良の形態
[0021] 一般式 (I)中、 Arに置換可能である R3は、ハロゲン (フッ素、塩素、臭素、ヨウ素)又は トリフルォロメチルが好ましぐ中でも、フッ素又は塩素が好ましい。置換数は、 1〜3 が好ましい。
[0022] R1は、水素、メチル又はェチルが好ましぐ中でも、水素が好ましい。
[0023] Yは、 NR4—、 一 O 、 一 S 又は一 S(=0)—が好ましぐ中でも、 0—又は一 S(
2
=0)一が好ましい。
2
[0024] R4は、水素、炭素数 1〜6のアルキル(例えば、メチル、ェチル、 1 プロピル、 2- プロピル、 1ーブチル、 2—ブチル、 2—メチルプロピル、 1 ペンチル、 1一へキシル など)、ヘテロァリール(例えば、ピリジンー2 ィル、ピリジンー3 ィル、ピリジンー4 —ィル、ピリミジン— 2—ィル、ピリミジン— 4—ィル、ビラジ-ルなど)、ァリールで置換 された炭素数 1〜2のアルキル(例えば、ベンジル、フエ-ルェチル(2—フエ-ルェ チル)など)、 C(=0)— R5 (例えば、ァセチル、プロピオ-ル、ベンゾィル、ニコチニ ル、イソニコチュルなど)、 CH— C(=0)— R5 (例えば、ァセトニル、フエナシルなど)
2
又は— SO— R5 (例えば、メチルスルホ -ル、ェチルスルホ -ル、 1—プロピルスルホ
2
-ル、フエ-ルスルホ -ル、ピリジン— 4—ィルスルホ-ルなど)、中でも、水素、メチ ル、ェチル、ピリジンー2 ィル、ピリジンー3 ィル、ピリジンー4 ィル、ピリミジン— 2 ィル、ピリミジン— 4—ィル、ベンジル、ァセチル、ベンゾィル、ニコチュル、イソ- コチニノレ、フエナシノレ、メチノレスノレホ-ノレ、ェチノレスノレホ-ノレ、フエ-ノレスノレホニノレが 好ましい。
[0025] m及び nは、 m及び nの合計が 2、 3又は 4が好ましぐ中でも、 m及び nが共に 2である 場合が好ましい。
[0026] R2は、単環性のァリール又はへテロ原子が 1から 3個、好ましくは 1又は 2個のへテロ ァリール (ここで、ァリール又はへテロアリールは、 R6又は R7力 選択される任意の 1〜 3個の置換基で置換されていてもよい)が好ましぐ下記の構造で示されるァリール又 はへテロァリール (ここで、ァリール又はへテロアリールは、 R6又は R7力 選択される 任意の 1〜3個の置換基で置換されて 、てもよ 、)がより好ま 、。
[化 2]

[化 3]

[0028] R は、水素、メチル、ェチル、 2—メトキシェチル、 2 エトキシェチル又は 2 モル ホリノエチルが好ましい。
[0029] R6は、炭素数 1〜6、好ましくは炭素数 1〜3のアルキル(1〜5個のハロゲンで置換 されていてもよい)(例えば、メチル、ェチル、 1 プロピル、 2—プロピル、 2—メチル —2 プロピル、トリフルォロメチル、 2, 2, 2 トリフルォロェチル、 3, 3, 3 トリフル ォロプロピル、 4, 4, 4 トリフルォロブチルなど)、炭素数 1〜6のアルキルォキシ(1 〜5個のハロゲンで置換されていてもよい)(例えば、メトキシ、エトキシ、 2 プロポキ シ、トリフルォロメトキシ、 2, 2, 2 トリフルォロエトキシ、 3, 3, 3 トリフルォロプロボ キシ、 4, 4, 4—トリフルォロブトキシなど)、炭素数 3〜8、好ましくは炭素数 3〜6のシ クロアルキル(シクロプロピル、シクロブチル、シクロペンチル、シクロへキシル)、へテ ロアリールォキシ(例えば、ピリジンー2 ィルォキシ、ピリジンー3 ィルォキシ、ピリ ジンー4 ィルォキシ、ピリミジン 2 ィルォキシ、ピリミジンー4 ィルォキシ、ビラ ジ-ルォキシなど)、ヒドロキシ、ハロゲン(フッ素、塩素、臭素、ヨウ素)、シァ入ー0 -(CH )q— R8 (例えば、 2—メトキシエトキシ、 2—エトキシエトキシ、 2—(2—プロポキ
2
シ)エトキシ、 3—メトキシプロボキシ、 3 エトキシプロポキシ、 3—(2 プロポキシ)プ ロポキシ、 2- (2—メトキシェトキシ)エトキシ、 2- (3—メトキシプロボキシ)エトキシ、 2- (2, 2, 2 卜!;フノレ才 Pェ卜キシ)ェ卜キシ、 2- (3, 3, 3 卜!;フノレ才 Pプ Pポキシ) エトキシ、 3—(2—メトキシェトキシ)プロポキシ、 3—(3—メトキシプロポキシ)プロボ キシ、 3- (2, 2, 2 HJフノレ才 Pェ卜キシ)プ Pポキシ、 3—(3, 3, 3 卜!;フノレ才 Pプ ロポキシ)プロポキシ、 2 フエノキシエトキシ、 3 フエノキシプロポキシ、 2 (ピリジ ン 2—ィルォキシ)エトキシ、 2—(ピリミジン一 2—ィルォキシ)エトキシ、 2—(ピリミ
ジンー4 ィルォキシ)エトキシ、 2—ビラジニルォキシエトキシ、 3 (ピリジンー2—ィ ルォキシ)プロポキシ、 3 (ピリミジン一 2 ィルォキシ)プロポキシ、 3 (ピリミジン一 4 ィルォキシ)プロポキシ、 3 ビラジニルォキシプロポキシ、 2 シァノエトキシ、 3 シァノプロポキシ、シクロプロピルォキシ、シクロブチルォキシ、シクロペンチルォキ シ、シクロへキシル才キシ、 2H—テトラヒドロピランー4 ィルォキシ、ォキセタン 3 ィルォキシ、ピぺリジン 4 ィルォキシ、 1ーメチルビペリジン 4 ィルォキシ、 チアンー 4 ィルォキシ、チアンー 1, 1ージォキシドー 4 ィルォキシ、シクロプロピ ルメトキシ、シクロブチルメトキシ、シクロペンチルメトキシ、シクロへキシノレメトキシ、(2 H—テトラヒドロピラン一 4—ィル)メトキシ、(ォキセタン一 3—ィル)メトキシ、(ピベリジ ン一 4—ィル)メトキシ、(1—メチルビペリジン一 4—ィル)メトキシ、(チアン一 4—ィル )メトキシ、(チアン一 1, 1—ジォキシド一 4—ィル)メトキシ、 2— (シクロプロピル)エト キシ、 2—(シクロブチル)エトキシ、 2—(シクロペンチル)エトキシ、 2—(シクロへキシ ル)エトキシ、 2— (2H—テトラヒドロピラン一 4—ィル)エトキシ、 2— (ォキセタン一 3— ィル)エトキシ、 2— (ピペリジン一 4—ィル)エトキシ、 2— (1—メチルビペリジン一 4— ィル)エトキシ、 2— (チアン— 4—ィル)エトキシ、 2— (チアン— 1, 1—ジォキシド— 4 —ィル)エトキシ、 3— (シクロプロピル)プロポキシ、 3— (シクロブチル)プロポキシ、 3 (シクロペンチル)プロポキシ、 3 (シクロへキシル)プロポキシ、 3—(2H—テトラヒ ドロピラン一 4—ィル)プロポキシ、 3— (ォキセタン一 3—ィル)プロポキシ、 3— (ピぺ リジン— 4—ィル)プロポキシ、 3— (1—メチルビペリジン— 4—ィル)プロポキシ、 3— (チアンー4 ィル)プロポキシ、 3—(チアン—1, 1ージォキシドー 4 ィル)プロポキ シ、 2—アミノエトキシ、 2—(メチルァミノ)エトキシ、 2—(ジメチルァミノ)エトキシ、 2— (2—メトキシェチルァミノ)エトキシ、 2—(3—メトキシプロピルァミノ)エトキシ、 2—(2 , 2, 2—トリフルォロェチルァミノ)エトキシ、 2—モルホリノエトキシ、 2—チオモルホリ ノエトキシ、 2—(チオモルホリン— 4, 4ージォキシドー 1 ィル)エトキシ、 2 ピペリ ジノエトキシ、 2 ピロリジノエトキシ、 2— (4, 4 ジフルォロピペリジン一 1—ィル)ェ トキシ、 2— (4—メチルビペラジン一 1—ィル)エトキシ、 2— (2—ォキソピロリジン一 1 —ィル)エトキシ、 2— (2—ォキソモルホリン— 1—ィル)エトキシ、 2— (4—メチル—2 ーォキソピペラジン 1 ィル)エトキシ、 3 ァミノプロポキシ、 3—(メチルァミノ)プロ
ポキシ、 3 (ジメチルァミノ)プロポキシ、 3—(2—メトキシェチルァミノ)プロポキシ、 3 — (3—メトキシプロピルァミノ)プロポキシ、 3— (2, 2, 2 トリフルォロェチルァミノ) プロポキシ、 3—モルホリノプロボキシ、 3—チオモルホリノプロボキシ、 3—(チオモル ホリン一 4, 4ージォキシドー 1 ィル)プロポキシ、 3 ピペリジノプロポキシ、 3 ピロ リジノプロポキシ、 3— (4, 4ージフルォロピペリジン 1 ィル)プロポキシ、 3—(4 メチルビペラジン 1 ィル)プロポキシ、 3一(2 ォキソピロリジン 1 ィル)プロボ キシ、 3—(2 ォキソモルホリン 1 ィル)プロポキシ、 3—(4ーメチルー 2 ォキソ ピぺラジン 1 ィル)プロポキシ、ァセチルメトキシ、ベンゾィルメトキシ、ニコチュル メトキシ、イソニコチニルメトキシ、 2—ァセチルエトキシ、 2—ベンゾィルエトキシ、 2— ニコチニルエトキシ、 2 イソニコチニルエトキシ、 3 ァセチルプロポキシ、 3 ベン ゾィルプロポキシ、 3—ニコチニルプロポキシ、 3—イソニコチニルプロポキシ、(ァミノ カルボ-ル)メトキシ、(メチルァミノカルボ-ル)メトキシ、(ジメチルァミノカルボ-ル) メトキシ、(ェチルァミノカルボ-ル)メトキシ、(ジェチルァミノカルボ-ル)メトキシ、(2 —メトキシェチルァミノカルボニル)メトキシ、(2, 2, 2—トリフルォロェチルァミノカル ボ -ル)メトキシ、(モルホリノカルボ-ル)メトキシ、(ピペリジノカルボ-ル)メトキシ、( ピロリジノカルボ-ル)メトキシ、(4ーメチルビペラジン 1ーィルカルボ-ル)メトキシ 、 2—(ァミノカルボ-ル)エトキシ、 2—(メチルァミノカルボ-ル)エトキシ、 2—(ジメチ ルァミノカルボ-ル)エトキシ、 2—(ェチルァミノカルボ-ル)エトキシ、 2—(ジェチル ァミノカルボ-ル)エトキシ、 2—(2—メトキシェチルァミノカルボ-ル)エトキシ、 2— ( 2, 2, 2—トリフルォロェチルァミノカルボ-ル)エトキシ、 2— (モルホリノカルボ-ル) エトキシ、 2—(ピペリジノカルボ-ル)エトキシ、 2—(ピロリジノカルボ-ル)エトキシ、 2—(4ーメチルビペラジン 1ーィルカルボ-ル)エトキシ、 3 (ァミノカルボ-ル)プ ロポキシ、 3—(メチルァミノカルボ-ル)プロポキシ、 3—(ジメチルァミノカルボ-ル) プロポキシ、 3—(ェチルァミノカルボ-ル)プロポキシ、 3—(ジェチルァミノカルボ- ル)プロポキシ、 3—(2—メトキシェチルァミノカルボ-ル)プロポキシ、 3—(2, 2, 2 トリフルォロェチルァミノカルボ-ル)プロポキシ、 3—(モルホリノカルボ-ル)プロ ポキシ、 3—(ピペリジノカルボ-ル)プロポキシ、 3—(ピロリジノカルボ-ル)プロポキ シ、 3—(4ーメチルビペラジン 1ーィルカルボ-ル)プロポキシ、 2 (メチルチオ)
エトキシ、 3 (メチルチオ)プロポキシ、 2 (メチルスルホ -ル)エトキシ、 3 (メチル スルホ -ル)プロポキシなど)、—SO— R5 (例えば、メチルスルホ -ル、ェチルスルホ
2
ニノレ、 1—プロピノレスノレホ -ノレ、フエ-ノレスノレホニノレ、ピリジン一 4—イノレスノレホ -ノレな ど)又は一 NR9R9' (例えば、アミ入メチルァミノ、ジメチルアミ入ェチルァミノ、ジェチ ルァミノ、ェチルメチルアミ入モルホリノ、ピペラジ入 N—メチルビペラジ入 N ェチ ルビペラジ入ピペリジ入ピロリジノなど)が好ましい。また、隣接する 2個の R6がー緒 になってメチレンジォキシ、トリメチレン又はテトラメチレンとなる場合も好ましい。中で も、メチル、ェチル、 2—プロピル、 2—メチルー 2—プロピル、トリフルォロメチル、メト キシ、ェ卜キシ、卜!;フノレ才 Pメ卜キシ、 2, 2, 2 卜!;フノレ才 Pェ卜キシ、 3, 3, 3 卜!;フ ルォロプロポキシ、 4, 4, 4 トリフルォロブトキシ、シクロプロピル、ピラジュルォキシ 、ヒドロキシ、フッ素、塩素、シァ入 2—メトキシエトキシ、 2—エトキシエトキシ、 2- (2 プロポキシ)エトキシ、 2—(2—メトキシエトキシ)エトキシ、 2—(2, 2, 2—トリフノレオ 口エトキシ)エトキシ、 2- (3, 3, 3 トリフルォロプロポキシ)エトキシ、 3—メトキシプ ロポキシ、 3 エトキシプロポキシ、 2 フエノキシエトキシ、 2 シァノエトキシ、 2 モ ルホリノエトキシ、 2—(チオモルホリン— 4, 4ージォキシドー 1 ィル)エトキシ、 2— ピベリジノエトキシ、 2- (4, 4 ジフルォロピペリジン— 1—ィル)エトキシ、 2— (4— メチルビペラジン 1 ィル)エトキシ、 3 モルホリノプロボキシ、 2 (ジメチルァミノ )ェトキシ、 2- (2—メトキシェチルァミノ)エトキシ、 2—(3—メトキシプロピルァミノ)ェ トキシ、 2— (2, 2, 2—トリフルォロェチルァミノ)エトキシ、 2— (2—ォキソピロリジン —1—ィル)エトキシ、 2— (2—ォキソモルホリン— 1—ィル)エトキシ、 2— (4—メチル —2—ォキソピペラジン一 1—ィル)エトキシ、(ジメチルァミノカルボ-ル)メトキシ、(ジ ェチルァミノカルボ-ル)メトキシ、(2—メトキシェチルァミノカルボ-ル)メトキシ、(2, 2, 2—トリフルォロェチルァミノカルボ-ル)メトキシ、(ピペリジノカルボ-ル)メトキシ 、(モルホリノカルボ-ル)メトキシ、(4ーメチルビペラジン 1ーィルカルボ-ル)メト キシ、 2—(ジメチルァミノカルボ-ル)エトキシ、 2—(2—メトキシェチルァミノカルボ -ル)エトキシ、 2— (2, 2, 2—トリフルォロェチルァミノカルボ-ル)エトキシ、 2H— テトラヒドロピラン一 4—ィルォキシ、 2— (2H—テトラヒドロピラン一 4—ィル)エトキシ 、 2—(メチルチオ)エトキシ、 2—(メチルスルホ -ル)エトキシ、メチルスルホ -ル、ジ
メチルァミノ又はモルホリノが好ましい。隣接する 2個の R6が一緒になつてメチレンジ ォキシ又はテトラメチレンとなる場合も特に好ましい例である。
[0030] R7は、フエ-ル、ピリジン一 2—ィル、ピリジン一 3—ィル、ピリジン一 4—ィル、ピリジ ンー 1 ォキシドー 2 ィル、ピリジン 1 ォキシドー 3 ィル、ピリジン 1 ォキシ ドー 4—ィノレ、ピロール一 1—ィル、ピロール一 2—ィル、 1—メチルピロール一 2—ィ ル、ピロ一ルー 3 ィル、 1 メチルピロ一ルー 3 ィル、フランー2 ィル、フランー3 ィル、チォフェン 2 ィル、チォフェン 3 ィル、イミダゾールー 1 ィル、イミダ ゾール 2 ィル、 1 メチルイミダゾール 2 ィル、イミダゾール 4 ィル、 1ーメ チルイミダゾールー 4 ィル、 1 メチルイミダゾールー 5 ィル、ピラゾールー 1ーィ ル、ピラゾールー 3 ィル、 1ーメチルピラゾールー 3 ィル、 2—メチルピラゾールー 3 ィル、ピラゾールー 4 ィル、 1ーメチルピラゾールー 4 ィル、ォキサゾールー 2 ィル、ォキサゾールー 4 ィル、ォキサゾールー 5 ィル、チアゾールー 2 ィル、 チアゾールー 4ーィル又はチアゾールー 5—ィルが好ましぐ中でも、フエ-ル、ピリジ ンー4 ィル、ピリジン 1 ォキシドー 4 ィル、ピロ一ルー 1 ィル、イミダゾール 1 ィル、 1 メチルイミダゾールー 5 ィル、ピラゾールー 1 ィル、ピラゾールー 4 ィル、チアゾールー 2 ィル、チアゾールー 4ーィル又はチアゾールー 5 ィルが 好ましい。
[0031] R11は、炭素数 1〜6、好ましくは炭素数 1〜3のアルキル(例えば、メチル、ェチル、 1 プロピル、 2—プロピルなど)、ハロゲン (フッ素、塩素、臭素、ヨウ素)、炭素数 1〜 6、好ましくは炭素数 1〜3のアルキルォキシ(例えば、メトキシ、エトキシ、 1—プロボ キシなど)、トリフルォロメチル、—SO—R5 (例えば、メチルスルホ -ル、ェチルスルホ
2
ニノレ、 1—プロピノレスノレホ -ノレ、フエ-ノレスノレホニノレ、ピリジン一 4—イノレスノレホ -ノレな ど)又は一 NR9R9' (例えば、アミ入メチルァミノ、ジメチルアミ入ェチルァミノ、ジェチ ルァミノ、ェチルメチルアミ入モルホリノ、ピペラジ入 N—メチルビペラジ入 N ェチ ルビペラジ入ピペリジ入ピロリジノなど)が好ましぐ中でも、メチル、ェチル、フッ素、 塩素、メトキシ、エトキシ、トリフルォロメチル、メチルスルホニル、モルホリノ又はピペリ ジノが好ましい。
[0032] Xは、原子価結合、メチレン又はエチレンが好ましぐ中でも原子価結合が好ましい。
[0033] 薬学的に許容される塩としては、酸付加塩の場合には、塩酸塩、硫酸塩、硝酸塩、 臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩、酢酸塩、トリフルォロ酢酸 塩、乳酸塩、クェン酸塩、シユウ酸塩、ダルタル酸塩、リンゴ酸塩、酒石酸塩、フマル 酸塩、マンデル酸塩、マレイン酸塩、安息香酸塩、フタル酸塩等の有機カルボン酸 塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、 p—トルエンス ルホン酸塩、カンファースルホン酸塩等の有機スルホン酸塩、ァスパラギン酸塩、グ ルタミン酸塩等の酸性アミノ酸塩等が挙げられ、中でも、塩酸塩、臭化水素酸塩、リン 酸塩、トリフルォロ酢酸塩、クェン酸、フマル酸、酒石酸塩、メタンスルホン酸塩等が 好ましいが、これらに限られるものではない。また、一般式 (I)にフエノール性水酸基な どの酸性置換基が存在する場合には塩基との塩も可能であり、その場合には、ナトリ ゥム塩、カリウム塩等のアルカリ金属塩、カルシウム塩、マグネシウム塩等のアルカリ 土類金属塩、アルミニウム塩、アンモ-ゥム塩、またトリメチルァミン塩、トリェチルアミ ン塩、ピリジン塩、ピコリン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノー ルァミン塩、ジシクロへキシルァミン塩、 N, N—ジベンジルエチレンジァミン塩等の有 機塩基塩、アルギニン塩、リジン塩、オル-チン塩等の塩基性アミノ酸塩等が挙げら れ、中でも、ナトリウム塩、カリウム塩、エタノールアミン塩等が好ましいが、これらに限 られるものではない。
[0034] 本発明の一般式 (I)の化合物のうち、次に示した Arが 2-フルオロフェ -ル、 R1が水 素、 Yがー NR4—、 R4がベンジル、 m及び nが共に 2、 Xが原子価結合、 R2がフエ-ル、 R7がフエニル、 R11がメチルスルホ -ルである化合物(実施例化合物 44のフリー塩基)
[化 4]

[0035] を(3R)— 3—ァミノ一 N— (1—ベンジル一 4— (2— (4—メチルスルホユルフェ-ル)
フエ-ル)ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミドと命名する。 また、本発明の一般式 (I)の化合物のうち、次に示した Arが 2, 4, 5 トリフルオロフ ェ -ル、 R1が水素、 Yが SO、 m及び nが共に 2、 Xが原子価結合、 R2がべンゾチアゾー
2
ルー 3 ォキシドー 2—ィルである化合物(実施例化合物 84のフリー塩基):
[化 5]

[0037] を(3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 3—ォキシド一 2—ィル) 1, 1 —ジォキソチアン一 4—ィル) 4— (2, 4, 5—トリフルオロフェ -ル)ブタナミドと命名 する。
[0038] また、本発明の一般式 (I)の化合物のうち、次に示した Arが 2, 4, 5 トリフルオロフ ェ -ル、 R1が水素、 Yがー 0—、 m及び nが共に 2、 Xが原子価結合、 R2が 1位にメチル 、 5位に 2—メトキシェトキシ基を置換基に持つベンゾイミダゾールー 2—ィルである化 合物(実施例化合物 141のフリー塩基):
[化 6]

[0039] を(3R)—3 アミノー 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (5— (2—メト キシェトキシ)一 1—メチルベンゾイミダゾールー 2—ィル) 2H—テトラヒドロピラン一 4 ィル)ブタナミドと命名する。
[0040] 本発明の化合物には、下記一般式 (II)及び (III)
[化 7]

[0041] [式中 Yは前記定義に同じ。 R3a、 R3b、 R3 ま水素又は前記 R3の定義に同じ。 R& R6bは 水素又は前記 R6の定義に同じ。 ]
[化 8]

[0042] [式中 Yは前記定義に同じ。 R3a、 R3b、 R3eは水素又は前記 R3の定義に同じ。 R6 R6bは 水素又は前記 R6の定義に同じ。 ]
で示される化合物も包含される。
[0043] 一般式 (II)及び (III)で示される化合物の具体例を表 1及び 2に示す。
[化 9]

[表 1]


0044


〔〕0046
o -n ■n -n -n -n -n -n -n 3
-π -η -π -n ■n "n -n -n m m ~n -n ~n -n ¾
刀
~n
w ω ω ω in o o 〇 o o O O O O 〇 O O O O o Ο Ο 〇 O O O O O O Ο O o O O o O O O 〇 〇 o O O 〇 O o O O O O O O o O O o ~<
〇 O O o O o O O O O O O 〇 O O Ο ο 〇 〇 〇 O O O 〇 ο O O O O 〇 〇 O 〇 O o 〇 〇 O O 〇 〇 〇 〇 o 〇 o O 〇 〇 o 〇 ≤:


0048
〔s I

0049
表 2

(表 2の :

前記一般式 (I)で示される、経口投与可能な糖尿病の治療又は予防剤の有効成分 として用いられる非環状アミンカルボキシアミド誘導体は、具体的には下記スキーム
に示す方法によって製造することができる力 これに限られるわけではない。下記ス キームにより示されな力つた方法に関しては、実施例において具体的に開示した。 一般式 (I)で示される化合物は、アミノ酸 (IV)とァミン (V)とを縮合して化合物 (VI) とし、化合物 (VI)の保護基 Pを除去することにより製造することができる (スキーム 1)。 アミノ酸 (IV)については、市販品より入手可能、又は Cole D. C.他著「テトラへドロン 」(Tetrahedron)、 1994年、 50卷、 p 9517-9582.; Cardillo G.他著「ケミカル'ソサイエ ティー'レビューズ」(Chemical Society Reviews) , 1996年、 25卷、 ρ 117- 128.; Juaristi
E.編「ェナンチォセレクティブ.シンセシス.ォブ. 13ーァミノ'ァシッズ」 (Enantioselec tive Synthesis of β -Amino Acids)、 1997年、 ワイリ^—— VHC (Wiley— VCH) .; Juaristi
E.他著「カレント'メディシナル 'ケミストリー」(Current Medicinal Chemistry)、 1999 年、 6卷、 p 983-1004.; Liu M.他著「テトラへドロン」(Tetrahedron)、 2002年、 58卷、 p
7991-8035. ;Angelaud R.他著「ジャーナル'ォブ 'オルガニック'ケミストリー」(Journal of Organic Chemistry)、 2005年、 70卷、 p 1946-1952.などに記載の方法により合成 し入手することが可能である。縮合の方法としては、一般的なペプチド合成法 (第 5版 実験化学講座 16有機化合物の合成 IV、 pp. 258-270など)、又は一般的なカルボ ン酸アミド合成法 (第 5版 実験化学講座 16有機化合物の合成 IV、 pp. 118-134.; Be nz G.著「コンプリへンシブ 'オノレガニック'シンセシス」(Comprehensive Organic Synt hesis)、 1991年、 6卷、 p.381- 417、パーガモン'プレス (Pergamon Press).; Bailey P.D. 他著「コンプリへンシブ ·オルガニック 'ファンクショナル ·グループ ·トランスフォーメー ンヨンス」 (し omprehensive Organic Functional Group Transformations)、 1991年、 5^ 、 p.257- 307、パーガモン'プレス (Pergamon Press).;田辺 他著「有機合成化学協会 誌」、 2004年、 62卷、 p 1249-1259. ;Montalbetti C.A.G.N.他著「テトラへドロン」(Tetr ahedron)、 2005年、 61卷、 10827-10852.など)により満足いく結果が得られる。保護 基 Pの除去は、一般的な脱保護法 (Green T.W.他著「プロテクティブ'グループス 'ィ ン.オルガニック.シンセシス」(Protective Groups in Organic Synthesis)、第 5版、 199 9年、ジョン 'ヮイリ一'アンド'サンズ (John Wiley & Sons) .など)を用いることにより、 一般式 (I)で示される化合物を製造することができる。スキーム中の Ar、
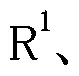 Y、 m、 n、 X、 R2は前記定義に同じであり、 Pは適当な保護基を表す。
[化 11]
Y、 m、 n、 X、 R2は前記定義に同じであり、 Pは適当な保護基を表す。
[化 11]

(I)
スキーム 1
更に、一般式 (I)で示される化合物において Yが SO又は SOである化合物 ( )は
2
、化合物 (VI' ) (ィ匕合物 (VI)において、 Yが Sである化合物)を酸ィ匕して化合物 (VI" )とした後、保護基 Pを除去することにより製造することも可能である (スキーム 2)。酸 ィ匕剤としては、過酢酸、過トリフルォロ酢酸、 m—クロ口過安息香酸 (以下、 mCPBA と略す)などの過酸、 tーブチルヒドロペルォキシド、タメンヒドロペルォキシド、ビス(ト リメチルシリル)ペルォキシド、ジメチルジォキシラン、過酸化ベンゾィル等の有機過 酸化物、過酸化水素、過硫酸カリウム、過ヨウ素酸ナトリウム等の無機過酸化物等を 用いることが可能であるが、多くの場合、 mCPBAで良好な結果が得られる。通常、 1 〜5当量、好ましくは 1〜3当量の酸化剤を用いることにより、満足な結果が得られる。 溶媒は、メタノール、エタノール等のアルコール系溶媒、ジクロロメタン、クロ口ホルム 等のハロゲン系溶媒、酢酸、トリフルォロ酢酸 (以下、 TFAと略す)等の有機カルボン 酸溶媒の他、アセトン、水等も使用可能である。反応温度は、— 100°C〜100°Cで実 施可能であるが、 0°C〜30°Cで良好な結果を与える場合が多い。反応は 1日以内に 完結することが多ぐ通常、 1時間以内に反応は完結する。また、過ルテニウム (VII)酸 テトラ- n-プロピルアンモ-ゥム(以下、 TPAPと略す)も有効な酸化剤の一例である。 TPAPを等量以上用いて反応を実施することも可能である力 通常、触媒量の TPA Pと N—メチルモルホリン N—ォキシド(以下、 NMOと略す)などの共酸化剤を用い ることで満足な結果が得られる。 TP APの触媒量としては、 0. 001モル%〜90モル %で実施可能であるが、 0. 1モル%〜50モル%で良好な結果を与える場合が多い
。共酸化剤の量は、 1〜50当量で実施可能であるが、通常、 1〜10当量で良好な結 果を与える。溶媒は、ジクロロメタン、クロ口ホルム等のハロゲン系溶媒、ァセトニトリル 、プロピオ-トリルなどの-トリル系溶媒、 N, N—ジメチルホルムアミド(以下、 DMFと 略す)及びこれら溶媒の混合物の使用が可能であるが、多くの場合、ジクロロメタン、 ァセトニトリル、 DMFで良好な結果が得られる。反応温度は 0°C〜50°Cで実施可能 であるが、通常、室温で良好な結果が得られる。反応は、 1時間〜 3日で完結すること が多い。反応が遅い場合には、反応時間を長くするよりも、酸化剤又は/及び共酸ィ匕 剤を追加する方が良好な結果が得られる場合がある。また、モレキュラーシーブス( 以下、 MSと略す)の添加も、有効な手段である。スキーム中の Ar、 P、
 m、 n、 p、 X 、 R2は前記定義に同じである。
m、 n、 p、 X 、 R2は前記定義に同じである。
[化 12]

(VI') (VI")
スキーム 2
[0054] アミノ酸 (IV)については、前述の通り、市販品より入手可能、又は文献記載の方法 により合成することにより入手可能である力 以下に、ラセミ体のアミノ酸 (IV)及び光 学活性なアミノ酸 (IV' )の具体的な合成法を示す (スキーム 3、 4)。
[0055] アミノ酸 (IV)は、各々市販品より入手可能である化合物 (VII)とブロモ酢酸エステ ル (VIII)とを反応させて化合物 (IX)とし、化合物 (IX)を還元して得られるアミノ酸ェ ステル (X)のァミノ基に保護基 Pの導入、エステルの加水分解を行うことにより製造可 能である (スキーム 3)。化合物(IX)の製造は、 Kishi Y.他著「ジャーナル'ォブ 'オル ガ-ック.ケミストリー」(Journal of Organic Chemistry)、 1983年、 48卷、 p 3833-3835. に記載のェナミノエステルの一般的合成法などに記載の方法により実施可能である
。化合物(IX)の還元反応は、 Palmieri G.他著「ジャーナル'ォブ 'オルガニック'ケミ ストリー」(Journal of Organic Chemistry)ゝ 1994年、 59卷、 p 5328-5335.などに記載 の方法により実施可能である。ァミノ基への保護基 Pの導入及びエステルの加水分解
は、一般的な方法(Green T.W.他著「プロテクティブ'グループス'イン'オルガニック 'シンセシス」 (Protective Groups in Organic Synthesis)、第 5版、 1999年、ジョン'ワイ リ一'アンド'サンズ (John Wiley & Sons) .など)により実施可能である。スキーム中の Ar、 Pは前記定義に同じであり、 R13は、炭素数 1〜6のアルキル又はベンジルを表す
[化 13]
0 NH2 0
Ar ^C + Brゝ
OR 13 Ar、 J* OR13
(VII) (VIII) (IX)

(X) (IV)
スキーム 3 光学活性なアミノ酸 (IV' )は、各々市販品より入手可能である化合物 (VII)とブロモ 酢酸エステル (VIII)とを反応させて化合物 (XI)とし、エステルの加水分解により得ら れる化合物 (ΧΠ)を不斉還元することにより、化合物 (ΧΠΙ)に変換する。これをアミド 化した後、環化反応を実施して得られたラタタム (XV)を開環反応、続いてベンジル ォキシ基を除去してアミノ酸エステル (Χ' )とした後、ァミノ基への保護基 Ρの導入及 びエステルの加水分解を実施することにより製造可能である (スキーム 4)。化合物 (X
I)の製造は、 Kishi Y.他著「ジャーナル'ォブ 'オルガニック'ケミストリー」(Journal of Organic Chemistry)、 1983年、 48卷、 p 3833-3835.に記載の j8—ケトエステルの一 般的合成法などに記載の方法により実施可能である。エステルの加水分解によるィ匕 合物 (ΧΠ)の製造は、一般的な方法 (Green T.W.他著「プロテクティブ ·グループス' イン'オルガニック 'シンセシス」(Protective Groups in Organic Synthesis)、第 5版、 1 999年、ジョン'ヮイリ一'アンド'サンズ (John Wiley & Sons) .など)により実施可能で ある。不斉還元反応は、 Wang Z.他著「テトラへドロン'アシンメトリー」(Tetrahedron A symmetry) , 1999年、 10卷、 p 225-228.などに記載の方法により実施可能である。ス キーム中の化合物 (ΧΙΠ)とは逆の立体構造を有する化合物の製造も可能であり、こ
の場合、スキーム中のアミノ酸 (IV ' )とは逆の立体構造を有するアミノ酸の製造が可 能となる。アミドィ匕反応は、一般的なカルボン酸アミド合成法 (第 5版 実験化学講座 16有機化合物の合成 IV、 pp. 118-134.; Benz G.著「コンプリヘンシブ'オルガニック 'シンセシス」 (Comprehensive Organic Synthesis) , 1991年、 6卷、 ρ.381- 417、パーガ モン'プレス (Pergamon Press).; Bailey P.D.他著「コンプリへンシブ 'オルガニック'フ アンクショナノレ'グノレープ'トランスフォーメーションズ」 (Comprehensive Organic Funct ional Group Transformations)、 1991年、 5卷、 p.257- 307、パーガモン ·プレス (Pergam on Press).;田辺 他著「有機合成化学協会誌」、 2004年、 62卷、 p 1249-1259.; Mont albetti C.A.G.N.他著「テトラへドロン」(Tetrahedron)、 2005年、 61卷、 10827-10852. など)により、環化反応は、一般的な光延反応条件 (Mitsunobu 0.著「シンセシス」 (S ynthesis)、 1981年、 p 1- 28. ; Hughes D.L.著「オルガニック'リアクションズ」(Organic Reactions) , 1992年、 42卷、 ρ 335-656.など)により実施可能である。ラタタムの開環 反応は、アルコール R13OHのリチウム塩、カリウム塩、ナトリウム塩等で実施可能であ る力 通常、ナトリウム塩で良好な結果が得られる。アルコールの塩は、市販されてい るものを用いてもよいし、使用前に調製してもよぐアルコール塩としては、ナトリウムメ トキシド、ナトリウムエトキシドの場合、良好な結果が得られる。アルコールの塩の量は 、 1〜: L00当量で実施可能である力 好ましくは 1〜10当量、より好ましくは、 1〜5当 量である。溶媒は、メタノール、エタノール、イソプロピルアルコール等のアルコール 系溶媒、テトラヒドロフラン(以下、 THFと略す)、 1, 4 ジォキサン等のエーテル系 溶媒、ジクロロメタン、ジクロロェタン等のハロゲン系溶媒、 DMF等を用いることがで きるが、通常、アルコール系溶媒で満足な結果が得られる。用いるアルコールの塩と 異なった種類のアルコール系溶媒を用いることも可能ではあるが、同一のアルコール 系溶媒を用いる方が良好な結果が得られる。反応温度は 0°Cから溶媒の還流温度で 実施可能であるが、室温で良好な結果が得られる場合が多い。反応は 1日以内に完 結することが多ぐ通常、 1時間以内に反応は完結する。ベンジルォキシ基の除去は 、 ノラジウム触媒を用いた接触水素添加反応により実施可能である。水素圧は、 1〜 10気圧で実施可能である力 通常、 1〜3気圧で良好な結果が得られる。パラジウム の量は、 0. 001モル%〜1000モル%で実施可能であるが、通常、 0. 1モル%〜10
0モル%で良好な結果が得られる。溶媒は、メタノール、エタノール等のアルコール 系溶媒を用いると、満足な結果が得られる場合が多い。反応温度は、 0〜100°Cで 実施可能であるが、通常は室温で実施される。反応は 1日以内に完結することが多く 、通常、 1時間以内に反応は完結する。接触水素添加反応の他に、 Naito T.他著「 ジャーナル'ォブ 'オルガニック'ケミストリー」(Journal of Organic Chemistry)、 2000 年、 65卷、 p 176-185.などに記載のモリブデンへキサカルボ-ルを用いる方法によつ ても実施可能である。ァミノ基への保護基 Pの導入及びエステルの加水分解は、一般 的な方法(Gren T.W.他著「プロテクティブ'グループス'イン'オルガニック'シンセシ ス」(Protective Groups in Organic Synthesis)、第 5版、 1999年、ジョン 'ワイリー'アン ド 'サンズ (John Wiley & Sons) .など)により実施可能である。スキーム中の Ar、 P、 R13 は前記定義に同じである。
[化 14]
Ar^CN + Br A0Ri3 ^ - Ar A 0Ri3 ^
(VII) (VIII) (ΧΙ)

(XII) (XII I) (XIV)
BnQ Ρゝ
Ν 0 ΝΗ2 Ο ΝΗ 0
(XV) (Χ') (IV) スキーム 4
[0057] ァミン (XX) (ァミン (V)にお!/、て、 R1が水素である化合物)は、主に方法 Α〜Εによ り製造可能である。
[0058] (方法 Α) (スキーム 5)
ァミン (XX)は、市販品で入手可能なケトン (XVI)と有機金属化合物 (XVII)との反 応により得られるアルコール (XVIII)を、 Krimen L.I.他著「オルガニック'リアクション ズ」 (Organic Reactions)、 1969年、 17卷、 p 213- 325.; Bishop R.著「コンプリへンシ
ブ 'オルガニック'シンセシス」(Comprehensive Organic Synthesis)、 1991年、 6卷、 p.2 61-300、パーガモン'プレス (Pergamon Press).などに記載の方法により化合物(XIX )へと導き、化合物 (XIX)力 ァシル基を除去することにより製造可能である。ケトン( XVI)と有機金属化合物 (XVII)との反応において、 Mはリチウム、マグネシウム、アル ミニゥム、亜鉛、銅、チタン、セリウム等の金属を表し、通常、リチウム、マグネシウムで 良好な結果が得られる。溶媒は、ジェチルエーテル、 THF、 1, 4 ジォキサン等の エーテル系溶媒、ベンゼン、トルエン等の芳香族炭化水素系溶媒等を用いることが できるが、通常、 THFで良好な結果が得られる。反応温度は、— 100°C〜100°Cで 実施可能であるが、通常、 78°C〜30°Cで良好な結果が得られる。有機金属化合 物 (XVII)はケトン (XVI)に対して、通常 1〜2当量用いることで良好な結果が得られ る力 それ以上の量を用いること、あるいは、反応が完結するまで逐次追加することも 可能である。反応は 1日以内に完結することが多ぐ通常、 1時間以内に反応は完結 する。化合物 (XIX)力もァシル基を除去する反応は、一般的な手法 (Green T.W.他 著「プロテクティブ.グループス.イン.オルガニック.シンセシス」(Protective Groups in Organic Synthesis)、第 5版、 1999年、ジョン'ワイリ^ ~ ·アンド'サンズ (John Wiley & S ons) .など)又は WO97/32880; Lee G.T.他著「シンセティック 'コミュニケーションズ」 (Synthetic Communications) , 1998年、 28卷、 ρ 4009-4018.などに記載の方法により 実施可能である。また、 R14がクロロメチル基の場合には、 Jirgensons A.他著「シンセ シス」(Synthesis)、 2000年、 p 1709-1711.などに記載の方法により実施することも可 能である。スキーム中、 Y、 m、 n、 X、 R2は前記定義に同じであり、 R14は 1個のハロゲン で置換されて 、てもよ 、炭素数 1〜3のアルキルを表す。
[化 15]

スキーム 5
(方法 Β) (スキーム 6)
ァミン (XX)は、ケトン (XVI)をベンジルォキシァミン'塩酸塩によりォキシム化し、得
られたォキシム (XXI)と有機金属化合物 (XVII)との反応により化合物 (XXII)に導き 、化合物 (ΧΧΠ)のベンジルォキシ基を除去することにより製造可能である。ォキシム 化反応は、塩基存在下、市販品で入手可能なケトン (XVI)と市販品で入手可能なベ ンジルォキシアミン'塩酸塩とで実施可能である。塩基としては、ピリジン、トリェチル ァミン (以下、 TEAと略す)等の有機塩基、酢酸ナトリウム、酢酸カリウム等の有機力 ルボン酸塩等を用いることができる。塩基の量は、 1当量力 溶媒量まで用いることが 可能である。ベンジルォキシァミン ·塩酸塩の量は、 1〜10当量で実施可能であるが 、通常、 1〜3当量で良好な結果が得られる。溶媒は、通常、メタノール、エタノール 等のアルコール系溶媒が用いられ、水を共溶媒として用いることも可能である。塩基 を溶媒として用いることも可能である。反応温度は、 0°Cから溶媒の還流温度で実施 可能であり、好ましくは室温力 溶媒の還流温度である。反応は 1日以内に完結する ことが多ぐ通常、 5時間以内に反応は完結する。ォキシム (XXI)と有機金属化合物 (XVII)との反応において、 Mはリチウム、マグネシウム、アルミニウム、亜鉛、銅、チタ ン、セリウム等の金属を表し、通常、リチウム、マグネシウムで良好な結果が得られる。 溶媒は、ジェチルエーテル、 THF、 1, 4 ジォキサン等のエーテル系溶媒、ベンゼ ン、トルエン等の芳香族炭化水素系溶媒等を用いることができるが、通常、ジェチル エーテルで良好な結果が得られる。反応温度は、— 100°C〜100°Cで実施可能であ る力 通常、 78°C〜30°Cで良好な結果が得られる。有機金属化合物 (XVII)はォ キシム (XXI)に対して、通常 1〜2当量用いることで良好な結果が得られる力 それ 以上の量を用いること、あるいは、反応が完結するまで逐次追加することも可能であ る。反応は 1日以内に完結することが多ぐ通常、 1時間以内に反応は完結する。ベ ンジルォキシ基の除去は、ノラジウム触媒を用いた接触水素添加反応により実施可 能である。水素圧は、 1〜10気圧で実施可能である力 通常、 1〜3気圧で良好な結 果が得られる。パラジウムの量は、 0. 001モル%〜1000モル0 /0で実施可能である 1S 通常、 0. 1モル%〜100モル%で良好な結果が得られる。溶媒は、メタノール、 エタノール等のアルコール系溶媒を用いると、満足な結果が得られる場合が多い。反 応温度は、 0〜: L00°Cで実施可能である力 通常は室温で実施される。反応は 1日 以内に完結することが多ぐ通常、 1時間以内に反応は完結する。接触水素添加反
応の他に、 Naito T.他著「ジャーナル'ォブ 'オルガニック 'ケミストリー」(Journal of 0 rganic Chemistry)、 2000年、 65卷、 p 176-185.などに記載のモリブデンへキサカル ボニルを用いる方法によっても実施可能である。スキーム中、 Y、 m、 n、 X、 R2は前記 定義に同じである。
[化 16]
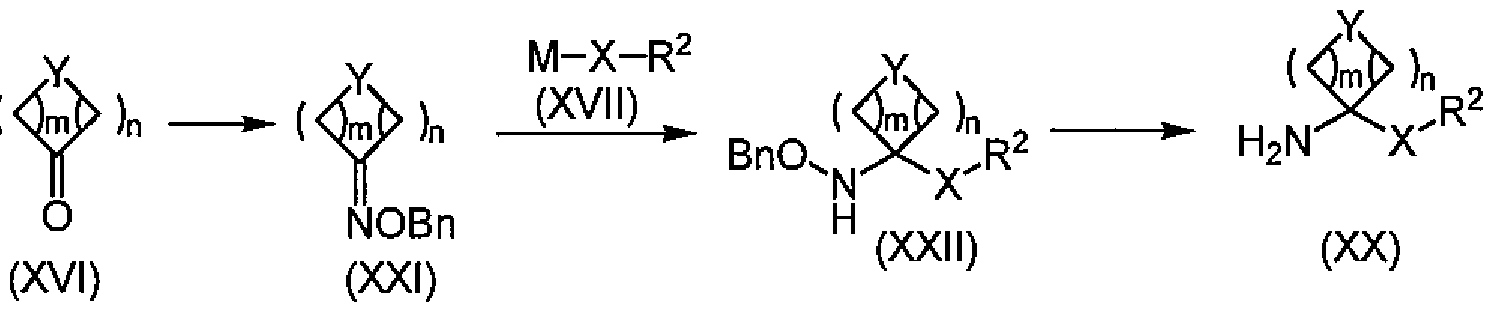
スキーム 6
(方法 C) (スキーム 7)
ァミン (XX)は、 Ellman J.A.他著「ァカウンツ'ォブ'ケミカル'リサーチ」(Accounts o f Chemical Research)、 2002年、 35卷、 p 984-995.などに記載の方法により、市販品 で入手可能なケトン (XVI)から、化合物 (ΧΧΙΠ)、化合物 (XXIV)を経て製造可能で ある。スキーム中、 Y、 m、 n、 M、 X、 R2は前記定義に同じである。
[化 17]

スキーム 7
前記の方法 A〜Cにおいて用いた有機金属化合物 (XVII)は、通常用いられるハロ ゲン 金属交換反応又は脱プロトンィ匕反応 (Tomooka K.他著「メイン 'グループ'メタ ノレズ 'イン'オノレガニック'シンセシス」(Main Group Metals in Organic Synthesis) , 20 04年、 1卷、 p 1-34.ワイリー (Weilly). ; Oshima K.他著「メイン 'グループ'メタルズ 'ィ ン'オノレガニック'シンセシス」(Main Group Metals in Organic Synthesis)、 2004年、 1 卷、 p 51-154.ワイリー (Weilly). ; Katritzky A.R.他著「コンプリへンシブ 'ヘテロサイク リック.ケミストリー」(Comprehensive Heterocyclic Chemistry)、 1984年、 5卷、 p 39-11
0.パーガモン'プレス (Pergamon Press).など)により製造可能である。有機金属化合 物 (XVII)の製造に用いられる化合物 (XXV)は、市販品で入手可能であるか、市販 されていない場合には、スキーム 8〜: L 1示す方法により合成し入手可能である。ここ で、 X、 R2は前記定義に同じであり、 Zは水素又はハロゲンを表す。
[化 18]
Z-X-R2
(XXV) 化合物(XXV' ) (ィ匕合物(XXV)において、 R2がチアゾールー 2—ィル、 Zが水素、 Xが原子価結合である化合物)は、市販品として入手可能なプロモケトン (XXVII)、 又は市販品として入手可能なケトン (XXVI)のブロモ化により得られたブロモケトン( XXVII)を、チォ尿素又はチォォキサム酸ェチルにより環化してチアゾール (XXVIII )とし、チアゾール (XXVIII)力も R15を除去することにより製造可能である (スキーム 8) 。ブロモケトン(XXVII)の製造は、 Jaeques J.他著「オルガニック 'シンセシス」(Organ ic Synthesis)、 1973年、 53卷、 p 111- 115. ; Bloch R.著「シンセシス」(Synthesis)、 197 8年、 p 140-142. ; Kajigaeshi S.他著「ブレティン'ォブ'ザ'ケミカル'ソサイエティ一' ォブ 'ジャパン」(Bulletin of the Chemical Society of Japan)、 1987年、 60卷、 p 1159- 1160. ; Sircar I.他著「テトラへドロン'レターズ」(Tetrahedron Letters) , 1988年、 29卷 、 ρ 6835- 6838. ; Tanemura K.他著「ケミカル'コミュニケーションズ」(Chemical Comm unications)、 2004年、 p 470-471.などに記載の方法により実施可能である。環化反 応において、チォ尿素又はチォォキサム酸ェチルを用いたとき、 R15は各々、アミノ基 又はエトキシカルボニル基となる。チォ尿素又はチォォキサム酸ェチルは、 0. 1〜1 0当量、好ましくは 0. 5〜2当量用いることにより、良好な結果が得られる。溶媒は、メ タノール、エタノールなどのアルコール系溶媒、アセトン、 DMFなどの非プロトン性極 性溶媒が好んで用いられる。反応温度は、 0°Cから溶媒の還流温度までで実施可能 であるが、通常、室温から溶媒の還流温度までで良好な結果が得られる。反応は 1日 以内に完結することが多ぐ通常、 3時間以内に反応は完結する。また、環化反応時 、必要に応じてヨウ化ナトリウム、ヨウ化カリウム、ヨウ化テトラプチルアンモニゥムなど の無機塩や重曹、 TEA,コリジンなど塩基を添加剤として加えると良好な結果が得ら
れることがある。チアゾール (xxvm)の R15を除去する反応において、 R15がァミノ基 である場合は、 Haddock E.他著「ジャーナル'ォブ'ケミカル'ソサイエティ一'シー」( Journal of Chemical Society C)、 1971年、 p 3994— 3999. ; Cadogan J.I.G.他著「ジャ ーナル 'ォブ 'ケミカル 'ソサイエティ一,パーキン 'トランスアクション 1」 (Journal of Ch emical Society, Parkin Transaction 1)、 1973年、 p 541-542. ; Doyle M.P.他著「ジャ 一ナル'ォブ'オルガニック'ケミストリー」(Journal of Organic Chemistry) , 1977年、 4 2卷、 ρ 3494-3498. ; Chedekel M.R.他著「ジャーナル'ォブ 'オルガニック'ケミストリ 一」(Journal of Organic Chemistry)ゝ 1984年、 49卷、 p 997- 1000. ; Elguero J.他著「 ジャーナル'ォブ'ケミカル'ソサイエティ一,パーキン 'トランスアクション 1」 (Journal of Chemical Society, Parkin Transaction 1)、 1993年、 p 2227-2232.などに記載の方 法により実施可能である。 R15がエトキシカルボ-ル基である場合、チアゾール (XXVI Π)のエトキシカルボ-ル基は、一般的な手法(Green T.W.他著「プロテクティブ 'グ ノレープス'イン'オノレガニック'シンセシス」 (Protective Groups in Organic Synthesis) 、第 5版、 1999年、ジョン 'ヮイリ一 ·アンド'サンズ (John Wiley & Sons) .など)によりカロ 水分解した後、塩酸、臭化水素酸などの酸を用いた脱炭酸により実施することが可 能である。脱炭酸反応において、反応に用いる酸を溶媒としてもよいし、 THF、 1, 4 ジォキサン等のエーテル系溶媒を共溶媒に用いてもよい。濃塩酸と 1, 4 ジォキ サンは好ましい組み合わせの 1つである。反応温度は、室温から溶媒の還流温度ま でで実施可能であるが、通常、溶媒の還流温度で好ましい結果が得られる。反応は 1日以内に完結することが多ぐ通常、 1時間以内に反応は完結する。スキーム中、 R7 は前記定義に同じであり、 R15はァミノ又はエトキシカルボニルを表す。
[化 19]
0
人 7 B
(XXVI)

スキーム 8
化合物(XXV,, ) (化合物(XXV)にお!/、て、 R2がべンゾチアゾールー 2 ィル、 Ζが 水素、 Xが原子価結合である化合物)は、市販品として入手可能なベンゾチアゾール
(XXXII)のアミノ基を除去することにより製造可能である (スキーム 9)。ベンゾチアゾ ール (XXXII)が市販品として入手不可能な場合には、市販品として入手可能なァ- リン (XXIX)をイソチオシアナート (XXX)に変換し、これをチォ尿素 (XXXI)とした後 (イソチオシアナート (XXX)又はチォ尿素 (XXXI)が市販品として入手可能である場 合には、これらから製造を実施することも可能である)、環化することにより製造可能 である。イソチオシアナ一ト (XXX)の製造は、ァ-リン (XXIX)とチォカルボ-ル化試 薬との反応により実施可能である。チォカルボ-ルイ匕試薬の好ましい例としては、チ ォホスゲン、チォカルボ-ルジイミダゾール(以下、 TCDIと略す)、 1, 1,ーチォカル ボニルジー 2 (1H)—ピリジノン(以下、 TCDPと略す)等が挙げられる。チォカルボ- ル化試薬の量は、 1〜10当量で実施可能である力 通常、 1〜3当量で好ましい結 果が得られる。溶媒は、 THF、 1, 4 ジォキサン等のエーテル系溶媒、ジクロロメタ ン、クロ口ホルム等のハロゲン系溶媒、ベンゼン、トルエン等の芳香族炭化水素系溶 媒等が用いられるが、通常、 THF、ジクロロメタン等で満足な結果が得られる。反応 温度は、— 100°C〜100°Cで実施可能であるが、— 20°C〜室温で満足な結果が得 られることが多い。反応は 1日以内に完結することが多ぐ通常、 3時間以内に反応は 完結する。チォ尿素 (XXXI)の製造は、イソチオシアナート (XXX)とアンモニアを反 応させることにより実施可能である。アンモニアは、ガスを用いてもよいし、アンモニア 水、メタノール、エタノール、 2 プロパノール、 1, 4 ジォキサン等のアンモニア溶 液を用いてもょ 、が、アンモニア溶液を用いる方法は簡便かつ良好な結果が得られ る方法である。アンモニアの量は特に制限はないが、通常、 1〜: LO当量で満足な結 果が得られる。溶媒は、 THF、 1, 4 ジォキサン等のエーテル系溶媒、ジクロロメタ ン、クロ口ホルム等のハロゲン系溶媒、ベンゼン、トルエン等の芳香族炭化水素系溶 媒等が用いることができる力 通常、 THF、ジクロロメタン等で満足な結果が得られる 。反応温度は、— 100°C〜100°Cで実施可能である力 — 20°C〜室温で満足な結 果が得られることが多い。反応は 1日以内に完結することが多ぐ通常、 3時間以内に 反応は完結する。チォ尿素(XXXI)は、 Meckler H.他著「シンセシス」 (Synthesis)、 2 000年、 p 1569-1574.などに記載の方法により、ァ-リン (XXIX)力 直接製造するこ とも可能である。環化反応は、 Allen C.F.H.他著「オルガニック'シンセシス」(Organic
Synthesis) , 1942年、 22卷、 p 16- 18. ; Chedekel M.R.他著「ジャーナル'ォブ 'オル ガ-ック.ケミストリー」(Journal of Organic Chemistry)ゝ 1984年、 49卷、 p 997-1000. などの方法により、実施可能である。チアゾール (XXXII)のァミノ基の除去は、 Hadd ock E.他著「ジャーナル'ォブ'ケミカル'ソサイエティ^ ~ ·シー」(Journal of Chemical Society C)、 1971年、 p 3994- 3999. ; Cadogan JIG.他著「ジャーナル'ォブ 'ケミカル .ソサイエティ一,パーキン 'トランスアクション 1」 (Journal of Chemical Society, Parkin
Transaction 1)、 1973年、 p 541-542. ; Doyle M.P.他著「ジャーナル'ォブ 'オルガ- ック 'ケミストリー」(Journal of Organic Chemistry) , 1977年、 42卷、 ρ 3494- 3498.; Che dekel M.R.他著「ジャーナル'ォブ 'オルガニック'ケミストリー」(Journal of Organic C hemistry)、 1984年、 49卷、 p 997- 1000. ; Elguero J.他著「ジャーナル'ォブ'ケミカル' ソサイエティー,パーキン 'トランスアクション 1」 (Journal of Chemical Society, Parkin Transaction 1)、 1993年、 p 2227-2232.などに記載の方法により、実施可能である。 スキーム中、 R16は R6又は R7 (R6、 R7は前記定義に同じ)を表し、 Xは 0、 1、 2又は 3を表 す。
[化 20]

スキーム 9
化合物(XXV, " ) (ィ匕合物(XXV)において、 R2が 1, 3, 4—ォキサジァゾ一ルー 2 ィル、 Ζが水素、 Xが原子価結合である化合物)は、 Hiremath U.S.他著「ジャーナ ノレ 'ォブ 'ケミカノレ'リサーチ,シノプセス」 (Journal of Chemical Research, Synopses)、 1994年、 p 502-503. ; Shyadligeri A.S.他著「インディアン 'ジャーナル'ォブ 'ケミストリ 一,セクション B :オルガニック 'アンド'メディシナル 'ケミストリー」(Indian Journal of C hemistry, Section B: Organic and Medicinal Chemistry)、 1995年、 34卷、 p 1059—106
5.などに記載の方法により、市販品として入手可能なヒドラジド (ΧΧΧΠΙ)力も製造可 能である (スキーム 10)。スキーム中、 R7は前記定義に同じである。
[化 21]
H?N

スキーム 1 0
[0065] 置換基 R6又は R7を持つ化合物 (XXV)は、上述のようにこれら置換基を持つ化合物 を原料にして製造することができるが、後から導入することも可能である。後から導入 する方法の 1例をスキーム 11に示す。他にも、一般的な鈴木カップリング条件(Suzuk i A.他著「ケミカル 'レビュー」(Chemical Review)、 1995年、 95卷、 p 2457-2483. ; Suz uki A.著「メタル一キヤタラィズド 'クロス一カップリング'リアクションズ」(Meta卜 Cataly zed Cross-Coupling Reactions)、 1998年、 p 49—97.ワイリー(Weiley) .など)、一般的 な Stilleカップリング条件(Stille J.K.著「アンゲバンテ 'へミ一'インターナショナル'ェ 7"インヨン' ン 'イングリツンュ」 (Angewante Chemie International Edition in Englisn 、 1986年、 25卷、 p 508-524. ; Farina V.他著「オルガニック'リアクションズ」(Organic Reactions) , 1997年、 50卷、 ρ 1-652.; Mitchell T.N.著「メタル キヤタラィズド 'クロス —カップリング'リアクションズ」(Meta卜 Catalyzed Cross-Coupling Reactions)、 1998 年、 p 167-202.ワイリー(Weiley) .など)、一般的な Buchwald-Hartwigカップリング条 件(Buchwald S.L.他著「ァカウンツ'ォブ'ケミカル'リサーチ」(Accounts of Chemical Research)、 1998年、 31卷、 p 805- 818. ; Hartwig J.F.著「アンゲバンテ 'へミ^ ~ ·イン ターナショナノレ'エアイシヨン 'イン'イングリッシュ」 (Angewante Chemie International Edition in English)、 1998年、 37卷、 p 2046-2067. ; Buchwald S.L.他著「ジャーナル' ォブ 'アメリカン 'ケミカノレ 'ソサイエティ一」(Journal of American Chemical Society)、 2 001年、 123卷、 p 7727-7729. ; Buchwalb S.L.他著「ジャーナル'ォブ 'オルガニック' ケミストリー」(Journal of Organic Chemistry)、 2004年、 69卷、 p 5578-5587.など)等 によっても製造可能である力 これらの具体例ついては、実施例において開示する。
[0066] ベンゾチアゾール(XXV,,,,)は、 Haddock E.他著「ジャーナル'ォブ'ケミカル'ソ
サイエティ^ ~ ·シー」(Journal of Chemical Society C)、 1971年、 p 3994— 3999. ; Cadog an JIG.他著「ジャーナル'ォブ'ケミカル'ソサイエティ一,パーキン 'トランスァクショ ン 1」 Ooumal ofし hemicai Society, Parkin Transaction 1)、 1973年、 p 541-542. ; Doyl e M.P.他著「ジャーナル'ォブ 'オルガニック'ケミストリー」(Journal of Organic Chemi stry)、 1977年、 42卷、 p 3494-3498. ; Chedekel M.R.他著「ジャーナル'ォブ 'オルガ ニック.ケミストリー」(journai 0f Organic Chemistry)ゝ 1984年、 49卷、 p 997- 1000. ; Elg uero J.他著「ジャーナル'ォブ'ケミカル'ソサイエティ一,パーキン 'トランスアクション 1」(Journal of Chemical Society, Parkin Transaction 1)、 1993年、 p 2227-2232.など に記載の方法により、市販品で入手可能であるべンゾチアゾール (ΧΧΧΙΓ )からアミ ノ基を除去し、続いて、通常の方法 (Green T.W.他著「プロテクティブ'グループス' イン'オルガニック 'シンセシス」(Protective Groups in Organic Synthesis)、第 5版、 1 999年、ジョン 'ヮイリ一 ·アンド'サンズ (John Wiley & Sons) .など)によりメチル基を除 去することにより、製造可能である。化合物 (XXV' ' ' ' ' )は、一般的なフエノール性 水酸基のアルキル化条件(Green T.W.他著「プロテクティブ'グループス'イン'オル ガ-ック.シンセシス」 (Protective Groups in Organic Synthesis)、第 5版、 1999年、ジ ヨン'ワイリー'アンド'サンズ (John Wiley & Sons) .など)又は一般的な光延反応条件 (Mitsunobu 0.著「シンセシス」 (Synthesis) , 1981年、 p 1- 28. ; Hughes D.L.著「ォノレ ガ-ック.リアクションズ」(Organic Reactions) , 1992年、 42卷、 ρ 335- 656.など)により 、ベンゾチアゾール (XXV,,, ' )力 製造可能である。スキーム中、 R17は、炭素数 1〜 6のアルキル又は (CH )q— R8 (q、 R8は前記定義に同じ)を表す。
2
[化 22]

(XXXI |') (XXV"") (XXV"") スキーム 1 1
(方法 D) (スキーム 12)
ァミン (XX,)(ァミン (XX)において、 R2がべンゾイミダゾールー 2—ィル、 Xが原子 価結合である化合物)は、市販品として入手可能なケトン (XVI)を一般的な Strecker
反応、その後のシァノ基の加水分解(Steiger R.E.著「オルガニック'シンセシス」(Or ganic Synthesis)、 1942年、 22卷、 p 13-15.など)により、アミノ酸 (XXXIV)に導き、ァ ミノ酸 (XXXIV)のァミノ基に Fmoc基を導入した後、これとフエ-レンジァミン (XXX VI)とでアミドィ匕反応を行い、続いて環化反応を実施した後、 Fmoc基を除去すること により製造可能である。 Fmoc基の導入は、 McLaughlm M.他著「テトラへドロン'レタ ーズ」(Tetrahedron Letters)、 1997年、 38卷、 p 4013-4016.などに記載の方法により 、実施可能である。アミノ酸 (XXXIV)とフエ-レンジァミン (XXXVI)とのアミド化反応 は、一般的なカルボン酸アミド合成法 (第 5版 実験化学講座 16有機化合物の合成 I V、 pp. 118-134.; Benz G.著「コンプリヘンシブ'オルガニック'シンセシス」(Compreh ensive Organic Synthesis)、 1991年、 6卷、 p.381- 417、パーガモン'プレス (Pergamon Press).; Bailey P.D.他著「コンプリへンシブ 'オルガニック'ファンクショナル 'グルー フ° 'トフンスフォ ~~メ ~~シヨンズ」 (Comprehensive Organic Functional Group Transfer mations)、 1991年、 5卷、 p.257- 307、パーガモン'プレス (Pergamon Press).; 田辺 他 著「有機合成化学協会誌」、 2004年、 62卷、 p 1249-1259. ;Montalbetti C.A.G.N.他 著「テトラへドロン」(Tetrahedron)、 2005年、 61卷、 10827- 10852.など)〖こより、実施可 能である。環化反応は酸を用いて実施する力 酸としては、ギ酸、酢酸、プロピオン 酸、トリフルォロ酢酸などの有機カルボン酸、塩酸、臭化水素酸などの無機酸、メタン スルホン酸、 P-トルエンスルホン酸などのスルホン酸、ポリリン酸等を用いることができ る力 通常、有機カルボン酸で満足な結果が得られる。酸は、溶媒量用いることが好 ましい。反応温度は、室温力 溶媒の還流温度で実施可能であるが、好ましくは、 50 °Cから溶媒の還流温度である。反応は 3日以内に完結することが多ぐ通常、 1日以 内に反応は完結する。 Fmoc基の除去は、一般的な方法 (Green T.W.他著「プロテ クティブ'グノレープス'イン'オノレガニック'シンセシス」(Protective Groups in Organic Synthesis)、第 5版、 1999年、ジョン'ワイリ一'アンド'サンズ (John Wiley & Sons) .な ど)により、実施可能である。スキーム中、 Y、 m、 n、 R12、 R16、 xは前記定義に同じであ る。
[化 23]

(XVI) (XXXIV) (XXXV) (XXXVI)

スキーム 1 2
[0068] フエ-レンジァミン (XXXVI)は、巿販品として入手可能な場合もある力 入手不可 能な場合には合成することにより入手可能である。フエ-レンジァミン (XXXVI)の製 造法の 1例をスキーム 13に示す。フエ-レンジァミン (XXXVI)は、市販品として入手 可能な-トロア-リン(XXXVII)のァミノ基に Voskresens S.他著「シンセティック 'コ ミュ-ケーシヨンズ」(Synthetic Communications)、 2000年、 30卷、 p 3523-3526.など に記載の方法により R12を導入し、得られた-トロア-リン (XXXVIII)の-トロ基を Sna dler S.R.他著「オルガニック'ファンクショナル 'グループ'プレパレーションズ」(Orga nic Functional Group Preparations)、第 2版、 1983年、 p 405-411.アカデミック'プレ ス (Academic Press) .などに記載の方法により還元することにより、製造可能である。 スキーム中、 R12、 R16、 xは前記定義に同じである。
[化 24]
H ^ 6)x

(XXXVII) (XXXVIII) (XXXVI)
スキーム 1 3
[0069] (方法 E) (スキーム 14)
ァミン (XX,,)(ァミン (XX)において、 R2がチアゾールー 4 ィル、 Xが原子価結合 である化合物)は、市販品として入手可能なアミノ酸エステル (XXXIX)を Chen P.他 著「テトラへドロン'レターズ」(Tetrahedron Letters) , 1997年、 38卷、 ρ 3175-3178.な
どに記載の方法により化合物 (XL)とし、化合物 (XL)とチオアミド (XLI)とを反応させ て環化した後、 Boc基を除去することにより製造可能である。チオアミド (XLI)は巿販 品として入手可能な場合もあるし、市販品として入手不可能な場合には、 Taylor E.C. 他著「ジャーナル'ォブ 'アメリカン 'ケミカル'ソサイエティ一」(Journal of American C hemical Society), 1960年、 82卷、 p 2656-2657.などに記載の方法により、市販品とし て入手可能な-トリル R16—CNより製造し入手可能である。環化反応において、化合 物(XL)はチオアミド (XLI)に対して 0. 1〜10当量用いることができる力 通常、 0. 5 〜2当量で満足な結果が得られる。溶媒は、メタノール、エタノールなどのアルコール 系溶媒、アセトン、 DMFなどの非プロトン性極性溶媒が好んで用いられる。反応温 度は、 0°C力 溶媒の還流温度までで実施可能である力 通常、室温から溶媒の還 流温度までで良好な結果が得られる。反応は 1日以内に完結することが多ぐ通常、 3時間以内に反応は完結する。また、環化反応時、必要に応じてヨウ化ナトリウム、ョ ゥ化カリウム、ヨウ化テトラブチルアンモ -ゥムなどの無機塩や重曹、 TEA、コリジンな ど塩基を添加剤として加えると良好な結果が得られることがある。 Boc基の除去は、 一般的な方法(Green T.W.他著「プロテクティブ'グループス'イン'オルガニック'シ ンセシス」 (Protective Groups in Organic Synthesis)、第 5版、 1999年、ジョン.ワイリー •アンド'サンズ (John Wiley & Sons) .など)により実施可能である。スキーム中、 Y、 m 、 n、 R13、 R16は前記定義に同じである。
[化 25]

本発明の化合物の糖尿病治療効果及び血糖低下効果は、例えば、武内浩ニ他 著「日本薬理学雑誌」、 2006年、 128卷、 p 37-41.などに記載の実験動物を用いた糖 負荷試験(Oral Glucose Tolerance Test.以下、 OGTT試験と略す)により確認する ことが可能であるが、この方法に限られるわけではない。また、実験動物としては、マ
ウス、ラット、ィヌ、サル等の正常動物又は糖尿病モデル動物(例えば、 Winzell M.S. 他著「ダイアベテイス」(Diabetes)、 2004年、 53卷、 p S215-S219.などに記載の肥満 糖尿病モデル(Diet Induced Obesity Model.以下、 DIOモデルと略す)等を用いるこ とが可能である力 これらに限られるわけではない。
[0071] 本発明の化合物を糖尿病治療薬などの薬剤として臨床で使用する際には、薬剤は フリー体又はその塩自体でもよぐまた賦形剤、安定化剤、保存剤、緩衝剤、溶解補 助剤、乳化剤、希釈剤、等張化剤などの添加剤が適宜混合されていてもよい。投与 形態としては、錠剤 'カプセル剤 ·顆粒剤 ·散剤 ·シロップ剤など〖こよる経口剤の他、 注射剤 ·坐剤'液剤などによる非経口剤、あるいは軟膏剤 ·クリーム剤 ·貼付剤などに よる局所投与等も可能である。これらの製剤は、一般的に知られている製法によって 作ることができる。本発明の糖尿病の治療又は予防剤は前記有効成分を 0.00001〜9 0重量%、より好ましくは 0.0001〜70重量%含有することが望ましい。その使用量は症 状、年齢、体重、投与方法等に応じて適宜選択されるが、成人に対して、経口剤の 場合、有効成分量として 1日 1 μ g〜10gであり、それぞれ 1回又は数回に分けて投与 することができる。
[0072] 本発明の一般式 (I)で示されたィ匕合物及びその塩は、糖尿病の治療又は予防剤 及び血糖低下剤として用いることができる。また、本発明の一般式 (I)で示されたィ匕 合物及びその塩は、その他の糖尿病治療剤 (以下、併用薬剤と略す)と組み合わせ て用いることができる。この際、本発明の一般式 (I)で示された化合物及びその塩と 併用薬剤との投与時期は限定されず、これらを投与対象に対し、同時に投与してもよ いし、時間差をおいて投与してもよい。併用薬剤の投与量は、臨床上用いられている 用量を基準として適宜選択することができる。また、本発明の一般式 (I)で示された化 合物及びその塩と併用薬剤との配合比は、投与対象、投与ルート、症状、組み合わ せなどにより適宜選択することができる。併用薬剤としては、インスリン製剤 (超速効型 インスリン製剤、速効型インスリン製剤、混合型インスリン製剤、中間型インスリン製剤 、持続型インスリン製剤、持効型溶解インスリン製剤、経肺インスリン製剤、経口インス リン製剤など)、インスリン抵抗性改善剤(ピオグリタゾン (Pioglitazone)、ロシグリタゾン (Rosiglitazone)、ネトグリタゾン (Netoglitazon)、ファルグリタザール (Farglitazar)、リボ
グリタゾン (Rivoglitazone)、ォキセグリタザール (Oxeglitazar)、ナベグリタザール (Naveg litazar),バラグリタゾン (Balaglitazone)、 ONO- 5129、 AVE- 0847、 LBM- 642、 CKD- 50 1、 AVE-5376など)、 oc—ダルコシダーゼ阻害剤(ァカルボース(Acarbose)、ボグリボ ース(Voglibose)、ミグリトール(Miglitol)、エミグリテート (Emiglitate)など)、ビグアナィ ド剤(メトホルミン(Metformin)、ブホルミン(Buformin)など)、スルホ -ルゥレア剤(トル ブタミド(Tolbutamide)、ァセトへキサミド(Acetohexamide)、クロルプロパミド(Chlorpr opamide)、トラザミド(Tolazamide)、グリクロビラミド(Glyclopyramide)、グリブゾール(G lybuzole)、グリベンクラミド(Glibenclamide)、グリクラジド(Gliclazide)、グリメピリド(Gli mepiride)、グリピジド (Glipizide)、グリキドン (Gliquidone)など)、速効型インスリン分泌 促進剤(ナテグリニド(Nateglinide)、レパグリニド(Repaglinide)、ミチグリニド(Mitiglini de)など)、 GLP-1作動薬(エタセナチド (Exenatide)、リラグルチド(Liraglutide) ZP-10A 、 BIM-51077など)、アミリン作動薬(プラムリンチド(Pramlintide)など)、 DPP-IV阻害 剤(ビルダグリプチン (Vildagliptin)、シタグリブチン(Sitagliptin)、サクサグリプチン(Sa xagliptin)、ァログリプチン (Alogliptin)、デナグリプチン (Denagliptin)、 P93/01、 TA- 66 66、 GRC- 8200、 PHX- 1149、 MP- 513、 TS- 021、 KRP- 104、 SSR- 162369、 ALS- 2- 042 6、 ABT-341など)、 β 作動薬(ソラベダロン (Solabegron)、 KRP- 204、 YM-178など)、
3
フルクトース一 1 , 6—ビスホスファターゼ阻害剤(MB- 6322、 MB- 07803など)、 SGLT ( somum- dependent renal glucose transporter)阻舍: ^(J (セルグリフロンン (¾ergliflozin八 AVE— 2268、 GSK— 189075、 TS— 033、 KGA— 2727、 SAR— 7226など)、 11 β— HSD1 (11 β -hydroxysteroid dehydrogenase 1)阻害剤(BVT- 3498、 AMG- 221、 INCB- 13739など )、 PTP-1B (protein tyrosine phosphatase- IB)阻害剤(ISIS- 113715、 JTT- 551など)、 GSK3 β (glycogen synthase kinase 3 β )阻害剤(SAR- 502250など)、グルカゴン拮抗 薬(BAY-27-9955、 NN- 2501など)、グリコーゲン ホスホリラーゼ阻害剤(ィソファゴミ ン (Isofagomine)、 PSN- 357など)、 CPT1 (カル-チン 0—パルミトイルトランスフェラー ゼ 1)阻害剤 (テグリカール (Teglicar)など)、ダルココルチコイド拮抗薬 (ミフエブリストン (Mifepristone), KB- 3305など)、 CB1 (カンナビノイド 1)拮抗薬(リモナバント (Rimonab ant),スリナバント (Surinabant)、 MK- 0364、 AVE- 1625など)などが挙げられる。また、 糖尿病合併症 (腎症、網膜症、神経症)の治療薬も併用薬剤として使用可能である。
併用可能な合併症の治療薬としては、 ACE (angiotensin 1 converting enzyme)阻害 剤(カプトプリル (Captopril)、リシノプリル (Lisinopril)、イミダプリル (Imidapril)など)、 AT 1 (angiotensin 1)括抗薬(ィノレべサノレタン (Irbesartan)、ロサノレタン (Losartan)、テノレミサ ノレタン (Telmisartan)、オノレメサノレタン (01mesartan)、カンデサノレタン (Candesartan)など )、エンドセリン拮抗薬(ァボセンタン (Avosentan)など)、 PKC jS (プロテインキナーゼ C β )阻害剤(ルボキスタウリン (Ruboxistaurin)など)、モノクローナル抗体(ベバシズマ ブ (Bevacizumab)など)、レチリン (Retilin)、アルドース還元酵素阻害剤(ェパレスタツト( Epalrestat)、フィダレスタツト (Fidarestat)、ラ-レスタツト (Ranirestat)など)、抗てんかん 薬(プレギヤパリン (Pregabalin)など)、抗酸化剤 ( a -リポイツク酸( a - lipoic acid)、ァ セチル L カル-チン塩酸塩 (acety卜 L- carnitine hydrochloride)など)、 VR1 (バニ ロイド 1)拮抗薬(カプサイシン (Capsaicin)など)、 NMDA拮抗薬(デキストロメトルファ ン (Dextromethorphan)、ロコサミド (Locosamide)、メマンチン (Memantine)など)、ピリド キサミン (Pyridoxamine)、牛車腎気丸 (Goshajinkigan)などが挙げられる。
図面の簡単な説明
[0073] [図 1]実施例化合物 8の血糖上昇抑制作用を示す図である。
[図 2]実施例化合物 60の血糖上昇抑制作用を示す図である。
[図 3]実施例化合物 73の血糖上昇抑制作用を示す図である。
[図 4]実施例化合物 75の血糖上昇抑制作用を示す図である。
[図 5]実施例化合物 121の血糖上昇抑制作用を示す図である。
[図 6]実施例化合物 122の血糖上昇抑制作用を示す図である。
[図 7]実施例化合物 125の血糖上昇抑制作用を示す図である。
[図 8]比較化合物 1及び 2の血糖上昇抑制作用を示す図である。
[図 9]糖尿病モデルマウスにおける、実施例化合物 60の血糖上昇抑制作用を示す 図である。
[0074] 本明細書は、本願の優先権の基礎である特願 2005— 345748の明細書及び Z 又は図面に記載された内容を包含する。
実施例
[0075] 以下、実施例を挙げて本発明を具体的に説明する。
[0076] 実施例 1
(3R)— 3 ァミノ一 N— (1—ベンジル一 4 フエ-ルビペリジン一 4—ィル) 4— (2 フルオロフェ -ル)ブタナミド ·二塩酸塩 (化合物 1)の合成
ステップ 1 1 ベンジル 4 フエ-ルビペリジン 4 オール(参考化合物 1 )の合 成
アルゴン雰囲気下、フエ-ルリチウム Zシクロへキサンージェチルエーテル溶液(1 . 05M, 5. OmL, 5. 3mmol)を氷冷し、 1—ベンジル— 4 ピぺリドン(0. 5mL, 2. 7mmol)の THF (3mL)溶液を滴下した。滴下終了後、氷浴を外し室温で攪拌し、 6 時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、 乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 382mg (53%)を得た。
[0077] ステップ 2 N—(l ベンジル一 4—フエ-ルビペリジン一 4—ィル)ァセタミド(参考 化合物 2)の合成
1—ベンジル— 4—フエ-ルビペリジン— 4—オール(382mg, 1. 4mmol)のァセト 二トリル(3mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(1. 6mL)をゆっくりと滴下 した。滴下終了後、氷浴を外し室温で攪拌し、 7時間後、氷水に注いだ。水層を水酸 化ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、有機層を飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 350mg (79%)を得た。
[0078] ステップ 3 1—べンジルー 4 フエ-ルビペリジン 4ーァミン(参考化合物 3)の合 成
アルゴン雰囲気下、 N— (1—ベンジル— 4—フエ-ルビペリジン— 4—ィル)ァセタ ミド(lOlmg, 0. 33mmol)を THF (0. 2mL)に懸濁し、チタンテトライソプロポキシド (96 ^ L, 0. 32mmol)、ジフエ-ルシラン(0. 15mL, 0. 81mmol)を加え、室温で 攪拌した。 12時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液をカロ えて激しく攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽出 し、有機層を合わせて飽和食塩水で洗浄後、乾燥し濃縮した。粗生成物として得ら れた表題化合物は、精製することなぐ次のステップの反応に用いた。
[0079] ステップ 4 (3R)— N— (1—ベンジル— 4—フエ-ルビペリジン— 4—ィル)—4— (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド( 参考化合物 4)の合成
アルゴン雰囲気下、 1 ベンジル 4 フエ-ルビペリジン 4 ァミンの粗生成物 をジクロロメタン(3mL)に溶解し、 (3R)—4— (2 フルオロフェ -ル) 3— (2—メ チルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. lmmol)、 1ーヒドロキシべ ンゾトリアゾール(以下、 HOBtと略す)(20mg, 0. 15mmol)、 N ェチルモルホリ ン(以下、 NEMと略す)(14 /z L, 0. l lmmol)、 1ーェチルー 3—(3 ジメチルアミ ノプロピル)カルボジイミド(以下、 EDCIと略す)'塩酸塩(23mg, 0. 12mmol)をカロ え、室温で攪拌した。 12時間後、飽和炭酸水素ナトリウム水溶液を加えクロ口ホルム で抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 60mg (定量的)を得た。
[0080] ステップ 5 (3R)— 3 ァミノ一 N—(l ベンジル一 4—フエ-ルビペリジン一 4—ィ ル) 4一(2 フルオロフヱニル)ブタナミド '二塩酸塩(ィ匕合物 1)の合成
アルゴン雰囲気下、 (3R)— N— (1—ベンジル— 4 フエ-ルビペリジン— 4—ィル ) 4一(2 フルオロフェ -ル)一 3—(2—メチルー 2 プロポキシカルボ-ルァミノ) ブタナミド(60mg, 0. lmmol)を酢酸ェチル(lmL)に溶解し、 4M塩化水素 Zl, 4 —ジォキサン溶液 (2mL)を加え、室温で攪拌した。 12時間後、飽和炭酸水素ナトリ ゥム水溶液を加え酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮 した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、精製物をメタ ノール溶液とし、 10%塩ィ匕水素 Zメタノール溶液を加えて塩ィ匕し、これを濃縮し、表 題ィ匕合物 1 lmg (22%)を得た。
[0081] 実施例 2
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (1—メチル 4 フエ-ルビ ペリジンー4 ィル)ブタナミド '二塩酸塩 (ィ匕合物 2)の合成
ステップ 1 1 メチル 4 フエ-ルビペリジン 4 オール(参考化合物 5)の合成 アルゴン雰囲気下、フエ-ルリチウム Zシクロへキサンージェチルエーテル溶液(1 . 05M, 7. 7mL, 8. lmmol)を THF (5mL)で希釈し、氷冷下、 1—メチル—4 ピ
ペリドン(0. 5mL, 4. lmmol)の THF (3mL)溶液を滴下した。滴下終了後氷浴を 外し室温で攪拌し、 1. 5時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチ ルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 877mg (97%)を得た。
[0082] ステップ 2 N—(l—メチルー 4 フエ-ルビペリジンー4 ィル)ァセタミド(参考化合 物 6)の合成
1—メチル 4—フエ-ルビペリジン— 4—オール(300mg, 1. 6mmol)のァセトニ トリル(2. 8mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(1. 8mL)をゆっくりと滴下 した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷水に注いだ。水層を水酸 化ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、有機層を飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 233mg (64%)を得た。
[0083] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)— N—(l—メチル—4—フエ-ルビぺ リジンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参 考化合物 7)の合成
アルゴン雰囲気下、 N— (1—メチル—4—フエ-ルビペリジン— 4—ィル)ァセタミド (58mg, 0. 25mmol)を THF (0. lmL)に懸濁し、チタンテトライソプロポキシド(75 ^ L, 0. 25mmol)、ジフエ-ルシラン(0. 12mL, 0. 65mmol)を加え、室温で攪拌 した。 12時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液を加えて激 しく攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽出した。 有機層を合わせて飽和食塩水で洗浄後、乾燥し濃縮した。アルゴン雰囲気下、得ら れた粗生成物をジクロロメタン(3mL)に溶解し、(3R)—4— (2 フルオロフヱ-ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(31mg, 0. 10mmol)、H OBt (20mg, 0. 15mmol)、 ΝΕΜ (14 Ι^, 0. l lmmol)、 EDCI-塩酸塩(23mg , 0. 12mmol)を加え、室温で攪拌した。 17時間後、飽和炭酸水素ナトリウム水溶液 を加えクロ口ホルムで抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 49mg (定量的)を得た。
[0084] ステップ 4 (3R)— 3 アミノー 4— (2 フノレオ口フエ-ノレ)一 N—(l—メチノレ一 4—
フエ-ルビペリジン 4 ィル)ブタナミド '二塩酸塩 (化合物 2)の合成 アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (1—メチル—4 フ ェ-ルビペリジン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブ タナミド (49mg, 0. lOmmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し、 室温で 3時間攪拌した後濃縮し、表題ィ匕合物 31mg (67%)を得た。
[0085] 実施例 3
(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N—(l—メチル 4 フ ェ-ルビペリジン 4 ィル)ブタナミド '二塩酸塩 (化合物 3)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (1—メチル 4 フ ェ-ルビペリジン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブ タナミド (参考化合物 8)の合成
アルゴン雰囲気下、 N— (1—メチル—4—フエ-ルビペリジン— 4—ィル)ァセタミド (参考化合物 6) (HOmg, 0. 47mmol)を THF (0. lmL)に懸濁し、チタンテトライ ソプロボキシド(0. 15mL, 0. 51mmol)、ジフエ-ルシラン(0. 22mL, 1. 2mmol) を加え、室温で攪拌した。 12時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウ ム水溶液を加えて激しく攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェ チルにて抽出した。有機層を合わせて飽和食塩水で洗浄後、乾燥し濃縮した。アル ゴン雰囲気下、得られた粗生成物をジクロロメタン(5mL)に溶解し、 (3R)—4— (2, 4, 5 トリフルオロフヱ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪 酸(83mg, 0. 25mmol)、 HOBt (51mg, 0. 38mmol)、 ΝΕΜ (35 L, 0. 28m mol)、EDCI'塩酸塩(59mg, 0. 31mmol)を加え、室温で攪拌した。 12時間後、 飽和炭酸水素ナトリウム水溶液を加えクロ口ホルムで抽出し、乾燥後濃縮した。得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 81mg (64 %)を得た。
[0086] ステップ 2 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N—(l—メチ ルー 4 フエ二ルビペリジン 4 ィル)ブタナミド ·二塩酸塩 (化合物 3)の合成 アルゴン雰囲気下、 (3R)— 4— (2, 4, 5—トリフルオロフヱ-ル)— N—(l—メチル —4 フエ-ルビペリジン— 4—ィル)—3— (2—メチル—2 プロポキシカルボ-ル
ァミノ)ブタナミド(81mg, 0. 16mmol)を 10%塩化水素 Zメタノール溶液(3mL)に 溶解し、室温で 3時間攪拌した後濃縮し、表題ィ匕合物 59mg (77%)を得た。
[0087] 実施例 4
(3R)—3 ァミノ一 4— (2 フノレオ口フエ-ノレ) N— (4 フエ-ノレ一 2H—テトラヒ ドロピラン 4 ィル)ブタナミド ·塩酸塩 (化合物 4)の合成
ステップ 1 4 フエ-ル 2H—テトラヒドロピラン 4 オール(参考化合物 9)の合 成
アルゴン雰囲気下、フエ-ルリチウム Zシクロへキサンージェチルエーテル溶液(1 . 05M, 10. 3mL, 10. 8mmol)を THF (5mL)で希釈した溶液を氷冷し、 2H—テト ラヒドロピランー4 オン(0. 5mL, 5. 4mmol)の THF (5mL)溶液を滴下した。滴 下終了後、氷浴を外し室温で攪拌し、 3時間後、蒸留水を加えた。酢酸ェチルで抽 出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 827mg (86%)を得た。
[0088] ステップ 2 N— (4 フエ-ル一 2H—テトラヒドロピラン一 4—ィル)ァセタミド(参考化 合物 10)の合成
4—フエ-ルー 2H—テトラヒドロピラン一 4—オール(307mg, 1. 7mmol)のァセト 二トリル(2. 8mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(1. 9mL)をゆっくりと滴 下した。滴下終了後、氷浴を外し室温で攪拌し、 12時間後、氷水に注いだ。水層を 水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、有機層を 飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 63mg (17%)を得た。
[0089] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ) N— (4 フエ-ル一 2H—テトラヒドロピラン一 4—ィル)ブタナミド( 参考化合物 11)の合成
アルゴン雰囲気下、 N— (4—フエ-ルー 2H—テトラヒドロピラン一 4—ィル)ァセタミ ド(63mg, 0. 29mmol)を THF (0. ImL)に懸濁し、チタンテトライソプロポキシド(8 0. 29mmol)、ジフエ-ルシラン(0. 13mL, 0. 70mmol)を加え、室温で攪 拌した。 14時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液を加えて
激しく攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽出した 。有機層を合わせて飽和食塩水で洗浄後、乾燥し濃縮した。アルゴン雰囲気下、得 られた粗生成物をジクロロメタン(3mL)に溶解し、 (3R)—4— (2—フルオロフェ-ル ) - 3- (2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. lmmol)、 H OBt (20mg, 0. 15mmol)、 ΝΕΜ (14 Ι^, 0. l lmmol)、 EDCI'塩酸塩(24mg , 0. 13mmol)を加え、室温で攪拌した。 12時間後、飽和炭酸水素ナトリウム水溶液 を加えクロ口ホルムで抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 43mg (93%)を得た。
[0090] ステップ 4 (3R)— 3 アミノー 4— (2 フノレオ口フエ-ノレ)一 N— (4 フエ-ノレ一 2 H—テトラヒドロピラン 4 ィル)ブタナミド ·塩酸塩 (化合物 4)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—フエ-ルー 2H—テトラヒドロピラン一 4—ィル) ブタナミド (42mg, 0. lmmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し、 室温で 13時間攪拌した後濃縮し、表題ィ匕合物 30mg (83%)を得た。
[0091] 実施例 5
(3R)—3 アミノー 4— (2 フノレオ口フエ-ノレ) N— (4 フエ-ノレチアン一 4—ィ ル)ブタナミド ·塩酸塩 (化合物 5)の合成
ステップ 1 4 フエ二ルチアン— 4 オール(参考化合物 12)の合成
アルゴン雰囲気下、フエ-ルリチウム Zシクロへキサンージェチルエーテル溶液(1 . 05M, 9. 5mL, 10. Ommol)を THF (5mL)で希釈した溶液を氷冷し、 4—ォキソ チアン(582mg, 5. Ommol)の THF (5mL)溶液を滴下した。滴下終了後、氷浴を 外し室温で攪拌し、 2. 5時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層を 飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 569mg (58%)を得た。
[0092] ステップ 2 N—(4 フエ-ルチアンー 4 ィル)ァセタミド(参考化合物 13)の合成
4 フエ-ルチアンー4 オール(307mg, 1. 6mmol)のァセトニトリル(3mL)溶 液を氷冷し、激しく攪拌しながら濃硫酸(1. 9mL)をゆっくりと滴下した。滴下終了後 、氷浴を外し室温で攪拌し、 12時間後、氷水に注いだ。水層を水酸ィ匕ナトリウム水溶
液にてアルカリ性にした後、酢酸ェチルにて抽出し、有機層を飽和食塩水で洗浄後 、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 310mg (76%)を得た。
[0093] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) -N- (4 フエ-ルチアンー4 ィル)ブタナミド(参考化合物 14)の 合成
アルゴン雰囲気下、 N— (4 フエ-ルチアン— 4—ィル)ァセタミド(60mg, 0. 25 mmol)を THF (0. lmL)に懸濁し、チタンテトライソプロポキシド(76 L, 0. 26m mol)、ジフエ-ルシラン(0. 12mL, 0. 65mmol)を加え、室温で攪拌した。 13時間 後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液を加えて激しく攪拌した 。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽出した。有機層を合 わせて飽和食塩水で洗浄後、乾燥し濃縮した。アルゴン雰囲気下、得られた粗生成 物をジクロロメタン(3mL)に溶解し、 (3R)—4— (2—フルオロフヱ-ル) 3— (2— メチル—2 プロポキシカルボ-ルァミノ)酪酸(40mg, 0. 13mmol)、 HOBt (28m g, 0. 21mmol)、 NEM l^ L, 0. 15mmol)、 EDCI-塩酸塩(31mg, 0. 16mm ol)を加え、室温で攪拌した。 13時間後、飽和炭酸水素ナトリウム水溶液を加えクロ口 ホルムで抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 60mg (94%)を得た。
[0094] ステップ 4 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4—フエ-ルチア ン 4 ィル)ブタナミド ·塩酸塩 (化合物 5)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—フエ-ルチアンー4 ィル)ブタナミド(26mg, 0. 06mmol)を 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、室温で 16時間攪 拌した後濃縮し、表題ィ匕合物 19mg (84%)を得た。
[0095] 実施例 6
(3R)—3 アミノー 4— (2 フノレオ口フエ-ノレ) N— (4 フエ-ノレ一 1, 1—ジォキ ソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 6)の合成
ステップ 1 (3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシ力
ルボニルァミノ) -N- (4 フエ-ルー 1, 1—ジォキソチアン一 4—ィル)ブタナミド( 参考化合物 15)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ) N—(4 フ 二ルチアンー 4 ィル)ブタナミド(参考化合物 14) (33mg, 0 . 07mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA ( >65%) (38mg, >0. 14mmol) ( >65%とは、 mCPBAの純度を表す。計算上、 65%純度の mCPB A38mgは 0. 14mmolとなる。 mCPBAの純度が〉 65%であるため、 >65%純度の mCPBA38mgは、正確なモル数は不明だ力 >0. 14mmolと見積もることができる 。 >0. 14mmolとはこのことを意味しており、以下も同様の意味で用いる。)を 2回に 分けて添加した。 2時間後、チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽 和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 30mg (85 %)を得た。
[0096] ステップ 2 (3R)— 3 アミノー 4— (2 フノレオ口フエ-ノレ)一 N— (4 フエ-ノレ一 1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 6)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—フエ-ルー 1, 1—ジォキソチアン— 4—ィル) ブタナミド(30mg, 0. 06mmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し 、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 24mg (92%)を得た。
[0097] 実施例 7
(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4 フエ-ルチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 7)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N—(4 フエ-ルチアンー 4 ィル)ブタナミド(参考化合 物 16)の合成
アルゴン雰囲気下、 N—(4 フエ-ルチアンー 4 ィル)ァセタミド(参考化合物 13 ) (181mg, 0. 77mmol)を THF (0. 3mL)に懸濁し、チタンテトライソプロポキシド( 0. 23mL, 0. 77mmol)、ジフエ-ルシラン(0. 36mL, 1. 94mmol)を加え、室温
で攪拌した。 20時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液を 加えて激しく攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽 出した。有機層を合わせて飽和食塩水で洗浄後、乾燥し濃縮した。アルゴン雰囲気 下、得られた粗生成物をジクロロメタン(10mL)に溶解し、 (3R) -4- (2, 4, 5 トリ フルオロフェ -ル) - 3- (2—メチルー 2—プロポキシカルボ-ルァミノ)酪酸(201m g, 0. 6mmol)、 HOBt (123mg, 0. 91mmol) , NEM (84 μ L, 0. 66mmol)、E DC 塩酸塩(139mg, 0. 73mmol)をカ卩え、室温で攪拌した。 13時間後、飽和炭 酸水素ナトリウム水溶液を加えクロ口ホルムで抽出し、乾燥後濃縮した。得られた粗 生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 287mg (93%)を 得た。
[0098] ステップ 2 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4 フエ 二ルチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 7)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4 フエ-ルチアンー4 ィル)ブタナミド
(32mg, 0. 06mmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し、室温で 1
2時間攪拌した後濃縮し、表題ィ匕合物 20mg (69%)を得た。
[0099] 実施例 8
(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4 フエ-ル一 1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 8)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4 フエ二ルー 1, 1—ジォキソチアン— 4—ィル)ブ タナミド (参考化合物 17)の合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N—(4 フエ-ルチアンー 4 ィル)ブタナミド(参考化合物 16) (2 45mg, 0. 48mmol)をジクロロメタン(20mL)に溶解し、氷冷下、 mCPBA( >65% ) (306mg, > 1. 2mmol)を 2回に分けて添カ卩した。 4時間後、チォ硫酸ナトリウム水 溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホル ムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー
で精製し、表題ィ匕合物 225mg (86%)を得た。
[0100] ステップ 2 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4 フエ 二ルー 1 , 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 8)の合成 アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル — 2 プロポキシカルボ-ルァミノ) N— (4 フエ-ル—1, 1—ジォキソチアン 4 ィル)ブタナミド(222mg, 4. lmmol)を 10%塩化水素 Zメタノール溶液(10mL) に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 185mg (94%)を得た。
[0101] 実施例 9
(3R) 3 アミノー 4一(2 フルオロフヱ-ル) N— (4—(3 フルオロフヱ-ル) — 1, 1 ジォキソチアン— 4 ィル)ブタナミド ·塩酸塩 (化合物 9)の合成
ステップ 1 4一(3 フルオロフヱニル)チアンー 4一オール(参考化合物 18)の合成 アルゴン雰囲気下、 1ーブロモー 3 フルォロベンゼン(753mg、 4. 30mmol)の T HF (15mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 39M, 3. 4mL, 4. 73mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30mmol )の THF (lOmL)溶液をカ卩え、 78°Cで 2. 5時間撹拌した後、更に室温に昇温さ せて撹拌した。 4時間後、飽和塩ィ匕アンモ-ゥム水溶液を加えて酢酸ェチルで抽出 し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合物 565mg (62%)を得た。
[0102] ステップ 2 N—(4一(3 フルオロフェ -ル)チアンー 4 ィル)ァセタミド(参考化合 物 19)の合成
アルゴン雰囲気下、 4 (3 フルオロフェ -ル)チアンー 4 オール(500mg, 2. 3 6mmol)のァセトニトリル (4mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(2. 6mL) をゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 12時間後、氷水に注い だ。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し 、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 493mg (83%)を得た。
[0103] ステップ 3 4—(3 フルオロフェ -ル)チアンー 4ーァミン(参考化合物 20)の合成 アルゴン雰囲気下、 N— (4—(3—フルオロフェ -ル)チアンー 4 ィル)ァセタミド(
490mg, 1. 93mmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 53mL, 1. 93mmol)、ジフエ-ルシラン(0. 90mL, 2. 50mmol)をカ卩えて室温で撹拌した 。 12時間後、酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた不溶物 をセライト濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗浄後、 乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題化合物 178mg (44%)を得た。
[0104] ステップ 4 (3R)—4 (2 フルオロフェ -ル)—N— (4— (3 フルオロフェ -ル) チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参 考化合物 21)の合成
アルゴン雰囲気下、 4 (3 フルオロフェ -ル)チアンー 4ーァミン(37mg, 0. 18 mmol)の DMF (2. 5mL)溶液に(3R)— 4一(2 フルオロフヱニル) 3—(2—メ チルー 2 プロポキシカルボ-ルァミノ)酪酸(50mg, 0. 17mmol)、 0—(7 ァザ ベンゾトリアゾール 1—ィル) 1, 1, 3, 3, —テトラメチルゥロニゥム へキサフルォ 口フォスファイト(以下、 HATUと略す)(77mg, 0. 20mmol)、ジイソプロピルェチル ァミン(以下、 DIPEAと略す)(0. 14mL, 0. 84mmol)をカ卩えて室温で撹拌した。 2 4時間後、飽和炭酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、飽和食塩 水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で精製し、表題ィ匕合物 77mg (92%)を得た。
[0105] ステップ 5 (3R)—4 (2 フルオロフェ -ル)—N— (4— (3 フルオロフェ -ル)
- 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)ブタナミド (参考化合物 22)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4—(3 フルオロフヱ-ル)チアンー 4 ーィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(74mg, 0. 1 5mmol)のジクロロメタン(3mL)溶液を氷冷し、 mCPBA( >65%) (65mg, >0. 2 4mmol)を 2回に分けて加え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水溶液を 加えて攪拌した後ジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶液 及び飽和食塩水で洗浄し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 44mg (56%)を得た。
[0106] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)— N— (4— (3 フルォロ フエ-ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 9)の合成 アルゴン雰囲気下、 (3R)— 4一(2 フルオロフヱ-ル) N—(4一(3 フルォロ フエ二ル)一 1, 1—ジォキソチアン一 4—ィル) 3— (2—メチル 2 プロポキシ力 ルポ-ルァミノ)ブタナミド(44mg, 0. 08mmol)を 10%塩化水素 Zメタノール溶液( 3mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 39mg (定量的)を得 た。
[0107] 実施例 10
(3R) 3 アミノー 4一(2 フノレオ口フエ-ノレ) N— (4—(4ーフノレオ口フエ-ノレ) — 1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 10)の合成 ステップ 1 4一(4一フルオロフヱニル)チアンー 4一オール(参考化合物 23)の合成 アルゴン雰囲気下、 1ーブロモー 4 フルォロベンゼン(753mg, 4. 30mmol)の T HF (15mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 39M, 3. 4mL, 4. 73mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30mmol )の THF (lOmL)溶液を加え、 78°Cで 20分撹拌した後、更に室温に昇温させて 撹拌した。 3. 5時間後、飽和塩ィ匕アンモ-ゥム水溶液を加えて酢酸ェチルで抽出し 、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル力 ラムクロマトグラフィーで精製後、ジェチルエーテルで洗浄することにより表題化合物 565mg (62%)を得た。
[0108] ステップ 2 N—(4一(4 フルオロフェ -ル)チアンー 4 ィル)ァセタミド(参考化合 物 24)の合成
アルゴン雰囲気下、 4 (4 フルオロフェ -ル)チアンー 4 オール(300mg, 1. 4 lmmol)のァセトニトリル (4mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(3mL)を ゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 12時間後、氷水に注いだ 。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、 乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 540mg (定量的)を得た。
[0109] ステップ 3 4—(4 フルオロフヱ-ル)チアンー 4ーァミン(参考化合物 25)の合成
アルゴン雰囲気下、 N— (4—(4 フルオロフェ -ル)チアンー 4 ィル)ァセタミド( 530mg, 2. O5mmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 53mL, 1. 93mmol)、ジフエ-ルシラン(0. 90mL, 2. 50mmol)をカ卩えて室温で撹拌した 。 12時間後、酢酸ェチル、飽和炭酸水素ナトリウム水溶液を加え、生じた不溶物をセ ライト濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥 し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 220mg (51%)を得た。
[0110] ステップ 4 (3R)—4—(2 フノレオ口フエ-ノレ)一N—(4一(4ーフノレオ口フエ-ノレ) チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参 考化合物 26)の合成
アルゴン雰囲気下、 4 (4 フルオロフェ -ル)チアンー 4ーァミン(37mg, 0. 18 mmol)の DMF (2. 5mL)溶液に、(3R)—4 (2 フルオロフヱ-ル)ー3—(2—メ チルー 2 プロポキシカルボ-ルァミノ)酪酸(50mg, 0. 17mmol)、 HATU (77mg , 0. 20mmol)、 DIPEA(0. 14mL, 0. 84mmol)をカ卩えて室温で撹拌した。 24時 間後、飽和炭酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 57mg (69%)を得た。
[0111] ステップ 5 (3R)—4—(2 フノレオ口フエ-ノレ) N—(4一(4ーフノレオ口フエ-ノレ)
- 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)ブタナミド (参考化合物 27)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4—(4 フルオロフヱ-ル)チアンー 4 ーィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(57mg, 0. 1 2mmol)のジクロロメタン(3mL)溶液を氷冷し、 mCPBA( >65%) (50mg、 >0. 1 9mmol)を 2回に分けて加え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水溶液を 加えて攪拌しジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶液及び 飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 33mg (54%)を得た。
[0112] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (4 フルォロ
フエ-ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 10)の合成 アルゴン雰囲気下、 (3R)— 4一(2 フルオロフヱ-ル) N—(4一(4 フルォロ フエ二ル)一 1, 1—ジォキソチアン一 4—ィル) 3— (2—メチル 2 プロポキシ力 ルポ-ルァミノ)ブタナミド(33mg, 0. 06mmol)を 10%塩化水素 Zメタノール溶液( 3mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 30mg (定量的)を得 た。
[0113] 実施例 11
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (2, 3 ジフルオロフェ- ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 11)の合成 ステップ 1 4一(2, 3 ジフルオロフェ -ル)チアンー 4 オール(参考化合物 28)の 合成
アルゴン雰囲気下、 1—ブロモ 2, 3 ジフルォロベンゼン(83 lmg, 4. 30mmol )の THF (15mL)溶液を—78°Cに冷却し、 n ブチルリチウム Zへキサン溶液(1. 3 9M, 3. 4mL, 4. 73mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30 mmol)の THF (lOmL)溶液を加え、 78°Cで 2. 5時間撹拌した後、更に室温に昇 温させて撹拌した。 2時間後、飽和塩ィ匕アンモ-ゥム水溶液をカ卩えて酢酸ェチルで 抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 430mg (43%)を得た。
[0114] ステップ 2 N— (4— (2, 3 ジフルオロフェ -ル)チアン— 4—ィル)ァセタミド(参考 化合物 29)の合成
アルゴン雰囲気下、 4 (2, 3 ジフルオロフェ -ル)チアンー 4 オール(290mg , 1. 26mmol)のァセトニトリル(2. 5mL)溶液を氷冷し、激しく攪拌しながら濃硫酸( 1. 5mL)をゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷 水に注いだ。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルに て抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 297mg (87%)を得た。
[0115] ステップ 3 4—(2, 3 ジフルオロフェ -ル)チアンー 4ーァミン(参考化合物 30)の 合成
アルゴン雰囲気下、 N— (4— (2, 3 ジフルオロフェ -ル)チアンー 4 ィル)ァセ タミド(297mg, 1. lOmmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 3 2mL, 1. lOmmol)、ジフエ-ルシラン(0. 51mL, 2. 70mmol)を加えて室温で撹 拌した。 17時間後、酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた 不溶物をセライト濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗 浄し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題ィ匕合物 100mg (40%)を得た。
[0116] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 3 ジフルオロフェ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合物 31)の合成
アルゴン雰囲気下、 4— (2, 3 ジフルオロフェ -ル)チアン— 4 ァミン(65mg, 0 . 28mmol)の DMF (3mL)溶液に(3R)— 4一(2 フルオロフヱニル)一 3—(2—メ チル— 2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 27mmol)、 HATU (123 mg, 0. 32mmol)、 DIPEA (0. 23mL, 1. 35mmol)を加えて室温で撹拌した。 24 時間後、飽和炭酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽 和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題化合物 110mg (81 %)を得た。
[0117] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 3 ジフルオロフェ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 32)の合成
アルゴン雰囲気下、 (3R)— 4— (2 フルオロフヱ-ル)— N— (4— (2, 3 ジフル オロフェ -ル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ )ブタナミド(l lOmg, 0. 22mmol)のジクロロメタン(4mL)溶液を氷冷し、 mCPBA ( >65%) (93mg、 >0. 35mmol)を 2回に分けて加え、撹拌した。 2時間後、飽和チ ォ硫酸ナトリウム水溶液を加えてジクロロメタンで抽出し、有機層を飽和炭酸水素ナト リウム水溶液及び飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 66mg (55%)を得た。
[0118] ステップ 6 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (2, 3 ジフ
ルォロフエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 11) の合成
アルゴン雰囲気下、 (3R)— 4— (2 フルオロフヱ-ル)— N— (4— (2, 3 ジフル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(66mg, 0. 12mmol)を 10%塩化水素 Zメタノール 溶液 (4mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 60mg (定量 的)を得た。
[0119] 実施例 12
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (2, 4 ジフルオロフェ- ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 12)の合成 ステップ 1 4一(2, 4 ジフルオロフェ -ル)チアンー 4 オール(参考化合物 33)の 合成
アルゴン雰囲気下、 1—ブロモ 2, 4 ジフルォロベンゼン(83 lmg, 4. 30mmol )の THF (20mL)溶液を—78°Cに冷却し、 n ブチルリチウム Zへキサン溶液(1. 3 9M, 3. 4mL, 4. 73mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30 mmol)の THF (lOmL)溶液を加え、 78°Cで 2. 5時間撹拌した後、更に室温に昇 温させて撹拌した。 2時間後、飽和塩ィ匕アンモ-ゥム水溶液をカ卩えて酢酸ェチルで 抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 996mg (81%)を得た。
[0120] ステップ 2 N— (4— (2, 4 ジフルオロフェ -ル)チアン— 4—ィル)ァセタミド(参考 化合物 34)の合成
アルゴン雰囲気下、 4 (2, 4ージフルオロフェ -ル)チアンー 4 オール(996mg , 4. 33mmol)のァセトニトリル(7mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(5 mL)をゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷水に 注いだ。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽 出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題化合物 1. 12g (95%)を得た。
[0121] ステップ 3 4—(2, 4 ジフルオロフェ -ル)チアンー 4ーァミン(参考化合物 35)の
合成
アルゴン雰囲気下、 N— (4— (2, 4ージフルオロフェ -ル)チアンー 4 ィル)ァセ タミド(1. 12g, 4. 13mmol)の THF (2mL)溶液にチタンテトライソプロポキド(1. 2 2mL, 4. 13mmol)、ジフエ-ルシラン(1. 92mL, 10. 3mmol)を加えて室温で撹 拌した。 19時間後、酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた 不溶物をセライト濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗 浄し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題ィ匕合物 129mg (14%)を得た。
[0122] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 4 ジフルオロフェ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合物 36)の合成
アルゴン雰囲気下、 4— (2, 4 ジフルオロフェ -ル)チアン— 4 ァミン(65mg, 0 . 28mmol)の DMF (3mL)溶液に、(3R)— 4一(2 フルオロフヱニル)一 3—(2— メチル—2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 27mmol)、 HATU (123 mg, 0. 32mmol)、 DIPEA (0. 23mL, 1. 35mmol)を加えて室温で撹拌した。 24 時間後、飽和炭酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽 和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 125mg (89%)を得た。
[0123] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 4 ジフルオロフェ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 37)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4— (2, 4ージフルオロフヱ-ル)チアン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(125mg , 0. 25mmol)のジクロロメタン(4mL)溶液を氷冷し、 mCPBA ( >65%) (93mg、 >0. 35mmol)を 2回に分けてカ卩え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水 溶液を加えて攪拌しジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶 液及び飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 61mg (45%)を得た。
[0124] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (2, 4 ジフ ルォロフエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 12) の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (2, 4 ジフル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(6 lmg, 0. l lmmol)を 10%塩化水素 Zメタノール 溶液 (4mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 56mg (定量 的)を得た。
[0125] 実施例 13
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (2, 5 ジフルオロフェ- ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 13)の合成 ステップ 1 4一(2, 5 ジフルオロフェ -ル)チアンー 4 オール(参考化合物 38)の 合成
アルゴン雰囲気下、 1—ブロモ 2, 5 ジフルォロベンゼン(83 lmg, 4. 30mmol )の THF (15mL)溶液を—78°Cに冷却し、 n ブチルリチウム Zへキサン溶液(1. 3 9M, 3. 4mL, 4. 73mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30 mmol)の THF (lOmL)溶液を加え、 78°Cで 2. 5時間撹拌した後、更に室温に昇 温させて撹拌した。 2時間後、飽和塩ィ匕アンモ-ゥム水溶液をカ卩えて酢酸ェチルで 抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 552mg (56%)を得た。
[0126] ステップ 2 N— (4— (2, 5 ジフルオロフェ -ル)チアン— 4—ィル)ァセタミド(参考 化合物 39)の合成
アルゴン雰囲気下、 4 (2, 5 ジフルオロフェ -ル)チアンー 4 オール(302mg , 1. 31mmol)のァセトニトリル(2. 5mL)溶液を氷冷し、激しく攪拌しながら濃硫酸( 1. 4mL)をゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 18時間後、氷 水に注いだ。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルに て抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 344mg (97%)を得た。
[0127] ステップ 3 4—(2, 5 ジフルオロフェ -ル)チアンー 4ーァミン(参考化合物 40)の 合成
アルゴン雰囲気下、 N—(4—(2, 5 ジフルオロフェ -ル)チアンー 4 ィル)ァセ タミド(340mg, 1. 25mmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 3 7mL, 1. 25mmol)、ジフエ-ルシラン(0. 58mL, 3. 13mmol)を加えて室温で撹 拌した。 19時間後、酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた 不溶物をセライト濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗 浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題ィ匕合物 69mg (24%)を得た。
[0128] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 5 ジフルオロフェ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合物 41)の合成
アルゴン雰囲気下、 4— (2, 5 ジフルオロフェ -ル)チアン— 4 ァミン(65mg, 0 . 28mmol)の DMF (3mL)溶液に、(3R)— 4一(2 フルオロフヱニル)一 3—(2— メチル—2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 27mmol)、 HATU (123 mg, 0. 32mmol)、 DIPEA (0. 23mL, 1. 35mmol)を加えて室温で撹拌した。 24 時間後、飽和炭酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽 和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 62mg (52%)を得た。
[0129] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 5 ジフルオロフェ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 42)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4— (2, 5 ジフルオロフヱ-ル)チアン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(62mg, 0. 12mmol)のジクロロメタン(4mL)溶液を氷冷し、 mCPBA ( >65%) (52mg、 > 0. 20mmol)を 2回に分けてカ卩え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水溶 液を加えて攪拌し、ジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶液 及び飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムク
口マトグラフィ一で精製し、表題ィ匕合物 22mg (34%)を得た。
[0130] ステップ 6 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (2, 5 ジフ ルォロフエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 13) の合成
アルゴン雰囲気下、 (3R)— 4— (2 フルオロフヱ-ル)— N— (4— (2, 5 ジフル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(22mg, 0. 04mmol)を 10%塩化水素 Zメタノール 溶液 (4mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 58mg (定量 的)を得た。
[0131] 実施例 14
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (2, 6 ジフルオロフェ- ル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 14)の合成 ステップ 1 4一(2, 6 ジフルオロフェ -ル)チアンー 4 オール(参考化合物 43)の 合成
アルゴン雰囲気下、 1—ブロモ 2, 6 ジフルォロベンゼン(83 lmg, 4. 30mmol )の THF (15mL)溶液を—78°Cに冷却し、 n ブチルリチウム Zへキサン溶液(1. 3 9M, 3. 4mL, 4. 73mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30 mmol)の THF (lOmL)溶液を加え、 78°Cで 2. 5時間撹拌した後、更に室温に昇 温させて撹拌した。 2時間後、飽和塩ィ匕アンモ-ゥム水溶液をカ卩えて酢酸ェチルで 抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 65 lmg (66%)を得た。
[0132] ステップ 2 N— (4— (2, 6 ジフルオロフェ -ル)チアン一 4—ィル)ァセタミド(参考 化合物 44)の合成
アルゴン雰囲気下、 4 (2, 6 ジフルオロフェ -ル)チアンー 4 オール(650mg , 1. 26mmol)のァセトニトリル (4. 5mL)溶液を氷冷し、激しく攪拌しながら濃硫酸( 4. 5mL)をゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷 水に注いだ。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルに て抽出し、乾燥後縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで
精製し、表題ィ匕合物 703mg (92%)を得た。
[0133] ステップ 3 4—(2, 6 ジフルオロフェ -ル)チアンー 4ーァミン(参考化合物 45)の 合成
アルゴン雰囲気下、 N— (4— (2, 6 ジフルオロフェ -ル)チアンー 4 ィル)ァセ タミド(650mg, 2. 40mmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 7 lmL, 2. 40mmol)、ジフエ-ルシラン(1. 10mL, 6. OOmmol)を加えて室温で撹 拌した。 16時間後、酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた 不溶物をセライト濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗 浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題ィ匕合物 220mg (40%)を得た。
[0134] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 6 ジフルオロフェ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合物 46)の合成
アルゴン雰囲気下、 4— (2, 6 ジフルオロフェ -ル)チアン— 4 ァミン(65mg, 0 . 28mmol)の DMF (3mL)溶液に、(3R)— 4一(2 フルオロフヱニル)一 3—(2— メチル—2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 27mmol)、 HATU (123 mg, 0. 32mmol)、 DIPEA (0. 23mL, 1. 35mmol)を加えて室温で撹拌した。 24 時間後、飽和炭酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽 和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 118mg (86%)を得た。
[0135] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (2, 6 ジフルオロフェ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 47)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4— (2, 6 ジフルオロフヱ-ル)チアン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(117mg , 0. 23mmol)のジクロロメタン(4mL)溶液を氷冷し、 mCPBA ( >65%) (93mg、 >0. 35mmol)を 2回に分けてカ卩え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水 溶液を加えて攪拌し、ジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶
液及び飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 52mg (42%)を得た。
[0136] ステップ 6 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (2, 6 ジフ ルォロフエ-ル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (ィ匕合物 14) の合成
アルゴン雰囲気下、 (3R)— 4— (2 フルオロフヱ-ル)— N— (4— (2, 6 ジフル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(22mg, 0. 04mmol)を 10%塩化水素 Zメタノール 溶液 (4mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 23mg (定量 的)を得た。
[0137] 実施例 15
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (3, 5 ジフルオロフェ- ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 15)の合成 ステップ 1 4一(3, 5 ジフルオロフェ -ル)チアンー 4 オール(参考化合物 48)の 合成
アルゴン雰囲気下、 1—ブロモ 3, 5 ジフルォロベンゼン(83 lmg, 4. 30mmol )の THF (15mL)溶液を—78°Cに冷却し、 n ブチルリチウム Zへキサン溶液(1. 3 9M, 3. 4mL, 4. 73mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30 mmol)の THF (lOmL)溶液を加え、 78°Cで 2. 5時間撹拌した後、更に室温に昇 温させて撹拌した。 2時間後、飽和塩ィ匕アンモ-ゥム水溶液をカ卩えて酢酸ェチルで 抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 280mg (28%)を得た。
[0138] ステップ 2 N— (4— (3, 5 ジフルオロフェ -ル)チアン— 4—ィル)ァセタミド(参考 化合物 49)の合成
アルゴン雰囲気下、 4 (3, 5 ジフルオロフェ -ル)チアンー 4 オール(270mg , 1. 17mmol)のァセトニトリル(2. 5mL)溶液を氷冷し、激しく攪拌しながら濃硫酸( 1. 5mL)をゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷 水に注いだ。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルに
て抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 315mg (99%)を得た。
[0139] ステップ 3 4—(3, 5 ジフルオロフェ -ル)チアンー 4ーァミン(参考化合物 50)の 合成
アルゴン雰囲気下、 N— (4— (3, 5—ジフルオロフェ -ル)チアンー 4 ィル)ァセ タミド(185mg, 0. 68mmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 2 OmL, 0. 68mmol)、ジフエ-ルシラン(0. 32mL, 0. 99mmol)を加えて室温で撹 拌した。 15時間後、酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた 不溶物をセライト濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗 浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題ィ匕合物 282mg (定量的)を得た。
[0140] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)— N— (4— (3, 5 ジフルオロフェ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合物 51)の合成
アルゴン雰囲気下、 4— (3, 5 ジフルオロフェ -ル)チアン— 4 ァミン(50mg, 0 . 22mmol)の DMF (3mL)溶液に、(3R)— 4一(2 フルオロフヱニル)一 3—(2— メチルー 2 プロポキシカルボ-ルァミノ)酪酸(62mg, 0. 21mmol)、 HATU (95 mg, 0. 25mmol)、 DIPEA (0. 18mL, 1. OOmmol)を加えて室温で撹拌した。 24 時間後、飽和炭酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽 和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 116mg (定量的)を得た。
[0141] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)— N— (4— (3, 5 ジフルオロフェ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 52)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4— (3, 5 ジフルオロフヱ-ル)チアン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(116mg , 0. 23mmol)のジクロロメタン(4mL)溶液を氷冷し、 mCPBA ( >65%) (93mg、 >0. 35mmol)を 2回に分けてカ卩え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水
溶液を加えてジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶液及び 飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 81mg (65%)を得た。
[0142] ステップ 6 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (3, 5 ジフ ルォロフエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 15) の合成
アルゴン雰囲気下、 (3R)— 4— (2 フルオロフヱ-ル)— N— (4— (3, 5 ジフル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(8 lmg, 0. 15mmol)を 10%塩化水素 Zメタノール 溶液 (4mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 75mg (定量 的)を得た。
[0143] 実施例 16
(3R)—3 ァミノ— 4— (2 フルオロフェ -ル) N— (4— (m—トリル)— 1, 1—ジ ォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 16)の合成
ステップ 1 4一(m トリル)チアンー 4 オール(参考化合物 53)の合成
アルゴン雰囲気下、 3 ブロモトルエン(736mg, 4. 30mmol)の THF (15mL)溶 液を— 78。Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 39M, 3. 4mL, 4. 7 3mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30mmol)の THF (10m L)溶液を加え、 78°Cで 2. 5時間撹拌した後、更に室温に昇温させて撹拌した。 2 時間後、飽和塩ィ匕アンモ-ゥム水溶液を加えて酢酸ェチルで抽出し、有機層を飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 579mg (65%)を得た。
[0144] ステップ 2 N— (4— (m—トリル)チアンー 4 ィル)ァセタミド(参考化合物 54)の合 成
アルゴン雰囲気下、 4— (m—トリル)チアン— 4—オール(578mg, 2. 77mmol)の ァセトニトリル (4. 5mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(3mL)をゆっくりと 滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷水に注いだ。水層を 水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、乾燥後濃
縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 683mg (99%)を得た。
[0145] ステップ 3 4—(m トリル)チアンー 4ーァミン(参考化合物 55)の合成
アルゴン雰囲気下、 N— (4— (m—トリル)チアン— 4—ィル)ァセタミド(683mg, 2 . 74mmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 81mL, 2. 74mm ol)、ジフエ-ルシラン(1. 27mL, 6. 85mmol)をカ卩えて室温で撹拌した。 18時間 後、酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた不溶物をセライト 濾過により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃 縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 262mg (46%)を得た。
[0146] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (m—トリル)チアン— 4—ィル)ブタナミド(参考化合物 56 )の合成
アルゴン雰囲気下、 4— (m—トリル)チアン— 4 ァミン(59mg, 0. 28mmol)の D MF (3mL)溶液に、 (3R)— 4一(2 フルオロフヱニル) 3—(2—メチルー 2 プロ ポキシカルボ-ルァミノ)酪酸(80mg, 0. 27mmol)、 HATU (123mg, 0. 32mmo 1)、DIPEA(0. 23mL, 1. 35mmol)をカ卩えて室温で撹拌した。 24時間後、飽和炭 酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄 後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 99mg (76%)を得た。
[0147] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ) N— (4— (m—トリル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミ ド (参考化合物 57)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)— N— (4— (m—トリル)チアン— 4—ィル)ブタナミド(99mg, 0. 20mmol)の ジクロロメタン(4mL)溶液を氷冷し、 mCPBA( >65%) (93mg、 >0. 35mmol)を 2回に分けて加え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水溶液を加えてジク ロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄
後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 72mg (42%)を得た。
[0148] ステップ 6 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (m—トリル) — 1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 16)の合成 アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4— (m—トリル)— 1, 1—ジォキソチアン— 4—ィ ル)ブタナミド(72mg, 0. 08mmol)を 10%塩化水素 Zメタノール溶液(4mL)に溶 解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 66mg (定量的)を得た。
[0149] 実施例 17
(3R)—3 ァミノ— 4— (2 フルオロフェ -ル) N— (4— (p トリル)— 1, 1—ジォ キソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 17)の合成
ステップ 1 4一(p—トリル)チアンー 4一オール(参考化合物 58)の合成
アルゴン雰囲気下、 4 ブロモトルエン(736mg, 4. 30mmol)の THF (15mL)溶 液を— 78。Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 39M, 3. 4mL, 4. 7 3mmol)を加えた。 10分後、 4—ォキソチアン(500mg, 4. 30mmol)の THF (10m L)溶液を加え、 78°Cで 2. 5時間撹拌した後、更に室温に昇温させて撹拌した。 2 時間後、飽和塩ィ匕アンモ-ゥム水溶液を加えて酢酸ェチルで抽出し、有機層を飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 664mg (74%)を得た。
[0150] ステップ 2 N— (4— (p—トリル)チアンー 4 ィル)ァセタミド(参考化合物 59)の合 成
アルゴン雰囲気下、 4— (p トリル)チアン— 4—オール(664mg, 3. 19mmol)の ァセトニトリル(5mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(3. 4mL)をゆっくりと 滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷水に注いだ。水層を 水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、乾燥後濃 縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 572mg (72%)を得た。
[0151] ステップ 3 4—(p—トリル)チアンー 4ーァミン(参考化合物 60)の合成
アルゴン雰囲気下、 N— (4— (p トリル)チアン— 4—ィル)ァセタミド(391mg, 1. 57mmol)の THF (lmL)溶液にチタンテトライソプロポキド(0. 47mL, 1. 57mmol )、ジフエ-ルシラン(0. 73mL, 3. 93mmol)を加えて室温で撹拌した。 17時間後、 酢酸ェチル及び飽和炭酸水素ナトリウム水溶液を加え、生じた不溶物をセライト濾過 により除去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した 。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 215 mg (94%)を得た。
[0152] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (p トリル)チアン— 4—ィル)ブタナミド(参考化合物 61 )の合成
アルゴン雰囲気下、 4— (p トリル)チアン— 4 ァミン(65mg, 0. 28mmol)の D MF (3mL)溶液に、(3R)— 4一(2 フルオロフヱニル) 3—(2—メチルー 2 プロ ポキシカルボ-ルァミノ)酪酸(80mg, 0. 27mmol)、 HATU (123mg, 0. 32mmo 1)、DIPEA(0. 23mL, 1. 35mmol)をカ卩えて室温で撹拌した。 24時間後、飽和炭 酸水素ナトリウム水溶液を加えて酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄 後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 108mg (82%)を得た。
[0153] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (p トリル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミ ド (参考化合物 62)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)— N— (4— (p トリル)チアン— 4—ィル)ブタナミド(107mg, 0. 22mmol) のジクロロメタン(4mL)溶液を氷冷し、 mCPBA( >65%) (93mg、 >0. 35mmol) を 2回に分けて加え、撹拌した。 2時間後、飽和チォ硫酸ナトリウム水溶液を加えて攪 拌し、ジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶液及び飽和食 塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合物 89mg (78%)を得た。
[0154] ステップ 6 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (p トリル)一
1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 17)の合成 アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4— (p トリル)— 1, 1—ジォキソチアン— 4—ィ ル)ブタナミド(89mg, 0. 17mmol)を 10%塩化水素 Zメタノール溶液(4mL)に溶 解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 80mg (定量的)を得た。
[0155] 実施例 18
(3R)—3 アミノー 4— (2 フノレオ口フエ-ノレ) N—メチノレ一 N— (4 フエ-ノレ一 1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 18)の合成
ステップ 1 N—メチルー N— (4 フエ-ルチアン 4 ィル)ァセタミド(参考化合物 63)の合成
アルゴン雰囲気下、 N—(4 フエ-ルチアンー 4 ィル)ァセタミド(参考化合物 13 ) (640mg, 2. 72mmol)の DMF (lOmL)溶液を氷冷し、 60%水素化ナトリウム(21 8mg, 5. 44mmol)を加えて撹拌した。 10分後、ヨウ化メチル(0. 19mL, 2. 99mm ol)を滴下した後、氷浴を外して室温で撹拌した。 3時間後、蒸留水を加えてジェチ ルエーテルで抽出し、有機層を蒸留水及び飽和食塩水で洗浄後、乾燥し濃縮した。 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 534m g (79%)を得た。
[0156] ステップ 2 N—メチルー 4 フエ-ルチアンー 4ーァミン(参考化合物 64)の合成 アルゴン雰囲気、 N—メチル N— (4 フエ-ルチアン— 4—ィル)ァセタミド(530 mg, 2. 13mmol)にチタンテトライソプロポキド(0. 63mL, 2. 13mmol)、ジフエ- ルシラン(0. 99mL, 5. 31mmol)をカ卩えて室温で撹拌した。 12時間後、酢酸ェチ ル及び飽和炭酸水素ナトリウム水溶液を加え、生じた不溶物をセライト濾過により除 去した。濾液を酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られ た粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 324mg (73 %)を得た。
[0157] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)— N—メチル—3— (2—メチル—2— プロポキシカルボ-ルァミノ) N— (4—フエ-ルチアンー4 ィル)ブタナミド(参考 化合物 65)の合成
アルゴン雰囲気下、 N—メチル—4 フエ-ルチアン— 4 ァミン(59mg, 0. 28m mol)の DMF (2mL)溶液に、(3R)— 4一(2 フルオロフヱニル) 3—(2 メチル —2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 27mmol)、 HATU (123mg, 0 . 32mmol)、 DIPEA (0. 23mL, 1. 35mmol)をカ卩えて室温で撹拌した。 24時間 後、蒸留水を加えてジェチルエーテルで抽出し、有機層を蒸留水及び飽和食塩水 で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 84mg (64%)を得た。
[0158] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)— N—メチル—3— (2—メチル—2— プロポキシカルボ-ルァミノ) N— (4—フエ-ルー 1, 1—ジォキソチアン— 4—ィル )ブタナミド (参考化合物 66)の合成
(3R)—4— (2 フルオロフェ -ル) N—メチル 3— (2—メチル—2 プロポキ シカルボ-ルァミノ)— N— (4 フエ-ルチアン— 4—ィル)ブタナミド(84mg, 0. 17 mmol)のジクロロメタン(4mL)溶液を氷冷し、 mCPBA( >65%) (74mg、 >0. 28 mmol)を 2回に分けて加え、撹拌した。 2. 5時間後、飽和チォ硫酸ナトリウム水溶液 を加えて攪拌し、ジクロロメタンで抽出し、有機層を飽和炭酸水素ナトリウム水溶液及 び飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 90mg (定量的)を得た。
[0159] ステップ 5 (3R)— 3 アミノー 4— (2 フノレオ口フエ-ノレ)一 N—メチノレ一 N— (4— フエ-ル—1, 1 ジォキソチアン— 4 -ィル)ブタナミド ·塩酸塩 (化合物 18)の合成 アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N—メチル 3— (2—メ チル 2 プロポキシカルボ-ルァミノ) N— (4 フエニル—1, 1—ジォキソチア ンー 4 ィル)ブタナミド(90mg, 0. 17mmol)を 10%塩化水素 Zメタノール溶液(4 mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 70mg (定量的)を得 た。
[0160] 実施例 19
(3R)—3 ァミノ一 4— (3 クロ口フエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ)一 1
, 1 -ジォキソチアン— 4 -ィル)ブタナミド ·塩酸塩 (化合物 19)の合成
ステップ 1 4一(2 フルオロフヱニル)チアンー 4一オール(参考化合物 67)の合成
アルゴン雰囲気下、 2 フルォロブ口モベンゼン(3. 91mL, 36. Ommol)の THF (50mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 21 . 6mL, 34. 4mmol)を滴下した。 20分後、 4—ォキソチアン(4. OOg, 34. 4mmol )の THF (25mL)溶液を滴下した。滴下終了 30分後、室温までゆっくりと昇温し、 30 分後、塩ィ匕アンモ-ゥム水溶液を加えた。酢酸ェチルで抽出し、有機層を蒸留水、 飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で粗精製し、表題化合物 5. 10g (約 65%)を得た。
[0161] ステップ 2 N—(4一 (2 フルオロフェ -ル)チアンー 4一ィル)ァセタミド(参考化合 物 68)の合成
4 (2 フルオロフェ -ル)チアンー 4 オールの粗精製物(5. Og,約 23. 6mmo 1)のァセトニトリル(37mL)溶液を— 10°Cに冷却し、濃硫酸(31. 2mL)を滴下した。 30分間撹拌後室温まで昇温し、更に 10分間撹拌した。反応液を氷の入ったアンモ ユア水にあけ、酢酸ェチルにて抽出した。有機層を蒸留水、飽和食塩水にて洗浄後 、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製 し、表題化合物 5. 10g (約 85%)を得た。
[0162] ステップ 3 4—(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化合物 69) の合成
アルゴン雰囲気下、 N— (4—(2 フルオロフェ -ル)チアンー 4 ィル)ァセタミド( 5. OOg, 19. 7mmol)を THF (3mL)に懸濁し、チタンテトライソプロポキシド(5. 85 mL, 19. 7mmol)、ジフエ-ルシラン(5. 50mL, 29. 6mmol)を加え、 15時間撹 拌した。反応液に酢酸ェチルを加え希釈した後、飽和炭酸水素ナトリウム水溶液を 加えさらに 30分間撹拌した。セライト濾過後、濾液に蒸留水を加え酢酸ェチルにて 抽出した。有機層を蒸留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗 生成物をシリカゲルカラムクロマトグラフィーで粗精製した後、ジェチルエーテル (35 mL)に溶解し、そこへ 2N塩ィ匕水素/ジェチルエーテル溶液を滴下して塩ィ匕し、生じ た沈殿を濾過することにより、表題化合物 4. 3g (88%)を得た。
[0163] ステップ 4 (3R)— 4— (3 クロ口フエ-ル)一 N— (4— (2 フルオロフェ -ル)チア ンー4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考
化合物 70)の合成
アルゴン雰囲気下、 4一(2 フルオロフェ -ル)チアンー 4 アミン'塩酸塩(85mg , 0. 34mmol)を DMF (6mL)に溶解し、(3R)—4— (3 クロ口フエ-ル)一 3— (2 —メチルー 2 プロポキシカルボ-ルァミノ)酪酸(150mg, 0. 48mmol)、 HATU ( 220mg, 0. 58mmol) , DIPEA (238 μ L, 1. 44mmol)を加え、室温で攪拌した。 14時間後、反応液を蒸留水にあけ、ジェチルエーテルで抽出した。有機層を蒸留水 及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 151mg (87%)を得た。
[0164] ステップ 5 (3R)—4 (3 クロ口フエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ) 1 , 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ )ブタナミド (参考化合物 71)の合成
(3R)—4— (3 クロ口フエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ)チアン一 4—ィ ノレ)一 3— (2—メチノレ一 2 プロポキシカノレボニノレアミノ)ブタナミド(150mg, 0. 30 mmol)をジクロロメタン(8mL)に溶解し、氷冷下、 mCPBA( >65%) (128mg, >0 . 48mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌した。チォ硫酸ナトリウ ム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口 ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合 148mg (95%)を得た。
[0165] ステップ 6 (3R)— 3 アミノー 4— (3 クロ口フエ-ル)一 N— (4— (2 フルオロフ ェ-ル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 19)の合成 アルゴン雰囲気下、 (3R) -4- (3 クロ口フエ-ル) N— (4—(2 フルオロフェ -ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ ニルァミノ)ブタナミド(148mg, 0. 28mmol)を 10%塩化水素 Zメタノール溶液(3m L)、クロ口ホルム(3mL)に溶解し、室温で 14時間攪拌した後濃縮し、表題化合物 1 29mg (98%)を得た。
[0166] 実施例 20
(3R) 3 アミノー 4一(2 フルオロフヱ-ル) N— (4—(2 フルオロフヱ-ル) — 1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 20)の合成
ステップ 1 (3R)—4一(2 フルオロフェ -ル)—N— (4—(2 フルオロフェ -ル) チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参 考化合物 72)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (177mg, 0. 71mmol)を DMF (8mL)に溶解し、(3R)— 4— (2 フルォ 口フエ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(300mg, 1. OOmmol)、 HATU (456mg, 1. 20mmol) , DIPEA(496 μ L, 3. OOmmol)をカロ え、室温で攪拌した。 14時間後、反応液を蒸留水にあけ、ジェチルエーテルで抽出 した。有機層を蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 265mg (約 76%)を 得た。
[0167] ステップ 2 (3R)—4 (2 フルオロフェ -ル)—N— (4— (2 フルオロフェ -ル)
- 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)ブタナミド (参考化合物 73)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4—(2 フルオロフヱ-ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(260mg, 0. 53mmol)をジクロロメタン(8mL)に溶解し、氷冷下、 mCPBA( >65%) (229mg, >0. 86mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌した。チォ硫酸ナ トリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。 クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合 205mg (74%)を得た。
[0168] ステップ 3 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (2 フルォロ フエ-ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 20)の合成 アルゴン雰囲気下、 (3R)— 4一(2 フルオロフヱ-ル)一 N—(4一(2 フルォロ フエ二ル)一 1, 1—ジォキソチアン一 4—ィル) 3— (2—メチル 2 プロポキシ力 ルポ-ルァミノ)ブタナミド(40mg, 0. 08mmol)を 10%塩化水素 Zメタノール溶液( 2mL)、クロ口ホルム(lmL)に溶解し、室温で 14時間攪拌した後濃縮し、表題化合 物 35mg (99%)を得た。
[0169] 実施例 21
(3R)—3 アミノー 4— (2 クロ口フエ-ノレ) -N- (4— (2 フノレオ口フエ-ノレ)一 1 , 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 21)の合成
ステップ 1 (3R)—4— (2 クロ口フエ-ル) N— (4— (2 フルオロフェ -ル)チア ンー4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考 化合物 74)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4一アミン'塩酸塩(参考化 合物 69) (71mg, 0. 33mmol)を DMF (6mL)に溶解し、(3R)—4— (2 クロロフ ェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(lOOmg, 0. 32m mol)、 HATU (146mg, 0. 38mmol) , DIPEA(165 μ L, 1. OOmmol)を加え、 室温で攪拌した。 14時間後、反応液を蒸留水にあけ、ジェチルエーテルで抽出した 。有機層を蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 1 lOmg (68%)を得た。
[0170] ステップ 2 (3R)—4— (2 クロ口フエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ) - 1 , 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ )ブタナミド (参考化合物 75)の合成
(3R)—4— (2 クロ口フエ-ノレ) -N- (4— (2 フノレオ口フエ-ノレ)チアン一 4—ィ ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(105mg, 0. 21 mmol)をジクロロメタン(14mL)に溶解し、氷冷下、 mCPBA( >65%) (93mg, >0 . 35mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌した。チォ硫酸ナトリウ ム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口 ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合 11 lmg (定量的)を得た。
[0171] ステップ 3 (3R)— 3 アミノー 4— (2 クロ口フエ-ル)一 N— (4— (2 フルオロフ ェ-ル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 21)の合成 アルゴン雰囲気下、 (3R) -4- (2 クロ口フエ-ル) N— (4—(2 フルオロフェ -ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ ニルァミノ)ブタナミド(l lOmg, 0. 20mmol)を 10%塩化水素 Zメタノール溶液(3m
L)、クロ口ホルム(3mL)に溶解し、室温で 14時間攪拌した後濃縮し、表題化合物 9 lmg (94%)を得た。
[0172] 実施例 22
(3R) 3 アミノー 4一(4 クロ口フエ-ノレ) N—(4一(2 フノレオ口フエ-ノレ) 1 , 1 -ジォキソチアン— 4 ィル)ブタナミド ·塩酸塩 (化合物 22)の合成
ステップ 1 (3R)—4— (4 クロ口フエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ)チア ンー4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考 化合物 76)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (71mg, 0. 33mmol)を DMF (6mL)に溶解し、(3R)—4— (4 クロロフ ェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(lOOmg, 0. 32m mol)、 HATU (146mg, 0. 38mmol) , DIPEA(165 μ L, 1. OOmmol)を加え、 室温で攪拌した。 14時間後、反応液を蒸留水にあけ、ジェチルエーテルで抽出した 。有機層を蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 120mg (74%)を得た。
[0173] ステップ 2 (3R) -4- (4 クロ口フエ-ノレ) N— (4—(2 フノレオ口フエ-ノレ) 1 , 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ )ブタナミド (参考化合物 77)の合成
(3R)—4— (4 クロ口フエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ)チアン一 4—ィ ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(115mg, 0. 23 mmol)をジクロロメタン(14mL)に溶解し、氷冷下、 mCPBA( >65%) (102mg, > 0. 38mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌した。チォ硫酸ナトリ ゥム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ 口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合 124mg (定量的)を得た。
[0174] ステップ 3 (3R)— 3 アミノー 4— (4 クロ口フエ-ル)一 N— (4— (2 フルオロフ ェ-ル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 22)の合成 ァノレゴン雰囲気下、(3R) -4- (4 クロ口フエ-ノレ) N— (4—(2 フノレオロフェ
-ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ ニルァミノ)ブタナミド(124mg, 0. 23mmol)を 10%塩化水素 Zメタノール溶液(3m L)、クロ口ホルム(3mL)に溶解し、室温で 14時間攪拌した後濃縮し、表題化合物 1 04mg (95%)を得た。
[0175] 実施例 23
(3R)—3 アミノー 4— (2, 4 ジクロロフエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ )— 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 23)の合成 ステップ 1 (3R)—4— (2, 4 ジクロロフエ-ル) N— (4— (2 フルオロフェ-ル )チアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド( 参考化合物 78)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (52mg, 0. 24mmol)を DMF (5mL)に溶解し、(3R)— 4— (2, 4 ジクロ 口フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 2 3mmol)、 HATU (105mg, 0. 28mmol)、 DIPEA (114 L, 0. 69mmol)を加え 、室温で攪拌した。 14時間後、反応液に蒸留水を加えた。析出した固体を濾取した 後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 97mg ( 約 78%)を得た。
[0176] ステップ 2 (3R)— 4— (2, 4 ジクロロフエ-ノレ)一 N— (4— (2 フノレオ口フエ-ノレ )—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)ブタナミド (参考化合物 79)の合成
(3R)—4— (2, 4 ジクロロフエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ)チアン一 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(90mg, 0. 17mmol)をジクロロメタン(12mL)に溶解し、氷冷下、 mCPBA ( >65%) (200mg , >0. 75mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ硫酸 ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた 。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合 lOOmg (定量的)を得た。
[0177] ステップ 3 (3R)— 3 アミノー 4— (2, 4 ジクロロフエ二ル)一 N— (4— (2 フル
オロフェ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 23)の 合成
アルゴン雰囲気下、(3R)—4— (2, 4 ジクロロフヱ-ル) N— (4— (2 フルォ 口フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロポキシ カルボ-ルァミノ)ブタナミド(lOOmg, 0. 20mmol)を 10%塩化水素 Zメタノール溶 液(3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、表題ィ匕 合物 87mg (98%)を得た。
[0178] 実施例 24
(3R) 3 アミノー 4 (3, 4ージクロ口フエ-ノレ) N—(4一(2 フノレオ口フエ-ノレ )— 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 24)の合成 ステップ 1 (3R)—4— (3, 4 ジクロロフエ-ル) N— (4— (2 フルオロフェ-ル )チアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド( 参考化合物 80)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (52mg, 0. 24mmol)を DMF (5mL)に溶解し、(3R)— 4— (3, 4 ジクロ 口フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 2 3mmol)、 HATU (105mg, 0. 28mmol)、 DIPEA (114 L, 0. 69mmol)を加え 、室温で攪拌した。 14時間後、反応液に蒸留水を加えた。析出した固体を濾取した 後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 84mg ( 約 67%)を得た。
[0179] ステップ 2 (3R)— 4— (3, 4 ジクロロフエ-ノレ)一 N— (4— (2 フノレオ口フエ-ノレ )—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)ブタナミド (参考化合物 81)の合成
(3R) -4- (3, 4ージクロ口フエ-ノレ) N— (4—(2 フノレオ口フエ-ノレ)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(80mg, 0. 15mmol)をジクロロメタン(12mL)に溶解し、氷冷下、 mCPBA ( >65%) (200mg , >0. 75mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ硫酸 ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた
。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合 75mg (88%)を得た。
[0180] ステップ 3 (3R)— 3 アミノー 4— (3, 4 ジクロロフエ-ル)一 N— (4— (2 フル オロフェ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 24)の 合成
アルゴン雰囲気下、(3R)—4— (3, 4 ジクロロフヱ-ル) N— (4— (2 フルォ 口フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロポキシ カルボ-ルァミノ)ブタナミド(75mg, 0. 13mmol)を 10%塩化水素 Zメタノール溶 液(3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、表題ィ匕 合物 61mg (92%)を得た。
[0181] 実施例 25
(3R)—3 アミノー N— (4— (3 クロ口フエ-ノレ)一 1, 1—ジォキソチアン一 4—ィ ル) 4一(2 フルオロフヱニル)ブタナミド '塩酸塩(ィ匕合物 25)の合成
ステップ 1 4一(3 クロロフヱニル)チアンー 4 オール(参考化合物 82)の合成 アルゴン雰囲気下、 1—クロ口 3 ョードベンゼン(1. lmL, 8. 7mmol)の THF ( 20mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 5. 4 mL, 8. 6mmol)を滴下した。 20分後、 4—ォキソチアン(1. Og, 8. 6mmol)の TH F (10mL)溶液を滴下した。滴下終了 30分後、室温までゆっくりと昇温し、 30分後、 塩ィ匕アンモ-ゥム水溶液を加えた。酢酸ェチルで抽出し、有機層を蒸留水、飽和食 塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題化合物 1. 5g (76%)を得た。
[0182] ステップ 2 N—(4一(3 クロ口フエ-ル)チアンー 4 ィル)ァセタミド(参考化合物 8 3)の合成
4— (3 クロ口フエ-ノレ)チアン一 4—ォーノレ(1. 45g, 6. 30mmol)のァセトニトリ ル(l lmL)溶液を— 5°Cに冷却し、ゆっくりと濃硫酸(7. 4mL)を滴下した。 30分間 撹拌後室温まで昇温し、さらに 1時間撹拌した。反応液を氷の入ったアンモニア水に あけ、酢酸ェチルにて抽出した。有機層を蒸留水、飽和食塩水にて洗浄後、乾燥し 濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕
合物 1. 38g (81%)を得た。
[0183] ステップ 3 4—(3 クロロフヱ-ル)チアンー 4ーァミン(参考化合物 84)の合成
アルゴン雰囲気下、 N— (4—(3 クロロフヱ-ル)チアンー 4 ィル)ァセタミド(62 Omg, 2. 30mmol)を THF (2mL)に懸濁し、チタンテトライソプロポキシド(678 L , 2. 30mmol)、ジフエ-ルシラン(640 /z L, 3. 54mmol)をカ卩え、室温で 15時間撹 拌した。酢酸ェチルを加え希釈した後、飽和炭酸水素ナトリウム水溶液を加え更に 3 0分間撹拌した。セライト濾過後、濾液を蒸留水にあけ酢酸ェチルにて抽出した。有 機層を蒸留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリ 力ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 320mg (61%)を得た。
[0184] ステップ 4 (3R)—N— (4— (3 クロ口フエ-ル)チアン— 4—ィル)—4— (2 フル オロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考 化合物 85)の合成
アルゴン雰囲気下、 4— (3 クロ口フエ-ル)チアン— 4 ァミン(120mg, 0. 45m mol)を DMF (4mL)に溶解し、(3R)—4一(2 フルオロフヱ-ル)ー3—(2—メチ ルー 2 プロポキシカルボ-ルァミノ)酪酸(119mg, 0. 40mmol)、 HATU (190m g, 0. 50mmol) , DIPEA(198 μ L, 1. 20mmol)を加え、室温で攪拌した。 15時 間後、反応液を蒸留水にあけジェチルエーテルにて抽出した。有機層を蒸留水、飽 和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 190mg (94%)を得た。
[0185] ステップ 5 (31¾—?^ー(4ー(3—クロロフェ-ル)ー1, 1ージォキソチァンー4ーィル ) 4一(2 フルオロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ) ブタナミド (参考化合物 86)の合成
(3R)— N— (4— (3 クロ口フエ-ノレ)チアン一 4—ィノレ) 4— (2 フノレオ口フエ- ノレ)一 3— (2—メチノレ一 2 プロポキシカノレボニノレアミノ)ブタナミド(190mg, 0. 38 mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (259mg, >0 . 72mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ硫酸ナトリ ゥム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ 口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ
フィ一で精製し、表題ィ匕合 185mg (90%)を得た。
[0186] ステップ 6 (3R)— 3 アミノー N— (4— (3 クロ口フエ-ル)一 1, 1—ジォキソチア ンー 4 ィル) 4一(2 フルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 25)の合成 アルゴン雰囲気下、(3R)— N— (4— (3 クロ口フエ-ル)— 1, 1—ジォキソチアン 4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ ニルァミノ)ブタナミド(180mg, 0. 33mmol)を 10%塩化水素 Zメタノール溶液(4m L)、クロ口ホルム(2mL)に溶解し、室温で 15時間攪拌した後濃縮し、表題化合物 1 52mg (96%)を得た。
[0187] 実施例 26
(3R)— 3 ァミノ一 N— (4— (4 トリフルォロメチルフエ-ル)一 1, 1—ジォキソチア ンー4ーィル)ー4ー(2, 4, 5 トリフルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 26) の合成
ステップ 1 4— (4 トリフルォロメチルフエ-ル)チアン— 4—オール(参考化合物 87 )の合成
アルゴン雰囲気下、 1—ブロモ—4 トリフルォロメチルベンゼン(2. 03g, 9. 04m mol)の THF (40mL)溶液を― 78°Cに冷却し、 n -ブチルリチウム Zへキサン溶液( 1. 59M, 5. 30mL, 8. 44mmol)を滴下した。 15分後、 4—ォキソチアン(0. 70g, 6. O3mmol)の THF (8mL)溶液を滴下した。滴下終了 30分後、室温までゆっくりと 昇温し、 30分後、塩ィ匕アンモ-ゥム水溶液を加え酢酸ェチルで抽出し、有機層を蒸 留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題化合物 1. 69g (定量的)を得た。
[0188] ステップ 2 N— (4— (4 トリフルォロメチルフエ-ル)チアン— 4—ィル)ァセタミド( 参考化合物 88)の合成
4 (4 トリフルォロメチルフエ-ル)チアンー 4 オール(1. 5g, 5. 7mmol)のァ セトニトリル(9mL)溶液を— 10°Cに冷却し、ゆっくりと濃硫酸(7. 5mL)を滴下した。 30分間撹拌後室温まで昇温し、さらに 10分間撹拌した。反応液を氷の入ったアンモ ユア水にあけ、酢酸ェチルにて抽出し、有機層を蒸留水、飽和食塩水にて洗浄後、 乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、
表題化合物 1. 2g (71%)を得た。
[0189] ステップ 3 4—(4 トリフルォロメチルフエ-ル)チアンー 4ーァミン(参考化合物 89) の合成
アルゴン雰囲気下、 N— (4—(4 トリフルォロメチルフエ-ル)チアンー 4 ィル)ァ セタミド(1. 20g, 3. 96mmol)を THF (3mL)に懸濁し、チタンテトライソプロポキシ ド(1. 17mL, 3. 96mmol)、ジフエ-ノレシラン(1. 10mL, 5. 94mmol)をカ卩え、 15 時間撹拌した。酢酸ェチルを加え希釈した後、飽和炭酸水素ナトリウム水溶液を加え 更に 30分間撹拌した。セライト濾過後、濾液を蒸留水にあけ酢酸ェチルにて抽出し 、有機層を蒸留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 913mg (88%)を得た。
[0190] ステップ 4 (3R)—N— (4— (4 トリフルォロメチルフエ-ル)チアン— 4—ィル)—4 一(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド (参考化合物 90)の合成
アルゴン雰囲気下、 4— (4—トリフルォロメチルフエ-ル)チアン— 4—ァミン(75mg , 0. 29mmol)を DMF (4mL)に溶解し、(3R)—4—(2, 4, 5 トリフルオロフヱ-ル )ー3—(2—メチルー2—プロポキシカルボ-ルァミノ)酪酸(801118, 0. 24mmol)、 HATU (119mg, 0. 31mmol)、 DIPEA (79 L, 0. 48mmol)をカ卩え、室温で攪 拌した。 15時間後、反応液を蒸留水にあけジェチルエーテルにて抽出し、有機層を 蒸留水、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 116mg (84%)を得た。
[0191] ステップ 5 (3R)—N— (4— (4 トリフルォロメチルフエ-ル)一 1, 1—ジォキソチア ンー 4 ィル) 4一(2, 4) 5 トリフルオロフェ-ル)ー3—(2—メチルー 2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 91)の合成
(3R)— N— (4— (4 トリフルォロメチルフエ-ル)チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナ ミド(l lOmg, 0. 19mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA ( > 65%) (82mg, >0. 31mmol)を添カ卩した。 5分後氷浴を外し、室温で 13時間攪拌 した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム
水溶液を加えクロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合 90mg (78%)を得た。
[0192] ステップ 6 (3R)— 3 ァミノ一 N— (4— (4 トリフルォロメチルフエ-ル)一 1, 1— ジォキソチアンー4ーィル)ー4ー(2, 4, 5 トリフルオロフヱ-ル)ブタナミド '塩酸塩 (化合物 26)の合成
アルゴン雰囲気下、(3R)— N— (4— (4 トリフルォロメチルフエ-ル)— 1, 1—ジ ォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(180mg, 0. 33mmol)を 10%塩化水素 Zメタノール溶液 (4mL)、クロ口ホルム(2mL)に溶解し、室温で 15時間攪拌した後 濃縮し、表題ィ匕合物 75mg (93%)を得た。
[0193] 実施例 27
(3R)— 3 ァミノ一 N— (4— (3 トリフルォロメチルフエ-ル)一 1, 1—ジォキソチア ンー4ーィル)ー4ー(2, 4, 5 トリフルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 27) の合成
ステップ 1 4一(3 トリフルォロメチルフエ-ル)チアンー 4 オール(参考化合物 92 )の合成
アルゴン雰囲気下、 1—ブロモ—3—トリフルォロメチルベンゼン(2. 03g, 9. 04m mol)の THF (40mL)溶液を― 78°Cに冷却し、 n -ブチルリチウム Zへキサン溶液( 1. 59M, 5. 30mL, 8. 44mmol)を滴下した。 15分後、 4—ォキソチアン(0. 70g, 6. O3mmol)の THF (8mL)溶液を滴下した。滴下終了 30分後、室温までゆっくりと 昇温し、 30分後、塩ィ匕アンモ-ゥム水溶液を加え酢酸ェチルで抽出し、有機層を蒸 留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題化合物 1. 55g (98%)を得た。
[0194] ステップ 2 N— (4— (3 トリフルォロメチルフエ-ル)チアン— 4—ィル)ァセタミド( 参考化合物 93)の合成
4 (3 トリフルォロメチルフエ-ル)チアンー 4 オール(1. 5g, 5. 7mmol)のァ セトニトリル(9mL)溶液を— 10°Cに冷却し、ゆっくりと濃硫酸(7. 5mL)を滴下した。 30分間撹拌後室温まで昇温し、さらに 10分間撹拌した。反応液を氷の入ったアンモ
ユア水にあけ、酢酸ェチルにて抽出し、有機層を蒸留水、飽和食塩水にて洗浄後、 乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題化合物 1. 3g (76%)を得た。
[0195] ステップ 3 4— (3 トリフルォロメチルフエ-ル)チアン— 4 ァミン(参考化合物 94) の合成
アルゴン雰囲気下、 N— (4— (3—トリフルォロメチルフヱ-ル)チアン— 4—ィル)ァ セタミド(1. 20g, 3. 96mmol)を THF (3mL)に懸濁し、チタンテトライソプロポキシ ド(1. 17mL, 3. 96mmol)、ジフエ-ノレシラン(1. 10mL, 5. 94mmol)をカ卩え、 15 時間撹拌した。酢酸ェチルを加え希釈した後、飽和炭酸水素ナトリウム水溶液を加え 更に 30分間撹拌した。セライト濾過後、濾液を蒸留水にあけ酢酸ェチルにて抽出し 、有機層を蒸留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 840mg (71%)を得た。
[0196] ステップ 4 (3R)—N— (4— (3 トリフルォロメチルフエ-ル)チアン— 4—ィル)—4 一(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド (参考化合物 95)の合成
アルゴン雰囲気下、 4— (3—トリフルォロメチルフエ-ル)チアン— 4—ァミン(75mg , 0. 29mmol)を DMF (4mL)に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフェ- ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(80mg, 0. 24mmol) 、 HATU (119mg, 0. 31mmol) , DIPEA(79 μ L, 0. 48mmol)を加え、室温で 攪拌した。 15時間後、反応液を蒸留水にあけジェチルエーテルにて抽出し、有機層 を蒸留水、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル力 ラムクロマトグラフィーで精製し、表題ィ匕合物 90mg (65%)を得た。
[0197] ステップ 5 (3R)—N— (4— (3 トリフルォロメチルフエ-ル)一 1, 1—ジォキソチア ンー 4 ィル) 4一(2, 4) 5 トリフルオロフェ-ル)ー3—(2—メチルー 2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 96)の合成
(3R)— N— (4— (3 トリフルォロメチルフエ-ル)チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナ ミド(80mg, 0. 14mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA ( >6
5%) (60mg, >0. 23mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌し た。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水 溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合 40mg (47%)を得た。
[0198] ステップ 6 (3R)— 3 ァミノ一 N— (4— (3 トリフルォロメチルフエ-ル)一 1, 1— ジォキソチアンー4ーィル)ー4ー(2, 4, 5 トリフルオロフヱ-ル)ブタナミド '塩酸塩 (化合物 27)の合成
アルゴン雰囲気下、 (3R)—N— (4— (3 トリフルォロメチルフエ-ル) 1, 1ージ ォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(40mg, 0. 06mmol)を 10%塩化水素 Zメタノール溶液 (4mL)、クロ口ホルム(2mL)に溶解し、室温で 15時間攪拌した後 濃縮し、表題ィ匕合物 35mg (定量的)を得た。
[0199] 実施例 28
3 アミノー 4— (2 クロ口一 6 フノレオ口フエ-ノレ) N— (4— (2 フノレオ口フエ- ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 28)の合成 ステップ 1 3 アミノー 4一(2 クロロー 6 フルオロフヱ-ル)酪酸ェチル(参考化 合物 97)の合成
アルゴン雰囲気下、活性化亜鉛(3. 28g, 50mmol)の THF (30mL)懸濁液の加 熱還流下、ブロモ酢酸ェチル (4. 4mL, 40mmol)を少量ずつ滴下した。反応が開 始し、反応液が緑色を呈し始めたところで加熱及び滴下を停止し、 2 クロ口 6 フ ルオロフヱ-ルァセトニトリル(1. 7g, lOmmol)を加えた。再度加熱還流させた後、 残りのブロモ酢酸ェチルをゆっくりと滴下した。滴下終了 10分後、反応液を放冷し、 THF (30mL)、炭酸カリウム水溶液を加え、更に 30分間攪拌した。二層になった反 応液の上澄み液をデカンテーシヨンした後、有機層を乾燥し濃縮し、得られた粗生成 物をシリカゲルカラムクロマトグラフィーで粗精製した。アルゴン雰囲気下、酢酸(15 ml)に室温にて水素化ホウ素ナトリウム(1. Olg, 26. 7mmol)をゆっくりと加え、 30 分間攪拌した反応液に、粗精製物の酢酸 (3mL)溶液を室温で加え 6時間攪拌した 。濃縮後、残渣をジクロロメタンに溶解し、蒸留水、飽和炭酸水素ナトリウム水溶液、
飽和食塩水にて洗浄した。有機層を乾燥後濃縮し、得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題化合物 1. 22g (47%)を得た。
[0200] ステップ 2 4— (2 クロ口一 6 フノレオ口フエ-ノレ)一 3— (2—メチノレ一 2 プロポキ シカルボニルァミノ)酪酸ェチル (参考化合物 98)の合成
アルゴン雰囲気下、 3 アミノー 4一(2 クロロー 6 フルオロフェ -ル)酪酸ェチル (1. 2g, 4. 7mmol)、ジ— tret—ブチル ジカルボナート(1. 4mL, 6. lmmol)を 蒸留水(18mL)、 1, 4 ジォキサン(lOmL)に溶解し、炭酸ナトリウム(1. 3g, 12m mol)を加え、 3時間室温にて攪拌した。反応液を蒸留水にあけ酢酸ェチルにて抽出 後、有機層を希塩酸、蒸留水、飽和食塩水にて洗浄し、乾燥後濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1. 5g (89%)を 得た。
[0201] ステップ 3 4— (2 クロ口一 6 フノレオ口フエ-ノレ)一 3— (2—メチノレ一 2 プロポキ シカルボニルァミノ)酪酸 (参考化合物 99)の合成
アルゴン雰囲気下、 4— (2 クロ口 6 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸ェチル(1. 5g, 4. 2mol)のメタノール(13mL) 、 THF (4mL)溶液に、 IN水酸ィ匕ナトリウム水溶液(5mL)を加え 24時間攪拌した。 1N塩酸を加え弱酸性とした後さらに 3時間攪拌した。析出した白色固体を濾取し、 乾燥させて表題ィ匕合物 1. 3g (89%)を得た。
[0202] ステップ 4 4— (2 クロ口一 6 フノレオ口フエ-ノレ)一 N— (4— (2 フノレオ口フエ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合物 100)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (90mg, 0. 36mmol)を DMF (7mL)に溶解し、 4— (2 クロ口 6 フル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(120mg, 0 . 36mmol)、 HATU (165mg, 0. 43mmol) , DIPEA(179 μ L, 1. 08mmol)を 加え、室温で攪拌した。 14時間後、反応液に蒸留水を加え、析出した固体を濾取し た後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 175 mg (93%)を得た。
[0203] ステップ 5 4— (2 クロ口一 6 フノレオ口フエ-ノレ)一 N— (4— (2 フノレオ口フエ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 101)の合成
4— (2 クロ口一 6 フルオロフェ -ル) N— (4— (2 フルオロフェ -ル)チアン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(170mg , 0. 32mmol)をジクロロメタン(7mL)に溶解し、氷冷下、 mCPBA( >65%) (200 mg, >0. 75mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ 硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加 え、クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合 143mg (80%)を得た。
[0204] ステップ 6 3 アミノー 4— (2 クロ口一 6 フノレオ口フエ-ノレ)一 N— (4— (2 フノレ オロフェ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 28)の 合成
アルゴン雰囲気下、 4— (2 クロ口一 6 フルオロフェ -ル) N— (4— (2 フル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(140mg, 0. 25mmol)を 10%塩化水素 Zメタノール 溶液(3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、表題 化合物 116mg (94%)を得た。
[0205] 実施例 29
3 アミノー 4一(2 クロロー 4ーフノレ才ロフエ二ノレ) N— (4—(2 フノレ才ロフエ二 ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 29)の合成 ステップ 1 3 アミノー 4一(2 クロロー 4 フルオロフヱ-ル)酪酸ェチル(参考化 合物 102)の合成
アルゴン雰囲気下、活性化亜鉛(1. 64g, 25mmol)の THF (15mL)懸濁液の加 熱還流下、ブロモ酢酸ェチル(2. 2mL, 20mmol)を少量ずつ滴下した。反応が始 まり、反応液が緑色を呈し始めたところで加熱及び滴下を停止し、 2 クロロー 4ーフ ルォロフエ-ルァセトニトリル(850mg, 5. Ommol)をカ卩えた。再度加熱還流させた 後、残りのブロモ酢酸ェチルをゆっくりと滴下した。滴下終了 30分後、反応液を放冷
し、 THF (15mL)、炭酸カリウム水溶液を加えそのまま更に 30分間攪拌した。二層 になった反応液の上澄み液をデカンテーシヨンした後、 THFにて水層を洗浄した。 有機層を乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗 精製した。アルゴン雰囲気下、酢酸(10ml)に室温にて水素化トリァセトキシホウ素ナ トリウム(3. 06g, 14. 4mmol)をゆっくりと加え、 10分間攪拌した反応液に、粗精製 物の酢酸(2mL)溶液を室温で加え 14時間攪拌した。濃縮後、内容物をジクロ口メタ ンに溶解し、蒸留水、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄した。有 機層を乾燥後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 368mg (28%)を得た。
[0206] ステップ 2 4— (2 クロ口— 4 フルオロフェ-ル)—3— (2—メチル—2 プロポキ シカルボニルァミノ)酪酸ェチル (参考化合物 103)の合成
アルゴン雰囲気下、 3 アミノー 4一(2 クロロー 4 フルオロフェ -ル)酪酸ェチル (360mg, 1. 3911111101)、ジー^61;—ブチノレ ジカノレボナート(416mL, 1. 81mmo 1)を蒸留水(6mL)、 1, 4 ジォキサン(3mL)に溶解し、炭酸ナトリウム(383mg, 3 . 61mmol)を加え、 13時間室温にて攪拌した。反応液を蒸留水にあけ酢酸ェチル にて抽出後、有機層を希塩酸、蒸留水、飽和食塩水にて洗浄した。有機層を乾燥後 濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 461mg (92%)を得た。
[0207] ステップ 3 4— (2 クロ口一 4 フルオロフェ-ル)一3— (2—メチル 2 プロポキ シカルボニルァミノ)酪酸 (参考化合物 104)の合成
アルゴン雰囲気下、 4— (2 クロ口— 4 フルオロフェ -ル) - 3- (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸ェチル(460mg, 1. 28mol)のメタノール(3mL ) , THF (1. 5mL)溶液に、 IN水酸ィ匕ナトリウム水溶液(1. 5mL)をカ卩ぇ 24時間攪 拌した。 1N塩酸を加え弱酸性とした後さらに 3時間攪拌した。析出した白色固体を濾 取し、乾燥させ、クロ口ホルム、メタノール、へキサンより結晶化し、表題ィ匕合物 399m g (93%)を得た。
[0208] ステップ 4 4— (2 クロ口一 4 フノレオ口フエ-ノレ)一 N— (4— (2 フノレオ口フエ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド
(参考化合物 105)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (90mg, 0. 36mmol)を DMF (7mL)に溶解し、 4— (2 クロ口— 4 フル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(120mg, 0 . 36mmol)、 HATU (165mg, 0. 43mmol) , DIPEA(179 μ L, 1. 08mmol)を 加え、室温で攪拌した。 14時間後、反応液に蒸留水を加え、析出した固体を濾取し た後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 124 mg (66%)を得た。
[0209] ステップ 5 4— (2 クロ口一 4 フノレオ口フエ-ノレ)一 N— (4— (2 フノレオ口フエ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 106)の合成
4— (2 クロ口一 4 フノレオ口フエ-ノレ) N— (4— (2 フノレオ口フエ-ノレ)チアン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(120mg , 0. 23mmol)をジクロロメタン(7mL)に溶解し、氷冷下、 mCPBA( >65%) (95m g, 0. > 36mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ硫 酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加え クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題化合 115mg (90%)を得た。
[0210] ステップ 6 3 アミノー 4— (2 クロ口一 4 フノレオ口フエ-ノレ)一 N— (4— (2 フノレ オロフェ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 29)の 合成
アルゴン雰囲気下、 4— (2 クロ口一 4 フルオロフェ -ル) N— (4— (2 フル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(115mg, 0. 25mmol)を 10%塩化水素 Zメタノール 溶液(3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、表題 化合物 97mg (95%)を得た。
[0211] 実施例 30
3 アミノー 4— (2 クロ口一 3, 6 ジフノレオ口フエ-ノレ) N— (4— (2 フノレオロフ
ェ-ル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 30)の合成 ステップ 1 3 アミノー 4 (2 クロロー 3, 6 ジフルオロフェ -ル)酪酸ェチル(参 考化合物 107)の合成
アルゴン雰囲気下、活性化亜鉛(1. 95g, 34. 7mmol)の THF (35mL)懸濁液の 加熱還流下、ブロモ酢酸ェチル(3. 06mL, 27. 7mmol)を少量ずつ滴下した。反 応が始まり、反応液が緑色を呈し始めたところで加熱及び滴下を停止し、 2—クロ口 - 3, 6 ジフルオロフェ-ルァセトニトリル(1. 30g, 6. 93mmol)を加えた。再度カロ 熱還流させた後、残りのブロモ酢酸ェチルをゆっくりと滴下した。滴下終了 30分後、 反応液を放冷し、 THF (30mL)、炭酸カリウム水溶液を加え更に 20分間攪拌した。 二層になった反応液の上澄み液をデカンテーシヨンした後、 THFにて水層を洗浄し た。有機層を乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー で粗精製した。アルゴン雰囲気下、酢酸(15ml)に室温にて水素化ホウ素ナトリウム( 918mg, 24. 3mmol)をゆっくりと加え、 30分間攪拌した反応液に、粗精製物の酢 酸(5mL)溶液を室温で加え 7時間攪拌した。濃縮後、内容物をジクロロメタンに溶解 し、蒸留水、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄した。有機層を乾 燥後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 1. 30g (47%)を得た。
[0212] ステップ 2 4— (2 クロ口一 3, 6 ジフノレオ口フエ-ノレ)一 3— (2—メチノレ一 2 プロ ポキシカルボ-ルァミノ)酪酸ェチル (参考化合物 108)の合成
アルゴン雰囲気下、 3 アミノー 4 (2 クロロー 3, 6 ジフルオロフェ -ル)酪酸 ェチル(1. 30g, 4. 68mmol)、ジ— tret—ブチル ジカルボナート(1. 33g, 6. 08 mmol)を 1, 4 ジォキサン(8mL)に溶解し、 1N炭酸ナトリウム水溶液(12mL, 12 mmol)を加え、 13時間室温にて攪拌した。反応液を蒸留水にあけ酢酸ェチルにて 抽出後、有機層を希塩酸、蒸留水、飽和食塩水にて洗浄した。有機層を乾燥後濃縮 し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1. 45g (82%)を得た。
[0213] ステップ 3 4— (2 クロ口一 3, 6 ジフノレオ口フエ-ノレ)一 3— (2—メチノレ一 2 プロ ポキシカルボニルァミノ)酪酸 (参考化合物 109)の合成
アルゴン雰囲気下、 4一(2 クロロー 3, 6 ジフルオロフェ -ル)一 3—(2 メチル 2 プロポキシカルボ-ルァミノ)酪酸ェチル(1. 40g, 3. 71mol)のメタノール(6 mL)、 THF (4mL)溶液に、 IN水酸ィ匕ナトリウム水溶液(6mL)をカロえ 14時間攪拌 した。 1N塩酸を加え弱酸性とした後、更に 3時間攪拌した。析出した白色固体を濾 取し、乾燥させ表題ィ匕合物 1. 17g (90%)を得た。
[0214] ステップ 4 4— (2 クロ口一 3, 6 ジフノレオ口フエ-ノレ)一 N— (4— (2 フノレオロフ ェ -ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタ ナミド (参考化合物 110)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (90mg, 0. 36mmol)を DMF (8mL)に溶解し、 4— (2 クロ口 3, 6— ジフルオロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(126 mg, 0. 36mmol)、 HATU (165mg, 0. 43mmol) , DIPEA(179 μ L, 1. 08mm ol)を加え、室温で攪拌した。 14時間後、反応液に蒸留水を加え、析出した固体を濾 取した後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1 75mg (90%)を得た。
[0215] ステップ 5 4— (2 クロ口一 3, 6 ジフノレオ口フエ-ノレ)一 N— (4— (2 フノレオロフ ェニル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 111)の合成
4— (2 クロ口 3, 6 ジフルオロフェ -ル) N— (4— (2 フルオロフェ -ル)チ アンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(170 mg, 0. 31mmol)をジクロロメタン(10mL)に溶解し、氷冷下、 mCPBA ( >65%) ( 134mg, >0. 50mmol)を添カ卩した。 5分後氷浴を外し、室温で 13時間攪拌した。 チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶 液を加えクロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合 139mg (78%)を得た。
[0216] ステップ 6 3 アミノー 4— (2 クロ口一 3, 6 ジフルオロフェ-ル)一 N— (4— (2 フルオロフェ-ル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (ィ匕合物 30)の合成
アルゴン雰囲気下、 4一(2 クロロー 3, 6 ジフルオロフヱ-ル) N— (4—(2— フルオロフェ-ル)—1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)ブタナミド(135mg, 0. 25mmol)を 10%塩化水素 Zメタノ ール溶液(3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、 表題化合物 116mg (97%)を得た。
実施例 31
3 アミノー 4— (3 トリフルォロメチルフエ-ル) N— (4— (2 フルオロフェ -ル) — 1, 1 ジォキソチアン— 4 -ィル)ブタナミド ·塩酸塩 (化合物 31 )の合成 ステップ 1 4— (3 トリフルォロメチルフエ-ル)—3— (2—メチル—2 プロポキシ カルボニルァミノ)酪酸ェチル (参考化合物 112)の合成
アルゴン雰囲気下、活性化亜鉛(2. 28g, 40. 5mmol)の THF (50mL)懸濁液の 加熱還流下、ブロモ酢酸ェチル(3. 57mL, 32. 4mmol)を少量ずつ滴下した。反 応が始まり、反応液が緑色を呈し始めたところで加熱及び滴下を停止し、 3—トリフル ォロメチルフエ-ルァセトニトリル(1. 50g, 8. lOmmol)を加えた。再度加熱還流さ せた後、残りのブロモ酢酸ェチルをゆっくりと滴下した。滴下終了 30分後、反応液を 放冷し、 THF (30mL)、炭酸カリウム水溶液を加えそのまま更に 20分間攪拌した。 二層になった反応液の上澄み液をデカンテーシヨンした後、 THFにて水層を洗浄し た。有機層を乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー で粗精製した。アルゴン雰囲気下、酢酸 (20ml)に室温にて水素化ホウ素ナトリウム( 918mg, 24. 3mmol)をゆっくりと加え、 30分間攪拌した反応液に、粗精製物の酢 酸(5mL)溶液を室温で加え 15時間攪拌した。濃縮後、内容物をジクロロメタンに溶 解し、蒸留水、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄した。有機層を 乾燥後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した 。アルゴン雰囲気下、得られた粗精製物、ジ—tret ブチル ジカルボナート(1. 18 g, 5. 43mmol)を 1, 4 ジォキサン(15mL)、蒸留水(25mL)に溶解し、 1N炭酸 ナトリウム水溶液(l lmL, l lmmol)を加え、 13時間室温にて攪拌した。反応液を蒸 留水にあけ酢酸ェチルにて抽出し、有機層を希塩酸、蒸留水、飽和食塩水にて洗浄 した。有機層を乾燥後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィ
一で精製し、表題化合物 1. 40g (46%)を得た。
[0218] ステップ 2 4— (3 トリフルォロメチルフエ-ル)— 3— (2—メチル—2 プロポキシ カルボニルァミノ)酪酸 (参考化合物 113)の合成
アルゴン雰囲気下、 4— (3 トリフルォロメチルフエ-ル)—3— (2—メチル—2— プロポキシカルボ-ルァミノ)酪酸ェチル(590mg, 1. 18mol)のメタノール(6mL)、 THF (4mL)溶液に IN水酸ィ匕ナトリウム水溶液 (6mL)をカ卩ぇ 14時間攪拌した。 1N 塩酸を加え弱酸性とした後、更に 3時間攪拌した。析出した白色固体を濾取し、乾燥 させ表題ィ匕合物 507g (93%)を得た。
[0219] ステップ 3 4—(3 トリフルォロメチルフエ-ル)—N— (4— (2 フルオロフェ -ル) チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参 考化合物 114)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (90mg, 0. 36mmol)を DMF (8mL)に溶解し、 4— (3 トリフルォロメチ ルフエ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(126mg, 0. 36mmol)、 HATU (165mg, 0. 43mmol) , DIPEA(179 μ L, 1. 08mmol)をカロ え、室温で攪拌した。 14時間後、反応液に蒸留水を加え、析出した固体を濾取した 後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 163mg (84%)を得た。
[0220] ステップ 4 4 (3 トリフルォロメチルフエ-ル)—N— (4— (2 フルオロフェ -ル)
- 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)ブタナミド (参考化合物 115)の合成
4一(3 トリフルォロメチルフエ-ル) N— (4—(2 フルオロフェ -ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(200mg, 0. 37mmol)をジクロロメタン(10mL)に溶解し、氷冷下、 mCPBA ( >65%) (160mg , >0. 60mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ硫酸 ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた 。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合 81mg (38%)を得た。
[0221] ステップ 5 3 ァミノ一 4— (3 トリフルォロメチルフエ-ル)一 N— (4— (2 フルォ 口フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 31)の合 成
アルゴン雰囲気下、 4一(3 トリフルォロメチルフエ-ル)一 N—(4一(2 フルォロ フエ二ル)一 1, 1—ジォキソチアン一 4—ィル) 3— (2—メチル 2 プロポキシ力 ルポ-ルァミノ)ブタナミド(80mg, 0. 14mmol)を 10%塩化水素 Zメタノール溶液( 3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、表題化合 物 67mg (94%)を得た。
[0222] 実施例 32
3 アミノー 4— (3 クロ口一 6 フノレオ口フエ-ノレ) N— (4— (2 フノレオ口フエ- ル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 32)の合成 ステップ 1 3 アミノー 4一(3 クロロー 6 フルオロフヱ-ル)酪酸ェチル(参考化 合物 116)の合成
アルゴン雰囲気下、活性化亜鉛(2. 16g, 38. 3mmol)の THF (40mL)懸濁液の 加熱還流下、ブロモ酢酸ェチル(3. 38mL, 30. 7mmol)を少量ずつ滴下した。反 応が始まり、反応液が緑色を呈し始めたところで加熱及び滴下を停止し、 3—クロ口 —6 フルオロフェ-ルァセトニトリル(1. 30g, 7. 66mmol)を加えた。再度加熱還 流させた後、残りのブロモ酢酸ェチルをゆっくりと滴下した。滴下終了 30分後放冷し 、 THF (30mL)、炭酸カリウム水溶液を加えそのまま更に 20分間攪拌した。二層に なった反応液の上澄み液をデカンテーシヨンした後、 THFにて水層を洗浄した。有 機層を乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精 製した。アルゴン雰囲気下、酢酸(35ml)に室温にて水素化ホウ素ナトリウム(1. 16g , 30. 6mmol)をゆっくりと加え、 30分間攪拌した反応液に、粗精製物の酢酸(5mL )溶液を室温で加え 15時間攪拌した。濃縮後、内容物をジクロロメタンに溶解し、蒸 留水、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄した。有機層を乾燥後 濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 810mg (46%)を得た。
[0223] ステップ 2 4— (3 クロ口一 6 フノレオ口フエ-ノレ)一 3— (2—メチノレ一 2 プロポキ
シカルボニルァミノ)酪酸ェチル (参考化合物 117)の合成
アルゴン雰囲気下、 3 アミノー 4一(3—クロロー 6—フルオロフェ -ル)酪酸ェチル (800mg, 2. 88mmol)、ジ— tret—ブチノレ ジカノレボナート(818mg, 3. 75mmol )を 1, 4 ジォキサン(8mL)に溶解し、 1N炭酸ナトリウム水溶液(7. 2mL, 7. 2mm ol)を加え、 62時間室温にて攪拌した。反応液を蒸留水にあけ酢酸ェチルにて抽出 後、有機層を希塩酸、蒸留水、飽和食塩水にて洗浄した。有機層を乾燥後濃縮し、 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1. 03 g (95%)を得た。
[0224] ステップ 3 4— (3 クロ口一 6 フノレオ口フエ-ノレ)一 3— (2—メチノレ一 2 プロポキ シカルボニルァミノ)酪酸 (参考化合物 118)の合成
アルゴン雰囲気下、 4— (3 クロ口 6 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸ェチル(490mg, 1. 36mol)のメタノール(9mL )、 THF (4mL)溶液に IN水酸化リチウム水溶液 (4mL)をカ卩ぇ 14時間攪拌した。 1 N塩酸を加え弱酸性とした後、更に 3時間攪拌し、析出した白色固体を濾取し、乾燥 させ表題ィ匕合物 377mg (75%)を得た。
[0225] ステップ 4 4— (3 クロ口一 6 フノレオ口フエ-ノレ)一 N— (4— (2 フノレオ口フエ- ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合物 119)の合成
アルゴン雰囲気下、 4一(2 フルオロフヱ-ル)チアンー 4 アミン'塩酸塩(参考化 合物 69) (82mg, 0. 33mmol)を DMF (8mL)に溶解し、 4— (3 クロ口一 6 フル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(lOOmg, 0 . 30mmol)、 HATU (397mg, 0. 36mmol) , DIPEA(397 μ L, 2. 40mmol)を 加え、室温で攪拌した。 14時間後、反応液に蒸留水を加え、析出した固体を濾取し た後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 119 mg (76%)を得た。
[0226] ステップ 5 4— (3 クロ口一 6 フノレオ口フエ-ノレ)一 N— (4— (2 フノレオ口フエ- ル)—1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 120)の合成
4— (3 クロ口一 6 フルオロフェ -ル) -N- (4— (2 フルオロフェ -ル)チアン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(115mg , 0. 22mmol)をジクロロメタン(lOmL)に溶解し、氷冷下、 mCPBA( >65%) (95 mg, >0. 36mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ 硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加 えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合 98mg (80%)を得た。
[0227] ステップ 6 3 アミノー 4一(3 クロロー 6 フノレオ口フエ-ノレ) N—(4一(2 フノレ オロフェ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 32)の 合成
アルゴン雰囲気下、 4— (3 クロ口一 6 フルオロフェ -ル) N— (4— (2 フル オロフェ-ル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(98mg, 0. 14mmol)を 10%塩化水素 Zメタノール 溶液(3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、表題 化合物 81mg (95%)を得た。
[0228] 実施例 33
3 アミノー 4— (3 クロ口一 2, 6 ジフノレオ口フエ-ノレ) N— (4— (2 フノレオロフ ェ-ル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 33)の合成 ステップ 1 4— (3 クロ口 2, 6 ジフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)酪酸ェチル (参考化合物 121)の合成
アルゴン雰囲気下、活性化亜鉛(1. 95g, 34. 7mmol)の THF (40mL)懸濁液の 加熱還流下、ブロモ酢酸ェチル(3. 06mL, 27. 7mmol)を少量ずつ滴下した。反 応が始まり、反応液が緑色を呈し始めたところで加熱及び滴下を停止し、 3—クロ口 —2, 6 ジフルオロフェ-ルァセトニトリル(1. 30g, 6. 93mmol)を加えた。再度カロ 熱還流させた後、残りのブロモ酢酸ェチルをゆっくりと滴下した。滴下終了 30分後放 冷し、 THF (30mL)、炭酸カリウム水溶液を加えそのまま更に 20分間攪拌した。二 層になった反応液の上澄み液をデカンテーシヨンした後、 THFにて水層を洗浄した 。有機層を乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで
粗精製した。アルゴン雰囲気下、酢酸 (35ml)に室温にて水素化ホウ素ナトリウム(1 . 16g, 30. 6mmol)をゆっくりと加え、 30分間攪拌した反応液に、粗精製物の酢酸 ( 5mL)溶液を室温で加え 15時間攪拌した。濃縮後、内容物をジクロロメタンに溶解し 、蒸留水、飽和炭酸水素ナトリウム水溶液、飽和食塩水にて洗浄した。有機層を乾燥 後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。ァ ルゴン雰囲気下、得られた粗精製物、ジ—tret ブチル ジカルボナート(400mg, 1. 83mmol)を 1, 4 ジォキサン(8mL)に溶解し、 1N炭酸ナトリウム水溶液 (4. 2 mL, 4. 2mmol)を加え、 14時間室温にて攪拌した。反応液を蒸留水にあけ酢酸ェ チルにて抽出後、有機層を希塩酸、蒸留水、飽和食塩水にて洗浄した。有機層を乾 燥後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 493mg (20%)を得た。
[0229] ステップ 2 4— (3 クロ口一 2, 6 ジフノレオ口フエ-ノレ)一 N— (4— (2 フノレオロフ ェ -ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタ ナミド (参考化合物 122)の合成
アルゴン雰囲気下、 4一(3 クロロー 2, 6 ジフルオロフェ -ル) 3—(2 メチル 2 プロポキシカルボ-ルァミノ)酪酸ェチル(490mg, 1. 30mol)のメタノール(9 mL)、 THF (4mL)溶液に IN水酸化リチウム水溶液 (4mL)をカ卩ぇ 14時間攪拌した 。 1N塩酸を加え弱酸性とした後、更に 3時間攪拌した。析出した白色固体を濾取し、 乾燥させ、得られた粗生成物 382mgのうち 105mgを、アルゴン雰囲気下、 DMF (8 mL)に溶解し、 4— (2—フルオロフェ -ル)チアン— 4—ァミン '塩酸塩 (参考化合物 69) (82mg, 0. 33mmol)、 HATU (397mg, 0. 36mmol) , DIPEA(397 μ L, 2 . 40mmol)を加え、室温で攪拌した。 14時間後、反応液に蒸留水を加えた。析出し た固体を濾取した後、得られた固体をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 164mg (83%)を得た。
[0230] ステップ 3 4— (3 クロ口一 2, 6 ジフノレオ口フエ-ノレ)一 N— (4— (2 フノレオロフ ェニル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 123)の合成
4— (3 クロ口 2, 6 ジフルオロフェ -ル) N— (4— (2 フルオロフェ -ル)チ
アンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(160 mg, 0. 29mmol)をジクロロメタン(10mL)に溶解し、氷冷下、 mCPBA ( >65%) ( 127mg, >0. 48mmol)を添カ卩した。 5分後氷浴を外し、室温で 13時間攪拌した。 チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶 液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合 107mg (85%)を得た。
[0231] ステップ 4 3 アミノー 4— (3 クロ口一 2, 6 ジフルオロフェ-ル)一 N— (4— (2 フルオロフェ-ル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (ィ匕合物 33)の合成
アルゴン雰囲気下、 4一(3 クロロー 2, 6 ジフルオロフェ -ル) N— (4—(2— フルオロフェ-ル)—1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)ブタナミド(107mg, 0. 19mmol)を 10%塩化水素 Zメタノ ール溶液(3mL)、クロ口ホルム (4mL)に溶解し、室温で 15時間攪拌した後濃縮し、 表題ィ匕合物 96mg (99%)を得た。
[0232] 実施例 34
(3R)—3 アミノー 4— (2 フノレオ口フエ-ノレ) N— (4 フエ-ノレピぺリジン一 4— ィル)ブタナミド '二塩酸塩 (化合物 34)の合成
ステップ 1 (3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4 フエ-ルビペリジン 4 ィル)ブタナミド(参考化合物 12 4)の合成
(3R)— N— (1—ベンジル— 4—フエ-ルビペリジン— 4—ィル)—4— (2 フルォ 口フエ-ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化 合物 4) (143mg, 0. 31mmol)をエタノール(4mL)に溶解し、 5%パラジウム Z炭 素(50mg)を加え、水素雰囲気下で 14時間攪拌した後、濾過した。濾液を濃縮し、 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 121m g (85%)を得た。
[0233] ステップ 2 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4 フエ-ルビぺ リジンー4 ィル)ブタナミド '二塩酸塩 (化合物 34)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—フエ-ルビペリジン— 4—ィル)ブタナミド(50 mg, 0. l lmmol)を 10%塩化水素 Zメタノール溶液(2mL)に溶解し、室温で 3時 間攪拌した後濃縮し、表題ィ匕合物 42mg (89%)を得た。
[0234] 実施例 35
(3R)—3 ァミノ— 4— (2 フルオロフェ -ル) N— (1—メチルスルホ -ル— 4— フエ-ルビペリジン 4 ィル)ブタナミド '塩酸塩 (ィ匕合物 35)の合成
ステップ 1 (3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (1—メチルスルホ-ルー 4—フエ-ルビペリジン— 4—ィル)ブ タナミド (参考化合物 125)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—フエ-ルビペリジン— 4—ィル)ブタナミド(参 考化合物 124) (70mg, 0. 15mmol)をジクロロメタン(4mL)に溶解し、 TEA(43 L, 0. 31mmol)、塩化メチルスルホ -ル(18mg, 0. 15mmol)を加え、室温で 3時 間攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機 層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 55mg (67%)を得た。
[0235] ステップ 2 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N—(l—メチルスルホ 二ルー 4 フエ-ルビペリジン 4 ィル)ブタナミド '塩酸塩(ィ匕合物 35)の合成 アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (1—メチルスルホ-ルー 4—フエ-ルビペリジン— 4 ィル)ブタナミド(55mg, 0. lOmmol)を 10%塩化水素 Zメタノール溶液(3mL) に溶解し、室温で 9時間攪拌した後濃縮し、表題ィ匕合物 51mg (定量的)を得た。
[0236] 実施例 36
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (1— (ピリミジン一 2—ィル) 4 フエ二ルビペリジン 4 ィル)ブタナミド ·二塩酸塩 (化合物 36)の合成 ステップ 1 1—ベンジル— 4— (2—メチル—2 プロポキシカルボ-ルァミノ)—4— フエ二ルビペリジン(参考化合物 126)の合成
アルゴン雰囲気下、 1 ベンジル 4 フエ-ルビペリジン 4ーァミン(参考化合 物 3)の粗生成物(1. Og,約 3. 7mmol)のジクロロメタン溶液(20mL)に、 DIPEA( 1. 6mL, 9. 4mmol)、ジー tert—ブチノレ ジカノレボナート(0. 9g, 4. lmmol)をカロ え、室温で 15時間撹拌した。濃縮して得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題化合物 1. 4g (定量的)を得た。
[0237] ステップ 2 4— (2—メチル—2 プロポキシカルボ-ルァミノ)— 4—フエ-ルビペリ ジン (参考化合物 127)の合成
1—ベンジル— 4— (2—メチル—2 プロポキシカルボ-ルァミノ)—4—フエ-ルビ ペリジン(920mg、 2. 5mmol)をエタノール(10mL)に溶解し、 5%パラジウム Z炭 素(500mg)を加え、水素雰囲気下で 24時間激しく攪拌した後、濾過した。濾液を濃 縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 385mg (55%)を得た。
[0238] ステップ 3 4— (2—メチル—2 プロポキシカルボ-ルァミノ)— 4—フエ-ルー 1—( ピリミジン 2 ィル)ピぺリジン(参考化合物 128)の合成
アルゴン雰囲気下、 4一(2—メチルー 2 プロポキシカルボ-ルァミノ)ー4 フエ- ルピペリジン(lOOmg, 0. 36mmol)のトルエン溶液(5mL)に、 TEA(100mL, 0. 7 2mmol)、 2 クロ口ピリミジン(41mg, 0. 36mmol)をカ卩え、 15時間加熱還流した。 放冷後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表 題ィ匕合物 67mg (52%)を得た。
[0239] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (1— (ピリミジン一 2—ィル) 4—フエ-ルビペリジン一 4—ィ ル)ブタナミド (参考化合物 129)の合成
アルゴン雰囲気下、 4一(2—メチルー 2 プロポキシカルボ-ルァミノ)ー4 フエ- ルー 1 (ピリミジン— 2 ィル)ピぺリジン(67mg, 0. 19mmol)を 10%塩化水素 Z メタノール溶液(3mL)に溶解し、室温で 13時間攪拌した。炭酸カリウム水溶液をカロ え酢酸ェチルで抽出し、有機層を乾燥後濃縮した。得られた粗生成物をジクロ口メタ ン(3mL)に溶解し、(3R)— 4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)酪酸(30mg, 0. 10mmol)、 HOBt (21mg, 0. 15mmol)
、DIPEA(19mL, 0. l lmmol)、 EDCI'塩酸塩(23mg, 0. 12mmol)をカ卩え、室 温で攪拌した。 21時間後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合物 35mg (66%)を得た。
[0240] ステップ 5 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N—(l— (ピリミジン一 2 ィル) 4 フエ-ルビペリジン 4 ィル)ブタナミド ·二塩酸塩 (化合物 36)の合 成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (1— (ピリミジン— 2—ィル)—4—フエ-ルビベリジ ンー 4 ィル)ブタナミド(35mg, 0. 066mmol)を 10%塩化水素 Zメタノール溶液( 3mL)に溶解し、室温で 5時間攪拌した後濃縮し、表題ィ匕合物 22mg (66%)を得た
[0241] 実施例 37
(3R)— N— (1—ァセチル— 4 フエ-ルビペリジン— 4—ィル)—3 ァミノ— 4— (2 フルオロフェ -ル)ブタナミド ·塩酸塩 (化合物 37)の合成
ステップ 1 1一ァセチルー 4一(2—メチルー 2 プロポキシカルボニルァミノ) -4- フエ二ルビペリジン(参考化合物 13)の合成
アルゴン雰囲気下、 4一(2—メチルー 2 プロポキシカルボ-ルァミノ)ー4 フエ- ルビペリジン(参考化合物 127) (80mg, 0. 29mmol)のピリジン溶液(3mL)に、無 水酢酸(27mL, 0. 29mmol)をカ卩え、室温で撹拌した。 23時間後濃縮し、得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 58mg (63%) を得た。
[0242] ステップ 2 1 ァセチルー 4 フエ-ルビペリジンー4ーァミン(参考化合物 131)の 合成
アルゴン雰囲気下、 1—ァセチルー 4— (2—メチル—2 プロポキシカルボ-ルアミ ノ) 4 フエ-ルビペリジン(58mg, 0. 18mmol)を 10%塩化水素 Zメタノール溶 液 (3mL)に溶解し、室温で 14時間攪拌した後濃縮した。飽和炭酸水素ナトリウム水 溶液を加え、酢酸ェチルで抽出し、有機層を乾燥後濃縮し、表題ィ匕合物 34mg (86 %)を得た。
[0243] ステップ 3 (3R)— N— (1—ァセチル— 4—フエ-ルビペリジン— 4—ィル)—4— (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド( 参考化合物 132)の合成
アルゴン雰囲気下、 1—ァセチルー 4 フエ-ルビペリジン— 4 ァミン(34mg, 0. 16mmol)をジクロロメタン(4mL)に溶解し、(3R)—4— (2 フルオロフェ -ル) 3 一(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. 10mmol)、HOB t (21mg, 0. 15mmol)、 DIPEA (53mL, 0. 30mmol)、 EDCI-塩酸塩(23mg, 0 . 12mmol)を加え、室温で攪拌した。 12時間後濃縮し、得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 47mg (93%)を得た。
[0244] ステップ 4 (3R)— N— (1—ァセチル— 4—フエ-ルビペリジン— 4—ィル) 3 ァ ミノ— 4— (2 フルオロフェ -ル)ブタナミド ·塩酸塩 (化合物 37)の合成
アルゴン雰囲気下、 (3R)— N— (1—ァセチル— 4 フエ-ルビペリジン— 4—ィル )— 4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) ブタナミド(47mg, 0. 093mmol)を 10%塩化水素 Zメタノール溶液(4mL)に溶解 し、室温で 19時間攪拌した後濃縮し、表題ィ匕合物 21mg (52%)を得た。
[0245] 実施例 38
(3R)—3 アミノー 4— (2 フノレオ口フエ-ノレ) N— (1—フエナシノレ一 4 フエ- ルビペリジン 4 ィル)ブタナミド ·塩酸塩 (化合物 38)の合成
ステップ 1 4一(2—メチルー 2 プロポキシカルボニルァミノ) 1 フエナシルー4 フエ二ルビペリジン(参考化合物 133)の合成
アルゴン雰囲気下、 4一(2—メチルー 2 プロポキシカルボ-ルァミノ)ー4一フエ- ルビペリジン(参考化合物 127) (80mg, 0. 29mmol)の THF溶液(3mL)に、 DIP EA (200mL、 1. 2mmol)、臭化フエナシル(58mg, 0. 29mmol)を加え、室温で撹 拌した。 23時間後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題ィ匕合物 51mg (44%)を得た。
[0246] ステップ 2 1 フエナシルー4 フエ-ルビペリジンー4ーァミン(参考化合物 134) の合成
アルゴン雰囲気下、 4一(2—メチルー 2 プロポキシカルボ-ルァミノ) 1 フエナ
シルー 4 フエ-ルビペリジン(51mg, 0. 13mmol)を 10%塩化水素 Zメタノール 溶液 (3mL)に溶解し、室温で 14時間攪拌した後濃縮した。飽和炭酸水素ナトリウム 水溶液を加え、酢酸ェチルで抽出し、有機層を乾燥後濃縮し、表題ィ匕合物 32mg (8 5%)を得た。
[0247] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (1—フエナシル— 4—フエ-ルビペリジン— 4—ィル)ブタナミ ド (参考化合物 135)の合成
アルゴン雰囲気下、 1—フエナシル— 4 フエ-ルビペリジン— 4 ァミン(32mg, 0 . l lmmol)をジクロロメタン(4mL)に溶解し、(3R)—4 (2 フルオロフヱ-ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. 10mmol)、HO Bt (21mg, 0. 15mmol)、 DIPEA(53mL, 0. 30mmol)、 EDCI-塩酸塩(23mg, 0. 12mmol)を加え、室温で攪拌した。 12時間後濃縮し、得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 15mg (26%)を得た。
[0248] ステップ 4 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N—(l フエナシル一 4 フエ二ルビペリジン 4 ィル)ブタナミド ·塩酸塩 (化合物 38)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (1—フエナシル— 4—フエ-ルビペリジン— 4—ィ ル)ブタナミド(15mg, 0. 027mmol)を 10%塩化水素 Zメタノール溶液(4mL)に溶 解し、室温で 19時間攪拌した後濃縮し、表題化合物 6. 7mg (46%)を得た。
[0249] 実施例 39
(3R)— 3 ァミノ一 N— (1—ベンジル一 4— (2 フルオロフェ -ル)ピぺリジン一 4 ィル) 4一(2 フルオロフヱニル)ブタナミド '二塩酸塩(ィ匕合物 39)の合成 ステップ 1 1一べンジルー 4一(2 フルオロフェ -ル)ピぺリジンー4 オール(参考 化合物 136)の合成
アルゴン雰囲気下、 1—ブロモ 2 フルォロベンゼン(925mg, 5. 3mmol)のジ ェチルエーテル溶液(20mL)を 78°Cに冷却し、 n ブチルリチウム Zへキサン溶 液(2. 55M, 2. 5mL, 6. 3mmol)を滴下した。 40分間撹拌した後、 1一べンジル 4ーピペリドン(1. 0g, 5. 3mmol)のジェチルエーテル溶液(5mL)を滴下した。
滴下終了後室温までゆっくり昇温し、 6時間後蒸留水を加え、酢酸ェチルで抽出し、 有機層を乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題化合物 1. lg (74%)を得た。
[0250] ステップ 2 N—(l ベンジル一 4— (2 フルオロフェ -ル)ピぺリジン一 4—ィル)ァ セタミド (参考化合物 137)の合成
1—ベンジル— 4— (2—フルオロフェ -ル)ピぺリジン— 4—オール(1. lg, 3. 9m mol)のァセトニトリル溶液 (8mL)を氷冷し、激しく攪拌しながら濃硫酸 (4. 7mL)を ゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間後、氷水に注いだ 。水層を 1N水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し 、有機層を乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 884mg (69%)を得た。
[0251] ステップ 3 (3R)— N— (1—ベンジル一 4— (2 フルオロフェ -ル)ピぺリジン一 4— ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド (参考化合物 138)の合成
アルゴン雰囲気下、 N— (1—ベンジル— 4— (2 フルオロフェ -ル)ピぺリジン— 4 ィル)ァセタミド(880mg, 2. 7mmol)を THF (0. lmL)に懸濁し、チタンテトライ ソプロボキシド(800 /z L, 2. 7mmol)、ジフエ-ルシラン(750 /z L, 4. Ommol)をカロ え、室温で攪拌した。 18時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水 溶液を加えて激しく攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチル にて抽出した。有機層を合わせて乾燥後濃縮し、得られた粗生成物 773mgのうち 1 OOmgを、アルゴン雰囲気下、ジクロロメタン(4mL)に溶解し、(3R)—4— (2—フル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. 10mmol)、 HOBt (21mg, 0. 15mmol)、 DIPEA(53mL, 0. 30mmol)、EDC 塩酸塩(23mg, 0. 12mmol)を加え、室温で攪拌した。 12時間後濃縮し、得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 50mg (87%) を得た。
[0252] ステップ 4 (3R)— 3 ァミノ一 N—(l ベンジル一 4— (2 フルオロフェ -ル)ピぺ リジン 4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '二塩酸塩(ィ匕合物 39)の
合成
アルゴン雰囲気下、 (3R) -N- (1—ベンジル— 4— (2 フルオロフヱ-ル)ピペリ ジンー4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシ力 ルポ-ルァミノ)ブタナミド(50mg, 0. 088mmol)を 10%塩化水素 Zメタノール溶液 (4mL)に溶解し、室温で 19時間攪拌した後濃縮し、表題ィ匕合物 3 lmg (66%)を得 た。
[0253] 実施例 40
(3R)— 3 ァミノ一 N— (1—ベンジル一 4— (3— (ピロール一 1—ィル)フエ-ル)ピ ペリジン 4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '二塩酸塩(ィ匕合物 40) の合成
ステップ 1 1一べンジルー 4一(3 ョードフエ-ル)ピぺリジンー4 オール(参考化 合物 139)の合成
アルゴン雰囲気下、 1, 3ージョードベンゼン(5. 2g, 16mmol)の THF溶液(50m L)を—78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 55M, 6. 2mL, 16 mmol)を滴下した。 10分間撹拌した後、 1—ベンジル一 4 ピぺリドン(3. 0g, 16m mol)の THF溶液(30mL)を滴下した。滴下終了後、 1時間撹拌した後、室温までゆ つくり昇温し、 16時間後蒸留水を加え、酢酸ェチルで抽出した。有機層を乾燥後濃 縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 5. 3g (85%)を得た c
[0254] ステップ 2 N—(l ベンジル一 4— (3 ョードフエ-ル)ピぺリジン一 4—ィル)ァセ タミド (参考化合物 140)の合成
アルゴン雰囲気下、 1—ベンジル— 4— (3—ョードフエ-ル)ピぺリジン— 4—ォー ル(5. 3g, 13mmol)のァセトニトリル溶液(27mL)を氷冷し、激しく攪拌しながら濃 硫酸(16mL)をゆっくりと滴下した。滴下終了後氷浴を外し室温で攪拌し、 16時間 後氷水に注いだ。水層を 1N水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸 ェチルにて抽出し、有機層を乾燥後濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題化合物 5. 0g (85%)を得た。
[0255] ステップ 3 N—(l ベンジル一 4— (3— (ピロール一 1—ィル)フエ-ル)ピぺリジン
—4—ィル)ァセタミド (参考化合物 141)の合成
アルゴン雰囲気下、 N— (1—ベンジル— 4— (3 ョードフエ-ル)ピぺリジン— 4— ィル)ァセタミド(200mg, 0. 46mmol)を 1, 4 ジォキサン(3mL)に溶解し、ピロ一 ル(31mg, 0. 46mmol)、ヨウ化銅(I) (4mg, 0. 023mmol)、 trans— N, N,—ジ メチル 1, 2 シクロへキサンジァミン(13mg, 0. 092mmol)、リン酸カリウム(258 mg, 0. 97mmol)を加え、 110°Cで 15時間撹拌した。放冷後固体を濾過し、濾液を 濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 125mg (73%)を得た。
[0256] ステップ 4 1—ベンジル— 4— (3— (ピロール— 1—ィル)フエ-ル)ピぺリジン— 4— ァミン (参考化合物 142)の合成
アルゴン雰囲気下、 N— (1—べンジルー 4一(3 (ピロ一ルー 1 ィル)フエ-ル) ピぺリジン— 4—ィル)ァセタミド(125mg, 0. 34mmol)を THF (0. 05mL)に懸濁 し、チタンテトライソプロポキシド(99 L, 0. 34mmol)、ジフエ-ルシラン(94 /z L, 0. 50mmol)を加え、室温で攪拌した。 15時間後、酢酸ェチルにて希釈し、飽和炭 酸水素ナトリウム水溶液を加えて激しく攪拌した。生じた白色固体を濾別し、濾液を 分液後、酢酸ェチルにて抽出した。有機層を合わせて乾燥後濃縮して得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 116mg (定量的)を 得た。
[0257] ステップ 5 (3R)— N— (1—ベンジル一 4— (3— (ピロール一 1—ィル)フエ-ル)ピ ペリジン一 4一ィル)ー4一(2 フルオロフェ -ル)一 3—(2—メチルー 2 プロポキ シカルボニルァミノ)ブタナミド (参考化合物 143)の合成
アルゴン雰囲気下、 1一べンジルー 4一(3 (ピロ一ルー 1 ィル)フエ-ル)ピペリ ジン一 4 ァミン(116mg, 0. 35mmol)をジクロロメタン(3mL)に溶解し、(3R)— 4 一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸 (30mg, 0. 10mmol)、 HOBt (21mg, 0. 15mmol)、 NEM (13mg, 0. l lmmol ;)、 EDCI'塩酸塩(23mg, 0. 12mmol)をカ卩え、室温で攪拌した。 16時間後濃縮し 、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 14m g (22%)を得た。
[0258] ステップ 6 (3R)— 3 ァミノ一 N— (1—ベンジル一 4— (3— (ピロール一 1—ィル) フエ-ル)ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '二塩酸塩( 化合物 40)の合成
アルゴン雰囲気下、 (3R)— N— (1—ベンジル— 4— (3— (ピロール— 1—ィル)フ ェ -ル)ピぺリジン— 4—ィル)—4— (2 フルオロフェ -ル)—3— (2—メチル—2— プロポキシカルボ-ルァミノ)ブタナミド(14mg, 0. 022mmol)を 10%塩化水素 Zメ タノール溶液 (4mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題化合物 6. lm g (47%)を得た。
[0259] 実施例 41
(3R)— 3 アミノー N— (1—ベンジル— 4— (3— (イミダゾールー 1—ィル)フエ-ル )ピペリジンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '三塩酸塩 (ィ匕合物 4 1)の合成
ステップ 1 N— (1 ベンジル一 4— (3— (イミダゾール一 1—ィル)フエ-ル)ピペリ ジン 4 ィル)ァセタミド(参考化合物 144)の合成
アルゴン雰囲気下、 N— (1—ベンジル— 4— (3 ョードフエ-ル)ピぺリジン— 4— ィル)ァセタミド(参考化合物 140) (400mg, 0. 92mmol)を 1, 4 ジォキサン(4m L)に溶解し、イミダゾーノレ(62mg, 0. 92mmol)、ヨウィ匕銅(I) (9mg, 0. 047mmol )、 1, 10 フエナン卜口リン(166mg, 0. 092mmol)、炭酸セシウム(600mg, 1. 8 mmol)を加え、 110°Cで 15時間撹拌した。放冷後固体を濾過し、濾液を濃縮して得 られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 217mg ( 63%)を得た。
[0260] ステップ 2 1 ベンジル— 4— (3— (イミダゾール— 1—ィル)フエ-ル)ピぺリジン— 4 ァミン (参考化合物 145)の合成
アルゴン雰囲気下、 N— (1—ベンジル— 4— (3— (イミダゾールー 1—ィル)フエ- ル)ピぺリジン一 4—ィル)ァセタミド(217mg, 0. 58mmol)を THF (0. 05mL)に懸 濁し、チタンテトライソプロポキシド(172 L, 0. 58mmol)、ジフエ-ルシラン(172 μ 0. 87mmol)を加え、室温で攪拌した。 3時間後、酢酸ェチルにて希釈し、飽 和炭酸水素ナトリウム水溶液を加えて激しく攪拌した。生じた白色固体を濾別し、濾
液を分液後、酢酸ェチルにて抽出した。有機層を合わせて乾燥後濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 20mg (10%) を得た。
[0261] ステップ 3 (3R)—N—(1—べンジルー 4ー(3—(ィミダゾールー1ーィル)フェ-ル )ピペリジン— 4—ィル)—4— (2 フルオロフェ -ル)—3— (2—メチル—2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 146)の合成
アルゴン雰囲気下、 1 ベンジル— 4— (3- (イミダゾール— 1—ィル)フエ-ル)ピ ペリジン一 4 ァミン(31mg, 0. 093mmol)をジクロロメタン(3mL)に溶解し、(3R) —4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) 酪酸(30mg, 0. 10mmol)、 HOBt (21mg, 0. 15mmol)、 NEM (13mg, 0. 11m mol)、EDCI'塩酸塩(23mg, 0. 12mmol)を加え、室温で攪拌した。 12時間後濃 縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 5. 0mg (8%)を得た。
[0262] ステップ 4 (3R)— 3 ァミノ一 N— (1—ベンジル一 4— (3— (イミダゾール一 1—ィ ル)フエ-ル)ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '三塩酸 塩 (ィ匕合物 41)の合成
アルゴン雰囲気下、 (3R) N— (1—べンジルー 4一(3 (イミダゾールー 1ーィル )フエ-ル)ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(5. Omg, 0. 0082mmol)を 10%塩化水 素 Zメタノール溶液 (4mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題化合物 0. 9mg (l. 4%)を得た。
[0263] 実施例 42
(3R)— 3 ァミノ一 N— (1—ベンジル一 4— (3— (ピラゾール一 1—ィル)フエ-ル) ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '三塩酸塩 (ィ匕合物 42 )の合成
ステップ 1 N— ( 1 ベンジル 4— ( 3 (ピラゾール 1 ィル)フエ-ル)ピベリジ ン 4 ィル)ァセタミド (参考化合物 147)の合成
アルゴン雰囲気下、 N— (1—ベンジル— 4— (3 ョードフエ-ル)ピぺリジン— 4—
ィル)ァセタミド(参考化合物 140) (200mg, 0. 46mmol)を 1, 4 ジォキサン(3m L)に溶解し、ピラゾーノレ(31mg, 0. 46mmol)、ヨウィ匕銅(I) (4mg, 0. 023mmol) 、 trans— N, N,一ジメチル 1, 2 シクロへキサンジァミン(13mg, 0. 092mmol) 、リン酸カリウム(258mg, 0. 97mmol)をカ卩え、 110°Cで 15時間撹拌した。放冷後 固体を濾過し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で精製し、表題ィ匕合物 165mg (96%)を得た。
[0264] ステップ 2 1 ベンジル一 4— (3- (ピラゾール一 1—ィル)フエ-ル)ピぺリジン一 4 ーァミン (参考化合物 148)の合成
アルゴン雰囲気下、 N— (1—べンジルー 4一(3 (ピラゾールー 1 ィル)フエ-ル )ピペリジン— 4—ィル)ァセタミド(165mg, 0. 44mmol)を THF (0. 05mL)に懸濁 し、チタンテトライソプロポキシド(130 L, 0. 44mmol)、ジフエ-ルシラン(123 L, 0. 66mmol)を加え、室温で攪拌した。 15時間後、酢酸ェチルにて希釈し、飽和 炭酸水素ナトリウム水溶液を加えて激しく攪拌した。生じた白色固体を濾別し、濾液 を分液後、酢酸ェチルにて抽出した。有機層を合わせて乾燥し、濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 103mg (71%) を得た。
[0265] ステップ 3 (3R)— N— (1—ベンジル一 4— (3— (ピラゾール一 1—ィル)フエ-ル) ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキ シカルボニルァミノ)ブタナミド (参考化合物 149)の合成
アルゴン雰囲気下、 1一べンジルー 4一(3 (ピラゾールー 1 ィル)フエ-ル)ピぺ リジン一 4 ァミン(103mg, 0. 31mmol)をジクロロメタン(3mL)に溶解し、(3R)— 4一(2 フルオロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪 酸(30mg, 0. 10mmol)、 HOBt (21mg, 0. 15mmol)、 NEM (13mg, 0. 11mm ol)、EDCI'塩酸塩(23mg, 0. 12mmol)をカ卩え、室温で攪拌した。 16時間後濃縮 し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 27 mg (43%)を得た。
[0266] ステップ 4 (3R)— 3 ァミノ一 N— (1—ベンジル一 4— (3— (ピラゾール一 1—ィル )フエ-ル)ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '三塩酸塩
(化合物 42)の合成
アルゴン雰囲気下、 (3R) -N- (1—ベンジル— 4— (3- (ピラゾール— 1—ィル) フエ-ル)ピぺリジン— 4—ィル)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)ブタナミド(27mg, 0. 044mmol)を 10%塩化水素 Zメタノール溶液 (4mL)に溶解し、室温で 3時間攪拌した。飽和炭酸水素ナトリウム 水溶液を加え酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した 。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 (フリ 一塩基) 1. 9mg (8%)を得た。表題ィ匕合物(フリー塩基)をメタノール溶液とし、 10% 塩ィ匕水素 Zメタノール溶液を加えて塩ィ匕し、これを濃縮して表題化合物(三塩酸塩) を得た。
[0267] 実施例 43
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (2 フルオロフ ェ-ル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 43)の合成 ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (2 フルオロフ ェ -ル)チアン— 3— (2—メチル—2 プロポキシカルボ-ルァミノ)—4—ィル)ブタ ナミド (参考化合物 150)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル —2 プロポキシカルボ-ルァミノ)酪酸(40mg, 0. 12mmol)のァセトニトリル溶液( 4mL)を氷冷し、 N—メチルイミダゾール(30mg, 0. 36mmol)、塩化 p—トルエンス ルホ-ル(29mg, 0. 15mmol)をカ卩え、室温で 30分間攪拌した。氷冷下、 4— (2— フルオロフヱ-ル)チアンー 4ーァミン(参考化合物 69) (フリー塩基)(38mg, 0. 18 mmol)を加え、 30分間撹拌した後、室温までゆっくりと昇温した。 2時間後濃縮し、 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 63mg (定量的)を得た。
[0268] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (2 フルオロフ ェニル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 151)の合成
(3R) -4- (2, 4, 5 トリフルオロフェ -ル) N— (4—(2 フルオロフェ -ル)チ
アンー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ー4 ィル)ブタナミド(63 mg, 0. 12mmol)をジクロロメタン(4mL)に溶解し、氷冷下、 mCPBA ( >65%) (7 7mg, >0. 29mmol)を 3回に分けて添加し、 3時間攪拌した。チォ硫酸ナトリウム水 溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホル ムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 68mg (定量的)を得た。
[0269] ステップ 3 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (2 フルオロフェ-ル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (ィ匕合物 43)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (2— フルオロフェ-ル)—1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)ブタナミド(68mg, 0. 12mmol)を 10%塩化水素 Zメタノー ル溶液 (4mL)に溶解し、室温で 2時間攪拌した後濃縮し、表題ィ匕合物 47mg (78% )を得た。
[0270] 実施例 44
(3R)— 3 ァミノ一 N— (1—ベンジル一 4— (3— (4—メチルスルホユルフェ-ル)フ ェ -ル)ピぺリジン 4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '二塩酸塩(ィ匕 合物 44)の合成
ステップ 1 N— (1—ベンジル— 4— (3— (4—メチルチオフエ-ル)フエ-ル)ピペリ ジン― 4 ィル)ァセタミド(参考化合物 152)の合成
アルゴン雰囲気下、 N— (1—ベンジル— 4— (3 ョードフエ-ル)ピぺリジン— 4— ィル)ァセタミド(参考化合物 140) (400mg, 0. 92mmol)を 1, 4 ジォキサン(6m L)に溶解し、 4— (メチルチオ)フ -ルボロン酸(155mg, 0. 92mmol)、テトラキス (トリフエ-ルホスフィン)パラジウム(106mg, 0. 092mmol)、リン酸カリウム(736mg , 2. 8mmol)をカ卩え、 110°Cで 15時間撹拌した。放冷後固体を濾過し、濾液を濃縮 して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 36 8mg (93%)を得た。
[0271] ステップ 2 N—(l ベンジル— 4— (3— (4—メチルスルホユルフェ-ル)フエ-ル)
ピぺリジン— 4—ィル)ァセタミド(参考化合物 153)の合成
N— (1—ベンジル— 4— (3- (4—メチルチオフエ-ル)フエ-ル)ピぺリジン— 4— ィル)ァセタミド(400mg, 0. 93mmol)をジクロロメタン(10mL)に溶解し、氷冷下、 mCPBA( >65%) (345mg, > 1. 3mmol)を 3回に分けて添カロし、 3時間攪拌した 。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶 液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合物 242mg (56%)を得た。
[0272] ステップ 3 1—ベンジル— 4— (3— (4—メチルスルホユルフェ-ル)フエ-ル)ピペリ ジン— 4—ァミン (参考化合物 154)の合成
アルゴン雰囲気下、 N— (1—べンジルー 4一(3—(4ーメチルスルホユルフェ-ル) フエ-ル)ピぺリジン— 4—ィル)ァセタミド(242mg, 0. 52mmol)を THF (0. 05mL )に懸濁し、チタンテトライソプロポキシド(154 L, 0. 52mmol)、ジフエ-ルシラン( 146 0. 78mmol)をカ卩え、室温で攪拌した。 16時間後、酢酸ェチルにて希釈 し、飽和炭酸水素ナトリウム水溶液を加えて激しく攪拌した。生じた白色固体を濾別 し、濾液を分液後、酢酸ェチルにて抽出した。有機層を合わせて乾燥し、濃縮して得 られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 146mg ( 66%)を得た。
[0273] ステップ 4 (3R)—N—(1—べンジルー 4ー(3—(4ーメチルスルホユルフェ-ル)フ ェ -ル)ピぺリジン— 4—ィル)—4— (2—フルオロフェ -ル)—3— (2—メチル—2— プロポキシカルボニルァミノ)ブタナミド (参考化合物 155)の合成
アルゴン雰囲気下、 (3R)—4— (2—フルオロフヱ-ル)—3— (2—メチル—2—プ 口ポキシカルボ-ルァミノ)酪酸(30mg, 0. lOmmol)のァセトニトリル溶液(4mL)を 氷冷し、 N—メチルイミダゾール(25mg, 0. 30mmol)、塩化 p—トルエンスルホ-ル (24mg, 0. 13mmol)を加え、室温で 30分間攪拌した。氷冷下、 1—ベンジル— 4 - (3- (4—メチルスルホユルフェ-ル)フエ-ル)ピぺリジン— 4—ァミン(64mg, 0. 15mmol)を加え、 30分間撹拌した後、室温までゆっくりと昇温した。 17時間後濃縮 し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 71 mg (定量的)を得た。
[0274] ステップ 5 (3R)— 3—ァミノ一 N—(l ベンジル一 4— (3— (4—メチルスルホ -ル フエ-ル)フエ-ル)ピぺリジンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド- ニ塩酸塩 (化合物 44)の合成
アルゴン雰囲気下、 (3R) -N- (1—ベンジル— 4— (3- (4—メチルスルホ -ルフ ェ -ル)フエ-ル)ピぺリジン 4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(71mg, 0. lOmmol)を 10%塩 化水素 Zメタノール溶液 (4mL)に溶解し、室温で 18時間攪拌した後濃縮し、表題 化合物 68mg (82%)を得た。
[0275] 実施例 45
(3R)—3 アミノー 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (3— (イミダゾ 一ルー 1 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '二塩酸塩( 化合物 45)の合成
ステップ 1 4一(3 ョードフエニル)チアンー 4一オール(参考化合物 156)の合成 アルゴン雰囲気下、 1, 3ージョードベンゼン(5. 7g, 17mmol)の THF (50mL)溶 液を 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 55M, 6. 8mL, 17m mol)を滴下した。 10分後、 4ーォキソチアン(2g, 17mmol)の THF (30mL)溶液を 滴下した。滴下終了後、室温までゆっくりと昇温し、 2時間後、蒸留水を加え、酢酸ェ チルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 4. 7g(85%)を得た。
[0276] ステップ 2 4—(3 ョードフエ-ル)チアンー 4ーァミン(参考化合物 157)の合成
4— (3 ョードフエ-ル)チアン— 4—オール(4. 7g, 15mmol)のクロロアセト-トリ ル (6. 7g)、酢酸(7. 2mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(7. 2mL)をゆ つくりと滴下した。滴下終了後、氷浴を外し室温で攪拌し、 5時間後、氷冷して氷を加 えた。生じた白色固体を濾過し、蒸留水、飽和炭酸水素ナトリウム水溶液、蒸留水の 順に洗浄した。得られた粗生成物(5. 8g)をエタノール (90mL)、酢酸(18mL)に溶 解し、チォ尿素(1. 3g, 18mmol)をカ卩えて加熱還流した。 12時間後放冷し、炭酸力 リウム水溶液を加え酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し 濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕
合物 1. 8g (38%)を得た。
[0277] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (3— (イミダゾー ルー 1—ィル)フエ-ル)チアン— 4—ィル)—3— (2—メチル—2 プロポキシカルボ -ルァミノ)ブタナミド (参考化合物 158)の合成
アルゴン雰囲気下、 4— (3 ョードフエ-ル)チアン— 4 ァミン(lOOmg, 0. 31m mol)、イミダゾール(43mg, 0. 63mmol)、ヨウィ匕銅(I) (6mg, 0. O3mmol)、 1, 1 0 フエナント口リン(56mg, 0. 31mmol)、炭酸セシウム(204mg, 0. 63mmol)を 1, 4 ジォキサン(2mL)に溶解し、 110°Cで 15時間撹拌した。放冷後固体を濾別 し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製 し、 4— (3— (イミダゾール— 1—ィル)フエ-ル)チアン— 4 ァミンの粗精製物(94 mg)を得た。アルゴン雰囲気下、(3R)— 4— (2, 4, 5—トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(121mg, 0. 36mmol)をァセト 二トリル(4mL)に溶解し、 N—メチルイミダゾール(90mg, 1. lmmol)を加え、氷冷 下、塩化 p—トルエンスルホ-ル(86mg, 0. 45mmol)をカ卩え、室温で 30分間攪拌 した。氷冷下、 4— (3— (イミダゾール— 1—ィル)フエ-ル)チアン— 4 ァミンの粗精 製物(94mg)を加え、室温までゆっくりと昇温した。 12時間後濃縮し、得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 50mg (28%)を得た
[0278] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (3— (イミダゾー ル一 1 ィル)フエ-ル) 1, 1 ジォキソチアン 4 ィル) 3— ( 2 メチル 2— プロポキシカルボニルァミノ)ブタナミド (参考化合物 159)の合成
(3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (3— (イミダゾールー 1— ィル)フエ-ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルアミ ノ)ブタナミド(50mg, 0. 087mmol)をジクロロメタン(4mL)に溶解し、氷冷下、 mC PBA( >65%) (46mg, >0. 17mmol)を 5回に分けて添カ卩した。 4時間後、チォ硫 酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加え た。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 36mg (68%)を得た。
[0279] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (3 - (イミダゾールー 1—ィル)フエ-ル) 1, 1 ジォキソチアン— 4—ィル)ブタナミド •二塩酸塩 (化合物 45)の合成
アルゴン雰囲気下、(3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(3— ( イミダゾールー 1—ィル)フエ-ル)一 1, 1—ジォキソチアン一 4—ィル) 3— (2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(36mg, 0. O59mmol)を 10%塩 化水素 Zメタノール溶液 (6mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題 化合物 27mg (73%)を得た。
[0280] 実施例 46
(3R) 3 アミノー 4一(2 フルオロフェ -ル)一 N— (4—(3 (ピリジンー4ーィノレ )フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '二塩酸塩 (ィ匕合物 46)の 合成
ステップ 1 (3R) -4- (2 フルオロフェ -ル) N— (4—(3 ョードフエ-ル)チア ンー4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考 化合物 160)の合成
アルゴン雰囲気下、(3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)酪酸(lOOmg, 0. 34mmol)をァセトニトリル(4mL)に溶 解し、 N—メチルイミダゾール(83mg, 1. Ommol)をカ卩え、氷冷下、塩化 p トルエン スルホニル(80mg, 0. 42mmol)をカ卩え、室温で 30分間攪拌した。氷冷下、 4— (3 ョードフエ-ル)チアンー 4ーァミン(参考化合物 157) (107mg, 0. 34mmol)を 加え、室温までゆっくりと昇温した。 12時間後濃縮し、得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合物 153mg (76%)を得た。
[0281] ステップ 2 (31¾—4ー(2—フルォロフェ-ル)ー?^ー(4ー(3—ョードフェ-ル)ー1 , 1ージォキソチアンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ )ブタナミド (参考化合物 161)の合成
(3R) -4- (2 フルオロフヱ-ル) N— (4—(3 ョードフエ-ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(153mg, 0. 2 56mmol)をジクロロメタン(6mL)に溶解し、氷冷下、 mCPBA( >65%) (186mg,
>0. 7mmol)を添加した。 3時間後、チォ硫酸ナトリウム水溶液を加えて激しく攪拌 した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮 して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 10 6mg (66%)を得た。
[0282] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (3— (ピリジン一 4—ィル)フエ-ル)一 1, 1—ジォキソチ アン— 4 ィル)ブタナミド (参考化合物 162)の合成
アルゴン雰囲気下、(3R)—4— (2 フルオロフヱ-ル) N— (4— (3 ョードフエ -ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ -ルァミノ)ブタナミド(50mg, 0. O79mmol)を 1, 4 ジォキサン(6mL)に溶解し、 4- (4, 4, 5, 5—テトラメチルー 1, 3, 2 ジォキサボロランー2 ィル)ピリジン(16 mg, 0. 079mmol)、炭酸セシウム(78mg, 0. 24mmol)、 [1, 1,一ビス(ジフエ- ルホスフイノ)フエ口セン]ジクロロパラジウム(6mg, 0. 008mmol)を加え、 80°Cで 2 0時間攪拌した。放冷後濾過し、濾液を濃縮して得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 42mg (91%)を得た。
[0283] ステップ 4 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (3— (ピリジ ンー4 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '二塩酸塩 (ィ匕 合物 46)の合成
アルゴン雰囲気下、(3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4— (3— (ピリジン— 4—ィル)フエ-ル)— 1, 1— ジォキソチアンー4 ィル)ブタナミド(42mg, 0. 072mmol)を 10%塩化水素 Zメタ ノール溶液 (6mL)に溶解し、室温で 14時間攪拌した後濃縮し、表題ィ匕合物 24mg ( 61%)を得た。
[0284] 実施例 47
(3R)—3 ァミノ— 4— (2 フルオロフェ -ル) N— (4— (3— (チアゾール—2— ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 47) の合成
ステップ 1 2 (トリブチルスタ-ル)チアゾール(参考化合物 163)の合成
アルゴン雰囲気下、 THF (24mL)に n ブチルリチウム Zへキサン溶液(2. 55M , 2. 5mL, 6. 5mmol)を加え、 78。Cに冷却した。チアゾール(500mg, 5. 9mm ol)の THF溶液(15mL)を 20分間かけて滴下し、 1時間撹拌した。塩化トリプチルス ズ(1. 8mL, 6. 5mmol)の THF溶液(9mL)を 15分間かけて滴下し、 1時間撹拌し た後、飽和炭酸水素ナトリウム水溶液、ジェチルエーテルを加え、室温で 2時間撹拌 した。分液後、有機層を乾燥し濃縮し、粗生成物として、表題化合物 2. 4gを得た。
[0285] ステップ 2 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) -N- (4— (3- (チアゾール—2—ィル)フエ-ル)— 1, 1—ジォキ ソチアン 4 ィル)ブタナミド (参考化合物 164)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (3 ョードフエ -ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ -ルァミノ)ブタナミド(参考化合物 161) (70mg, 0. l lmmol)をァセトニトリル(2m L)に溶解し、 2 (トリブチルスタ-ル)チアゾールの粗生成物(83mg,約 0. 22mm ol)、テトラキス(トリフエ-ルホスフィン)パラジウム(38mg, 0. 033mmol)をカ卩え、 80 °Cで 12時間撹拌した。放冷後、濃縮して得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 17mg (25%)を得た。
[0286] ステップ 3 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (3— (チアゾ 一ルー 2—ィル)フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩(ィ匕 合物 47)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(3 (チアゾールー 2 ィル)フエ-ル)—1, 1 ージォキソチアンー4 ィル)ブタナミド(17mg, 0. 028mmol)を 10%塩化水素 Z メタノール溶液 (4mL)に溶解し、室温で 2時間攪拌した後濃縮し、表題ィ匕合物 12m g (79%)を得た。
[0287] 実施例 48
(3R)—3 ァミノ— 4— (2 フルオロフェ -ル) N— (4— (3— (チアゾール—5— ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 48) の合成
ステップ 1 5 (トリブチルスタ-ル) 2 トリメチルシリルチアゾール(参考化合物 1 65)の合成
アルゴン雰囲気下、 n—ブチルリチウム Zへキサン溶液(2. 55M, 1. 4mL, 3. 5m mol)を THF (15mL)で希釈し、 78°Cに冷却した後、 2 トリメチルシリルチアゾー ル(500mg, 3. 2mmol)の THF溶液(6mL)を 20分間かけて滴下し、 1時間撹拌し た。塩化トリブチルスズ(0. 86mL, 3. 2mmol)の THF溶液(5mL)を 10分間かけて 滴下し、 1時間撹拌した。飽和炭酸水素ナトリウム水溶液、ジェチルエーテルを加え、 室温で 2時間撹拌した。分液し、有機層を乾燥後濃縮し、粗生成物として、表題化合 物 1. 4gを得た。
[0288] ステップ 2 5 (トリブチルスタ-ル)チアゾール(参考化合物 166)の合成
アルゴン雰囲気下、 5— (トリブチルスタ-ル)—2 トリメチルシリルチアゾールの粗 生成物(1. 4g)を THF (lOmL)に溶解し、 1N塩酸(lmL)をカ卩え、室温で 1時間攪 拌した。ジェチルエーテルで抽出し、有機層を乾燥後濃縮し、粗生成物として、表題 化合物 1. 2gを得た。
[0289] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) -N- (4— (3- (チアゾール—5—ィル)フエ-ル)— 1, 1—ジォキ ソチアン— 4 -ィルブタナミド (参考化合物 167)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (3 ョードフエ -ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ ニルァミノ)ブタナミド(参考化合物 161) (70mg, 0. l lmmol)を DMF (2mL)に溶 解し、 5— (トリブチルスタ-ル)チアゾールの粗生成物(125mg、約 0. 33mmol)、テ トラキス(トリフエ-ルホスフィン)パラジウム(64mg, 0. 056mmol)を加え、 80°Cで 1 8時間撹拌した。放冷後、濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 l lmg (16%)を得た。
[0290] ステップ 4 (3R)— 3 ァミノ— 4— (2 フルオロフェ-ル)— N— (4— (3— (チアゾ 一ルー 5—ィル)フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩(ィ匕 合物 48)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ
口ポキシカルボ-ルァミノ) N— (4—(3 (チアゾールー 5 ィル)フエ-ル)—1, 1 —ジォキソチアン— 4—ィルブタナミド(l lmg, 0. 018mmol)を 10%塩化水素 Zメ タノール溶液 (4mL)に溶解し、室温で 2時間攪拌した後濃縮し、表題化合物 6. 9m g (73%)を得た。
[0291] 実施例 49
(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (4— (3— (ピラゾールー 4—ィ ル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 49)の 合成
ステップ 1 4 ョード 1—トリチルピラゾール(参考化合物 168)の合成
アルゴン雰囲気下、 4ーョードピラゾール(1. Og, 5. 2mmol)のピリジン溶液(12m L)に塩ィ匕トリチル(1. 7g, 6. 2mmol)、4— (N, N ジメチルァミノ)ピリジン(126m g, 1. Ommol)をカ卩え、 80°Cで 18時間撹拌した。放冷後、蒸留水を加えジェチルェ 一テル Zジクロロメタン =6Zlにより抽出し、有機層を乾燥後濃縮した。得られた固 体をジェチルエーテルで洗净し、乾燥した。粗生成物として、表題化合物 2. lg (92 %)を得た。
[0292] ステップ 2 4 トリブチルスタ-ルー 1 トリチルピラゾール(参考化合物 169)の合成 アルゴン雰囲気下、 THF (30mL)に n ブチルリチウム Zへキサン溶液(2. 55M , 1. OmL, 2. 5mmol)をカ卩え、—78°Cに冷却した。 4 ョード—1—トリチルピラゾ ール(1. Og,約 2. 3mmol)の THF溶液(6mL)を 20分間かけて滴下し、 1時間撹拌 した。塩化トリブチルスズ(0. 62mL, 2. 3mmol)の THF溶液(6mL)を 10分間かけ て滴下し、 30分間撹拌した後、室温まで昇温し、 2時間撹拌した。塩ィ匕アンモ-ゥム 水溶液を加えて、ジェチルエーテルで抽出し、有機層を乾燥後濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1. 2g (84%)を 得た。
[0293] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (3— (1—トリチルピラゾール— 4—ィル)フエ-ル)— 1, 1 ジォキソチアン 4 ィル)ブタナミド (参考化合物 170)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (3 ョードフエ
-ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ -ルァミノ)ブタナミド(参考化合物 161) (lOOmg, 0. 16mmol)、 4 トリブチルスタ 二ルー 1 トリチルピラゾール(133mg, 0. 22mmol)をァセトニトリル(2mL)に溶解 し、 15分間撹拌した後、 10%パラジウム Z炭素(lOmg)、ヨウ化銅 (I) (3mg, 0. 01 6mmol)、トリフエ-ル砒素(lOmg, 0. 032mmol)をカ卩え、 80°Cで 72時間撹拌した 。放冷後、濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 71mg (55%)を得た。
[0294] ステップ 4 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (3—(ビラゾ 一ルー 4—ィル)フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩(ィ匕 合物 49)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4— (3— (1—トリチルピラゾール— 4—ィル)フエ -ル)ー1, 1ージォキソチアンー4 ィル)ブタナミド(77mg, 0. 095mmol)を 10% 塩ィ匕水素 Zメタノール溶液(10mL)に溶解し、室温で 7時間攪拌した後、炭酸力リウ ム水溶液を加え、クロ口ホルムで抽出した。有機層を乾燥、濃縮し、得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物(フリー塩基) 10mg (2 2%)を得た。表題ィ匕合物 (フリー塩基)をメタノールに溶解し、 10%塩ィ匕水素 Zメタノ ール溶液を加えて塩化し、濃縮して表題化合物(二塩酸塩)を得た。
[0295] 実施例 50
(3R)—3 アミノー 4— (2 フルオロフヱ-ル) N— (4— (3— (1—メチルイミダゾ 一ルー 5 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '二塩酸塩( 化合物 50)の合成
ステップ 1 5 トリブチルスタ-ル— 1—メチルイミダゾール(参考化合物 171)の合 成
アルゴン雰囲気下、 5 ブロモ—1—メチルイミダゾール(1. Og, 6. 2mmol)の TH F溶液(60mL)に臭化工チルマグネシウム ZTHF溶液(1. 0M, 7. 5mL, 7. 5mm ol)をゆっくり滴下し、 2時間撹拌した。塩化トリブチルスズ(2. lmL, 7. 8mmol)を 滴下し、 15時間撹拌した。塩ィ匕アンモ-ゥム水溶液をカ卩え、ジェチルエーテルで抽
出し、有機層を蒸留水で洗浄した後、乾燥し濃縮した。得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合物 640mg (28%)を得た。
[0296] ステップ 2 (3R)— 4— (2 フルオロフェ-ル)— N— (4— (3— (1—メチルイミダゾ 一ルー 5—ィル)フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 172)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (3 ョードフエ -ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ -ルァミノ)ブタナミド(参考化合物 161) (lOOmg, 0. 16mmol)、 5 トリブチルスタ -ル— 1—メチルイミダゾール(71mg, 0. 19mmol)をァセトニトリル(3mL)に溶解 し、 15分間撹拌した後、テトラキス(トリフエ-ルホスフィン)パラジウム(55mg, 0. 04 8mmol)を加え、 80°Cで 40時間撹拌した。放冷後、濃縮して得られた粗生成物をシ リカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 35mg (38%)を得た。
[0297] ステップ 3 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (3— (1—メ チルイミダゾールー 5 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミ ド ·二塩酸塩 (化合物 50)の合成
アルゴン雰囲気下、 (3R) -4- (2 フルオロフヱ-ル) N— (4—(3—(1ーメチ ルイミダゾ一ルー 5—ィル)フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)—3— (2— メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(35mg, 0. 060mmol)を 10% 塩ィ匕水素 Zメタノール溶液 (6mL)に溶解し、室温で 2時間攪拌した後濃縮し、表題 化合物 28mg (79%)を得た。
[0298] 実施例 51
(3R)—3 アミノー 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (3— (チアゾー ルー 5 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合 物 51)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (3 ョードフエ -ル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナ ミド (参考化合物 173)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル
2 プロポキシカルボ-ルァミノ)酪酸(522mg, 1. 6mmol)をァセトニトリル(4mL )に溶解し、 N—メチルイミダゾール(386mg, 4. 7mmol)を加え、氷冷下、塩化 p— トルエンスルホ-ル(328mg, 1. 7mmol)をカ卩え、室温で 30分間攪拌した。氷冷下 、 4 (3 ョードフエ-ル)チアンー 4ーァミン(参考化合物 157) (500mg, 1. 6mm ol)を加え、室温までゆっくりと昇温した。 12時間後、濃縮し、得られた粗生成物をシ リカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 702mg (71%)を得た。
[0299] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (3 ョードフエ -ル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ -ルァミノ)ブタナミド (参考化合物 174)の合成
(3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (3 ョードフエ-ル)チア ンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(702m g, 1. Immol)をジクロロメタン(20mL)に溶解し、氷冷下、 mCPBA( >65%) (764 mg, > 2. 9mmol)を 2回に分けて添カ卩した。 3時間後、チォ硫酸ナトリウム水溶液を 加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽 出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 559mg (76%)を得た。
[0300] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)—N— (4— (3 (チアゾールー 5 ィル)フエ-ル)—1, 1 -ジォキソチアン 4 ィル)ブタナミド (参考化合物 175)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (3— ョードフエ-ル)— 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プロポキ シカルボ-ルァミノ)ブタナミド(259mg, 0. 39mmol)、 5— (トリブチルスタ-ル)チ ァゾール(参考化合物 166) (291mg, 0. 78mmol)をァセトニトリル(6mL)に溶解し 、 15分間激しく撹拌した後、テトラキス(トリフエ-ルホスフィン)パラジウム (45mg, 0. O39mmol)を加え、 80°Cで 15時間撹拌した。放冷後固体を濾別し、濾液を濃縮し て得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 89m g (37%)を得た。
[0301] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (3
(チアゾールー 5 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド' 塩酸塩 (ィ匕合物 51)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ)—N— (4— (3 (チアゾールー 5 ィル)フエ- ル)ー1, 1ージォキソチアンー4 ィル)ブタナミド(89mg, 0. 14mmol)を 10%塩 化水素 Zメタノール溶液 (6mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題ィ匕 合物 82mg (定量的)を得た。
[0302] 実施例 52
(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (4— (4— (チアゾール 5— ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 52) の合成
ステップ 1 4一 (4一ョードフエニル)チアンー 4一オール(参考化合物 176)の合成 アルゴン雰囲気下、 1, 4ージョードベンゼン(5. 7g, 17mmol)の THF (50mL)溶 液を 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 55M, 6. 8mL, 17m mol)を滴下した。 30分後、 4ーォキソチアン(2. Og, 17mmol)の THF (30mL)溶 液を滴下した。滴下終了後、室温までゆっくりと昇温し、 4時間後、蒸留水を加えクロ 口ホルム及び酢酸ェチルで抽出し、有機層を合わせて飽和食塩水にて洗浄後、乾 燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表 題化合物 4. 5g(82%)を得た。
[0303] ステップ 2 4—(4 ョードフエ-ル)チアンー 4ーァミン(参考化合物 177)の合成
4— (4 ョードフエ-ル)チアン— 4—オール(4. 5g, 14mmol)のクロロアセト-トリ ル (6. 4g)、酢酸(7. 2mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(7. 2mL)をゆ つくりと滴下した。滴下終了後、氷浴を外し室温で攪拌し、 5時間後、氷水を加えた。 生じた白色固体を濾過し、蒸留水、飽和炭酸水素ナトリウム水溶液、蒸留水の順に 洗浄した。得られた粗生成物をエタノール (90mL)、酢酸(18mL)に溶解し、チォ尿 素(1. 3g, 18mmol)を加えて加熱還流した。 12時間後放冷し、炭酸カリウム水溶液 を加え、酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 2. 5g (
55%)を得た。
[0304] ステップ 3 4—(4 (チアゾールー 5 ィル)フエ-ル)チアンー 4ーァミン(参考化合 物 178)の合成
アルゴン雰囲気下、 4— (4 ョードフエ-ル)チアン— 4 ァミン(200mg, 0. 63m mol)、 5 (トリブチルスタ-ル)チアゾール(参考化合物 166) (234mg, 0. 63mm ol)をァセトニトリル(3mL)に溶解し、 15分間撹拌した後、テトラキス(トリフエ-ルホス フィン)パラジウム(72mg, 0. 063mmol)を加え、 80°Cで 20時間撹拌した。放冷後 固体を濾別し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で精製し、表題ィ匕合物 127mg (73%)を得た。
[0305] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (4— (チアゾール—5—ィル)フエ-ル)チアン— 4—ィル )ブタナミド (参考化合物 179)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)酪酸(136mg, 0. 46mmol)をァセトニトリル(4mL)に溶 解し、 N—メチルイミダゾール(113mg, 1. 4mmol)を加え、氷冷下、塩化 p トルェ ンスルホ-ル(88mg, 0. 46mmol)を加え、室温で 1時間攪拌した。氷冷下、 4一(4 —(チアゾール—5—ィル)フエ-ル)チアン— 4 ァミン(127mg, 0. 46mmol)のジ クロロメタン溶液(10mL)を加え、室温までゆっくりと昇温した。 20時間後、濃縮し、 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 127m g (50%)を得た。
[0306] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (4— (チアゾール—5—ィル)フエ-ル)— 1, 1—ジォキ ソチアン— 4 -ィル)ブタナミド (参考化合物 180)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ) N— (4—(4 (チアゾールー 5—ィル)フエ-ル)チアンー 4 ィル)ブタナミ ド(127mg, 0. 23mmol)をジクロロメタン(6mL)に溶解し、氷冷下、 mCPBA( >6 5%) (208mg, >0. 78mmol)を 2回に分けて添カロした。 4時間後、チォ硫酸ナトリ ゥム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ
口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 88mg (65%)を得た。
[0307] ステップ 6 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (4— (チアゾ 一ルー 5—ィル)フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩(ィ匕 合物 52)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(4 (チアゾールー 5 ィル)フエ-ル)—1, 1 ージォキソチアンー4 ィル)ブタナミド(86mg, 0. 15mmol)を 10%塩化水素 Zメ タノール溶液 (6mL)に溶解し、室温で 2時間攪拌した後濃縮し、表題ィ匕合物 74mg ( 定量的)を得た。
[0308] 実施例 53
(3R)—3—ァミノ一 4— (3—クロ口フエ-ル) N— (4— (3— (チアゾール 5—ィル )フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 53)の合 成
ステップ 1 4一(3 (チアゾールー 5 ィル)フエ-ル)チアンー 4ーァミン(参考化合 物 181)の合成
アルゴン雰囲気下、 4一(3 ョードフエニル)チアンー 4ーァミン(参考化合物 157) (400mg, 1. 3mmol)、 5 (トリブチルスタ-ル)チアゾール(参考化合物 166) (46 8mg, 1. 3mmol)をァセトニトリル(10mL)に溶解し、 15分間撹拌した後、テトラキス (トリフエ-ルホスフィン)パラジウム(144mg, 0. 13mmol)をカロえ、 80°Cで 12時間 撹拌した。放冷後固体を濾別し、濾液を濃縮して得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 204mg (59%)を得た。
[0309] ステップ 2 (3R)— 4— (3 クロ口フエ-ノレ)一 3— (2—メチノレ一 2 プロポキシ力ノレ ボ-ルァミノ) N— (4—(3—(チアゾールー 5—ィル)フエ-ル)チアンー 4 ィル) ブタナミド (参考化合物 182)の合成
アルゴン雰囲気下、 (3R)—4— (3 クロ口フエ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)酪酸(116mg, 0. 37mmol)のァセトニトリル(3mL)、ジクロ ロメタン(4mL)混合溶液に、 N—メチルイミダゾール(91mg, 1. lmmol)をカ卩え、氷
冷下、塩化 p—トルエンスルホ-ル(70mg, 0. 37mmol)をカ卩え、室温で 1時間攪拌 した。氷冷下、 4— (3- (チアゾール—5—ィル)フエ-ル)チアン— 4 ァミン(102m g, 0. 37mmol)のジクロロメタン溶液(2mL)をカ卩え、室温までゆっくりと昇温した。 2 0時間後、濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 152mg (72%)を得た。
[0310] ステップ 3 (3R)—4— (3 クロ口フエ-ノレ)ー3—(2—メチノレー 2 プロポキシ力ノレ ボ-ルァミノ) N— (4— (3— (チアゾール 5—ィル)フエ-ル)一 1, 1—ジォキソ チアン— 4—ィル)ブタナミド (参考化合物 183)の合成
(3R)—4— (3 クロ口フエ-ル)—3— (2—メチル—2 プロポキシカルボ-ルアミ ノ) N— (4—(3—(チアゾールー 5—ィル)フエ-ル)チアンー 4 ィル)ブタナミド( 152mg, 0. 27mmol)をジクロロメタン(6mL)に溶解し、氷冷下、 mCPBA( >65% ) (183mg, >0. 73mmol)を添カロした。 4時間後、チォ硫酸ナトリウム水溶液をカロえ て激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し 、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 104mg (65%)を得た。
[0311] ステップ 4 (3R)—3 アミノー 4 (3 クロ口フエニル) N— (4— (3 (チアゾー ルー 5 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合 物 53)の合成
アルゴン雰囲気下、 (3R)—4— (3 クロ口フエ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)—N— (4— (3 (チアゾールー 5 ィル)フエ-ル)—1, 1 ージォキソチアンー4 ィル)ブタナミド(104mg, 0. 15mmol)を 10%塩化水素 Z メタノール溶液 (6mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題化合物 94 mg (定量的)を得た。
[0312] 実施例 54
(3R)—3 アミノー 4— (2, 4 ジクロロフエ-ル) N— (4— (3— (チアゾーノレ一 5 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 54 )の合成
ステップ 1 (3R)—4— (2, 4 ジクロロフエ-ル)—3— (2—メチル—2 プロポキシ
カルボ-ルァミノ) N— (4— (3— (チアゾール 5—ィル)フエ-ル)チアン一 4—ィ ル)ブタナミド (参考化合物 184)の合成
アルゴン雰囲気下、 (3R)—4—(2, 4 ジクロ口フエ-ル)ー3—(2—メチルー 2— プロポキシカルボ-ルァミノ)酪酸(128mg, 0. 37mmol)のァセトニトリル(3mL)、 ジクロロメタン(4mL)混合溶液に、 N—メチルイミダゾール(9 lmg, 1. lmmol)を加 え、氷冷下、塩化 ρ トルエンスルホ-ル(70mg, 0. 37mmol)をカ卩え、室温で 1時 間攪拌した。氷冷下、 4— (3— (チアゾール—5—ィル)フエ-ル)チアン— 4—ァミン (参考化合物 181) (102mg, 0. 37mmol)のジクロロメタン溶液(2mL)をカ卩え、室 温までゆっくりと昇温した。 20時間後濃縮し、得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 169mg (75%)を得た。
[0313] ステップ 2 (3R)—4—(2, 4 ジクロ口フエ二ノレ)ー3—(2—メチノレー 2 プロポキシ カルボ-ルァミノ) N— (4— (3— (チアゾール 5—ィル)フエ-ル)一 1, 1—ジォ キソチアン— 4—ィル)ブタナミド (参考化合物 185)の合成
(3R)—4— (2, 4 ジクロロフエ-ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ) N— (4—(3—(チアゾールー 5—ィル)フエ-ル)チアンー 4 ィル)ブタ ナミド(169mg, 0. 28mmol)をジクロロメタン(6mL)に溶解し、氷冷下、 mCPBA( >65%) (192mg, >0. 72mmol)を添カ卩した。 4時間後、チォ硫酸ナトリウム水溶 液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルム で抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題ィ匕合物 115mg (65%)を得た。
[0314] ステップ 3 (3R)— 3 アミノー 4— (2, 4 ジクロロフエ二ル)一 N— (4— (3— (チア ゾールー 5 ィル)フエ-ル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 54)の合成
アルゴン雰囲気下、 (3R)—4—(2, 4 ジクロ口フエ-ル)ー3—(2—メチルー 2— プロポキシカルボ-ルァミノ) N— (4—(3 (チアゾールー 5 ィル)フエ-ル) 1 , 1—ジォキソチアン— 4—ィル)ブタナミド(115mg, 0. 18mmol)を 10%塩化水素 Zメタノール溶液 (6mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題化合物 1 04mg (定量的)を得た。
[0315] 実施例 55
(3R) 3 アミノー 4一(2 フルオロフヱ-ル)一 N— (4—(3—(2—メチルチアゾ 一ルー 5—ィル)フエ-ル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩(ィ匕 合物 55)の合成
ステップ 1 5 トリブチルスタ-ル—2—メチルチアゾール(参考化合物 186)の合成 アルゴン雰囲気下、 THF (40mL)に n ブチルリチウム Zへキサン溶液(2. 55M , 4. 4mL, l lmmol)を加え、 78。Cに冷却した。 2—メチルチアゾール(1. Og, 10 mmol)の THF溶液(15mL)を 50分間かけて滴下し、 1時間撹拌した。塩ィ匕トリプチ ルスズ(2. 7mL, lOmmol)の THF溶液(15mL)を 20分間かけて滴下し、 1時間撹 拌した。飽和炭酸水素ナトリウム水溶液を加えて、室温まで昇温し、ジェチルエーテ ルで抽出し、有機層を乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題化合物 3. 2g (81%)を得た。
[0316] ステップ 2 4—(3— (2—メチルチアゾールー 5 ィル)フエ-ル)チアゾールー 4 ァミン (参考化合物 187)の合成
アルゴン雰囲気下、 4一(3 ョードフエニル)チアンー 4ーァミン(参考化合物 157) (200mg, 0. 63mmol)、 5 トリブチルスタ-ル—2—メチルチアゾール(243mg, 0. 63mmol)をァセトニトリル (4mL)に溶解し、 15分間撹拌した後、テトラキス(トリフ ェ-ルホスフィン)パラジウム(72mg, 0. O63mmol)をカ卩え、 80°Cで 14時間撹拌し た。放冷後固体を濾別し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 114mg (63%)を得た。
[0317] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ)—N— (4— (3—(2—メチルチアゾールー 5 ィル)フエ-ル)チアン 4 ィル)ブタナミド (参考化合物 188)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)酪酸(117mg, 0. 39mmol)のァセトニトリル溶液(3mL) に、 N—メチルイミダゾール(97mg, 1. 2mmol)をカ卩え、氷冷下、塩化 p—トルエンス ルホ-ル(74mg, 0. 39mmol)をカ卩え、室温で 1時間攪拌した。氷冷下、 4— (3— ( 2—メチルチアゾールー 5 ィル)フエ-ル)チアゾールー 4ーァミン(114mg, 0. 39
mmol)のジクロロメタン溶液(2mL)を加え、室温までゆっくりと昇温した。 20時間後 、濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕 合物 124mg (55%)を得た。
[0318] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) -N- (4- (3- (2—メチルチアゾールー 5 ィル)フエ-ル)—1, 1 -ジォキソチアン― 4 ィル)ブタナミド (参考化合物 189)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ) N— (4—(3—(2—メチルチアゾールー 5 ィル)フエ-ル)チアンー 4ーィ ル)ブタナミド(124mg, 0. 22mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mC PBA( >65%) (157mg, >0. 59mmol)を添カ卩した。 2時間後、チォ硫酸ナトリウム 水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロロホ ルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で精製し、表題ィ匕合物 74mg (57%)を得た。
[0319] ステップ 5 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (3— (2—メ チルチアゾールー 5 ィル)フエ-ル)—1, 1ージォキソチアン 4 ィル)ブタナミド •塩酸塩 (ィ匕合物 55)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(3—(2—メチルチアゾールー 5 ィル)フエ- ル)ー1, 1ージォキソチアンー4 ィル)ブタナミド(74mg, 0. 12mmol)を 10%塩 化水素 Zメタノール溶液 (6mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題ィ匕 合物 41mg (61%)を得た。
[0320] 実施例 56
(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (4— (3— (2 ピペリジノチア ゾールー 5 ィル)フエ-ル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 56)の合成
ステップ 1 2 ピペリジノチアゾール(参考化合物 190)の合成
アルゴン雰囲気下、 2 ブロモチアゾール(1. 5g, 9. Immol)の THF (lOmL)溶 液に TEA(3. 8mL, 27mmol)、ピペリジン(779mg, 9. Immol)を加え、 100°Cで
4時間撹拌した。さらにピぺリジン(3. 9g, 45mmol)を加え、 100°Cで 8時間撹拌し た後放冷し、蒸留水を加えて、酢酸ェチルで抽出し、有機層を乾燥後濃縮した。得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 316mg (2 1%)を得た。
[0321] ステップ 2 5 トリブチルスタ-ル—2 ピペリジノチアゾール(参考化合物 191)の 合成
アルゴン雰囲気下、 THF (8mL)に n ブチルリチウム Zへキサン溶液(2. 55M, 0. 81mL, 2. lmmol)を加え、 78°Cに冷却した。 2 ピペリジノチアゾール(316 mg, 1. 9mmol)の THF溶液(3mL)を 20分間かけて滴下し、 1時間撹拌した。塩化 トリブチルスズ(0. 51mL, 1. 9mmol)の THF溶液(3mL)を 20分間かけて滴下し、 1時間撹拌した。飽和炭酸水素ナトリウム水溶液を加えて、室温まで昇温し、ジェチ ルエーテルで抽出し、有機層を乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 520mg (61%)を得た。
[0322] ステップ 3 4—(3— (2 ピペリジノチアゾールー 5 ィル)フエ-ル)チアンー 4ーァ ミン (参考化合物 192)の合成
アルゴン雰囲気下、 4一(3 ョードフ ニル)チアンー 4ーァミン(参考化合物 157) ( 200mg, 0. 63mmol)、 5 トリブチルスタ-ル—2 ピペリジノチアゾール(520mg , 1. lmmol)をァセトニトリル (4mL)に溶解し、 15分間撹拌した後、テトラキス(トリフ ェ-ルホスフィン)パラジウム(72mg, 0. 063mmol)をカ卩え、 80°Cで 14時間撹拌し た。放冷後固体を濾別し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 192mg (85%)を得た。
[0323] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (3— (2 ピペリジノチアゾール—5—ィル)フエ-ル)チ アン— 4 ィル)ブタナミド (参考化合物 193)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)酪酸(159mg, 0. 53mmol)のァセトニトリル溶液(3mL) に、 N—メチルイミダゾール(132mg, 1. 6mmol)をカ卩え、氷冷下、塩化 p トルエン スルホ-ル(102mg, 0. 53mmol)をカ卩え、室温で 1時間攪拌した。氷冷下、 4 (3
— (2 ピペリジノチアゾール—5—ィル)フエ-ル)チアン— 4 ァミン(192mg, 0. 5 3mmol)のジクロロメタン溶液(2mL)を加え、室温までゆっくりと昇温した。 20時間後 、濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕 合物 206mg (82%)を得た。
[0324] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ)—N— (4— (3—(2 ピペリジノチアゾールー 5 ィル)フエ-ル) 1, 1 ジォキソチアン 4 ィル)ブタナミド (参考化合物 194)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ) N— (4—(3—(2 ピペリジノチアゾールー 5 ィル)フエ-ル)チアンー 4 ィル)ブタナミド(206mg, 0. 32mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 m CPBA( >65%) (233mg, >0. 88mmol)を添カ卩した。 2時間後、チォ硫酸ナトリウ ム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口 ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合物 46mg (32%)を得た。
[0325] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (3— (2 ピ ペリジノチアゾールー 5 ィル)フエ-ル)—1, 1ージォキソチアンー4 ィル)ブタナ ミド ·塩酸塩 (化合物 56)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(3—(2 ピペリジノチアゾールー 5 ィル)フ ェニル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド(47mg, 0. 069mmol)を 10 %塩ィ匕水素 Zメタノール溶液 (6mL)に溶解し、室温で 3時間攪拌した後濃縮し、表 題ィ匕合物 31mg (71%)を得た。
[0326] 実施例 57
(3R)—3 アミノー 4— (2 フルオロフヱ-ル) N— (4— (チアゾール—2—ィル)
— 1, 1 ジォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 57)の合成 ステップ 1 4 (チアゾールー 2 ィル)チアンー 4 オール(参考化合物 195)の合 成
アルゴン雰囲気下、チアゾール(0. 2mL, 2. 811111101)の丁11 (3111 溶液をー78
°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 57M, 1. 8mL, 2. 8mmol)を 滴下した。 20分後、 4ーォキソチアン(301mg, 2. 6mmol)の THF (3mL)溶液を滴 下した。滴下終了後、室温までゆっくりと昇温し、 3時間後、飽和炭酸水素ナトリウム 水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃 縮した。得られた粗生成物を酢酸ェチルに加熱溶解後放冷して結晶化させ、表題ィ匕 合物 241mg (46%)を得た。
[0327] ステップ 2 N—(4 (チアゾールー 2 ィル)チアンー 4 ィル)ァセタミド(参考化合 物 196)の合成
4— (チアゾール—2—ィル)チアン— 4—オール(200mg, 0. 99mmol)のァセトリ トリル(1. 7mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(1. ImL)をゆっくりと滴下 した。滴下終了後、氷浴を外し室温で攪拌し、 12時間後、氷水に注いだ。水層を水 酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、有機層を飽 和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 42mg (17%)を得た。
[0328] ステップ 3 4 (チアゾールー 2 ィル)チアンー 4ーァミン(参考化合物 197)の合成 アルゴン雰囲気下、 N— (4 (チアゾールー 2 ィル)チアンー 4 ィル)ァセタミド (70mg, 0. 29mmol)を THF (0. 2mL)に懸濁し、チタンテトライソプロポキシド(86 /z L, 0. 29mmol)、ジフエ-ルシラン(0. 15mL, 0. 76mmol)をカ卩え、室温で攪拌 した。 15時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液を加えて激 しく攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽出した。 有機層を合わせて飽和食塩水で洗浄後、乾燥し濃縮し、得られた組成生物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 12mg (21%)を得た。
[0329] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (チアゾール—2—ィル)チアン— 4—ィル)ブタナミド(参 考化合物 198)の合成
アルゴン雰囲気下、 4— (チアゾール—2—ィル)チアン— 4 ァミン(12mg, 0. 06 mmol)を DMF (3mL)に溶解し、(3R)—4一(2 フルオロフヱ-ル)ー3—(2—メ チル— 2 プロポキシカルボ-ルァミノ)酪酸(18mg, 0. 06mmol)、 ΝΕΜ (12 Ι^
, 0. 09mmol)、 HATU (25mg, 0. 07mmol)をカ卩え、室温で攪拌した。 15時間後 、飽和炭酸水素ナトリウム水溶液を加えジェチルエーテルで抽出し、蒸留水及び飽 和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 28mg (96%)を得た。
[0330] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ) -N- (4— (チアゾール 2—ィル) 1, 1—ジォキソチアン一 4— ィル)ブタナミド (参考化合物 199)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ) N— (4 (チアゾールー 2 ィル)チアンー 4 ィル)ブタナミド(28mg, 0. 06mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (29mg, > 0. l lmmol)を 2回に分けて添カ卩した。 3時間後、チォ硫酸ナトリウム水溶液を加えて 激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、 乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表 題ィ匕合物 1 lmg (37%)を得た。
[0331] ステップ 6 (3R)— 3 ァミノ— 4— (2 フルオロフェ-ル)— N— (4— (チアゾール
2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 57)の合 成
アルゴン雰囲気下、(3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(チアゾールー 2—ィル)—1, 1ージォキソチ アン 4 ィル)ブタナミド(1 lmg, 0. 02mmol)を 10%塩化水素 Zメタノール溶液( 3mL)に溶解し、室温で 5時間攪拌した後濃縮し、表題ィ匕合物 8mg (84%)を得た。
[0332] 実施例 58
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (ピリジン一 2—ィル) 1 , 1ージォキソチアンー4 ィル)ブタナミド '二塩酸塩 (化合物 58)の合成
ステップ 1 4ーォキソチアン O ベンジルォキシム(参考化合物 200)の合成 アルゴン雰囲気下、 4ーォキソチアン(500mg, 4. 30mmol)の 16%含水メタノー ル(60mL)溶液に O べンジロシアミン ·塩酸塩(1. 37g, 8. 60mmol)、酢酸ナトリ ゥム(1. 76g, 21. 5mmol)をカ卩え、加熱還流した。 2時間後放冷し、蒸留水を加え
て酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1. 95g (定量的 )を得た。
[0333] ステップ 2 N ベンジルォキシ 4— (ピリジン— 2—ィル)チアン— 4 ァミン(参考化 合物 201)の合成
アルゴン雰囲気下、 2 ブロモピリジン(890mg, 5. 60mmol)のジェチルエーテ ル(10mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 60M, 4. 2mL, 6. 80mmol)を加えた。 10分間撹拌後、 4—ォキソチアン O ベンジル ォキシム(500mg, 2. 30mmol)のジェチルエーテル(10mL)溶液を滴下し、その 後ゆっくりと 0°Cまで昇温した。 5時間後、飽和食塩水をカ卩えてジクロロメタンで抽出し 、有機層を飽和塩ィ匕アンモ-ゥム水溶液及び飽和食塩水で洗浄後、乾燥し濃縮した 。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 512 mg (75%)を得た。
[0334] ステップ 3 4—(ピリジン 2 ィル)チアンー 4ーァミン(参考化合物 202)の合成 アルゴン雰囲気下、 N ベンジルォキシ 4— (ピリジン— 2—ィル)チアン— 4 アミ ン(300mg, 1. OOmmol)のァセトニトリル(17mL)溶液に蒸留水(lmL)、モリブデ ンへキサカルボニル(264mg, 1. OOmmol)をカ卩え、加熱還流した。 3時間後放冷し 、飽和炭酸水素ナトリウム水溶液を加えてジクロロメタンで抽出し、有機層を飽和食塩 水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で精製し、表題ィ匕合物 60mg (31%)を得た。
[0335] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) -N- (4 (ピリジン 2 ィル)チアンー 4 ィル)ブタナミド(参考 化合物 203)の合成
アルゴン雰囲気下、 4— (ピリジン— 2—ィル)チアン— 4 ァミン(60mg, 0. 31mm ol)の DMF (4mL)溶液に(3R)— 4一(2 フルオロフヱニル) 3—(2—メチルー 2 —プロポキシカルボ-ルァミノ)酪酸(88mg, 0. 30mmol)、 HATU (135mg, 36m mol)、 DIPEA (0. 26mL, 1. 48mmol)をカ卩えて室温で撹拌した。 24時間後、蒸 留水を加えてジェチルエーテルで抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃
縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 119mg (85%)を得た。
[0336] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (ピリジン一 2—ィル) 1 , 1—ジォキソチアン一 4—ィル )ブタナミド (参考化合物 204)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4 (ピリジンー2 ィル)チアンー 4 ィル)ブタナ ミド(33mg, 0. 07mmol)のァセトニトリル(2mL)溶液に、 TPAP ( 1. 2mg, 3. 5 μ mol)、 NMO (29mg, 0. 21mmol)、粉末 MS4A (3. Omg)をカ卩えて室温で撹拌し た。 12時間後反応液を濃縮し、酢酸ェチルを加えてセライト濾過し、再度これを濃縮 した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1 6mg (44%)を得た。
[0337] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (ピリジン一 2 —ィル) 1 , 1—ジォキソチアン一 4—ィル)ブタナミド '二塩酸塩 (ィ匕合物 58)の合成 アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4— (ピリジン— 2—ィル)—1 , 1—ジォキソチアン 4 ィル)ブタナミド(15mg, 0. O3mmol)を 10%塩化水素 Zメタノール溶液(6m L)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕合物 lOmg (定量的)を得た
[0338] 実施例 59
(3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1 , 1—ジォキソチア ンー 4 ィル) 4一(2 フルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 59)の合成 ステップ 1 2—メチル N— (チアン一 4—イリデン) 2 プロパンスルフィンイミド( 参考化合物 205)の合成
アルゴン雰囲気下、 4ーォキソチアン(239mg, 2. Ommol)、 2—メチルー 2 プロ パンスルフィンアミド(純度 97%) (291mg, 2. 3mmol)の THF ( lOmL)溶液にチタ ンテトラエトキシド (0. 85mL, 4. lmmol)をカ卩え、加熱還流した。 30分後放冷し、飽 和食塩水、酢酸ェチルを加えて激しく攪拌した。生じた白色個体を濾別し、酢酸ェチ
ルで抽出した。有機層を乾燥後濃縮し、粗生成物として表題ィ匕合物 410mgを得た。 特に精製することなぐ次の反応に用いた。
[0339] ステップ 2 4 (ベンゾチアゾールー 2 ィル)チアンー 4 アミン'塩酸塩(参考化合 物 206)合成
アルゴン雰囲気下、ベンゾチアゾール(49 L, 0. 45mmol)のトルエン(2mL)溶 液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 57M, 0. 32mL, 0. 50mmol)を滴下した。 15分後、 2—メチル—N— (チアン— 4—イリデン)—2 プロ パンスルフィンイミドの粗生成物(66mg,約 0. 3mmol)のトルエン(2mL)溶液を滴 下した。滴下終了後、室温までゆっくりと昇温し、 6時間後、飽和炭酸水素ナトリウム 水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃 縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。ァルゴ ン雰囲気下、得られた粗精製物をメタノール (2mL)に溶解し、 10%塩ィ匕水素 Zメタ ノール (2mL)を加え、室温で 30分間攪拌した後濃縮し、粗生成物として、表題化合 物 28mgを得た。
[0340] ステップ 3 (3R)—N— (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4 一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタ ナミド (参考化合物 207)の合成
アルゴン雰囲気下、 4 (ベンゾチアゾールー 2 ィル)チアンー 4 アミン'塩酸塩 の粗生成物(28mg,約 0. lmmol)をジクロロメタン(3mL)に懸濁し、(3R)— 4— (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(29 mg, 0. lmmol)、 HOBt (21mg, 0. 16mmol) , NEM (32 μ L, 0. 25mmol)、E DCI'塩酸塩(23mg, 0. 12mmol)をカ卩え、室温で攪拌した。 24時間後、飽和炭酸 水素ナトリウム水溶液を加えクロ口ホルムで抽出し、乾燥後濃縮した。得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 35mg (約 22%)を 得た。
[0341] ステップ 4 (3R)— N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン
4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ -ルァミノ)ブタナミド (参考化合物 208)の合成
(3R) -N- (4- (ベンゾチアゾールー 2 ィル)チアンー 4 ィル)ー4一(2 フル オロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(35mg , 0. 07mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (39m g, >0. 15mmol)を 2回に分けて添カ卩した。 4時間後、チォ硫酸ナトリウム水溶液を 加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽 出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 32mg (86%)を得た。
[0342] ステップ 5 (3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジ ォキソチアンー4 ィル) -4- (2 フルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 59 )の合成
アルゴン雰囲気下、(3R)— N— (4— (ベンゾチアゾール—2—ィル)—1, 1—ジォ キソチアン— 4—ィル)—4— (2 フルオロフェ -ル)—3— (2—メチル—2—プロボ キシカルボ-ルァミノ)ブタナミド(32mg, 0. 06mmol)を 10%塩化水素 Zメタノール 溶液 (5mL)に溶解し、室温で 5時間攪拌した後濃縮し、表題ィ匕合物 25mg (88%) を得た。
[0343] 実施例 60
(3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジォキソチア ンー4ーィル)ー4ー(2, 4, 5 トリフルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 60) の合成
ステップ 1 (3R)— N— (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4 一(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド (参考化合物 209)の合成
アルゴン雰囲気下、 4 (ベンゾチアゾールー 2 ィル)チアンー 4 アミン'塩酸塩 (参考化合物 206)の粗生成物(46mg,約 0. 16mmol)を DMF (4mL)に溶解し、 ( 3R) -4- (2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカル ボ-ルァミノ)酪酸(53mg, 0. 16mmol)、 ΝΕΜ (67 Ι^, 0. 53mmol)、 HATU (6 7mg, 0. 18mmol)を加え、室温で攪拌した。 14時間後、飽和炭酸水素ナトリウム水 溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥
し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 81mg (約 90%)を得た。
[0344] ステップ 2 (3R)— N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン —4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキ シカルボニルァミノ)ブタナミド (参考化合物 210)の合成
(3R) -N- (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミ ド(81mg, 0. 14mmol)をジクロロメタン(10mL)に溶解し、氷冷下、 mCPBA( >6 5%) (118mg, >0. 44mmol)を 2回に分けて添カ卩した。 3. 5時間後、チォ硫酸ナト リウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。ク ロロホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグ ラフィ一で精製し、表題ィ匕合物 69mg (81%)を得た。
[0345] ステップ 3 (3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジ ォキソチアン— 4—ィル)—4— (2, 4, 5—トリフルオロフェ -ル)ブタナミド '塩酸塩( 化合物 60)の合成
アルゴン雰囲気下、(3R)— N— (4— (ベンゾチアゾール—2—ィル)—1, 1—ジォ キソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(94mg, 0. 16mmol)を 10%塩化水素 Z メタノール溶液(10mL)に溶解し、室温で 2時間攪拌した後濃縮し、表題化合物 79 mg (94%)を得た。
[0346] 実施例 61
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (6—メトキシベンゾチア ゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 61 )の合成
ステップ 1 6—メトキシベンゾチアゾール(参考化合物 211)の合成
2 アミノー 6—メトキシベンゾチアゾール(1. Og, 5. 55mmol)のホスフィン酸(20 mL)溶液を— 15°Cに冷却し、亜硝酸ナトリウム(2. 68g, 38. 8mmol)の蒸留水(3 mL)溶液をゆっくりと滴下した。 6時間後、蒸留水を加えて希釈した後飽和炭酸水素
ナトリウム水溶液を加えて中和した。酢酸ェチルで抽出し、有機層を飽和食塩水にて 洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題ィ匕合物 717mg (78%)を得た。
[0347] ステップ 2 N— (4— (6—メトキシベンゾチアゾールー 2 ィル)チアンー 4 ィル) 2—メチル - 2-プロパンスルフィンアミド(参考化合物 212)の合成
アルゴン雰囲気下、 6—メトキシベンゾチアゾール(217mg, 1. 3mmol)の THF (5 mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 74 mL, 1. 2mmol)を滴下した。 15分後、 2—メチルー N—(チアンー4 イリデン)ー2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(237mg,約 1. lmmol) の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 2. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 104mg (約 25%)を得た。
[0348] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (6—メトキシベンゾチアゾ 一ルー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド (参考化合物 213)の合成
アルゴン雰囲気下、 N— (4—(6—メトキシベンゾチアゾールー 2 ィル)チアンー 4 —ィル)一2—メチル 2 プロパンスルフィンアミド(23mg, 0. 06mmol)をメタノー ル(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 2時間攪拌し た。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R) —4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) 酪酸(18mg, 0. 06mmol) , ΝΕΜ (27 μ L, 0. 21mmol)、 HATU (25mg, 0. 07 mmol)を加え、室温で攪拌した。 15時間後、飽和炭酸水素ナトリウム水溶液を加え ジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 31mg (93%)を得た。
[0349] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (6—メトキシベンゾチアゾ 一ルー 2 ィル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2—プロボ
キシカルボニルァミノ)ブタナミド (参考化合物 214)の合成
(3R)—4— (2 フルオロフェ -ル) N— (4— (6—メトキシベンゾチアゾール 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタ ナミド(31mg, 0. 06mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( > 65%) (38mg, >0. 14mmol)を 2回に分けて添カ卩した。 4時間後、チォ硫酸ナトリ ゥム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ 口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合 22mg (67%)を得た。
[0350] ステップ 5 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (6—メトキシ ベンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (化合物 61)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (6—メトキシべ ンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(22mg, 0. 04mmol)を 10%塩化水素 Zメタノール溶液 (5mL)に溶解し、室温で 4時間攪拌した後濃縮し、表題化合物 18 mg (92%)を得た。
[0351] 実施例 62
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (6 ヒドロキシベンゾチ ァゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 62)の合成
ステップ 1 (3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (6—メトキシ ベンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド(ィ匕合物 61) (フリー塩基)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (6—メトキシべ ンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 214) (68mg, 0. l lmmol) をジクロロメタン(5mL)に溶解し、 78°Cに冷却して、 1. OM三臭化ホウ素 Zジクロ ロメタン溶液(0. 35mL, 0. 35mmol)を滴下した。 4. 5時間後、クロ口ホルムで希釈
し、氷冷下、飽和炭酸水素ナトリウム水溶液をゆっくりと加えた。分液した後、クロロホ ルムで抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合 39mg (69%)を得た。
[0352] ステップ 2 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (6 ヒドロキ シベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸 塩 (ィ匕合物 62)の合成
アルゴン雰囲気下、 (3R)—3 ァミノ— 4— (2 フルオロフヱ-ル) N— (4— (6 ーメトキシベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミ ド(39mg, 0. 08mmol)をジクロロメタン(3mL)に溶解し、氷冷下、 1. OM三臭化ホ ゥ素 Zジクロロメタン溶液(0. 24mL, 0. 24mmol)を滴下した。 3. 5時間後、更に三 臭化ホウ素 Zジクロロメタン溶液 (0. 24mL, 0. 24mmol)を滴下し、 2時間攪拌した 。反応液をクロ口ホルムで希釈し、氷冷下、飽和炭酸水素ナトリウム水溶液をゆっくり と加えた。分液した後、クロ口ホルムで抽出し、乾燥後濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題化合物(フリー塩基) 27mg (71%) を得た。表題ィ匕合物(フリー塩基)をメタノールに溶解し、 10%塩ィ匕水素 Zメタノール 溶液を滴下して塩化した。この溶液を濃縮することにより、表題ィ匕合物 (塩酸塩) 21m gを得た。
[0353] 実施例 63
(3R)—3 ァミノ一 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (6—メトキシべ ンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 63)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6—メトキシべ ンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシ力 ルポニルァミノ)ブタナミド (参考化合物 215)の合成
アルゴン雰囲気下、 N— (4—(6—メトキシベンゾチアゾールー 2 ィル)チアンー 4 ーィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 212) (104mg, 0. 27mmol)をメタノール(5mL)に溶解し、 10%塩化水素 Zメタノール(5mL)を加え、 室温で 1時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (5m
L)に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プ 口ポキシカルボ-ルァミノ)酪酸(92mg, 0. 28mmol)、 NEM (0. 12mL, 0. 94m mol)、 HATU (114mg, 0. 30mmol)をカ卩え、室温で攪拌した。 17時間後、飽和炭 酸水素ナトリウム水溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水 にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で精製し、表題ィ匕合物 127mg (79%)を得た。
[0354] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6—メトキシべ ンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 216)の合成
(3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6—メトキシベンゾチアゾ 一ルー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド(127mg, 0. 21mmol)をジクロロメタン(lOmL)に溶解し、氷冷下、 mCPBA( >65%) (116mg, >0. 44mmol)を 2回に分けて添カ卩した。 4時間後、 チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶 液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合 120mg (90%)を得た。
[0355] ステップ 3 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 ーメトキシベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミ ド '塩酸塩 (ィ匕合物 63)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (6— メトキシベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3— (2 —メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(120mg, 0. 19mmol)を 10 %塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 13時間攪拌した後濃縮し、 表題ィ匕合物 106mg (98%)を得た。
[0356] 実施例 64
(3R)—3 アミノー 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (6 ヒドロキシ ベンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (化合物 64)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (6— メトキシベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 216) (lOOmg, 0. 16mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 12時間攪 拌した。飽和炭酸水素ナトリウム水溶液を加え、クロ口ホルムにて抽出し、乾燥後濃縮 した。得られた粗生成物を、アルゴン雰囲気下、ジクロロメタン(3mL)に溶解し、氷冷 下、 1. OM三臭化ホウ素 Zジクロロメタン溶液(0. 91mL, 0. 91mmol)を滴下した。 3時間後、アンモニア水をゆっくりと加えた後、クロ口ホルムで抽出し、乾燥後濃縮し た。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 (フ リー塩基) 50mg (64%)を得た。表題ィ匕合物(フリー塩基)をメタノールに溶解し、 10 %塩ィ匕水素 Zメタノール溶液を滴下して塩ィ匕した。この溶液を濃縮することにより、表 題化合物 (塩酸塩) 50mgを得た。
[0357] 実施例 65
(3R)— 3 ァミノ一 N— (4— (6 クロ口べンゾチアゾール 2—ィル) 1, 1—ジォ キソチアン 4 ィル) 4一(2 フルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 65) の合成
ステップ 1 6 クロ口べンゾチアゾール(参考化合物 217)の合成
2 ァミノ一 6 クロ口べンゾチアゾール(1. Og, 5. 4mmol)を酢酸(20mL)、 50 %硫酸(lOmL)に懸濁し、 15°Cに冷却した。亜硝酸ナトリウム (450mg, 6. 5mm ol)の蒸留水(1. 5mL)溶液をゆっくりと滴下し、 40分間攪拌した。酸化銅 (I) (429 mg, 3. Ommol)をホスフィン酸(30mL)に懸濁し、 15°Cに冷却したところに、反 応液を加え、 20時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温)。蒸留水 で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、セライト濾過した。濾液を酢 酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシ リカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 599mg (65%)を得た。
[0358] ステップ 2 (3R)—N— (4— (6 クロ口べンゾチアゾール—2—ィル)チアン— 4—ィ ル)— 4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルアミ ノ)ブタナミド (参考化合物 218)の合成
アルゴン雰囲気下、 6 クロ口べンゾチアゾール(95mg, 0. 56mmol)の THF (5m L)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 57M, 0. 31mL , 0. 49mmol)を滴下した。 15分後、 2—メチル—N— (チアン— 4—イリデン)—2— プロパンスルフィンイミド(参考化合物 205)の粗生成物(102mg,約 0. 46mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 2時間後、飽 和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水 にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール (2mL)に溶解し 、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 19時間攪拌した。アルゴン雰囲 気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)—4— (2—フルォ 口フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(22mg, 0. 0 8mmol)、 ΝΕΜ (32 Ι^, 0. 25mmol)、 HATU (31mg, 0. 08mmol)を加え、室 温で攪拌した。 16時間後、飽和炭酸水素ナトリウム水溶液を加えジェチルエーテル で抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 35mg (約 14%)を得た。
[0359] ステップ 3 (3R)—N— (4— (6 クロ口べンゾチアゾールー 2—ィル) 1, 1ージォ キソチアン— 4—ィル)—4— (2 フルオロフェ -ル)—3— (2—メチル—2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 219)の合成
(3R)— N— (4— (6 クロ口べンゾチアゾール—2—ィル)チアン— 4—ィル)—4— (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミ ド(35mg, 0. 06mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65 %) (39mg, >0. 15mmol)を添カ卩した。 2. 5時間後、チォ硫酸ナトリウム水溶液を 加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽 出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合 30mg (81%)を得た。
[0360] ステップ 4 (3R)— 3 ァミノ一 N— (4— (6 クロ口べンゾチアゾール 2—ィル)一 1, 1ージォキソチアンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '塩酸塩( 化合物 65)の合成
アルゴン雰囲気下、(3R) -N- (4— (6 クロ口べンゾチアゾール—2—ィル)—1 , 1ージォキソチアンー4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(30mg, 0. 05mmol)を 10%塩化水素 Z メタノール溶液(5mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題ィ匕合物 27m g (定量的)を得た。
[0361] 実施例 66
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (4—メチルベンゾチアゾ 一ルー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 66) の合成
ステップ 1 4 メチルベンゾチアゾール(参考化合物 220)の合成
2 アミノー 4 メチルベンゾチアゾール(1. Og, 6. lmmol)を 50%硫酸(lOmL) 、 85%リン酸(lOmL)に懸濁し、 15°Cに冷却した。亜硝酸ナトリウム(506mg, 7. 3mmol)の蒸留水(1. 5mL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、—1 5°Cに冷却したホスフィン酸(30mL)を加え、 16時間攪拌した (氷浴中の氷が溶ける に従い、次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し 、酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 481mg (53%)を得た。
[0362] ステップ 2 (3R)—4 (2 フルオロフェ -ル)—N— (4— (4 メチルベンゾチアゾ 一ルー 2 ィル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 221)の合成
アルゴン雰囲気下、 4 メチルベンゾチアゾール(83mg, 0. 56mmol)の THF (5 mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 57M, 0. 32 mL, 0. 50mmol)を滴下した。 20分後、 2—メチル—N— (チアン— 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(lOOmg,約 0. 46mmol )の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 2. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール(2mL)に溶
解し、 10%塩ィ匕水素 Zメタノール(2mL)をカ卩え、室温で 6時間攪拌した。アルゴン雰 囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)—4— (2—フル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(14mg, 0. O5mmol)、 ΝΕΜ (21 Ι^, 0. 17mmol)、 HATU (20mg, 0. 05mmol)を加え、 室温で攪拌した。 12時間後、飽和炭酸水素ナトリウム水溶液を加えジェチルエーテ ルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮して得られた粗生成 物をシリカゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物をジクロロメ タン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (24mg, >0. 09mmol)を添カロ した。 5分後氷浴を外し、室温で 1時間攪拌した。チォ硫酸ナトリウム水溶液を加えて 激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、 乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表 題ィ匕合 17mg (約 7%)を得た。
[0363] ステップ 3 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (4—メチルベ ンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 66)の合成
アルゴン雰囲気下、 (3R) -4- (2 フルオロフヱ-ル) N— (4—(4 メチルベ ンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(17mg, 0. 03mmol)を 10%塩化水素 Zメタノール溶液 (3mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題化合物 1 4mg (93%)を得た。
[0364] 実施例 67
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (6—メチルベンゾチアゾ 一ルー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 67) の合成
ステップ 1 6 メチルベンゾチアゾール(参考化合物 222)の合成
2 アミノー 6—メチルベンゾチアゾール(1. Og, 6. lmmol)を TFA(10mL)、 50 %硫酸(lOmL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(507mg, 7. 3mm ol)の蒸留水(1. 5mL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、 15°Cに
冷却したホスフィン酸(30mL)を加え、 13時間攪拌した (氷浴中の氷が溶けるに従い 、次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸 ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 530mg (58%)を得た。
[0365] ステップ 2 (3R)—4 (2 フルオロフェ -ル) N— (4— (6 メチルベンゾチアゾ 一ルー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド (参考化合物 223)の合成
アルゴン雰囲気下、 6 メチルベンゾチアゾール(85mg, 0. 57mmol)の THF (5 mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 57M, 0. 32 mL, 0. 50mmol)を滴下した。 20分後、 2—メチル—N— (チアン— 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(lOOmg,約 0. 46mmol )の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 2. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール(2mL)に溶 解し、 10%塩ィ匕水素 Zメタノール(2mL)をカ卩え、室温で 5. 5時間攪拌した。ァルゴ ン雰囲気下、濃縮して得られた粗生成物を DMF (4mL)に溶解し、(3R)— 4一(2— フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(19mg , 0. 06mmol) , ΝΕΜ (28 μ L, 0. 22mmol)、 HATU (27mg, 0. 07mmol)をカロ え、室温で攪拌した。 12時間後、飽和炭酸水素ナトリウム水溶液を加えジェチルェ 一テルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗 生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 22mg (約 8%)を 得た。
[0366] ステップ 3 (3R)—4 (2 フルオロフェ -ル)—N— (4— (6 メチルベンゾチアゾ 一ルー 2 ィル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 224)の合成
(3R) -4- (2 フルオロフェ -ル) N— (4—(6 メチルベンゾチアゾールー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタ
ナミド(22mg, 0. 04mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( > 65%) (25mg, >0. O9mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌し た。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水 溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合 17mg (75%)を得た。
[0367] ステップ 4 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (6—メチルベ ンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 67)の合成
アルゴン雰囲気下、 (3R) -4- (2 フルオロフヱ-ル) N— (4—(6 メチルベ ンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(17mg, 0. O3mmol)を 10%塩化水素 Zメタノール溶液 (3mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題化合物 14 mg (93%)を得た。
[0368] 実施例 68
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) N— (4— (4—メトキシベンゾチア ゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 68 )の合成
ステップ 1 4ーメトキシベンゾチアゾール(参考化合物 225)の合成
2 アミノー 4—メトキシベンゾチアゾール(502mg, 2. 8mmol)を TFA(5mL)、 5 0%硫酸(5mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(231mg, 3. 3mm ol)の蒸留水(lmL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、 15°Cに冷 却したホスフィン酸(15mL)を加え、 13時間攪拌した (氷浴中の氷が溶けるに従!、、 次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸ェ チルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合物 155mg (34%)を得た。
[0369] ステップ 2 N— (4— (4—メトキシベンゾチアゾールー 2 ィル)チアンー 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考化合物 226)の合成
アルゴン雰囲気下、 4ーメトキシベンゾチアゾール(90mg, 0. 54mmol)の THF (5
mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 31 mL, 0. 49mmol)を滴下した。 20分後、 2—メチル—N— (チアン— 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(lOlmg,約 0. 46mmol )の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 4. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 48mg (約 27%)を得た。
[0370] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (4—メトキシベンゾチアゾ 一ルー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド (参考化合物 227)の合成
アルゴン雰囲気下、 N— (4—(4ーメトキシベンゾチアゾールー 2 ィル)チアンー 4 —ィル)一2—メチル 2 プロパンスルフィンアミド(48mg, 0. 12mmol)をメタノー ル(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 3. 5時間攪 拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、 ( 3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルアミ ノ)酪酸(36mg, 0. 12mmol)、 ΝΕΜ (50 Ι^, 0. 40mmol)、 HATU (50mg, 0. 13mmol)を加え、室温で攪拌した。 18時間後、飽和炭酸水素ナトリウム水溶液をカロ ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した 。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 51m g (75%)を得た。
[0371] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (4—メトキシベンゾチアゾ 一ルー 2 ィル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 228)の合成
(3R) -4- (2 フルオロフェ -ル) N— (4—(4ーメトキシベンゾチアゾールー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタ ナミド(51mg, 0. 09mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( > 65%) (50mg, >0. 19mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌し た。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水
溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合 43mg (80%)を得た。
[0372] ステップ 5 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (4—メトキシ ベンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (化合物 68)の合成
アルゴン雰囲気下、 (3R) -4- (2 フルオロフヱ-ル) N— (4— (4—メトキシべ ンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(43mg, 0. 07mmol)を 10%塩化水素 Zメタノール溶液(5mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題化合物 37 mg (96%)を得た。
[0373] 実施例 69
(3R)— 3 ァミノ— N— (4— (6 フルォロベンゾチアゾール—2—ィル)—1, 1—ジ ォキソチアンー4 ィル) -4- (2 フルオロフェ -ル)ブタナミド '塩酸塩(ィ匕合物 69 )の合成
ステップ 1 6 フルォロベンゾチアゾール(参考化合物 229)の合成
2 アミノー 6 フルォロベンゾチアゾール(503mg, 3. Ommol)を TFA(5mL)、 50%硫酸(5mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(247mg, 3. 6m mol)の蒸留水(lmL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、 15°Cに 冷却したホスフィン酸(15mL)を加え、 13時間攪拌した (氷浴中の氷が溶けるに従!、 、次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸 ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 204mg (45%)を得た。
[0374] ステップ 2 N— (4— (6 フルォロベンゾチアゾールー 2 ィル)チアンー 4 ィル)
2 メチル 2 プロパンスルフィンアミド(参考化合物 230)の合成
アルゴン雰囲気下、 6 フルォロベンゾチアゾール(84mg, 0. 55mmol)の THF ( 5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 31 mL, 0. 49mmol)を滴下した。 15分後、 2—メチル—N— (チアン— 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(lOOmg,約 0. 46mmol
)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 4. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合物 41mg (約 24%)を得た。
[0375] ステップ 3 (3R)—N— (4— (6 フルォロベンゾチアゾールー 2 ィル)チアンー 4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)ブタナミド (参考化合物 231)の合成
アルゴン雰囲気下、 N—(4一(6 フルォロベンゾチアゾールー 2 ィル)チアンー 4—ィル) 2—メチル 2 プロパンスルフィンアミド(41mg, 0. l lmmol)をメタノ ール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 16時間攪 拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、 ( 3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルアミ ノ)酪酸(33mg, 0. l lmmol) , NEM (45 ^ L, 0. 35mmol)、 HATU (45mg, 0. 12mmol)を加え、室温で攪拌した。 17時間後、飽和炭酸水素ナトリウム水溶液をカロ ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮して 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 49mg (82%)を得た。
[0376] ステップ 4 (31¾—?^ー(4ー(6—フルォロべンゾチァゾールー2—ィル)ー1, 1ージ ォキソチアン— 4—ィル)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)ブタナミド (参考化合物 232)の合成
(3R) N— (4—(6 フルォロベンゾチアゾールー 2 ィル)チアンー 4 ィル) 4一(2 フルオロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタ ナミド(49mg, 0. 09mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( > 65%) (47mg, >0. 19mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌し た。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水 溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合 40mg (78%)を得た。
[0377] ステップ 5 (3R)— 3 ァミノ一 N— (4— (6 フルォロベンゾチアゾール 2—ィル)
- 1, 1ージォキソチアンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '塩酸塩 (化合物 69)の合成
アルゴン雰囲気下、(3R) -N- (4— (6—フルォロベンゾチアゾール—2—ィル) - 1, 1ージォキソチアンー4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2 メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(40mg, 0. 07mmol)を 10%塩化水 素 Zメタノール溶液 (5mL)に溶解し、室温で 3時間攪拌した後濃縮し、表題化合物 35mg (98%)を得た。
[0378] 実施例 70
(3R)—3 アミノー N— (4— (4 クロ口べンゾチアゾール 2—ィル) 1, 1—ジォ キソチアン 4 ィル) 4一(2 フルオロフェ -ル)ブタナミド '塩酸塩 (ィ匕合物 70) の合成
ステップ 1 4 クロ口べンゾチアゾール(参考化合物 233)の合成
2 アミノー 4 クロ口べンゾチアゾール(1. Og, 5. 4mmol)を酢酸(20mL)、 50 %硫酸(lOmL)に懸濁し、 15°Cに冷却した。亜硝酸ナトリウム (448mg, 6. 5mm ol)の蒸留水(1. 5mL)溶液をゆっくりと滴下し、 30分間攪拌した。酸化銅 (I) (426 mg, 3. Ommol)をホスフィン酸(30mL)に懸濁し、 15°Cに冷却したところに、反 応液を加え、 22時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温)。蒸留水 で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、セライト濾過した。濾液を酢 酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシ リカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 378mg (41%)を得た。
[0379] ステップ 2 (3R)—N— (4— (4 クロ口べンゾチアゾール 2—ィル)チアン一 4—ィ ル)— 4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルアミ ノ)ブタナミド (参考化合物 234)の合成
アルゴン雰囲気下、 4 クロ口べンゾチアゾール(93mg, 0. 55mmol)の THF (5m L)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 31mL , 0. 49mmol)を滴下した。 15分後、 2—メチル—N— (チアン— 4—イリデン)—2— プロパンスルフィンイミド(参考化合物 205)の粗生成物(106mg,約 0. 48mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 4時間後、飽
和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水 にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール (2mL)に溶解し 、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 18時間攪拌した。アルゴン雰囲 気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)—4— (2—フルォ 口フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(29mg, 0. 1 Ommol) , ΝΕΜ (40 μ L, 0. 32mmol)、 HATU (40mg, 0. l lmmol)を加え、室 温で攪拌した。 18時間後、飽和炭酸水素ナトリウム水溶液を加えジェチルエーテル で抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 45mg (約 17%)を得た。
[0380] ステップ 3 (3R)— N— (4— (4 クロ口べンゾチアゾール 2—ィル) 1, 1—ジォ キソチアン— 4—ィル)—4— (2 フルオロフェ -ル)—3— (2—メチル—2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 235)の合成
(3R)— N— (4— (4 クロ口べンゾチアゾーノレ一 2—ィノレ)チアン一 4—ィノレ) 4— (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミ ド(43mg, 0. 08mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65 %) (50mg, >0. 16mmol)を添加した。 5分後氷浴を外し、室温で 1時間攪拌した 。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶 液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合 34mg (71%)を得た。
[0381] ステップ 4 (3R)— 3 アミノー N— (4— (4 クロ口べンゾチアゾール 2—ィル)一 1, 1ージォキソチアンー4 ィル)ー4一(2 フルオロフェ -ル)ブタナミド '塩酸塩( 化合物 70)の合成
アルゴン雰囲気下、 (3R)— N— (4— (4 クロ口べンゾチアゾール—2—ィル)—1 , 1ージォキソチアンー4 ィル)ー4一(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(34mg, 0. 06mmol)を 10%塩化水素 Z メタノール溶液(5mL)に溶解し、室温で 13時間攪拌した後濃縮し、表題化合物 30 mg (98%)を得た。
[0382] 実施例 71
(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (4— (5, 6 ジメチルベンゾ チアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (ィ匕合 物 71)の合成
ステップ 1 5, 6 ジメチルベンゾチアゾール(参考化合物 236)の合成
2 アミノー 5, 6 ジメチルベンゾチアゾール(1. Og, 5. 6mmol)を TFA (lOmL) 、 50%硫酸(lOmL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム (467mg, 6. 8 mmol)の蒸留水(1. 5mL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、—15 °Cに冷却したホスフィン酸(30mL)を加え、 14時間攪拌した (氷浴中の氷が溶けるに 従い、次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、 酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 484mg (53%)を得た。
[0383] ステップ 2 N— (4— (5, 6 ジメチルベンゾチアゾールー 2 ィル)チアンー 4ーィル )一 2—メチルー 2 プロパンスルフィンアミド(参考化合物 237)の合成
アルゴン雰囲気下、 5, 6 ジメチルベンゾチアゾール(9 lmg, 0. 55mmol)の TH F (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 31mL, 0. 49mmol)を滴下した。 15分後、 2—メチル N— (チアン一 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(108mg,約 0. 49m mol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 4. 5 時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽 和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 34mg (約 18%)を得た。
[0384] ステップ 3 (3R)—4— (2 フルオロフェ -ル)—N— (4— (5, 6 ジメチルベンゾチ ァゾールー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 238)の合成
アルゴン雰囲気下、 N— (4— (5, 6 ジメチルベンゾチアゾールー 2 ィル)チアン —4—ィル)—2—メチル—2 プロパンスルフィンアミド(34mg, 0. O9mmol)をメタ ノール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 3時間攪
拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、 ( 3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルアミ ノ)酪酸(27mg, 0. 09mmol)、 ΝΕΜ (37 Ι^, 0. 29mmol)、 HATU (37mg, 0. lOmmol)を加え、室温で攪拌した。 4. 5時間後、飽和炭酸水素ナトリウム水溶液を 加えジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮し て得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 33m g (66%)を得た。
[0385] ステップ 4 (3R)—4— (2 フルオロフェ -ル)—N— (4— (5, 6 ジメチルベンゾチ ァゾールー 2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)ブタナミド (参考化合物 239)の合成
(3R)—4— (2 フルオロフェ -ル) N— (4— (5, 6 ジメチルベンゾチアゾール 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ) ブタナミド(33mg, 0. 06mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA ( >65%) (32mg, >0. 12mmol)を添カ卩した。 5分後氷浴を外し、室温で 1. 5時間 攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリ ゥム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシ リカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 31mg (90%)を得た。
[0386] ステップ 5 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (5, 6 ジメ チルベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩 酸塩 (化合物 71)の合成
アルゴン雰囲気下、(3R)— 4— (2 フルオロフヱ-ル)— N— (4— (5, 6 ジメチ ルベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ー3—(2—メチ ルー 2 プロポキシカルボ-ルァミノ)ブタナミド(31mg, 0. 05mmol)を 10%塩ィ匕 水素 Zメタノール溶液(5mL)に溶解し、室温で 12時間攪拌した後濃縮し、表題ィ匕 合物 24mg (87%)を得た。
[0387] 実施例 72
(3R)— 3 ァミノ一 N— (4— (6 シァノベンゾチアゾール 2—ィル) 1, 1—ジォ キソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ブタナミド '塩酸塩(ィ匕
合物 72)の合成
ステップ 1 N—(4 シァノフ ニル)チォ尿素(参考化合物 240)の合成
4—シァノア-リン(497mg, 4. lmmol)を 4M塩酸(8. 5mL)に溶解し、チオシァ ン酸アンモ-ゥム(2. 25g, 30. Ommol)をカ卩え、加熱還流した。 18時間後放冷し、 酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を メタノール含有ジクロロメタンにより洗浄し、表題ィ匕合物 324mg (43%)を得た。
[0388] ステップ 2 6 シァノベンゾチアゾール(参考化合物 241)の合成
N— (4 シァノフエ-ル)チォ尿素(320mg, 1. 8mmol)をクロ口ホルム(10mL) に懸濁し、氷冷下、臭素(0. 21mL, 4. lmmol)をゆっくりと滴下した。滴下終了後、 1時間室温で攪拌した後、 6. 5時間加熱還流した。生成した固体を濾別し、濾物をメ タノールに懸濁した。ここに飽和炭酸水素ナトリウム水溶液をカ卩え、酢酸ェチルで抽 出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を TFA (2. 5mL) 、 50%硫酸(2. 5mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(124mg, 1. 8mmol)の蒸留水(0. 3mL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、—1 5°Cに冷却したホスフィン酸(10mL)を加え、 18時間攪拌した (氷浴中の氷が溶ける に従い、次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し 、酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 68mg (24%)を得た。
[0389] ステップ 3 N— (4— (6 シァノベンゾチアゾールー 2 ィル)チアンー 4ーィル)ー2 ーメチルー 2 プロパンスルフィンアミド(参考化合物 242)の合成
アルゴン雰囲気下、 6 シァノベンゾチアゾール(68mg, 0. 42mmol)の THF (5 mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 25 mL, 0. 40mmol)を滴下した。 10分後、 2—メチルー N—(チアンー4 イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(85mg,約 0. 39mmol) の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 3. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 14mg (約 10%)を得た。
[0390] ステップ 4 (3R)—N— (4— (6 シァノベンゾチアゾール—2—ィル)チアン— 4— ィル)ー4ー(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 243)の合成
アルゴン雰囲気下、 N— (4—(6 シァノベンゾチアゾールー 2 ィル)チアンー 4 —ィル)一2—メチル 2 プロパンスルフィンアミド(14mg, 0. 04mmol)をメタノー ル(lmL)に溶解し、 10%塩ィ匕水素 Zメタノール(2mL)をカ卩え、室温で 1. 5間攪拌 した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (2mL)に溶解し、 (3R )一 4一(2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ)酪酸(13mg, 0. 04mmol)、 NEM T /z L, 0. 13mmol)、 HATU ( 17m g, 0. 04mmol)をカロえ、室温で攪拌した。 9. 5時間後、(3R)— 4— (2, 4, 5 トリフ ルォロフエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(13mg, 0. 04mmol)、 NEM T /z L, 0. 13mmol)、 HATU (17mg, 0. 04mmol)を追カロ し、更に 6時間攪拌した。飽和炭酸水素ナトリウム水溶液を加えジェチルエーテルで 抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 20mg (90%)を得た。
[0391] ステップ 5 (3R)— N— (4— (6 シァノベンゾチアゾール 2—ィル) 1, 1—ジォ キソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 244)の合成
(3R)— N— (4— (6—シァノベンゾチアゾール—2—ィル)チアン— 4—ィル)—4 一(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド(20mg, 0. 03mmol)をジクロロメタン(3mL)に溶解し、氷冷下、 mC PBA( >65%) (21mg, >0. 08mmol)を添カ卩した。 5分後氷浴を外し、室温で 30 分間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素 ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 15mg (71%)を得た。
[0392] ステップ 6 (3R)— 3 ァミノ一 N— (4— (6 シァノベンゾチアゾール 2—ィル)一 1, 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブタナミド' 塩酸塩 (ィ匕合物 72)の合成
アルゴン雰囲気下、(3R) -N- (4— (6 シァノベンゾチアゾール—2—ィル)—1 , 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(15mg, 0. 02mmol)を 10%塩 化水素 Zメタノール溶液(3mL)に溶解し、室温で 13時間攪拌した後濃縮し、表題 化合物 12mg (89%)を得た。
[0393] 実施例 73
(3R)— 3 ァミノ一 N— (4— (6 トリフルォロメトキシベンゾチアゾール 2—ィル) —1, 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブタナミ ド '塩酸塩 (ィ匕合物 73)の合成
ステップ 1 N—(4 トリフルォロメトキシフヱニル)チォ尿素(参考化合物 245)の合 成
4 トリフルォロメトキシァ-リン(0. 5mL, 3. 7mmol)を 4M塩酸(8. 5mL)に溶解 し、チォシアン酸アンモ-ゥム(723mg, 7. 4mmol)をカ卩え、加熱還流した。 18. 5 時間後放冷し、蒸留水で希釈し、クロ口ホルム Zエタノール =3Zlで抽出し、 1M塩 酸で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で精製し、表題ィ匕合物 438mg (50%)を得た。
[0394] ステップ 2 6 トリフルォロメトキシベンゾチアゾール(参考化合物 246)の合成
N— (4 トリフルォロメトキシフエ-ル)チォ尿素(884mg, 3. 7mmol)をクロ口ホル ム(10mL)に懸濁し、氷冷下、臭素(0. 42mL, 8. 2mmol)のクロ口ホルム(5mL) 溶液をゆっくりと滴下した。滴下終了後、 30分間室温で攪拌した後、 3. 5時間加熱 還流した。生成した固体を濾別し、濾物をメタノールに懸濁した。ここに飽和炭酸水 素ナトリウム水溶液を加え、酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮 した。得られた粗生成物を TFA(5mL)、 50%硫酸(5mL)に溶解し、 15°Cに冷却 した。亜硝酸ナトリウム(180mg, 2. 6mmol)の蒸留水(0. 5mL)溶液をゆっくりと滴 下し、 30分間攪拌した。ここ〖こ、 15°Cに冷却したホスフィン酸(lOmL)をカ卩え、 12 時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温)。蒸留水で希釈後、氷冷 下水酸ィ匕ナトリウム水溶液にて中和し、酢酸ェチルで抽出し、飽和食塩水で洗浄後 、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、
表題化合物 136mg ( 17%)を得た。
[0395] ステップ 3 N— (4— (6 トリフルォロメトキシベンゾチアゾール 2—ィル)チアン一 4 ィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 247)の合成 アルゴン雰囲気下、 6 トリフルォロメトキシベンゾチアゾール(136mg, 0. 62mm ol)の THF (5mL)溶液を― 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液( 1. 59M, 0. 35mL, 0. 57mmol)を滴下した。 20分後、 2—メチル N— (チアン一 4 イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物( 113mg, 約 0. 52mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇 温し、 2時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有 機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 76mg (約 34%)を得た。
[0396] ステップ 4 (3R)—N— (4— (6 トリフルォロメトキシベンゾチアゾール—2—ィル) チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)ブタナミド(参考化合物 248)の合成
アルゴン雰囲気下、 N— (4— (6—トリフルォロメトキシベンゾチアゾール—2—ィル )チアン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(76mg, 0. 17mmol )を 10%塩ィ匕水素 Zメタノール(3mL)に溶解し、室温で 30分間攪拌した。アルゴン 雰囲気下、濃縮して得られた粗生成物を DMF (5mL)に溶解し、(3R)—4— (2, 4, 5 トリフルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸( 58mg, 0. 17mmol)、 ΝΕΜ (77 Ι^, 0. 61mmol)、 HATU (74mg, 0. 19mmol )を加え、室温で攪拌した。 13時間後、飽和炭酸水素ナトリウム水溶液を加えジェチ ルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られ た粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 lOlmg (89 %)を得た。
[0397] ステップ 5 (3R)—N— (4— (6 トリフルォロメトキシベンゾチアゾール 2—ィル) —1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 249)の合成
(3R)— N— (4— (6—トリフルォロメトキシベンゾチアゾール—2—ィル)チアン— 4
—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ)ブタナミド(lOlmg, 0. 16mmol)をジクロロメタン(5mL)に溶解し、 氷冷下、 mCPBA( >65%) (91mg, >0. 34mol)を添カ卩した。 5分後氷浴を外し、 室温で 30分間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和 炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 89mg (84%)を 得た。
[0398] ステップ 6 (3R)— 3 ァミノ一 N— (4— (6 トリフルォロメトキシベンゾチアゾール — 2—ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ- ル)ブタナミド ·塩酸塩 (化合物 73)の合成
アルゴン雰囲気下、(3R)— N— (4— (6—トリフルォロメトキシベンゾチアゾール— 2—ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(89mg, 0. 13mmol )を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 19時間攪拌した後濃 縮し、表題ィ匕合物 76mg (94%)を得た。
[0399] 実施例 74
(3R)— 3 ァミノ一 N— (4— (5, 7 ジクロロべンゾチアゾール 2—ィル) 1, 1— ジォキソチアンー4ーィル)ー4ー(2, 4, 5 トリフルオロフヱ-ル)ブタナミド '塩酸塩 (化合物 74)の合成
ステップ 1 5, 7 ジクロ口べンゾチアゾール(参考化合物 250)の合成
アルゴン雰囲気下、 3, 5 ジクロロフエ-ルイソチオシアナ一ト(1. Og, 4. 9mmol )を THF (lOmL)に溶解し、氷冷下、 7Nアンモニア Zメタノール溶液(2. lmL, 14 . 7mmol)を加え、室温で 3時間攪拌した。酢酸ェチルで抽出し、飽和食塩水で洗浄 後、乾燥し濃縮した。得られた粗生成物をクロ口ホルム(15mL)に懸濁し、氷冷下、 臭素(0. 54mL, 10. 5mmol)のクロ口ホルム(5mL)溶液をゆっくりと滴下した。滴 下終了後、 30分間室温で攪拌した後、 3. 5時間加熱還流した。生成した固体を濾 別し、濾物をメタノールに懸濁した。ここに飽和炭酸水素ナトリウム水溶液を加え、酢 酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を TF
A (10mL)、 50%硫酸(lOmL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(31 9mg, 4. 8mmol)の蒸留水(0. 7mL)溶液をゆっくりと滴下し、 30分間攪拌した。こ こに、 15°Cに冷却したホスフィン酸(10mL)をカ卩え、 14時間攪拌した (氷浴中の氷 が溶けるに従い、次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液に て中和し、酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 383mg (38%) を得た。
[0400] ステップ 2 N— (4— (5, 7 ジクロ口べンゾチアゾールー 2 ィル)チアンー 4ーィル )一 2 メチル - 2-プロパンスルフィンアミド(参考化合物 251)の合成
アルゴン雰囲気下、 5, 7 ジクロロべンゾチアゾール(112mg, 0. 55mmol)の T HF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0 . 31mL, 0. 49mmol)を滴下した。 10分後、 2—メチル—N— (チアン— 4—イリデ ン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(105mg,約 0. 48 mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 2. 5時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を 飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 64mg (約 14%)を得た。
[0401] ステップ 3 (3R)—N— (4— (5, 7 ジクロロべンゾチアゾール 2—ィル)チアン一 4 ィル) 4一(2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ カルボニルァミノ)ブタナミド (参考化合物 252)の合成
アルゴン雰囲気下、 N— (4— (5, 7 ジクロ口べンゾチアゾールー 2 ィル)チアン —4—ィル)—2—メチル—2 プロパンスルフィンアミド(64mg, 0. 15mmol)をメタ ノール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 1時間攪 拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、 ( 3R) -4- (2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカル ボ-ルァミノ)酪酸(50mg, 0. 15mmol)、 ΝΕΜ (63 Ι^, 0. 50mmol)、 HATU (6 2mg, 0. 17mmol)を加え、室温で攪拌した。 19時間後、飽和炭酸水素ナトリウム水 溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥
し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 70mg (74%)を得た。
[0402] ステップ 4 (3R)— N—(4— (5, 7 ジクロロべンゾチアゾール 2—ィル) 1, 1— ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル —2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 253)の合成
(3R) -N- (4— (5, 7 ジクロロべンゾチアゾール—2—ィル)チアン— 4—ィル) -4- (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボュ ルァミノ)ブタナミド(70mg, 0. l lmmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (66mg, >0. 25mol)を添カ卩した。 5分後氷浴を外し、室温で 3 0分間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水 素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 64mg (87%)を得た。
[0403] ステップ 5 (3R)— 3 ァミノ一 N— (4— (5, 7 ジクロロべンゾチアゾール 2—ィ ル)一 1, 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブタ ナミド ·塩酸塩 (化合物 74)の合成
アルゴン雰囲気下、 (3R)—N—(4—(5, 7 ジクロ口べンゾチアゾールー 2—ィル ) - 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3—( 2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(64mg, 0. lOmmol)を 10 %塩ィ匕水素 Zメタノール溶液 (5mL)に溶解し、室温で 30分間攪拌した後濃縮し、 表題ィ匕合物 53mg (92%)を得た。
[0404] 実施例 75
(3R)—3 ァミノ一 4一(2, 4, 5 トリフノレオ口フエ-ル) N— (4— (5—メトキシべ ンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 75)の合成
ステップ 1 6 ブロモー 5—メトキシベンゾチアゾール(参考化合物 254)の合成 アルゴン雰囲気下、 3—メトキシフエ-ルイソチオシアナ一ト(1. Og, 6. lmmol)を THF (lOmL)に溶解し、氷冷下、 7Nアンモニア Zメタノール溶液(2. 6mL, 18. 2 mmol)を加え、室温で 3時間攪拌した。酢酸ェチルで抽出し、飽和食塩水で洗浄後
、乾燥し濃縮した。得られた粗生成物をクロ口ホルム(70mL)に懸濁し、氷冷下、臭 素(0. 66mL, 12. 9mmol)のクロ口ホルム(5mL)溶液をゆっくりと滴下した。滴下 終了後、 1時間室温で攪拌し、生成した固体を濾別して得られた濾物をメタノールに 懸濁した。ここに飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を TFA(10mL)、 50%硫酸( lOmL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(389mg, 5. 6mmol)の蒸 留水(0. 8mL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、 15°Cに冷却し たホスフィン酸(lOmL)を加え、 13時間攪拌した (氷浴中の氷が溶けるに従い、次第 に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸ェチル で抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル力 ラムクロマトグラフィーで精製し、表題ィ匕合物 650mg (43%)を得た。
[0405] ステップ 2 5—メトキシベンゾチアゾール(参考化合物 255)の合成
アルゴン雰囲気下、 6 ブロモー 5—メトキシベンゾチアゾール(650mg, 2. 7mm ol)の THF (lOmL)溶液を― 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液( 1 . 59M, 5. OmL, 8. Ommol)を滴下した。 10分後、蒸留水を加え、酢酸ェチルで 抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリ 力ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 325mg (74%)を得た。
[0406] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5—メトキシべ ンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシ力 ルポニルァミノ)ブタナミド (参考化合物 256)の合成
アルゴン雰囲気下、 5—メトキシベンゾチアゾール(90mg, 0. 54mmol)の THF (5 mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 31 mL, 0. 49mmol)を滴下した。 10分後、 2—メチルー N—(チアンー4 イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(lOlmg,約 0. 46mmol )の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 2. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール(2mL)に溶
解し、 10%塩ィ匕水素 Zメタノール (4mL)をカ卩え、室温で 1. 5時間攪拌した。ァルゴ ン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)— 4 (2, 4, 5 トリフルオロフヱ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪 酸(31mg, 0. O9mmol)、 ΝΕΜ (39 Ι^, 0. 30mmol)、 HATU (39mg, 0. 10m mol)を加え、室温で攪拌した。 16. 5時間後、飽和炭酸水素ナトリウム水溶液を加え ジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 42mg (約 14%)を得た。
[0407] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5—メトキシべ ンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 257)の合成
(3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (5—メトキシベンゾチアゾ 一ルー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド(42mg, 0. 07mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mC PBA( >65%) (45mg, >0. 17mol)を添カ卩した。 5分後氷浴を外し、室温で 30分 間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナ トリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 32mg (72%)を得た。
[0408] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5 ーメトキシベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミ ド '塩酸塩 (ィ匕合物 75)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (5— メトキシベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(32mg, 0. O5mmol)を 10% 塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 13時間攪拌した後濃縮し、表 題ィ匕合物 26mg (90%)を得た。
[0409] 実施例 76
(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (2H— 1, 3—
ジォキソレン [4, 5—f]ベンゾチアゾールー 6—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミド ·塩酸塩 (化合物 76)の合成
ステップ 1 2H— 1, 3 ジォキソレン [4, 5— f]ベンゾチアゾール(参考化合物 258) の合成
アルゴン雰囲気下、 2H べンゾ [d] l, 3 ジォキソレンー5 ィルァミン(504mg , 3. 7mmol)をジクロロメタン(lOmL)に溶解し、 TCDP (875mg, 3. 7mmol)をカロ え、室温で 2時間攪拌した。蒸留水を加え、クロ口ホルムで抽出し、乾燥後濃縮した。 得られた粗生成物を、アルゴン雰囲気下、 THF (6mL)に溶解し、氷冷下、 7Nアン モ-ァ Zメタノール溶液(1. 5mL, 10. 5mmol)をカ卩え、室温で 1. 5時間攪拌した。 生成した固体を濾別して得られた粗生成物をクロ口ホルム(lOmL)に懸濁し、氷冷下 、臭素(0. 3mL, 5. 9mmol)のクロ口ホルム(lmL)溶液をゆっくりと滴下した。滴下 終了後、 4時間室温で攪拌し、生成した固体を濾別して得られた濾物をメタノールに 懸濁した。ここに飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を TFA(5mL)、 50%硫酸(5 mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(208mg, 3. Ommol)の蒸留 水(0. 8mL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、 15°Cに冷却した ホスフィン酸(lOmL)を加え、 13時間攪拌した (氷浴中の氷が溶けるに従い、次第に 昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸ェチルで 抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 177mg (27%)を得た。
ステップ 2 2—メチルー?^ー(4ー(211—1, 3 ジォキソレン [4, 5—f]ベンゾチアゾ 一ルー 6 ィル)チアンー 4ーィル)ー2 プロパンスルフィンアミド(参考化合物 259) の合成
アルゴン雰囲気下、 2H—1, 3 ジォキソレン [4, 5—f]ベンゾチアゾール(98mg , 0. 55mmol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサ ン溶液(1. 59M, 0. 31mL, 0. 49mmol)を滴下した。 10分後、 2—メチル N— ( チアン— 4—イリデン)—2 プロパンスルフィンイミド (参考化合物 205)の粗生成物( 103mg,約 0. 47mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆ
つくりと昇温し、 3時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽 出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 45mg (約 24%)を得た。
[0411] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカノレポ-ノレァミノ) Ν—(4—(2Η—1, 3 ジォキソレン [4, 5—f]ベンゾチア ゾールー 6 ィル)チアンー 4 ィル)ブタナミド(参考化合物 260)の合成
アルゴン雰囲気下、 2—メチルー N— (4— (2H—1, 3 ジォキソレン [4, 5—f]ベ ンゾチアゾール—6—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(45mg , 0. l lmmol)をメタノール(2mL)に溶解し、 10%塩化水素 Zメタノール(4mL)を 加え、室温で 1. 5時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)—4 (2, 4, 5 トリフルオロフヱ-ル)ー3—(2—メチ ルー 2 プロポキシカルボ-ルァミノ)酪酸(37mg, 0. l lmmol) , NEM (46 ^ L, 0 . 36mmol)、HATU (47mg, 0. 12mmol)をカ卩え、室温で攪拌した。 17. 5時間後 、飽和炭酸水素ナトリウム水溶液を加えジェチルエーテルで抽出し、蒸留水及び飽 和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 60mg (89%)を得た。
[0412] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカノレポ-ノレァミノ) Ν—(4—(2Η—1, 3 ジォキソレン [4, 5—f]ベンゾチア ゾールー 6 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド(参考化合物 261) の合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ)—Ν—(4—(2Η—1, 3 ジォキソレン [4, 5—f]ベンゾチアゾール —6—ィル)チアン一 4—ィル)ブタナミド(58mg, 0. 22mmol)をジクロロメタン(5m L)に溶解し、氷冷下、 mCPBA( >65%) (45mg, >0. 17mol)を添カ卩した。 5分後 氷浴を外し、室温で 1. 5時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪 拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃 縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 44 mg (70%)を得た。
[0413] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (2H - 1, 3 ジォキソレン [4, 5— f]ベンゾチアゾール 6—ィル) 1, 1—ジォキソチア ン 4 ィル)ブタナミド ·塩酸塩 (化合物 76)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル —2 プロポキシカルボ-ルァミノ)— N— (4— (2H—1, 3 ジォキソレン [4, 5— f] ベンゾチアゾールー 6—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド(44mg , 0. 07mmol)を 10%塩化水素 Zメタノール溶液(5mL)に溶解し、室温で 13時間 攪拌した後濃縮し、表題ィ匕合物 39mg (98%)を得た。
[0414] 実施例 77
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2—メトキ シエトキシ)ベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナ ミド ·塩酸塩 (化合物 77)の合成
ステップ 1 6 ヒドロキシベンゾチアゾール(参考化合物 262)の合成
6—メトキシベンゾチアゾール(参考化合物 211) (293mg, 1. 8mmol)を臭化水素 酸(5mL)に懸濁し、加熱還流した。 3. 5時間後放冷し、蒸留水を加え、酢酸ェチル 及びクロ口ホルムで抽出し、飽和食塩水で洗浄後、乾燥し濃縮することにより、表題 化合物 172mg (64%)を得た。
[0415] ステップ 2 6—(2—メトキシエトキシ)ベンゾチアゾール(参考化合物 263)の合成 アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(172mg,約 1. lmmol)を D MF (5mL)に溶解し、臭化 2—メトキシェチル(0. 14mL, 1. 5mmol)、炭酸カリウム (318mg, 2. 3mmol)を加え、室温で攪拌した。 24時間後、水酸化ナトリウム水溶液 を加え、ジェチルエーテルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮 した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1 55mg (65%)を得た c
[0416] ステップ 3 N— (4— (6— (2—メトキシエトキシ)ベンゾチアゾールー 2 ィル)チアン
4 ィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 264)の合成 アルゴン雰囲気下、 6—(2—メトキシエトキシ)ベンゾチアゾール(114mg, 0. 54m mol)の THF (5mL)溶液を― 78°Cに冷却し、 n -ブチルリチウム Zへキサン溶液( 1
. 59M, 0. 31mL, 0. 49mmol)を滴下した。 10分後、 2—メチル—N— (チアン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(102mg ,約 0. 46mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと 昇温し、 2時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、 有機層を飽和食塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲル力 ラムクロマトグラフィーで精製し、表題ィ匕合物 25mg (約 13%)を得た。
[0417] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6— (2—メトキ シエトキシ)ベンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2— プロポキシカルボニルァミノ)ブタナミド (参考化合物 265)の合成
アルゴン雰囲気下、 N— (4— (6- (2—メトキシェトキシ)ベンゾチアゾール—2—ィ ル)チアン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(25mg, 0. 06m mol)をメタノール(2mL)に溶解し、 10%塩化水素 Zメタノール(4mL)をカ卩え、室温 で 1時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL) に溶解し、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プロ ポキシカルボ-ルァミノ)酪酸(19mg, 0. 06mmol) , ΝΕΜ (24 μ L, 0. 19mmol) 、 HATU (24mg, 0. 06mmol)をカ卩え、室温で攪拌した。 14時間後、(3R)—4— ( 2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) 酪酸(20mg, 0. 06mmol) , ΝΕΜ (24 ^ L, 0. 19mmol)、 HATU (24mg, 0. 06 mmol)を追加し、更に 5時間攪拌した。飽和炭酸水素ナトリウム水溶液を加えジェチ ルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られ た粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 32mg (84% )を得た。
[0418] ステップ 5 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6— (2—メトキ シエトキシ)ベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3— (2 メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 266)の合成
(3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6— (2—メトキシェトキシ )ベンゾチァゾ一ルー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシ カルボ-ルァミノ)ブタナミド(3 lmg, 0. 05mmol)をジクロロメタン(5mL)に溶解し、
氷冷下、 mCPBA( >65%) (30mg, >0. l lmol)を添カ卩した。 5分後氷浴を外し、 室温で 1時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和 炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 27mg (80%)を 得た。
[0419] ステップ 6 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
- (2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン一 4 ィル)ブタナミド '塩酸塩 (ィ匕合物 77)の合成
アルゴン雰囲気下、(3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(6— ( 2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン一 4—ィ ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(27mg, 0. 04m mol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 15時間攪拌した後 濃縮し、表題ィ匕合物 24mg (98%)を得た。
[0420] 実施例 78
(3R)—3 アミノー 4— (5 クロ口一 2 フノレオ口フエ-ノレ) N— (4— (6— (2—メト キシエトキシ)ベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタ ナミド ·塩酸塩 (化合物 78)の合成
ステップ 1 4— (5—クロ口 2 フルオロフェ -ル)—3—ォキソ酪酸ェチル(参考化 合物 267)の合成
アルゴン雰囲気下、活性化亜鉛(4. 99g, 88. 5mmol)の THF (90mL)懸濁液の 加熱還流下、ブロモ酢酸ェチル(7. 80mL, 70. 7mmol)を少量ずつ滴下した。反 応が始まり、反応液が緑色を呈し始めたところで加熱及び滴下を停止し、 5—クロ口 —2 フルオロフェ-ルァセトニトリル(3. Og, 17. 7mmol)を加えた。再度加熱還流 させた後、残りのブロモ酢酸ェチルをゆっくりと滴下した。滴下終了 30分後放冷し、 3 N塩酸(30mL)を加えそのままさらに 2時間攪拌した。濃縮後、酢酸ェチルにて抽出 し、有機層を蒸留水、飽和食塩水にて洗浄し、乾燥後濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 3. 45g (75%)を得た。
[0421] ステップ 2 4—(5 クロロー 2 フルオロフヱ-ル) 3 ォキソ酪酸(参考化合物 26
8)の合成
アルゴン雰囲気下、 4— (5—クロ口— 2—フルオロフェ -ル)—3—ォキソ酪酸ェチ ル(1. 35g, 5. 23mmol)の酢酸(8mL)溶液に、室温にて濃塩酸(1. 31mL)をカロ えそのまま 30時間攪拌した。氷冷下、蒸留水(15mL)を加えた後、酢酸ェチルで抽 出し、有機層を蒸留水、飽和食塩水にて洗浄し、乾燥後濃縮した。 20°C冷却して 析出してきた結晶を濾別し、得られた固体をへキサン:酢酸ェチル(10 : 1)にて洗浄 し、 目的物 530mg (44%)を得た。
[0422] ステップ 3 (3S)—4— (5 クロロー 2 フルオロフェ-ル)ー3 ヒドロキシ酪酸(参 考化合物 269)の合成
アルゴン雰囲気下、 4— (5 クロ口 2 フルオロフェ -ル)—3—ォキソ酪酸(510 mg, 2. 21mmol)のジクロロメタン(5mL)懸濁液を— 40°Cに冷却し、 TEA(308 L, 2. 21mmol)を加えた。得られた溶液に、(+ )—B クロロジイソピノカンフエ-ル ボラン(851mg, 2. 65mmol)のジクロロメタン(4mL)溶液をゆっくりと加え、 5°Cま で昇温し、そのまま 15時間攪拌した。蒸留水、 1N水酸ィ匕ナトリウムを加え、 2—メチ ル— 2—プロピル メチル エーテルにて抽出し、有機層を蒸留水にて洗浄した。水 層を合わせて 1N塩酸を加え酸性にし、酢酸ェチルにて抽出を行い、有機層を蒸留 水で洗浄後、乾燥し濃縮し、粗生成物として表題ィ匕合物 510mg (約 99%)を得た。こ の反応を繰り返す事によって得た表題ィ匕合物を、次の反応に用いた。
[0423] ステップ 4 (3S)—N ベンジルォキシ 4— (5 クロ口一 2 フルオロフェ-ル)一3 ーヒドロキシブタナミドの合成 (参考化合物 270)の合成
アルゴン雰囲気下、 (3S)—4— (5 クロ口一 2 フルオロフェ -ル) 3 ヒドロキ シ酪酸(1. 70g,約 7. 31mmol)の THF (36mL)、蒸留水(9mL)溶液に、室温に てべンジルォキシァミン'塩酸塩(2. 33g, 14. 6mmol)、 EDCI (2. 80g, 14. 6m mol)を加え、 15時間攪拌した。濃縮後、蒸留水を加え酢酸ヱチルにて抽出し、有機 層を希塩酸、蒸留水、飽和食塩水にて洗浄し、乾燥後濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーにて精製し表題ィ匕合物 2. 04g (約 83%)得た。
[0424] ステップ 5 (3R)—N ベンジルォキシ 4— (5 クロ口一 2 フルォロベンジル)ァゼ チジン 2 オン (参考化合物 271)の合成
アルゴン雰囲気下、 (3S)—N ベンジルォキシ 4— (5 クロ口 2 フルオロフェ -ル) 3 ヒドロキシブタナミド(2. 00g, 5. 29mmol)、トリフエ-ルホスフィン(1. 7 lg, 6. 51mmol)のトルエン(30mL)懸濁液に、氷冷下、ァゾジカルボン酸ジイソプ 口ピル(以下、 DIADと略す)(1. 28mL, 6. 51mmol)をカ卩え、室温にて 13時間攪 拌した。濃縮して得られた残渣をシリカゲルカラムクロマトグラフィーで精製した。これ を、メタノール力も再結晶(一 40°Cまで冷却)し、表題化合物 1. 69g (89%)を得た。
[0425] ステップ 6 (3R)— 3—ペンジノレオキシァミノ一 4— (5 クロ口一 2 フノレオ口フエ- ル)酪酸メチル (参考化合物 272)の合成
アルゴン雰囲気下、 (3R)—N ベンジルォキシ 4— (5 クロ口 2 フルォロベン ジル)ァゼチジン— 2—オンのメタノール(20mL)溶液に、ナトリウム(154mg, 6. 71 mmol)、メタノール(5mL)より調製したナトリウムメトキシド/メタノール溶液を室温に て加えた。 30分後反応液を蒸留水にあけ、酢酸ェチルにて抽出し、塩化アンモ-ゥ ム水溶液、蒸留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題化合物 1. 62g (89%)を得た。
[0426] ステップ 7 (3R)— 4— (5 クロ口一 2 フノレオ口フエ-ノレ)一 3— (2—メチノレ一 2— プロポキシカルボニルァミノ)酪酸メチル (参考化合物 273)の合成
アルゴン雰囲気下、 (3R)—3 ベンジルォキシァミノ一 4— (5—クロ口一 2 フル オロフェ -ル)酪酸メチル(1. 50g, 4. 26mmol)のァセトニトリル(45mL)、蒸留水( 4mL)溶液に、室温にてモリブデンへキサカルボ-ル(1. 24g, 4. 70mmol)を加え 、 2. 5時間加熱還流した。放冷後セライト濾過し、濾液に 1N塩酸を加えて酸性としジ ェチルエーテルで洗净した。水層を飽和炭酸水素ナトリウム水溶液にてアルカリ性に し、酢酸ェチルにて抽出し、有機層を蒸留水、飽和食塩水にて洗浄した後、乾燥し 濃縮した。アルゴン雰囲気下、得られた粗生成物を THF (20mL)に溶解し、 TEA (2 . 09mL, 15mmol)、ジ— tret—ブチル ジカルボナート(1. 21g, 5. 45mmol)を 加え、室温で 13時間攪拌した。希塩酸を加え 20分攪拌した後蒸留水にあけ、酢酸 ェチルで抽出し、有機層を蒸留水、飽和食塩水にて洗浄後、乾燥し濃縮した。得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 950mg (6 4%)を得た。
[0427] ステップ 8 (3R)— 4— (5 クロ口一 2 フノレオ口フエ-ノレ)一 3— (2—メチノレ一 2— プロポキシカルボニルァミノ)酪酸 (参考化合物 274)の合成
アルゴン雰囲気下、 (3R)—4— (5 クロ口一 2 フルオロフェ -ル) 3— (2—メチ ルー 2 プロポキシカルボ-ルァミノ)酪酸メチル(950mg, 2. 75mmol)の THF (1 OmL)溶液に、室温にて水酸化リチウム(5. 5mmol)の蒸留水(2mL)溶液、メタノー ル(5mL)をカ卩え、 14時間攪拌した。 0°Cに冷却し、 1N塩酸をカ卩え、析出した白色固 体を濾取し乾燥した後、へキサン:酢酸ェチル (6 : 2)にて再結晶を行い、表題化合 物 844mg (93%)を得た。
[0428] ステップ 9 (3R)— 4— (5 クロ口一 2 フノレオ口フエ-ノレ)一 N— (4— (6— (2—メト キシエトキシ)ベンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 275)の合成
アルゴン雰囲気下、 N— (4—(6—(2—メトキシェトキシ)ベンゾチアゾールー 2—ィ ル)チアンー 4ーィル)ー2—メチルー 2 プロパンスルフィンアミド(参考化合物 264) (424mg, 0. 99mmol)をメタノール(5mL)に溶解し、 10%塩化水素 Zメタノール( 15mL)をカ卩え、室温で 1. 5時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗 生成物を DMF (lOmL)に溶解し、 (3R)—4— (5 クロ口一 2 フルオロフェ -ル) 3—(2—メチルー2—プロポキシカルボ-ルァミノ)酪酸(1641118, 0. 49mmol)、 NEM (0. 22mL, 1. 7mmol)、 HATU (207mg, 0. 54mmol)を加え、室温で攪 拌した。 12時間後、蒸留水を力卩ぇジェチルエーテルで抽出し、飽和食塩水にて洗浄 後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 220mg (71%)を得た。
[0429] ステップ 10 (3R)— 4— (5 クロ口一 2 フルオロフェ-ル)一 N— (4— (6— (2—メ トキシエトキシ)ベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 276)の合 成
(3R) -4- (5 クロロー 2 フノレオ口フエ-ノレ) N— (4—(6—(2—メトキシェトキ シ)ベンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキ シカルボ-ルァミノ)ブタナミド(220mg, 0. 34mmol)をジクロロメタン(10mL)に溶
解し、氷冷下、 mCPBA ( >65%) (224mg, >0. 84mmol)を添カ卩した。 5分後氷 浴を外し、室温で 30分間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌し た後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮し て得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 180m g (78%)を得た。
[0430] ステップ 11 (3R)— 3 アミノー 4— (5 クロ口一 2 フノレオ口フエ-ノレ)一 N— (4—
(6— (2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン一 4 ィル)ブタナミド ·塩酸塩 (化合物 78)の合成
アルゴン雰囲気下、 (3R)—4— (5 クロ口 2 フルオロフェ -ル) N— (4— (6 - (2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン一 4 ィル) - 3- (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(180mg, 0. 27mmol)を 10%塩ィ匕水素 Zメタノール溶液(10mL)に溶解し、室温で 1時間攪拌 した後濃縮し、表題ィ匕合物 164mg (定量的)を得た。
[0431] 実施例 79
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2—メチ ルー 2 プロピル)ベンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル) ブタナミド ·塩酸塩 (化合物 79)の合成
ステップ 1 6—(2—メチルー 2 プロピル)ベンゾチアゾール(参考化合物 277)の合 成
アルゴン雰囲気下、 4 (2—メチルー 2 プロピル)ァ-リン(505mg, 3. 4mmol) をジクロロメタン(10mL)に溶解し、 TCDl (612mg, 3. 4mmol)を加え、室温で 2. 5時間攪拌した。蒸留水を加え、クロ口ホルムで抽出し、乾燥後濃縮した。得られた粗 生成物 645mgのうち 396mgを、アルゴン雰囲気下、 THF (5mL)に溶解し、氷冷下 、 7Nアンモニア Zメタノール溶液(0. 9mL, 6. 3mmol)を加え、室温で 1. 5時間攪 拌した。酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗 生成物をクロ口ホルム(10mL)に懸濁し、氷冷下、臭素(0. 21mL, 4. lmmol)のク ロロホルム(2mL)溶液をゆっくりと滴下した。滴下終了後、 10分間室温で攪拌した 後、加熱還流した。 4時間後放冷し、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェ
チルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を TFA (5 mL)、 50%硫酸(5mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(154mg, 2. 2mmol)の蒸留水(0. 5mL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、 — 15°Cに冷却したホスフィン酸(10mL)を加え、 12時間攪拌した (氷浴中の氷が溶 けるに従い、次第に昇温)。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中 和し、酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 208mg (57%)を得 た。
[0432] ステップ 2 2—メチルー N— (4— (6— (2—メチルー 2 プロピル)ベンゾチアゾール
2 ィル)チアン 4 ィル) 2 プロパンスルフィンアミド(参考化合物 278)の合 成
アルゴン雰囲気下、 6—(2—メチルー2—プロピル)べンゾチァゾール(1061118, 0 . 55mmol)の THF (5mL)溶液を—78°Cに冷却し、 n—ブチルリチウム Zへキサン 溶液(1. 59M, 0. 31mL, 0. 49mmol)を滴下した。 10分後、 2—メチル—N— (チ アン 4 イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物( 1 Olmg,約 0. 46mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆつ くりと昇温し、 3時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出 し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合物 26mg (約 14%)を得た。
[0433] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2—メチルー 2 プロピル)ベンゾチアゾ 一ルー 2 ィル)チアンー 4 ィル)ブタナミド(参考化合物 279)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(6—(2—メチルー 2 プロピル)ベンゾ チアゾールー 2 ィル)チアンー 4 ィル) 2 プロパンスルフィンアミド(26mg, 0. 06mmol)をメタノール (2mL)に溶解し、 10%塩化水素 Zメタノール(4mL)を加え、 室温で 1時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3m L)に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プ 口ポキシカルボ-ルァミノ)酪酸(20mg, 0. 06mmol)、 ΝΕΜ (25 Ι^, 0. 20mmol
)、 HATU (27mg, 0. 07mmol)をカ卩え、室温で攪拌した。 16. 5時間後、飽和炭酸 水素ナトリウム水溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水に て洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 31mg (83%)を得た。
[0434] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2—メチルー 2 プロピル)ベンゾチアゾ 一ルー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド(参考化合物 280)の 合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (6— (2—メチル—2 プロピル)ベンゾチアゾール—2 —ィル)チアン一 4—ィル)ブタナミド(31mg, 0. 05mmol)をジクロロメタン(7mL)に 溶解し、氷冷下、 mCPBA ( >65%) (32mg, >0. 12mmol)を添カ卩した。 5分後氷 浴を外し、室温で 1時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した 後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 28mg (80%)を得た。
[0435] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 一(2—メチルー 2—プロピル)ベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 79)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル —2 プロポキシカルボ-ルァミノ) N— (4— (6— (2—メチル—2 プロピル)ベン ゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミド(28mg, 0. 04mmol)を 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、室温で 2時間攪拌し た後濃縮し、表題ィ匕合物 22mg (87%)を得た。
[0436] 実施例 80
(3R)— 3 ァミノ— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (5, 6, 7 トリメ トキシベンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド' 塩酸塩 (ィ匕合物 80)の合成
ステップ 1 4ーブロモー 5, 6, 7 トリメトキシベンゾチアゾール(参考化合物 281)の 合成
アルゴン雰囲気下、 3, 4, 5 トリメトキシァ-リン(3. Og, 16. 4mmol)をジクロロメ タン(30mL)に溶解し、 TCDI (3. 9g, 19. 7mmol)を加え、室温で 2. 5時間攪拌し た。蒸留水を加え、クロ口ホルムで抽出し、乾燥後濃縮した。アルゴン雰囲気下、得ら れた粗生成物を THF (lOmL)に溶解し、氷冷下、 7Nアンモニア Zメタノール溶液( 6. 2mL, 43. 3mmol)を加え、室温で 3時間攪拌した。生成した固体を濾別すること により得られた粗生成物 3. 12gのうち 1. Ogをクロ口ホルム(50mL)に懸濁し、氷冷 下、臭素(0. 45mL, 8. 8mmol)のクロ口ホルム(5mL)溶液をゆっくりと滴下した。 滴下終了後、生成した固体を直ちに濾別し、濾物をメタノールに懸濁した。ここに飽 和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、飽和食塩水で洗浄後、 乾燥し濃縮した。得られた粗生成物を TFA(10mL)、 50%硫酸(10mL)に溶解し、 — 15°Cに冷却した。亜硝酸ナトリウム(170mg, 2. 5mmol)の蒸留水(1. OmL)溶 液をゆっくりと滴下し、 30分間攪拌した。ここ〖こ、 15°Cに冷却したホスフィン酸(20 mL)を加え、 11時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温)。蒸留水 で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸ェチルで抽出し、飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 279mg (17%)を得た。
ステップ 2 5, 6, 7 トリメトキシベンゾチアゾール(参考化合物 282)の合成
アルゴン雰囲気下、 4 ブロモ—5, 6, 7 トリメトキシベンゾチアゾール(279mg, 0. 92mmol)の THF (5mL)溶液を—78°Cに冷却し、 n—ブチルリチウム Zへキサン 溶液(1. 59M, 1. 5mL, 2. 4mmol)を滴下した。 10分後、蒸留水を加え、酢酸ェ チルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 191mg (92%)を得た ステップ 3 N— (4— (5, 6, 7 トリメトキシベンゾチアゾール 2—ィル)チアン一 4 ィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 283)の合成 アルゴン雰囲気下、 5, 6, 7 トリメトキシベンゾチアゾール(190mg, 0. 84mmol)
の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59 M, 0. 48mL, 0. 76mmol)を滴下した。 10分後、 2—メチル—N— (チアン— 4—ィ リデン)—2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(158mg,約 0 . 72mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し 、 1. 5時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機 層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 80mg (約 25%)を得た。
[0439] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6, 7 トリメ トキシベンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 284)の合成
アルゴン雰囲気下、 N— (4— (5, 6, 7 トリメトキシベンゾチアゾールー 2 ィル) チアン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(109mg, 0. 25mmo 1)をメタノール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール(5mL)をカ卩え、室温で 1 . 5時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に 溶解し、 (3R) -4- (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル—2—プロボ キシカルボ-ルァミノ)酪酸(87mg, 0. 26mmol)、 ΝΕΜ (109 Ι^, 0. 86mmol)、 HATU (109mg, 0. 29mmol)を加え、室温で攪拌した。 13. 5時間後、飽和炭酸 水素ナトリウム水溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水に て洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 135mg (79%)を得た。
[0440] ステップ 5 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6, 7 トリメ トキシベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ー3—(2— メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 285)の合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6, 7 トリメトキシベン ゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカル ボ-ルァミノ)ブタナミド(135mg, 0. 21mmol)をジクロロメタン(5mL)に溶解し、氷 冷下、 mCPBA( >65%) (147mg, >0. 55mmol)を添カ卩した。 5分後氷浴を外し 、室温で 3時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和
炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 48mg (33%)を 得た。
[0441] ステップ 6 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6, 7 トリメトキシベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン一 4—ィル) ブタナミド ·塩酸塩 (化合物 80)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (5, 6 , 7 トリメトキシベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)― 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(48mg, 0. 07mmol) を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 1時間攪拌した後濃縮し 、表題ィ匕合物 43mg (99%)を得た。
[0442] 実施例 81, 82
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—メトキシー 5, 7 ジメチルベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブ タナミド '塩酸塩 (ィ匕合物 81)及び(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6—メトキシ— 5, 7—ジメチルベンゾチアゾール—3—ォキシドー 2—ィル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 82)の合成 ステップ 1 4ーメトキシー 3, 5 ジメチルァ-リン(参考化合物 286)の合成
アルゴン雰囲気下、 2, 6 ジメチルー 4 -トロフエノール(3. Og, 17. 6mmol)を DMF (80mL)に溶解し、ヨウ化メチル(1. 3mL, 20. 9mmol)、炭酸カリウム(4. 89 g, 35. 4mmol)をカ卩え、室温で攪拌した。 6. 5時間後、蒸留水を加えてジェチルェ 一テルで抽出し、蒸留水及び飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生 成物を、アルゴン雰囲気下、 THF (18mL)、メタノール(30mL)に溶解し、蒸留水(3 6mL)をカ卩えて懸濁した。ここに塩化アンモ-ゥム(3. 24g, 60. 6mmol)を加え、 60 °Cに加熱した。鉄粉(2. 90g, 51. 9mmol)をカ卩えた後、 12時間、加熱攪拌した。放 冷後、セライト濾過し、濾液に飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで 抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題化合物 2. 18g (82%)を得た。
[0443] ステップ 2 4 ブロモー 6—メトキシ—5, 7 ジメチルベンゾチアゾール(参考化合 物 287)の合成
アルゴン雰囲気下、 4—メトキシ— 3, 5 ジメチルァ-リン(2. 12g, 14. Ommol) をジクロロメタン(20mL)に溶解し、 TCDI (2. 94g, 14. 8mmol)をカ卩え、室温で 2. 5時間攪拌した。蒸留水を加え、クロ口ホルムで抽出し、乾燥後濃縮した。アルゴン雰 囲気下、得られた粗生成物を、 THF (6mL)に溶解し、氷冷下、 7Nアンモニア Zメタ ノール溶液(6. OmL, 42. Ommol)を加え、室温で 3時間攪拌した。生成した固体を 濾別して得られた粗生成物 2. 85gのうち 1. 02gをクロ口ホルム(50mL)に懸濁し、 氷冷下、臭素(0. 55mL, 10. 7mmol)のクロ口ホルム(3mL)溶液をゆっくりと滴下 した。室温で 30分間攪拌した後、生成した固体を濾別し、濾物をメタノールに懸濁し た。ここに飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、飽和食塩水 で洗浄後、乾燥し濃縮した。得られた粗生成物を TFA(10mL)、 50%硫酸(10mL) に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(175mg, 2. 5mmol)の蒸留水(1 . OmL)溶液をゆっくりと滴下し、 30分間攪拌した。ここに、 15°Cに冷却したホスフ イン酸(20mL)を加え、 16時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温) 。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸ェチルで抽出 し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 292mg (約 22%)を得た。
[0444] ステップ 3 6—メトキシ—5, 7 ジメチルベンゾチアゾール(参考化合物 288)の合 成
アルゴン雰囲気下、 4ーブロモー 6—メトキシ—5, 7 ジメチルベンゾチアゾール(2 92mg, 1. lmmol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Z へキサン溶液(1. 59M, 2. lmL, 3. 3mmol)を滴下した。 10分後、蒸留水をカロえ 、酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られ た粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 180mg (87 %)を得た。
[0445] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6—メトキシ一 5 , 7 ジメチルベンゾチアゾール 2 ィル)チアンー 4 ィル) 3—( 2 メチル 2
プロポキシカルボニルァミノ)ブタナミド (参考化合物 289)の合成
アルゴン雰囲気下、 6—メトキシ—5, 7 ジメチルベンゾチアゾール(180mg, 0. 9 3mmol)の THF (5mL)溶液を一 78°C〖こ冷却し、 n—ブチルリチウム Zへキサン溶 液(1. 59M, 0. 56mL, 0. 89mmol)を滴下した。 10分後、 2—メチル—N— (チア ン― 4 イリデン) - 2-プロパンスルフィンイミド(参考化合物 205)の粗生成物(193 mg,約 0. 88mmol)の THF (3mL)溶液を滴下した。滴下終了後、室温までゆっくり と昇温し、 2. 5時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出 し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで粗精製した。得られた粗精製物を、アルゴン雰囲気下、 10%塩ィ匕水素 Zメタノール(5mL)に溶解し、室温で 30分間攪拌した。アルゴン雰 囲気下、濃縮して得られた粗生成物を DMF (5mL)に溶解し、(3R)—4— (2, 4, 5 —トリフルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(9 Omg, 0. 27mmol) , NEM d ie ^ L, 0. 93mmol)、 HATU (115mg, 0. 30mm ol)を加え、室温で攪拌した。 12時間後、飽和炭酸水素ナトリウム水溶液を加えジェ チルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 163mg (約 28%)を得た。
ステップ 5 (3R)—4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6—メトキシ一 5 , 7 ジメチルベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3 一(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 290)及び( 3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6—メトキシ一 5, 7 ジメチ ルベンゾチアゾール—3—ォキシドー 2—ィル)—1, 1—ジォキソチアン— 4—ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 291)の 合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6—メトキシ一 5, 7 ジメ チルベンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2—プロボ キシカルボ-ルァミノ)ブタナミド(163mg, 0. 26mmol)をジクロロメタン(7mL)に溶 解し、氷冷下、 mCPBA ( >65%) (242mg, >0. 91mmol)を添カ卩した。 5分後氷
浴を外し、室温で 2. 5時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌 した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮 して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合を各 々 28mg (16%)及び 69mg (41%)得た。
[0447] ステップ 6 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 ーメトキシ 5, 7 ジメチルベンゾチアゾールー 2—ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 81)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (6— メトキシ 5, 7 ジメチルベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ーィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(28mg, 0. 0 4mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 4. 5時間攪拌 した後濃縮し、表題ィ匕合物 23mg (91%)を得た。
[0448] ステップ 6, (3R)— 3 ァミノ— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (6 ーメトキシ 5, 7 ジメチルベンゾチアゾールー 3 ォキシドー 2—ィル) 1, 1ージ ォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 82)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (6— メトキシー 5, 7 ジメチルベンゾチアゾールー 3 ォキシドー 2 ィル) 1, 1ージォ キソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミ ド(69mg, 0. lOmmol)を 10%塩化水素 Zメタノール溶液(5mL)に溶解し、室温で 4. 5時間攪拌した後濃縮し、表題ィ匕合物 59mg (94%)を得た。
[0449] 実施例 83
(3R)—3 ァミノ一 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (6— (2 モノレ ホリノエトキシ)ベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブ タナミド '二塩酸塩 (ィ匕合物 83)の合成
ステップ 1 6—(2 モルホリノエトキシ)ベンゾチアゾール(参考化合物 292)の合成 アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (560mg, 3 . 7mmol)を DMF (lOmL)に溶解し、塩化 2 モルホリノェチル '塩酸塩(83 lmg, 4. 5mmol)、炭酸カリウム(1. 54g, 11. lmmol)を加え、室温で攪拌した。 17時間
後、炭酸カリウム(750mg, 5. 4mmol)、ヨウ化カリウム(309mg, 1. 9mmol)を加え 、更に 26時間攪拌した。蒸留水を加え、ジェチルエーテルで抽出し、有機層を飽和 食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグ ラフィ一で精製し、表題ィ匕合物 454mg (46%)を得た。
[0450] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2 モルホリノエトキシ)ベンゾチアゾール —2—ィル)チアン— 4—ィル)ブタナミド (参考化合物 293)の合成
アルゴン雰囲気下、 6—(2—モルホリノェトキシ)べンゾチァゾール(16211^, 0. 6 lmmol)の THF (5mL)溶液を一 78°C〖こ冷却し、 n—ブチルリチウム Zへキサン溶 液(1. 59M, 0. 36mL, 0. 57mmol)を滴下した。 10分後、 2—メチル—N— (チア ン― 4 イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(113 mg,約 0. 52mmol)の THF (3mL)溶液を滴下した。滴下終了後、室温までゆっくり と昇温し、 2. 5時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出 し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで粗精製した。得られた粗精製物を、アルゴン雰囲気下、 メタノール(2mL)に溶解し、 10%塩化水素 Zメタノール(4mL)をカ卩え、室温で 1. 5 時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶 解し、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキ シカルボ-ルァミノ)酪酸(72mg, 0. 22mmol) , ΝΕΜ (84 μ L, 0. 66mmol)、H ATU (84mg, 0. 22mmol)を加え、室温で攪拌した。 7時間後、飽和炭酸水素ナト リウム水溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後 、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 98mg (約 27%)を得た。
[0451] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2 モルホリノエトキシ)ベンゾチアゾール —2—ィル)—1, 1—ジォキソチアン— 4—ィル)ブタナミド (参考化合物 294)の合成 アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6—(2 モルホリノエトキシ)ベンゾ
チアゾールー 2 ィル)チアンー 4 ィル)ブタナミド(98mg, 0. 14mmol)をァセトニ トリル(5mL)に溶解し、粉末 MS4A(15mg)、 NMO (50mg, 0. 43mmol)のァセト 二トリル (4mL)溶液、 TPAP (3. 3mg, 9. 4 mol)を加え、室温で 12時間攪拌した 。反応液をセライト濾過し、濾液を濃縮した。得られた粗生成物を再びァセトニトリル( 5mL)に溶解し、粉末 MS4A(60mg)、 NMO (60mg, 0. 51mmol)のァセトニトリ ル(2mL)溶液、 TPAP (13mg, 37 mol)を加え、室温で 10時間攪拌した。反応 液をセライト濾過し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合 23mg (22%)を得た。
[0452] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
- (2—モルホリノエトキシ)ベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4 ィル)ブタナミド '二塩酸塩 (化合物 83)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6—(2 モルホリノエトキシ)ベンゾ チアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミド(23mg, 0. 03 mmol)を 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、室温で 1時間攪拌した 後濃縮し、表題ィ匕合物 19mg (86%)を得た。
[0453] 実施例 84
(3R)— 3 ァミノ— N— (4— (ベンゾチアゾール—3—ォキシド— 2—ィル)—1, 1— ジォキソチアンー4ーィル)ー4ー(2, 4, 5 トリフルオロフヱ-ル)ブタナミド '塩酸塩 (化合物 84)の合成
ステップ 1 (3R)— N— (4— (ベンゾチアゾール—3—ォキシド— 2—ィル)—1, 1— ジォキソチアンー4ーィル)ー4ー(2, 4, 5 フルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 295)の合成
(3R)— N— (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミ ド(参考化合物 209) (196mg, 0. 35mmol)をジクロロメタン(10mL)に溶解し、氷 冷下、 mCPBA( >65%) (460mg, > 1. 7mmol)を添カ卩した。 5分後氷浴を外し、 室温で 11時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和
炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 79mg (37%) を得た。
[0454] ステップ 2 (3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 3—ォキシド一 2—ィ ル)— 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ブタ ナミド ·塩酸塩 (化合物 84)の合成
アルゴン雰囲気下、(3R) -N- (4— (ベンゾチアゾール—3—ォキシド— 2—ィル ) - 1, 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 フルオロフェ-ル)一3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(97mg, 0. 25mmol)を 10% 塩ィ匕水素 Zメタノール溶液 (5mL)に溶解し、室温で 1時間攪拌した。飽和炭酸水素 ナトリウム水溶液を加え、クロ口ホルム Zエタノール =3Zlで抽出し、乾燥後濃縮し た。得られた粗生成物をシリカゲルクロマトグラフィーで精製し、表題ィ匕合物 (フリー塩 基) 59mg (46%)を得た。これをメタノールに溶解し、 10%塩ィ匕水素 Zメタノールを 滴下して塩ィ匕し、濃縮して表題化合物 (塩酸塩) 59mgを得た。
[0455] 実施例 85
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6 ジメチルァ ミノべンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '二 塩酸塩 (ィ匕合物 85)の合成
ステップ 1 6 ジメチルァミノべンゾチアゾール(参考化合物 296)の合成
アルゴン雰囲気下、 6 ァミノべンゾチアゾール(300mg, 2. Ommol)をァセトニト リル(30mL)に溶解し、ホルマリン(666mg, 8. Ommol)、シァノ水素化ホウ素ナトリ ゥム(350mg, 5. 57mmol)をカ卩え、室温で 2時間攪拌した。 1N塩化水素を加えて 撹拌したのち、アンモニア水を加えて濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 152mg (43%)を得た。
[0456] ステップ 2 2—メチル—N— (4— (6 ジメチルァミノべンゾチアゾール—2—ィル) チアンー 4 ィル) 2 プロパンスルフィンアミド(参考化合物 297)の合成
アルゴン雰囲気下、 6 ジメチルァミノべンゾチアゾール(178mg, 1. Ommol)の T HF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 64M, 0
. 38mL, 1. Ommol)を滴下した。 30分後、 2—メチル N— (チアン一 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(241mg,約 1. lmmo 1)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 3時間後 、蒸留水を加えた。クロ口ホルムで抽出し、有機層を乾燥後濃縮した。得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 108mg (約 27%)を 得た。
[0457] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (6 ジメチルァ ミノべンゾチアゾールー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキ シカルボニルァミノ)ブタナミド (参考化合物 298)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(6 ジメチルァミノべンゾチアゾールー 2—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(108mg, 0. 27mmol) を 10%塩ィ匕水素 Zメタノール (6mL)に溶解し、室温で 2時間攪拌した。アルゴン雰 囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)—4— (2, 4, 5 —トリフルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(7 2mg, 0. 22mmol) , DIPEA ( 131 ^ L, 0. 75mmol)、 HATU (107mg, 0. 28m mol)を加え、室温で攪拌し、 15時間後、濃縮した。得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合物 107mg (81%)を得た。
[0458] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (6 ジメチルァ ミノべンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ー3—(2—メチ ル— 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 299)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (6— ジメチルァミノべンゾチアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 —プロポキシカルボ-ルァミノ)ブタナミド(107mg, 0. 18mmol)をァセトニトリル(1 OmL)に溶解し、粉末 MS4A(17mg)、 NMO (62mg, 0. 53mmol)をカ卩え、 5分間 室温で撹拌した。 TPAP (3mg, 0. 0085mmol)をカ卩えて 12時間撹拌したのち、セ ライトで濾過した。濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合 31mg (27%)を得た。
[0459] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
ージメチルァミノべンゾチアゾールー 2—ィル) 1, 1ージォキソチアン 4 ィル)ブ タナミド '二塩酸塩 (ィ匕合物 85)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (6— ジメチルァミノべンゾチアゾールー 2—ィル) 1, 1ージォキソチアン 4 ィル) 3 一(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(31mg, 0. 049mmol) を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 9時間攪拌した後濃縮し 、表題ィ匕合物 25mg (83%)を得た。
[0460] 実施例 86
(3R)—3 アミノー 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (6— (ピリジン 4 ィル)ベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナ ミド ·二塩酸塩 (化合物 86)の合成
ステップ 1 6 ブロモベンゾチアゾール(参考化合物 300)の合成
2 アミノー 6 ブロモベンゾチアゾール(600mg, 2. 6mmol)を TFA(8mL)、 50 %硫酸(8mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(216mg, 3. lmmol )の蒸留水(0. 5mL)溶液をゆっくりと滴下し、 30分間攪拌した後、ホスフィン酸(16 mL)を加え、 13時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温)。氷冷下、 1N水酸ィ匕ナトリウム水溶液にて中和し、クロ口ホルムで抽出し、有機層を乾燥後濃縮 した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 3 79mg (68%)を得た Q
[0461] ステップ 2 6 (ピリジン 4 ィル)ベンゾチアゾール(参考化合物 301)の合成
アルゴン雰囲気下、 6 ブロモベンゾチアゾール(379mg, 1. 8mmol)の 1, 4ージ ォキサン溶液(6mL)に、 4一(4, 4, 5, 5—テトラメチルー 1, 3, 2 ジォキサボロラ ン— 2—ィル)ピリジン(363mg, 1. 8mmol)、炭酸セシウム(1. 7g, 5. 3mmol)、 [ 1, 1,-ビス(ジフエ-ルホスフイノ)フエ口セン]ジクロロパラジウム(145mg, 0. 18mm ol)を加え、 80°Cで 14時間攪拌した。放冷後濾過し、濾液を濃縮して得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 240mg (64%)を得 た。
[0462] ステップ 3 2—メチルー N— (4— (6 (ピリジンー4 ィル)ベンゾチアゾールー 2—
ィル)チアン 4 ィル) - 2-プロパンスルフィンアミド(参考化合物 302)の合成 アルゴン雰囲気下、 6 (ピリジンー4 ィル)ベンゾチアゾール(318mg, 1. 5mm ol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 64M, 0. 57mL, 1. 5mmol)を滴下した。 30分後、 2—メチル N— (チアン一 4— イリデン)—2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(329mg,約 1. 5mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し 、 15時間後、蒸留水を加えた。クロ口ホルム、酢酸ェチルで抽出し、有機層を乾燥後 濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕 合物 227mg (約 35%)を得た。
[0463] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6 (ピリジンー4 ィル)ベンゾチアゾールー 2 ィル)チアンー 4 ィル)ブタナミド (参考化合物 303)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(6 (ピリジンー4 ィル)ベンゾチアゾ 一ルー 2—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(227mg, 0. 53m mol)を 10%塩ィ匕水素 Zメタノール (6mL)に溶解し、室温で 2時間攪拌した。ァルゴ ン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)— 4 (2, 4, 5 トリフルオロフヱ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪 酸(138mg, 0. 41mmol)、 ϋΙΡΕΑ (252 Ι^ 1. 45mmol)、 HATU (205mg, 0 . 54mmol)を加え、室温で攪拌し、 11時間後、濃縮した。得られた粗生成物をシリ 力ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 74mg (28%)を得た。
[0464] ステップ 5 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6 (ピリジンー4 ィル)ベンゾチアゾールー 2 —ィル) 1, 1—ジォキソチアン— 4—ィル)ブタナミド (参考化合物 304)の合成 アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6 (ピリジンー4 ィル)ベンゾチア ゾールー 2 ィル)チアンー 4 ィル)ブタナミド(74mg, 0. 12mmol)をァセトニトリ ル(7mL)に溶解し、粉末 MS4A(12mg)、 NMO (41mg, 0. 35mmol)をカ卩え、 5 分間室温で撹拌した。 TPAP (2mg, 0. 006mmol)をカ卩えて 18時間撹拌したのち、
セライトで濾過した。濾液を濃縮して得られた粗生成物をァセトニトリル(7mL)に溶 解し、粉末 MS4A(12mg)、 NMO (41mg, 0. 35mmol)をカ卩え、 5分間室温で撹 拌した。 TPAP (2mg, 0. 006mmol)をカ卩えて 13時間撹拌した後、セライトで濾過し た。濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し 、表題ィ匕合 43mg (55%)を得た。
[0465] ステップ 6 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 (ピリジンー4 ィル)ベンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '二塩酸塩 (化合物 86)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6 (ピリジンー4 ィル)ベンゾチア ゾールー 2 ィル) 1, 1—ジォキソチアン一 4—ィル)ブタナミド(43mg, 0. 063m mol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 9時間攪拌した後 濃縮し、表題ィ匕合物 38mg (93%)を得た。
[0466] 実施例 87
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6 モルホリノ ベンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '二塩酸 塩 (ィ匕合物 87)の合成
ステップ 1 6 モルホリノべンゾチアゾール(参考化合物 305)の合成
アルゴン雰囲気下、 6 ァミノべンゾチアゾール(500mg, 3. 3mmol)をトルエン( 50mL)に溶解し、 DIPEA(8. 7mL, 50mmol)、ビス(2 ブロモェチル)エーテル( 3. 9g, 17mmol)を加え、 12時間加熱還流した。放冷後濃縮して得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 648mg (88%)を得た。
[0467] ステップ 2 2—メチルー N— (4— (6 モルホリノべンゾチアゾールー 2 ィル)チア ン 4 ィル) 2 プロパンスルフィンアミド(参考化合物 306)の合成
アルゴン雰囲気下、 6 モルホリノべンゾチアゾール(330mg, 1. 5mmol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 64M, 0. 5 7mL, 1. 5mmol)を滴下した。 30分後、 2—メチル—N— (チアン— 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(329mg,約 1. 5mmol)
の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 8時間後、 蒸留水を加えた。クロ口ホルムで抽出し、有機層を乾燥後濃縮した。得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 172mg (約 26%)を得 た。
[0468] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6 モルホリノべンゾチアゾールー 2 ィル)チ アン— 4—ィル)ブタナミド (参考化合物 307)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(6 モルホリノべンゾチアゾールー 2— ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(172mg, 0. 39mmol)を 10 %塩ィ匕水素 Zメタノール (6mL)に溶解し、室温で 2時間攪拌した。アルゴン雰囲気 下、濃縮して得られた粗生成物を DMF (3mL)に溶解し、(3R)—4— (2, 4, 5 トリ フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(131m g, 0. 39mmol) , DIPEA(300 μ L, 1. 8mmol)、 HATU (194mg, 0. 51mmol) を加え、室温で攪拌し、 12時間後濃縮し、得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 lOlmg (40%)を得た。
[0469] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6 モルホリノべンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド (参考化合物 308)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6 モルホリノべンゾチアゾールー 2 —ィル)チアン— 4—ィル)ブタナミド(lOlmg, 0. 16mmol)をァセトニトリル(8mL) に溶解し、粉末 MS4A (16mg)、 NMO (55mg, 0. 47mmol)をカ卩え、 5分間室温 で撹拌した。 TPAP (3mg, 0. 0078mmol)をカ卩えて 15時間撹拌したのち、セライト で濾過した。濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合 45mg (43%)を得た。
[0470] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 モルホリノべンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタ ナミド '二塩酸塩 (ィ匕合物 87)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6 モルホリノべンゾチアゾールー 2 —ィル)一 1, 1—ジォキソチアン一 4—ィル)ブタナミド(45mg, 0. 066mmol)を 10 %塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 5時間攪拌した後濃縮し、表 題ィ匕合物 41mg (95%)を得た。
[0471] 実施例 88
(3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジォキソチア ンー 4一ィル)一 4一 (3—クロロフヱ-ル)ブタナミド '塩酸塩(ィ匕合物 88)の合成 ステップ 1 (3R)— N— (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4 一(3 クロ口フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミ ド (参考化合物 309)の合成
アルゴン雰囲気下、 4 (ベンゾチアゾールー 2 ィル)チアンー 4 アミン'塩酸塩 (参考化合物 206)の粗生成物(90mg,約 0. 31mmol)を DMF (5mL)に溶解し、 ( 3R)—4— (3 クロ口フエ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) 酪酸(lOOmg, 0. 32mmol)、 HATU (146mg, 0. 38mmol) , DIPEA(154 μ L, 0. 93mmol)を加え、室温で攪拌した。 14時間後、反応液に蒸留水を力卩ぇジェチル エーテルにて抽出した。有機層を蒸留水、飽和食塩水で洗浄後、乾燥し濃縮して得 られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 134mg ( 約 79%)を得た。
[0472] ステップ 2 (3R)— N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン —4—ィル)—4— (3 クロ口フエ-ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 310)の合成
(3R)— N— (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4— (3—クロ 口フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(120mg , 0. 22mmol)をジクロロメタン(lOmL)に溶解し、氷冷下、 mCPBA( >65%) (11 4mg, >0. 43mmol)を添加した。 5分後氷浴を外し、室温で 13時間攪拌した。チォ 硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加 えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムク
口マトグラフィ一で精製し、表題ィ匕合 108mg (85%)を得た。
[0473] ステップ 3 (3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジ ォキソチアンー4 ィル) -4- (3—クロ口フエ-ル)ブタナミド '塩酸塩 (ィ匕合物 88) の合成
アルゴン雰囲気下、(3R)— N— (4— (ベンゾチアゾール—2—ィル)—1, 1—ジォ キソチアン一 4—ィル) 4— (3 クロ口フエニル) 3— (2—メチル 2 プロポキシ カルボ-ルァミノ)ブタナミド(108mg, 0. 19mmol)を 10%塩化水素 Zメタノール溶 液 (4mL)、クロ口ホルム(5mL)に溶解し、室温で 14時間攪拌した後濃縮し、表題ィ匕 合物 94mg (96%)を得た。
[0474] 実施例 89
(3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジォキソチア ンー4 ィル)ー4 (2, 4ージクロロフヱ-ル)ブタナミド '塩酸塩(ィ匕合物 89)の合成 ステップ 1 (3R)— N— (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4 - (2, 4 ジクロロフエ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)ブ タナミド (参考化合物 311)の合成
アルゴン雰囲気下、 4 (ベンゾチアゾールー 2 ィル)チアンー 4 アミン'塩酸塩 (参考化合物 206)の粗生成物(80mg,約 0. 28mmol)を DMF (4mL)に溶解し、 (3 R)—4— (2, 4 ジクロロフエ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァ ミノ)酪酸(104mg, 0. 27mmol)、 HATU (128mg, 0. 34mmol)、 DIPEA(139 n 0. 84mmol)を加え、室温で攪拌した。 63時間後、反応液に蒸留水を加えジ ェチルエーテルにて抽出し、有機層を蒸留水、飽和食塩水で洗浄後、乾燥し濃縮し た。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 11 5mg (約 71%)を得た。
[0475] ステップ 2 (3R)— N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン —4—ィル)—4— (2, 4 ジクロロフエ-ル)—3— (2—メチル—2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 312)の合成
(3R)— N— (4— (ベンゾチアゾール—2—ィル)チアン— 4—ィル)—4— (2, 4— ジクロロフエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(11
5mg, 0. 20mmol)をジクロロメタン(8mL)に溶解し、氷冷下、 mCPBA ( >65%) ( 104mg, >0. 39mmol)を添カ卩した。 5分後氷浴を外し、室温で 13時間攪拌した。 チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶 液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合 65mg (53%)を得た。
[0476] ステップ 3 (3R)— 3 ァミノ一 N— (4— (ベンゾチアゾール 2—ィル) 1, 1—ジ ォキソチアンー4 ィル) -4- (2, 4ージクロ口フエ-ル)ブタナミド '塩酸塩 (ィ匕合物 89)の合成
アルゴン雰囲気下、(3R)— N— (4— (ベンゾチアゾール—2—ィル)—1, 1—ジ ォキソチアン一 4—ィル) 4— (2, 4 ジクロロフエ-ル) 3— (2—メチル 2 プ 口ポキシカルボ-ルァミノ)ブタナミド(65mg, 0. l lmmol)を 10%塩化水素 Zメタノ ール溶液(3mL)、クロ口ホルム(lmL)に溶解し、室温で 4時間攪拌した後濃縮し、 表題ィ匕合物 60mg (定量的)を得た。
[0477] 実施例 90
(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6 ジメトキ シベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸 塩 (ィ匕合物 90)の合成
ステップ 1 3, 4 ジメトキシフエ-ルイソチオシアナート(参考化合物 313)の合成 アルゴン雰囲気下、 3, 4 ジメトキシァ-リン(3. 06g, 20mmol)をジクロロメタン(
40mL)に溶解し、 TCDI (3. 56g, 20mmol)を加え室温で撹拌した。 13時間後、反 応液を濃縮して得た粗生成物をシリカゲルカラムクロマトグラフィーで精製して、表題 化合物 3. l lg (80%)を得た。
[0478] ステップ 2 N (3, 4 ジメトキシフ -ル)チォ尿素(参考化合物 314)の合成
アルゴン雰囲気下、 3, 4 ジメトキシフエ-ルイソチオシアナート(3. l lg, 16mmo
1)を THF (32mL)に溶解し、氷冷下、 7Nアンモニア Zメタノール溶液(6. 9mL, 48 mmol)を加え、室温で攪拌した。 5時間後、反応液を濃縮して表題化合物 3. 38g ( 定量的)を得た。
[0479] ステップ 3 2 アミノー 5, 6 ジメトキシベンゾチアゾール(参考化合物 315)の合成
アルゴン雰囲気下、 N— (3, 4 ジメトキシフヱ-ル)チォ尿素(3. 38g, 16mmol) をジクロロメタン(80mL)に懸濁し、氷冷下、塩化スルフリル(1. 4mL, 18mmol)を 滴下した。室温で 2時間撹拌した後、飽和炭酸水素ナトリウム水溶液を加え酢酸ェチ ルで抽出し、有機層を乾燥し濃縮して表題ィ匕合物 2. 57g (76%)を得た。
[0480] ステップ 4 5, 6 ジメトキシベンゾチアゾール(参考化合物 316)の合成
2 アミノー 5, 6 ジメトキシベンゾチアゾール(2. 75g, 12mmol)を TFA(18mL )、 50%硫酸水溶液(18mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(926 mg, 13mmol)の蒸留水(2. 8mL)溶液を滴下し、 15°Cで 30分間撹拌した後、ホ スフイン酸(30mL)を滴下し、 15°Cで 4時間撹拌した。反応液を 1N水酸化ナトリウ ム水溶液と酢酸ェチルの混合液に移し、分液した。有機層をセライト濾過し、得られ た溶液を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題化合物 1. 26g (53%)を得た。
[0481] ステップ 5 N— (4— (5, 6 ジメトキシベンゾチアゾールー 2 ィル)チアンー 4ーィ ル) 2 メチル 2 プロパンスルフィンアミド(参考化合物 317)の合成
アルゴン雰囲気下、 5, 6 ジメトキシベンゾチアゾール(195mg, 1. Ommol)の T HF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0 . 63mL, 1. Ommol)を滴下した。 30分後、 2—メチル N— (チアン一 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(329mg,約 1. 5mmo 1)の THF (3mL)溶液を滴下し、 1. 5時間かけて 0°Cまで昇温した。蒸留水を加え、 酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 146mg (約 35 %)を得た。
[0482] ステップ 6 4— (5, 6 ジメトキシベンゾチアゾール 2—ィル)チアン一 4—ァミン' 塩酸塩 (参考化合物 318)の合成
アルゴン雰囲気下、 N— (4— (5, 6 ジメトキシベンゾチアゾールー 2 ィル)チア ン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(145mg, 0. 35mmol)を メタノール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(2mL)をカ卩え、室温で 2時間撹拌した。反応液を濃縮し、減圧下で乾燥して表題ィ匕合物 121mg (定量的)
を得た。
[0483] ステップ 7 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6 ジメトキ シベンゾチアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ー3—(2—メチ ルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 319)の合成
アルゴン雰囲気下、 4— (5, 6 ジメトキシベンゾチアゾール—2—ィル)チアン— 4 —アミン'塩酸塩(121mg, 0. 35mmol)を DMF (3mL)に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪 酸(117mg, 0. 35mmol)、 HATU (133mg, 0. 35mmol) , TEA (148 μ L, 1. 0 5mmol)を加え、室温で攪拌した。 13時間後、蒸留水を力卩ぇジェチルエーテルで抽 出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をジクロ ロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (240mg, >0. 89mmol)を 加え、室温で撹拌した。 2時間後、クロ口ホルム、アンモニア水、蒸留水を加え、分液 した。有機層をチォ硫酸ナトリウム水溶液で洗浄し、乾燥後濃縮して得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 150mg (65%)を得た。
[0484] ステップ 8 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6 ジメトキシベンゾチアゾールー 2—ィル) 1, 1ージォキソチアン 4 ィル)ブタ ナミド ·塩酸塩 (化合物 90)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (5, 6 —ジメトキシベンゾチアゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3— (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(150mg, 0. 23mmol)をメ タノール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(6mL)を加え、室温で撹 拌した。 4. 5時間後、反応液を濃縮し、表題ィ匕合物 135mg (定量的)を得た。
[0485] 実施例 91
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2 フエノ キシエトキシ)ベンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブナ タミド '塩酸塩 (ィ匕合物 91)の合成
ステップ 1 6—(2 フ ノキシエトキシ)ベンゾチアゾール(参考化合物 320)の合成 アルゴン雰囲気下、 1—ブロモ 2 フエノキシェタン(362mg, 1. 8mmol)を DM
F (4mL)に溶解し、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (181mg, 1. 2mmol)、炭酸カリウム(331mg, 2. 4mmol)を加え、室温で撹拌した。 18時間後、 蒸留水を加え、酢酸ェチルで抽出し、有機層を蒸留水、飽和食塩水で洗浄し、乾燥 後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して、表題 化合物 264mg (81 %)を得た。
[0486] ステップ 2 2—メチルー N— (4— (6— (2 フエノキシエトキシ)ベンゾチアゾールー 2 ィル)チアンー 4 ィル) 2 プロパンスルフィンアミド(参考化合物 321)の合成 アルゴン雰囲気下、 6—(2 フエノキシエトキシ)ベンゾチアゾール(264mg, 1. 0 mmol)の THF (lOmL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶 液(1. 58M, 0. 65mL, 1. Ommol)を滴下し、 30分間撹拌した。 2—メチル N— ( チアン— 4—イリデン)—2 プロパンスルフィンイミド (参考化合物 205)の粗生成物( 219mg,約 1. Ommol)の THF (5mL)溶液を滴下し、 2時間かけて 10°Cまで昇温し た。蒸留水を加え酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃 縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 86mg (約 18%)を得た。
[0487] ステップ 3 4— (6— (2 フエノキシエトキシ)ベンゾチアゾール—2—ィル)チアン— 4 ァミン'塩酸塩 (参考化合物 322)の合成
アルゴン雰囲気下、 2—メチル N— (4— (6- (2 フエノキシエトキシ)ベンゾチア ゾール—2—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(86mg, 0. 18 mmol)を 10%塩ィ匕水素メタノール溶液 (4mL)に溶解し、室温で 2時間撹拌した。反 応液を濃縮し、減圧下で乾燥して表題ィ匕合物 74mg (定量的)を得た。
[0488] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
- (2—フエノキシエトキシ)ベンゾチアゾール 2—ィル) 1, 1—ジォキソチアン一 4 ィル)ブナタミド '塩酸塩 (ィ匕合物 91)の合成
アルゴン雰囲気下、 4 (6- (2 フエノキシエトキシ)ベンゾチアゾールー 2—ィル )チアン— 4 ァミン.塩酸塩(74mg, 0. 18mmol)を DMF (3mL)に溶解し、(3R) -4- (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ)酪酸(71mg, 0. 21mmol)、 HATU (84mg, 0. 21mmol)、 ΤΕΑ(76 L
, 0. 54mmol)を加え、室温で攪拌した。 10時間後、蒸留水を加えジェチルエーテ ルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物 をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (134mg, >0. 50m mol)を加え、室温で撹拌した。 1. 5時間後、アンモニア飽和クロ口ホルムをカ卩え、不 溶物を濾別し、濾液を濃縮した。これをシリカゲルカラムクロマトグラフィーで粗精製し て得られた粗精製物を、アルゴン雰囲気下、 10%塩ィ匕水素メタノール溶液 (4mL)に 溶解し、室温で撹拌した。 2時間後、飽和炭酸水素ナトリウム水溶液を加え、クロロホ ルムで抽出し、有機層を乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製して、表題ィ匕合物(フリー体) 54mg (49%)を得た。表題化合物 (フリー体)を酢酸ェチルに溶解し、 10%塩ィ匕水素 Zメタノール (0. lmL)をカ卩えて 塩化し、濃縮することにより、表題ィ匕合物 (塩酸塩)を得た。
[0489] 実施例 92
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (6 ピラジュル ォキシベンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブナタミド' 塩酸塩 (ィ匕合物 92)の合成
ステップ 1 6 ビラジ-ルォキシベンゾチアゾール(参考化合物 323)の合成 アルゴン雰囲気下、クロロビラジン(206mg, 1. 80mmol)を DMF (6mL)に溶解 し、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (227mg, 1. 50mmol)、炭 酸セシウム(831mg, 2. 55mmol)を加え、 70°Cで撹拌した。 15時間後放冷し、蒸 留水を加えジェチルエーテルで抽出した。有機層を蒸留水、飽和食塩水で洗浄し、 乾燥後濃縮して、表題ィ匕合物 308mg (90%)を得た。
[0490] ステップ 2 2—メチルー N— (4— (6 ピラジュルォキシベンゾチアゾールー 2—ィル )チアン 4 ィル) 2 プロパンスルフィンアミド(参考化合物 324)の合成
アルゴン雰囲気下、 6 ピラジュルォキシベンゾチアゾール(229mg, 1. Ommol) の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 58 M, 0. 67mL. 1. lmmol)を滴下し、 15分間撹拌した。 2—メチル—N— (チアン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(219mg ,約 1. Ommol)の THF (5mL)溶液を滴下し、 2時間かけて 10°Cまで昇温した。蒸
留水を加え酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した
。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 23m g (約 5%)を得た。
[0491] ステップ 3 4—(6 ピラジュルォキシベンゾチアゾールー 2 ィル)チアンー 4 アミ ン '塩酸塩 (参考化合物 325)の合成
アルゴン雰囲気下、 2—メチルー N—(4一(6 ピラジュルォキシベンゾチアゾール —2—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(23mg, 0. O51mmol )を 10%塩ィ匕水素 Zメタノール溶液(2mL)に溶解し、室温で 2時間撹拌した。反応 液を濃縮し、減圧下で乾燥して表題ィ匕合物 20mg (定量的)を得た。
[0492] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)—N— (4—(6 ピラジュルォキシベンゾチアゾールー 2— ィル)—1, 1—ジォキソチアン— 4—ィル)ブタナミド (参考化合物 326)の合成 アルゴン雰囲気下、 4一(6 ピラジュルォキシベンゾチアゾールー 2 ィル)チアン — 4 ァミン'塩酸塩(20mg, 0. O51mmol)を DMF (2mL)に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ )酪酸(18mg, 0. 054mmol)、 HATU (21mg, 0. 054mmol) , TEA (20 μ L, 0. 14mmol)を加え、室温で攪拌した。 10時間後、蒸留水を加え酢酸ェチルで抽出し 、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物を、アルゴン 雰囲気下、ジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (44mg, > 0. 17mmol)を加え、室温で撹拌した。 1. 5時間後、アンモニア飽和クロ口ホルムを 加え、不溶物を濾別し、濾液を濃縮した。これを薄層クロマトグラフィーで精製して、 表題ィ匕合物 28mg (80%)を得た。
[0493] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 —ビラジニルォキシベンゾチアゾールー 2—ィル)—1, 1ージォキソチアンー4ーィル )ブナタミド '塩酸塩 (ィ匕合物 92)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ)—N— (4—(6—ビラジ-ルォキシベンゾチアゾ 一ルー 2—ィル)—1, 1—ジォキソチアン— 4—ィル)ブタナミド(28mg, 0. 040mm
ol)を 10%塩ィ匕水素 Zメタノール溶液(2mL)に溶解し、室温で撹拌した。 2時間後、 飽和炭酸水素ナトリウム水溶液を加えクロ口ホルムで抽出し、有機層を乾燥後濃縮し た。得られた粗生成物を薄層クロマトグラフィーで精製して、表題ィ匕合物 (フリー体) 1 9mg (59%)を得た。表題ィ匕合物(フリー体)を酢酸ェチルに溶解し、 10%塩化水素 Zメタノール溶液 (0. lmL)を加えて塩ィ匕し、濃縮することにより、表題化合物 (塩酸 塩)を得た。
[0494] 実施例 93
(3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (4, 5, 6, 7—テトラヒドロ ベンゾチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (化合物 93)の合成
ステップ 1 2 アミノー 4, 5, 6, 7—テトラヒドロべンゾチアゾール(参考化合物 327) の合成
アルゴン雰囲気下、 2 クロロシクロへキサノン(1. l lg, 8. Ommol)をエタノール( 20mL)に溶解し、チォ尿素(913mg, 12mmol)をカ卩え、加熱還流した。 13時間後 放冷し、濃縮した。これに蒸留水、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチ ルで抽出し、有機層を乾燥後濃縮して表題ィ匕合物 1. 13g (収率 74%)を得た。
[0495] ステップ 2 4, 5, 6, 7—テトラヒドロベンゾチアゾール(参考化合物 328)の合成
2 ァミノ一 4, 5, 6, 7—テトラヒドロべンゾチアゾール(626g, 4. 5mmol)を TFA( 9mL)、 50%硫酸(9mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(276mg , 4. Ommol)の蒸留水(1. 5mL)溶液を滴下し、 15°Cで 30分間撹拌した後、ホス フィン酸(10mL)を滴下し、 15°Cで 64時間撹拌した (氷浴中の氷が溶けるに従い 、次第に昇温)。反応液を飽和炭酸水素ナトリウム水溶液と酢酸ェチルの混合液に移 し、分液した。有機層をセライトろ過し、得られた溶液を飽和食塩水にて洗浄後、乾 燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 138mg (22%)を得た。
[0496] ステップ 3 N— (4— (4, 5, 6, 7—テトラヒドロべンゾチアゾールー 2 ィル)チアン
4 ィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 329)の合成 アルゴン雰囲気下、 4, 5, 6, 7—テトラヒドロべンゾチアゾール(138mg, 0. 99mm
ol)を THF (5mL)に溶解し、 78°Cに冷却した。 n—ブチルリチウム Zへキサン溶 液(1. 60M, 0. 62mL, 0. 99mmol)を滴下し、 1時間撹拌した。 2—メチル N— ( チアン— 4—イリデン)—2 プロパンスルフィンイミド (参考化合物 205)の粗生成物( 219mg,約 1. Ommol)の THF (3mL)溶液を滴下し、 1時間かけて 0°Cまで昇温し た。蒸留水、酢酸ェチル、飽和食塩水を加え、分液した。有機層を飽和食塩水で洗 浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 142mg (約 40%)を得た。
[0497] ステップ 4 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (4, 5, 6, 7 ーテトラヒドロべンゾチアゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタ ナミド '塩酸塩 (ィ匕合物 93)の合成
アルゴン雰囲気下、 N—(4— (4, 5, 6, 7—テトラヒドロべンゾチアゾールー 2—ィ ル)チアン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(67mg, 0. 40m mol)をメタノール (2mL)に溶解し、 10%塩化水素 Zメタノール溶液(3mL)を加え、 室温で 1. 5時間撹拌した。反応液を濃縮し、減圧下で乾燥して得た粗生成物 116m gのうち 31mgを、アルゴン雰囲気下、 DMF (3mL)に溶解し、(3R)—4— (2 フル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. 10mmol)、 HATU (40mg, 0. l lmmol)、 ΤΕΑ(42 L, 0. 30mmol)を加え、室 温で 12時間撹拌した。蒸留水を加えジェチルエーテルで抽出し、有機層を飽和食 塩水で洗浄した後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で粗精製した。得られた粗精製物を、ジクロロメタン(3mL)に溶解し、氷冷下、 mCPBA( >65%) (51mg, >0. 19mmol)を加え、室温で 1時間撹拌した。アンモ ユア飽和クロ口ホルム(3mL)を加え、生成した沈殿を濾別し、濾液を濃縮して得られ た粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物を 、アルゴン雰囲気下、 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、室温で 16 時間撹拌した後濃縮して表題ィ匕合物 29mg (56%)を得た。
[0498] 実施例 94
(3R) 3 アミノー 4一(2 フルオロフェ -ル) N— (4—(4 フエ-ルチアゾール 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 94)の合
成
ステップ 1 4 フエ二ルチアゾール(参考化合物 330)の合成
アルゴン雰囲気下、 2 ブロモアセトフヱノン(2. Og, 10. Ommol)、チォ尿素(80 lmg, 10. 5mmol)のエタノール(50mL)溶液を 2時間加熱還流した。放冷後、飽 和炭酸水素ナトリウム水溶液を加え酢酸ェチルで抽出し、飽和食塩水で洗浄した後 、乾燥し濃縮した。得られた粗生成物 1. 75gのうち 889mgを TFA (10mL)、 50% 硫酸(lOmL)に溶解し、—15°Cに冷却して亜硝酸ナトリウム (420mg, 6. lmmol) の蒸留水(1. 5mL)溶液をゆっくりと滴下した。 30分後、冷却したホスフィン酸(30m L)を加えて 18時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温)。蒸留水で 希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、酢酸ェチルで抽出した。有機 層を飽和食塩水で洗浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 287mg (約 35%)を得た。
[0499] ステップ 2 2—メチルー N— (4— (4 フエ-ルチアゾールー 2 ィル)チアンー 4 ィル) 2 プロパンスルフィンアミド(参考化合物 331)の合成
アルゴン雰囲気下、 4 フエ-ルチアゾール(195mg, 1. 2mmol)の THF (5mL) 溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 57M, 0. 68mL, 1. lmmol)を滴下した。 10分後、 2—メチル—N— (チアン— 4—イリデン)—2 プ 口パンスルフィンイミド(参考化合物 205)の粗生成物(218mg,約 0. 99mmol)の T HF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 5時間後、飽和 炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水に て洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 96mg (約 25%)を得た。
[0500] ステップ 3 4—(4 フエ-ルチアゾールー 2 ィル)チアンー 4ーァミン '塩酸塩(参 考化合物 332)の合成
アルゴン雰囲気下、 2—メチルー N—(4一(4 フエ-ルチアゾールー 2 ィル)チ アン一 4—ィル) 2 プロパンスルフィンアミド(36mg, 0. O9mmol)をメタノール(2 mL)に溶解し、 10%塩ィ匕水素 Zメタノール(2mL)をカ卩え、室温で 2時間攪拌し、濃 縮することにより、粗生成物として表題ィ匕合物 30mgを得た。
[0501] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ) N— (4— (4 フエ-ルチアゾール—2—ィル)チアン— 4—ィル) ブタナミド (参考化合物 333)の合成
アルゴン雰囲気下、 4一(4 フエ-ルチアゾールー 2 ィル)チアンー 4ーァミン' 塩酸塩の粗生成物(30mg,約 0. lmmol)を DMF (3mL)に溶解し、(3R)—4—(2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(29 mg, 0. lmmol) , NEM (33 ^ L, 0. 32mmol)、 HATU (42mg, 0. 1 lmmol)を 加え、室温で攪拌した。 14時間後、飽和炭酸水素ナトリウム水溶液を加えジェチル エーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 41mg (約 76% )を得た。
[0502] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4— (4 フエ-ルチアゾール—2—ィル)—1, 1—ジォキソ チアン— 4—ィル)ブタナミド (参考化合物 334)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ) N— (4—(4 フエ-ルチアゾールー 2 ィル)チアンー 4 ィル)ブタナミド (9mg, 0. 016mmol)をジクロロメタン(3mL)に溶解し、氷冷下、 mCPBA ( >65% ) (lOmg, >0. O38mmol)を添カ卩した。 4時間後、チォ硫酸ナトリウム水溶液をカロえ て激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し 、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 7mg (74%)を得た。
[0503] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (4 フエ-ル チアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (ィ匕合 物 94)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(4 フエ-ルチアゾールー 2—ィル)—1, 1 ジォキソチアンー4 ィル)ブタナミド(7mg, 0. 012mmol)を 10%塩化水素 Zメタノ ール溶液 (3mL)に溶解し、室温で 4時間攪拌した後濃縮し、表題ィ匕合物 6mg (96
%)を得た。
[0504] 実施例 95, 96
(3R) 3 アミノー 4一(2 フルオロフェ -ル) N— (4—(4 フエ-ルチアゾール —2—ィル)—1—ォキソチアン— 4—ィル)ブタナミド (低極性) '塩酸塩 (化合物 95) 及び(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (4— (4 フエ-ルチアゾ 一ルー 2—ィル)ー1ーォキソチアンー4 ィル)ブタナミド (高極性) '塩酸塩 (化合物 96)の合成
ステップ 1 (3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ) N— (4— (4—フエ-ルチアゾール—2—ィル)—1—ォキソチアン —4—ィル)ブタナミド (低極性)(参考化合物 335)及び(3R)—4— (2—フルオロフ ェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) N— (4— (4 フエ- ルチアゾール—2—ィル)—1—ォキソチアン— 4—ィル)ブタナミド(高極性)(参考化 合物 336)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ) N— (4—(4 フエ-ルチアゾールー 2 ィル)チアンー 4 ィル)ブタナミド (参考化合物 333) (25mg, 0. 045mmol)をジクロロメタン(3mL)に溶解し、氷冷下 、 mCPBA( >65%) (lOmg, >0. O38mmol)を添カ卩した。 10分後、チォ硫酸ナト リウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。ク ロロホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグ ラフィ一で精製し、表題化合物を各々 9mg (35%)及び 6mg (23%)得た。
[0505] ステップ 2 ( 3R)— 3 アミノー 4ー( 2 フノレオ口フエ-ノレ) N— (4— (4 フエ-ノレ チアゾール—2—ィル)—1—ォキソチアン— 4—ィル)ブタナミド (低極性) '塩酸塩( 化合物 95)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(4 フエ-ルチアゾールー 2—ィル) 1ーォ キソチアンー4 ィル)ブタナミド(低極性)(9mg, 0. 016mmol)を 10%塩化水素 Z メタノール溶液(3mL)に溶解し、室温で 10時間攪拌した後濃縮し、表題化合物 7m g (88%)を得た。
[0506] ステップ 2, (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (4 フエ- ルチアゾールー 2 ィル) 1ーォキソチアン 4 ィル)ブタナミド(高極性) '塩酸塩 (化合物 96)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(4 フエ-ルチアゾールー 2—ィル) 1ーォ キソチアン 4 ィル)ブタナミド(高極性)(6mg, 0. OlOmmol)を 10%塩化水素 Z メタノール溶液(3mL)に溶解し、室温で 10時間攪拌した後濃縮し、表題化合物 4m g (75%)を得た。
[0507] 実施例 97
(3R) 3 アミノー 4一(2 フルオロフェ -ル) N— (4—(4 フエ-ルチアゾール —2—ィル)チアン一 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 97)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4—(4 フエ-ルチアゾールー 2 ィル)チアンー 4—ィル)ブタナミド(参考化合物 333) (8mg, 0. 014mmol)を 10%塩化水素 Zメタ ノール溶液 (3mL)に溶解し、室温で 4時間攪拌した後濃縮し、表題ィ匕合物 6mg (85 %)を得た。
[0508] 実施例 98
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (4 フエ-ルチ ァゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 98)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(4 フエ-ルチアゾールー 2 ィル)チアンー 4 ィル)ブタナミド (参考化合物 337)の合成
アルゴン雰囲気下、 4一(4 フエ-ルチアゾールー 2 ィル)チアンー 4ーァミン' 塩酸塩(参考化合物 332)の粗生成物(66mg,約 0. 21mmol)を DMF (5mL)に溶 解し、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキ シカルボ-ルァミノ)酪酸(70mg, 0. 21mmol)、 ΝΕΜ (88 Ι^, 0. 69mmol)、H ATU (88mg, 0. 23mmol)を加え、室温で攪拌した。 11時間後、飽和炭酸水素ナ
トリウム水溶液を加えジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄 後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 103mg (約 84%)を得た。
[0509] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4— (4 フエ-ルチアゾール—2—ィル)—1, 1— ジォキソチアンー4 ィル)ブタナミド (参考化合物 338)の合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ) N— (4— (4—フエ-ルチアゾール—2—ィル)チアン— 4—ィル) ブタナミド(103mg, 0. 17mmol)をジクロロメタン(10mL)に溶解し、氷冷下、 mCP BA( >65%) (106mg, >0. 40mmol)を 2回に分けて添カ卩した。 2. 5時間後、チォ 硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加 えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 95mg (88%)を得た。
[0510] ステップ 3 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (4 フエ-ルチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩 酸塩 (化合物 98)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル —2 プロポキシカルボ-ルァミノ) N— (4— (4 フエ-ルチアゾール—2—ィル) - 1, 1ージォキソチアンー4 ィル)ブタナミド(95mg, 0. 15mmol)を 10%塩化水 素 Zメタノール溶液(10mL)に溶解し、室温で 13時間攪拌した後濃縮し、表題化合 物 79mg (93%)を得た。
[0511] 実施例 99
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) -N- (4— (4— (3—メトキシフエ- ル)チアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 99)の合成
ステップ 1 2 アミノー 4一(3—メトキシフヱ-ル)チアゾール ·塩酸塩(参考化合物 3 39)の合成
アルゴン雰囲気下、 3—メトキシァセトフエノン(1. 4mL, 10. 2mmol)を THF (10
mL)に溶解し、三臭化フエ-ルトリメチルアンモ -ゥム(3. 84g、 10. 2mmol)を少量 ずつ加え、室温で攪拌した。 23時間後、エタノール(20mL)、チォ尿素(778mg, 1 0. 2mmol)を加えて、加熱還流した。 2時間後放冷し、飽和炭酸水素ナトリウム水溶 液を加え酢酸ェチルで抽出し、飽和食塩水で洗浄した後、乾燥し濃縮した。得られ た粗生成物をメタノールに溶解し、 10%塩ィ匕水素 Zメタノールを滴下して塩ィ匕した。 ここに酢酸ェチルを加え、生じた沈殿を濾別し、表題化合物 1. 03g (42%)を得た。
[0512] ステップ 2 4—(3—メトキシフヱ-ル)チアゾール(参考化合物 340)の合成
2 アミノー 4— (3—メトキシフエ-ル)チアゾール '塩酸塩(499mg, 2. lmmol)を 酢酸(9mL)、 50%硫酸(15mL)に溶解し、 15°Cに冷却して亜硝酸ナトリウム(17 2mg, 2. 5mmol)の蒸留水(0. 5mL)溶液をゆっくりと滴下した。 30分後、この反応 液を—15°Cに冷却した酸化銅(I) (164mg, 1. lmmol)のホスフィン酸(30mL)懸 濁液に加えて 12時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温)。蒸留水 で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、セライト濾過した。濾液を酢 酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシ リカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 150mg (37%)を得た。
[0513] ステップ 3 N— (4— (4— (3—メトキシフエ-ル)チアゾール—2—ィル)チアン— 4— ィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 341)の合成
アルゴン雰囲気下、 4一(3—メトキシフエ-ル)チアゾール(87mg, 0. 45mmol)の THF (3mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M , 0. 26mL, 0. 41mmol)を滴下した。 30分後、 2—メチル N— (チアン一 4—イリ デン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(68mg,約 0. 3 lmmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 3 . 5時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層 を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 54mg (約 42%)を得た。
[0514] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (4— (3—メトキシフエ- ル)チアゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 342)の合成
アルゴン雰囲気下、 N— (4—(4一(3—メトキシフエ-ル)チアゾールー 2 ィル)チ アン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(54mg, 0. 13mmol)を メタノール(5mL)に溶解し、 10%塩化水素 Zメタノール(5mL)をカ卩え、室温で 2時 間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (4mL)に溶解 し、 (3R)—4— (2 フルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ)酪酸(45mg, 0. 15mmol)、 ΝΕΜ (55 Ι^, 0. 43mmol)、 HATU (55m g, 0. 14mmol)を加え、室温で攪拌した。 15時間後、飽和炭酸水素ナトリウム水溶 液を加えジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥し 濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕 合物 70mg (91%)を得た。
[0515] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)一 N— (4— (4— (3—メトキシフエ- ル)チアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 343)の合成
(3R)—4— (2 フルオロフヱ-ル) N— (4— (4— (3—メトキシフヱ-ル)チアゾ 一ルー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド(70mg, 0. 12mmol)をジクロロメタン(7mL)に溶解し、氷冷下、 mC PBA( >65%) (74mg, >0. 28mmol)を 2回に分けて添カ卩した。 3時間後、チォ硫 酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加え た。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 64mg (87%)を得た。
[0516] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (4— (3—メト キシフエ-ル)チアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド' 塩酸塩 (ィ匕合物 99)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (4— (3—メトキ シフエ-ル)チアゾール 2—ィル) 1, 1—ジォキソチアン一 4—ィル) 3— (2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(64mg, 0. lOmmol)を 10%塩 化水素 Zメタノール溶液(10mL)に溶解し、室温で 14時間攪拌した後濃縮し、表題 化合物 52mg (91%)を得た。
[0517] 実施例 100
(3R) 3 アミノー 4一(2 フルオロフェ -ル) N—(4一(4 (ピリジン 1ーォキ シドー 4 ィル)チアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミ ド ·塩酸塩 (化合物 100)の合成
ステップ 1 2 アミノー 4 (ピリジン 4 ィル)チアゾール '二塩酸塩(参考化合物 344)の合成
アルゴン雰囲気下、 4 ァセチルビリジン(0. 5mL, 4. 5mmol)を THF (lOmL) に溶解し、三臭化フエ-ルトリメチルアンモ -ゥム(1. 86g、4. 9mmol)を少量ずつ 加え、加熱還流した。 14時間後放冷し、エタノール(20mL)、チォ尿素(362mg, 4 . 8mmol)を加えて、加熱還流した。 3. 5時間後放冷し、飽和炭酸水素ナトリウム水 溶液を加え酢酸ェチルで抽出し、飽和食塩水で洗浄した後、乾燥し濃縮した。得ら れた粗生成物をメタノールに懸濁し、 10%塩ィ匕水素 Zメタノールを滴下して塩ィ匕した 。ここに酢酸ェチルを加え、生じた沈殿を濾別し、表題ィ匕合物 353mg (37%)を得た
[0518] ステップ 2 4 (ピリジン 4 ィル)チアゾール(参考化合物 345)の合成
2 アミノー 4— (ピリジン— 4—ィル)チアゾール '二塩酸塩(353mg, 1. 7mmol) を酢酸(2. 5mL)、 50%硫酸(2. 5mL)に溶解し、 15°Cに冷却して亜硝酸ナトリウ ム(140mg, 2. Ommol)の蒸留水(0. 5mL)溶液をゆっくりと滴下した。 30分後、こ の反応液を—15°Cに冷却した酸化銅(I) (1631mg, 0. 91mmol)のホスフィン酸(1 OmL)懸濁液に加えて 13時間攪拌した (氷浴中の氷が溶けるに従い、次第に昇温) 。蒸留水で希釈後、氷冷下水酸ィ匕ナトリウム水溶液にて中和し、セライト濾過した。濾 液を酢酸ェチルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 154mg (58%)を得た
[0519] ステップ 3 2—メチルー N— (4— (4 (ピリジンー4 ィル)チアゾールー 2 ィル) チアン 4 ィル) 2 プロパンスルフィンアミド(参考化合物 346)の合成
アルゴン雰囲気下、 4 (ピリジンー4 ィル)チアゾール(117mg, 0. 72mmol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 57M
, 0. 42mL, 0. 66mmol)を滴下した。 15分後、 2—メチル N— (チアン一 4—イリ デン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(102mg,約 0. 46mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 3. 5時間後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機 層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 29mg (約 16%)を得た。
[0520] ステップ 4 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ)—N— (4— (4 (ピリジン 4 ィル)チアゾールー 2 ィル)チアン —4—ィル)ブタナミド (参考化合物 347)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(4 (ピリジンー4 ィル)チアゾールー 2—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(29mg, 0. 08mmol)を メタノール(2mL)に溶解し、 10%塩化水素 Zメタノール(2mL)をカ卩え、室温で 3. 5 時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (5mL)に溶 解し、 (3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ -ルァミノ)酪酸(23mg, 0. 08mmol) , ΝΕΜ (44 μ L, 0. 35mmol)、 HATU (32 mg, 0. 08mmol)を加え、室温で攪拌した。 12. 5時間後、飽和炭酸水素ナトリウム 水溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾 燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表 題ィ匕合物 32mg (76%)を得た。
[0521] ステップ 5 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルボニルァミノ) N— (4— (4— (ピリジン— 1—ォキシドー 4—ィル)チアゾール—2 —ィル) 1, 1—ジォキソチアン— 4—ィル)ブタナミド (参考化合物 348)の合成
(3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ル ァミノ)—N— (4— (4 (ピリジン 4 ィル)チアゾールー 2 ィル)チアンー 4ーィ ル)ブタナミド(32mg, 0. 06mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCP BA( >65%) (35mg, >0. 13mmol)を 2回に分けて添カ卩した。 6. 5時間後、チォ 硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加 えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムク
口マトグラフィ一で精製し、表題ィ匕合物 19mg (55%)を得た。
[0522] ステップ 6 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (4— (ピリジ ン一 1—ォキシド一 4—ィル)チアゾール 2—ィル) 1, 1—ジォキソチアン一 4—ィ ル)ブタナミド ·塩酸塩 (ィ匕合物 100)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4— (4— (ピリジン— 1—ォキシドー 4—ィル)チア ゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)ブタナミド(19mg, 0. 03mm ol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 13時間攪拌した後 濃縮し、表題ィ匕合物 17mg (94%)を得た。
[0523] 実施例 101
(3R) 3 アミノー 4一(2 フノレオ口フエ-ノレ) N—(4一(4一(4ーフノレオ口フエ- ル)チアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 101)の合成
ステップ 1 4一(4 フルオロフヱニル)チアゾール(参考化合物 349)の合成
2 アミノー 4— (4 フルオロフェ -ル)チアゾール(1. 05g, 5. Ommol)を TFA(1 OmL)、 50%硫酸(10mL)に溶解し、 15°Cに冷却した。これに亜硝酸ナトリウム (4 14mg, 6. Ommol)の蒸留水(1. 5mL)溶液を滴下した。 15°Cで 30分間撹拌し た後、ホスフィン酸(20mL)を加え、—15°Cで 12時間撹拌した (氷浴中の氷が溶け るに従い、次第に昇温)。 1N水酸ィ匕ナトリウム水溶液を加え、酢酸ェチルで抽出した 。有機層をセライトろ過し、得られた溶液を飽和食塩水にて洗浄後、乾燥し濃縮した 。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 235 mg (24%)を得た。
[0524] ステップ 2 N— (4— (4— (4 フルオロフェ -ル)チアゾールー 2 ィル)チアンー 4 ィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 350)の合成 アルゴン雰囲気下、 4一(4 フルオロフヱ-ル)チアゾール(397mg, 2. Ommol) を THF (15mL)に溶解し、 78°Cに冷却した。 n—ブチルリチウム Zへキサン溶液( 1. 59M, 1. 3mL, 2. Ommol)を滴下し、 30分間撹拌した。これに 2—メチル—N - (チアン— 4—イリデン)—2 プロパンスルフィンイミド (参考化合物 205)の粗生成
物(405mg,約 1. 8mmol)の THF (5mL)溶液を滴下し、 78°Cで 30分間撹拌し た。蒸留水、酢酸ェチル、飽和食塩水を加えて室温まで昇温し、分液した。有機層を 飽和食塩水で洗浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 248mg (約 35%)を得た。
[0525] ステップ 3 4—(4— (4 フルオロフェ -ル)チアゾールー 2 ィル)チアンー 4 アミ ン '塩酸塩 (参考化合物 351)の合成
アルゴン雰囲気下、 N—(4一(4一(4 フルオロフヱ-ル)チアゾールー 2 ィル) チアン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(247mg, 0. 62mmo 1)をメタノール(5mL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(5mL)をカ卩え、室 温で 30分間撹拌した。反応液を濃縮し、減圧下で乾燥して表題ィ匕合物 200mg (98 %)を得た。
[0526] ステップ 4 (3R)—4—(2 フノレオ口フエ-ノレ) N—(4一(4一(4ーフノレオ口フエ- ル)チアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 352)
アルゴン雰囲気下、 4一(4一(4 フルオロフェ -ル)チアゾールー 2 ィル)チアン — 4 アミン'塩酸塩(112mg, 0. 34mmol)を DMF (7mL)に溶解し、(3R)— 4— (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(1 Olmg, 0. 34mmol)、 HATU (142mg, 0. 37mmol) , TEA (143 μ L, 1. 02mm ol)を加え、室温で 14時間撹拌した。蒸留水を加え、ジェチルエーテルで抽出し、有 機層を蒸留水、飽和食塩水で洗浄した後、乾燥し濃縮した。得られた粗生成物をジ クロロメタン(10mL)に溶解し、 0°Cに冷却した。これに mCPBA ( >65%) (220mg, >0. 83mmol)を加え、室温で 1時間撹拌した。アンモニア飽和クロ口ホルムをカロえ、 生成した沈殿を濾過により除去し、濾液を濃縮した。得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合物 165mg (80%)を得た。
[0527] ステップ 5 (3R)— 3 アミノー 4— (2 フルオロフェ-ル)一 N— (4— (4— (4 フ ルォロフエ-ル)チアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミ ド '塩酸塩 (ィ匕合物 101)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (4— (4 フル
オロフェ -ル)チアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ー3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(25mg, 0. 041mmol)をメタ ノール(lmL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(2mL)をカ卩え、室温で 30 分間撹拌した。これを濃縮して表題ィ匕合物 21mg (95%)を得た。
[0528] 実施例 102
(3R)— 3 ァミノ一 N— (4— (4 シクロプロピル一 5— (2 フルオロフェ -ル)チア ゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ー4一(2 フルオロフェ-ル )ブタナミド '塩酸塩 (ィ匕合物 102)の合成
ステップ 1 1—シクロプロピル一 2— (2—フルオロフェ -ル)ェタン一 1—オン(参考 化合物 353)の合成
滴下ロートを装着した三口フラスコにマグネシウム(1. 35g, 55. 5mmol)を加え、 アルゴン雰囲気下で 1時間撹拌した。ジェチルエーテル(30mL)、 1, 2 ジブロモ ェタン(10 L)をカ卩えた後、氷冷下、 1ーブロモメチルー 2—フルォロベンゼン(10g , 52. 9mmol)のジェチルエーテル(50mL)溶液を 30分かけて滴下した。 0°Cで 1 時間、室温で 30分間撹拌し、 2 フルォ口べンジルマグネシウムブロミドのジェチル エーテル溶液を調製した。アルゴン雰囲気下、シクロプロパンカルボ-トリル(3. 66g , 52. 9mmoL)のジェチルエーテル(50mL)溶液を氷冷下、調製した 2 フルォロ ベンジルマグネシウムブロミドのジェチルエーテル溶液を滴下した。 0°Cで 1時間撹 拌した後、 1N塩酸(50mL)、蒸留水を加え、酢酸ェチルで抽出した。有機層を飽和 食塩水で洗浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題化合物 3. 36 g(36%)を得た。
[0529] ステップ 2 2 ブロモー 1ーシクロプロピノレー 2—(2 フノレオ口フエ-ノレ)エタンー 1 オン (参考化合物 354)の合成
アルゴン雰囲気下、 1—シクロプロピル— 2— (2—フルオロフェ -ル)ェタン— 1— オン(3. 36g, 18. 9mmol)を四塩化炭素(50mL)に溶解し、 N ブロモスクシンイミ ド(3. 7g, 21mmol)、過酸ィ匕べンゾィノレ(305mg, 0. 95mmol)をカロえ、カロ熱還流 した。 10時間後、放冷し、トルエンを加えた。不溶物を濾別し、得られた濾液を濃縮 することにより得た粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合
物 3. 09g (64%)を得た。
[0530] ステップ 3 4 シクロプロピルー5—(2 フルオロフェ -ル)チアゾールー 2 カルボ ン酸ェチル (参考化合物 355)の合成
アルゴン雰囲気下、 2—ブロモー 1ーシクロプロピル 2—(2—フルオロフェ -ル) ェタン一 1—オン(670mg, 2. 6mmol)をエタノールに溶解し、チォォキサム酸ェチ ル(382mg, 2. 9mmol)、ピリジン(317 /z L, 3. 9mmol)を加え、カロ熱還流した。 3 0分後放冷し、 1N塩酸、蒸留水を加え、酢酸ヱチルで抽出し、乾燥後濃縮した。得 られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 593mg ( 78%)を得た。
[0531] ステップ 4 4ーシクロプロピル 5—(2 フルオロフェ -ル)チアゾール(参考化合物
356)の合成
アルゴン雰囲気下、 4ーシクロプロピルー5—(2 フルオロフェ -ル)チアゾールー 2—力ルボン酸ェチル(593mg, 2. Ommol)をエタノール(20mL)に溶解し、蒸留 水(lmL)、水酸ィ匕ナトリウム(277mg, 6. 9mmol)を加え、室温で撹拌した。 1. 5時 間後、 1N塩酸 (8mL)、蒸留水を加え、酢酸ェチルで抽出した。有機層を飽和食塩 水で洗浄し、乾燥後濃縮して得られた粗生成物を 1, 4 ジォキサン(15mL)に溶解 し、濃塩酸(1. 5mL)を加え、加熱還流した。 1時間後放冷し、蒸留水を加え、酢酸 ェチルで抽出した。有機層を飽和食塩水で洗浄し、乾燥後濃縮して得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 267mg (64%)を得た
[0532] ステップ 5 N— (4— (4 シクロプロピルー5—(2 フルオロフェ -ル)チアゾールー 2 ィル)チアン 4 ィル) - 2 メチル - 2-プロパンスルフィンアミド(参考化合物
357)の合成
アルゴン雰囲気下、 4ーシクロプロピルー5—(2 フルオロフェ -ル)チアゾール(2 19mg, 1. Ommol)を THF (7mL)に溶解し、 78°Cに冷却した。 n—ブチルリチウ ム Zへキサン溶液(1. 60M, 0. 63mL, 1. Ommol)を滴下し、 40分間撹拌した。 2 —メチルー N— (チアン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合物 2 05)の粗生成物(263mg,約 1. 2mmol)の THF (3mL)溶液を滴下し、 40°Cで 4
0分間撹拌した。蒸留水、酢酸ェチル、飽和食塩水を加えて室温まで昇温し、分液し た。有機層を飽和食塩水で洗浄し、乾燥後濃縮して得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合物 146mg (約 33%)を得た。
[0533] ステップ 6 (3R)— 3 アミノー N— (4— (4 シクロプロピノレー 5— (2 フノレオロフェ -ル)チアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ー4一(2 フルォ 口フエ-ル)ブタナミド ·塩酸塩 (ィ匕合物 102)の合成
アルゴン雰囲気下、 N— (4—(4ーシクロプロピルー5—(2 フルオロフェ -ル)チ ァゾール - 2 ィル)チアン 4 ィル) - 2 メチル - 2-プロパンスルフィンアミド( 146mg, 0. 33mmol)をメタノール(2mL)に溶解し、 10%塩化水素 Zメタノール溶 液(2mL)を加え、室温で 1時間撹拌した。反応液を濃縮し、減圧下で乾燥して得た 粗生成物を、アルゴン雰囲気下、 DMF (2mL)に溶解し、(3R)—4— (2 フルォロ フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(43mg, 0. 14 mmol)、 HATU (55mg, 0. 14mmol)、 ΤΕΑ(55 L, 0. 39mmol)を加え、室温 で 5. 5時間撹拌した。蒸留水を加え、ジェチルエーテルで抽出し、有機層を蒸留水 、飽和食塩水で洗浄した後、乾燥し濃縮した。得られた粗生成物を、アルゴン雰囲気 下、ジクロロメタン(4mL)に溶解し、氷冷下、 mCPBA( >65%) (67mg, >0. 25m mol)を加え、室温で 1時間撹拌した。アンモニア飽和クロ口ホルムをカ卩え、生成した 沈殿を濾別し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール (0. 5mL)、 T HF (0. 5mL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(lmL)をカ卩え、室温で 4時 間撹拌した後濃縮して表題ィ匕合物 52mgを得た (69%)。
[0534] 実施例 103
(3R)—3 アミノー 4— (3 クロ口フエ-ノレ) N— (4 フエ-ノレチアゾーノレ一 2—ィ ル)— 1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 103)の合成 アルゴン雰囲気下、 4一(4 フエ-ルチアゾールー 2 ィル)チアンー 4ーァミン' 塩酸塩(参考化合物 332)の粗生成物(44mg,約 0. 14mmol)を DMF (2mL)に溶 解し、 (3R)—4— (3 クロ口フエ-ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ)酪酸(45mg, 0. 14mmol)、 HATU (55mg, 0. 14mmol)、 ΤΕΑ(55 L
, 0. 39mmol)をカ卩え、室温で 3時間撹拌した。蒸留水を加え、ジェチルエーテルで 抽出し、有機層を蒸留水、飽和食塩水で洗浄した後、乾燥し濃縮した。得られた粗 生成物をジクロロメタン(4mL)に溶解し、氷冷下、 mCPBA( >65%) (67mg, >0. 25mmol)をカ卩え、室温で 1時間撹拌した。アンモニア飽和クロ口ホルムをカ卩え、生成 した沈殿を濾別し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール (0. 5mL) に溶解し、 10%塩ィ匕水素 Zメタノール溶液(lmL)を加え、室温で 4時間撹拌した。 飽和炭酸水素ナトリウム水溶液を加え酢酸ェチルで抽出し、有機層を飽和食塩水で 洗浄後、乾燥し濃縮した。得られた粗生成物を薄層クロマトグラフィーで精製し、精製 物をメタノール溶液とし、 10%塩ィ匕水素 Zメタノール溶液を加えて、塩酸塩とした。こ れを濃縮し、表題ィヒ合物 49mg (約 42%)を得た。
実施例 104
(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (4, 5 ジフエ-ルチアゾール —2—ィル)—1, 1—ジォキソチアン— 4—ィル)ブタナミド '塩酸塩 (ィ匕合物 104)の 合成
ステップ 1 4, 5 ジフエ-ルチアゾール(参考化合物 358)の合成
アルゴン雰囲気下、 2 ブロモ—1, 2 ジフエ-ルエタノン(386mg, 1. 5mmol) をエタノール(15mL)に溶解し、チォォキサム酸ェチル(200mg, 1. 5mmol)をカロ え、加熱還流した。 18時間後放冷し、蒸留水、飽和炭酸水素ナトリウム水溶液を加え 酢酸ェチルで抽出し、有機層を乾燥後濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで粗精製し、得られた粗精製物を、アルゴン雰囲気下、エタノー ル(5mL)に溶解し、蒸留水(lmL)、水酸化ナトリウム(200mg, 5. Ommol)を加え 、室温で撹拌した。 8時間後、 1N塩酸(7mL)、蒸留水を加え、酢酸ェチルで抽出し た。有機層を飽和食塩水で洗浄し、乾燥後濃縮して得られた粗生成物を、アルゴン 雰囲気下、 1, 4 ジォキサン(7mL)に溶解し、濃塩酸 (0. 7mL)をカ卩え、加熱還流 した。 4時間後放冷し、蒸留水を加え、酢酸ェチルで抽出し、有機層を飽和食塩水で 洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題ィ匕合物 168mg (49%)を得た。
[0536] ステップ 2 2—メチルー N— (4— (4, 5 ジフエ-ルチアゾールー 2 ィル)チアン 4 ィル) 2 プロパンスルフィンアミド(参考化合物 359)の合成
アルゴン雰囲気下、 4, 5 ジフエ-ルチアゾール(168mg, 0. 71mmol)を THF ( 5mL)に溶解し、 78°Cに冷却した。 n—ブチルリチウム/へキサン溶液(1. 60M, 0. 44mL, 0. 71mmol)を滴下し、 45分間撹拌した。 2—メチル—N— (チアン— 4 イリデン) 2 プロパンスルフィンイミド(参考化合物 205) (219mg,約 1. Ommo 1)の THF (3mL)溶液を滴下し、 1. 5時間かけて 10°Cまで昇温した。蒸留水、酢酸 ェチル、飽和食塩水を加え、分液し、有機層を飽和食塩水で洗浄後、乾燥し濃縮し た。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 67 mg (約 21%)を得た。
[0537] ステップ 3 (3R)— 4— (2 フルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ)—N—(4— (4, 5 ジフエ-ルチアゾールー 2—ィル) 1, 1ージォ キソチアン— 4—ィル)ブタナミド (参考化合物 360)の合成
アルゴン雰囲気下、 2—メチルー N— (4— (4, 5 ジフエ-ルチアゾールー 2—ィ ル)チアン一 4—ィル) 2 プロパンスルフィンアミド(67mg, 0. 15mmol)をメタノ ール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(2mL)をカ卩え、室温で 2時 間撹拌した。反応液を濃縮し、減圧下で乾燥して得た粗生成物を、アルゴン雰囲気 下、 DMF (4mL)に溶解し、(3R)—4一(2 フルオロフヱ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(54mg, 0. 14mmol)、 HATU (56mg, 0. 1 5mmol)、 TEA (59 μ L, 0. 42mmol)を加え、室温で 20時間撹拌した。蒸留水を 加えジェチルエーテルで抽出し、有機層を飽和食塩水で洗浄した後、乾燥し濃縮し た。得られた粗生成物をジクロロメタン(3mL)に溶解し、氷冷下、 mCPBA( >65%) (52mg, >0. 20mmol)を加え、室温で 3時間撹拌した。アンモニア飽和クロ口ホル ム(2mL)を加え、生成した沈殿を濾別し、濾液を濃縮して得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 50mg (51%)を得た。
[0538] ステップ 4 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (4, 5 ジフ ェ-ルチアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩 (化合物 104)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ) N— (4— (4, 5 ジフエ-ルチアゾールー 2—ィル) 1 , 1—ジォキソチアン一 4—ィル)ブタナミド(50mg, 0. 075mmol)をメタノール(lm L)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(2mL)をカ卩え、室温で 4時間撹拌した 後濃縮し、表題ィ匕合物 45mg (定量的)を得た。
[0539] 実施例 105
(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (5—メチル—4 フエ-ルチ ァゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕合物 105)の合成
ステップ 1 5 メチル 4 フエ-ルチアゾール 2 カルボン酸ェチル(参考化合 物 361)の合成
アルゴン雰囲気下、 2 ブロモ—2—メチルァセトフエノン(426mg, 2. Ommol)を エタノール(20mL)に溶解し、チォォキサム酸ェチル(267mg, 2. Ommol)をカロえ、 加熱還流した。 27時間後放冷し、蒸留水を加え酢酸ェチルで抽出し、有機層を飽和 食塩水で洗浄した後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 284mg (57%)を得た。
[0540] ステップ 2 5—メチルー 4 フエ-ルチアゾールー 2—力ルボン酸(参考化合物 362) の合成
アルゴン雰囲気下、 5ーメチルー 4 フエ-ルチアゾールー 2—力ルボン酸ェチル( 284mg, 1. 15mmol)をエタノール(lOmL)に溶解し、蒸留水(lmL)、水酸化ナト リウム(223mg)を加え、室温で撹拌した。 8時間後、 1N塩酸(lOmL)、蒸留水をカロ え、酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄した後、乾燥し濃縮して表題 化合物 244mg (97%)を得た。
[0541] ステップ 3 5—メチルー 4 フエ-ルチアゾール(参考化合物 363)の合成
アルゴン雰囲気下、 5ーメチルー 4 フエ-ルチアゾールー 2—力ルボン酸(244m g, 1. l lmmol)を 1, 4 ジォキサン(lOmL)に溶解し、濃塩酸(lmL)をカ卩え、加熱 還流した。 2時間後放冷し、反応液を飽和炭酸水素ナトリウム水溶液と酢酸ェチルの 混合液に加え、分液した。水層を酢酸ェチルで抽出し、有機層を集めて飽和食塩水
で洗浄し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 165mg (86%)を得た。
[0542] ステップ 4 2—メチルー N— (4— (5—メチルー 4 フエ-ルチアゾールー 2 ィル) チアン 4 ィル) 2 プロパンスルフィンアミド(参考化合物 364)の合成
アルゴン雰囲気下、 5—メチル 4 フエ-ルチアゾール(165mg, 0. 94mmol) を THF (5mL)に溶解し、 78°Cに冷却した。 n—ブチルリチウム Zへキサン溶液(1 . 60M, 0. 59mL, 0. 94mmol)を滴下し、 1時間撹拌した。 2—メチル—N— (チア ン― 4 イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(219 mg,約 1. Ommol)の THF (3mL)溶液を滴下し、 1時間かけて 0°Cまで昇温した。蒸 留水、飽和食塩水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、 乾燥後濃縮したて得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 172mg (約 46%)を得た。
[0543] ステップ 5 (3R)— 3 ァミノ一 4— (2 フノレオ口フエ-ノレ)一 N— (5—メチノレ一 4— フエ-ルチアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸 塩 (ィ匕合物 105)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(5—メチルー 4 フエ-ルチアゾールー 2—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(172mg, 0. 44mmol) をメタノール(2mL)に溶解し、 10%塩ィ匕水素メタノール溶液(3mL)をカ卩え、室温で 1時間撹拌した。反応液を濃縮し、減圧下で乾燥して得られた粗生成物 135mgのう ち 34mgを、アルゴン雰囲気下、 DMF (3mL)に溶解し、(3R)—4— (2 フルオロフ ェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. 10m mol)、 HATU (40mgゝ 0. 1 lmmol) , TEA (42 μ L,0. 30mmol)を加え、室温で 1 2時間撹拌した。蒸留水を加えジェチルエーテルで抽出し、有機層を飽和食塩水で 洗浄した後、乾燥し濃縮した。得られた粗生成物をジクロロメタン(3mL)に溶解し、 氷冷下、 mCPBA( >65%) (51mg, >0. 19mmol)をカ卩え、室温で 45分間撹拌し た。アンモニア飽和クロ口ホルム(3mL)を加え、生成した沈殿を濾別し、濾液を濃縮 して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。得られた 粗精製物を、アルゴン雰囲気下、 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、
室温で 16時間撹拌した後濃縮して表題ィ匕合物 38mg (67%)を得た。
[0544] 実施例 106
(3R)—3 ァミノ一 4— (2 フルオロフェ -ル) -N- (4— (4— (4—メトキシフエ- ル)チアゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '塩酸塩( 化合物 106)の合成
ステップ 1 4一(4ーメトキシフエ-ル)チアゾールー 2—力ルボン酸ェチル(参考化合 物 365)の合成
アルゴン雰囲気下、 2 ブロモ—4,—メトキシァセトフエノン(343mg, 1. 5mmol) 、チォォキサム酸ェチル(200mg, 1. 5mmol)をエタノール(15mL)に溶解し、カロ 熱還流した。 20分後放冷し、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで 抽出した。有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 297mg (75%)を得た。
[0545] ステップ 2 4—(4ーメトキシフヱ-ル)チアゾール(参考化合物 366)の合成
4— (4—メトキシフエ-ル)チアゾール—2—カルボン酸ェチル(297mg, 1. lmm ol)のエタノール(15mL)溶液に水酸化ナトリウム(200mg、 5. Ommol)、蒸留水(3 mL)をカ卩え、室温で攪拌した。 2時間後、 1N塩酸 (8mL)をカ卩えた後、酢酸ェチルに て抽出した。有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。アルゴン雰囲気下 、得られた粗生成物を、 4一(4ーメトキシフエ-ル)チアゾールー 2—力ルボン酸の粗 生成物を 1, 4 ジォキサン(l lmL)、濃塩酸(lmL)に溶解し、加熱還流した。 5時 間後放冷し、蒸留水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水にて洗浄 後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 214mg (定量的)を得た。
[0546] ステップ 3 N— (4— (4— (4—メトキシフエ-ル)チアゾールー 2 ィル)チアンー 4 ィル) 2—メチルー 2 プロパンスルフィンアミド(参考化合物 367)の合成
アルゴン雰囲気下、 4 (4ーメトキシフエ-ル)チアゾール(214mg, 0. 56mmol) の THF (1. 3mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 60M, 0. 35mL, 0. 56mmol)をゆっくりと滴下し、 1時間攪拌した。 2—メチノレ一 N - (チアン— 4—イリデン)—2 プロパンスルフィンイミド (参考化合物 205)の粗生成
物(122mg,約 0. 56mmol)の THF (1. 5mL)溶液を滴下し、 0°Cまでゆっくり昇温 した。 1時間後、メタノールと蒸留水を加え、酢酸ェチルで抽出した。有機層を飽和食 塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 41mg (約 18%)を得た。
[0547] ステップ 4 (3R)—4 (2 フノレオ口フエ-ノレ) N— (4— (4— (4—メトキシフエ- ル)チアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 368)の合成
N- (4一(4一(4ーメトキシフエ-ル)チアゾールー 2 ィル)チアンー 4 ィル)ー2 ーメチルー 2 プロパンスルフィンアミド(41mg, 0. lmmol)を 10%塩化水素/メタノ ール溶液 (0. 75mL)に溶解し、室温で 1時間攪拌した。濃縮して得られた粗生成物 を、アルゴン雰囲気下、 DMF (3mL)に溶解し、(3R)—4— (2—フルオロフヱ-ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(30mg, 0. lmmol) , Η ATU (38mg, 0. lmmol)、 TEA (0. 042mL, 0. 3mmol)を加え、室温で攪拌し た。 15時間後、蒸留水を加え、ジェチルエーテルで抽出した。有機層を飽和食塩水 にて洗浄後、乾燥し濃縮した。得られた粗生成物をジクロロメタン(lmL)に溶解し、 mCPBA( >65%) (48mg、 >0. 18mmol)を 2回に分けて加え、室温で 20分間攪 拌した。飽和炭酸水素ナトリウム水溶液、飽和チォ硫酸ナトリウム水溶液を加え激しく 攪拌した後、ジェチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、乾燥 し濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕 合物 13mg (22%)を得た。
[0548] ステップ 5 (3R)—3 アミノー 4 (2 フルオロフェ -ル) N— (4— (4— (4—メト キシフエ-ル)チアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド' 塩酸塩 (ィ匕合物 106)の合成
アルゴン雰囲気下、 (3R)—4— (2 フルオロフヱ-ル) N— (4— (4— (4—メトキ シフエ-ル)チアゾール 2—ィル) 1, 1—ジォキソチアン一 4—ィル) 3— (2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(13mg, 0. 02mmol)を 10%塩 化水素 Zメタノール溶液 (0. 3mL)に溶解し、室温で攪拌した。 17時間後、飽和炭 酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗
浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 (フリー塩基) 7. 5mg (80%)を得た。表題ィ匕合物 (フリー塩基)をメタ ノールに溶解し、 10%塩ィ匕水素 Zメタノール溶液を滴下して塩ィ匕した。この溶液を濃 縮することにより、表題ィ匕合物 (塩酸塩) 7mgを得た。
[0549] 実施例 107
(3R)—3 アミノー N— (4— (4— (4 クロ口フエ-ル)チアゾール 2—ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ルブタナミド '塩酸 塩 (ィ匕合物 107)の合成
ステップ 1 4一(4ークロロフヱ-ル)チアゾールー 2—力ルボン酸ェチル(参考化合 物 369)の合成
アルゴン雰囲気下、 2 ブロモ—4,—クロロアセトフエノン(368mg, 1. 5mmol)、 チォォキサム酸ェチル(200mg, 1. 5mmol)をエタノール(15mL)に溶解し、加熱 還流した。 20分後放冷し、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽 出した。有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリ 力ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 397mg (97%)を得た。
[0550] ステップ 2 4—(4ークロロフヱ-ル)チアゾール(参考化合物 370)の合成
4一(4一クロ口フエ-ル)チアゾールー 2一力ルボン酸ェチル(397mg, 1. 5mmol )をエタノール(15mL)に溶解し、水酸ィ匕ナトリウム(200mg, 6. 7mmol)、蒸留水( 3mL)を加え、室温で攪拌した。 2時間後、 1N塩酸 (8mL)を加え濃縮した後、酢酸 ェチルにて抽出し、乾燥後濃縮した。得られた粗生成物を、アルゴン雰囲気下、濃塩 酸(1. 5mL)、 1, 4 ジォキサン(15mL)に溶解し、加熱還流した。 5時間後放冷し 、蒸留水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水にて洗浄後、乾燥し 濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕 合物 263mg (91%)を得た。
[0551] ステップ 3 4— (4— (4 クロ口フエ-ル)チアゾール—2—ィル)チアン— 4 アミン( 参考化合物 371)の合成
アルゴン雰囲気下、 4一(4 クロ口フエ-ル)チアゾール(263mg, 1. 34mmol)の ジェチルエーテル (5mL)溶液を一 78°C〖こ冷却し、 n—ブチルリチウム Zへキサン溶
液(1. 60M, 0. 84mL, 1. 34mmol)をゆっくりと滴下した。滴下終了後— 78。Cで 1 時間攪拌し、 4—ォキソチアン O ベンジルォキシム(参考化合物 200) (445mg, 1. 61mmol)のジェチルエーテル(1. 7mL)溶液を滴下し、 0°Cまでゆっくり昇温し た。 1時間後、メタノールと蒸留水を加え、酢酸ェチルで抽出し、有機層を飽和食塩 水にて洗浄後、乾燥し濃縮した。得られた粗生成物を、アルゴン雰囲気下、ァセトニ トリル(21mL)、蒸留水(1. 34mL)に溶解し、モリブデンへキサカルボ-ル(353mg , 1. 34mmol)を加え、加熱還流した。 2時間後放冷し、反応溶液をセライト濾過し、 濾液に飽和炭酸水素ナトリウム水溶液を加え、ジクロロメタンで抽出した。有機層を飽 和食塩水にて洗浄後、乾燥し濃縮し、得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 35mg (8%)を得た。
[0552] ステップ 4 (3R)—N— (4— (4— (4 クロ口フエ-ノレ)チアゾーノレ一 2—ィノレ)一 1, 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 372)の合成
アルゴン雰囲気下、 4一(4一(4 クロ口フエ-ル)チアゾールー 2 ィル)チアンー 4ーァミン(35mg, 0. l lmmol)を DMF (3mL)に溶解し、 (3R)— 4一(2, 4, 5 ト リフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(33m g, 0. l lmmol)、 HATU (42mg, 0. l lmmol) , TEA(0. 047mL, 0. 33mmol) を加え、室温で攪拌した。 16時間後、蒸留水を加え、ジェチルエーテルで抽出し、 有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をジクロ口メタ ン(1. lmL)に溶解し、 mCPBA( >65%) (36mg、 >0. 14mmol)を 2回に分けて 加え、室温で攪拌した。 20分後、飽和炭酸水素ナトリウム水溶液、飽和チォ硫酸ナト リウム水溶液を加えて激しく攪拌し、ジェチルエーテルで抽出した。有機層を飽和食 塩水にて洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合物 14mg (19%)を得た。
[0553] ステップ 5 (3R)— 3 アミノー N— (4— (4— (4 クロ口フエ-ル)チアゾール 2— ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ブ タナミド ·塩酸塩 (ィ匕合物 107)の合成
アルゴン雰囲気下、 (3R)— N— (4— (4— (4—クロロフヱ-ル)チアゾール—2—ィ
ル)— 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3 一(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(14mg, 0. 02mmol)を 10%塩ィ匕水素 Zメタノール溶液 (0. 4mL)に溶解し、室温で攪拌した。 6時間後、飽 和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出した。有機層を飽和食塩水 で洗浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題化合物 (フリー塩基) 5mg (42%)を得た。表題化合物 (フリー塩基)をメ タノールに溶解し、 10%塩ィ匕水素 Zメタノール溶液を滴下して塩ィ匕した。この溶液を 濃縮することにより、表題ィ匕合物 (塩酸塩) 5mgを得た。
[0554] 実施例 108
(3R)— 3 ァミノ— N— (4— (4— (4 トリフルォロメチルフエ-ル)チアゾール—2 —ィル)— 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル) ブタナミド ·塩酸塩 (ィ匕合物 108)の合成
ステップ 1 4一(4 トリフルォロメチルフエ-ル)チアゾール(参考化合物 373)の合 成
4 トリフルォロメチルァセトフエノン(995mg, 5. 3mmol)の THF (lOmL)溶液に 三臭化フエ-ルトリメチルアンモ -ゥム(2. 19g, 5. 8mmol)を加え、室温で攪拌した 。 18時間後、エタノール(20mL)、チォ尿素(427mg, 5. 6mmol)を加え加熱還流 した。 4時間後放冷し、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、 有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を 50%硫酸( 6. 2mL)、TFA(6. 2mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(260mg , 3. 76mmol)の蒸留水(0. 9mL)溶液をゆっくりと加え、 30分間撹拌した。ホスフィ ン酸(12mL)を加え、 17時間後(氷浴中の氷が溶けるに従い、次第に昇温)、 1N水 酸ィ匕ナトリウム水溶液で反応液をアルカリ性とした。酢酸ェチルで抽出し、有機層を 飽和食塩水にて洗浄後、乾燥し濃縮し、得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 290mg (40%)を得た。
[0555] ステップ 2 4—(4一(4 トリフルォロメチルフエ-ル)チアゾールー 2 ィル)チアン
4ーァミン (参考化合物 374)の合成
アルゴン雰囲気下、 4— (4 トリフルォロメチルフエ-ル)チアゾール(290mg, 1.
26mmol)のジェチルエーテル(4. 3mL)溶液を—78°Cに冷却し、 n—ブチルリチウ ム Zへキサン溶液(1. 59M, 0. 79mL, 1. 26mmol)をゆっくりと滴下した。滴下終 了後— 78°Cで 1時間攪拌し、 4—ォキソチアン O ベンジルォキシム(参考化合物 200) (420mg, 1. 51mmol)のジェチルエーテル(2mL)溶液を加え、 0°Cまでゆつ くり昇温した。 1時間後、メタノールと蒸留水を加え、酢酸ェチルで抽出し、有機層を 飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を、アルゴン雰囲気下 、ァセトニトリル(20mL)、蒸留水(1. 26mL)に溶解し、モリブデンへキサカルボ-ル (332mg, 1. 26mmol)を加え、加熱還流した。 3時間後放冷し、反応溶液をセライト 濾過し、濾液に飽和炭酸水素ナトリウム水溶液を加え、ジクロロメタンで抽出した。有 機層を飽和食塩水にて洗浄後、乾燥し濃縮し、得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 160mg (37%)を得た。
[0556] ステップ 3 (3R)—N— (4— (4— (4 トリフルォロメチルフエ-ル)チアゾールー 2— ィル)チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 375)の合成
アルゴン雰囲気下、 4— (4— (4—トリフルォロメチル)フエ-ルチアゾール—2—ィ ル)チアン— 4 ァミン(160mg, 0. 46mmol)を DMF (12mL)に溶解し、(3R)— 4 一(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)酪酸(138mg, 0. 46mmol)、 HATU (177mg, 0. 46mmol)、 TEA(0. 195 mL, 1. 39mmol)をカ卩え、室温で攪拌した。 17時間後、蒸留水を加え、ジェチルェ 一テルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 44mg (14%)を得た
[0557] ステップ 4 (3R)—N— (4— (4— (4 トリフルォロメチルフエ-ル)チアゾールー 2— ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3 — (2—メチル—2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 376)の合 成
(3R)— N—(4一(4一(4 トリフルォロメチルフエ-ル)チアゾールー 2 ィル)チ アン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ
ポキシカルボ-ルァミノ)ブタナミド(44mg, 0. 06mmol)をジクロロメタン(0. 66mL )に溶解し、 mCPBA( >65%) (24mg、 >0. O9mmol)を 2回に分けて加え、室温 で攪拌した。 20分後、飽和炭酸水素ナトリウム水溶液、飽和チォ硫酸ナトリウム水溶 液を加えて激しく攪拌し、ジェチルエーテルで抽出した。有機層を飽和食塩水にて 洗浄後、乾燥し濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題ィ匕合物 37mg (80%)を得た。
[0558] ステップ 5 (3R)— 3 ァミノ一 N— (4— (4— (4 トリフルォロメチルフエ-ル)チア ゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルォロ フエニル)ブタナミド ·塩酸塩 (ィ匕合物 108)の合成
(3R)—N— (4— (4—(4 トリフルォロメチルフエ-ル)チアゾールー 2—ィル) 1 , 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(37mg, 0. 06mmol)を 10%塩 化水素 Zメタノール溶液 (0. 5mL)に溶解し、室温で攪拌した。 15時間後、飽和炭 酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗 浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 (フリー塩基) 21mg (68%)を得た。表題ィ匕合物 (フリー塩基)をメタノ ールに溶解し、 10%塩ィ匕水素 Zメタノール溶液を滴下して塩ィ匕した。この溶液を濃 縮することにより、表題ィ匕合物 (塩酸塩) 2 lmgを得た。
[0559] 実施例 109
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(4一(2—メトキ シフエ-ル)チアゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド' 塩酸塩 (ィ匕合物 109)の合成
ステップ 1 4一(2—メトキシフヱニル)チアゾール(参考化合物 377)の合成
2 ブロモ 2, 一メトキシァセトフエノン(lg, 4. 4mmol)のエタノール(20mL)溶 液にチォ尿素(353mg, 4. 6mmol)をカ卩ぇ加熱還流した。 3時間後放冷し、飽和炭 酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、有機層を飽和食塩水にて洗 浄後、乾燥し濃縮した。得られた粗生成物を 50%硫酸(5mL)、 TFA(5mL)に溶解 し、 15°Cに冷却した。亜硝酸ナトリウム(403mg, 5. 82mmol)の蒸留水(0. 8mL
)溶液をゆっくりと加え、 30分間撹拌した。ホスフィン酸(20mL)を加え、 16時間後( 氷浴中の氷が溶けるに従い、次第に昇温)、 1N水酸化ナトリウム水溶液で反応液を アルカリ性とした。酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃 縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 537mg (64%)を得た。
ステップ 2 (3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (4 一(2—メトキシフエ-ル)チアゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル) ブタナミド '塩酸塩 (ィ匕合物 109)の合成
アルゴン雰囲気下、 4 (2—メトキシフエ-ル)チアゾール(537mg, 2. 8mmol)の ジェチルエーテル (lOmL)溶液を 78°Cに冷却し、 n ブチルリチウム Zへキサン 溶液(1. 60M, 1. 75mL, 2. 8mmol)をゆっくりと滴下した。滴下終了後— 78。Cで 1時間攪拌し、 4ーォキソチアン O ベンジルォキシム(参考化合物 200) (698mg , 3. 3mmol)のジェチルエーテル(4mL)溶液を加え、 0°Cまでゆっくり昇温した。 1 時間後、メタノールと蒸留水を加え、酢酸ェチルで抽出し、有機層を飽和食塩水にて 洗浄後、乾燥し濃縮した。得られた粗生成物を、アルゴン雰囲気下、ァセトニトリル (4 2mL)、蒸留水(2. 8mL)に溶解し、モリブデンへキサカルボ-ル(739mg, 2. 8m mol)を加え、加熱還流した。 2時間後放冷し、反応溶液をセライト濾過し、濾液に飽 和炭酸水素ナトリウム水溶液を加え、ジクロロメタンで抽出した。有機層を飽和食塩水 にて洗浄後、乾燥し濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー で粗精製した。得られた粗精製物を、アルゴン雰囲気下、 DMF (16mL)に溶解し、 ( 3R) -4- (2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカル ボ-ルァミノ)酪酸(181mg, 0. 61mmol)、 HATU (232mg, 0. 61mmol)、TEA (0. 257mL, 1. 83mmol)をカ卩え、室温で攪拌した。 16時間後、蒸留水を加え、ジ ェチルエーテルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物 (113mg, 0. 19mmol)をジクロロメタン(1. 9mL)に溶解し、 mCPBA( >65%) (9 6mg、 >0. 36mmol)を 2回に分けてカ卩え、室温で攪拌した。 40分後、飽和炭酸水 素ナトリウム水溶液、飽和チォ硫酸ナトリウム水溶液を加えて激しく攪拌し、ジェチル
エーテルで抽出した。有機層を飽和食塩水にて洗浄後、乾燥し濃縮して得られた粗 生成物をシリカゲルカラムクロマトグラフィーで粗精製した。アルゴン雰囲気下、得ら れた粗精製物(63mg, 0. lmmol)を 10%塩ィ匕水素 Zメタノール溶液(1. 8mL)に 溶解し、室温で攪拌した。 6時間後、濃縮して表題ィ匕合物 63mg (4%)を得た。
[0561] 実施例 110
(3R)— 3 ァミノ— N— (4— (4— (3 トリフルォロメチルフエ-ル)チアゾール—2 —ィル)— 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル) ブタナミド ·塩酸塩 (化合物 110)の合成
ステップ 1 4— (3—トリフルォロメチルフエ-ル)チアゾール(参考化合物 378)の合 成
3 トリフルォロメチルァセトフエノン(lg, 5. 3mmol)の THF (lOmL)溶液に三臭 化フエ-ルトリメチルアンモ -ゥム(2. 2g, 5. 8mmol)を加え、室温で攪拌した。 2時 間後、エタノール(20mL)、チォ尿素(433mg, 5. 6mmol)をカ卩ぇ加熱還流した。 3 時間後放冷し、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、有機層 を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物を 50%硫酸(5. 7m L)、TFA(5. 7mL)に溶解し、 15°Cに冷却した。亜硝酸ナトリウム(236mg, 3. 4 2mmol)の蒸留水(0. 8mL)溶液ゆっくりとを加え、 30分間撹拌した。ホスフィン酸( l lmL)を加え、 17時間後(氷浴中の氷が溶けるに従い、次第に昇温)、 1N水酸ィ匕 ナトリウム水溶液で反応液をアルカリ性とした。酢酸ェチルで抽出し、有機層を飽和 食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグ ラフィ一で精製し、表題ィ匕合物 300mg (25%)を得た。
[0562] ステップ 2 (3R)—N— (4— (4— (3 トリフルォロメチルフエ-ル)チアゾール—2— ィル)チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 379)の合成
アルゴン雰囲気下、 4— (3—トリフルォロメチルフエ-ル)チアゾール(298mg, 1. 3mmol)の THF (4mL)溶液を—78°Cに冷却し、 n—ブチルリチウム Zへキサン溶 液(1. 59M, 0. 82mL, 1. 3mmol)をゆっくりと滴下した。滴下終了後— 78。Cで 30 分間攪拌し、 2—メチル N— (チアン— 4—イリデン)—2 プロパンスルフィンイミド
(参考化合物 205)の粗生成物(285mg,約 1. 3mmol)の THF (2. 5mL)溶液を加 え、 0°Cまでゆっくり昇温した。 1時間後、メタノールと蒸留水をカ卩え、酢酸ェチルで抽 出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物を 10% 塩化水素 Z メタノール溶液(1. 86mL)に溶解し、室温で 1時間攪拌した。濃縮して得られた粗生 成物を、アルゴン雰囲気下、 DMF (6. 5mL)に溶解し、(3R)—4— (2, 4, 5—トリフ ルォロフエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(74mg, 0. 25mmol)、 HATU (95mg, 0. 25mmol)、 TEA(0. 105mL, 0. 75mmol)を 加え、室温で攪拌した。 16時間後蒸留水を加え、ジェチルエーテルで抽出し、有機 層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラム クロマトグラフィーで精製し、表題ィ匕合物 103mg (約 12%)を得た。
[0563] ステップ 3 (3R)—N— (4— (4— (3 トリフルォロメチルフエ-ル)チアゾール—2— ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3 — (2 メチル 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 380)の合 成
(3R)— N— (4— (4— (3—トリフルォロメチルフエ-ル)チアゾール—2—ィル)チ アン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ)ブタナミド(103mg, 0. 16mmol)をジクロロメタン(1. 56m L)に溶解し、 mCPBA ( >65%) (112mg、 >0. 42mmol)を 2回に分けて加え、室 温で攪拌した。 40分後、飽和炭酸水素ナトリウム水溶液、飽和チォ硫酸ナトリウム水 溶液を加えて激しく攪拌し、ジェチルエーテルで抽出した。有機層を飽和食塩水に て洗浄後、乾燥し濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題ィ匕合物 37mg (35%)を得た。
[0564] ステップ 4 (3R)— 3 ァミノ— N— (4— (4— (3 トリフルォロメチルフエ-ル)チア ゾール—2—ィル)—1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルォロ フエニル)ブタナミド ·塩酸塩 (化合物 110)の合成
アルゴン雰囲気下、(3R)— N— (4— (4— (3—トリフルォロメチルフエ-ル)チアゾ ール一 2—ィル) 1, 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフ
ェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(37mg, 0. 0 5mmol)を 10%塩ィ匕水素 Zメタノール溶液(lmL)に溶解し、室温で攪拌した。 14 時間後、濃縮して表題ィ匕合物 19mg (57%)を得た。
[0565] 実施例 111
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (1—メチル 4 —フエ-ルイミダゾ一ルー 2—ィル) 1, 1—ジォキソチアン一 4—ィル)ブタナミドニ塩酸塩 (化合物 111)の合成
ステップ 1 1ーメチルー 4 フエ-ルイミダゾール(参考化合物 381)の合成
4—フエ-ルー 1H—イミダゾール(576mg, 4mmol)の DMF (2mL)溶液に 60% 水素化ナトリウム(160mg、 4mmol)を加え、室温で攪拌した。 10分後、ヨウ化メチル (0. 3mL、4. 8mmol)を加え、 1時間撹拌し、飽和塩化アンモ-ゥム水溶液を加え、 酢酸ェチルで抽出した。有機層を飽和食塩水にて洗浄後、乾燥し濃縮し、得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 377mg (59%) を得た。
[0566] ステップ 2 2—メチル—N— (4— (1—メチル—4 フエ-ルイミダゾ一ルー 2—ィル) チアンー 4 ィル) 2 プロパンスルフィンアミド(参考化合物 382)の合成
アルゴン雰囲気下、 1—メチル 4 フエ-ルイミダゾール(377mg, 2. 4mmol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 60M , 1. 5mL, 2. 4mmol)をゆっくりと滴下した。滴下終了後— 78°Cで 20分間攪拌し、 2—メチル N— (チアン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(522mg,約 2. 4mmol)の THF (6mL)溶液を加え、 0°Cまでゆつ くり昇温した。 2時間後、メタノールと蒸留水を加え、酢酸ェチルで抽出し、有機層を 飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロ マトグラフィ一で精製し、表題ィ匕合物 300mg (33%)を得た。
[0567] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1—メチル 4 —フエ-ルイミダゾール一 2—ィル) 1, 1—ジォキソチアン一 4—ィル) 3— (2—メ チル— 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 383)の合成
2—メチル N— (4— (1—メチル—4 フエ-ルイミダゾ一ルー 2—ィル)チアン—
4—ィル)—2 プロパンスルフィンアミド(300mg, 0. 8mmol)を 10% 塩化水素 Z メタノール溶液 (6mL)に溶解し、室温で 1時間攪拌した。濃縮して得られた粗生成物 を、アルゴン雰囲気下、 DMF (21mL)に溶解し、(3R)—4— (2, 4, 5—トリフルォロ フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(265mg, 0. 8 mmol)、 HATU (302mg, 0. 8mmol)、 TEA(0. 335mL, 2. 4mmol)を加え、室 温で攪拌した。 14時間後、蒸留水を加え、ジェチルエーテルで抽出し、有機層を飽 和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で粗精製した。得られた粗精製物をジクロロメタン(5. lmL)に溶解し、 m CPBA( >65%) (220mg, >0. 83mmol)を 2回に分けて加え、室温で攪拌した。 40分後、飽和炭酸水素ナトリウム水溶液、飽和チォ硫酸ナトリウム水溶液を加えて激 しく攪拌し、ジェチルエーテルで抽出した。有機層を飽和食塩水にて洗浄後、乾燥し 濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 79mg (25%)を得た。
[0568] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1 —メチルー 4 フエ-ルイミダゾ一ルー 2—ィル)—1, 1—ジォキソチアン— 4—ィル) ブタナミド ·二塩酸塩 (化合物 111)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (1— メチル—4 フエ-ルイミダゾ一ルー 2—ィル)—1, 1—ジォキソチアン— 4—ィル) - 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(25mg, 0. 04mmol) を 10%塩ィ匕水素 Zメタノール溶液(1. 2mL)に溶解し、室温で攪拌した。 15時間後 、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出した。有機層を飽和食塩 水で洗浄し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題化合物 (フリー塩基) 7mg (32%)を得た。表題化合物 (フリー塩基)を メタノールに溶解し、 10%塩ィ匕水素 Zメタノール溶液を滴下して塩ィ匕した。この溶液 を濃縮することにより、表題化合物(二塩酸塩) 5mgを得た。
[0569] 実施例 112
(3R)—3 アミノー 4— (2 フルオロフェ -ル) N— (4— (1—メチルベンゾイミダ ゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミド '二塩酸塩 (ィ匕合物
112)の合成
ステップ 1 (3R)— 4— (2 フルオロフェ -ル) N— (4— (1—メチルベンゾイミダゾ 一ルー 2 ィル) - 3- (2—メチル—2 プロポキシカルボ-ルァミノ)チアン— 4—ィ ル)ブタナミド (参考化合物 384)の合成
アルゴン雰囲気下、 1 メチルベンゾイミダゾール(57mg, 0. 43mmol)の THF (3 mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(1. 59M, 0. 25 mL, 0. 40mmol)を滴下した。 30分後、 2—メチル—N— (チアン— 4—イリデン) 2 プロパンスルフィンイミド(参考化合物 205)の粗生成物(64mg,約 0. 29mmol) の THF (3mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し、 3. 5時間 後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し、有機層を飽和食 塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で粗精製し、 2—メチル N— (4— (1—メチルベンゾイミダゾール— 2—ィル) チアン— 4—ィル)—2 プロパンスルフィンアミドの粗精製物 5mgを得た。この反応 を繰り返して得た 2—メチルー N—(4一 (1一メチルベンゾイミダゾールー 2 ィル)チ アン— 4—ィル)—2 プロパンスルフィンアミドの粗精製物 18mgを、アルゴン雰囲気 下、メタノール(2mL)に溶解し、 10%塩ィ匕水素 Zメタノール(2mL)を加え、室温で 2 時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶 解し、 (3R) -4- (2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ -ルァミノ)酪酸(16mg, 0. 05mmol)、 ΝΕΜ (23 Ι^, 0. 18mmol)、 HATU (21 mg, 0. 06mmol)を加え、室温で攪拌した。 14時間後、飽和炭酸水素ナトリウム水 溶液を力卩ぇジェチルエーテルで抽出し、蒸留水及び飽和食塩水にて洗浄後、乾燥 し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 16mg (約 3%)を得た。
ステップ 2 (3R) -4- (2 フルオロフェ -ル)—N— (4—(1 メチルベンゾイミダゾ 一ルー 2 ィル) 1, 1—ジォキソチアン— 4—ィル)—3— (2—メチル—2—プロボ キシカルボニルァミノ)ブタナミド (参考化合物 385)の合成
(3R) -4- (2 フルオロフェ -ル) N— (4—(1 メチルベンゾイミダゾールー 2 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)チアンー 4 ィル)ブタ
ナミド(16mg, 0. O3mmol)をジクロロメタン(3mL)に溶解し、氷冷下、 mCPBA( > 65%) (22mg, >0. 08mmol)を 2回に分けて添カ卩した。 2. 5時間後、チォ硫酸ナト リウム水溶液を加えて激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。ク ロロホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグ ラフィ一で精製し、表題ィ匕合物 12mg (71%)を得た。
[0571] ステップ 3 (3R)— 3 ァミノ一 4— (2 フルオロフェ-ル)一 N— (4— (1—メチルベ ンゾイミダゾールー 2 ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド '二塩酸 塩 (ィ匕合物 112)の合成
アルゴン雰囲気下、 (3R) -4- (2 フルオロフヱ-ル) N— (4—(1 メチルベ ンゾイミダゾール 2 ィル)—1, 1 ジォキソチアン 4 ィル) 3— ( 2 メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(12mg, 0. 02mmol)を 10%塩化水 素 Zメタノール溶液 (5mL)に溶解し、室温で 4時間攪拌した後濃縮し、表題化合物 10mg (88%)を得た。
[0572] 実施例 113
(3R)—3 アミノー N— (4— (6 クロ口一 1—メチルベンゾイミダゾールー 2—ィル) —1, 1—ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブタナミ ド ·二塩酸塩 (化合物 113)の合成
ステップ 1 4ーシァノチアン 4ーァミン(参考化合物 386)の合成
4ーォキソチアン(10g, 86mmol)のエタノール(45mL)、蒸留水(95mL)混合溶 液に、塩化アンモ-ゥム(4. 8g, 90mmol)、シアン化カリウム(5. 9g, 90mmol)を 加え、室温で 17時間攪拌した。 1N塩酸をカ卩え、ジクロロメタンで洗浄した後、水層に 1N水酸ィ匕ナトリウム水溶液をカロえアルカリ性にして、ジクロロメタンで抽出し、有機層 を乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し 、表題化合物 9. 9g (81%)を得た。
[0573] ステップ 2 4 アミノチアン 4一力ルボン酸 ·塩酸塩(参考化合物 387)の合成
4 シァノチアン— 4 ァミン(1. 6g、 l lmmol)を濃塩酸(16mL)に溶解し、 100 °Cで撹拌した。 7時間後、 0°Cまで冷却し 1時間撹拌した。得られた懸濁液をろ過し、 固体を 2—プロパノール、ジェチルエーテルで洗浄し、減圧下で乾燥することにより、
表題化合物 2. lg (93%)を得た。
[0574] ステップ 3 4—(9 フルォレニルメトキシカルボ-ルァミノ)チアンー 4一力ルボン酸( 参考化合物 388)の合成
アルゴン雰囲気下、 4 アミノチアン 4一力ルボン酸 ·塩酸塩(4. 9g, 25mmol) をジクロロメタン(150mL)に溶解し、 DIPEA(14mL, 87mmol)を滴下した。室温 で 30分間撹拌した後、クロロトリメチルシラン(6. 3mL, 50mmol)を滴下した。室温 で 30分間撹拌した後、加熱還流した。 3時間後、 10°Cに冷却し、塩化(9 フルォ レニルメトキシカルボ-ル)(以下、 Fmoc— C1と略す)(6. Og, 23mmol)を一気に加 え、室温までゆっくり昇温し、 18時間撹拌した。濃縮し、飽和炭酸水素ナトリウム水溶 液を加え、ジェチルエーテルで抽出した。水層を氷冷し、 3N塩酸を加え酢酸ェチル で抽出し、有機層を乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグ ラフィ一で精製し、表題化合物 5. 9g (62%)を得た。
[0575] ステップ 4 5 クロロー N—メチルー 2 -トロア-リン(参考化合物 389)の合成
5 クロ口一 2 -トロア-リン(1. Og, 6. Ommol)のトルエン溶液(4mL)に、テトラ — n—ブチルアンモ-ゥム'硫酸水素塩(123mg, 0. 4mmol)、 50%水酸化ナトリウ ム水溶液(1. 5mL)、硫酸ジメチル(627 L, 6. 6mmol)を加え、室温で 15時間攪 拌した。蒸留水を加えクロ口ホルムで抽出し、有機層を乾燥後濃縮した。得られた粗 生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 731mg (65%)を 得た。
[0576] ステップ 5 4— (6 クロ口一 1—メチルベンゾイミダゾールー 2—ィル) 4— (9 フ ルォレニルメトキシカルボニルァミノ)チアン(参考化合物 390)の合成
アルゴン雰囲気下、 5—クロ口— N—メチル 2 -トロア-リン(731mg, 3. 9mm ol)のエタノール(10mL)、 THF (6mL)、蒸留水(2mL)混合溶液に、塩化アンモニ ゥム(1. lg, 20mmol)、鉄粉(1. lg, 20mmol)を加え、加熱還流した。 12時間後 放冷し、セライトで濾過し、濾液を 1N水酸ィ匕ナトリウム水溶液で中和した後クロ口ホル ムで抽出し、有機層を乾燥後濃縮した。得られた粗生成物 558mgのうち 300mgを 、アルゴン雰囲気下、 DMF (6mL)に溶解し、 4一(9 フルォレニルメトキシカルボ- ルァミノ)チアンー 4一力ルボン酸(参考化合物 388) (272mg, 0. 71mmol)、HAT
U (350mg, 0. 92mmol)をカ卩え、室温で攪拌した。 15時間後、蒸留水をカ卩えてジ ェチルエーテルで抽出し、飽和食塩水で洗浄後乾燥し濃縮し、得られた粗生成物を シリカゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物を、アルゴン雰 囲気下、酢酸(lOmL)に溶解し、 80°Cで 15時間攪拌した後、放冷し濃縮した。得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 73mg (19 %)を得た。
[0577] ステップ 6 4— (6 クロ口一 1—メチルベンゾイミダゾールー 2—ィル)チアン一 4 ァ ミン (参考化合物 391)の合成
アルゴン雰囲気下、 4— (6—クロ口— 1—メチルベンゾイミダゾールー 2—ィル)—4 — (9 フルォレ -ルメトキシカルボ-ルァミノ)チアン(73mg, 0. 15mmol)を DMF (3mL)に溶解し、ピぺリジン (0. 5mL)をカ卩え、室温で 2時間撹拌した。蒸留水をカロ ぇジェチルエーテルで抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラ ムクロマトグラフィーで精製し、表題ィ匕合物 29mg (69%)を得た。
[0578] ステップ 7 (3R)—N— (4— (6 クロ口 1—メチルベンゾイミダゾールー 2—ィル) チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)ブタナミド (参考化合物 392)の合成
アルゴン雰囲気下、 4一(6 クロロー 1 メチルベンゾイミダゾールー 2 ィル)チア ン— 4 ァミン(29mg, 0. lOmmol)を DMF (3mL)に溶解し、(3R)— 4— (2, 4, 5 —トリフルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(3 4mg, 0. lOmmol) , DIPEA (44 μ L, 0. 25mmol)、 HATU (50mg, 0. 13mmo 1)を加え、室温で攪拌した。 12時間後、蒸留水をカ卩えてジェチルエーテルで抽出し 、飽和食塩水で洗浄後乾燥し濃縮し、得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 42mg (70%)を得た。
[0579] ステップ 8 (3R)—N— (4— (6 クロ口 1—メチルベンゾイミダゾールー 2—ィル) —1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2 —メチル— 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 393)の合成 アルゴン雰囲気下、 (3R)— N— (4— (6 クロ口 1—メチルベンゾイミダゾール— 2—ィル)チアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル
2 プロポキシカルボ-ルァミノ)ブタナミド(42mg, 0. 071mmol)をジクロロメタン (4mL)に溶解し、氷冷下、 mCPBA ( >65%) (32mg, >0. 12mmol)を添カ卩し、 2 時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽和炭酸水素 ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 9mg (20%)を得た。
[0580] ステップ 9 (3R)—3 アミノー N— (4— (6 クロロー 1 メチルベンゾイミダゾール — 2—ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ- ル)ブタナミド ·二塩酸塩 (化合物 113)の合成
アルゴン雰囲気下、(3R)— N— (4— (6 クロ口 1—メチルベンゾイミダゾール— 2—ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(9mg, 0. 014mmol )を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 2時間攪拌した後濃縮 し、表題ィ匕合物 9mg (定量的)を得た。
[0581] 実施例 114
(3R)— 3 ァミノ— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (1, 5, 7 トリメ チルベンゾイミダゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミドニ塩酸塩 (化合物 114)の合成
ステップ 1 N, 2, 4 トリメチル—6— -トロア-リン(参考化合物 394)の合成
2, 4 ジメチル— 6 トロア-リン(2. Og, 12. Ommol)のトルエン溶液(20mL) に、テトラー n—ブチルアンモ-ゥム'硫酸水素塩(490mg, 1. 4mmol)、 50%水酸 化ナトリウム水溶液(3mL)、硫酸ジメチル(1. 2mL, 13mmol)をカ卩え、室温で 15時 間攪拌した。蒸留水を加えクロ口ホルムで抽出し、有機層を乾燥後濃縮した。得られ た粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 1. 3g (59%) を得た。
[0582] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1, 5, 7 トリメ チルベンゾイミダゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ー3—(2— メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 395)の合成
N, 2, 4 トリメチル—6 トロア-リン(1. 3g, 7. Ommol)のエタノール溶液(10
mL)に、 10%パラジウム Z炭素(250mg)をカ卩え、水素雰囲気下、室温で 12時間撹 拌した。濾過後、濾液を濃縮して得られた粗生成物を、アルゴン雰囲気下、 DMF (1 OmL)に溶解し、 4一(9 フルォレニルメトキシカルボ-ルァミノ)チアンー 4 カルボ ン酸(参考ィ匕合物 388) (500mg, 1. 3mmol)、 HATU (644mg, 1. 7mmol)をカロ え、室温で攪拌し、 18時間後、蒸留水をカ卩えてジェチルエーテルで抽出し、飽和食 塩水で洗浄後乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で粗精製し、得られた粗精製物を、アルゴン雰囲気下、酢酸(10mL)に溶解し、 8 0°Cで 48時間攪拌した後、放冷し濃縮した。得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、 DMF (3m L)に溶解し、ピぺリジン (0. 5mL)を加え、室温で 15時間撹拌した。濃縮して得られ た粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物を 、アルゴン雰囲気下、 DMF (3mL)に溶解し、(3R)—4— (2, 4, 5 トリフルオロフ ェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(lOOmg, 0. 30m mol)、 DIPEA /z L, 0. 75mmol)、 HATU (137mg, 0. 36mmol)を加え、 室温で攪拌し、 15時間後、蒸留水をカ卩えてジェチルエーテルで抽出し、飽和食塩水 で洗浄後乾燥し濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗 精製した。得られた粗精製物を、ジクロロメタン (4mL)に溶解し、氷冷下、 mCPBA( >65%) (l lOmg, >0. 41mmol)を添カ卩し、 3時間攪拌した。チォ硫酸ナトリウム水 溶液を加えて激しく攪拌した後、炭酸カリウム水溶液を加えた。クロ口ホルムで抽出し 、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 31mg (4%)を得た。
ステップ 3 (3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (1, 5, 7 トリメチルベンゾイミダゾール一 2—ィル) 1, 1—ジォキソチアン一 4—ィル) ブタナミド ·二塩酸塩 (化合物 114)の合成
アルゴン雰囲気下、(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (1, 5 , 7 トリメチルベンゾイミダゾール— 2—ィル)—1, 1—ジォキソチアン— 4—ィル)— 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(31mg, 0. 050mmol) を 10%塩ィ匕水素 Zメタノール溶液 (6mL)に溶解し、室温で 12時間攪拌した後濃縮
し、表題ィ匕合物 26mg (87%)を得た。
[0584] 実施例 115
(3R)— 3 ァミノ一 N— (4— (1H ベンゾイミダゾール一 2—ィル) 1, 1—ジォキ ソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフヱ-ル)ブタナミド '塩酸塩 (ィ匕合 物 115)の合成
ステップ 1 4— (1H ベンゾイミダゾールー 2—ィル) 4— (9 フルォレ -ルメトキ シカルボニルァミノ)チアン(参考化合物 396)の合成法
アルゴン雰囲気下、 o フエ-レンジァミン(226mg, 2. O9mmol)を DMF (5mL) に溶解し、 4一(9 フルォレニルメトキシカルボ-ルァミノ)チアンー 4一力ルボン酸( 参考ィ匕合物 388) (400mg, 1. 04mmol)、 HATU (514mg, 1. 35mmol)、DIPE A OO ^ L, 0. 19mmol)をカ卩ぇ室温で 16時間攪拌した。反応液に蒸留水を加え、 析出してきた固体を濾取し、乾燥後、クロ口ホルム、メタノール、へキサンから結晶化 し、得られた粗生成物 450mgのうち 330mgを、アルゴン雰囲気下、酢酸(12mL)に 溶解し、 80°Cにて 2時間攪拌した。放冷後濃縮し、 THF、酢酸ェチル、へキサンから 結晶化を行 、、表題ィ匕合物 275mg (79%)を得た。
[0585] ステップ 2 4—(1Η べンゾイミダゾールー 2 ィル)チアンー 4ーァミン(参考化合 物 397)の合成
アルゴン雰囲気下、 4一(1H べンゾイミダゾールー 2 ィル)ー4一(9 フルォレ -ルメトキシカルボ-ルァミノ)チアン(275mg, 0. 60mmol)の DMF (5ml)溶液に ピペリン (0. 2mL)を加え室温にて 30分間攪拌した。反応液を 1N塩酸にあけ、酢酸 ェチルにて 2回洗浄した。得られた水層に、 1N水酸ィ匕ナトリウム水溶液をカ卩ぇアル力 リ性にした後、酢酸ェチルにて抽出した。有機層を乾燥し濃縮し、得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 95mg (68%)を得た。
[0586] ステップ 3 (3R)—N— (4—(1H べンゾイミダゾールー 2 ィル)チアンー 4ーィル )一 4一(2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ)ブタナミド (参考化合物 398)の合成
アルゴン雰囲気下、 4— (1H—ベンゾイミダゾールー 2—ィル)チアン— 4 アミン( 95mg, 0. 41mmol)を DMF (lOmL)に溶解し、(3R)— 4— (2, 4, 5 トリフルォロ
フエ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(147mg, 0. 44 mmol)、 HATU (198mg, 0. 52mmol)、 DIPEA (198 L, 1. 20mmol)を加え、 室温で 14時間攪拌した。反応液を蒸留水にあけ、ジェチルエーテルで抽出し、有機 層を蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリ 力ゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 135mg (60%)を得た。
[0587] ステップ 4 (31¾—?^ー(4ー(111ーべンゾィミダゾールー2—ィル)ー1, 1ージォキ ソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2— プロポキシカルボニルァミノ)ブタナミド (参考化合物 399)の合成
(3R)— N— (4— (1H ベンゾイミダゾール— 2—ィル)チアン— 4—ィル)—4— ( 2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) ブタナミド(130mg, 0. 24mmol)をジクロロメタン(7mL)、酢酸ェチル(15mL)に 溶解し、氷冷下、 mCPBA ( >65%) (90mg, >0. 49mmol)を添カ卩した。 5分後氷 浴を外し、室温で 14時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌し た後、飽和炭酸水素ナトリウム水溶液を加えクロ口ホルムで抽出し、乾燥後濃縮して 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 93mg (6 8%)を得た。
[0588] ステップ 5 (3R)— 3 ァミノ— N— (4— (1H ベンゾイミダゾール— 2—ィル) 1, 1—ジォキソチアン— 4—ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ブタナミド '塩 酸塩 (ィ匕合物 115)の合成
アルゴン雰囲気下、(3R)— N— (4— (1H ベンゾイミダゾール— 2—ィル) 1, 1 —ジォキソチアン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチ ルー 2 プロポキシカルボ-ルァミノ)ブタナミド(90mg, 0. 16mmol)を 10%塩ィ匕 水素 Zメタノール溶液(5mL)に溶解し、室温で 13時間攪拌した後濃縮し、表題ィ匕 合物 75mg (94%)を得た。
[0589] 実施例 116
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (1—メチルベン ゾイミダゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナミド '塩酸塩 (ィ匕 合物 116)の合成
ステップ 1 4— (1—メチルベンゾイミダゾールー 2—ィル)チアン— 4 ァミン(参考 化合物 400)の合成
アルゴン雰囲気下、 N—メチル—1, 2 フエ-レンジァミン(266 L, 2. 34mmol )を DMF (5mL)に溶解し、 4— (9—フルォレニルメトキシカルボ-ルァミノ)チアン— 4—カルボン酸(参考ィ匕合物 388) (300mg, 0. 78mmol)、 HATU (387mg, 1. 0 2mmol)、 DIPEA(30 L, 0. 19mmol)を加え室温で 14時間攪拌した。反応液に 蒸留水を加え、析出してきた固体を濾取し、得られた個体を乾燥した。アルゴン雰囲 気下、得られた粗生成物を酢酸(15mL)に溶解し、 80°Cにて 10時間攪拌した。放 冷後濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。 アルゴン雰囲気下、得られた粗精製物 285mgのうち 275mgを DMF溶液 (4mL)に 溶解し、ピぺリジン (0. 15mL)を加え室温にて 30分間攪拌した。反応液を 1N塩酸 にあけ、酢酸ェチルにて 2回洗浄し、得られた水層に、水酸化ナトリウム水溶液をカロ えアルカリ性にした後、酢酸ェチルにて抽出した。有機層を乾燥し濃縮し、得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 127mg (61%) を得た。
[0590] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1—メチルベン ゾイミダゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 401)の合成
アルゴン雰囲気下、 4— (1—メチルベンゾイミダゾールー 2—ィル)チアン— 4 アミ ン(lOOmg, 0. 40mmol)を DMF (lOmL)に溶解し、(3R)— 4— (2, 4, 5 卜リフ ルォロフエ-ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(135mg , 0. 40mmol)、 HATU (200mg, 0. 53mmol) , DIPEA(167 μ L, 1. Olmmol) を加え室温で 14時間攪拌した。反応液を蒸留水にあけ、ジェチルエーテルで抽出し 、得られた有機層を蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 175mg (78%) を得た。
[0591] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1—メチルベン ゾイミダゾール 2 ィル)—1, 1 ジォキソチアン 4 ィル) 3— ( 2 メチル 2
プロポキシカルボニルァミノ)ブタナミド (参考化合物 402)の合成
(3R) -4- (2, 4, 5 トリフルオロフェ -ル) N— (4—(1 メチルベンゾイミダゾ 一ルー 2 ィル)チアンー 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)ブタナミド(170mg, 0. 30mmol)をジクロロメタン(7mL)、酢酸ェチル(10mL) に溶解し、氷冷下、 mCPBA( >65%) (115mg, >0. 43mmol)を添カ卩した。 5分 後氷浴を外し、室温で 14時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪 拌した後、飽和炭酸水素ナトリウム水溶液を加え、クロ口ホルムで抽出し、乾燥後濃 縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 14 0mg (78%)を得た。
[0592] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1 メチルベンゾイミダゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナミ ド ·塩酸塩 (化合物 116)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (1— メチルベンゾイミダゾール 2 ィル)—1, 1 ジォキソチアン 4 ィル) 3— ( 2 —メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(130mg, 0. 22mmol)を 10 %塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 13時間攪拌した後濃縮し、 表題ィ匕合物 114mg (92%)を得た。
[0593] 実施例 117
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(5—メトキシー 1 メチルベンゾイミダゾールー 2—ィル)—1, 1ージォキソチアン 4 ィル)ブタナ ミド ·塩酸塩 (化合物 117)の合成
ステップ 1 4ーメトキシ—N—メチルー 2 -トロア-リン(参考化合物 403)の合成 アルゴン雰囲気下、 4—メトキシ— 2 -トロア-リン(1. 5g, 7. 3mmol)のトルエン (4ml)溶液に、 50%水酸ィ匕ナトリウム水溶液(2mL)、硫酸ジメチル (0. 76mL)、テ トラー n—ブチルアンモ -ゥム '硫酸水素塩(148mg, 0. 4mmol)をカ卩えた。室温に て 5時間攪拌した後、 1N塩酸にあけ、ジクロロメタンにて抽出後、乾燥し濃縮した。得 られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 600mg ( 45%)を得た。
[0594] ステップ 2 4— (9 フルォレニルメトキシカルボ-ルァミノ)—4— (5—メトキシ— 1 メチルベンゾイミダゾールー 2 ィル)チアン(参考化合物 404)の合成
4—メトキシ一 N—メチル 2 -トロア-リン(600mg, 3. 3mmol)メタノール(5m L)溶液に 10%パラジウム Z炭素(50mg)を加え、水素雰囲気下、室温で 14時間攪 拌した。濾過後、濾液を濃縮し、得られた粗生成物 470mgのうち 198mgを、ァルゴ ン雰囲気下、 DMF (5mL)に溶解し、 4一(9 フルォレニルメトキシカルボ-ルァミノ )チアンー4一力ルボン酸(参考化合物 388) (250mg, 0. 65mmol)、 HATU (322 mg, 0. 85mmol)、 DIPEA (32 /z L, 0. 20mmol)をカ卩ぇ室温で攪拌した。 16時間 後、反応液を蒸留水にあけ、ジェチルエーテルで抽出し、蒸留水及び飽和食塩水に て洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで 粗精製した。アルゴン雰囲気下、得られた粗精製物(250mg,約 0. 45mmol)を酢 酸(12mL)に溶解し、 80°Cにて 15時間攪拌した。放冷後濃縮し、得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 220mg (約 65%)を得た
[0595] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5—メトキシ一 1 メチルベンゾイミダゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プ 口ポキシカルボ-ルァミノ)ブタナミド(参考化合物 405)の合成
アルゴン雰囲気下、 4一(9 フルォレニルメトキシカルボ-ルァミノ) -4- (5—メト キシ— 1—メチルベンゾイミダゾールー 2—ィル)チアン(240mg, 0. 48mmol)の D MF溶液 (4mL)にピペリジン (0. 4mL)を加え室温にて 30分間攪拌した。反応液を 1N塩酸にあけ、酢酸ェチルにて 2回洗浄し、得られた水層に 1N水酸ィ匕ナトリウム水 溶液を加えアルカリ性にした後、酢酸ェチルにて抽出した。有機層を乾燥し濃縮し、 得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。アルゴン雰囲 気下、得られた粗精製物を DMF (7mL)に溶解し、(3R)—4— (2, 4, 5—トリフルォ 口フエ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(153mg, 0. 46mmol)、 HATU (229mg, 0. 6mmol) , DIPEA (228 μ L, 1. 38mmol)を加え 室温で 14時間攪拌した。反応液を蒸留水にあけ、ジェチルエーテルで抽出し、得ら れた有機層を蒸留水及び飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生
成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 165mg (55%)を得 た。
[0596] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5—メトキシ一 1
-メチルベンゾイミダゾール 2 ィル)—1, 1 ジォキソチアン 4 ィル) 3— ( 2 メチル - 2-プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 406)の合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5—メトキシ一 1—メチル ベンゾイミダゾールー 2 ィル)チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシ カルボ-ルァミノ)ブタナミド(155mg, 0. 26mmol)をジクロロメタン(5mL)に溶解し 、氷冷下、 mCPBA ( >65%) (103mg, >0. 39mol)を添カ卩した。 5分後氷浴を外 し、室温で 2時間攪拌した。チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、飽 和炭酸水素ナトリウム水溶液を加えた。クロ口ホルムで抽出し、乾燥後濃縮して得ら れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合 135mg (83 %)を得た。
[0597] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5 ーメトキシ 1 メチルベンゾイミダゾールー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミド ·塩酸塩 (化合物 117)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (5— メトキシ一 1—メチルベンゾイミダゾール一 2—ィル) 1, 1—ジォキソチアン一 4—ィ ノレ)一 3— (2—メチノレ一 2 プロポキシカノレボニノレアミノ)ブタナミド(120mg, 0. 19 mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 13時間攪拌した 後濃縮し、表題ィ匕合物 100mg (75%)を得た。
[0598] 実施例 118
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (5 フエ-ル一 1, 3, 4 ォキサジァゾ一ルー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミ ド · 1Z2フマル酸塩 (化合物 118)の合成
ステップ 1 2 フエ-ルー 1, 3, 4ーォキサジァゾール(参考化合物 407)の合成 アルゴン雰囲気下、ベンズヒドラジド(2. 04g, 15mmol)をオルトギ酸トリェチル(3 OmL)に溶解し、 12時間加熱還流した。放冷後濃縮して得られた粗生成物をシリカ
ゲルカラムクロマトグラフィーで精製して、表題化合物 2. 17g (99%)を得た。
[0599] ステップ 2 2—メチルー N— (4— (5 フエ-ルー 1, 3, 4 ォキサジァゾ一ルー 2— ィル)チアン 4 ィル) 2 プロパンスルフィンアミド(参考化合物 408)の合成 アルゴン雰囲気下、 2 フエ-ルー 1, 3, 4ーォキサジァゾール(292mg, 2. Omm ol)を THF (lOmL)に溶解し、 78°Cに冷却した。 n—ブチルリチウム Zへキサン溶 液(1. 60M, 1. 3mL, 2. Ommol)を滴下し、 20。Cで 45分間撹拌した。 2—メチ ルー N— (チアン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合物 205)の 粗生成物(438mg,約 2. Ommol)の THF (5mL)溶液を—78°Cで滴下し、 1時間か けて 0°Cまで昇温した。蒸留水、酢酸ェチルを加え分液し、有機層を飽和食塩水で 洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精 製し、表題ィ匕合物 102mg (約 14%)を得た。
[0600] ステップ 3 4—(5 フエ-ルー 1, 3, 4 ォキサジァゾ一ルー 2 ィル)チアンー 4 ァミン ·塩酸塩 (参考化合物 409)の合成
アルゴン雰囲気下、 2—メチル—N— (4— (5—フエ-ルー 1, 3, 4—ォキサジァゾ 一ルー 2—ィル)チアン— 4—ィル)—2 プロパンスルフィンアミド(102mg, 0. 28m mol)をメタノール(1. 5mL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(3mL)をカロ え、室温で 30分間撹拌した。反応液を濃縮し、減圧下で乾燥して表題ィ匕合物 79mg (95%)を得た。
[0601] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5 フエ二ルー 1, 3, 4 ォキサジァゾ一ルー 2—ィル) 1, 1ージォキソチアンー4 ィル)ブタナミド · 1Z2フマル酸塩 (化合物 118)の合成
アルゴン雰囲気下、 4 (5 フエ-ルー 1, 3, 4 ォキサジァゾ一ルー 2 ィル)チ アン— 4 アミン'塩酸塩(45mg、 0. 15mmol)を DMF (3mL)に溶解し、(3R)— 4 一(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)酪酸(50mg, 0. 15mmol)、 HATU (60mg, 0. 16mmol)、 ΤΕΑ(63 L, 0. 45mmol)をカ卩え、室温で 10時間撹拌した。蒸留水を力卩ぇジェチルエーテルで抽出 し、有機層を飽和食塩水で洗浄した後、乾燥し濃縮した。得られた粗生成物をジクロ ロメタン(10mL)に溶解し、氷冷下、 mCPBA( >65%) (82mg, >0. 31mmol)を
加え、室温で 45分間撹拌した。アンモニア飽和クロ口ホルム(5mL)を加え、生成した 沈殿を濾別し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィ 一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、メタノール (2mL)に溶 解し、 10%塩ィ匕水素 Zメタノール溶液 (4mL)を加え、室温で撹拌した。 5時間後、 飽和炭酸水素ナトリウム水溶液を加えてクロ口ホルムで抽出し、乾燥後濃縮した。得 られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して、表題化合物(フリー 体) 43mg (56%)を得た。表題ィ匕合物(フリー体)を酢酸ェチルに溶解し、フマル酸( 5. Omg)のメタノール溶液を加えて塩ィ匕し、濃縮することにより表題ィ匕合物(1Z2フ マル酸塩)を得た。
[0602] 実施例 119
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (5— (3 フル オロフェニル)ー1, 3, 4 ォキサジァゾ一ルー 2—ィル) 1, 1ージォキソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 119)の合成
ステップ 1 2—(3 フルオロフェ-ル) 1, 3, 4—ォキサジァゾール(参考化合物 4
10)の合成
アルゴン雰囲気下、 3 フルォロベンズヒドラジド(340mg, 2. 2mmol)をオルトギ 酸トリェチル(5mL)に溶解し、 12時間加熱還流した。放冷後濃縮して得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製して、表題ィ匕合物 337mg (93%)を 得た。
[0603] ステップ 2 N— (4— (5— (3 フルオロフェ-ル) 1, 3, 4 ォキサジァゾ一ルー 2 ィル)チアン 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考化合物 4
11)の合成
2— (3 フルオロフェ-ル)—1, 3, 4—ォキサジァゾール(164mg, 1. Ommol)を THF (lOmL)に溶解し、 78°Cに冷却した。 n—ブチルリチウム Zへキサン溶液(1 . 60M, 0. 63mL, 1. Ommol)を滴下し、 0。Cで 1時間撹拌した。 2—メチル N— ( チアン— 4—イリデン)—2 プロパンスルフィンイミド (参考化合物 205)の粗生成物( 329mg,約 1. 5mmol)の THF (6mL)溶液を— 78°Cで滴下し、 1時間かけて 10°C まで昇温した。蒸留水を加え酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄後、
乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表 題ィ匕合物 102mg (約 27%)を得た。
[0604] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (5— (3 フル オロフェニル)ー1, 3, 4 ォキサジァゾ一ルー 2—ィル) 1, 1ージォキソチアン 4 ィル) - 3- (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (参考化合 物 412)の合成
アルゴン雰囲気下、 N— (4—(5—(3 フルオロフェ-ル)—1, 3, 4ーォキサジァ ゾール 2 ィル)チアン— 4 ィル) 2 メチル 2 プロパンスルフィンアミド( 10 2mg, 0. 27mmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し、室温で 1時 間撹拌した。反応液を濃縮し、減圧下で乾燥して得られた粗生成物 73mgのうち 47 mgを、アルゴン雰囲気下、 DMF (3mL)に溶解し、(3R)—4— (2, 4, 5—トリフルォ 口フエ-ル)— 3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(50mg, 0. 1 5mmol)、 HATU (57mg, 0. 15mmol)、 ΤΕΑ(63 L, 0. 45mmol)を加え、室 温で 10時間撹拌した。蒸留水を加えジェチルエーテルで抽出し、有機層を飽和食 塩水で洗浄した後、乾燥し濃縮した。得られた粗生成物(93mg)をジクロロメタン (4 mL)に溶解し、氷冷下、 mCPBA ( >65%) (88mg, >0. 33mmol)をカ卩え、室温 で 1. 5時間撹拌した。アンモニア飽和クロ口ホルム(5mL)をカ卩え、生成した沈殿を濾 別し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 54mg (47%)を得た。
[0605] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5 一(3 フルオロフェ-ル) 1, 3, 4 ォキサジァゾ一ルー 2—ィル) 1, 1 ジォキ ソチアン— 4 ィル)ブタナミド ·塩酸塩 (化合物 119)の合成
アルゴン雰囲気下、(3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(5— ( 3 フルオロフェ-ル)—1, 3, 4—ォキサジァゾール— 2—ィル)—1, 1—ジォキソ チアンー 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(5 4mg, 0. 086mmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し、室温で撹 拌した。 3時間後、クロ口ホルム、飽和炭酸水素ナトリウム水溶液を加えて分液し、水 層をクロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物を薄層クロマトグラフィ
一で精製し、表題化合物 (フリー体) 25mg (55%)を得た。表題化合物 (フリー体)を 酢酸ェチルに溶解し、 10%塩ィ匕水素 Zメタノール溶液 (0. lmL)をカ卩えて塩ィ匕し、 濃縮することにより表題化合物 (塩酸塩)を得た。
[0606] 実施例 120
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(5—(3—メトキ シフエ二ル)一 1, 3, 4—ォキサジァゾール一 2—ィル) 1, 1—ジォキソチアン一 4 ィル)ブタナミド ·塩酸塩 (化合物 120)の合成
ステップ 1 2—(3—メトキシフエ-ル) 1, 3, 4ーォキサジァゾール(参考化合物 41 3)の合成
アルゴン雰囲気下、 3—メトキシベンズヒドラジド(366mg, 2. 2mmol)をオルトギ酸 トリェチル(5mL)に溶解し、 12時間加熱還流した。放冷後濃縮して得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製して、表題ィ匕合物 356mg (92%)を得 た。
[0607] ステップ 2 N— (4— (5— (3—メトキシフエ-ル) 1, 3, 4 ォキサジァゾ一ルー 2 ィル)チアン 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考化合物 4 14)の合成
アルゴン雰囲気下、 2—(3—メトキシフェ-ル)ー1, 3, 4ーォキサジァゾール(176 mg, 1. Ommol)を THF (lOmL)に溶解し、 78°Cに冷却した。 n—ブチルリチウム Zへキサン溶液(1. 60M, 0. 63mL, 1. Ommol)を滴下し、 0°Cで 1時間撹拌した 。 2—メチル N— (チアン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合 物 205)の粗生成物(329mg,約 1. 5mmol)の THF (6mL)溶液を 78°Cで滴下 し、 1時間かけて 10°Cまで昇温した。蒸留水を加え酢酸ェチルで抽出し、有機層を 飽和食塩水で洗浄し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 43mg (約 11%)を得た。
[0608] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5— (3—メトキ シフエ二ル)一 1, 3, 4—ォキサジァゾール一 2—ィル) 1, 1—ジォキソチアン一 4 ィル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 415)の合成
アルゴン雰囲気下、 N— (4— (5- (3—メトキシフエ-ル)—1, 3, 4 ォキサジァゾ 一ルー 2—ィル)チアン— 4—ィル)—2—メチル—2 プロパンスルフィンアミド(43m g, 0. l lmmol)を 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、室温で 1時間 撹拌した。反応液を濃縮し、減圧下で乾燥して得られた粗生成物を、アルゴン雰囲 気下、 DMF (3mL)に溶解し、(3R)—4— (2, 4, 5 トリフルオロフヱ-ル)—3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)酪酸(33mg, 0. 10mmol)、 HATU (3 8mg, 0. 10πιπιο1)、ΤΕΑ(42 /^, 0. 30mmol)を加え、室温で 10時間撹拌した。 蒸留水を加えジェチルエーテルで抽出し、有機層を飽和食塩水で洗浄した後、乾燥 し濃縮した。得られた粗生成物を、ジクロロメタン (4mL)に溶解し、氷冷下、 mCPBA ( >65%) (60mg, >0. 23mol)をカ卩え、室温で 1. 5時間撹拌した。アンモニア飽和 クロ口ホルム(5mL)を加え、生成した沈殿を濾別し、濾液を濃縮して得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 33mg (48%)を得た。
[0609] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5 — (3—メトキシフエ-ル)— 1, 3, 4—ォキサジァゾール— 2—ィル)—1, 1—ジォキ ソチアン 4 ィル)ブタナミド ·塩酸塩 (化合物 120)の合成
アルゴン雰囲気下、(3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(5— ( 3—メトキシフエニル) 1, 3, 4 ォキサジァゾ一ルー 2—ィル) 1, 1ージォキソチ アンー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(33 mg, 0. 052mmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し、室温で撹 拌した。 3時間後、クロ口ホルム、飽和炭酸水素ナトリウム水溶液を加えて分液し、水 層をクロ口ホルムで抽出した。有機層を乾燥後濃縮して得た粗生成物を薄層クロマト グラフィ一で精製し、表題ィ匕合物 (フリー体) 15mg (52%)を得た。表題ィ匕合物 (フリ 一体)を酢酸ェチルに溶解し、 10%塩ィ匕水素 Zメタノール溶液 (0. lmL)をカ卩えて 塩化し、濃縮することにより表題ィ匕合物 (塩酸塩)を得た。
[0610] 実施例 121
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2—メトキ シエトキシ)ベンゾチアゾール 2—ィル) - 2H—テトラヒドロピラン一 4—ィル)ブタナ ミド ·塩酸塩 (化合物 121)の合成
ステップ 1 2—メチル N— (2H—テトラヒドロピラン一 4—イリデン) 2 プロパン スルフィンイミド (参考化合物 416)の合成
アルゴン雰囲気下、チタンテトラエトキシド(2. 74g, 12. Ommol)の THF (32mL) 溶液に、 2—メチル—2 プロパンスルフィンアミド(純度 97%) (728mg, 6. Ommol )の THF (lOmL)溶液をカ卩えた後、 2H—テトラヒドロピラン一 4—オン(601mg, 6. Ommol)を加え、 80°Cで 1時間撹拌した。氷冷し、飽和炭酸水素ナトリウム水溶液(1 OmL)、酢酸ェチル (40mL)を加えて 5分間攪拌した。生じた黄白色個体を濾別し、 蒸留水を加えて酢酸ェチルで抽出した。有機層を乾燥後濃縮し、粗生成物として表 題ィ匕合物 944mgを得た。特に精製することなぐ次の反応に用いた。
[0611] ステップ 2 N— (4— (6— (2—メトキシエトキシ)ベンゾチアゾールー 2—ィル)—2H —テトラヒドロピラン一 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考化 合物 417)の合成
アルゴン雰囲気下、 6—(2—メトキシエトキシ)ベンゾチアゾール(参考化合物 263) (209mg, 1. Ommol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 64M, 0. 38mL, 1. Ommol)を滴下した。 30分後、 2—メチル -N- (2H—テトラヒドロピラン一 4—イリデン) 2 プロパンスルフィンイミドの粗生 成物(160mg,約 0. 8mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温ま でゆっくりと昇温し、 4時間後、蒸留水を加え、酢酸ェチルで抽出し、有機層を飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 63mg (約 15%)を得た。
[0612] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6— (2—メトキ シエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 3— ( 2 メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 418)の合成 アルゴン雰囲気下、 N— (4— (6— (2—メトキシェトキシ)ベンゾチアゾール—2—ィ ル) 2H—テトラヒドロピラン一 4—ィル) 2—メチル 2 プロパンスルフィンアミド (63mg, 0. 15mmol)を 10%塩化水素 Zメタノール(6mL)に溶解し、室温で 12時 間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解 し、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ
カルボ-ルァミノ)酪酸(5 lmg, 0. 15mmol) , DIPEA(100 μ L, 0. 57mmol)、H ATU (76mg, 0. 20mmol)を加え、室温で攪拌し、 16時間後、蒸留水をカ卩えてジ ェチルエーテルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 64mg (67%)を得た。
[0613] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
- (2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4 —ィル)ブタナミド '塩酸塩 (ィ匕合物 121)の合成
アルゴン雰囲気下、(3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(6— ( 2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィ ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(64mg, 0. 10m mol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 5時間攪拌した後 濃縮し、表題ィ匕合物 52mg (92%)を得た。
[0614] 実施例 122
(3R)—3 アミノー 4— (5 クロ口一 2 フノレオ口フエ-ノレ) N— (4— (6— (2—メト キシエトキシ)ベンゾチアゾール 2—ィル) - 2H—テトラヒドロピラン一 4—ィル)ブタ ナミド ·塩酸塩 (化合物 122)の合成
ステップ 1 (3R)—4— (5 クロ口一 2 フノレオ口フエ-ノレ) -N- (4— (6— (2—メト キシエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) - 3 一(2 メチル一 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 419)の合 成
アルゴン雰囲気下、 N— (4— (6- (2—メトキシェトキシ)ベンゾチアゾールー 2—ィ ル) 2H—テトラヒドロピラン一 4—ィル) 2—メチル 2 プロパンスルフィンアミド (参考化合物 417) (194mg, 0. 47mmol)を 10%塩化水素 Zメタノール(6mL)に 溶解し、室温で 1時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を D MF (3mL)に溶解し、(3R)—4— (5 クロ口一 2 フノレオ口フエ-ノレ) 3— (2—メ チルー 2 プロポキシカルボニルァミノ)酪酸(参考化合物 274) (156mg, 0. 47m mol)、 ϋΙΡΕΑ (205 ]^ 1. 2mmol)、 HATU (215mg, 0. 57mmol)を加え、室 温で攪拌し、 15時間後、濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ
フィ一で精製し、表題ィ匕合物 149mg (51%)を得た。
[0615] ステップ 2 (3R)— 3 アミノー 4 (5 クロロー 2 フノレオ口フエ-ノレ) N— (4—(
6— (2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4 ィル)ブタナミド ·塩酸塩 (化合物 122)の合成
アルゴン雰囲気下、(3R)—4— (5 クロ口 2 フルオロフェ -ル) N— (4— (6
- (2—メトキシエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4 ィル) - 3- (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(149mg, 0.
24mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 5時間攪拌し た後濃縮し、表題ィ匕合物 133mg (定量的)を得た。
[0616] 実施例 123
(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6 ジメトキ シベンゾチアゾール 2 ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミド ·塩酸 塩 (ィ匕合物 123)の合成
ステップ 1 N—(4—(5, 6 ジメトキシベンゾチアゾーノレー2—ィノレ)一2H—テトラヒ ドロピラン 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考化合物 420) の合成
アルゴン雰囲気下、 5, 6 ジメトキシベンゾチアゾール(参考化合物 316) (390mg , 2. Ommol)の THF (lOmL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサ ン溶液(1. 58M, 1. 33mL, 2. lmmol)を滴下し、 30分間撹拌した。 2—メチル— N— (2H—テトラヒドロピラン— 4—イリデン)—2 プロパンスルフィンイミド(参考化 合物 416)の粗生成物(326mg,約 1. 6mmol)の THF (4mL)溶液を滴下し、—20 °Cで撹拌した。 2時間後、蒸留水を加え、酢酸ェチルで抽出し、有機層を飽和食塩 水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合物 349mg (約 55%)を得た。
[0617] ステップ 2 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6 ジメトキ シベンゾチアゾールー 2 ィル) 2H—テトラヒドロピランー4 ィル)ー3—(2—メチ ルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 421)の合成
アルゴン雰囲気下、 N— (4— (5, 6 ジメトキシベンゾチアゾールー 2 ィル)—2
H—テトラヒドロピラン 4 ィル) - 2 メチル - 2-プロパンスルフィンアミド(347m g, 0. 87mmol)を 10%塩化水素 Zメタノール溶液(lOmL)に溶解し、室温で 2時間 撹拌した。反応液を濃縮し、減圧下で乾燥して得た粗生成物 327mgのうち 73mgを 、アルゴン雰囲気下、 DMF (4mL)に溶解し、(3R)—4— (2, 4, 5 トリフルオロフ ェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(67mg, 0. 20m mol)、 HATU (84mg, 0. 22mmol)、 ΤΕΑ(84 L, 0. 60mmol)を加え、室温で 攪拌した。 17時間後、蒸留水を加えジェチルエーテルで抽出し、有機層を飽和食塩 水にて洗浄後、乾燥し濃縮した。これをシリカゲルカラムクロマトグラフィーで精製して 、表題ィ匕合物 71mg (60%)を得た。
[0618] ステップ 3 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5, 6 -ジメトキシベンゾチアゾール 2 ィル) 2H テトラヒドロピラン一 4 ィル)ブタ ナミド ·塩酸塩 (化合物 123)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (5, 6 —ジメトキシベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 3 一(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(71mg, 0. 12mmol)を メタノール(lmL)に溶解し、 10%塩ィ匕水素 Zメタノール溶液(3mL)をカ卩え、室温で 撹拌した。 2. 5時間後、反応液を濃縮し、表題ィ匕合物 63mg (99%)を得た。
[0619] 実施例 124
(3R)—3 ァミノ一 4— (5 クロ口一 2 フノレオ口フエ-ノレ) N— (4— (5, 6 ジメト キシベンゾチアゾール - 2 ィル) - 2H—テトラヒドロピラン 4 ィル)ブタナミド ·塩 酸塩 (ィ匕合物 124)の合成
ステップ 1 (3R)—4— (5 クロ口一 2 フノレオ口フエ-ノレ) -N- (4— (5, 6 ジメト キシベンゾチアゾーノレ 2—ィノレ) 2H—テトラヒドロピランー4ーィノレ) - 3- (2—メ チルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 422)の合成
アルゴン雰囲気下、 N— (4— (5, 6 ジメトキシベンゾチアゾールー 2 ィル)ー2 H—テトラヒドロピラン 4 ィル) - 2 メチル - 2-プロパンスルフィンアミド(参考化 合物 420) (347mg, 0. 87mmol)を 10%塩化水素 Zメタノール溶液(10mL)に溶 解し、室温で 2時間撹拌した。反応液を濃縮し、減圧下で乾燥して得た粗生成物 32
7mgのうち 73mgを、アルゴン雰囲気下、 DMF (4mL)に溶解し、(3R)—4— (5—ク ロロ 2 フルオロフェ -ル) - 3- (2—メチル—2 プロポキシカルボ-ルァミノ)酪 酸(参考ィ匕合物 274) (66mg, 0. 20mmol)、 HATU (84mg, 0. 22mmol)、TEA (84 ^ L, 0. 60mmol)をカ卩え、室温で攪拌した。 17時間後、蒸留水を力卩ぇジェチル エーテルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗 生成物をシリカゲルカラムクロマトグラフィーで精製して、表題ィ匕合物 73mg (63%)を 得た。
[0620] ステップ 2 (3R)— 3 アミノー 4 (5 クロロー 2 フノレオ口フエ-ノレ) N— (4—( 5, 6 ジメトキシベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) ブタナミド ·塩酸塩 (化合物 124)の合成
アルゴン雰囲気下、 (3R)—4— (5 クロ口 2 フルオロフェ -ル) N— (4— (5 , 6 ジメトキシベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(73mg、 0. 12mmol) をメタノール(lmL)に溶解し、 10%塩ィ匕水素メタノール溶液(3mL)をカ卩え、室温で 撹拌した。 2. 5時間後、反応液を濃縮し、表題ィ匕合物 65mg (定量的)を得た。
[0621] 実施例 125
(3R) 3 アミノー 4一(5 クロロー 2 フノレオ口フエ-ノレ) N—(4一(6—(2 モ ルホリノエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) ブナタミド ·二塩酸塩 (化合物 125)の合成
ステップ 1 2—メチルー N— (4—(6—(2 モルホリノエトキシ)ベンゾチアゾールー 2 ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィンアミド(参考 化合物 423)の合成
アルゴン雰囲気下、 6—(2 モルホリノエトキシ)ベンゾチアゾール(参考化合物 29 2) (186mg, 0. 705mmol)の THF (7mL)溶液を— 78°Cに冷却し、 n—ブチルリチ ゥム Zへキサン溶液(1. 58M, 0. 49mL, 0. 78mmol)を滴下し、 30分間撹拌した 。 2—メチル N— (2H—テトラヒドロピラン一 4—イリデン) 2 プロパンスルフィン イミド(参考化合物 416)の粗生成物(136mg,約 0. 67mmol)の THF (3mL)溶液 を滴下し、 2時間かけて— 20°Cまで昇温した。蒸留水を加え酢酸ェチルで抽出し、
有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製し、表題ィ匕合物 201mg (約 64%)を得た。
[0622] ステップ 2 (3R)— 3 アミノー 4 (5 クロロー 2 フノレオ口フエ-ノレ) N— (4—( 6— (2 モルホリノエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン - 4 ィル)ブナタミド ·二塩酸塩 (化合物 125)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(6—(2 モルホリノエトキシ)ベンゾチア ゾールー 2 ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィンアミ ド(201mg, 0. 43mmol)を 10%塩化水素 Zメタノール溶液(4mL)に溶解し、室温 で 2時間撹拌した。反応液を濃縮し、減圧下で乾燥して得られた粗生成物 212mgの うち 141mgを、アルゴン雰囲気下、 DMF (4mL)に溶解し、(3R)—4— (5 クロ口 2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸( 参考ィ匕合物 274) (77mg, 0. 23mmol)、 HATU (98mg, 0. 26mmol)、 TEA(14 3 /z L, 1. 02mmol)を加え、室温で攪拌した。 12時間後、蒸留水をカ卩ぇ酢酸ェチル で抽出し、有機層を蒸留水で洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで粗精製した。得られた粗精製物を、アルゴン雰囲気下、 10%塩化水素メタノール溶液(2mL)に溶解し、室温で撹拌した。 2時間後、飽和炭 酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、有機層を乾燥後濃縮した。得 られた粗生成物を薄層クロマトグラフィーで精製して、表題化合物 (フリー体) 27mg ( 16%)を得た。表題ィ匕合物(フリー体)を酢酸ェチルに溶解し、 10%塩ィ匕水素 Zメタ ノール溶液 (0. lmL)を加えて塩ィ匕し、濃縮することにより、表題化合物 (塩酸塩)を 得た。
[0623] 実施例 126
(3R)—3 ァミノ一 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (6— (2 モノレ ホリノエトキシ)ベンゾチアゾーノレ 2—ィノレ) 2H—テトラヒドロピランー4 ィル)ブ ナタミド ·二塩酸塩 (化合物 126)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2 モルホリノエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピランー4 ィル)ブタナミド(参考化合物 424)の合成
アルゴン雰囲気下、 2—メチルー N— (4—(6—(2 モルホリノエトキシ)ベンゾチア ゾールー 2 ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィンアミ ド(参考化合物 423) (201mg, 0. 43mmol)を 10%塩化水素 Zメタノール溶液(4m L)に溶解し、室温で 2時間撹拌した。反応液を濃縮し、減圧下で乾燥して得られた 粗生成物 212mgのうち 65mgを、アルゴン雰囲気下、 DMF (2mL)に溶解し、(3R) -4- (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ- ルァミノ)酪酸(59mg, 0. 18mmol)、 HATU (67mg, 0. 18mmol)、 DIPEA(112 n 0. 64mmol)を加え、室温で攪拌した。 12時間後、蒸留水を加え酢酸ェチル で抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシ リカゲルカラムクロマトグラフィーで精製して、表題ィ匕合物 46mg (51%)を得た。
[0624] ステップ 2 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
- (2—モルホリノエトキシ)ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4 ィル)ブナタミド ·二塩酸塩 (化合物 126)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6—(2 モルホリノエトキシ)ベンゾ チアゾールー 2—ィル) 2H—テトラヒドロピランー4 ィル)ブタナミド(45mg, 0. 0 66mmol)を 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、室温で撹拌した。 12 時間後、飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出し、有機層を乾 燥後縮した。得られた粗生成物を薄層クロマトグラフィーで精製して、表題化合物 (フ リー体) 38mg (定量的)を得た。表題ィ匕合物(フリー体)を酢酸ェチルに溶解し、 10 %塩ィ匕水素 Zメタノール溶液 (0. lmL)を加えて塩ィ匕し、濃縮することにより、表題 化合物 (塩酸塩)を得た。
[0625] 実施例 127
(3R)—3 ァミノ一 4— (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4— (6—メトキシべ ンゾチアゾール 2 ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミド ·塩酸塩( 化合物 127)の合成
ステップ 1 N— (4— (6—メトキシベンゾチアゾール 2—ィル) 2H—テトラヒドロピ ラン 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考化合物 425)の合
成
アルゴン雰囲気下、 6—メトキシベンゾチアゾール(参考化合物 211) (1. 49g, 9. Ommol)の THF (45mL)溶液を—78°Cに冷却し、 n—ブチルリチウム Zへキサン溶 液(1. 58M, 5. 70mL, 9. Ommol)を滴下し、 30分間撹拌した。 2—メチル N— ( 2H テトラヒドロピラン - 4—イリデン) - 2-プロパンスルフィンイミド(参考化合物 41 6)の粗生成物(1. 83g,約 9. Ommol)の THF (lOmL)溶液を滴下し、 20°Cで撹 拌した。 1. 5時間後、蒸留水、飽和食塩水を加え、酢酸ェチルで抽出し、有機層を 飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 380mg (約 11%)を得た。
[0626] ステップ 2 4— (6—メトキシベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン
4 ァミン ·塩酸塩 (参考化合物 426)の合成
アルゴン雰囲気下、 N— (4—(6—メトキシベンゾチアゾールー 2 ィル)—2H—テ トラヒドロピラン一 4—ィル) 2—メチル 2 プロパンスルフィンアミド(380mg, 1. O3mmol)を 10%塩ィ匕水素 Zメタノール溶液(10mL)に溶解し、室温で 2時間撹拌 した。反応液を濃縮し、減圧下で乾燥して表題ィ匕合物 309mg (定量的)を得た。
[0627] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6—メトキシべ ンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) - 3- (2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 427)の合成
アルゴン雰囲気下、 4一(6—メトキシベンゾチアゾールー 2 ィル)—2H—テトラヒ ドロピラン— 4 アミン'塩酸塩(307mg, 1. 02mmol)を DMF (5mL)に溶解し、 (3 R) -4- (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ -ルァミノ)酪酸(340mg, 1. 02mmol)、 HATU (388mg, 1. 02mmol)、 DIPEA (532 /z L, 3. 06mmol)をカ卩え、室温で攪拌した。 10時間後、蒸留水を加え酢酸ェ チルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製して、表題ィ匕合物 489mg (83%)を得 た。
[0628] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 ーメトキシベンゾチアゾーノレ 2—ィノレ) 2Hーテトラヒドロピラン 4 ィル)ブタナミ
ド ·塩酸塩 (化合物 127)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (6— メトキシベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(488mg, 0. 84mmol)を 10 %塩ィ匕水素 Zメタノール溶液(10mL)に溶解し、室温で撹拌した。 3時間後、反応液 を濃縮し、表題ィ匕合物 434mg (定量的)を得た。
[0629] 実施例 128
(3R)—3 アミノー N— (4— (6— (3 エトキシプロポキシ)ベンゾチアゾール—2— ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブ タナミド ·塩酸塩 (化合物 128)の合成
ステップ 1 6—(3 エトキシプロポキシ)ベンゾチアゾール(参考化合物 428)の合成 アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (228mg, 1 . 5mmol)を THF (6mL)に溶解し、トリフエ-ルホスフィン(440mg, 1. 7mmol)、D IAD (335 ^ L, 1. 7mmol)、 3 エトキシ一 1—プロノ ノーノレ(190 , 1. 6mmol )を加え、室温で撹拌した。 17時間後反応液を濃縮し、シリカゲルカラムクロマトダラ フィ一で精製して表題ィ匕合物 316mg (89%)を得た。
[0630] ステップ 2 N— (4— (6— (3 エトキシプロポキシ)ベンゾチアゾールー 2 ィル) 2H テトラヒドロピラン 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考 化合物 429)の合成
アルゴン雰囲気下、 6—(3 エトキシプロポキシ)ベンゾチアゾール(316mg, 1. 3 3mmol)の THF ( 10mL)溶液を 78°Cに冷却し、 n ブチルリチウム Zへキサン溶 液(1. 58M, 0. 84mL,l. 3mmol)を滴下し、 30分間撹拌した。 2—メチル N— ( 2H テトラヒドロピラン - 4—イリデン) - 2-プロパンスルフィンイミド(参考化合物 41 6)の粗生成物(223mg,約 1. lOmmol)の THF (3mL)溶液を滴下し、 1時間かけ て— 30°Cまで昇温した。蒸留水を加え酢酸ェチルで抽出し、有機層を飽和食塩水 で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 195mg (約 25%)を得た。
[0631] ステップ 3 (3R)—N— (4— (6— (3 エトキシプロポキシ)ベンゾチアゾール—2—
ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 430)の合 成
アルゴン雰囲気下、 N— (4—(6—(3 エトキシプロポキシ)ベンゾチアゾールー 2 —ィル) 2H—テトラヒドロピラン一 4—ィル) 2—メチル 2 プロパンスルフィン アミド(195mg, 0. 44mmol)をメタノール(2mL)に溶解し、 10%塩化水素 Zメタノ ール溶液 (9mL)を加え、室温で 3時間撹拌した。反応液を濃縮し、減圧下で乾燥し て得られた粗生成物を、アルゴン雰囲気下、 DMF (5mL)に溶解し、(3R)—4 (2 , 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ) 酪酸(150mg, 0. 45mmol)、 HATU (192mg, 0. 50mmol) , DIPEA(250 μ L, 1. 47mmol)を加え、室温で攪拌した。 15時間後、蒸留水を加え酢酸ェチルで抽出 し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル カラムクロマトグラフィーで精製して、表題ィ匕合物 240mg (84%)を得た。
[0632] ステップ 4 (3R)—3 アミノー N— (4— (6— (3 エトキシプロポキシ)ベンゾチアゾ ール一 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルォロ フエニル)ブタナミド ·塩酸塩 (化合物 128)の合成
アルゴン雰囲気下、(3R)— N— (4— (6- (3—エトキシプロポキシ)ベンゾチアゾ ール一 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルォロ フエ-ル) - 3- (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(97mg, 0 . 15mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で撹拌した。 2 時間後反応液を濃縮し、蒸留水を加えて酢酸ェチルで抽出し、有機層を飽和食塩 水で洗浄した後、乾燥し濃縮した。得られた粗生成物を薄層クロマトグラフィーで精 製して、表題ィ匕合物(フリー体) 60mg (73%)を得た。表題化合物(フリー体)を酢酸 ェチルに溶解し、 10%塩ィ匕水素 Zメタノール溶液 (0. lmL)をカ卩えて塩ィ匕し、濃縮 することにより表題ィ匕合物 (塩酸塩)を得た。
[0633] 実施例 129
(3R)—3—アミノー N— (4— (6— ( (ジェチルァミノカルボ-ル)メトキシ)ベンゾチア ゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルォ
口フエ-ル)ブタナミド ·塩酸塩 (化合物 129)の合成
ステップ 1 4— (6—ヒドロキシベンゾチアゾール 2—ィル) 2H—テトラヒドロビラ ンー 4ーァミン (参考化合物 431)の合成
4— (6—メトキシベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4 アミ ン'塩酸塩 (参考化合物 426)を飽和炭酸水素ナトリウム水溶液、酢酸ェチルで処理 することにより、フリー塩基とした。アルゴン雰囲気下、 4— (6—メトキシベンゾチアゾ 一ルー 2 ィル)—2H—テトラヒドロピラン— 4 ァミン(参考化合物 426) (フリー塩 基)(53mg, 0. 20mmol)を DMF (2mL)に溶解し、 2 プロパンチオール(93 L , 1. Ommol)、カリウム tert—ブトキシド(90mg, 0. 8mmol)を加え、 135。Cで撹拌 した。 3時間後放冷し、蒸留水、 1N塩酸 (0. 8mL)をカ卩えて中和し、酢酸ェチルで抽 出し、有機層を蒸留水、飽和食塩水で洗浄後、乾燥し濃縮して表題化合物の粗生 成物 47mgを得た。
[0634] ステップ 2 4—(6 ((ジェチルァミノカルボ-ル)メトキシ)ベンゾチアゾールー 2—ィ ル) 2H—テトラヒドロピラン 4ーァミン(参考化合物 432)の合成
アルゴン雰囲気下、 4一(6 ヒドロキシベンゾチアゾールー 2 ィル)—2H—テトラ ヒドロピラン— 4 ァミンの粗生成物(23. 5mg)を THF (6mL)に溶解し、トリフエニル ホスフィン(27mg, 0. 103mmol) , DIAD (20 μ L, 0. 103mmol)、N, N ジェチ ルー 2 ヒドロキシァセタミド(13 L, 0. 103mmol)をカ卩え、室温で撹拌した。 16時 間後反応液を濃縮し、得られた粗生成物を薄層クロマトグラフィーで精製し、表題ィ匕 合物 24mg (約 70%)を得た。
[0635] ステップ 3 (3R)— N— (4— (6— ( (ジェチルァミノカルボ-ル)メトキシ)ベンゾチア ゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルォ 口フエ-ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化 合物 433)の合成
アルゴン雰囲気下、 4一(6—((ジェチルァミノカルボ-ル)メトキシ)ベンゾチアゾー ル一 2—ィル) 2H—テトラヒドロピラン一 4 ァミン(24mg, 0. 066mmol)を DMF (2mL)に溶解し、 (3R) -4- (2, 4, 5 トリフルオロフヱ-ル)ー3—(2—メチルー 2 プロポキシカノレポ-ノレァミノ)酪酸(27mg, 0. 080mmol)、 HATU (30mg, 0.
080mmol) , DIPEA(34 μ L, 0. 20mmol)をカ卩え、室温で攪拌した。 9時間後、蒸 留水、飽和食塩水を加え、酢酸ェチルで抽出し、有機層を蒸留水で洗浄後、乾燥し 濃縮した。得られた粗生成物を薄層クロマトグラフィーで精製して、表題ィ匕合物 39m g (87%)を得た。
[0636] ステップ 4 (3R)— 3 ァミノ一 N— (4— (6— ( (ジェチルァミノカルボ-ル)メトキシ) ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5— トリフルオロフェ -ル)ブタナミド ·塩酸塩 (化合物 129)の合成
アルゴン雰囲気下、 (3R)— N— (4—(6—((ジェチルァミノカルボ-ル)メトキシ) ベンゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5— トリフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド (39mg, 0. O57mmol)を 10%塩化水素 Zメタノール溶液(3mL)に溶解し、室温で 撹拌した。 11時間後反応液を濃縮し、減圧下乾燥することにより表題ィ匕合物 34mg ( 97%)を得た。
[0637] 実施例 130
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2—(2— ォキソピロリジン一 1—ィル)エトキシ)ベンゾチアゾール 2—ィル) - 2H—テトラヒド 口ピラン 4 ィル)ブタナミド ·塩酸塩 (化合物 130)
アルゴン雰囲気下、 4一(6 ヒドロキシベンゾチアゾールー 2 ィル)—2H—テトラ ヒドロピランー4ーァミン(参考化合物 431)の粗生成物(23. 5mg)を THF (6mL)に 溶解し、トリフエ-ルホスフィン(27mg, 0. 103mmol) , DIAD (20 μ L, 0. 103mm ol)、 1— (2 ヒドロキシェチル) 2 ピロリドン(12 L, 0. 103mmol)をカ卩え、室 温で撹拌した。 16時間後反応液を濃縮し、得られた粗生成物を薄層クロマトグラフィ 一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、 DMF (2mL)に溶解し、 (3R) -4- (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカル ボ-ルァミノ)酪酸(29mg, 0. 087mmol)、 HATU (33mg, 0. 087mmol)、 DIPE A OO ^ L, 0. 17mmol)をカ卩え、室温で攪拌した。 9時間後、蒸留水、飽和食塩水 を加え、酢酸ェチルで抽出し、有機層を蒸留水で洗浄後、乾燥し濃縮して得られた 粗生成物を薄層クロマトグラフィーで粗精製した。得られた粗生成物を、アルゴン雰
囲気下、 10%塩ィ匕水素 Zメタノール溶液(3mL)に溶解し、室温で撹拌した。 11時 間後反応液を濃縮し、表題ィ匕合物 19mg (約 63%)を得た。
[0638] 実施例 131
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2H—テト ラヒドロピラン一 4 ィルォキシ)ベンゾチアゾール - 2-ィル) 2H—テトラヒドロビラ ン― 4 ィル)ブタナミド ·塩酸塩 (化合物 131)の合成
ステップ 1 6—(2H—テトラヒドロピランー4 ィルォキシ)ベンゾチアゾール(参考化 合物 434)の合成
アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (363mg, 2 . 4mmol)を THF (12mL)に溶解し、トリフエ-ルホスフィン(682mg, 2. 6mmol)、 ァゾジカルボン酸ジェチル(40%トルエン溶液、 1. 13mL)、 2H—テトラヒドロピラン —4—オール(248 L, 2. 6mmol)をカ卩え、室温で撹拌した。 24時間後、反応液に 1N水酸ィ匕ナトリウム水溶液(10mL)をカ卩え、酢酸ェチルで抽出し、有機層を飽和炭 酸水素ナトリウム水溶液で洗浄した後、乾燥し濃縮した。得られた粗生成物をシリカ ゲルカラムクロマトグラフィーで精製して表題ィ匕合物 259mg (46%)を得た。
[0639] ステップ 2 N— (4— (6— (2H—テトラヒドロピランー4 ィルォキシ)ベンゾチアゾー ルー 2 ィル) 2H—テトラヒドロピラン一 4—ィル) 2—メチル 2 プロパンスル フィンアミド (参考化合物 435)の合成
アルゴン雰囲気下、 6—(2H—テトラヒドロピランー4 ィルォキシ)ベンゾチアゾー ル(259mg, 1. lOmmol)の THF (7mL)溶液を— 78°Cに冷却し、 n—ブチルリチウ ム/へキサン溶液(1. 58M, 0. 696mL, 1. lOmmol)を滴下し、 30分間撹拌した 。 2—メチル N— (2H—テトラヒドロピラン一 4—イリデン) 2 プロパンスルフィン イミド(参考化合物 416)の粗生成物(208mg,約 1. 02mmol)の THF (3mL)溶液 を滴下し、 1. 5時間かけて— 10°Cまで昇温した。蒸留水を加え酢酸ェチルで抽出し 、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル力 ラムクロマトグラフィーで精製し、表題ィ匕合物 90mg (約 20%)を得た。
[0640] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (6— (2H—テト ラヒドロピラン一 4 ィルォキシ)ベンゾチアゾール 2 ィル) 2H—テトラヒドロビラ
ンー4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考 化合物 436)の合成
アルゴン雰囲気下、 N— (4—(6—(2H—テトラヒドロピランー4 ィルォキシ)ベン ゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 2—メチル 2 プ 口パンスルフィンアミド(90mg, 0. 21mmol)を 10%塩化水素 Zメタノール溶液(7m L)に溶解し、室温で 2時間撹拌した。反応液を濃縮し、減圧下で乾燥して得られた 粗生成物をアルゴン雰囲気下、 DMF (8mL)に溶解し、(3R)— (2, 4, 5 トリフル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ー4酪酸(75mg , 0. 23mmol)、 HATU (86mg, 0. 23mmol) , DIPEA(107 μ L, 90. 615mmol )を加え、室温で攪拌した。 19時間後、蒸留水を加え酢酸ェチルで抽出し、有機層 を蒸留水、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲル力 ラムクロマトグラフィーで精製して、表題ィ匕合物 88mg (66%)を得た。
[0641] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
- (2H—テトラヒドロピラン一 4—ィルォキシ)ベンゾチアゾール 2—ィル) 2H— テトラヒドロピラン 4 ィル)ブタナミド ·塩酸塩 (化合物 131)の合成
アルゴン雰囲気下、(3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(6— ( 2H テトラヒドロピラン 4 ィルォキシ)ベンゾチアゾール 2 ィル) 2H テト ラヒドロピランー4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナ ミド(87mg, 0. 13mmol)を 10%塩化水素 Zメタノール溶液(5mL)に溶解し、室温 で撹拌した。 11時間後反応液を濃縮し、減圧下乾燥することにより表題ィ匕合物 78m g (定量的)を得た。
[0642] 実施例 132
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2—(2— プロポキシ)エトキシ)ベンゾチアゾールー 2—ィル) 2H—テトラヒドロピランー4ーィ ル)ブタナミド ·塩酸塩 (化合物 132)の合成
ステップ 1 6—(2—(2 プロポキシ)エトキシ)ベンゾチアゾール(参考化合物 437) の合成
アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (181mg, 1
. 2mmol)を THF (6mL)に溶解し、トリフエ-ルホスフィン(359mg、 1. 3mmol)、 D IAD (276 ;z :L、 1. 3mmol)、 2— (2 プロポキシ)エタノーノレ(152 L, 1. 3mmol )を加え、室温で撹拌した。 3. 5時間後反応液を濃縮し、シリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 169mg (59%)を得た。
[0643] ステップ 2 2—メチルー N— (4— (6— (2— (2 プロポキシ)エトキシ)ベンゾチアゾ 一ルー 2 ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィンアミド (参考化合物 438)の合成
アルゴン雰囲気下、 6—(2—(2 プロポキシ)エトキシ)ベンゾチアゾール(167mg , 0. 70mmol)の THF (7mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサ ン溶液(1. 58M, 0. 47mL, 0. 74mmol)を滴下し、 30分間撹拌した。 2—メチル — N— (2H—テトラヒドロピラン— 4—イリデン)—2 プロパンスルフィンイミド(参考 化合物 416)の粗生成物(17711^,約 0. 88mmol)の THF (2mL)溶液を滴下し、 2 時間かけて 10°Cまで昇温した。蒸留水を加え酢酸ェチルで抽出し、有機層を飽和 食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 25mg (約 8%)を得た。
[0644] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2—(2 プロポキシ)エトキシ)ベンゾチア ゾールー 2—ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミド(参考化合物 439) の合成
アルゴン雰囲気下、 2—メチルー N— (4—(6—(2—(2 プロポキシ)エトキシ)ベ ンゾチアゾーノレ一 2—ィノレ) 2H—テトラヒドロピラン一 4—ィノレ) 2 プロパンスル フィンアミド(25mg, 0. 057mmol)をメタノール(lmL)に溶解し、 10%塩化水素 Z メタノール溶液(3mL)を加え、室温で撹拌した。 3. 5時間後反応液を濃縮し、減圧 下で乾燥して得られた粗生成物を、アルゴン雰囲気下、 DMF (3mL)に溶解し、 (3 R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシカルボ -ルァミノ)酪酸(21mg, 0. 063mmol)、 HATU (28mg, 0. 073mmol)、 DIPEA (34 μ L·, 0. 20mmol)をカ卩え、室温で攪拌した。 21時間後、蒸留水、飽和食塩水を 加え、酢酸ェチルで抽出し、有機層を蒸留水で洗浄後、乾燥し濃縮した。得られた
粗生成物をシリカゲルカラムクロマトグラフィーで精製して、表題ィ匕合物 33mg (89% )を得た。
[0645] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
(2- (2—プロポキシ)エトキシ)ベンゾチアゾールー 2—ィル)—2H—テトラヒドロ ピラン― 4 ィル)ブタナミド ·塩酸塩 (化合物 132)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6—(2—(2 プロポキシ)エトキシ) ベンゾチアゾールー 2—ィル) 2H—テトラヒドロピランー4 ィル)ブタナミド(33mg , 0. O51mmol)を 10%塩化水素 Zメタノール溶液 (4mL)に溶解し、室温で撹拌し た。 15時間後反応液を濃縮し、減圧下乾燥することにより表題ィ匕合物 30mg (定量的 )を得た。
[0646] 実施例 133
(3R) 3 アミノー N— (4—(6—(2 エトキシエトキシ)ベンゾチアゾーノレ 2—ィ ル)一 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブタ ナミド ·塩酸塩 (化合物 133)の合成
ステップ 1 6—(2 エトキシエトキシ)ベンゾチアゾール(参考化合物 440)の合成 アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (180mg, 1 . 2mmol)の THF溶液(6mL)に、トリフエ-ルホスフィン(375mg, 1. 4mmol)、 DI AD (282mg, 1. 4mmol)、 2 エトキシエタノール(129mg, 1. 4mmol)を加え、 室温で 14時間攪拌した。濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で精製し、表題ィ匕合物 197mg (64%)を得た。
[0647] ステップ 2 N— (4— (6— (2 エトキシエトキシ)ベンゾチアゾールー 2—ィル)—2H —テトラヒドロピラン一 4 ィル) 2 メチル 2 プロパンスルフィンアミド(参考化 合物 441)の合成
アルゴン雰囲気下、 6—(2 エトキシエトキシ)ベンゾチアゾール(197mg, 0. 88 mmol)の THF (5mL)溶液を— 78°Cに冷却し、 n ブチルリチウム Zへキサン溶液( 2. 64M, 0. 41mL, 1. lmmol)を滴下した。 30分後、 2—メチル N— (2H—テト ラヒドロピラン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合物 416)の粗
生成物(244mg,約 1. 2mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温 までゆっくりと昇温し、 12時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層を 飽和食塩水で洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 45mg (約 12%)を得た。
[0648] ステップ 3 (3R)—N— (4— (6— (2 エトキシエトキシ)ベンゾチアゾール—2—ィ ル)一 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3 一(2 メチル 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 442)の合 成
アルゴン雰囲気下、 N— (4— (6- (2 エトキシエトキシ)ベンゾチアゾールー 2— ィル) 2H—テトラヒドロピラン一 4—ィル) 2—メチル 2 プロパンスルフィンアミ ド(45mg, 0. l lmmol)を 10%塩化水素 Zメタノール(6mL)に溶解し、室温で 2時 間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)に溶解 し、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ カルボ-ルァミノ)酪酸(35mg, 0. 1 lmmol) , DIPEA (46 μ L, 0. 26mmol)、HA TU (48mg, 0. 13mmol)をカ卩え、室温で攪拌し、 12時間後、蒸留水を加えてジェ チルエーテルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物 をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 19mg (28%)を得た。
[0649] ステップ 4 (3R)— 3 アミノー N— (4— (6— (2 エトキシエトキシ)ベンゾチアゾー ル一 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブタナミド ·塩酸塩 (化合物 133)の合成
アルゴン雰囲気下、 (3R)— N— (4— (6— (2 エトキシエトキシ)ベンゾチアゾー ル一 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(19mg, 0. 03 Ommol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 2時間攪拌した 後濃縮し、表題ィ匕合物 l lmg (65%)を得た。
[0650] 実施例 134
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2 (ジメ チルァミノ)エトキシ)ベンゾチアゾール 2 ィル) 2H テトラヒドロピラン一 4 ィ
ル)ブタナミド ·二塩酸塩 (化合物 134)の合成
ステップ 1 2—メチルー N— (4— (6- (2 (ジメチルァミノ)エトキシ)ベンゾチアゾ 一ルー 2 ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィンアミド (参考化合物 443)の合成
アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (180mg, 1 . 2mmol)の THF溶液(6mL)に、トリフエ-ルホスフィン(375mg, 1. 4mmol)、DI AD (282mg, 1. 4mmol)、 2— (ジメチルァミノ)エタノール(127mg, 1. 4mmol)を 加え、室温で 14時間攪拌した。濃縮して得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で粗精製た。得られた粗精製物を、アルゴン雰囲気下、 THF (5mL)に 溶解して 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶液(2. 64M, 0. 57mL , 1. 5mmol)を滴下した。 30分後、 2—メチル N— (2H—テトラヒドロピラン一 4— イリデン) 2 プロパンスルフィンイミド(参考化合物 416)の粗生成物(305mg,約 1. 5mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温までゆっくりと昇温し 、 12時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄後 、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 205mg (約 48%)を得た。
ステップ 2 (3R)—4— (2, 4, 5 トリフルオロフヱ-ル) N— (4— (6— (2— (ジメ チルァミノ)エトキシ)ベンゾチアゾール 2 ィル) 2H テトラヒドロピラン一 4 ィ ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 444 )の合成
アルゴン雰囲気下、 2—メチルー N—(4一(6—(2 (ジメチルァミノ)エトキシ)ベン ゾチアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフ インアミド(205mg, 0. 48mmol)を 10%塩化水素 Zメタノール(6mL)に溶解し、室 温で 2時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL )に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プ 口ポキシカルボ-ルァミノ)酪酸(161mg, 0. 48mmol) , DIPEA(294 μ L, 1. 7m mol)、 HATU (220mg, 0. 58mmol)をカ卩え、室温で攪拌し、 12時間後、蒸留水を 加えてジェチルエーテルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られ
た粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 159mg (52 %)を得た。
[0652] ステップ 3 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
(2- (ジメチルァミノ)エトキシ)ベンゾチアゾールー 2—ィル) 2H—テトラヒドロピ ラン 4 ィル)ブタナミド ·二塩酸塩 (化合物 134)の合成
アルゴン雰囲気下、 (3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(6— ( 2—(ジメチルァミノ)エトキシ)ベンゾチアゾールー 2—ィル) 2H—テトラヒドロピラン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(159mg , 0. 25mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 2時間攪 拌した後濃縮し、表題ィ匕合物 137mg (90%)を得た。
[0653] 実施例 135
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(3 モノレ ホリノプロボキシ)ベンゾチアゾーノレ 2—ィノレ) 2Hーテトラヒドロピラン 4ーィノレ) ブタナミド ·二塩酸塩 (化合物 135)の合成
ステップ 1 6—(3 モルホリノプロボキシ)ベンゾチアゾール(参考化合物 445)の合 成
アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (180mg, 1 . 2mmol)の THF溶液(6mL)に、トリフエ-ルホスフィン(375mg, 1. 4mmol)、DI AD (282mg, 1. 4mmol)、 3 モルホリノ— 1—プロノ V—ル(208mg, 1. 4mmol )を加え、室温で 14時間攪拌した。濃縮して得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 246mg (75%)を得た。
[0654] ステップ 2 2—メチルー N— (4— (6— (3 モルホリノプロボキシ)ベンゾチアゾール —2—ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィンアミド(参 考化合物 446)の合成
アルゴン雰囲気下、 6—(3 モルホリノプロボキシ)ベンゾチアゾール(246mg, 0. 89mmol)の THF (5mL)溶液を— 78°Cに冷却し、 n—ブチルリチウム Zへキサン溶 液(2. 64M, 0. 41mL, 1. lmmol)を滴下した。 30分後、 2—メチル N— (2H— テトラヒドロピラン 4 イリデン) 2 プロパンスルフィンイミド(参考化合物 416)の
粗生成物(244mg,約 1. 2mmol)の THF (5mL)溶液を滴下した。滴下終了後、室 温までゆっくりと昇温し、 12時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層 を飽和食塩水で洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 33mg (約 8%)を得た。
[0655] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(3—モルホリノプロボキシ)ベンゾチアゾー ルー 2 ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミド(参考化合物 447)の合 成
アルゴン雰囲気下、 2—メチルー N— (4—(6—(3 モルホリノプロボキシ)ベンゾ チアゾール 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィ ンアミド(33mg, 0. O69mmol)を 10%塩化水素 Zメタノール(6mL)に溶解し、室 温で 2時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL )に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プ 口ポキシカルボ-ルァミノ)酪酸(23mg, 0. 069mmol) , DIPEA(30 μ L, 0. 2mm ol)、 HATU (31mg, 0. 082mmol)を加え、室温で攪拌し、 12時間後、蒸留水を加 えてジェチルエーテルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 27mg (56%) を得た。
[0656] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 一(3 モルホリノプロボキシ)ベンゾチアゾールー 2 ィル)一2H—テトラヒドロピラン - 4 ィル)ブタナミド ·二塩酸塩 (化合物 135)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6—(3 モルホリノプロボキシ)ベン ゾチアゾールー 2 ィル) 2H—テトラヒドロピランー4 ィル)ブタナミド(27mg, 0. 038mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 2時間攪拌 した後濃縮し、表題ィ匕合物 20mg (79%)を得た。
[0657] 実施例 136
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2—メチ
ルチオエトキシ)ベンゾチアゾール - 2-ィル) 2H テトラヒドロピラン一 4 ィル) ブタナミド ·塩酸塩 (化合物 136)の合成
ステップ 1 6—(2—メチルチオエトキシ)ベンゾチアゾール(参考化合物 448)の合 成
アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (180mg, 1 . 2mmol)のァセトニトリル溶液(20mL)に、炭酸カリウム(1. 6g, 12mmol)、 2 ク ロロェチルメチルスルフイド(659mg, 6. Ommol)を加え、 14時間加熱還流した。放 冷後、蒸留水を加えて酢酸ェチルで抽出し、乾燥後濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 219mg (82%)を得た。
[0658] ステップ 2 2—メチルー N—(4一(6—(2—メチルチオエトキシ)ベンゾチアゾールー 2 ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィンアミド(参考 化合物 449)の合成
アルゴン雰囲気下、 6—(2—メチルチオエトキシ)ベンゾチアゾール(219mg, 0. 9 7mmol)の THF (5mL)溶液を—78°Cに冷却し、 n—ブチルリチウム Zへキサン溶 液(2. 64M, 0. 57mL, 1. 5mmol)を滴下した。 30分後、 2—メチル N— (2H— テトラヒドロピラン 4 イリデン) 2 プロパンスルフィンイミド(参考化合物 416)の 粗生成物(305mg,約 1. 5mmol)の THF (5mL)溶液を滴下した。滴下終了後、室 温までゆっくりと昇温し、 12時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層 を飽和食塩水で洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で精製し、表題ィ匕合物 191mg (約 46%)を得た。
[0659] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2—メチルチオエトキシ)ベンゾチアゾー ルー 2 ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミド(参考化合物 450)の合 成
アルゴン雰囲気下、 2—メチルー N—(4一(6—(2—メチルチオエトキシ)ベンゾチ ァゾール一 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 2 プロパンスルフィン アミド(191mg, 0. 45mmol)を 10%塩化水素 Zメタノール(6mL)に溶解し、室温 で 2時間攪拌した。アルゴン雰囲気下、濃縮して得られた粗生成物を DMF (3mL)
に溶解し、 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチル 2 プロ ポキシカルボ-ルァミノ)酪酸(149mg, 0. 45mmol) , DIPEA(272 μ L, 1. 6mm ol)、 HATU (203mg, 0. 54mmol)をカ卩え、室温で攪拌し、 12時間後、蒸留水を加 えてジェチルエーテルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 40mg (14%) を得た。
[0660] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 一(2—メチルチオエトキシ)ベンゾチアゾールー 2—ィル) 2H—テトラヒドロピラン - 4 ィル)ブタナミド ·塩酸塩 (化合物 136)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6—(2—メチルチオエトキシ)ベンゾ チアゾールー 2—ィル) 2H—テトラヒドロピランー4 ィル)ブタナミド(20mg, 0. 0 38mmol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 2時間攪拌し た後濃縮し、表題ィ匕合物 18mg (定量的)を得た。
[0661] 実施例 137
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(2—メチ ルスルホ -ルエトキシ)ベンゾチアゾール 2 ィル) 2H テトラヒドロピラン一 4— ィル)ブタナミド ·塩酸塩 (化合物 137)の合成
ステップ 1 (3R)—4— (2, 4, 5 トリフルオロフェ -ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (4—(6—(2—メチルスルホ -ルエトキシ)ベンゾチア ゾールー 2—ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミド(参考化合物 451) の合成
(3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロポキシ力 ルポ-ルァミノ) N— (4—(6—(2—メチルチオエトキシ)ベンゾチアゾールー 2—ィ ル)— 2H—テトラヒドロピラン— 4—ィル)ブタナミド(参考化合物 450) (108mg, 0. 1 7mmol)をジクロロメタン(5mL)に溶解し、氷冷下、 mCPBA( >65%) (61mg, >0 . 23mol)を 3回に分けて添加し、 2時間攪拌した。チォ硫酸ナトリウム水溶液を加え て激しく攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで抽出し
、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 59mg (52%)を得た。
[0662] ステップ 2 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6
- (2—メチルスルホ -ルエトキシ)ベンゾチアゾール—2—ィル)—2H—テトラヒドロ ピラン― 4 ィル)ブタナミド ·塩酸塩 (化合物 137)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボ-ルァミノ) N— (4—(6—(2—メチルスルホ -ルエトキシ) ベンゾチアゾールー 2—ィル) 2H—テトラヒドロピランー4 ィル)ブタナミド(59mg
, 0. 088mmol)を 10%塩化水素 Zメタノール溶液(5mL)に溶解し、室温で 2時間 攪拌した後濃縮し、表題ィ匕合物 36mg (68%)を得た。
[0663] 実施例 138
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(6—(3—メトキ シプロボキシ)ベンゾチアゾールー 2 ィル) 2H—テトラヒドロピランー4 ィル)ブ タナミド ·塩酸塩 (化合物 138)の合成
ステップ 1 6—(3—メトキシプロボキシ)ベンゾチアゾール(参考化合物 452)の合成 アルゴン雰囲気下、 6 ヒドロキシベンゾチアゾール(参考化合物 262) (180mg, 1 . 2mmol)の THF溶液(6mL)に、トリフエ-ルホスフィン(375mg, 1. 4mmol)、DI AD (282mg, 1. 4mmol)、 3—メトキシ一 1—プロパノール(129mg, 1. 4mmol)を 順次加え、室温で 14時間攪拌した。濃縮して得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 252mg (95%)を得た。
[0664] ステップ 2 4—(6— (3—メトキシプロボキシ)ベンゾチアゾールー 2—ィル)—2H— テトラヒドロピラン 4 ァミン ·塩酸塩 (参考化合物 453)の合成
アルゴン雰囲気下、 6—(3—メトキシプロボキシ)ベンゾチアゾール(252mg, 1. 1 mmol)の THF (5mL)溶液を— 78°Cに冷却し、 n ブチルリチウム Zへキサン溶液( 2. 64M, 0. 62mL, 1. 6mmol)を滴下した。 30分後、 2—メチル N— (2H—テト ラヒドロピラン— 4—イリデン)—2 プロパンスルフィンイミド(参考化合物 416)の粗 生成物(333mg,約 1. 6mmol)の THF (5mL)溶液を滴下した。滴下終了後、室温 までゆっくりと昇温し、 12時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層を
飽和食塩水で洗浄後、乾燥し濃縮して得られた粗生成物をシリカゲルカラムクロマト グラフィ一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、 10%塩化水素 Zメタノール (6mL)に溶解し、室温で 2時間攪拌後濃縮して、表題ィ匕合物 180mg ( 約 44%)を得た。
[0665] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6— (3—メトキ シプロボキシ)ベンゾチアゾールー 2 ィル) 2H—テトラヒドロピランー4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 454)の合 成
アルゴン雰囲気下、 4一(6—(3—メトキシプロボキシ)ベンゾチアゾールー 2—ィル )— 2H—テトラヒドロピラン一 4—ァミン '塩酸塩を DMF (3mL)に溶解し、(3R)— 4 一(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァ ミノ)酪酸(167mg, 0. 50mmol) , DIPEA(306 μ L, 1. 76mmol)、 HATU (229 mg, 0. 60mmol)を加え、室温で攪拌し、 12時間後、蒸留水をカ卩えてジェチルエー テルで抽出し、飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合物 126mg (39%)を得た。
[0666] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (6 一(3—メトキシプロボキシ)ベンゾチアゾールー 2 ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミド ·塩酸塩 (化合物 138)の合成
アルゴン雰囲気下、 (3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(6— ( 3—メトキシプロボキシ)ベンゾチアゾーノレ 2—ィノレ) 2Hーテトラヒドロピラン 4 ィル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(126mg, 0. 2 Ommol)を 10%塩ィ匕水素 Zメタノール溶液(5mL)に溶解し、室温で 2時間攪拌した 後濃縮し、表題ィ匕合物 98mg (86%)を得た。
[0667] 実施例 139
(3R)—3 ァミノ一 4— (2, 4, 5 トリフルオロフェ -ル) N— (4— (1—メチルベン ゾイミダゾール一 2—ィル) 2H—テトラヒドロピラン一 4—ィル)ブタナミド '二塩酸塩 (化合物 139)の合成
ステップ 1 4ーシァノー 2H—テトラヒドロピラン 4ーァミン(参考化合物 455)の合成
2H—テトラヒドロピラン一 4—オン(1. Og, lOmmol)のエタノール(5mL)、蒸留水 (10mL)混合溶液に、塩化アンモ-ゥム(561mg, lOmmol)、シアン化カリウム(68 3mg, lOmmol)をカ卩え、室温で 17時間攪拌した。 1N水酸化ナトリウム水溶液をカロ え、クロ口ホルムで抽出し、有機層を乾燥後濃縮した。得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合物 760mg (60%)を得た。
[0668] ステップ 2 4— (9—フルォレニルメトキシカルボ-ルァミノ)一 2H—テトラヒドロピラン
4一力ルボン酸 (参考化合物 456)の合成
4 シァノ 2H—テトラヒドロピラン一 4 ァミン(300mg, 2. 4mmol)の蒸留水(8 mL)溶液に、水酸ィ匕カリウム(667mg, 12mmol)をカ卩え、加熱還流した。 12時間後 放冷し、氷冷下、 1N塩ィ匕水素で中和し、濃縮した。アルゴン雰囲気下、得られた粗 生成物をジクロロメタン(20mL)に溶解し、 DIPEA(1. 4mL, 8. 3mmol)を滴下し た。室温で 30分間撹拌した後、クロロトリメチルシラン(600 L, 4. 8mmol)を滴下 し、室温で 30分間撹拌した後、加熱還流した。 3時間後、—15°Cに冷却し、 Fmoc- Cl(472mg, 2. 2mmol)を一気に加え、室温までゆっくり昇温しながら、 18時間撹 拌した。濃縮し、飽和炭酸水素ナトリウム水溶液を加え、ジェチルエーテルで抽出し た。水層を氷冷し、 1N塩酸を加え酢酸ェチルで抽出し、有機層を乾燥後濃縮して得 られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 167mg ( 19%)を得た。
[0669] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)— N— (4— (1—メチルベン ゾイミダゾールー 2—ィル) 2H—テトラヒドロピラン一 4—ィル) - 3- (2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 457)の合成
アルゴン雰囲気下、 4一(9 フルォレニルメトキシカルボ-ルァミノ)—2H—テトラ ヒドロピランー4一力ルボン酸(167mg, 0. 46mmol)の DMF溶液(6mL)に、 N—メ チル— 1, 2 フエ-レンジァミン(56mg, 0. 46mmol)、 HATU (225mg, 0. 59m mol)を加え、室温で攪拌した。 12時間後、蒸留水をカ卩えてジェチルエーテルで抽 出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精 製した。得られた粗精製物を、アルゴン雰囲気下、酢酸(lOmL)に溶解し、 80°Cで 6 0時間攪拌した後放冷し、濃縮した。得られた粗生成物をシリカゲルカラムクロマトグ
ラフィ一で粗精製し、得られた粗精製物を、アルゴン雰囲気下、 DMF (3mL)に溶解 し、ピぺリジン (0. ImL)をカ卩え、室温で 15時間撹拌した。濃縮して得られた粗生成 物をシリカゲルカラムクロマトグラフィーで粗精製した。アルゴン雰囲気下、得られた 粗精製物を DMF (4mL)に溶解し、(3R)— 4一(2, 4, 5 トリフルオロフヱニル) 3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸(19mg, 0. 056mmol)、DI ΡΕΑ (25 /ζ Ι^, 0. 14mmol)、 HATU (28mg, 0. 074mmol)を加え、室温で攪拌 し、 15時間後、蒸留水をカ卩えてジェチルエーテルで抽出し、飽和食塩水で洗浄後乾 燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表 題ィ匕合物 20mg (65%)を得た。
[0670] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1 メチルベンゾイミダゾール 2 ィル) 2H—テトラヒドロピラン 4 ィル)ブタナミ ド ·二塩酸塩 (化合物 139)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— N— (4— (1— メチルベンゾイミダゾールー 2—ィル) 2H—テトラヒドロピラン一 4—ィル) 3— (2 ーメチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(20mg, 0. 037mmol)を 10 %塩ィ匕水素 Zメタノール溶液 (6mL)に溶解し、室温で 12時間攪拌した後濃縮し、 表題ィ匕合物 18mg (95%)を得た。
[0671] 実施例 140
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(1— (2—メトキ シェチル)ベンゾイミダゾールー 2—ィル)—1, 1ージォキソチアンー4 ィル)ブタナ ミド ·二塩酸塩 (ィ匕合物 140)の合成
ステップ 1 N— (2—メトキシェチル) 1, 2 フエ-レンジァミン(参考化合物 458) の合成
アルゴン雰囲気下、 2 フルォ口-トロベンゼン(1. Og, 7. lmmol)のジクロ口メタ ン溶液(30mL)に、 2—メトキシェチルァミン(3. 2g, 43mmol)、 TEA(12mL, 85 mmol)を加え、加熱還流した。 15時間後放冷し、蒸留水をカ卩えてクロ口ホルムで抽 出し、有機層を乾燥後濃縮した。得られた粗生成物をエタノール(10mL)に溶解し、 10%パラジウム Z炭素(250mg)をカ卩え、水素雰囲気下、室温で撹拌した。 15時間
後濾過し、濾液を濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題化合物 1. lg (91%)を得た。
[0672] ステップ 2 4— (9 フルォレニルメトキシカルボ-ルァミノ)— 4— (1— (2—メトキシ ェチル)ベンゾイミダゾールー 2 ィル)チアン(参考化合物 459)の合成
アルゴン雰囲気下、 4一(9 フルォレニルメトキシカルボ-ルァミノ)チアンー 4一力 ルボン酸(参考化合物 388) (600mg, 1. 6mmol)の DMF (lOmL)溶液に、 N—(2 —メトキシェチル)一 1, 2 フエ-レンジァミン(260mg, 1. 6mmol)、 HATU (714 mg, 1. 9mmol) , DIPEA(681 μ L, 3. 9mmol)をカ卩え、室温で攪拌した。 12時間 後、蒸留水を加えてジェチルエーテルで抽出し、有機層を乾燥、濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物を、 アルゴン雰囲気下、酢酸(lOmL)に溶解し、 80°Cで 48時間攪拌した後放冷し、濃 縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合 物 87mg (17%)を得た。
[0673] ステップ 3 4—(1一(2—メトキシェチル)ベンゾイミダゾールー 2 ィル)チアンー 4 ーァミン (参考化合物 460)の合成
アルゴン雰囲気下、 4一(9 フルォレニルメトキシカルボ-ルァミノ) -4- (1— (2 —メトキシェチル)ベンゾイミダゾールー 2—ィル)チアン(87mg, 0. 17mmol)を D MF (3mL)に溶解し、ピぺリジン (0. 2mL)をカ卩え、室温で 12時間撹拌した。濃縮し て得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 37m g (73%)を得た。
[0674] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1— (2—メトキ シェチル)ベンゾイミダゾールー 2—ィル)—1, 1—ジォキソチアン— 4—ィル)—3— (2 メチル 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 461)の合成 アルゴン雰囲気下、 4一(1— (2—メトキシェチル)ベンゾイミダゾールー 2 ィル)チ アン— 4 ァミン(37mg, 0. 12mmol)を DMF (4mL)に溶解し、(3R)— 4— (2, 4 , 5 トリフルオロフェ -ル)—3— (2—メチル—2 プロポキシカルボ-ルァミノ)酪酸 (41mg, 0. 12mmol) , DIPEA (53 μ L, 0. 31mmol)、 HATU (56mg, 0. 15m mol)を加え、室温で攪拌し、 12時間後、蒸留水をカ卩えてジェチルエーテルで抽出し
、有機層を乾燥、濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで 粗精製した。得られた粗精製物を、アルゴン雰囲気下、ジクロロメタン (4mL)に溶解 し、氷冷下、 mCPBA ( >65%) (62mg, >0. 23mmol)を添カ卩し、 3時間攪拌した。 チォ硫酸ナトリウム水溶液を加えて激しく攪拌した後、炭酸カリウム水溶液を加えた。 クロ口ホルムで抽出し、乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマト グラフィ一で精製し、表題ィ匕合物 41mg (53%)を得た。
[0675] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (1
- (2—メトキシェチル)ベンゾイミダゾールー 2—ィル)—1, 1—ジォキソチアン— 4 —ィル)ブタナミド ·二塩酸塩 (ィ匕合物 140)の合成
アルゴン雰囲気下、 (3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(1一( 2—メトキシェチル)ベンゾイミダゾールー 2—ィル) 1, 1—ジォキソチアン一 4—ィ ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(41mg, 0. 064 mmol)を 10%塩ィ匕水素 Zメタノール溶液 (6mL)に溶解し、室温で 12時間攪拌した 後濃縮し、表題ィ匕合物 32mg (80%)を得た。
[0676] 実施例 141
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (4—(5—(2—メトキ シェトキシ)一 1—メチルベンゾイミダゾールー 2—ィル) 2H—テトラヒドロピラン一 4 —ィル)ブタナミド '二塩酸塩 (ィ匕合物 141)の合成
ステップ 1 4— (2—メトキシェトキシ)—N—メチル 2 -トロア-リン(参考化合物 462)の合成
アルゴン雰囲気下、 4 アミノー 3 -トロフエノール(1. Og, 6. 5mmol)のアセトン 溶液(20mL)に、臭化 2—メトキシェチル (4. 5g, 32mmol)、炭酸カリウム(4. 5g, 32mmol)をカ卩え、加熱還流した。 15時間後放冷し、蒸留水をカ卩えて、クロ口ホルム で抽出し、有機層を乾燥後濃縮して得られた粗生成物をシリカゲルカラムクロマトダラ フィ一で粗精製した。得られた粗精製物 2. Ogのうち 1. Ogをトルエン(lOmL)に溶解 し、テトラ— n—ブチルアンモ-ゥム'硫酸水素塩(160mg, 0. 47mmol)、 50%水酸 化ナトリウム水溶液(2mL)、硫酸ジメチル(0. 6mL, 6. 3mmol)を加え、室温で 14 時間攪拌した。蒸留水を加え酢酸ェチルで抽出し、有機層を乾燥後濃縮した。得ら
れた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 680mg (6 4%)を得た。
[0677] ステップ 2 4—(5— (2—メトキシェトキシ)ー1 メチルベンゾイミダゾールー 2—ィル )—2H—テトラヒドロピラン— 4 ァミン (参考化合物 463)の合成
4— (2—メトキシェトキシ)一 N—メチル 2 -トロア-リン(680mg, 3. Ommol) のエタノール溶液(20mL)に、 10%パラジウム Z炭素(lOOmg)をカ卩え、水素雰囲 気下、室温で撹拌した。 15時間後濾過し、濾液を濃縮して得られた粗生成物をシリ 力ゲルカラムクロマトグラフィーで粗精製した。得られた粗精製物 367mgのうち 111m gを、アルゴン雰囲気下、 DMF (lOmL)に溶解し、 4— (9—フルォレニルメトキシカ ルポ-ルァミノ) 2H—テトラヒドロピランー4一力ルボン酸(参考化合物 456) (208 mg, 0. 57mmol)、 HATU (258mg, 0. 68mmol) , DIPEA(246 μ L, 1. 4mmol )を加え、室温で攪拌した。 15時間後、蒸留水をカ卩えてジェチルエーテルで抽出し、 有機層を乾燥、濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗 精製した。得られた粗精製物を、アルゴン雰囲気下、酢酸(lOmL)に溶解し、 80°C で 24時間攪拌した後放冷し、濃縮した。得られた粗生成物をシリカゲルカラムクロマ トグラフィ一で粗精製した。得られた粗精製物を、アルゴン雰囲気下、 DMF (3mL) に溶解し、ピぺリジン (0. 2mL)をカ卩え、室温で 12時間撹拌した。濃縮して得られた 粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 44mg (16%) を得た。
[0678] ステップ 3 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5— (2—メトキ シェトキシ)一 1—メチルベンゾイミダゾールー 2—ィル) 2H—テトラヒドロピラン一 4 ィル)ー3—(2—メチルー 2 プロポキシカルボニルァミノ)ブタナミド (参考化合物 464)の合成
アルゴン雰囲気下、 4一(5—(2—メトキシェトキシ) 1 メチルベンゾイミダゾール — 2—ィル) 2H—テトラヒドロピラン一 4 ァミン(44mg, 0. 14mmol)を DMF (4 mL)に溶解し、 (3R) -4- (2, 4, 5 トリフルオロフヱ-ル)ー3—(2—メチルー 2— プロポキシカルボ-ルァミノ)酪酸(48mg, 0. 14mmol) , DIPEA(62 μ L, 0. 36m mol)、 HATU (65mg, 0. 17mmol)をカ卩え、室温で攪拌し、 12時間後、蒸留水を
加えてジェチルエーテルで抽出し、有機層を乾燥、濃縮した。得られた粗生成物を シリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 34mg (38%)を得た。
[0679] ステップ 4 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4— (5
- (2—メトキシェトキシ)一 1—メチルベンゾイミダゾールー 2—ィル) 2H—テトラヒ ドロピラン一 4—ィル)ブタナミド '二塩酸塩 (ィ匕合物 141)の合成
アルゴン雰囲気下、 (3R) -4- (2, 4, 5 トリフルオロフヱ-ル) N— (4—(5— ( 2—メトキシェトキシ)一 1—メチルベンゾイミダゾールー 2—ィル) 2H—テトラヒドロ ピラン 4 ィル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(34 mg, 0. O55mmol)を 10%塩化水素 Zメタノール溶液(6mL)に溶解し、室温で 12 時間攪拌した後濃縮し、表題ィ匕合物 28mg (85%)を得た。
[0680] 実施例 142
(3R) 3 アミノー 4 (2, 4, 5 トリフノレオ口フエ-ノレ) N— (2—(2—(4ーメチ ルスルホ -ルフエ-ル)チアゾールー 4 ィル) 1H ジヒドロインデン 2 ィル) ブタナミド ·塩酸塩 (ィ匕合物 142)の合成
ステップ 1 2—(2—メチルー 2—プロポキシカルボ-ルァミノ) 1H—ジヒドロインデ ンー 2 カルボン酸メチル(参考化合物 465)の合成
アルゴン雰囲気下、 2—(2—メチルー 2—プロポキシカルボ-ルァミノ) 1H—ジヒ ドロインデンー 2—力ルボン酸(500mg, 1. 80mmol)の DMF (3mL)溶液に炭酸水 素カリウム(361mg, 3. 60mmol)、ョウイ匕メチル(0. 22mL, 3. 53mmol)を加え、 室温にて 16時間撹拌した。反応液を蒸留水にあけ、酢酸ェチルにて抽出し、有機層 を蒸留水で洗浄した後、乾燥し濃縮した。得られた粗生成物をへキサン、クロ口ホル ムから再結化し、表題ィ匕合物を 426mg (81%)得た。
[0681] ステップ 2 2 クロロメチルカルボ-ルー 2—(2—メチルー 2 プロポキシカルボ- ルァミノ) 1H ジヒドロインデン(参考化合物 466)の合成
アルゴン雰囲気下、 2—(2—メチルー 2—プロポキシカルボ-ルァミノ) 1H—ジヒ ドロインデン一 2—カルボン酸メチル(400mg, 1. 37mmol)、クロロヨ一ドメタン(0. 40mL, 5. 49mmol)の THF (12mL)溶液を— 78°Cに冷却し、リチウムジイソプロピ ルアミド(6. 8mmol)の THF (lOmL)溶液を滴下した。そのまま、 20分間撹拌後、
酢酸(2. lmL)の THF (lOmL)をゆっくりと加えた。 10分後、飽和塩化アンモ -ゥム 水溶液を加えた後、反応液を室温まで昇温し、酢酸ェチルにて抽出後、有機層を飽 和炭酸水素ナトリウム水溶液、チォ硫酸ナトリウム水溶液にて洗浄し乾燥後濃縮した 。得られた粗生成物を、シリカゲルカラムクロマトグラフィーにて精製し、表題化合物 3 04mg (72%)を得た。
[0682] ステップ 3 2—(2—メチルー 2 プロポキシカルボ-ルァミノ)ー2—(2— (4—メチル スルホユルフェ-ル)チアゾールー 4 ィル) 1H ジヒドロインデン(参考化合物 46 7)の合成
アルゴン雰囲気下、 2—クロロメチルカルボ-ルー 2—(2—メチルー 2—プロポキシ カルボ-ルァミノ)一 1H ジヒドロインデン(lOOmg, 0. 32mmol)をエタノール(4m L)に溶解し、 4— (メチルスルホ -ル)ベンゾチオアミド(11 lmg, 0. 52mmol)、ヨウ ィ匕カリウム(107mg, 0. 77mmol)、炭酸水素ナトリウム(30mg, 0. 36mmol)を加 え 70°Cにて 4時間撹拌した。放冷後、不溶を濾別し、濾液に蒸留水を加え酢酸ェチ ルにて抽出した。有機層を乾燥後濃縮し、得られた粗生成物をシリカゲルカラムクロ マトグラフィ一にて精製し、表題ィ匕合物 55mg (36%)を得た。
[0683] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ ポキシカルボ-ルァミノ) N— (2—(2—(4ーメチルスルホユルフェ-ル)チアゾー ルー 4 ィル) 1H ジヒドロインデンー2 ィル)ブタナミド(参考化合物 468)の合 成
アルゴン雰囲気下、 2—(2—メチルー 2—プロポキシカルボ-ルァミノ) 2—(2— ( 4—メチルスルホユルフェ-ル)チアゾール—4—ィル) 1H ジヒドロインデンを 10 %塩ィ匕水素 Zメタノール(2mL)に溶解し、 18時間撹拌した。濃縮して得られた粗生 成物を、アルゴン雰囲気下、 DMF (lmL)に溶解し、(3R)—4— (2, 4, 5 トリフル オロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(16mg, 0. O5mmol)、 TEA (30 μ L, 0. 15mmol)、 PyBOP (PyBOPとは、ベンゾトリァゾー ル一 1—ィル -ォキシ一トリス一ピロリジノ一ホスホ-ゥム へキサフルオロフォスフエ イトを意味する) (37mg, 0. 075mmol)を加え 23時間撹拌した。濃縮後、飽和炭酸 水素ナトリウム水溶液を加え、酢酸ェチル: THF = 3Zlにて抽出した。有機層を蒸
留水、飽和塩化アンモニゥム水溶液にて洗浄後、乾燥し濃縮した。得られた粗生成 物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 32mg (99%)を得た。
[0684] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (2— (2 一 (4ーメチルスルホユルフェ-ル)チアゾールー 4一ィル)一 1H ジヒドロインデンー
2 ィル)ブタナミド ·塩酸塩 (ィ匕合物 142)の合成
アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル
—2 プロポキシカルボ-ルァミノ) N— (2— (2— (4—メチルスルホユルフェ-ル) チアゾールー 4 ィル) 1H ジヒドロインデン 2 ィル)ブタナミド(32mg, 0. 05 mmol)を塩酸メタノール(2mL)に溶解し、 4時間撹拌した後濃縮し、 目的物を 29mg
(99%)得た。
[0685] 実施例 143
(3R)— 3 ァミノ一 N— (1, 4 ジベンジルピペリジン一 4—ィル) 4— (2 フルォ 口フエ-ル)ブタナミド ·二塩酸塩 (ィ匕合物 143)の合成
ステップ 1 1, 4ージベンジルピペリジン 4 オール(参考化合物 469)の合成 アルゴン雰囲気下、塩化べンジルマグネシウム ZTHF溶液(1. 03M, 3mL, 3. 1 mmol)を THF (3mL)で希釈し、氷冷下、 1—ベンジル— 4 ピぺリドン(0. 52mL, 2. 8mmol)の THF (2mL)溶液を滴下した。滴下終了後、氷浴を外し室温で攪拌し 、 10時間後、蒸留水を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄 後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製 し、表題ィ匕合物 382mg (48%)を得た。
[0686] ステップ 2 N—(l, 4 ジベンジルピペリジンー4 ィル)ァセタミド(参考化合物 470 )の合成
1, 4 ジベンジルピペリジン— 4—オール(363mg, 1. 3mmol)のァセトニトリル( 3mL)溶液を氷冷し、激しく攪拌しながら濃硫酸(2. OmL)をゆっくりと滴下した。滴 下終了後、氷浴を外し室温で攪拌し、 6時間後、氷水に注いだ。水層を水酸化ナトリ ゥム水溶液にてアルカリ性にした後、酢酸ェチルにて抽出し、有機層を飽和食塩水 で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー で精製し、表題ィ匕合物 118mg (28%)を得た。
[0687] ステップ 3 (3R)— N— (1, 4 ジベンジルピペリジン— 4—ィル)—4— (2 フルォ 口フエ-ル) 3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化 合物 471)の合成
アルゴン雰囲気下、 N— (1, 4 ジベンジルピペリジン— 4—ィル)ァセタミド(104 mg, 0. 32mmol)を THF (0. 2mL)に懸濁し、チタンテトライソプロポキシド(96 L , 0. 32mmol)、ジフエ-ルシラン(0. 15mL, 0. 81mmol)をカ卩え、室温で攪拌した 。 12時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液を加えて激しく 攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽出し、有機 層を合わせて飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をジクロロメ タン(3mL)に溶解し、 (3R)—4— (2 フルオロフヱ-ル)—3— (2—メチル—2 プ 口ポキシカルボ-ルァミノ)酪酸(29mg, 0. lmmol)、 HOBt (20mg, 0. 15mmol) 、ΝΕΜ (14 Ι^, 0. l lmmol)、 EDCI'塩酸塩(23mg, 0. 12mmol)を加え、室温 で攪拌した。 12時間後、飽和炭酸水素ナトリウム水溶液を加えクロ口ホルムで抽出し 、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、 表題ィ匕合物 56mg (定量的)を得た。
[0688] ステップ 4 (3R)— 3 ァミノ一 N— (1, 4 ジベンジルピペリジン一 4—ィル) 4— ( 2 フルオロフヱ-ル)ブタナミド ·二塩酸塩(ィ匕合物 143)の合成
アルゴン雰囲気下、 (3R)— N— (1, 4 ジベンジルピペリジン— 4—ィル)—4— ( 2 フルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミ ド(56mg, 0. lmmol)を酢酸ェチル(lmL)に溶解し、 4M塩化水素 Zl, 4 ジォ キサン溶液 (2mL)を加え、室温で攪拌した。 12時間後、飽和炭酸水素ナトリウム水 溶液を加え酢酸ェチルで抽出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。 得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題化合物 (フリー 塩基) 31mg (67%)を得た。表題ィ匕合物(フリー塩基)をメタノール溶液とし、 10%塩 化水素 Zメタノール溶液を加えて塩ィ匕し、これを濃縮し、表題ィ匕合物(二塩酸塩) 31 mg¾ ^守に。
[0689] 実施例 144
(3R)— 3 ァミノ一 N— (1—ベンジル一 4— (2 フエ-ルェチル)ピぺリジン一 4—
ィル)—4— (2, 4, 5 トリフルオロフェ -ル)ブタナミド '二塩酸塩(ィ匕合物 144)の合 成
ステップ 1 1一べンジルー 4一(2 フエ-ルェチル)ピぺリジンー4 オール(参考 化合物 472)の合成
アルゴン雰囲気下、塩化(2—フエ-ルェチル)マグネシウム ZTHF溶液(1. 0M, 5mL, 5. Ommol)をとり、氷冷下、 1—ベンジル一 4 ピぺリドン(0. 5mL, 2. 7mm ol)の THF (5mL)溶液を滴下した。滴下終了後、氷浴を外し室温で攪拌し、 5時間 後、蒸留水を加えた。酢酸ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥 し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、表題 化合物 198mg (25%)を得た。
[0690] ステップ 2 N—(l—べンジルー 4 (2 フエ-ルェチル)ピぺリジンー4 ィル)ァ セタミド (参考化合物 473)の合成
1—ベンジル— 4— (2—フエ-ルェチル)ピぺリジン— 4—オール(198mg, 0. 67 mmol)のァセトニトリル(1. lmL)溶液を氷冷し、激しく攪拌しながら濃硫酸 (0. 75 mL)をゆっくりと滴下した。滴下終了後、氷浴を外し室温で攪拌し、 4時間後、氷水に 注いだ。水層を水酸ィ匕ナトリウム水溶液にてアルカリ性にした後、酢酸ェチルにて抽 出し、有機層を飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲ ルカラムクロマトグラフィーで精製し、表題ィ匕合物 128mg (57%)を得た。
[0691] ステップ 3 (3R)— N— (1—ベンジル— 4— (2 フエ-ルェチル)ピぺリジン— 4— ィル)ー4ー(2, 4, 5 トリフルオロフェ-ル)ー3—(2—メチルー 2 プロポキシカル ボニルァミノ)ブタナミド (参考化合物 474)の合成
アルゴン雰囲気下、 N— (1—べンジルー 4一(2 フエ-ルェチル)ピぺリジンー4 ーィノレ)ァセタミド(116mg, 0. 34mmol)に、チタンテトライソプロポキシド(123 L , 0. 42mmol)、ジフエ-ルシラン(0. 50mL, 2. 7mmol)をカ卩え、室温で攪拌した。 12時間後、酢酸ェチルにて希釈し、飽和炭酸水素ナトリウム水溶液を加えて激しく 攪拌した。生じた白色固体を濾別し、濾液を分液後、酢酸ェチルにて抽出し、有機 層を合わせて飽和食塩水で洗浄後、乾燥し濃縮した。得られた粗生成物をジクロロメ タン(5mL)に溶解し、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチ
ルー 2—プロポキシカルボ-ルァミノ)酪酸(58mg, 0. 17mmol)、 HOBt (35mg, 0 . 26mmol) , NEM (24 ^ L, 0. 19mmol)、 EDCI'塩酸塩(40mg, 0. 21mmol) を加え、室温で攪拌した。 12時間後、飽和炭酸水素ナトリウム水溶液を加えクロロホ ルムで抽出し、乾燥後濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフ ィ一で精製し、表題ィ匕合物 70mg (66%)を得た。
[0692] ステップ 4 (3R)— 3—ァミノ一 N—(l—ベンジル一 4— (2—フエ-ルェチル)ピペリ ジン— 4—ィル)—4— (2, 4, 5—トリフルオロフェ -ル)ブタナミド '二塩酸塩(ィ匕合物 144)の合成
アルゴン雰囲気下、 (3R)— N— (1—ベンジル— 4— (2—フエ-ルェチル)ピペリ ジン— 4—ィル)—4— (2, 4, 5—トリフルオロフェ-ル)—3— (2—メチル—2—プロ ポキシカルボ-ルァミノ)ブタナミド(70mg, 0. l lmmol)を 10%塩化水素 Zメタノー ル溶液(5mL)に溶解し、室温で 13時間攪拌した後、濃縮し、表題ィ匕合物 50mg (75 %)を得た。
[0693] 以下、参考例 1、 2において、特許文献 3の請求範囲に含まれる化合物(比較化合 物 1)及び特許文献 4の請求範囲に含まれる化合物(比較化合物 2)の具体的な合成 法を示す。
[化 26]

比較化合物 1
[化 27]

比較化合物 2
[0694] 参考例 1
(3R)— 3 ァミノ一 N— (4— (N ベンジル一 N—メチルァミノカルボ-ル) 2H— テトラヒドロピラン一 4—ィル) 4— (2, 4, 5—トリフルオロフヱ-ル)ブタナミド '塩酸 塩 (比較化合物 1)の合成
ステップ 1 (3R)— N— (4— (N ベンジル一 N—メチルァミノカルボ-ル) 2H— テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— (2—メチ ルー 2 プロポキシカルボ-ルァミノ)ブタナミド(参考化合物 475)の合成
アルゴン雰囲気下、 N—ベンジルメチルァミン(55mg, 0. 43mmol)を DMF (4m L)に溶解し、 4— (9—フルォレニルメトキシカルボ-ルァミノ) 2H—テトラヒドロビラ ンー4一力ルボン酸(参考化合物 456) (150mg, 0. 41mmol)、 HATU (163mg, 0. 43mmol) , DIPEA (142 μ L, 0. 82mmol)をカ卩え、室温で攪拌した。 12時間 後、反応液に蒸留水を加え、酢酸ェチルで抽出した。有機層を蒸留水、飽和食塩水 で洗浄した後、乾燥し濃縮した。アルゴン雰囲気下、得られた粗生成物を DMF (3m L)に溶解し、ピぺリジン(lmL)を加え、室温で撹拌した。 2時間後、反応液に蒸留水 を加え、酢酸ェチルで抽出した。有機層を蒸留水、飽和食塩水で洗浄した後、乾燥 し濃縮して得られた粗生成物をシリカゲルカラムクロマトグラフィーで粗精製した。ァ ルゴン雰囲気下、得られた粗精製物を DMF (3mL)に溶解し、(3R)— 4— (2, 4, 5 トリフルオロフェ -ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(1 02mg, 0. 31mmol)、 HATU (116mg, 0. 31mmol) , DIPEA(97 μ L, 0. 56m mol)を加え、室温で攪拌した。 16時間後、蒸留水、飽和食塩水を加え、酢酸ェチル で抽出し、有機層を蒸留水で洗浄後、乾燥し濃縮した。酢酸ェチルカゝら再結晶して 表題化合物 120mg (48%)を得た。
ステップ 2 (3R)— 3 ァミノ一 N— (4— (N ベンジル一 N—メチルァミノカルボ- ル)一 2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ -ル)ブタ ナミド'塩酸塩 (比較化合物 1 )の合成
アルゴン雰囲気下、(3R)— N— (4— (N ベンジル— N—メチルァミノカルボ-ル )—2H—テトラヒドロピラン一 4—ィル) 4— (2, 4, 5 トリフルオロフェ-ル)一3— ( 2—メチルー 2 プロポキシカルボ-ルァミノ)ブタナミド(118mg, 0. 21mmol)を 10 %塩ィ匕水素メタノール溶液 (4mL)に溶解し、室温で撹拌した。 4時間後、反応液を
濃縮し、減圧下乾燥することにより表題ィ匕合物 105mg (定量的)を得た。
[0696] 参考例 2
(3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4—メチル 2H— テトラヒドロピランー4 ィル)ブタナミド '塩酸塩 (比較ィ匕合物 2)の合成
ステップ 1 2H—テトラヒドロピランー4 オン O ベンジルォキシム(参考化合物 4 76)の合成
2H—テトラヒドロピラン一 4—オン(300mg、 3. OOmmol)とベンジルォキシァミン. 塩酸塩(958mg, 6. Ommol)と酢酸ナトリウム(1. 23g, 15. Ommol)にメタノーノレ( 36mL)と蒸留水(6mL)を加え、 1時間加熱還流した。放冷後、蒸留水を加え、酢酸 ェチルで抽出し、有機層を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生 成物をシリカゲルカラムクロマトグラフィーで精製し、表題ィ匕合物 540mg (88%)を得 た。
[0697] ステップ 2 N ベンジルォキシ 4—メチル 2H—テトラヒドロピラン一 4 ァミン(参 考化合物 477)の合成
アルゴン雰囲気下、 2H—テトラヒドロピラン一 4—オン O ベンジルォキシム(450 mg、 2. 19mmol)のジェチルエーテル(5mL)溶液を 0°Cに冷却し、メチルリチウム Zジェチルエーテル溶液(1. 04M, 12. 64mL, 13. lmmol)をゆっくりと滴下した 。滴下終了後室温で 20分間攪拌し、蒸留水を加え、酢酸ェチルで抽出した。有機層 を飽和食塩水にて洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムク 口マトグラフィ一で精製し、表題ィ匕合物 247mg (51%)を得た。
[0698] ステップ 3 4—メチル 2H—テトラヒドロピラン一 4 ァミン '塩酸塩 (参考化合物 47 8)の合成
N—ベンジルォキシ 4—メチル 2H—テトラヒドロピラン一 4 ァミン(100mg、 0. 4 5mmol)をメタノール(12mL)に溶解し、 10%パラジウム/炭素(116mg)を加え、水 素雰囲気下、室温で攪拌した。 3時間後、反応溶液をセライトで濾過し、 10%塩ィ匕水 素 Zメタノール溶液 (3mL)を加えて塩ィ匕した後濃縮し、表題ィ匕合物 28mg (41%)を 得た。
[0699] ステップ 4 (3R)— 4— (2, 4, 5 トリフルオロフェ-ル)—3— (2—メチル—2 プロ
ポキシカルボ-ルァミノ) N— (4—メチルー 2H—テトラヒドロピラン 4 ィル)ブタ ナミド (参考化合物 479)の合成
アルゴン雰囲気下、 4ーメチルー 2H—テトラヒドロピランー4ーァミン '塩酸塩(28m g, 0. 18mmol)を DMF (5mL)に溶解し、 (3R)— 4— (2, 4, 5 トリフルオロフェ- ル)ー3—(2—メチルー 2 プロポキシカルボ-ルァミノ)酪酸(62mg, 0. 18mmol) 、 HATU (70mg, 0. 18mmol)、 DIPEA (0. 096mL, 0. 55mmol)を加え、 14時 間室温で攪拌した。蒸留水を加え、酢酸ェチルで抽出し、有機層を飽和食塩水にて 洗浄後、乾燥し濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィーで 精製し、表題ィ匕合物 50mg (63%)を得た。
[0700] ステップ 5 (3R)— 3 ァミノ一 4— (2, 4, 5 トリフルオロフェ-ル)一 N— (4—メチ ルー 2H テトラヒドロピラン 4 ィル)ブタナミド ·塩酸塩 (比較化合物 2)の合成 アルゴン雰囲気下、 (3R)— 4— (2, 4, 5 トリフルオロフヱ-ル)— 3— (2—メチル 2 プロポキシカルボニルァミノ) N— (4—メチルー 2H—テトラヒドロピランー4 ィル)ブタナミド(50mg、 0. l lmmol)を 10%塩化水素 Zメタノール溶液(1. 5mL )に溶解し、室温で 1時間攪拌した後濃縮し、表題ィ匕合物 43mg (定量的)を得た。
[0701] 以上の実施例及び参考例で挙げた本発明の化合物、比較化合物及び参考化合 物の物性データを以下の表 3〜72に示す。
[表 3]

[表 4]

[表 5]
 6]
6]

表 7

[表 8]
表 8

[表 9]
表 9

[表 10]
688/v:s£90sfcl>d 663/ 8ί6ε90/-00ί OAV

〕

[表 12]
〔




¾〔s

表 17

[表 18]
¾〔〕19

表 19

[表 20]
重〔

o

&

墜〔


[表 25]

[表 26]

^
¾〔¾2


»28

»29

»30




S4

[表 36]

¾〔〕38

7

〔¾S4

^
¾〔£





[表 45]

環〕


S

[表 49]
S〕50

0


[表 53]


4

[表 56]


[表 58]
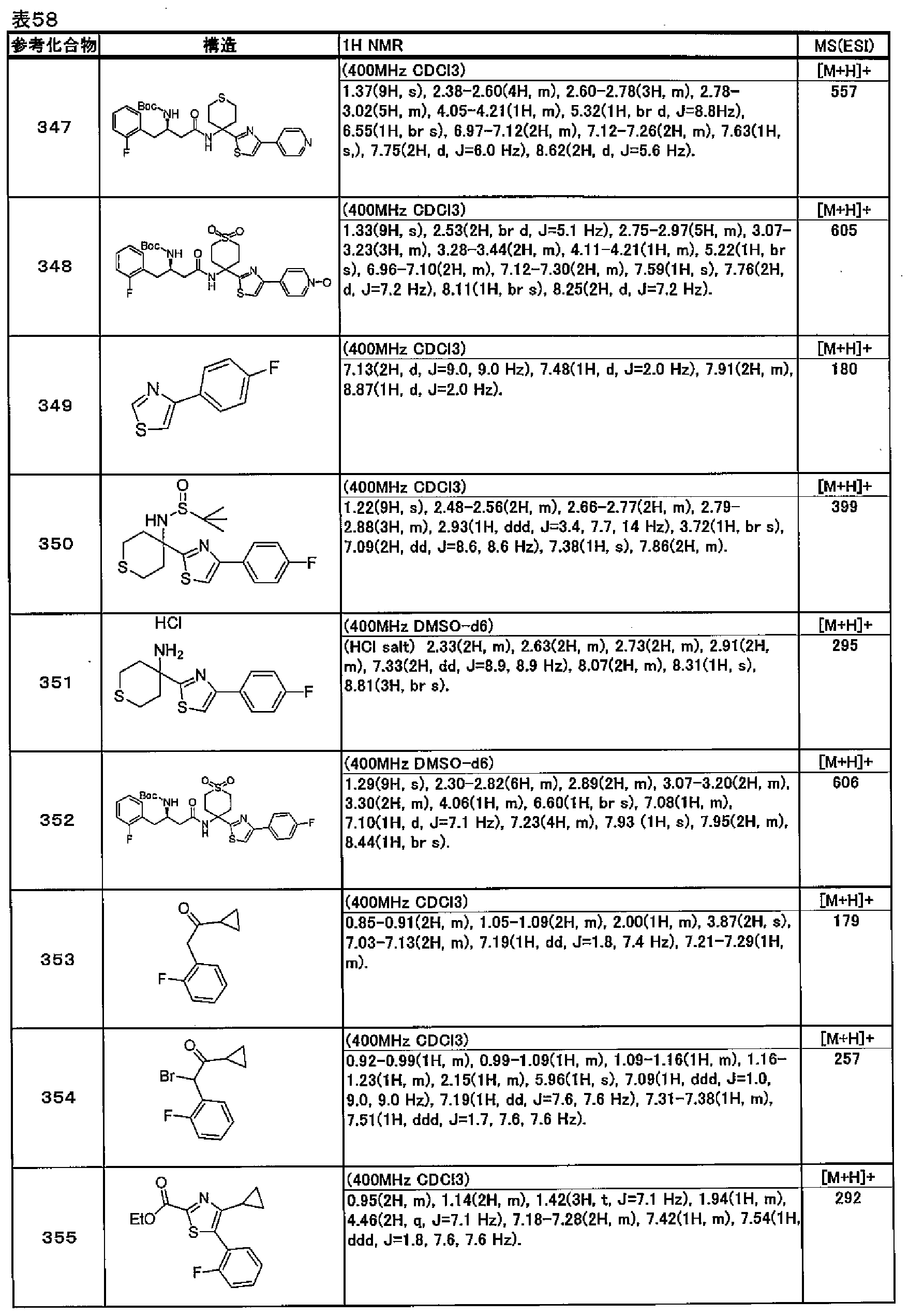


»60
〔〕 62


¾2

表 64

[表 65]

[表 66]

[表 67]


[表 69]

表 70

[表 71]

[表 72]
表 72

[0702] 実施例 145
DPP-IV阻害活性評価
(1) PP- IV粗酵素溶液はヒト結腸腺癌由来細胞株 Caco - 2より調製した。 Caco - 2 に存在する DPP-IVにつ!/、ては、すでに Laurentらによって報告されて!、る (「ジャー ナル 'ォブ 'セル'サイエンス」(Journal of Cell Science)、 1995年、 108卷、 p 2109-212 l.) o Caco— 2細胞は、 10%FBS (牛胎児血清、ジエーアールエイチバイオサイェン ス社製)を含有する D— MEM培地 (ギブコ社製)で培養した。細胞抽出液は、培地を 除いて集めた細胞を 0. 5%Nonidet— P40を含む 10mMトリス塩酸緩衝液(pH8. 0)に浸し、氷浴中で抽出し、 18500gで 1時間遠心した上清を分離して調製した。
[0703] (2) Caco— 2由来の DPP-IV阻害活性の測定反応は、クボタらの方法(「タリ-カル ·
アンド'ェクスペリメンタノレ'イミュノロジィ」 (Clinical and Experimental Immunology) , 1 992年、 89卷、 ρ 192-197.)に準じて、 96穴平底プレートを用いて 37°Cで実施した。 化合物、前記(1)で調製した Caco— 2細胞抽出液の 5 /z gタンパク質量及び基質 (H - Ala - Pro - AFC ( AFCとは、 7—ァミノ一 4—トリフルォロメチルクマリンを意味す る)、フナコシ)最終濃度 40 /z Mを 25mMトリス—塩酸緩衝液 (pH7. 4)、 140mM塩 化ナトリウム、 10mM塩ィ匕カリウム、 1%ゥシ血清アルブミン水溶液 150 Lに希釈し 、 37°Cで酵素反応を開始した。 10分後 25%酢酸水溶液 10 Lを添加して反応を停 止させた。蛍光プレートリーダーを用いて、励起波長 380nm、測定波長 485nmにお ける蛍光強度を測定した。基質溶液添加前にあらカゝじめ 25%酢酸水溶液を添加して 反応を停止させたバックグラウンドゥエルと化合物を添カ卩しないコントロールゥエルの 蛍光強度の差を 100%とし、化合物添加ゥエルの蛍光強度を内挿し、化合物添加時の 残存酵素活性を相対値として算出した。複数濃度の化合物添加時の相対残存酵素 活性値から、酵素活性を 50%阻害する化合物濃度を IC 値として算出した。結果を
50
表 73に示す。表 73に示した実施例化合物は、強力な DPP-IV阻害活性を示した。
[表 73]
表 73

血糖上昇抑制作用
16時間以上絶食した ICR系雄性マウス(6〜8週齢、チヤ一ルスリバ一)の血糖値を 測定した後、実施例化合物を蒸留水に溶解し、 10又は 30mgZkg (実施例化合物 8 は、 1、 3及び lOmgZkg)の用量で経口投与し、投与 30分後にグルコース(2gZkg )を経口投与した。グルコース負荷直前、グルコース負荷後 20分、 40分、 1時間、 2 時間の血液の血糖値を簡易型血糖測定装置 (メディセンスプレシジョン Q · I · D、アポ ットジャパン)にて測定した。結果を図 1〜7に示す。実施例化合物 8、 60、 73、 75、 1 21、 122及び 125は、経口投与において血糖低下作用を示した。
[0705] 比較例
血糖上昇抑制作用
実施例 146と同様の手法により、比較化合物 1 (特許文献 3の請求範囲に含まれる 化合物)及び比較化合物 2 (特許文献 4の請求範囲に含まれる化合物)の血糖上昇 抑制作用を評価した。結果を図 8に示す。比較ィヒ合物 1及び 2は、経口投与におい て血糖低下作用を示さな力つた。
[0706] 実施例 147
病態モデルに対する作用
C57BL/6N系雄性マウス (4週齢、チャールズリバ一)を 1週間通常飼料給餌で飼育 後 60%fatを含む高脂肪飼料(固形飼料、リサーチダイエット社)に切り替え、 18週間飼 育した肥満糖尿病マウスを用いて血糖上昇抑制作用を実施例 146と同様に測定し た。 16時間以上絶食したこのマウスの血糖値を測定した後、実施例化合物 60を蒸 留水に溶解し、 lOmgZkgの用量で経口投与し、投与 30分後にグルコース(2gZk g)を経口投与した。グルコース負荷直前、グルコース負荷後 20分、 40分、 1時間、 2 時間の血液の血糖値を簡易型血糖測定装置 (メディセンスプレシジョン Q · I · D、アポ ットジャパン)にて測定した。
[0707] 結果を図 9に示す。実施例化合物 60は、肥満糖尿病マウスにおいても血糖値上昇 抑制作用を示した。
[0708] 本明細書中で引用した全ての刊行物、特許及び特許出願をそのまま参考として本 明細書中にとり入れるものとする。
Claims
請求の範囲
[1] 一般式 (I)
[化 1]

(I)
[式中、 Arは、任意の 1〜5個の R3で置換されていてもよいフエ-ルを表し、
R3は、ハロゲン、ヒドロキシ、シァ入炭素数 1〜6のアルキル又は炭素数 1〜6のアル キルォキシを表し(ここで、アルキル又はアルキルォキシは 1〜5個のハロゲンで置換 されていてもよい)、
R1は、水素又は炭素数 1〜6のアルキルを表し(ここでアルキルは、任意の 1〜5個の ハロゲン又はヒドロキシで置換されて 、てもよ 、)、
Yは、 NR4—、 一 0—、 一 S(=0)p 又は。一フエ-レンを表し、
R4は、水素、炭素数 1〜6のアルキル、ァリール、ヘテロァリール、ァリールで置換され た炭素数 1〜3のアルキル、ヘテロァリールで置換された炭素数 1〜3のアルキル、 C(=0)— R5、 一 CH— C(=0)— R5又は一 SO— R5を表し、
2 2
R5は、炭素数 1〜6のアルキル、ァリール又はへテロアリールを表し、
pは、 0、 1又は 2を表し、
m及び nは、各々独立して、 1、 2又は 3を表し、
R2は、 R6又は R7力も選択される任意の 1〜3個の置換基で置換されていてもよいァリ ール又はへテロアリールを表し、
R6は、炭素数 1〜6のアルキル、炭素数 1〜6のアルキルォキシ(ここで、アルキル又 はアルキルォキシは 1〜5個のハロゲンで置換されていてもよい)、シクロアルキル、 ァリールォキシ、ヘテロァリールォキシ、ヒドロキシ、ハロゲン、シァ入 0— (CH )q
2
— R8、 —SO— R5又は— NR 9'を表す力、 2個の隣接する R6が一緒になつてメチレン
2
ジォキシ又は炭素数 3又は 4のアルキレンを表し、
R8は、炭素数 1〜3のアルキルォキシ(ここで、アルキルォキシは 1個の炭素数 1〜3
のアルキルォキシ又は 1〜5個のハロゲンで置換されていてもよい)、ァリールォキシ 、ヘテロァリールォキシ、シァ入シクロアルキル(ここで、シクロアルキルの 1個のメチ レンは、— 0—、— NR9 又は— S(=0)p のいずれかで置換されていてもよい)、 N — (C=0)— R5、— (C=0)— NR9R9'又は— S(=0)p— R5を表し、 R9及び R9'は、各々独立して、水素、炭素数 1〜6のアルキル(ここで、アルキルは 1個 の炭素数 1〜3のアルキルォキシ又は 1〜5個のハロゲンで置換されていてもよい)を 表すか、又は R9、 R9'及び窒素が一緒になつてピロリジン、ピぺリジン (ここで、ピロリジ ン又はピペリジンは 1〜5個のハロゲンで置換されていてもよい)、ピぺラジン、 N—ァ ルキルピペラジン(ここでアルキルの炭素数は 1〜6である)、モルホリン、チオモルホ リン又はチオモルホリン 4, 4 ジォキシドを表し、
R1Q及び R1Q'は、各々独立して、水素、炭素数 1〜6のアルキル(1個の炭素数 1〜3の アルキルォキシ又は 1〜5個のハロゲンで置換されていてもよい)を表すか、又は R1Q、 R1Q'、窒素が一緒になつてピロリジン、ピぺリジン(ここで、ピロリジン又はピぺリジンは 1 〜5個のハロゲンで置換されていてもよい)、ピぺラジン、 N—アルキルピぺラジン(こ こでアルキルの炭素数は 1〜6である)、モルホリン、チオモルホリン又はチオモルホリ ンー 4, 4 ジォキシドを表し(ここで、ピロリジン、ピぺリジン、ピぺラジン、 N—アルキ ルビペラジン、モルホリン、チオモルホリン又はチオモルホリン 4, 4 ジォキシドの 1個のメチレンは、ォキソで置換されていてもよい)、
qは、 0、 1、 2又は 3を表し、
R7は、ァリール又はへテロアリールを表し(ここで、ァリール又はへテロアリールは、任 意に選択される 1〜3個の R11で置換されていてもよい)、
R11は、炭素数 1〜3のアルキル、ハロゲン、炭素数 1〜6のアルキルォキシ、トリフルォ ロメチル、 -SO— R5又は一 NR9R9'を表し、
2
Xは、原子価結合又は炭素数 1〜6のアルキレンを表す。 ]
で示される非環状アミンカルボキシアミド誘導体又はその薬学的に許容される塩。
[2] Xが原子価結合である請求項 1記載の非環状アミンカルボキシアミド誘導体又はそ の薬学的に許容される塩。
[3] R3がハロゲンである請求項 1又は 2記載の非環状アミンカルボキシアミド誘導体又は
その薬学的に許容される塩。
[4] R1が水素である請求項 1から 3の!、ずれ力 1項に記載の非環状アミンカルボキシアミ ド誘導体又はその薬学的に許容される塩。
[5] Yが— 0—又は— S(=0)p— (ここで、 pは前記定義に同じ)である請求項 1から 4のい ずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又はその薬学的に許容さ れる塩。
[6] m及び nが共に 2である請求項 1から 5の!、ずれ力 1項に記載の非環状アミンカルボ キシアミド誘導体又はその薬学的に許容される塩。
[7] R2が、 R6又は R7力も選択される任意の 1〜3個の置換基で置換されていてもよいァリ ール (ここで、 R6及び R7は前記定義に同じ)である請求項 1から 6のいずれか 1項に記 載の非環状アミンカルボキシアミド誘導体又はその薬学的に許容される塩。
[8] R2が、 R6又は R7力も選択される任意の 1〜3個の置換基で置換されていてもよいフエ
-ル (ここで、 R6及び R7は前記定義に同じ)である請求項 7記載の非環状アミンカルボ キシアミド誘導体又はその薬学的に許容される塩。
[9] R2が、 R6又は R7力 選択される任意の 1〜3個の置換基で置換されていてもよいへ テロアリール (ここで、 R6及び R7は前記定義に同じ)である請求項 1から 6のいずれか 1 項に記載の非環状アミンカルボキシアミド誘導体又はその薬学的に許容される塩。
[10] R2が、 R6又は R7力も選択される任意の 1〜3個の置換基で置換されていてもよい、下 記の構造であるへテロアリール(ここで、 R12は、水素、炭素数 1〜6のアルキル又は— (CH R7及び R8は前記定義に同じ)である請求項 9記載の非環状

ァミンカルボキシアミド誘導体又はその薬学的に許容される塩。
[化 2]
 請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は その薬学的に許容される塩を有効成分として含有する医薬組成物。
請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は その薬学的に許容される塩を有効成分として含有する医薬組成物。
請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は
その薬学的に許容される塩を有効成分として含有する糖尿病の治療又は予防剤。
[13] 請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は その薬学的に許容される塩の糖尿病の治療又は予防剤の製造のための使用。
[14] 請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は その薬学的に許容される塩の有効量を患者に投与することを含む糖尿病の治療又 は予防方法。
[15] 請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は その薬学的に許容される塩を有効成分として含有する血糖低下剤。
[16] 請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は その薬学的に許容される塩の血糖低下剤の製造のための使用。
[17] 請求項 1から 10のいずれ力 1項に記載の非環状アミンカルボキシアミド誘導体又は その薬学的に許容される塩の有効量を患者に投与することを含む血糖低下方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005345748 | 2005-11-30 | ||
| JP2005-345748 | 2005-11-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007063928A1 true WO2007063928A1 (ja) | 2007-06-07 |
Family
ID=38092262
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/323889 Ceased WO2007063928A1 (ja) | 2005-11-30 | 2006-11-30 | 新規な非環状アミンカルボキシアミド誘導体及びその塩 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007063928A1 (ja) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2010114291A2 (ko) | 2009-03-30 | 2010-10-07 | 동아제약 주식회사 | 디펩티딜 펩티다아제-ⅳ 저해제 및 중간체의 개량된 제조방법 |
| WO2010114292A2 (ko) | 2009-03-30 | 2010-10-07 | 동아제약 주식회사 | 디펩티딜 펩티다아제-ⅳ 저해제 및 중간체의 개량된 제조방법 |
| US20110059970A1 (en) * | 2007-09-13 | 2011-03-10 | Ipsen Pharma S.A.S. | 4-phenyl-1,3-thiazoles and 4-phenyl-1,3-oxazoles derivatives as cannabinoid receptor ligands |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012035549A2 (en) | 2010-09-13 | 2012-03-22 | Panacea Biotec Ltd | An improved process for the synthesis of beta amino acid derivatives |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| JP2015044845A (ja) * | 2008-01-17 | 2015-03-12 | 田辺三菱製薬株式会社 | Sglt阻害剤及びdpp4阻害剤からなる併用療法 |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004168772A (ja) * | 2002-11-06 | 2004-06-17 | Takeda Chem Ind Ltd | 受容体調節剤 |
| JP2004535445A (ja) * | 2001-06-27 | 2004-11-25 | スミスクライン ビーチャム コーポレーション | ジペプチジルペプチダーゼ阻害剤としてのフルオロピロリジン類 |
| JP2004535433A (ja) * | 2001-06-20 | 2004-11-25 | メルク エンド カムパニー インコーポレーテッド | 糖尿病治療用のジペプチジルペプチダーゼ阻害薬 |
| WO2005040095A1 (en) * | 2003-10-16 | 2005-05-06 | Astrazeneca Ab | Inhibitors of dipeptidyl peptidase iv |
| WO2005095343A1 (en) * | 2004-03-05 | 2005-10-13 | Santhera Pharmaceuticals (Schweiz) Gmbh | Dpp-iv inhibitors |
-
2006
- 2006-11-30 WO PCT/JP2006/323889 patent/WO2007063928A1/ja not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004535433A (ja) * | 2001-06-20 | 2004-11-25 | メルク エンド カムパニー インコーポレーテッド | 糖尿病治療用のジペプチジルペプチダーゼ阻害薬 |
| JP2004535445A (ja) * | 2001-06-27 | 2004-11-25 | スミスクライン ビーチャム コーポレーション | ジペプチジルペプチダーゼ阻害剤としてのフルオロピロリジン類 |
| JP2004168772A (ja) * | 2002-11-06 | 2004-06-17 | Takeda Chem Ind Ltd | 受容体調節剤 |
| WO2005040095A1 (en) * | 2003-10-16 | 2005-05-06 | Astrazeneca Ab | Inhibitors of dipeptidyl peptidase iv |
| WO2005095343A1 (en) * | 2004-03-05 | 2005-10-13 | Santhera Pharmaceuticals (Schweiz) Gmbh | Dpp-iv inhibitors |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US20110059970A1 (en) * | 2007-09-13 | 2011-03-10 | Ipsen Pharma S.A.S. | 4-phenyl-1,3-thiazoles and 4-phenyl-1,3-oxazoles derivatives as cannabinoid receptor ligands |
| JP2015044845A (ja) * | 2008-01-17 | 2015-03-12 | 田辺三菱製薬株式会社 | Sglt阻害剤及びdpp4阻害剤からなる併用療法 |
| EP2669266A1 (en) | 2009-03-30 | 2013-12-04 | Dong-A Pharmaceutical Co., Ltd. | Improved Method for Manufacturing Dipeptidyl Peptidase-IV Inhibitor and Intermediate |
| WO2010114291A2 (ko) | 2009-03-30 | 2010-10-07 | 동아제약 주식회사 | 디펩티딜 펩티다아제-ⅳ 저해제 및 중간체의 개량된 제조방법 |
| WO2010114292A2 (ko) | 2009-03-30 | 2010-10-07 | 동아제약 주식회사 | 디펩티딜 펩티다아제-ⅳ 저해제 및 중간체의 개량된 제조방법 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012035549A2 (en) | 2010-09-13 | 2012-03-22 | Panacea Biotec Ltd | An improved process for the synthesis of beta amino acid derivatives |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007063928A1 (ja) | 新規な非環状アミンカルボキシアミド誘導体及びその塩 | |
| EP2906553B1 (en) | Orexin receptor antagonists which are [ortho bi (hetero )aryl]-[2-(meta bi (hetero)aryl)-pyrrolidin-1-yl]-methanone derivatives | |
| US7943645B2 (en) | Piperidine compounds for use as orexin receptor antagonist | |
| AU2014270152B9 (en) | Heterocyclic derivates | |
| CN111757875A (zh) | 作为心肌节抑制剂的二氢苯并呋喃和茚类似物 | |
| TW200825071A (en) | Indole compound | |
| CN102239152B (zh) | 新型酰胺衍生物及其作为药物的用途 | |
| JPH10511687A (ja) | グリコーゲンホスホリラーゼ抑制剤としての置換されたn−(インドール−2−カルボニル)−グリシンアミド類および誘導体 | |
| CN108200760A (zh) | 作为rsv抑制剂的苯并二氮杂*衍生物 | |
| CN109641843A (zh) | 作为apj激动剂的4-羟基-3-磺酰基吡啶-2(1h)-酮 | |
| CN120957993A (zh) | 杂环glp-1激动剂 | |
| CN109195963A (zh) | 作为apj激动剂的6-羟基-4-氧代-1,4-二氢嘧啶-5-甲酰胺 | |
| EP2220081A2 (en) | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient | |
| CN111434655A (zh) | 溶血磷脂酸受体拮抗剂及其制备方法 | |
| WO2002062792A9 (fr) | Inhibiteur de jnk | |
| IL207349A (en) | Pyridazinone glucokinase plants, their preparation and their use in the preparation of diabetic drugs | |
| KR20060021890A (ko) | 키나제 저해제로서의 치환된 인다졸릴(인돌릴)말레이미드유도체 | |
| CN103339111A (zh) | 双环乙酰基-CoA羧化酶抑制剂 | |
| CN112384500A (zh) | 免疫调节剂及其组合物和制备方法 | |
| JP7600228B2 (ja) | キナーゼ阻害剤としてのインダゾールカルボキサミド | |
| CN114728170A (zh) | 对核受体具有活性的化合物 | |
| WO2012041860A1 (en) | Benzazole derivatives as histamine h4 receptor ligands | |
| CN101679404A (zh) | 哌啶-酰胺衍生物 | |
| CN111630058A (zh) | 吡喃葡萄糖基衍生物及其用途 | |
| MX2012010668A (es) | 3-oxo-3,9-dihidro-1h-cromeno[2, 3-c]pirrolos como activadores de glucocinasa. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06833692 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |