WO2007099896A1 - 核酸保護基の脱離方法 - Google Patents
核酸保護基の脱離方法 Download PDFInfo
- Publication number
- WO2007099896A1 WO2007099896A1 PCT/JP2007/053490 JP2007053490W WO2007099896A1 WO 2007099896 A1 WO2007099896 A1 WO 2007099896A1 JP 2007053490 W JP2007053490 W JP 2007053490W WO 2007099896 A1 WO2007099896 A1 WO 2007099896A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- acid derivative
- chemical
- following general
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to an ether-type protecting group that protects the 2′-position hydroxyl group of each ribose of an oligonucleic acid derivative and is removable under neutral conditions, such as 2-cyanethoxymethyl (hereinafter referred to as “2-canoethoxymethyl”).
- CEM group is related to a method for detaching efficiently and reproducibly.
- Oligoribonucleic acid is well known to be useful as an RNA probe for gene analysis, RNA drug material (antisense RNA, ribozyme, gene expression control using RNAi), artificial enzyme, and aptamer. is there.
- Non-patent Document 1 a phosphoramidite complex compound in which the 2′-position hydroxyl group of ribose is protected by substitution with a removable CEM group under neutral conditions is known.
- the solid support and protecting groups for each substituent are prepared from the oligo RNA. Need to be removed.
- One of the steps to remove the strong protecting group is to remove the ether-type protecting group that can be removed under neutral conditions, protecting the 2 'hydroxyl group of each ribose of the oligo RNA.
- TBAF tetrabutyl ammonium fluoride
- THF tetrahydrofuran
- Non-Patent Document 1 Oki et al., ORGANIC LETTERS, Vol. 7, 3477 (2005)
- the purpose of the present invention is mainly to remove the ether-type protecting group that protects the 2'-position hydroxyl group of each ribose of an oligonucleic acid derivative and can be removed under neutral conditions, with good reproducibility and efficiency. It is to provide a method of separating. Means for solving the problem
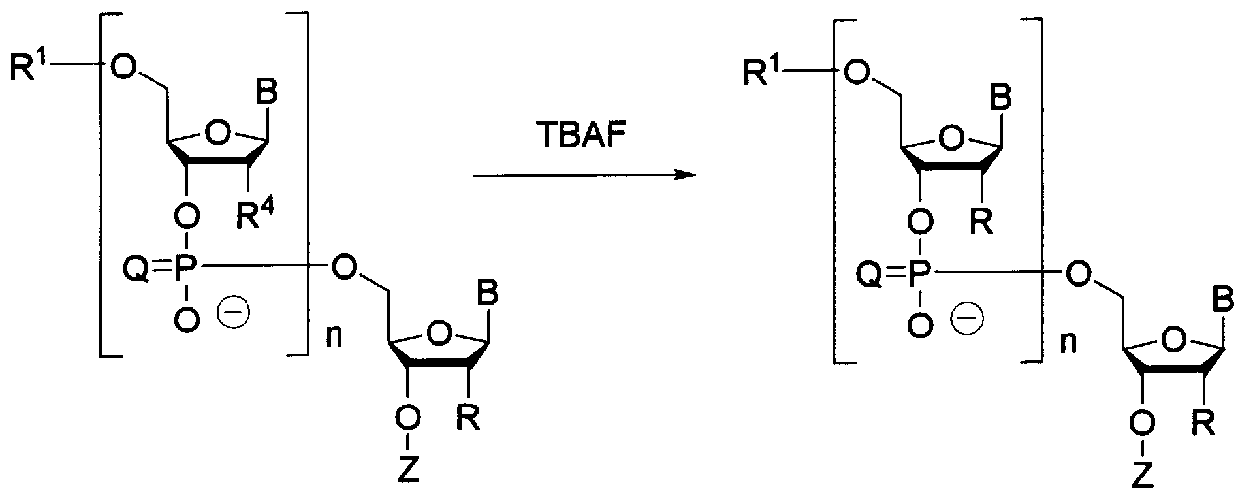
- the present inventor acts TBAF on the oligonucleic acid derivative represented by the following general formula (10) to protect the 2′-position hydroxyl group of each ribose,
- a sulfoxide solvent, an amide solvent, or a mixed solvent thereof, which may contain THF is reacted. It has been found that by using it as a solvent, an oligonucleic acid derivative represented by the following general formula (11) can be produced efficiently, and the present invention has been completed.
- each B independently represents a nucleobase or a modified form thereof.
- n represents an integer in the range of 1 to 200.
- n is preferably an integer within the range of 10 to 100, and more preferably an integer within the range of 15 to 50.
- Each Q independently represents O or S.
- Each R is independently H, hydroxyl group, halogen, alkoxy, alkylthio, alkylamino, dialkylamino, alkenyloxy, alkenylthio, alkenylamino, dialkenylamidoalkynoxy, alkynylthio, alkyl. Represents -ramino, dialuylamino or alkoxyalkyloxy, at least one of which represents a hydroxyl group.
- Z represents H, a phosphate group or a thiophosphate group.
- R 1 represents a substituent represented by the following general formula (3).
- R ′ ⁇ R ′′ are the same or different and each represents hydrogen or alkoxy.
- Each R 4 independently represents H, halogen, alkoxy, alkylthio, alkylamine-containing dialkylamino, alkenyloxy, An alkenylthio, an alkenylamino, a dialkenylamino, an alkynyloxy, an alkynylthio, an alkynylamino, a dialkynylamino-substituted alkoxyalkyloxy, or a substituent represented by the following general formula (4).
- WG 1 represents an electron-withdrawing group.
- the nucleobase represented by B is not particularly limited, and examples thereof include pyrimidine bases such as cytosine, uracil and thymine, and purine bases such as adenine and guanine.
- the “modified product” of B is a group in which a nucleobase is substituted with an arbitrary substituent.
- substituents include halogen, acyl, alkyl, arylalkyl, alkoxy, alkoxyalkyl, hydroxy , Monoalkyl-containing dialkylamino, carboxy, and nitro-containing nitro, and 13 of them are substituted at an arbitrary position.
- halogen related to the modified form of B include fluorine, chlorine, bromine and iodine.
- alkyl related to the modified form of B include linear or branched alkyl having 1 to 5 carbon atoms. Specific examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec butyl, tert-butyl, n pentyl, isopentyl, neopentyl, tert pentyl and the like.
- substituent that may be substituted with the alkyl include halogen, alkyl, alkoxy, and nitro-containing nitro, and 1 to 3 of these are substituted at an arbitrary position. Also good.
- alkyl part of “arylalkyl”, “alkoxyalkyl”, “monoalkylamino” and “dialkylamino” in the modified form of B can be the same as the above “alkyl”.
- alkoxy related to the modified form of B include linear or branched alkoxy having 1 to 4 carbon atoms. Specific examples include methoxy, ethoxy, n-propoxy, isopropoxy, n butoxy, isobutoxy, sec butoxy, tert-butoxy and the like. Of these, those having 1 to 3 carbon atoms are preferred, and methoxy is particularly preferred.
- alkoxy part of the “alkoxyalkyl” related to the modified form of B can be the same as the above “alkoxy”.
- aryl of “aryl alkyl” related to the modified form of B include aryl having 6 to 12 carbon atoms. Specifically, for example, phenyl, 1-naphthyl, 2-naphthyl, biphenyl and the like can be mentioned. Examples of the substituent which may be substituted with the aryl include halogen, alkyl, alkoxy, and nitro-containing nitro, and 1 to 3 of these may be substituted at an arbitrary position. .
- alkylogen examples of the “alkylogen”, “aryl”, “alkylene”, “alkyl” and “alkoxy” as substituents of “alkyl” in the modified form of B can be the same as those described above.
- Norogen”, “alkoxy”, “alkylamino” or “dialkylamino” relating to R and R 4 may be the same as those relating to the modified form of B.
- Alkoxyalkyloxy according to R and R 4 , “alkyl” part of “alkylthio” and Examples thereof include the same “alkyl” according to the modified form of B.
- Examples of the “alkoxy” part of the “alkoxyalkyloxy” according to R and R 4 include the same “alkoxy” as the modified B.
- alkyl part of “alkyloxy”, “alkylthio”, “alkylamino” and “dialkylamino” according to R and R 4 include, for example, linear or branched And an alkyl having 2 to 4 carbon atoms. Specifically, for example, ethur, 2-probule, 1-butul and the like can be mentioned.
- alkoxy examples include the same “alkoxy” as B.
- Examples of the “electron-withdrawing group” according to WG 1 include silane-containing nitro, alkylsulfol, arylsulfol, and halogen. Of these, Ciano is preferred.
- Examples of the “alkyl” part of the “alkylsulfol” according to WG 1 include the same “alkyl” as in B above.
- Examples of the “aryl” part of “aryl reel” according to WG 1 include the same as the “aryl” in B.
- sulfoxide solvent examples include compounds represented by the following general formula (I). Specific examples include dimethyl sulfoxide (hereinafter referred to as “DMSO”), ethyl methyl sulfoxide, and the like. Of these, DMSO is suitable.
- DMSO dimethyl sulfoxide
- amide solvent examples include compounds represented by the following general formula ( ⁇ ). Specifically, N, N-dimethylformamide (hereinafter referred to as “DMF”), N, N-jetylformamide, N, N-dimethylacetamide, N, N-jetylacetamide, N— And methylpyrrolidone. Of these, DMF is appropriate.
- R a and R b are the same or different and represent alkyl.
- R e and R d are the same or different and each represents an alkyl;
- R e represents a force representing hydrogen or alkyl; or
- R d represents an alkyl, and
- R e and R e are adjacent nitrogen and carbon atoms. It represents a 5- or 6-membered saturated cyclic amide group formed together with.
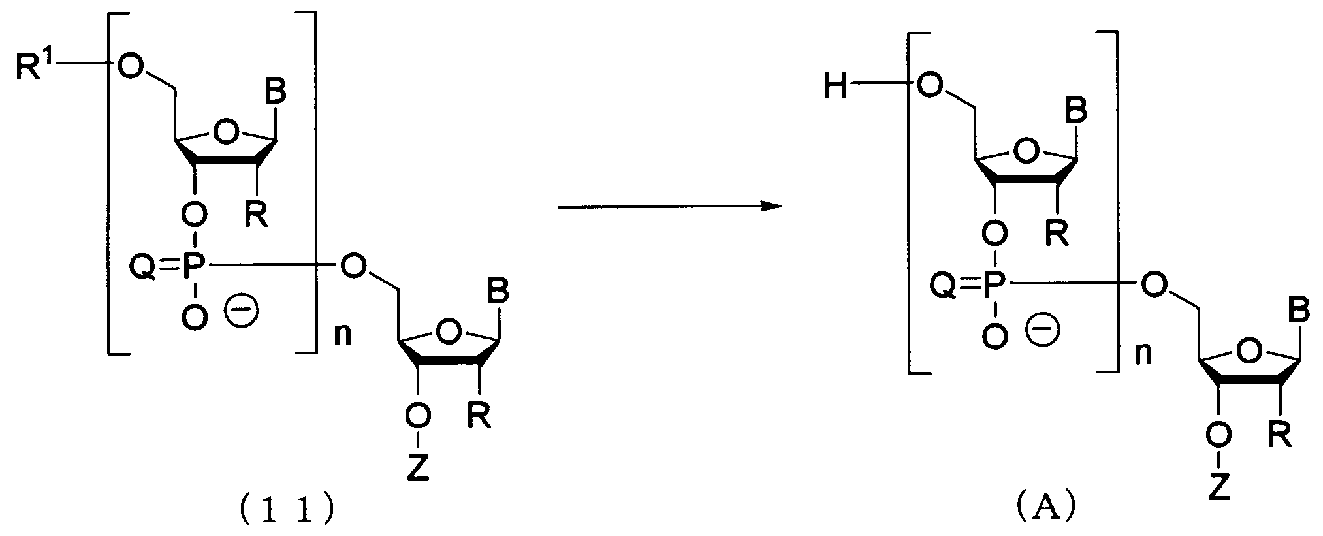
- the oligonucleic acid derivative represented by the following general formula (10) is used for TBAF to protect the 2′-hydroxyl group of each ribose and desorbed under neutral conditions.
- a sulfoxide-based solvent or an amide-based solvent, which may contain THF, or a mixed solvent thereof is used as a reaction solvent.
- a method for producing an oligo RNA represented by the following general formula (A) (hereinafter referred to as “oligo RNA (A)”) comprising a step of producing an oligonucleic acid derivative represented by the general formula (11): Can be mentioned.
- each B, each Q, each R, and each R 4 are independently the same as defined above.
- n, Z is as defined above.
- each B, each Q, and each R are independently the same as defined above.
- n and Z are as defined above.
- phosphoramidite toy compound (B) As a ribonucleic acid derivative used for the production of the above-mentioned oligo RNA (A), a phosphoramidite toy compound represented by the following general formula (B) (hereinafter referred to as “phosphoramidite toy compound (B)”) , U.).
- Bz represents a nucleobase which may have a protecting group or a modified form thereof.
- WG 1 has the same meaning as described above.
- WG 2 represents an electron withdrawing group.
- R 2a and R 2b are the same or different and represent a force representing alkyl or a 5- to 6-membered saturated amino ring group formed by R 2a and R 2b together with an adjacent nitrogen atom.
- the powerful saturated amino ring group may have one oxygen atom or sulfur atom as a ring constituent atom in addition to the nitrogen atom.
- nucleobase relating to Bz is not particularly limited as long as it is used for nucleic acid synthesis, and examples thereof include pyrimidine bases such as cytosine and uracil, and purine bases such as adenine and guanine. it can.
- nucleobase is preferably protected from a nucleic acid base having an amino group, for example, adenine, guanine, and cytosine, wherein the amino group is protected.
- the “protecting group for amino group” is not particularly limited as long as it is used as a protecting group for nucleic acids.
- benzoyl, 4-methoxybenzoyl, acetyl, propionyl examples include butyryl, isobutyryl, phenylacetyl, phenoxyacetyl, 4 t tert butyl phenoxy acetyl, 4 isopropyl phenoxy acetyl, (dimethylamino) methylene, and the like.
- the “modified product” of Bz is a group in which the nucleobase is substituted with an arbitrary substituent.
- substituents related to the “modified product” of Bz include halogen, acyl, alkyl, arylalkyl, Examples thereof include alkoxy, alkoxyalkyl, hydroxy, amino-containing monoalkylamino-containing dialkylamino, carboxy, and cyanated nitro, and 1 to 3 of them are substituted at any position.
- Examples of the “alkyl” relating to R 2a and R 2b include the same “alkyl” relating to the modified form of B.
- Examples of the “5- to 6-membered saturated amino ring group” related to R 2a and R 2b include pyrrolidine-1-yl, piperidine 1-yl, morpholine 1-yl, and thiomorpholine 1-yl. be able to.
- Examples of the “electron withdrawing group” according to WG 2 include the same as the electron withdrawing group of WG 1 .
- the phosphoramidite compound (B) is a phosphoramidite compound having an ether-type protecting group that can be removed under a neutral condition at the 2′-position hydroxyl group.
- the group introduced into the hydroxyl group at the 2 ′ position is a linear substituent, and the three-dimensional structure around the phosphorus atom bonded to the hydroxyl group at the 3 ′ position is crowded.
- the condensation reaction proceeds in a very short time, and the condensation yield has a characteristic that it is slightly different.
- oligo DNA means an oligonucleic acid that only has deoxyribonucleic acid (DNA)! Uh.
- oligo RNA refers to oligonucleic acid such as ribonucleic acid (RNA) and deoxyribonucleic acid (DNA), and at least one refers to an oligonucleic acid containing ribonucleic acid (RNA).
- the raw material when the raw material has a substituent that affects the reaction (for example, hydroxy, amide-containing carboxy), the raw material is intensively protected with an appropriate protecting group according to a known method. The reaction is performed later. The protecting group can be finally removed according to a known method such as catalytic reduction, alkali treatment, acid treatment and the like.
- a substituent that affects the reaction for example, hydroxy, amide-containing carboxy
- the phosphoramidite toy compound (B) can be produced as follows.
- the phosphoramidaitoy compound (B) can be produced from a known compound or an intermediate that can be easily produced, for example, by performing the operations of the following steps a to h.
- An ether-type protecting group that is eliminated under neutral conditions is introduced into the 2′-position hydroxyl group by allowing an alkylating reagent to act on the ribonucleic acid derivative represented by the following general formula (12).
- alkylating reagent for example, an ether compound represented by the following general formula (14) can be mentioned.
- L represents a halogen, an arylthio group, an alkyl sulfoxide group or an alkylthio group.
- WG 1 has the same meaning as described above.
- ether compound (14) examples include the following compounds 1 and 2.
- the ether compound (14) is a novel alkyl reagent that can introduce an ether-type substituent that can be eliminated under neutral conditions into the 2′-position hydroxyl group under basic conditions. It is useful as a reagent for producing compound (B).
- the ether compound (14) can be produced by carrying out the following Step 1 to Step 4.
- R 3 represents alkyl or aryl.
- Compound (16) is an ethereal compound (14) in which L is an alkylthio group.
- Examples of the “alkyl” related to R 3 include the same “alkyl” related to the modified form of B.
- examples of the alkylthiomethylating reagent include a mixed solution of dimethyl sulfoxide, acetic anhydride and acetic acid.
- the amount of “dimethyl sulfoxide” used is suitably 10 to 200-fold mol amount, preferably 20 to 100-fold mol amount based on the mol amount of Compound (15).
- the amount of “acetic acid” used is suitably 10 to 150 times the molar amount, preferably 20 to 100 times the molar amount of the compound (15).
- the amount of “hydrous acetic acid” used is appropriately 10 to 150 times the molar amount of the compound (15), preferably 20 to L00 times the molar amount.
- the reaction temperature is suitably from 0 ° C to 100 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 1 to 48 hours is appropriate.
- R 3 has the same meaning as described above.
- X 2 represents halogen.
- Compound (17) is a compound in which L in the etheric compound (14) is halogen.
- Examples of the “norogen” according to X 2 include the same “norogen” according to the modified form of B.
- This step can be performed by a known method (for example, T. Benneche et al., Synthesis 762 (1983)).
- the solvent to be used is not particularly limited as long as it does not participate in the reaction.
- 1S Examples include halogenated hydrocarbons such as dichloromethane, chlorophenol, carbon tetrachloride, and 1,2-dichloroethane.
- Examples of the halogenating reagent include salt, sulfuryl and phosphorus oxychloride.
- the amount of the “halogen reagent” used is suitably 0.8 to 20 times the molar amount, preferably 1 to 10 times the molar amount of the compound (16).
- the reaction temperature is suitably from 0 ° C to 100 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- Process 3 A process in which compound (17) is alliated to produce a compound represented by the following general formula (18).
- X 2 has the same meaning as described above.
- R 3a represents a reel.
- Compound (18) is a compound in which L in the etheric compound (14) is an arylthio group.
- Examples of the “aryl” related to R 3a include the same “aryl” related to the modified form of B.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane and acetonitrile.
- the arylthiolation reagent for example, thienol and 4-methylbenzene thiol can be mentioned.
- the amount of the “arylthioy reagent” used is suitably 0.8 to 20 times the molar amount, preferably 1 to 5 times the molar amount of the compound (17).
- the reaction temperature is 0 A temperature of ° C to 100 ° C is appropriate.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc., but 1 to 48 hours is usually appropriate.
- R 3 has the same meaning as described above.
- Compound (19) is a compound in which L in the ether compound (14) is an alkyl sulfoxide group.
- Examples of the “alkyl” related to R 3 include the same “alkyl” related to the modified form of B.
- This step can be performed by a known method.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, chloroform, and methanol.
- Examples of the oxidizing agent include methacroperoxybenzoic acid, metaperiodic acid salt, and hydrogen peroxide.
- the amount of the “oxidant” to be used is appropriately 0.8 to 10 times, preferably 1 to 2 times the amount of the compound (16).
- the reaction temperature is suitably from 0 ° C to 100 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 1 to 48 hours is appropriate.
- This step can be carried out according to a known method by allowing an alkylating reagent and a base to act on a ribonucleic acid derivative (12) which is available as a commercial product or can be synthesized according to a method described in the literature.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include halogenated hydrocarbons such as dichloromethane, chlorophenol, carbon tetrachloride, 1,2-dichloroethane.
- the amount of the “alkylating reagent” to be used is suitably 0.8 to 20 times the molar amount, preferably 1 to: LO times the molar amount of the ribonucleic acid derivative (12).
- an alkylating reagent can be allowed to act on the ribonucleic acid derivative (12) after passing through an intermediate produced by reacting a metal reagent and a base.
- a metal reagent is disodium dibutyltin.
- the amount of the “metal reagent” used is suitably 0.8 to 20 times the molar amount, preferably 1 to 10 times the molar amount of the ribonucleic acid derivative (12).
- base examples include pyridine, 2,6-dimethylpyridine, 2,4,6-trimethylpyridine, N-methylimidazole, triethylamine, tributylamine, N, N-diisopropylethylamine, 1, 8 —Diazabicyclo [5. 4. 0] — 7—Undecene and other organic bases.
- the amount of the “base” used is suitably 0.8 to 20 times, preferably 1 to 10 times the molar amount of the ribonucleic acid derivative (12).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc., but usually 30 minutes to 24 hours is appropriate.
- This step is carried out according to a known method (for example, M. Matteucci, Tetrahedron Letters, Vol. 31, 2385 (1990)), and can be obtained as a commercial product or synthesized according to the method described in the literature (12) It can be carried out by reacting an alkylating reagent, an acid and a halogenating agent for the sulfur atom.
- the amount of the “alkylating reagent” used is suitably 0.8 to 5 times the molar amount, preferably 1 to 3 times the molar amount of the ribonucleic acid derivative (12).
- the “acid” examples include trifluoromethanesulfonic acid, silver trifluoromethanesulfonate, and trimethylsilyl trifluoromethanesulfonate.
- the amount of the “acid” used is suitably 0.01 to 20 times the molar amount, preferably 0.02 to L0 times the molar amount of the ribonucleic acid derivative (12).
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, but for example, dichloromethane, chloroform, tetrachlorocarbon, 1,2-dichloroethane, benzene, toluene, xylene, THF, acetonitrile, or any of these A mixed solvent can be mentioned.
- halogenating agent for sulfur atom examples include N-bromosuccinimide (NBS) and N-iodosuccinimide (NIS).
- NBS N-bromosuccinimide
- NIS N-iodosuccinimide
- the amount of the “halogenating agent for sulfur atom” is suitably 0.8 to 10 times, preferably 1 to 5 times the amount of the ribonucleic acid derivative (12).
- the reaction temperature is suitably -78 ° C to 30 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 5 hours is appropriate.
- the ribonucleic acid derivative (12) which is available as a commercial product or can be synthesized according to a method described in the literature, is made to work with an alkyl reagent, an acid anhydride, and a base according to a known method. This can be implemented.
- the amount of the “alkyly reagent” used is suitably 0.8 to 5 times, preferably 1 to 3 times the amount of the ribonucleic acid derivative (12).
- the “acid anhydride” include trifluoromethanesulfonic acid anhydride and anhydrous acetic acid.
- the amount of the “anhydride” used is suitably 0.01 to 20 times the molar amount, preferably 0.02 to 10 times the molar amount of the ribonucleic acid derivative (12).
- the Examples of the base include tetramethylurea and collidine.
- the amount of the “base” used is suitably 0.01 to 20 times the molar amount of the ribonucleic acid derivative (12), preferably 0.02 to LO times the molar amount.
- the solvent used is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, and any mixed solvent thereof.
- the reaction temperature is
- reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 24 hours is appropriate.
- a step of isolating and purifying the ribonucleic acid derivative (13) produced in step a This step can be isolated and purified by using a usual separation and purification means such as thin layer chromatography, silica gel chromatography, etc. from the mixture produced in step a.
- an ether-type protecting group that is eliminated under neutral conditions can be removed at the 2′-position by allowing an alkyl reagent to act on the ribonucleic acid derivative represented by the following general formula (20).
- A represents a silicon substituent represented by the following general formula (22a) or (22b).
- R 6 represents alkyl.
- Examples of the “alkyl” related to R 6 include the same “alkyl” related to the modified form of B.
- alkylating reagent examples include the same as described above.
- This step can be performed by reacting an alkylating reagent and a base with a ribonucleic acid derivative (20) which is available as a commercial product or can be synthesized according to a method described in the literature, according to a known method.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include halogenated hydrocarbons such as dichloromethane, chlorophenol, carbon tetrachloride, 1,2-dichloroethane, and the like.
- the amount of the “alkylating reagent” used is suitably 0.8 to 20 times the molar amount, preferably 1 to LO times the molar amount of the ribonucleic acid derivative (20).
- an alkylating reagent can be allowed to act after an intermediate produced by reacting the ribonucleic acid derivative (20) with a metal reagent and a base.
- metal reagents examples include dibutyltin dichloride and tert-butylmagnesium chloride.
- the amount of the “metal reagent” to be used is suitably 0.8 to 20 times, preferably 1 to 10 times, the molar amount of the ribonucleic acid derivative (20).
- Base includes pyridine, 2,6-dimethylpyridine, 2,4,6-trimethylpyridine, N-methylimidazole, triethylamine, tributylamine, N, N-dipropylethylamine, 1 , 8-diazabicyclo [5. 4. 0]-7-undecene and other organic bases.
- the amount of the “base” used is suitably 0.8 to 20 times the molar amount, preferably 1 to 10 times the molar amount of the ribonucleic acid derivative (20).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- alkylating reagents and halo for acid and sulfur atoms It can be carried out by reacting with a genating agent.
- the amount of the “alkylating reagent” to be used is appropriately 0.8 to 5 times, preferably 1 to 3 times the amount of the ribonucleic acid derivative (20).
- the “acid” include trifluoromethanesulfonic acid, silver trifluoromethanesulfonate, and trimethylsilyl trifluoromethanesulfonate.
- the amount of the “acid” used is suitably from 0.01 to 20 times the molar amount of the ribonucleic acid derivative (20), preferably from 0.02 to L0 times the molar amount.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction. For example, dichloromethane, chloroform, tetrasalt-carbon, 1,2-dichloroethane, benzene, toluene, xylene, THF, acetonitrile, or any of these A mixed solvent can be mentioned.
- Examples of the “halogenating agent for sulfur atom” used in this step include N-bromosuccinimide (NBS) and N-iodosuccinimide (NIS).
- the amount of the “halogenating agent for sulfur atom” is suitably 0.8 to 10 times, preferably 1 to 5 times the molar amount of the ribonucleic acid derivative (20). .
- the reaction temperature is suitably -78 ° C to 30 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 5 hours is appropriate.
- a ribonucleic acid derivative (20) which is available as a commercial product or can be synthesized according to a method described in the literature, according to a known method, causes an alkyl reagent, an acid anhydride and a base to work.
- the amount of the “alkyly reagent” used is suitably 0.8 to 5 times, preferably 1 to 3 times the amount of the ribonucleic acid derivative (20).
- the “acid anhydride” include trifluoromethanesulfonic acid anhydride and anhydrous acetic acid.
- the amount of the “acid anhydride” used is suitably 0.01 to 20 times the molar amount, preferably 0.02 to 10 times the molar amount of the ribonucleic acid derivative (20).
- the base include tetramethylurea and collidine.
- the amount of the “base” used is suitably 0.01 to 20 times the molar amount of the ribonucleic acid derivative (20), preferably 0.02 to L0 times the molar amount.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction. For example, dichloromethane, chloroform, carbon tetrachloride, 1, 2 Dichloroethane or any mixed solvent thereof.
- the reaction temperature is
- reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 24 hours is appropriate.
- a step of producing a ribonucleic acid derivative represented by the following general formula (23) by reacting the ribonucleic acid derivative (20) with dimethyl sulfoxide, acetic acid and acetic anhydride.
- This step is performed according to a known method by allowing dimethyl sulfoxide, acetic acid and acetic anhydride to act on a ribonucleic acid derivative (20) which is available as a commercial product or can be synthesized according to a method described in the literature. be able to.
- the amount of “dimethyl sulfoxide” used is suitably 10 to 200 times, preferably 20 to 100 times the molar amount of the ribonucleic acid derivative (20).
- the amount of “acetic acid” used is suitably 10 to 150 times the molar amount, preferably 20 to L00 times the molar amount of the ribonucleic acid derivative (20).
- the amount of “acetic anhydride” used is suitably 10 to 150 times, preferably 20 to 100 times, the molar amount of the ribonucleic acid derivative (20).
- the reaction temperature is suitably 10 ° C to 50 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- step d the ribonucleic acid derivative (23) produced in the step d is allowed to react with an alcohol compound represented by the following general formula (24), an acid, and a halogenating agent for a sulfur atom to neutralize the ribonucleic acid derivative (23).
- Equation (21), (23) and (24), A ⁇ Bz, WG 1 have the same meanings as described above.
- This step can be performed by reacting the ribonucleic acid derivative (23) with an alcohol compound (24), an acid, and a halogenating agent for a sulfur atom according to a known method.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction. For example, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, benzene, toluene, xylene, THF, acetonitrile, or any mixture thereof. Mention may be made of solvents.
- the amount of “alcohol compound (24)” used is suitably 0.8 to 20 times the molar amount, preferably 1 to 10 times the molar amount of the ribonucleic acid derivative (23).
- Examples of the “acid” include trifluoromethanesulfonic acid, silver trifluoromethanesulfonate, and trimethylsilyl trifluoromethanesulfonate.
- Examples of the “halogenating agent for sulfur atom” include N-bromosuccinimide (NBS) and N-odosuccinimide (NIS).
- the amount of the “halogenating agent for sulfur atom” is suitably 0.1 to 20 times the molar amount of the ribonucleic acid derivative (23), preferably 0.2 to 10 times the molar amount. is there.
- the reaction temperature is suitably from -100 ° C to 20 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 12 hours is appropriate.
- the ribonucleic acid derivative (21) is dissolved in an organic solvent, and a fluorinating agent alone or a fluorinating agent and an acid (for example, acetic acid, hydrochloric acid, sulfuric acid) are reacted as a mixed reagent in an arbitrary mixing ratio.
- a fluorinating agent alone or a fluorinating agent and an acid for example, acetic acid, hydrochloric acid, sulfuric acid
- an acid for example, acetic acid, hydrochloric acid, sulfuric acid
- Examples of the “fluorinating agent” that can be used in this step include ammonium fluoride, TBAF, triethylamine trihydrofluoride, and hydrogen fluoride pyridine.
- the amount of the “fluorinating agent” to be used is suitably 0.1 to 20 times the molar amount, preferably 0.2 to 10 times the molar amount of the ribonucleic acid derivative (21).
- the reaction temperature is suitably from 0
- the mixing ratio of the fluorinating agent and the acid in the mixed reagent is suitably from 1: 2 to 1: 0.1 (fluorinating agent: acid), preferably from 1: 1.2 to 1: 1.
- Examples of the “norogen” relating to X 3 include the same “norogen” relating to the modified form of B.
- This step can be performed by reacting ribonucleic acid derivative (25) coconut R 3 according to a known method.
- the amount of R 3 used is suitably 0.8 to 20 times the molar amount, preferably 1 to 10 times the molar amount of the ribonucleic acid derivative (25).
- the solvent used is not particularly limited as long as it does not participate in the reaction, and examples thereof include acetonitrile and THF.
- Base includes pyridine, 2, 6-dimethyl pyridine, 2, 4, 6-trimethyl Organic bases such as pyridine, N-methylimidazole, triethylamine, tributylamine, N, N-diisopropylethylamine, 1,8-diazabicyclo [5.4.0] -7-undecene it can.
- the amount of the “base” to be used is suitably 0.8 to 20 times, preferably 1 to 10 times, the molar amount of the ribonucleic acid derivative (25).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- (8) Process h is pyridine, 2, 6-dimethyl pyridine, 2, 4, 6-trimethyl Organic bases such as pyridine, N-methylimidazole, triethylamine, tributylamine, N, N-diisopropy
- step b or step f By reacting the ribonucleic acid derivative (13) produced in step b or step f with a phosphoramidite reagent and, if necessary, an activator, the 3′-position hydroxyl group is phosphoramidite-phosphorylated.
- Examples of the “phosphoramidite candy reagent” include compounds represented by the following general formulas (26a) and (26b).
- R 2a , R 2b and WG 2 have the same meanings as described above.
- X 1 represents halogen
- Examples of the “norogen” relating to X 1 include the same “norogen” relating to the above-mentioned modified form of ⁇ .
- This step is a reaction in which a phosphoramidite reagent is allowed to act on the liponucleic acid derivative (13) to phosphorylate the 3′-position hydroxyl group, and can be performed according to a known method. An activator can also be used as needed.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include acetonitrile and THF.
- the amount of the “phosphoramidite candy reagent” used is suitably 0.8 to 20 times, preferably 1 to 10 times, the molar amount of the ribonucleic acid derivative (13).
- the “activator” include 1H-tetrazole, 5-ethylthiotetrazole, 5-benzilmer force, Pto 1H-tetrazole, 4,5-dichloroimidazole, 4,5-disyanoimidazole, and benzotriazole. Mention may be made of triflate, imidazole triflate, pyridinium triflate, N, N-diisopropylethylamine, 2,4,6-collidine ZN-methylimidazole.
- the amount of the “activator” to be used is suitably 0.8 to 20 times, preferably 1 to 10 times the molar amount of the ribonucleic acid derivative (13).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 30 minutes to 24 hours is appropriate.
- the produced phosphoramidaito compound (B) can be obtained by means known per se, for example, concentration, liquid conversion, phase transfer, solvent extraction, crystallization, recrystallization, fractional distillation, chromatography. It can be separated and purified by the first class.
- each B, each Q, and each R are independently the same as defined above.
- n and Z are as defined above.
- the oligo RNA (A) can be produced by a force that can be performed according to a known method.For example, by performing the following steps A to H, the nucleic acid module is gradually moved from 3 ′ to 5 ′. This can be done by condensing a nomer compound.
- those other than the phosphoramidite compound (B) are not particularly limited as long as they are generally used for the synthesis of oligo RNA or oligo DNA.
- all steps can be produced using a manual or a commercially available DNA automatic synthesizer. It is desirable to use an automatic synthesizer in order to simplify the operation method by using an automatic synthesizer and to ensure the accuracy of synthesis.
- those other than the nucleic acid monomer compound are not particularly limited as long as they are generally used for the synthesis of oligo DNA or oligo RNA.
- the phosphoramidite compound (B) is used at least once as a nucleic acid monomer compound, so that at least one of each scale is a hydroxyl group. (A) can be manufactured. Further, for example, in Step B described later, by using the phosphoramidite compound (B) as the nucleic acid monomer compound, an oligo RNA (A) in which each R is a hydroxyl group can be produced.
- n and R 1 are as defined above.
- Each Q, each R 4 and each WG 2 are independently the same as above.
- Each Bx independently represents a nucleobase which may have a protecting group or a modified form thereof.
- E represents isacyl or a substituent represented by the following general formula (5).
- E 1 represents a single bond or a substituent represented by the following general formula (6).
- Q and WG 2 are as defined above.
- T is H, acyloxy, halogen, alkoxy, alkylthio, alkylamino, dialkylamino, alkenyloxy, alkenylthio, alkenylamino, dialkenyl-containing alkynyloxy, alkynylthio, alkynylamino, dialkynylamino-containing alcohol
- Xyloxyloxy represents a substituent represented by the general formula (4) or a substituent represented by the general formula (5). However, either E or T represents the substituent (5).
- nucleobase relating to Bx is not particularly limited as long as it is used for nucleic acid synthesis, and examples thereof include pyrimidine bases such as cytosine, uracil and thymine, and purine bases such as adenine and guanine. .
- nucleobase relating to Bx is preferably a protected nucleic acid base having an amino group, such as adenine, guanine, and cytosine, wherein the amino group is preferably protected.
- the “protecting group” is not particularly limited as long as it is used as a protecting group for nucleic acids, and specifically includes, for example, benzoyl, 4-methoxybenzoyl, acetyl, propionyl, butyryl, isobutyryl, phenyl. Ruacetyl, phenoxyacetyl, 4 t ert butyl phenoxy acetyl, 4 isopropyl phenoxy acetyl, (dimethylamino) methylene.
- the “modified product” of Bx is a group in which the nucleobase is substituted with an arbitrary substituent.
- substituents related to the “modified product” of Bx include halogen, acyl, alkyl, arylalkyl, alkoxy. , Alkoxyalkyl, hydroxy, amino-containing monoalkylamino-containing dialkylamino, carboxy, and cyanated nitro, and 1 to 3 of them are substituted at any position.
- Examples of the “acyl” related to E include the same “acyl” related to the modified form of B.
- acyl part related to the “acyloxy” of the Ding include the same “acyl” related to the modified form of B.
- Examples of “norogen”, “alkoxy”, “alkylamino” and “dialkylamino” related to T include the same as those related to the modified form of B. May be the same as the “alkyl” related to the modified form of B.
- Examples of the “alkoxy” part of the “alkoxyalkyloxy” related to T include the same “alkoxy” related to the modified B.
- alkynyl part of “alkyloxy”, “alkylthio”, “alkylamino”, and “dialkylamino” related to T should be the same as the “alkyl” related to R above. It is out.
- the “alkylamino”, “alkenylamino”, and “alkynylamino” related to T are protected!
- the protective groups that can be used are not particularly limited as long as they are used as protective groups for the amino groups.
- trifluoroacetyl is preferred.
- B represents the substituent (5).
- R 2 represents acyloxy.
- R 4a is H, acyloxy, halogen, alkoxy, alkylthio, alkylamino, dialkylamino, alkenyloxy, alkenylthio, alkenylamino, dialkenylamino, alkynyloxy, alkynylthio, alkylamino, Represents a dialkylamyl-containing alkoxyalkyloxy or substituent (4)
- acyl related to “acyloxy” of R 2 and R 4a
- examples of the “acyl” related to the modified form of B include the same “acyl” related to the modified form of B.
- Examples of “norogen”, “alkoxy”, “alkylamino” and “dialkylamino” relating to R 4a include the same as those relating to the modified form of B. Examples thereof include the same “alkyl” related to the modified form of B.
- alkoxy part of the “alkoxyalkyloxy” according to R 4a examples include the same “alkoxy” as the modified B.
- the “alkellyl” part of “alkylene”, “alkylene”, “alkylene” and “geluceruamino” relating to R is the same as “alkenyl” relating to said R. That's right.
- alkyl part of “alkyloxy”, “alkylthio”, “alkylamino” and “dialkylamino” relating to R 4a is the same as “alkynyl” relating to said R. It can be done.
- Alkylamino”, “alkylkeamino” and “alkylamino” according to R 4a are not particularly limited as long as they can be protected as long as they are used as protective groups for the amino groups.
- trifluoroacetyl is preferred.
- solid phase carriers examples include controlled pore glass (CPG), oxalylated-constant glass (eg, Alul et al., Nucleic Acids Research, Vol. 19, 15 27 (1991)). ), TentaGel support-aminopolyethylene glycol derivative support (see, eg, Wright et al., Tetrahedron Letters, Vol. 34, 3373 (1993)),
- linker examples include 3aminopropyl, succiol, 2,2′-diethanol sulfol, and long chain alkylamino (LCAA).
- the nucleic acid derivative (27a) and the nucleic acid derivative (27b) are nucleic acid derivatives produced according to a known method or supported on a solid phase carrier that can be obtained as a commercial product. Examples thereof include nucleic acid derivatives represented by formulas (28) and (29).
- B WG 2 has the same meaning as described above.
- the nucleic acid derivatives (28) and (29) in which R 4 is the substituent (4) can be produced from the phosphoramidite compound (B) according to a known method.
- the “acid” that can be used in this step include trifluoroacetic acid, dichloroacetic acid, and trichloroacetic acid.
- the acid that can be used in this step can be used after diluting with a suitable solvent so as to have a concentration of 1 to 5%.
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, acetonitrile, water, and any mixed solvent thereof.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of oligonucleic acid derivative (1), the type of acid used, the reaction temperature, etc., but usually 1 minute to 1 hour is appropriate.
- the amount of the reagent to be used is suitably 0.8 to: LOO-fold molar amount, preferably 1 to 10-fold molar amount with respect to the (oligo) nucleic acid derivative supported on the solid phase carrier.
- the (oligo) nucleic acid derivative (2) produced in step A is condensed with a nucleic acid monomer compound using an activator to produce an oligonucleic acid derivative represented by the following general formula (7).
- each B, each Q, each R 4 , and each WG 2 are independently the same as defined above.
- n and R ⁇ T are as defined above. This process is carried on a solid support! It can be carried out by allowing a nucleic acid monomer compound and an activator to act on the oligonucleic acid derivative.
- nucleic acid monomer compound examples include a phosphoramidite compound (B) or a nucleic acid derivative represented by the following general formula (30).
- RR 2a , R 2b , R 4a and WG 2 are as defined above.
- B has a protecting group
- V may be a nucleobase or a modified form thereof.
- nucleobase is not particularly limited as long as it is used for nucleic acid synthesis.
- Examples thereof include pyrimidine bases such as cytosine, uracil and thymine, and purine bases such as adenine and guanine.
- Nucleobase according to B is a nucleobase having an amino group, whether it is protected or not.
- adenine, guanine, and cytosine preferably have a protected amino group.
- the “protecting group for amino group” is not particularly limited as long as it is used as a protecting group for nucleic acids. Specifically, for example, benzoyl, 4-methoxybenzoyl, acetyl, propionyl, butyryl , Isobutyryl, phenylacetyl, phenoxyacetyl, 4 tert butyl phenoxyacetyl, 4 isopropylphenoxyacetyl, and (dimethylamino) methylene.
- the “modified form” of B is a group in which the nucleobase is substituted with an arbitrary substituent.
- substituent related to the “modified product” examples include halogen, acyl, alkyl, arylalkyl, alkoxy, alkoxyalkyl, hydroxy, amide-containing monoalkylamino-alkylalkylamino, carboxy, and cyano-containing nitro. 1 to 3 are substituted at any position.
- Examples of the “activator” include the same as described above.
- the reaction solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include acetonitrile and THF.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of oligonucleic acid derivative (2), the type of activator used, the reaction temperature, etc., but usually 1 minute to 1 hour is appropriate.
- the amount of the reagent to be used is suitably 0.8 to: LOO-fold molar amount, preferably 1 to 10-fold molar amount with respect to the oligonucleic acid derivative supported on the solid phase carrier.
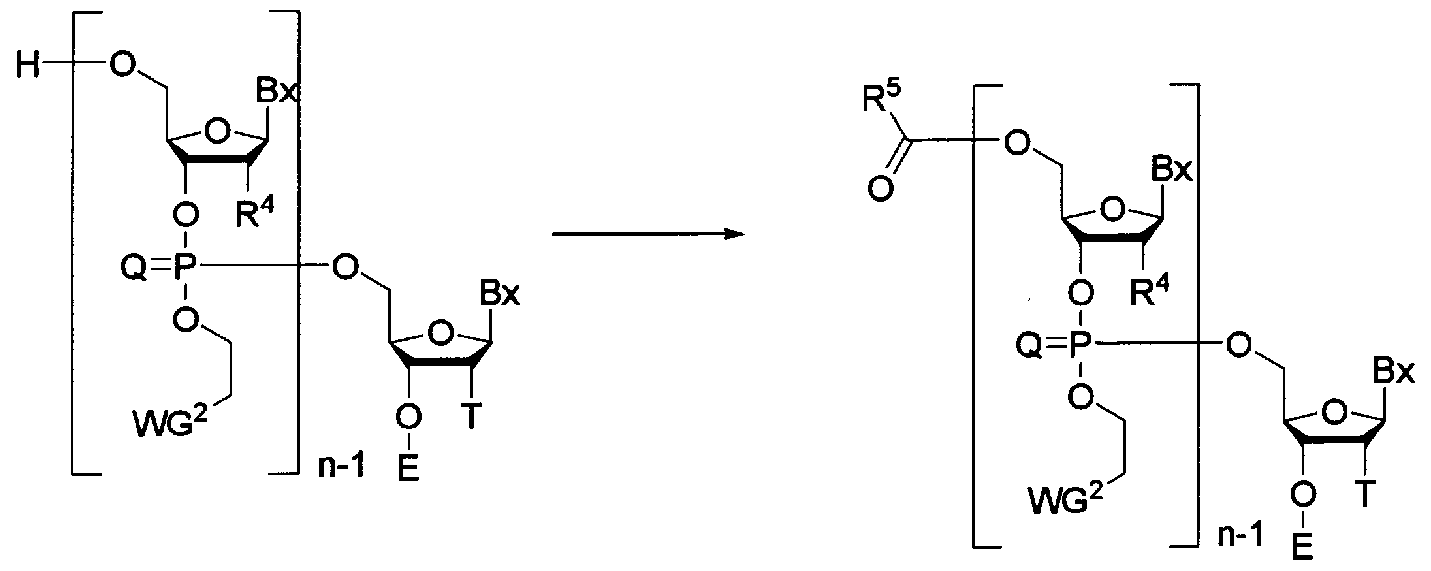
- step B The step of capping the hydroxyl group at the 5-position of the (oligo) nucleic acid derivative (2) that has not been reacted in step B.
- each B, each Q, each R 4, each WG 2 are each independently as defined above
- R 5 represents methyl, phenoxymethyl, or tert-butylphenoxymethyl.
- This step is a reaction for protecting the hydroxyl group at the 5 ′ position, which has not been reacted in Step B, and can be carried out by acting a capping agent on the oligonucleic acid derivative supported on the solid phase carrier. .
- the “capping agent” examples include acetic anhydride, phenoxyacetic anhydride or tert-butylphenoxyacetic anhydride.
- the capping agent is from 0.05. It can also be used after diluting with a suitable solvent so as to achieve a proper degree.
- the solvent is not particularly limited as long as it is not involved in the reaction, and examples thereof include pyridine, dichloromethane, acetonitrile, THF, and any mixed solvent thereof.
- 4-dimethylaminopyridine or N-methylimidazole can be used as a “reaction accelerator” as necessary.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of oligonucleic acid derivative (2), the type of capping agent used, the reaction temperature, etc., but usually 1 to 30 minutes is appropriate.
- the amount of the reagent to be used is suitably 0.8 to: LOO-fold molar amount, preferably 1 to 10-fold molar amount with respect to the oligonucleic acid derivative supported on the solid support.
- each B, each Q, each R 4, each WG 2 are each independently as defined above
- This step is a reaction for converting trivalent phosphorus to pentavalent phosphorus using an oxidizing agent, and can be performed by allowing an oxidizing agent to act on the oligonucleic acid derivative supported on the solid phase carrier. I'll do it.
- iodine or tert-butyl hydroperoxide can be used as the “oxidant”, for example.
- the oxidant should have a concentration of 0.05-2M.
- the solvent used in the reaction is not particularly limited as long as it does not participate in the reaction, and examples thereof include pyridine, THF, water, and any mixed solvent thereof.
- iodine Z water Z pyridine THF or iodine Z pyridine acetic acid or a peroxide (such as tert butyl hydroperoxide Z methylene chloride) can be used.
- oxidizing phosphorus with sulfur as an “oxidant”, for example, sulfur, Beaucage reagent (3H-1,2, benzodiol-3 on 1,1 dioxide), 3 amino-1,2,4 dithiazole —5 Thion (ADTT) can be used.
- the oxidizing agent can be used by diluting with an appropriate solvent so as to have a concentration of 0.01 to 2M.
- the solvent used in the reaction is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, acetonitrile, pyridine, and any mixed solvent thereof.
- the reaction temperature is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of oligonucleic acid derivative (7), the type of oxidizing agent used, the reaction temperature, etc., but usually 1 to 30 minutes is appropriate.
- the amount of the reagent to be used is suitably 0.8 to 100 times, preferably 10 to 50 times the molar amount of the oligonucleic acid derivative supported on the solid phase carrier.
- step D the oligonucleic acid derivative (9) produced in this step is excised from the solid phase carrier force to remove each nucleobase and the protecting group for each phosphate group.
- each B, each Q, each R 4, each WG 2 are each independently a
- the cleaving step is a reaction in which oligo RNA having a desired chain length is removed from the solid phase carrier and the linker by a cleaving agent, and the cleaving agent is added to a solid carrier on which the oligonucleic acid derivative having the desired chain length is supported. Can be implemented. In this step, the protecting group at the nucleobase portion can be removed.
- Examples of the “cutting agent” include concentrated aqueous ammonia and methylamine.
- the “cleaving agent” that can be used in this step can be used by diluting with, for example, water, methanol, ethanol, isopropyl alcohol, acetonitrile, THF, or any mixed solvent thereof. Of these, ethanol is preferred.
- the reaction temperature is suitably from 15 ° C to 75 ° C, preferably from 15 ° C to 30 ° C, more preferably from 18 ° C to 25 ° C.
- the deprotection reaction time varies depending on the type of oligonucleic acid derivative (9), reaction temperature, etc., and a suitable force is 10 minutes to 30 hours, preferably 30 minutes to 24 hours, more preferably 1 to 4 hours It is.
- the concentration of hydroxyammonium hydroxide in the solution used for deprotection is suitably 20-30% by weight, preferably 25-30% by weight, more preferably 28-30% by weight. is there.
- the amount of the reagent to be used is suitably 1 to: 00 times the molar amount, preferably 10 to 50 times the molar amount of the oligonucleic acid derivative supported on the solid phase carrier.
- a sulfoxide system that may contain THF
- each B, each Q, each R, and each R 4 are independently the same as defined above.
- n and R ⁇ Z are as defined above.
- This step can be performed by allowing TBAF to act on the oligonucleic acid derivative (10).
- the amount of TBAF to be used is suitably 1 to 500-fold mol amount, preferably 5 to 10-fold mol amount based on the protecting group to be removed.
- a sulfoxide solvent, an amide solvent, or a mixed solvent thereof, which may contain THF can be used.
- the amount of THF used in the sulfoxide solvent or amide solvent or these mixed solvents is 0 to 95%. Is suitable, preferably 0 to 50%.
- the amount of ⁇ sulfoxide solvent or amide solvent that may contain THF, or a mixed solvent thereof (reaction solvent) '' varies depending on the type of oligonucleic acid derivative (10), the reaction solvent used, etc. 0.8 to: L00 times the molar amount is appropriate for TBAF, and preferably 1 to 10 times the molar amount.
- the reaction temperature varies depending on the type of oligonucleic acid derivative (10) and the reaction solvent used, but 20 ° C. to 80 ° C. is preferable.
- the reaction time varies depending on the oligonucleic acid derivative (10), the reaction solvent to be used, the reaction temperature, etc., but usually 1 to 100 hours is appropriate.
- nitroalkane examples include a linear 1- to 6-troalkane having carbon atoms. Specific examples include nitromethane.
- alkylamine examples include linear alkylamines having 1 to 6 carbon atoms. Specific examples include methylamine, ethylamine, n-propylamine, n-butylamine, n-pentylamine, and n-hexylamine.
- Examples of “amidine” include benzamidine and formamidine.
- Examples of the “thiol” include linear thiols having 1 to 6 carbon atoms. Specific examples include methanethiol, ethanethiol, 1 propanethiol, 1 butanethiol, 1 pentanethiol, and 1 hexanethiol.
- Thiol derivatives "" Can include, for example, alcohols or ethers having the same or different linear alkyl thiol group having 1 to 6 carbon atoms.
- 2-mercaptoethanol 4 mercapto 1-butanol, 6 mercapto 1 monohexanol, mercaptomethyl ether, 2 mercaptoethyl ether, 3 mercaptopropyl ether, 4 mercaptobutyl ether, 5-mercaptopentyl ether
- Mention may be made of 6-mercaptohexyl ether.
- the amount of “acrylonitrile scavenger” used depends on the type of oligonucleic acid derivative (10), etc.
- the 2′-position hydroxyl group of each ribose of the oligonucleic acid derivative (10) is protected.
- 0.1 to 500-fold molar amount is appropriate with respect to methyl, and preferably 1 to 10-fold molar amount.
- each B, each Q, and each R are independently the same as defined above.
- n and Z are as defined above.
- This step is a reaction that finally removes the protecting group of the 5 'hydroxyl group of the oligo RNA, and can be carried out by allowing an acid to act on the oligo RNA cleaved by the solid carrier.
- the “acid” that can be used include trichlorodiacetic acid, dichloroacetic acid, and acetic acid.
- the acid that can be used in this step can also be diluted with an appropriate solvent.
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, acetonitrile, water, a buffer solution having a pH of 2 to 5 or any mixed solvent thereof. You can.
- the buffer solution include an acetate buffer solution.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of oligonucleic acid derivative (11), the type of acid used, the reaction temperature, etc., but usually 1 minute to 1 hour is appropriate.
- the amount of the reagent to be used is suitably 0.8 to 100 times, preferably 1 to 10 times the molar amount of the oligonucleic acid derivative supported on the solid phase carrier.
- a step of separating and purifying the oligo RNA (A) produced in step G is a step of separating and purifying the oligo RNA (A) produced in step G.
- the “separation and purification step” refers to usual separation and purification means from the above reaction mixture, such as extraction, concentration, neutralization, filtration, centrifugation, recrystallization, C-force C reverse phase column chromatography, C
- the desired oligo RNA is isolated and purified by using means such as column chromatography, gel filtration column chromatography, high performance liquid chromatography, dialysis, and ultrafiltration.
- Examples of the “elution solvent” include acetonitrile, methanol, ethanol, isopropyl alcohol, water alone, or a mixed solvent in an arbitrary ratio.
- examples of additives include sodium phosphate, potassium phosphate, sodium chloride salt, potassium salt salt, ammonium acetate, triethylammonium acetate, sodium acetate, acetic acid lithium, Tris-HCl, Ethylenediamine tetraacetic acid can be added at a concentration of lmM to 2M, and the pH of the solution can be adjusted in the range of 1 to 9.
- an oligo RNA (A) having a desired chain length can be produced.
- compound (27a) in which R 4a is substituent (4), nucleic acid derivative (27a) in which R 4a is H or acyloxy, or A nucleic acid derivative (27b) where R 2 is acyl can be used.
- nucleic acid derivative (27a) in which R 4a is H or acyloxy or a nucleic acid derivative (27b) in which R 2 is acyloxy is used as a starting material
- at least one of the phosphonic acid monomer compounds of the present invention is used as the nucleic acid monomer compound. It is necessary to use Loamidaitoi compound.
- reaction solution was added to a saturated aqueous sodium hydrogen carbonate solution, extracted with methylene chloride, dried over anhydrous magnesium sulfate, the solvent was distilled off, and the resulting mixture was purified by 30 g silica gel column chromatography.
- '—O— (4, 4, -dimethoxytrityl) 2 —O— (2 cyanoethoxymethyl) uridine was obtained (19 7 mg; yield 34%).
- Step 2 5'—O— (4.4′-Dimethoxytrityl) 2 ′ ⁇ 0- (2 Cyanethoxymethyl) uridine 3, 1 O— (2 Cyanethyl N. N diisopropyl phosphoramidite)
- the reaction solution was filtered through Celite and washed with methylene chloride, and then the organic phase was washed with 1M aqueous sodium hydrogen thiosulfate solution, washed with saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous magnesium sulfate and evaporated. did.
- the obtained residue was purified by thin-layer chromatography, and 3 ', 5, —O— (tetraisopropyldisiloxane—1,3 diyl) —2, —O— (2-cyanethoxymethyl) uridine was purified. Obtained (150 mg; 85% yield).
- Step 1 Making acetylyl-5, -0- (4.4, -dimethoxytrityl) -2, -0- (2-cyanethoxymethyl) cytidine
- N 4 acetylyl 5, -0- (4, 4, -dimethoxytrityl) cytidine 588 mg (lmmol) is dissolved in 1,2-dichloroethane 4 mL, and diisopropylethylamine 452 mg (3.5 mm ol) is prepared. Then, 365 mg (l.2 mmol) of dibutyltin dichloride was added, and the mixture was reacted at room temperature for 1 hour. Thereafter, the temperature was raised to 80 ° C., and 155.4 mg (l. 3 mmol) of chloromethyl 2-cyanoethyl ether was added dropwise, followed by stirring for 60 minutes.
- reaction mixture was extracted with saturated aqueous sodium hydrogen carbonate solution with methylene chloride, dried over anhydrous magnesium sulfate, and the solvent was distilled off.
- the resulting mixture was subjected to 30 g silica gel column chromatography. Purification gave N 4 -acetyl- 5,5-0- (4,4, -dimethoxytrityl) -2, -0- (2-cyanethoxymethyl) cytidine (219 mg; yield 35%).
- N 4 acetyleno 3,5,1 O tetraisopropyldisiloxane 1,3 diyl
- cytidine 1.
- THFlOmL in a mixed solvent 975 mg (3.79 mmol) of silver trifluoromethanesulfonate was added, and molecular sieve 4A was added and dried. Under ice cooling, 370 mg (2.08 mmol) of N bromosuccinimide was added, and the reaction vessel was shielded from light and stirred for 10 minutes.
- the filtrate was concentrated with an evaporator and the residue was dissolved in ethyl acetate and separated into a saturated aqueous sodium bicarbonate solution.
- the organic phase was washed with a saturated sodium chloride aqueous solution, dried over anhydrous magnesium sulfate, and the solvent was distilled off.
- the resulting residue was purified by silica gel chromatography to obtain the target compound (15 g; yield 83%).
- Step 1 N 2 —Acetyl-5, —O— (4.4, -dimethoxytrityl) -2 ′ -0-
- N 2 -acetylyl 5, -0- (4, 4, -dimethoxytrityl) guanosine 627 mg (l mmol) is dissolved in 1,2-dichloroethane 4 mL, and diisopropyl etheramine 452 mg (3.5 mm ol) is added, Subsequently, 365 mg (l.2 mmol) of dibutyltin dichloride was added, and the mixture was reacted at room temperature for 1 hour. Thereafter, the temperature was raised to 80 ° C., and 155.4 mg (l.3 mmol) of chloromethyl 2-cyanoethyl ether was added dropwise, and the mixture was stirred for 60 minutes.
- Step 1 N s —acetylyl-5, —O— (4.4, -dimethoxytrityl) -2′-0
- Og (36. Om mol) is dissolved in 170 mL of 1,2-dichloroethane and 16.3 g (126 mmol) of diisopropylethylamine Then, 12. lg (39.7 mmol) of dibutyltin dichloride was added and reacted at room temperature for 1 hour. Thereafter, the mixture was stirred at 80 ° C. for 15 minutes, and 4.30 g (36. Ommol) of chloromethyl 2-cyanoethyl ether was added dropwise, followed by stirring for 30 minutes.
- reaction solution was added to a saturated aqueous sodium hydrogen carbonate solution, extracted with methylene chloride, dried over anhydrous magnesium sulfate, evaporated, and the resulting mixture was purified by silica gel column chromatography. 6 -Acetyl-1,5-0- (4,4,1-dimethoxytrityl) -2, -O- (2-cyanethoxymethyl) adenosine was obtained. (7. 47g; Yield 33%)
- Step 2 N a —Acetyl— 5′— O— (4.4′-Dimethoxytrityl) —2 ′ ⁇ 0- (2-Cyanethoxymethyl) adenosine 3, 1 O— (2-Cyanoethyl N. N-diisopropyl Phosphoramidite)
- N 2 full enoki Xia cetyl over 5'- O- (4, 4'- dimethoxytrityl) was dissolved guanosine 720mg of (Lmmol) in 1, 2 Jikuroroetan 4 mL, diisopropyl E chill ⁇ Min 452mg of (3. 5 mmol) was added, Next, 365 mg (l.2 mmol) of dibutyltin dichloride was added and reacted at room temperature for 1 hour. Thereafter, the temperature was raised to 80 ° C., and 55.4 mg (l.3 mmol) of chloromethyl 2-cyanoethyl ether was added dropwise and stirred as it was for 60 minutes.
- Step 2 N 2 —Phenoxyacetyl- 5′—O— (4.4′-dimethoxytrityl) 2 ′ ⁇ 0- (2-Cyanethoxymethyl) guanosine 3′—O— (2-Cyanoethyl N. N-Diisopropyl Mouth Pill Phosphoramidite Making
- N 2 phenoxycetyl-5, -0- (4, 4, -dimethoxytrityl) — 2, 1, O— (2 cyanoethoxymethyl) guanosine obtained in Step 1 was mixed with methyl chloride.
- Dissolve in 4 mL of Len add 128.8 mg (0.996 mmol) of diisopropylethylamine, add 14-1.5 mg (0.598 mmol) of 2-cyanoethyl N, N-diisopropylchlorophosphoramidite, and react at room temperature for 1 hour. . After the reaction, the solvent was distilled off, and the resulting mixture was purified by 3 Og silica gel column chromatography to obtain the target compound (316 mg; yield 79%).
- New 2 full enoki Xia cetyl-2, -0- (2 Xia Roh ethoxymethyl) guanosine 660 mg (1. 32 mmol) was dried for 30 minutes by azeotropic vacuum pump with pyridine. After dissolving in 9 mL of THF, 2. lg (26.4 mmol) of pyridine and 600 mg of molecular sieves 4A were added under an argon atmosphere and stirred for 10 minutes. To this, 540 mg (l. 58 mmol) of 4,4,1-dimethoxytrityl chloride was added in 3 portions every 1 hour, and the mixture was further stirred for 1 hour.
- Step 1 N a —Acetyl 1 3 ′. 5′— O— (Tetraisopropyldisiloxane 1 1.3 Dil) 2′— O Preparation of methylthiomethyladenosine
- Step 2 N a Asechiru 3 ,. 5'-O- (tetraisopropyldisiloxane 1.3 Jie Le) 2'O-(2-Xia Bruno ethoxymethyl) Preparation of Adenosine
- reaction mixture was cooled and neutralized by adding triethylamine, diluted with methylene chloride, washed with aqueous sodium thiosulfate and saturated aqueous sodium bicarbonate, dried over anhydrous sodium sulfate, and evaporated.
- the resulting mixture was purified by silica gel column chromatography to obtain the target compound. (722 mg; 71% yield).
- nucleic acid monomer compounds 5 —O— (4,4, -dimethoxytrityl) -2, —O— (2—cyanethoxymethyl) uridine 3,1 O— (2 cyanethyl N, N diisopropyl phosphoramidite) Tetrazole was used as the condensation catalyst, iodine solution was used as the oxidizing agent, and acetic anhydride and N-methylimidazole solution were used as the cabbing solution.
- Nucleic acid monomer After condensing a single compound 20 times, using a 10M methylamine solution in ethanol as a cleaving agent, cleaving from the CPG solid phase carrier at room temperature for 1-2 hours and removing the protecting group at each phosphate site Reaction was performed. The reaction mixture was concentrated under reduced pressure, unnecessary peaks were removed with a reverse phase column (ODS), and then purified using an elution solvent (acetonitrile—50 mM triethylamine—acetic acid buffer). The residue was concentrated under reduced pressure and then reacted with 1M TBAF in THF for 1 hour at room temperature to remove the hydroxyl protecting group at the 2 ′ position.
- ODS reverse phase column
- the protecting group at the 5 ′ end was removed with 80% acetic acid (10 minutes at room temperature). After concentration under reduced pressure, the aqueous layer was washed with ether to obtain a high-purity target compound without purification.
- a commercially available 5 '-0- (4,4'-dimethoxytrityl) thymidine-supported CPG solid phase carrier (22 mg, 1 ⁇ mol) is placed in a column with a glass filter, and an automatic nucleic acid synthesizer (Exp edite TM: (Appliedio Systems) was used to synthesize the oligo RNA of the title compound.
- nucleic acid monomer compounds 5, —O— (4,4, -dimethoxytrityl) -2, —O— (2—cyanethoxymethyl) uridine 3,1 O— (2 cyanethyl N, N diisopropyl phosphoramidite) , N 4 -Acetyl-5, -0- (4, 4, -dimethoxytrityl) -2,-O- (2 cyanoethoxymethyl) cytidine 3, -O- (2 cyanoethinole N, N diisopropyl phosphoramidite ), N 6- acetyl--5, —O— (4, 4, -dimethoxytrityl) — 2, —O— (2 cyanoethoxymethinole) adenosine 3, —O— (2 cyanoethinole N, N diisopropyl phosphoramidite), N 2 —phenoxyacetylyl 5, -0- (4, 4, —dimeth
- Liquid feeding unit LC-10AT (manufactured by Shimadzu Corporation)
- DNAPac PA100 ⁇ 4mm ⁇ x250mm> DIONEX
- Solution B 25 mM Tris—HC 1 buffer containing 10% acetonitrile and 700 mM sodium perchlorate
- UV-visible spectrometer detection wavelength 260nm
- oligo RNA resin was cleaved from the CPG solid support at 40 ° C for 4 hours using 3 mL of concentrated ammonia / ethanol mixture (3: 1) as a cleaving agent.
- the elimination reaction of the protecting group at each phosphate site and the removal of the protecting group of the base were performed.
- a reaction was carried out at room temperature under the deprotection conditions shown in Table 2 below to remove the protecting group at the 2′-position hydroxyl group of each ribose.
- 1 ⁇ L of -tromethane was added to 100 mol of TBAF.
- the amount of TB AF used and the amount of reaction solvent used could be reduced.
- the amount of TBAF used could be reduced to about 1Z5 compared to when THF was used as the reaction solvent. This clearly reduces the amount of expensive TBAF used and the amount of ethanol used to precipitate the final product.
- a sulfoxide solvent, an amide solvent, or a mixed solvent thereof as a reaction solvent in the step of removing TB2's hydroxyl group protecting group by TBAF.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Saccharide Compounds (AREA)
Description

























Claims

















Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008502759A JP5187189B2 (ja) | 2006-02-27 | 2007-02-26 | 核酸保護基の脱離方法 |
| EP07714922.7A EP1995253B1 (en) | 2006-02-27 | 2007-02-26 | Method for detaching protecting group on nucleic acid |
| CN2007800138626A CN101426805B (zh) | 2006-02-27 | 2007-02-26 | 核酸保护基的脱去方法 |
| CA002643108A CA2643108A1 (en) | 2006-02-27 | 2007-02-26 | Method for removing a protecting group for nucleic acids |
| US12/280,764 US8158775B2 (en) | 2006-02-27 | 2007-02-26 | Method for detaching protecting group on nucleic acid |
| KR1020087022894A KR101345465B1 (ko) | 2006-02-27 | 2008-09-19 | 핵산 보호기의 탈리 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006050381 | 2006-02-27 | ||
| JP2006-050381 | 2006-02-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007099896A1 true WO2007099896A1 (ja) | 2007-09-07 |
Family
ID=38459003
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/053490 Ceased WO2007099896A1 (ja) | 2006-02-27 | 2007-02-26 | 核酸保護基の脱離方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8158775B2 (ja) |
| EP (1) | EP1995253B1 (ja) |
| JP (1) | JP5187189B2 (ja) |
| KR (1) | KR101345465B1 (ja) |
| CN (1) | CN101426805B (ja) |
| CA (1) | CA2643108A1 (ja) |
| WO (1) | WO2007099896A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008090829A1 (ja) * | 2007-01-22 | 2008-07-31 | Nippon Shinyaku Co., Ltd. | リボ核酸化合物の製造方法 |
| JPWO2021070494A1 (ja) * | 2019-10-11 | 2021-04-15 | ||
| JPWO2023182274A1 (ja) * | 2022-03-23 | 2023-09-28 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008064082A2 (en) * | 2006-11-20 | 2008-05-29 | Integrated Dna Technologies Inc. | Methods for rna desilylation |
| JP7423533B2 (ja) * | 2018-09-07 | 2024-01-29 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| EP3805242A1 (en) * | 2019-10-07 | 2021-04-14 | Sterna Biologicals GmbH & Co. KG | Method for the production of a catalytically active dna molecule having improved activity and its use in a method of treating asthma |
| KR102938084B1 (ko) * | 2019-10-08 | 2026-03-11 | 스미또모 가가꾸 가부시끼가이샤 | 배당체 화합물의 제조 방법 |
| CN110922434B (zh) * | 2019-12-05 | 2021-03-23 | 武汉金开瑞生物工程有限公司 | 一种脱氧核苷酸引物合成方法 |
| JP7719788B2 (ja) * | 2020-09-24 | 2025-08-06 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| CN115873056B (zh) * | 2022-08-04 | 2025-05-16 | 百力格生物科技(上海)股份有限公司 | 使用混合脱保护剂合成rna核酸的方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0856679A (ja) * | 1994-07-18 | 1996-03-05 | Hoechst Ag | Rna切断性又はrna結合性オリゴヌクレオチド |
| JP2002501931A (ja) * | 1998-02-02 | 2002-01-22 | プリンストン ユニバーシティ | MurGの基質類似体、その製造方法およびそれを用いたアッセイ |
| WO2005023828A1 (ja) * | 2003-09-02 | 2005-03-17 | Takeshi Wada | リボヌクレオチド又はリボヌクレオチド誘導体の製造方法 |
| WO2006022323A1 (ja) * | 2004-08-26 | 2006-03-02 | Nippon Shinyaku Co., Ltd. | ホスホロアミダイト化合物及びオリゴrnaの製法 |
| WO2006095739A1 (ja) * | 2005-03-09 | 2006-09-14 | Tokyo Institute Of Technology | リボヌクレオシドの2’水酸基の脱保護方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2007097447A1 (ja) * | 2006-02-27 | 2009-07-16 | 日本新薬株式会社 | 核酸保護基の脱離方法 |
| JPWO2007097446A1 (ja) * | 2006-02-27 | 2009-07-16 | 日本新薬株式会社 | オリゴ核酸のキャッピング法 |
-
2007
- 2007-02-26 US US12/280,764 patent/US8158775B2/en active Active
- 2007-02-26 CN CN2007800138626A patent/CN101426805B/zh not_active Expired - Fee Related
- 2007-02-26 WO PCT/JP2007/053490 patent/WO2007099896A1/ja not_active Ceased
- 2007-02-26 CA CA002643108A patent/CA2643108A1/en not_active Abandoned
- 2007-02-26 EP EP07714922.7A patent/EP1995253B1/en active Active
- 2007-02-26 JP JP2008502759A patent/JP5187189B2/ja active Active
-
2008
- 2008-09-19 KR KR1020087022894A patent/KR101345465B1/ko active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0856679A (ja) * | 1994-07-18 | 1996-03-05 | Hoechst Ag | Rna切断性又はrna結合性オリゴヌクレオチド |
| JP2002501931A (ja) * | 1998-02-02 | 2002-01-22 | プリンストン ユニバーシティ | MurGの基質類似体、その製造方法およびそれを用いたアッセイ |
| WO2005023828A1 (ja) * | 2003-09-02 | 2005-03-17 | Takeshi Wada | リボヌクレオチド又はリボヌクレオチド誘導体の製造方法 |
| WO2006022323A1 (ja) * | 2004-08-26 | 2006-03-02 | Nippon Shinyaku Co., Ltd. | ホスホロアミダイト化合物及びオリゴrnaの製法 |
| WO2006095739A1 (ja) * | 2005-03-09 | 2006-09-14 | Tokyo Institute Of Technology | リボヌクレオシドの2’水酸基の脱保護方法 |
Non-Patent Citations (9)
| Title |
|---|
| ALUL ET AL., NUCLEIC ACIDS RESEARCH, vol. 19, 1991, pages 1527 |
| M. MATTEUCCI, TETRAHEDRON LETTERS, vol. 31, 1990, pages 2385 |
| OHGI ET AL., ORGANIC LETTERS, vol. 7, 2005, pages 3477 |
| OHGI T. ET AL.: "A new RNA synthesis method with a 2'-O-(2-cyanoethoxymethyl) protecting group", ORG. LETT., vol. 7, no. 16, 2005, pages 3477 - 3480, XP003009345 * |
| RAMOS L.A. ET AL.: "Carboxymethylation of cellulose in the new solvent dimethyl sulfoxide/tetrabutylammonium fluoride", CARBOHYDRATE POLYMERS, vol. 60, no. 2, 2005, pages 259 - 267, XP004844346 * |
| See also references of EP1995253A4 * |
| SINHA N.D. ET AL.: "Polymer support oligonucleotide synthesis XVIII: use of beta-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product", NUCLEIC ACIDS RES., vol. 12, no. 11, 1984, pages 4539 - 4557, XP002134520 * |
| T. BENNECHE ET AL., SYNTHESIS, 1983, pages 762 |
| WRIGHT ET AL., TETRAHEDRON LETTERS, vol. 34, 1993, pages 3373 |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008090829A1 (ja) * | 2007-01-22 | 2008-07-31 | Nippon Shinyaku Co., Ltd. | リボ核酸化合物の製造方法 |
| JPWO2021070494A1 (ja) * | 2019-10-11 | 2021-04-15 | ||
| WO2021070494A1 (ja) * | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| JP7640461B2 (ja) | 2019-10-11 | 2025-03-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| US12509483B2 (en) | 2019-10-11 | 2025-12-30 | Sumitomo Chemical Company, Limited | Method for producing nucleic acid oligomers |
| JPWO2023182274A1 (ja) * | 2022-03-23 | 2023-09-28 | ||
| US20250145655A1 (en) * | 2022-03-23 | 2025-05-08 | Sumitomo Chemical Company, Limited | Method for producing nucleic acid oligomer |
| US12509485B2 (en) * | 2022-03-23 | 2025-12-30 | Sumitomo Chemical Company, Limited | Method for producing nucleic acid oligomer |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1995253B1 (en) | 2016-12-21 |
| KR101345465B1 (ko) | 2013-12-27 |
| EP1995253A4 (en) | 2013-04-24 |
| US20090149645A1 (en) | 2009-06-11 |
| CA2643108A1 (en) | 2007-09-07 |
| EP1995253A1 (en) | 2008-11-26 |
| JP5187189B2 (ja) | 2013-04-24 |
| CN101426805B (zh) | 2012-07-18 |
| JPWO2007099896A1 (ja) | 2009-07-16 |
| US8158775B2 (en) | 2012-04-17 |
| CN101426805A (zh) | 2009-05-06 |
| KR20080106441A (ko) | 2008-12-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006022323A1 (ja) | ホスホロアミダイト化合物及びオリゴrnaの製法 | |
| WO2007099896A1 (ja) | 核酸保護基の脱離方法 | |
| WO2007097447A1 (ja) | 核酸保護基の脱離方法 | |
| WO2007097446A1 (ja) | オリゴ核酸のキャッピング法 | |
| JP5168145B2 (ja) | 核酸保護基の導入方法 | |
| RU2415862C2 (ru) | Производное фосфорамидита и способ получения олиго-рнк |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2008502759 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2643108 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4426/CHENP/2008 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087022894 Country of ref document: KR |
|
| REEP | Request for entry into the european phase |
Ref document number: 2007714922 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007714922 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780013862.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12280764 Country of ref document: US |