JP7640461B2 - 核酸オリゴマーの製造方法 - Google Patents
核酸オリゴマーの製造方法 Download PDFInfo
- Publication number
- JP7640461B2 JP7640461B2 JP2021550425A JP2021550425A JP7640461B2 JP 7640461 B2 JP7640461 B2 JP 7640461B2 JP 2021550425 A JP2021550425 A JP 2021550425A JP 2021550425 A JP2021550425 A JP 2021550425A JP 7640461 B2 JP7640461 B2 JP 7640461B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- group
- nucleic acid
- acid oligomer
- protecting group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Saccharide Compounds (AREA)
Description

G4は、水素原子または水酸基の保護基を表し、
G5は、アンモニウムイオン、アルキルアンモニウムイオン、アルカリ金属イオン、水素イオンまたはヒドロキシアルキルアンモニウムイオンを表し、
BCは、それぞれ独立して同一又は相異なる核酸塩基を表し、
Rは、それぞれ独立して同一又は相異なって、水素原子、フッ素原子またはOQ基を表し、
Qは、それぞれ独立して同一又は相異なって、tert-ブチルジメチルシリル基、メチル基、2-メトキシエチル基、リボースの4’位の炭素原子と結合しているメチレン基、リボースの4’位の炭素原子と結合しているエチレン基、リボースの4’位の炭素原子と結合しているエチリデン基、または式(1):

RaおよびRbは同一又は相異なって、メチル基、エチル基または水素原子を表し、
*印のついた結合は、OQ基の酸素原子との結合であることを表し、
nは1~5の何れかの整数を表す。
ただしRaおよびRbが同時に水素原子を表すことはない。)
の保護基を表し、
Yは、それぞれ独立して同一又は相異なって、酸素原子または硫黄原子を表し、
mは、2以上200までの何れかの整数を表し、
WおよびXは、下記の(a)または(b)のいずれかで定義され、
(a)Wが水酸基であるときは、Xは前記R基と同じ定義である。
(b)Xが水酸基であるときは、WはOV基を表し、
Vは、tert-ブチルジメチルシリル基、または前記式(1)の基を表す。
ただし、前記R、WおよびXのうち少なくとも一つの基は、前記式(1)の保護基で保護された水酸基を表す。そして
mが3以上の整数のとき、式(3)で示される核酸オリゴマーは、それぞれの5’末端と3’末端のヌクレオチドの間のp個(ただし、pは、式:m-1>pを満たす正の整数である。)のヌクレオチドの代わりに、非ヌクレオチドリンカーが組み込まれていてもよい核酸オリゴマーである。)
で示される核酸オリゴマーを、フッ化物イオンと接触させることを特徴とする、式(4):

R’は、それぞれ独立して同一又は相異なって、水酸基、水素原子、フッ素原子、メトキシ基、2-メトキシエチル基、またはOQ’基を表し、
Q’は、それぞれ独立して同一又は相異なって、リボースの4’位の炭素原子と結合しているメチレン基、4’位の炭素原子と結合しているエチレン基、または4’位の炭素原子と結合しているエチリデン基を表し、
式(4)の置換基G4、G5、Y、Bcおよびmの定義は、前記式(3)における定義と同じであり、
W0は、水酸基であり、
X0は、前記R’基と同じ定義である。
そして、mが3以上の整数のとき、式(4)で示される核酸オリゴマーは、それぞれの5’末端と3’末端のヌクレオチドの間のp個(ただし、pは、式:m-1>pを満たす正の整数である。)のヌクレオチドの代わりに、非ヌクレオチドリンカーが組み込まれていてもよい核酸オリゴマーである。)
で示される核酸オリゴマーの製造方法(以下、本明細書中、「本発明の製造方法」と呼称する)。
項2. 非ヌクレオチドリンカーが、アミノ酸骨格からなるリンカーである、前記項1に記載の製造方法。
項3. アミノ酸骨格からなるリンカーが、下記式(A14-1)、(A14-2)または式(A14-3)の構造を有するリンカーである、前記項2に記載の製造方法。

項4. Wが水酸基であり、XがR基であり、W0が水酸基であり、そしてX0がR’基である、前記項1~3の何れか1項に記載の製造方法。
項5. フッ化物イオン源が、フッ化テトラアルキルアンモニウムである、前記項1~4の何れか1項に記載の製造方法。
項6. フッ化テトラアルキルアンモニウムが、フッ化テトラ-n-ブチルアンモニウム(TBAF)である、前記項1~5の何れか1項に記載の製造方法。
項7. 酸素濃度が10%以下の不活性ガス雰囲気下で反応を行う、前記項1~6の何れか1項に記載の製造方法。
項8. 酸素濃度が5%以下の不活性ガス雰囲気下で反応を行う、前記項1~6の何れか1項に記載の製造方法。
項9. 酸素濃度が0%の不活性ガス雰囲気下で反応を行う、前記項1~6の何れか1項に記載の製造方法。
項10. 式(3)で示される核酸オリゴマーにフッ化物イオンの全量を添加するために要する時間が30分以上である、前記項1~9の何れか1項に記載の製造方法。
項11. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が5%以上である、前記項1~10の何れか1項に記載の製造方法。
項12. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が10%以上である、前記項1~10の何れか1項に記載の製造方法。
項13. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が20%以上である、前記項1~10の何れか1項に記載の製造方法。
項14. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が30%以上である、前記項1~10の何れか1項に記載の製造方法。
項15. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が40%以上である、前記項1~10の何れか1項に記載の製造方法。
項16. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が50%以上である、前記項1~10の何れか1項に記載の製造方法。
項17. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が60%以上である、前記項1~10の何れか1項に記載の製造方法。
項18. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が70%以上である、前記項1~10の何れか1項に記載の製造方法。
項19. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が80%以上である、前記項1~10の何れか1項に記載の製造方法。
項20. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が90%以上である、前記項1~10の何れか1項に記載の製造方法。
項21. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が95%以上である、前記項1~10の何れか1項に記載の製造方法。
項22. 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が100%である、前記項1~10の何れか1項に記載の製造方法。
かかる添加は、式(3)で示される核酸オリゴマーを含む溶液の液表面または液中に5分以上かけて滴下することが好ましく、10分以上かけて滴下することがより好ましく、15分以上かけて滴下することがより好ましく、30分以上かけて滴下することがより好ましく、更に好ましくは1時間以上かけて滴下することである。
フッ化テトラアルキルアンモニウムとしては、テトラブチルアンモニウムフルオリド、およびテトラメチルアンモニウムフルオリド等が挙げられる。中でもテトラブチルアンモニウムフルオリド(TBAF)がより好ましい。
使用するフッ化物イオンの量は、通常、除去される保護基1モル当たり1~1000モル、好ましくは1~500モル、より好ましくは2~200モル、更に好ましくは4~100モルである。

で示される化合物。
更に、脱保護反応の時間は、使用する脱保護剤の種類、反応の温度によって異なるが、通常、1時間~100時間、好ましくは、1~24時間、より好ましくは2~12時間、より好ましくは3~10時間、より好ましくは3~8時間、更に好ましくは3~6時間である。
尚、反応の温度は、任意のタイミングで変更してもよく、また、式(1)で示される水酸基の保護基を脱保護する脱保護剤は、任意のタイミングで追加してもよい。
ここで、本発明の製法で使用する不活性ガスとは、窒素ガス、アルゴンガス、ヘリウムガス、二酸化炭素を挙げられるが、これらに限定されない。好ましくは、窒素ガスまたはアルゴンガスを挙げられる。
酸素濃度は、好ましくは15%以下、より好ましくは10%以下、より好ましくは5%以下がよく、更により好ましくは0%である。



式中、
置換基Baは、それぞれ独立して同一又は相異なる保護されていてもよい核酸塩基を表し、
G4およびYは、前記の式(3)において定義されたとおりであり、
G2は、それぞれ独立して同一又は相異なるリン酸の保護基を表し、
X1がOZを表すとき、W1はOV基を表し、
Vは、tert-ブチルジメチルシリル基、または前記式(1)の基を表す。
X1がR基を表すとき、W1はOZで表される基を表し、
Zは、固相担体、および固相担体と核酸オリゴマーの3’末端のリボースの2’位もしくは3’位の水酸基の酸素原子とをつなぐ連結部からなる基を表す。

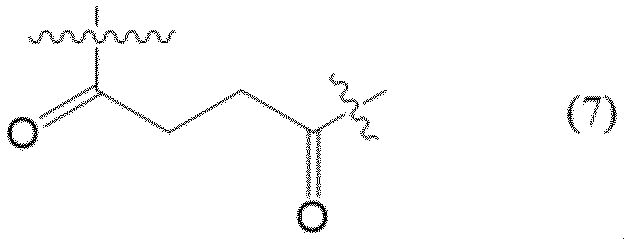
スペーサー(Sp)は、例えば、下記式(7)に示す構造式を有するものが例示される。
Solid supportとしては、無機多孔質担体や有機系樹脂担体などが挙げられる。無機多孔質担体には、例えば、Controlled pore Glass(CPG)およびゼオライトが挙げられる。有機系樹脂担体には、例えば、ポリスチレンからなる担体が挙げられる。



Rは、水素原子、フッ素原子、またはOQ基を表し、
Qは、tert-ブチルジメチルシリル基、メチル基、2-メトキシエチル基、4’の炭素原子と結合したメチレン基、4’の炭素原子と結合したエチレン基、4’の炭素原子と結合したエチリデン基、または前記式(1)で示される保護基を表し、
Baは、保護されていてもよい核酸塩基を表し、
G1は、水酸基の保護基を表し、
G2は、リン酸の保護基を表し、
G3は、アルキル基、または互いにその末端で結合して環状構造を表す。)

R4は、水素原子、メチル基、フェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、4-イソプロピルフェノキシアセチル基、フェニルアセチル基、アセチル基又はベンゾイル基を表し、
R5は、水素原子、アセチル基、イソブチリル基又はベンゾイル基を表し、
R6は、水素原子、フェノキシアセチル基、4-tert-ブチルフェノキシアセチル基、4-イソプロピルフェノキシアセチル基、フェニルアセチル基、アセチル基又はイソブチリル基を表し、
R7は、2-シアノエチル基を表し、
R8は、水素原子、メチル基、ベンゾイル基、4-メトキシベンゾイル基又は4-メチルベンゾイル基を表し、
R9は、ジメチルアミノメチレン基を表す。)
のいずれかで表される基を表す。


R4’は、水素原子、またはメチル基を表し、
R5’は、水素原子、またはアセチル基を表し、
R6’は、水素原子を表し、
R8’は、水素原子、メチル基を表す。)
リボースの2’位の水酸基が式(1)で示される保護基で保護されたアミダイト化合物は、例えば国際公開第2019/208571号公報および特願2018-083148号公報およびPCT/JP2019/017249号公報に記載の以下のスキーム3および4に従って製造することができる。


RaおよびRbは同一または相異なって、メチル基、エチル基又は水素原子を表し、ただし、RaおよびRbが同時に水素原子を表すことはない;
Rcは、メチル基又はエチル基であり、
Baは、保護されていてもよい核酸塩基を表し、
nは、1~5の何れかの整数を表し、
Gは、式中に示す水酸基の保護基を表し
G1およびG2は、それぞれ独立して同一または相異なって、水酸基の保護基を表し、そして、
G3は、同一または相異なってアルキル基を表す。)
次に、スキーム4に示す通り、化合物4を酸化剤の存在下、化合物5(これは、スキーム3に記載する方法に準じて製造する)と反応させて、化合物6(リボースの2’位の水酸基が式(1)で示される保護基で保護されている)を得る。次いで、該化合物6を脱保護して、化合物7を得る。該化合物7の水酸基を保護基G1を用いて選択的に保護して、化合物8を得る。また、該化合物8をホスホロジアミダイト化合物9と反応させて、所望するアミダイト化合物10を製造する。
スキーム3の反応において、
シアン化物イオンとしては、例えば、シアン化ナトリウム、シアン化カリウム、シアン化銅、トリメチルシリルシアニドなどに由来するシアン化物イオンを使用することができる。

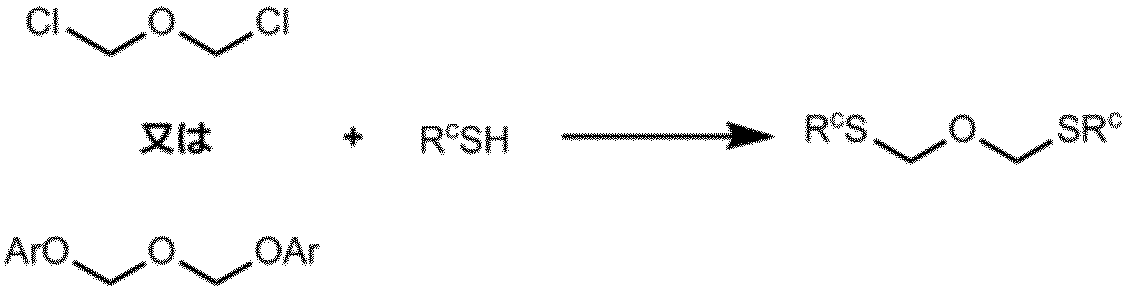
スキーム4の反応において、
工程1(エーテル化工程)
化合物6を製造する工程は、化合物4を化合物5と反応させて実施される。この反応は、通常、酸化剤を添加して実施される。この工程において用いる酸化剤は、特に限定されないが、N-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨードスクシンイミド、ヨウ素、1,3-ジヨード-5、5‘-ジメチルヒダントイン、臭素および塩素からなる群から選択される少なくとも一つであることが好ましい。
前記工程1で得られた化合物6は、脱保護反応に供して化合物7に変換される。脱保護工程は、公知の方法で実施できるが、典型的には、溶媒中、フッ化水素/トリエチルアミンあるいはフッ化水素/ピリジンを作用させ、脱保護することができる。
前記工程で得られた化合物7は、保護工程に供され、保護基の導入は、公知の方法で実施できるが、典型的には、ピリジン中、化合物7に4,4’-ジメトキシトリチルクロリドを反応させて保護基が導入され、化合物8が製造される。
この工程は前記工程で得られた化合物8に、化合物9を反応させることによって実施される。典型的には、ジイソプロピルアンモニウムテトラゾリドの存在下、化合物9として2-シアノエチル-N,N,N’,N’-テトライソプロピルホスホロジアミダイトを反応させて行われ、アミダイト化合物10が製造される。アミダイト化は、特許第5554881号公報の実施例2~5に記載された方法に準じて行うことができる。
非ヌクレオチドリンカーとしては、アミノ酸骨格からなるリンカー(例えば、特許第5157168号公報または特許第5554881号公報に記載されたアミノ酸骨格からなるリンカー)が例示される。具体的には、非限定的な例として、例えば、式(A14-1)、(A14-2)もしくは(A14-3)(例えば、特許第5555346号公報または特許第5876890号公報に記載)で表されるリンカーが例示される。これらのリンカー以外に国際公開第2012/005368号公報、国際公開第2018/182008号公報または国際公開第2019/074110号公報に記載のリンカーが例示される。

切り出し工程は、所望の鎖長の核酸オリゴマーを、切り出し剤として濃アンモニア水を用いて実施した。
本明細書において、「核酸伸長反応」とは、ホスホジエステル結合を介して、ヌクレオチドを順次結合させることにより、オリゴヌクレオチドを伸長させる反応を意味する。核酸伸長反応は、一般的なホスホロアミダイト法の手順に従い行うことができる。核酸伸長反応は、ホスホロアミダイト法を採用する核酸自動合成装置等を用いて行ってもよい。
亜リン酸基をリン酸基に変換する場合には、「酸化剤」として、例えば、ヨウ素、あるいはtert-ブチルヒドロペルオキシドや過酸化水素などの過酸、あるいは(10-カンファースルホニル)オキサジリジン(CSO)を使用することができる。該酸化剤は、0.005~2Mの濃度になるように適当な溶媒で希釈して使用することができる。反応に使用する溶媒としては、反応を妨げないものであればよく、特に限定されないが、ピリジン、THF、水、アセトニトリル、1-メチルイミダゾール(NMI)又はこれらの2つ以上の任意の混合溶媒を挙げることができる。例えば、ヨウ素/水/ピリジン/アセトニトリル、あるいはヨウ素/水/ピリジン、あるいはヨウ素/水/ピリジン/アセトニトリル/NMI、あるいはヨウ素/水/ピリジン/THF、あるいはヨウ素/水/ピリジン/THF/NMI、あるいはCSO/アセトニトリル、あるいはヨウ素/ピリジン-酢酸や過酸(tert-ブチルヒドロペルオキシド/メチレンクロリド)を用いることができる。
例えば、Xiulong, Shenら著、Nucleic Acids Research, 2018, Vol. 46, No.46, 1584-1600、およびDaniel O'Reillyら著、Nucleic Acids Research, 2019, Vol. 47, No.2, 546-558には、様々なヌクレオシドの例が記載されている。
以下、配列の説明中、Uはウリジンを、Cはシチジンを、Aはアデノシンを、またはGはグアノシンを示す。
国際公開第2019/060442号に記載されている、下記の配列(B)および(C)を有する核酸オリゴマーを挙げられる。
配列(B):5’-AUGGAAUmACUCUUGGUUmACdTdT-3’(Antisense)(配列番号3) 21mer
配列(C):5’-GUmAACmCmAAGAGUmAUmUmCmCmAUmdTdT-3’(Sense)(配列番号4) 21mer
配列(B)および(C)中、Umは2'-O-メチルウリジンを、Cmは2'-O-メチルシチジンを、またdTはチミジンを示す。
Daniel O'Reillyら著、Nucleic Acids Research, 2019, Vol. 47, No.2, 546-558頁に記載されている核酸オリゴマー(553頁参照)が挙げられる。典型例として、下記の配列(D)を有する核酸オリゴマーを挙げられる。
配列(D):5’-AGAGCCAGCCUUCUUAUUGUUUUAGAGCUAUGCUGU-3’(配列番号5) 36mer
JP4965745に記載されている核酸オリゴマーが挙げられる。典型例として、下記の配列(E)を有する核酸オリゴマーを挙げられる。
配列(E):5’-CCAUGAGAAGUAUGACAACAGCC-P-GGCUGUUGUCAUACUUCUCAUGGUU-3’ 49mer。CCAUGAGAAGUAUGACAACAGCC(配列番号6)、GGCUGUUGUCAUACUUCUCAUGGUU(配列番号7)。
配列(E)中、”P”は、以下の式(A5)において波線で区切られる部分構造で示される。
Nucleic Acids Research, 2019, Vol. 47, No. 2: 547頁に記載されている、下記の配列(F)を有する核酸オリゴマーを挙げられる。
配列(F):5’-ACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU-3’(配列番号8) 67mer
JP 2015-523856, 173頁に記載されている、下記の配列(G)を有する核酸オリゴマーを挙げられる。
配列(G):5’-GUUUUCCCUUUUCAAAGAAAUCUCCUGGGCACCUAUCUUCUUAGGUGCCCUCCCUUGUUUAAACCUGACCAGUUAACCGGCUGGUUAGGUUUUU-3’(配列番号9) 94mer
JP 2017-537626に記載されている核酸オリゴマーが挙げられる。典型例として、下記の配列(F)、(G)、(H)、(J)を有する核酸オリゴマーを挙げられる。
配列(F):5’-AGUCCUCAUCUCCCUCAAGCGUUUUAGAGCUAGUAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUU-3’(配列番号10) 100mer
配列(G):5’-GCAGAUGUAGUGUUUCCACAGUUUAAGAGCUAUGCUGGAAACAGCAUAGCAAGUUUAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUUUUU-3’(配列番号11) 113mer
配列(H):5’-dAdGdTdCdCdTdCdAdTdCdTdCdCdCdTdCdAdAdGdCGUUUAAGAGCUAUGCUGGUAACAGCAUAGCAAGUUUAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUUUUU -3’(配列番号12) 113mer
配列(H)中、dTはチミジンを、dCは2'-デオキシシチジンを、dAは2'-デオキシアデノシンを、またdGは2'-デオキシグアノシンを示す。
配列(J):5’-AmsGmsUmsCCUCAUCUCCCUCAAGCGUUUAAGAGCUAUGCUGGUAACAGCAUAGCAAGUUUAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUUmsUmsUmsU-3’(配列番号13) 113mer
配列(J)中、Umは2'-O-メチルウリジンを、Amは2'-O-メチルアデノシンを、Gmは2'-O-メチルグアノシンを、またsはホスホロチオエート修飾を示す。
以下の試験で用いた各測定方法を以下に示す。
固相合成後のオリゴヌクレオチド粗生成物の純度の測定は、HPLCにより行った。粗生成物をHPLC(波長260nm、カラムACQUITY UPLC Oligonucleotide BEH C18, 2.1mm×100mm, 1.7μm)によって各成分に分離し、得られたクロマトグラムの総面積値における主生成物の面積値からオリゴヌクレオチドの純度を算出した。

前記粗生成物のOD260を測定した。OD260とは1mL溶液(pH=7.5)における10mm光路長あたりのUV260nmの吸光度を表す。一般的にRNAでは1OD=40μgであることが知られていることから、前記OD260の測定値に基づき、収量を算出した。さらに、固相担体の単位体積当たりの収量を算出した。実施例1~5及び比較例1、2については実施例1の収量に対する相対収量を求めた。
反応系の雰囲気(気相)の酸素濃度はIIJIMA ELECTRONICS CORP.製のPACK KEEPER(Residual Oxygen Meter)を用いて測定した。酸素濃度測定前には空気中及び純窒素中酸素濃度の測定により装置を校正後、装置に付属の針をセプタムなどで蓋をしたフラスコなどの容器に突き刺し、系中気相部分の酸素濃度を測定した。酸素濃度の測定値はリアルタイムで表示され、測定値が安定した所をその雰囲気の酸素濃度とした。
配列(I):5’-AGCAGAGUACACACAGCAUAUACC-P-GGUAUAUGCUGUGUGUACUCUGCUUC-P-G-3’(配列番号1、2) 53mer
前記配列(I)において、”A”は、以下の式(A1)において波線で区切られる部分構造で示される。”C”は、以下の式(A2)において波線で区切られる部分構造で示される。”G”は、以下の式(A3)において波線で区切られる部分構造で示される。Uは、以下の式(A4)において波線で区切られる部分構造で示される。”P”は、以下の式(A5)において波線で区切られる部分構造で示される。なお、5‘末端の”A”は、以下の式(A6)において波線で区切られる部分構造で示される。また、3‘末端の”G”は、以下の式(A7)において波線で区切られる部分構造で示される。但し、構造式中のリン酸基は塩であってもよい。
AGCAGAGUAC ACACAGCAUA UACC(配列番号1)
GGUAUAUGCU GUGUGUACUC UGCUUC(配列番号2)












また、以下の実施例および比較例中に記載するグアノシン誘導体とは、下記の構造式で示される化合物を意味する。下記構造式において図示されたサークルは、CPGを模式的に示すものである。

78.20μmolのグアノシン誘導体を担持したCPGと、式(A8)、式(A9)、式(A10)、式(A11)、または式(A12)に示すアミダイトとを用いて、配列(I)の固相合成をAKTA oligopilot plus100により実施した。その後、16.46μmol分のオリゴヌクレオチドを担持したCPG担体を採取し、アンモニア水を用いてオリゴヌクレオチドを固相担体から遊離させた。次いで遊離オリゴヌクレオチドを6.6mLのジメチルスルホキシドに溶解後に、1.97μmol分の溶液を容量100mL、口径29mmナス型フラスコに採取し、更にそこにニトロメタン10.6μLと直径15mmの撹拌子を入れた後、口径29mmのセプタムにて蓋をして密閉した。更に系内に窒素ボンベから窒素を吹き付ける針と、吹き付けた窒素を抜くための針、更にOxygen Meterの測定用針をセプタムに突き刺し、窒素をフローすることで系内を窒素に置換し、気相中の酸素濃度を0%とした。ここで、気相中の酸素濃度は、前記測定方法3に記載の方法を用いて測定した。更にモレキュラーシーブ4Aにて脱水処理を施した1Mのフッ化テトラ-n-ブチルアンモニウム(TBAF)のジメチルスルホキシド溶液1.10mL(TBAFの量は保護基1モル当たり10.2モル)をスターラーによる攪拌下33℃でオリゴヌクレオシド溶液表面にKDScientific社のシリンジポンプを用いて1時間かけて滴下し、混合物を4時間保温することで2’-PMM保護基の脱保護を行った。粗生成物は沈殿操作により得た。収量は14.1mg、純度は62%であった。得られた粗生成物について、前記測定方法1に記載の方法を用いて、オリゴヌクレオチドの純度を測定し、また、前記測定方法2に記載の方法を用いて、オリゴヌクレオチドの収量を測定した。
78.20μmolのグアノシン誘導体を担持したCPGと、式(A8)、式(A9)、式(A10)、式(A11)、または式(A12)に示すアミダイトとを用いて、配列(I)の固相合成をAKTA oligopilot plus100により実施した。その後、19.91μmol分のオリゴヌクレオチドを担持したCPG担体を採取し、アンモニア水を用いてオリゴヌクレオチドを固相担体から遊離させた。次いで遊離オリゴヌクレオシドを8.0mLのジメチルスルホキシドに溶解後に、3.03μmol分の溶液を容量100mL、口径29mmナス型フラスコに採取し、更にそこにニトロメタン15.9μLと直径15mmの撹拌子を入れた後、口径29mmのセプタムにて蓋をして密閉した。更に系内に窒素ボンベから窒素を吹き付ける針と、吹き付けた窒素を抜くための針、更にOxygen Meterの測定用針をセプタムに突き刺し、窒素をフローすることで系内を窒素に置換し、気相中の酸素濃度を0%とした。気相中の酸素濃度は、実施例1と同様に測定することにより確認した。更にモレキュラーシーブ4Aにて脱水処理を施した1Mのフッ化テトラ-n-ブチルアンモニウム(TBAF)のジメチルスルホキシド溶液1.66mL(TBAFの量は保護基1モル当たり10.0モル)をスターラーによる攪拌下33℃でオリゴヌクレオシド溶液表面にシリンジを用いて1分以内に流入し、混合物を4時間保温することで2’-PMM保護基の脱保護を行った。粗生成物は沈殿操作により得た。収量は21.7mg、純度は60%であった。得られた粗生成物について、実施例1と同様に、オリゴヌクレオチドの純度および収量を測定した。
78.20μmolのグアノシン誘導体を担持したCPGと、式(A8)、式(A9)、式(A10)、式(A11)、または式(A12)に示すアミダイトとを用いて、配列(I)の固相合成をAKTA oligopilot plus100により実施した。その後、16.46μmol分のオリゴヌクレオチドを担持したCPG担体を採取し、アンモニア水を用いてオリゴヌクレオチドを固相担体から遊離させた。次いで遊離オリゴヌクレオシドを6.6mLのジメチルスルホキシドに溶解後に、1.00μmol分の溶液を容量100mL口径29mmナス型フラスコに採取し、更にそこにニトロメタン5.3μLと直径15mmの撹拌子を入れた後、口径29mmのセプタムにて蓋をして密閉した。更に系内に窒素ボンベから窒素を吹き付ける針と、吹き付けた窒素を抜くための針、更にOxygen Meterの測定用針をセプタムに突き刺し、窒素をフローすることで系内を窒素に置換し、気相中の酸素濃度を5%とした。気相中の酸素濃度は、実施例1と同様に測定することにより確認した。更にモレキュラーシーブ4Aにて脱水処理を施した1Mのフッ化テトラ-n-ブチルアンモニウム(TBAF)のジメチルスルホキシド溶液0.56mL(TBAFの量は保護基1モル当たり10.2モル)をスターラーによる攪拌下33℃でオリゴヌクレオシド溶液表面にシリンジを用いて1分以内に流入し、混合物を4時間保温することで2’-PMM保護基の脱保護を行った。粗生成物は沈殿操作により得た。収量は7.1mg、純度は60%であった。得られた粗生成物について、実施例1と同様に、オリゴヌクレオチドの純度および収量を測定した。
78.20μmolのグアノシン誘導体を担持したCPGと、式(A8)、式(A9)、式(A10)、式(A11)、または式(A12)に示すアミダイトとを用いて、配列(I)の固相合成をAKTA oligopilot plus100により実施した。その後、16.46μmol分のオリゴヌクレオチドを担持したCPG担体を採取し、アンモニア水を用いてオリゴヌクレオチドを固相担体から遊離させた。次いで遊離オリゴヌクレオシドを6.6mLのジメチルスルホキシドに溶解後に、0.98μmol分の溶液を容量100mL口径29mmナス型フラスコに採取し、更にそこにニトロメタン5.3μLと直径15mmの撹拌子を入れた後、口径29mmのセプタムにて蓋をして密閉した。更に系内に窒素ボンベから窒素を吹き付ける針と、吹き付けた窒素を抜くための針、更にOxygen Meterの測定用針をセプタムに突き刺し、窒素をフローすることで系内を窒素に置換し、気相中の酸素濃度を10%とした。気相中の酸素濃度は、実施例1と同様に測定することにより確認した。更にモレキュラーシーブ4Aにて脱水処理を施した1Mのフッ化テトラ-n-ブチルアンモニウム(TBAF)のジメチルスルホキシド溶液0.56mL(TBAFの量は保護基1モル当たり10.4モル)をスターラーによる攪拌下33℃でオリゴヌクレオシド溶液表面にシリンジを用いて1分以内に流入し、混合物を4時間保温することで2’-PMM保護基の脱保護を行った。粗生成物は沈殿操作により得た。収量は6.9mg、純度は54%であった。得られた粗生成物について、実施例1と同様に、オリゴヌクレオチドの純度および収量を測定した。
78.20μmolのグアノシン誘導体を担持したCPGと、式(A8)、式(A9)、式(A10)、式(A11)、または式(A12)に示すアミダイトとを用いて、配列(I)の固相合成をAKTA oligopilot plus100により実施した。その後、16.46μmol分のオリゴヌクレオチドを担持したCPG担体を採取し、アンモニア水を用いてオリゴヌクレオチドを固相担体から遊離させた。次いで遊離オリゴヌクレオシドを6.6mLのジメチルスルホキシドに溶解後に、1.00μmol分の溶液を容量100mL口径29mmナス型フラスコに採取し、更にそこにニトロメタン5.3μLと直径15mmの撹拌子を入れた後、口径29mmのセプタムにて蓋をして密閉した。更に系内に窒素ボンベから窒素を吹き付ける針と、吹き付けた窒素を抜くための針、更にOxygen Meterの測定用針をセプタムに突き刺し、窒素をフローすることで系内を窒素に置換し、気相中の酸素濃度を15%とした。気相中の酸素濃度は、実施例1と同様に測定することにより確認した。更にモレキュラーシーブ4Aにて脱水処理を施した1Mのフッ化テトラ-n-ブチルアンモニウム(TBAF)のジメチルスルホキシド溶液0.56mL(TBAFの量は保護基1モル当たり10.2モル)をスターラーによる攪拌下33℃でオリゴヌクレオシド溶液表面にシリンジを用いて1分以内に流入し、混合物を4時間保温することで2’-PMM保護基の脱保護を行った。粗生成物は沈殿操作により得た。収量は7.1mg、純度は46%であった。得られた粗生成物について、実施例1と同様に、オリゴヌクレオチドの純度および収量を測定した。
78.20μmolのグアノシン誘導体を担持したCPGと、式(A8)、式(A9)、式(A10)、式(A11)、または式(A12)に示すアミダイトとを用いて、配列(I)の固相合成をAKTA oligopilot plus100により実施した。その後、19.91μmol分を採取し、アンモニア水を用いてオリゴヌクレオチドを固相担体から遊離させた。次いで遊離オリゴヌクレオシドを8.0mLのジメチルスルホキシドに溶解後に、1.01μmol分を容量100mL口径29mmナス型フラスコに採取し、更にそこにニトロメタン5.3μLと直径15mmの撹拌子を入れた後、口径29mmのセプタムにて蓋をして密閉した。Oxygen Meterの測定用針をセプタムに突き刺し、系内気相中の酸素濃度を実施例1と同様にして測定した所、酸素濃度は21%であった。更にモレキュラーシーブ4Aにて脱水処理を施した1Mのフッ化テトラ-n-ブチルアンモニウム(TBAF)のジメチルスルホキシド溶液0.56mL(TBAFの量は保護基1モル当たり10.1モル)をスターラーによる攪拌下33℃でオリゴヌクレオシド溶液表面にシリンジを用いて1分以内に流入し、混合物を4時間保温することで2’-PMM保護基の脱保護を行った。粗生成物は沈殿操作により得た。収量は7.2mg、純度は41%であった。得られた粗生成物について、実施例1と同様に、オリゴヌクレオチドの純度および収量を測定した。
78.20μmolのグアノシン誘導体を担持したCPGと、式(A8)、式(A9)、式(A10)、式(A11)、または式(A12)に示すアミダイトとを用いて、配列(I)の固相合成をAKTA oligopilot plus100により実施した。その後、16.46μmol分のオリゴヌクレオチドを担持したCPG担体を採取し、アンモニア水を用いてオリゴヌクレオチドを固相担体から遊離させた。次いで遊離オリゴヌクレオシドを6.6mLのジメチルスルホキシドに溶解後に、1.96μmol分の溶液を容量100mL口径29mmナス型フラスコに採取し、更にそこにニトロメタン10.6μLと直径15mmの撹拌子を入れた後、口径29mmのセプタムにて蓋をして密閉した。Oxygen Meterの測定用針をセプタムに突き刺し、系内気相中の酸素濃度を実施例1と同様にして測定した所、酸素濃度は21%であった。更にモレキュラーシーブ4Aにて脱水処理を施した1Mのフッ化テトラ-n-ブチルアンモニウム(TBAF)のジメチルスルホキシド溶液1.10mL(TBAFの量は保護基1モル当たり10.2モル)をスターラーによる攪拌下33℃でオリゴヌクレオシド溶液表面にKDScientific社のシリンジポンプを用いて1時間かけて滴下し、混合物を4時間保温することで2’-PMM保護基の脱保護を行った。粗生成物は沈殿操作により得た。収量は14.2mg、純度は47%であった。得られた粗生成物について、実施例1と同様に、オリゴヌクレオチドの純度および収量を測定した。

PMMアミダイトの製造
1)3-ヒドロキシブタンニトリルの製造



1H-NMR (CDCl3): δ 4.93-4.84 (m, 2H) 4.75(s, 2H) 4.06-4.00 (m, 1H) 2.57 (t, 2H), 2.17(s, 3H) 1.35 (d, 3H)

核酸塩基部分がウラシルであるPMMアミダイトUの製造例を以下に示す。ウラシル以外の核酸塩基を有するPMMアミダイトについても同様の方法で製造することができる。




Claims (19)
- 酸素濃度が15%以下の不活性ガス雰囲気下で、式(3):

G4は、水素原子または水酸基の保護基を表し、保護基を表す場合は以下の基を表し、
(式中、R 1 、R 2 及びR 3 は同一又は相異なって水素又はアルコキシ基を表す。)
G5は、アンモニウムイオン、アルキルアンモニウムイオン、アルカリ金属イオン、水素イオンまたはヒドロキシアルキルアンモニウムイオンを表し、
BCは、それぞれ独立して同一又は相異なる核酸塩基を表し、
Rは、それぞれ独立して同一又は相異なって、水素原子、フッ素原子またはOQ基を表し、
Qは、それぞれ独立して同一又は相異なって、tert-ブチルジメチルシリル基、メチル基、2-メトキシエチル基、リボースの4’位の炭素原子と結合しているメチレン基、リボースの4’位の炭素原子と結合しているエチレン基、リボースの4’位の炭素原子と結合しているエチリデン基、または式(1):

RaおよびRbは同一又は相異なって、メチル基、エチル基または水素原子を表し、
*印のついた結合は、OQ基の酸素原子との結合であることを表し、
nは1~5の何れかの整数を表す。
ただしRaおよびRbが同時に水素原子を表すことはない。)
の保護基を表し、
Yは、それぞれ独立して同一又は相異なって、酸素原子または硫黄原子を表し、
mは、2以上200までの何れかの整数を表し、
WおよびXは、下記の(a)または(b)のいずれかで定義され、
(a)Wが水酸基であるときは、Xは前記R基と同じ定義である。
(b)Xが水酸基であるときは、WはOV基を表し、
Vは、tert-ブチルジメチルシリル基、または前記式(1)の基を表す。
ただし、前記R、WおよびXのうち少なくとも一つの基は、前記式(1)の保護基で保護された水酸基を表す。そして
mが3以上の整数のとき、式(3)で示される核酸オリゴマーは、それぞれの5’末端と3’末端のヌクレオチドの間のp個(ただし、pは、式:m-1>pを満たす正の整数である。)のヌクレオチドの代わりに、非ヌクレオチドリンカーが組み込まれていてもよい核酸オリゴマーである。)
で示される核酸オリゴマーを、フッ化物イオンと接触させることを特徴とする、式(4):

R’は、それぞれ独立して同一又は相異なって、水酸基、水素原子、フッ素原子、メトキシ基、2-メトキシエチル基、またはOQ’基を表し、
Q’は、それぞれ独立して同一又は相異なって、リボースの4’位の炭素原子と結合しているメチレン基、4’位の炭素原子と結合しているエチレン基、または4’位の炭素原子と結合しているエチリデン基を表し、
式(4)の置換基G4、G5、Y、Bcおよびmの定義は、前記式(3)における定義と同じであり、
W0は、水酸基であり、
X0は、前記R’基と同じ定義である。
そして、mが3以上の整数のとき、式(4)で示される核酸オリゴマーは、それぞれの5’末端と3’末端のヌクレオチドの間のp個(ただし、pは、式:m-1>pを満たす正の整数である。)のヌクレオチドの代わりに、非ヌクレオチドリンカーが組み込まれていてもよい核酸オリゴマーである。)
で示される核酸オリゴマーの製造方法であって、
該フッ化物イオンのフッ化物イオン源が、フッ化テトラアルキルアンモニウムであり、
該非ヌクレオチドリンカーが、アミノ酸骨格からなるリンカーであり、
該アミノ酸骨格からなるリンカーが、下記式(A14-1)、式(A14-2)または式(A14-3)の構造を有するリンカーである、方法。
(式中、5’および3’は、核酸オリゴマーの5’末端側および3’末端側をそれぞれ示す。)
- Wが水酸基であり、XがR基であり、W0が水酸基であり、そしてX0がR’基である、請求項1に記載の製造方法。
- フッ化テトラアルキルアンモニウムが、フッ化テトラ-n-ブチルアンモニウム(TBAF)である、請求項1または2に記載の製造方法。
- 酸素濃度が10%以下の不活性ガス雰囲気下で反応を行う、請求項1~3の何れか1項に記載の製造方法。
- 酸素濃度が5%以下の不活性ガス雰囲気下で反応を行う、請求項1~3の何れか1項に記載の製造方法。
- 酸素濃度が0%の不活性ガス雰囲気下で反応を行う、請求項1~3の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーにフッ化物イオンの全量を添加するために要する時間が30分以上である、請求項1~6の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が5%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が10%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が20%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が30%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が40%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が50%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が60%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が70%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が80%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が90%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が95%以上である、請求項1~7の何れか1項に記載の製造方法。
- 式(3)で示される核酸オリゴマーのR、WおよびXのうち、前記式(1)の保護基の割合が100%である、請求項1~7の何れか1項に記載の製造方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019187930 | 2019-10-11 | ||
| JP2019187930 | 2019-10-11 | ||
| PCT/JP2020/032020 WO2021070494A1 (ja) | 2019-10-11 | 2020-08-25 | 核酸オリゴマーの製造方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JPWO2021070494A1 JPWO2021070494A1 (ja) | 2021-04-15 |
| JPWO2021070494A5 JPWO2021070494A5 (ja) | 2023-08-04 |
| JP7640461B2 true JP7640461B2 (ja) | 2025-03-05 |
Family
ID=75437077
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021550425A Active JP7640461B2 (ja) | 2019-10-11 | 2020-08-25 | 核酸オリゴマーの製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US12509483B2 (ja) |
| EP (1) | EP4043473A4 (ja) |
| JP (1) | JP7640461B2 (ja) |
| KR (1) | KR20220079832A (ja) |
| CN (1) | CN114502566B (ja) |
| WO (1) | WO2021070494A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022064908A1 (ja) * | 2020-09-24 | 2022-03-31 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20230312635A1 (en) * | 2020-01-29 | 2023-10-05 | Sumitomo Chemical Company, Limited | Method for producing nucleic acid oligomer |
| CN118460647B (zh) * | 2024-07-10 | 2025-01-28 | 凯莱英医药集团(天津)股份有限公司 | 一种Patisiran的制备方法 |
| WO2026048956A1 (ja) * | 2024-08-29 | 2026-03-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2026071145A1 (ja) * | 2024-09-30 | 2026-04-02 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007099896A1 (ja) | 2006-02-27 | 2007-09-07 | Nippon Shinyaku Co., Ltd. | 核酸保護基の脱離方法 |
| WO2011034072A1 (ja) | 2009-09-16 | 2011-03-24 | 株式会社キラルジェン | Rna及びその誘導体合成のための新規保護基 |
| WO2013027843A1 (ja) | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| WO2018070543A1 (ja) | 2016-10-14 | 2018-04-19 | 株式会社ボナック | 新規な配糖体化合物及びその製造方法 |
| WO2019208571A1 (ja) | 2018-04-24 | 2019-10-31 | 住友化学株式会社 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6077709A (en) | 1998-09-29 | 2000-06-20 | Isis Pharmaceuticals Inc. | Antisense modulation of Survivin expression |
| DK1149109T3 (da) | 1999-02-05 | 2010-01-11 | Ge Healthcare Bio Sciences | Fremgangsmåde til afbeskyttelse af oligonukleotider |
| US6261840B1 (en) | 2000-01-18 | 2001-07-17 | Isis Pharmaceuticals, Inc. | Antisense modulation of PTP1B expression |
| WO2005007825A2 (en) * | 2003-07-12 | 2005-01-27 | Isis Pharmaceuticals, Inc. | Modulation of aminopeptidase n expression |
| ES2386709T3 (es) | 2004-08-26 | 2012-08-27 | Nippon Shinyaku Co., Ltd. | Compuesto de fosforamidita y método para producir un oligo-ARN |
| WO2008049085A1 (en) | 2006-10-18 | 2008-04-24 | Isis Pharmaceuticals, Inc. | Antisense compounds |
| HUE037500T2 (hu) | 2010-07-08 | 2018-08-28 | Bonac Corp | Egyszálú nukleinsav molekula a génexpresszió szabályozására |
| ES2527660T3 (es) | 2010-08-03 | 2015-01-28 | Bonac Corporation | Molécula de ARN monocatenario que tiene esqueleto alicíclico que contiene nitrógeno |
| EP3241902B1 (en) | 2012-05-25 | 2018-02-28 | The Regents of The University of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| JP6459852B2 (ja) | 2014-08-29 | 2019-01-30 | 住友化学株式会社 | エーテル化合物の製造方法 |
| US10900034B2 (en) | 2014-12-03 | 2021-01-26 | Agilent Technologies, Inc. | Guide RNA with chemical modifications |
| JP6787398B2 (ja) | 2016-04-26 | 2020-11-18 | 住友化学株式会社 | 一本鎖核酸分子用モノマーの製造方法 |
| JP6803730B2 (ja) | 2016-11-22 | 2020-12-23 | 旭サナック株式会社 | 静電塗装装置 |
| EP3604528A4 (en) | 2017-03-31 | 2021-01-13 | Bonac Corporation | CYCLIC NUCLEIC ACID MOLECULE WITH GENE EXPRESSION CONTROL FUNCTION |
| KR20250046363A (ko) | 2017-09-19 | 2025-04-02 | 알닐람 파마슈티칼스 인코포레이티드 | 트랜스타이레틴(ttr) 매개 아밀로이드증을 치료하기 위한 조성물 및 방법 |
| US20210188895A1 (en) | 2017-10-13 | 2021-06-24 | Bonac Corporation | Single-stranded nucleic acid molecule, and production method therefor |
| JP7207682B2 (ja) | 2018-04-26 | 2023-01-18 | 株式会社サンセイアールアンドディ | 遊技機 |
| EP4114384A1 (en) | 2020-03-06 | 2023-01-11 | Pfizer Inc. | Methods of inhibiting sars-cov-2 replication and treating coronavirus disease 2019 |
| JP7719788B2 (ja) * | 2020-09-24 | 2025-08-06 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
-
2020
- 2020-08-25 US US17/767,825 patent/US12509483B2/en active Active
- 2020-08-25 CN CN202080070607.0A patent/CN114502566B/zh active Active
- 2020-08-25 WO PCT/JP2020/032020 patent/WO2021070494A1/ja not_active Ceased
- 2020-08-25 JP JP2021550425A patent/JP7640461B2/ja active Active
- 2020-08-25 EP EP20875506.6A patent/EP4043473A4/en active Pending
- 2020-08-25 KR KR1020227010696A patent/KR20220079832A/ko active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007099896A1 (ja) | 2006-02-27 | 2007-09-07 | Nippon Shinyaku Co., Ltd. | 核酸保護基の脱離方法 |
| WO2011034072A1 (ja) | 2009-09-16 | 2011-03-24 | 株式会社キラルジェン | Rna及びその誘導体合成のための新規保護基 |
| WO2013027843A1 (ja) | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| WO2018070543A1 (ja) | 2016-10-14 | 2018-04-19 | 株式会社ボナック | 新規な配糖体化合物及びその製造方法 |
| WO2019208571A1 (ja) | 2018-04-24 | 2019-10-31 | 住友化学株式会社 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| OHGI,T. et al.,Synthetic Method with a 2'-O-(2-Cyanoethoxymethyl) Protecting Group,Organic Letters,2005年,Vol.7, No.16,pp.3477-3480,DOI 10.1021/ol051151f |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022064908A1 (ja) * | 2020-09-24 | 2022-03-31 | ||
| JP7719788B2 (ja) | 2020-09-24 | 2025-08-06 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20220079832A (ko) | 2022-06-14 |
| EP4043473A4 (en) | 2024-03-27 |
| JPWO2021070494A1 (ja) | 2021-04-15 |
| EP4043473A1 (en) | 2022-08-17 |
| US12509483B2 (en) | 2025-12-30 |
| WO2021070494A1 (ja) | 2021-04-15 |
| US20240116977A1 (en) | 2024-04-11 |
| CN114502566A (zh) | 2022-05-13 |
| CN114502566B (zh) | 2025-02-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7640461B2 (ja) | 核酸オリゴマーの製造方法 | |
| JP7719788B2 (ja) | 核酸オリゴマーの製造方法 | |
| JP7777452B2 (ja) | 核酸オリゴマーの製造方法 | |
| IL108467A (en) | Modified oligodeoxyribo-nucleotides their preparation and their therapeutic use | |
| JP7759323B2 (ja) | 核酸オリゴマーの製造方法 | |
| JP7698631B2 (ja) | 核酸オリゴマーの製造方法 | |
| ES2981209T3 (es) | Proceso de preparación de oligómero de ácido nucleico | |
| WO2024019137A1 (ja) | オリゴヌクレオチドの製造方法 | |
| JP5168145B2 (ja) | 核酸保護基の導入方法 | |
| WO2024024873A1 (ja) | チオ化溶液 | |
| WO2023054350A1 (ja) | 精製ジクロロ酢酸の製造方法 | |
| JP7825064B2 (ja) | オリゴヌクレオチドの製造方法 | |
| WO2025229953A1 (ja) | オリゴヌクレオチドの製造方法 | |
| WO2024143276A1 (ja) | オリゴヌクレオチドの製造方法 | |
| WO2026071145A1 (ja) | 核酸オリゴマーの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230727 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230727 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20241001 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20241118 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20250218 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20250220 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7640461 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |