WO2007110171A1 - Substituierte n-benzyl-n-phenylbenzolsulfonamide zur behandlung von virusinfektionen - Google Patents
Substituierte n-benzyl-n-phenylbenzolsulfonamide zur behandlung von virusinfektionen Download PDFInfo
- Publication number
- WO2007110171A1 WO2007110171A1 PCT/EP2007/002441 EP2007002441W WO2007110171A1 WO 2007110171 A1 WO2007110171 A1 WO 2007110171A1 EP 2007002441 W EP2007002441 W EP 2007002441W WO 2007110171 A1 WO2007110171 A1 WO 2007110171A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- halogen
- cyano
- alkyl
- substituents
- trifluoromethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(c1cc(N)cc(*)c1*)=O Chemical compound *C(c1cc(N)cc(*)c1*)=O 0.000 description 1
- UENPPHAHQSTSQV-UHFFFAOYSA-N CNc1cccc(S(N(Cc(c(Cl)ccc2)c2Cl)c2cccc(C(O)=O)c2)(=O)=O)c1 Chemical compound CNc1cccc(S(N(Cc(c(Cl)ccc2)c2Cl)c2cccc(C(O)=O)c2)(=O)=O)c1 UENPPHAHQSTSQV-UHFFFAOYSA-N 0.000 description 1
- IURZBWKESDIYEC-UHFFFAOYSA-N COc(ccc(N(Cc(c(Cl)ccc1)c1Cl)S(c1cc(C#N)ccc1)(=O)=O)c1)c1C(O)=O Chemical compound COc(ccc(N(Cc(c(Cl)ccc1)c1Cl)S(c1cc(C#N)ccc1)(=O)=O)c1)c1C(O)=O IURZBWKESDIYEC-UHFFFAOYSA-N 0.000 description 1
- QBEOMAFEHSMKFX-UHFFFAOYSA-N Cc1cc(S(N(Cc(c(Cl)ccc2)c2Cl)c2cc(C(OC)=O)ccc2)(=O)=O)cc(CO)c1 Chemical compound Cc1cc(S(N(Cc(c(Cl)ccc2)c2Cl)c2cc(C(OC)=O)ccc2)(=O)=O)cc(CO)c1 QBEOMAFEHSMKFX-UHFFFAOYSA-N 0.000 description 1
- RILMIWZUBANRNS-UHFFFAOYSA-N N#Cc1cccc(S(N(Cc(c(Cl)ccc2)c2Cl)c2cccc(C(NC3CCC3)=O)c2)(=O)=O)c1 Chemical compound N#Cc1cccc(S(N(Cc(c(Cl)ccc2)c2Cl)c2cccc(C(NC3CCC3)=O)c2)(=O)=O)c1 RILMIWZUBANRNS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/235—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group
- A61K31/24—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group having an amino or nitro group
- A61K31/245—Amino benzoic acid types, e.g. procaine, novocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/275—Nitriles; Isonitriles
- A61K31/277—Nitriles; Isonitriles having a ring, e.g. verapamil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/365—Lactones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system having sulfur as a ring hetero atom, e.g. ticlopidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
- A61K31/606—Salicylic acid; Derivatives thereof having amino groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/21—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/82—Benzo [b] furans; Hydrogenated benzo [b] furans with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
- C07D307/84—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D307/85—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 2
Definitions
- the invention relates to substituted N-benzyl-N-phenylbenzenesulfonamides and processes for their preparation, their use for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of diseases, in particular for use as antiviral agents, especially against hepatitis C viruses.
- HCV hepatitis C virus
- Interferon-alpha IFN- ⁇
- EFTSI interferon
- Interferon therapy with a long treatment duration of at least six months is very often associated with serious side effects (eg, leukopenia, thrombocytopenia, retinopathy, thyroiditis, acute pancreatitis, depression), which severely limit the patients' quality of life.
- side effects eg, leukopenia, thrombocytopenia, retinopathy, thyroiditis, acute pancreatitis, depression
- ribavirin is approved.
- This combination therapy results in improved efficacy, but does not improve the IFN-associated side-effect profile, and it also associates with ribavirin side effects (eg, hemolytic anemia).
- pegylated forms of IFN such as PEG-Intron ® and Pegasys ® can be at least partially mitigated these undesirable side effects.
- the hepatitis C virus is the only member of the genus Hepacivirus within the family Flaviviridae. There are at least 6 genotypes and a variety of subtypes. The virus is surrounded by a shell and has as a genome a positive single strand of viral RNA. The length of the viral RNA genome is approximately 9500 nucleotides. Propagation of the viral genome and translation into protein is mediated by RNA structures located at the beginning and end of the genome (5 'untranslated region, 3' untranslated region). The genome has a single reading frame (ORF) encoding a polyprotein (about 3000 amino acids).
- ORF single reading frame
- HCV structural and non-structural proteins
- HCV encodes a capsid protein (c) and two envelope proteins (El and E2).
- a small protein (p7) could be a so-called viroporin, which is essential for the infectivity of the mature virus particle.
- the mature NS proteins include the NS2, NS3, NS4A, NS4B, NS5A and NS5B proteins. For their cleavage from the polyprotein two viral proteases are responsible.
- the poorly characterized NS2 / 3 protease is likely a cysteine protease that cleaves the NS2 NS3 site.
- the second protease (NS3 / 4A protease) is a serine protease whose catalytic activity is contained in the N-terminal part of the multifunctional NS3 protein and which requires the small NS4A protein as a cofactor. It catalyzes all proteolytic cleavages down the NS3 amino acid sequence, ie NS3 NS4A proteolysis as well as the NS4A NS4B, NS4B NS5A and NS5A NS5B digests.
- the NS4A protein may also have a role in membrane localization of NS3 and other NS proteins.
- NS3 and NS4A The complex formation between NS3 and NS4A is presumably a prerequisite for protein processing and increases the proteolytic activities in relation to all interfaces.
- the NS3 protein also has NTPase and helicase activity located in the C-terminal domain.
- NS5B is an RNA-dependent RNA polymerase that plays a key role in HCV replication. Very little is known about the functions of the NS4B and NS5A proteins. However, NS5A involvement in clinical resistance to interferon is discussed.
- Hepatitis C closely related viruses such as the GBV B virus, which infects New World monkeys, or the BVDV (Bovine Viral Diarrhea Virus) are often used as model viruses to study certain aspects of the virus life cycle.
- GBV B virus which infects New World monkeys
- BVDV Bovine Viral Diarrhea Virus
- Structurally similar compounds are described for example in WO 01/56989 for the treatment and / or prophylaxis of thrombosis and in WO 2004/026823 are described as estrogen receptor ligands.
- the invention relates to the use of the compounds of the formula
- R 1 is hydroxy or -NR y R lü , in which
- R 9 and R 10 independently of one another represent hydrogen, (C 1 -C 6 -alkyl, benzyl, 2-phenylethyl or (C 3 -C 6 ) -cycloalkyl,
- benzyl and 2-phenylethyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from
- R 2 is hydrogen, halogen, cyano, hydroxyl, amino, trifluoromethyl, trifluoromethoxy, (C r C 4 ) alkyl, (Ci-C 4 ) alkoxy, (C] -C 4 ) alkylamino or a bonded via a nitrogen atom 5- to 7-membered heterocyclyl,
- alkyl, alkoxy and alkylamino may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl and trifluoromethoxy,
- heterocyclyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C, -C 4 ) alkyl, (C r C 4) -alkoxy and (C r C 4) alkylamino,
- R 1 and R 2 are linked via a * -O-CH 2 - # chain and form a ring,
- R 3 is hydrogen, halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C 1 - C 4) alkyl, (Ci-C 4) alkoxy, (C 1 -C 4) alkylamino or via a nitrogen atom bound 5- to 7-membered heterocyclyl, where alkyl, alkoxy and alkylamino may be substituted by 1 to 3 substituents, the substituents being selected independently of one another from the group consisting of halogen, cyano, hydroxyl, amino, trifluoromethyl and trifluoromethoxy,
- heterocyclyl can be substituted with 1 to 3 substituents, whereby the substituents are independently selected oxy from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, Trifluormeth-, (C r C4) alkyl, (Ci- C 4 ) alkoxy and (C 1 -C 4 ) -alkylamino,
- R 4 is halogen, cyano, methyl, ethyl or cyclopropyl
- R 5 is hydrogen, (C, -C 4) alkyl, (C 2 -C 4) alkenyl, (C r C4) alkoxy, (C r C4) alkylthio, (C 1 - C 4) Alkylamino, 5- to 10-membered heteroaryl or R 1 '-Y-CH 2 -,
- heteroaryl may be substituted by 1 to 3 substituents, the substituents being independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl,
- alkyl, alkenyl, alkoxy, alkylthio and alkylamino may be substituted with a substituent, wherein the substituent is selected from the group consisting of phenyl and 5- to 10-membered heteroaryl,
- phenyl and heteroaryl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl, aminosulfonyl, (C r C 4 ) alkyl , (C r C 4) alkoxy and (C r C 4) alkylamino,
- Y is an oxygen atom, a sulfur atom or -N (R 12 ) -,
- R 12 is hydrogen, (C 1 -C 4 ) -alkyl or (C 3 -C 6 ) -cycloalkyl
- R 1 ' is 5- to 10-membered heteroaryl
- heteroaryl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, Trifluormeth- oxy, aminocarbonyl, aminosulfonyl, (C r C 4 ) alkyl , (Ci-C 4) -alkoxy and (C 1 -C 4) - alkylamino,
- R 6 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl, trifluoromethoxy or 2-cyanoethen-1-yl,
- R 7 is hydrogen, (C r C4) alkyl, (C 2 -C 4) alkenyl, (C r C4) alkoxy, (C, -C 4) -alkylthio or (Ci-C 4) - Alkylamino stands,
- alkyl, alkenyl, alkoxy, alkylthio and alkylamino may be substituted with a substituent, wherein the substituent is selected from the group consisting of phenyl and 5- to 10-membered heteroaryl,
- phenyl and heteroaryl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl, aminosulfonyl, (Ci-C 4 ) alkyl , (C r C 4) alkoxy and (C r C 4) alkylamino,
- R 8 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl or trifluoromethoxy
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts, compounds mentioned below as examples (e) and their salts, solvates and solvates of the salts, as far as those of the formula (I) are concerned,
- the compounds mentioned below are not already salts, solvates and solvates of the salts.
- the compounds of the invention may exist in stereoisomeric forms (enantiomers, diastereomers).
- the invention therefore relates to the Enan- tiomeren or diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner.
- the present invention encompasses all tautomeric forms.
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. However, also included are salts which are not suitable for pharmaceutical applications themselves but can be used, for example, for the isolation or purification of the compounds according to the invention.
- Physiologically acceptable salts of the compounds of the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid, acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid acetic acid, trifluoroacetic acid, propionic acid
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having from 1 to 16 carbon atoms, for example and preferably, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salt
- solvates are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water.
- Alkylthio and alkylamino stand for a linear or branched alkyl radical with generally 1 to 6 ("C 1 -C 6 -alkyl”), preferably 1 to 4, particularly preferably 1 to 3 carbon atoms, by way of example and preferably methyl, ethyl, n-butyl Propyl, isopropyl, tert -butyl, n -pentyl and n -hexyl.
- Alkoxy is exemplified and preferably methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, tert-butoxy, n-pentoxy and n-hexoxy.
- Alkylthio is exemplified and preferably methyltbio, ethyltbio, n-propylthio, isopropylthio, n-butylthio, tert-butylthio, n-pentylthio and n-hexylthio.
- Alkylamino represents an alkylamino radical having one or two (independently selected) alkyl substituents, by way of example and by way of preference methylamino, ethylamino, n-propylamino, isopropylamino, tert-butylamino, n-pentylamino, n-hexylamino, N, N-dimethylamino , N, N-diethylamino, N-ethyl-N-methylamino, N-methyl-Nn-propylamino, N-isopropyl-N-n-propylamino, N-tert-butyl-N-methylamino, N-ethyl-Nn pentylamino and Nn-hexyl-N-methylamino.
- C 1 -C 3 -alkylamino is, for example, a monoalkylamino radical having 1 to 3 carbon atoms or
- Alkenyl represents a straight-chain or branched alkenyl radical having 2 to 6 carbon atoms. Preference is given to a straight-chain or branched alkenyl radical having 2 to 4, particularly preferably 2 to 3, carbon atoms. Examples which may be mentioned are: vinyl, allyl, n-prop-1-en-1-yl and n-but-2-en-1-yl.
- Cycloalkyl represents a cycloalkyl group having usually 3 to 6, preferably 3 to 5 carbon atoms. Exemplary and preferably for cycloalkyl are called cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- 5- to 7-membered heterocyclic is in the context of the invention for a monocyclic, saturated or partially unsaturated heterocycle having up to three heteroatoms from the series N, O and / or S, which is linked via a ring carbon atom or a nitrogen atom of the heterocycle and the optionally substituted oxo.
- Exemplary and preferred are: tetrahydrofuryl, dihydrofuryl, imidazolidinyl, thiolanyl, dioxolanyl, pyrrolidinyl, pyrrolinyl, tetrahydro-2H-pyranyl, dihydropyranyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, tetrahydro-2H-thiopyranyl, oxidotetrahydro-2H-thiopyranyl, 1, 1-Dioxide-tetrahydro-2H-thiopyranyl, tetrahydrothienyl and 1,4-diazepanyl.
- Heteroaryl is an aromatic, mono- or bicyclic radical having usually 5 to 10, preferably 5 to 6 ring atoms and up to 5, preferably up to 4 heteroatoms from the series S, O and N, where a nitrogen atom is also an N- Oxide, by way of example and with preference thienyl, furyl, pyrrolyl, thiazolyl, oxazolyl, oxadiazolyl, pyrazolyl, imidazolyl, pyridyl, Pyrimidyl, pyridazinyl, pyrazinyl, indolyl, indazolyl, benzofuranyl, benzothiophenyl, quinolinyl, isoquinolinyl, benzoxazolyl, benzimidazolyl.
- Halogen is fluorine, chlorine, bromine and iodine, preferably fluorine and chlorine.
- the invention also provides the use of the compounds of the formula (I) in which
- R 1 is hydroxy or -NR 9 R 10 ,
- R 9 and R 10 independently of one another represent hydrogen, (C 1 -C 6 ) -alkyl, benzyl or
- benzyl and 2-phenylethyl can be substituted by 1 to 3 substituents, where the substituents are selected independently of one another from the group consisting of halogen, cyano, hydroxyl, amino, trifluoromethyl, trifluoromethoxy, (C 1 -C 4 ) -alkyl, ( C r C 4 ) alkoxy and (C 1 -C 4 ) alkylamino,
- R 2 is hydrogen, halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C] -C 4 ) -alkyl, (C r C 4 ) -alkoxy, (Ci-C 4 ) -alkylamino or a bonded via a nitrogen atom 5- to 7-membered heterocyclyl,
- alkyl, alkoxy and alkylamino may be substituted by 1 to 3 substituents, the substituents being selected independently of one another from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl and trifluoromethoxy,
- heterocyclyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C r C 4 ) alkyl, (C r C 4 ) alkoxy and (C r C 4) alkylamino,
- R 1 and R 2 are linked via a * -O-CH 2 - # chain and form a ring
- R 3 is hydrogen, halogen, cyano, hydroxyl, amino, trifluoromethyl, trifluoromethoxy, (C 1 -C 4 ) -alkyl, (C r C 4 ) -alkoxy, (Ci-C 4 ) -alkylamino or a bonded via a nitrogen atom 5- to 7-membered heterocyclyl,
- alkyl, alkoxy and Alkyla ⁇ üno may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl and trifluoromethoxy,
- heterocyclyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C r C 4 ) alkyl, (C r C 4 ) alkoxy and (C r C 4) alkylamino,
- R 4 is halogen, cyano, methyl, ethyl or cyclopropyl
- R 5 is hydrogen, (C r C4) alkyl, (C 2 -C 4) alkenyl, (C, -C 4) alkoxy, (C, -C 4) alkylthio, (C 1 - C 4 ) -Alkylamino or R 11 -Y-CH 2 -,
- alkyl, alkenyl, alkoxy, alkylthio and alkylamino may be substituted with a substituent, wherein the substituent is selected from the group consisting of phenyl and 5- to 10-membered heteroaryl,
- phenyl and heteroaryl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl,
- Y is an oxygen atom, a sulfur atom or -N (R 12 ) -
- R 12 (4 C r C) alkyl or (C 3 -C 6) -cycloalkyl stands for hydrogen
- R 11 is 5- to 10-membered heteroaryl
- heteroaryl may be substituted with 1 to 3 substituents, wherein the
- Substituents independently of one another are selected from the group consisting of halogen, cyano, hydroxyl, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl, aminosulfonyl, (C 1 -C 4 ) -alkyl, (C 1 -C 4 ) -alkoxy and (C) -C 4 ) - alkylamino,
- R 6 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl, trifluoromethoxy or 2-cyanoethen-1-yl,
- R 7 is hydrogen, (C r C4) alkyl, (C 2 -C 4) alkenyl, (C r C4) alkoxy, (C r C4) -alkylthio or (Ci-C 4) alkylamino stands,
- alkyl, alkenyl, alkoxy, alkylthio and alkylamino may be substituted with a substituent, wherein the substituent is selected from the group consisting of phenyl and 5- to 10-membered heteroaryl,
- phenyl and heteroaryl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl, aminosulfonyl, (Ci-C 4 ) alkyl , (C 1 -C 4 ) -alkoxy and (C 1 -C 4 ) -alkylamino,
- R 8 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl or trifluoromethoxy
- the invention also provides the use of the compounds of the formula (I) in which
- R 1 is hydroxy or -NR 9 R 10 , in which
- R 9 and R 10 are independently hydrogen, (Ci-C 6) -alkyl, Ben2yl, 2-phenylethyl or (C 3 -C 6) -cycloalkyl stand,
- benzyl and 2-phenylethyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from
- R 2 is hydrogen, halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C 1 - C 4) alkyl, (C] -C4) alkoxy, (Ci-C 4) alkylamino or via a nitrogen atom bound 5- to 7-membered heterocyclyl,
- alkyl, alkoxy and alkylamino may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl and trifluoromethoxy,
- heterocyclyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C r C 4 ) alkyl, (C r C 4 ) -Alkoxy and (Q-GO-alkylamino,
- R 1 and R 2 are linked via a * -O-CH 2 - # chain and form a ring,
- R 3 is hydrogen, (C 1 -C 4 ) -alkylamino or a 5- to 7-membered heterocyclyl bonded via a nitrogen atom,
- heterocyclyl can be substituted by 1 to 3 substituents, where the substituents are selected independently of one another from the group oxy consisting of halogen, cyano, hydroxy, amino, Trifluo ⁇ nethyl, Trifluo ⁇ neth-, (C r C4) alkyl, (C, -C 4) -alkoxy and (C r C 4) alkylamino,
- R 4 is halogen, cyano, methyl, ethyl or cyclopropyl
- R 5 is hydrogen, 5- to 10-membered heteroaryl or R n is -Y-CH 2 -,
- heteroaryl can be substituted by 1 to 3 substituents, the substituents being selected independently of one another from the group consisting of halogen, cyano, hydroxyl, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl, aminosulfonyl, (C 1 -C 4 ) -alkyl, ( C 1 -C 4 -alkoxy and (C 1 -C 4 ) -alkylamino,
- Y is an oxygen atom, a sulfur atom or -N (R 12 ) -,
- R 12 is hydrogen, (C 1 -C 4 ) -alkyl or (C 3 -C 6 ) -cycloalkyl,
- R 11 is 5- to 10-membered heteroaryl
- heteroaryl may be substituted with 1 to 3 substituents, wherein the
- Substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl, aminosulfonyl, (Ci-C 4 ) alkyl, (Ci-C 4 ) alkoxy and (Ci-C 4 ) - alkylamino,
- R 6 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl, trifluoromethoxy or 2-cyanoethen-1-yl,
- R 7 is hydrogen, (C r C4) alkyl, (C r C4) alkoxy, (C, -C 4) -alkylthio or (C, -C 4) alkyl amino group,
- R 8 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl or trifluoromethoxy
- R 1 is hydroxy or -NR 9 R 10 ,
- R 9 and R 10 independently of one another represent hydrogen, (C 1 -C 6 ) -alkyl, benzyl or 2-phenylethyl,
- benzyl and 2-phenylethyl may be substituted with 1 to 3 substituents, whereby the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C r C4) alkyl, ( Ci-C 4) -alkoxy and (C r C 4) alkylamino,
- R 2 is hydrogen, halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C] - C 4) alkyl, (Ci-C 4) alkoxy, (C 1 -C 4) alkylamino or via a nitrogen atom bound 5- to 7-membered heterocyclyl,
- alkyl, alkoxy and alkylamino may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl and trifluoromethoxy,
- heterocyclyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C r C 4 ) alkyl, (C r C 4 ) Alkoxy and (C 1 -C 4 ) -alkylamino,
- R 1 and R 2 are linked via a * -O-CH 2 - # chain and form a ring,
- R 3 is hydrogen, (C 1 -C 4 ) -alkylamino or a 5- to 7-membered heterocyclyl bonded via a nitrogen atom,
- heterocyclyl can be substituted by 1 to 3 substituents, where the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (C 1 -C 4 ) -alkyl, (C r C 4 ) Alkoxy and (C 1 -C 4 ) -alkylamino,
- R 4 is halogen, cyano, methyl, ethyl or cyclopropyl
- R 5 is hydrogen or R ⁇ -Y-CH 2 -
- Y is an oxygen atom, a sulfur atom or -N (R 12 ) -,
- R 12 (4 C r C) alkyl or (C 3 -C 6) -cycloalkyl stands for hydrogen
- R 11 is 5- to 10-membered heteroaryl
- heteroaryl may be substituted by 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, aminocarbonyl, aminosulfonyl, (C] -C 4 ) - alkyl, (Ci-C 4) alkoxy and (Ci-C4) - alkylamino,
- R 6 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl, trifluoromethoxy or 2-cyanoethen-1-yl,
- R 7 is hydrogen, (C r C4) alkyl, (C r C4) alkoxy, (C r C4) -alkylthio or (C, -C 4) amino alkyl,
- R 8 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl or trifluoromethoxy
- the invention also provides the use of the compounds of the formula (I) in which R 1 is hydroxy or -NR 9 R 10 ,
- R 9 and R 10 are independently hydrogen or (C r C 6) -alkyl
- R 2 is hydrogen, halogen, cyano, hydroxy, amino, trifluoromethyl, trifluoromethoxy, (Cr C4) alkyl, (C] -C4) alkoxy, (Ci-C 4) alkylamino or a bonded via a nitrogen atom 5 - to 7-membered heterocyclyl,
- R 1 and R 2 are linked via a * -O-CH 2 - # chain and form a ring,
- R 3 is hydrogen
- R 4 is halogen, cyano, methyl, ethyl or cyclopropyl
- R 5 is hydrogen
- R 6 represents halogen, cyano, nitro, methyl, trifluoromethyl, trifluoromethoxy or 2-cyanoethen-1-yl
- R 7 is hydrogen
- R 8 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl or trifluoromethoxy
- the invention furthermore relates to compounds of the formula (I) in which
- R 1 is hydroxy or -NR 9 R 10 ,
- R 9 and R 10 independently of one another represent hydrogen or (C 1 -C 6) -alkyl
- R 2 is hydrogen, halogen, cyano, hydroxy, Arnino, Trifluo ⁇ nethyl, trifluoromethoxy, (C 1 - C 4) alkyl, (C 1 -C 4 ⁇ -alkoxy, (C 1 -C 4) or via a -Alkyiamino Nitrogen atom bound 5- to 7-membered heterocyclyl,
- R 1 and R 2 are linked via a * -O-CH 2 - # chain and form a ring,
- R 3 is hydrogen
- R 4 is halogen, cyano, methyl, ethyl or cyclopropyl
- R 5 is hydrogen
- R 6 is halogen, cyano, nitro, methyl, trifluoromethyl, trifluoromethoxy or 2-cyanoethen-1-yl,
- R 7 is hydrogen
- R 8 is halogen, cyano, nitro, methyl, ethyl, trifluoromethyl or trifluoromethoxy
- the invention also relates to compounds of the formula (I) in which R 1 is hydroxyl.
- the invention also relates to compounds of formula (I) in which R 1 is -NR 9 R 10, wherein R 9 and R 10 represent hydrogen or independently of one another (C r C6) alkyl.
- the invention also relates to compounds of the formula (I) in which R 1 and R 2 are linked via an * -O-CH 2 - # chain and form a ring, where * the attachment site to the carbonyl carbon atom and "the attachment site to the Phenyl ring is.
- the invention further provides a process for the preparation of the compounds of the formula (I), where compounds of the formula
- R> 1, r R> 2 , DR 3 , r R> 4 and j ⁇ R> 5 have the abovementioned meaning
- R, R and R have the abovementioned meaning
- X 1 is halogen, preferably chlorine or bromine
- the reaction is generally carried out with a base in inert solvents, preferably in a temperature range from room temperature to reflux of the solvent at atmospheric pressure.
- Inert solvents are, for example, halogenated hydrocarbons such as methylene chloride, trichloromethane or 1,2-dichloroethane, ethers such as dioxane, tetrahydrofuran or 1,2-dimethoxyethane, or other solvents such as acetone, dimethylformamide, dimethylacetamide, 2-butanone or acetonitrile, preferably tetrahydrofuran, methylene chloride , Acetone, 2-butanone, acetonitrile, dimethylformamide or 1,2-dimethoxyethane.
- halogenated hydrocarbons such as methylene chloride, trichloromethane or 1,2-dichloroethane
- ethers such as dioxane, tetrahydrofuran or 1,2-dimethoxyethane
- other solvents such as acetone, dimethylformamide, dimethylacetamide, 2-butanone or acetonitrile,
- bases are alkali metal carbonates, such as cesium carbonate, sodium or potassium carbonate, or sodium or potassium methoxide, or sodium or potassium ethanolate or potassium tert. butylate, or amides such as sodium amide, lithium bis (trimethylsilyl) amide or lithium diisopropyl amide, or organometallic compounds such as butyllithium or phenyllithium, or amine bases such as triethylamine or diisopropylethylamine, other bases such as sodium hydride or DBU, preferably sodium hydride or diisopropylethylamine.
- alkali metal carbonates such as cesium carbonate, sodium or potassium carbonate, or sodium or potassium methoxide, or sodium or potassium ethanolate or potassium tert. butylate, or amides such as sodium amide, lithium bis (trimethylsilyl) amide or lithium diisopropyl amide, or organometallic compounds such as butyllithium or phenyllithium
- R 1 , R 2 and R 3 have the abovementioned meaning
- R 4 and R 5 have the abovementioned meaning
- the reaction is generally carried out with a base in inert solvents, preferably in a temperature range from room temperature to the reflux of the solvent under atmospheric pressure.
- Inert solvents are, for example, halogenated hydrocarbons, such as methylene chloride, trichloromethane or 1,2-dichloroethane, ethers, such as dioxane, tetrahydrofuran or 1,2-dimethoxyethane, or other solvents such as acetone, dimethylformamide, dimethylacetamide, 2-butanone or acetonitrile, preferably tetrahydrofuran, methylene chloride, acetone, 2-butanone, acetonitrile, dimethylformamide or 1,2-dimethoxyethane.
- halogenated hydrocarbons such as methylene chloride, trichloromethane or 1,2-dichloroethane
- ethers such as dioxane, tetrahydrofuran or 1,2-dimethoxyethane
- other solvents such as acetone, dimethylformamide, dimethylacetamide, 2-butanone or acetonitrile
- bases examples include alkali metal carbonates such as cesium carbonate, sodium or potassium carbonate, or amides such as lithium bis (trimethylsilyl) amide or lithium diisopropylamide, or amine bases such as triethylamine or diisopropylethylamine, other bases such as sodium hydride, DBU or pyridine, preferably pyridine or diisopropylethylamine.
- alkali metal carbonates such as cesium carbonate, sodium or potassium carbonate
- amides such as lithium bis (trimethylsilyl) amide or lithium diisopropylamide
- amine bases such as triethylamine or diisopropylethylamine
- other bases such as sodium hydride, DBU or pyridine, preferably pyridine or diisopropylethylamine.
- radical R 1 is hydroxyl, this is part of a carboxylic acid ester function during the reaction.
- the compounds of the invention show an unpredictable, valuable spectrum of activity against hepatitis C viruses.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of diseases, especially of infections with viruses, in particular the viruses mentioned above, and the infectious diseases caused thereby.
- a virus infection is understood below to mean both an infection with a virus and an infection caused by a virus.
- HCV infections in ATDS patients and patients who are infected with HTV (human immunodeficiency virus) (a co-infection of HTV with Hepatitis C leads to a rapid worsening of the clinical picture);
- HCV infections in patients infected with HBV (hepatitis B virus) or other hepatotrophic viruses (eg hepatitis A virus, hepatitis G virus);
- viruses related to HCV e.g. Fever virus, dengue virus, West Nile virus, early summer meningoencephalitis virus,
- hepatitis C Treatment of materials or biological agents to prevent or reduce the transmission of hepatitis C (e.g., in blood and blood products, blood donation items or surgical instruments).
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- the compounds according to the invention are preferably used for the preparation of medicaments which are suitable for the prophylaxis and / or treatment of infections with hepatitis C virus or other members of the family Flaviviridae.
- Another object of the present invention is the use of compounds of the invention alone or in combination with other agents for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- the present invention furthermore relates to medicaments which comprise at least one compound according to the invention, preferably together with interferon (pegylated or non-pegylated) or with ribavirin or with one or more anti-HCV agents or with a combination thereof. and their use for the purposes mentioned above.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an antiviral effective amount of the compounds of the invention.
- the present invention furthermore relates to a method for treating an HCV infection by administering an effective amount of at least one of the compounds according to the invention, a pharmacologically acceptable salt, solvate or solvate of a salt thereof or of a medicament as described above, alone or together with interferon ( pegylated or non-pegylated) or with immunomodulators (for example, ribavirin or viramidine) or with one or more anti-HCV agents or with a combination thereof, which may be administered together or separately.
- interferon pegylated or non-pegylated
- immunomodulators for example, ribavirin or viramidine
- the present invention furthermore relates to a method for the prophylaxis of an HCV infection by administering an effective amount of at least one of the compounds according to the invention, a pharmacologically acceptable salt, solvate or solvate of a salt thereof or of a medicament as described above, alone or together with interferon ( pegylated or non-pegylated) or with immunomodulators (for example ribavirin or viramidine) or with one or more anti-HCV agents or with a combination thereof, which can be administered together or separately.
- interferon pegylated or non-pegylated
- immunomodulators for example ribavirin or viramidine
- Medicaments of the present invention may contain one or more additional active agents, preferably selected from the group of antiviral agents, immunomodulatory agents, HCV protease inhibitors, HCV polymerase inhibitors, inhibitors of another target in the HCV life cycle, HIV inhibitors, HAV inhibitors and HBV inhibitors. Examples of such agents are listed and explained below.
- ribavirin and amantadine antiviral agents
- class I interferons class Ü interferons and pegylated interferons
- antiviral agents antiviral agents
- class I interferons class Ü interferons and pegylated interferons
- IRES inhibitors of a other targets in the HCV life cycle
- nucleoside inhibitors non-nucleoside inhibitors
- protease inhibitors fusion inhibitors and integrase inhibitors of HTV (HIV inhibitors) or agents that inhibit HBV DNA polymerase, or hepatitis B vaccines (HBV) inhibitors.
- HTV HBV inhibitors
- HBV hepatitis B vaccines
- the present invention thus also relates to a combination therapy in which at least one of the compounds according to the invention or a pharmacologically acceptable salt, solvate or solvate of a salt thereof together with at least one additional agent selected from the group of antiviral agents, immunomodulatory agents, HCV protease inhibitors , HCV polymerase inhibitors, inhibitors of another target in the HCV life cycle, HTV inhibitors, HAV inhibitors and HBV inhibitors.
- the additional agents may be combined with the compounds of the present invention to form a single pharmaceutical dosage form. Alternatively, these additional agents can be administered separately. Such additional agents may be administered before, during or after the administration of a compound of the invention or a pharmacologically acceptable salt, solvate or solvate of a salt thereof.
- anti-viral agent means an agent that inhibits the formation and / or replication of a virus. This includes agents that interfere with host or virus mechanisms necessary for the formation and / or replication of a virus. Anti-viral agents are e.g. Ribavirin, Amantadine, VX-497 (Merimepodib, Vertex Pharmaceuticals), Viramidine, Ceplene (Maxamine), XTL-001 and XTL-002 (XTL-Biopharmaceuticals).
- anti-HCV agent means an agent that reduces or prevents hepatitis C-related disease symptoms.
- Such an agent may be an anti-viral agent, an immunomodulatory agent, an HCV protease inhibitor, an HCV polymerase inhibitor or an inhibitor of another target in the HCV life cycle.
- immunomodulatory agent means an agent that enhances the immune response or restricts harmful immune responses.
- Immunomodulatory agents include class I interferons (such as alpha, beta, delta and omega interferons, tau interferons, consensus interferons and asialo interferons), class Ü interferons (such as gamma interferons) and pegylated interferons, as well Substances like levovirin.
- HCV protease inhibitor means an agent that inhibits the function of the HCV NS2 / 3 cysteine protease or the NS3 / 4A serine protease.
- HCV NS3 / 4A serine protease inhibitors are eg BILN 2061 (Boehringer Ingelheim), or VX-950 / LY-570310 (Vertex / Eli Lilly).
- HCV polymerase inhibitor means an agent that inhibits the function of HCV polymerase. This includes e.g. Inhibitors of HCV NS5B polymerase. HCV polymerase inhibitors include non-nucleosides, for example compounds described in WO
- HCV polymerase inhibitors include nucleoside analogs, for example compounds described in WO 01/90121 (Idenix), WO 02/069903 (Biochryst
- Polymerase inhibitors are JTK-002, JTK-003 and JTK-109 (Japan Tobacco).
- inhibitor of another target in the HCV life cycle means an agent that inhibits the formation and / or replication of HCV other than by inhibiting the function of an HCV protease or HCV polymerase. This includes agendas that interfere with host or HCV mechanisms necessary for the formation and / or replication of HCV.
- Inhibitors of another target in the HCV life cycle include agents that inhibit, for example, a helicase or an IRES as a target.
- a specific example of an inhibitor of another target in the HCV life cycle is ISIS-14803 (ISIS Pharmaceuticals).
- HTV inhibitor means an agent that inhibits the formation and / or replication of HTV. This includes agents that interfere with host or HTV mechanisms necessary for the formation and / or replication of HTV.
- HTV inhibitors include e.g. nucleoside inhibitors, non-nucleoside inhibitors, protease inhibitors, fusion inhibitors and integrase inhibitors.
- HAV inhibitor means an agent that inhibits the formation and / or replication of HAV. This includes agents that interfere with mechanisms of the host or HAV necessary for the formation and / or replication of HAV.
- HAV inhibitors include Hepatitis A vaccines, for example, [for example, Havrix ® (GSK), VAQTA ® (Merck), Avaxim ® (Aventis Pasteur)] a.
- HBV inhibitor means an agent that inhibits the formation and / or replication of HBV. This includes agents that interfere with host or HBV mechanisms necessary for the formation and / or replication of HBV.
- HBV inhibitors include agents that inhibit HBV DNA polymerase, or hepatitis B vaccines.
- HBV inhibitors include Lamivudine (Epivir-HBV ®), Adefovir Dipivoxil, Entecavir, FTC (Coviracil ®), DAPD (DXG), L-FMAU (Clevudine ®), AM365 (Amrad), Ldt (Telbivudine), monoval -LdC (valtorcitabine), BAY 41-4109 (Bayer), ACH-126.443 (L-Fd4C) (Achillion), MCC 4 78 (Eli Lilly), Racivir (RCV), fluoro-L and D nucleosides, Robustaflavone, ICN2001-3 (ICN), Barn 205 (Novelos), XTL-001 (XTL), imino-sugar (Nony-DNJ) (Synergy), HepBzyme, as well as immunomodulatory products such as Interferon alpha-2b, HE2000 (Hollis-Eden)
- class I interferons means an interferon selected from a group of interferons which all bind to the type I receptor. This includes natural and synthetically produced class I interferons. Examples of class I interferons are alpha, beta and omega interferons, tau interferons, consensus interferons and asialo interferons.
- class Ü interferons means an interferon selected from a group of interferons which all bind to the type II receptor.
- class H interferons are gamma interferons.
- treatment means the administration of a drug according to the present invention to alleviate or eliminate the disease symptoms of hepatitis C and / or to reduce the amount of virus.
- prophylaxis means the administration of a drug according to the present invention after infection with HCV, but before the onset of disease symptoms and / or prior to the detection of HCV in the blood.
- the compounds according to the invention can act systemically and / or locally.
- they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds of the invention in crystalline and / or amorphised and / or dissolved Contain tablets such as tablets (uncoated or coated tablets, for example, with enteric or delayed-dissolving or insoluble coatings that control the release of the compound of the invention), in the oral cavity rapidly disintegrating tablets or films / wafers, films / lyophilisates, capsules (for example Hard or soft gelatin capsules), dragees, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- Contain tablets such as tablets (uncoated or coated tablets, for example, with enteric or delayed-dissolving or insoluble coatings that control the release of the compound of the invention)
- in the oral cavity rapidly disintegrating tablets or films / wafers, films / lyophilisates, capsules (for example Hard or soft gelatin capsules), dragees, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally).
- a resorption step e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar
- absorption e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally.
- suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicines including powder inhalers, nebulizers
- nasal drops solutions, sprays
- lingual, sublingual or buccal tablets to be applied, films / wafers or capsules, suppositories, ear or ophthalmic preparations, vaginal capsules, aqueous suspensions (lotions, shake mixtures), lipophilic suspensions, ointments, creams, transdermal therapeutic systems, milk, pastes, foams, powdered litter , Implants or stents.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitol oleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- Stabilizers eg antioxidants such as ascorbic acid
- dyes eg inorganic pigments such as iron oxides
- flavor and / or odoriferous include, among others.
- Excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecy
- compositions containing at least one compound of the invention usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the dosage is about 0.01 to 50 mg / kg, preferably 0.1 to 10 mg / kg body weight.
- Method 1 HPLC, preparative separation: Column: CromSil Cl 8, 250 mm x 30 mm; Eluent A: water, eluent B: acetonitrile; Gradient: 3 min 20% B - »31 min 90% B -> 34 min 90% B -» 34.01 min 20% B; Running time: 38 min; Flow: 50 ml / min; UV detection: 210 nm.
- Method 2 Device Type MS: Micromass Quattro LCZ; Device type HPLC: Agilent series 1100; Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A -> 2.5 min 30% A -> 3.0 min 5% A -> 4.5 min 5% A; Flow: 0.0 min 1 ml / min, 2.5 min / 3.0 min / 4.5 min 2 ml / min; Oven: 50 ° C; UV detection: 208-400 nm.
- Method 3 Device Type MS: Micromass ZQ; Device type HPLC: HP 1100 Series; UV DAD; Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A -> 2.5 min 30% A -> 3.0 min 5% A -> 4.5 min 5% A; Flow: 0.0 min 1 ml / min, 2.5 min / 3.0 min / 4.5 min. 2 ml / min; Oven: 50 ° C .; UV detection: 210 nm.
- Method 4 Device Type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2795; Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A - »2.5 min 30% A -> 3.0 min 5% A -> 4.5 min 5% A; Flow: 0.0 min 1 ml / min, 2.5 min / 3.0 min / 4.5 min 2 ml / min; Oven: 50 ° C .; UV detection: 210 nm.
- Method 5 Device Type MS: Waters ZQ 2000; Device type HPLC: Agilent 1100, 2-post circuit, autosampler: HTC PAL; Column: YMC-ODS-AQ, 50 mm x 4.6 mm, 3.0 ⁇ m; Eluent A: water + 0.1% formic acid, eluent B: acetonitrile + 0.1% formic acid; Gradient: 0.0 min 100% A -> 0.2 min 95% A - »1.8 min 25% A -» 1.9 min 10% A -> 2.0 min 5% A -> 3.2 min 5% A - »3.21 min 100% A - > 3.35 min 100% A; Oven: 40 ° C; Flow: 3.0 ml / min; UV detection: 210 nm.
- Method 6 Method: Device Type MS: Micromass ZQ; Device type HPLC: HP 1100 Series; UV DAD; Column: Phenomenex Gemini 3 ⁇ 30 mm x 3.00 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A ⁇ > 2.5 min 30% A -> 3.0 min 5% A -> 4.5 min 5% A; Flow: 0.0 min 1 ml / min, 2.5 min / 3.0 min / 4.5 min. 2 ml / min; Oven: 50 ° C .; UV detection: 210 nm.
- Method 7 Method: Instrument: Micromass Quattro LCZ with HPLC Agilent Series 1100; Column: Phenomenex Onyx Monolithic Cl 8, 100 mm x 3 mm.
- Eluent A 1 liter of water + 0.5 ml 50% formic acid
- eluent B 1 liter acetonitrile + 0.5 ml 50% formic acid
- Flow 2 ml / min
- Oven 40 ° C
- UV detection 208-400 ⁇ m.
- the suspension is stirred at 0 ° C. for 2 h and at RT for 1 h, concentrated to 20 ml and diluted with 100 ml of toluene. The mixture is then filtered through Celite, concentrated and dried under high vacuum. The resulting sulfonyl chloride is dissolved in 200 ml of acetonitrile, mixed with 4.88 ml (60.3 mmol) of pyridine and 5.47 g (36.2 mmol) of methyl 3-aminobenzoate and stirred at RT for 1 h.
- the solution is concentrated, admixed with ethyl acetate and washed with 1 M hydrochloric acid, saturated sodium bicarbonate solution and saturated sodium chloride solution, dried over sodium sulfate, filtered and concentrated.
- the residue is chromatographed on silica gel with a gradient of 0% to 30% ethyl acetate in cyclohexane and the desired product is obtained in a yield of 10.1 g (77% of theory).
- Example IA To a solution of 4.80 g (15.3 mmol) of Example IA in 100 ml of DMF 733 mg (18.3 mmol) of a 60 percent. Dispersion of sodium hydride in mineral oil and 229 mg (1.53 mmol) of sodium iodide. The mixture is stirred for 1 h at 70 0 C, then treated with 4.03 g (16.8 mmol) of 2,6-dichlorobenzyl bromide and stirred at 70 0 C for a further 2 h. The batch is stirred into 500 ml of water, the solid is filtered off, washed with water and dried under high vacuum. 7.24 g (100% of theory) of the desired product are obtained.
- Example IA To a solution of 50 mg (0.159 mmol) of Example IA in 4 ml of DMF are added 25 mg (0.159 mmol) of 2,6-dimethylbenzyl chloride, 25 mg (0.191 mmol) of diisopropylethylamine and 6 mg (0.016 mmol) of tetrabutylammonium iodide. The mixture is stirred for 3 days at RT, filtered and separated by preparative HPLC (Method 1). 34 mg (49% of theory) of the desired product are isolated.
- Example IA To a solution of 50 mg (0.159 mmol) of Example IA in 4 ml of DMF are added 0.159 mmol of a substituted benzyl halide, 25 mg (0.191 mmol) of diisopropylethylamine and 6 mg (0.016 mmol) of tetrabutylammonium iodide. The mixture is stirred for 20 h at 60 0 C, filtered and separated by preparative HPLC (Method 1).
- Examples 3 to 9 are prepared in an analogous manner according to the general procedure [A] from the corresponding starting compounds.
- Examples 12 to 14 are prepared in an analogous manner according to the general procedure [B] from the corresponding starting compounds.
- Example 4A A solution of 1 g (1.93 mmol) of Example 4A in 18 ml of dichloromethane and 2 ml of trifluoroacetic acid is stirred for 30 min at RT and then concentrated. 200 mg of 1084 mg of the crude product obtained are stirred with diethyl ether, filtered off with suction through a frit and dried. 63 mg (7% of theory) of the desired product are isolated.
- Example 17 is prepared in an analogous manner according to the general procedure [C] from the corresponding starting compounds.
- Example 1 A solution of 500 mg (1.056 mmol) of Example 1 in 15 ml of dioxane is mixed with 1.37 ml (1.37 mmol) of a 1 M aqueous lithium hydroxide solution and stirred at RT for 18 h. The solution will be concentrated and separated by preparative HPLC (Method 1). 278 mg (54% of theory) of the desired product are isolated.
- the hepatitis C virus RNA can be propagated in cell culture in the so-called replicon system. Inhibition of the replication cycle of the hepatitis C virus by the substance to be tested can therefore be simulated in the replicon system.
- the replicon system are genome parts of HCV or whole genomes of HCV, which are brought into cell lines (here HuH-7 cells) of human origin. By inserting a selection marker, it is possible to obtain stable cell lines which under selection pressure multiply genomic or subgenomic RNA from HCV [Lohmann et al., Science 285, 110-113 (1999); Blight et al., Science 290, 1972-1974 (2000)].
- the HuH5-2 cells used here harbor a selectable, luciferase-bearing, cell culture-adapted replicon as described in EP 1 043 399 described.
- the cells are cultured in Dulbecco's Modified Earle's Medium (DMEM) with 10% fetal calf serum (FCS), 1% Pen / Strep, 1% NEAA, 1% L-glutamine and 250 ⁇ g / ml Geneticin (G418).
- DMEM Dulbecco's Modified Earle's Medium
- FCS fetal calf serum
- Pen / Strep fetal calf serum
- NEAA fetal calf serum
- L-glutamine 250 ⁇ g / ml Geneticin
- test compounds are prepared as a 50 mM stock solution in DMSO.
- the substances are subsequently diluted in DMEM serially in suitable stages (eg 100, 30, 10, 3 ⁇ M, etc.). This is followed by the addition of the trypsinized, resuspended in medium cells.
- the final concentrations of the test substances in the cell culture cavities is for example 100 ⁇ M to 0.0001 ⁇ M.
- Interferon-alpha serves as reference substance in concentrations from 100 IU / ml to 0.01 IU / ml. Only DMSO-treated cells serve as reference (placebo control).
- the plates are then incubated at 37 ° C under 5% CO 2 for 4 days. This is followed by the different measurements or the quantification of the HCV replicon RNA.
- the above test is set up in a transparent cell culture plate. The qualitative evaluation is done visually under the microscope. In this case, the NOEC value (no observed effect concentratiori) is determined, which corresponds to the lowest substance dilution stage at which no substance-related cytopathic effects or other impairment of cell growth can be detected.
- Alamar Blue is a water-soluble redox indicator, which is reduced depending on the metabolic activity of the cells to be examined.
- the Alamar Blue test is considered quantitative
- the gene for the enzyme luciferase from Photinus pyralis as reporter gene is inserted into the HCV replicon HuH5-2.
- lysis buffer 3 Triton X-100, 11.5% glycerol, 2 mM DTT, 25 mM Na 2 HPO 4 , 25 mM Tris HCl, pH 7.8
- luciferase substrate buffer 20 mM Tris / HCl, 20 mM Tricine, 2.67 mM MgSO 4, 0.1 mM EDTA, 33.3 mM DTT, 0:27 mM coenzyme a, 0:47 mM luciferin, 0:53 mM ATP, pH 7.8
- the photons are measured in a period of 10 seconds to 60 seconds.
- CC 50 substance concentration in ⁇ M at which the Alamar Blue fluorescence decreases by 50% compared to the untreated control;
- SI (selectivity index ) CC 5O / EC 5 o.
- RNA which amplify subgenomic HCV RNA are propagated in transparent 96-well plates in DMEM / 10% FCS without addition of G418 in the presence of the substances in suitable dilution stages as described above (see a.). After 4 days of incubation, the medium is discarded, the cells are washed once with PBS and harvested to isolate the total RNA with the Qiagen RNeasy 96 Kit (Order No. 74181) according to the manufacturer's instructions. After RNA isolation, elution is carried out in 50 ⁇ l of RNase-free water. The RNA is stored at -80 0C. Using TaqMan ® assays (Fa. Applied Biosystems) determining the amount of HCV RNA contained.
- the primers and gene probes used bind to the conserved 5'-untranslated region of the viral genome (primer for coding DNA strand: aatgcctggagatttgggc; primer in the opposite direction: tttcgcgacccaacactactc; probe: 6-carboxyfluorine-tgcccccgcgagactgcatagc-N, N, N ', N 'tetramethyl-6-carboxyrhodamine).
- the expression of a cell-specific gene is determined (TaqMan Ribosomal RNA Control Reagents, Applied Biosystems P / N 4308329).
- the kit Platinum ® Quantitative RT-PCR The ⁇ noscript TM one step system from the company. Invitrogen (part no. 12267-019) is used. The reaction takes place in a final volume of 25 ⁇ l with 1 ⁇ l sample volume. The reaction conditions are: 30 min incubation at 5O 0 C, followed by 5 min incubation at 95 0 C. Following this step, the actual amplification phase followed with 40 repetitions of the following steps: 15 sec incubation at 95 ° C followed by 1 min of incubation at 60 0 C The measurement and evaluation takes place in an Applied Biosystems Abi Prism 7700 Sequence Detection device.
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- Composition 100 mg of the compound of Example 1, 50 mg of lactose (monohydrate), 50 mg of corn starch (native), 10 mg of polyvinylpyrrolidone (PVP 25) (BASF, Ludwigshafen, Germany) and 2 mg of magnesium stearate.
- Preparation The mixture of active ingredient, lactose and starch is granulated with a 5% solution (m / m) of the PVP in water. The granules, after drying with the magnesium stearate for 5 min. mixed. This mixture is compressed with a conventional tablet press (for the tablet format see above). As a guideline for the compression, a pressing force of 15 kN is used.
- Composition 1000 mg of the compound of Example 1, 1000 mg of ethanol (96%), 400 mg of rhodigel (xanthan gum from FMC, Pennsylvania, USA) and 99 g of water.
- a single dose of 100 mg of the compound according to the invention corresponds to 10 ml of oral suspension.
- Composition 500 mg of the compound according to the invention, 2.5 g of polysorbate and 97 g of polyethylene glycol 400. A single dose of 100 mg of the compound according to the invention corresponds to 20 g of oral solution.
- the compound of the invention is dissolved in a concentration below saturation solubility in a physiologically acceptable solvent (e.g., isotonic saline, glucose solution 5% and / or PEG 400 solution 30%).
- a physiologically acceptable solvent e.g., isotonic saline, glucose solution 5% and / or PEG 400 solution 30%.
- the solution is sterile filtered and filled into sterile and pyrogen-free injection containers.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Die Erfindung betrifft substituierte N-Benzyl-N-phenylbenzolsulfonamide und Verfahren zu ihrer Herstellung, ihre Verwendung zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, insbesondere zur Verwendung als antivirale Mittel, insbesondere gegen Hepatitis C Viren.
Description
SUBSTITUIERTE N-BENZYL-N-PHENYLBEN20LSULF0NAMIDE ZUR BEHANDLUNG VON VIRUSINFEKTIONEN
Die Erfindung betrifft substituierte N-Benzyl-N-phenylbenzolsulfonamide und Verfahren zu ihrer Herstellung, ihre Verwendung zur Behandlung und/Oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, insbesondere zur Verwendung als antivirale Mittel, insbesondere gegen Hepatitis C Viren.
Infektionen mit dem Hepatitis C-Virus (HCV) sind weltweit die Hauptursache für Nicht- A/Nicht-B Hepatitis-Erkrankungen. Nach Schätzungen sind etwa 170 Millionen Menschen weltweit mit dem Virus infiziert. Die Infektion mit HCV erfolgt vor allem parenteral, beispielsweise durch Bluttransfusion oder durch die Gabe von Medikamenten aus Blutprodukten. Bei einem hohen Prozentsatz der Virusträger führt dies zu einer chronischen Hepatitis C-Erkrankung. Insgesamt sind rund 3% der Weltbevölkerung chronisch mit dem Hepatitis C Virus infiziert. Für diese Gruppe an Infizierten besteht ein erhöhtes Risiko, in der Folge an lebensbedrohlichen Lebererkrankungen wie Leberzirrhose, hepatozellulärem Karzinom oder terminalem Leberversagen zu versterben. Hepatitis C-Infektion ist die häufigste Ursache für eine Lebertransplantation. Noch nicht völlig geklärt sind die Mechanismen, wie es zum Persistieren der Virusinfektion und zur hohen Rate an daraus resultierenden ernsten Lebererkrankungen kommt. Es ist unbekannt, wie das Virus mit dem menschlichen Immunsystem interagiert und die Immunabwehr überwindet. Die Rolle der zellulären wie der humoralen Immunantworten beim Schutz gegen HCV-Infektion ist noch nicht verstanden. Es wurde berichtet, dass Immunglobuline zum prophylaktischen Schutz gegen transfusionsbedingte virale Hepatitis eingesetzt wurden; allerdings wird der Einsatz von Immunglobulinen für diesen Zweck gegenwärtig vom Center for Disease Control nicht empfohlen. Das Fehlen einer effizienten Immunantwort steht bislang der Etablierung eines Impfstoffes im Wege, ebenso wie einer Prophylaxe, die nach Kontakt mit dem Virus eingesetzt werden könnte. In der näheren Zukunft werden daher hauptsächlich antivirale Prinzipien eine Rolle in der Bekämpfung des Hepatitis C-Virus spielen.
m verschiedenen klinischen Studien wurden Substanzen mit dem Ziel untersucht, HCV- Infektionen in Patienten mit chronischer Hepatitis wirkungsvoll zu therapieren. In diesen Studien kam Interferon-alpha (IFN-α), in Alleingabe oder in Kombination mit anderen antiviralen Wirkstoffen, zum Einsatz. Diese Untersuchungen haben gezeigt, dass eine erhebliche Anzahl an Patienten auf diese Therapie nicht anspricht, und dass ein großer Teil derjenigen, bei denen Interferon-alpha eine Wirkung zeigt, nach Absetzen der Substanz Rückfälle erleiden.
Bis vor kurzem war die Behandlung mit Interferon (EFTSI) die einzige Therapieform mit klinisch nachgewiesener Wirksamkeit bei chronischer Hepatitis C-Erkrankung. Der Anteil der Patienten mit nachhaltigem Therapieerfolg ist jedoch gering. Die Interferon-Therapie mit einer langen Behandlungsdauer von mindestens sechs Monaten ist sehr häufig mit ernsten Nebenwirkungen (z.B. Leukopenie, Thrombopenie, Retinopathie, Thyroiditis, akute Pankreatitis, Depression) verbunden, die die Lebensqualität der Patienten erheblich einschränken. Neben dieser Monotherapie ist die Kombination von Interferon mit der antiviralen Substanz Ribavirin zugelassen. Diese Kombinationstherapie führt zu einer verbesserten Wirksamkeit, verbessert aber nicht das mit IFN assoziierte Nebenwirkungsprofil, zudem sind auch mit Ribavirin Nebenwirkungen (z. B. hämolytische Anämie) assoziiert. Durch den Einsatz von pegylierten Formen des IFN wie PEG- Intron® oder Pegasys® können diese unerwünschten Nebeneffekte zumindest teilweise abgemildert werden. Nach wie vor gibt es jedoch eine Großzahl von Patienten, die auf diese Therapie nicht ansprechen. Bei dem HCV Genotyp Ib versagt die beschriebene Kombinationstherapie bei rund der Hälfte der Patienten. Daher besteht weiterhin ein großer Bedarf an oral anwendbaren antiviralen Wirkstoffen, mit denen die Einschränkungen der bislang etablierten Therapieformen überwunden werden können (S.-L. Tan et al., Nature Rev. Drug Discov. 2002, 1, 867-881).
Der Hepatitis C-Virus (HCV) ist der einzige Vertreter des Genus Hepacivirus innerhalb der Familie der Flaviviridae. Man unterscheidet mindestens 6 Genotypen und eine Vielzahl von Subtypen. Das Virus ist von einer Hülle umgeben und besitzt als Genom einen Positiv-Einzelstrang an viraler RNA. Die Länge des viralen RNA-Genoms beträgt ca. 9500 Nucleotide. Die Vermehrung des viralen Genoms und die Translation in Protein wird von RNA-Strukturen, welche am Anfang und am Ende des Genoms liegen vermittelt (5' nicht-translatierte Region, 3' nicht-translatierte Region). Das Genom hat einen einzigen Leserahmen (open reading frame, ORF), der für ein Polyprotein (ca. 3000 Aminosäuren) kodiert. Dieses wird in einer infizierten Zelle durch virale und Wirtszelleinegene -Erßyme (Proteasen) in Struktur- und Nichtstruktur (NS)-proteine gespalten. HCV kodiert für ein Kapsidprotein (c) und zwei Hüllproteine (El und E2). Ein kleines Protein (p7) könnte ein sogenanntes Viroporin sein, welches für die Infektiosität des reifen Viruspartikels essentiell ist. Zu den reifen NS-Proteinen zählen die Proteine NS2, NS3, NS4A, NS4B, NS5A und NS5B. Für ihre Abspaltung aus dem Polyprotein sind zwei virale Proteasen verantwortlich. Bei der nur gering charakterisierten NS2/3 -Protease handelt es sich wahrscheinlich um eine Cysteinprotease, die die NS2-NS3-Schnittstelle spaltet. Die zweite Protease (NS3/4A-Protease) ist eine Serinprotease, dessen katalytische Aktivität im N-terminalen Teil des multifunktionalen NS3- Proteins enthalten ist und das kleine NS4A Protein als Kofaktor benötigt. Sie katalysiert alle proteolytischen Spaltungen abwärts der NS3 -Aminosäuresequenz, d.h. die NS3-NS4A-Proteolyse ebenso wie die Spaltungen an den Stellen NS4A-NS4B, NS4B-NS5A und NS5A-NS5B.
Das NS4A-Protein besitzt möglicherweise neben der Funktion als Kofaktor der NS3-Protease auch noch eine Aufgabe zur Membranlokalisierung von NS3 und anderen NS-Proteinen. Die Komplexbildung zwischen NS3 und NS4A ist vermutlich eine Grundvoraussetzung für die Protein- prozessierung und erhöht die proteolytischen Aktivitäten bezogen auf alle Schnittstellen. Das NS3- Protein besitzt außerdem Aktivität als NTPase und Helikase, welche in der C-terminalen Domäne lokalisiert sind. NS5B ist eine RNA-abhängige RNA-Polymerase, die entscheidend an der HCV- Replikation beteiligt ist. Über die Funktionen der NS4B- und NS5A-Proteine ist sehr wenig bekannt. Für NS5A wird jedoch eine Beteiligung an der klinischen Resistenz gegenüber Interferon diskutiert.
Dem Hepatitis C nah verwandte Viren wie beispielsweise das GBV B-Virus, welches Neuweltaffen infiziert, oder das BVDV (Bovines Virale Diarrhoe Virus) werden häufig als Modellviren verwendet, um bestimmte Aspekte des Viruslebenszyklus zu untersuchen.
Eine Aufgabe der vorliegenden Erfindung ist es daher, neue Verbindungen mit gleicher oder verbesserter antiviraler Wirkung zur Behandlung und/oder Prophylaxe von viralen Infektionskrank- heiten bei Menschen und Tieren, insbesondere von Hepatitis C und deren Folgen, zur Verfügung zu stellen.
Strukturell ähnliche Verbindungen sind beispielsweise in WO 01/56989 zur Behandlung und/oder Prophylaxe von Thrombose und in WO 2004/026823 sind als Estrogen-Rezeptor Liganden beschrieben.
Überraschenderweise wurde gefunden, dass die in der vorliegenden Erfindung beschriebenen substituierten N-Benzyl-N-phenylbenzolsulfonamide antiviral wirksam sind.
Gegenstand der Erfindung ist die Verwendung der Verbindungen der Formel

R1 für Hydroxy oder -NRyRlü steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff, (Ci-CβJ-Alkyl, Benzyl, 2-Phenylethyl oder (C3-C6)-Cycloalkyl stehen,
wobei Benzyl und 2-Phenylethyl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der
Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Ci-C4)-Alkyl, (C1-C)-AIkOXy und (C,-C4)-Alkylamino,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Cr C4)-Alkyl, (Ci-C4)-Alkoxy, (C]-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylamino substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl und Trifluormethoxy,
und
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (C,-C4)-Alkyl, (CrC4)-Alkoxy und (CrC4)-Alkylamino,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
# die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (C1- C4)-Alkyl, (Ci-C4)-Alkoxy, (C1-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylamino substituiert sein können mit 1 bis 3 Sub- stituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl und Trifluoπnethoxy,
und
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormeth- oxy, (CrC4)-Alkyl, (Ci-C4)-Alkoxy und (C,-C4)-Alkylamino,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff, (C,-C4)-Alkyl, (C2-C4)-Alkenyl, (CrC4)-Alkoxy, (CrC4)-Alkylthio, (C1- C4)-Alkylamino, 5- bis 10-gliedriges Heteroaryl oder R1 '-Y-CH2- steht,
wobei Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl,
Aminosulfonyl, (C]-C4)-Alkyl, (C]-C4)-Alkoxy und (Ci-C4)-Alkylamino,
und
wobei Alkyl, Alkenyl, Alkoxy, Alkylthio und Alkylamino substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe, bestehend aus Phenyl und 5- bis 10-gliedriges Heteroaryl,
worin Phenyl und Heteroaryl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (CrC4)-Alkyl, (CrC4)-Alkoxy und (CrC4)-Alkylamino,
wobei
Y für ein Sauerstoffatom, ein Schwefelatom oder -N(R12)- steht,
wobei
R12 für Wasserstoff, (C,-C4)-Alkyl oder (C3-C6)-Cycloalkyl steht,
R1 ' für 5- bis 10-gliedriges Heteroaryl steht,
worin Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormeth- oxy, Aminocarbonyl, Aminosulfonyl, (CrC4)-Alkyl, (Ci-C4)-Alkoxy und (C1-C4)- Alkylamino,
R6 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl, Trifluormethoxy oder 2-Cyanoethen-l-yl steht,
R7 für Wasserstoff, (CrC4)-Alkyl, (C2-C4)-Alkenyl, (CrC4)-Alkoxy, (C,-C4)-Alkylthio oder (Ci-C4)-Alkylamino steht,
wobei Alkyl, Alkenyl, Alkoxy, Alkylthio und Alkylamino substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe, bestehend aus Phenyl und 5- bis 10-gliedriges Heteroaryl,
worin Phenyl und Heteroaryl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (Ci-C4)-Alkyl, (CrC4)-Alkoxy und (CrC4)-Alkylamino,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze,
zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von viralen Erkrankungen.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, nachfolgend als Ausführungsbeispiel(e) genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung betrifft deshalb die Enan-
tiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfϊndungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind aber auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind aber beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfϊndungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasser- stoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsäure, Propionsäure, Milchsäure, Weinsäure, Apfelsäure, Zitronensäure, Fumarsäure, Malein- säure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclo-hexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungs- mittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt.
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Alkyl per se und "Alk" und "Alkyl" in Alkoxy. Alkylthio und Alkylamino stehen für einen linearen oder verzweigten Alkylrest mit in der Regel 1 bis 6 („Ci-C6-Alkyl"), vorzugsweise 1 bis 4, besonders bevorzugt 1 bis 3 Kohlenstoffatomen, beispielhaft und vorzugsweise für Methyl, Ethyl, n-Propyl, Isopropyl, tert.-Butyl, n-Pentyl und n-Hexyl.
Alkoxy steht beispielhaft und vorzugsweise für Methoxy, Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy, tert-Butoxy, n-Pentoxy und n-Hexoxy.
Alkylthio steht beispielhaft und vorzugsweise für Methyltbio, Ethyltbio, n-Propylthio, Isopropylthio, n-Butylthio, tert.-Butylthio, n-Pentylthio und n-Hexylthio.
Alkylamino steht für einen Alkylaminorest mit einem oder zwei (unabhängig voneinander gewählten) Alkylsubstituenten, beispielhaft und vorzugsweise für Methylamino, Ethylamino, n- Propylamino, Isopropylamino, tert.-Butylamino, n-Pentylamino, n-Hexylamino, N,N-Dimethyl- amino, N,N-Diethylamino, N-Ethyl-N-methylamino, N-Methyl-N-n-propylamino, N-Isopropyl-N- n-propylamino, N-tert.-Butyl-N-methylamino, N-Ethyl-N-n-pentylamino und N-n-Hexyl-N-methyl- amino. Ci-C3-Alkylamino steht beispielsweise für einen Monoalkylaminorest mit 1 bis 3 Kohlenstoffatomen oder für einen Dialkylaminorest mit jeweils 1 bis 3 Kohlenstoffatomen pro Alkyl- substituent.
Alkenyl steht für einen geradkettigen oder verzweigten Alkenylrest mit 2 bis 6 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger oder verzweigter Alkenylrest mit 2 bis 4, besonders bevorzugt mit 2 bis 3 Kohlenstoffatomen. Beispielsweise und vorzugsweise seien genannt: Vinyl, Allyl, n-Prop-1-en- 1-yl und n-But-2-en-l-yl.
Cvcloalkyl steht für eine Cycloalkylgruppe mit in der Regel 3 bis 6, bevorzugt 3 bis 5 Kohlenstoffatomen. Beispielhaft und vorzugsweise für Cycloalkyl sind genannt Cyclopropyl, Cyclobutyl, Cyclopentyl und Cyclohexyl.
5- bis 7-gliedriges Heterocvclyl steht im Rahmen der Erfindung für einen monoeyclischen, gesättigten oder partiell ungesättigten Heterocyclus mit bis zu drei Heteroatomen aus der Reihe N, O und/oder S, der über ein Ringkohlenstoffatom oder ein Stickstoffatom des Heterocyclus verknüpft ist und der gegebenenfalls Oxo substituiert ist. Beispielhaft und vorzugsweise seien genannt: Tetrahydrofuryl, Dihydrofuryl, Imidazolidinyl, Thiolanyl, Dioxolanyl, Pyrrolidinyl, Pyrrolinyl, Tetrahydro-2H-pyranyl, Dihydropyranyl, Piperidinyl, Piperazinyl, Morpholinyl, Thiomorpholinyl, Tetrahydro-2H-thiopyranyl, Oxidotetrahydro-2H-thiopyranyl, 1 , 1 -Dioxidotetrahydro-2H-thio- pyranyl, Tetrahydrothienyl und 1,4-Diazepanyl.
Heteroaryl steht für einen aromatischen, mono- oder bicyclischen Rest mit in der Regel 5 bis 10, vorzugsweise 5 bis 6 Ringatomen und bis zu 5, vorzugsweise bis zu 4 Heteroatomen aus der Reihe S, O und N, wobei ein Stickstoffatom auch ein N-Oxid bilden kann, beispielhaft und vorzugsweise für Thienyl, Furyl, Pyrrolyl, Thiazolyl, Oxazolyl, Oxadiazolyl, Pyrazolyl, Imidazolyl, Pyridyl,
Pyrimidyl, Pyridazinyl, Pyrazinyl, Indolyl, Indazolyl, Benzofuranyl, Benzothiophenyl, Chinolinyl, Isochinolinyl, Benzoxazolyl, Benzimidazolyl.
Halogen steht für Fluor, Chlor, Brom und Jod, bevorzugt Fiuor und Chlor.
In den Formeln der Gruppe, die für R1 und R2 stehen kann, steht der Endpunkt der Linie, neben der jeweils ein * oder # steht, nicht für ein Kohlenstoffatom beziehungsweise eine CH2-Gruppe sondern ist Bestandteil der Bindung zu dem Atom, an das R1 bzw. R2 gebunden ist.
Gegenstand der Erfindung ist auch die Verwendung der Verbindungen der Formel (I), in welcher
R1 für Hydroxy oder -NR9R10 steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff, (Ci-C6)-Alkyl, Benzyl oder
2-Phenylethyl stehen,
wobei Benzyl und 2-Phenylethyl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Ci-C4)-Alkyl, (CrC4)-Alkoxy und (CrC^-Alkylamino,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (C]- C4)-Alkyl, (CrC4)-Alkoxy, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylamino substituiert sein können mit 1 bis 3 Sub- stituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl und Trifluormethoxy,
und
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (CrC4)-Alkyl, (CrC4)-Alkoxy und (CrC4)-Alkylamino,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
# die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (C1- C4)-Alkyl, (CrC4)-Alkoxy, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylaπüno substituiert sein können mit 1 bis 3 Sub- stituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl und Trifluormethoxy,
und
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (CrC4)-Alkyl, (CrC4)-Alkoxy und (CrC4)-Alkylamino,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff, (CrC4)-Alkyl, (C2-C4)-Alkenyl, (C,-C4)-Alkoxy, (C,-C4)-Alkylthio, (C1- C4)-Alkylamino oder R11 -Y-CH2- steht,
wobei Alkyl, Alkenyl, Alkoxy, Alkylthio und Alkylamino substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe, bestehend aus Phenyl und 5- bis 10-gliedriges Heteroaryl,
worin Phenyl und Heteroaryl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl,
Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (CrC4)-Alkyl, (C]-C4)-Alkoxy und (CrC4)-Alkylamino,
wobei
Y für ein Sauerstoffatom, ein Schwefelatom oder -N(R12)- steht,
wobei
R12 für Wasserstoff, (CrC4)-Alkyl oder (C3-C6)-Cycloalkyl steht,
R11 für 5- bis 10-gliedriges Heteroaryl steht,
worin Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die
Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormeth- oxy, Aminocarbonyl, Aminosulfonyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (C]-C4)- Alkylamino,
R6 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl, Trifluormethoxy oder 2-Cyanoethen-l-yl steht,
R7 für Wasserstoff, (CrC4)-Alkyl, (C2-C4)-Alkenyl, (CrC4)-Alkoxy, (CrC4)-Alkylthio oder (Ci-C4)-Alkylamino steht,
wobei Alkyl, Alkenyl, Alkoxy, Alkylthio und Alkylamino substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe, bestehend aus Phenyl und 5- bis 10-gliedriges Heteroaryl,
worin Phenyl und Heteroaryl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylamino,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze,
zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von viralen Erkrankungen.
Gegenstand der Erfindung ist auch die Verwendung der Verbindungen der Formel (I), in welcher
R1 für Hydroxy oder -NR9R10 steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff, (Ci-C6)-Alkyl, Ben2yl, 2-Phenylethyl oder (C3-C6)-Cycloalkyl stehen,
wobei Benzyl und 2-Phenylethyl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der
Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Ci-C4)-Alkyl, (CrC4)-Alkoxy und (CrC4)-Alkylamino,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (C1- C4)-Alkyl, (C]-C4)-Alkoxy, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebun- denes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylamino substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl und Trifluormethoxy,
und
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (CrC4)-Alkyl, (CrC4)-Alkoxy und (Q-GO-Alkylamino,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
# die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff, (C,-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7- gliedriges Heterocyclyl steht,
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe,
bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluoπnethyl, Trifluoπneth- oxy, (CrC4)-Alkyl, (C,-C4)-Alkoxy und (CrC4)-Alkylamino,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff, 5- bis 10-gliedriges Heteroaryl oder Rn-Y-CH2- steht,
wobei Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylamino,
und
wobei
Y für ein Sauerstoffatom, ein Schwefelatom oder -N(R12)- steht,
wobei
R12 für Wasserstoff, (C, -C4)-Alkyl oder (C3-C6)-Cycloalkyl steht,
R11 für 5- bis 10-gliedriges Heteroaryl steht,
worin Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die
Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)- Alkylamino,
R6 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl, Trifluormethoxy oder 2- Cyanoethen-1-yl steht,
R7 für Wasserstoff, (CrC4)-Alkyl, (CrC4)-Alkoxy, (C,-C4)-Alkylthio oder (C,-C4)-Alkyl- amino steht,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze,
zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von viralen Erkrankungen.
Gegenstand der Erfindung ist auch die Verwendung der Verbindungen der Formel (I), in welcher
R1 für Hydroxy oder -NR9R10 steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff, (Ci-C6)-Alkyl, Benzyl oder 2-Phenylethyl stehen,
wobei Benzyl und 2-Phenylethyl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (CrC4)-Alkyl, (Ci-C4)-Alkoxy und (CrC4)-Alkylamino,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (C]- C4)-Alkyl, (Ci-C4)-Alkoxy, (C1-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylamino substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl und Trifluormethoxy,
und
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (CrC4)-Alkyl, (CrC4)-Alkoxy und (Ci-C4)-Alkylamino,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
# die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7- gliedriges Heterocyclyl steht,
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormeth- oxy, (CrC4)-Alkyl, (CrC4)-Alkoxy und (Ci-C4)-Alkylamino,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff oder Rπ-Y-CH2- steht,
wobei
Y für ein Sauerstoffatom, ein Schwefelatom oder -N(R12)- steht,
wobei
R12 für Wasserstoff, (CrC4)-Alkyl oder (C3-C6)-Cycloalkyl steht,
R11 für 5- bis 10-gliedriges Heteroaryl steht,
worin Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormeth- oxy, Aminocarbonyl, Aminosulfonyl, (C]-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)- Alkylamino,
R6 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl, Trifluormethoxy oder 2- Cyanoethen-1-yl steht,
R7 für Wasserstoff, (CrC4)-Alkyl, (CrC4)-Alkoxy, (CrC4)-Alkylthio oder (C,-C4)-Alkyl- amino steht,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze,
zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von viralen Erkrankungen.
Gegenstand der Erfindung ist auch die Verwendung der Verbindungen der Formel (I), in welcher
R1 für Hydroxy oder -NR9R10 steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff oder (CrC6)-Alkyl stehen,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Cr C4)-Alkyl, (C]-C4)-Alkoxy, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
# die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff steht,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff steht,
R6 für Halogen, Cyano, Nitro, Methyl, Trifluormethyl, Trifluormethoxy oder 2-Cyanoethen-l- yl steht,
R7 für Wasserstoff steht,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze,
zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von viralen Erkrankungen.
Gegenstand der Erfindung sind weiterhin Verbindungen der Formel (I), in welcher
R1 für Hydroxy oder -NR9R10 steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff oder (C1-Ce)-AIlCyI stehen,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Arnino, Trifluoπnethyl, Trifluormethoxy, (C1- C4)-Alkyl, (C1-C4^-AIkOXy, (C 1-C4)-Alkyiamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
* die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff steht,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff steht,
R6 für Halogen, Cyano, Nitro, Methyl, Trifiuormethyl, Trifluormethoxy oder 2-Cyanoethen-l- yl steht,
R7 für Wasserstoff steht,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifiuormethyl oder Trifluormethoxy steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Gegenstand der Erfindung sind auch Verbindungen der Formel (I), in welcher R1 für Hydroxy steht.
Gegenstand der Erfindung sind auch Verbindungen der Formel (I), in welcher R1 für -NR9R10 steht, wobei R9 und R10 unabhängig voneinander für Wasserstoff oder (CrC6)-Alkyl stehen.
Gegenstand der Erfindung sind auch Verbindungen der Formel (I), in welcher R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden, wobei * die Anknüpfstelle an das Carbonylkohlenstoffatom und " die Anknüpfstelle an den Phenylring ist.
Gegenstand der Erfindung ist weiterhin ein Verfahren zur Herstellung der Verbindungen der Formel (I), wobei Verbindungen der Formel

R > 1 , r R>2 , D R3 , r R> 4 und j τ R> 5 die oben angegebene Bedeutung haben,
mit Verbindungen der Formel

R , R und R die oben angegebene Bedeutung haben, und
X1 für Halogen, bevorzugt Chlor oder Brom, steht,
umgesetzt werden.
Die Umsetzung erfolgt im Allgemeinen mit einer Base in inerten Lösungsmitteln, bevorzugt in einem Temperaturbereich von Raumtemperatur bis zum Rückfluss der Lösungsmittel bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Methylenchlorid, Trichlormethan oder 1 ,2-Dichlorethan, Ether wie Dioxan, Tetrahydrofuran oder 1,2-Dimeth- oxyethan, oder andere Lösemittel wie Aceton, Dimethylformamid, Dimethylacetamid, 2-Butanon oder Acetonitril, bevorzugt Tetrahydrofuran, Methylenchlorid, Aceton, 2-Butanon, Acetonitril, Dimethylformamid oder 1,2-Dimethoxyethan.
Basen sind beispielsweise Alkalicarbonate wie Cäsiumcarbonat, Natrium- oder Kaliumcarbonat, oder Natrium- oder Kaliummethanolat, oder Natrium- oder Kaliumethanolat oder Kalium-tert.-
butylat, oder Amide wie Natriumamid, Lithium-bis-(trimethylsilyl)amid oder Lithiumdiisopropyl- amid, oder metallorganische Verbindungen wie Butyllithium oder Phenyllithium, oder Aminbasen wie Triethylamin oder Diisopropylethylamin, andere Basen wie Natriumhydrid oder DBU, bevorzugt ist Natriumhydrid oder Diisopropylethylamin.
Ist der Rest R1 Hydroxy, so ist dieser während der Umsetzung Bestandteil einer Carbonsäureesterfunktion. Nach der Umsetzung wird der Carbonsäureester nach dem Fachmann bekannten Bedingungen zur Carbonsäure gespalten, und es entstehen Verbindungen der Formel (I).
Die Verbindungen der Formel (II) sind bekannt oder können hergestellt werden, indem Verbindungen der Formel

R1, R2 und R3 die oben angegebene Bedeutung haben,
mit Verbindungen der Formel

R4 und R5 die oben angegebene Bedeutung haben,
umgesetzt werden.
Die Umsetzung erfolgt im Allgemeinen mit einer Base in inerten Lösungsmitteln, bevorzugt in einem Temperaturbereich von Raumtemperatur bis zum Rückfluss der Lösungsmittel bei Normal- druck.
Inerte Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Methylenchlorid, Tri- chlormethan oder 1 ,2-Dichlorethan, Ether wie Dioxan, Tetrahydrofuran oder 1 ,2-Dimethoxyethan,
oder andere Lösemittel wie Aceton, Dimethylformamid, Dimethylacetamid, 2-Butanon oder Acetonitril, bevorzugt Tetrahydrofuran, Methylenchlorid, Aceton, 2-Butanon, Acetonitril, Dimethylformamid oder 1,2-Dimethoxyethan.
Basen sind beispielsweise Alkalicarbonate wie Cäsiumcarbonat, Natrium- oder Kaliumcarbonat, oder Amide wie Lithium-bis-(trimethylsilyl)amid oder Lithiumdiisopropylamid, oder Aminbasen wie Triethylamin oder Diisopropylethylamin, andere Basen wie Natriumhydrid, DBU oder Pyridin, bevorzugt ist Pyridin oder Diisopropylethylamin.
Ist der Rest R1 Hydroxy, so ist dieser während der Umsetzung Bestandteil einer Carbonsäureesterfunktion.
Die Verbindungen der Formeln (DI), (IV) und (V) sind bekannt oder lassen sich nach bekannten Verfahren aus den entsprechenden Edukten synthetisieren.
Die Herstellung der erfindungsgemäßen Verbindungen kann durch folgendes Syntheseschema verdeutlicht werden.
Syntheseschema:

Pyridin, Acetonitril



Diisopropylethylamin Acetonitril

Die erfindungsgemäßen Verbindungen zeigen ein nicht vorhersehbares, wertvolles Wirkspektrum gegenüber Hepatitis C-Viren.
Weiterer Gegenstand der vorliegenden Erfindung ist der Einsatz der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Krankheiten, vor allem von Infektionen mit Viren, insbesondere den vorstehend genannten Viren, und den dadurch hervorgerufenen Infektionskrankheiten. Unter einer Virusinfektion wird nachfolgend sowohl eine Infektion mit einem Virus als auch eine durch eine Infektion mit einem Virus hervorgerufene Krankheit verstanden.
Als Indikationsgebiete können beispielsweise genannt werden:
1. Behandlung von akuten und chronischen Hepatitis C-Infektionen und die Vermeidung, Linderung oder Eliminierung der Begleiterscheinungen und Folgeschäden wie beispielsweise Leberfibrose, Leberzirrhose, Leberkrebs (hepatozelluläres Karzinom) und/oder die Verminderung der Anzahl der viralen Genomkopien in einem Patienten;
2. Behandlung und Prophylaxe bei Organtransplantationspatienten, insbesondere bei einer Hepatitis C-Infektion des Organspenders oder Organempfängers;
3. Behandlung von HCV-Infektionen bei ATDS-Patienten und Patienten, welche mit HTV (humanes Immunodefizienz- Virus) infiziert sind (eine Co-Infektion von HTV mit Hepa- titis C führt zu einer raschen Verschlechterung des Krankheitsbildes);
4. Behandlung von HCV-Infektionen bei Patienten, welche mit HBV (Hepatitis B-Virus) oder anderen hepatotrophen Viren (beispielsweise Hepatitis A-Virus, Hepatitis G- Virus) infiziert sind;
5. Behandlung von Erkrankungen mit Viren, welche mit HCV verwandt sind, wie z.B. GeIb- fieber-Virus, Dengue-Virus, West Nil-Virus, Frühsommermeningoenzephalitis-Virus,
Japanisches Enzephalitis-Virus;
6. Behandlung von Säugetieren mit verwandten animalen Viren wie beispielsweise Pesti- viren;
7. Behandlung von Materialien oder biologischen Agenden, um eine Übertragung von Hepatitis C zu vermeiden oder eine solche Gefahr zu verringern (z.B. bei Blut und Blutprodukten, Blutspende-Utensilien oder chirurgischen Instrumenten).
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Bevorzugt werden die erfindungsgemäßen Verbindungen zur Herstellung von Arzneimitteln verwendet, die zur Prophylaxe und/oder Behandlung von Infektionen mit Hepatitis C-Virus oder anderen Mitgliedern der Familie der Flaviviridae geeignet sind.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Ver- bindungen allein oder in Kombination mit anderen Wirkstoffen zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, vorzugsweise zusammen mit Interferon (pegyliert oder nicht-pegy- liert) oder mit Ribavirin oder mit einem oder mehreren anti-HCV-Agentien oder mit einer Kom- bination hiervon, enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer antiviral wirksamen Menge der erfindungsgemäßen Verbindungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung einer HCV- Infektion durch Gabe einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen, eines pharmakologisch unbedenklichen Salzes, Solvates oder Solvates eines Salzes davon oder eines wie oben beschriebenen Arzneimittels, allein oder zusammen mit Interferon (pegyliert oder nicht-pegyliert) oder mit Immunmodulatoren (beispielsweise Ribavirin oder Viramidin) oder mit einem oder mehreren anti-HCV-Agentien oder mit einer Kombination hiervon, die zusammen oder getrennt verabreicht werden können.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Prophylaxe einer HCV- Infektion durch Gabe einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen, eines pharmakologisch unbedenklichen Salzes, Solvates oder Solvates eines Salzes davon oder eines wie oben beschriebenen Arzneimittels, allein oder zusammen mit Interferon (pe- gyliert oder nicht-pegyliert) oder mit Immunmodulatoren (beispielsweise Ribavirin oder Viramidin) oder mit einem oder mehreren anti-HCV-Agentien oder mit einer Kombination hiervon, die zusammen oder getrennt verabreicht werden können.
Arzneimittel der vorliegenden Erfindung können eines oder mehrere zusätzliche aktive Agentien enthalten, vorzugsweise ausgewählt aus der Gruppe antivirale Agentien, immunomodulatorische Agentien, HCV Protease-Inhibitoren, HCV Polymerase-Inhibitoren, Inhibitoren eines anderen Targets im HCV-Lebenszyklus, HIV-Inhibitoren, HAV-Inhibitoren und HBV-Inhibitoren. Beispiele solcher Agentien sind nachstehend aufgeführt und erläutert.
Bevorzugte Beispiele einiger dieser Agentien sind Ribavirin und Amantadin (antivirale Agentien), Klasse I-Interferone, Klasse Ü-Interferone und pegylierte Interferone (immunomodulatorische Agentien), Inhibitoren von HCV NS5B-Polymerase, HCV NS3-Helicase, HCV Protease oder IRES (Inhibitoren eines anderen Targets im HCV-Lebenszyklus), nukleosidische Inhibitoren, nicht- nukleosidische Inhibitoren, Protease-Inhibitoren, Fusions-Inhibitoren und Integrase-Inhibitoren von HTV (HIV-Inhibitoren) oder Agentien, die die HBV DNA-Polymerase inhibieren, oder Hepatitis B-Impfstoffe (HBV-Inhibitoren).
Gegenstand der vorliegenden Erfindung ist somit auch eine Kombinationstherapie, bei der mindestens eine der erfindungsgemäßen Verbindungen oder ein pharmakologisch unbedenkliches Salz, Solvat oder Solvat eines Salzes davon zusammen mit mindestens einem zusätzlichen Agens, ausgewählt aus der Gruppe antivirale Agentien, immunomodulatorische Agentien, HCV Protease- Inhibitoren, HCV Polymerase-Inhibitoren, Inhibitoren eines anderen Targets im HCV-Lebenszyklus, HTV-Inhibitoren, HAV-Inhibitoren und HBV-Inhibitoren, verabreicht werden. Die zusätzlichen Agentien können mit den erfindungsgemäßen Verbindungen zu einer einzigen pharmazeu- tischen Dosierungsform vereinigt werden. Alternativ können diese zusätzlichen Agentien separat verabreicht werden. Solche zusätzlichen Agentien können vor, während oder nach der Gabe einer erfindungsgemäßen Verbindung oder eines pharmakologisch unbedenklichen Salzes, Solvates oder Solvates eines Salzes davon verabreicht werden.
Definitionen:
Der Tema "anti-virales Agens" meint ein Agens, das die Bildung und/oder Replikation eines Virus inhibiert. Das schließt Agentien ein, die in Mechanismen des Wirts oder des Virus eingreifen, die für die Bildung und/oder Replikation eines Virus notwendig sind. Anti-virale Agentien sind z.B. Ribavirin, Amantadin, VX-497 (Merimepodib, Vertex Pharmaceuticals), Viramidin, Ceplene (Maxamine), XTL-001 und XTL-002 (XTL-Biopharmaceuticals).
Der Term "anti-HCV-Agens" meint ein Agens, das Hepatitis C-bedingte Krankheitssymptome vermindert oder verhindert. Ein solches Agens kann ein anti-virales Agens, ein immunomodula- torisches Agens, ein HCV Protease-Inhibitor, ein HCV Polymerase-Inhibitor oder ein Inhibitor eines anderen Targets im HCV-Lebenszyklus sein.
Der Term "immunomodulatorisches Agens" meint ein Agens, das die Immunantwort verstärkt oder schädliche Immunreaktionen eindämmt. Immunomodulatorische Agentien sind z.B. Klasse I-Interferone (wie alpha-, beta-, delta- und omega-Interferone, tau-Interferone, consensus-Interferone und asialo-Interferone), Klasse Ü-Interferone (wie gamma-Interferone) und pegylierte Interferone sowie Substanzen wie Levovirin.
Der Term "HCV Protease-Inhibitor" meint ein Agens, das die Funktion der HCV NS2/3-Cystein- protease oder der NS3/4A-Serinprotease inhibiert. HCV NS3/4A-Serinprotease-Inhibitoren sind z.B. BILN 2061 (Boehringer Ingelheim), oder VX-950 /LY-570310 (Vertex/Eli Lilly).
Der Term "HCV Polymerase-Inhibitor" meint ein Agens, das die Funktion der HCV-Polymerase inhibiert. Dies schließt z.B. Inhibitoren der HCV NS5B-Polymerase ein. HCV Polymerase-Inhibi- toren schließen Nicht-Nukleoside ein, beispielsweise Verbindungen, die beschrieben sind in WO
02/100846 und WO 02/100851 (Shire), WO 01/85172 und WO 02/098424 (GSK), WO 00/06529 und WO 02/06246 (Merck), WO 01/47883 und WO 03/000254 (Japan Tobacco) und EP 1 256 628
(Agouron). Außerdem schließen HCV Polymerase-Inhibitoren Nukleosid- Analoga ein, beispiels- weise Verbindungen, die beschrieben sind in WO 01/90121 (Idenix), WO 02/069903 (Biochryst
Pharmaceuticals), WO 02/057287 und WO 02/057425 (Merck/Isis). Weitere Beispiele für HCV
Polymerase-Inhibitoren sind JTK-002, JTK-003 und JTK-109 (Japan Tobacco).
Der Term "Inhibitor eines anderen Targets im HCV-Lebenszyklus" meint ein Agens, das die Bildung und/oder Replikation von HCV auf andere Weise als durch die Inhibition der Funktion einer HCV Protease oder der HCV Polymerase inhibiert. Das schließt Agenden ein, die in Mechanismen des Wirts oder von HCV eingreifen, die für die Bildung und/oder Replikation von HCV notwendig sind. Inhibitoren eines anderen Targets im HCV-Lebenszyklus schließen Agentien ein, die beispielsweise eine Helicase oder eine IRES als Target inhibieren. Ein spezifisches Beispiel für einen Inhibitor eines anderen Targets im HCV-Lebenszyklus ist ISIS-14803 (ISIS-Pharma- ceuticals).
Der Term "HTV-Inhibitor" meint ein Agens, das die Bildung und/oder Replikation von HTV inhibiert. Das schließt Agentien ein, die in Mechanismen des Wirts oder von HTV eingreifen, die für die Bildung und/oder Replikation von HTV notwendig sind. HTV-Inhibitoren schließen z.B. nukleo- sidische Inhibitoren, nicht-nukleosidische Inhibitoren, Protease-Inhibitoren, Fusions-Inhibitoren und Integrase-Inhibitoren ein.
Der Term "HAV-Inhibitor" meint ein Agens, das die Bildung und/oder Replikation von HAV inhibiert. Das schließt Agentien ein, die in Mechanismen des Wirts oder von HAV eingreifen, die für die Bildung und/oder Replikation von HAV notwendig sind. HAV-Inhibitoren schließen z.B. Hepatitis A-Impfstoffe [z.B. Havrix® (GSK), VAQTA® (Merck), Avaxim® (Aventis Pasteur)] ein.
Der Term "HBV-Inhibitor" meint ein Agens, das die Bildung und/oder Replikation von HBV inhibiert. Das schließt Agentien ein, die in Mechanismen des Wirts oder von HBV eingreifen, die für die Bildung und/oder Replikation von HBV notwendig sind. HBV-Inhibitoren schließen z.B. Agentien ein, die die HBV DNA-Polymerase inhibieren, oder Hepatitis B-Impfstoffe. Spezifische
Beispiele von HBV-Inhibitoren sind: Lamivudine (Epivir-HBV®), Adefovir Dipivoxil, Entecavir, FTC (Coviracil®), DAPD (DXG), L-FMAU (Clevudine®), AM365 (Amrad), Ldt (Telbivudine), monoval-LdC (Valtorcitabine), BAY 41-4109 (Bayer), ACH-126,443 (L-Fd4C) (Achillion), MCC478 (Eli Lilly), Racivir (RCV), Fluoro-L- und D-Nukleoside, Robustaflavone, ICN2001-3 (ICN), Barn 205 (Novelos), XTL-001 (XTL), Imino-Zucker (Nony-DNJ) (Synergy), HepBzyme, sowie immunomodulatorische Produkte wie z.B. Interferon alpha-2b, HE2000 (Hollis-Eden), Theradigm (Epimmune), EHT899 (Enzo Biochem), Thymosin alpha-1 (Zadaxin®), HBV DNA Vaccine (PowderJect), HBV DNA Vaccine (Jefferon Center), HBV Antigen (OraGen), BayHep B® (Bayer), Nabi-HB® (Nabi) und Anti-hepatitis B (Cangene), sowie HBV-Impfstoffe wie z.B. Engerix B, Recombivax HB, GenHevac B, Hepacare, Bio-Hep B, TwinRix, Comvax, Hexavac.
Der Term "Klasse I-Interferone" meint ein Interferon ausgewählt aus einer Gruppe von Inter- feronen, die alle an den Rezeptor Typ I binden. Das schließt natürliche und synthetisch hergestellte Klasse I-Interferone ein. Beispiele von Klasse I-Interferonen sind alpha-, beta- und omega-Inter- ferone, tau-Interferone, consensus-Interferone und asialo-Interferone.
Der Term "Klasse Ü-Interferone" meint ein Interferon ausgewählt aus einer Gruppe von Inter- feronen, die alle an den Rezeptor Typ II binden. Beispiele von Klasse H-Interferonen sind gamma- Interferone.
Der Term "Behandlung" meint die Verabreichung eines Arzneimittels entsprechend der vorliegenden Erfindung, um die Krankheitssymptome von Hepatitis C zu lindem oder zu eliminieren und/oder um die Virenmenge zu reduzieren.
Der Term "Prophylaxe" meint die Verabreichung eines Arzneimittels entsprechend der vorliegenden Erfindung nach der Infektion mit HCV, aber vor dem Auftreten von Krankheitssymptomen und/oder vor der Detektion von HCV im Blut.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende schnell und/oder modifiziert die erfindungsgemäßen Verbindungen abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/ oder amorphisierter und/oder gelöster
Form enthalten, wie z.B. Tabletten (nichtüberzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die paren- terale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen, -sprays; lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augenpräparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme, Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Die erfϊndungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharma- zeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Laktose, Mannitol), Lösungsmittel (z.B. flüssige PoIy- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispiels- weise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und / oder Geruchskorrigentien.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei intravenöser Applikation Mengen von etwa 0.001 bis 20 mg/kg, vorzugsweise etwa 0.01 bis 5 mg/kg Körpergewicht zur Erzielung
wirksamer Ergebnisse zu verabreichen, und bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 50 mg/kg, vorzugsweise 0.1 bis 10 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Verwendete Abkürzungen:
AiBN a, α'-Azobis(isobutyronitrü)
CDCl3 Deuterochloroform
DC Dünnschichtchromatographie
DCI direkte chemische Ionisation (bei MS)
DCM Dichlormethan
DIEA N,N-Diisopropylethylamin (Hünig Base)
DMAP 4-N,N-Dimethylaminopyridin
DMSO Dimethylsulfoxid
DMF N,N '-Dimethylformamid d. Th. der Theorie
EE Ethylacetat (Essigsäureethylester)
EI Elektronenstoß-Ionisation (bei MS)
ESI Elektrospray-Ionisation (bei MS)
Fp. Schmelzpunkt ges. gesättigt h Stunde
HPLC Hochdruck-, Hochleistungsflüssigchromatograpbie konz. konzentriert
LC-MS Flüssigchromatographie-gekoppelte Massenspektroskopie
MS Massenspektroskopie
NMR Kernresonanzspektroskopie präp. präparative proz. prozentig
RP-HPLC Reverse Phase HPLC
RT Raumtemperatur
Rt Retentionszeit (bei HPLC)
THF Tetrahydrofuran
Allgemeine LCMS- und HPLC-Methoden:
Methode 1 (HPLC, präparative Trennung): Säule: CromSil Cl 8, 250 mm x 30 mm; Eluent A: Wasser, Eiuent B: Acetonitril; Gradient: 3 min 20%B -» 31 min 90%B -> 34 min 90%B -» 34.01 min 20%B; Laufzeit: 38 min; Fluss: 50 ml/min; UV-Detektion: 210 nm.
Methode 2 (LC-MS): Gerätetyp MS: Micromass Quattro LCZ; Gerätetyp HPLC: Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A -> 2.5 min 30%A -> 3.0 min 5%A -> 4.5 min 5%A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 50°C; UV-Detektion: 208- 400 nm.
Methode 3 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A -> 2.5 min 30%A -> 3.0 min 5%A -> 4.5 min 5%A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min. 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 4 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A -» 2.5 min 30%A -> 3.0 min 5%A -> 4.5 min 5%A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 5 (LC-MS): Gerätetyp MS: Waters ZQ 2000; Gerätetyp HPLC: Agilent 1100, 2-Säulen- Schaltung, Autosampier: HTC PAL; Säule: YMC-ODS-AQ, 50 mm x 4.6 mm, 3.0 μm; Eluent A: Wasser + 0.1% Ameisensäure, Eluent B: Acetonitril + 0.1% Ameisensäure; Gradient: 0.0 min 100%A -> 0.2 min 95%A -» 1.8 min 25%A -» 1.9 min 10%A -> 2.0 min 5%A -> 3.2 min 5%A -» 3.21 min 100%A -> 3.35 min 100%A; Ofen: 400C; Fluss: 3.0 ml/min; UV-Detektion: 210 nm.
Methode 6 (LC-MS): Methode: Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A ~> 2.5 min 30%A -> 3.0 min 5%A -> 4.5 min 5%A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min. 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 7 (LC-MS): Methode: Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Onyx Monolithic Cl 8, 100 mm x 3 mm. Eluent A: 1 1 Wasser + 0.5 ml
50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A ^ 2 min 65%A -> 4.5 min 5%A -» 6 min 5%A; Fluss: 2 ml/min; Ofen: 400C; UV- Detektion: 208- 400 πm.
Ausgangsverbindungen
Beispiel IA
3-Cyano-N-(3-oxo-l,3-dihydro-2-benzofuran-5-yl)benzolsulfonamid

Eine Lösung von 7 g (46.9 mmol) 6-Amino-l,3-dihydroisobenzofuran-l-on und 9.46 g (46.9 mmol) 3-Cyanobenzolsulfonsäurechlorid in 420 ml Acetonitril wird mit 5.57 g (70.4 mmol) Pyridin versetzt und 2 h bei RT gerührt. Der Ansatz wird mit Wasser versetzt und der ausgefallene Feststoff abgesaugt. Das Filtrat wird auf ca. zwei Drittel eingeengt und der ausgefallene Feststoff abgesaugt. Nach Trocknen der vereinten Feststoffe werden 11.53 g (78% d. Th.) des gewünschten Produktes erhalten.
LC-MS (Methode 3): R, = 1.96 min.
MS (ESIneg): m/z = 313 (M-H)".
1H-NMR (400 MHz, DMSO-(I6): δ = 5.32 (s, 2H), 7.48 (s, IH), 7.49 (d, IH), 7.59 (d, IH), 7.79 (t, IH), 8.05 (d, IH), 8.12 (d, IH), 8.23 (s, IH), 10.90, (s, IH).
Beispiel 2A
Methyl-5-[[(3-cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]-2-methoxybenzoat

Eine Lösung von 100 mg (0.552 mmol) Methyl-5-amino-2-methoxybenzoat und 111 mg (0.552 mmol) 3-Cyanobenzolsulfonsäurechlorid in 3 ml Acetonitril wird mit 52 mg (0.662 mmol) Pyridin
versetzt und 2 h bei RT gerührt. Dann werden 132 mg (0.552 mmol) 2,6-Dichlorbenzylbromid und 178 mg (1.38 mmol) Diisopropylethylamin zugegeben und 3 h bei 70°C gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und 74 mg (27% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode 2): R4 = 2.65 min.
MS (ESIpos): m/z = 505 (M+H)+.
1H-NMR (400 MHz, DMSOA): δ = 3.71 (s, 3H), 3.75 (s, 3H), 5.05 (s, 2H), 6.96-7.10 (m, 3H), 7.26 (t, IH), 7.36 (d, 2H), 7.85 (t, IH), 7.94 (d, IH), 8.11 (s, IH), 8.24 (d, IH).
Beispiel 3A
Methyl-2-chlor-5-[[(3-cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]benzoat

Eine Lösung von 100 mg (0.539 mmol) Methyl-5-amino-2-chlorbenzoat und 109 mg (0.539 mmol) 3-Cyanobenzolsulfonsäurechlorid in 3 ml Acetonitril wird mit 51 mg (0.647 mmol) Pyridin versetzt und 2 h bei RT gerührt. Dann werden 129 mg (0.539 mmol) 2,6-Dichlorbenzylbromid und 174 mg (1.35 mmol) Diisopropylethylamin zugegeben und 3 h bei 700C gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und 67 mg (24% d. Th.) des gewünschten Produkts isoliert.
LC-MS (Methode 4): R, = 2.67 min.
MS (ESIpos): m/z = 509 (M+H)+.
1H-NMR (400 MHz, DMSO-Cl6): δ = 3.81 (s, 3H), 5.07 (s, 2H), 7.14 (dd, IH), 7.27-7.34 (m, 2H), 7.38 (d, 2H), 7.49 (d, IH), 7.85 (t, IH), 7.94 (d, IH), 8.18 (s, IH), 8.26 (d, IH).
Beispiel 4A
tert.-Butyl-3-[[(3-cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]benzoat

Eine Lösung von 50 mg (0.26 mmol) tert.-Butyl-3-aminobenzoat und 52 mg (0.26 mmol) 3- Cyanobenzolsulfonsäurechlorid in 3 ml Acetonitril wird mit 25 mg (0.31 mmol) Pyridin versetzt und 2 h bei RT gerührt. Dann werden 62 mg (0.26 mmol) 2,6-Dichlorbenzylbromid und 84 mg (0.65 mmol) Diisopropylethylamin zugegeben und 3 h bei 700C gerührt. Anschließend wird der Ansatz über präparative HPLC getrennt (Methode 1) und 49 mg (37% d. Th.) des gewünschten Produkts isoliert.
LC-MS (Methode 3): R1 = 3.18 min.
MS (ESIpos): m/z = 517 (M+H)+.
1H-NMR (400 MHz, DMSO-Cl6): δ = 1.48 (s, 9H), 5.08 (s, 2H), 7.18-7.28 (m, 3H), 7.32-7.40 (m, 3H), 7.38 (d, 2H), 7.78 (d, IH), 7.88 (t, IH), 7.98 (d, IH), 8.10 (s, IH), 8.28 (d, IH).
Beispiel 5A
l-Brom-3-(brommethyl)-5-methylbenzol

Eine Lösung von 5 g (27 mmol) 5-Brom-m-xylol, 5.29 g (29.7 mmol) und 0.05 g (0.3 mmol) AiBN in 100 ml Chlorbenzol wird 1.5 h auf 1000C erhitzt. Die erhaltene Suspension wird über Kieselgel abgesaugt und der Filterkuchen mit DCM gewaschen. Das Filtrat wird zweimal mit IM Natriumthiosulfat-Lösung und einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über
Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird fraktioniert destilliert und das gewünschte Produkt mit einer Ausbeute von 4.95 g (69% d. Th.) erhalten.
GCMS (EI): m/z = 264
1H-NMR (400 MHz, DMSO-Cl6): δ = 2.3 (s, 3H), 4.65 (s, 2H), 7.27 (s, IH), 7.36 (s, IH), 7.47 (s, IH).
Beispiel 6A
l-Brom-3-(tert-butoxymethyl)-5-methylbenzol
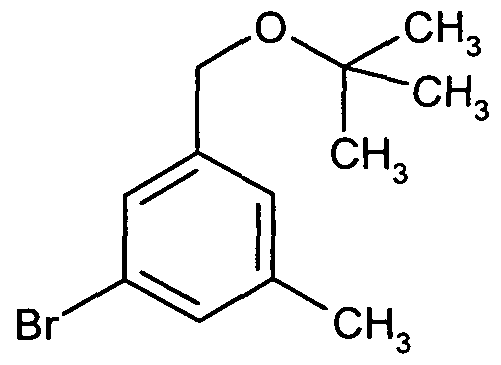
Zu einer Lösung von 15 g (56.8 mmol) l-Brom-3-(brommethyl)-5-methylbenzol (Beispiel 5A) in 150 ml THF werden bei 00C 9.57 g (85.2 mmol) Kalium-tert.-butylat zugegeben. Die cremefarbene Suspension wird 30 min bei 00C gerührt und dann mit Wasser versetzt. Das Gemisch wird mit Ethylacetat extrahiert, die organischen Phasen über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird über Kieselgel mit 0% bis 3% Etbylacetat/Cyclohexan chromatographiert. Es werden 10.96 g (75% d. Th.) des gewünschten Produktes erhalten.
GCMS (EI): m/z = 256
1H-NMR (400 MHz, DMSO-dö): δ = 1.22 (s, 9H), 2.28 (s, 3H), 4.35 (s, 2H), 7.1 (s, IH), 7.27 (s, IH).
Beispiel 7A
Methyl-3 -( { [3 -(tert-butoxymethyl)-5 -methylphenyl] sulfonyl } amino)benzoat

LC-MS (Methode 6) : R, = 2.63 min.
MS (ESIneg): m/z = 390 (M+H)\
Beispiel 8A
Memyl-3-[{[3-(tert-butoxymetiιyl)-5-methylphenyl]sulfonyl}(2,6-dichlorbenzyl)amino]benzoat

Zu einer Lösung von 10.1 g (25.8 mmol) Methyl-3-({[3-(tert-butoxymethyl)-5-methylphenyI]- sulfonyl}amino)benzoat (Beispiel 7A) in 100 ml Acetonitril werden 6.4 g (26.7 mmol) 2,6- Dichlorbenzylbromid und 6.3 ml (36.4 mmol) DEEA zugegeben und das Gemisch 16 h bei RT gerührt. Die Lösung wird eingeengt und der Rückstand über Kieselgel mit einem Gradienten von 0% bis 20% Ethylacetat in Cyclohexan chromatographiert. Es werden 13.5 g (95% d. Th.) des gewünschten Produktes erhalten.
LC-MS (Methode 6): R, = 3.30 min.
MS (ESIpos): m/z = 550 (M+H)+.
1H-NMR (400 MHz, DMSO-(I6): δ = 1.19 (s, 9H), 2.37 (s, 3H), 3.8 (s, 3H), 4.44 (s, 2H), 4.98 (s, 2H), 7.16 (d, IH), 7.23 (t, IH), 7.3-7.41 (m, 5H), 7.48 (d, 2H), 7.83 (d, IH).
Beispiel 9A
Methyl-3-[{[3-(hydroxymethyl)-5-methylphenyl]sulfonyl}(2,6-dichlorbenzyl)amino]benzoat

Eine Lösung von 13.5 g (24.5 mmol) Methyl-3-[{[3-(tert-butoxymethyl)-5-methylphenyl]- sulfonyl}(2,6-dichlorbenzyl)amino]benzoat (Beispiel 8A) in 210 ml 30 %iger TFA in DCM wird 3 h bei RT gerührt und dann eingeengt. Der Rückstand wird in 60 ml THF gelöst und mit 12 ml einer IM Lithiumhydroxid-Lösung versetzt. Nach 2 h Rühren bei RT wird mit gesättigter Natriumhydrogencarbonat-Lösung versetzt und mit Ethylacetat extrahiert. Die organische Phase wird mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Der Rückstand wird in Methanol digeriert, abfiltriert und das erhaltene Produkt getrocknet. Das Filtrat wird eingeengt und über Kieselgel mit einem Gradienten von 0% bis 40% Ethylacetat in Cyclohexan chromatographiert. Zusammen werden 10.5 g (87% d. Th.) des gewünschten Produktes erhalten.
LC-MS (Methode 6): Rt = 2.57 min.
MS (ESIpos): m/z = 494 (M+H)+.
1H-NMR (400 MHz, DMSOd6): δ = 2.37 (s, 3H), 3.8 (s, 3H), 4.54 (d, 2H), 4.98 (s, 2H), 5.39 (t, IH), 7.13 (d, IH), 7.23 (t, IH), 7.3-7.41 (m, 4H), 7.45 (d, 2H), 7.5 (s, IH), 7.83 (d, IH).
Beispiel IQA
Methyl-3-[{[3-(brommethyl)-5-methylphenyl]sulfonyl}(2,6-dichlorbenzyl)amino]benzoat

Zu einer Lösung von 30 g (60.7 mmol) Methyl-3-[{[3-(hydroxymethyl)-5-methylphenyl]sulfonyl}- (2,6-dichlorbenzyl)amino]benzoat (Beispiel 9A) in 750 ml Toluol werden 8.65 ml (91 mmol) Phoshphortribromid zugegeben und der Ansatz 18 h unter Rückfluss erhitzt. Anschließend wird auf RT abgekühlt und vorsichtig mit Wasser versetzt. Die organische Phase wird abgetrennt, mit gesättigter Natriumhydrogencarbonat-Lösung und gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert, eingeengt und am Hochvakuum getrocknet. Es werden 35 g (94% d. Th.) des gewünschten Produktes erhalten.
LC-MS (Methode 7): R1 = 4.34 min.
MS (ESIpos): m/z = 556 (M+H)+.
1H-NMR (400 MHz, DMSOd6): δ = 2.39 (s, 3H), 3.81 (s, 3H), 4.75 (s, 2H), 5.01 (s, 2H), 7.14 (d, IH), 7.25 (t, IH), 7.31-7.41 (m, 3H), 7.45 (s, 2H), 7.5 (s, IH), 7.66 (s, IH), 7.82 (d, IH).
Ausführunesbeispiele
Beispiel 1
3-Cyano-N-(2,6-dichlorbenzyl)-N-(3-oxo-l,3-dihydro-2-benzofuran-5-yl)benzolsulfonamid

Zu einer Lösung von 4.80 g (15.3 mmol) Beispiel IA in 100 ml DMF werden 733 mg (18.3 mmol) einer 60 proz. Dispersion von Natriumhydrid in Mineralöl und 229 mg (1.53 mmol) Natriumiodid gegeben. Das Gemisch wird 1 h bei 700C gerührt, dann mit 4.03 g (16.8 mmol) 2,6- Dichlorbenzylbromid versetzt und weitere 2 h bei 700C gerührt. Der Ansatz wird in 500 ml Wasser eingerührt, der Feststoff abfiltriert, mit Wasser gewaschen und im Hochvakuum getrocknet. Es werden 7.24 g (100% d. Th.) des gewünschten Produktes erhalten.
LC-MS (Methode 2): Rt = 2.58 min.
MS (ESIpos): m/z = 473 (M+H)+.
1H-NMR (400 MHz, DMSOd6): δ = 5.16 (s, 2H), 5.38 (s, 2 H), 7.27 (t, 3H), 7.33-7.38 (m, 4H), 7.57 (d, IH), 7.82 (t, IH), 7.88 (d, IH), 8.20 (s, IH), 8.25 (d, IH).
Beispiel 2
3-Cyano-N-(2,6-dimethylbenzyl)-N-(3-oxo-l,3-dihydro-2-benzofuran-5-yl)benzolsulfonamid

LC-MS (Methode 3): R1 = 2.63 min.
MS (ESIpos): m/z - 433 (M+H)+.
1H-NMR (400 MHz, DMSOd6): δ = 2.21 (s, 6H), 4.98 (s, 2H), 5.37 (s, 2 H), 6.86 (d, 2H), 6.97 (t, IH), 7.30 (d, IH), 7.36 (s, IH), 7.53 (d, IH), 7.82 (t, IH), 7.88 (d, IH), 8.19 (s, IH), 8.25 (d, IH).
Allgemeine Arbeitsvorschrift [Al ; AIkylierun2 von Sulfonamiden
Zu einer Lösung von 50 mg (0.159 mmol) Beispiel IA in 4 ml DMF werden 0.159 mmol eines substituierten Benzyllialogenids, 25 mg (0.191 mmol) Diisopropylethylamin und 6 mg (0.016 mmol) Tetrabutylammoniumiodid gegeben. Der Ansatz wird 20 h bei 600C gerührt, filtriert und über präparative HPLC (Methode 1) getrennt.
Die Beispiele 3 bis 9 werden in analoger Weise nach der allgemeinen Arbeitsvorschrift [A] aus den entsprechenden Ausgangsverbindungen hergestellt.


Beispiel 10
N-(2,6-Dichlorbenzyl)-3-methoxy-N-(3-oxo-l,3-dihydro-2-benzofuran-5-yl)benzolsulfonamid
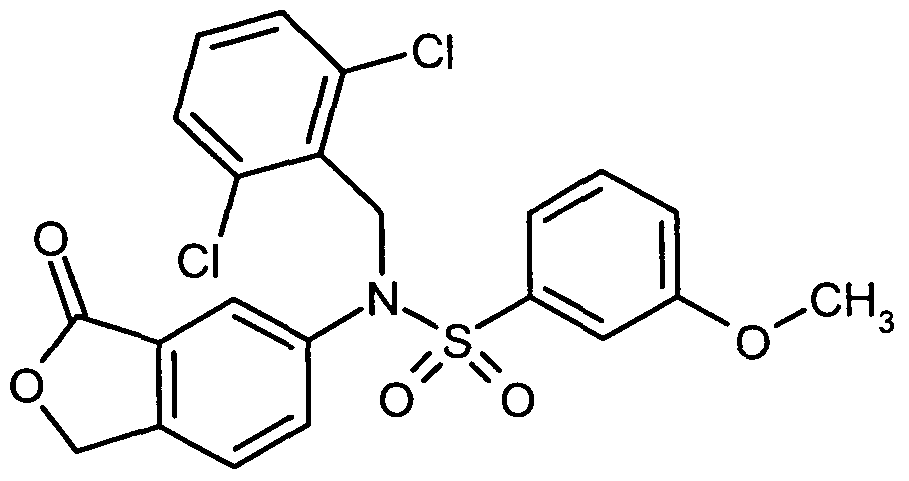
LC-MS (Methode 3): R, = 2.70 min.
MS (ESIpos): m/z = 478 (M+H)+.
1H-NMR (400 MHz, DMSOd6): δ = 3.80 (s, 3H), 5.07 (s, 2H), 5.38 (s, 2 H), 7.12 (s, IH), 7.18- 7.38 (m, 7H), 7.56 (t, IH).
Beispiel 11
3-Chlor-N-(2,6-dichlorbenzyl)-N-(3-oxo-l,3-dihydro-2-benzofuran-5-yl)benzolsulfonamid

Eine Lösung von 50 mg (0.335 mmol) 6-Amino-l,3-dihydroisobenzofuran-l-on und 71 mg (0.335 mmol) 3-Chlor-benzolsulfonsäurechlorid in 2 ml Acetonitril wird mit 32 mg (0.402 mmol) Pyridin versetzt und 24 h bei RT gerührt. Dann werden 80 mg (0.335 mmol) 2,6-Dichlorbenzylbromid und 108 mg (0.838 mmol) Diisopropylethylamin zugegeben und 18 h bei 700C gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und 88 mg (54% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode 3): Rt = 2.90 min.
MS (ESIpos): m/z = 482 (M+H)+.
1H-NMR (400 MHz, DMSO-O6): δ = 5.12 (s, 2H), 5.38 (s, 2 H), 7.26 (t, IH), 7.30-7.40 (m, 4H), 7.58 (t, 2H), 7.65 (m, 2H), 7.87 (d, IH).
Allgemeine Arbeitsvorschrift [Bl: Sulfonylierung und nachfolgende Alkylierung von 6- Amino-1.3-dihvdroisobenzofuran-l-on
Eine Lösung von 50 mg (0.335 mmol) 6-Amino-l,3-dihydroisobenzofuran-l-on und 0.335 mmol substituiertem Benzolsulfonsäurechlorid in 2 ml Acetonitril wird mit 32 mg (0.402 mmol) Pyridin versetzt und 24 h bei RT gerührt. Dann werden 80 mg (0.335 mmol) 2,6-Dichlorbenzylbromid und 108 mg (0.838 mmol) Diisopropylethylamin zugegeben und 18 h bei 700C gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und das gewünschte Produkt isoliert.
Die Beispiele 12 bis 14 werden in analoger Weise nach der allgemeinen Arbeitsvorschrift [B] aus den entsprechenden Ausgangsverbindungen hergestellt.

3 -[ [(3 -Cyanophenyl) sulfonyl] (2,6 -dichlorben2yl)amino]benzoesäure

Eine Lösung von 1 g (1.93 mmol) Beispiel 4A in 18 ml Dichloπnethan und 2 ml Trifluoressigsäure wird 30 min bei RT gerührt und dann eingeengt. 200 mg von 1084 mg des erhaltenen Rohprodukts werden mit Diethylether verrührt, über eine Fritte abgesaugt und getrocknet. Es werden 63 mg (7% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode 2): R, = 2.48 min.
MS (ESIpos): m/z = 461 (M+H)+.
1H-NMR (400 MHz, DMSOd6): δ = 5.10 (s, 2 H), 7.17 (d, IH), 7.25 (t, IH), 7.31-7.39 (m, 3H), 7.43 (s, IH), 7.83 (q, 2H), 7.92 (d, IH), 8.13 (s, IH), 8.25 (d, IH), 13.12 (br, IH).
Beispiel 16
5-[[(3-Cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]-2-methoxybenzoesäure

Eine Lösung von 59 mg (0.117 mmol) Beispiel 2A in 3 ml THF und 3 Tropfen Methanol wird mit 0.152 ml (0.152 mmol) einer 1 M wässrigen Natriumhydroxid-Lösung versetzt und 3 h bei 500C gerührt. Nach Trennung des Ansatzes über präparative HPLC (Methode 1) werden 16 mg (28% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode 4): R, = 2.20 min.
MS (ESIpos): m/z = 491 (M+H)+.
1H-NMR (400 MHz, DMSO-Cl6): δ = 3.60 (s, 3H), 5.02 (s, 2 H), 6.56 (d, IH), 6.63 (d, IH), 6.78 (s, IH), 7.25 (t, IH), 7.34 (d, 2H), 7.81 (t, IH), 7.89 (d, IH), 8.12 (s, IH), 8.22 (d, IH).
Allgemeine Arbeitsvorschrift [Cl : Verseifung von Carbonsäureestern
Eine Lösung von 0.1 mmol Carbonsäureester in 3 ml THF und 3 Tropfen Methanol wird mit 0.13 ml (0.13 mmol) einer 1 M wässrigen Natriumhydroxid-Lösung versetzt und 3 h bei 500C gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und das gewünschte Produkt isoliert.
Das Beispiel 17 wird in analoger Weise nach der allgemeinen Arbeitsvorschrift [C] aus den entsprechenden Ausgangsverbindungen hergestellt.

Beispiel 18
5-[[(3-Cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]-2-(hydroxymethyl)benzoesäure
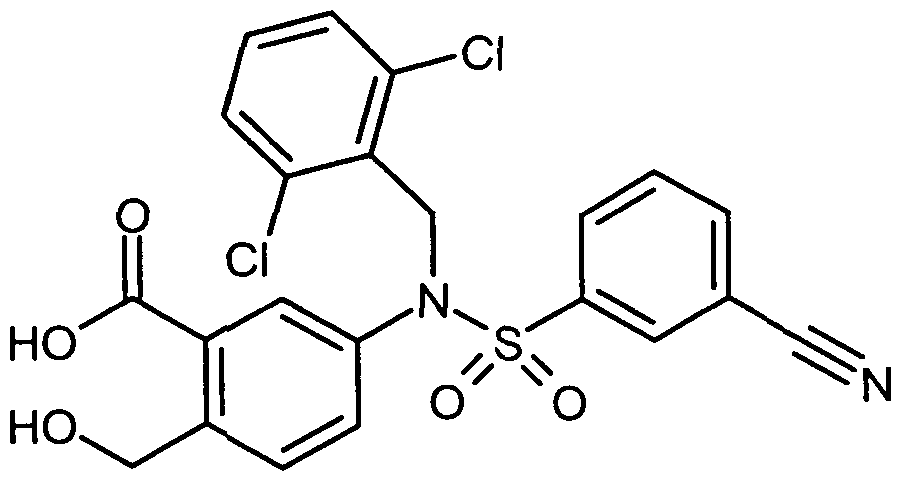
Eine Lösung von 500 mg (1.056 mmol) Beispiel 1 in 15 ml Dioxan wird mit 1.37 ml (1.37 mmol) einer 1 M wässrigen Lithiumhydroxid-Lösung versetzt und 18 h bei RT gerührt. Die Lösung wird
eingeengt und über präparative HPLC (Methode 1) getrennt. Es werden 278 mg (54% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode 3): R, = 2.48 min.
MS (ESIpos): m/z = 491 (M+H)+.
1H-NMR (400 MHz, DMSO-O6): δ = 4.33 (d, 2H), 5.07 (s, 2 H), 6.66 (dd, IH), 6.96 (2, IH), 7.23 (t, IH), 7.32 (d, 2H), 7.40 (d, IH), 7.82 (m, 2H), 7.89 (d, IH), 8.12 (s, IH), 8.21 (d, IH).
Beispiel 19
3-[[(3-Cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]benzamid

Eine Lösung von 500 mg (3.67 mmol) 3-Aminobenzamid und 740 mg (3.67 mmol) 3- Cyanobenzolsulfonsäurechlorid in 30 ml Acetonitril wird mit 349 mg (4.41 mmol) Pyridin versetzt und 2 h bei RT gerührt. Dann werden 881 mg (3.67 mmol) Dichlorbenzylbromid und 1.19 g (9.18 mmol) Diisopropylethylamin zugegeben und 3 h bei 700C gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und 360 mg (21% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode3): R, = 2.36 min.
MS (ESIpos): m/z = 460 (M+H)+.
1H-NMR (400 MHz, DMSO-Cl6): δ = 5.08 (s, 2H), 7.07 (d, IH), 7.25 (t, IH), 7.28-7.42 (m, 4H), 7.50 (s, IH), 7.27-7.85 (m, 2H), 7.88-7.95 (m, 2H), 8.12 (s, IH), 8.24 (d, IH).
Beispiel 20
3-[[(3-Cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]-N-methylbenzamid

Eine Lösung von 50 mg (0.33 mmol) 3-Amino-N-methylbenzamid und 67 mg (0.33 mmol) 3- Cyanobenzolsulfonsäurechlorid in 3 ml Acetonitril wird mit 32 mg (0.4 mmol) Pyridin versetzt und 2 h bei RT gerührt. Dann werden 80 mg (0.33 mmol) 2,6-Dichlorbenzylbromid und 108 mg (0.832 mmol) Diisopropylethylamin zugegeben und 3 h bei 700C gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und 48 mg (30% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode 3): R, = 2.45 min.
MS (ESIpos): m/z = 474 (M+H)+.
1H-NMR (400 MHz, DMSO-Cl6): δ = 2.72 (d, 3H), 5.08 (s, 2H), 7.03 (d, IH), 7.25 (t, IH), 7.28- 7.42 (m, 3H), 7.47 (s, IH), 7.74 (d, IH), 7.82 (t, IH), 7.90 (d, IH), 8.12 (s, IH), 8.24 (d, IH), 8.40 (q, IH).
Beispiel 21
3-[[(3-Cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]-N-cyclobutylbenzamid

Eine Lösung von 100 mg (0.217 mmol) 3-[[(3-Cyanophenyl)sulfonyl](2,6-dichlorbenzyl)amino]- benzoesäure (Beispiel 15) in 1.5 ml DMF wird mit 94 μl (0.542 mmol) DIEA und 84 mg (0.26
mmol) TBTU versetzt und 1 h bei RT gerührt. Dann werden 22 μl (0.26 mmol) Cyclobutylamin zugegeben und 18 h bei RT gerührt. Anschließend wird der Ansatz über präparative HPLC (Methode 1) getrennt und 74 mg (66% d. Th.) des gewünschten Produktes isoliert.
LC-MS (Methode 7): Rt = 3.85 min.
MS (ESIpos): m/z = 514 (M+H)+.
1H-NMR (400 MHz, DMSOd6): δ = 1.6-1.7 (m, 2H), 1.97-2.09 (m, 2H), 2.13-2.22 (m, 2H), 4.3- 4.4 (m, IH), 5.09 (s, 2H), 7.02 (d, IH), 7.25 (t, IH), 7.28-7.38 (m, 3H), 7.48 (s, IH), 7.78 (d, IH), 7.82 (t, IH), 7.90 (d, IH), 8.1 (s, IH), 8.24 (d, IH), 8.52 (d, IH).
Beispiel 22
3-[{[3-(3-AjnmotMeno[2,3-b]pyridm-2-yl)-5-memylphenyl]sulfonyl}(2,6-dicUorberizyl)arnino]- benzoesäure

Zu einer Lösung von 200 mg (0.36 mmol) Methyl-3-[{[3-(brommethyl)-5-methylphenyl]sulfonyl}- (2,6-dichlorbenzyl)amino]benzoat (Beispiel 10A) in 10 ml DMF werden 49 mg (0.36 mmol) 2- Mercaptonicotinonitril und 45 mg (0.54 mmol) Natriumhydrogencarbonat gegeben. Der Ansatz wird 18 h bei RT gerührt, dann mit 1.8 ml 1 M Lithiumhydroxid-Lösung versetzt und 2 h bei 600C gerührt. Nach Abkühlen auf RT wird mit 1 M Salzsäure neutralisiert und eingeengt. Nach Trennung des Rückstands über präparative HPLC (Methode 1) werden 90 mg (47% d. Th.) des Produktes erhalten.
LC-MS (Methode 7): R, = 4.03 min.
MS (ESIpos): m/z = 598 (M+H)+.
1H-NMR (400 MHz, DMSO-U6): δ = 2.36 (s, 3H), 4.6 (s, 2H), 4.93 (s, 2H), 7.0 (d, IH), 7.2-7.33 (m, 6H), 7.42 (s, IH), 7.59 (s, IH), 7.63 (s, IH), 7.75 (d, IH), 8.22 (d, IH), 8.69 (s, IH).
B. Bewertung der physiologischen Wirksamkeit
Abkürzungen:
DMSO Dimethylsulfoxid
DMEM Dulbecco's Modified Earle's Medium
FKS Fötales Kälberserum
G418 Geneticiα
ECso Effektive Konzentration 50
IC50 Inhibitorische Konzentration 50
CC50 Cytotoxische Konzentration 50
NOEC no observed effect concentration
Pen Penicillin
Strep Streptomycin min. Minuten
NEAA Nichtessentielle Aminosäuren
PBS Phosphate buffered sahne (Phosphatpuffer) h Stunden sec Sekunden
RT Raumtemperatur
Die in vitro-Wirkung der erfϊndungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
1. Messung der Substanzaktivität in einem zellulären HCV-RNA-Replikationsassay ("Replikon-Svstem")
Die Hepatits C Virus-RNA kann in Zellkultur im sogenannten Replikonsystem vermehrt werden. Eine Hemmung des Replikationszyklus des Hepatitis C Virus durch die zu testende Substanz kann daher in dem Replikonsystem simuliert werden. Bei dem Replikonsystem handelt es sich um Genomteile des HCV oder um gesamte Genome von HCV, welche in Zelllinien (hier HuH-7 Zellen) menschlichen Ursprungs verbracht werden. Durch Einfügen eines Selektionsmarkers können stabile Zelllinien gewonnen werden, welche unter Selektionsdruck genomische oder subgenomische RNA von HCV vermehren [Lohmann et al., Science 285, 110-113 (1999); Blight et al., Science 290, 1972-1974 (2000)]. Die hier verwendeten HuH5-2 Zellen beherbergen ein selektionierbares, Luciferase tragendes, zellkulturadaptiertes Replikon wie in EP 1 043 399
beschrieben. Die Zellen werden in Dulbecco's Modified Earle's Medium (DMEM) mit 10% fötalem Kälberserum (FKS), 1% Pen/Strep, 1% NEAA, 1% L-Glutamin und 250 μg/ml Geneticin (G418) kultiviert. Für die Durchfuhrung der unten beschriebenen Assays werden die Zellen zunächst trypsiniert, mit dem oben beschriebenen DMEM-Medium ohne G418 resuspendiert und in 96-well-Platten eingesät. In Abhängigkeit vom verwendeten Auslese- oder Testverfahren werden transparente oder weiße für die Zellkultur beschichtete Testplatten verwendet.
a) Zubereitung der Testsubstanzen:
Die Testverbindungen werden als 50 mM-Stammlösung in DMSO angesetzt. Für die Bestimmung der EC50- Werte werden die Substanzen anschließend in DMEM seriell in geeigneten Stufen (z.B. 100, 30, 10, 3 μM usw.) verdünnt. Anschließend erfolgt die Zugabe der trypsinierten, in Medium resuspendierten Zellen. Die Endkonzentrationen der Testsubstanzen in den Zellkulturkavitäten beträgt beispielsweise 100 μM bis 0.0001 μM. Interferon-alpha dient als Referenzsubstanz in Konzentrationen von 100 IU/ml bis 0,01 IU/ml. Nur mit DMSO behandelte Zellen dienen als Referenz (Placebokontrolle). Die Platten werden dann bei 37°C unter 5% CO2 für 4 Tage inkubiert. Im Anschluss daran erfolgen die unterschiedlichen Messungen bzw. die Quantifizierung der HCV-Replikon-RNA.
b) Zytotoxizitätstest (visuell):
Für die Bewertung der Zytotoxizität der Testsubstanzen auf HuH5-2 Zellen wird der obige Test in einer transparenten Zellkulturplatte angesetzt. Die qualitative Auswertung erfolgt visuell unter dem Mikroskop. Ermittelt wird hierbei der NOEC-Wert (no observed effect concentratiori), welcher der niedrigsten Substanzverdünnungsstufe entspricht, bei der keine Substanz-bedingten cytopathischen Effekte oder sonstige Beeinträchtigungen des Zellwachstums erkennbar sind.
c) Alamar Blue-Test (quantitativer Zytotoxizitätstest):
Alamar Blue ist ein wasserlöslicher Redox-Indikator, der in Abhängigkeit von der metabolischen Aktivität der zu untersuchenden Zellen reduziert wird. Der Alamar Blue-Test wird als quantitativer
Zytotoxizitätsassay verwendet. Hierfür werden die Zellen mit den entsprechenden Testsubstanzen
(siehe oben) in weiße 96-well-Zellkulturplatten eingesät und entsprechend bei 37°C unter 5% CO2 für 4 Tage inkubiert. 5 Stunden vor der Messung erfolgt die Zugabe von 1/10 des Kulturvolumens
Alamar Blue pro well. Anschließend wird die Fluoreszenz bei einer Emissionswellenlänge von 544 nm und bei einer Extinktionswellenlänge von 590 nm gemessen. Soll im Anschluss an diesen Test auf der gleichen Platte noch die Chemolumineszenzmessung erfolgen, wird die Farbstofflösung von den Zellen abgesaugt und entsprechend weiter behandelt (siehe d.).
d) Messung der HCV-RNA-Menge durch Ermittlung der Aktivität eines Reportergens:
In das HCV-Replicon-HuH5-2 ist das Gen für das Enzym Luciferase aus Photinus pyralis als Reportergen eingefügt. Nach Zugabe eines Gemisches bestehend aus gleichem Volumen an Lysispuffer (3 % Triton X-100, 11,5 % Glycerin, 2 mM DTT, 25 mM Na2HPO4, 25 mM Tris HCl, pH 7.8) und Luciferase-Substratpuffer (20 mM Tris/HCl, 20 mM Tricine, 2.67 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 0.27 mM Coenzym A, 0.47 mM Luciferin, 0.53 mM ATP, pH 7.8) zu den Zellen erfolgt die Chemolumineszenzmessung in einem Luminometer. Üblicherweise werden die Photonen in einem Zeitraum von 10 sec bis 60 sec gemessen.
Erhaltene Werte werden durch Kurvenanalysen (sigmoidale Dosis-Wirkungskurven mit variablem Kurvenverlauf; GraphPad Prism Version 4.02 für Windows, GraphPad Software Inc.) ausgewertet. Die folgenden Daten können von den Testplatten ermittelt werden:
NOEC (siehe b.)
CC50 = Substanzkonzentration in μM, bei der die Alamar Blue-Fluoreszenz im Vergleich zxir unbehandelten Kontrolle um 50% abnimmt;
EC5O ~ Substanzkonzentration, bei der die Luciferaseaktivität im Vergleich zur Placebokontrolle um 50% abnimmt;
SI (Selektivitätsindex) = CC5O/EC5o.
Repräsentative Wirkdaten aus diesem Test sind in der folgenden Tabelle A aufgeführt:
Tabelle A


e) Messung der Substanzaktivität mittels direkter Messung der HCV-Genommenge:
Zellen, welche subgenomische HCV-RNA vermehren, werden wie oben beschrieben in transparenten 96-well Platten in DMEM / 10% FKS ohne Zusatz von G418 in Gegenwart der Substanzen in geeigneten Verdünnungsstufen vermehrt (siehe a.). Nach 4 Tagen Inkubation wird das Medium verworfen, die Zellen einmal mit PBS gewaschen und zur Isolation der Gesamt-RNA mit dem Qiagen RNeasy 96 Kit (Bestell-Nr. 74181) nach Anleitung des Herstellers geerntet. Nach RNA-Isolation erfolgt die Elution in 50 μl RNase-freiem Wasser. Die RNA wird bei -800C gelagert. Mit Hilfe von TaqMan®-Assays (Fa. Applied Biosystems) wird die enthaltene Menge an HCV-RNA bestimmt. Die verwendeten Primer und Gensonden binden an die konservierte 5'- nichttranslatierte Region des Virusgenoms (primer für kodierenden DNA-Strang: aatgcctggagatttgggc; primer in Gegenrichtung: tttcgcgacccaacactactc; Gensonde: 6-Carboxy- fluorescin-tgcccccgcgagactgcatagc-N,N,N',N'-tetramethyl-6-carboxyrhodamine). Zur Standardisierung der eingesetzten Probe wird die Expression eines zelleigenen Genes bestimmt (TaqMan Ribosomal RNA Control Reagents, Fa. Applied Biosystems P/N 4308329). Für die Reaktion wird der Kit Platinum® Quantitative RT-PCR Theπnoscript™ one Step System der Fa. Invitrogen (Bestell-Nr. 12267-019) verwendet. Die Reaktion findet in einem Endvolumen von 25 μl mit 1 μl Probenvolumen statt. Die Reaktionsbedingungen sind: 30 min Inkubation bei 5O0C, danach 5 min Inkubation bei 950C. Nach diesem Schritt folgt die eigentliche Amplifikationsphase mit 40 Wiederholungen folgender Schritte: 15 sec Inkubation bei 95°C gefolgt von 1 min Inkubation bei 600C. Die Messung und Auswertung erfolgt in einem Applied Biosystems Abi Prism 7700 Sequence Detection-Gerät. Mit Hilfe der erhaltenen CT-Werte aus den Reaktionen für das Zielgen (hier: HCV) und das zelluläre Referenzgen (hier: 18s RNA) wird die relative Expression errechnet wie beschrieben unter: Abi Prism 7700 Sequence Detection System User Bulletin #2: Relative Quantification of Gene expression (P/N 4303859). Zur Ermittlung der EC5o-Konzentrationen
werden die erhaltenen Werte durch Kurvenanalysen (sigmoidale Dosis-Wirkungskurven mit variablem Kurvenverlauf; GraphPad Prism Version 4.02 für Windows, GraphPad Software Inc.) ausgewertet.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Tablette:
Zusammensetzung: 100 mg der Verbindung von Beispiel 1, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung: Die Mischung aus Wirkstoff, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat für 5 min. gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung: 1000 mg der Verbindung von Beispiel 1, 1000 mg Ethanol (96%), 400 mg Rhodigel (Xanthan gum der Fa. FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfϊndungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung: Das Rhodigel wird in Ethanol suspendiert, der Wirkstoff wird der Suspension zuge- fügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluss der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung: 500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g PoIy- ethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung: Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.
Claims
1. Verwendung einer Verbindung der Formel

in welcher
R1 für Hydroxy oder -NR9R10 steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff, (Ci-C6)-Alkyl, Benzyl, 2- Phenylethyl oder (C3-C6)-Cycloalkyl stehen,
wobei Benzyl und 2-Phenylethyl substituiert sein können mit 1 bis 3 Sub- stituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Ci-C4)-Alkyl, (CrC4)-Alkoxy und (C1- C4)-Alkylamino,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Ci-C4)-Alkyl, (C]-C4)-Alkoxy, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylamino substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl und Trifluormethoxy,
und wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (CrC4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylamino,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
# die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (CrC4)-Alkyl, (CrC4)-Alkoxy, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
wobei Alkyl, Alkoxy und Alkylamino substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausge- wählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy,
Amino, Trifluormethyl und Trifluormethoxy,
und
wobei Heterocyclyl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl,
Trifluormethoxy, (C,-C4)-Alkyl, (C]-C4)-Alkoxy und (CrC4)-Alkylamino,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff, (CrC4)-Alkyl, (C2-C4) -Alkenyl, (CrC4)-Alkoxy, (C1-C4)- Alkylthio, (CrC4)-Alkylamino, 5- bis 10-gliedriges Heteroaryl oder Rn-Y-CH2- steht,
wobei Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (CrC4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-CO-Alkylamino,
und
wobei Alkyl, Alkenyl, Alkoxy, Alkylthio und Alkylamino substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe, bestehend aus Phenyl und 5- bis 10-gliedriges Heteroaryl,
worin Phenyl und Heteroaryl substituiert sein können mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (C1-
C4)-Alkyl, (CrC4)-Alkoxy und (Ci-C4)-Alkylamino,
wobei
Y für ein Sauerstoffatom, ein Schwefelatom oder -N(R12)- steht,
wobei
R12 für Wasserstoff, (Ci-C4)-Alkyl oder (C3-C6)-Cycloalkyl steht,
R11 für 5- bis 10-gliedriges Heteroaryl steht,
worin Heteroaryl substituiert sein kann mit 1 bis 3 Substituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (Ci-C4)-Alkyl, (C]-C4)-
Alkoxy und (Ci-C4)-Alkylamino,
R6 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl, Trifluormethoxy oder 2- Cyanoethen-1-yl steht,
R7 für Wasserstoff, (CrC4)-Alkyl, (C2-C4)-Alkenyl, (C1-Q)-AIkOXy, (CrC4)-Alkyl- thio oder (C1-C4)-Alkylamino steht,
wobei Alkyl, Alkenyl, Alkoxy, Alkylthio und Alkylamino substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe, bestehend aus Phenyl und 5- bis 10-gliedriges Heteroaryl, worin Phenyl und Heteroaryl substituiert sein können mit 1 bis 3 Sub- stituenten, wobei die Substituenten unabhängig voneinander ausgewählt werden aus der Gruppe, bestehend aus Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, Aminocarbonyl, Aminosulfonyl, (Q- C4)-Alkyl, (CrC4)-Alkoxy und (C1-C4)-Alkylamino,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht,
oder eines ihrer Salze, ihrer Solvate oder der Solvate ihrer Salze,
zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von viralen Erkrankungen.
2. Verwendung einer Verbindung nach Anspruch 1, dadurch gekennzeichnet, dass
R1 für Hydroxy oder -NR9R10 steht,
wobei
R9 und R10 unabhängig voneinander für Wasserstoff oder (C]-C6)-Alkyl stehen,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy, (CrC4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
oder
R1 und R2 über eine *-O-CH2-# Kette verbunden sind und einen Ring bilden,
wobei
* die Anknüpfstelle an das Carbonylkohlenstoffatom und
" die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff steht,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff steht, R6 für Halogen, Cyano, Nitro, Methyl, Trifluormethyl, Trifluormethoxy oder 2-Cyanoethen-l-yl steht,
R7 für Wasserstoff steht,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht.
3. Verbindung der Formel

in welcher
R1 für Hydroxy oder -NR9R10 steht,
wobei
Ry und R .110U unabhängig voneinander für Wasserstoff oder (CrC6)-Alkyl stehen,
R2 für Wasserstoff, Halogen, Cyano, Hydroxy, Arnino, Trifluormethyl, Trifluormeth- oxy, (Ci-C4)-Alkyl, (CrC4)-Alkoxy, (Ci-C4)-Alkylamino oder ein über ein Stickstoffatom gebundenes 5- bis 7-gliedriges Heterocyclyl steht,
oder
R1 und Rz über eine *-O-CH2-s Kette verbunden sind und einen Ring bilden,
wobei
die Anknüpfstelle an das Carbonylkohlenstoffatom und
die Anknüpfstelle an den Phenylring ist,
R3 für Wasserstoff steht,
R4 für Halogen, Cyano, Methyl, Ethyl oder Cyclopropyl steht,
R5 für Wasserstoff steht,
R6 für Halogen, Cyano, Nitro, Methyl, Trifluormethyl, Trifiuormethoxy oder 2- Cyanoethen-1-yl steht, R7 für Wasserstoff steht,
R8 für Halogen, Cyano, Nitro, Methyl, Ethyl, Trifluormethyl oder Trifluormethoxy steht,
oder eines ihrer Salze, ihrer Solvate oder der Solvate ihrer Salze.
4. Verfahren zur Herstellung einer Verbindung der Formel (I) nach Anspruch 1, dadurch gekennzeichnet, dass eine Verbindung der Formel

in welcher
R > 1 , r R>2 , n R3 , T R>4 und R die in Anspruch 1 angegebene Bedeutung haben,
mit einer Verbindung der Formel

in welcher
R6, R7 und R8 die in Anspruch 1 angegebene Bedeutung haben, und
X1 für Halogen, bevorzugt Chlor oder Brom, steht,
umgesetzt wird.
5. Verbindung nach Anspruch 3 zur Behandlung und/oder Prophylaxe von Krankheiten.
6. Verwendung einer Verbindung nach Anspruch 3 zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Krankheiten.
7. Verwendung einer Verbindung nach Anspruch 3 zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Virusinfektionen.
8. Verwendung nach einem der Ansprüche 1, 2 oder 7, dadurch gekennzeichnet, dass die Virusinfektion eine Infektion mit dem Hepatitis C Virus ist.
9. Arzneimittel enthaltend eine Verbindung, wie in Anspruch 3 definiert, in Kombination mit einem weiteren Wirkstoff.
10. Arzneimittel enthaltend eine Verbindung nach Anspruch 3 in Kombination mit einem inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoff.
11. Arzneimittel nach Anspruch 10 zur Behandlung und/oder Prophylaxe von Virusinfek- tionen.
12. Verfahren zur Bekämpfung von Virusinfektionen in Menschen und Tieren durch Verabreichung einer antiviral wirksamen Menge mindestens einer Verbindung nach Anspruch 3, eines Arzneimittels nach Anspruch 9 oder 10 oder eines nach einem der Ansprüche 1, 2, 6 oder 7 erhaltenen Arzneimittels.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102006013957A DE102006013957A1 (de) | 2006-03-27 | 2006-03-27 | Substituierte N-Benzyl-N-phenylbenzolsulfonamide |
| DE102006013957.7 | 2006-03-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007110171A1 true WO2007110171A1 (de) | 2007-10-04 |
Family
ID=38180503
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/002441 Ceased WO2007110171A1 (de) | 2006-03-27 | 2007-03-20 | Substituierte n-benzyl-n-phenylbenzolsulfonamide zur behandlung von virusinfektionen |
Country Status (2)
| Country | Link |
|---|---|
| DE (1) | DE102006013957A1 (de) |
| WO (1) | WO2007110171A1 (de) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010023448A1 (en) * | 2008-08-29 | 2010-03-04 | Xention Limited | Novel potassium channel blockers |
| WO2010139953A1 (en) | 2009-06-04 | 2010-12-09 | Xention Limited | Compounds |
| WO2010139967A1 (en) | 2009-06-04 | 2010-12-09 | Xention Limited | Dihydroindole and tetrahydroisoquinoline derivatives useful as potassium channel inhibitors |
| CN102395273A (zh) * | 2009-02-27 | 2012-03-28 | 西佳技术公司 | 用于治疗和预防登革热病毒感染的噻吩并吡啶衍生物 |
| US8258138B2 (en) | 2008-08-29 | 2012-09-04 | Xention Limited | Potassium channel blockers |
| US8372840B2 (en) | 2008-08-29 | 2013-02-12 | Xention Limited | Potassium channel blockers |
| US12398097B2 (en) | 2019-07-29 | 2025-08-26 | Vanderbilt University | WDR5-MYC inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004026823A1 (en) * | 2002-09-20 | 2004-04-01 | Pfizer Products Inc. | Amide and sulfonamide ligands for the estrogen receptor |
| WO2004092146A2 (en) * | 2003-04-14 | 2004-10-28 | The Institutes For Pharmaceutical Discovery, Llc | N- (((((1,3-thiazol-2-yl) amino) carbonyl) phenyl) sulfonyl) phenylalanine derivatives and related compounds for the treatment of diabetes |
| WO2006072348A2 (de) * | 2004-12-22 | 2006-07-13 | Aicuris Gmbh & Co. Kg | Alkinyl-substituierte thiophene |
-
2006
- 2006-03-27 DE DE102006013957A patent/DE102006013957A1/de not_active Withdrawn
-
2007
- 2007-03-20 WO PCT/EP2007/002441 patent/WO2007110171A1/de not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004026823A1 (en) * | 2002-09-20 | 2004-04-01 | Pfizer Products Inc. | Amide and sulfonamide ligands for the estrogen receptor |
| WO2004092146A2 (en) * | 2003-04-14 | 2004-10-28 | The Institutes For Pharmaceutical Discovery, Llc | N- (((((1,3-thiazol-2-yl) amino) carbonyl) phenyl) sulfonyl) phenylalanine derivatives and related compounds for the treatment of diabetes |
| WO2006072348A2 (de) * | 2004-12-22 | 2006-07-13 | Aicuris Gmbh & Co. Kg | Alkinyl-substituierte thiophene |
Non-Patent Citations (1)
| Title |
|---|
| DATABASE REGISTRY 2 March 2004 (2004-03-02), XP002440508, Database accession no. RN 656815-36-0 * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010023448A1 (en) * | 2008-08-29 | 2010-03-04 | Xention Limited | Novel potassium channel blockers |
| US9073834B2 (en) | 2008-08-29 | 2015-07-07 | Xention Limited | Potassium channel blockers |
| US8673901B2 (en) | 2008-08-29 | 2014-03-18 | Xention Limited | Potassium channel blockers |
| JP2012500836A (ja) * | 2008-08-29 | 2012-01-12 | ゼンション・リミテッド | 新規なカリウムチャネルブロッカー |
| US8372840B2 (en) | 2008-08-29 | 2013-02-12 | Xention Limited | Potassium channel blockers |
| US8258138B2 (en) | 2008-08-29 | 2012-09-04 | Xention Limited | Potassium channel blockers |
| EP2400845A4 (de) * | 2009-02-27 | 2012-12-19 | Siga Technologies Inc | Thienopyridinderivate zur behandlung und vorbeugung von dengue-virusinfektionen |
| CN102395273A (zh) * | 2009-02-27 | 2012-03-28 | 西佳技术公司 | 用于治疗和预防登革热病毒感染的噻吩并吡啶衍生物 |
| AU2010218152B2 (en) * | 2009-02-27 | 2015-04-09 | Siga Technologies, Inc. | Thienopyridine derivatives for the treatment and prevention of Dengue virus infections |
| US9301949B2 (en) | 2009-02-27 | 2016-04-05 | Siga Technologies, Inc. | Thienopyridine derivatives for the treatment and prevention of dengue virus infections |
| US8399481B2 (en) | 2009-06-04 | 2013-03-19 | Xention Ltd | Compounds |
| US8426442B2 (en) | 2009-06-04 | 2013-04-23 | Xention Ltd | Compounds |
| WO2010139967A1 (en) | 2009-06-04 | 2010-12-09 | Xention Limited | Dihydroindole and tetrahydroisoquinoline derivatives useful as potassium channel inhibitors |
| WO2010139953A1 (en) | 2009-06-04 | 2010-12-09 | Xention Limited | Compounds |
| US12398097B2 (en) | 2019-07-29 | 2025-08-26 | Vanderbilt University | WDR5-MYC inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102006013957A1 (de) | 2007-10-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE10359791A1 (de) | Substituierte Thiophene | |
| DE112021000413B4 (de) | Funktionalisierte Peptide als antivirale Wirkstoffe | |
| TWI250165B (en) | Hepatitis C inhibitor tri-peptides | |
| DE60312824T2 (de) | Tripeptide zur inhibierung von ns3 (hepatitis c) umfassend einen hydroxyprolinrest, die durch etherbindung an einen substituiertes chinolin gebunden sind | |
| DE102004061746A1 (de) | Alkinyl-substituierte Thiophene | |
| DE60315420T2 (de) | Heterocyclische tripeptide als hepatitis c-inhibitoren | |
| CN107820496B (zh) | 用于治疗和预防乙型肝炎病毒感染的新的四环4-氧代-吡啶-3-甲酸衍生物 | |
| EP1730110B1 (de) | Schwefelverbindungen als inhibitoren der ns3-serinprotease des hepatitis-c-virus | |
| CN105001225B (zh) | 脂质合成的杂环调节剂 | |
| EP2334683B1 (de) | Gesättigte bicyclische heterocyclische derivate als smo-antagonisten | |
| TW201408669A (zh) | Hcv ns3蛋白酶抑制劑 | |
| CA2557322A1 (en) | Inhibitors of hepatitis c virus ns3 protease | |
| WO2007110171A1 (de) | Substituierte n-benzyl-n-phenylbenzolsulfonamide zur behandlung von virusinfektionen | |
| CN106459032A (zh) | 治疗和预防乙型肝炎病毒感染的新的二氢喹嗪酮类 | |
| ZA200604638B (en) | Inhibitors of hepatitis C virus NS3/NS4A serine protease | |
| CA2557247A1 (en) | Compounds as inhibitors of hepatitis c virus ns3 serine protease | |
| TW200827368A (en) | Amido anti-viral compounds | |
| TW200817405A (en) | Pyrro[1,2-b]pyridazinone compounds | |
| TW200817413A (en) | Polycyclic viral inhibitors | |
| WO2006072348A2 (de) | Alkinyl-substituierte thiophene | |
| JP2021529159A (ja) | N−置換テトラヒドロチエノピリジン誘導体及びその使用 | |
| WO2010082044A1 (en) | Unsaturated bicyclic heterocyclic derivatives as smo antagonists | |
| DE60307804T2 (de) | Pyranon- und pyrandioninhibitoren der rna-abhängigen rna-polymerase des hepatitis-c-virus | |
| EP1966218B1 (de) | Neue, zyklisch substituierte furopyrimidin-derivate und ihre verwendung zur behandlung von kardiovaskulären erkrankungen | |
| CN106470974B (zh) | 用于治疗病毒感染的化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07723406 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07723406 Country of ref document: EP Kind code of ref document: A1 |