明 細 書
ポリカーボネート共重合体及びその製造方法
技術分野
[0001] 本発明は、バイオマス資源であるデンプンなどの糖質力 誘導することができる構 成単位を含有する、耐熱性、成形性、及び機械的強度に優れ、かつ屈折率が小さく 、アッベ数が大きいという優れた光学特性を有するポリカーボネート共重合体と、その 製造方法に関する。
背景技術
[0002] ポリカーボネートは一般的に石油資源力 誘導される原料を用いて製造される。し 力しながら、近年、石油資源の枯渴が危惧されており、植物などのバイオマス資源か ら得られる原料を用いたポリカーボネートの提供が求められている。また、二酸化炭 素排出量の増加、蓄積による地球温暖化が、気候変動などをもたらすことが危惧され ていることからも、使用後の廃棄処分をしてもカーボン-ユートラルな、植物由来モノ マーを原料としたポリカーボネートの開発が求められている。
[0003] 従来、植物由来モノマーとしてイソソルビドを使用し、炭酸ジフエ-ルとのエステル 交換により、ポリカーボネートを得ることが提案されている(例えば、特許文献 1参照) 。し力しながら、得られたポリカーボネートは、褐色であり、満足できるものではない。 また、イソソルビドと他のジヒドロキシ化合物との共重合ポリカーボネートとして、ビスフ ェノール Aを共重合したポリカーボネートが提案されており(例えば、特許文献 2参照 )、更に、イソソルビドと脂肪族ジオールとを共重合することにより、イソソルビドからな るホモポリカーボネートの剛直性を改善する試みがなされている(例えば、特許文献 3参照)。
[0004] 一方、脂環式ジヒドロキシィ匕合物である 1 , 4 シクロへキサンジメタノールを重合し たポリカーボネートとしては、多数提案されているが(例えば、特許文献 4、 5)これら のポリカーボネートの分子量は高々 4000程度と低分子量のものであり、このため、ガ ラス転移温度が低 、ものが多!、。
[0005] このようにイソソルビドを用いたポリカーボネートは、多数提案されている力 イソソ
ルビドと脂環式ジヒドロキシ化合物とを共重合したポリカーボネートは報告されておら ず、また、屈折率、アッベ数などの光学定数も開示されていない。
特許文献 1: GB1079686号公報
特許文献 2:特開昭 56 - 55425号公報
特許文献 3: WO2004Z111106公報
特許文献 4:特開平 6— 145336号公報
特許文献 5:特公昭 63— 12896号公報
発明の開示
発明が解決しょうとする課題
[0006] 特許文献 1〜5に記載されているポリカーボネートは、石油原料由来の従来の芳香 族ポリカーボネートに比べ、耐熱性、透明性の点で不十分であり、光学材料や成形 材料に用いることが困難であった。このため、芳香族ポリカーボネートの高い耐熱性 と透明性を維持しながら、屈折率が小さぐアッベ数が小さい高透明性のポリカーボ ネートの開発が望まれている。
[0007] 本発明の目的は、上記従来の問題点を解消し、機械的強度に優れ、耐熱性があり 、屈折率が小さぐアッベ数が大きぐ複屈折が小さぐ透明性に優れた、植物由来の 構成単位を含むポリカーボネート共重合体を提供することにある。
課題を解決するための手段
[0008] 本発明者は、上記課題を解決するべぐ鋭意検討を重ねた結果、下記一般式(1) で表されるジヒドロキシィ匕合物と脂環式ジヒドロキシィ匕合物とから得られるポリカーボ ネート共重合体が、機械的強度に優れ、耐熱性があり、屈折率が小さぐアッベ数が 大きぐ複屈折が小さぐ透明性に優れていることを見出し、本発明に到達した。 即ち、本発明の要旨は下記 [1]〜[13]に存する。
[0009] [1] 下記一般式(1)で表されるジヒドロキシ化合物に由来する構成単位と脂環式ジ ヒドロキシィ匕合物に由来する構成単位とを含むポリカーボネート共重合体であって、 アッベ数が 50以上であり、且つ 5%熱減量温度が 340°C以上であることを特徴とする ポリカーボネート共重合体。
[0010] [2] 下記一般式(1)で表されるジヒドロキシィ匕合物に由来する構成単位と脂環式ジ ヒドロキシィ匕合物に由来する構成単位とを含むポリカーボネート共重合体であって、 該共重合体を構成する全ジヒドロキシィ匕合物に対する、下記一般式(1)で表されるジ ヒドロキシィ匕合物と脂環式ジヒドロキシィ匕合物の割合が 90モル%以上であることを特 徴とするポリカーボネート共重合体。
[化 2]
[0011] [3] ポリカーボネート共重合体を構成する全ジヒドロキシィ匕合物に対する、前記一 般式(1)で表されるジヒドロキシィ匕合物と脂環式ジヒドロキシィ匕合物の割合が 90モル
%以上である [1]に記載のポリカーボネート共重合体。
[0012] [4]前記脂環式ジヒドロキシ化合物力 5員環構造、又は 6員環構造を含むことを特 徴とする [1]な 、し [3]の 、ずれかに記載のポリカーボネート共重合体。
[0013] [5] 前記脂環式ジヒドロキシ化合物に含まれる炭素原子数が 30以下である [4]に 記載のポリカーボネート共重合体。
[0014] [6] 前記脂環式ジヒドロキシィ匕合物力 シクロへキサンジメタノール、トリシクロデカン ジメタノール、ァダマンタンジオール、及びペンタシクロペンタデカンジメタノールより なる群力 選ばれる少なくとも一種の化合物であることを特徴とする [5]に記載のポリ カーボネート共重合体。
[0015] [7] 光弾性係数が 20 X 10_ 12Pa_1以下であることを特徴とする [1]ないし [6]のい ずれかに記載のポリカーボネート共重合体。
[0016] [8] アイゾット衝撃強度が 30jZm2以上であることを特徴とする [1]ないし [7]のい ずれかに記載のポリカーボネート共重合体。
[0017] [9] 110°Cでの単位面積あたりのフエノール成分以外の発生ガス量が 5ngZcm2以 下であることを特徴とする [1]な 、し [8]の 、ずれかに記載のポリカーボネート共重合 体。
[0018] [10] 前記一般式(1)で表されるジヒドロキシィ匕合物に由来する構成単位として、ィ ソソルビド、イソマン-ド、及びイソイデットよりなる群力 選ばれる少なくとも 1種に由 来する構成単位を含むことを特徴とする [ 1]な 、し [9]の 、ずれか〖こ記載のポリカー ボネート共重合体。
[0019] [11] フエノールと 1, 1, 2, 2—テトラクロロェタンの重量比 1 : 1溶液での 20。C±0.
1°Cにおける濃度 1. OOgZdlの還元粘度が 0. 40dlZg以上であることを特徴とする
[1]ないし [10]のいずれかに記載のポリカーボネート共重合体。
[0020] [12] 下記一般式(1)で表されるジヒドロキシィ匕合物と脂環式ジヒドロキシ化合物とを 重合触媒の存在下、炭酸ジエステルと反応させることを特徴とする [ 1 ]ないし [ 11 ]の いずれかに記載のポリカーボネート共重合体の製造方法。
[0021] [13] 重合触媒として、アルカリ金属化合物及び Z又はアルカリ土類金属化合物を 用いることを特徴とする [12]に記載のポリカーボネート共重合体の製造方法。
発明の効果
[0022] 本発明のポリカーボネート共重合体は、熱安定性が高ぐ屈折率が低ぐアッベ数 が大きぐ光学的異方性が小さい。また、機械的強度に優れ、用途に応じてガラス転 移温度を 45°Cから 155°Cまで調整できるので、柔軟性が必要な、フィルム、シート分 野、耐熱性が必要な、ボトル、容器分野、さら〖こは、カメラレンズ、ファインダーレンズ
、 CCDや CMOS用レンズなどのレンズ用途、液晶やプラズマディスプレイなどに利 用される位相差フィルム、拡散シート、偏光フィルムなどのフィルム、シート、光デイス ク、光学材料、光学部品、色素、電荷移動剤等を固定化するバインダー用途といつ た幅広 、分野への材料提供が可能である。
図面の簡単な説明
[0023] [図 1]図 1は、実施例 1で製造されたポリカーボネート共重合体の NMRチャートを示 す図である。
[図 2]図 2は、実施例 26で製造されたポリカーボネート共重合体の NMRチャートを示 す図である。
発明を実施するための最良の形態
[0024] 以下に本発明の実施の形態を詳細に説明するが、以下に記載する構成要件の説 明は、本発明の実施態様の一例 (代表例)であり、本発明はその要旨を超えない限り
、以下の内容に限定されない。
[0025] 本発明のポリカーボネート共重合体は、下記一般式(1)で表されるジヒドロキシ化 合物に由来する構成単位と脂環式ジヒドロキシィ匕合物に由来する構成単位とを含む ことを特徴とするものである。
[0026] [化 4]
[0027] 本発明において、上記一般式(1)で表されるジヒドロキシィ匕合物としては、立体異 性体の関係にある、イソソルビド、イソマン-ド、イソイデットが挙げられ、これらは 1種 を単独で用いても良ぐ 2種以上を組み合わせて用 、ても良 、。
[0028] これらのジヒドロキシィ匕合物のうち、資源として豊富に存在し、容易に入手可能な種 々のデンプン力 製造されるソルビトールを脱水縮合して得られるイソソルビド力 入 手及び製造のし易さ、光学特性、成形性の面力も最も好ましい。
[0029] なお、イソソルビドは酸素によって徐々に酸化されやすいので、保管や、製造時の 取り扱いの際には、酸素による分解を防ぐため、水分が混入しないようにし、また、脱 酸素剤を用いたり、窒素雰囲気下にしたりすることが肝要である。イソソルビドが酸ィ匕 されると、蟻酸をはじめとする分解物が発生する。例えば、これら分解物を含むイソソ ルビドを用いてポリカーボネートを製造すると、得られるポリカーボネートに着色が発 生したり、物性を著しく劣化させる原因となる。また、重合反応に影響を与え、高分子 量の重合体が得られないこともあり、好ましくない。また、蟻酸の発生を防止するような 安定剤を添加してあるような場合、安定剤の種類によっては、得られるポリカーボネ ートに着色が発生したり、物性を著しく劣化させたりする。安定剤としては還元剤や制 酸剤が用いられる力 還元剤としては、ナトリウムポロハイドライド、リチウムポロハイド ライドなどがあげられ、制酸剤としては水酸ィ匕ナトリウム等のアルカリが挙げられるが、 このようなアルカリ金属塩の添カ卩は、アルカリ金属が重合触媒ともなるので、過剰に添 加し過ぎると重合反応を制御できなくなり、好ましくな 、。
[0030] 酸ィ匕分解物を含まないイソソルビドを得るために、必要に応じてイソソルビドを蒸留 しても良い。また、イソソルビドの酸ィ匕や、分解を防止するために安定剤が配合されて いる場合も、必要に応じて、イソソルビドを蒸留しても良い。これらの場合イソソルビド の蒸留は単蒸留であっても、連続蒸留であっても良ぐ特に限定されない。雰囲気は アルゴンや窒素などの不活性ガス雰囲気にした後、減圧下で蒸留を実施する。
[0031] このようなイソソルビドの蒸留を行うことにより、本発明では蟻酸含有量が 20PPM以 下、特に 5PPM以下であるような高純度のイソソルビドを用いることが好ましい。 なお、イソソルビド中の蟻酸含有量の測定方法は、実施例の項で後述する通りであ る。
[0032] 一方、本発明に使用できる、脂環式ジヒドロキシィ匕合物としては、特に限定されない 力 通常、 5員環構造又は 6員環構造を含む化合物を用いる。また、 6員環構造は共 有結合によって椅子形もしくは舟形に固定されていてもよい。脂環式ジヒドロキシィ匕 合物が 5員環、 6員環構造であることにより、得られるポリカーボネートの耐熱性を高く することができる。脂環式ジヒドロキシィ匕合物に含まれる炭素原子数は通常 70以下で あり、好ましくは 50以下、さらに好ましくは 30以下である。この値が大きくなるほど、耐
熱性が高くなるが、合成が困難になったり、精製が困難になったり、コストが高価だつ たりする。炭素原子数が小さくなるほど、精製しやすぐ入手しやすくなる。
[0033] 本発明で用いる 5員環構造又は 6員環構造を含む脂環式ジヒドロキシィ匕合物として は、具体的には、下記一般式 (Π)又は (III)で表される脂環式ジヒドロキシィ匕合物が挙 げられる。
HOCH R1— CH OH (II)
2 2
HO - R2 - OH (III)
(式 (Π) , (III)中、 R1, R2は、炭素数 4〜20のシクロアルキル基、又は炭素数 6〜20 のシクロアルコキシル基を表す。 )
[0034] 上記一般式 (Π)で表される脂環式ジヒドロキシィ匕合物であるシクロへキサンジメタノ ールとしては、一般式 (Π)において、 R1が下記一般式 (Ila) (式中、 R3は炭素数 1〜1 2のアルキル基を表す。)で示される種々の異性体を包含する。このようなものとして は、具体的には、 1, 2 シクロへキサンジメタノール、 1, 3 シクロへキサンジメタノ ール、 1, 4ーシクロへキサンジメタノールなどが挙げられる。
[化 5]
上記一般式 (π)で表される脂環式ジヒドロキシィ匕合物であるトリシクロデカンジメタノ ール、ペンタシクロペンタデカンジメタノールとしては、一般式(Π)において、 R1が下 記一般式 (lib) (式中、 nは 0又は 1で表す。)で表される種々の異性体を包含する。
上記一般式 (Π)で表される脂環式ジヒドロキシィ匕合物であるデカリンジメタノール又 は、トリシクロテトラデカンジメタノールとしては、一般式 (Π)において、 R1が下記一般 式 (lie) (式中、 mは 0、又は 1を表す。)で表される種々の異性体を包含する。このよう なものとしては、具体的には、 2, 6—デカリンジメタノール、 1, 5—デカリンジメタノー ル、 2, 3—デカリンジメタノールなどが挙げられる。
[化 7]
また、上記一般式 (π)で表される脂環式ジヒドロキシィ匕合物であるノルボルナンジメ タノールとしては、一般式 (Π)において、 R1が下記一般式 (lid)で表される種々の異 性体を包含する。このようなものとしては、具体的には、 2, 3—ノルボルナンジメタノ ール、 2, 5—ノルボルナンジメタノールなどが挙げられる。
[化 8]
d i d )
一般式 (Π)で表される脂環式ジヒドロキシィ匕合物であるァダマンタンジメタノールと しては、一般式 (Π)において、 R
1が下記一般式 (lie)で表される種々の異性体を包含 する。このようなものとしては、具体的には、 1, 3—ァダマンタンジメタノールなどが挙 げられる。
[化 9]
また、上記一般式 (ΠΙ)で表される脂環式ジヒドロキシィ匕合物であるシクロへキサン ジオールは、一般式 (III)において、 R2が下記一般式 (Ilia) (式中、 R3は炭素数 1〜1 2のアルキル基で表される。)で表される種々の異性体を包含する。このようなものとし ては、具体的には、 1, 2—シクロへキサンジオール、 1, 3—シクロへキサンジオール 、 1, 4ーシクロへキサンジオール、 2—メチルー 1, 4ーシクロへキサンジオールなど が挙げられる。
[化 10]
上記一般式 (in)で表される脂環式ジヒドロキシィ匕合物であるトリシクロデカンジォー ル、ペンタシクロペンタデカンジオールとしては、一般式 (ΠΙ)において、 R2が下記一
般式 (nib) (式中、 nは 0又は 1で表す。)で表される種々の異性体を包含する。
[化 11]
上記一般式 (in)で表される脂環式ジヒドロキシィ匕合物であるデカリンジオール又は
、トリシクロテトラデカンジオールとしては、一般式 (ΠΙ)において、 R2が下記一般式 (II Ic) (式中、 mは 0、又は 1を表す。)で表される種々の異性体を包含する。このようなも のとしては、具体的には、 2, 6 デカリンジオール、 1, 5 デカリンジオール、 2, 3— デカリンジオールなどが用いられる。
[化 12]
上記一般式 (ΠΙ)で表される脂環式ジヒドロキシィ匕合物であるノルボルナンジオール としては、一般式 (III)において、 R2が下記一般式 (Hid)で表される種々の異性体を 包含する。このようなものとしては、具体的には、 2, 3 ノルボルナンジオール、 2, 5 ノルボルナンジオールなどが用いられる。
上記一般式 (in)で表される脂環式ジヒドロキシィ匕合物であるァダマンタンジオール としては、一般式 (ΠΙ)において、 R2が下記一般式 (Hie)で表される種々の異性体を 包含する。このようなものとしては具体的には、 1, 3—ァダマンタンジオールなどが用 いられる。
[化 14]
[0044] 上述した脂環式ジヒドロキシィ匕合物の具体例のうち、特に、シクロへキサンジメタノ ール類、トリシクロデカンジメタノール類、ァダマンタンジオール類、ペンタシクロペン タデカンジメタノール類が好ましぐ入手のしゃすさ、取り扱いのしゃすさという観点か ら、 1, 4ーシクロへキサンジメタノール、 1, 3—シクロへキサンジメタノール、 1, 2—シ クロへキサンジメタノール、トリシクロデカンジメタノールが好まし 、。
[0045] なお、上記例示化合物は、本発明に使用し得る脂環式ジヒドロキシ化合物の一例 であって、何らこれらに限定されるものではない。これらの脂環式ジオールィ匕合物は 、 1種を単独で用いても良ぐ 2種以上を混合して用いても良い。
[0046] 本発明のポリカーボネート共重合体における一般式(1)で表されるジヒドロキシィ匕 合物に由来する構成単位と脂環式ジヒドロキシ化合物に由来する構成単位との含有
割合については、任意の割合で選択できる力 一般式(1)で表されるジヒドロキシ化 合物に由来する構成単位:脂環式ジヒドロキシ化合物に由来する構成単位 = 1 : 99 〜99: 1 (モル%)、特に一般式(1)で表されるジヒドロキシィ匕合物に由来する構成単 位:脂環式ジヒドロキシィ匕合物に由来する構成単位 = 10: 90-90: 10 (モル%)であ ることが好ま U、。上記範囲よりも一般式(1)で表されるジヒドロキシィ匕合物に由来す る構成単位が多く脂環式ジヒドロキシィ匕合物に由来する構成単位が少ないと着色し やすくなり、逆に一般式( 1 )で表されるジヒドロキシィ匕合物に由来する構成単位が少 なく脂環式ジヒドロキシィ匕合物に由来する構成単位が多いと分子量が上がりに《な る傾向がある。
なお、本発明のポリカーボネート共重合体は、一般式(1)で表されるジヒドロキシィ匕 合物及び脂環式ジヒドロキシィ匕合物以外のジヒドロキシィ匕合物(以下「その他のジヒド 口キシィ匕合物」と称す場合がある。 )に由来する構成単位が含まれていても良ぐこの 場合、その他のジヒドロキシ化合物としては、エチレングリコール、 1, 3 プロパンジ オール、 1, 2 プロパンジオール、 1, 4 ブタンジオール、 1, 3 ブタンジオール、 1, 2 ブタンジオール、 1, 5 ヘプタンジオール、 1, 6 へキサンジオールのなど の脂肪族ジヒドロキシィ匕合物、ジエチレングリコール、トリエチレングリコール、テトラエ チレングリコールなどのォキシアルキレングリコール類、 2, 2 ビス(4ーヒドロキシフ ェ -ル)プロパン [ =ビスフエノール A]、 2, 2 ビス(4 ヒドロキシ一 3, 5 ジメチル フエ-ル)プロパン、 2, 2 ビス(4ーヒドロキシ—3, 5 ジェチルフエ-ル)プロパン、 2, 2 ビス(4 ヒドロキシ一(3, 5 ジフエ-ル)フエ-ル)プロパン、 2, 2 ビス(4— ヒドロキシ一 3, 5 ジブロモフエ-ル)プロパン、 2, 2 ビス(4 ヒドロキシフエ-ル) ペンタン、 2, 4'—ジヒドロキシ一ジフエ二ルメタン、ビス(4 ヒドロキシフエニル)メタン 、ビス(4 ヒドロキシ一 5 -トロフエ-ル)メタン、 1, 1—ビス(4 ヒドロキシフエ-ノレ )ェタン、 3, 3 ビス(4 ヒドロキシフエ-ル)ペンタン、 1, 1—ビス(4 ヒドロキシフエ -ル)シクロへキサン、ビス(4 ヒドロキシフエ-ル)スルホン、 2, 4'—ジヒドロキシジ フエニルスルホン、ビス(4—ヒドロキシフエ-ル)スルフイド、 4, 4'—ジヒドロキシジフ ェニルエーテル、 4, 4'ージヒドロキシー 3, 3,ージクロロジフエニルエーテル、 4, 4, —ジヒドロキシ一 2, 5 ジエトキシジフエ-ルエーテル、 9, 9 ビス(4— (2 ヒドロキ
シエトキシ)フエ-ル)フルオレン、 9, 9—ビス(4— (2—ヒドロキシエトキシ一 2—メチ ル)フエ-ル)フルオレン、 9, 9—ビス(4—ヒドロキシフエ-ル)フルオレン、 9, 9—ビス (4ーヒドロキシエー 2—メチルフエ-ル)フルオレン、等の芳香族ビスフエノール類な どの 1種又は 2種以上が挙げられる。
[0048] これらのその他のジヒドロキシィ匕合物を用いることにより、柔軟性の改善、耐熱性の 向上、成形性の改善などの効果を得ることもできる力 その他のジヒドロキシィ匕合物に 由来する構成単位の含有割合が多過ぎると本来の光学特性の性能を低下させたり することがあるため、本発明のポリカーボネート共重合体においては、ポリカーボネー ト共重合体を構成する全ジヒドロキシィ匕合物に対する一般式(1)で表されるジヒドロキ シ化合物と脂環式ジヒドロキシィ匕合物の合計の割合が 90モル%以上であることが好 ましぐ特に、本発明のポリカーボネート共重合体はジヒドロキシィ匕合物として一般式 (1)で表されるジヒドロキシィ匕合物と脂環式ジヒドロキシィ匕合物のみで構成されること が好ましい。
[0049] また、本発明のポリカーボネート共重合体の重合度は、溶媒としてフエノールと 1, 1 , 2, 2, ーテトラクロロェタンの重量比 1 : 1の混合溶液を用い、ポリカーボネート共重 合体濃度を 1. OOgZdlに精密に調整し、温度 20. 0°C±0. 1°Cで測定した還元粘 度(以下、単に「ポリカーボネート共重合体の還元粘度」と称す。)として、 0. 40dl/g 以上、特に 0. 40dlZg以上で 2. OdlZg以下であるような重合度であることが好まし V、。このポリカーボネート共重合体還元粘度が極端に低 、ものではレンズ等に成形し た時の機械的強度が弱い。また、ポリカーボネート共重合体の還元粘度が大きくなる と、成形する際の流動性が低下し、サイクル特性を低下させ、成形品の複屈折率が 大きくなり易い傾向がある。従って、本発明のポリカーボネート共重合体の還元粘度 は 0. 40dlZg以上 2. OdlZg以下、特に 0. 45dlZg以上 1. 5dlZg以下の範囲内 であることが好ましい。
[0050] また、本発明のポリカーボネート共重合体のアッベ数は、 50以上が好ましぐ特に 好ましくは 55以上である。この値が大きくなるほど、屈折率の波長分散が小さくなり、 例えば、単レンズで使用した場合の色収差が小さくなり、より鮮明な画像が得やすく なる。アッベ数力 、さくなるほど屈折率の波長分散が大きくなり、単レンズで使用した
場合、色収差が大きくなり、画像のぼけの度合いが大きくなる。
[0051] また、本発明のポリカーボネート共重合体の 5%熱減量温度は 340°C以上が好まし ぐ特に好ましくは 345°C以上である。 5%熱減量温度が大きいほど、熱安定性が高 くなり、より高温での使用に耐えるものとなり、小さくなるほど、熱安定性が低くなり、高 温での使用がしに《なる。また、製造時の制御許容幅が狭くなり作りに《なる。また 、製造温度も高くでき、より製造時の制御幅が広くできるので、製造し易くなる。
[0052] また、本発明のポリカーボネート共重合体の光弾性係数は、 40 X 10_12Pa_1以下 であることが好ましぐ更に好ましくは 20 X 10_12Pa_1以下である。光弾性係数の値 が高 、と、溶融押出や溶液キャスト法等で製膜したフィルムの位相差の値が大きくな り、これを延伸した場合、張力のわずかな振れにより、フィルム面内の位相差のばら つきがさらに大きくなる。またこのような位相差フィルムを貼合する場合、貼合時の張 力により所望する位相差がずれてしまうば力りでなぐ貼合後の偏光板の収縮等によ り、位相差値が変化しやすい。光弾性係数が小さいほど位相差のばらつきが小さくな る。
[0053] また、本発明のポリカーボネート共重合体はアイゾット衝撃強度が 30jZm2以上で あることが好ましい。アイゾット衝撃強度が大きい程、成形体の強度が高くなり、こわれ に《なる。
[0054] また、本発明のポリカーボネート共重合体は、 110°Cでの単位面積あたりのフエノー ル成分以外の発生ガス量 (以下、単に「発生ガス量」と称す場合がある。)が 5ngZc m2以下であることが好ましぐまた、一般式(1)で表わされるジヒドロキシィ匕合物以外 のジヒドロキシィ匕合物由来の発生ガス量は 0. 5ngZcm2以下であることがより好まし い。この発生ガス量が少ない程、発生ガスの影響を嫌う用途、例えば、半導体などの 電子部品を保管する用途、建物の内装材用途、家電製品などの筐体などに適用す ることがでさる。
[0055] なお、本発明のポリカーボネート共重合体のアッベ数、 5%熱減量温度、光弾性係 数、アイゾット衝撃強度、発生ガス量の測定方法は、具体的には後述の実施例の項 で示す通りである。
[0056] また、本発明のポリカーボネート共重合体は、示差走査熱量測定 (DSC)を行った
とき、単一のガラス転移温度を与えるが、本発明のポリカーボネート共重合体は、一 般式(1)で表されるジヒドロキシィ匕合物と脂環式ジヒドロキシィ匕合物の種類や配合比 を調整することで、そのガラス転移温度を、用途に応じて、 45°C程度から 155°C程度 まで任意のガラス転移温度を持つ重合体として得ることができる。
[0057] 例えば、柔軟性が必要とされるフィルム用途では、ポリカーボネート共重合体のガラ ス転移温度が 45°C以上、例えば 45〜100°Cに、また、ある程度の耐熱性が求められ るボトルやパックと 、つた成形体用途では、ポリカーボネート共重合体のガラス転移 温度は 90°C以上、例えば 90〜130°Cに調整することが好ましい。さらにガラス転移 温度が 120°C以上であると、レンズ用途に好適となる。即ち、このようなガラス転移温 度を有するものであれば、温度 85°C、相対湿度 85%といった高温高湿度下におい ても変形が起こりにくく、レンズの面精度のバラツキが少な 、ので好まし!/、。
[0058] 本発明のポリカーボネート共重合体は、一般に用いられる重合方法で製造すること ができ、その重合方法は、ホスゲンを用いた溶液重合法、炭酸ジエステルと反応させ る溶融重合法のいずれの方法でも良いが、重合触媒の存在下に、一般式(1)で表さ れるジヒドロキシィ匕合物と脂環式ジヒドロキシィ匕合物と、必要に応じて用いられるその 他のジヒドロキシィ匕合物とを、より環境への毒性の低 、炭酸ジエステルと反応させる 溶融重合法が好ましい。
[0059] この溶融重合法で用いられる炭酸ジエステルとしては、通常、下記一般式(2)で表 されるものが挙げられる。
[0060] [化 15]
o
A-O-C-O-A' (2)
(一般式(2)において、 A及び A'は、置換基を有していても良い炭素数 1〜18の脂 肪族基又は置換基を有していても良い芳香族基であり、 A及び A'は同一であっても 異なっていても良い。 )
[0061] 上記一般式(2)で表される炭酸ジエステルとしては、例えば、ジフエ-ルカーボネ ート、ジトリルカーボネートに代表される置換ジフエ-ルカーボネート、ジメチルカーボ
ネート、ジェチルカーボネート及びジー t ブチルカーボネート等が例示される力 特 に好ましくはジフエ-ルカーボネート及び置換ジフエ-ルカーボネートが挙げられる。 これらの炭酸ジエステルは、 1種を単独で用いても良ぐ 2種以上を混合して用いても 良い。
[0062] 炭酸ジエステルは、反応に用いる全ジヒドロキシ化合物に対して、 0. 90〜: L 10の モル比率で用いることが好ましぐさらに好ましくは、 0. 96〜: L . 04のモル比率である 。このモル比が 0. 90より小さくなると、製造されたポリカーボネート共重合体の末端 O H基が増加して、ポリマーの熱安定性が悪ィ匕したり、所望する高分子量体が得られな 力つたりする。また、このモル比が 1. 10より大きくなると、同一条件下ではエステル交 換反応の速度が低下したり、所望とする分子量のポリカーボネート共重合体の製造 が困難となるばかりか、製造されたポリカーボネート共重合体中の残存炭酸ジエステ ル量が増加し、この残存炭酸ジエステル力 成形時、又は成形品の臭気の原因とな り好ましくない。
[0063] なお、一般式(1)で表されるジヒドロキシィ匕合物と、脂環式ジヒドロキシ化合物と、必 要に応じて用いられるその他のジヒドロキシィ匕合物との使用割合は、本発明のポリ力 ーボネート共重合体を構成する各ジヒドロキシ化合物に由来する構成単位の割合と して前述した通りである。
[0064] また、溶融重合における重合触媒 (エステル交換触媒)としては、アルカリ金属化合 物及び Z又はアルカリ土類金属化合物が使用される。アルカリ金属化合物及び Z又 はアルカリ土類金属化合物と共に補助的に、塩基性ホウ素化合物、塩基性リン化合 物、塩基性アンモニゥム化合物、アミン系化合物等の塩基性化合物を併用することも 可能である力 アルカリ金属化合物及び Z又はアルカリ土類金属化合物のみを使用 することが特に好ましい。
[0065] 重合触媒として用いられるアルカリ金属化合物としては、例えば、水酸ィ匕ナトリウム 、水酸ィ匕カリウム、水酸化リチウム、水酸ィ匕セシウム、炭酸水素ナトリウム、炭酸水素力 リウム、炭酸水素リチウム、炭酸水素セシウム、炭酸ナトリウム、炭酸カリウム、炭酸リチ ゥム、炭酸セシウム、酢酸ナトリウム、酢酸カリウム、酢酸リチウム、酢酸セシウム、ステ アリン酸ナトリウム、ステアリン酸カリウム、ステアリン酸リチウム、ステアリン酸セシウム
、水素化ホウ素ナトリウム、水素化ホウ素カリウム、水素化ホウ素リチウム、水素化ホウ 素セシウム、フエ-ル化ホウ素ナトリウム、フエ-ル化ホウ素カリウム、フエ-ル化ホウ 素リチウム、フ ニル化ホウ素セシウム、安息香酸ナトリウム、安息香酸カリウム、安息 香酸リチウム、安息香酸セシウム、リン酸水素 2ナトリウム、リン酸水素 2カリウム、リン 酸水素 2リチウム、リン酸水素 2セシウム、亜リン酸水素 2ナトリウム、亜リン酸水素カリ ゥム、亜リン酸水素 2リチウム、亜リン酸水素 2セシウム、フエ-ルリン酸 2ナトリウム、フ ェ-ルリン酸 2カリウム、フエ-ルリン酸 2リチウム、フエ-ルリン酸 2セシウム、ナトリウム 、カリウム、リチウム、セシウムのアルコレート、フエノレート、ビスフエノール Aの 2ナトリ ゥム塩、 2カリウム塩、 2リチウム塩、 2セシウム塩等が挙げられる。
[0066] また、アルカリ土類金属化合物としては、例えば、水酸ィ匕カルシウム、水酸化バリウ ム、水酸化マグネシウム、水酸化ストロンチウム、炭酸水素カルシウム、炭酸水素バリ ゥム、炭酸水素マグネシウム、炭酸水素ストロンチウム、炭酸カルシウム、炭酸バリゥ ム、炭酸マグネシウム、炭酸ストロンチウム、酢酸カルシウム、酢酸バリウム、酢酸マグ ネシゥム、酢酸ストロンチウム、ステアリン酸カルシウム、ステアリン酸バリウム、ステアリ ン酸マグネシウム、ステアリン酸ストロンチウム等が挙げられる。
[0067] これらのアルカリ金属化合物及び Z又はアルカリ土類金属化合物は 1種を単独で 用いても良ぐ 2種以上を併用しても良い。
[0068] またアルカリ金属化合物及び Z又はアルカリ土類金属化合物と併用される塩基性 ホウ素化合物の具体例としては、テトラメチルホウ素、テトラエチルホウ素、テトラプロ ピルホウ素、テトラブチルホウ素、トリメチルェチルホウ素、トリメチルベンジルホウ素、 トリメチルフエ-ルホウ素、トリェチルメチルホウ素、トリェチルベンジルホウ素、トリエ チルフエ-ルホウ素、トリブチルベンジルホウ素、トリブチルフエ-ルホウ素、テトラフエ -ルホウ素、ベンジルトリフエ-ルホウ素、メチルトリフエ-ルホウ素、ブチルトリフエ- ルホウ素等のナトリウム塩、カリウム塩、リチウム塩、カルシウム塩、ノ リウム塩、マグネ シゥム塩、あるいはストロンチウム塩等が挙げられる。
[0069] 塩基性リンィ匕合物としては、例えば、トリェチルホスフィン、トリ一 n—プロピルホスフ イン、トリイソプロピルホスフィン、トリ一 n—ブチルホスフィン、トリフエニルホスフィン、ト リブチルホスフィン、あるいは四級ホスホ-ゥム塩等が挙げられる。
[0070] 塩基性アンモ-ゥム化合物としては、例えば、テトラメチルアンモ-ゥムヒドロキシド、 テトラエチルアンモ-ゥムヒドロキシド、テトラプロピルアンモ-ゥムヒドロキシド、テトラ ブチルアンモ-ゥムヒドロキシド、トリメチルェチルアンモ-ゥムヒドロキシド、トリメチル ベンジルアンモ-ゥムヒドロキシド、トリメチルフエ-ルアンモ-ゥムヒドロキシド、トリエ チルメチルアンモ-ゥムヒドロキシド、トリェチルベンジルアンモ-ゥムヒドロキシド、トリ ェチルフエ-ルアンモ-ゥムヒドロキシド、トリブチルベンジルアンモ-ゥムヒドロキシド 、トリブチルフエ-ルアンモ-ゥムヒドロキシド、テトラフエ-ルアンモ-ゥムヒドロキシド 、ベンジルトリフエ-ルアンモ-ゥムヒドロキシド、メチルトリフエ-ルアンモ-ゥムヒドロ キシド、ブチルトリフエ-ルアンモ-ゥムヒドロキシド等が挙げられる。
[0071] アミン系化合物としては、例えば、 4 アミノビリジン、 2 アミノビリジン、 N, N ジメ チルー 4 アミノビリジン、 4ージェチルァミノピリジン、 2 ヒドロキシピリジン、 2—メト キシピリジン、 4ーメトキシピリジン、 2 ジメチルァミノイミダゾール、 2—メトキシイミダ ゾール、イミダゾール、 2—メルカプトイミダゾール、 2—メチルイミダゾール、ァミノキノ リン等が挙げられる。
[0072] これらの塩基性ィ匕合物も 1種を単独で用いても良ぐ 2種以上を併用しても良い。
[0073] 上記重合触媒の使用量は、アルカリ金属化合物及び Z又はアルカリ土類金属化合 物を用いる場合、反応に用いる全ジヒドロキシィ匕合物 1モルに対して、金属換算量と して、通常、 0. 01〜: LOO /zモノレの範囲内で用!/、、好ましく ίま 0. 05〜50 モノレの範 囲内であり、さらに好ましくは 0. 1〜: LO /zモルの範囲内である。重合触媒の使用量 が少なすぎると、所望の分子量のポリカーボネート共重合体を製造するのに必要な 重合活性が得られず、一方、重合触媒の使用量が多すぎると、得られるポリカーボネ ート共重合体の色相が悪ィ匕し、副生成物が発生したりして流動性の低下やゲルの発 生が多くなり、 目標とする品質のポリカーボネート共重合体の製造が困難になる。
[0074] このような本発明のポリカーボネート共重合体の製造に当たり、前記一般式 (I)で表 されるジヒドロキシィ匕合物は、固体として供給しても良いし、加熱して溶融状態として 供給しても良いし、水溶液として供給しても良い。
[0075] 一方、脂環式ジヒドロキシィ匕合物も、固体として供給しても良いし、加熱して溶融状 態として供給しても良いし、水に可溶なものであれば、水溶液として供給しても良い。
その他のジヒドロキシィ匕合物につ 、ても同様である。
[0076] これらの原料ジヒドロキシ化合物を溶融状態や、水溶液で供給すると、工業的に製 造する際、計量や搬送がしゃす V、と V、う利点がある。
[0077] 本発明にお 、て、一般式 (I)で表されるジヒドロキシィ匕合物と脂環式ジヒドロキシィ匕合 物と必要に応じて用いられるその他のジヒドロキシィ匕合物とを重合触媒の存在下で炭 酸ジエステルと反応させる方法は、通常、 2段階以上の多段工程で実施される。具体 的には、第 1段目の反応は 140〜240°C、好ましくは 150〜220°Cの温度で 0. 1〜1 0時間、好ましくは 0. 5〜3時間実施される。第 2段目以降は、反応系の圧力を第 1段 目の圧力力も徐々に下げながら反応温度を上げていき、同時に発生するフエノール を反応系外へ除きながら、最終的には反応系の圧力が 200Pa以下で、 180〜280 °Cの温度範囲のもとで重縮合反応を行う。
[0078] この重縮合反応における減圧において、温度と反応系内の圧力のバランスを制御 することが重要である。特に、温度、圧力のどちらか一方でも早く変化させすぎると、 未反応のモノマーが留出し、ジヒドロキシィ匕合物と炭酸ジエステルのモル比を狂わせ 、重合度が低下することがある。例えば、ジヒドロキシィ匕合物としてイソソルビドと 1, 4 —シクロへキサンジメタノールを用いる場合は、全ジヒドロキシィ匕合物に対し、 1, 4- シクロへキサンジメタノールのモル比が 50モル0 /0以上の場合は、 1, 4ーシクロへキサ ンジメタノールがモノマーのまま留出しゃすくなるので、反応系内の圧力が 13kPa程 度の減圧下で、温度を 1時間あたり 40°C以下の昇温速度で上昇させながら反応させ 、さらに、 6. 67kPa程度までの圧力下で、温度を 1時間あたり 40°C以下の昇温速度 で上昇させ、最終的に 200Pa以下の圧力で、 200から 250°Cの温度で重縮合反応 を行うと、十分に重合度が上昇したポリカーボネート共重合体が得られるため、好まし い。
[0079] また、全ジヒドロキシ化合物に対し、 1, 4ーシクロへキサンジメタノールのモル比が 5 0モル%より少なくなつた場合、特に、モル比が 30モル%以下となった場合は、 1, 4 ーシクロへキサンジメタノールのモル比が 50モル%以上の場合と比べて、急激な粘 度上昇が起こるので、例えば、反応系内の圧力が 13kPa程度の減圧下までは、温度 を 1時間あたり 40°C以下の昇温速度で上昇させながら反応させ、さらに、 6. 67kPa
程度までの圧力下で、温度を 1時間あたり 40°C以上の昇温速度、好ましくは 1時間あ たり 50°C以上の昇温速度で上昇させながら反応させ、最終的に 200Pa以下の減圧 下、 200から 280°Cの温度で重縮合反応を行うと、十分に重合度が上昇したポリカー ボネート共重合体が得られるため、好ましい。
[0080] 反応の形式は、バッチ式、連続式、ある 、はバッチ式と連続式の組み合わせの 、 ずれの方法でもよい。
[0081] 本発明のポリカーボネート共重合体を溶融重合法で製造する際に、着色を防止す る目的で、リン酸化合物や亜リン酸化合物又はこれらの金属塩を重合時に添加する ことができる。
[0082] リン酸化合物としては、リン酸トリメチル、リン酸トリェチル等のリン酸トリアルキルの 1 種又は 2種以上が好適に用いられる。これらは、反応に用いる全ジヒドロキシィ匕合物 に対して、 0. 0001モル%以上 0. 005モル%以下添加することが好ましぐさらに好 ましくは 0. 0003モル%以上 0. 003モル%以下添加することが好ましい。リン化合物 の添加量が上記下限より少ないと、着色防止効果が小さぐ上記上限より多いと、へ ィズが高くなる原因となったり、逆に着色を促進させたり、耐熱性を低下させたりする
[0083] 亜リン酸ィ匕合物を添加する場合は、下記に示す熱安定剤を任意に選択して使用で きる。特に、亜リン酸トリメチル、亜リン酸トリェチル、トリスノ -ルフエ-ルホスフアイト、 トリメチルホスフェート、トリス(2, 4—ジ— tert—ブチルフエ-ル)ホスファイト、ビス(2 , 4ージー tert—ブチルフエ-ル)ペンタエリスリトールジホスファイトの 1種又は 2種以 上が好適に使用できる。これらの亜リン酸ィ匕合物は、反応に用いる全ジヒドロキシィ匕 合物に対して、 0. 0001モル%以上 0. 005モル%以下添加することが好ましぐさら に好ましくは 0. 0003モル%以上 0. 003モル%以下添加することが好ましい。亜リン 酸ィ匕合物の添加量が上記下限より少ないと、着色防止効果が小さぐ上記上限より 多いと、八ィズが高くなる原因となったり、逆に着色を促進させたり、耐熱性を低下さ せたりすることちある。
[0084] リン酸ィ匕合物と亜リン酸ィ匕合物又はこれらの金属塩は併用して添加することができ る力 その場合の添加量はリン酸ィ匕合物と亜リン酸ィ匕合物又はこれらの金属塩の総
量で、先に記載した、全ジヒドロキシ化合物に対して、 0. 0001モル%以上 0. 005モ ル%以下とすることが好ましぐさらに好ましくは 0. 0003モル%以上 0. 003モル% 以下である。この添加量が上記下限より少ないと、着色防止効果が小さぐ上記上限 より多いと、八ィズが高くなる原因となったり、逆に着色を促進させたり、耐熱性を低 下させたりすることちある。
[0085] なお、リン酸化合物、亜リン酸ィ匕合物の金属塩としては、亜鉛塩が好ましぐこのリン 酸亜鉛塩の中でも、ステアリルリン酸亜鉛塩などの長鎖アルキルリン酸亜鉛塩が好ま しい。
[0086] また、このようにして製造された本発明のポリカーボネート共重合体には、成形時等 における分子量の低下や色相の悪ィ匕を防止するために熱安定剤を配合することが できる。
[0087] 力かる熱安定剤としては、亜リン酸、リン酸、亜ホスホン酸、ホスホン酸及びこれらの エステル等が挙げられ、具体的には、トリフエ-ルホスフアイト、トリス(ノユルフェ-ル) ホスファイト、トリス(2, 4 ジ一 tert—ブチルフエ-ル)ホスファイト、トリデシルホスフ アイト、トリオクチルホスフアイト、トリオクタデシルホスフアイト、ジデシルモノフエ-ルホ スフアイト、ジォクチルモノフエ-ルホスフアイト、ジイソプロピルモノフエ-ルホスフアイ ト、モノブチルジフエ-ルホスフアイト、モノデシルジフエ-ルホスフアイト、モノォクチ ルジフエ-ルホスフアイト、ビス(2, 6 ジ tert—ブチルー 4 メチルフエ-ル)ペン タエリスリトールジホスフアイト、 2, 2—メチレンビス(4, 6 ジ一 tert—ブチルフエ- ル)ォクチルホスファイト、ビス(ノユルフェ-ル)ペンタエリスリトールジホスフアイト、ビ ス(2, 4 ジ tert ブチルフエ-ル)ペンタエリスリトールジホスフアイト、ジステアリ ノレペンタエリスリトールジホスフアイト、トリブチノレホスフェート、トリェチノレホスフェート、 トリメチルホスフェート、トリフエ-ルホスフェート、ジフエ-ルモノオルソキセ -ルホスフ エート、ジブチノレホスフェート、ジォクチノレホスフェート、ジイソプロピノレホスフェート、 4 , 4'—ビフエ-レンジホスフィン酸テトラキス(2, 4 ジ一 tert—ブチルフエ-ル)、ベ ンゼンホスホン酸ジメチル、ベンゼンホスホン酸ジェチル、ベンゼンホスホン酸ジプロ ピル等が挙げられる。なかでも、トリスノユルフェ-ルホスフアイト、トリメチルホスフエ一 ト、トリス(2, 4 ジ一 tert ブチルフエ-ル)ホスファイト、ビス(2, 4 ジ一 tert ブ
チルフエ-ル)ペンタエリスリトールジホスフアイト、ビス(2, 6 ジ—tert—ブチルー 4 メチルフエ-ル)ペンタエリスリトールジホスフアイト、及びベンゼンホスホン酸ジメチ ルが好ましく使用される。
[0088] これらの熱安定剤は、 1種を単独で用いても良ぐ 2種以上を併用しても良い。
[0089] 力かる熱安定剤は、溶融重合時に添加した添加量に加えて更に追加で配合するこ とができる。即ち、適当量の亜リン酸化合物やリン酸化合物を配合して、ポリカーボネ ート共重合体を得た後に、後に記載する配合方法で、さらに亜リン酸化合物を配合 すると、重合時のヘイズの上昇、着色、及び耐熱性の低下を回避して、さらに多くの 熱安定剤を配合でき、色相の悪ィ匕の防止が可能となる。
[0090] これらの熱安定剤の配合量は、ポリカーボネート共重合体を 100重量部とした場合 、 0. 0001〜1重量咅力 S好まし <、 0. 0005〜0. 5重量咅力 Sより好まし <、 0. 001〜0 . 2重量部が更に好ましい。
[0091] また、本発明のポリカーボネート共重合体には、酸化防止の目的で通常知られた 酸化防止剤を配合することもできる。
[0092] 力かる酸ィ匕防止剤としては、例えばペンタエリスリトールテトラキス(3—メルカプトプ 口ピオネート)、ペンタエリスリトールテトラキス(3—ラウリルチオプロピオネート)、ダリ セロール 3—ステアリルチオプロピオネート、トリエチレングリコール ビス [3— (3 —tert—ブチルー 5—メチルー 4ーヒドロキシフエ-ル)プロピオネート]、 1, 6 へキ サンジオール—ビス [3— (3, 5—ジ— tert ブチル—4—ヒドロキシフエ-ル)プロピ ォネート]、ペンタエリスリトールーテトラキス [3— (3, 5—ジ tert—ブチルー 4ーヒド ロキシフエ-ル)プロピオネート]、ォクタデシルー 3— (3, 5—ジ一 tert—ブチル 4 —ヒドロキシフエ-ル)プロピオネート、 1, 3, 5 トリメチルー 2, 4, 6 トリス(3, 5— ジ一 tert ブチル 4—ヒドロキシベンジル)ベンゼン、 N, N へキサメチレンビス( 3, 5—ジ tert—ブチルー 4ーヒドロキシーヒドロシンナマイド)、 3, 5—ジ tert— ブチルー 4ーヒドロキシ一べンジルホスホネートージェチルエステル、トリス(3, 5—ジ —tert—ブチル 4—ヒドロキシベンジル)イソシァヌレート、 4, 4,一ビフエ-レンジ ホスフィン酸テトラキス(2, 4 ジ一 tert—ブチルフエ-ル)、 3, 9 ビス { 1, 1—ジメ チルー 2— [ β一(3— tert—ブチルー 4ーヒドロキシー5 メチルフエ-ル)プロピオ
-ルォキシ]ェチル }— 2, 4, 8, 10—テトラオキサスピロ(5, 5)ゥンデカン等の 1種 又は 2種以上が挙げられる。
[0093] これら酸化防止剤の配合量は、ポリカーボネートを 100重量部とした場合、 0. 000
1〜0. 5重量部が好ましい。
[0094] また、本発明のポリカーボネート共重合体には、溶融成形時の金型からの離型性を より向上させるために、本発明の目的を損なわない範囲で離型剤を配合することも可 能である。
[0095] 力かる離型剤としては、一価又は多価アルコールの高級脂肪酸エステル、高級脂 肪酸、パラフィンワックス、蜜蝌、ォレフィン系ワックス、カルボキシ基及び/又はカル ボン酸無水物基を含有するォレフイン系ワックス、シリコーンオイル、オルガノポリシ口 キサン等が挙げられる。
[0096] 高級脂肪酸エステルとしては、炭素原子数 1〜20の一価又は多価アルコールと炭 素原子数 10〜30の飽和脂肪酸との部分エステル又は全エステルが好ま 、。力か る一価又は多価アルコールと飽和脂肪酸との部分エステル又は全エステルとしては 、ステアリン酸モノグリセリド、ステアリン酸ジグリセリド、ステアリン酸トリグリセリド、ステ アリン酸モノソルビテート、ステアリン酸ステアリル、ベへニン酸モノグリセリド、ベへ- ン酸ベへ-ル、ペンタエリスリトールモノステアレート、ペンタエリスリトールテトラステ ァレート、ペンタエリスリトールテトラペラルゴネート、プロピレングリコールモノステアレ ート、ステアリルステアレート、ノ レミチルパルミテート、ブチルステアレート、メチルラ ゥレート、イソプロピルパルミテート、ビフエ-ルビフエネート、ソルビタンモノステアレ ート、 2—ェチルへキシルステアレート等が挙げられる。
[0097] なかでも、ステアリン酸モノグリセリド、ステアリン酸トリグリセリド、ペンタエリスリトール テトラステアレート、ベへニン酸べへニルが好ましく用いられる。
[0098] 高級脂肪酸としては、炭素原子数 10〜30の飽和脂肪酸が好ま 、。力かる脂肪酸 としては、ミリスチン酸、ラウリン酸、パルミチン酸、ステアリン酸、ベへニン酸などが挙 げられる。
[0099] これらの離型剤は、 1種を単独で用いても良ぐ 2種以上を混合して用いても良い。
[0100] 力かる離型剤の配合量は、ポリカーボネートを 100重量部とした場合、 0. 01〜5重
量部が好ましい。
[0101] また、本発明のポリカーボネート共重合体には、本願発明の目的を損なわない範囲 で、光安定剤を配合することができる。
[0102] 力かる光安定剤としては、例えば 2— (2,一ヒドロキシ一 5,一 tert—ォクチノレフエ- ル)ベンゾトリァゾール、 2—(3— tert—ブチルー 5—メチルー 2 ヒドロキシフエ-ル )—5 クロ口べンゾトリァゾール、 2— (5—メチル 2 ヒドロキシフエ-ル)ベンゾトリ ァゾール、 2— [2 ヒドロキシ— 3, 5 ビス( α , a—ジメチルベンジル)フエ-ル] 2H ベンゾトリァゾール、 2, 2,一メチレンビス(4—タミル 6 ベンゾトリァゾールフ ェ-ル)、 2, 2,一p—フエ-レンビス(1, 3 べンゾォキサジンー4 オン)等が挙げ られる。
[0103] これらの光安定剤は、 1種を単独で用いても良ぐ 2種以上を併用しても良い。
[0104] 力かる光安定剤の配合量は、ポリカーボネート共重合体を 100重量部とした場合、
0. 01〜2重量部が好ましい。
[0105] また、本発明のポリカーボネート共重合体には、重合体や紫外線吸収剤に基づくレ ンズの黄色味を打ち消すためにブルーイング剤を配合することができる。ブルーイン グ剤としては、ポリカーボネート榭脂、ポリカーボネート榭脂に使用されるものであれ ば、特に支障なく使用することができる。一般的にはアンスラキノン系染料が入手容 易であり好ましい。
[0106] 具体的なブルーイング剤としては、例えば一般名 Solvent Violet 13 [CA. No (力 ラーインデックス No) 60725]、一般名 Solvent Violet31 [CA. No 68210、一般 名 Solvent Violet33 [CA. No 60725 "—般名 Solvent Blue94 [CA. No 6 1500]、一般名 Solvent Violet36 [CA. No 68210]、一般名 Solvent Blue 97 [バイエル社製「マクロレックスバイオレット RR」]及び一般名 Solvent Blue45 [CA . No61110]が代表例として挙げられる。
[0107] これらのブルーイング剤は、 1種を単独で用いても良ぐ 2種以上を併用しても良い
[0108] これらブルーイング剤は、通常、ポリカーボネート共重合体を 100重量部とした場合 、 0. 1 X 10一4〜 2 X 10_4重量部の割合で配合される。
[0109] 本発明のポリカーボネート共重合体と上述のような各種の添加剤との配合は、例え ばタンブラ一、 V型ブレンダー、スーパーミキサー、ナウターミキサー、バンバリ一ミキ サー、混練ロール、押出機等で混合する方法、あるいは上記各成分を例えば塩化メ チレンなどの共通の良溶媒に溶解させた状態で混合する溶液ブレンド方法などがあ る力 これは特に限定されるものではなぐ通常用いられるポリマーブレンド方法であ ればどのような方法を用いてもょ 、。
[0110] こうして得られる本発明のポリカーボネート共重合体或いは、これに各種添加剤を 添加してなるポリカーボネート共重合体組成物は、そのまま、又は溶融押出機でー且 ペレット状にしてから、射出成形法、押出成形法、圧縮成形法等の通常知られている 方法で成形物にすることができる。
[0111] 本発明のポリカーボネート共重合体の混和性を高めて安定した離型性や各物性を 得るためには、溶融押出において単軸押出機、二軸押出機を使用するのが好ましい 。単軸押出機、二軸押出機を用いる方法は、溶剤等を用いることがなぐ環境への負 荷が小さぐ生産性の点からも好適に用いることができる。
[0112] 押出機の溶融混練温度は、本発明のポリカーボネート共重合体のガラス転移温度 に依存するが、本発明のポリカーボネート共重合体のガラス転移温度が 90°Cより低 い場合は、押出機の溶融混練温度は通常 130°Cから 250°C、好ましくは 150から 24 0°Cである。溶融混練温度が 130°Cより低い温度であると、ポリカーボネート共重合体 の溶融粘度が高ぐ押出機への負荷が大きくなり、生産性が低下する。 250°Cより高 いと、ポリカーボネート共重合体の溶融粘度が低くなり、ペレットを得にくくなり、生産 性が低下する。
[0113] また、本発明のポリカーボネート共重合体のガラス転移温度が 90°C以上の場合は 、押出機の溶融混練温度は通常 200から 300°C、好ましくは 220°Cから 260°Cである 。溶融混練温度が 200°Cより低い温度であると、ポリカーボネート共重合体の溶融粘 度が高ぐ押出機への負荷が大きくなり、生産性が低下する。 300°Cより高いと、ポリ カーボネート共重合体の劣化が起こりやすくなり、ポリカーボネート共重合体の色が 黄変したり、分子量が低下するため強度が劣化したりする。
[0114] 押出機を使用する場合、押出時にポリカーボネート共重合体の焼け、異物の混入
を防止するため、フィルターを設置することが望ましい。フィルターの異物除去の大き さ(目開き)は、求められる光学的な精度依存するが、 100 m以下が好ましい。特に 、異物の混入を嫌う場合は、 40 m以下、さらには 10 m以下が好ましい。
[0115] ポリカーボネート共重合体の押出は、押出後の異物混入を防止するために、タリー ンルーム中で実施することが望ましい。
[0116] また、押出されたポリカーボネート共重合体を冷却しチップ化する際は、空冷、水冷 等の冷却方法を使用するのが好ましい。空冷の際に使用する空気は、へパフィルタ 一等で空気中の異物を事前に取り除!/、た空気を使用し、空気中の異物の再付着を 防ぐのが望ましい。水冷を使用する際は、イオン交換榭脂等で水中の金属分を取り 除き、さらにフィルタ一にて、水中の異物を取り除いた水を使用することが望ましい。 用いるフィルターの大きさ(目開き)は種々ある力 10〜0. 45 mのフィルターのも のが好ましい。
[0117] 本発明のポリカーボネート共重合体を用いたレンズの成形には、射出成形機や、射 出圧縮成形機が適合し、この際の成形条件としては、特に金型表面温度と榭脂温度 が重要である。これらの成形条件は、ポリカーボネート共重合体の組成及び重合度 などにより一概に規定できないが、金型表面温度は 30°C以上 170°C以下が好ましく 、また、この時の榭脂温度は 220°C以上 290°C以下となるようにするのが良い。金型 表面温度が 30°C以下の場合には、榭脂の流動性と転写性が共に悪ぐ射出成形時 に応力歪が残って、複屈折率が大きくなる傾向があり、また、金型温度が 170°C以上 の場合、転写性は良いが、離型時に変形し易い。また、榭脂温度が 290°C以上の場 合は榭脂の分解が起こり易ぐ成形品の強度低下、着色の原因となる。また、成形サ イタルも延びるので経済的でな 、。
[0118] 本発明のポリカーボネート共重合体力も光学材料、光学部品を成形する場合には 、原料の投入工程を始め、重合工程、得られた共重合体を冷媒中に押し出してペレ ット状又はシート状にする工程では、塵埃等が入り込まないように留意して行う事が望 まれる。このクリーン度は、通常コンパクトディスク用の場合にはクラス 1000以下であ り、更に高度な情報記録用の場合にはクラス 100以下である。
[0119] 本発明のポリカーボネート共重合体は例えば、芳香族ポリカーボネート、芳香族ポ
リエステル、脂肪族ポリエステル、ポリアミド、ポリスチレン、ポリオレフイン、アクリル、ァ モルファスポリオレフイン、 ABS、 ASなど、の合成樹脂、ポリ乳酸、ポリブチレンスクシ ネートなどの生分解性榭脂、ゴムなどと混練して、ポリマーァロイとしても用いることも できる。
[0120] 以下、実施例により本発明を更に詳細に説明するが、本発明は、その要旨を超え ない限り、以下の実施例により限定されるものではない。
以下において、ポリカーボネート共重合体の物性ないし特性の評価は次の方法に より行った。
[0121] (1)屈折率及びアッベ数
アッベ屈折計(ァタゴ社製「DR— M4」)で、波長 656nm (C線)、 589nm (d線) 54 6nm (e線)、 486nm (F線)の干渉フィルターを用いて、各波長の屈折率、 nC、 nD、 ne、 nFを測定した。
測定試料は榭脂を 160〜200°Cでプレス成形し、厚み 80 μ m力 500 μ mのフィ ルムを作製し、得られたフィルムを幅約 8mm、長さ 10から 40mmの短冊状に切り出 し、測定試験片とした。
測定は、界面液として 1—ブロモナフタレンを用い、 20°Cで行った。
アッベ数 V dは次の式で計算した。
v d= (l -nD) / (nC-nF)
アッベ数が大きいほど、屈折率の波長依存性が小さくなり、例えば単レンズにした 際の波長による焦点のずれが小さくなる。
[0122] (2)ガラス転移温度 (Tig)
示差走査熱量計 (メトラー社製「DSC822」)に試料約 lOmgを用いて、 10°CZmin の昇温速度で加熱して測定し、 JIS K 7121 (1987)に準拠して、低温側のベース ラインを高温側に延長した直線と、ガラス転移の階段状変化部分の曲線の勾配が最 大になるような点で引いた折線との交点の温度である、補外ガラス転移開始温度 Tig を求めた。
[0123] (3)カラー
カラーメーター(日本電色社製「300A」)を用いて、チップカラーを測定した。
ガラスセルに、チップを所定量入れ、反射測定で測定し、 b値を測定した。
この数値力^に近いほど、黄色みが小さい。
[0124] (4)還元粘度
中央理化製 DT— 504型自動粘度計にてウベローデ型粘度計を用い、溶媒として 、フエノールと 1, 1, 2, 2—テトラクロロェタンの 1 : 1混合溶媒を用い、温度 20. 0°C ±0. 1°Cで測定した。濃度は 1. OOgZdlになるように、精密に調整した。
サンプルは 120°Cで攪拌しながら、 30分で溶解し、冷却後測定に用いた。 溶媒の通過時間 t0、溶液の通過時間 tから相対粘度 η relを求め、
η rel = t/ 10、g ' cm— 1 ' sec—丄ノ
相対粘度 η relから比粘度 η spを求めた。
η sp = ( η — η 0) η 0 = η rel— 1
比粘度 η spを濃度 c gZdlで割って還元粘度 (換算粘度) r? redを求めた。
η rea = η sp/ c
この数値が高 、ほど分子量が大き!/、。
[0125] (5) 5%熱減量温度
セイコー電子製「TG— DTA」(SSC— 5200、 TG/DTA220)を用い、資料 10m gをアルミニウム製容器に載せ、窒素雰囲気下 (窒素流量 200mlZ分)で昇温速度 1 0°CZ分で 30°Cから 450°Cまで測定し、 5%重量が減少した際の温度を求めた。 この温度が高いほど、熱分解しにくい。
[0126] (6)アイゾット衝撃強度
カスタム ·サイエンティフィック(Custom Scientific)社製ミニマックス射出成形機「 CS— 183MMXJを用!ヽて、温度 240力ら 300。Cで、長さ 31. 5mm、幅 6. 2mm, 厚さ 3. 2mmの試験片を射出成形し、深さ 1. 2mmのノッチをノッチングマシンで付け 、試験片とした。
この試験片について、カスタム 'サイエンティフィック社製ミニマックスアイゾット衝撃 試験機「CS— 183TI型」を用いて、 23°Cにおけるノッチ付きのアイゾット衝撃強度を 測定した。
この数値が大きいほど、耐衝撃強度が大きぐ割れにくい。
[0127] (7)引張試験
上記射出成形機を用いて温度 240°Cから 300°Cで、平行部長さ 9mm、平行部直 径 1. 5mmの引張試験片を射出成形し、カスタム 'サイテンティフィック社製引張試験 機「CS— 183TE型」を用いて、引張速度 lcmZ分の条件で引張試験を行い、降伏 時伸び、引張降伏強さ、引張降伏弾性率、及び破断時伸びそれぞれ測定した。 それぞれの数値が大きいほど、強さ、伸びがある。
[0128] (8) NMR
溶媒として、重クロ口ホルムを用い、 Varian社製「Unity InovaJにて、共鳴周波数 500MHz,フリップ角 45° 、測定温度 25°Cにて、 H—NMRを測定した。
[0129] (9)光弾性係数
<サンプル作製 >
80°Cで 5時間真空乾燥をしたポリカーボネート榭脂サンプル 4. Ogを、幅 8cm、長 さ 8cm、厚さ 0. 5mmのスぺーサーを用いて、熱プレスにて熱プレス温度 200〜250 °Cで、予熱 1〜3分、圧力 20MPaの条件で 1分間加圧後、スぺーサーごと取り出し、 水管冷却式プレスで圧力 20MPaで 3分間加圧冷却してシートを作製した。このシー トから幅 5mm、長さ 20mmにサンプルを切り出した。
<測定 >
He— Neレーザー、偏光子、補償板、検光子、及び光検出器力 なる複屈折測定 装置と振動型粘弾性測定装置 (レオロジ一社製 DVE— 3)を組み合わせた装置を用 いて測定した。(詳細は、日本レオロジ一学会誌 Vol. 19, p93— 97 (1991)を参照 o )
切り出したサンプルを粘弾性測定装置に固定し、 25°Cの室温で貯蔵弾性率 E,を 周波数 96Hzにて測定した。同時に、出射されたレーザー光を偏光子、試料、補償 板、検光子の順に通し、光検出器 (フォトダイオード)で拾い、ロックインアンプを通し て角周波数 ω又は 2 ωの波形につ!、て、その振幅とひずみに対する位相差を求め、 ひずみ光学係数 Ο'を求めた。このとき、偏光子と検光子の方向は直交し、またそれ ぞれ、試料の伸長方向に対して π Ζ4の角度をなすように調整した。
光弾性係数 Cは、貯蔵弾性率 E'とひずみ光学係数 Ο'を用いて次式より求めた。
C = 0 ' /E '
[0130] ( 10)発生ガス量
<サンプル作製 >
100°Cで 5時間真空乾燥をしたポリカーボネート榭脂サンプル 8gを、幅 8cm、長さ 8c m、厚さ 0. 5mmのスぺーサーを用いて、熱プレスにて熱プレス温度 200〜250°Cで 、予熱 1〜3分、圧力 20MPaの条件で 1分間加圧後、スぺーサーごと取り出し、水管 冷却式プレスを用いて圧力 20MPaで 3分間加圧冷却しシートを作製した。このシー トから幅 lcm長さ 2cmのサンプルを切り出した。厚さは lmmであった。
<測定 >
加熱脱着 ガスクロマトグラフ Z質量分析法 (TDS GCZMS)にて発生ガスを測 定した。測定装置として、 GERSTEL社製 TDS2を用い、加熱脱着温度を 250°C、 10分、トラップ温度を—130°C、で実施した。
サンプルをガラスチャンバ一に入れ、 110°Cで 30分間、ヘリウム 60mLZ分で発生 するガスを捕集管 Tenax— TAで捕集した。
GC/MSとして Agilent社製 HP6890/5973N,カラムとして HP— VOC 0. 3 2 X 60m 1. 8 μ mdfを用い、 40°C、 5分保持した後、 8°CZ分で 280°Cまで昇温後 、 280°Cで 25分保持して、測定した。キャリアガスとしてヘリウム 1. 3mLZ分で測定 した。
ガス発生量は製造時に留出するフエノール及びフエノールに由来するべンズアル デヒドを除いた単位面積当たりのトータル発生量としてトルエンによる換算値にて求 めた。
[0131] ( 11)鉛筆硬度
測定装置として、新東科学製 表面測定機 トライポギア タイプ 14DRを用い、 JIS — K5600に準拠して測定した。
荷重 750g
測定スピード 30mmZmin
測定距離 7mm
鉛筆として三菱鉛筆製 UNI を用いた。
鉛筆硬度としては 4H, 3H, 2H, H, F, HB, B、 2B, 3B, 4Bを用いた。
5回測定し、 2回以上、傷がついた鉛筆硬度のひとつ柔らかい硬度を測定物質の鉛 筆硬度とした。
[0132] (12)蟻酸の定量
イソソルビドを純水で 100倍希釈してイオンクロマトグラフ Dionexy社製 DX—5
00型で測定した。
なお、反応に用いたイソソルビドはロケットフルーレネ土製、三光化学社製、 1, 4ーシ クロへキサンジメタノールはイーストマン社製、炭酸セシウムは和光純薬社製、ジフエ
-ルカーボネートは三菱ィ匕学 (株)製、トリシクロデカンジメタノールはセラ-一ズ社製 、ペンタシクロデカンジメタノールはセラニーズ社製、 1, 3 ァダマンタンジオールは アルドリッチ社製、 1, 4 ブタンジオールは三菱ィ匕学社製、 1, 6 へキサンジオール は和光純薬社製、 9, 9 ビス一(4一(2 ヒドロキシエトキシ)フエ-ル)フルオレンは 大阪ガスケミカル社製である。
[0133] また、以下の実施例の記載の中で用いた化合物の略号は次の通りである。
ISOB:イソソルビド
1, 4— CHDM : 1, 4 シクロへキサンジメタノール
TCDDM:トリシクロデカンジメタノール
PCPDM:ペンタシクロペンタデカンジメタノール
1, 4 BG : 1, 4 ブタンジオール
1, 6— HD : 1, 6 へキサンジオール
BHEPF : 9, 9—ビス一(4— (2—ヒドロキシエトキシ)フエ-ル)フルオレン BCF : 9, 9—ビスクレゾーノレフノレ才レン
DPC:ジフエ-ルカーボネート
[0134] 〈実施例 1〉
イソソノレビド 27. 7重量部(0. 516モノレ)に対して、 1, 4 シクロへキサンジメタノー ル(以下「1, 4— CHDM」と略記する。) 13. 0重量部(0. 246モル)、ジフヱ-ルカ ーボネート(以下「DPC」と略記する。) 59. 2重量部(0. 752モル)、及び触媒として 、炭酸セシウム 2. 21 X 10—4重量部(1. 84 X 10—6モル)を反応容器に投入し、窒素
雰囲気下にて、反応の第 1段目の工程として、加熱槽温度を 150°Cに加熱し、必要 に応じて攪拌しながら、原料を溶解させた (約 15分)。
次いで、圧力を常圧から 13. 3kPaにし、加熱槽温度を 190°Cまで 1時間で上昇さ せながら、発生するフ ノールを反応容器外へ抜き出した。
[0135] 反応容器全体を 190°Cで 15分保持した後、第 2段目の工程として、反応容器内の 圧力を 6. 67kPaとし、加熱槽温度を 230°Cまで、 15分で上昇させ、発生するフエノ ールを反応容器外へ抜き出した。攪拌機の攪拌トルクが上昇してくるので、 8分で 25 0°Cまで昇温し、さらに発生するフエノールを取り除くため、反応容器内の圧力を 0. 2 OOkPa以下に到達させた。所定の攪拌トルクに到達後、反応を終了し、生成した反 応物を水中に押し出して、ポリカーボネート共重合体のペレットを得た。
[0136] 得られたポリカーボネート共重合体の還元粘度は 1. 007dlZg、ガラス転移温度 Ti gは 124°C、カラー b値は 8. 8であった。これらの結果を表 1に示す。
また、このポリカーボネート共重合体を 245°Cで、金型温度 90°Cで成形し、長さ 31 . 5mm、幅 6. 2mm、厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1. 5m mの引張試験片を得た。これらの試験片を用いて、機械物性の評価を行った結果、 引張降伏強さ 84MPa、引張降伏弾性率 748MPa、降伏時伸び 16%、破断時伸び 30%、アイゾット衝撃強度 227jZm2であった。これらの結果を表 2に示す。
更に、このポリカーボネート共重合体を 200°Cでプレスし、厚さ約 200 mのフィル ムに成形したときの d線の屈折率は 1. 4992、アッベ数は 58であった。これらの結果 を表 3へ示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 344°C であった。この結果を表 4に示す。
また、発生ガス量を調べたところ、フエノール成分以外の発生ガス量は 3. 7ng/c m2で、一般式(1)で表されるジヒドロキシィ匕合物を除 、たジヒドロキシィ匕合物由来の 発生ガスは検出されな力つた。この結果を表 6に示す。
また、このポリカーボネート共重合体の NMRチャートを図 1に示す。
[0137] 〈実施例 2〉
イソソルビド 31. 8重量部(0. 458モル)、 1, 4-CHDM8. 7重量部(0. 127モル
) , DPC59. 5重量部(0. 583モル)、触媒として、炭酸セシウム 2. 22 X 10—4重量部 (1. 43 X 10—6モル)を反応容器に投入し、窒素雰囲気下にて、反応の第 1段目のェ 程として、加熱槽温度を 150°Cに加熱し、必要に応じて攪拌しながら、原料を溶解さ せた (約 15分)。
次いで、圧力を常圧から 13. 3kPaにし、加熱槽温度を 190°Cまで 1時間で上昇さ せながら、発生するフ ノールを反応容器外へ抜き出した。
[0138] 反応容器全体を 190°Cで 15分保持した後、第 2段目の工程として、反応容器内の 圧力を 6. 67kPaとし、加熱槽温度を 240°Cまで、 20分で上昇させ、発生するフエノ ールを反応容器外へ抜き出した。攪拌機の攪拌トルクが上昇してくるので、さらに発 生するフエノールを取り除くため、反応容器内の圧力を 0. 200kPa以下に到達させ た。所定の攪拌トルクに到達後、反応を終了し、生成した反応物を水中に押し出して 、ポリカーボネート共重合体のペレットを得た。
[0139] 得られたポリカーボネート共重合体の還元粘度は 0. 757dl/g,ガラス転移温度 Ti gは 133°C、カラー b値は 8. 2であった。これらの結果を表 1に示す。
更に、このポリカーボネート共重合体を 200°Cでプレスし、厚さ約 200 mのフィル ムに成形したときの d線の屈折率は 1. 5004、アッベ数は 57であった。これらの結果 を表 3に示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 343°C であった。この結果を表 4に示す。
また、光弾性係数を測定したところ 20 X 10_ 12Pa_1であった。この結果をガラス転 移温度の値と共に、表 5に示す。
[0140] 〈実施例 3〉
実施 f列 2【こお!ヽて、イソソノレビ、ド 35. 9重量咅 (0. 674モノレ)、 1, 4-CHDM4. 4 重量部(0. 083モル)、 DPC59. 7重量部(0. 764モル)、触媒として、炭酸セシウム 2. 22 X 10—4重量部(1. 87 X 1CT6モル)に変更した以外は、同様に実施した。
[0141] 得られたポリカーボネート共重合体の還元粘度は 0. 712dlZg、ガラス転移温度 Ti gは 148°C、カラー b値は 9. 1であった。これらの結果を表 1に示す。
更に、このポリカーボネート共重合体を 200°Cでプレスし、厚さ約 200 mのフィル
ムに成形したときの d線の屈折率は 1. 5014、アッベ数は 57であった。これらの結果 を表 3に示す。
[0142] 〈実施例 4〉
実施 f列: Uこお ヽて、イソソノレヒ、、ド 19. 7重量咅 (0. 363モノレ)、 1, 4-CHDM21. 6 重量部(0. 404モル)、 DPC58. 8重量部(0. 741モル)、触媒として、炭酸セシウム 2. 19 X 10—4重量部(1. 82 X 1CT6モル)に変更した以外は、同様に実施した。
[0143] 得られたポリカーボネート共重合体の還元粘度は 1. 196dlZg、ガラス転移温度 Ti gは 101°C、カラー b値は 7. 7であった。これらの結果を表 1に示す。
また、このポリカーボネート共重合体を温度 245°Cで、金型温度 80°Cで成形し、長 さ 31. 5mm,幅 6. 2mm,厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1 . 5mmの引張試験片を得た。これらの試験片を用いて、機械物性の評価を行った結 果、引張降伏強さ 66MPa、引張降伏弾性率 595MPa、降伏時伸び 16%、破断時 伸び 27%、アイゾット衝撃強度 293jZm2であった。これらの結果を表 2に示す。 更に、このポリカーボネート共重合体を 200°Cでプレスし、厚さ約 200 mのフィル ムに成形したときの d線の屈折率は 1. 4993、アッベ数は 61であった。結果を表 3に 示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 345°C であった。この結果を表 4に示す。
[0144] 〈実施例 5〉
イソソルビド 15. 7重量部(0. 288モル)に対して、 1, 4-CHDM25. 8重量部(0 . 480モル)、 DPC58. 6重量部(0. 734モル)、及び触媒として、炭酸セシウム 2. 1 8 10_4重量部(1. 80 X 10—6モル)を反応容器に投入し、窒素雰囲気下にて、反 応の第 1段目の工程として、加熱槽温度を 150°Cに加熱し、必要に応じて、攪拌しな がら、原料を溶解させた (約 15分)。
次いで、圧力を常圧から 13. 3kPaにし、加熱槽温度を 190°Cまで 1時間で上昇さ せながら発生するフエノールを反応容器外へ抜き出し、 190°Cで 30分保持した後、 第 2段目の工程として、反応容器内の圧力を 6. 67kPaとし、加熱槽温度を 240°Cま で、 45分で上昇させ、発生するフエノールを反応容器外へ抜き出した。攪拌機の攪
拌トルクが上昇してくる力 さらに発生するフエノールを取り除くため、反応容器内の 圧力を 0. 200kPa以下に到達させた。所定の攪拌トルクに到達後反応を終了し、反 応物を水中に押し出してペレットを得た。
[0145] 得られたポリカーボネート共重合体の還元粘度は 1. 186dlZg、ガラス転移温度 Ti gは 89°C、カラー b値は 5. 1であった。これらの結果を表 1に記す。
更に、このポリカーボネート共重合体を温度 245°Cで、金型温度 70°Cで成形し、長 さ 31. 5mm,幅 6. 2mm,厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1 . 5mmの引張試験片を得た。これらの試験片を用いて、機械物性の評価を行った結 果、引張降伏強さ 59MPa、引張降伏弾性率 541MPa、降伏時伸び 15%、破断時 伸び 70%、アイゾット衝撃強度 78 jZm2であった。これらの結果を表 2に記す。 また、このポリカーボネート共重合体を、 200°Cでプレスし、厚さ約 200 μ mのフィル ムを得た。 d線の屈折率は 1. 4993、アッベ数は 62であった。これらの結果を表 3に 記す。
[0146] 〈実施例 6〉
実施 f列 2【こお!ヽて、イソソノレヒ、、ド 27. 7重量咅 (0. 516モノレ)、 1, 4-CHDM13. 0 重量部(0. 246モル)、 DPC59. 2重量部(0. 752モル)、触媒として、炭酸セシウム 2. 21 X 10—4重量部(1. 84 X 10—6モル)に変更した以外は、同様に実施した。 得られたポリカーボネート共重合体の還元粘度は 0. 979dlZg、ガラス転移温度 Ti gは 124°C、カラー b値は 9. 5であった。これらの結果を表 1に記す。
更に、このポリカーボネート共重合体を温度 245°Cで、金型温度 90°Cで成形し、長 さ 31. 5mm,幅 6. 2mm,厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1 . 5mmの引張試験片を得た。これらの試験片を用いて、機械物性の評価を行った結 果、引張降伏強さ 78MPa、引張降伏弾性率 691MPa、降伏時伸び 16%、破断時 伸び 47%、アイゾット衝撃強度 18 jZm2であった。これらの結果を表 2に記す。 また、鉛筆硬度は Hであった。この結果を表 7に示す。
[0147] 〈実施例 7〉
実施 f列 2【こお!ヽて、イソソノレヒ、、ド 27. 7重量咅 (0. 516モノレ)、 1, 4-CHDM13. 0 重量部(0. 246モル)、 DPC59. 2重量部(0. 752モル)、触媒を、水酸化ナトリウム
8. 7 X 10_5重量部(5. 9 X 10_6モル)に変更した以外は同様に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 965dl/g,ガラス転移温度 Ti gは 123°C、カラー b値は 9. 4であった。これらの結果を表 1に記す。
[0148] 〈実施例 8〉
実施 f列 2【こお!ヽて、イソソノレヒ、、ド 28. 2重量咅 (0. 516モノレ)、 1, 4-CHDM13. 3 重量部(0. 246モル)、 DPC58. 5重量部(0. 730モル)、触媒として、炭酸セシウム 2. 25 X 10—4重量部(1. 84 X 10—6モル)に変更した以外は、同様に実施した。
[0149] 得られたポリカーボネート共重合体の還元粘度は 0. 496dlZg、ガラス転移温度 Ti gは 122°C、カラー b値は 9. 6であった。これらの結果を表 1に記す。
また、鉛筆硬度は Hであった。この結果を表 7に示す。
[0150] 〈実施例 9〉
実施 f列 2【こお!ヽて、イソソノレヒ、、ド 27. 7重量咅 (0. 516モノレ)、 1, 4-CHDM13. 0 重量部(0. 246モル)、 DPC59. 2重量部(0. 752モル)、触媒を、炭酸セシウム 2. 21 X 10_5重量部(1. 84 X 10_7モル)に変更した以外は同様に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 910dlZg、ガラス転移温度 Ti gは 124°C、カラー b値は 9. 8であった。これらの結果を表 1に記す。
[0151] 〈実施例 10〉
実施 f列 2【こお!ヽて、イソソノレヒ、、ド 27. 7重量咅 (0. 516モノレ)、 1, 4-CHDM13. 0 重量部(0. 246モル)、 DPC59. 2重量部(0. 752モル)、触媒として、炭酸セシウム 2. 21 X 10—3重量部(1. 84 X 10—5モル)に変更した以外は同様に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 980dlZg、ガラス転移温度 Ti gは 124°C、カラー b値は 8. 3であった。これらの結果を表 1に記す。
[0152] 〈実施例 11〉
実施 f列 2【こお!ヽて、イソソノレヒ、、ド 27. 7重量咅 (0. 516モノレ)、 1, 4-CHDM13. 0 重量咅 (0. 246モノレ)、 DPC59. 2重量咅 (0. 752モノレ)に変更し、原料ととちに、熱 安定剤「PEP— 36」(旭電化製、ビス(2, 6 ジ—tert—ブチルー 4 メチルフエ-ル )ペンタエリスリトールジホスフアイト) 0. 096重量部を反応容器に投入して重合させ た以外は同様に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 975dl/g,ガラス転移温度 Ti gは 124°C、カラー b値は 7. 2であった。これらの結果を表 1に記す。
[0153] 〈実施例 12〉
実施 f列 2【こお!ヽて、イソソノレヒ、、ド 19. 7重量咅 (0. 363モノレ)、 1, 4-CHDM21. 6 重量部(0. 404モル)、 DPC58. 8重量部(0. 741モル)、触媒として、炭酸セシウム 2. 19 10_4重量部(1. 82 X 10—6モル)に変更し、原料とともに、熱安定剤「PEP 36」(旭電化製、ビス(2, 6 ジー tert—ブチルー 4 メチルフエ-ル)ペンタエリス リトールジホスフアイト) 0. 096重量部を反応容器に投入し、重合させた以外は同様 に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 850dlZg、ガラス転移温度 Ti gは 100°C、カラー b値は 3. 6であった。これらの結果を表 1に記す。
[0154] [比較例 1]
イソソルビド 40. 1重量部(0. 581モル)に対して、 DPC59. 9重量部(0. 592モル 、触媒として、炭酸セシウム 2. 23 X 10_4重量部(1. 45 X 10_6モル)を反応容器に 投入し、攪拌しながら、室温から 150°Cに加熱して溶解をした (約 15分)。
次いで、圧力を常圧から 13. 3kPaにし、 190°Cまで 1時間で上昇させながら発生 するフエノールを系外へ抜き出した。 190°Cで 15分保持した後、反応器内圧力を 6. 67kPaとし、加熱槽温度を 230°Cまで、 15分で上昇させ、発生するフエノールを抜い た。攪拌トルクが上昇してくるので、 8分で 250度まで昇温し、さらに発生するフエノー ルを取り除くため、真空度を 0. 200kPa以下に到達させた。所定の攪拌トルクに到達 後反応を終了し、反応物を水中に押し出してペレットを得ようとした力 押し出せなか つたので、固まりで取り出した。
[0155] 得られたポリカーボネート共重合体の還元粘度は 0. 679dl/g,ガラス転移温度は 160°C、カラー b値は 13. 0であり、実施例のものに比べ、 b値が高ぐ褐色に着色し たものだった。これらの結果を表 1に記す。
更に、このポリカーボネート共重合体を 265°Cで成形し、長さ 31. 5mm、幅 6. 2m m、厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1. 5mmの引張試験片の 採取を試みたが、溶融粘度が高ぐまた、着色が激しぐ発泡も激しく成型品の歩留
まりが悪力つた。これらの試験片を用いて、機械物性の評価を行った結果、引張降伏 強さ 105MPa、引張降伏弾性率 353MPa、降伏時伸び 17%、破断時伸び 31%、ァ ィゾット衝撃強度 l ljZm2であり、アイゾット衝撃強度が実施例に比べ、著しく低いこ とが分力つた。これらの結果を表 2に記す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 339°C であり、実施例のものに比べて低いことが分かった。この結果を表 4に記す。
なお、このポリカーボネート共重合体を、 200°Cでプレスし、厚さ約 200 μ mのフィ ルムを得た。このフィルムははさみで切るとひび割れができ、もろかった。
[0156] [比較例 2]
1, 4-CHDM42. 3重量部(0. 776モル)、 DPC57. 7重量部(0. 712モル)、触 媒として、炭酸セシウム 2. 15 X 10—4重量部(1. 75 X 10—6モル)を反応容器に投入 し、窒素雰囲気下にて、反応の第 1段目の工程として、加熱槽温度を 150°Cに加熱 し、必要に応じて攪拌しながら、原料を溶解させた (約 15分)。
次いで、圧力を常圧から 13. 3kPaに 3分で減圧し、保持した。加熱槽温度を 190 °Cまで 60分で上昇させながら、発生するフエノールを反応容器外へ抜き出した。 反応容器全体を 190°Cで 15分保持した後、第 2段目の工程として、反応容器内の 圧力を 6. 67kPaとし、加熱槽温度を 220°Cまで、 45分で上昇させ、発生するフエノ ールを反応容器外へ抜き出した。攪拌機の攪拌トルクが上昇してくるが、さらに発生 するフ ノールを取り除くため、反応容器内の圧力を 0. 200kPa以下に到達させた。 所定の攪拌トルクに到達後、反応を終了し、生成した反応物を水中に押し出して、 ポリカーボネート共重合体のペレットを得た。
得られたポリカーボネート共重合体の還元粘度は 0. 662dlZg、ガラス転移温度は 40°C、カラー b値は 4. 5で、ガラス転移温度が低いので、団子状になり、チップ化し にく力つた。これらの結果を表 1に示す。
[0157] [比較例 3]
市販の芳香族ポリカーボネート榭脂「ユーピロン H4000」(三菱エンジニアリングプ ラスチック社製、還元粘度 0. 456dl/g)を 280°Cで成形し、長さ 31. 5mm、幅 6. 2 mm、厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1. 5mmの引張試験片
を得た。
これらの試験片を用いて、機械物性の評価を行った結果、引張降伏強さ 63MPa、 引張降伏弾性率 629MPa、降伏時伸び 13%、破断時伸び 74%、アイゾット衝撃強 度 6jZm2であった。これらの結果を表 2に示す。
更に、この芳香族ポリカーボネート榭脂を 200°Cでプレスし、厚さ約 200 mのフィ ルムを得た。 d線の屈折率は 1. 5828、アッベ数は 30であった。結果を表 3に示す。 また、光弾性係数を測定したところ 72 X 10_ 12Pa_1であった。この結果をガラス転 移温度の値と共に、表 5に示す。
また、鉛筆硬度は 2Bであった。この結果を表 7に示す。
[0158] [比較例 4]
市販の芳香族ポリカーボネート榭脂「ユーピロン S2000」(三菱エンジニアリングプ ラスチック社製、還元粘度 0. 507dl/g)を 280°Cで成形し、長さ 31. 5mm、幅 6. 2 mm、厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1. 5mmの引張試験片 を得た。
これらの試験片を用いて、機械物性の評価を行った結果、引張降伏強さ 63MPa、 引張降伏弾性率 565MPa、降伏時伸び 13%、破断時伸び 85%、高いアイゾット衝 撃強度 641jZm2であった。これらの結果を表 2に示す。
[0159] [比較例 5]
市販のポリ乳酸「レイシァ H— 440」(三井化学社製)について窒素雰囲気下での 5 %熱減量温度を測定したところ、 320°Cであった。この結果を表 4に示す。
[0160] 〈実施例 13〉
イソソルビド 26. 9重量部(0. 483モル)に対して、トリシクロデカンジメタノール(以 下「TCDDM」と略記する。) 15. 8重量部(0. 211モル)、 DPC57. 4重量部(0. 70 9モル)、及び触媒として、炭酸セシウム 2. 14 X 10_4重量部(1. 73 X 10_6モル)を 反応容器に投入し、窒素雰囲気下にて、反応の第 1段目の工程として、加熱槽温度 を 150°Cに加熱し、必要に応じて攪拌しながら、原料を溶解させた (約 15分)。
次いで、圧力を常圧から 13. 3kPaに 40分で減圧し、加熱槽温度を 190°Cまで 40 分で上昇させながら、発生するフエノールを反応容器外へ抜き出した。
[0161] 反応容器全体を 190°Cで 15分保持した後、第 2段目の工程として、加熱槽温度を 220°Cまで、 30分で上昇させた。昇温に入ってから 10分後に、反応容器内の圧力を 30分で 0. 200kPa以下とし、発生するフ ノールを溜出させた。所定の攪拌トルクに 到達後、反応を終了し、生成した反応物を水中に押し出して、ポリカーボネート共重 合体のペレットを得た。
[0162] 得られたポリカーボネート共重合体の還元粘度は 0. 640dlZg、ガラス転移温度 Ti gは 126°C、カラー b値は 4. 6であった。これらの結果を表 1に示す。
また、このポリカーボネート共重合体を 245°Cで、金型温度 90°Cで成形し、長さ 31 . 5mm、幅 6. 2mm、厚さ 3. 2mmの試験片と平行部長さ 9mm、平行部直径 1. 5m mの引張試験片を得た。これらの試験片を用いて、機械物性の評価を行った結果、 引張降伏強さ 89MPa、引張降伏弾性率 834MPa、降伏時伸び 15%、破断時伸び 76%、アイゾット衝撃強度 48jZm2であった。これらの結果を表 2に示す。
更に、このポリカーボネート共重合体を 200°Cでプレスし、厚さ約 200 mのフィル ムに成形したときの d線の屈折率は 1. 5095、アッベ数は 62であった。これらの結果 を表 3に示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 348°C であった。この結果を表 4に示す。
また、光弾性係数を測定したところ 9 X 10_ 12Pa_1であった。この結果をガラス転移 温度の値と共に、表 5に示す。
また、発生ガス量を調べたところ、フエノール成分以外の発生ガス量は 4. 5ng/c m2で、一般式(1)で表されるジヒドロキシィ匕合物を除くジヒドロキシィ匕合物由来の発 生ガスは検出されな力つた。この結果を表 6に示す。
また、鉛筆硬度は Fであった。この結果を表 7に示す。
[0163] 〈実施例 14〉
イソソルビド 35. 5重量部(0. 660モル)に対して、 TCDDM5. 4重量部(0. 075 モル)、 DPC59. 0重量部(0. 748モル)、及び触媒として、炭酸セシウム 2. 20 X 10 _4重量部(1. 83 X 10—6モル)に変更した以外は、実施例 13と同様に実施した。
[0164] 得られたポリカーボネート共重合体の還元粘度は 0. 546dlZg、ガラス転移温度 Ti
gは 144°C、カラー b値は 6. 4であった。これらの結果を表 1に記す。
機械物性の評価を行った結果、引張降伏強さ 106MPa、引張降伏弾性率 872MP a、降伏時伸び 16%、破断時伸び 26%、アイゾット衝撃強度 65jZm2であった。これ らの結果を表 2に示す。
また、フィルムに成形したときの d線の屈折率は 1. 5052、アッベ数は 60であった。 これらの結果を表 3へ示す。
[0165] 〈実施例 15〉
イソソルビド 31. 1重量部(0. 569モル)に対して、 TCDDM10. 7重量部(0. 145 モル)、 DPC58. 2重量部(0. 725モル)、及び触媒として、炭酸セシウム 2. 17 X 10 _4重量部(1. 78 X 10—6モル)に変更した以外は、実施例 13と同様に実施した。
[0166] 得られたポリカーボネート共重合体の還元粘度は 0. 644dlZg、ガラス転移温度 Ti gは 136°C、カラー b値は 2. 8であった。これらの結果を表 1に記す。
機械物性の評価を行った結果、引張降伏強さ 107MPa、引張降伏弾性率 934MP a、降伏時伸び 16%、破断時伸び 39%、アイゾット衝撃強度 58jZm2であった。これ らの結果を表 2に示す。
また、フィルムに成形したときの d線の屈折率は 1. 5090、アッベ数は 61であった。 これらの結果を表 3へ示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 344°C であった。この結果を表 4に示す。
[0167] 〈実施例 16〉
イソソルビド 22. 7重量部(0. 403モル)に対して、 TCDDM20. 7重量部(0. 274 モル)、 DPC56. 6重量部 (0. 684モル)、及び触媒として、炭酸セシウム 2. 11 X 10 _4重量部(1. 68 X 1CT6モル)に変更した以外は、実施例 13と同様に実施した。 得られたポリカーボネート共重合体の還元粘度は 0. 637dlZg、ガラス転移温度 Ti gは 118°C、カラー b値は 2. 3であった。これらの結果を表 1に記す。
また、フィルムに成形したときの d線の屈折率は 1. 5135、アッベ数は 58であった。 これらの結果を表 3へ示す。
また、光弾性係数を測定したところ 7 X 10_ 12Pa_1であった。この結果をガラス転移
温度の値と共に、表 5に示す。
[0168] 〈実施例 17〉
イソソルビド 18. 7重量部(0. 327モル)に対して、 TCDDM25. 6重量部(0. 333 モル)、 DPC55. 8重量部(0. 666モル)、及び触媒として、炭酸セシウム 2. 08 X 10 _4重量部(1. 63 X 1CT6モル)に変更した以外は、実施例 13と同様に実施した。
[0169] 得られたポリカーボネート共重合体の還元粘度は 0. 785dl/g,ガラス転移温度 Ti gは 110°C、カラー b値は 4. 7であった。これらの結果を表 1に記す。
機械物性の評価を行った結果、引張降伏強さ 79MPa、引張降伏弾性率 807MPa 、降伏時伸び 13%、破断時伸び 18%、アイゾット衝撃強度 58jZm2であった。これら の結果を表 2に示す。
また、フィルムに成形したときの d線の屈折率は 1. 5157、アッベ数は 60であった。 これらの結果を表 3へ示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 349°C であった。この結果を表 4に示す。
[0170] 〈実施例 18〉
イソソルビド 14. 7重量部(0. 257モル)に対して、 TCDDM30. 3重量部(0. 394 モル)、 DPC55. 0重量部(0. 656モル)、及び触媒として、炭酸セシウム 2. 05 X 10 _4重量部(1. 61 X 10_6モル)を窒素雰囲気下にて、反応の第 1段目の工程として、 加熱槽温度を 150°Cに加熱し、必要に応じて攪拌しながら、原料を溶解させた (約 1 5分)。
次いで、圧力を常圧から 13. 3kPaに 3分で減圧し、加熱槽温度を 190°Cまで 60分 で上昇させながら、発生するフエノールを反応容器外へ抜き出した。
[0171] 反応容器全体を 190°Cで 15分保持した後、第 2段目の工程として、反応容器内の 圧力を 6. 67kPaとし、加熱槽温度を 240°Cまで、 45分で上昇させ、発生するフエノ ールを反応容器外へ抜き出した。さらに発生するフ ノールを取り除くため、反応容 器内の圧力を 0. 200kPa以下に到達させた。所定の攪拌トルクに到達後、反応を終 了し、生成した反応物を水中に押し出して、ポリカーボネート共重合体のペレットを得
[0172] 得られたポリカーボネート共重合体の還元粘度は 0. 672dl/g,ガラス転移温度 Ti gは 102°C、カラー b値は 9. 2であった。これらの結果を表 1に記す。
機械物性の評価を行った結果、引張降伏強さ 76MPa、引張降伏弾性率 850MPa 、降伏時伸び 12%、破断時伸び 31%、アイゾット衝撃強度 40j/m2であった。これら の結果を表 2に示す。
また、フィルムに成形したときの d線の屈折率は 1. 5185、アッベ数は 58であった。 これらの結果を表 3へ示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 352°C であった。この結果を表 4に示す。
[比較例 6]
TCDDM47. 8重量部(0. 586モル)、 DPC58. 2重量部(0. 585モル)、触媒と して、炭酸セシウム 1. 95 X 10—4重量部(1. 44 X 10—6モル)を反応容器に投入し、 窒素雰囲気下にて、反応の第 1段目の工程として、加熱槽温度を 150°Cに加熱し、 必要に応じて攪拌しながら、原料を溶解させた (約 15分)。
次いで、圧力を常圧から 13. 3kPaに 40分で減圧しながら、加熱槽温度を 190°Cま で 40分で上昇させた。発生するフ ノールを反応容器外へ抜き出した。
反応容器全体を 190°Cで 15分保持した後、第 2段目の工程として、加熱槽温度を 220°Cまで、 30分で上昇させた。昇温に入ってから 10分後に、反応容器内の圧力を 30分で 0. 200kPa以下とし、発生するフ ノールを溜出させた。所定の攪拌トルクに 到達後、反応を終了し、生成した反応物を水中に押し出して、ポリカーボネート共重 合体のペレットを得た。
得られたポリカーボネート共重合体の還元粘度は 0. 899dlZg、ガラス転移温度は 73°C、カラー b値は 3. 9であった。これらの結果を表 1に示す。
[0173] 〈実施例 19〉
イソソルビド 25. 6重量部(0. 339モル)に対して、ペンタシクロペンタデカンジメタノ ール(以下「PCPDM」と略記する。) 19. 7重量部(0. 145モル)、 DPC54. 7重量 部(0. 494モル)、及び触媒として、炭酸セシウム 2. 04 X 10—4重量部(1. 21 X 10" 6モル)を反応容器に投入し、窒素雰囲気下にて、反応の第 1段目の工程として、カロ
熱槽温度を 150°Cに加熱し、必要に応じて攪拌しながら、原料を溶解させた (約 15 分)。
次いで、圧力を常圧から 13. 3kPaに 40分で減圧し、加熱槽温度を 220°Cまで 70 分で上昇させながら、発生するフエノールを反応容器外へ抜き出した。
[0174] 反応容器全体を 220°Cで 10分保持した後、第 2段目の工程として、加熱槽温度を 240°Cまで、 20分で上昇させながら、反応容器内の圧力を 30分で 0. 200kPa以下 とし、発生するフエノールを溜出させた。所定の攪拌トルクに到達後、反応を終了し、 生成した反応物を水中に押し出して、ポリカーボネート共重合体のペレットを得た。
[0175] 得られたポリカーボネート共重合体の還元粘度は 0. 730dlZg、ガラス転移温度 Ti gは 149°C、カラー b値は 8. 4であった。これらの結果を表 1に示す。
更に、このポリカーボネート共重合体を 200°Cでプレスし、厚さ約 200 mのフィル ムに成形したときの d線の屈折率は 1. 5194、アッベ数は 60であった。これらの結果 を表 3へ示す。
また、このポリカーボネート共重合体の窒素雰囲気下での 5%熱減量温度は 347°C であった。この結果を表 4に示す。
また、光弾性係数を測定したところ 8 X 10_ 12Pa_1であった。この結果をガラス転移 温度の値と共に、表 5に示す。
[0176] 〈実施例 20〉
イソソルビド 54. 7重量部(0. 374モル)に対して、ァダマンタンジメタノール 31. 5 重量部(0. 161モル)、 DPC116. 8重量部(0. 545モル)、及び触媒として、炭酸セ シゥム 6. 12 X 10—4重量部 (4. 84 X 1CT6モル)を反応容器に投入し、窒素雰囲気 下にて、反応の第 1段目の工程として、加熱槽温度を 150°Cに加熱し、必要に応じて 攪拌しながら、原料を溶解させた (約 15分)。
次いで、圧力を常圧から 13. 3kPaに 40分で減圧し、加熱槽温度を 220°Cまで 70 分で上昇させながら、発生するフエノールを反応容器外へ抜き出した。
[0177] 反応容器全体を 220°Cで 10分保持した後、第 2段目の工程として、加熱槽温度を 230°Cまで、 20分で上昇させながら、反応容器内の圧力を 30分で 0. 200kPa以下 とし、発生するフエノールを溜出させた。所定の攪拌トルクに到達後、反応を終了し、
生成した反応物を水中に押し出して、ポリカーボネート共重合体のペレットを得た。
[0178] 得られたポリカーボネート共重合体の還元粘度は 0. 409dlZg、ガラス転移温度 Ti gは 125°C、カラー b値は 14. 8であった。これらの結果を表 1に示す。
[0179] 〈実施例 21〉
実施例 20において、イソソルビド 54. 5重量部(0. 373モル)に対して、ビスシクロ へキサンジオール 31. 7重量部(0. 160モル)、 DPC116. 4重量部(0. 543モル)、 及び触媒として、炭酸セシウム 2. 04 X 10_4重量部(1. 21 X 10_6モル)に変更した 以外は、同様の方法で実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 260dlZg、ガラス転移温度 Ti gは 125°C、カラー b値は 8. 6であった。これらの結果を表 1に示す。
[0180] 〈実施例 22〉
イソソルビド 11. 7重量部(0. 165モル)に対して、 1, 4-CHDM30. 0重量部(0 . 428モル)、 DPC58. 3重量部(0. 561モル)、及び触媒として、炭酸セシウム 2. 1 8 X 10_4重量部(1. 38 X 10_6モル)を反応容器に投入し、窒素雰囲気下にて、反 応の第 1段目の工程として、加熱槽温度を 150°Cに加熱し、必要に応じて、攪拌しな がら、原料を溶解させた (約 15分)。
次いで、圧力を常圧から 13. 3kPaにし、加熱槽温度を 190°Cまで 1時間で上昇さ せながら発生するフエノールを系外へ抜き出し、 190°Cで 30分保持した後、第 2段目 の工程として、反応容器内の圧力を 6. 67kPaとし、加熱槽温度を 220°Cまで、 45分 で上昇させ、発生するフエノールを抜き出した。攪拌トルクが上昇してくる力 さらに発 生するフエノールを取り除くため、反応容器内の圧力を 0. 200kPa以下に到達させ た。所定の攪拌トルクに到達後反応を終了し、反応物を水中に押し出してペレットを 得た。
[0181] 得られたポリカーボネート共重合体の還元粘度は 0. 979dlZg、ガラス転移温度は 74°C、カラー b値は 4. 7であった。これらの結果を表 1に記す。
更に、このポリカーボネート共重合体を、 200°Cでプレスし、厚さ約 200 mのフィ ルムを得た。 d線の屈折率は 1. 5002、アッベ数は 56であった。これらの結果を表 3 に記す。
[0182] 〈実施例 23〉
実施 f列 22【こお!ヽて、イソソノレヒ、、ド 7. 8重量咅 (0. 142モノレ)、 1, 4-CHDM34. 1 重量部(0. 631モル)、 DPC58. 1重量部(0. 723モル)、触媒として、炭酸セシウム 2. 17 X 10—4重量部(1. 77 X 1CT6モル)に変更した以外は、同様に実施した。 得られたポリカーボネート共重合体の還元粘度は 1. 159dl/g,ガラス転移温度 Ti gは 63°C、カラー b値は 2. 9であった。これらの結果を表 1に記す。
更に、このポリカーボネート共重合体を、 200°Cでプレスし、厚さ約 200 mのフィ ルムを得た。 d線の屈折率は 1. 5024、アッベ数は 56であった。これらの結果を表 3 に記す。
[0183] 〈実施例 24〉
実施 f列 22【こお!ヽて、イソソノレヒ、、ド 3. 9重量咅 (0. 070モノレ)、 1, 4-CHDM38. 2 重量部(0. 703モル)、 DPC57. 9重量部(0. 717モル)、触媒として、炭酸セシウム 2. 16 X 10—4重量部(1. 76 X 10—6モル)に変更した以外は、同様に実施した。 得られたポリカーボネート共重合体の還元粘度は 0. 670dlZg、ガラス転移温度 Ti gは 51°C、カラー b値は 2. 8であった。これらの結果を表 1に記す。
[0184] 〈実施例 25〉
実施 f列 22【こお!ヽて、イソソノレヒ、、ド 1. 9重量咅 (0. 035モノレ)、 1, 4-CHDM40. 3 重量部(0. 740モル)、 DPC57. 8重量部(0. 715モル)、触媒として、炭酸セシウム 2. 15 X 10—4重量部(1. 75 X 10—6モル)に変更した以外は、同様に実施した。 得られたポリカーボネート共重合体の還元粘度は 0. 640dlZg、ガラス転移温度 Ti gは 45°C、カラー b値は 3. 0であった。これらの結果を表 1に記す。
[0185] [比較例 7]
実施 f列 13【こお!ヽて、イソソノレビ、ド 85. 61重量咅 (0. 585モノレ)、 1, 4 ブタンジ才 ール(以下「1, 4— BG」と略記する。) 22. 6重量部(0. 251モル)、 DPC166. 8重 量部(0. 779モル)、触媒として、炭酸セシウム 1. 08 X 10—4重量部(0. 87 X 10"6 モル)に変更した以外は同様に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 568dlZg、ガラス転移温度は 116°C、カラー b値は 12. 4であった。これらの結果を表 1に示す。
また、光弾性係数を測定したところ 23 X 10_"Pa_1であった。この結果をガラス転 移温度の値と共に、表 5に示す。
また、発生ガス量を調べたところ、フエノール成分以外の発生ガス量は 10. Ong/c m2で、一般式(1)で表されるジヒドロキシィ匕合物を除くジヒドロキシィ匕合物由来の発 生ガスとして、テトラヒドロフラン (THF)が 2. Ong/cm2検出された。この結果を表 6 に示す。
[0186] [比較例 8]
実施 f列 13【こお!ヽて、イソソノレビ、ド 81. 22 (0. 556モノレ)、 1, 6—へキサンジ才一ノレ (以下「1, 6— HD」と略記する。) 28. 2重量部(0. 239モル)、 DPC161. 6重量部( 0. 754モル)、触媒として、炭酸セシウム 1. 08 X 10—4重量部(0. 87 X 10—6モル) に変更した以外は同様に実施した。
得られたポリカーボネート共重合体の還元粘度は 1. 063dlZg、ガラス転移温度は 85°C、カラー b値は 8. 9であった。これらの結果を表 1に示す。
また、光弾性係数を測定したところ 20 X 10_ 12Pa_1であった。この結果をガラス転 移温度の値と共に、表 5に示す。
また、発生ガス量を調べたところ、フエノール成分以外の発生ガス量は 11. Ong/c m2で、一般式(1)で表されるジヒドロキシィ匕合物を除くジヒドロキシィ匕合物由来の発 生ガスとして、シクロへキサジェン、シクロへキシルフェニルエーテルが 5. 6ng/cm2 検出された。この結果を表 6に示す。
また、鉛筆硬度は HBであった。この結果を表 7に示す。
[0187] [比較例 9]
実施 f列 7【こお!ヽて、イソソノレヒ、、ド 42. 6 (0. 292モノレ)、 TCDDM25. 6重量咅 (0. 1 30モル)、 9, 9—ビス一(4— (2—ヒドロキシエトキシ)フエ-ル)フルオレン(以下 BH EPFと略す) 46. 7重量咅 (0. 106モノレ) DPC127. 6重量咅 (0. 596モノレ)、 虫媒と して、水酸ィ匕ナトリウム 8. 7 X 10—5重量部(5. 9 X 1CT6モル)に変更した以外は同様 に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 659dl/g,ガラス転移温度は 121°C、カラー b値は 9. 2であった。これらの結果を表 1に示す。
更に、このポリカーボネート共重合体を条件 200°Cでプレスし、厚さ 200 mのフィ ルムを得た。 d線の屈折率は 1. 5410、アッベ数は 42であった。これらの結果を表 3 に記す。
[0188] [比較例 10]
実施 f列 13【こお!ヽて、イソソノレビ、ド 63. 84 (0. 437モノレ)、 9, 9 ヒ、、スクレ、ノーノレフノレ オレン(以下 BCFと略す) 27. 6重量部(0. 0729モル)、 1, 4 ブタンジオール 19. 7重量部(0. 219モル)、触媒として、炭酸セシウム 1. 08 X 10—4重量部(0. 87 X 10 _6モル)に変更した以外は同様に実施した。
得られたポリカーボネート共重合体の還元粘度は 0. 464dlZg、ガラス転移温度は 129°C、カラー b値は 8. 3であった。これらの結果を表 1に示す。
また、光弾性係数を測定したところ 23 X 10_ 12Pa_1であった。この結果をガラス転 移温度の値と共に、表 5に示す。
[0189] 〈実施例 26〉
イソソルビド 26. 8重量部(18. 70モル)に対して、 TCDDM15. 4重量部(8. 02 モル)、 DPC57. 7重量部(26. 72モル)、及び触媒として、炭酸セシウム 2. 14 X 10 _4重量部(6. 68 X 10—5モル)を反応容器に投入し、窒素雰囲気下にて、反応の第 1 段目の工程として、加熱槽温度を 150°Cに加熱し、必要に応じて攪拌しながら、原料 を溶解させた (約 60分)。
次いで、圧力を常圧から 13. 3kPaに 40分で減圧し、加熱槽温度を 190°Cまで 40 分で上昇させながら、発生するフエノールを反応容器外へ抜き出した。
[0190] 反応容器全体を 190°Cで 15分保持した後、第 2段目の工程として、加熱槽温度を 220°Cまで、 30分で上昇させた。 220°Cなつてから 10分後に、反応容器内の圧力を 30分で 0. 200kPa以下とし、発生するフ ノールを溜出させた。所定の攪拌トルクに 到達後、反応を終了し、生成した反応物を水中に押し出して、ポリカーボネート共重 合体のペレットを得た。
[0191] 得られたポリカーボネート共重合体の還元粘度は 0. 506dlZg、ガラス転移温度 Ti gは 126°C、カラー b値は 10. 0であった。これらの結果を表 8に示す。
また、このポリカーボネート共重合体の NMRチャートを図 2に示す。
〈実施例 27〉
イソソルビド 26. 9重量部(19. 88モル)に対して、 CHDM12. 7重量部(9. 47モ ル)、 DPC60. 4重量部(30. 39モル)、及び触媒として、炭酸セシウム 2. 15 X 10"4 重量部(7. 10 X 10_5モル)以外は実施例 26と同様にして実施した。得られたポリ力 ーボネート共重合体の還元粘度は 0. 621dlZg、ガラス転移温度 Tigは 123°C、カラ 一 b値は 11. 0であった。これらの結果を表 8に示す。
イソソルビドの蒸留
イソソルビド約 1. 3kgをアルゴン気流下、 2Lフラスコに入れ、フラスコにクライゼン 管を付け、フラクションカッターを通して、受器を装着した。配管など各部固化しない よう、保温をした。減圧を徐々に開始後、加温を行い、内温約 100°Cで溶解した。そ の後、内温 160°Cにて溜出開始。このとき圧力は 133— 266Paであった。初溜を取 つた後、内温 160— 170°C、塔頂温度 150— 157°C、 133Paで蒸留を実施した。蒸 留終了後、アルゴンを入れ常圧に戻した。得られた蒸留品をアルゴン気流下で冷却 粉砕し、イソソルビドを得た。アルミラミネート袋にアルゴン気流下でシールし、保管し た。
〈実施例 28〜29〉
実施例 13において、 ISOBを予め蒸留し、蟻酸含有量が表 8に示す量となった IS OBを用いたこと以外は、同様に実施した。
得られたポリカーボネート共重合体の還元粘度、ガラス転移温度、カラー b値の測 定結果を表 8に示す。
〈実施例 30〜31〉
実施例 2において、 ISOBを予め蒸留し、蟻酸含有量が表 8に示す量となった ISO Bを用いたこと以外は、同様に実施した。
得られたポリカーボネート共重合体の還元粘度、ガラス転移温度、カラー b値の測 定結果を表 8に示す。
(参考例 1〜3)
実施例 13において、 ISOBを予め蒸留せず、蟻酸含有量が表 8に示す量である IS OBを用いたこと以外は、同様に実施した。
フエノールの溜出はあるが、反応液は次第に着色し、トルク上昇が見られず、ポリマ 一は得られなかった。
(参考例 4)
実施例 2において、 ISOBを予め蒸留せず、蟻酸含有量が表 8に示す量である ISO Bを用いたこと以外は、同様に実施した。
フエノールの溜出はあるが、反応液は次第に着色し、トルク上昇が見られず、ポリマ 一は得られなかった。
[表 1]
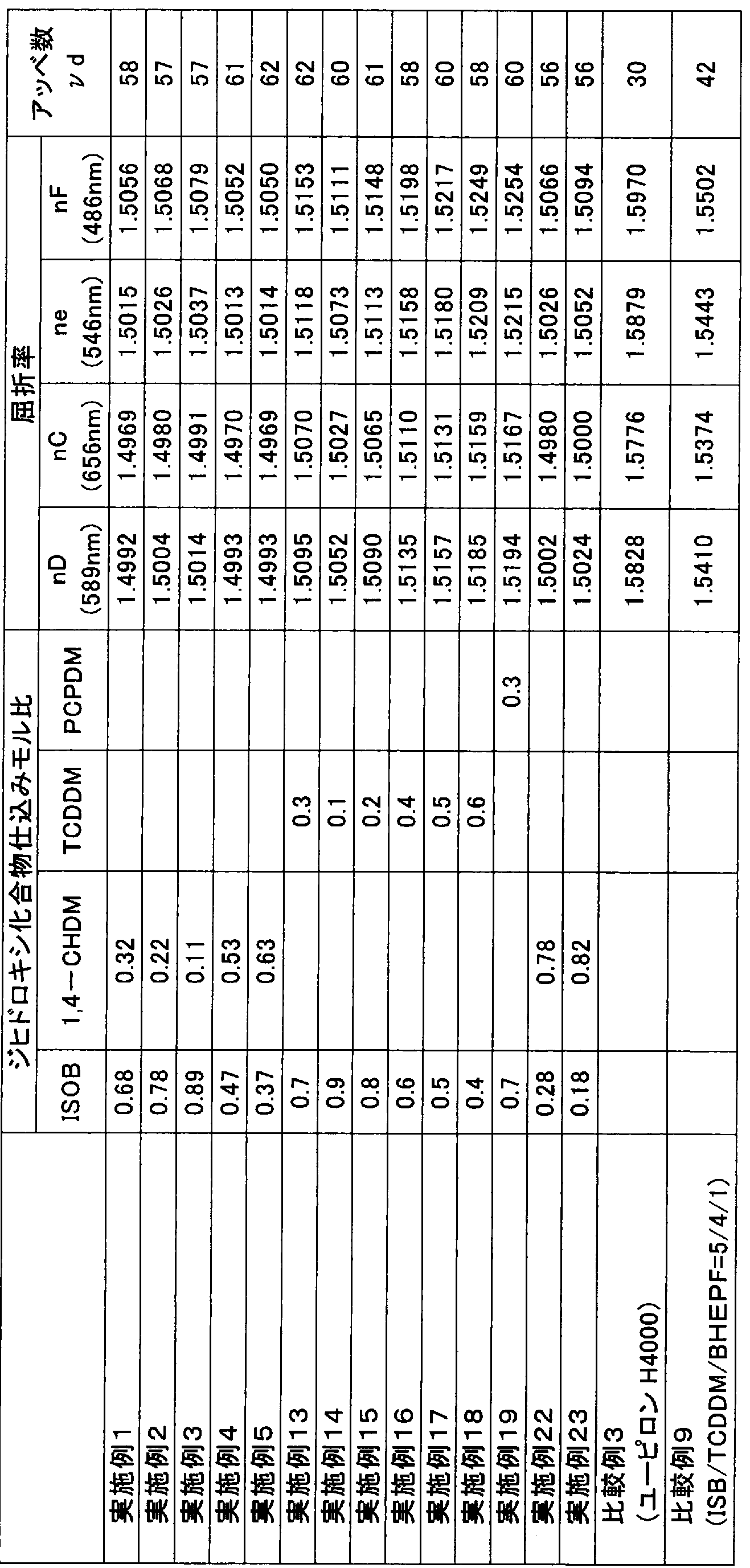
SJ)嗨¾娜 Ι ¾ί6 I2
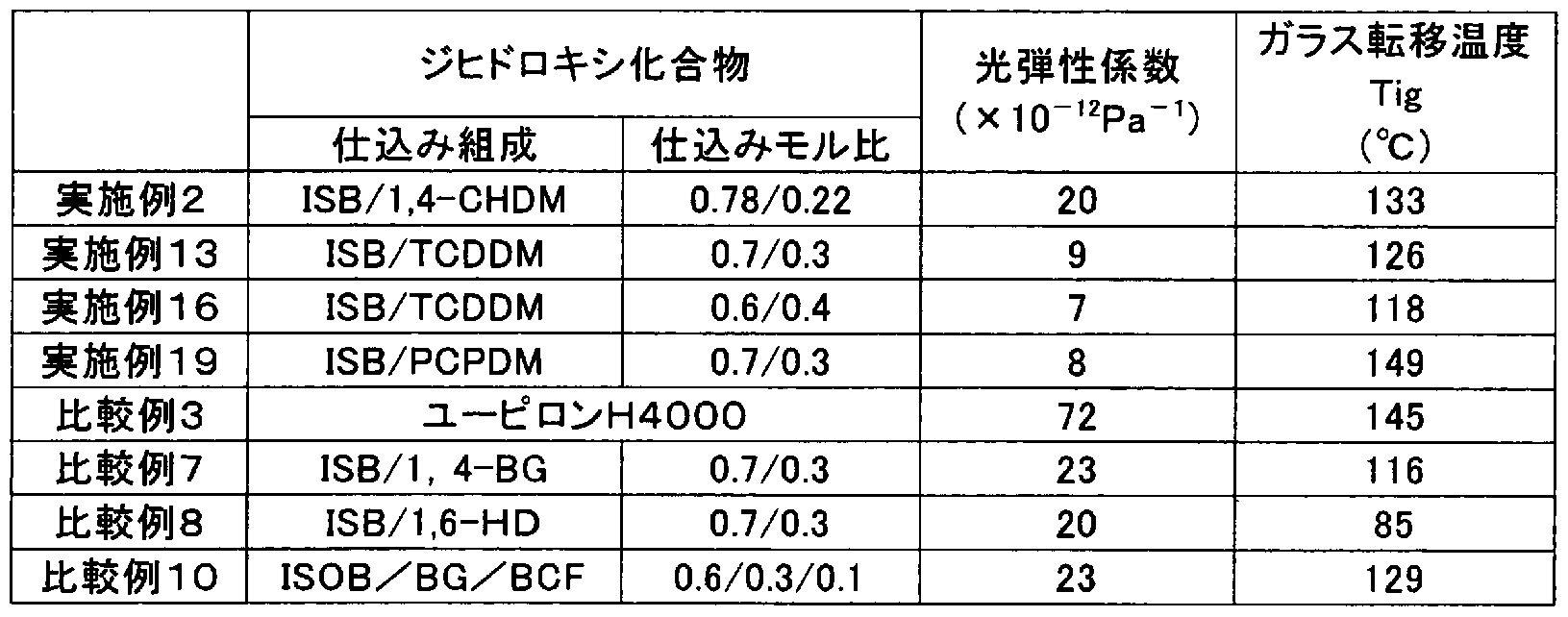
注 1 ) 全ジヒドロキシ化^ Πモルに射る βとして C ¾H¾単位 jUモル) ¾2〉 PEP— 36添力 :難例 15 16 0.096¾M|5
L£0Z90/L00Zd /lDd 39 ^098Μ/.00Ζ OAV
[ε挲] [36 TO]
L£0Z90/L00Zd /lDd Ρ9 ^098Μ/.00Ζ OAV
1 ) THF
2)シクロへキサジェン、シクロへキシルフヱニルエーテル
[0199] [表 7]
[0200] [表 8]
ガラス
ISOB中の
ISOBの蒸留の 兀粘度 転移温度
蟻酸量 カラ一 b 有無 (dl/g) Tig
(PPM)
(°C)
実施例 26 無し 5 0.506 126 10.0 実施例 27 無し 5 0.621 123 1 1.0 実施例 28 有り 3 0.510 126 4.5 実施例 29 有り 2 0.640 26 3.7 実施例 30 有り 3 0.658 123 7.0 実施例 31 有り 2 0.590 123 6.5 参考例 1 無し 400 重合せず ― ―
参考例 2 無し 50 重合せず ― ―
参考例 3 無し 20 重合せず ― ―
参考例 4 無し 50 重合せず ― ― 表 2から、本発明のポリカーボネート共重合体は、市販のポリカーボネートと同等以 上の引張降伏強さ、引張降伏弾性率、降伏時伸びを示し、高いアイゾット衝撃強度 を持つことが分かる。
表 3から、本発明のポリカーボネート共重合体は、市販のポリカーボネートや従来の ポリカーボネートに比べ、屈折率が小さぐアッベ数が大きいことが分かる。
表 4から、本発明のポリカーボネート共重合体は、市販のポリ乳酸や従来のポリカー ボネートに比べ、熱安定性が高いことが分かる。
以上の結果から、本発明のポリカーボネート共重合体は、機械的強度に優れ、耐 熱性が良好で、屈折率が小さぐアッベ数が大きぐ透明性に優れ、光学材料や、各 種の成形材料に好適に用いることができることが分かる。
表 5から、本発明のポリカーボネート共重合体は光弾性係数が小さぐフィルムや、 レンズなどの光学材料に好適に用いることが、出来ることが分かる。
表 6から、脂環式ジヒドロキシィ匕合物を共重合した本発明のポリカーボネートは、発 生するガスが脂肪族ジオールを共重合したポリカーボネートより少ないことが分かる。 即ち、ジヒドロキシ化合物として、 1, 4 ブタンジオール、 1, 6 へキサンジオールの ような脂肪族ジオールを用いた際には、これらのジオール由来の環状エーテル等の ガスの発生が見られる力 シクロへキサンジメタノール、トリシクロデカンジメタノール のような脂環式ジオールを用いた際にはこのような発生ガスがほとんどない。そのた
め、脂環式ジオール含有ポリカーボネートは、光学フィルム等の家電製品等に用い た際に環境への影響が少ないことが分力る。
表 7から、本発明のポリカーボネート共重合体は、鉛筆硬度が高ぐ表面硬度が高く 、表面の傷つきを嫌う、フィルム用途や、筐体などの構造材料用途に好適に用いるこ とができることが分力る。
表 8から、蒸留などにより蟻酸を除去したイソソルビドを使用することで、より着色の 少な 、、ポリカーボネート共重合体が得られることがわかる。
本発明を詳細にまた特定の実施態様を参照して説明したが、本発明の精神と範囲 を逸脱することなく様々な変更や修正を加えることができることは当業者にとって明ら かである。
本出願は、 2006年 6月 19日出願の日本特許出願 (特願 2006— 168929)に基づ くものであり、その内容はここに参照として取り込まれる。
産業上の利用可能性
本発明のポリカーボネート共重合体は、柔軟性が必要なフィルム、シート分野、耐 熱性が必要なボトル、容器分野、衝撃強度が要求される種々の構造材料、さらには、 カメラレンズ、ファインダーレンズ、 CCDや CMOS用レンズなどのレンズ用途、液晶 やプラズマディスプレイなどに利用される位相差フィルム、拡散シート、偏光フィルム などのフィルム、シート、光ディスク、フィルム、シート、光学材料、光学部品、色素や 電荷移動剤等を固定ィ匕するバインダーなどの用途への使用に適して 、る。