WO2008010585A1 - Process for producing composite metal oxide - Google Patents
Process for producing composite metal oxide Download PDFInfo
- Publication number
- WO2008010585A1 WO2008010585A1 PCT/JP2007/064388 JP2007064388W WO2008010585A1 WO 2008010585 A1 WO2008010585 A1 WO 2008010585A1 JP 2007064388 W JP2007064388 W JP 2007064388W WO 2008010585 A1 WO2008010585 A1 WO 2008010585A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- metal oxide
- oxide
- metal
- basic
- aqueous solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/63—Platinum group metals with rare earths or actinides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B13/00—Oxygen; Ozone; Oxides or hydroxides in general
- C01B13/14—Methods for preparing oxides or hydroxides in general
- C01B13/36—Methods for preparing oxides or hydroxides in general by precipitation reactions in aqueous solutions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/74—General processes for purification of waste gases; Apparatus or devices specially adapted therefor
- B01D53/86—Catalytic processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B13/00—Oxygen; Ozone; Oxides or hydroxides in general
- C01B13/14—Methods for preparing oxides or hydroxides in general
- C01B13/36—Methods for preparing oxides or hydroxides in general by precipitation reactions in aqueous solutions
- C01B13/363—Mixtures of oxides or hydroxides by precipitation
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B33/00—Silicon; Compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/206—Rare earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/30—Silica
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/70—Catalysts, in general, characterised by their form or physical properties characterised by their crystalline properties, e.g. semi-crystalline
- B01J35/735—Pyrochlore-type A2B2O7
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/024—Multiple impregnation or coating
- B01J37/0242—Coating followed by impregnation
Definitions
- the present invention relates to a method for producing a composite metal oxide, particularly a composite metal oxide catalyst support.
- composite metal oxides have been developed and used in many applications such as catalysts, catalyst carriers, adsorbents, electrodes, magnetic materials, and electronic parts because of their unique properties.
- composite metal oxides of ceria and zirconia are generally known.
- This ceria-zirconia composite oxide has the SC ability (oxygen storage capacity) of ceria, that is, oxygen is stored when the oxygen concentration in the exhaust gas is high, and oxygen is stored when the oxygen concentration in the exhaust gas is low.
- SC ability oxygen storage capacity
- a single ceria while providing a releasing property Can have significantly improved heat resistance.
- an aqueous salt solution containing a cerium salt and a zirconium salt is provided, and by making the aqueous salt solution basic, the ceria-zirconia composite oxide is provided.
- a coprecipitation method in which a precursor of a composite oxide, particularly a ceria-zirconia composite hydroxide, is precipitated, and this precursor is dried and calcined is performed.
- Japanese Patent Application Laid-Open No. 5-49 8 6 4 proposes to use a catalyst composed of a single-cell silicate as an exhaust gas purification catalyst other than noble metal-supported alumina.
- the metallosilicate is a compound having a structure in which at least a part of aluminum in the zeolite is substituted with another element.
- an element that substitutes aluminum in zeolite it is considered that an element having an ion radius similar to that of aluminum is preferable, and specifically, iron, gallium, zinc, lanthanum, and the like can be given. Yes.
- an alkali metal type metal mouth silicate is produced by hydrothermal treatment of a silica source and an alkali metal source, and the alkali metal of the alkali metal type metal mouth silicate is used.
- This metallosilicate gate is manufactured by ion exchange with a desired metal.
- titania - Jirukonia composite oxide, lanthanum, for N_ ⁇ x storage reduction catalyst including an element selected Ri by the group consisting of neodymium and praseodymium A carrier is disclosed.
- titania is prepared by a general coprecipitation method in which zirconium tetrachloride is mixed with titanium tetrachloride and zirconium is added dropwise to perform coprecipitation.
- zirconium tetrachloride is mixed with titanium tetrachloride and zirconium is added dropwise to perform coprecipitation.
- zirconium tetrachloride is mixed with titanium tetrachloride and zirconium is added dropwise to perform coprecipitation.
- a salt solution such as lanthanum nitrate solution
- the lanthanide element present in zirconia has an anchor effect, that is, the effect of suppressing the movement of rhodium on the zirconia surface. Suppressing thin ring is disclosed.
- a method for producing a composite metal oxide a plurality of kinds of powdered metal oxides are mixed and fired at a very high temperature.
- the metal oxide particles are immersed in a metal salt solution and dried.
- a method of baking at a high temperature and a so-called coprecipitation method are generally known.
- the coprecipitation method is preferable in that a composite metal oxide having a uniform composition can be obtained and a composite metal oxide can be obtained by firing at a relatively low temperature. .
- microemulsion method water is dispersed in a hydrophobic solvent together with an appropriate surfactant to obtain a dispersion in which minute water droplets are dispersed in the hydrophobic solvent. Thereafter, a water-soluble metal salt is added to the dispersion, and the metal hydroxide is precipitated in the fine water droplets dispersed in the hydrophobic solvent.
- a composite metal oxide having a minute secondary particle diameter corresponding to the amount of metal element contained in minute water droplets can be obtained.
- the present invention provides a method for producing a composite metal oxide, which can easily obtain a composite metal oxide containing a plurality of types of metals having different properties at a low cost. Disclosure of the invention
- the method of the present invention for producing a composite metal oxide comprises a base selected from the group consisting of an acid metal oxide having an electronegativity of more than 2.80 as a metal oxide, and a rare earth and an alkaline earth metal.
- This is a method for producing a composite metal oxide with a basic metal oxide which is an oxide of a conductive metal.
- This method of the present invention comprises the following steps (a) to (c):
- the pH of the aqueous solution is adjusted to 0.1 to 5 mol%, particularly 0.1 to 1.5 mol%, more particularly 0 to 0.1% of a portion of the basic metal, e.g.
- the hydroxide of the basic metal is dissolved and re-precipitated by the equilibrium between the hydroxide of the basic metal and the ion of the metal. Can be preferentially re-deposited or rearranged on the surface of the acidic metal oxide colloidal particles. Therefore, according to the method of the present invention, a composite metal oxide precursor having primary particles based on colloidal particles of acidic metal oxide is obtained, and such composite metal oxide precursor is dried and calcined. Thus, a composite metal oxide having primary particles based on colloidal particles of acidic metal oxide can be obtained.
- One embodiment of the method of the present invention is to further increase the pH of the aqueous solution after step (a) and before step (b) to be greater than the pH maintained in step (b). Including.
- a majority of the basic metal is precipitated as a hydroxide before step (b), so that in step (b) the colloid of acidic metal oxide.
- the distance between these components can be reduced so that the particles and the hydroxide of the basic metal together form a precipitate. According to this, it is possible to promote the rearrangement of the basic metal hydroxide on the surface of the colloidal particle of the acidic metal oxide.
- the predetermined time in step (b) is 1 hour to 5 days, for example, 10 hours to 3 days.
- the pH of the aqueous solution is further set larger than the pH maintained in the step (b). Including.
- the basic metal hydroxide is surely precipitated, and the structure of the resulting precursor is strengthened, and this structure is retained during drying and firing. Can be.
- the acidic metal is selected from the group consisting of silica, titania, tungsten oxide, molybdenum oxide and niobium oxide, particularly silica and titania.
- the basic metal is selected from the group consisting of rare earths, particularly yttrium, lanthanum, praseodymium and neodymium.
- the composite metal oxide to be produced is a composite metal oxide catalyst support, in particular, a composite metal oxide catalyst support particle.
- the composite metal oxide catalyst support is produced by the method of the present invention.
- the resulting composite metal oxide catalyst support can have relatively neutral properties as a whole catalyst support.
- the composite metal oxide catalyst support obtained by the method of the present invention provides high heat resistance by forming a composite oxide, and locally strong in the basic metal oxide portion. Providing basicity, and no or acidic metal oxidation It can provide locally strong acidity in the object part.
- the composite metal oxide of the present invention is produced by the method of the present invention for producing a composite metal oxide.
- the exhaust gas purifying catalyst of the present invention comprises a noble metal supported on a composite metal oxide catalyst carrier produced by the method of the present invention for producing a composite metal oxide catalyst carrier.
- FIG. 1 is a conceptual diagram showing the mechanism of the method of the present invention.
- Fig. 2 is a diagram showing the See evening potential curve of lanthanum hydroxide.
- Fig. 3 is a graph showing the z-potential curve of titania particles.
- FIG. 4 is a diagram for explaining the pH range maintained in step (b) of the method of the present invention.
- the method of the present invention for producing a composite metal oxide of an acidic metal oxide and a basic metal oxide includes the following steps (a) to (c):
- the basic metal (A) has an equilibrium as shown by the following formula (I), that is, an ion of the basic metal ( There is an equilibrium between A a + ) and a precipitate of this metal hydroxide (A (OH) a ):
- A is a basic metal, and a is the valence of the basic metal A).
- the basic metal hydroxides obtained here can generally have a positive potential.
- basic metal hydroxides generally have a zeta-potential curve as shown in FIG. 2 for yttrium hydroxide.
- colloidal particles of metal oxide have a positive zeta potential in a relatively acidic solution, and this zeta potential decreases as PH increases, so that a relatively basic solution. There is a negative potential in the inside. That is, the metal oxide colloidal particles generally have a zeta potential curve as shown in FIG. 3 for the titania colloidal particles.
- the basic metal hydroxide having a positive zeta potential is preferentially re-applied to the surface of the colloidal particle of the acidic metal oxide having a negative zeta potential.
- Precipitate That is, for example, as shown in FIG.
- a composite metal oxide precursor having primary particles based on colloidal particles of acidic metal oxide can be obtained by such a mechanism.
- acidic metal oxide means a metal oxide having an electronegativity of more than 2.80 as a metal oxide.
- this “electronegativity as a metal oxide” is a weighting of the electronegativity due to poling of the metal elements and oxygen constituting the metal oxide according to the ratio of these elements contained in the metal oxide. The average value.
- acidic metal means a metal constituting an acidic metal oxide.
- the electronegativity as a metal oxide of silica (S i ⁇ 2) is calculated as follows:
- Table 1 below shows examples of electronegativity as metal oxides calculated as above.
- the large electronegativity as the metal oxide means that the metal oxide has a relatively acidic property
- the low electronegativity as the metal oxide means that the metal oxide is It means having a relatively basic property
- the oxygen used for the calculation of the electronegativity of the metal oxide is 3.44.
- the metal electronegativity is as follows. .
- Colloidal particles of acidic metal oxide used in the method of the present invention are generally used.
- the isoelectric point of the colloidal particles can be adjusted by the surface modification of the colloidal particles, particularly the surface modification of the colloidal particles by an organic compound.
- the colloidal particles include substances obtained by hydrolysis and condensation of metal alkoxides, acetyl cettonates, acetates, nitrates, and the like.
- a colloid solution (sol) such as a silica colloid solution or a titania colloid solution is a known material, and a commercially available one can also be obtained.
- the metal oxide selected from the group consisting of acidic metal oxides used in the catalyst support for example, silica, titania, tungsten oxide, molybdenum oxide and niobium oxide. Products, especially siri force and / or titania.
- basic metal means a metal selected from the group consisting of rare earths and alkaline earth metals.
- basic metal oxide means an oxide of a basic metal.
- Examples of the basic metal salt include any salt that can be dissolved in the aqueous solution used in the present invention. Accordingly, examples of basic metal salts used in the present invention include inorganic acid salts such as nitrates and organic acid salts such as acetates.
- the basic metal is preferably a metal element that accepts electrons from a noble metal element, such as a rare earth element.
- a noble metal element such as a rare earth element.
- the rare earth element has a young atomic number, and
- Elements that form ions in the 4f, 4d or 5d orbitals that have vacancies or vacancies are particularly preferred.
- a noble metal as a catalyst metal is supported on a catalyst support of a composite metal oxide having such an oxide of a metal element
- the noble metal element particularly the electrons of platinum, can be coordinated to the composite metal oxide support, thereby suppressing the noble metal synthesizing during the use of the catalyst.
- the basic metal is electron-accepting to accept electrons from a noble metal element and has a valence by an oxidation-reduction reaction during use of the catalyst.
- a noble metal as a catalyst metal is supported on a composite metal oxide catalyst carrier having an oxide of such a metal element, it is possible to better suppress the noble metal synthesizing during use of the catalyst. . This is the case with the oxides of elements whose valence changes due to the redox reaction during the use of the catalyst, such as ceria, which is preferably used for OSC ability.
- a complex oxide of an acidic metal oxide and a basic metal oxide is formed in an arbitrary combination and at an arbitrary ratio regardless of the difference in properties between the acidic metal and the basic metal.
- the acidic metal is selected from the group consisting of silica, titania, tungsten oxide, molybdenum oxide and niobium oxide, particularly silica and titania, and is basic.
- the metal is selected from the group consisting of rare earths, especially yttrium, lanthanum, praseodymium and neodymium, more particularly lanthanum and neodymium.
- the catalyst support of the present invention a basic metal oxide and an acidic
- the catalyst support as a whole can have relatively neutral properties.
- the catalyst carrier for the exhaust gas purification catalyst is alumina
- metal oxides with a higher electronegativity than alumina can be considered acidic, and metal oxides with a lower electronegativity than alumina can be considered basic. .
- the catalyst carrier for the exhaust gas purification catalyst has a relatively neutral electronegativity similar to alumina.
- the electronegativity of the composite metal oxide with respect to the ratio of the silica in the acidic metal oxide (S i ⁇ 2) and lanthanum oxide as a basic metal oxide (L a 2 O 3) It is shown in Table 2 below.
- the electronegativity of alumina (A l 2 0 3 ) is 2.71.
- Table 2 Electronegativity of lanthanum oxide silica composite oxide
- step (b) that is, a part of the basic metal is dissolved in the aqueous solution, the remainder of the basic metal is precipitated as a hydroxide and has a positive zeta potential, and the colloid of the acidic metal oxide
- the pH which has a negative surface potential without dissolving the particles, depends on the composition, temperature, concentration, etc. of the aqueous solution used and can be determined based on experiments.
- the basic metal is a rare earth
- a part of the basic metal is dissolved in the aqueous solution, and the remainder of the basic metal is precipitated as a hydroxide, and has a positive voltage.
- a pH of 7-9 can be mentioned.
- FIG. 4 shows a pH range (L a) in which a portion of lanthanum, which is a basic metal, precipitates as a hydroxide and has a positive zeta potential.
- pH in the step (b) can be expressed as a pH range X in FIG.
- pH range X 0.1 to 5 mol% of the basic metal is dissolved in the aqueous solution as ions. This percentage is determined by filtering the precipitate in equilibrium It can be known by measuring the amount of basic metal contained in or the amount of basic metal contained in the filtrate.
- step (b) If the pH in this step (b) is excessively large, so that substantially all of the basic metal is precipitated as hydroxide, the basic metal hydroxide is converted into an acidic metal. It takes a very long time to rearrange the particles on the surface of the particles, and it is difficult to achieve the rearrangement. In addition, when pH in the step (b) is excessively large, the colloidal particles of the acidic metal oxide may dissolve and have no negative potential. If the pH in step (b) is too small so that the basic metal does not precipitate as a hydroxide, the basic metal hydroxide will re-apply to the surface of the acidic metal oxide colloidal particles. Not placed.
- the time for which the solution is maintained at the predetermined pH in step (b) depends on the temperature, concentration and properties of the aqueous solution, in particular the pH of the aqueous solution, and the concentration and solubility of the basic metal intended to be rearranged. To do.
- the time for maintaining the solution at a predetermined pH in the step (b) can be, for example, 1 hour to 5 days.
- the basic metal is added before step (b) by increasing the pH of the aqueous solution to be greater than the pH maintained in step (b).
- the pH can be arbitrarily determined according to the nature of the basic metal, the temperature of the aqueous solution, and the like. For example, as this pH, a pH that is 1 or more or 2 or more larger than the pH maintained in step (b) can be selected.
- the aqueous solution of step (b) can be changed to a base by increasing the pH of the aqueous solution to be higher than the pH maintained in step (b).
- This pH can be arbitrarily determined according to the nature of the basic metal, the temperature of the aqueous solution, etc., in order to ensure the precipitation of the basic metal hydroxide. For example, this pH is maintained in step (b).
- the pH can be selected to be 1 or more or 2 or more larger than the pH.
- a composite metal oxide having primary particles based on acidic metal oxide colloidal particles can be obtained by drying and calcining the obtained composite metal oxide precursor.
- Removal of the dispersion medium from the composite metal oxide precursor and drying can be performed by any method and at any temperature. This can be accomplished, for example, by placing the composite metal oxide precursor in an oven at 120. Thus, the raw material obtained by removing and drying the dispersion medium from the composite metal oxide precursor can be fired to obtain a composite metal oxide. This calcination can be performed at a temperature generally used in the synthesis of metal oxides, for example, a temperature of 500 to 800.
- the exhaust gas purifying catalyst of the present invention can be obtained by supporting a noble metal such as platinum on a composite metal oxide catalyst support manufactured by the method of the present invention for manufacturing a composite metal oxide catalyst support.
- the loading of the noble metal can be achieved by any method. For example, in the case of loading platinum, a mixed metal oxide is impregnated with a dinitrodiammine platinum nitrate aqueous solution, dried and calcined, and 0.5-2% by weight of platinum is loaded.
- the supported exhaust gas purifying catalyst of the present invention can be manufactured.
- loading of the NO x storage material such as barium is a solution containing a salt of these metals, such as acetic acid Bariumu solution This can be achieved by impregnating the catalyst support in a catalyst support, drying and calcining.
- a salt of these metals such as acetic acid Bariumu solution
- a lanthanum oxide-silica composite metal oxide catalyst support was produced as follows.
- Silica sol (manufactured by Nissan Chemical Co., Ltd., Snowtex NX S, average particle size of about 5 nm) was diluted with distilled water to obtain a diluted colloidal silica sol.
- a commercially available lanthanum nitrate aqueous solution was diluted with distilled water to obtain a diluted lanthanum nitrate aqueous solution.
- the diluted colloidal silica sol and the diluted lanthanum nitrate aqueous solution were mixed to obtain 10 L of an acidic (pH 2.5 to 3.0) mixed solution containing colloidal silica and lanthanum nitrate.
- lanthanum (L a): caine (S i) (molar ratio) was 62.5: 37.5.
- the target yield was 3500 g.
- aqueous ammonia was added to bring the pH to 8.5 and maintained for 2 days (at this pH, 1% is dissolved in the solution). Thereafter, ammonia water was further added to adjust the pH to 10 to obtain a precipitate. The precipitate thus obtained is filtered, washed with a small amount of dilute aqueous ammonia with a pH of 0, filtered again, dried overnight and calcined at 800 for 5 hours.
- a lanthanum oxide-silica composite metal oxide catalyst support was obtained.
- a lanthanum oxide-silica composite metal oxide catalyst support was produced as follows.
- a lanthanum oxide-titania composite metal oxide catalyst support was produced as follows.
- a titania sol (Ishihara Sangyo, STS-1100) was diluted with distilled water to obtain a diluted titania sol.
- a commercially available lanthanum nitrate aqueous solution was diluted with distilled water to obtain a diluted lanthanum nitrate aqueous solution.
- the diluted titania sol and the diluted lanthanum nitrate aqueous solution were mixed to obtain a mixed solution 10 L (pH is about 3) containing colloidal titania and lanthanum nitrate.
- lanthanum (L a): titanium (T i) (element molar ratio) 1: 1.
- the target yield was 40 g.
- Titania sol (STS-1100, manufactured by Ishihara Sangyo Co., Ltd.) was diluted with distilled water to obtain a diluted titania sol.
- a commercially available aqueous solution of yttrium nitrate was diluted with distilled water to obtain a diluted aqueous solution of yttrium nitrate.
- Diluted titania sol and diluted aqueous solution of yttrium nitrate were mixed to obtain 10 L (pH is about 3) of a mixed solution containing colloidal titania and yttrium nitrate.
- yttrium (Y): titanium (T i) (molar ratio) 1: 1.
- the target yield is 4. Met.
- Lanthanum oxide-silica composite metal oxidation was carried out in the same manner as in Example 1 except that the mixed solution containing colloidal silica and lanthanum nitrate was not maintained at basic pH. Material A catalyst carrier was produced.
- a mixed solution containing colloidal silica and lanthanum nitrate was maintained in the same manner as in Example 1 except that lanthanum was maintained at basic pH where substantially all of the lanthanum precipitated as hydroxide.
- 10 L of a mixed solution containing colloidal silica and lanthanum nitrate was obtained in the same manner as in Example 1 in which the lanthanum oxide-silica composite metal oxide catalyst support was produced.
- Aqueous ammonia was added to the mixed solution to adjust the pH to 10 to obtain a precipitate, and this solution was kept for 2 days (at this pH, substantially all of the lanthanum was in water at equilibrium. It is deposited as an oxide, and lanthanum is substantially not dissolved in the solution). Thereafter, the precipitate thus obtained was filtered in the same manner as in Example 1, washed with dilute aqueous ammonia having a pH of 0, filtered again, and dried for a whole day and night. And calcining for 5 hours to obtain a lanthanum oxide monosilica composite metal oxide catalyst support.
- lanthanum oxide-silica composite metal oxide catalyst support was produced by a coprecipitation method as follows. Dilute the aqueous sodium solution with distilled water and dilute A solution of aqueous solution was obtained. Further, a dilute lanthanum nitrate aqueous solution was obtained by diluting a commercially available lanthanum nitrate aqueous solution with distilled water. When dilute aqueous sodium silicate solution was mixed with dilute aqueous lanthanum nitrate solution, precipitation occurred immediately.
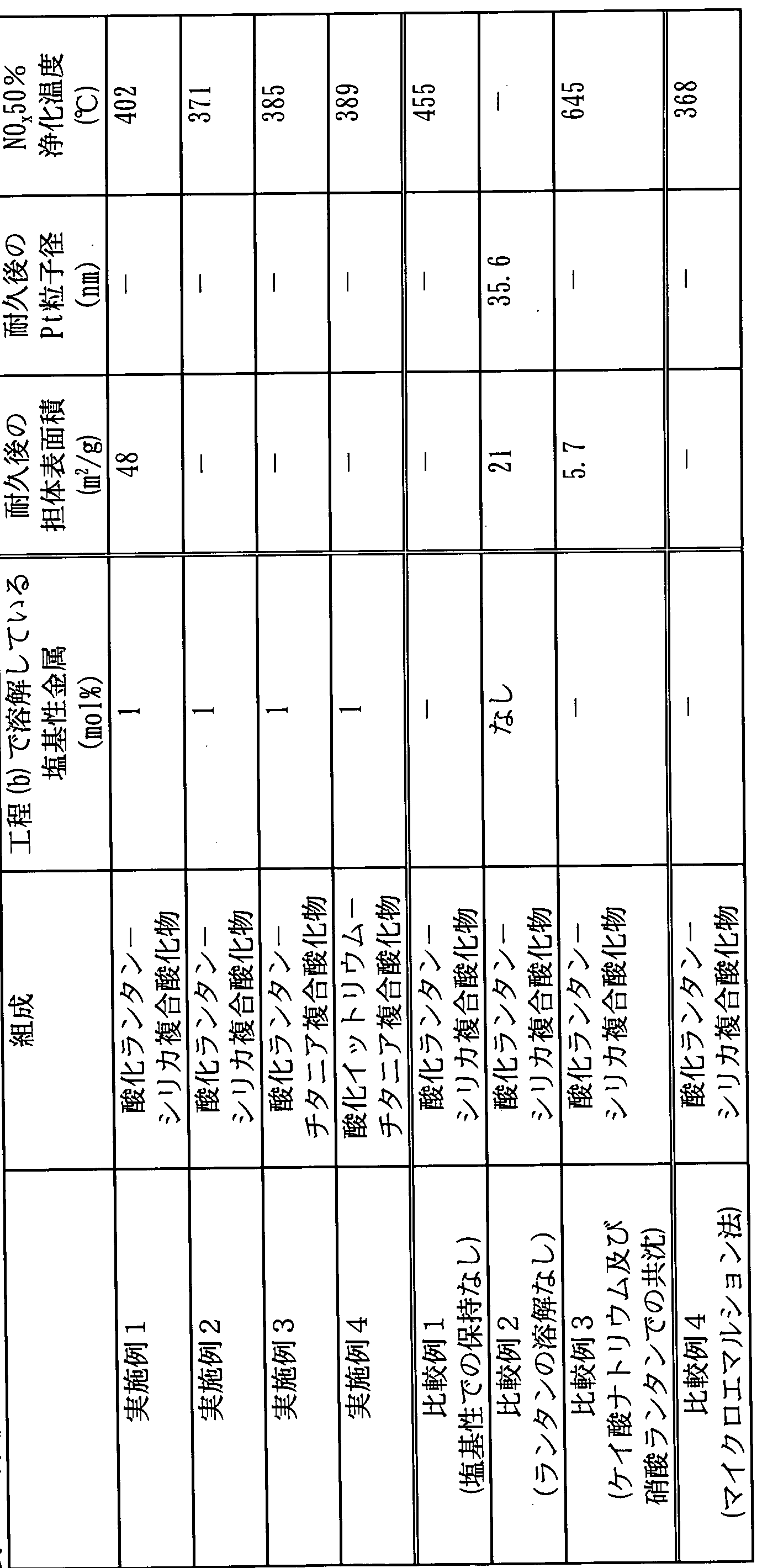
- the composite metal oxide of Comparative Example 3 obtained in this way was clearly different in aggregation state from the composite oxides obtained in Examples 1 and 2 and Comparative Example 1, and the apparent volume was 1 node. It was about 7 times smaller. Further, the specific surface area of the composite metal oxide of Comparative Example 3 is 5.7 m 2 Z g, and this surface area is considered to be insufficient as a composite metal oxide for a catalyst support.
- a lanthanum oxide monosilica composite metal oxide catalyst support was produced as follows.
- TEOS tetraethoxysilane
- the agglomerates thus obtained were taken out, the adhering surfactant was washed with alcohol, dried and calcined to obtain a lanthanum oxide-silica composite metal oxide catalyst carrier.
- Each of the composite metal oxide catalyst supports of Examples and Comparative Examples was coated on a monolith honeycomb substrate in an amount of 120 g substrate_L using a slurry, dried and fired. Thereafter, 1.2 g of platinum / base material-L was supported on the monolith honeycomb substrate having the composite metal oxide catalyst carrier of the example and the comparative example to obtain exhaust gas purification catalysts of the example and the comparative example. .
- the exhaust gas purifying catalysts of Examples and Comparative Examples obtained in this manner were heated at 80 ° C. for 2 hours in the air to be durable.
- Evaluation gas composition Table 4 shows the evaluation results for the specific surface area of the support, the particle size of the platinum supported, and the N 0 x 50% purification temperature.
- the catalyst carrier of the example of the present invention was obtained by the microemulsion method (Comparative Example 4), although it was obtained by a relatively easy production method. It is understood that it has a performance corresponding to the catalyst support.
- the mixed solution containing colloidal silica and lanthanum nitrate is not maintained with basic pH (Comparative Example 1), the lanthanum is substantially all hydroxides. In the case where the basic pH precipitated was maintained (Comparative Example 2), the effect as in the example of the present invention was not obtained.
- Example 2 when comparing the catalyst carriers of Examples 1 and 2 having the same composition, better results were obtained in Example 2 in which the solution was made relatively basic before maintaining the basic pH. It is understood that
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Materials Engineering (AREA)
- Inorganic Chemistry (AREA)
- Environmental & Geological Engineering (AREA)
- Biomedical Technology (AREA)
- Health & Medical Sciences (AREA)
- Analytical Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Catalysts (AREA)
- Exhaust Gas Treatment By Means Of Catalyst (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Silicon Compounds (AREA)
Description



Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/373,803 US8298983B2 (en) | 2006-07-18 | 2007-07-17 | Production process of composite metal oxide |
| EP07791125.3A EP2050716B1 (en) | 2006-07-18 | 2007-07-17 | Process for producing composite metal oxide |
| CN200780026894XA CN101489921B (zh) | 2006-07-18 | 2007-07-17 | 复合金属氧化物的制造方法 |
| KR1020087030674A KR101057293B1 (ko) | 2006-07-18 | 2007-07-17 | 복합 금속 산화물의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006-195668 | 2006-07-18 | ||
| JP2006195668A JP4710744B2 (ja) | 2006-07-18 | 2006-07-18 | 複合金属酸化物の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008010585A1 true WO2008010585A1 (en) | 2008-01-24 |
Family
ID=38956915
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/064388 Ceased WO2008010585A1 (en) | 2006-07-18 | 2007-07-17 | Process for producing composite metal oxide |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8298983B2 (ja) |
| EP (1) | EP2050716B1 (ja) |
| JP (1) | JP4710744B2 (ja) |
| KR (1) | KR101057293B1 (ja) |
| CN (1) | CN101489921B (ja) |
| WO (1) | WO2008010585A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5311097B2 (ja) * | 2008-03-25 | 2013-10-09 | 株式会社豊田中央研究所 | 複合酸化物、それを用いた排ガス浄化用触媒及び複合酸化物の製造方法 |
| GB0822633D0 (en) * | 2008-12-11 | 2009-01-21 | Novartis Ag | Formulation |
| EP2602022A4 (en) | 2010-08-05 | 2017-09-06 | DOWA Electronics Materials Co., Ltd. | Method for producing catalyst composition, catalyst composition, diesel particulate filter using same, and exhaust gas purification system |
| DE102010050312A1 (de) * | 2010-11-03 | 2012-05-03 | Süd-Chemie AG | Ammoniak-Oxidationskatalysator mit geringer N2O Nebenproduktbildung |
| EP2989072A2 (en) | 2013-04-24 | 2016-03-02 | Saudi Basic Industries Corporation | Production of products from natural resources |
| WO2018151289A1 (ja) * | 2017-02-20 | 2018-08-23 | 株式会社キャタラー | 排ガス浄化用触媒 |
| JP6812995B2 (ja) * | 2018-02-16 | 2021-01-13 | 京セラドキュメントソリューションズ株式会社 | 正帯電性トナー |
| CN110833831B (zh) * | 2019-11-07 | 2022-11-01 | 上海纳米技术及应用国家工程研究中心有限公司 | 铬钴基一氧化氮常温常压催化剂的制备方法及其产品和应用 |
| CN114551910B (zh) * | 2022-02-25 | 2023-09-22 | 内蒙古科技大学 | 一种复合稀土氧化物及其制备方法和应用 |
| CN116943619A (zh) * | 2023-07-30 | 2023-10-27 | 南京大学 | 一种树脂基双金属氧化物复合纳米材料、制备方法和应用 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01201023A (ja) * | 1987-10-09 | 1989-08-14 | Rhone Poulenc Chim | 安定化された特性を有する酸化チタン |
| JPH0549864A (ja) | 1991-01-08 | 1993-03-02 | Sekiyu Sangyo Kasseika Center | 排ガスの浄化方法 |
| JPH06198174A (ja) * | 1992-12-29 | 1994-07-19 | Tonen Corp | 固体酸物質及びそれを含む流動接触分解用触媒組成物 |
| JPH08266865A (ja) | 1995-03-30 | 1996-10-15 | Toyota Motor Corp | ディーゼルエンジン用排ガス浄化触媒 |
| JP2001314763A (ja) | 2000-05-10 | 2001-11-13 | Johnson Matthey Japan Inc | NOx吸蔵還元型触媒用支持材とそれを用いたNOx吸蔵還元型触媒 |
| JP2002282692A (ja) | 2001-03-26 | 2002-10-02 | Toyota Motor Corp | 排ガス浄化用触媒 |
| JP2005518929A (ja) * | 2002-02-28 | 2005-06-30 | エクソンモービル・ケミカル・パテンツ・インク | 分子篩組成物、それらの触媒、それらの製造、及び変換法における使用 |
| JP2005254047A (ja) * | 2004-03-09 | 2005-09-22 | Toyota Motor Corp | 排ガス浄化触媒並びに、金属酸化物粒子及びその製造方法 |
| JP2005313024A (ja) * | 2004-04-27 | 2005-11-10 | Toyota Motor Corp | 内燃機関の排ガス浄化用触媒 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE8400426L (sv) | 1984-01-30 | 1985-07-31 | Eka Ab | Sett att framstella fina partiklar med en ytbeleggning av metall eller metallforening, serskilt ett katalytiskt aktivt emne |
| US5021392A (en) * | 1987-09-18 | 1991-06-04 | American Cyanamid Company | High porosity titania-zirconia catalyst support prepared by a process |
| US5466646A (en) * | 1992-08-18 | 1995-11-14 | Worcester Polytechnic Institute | Process for the preparation of solid state materials and said materials |
| JP2958873B2 (ja) | 1998-03-23 | 1999-10-06 | 株式会社次世代排ガス触媒研究所 | 排ガス浄化触媒およびその製造方法 |
| JP4123644B2 (ja) | 1999-06-22 | 2008-07-23 | トヨタ自動車株式会社 | 排ガス浄化触媒 |
| CN104096594A (zh) | 2004-04-27 | 2014-10-15 | 丰田自动车株式会社 | 制造金属氧化物粒子的方法及废气净化用催化剂 |
-
2006
- 2006-07-18 JP JP2006195668A patent/JP4710744B2/ja not_active Expired - Fee Related
-
2007
- 2007-07-17 WO PCT/JP2007/064388 patent/WO2008010585A1/ja not_active Ceased
- 2007-07-17 CN CN200780026894XA patent/CN101489921B/zh not_active Expired - Fee Related
- 2007-07-17 US US12/373,803 patent/US8298983B2/en not_active Expired - Fee Related
- 2007-07-17 EP EP07791125.3A patent/EP2050716B1/en not_active Ceased
- 2007-07-17 KR KR1020087030674A patent/KR101057293B1/ko not_active Expired - Fee Related
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01201023A (ja) * | 1987-10-09 | 1989-08-14 | Rhone Poulenc Chim | 安定化された特性を有する酸化チタン |
| JPH0549864A (ja) | 1991-01-08 | 1993-03-02 | Sekiyu Sangyo Kasseika Center | 排ガスの浄化方法 |
| JPH06198174A (ja) * | 1992-12-29 | 1994-07-19 | Tonen Corp | 固体酸物質及びそれを含む流動接触分解用触媒組成物 |
| JPH08266865A (ja) | 1995-03-30 | 1996-10-15 | Toyota Motor Corp | ディーゼルエンジン用排ガス浄化触媒 |
| JP2001314763A (ja) | 2000-05-10 | 2001-11-13 | Johnson Matthey Japan Inc | NOx吸蔵還元型触媒用支持材とそれを用いたNOx吸蔵還元型触媒 |
| JP2002282692A (ja) | 2001-03-26 | 2002-10-02 | Toyota Motor Corp | 排ガス浄化用触媒 |
| JP2005518929A (ja) * | 2002-02-28 | 2005-06-30 | エクソンモービル・ケミカル・パテンツ・インク | 分子篩組成物、それらの触媒、それらの製造、及び変換法における使用 |
| JP2005254047A (ja) * | 2004-03-09 | 2005-09-22 | Toyota Motor Corp | 排ガス浄化触媒並びに、金属酸化物粒子及びその製造方法 |
| JP2005313024A (ja) * | 2004-04-27 | 2005-11-10 | Toyota Motor Corp | 内燃機関の排ガス浄化用触媒 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2050716A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2050716A4 (en) | 2010-11-17 |
| EP2050716A1 (en) | 2009-04-22 |
| KR101057293B1 (ko) | 2011-08-16 |
| US20090264286A1 (en) | 2009-10-22 |
| JP2008024529A (ja) | 2008-02-07 |
| CN101489921A (zh) | 2009-07-22 |
| CN101489921B (zh) | 2012-11-28 |
| KR20090016710A (ko) | 2009-02-17 |
| JP4710744B2 (ja) | 2011-06-29 |
| EP2050716B1 (en) | 2013-06-05 |
| US8298983B2 (en) | 2012-10-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008010585A1 (en) | Process for producing composite metal oxide | |
| TWI417137B (zh) | 在無釩可移動觸媒中之觸媒促進劑 | |
| JP4006976B2 (ja) | 複合酸化物粉末とその製造方法及び触媒 | |
| CN101484241B (zh) | 废气净化催化剂 | |
| EP2039425B1 (en) | Process for production of an exhaust gas clean-up catalyst | |
| CN108355649B (zh) | 通过捕集活动铂族金属(pgm)催化剂物质的抗烧结稳定催化剂体系 | |
| WO2002066155A1 (en) | Exhaust gas clarification catalyst | |
| JP4325648B2 (ja) | 触媒担体及び排ガス浄化用触媒 | |
| CN101351267B (zh) | 催化剂载体颗粒、废气净化催化剂及它们的制造方法 | |
| JP3997783B2 (ja) | 触媒担体の製造方法 | |
| JP2003073123A (ja) | 複合酸化物とその製造方法及び排ガス浄化用助触媒 | |
| JP4432588B2 (ja) | 触媒及び触媒の製造方法 | |
| JP4770959B2 (ja) | 触媒担体及び排ガス浄化用触媒 | |
| JP2004321847A (ja) | 触媒担体及びその製造方法と触媒及び排ガス浄化方法 | |
| JP7262975B2 (ja) | セリア・ジルコニア系複合酸化物酸素吸収放出材料および排ガス浄化触媒 | |
| JP2009078203A (ja) | 排ガス浄化用触媒材、同触媒材の製造方法、及び同触媒材を用いた触媒 | |
| JP6640755B2 (ja) | 貴金属フリー触媒組成物 | |
| JP5019019B2 (ja) | 排ガス浄化用触媒担体、それを用いた排ガス浄化用触媒及び排ガス浄化方法 | |
| JP5229096B2 (ja) | 排ガス浄化用触媒 | |
| WO2006033168A1 (ja) | 排ガス浄化用触媒、および排ガス浄化用装置 | |
| JP2007229715A (ja) | 触媒担体及び触媒 | |
| JP2006223949A (ja) | 排ガス浄化用触媒及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780026894.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07791125 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 12373803 Country of ref document: US Ref document number: 2007791125 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) |