WO2009043577A2 - 19-nor-progesterone zur kontrazeption - Google Patents
19-nor-progesterone zur kontrazeption Download PDFInfo
- Publication number
- WO2009043577A2 WO2009043577A2 PCT/EP2008/008342 EP2008008342W WO2009043577A2 WO 2009043577 A2 WO2009043577 A2 WO 2009043577A2 EP 2008008342 W EP2008008342 W EP 2008008342W WO 2009043577 A2 WO2009043577 A2 WO 2009043577A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- compound
- pharmaceutical composition
- acid
- compound according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/18—Feminine contraceptives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
Definitions
- the invention relates to compounds of the general formula (I)
- An object of the invention is to provide compounds which are useful as pharmaceutical active ingredients and have advantages over conventional pharmaceutical agents.
- the pharmaceutical agents should be particularly suitable for contraception.
- the compounds of the general formula (I) have affinity for the human progesterone receptor and are therefore particularly suitable as pharmaceutical active substances, for example for hormone replacement therapy or for contraception.
- An object of the invention relates to a compound of general formula (I) in which
- R 1 and R 2 are each independently -H 1 -OH or -OCO-R 3 ;
- R 3 , R 4 , R 5 , R 6 and R 7 are each independently of one another -H or a linear or branched hydrocarbon radical having 1 to 12 carbon atoms, where the hydrocarbon radical is unsubstituted or optionally having 1, 2, 3, 4 or 5 Substituents substituted is independently selected from the group consisting of -F, -Cl, -Br, -I and -OH; or their pharmaceutically acceptable salts and / or solvates.
- A is a phosphorus atom
- q O
- the phosphorus atom has the oxidation number III and the compound of the general formula (I) is a phosphonate.
- the tautomeric form is not taken into account in the general formula (I), but a person skilled in the art recognizes that, depending on the meaning of R 6 and R 7 , the tautomeric form may also be present.
- A is a sulfur atom
- q 1, then the sulfur atom has the oxidation number IV and the compound of the general formula (I) is a sulfite.
- q 2 the sulfur atom has the oxidation number VI and the compound of the general formula (I) is a sulfate.
- hydrocarbon radical may have at least one double bond and / or one triple bond, preferably 1, 2 or 3 double bonds and / or triple bonds.
- R 1 and R 2 are each independently -H, -OH or -OCO-R 3 ;
- R 4 and R 5 are each independently -C 1-6 alkyl, preferably methyl, and
- R 3 , R 6 and R 7 are each as defined above;
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 and R 7 are each as defined above; in each case optionally in the form of appropriate pharmaceutically acceptable salts and / or corresponding solvates.
- R 4 and R 5 are each independently of one another C 1-4 alkyl, preferably methyl; and the remaining radicals are as defined above; in each case optionally in the form of appropriate pharmaceutically acceptable salts and / or corresponding solvates.
- R 1 is H and R 2 is -OH or -OCO-C 1-4 alkyl; or R 1 is -OH or -OCO-C 1-6 alkyl and R 2 is -H; and the remaining radicals are as defined above; in each case optionally in the form of appropriate pharmaceutically acceptable salts and / or corresponding solvates.
- B, B 1 , B ", C, C and C" are a radical selected from the group consisting of
- a pharmaceutically acceptable cation is preferably a cation which is monovalent (1 positive charge), divalent (2 positive charges) or trivalent (3 positive charges) and which is physiologically generally harmless.
- the cation is derived from an organic or inorganic base.
- the cation is a metal cation, preferably selected from the group consisting of cations of main group metals, in particular alkali metals and alkaline earth metals, and transition metals.
- the cation is an organic cation, preferably a quaternary ammonium compound N + RR 1 R 11 R 1 ", where R, R 1 , R" and R 1 "are preferably independently of one another -H or - are C 8 alkyl, in each case optionally substituted by a radical -OH, or at least two of the radicals R, R 1 , R “and R” 1 form a saturated, unsaturated or aromatic four-, five-, six- or seven-membered ring.
- organic cations derived from organic bases are the protonated forms of ammonia, ethylenediamine, ethanolamine, 1H-imidazole, diethylamine, piperazine, deanol, diethanolamine, pyrrolidine, betaine, 2- (diethylamino) ethanol, tropethamine , Choline, morpholine, lysine, triethanolamine, L-arginine, N-methylglucamine, betethamine, benzathine, hydrabamine, etc.
- H + Li + , Na + , K + , Mg 2+ , Ca 2+ , Al 3+ , Mn 2+ , Fe 2+ , Fe 3+ , Co 2+ , Co 3+ , Ni 2+, Cu +, Cu 2+, Ag +, Zn 2+, NH 4 +, N (C 1 -C 8 -alkyl) 4 +, triethanolammonium, fr / s (hydroxymethyl) aminomethane + and pyridinium called + .
- PH Steel et al. Handbook of Pharmaceutical Salts, Properties, Selection and Use, Wiley-VCH.
- M n + is a pharmaceutically acceptable cation selected from the group consisting of Li + , Na + , K + , NH 4 + , Ag + , Mg 2+ , Ca 2+ , Fe 2+ , Fe 3+ , Al 3+ and pyridinium + ; in each case in the form of appropriate solvates.
- M n + is a pharmaceutically acceptable cation selected from the group consisting of Li + , Na + , K + , NH 4 + , Ag + , Mg 2+ , Ca 2+ , Fe 2+ , Fe 3+ , Al 3+ and pyridinium + ; in each case in the form of appropriate solvates.
- Particularly preferred is a compound selected from the group consisting of
- Another object of the invention is a process for the preparation of compounds of general formulas (I), (IA), (IB), (IB 1 ), (1-B "), (1-C) 1 (IC) and ( 1-C ") comprising (a) the reaction of a compound of general formula (II)
- R 1 , R 2 , R 4 and R 5 are each as defined above, with a halide or anhydride of sulfurous acid, sulfuric acid, phosphorous acid or phosphoric acid, preferably in a suitable solvent, preferably at a temperature of - 20 0 C to 100 0 C, more preferably at a temperature of 0 0 C to 100 0 C, more preferably at a temperature of 5 ° C to 70 0 C, most preferably at a temperature of 10 0 C to 50 0 C and in particular at a temperature of 15 ° C to 25 ° C.
- the sulfuric acid anhydride is complexed with pyridine ⁇ pyridine • SO 3 ), with dimethylformamide ⁇ (HCON (CH 3 ) 2 ⁇ SO 3 ), with N-ethyldiisopropylamine ⁇ [(CH 3 ) 2 CH] 2 NCH 2 CH 3 ⁇ SO 3 J, with triethylamine ⁇ (CH 3 CH 2 ) 3 N • SO 3 ) or with trimethylamine ⁇ (CH 3 ) 3 N • SO 3 ).
- the anhydride of the sulfuric acid is complexed with pyridine.
- the process according to the invention preferably comprises the conversion of the product obtained in step (a) into a (different) pharmaceutically acceptable salt.
- a salt into another salt metalthesis
- the invention further relates to compounds of the general formulas (I), (I-A), (IB), (IB 1 ), (1-B “), (IC), (IC 1 ) and (IC 11 ) ( IB), (IB 1 ), (1-B "), (IC), (IC 1 ) or (IC 11 ) obtainable by the above-mentioned method.
- the intermediate and end products obtained according to the above-described reactions can each be purified and / or isolated, if desired and / or required, by customary methods known to those skilled in the art. Suitable purification methods are, for example, extraction methods and chromatographic methods.
- the compounds of the general formula (I) according to the invention and, if appropriate, corresponding stereoisomers and in each case the corresponding pharmaceutically / physiologically tolerable salts and solvates appear toxicologically harmless.
- these compounds may have a higher half-life than, for example, trimegestone, which is why these compounds are particularly useful as pharmaceutical agents in pharmaceutical compositions or for contraception.
- the abovementioned binding affinity for the human progesterone receptor is hereby preferably determined according to EP-A 808 845 or as described in I. Lacroix et al., Bioorganic & Medicinal Chemistry, 1999, 7, 2329-2341.
- Another object of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising at least one compound of general formula (I), in each case optionally in the form of appropriate pharmaceutically acceptable salts and / or corresponding solvates.
- the pharmaceutical composition according to the invention may comprise one or more salts of one or more of these compounds.
- the pharmaceutical composition contains one or more pharmaceutically acceptable excipients.
- the amount of the compound of the general formula (I) in the pharmaceutical composition of the present invention is preferably at least 100 ⁇ g, more preferably at least 200 ⁇ g, still more preferably at least 300 ⁇ g, most preferably at least 400 ⁇ g and especially at least 500 ⁇ g.
- the amount of the compound of general formula (I) in the pharmaceutical composition of the invention is in the range of 500 ⁇ g to 3,000 ⁇ g, more preferably 510 to 2,500 ⁇ g, even more preferably 525 to 2,000 ⁇ g, most preferably 550 to 1,500 ⁇ g and in particular from 600 to 900 ⁇ g.
- the amount of the compound of general formula (I) in the pharmaceutical composition of the invention corresponds to a trimegestone equivalent dose of at least 100 ⁇ g, more preferably at least 200 ⁇ g, even more preferably at least 300 ⁇ g, most preferably at least 400 ⁇ g, and most preferably at least 500 ⁇ g.
- the amount of the compound of general formula (I) in the pharmaceutical composition of the invention corresponds to an equivalent dose of trimegestone in the range of 500 ⁇ g to 3,000 ⁇ g, more preferably 510 to 2,500 ⁇ g, even more preferably 525 to 2,000 ⁇ g most preferably from 550 to 1500 micrograms and especially from 600 to 900 micrograms.
- the equivalent dose of the compound of the general formula (I) in comparison with trimegestone is chosen so that the gestagenic activity corresponds to that which would cause the administration of trimegestone in the stated amount. Suitable methods for determining the equivalent dose are known to those skilled in the art.
- the pharmaceutical composition preferably contains the at least one compound of the general formula (I) in an amount of 0.001 to 99.999% by weight, more preferably 0.1 to 99.9% by weight, still more preferably 0.5 to 75% by weight. -%, most preferred 1, 0 to 50 wt .-% and in particular 2.0 to 25 wt .-%, each based on the total weight of the pharmaceutical composition.
- the pharmaceutical composition additionally comprises at least one progestin, which is preferably selected from the group consisting of allylestrenol, chlormadinone, cyproterone, danazol, demegestone, desogestrel, dienogest, drospir renon, dydrogesterone, ethisterone, etynodiol, gestodene, gestonorone, hydroxyprogesterone, levonorgestrel, lynestrenol, medroxyprogesterone, medrogestone, megestrol, methylestronolone, methylnortestosterone, nomegestrol, norethisterone, norethynodrel, norgestrel, norgestimate, progesterone, promegestone, tibolone, trimegestone, 1 ⁇ -hydroxytrimegestone, desogestrel, dienogest, drospir renon,
- esters of the progestagens listed above are acetates (e.g., chlormadinone acetate, medroxyprogesterone acetate, megestrol acetate, norethisterone acetate), capronates (e.g., hydroxy progesterone capronate), and enantates (e.g., norethisterone antidote).
- acetates e.g., chlormadinone acetate, medroxyprogesterone acetate, megestrol acetate, norethisterone acetate
- capronates e.g., hydroxy progesterone capronate
- enantates e.g., norethisterone antidote
- the amount of additional gestagen corresponds to an equivalent dose of 100 to 5,000 ⁇ g, more preferably 250 to 4,000 ⁇ g, even more preferably 500 to 3,500 ⁇ g, most preferably 750 to 3,000 ⁇ g and especially 1,000 to 2,500 ⁇ g chlormadinone acetate.
- the equivalent dose to chlormadinone acetate can be realized by an equivalent amount of any suitable gestagen, the amount chosen to be such that the progestational activity is that which would cause the administration of chlormadinone acetate in the amount indicated. It is also possible that two or more different gestagens are used in an amount which corresponds to the total equivalent dose equivalent. Suitable methods for determining the equivalent dose are known to those skilled in the art.
- the pharmaceutical composition additionally contains at least one estrogen, preferably selected from the group consisting of chlorotrian iron, dienestrol, diethylstilbestrol, estradiol (17 ⁇ -estradiol), estriol, estrone, ethinylestradiol, estradiol benzoate, hexestrol, mestranol, methyl stearate, methylestre - nol, Promestrien and conjugated estrogens or their pharmaceutically acceptable esters, such as valerates, with particular preference as additional ⁇ stro- genkomponente ethinylestradiol or a combination of ethinylestradiol and estradiol (17ß-estradiol) are.
- at least one estrogen preferably selected from the group consisting of chlorotrian iron, dienestrol, diethylstilbestrol, estradiol (17 ⁇ -estradiol), estriol, estrone, ethinylestradiol, estradiol benzoate,
- the pharmaceutical composition contains a combination of at least one compound of the general formula (I) and at least one additional of the above listed gestagens and / or at least one of the estrogens listed above.
- the pharmaceutical composition contains a combination of at least one compound of general formula (I) and ethinyl estradiol or a combination of at least one compound of general formula (I) and a combination of ethinylestradiol and estradiol (17 ⁇ -estradiol).
- the pharmaceutical composition may be liquid (e.g., solution, dispersion, suspension, emulsion), pasty or solid (e.g., powder, granules). Preferably, it is solid.
- the pharmaceutical composition preferably additionally comprises at least one iron-containing preparation, folic acid and / or folinic acid.
- iron-containing preparations are iron (II) preparations, e.g. Iron (II) sulfate, ferrous carbonate, ferrous chloride, ferrous tartrate, ferrous gluconate, ferrous aspartate, ferrous glycine sulfate, ferrous fumarate, iron (II) II) ascorbate, iron (II) iodate, iron (II) succinate and ammonium iron (II) sulfate; and iron (III) preparations, e.g.
- iron (II) preparations e.g. Iron (II) sulfate, ferrous carbonate, ferrous chloride, ferrous tartrate, ferrous gluconate, ferrous aspartate, ferrous glycine sulfate, ferrous fumarate, iron (II) II) ascorbate, iron (II) iodate, iron (II) succinate and ammonium iron (II) sulfate; and iron (III) preparation
- the folic acid or its derivative is preferably present in free form or as a salt, for example as calcium folate.
- the pharmaceutical composition in each case optionally in the form of corresponding pharmaceutically acceptable salts and / or corresponding solvates and optionally a further progestogen and / or at least one estrogen, preferably additionally contains one or more excipients which preferably selected from the group consisting of salt formers, buffers, emulsifiers, embedding agents, thickeners, penetration promoters, film formers, binders, lubricants, surface-active agents, plasticizers, disintegrants, solvents, humectants, gelling agents, preservatives, stabilizers (reducing agents, antioxidants), Mold release agents, fillers, lubricants, chelating agents, flavor additives, fragrances and dyes.
- excipients which preferably selected from the group consisting of salt formers, buffers, emulsifiers, embedding agents, thickeners, penetration promoters, film formers, binders, lubricants, surface-active agents, plasticizers, disintegrants, solvents, hume
- Suitable buffers which may be used for the pharmaceutical composition are known to those skilled in the art.
- succinic acid, citric acid, lactic acid, phosphoric acid, trisodium phosphate, disodium hydrogen phosphate, sodium dihydrogen phosphate, sodium carbonate, sodium bicarbonate and combinations of lactic acid with sodium hydroxide can be used as the buffer.
- the buffer which is preferably a mixture of citric acid and disodium hydrogen phosphate
- the pH is preferably adjusted to 2.0-5.5.
- Emulsifiers are preferably added in amounts such that they allow a uniform mixing of the components of the pharmaceutical composition according to the invention.
- Conventional emulsifiers preferably comprise anionic, cationic and / or nonionic surfactants.
- emulsifiers include preferably potassium stearate, sodium stearate, ammonium stearate, triethanolamine stearate, glycerol monostearate, sodium lauryl-5-sulfate, sodium acetyl sulfate, N- (stearoyl-colamino-formylmethyl) -pyridium, N-soy-N-ethyl-morpholinium ethosulfate, alkyl-dimethyl- benzyl ammonium chloride, diisobutylphenoxyethoxyethyldimethylbenzylammonium chloride, acetylpyridium chloride, monostearate, polyoxyethylene stearate, polyoxyethylene sorbitan
- embedding agents are carnauba wax, montanglycol wax, stearic palmitic acid, glycerol trioleate and cetylstearyl alcohol.
- Thickening agents which may preferably be included in the composition of the invention include, for example, candelilla, carnauba and microcrystalline waxes, carbomer and polyethylene thickeners.
- the thickening agent is preferably used in an amount of 0.5 to 2 wt .-%, based on the total weight of the pharmaceutical composition according to the invention.
- Suitable penetration promoters in the sense of the description preferably comprise penetration promoters which are selected from the group comprising acid amides and amines. It is particularly preferred to use urea as the penetration promoter.
- the penetration promoter is preferably used in an amount of 0.5 to 10 wt .-%, based on the total weight of the pharmaceutical composition according to the invention.
- film formers are shellac, methylcellulose, hydroxypropylmethylcellulose (HPMC), hydroxypropylcellulose, hydroxyethylcellulose, ethylcellulose, polyacrylates and polymethacrylates.
- Binders impart cohesive properties to a pharmaceutical composition and, for example, improve granularity.
- Suitable binders are e.g. Hydroxypropylcellulose, starch, cellulose ethers, polyvinylpyrrolidone (povidone), hydroxypropylmethylcellulose, gelatin and sugars, e.g. Sucrose and glucose syrup.
- the binders may account for preferably from 0.5 to 5.0% by weight, based on the total weight of the pharmaceutical composition.
- Lubricants are added to a pharmaceutical composition to improve their rheology during granulation, to prevent the composition from sticking to the devices for granulation, to reduce interparticle friction, and to aid in the release of the tablets from the molds facilitate.
- Suitable lubricants are e.g. Talc, long-chain fatty acids such as stearic acid and palmitic acid, their salts such as magnesium stearate and calcium stearate, polyethylene glycol and hydrogenated vegetable oils.
- the lubricants may be from about 0.25% to about 3.0% by weight, based on the total weight of the pharmaceutical composition.
- Lubricants are usually distinguished from flow improvers which are added to the composition after granulation and before tableting to prevent clumping of the granules.
- a suitable flow improver is e.g. colloidal silica.
- the flow improvers can account for preferably from 0.1 to 3.0% by weight, based on the total weight of the pharmaceutical composition.
- nonionic surfactants are used for the pharmaceutical composition, preferably sorbitan esters such as sorbitan monolaurate, sorbitan monooleate, sorbitan monoisostearate; Polyoxyethylene sorbitan esters such as polyoxyethylene sorbitan monoisostearate, polyoxyethylene sorbitan monolaurate, polyoxyethylene sorbitan monooleate; Glycerol esters, such as glycerol monoisostearate, glycerol monomyristate; Polyoxyethylene glycerol ethers such as polyoxyethylene glycerol monoisostearate, polyoxyethylene glycerol monomyristate; Polyglycerol fatty acid esters, such as diglycerol monostearate, decaglycerol decaisostearate, diglycerol diisostearate
- Anionic surfactants preferably include such amounts as diethanolamine salt, triethanolamine salt, amino acid salt, sodium salt, potassium salt; higher fatty acids such as oleic acid, stearic acid, isostearic acid, palmitic acid, myristic acid, ether carboxylic acid alkaline salts and N-acylamino acid salts.
- the surfactant is preferably used in an amount of 0.1 to 10% by weight based on the total weight of the pharmaceutical composition of the present invention.
- Plasticizers may also be included in the pharmaceutical composition of the present invention.
- Conventional plasticizers are preferably selected from the group comprising oils and waxes, silicone oils, triglyceride esters, acetoglyceride esters, ethoxylated glycerides, alkyl esters, alkenyl esters, fatty acids, fatty alcohols, fatty alcohol esters, lanolin and its derivatives, polyhydric alcohols and their ethers, polyhydric alcohol esters, wax esters, Beeswax derivatives, vegetable waxes, phospholipids, sterols and amides.
- the plasticizers are preferably used in an amount of 1 to 25 wt .-%, based on the total weight of the composition according to the invention.
- Disintegrators are added to a pharmaceutical composition to promote disintegration of a tablet made from the composition.
- Suitable disintegrating agents are, for example, modified or unmodified starch (eg corn starch, wheat starch, potato starch, etc.), clay minerals, cross-linked polyvinylpyrrolidone, modified or unmodified cellulose (eg low substituted sodium carboxymethylcellulose), gum or algins.
- the tablet disintegrants can a weight proportion of preferably 5.0 to 50 wt .-%, more preferably 5.0 to 15 wt .-% make based on the total weight of the pharmaceutical composition.
- Solvents may also be included in the pharmaceutical composition of the invention, e.g. Water, ethanol, mixtures of water and ethanol, propylene glycol or glycerol.
- the pharmaceutical composition is solvent-free, i. it has a (residual) moisture of less than 10% by weight, more preferably less than 5.0% by weight, even more preferably less than 2.0% by weight, most preferably less than 1.0% by weight. % and in particular less than 0.5 wt .-% based on the total weight of the pharmaceutical composition.
- a humectant is glycerine.
- Gel formers suitable for the compositions of the invention preferably comprise natural or synthetic polymers.
- Natural polymers are preferably selected from the group comprising agar-agar, alginic acid, alginate, amidated pectin, propylene glycol alginate, carbomer, carrageenan, casein, dammar gum, dextrins, furcellaran, gelatin, guar gum, guar gum, gellan, ghatti gum, gum arabic , Spruce juice, locust bean gum, karaoke gum, keratin, konjac flour, L-HPC, locust bean gum, mastic, pectin, shellac, starch (if modified), tara gum, tragacanth, xanthan gum and their derivatives.
- Preferred synthetic polymers which can be used as gelling agents for the composition according to the invention are selected from the group comprising acrylic acid polymers, carbomers, polyacrylamides and alkylene oxide polymers.
- the gelling agents are preferably used in an amount of 0.1 to 5 wt .-%, based on the total weight of the composition according to the invention.
- Preservatives may also be included in the pharmaceutical composition of the invention.
- preservatives are alcohols (eg ethanol, chlorobutanol, phenylethyl alcohol or benzyl alcohol), acids (eg sorbic acid or benzoic acid), phenol derivatives (eg phenol, chresol or chlorocresol) or organo-mercury compounds such as phenylmercurinitrate or thiomersal.
- the preservatives are preferably used in an amount of 0.001 to 15% by weight, based on the total weight of the pharmaceutical composition of the invention.
- stabilizers antioxidants and / or reducing agents can be used.
- Particularly preferred for the pharmaceutical composition is at least one reducing agent selected from the group comprising sulfites such as sodium sulfite, potassium sulfite, ammonium sulfite, sodium hydrogen sulfite, potassium hydrogen sulfite, sodium bisulfite, calcium sulfite, calcium hydrogen sulfite, potassium bisulfite, sodium bisulfite, ammonium bisulfite, sodium metabisulfite, potassium metabisulfite; Mercaptocarboxylic acids, such as 2-mercaptopropionic acid, 3-mercaptopropionic acid, mercapto-succinic acid, thioglycolic acid, ammonium thioglycolate, sodium thioglycolate, L-cysteine, dimercapto-adipic acid; Mercapto-amines such as L-cysteine ethyl ester, L-cysteine methyl ester, N-acetyl-L-cysteine,
- At least one antioxidant is selected from the group comprising ascorbic acid (vitamin C), sodium L-ascorbate, calcium L-ascorbate, ascorbyl palmitate, butylhydroxyanisole, butylhydroxytoluene, calcium di-sodium EDTA, Propyl gallate, octyl gallate, dodecyl gallate (lauryl gallate), isoascorbic acid, sodium isoascorbate, lecithin, lactic acid, polyphosphate, sulfur dioxide, selenium, tocopherol (vitamin E) 1 ⁇ -tocopherol, ⁇ -tocopherol, ⁇ -tocopherol, stannous chloride, citric acid, sodium citrate and potassium citrate.
- the antioxidant is preferably used in an amount of 0.001 to 2% by weight based on the total weight of the pharmaceutical composition of the present invention.
- Fillers increase the bulk and volume of a pharmaceutical composition.
- suitable fillers include lactose, mannitol, sorbitol, cellulose, microcrystalline cellulose, XyNt, dextrose, fructose, starch, calcium carbonate (E 170), calcium phosphate, NaCaPO 4 , magnesium carbonate, sucrose and mixtures thereof.
- the fillers can one Weight proportion of preferably 70 to 95 wt .-% based on the total weight of the pharmaceutical composition.
- lubricants are stearic acid, magnesium stearate, calcium stearate and zinc stearate.
- chelating agents are citric acid, phenylalanine, sodium calcium edetate and disodium edetate (EDTA-Na 2 ).
- the pharmaceutical composition may preferably contain at least one perfume and / or one dye.
- the pharmaceutical composition as perfume or flavor additive particularly preferably contains at least one natural or nature-identical compound selected from the group comprising anethole, benzaldehyde, benzyl acetate, benzyl alcohol, benzyl formate, isobutyl acetate, camphene, neral, citronellal, citronellol, citronellyl acetate, para-cymene, Decanal, dihydrolinalool, dihydromyrcenol, dimethylphenylcarbinol, eucalyptol, geraniol, geranylacetate, geranylnitrile, cis-3-hexenylacetate, hydroxycitronellal, limonene, linalool, linalooloxide, linalylacetate, linalylpropionate, methylanthranilate, alpha-methylionone
- At least one naturally occurring mixture of fragrances or aroma additives can also be used.
- at least one perfume or flavor additive mixture is selected from the group comprising rosemary oil, sandalwood oil, violet oil, lemon grass oil, lavender flower oil, eucalyptus oil, peppermint oil, camomile oil, clove leaf oil, cinnamon oil, thyme oil, tea tree oil, cajeput oil, niaouli oil, manuka oil, citrus oil, Mountain pine oil, jasmine oil, geranium oil, caraway oil, pine needle oil, bergamot oil, turpentine oil, linalol oil, Blood orange oil, Cypressenöl, Edelfannenöl, fennel oil, grapefruit oil, ginger oil, pine oil, lavandin oil, lime oil, tangerine oil, lemon balm oil, myrrh oil, patchouli oil, rosewood oil and thuja oil.
- the perfume or flavor additive is preferably used in an amount of 0.001 to 2 w
- Preferred dyes for the pharmaceutical composition include:
- A) inorganic and organic pigments such as titanium oxide, zirconium oxide, cerium oxide, zinc oxide, iron oxide, Prussian blue, carbon black, calcium lake and aluminum lake.
- fat-soluble dyes such as Sudan Red, DC Red 17, DC Green 6, Beta Caroten, Soybean Oil, Sudan Brown, DC Yellow 11, DC Violet 2, DC Orange 5 and Quinoline Yellow.
- water-soluble dyes such as iron sulfates, (Rhodamine), methylene blue and natural dyes.
- the dye is preferably used in an amount of 0.001 to 2 wt .-%, based on the total weight of the pharmaceutical composition according to the invention.
- the pharmaceutical composition according to the invention contains the following excipients in the following preferred amounts (percentages are based on the total weight of the pharmaceutical composition ):
- compositions according to the invention are carried out by means of the usual means, devices, methods and processes known from the prior art, as described for example in "Remington's Pharmaceutical Sciences", published by AR Gennaro, 17th edition, Mack Publishing Company, Easton, Pa , 1985, especially in Part 8, Chapters 76-93.
- Another object of the invention relates to a pharmaceutical dosage form comprising the pharmaceutical composition described above.
- the pharmaceutical composition of the invention as a liquid, semi-solid or solid dosage form, for example in the form of suspensions, ointments, creams, lotions, gels, emulsions, tablets, capsules, dragees, powders, suppositories, patches, vaginal rings, vaginal rods, implants, Intrauterinpessaren, Hormone spirals, syringes or depot vials are present.
- the dosage form according to the invention is preferably present as a tablet, tablet, dragee, capsule, pellet formulation, suppository, transdermal patch or vaginal ring. Suitable embodiments are known in principle to the person skilled in the art.
- the pharmaceutical composition is formulated as a solid dosage form, it can also be present in multiparticulate form, preferably in the form of microtablets, microcapsules, micropellets, construction pellets, granules, extrudates, spheroids, pearls or pellets, optionally filled into capsules or ( Film) tablets are pressed, wherebynickompaktierept are possible.
- the pharmaceutical composition is formulated as a liquid or pasty dosage form, it can be used, for example, as a liquid, foam, cream, gel, paste, balm, spray, ointment, lotion, conditioner, tonic, tincture, milk, mus, powder for dissolution, emulsion (oil-in-water, water-in-oil), serum, oil, shampoo, suspension, such as liposomes or nanosomes, or as a dispersion.
- emulsion oil-in-water, water-in-oil
- serum oil
- shampoo suspension
- suspension such as liposomes or nanosomes, or as a dispersion.
- the dosage form according to the invention is preferably formulated for administration once, twice or three times a day, preferably for oral administration.
- a dosage form, preferably for oral administration containing the pharmaceutical composition described above, which is provided in the form of daily units.
- the dosage form according to the invention can release the compound of general formula (I) immediately (immediate release) or controlled (controlled release). If the release is controlled, it may, for example, be delayed (delayed release), sustained release (pulsed release) or pulsed release (repeat action release). If the dosage form according to the invention contains auxiliaries, these correspond to the abovementioned excipients, which can also be used for the formulation of the pharmaceutical composition according to the invention.
- the dosage form according to the invention preferably contains in the form of daily units, preferably for oral administration, the compound of the general formula in an amount of 0.001 to 99.999% by weight, more preferably 0.1 to 99.9% by weight more preferably 0.5 to 75 wt .-%, most preferably 1, 0 to 50 wt .-% and in particular 2.0 to 25 wt .-%, each based on the total weight of the dosage form.
- the dose of the compound of the general formula (I) contained in the dosage form according to the invention is preferably at least 100 ⁇ g, more preferably at least 200 ⁇ g, even more preferably at least 300 ⁇ g, most preferably at least 400 ⁇ g and in particular at least 500 ⁇ g.
- the dosage of the compound of general formula (I) in the dosage form of the invention is in the range of 500 ⁇ g to 3,000 ⁇ g, more preferably 510 to 2,500 ⁇ g, even more preferably 525 to 2,000 ⁇ g, most preferably 550 to 1,500 ⁇ g and in particular from 600 to 900 ⁇ g.
- the compound of the general formula (I) is present in an amount such that its dose corresponds to the equivalent dose of trimegestone in the range defined above.
- the dose of the estrogen in the dosage form preferably corresponds to an equivalent dose of 5.0 to 55 ⁇ g, more preferably 10 to 50 ⁇ g, more preferably 15 to 48 ⁇ g, most preferably 20 to 45 ⁇ g and in particular 22 to 40 ⁇ g ethinyl estradiol. If several estrogens are present, then their total dose preferably corresponds to the aforementioned equivalent doses.
- the equivalent dose to ethinylestradiol may be realized by an equivalent amount of any suitable estrogen, the amount being chosen such that the estrogenic activity, preferably the ovulation inhibition, is that which would cause the administration of ethinyl estradiol in the indicated amount. It is also possible that two or more different estrogens, for example ethinylestradiol in combination with estradiol, are used in an amount which corresponds in total to the stated equivalent dose. Suitable methods for determining the equivalent dose are known to those skilled in the art.
- the compound of the general formula (I) is present in an amount such that its dose corresponds to the equivalent dose X of trimegestone in the range defined above.
- the compound of the general formula (I) is present in an amount such that its dose corresponds to the equivalent dose X of trimegestone in the range defined above.
- Another object of the invention relates to a cosmetic composition containing at least one compound of general formula (I).
- the cosmetic composition of the care of the skin and / or hair, preferably by topical application.
- the usual auxiliaries of such compositions are preferably suitable.
- the above-mentioned adjuvants may be used, which may also be included in the pharmaceutical compositions of the invention.
- These excipients are physiologically compatible and the amounts of the respective components are preferably selected so that the cosmetic composition according to the invention is in conformity with EU Cosmetics Directive 76/768 / EEC or EU Directive 95/17 / EC.
- excipients and the amounts to be used depend on whether the pharmaceutical or cosmetic composition according to the invention is to be administered orally, topically, subcutaneously, parenterally, intradermally, vaginally or locally, with one oral, vaginal, subcutaneous, or transdermal Application is particularly preferred. Orally or percutaneously applicable preparation forms may also release the compound of general formula (I) with a delay.
- Another object of the invention relates to a method for contraception comprising the preferably oral administration of at least one compound of general formula (I) or the above-described pharmaceutical composition or the above-described pharmaceutical dosage form to a woman of childbearing age at least 21, preferably 21 bis 26, more preferably 22 to 25, and most preferably 23 or 24 consecutive days of a preferably 28-day menstrual cycle beginning on day 1 of the menstrual cycle, wherein at least one, preferably at least 2, more preferably at least 5, more preferably at least 8, most preferably at least 14 and especially on all of the at least 21 consecutive days, the daily dosage of the compound of general formula (I) in the range of 500 ⁇ g to 3,000 ⁇ g, more preferably 510 to 2,500 ⁇ g, even more preferably 525 to 2,000 ⁇ g, most preferably from 550 to 1,500 ⁇ g and in particular from 600 to 900 ⁇ g.
- the daily dosage of the compound of the general formula (I) is such that its dose corresponds to the equivalent dose of trimesterone in the range defined above.
- At least one, preferably all of the at least 21 consecutive days is administered at least one compound of the general formula (I) in combination with at least one estrogen, the estrogen preferably being selected from the group consisting of from chlorotrian iron, dienestrol, diethylstilbestrol, estradiol (17 ⁇ -estradiol), estriol, estrone, ethinyl estradiol, estradiol benzoate, hexestrol, mestranol, methyl styrene, methylestrenol, promestins and conjugated estrogens or their pharmaceutically acceptable esters, such as valerates.
- the estrogen preferably being selected from the group consisting of from chlorotrian iron, dienestrol, diethylstilbestrol, estradiol (17 ⁇ -estradiol), estriol, estrone, ethinyl estradiol, estradiol benzoate, hexestrol, mestranol, methyl styrene, methylestreno
- the additional estrogen component comprises ethinylestradiol or a combination of ethinylestradiol and estradiol (17 ⁇ -estradiol), wherein the amount of the estrogen component is preferably at an equivalent dose of 5.0 to 55 ⁇ g, more preferably 10 to 50 ⁇ g, even more preferably 15 to 48 ⁇ g , most preferably 20 to 45 ⁇ g and in particular 22 to 40 ⁇ g ethinyl estradiol. If several estrogens are used, their daily total dosage preferably corresponds to the abovementioned equivalent doses.
- the administration is in each case in combination with at least one estrogen.
- the compound of the general formula (I) is present in an amount such that its dose corresponds to the equivalent dose A1, A2 or A3 of trimegestone in the range defined above.
- the administration of the at least one compound of general formula (I) does not take place on all days of the preferably 28-day menstrual cycle. Rather, it is preferable that on the days following the at least 21 consecutive days,
- the menstrual cycle is terminated by the withdrawal bleeding, so that a new menstrual cycle can begin.
- the menstrual cycle is 28 days.
- the menstrual cycle is longer than 28 days.
- administration of the at least one compound of general formula (I) is preferably on more than 28 consecutive days.
- (uninterrupted) administration of the at least one compound of general formula (I) in this embodiment is at least 42 or 56, more preferably at least 63, even more preferably at least 84, most preferably at least 105 or 112 and especially at least 126 or 140 consecutive days so that no triggering of withdrawal bleeding is intended within this period.
- the contiguous period in which daily administration of the at least one compound of the general formula (I) is carried out may also be longer according to the invention.
- the at least one compound of the general formula (I) is administered in combination with at least one further progestogen on at least one of the at least 21 consecutive days.
- the gestagens used in such a combination as well as their respective respective dosages correspond to those of the abovementioned pharmaceutical composition according to the invention.
- the method according to the invention is carried out during at least one menstrual cycle.
- the method according to the invention is preferably carried out during several, in particular during at least 6 consecutive menstrual cycles.
- a further aspect of the invention relates to a kit comprising at least one of the administration forms according to the invention described above.
- the kit according to the invention is preferably designed for administration of the dosage forms contained therein once daily.
- the kit is preferably composed in such a way that the contraceptive method according to the invention described above can be carried out with the aid of the dosage forms according to the invention contained in the kit without it being necessary to procure further administration forms according to the invention which are not contained in the kit.
- the kit preferably contains one administration form for one day, since the administration is preferably carried out once a day.
- the kit of the invention preferably comprises at least as many dosage forms of the invention as are required to administer at least one compound of general formula (I) on at least 21 consecutive days of a 28-day menstrual cycle 1 . If the at least one compound of general formula (I) is administered in less than 28 days, the kit according to the invention for the remaining days until the expiration of the 28 days of the menstrual cycle, either no dosage forms, iron-containing preparations, folic acid-containing preparations or placebos, preferably an iron-containing preparation. It is necessary that at least one of the dosage forms of the kit according to the invention is a dosage form according to the invention.
- the menstrual cycle is prolonged, i. If it is more than 28 days, then the number of dosage forms containing is increased accordingly in the kit according to the invention, again at least one of the dosage forms containing a dosage form according to the invention described above.
- the kit according to the invention comprises all dosage forms which are required for administration of at least one compound of general formula (I) for at least one or two, more preferably at least three, even more preferably at least four, most preferably at least five and in particular at least six menstrual cycles ,
- the kit according to the invention is designed for single- or multi-phase administration of at least one compound of the general formula (I) in combination with an estrogen.
- the menstrual cycle is preferably 28- day.
- the daily dosage of the at least one compound of general formula (I) and the estrogen is constant on all days within one phase and different on two consecutive days of different phases.
- Preferred embodiments Nos. 1, 2 ⁇ 2 2, 3i, 3 2, 3 3, ⁇ ⁇ and 4 2 of the kit of the invention comprise a total of 21-25 dosage forms according to the invention, which, depending on the number of phases of the at least one compound of the invention of the general formula (I) in the dosages A1, A2, A3 and at least one estrogen in the dosage B according to the following table:
- the compound of the general formula (I) is present in an amount such that its dose corresponds to the equivalent dose A1, A2 or A3 of trimegestone in the range defined above.
- the compound of general formula (I) is preferably used in combination with ethinylestradiol or in combination with ethinylestradiol and estradiol (17 ⁇ -estradiol).
- the daily dosage of the compound of general formula (I) and the estrogen is constant on all days within one phase and different on two consecutive days of different phases.
- the administration of at least one compound of the general formula (I) 1, if appropriate in combination with an estrogen and / or another progestin, if appropriate for a period of more than 28 days, can also be carried out for therapeutic reasons, such as for treatment and / or prevention of at least one of the conditions selected from the group consisting of rosacea; Psoriasis; Circulatory disorders; Dysmenorrhea (regular symptoms); menstrual cycle-dependent diseases such as endometriosis, polycystic ovarian syndrome (PCOS), uterine myomatosus, functional cysts, menstrual cycle-dependent mood swings, premenstrual dysphoric disorder (PMDD), premenstrual syndrome (PMS) and headache / migraine; menstrual cycle-related illnesses such as epilepsy, multiple sclerosis, diabetes mellitus, depression, schizophrenia, asthma and Parkinson's disease; and androgen-induced disorders such as seborrhoea, acne, androgenetic alopecia and hirsut
- Another object of the invention therefore relates to the treatment and / or prevention of at least one of the complaints or diseases selected from the group consisting of rosacea; Psoriasis; Circulatory disorders; Dysmenorrhea (regular symptoms); menstrual cycle-dependent diseases such as endometriosis, polycystic ovarian syndrome (PCOS), uterine myomatosus, functional cysts, menstrual cycle-dependent mood swings, premenstrual dysphoric disorder (PMDD), premenstrual syndrome (PMS) and headache / migraine; menstrual cycle-related diseases such as epilepsy, multiple sclerosis, diabetes mellitus, depression, schizophrenia, asthma and Parkinson's disease; and androgen-induced disorders such as seborrhoea, acne, androgenetic alopecia and hirsutism by the pharmaceutical composition or dosage form according to the invention, which is particularly suitable for contraception.
- the complaints or diseases selected from the group consisting of rosacea; Psoriasis
- Another object of the invention relates to the use of a compound of general formula (I) as a medicament. Another object of the invention relates to the use of at least one compound of general formula (I), optionally in combination with at least one estrogen and / or another gestagen, for the preparation of a dosage form for hormone replacement therapy (HRT) described above.
- HRT hormone replacement therapy
- Another object of the invention relates to the use of at least one compound of general formula (I), optionally in combination with at least one estrogen and / or another progestin, for the preparation of a dosage form described above for the treatment and / or prevention of at least one of Complaints or diseases selected from the group consisting of rosacea; Psoriasis; Circulatory disorders; Dysmenorrhea (regular symptoms); menstrual cycle-dependent diseases such as endometriosis, polycystic ovarian syndrome (PCOS), uterine myomatosus, functional cysts, menstrual cycle-dependent mood swings, premenstrual dysphoric disorder (PMDD), premenstrual syndrome (PMS) and headache / migraine; menstrual cycle-related diseases such as epilepsy, multiple sclerosis, diabetes mellitus, depression, schizophrenia, asthma and Parkinson's disease; and androgen-induced disorders such as seborrhoea, acne, androgenetic alopecia and hirsutism
- Trimegestone sulfate - pyridinium salt was prepared directly from trimegestone (1) by reaction with sulfur trioxide (as pyridine complex; (2)) in pyridine:
- the reagents sulfur trioxide pyridine complex and pyridine were purchased from Sigma-Aldrich and used in the synthesis without further purification.
- trimegestone sulfate for the human progesterone receptor was determined experimentally and compared to Trimegeston's 6 ⁇ -OH and 1 ⁇ -OH metabolites (see EP-A 808 845). The results are summarized in the following table:
- trimegestone sulfate according to the invention has a relative affinity to the progesterone receptor of 40% of the affinity of natural progesterone.
- Trimegestone sulfate has been subjected to the McPhail test, a test for progesterone and related substances.
- Non-sexually mature female rabbits are treated with 150 IU estrone for a period of 6 days.
- the material to be tested is then administered in five daily subcutaneous dosages.
- the progestational proliferation of the endometrium is observed and the results are estimated on a scale of 0 to 4.0.
- the amount required to produce an average result is considered equivalent to 0.25 mg progesterone.
- trimegestone sulfate has a dose-dependent, potent transforming effect on the endometrium in the McPhail test which is specific for progestational effects:
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Reproductive Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Endocrinology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Gynecology & Obstetrics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Die Erfindung betrifft Verbindungen der allgemeinen Formel (I). Verfahren zu deren Herstellung, pharmazeutische, kosmetische Zusammensetzungen oder Darreichungsformen, welche diese Verbindungen enthalten, sowie Verfahren zur Kontrazeption durch Verabreichung dieser Verbindungen.
Description
19-nor-Progesterone zur Kontrazeption
Die Erfindung betrifft Verbindungen der allgemeinen Formel (I)

Verfahren zu deren Herstellung, pharmazeutische, kosmetische Zusammensetzungen oder Darreichungsformen, welche diese Verbindungen enthalten, Verfahren zur Kontrazeption durch Verabreichung dieser pharmazeutischen Zusammensetzungen, sowie die Verwendung dieser Verbindungen zur Herstellung von Arzneimitteln.
Eine der meist angewendeten Methoden der Empfängnisverhütung (Kontrazeption) stellt die Verabreichung bzw. Applikation von Steroidhormonen wie Östrogenen in Kombination mit Gestagenen dar. Viele der heutzutage eingesetzten, kommerziell erhältlichen, hormonellen Kontrazeptiva weisen jedoch häufig Nebenwirkungen auf, welche dazu führen können, dass die Einnahme des Kontrazeptivums unterbrochen werden muss und somit keine effiziente Therapie bzw. Empfängnisverhütung mehr gewährleistet ist.
Eine Aufgabe der Erfindung besteht darin, Verbindungen zur Verfügung zu stellen, welche sich als pharmazeutische Wirkstoffe eignen und Vorteile gegenüber herkömmlichen pharmazeutischen Wirkstoffen aufweisen. Die pharmazeutischen Wirkstoffe sollten insbesondere zur Kontrazeption geeignet sein.
Diese Aufgabe wird durch den Gegenstand der Patentansprüche gelöst.
Es wurde überraschend gefunden, dass die Verbindungen der allgemeinen Formel (I) Affinität zum humanen Progesteronrezeptor aufweisen und daher insbesondere als pharmazeutische Wirkstoffe geeignet sind, beispielsweise zur Hormonersatz-Therapie oder zur Kontrazeption.
Ein Gegenstand der Erfindung betrifft eine Verbindung der allgemeinen Formel (I)
 wobei
wobei
A entweder ein Schwefelatom ist, p = 0 und q = 1 oder 2 ist; oder ein
Phosphoratom ist und p = 1 und q = 0 oder 1 ist;
R1 und R2 jeweils unabhängig voneinander -H1 -OH oder -OCO-R3 sind;
R3, R4, R5, R6 und R7 jeweils unabhängig voneinander jeweils -H oder ein linearer oder verzweigter Kohlenwasserstoffrest mit 1 bis 12 Kohlenstoffatomen sind, wobei der Kohlenwasserstoffrest unsubstituiert oder mit ggf. 1, 2, 3, 4 oder 5 Substituenten substituiert ist unabhängig voneinander ausgewählt aus der Gruppe bestehend aus -F, -Cl, -Br, -I und -OH; oder deren pharmazeutisch verträgliche Salze und/oder Solvate.
Sofern zum Zwecke der Beschreibung auf Verbindungen der allgemeinen Formel (I) verwiesen wird, sind die pharmazeutisch verträglichen Salze oder Solvate mit umfasst, auch wenn diese nicht jeweils ausdrücklich erwähnt werden.
Ist A ein Phosphoratom, so ist p = 1 und q = O oder 1. Ist q = O, so weist das Phosphoratom die Oxidationszahl III auf und es handelt es sich bei der Verbindung der allgemeinen Formel (I) um ein Phosphonat. Aus Gründen der Übersichtlichkeit ist in der allgemeinen Formel (I) die tautomere Form nicht berücksichtigt, ein Fachmann erkennt jedoch, dass je nach Bedeutung von R6 und R7 auch die tautomere Form vorliegen kann. Ist q = 1 , so weist das Phosphoratom die Oxidationszahl IV auf und es handelt es sich bei der Verbindung der allgemeinen Formel (I) um ein Phosphat.
Ist A ein Schwefelatom, so ist p = O und q = 1 oder 2. Ist q = 1 , so weist das Schwefelatom die Oxidationszahl IV auf und es handelt sich bei der Verbindung der allgemeinen Formel (I) um ein Sulfit. Ist q = 2, so weist das Schwefelatom die Oxidationszahl VI auf und es handelt sich bei der Verbindung der allgemeinen Formel (I) um ein Sulfat.
Unter einem linearen oder verzweigten Kohlenwasserstoffrest ist im Sinne der Beschreibung ein azyklischer, gesättigter (= Alkyl) bzw. einfach oder mehrfach ungesättigter (z.B. = Alkenyl bzw. Alkinyl) Kohlenwasserstoffrest zu verstehen. Ist der Kohlenwasserstoffrest ungesättigt, so kann er wenigstens eine Doppelbindung und/oder eine Dreifachbindung, bevorzugt 1 , 2 oder 3 Doppelbindungen und/oder Dreifachbindungen aufweisen. Als geeignete lineare oder verzweigte, gesättigte oder ungesättigte Kohlenwasserstoffreste seien beispielhaft Methyl, Ethyl, n-Propyl, /so-Propyl, /7-Butyl, /so-Butyl, 2-Butyl, terf-Butyl, n-Pentyl, neo-Pentyl, /7-Hex- yl, π-Heptyl, π-Octyl, /7-Nonyl, n-Decyl, Ethenyl, 1-Propenyl, 2-Propenyl, 1-Butenyl, 2-Butenyl, 3-Butenyl, 1-Pentenyl, 2-Pentenyl, 3-Pentenyl, 4-Pentenyl, Hexenyl, -CH=CH-CH=CH-CH3 und -CH2-CH2-CH=CH2 genannt.
Bevorzugt sind Verbindungen der allgemeinen Formel (I), wobei
A ein Schwefelatom mit p = 0 und q = 2 ist;
R1 und R2 jeweils unabhängig voneinander -H, -OH oder -OCO-R3 sind; und
R4 und R5 jeweils unabhängig voneinander -C1-6-Alkyl, vorzugsweise Methyl, sind, und
R3, R6 und R7 jeweils die vorstehend genannte Bedeutung haben;
jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate.
Bevorzugt sind Verbindungen der allgemeinen Formel (I-A),

wobei p, q, R1, R2, R3, R4, R5, R6 und R7 jeweils die vorstehend genannte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate.
Besonders bevorzugt sind Verbindungen mit der allgemeinen Formel (I) oder (I-A), wobei R1 -H ist; und die übrigen Reste die vorstehend aufgeführte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/ oder entsprechender Solvate.
Weiterhin bevorzugt sind Verbindungen mit der allgemeinen Formel (I) oder (I-A), wobei R2 - H ist; und die übrigen Reste die vorstehend aufgeführte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate. Insbesondere bevorzugt sind Verbindungen mit der allgemeinen Formel (I) oder (I-A), wobei sowohl R1 als auch R2 -H ist; und die übrigen Reste die vorstehend aufgeführte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/ oder entsprechender Solvate.
Bevorzugt sind auch Verbindungen mit der allgemeinen Formel (I) oder (I-A), wobei R4 -Ci-6- Alkyl, vorzugsweise Methyl ist; und die übrigen Reste die vorstehend aufgeführte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate. Weiterhin bevorzugt sind Verbindungen mit der allgemeinen Formel (I) oder (I-A), wobei R5 -C1-6-Alkyl, vorzugsweise Methyl ist; und die übrigen Reste die vorstehend aufgeführte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate. Insbesondere bevorzugt sind Verbindungen mit der allgemeinen Formel (I) oder (I-A), wobei sowohl R4 als auch R5 jeweils unabhängig voneinander -C^-Alkyl, vorzugsweise Methyl sind; und die übrigen Reste die vorstehend aufgeführte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate.
Weiterhin bevorzugt sind Verbindungen mit der allgemeinen Formel (I) oder (I-A), wobei R1 - H und R2 -OH oder -OCO-C^-Alkyl ist; oder R1 -OH oder -OCO-C1-6-Alkyl und R2 -H ist; und die übrigen Reste die vorstehend aufgeführte Bedeutung haben; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate.
Weiterhin bevorzugt sind Verbindungen mit der allgemeinen Formel (I-B), (I-B1), (1-B"), (I-C),
(I-C) oder (1-C")
/|_QJ

 wobei B, B1, B", C, C und C" für einen Rest stehen, der ausgewählt ist aus der Gruppe bestehend aus
wobei B, B1, B", C, C und C" für einen Rest stehen, der ausgewählt ist aus der Gruppe bestehend aus

und wobei Mn+ ein pharmazeutisch verträgliches Kation mit n = 1 , 2 oder 3 ist; jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate.
Unter einem pharmazeutisch verträglichen Kation ist im Sinne der Beschreibung vorzugsweise ein Kation zu verstehen, welches monovalent (1 positive Ladung), divalent (2 positive Ladungen) oder trivalent (3 positive Ladungen) ist und physiologisch allgemein unbedenklich ist. Bevorzugt leitet sich das Kation von einer organischen oder anorganischen Base ab.
In einer bevorzugten Ausführungsform handelt es sich bei dem Kation um ein Metallkation, vorzugsweise ausgewählt aus der Gruppe bestehend aus Kationen von Hauptgruppenmetallen, insbesondere Alkalimetallen und Erdalkalimetallen, und Übergangsmetallen.
In einer anderen bevorzugten Ausführungsform handelt es sich bei dem Kation um ein organisches Kation, vorzugsweise um eine quartäre Ammoniumverbindung N+RR1R11R1", wobei R, R1, R" und R1" vorzugsweise unabhängig voneinander -H oder -d-C8-Alkyl sind,
jeweils ggf. mit einem Rest -OH substituiert, oder zumindest zwei der Reste R, R1, R" und R"1 einen gesättigten, ungesättigten oder aromatischen vier-, fünf-, sechs- oder siebengliedrigen Ring bilden. Andere organische Kationen, welche sich von organischen Basen ableiten, sind die protonierten Formen von Ammoniak, Ethylendiamin, Ethanolamin, 1H-lmidazol, Diethyl- amin, Piperazin, Deanol, Diethanolamin, Pyrrolidin, Betain, 2-(Diethylamino)ethanol, Tro- methamin, Cholin, Morpholin, Lysin, Triethanolamin, L-Arginin, N-Methylglucamin, Ben- ethamin, Benzathin, Hydrabamin, etc.
Beispielhaft seien als pharmazeutisch verträgliche Kationen H+, Li+, Na+, K+, Mg2+, Ca2+, Al3+, Mn2+, Fe2+, Fe3+, Co2+, Co3+, Ni2+, Cu+, Cu2+, Ag+, Zn2+, NH4 +, N(C1-C8-AIKyI)4 +, Triethanol- ammonium, fr/s-(Hydroxymethyl)aminomethan+ und Pyridinium+ genannt. Bezüglich weiterer Einzelheiten kann beispielsweise vollumfänglich verwiesen werden auf P. H. Stahl et al., Handbook of Pharmaceutical Salts, Properties, Selection and Use, Wiley-VCH.
Besonders bevorzugt sind Verbindungen ausgewählt aus der Gruppe bestehend aus

wobei Mn+ ein pharmazeutisch verträgliches Kation ist, das ausgewählt ist aus der Gruppe bestehend aus Li+, Na+, K+, NH4 +, Ag+, Mg2+, Ca2+, Fe2+, Fe3+, Al3+ und Pyridinium+; jeweils ggf. in Form entsprechender Solvate.
Besonders bevorzugt ist eine Verbindung ausgewählt aus der Gruppe bestehend aus

wobei Mn+ ein pharmazeutisch verträgliches Kation ist, das ausgewählt ist aus der Gruppe bestehend aus Li+, Na+, K+, NH4 +, Ag+, Mg2+, Ca2+, Fe2+, Fe3+, Al3+ und Pyridinium+; jeweils ggf. in Form entsprechender Solvate.
Besonders bevorzugt ist eine Verbindung ausgewählt aus der Gruppe bestehend aus

1/2
[3] [4]
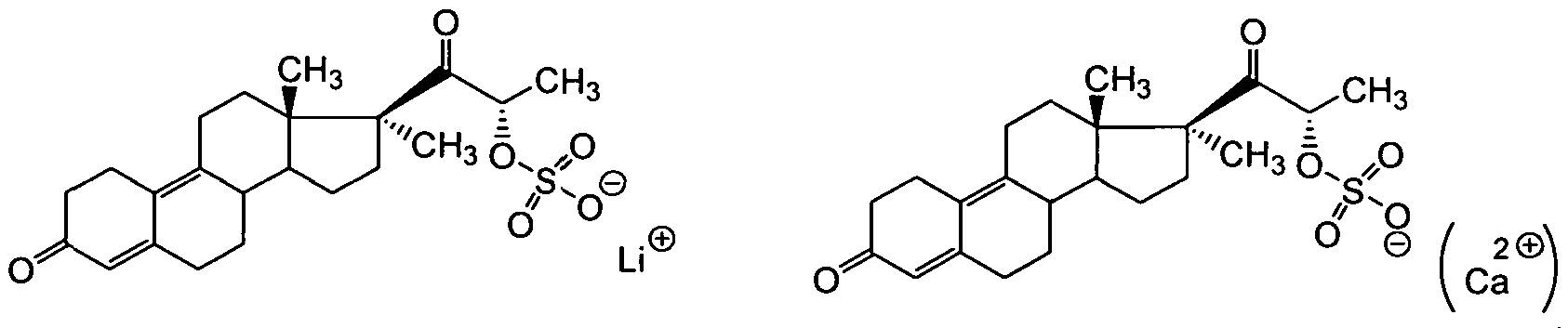
1/2
[5] [6]

[7] [8]
jeweils ggf. in Form entsprechender Solvate.
Insbesondere bevorzugt ist die Verbindung

[9]
jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate.
Ein weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung von Verbindungen der allgemeinen Formeln (I), (I-A), (I-B), (I-B1), (1-B"), (1-C)1 (I-C) und (1-C") umfassend (a) die Umsetzung einer Verbindung der allgemeinen Formel (II)

wobei R1, R2, R4 und R5 jeweils die vorstehend angegebene Bedeutung haben, mit einem Halogenid oder Anhydrid der Schwefligen Säure, der Schwefelsäure, der Phosphorigen Säure oder der Phosphorsäure, vorzugsweise in einem geeigneten Lösemittel, vorzugsweise bei einer Temperatur von -200C bis 1000C, bevorzugter bei einer Temperatur von 00C bis 1000C, noch bevorzugter bei einer Temperatur von 5°C bis 700C am bevorzugtesten bei einer Temperatur von 100C bis 500C und insbesondere bei einer Temperatur von 15°C bis 25°C.
In einer bevorzugten Ausführungsform ist das Anhydrid der Schwefelsäure komplexiert mit Pyridin {Pyridin • SO3), mit Dimethylformamid {(HCON(CH3)2 ■ SO3), mit N-Ethyldiisopropyl- amin {[(CH3)2CH]2NCH2CH3 ■ SO3J, mit Triethylamin {(CH3CH2)3N • SO3) oder mit Trimethyl- amin {(CH3)3N • SO3). Besonders bevorzugt ist das Anhydrid der Schwefelsäure mit Pyridin komplexiert.
Bevorzugt umfasst das erfindungsgemäße Verfahren als weiteren Schritt (b) die Überführung des in Schritt (a) erhaltenen Produkts in ein (anderes) pharmazeutisch verträgliches Salz. Methoden zur Überführung eines Salzes in ein anderes Salz (Metathese) sind dem Fachmann bekannt.
Die vorstehend beschriebenen Umsetzungen können jeweils unter üblichen, dem Fachmann geläufigen Bedingungen, beispielsweise in Hinblick auf Druck oder Reihenfolge der Zugabe der Komponenten durchgeführt werden. Ggf. kann die gemäß den jeweiligen Bedingungen optimale Verfahrensführung vom Fachmann durch einfache Vorversuche ermittelt werden.
Ein weiterer Gegenstand der Erfindung betrifft Verbindungen der allgemeinen Formeln (I), (I- A), (I-B), (I-B1), (1-B"), (I-C), (I-C1) und (I-C11) (I-B), (I-B1), (1-B"), (I-C), (I-C1) oder (I-C11), erhältlich durch das vorstehend genannte Verfahren.
Die nach den vorstehend beschriebenen Umsetzungen erhaltenen Zwischen- und Endprodukte können jeweils, falls gewünscht und/oder erforderlich, nach üblichen, dem Fachmann bekannten Methoden gereinigt und/oder isoliert werden. Geeignete Reinigungsverfahren sind beispielsweise Extraktionsverfahren und chromatographische Verfahren.
Sämtliche der vorstehend beschriebenen Verfahrensschritte sowie jeweils auch die Reinigung und/oder Isolierung von Zwischen- oder Endprodukten können teilweise oder vollständig unter einer Inertgasatmosphäre, vorzugsweise unter Stickstoffatmosphäre, durchgeführt werden.
Die erfindungsgemäßen Verbindungen der allgemeinen Formel (I), (I-A), (I-B), (I-B1), (1-B"), (I-B111), (I-C), (1-C") und (1-C") - welche nachfolgend als Verbindungen der allgemeinen Formel (I) bezeichnet werden - sowie ggf. jeweils entsprechende Stereoisomere können nach üblichen, dem Fachmann bekannten Verfahren in Form entsprechender Salze, insbesondere in Form entsprechender physiologisch verträglicher Salze erhalten werden.
Die erfindungsgemäßen Verbindungen der allgemeinen Formel (I) sowie ggf. entsprechende Stereoisomere und jeweils deren pharmazeutisch/physiologisch verträgliche Salze können nach üblichen, dem Fachmann bekannten Verfahren auch in Form ihrer Solvate, insbesondere in Form ihrer Hydrate erhalten werden.
Die erfindungsgemäßen Verbindungen der allgemeinen Formel (I) und ggf. entsprechende Stereoisomere sowie jeweils die entsprechenden pharmazeutisch/ physiologisch verträglichen Salze und Solvate erscheinen toxikologisch unbedenklich. Darüber hinaus können diese Verbindungen eine höhere Halbwertszeit als beispielsweise Trimegeston aufweisen, weshalb sich diese Verbindungen besonders als pharmazeutische Wirkstoffe in pharmazeutischen Zusammensetzungen oder zur Kontrazeption eignen.
In einer bevorzugten Ausführungsform weisen die erfindungsgemäßen Verbindungen der Formel (I) eine relative Bindungsaffinität zum humanen Progesteron-Rezeptor von mindestens 10%, bevorzugter mindestens 15%, noch bevorzugter mindestens 20%, am bevorzugtesten mindestens 25%, mindestens 30%, mindestens 35%, mindestens 40%, mindestens 45%, mindestens 50% oder mindestens 55% und insbesondere mindestens 60%, mindestens 65%, mindestens 70%, mindestens 75%, mindestens 80%, mindestens 85%, mindestens 90%, oder mindestens 95% auf, wobei als Referenzsubstanz zur Bindung an den humanen Progesteronrezeptor Progesteron eingesetzt wird (= 100%-Wert). Die
vorstehend genannte Bindungsaffinität zum humanen Progesteronrezeptor wird hierbei vorzugsweise gemäß EP-A 808 845 oder wie in I. Lacroix et al., Bioorganic & Medicinal Chemistry, 1999, 7, 2329-2341 bestimmt.
Ein weiterer Gegenstand der Erfindung betrifft eine pharmazeutische Zusammensetzung enthaltend wenigstens eine Verbindung der allgemeinen Formel (I), jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate. Die erfindungsgemäße pharmazeutische Zusammensetzung kann eines oder mehrere Salze einer oder mehrerer dieser Verbindungen aufweisen.
Bevorzugt enthält die pharmazeutische Zusammensetzung einen oder mehrere pharmazeutisch verträglicher Hilfsstoffe. Die Menge der Verbindung der allgemeinen Formel (I) in der erfindungsgemäßen, pharmazeutischen Zusammensetzung beträgt bevorzugt mindestens 100 μg, bevorzugter mindestens 200 μg, noch bevorzugter mindestens 300 μg, am bevorzugtesten mindestens 400 μg und insbesondere mindestens 500 μg. In einer bevorzugten Ausführungsform liegt die Menge der Verbindung der allgemeinen Formel (I) in der erfindungsgemäßen, pharmazeutischen Zusammensetzung im Bereich von 500 μg bis 3.000 μg, bevorzugter von 510 bis 2.500 μg, noch bevorzugter von 525 bis 2.000 μg, am bevorzugtesten von 550 bis 1.500 μg und insbesondere von 600 bis 900 μg. In einer anderen bevorzugten Ausführungsform entspricht die Menge der Verbindung der allgemeinen Formel (I) in der erfindungsgemäßen, pharmazeutischen Zusammensetzung einer Äquivalentdosis von Trimegeston von mindestens 100 μg, bevorzugter mindestens 200 μg, noch bevorzugter mindestens 300 μg, am bevorzugtesten mindestens 400 μg und insbesondere mindestens 500 μg. In einer bevorzugten Ausführungsform entspricht die Menge der Verbindung der allgemeinen Formel (I) in der erfindungsgemäßen, pharmazeutischen Zusammensetzung einer Äquivalentdosis von Trimegeston im Bereich von 500 μg bis 3.000 μg, bevorzugter von 510 bis 2.500 μg, noch bevorzugter von 525 bis 2.000 μg, am bevorzugtesten von 550 bis 1.500 μg und insbesondere von 600 bis 900 μg.
Die Äquivalentdosis der Verbindung der allgemeinen Formel (I) im Vergleich zu Trimegeston wird dabei so gewählt, dass die gestagene Aktivität derjenigen entspricht, welche die Verabreichung von Trimegeston in der angegebenen Menge hervorrufen würde. Geeignete Verfahren zur Bestimmung der Äquivalentdosis sind dem Fachmann bekannt.
Vorzugsweise enthält die pharmazeutische Zusammensetzung die wenigstens eine Verbindung der allgemeinen Formel (I) bevorzugt in einer Menge von 0,001 bis 99,999 Gew.-%, bevorzugter 0,1 bis 99,9 Gew.-%, noch bevorzugter 0,5 bis 75 Gew.-%, am bevorzugtesten
1 ,0 bis 50 Gew.-% und insbesondere 2,0 bis 25 Gew.-%, jeweils bezogen auf das Gesamtgewicht der pharmazeutischen Zusammensetzung.
In einer bevorzugten Ausführungsform enthält die pharmazeutische Zusammensetzung neben den erfindungsgemäßen Verbindungen der allgemeinen Formel (I) zusätzlich wenigstens ein Gestagen, das vorzugsweise ausgewählt ist aus der Gruppe bestehend aus AIIyI- estrenol, Chlormadinon, Cyproteron, Danazol, Demegeston, Desogestrel, Dienogest, Drospi- renon, Dydrogesteron, Ethisteron, Etynodiol, Gestoden, Gestonoron, Hydroxyprogesteron, Levonorgestrel, Lynestrenol, Medroxyprogesteron, Medrogeston, Megestrol, Methylestre- nolon, Methylnortestosteron, Nomegestrol, Norethisteron, Norethynodrel, Norgestrel, Nor- gestimat, Progesteron, Promegeston, Tibolon, Trimegeston, 1ß-Hydroxytrimegeston und 6ß- Hydroxytrimegeston. Bevorzugte pharmazeutisch verträgliche Ester der vorstehend aufgeführten Gestagene sind Acetate (z.B. Chlormadinonacetat, Medroxyprogesteronacetat, Megestrolacetat, Norethisteronacetat), Capronate (z.B. Hydroxyprogesteroncapronat) und Enantate (z.B. Norethisteronenantat).
Bevorzugt entspricht die Menge des zusätzlichen Gestagens einer Äquivalentdosis von 100 bis 5.000 μg, bevorzugter 250 bis 4.000 μg, noch bevorzugter 500 bis 3.500 μg, am bevorzugtesten 750 bis 3.000 μg und insbesondere 1.000 bis 2.500 μg Chlormadinonacetat.
Die Äquivalentdosis zu Chlormadinonacetat kann durch eine äquivalente Menge jedes geeigneten Gestagens verwirklicht werden, wobei die Menge dabei so gewählt wird, dass die gestagene Aktivität derjenigen entspricht, welche die Verabreichung von Chlormadinonacetat in der angegebenen Menge hervorrufen würde. Es ist auch möglich dass zwei oder mehrere unterschiedliche Gestagene in einer Menge eingesetzt werden, die insgesamt der angegebenen Äquivalentdosis entspricht. Geeignete Verfahren zur Bestimmung der Äquivalentdosis sind dem Fachmann bekannt.
In einer bevorzugten Ausführungsform enthält die pharmazeutische Zusammensetzung zusätzlich wenigstens ein Östrogen, das vorzugsweise ausgewählt ist aus der Gruppe bestehend aus Chlorotrianisen, Dienestrol, Diethylstilbestrol, Estradiol (17ß-Estradiol), Estriol, Estron, Ethinylestradiol, Estradiolbenzoat, Hexestrol, Mestranol, Methallenestril, Methylestre- nol, Promestrien und konjugierten Östrogenen bzw. deren pharmazeutisch verträglichen Estern, wie beispielsweise Valerate, wobei insbesondere bevorzugt als zusätzliche Östro- genkomponente Ethinylestradiol bzw. eine Kombination aus Ethinylestradiol und Estradiol (17ß-Estradiol) sind.
In einer bevorzugten Ausführungsform enthält die pharmazeutische Zusammensetzung eine Kombination aus wenigstens einer Verbindung der allgemeinen Formel (I) und wenigstens ein zusätzliches der vorstehend aufgeführten Gestagene und/oder wenigstens eines der vorstehend aufgeführten Östrogene. Besonders bevorzugt enthält die pharmazeutische Zusammensetzung eine Kombination aus wenigstens einer Verbindung der allgemeinen Formel (I) und Ethinylestradiol oder eine Kombination aus wenigstens einer Verbindung der allgemeinen Formel (I) und einer Kombination aus Ethinylestradiol und Estradiol (17ß-Estradiol).
Die pharmazeutische Zusammensetzung kann flüssig (z.B. Lösung, Dispersion, Suspension, Emulsion), pastös oder fest (z.B. Pulver, Granulat) sein. Vorzugsweise ist sie fest.
Bevorzugt enthält die pharmazeutische Zusammensetzung neben wenigstens einer Verbindung der allgemeinen Formel (I) und ggf. zumindest einem Östrogen und/oder zumindest einem weiteren Gestagen zusätzlich wenigstens ein eisenhaltiges Präparat, Folsäure und/oder Folinsäure.
Beispiele für eisenhaltige Präparate sind Eisen(ll)-Präparate, wie z.B. Eisen(ll)sulfat, Eisen(ll)carbonat, Eisen(ll)chlorid, Eisen(ll)tartrat, Eisen(ll)gluconat, Eisen(ll)aspartat, Eisen(ll)glycinsulfat, Eisen(ll)fumarat, Eisen(ll)ascorbat, Eisen(ll)iodat, Eisen(ll)succinat und Ammoniumeisen(ll)sulfat; und Eisen(lll)-Präparate, wie z.B. Eisen(lll)-Natriumcitrat, Eisen(lll)oxid-Sachharose-Komplex, Natriumferedetat, Eisen(lll)hydroxid, Dextriferron, Eisen(lll)citrat, Chondroitinsulfat-Eisen(lll)-Komplex, Eisen(lll)-Acetyltransferrin, Eisen(lll)- Proteinsuccinylat und Kalium-Eisen(lll)-Phosphat-Citrat-Komplex.
Die Folsäure bzw. deren Derivat liegt dabei bevorzugt in freier Form oder als Salz, beispielseise als Calciumfolat vor.
Neben wenigstens einer Verbindung der allgemeinen Formel (I), jeweils ggf. in Form entsprechender pharmazeutisch verträglicher Salze und/oder entsprechender Solvate und ggf. einem weiteren Gestagen und/oder wenigstens einem Östrogen, enthält die pharmazeutische Zusammensetzung bevorzugt zusätzlich einen oder mehrere Hilfsstoffe, die vorzugsweise ausgewählt werden aus der Gruppe bestehend aus Salzbildnern, Puffern, Emulga- toren, Einbettungsmittel, Verdickungsmitteln, Penetrationspromotoren, Filmbildnern, Bindemitteln, Gleitmitteln, oberflächenaktiven Stoffen, Weichmachern, Zerfallsbeschleunigern, Lösungsmitteln, Befeuchtungsmitteln, Gelbildnern, Konservierungsmitteln, Stabilisatoren (Reduktionsmitteln, Antioxidantien), Formtrennmitteln, Füllmitteln, Schmiermittel, Chelat- bildnem, Aromazusätzen, Duftstoffen und Farbstoffen.
Geeignete Puffer, die für die pharmazeutische Zusammensetzung verwendet werden können, sind dem Fachmann bekannt. So kann als Puffer beispielsweise Bernsteinsäure, Zitronensäure, Milchsäure, Phosphorsäure, Trinatriumphosphat, Dinatriumhydrogenphosphat, Natriumdihydrogenphosphat, Natriumcarbonat, Natriumhydrogencarbonat, sowie Kombinationen aus Milchsäure mit Natriumhydroxid eingesetzt werden. Mit dem Puffer, der vorzugsweise eine Mischung aus Zitronensäure und Dinatriumhydrogenphosphat ist, wird vorzugsweise der pH-Wert auf 2,0-5,5 eingestellt.
Emulgatoren werden vorzugsweise in solchen Mengen beigegeben, dass sie ein gleichmäßiges Vermischen der Komponenten der erfindungsgemäßen, pharmazeutischen Zusammensetzung ermöglichen. Übliche Emulgatoren umfassen vorzugsweise anionische, kationische und/oder nichtionische Tenside. Beispiele für derartige Emulgatoren umfassen vorzugsweise Kaliumstearat, Natriumstearat, Ammoniumstearat, Triethanolaminstearat, Glycerin Monostearat, Natriumlauryl-5-sulfat, Natriumacetylsulfat, N-(stearoyl-colamino formylmethyl)-pyridium, N-soja-N-ethyl morpholinium ethosulfat, Alkyl-dimethyl-benzyl ammoniumchlorid, Diisobutylphenoxyethoxyethyl-dimethyl-benzyl- ammoniumchlorid, Acetyl- pyridiumchlorid, Monostearat, Polyoxyethylenstearat, Polyoxyethylensorbitan-monostearat, Sorbitan, Propylenglycol-monostearat und/oder ethoxyliertes Lanolin. Der Emulgator wird vorzugsweise in einer Menge von 0,1 bis 10 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung eingesetzt.
Beispiele für Einbettungsmittel sind Camaubawachs, Montanglycolwachs, Stearinpalmitin- säure, Glyceroltrioleat und Cetylstearylalkohol.
Verdickungsmittel, die vorzugsweise in der erfindungsgemäßen Zusammensetzung enthalten sein können, umfassen beispielsweise Candelilla, Camauba- und mikrokristalline Wachse, Carbomer und Polyethylen-Verdicker. Das Verdickungsmittel wird vorzugsweise in einer Menge von 0,5 bis 2 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung eingesetzt.
Geeignete Penetrationspromotoren im Sinne der Beschreibung umfassen vorzugsweise Penetrationspromotoren, die ausgewählt sind aus der Gruppe umfassend Säureamide und Amine. Besonders bevorzugt wird als Penetrationspromotor Harnstoff eingesetzt. Der Penetrationspromotor wird vorzugsweise in einer Menge von 0,5 bis 10 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung eingesetzt.
Beispiele für Filmbildner sind Schellack, Methylcellulose, Hydroxypropylmethylcellulose (HPMC), Hydroxypropylcellulose, Hydroxyethylcellulose, Ethylcellulose, Polyacrylate und Polymethacrylate.
Bindemittel verleihen einer pharmazeutischen Zusammensetzung kohäsive Eigenschaften und verbessern beispielsweise die Granulierbarkeit. Geeignete Bindemittel sind z.B. Hydroxypropylcellulose, Stärke, Celluloseether, Polyvinylpyrrolidon (Povidon), Hydroxypropylmethylcellulose, Gelatine und Zucker, z.B. Saccharose und Glucosesirup. Die Bindemittel können einen Gewichtsanteil von bevorzugt 0,5 bis 5,0 Gew.-% ausmachen bezogen auf das Gesamtgewicht der pharmazeutischen Zusammensetzung.
Gleitmittel werden einer pharmazeutischen Zusammensetzung zugesetzt, um ihr Fließverhalten bei der Granulierung zu verbessern, um das Anhaften der Zusammensetzung an den Geräten zur Granulierung bzw. Verpressung zu verhindern, um die Reibung zwischen den Partikeln zu verringern und um das Herauslösen der Tabletten aus den Pressformen zu erleichtern. Geeignete Gleitmittel sind z.B. Talk, langkettige Fettsäuren wie Stearinsäure und Palmitinsäure, deren Salze wie Magnesiumstearat und Calciumstearat, Polyethylenglykol und hydrierte pflanzliche Öle. Die Gleitmittel können einen Gewichtsanteil von bevorzugt 0,25 bis 3,0 Gew.-% ausmachen bezogen auf das Gesamtgewicht der pharmazeutischen Zusammensetzung.
Von den Gleitmitteln werden üblicherweise Fließverbesserer unterschieden, welche der Zusammensetzung nach der Granulierung und vor der Tablettierung zugesetzt werden, um das Verklumpen der Granulen zu verhindern. Ein geeigneter Fließverbesserer ist z.B. kolloidales Siliziumdioxid. Die Fließverbesserer können einen Gewichtsanteil von bevorzugt 0,1 bis 3,0 Gew.-% ausmachen bezogen auf das Gesamtgewicht der pharmazeutischen Zusammensetzung.
Weiterhin können auch oberflächenaktive Stoffe (Tenside), die nichtionische, kationische, anionische und/oder ampholytische Eigenschaften besitzen, enthalten sein. Bevorzugt werden nichtionische oberflächenaktive Stoffe für die pharmazeutische Zusammensetzung verwendet, die vorzugsweise Sorbitanester, wie Sorbitan-monolaurat, Sorbitan-monooleat, Sorbitan-monoisostearat; Polyoxyethylen-sorbitanester, wie Polyoxyethylen-sorbitan-mono- isostearat, Polyoxyethylen-sorbitan-monolaurat, Polyoxyethylen-sorbitanmonooleat; Glycerinester, wie Glycerin-monoisostearat, Glycerin-monomyristat; Polyoxyethylenglycerin- ether, wie Polyoxyethylenglycerin monoisostearat, Polyoxyethylenglycerin-monomyristat;
Polyglycerinfettsäureester, wie Diglycerin-monostearat, Decaglycerin-decaisostearat, Diglycerin-diisostearat; Glycerinfettsäureester, wie Glycerin-monocaprat, Glycerin-mono- laurat, Glycerin-monomyristat, Glycerin-monopalmitat, Glycerin-monooleat, Glycerin-mono- stearat, Glycerin-monolinoleät, Glycerin-monoisostearat, Glycerin-monodilinoleat, Glycerin- monodhcaprat; Polyoxyethylenglycerinfettsäureester, wie Polyoxyethylenglycerin-mono- myristat, Polyoxyethylenglycerin-monooleat, Polyoxyethylenglycerin-monostearat; Polyoxy- ethylen-verzweigte Alkylether, wie Polyoxyethylen-octyl-dodecylalkohol, Polyoxyethylen-2- decyl-tetradecylalkohol; Polyoxyethylenalkylether, wie Polyoxyethylen-oleyl-alkoholether, Polyoxyethylen-acetylalkoholether; Polyoxyethylen-hydrogenierte Castorölfettsäureester, wie Polyoxyethylen-hydrogeniertes Castoröl, Polyoxyethylen-dihydrocholesterolether, Polyoxy- ethylen-hydrogeniertes Castoröl isostearat und/oder Polyoxyethylenalkylarylether, wie Polyoxyethylen-octylphenolether umfassen.
Anionische oberflächenaktive Stoffe umfassen vorzugsweise Satze, wie Diethanolaminsalz, Triethanolaminsalz, Aminosäurensalz, Natriumsalz, Kaliumsalz; höherer Fettsäuren, wie Ölsäure, Stearinsäure, Isostearinsäure, Palmitinsäure, Myristinsäure, Ethercarbonsäure- alkaline Salze und N-Acylaminosäurensalze. Der oberflächenaktive Stoff wird vorzugsweise in einer Menge von 0,1 bis 10 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung eingesetzt.
Weichmacher können ebenfalls in der erfindungsgemäßen, pharmazeutischen Zusammensetzung enthalten sein. Übliche Weichmacher sind vorzugsweise ausgewählt aus der Gruppe umfassend Öle und Wachse, Silikonöle, Triglyceridester, Azetoglyceridester, ethoxylierte Glyceride, Alkylester, Alkenylester, Fettsäuren, Fettalkohole, Fettalkoholester, Lanolin und dessen Derivate, mehrfach hydrierte Alkohole und deren Ether, mehrfach hydrierte Alkoholester, Wachsester, Bienenwachs-Derivate, pflanzliche Wachse, Phospho- lipide, Sterole und Amide. Die Weichmacher werden vorzugsweise in einer Menge von 1 bis 25 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen Zusammensetzung eingesetzt.
Zerfallsbeschleuniger (Tablettensprengmittel) werden einer pharmazeutischen Zusammensetzung zugesetzt, um den Zerfall einer aus der Zusammensetzung hergestellten Tablette zu begünstigen. Geeignete Tablettensprengmittel sind z.B. modifizierte oder unmodifizierte Stärke (z.B. Maisstärke, Weizenstärke, Kartoffelstärke, etc.), Tonmineralien, quervemetztes Polyvinylpyrrolidon, modifizierte oder unmodifizierte Cellulose (z.B. niedrig substituierte Natriumcarboxymethylcellulose), Gummi oder Algine. Die Tablettensprengmittel können
einen Gewichtsanteil von bevorzugt 5,0 bis 50 Gew.-%, bevorzugter 5,0 bis 15 Gew.-% ausmachen bezogen auf das Gesamtgewicht der pharmazeutischen Zusammensetzung.
Lösungsmittel können auch in der erfindungsgemäßen pharmazeutischen Zusammensetzung enthalten sein, z.B. Wasser, Ethanol, Mischungen aus Wasser und Ethanol, Propylenglykol oder Glycerol. Bevorzugt ist die pharmazeutische Zusammensetzung jedoch frei von Lösungsmitteln, d.h. sie weist eine (Rest-)Feuchte von weniger als 10 Gew.-%, bevorzugter weniger als 5,0 Gew.-%, noch bevorzugter weniger als 2,0 Gew.-%, am bevorzugtesten weniger als 1 ,0 Gew.-% und insbesondere weniger als 0,5 Gew.-% auf bezogen auf das Gesamtgewicht der pharmazeutischen Zusammensetzung.
Ein Beispiel für ein Befeuchtungsmittel ist Glycerin.
Für die erfindungsgemäßen Zusammensetzungen geeignete Gelbildner umfassen vorzugsweise natürliche oder synthetische Polymere. Natürliche Polymere sind vorzugsweise ausgewählt aus der Gruppe umfassend Agar-Agar, Alginsäure, Alginat, amidiertes Pektin, Propylenglycolalginat, Carbomer, Carrageenan, Casein, Dammar-Gummi, Dextrine, Furcellaran, Gelatine, Guargummi, Guarkernmehl, Gellan, Ghatti-Gummi, Gummi arabicum, Gummi aus Fichtensaft, Johannisbrotkernmehl, Karayagummi, Keratin, Konjakmehl, L-HPC, Locust Bean Gum, Mastix, Pektin, Schellack, (ggf. modifizierte) Stärke, Tarakernmehl, Traganth, Xanthangummi und deren Derivate. Bevorzugte synthetische Polymere, die als Geliermittel für die erfindungsgemäße Zusammensetzung eingesetzt werden können sind ausgewählt aus der Gruppe umfassend Acrylsäurepolymere, Carbomer, Polyacrylamide und Alkylenoxidpolymere. Die Gelbildner werden vorzugsweise in einer Menge von 0,1 bis 5 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen Zusammensetzung eingesetzt.
Konservierungsmittel können auch in der erfindungsgemäßen pharmazeutischen Zusammensetzung enthalten sein. Beispiele für Konservierungsmittel sind Alkohole (z.B. Ethanol, Chlorbutanol, Phenylethylalkohol oder Benzylalkohol), Säuren (z.B. Sorbinsäure oder Benzoesäure), Phenolderivate (z.B. Phenol, Chresol oder Chlorkresol) oder Organo- Quecksilber-Verbindungen wie beispielsweise Phenylmercurinitrat oder Thiomersal. Die Konservierungsmittel werden vorzugsweisein einer Menge von 0,001 bis 15 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung verwendet.
Als Stabilisatoren können Antioxidantien und/oder Reduktionsmittel eingesetzt werden. Insbesondere bevorzugt wird für die pharmazeutische Zusammensetzung wenigstens ein Reduktionsmittel ausgewählt aus der Gruppe, umfassend Sulfite, wie Natriumsulfit, Kaliumsulfit, Ammoniumsulfit, Natriumhydrogensulfit, Kaliumhydrogensulfit, Natriumbisulfit, Calcium- sulfit, Calciumhydrogensulfit, Kaliumbisulfit, Natriumdisulfit, Ammoniumhydrogensulfit, Natriummetabisulfit, Kaliummetabisulfit; Mercaptocarbonsäuren, wie 2-Mercaptopropion- säure, 3-Mercaptopropionsäure, Mercapto-Bernsteinsäure, Thioglycolsäure, Ammonium- thioglycolat, Natrium-thioglycolat, L-Cystein, Dimercapto-Adipinsäure; Mercapto-amine, wie L-Cysteinethylester, L-Cysteinmethylester, N-acetyl-L-cystein, Cysteamin; Mercaptoamide, wie Thioglycolamid, N-hydroxyethyl-mercapto-acetamid, N-methyl-mercapto-acetamid, 2- Mercapto-propionamid; Hydroxide, wie Guanidinhydroxid, Natriumhydroxid; Alkohole und Diole, wie Resorcinol, Thioglycerin, Glycerolmonothioglycolat, Glycolthioglycolat; Di-thio- Verbindungen, wie Dihydroliponsäure, Natrium-dihydroliponsäure, Dithiothreitol, 1 ,3-dithio- propanol; Lithiumchlorid, Tris(hydroxymethyl)phosphin, Thioglycolhydrazid, 2-Mercapto- ethansulfonsäure, Homocysteinthiolacton, Polythiolpolymere, Salze von Hydrogensulfids, Amine in alkalischer Lösung, Salze von Hydrogencyanid, Borohydrid, Dithionit, Estersalze von Sulphoxylaten, Ameisensäure, Oxalsäure, Diazolidinyl-Hamstoff, lodopropynylbutyl- carbamat, Chloromethylisotiazolinon, Methylisothiazolinon, Butylparaben, Ethylparaben, Methylparaben, Propylparaben, Isobutylparaben und Phenoxyethanol. Die Reduktionsmittel werden vorzugsweisein einer Menge von 0,001 bis 2 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung verwendet.
Vorzugsweise wird als Antioxidantien-Komponente für die pharmazeutische Zusammensetzung wenigstens ein Antioxidans ausgewählt aus der Gruppe umfassend Ascorbinsäure (Vitamin C), Natrium-L-Ascorbat, Calcium-L-Ascorbat, Ascorbylpalmitat, Butylhydroxyanisol, Butylhydroxytoluol, Calcium-Di-natrium-EDTA, Propylgallat, Octylgallat, Dodecylgallat (Laurylgallat), Isoascorbinsäure, Natriumisoascorbat, Lecithin, Milchsäure, Polyphosphat, Schwefeldioxid, Selen, Tocopherol (Vitamin E)1 α-Tocopherol, γ-Tocopherol, δ-Tocopherol, Zinn-Il-Chlorid, Zitronensäure, Natriumeitrat und Kaliumeitrat verwendet. Das Antioxidans wird vorzugsweise in einer Menge von 0,001 bis 2 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung verwendet.
Füllmittel erhöhen die Masse und das Volumen einer pharmazeutischen Zusammensetzung. Geeignete Füllmittel sind z.B. Laktose, Mannit, Sorbit, Cellulose, mikrokristalline Cellulose, XyNt, Dextrose, Fructose, Stärke, Calciumcarbonat (E 170), Calciumphosphat, NaCaPO4, Magnesiumcarbonat, Sucrose und Mischungen daraus. Die Füllmittel können einen
Gewichtsanteil von bevorzugt 70 bis 95 Gew.-% ausmachen bezogen auf das Gesamtgewicht der pharmazeutischen Zusammensetzung.
Beispiele für Schmiermittel sind Stearinsäure, Magnesiumstearat, Calciumstearat und Zinkstearat.
Beispiele für Chelatbildner sind Zitronensäure, Phenylalanin, Natriumcalciumedetat und Dinatriumedetat (EDTA-Na2).
Die pharmazeutische Zusammensetzung kann vorzugsweise wenigstens einen Duftstoff und/oder einen Farbstoff enthalten. Besonders bevorzugt enthält die pharmazeutische Zusammensetzung als Duftstoff bzw. Aromazusatz wenigstens eine natürliche oder naturidentische Verbindung ausgewählt aus der Gruppe umfassend Anethol, Benzaldehyd, Benzylacetat, Benzylalkohol, Benzylformiat, iso-Bomylacetat, Camphen, Neral, Citronellal, Citronellol, Citronellylacetat, para-Cymen, Decanal, Dihydrolinalool, Dihydromyrcenol, Dimethylphenylcarbinol, Eukalyptol, Geraniol, Geranylacetat, Geranylnitril, cis-3-Hexenyl- acetat, Hydroxycitronellal, Limonen, Linalool, Linalooloxid, Linalylacetat, Linalylpropionat, Methylanthranilat, alpha-Methylionon, Methylnonylacetaldehyd, Methylphenylcarbinylacetat, Menthon, iso-Menthon, Myrcen, Myrcenylacetat, Myrcenol, Nerol, Nerylacetat, Nonylacetat, Phenylethylalkohol, alpha-Pinen, beta-Pinen, gamma-Terpinen, alpha-Terpinol, beta- Terpinol, Terpinylacetat, para-tert-Butylcyclohexylacetat, alpha-Amylzimtaldehyd, Amyl- salicylat, Caryophyllen, Cedren, Zimtalkohol, Dimethylbenzylcarbinylacetat, Ethylvanillin, Eugenol, iso-Eugenol, Tricyclodecenylacetat, Piperonal, 3-cis-Hexenylsalicylat, Hexylsali- cylat, Lilial, gamma-Methylionon, Nerolidol, Patschulialkohol, Phenylhexanol, beta-Selinen, Trichlormethylphenylcarbinylacetat, Triethylcitrat, Vanillin, Dimethoxybenzaldehyd, Benzo- phenon, Ethylenbrassylat, Galaxolid, Hexylzimtaldehyd, Lyral, Methylcedrylon, Methyl-beta- naphthylketon, Moschusketon, Phenylethylphenylacetat, Ambrettolid, Cyclohexylsalicylat, delta-Nonalacton, delta-Undecalacton, Dodecalacton, Ethylundecylenat, Exaltolid, gamma- Undecalacton, Hexadecanolid, Myristicin und Moschusxylol.
Als Duftstoff bzw. Aromazusatz für die pharmazeutische Zusammensetzung kann auch wenigstens eine natürlich vorkommende Mischung von Duftstoffen bzw. Aromazusätzen verwendet werden. Insbesondere eignet sich wenigstens ein Duftstoff- bzw. Aromazusatz- Mischung ausgewählt aus der Gruppe umfassend Rosmarinöl, Sandelholzöl, Veilchenöl, Zitroneng rasöl, Lavendelblütenöl, Eukalyptusöl, Pfefferminzöl, Kamillenöl, Nelkenblätteröl, Zimtöl, Thymianöl, Teebaumöl, Cajeputöl, Niaouliöl, Manukaöl, Zitrusöl, Latschenkiefemöl, Jasminöl, Geraniumöl, Kümmelöl, Fichtennadelöl, Bergamotteöl, Terpentinöl, Linalolöl,
Blutorangenöl, Cypressenöl, Edeltannenöl, Fenchelöl, Grapefruitöl, Ingweröl, Kiefernadelöl, Lavandinöl, Limetteöl, Mandarinenöl, Melissenöl, Myrrhenöl, Patchouliöl, Rosenholzöl und Thujaöl genannt. Der Duftstoff bzw. Aromazusatz wird vorzugsweise in einer Menge von 0,001 bis 2 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung eingesetzt.
Bevorzugt verwendete Farbstoffe für die pharmazeutische Zusammensetzung umfassen:
A) anorganische und organische Pigmente, wie beispielsweise Titanoxid, Zirkonoxid, Ceroxid, Zinkoxid, Eisenoxid, Preußisch-Blau, Kohleschwärze, Calciumlake und Aluminiumlake.
B) fettlösliche Farbstoffe, wie beispielsweise Sudanrot, DC Red 17, DC Green 6, Beta- Caroten, Sojaöl, Sudanbraun, DC Yellow 11 , DC Violet 2, DC Orange 5 und Quinoline Yellow.
C) wasserlösliche Farbstoffe, wie beispielsweise Eisensulfate, (Rhodamine), Methylenblau und natürliche Farbstoffe.
Der Farbstoff wird vorzugsweise in einer Menge von 0,001 bis 2 Gew.-%, bezogen auf das Gesamtgewicht der erfindungsgemäßen, pharmazeutischen Zusammensetzung eingesetzt.
In einer bevorzugten Ausführungsform enthält die erfindungsgemäße pharmazeutische Zusammensetzung neben wenigstens einer Verbindung der allgemeinen Formel (I) und ggf. zumindest einem Östrogen und/oder zumindest einem weiteren Gestagen folgende Hilfs- stoffe in folgenden bevorzugten Mengen (Prozentangaben sind auf das Gesamtgewicht der pharmazeutische Zusammensetzung bezogen):

Die Herstellung der erfindungsgemäßen pharmazeutischen Zusammensetzung erfolgt mittels der üblichen, aus dem Stande der Technik bekannten Mitteln, Vorrichtungen, Methoden und Verfahren, wie sie beispielsweise in "Remington's Pharmaceutical Sciences", Herausgeber A.R. Gennaro, 17. Auflage, Mack Publishing Company, Easton, Pa, 1985, insbesondere in Teil 8, Kapitel 76 bis 93 beschrieben sind.
Ein weiterer Gegenstand der Erfindung betrifft eine pharmazeutische Darreichungsform umfassend die vorstehend beschriebene pharmazeutische Zusammensetzung.
So kann die erfindungsgemäße pharmazeutische Zusammensetzung als flüssige, halbfeste oder feste Darreichungsform, beispielsweise in Form von Suspensionen, Salben, Cremes, Lotionen, Gelen, Emulsionen, Tabletten, Kapseln, Dragees, Pulvern, Suppositorien, Pflastern, Vaginalringen, Vaginalstäbchen, Implantaten, Intrauterinpessaren, Hormonspiralen, Spritzen oder Depot-Ampullen vorliegen. Die erfindungsgemäße Darreichungsform liegt vorzugsweise als Tablette, Filmtablette, Dragee, Kapsel, Pelletformulierung, Suppositorium, Transdermalpflaster oder Vaginalring vor. Geeignete Ausführungsformen sind dem Fachmann grundsätzlich bekannt.
Sofern die pharmazeutische Zusammensetzung als feste Darreichungsform formuliert ist, kann diese auch in multipartikulärer Form, bevorzugt in Form von Mikrotabletten, Mikro- kapseln, Mikropellets, Aufbaupellets, Granulaten, Extrudaten, Sphäroiden, Perlen oder Pellets vorliegen, ggf. in Kapseln abgefüllt oder zu (Film-)Tabletten verpresst sein, wobei auch Trockenkompaktierungen möglich sind.
Sofern die pharmazeutische Zusammensetzung als flüssige oder pastöse Darreichungsform formuliert ist, kann diese beispielsweise als Flüssigkeit, Schaum, Creme, Gel, Paste, Balsam, Spray, Salbe, Lotion, Spülung („Conditioner"), Tonikum, Tinktur, Milch, Mus, Pulver zum Auflösen, Emulsion (Öl-in-Wasser, Wasser-in-ÖI), Serum, Öl, Shampoo, Suspension, wie Liposome oder Nanosome, oder als Dispersion vorliegen.
Die erfindungsgemäße Darreichungsform ist bevorzugt zur einmal, zweimal oder dreimal täglichen Verabreichung konfektioniert, vorzugsweise zur oralen Verabreichung. Besonders bevorzugt ist eine Darreichungsform, vorzugsweise zur oralen Verabreichung, enthaltend die vorstehend beschriebene pharmazeutische Zusammensetzung, welche in Form von Tageseinheiten zur Verfügung gestellt wird.
Die erfindungsgemäße Darreichungsform kann die Verbindung der allgemeinen Formel (I) sofort (immediate release) oder kontrolliert (controlled release) freisetzen. Erfolgt die Freisetzung kontrolliert, so kann sie beispielsweise zeitlich verzögert (delayed release), hinhaltend (sustained release) oder gepulst (pulsed release, repeat action release) erfolgen.
Sofern die erfindungsgemäße Darreichungsform Hilfsstoffe beinhaltet, so entsprechen diese den vorstehend aufgeführten Hilfsstoffen, welche auch für die Formulierung der erfindungsgemäßen pharmazeutischen Zusammensetzung eingesetzt werden können.
In einer bevorzugten Ausführungsform enthält die erfindungsgemäße Darreichungsform vorzugsweise in Form von Tageseinheiten, vorzugsweise zur oralen Verabreichung, die Verbindung der allgemeinen Formel in einer Menge von 0,001 bis 99,999 Gew.-%, bevorzugter 0,1 bis 99,9 Gew.-%, noch bevorzugter 0,5 bis 75 Gew.-%, am bevorzugtesten 1 ,0 bis 50 Gew.-% und insbesondere 2,0 bis 25 Gew.-%, jeweils bezogen auf das Gesamtgewicht der Darreichungsform.
Die in der erfindungsgemäßen Darreichungsform enthaltene Dosis der Verbindung der allgemeinen Formel (I) beträgt bevorzugt mindestens 100 μg, bevorzugter mindestens 200 μg, noch bevorzugter mindestens 300 μg, am bevorzugtesten mindestens 400 μg und insbesondere mindestens 500 μg. In einer bevorzugten Ausführungsform liegt die Dosis der Verbindung der allgemeinen Formel (I) in der erfindungsgemäßen Darreichungsform im Bereich von 500 μg bis 3.000 μg, bevorzugter von 510 bis 2.500 μg, noch bevorzugter von 525 bis 2.000 μg, am bevorzugtesten von 550 bis 1.500 μg und insbesondere von 600 bis 900 μg. In einer bevorzugten Ausführungsform liegt die Verbindung der allgemeinen Formel (I) in einer solchen Menge vor, dass ihre Dosis der Äquivalentdosis von Trimegeston im vorstehend definierten Bereich entspricht.
Liegt die Verbindung der allgemeinen Formel (I) in Kombination mit wenigstens einem Östrogen vor, vorzugsweise mit Ethinylestradiol, so entspricht die Dosis des Östrogens in der Darreichungsform vorzugsweise einer äquivalenten Dosis von 5,0 bis 55 μg, bevorzugter 10 bis 50 μg, noch bevorzugter 15 bis 48 μg, am bevorzugtesten 20 bis 45 μg und insbesondere 22 bis 40 μg Ethinylestradiol. Sind mehrere Östrogene enthalten, so entspricht deren Gesamtdosis vorzugsweise den vorstehend genannten Äquivalentdosen.
Die Äquivalentdosis zu Ethinylestradiol kann durch eine äquivalente Menge jedes geeigneten Östrogens verwirklicht werden, wobei die Menge dabei so gewählt wird, dass die Östrogene Aktivität, vorzugsweise die Ovulationsinhibition, derjenigen entspricht, welche die Verabreichung von Ethinylestradiol in der angegebenen Menge hervorrufen würde. Es ist auch möglich, dass zwei oder mehrere unterschiedliche Östrogene, beispielsweise Ethinylestradiol in Kombination mit Estradiol, in einer Menge eingesetzt werden, die insgesamt der angegebenen Äquivalentdosis entspricht. Geeignete Verfahren zur Bestimmung der Äquivalentdosis sind dem Fachmann bekannt.
Besonders bevorzugte Ausführungsformen für Kombinationen einer täglichen Dosierung X von wenigstens einer Verbindung der allgemeinen Formel (I) mit der täglichen Dosierungen Y von Ethinylestradiol, welche in der erfindungsgemäßen pharmazeutischen Darreichungsform enthalten sein können, sind in nachfolgender Tabelle zusammengefasst:
Verbindung der Ethinylestradiol allgemeinen Formel (I)
500 ≤ X ≤ 3.000 μg 10 μg ≤ Y ≤ 50 μg
510 ≤ X ≤ 2.500 μg 12 μg ≤ Y ≤ 48 μg
525 ≤ X ≤ 2.000 μg 15 μg < Y ≤ 45 μg
550 ≤ X ≤ 1.500 μg 18 μg ≤ Y ≤ 42 μg
600 ≤ X ≤ 900 μg 20 μg ≤ Y ≤ 40 μg
In einer bevorzugten Ausführungsform liegt die Verbindung der allgemeinen Formel (I) in einer solchen Menge vor, dass ihre Dosis der Äquivalentdosis X von Trimegeston im vorstehend definierten Bereich entspricht.
Besonders bevorzugte Ausführungsformen für Kombinationen der täglichen Dosierung X von wenigstens einer Verbindung der allgemeinen Formel (I) mit der täglichen Dosierungen Y von Ethinylestradiol und der täglichen Dosierung Z von Estradiol (17ß-Estradiol), welche in der erfindungsgemäßen Darreichungsform enthalten sein können, sind in nachfolgender Tabelle zusammengefasst:

In einer bevorzugten Ausführungsform liegt die Verbindung der allgemeinen Formel (I) in einer solchen Menge vor, dass ihre Dosis der Äquivalentdosis X von Trimegeston im vorstehend definierten Bereich entspricht.
Ein weiterer Gegenstand der Erfindung betrifft eine kosmetische Zusammensetzung, welche wenigstens eine Verbindung der allgemeinen Formel (I) enthält. Bevorzugt dient die kosmetische Zusammensetzung der Pflege der Haut und/oder der Haare, vorzugsweise durch topische Anwendung.
Als kosmetische Hilfsstoffe sind vorzugsweise die üblichen Hilfsstoffe solcher Zusammensetzungen geeignet. In diesem Zusammenhang kann beispielsweise vollumfänglich ver-
wiesen werden auf HP. Fiedler, Lexikon der Hilfsstoffe für Pharmazie, Kosmetik und angrenzende technische Gebiete, Editio Cantor Aulendorff, 2002. Vorzugsweise können die vorstehend aufgeführten Hilfsstoffe zum Einsatz kommen, welche auch in der erfindungsgemäßen pharmazeutischen Zusammensetzungen enthalten sein können. Diese Hilfsstoffe sind physiologisch verträglich und die Mengen der jeweiligen Komponenten sind vorzugsweise so gewählt, dass die erfindungsgemäße, kosmetische Zusammensetzung konform mit der EU Kosmetikrichtlinie 76/768/EEC bzw. der EU-Richtlinie 95/17/EC ist.
Die Auswahl der Hilfsstoffe sowie deren einzusetzende Mengen hängt davon ab, ob die erfindungsgemäße pharmazeutische bzw. kosmetische Zusammensetzung oral, topisch, subkutan, parenteral, intradermal, vaginal oder lokal appliziert bzw. verabreicht werden soll, wobei eine orale, vaginale, subkutane, oder transdermalen Anwendung besonders bevorzugt ist. Oral oder perkutan anwendbare Zubereitungsformen können die Verbindung der allgemeinen Formel (I) auch verzögert freisetzen.
Ein weiterer Gegenstand der Erfindung betrifft ein Verfahren zur Kontrazeption umfassend die vorzugsweise orale Verabreichung wenigstens einer Verbindung der allgemeinen Formel (I) bzw. der vorstehend beschriebenen pharmazeutischen Zusammensetzung oder der vorstehend beschriebenen pharmazeutischen Darreichungsform an eine Frau im gebärfähigen Alter an wenigstens 21 , bevorzugt 21 bis 26, bevorzugter 22 bis 25 und am bevorzugtesten 23 oder 24 aufeinander folgenden Tagen eines vorzugsweise 28-tägigen Menstruationszyklus', beginnend am Tag 1 des Menstruationszyklus', wobei an zumindest einem, bevorzugt zumindest an 2, bevorzugter zumindest an 5, noch bevorzugter zumindest an 8, am bevorzugtesten zumindest an 14 und insbesondere an allen der wenigstens 21 aufeinander folgenden Tage die tägliche Dosierung der Verbindung der allgemeinen Formel (I) im Bereich von 500 μg bis 3.000 μg, bevorzugter von 510 bis 2.500 μg, noch bevorzugter von 525 bis 2.000 μg, am bevorzugtesten von 550 bis 1.500 μg und insbesondere von 600 bis 900 μg liegt.
In einer bevorzugten Ausführungsform beträgt die tägliche Dosierung der Verbindung der allgemeinen Formel (I) eine solche Menge, dass ihre Dosis der Äquivalentdosis von Trime- geston im vorstehend definierten Bereich entspricht.
In einer bevorzugten Ausführungsform des erfindungsgemäßen Verfahrens wird an zumindest einem, vorzugsweise an allen der wenigstens 21 aufeinander folgenden Tage wenigstens eine Verbindung der allgemeinen Formel (I) in Kombination mit zumindest einem Östrogen verabreicht, wobei das Östrogen vorzugsweise ausgewählt ist aus der Gruppe beste-
hend aus Chlorotrianisen, Dienestrol, Diethylstilbestrol, Estradiol (17ß-Estradiol), Estriol, Estron, Ethinylestradiol, Estradiolbenzoat, Hexestrol, Mestranol, Methallenestril, Methyl- estrenol, Promestrien und konjugierten Östrogenen bzw. deren pharmazeutisch verträglichen Estern, wie beispielsweise Valerate. Insbesondere bevorzugt umfasst die zusätzliche Östrogenkomponente Ethinylestradiol bzw. eine Kombination aus Ethinylestradiol und Estradiol (17ß-Estradiol), wobei die Menge der Östrogenkomponente vorzugsweise einer äquivalenten Dosis von 5,0 bis 55 μg, bevorzugter 10 bis 50 μg, noch bevorzugter 15 bis 48 μg, am bevorzugtesten 20 bis 45 μg und insbesondere 22 bis 40 μg Ethinylestradiol entspricht. Werden mehrere Östrogene eingesetzt, so entspricht deren tägliche Gesamtdosierung vorzugsweise den vorstehend genannten Äquivalentdosen.
In einer besonders bevorzugten Ausführungsform des erfindungsgemäßen Verfahrens erfolgt an keinem Tag der zumindest 21 aufeinander folgenden Tage die Verabreichung eines Östrogens ohne die Verabreichung von wenigstens einer erfindungsgemäßen Verbindung der Formel (I).
In einer bevorzugten Ausführungsform des erfindungsgemäßen Verfahrens ist an jedem der wenigstens 21 , bevorzugter wenigstens 22, noch bevorzugter wenigstens 23, am bevorzugtesten wenigstens 24 und insbesondere wenigstens 25 aufeinander folgenden Tage die tägliche Dosierung der Verbindung der Formel (I) gleich (= einphasiges Schema), wobei die Verabreichung vorzugsweise jeweils in Kombination mit zumindest einem Östrogen erfolgt.
In einer anderen bevorzugten Ausführungsform des erfindungsgemäßen Verfahrens werden die wenigstens 21 , bevorzugter wenigstens 22, noch bevorzugter wenigstens 23, am bevorzugtesten wenigstens 24 und insbesondere wenigstens 25 aufeinander folgenden Tage in zwei, drei oder mehr Gruppen von Tagen aufgeteilt, wobei an allen Tagen innerhalb einer Gruppe die tägliche Dosierung der Verbindung der allgemeinen Formel (I) gleich ist, jedoch an aufeinander folgenden Tagen unterschiedlicher Gruppen die tägliche Dosierung der Verbindung der allgemeinen Formel (I) unterschiedlich ist (= mehrphasiges Schema), und wobei die Verabreichung vorzugsweise jeweils in Kombination mit zumindest einem Östrogen erfolgt.
Bevorzugte Schemata sind in nachfolgender Tabelle aufgeführt, wobei die tägliche Dosis der Verbindung der allgemeinen Formel (I) A1 , A2 bzw. A3 und die tägliche Dosis des zumindest einen Östrogens B ist:

Die jeweiligen Wertebereiche der Dosierungen für die jeweiligen Kombinationen von A1 , A2, A3 und B für jede einzelne dieser Ausführungsformen Nr. 1 , 2!, 22, 3i, 32, 33, 4τ und A2 sind nachfolgender Tabelle zu entnehmen, wobei die Dosierung B des zumindest einen Östrogens als Äquivalentdosis zu Ethinylestradiol angegeben ist:

In einer bevorzugten Ausführungsform liegt die Verbindung der allgemeinen Formel (I) in einer solchen Menge vor, dass ihre Dosis der Äquivalentdosis A1 , A2 bzw. A3 von Trimegeston im vorstehend definierten Bereich entspricht.
In einer bevorzugten Ausführungsform des erfindungsgemäßen Verfahrens erfolgt die Verabreichung der wenigstens einen Verbindung der allgemeinen Formel (I) nicht an allen Tagen des vorzugsweise 28-tägigen Menstruationszyklus'. Vielmehr ist es bevorzugt, dass an den Tagen, welche sich an die wenigstens 21 aufeinander folgenden Tage anschließen,
- ein Placebo,
- ein eisenhaltiges pharmazeutisch verträgliches Präparat oder
- ein folsäurehaltiges Präparat verabreicht wird;
- oder überhaupt keine Verabreichung erfolgt.
Auf diese Weise wird erreicht, dass der Menstruationszyklus durch die Entzugsblutung beendet wird, so dass ein neuer Menstruationszyklus beginnen kann. Bevorzugt ist der Menstruationszyklus 28-tägig.
Gemäß einer anderen bevorzugten Ausführungsform des erfindungsgemäßen Verfahrens ist es jedoch auch möglich, dass der Menstruationszyklus länger als 28-tägig ist. Dies kann erfindungsgemäß dadurch erreicht werden, dass die Absetzung der wenigstens einen Verbindung der allgemeinen Formel (I) (und ggf. zumindest eines Östrogens und/oder zumindest eines weiteren Gestagens) erst zu einem späteren Zeitpunkt erfolgt, so dass die Entzugsblutung auch erst zu einem späteren Zeitpunkt stattfindet und damit der Menstruationszyklus auch erst zu einem späteren Zeitpunkt endet. In dieser Ausführungsform erfolgt die Verabreichung der wenigstens einen Verbindung der allgemeinen Formel (I) bevorzugt an mehr als 28 aufeinander folgenden Tagen.
Bevorzugt erfolgt die (ununterbrochene) Verabreichung der wenigstens einen Verbindung der allgemeinen Formel (I) bei dieser Ausführungsform an wenigstens 42 oder 56, bevorzugter wenigstens 63, noch bevorzugter wenigstens 84, am bevorzugtesten wenigstens 105 oder 112 und insbesondere wenigstens 126 oder 140 aufeinander folgenden Tagen, so dass innerhalb dieses Zeitraums keine Auslösung der Entzugsblutung beabsichtigt ist. Der zusammenhängende Zeitraum, in dem eine tägliche Verabreichung der wenigstens einen Verbindung der allgemeinen Formel (I) erfolgt, kann erfindungsgemäß auch noch länger sein. So ist es grundsätzlich möglich, eine Verabreichung der wenigstens einen Verbindung der allgemeinen Formel (I) über ein Jahr oder mehrere Jahre hinweg an allen aufeinander folgenden Tagen vorzunehmen, ohne dass es zu einer Entzugsblutung kommt.
In einer bevorzugten Ausführungsform des erfindungsgemäßen Verfahrens wird zumindest an einem der wenigstens 21 aufeinander folgenden Tage die wenigstens eine Verbindung der allgemeinen Formel (I) in Kombination mit zumindest einem weiteren Gestagen verabreicht. Die bei einer derartigen Kombination eingesetzten Gestagene sowie deren entsprechende, jeweilige Dosierungen entsprechen jenen der vorstehend aufgeführten, erfindungsgemäßen pharmazeutischen Zusammensetzung.
Das erfindungsgemäße Verfahren wird während mindestens eines Menstruationszyklus' durchgeführt. Bevorzugt wird das erfindungsgemäße Verfahren während mehrerer, insbesondere während mindestens 6 aufeinander folgenden Menstruationszyklen durchgeführt.
Ein weiterer Aspekt der Erfindung betrifft ein Kit umfassend zumindest eine der vorstehend beschriebenen, erfindungsgemäßen Darreichungsformen. Bevorzugt ist das erfindungsgemäße Kit zur jeweils einmal täglichen Verabreichung der darin enthaltenen Darreichungsformen ausgelegt.
Das Kit ist bevorzugt so zusammengestellt, dass mit Hilfe der in dem Kit enthaltenen, erfindungsgemäßen Darreichungsformen das vorstehend beschriebene, erfindungsgemäße Verfahren zur Kontrazeption durchgeführt werden kann, ohne dass weitere, erfindungsgemäße Darreichungsformen, welche nicht in dem Kit enthalten sind, beschafft werden müssen. Bevorzugt enthält das Kit für einen Tag jeweils eine Darreichungsform, da die Verabreichung bevorzugt einmal täglich erfolgt.
Ist der Menstruationszyklus 28-tägig, so umfasst das erfindungsgemäße Kit bevorzugt wenigstens so viele erfindungsgemäße Darreichungsformen wie erforderlich sind, um wenigstens eine Verbindung der allgemeinen Formel (I) an wenigstens 21 aufeinander folgenden Tagen eines 28-tägigen Menstruationszyklus1 zu verabreichen. Wird die wenigstens eine Verbindung der allgemeinen Formel (I) an weniger als 28 Tagen verabreicht, so kann das erfindungsgemäße Kit für die verbleibenden Tage bis zum Ablauf der 28 Tage des Menstruationszyklus' entweder überhaupt keine Darreichungsformen, eisenhaltige Präparate, folsäurehaltige Präparate oder Placebos, vorzugsweise ein eisenhaltiges Präparat, enthalten. Dabei ist es erforderlich, dass zumindest eine der Darreichungsformen des erfindungsgemäßen Kits eine erfindungsgemäße Darreichungsform ist.
Ist der Menstruationszyklus verlängert, d.h. ist er mehr als 28-tägig, so ist in dem erfindungsgemäßen Kit die Anzahl der enthaltenden Darreichungsformen entsprechend vergrößert, wobei wiederum wenigstens eine der enthaltenden Darreichungsformen eine vorstehend beschriebene erfindungsgemäße Darreichungsform ist.
In einer bevorzugten Ausführungsform umfasst das erfindungsgemäße Kit alle Darreichungsformen, welche zur Verabreichung von wenigstens einer Verbindung der allgemeinen Formel (I) für mindestens einen oder zwei, bevorzugter mindestens drei, noch bevorzugter mindestens vier, am bevorzugtesten mindestens fünf und insbesondere mindestens sechs Menstruationszyklen erforderlich sind.
In einer bevorzugten Ausführungsform ist das erfindungsgemäße Kit für eine ein- oder mehrphasige Verabreichung von wenigstens einer Verbindung der allgemeinen Formel (I) in Kombination mit einem Östrogen ausgelegt. Der Menstruationszyklus ist dabei bevorzugt 28-
tägig. Bei den zwei-, drei und vierphasigen Schemata ist die tägliche Dosierung der wenigstens einen Verbindung der allgemeinen Formel (I) und des Östrogens an allen Tagen innerhalb einer Phase jeweils konstant und an zwei aufeinanderfolgenden Tagen verschiedener Phasen verschieden.
Bevorzugte Ausführungsformen Nr. 1 , 2^ 22, 3i, 32, 33, ΛΛ und 42 des erfindungsgemäßen Kits umfassen insgesamt 21-25 erfindungsgemäße Darreichungsformen, wobei diese je nach Anzahl der Phasen die wenigstens eine erfindungsgemäße Verbindung der allgemeinen Formel (I) in den Dosierungen A1 , A2, A3 und zumindest ein Östrogen in der Dosierung B gemäß nachfolgender Tabelle enthalten:
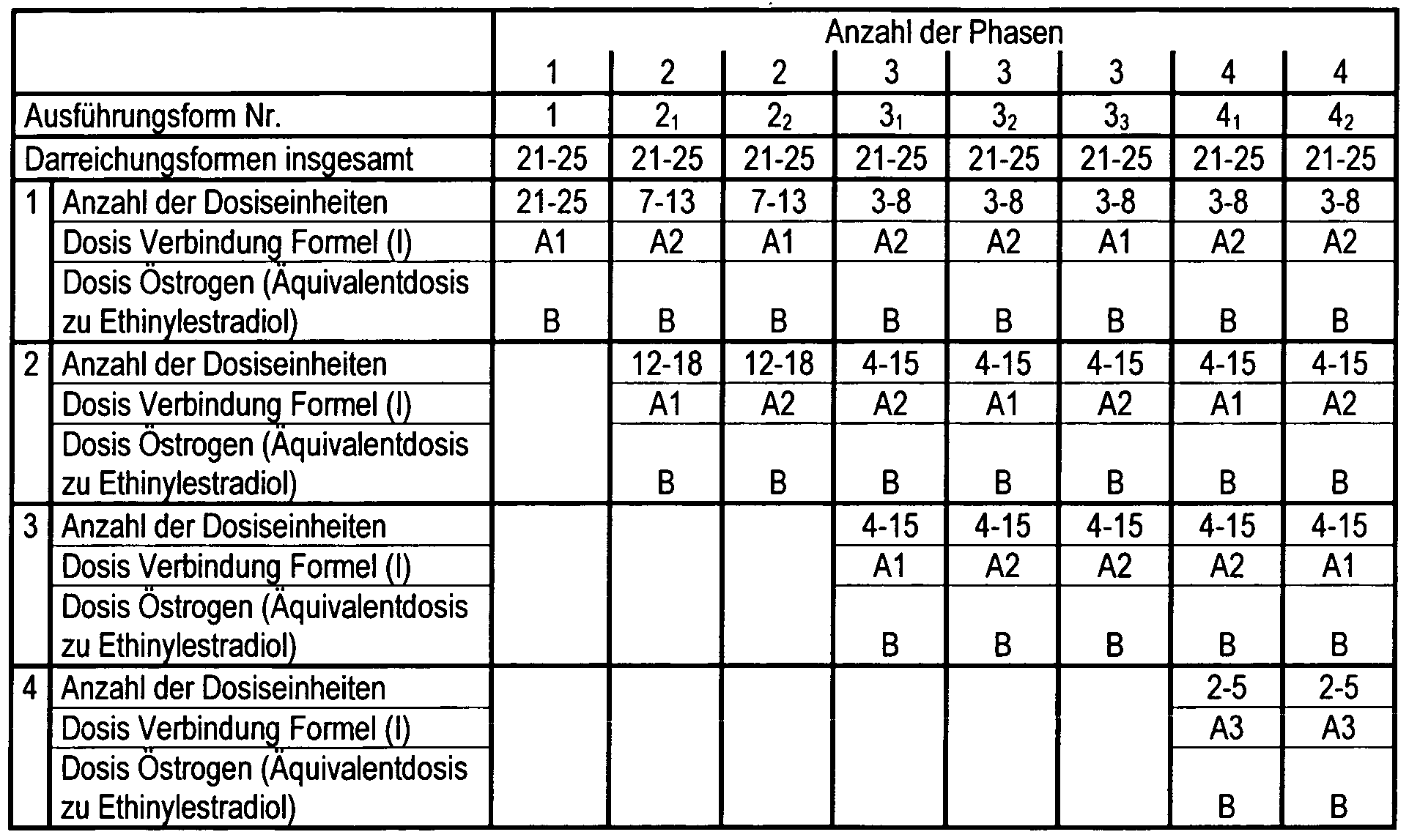
Die jeweiligen Wertebereiche der Dosierungen für die jeweiligen Kombinationen von A1 , A2, A3 und B für jede einzelne dieser Ausführungsformen Nr. 1 , 21 f 22, 3^ 32, 33l 4^ und 42 sind nachfolgender Tabelle zu entnehmen, wobei die Dosierung B des zumindest einen Östrogens als Äquivalentdosis zu Ethinylestradiol angegeben ist:

In einer bevorzugten Ausführungsform liegt die Verbindung der allgemeinen Formel (I) in einer solchen Menge vor, dass ihre Dosis der Äquivalentdosis A1 , A2 bzw. A3 von Trimegeston im vorstehend definierten Bereich entspricht.
Bevorzugt wird die Verbindung der allgemeinen Formel (I) in Kombination mit Ethinylestradiol oder in Kombination mit Ethinylestradiol und Estradiol (17ß-Estradiol) eingesetzt.
Bei den zwei-, drei und vierphasigen Schemata ist die tägliche Dosierung der Verbindung der allgemeinen Formel (I) und des Östrogens an allen Tagen innerhalb einer Phase jeweils konstant und an zwei aufeinanderfolgenden Tagen verschiedener Phasen verschieden.
Die Einnahme von wenigstens einer Verbindung der allgemeinen Formel (I)1 ggf. in Kombination mit einem Östrogen und/oder einem weiteren Gestagen, ggf. für einen Zeitraum von mehr als 28 Tagen, kann auch aus therapeutischen Gründen erfolgen, wie z.B. zur Behandlung und/oder Vorbeugung von wenigstens einer der Beschwerden bzw. Erkrankungen ausgewählt aus der Gruppe bestehend aus Rosazea; Psoriasis; Blutungsstörungen; Dysmenorrhoe (Regelbeschwerden); menstruationszyklusabhängigen Erkrankungen, wie Endometriose, polyzystischem, ovariellen Syndrom (PCOS), Uterus myomatosus, funktionellen Zysten, menstruationszyklusabhängigen Stimmungsschwankungen, prämenstrueller dysphorischer Störung (PMDD), prämenstruellem Syndrom (PMS) und Kopfschmerzen/Migräne; menstrua- tionszyklusbeeinflussten Erkrankungen, wie Epilepsie, Multiple Sklerose, Diabetes mellitus, Depression, Schizophrenie, Asthma und Morbus Parkinson; und androgeninduzierten Störungen, wie Seborrhoe, Akne, androgenetischer Alopezie und Hirsutismus.
Ein weiterer Gegenstand der Erfindung betrifft daher die Behandlung und/oder Vorbeugung von wenigstens einer der Beschwerden bzw. Erkrankungen ausgewählt aus der Gruppe bestehend aus Rosazea; Psoriasis; Blutungsstörungen; Dysmenorrhoe (Regelbeschwerden); menstruationszyklusabhängigen Erkrankungen, wie Endometriose, polyzystischem, ovariellen Syndrom (PCOS), Uterus myomatosus, funktionellen Zysten, menstruationszyklusabhängigen Stimmungsschwankungen, prämenstrueller dysphorischer Störung (PMDD), prämenstruellem Syndrom (PMS) und Kopfschmerzen/Migräne; menstruationszyklusbeein- flussten Erkrankungen, wie Epilepsie, Multiple Sklerose, Diabetes mellitus, Depression, Schizophrenie, Asthma und Morbus Parkinson; und androgeninduzierten Störungen, wie Seborrhoe, Akne, androgenetischer Alopezie und Hirsutismus durch die erfindungsgemäße pharmazeutische Zusammensetzung bzw. Darreichungsform, welche sich insbesondere auch zur Kontrazeption eignet.
Ein weiterer Gegenstand der Erfindung betrifft die Verwendung einer Verbindung der allgemeinen Formel (I) als Medikament.
Ein weiterer Gegenstand der Erfindung betrifft die Verwendung von wenigstens einer Verbindung der allgemeinen Formel (I), ggf. in Kombination mit wenigstens einem Östrogen und/ oder einem weiteren Gestagen, zur Herstellung einer vorstehend beschriebenen Darreichungsform zur Hormonersatz-Therapie (HRT).
Ein weiterer Gegenstand der Erfindung betrifft die Verwendung von wenigstens einer Verbindung der allgemeinen Formel (I), ggf. in Kombination mit wenigstens einem Östrogen und/ oder einem weiteren Gestagen, zur Herstellung einer vorstehend beschriebenen Darreichungsform zur Behandlung und/oder Vorbeugung von wenigstens einer der Beschwerden bzw. Erkrankungen ausgewählt aus der Gruppe bestehend aus Rosazea; Psoriasis; Blutungsstörungen; Dysmenorrhoe (Regelbeschwerden); menstruationszyklusabhängigen Erkrankungen, wie Endometriose, polyzystischem, ovariellen Syndrom (PCOS), Uterus myomatosus, funktionellen Zysten, menstruationszyklusabhängigen Stimmungsschwankungen, prämenstrueller dysphorischer Störung (PMDD), prämenstruellem Syndrom (PMS) und Kopfschmerzen/Migräne; menstruationszyklusbeeinflussten Erkrankungen, wie Epilepsie, Multiple Sklerose, Diabetes mellitus, Depression, Schizophrenie, Asthma und Morbus Parkinson; und androgeninduzierten Störungen, wie Seborrhoe, Akne, androgenetischer Alopezie und Hirsutismus.
Die folgenden Beispiele dienen zur näheren Erläuterung der Erfindung, sind jedoch nicht einschränkend auszulegen.
Trimeqeston-Sulfat - Pyridiniumsalz:
Die Synthese wurde angelehnt an das in der DE 1 152 105 beschriebene Verfahren für 17α- Hydroxyprogesteron durchgeführt. Trimegestonsulfat - Pyridiniumsalz wurde direkt aus Trimegeston (1 ) durch Umsetzung mit Schwefeltrioxid (als Pyridinkomplex; (2)) in Pyridin hergestellt:

Durchführung:
Trimegeston (3,18 g; 9,29 mmol) und Schwefeltrioxid-Pyridinkomplex (1 ,63 g; 10,24 mmol) wurden in Pyridin (16,5 ml) gelöst. Der Ansatz wurde zwei Stunden bei Raumtemperatur gerührt. Die hiernach durchgeführte Reaktionskontrolle per Dünnschichtchromatographie (Kieselgel; Essigester/n-Hexan/Methanol: 2/2/1 ) zeigte vollständigen Umsatz an. Das Reaktionsgemisch wurde mit 0,1 ml Wasser versetzt und 30 Minuten gerührt. Anschließend wurde das Reaktionsgemisch mit 165 ml Diethylether versetzt und weitere zwei Stunden bei Raumtemperatur gerührt. Der ausgefallene Feststoff wurde über eine Glasfilterfritte abgesaugt, gründlich mit Diethylether gewaschen und zunächst an der Luft (ca. 1 Stunde) und anschließend im Hochvakuum (4 Stunden) getrocknet.
Ausbeute: 4,51 g (97,0 %) beigefarbener Feststoff, [α]D = -166,1 (c = 1 ,0; MeOH)
1H-NMR (400 MHz. DMSO-dg) i b = 0,77 (s, 3H); 1 ,11 (s, 3H); 1 ,09-1 ,39 (m, 3H); 1,28 (d, J = 6,5 Hz, 3H); 1 ,48-1 ,60 (m, 2H); 1 ,62-1 ,73 (m,1 H); 1,81-1 ,89 (m, 1 H); 2,01-2,18 (m, 2H); 2,20-2,59 (m, 7H); 2,78 (dd, J = 15,6 / 3,5 Hz, 1 H); 2,89 (dt, J = 14,6 / 5,0 Hz, 1 H); 4,93 (q, J = 6,5 Hz, 1 H); 5,57 (s, 1 H); 8,12 (dd, J = 7,5 / 6,8 Hz, 2H); 8,65 (dt, J = 7,5 / 1 ,5 Hz, 1 H); 8,96 (dd, J = 6,5 / 1 ,5 Hz, 2H) ppm.
13C-NMR (100 MHz. DMSO-dgh δ = 14,94; 18,33; 20,98; 23,46; 25,04; 25,17; 27,18; 30,12; 30,60; 31 ,91 ; 36,63; 38,85; 44,63;
50,34; 59,93; 72,71 ; 121 ,25;124,55;127,16; 142,24; 146,18; 156,57; 197,83; 211 ,16 ppm.
Die relative Affinität von Trimegeston-Sulfat an den humanen Progesteron-Rezeptor wurde experimentell bestimmt und mit den 6ß-OH- und den 1ß-OH-Metaboliten von Trimegeston (vgl. EP-A 808 845) verglichen. Die Ergebnisse sind in nachfolgender Tabelle zusammengestellt:

Wie die Daten belegen, weist das erfindungsgemäße Trimegeston-Sulfat eine relative Affinität an den Progesteron-Rezeptor von 40% der Affinität des natürlichen Progesterons auf.
McPhail Test
Trimegeston-Sulfat wurde dem McPhail Test unterzogen, einem Test für Progesteron und verwandte Substanzen. Nicht-geschlechtsreife weibliche Kaninchen werden mit 150 IU Estron für eine Dauer von 6 tagen behandelt. Das zu testende Material wird dann in fünf täglichen subkutanen Dosierungen verabreicht. Die progestationale Proliferation des Endometriums wird beobachtet und die Ergebnisse werden auf einer Skala von 0 bis 4.0 geschätzt. Diejenige Menge, welche erforderlich ist, um ein durchschnittliches Ergebnis zu bewirken, wird als Äquivalentdosis zu 0,25 mg Progesteron angesehen.
Wie durch die in der nachfolgenden Tabelle zusammengefassten experimentellen Daten belegt wird, weist Trimegeston-Sulfat eine dosisabhängige, potente transformierende Wirkung auf das Endometrium im McPhail Test auf, welcher für progestationale Wirkungen spezifisch ist:

Claims
1. Verbindung der allgemeinen Formel (I)

A entweder ein Schwefelatom ist, p = 0 und q = 1 oder 2 ist; oder ein
Phosphoratom ist und p = 1 und q = 0 oder 1 ist;
R1 und R2 jeweils unabhängig voneinander -H, -OH oder -OCO-R3 sind;
R3, R4, R5, R6 und R7 jeweils unabhängig voneinander jeweils -H oder ein linearer oder verzweigter Kohlenwasserstoffrest mit 1 bis 12 Kohlenstoffatomen sind, wobei der Kohlenwasserstoffrest unsubstituiert oder mit ggf. 1 , 2, 3, 4 oder 5 Substituenten substituiert ist jeweils unabhängig voneinander ausgewählt aus der Gruppe bestehend aus -F, -Cl, -Br, -I und -OH; oder deren pharmazeutisch verträgliche Salze und/oder Solvate.
2. Verbindung nach Anspruch 1 , dadurch gekennzeichnet, dass
A ein Schwefelatom mit p = O und q = 2 ist;
R1 und R2 jeweils unabhängig voneinander -H, -OH oder -OCO-R3 sind; und
R4 und R5 jeweils unabhängig voneinander -C1-6-Alkyl ist.
3. Verbindung nach Anspruch 1 oder 2 mit der allgemeinen Formel (I-A)

4. Verbindung nach einem der vorstehenden Ansprüche, dadurch gekennzeichnet, dass R1 und/oder R2 -H ist.
5. Verbindung nach einem der vorstehenden Ansprüche, dadurch gekennzeichnet, dass R4 und/oder R5 -C1-6-AIkVl ist.
6. Verbindung nach einem der vorstehenden Ansprüche, dadurch gekennzeichnet, dass R1 -H und R2 -OH oder -OCO-C1-6-Alkyl ist; oder R1 -OH oder -OCO-C1-6-Alkyl und R2 -H ist.
7. Verbindung nach einem der vorstehenden Ansprüche ausgewählt aus der Gruppe bestehend aus

wobei Mn+ ein pharmazeutisch verträgliches Kation und n 1 , 2 oder 3 ist.
8. Verfahren zur Herstellung einer Verbindung nach einem der Ansprüche 1 bis 7 umfassend den Schritt
(a) Umsetzen einer Verbindung der allgemeinen Formel (II)

wobei R1, R2, R4 und R5 jeweils die Bedeutung definiert wie in einem der Ansprüche 1 bis 7 haben,
mit einem Halogenid oder Anhydrid der Schwefligen Säure, der Schwefelsäure, der Phosphorigen Säure oder der Phosphorsäure.
9. Verfahren nach Anspruch 8, dadurch gekennzeichnet, dass das Anhydrid der Schwefelsäure mit Pyridin komplexiert ist.
10. Pharmazeutische Zusammensetzung enthaltend eine Verbindung nach einem der Ansprüche 1 bis 7.
11. Zusammensetzung nach Anspruch 10, dadurch gekennzeichnet, dass sie zusätzlich ein Östrogen enthält.
12. Zusammensetzung nach Anspruch 11 , dadurch gekennzeichnet, dass das Östrogen Ethinylestradiol ist.
13. Zusammensetzung nach einem der Ansprüche 10 bis 12, dadurch gekennzeichnet, dass die Menge des Östrogens einer äquivalenten Dosis von 5,0 bis 55 μg Ethinylestradiol entspricht.
14. Zusammensetzung nach einem der Ansprüche 10 bis 13, dadurch gekennzeichnet, dass sie zusätzlich einen oder mehrere Hilfsstoffe enthält ausgewählt aus der Gruppe bestehend aus Salzbildnern, Puffern, Emulgatoren, Einbettungsmittel, Verdickungsmitteln, Penetrationspromotoren, Filmbildnern, Bindemitteln, Gleitmitteln, oberflächenaktiven Stoffen, Weichmachern, Zerfallsbeschleunigern, Lösungsmitteln, Befeuchtungsmitteln, Gelbildnern, Konservierungsmitteln, Stabilisatoren, Formtrennmitteln, Füllmitteln, Schmiermittel, Chelatbildnern, Aromazusätzen, Duftstoffen und Farbstoffen.
15. Darreichungsform umfassend eine pharmazeutische Zusammensetzung nach einem der Ansprüche 10 bis 14.
16. Darreichungsform nach Anspruch 15, dadurch gekennzeichnet, dass sie zur einmal täglichen Verabreichung konfektioniert ist.
17. Darreichungsform nach Anspruch 15 oder 16, dadurch gekennzeichnet, dass sie zur oralen Verabreichung konfektioniert ist.
18. Verfahren zur Kontrazeption umfassend die Verabreichung einer Darreichungsform nach einem der Ansprüche 15 bis 17 an Frauen im gebärfähigen Alter an wenigstens 21 aufeinander folgenden Tagen, beginnend am Tag 1 des Menstruationszyklus1.
19. Verfahren nach Anspruch 18, dadurch gekennzeichnet, dass die Verabreichung oral erfolgt.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102007047608.8 | 2007-10-04 | ||
| DE102007047608A DE102007047608A1 (de) | 2007-10-04 | 2007-10-04 | 19-nor-Progesterone zur Kontrazeption |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009043577A2 true WO2009043577A2 (de) | 2009-04-09 |
| WO2009043577A3 WO2009043577A3 (de) | 2009-06-25 |
Family
ID=40377693
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/008342 Ceased WO2009043577A2 (de) | 2007-10-04 | 2008-10-02 | 19-nor-progesterone zur kontrazeption |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20090093448A1 (de) |
| DE (1) | DE102007047608A1 (de) |
| WO (1) | WO2009043577A2 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10568900B1 (en) | 2013-03-15 | 2020-02-25 | Suzanne Janine Paxton-Pierson | Androgen effectors |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012058463A2 (en) * | 2010-10-27 | 2012-05-03 | Dignity Health | Trimegestone (tmg) for treatment of preterm birth |
| US9737609B2 (en) * | 2014-08-20 | 2017-08-22 | Professional Compounding Centers Of America (Pcca) | Natural suspending agent including a synergistic blend of xanthan gum and konjac powder for oral pharmaceutical suspensions |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1152105B (de) | 1961-11-15 | 1963-08-01 | Schering Ag | Verfahren zur Herstellung eines wasserloeslichen Gestagens |
| FR2749012B1 (fr) | 1996-05-22 | 1998-08-07 | Hoechst Marion Roussel Inc | Nouveaux steroides 1 ou 6-hydroxyles, leur procede de preparation, leur application a titre de medicament et les compositions pharmaceutiques les renfermant |
| US6667050B1 (en) * | 1999-04-06 | 2003-12-23 | Galen (Chemicals) Limited | Chewable oral contraceptive |
| DE102005034498A1 (de) * | 2005-07-20 | 2007-01-25 | Grünenthal GmbH | Orale Kontrazeption mit Trimegeston |
-
2007
- 2007-10-04 DE DE102007047608A patent/DE102007047608A1/de not_active Withdrawn
-
2008
- 2008-10-02 WO PCT/EP2008/008342 patent/WO2009043577A2/de not_active Ceased
- 2008-10-02 US US12/244,480 patent/US20090093448A1/en not_active Abandoned
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10568900B1 (en) | 2013-03-15 | 2020-02-25 | Suzanne Janine Paxton-Pierson | Androgen effectors |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102007047608A1 (de) | 2009-04-09 |
| US20090093448A1 (en) | 2009-04-09 |
| WO2009043577A3 (de) | 2009-06-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0188749B1 (de) | Fumarsäurederivate, Verfahren zu ihrer Herstellung und sie enthaltende pharmazeutische Zubereitungen | |
| EP0775155B1 (de) | Estra-1,3,5(10)-trien-derivate, verfahren zu ihrer herstellung und diese verbindungen enthaltende pharmazeutische zusammensetzungen | |
| WO1999042109A1 (de) | Kombinationspräparat aus östrogen und antiöstrogen | |
| DE19540233B4 (de) | Sulfamat-Derivate von 1,3,5(10)-Estratrien-Derivaten, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende pharmazeutische Zusammensetzungen | |
| DE69910068T2 (de) | 17beta-acyl-17alpha-propynyl-11beta-arylsteroide und ihre derivate mit agonistischen oder antagonistischen hormonalen aktivitäten | |
| WO2009043577A2 (de) | 19-nor-progesterone zur kontrazeption | |
| EP1525215A1 (de) | Progesteronrezeptormodulatoren mit erhöhter antigonadotroper aktivität für die weibliche fertilitätskontrolle und hormonersatztherapie | |
| EP1753408A1 (de) | Hormonales kontrazeptivum enthaltend eine kombination aus ethinylestradiol und chlormadinonacetat | |
| WO2001032680A2 (de) | 18-nor-steroide als selektiv wirksame estrogene | |
| EP0153270A2 (de) | 1alpha, 2alpha-Methylen-6-methylen- und 6alpha-methyl-pregnene, Verfahren zu deren Herstellung und diese enthaltende pharmazeutische Präparate | |
| EP1517914A2 (de) | 9-alpha-substituierte estratriene als selektiv wirksame estrogene | |
| DE60031557T2 (de) | Steroide, ihre herstellung , pharmazeutische zusammensetzungen mit ihnen und verwendungen dieser verbindungen | |
| DE4326240A1 (de) | 15,15-Dialkyl-substituierte Derivate des Estradiols | |
| DE60131253T2 (de) | 6-oxygenierte steroid-estrogene mit aromatischen a- und b-ringen | |
| WO2009124702A1 (de) | Estra-1,3,5(10)-trien-derivate zur kontrazeption | |
| EP1423407B1 (de) | 4-halogenierte 17-methylensteroide, verfahren zu deren herstellung und diese verbindungen enthaltende pharmazeutische zusammensetzungen | |
| EP0942920B1 (de) | Steroidester, verfahren zu ihrer herstellung und ihre pharmazeutische verwendung | |
| DE1902641C3 (de) | 3-Cyclopentyloxysteroide, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| DE69810617T2 (de) | B-ring estratriendiol-sulfate | |
| EP1490391B1 (de) | 11beta-langkettig-substituierte 19-nor-17alpha-pregna-1,3,5(10)-trien-17beta-ole mit einem 21,16alpha-laktonring | |
| DE1952461C3 (de) | 17 alpha-Acetoxy-7 alpha-alkylthioprogesterone Verfahren zu ihrer Herstellung und diese enthaltende Arzneimittel | |
| WO1998024803A9 (de) | Steroidester, verfahren zu ihrer herstellung und ihre pharmazeutische verwendung | |
| DE1620102C3 (de) | 3-Oxime der Androsten- und Gonenreihe, Verfahren zu ihrer Herstellung und diese enthaltende pharmazeutische Mittel | |
| EP1737880A1 (de) | 17 alpha-fluor 17 beta-hydroximinomethyl-steroide, ein verfahren zu deren herstellung und diese verbindung enthaltende pharmazeutische zusammensetzungen | |
| EP1490393A1 (de) | 11beta-substituierte 19-nor-17alpha-pregna-1,3,5(10)-trien-17beta-ole mit einem 21,16alpha-laktonring |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08802748 Country of ref document: EP Kind code of ref document: A2 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08802748 Country of ref document: EP Kind code of ref document: A2 |