WO2009055674A1 - Pyrrolopyrimidine alkynyl compounds and methods of making and using same - Google Patents
Pyrrolopyrimidine alkynyl compounds and methods of making and using same Download PDFInfo
- Publication number
- WO2009055674A1 WO2009055674A1 PCT/US2008/081119 US2008081119W WO2009055674A1 WO 2009055674 A1 WO2009055674 A1 WO 2009055674A1 US 2008081119 W US2008081119 W US 2008081119W WO 2009055674 A1 WO2009055674 A1 WO 2009055674A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- mmol
- group
- independently
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- RSEPYQZONPBLLQ-UHFFFAOYSA-N Brc1ccccc1N1CCCC1 Chemical compound Brc1ccccc1N1CCCC1 RSEPYQZONPBLLQ-UHFFFAOYSA-N 0.000 description 1
- QMZMUKNIECQCBY-UHFFFAOYSA-N CC(C)[n]1c(c(C#Cc2ncnc3c2cc[nH]3)ccc2)c2c(Cl)c1 Chemical compound CC(C)[n]1c(c(C#Cc2ncnc3c2cc[nH]3)ccc2)c2c(Cl)c1 QMZMUKNIECQCBY-UHFFFAOYSA-N 0.000 description 1
- SRABAWNVMWCZHQ-UHFFFAOYSA-N CC(C)c1ccccc1C#Cc1nc(Cl)nc2c1cc[nH]2 Chemical compound CC(C)c1ccccc1C#Cc1nc(Cl)nc2c1cc[nH]2 SRABAWNVMWCZHQ-UHFFFAOYSA-N 0.000 description 1
- GWCTYLHBCKXUMP-UHFFFAOYSA-N CCc1nc(cccc2Br)c2[n]1C(C)C Chemical compound CCc1nc(cccc2Br)c2[n]1C(C)C GWCTYLHBCKXUMP-UHFFFAOYSA-N 0.000 description 1
- YYFGRKSHLFFCQA-UHFFFAOYSA-N CCc1nc(cccc2C#C[Si](C)(C)C)c2[n]1C(C)C Chemical compound CCc1nc(cccc2C#C[Si](C)(C)C)c2[n]1C(C)C YYFGRKSHLFFCQA-UHFFFAOYSA-N 0.000 description 1
- KNLGKCPOEVVTDY-UHFFFAOYSA-N CNc(cc1)ccc1N1CCN(C)CC1 Chemical compound CNc(cc1)ccc1N1CCN(C)CC1 KNLGKCPOEVVTDY-UHFFFAOYSA-N 0.000 description 1
- PTLSPMFOAUSYPU-UHFFFAOYSA-N CNc(cc1)ccc1N1CCNCC1 Chemical compound CNc(cc1)ccc1N1CCNCC1 PTLSPMFOAUSYPU-UHFFFAOYSA-N 0.000 description 1
- DAPOUPJYRMRXGW-UHFFFAOYSA-N CNc(cc1)ccc1N1CC[O](C2)C2CC1 Chemical compound CNc(cc1)ccc1N1CC[O](C2)C2CC1 DAPOUPJYRMRXGW-UHFFFAOYSA-N 0.000 description 1
- ZMDDOSWQDIJCRL-UHFFFAOYSA-N CNc(cc1)ccc1OCCN1CCCC1 Chemical compound CNc(cc1)ccc1OCCN1CCCC1 ZMDDOSWQDIJCRL-UHFFFAOYSA-N 0.000 description 1
- KWSBIEFIWQOXTF-UHFFFAOYSA-N Cc1ncc[n]1Cc(cc1)ccc1N Chemical compound Cc1ncc[n]1Cc(cc1)ccc1N KWSBIEFIWQOXTF-UHFFFAOYSA-N 0.000 description 1
- DRYFDUUAYSVNSN-UHFFFAOYSA-N Nc1ccc(CN2CCCCC2)cc1 Chemical compound Nc1ccc(CN2CCCCC2)cc1 DRYFDUUAYSVNSN-UHFFFAOYSA-N 0.000 description 1
- CYZMCVUHENKFKR-UHFFFAOYSA-N O=S(C(F)(F)F)(Oc1ccccc1C1CCCC1)=O Chemical compound O=S(C(F)(F)F)(Oc1ccccc1C1CCCC1)=O CYZMCVUHENKFKR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Definitions
- Protein kinases are enzymes that play key roles in signaling pathways since they catalyze the phosphorylation of specific residues leading to the transduction of extra and intra cellular signals, including the action of cytokines on their receptors, growth factors, communication with the nuclei and the triggering of various biological events. In normal cellular physiology, cell cycle control, cell growth, differentiation, apoptosis, mobility, mitogenesis, and various other structural and functional events appear to be mediated by kinases.
- Aberrant kinase activity has been implicated in many diseases including cancers, in immunological and auto-immune disorders, in diabetes, fibrosis of the liver and kidney, atherosclerosis and in ocular diseases. Inhibition of such kinase activity may be beneficial in e.g., the treatment of such diseases.
- the Janus kinases are cellular kinases and consist of four members - JAKl, JAK2, JAK3 and TYK2.
- the JAKs may play a crucial role in regulating cell behavior induced by a number of cytokines.
- compounds which modulate the activity of the JAKs have potential utility in several indications driven by a dysregulation of signaling pathways normally associated with cytokine regulation. This includes immune and inflammatory diseases in which dysregulated cytokine pathways are thought to play a roles.
- somatic mutations in the hematopoietic system leading to activation of the JAK pathway has been linked to the myeloproliferative disorders, of cells proliferation and in several cells related to several kinds of immune function.
- JAK kinases have been implicated in ocular diseases such as Age Related Macular Degeneration (AMD), diabetic macular edema (DME) and proliferative diabetic retinopathy (PDR).
- AMD Age Related Macular Degeneration
- DME diabetic macular edema
- PDR proliferative diabetic retinopathy
- novel compounds that may inhibit and/or modulate JAK, for example, JAK2.
- the disclosed compounds may inhibit or modulate one or more of the JAK family, e.g, JAKl, JAK2, JAK3, and/or TYK2, and/or may inhibit or modulate KDR.
- Treatment or amelioration of disease states and pathological conditions that implicate JAK, e.g. JAK2, pathways are contemplated herein, and such treatment comprises administering one or more of the disclosed compounds, such as those recited in Formulas I, Ha, lib, lie or Hd, or administering a composition as described herein comprising a disclosed compound.
- disclosed compounds may have a IC 50 against a JAK of less than about 500 nM.
- the present disclosure is directed in part towards novel compounds and compositions that modulate or inhibit JAK and methods of making and using the same.
- the disclosed compounds may inhibit or modulate one or more of the JAK family, e.g, JAKl, JAK2, JAK3, and/or TYK2, and/or may inhibit or modulate KDR.
- JAK family e.g, JAKl, JAK2, JAK3, and/or TYK2
- KDR KDR
- therapeutic agent refers to any chemical moiety that is a biologically, physiologically, or pharmacologically active substance that acts locally or systemically in a subject.
- therapeutic agents also referred to as "drugs”
- drug are described in well-known literature references such as the Merck Index, the Physicians Desk Reference, and The Pharmacological Basis of Therapeutics, and they include, without limitation, medicaments; vitamins; mineral supplements; substances used for the treatment, prevention, diagnosis, cure or mitigation of a disease or illness; substances which affect the structure or function of the body; or pro-drugs, which become biologically active or more active after they have been placed in a physiological environment.
- therapeutic effect is art-recognized and refers to a local or systemic effect in animals, particularly mammals, and more particularly humans caused by a pharmacologically active substance.
- the term thus means any substance intended for use in the diagnosis, cure, mitigation, treatment or prevention of disease or in the enhancement of desirable physical or mental development and/or conditions in an animal or human.
- therapeutically-effective amount means that amount of such a substance that produces some desired local or systemic effect at a reasonable benefit/risk ratio applicable to any treatment.
- the therapeutically effective amount of such substance will vary depending upon the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the manner of administration and the like, which can readily be determined by one of ordinary skill in the art.
- compositions of the present invention may be administered in a sufficient amount to produce a at a reasonable benefit/risk ratio applicable to such treatment.
- modulation is art-recognized and refers to up regulation (i.e., activation or stimulation), down regulation (i.e., inhibition or suppression) of a response, or the two in combination or apart.
- a "patient,” “subject” or “host” to be treated by the subject method may mean either a human or non-human animal.
- the term “treating” is art-recognized and refers to curing as well as ameliorating at least one symptom of any condition or disease.
- the term “prodrug” is art-recognized and is intended to encompass compounds which, under physiological conditions, are converted into the agents of the present invention.
- a common method for making a prodrug is to select moieties which are hydrolyzed under physiological conditions to provide the desired compound. In other embodiments, the prodrug is converted by an enzymatic activity of the host animal or the target organ or cell.
- alkyl is art-recognized, and includes saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups.
- a straight chain or branched chain alkyl has about 30 or fewer carbon atoms in its backbone (e.g., C 1 -C 3 O for straight chain, C3-C30 for branched chain), and alternatively, about 20 or fewer, e.g. from 1 to 6 carbons.
- cycloalkyls have from about 3 to about 10 carbon atoms in their ring structure, and alternatively about 5, 6 or 7 carbons in the ring structure.
- alkyl is also defined to include halosubstituted alkyls.
- alkyl (or “lower alkyl”) includes “substituted alkyls”, which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- Such substituents may include, for example, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety.
- a carbonyl such as a carboxyl, an alkoxy
- the moieties substituted on the hydrocarbon chain may themselves be substituted, if appropriate.
- the substituents of a substituted alkyl may include substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl (including phosphonate and phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), -CN and the like. Exemplary substituted alkyls are described below.
- Cycloalkyls may be further substituted with alkyls, alkenyls, alkoxys, alkylthios, aminoalkyls, carbonyl- substituted alkyls, -CN, and the like.
- aralkyl is art-recognized and refers to an alkyl group substituted with an aryl group (e.g., an aromatic or heteroaromatic group).
- alkenyl and alkynyl are art-recognized and refer to unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- alkylene refers to an organic radical formed from an unsaturated aliphatic hydrocarbon and can be substituted analogous to the alkyls described above;
- alkenylene denotes an acyclic carbon chain which includes a carbon-to-carbon double bond, and can be optionally substituted analogous to the alkyls as described above.
- lower alkyl refers to an alkyl group, as defined above, but having from one to about ten carbons, alternatively from one to about six carbon atoms in its backbone structure.
- lower alkenyl and “lower alkynyl” have similar chain lengths.
- heteroatom is art-recognized and refers to an atom of any element other than carbon or hydrogen. Illustrative heteroatoms include boron, nitrogen, oxygen, phosphorus, sulfur and selenium.
- aryl is art-recognized and refers to 5-, 6- and 7-membered single-ring aromatic groups that may include from zero to four heteroatoms, for example, benzene, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, triazole, pyrazole, pyridine, pyrazine, pyridazine and pyrimidine, and the like.
- aryl groups having heteroatoms in the ring structure may also be referred to as "heteroaryl” or “heteroaromatics.”
- the aromatic ring may be substituted at one or more ring positions with such substituents as described above, for example, halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, -CF3, -CN, or the like.
- aryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, e.g., the other cyclic rings may be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls.
- heterocyclyl or “heterocyclic group” are art-recognized and refer to 3- to about 10-membered ring structures, alternatively 3- to about 7-membered rings, whose ring structures include one to four heteroatoms. Heterocycles may also be polycycles.
- Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxanthene, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, o
- the heterocyclic ring may be substituted at one or more positions with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
- substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl,
- polycyclyl or “polycyclic group” are art-recognized and refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings". Rings that are joined through non-adjacent atoms are termed "bridged" rings.
- Each of the rings of the polycycle may be substituted with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, - CN, or the like.
- the term "carbocycle” is art-recognized and refers to an aromatic or non-aromatic ring in which each atom of the ring is carbon.
- nitro is art-recognized and refers to -NO2; the term “halogen” is art- recognized and refers to -F, -Cl, -Br or -I; the term “sulfhydryl” is art-recognized and refers to -SH; the term “hydroxyl” means -OH; and the term “sulfonyl” is art-recognized and refers to - SO2 " .
- amine and “amino” are art-recognized and refer to both unsubstituted and substituted amines, e.g., a moiety that may be represented by the general formulas:
- R50, R51 and R52 each independently represent a hydrogen, an alkyl, an alkenyl, - (CH2)m-R61, or R50 and R51, taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure;
- R61 represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocycle or a polycycle; and
- m is zero or an integer in the range of 1 to 8.
- only one of R50 or R51 may be a carbonyl, e.g., R50, R51 and the nitrogen together do not form an imide.
- R50 and R51 each independently represent a hydrogen, an alkyl, an alkenyl, or -(CH2)m- R61.
- alkylamine includes an amine group, as defined above, having a substituted or unsubstituted alkyl attached thereto, i.e., at least one of R50 and R51 is an alkyl group.
- amino is art recognized as an amino- substituted carbonyl and includes a moiety that may be represented by the general formula:
- acylamino is art-recognized and refers to a moiety that may be represented by the general formula:
- R50 is as defined above
- R54 represents a hydrogen, an alkyl, an alkenyl or - (CH 2 ) m -R61, where m and R61 are as defined above.
- alkylthio refers to an alkyl group, as defined above, having a sulfur radical attached thereto.
- the "alkylthio" moiety is represented by one of -S-alkyl, -S-alkenyl, -S-alkynyl, and -S-(CH2)m-R61, wherein m and R61 are defined above.
- Representative alkylthio groups include methylthio, ethyl thio, and the like.
- carbonyl is art recognized and includes such moieties as may be represented by the general formulas:
- X50 is a bond or represents an oxygen or a sulfur
- R55 and R56 represents a hydrogen, an alkyl, an alkenyl, -(CH 2 ) m -R61or a pharmaceutically acceptable salt
- R56 represents a hydrogen, an alkyl, an alkenyl or -(CH 2 ) m -R61, where m and R61 are defined above.
- X50 is an oxygen and R55 or R56 is not hydrogen
- the formula represents an "ester”.
- X50 is an oxygen
- R55 is as defined above, the moiety is referred to herein as a carboxyl group, and particularly when R55 is a hydrogen, the formula represents a "carboxylic acid".
- X50 is an oxygen, and R56 is hydrogen
- the formula represents a "formate".
- the oxygen atom of the above formula is replaced by sulfur
- the formula represents a "thiolcarbonyl” group.
- X50 is a sulfur and R55 or R56 is not hydrogen
- the formula represents a "thiolester.”
- X50 is a sulfur and R55 is hydrogen
- the formula represents a "thiolcarboxylic acid.”
- X50 is a sulfur and R56 is hydrogen
- the formula represents a "thiolformate.”
- X50 is a bond, and R55 is not hydrogen
- the above formula represents a "ketone” group.
- X50 is a bond, and R55 is hydrogen
- the above formula represents an "aldehyde” group.
- each expression e.g. alkyl, m, n, and the like, when it occurs more than once in any structure, is intended to be independent of its definition elsewhere in the same structure.
- compositions of the present invention may exist in particular geometric or stereoisomeric forms.
- polymers of the present invention may also be optically active.
- the present invention contemplates all such compounds, including cis- and trans-isomers, R- and S-enantiomers, diastereomers, (D)-isomers, (L)- isomers, the racemic mixtures thereof, and other mixtures thereof, as falling within the scope of the invention.
- Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
- a particular enantiomer of compound of the present invention may be prepared by asymmetric synthesis, or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers.
- the molecule contains a basic functional group, such as amino, or an acidic functional group, such as carboxyl, diastereomeric salts are formed with an appropriate optically- active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means well known in the art, and subsequent recovery of the pure enantiomers.
- substitution or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction.
- substituted is also contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds.
- Illustrative substituents include, for example, those described herein above.
- the permissible substituents may be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. This invention is not intended to be limited in any manner by the permissible substituents of organic compounds.
- the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 67 th Ed., 1986-87, inside cover.
- the term "hydrocarbon” is contemplated to include all permissible compounds having at least one hydrogen and one carbon atom.
- the permissible hydrocarbons include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic organic compounds that may be substituted or unsubstituted.
- compositions of the present invention refers to the relatively non-toxic, inorganic and organic acid addition salts of compounds, including, for example, those contained in compositions of the present invention.
- pharmaceutically acceptable carrier refers to a pharmaceutically-acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting any subject composition or component thereof from one organ, or portion of the body, to another organ, or portion of the body.
- a pharmaceutically-acceptable material such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting any subject composition or component thereof from one organ, or portion of the body, to another organ, or portion of the body.
- Each carrier must be “acceptable” in the sense of being compatible with the subject composition and its components and not injurious to the patient.
- materials which may serve as pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide;
- systemic administration refers to the administration of a subject composition, therapeutic or other material other than directly into the central nervous system, such that it enters the patient's system and, thus, is subject to metabolism and other like processes, for example, subcutaneous administration.
- ocular administration refers to the administration of a subject composition, therapeutic or other material on or into the eye, including topical and parenteral administration.
- inhalation administration or “administered by inhalation” refers to administration of a subject composition, therapeutic or other material by a pulmonary route, e.g. aerosol inhalation or nasal administration.
- parenteral administration and “administered parenterally” are art- recognized and refer to modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intra-articulare, subcapsular, subarachnoid, intraspinal, and intrasternal injection and infusion.
- R 1 of Formula I is an aryl or heteroaryl optionally substituted on a ring carbon by one, two, three, four, or five substituents, ( e.g. when R 1 is monocyclic), or even six, seven or more substitutents (e.g.
- R 1 when R 1 is multicylic), each independently selected from the group consisting of hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF 3 , ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N-alkylsulphamoyl, N-alkylcarbamoyl, -O-alkyl, -S- alkyl, or -R 13 R 11 .
- Rl can be optionally substituted phenyl, monocyclic heteroaryl, or bicyclic heteroaryl.
- R 1 can be selected, in some embodiments, from the group consisting of optionally substituted indole, optionally substituted benzoimidazole, and optionally substituted phenyl.
- R 4 can be H, NH 2 , -NHR 10 or -NH-alkyl.
- R 4 can be H.
- R 1O is phenyl or pyridinyl, wherein R 10 is optionally substituted on a ring carbon is optionally substituted on a ring carbon by one, two, or three substituents each independently selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, -N-alkyl-amino, carbamoyl, sulphamoyl, CF 3 , ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, -OR 11 , -OR 12 R 11 , or -R 12 R 11 .
- R 1O is phenyl.
- R 11 is independently selected from aryl, heteroaryl, cycloalkyl and heterocycloalkyl, wherein R 11 can be optionally substituted by one to four substituents each independently selected from with halo, alkyl, carbonyl, hydroxyl, alkyl-hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, CF 3 , ureido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, aryl, cycloalkyl, heteroaryl, or heterocycloalkyl;
- R 12 is alkylene or a bond
- R 13 is alkylene, alkenylene, -C(O)-, or a bond.
- the pharmaceutically acceptable salts , prodrugs, N-oxides, diastereomers and hydrates of disclosed compounds such as those represented by formula I is also contemplated.
- R 1 can be selected from:
- R 5 is selected from H, halo, hydroxyl, nitro, cyano, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, -O-alkyl, -S-alkyl, or optionally substituted monocyclic aryl or optionally substituted monocyclic heteraryl.
- Re independently for each occurrence, is selected from H, C 1 - S straight chain alkyl, or C 1 - 5 branched alkyl; and R 7 , independently for each occurrence is selected from H, C 1 - S straight chain alkyl, C 1-4 branched alkyl, cyano, carboxyl, acyl, aldehyde, oxyalkylnitrile, or alkylamine.
- R 1 can be represented by
- R 5 is independently, for each occurrence, selected from the group consisting of H, halo, cyano, nitro, C 1 - S straight chain alkyl, C 1 - S branched alkyl, -O-alkyl, -S- alkyl, or -C(O)-alkyl.
- R 1 can be represented by:
- Re independently for each occurrence, is selected from H, C 1 - S straight chain alkyl, or C 1 - 5 branched alkyl;
- R 7 independently for each occurrence is selected from H, C 1 - S straight chain alkyl,
- R 1 can be represented by:
- R 6 independently for each occurrence, is selected from H, C 1- 5 straight chain alkyl, or C 1-5 branched alkyl; and R 7 , independently for each occurrence is selected from H, C 1-5 straight chain alkyl, C 1-4 branched alkyl, cyano, carboxyl, acyl, or aldehyde.
- R 1O for example, can be represented in some embodiments by:
- X is N or CRg; with, for example,
- Rg and Rg independently for each occurrence chosen from H, heterocycle, -O- heterocycle, -alkylene-heterocycle, or -O-alkylene-heterocycle, wherein said heterocycle for each occurrence is optionally substituted with one to three substituents each independently selected from halo, alkyl, carbonyl, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N- alkylsulphamoyl, and N-alkylcarbamoyl.
- the heterocycle, or the heterocycle of: O-heterocycle, -alkylene-heterocycle, or -O-alkylene-heterocycle can beis substituted with methyl.
- R 8 can be H and R 9 can be heterocycle or -O-alkylene-heterocycle, wherein said heterocycle is chosen from: pyrrolidinyl, piperazinyl, imidazoyl, piperdinyl, or morpholinyl.
- Rg is H and Rg is selected from the group consisting of: methylpiperazine, piperazine, or 2-pyrrolidin-lylethoxy. In some embodiments, at least one occurrence of Rg is H.
- R 4 is H or -NHR 10 , for example, R 4 is H, or R 4 can be -N-phenyl, wherein the phenyl is optionally substituted.
- R 5 is independently selected for each occurence from the group consisting of H, halo, hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF 3 , ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N-alkylsulphamoyl, N-alkylcarbamoyl, -O-alkyl, -S-alkyl, -O-CF 3 , or - R 13 Rn.
- R 6 independently for each occurrence, is selected from H or alkyl;
- R 7 independently for each occurrence is selected from H, halo, hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF 3 , ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N- alkylsulphamoyl, N-alkylcarbamoyl;
- R 1O is phenyl or pyridinyl, wherein R 1O is optionally substituted on a ring carbon is optionally substituted on a ring carbon by one, two, or three substituents each independently selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, -N-alkyl-amino, carbamoyl, sulphamoyl, CF 3 , ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, -OR 11 , -OR 12 Rn, or -R 12 R 11 ; and
- R 11 is independently selected from aryl, heteroaryl, cycloalkyl and heterocycloalkyl, wherein R 11 can be optionally substituted by one to four substituents each independently selected from with halo, alkyl, carbonyl, hydroxyl, alkyl-hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, CF 3 , ureido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, aryl, -N-alkyl-, cycloalkyl, heteroaryl, or heterocycloalkyl;
- R 12 is alkylene or a bond
- R 13 is alkylene, -SO 2 -, or a bond; or Pharmaceutically acceptable salts , prodrugs, N-oxides, diastereomers and hydrates thereof.
- R 4 may be, for example, selected from the group consisting of:
- exemplary compounds can be represented by:
- R 5 for each occurrence, is selected from the group consisting of: H, halo, alkyl, nitro, -O-alkyl, -S-alkyl, phenyl, or heterocycle, wherein said phenyl or heterocycle is optionally substituted with one to three substituents each independently selected from halo, alkyl, carbonyl, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, and N- alkylcarbamoyl.
- Other exemplary compounds can be represented by: H, halo, alkyl, nitro, -O-alkyl, -S-alkyl, phenyl, or heterocycle, wherein said phenyl or heterocycle is optionally substituted with one to three substituents each
- R 6 independently for each occurrence, is selected from H or branched alkyl.
- R 7 can be, for example, independently for each occurrence is selected from H or alkyl.
- Other exemplary compounds can be represented by:
- R 6 is H or branched alkyl.
- compositions that include the disclosed compounds and a pharmaceutically acceptable carrier.
- any compositions of the present invention will vary depending on the symptoms, age and body weight of the patient, the nature and severity of the disorder to be treated or prevented, the route of administration, and the form of the subject composition. Any of the subject formulations may be administered in a single dose or in divided doses. Dosages for the compositions of the present invention may be readily determined by techniques known to those of skill in the art or as taught herein. [0066] In certain embodiments, the dosage of the subject compounds will generally be in the range of about 0.01 ng to about 10 g per kg body weight, specifically in the range of about 1 ng to about 0.1 g per kg, and more specifically in the range of about 100 ng to about 10 mg per kg.
- An effective dose or amount, and any possible affects on the timing of administration of the formulation may need to be identified for any particular composition of the present invention. This may be accomplished by routine experiment as described herein, using one or more groups of animals (preferably at least 5 animals per group), or in human trials if appropriate.
- the effectiveness of any subject composition and method of treatment or prevention may be assessed by administering the composition and assessing the effect of the administration by measuring one or more applicable indices, and comparing the post-treatment values of these indices to the values of the same indices prior to treatment.
- the health of the patient may be monitored by measuring one or more of the relevant indices at predetermined times during the treatment period.
- Treatment including composition, amounts, times of administration and formulation, may be optimized according to the results of such monitoring.
- the patient may be periodically reevaluated to determine the extent of improvement by measuring the same parameters.
- Adjustments to the amount(s) of subject composition administered and possibly to the time of administration may be made based on these reevaluations.
- Treatment may be initiated with smaller dosages which are less than the optimum dose of the compound. Thereafter, the dosage may be increased by small increments until the optimum therapeutic effect is attained. [0071]
- the use of the subject compositions may reduce the required dosage for any individual agent contained in the compositions because the onset and duration of effect of the different agents may be complimentary.
- Toxicity and therapeutic efficacy of subject compositions may be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 and the ED50.
- the data obtained from the cell culture assays and animal studies may be used in formulating a range of dosage for use in humans.
- the dosage of any subject composition lies preferably within a range of circulating concentrations that include the ED50 with little or no toxicity.
- the dosage may vary within this range depending upon the dosage form employed and the route of administration utilized.
- the therapeutically effective dose may be estimated initially from cell culture assays.
- compositions of the present invention may be administered by various means, depending on their intended use, as is well known in the art.
- compositions of the present invention may be formulated as tablets, capsules, granules, powders or syrups.
- formulations of the present invention may be administered parenterally as injections (intravenous, intramuscular or subcutaneous), drop infusion preparations, suppositories or administration intranasally (for example, to deliver a dosage to the brain via the nose or to deliver a dosage to the nose directly) or by inhalation (e.g. to treat a condition of the respiratory tract or to pretreat or vaccinate via the respiratory tract).
- compositions of the present invention may be formulated as eyedrops or eye ointments. These formulations may be prepared by conventional means, and, if desired, the compositions may be mixed with any conventional additive, such as an excipient, a binder, a disintegrating agent, a lubricant, a corrigent, a solubilizing agent, a suspension aid, an emulsifying agent or a coating agent.
- any conventional additive such as an excipient, a binder, a disintegrating agent, a lubricant, a corrigent, a solubilizing agent, a suspension aid, an emulsifying agent or a coating agent.
- compositions of the subject invention may be suitable for oral, nasal, topical (including buccal and sublingual), rectal, vaginal, aerosol and/or parenteral administration.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy.
- the amount of composition that may be combined with a carrier material to produce a single dose vary depending upon the subject being treated, and the particular mode of administration.
- Methods of preparing these formulations include the step of bringing into association compositions of the present invention with the carrier and, optionally, one or more accessory ingredients.
- the formulations are prepared by uniformly and intimately bringing into association agents with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- Formulations suitable for oral administration may be in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in- water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia), each containing a predetermined amount of a subject composition thereof as an active ingredient.
- Compositions of the present invention may also be administered as a bolus, electuary, or paste.
- the subject composition is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example,
- compositions may also comprise buffering agents.
- Solid compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface- active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine a mixture of the subject composition moistened with an inert liquid diluent. Tablets, and other solid dosage forms, such as dragees, capsules, pills and granules, may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, cyclodextrins and mixtures thereof.
- inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing
- Suspensions in addition to the subject composition, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- Formulations for rectal or vaginal administration may be presented as a suppository, which may be prepared by mixing a subject composition with one or more suitable non- irritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the body cavity and release the active agent.
- suitable non- irritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the body cavity and release the active agent.
- Formulations which are suitable for vaginal administration also include pessaries, tampons, creams, gels, pastes, foams or spray formulations containing such carriers as are known in the art to be appropriate.
- Dosage forms for transdermal administration of a subject composition includes powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants.
- the active component may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants which may be required.
- the ointments, pastes, creams and gels may contain, in addition to a subject composition, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- Powders and sprays may contain, in addition to a subject composition, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances.
- Sprays may additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
- compositions and compounds of the present invention may alternatively be administered by aerosol. This is accomplished by preparing an aqueous aerosol, liposomal preparation or solid particles containing the compound.
- a non-aqueous (e.g., fluorocarbon propellant) suspension could be used.
- Sonic nebulizers may be used because they minimize exposing the agent to shear, which may result in degradation of the compounds contained in the subject compositions.
- an aqueous aerosol is made by formulating an aqueous solution or suspension of a subject composition together with conventional pharmaceutically acceptable carriers and stabilizers.
- the carriers and stabilizers vary with the requirements of the particular subject composition, but typically include non-ionic surfactants (Tweens, Pluronics, or polyethylene glycol), innocuous proteins like serum albumin, sorbitan esters, oleic acid, lecithin, amino acids such as glycine, buffers, salts, sugars or sugar alcohols. Aerosols generally are prepared from isotonic solutions.
- Dosages for administration by nasal delivery e.g.
- compositions for inhalation and/or delivery to the nose may contain from 1% to 20% by weight of a penetrator enhancer (for example, surfactants, e.g. sugar esters, sugar ethers, carbohydrate esters) which may allow enhanced nose permeability of the active agent.
- a penetrator enhancer for example, surfactants, e.g. sugar esters, sugar ethers, carbohydrate esters
- Dosages for administration by inhalation or by delivered to or via the lung can be applied as mists/sprays (aqueous or nonaqueous), aerosols (liquids, suspensions or dry powders),liquids or suspensions (aqueous or nonaqueous), powders, or combinations thereof.
- Such delivery can be achieved by commercially available devices such as 1) nebulizers, 2) metered dose inhalers, 3) dry powder inhalers, 4) soft mist inhalers, or by instillation or insufflation, or other mechanisms and/or devices known in the art.
- compositions of this invention may take the form of solutions, gels, ointments, suspensions or solid inserts, formulated so that a unit dosage comprises a therapeutically effective amount of the active component or some multiple thereof in the case of a combination therapy.
- compositions of this invention suitable for parenteral administration comprise a subject composition in combination with one or more pharmaceutically- acceptable sterile isotonic aqueous or non-aqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
- aqueous and non-aqueous carriers examples include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate and cyclodextrins.
- polyols such as glycerol, propylene glycol, polyethylene glycol, and the like
- vegetable oils such as olive oil
- injectable organic esters such as ethyl oleate and cyclodextrins.
- Proper fluidity may be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
- Treatment or amelioration of disease states and pathological conditions that implicate JAK, e.g. JAK2, pathways comprises administering one or more of the disclosed compounds, such as those recited in Formulas I, Ha, lib, Hc, or Hd, or a composition as described herein comprising a disclosed compound.
- the disclosed compounds may inhibit or modulate one or more of the JAK family, e.g, JAKl, JAK2, JAK3, and/or TYK2, and/or may inhibit or modulate KDR.
- the disclosed compounds may for example inhibit JAK2 but may not substantially modulate JAK3 and/or KDR.
- Methods of treating a patient in need thereof e.g. suffering from a disease where inihibition of kinases are useful, for example, immunological and autoimmune disorders, inflammatory disease, diabetes, fibrosis of the liver and/or kidney, atherosclerosis, and ocular diseases are contemplated.
- JAKs appear to play a crucial role in regulating cell behavior induced by a number of cytokines
- treatment of indications driven by a dysregulation of signaling pathways normally associated with cytokine regulation may includecompounds which modulate the activity of the JAKs, such as those recited in Formulas I, Ha, lib, Hc, or Hd is contemplated, such as the treatment of immune and inflammatory diseases, e.g.
- RA rheumatoid arthritis
- COPD chronic obstructive pulmonary disease
- Somatic mutations in the hematopoietic system leading to activation of the JAK pathway has been linked to the myeloproliferative disorders polycythemia vera, essential thrombocythemia and myeloid metaplasia with myelofibrosis.
- upregulation of the JAK pathway may contribute to the myeloproliferative disorders chronic myelogenous leukemia, chronic myelomomocytic leukemia, thallasemia gravis, hypereosinophilic syndrome, and systemic mast cell disease.
- methods for treating cancers e.g.
- cancers are associated with activation of Janus kinases including acute myeloid leukemia, hepatocellular carcinoma, multiple myeloma, Hodgkin's lymphomas and T cell leukemia/lymphoma , wherein the method includes administrating a disclosed compounds.
- ALD Age Related Macular Degeneration
- DME diabetic macular edema
- PDR proliferative diabetic retinopathy
- a method of treating an ocular or other disease includes administration of a disclosed compound that modulates JAK and in some embodiments, inhibits VEGFr. may also be an advantage.
- Also contemplated herein is a method for treating or ameliorating transplant rejection that includes administering an instantly disclosed compound.
- a method for treating or ameliorating rheumatoid arthritis that includes administering an instantly disclosed compound is contemplated.
- Dysregulation in the hematopoietic stem cells of the myeloid compartment may lead to related myeloproliferative disorders (MPDs) including polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF), and to acute myeloid leukemia (AML) Underlying each of these myeloid diseases may be a cytokine-independent activation of molecular signaling pathways critical for the proliferation and aberrant survival of the cells associated with the disease's pathology.
- MPDs myeloproliferative disorders
- PV polycythemia vera
- ET essential thrombocythemia
- MF myelofibrosis
- AML acute myeloid leukemia
- JAK2 V617F Janus kinase 2
- JAK2 T875N tyrosine kinase mutations
- JAK2 activation leads to phosphorylation of signal transducer and activator of transcription (STAT) proteins, transcription factors that stimulate the cell's genetic machinery to induce proliferation and prevent apoptosis.
- STAT signal transducer and activator of transcription
- AML features ligand-independent activation of the JAK-STAT pathway in the majority of patients. Although there is no predominant known mutation that leads to activation of the JAK-STAT pathway in AML, approximately 30% of AML patients appear to have this activation mediated through mutations in the FMS-like receptor tyrosine kinase 3 (FLT3).
- FLT3 FMS-like receptor tyrosine kinase 3
- Methods of treating a patient suffering from acute leukaemias, myeloid and lymphoid malignancies or myeloproliferative disorders such as polycythemia vera, myelofibrosis and essential thrombocythemia are contemplated and may comprise administering an effective amount of a disclosed compound, such as those recited in Formulas I, Ha, lib, lie or Hd, or a composition comprising a disclosed compounds.
- a method of treatment of AML, PV, ET and MT for example, in patients with mutations in FLT3, comprising administering a disclosed compound, e.g. a compound of Formulas I, Ha, lib, lie or Hd.
- Treatment of other cancers comprising administering an effective amount of a disclosed compound.
- the treatment of cancers can include, but are not limited to, an alimentary/gastrointestinal tract cancer, colon cancer, liver cancer, skin cancer, breast cancer, ovarian cancer, prostate cancer, leukemia (including acute myelogenous leukemia and chronic myelogenous leukemia), kidney cancer, lung cancer, muscle cancer, bone cancer, bladder cancer or brain cancer.
- Examples of some additional diseases and disorders that can be treated using a disclosed include cell mediated hypersensitivity (allergic contact dermatitis, hypersensitivity pneumonitis), rheumatic diseases (e.g., systemic lupus erythematosus (SLE), juvenile arthritis, Sjogren's Syndrome, scleroderma, polymyositis, ankylosing spondylitis, psoriatic arthritis), viral diseases (Epstein Barr Virus, Hepatitis B, Hepatitis C, HIV, HTLVl, Vaicella-Zoster Virus, Human Papilloma Virus), food allergy, cutaneous inflammation, and immune suppression induced by solid tumors.
- SLE systemic lupus erythematosus
- rheumatic diseases e.g., systemic lupus erythematosus (SLE), juvenile arthritis, Sjogren's Syndrome, scleroderma, polymyositis, ankylosing spondylitis
- Reverse-phase HPLC chromatography was carried out on Gilson 215 liquid handler equipped with Waters SymmetryShieldTM RP18 7 ⁇ m (40 x 100mm) Prep-Pak cartridge.
- Mobile phase consisted of standard acetonitrile (ACN) and DI Water, each with 0.1% TFA added. Purification was carried out at a flow rate of 4OmL/ min.
- NMR spectra 1 H Nuclear magnetic resonance spectra were recorded at 500 MHz.
- reaction mixture was heated for 2 h at reflux and cooled to room temperature.
- the resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 90:10 gradient) to afford the title compound as yellow oil (0.18 g, 80%).
- the aqueous was extracted with EtOAc (3 x 30 mL). Combined organic layer was dried (Na 2 SO 4 ). The solvent was removed in vacuo.
- the crude product was purified by using ⁇ PLC. The ⁇ PLC fractions containing product were combined and neutralized with saturated NaHCO 3 (30 mL) and extracted with EtOAc (2 x 30 mL). The organic layers were combined and dried (Na 2 SO 4 ). The solvent was removed in vacuo. The title compound (12 mg, 4%) was afforded as a yellow solid.
- the aqueous was extracted with EtOAc (3 x 30 mL). Combined organic layers were dried (Na 2 SO 4 ). The solvent was removed in vacuo.
- the crude product was purified by using ⁇ PLC. The ⁇ PLC fractions containing product were combined and neutralized with saturated NaHCO 3 (30 mL) and extracted with EtOAc (2 x 30 mL). The organic layers were combined and dried (Na 2 SO 4 ). The solvent was removed in vacuo. The title compound (19 mg, 6%) was afforded as a yellow solid.
- the aqueous layer was extracted with EtOAc (3 x 20 rnL). Combined organic layers were dried (Na 2 SO 4 ). The solvent was removed in vacuo.
- the crude product was purified by using HPLC. The HPLC fractions containing product were combined and neutralized with saturated NaHCO 3 (30 mL) and extracted with EtOAc (2 x 30 mL). The organic layers were combined and dried (Na 2 SO 4 ). The solvent was removed in vacuo. The title compound (7 mg, 7%) was afforded as a yellow solid.
- Trifluoromethylsulfonic anhydride was added to a solution of 2-cyclopentylphenol (200 mg, 1.23 mmol), in pyridine (12 rnL) at 0 0 C under constant stirring.
- the reaction mixture was stirred at room temperature for 1 h, diluted with ethyl acetate (100 mL), washed with water (100 mL), IN hydrochloric acid (2 X 50 mL), brine (100 mL), dried (Na 2 SO 4 ), filtered, concentrated and purified using flash chromatography (SiO 2 , hexanes-ethyl acetate) to afford the title compound as a colorless liquid (342 mg, 94%).
- Argon was bubbled into a solution of 14 (100 mg, 0.47 mmol), 27 (209 mg, 0.71 mmol), PdCl 2 (PPh 3 ) 2 (63 mg, 0.09 mmol), cuprous iodide (20 mg, 0.1 mmol), diisopropylethylamine (0.7 mL), tetrabutylammoniumfluoride (123 mg, 0.47 mmol) in dimethyl formamide (4 mL) for 5 min.
- the reaction vessel was sealed, and heated at 150 0 C for 30 min in a microwave reactor.
- the filtrate was concentrated and the residue purified by ⁇ PLC.
- the fractions were combined and poured into saturated NaHCO 3 solution (30 mL).
- the combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na 2 SO 4 and filtered.
- the filtrate was concentrated and the residue re-dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as an off white solid (20 mg, 31%).
- IC 50 values for compounds were determined using a luminescence -based kinase assay with recombinant JAK2, JAK3 and KDR (VEGF r2 ) obtained from Invitrogen.
- JAK2, JAK3 and KDR VEGF r2
- Each well contained 40 ⁇ L of buffer consisting of 40 mM Tris buffer, pH 7.4, containing 50 mM MgCl 2 , 800 ⁇ M EGTA, 350 ⁇ M Triton X-100, 2 mM ⁇ - mercaptoethanol, 250 ⁇ M peptide substrate and an appropriate amount of either JAK2, JAK3 or KDR (75 - 25 ng/well) such that the assay was linear over 60 min.
- the final concentrations of compounds for IC 50 value determinations ranged from 10 to 0.001 ⁇ M by adding the appropriate amount of compound in 2.5 ⁇ L of DMSO; the DMSO present in each assay was constant at 5%.
- the reaction was initiated by the addition of 10 ⁇ L of ATP to a final assay concentration of 3 ⁇ M. After the reaction had proceeded for 60 min, 50 ⁇ L of Kinase-Glo reagent (Promega) was added to terminate the reaction. This solution was then allowed to proceed for an additional 10 min to maximize the luminescence reaction. Values were then measured using an Ultra 384 instrument (Tecan) set for luminosity measurements. Two control reactions were also ran: one reaction containing no compound and the second containing neither inhibitor nor peptide substrate. IC 50 values were derived from experimental data using the non-linear curve fitting capabilities of Prism (Version 4; GraphPad Software). The results, expressed as IC 50 , are presented in Table 1.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Provided herein are pyrrolopyrimide alkynyl compounds, and methods of making and using the same. Such compounds may be used in inflammatory or myeloproliferative disorders. The disclosure also provides for treating cancer.
Description
PYRROLOPYRIMIDINE ALKYNYL COMPOUNDS AND METHODS OF MAKING
AND USING SAME
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of priority under 35 U.S.C. § 119(e) to U.S. Provisional Application No. 60/982,829, filed October 26, 2007, the entire contents of which are incorporated herein by reference.
BACKGROUND [0002] Protein kinases are enzymes that play key roles in signaling pathways since they catalyze the phosphorylation of specific residues leading to the transduction of extra and intra cellular signals, including the action of cytokines on their receptors, growth factors, communication with the nuclei and the triggering of various biological events. In normal cellular physiology, cell cycle control, cell growth, differentiation, apoptosis, mobility, mitogenesis, and various other structural and functional events appear to be mediated by kinases.
[0003] Aberrant kinase activity has been implicated in many diseases including cancers, in immunological and auto-immune disorders, in diabetes, fibrosis of the liver and kidney, atherosclerosis and in ocular diseases. Inhibition of such kinase activity may be beneficial in e.g., the treatment of such diseases.
[0004] The Janus kinases (JAKs) are cellular kinases and consist of four members - JAKl, JAK2, JAK3 and TYK2. The JAKs may play a crucial role in regulating cell behavior induced by a number of cytokines. As such, compounds which modulate the activity of the JAKs have potential utility in several indications driven by a dysregulation of signaling pathways normally associated with cytokine regulation. This includes immune and inflammatory diseases in which dysregulated cytokine pathways are thought to play a roles. In addition, somatic mutations in the hematopoietic system leading to activation of the JAK pathway has been linked to the myeloproliferative disorders, of cells proliferation and in several cells related to several kinds of immune function. Through the angiogenic role of JAK2 downstream of EPO receptors, JAK kinases have been implicated in ocular diseases such as
Age Related Macular Degeneration (AMD), diabetic macular edema (DME) and proliferative diabetic retinopathy (PDR).
[0005] Accordingly, there is a need to develop compounds useful as modulators of kinases, particularly, JAK kinase, given the inadequate treatments available for the aforementioned diseases where the JAK signaling pathway is dysregulated, or recruited directly or indirectly.
SUMMARY
[0006] Provided herein are novel compounds that may inhibit and/or modulate JAK, for example, JAK2. In some embodiments, the disclosed compounds may inhibit or modulate one or more of the JAK family, e.g, JAKl, JAK2, JAK3, and/or TYK2, and/or may inhibit or modulate KDR. Treatment or amelioration of disease states and pathological conditions that implicate JAK, e.g. JAK2, pathways are contemplated herein, and such treatment comprises administering one or more of the disclosed compounds, such as those recited in Formulas I, Ha, lib, lie or Hd, or administering a composition as described herein comprising a disclosed compound. For example, disclosed compounds may have a IC50 against a JAK of less than about 500 nM.
[0007] Also contemplated herein are methods of treating myeloproliferative disorders such as polycythemia vera, myelofibrosis, and essential thrombocythemia by administering disclosed compounds. Additionally, method of treating afflication such as cancer and/or inflammation are contemplated.
DETAILED DESCRIPTION
[0008] The present disclosure is directed in part towards novel compounds and compositions that modulate or inhibit JAK and methods of making and using the same.
[0009] In some embodiments, the disclosed compounds may inhibit or modulate one or more of the JAK family, e.g, JAKl, JAK2, JAK3, and/or TYK2, and/or may inhibit or modulate KDR.
[0010] Before further description of the present invention, certain terms employed in the specification, examples and appended claims are collected here. These definitions should be read in light of the remainder of the disclosure and understood as by a person of skill in the art.
Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by a person of ordinary skill in the art.
[0011] The term "therapeutic agent" is art-recognized and refers to any chemical moiety that is a biologically, physiologically, or pharmacologically active substance that acts locally or systemically in a subject. Examples of therapeutic agents, also referred to as "drugs", are described in well-known literature references such as the Merck Index, the Physicians Desk Reference, and The Pharmacological Basis of Therapeutics, and they include, without limitation, medicaments; vitamins; mineral supplements; substances used for the treatment, prevention, diagnosis, cure or mitigation of a disease or illness; substances which affect the structure or function of the body; or pro-drugs, which become biologically active or more active after they have been placed in a physiological environment.
[0012] The term "therapeutic effect" is art-recognized and refers to a local or systemic effect in animals, particularly mammals, and more particularly humans caused by a pharmacologically active substance. The term thus means any substance intended for use in the diagnosis, cure, mitigation, treatment or prevention of disease or in the enhancement of desirable physical or mental development and/or conditions in an animal or human. The phrase "therapeutically-effective amount" means that amount of such a substance that produces some desired local or systemic effect at a reasonable benefit/risk ratio applicable to any treatment. The therapeutically effective amount of such substance will vary depending upon the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the manner of administration and the like, which can readily be determined by one of ordinary skill in the art. For example, certain compositions of the present invention may be administered in a sufficient amount to produce a at a reasonable benefit/risk ratio applicable to such treatment. [0013] The term "modulation" is art-recognized and refers to up regulation (i.e., activation or stimulation), down regulation (i.e., inhibition or suppression) of a response, or the two in combination or apart.
[0014] A "patient," "subject" or "host" to be treated by the subject method may mean either a human or non-human animal. [0015] The term "treating" is art-recognized and refers to curing as well as ameliorating at least one symptom of any condition or disease.
[0016] The term "prodrug" is art-recognized and is intended to encompass compounds which, under physiological conditions, are converted into the agents of the present invention. A common method for making a prodrug is to select moieties which are hydrolyzed under physiological conditions to provide the desired compound. In other embodiments, the prodrug is converted by an enzymatic activity of the host animal or the target organ or cell.
[0017] The term "alkyl" is art-recognized, and includes saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups. In certain embodiments, a straight chain or branched chain alkyl has about 30 or fewer carbon atoms in its backbone (e.g., C1-C3O for straight chain, C3-C30 for branched chain), and alternatively, about 20 or fewer, e.g. from 1 to 6 carbons. Likewise, cycloalkyls have from about 3 to about 10 carbon atoms in their ring structure, and alternatively about 5, 6 or 7 carbons in the ring structure. The term "alkyl" is also defined to include halosubstituted alkyls.
[0018] Moreover, the term "alkyl" (or "lower alkyl") includes "substituted alkyls", which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone. Such substituents may include, for example, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain may themselves be substituted, if appropriate. For instance, the substituents of a substituted alkyl may include substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl (including phosphonate and phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), -CN and the like. Exemplary substituted alkyls are described below. Cycloalkyls may be further substituted with alkyls, alkenyls, alkoxys, alkylthios, aminoalkyls, carbonyl- substituted alkyls, -CN, and the like. [0019] The term "aralkyl" is art-recognized and refers to an alkyl group substituted with an aryl group (e.g., an aromatic or heteroaromatic group).
[0020] The terms "alkenyl" and "alkynyl" are art-recognized and refer to unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively. The term "alkylene" refers to an organic radical formed from an unsaturated aliphatic hydrocarbon and can be substituted analogous to the alkyls described above; "alkenylene" denotes an acyclic carbon chain which includes a carbon-to-carbon double bond, and can be optionally substituted analogous to the alkyls as described above.
[0021] Unless the number of carbons is otherwise specified, "lower alkyl" refers to an alkyl group, as defined above, but having from one to about ten carbons, alternatively from one to about six carbon atoms in its backbone structure. Likewise, "lower alkenyl" and "lower alkynyl" have similar chain lengths.
[0022] The term "heteroatom" is art-recognized and refers to an atom of any element other than carbon or hydrogen. Illustrative heteroatoms include boron, nitrogen, oxygen, phosphorus, sulfur and selenium. [0023] The term "aryl" is art-recognized and refers to 5-, 6- and 7-membered single-ring aromatic groups that may include from zero to four heteroatoms, for example, benzene, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, triazole, pyrazole, pyridine, pyrazine, pyridazine and pyrimidine, and the like. Those aryl groups having heteroatoms in the ring structure may also be referred to as "heteroaryl" or "heteroaromatics." The aromatic ring may be substituted at one or more ring positions with such substituents as described above, for example, halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, -CF3, -CN, or the like. The term "aryl" also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, e.g., the other cyclic rings may be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls.
[0024] The terms ortho, meta and para are art-recognized and refer to 1,2-, 1,3- and 1,4- disubstituted benzenes, respectively. For example, the names 1,2-dimethylbenzene and ortho- dimethylbenzene are synonymous.
[0025] The terms "heterocyclyl" or "heterocyclic group" are art-recognized and refer to 3- to about 10-membered ring structures, alternatively 3- to about 7-membered rings, whose ring structures include one to four heteroatoms. Heterocycles may also be polycycles. Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxanthene, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, oxolane, thiolane, oxazole, piperidine, piperazine, morpholine, lactones, lactams such as azetidinones and pyrrolidinones, sultams, sultones, and the like. The heterocyclic ring may be substituted at one or more positions with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
[0026] The terms "polycyclyl" or "polycyclic group" are art-recognized and refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings". Rings that are joined through non-adjacent atoms are termed "bridged" rings. Each of the rings of the polycycle may be substituted with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, - CN, or the like. [0027] The term "carbocycle" is art-recognized and refers to an aromatic or non-aromatic ring in which each atom of the ring is carbon.
[0028] The term "nitro" is art-recognized and refers to -NO2; the term "halogen" is art- recognized and refers to -F, -Cl, -Br or -I; the term "sulfhydryl" is art-recognized and refers to -SH; the term "hydroxyl" means -OH; and the term "sulfonyl" is art-recognized and refers to - SO2".
[0029] The terms "amine" and "amino" are art-recognized and refer to both unsubstituted and substituted amines, e.g., a moiety that may be represented by the general formulas:
R50
R50
I +
-N -N- -R53
\
R51 R52
wherein R50, R51 and R52 each independently represent a hydrogen, an alkyl, an alkenyl, - (CH2)m-R61, or R50 and R51, taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure; R61 represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocycle or a polycycle; and m is zero or an integer in the range of 1 to 8. In certain embodiments, only one of R50 or R51 may be a carbonyl, e.g., R50, R51 and the nitrogen together do not form an imide. In other embodiments, R50 and R51 (and optionally R52) each independently represent a hydrogen, an alkyl, an alkenyl, or -(CH2)m- R61. Thus, the term "alkylamine" includes an amine group, as defined above, having a substituted or unsubstituted alkyl attached thereto, i.e., at least one of R50 and R51 is an alkyl group.
[0030] The term "amido" is art recognized as an amino- substituted carbonyl and includes a moiety that may be represented by the general formula:

wherein R50 and R51 are as defined above. Certain embodiments of the amide in the present invention will not include imides which may be unstable.
[0031] The term "acylamino" is art-recognized and refers to a moiety that may be represented by the general formula:
O
-N- -R54
R50
wherein R50 is as defined above, and R54 represents a hydrogen, an alkyl, an alkenyl or - (CH2)m-R61, where m and R61 are as defined above.
[0032] The term "alkylthio" refers to an alkyl group, as defined above, having a sulfur radical attached thereto. In certain embodiments, the "alkylthio" moiety is represented by one of -S-alkyl, -S-alkenyl, -S-alkynyl, and -S-(CH2)m-R61, wherein m and R61 are defined above. Representative alkylthio groups include methylthio, ethyl thio, and the like.
[0033] The term "carbonyl" is art recognized and includes such moieties as may be represented by the general formulas:

wherein X50 is a bond or represents an oxygen or a sulfur, and R55 and R56 represents a hydrogen, an alkyl, an alkenyl, -(CH2)m-R61or a pharmaceutically acceptable salt, R56 represents a hydrogen, an alkyl, an alkenyl or -(CH2)m-R61, where m and R61 are defined above. Where X50 is an oxygen and R55 or R56 is not hydrogen, the formula represents an "ester". Where X50 is an oxygen, and R55 is as defined above, the moiety is referred to herein as a carboxyl group, and particularly when R55 is a hydrogen, the formula represents a "carboxylic acid". Where X50 is an oxygen, and R56 is hydrogen, the formula represents a "formate". In general, where the oxygen atom of the above formula is replaced by sulfur, the formula represents a "thiolcarbonyl" group. Where X50 is a sulfur and R55 or R56 is not
hydrogen, the formula represents a "thiolester." Where X50 is a sulfur and R55 is hydrogen, the formula represents a "thiolcarboxylic acid." Where X50 is a sulfur and R56 is hydrogen, the formula represents a "thiolformate." On the other hand, where X50 is a bond, and R55 is not hydrogen, the above formula represents a "ketone" group. Where X50 is a bond, and R55 is hydrogen, the above formula represents an "aldehyde" group.
[0034] The definition of each expression, e.g. alkyl, m, n, and the like, when it occurs more than once in any structure, is intended to be independent of its definition elsewhere in the same structure.
[0035] Certain compounds contained in compositions of the present invention may exist in particular geometric or stereoisomeric forms. In addition, polymers of the present invention may also be optically active. The present invention contemplates all such compounds, including cis- and trans-isomers, R- and S-enantiomers, diastereomers, (D)-isomers, (L)- isomers, the racemic mixtures thereof, and other mixtures thereof, as falling within the scope of the invention. Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
[0036] If, for instance, a particular enantiomer of compound of the present invention is desired, it may be prepared by asymmetric synthesis, or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers. Alternatively, where the molecule contains a basic functional group, such as amino, or an acidic functional group, such as carboxyl, diastereomeric salts are formed with an appropriate optically- active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means well known in the art, and subsequent recovery of the pure enantiomers. [0037] It will be understood that "substitution" or "substituted with" includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction.
[0038] The term "substituted" is also contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds. Illustrative substituents include, for example, those described herein above. The permissible substituents may be one or more and the same or different for appropriate organic compounds. For purposes of this invention, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. This invention is not intended to be limited in any manner by the permissible substituents of organic compounds.
[0039] For purposes of this invention, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 67th Ed., 1986-87, inside cover. Also for purposes of this invention, the term "hydrocarbon" is contemplated to include all permissible compounds having at least one hydrogen and one carbon atom. In a broad aspect, the permissible hydrocarbons include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic organic compounds that may be substituted or unsubstituted.
[0040] The term "pharmaceutically- acceptable salts" is art-recognized and refers to the relatively non-toxic, inorganic and organic acid addition salts of compounds, including, for example, those contained in compositions of the present invention.
[0041] The term "pharmaceutically acceptable carrier" is art-recognized and refers to a pharmaceutically-acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting any subject composition or component thereof from one organ, or portion of the body, to another organ, or portion of the body. Each carrier must be "acceptable" in the sense of being compatible with the subject composition and its components and not injurious to the patient. Some examples of materials which may serve as pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed
oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid; (16) pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) phosphate buffer solutions; and (21) other non-toxic compatible substances employed in pharmaceutical formulations.
[0042] The terms "systemic administration," "administered systemically," "peripheral administration" and "administered peripherally" are art-recognized and refer to the administration of a subject composition, therapeutic or other material other than directly into the central nervous system, such that it enters the patient's system and, thus, is subject to metabolism and other like processes, for example, subcutaneous administration.
[0043] The term "ocular administration" refers to the administration of a subject composition, therapeutic or other material on or into the eye, including topical and parenteral administration. [0044] "Inhalation administration" or "administered by inhalation" refers to administration of a subject composition, therapeutic or other material by a pulmonary route, e.g. aerosol inhalation or nasal administration.
[0045] The terms "parenteral administration" and "administered parenterally" are art- recognized and refer to modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intra-articulare, subcapsular, subarachnoid, intraspinal, and intrasternal injection and infusion.
Compounds
[0046] Provided herein, in part, is a compound according to formula I:

I [0047] In general, R1 of Formula I is an aryl or heteroaryl optionally substituted on a ring carbon by one, two, three, four, or five substituents, ( e.g. when R1 is monocyclic), or even six, seven or more substitutents (e.g. when R1 is multicylic), each independently selected from the group consisting of hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF3, ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N-alkylsulphamoyl, N-alkylcarbamoyl, -O-alkyl, -S- alkyl, or -R13R11. In some embodiments, Rl can be optionally substituted phenyl, monocyclic heteroaryl, or bicyclic heteroaryl. For example, R1 can be selected, in some embodiments, from the group consisting of optionally substituted indole, optionally substituted benzoimidazole, and optionally substituted phenyl. R4 can be H, NH2, -NHR10 or -NH-alkyl. For example, R4 can be H.
[0048] R1O is phenyl or pyridinyl, wherein R10 is optionally substituted on a ring carbon is optionally substituted on a ring carbon by one, two, or three substituents each independently selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, -N-alkyl-amino, carbamoyl, sulphamoyl, CF3, ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, -OR11, -OR12R11, or -R12R11. In some embodiments, R1O is phenyl.
[0049] R11 is independently selected from aryl, heteroaryl, cycloalkyl and heterocycloalkyl, wherein R11 can be optionally substituted by one to four substituents each independently selected from with halo, alkyl, carbonyl, hydroxyl, alkyl-hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, CF3, ureido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy,
alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, aryl, cycloalkyl, heteroaryl, or heterocycloalkyl;
R12 is alkylene or a bond;
R13 is alkylene, alkenylene, -C(O)-, or a bond. The pharmaceutically acceptable salts , prodrugs, N-oxides, diastereomers and hydrates of disclosed compounds such as those represented by formula I is also contemplated.
[0050] In some embodiments, R1 can be selected from:

wherein
R5, independently for each occurrence, is selected from H, halo, hydroxyl, nitro, cyano, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, -O-alkyl, -S-alkyl, or optionally substituted monocyclic aryl or optionally substituted monocyclic heteraryl.
Re, independently for each occurrence, is selected from H, C1-S straight chain alkyl, or C1 -5 branched alkyl; and
R7, independently for each occurrence is selected from H, C1-S straight chain alkyl, C1-4 branched alkyl, cyano, carboxyl, acyl, aldehyde, oxyalkylnitrile, or alkylamine.
[0051] For example, R1 can be represented by

wherein, for example, R5 is independently, for each occurrence, selected from the group consisting of H, halo, cyano, nitro, C1-S straight chain alkyl, C1-S branched alkyl, -O-alkyl, -S- alkyl, or -C(O)-alkyl. For example, R5 can be CF3, -0-CF3, -C-C(=C)-OH, -C(O)-alkyl-CN.
[0052] In other embodiments R1 can be represented by:

and wherein
Re, independently for each occurrence, is selected from H, C1-S straight chain alkyl, or C1 -5 branched alkyl; and
R7, independently for each occurrence is selected from H, C1-S straight chain alkyl,
C1-4 branched alkyl, halo, cyano, carboxyl, acyl, oxyalkylnitrile, or aldehyde.
[0053] In yet another embodiment, R1 can be represented by:

wherein, for example, R6, independently for each occurrence, is selected from H, C1- 5 straight chain alkyl, or C1-5 branched alkyl; and R7, independently for each occurrence is selected from H, C1-5 straight chain alkyl, C1-4 branched alkyl, cyano, carboxyl, acyl, or aldehyde. [0054] R1O, for example, can be represented in some embodiments by:
[0055]

wherein X is N or CRg; with, for example,
Rg and Rg, independently for each occurrence chosen from H, heterocycle, -O- heterocycle, -alkylene-heterocycle, or -O-alkylene-heterocycle, wherein said heterocycle for each occurrence is optionally substituted with one to three substituents each independently selected from halo, alkyl, carbonyl, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N- alkylsulphamoyl, and N-alkylcarbamoyl. For example the heterocycle, or the heterocycle of: O-heterocycle, -alkylene-heterocycle, or -O-alkylene-heterocycle, can beis substituted with methyl. For example, R8 can be H and R9 can be heterocycle or -O-alkylene-heterocycle, wherein said heterocycle is chosen from: pyrrolidinyl, piperazinyl, imidazoyl, piperdinyl, or morpholinyl. In another exemplary embodiment, Rg is H and Rg is selected from the group
consisting of: methylpiperazine, piperazine, or 2-pyrrolidin-lylethoxy. In some embodiments, at least one occurrence of Rg is H.
[0056] Also contemplated herein are the compounds:





wherein
R4 is H or -NHR10, for example, R4 is H, or R4 can be -N-phenyl, wherein the phenyl is optionally substituted.
[0058] R5 is independently selected for each occurence from the group consisting of H, halo, hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF3, ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N-alkylsulphamoyl, N-alkylcarbamoyl, -O-alkyl, -S-alkyl, -O-CF3, or - R13Rn.
[0059] R6, independently for each occurrence, is selected from H or alkyl;
R7, independently for each occurrence is selected from H, halo, hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF3, ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N- alkylsulphamoyl, N-alkylcarbamoyl;
R1O is phenyl or pyridinyl, wherein R1O is optionally substituted on a ring carbon is optionally substituted on a ring carbon by one, two, or three substituents each independently selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, -N-alkyl-amino, carbamoyl, sulphamoyl, CF3, ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, -OR11, -OR12Rn, or -R12R11; and
R11 is independently selected from aryl, heteroaryl, cycloalkyl and heterocycloalkyl, wherein R11 can be optionally substituted by one to four substituents each independently selected from with halo, alkyl, carbonyl, hydroxyl, alkyl-hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, CF3, ureido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, aryl, -N-alkyl-, cycloalkyl, heteroaryl, or heterocycloalkyl;
R12 is alkylene or a bond;
R13 is alkylene, -SO2-, or a bond; or Pharmaceutically acceptable salts , prodrugs, N-oxides, diastereomers and hydrates thereof.
[0060] R4 may be, for example, selected from the group consisting of:


[0061] For example, exemplary compounds can be represented by:

wherein R5, for each occurrence, is selected from the group consisting of: H, halo, alkyl, nitro, -O-alkyl, -S-alkyl, phenyl, or heterocycle, wherein said phenyl or heterocycle is optionally substituted with one to three substituents each independently selected from halo, alkyl, carbonyl, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, and N- alkylcarbamoyl.
[0062] Other exemplary compounds can be represented by

wherein R6, independently for each occurrence, is selected from H or branched alkyl. R7, can be, for example, independently for each occurrence is selected from H or alkyl. [0063] Other exemplary compounds can be represented by:

Also contemplated herein are the compounds: or pharmaceutically acceptable salts, prodrugs, N-oxides, and hydrates thereof.
[0064] Contemplated herein also compositions that include the disclosed compounds and a pharmaceutically acceptable carrier.
Dosages
[0065] The dosage of any compositions of the present invention will vary depending on the symptoms, age and body weight of the patient, the nature and severity of the disorder to be treated or prevented, the route of administration, and the form of the subject composition. Any of the subject formulations may be administered in a single dose or in divided doses. Dosages for the compositions of the present invention may be readily determined by techniques known to those of skill in the art or as taught herein.
[0066] In certain embodiments, the dosage of the subject compounds will generally be in the range of about 0.01 ng to about 10 g per kg body weight, specifically in the range of about 1 ng to about 0.1 g per kg, and more specifically in the range of about 100 ng to about 10 mg per kg. [0067] An effective dose or amount, and any possible affects on the timing of administration of the formulation, may need to be identified for any particular composition of the present invention. This may be accomplished by routine experiment as described herein, using one or more groups of animals (preferably at least 5 animals per group), or in human trials if appropriate. The effectiveness of any subject composition and method of treatment or prevention may be assessed by administering the composition and assessing the effect of the administration by measuring one or more applicable indices, and comparing the post-treatment values of these indices to the values of the same indices prior to treatment.
[0068] The precise time of administration and amount of any particular subject composition that will yield the most effective treatment in a given patient will depend upon the activity, pharmacokinetics, and bioavailability of a subject composition, physiological condition of the patient (including age, sex, disease type and stage, general physical condition, responsiveness to a given dosage and type of medication), route of administration, and the like. The guidelines presented herein may be used to optimize the treatment, e.g., determining the optimum time and/or amount of administration, which will require no more than routine experimentation consisting of monitoring the subject and adjusting the dosage and/or timing.
[0069] While the subject is being treated, the health of the patient may be monitored by measuring one or more of the relevant indices at predetermined times during the treatment period. Treatment, including composition, amounts, times of administration and formulation, may be optimized according to the results of such monitoring. The patient may be periodically reevaluated to determine the extent of improvement by measuring the same parameters.
Adjustments to the amount(s) of subject composition administered and possibly to the time of administration may be made based on these reevaluations.
[0070] Treatment may be initiated with smaller dosages which are less than the optimum dose of the compound. Thereafter, the dosage may be increased by small increments until the optimum therapeutic effect is attained.
[0071] The use of the subject compositions may reduce the required dosage for any individual agent contained in the compositions because the onset and duration of effect of the different agents may be complimentary.
[0072] Toxicity and therapeutic efficacy of subject compositions may be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 and the ED50.
[0073] The data obtained from the cell culture assays and animal studies may be used in formulating a range of dosage for use in humans. The dosage of any subject composition lies preferably within a range of circulating concentrations that include the ED50 with little or no toxicity. The dosage may vary within this range depending upon the dosage form employed and the route of administration utilized. For compositions of the present invention, the therapeutically effective dose may be estimated initially from cell culture assays.
Formulations
[0074] The compositions of the present invention may be administered by various means, depending on their intended use, as is well known in the art. For example, if compositions of the present invention are to be administered orally, they may be formulated as tablets, capsules, granules, powders or syrups. Alternatively, formulations of the present invention may be administered parenterally as injections (intravenous, intramuscular or subcutaneous), drop infusion preparations, suppositories or administration intranasally (for example, to deliver a dosage to the brain via the nose or to deliver a dosage to the nose directly) or by inhalation (e.g. to treat a condition of the respiratory tract or to pretreat or vaccinate via the respiratory tract). For application by the ophthalmic mucous membrane route, compositions of the present invention may be formulated as eyedrops or eye ointments. These formulations may be prepared by conventional means, and, if desired, the compositions may be mixed with any conventional additive, such as an excipient, a binder, a disintegrating agent, a lubricant, a corrigent, a solubilizing agent, a suspension aid, an emulsifying agent or a coating agent.
[0075] In formulations of the subject invention, wetting agents, emulsifiers and lubricants, such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, release agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants may be present in the formulated agents.
[0076] Subject compositions may be suitable for oral, nasal, topical (including buccal and sublingual), rectal, vaginal, aerosol and/or parenteral administration. The formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. The amount of composition that may be combined with a carrier material to produce a single dose vary depending upon the subject being treated, and the particular mode of administration.
[0077] Methods of preparing these formulations include the step of bringing into association compositions of the present invention with the carrier and, optionally, one or more accessory ingredients. In general, the formulations are prepared by uniformly and intimately bringing into association agents with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
[0078] Formulations suitable for oral administration may be in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in- water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia), each containing a predetermined amount of a subject composition thereof as an active ingredient. Compositions of the present invention may also be administered as a bolus, electuary, or paste.
[0079] In solid dosage forms for oral administration (capsules, tablets, pills, dragees, powders, granules and the like), the subject composition is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example, acetyl alcohol and glycerol monostearate; (8) absorbents, such as kaolin and bentonite clay; (9) lubricants, such a talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof; and (10) coloring agents. In the case of capsules, tablets and pills, the compositions may also comprise buffering
agents. Solid compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
[0080] A tablet may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface- active or dispersing agent. Molded tablets may be made by molding in a suitable machine a mixture of the subject composition moistened with an inert liquid diluent. Tablets, and other solid dosage forms, such as dragees, capsules, pills and granules, may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art.
[0081] Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs. In addition to the subject composition, the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, cyclodextrins and mixtures thereof.
[0082] Suspensions, in addition to the subject composition, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof. [0083] Formulations for rectal or vaginal administration may be presented as a suppository, which may be prepared by mixing a subject composition with one or more suitable non- irritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the body cavity and release the active agent. Formulations which are suitable for vaginal administration also include pessaries, tampons,
creams, gels, pastes, foams or spray formulations containing such carriers as are known in the art to be appropriate.
[0084] Dosage forms for transdermal administration of a subject composition includes powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants. The active component may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants which may be required.
[0085] The ointments, pastes, creams and gels may contain, in addition to a subject composition, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
[0086] Powders and sprays may contain, in addition to a subject composition, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances. Sprays may additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
[0087] Compositions and compounds of the present invention may alternatively be administered by aerosol. This is accomplished by preparing an aqueous aerosol, liposomal preparation or solid particles containing the compound. A non-aqueous (e.g., fluorocarbon propellant) suspension could be used. Sonic nebulizers may be used because they minimize exposing the agent to shear, which may result in degradation of the compounds contained in the subject compositions.
[0088] Ordinarily, an aqueous aerosol is made by formulating an aqueous solution or suspension of a subject composition together with conventional pharmaceutically acceptable carriers and stabilizers. The carriers and stabilizers vary with the requirements of the particular subject composition, but typically include non-ionic surfactants (Tweens, Pluronics, or polyethylene glycol), innocuous proteins like serum albumin, sorbitan esters, oleic acid, lecithin, amino acids such as glycine, buffers, salts, sugars or sugar alcohols. Aerosols generally are prepared from isotonic solutions.
[0089] Dosages for administration by nasal delivery, e.g. delivered to or via the nasal cavity, can be applied as drops, ointments, gels, mists/sprays (aqueous or nonaqueous), aerosols (liquids, suspensions or dry powders), powders, or combinations thereof. Such delivery can be achieved by commercially available devices such as droppers, nasal sprayers, metered dose aerosols, or other mechanisms known in the art. Pharmaceutical formulations for inhalation and/or delivery to the nose, may contain from 1% to 20% by weight of a penetrator enhancer (for example, surfactants, e.g. sugar esters, sugar ethers, carbohydrate esters) which may allow enhanced nose permeability of the active agent.
[0090] Dosages for administration by inhalation or by delivered to or via the lung, can be applied as mists/sprays (aqueous or nonaqueous), aerosols (liquids, suspensions or dry powders),liquids or suspensions (aqueous or nonaqueous), powders, or combinations thereof. Such delivery can be achieved by commercially available devices such as 1) nebulizers, 2) metered dose inhalers, 3) dry powder inhalers, 4) soft mist inhalers, or by instillation or insufflation, or other mechanisms and/or devices known in the art. [0091] For topical ocular administration compositions of this invention may take the form of solutions, gels, ointments, suspensions or solid inserts, formulated so that a unit dosage comprises a therapeutically effective amount of the active component or some multiple thereof in the case of a combination therapy.
[0092] Pharmaceutical compositions of this invention suitable for parenteral administration comprise a subject composition in combination with one or more pharmaceutically- acceptable sterile isotonic aqueous or non-aqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
[0093] Examples of suitable aqueous and non-aqueous carriers which may be employed in the pharmaceutical compositions of the invention include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate and cyclodextrins. Proper fluidity may be maintained, for example, by the use of coating
materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
Methods
[0094] Treatment or amelioration of disease states and pathological conditions that implicate JAK, e.g. JAK2, pathways are contemplated herein, and such treatment comprises administering one or more of the disclosed compounds, such as those recited in Formulas I, Ha, lib, Hc, or Hd, or a composition as described herein comprising a disclosed compound. In some embodiments, the disclosed compounds may inhibit or modulate one or more of the JAK family, e.g, JAKl, JAK2, JAK3, and/or TYK2, and/or may inhibit or modulate KDR. In other embodiments, the disclosed compounds may for example inhibit JAK2 but may not substantially modulate JAK3 and/or KDR.
[0095] Methods of treating a patient in need thereof, e.g. suffering from a disease where inihibition of kinases are useful, for example, immunological and autoimmune disorders, inflammatory disease, diabetes, fibrosis of the liver and/or kidney, atherosclerosis, and ocular diseases are contemplated.
[0096] Because JAKs appear to play a crucial role in regulating cell behavior induced by a number of cytokines, treatment of indications driven by a dysregulation of signaling pathways normally associated with cytokine regulation may includecompounds which modulate the activity of the JAKs, such as those recited in Formulas I, Ha, lib, Hc, or Hd is contemplated, such as the treatment of immune and inflammatory diseases, e.g. rheumatoid arthritis (RA), psoriatic arthritis, asthma, systemic lupus erythematosus, inflammatory bowel disease, multiple sclerosis, type I diabetes mellitus, myasthenia gravis, thyroiditis, myocarditis, psoriasis, immunoglobulin nephropathies, uveitis, iritis, scleritis, conjunctivitis, graft versus host disease and dermatitis. Particularly, methods of treating asthma and/or chronic obstructive pulmonary disease (COPD) are contemplated.
[0097] Somatic mutations in the hematopoietic system leading to activation of the JAK pathway has been linked to the myeloproliferative disorders polycythemia vera, essential thrombocythemia and myeloid metaplasia with myelofibrosis. Similarly, upregulation of the JAK pathway may contribute to the myeloproliferative disorders chronic myelogenous leukemia, chronic myelomomocytic leukemia, thallasemia gravis, hypereosinophilic syndrome, and systemic mast cell disease. Specifically contemplated herein are methods for
treating cancers, e.g. cancers are associated with activation of Janus kinases including acute myeloid leukemia, hepatocellular carcinoma, multiple myeloma, Hodgkin's lymphomas and T cell leukemia/lymphoma , wherein the method includes administrating a disclosed compounds.
[0098] The angiogenic role of JAK2 downstream of the EPO receptor has been implicated in ocular diseases such as Age Related Macular Degeneration (AMD), diabetic macular edema (DME) and proliferative diabetic retinopathy (PDR), and treatement of one or more of these diseases is contemplated. In an embodiment, a method of treating an ocular or other disease is contemplated that includes administration of a disclosed compound that modulates JAK and in some embodiments, inhibits VEGFr. may also be an advantage. [0099] Also contemplated herein is a method for treating or ameliorating transplant rejection that includes administering an instantly disclosed compound.
[0100] In an embodiment, a method for treating or ameliorating rheumatoid arthritis that includes administering an instantly disclosed compound is contemplated.
[0101] Dysregulation in the hematopoietic stem cells of the myeloid compartment may lead to related myeloproliferative disorders (MPDs) including polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF), and to acute myeloid leukemia (AML) Underlying each of these myeloid diseases may be a cytokine-independent activation of molecular signaling pathways critical for the proliferation and aberrant survival of the cells associated with the disease's pathology. For example, a majority of PV, ET, and MF patients harbor an activating valine to phenylalanine point mutation at residue 617 in Janus kinase 2 (JAK2V617F) that known suggest is necessary and sufficient for myeloid expansion and the symptoms manifested by these diseases. Along with JAK2V617F, there are less prevalent tyrosine kinase mutations, all of which constitutively activate JAK2 in these 3 diseases such as JAK2T875N, exon 12 mutations in JAK2, and mutations in the upstream thrombopoietin receptor (MPLW515L/K). Without being bound by any theory, JAK2 activation leads to phosphorylation of signal transducer and activator of transcription (STAT) proteins, transcription factors that stimulate the cell's genetic machinery to induce proliferation and prevent apoptosis. Similarly, AML features ligand-independent activation of the JAK-STAT pathway in the majority of patients. Although there is no predominant known mutation that leads to activation of the JAK-STAT pathway in AML, approximately 30% of AML patients appear to have this activation mediated through mutations in the FMS-like receptor tyrosine
kinase 3 (FLT3). Methods of treating a patient suffering from acute leukaemias, myeloid and lymphoid malignancies or myeloproliferative disorders such as polycythemia vera, myelofibrosis and essential thrombocythemia are contemplated and may comprise administering an effective amount of a disclosed compound, such as those recited in Formulas I, Ha, lib, lie or Hd, or a composition comprising a disclosed compounds. In an embodiment, a method of treatment of AML, PV, ET and MT, for example, in patients with mutations in FLT3, comprising administering a disclosed compound, e.g. a compound of Formulas I, Ha, lib, lie or Hd.
[0102] Treatment of other cancers is contemplated comprising administering an effective amount of a disclosed compound. The treatment of cancers can include, but are not limited to, an alimentary/gastrointestinal tract cancer, colon cancer, liver cancer, skin cancer, breast cancer, ovarian cancer, prostate cancer, leukemia (including acute myelogenous leukemia and chronic myelogenous leukemia), kidney cancer, lung cancer, muscle cancer, bone cancer, bladder cancer or brain cancer. [0103] Also contemplated herein are methods of treating ocular neovasculariaztion, infantile haemangiomas; organ hypoxia, vascular hyperplasia, organ transplant rejection, lupus, multiple sclerosis, rheumatoid arthritis, psoriasis, Type 1 diabetes and complications from diabetes, inflammatory disease, acute pancreatitis, chronic pancreatitis, asthma, allergies, adult respiratory distress syndrome, cardiovascular disease, liver disease, other blood disorders, asthma, rhinitis, atopic, dermatitits, autoimmune tliryroid disorders, ulerative colitis, Crohn's disease, metastatic melanoma, Kaposi's sarcoma, multiple myeloma, conditions associated with cytokines, and other autoimmune diseases including glomerulonephritis,, scleroderma, chronic thyroiditis, Graves' disease, autoimmune gastritis, autoimmune hemolytic anemia, autoimmune neutropenia, thrombocytopenia, atopy (e.g., allergic asthma, atopic dermatitis, or allergic rhinitis), chronic active hepatitis, myasthenia graivs, multiple sclerosis, inflammatory bowel disease, graft vs host disease, neurodegenerative diseases including motor neuron disease, Alzheimer's disease, Parkinson's disease, amyotrophic lateral scelerosis, Huntington's disease, cerebral ischemia, or neurodegenerative disease caused by traumatic injury, strike, gluatamate neurtoxicity or hypoxia; ischemic/reperfusion injury in stroke, myocardial ischemica, renal ischemia, heart attacks, cardiac hypertrophy, atherosclerosis and
arteriosclerosis, organ hyoxia, and platelet aggregation. Such treatment includes administering an effective amount of a disclosed compound.
[0104] Examples of some additional diseases and disorders that can be treated using a disclosed include cell mediated hypersensitivity (allergic contact dermatitis, hypersensitivity pneumonitis), rheumatic diseases (e.g., systemic lupus erythematosus (SLE), juvenile arthritis, Sjogren's Syndrome, scleroderma, polymyositis, ankylosing spondylitis, psoriatic arthritis), viral diseases (Epstein Barr Virus, Hepatitis B, Hepatitis C, HIV, HTLVl, Vaicella-Zoster Virus, Human Papilloma Virus), food allergy, cutaneous inflammation, and immune suppression induced by solid tumors. [0105] The examples which follow are intended in no way to limit the scope of this invention but are provided to illustrate how to prepare and use compounds of the present invention. Many other embodiments of this invention will be apparent to one skilled in the art.
EXAMPLES General Methods [0106] All experiments were performed under anhydrous conditions (i.e. dry solvents) in an atmosphere of argon, except where stated, using oven-dried apparatus and employing standard techniques in handling air- sensitive materials. Aqueous solutions of sodium bicarbonate (NaHCO3) and sodium chloride (brine) were saturated. Analytical thin layer chromatography (TLC) was carried out on Merck Kieselgel 60 F254 plates with visualization by ultraviolet and/or anisaldehyde, potassium permanganate or phosphomolybdic acid dips. Reverse-phase HPLC chromatography was carried out on Gilson 215 liquid handler equipped with Waters SymmetryShield™ RP18 7μm (40 x 100mm) Prep-Pak cartridge. Mobile phase consisted of standard acetonitrile (ACN) and DI Water, each with 0.1% TFA added. Purification was carried out at a flow rate of 4OmL/ min. NMR spectra: 1H Nuclear magnetic resonance spectra were recorded at 500 MHz. Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, dd = doublet of doublets, m = multiplet, br s = broad singlet), coupling constant (J/Hz) and integration. Coupling constants were taken directly from the spectra and are uncorrected. Low resolution mass spectra: Electrospray (ES+) ionization was used. The protonated parent ion (M+H) or fragment of
highest mass is quoted. Analytical gradient consisted of 10% ACN in water ramping up to 100% ACN over 5 min unless otherwise stated.
Example 1 Preparation of 4-(o-Tolylethvnyl)-7H-pyrrolor23-<ilpyrimidine

[0107] To a solution of Pd(PPh3)2Cl2 (0.001 g, 1.6 X 10 v"3J mmol) and triphenylphosphine (0.9 mg, 3.3 X 103 mmol) in TΗF (0.25 mL) and triethylamine (0.4 mL) was added 4-chloro- 7H-pyrrolo[2,3-<i]pyrimidine (0.05 g, 0.33 mmol). The mixture was purged for 15 min with argon and added CuI (0.6 mg, 3.3 X 10"3 mmol) and 2-ethynyltoluene (0.04 g, 0.36 mmol). The reaction mixture was heated for 2 h at reflux and cooled to room temperature. The resulting mixture was concentrated and purified by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated to afford the title compound as a white solid (0.024 g, 32%). [0108] 1H NMR (500 MHz, DMSO-J6): δ 2.56 (s, 3H), 6.69 (dd, J = 3.4, 1.7 Hz, IH), 7.31 (td, J = 6.6, 1.9 Hz, IH), 7.41 (s, IH), 7.39-7.43 (m, IH), 7.68 (d, J= 7.8 Hz, IH), 7.70 (t, J = 2.8 Hz, IH), 8.76 (s, IH), 12.35 (br s, IH); MS (ES+): m/z 234 (M+H)+
Example 2 Preparation of 4-((2-Chlorophenyl)ethvnyl)-7H-pyrrolor2,3-<ilpyrimidine

Hz, IH), 7.55 (td, J = 7.5, 1.5 Hz, IH), 7.68 (dd, J = 8.1, 1.1 Hz, IH), 7.72 (dd, J= 3.4, 2.5 Hz, IH), 7.87 (dd, J = 7.7, 1.7 Hz, IH), 8.79 (s, IH), 12.40 (br s, IH)
[0111] MS (ES+): m/z 255 (M+H)+
Example 3 Preparation of 4-((2-(Trifluoromethyl)phenyl)ethynyl)-7H-pyrrolor2,3- Jlpyrimidine

[0112] To a solution of Pd(PPh3)2Cl2 (0.001 g, 1.6 X 10 mmol) and triphenylphosphine (0.9 mg, 3.3 X 10"3 mmol) in TΗF (0.25 mL) and triethylamine (0.4 mL) was added 4-chloro- 7H-pyrrolo[2,3-<i]pyrimidine (0.05 g, 0.33 mmol). The mixture was purged for 15 min with and added CuI (0.6 mg, 3.3 X 10" mmol) and l-ethynyl-2-(trifluoromethyl)benzene (0.06 g, 0.36 mmol). The reaction mixture was heated for 2 h at reflux and cooled to room temperature. The resulting mixture was concentrated and purified by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated to afford the title compound as a white solid (0.029 g, 31%).
[0113] 1H NMR (500 MHz, DMSO-J6): δ 6.65 (dd, J = 3.5, 1.7 Hz, IH), 7.71-7.74 (m, 2H), 7.81 (t, J = 7.5 Hz, IH), 7.92 (d, J= 7.8 Hz, IH), 8.01 (d, J= 7.6 Hz, IH), 8.80 (s, IH), 12.42 (br s, IH)
[0114] MS (ES+): m/z 288 (M+H)+
Example 4 Preparation of 2-((7H-Pyrrolor23-Jlpyrimidin-4-yl)ethvnyl)aniline

[0115] To a solution of Pd(PPh3)2Cl2 (0.001 g, 1.6 X 10"3 mmol) and triphenylphosphine (0.9 mg, 3.3 X 103 mmol) in THF (0.25 mL) and triethylamine (0.4 mL) was added 4-chloro- 7H-pyrrolo[2,3-<i]pyrimidine (0.05 g, 0.33 mmol). The mixture was purged for 15 min with argon and added CuI (0.6 mg, 3.3 X 10"3 mmol) and 3-ethynylaniline (0.04 g, 0.36 mmol). The reaction mixture was heated for 2 h at reflux and cooled to room temperature. The resulting mixture was concentrated and purified by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated to afford the title compound as a peach solid (0.014 g, 18%).
[0116] 1H NMR (500 MHz, DMSO-J6): δ 5.35 (s, 2H), 6.66 (dd, J = 3.4, 1.5 Hz, IH), 6.69 (dd, J = 7.5, 1.5 Hz, IH), 6.83 (dd, J= 7.6, 1.3 Hz, IH), 6.86 (t, J = 1.8 Hz, IH), 7.12 (t, J = 7.8 Hz, IH), 7.67 (dd, J = 3.3, 2.5 Hz, IH), 8.74 (s, IH), 12.32 (br s, IH)
[0117] MS (ES+): m/z 235 (M+H)+
Example 5 Preparation of 4-((2-Isopropylphenyl)ethvnyl)-7H-pyrrolor23-Jlpyrimidine

[0118] To a solution of Pd(PPh3)2Cl2 (0.001 g, 1.8 X 10"3 mmol) and triphenylphosphine (0.9 mg, 3.6 X 10"3 mmol) in TΗF (0.25 mL) and triethylamine (0.4 mL) was added l-iodo-2- isopropylbenzene (0.09 g, 0.36 mmol). The mixture was purged 15 min with and added CuI (0.7 mg, 3.6 X 103 mmol) and 4-ethynyl-7H-pyrrolo[2,3-d]pyrimidine (0.05 g, 0.35 mmol). The reaction mixture was heated for 2 h at reflux and cooled to room temperature. The resulting mixture was concentrated and purified by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated to afford the title compound as a white solid (0.04 g, 4%).
[0119] 1H NMR (500 MHz, DMSO-J6): δ 1.32 (d, J = 6.9 Hz, 6H), 3.57 (qn, J = 6.9 Hz, IH), 6.66 (dd, J = 3.7, 1.6 Hz, IH), 3.31 (td, J= 7.4, 1.6 Hz, IH), 7.45-7.49 (m, 2H), 7.67-7.71 (m, 2H), 8.75 (s, IH), 12.36 (br s, IH) [0120] MS (ES+): m/z 262 (M+H)+
Example 6 N-fert-Butyl-2-((trimethylsilyl)ethvnyl)benzenesulfonamide (1)

1
[0121] To a solution of Pd(PPh3)2Cl2 (0.002 g, 3.7 X 10 v"3J mmol) and triphenylphosphine (1.9 mg, 7.4 X 10"3 mmol) in THF (0.55 mL) and triethylamine (0.8 mL) was added N-tert- butyl-2-iodobenzenesulfonamide (0.25 g, 0.74 mmol). The mixture was purged for 15 min
with argon and added CuI (1.4 mg, 7.4 X 10~3 mmol) and trimethylsilylacetylene (0.19 g, 0.81 mmol). The reaction mixture was heated for 2 h at reflux and cooled to room temperature. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 90:10 gradient) to afford the title compound as yellow oil (0.18 g, 80%).
[0122] 1K NMR (500 MHz, DMSO-J6): δ 0.25 (s, 9H), 1.13 (s, 9H), 6.62 (s, IH), 7.55-7.59 (m, 2H), 7.60-7.66 (m, IH), 7.92-7.94 (m, IH)
Example 7 Preparation of jY-fe/t-Butyl-2-ethvnylbenzenesulfonamide (2)

[0123] To a solution of 1 (0.18 g, 0.57 mmol) in methanol (0.7 mL) was added potassium hydroxide (3.2 mg, 5.7 X 10" mmol). The solution was stirred 1 h at room temperature and concentrated to afford the title compound as a yellow solid (0.13 g, 93%) without further purification.
[0124] 1H NMR (500 MHz, DMSO-J6): δ 1.11 (s, 9H), 4.54 (s, IH), 7.50-7.57 (m, 3H), 7.63-7.70 (m, IH), 7.92-7.94 (m, IH)
Example 8 Preparation of 2-((7H-Pyrrolor23-Jlpyrimidin-4-yl)ethvnyl)-/V-fe/t- butylbenzenesulfonamide

[0125] To a solution of Pd(PPh3)2Cl2 (0.002 g, 2.3 X 10 mmol) and triphenylphosphine (1.4 mg, 5.3 X 10" mmol) in TΗF (0.35 mL) and triethylamine (0.5 mL) was added 4-chloro- 7H-pyrrolo[2,3-<i]pyrimidine (0.09 g, 0.58 mmol). The mixture was purged for 15 min with
argon and added CuI (1.0 mg, 5.3 X 10~3 mmol) and 2 (0.13 g, 0.53 mmol). The reaction mixture was heated for 2 h at reflux and cooled to room temperature. The resulting mixture was concentrated and purified by preparative HPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated to afford the title compound as a yellow solid (0.018 g, 10%).
[0126] 1H NMR (500 MHz, DMSO-J6): δ 1.18 (s, 9H), 6.84 (dd, J = 3.5, 1.8 Hz, IH), 7.33 (s, IH), 7.67-7.72 (m, 3H), 7.89-7.90 (m, IH), 8.03-8.05 (m, IH), 8.80 (s, IH), 12.28 (br s, IH) [0127] MS (ES+): m/z 355 (M+H)+
Example 9 Preparation of 2-((7H-Pyrrolor2,3-(ilpyrimidin-4-yl)ethvnyl)-N,N- diethylbenzenesulfonamide

[0129] 1H NMR (500 MHz, DMSO-J6): δ 1.01 (t, J = 7.1 Hz, 6H), 3.36 (q, J= 7.1 Hz, 4H), 6.86 (dd, / = 3.2, 1.6 Hz, IH), 7.69-7.79 (m, 3H), 7.97 (d, / = 7.9 Hz, IH), 7.99 (d, / = 7.2 Hz, IH), 8.79 (s, IH), 12.38 (br s, IH) [0130] MS (ES+): m/z 355 (M+H)+
Example 10 Preparation of l-Cvclohexyl-2-iodobenzene (3)

[0132] 1H NMR (500 MHz, CDCl3): δ 1.21-1.30 (m, IH), 1.33-1.53 (m, 4H), 1.76-1.90 (m, 5H), 2.77 (tt, J = 11.7, 3.1 Hz, IH), 6.86 (td, J = 7.7, 1.6 Hz, IH), 7.20 (dd, J= 7.8, 1.6 Hz, IH), 7.27 (q, J = 7.5 Hz, IH), 7.81 (d, J = 7.9 Hz, IH)
Example 11 Preparation of 4-((2-Cyclohexylphenyl)ethynyl)-7H-pyrrolor2,3-(ilpyrimidine

[0133] A solution of Pd(PPh3)2Cl2 (0.022 g, 0.035 mmol), 3 (0.12 g, 0.42 mmol) and triethylamine (0.1 mL, 0.7 mmol) in DMF (2 mL) was purged for 5 min with argon and added CuI (6.7 mg, 0.035 mmol) and 4-ethynyl-7H-pyrrolo[2,3-J]pyrimidine (0.05 g, 0.35 mmol). The reaction mixture was heated for 1 h at 70 0C and cooled to room temperature. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 40:60 gradient) to afford the title compound as yellow oil (0.97 g, 92%).
[0134] 1H NMR (500 MHz, DMSO-J6): δ 1.27-1.32 (m, IH), 1.39-1.55 (m, 8H), 1.74 (d, J = 13.6 Hz, IH), 1.87 (d, J = 13.6 Hz, 4H), 3.20 (t, J = 11.5 Hz, IH), 6.67 (dd, J = 3.4, 1.7 Hz, IH), 7.32 (t, J = 1.5 Hz, IH), 7.43 (d, J = 1.3 Hz, IH), 7.48 (q, J = 1.9 Hz, IH), 7.66 (d, J = 10.9 Hz, IH), 7.73 (t, J = 2.6 Hz, IH), 8.78 (s, IH), 12.31 (br s, IH)
[0135] MS (ES+): m/z 302 (M+H)+
Example 12 Preparation of 4-((2-Iodophenyl)ethvnyl)-7H-pyrrolor23-Jlpyrimidine
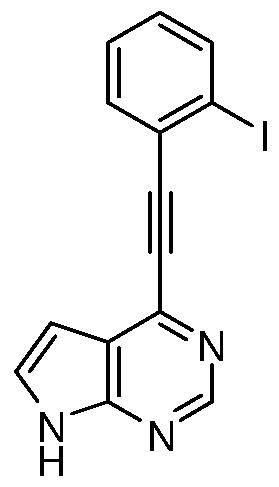
[0136] A solution of Pd(PPh3)2Cl2 (0.022 g, 0.035 mmol), 1,2-diiodobenzene (0.17 g, 0.52 mmol) 4-ethynyl-7H-pyrrolo[2,3-<i]pyrimidine (0.05 g, 0.35 mmol), and triethylamine (0.2 mL) in TΗF (1 mL) was purged for 5 min with argon and added CuI (6.7 mg, 0.035 mmol). The reaction mixture was heated overnight under reflux and cooled to room temperature. The resulting mixture was filtered through a pad of silica gel, concentrated and purified by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated to afford the title compound as a white solid (0.030 g, 25%).
[0137] 1H NMR (500 MHz, DMSO-J6): δ 6.86 (d, J = 2.7 Hz, IH), 7.26 (td, J = 7.8, 1.5 Hz, IH), 7.52 (t, J = 7.7 Hz, IH), 7.71-7.72 (m, IH), 7.78 (dd, J = 7.7, 1.5 Hz, IH), 8.03 (d, J = 7.8 Hz, IH), 8.78 (s, IH), 12.35 (br s, IH)
[0138] MS (ES+): m/z 346 (M+H)+
Example 13 Preparation of l,2-Bis((7H-pyrrolor23-<ilpyrimidin-4-yl)ethvnyl)benzene

[0140] 1H NMR (500 MHz, DMSO- J6): δ 6.90 (t, J = 2.3 Hz, 2H), 7.60 (t, J = 2.6 Hz, 2H), 7.65 (dd, J = 5.8, 3.3 Hz, 2H), 7.92 (dd, J = 5.7, 3.3 Hz, 2H), 8.82 (s, 2H)
[0141] MS (ES+): m/z 361 (M+H)+
Example 14 Preparation of l-(2-((7H-Pyrrolor23-^lpyrimidin-4-yl)ethvnyl)phenyl)ethanol

[0142] A solution of Pd(PPh3)2Cl2 (0.022 g, 0.035 mmol), 2-iodoacetophenone (0.10 g, 0.42 mmol) 4-ethynyl-7H-pyrrolo[2,3-J]pyrimidine (0.05 g, 0.35 mmol), and triethylamine (0.2 mL) in DMF (1 mL) was purged for 5 min with argon and added CuI (6.7 mg, 0.035 mmol). The reaction mixture was heated overnight at 70 0C and cooled to room temperature. The resulting mixture was concentrated and purified by silica gel chromatography (DCM/methanol 100:0 to 85:15 gradient) to afford the title compound as a yellow solid (0.009 g, 10%).
[0143] 1H NMR (500 MHz, DMSO-J6): δ 5.07 (d, J = 3.0 Hz, IH), 5.27 (d, J = 3.0 Hz, IH), 6.97 (dd, J = 3.5, 1.8 Hz, IH), 7.52 (dd, J= 3.4, 2.4 Hz, IH), 7.62 (d, J = 5.8 Hz, IH), 7.64 (d, J = 7.2 Hz, IH), 7.91-7.95 (m, IH), 8.15-8.18 (m, IH), 8.72 (s, IH), 11.99 (br s, IH)
[0144] MS (ES+): m/z 262 (M+H)+
Example 15 Preparation of 2-Chloro-4-((trimethylsilyl)ethynyl)-7H-pyrrolor2,3-(ilpyrimidine
(4)

[0145] To a solution of Pd(PPh3)2Cl2 (0.17 g, 0.27 mmol), 2,4-dichloro-7H-pyrrolo[2,3- d]pyrimidine (0.50 g, 2.66 mmol) in DMF (0.55 mL) was added trimethylsilylacetylene (0.31 g, 3.2 mmol), and triethylamine (0.74 mL, 5.3 mmol). The mixture was purged for 15 min with argon and added CuI (0.051 g, 0.27 mmol). The reaction mixture was heated for 30 min at reflux and cooled to room temperature. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 40:60 gradient) to afford the title compound as a pale yellow solid (0.21 g, 31%).
[0146] 1H NMR (500 MHz, DMSO-J6): δ 0.31 (s, 9H), 6.66 (d, J = 3.4 Hz, IH), 7.71 (d, J = 3.4 Hz, IH), 12.56 (br s, IH)
[0147] MS (ES+): m/z 250 (M+H)+
Example 16 Preparation of 2-Chloro-4-ethvnyl-7H-pyrrolor23-Jlpyrimidine (5)

[0148] To a solution of 4 (0.20 g, 0.8 mmol) in methanol (5 mL) was added potassium hydroxide (0.4 mg, 0.8 X 10" mmol). The solution was stirred 1 h at room temperature and concentrated to afford the title compound as a tan solid (0.15 g, 100%) without further purification.
[0149] 1H NMR (500 MHz, DMSO-J6): δ 5.00 (s, IH), 6.63 (t, J = 3.6 Hz, IH), 7.72 (d, J = 3.5 Hz, IH)
Example 17 Preparation of 2-Chloro-4-((2-isopropylphenyl)ethynyl)-7H-pyrrolor2,3- (jlpyrimidine

[0150] To a solution of Pd(PPh3)2Cl2 (0.049 g, 0.079 mmol), 2-iodoisopropylbenzene (0.29 g, 1.2 mmol) in DMF (3 mL) was added 5 (0.14 g, 0.79 mmol), and triethylamine (0.33 mL, 2.4 mmol). The mixture was purged for 15 min with argon and added CuI (0.051 g, 0.27 mmol). The reaction mixture was heated overnight at 80 0C and cooled to room temperature. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 70:30 gradient) to afford the title compound as orange oil (0.089 g, 38%).
[0151] 1H NMR (500 MHz, DMSO-J6): δ 1.32 (d, J = 6.9 Hz, 6H), 3.54 (qn, J = 6.9 Hz, IH), 6.69-6.70 (m, IH), 7.34 (dd, J = 7.6, 1.3 Hz, IH), 7.46 (d, J = 7.8 Hz, IH), 7.52 (dd, J = 8.0, 6.8 Hz, IH), 7.70 (d, J= 6.9 Hz, IH), 7.74-7.75 (m, IH), 12.59 (br s, IH)
[0152] MS (ES+): m/z 296 (M+H)+
Example 18 Preparation of ((2-Isopropylphenyl)ethvnyl)trimethylsilane (6)

[0153] A solution of Pd(PPh3)2Cl2 (0.26 g, 0.41 mmol), triethylamine (1.7 mL, 12.2 mmol), l-iodo-2-isopropylbenzene (1.0 g, 4.1 mmol), CuI (0.077 g, 0.41 mmol) and trimethylsilylacetylene (0.19 g, 0.81 mmol) in DMF (15 mL) was heated for 30 min at 80 0C. The resulting mixture was concentrated and purified by silica gel chromatography using hexanes to afford the title compound as yellow oil (0.85 g, 97%).
[0154] 1H NMR (500 MHz, DMSO-J6): δ 0.23 (s, 9H), 1.20 (d, J = 7.0 Hz, 6H), 3.35 (qn, J = 6.9 Hz, IH), 7.17 (td, J = 7.3, 1.8 Hz, IH), 7.31-7.39 (m, 3H)
Example 19 Preparation of l-Ethvnyl-2-isopropylbenzene (7)

[0155] To a solution of 6 (1.70 g, 7.9 mmol)) in methanol (30 mL) was added potassium hydroxide (4.4 mg, 7.9 X 10" mmol). The solution was stirred 1 h at room temperature and concentrated to afford the title compound as brown oil (0.63 g, 56%) without further purification.
[0156] 1H NMR (500 MHz, DMSO-J6): δ 1.21 (d, J= 6.9 Hz, 6H), 3.39 (qn, J= 6.9 Hz, IH), 4.34 (s, IH), 7.19 (t, J = 7.19 Hz, IH), 7.34-7.36 (m, 2H), 7.42 (d, J= 8.4 Hz, IH)
Example 20 Preparation of 2,4-Dichloro-7-tosyl-7H-pyrrolor23-<ilpyrimidine (8)

8
[0157] To a solution of 2,4-dichloro-7H-pyrrolo[2,3-J]pyrimidine (1.0 g, 5.3 mmol) in DMF (30 mL) was added NaH (60% in mineral oil, 0.21 g, 5.3 mmol). The mixture was stirred for 5 min at room temperature and added tosyl chloride (1.0 g, 5.3 mmol). The mixture was stirred for 1 h at room temperature, diluted with H2O (100 mL), and filtered. The solid was washed with H2O (20 mL), and dried under vacuum 5 h at 80 0C to afford the title compound as a yellow solid (1.6 g, 90%).
[0158] 1H NMR (500 MHz, DMSO-J6): δ 2.38 (s, 3H), 6.98 (d, J = 4.1 Hz, IH), 7.50 (d, J = 8.3 Hz, 2H), 8.03 (d, J = 8.5 Hz, 2H), 8.12 (d, J = 4.1 Hz, IH)
[0159] MS (ES+): m/z 342 (M+H)+
Example 21 Preparation of 2-Chloro-4-((2-isopropylphenyl)ethvnyl)-7-tosyl-7H-pyrrolor2,3- (jlpyrimidine (9)

[0160] A solution of Pd(PPh3 )2C12 (0.073 g, 0.12 mmol), 7 (0.19 g, 1.3 mmol), 8 (0.40 g, 1.2 mmol), and triethylamine (0.5 mL, 3.5 mmol) in DMF (5 mL) was purged for 5 min with argon and added CuI (0.022g, 0.12 mmol). The reaction mixture was heated for Ih at 80 0C and cooled to room temperature. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 90:10 gradient) to afford the title compound as a yellow solid (0.35 g, 67%).
[0161] 1H NMR (500 MHz, DMSO-J6): δ 1.29 (d, J= 6.9 Hz, 6H), 2.38 (s, 3H), 3.47 (qn, J = 6.8 Hz, IH), 7.01 (d, J = 4.1 Hz, IH), 7.32 (t, J = IA Hz, IH), 7.47 (t, J = 1.6 Hz, IH), 7.50 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 1.1 Hz, IH), 7.73 (d, J = 7.8 Hz, IH), 8.04 (d, J = 8.4 Hz, 2H), 8.12 (d, J = 4.0 Hz, IH) [0162] MS (ES+): m/z 450 (M+H)+
Example 22 Preparation of 4-((2-Isopropylphenyl)ethynyl)-N-(4-moφholinophenyl)-7-tosyl- 7H-pyrrolor2,3-(ilPyrimidin-2-amine (10)

10 [0163] A solution of 9 (0.027 g, 0.06 mmol) and 4-morpholinoaniline (0.021 g, 0.12 mmol) in ra-butanol (01.5 rnL) was heated overnight at reflux. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 65:35 gradient) to afford the title compound as a brown solid (0.0135 g, 38%).
[0164] 1K NMR (500 MHz, DMSO-J6): δ 1.29 (d, J = 6.9 Hz, 6H), 2.33 (s, 3H), 3.10 (t, J = 4.7 Hz, 4H), 3.48 (qn, J = 6.9 Hz, IH), 3.76 (t, J = 4.5 Hz, 4H), 6.75 (d, J = 4.0 Hz, IH), 6.99 (d, J = 9.1 Hz, 2H), 7.30 (td, J = 7.6, 1.3 Hz, IH), 7.39 (d, J= 8.3 Hz, 2H), 7.45 (d, J= 7.0 Hz, IH), 7.50 (q, J = 8.0 Hz, IH), 7.64-7.66 (m, 2H), 7.22 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 8.3 Hz, 2H), 9.77 (s, IH)
[0165] MS (ES+): m/z 592 (M+H)+
Example 23 Preparation of 4-((2-Isopropylphenyl)ethvnyl)-Λ^(4-morpholinophenyl)-7H- pyrrolor2,3-<ilpyrimidin-2-amine
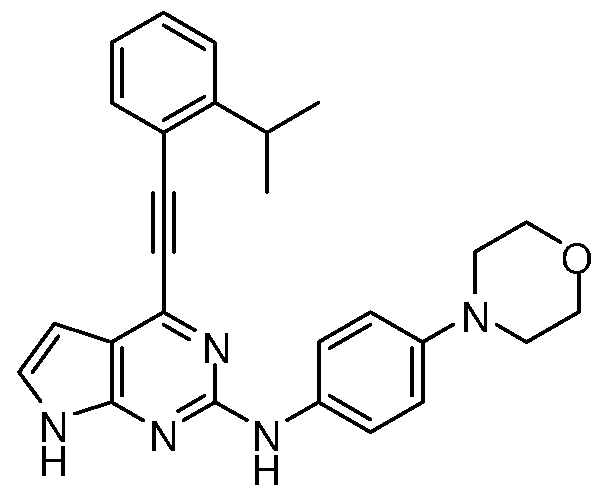
[0167] 1H NMR (500 MHz, DMSO-J6): δ 1.32 (d, J = 6.9 Hz, 6H), 3.02 (t, J = 4.7 Hz, 4H), 3.57 (qn, J = 6.9 Hz, IH), 3.74 (t, J = 4.6 Hz, 4H), 6.43 (dd, J = 3.6, 1.6 Hz, IH), 6.89 (d, J = 9.0 Hz, 2H), 7.24 (dd, J = 3.3, 2.4 Hz, IH), 7.30 (td, J = 7.7, 1.9 Hz, IH), 7.45-7.49 (m, 2H), 7.64 (d, J = 6.4 Hz, IH), 7.69 (d, J = 9.0 Hz, 2H), 9.25 (s, IH), 11.66 (s, IH)
[0168] MS (ES+): m/z 438 (M+H)+
Example 24 Preparation of 4-((2-Isopropylphenyl)ethynyl)-N-(4-(piperidin-4-yloxy)phenyl)- 7H-pyrrolor2,3-(ilPyrimidin-2-amine

[0169] In a microwave vial was sequentially added 9 (0.17 g, 0.37 mmol), Pd(OAc)2 (0.008 g, 0.037 mmol), DMF (3 mL), tert-butyl 4-(4-aminophenoxy)piperidine-l-carboxylate (0.13 g, 0.44 mmol), P(Z-Bu)3 (1 M in toluene, 0.073 mL, 0.73 mmol), and TEA (0.52 mL, 3.7 mmol). The reaction mixture was heated for 20 min at 180 0C in a Biotage microwave reactor. The resulting mixture was added 1 M tetrabutylammonium floride in TΗF (1 mL) and stirred for 30 min under reflux. The resulting mixture was separated by preparative ΗPLC. Fractions that contained the Boc protected intermediate were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was dried over MgSO4, concentrated, and redissolved in CH2Cl2. The solution was added trifluoroacetic acid (0.5 mL) and stirred overnight at room temperature. The resulting mixture was neutralized with saturated Na2CO3, extracted with EtOAc, washed with brine, dried over MgSO4, and concentrated to afford the title compound as a yellow solid (0.022 g, 14%).
[0170] 1H NMR (500 MHz, DMSO- J6): δ 1.32 (d, J = 6.9 Hz, 6H), 1.70- 1.75 (m, 2H), 2.03-2.05 (m, 2H), 2.89-2.95 (m, 2H), 3.08-3.17 (m, 2H), 3.54 (qn, J = 6.9 Hz, IH), 4.46-4.50 (m, IH), 6.44 (dd, J = 3.5, 1.7 Hz, IH), 6.92 (d, J = 9.1 Hz, IH), 7.27 (dd, J = 3.5, 2.1 Hz, IH), 7.31 (td, J= 8.5, 1.7 Hz, IH), 7.45-7.50 (m, 3H), 7.63 (d, J= 7.3 Hz, IH), 7.73 (d, J= 9.1 Hz, 2H), 9.36 (s, IH), 11.68 (s, IH)
[0171] MS (ES+): m/z 452 (M+H)+
Example 25 Preparation of 4-((2-Isopropylphenyl)ethvnyl)-ΛK4-((4-methylpiperazin-l- yl)methyl)phenyl)-7H-pyrrolor2,3-<ilpyrimidin-2-amine hydrochloride

[0172] In a microwave vial was sequentially added 9 (0.17 g, 0.37 mmol), Pd(OAc)2 (0.008 g, 0.037 mmol), DMF (3 mL), tert-butyl 4-(4-aminophenoxy)piperidine-l-carboxylate (0.13 g, 0.44 mmol), P(Z-Bu)3 (1 M in toluene, 0.073 mL, 0.73 mmol), and TEA (0.52 mL, 3.7 mmol). The reaction mixture was heated for 20 min at 180 0C in a Biotage microwave reactor. The resulting mixture was added 1 M tetrabutylammonium floride in TΗF (1 mL) and stirred for 30 min under reflux. The resulting mixture was separated by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was dried over MgSO4, added 4.0 M HCl in 1,4- dioxane (0.5 mL), and concentrated to afford the title compound as a yellow solid (0.016 g).
[0173] 1H NMR (500 MHz, DMSO-J6): δ 1.33 (d, J= 6.9 Hz, 6H), 2.78-2.82 (m, 4H), 3.56-3.62 (m, 5H), 4.30 (br s, 2H), 6.50 (dd, J = 3.5, 1.9 Hz, IH), 7.30 (td, J = 8.8, 1.9 Hz, IH), 7.35 (dd, J = 3.4, 2.3 Hz, IH), 7.46-7.48 (m, 2H), 7.51 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.2 Hz, IH), 7.95 (d, J = 8.5 Hz, 2H), 9.80 (s, IH), 11.88 (s, IH)
[0174] MS (ES+): m/z 465 (M+H)+
Example 26 Preparation of 4-((2-Isopropylphenyl)ethynyl)-/V-(4-(piperidin-l- ylmethyl)phenyl)-7H-pyrrolor23-(ilPyrimidin-2-amine hydrochloride

[0175] In a microwave vial was sequentially added 9 (0.17 g, 0.37 mmol), Pd(OAc)2 (0.008 g, 0.037 mmol), DMF (3 mL), 4-(piperidin-l-ylmethyl)aniline (0.08 g, 0.44 mmol), P(J-Bu)3 (1 M in toluene, 0.073 mL, 0.73 mmol), and TEA (0.52 mL, 3.7 mmol). The reaction mixture was heated for 20 min at 180 0C in a Biotage microwave reactor. The resulting mixture was added I M tetrabutylammonium floride in TΗF (1 mL) and stirred for 30 min under reflux. The resulting mixture was separated by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was dried over MgSO4, added 4.0 M HCl in 1,4-dioxane (0.5 mL), and concentrated to afford the title compound as a yellow solid (0.010 g). [0176] 1H NMR (500 MHz, DMSO-J6): δ 1.32 (d, J = 6.9 Hz, 6H), 1.54-1.80 (m, 6H),
2.76-2.82 (m, 2H), 3.24-3.30 (m, 2H), 3.56 (qn, J = 1.2 Hz, IH), 4.16 (d, J = 5.1 Hz, 2H), 6.50 (dd, J = 3.5, 1.7 Hz, IH), 3.31 (td, J= 9.0, 1.7 Hz, IH), 7.35-7.36 (m, IH), 7.48 (d, J= 8.7 Hz, 2H), 8.46-8.51 (m, 2H), 7.65 (d, J = 7.4 Hz, IH), 7.92 (d, J = 8.6 Hz, 2H), 9.78 (s, IH), 10.30 (s, IH), 11.89 (s, IH) [0177] MS (ES+): m/z 450 (M+H)+
Example 27 Preparation of 4-((2-Isopropylphenyl)ethynyl)-/V-(4-(pyridin-3-yl)phenyl)-7H- pyrrolor2,3-<ilpyrimidin-2-amine hydrochloride

[0178] In a microwave vial was sequentially added 9 (0.17 g, 0.37 mmol), Pd(OAc)2 (0.008 g, 0.037 mmol), DMF (3 mL), 4-(pyridine-3-yl)aniline (0.08 g, 0.44 mmol), P(J-Bu)3 (1 M in toluene, 0.073 mL, 0.73 mmol), and TEA (0.52 mL, 3.7 mmol). The reaction mixture was heated for 20 min at 180 0C in a Biotage microwave reactor. The resulting mixture was added I M tetrabutylammonium floride in TΗF (1 mL) and stirred for 30 min under reflux. The resulting mixture was separated by preparative ΗPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was dried over MgSO4, added 4.0 M HCl in 1,4-dioxane (0.5 mL), and concentrated to afford the title compound as a yellow solid (2.5 mg). [0179] 1H NMR (500 MHz, DMSO-J6): δ 1.34 (d, J = 7.0 Hz, 6H), 3.59 (qn, J = 7.1 Hz, IH), 6.52 (dd, J = 3.5, 1.7 Hz, IH), 7.32 (dd, J= 7.3, 1.8 Hz, IH), 7.38 (dd, J = 3.3, 2.2 Hz, IH), 7.46-7.51 (m, 2H), 7.66 (d, J = 7.3 Hz, IH), 7.85 (d, J = 8.8 Hz, 2H), 8.08 (d, J = 8.8 Hz, 2H), 8.78 (d, J = 5.9 Hz, IH), 8.81 (d, J = 8.4 Hz, IH), 9.22 (s, IH), 9.93 (s, IH), 11.93 (s, IH)
[0180] MS (ES+): m/z 430 (M+H)+
Example 28 Preparation of 7-Bromo-l-isopropyl-2-methyl-lH-indole (11)

11
[0181] A suspension of NaH (6.5 g, 379 mmol, 60% dispersion in mineral oil) in DMF (20 rnL) was slowly treated with a 10 rnL solution of 7-bromo-2-methyl- IH- indole (3 g, 14.3 mmol) in dry DMF. Once addition of indole was complete, 2-bromopropane (14 g, 114 mmol) was then added via syringe. Reaction was then heated to 90 0C for 13 h. Reaction was then cooled to room temperature and poured onto ethyl acetate (100 mL) and washed sequentially with water and brine. Organic phase was dried over sodium sulfate, filtered and evaporated. The crude product was purified by column chromatography to afford the desired product as clear oil (0.13 g, 4%).
Example 29 Preparation of 7-Bromo-l-isopropyl-lH-indole (12)
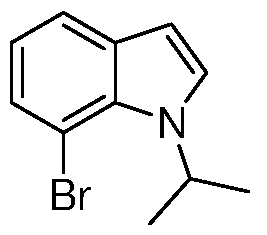
12
[0182] A suspension of NaH (3.05 g, 76 mmol, 60% dispersion in mineral oil) in DMF (25 mL) was slowly treated with a 5 mL solution of 7-bromo- IH- indole (2.5 g, 12.7 mmol) in dry DMF. Once addition of indole was complete, reaction was stirred at room temperature for 45 min. 2-Bromopropane (12.5 g, 102 mmol) was then added via syringe and reaction was stirred for 18 h. Reaction was then poured onto ethyl acetate (200 mL) and washed with water and brine. Organic phase was dried over sodium sulfate, filtered and evaporated. The crude product was purified by column chromatography to afford the desired product as clear oil (3.3 g, 99%).
Example 30 Preparation of 7-Bromo-l-isopropyl-lH-indole-3-carbaldehvde (13)
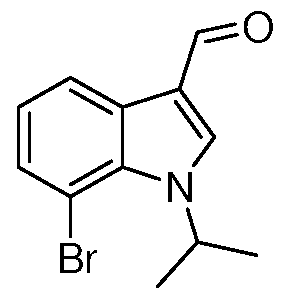
13
[0183] DMF (1 mL) was chilled to zero degrees in ice bath and treated dropwise with POCI3 (0.16 g, 1.05 mmol). Once addition Of POCl3 was complete, reaction mixture removed from ice bath and stirred for 5 min. 12 (0.248 g, 1.05 mmol) was then added via syringe. A
slight exotherm was observed and reaction solidified upon complete addition of indole. Reaction was then heated to 90 0C, stirred for 90 min and then cooled back down to room temperature. Solids quickly formed upon cooling. Reaction was then slowly diluted with ice water (10 mL) and neutralized with saturated sodium acetate (15 mL). pH was adjusted to -10 using 2N NaOH and resulting solids collected by vacuum filtration (0.24 g, 86%).
Example 31 Preparation of 4-Trimethylsilanylethvnyl-7H-pyrrolor23-<ilpyrimidine (14)

14
[0184] A mixture of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (10 g, 65 mmol), ethynyl- trimethyl-silane (9.5 g, 97 mmol), PdCl2(PPh3)2 (4.56 g, 6.5 mmol), CuI (1.2 g, 6.5 mmol) and TEA (195 mL) were suspended in anhydrous DMF (38 mL) and heated at 110 0C for 3 h. The reaction mixture was filtered while still hot and concentrated in vacuo. Resulting residue was taken up in EtOAc (100 mL) and washed with water and brine. Organic phase was evaporated and purified on silica gel column to afford the desired product as a beige solid (9 g, 64%).
Example 32 Preparation of 4-(l-Isopropyl-2-methyl-lH-indol-7-ylethynyl)-7H-pyrrolor2,3- Jlpyrimidine

[0186] 1H NMR (500 MHz, DMSO-J6): δ 1.59 (br s, 6H), 2.58 (br s, 3H), 6.35-6.38 (m, 2H), 6.66 (br s, IH), 7.08 (t, J = 7.6 Hz, IH), 7.47 (d, J = 6.0 Hz, IH), 7.60 (d, J = 7.7 Hz, IH), 7.72 (d, J = 3.4 Hz, IH), 8.78 (s, IH), 12.4 (s, IH)
[0187] MS (ES+): m/z 315 (M+H)+
Example 33 Preparation of l-Isopropyl-7-(7H-pyrrolor2,3-(ilpyrimidin-4-ylethynyl)-lH- indole- 3 -carbaldehyde

[0188] A mixture of 13 (0.132 g, 0.496 mmol), 14 (0.085 g, 0.397 mmol), PdCl2(PPh3)2 (0.028 g, 0.039 mmol) and TBAF (IM in TΗF) (1.2 mL, 1.2 mmol) were suspended in anhydrous TΗF (3 mL) and microwaved at 140 0C for 15 min. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and concentrated in vacuo. The crude product was purified by ΗPLC to afford the title compound (0.028 g, 22%).
[0189] 1H NMR (500 MHz, DMSO-J6): δ 1.63 (d, J = 6.5 Hz, 6H), 5.90-5.95 (m, IH), 6.70 (d, J = 3.2 Hz, IH), 7.37 (t, J = 7.8 Hz, IH), 7.72-7.74 (m, 2H), 8.32 (d, J = 7.9 Hz, IH), 8.65 (s, IH), 8.81 (s, IH), 9.99 (s, IH), 12.4 (s, IH)
[0190] MS (ES+): m/z 329 (M+H)+
Example 34 Preparation of l-(7-Bromo-l-isopropyl-lH-indol-3-yl)-ethanone (15)

15
[0191] A solution of ZnCl2 (0.181 g, 1.33 mmol) in DCM (4 rnL) was treated with acetyl chloride (0.109 g, 1.39 mmol). 12 (0.3 g, 1.27 mmol) was then added in one portion resulting in a pink solution. After stirring at room temperature for 4 h, reaction was diluted with DCM (25 mL) and washed with saturated NH4Cl. Solvents were removed and resulting residue purified by column chromatography to afford the title compound as a white solid (0.155 g, 44%).
Example 35 Preparation of l-ri-Isopropyl-7-(7H-pyrrolor23-Jlpyrimidin-4-ylethvnyl)-lH- indol- 3 - yll -ethanone

[0192] A mixture of 14 (0.132 g, 0.616 mmol), 15 (0.15 g, 0.536 mmol), PdCl2(PPh3)2 (0.038 g, 0.054 mmol) and TBAF (IM in TΗF) (1.6 mL, 1.6 mmol) were suspended in anhydrous TΗF (3 mL) and microwaved at 160 0C for 15 min. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and concentrated in vacuo. The crude product was purified by ΗPLC to afford the title compound (0.017 g, 9%).
[0193] 1H NMR (500 MHz, DMSO-J6): δ 1.63 (d, J = 6.7 Hz, 6H), 2.51 (s, 3 H), 5.93-5.98 (m, IH), 6.69-6.70 (m, IH), 7.32 (t, J = 1.6 Hz, IH), 7.67 (d, J = 1.3 Hz, IH), 7.73 (t, J = 2.7 Hz, IH), 8.42 (d, J = 7.9 Hz, IH), 8.60 (s, IH), 8.80 (s, IH), 12.4 (s, IH)
[0194] MS (ES+): m/z 343 (M+H)+
Example 36 Preparation of 4-(l-Isopropyl-lH-indol-7-ylethynyl)-7H-pyrrolor2,3- (jlpyrimidine
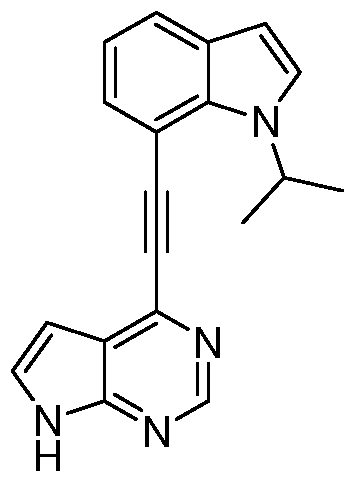
[0195] A mixture of 12 (0.242 g, 1.02 mmol), 4-ethynyl-7H-pyrrolo[2,3- d]pyrimidine (0.183 g, 1.28 mmol), PdCl2(PPh3)2 (0.072 g, 0.102 mmol), CuI (0.019g, 0.102 mmol) and TEA (3 mL) were suspended in anhydrous DMF (0.6 mL) and microwaved at 140 0C for 15 min. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and concentrated in vacuo. The crude product was purified by ΗPLC to afford the title compound (0.09 g, 3%).
[0196] 1H NMR (500 MHz, DMSO-J6): δ 1.55 (d, J = 6.7 Hz, 6H), 5.91-5.97 (m, IH), 6.63 (d, J = 3.3 Hz, IH), 6.66 (d, J = 3.5 Hz, IH), 7.12 (t, J = 7.7 Hz, IH), 7.54 (d, J = 7.4 Hz, IH), 7.67 (d, J = 3.3 Hz, IH), 7.71 (d, J = 3.5 Hz, IH), 7.74 (d, J = 6.8 Hz, IH), 8.79 (s, IH), 12.4 (s, IH)
[0197] MS (ES+): m/z 301 (M+H)+
Example 37 Preparation of l-Isopropyl-7-trimethylsilanylethvnyl-lH-indole (16)

16
[0198] A mixture of 12 (2.5 g, 11 mmol), ethynyl-trimethyl-silane (1.55 g, 16 mmol), PdCl2(PPh3)2 (0.74 g, 1.05 mmol), CuI (0.2 g, 1.05 mmol) and TEA (30 mL) were suspended in anhydrous DMF (6 mL) and heated at 110 0C for 3 h. The reaction mixture was filtered while still hot and concentrated in vacuo. Resulting residue was taken up in EtOAc (100 mL)
and washed with water and brine. Organic phase was evaporated and purified on silica gel column to afford the desired product as amber oil (0.617 g, 23%).
Example 38 Preparation of S-Bromo-l-isopropyl^-trimethylsilanylethvnyl- IH- indole (17)

[0199] A solution of 16 (0.55 g, 2.15 mmol) in acetonitrile (8 mL) was treated with NBS (0.422 g, 2.37 mmol) and microwaved at 100 0C for 10 min. Reaction solvents were removed and crude adsorbed onto Celite. The crude product was purified by column chromatography to afford the title compound as amber oil (0.72 g, 99%).
Example 39 Preparation of l-Isopropyl-7-trimethylsilanylethvnyl-lH-indole-3-carboxylic acid (18)

18
[0200] A solution of 17 (0.3 g, 0.9 mmol) in TΗF (5 mL) was chilled to -78 0C and treated slowly with nBuLi (1.6M in Ηexanes) (0.96 mL, 1.53 mmol). Several pieces of dry ice (CO2) were then added to the reaction mixture and subsequently allowed to come to room temperature. After 1 h, reaction was poured onto ice-cold IN HCl (-20 mL) and extracted with EtOAc (30 mL). Organic phase was dried and evaporated. Crude residue was purified by column chromatography to afford title compound as greenish oil that solidified upon standing (0.184 g, 68%).
Example 40 Preparation of l-Isopropyl-7-(7H-pyrrolor23-<ilpyrimidin-4-ylethynyl)-lH- indole-3-carboxylic acid
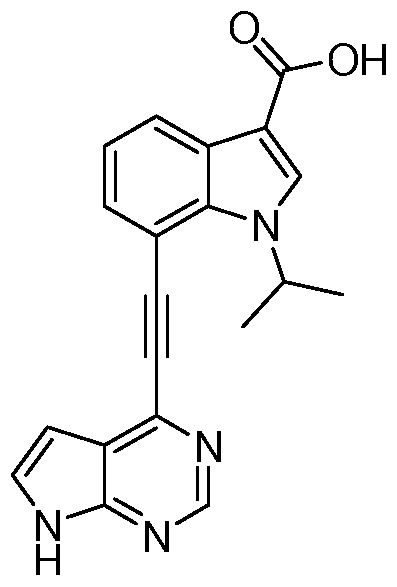
[0201] A mixture of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (0.071 g, 0.464 mmol), 18
(0.111 g, 0.371 mmol), PdCl2(PPh3)2 (0.026 g, 0.037 mmol) and TBAF (IM in TΗF) (1.1 mL, 1.1 mmol) were suspended in anhydrous TΗF (3 mL) and microwaved at 160 0C for 15 min. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and concentrated in vacuo. The crude product was purified by ΗPLC to afford the title compound (0.066 g, 52%).
[0202] 1H NMR (500 MHz, DMSO-J6): δ 1.60 (d, J = 6.7 Hz, 6H), 5.91-5.96 (m, IH), 6.70-6.71 (m, IH), 7.31 (t, J = 7.8 Hz, IH), 7.67 (d, J= 7.4 Hz, IH), 7.74 (t, J= 2.7 Hz, IH), 8.25 (d, J = 8.0 Hz, IH), 8.29 (s, IH), 8.82 (s, IH), 12.4 (s, IH)
[0203] MS (ES+): m/z 345 (M+H)+
Example 41 Preparation of 4-(2-Ethyl-phenylethvnyl)-7H-pyrrolor2,3-<ilpyrimidine
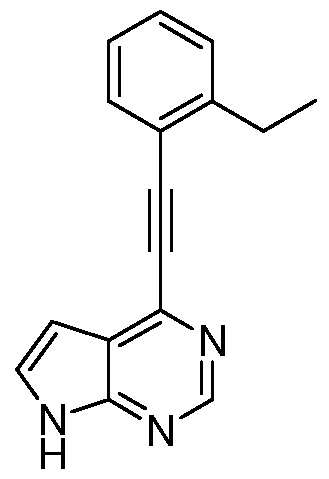
[0205] 1H NMR (500 MHz, DMSO-J6): δ 1.29 (t, J = 7.5 Hz, 3H), 2.93 (q, J = 7.6 Hz, 2H), 6.66-6.67 (m, IH), 7.32 (t, J= 7.5 Hz, IH), 7.40-7.47 (m, 2H), 7.67-7.70 (m, 2H), 8.77 (s, IH), 12.3 (s, IH)
[0206] MS (ES+): m/z 248 (M+H)+
Example 42 Preparation of l-Bromo-2-isopropoxy-benzene (19)

19
[0207] A solution of 2-bromo-phenol (2.8 g, 16 mmol) in acetone (60 mL) was treated with Cs2CO3 (11.6 g, 36 mmol), 2-bromopropane (8 g, 65 mmol) and refluxed for 2 h. Reaction solvents were removed and residue taken up in EtOAc (100 mL) and washed with water and brine. Organic phase was evaporated to afford the title compound as clear oil (2.9 g, 83%).
Example 43 Preparation of 4-(2-Isopropoxy-phenylethvnyl)-7H-pyrrolor2,3-<ilpyrimidine
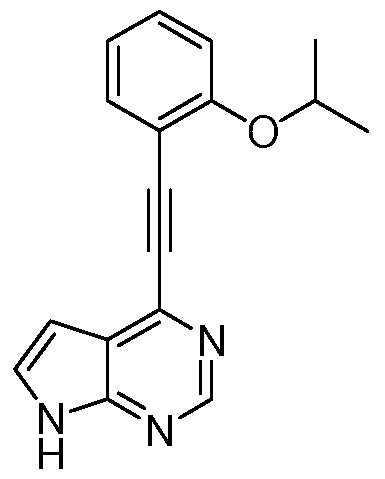
[0209] 1H NMR (500 MHz, DMSO-J6): δ 1.37 (d, J = 6.1 Hz, 6H), 4.75-4.80 (m, IH), 6.67 (d, J = 3.5 Hz, IH), 7.01 (t, J = 7.4 Hz, IH), 7.17 (d, J = 8.4 Hz, IH), 7.44-7.47 (m, IH), 7.61- 7.63 (m, IH), 7.69 (d, J = 3.5 Hz, IH ), 8.75 (s, IH), 12.3 (s, IH)
[0210] MS (ES+): m/z 278 (M+H)+
Example 44 Preparation of ri-Isopropyl-7-(7H-pyrrolor23-Jlpyrimidin-4-ylethvnyl)-lH- indol-3-ylmethyll-dimethyl-amine

[0211] l-Isopropyl-7-(7Η-pyrrolo[2,3-d]pyrimidin-4-ylethynyl)- lΗ-indole-3-carbaldehyde (0.055 g, 0.168 mmol) was suspended in anhydrous methanol (3 mL) and treated with HOAc (0.3 mL), dimethylamine (2M solution in THF) (0.4 mL, 0.335 mmol) and NaBH3CN (0.063 g, 0.672 mmol). This was then microwaved at 120 0C for 15 min. The reaction mixture was cooled to room temperature and syringe filtered. The crude product was purified by HPLC to afford the title compound (0.06 g, 10%).
[0212] 1H NMR (500 MHz, DMSO-J6): δ 1.54 (d, J= 6.6 Hz, 6H), 2.17 (s, 6H), 3.58 (s, 2H), 5.88-5.94 (m, IH), 6.67 (d, J = 3.5 Hz, IH), 7.11 (t, J = 7.5 Hz, IH), 7.53-7.55 (m, 2H), 7.71 (d, J = 3.5 Hz, IH), 7.81 (d, J = 8.0 Hz, IH), 8.78 (s, IH), 12.3 (s, IH)
[0213] MS (ES+): m/z 358 (M+H)+
Example 45 Preparation of 4-(2,6-Diethyl-phenylethvnyl)-7H-pyrrolor23-Jlpyrimidine

[0214] A mixture of 14 (0.1474 g, 0.684 mmol), 2-bromo-l,3-diethyl-benzene (0.218 g, 1.03 mmol), PdCl2(PPh3)2 (0.048 g, 0.068 mmol) and TBAF (IM in TΗF) (2 mL, 2 mmol) were suspended in anhydrous TΗF (1 mL) and microwaved at 160 0C for 15 min. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and concentrated in vacuo. The crude product was purified by column chromatography to afford the title compound (0.028 g, 15%).
[0215] 1H NMR (500 MHz, DMSO-J6): δ 1.28 (d, J = 7.5 Hz, 6H), 2.94 (q, J = 7.6 Hz, 4H), 6.61-6.62 (m, IH), 7.23 (d, J = 7.7 Hz, 2H), 7.35-7.38 (m, IH), 7.70-7.71 (m, IH), 8.78 (s, IH), 12.3 (s, IH)
[0216] MS (ES+): m/z 276 (M+H)+
Example 46 Preparation of 3-(7-Bromo-l-isopropyl-lH-indol-3-yl)-3-oxopropanenitrile (20)

[0217] A mixture of 12 (148 mg, 0.62 mmol) and 2-cyanoacetic acid (64 mg, 0.75 mmol) in acetic anhydride (5 mL) was heated under reflux for 30 min. The solvent was removed in vacuo and to the residue was added EtOAc (20 mL) and saturated NaHCO3 (20 mL). The mixture was sonicated and organic layer separated. The aqueous layer was extracted with EtOAc (10 mL x X). The combined organic layer was dried (Na2SO4). The solvent was removed in vacuo and the crude product used in the next step without further purification.
Example 47 Preparation of 3-(7-(2-(7H-Pyrrolor23-Jlpyrimidin-4-yl)ethynyl)-l-isopropyl- lH-indol-3-yl)-3-oxopropanenitrile

[0218] To a solution of 14 (183 mg, 0.85 mmol) in DMF (10 rnL) was added 20 (0.85 mmol) and 1.0 M solution of tetrabutylammonium fluoride in TΗF (3 rnL, 3 mmol). The solution was bubbled with argon for 1 min followed by adding dichlorobis(triphenylphosphine)-palladium (II) (Pd (Ph3P)2Cl2, 70 mg, 0.1 mmol). The reaction mixture was heated at 150 0C for 30 min under μ-wave. The hot solution was filtered and the solid was washed with EtOAc. The filtrate was washed with brine (50 mL). The aqueous was extracted with EtOAc (3 x 30 mL). Combined organic layer was dried (Na2SO4). The solvent was removed in vacuo. The crude product was purified by using ΗPLC. The ΗPLC fractions containing product were combined and neutralized with saturated NaHCO3 (30 mL) and extracted with EtOAc (2 x 30 mL). The organic layers were combined and dried (Na2SO4). The solvent was removed in vacuo. The title compound (12 mg, 4%) was afforded as a yellow solid.
[0219] 1H NMR (500 MHz, DMSO-J6): δ 1.62 (d, J = 6.5 Hz, 6H), 2.43 (s, 2H), 5.93-5.98 (m, IH), 6.70-6.72 (m, IH), 7.39 (br s, IH), 7.62 (br s, IH), 7.73-7.75 (m, 2H), 8.37 (d, J = 7.8 Hz, IH), 8.81 (s, IH), 12.37 (s, IH)
[0220] MS (ES+): m/z 368 (M+H)+
Example 48 Preparation of 7-Bromo-3-chloro-l-isopropyl-lH-indole (21)

21
[0221] A solution of 12 (243 mg, 1.02 mmol) in diethyl ether (10 rnL) was cooled in ice- water and added sulfuryl chloride (0.16 rnL, 2.0 mmol). The mixture was stirred for 10 min at 0 0C before adding water (10 mL). The organic layer was separated and aqueous layer extracted with EtOAc (10 mL x X). The combined organic layers were dried (Na2SO4). The solvent was removed in vacuo and the crude product used in the next step without further purification.
Example 49 Preparation of 4-(2-(3-Chloro-l-isopropyl-lH-indol-7-yl)ethvnyl)-7H- pyrrolor2,3-<ilpyrimidine

[0222] To a solution of 14 (215 mg, 1.0 mmol) in DMF (10 mL) was added 21 (1.0 mmol) and 1.0 M solution of tetrabutylammonium fluoride in TΗF (4 mL, 4 mmol). The solution was bubbled with argon for 1 min followed by adding dichlorobis(triphenylphosphine)- palladium (II) (Pd (Ph3P)2Cl2, 70 mg, 0.1 mmol). The reaction mixture was heated at 150 0C for 30 min under //-wave. The hot solution was filtered and the solid washed with EtOAc. The filtrate was washed with brine (50 mL). The aqueous was extracted with EtOAc (3 x 30 mL). Combined organic layers were dried (Na2SO4). The solvent was removed in vacuo. The crude product was purified by using ΗPLC. The ΗPLC fractions containing product were combined and neutralized with saturated NaHCO3 (30 mL) and extracted with EtOAc (2 x 30 mL). The organic layers were combined and dried (Na2SO4). The solvent was removed in vacuo. The title compound (19 mg, 6%) was afforded as a yellow solid.
[0223] 1H NMR (500 MHz, DMSO-J6): δ 1.55 (d, J = 6.6 Hz, 6H), 5.92-5.97 (m, IH), 6.68-6.69 (m, IH), 7.26 (t, J = 7.6 Hz, IH), 7.65-7.69 (m, 2H), 7.72-7.74 (m, IH), 7.92 (s, IH), 8.80 (s, IH), 12.41 (s, IH)
[0224] MS (ES+): m/z 335 (M+H)+
Example 50 Preparation of 7-Bromo-l-isopropyl-lH-indole-3-carbonitrile (22)

22
[0225] A mixture of 13 (133 mg, 0.5 mmol) and hydroxylamine hydrochloride (450 mg, 6.4 mmol) was heated at 150 0C for 4 h. After reaction mixture was cooled, EtOAc (20 mL) and water (20 mL) were added and sonicated. The organic layer was separated and aqueous layer extracted with EtOAc (10 mL x X). The combined organic layer was dried (Na2SO4). The solvent was removed in vacuo. The crude product was purified by flash column (SiO2/20% EtOAc in Ηexanes) and afforded title compound (76 mg, 58%) as yellow solid.
Example 51 Preparation of 7-(2-(7H-Pyrrolor23-<ilpyrimidin-4-yl)ethynyl)- 1-isopropyl- IH- indole- 3 -carbonitrile
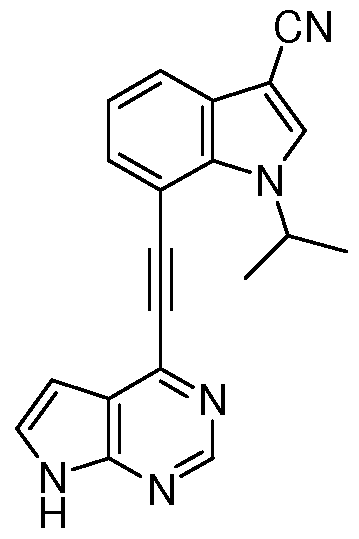
[0226] To a solution of 14 (62 mg, 0.3 mmol) in DMF (10 mL) was added 22 (76 mg, 0.3 mmol) and 1.0 M solution of tetrabutylammonium fluoride in TΗF (1.2 mL, 1.2 mmol). The solution was bubbled with argon for 1 min followed by adding dichlorobis(triphenyl- phosphine)palladium (II) (Pd (Ph3P)2Cl2, 21 mg, 0.03 mmol). The reaction mixture was heated at 150 0C for 30 min under //-wave. The hot solution was filtered and the solid washed with
EtOAc. The filtrate was washed with brine (50 rnL). The aqueous layer was extracted with EtOAc (3 x 20 rnL). Combined organic layers were dried (Na2SO4). The solvent was removed in vacuo. The crude product was purified by using HPLC. The HPLC fractions containing product were combined and neutralized with saturated NaHCO3 (30 mL) and extracted with EtOAc (2 x 30 mL). The organic layers were combined and dried (Na2SO4). The solvent was removed in vacuo. The title compound (7 mg, 7%) was afforded as a yellow solid.
[0227] 1H NMR (500 MHz, DMSO-J6): δ 1.60 (d, J = 6.7 Hz, 6H), 5.91-5.96 (m, IH), 6.70 (d, J = 3.5 Hz, IH), 7.39 (t, J = 7.6 Hz, IH), 7.74 (d, J = 3.5 Hz, IH), 7.77 (d, J = 7.5 Hz, IH), 7.84 (d, J = 7.5 Hz, IH), 8.65 (s, IH), 8.81 (s, IH), 12.41 (s, IH)
[0228] MS (ES+): m/z 326 (M+H)+
Example 52 Preparation of fe^Butyir(lZ)-r(fe/t-butoxycarbonyl)aminol { \ A-(A- methylpiperazin- 1 - vPphenyll amino I methylidenel carbamate (23)

23
[0229] A mixture of (fer^-butoxycarbonylimino-pyrazol-l-yl-methy^-carbamic acid tert- butyl ester (3.1 g, 10.0 mmol) and 4-(4-methylpiperazin-l-yl)benzenamine (1.9 g, 10.0 mmol) in DMF (10 mL) was stirred at room temperature for 17 h. The reaction mixture was concentrated and the residue taken in water (50 mL). The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as an off white solid. The material was used in the next step without purification.
Example 53 Preparation of (4-(4-Methylpiperazin-l-yl)phenyl)guanidine TFA salt (24)

24
[0230] A solution of 23 in 50% TFA/DCM (10 niL) was stirred at room temperature for 17 h. The solution was concentrated to afford the title compound (4.6 g, 99%), which was used in the next step without purification.
Example 54 Preparation of 2-(4-(4-Methylpiperazin-l-yl)phenylamino)-7H-pyrrolor2,3- (ilpyrimidin-4-ol (25)

25
[0231] To a microwave reaction tube was charged with 24 (2.3 g, 5.0 mmol), 2-cyano-4,4- diethoxy-butyric acid ethyl ester (2.3 g, 10.0 mmol) and NaOMe (25% by wt in MeOH; 12 mL) in EtOH (5 mL). The reaction tube was sealed and the solution irradiated with microwave at 160 0C for 30 min. After cooling to room temperature, the mixture was concentrated. The residue was taken up in water (10 mL) and the pH adjusted to 1 with 6M of HCl. The resulting solution was stirred at room temperature for 25 min and then the pH adjusted to 9 with concentrated 10% NaOH. The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as a brown solid (1.6 g, 96%). The material was used in the next step without purification.
Example 55 Preparation of 4-Chloro-Λ^(4-(4-methylpiperazin-l-yl)phenyl)-7H-pyrrolor23- Jlpyrimidin-2-amine (26)

[0232] A solution of 25 (1.6 g, 4.8 mmol) in POCl3 (5 mL) was heated at 120 0C for 20 min. After cooling to room temperature, the excess POCl3 was removed in vacuo. To the resulting dark residue was added ice water slowly and the pΗ was adjusted to 9-10 with 10% NaOH. The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as a brown solid (1.1 g, 69%). The material was used in the next step without purification.
Example 56 Preparation of N-(4-(4-Methylpiperazin-l-yl)phenyl)-4-(2-o-tolylethynyl)-7H- pyrrolor2,3-<ilpyrimidin-2-amine

[0233] To a solution of 26 (88 mg, 0.25 mmol) in DMF (5 rnL) was added Et3N (5 rnL), triphenylphosphine (26 mg, 0.1 mmol), CuI (10 mg, 0.05 mmol), and dichlorobis(triphenylphosphine)palladium (II) (Pd (Ph3P)2Cl2, 35 mg, 0.05 mmol). The solution was bubbled with argon for 1 min followed by adding l-ethynyl-2-methylbenzene (45 mg, 0.38 mmol). The reaction mixture was heated at 150 0C for 30 min under //-wave. The hot solution was filtered and the solid washed with EtOAc. The filtrate was washed with brine (50 mL). The aqueous layer was extracted with EtOAc (3 x 20 mL). Combined organic layers were dried (Na2SO4). The solvent was removed in vacuo. The crude product was purified by using ΗPLC. The ΗPLC fractions containing product were combined and neutralized with saturated NaHCO3 (30 mL) and extracted with EtOAc (2 x 30 mL). The organic layers were combined and dried (Na2SO4). The solvent was removed in vacuo. The residue was dissolved in MeOH (2 mL), and 4.0 M HCl solution (0.2 mL, 0.8 mmol) in dioxane was added. The solution was stirred for 5 min at room temperature and then solvent removed in vacuo. The residue was dissolved in MeOH (1 mL), and anhydrous Et2O (20 mL) was added. The solid was collected by centrifuging. The title compound (26 mg, 23%) was afforded as a yellow solid.
[0234] 1H NMR (500 MHz, DMSO-J6): δ 2.56 (s, 3H), 2.81 (d, J = 2.9 Hz, 3H), 3.06-3.11 (m, 2H), 3.14-3.20 (m, 2H), 3.48 (d, J = 11.5 Hz, 2H), 3.73 (d, J = 12.7 Hz, 2H), 6.54-6.55 (m, IH), 7.00 (d, J = 8.9 Hz, 2H), 7.31-7.35 (m, 2H), 7.40-7.46 (m, 2H), 7.66-7.69 (m, 3H), 9.55 (br s, IH), 10.93 (br s, IH), 11.94 (s, IH) [0235] MS (ES+): m/z 423 (M+H)+
Example 57 Preparation of 2-Cvclopentylphenyltrifluoromethanesulfonate (27)

[0236] Trifluoromethylsulfonic anhydride was added to a solution of 2-cyclopentylphenol (200 mg, 1.23 mmol), in pyridine (12 rnL) at 0 0C under constant stirring. The reaction mixture was stirred at room temperature for 1 h, diluted with ethyl acetate (100 mL), washed with water (100 mL), IN hydrochloric acid (2 X 50 mL), brine (100 mL), dried (Na2SO4), filtered, concentrated and purified using flash chromatography (SiO2, hexanes-ethyl acetate) to afford the title compound as a colorless liquid (342 mg, 94%).
[0237] 1H NMR (500 MHz, DMSO-J6): δ 1.50-1.60 (m, 2H), 1.62-1.68 (m, 2H), 1.77-1.83 (m, 2H), 2.00-2.03 (m, 2H), 3.14-3.21 (m, IH), 7.33 (dd, J = 8.2, 1.1 Hz, IH), 7.36-7.40 (m, IH), 7.44-7.48 (m, IH), 7.56 (dd, J = 7.8, 1.7 Hz, IH)
Example 58 Preparation of 2-Bromo-4-nitroisopropylbenzene (28)

[0238] Bromine (0.32 mL) was added drop wise over 5 min to a reaction mixture containing 4-nitroisopropylbenzene (1.0 g, 6.06 mmol), silver sulfate (1.04 g, 3.33 mmol), and concentrated sulfuric acid (5.5 mL) in water (0.61 mL) at room temperature under constant stirring. The reaction mixture was stirred for 2 h, and poured on ether (200 mL). The ether layer was separated, washed with brine (2 X 50 mL), dried (Na2SO4), filtered, concentrated, and purified on a flash chromatography (SiO2, hexanes) to afford the title compound as a pale yellow liquid (1.15 g, 18%). [0239] 1H NMR (500 MHz, DMSO-J6): δ 1.23 (d, J= 6.9 Hz, 6H), 3.31-3.37 (m, IH), 7.68 (d, J = 8.7 Hz, IH), 8.20 (dd, J = 8.6, 2.4 Hz, IH), 8.37 (d, J= 2.4 Hz, IH)
Example 59 Preparation of 2-Iodo-5-nitroisopropylbenzene (29)

29
[0240] A solution of 2-iodoisopropylbenzene (4.0 g, 16.3 mmol), concentrated sulfuric acid (4.6 rnL), and 60% nitric acid (1.5 g, 24.5 mmol) in chloroform (12 mL) was stirred at room temperature for 16 h. The reaction mixture was concentrated and purified using flash chromatography (SiO2, hexanes-ethyl acetate) to afford the title compound as a yellow liquid (1.7 g, 36%).
[0241] 1H NMR (500 MHz, DMSO-J6): δ 1.24 (d, J = 6.8 Hz, 6H), 3.17-3.23 (m, IH), 7.76 (dd, J = 6.6, 2.5 Hz, IH), 8.04 (d, J= 2.8 Hz, IH), 8.15 (d, J= 8.5 Hz, IH)
Example 60 Preparation of 4-r(2-cvclopentylphenyl)ethvnyll-7H-pyrrolor23-Jlpyrimidine

[0242] Argon was bubbled into a solution of 14 (100 mg, 0.47 mmol), 27 (209 mg, 0.71 mmol), PdCl2(PPh3)2 (63 mg, 0.09 mmol), cuprous iodide (20 mg, 0.1 mmol), diisopropylethylamine (0.7 mL), tetrabutylammoniumfluoride (123 mg, 0.47 mmol) in dimethyl formamide (4 mL) for 5 min. The reaction vessel was sealed, and heated at 1500C for 30 min in a microwave reactor. The reaction mixture was cooled, concentrated under reduced pressure, diluted with ethyl acetate (100 mL), filtered, concentrated, and purified on preparative ΗPLC to give the title compound as yellow solid (18 mg, 13%).
[0243] 1H NMR (500 MHz, DMSO-J6): δ 1.62-1.75 (m, 4H), 1.75-1.92 (m, 2H), 2.05-2.13 (m, 2H), 3.60-3.64 (m, IH), 6.63 (d, J = 2.7 Hz, IH), 7.28-7.33 (m, IH), 7.44-7.50 (m, 2H), 7.68 (d, J = 7.5 Hz, IH), 7.71 (dd, J= 3.3, 1.9 Hz, IH), 8.77 (s, IH), 12.36 (s, IH)
[0244] MS (ES+): m/z 288 (M+H)+
Example 61 Preparation of 4-{ r2-(l-Methylethyl)-5-nitrophenyllethynyl|-7H-pyrrolor2,3- (jlpyrimidine

[0245] Argon was bubbled into a solution of 14 (200 mg, 0.93 mmol), 28 (226 mg, 0.93 mmol), PdCl2(PPrIs)2 (133 mg, 0.19 mmol), cuprous iodide (38 mg, 0.2 mmol), triethylamine (1 mL), tetrabutylammoniumfluoride (243 mg, 0.93 mmol) in dimethyl formamide (7 mL) for 5 min. The reaction vessel was sealed and heated at 1400C for 60 min in a microwave reactor. The reaction mixture was cooled, concentrated under reduced pressure, diluted with ethyl acetate (100 mL), filtered, concentrated and purified on preparative ΗPLC to give the title compound as a yellow solid (45 mg, 16%).
[0246] 1H NMR (500 MHz, DMSO-J6): δ 1.36 (d, J= 6.9 Hz, 6H), 3.63-3.69 (m, IH), 6.77 (dd, J = 3.4, 1.6 Hz, IH), 7.73-7.78 (m, 2H), 8.30 (dd, J = 8.7, 2.4 Hz, IH), 8.50 (d, J= 2.5 Hz, IH), 8.81 (s, IH), 12.45 (s, IH) [0247] MS (ES+): m/z 307 (M+H)+
Example 62 Preparation of 4-{ r2-(l-Methylethyl)-4-nitrophenyllethynyl|-7H-pyrrolor2,3- Jlpyrimidine

[0248] Argon was bubbled into a solution of 14 (100 mg, 0.47 mmol), 29 (137 mg, 0.47 mmol), PdCl2(PPhS)2 (63 mg, 0.09 mmol), cuprous iodide (20 mg, 0.1 mmol), triethylamine (0.5 mL), tetrabutylammoniumfluoride (123 mg, 0.47 mmol) in dimethyl formamide (4.2 mL) for 5 min. The reaction vessel was sealed, and heated at 150 0C for 30 min in a microwave reactor. The reaction mixture was cooled, concentrated under reduced pressure, diluted with ethyl acetate (100 mL), filtered, concentrated and purified on preparative ΗPLC to give the title compound as a yellow solid (26 mg, 18%).
[0249] 1H NMR (500 MHz, DMSO-J6): δ 1.38 (d, J = 6.9 Hz, 6H), 3.61-3.67 (m, IH), 6.72 (d, J = 2.1 Hz, IH), 7.76 (t, J = 2.8 Hz, IH), 8.00 (d, J = 8.5 Hz, IH), 8.16 (dd, J = 8.5, 2.3 Hz, IH), 8.23 (d, J = 2.3 Hz, IH), 8.82 (s, IH), 12.45 (s, IH)
[0250] MS (ES+): m/z 307 (M+H)+
Example 63 Preparation of (2-Bromophenyl)(isopropyl)sulfane (30)

[0251] A 100 mL round bottom flask equipped with a stirbar and condenser was charged with 2-bromobenzenethiol (1.0 g, 5.3 mmol), acetone (25 mL), 2-bromopropane (0.55 mL, 5.8 mmol), and cesium carbonate (3.4 g, 10.4 mmol) sequentially. The resulting suspension was heated at 70 0C for 2 h, cooled, and the solids filtered. The filtrate was concentrated in vacuo to afford the title compound as a pale yellow oil (1.2 g, 98%).
[0252] 1H NMR (500 MHz, DMSO-J6) δ 1.29 (d, J = 6.7 Hz, 6H), 3.60 (qn, J = 6.6 Hz, IH), 7.13 (td, J = 7.6, 1.5 Hz, IH), 7.38 (td, J = 7.6, 1.3 Hz, IH), 7.44 (dd, J = 7.9, 1.6 Hz, IH), 7.62 (dd, J = 8.0, 1.3 Hz, IH)
Example 64 Preparation of 4-((2-(Isopropylthio)phenyl)ethvnyl)-7H-pyrrolor23-Jlpyrimidine

[0253] A mixture of 14 (107 mg, 0.5 mmol), 30 (225 mg, 1.0 mmol), Pd(PPh3)4Cl2 (16 mg, 0.02 mmol), and IM TBAF in TΗF (2.8 mL, 2.8 mmol) was heated in a sealed tube at 110 0C for 5.5 h. The solids were filtered, the filtrate was concentrated in vacuo, and the residue purified by ΗPLC. The fractions were neutralized with sat NaHCO3 and extracted with EtOAc. The organic layers were concentrated to afford the title compound as a white solid (15 mg, 10%).
[0254] 1H NMR (500 MHz, DMSO-J6) δ 1.33 (d, J= 6.6 Hz, 6H), 3.73 (qn, J= 6.6 Hz, IH), 6.78 (d, J = 3.4 Hz, IH), 7.31 (td, J= 7.5, 1.0 Hz, IH), 7.49 (td, J = 7.6, 1.2 Hz, IH), 7.54 (d, J = 9.9 Hz, IH), 7.72 (d, J = 5.7 Hz, 2H)
[0255] MS (ES+): m/z 294 (M+H)+
Example 65 Preparation of 4-Bromo-l-iodo-2-isopropylbenzene (31)

31
[0256] l-Iodo-2-isopropylbenzene (1.5 g, 6.1 mmol) was suspended with silver sulfate (1.0 g, 3.1 mmol), concentrated sulfuric acid (5.5 mL), water (0.6 mL), and bromine (0.3 mL, 6.4 mmol). The triphasic solution was stirred at room temperature for 1 h. The mixture was
poured into ether (150 niL) and water (75 rnL). The solids were filtered and the layers separated. The organic layer was concentrated in vacuo and the residue purified by preparatory HPLC to afford the title compound as a white solid (189 mg, 10%).
[0257] 1H NMR (500 MHz, DMSO-J6) δ 1.18 (d, J = 6.8 Hz, 6H), 3.07 (qn, J = 6.8 Hz, IH), 7.15 (dd, J = 8.4, 2.5 Hz, IH), 7.46 (d, J = 2.4 Hz, IH), 7.75 (d, J= SA Hz, IH) 2D NOE (500 MHz, DMSO-J6): crosspeak between δ 1.18 and 7.46
Example 66 Preparation of 4-((4-Bromo-2-isopropylphenyl)ethvnyl)-7H-pyrrolor2,3- Jlpyrimidine

[0258] A mixture of 14 (182 mg, 0.85 mmol), 31 (182 mg, 0.56 mmol), Pd(PPh3)4Cl2 (40 mg, 0.06 mmol), and IM TBAF in TΗF (3.5 mL, 3.5 mmol) was irradiated in the microwave at 160 0C for 15 min. The solids were filtered, the filtrate was concentrated in vacuo, and the residue purified by ΗPLC. The fractions were neutralized with sat NaHCO3 and extracted with EtOAc. The organic layers were concentrated in vacuo to afford the title compound as a yellow-brown solid (30 mg, 16%).
[0259] 1H NMR (500 MHz, DMSO-J6) δ 1.32 (d, J= 6.9 Hz, 6H), 3.53 (qn, J= 6.8 Hz, IH), 6.66 (d, J = 3.4 Hz, IH), 7.53 (dd, J = 8.3, 1.7 Hz, IH), 7.62-7.67 (m, 2H), 7.71 (d, J = 3.4 Hz, IH), 8.77 (s, IH)
[0260] MS (ES+): m/z 340/342 (M+H)+
Example 67 Preparation of tert-Buty\\( IZ)- [(fe/t-butoxycarbonyPaminol { r4-(2-pyrrolidin- 1 - ylethoxy)phenyll amino I methylidenel carbamate (32)

32
[0261] A mixture of (fer^-butoxycarbonylimino-pyrazol-l-yl-methy^-carbamic acid tert- butyl ester (5.0 g, 16.1 mmol) and 4-(2-pyrrolidin-l-yl-ethoxy)-phenylamine (4.0 g, 19.4 mmol) in DMF (10 mL) was stirred at room temperature for 17 h. The reaction mixture was concentrated and the residue taken in water (50 mL). The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as an off white solid (7 g, 97%). The material was used in the next step without purification.
[0262] MS (ES+): m/z 449 (M+H)+, 249 (M+l-2 Boc)+
Example 68 Preparation of Λ^r4-(2-Pyrrolidin-l-yl-ethoxy)-phenyll-guanidine TFA salt (33)

33
[0263] A solution of 32 (2.0 g, 4.5 mmol) in 50% TFA/DCM (10 mL) was stirred at room temperature for 17 h. The solution was concentrated to afford the title compound (quant, yield), which was used in the next step without purification.
[0264] MS (ES+): m/z 249 (M+H)+
Example 69 Preparation of 2-r4-(2-Pyrrolidin-l-yl-ethoxy)-phenylaminol-7H-pyrrolor2,3- (ilpyrimidin-4-ol (34)

34 [0265] To a microwave reaction tube was charged with 33 (1.6 g, 4.5 mmol), 2-cyano-4,4- diethoxy-butyric acid ethyl ester (1.7 g, 7.4 mmol) and NaOMe (25% by wt in MeOH; 10 mL) in EtOH (5 mL). The reaction tube was sealed and the solution irradiated with microwave at 160 0C for 30 min. After cooling to room temperature, the mixture was concentrated. The residue was taken up in water (10 mL) and the pH adjusted to 1 with 6M of HCl. The resulting solution was stirred at room temperature for 25 min and then the pH adjusted to 9 with concentrated NH4OH. The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as a brown solid (0.8 g, 52%). The material was used in the next step without purification.
[0266] MS (ES+): m/z 340 (M+H)+
Example 70 Preparation of (4-Chloro-7H-pyrrolor2,3-(ilpyrimidin-2-yl)-r4-(2-pyrrolidin-l-yl- ethoxy) -phenyll- amine (35)
35 [0267] A solution of 34 ( 1.4 g, 4.1 mmol) in POCl3 (5 mL) was heated at 120 0C for 20 min. After cooling to room temperature, the resulting dark solution was poured into ice water slowly and the pΗ adjusted to 9-10. The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as a brown solid (1.4 g, 95%). The material was used in the next step without purification. [0268] MS (ES+): m/z 358 (M+Η)+
Example 71 Preparation of 4-((2-Isopropylphenyl)ethynyl)-/V-(4-(2-(pyrrolidin- 1 - yl)ethoxy)phenyl)-7H-pyrrolor2,3-(ilPyrimidin-2-amine

[0269] A mixture of 7 (62 mg, 0.43 mmol), 35 (143 mg, 0.40 mmol), Pd(PPh3)4Cl2 (29 mg, 0.04 mmol), copper(I) iodide (8.8 mg, 0.05 mmol), and TEA (0.17 mL, 1.20 mmol) in DMF (1.6 mL) was heated at 80 0C for 25 min. The solids were filtered, the filtrate was concentrated in vacuo, and the residue purified by ΗPLC. The fractions were neutralized with sat NaHCO3 and extracted with EtOAc. The organic layers were concentrated in vacuo and the residue dissolved in MeOH. Concentrated HCl was added and the solvent and excess HCl removed in vacuo to afford the title compound as a red solid (67 mg, 33%).
[0270] 1H NMR (500 MHz, DMSO-J6) δ 1.16-1.26 (m, 2H), 1.32 (d, J = 6.9 Hz, 6H), 1.84- 1.94 (m, 2H), 1.98-2.08 (m, 2H), 3.05-3.18 (m, 2H), 3.52-3.65 (m, 2H, overlap with impurity), 4.33 (t, J = 4.8 Hz, 2H), 6.49 (s, IH), 6.98 (d, J = 8.9 Hz, 2H), 7.28-7.37 (m, 2H), 7.45-7.53 (m, 2H), 7.66 (d, J = 7.3 Hz, IH), 7.73 (d, J= 8.9 Hz, 2H), 9.49 (br s, IH), 10.90 (br s, IH), 11.85 (br s, IH)
[0271] MS (ES+): m/z 466 (M+H)+
Example 72 Preparation of l-Isopropyl-7-((trimethylsilyl)ethvnyl)-lH-indole-3-carbonitrile (36)

Example 73 Preparation of 7-Ethvnyl-l-isopropyl-lH-indole-3-carbonitrile (37)

37
[0273] To a solution of 36 (350 mg, 1.25 mmol) in TΗF (3 mL) was added IN TBAF in TΗF (2.0 mL, 2.0 mmol). Upon completion of addition, the reaction was checked by LCMS and found to be complete. The solution was concentrated in vacuo and the residue purified silica gel chromatography (0-100% EtOAc in hexanes) to afford the title compound as a yellow oil (217 mg, 83%). The compound was used directly in the next reaction.
Example 74 Preparation of l-Isopropyl-7-((2-(4-(2-(pyrrolidin-l-yl)ethoxy)phenylamino)-7H- pyrrolor2,3-(ilpyrimidin-4-yl)ethynyl)-lH-indole-3-carbonitrile

[0275] 1H NMR (500 MHz, DMSO-J6) δ 1.60 (d, J = 6.6 Hz, 6H), 1.68-1.77 (m, 4H), 2.65 (br s, 4H), 2.89 (br s, 2H), 4.06 (t, J = 5.7 Hz, 2H), 5.94 (qn, J = 6.6 Hz, IH), 6.46 (dd, J = 3.5, 1.8 Hz, IH), 6.89 (d, J = 9.1 Hz, 2H), 7.30 (dd, J = 3.5, 2.3 Hz, IH), 7.38 (t, J = 7.7 Hz, IH), 7.69-7.74 (m, 3H), 7.83 (dd, J= 7.9, 0.8 Hz, IH), 8.65 (s, IH), 9.28 (s, IH), 11.72 (s, IH)
[0276] MS (ES+): m/z 530 (M+H)+
Example 75 Preparation of l-Isopropyl-7-((2-(4-(4-methylpiperazin-l-yl)phenylamino)-7H- pyrrolor2,3-(ilPyrimidin-4-yl)ethvnyl)-lH-indole-3-carbonitrile

[0277] A mixture of 37 (123 mg, 0.59 mmol), 26 (100 mg, 0.29 mmol), Pd(PPh3)4Cl2 (22 mg, 0.03 mmol), copper(I) iodide (8.5 mg, 0.04 mmol), and TEA (0.12 mL, 0.88 mmol) in DMF (1.2 mL) was heated at 80 0C for Ih. The solids were filtered, the filtrate was concentrated in vacuo, and the residue purified by ΗPLC. The fractions were neutralized with sat NaHCO3 and extracted with EtOAc. The organic layers were concentrated in vacuo to afford the title compound as a yellow solid (29 mg, 19%).
[0278] 1H NMR (500 MHz, DMSO- J6) δ 1.60 (d, J = 6.6 Hz, 6H), 2.22 (s, 3H), 2.42-2.48 (m, 4H), 3.06-3.11 (m, 4H), 5.94 (qn, J = 6.6 Hz, IH), 6.45 (d, J = 1.7 Hz, IH), 6.88 (d, J = 8.9 Hz, 2H), 7.25-7.28 (m, IH), 7.38 (t, J = 7.7 Hz, IH), 7.65 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 7.3 Hz, IH), 7.83 (d, J= 7.9, IH), 8.65 (s, IH), 9.18 (s, IH), 11.69 (s, IH)
[0279] MS (ES+) : m/z 515 (M+H)+
Example 76 Preparation of 4-(2-(2-Ethyl-6-methylphenyl)ethynyl)-7H-pyrrolor2,3- Jlpyrimidine

[0280] A mixture of 4-ethynyl-7H-pyrrolo[2,3-d]pyrimidine (0.14 g, 1.0 mmol), l-ethyl-2- iodo-3-methylbenzene (0.29 g, 1.2 mmol), Pd(PPh3)2Cl2 (70 mg, 0.1 mmol), CuI ( 25 mg, 0.13 mmol), DIEA (1 mL) in anhydrous DMF (12 mL) was degassed with argon for 5 min and then refluxed in a sealed tube at 150 0C for 4 h. After cooling to room temperature, the cap was removed and the solvent removed by rotovap. The crude product was then purified by silica gel column with 10% CΗ3OΗ/CΗCI3 as an eluent to afford the title compound as a yellow solid (25 mg, 10%).
[0281] 1H NMR (500 MHz, CDCl3): δ 1.36 (t, J= 7.5 Hz, 3H), 3.02 (dd, J = 15.0, 7.5 Hz, 2H), 6.79 (s, IH), 7.10-7.20 (m, 2H), 7.25-7.30 (m, IH), 7.45 (br s, IH), 8.99 (br s, IH), 10.37 (br s, IH)
Example 77 Preparation of l-(2-Bromo-phenyl)-pyrrolidine (38)

38
[0282] 100 niL round-bottom flask was charged with 1,2-dibromobenzene (1.4 g, 6.0 mmol), Pd2(dba)3 (220 mg, 0.24 mmol) and (±)BINAP (300 mg, 0.48 mmol). 40 rnL of anhydrous dioxane were added, followed by pyrrolidine (0.51 g, 0.6 rnL, 7.18 mmol) and solid sodium tert-butoxide (577 mg, 6.0 mmol). The reaction mixture was purged with argon, brought to reflux and refluxed under the argon atmosphere for 15 min. Then the reaction mixture was cooled down to ambient temperature, filtered through a short pad of silica gel. The silica gel pad was washed with EtOAc. Combined organic solutions were concentrated in vacuo with ca. 15 g of silica gel. The loaded silica gel was taken to the ISCO system for further purification (80 g column, solid method, isocratic 10% EtOAc in hexanes, 40 min method). Fractions, containing the product, were combined and concentrated in vacuo to give the title compound as yellow oil (1.1 g, 81% yield).
Example 78 Preparation of 4-r(2-Pyrrolidin-l-ylphenyl)ethvnyll-7H-pyrrolor23-^lpyrimidine

[0283] 5 mL microwave vial was charged with 38 (452 mg, 2.0 mmol), PdCl2(PPh3)2 (70 mg, 0.1 mmol) and 1.0 M solution of TBAF in TΗF (6.0 mL, 6.0 mmol). The reaction mixture was purged with argon gas for 5 min, and then solid 14 (215 mg, 1.0 mmol) was added, the vial was sealed and placed in a heating block at 100 0C for 3 h. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (50 mL). The resulting solution was washed with saturated aqueous NaHCO3 (2 x 50 mL), brine (1 x 50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give red oil. The oil was purified by silica gel chromatography on the ISCO system (12 g column, 20 min method, 50% to 100% EtOAc gradient in hexanes). Fractions, containing the product, were combined and concentrated in vacuo. The residue was re-crystallized from 2 mL of EtOAc to give the title product as bright- yellow crystals (78 mg, 27% yield).
[0284] 1K NMR (500 MHz, DMSO-J6): δ 1.93-1.96 (m, 4H), 3.63-3.66 (m, 4H), 6.62 (dd, J = 3.4, 1.7 Hz, IH), 6.71 (t, J = 7.4 Hz, IH), 6.75 (d, J = 8.5 Hz, IH), 7.27 (dt, J = 7.3, 1.6 Hz, IH), 7.51 (dd, J = 7.7, 1.6 Hz, IH), 7.64 (t, J = 2.9 Hz, IH), 8.71 (s, IH), 12.26 (s, IH)
[0285] MS (ES+): m/z 287 (M+H)+
Example 79 Preparation of 4-{ r2-(2,5-Dimethyl-lH-pyrrol-l-yl)phenyllethynyl|-7H- pyrrolor2,3-<ilpyrimidine

[0286] 5 rnL microwave vial was charged with N-(2-bromo-phenyl)-2,5-dimethylpyrrole (250 mg, 1.0 mmol), PdCl2(PPh3)2 (35 mg, 0.05 mmol) and 1.0 M solution of TBAF in TΗF (3.0 mL, 3.0 mmol). The reaction mixture was purged with argon gas for 5 min, and then solid 14 (215 mg, 1.0 mmol) was added, the vial was sealed and placed in a heating block at 110 0C for 4 h. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (50 mL). The resulting solution was washed with saturated aqueous NaHCO3 (2 x 50 mL), brine (1 x 50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give brown oil. The oil was purified by silica gel chromatography on the ISCO system (12 g column, 20 min method, 50% to 100% EtOAc gradient in hexanes). Fractions, containing the product, were combined and concentrated in vacuo. The residue was triturated with 2 mL of Et2O, filtered and dried in vacuo to give the title product a light- yellow solid (36 mg, 64% yield). [0287] 1H NMR (500 MHz, DMSO-J6): δ 1.97 (s, 6H), 5.89 (s, 2H), 6.29 (dd, J = 3.5, 1.8 Hz, IH), 7.42 (dd, J = 7.8, 1.1 Hz, IH), 7.59-7.62 (m, 2H), 7.68 (dt, J = 7.7, 1.5 Hz, IH), 7.90 (dd, J = 7.8, 1.5 Hz, IH), 8.69 (s, IH), 12.27 (s, IH)
[0288] MS (ES+): m/z 313 (M+H)+
Example 80 Preparation of N-(2-Bromo-6-nitro-phenyl)acetamide (39)

Example 81 Preparation of N-(2-Bromo-6-nitro-phenyl)-N-isopropyl-acetamide (40)

[0291] 1H NMR (500 MHz, DMSO-J6): δ 2.04 (s, 3H), 7.43 (t, J = 8.1 Hz, IH), 7.94 (dd, J = 8.1, 1.0 Hz, IH), 8.05 (dd, J = 8.2, 1.1 Hz, IH), 10.16 (s, IH)
[0292] MS (ES+): m/z 260 (M+H)+
Example 82 Preparation of 3-Bromo-/V2-isopropyl -benzene- 1,2-diamine (41)

[0293] To a solution of SnCl2 x 2H2O (7.92 g, 35.1 mmol)) in 40 mL of concentrated HCl was added 40 (1.51 g, 5.01 mmol). The resulting solution was stirred at ambient temperature for 10 min, then brought to reflux and refluxed for 1 h. Then it was cooled down to ambient temperature, placed in an ice-acetone bath and 12 N NaOH was added dropwise very carefully until pH reached 14. The resulting grey suspension was extracted with EtOAc (5 x 100 mL). Combined EtOAc solutions were washed with brine (3 x 100 mL), dried over anhydrous
Na2SO4 and concentrated in vacuo to give the title product as yellow oil (1.08 g, 94% yield).
[0294] 1H NMR (500 MHz, DMSO-J6): δ 1.07 (d, J = 6.4 Hz, 6H), 3.29 (d, J= 10.0 Hz, IH), 3.39-3.44 (m, IH), 4.90 (s, 2H), 6.60-6.65 (m, 2H), 7.72 (dd, J = 6.9, 2.5 Hz, IH)
[0295] MS (ES+): m/z 230 (M+H)
Example 83 Preparation of 7-Bromo-l-isopropyl-lH-benzoimidazole (42)

[0296] To a solution of 41 (0.5 g, 2.18 mmol) in 30 rnL of EtOAc was added trimethyl orthoformate (2.31 g, 21.8 mmol), followed by p-toluenesulfonic acid mono-hydrate (42 mg, 0.218 mmol). The reaction mixture was left to stir at ambient temperature overnight. Then solvent was removed in vacuo to give yellow oil, which was taken to the ISCO system for purification (40 g column, 20 - 100% gradient of EtOAc in hexanes). Fractions, containing the product, were combined and concentrated in vacuo to give the title product as yellow oil (0.454 g, 87% yield).
[0297] 1H NMR (500 MHz, DMSO-J6): δ 1.56 (d, J = 6.6 Hz, 6H), 5.46 (septet, J = 6.7 Hz, IH), 7.13 (t, J= 7.9 Hz, IH), 7.45 (d, J = 7.8 Hz, IH), 7.68 (d, J = 8.2 Hz, IH), 8.51 (s, IH)
[0298] MS (ES+): m/z 240 (M+H)+
Example 84 Preparation of 7-Bromo-l-isopropyl-2-methyl-lH-benzoimidazole (43)

43
[0299] To a solution of 41 (0.5 g, 2.18 mmol) in 30 mL of EtOAc was added trimethyl orthoacetate (2.62 g, 21.8 mmol), followed byp-toluenesulfonic acid mono-hydrate (42 mg, 0.218 mmol). The reaction mixture was left to stir at ambient temperature overnight. Then solvent was removed in vacuo to give yellow oil, which was taken to the ISCO system for purification (40 g column, 20-100% gradient of EtOAc in hexanes). Fractions, containing the product, were combined and concentrated in vacuo to give the title product as yellow oil, which solidified upon standing into a yellow solid (0.481 g, 87% yield).
[0300] 1H NMR (500 MHz, DMSO-J6): δ 1.55 (d, J= 6.7 Hz, 6H), 2.65 (s, 3H), 5.92 (septet, J = 6.7 Hz, IH), 7.05 (t, J = 1.9 Hz, IH), 7.38 (d, J = 1.6 Hz, IH), 7.53 (d, J = 1.9 Hz, IH)
[0301] MS (ES+): m/z 254 (M+H)
Example 85 Preparation of 4-{ ri-(l-Methylethyl)-lH-benzimidazol-7-yllethynyl|-7H- pyrrolor2,3-<ilpyrimidine

[0302] 5 rnL microwave vial was charged with 42 (359 mg, 1.5 mmol), PdCl2(PPh3)2 (70 mg, 0.1 mmol) and 1.0 M solution of TBAF in TΗF (6.0 mL, 6.0 mmol). The reaction mixture was purged with argon gas for 5 min, and then solid 14 (215 mg, 1.0 mmol) was added, the vial was sealed and placed in a heating block at 130 0C for 1 h. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (100 mL). The resulting solution was washed with saturated aqueous NaHCO3 (2 x 50 mL), brine (1 x 50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give brown oil. The oil was purified by silica gel chromatography on the ISCO system (12 g column, 20 min method, 0% to 20% MeOH gradient in EtOAc). Fractions, containing the product, were combined and concentrated in vacuo. The residue was re-crystallized from 3 mL of EtOAc, filtered and dried in vacuo to give the title product a light- yellow solid (108 mg, 36% yield).
[0303] 1H NMR (500 MHz, DMSO-J6): δ 1.65 (d, J = 6.8 Hz, 6H), 5.89 (septet, J = 6.7 Hz, IH), 6.70 (d, J = 3.5 Hz, IH), 7.32 (d, J = 7.7 Hz, IH), 7.66 (d, J = 7.2 Hz, IH), 7.73 (d, J = 3.4 Hz, IH), 7.85 (d, J = 7.6 Hz, IH), 8.56 (s, IH), 8.80 (s, IH), 12.40 (s, IH) [0304] MS (ES+): m/z 302 (M+H)+
Example 86 Preparation of 4-{ r2-Methyl-l-(l-methylethyl)-lH-benzimidazol-7-yllethynyl|- 7H-pyrrolor23-<ilpyrimidine

[0305] 5 rnL microwave vial was charged with 43 (380 mg, 1.5 mmol), PdCl2(PPh3)2 (70 mg, 0.1 mmol) and 1.0 M solution of TBAF in TΗF (6.0 mL, 6.0 mmol). The reaction mixture was purged with argon gas for 5 min, and then solid 14 (215 mg, 1.0 mmol) was added, the vial was sealed and placed in a heating block at 130 0C for 1 h. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (100 mL). The resulting solution was washed with saturated aqueous NaHCO3 (2 x 50 mL), brine (1 x 50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give a brown residue. The residue was re- dissolved in 2 mL of DMF and purified by reverse-phase preparative HPLC in CH3CN/H2O system containing 0.1% of TFA. Fractions, containing the product, were combined and partitioned between EtOAc (50 mL) and saturated aqueous NaHCO3 (50 mL). The organic layer was washed with brine (1 x 30 mL), dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to give the title product as a yellow solid (72 mg, 23% yield).
[0306] 1H NMR (500 MHz, DMSO-J6): δ 1.65 (br s, 6H), 2.67 (s, 6H), 6.15 (br s, IH), 6.69 (s, IH), 7.25 (t, J = 7.7 Hz, IH), 7.60 (d, J = 7.1 Hz, IH), 7.71 (d, J= 7.9 Hz, IH), 7.73 (t, J = 2.9 Hz, IH), 8.80 (s, IH), 12.40 (s, IH)
[0307] MS (ES+): m/z 316 (M+H)+
Example 87 Preparation of 2-Chloro-4-{ ri-(l-methylethyl)-lH-benzimidazol-7-yllethynyl|- 7H-pyrrolor23-Jlpyrimidine (44)

[0308] 5 rnL microwave vial was charged with 42 (210 mg, 0.88 mmol), PdCl2(PPh3)2 (62 mg, 0.088 mmol) and 1.0 M solution of TBAF in TΗF (6.0 mL, 6.0 mmol). The reaction mixture was purged with argon gas for 5 min, and then solid 4 (220 mg, 0.88 mmol) was added, the vial was sealed and placed in a heating block at 130 0C for 1 h. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (100 mL). The resulting solution was washed with saturated aqueous NaHCO3 (3 x 50 mL), brine (1 x 50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo with ca. 5 g of silica gel. The loaded silica gel was taken to the ISCO system for purification by silica gel chromatography (12 g column, 20 min method, 5-30% MeOH gradient in EtOAc). Fractions, containing the product, were combined and concentrated in vacuo to give the title product a yellow solid (210 mg, 71% yield).
Example 88 Preparation of 4-{ ri-(l-Methylethyl)-lH-benzimidazol-7-yllethvnyl|-N-r4-(2- Pyrrolidin-l-ylethoxy)phenyll-7H-pyrrolor2,3-(ilPyrimidin-2-amine

[0310] 1H NMR (500 MHz, DMSO-J6): δ 1.65 (d, J = 6.7 Hz, 6H), 1.70 (br s, 4H), 2.59 (br s, 4H), 2.84 (br s, 2H), 4.06 (t, J = 5.9 Hz, 2H), 5.65 (septet, J = 6.7 Hz, IH), 6.45 (dd, J = 3.4, 1.7 Hz, IH), 6.88 (d, J = 9.0 Hz, 2H), 7.29 (d, J = 3.3 Hz, IH), 7.31 (t, J = 7.9 Hz, IH), 7.61 (d, J = 7.3 Hz, IH), 7.72 (d, J = 9.0 Hz, 2H), 7.84 (d, J = 8.0 Hz, IH), 8.57 (s, IH), 9.26 (s, IH), 11.77 (s, IH)
[0311] MS (ES+): m/z 506 (M+H)+
Example 89 Preparation of 4-{ ri-(l-Methylethyl)-lH-benzimidazol-7-yllethynyl|-N-r4-(4- methylpiperazin-l-yl)phenyll-7H-pyrrolor2,3-(ilPyrimidin-2-amine

[0312] 5 mL microwave vial was charged with 44 (101 mg, 0.3 mmol), 4-(4-methyl- piperazin-l-yl)-phenylamine (230 mg, 1.2 mmol), 6 mL of 2,2,2-trifluoroethanol and 0.2 mL of TFA. The vial was sealed and placed in a heating block at 105 0C for 48 h. Then the reaction mixture was cooled to ambient temperature, and solvent was removed in vacuo. The
residue was re-dissolved in 2 niL of DMF and purified by reverse-phase preparative HPLC in CH3CN/H2O system containing 0.1% of TFA. Fractions, containing the product, were combined and partitioned between EtOAc (50 mL) and saturated aqueous NaHCO3 (50 mL). The organic layer was washed with brine (1 x 30 mL), dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo to give the title product as a dirty- yellow solid (6.2 mg, 4% yield).
[0313] 1H NMR (500 MHz, DMSO-J6): δ 1.65 (d, J = 6.7 Hz, 6H), 2.22 (s, 3H), 2.46 (t, J = 4.8 Hz, 4H), 3.05 (t, J = 4.9 Hz, 4H), 2.59 (br s, 4H), 2.84 (br s, 2H), 4.06 (t, J= 5.9 Hz, 2H), 5.65 (septet, J= 6.7 Hz, IH), 6.44 (dd, J = 3.5, 1.7 Hz, IH), 6.88 (d, J= 9.1 Hz, 2H), 7.28 (dd, /= 3.5, 2.2 Hz, IH), 7.31 (t, J = 7.7 Hz, IH), 7.60 (d, J = 7.1 Hz, IH), 7.65 (d, J = 9.1 Hz, 2H), 7.84 (dd, J = 8.3, 0.8 Hz, IH), 8.57 (s, IH), 9.18 (s, IH), 11.71 (s, IH)
[0314] MS (ES+): m/z 491 (M+H)+
Example 90 Preparation of 2-Chloro-4-{ r2-methyl-l-(l-methylethyl)-lH-benzimidazol-7- yllethynyl|-7H-pyrrolor2,3-<ilpyrimidine (45)

45
[0315] 20 mL microwave vial was charged with 43 (0.74 g, 2.92 mmol), PdCl2(PPh3)2 (205 mg, 0.29 mmol) and 1.0 M solution of TBAF in TΗF (18 mL, 18.0 mmol). The reaction mixture was purged with argon gas for 5 min, and then solid 4 (0.73 g, 2.92 mmol) was added, the vial was sealed and placed in a heating block at 130 0C for 1 h. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (200 mL). The resulting solution was washed with saturated aqueous NaHCO3 (2 x 100 mL), brine (I x 100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo with ca. 15 g of silica gel. The loaded silica gel was taken to the ISCO system for purification by silica gel chromatography (80 g column, 40 min method, 5-30% MeOH gradient in EtOAc). Fractions, containing the product,
were combined and concentrated in vacuo to give the title product a light-brown solid (0.377 g, 38% yield).
Example 91 Preparation of 2-Chloro-4-{ r2-methyl-l-(l-methylethyl)-lH-benzimidazol-7- yllethvnyl|-7-r(4-methylphenyl)sulfonyll-7H-pyrrolor2,3-(ilPyrimidine (46)

46
[0316] To a solution of 45 (0.554 g, 1.58 mmol) in 20 mL of anhydrous DMF was added 60% suspension of NaH in mineral oil (0.127 g, 3.17 mmol). The mixture was stirred at ambient temperature for 10 min. Then p-toluenesulfonyl chloride (0.604 g, 3.17 mmol) was added all at once, and the reaction mixture was left to stir at ambient temperature for 1 h. Then the reaction mixture was quenched with saturated aqueous NH4CI (100 mL) and extracted with EtOAc (2 x 100 mL). The combined EtOAc solutions were washed with sat NaHCO3 (2 x 50 mL), brine (2 x 50 mL), dried over anhydrous Na2SO4 and filtered. Solvent was removed in vacuo with ca. 15 g of silica gel. The loaded silica gel was taken to the ISCO system for purification (40 g column, 5-30% MeOH gradient in EtOAc). Fractions, containing the product, were combined and concentrated in vacuo to give the title product a dark- yellow solid (0.754 g, 94% yield).
Example 92 Preparation of 4-{ r2-Methyl-l-(l-methylethyl)-lH-benzimidazol-7-yllethynyl|- N-(4-morpholin-4-ylphenyl)-7H-pyrrolor2,3-(ilPyrimidin-2-amine

[0317] 5 rnL microwave vial was charged with 46 (252 mg, 0.5 mmol), 4-(morpholine)- phenylamine (357 mg, 2.0 mmol), 5 mL of ra-butanol. The vial was sealed and placed in a heating block at 170 0C for 12 h. Then the reaction mixture was cooled to ambient temperature, and solvent was removed in vacuo to give a dark oily residue. The residue was treated with 6 mL of 1 M TBAF in TΗF at 60 0C for 30 min. Then the reaction mixture was diluted with 100 mL of EtOAc. This solution was washed with sat. NaHCO3 (4 x 50 mL), brine (2 x 50 mL), dried over anhydrous Na2SO4 and filtered and concentrated in vacuo. The residue was re-dissolved in 2 mL of DMF and purified by reverse-phase preparative HPLC in CH3CN/H2O system containing 0.1% of TFA. Fractions, containing the product, were combined and concentrated in vacuo to give the trifluoroacetic acid salt of the title product as dark-red oil (18 mg, 6% yield).
[0318] 1H NMR (500 MHz, DMSO-J6): δ 1.75 (br s, 6H), 2.91 (s, 3H), 3.15 (br s, 4H), 3.80 (t, J = 4.3 Hz, 4H), 6.28 (br s, IH), 6.50 (br s, IH), 7.05 (d, J = 8.3 Hz, 2H), 7.34 (s, IH), 7.59 (d, J = 7.6 Hz, IH), 7.75 (d, J = 7.8 Hz, 2H), 7.88 (br s, IH), 7.94 (d, J = 8.2 Hz, IH), 9.35 (s, IH), 11.79 (s, IH)
[0319] MS (ES+): m/z 492 (M+H)+
Example 93 Preparation of l-Isopropyl^-methyl^-trimethylsilanylethynyl-lH-benzimidazole
(47)

[0320] 100 rnL round-bottom flask was charged with 43 (4.7 g, 18.56 mmol), PdCl2(PPh3)2 (0.651 g, 0.93 mmol) and CuI (177 mg, 0.93 mmol). 30 mL of anhydrous TΗF were added, after 10 min of stirring followed by 50 mL of Et3N. The reaction mixture was purged with argon for 10 min. Then trimethylsilyl-acetylene (2.75 g, 1.97 mL, 27.85 mmol) was added. The reaction mixture was brought to reflux under argon atmosphere and refluxed for 3 h. Then it was cooled down to ambient temperature and concentrated in vacuo down to ca. 30 mL of the total volume. The resulting suspension was diluted with ca. 200 mL of EtOAc and filtered through a short pad of silica gel. The resulting reddish-brown solution was concentrated in vacuo with ca. 15 g of silica gel. The loaded silica gel was taken to the ISCO system for purification (80 g column, 50-100% EtOAc gradient in hexanes). Fractions, containing the product, were combined and concentrated in vacuo to give the title product as thick oil, which solidified in to a dark- yellow solid (3.44 g, 68% yield).
Example 94 Preparation of 4-{ r2-Methyl-l-(l-methylethyl)-lH-benzimidazol-7-yllethynyl|- N-r4-(2-pyrrolidin-l-ylethoxy)phenyll-7H-pyrrolor2,3-(ilPyrimidin-2-amine

[0322] 1H NMR (500 MHz, DMSO-J6): δ 1.60-1.81 (m, 10H), 2.54 (br s, 4H), 2.68 (s, 3H), 2.79 (t, J = 5.7 Hz, 2H), 4.03 (t, J = 6.0 Hz, 2H), 6.45 (br s, IH), 6.88 (d, J = 9.1 Hz, 2H), 7.25 (t, J = 7.8 Hz, IH), 7.29 (dd, J = 3.4, 2.2 Hz, IH), 7.54 (br s, IH), 7.70 (dd, J = 7.9, 0.8 Hz, IH), 7.73 (d, J = 9.1 Hz, 2H), 9.25 (s, IH), 11.70 (s, IH)
[0323] MS (ES+): m/z 520 (M+H)+
Example 95 Preparation of 4-{ r2-Methyl-l-(l-methylethyl)-lH-benzimidazol-7-yllethynyl|- N-r4-(4-methylpiperazin-l-yl)phenyll-7H-pyrrolor2,3-(ilPyrimidin-2-amine

[0324] 5 mL microwave vial was charged with 26 (343 mg, 1.0 mmol), PdCl2(PPh3)2 (70 mg, 0.1 mmol) and 1.0 M solution of TBAF in TΗF (6.0 mL, 6.0 mmol). The reaction mixture was purged with argon gas for 5 min, and then solid 47 (270 mg, 1.0 mmol) was added. The
vial was sealed and placed in a heating block at 130 0C for 1.5h, then left to stir at 70 0C overnight. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (100 mL). The resulting solution was washed with saturated aqueous NaHCO3 (4 x 50 mL), brine (2 x 50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography on the ISCO system (40 g column, 10 - 30% MeOH gradient in CH2Cl2) Fractions, containing the product, were combined and solvent was removed in vacuo. The residue was re-crystallized from 4 mL of 1:1 EtOAc/MeOH solution, centrifuged down and dried in vacuo to give the title product a bright- yellow solid (82 mg, 16% yield). [0325] 1H NMR (500 MHz, DMSO-J6): δ 1.68 (br s, 6H), 2.21 (s, 3H), 2.46 (t, J = 4.8 Hz, 4H), 2.68 (s, 3H), 3.05 (t, J = 4.8 Hz, 4H), 6.15 (br s, IH), 6.44 (br s, IH), 6.88 (d, J = 9.1 Hz, 2H), 7.25 (t, J= I. S Hz, IH), 7.28 (m, IH), 7.54 (d, J = 6.6 Hz, IH), 7.67 (d, J = 9.0 Hz, 2H), 7.70 (d, J = 8.0 Hz, IH), 9.18 (s, IH), 11.68 (s, IH)
[0326] MS (ES+): m/z 505 (M+H)+
Example 96 Preparation of 7-Bromo-2-ethyl-l-(l-methylethyl)-lH-benzimidazole (48)

[0327] To a solution of 41 ( 1.0 g, 4.36 mmol) in 80 mL of EtOAc was added trimethyl orthopropionate (7.7 g, 43.6 mmol), followed byp-toluenesulfonic acid mono-hydrate (83 mg, 0.436 mmol). The reaction mixture was left to stir at ambient temperature overnight. Then solvent was removed in vacuo to give yellow oil, which was taken to the ISCO system for purification (40 g column, 0-10% gradient of MeOH in CH2Cl2). Fractions, containing the product, were combined and concentrated in vacuo to give the title product as yellow oil, which solidified upon standing into a yellow solid (0.84 g, 72% yield).
Example 97 Preparation of 2-Ethyl-l-(l-methylethyl)-7-r(trimethylsilyl)ethynyll-lH- benzimidazole (49)

[0328] 100 rnL round-bottom flask was charged with 48 (0.84 g, 3.14 mmol), PdCl2(PPh3)2 (110 mg, 0.157 mmol) and CuI (30 mg, 0.157 mmol). 15 mL of anhydrous TΗF were added, after 10 min of stirring followed by 25 mL of Et3N. The reaction mixture was purged with argon for 10 min. Then trimethylsilyl-acetylene (0.62 g, 0.44 mL, 6.28 mmol) was added. The reaction mixture was brought to reflux under argon atmosphere and refluxed for 3 h. Then more PdCl2(PPh3)2 (110 mg, 0.157 mmol), CuI (30 mg, 0.157 mmol) and trimethylsilyl- acetylene (0.62 g, 0.44 mL, 6.28 mmol) were added and the reaction mixture was refluxed for additional 3 h. Then it was cooled down to ambient temperature and the reaction solution was centrifuged down. The resulting reddish-brown solution was concentrated in vacuo with ca. 15 g of silica gel. The loaded silica gel was taken to the ISCO system for purification (80 g column, 0-20% MeOH gradient in CH2Cl2). Fractions, containing the product, were combined and concentrated in vacuo to give the title product as dark-brown oil, which solidified into a dark- yellow solid (0.68 g, 76% yield).
Example 98 Preparation of 2-Ethyl-7-ethvnyl-l-(l-methylethyl)-lH-benzimidazole (50)

50
[0329] To a solution of 49 (440 mg, 1.55 mmol) in 6 Ml of a 1 : 1 mixture of TΗF/MeOΗ was added solid Cs2CO3 (504 mg, 1.55 mmol). The reaction mixture was left to stir at ambient temperature for 2 h. Then the resulting solution was concentrated in vacuo with ca. 5 g of silica gel. The loaded silica gel was taken to the ISCO system for purification (12 g column, 100% EtOAc as eluent). Fractions, containing the product, were combined and
concentrated in vacuo to give the title product as yellow oil, which solidified into a pale- yellow solid (320 mg, 98% yield).
Example 99 Preparation of 4-{ r2-Ethyl-l-(l-methylethyl)-lH-benzimidazol-7-yllethvnyl|-N- r4-(2-pyrrolidin-l-ylethoxy)phenyll-7H-pyrrolor23-(ilPyrimidin-2-amine

[0330] 5 rnL microwave vial was charged with 35 (300 mg, 0.838 mmol), 50 (267 mg, 1.26 mmol), PdCl2(PPh3)I (60 mg, 0.084 mmol) and CuI (16 mg, 0.084 mmol). 2 mL of anhydrous TΗF were added, after 10 min of stirring followed by 3 mL of Et3N. The reaction mixture was purged with argon for 10 min. Then the vial was capped and placed in a heating block at 130 0C for 1.5 h. Then the reaction mixture was cooled to ambient temperature, and diluted with EtOAc (50 mL). The resulting solution was washed with brine (2 x 50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The resulting reddish-brown oil was taken to the ISCO system for purification (40 g column, 10-30% MeOH gradient in CH2Cl2). The product came out at 20% MeOH as a very broad and small peak. Fractions, containing the product, were combined and concentrated in vacuo to give a dark-yellow solid (167 mg, 37.3% yield), which was re-crystallized from 4 mL of 1:1 EtOAc/MeOH solution, centrifuged down and dried in vacuo to give the title product as a light-yellow solid (75 mg, 16.7% yield). [0331] 1H NMR (500 MHz, DMSO-J6): δ 1.38 (br s, 3H), 1.60-1.81 (br s, 10H), 2.55 (br s, 4H), 2.80 (br s, 2H), 3.01 (br s, 2H), 4.04 (t, J = 5.9 Hz, 2H), 6.20 (br s, IH), 6.46 (br s, IH), 6.88 (d, / = 9.1 Hz, 2H), 7.25 (t, J = 7.8 Hz, IH), 7.29 (dd, J = 3.4, 2.2 Hz, IH), 7.56 (br s, IH), 7.72-7.75 (m, 3H), 9.26 (s, IH), 11.70 (s, IH)
[0332] MS (ES+): m/z 534 (M+H)+
Example 100 Preparation of 4-{ r2-(Trifluoromethoxy)phenyllethynyl|-7H-pyrrolor2,3- Jlpyrimidine

[0333] 5 rnL microwave vial was charged 4-ethynyl-7H-pyrrolo[2,3-d]pyrimidine (143 mg, 1.0 mmol), l-bromo-2-(trifluoromethoxy)benzene (241 mg, 1.0 mmol), PdCl2(PPhS)2 (35 mg, 0.05 mmol) and CuI (10 mg, 0.05 mmol). 3 mL of anhydrous CH3CN were added, after 10 min of stirring followed by 1.4 mL of Et3N. The reaction mixture was purged with argon for 5 min. Then the vial was capped and placed in a heating block at 60 0C for 18 h. Then it was cooled down to ambient temperature and concentrated in vacuo. The resulting black residue was re-dissolved in 3 mL of DMF and purified by reverse-phase preparative HPLC in CH3CN/H2O system containing 0.1% of TFA. Fractions, containing the product, were combined and partitioned between EtOAc (50 mL) and saturated aqueous NaHCO3 (50 mL). The organic layer was washed with brine (1 x 30 mL), dried over anhydrous Na2SO4 and filtered through a short silica gel plug. The filtrate was concentrated in vacuo to give the title product as a yellow solid (43 mg, 14% yield).
[0334] 1H NMR (500 MHz, DMSO-J6): δ 6.63 (d, J = 3.5 Hz, IH), 7.55 (dt, J = 7.5, 1.0 Hz, IH), 7.59 (d, J = 8.4 Hz, IH), 7.67 (dt, J = 8.4, 1.7 Hz, IH), 7.75 (d, J = 3.5 Hz, IH), 7.92 (dd, J = 7.7, 1.6 Hz, IH), 8.79 (s, IH), 12.42 (s, IH)
[0335] MS (ES+): m/z 304 (M+H)+
Example 101 Preparation of 4-(2-(2-Bromophenyl)ethvnyl)-7H-pyrrolor23-<ilpyrimidine

[0337] 1H NMR (500 MHz, DMSO-J6): δ 6.77 (d, J = 3.5 Hz, IH), 6.30 (td, J = 7.8, 2.0 Hz, IH), 7.52 (td, J = 7.3, 1.0 Hz, IH), 7.70-7.73 (m, IH), 7.84 (t, J = 7.5 Hz, IH), 7.85 (t, J -- 7.5 Hz, IH), 8.79 (s, IH)
[0338] MS (ES+): m/z 298/300 (M+H)+
Example 102 Preparation of 4-(2-(2-(Thiophen-2-yl)phenyl)ethvnyl)-7H-pyrrolor2,3- Jlpyrimidine
[0339] A suspension of 4-(2-(2-Bromophenyl)ethynyl)-7H-pyrrolo[2,3-d]pyrimidine (25 mg, 0.08 mmol), 2-thiopheneboronic acid (11 mg, 0.09 mmol), Pd(PPh3 )4 (9 mg, .008 mmol), aqueous Na2CO3 (0.2 ml of IM solution) in DME/DMF (95:5 v/v; 2 mL) was sealed in a microwave reaction vial and irradiated with microwave at 160 0C for 30 min. After cooling to room temperature, the cap was removed and the crude mixture triturated with DCM and water. The DCM layer was separated, dried (Na2SO4) and evaporated. The residue on purification with ΗPLC gave a fraction that was neutralized with aqueous sodium bicarbonate to afford the title compound (11 mg, 45%) as a brown solid.
[0340] 1H NMR (500 MHz, DMSO-J6): δ 6.51 (d, J = 3.5 Hz, IH), 7.21-7.26 (m, IH), 7.47 (t, J = 7.8 Hz, IH), 7.57 (t, J= 7.3 Hz, IH), 7.67 (d, J= 3.5 Hz, IH), 7.70-7.75 (m, 3H), 7.84- 7.87 (m, IH), 8.76 (s, IH), 12.20 (br s, IH)
[0341] MS (ES+): m/z 301 (M+H)+
Example 103 Preparation of 4-(2-(2-(Furan-3-yl)phenyl)ethynyl)-7H-pyrrolor2,3- Jlpyrimidine

[0342] A suspension of 4-(2-(2-Bromophenyl)ethynyl)-7H-pyrrolo[2,3-J]pyrimidine (15 mg, 0.05 mmol), 3-furanoboronic acid (6 mg, 0.06 mmol), Pd(PPh3)4 (2.3 mg, .003 mmol), aqueous Na2CO3 (0.2 ml of IM solution) in DMF (2 mL) was sealed in a microwave reaction vial and irradiated with microwave at 140 0C for 20 min. After cooling to room temperature, the cap was removed and the crude mixture triturated with DCM and water. The DCM layer was separated, dried (Na2SO4) and evaporated. The residue on purification with ΗPLC gave a fraction that was neutralized with aqueous sodium bicarbonate to afford the title compound (6 mg, 43%) as a brown solid.
[0343] 1H NMR (500 MHz, DMSO-J6): δ 6.60 (d, J = 3.2 Hz, IH), 7.13-7.16 (m, IH), 7.42 (t, J = 7.5 Hz, IH), 7.56 (t, J = 7.5 Hz, IH), 7.65-7.71 (m, 2H), 7.80-7.85 (m, 2H), 7.39-7.42 (m, IH), 8.78 (s, IH), 12.20 (br s, IH)
[0344] MS (ES+): m/z 286 (M+H)
Example 104 Preparation of 7-Bromo-l-isopropyl-lH-indazole (51)

Example 105 Preparation of 4-(2-(2-(Furan-3-yl)phenyl)ethvnyl)-7H-pyrrolor2,3- Jlpyrimidine

[0347] A mixture of 4-ethynyl-7H-pyrrolo[2,3- d]pyrimidine (24 mg, 0.16 mmol), 51 (40 mg, 0.16 mmol), PdCl2(PPh3)2 (11.7 mg, 0.016 mmol), CuI (3.0 mg, 0.016 mmol), TEA (0.117 mL, 0.835 mmol) in DMF (1 mL) were sealed in a microwave reaction vial and irradiated with microwave at 110 0C for 25 min. After cooling to room temperature, the residue on purification with ΗPLC gave a fraction that was neutralized with aqueous sodium bicarbonate to afford the title compound (18 mg, 37%) as a brown solid.
[0348] 1H NMR (500 MHz, DMSO-J6): δ 1.57 (s, 3H), 1.59 (s, 3H), 5.98-6. l(m, IH), 6.60 (d, J = 3.2 Hz, IH), 7.25 (t, J = 7.5 Hz, IH), 7.71-7.74 (m, IH), 7.80-7.85 (m, IH), 7.91-8.00 (m, IH), 8.26 (s, IH), 8.81 (s, IH), 12.45 (br s, IH)
[0349] MS (ES+): m/z 302 (M+H)+
Example 106 Preparation of 4-(2-(l-Isopropyl-lH-indazol-7-yl)ethynyl)-7H-pyrrolor2,3- (jlpyrimidine

[0350] A mixture of 16 (13 mg, 0.05 mmol), 4-chloro-7H-pyrrolo[2,3-d]pyrimidin-2-amine (8.0 mg, 0.05 mmol), PdCl2(PCy3)2 (0.74 mg, 0.001 mmol), TBAF (0.1 mL of IM TΗF solution, 0.1 mmol), Cs2CO3 (25 mg, 0.075 mmol), in DMSO (1 mL) were sealed in a microwave reaction vial and irradiated with microwave at 160 0C for 20 min. After cooling to room temperature, the residue was purified using ΗPLC to give a fraction that was neutralized with aqueous sodium bicarbonate to afford the title compound (6 mg, 36%) as a brown solid.
[0351] 1H NMR (500 MHz, DMSO-J6): δ 1.54 (d, J= 6.5 Hz, 6H), 5.98-6.10 (m, IH), 6.23 (br s, 2H), 6.32 (d, J = 3.7 Hz, IH), 6.61 (d, J = 3.5 Hz, IH), 7.10 (t, J = 7.5 Hz, IH), 7.15 (d, J = 3.5 Hz, IH), 7.44 (t, J = 7.5 Hz, IH), 7.65 (d, J = 3.2 Hz, IH), 7.70 (d, J = 8.0 Hz, IH), 11.35 (br s, IH) [0352] MS (ES+): m/z 316 (M+H)+
Example 107 Preparation of 2'-(7H-Pyrrolor2,3-(ilpyrimidin-4-ylethynyl)-biphenyl-3-sulfonic acid methylamide

[0353] To a microwave reaction tube was charged with 4-(2-(2-Bromophenyl)ethynyl)-7H- pyrrolo[2,3-d]pyrimidine (50 mg, 0.17 mmol), N-methyl-3-boronobenzenesulfonamide (45 mg, 0.21 mmol) and Pd(PPh3)4 (15 mg, 0.013 mmol). DMF (3 mL) was added to the above mixture followed by aqueous sodium carbonate (2 M; 0.5 mL, 1.0 mmol). The reaction tube was sealed and the suspension irradiated with microwave at 160 0C for 15 min. After cooling to room temperature, the mixture was filtered and the filtered solid washed with DCM. The filtrate was concentrated and the residue purified by ΗPLC. The fractions were combined and poured into saturated NaHCO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the residue re-dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as an off white solid (20 mg, 31%).
[0354] 1H NMR (500 MHz, DMSO-J6): δ 2.36 (d, J = 5.1, 3H), 6.14 (dd, J = 3.4, 1.6 Hz, IH), 7.51 (q, J = 5.1 Hz, IH), 7.51-7.60 (m, 3H), 7.64-7.68 (m, IH), 7.75 (t, J= 7.7 Hz, IH), 7.86 (d, J = 7.9 Hz, IH), 7.91 (d, J = 7.8 Hz, IH), 7.96 (d, J = 7.5 Hz, IH), 7.99 (t, J = U Hz, IH), 8.70 (s, IH), 12.30 (br s, IH)
[0355] MS (ES+): m/z 389 (M+H)+
Example 108 Preparation of (E)-tert-Buty\ (fe/t-butoxycarbonylamino)(4-((2-methyl-lH- imidazol- 1 - yl)methyl)phenylamino)methylenecarbamate (52)

52 [0356] A slurry of N^-bis^ert-butoxycarbony^-lH-pyrazole-l-carboxamidine (5.0 g, 16.1 mmol) and 4-(2-methylimidazol-l-ylmethyl)phenylamine (3.0 g, 16.1 mmol) in DMF (11 rnL) was stirred for 64 h. The resulting clear solution was poured into 100 rnL water and extracted with DCM (2x200 rnL) and ethyl acetate (1x100 rnL). The organic layers were combined and washed with brine (50 mL), dried (Na2SO4), and concentrated in vacuo. The residual material was partially purified by silica gel column chromatography (0-20% MeOH in DCM with 0.05% ammonium hydroxide) to afford the title compound as a slightly impure yellow oil (6.6g, 95%)
[0357] 1H NMR (500 MHz, DMSO-J6) δ 1.45 (br s, 18H), 2.23 (s, 3H), 5.10 (s, 2H), 6.75 (d, J = 0.8 Hz, IH), 7.08-7.16 (m, 3H), 7.52 (d, J = 8.4 Hz, 2H), 9.98 (s, IH), 11.39 (s, IH) [0358] MS (ES+): m/z 430 (M+H)+
Example 109 Preparation of l-(4-((2-Methyl-lH-imidazol-l-yl)methyl)phenyl)guanidine (53)

53 [0359] To a solution of 52 (6.1 g, 14.2 mmol) in DCM (35 mL) was slowly added trifluoroacetic acid (35 mL) and the resulting mixture was stirred for 1.5 h at which point the reaction was concentrated in vacuo to afford the title compound contaminated with DMF and trifluroracetic acid as a yellow oil. It was used as is for the next reaction.
[0360] MS (ES+): m/z 230 (M+Η)+
Example 110 Preparation of 2-r4-(2-Methyl-imidazol-l-ylmethyl)-phenylaminol-7H- pyrrolor2,3-(ilPyrimidin-4-ol (54)

[0361] To a microwave reaction tube was charged with 53 (1.1 g, 5.0 mmol), 2-cyano-4,4- diethoxy-butyric acid ethyl ester (1.7 g, 7.4 mmol) and NaOMe (25% by wt in MeOH; 10 mL). The reaction tube was sealed and the solution irradiated with microwave at 150 0C for 30 min. After cooling to room temperature, the mixture was concentrated. The residue was taken up in water (10 mL) and the pΗ adjusted to 1 with 6M of HCl. The resulting solution was stirred at room temperature for 25 min and then the pΗ adjusted to 9 with concentrated NH4OH. The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as a grey solid (1.5 g, 94%). The material was used in the next step without purification. [0362] H NMR (500 MHz, DMSO-J6) δ 2.25 (s, 3H), 5.07 (s, 2H), 6.29 (dd, J = 3.0, 2.2 Hz, IH), 6.75 (t, J = 2.7 Hz, IH), 6.76 (d, J = 0.7 Hz, IH), 7.09-7.15 (m, 3H), 7.60 (d, J = 8.5 Hz, 2H), 8.71 (s, IH), 10.28 (br s, IH), 11.36 (s, IH)
[0363] MS (ES+): m/z 321 (M+H)+
Example 111 Preparation of ((4-Chloro-7H-pyrrolor2,3-(ilpyrimidin-2-yl)-r4-(2-methyl- imidazol- 1 - ylmeth yl) -phenyll - amine (55)

55
[0364] A solution of 54 ( 1.4 g, 4.1 mmol) in POCl3 (5 niL) was heated at 120 0C for 30 min. After cooling to room temperature, the resulting dark solution was poured into ice water slowly and the pH adjusted to 9-10. The resulting solid was filtered, washed with water and dried under high vacuum to afford the title compound as a brown solid (1.1 g, 69%). The material was used in the next step without purification.
[0365] 1K NMR (500 MHz, DMSO-J6) δ 2.32 (s, 3H), 5.09 (s, 2H), 6.39 (dd, J = 3.3, 1.4 Hz, IH), 6.89 (d, J = 0.6 Hz, IH), 7.14 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 0.8 Hz, IH), 7.27 (dd, J= 3.3, 2.1 Hz, IH), 7.77 (d, J = 8.6 Hz, 2H), 9.71 (s, IH), 11.93 (s, IH)
[0366] MS (ES+): m/z 339 (M+H)+
Example 112 Preparation of l-Isopropyl-7-((2-(4-((2-methyl-lH-imidazol-l- yl)methyl)phenylamino)-7H-pyrrolor2,3-(ilpyrimidin-4-yl)ethvnyl)-lH-indole-3-carbonitrile
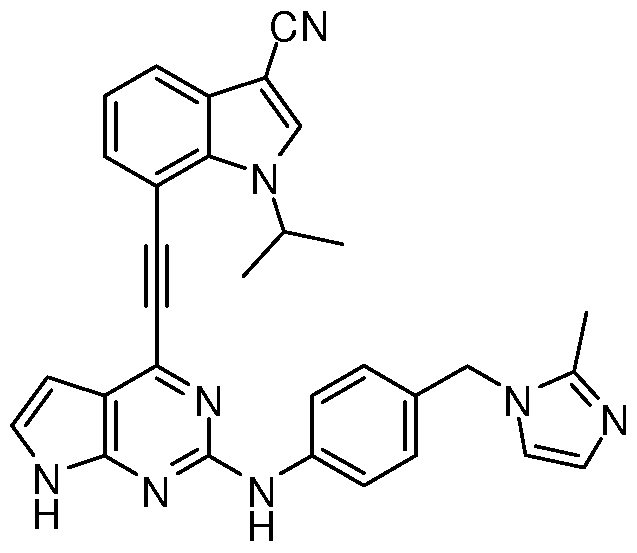
[0367] A mixture of 37 (245 mg, 1.18 mmol), 55 (199 mg, 0.59 mmol), Pd(PPh3)4Cl2 (40 mg, 0.06 mmol), copper(I) iodide (14 mg, 0.07 mmol), and TEA (0.25 mL, 1.76 mmol) in DMF (2.4 mL) was heated at 80 0C for 1 h. The solids were filtered, the filtrate was concentrated in vacuo, and the residue was purified by ΗPLC. The fractions were neutralized with sat NaHCO3 and extracted with EtOAc. The organic layers were concentrated in vacuo to afford the title compound as a yellow-brown solid (62 mg, 21%). [0368] 1H NMR (500 MHz, DMSO-J6) δ 1.59 (d, J= 6.6 Hz, 6H), 2.26 (s, 3H), 5.06 (s,
2H), 5.93 (qn, J = 6.6 Hz, IH), 6.50 (dd, J = 3.5, 1.8 Hz, IH), 6.75 (s, IH), 7.06-7.12 (m, 3H), 7.35 (dd, J = 3.4, 2.2 Hz, IH), 7.38 (t, J = 1.1 Hz, IH), 7.72 (dd, J = 7.3, 0.6 Hz, IH), 7.79- 7.85 (m, 3H), 8.66 (s, IH), 9.52 (s, IH), 11.79 (s, IH)
[0369] MS (ES+): m/z 429 (M+H)+
Example 113: Enzyme Assays
[0370] The IC50 values for compounds were determined using a luminescence -based kinase assay with recombinant JAK2, JAK3 and KDR (VEGFr2) obtained from Invitrogen. In white, flat-bottom, 96-well plates (Nunc) parallel assays were run at room temperature at a final volume of 50 μL. Each well contained 40 μL of buffer consisting of 40 mM Tris buffer, pH 7.4, containing 50 mM MgCl2, 800 μM EGTA, 350 μM Triton X-100, 2 mM β- mercaptoethanol, 250 μM peptide substrate and an appropriate amount of either JAK2, JAK3 or KDR (75 - 25 ng/well) such that the assay was linear over 60 min. The final concentrations of compounds for IC50 value determinations ranged from 10 to 0.001 μM by adding the appropriate amount of compound in 2.5 μL of DMSO; the DMSO present in each assay was constant at 5%. The reaction was initiated by the addition of 10 μL of ATP to a final assay concentration of 3 μM. After the reaction had proceeded for 60 min, 50 μL of Kinase-Glo reagent (Promega) was added to terminate the reaction. This solution was then allowed to proceed for an additional 10 min to maximize the luminescence reaction. Values were then measured using an Ultra 384 instrument (Tecan) set for luminosity measurements. Two control reactions were also ran: one reaction containing no compound and the second containing neither inhibitor nor peptide substrate. IC50 values were derived from experimental data using the non-linear curve fitting capabilities of Prism (Version 4; GraphPad Software). The results, expressed as IC50, are presented in Table 1.
Table 1. Data for compounds in nM














References
[0371] All publications and patents mentioned herein, including those items listed below, are hereby incorporated by reference in their entirety as if each individual publication or patent was specifically and individually incorporated by reference. In case of conflict, the present application, including any definitions herein, will control.
Equivalents
[0372] While specific embodiments of the subject invention have been discussed, the above specification is illustrative and not restrictive. Many variations of the invention will become apparent to those skilled in the art upon review of this specification. The full scope of the invention should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
[0373] Unless otherwise indicated, all numbers expressing quantities of ingredients, reaction conditions, and so forth used in the specification and claims are to be understood as being modified in all instances by the term "about." Accordingly, unless indicated to the contrary, the numerical parameters set forth in this specification and attached claims are approximations that may vary depending upon the desired properties sought to be obtained by the present invention.
Claims
1. A compound represented by Formula I:

I wherein R1 is an aryl or heteroaryl, wherein R1 is optionally substituted on a ring carbon by one to seven substituents each independently selected from the group consisting of halo, hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF3, ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N-alkylsulphamoyl, N-alkylcarbamoyl, -O-alkyl, -S-alkyl, or -R13R11;
R4 is selected from the group consisting of H, NH2, -NHR10 or -NH-alkyl; R1O is phenyl or pyridinyl, wherein R1O is optionally substituted on a ring carbon by one, two, or three substituents each independently selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, -N-alkyl-amino, carbamoyl, sulphamoyl, CF3, ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N- alkylsulphamoyl, N-alkylcarbamoyl, -OR11, -OR12Rn, or -R12R11; and
R11 is independently selected from the group consisting of aryl, heteroaryl, cycloalkyl and heterocycloalkyl, wherein R11 can be optionally substituted by one to four substituents each independently selected from with halo, alkyl, carbonyl, hydroxyl, alkyl-hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, CF3, ureido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, aryl, cycloalkyl, heteroaryl, or heterocycloalkyl;
R12 is alkylene or a bond; R13 is selected from the group consisting of alkylene, alkenylene, -C(O)-, -SO2-, or a bond; or
pharmaceutically acceptable salts , prodrugs, N-oxides, diastereomers and hydrates thereof.
2. The compound of claim 1, wherein R1 is selected from the group consisting of phenyl, monocyclic heteroaryl, or bicyclic heteroaryl.
3. The compound of claim 1 or 2, wherein R1 is selected from the group consisting of optionally substituted indole, optionally substituted benzoimidazole, and optionally substituted phenyl.
4. The compound of any one of claims 1-3, wherein R1 is selected from the group consisting of:

or
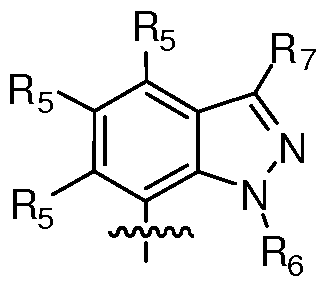
wherein
R5, independently for each occurrence, is selected from the group consisting of: H, halo, hydroxyl, nitro, cyano, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, -O-alkyl, -S-alkyl, or optionally substituted monocyclic aryl or optionally substituted monocyclic heteraryl;
Re, independently for each occurrence, is selected from the group consisting of: H, C1-5 straight chain alkyl, or C1-5 branched alkyl; and R7, independently for each occurrence is selected from the group consisting of H, C1-S straight chain alkyl, C1-4 branched alkyl, cyano, carboxyl, acyl, aldehyde, oxyalkylnitrile, -S- alkyl, -O-alkyl, or alkylamine.
5. The compound of any one of claims 1-3, wherein R1 is:

and wherein R5 is independently, for each occurrence, selected from the group consisting of H, halo, cyano, nitro, C1-5 straight chain alkyl, C1-5 branched alkyl, -O-alkyl, -S- alkyl, or -C(O)-alkyl.
6. The compound of any one of claims 1-3, wherein R1 is:

and wherein
Re, independently for each occurrence, is selected from the group consisting of: H, C1-5 straight chain alkyl, or C1-5 branched alkyl; and
R7, independently for each occurrence is selected from the group consisting of H, C1-S straight chain alkyl, C1-4 branched alkyl, halo, cyano, carboxyl, acyl, oxyalkylnitrile, -S-alkyl, - O-alkyl, or aldehyde.
7. The compound of any one of claims 1-3, wherein R1 is:

and wherein
R6, independently for each occurrence, is selected from the group consisting of H, C1-S straight chain alkyl, or C1-S branched alkyl; and
R7, independently for each occurrence is selected from the group consisting of H, C1-S straight chain alkyl, C1-4 branched alkyl, cyano, carboxyl, acyl, -S-alkyl, -O-alkyl, or aldehyde.
8. The compound of any one of claims 1-6, wherein R4 is H.
9. The compound of any one of claims 1-6, wherein R1O is phenyl.
10. The compound of claim 8, wherein R10 is represented by:

wherein:
X is N or CR8;
R8 and R9, independently for each occurrence, is chosen from H, heterocycle, -O- heterocycle, -alkylene-heterocycle, or -O-alkylene-heterocycle, wherein said heterocycle for each occurrence is optionally substituted with one to three substituents each independently selected from halo, alkyl, carbonyl, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N- alkylsulphamoyl, and N-alkylcarbamoyl.
11. The compound of claim 10, wherein said heterocycle is substituted with methyl.
12. The compound of claim 10, wherein R8 is H and R9 is a heterocycle or -O-alkylene- heterocycle, wherein said heterocycle is chosen from: pyrrolidinyl, piperazinyl, imidazoyl, piperdinyl, or morpholinyl.
13. The compound of claim 10, wherein R8 is H and R9 is selected from the group consisting of: methylpiperazine, piperazine, or 2-pyrrolidin-lylethoxy.
14. The compound of claim 10, wherein at least one R8 is H.
15. A compound represented by formula Ha, lib, Hc or Hd:

wherein
R4 is H or -NHR10; R5 is independently selected for each occurence from the group consisting of H, halo, hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF3, ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, N-alkylsulphamoyl, N-alkylcarbamoyl, -O-alkyl, -S-alkyl, -0-CF3, or - Ri3Rn;
Re, independently for each occurrence, is selected from H or alkyl;
R7, independently for each occurrence is selected from H, halo, hydroxyl, carbonyl, nitro, formyl, formamido, carboxy, amino, amido, acylamino, carbamoyl, sulphamoyl, alkyl, alkenyl, CF3, ureido, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, carbaldehyde oxime, -S-alkyl, -O-alkyl, N-alkylsulphamoyl, or N-alkylcarbamoyl;
Rio is phenyl or pyridinyl, wherein Ri0 is optionally substituted on a ring carbon is optionally substituted on a ring carbon by one, two, or three substituents each independently selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, -N-alkyl-amino, carbamoyl, sulphamoyl, CF3, ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, -ORn, -ORi2Rn, or 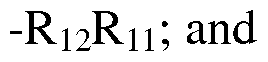
Rn is independently selected from aryl, heteroaryl, cycloalkyl and heterocycloalkyl, wherein Rn can be optionally substituted by one to four substituents each independently selected from with halo, alkyl, carbonyl, hydroxyl, alkyl-hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, CF3, ureido, carbamoyl, sulphamoyl, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, N-alkylcarbamoyl, aryl, cycloalkyl, heteroaryl, or heterocycloalkyl;
R12 is alkylene or a bond;
Ri3 is alkylene, or a bond; or
pharmaceutically acceptable salts , prodrugs, N-oxides, diastereomers and hydrates thereof.
16. The compound of claim 14, wherein R4 is H.
17. The compound of claim 14, wherein R4 is -N-phenyl, wherein the phenyl is optionally substituted.
18. The compound of claim 14 or 16, wherein R4 is selected from the group consisting of:


19. The compound of any one of claims 14-16, wherein the compound is represented by:

and wherein R5, for each occurrence, is selected from the group consisting of: H, halo, alkyl, nitro, -O-alkyl, -S-alkyl, phenyl, or heterocycle, wherein said phenyl or heterocycle is optionally substituted with one to three substituents each independently selected from halo, alkyl, carbonyl, hydroxyl, nitro, formyl, formamido, carboxy, amino, amido, carbamoyl, sulphamoyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, N-alkylsulphamoyl, and N- alkylcarbamoyl.
20. The compound of any one of claims 14-16, wherein the compound is represented by:

21. The compound of claim 19, wherein R7, independently for each occurrence is selected from H or alkyl.
22. The compound of any one of claims 14-16, wherein the compound is represented by:
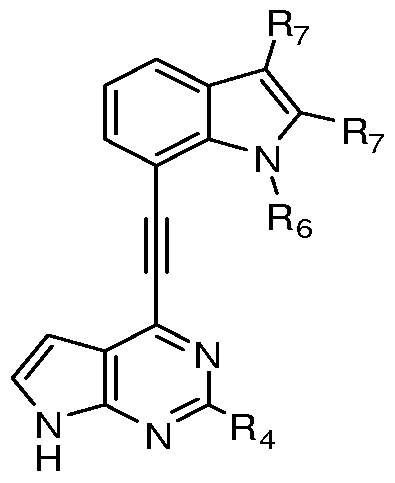
23. The compound of claim 21, wherein R7, independently for each occurrence is selected from H, halo, cyano, acetylaldehyde, ketone, carboxyl, oxyalkylnitrile -S-alkyl, -O-alkyl, or alkyl.
24. A compound selected from the group consisting of:



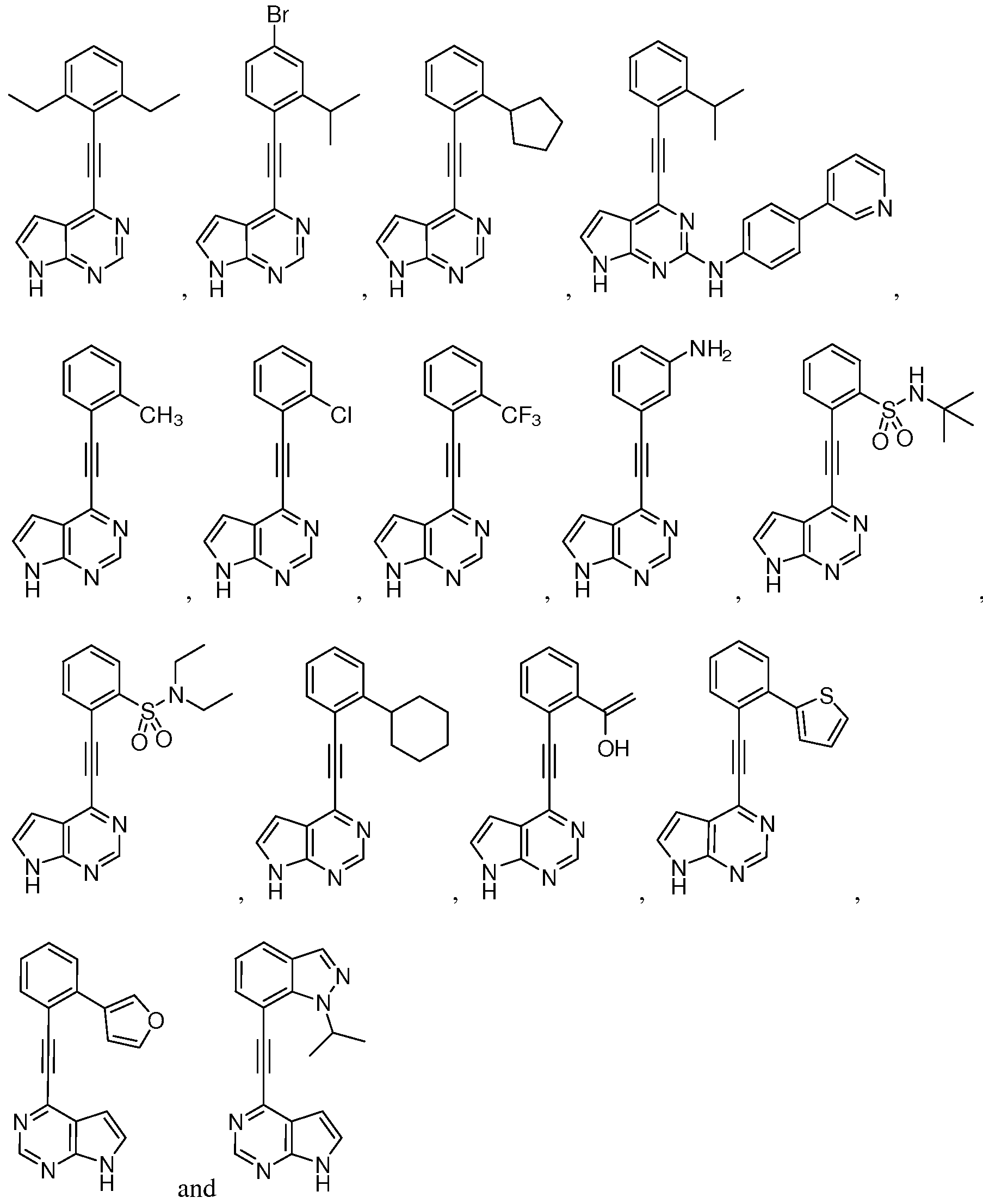
25. The compound of any one of claims 1-23, wherein said compound inhibits JAK.
26. The compound of any one of claims 1-24, wherein said compound inhibits one or more of: JAKl, JAK2. JAK3, and TYK2.
27. The compound of any one of claims 1-25, wherein said compound does not substantially modulate KDR.
28. A method for treating a myeloproliferative disorder in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
29. The method of claim 27, wherein the myeloproliferative disorder is one of: polycythemia vera, myelofibrosis, and essential thrombocythemia.
30. A method for treating polycythemia vera in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
31. A method for treating myelofibrosis in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
32. A method for treating essential thrombocythemia in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
33. A method for treating acute myeloid leukemia (AML)in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
34. A method of treating cancer in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
35. A method of treating an immune disorder and/or inflammation in a patient in need thererof, comprising administering an effective amount of a compound of any one of claims 1- 23.
36. A method of treating respiratory inflammation in a patient in need thererof, comprising administering an effective amount of a compound of any one of claims 1-23.
37. A method of treating asthma in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
38. A method of treating chronic obstructive pulmonary disease (COPD) in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
39. A method of rheumatoid arthritis (RA) in a patient in need thereof, comprising administering an effective amount of a compound of any one of claims 1-23.
40. A method for treating rheumatoid arthritis (RA), psoriatic arthritis, asthma, systemic lupus erythematosus, inflammatory bowel disease, multiple sclerosis, type I diabetes mellitus, myasthenia gravis, thyroiditis, myocarditis, psoriasis, immunoglobulin nephropathies, uveitis, iritis, scleritis, conjunctivitis, graft versus host disease or dermatitis, comprising administering an effective amount of a compound of any one of claims 1-23.
41. A method of treating age related macular degeneration, diabetic macular edema, and/or proliferative diabetic retinopathy, comprising administering an effective amount of a compound of any one of claims 1-23.
42. A composition comprising a compound of any one of claims 1-23 and a pharmaceutically acceptable excipient.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US98282907P | 2007-10-26 | 2007-10-26 | |
| US60/982,829 | 2007-10-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009055674A1 true WO2009055674A1 (en) | 2009-04-30 |
Family
ID=40303714
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/081119 Ceased WO2009055674A1 (en) | 2007-10-26 | 2008-10-24 | Pyrrolopyrimidine alkynyl compounds and methods of making and using same |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009055674A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110282056A1 (en) * | 2008-01-22 | 2011-11-17 | Merck Patent Gmbh | Protein kinase inhibitors and use thereof |
| WO2012045195A1 (en) * | 2010-10-09 | 2012-04-12 | Abbott Laboratories | Pyrrolopyrimidines as fak and alk inhibiters for treatment of cancers and other diseases |
| US8372971B2 (en) | 2004-08-25 | 2013-02-12 | Targegen, Inc. | Heterocyclic compounds and methods of use |
| US8481536B2 (en) | 2004-04-08 | 2013-07-09 | Targegen, Inc. | Benzotriazine inhibitors of kinases |
| WO2013160387A1 (en) | 2012-04-26 | 2013-10-31 | Bayer Cropscience Ag | Process for preparing n-(5-chloro-2-isopropylbenzyl)cyclopropanamine |
| CN108822145A (en) * | 2018-06-11 | 2018-11-16 | 广东工业大学 | A kind of sulfamide compound and its preparation method and application |
| CN109096178A (en) * | 2017-06-21 | 2018-12-28 | 肯塔基大学研究基金会 | Phenylethynyl-substituted benzenes and heterocycles for cancer treatment and applications thereof |
| EP4041241A1 (en) | 2019-09-27 | 2022-08-17 | Disc Medicine, Inc. | Methods for treating myelofibrosis and related conditions |
| US12365729B2 (en) | 2020-05-13 | 2025-07-22 | Disc Medicine, Inc. | Anti-hemojuvelin (HJV) antibodies for treating myelofibrosis |
| US12559454B2 (en) | 2019-07-17 | 2026-02-24 | 2692372 Ontario, Inc. | Benzenesulfonamide derivatives and uses thereof |
| US12583825B2 (en) | 2020-10-08 | 2026-03-24 | Redona Therapeutics, Inc. | Benzo[h]quinazolin-4-amine and thieno[3,2-h]quinazolin-4-amine derivatives for the treatment of cancer |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001042246A2 (en) * | 1999-12-10 | 2001-06-14 | Pfizer Products Inc. | PYRROLO[2,3-d]PYRIMIDINE COMPOUNDS |
-
2008
- 2008-10-24 WO PCT/US2008/081119 patent/WO2009055674A1/en not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001042246A2 (en) * | 1999-12-10 | 2001-06-14 | Pfizer Products Inc. | PYRROLO[2,3-d]PYRIMIDINE COMPOUNDS |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8481536B2 (en) | 2004-04-08 | 2013-07-09 | Targegen, Inc. | Benzotriazine inhibitors of kinases |
| US8372971B2 (en) | 2004-08-25 | 2013-02-12 | Targegen, Inc. | Heterocyclic compounds and methods of use |
| US20110282056A1 (en) * | 2008-01-22 | 2011-11-17 | Merck Patent Gmbh | Protein kinase inhibitors and use thereof |
| WO2012045195A1 (en) * | 2010-10-09 | 2012-04-12 | Abbott Laboratories | Pyrrolopyrimidines as fak and alk inhibiters for treatment of cancers and other diseases |
| WO2013160387A1 (en) | 2012-04-26 | 2013-10-31 | Bayer Cropscience Ag | Process for preparing n-(5-chloro-2-isopropylbenzyl)cyclopropanamine |
| CN109096178A (en) * | 2017-06-21 | 2018-12-28 | 肯塔基大学研究基金会 | Phenylethynyl-substituted benzenes and heterocycles for cancer treatment and applications thereof |
| CN109096178B (en) * | 2017-06-21 | 2022-12-16 | 肯塔基大学研究基金会 | Phenylethynyl-substituted benzenes and heterocycles for cancer treatment and applications thereof |
| CN108822145A (en) * | 2018-06-11 | 2018-11-16 | 广东工业大学 | A kind of sulfamide compound and its preparation method and application |
| CN108822145B (en) * | 2018-06-11 | 2020-10-23 | 广东工业大学 | A kind of sulfonamide compound and its preparation method and application |
| US12559454B2 (en) | 2019-07-17 | 2026-02-24 | 2692372 Ontario, Inc. | Benzenesulfonamide derivatives and uses thereof |
| EP4041241A1 (en) | 2019-09-27 | 2022-08-17 | Disc Medicine, Inc. | Methods for treating myelofibrosis and related conditions |
| US12365729B2 (en) | 2020-05-13 | 2025-07-22 | Disc Medicine, Inc. | Anti-hemojuvelin (HJV) antibodies for treating myelofibrosis |
| US12497452B2 (en) | 2020-05-13 | 2025-12-16 | Disc Medicine, Inc. | Anti-hemojuvelin (HJV) antibodies for treating myelofibrosis |
| US12583825B2 (en) | 2020-10-08 | 2026-03-24 | Redona Therapeutics, Inc. | Benzo[h]quinazolin-4-amine and thieno[3,2-h]quinazolin-4-amine derivatives for the treatment of cancer |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009055674A1 (en) | Pyrrolopyrimidine alkynyl compounds and methods of making and using same | |
| ES2902198T3 (en) | 1,4-Dissubstituted Pyridazine Analogs and Methods for Treating Conditions Related to SMN Deficiency | |
| EP3209656B1 (en) | Indole carboxamides compounds useful as kinase inhibitors | |
| CN102574857B (en) | Heterocyclic compounds as jak receptor and protein tyrosine kinase inhibitors | |
| EP3728207B1 (en) | Quinazolinones as parp14 inhibitors | |
| CN107531695B (en) | JAK inhibitors | |
| CN111315747B (en) | Dihydropyrazolopyrimidine compounds and their preparation methods and uses | |
| KR101727264B1 (en) | Imidazo[2,1-b][1,3,4]thiadiazole derivatives | |
| WO2009046416A1 (en) | Anilinopyrimidines as jak kinase inhibitors | |
| CN102471346B (en) | Tricyclic inhibitors of JAK | |
| JP2025505882A (en) | PARG inhibitors | |
| EA021504B1 (en) | 1-heterocyclyl-1,5-dihydropyrazolo[3,4-d]pyrimidin-4-one derivatives and their use as pde9a modulators | |
| KR20170002623A (en) | Pyrazolopyridines and pyrazolopyrimidines | |
| KR102345381B1 (en) | Carbazole carboxamide compounds useful as kinase inhibitors | |
| CN101595110A (en) | 7-substituted purine derivatives for immunosuppression | |
| JP2009502734A (en) | Fused heterocycles as Lck inhibitors | |
| JP2010523522A (en) | Pyrrolopyrimidine derivatives as JAK3 inhibitors | |
| CN102227422A (en) | Heterocyclic jak kinase inhibitors | |
| CN107207479B (en) | Substituted pyrimidines as phosphatidylinositol 3-kinase delta inhibitors and their applications | |
| WO2009049028A1 (en) | Pyrrolopyrimidine compounds and their use as janus kinase modulators | |
| CA2709710A1 (en) | Pyrazolo [1,5-a] pyrimidines useful as jak2 inhibitors | |
| KR20130004342A (en) | Pyrrolopyrazine derivatives and their use as jak and syk inhibitors | |
| WO2017000277A1 (en) | Substituted triazolo bicycliccompounds as pde2 inhibitors | |
| CN116444492A (en) | P38MAPK/MK2 pathway regulator, and composition, preparation method and application thereof | |
| WO2021099837A1 (en) | Adenosine receptor antagonist compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08843254 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08843254 Country of ref document: EP Kind code of ref document: A1 |