WO2009080248A1 - Substituierte 2-phenylpyrimidin-5-carbonsäuren und ihre verwendung - Google Patents
Substituierte 2-phenylpyrimidin-5-carbonsäuren und ihre verwendung Download PDFInfo
- Publication number
- WO2009080248A1 WO2009080248A1 PCT/EP2008/010698 EP2008010698W WO2009080248A1 WO 2009080248 A1 WO2009080248 A1 WO 2009080248A1 EP 2008010698 W EP2008010698 W EP 2008010698W WO 2009080248 A1 WO2009080248 A1 WO 2009080248A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- methyl
- alkyl
- trifluoromethyl
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1c(*)c(*)c(*)c(-c2nc(Cl)c(C(O*)=O)c(*)n2)c1 Chemical compound *c1c(*)c(*)c(*)c(-c2nc(Cl)c(C(O*)=O)c(*)n2)c1 0.000 description 8
- RMZXLQUPWCEUIM-UHFFFAOYSA-N CC(C)Cc1nc(-c(cc2)ccc2Cl)nc(OCC2CC2)c1C(O)=O Chemical compound CC(C)Cc1nc(-c(cc2)ccc2Cl)nc(OCC2CC2)c1C(O)=O RMZXLQUPWCEUIM-UHFFFAOYSA-N 0.000 description 1
- GVLNNVAHAVMELW-UHFFFAOYSA-N CC(C)Oc1c(C(O)=O)c(C)nc(-c(cc2)ccc2Cl)n1 Chemical compound CC(C)Oc1c(C(O)=O)c(C)nc(-c(cc2)ccc2Cl)n1 GVLNNVAHAVMELW-UHFFFAOYSA-N 0.000 description 1
- RBMOKAINBKGLRG-UHFFFAOYSA-N CCC(NC(c(cc1)ccc1Cl)=NC1CC(C)C)=C1C(OC)=O Chemical compound CCC(NC(c(cc1)ccc1Cl)=NC1CC(C)C)=C1C(OC)=O RBMOKAINBKGLRG-UHFFFAOYSA-N 0.000 description 1
- NHQKXSFPCHLSKS-UHFFFAOYSA-N CCCOc1c(C(OC)=O)c(C2CC2)nc(-c(cc2)ccc2Cl)n1 Chemical compound CCCOc1c(C(OC)=O)c(C2CC2)nc(-c(cc2)ccc2Cl)n1 NHQKXSFPCHLSKS-UHFFFAOYSA-N 0.000 description 1
- IADDNMQFYPFTRK-UHFFFAOYSA-N CCCc1nc(-c(cc2)ccc2Cl)nc(OCC(C)C)c1C(O)=O Chemical compound CCCc1nc(-c(cc2)ccc2Cl)nc(OCC(C)C)c1C(O)=O IADDNMQFYPFTRK-UHFFFAOYSA-N 0.000 description 1
- QTGFWPUFKNJDMM-UHFFFAOYSA-N CCOC(C(C(C)N=C(c(cc1)ccc1Cl)N1)C1=O)=O Chemical compound CCOC(C(C(C)N=C(c(cc1)ccc1Cl)N1)C1=O)=O QTGFWPUFKNJDMM-UHFFFAOYSA-N 0.000 description 1
- SELIUZVMQXQVPF-UHFFFAOYSA-N CCOC(c(c(C(C)C)nc(-c(cc1)ccc1Cl)n1)c1OC(C)C)=O Chemical compound CCOC(c(c(C(C)C)nc(-c(cc1)ccc1Cl)n1)c1OC(C)C)=O SELIUZVMQXQVPF-UHFFFAOYSA-N 0.000 description 1
- KBODNVIEHWOHJA-UHFFFAOYSA-N CCOC(c(c(C)nc(-c(cc1)ccc1Cl)n1)c1Cl)=O Chemical compound CCOC(c(c(C)nc(-c(cc1)ccc1Cl)n1)c1Cl)=O KBODNVIEHWOHJA-UHFFFAOYSA-N 0.000 description 1
- TZZOJZCNMUCDBK-UHFFFAOYSA-N CCc1nc(-c(cc2)ccc2Cl)nc(CC(C)C)c1C(OC)=O Chemical compound CCc1nc(-c(cc2)ccc2Cl)nc(CC(C)C)c1C(OC)=O TZZOJZCNMUCDBK-UHFFFAOYSA-N 0.000 description 1
- CUOWBEDLRKOMPP-UHFFFAOYSA-N COC(C(C(C1CC1)N=C(c(cc1)ccc1Cl)N1)C1=O)=O Chemical compound COC(C(C(C1CC1)N=C(c(cc1)ccc1Cl)N1)C1=O)=O CUOWBEDLRKOMPP-UHFFFAOYSA-N 0.000 description 1
- HWCVMEZURRVBIS-UHFFFAOYSA-N COC(C1=C(C2CC2)N=C(c(cc2)ccc2Cl)NC1=O)=O Chemical compound COC(C1=C(C2CC2)N=C(c(cc2)ccc2Cl)NC1=O)=O HWCVMEZURRVBIS-UHFFFAOYSA-N 0.000 description 1
- YJXURLJPBGRLNT-UHFFFAOYSA-N Cc1nc(-c(cc2)ccc2Cl)nc(OC2CCCC2)c1C(O)=O Chemical compound Cc1nc(-c(cc2)ccc2Cl)nc(OC2CCCC2)c1C(O)=O YJXURLJPBGRLNT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present application relates to novel substituted 2-phenylpyrimidine-5-carboxylic acid derivatives, processes for their preparation, their use for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of diseases, preferably for treatment and / or prophylaxis of cardiovascular diseases, especially dyslipidaemias, atherosclerosis and cardiac insufficiency.
- fibrates are currently the only treatment option for patients in these risk groups. They lower elevated triglycerides by 20-50%, lower LDL-C by 10-15%, change the LDL particle size from low density atherogenic LDL to normal dense and less atherogenic LDL and increase the HDL concentration by 10-15%.
- Fibrates act as weak agonists of the peroxisome proliferator-activated receptor (PPAR) - ⁇ (Nature 1990, 347, 645-50).
- PPAR-alpha is a nuclear receptor that regulates the expression of target genes by binding to DNA sequences in the promoter region of these genes [also called PPAR response elements (PPRE)].
- PPREs have been identified in a number of genes that encode proteins that regulate lipid metabolism.
- PPAR-alpha is highly expressed in the liver, and its activation leads, inter alia, to decreased VLDL production secretion and reduced apolipoprotein CHI (ApoCIII) synthesis. In contrast, the synthesis of apolipoprotein Al (ApoAl) is increased.
- a disadvantage of previously approved fibrates is their weak interaction with the receptor (EC 50 in the ⁇ M range), which in turn leads to the relatively low pharmacological effects described above.
- WO 99/41253 discloses substituted pyrimidines for the treatment of viral infections.
- WO 2004/111014 claims substituted pyrimidines for the treatment of cystic fibrosis.
- WO 2005/049573 and WO 2005/049606 describe substituted Pyrimidincarbonklar as Syntheseintermediate without biological activity.
- WO 2005/110416 discloses 4,5-disubstituted 2-arylpyrimidines as C5a receptor ligands for the treatment of inflammatory, immunological and cardiovascular diseases.
- WO 2006/124874 are under described other substituted pyrimidines for the treatment of cancer.
- WO 2006/097220 claims 4-phenoxy-2-phenylpyrimidinecarboxylic acids and in WO 2008/031500 and WO 2008/031501 4-phenoxy- or 2-phenoxynicotinic acids as PPAR-alpha modulators for the treatment of dyslipidemias and arteriosclerosis.
- the present invention relates to compounds of the general formula (I)
- R 1 is (C 3 -C 10) -alkyl, (C 3 -C 7 ) -cycloalkyl or -OR A ,
- (C 3 -C O) alkyl having 1 or 2 substituents independently selected from the group of fluorine, trifluoromethyl, hydroxy, (Ci-C 4) -alkoxy, trifluoromethoxy, and (C 3 -C 7) cycloalkyl may be substituted .
- R A is (C r C 10 ) -alkyl or (C 3 -C 7 ) -cycloalkyl
- (C 1 -C 10) -alkyl can be substituted by one substituent selected from the group consisting of hydroxyl, (C 1 -C 4 ) -alkoxy and (C 3 -C 7 ) -cycloalkyl,
- R 2 is (C 1 -C 4 ) -alkyl or cyclopropyl
- R 3 is hydrogen or fluorine
- R 4 is hydrogen, fluorine, chlorine, methyl or trifluoromethyl
- R 5 is hydrogen, halogen, nitro, cyano, trifluoromethyl, methyl, ethyl, trifluoromethoxy or methoxy,
- R 6 is hydrogen, fluorine, chlorine, methyl or trifluoromethyl
- R 3 , R 4 , R 5 and R 6 is different from hydrogen
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts comprising the compounds of the formulas below and their salts, solvates and solvates of the salts and of the formula (I) encompassed by formula (I), hereinafter referred to as exemplary compounds and their salts, solvates and solvates of the salts, as far as the compounds of formula (I), the compounds mentioned below are not already salts, solvates and solvates of the salts.
- the compounds of the invention may exist in stereoisomeric forms (enantiomers, diastereomers).
- the invention therefore includes the enantiomers or diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner.
- the present invention encompasses all tautomeric forms.
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. Also included are salts which are themselves unsuitable for pharmaceutical applications but can be used, for example, for the isolation or purification of the compounds of the invention.
- Physiologically acceptable salts of the compounds of the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid, acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid acetic acid, trifluoroacetic acid, propionic acid
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms, such as, by way of example and by way of illustration, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts
- solvates are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water. As solvates, hydrates are preferred in the context of the present invention.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs includes compounds which may themselves be biologically active or inactive, but during their residence time in the body are converted to compounds of the invention (for example metabolically or hydrolytically).
- the present invention also includes hydrolyzable ester derivatives of the carboxylic acids of the formula (I).
- esters which can be hydrolyzed in physiological media and in particular in vivo enzymatically or chemically to the free carboxylic acids.
- esters are linear or branched (Ci-C 6) -alkyl, wherein the alkyl group by hydroxy, (Ci-C 4) alkoxy, amino, mono (C 1 -C 4) -alkylamino and / or di- (C 1 -C 4 ) -alkylamino may be substituted.
- Particularly preferred are the methyl or ethyl esters of the compounds of formula (I).
- alkyl is a linear or branched alkyl radical having in each case the number of carbon atoms specified.
- alkyl is a linear or branched alkyl radical having in each case the number of carbon atoms specified.
- Cycloalkyl in the context of the invention is a monocyclic, saturated alkyl radical having 3 to 7 carbon atoms. Examples which may be mentioned by way of example include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- Alkoxy in the context of the invention is a linear or branched alkoxy radical having 1 to 4 carbon atoms. Examples which may be mentioned are: methoxy, ethoxy, n-propoxy, isopropoxy, 1-methylpropoxy, n-butoxy, isobutoxy and tert-butoxy.
- Halogen in the context of the invention includes fluorine, chlorine, bromine and iodine. Preference is given to chlorine or fluorine. If radicals are substituted in the compounds according to the invention, the radicals can, unless otherwise specified, be monosubstituted or polysubstituted. In the context of the present invention, the meaning is independent of each other for all radicals which occur repeatedly. Substitution with one, two or three identical or different substituents is preferred. Very particular preference is given to the substitution with a substituent.
- R 1 is (C 3 -C 8 ) -alkyl, cyclopropyl, cyclopentyl, cyclohexyl or -OR A ,
- (C 3 -C 8 ) -alkyl can be substituted by 1 or 2 substituents independently of one another selected from the group consisting of fluorine, trifluoromethyl, hydroxyl, methoxy, ethoxy and trifluoromethoxy,
- cyclopropyl, cyclopentyl and cyclohexyl can be substituted with 1 or 2 substituents independently of one another selected from the group fluorine, hydroxyl, oxo, methyl, ethyl, trifluoromethyl and methoxy,
- R A is methyl, ethyl, (C 3 -C 8 ) -alkyl, cyclopropyl, cyclopentyl or cyclohexyl,
- methyl and ethyl are substituted by a substituent selected from the group cyclopropyl, cyclopentyl and cyclohexyl,
- Substituents independently of one another can be substituted from the group fluorine, hydroxyl, oxo, methyl, ethyl, trifluoromethyl and methoxy,
- (C 3 -C 8 ) -alkyl can be substituted by 1 or 2 substituents independently of one another selected from the group consisting of hydroxyl, methoxy and ethoxy,
- cyclopropyl, cyclopentyl and cyclohexyl can be substituted with 1 or 2 substituents independently of one another selected from the group fluorine, hydroxyl, oxo, methyl, ethyl, trifluoromethyl and methoxy,
- R 2 is (C 1 -C 4 ) -alkyl, trifluoromethyl or cyclopropyl,
- R 3 is hydrogen
- R 4 is hydrogen or fluorine
- R 5 is hydrogen, fluorine, chlorine, trifluoromethyl or methyl
- R 6 is hydrogen or methyl
- R 1 is (C 3 -C 6 ) -alkyl, cyclopentyl, cyclohexyl, tetrahydrofuranyl or -OR A ,
- R A is methyl, (C 3 -C 6 ) -alkyl, cyclopropyl, cyclopentyl or cyclohexyl,
- methyl is substituted by a substituent selected from the group cyclopropyl, cyclopentyl and cyclohexyl,
- CyI may be substituted by 1 or 2 substituents independently of one another selected from the group consisting of hydroxy and methoxy,
- R 2 is methyl, ethyl, n-propyl, isopropyl, trifluoromethyl or cyclopropyl,
- R 3 is hydrogen
- R 4 is hydrogen or fluorine
- R 5 is hydrogen, chlorine or methyl
- R 6 is hydrogen or methyl
- R 1 is isopropyl, isobutyl, 1-methylpropyl, isopentyl, 1-methylbutyl or -OR A ,
- R A is isopropyl, iso-butyl, 1-methylpropyl, 1-methylbutyl or 3-methylbutyl,
- R 2 is methyl, ethyl or trifluoromethyl
- R 3 is hydrogen
- R 4 is hydrogen
- R 5 is hydrogen, chlorine or methyl
- R 6 is hydrogen or methyl
- R 5 and R 6 are other than hydrogen
- R 1 is isopropyl, isobutyl, 1-methylpropyl, isopentyl or 1-methylbutyl.
- R 1 is -OR A , where R A is isopropyl, isobutyl, 1-methylpropyl, 1-methylbutyl or 3-methylbutyl.
- Another object of the present invention is the compound 4-cyclopropyl-6- (methoxymethyl) -2- [4- (trifluoromethyl) phenyl] -pyrimidine-5-carboxylic acid for the prophylaxis and / or treatment of cardiovascular diseases.
- Another object of the present invention is the use of the compound 4-cyclopropyl-6- (methoxymethyl) -2- [4- (trifluoromethyl) phenyl] pyrimidine-5-carboxylic acid to
- the invention further provides a process for preparing the compounds of the formula (I) according to the invention, which comprises reacting a compound of the formula (II)
- R 2 , R 3 , R 4 , R 5 and R 6 each have the meanings given above,
- R 7 is (C 1 -C 4 ) -alkyl
- R ' A is -OR A
- R 1A , R 2 , R 3 , R 4 , R 5 , R 6 and R 7 each have the meanings given above,
- R 2 , R 3 , R 4 , R 5 , R 6 and R 7 each have the meanings given above,
- R IB is (C 3 -C 10) -alkyl or (C 3 -C 7 ) -cycloalkyl
- (C 3 -C 10) -alkyl can be substituted by one or two substituents independently of one another selected from the group consisting of fluorine, trifluoromethyl, hydroxyl, (CrQ) -alkoxy, trifluoromethoxy and (C 3 -C 7 ) -cycloalkyl,
- X 1 represents a group of the formula -B (OR 8 ) 2 or -ZnHaI
- Hal is halogen, in particular chlorine, bromine or iodine,
- R 8 is hydrogen or (C r C 4 ) alkyl
- both radicals R 8 together form a -C (CH 3 ) 2 -C (CH 3 ) 2 bridge
- R 1B , R 2 , R 3 , R 4 , R 5 , R 6 and R 7 each have the meanings given above,
- the compounds of the formula (II) can be prepared by reacting compounds of the formula (VI)
- R 2 , R 3 , R 4 , R 5 , R 6 and R 7 each have the meanings given above,
- R 2 , R 3 , R 4 , R 5 , R 6 and R 7 each have the meanings given above,
- the invention further provides a process for the preparation of the compounds of the formula (I) according to the invention in which R 1 is R 1B in which R 1B has the abovementioned meanings, characterized in that a compound of the formula (X)
- R 1B has the abovementioned meaning
- R 1B , R 2 , R 3 , R 4 , R 5 , R 6 and R 7 each have the meanings given above,
- R 1B , R 2 , R 3 , R 4 , R 5 , R 6 and R 7 each have the meanings given above,
- the compound 4-cyclopropyl-6- (methoxymethyl) -2- [4- (trifluoromethyl) phenyl] pyrimidine-5-carboxylic acid can be prepared analogously to the above-described process [B] or as described in WO 2005/049573.
- the reaction (EI) -> (HI) is carried out without solvent or optionally in an inert solvent suitable under the reaction conditions such as hydrocarbons such as benzene, toluene, xylene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide, dimethyl sulfoxide, N-methylpyrrolidone ( ⁇ MP) or acetonitrile. It is likewise possible to use mixtures of the solvents mentioned.
- the reaction preferably takes place without solvent.
- the reaction (ET) -> (IH) is generally carried out in a temperature range from 0 0 C to +160 0 C, preferably at +20 0 C to +120 0 C, optionally in a microwave.
- the reaction can be carried out at normal, elevated or at reduced pressure (eg from 0.5 to 5 bar). Generally, one works at normal pressure.
- Transition metal catalysts, catalyst ligands and auxiliary bases for the coupling reactions (V) + (III-B) ⁇ (IV-B) are known from the literature [cf. e.g. J. Hassan et al., Chem. Rev. JO2, 1359-1469 (2002)] and commercially available. Preference is given to using palladium or nickel catalysts.
- tetrakis (triphenylphosphine) palladium (0) is preferably used as the catalyst.
- Diethylene glycol dimethyl ether such as benzene, toluene, xylene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide, dimethyl sulfoxide, NN '
- DMPU Dimethylpropyleneurea
- ⁇ MP N-methylpyrrolidone
- pyridine pyridine
- acetonitrile acetonitrile or even water. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to using dimethylformamide or dioxane.
- the reactions can be carried out at normal, elevated or reduced pressure (eg from 0.5 to 5 bar).
- triphenylphosphine or tri-n-butylphosphine, 1, 2-bis (diphenylphosphino) ethane (DPPE), diphenyl (2-pyridyl) phosphine (Ph2P-Py), (p-dimethylaminophenyl ) diphenylphosphine (DAP-DP), tris (4-dimethylaminophenyl) phosphine (tris-DAP) and a suitable dialkyl azodicarboxylate such as diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD), di-tert-butyl azodicarboxylate, NNN'N ' -Tetramethylazodicarboxamide (TMAD), I, L '- (azodicarbonyl) - dipiperidine (ADDP) or 4,7-dimethyl-3,5,7-
- DPPE 1, 2-bis (diphen
- Inert solvents for the Mitsunobu reaction (ET) + (IQ-A) ⁇ (IV-A) are, for example, ethers such as tetrahydrofuran, diethyl ether, hydrocarbons such as benzene, toluene, xylene, halogenated hydrocarbons such as dichloromethane, dichloroethane or other solvents such as acetonitrile. It is likewise possible to use mixtures of the solvents mentioned. Preferably, THF is used.
- the Mitsunobu reaction (U) + (IH-A) -> • (IV-A) is generally carried out in a temperature range from -78 ° C to +180 0 C, preferably at 0 0 C to + 50 0 C, optionally in a microwave.
- the reactions can be carried out at normal, elevated or reduced pressure (eg from 0.5 to 5 bar).
- the hydrolysis of the carboxylic acid esters in process steps (IV-A) - »(IA) and (IV-B) -> (IB) is carried out by customary methods, if appropriate in a microwave, by treating the esters in acids or bases with inert solvents , Where the salts initially formed in the latter are converted into the free carboxylic acids by subsequent treatment with acid.
- the tert-butyl ester ester cleavage is preferably carried out with acids.
- Suitable inert solvents for the hydrolysis of the carboxylic acid esters are water or the organic solvents customary for ester cleavage. These include in particular alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert. Butanol, ethers such as diethyl ether, tetrahydrofuran, dioxane or glycol dimethyl ether, or other solvents such as acetone, acetonitrile, dichloromethane, dimethylformamide or dimethyl sulfoxide. It is likewise possible to use mixtures of the solvents mentioned.
- Suitable bases for the ester hydrolysis are the customary inorganic bases. These include in particular alkali or alkaline earth hydroxides such as sodium, lithium, potassium or barium hydroxide, or alkali or alkaline earth metal carbonates such as sodium, potassium or calcium carbonate. Preference is given to using sodium hydroxide or potassium hydroxide.
- Suitable acids for the ester cleavage are generally sulfuric acid, hydrogen chloride / hydrochloric acid, hydrogen bromide / hydrobromic acid, phosphoric acid, acetic acid, trifluoroacetic acid, Toluene sulfonic acid, methanesulfonic acid or trifluoromethanesulfonic acid or mixtures thereof, optionally with the addition of water. Hydrogen chloride or trifluoroacetic acid are preferred in the case of the tert-butyl esters and hydrochloric acid in the case of the methyl esters.
- the Esterspaltung is generally carried out in a temperature range of 0 0 C to +100 0 C, forthcoming Trains t at 0 0 C to +50 0 C.
- the reaction can be carried out at atmospheric, elevated or reduced pressure (for example from 0.5 to 5 bar).
- R 1A is -H, triphenylphosphine, DIAD, room temperature].
- the compounds according to the invention have valuable pharmacological properties and can be used for the prevention and treatment of diseases in humans and animals.
- the compounds according to the invention are highly effective PPAR-alpha modulators and moreover have increased metabolic stability. They are particularly useful for primary and / or secondary prevention and treatment of cardiovascular disorders caused by disorders in fatty acid and glucose metabolism.
- cardiovascular disorders include dyslipidemias (hypercholesterolemia, hypertriglyceridemia, increased levels postprandial plasma triglycerides, hypoalphalipoproteinemia, combined hyperlipidemias), arteriosclerosis and metabolic disorders (metabolic syndrome, hyperglycemia, insulin-dependent diabetes, non-insulin-dependent diabetes, gestational diabetes, hyperinsulinemia, insulin resistance, glucose intolerance, obesity and diabetic Late effects such as retinopathy, nephropathy and neuropathy).
- the compounds of the invention are particularly suitable for primary and / or secondary prevention and treatment of heart failure.
- cardiac insufficiency also encompasses more specific or related forms of disease such as right heart failure, left heart failure, global insufficiency, hypertension-induced heart failure, ischemic cardiomyopathy, dilated cardiomyopathy, congenital heart defects, valvular heart failure, cardiac insufficiency in heart valve defects, mitral valve stenosis, mitral valve insufficiency, aortic valve stenosis, aortic valve insufficiency , Tricuspid stenosis, tricuspid insufficiency, pulmonary valve stenosis, pulmonary valvular insufficiency, combined valvular heart failure, myocarditis, chronic myocarditis, acute myocarditis, viral myocarditis, diabetic heart failure, alcoholic cardiomyopathy, cardiac storage disorders, diastolic heart failure, and systolic heart failure.
- myocarditis chronic myocarditis, acute myocarditis, viral myo
- CAD-I independent cardiovascular disease risk factors
- hypertension ischemia, myocardial infarction, angina pectoris, myocardial insufficiency, restenosis, pulmonary hypertension, increased levels of fibrinogen and low density LDL, and elevated levels of plasminogen activator.
- Inhibitor 1 PAI-I
- the compounds according to the invention can also be used for the treatment and / or prevention of cancers such as, for example, skin cancer, breast cancer, brain tumors, head and neck tumors, liposarcomas, carcinomas of the eye, the gastrointestinal tract, the thyroid, the liver, the pancreas, the respiratory organs, the kidney, the ureter, the prostate, the genital tract and their distant metastases, and lyphomas, sarcomas and leukemias.
- cancers such as, for example, skin cancer, breast cancer, brain tumors, head and neck tumors, liposarcomas, carcinomas of the eye, the gastrointestinal tract, the thyroid, the liver, the pancreas, the respiratory organs, the kidney, the ureter, the prostate, the genital tract and their distant metastases, and lyphomas, sarcomas and leukemias.
- the compounds according to the invention can also be used for the treatment and / or prevention of micro- and macrovascular damage (vasculitis), reperfusion damage, arterial and venous thromboses, edema, diseases of the central nervous system and neurodegenerative disorders (stroke, Alzheimer's disease, Parkinson's disease).
- vasculitis micro- and macrovascular damage
- reperfusion damage arterial and venous thromboses
- edema diseases of the central nervous system and neurodegenerative disorders
- stroke Alzheimer's disease, Parkinson's disease.
- the activity of the compounds of the invention can be e.g. in vitro by the transactivation assay described in the Examples section.
- the efficacy of the compounds of the invention in vivo can be e.g. Check by the tests described in the example section.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prevention of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prevention of diseases, in particular the aforementioned diseases.
- Another object of the present invention is a method for the treatment and / or prevention of diseases, in particular the aforementioned diseases, using an effective amount of at least one of the compounds of the invention.
- Another object of the present invention are the compounds of the invention for use in a method for the treatment and / or prophylaxis of dyslipidaemias, arteriosclerosis and heart failure.
- the compounds of the invention may be used alone or as needed in combination with other agents.
- Another object of the present invention are pharmaceutical compositions containing at least one of the compounds of the invention and one or more other active ingredients, in particular for the treatment and / or prevention of the aforementioned diseases.
- Suitable combination active ingredients which may be mentioned by way of example are active substances which alter the metabolism of lipids, antidiabetics, blood pressure depressants, circulation-promoting and / or or antithrombotic agents and antioxidants, chemokine receptor antagonists, p38 kinase inhibitors, NPY agonists, orexin agonists, anorectics, PAF-AH inhibitors, anti-inflammatory drugs (COX inhibitors, LTB 4 receptor antagonists), analgesics (Aspirin), antidepressants and other psychotropic drugs.
- active substances which alter the metabolism of lipids, antidiabetics, blood pressure depressants, circulation-promoting and / or or antithrombotic agents and antioxidants, chemokine receptor antagonists, p38 kinase inhibitors, NPY agonists, orexin agonists, anorectics, PAF-AH inhibitors, anti-inflammatory drugs (COX inhibitors, LTB 4 receptor antagonists), analgesics (Aspirin),
- the present invention relates, in particular, to combinations of at least one of the compounds according to the invention with at least one lipid metabolism-altering active ingredient, an antidiabetic agent, a hypotensive agent and / or an antithrombotic agent.
- the compounds of the invention may preferably be with one or more
- the substances which modify the lipid metabolism by way of example and preferably from the group of HMG-CoA reductase inhibitors, inhibitors of HMG-CoA reductase expression, squalene synthesis inhibitors, ACAT inhibitors, LDL receptor inducers, cholesterol Anti-absorption agents, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, MTP inhibitors, lipase inhibitors, LpL activators, fibrates, niacin, CETP inhibitors, PPAR- ⁇ and / or PPAR- ⁇ agonists, RXR modulators, FXR modulators , LXR modulators, thyroid hormones and / or thyroid mimetics, ATP citrate lyase inhibitors, Lp (a) antagonists, cannabinoid receptor 1 antagonists, leptin receptor agonists, bombesin receptor agonists, histamine receptor Agonists as well as the antioxidants / free radical scavengers;
- Antidiabetic agents mentioned in the Red List 2004 / ⁇ , Chapter 12, and by way of example and preferably those from the group of sulfonylureas, biguanides, meglitinide derivatives,
- Glucosidase inhibitors oxadiazolidinones, thiazolidinediones, GLP 1 receptor agonists, glucagon antagonists, insulin sensitizers, CCK 1 receptor agonists, leptin receptor agonists, inhibitors of liver enzymes that stimulate gluconeogenesis and / or glycogenolysis involved, modulators of glucose uptake and the potassium channel opener, such as those disclosed in WO 97/26265 and WO 99/03861;
- hypotensive agents by way of example and preferably from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, beta-receptor blockers, alpha-receptor blockers, ECE inhibitors and the vasopeptidase inhibitors;
- Antithrombotic agents by way of example and preferably from the group of platelet aggregation inhibitors or anticoagulants;
- cGMP cyclic guanosine monophosphate
- cAMP cyclic adenosine monophosphate
- PDE phosphodiesterases
- sildenafil sildenafil
- Vardenafil tadalafil
- PDE 3 inhibitors such as milrinone
- Natriuretic peptides e.g. atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP, Nesiritide), C-type natriuretic peptide (CNP) and urodilatin;
- ABP atrial natriuretic peptide
- BNP B-type natriuretic peptide
- CNP C-type natriuretic peptide
- urodilatin urodilatin
- Calcium sensitizers such as by way of example and preferably levosimendan
- NO-independent, but heme-dependent guanylate cyclase stimulators in particular those described in WO 00/06568, WO 00/06569, WO 02/42301 and WO 03/095451
- Guanylate cyclase NO- and heme-independent activators in particular the compounds described in WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 and WO 02/070510;
- HNE human neutrophil elastase
- the signal transduction cascade inhibiting compounds such as tyrosine kinase inhibitors, especially sorafenib, imatinib, gefitinib and erlotinib; and or
- lipid metabolism-changing active compounds are preferably compounds from the group of HMG-CoA reductase inhibitors, squalene synthesis inhibitors, ACAT inhibitors, cholesterol absorption inhibitors, MTP inhibitors, lipase inhibitors, thyroid hormones and / or thyroid mimetics, niacin receptor Agonists, CETP inhibitors, PPAR-gamma agonists, PPAR delta agonists, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, antioxidants / radical scavengers, and the cannabinoid receptor 1 antagonists.
- HMG-CoA reductase inhibitors preferably compounds from the group of HMG-CoA reductase inhibitors, squalene synthesis inhibitors, ACAT inhibitors, cholesterol absorption inhibitors, MTP inhibitors, lipase inhibitors, thyroid hormones and / or thyroid mimetics, niacin receptor Agonists, CETP inhibitors, PPAR-gam
- the compounds according to the invention are administered in combination with an HMG-CoA reductase inhibitor from the class of statins, by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin, cerivastatin or pitavastatin ,
- an HMG-CoA reductase inhibitor from the class of statins, by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin, cerivastatin or pitavastatin ,
- the compounds according to the invention are administered in combination with a squalene synthesis inhibitor, such as by way of example and preferably BMS-188494 or TAK-475.
- a squalene synthesis inhibitor such as by way of example and preferably BMS-188494 or TAK-475.
- the compounds according to the invention are administered in combination with an ACAT inhibitor, such as by way of example and preferably melinamide, pactimibe, eflucimibe or SMP-797.
- an ACAT inhibitor such as by way of example and preferably melinamide, pactimibe, eflucimibe or SMP-797.
- the compounds according to the invention are administered in combination with a cholesterol absorption inhibitor such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- a cholesterol absorption inhibitor such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- the compounds according to the invention are administered in combination with an MTP inhibitor, such as by way of example and preferably implitapide or JTT-130.
- the compounds according to the invention are administered in combination with a lipase inhibitor, such as, for example and preferably, orlistat.
- a lipase inhibitor such as, for example and preferably, orlistat.
- the compounds according to the invention are administered in combination with a thyroid hormone and / or thyroid mimetic, such as by way of example and preferably D-thyroxine or 3,5,3'-triiodothyronine (T3).
- a thyroid hormone and / or thyroid mimetic such as by way of example and preferably D-thyroxine or 3,5,3'-triiodothyronine (T3).
- the compounds according to the invention are administered in combination with an agonist of the niacin receptor, such as by way of example and preferably niacin, Acipimox, A mecanical or Radecol.
- an agonist of the niacin receptor such as by way of example and preferably niacin, Acipimox, A mecanical or Radecol.
- the compounds according to the invention are administered in combination with a CETP inhibitor, such as, by way of example and by way of preference, torcetrapib, JTT-705 or CETP vaccine (Avant).
- the compounds according to the invention are administered in combination with a PPAR-gamma agonist, such as by way of example and preferably pioglitazone or rosiglitazone.
- a PPAR-gamma agonist such as by way of example and preferably pioglitazone or rosiglitazone.
- the compounds of the invention are administered in combination with a PPAR delta agonist such as, for example and preferably, GW-501516.
- a PPAR delta agonist such as, for example and preferably, GW-501516.
- the compounds according to the invention are administered in combination with a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- ASBT IBAT
- AZD-7806 S-8921
- AK-105 AK-105
- BARI-1741 AK-105
- SC-435 SC-635.
- the compounds according to the invention are administered in combination with an antioxidant / free-radical scavenger such as, by way of example and by way of preference, probucol, AGI-1067, BO-653 or AEOL-10150.
- an antioxidant / free-radical scavenger such as, by way of example and by way of preference, probucol, AGI-1067, BO-653 or AEOL-10150.
- the compounds of the invention are administered in combination with a cannabinoid receptor 1 antagonist, such as by way of example and preferably rimonabant or SR-147778.
- a cannabinoid receptor 1 antagonist such as by way of example and preferably rimonabant or SR-147778.
- Antidiabetic agents are preferably understood as meaning insulin and insulin derivatives as well as orally active hypoglycemic agents.
- Insulin and insulin derivatives here include both insulins of animal, human or biotechnological origin as well as mixtures thereof.
- the orally active hypoglycemic agents preferably include sulphonylureas, biguanides, meglitinide derivatives, glucosidase inhibitors and PPAR-gamma agonists.
- the compounds according to the invention are administered in combination with insulin.
- the compounds according to the invention are administered in combination with a sulphonylurea, such as, by way of example and by way of preference, tolbutamide, glibenclamide, glimepiride, glipizide or gliclazide.
- the compounds according to the invention are administered in combination with a biguanide, such as by way of example and preferably metformin.
- the compounds of the invention are administered in combination with a meglitinide derivative, such as by way of example and preferably repaglinide or nateglinide.
- a meglitinide derivative such as by way of example and preferably repaglinide or nateglinide.
- the compounds according to the invention are administered in combination with a glucosidase inhibitor, such as by way of example and preferably miglitol or acarbose.
- a glucosidase inhibitor such as by way of example and preferably miglitol or acarbose.
- the compounds according to the invention are administered in combination with a PPAR-gamma agonist, for example from the class of thiazolidinediones, such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- a PPAR-gamma agonist for example from the class of thiazolidinediones, such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- the blood pressure lowering agents are preferably understood as meaning compounds from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, beta-receptor blockers, alpha-receptor blockers and diuretics.
- the compounds of the invention are administered in combination with a diuretic, such as by way of example and preferably a loop diuretic such as furosemide, bumetanide or torsemide, or a thiazide or thiazide-like diuretic such as chlorothiazide or hydrochlorothiazide.
- a diuretic such as by way of example and preferably a loop diuretic such as furosemide, bumetanide or torsemide, or a thiazide or thiazide-like diuretic such as chlorothiazide or hydrochlorothiazide.
- the compounds according to the invention are administered in combination with an aldosterone or mineralocorticoid receptor antagonist, such as by way of example and preferably spironolactone or eplerenone.
- an aldosterone or mineralocorticoid receptor antagonist such as by way of example and preferably spironolactone or eplerenone.
- the compounds according to the invention are administered in combination with a vasopressin receptor antagonist, such as by way of example and preferably Conivaptan, tolvaptan, lixivaptan or SR-121463.
- a vasopressin receptor antagonist such as by way of example and preferably Conivaptan, tolvaptan, lixivaptan or SR-121463.
- the compounds according to the invention are used in combination with an organic nitrate or NO donor, such as by way of example and by way of example.
- an organic nitrate or NO donor such as by way of example and by way of example.
- the compounds according to the invention are used in combination with a positive inotropically active compound, such as by way of example and preferably cardiac glycosides (digoxin), beta-adrenergic and dopaminergic agonists such as isoproterenol, adrenaline, norepinephrine, dopamine or dobutamine, administered.
- a positive inotropically active compound such as by way of example and preferably cardiac glycosides (digoxin), beta-adrenergic and dopaminergic agonists such as isoproterenol, adrenaline, norepinephrine, dopamine or dobutamine, administered.
- the compounds according to the invention are administered in combination with a calcium antagonist, such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- a calcium antagonist such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- the compounds according to the invention are administered in combination with an angiotensin AQ antagonist, such as by way of example and preferably losartan, valsartan, candesartan, embusartan or telmisartan.
- angiotensin AQ antagonist such as by way of example and preferably losartan, valsartan, candesartan, embusartan or telmisartan.
- the compounds according to the invention are administered in combination with an ACE inhibitor such as, for example and preferably, enalapril, captopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- an ACE inhibitor such as, for example and preferably, enalapril, captopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- the compounds according to the invention are used in combination with a beta-receptor blocker, such as by way of example and preferably propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipranolol, nadolol, mepindolol, carazalol, Sotalol, metoprolol, betaxolol, celiprolol, bisoprolol, Carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol or bucine dolol administered.
- a beta-receptor blocker such as by way of example and preferably propranolol, atenolol, timolo
- the compounds according to the invention are administered in combination with an alpha-receptor blocker, such as by way of example and preferably prazosin.
- the compounds according to the invention are used in combination with an antisympathotonicum, such as by way of example and preferably reserpine, clonidine or alpha-methyl-dopa, or in combination with a potassium channel agonist such as, for example and preferably, minoxidil, diazoxide, dihydralazine or hydralazine.
- an antisympathotonicum such as by way of example and preferably reserpine, clonidine or alpha-methyl-dopa
- a potassium channel agonist such as, for example and preferably, minoxidil, diazoxide, dihydralazine or hydralazine.
- Antithrombotic agents are preferably understood as meaning compounds from the group of platelet aggregation inhibitors or anticoagulants.
- the compounds according to the invention are administered in combination with a platelet aggregation inhibitor, such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- the compounds according to the invention are administered in combination with a thrombin inhibitor, such as by way of example and preferably ximelagarran, melagatran, bivalirudin or Clexane.
- a thrombin inhibitor such as by way of example and preferably ximelagarran, melagatran, bivalirudin or Clexane.
- the compounds according to the invention are administered in combination with a GPUb / IHa antagonist, such as by way of example and preferably tirofiban or abciximab.
- the compounds according to the invention are used in combination with a factor Xa inhibitor, such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD No. 3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 or SSR-128428.
- a factor Xa inhibitor such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD No. 3112, YM-150, KFA-1982, EMD-503982, MCM
- the compounds according to the invention are administered in combination with heparin or a low molecular weight (LMW) heparin derivative.
- LMW low molecular weight
- the compounds according to the invention are administered in combination with a vitamin K antagonist, such as by way of example and preferably coumarin.
- HMG-CoA reductase inhibitors statins
- diuretics beta-receptor blockers
- organic nitrates and NO Donors ACE inhibitors
- angiotensin Aü antagonists aldosterone and mineralocorticoid receptor antagonists
- vasopressin receptor antagonists platelet aggregation inhibitors and anticoagulants, and their use for the treatment and / or prevention of the aforementioned diseases.
- the compounds according to the invention can be used for the treatment and / or prophylaxis of cancers alone or as needed in combination with other anti-tumor agents.
- the present invention particularly relates to combinations of at least one of the compounds according to the invention with at least one other anti-tumor active ingredient selected from the group consisting of alkylating agents, antimetabolites, of Plant derived anti-tumor agents, hormone therapy agents, topoisomerase inhibitors, camptothecin derivatives, kinase inhibitors, targeted drugs, antibodies, immunoconjugates, interferon and / or immunomodulators, antiangiogenic compounds, antisense RNA and RNA interference, and others Tumor medication.
- Alkylating agents such as chloromethine N-oxide, cyclophosphamide, ifosfamide, thiotepa, ranimustine, ⁇ imustin, temozolomide, altretamine, apaciquin, brostallicin, bendamustine, carmustine, estramustine, fotemustine, glufosfamide, mafosfamide and mitolactol;
- Platinum-coordinated alkylating agents such as, for example, cisplatin, carboplatin, eptaplatin, lobaplatin, ⁇ edaplatin, oxaliplatin and satraplatin;
- Antimetabolites such as methotrexate, 6-mercaptopurine riboside, mercaptopurine, 5-fluorouracil alone or in combination with leucovorin, tegafur, doxifluridine, carmofur, cytarabine, cytarabine ocfosfate, enocitabine, gemcitabine, fludarabine, 5-azacitidine, capecitabine, cladribine, clofarabine, Decitabine, eflornithine, ethynylcytidine, cytosine arabinoside, hydroxyurea, melphalan, ⁇ elarabine, ⁇ olatrexed, ocfosfit, disodium
- Premetrexed pentostatin, pelitrexol, raltitrexed, triapine, trimetrexate, vidarabine, vincristine, and vinorelbine;
- hormonal agents such as exemestane, lupron, anastrozole, doxercalciferol, fadrozole, formestan, 11-beta hydroxysteroid dehydrogenase-1 inhibitors, 17-alpha hydroxylase / 17,20 lyase inhibitors such as abiraterone acetate, 5-alpha reductase
- Inhibitors such as finasteride and epristeride, anti-estrogens such as tamoxifen citrate and fulvestrant, trelstar, toremifene, raloxifene, lasofoxifene, letrozole, anti-androgens such as bicalutamide, flutamide, mifepristone, ilutamide, Casodex and anti-progesterone, and combinations thereof;
- anti-estrogens such as tamoxifen citrate and fulvestrant, trelstar, toremifene, raloxifene, lasofoxifene, letrozole
- anti-androgens such as bicalutamide, flutamide, mifepristone, ilutamide, Casodex and anti-progesterone, and combinations thereof;
- plant-derived antitumor agents such as mitotic inhibitors such as epothilones (sagopilone, ixabepilone and epothilone B), vinblastine, vinflunine, docetaxel, and paclitaxel;
- mitotic inhibitors such as epothilones (sagopilone, ixabepilone and epothilone B), vinblastine, vinflunine, docetaxel, and paclitaxel;
- Cytotoxic topoisomerase inhibitors such as aclarubicin, doxorubicin, amonafide, belotecan, camptothecin, 10-hydroxycamptothecin, 9-aminocamptothecin, diflomotecan, irinotecan, topotecan, edotecarin, epimbicin, etoposide, exatecan, germantcan, lurtotecan, mitoxantrone, pirambicin, pixantrone, Rubitecan, Sobuzoxan, Tafluposide, and combinations thereof; • Immunological agents exemplified and preferably from the group of interferons such.
- Ibritumomab Imiquimod, Lenograstim, Lentinan, Melanoma Vaccine (Corixa), Molgramostim, Sargramostim, Tasonermin, Tecleukin, Thymalasin, Tositumomab, Vimlizine, Epratuzumab, Mitumomab, Oregovomab, Pemtumomab and Proveng;
- Immunomodulators such as Krestin, Lentinan, Sizofiran, Picibanil, ProMun and Ubenimex;
- Antiangiogenic compounds such as, for example, acitretin, aflibercept, angiostatin, aplidine, asentar, axitinib, recentin, bevacizumab, brivanib alaninate, cilengtide, combretastatin, DAST, endostatin, fenretinide, halofuginone, pazopanib, ranibizumab, rebarastat, Removab, Revlimid , Sorafenib, vatalanib, squalamine, sunitinib, telatinib, thalidomide, Ukrain and vitaxin;
- VEGF inhibitors such as sorafenib, DAST, bevacizumab, sunitinib, recentin, axitinib, aflibercept, telatinib, brivanib alaninate, vatalanib, pazopanib, and ranibizumab;
- antibodies such as trastuzumab, cetuximab, bevacizumab, rituximab, ticilimumab, ipilimumab, lumiliximab, catumaxomab, atacicept, orregovomab and alemtumab;
- EGFR (HERI) inhibitors such as cetuximab, panitumumab, vectibix, gefitinib, erlotinib and Zactima;
- HER2 inhibitors such as lapatinib, tratuzumab and pertuzumab;
- mTOR inhibitors such as temsirolimus, sirolimus / rapamycin and everolimus;
- CDK inhibitors such as roscovitine and flavopiridol; • Spindle assembly checkpoint inhibitors and targeted mitotic inhibitors such as PLK inhibitors, Aurora inhibitors (eg hesperadine), checkpoint kinase inhibitors and KSP inhibitors;
- HDAC inhibitors such as Panobinostat, Vorinostat, MS275, Belinostat and LBH589;
- Proteasome inhibitors such as bortezomib and carf ⁇ lzomib;
- Serine / threonine kinase inhibitors such as MEK inhibitors and Raf inhibitors such as sorafenib;
- Tyrosine kinase inhibitors such as dasatinib, nilotibib, DAST, bosutinib, sorafenib, bevacizumab, sunitinib, AZD2171, axitinib, aflibercept, telatinib, imatinib mesylate, brivanib alaninate, pazopanib, ranibizumab, vatalanib, cetuximab, panitumumab, vectibix, gefitinib, erlotinib , Lapatinib, tratuzumab, pertuzumab and c-kit inhibitors;
- Corticoids e.g. dexamethasone
- Thalidomide or thalidolide analogs e.g. lenalidomide
- Bcl-2 protein inhibitors such as Obatoclax, Oblimersen sodium and Gossypol;
- CD20 receptor antagonists such as rituximab
- Ribonucleotide reductase inhibitors such as gemcitabine
- Tumor necrosis apoptosis inducing ligand receptor 1 agonists such as Mapatumumab
- 5-hydroxytryptamine receptor antagonists such as rEV598, xaliprod, palonosetron hydrochloride, granisetron, zindol and AB-1001; • Integrin inhibitors including Alpha5-betal integrin inhibitors. E7820, JSM 6425, Volociximab and Endostatin;
- Androgen receptor antagonists including e.g. Nandrolone Decanoate, Fluoxymesterone, Android, Prost-Aid, Andromustine, Bicalutamide, Flutamide, Apo-cyproterone, Apo-Flutamide, Chlormadinone Acetate, Androcur, Tabi, Cyproterone Acetate and Nilutamide;
- aromatase inhibitors such as Anastrozole, letrozole, testolactone, exemestane, amino-glutethimide and formestane;
- the compounds of the invention may also be used to treat cancers associated with radiotherapy and / or surgery.
- compositions containing at least one compound of the invention usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the compounds according to the invention can act systemically and / or locally.
- they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds of the invention rapidly and / or modified donating application forms containing the inventive compounds in crystalline and / or amorphized and / or dissolved form, such as tablets (uncoated or coated Tablets, for example with enteric or delayed-dissolving or insoluble coatings, which control of the compound according to the invention), rapidly disintegrating tablets or films / wafers, films / lyophilisates, capsules (for example hard or soft gelatin capsules), dragees, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- tablets uncoated or coated Tablets, for example with enteric or delayed-dissolving or insoluble coatings, which control of the compound according to the invention
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenous, intraarterial, intracardiac, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscular, subcutaneous, intracutaneous, percutaneous, or intraperitoneal).
- a resorption step e.g., intravenous, intraarterial, intracardiac, intraspinal, or intralumbar
- absorption e.g., intramuscular, subcutaneous, intracutaneous, percutaneous, or intraperitoneal.
- parenteral administration are suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicaments including powder inhalers, nebulizers
- nasal drops solutions or sprays
- lingual, sublingual or buccal tablets films / wafers or capsules
- suppositories ear or ophthalmic preparations
- vaginal capsules aqueous suspensions (lotions, shake mixtures), lipophilic suspensions
- Ointments creams, transdermal therapeutic systems (eg patches), milk, pastes, foams, scattering powders, implants or stents.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitanoleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- Stabilizers eg antioxidants such as ascorbic acid
- dyes eg inorganic pigments such as iron oxides
- flavor and / or odoriferous include, among others.
- Excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl
- the dosage is about 0.01 to 100 mg / kg, preferably about 0.01 to 20 mg / kg and most preferably 0.1 to 10 mg / kg of body weight.
- UV ultraviolet spectrometry v / v volume-to-volume ratio (of a mixture)
- Method 1 Device Type MS: Micromass ZQ; Device type HPLC: HP 1100 Series; UV DAD; Column: Phenomenex Gemini 3 ⁇ 30 mm x 3.00 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A ⁇ 2.5 min 30% A ⁇ 3.0 min 5% A ⁇ 4.5 min 5% A; Flow: 0.0 min 1 ml / min ⁇ 2.5 min / 3.0 min / 4.5 min 2 ml / min; Oven: 50 ° C .; UV detection: 210 nm.
- Method 3 Device Type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2795; Column: Merck Chromolith SpeedROD RP-18e 100 x 4.6 mm; Eluent A: 1 liter of water + 0.5 ml of 50% formic acid; Eluent B: 1 liter acetonitrile + 0.5 ml 50% formic acid; Gradient: 0.0 min 10% B-> 7.0 min 95% B-> 9.0 min 95% B; Oven: 35 ° C; Flow: 0.0 min 1.0 ml / min -> 7.0 min 2.0 ml / min-> 9.0 min 2.0 ml / min; UV detection: 210 nm
- Method 4 Instrument: Micromass Quattro LCZ with HPLC Agilent Series 1100; Column: Phenomenex Synergi 2.5 ⁇ MAX-RP 100A Mercury 20 mm x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A -> 0.1 min 90% A ⁇ 3.0 min 5% A ⁇ »4.0 min 5% A -» 4.1 min 90% A; Flow: 2 ml / min; Oven: 50 ° C .; UV detection: 208-400 nm.
- Method 5 Instrument: Micromass Platform LCZ with HPLC Agilent Series 1100; Column: Thermo Hypersil GOLD 3 ⁇ 20 x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 100% A -> 0.2 min 100% A - »2.9 min 30% A -> 3.1 min 10% A -» 5.5 min 10% A; Oven: 50 ° C; Flow: 0.8 ml / min; UV detection: 210 nm.
- Method 6 Instrument: Micromass QuattroPremier with Waters UPLC Acquity; Column: Thermo Hypersil GOLD l, 9 ⁇ 50 x lmm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A -> 0.1 min 90% A ⁇ 1.5 min 10% A - »2.2 min 10% A; Oven: 50 ° C; Flow: 0.33 ml / min; UV detection: 210 nm.
- Method 7 Instrument: Micromass Quattro Micro MS with HPLC Agilent Series 1100; Column: Thermo Hypersil GOLD 3 ⁇ 20 x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 100% A ⁇ 3.0 min 10% A -> 4.0 min 10% A ⁇ 4:01 min 100% A (flow 2.5ml) -> 5:00 min 100% A furnace: 50 0 C; Flow: 2 ml / min; UV detection: 210 nm
- Example (150 mg, 0.448 mmol) of IA were taken up in 10 ml of benzene and 112 mg (0.493 mmol) of DDQ were added. The reaction was stirred overnight at reflux temperature. After cooling, the reaction was concentrated, taken up in water and then extracted with ethyl acetate. It was dried with magnesium sulfate and the solvent was removed by distillation under reduced pressure. The mixture was then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 107 mg (72% of theory) of the target compound.
- Example 4A A solution of 100 mg (about 0.342 mmol) Example 4A, 42 mg (0.478 mmol) of 3-methyl-1-butanol and 125 mg (0.478 mmol) of triphenylphosphine in 4 ml of THF was stirred for 20 minutes at room temperature. 96 mg (0.478 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for 3 h at room temperature. The mixture was purified by preparative HPLC without elution (eluent: acetonitrile / water, 90:10, isocratic). This gave 41 mg (46% of theory) of the target compound.
- Example 4A A solution of 100 mg (about 0.342 mmol) of Example 4A, 50 mg (0.478 mmol) of 4-methoxybutan-2-ol and 125 mg (0.478 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. 96 mg (0.478 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for 3 h at room temperature. The mixture was purified by preparative HPLC without further work-up (eluent: acetonitrile / water 90:10, isocratic). 40 mg (43% of theory) of the target compound were thus obtained.
- Example 4A A solution of 100 mg (about 0.342 mmol) of Example 4A, 28 mg (0.478 mmol) of isopropanol and 125 mg (0.478 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. 96 mg (0.478 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for 3 h at room temperature. The mixture was purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 42 mg (51% of theory) of the target compound.
- Example 4A A solution of 100 mg (about 0.342 mmol) of Example 4A, 28 mg (0.478 mmol) of 3- (trifluoromethyl) cyclohexanol and 125 mg (0.478 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. 96 mg (0.478 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was allowed to stand for 3 h Room temperature stirred. The mixture was purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 14 mg (13% of theory) of the target compound.
- Example 10A A solution of 92 mg (about 0.200 mmol) of Example 10A, 9 mg (0.201 mmol) of methanol and 74 mg (0.281 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. Then 57 mg (0.281 mmol) of diisopropyl azodicarboxylate were added and the reaction mixture was further stirred for 1.5 h at room temperature. The mixture was purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 26 mg (39% of theory) of the target compound.
- Example 4A A solution of 100 mg (about 0.342 mmol) of Example 4A, 34 mg (0.478 mmol) of cyclopropylmethanol and 125 mg (0.478 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. 96 mg (0.478 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for 3 h at room temperature. The mixture was purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 68 mg (81% of theory) of the target compound.
- Example 13A 940 mg (2.93 mmol) Example 13A were taken up in 50 ml benzene and treated with 731 mg (3.22 mmol) DDQ. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 -> 90:10). This gave 390 mg (41% of theory) of the target compound and 190 mg (21% of theory) of Example 7 directly by partial saponification.
- Example 15A were taken up in 40 ml benzene and treated with 784 mg (3.46 mmol) DDQ. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and then by means of preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 690 mg (58% of theory) of the target compound.
- Example 4A A solution of 100 mg (about 0.304 mmol) of Example 4A, 19 mg (0.426 mmol) of ethanol and 111 mg (0.426 mmol) of triphenylphosphine in 1.8 ml of THF was stirred for 20 minutes at room temperature. There were added 86 mg (0.426 mmol) of diisopropyl azodicarboxylate and then the reaction mixture was stirred for 2.5 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). 45 mg (58% of theory) of the target compound were thus obtained
- Example 4A A solution of 100 mg (about 0.304 mmol) of Example 4A, 36 mg (0.426 mmol) of (1-methylcyclopropyl) methanol and 111 mg (0.426 mmol) of triphenylphosphine in 1.8 ml of THF was stirred for 20 minutes at room temperature. 86 mg (0.426 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred at room temperature for 2.5 h. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). 53 mg (48% of theory) of the target compound were thus obtained
- Example 4A 1.5 g (5.124 mmol) of Example 4A in 19.1 ml (204.971 mmol) of phosphorus oxychloride were stirred at reflux temperature for 2 h. The reaction mixture was evaporated. The residue was treated with a 25% aqueous ammonium hydroxide solution, adjusted to pH 7 with 1N hydrochloric acid and then extracted with dichloromethane. The organic phase was dried over sodium sulfate and concentrated. This gave 1.38 g (87% of theory) of the target compound.
- Example 25A A solution of 60 mg (0.187 mmol) of Example 25A, 12 mg (0.262 mmol) of ethanol and 69 mg (0.262 mmol) of triphenylphosphine in 1.2 ml of THF was stirred for 20 minutes at room temperature. 53 mg (0.262 mmol) of diisopropyl azodicarboxylate were added and the reaction mixture was stirred at room temperature for 2.5 h. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water: 90/10, isocratic). This gave 35 mg (53% Th.) Of the target compound.
- Example 245 mg (0.706 mmol) of example 36A were taken up in 40 ml of benzene and 176 mg (0.777 mmol) of DDQ were added. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and the residue was taken up in about 5 ml of acetonitrile. The precipitate was filtered off and the remaining solution was then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 70 mg (26% of theory) of the target compound.
- Example 23A 100 mg (0.321 mmol) of Example 23A were dissolved in 2.5 ml of DMF. Subsequently, 1.28 ml (0.643 mmol) of 1-butylzinc bromide solution (0.5M in THF) and 18 mg (0.016 mmol) of tetrakis (triphenylphosphine) palladium (0) were added. The reaction mixture was stirred at room temperature overnight and allowed to stand at room temperature over the weekend. The reaction mixture was purified by preparative HPLC (eluent: acetonitrile / water, gradient 90:10, isocratic) without further work-up. 20 mg (19% of theory) of the target compound were obtained.
- preparative HPLC eluent: acetonitrile / water, gradient 90:10, isocratic
- Example 23 A 100 mg (0.321 mmol) of Example 23 A were dissolved in 2.5 ml of DMF. Subsequently, 1.28 ml (0.643 mmol) of 2-butylzinc bromide solution (0.5M in THF) and 18 mg (0.016 mmol) of tetrakis (triphenylphosphine) palladium (0) were added. The reaction mixture was stirred at room temperature overnight and allowed to stand at room temperature over the weekend. The reaction mixture was purified by preparative HPLC (eluent: acetonitrile / water, gradient 90:10, isocratic) without further work-up. 8 mg (8% of theory) of the target compound were obtained.
- preparative HPLC eluent: acetonitrile / water, gradient 90:10, isocratic
- Example 10A A solution of 100 mg (about 0.143 mmol) of Example 10A, 17 mg (0.201 mmol) of cyclopentanol and 53 mg (0.201 mmol) of triphenylphosphine in 1.4 ml of THF was stirred for 20 minutes at room temperature. 41 mg (0.201 mmol) of diisopropyl azodicarboxylate were added and the reaction mixture was stirred at room temperature overnight. The mixture was without further Workup purified by preparative HPLC (eluent: acetonitrile / water: 90:10, isocratic). 54 mg (85% th.) Of the target compound were thus obtained.
- Example 10A A solution of 100 mg (about 0.143 mmol) of Example 10A, 157 mg (0.201 mmol) of cyclopropylmethanol and 53 mg (0.201 mmol) of triphenylphosphine in 1.4 ml of THF was stirred for 20 minutes at room temperature. 53 mg (0.262 mmol) of diisopropyl azodicarboxylate were added and the reaction mixture was stirred at room temperature overnight. The mixture was purified by preparative HPLC without elution (eluent: acetonitrile / water: 90/10, isocratic). This gave 28 mg (52% of theory) of the target compound.
- Example 1OA A solution of 100 mg (about 0.143 mmol) of Example 1OA, 12 mg (0.201 mmol) of isopropanol and 53 mg (0.201 mmol) of triphenylphosphine in 1.4 ml of THF was stirred for 20 minutes at room temperature. 53 mg (0.262 mmol) of diisopropyl azodicarboxylate were added and the reaction mixture was stirred at room temperature overnight. The mixture was purified by preparative HPLC without elution (eluent: acetonitrile / water: 90/10, isocratic). This gave 44 mg (85% th.) Of the target compound.
- Example 45A A solution of 200 mg (about 0.341 mmol) of Example 45A, 28 mg (0.478 mmol) of isopropanol and 125 mg (0.478 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. 96 mg (0.487 mmol) of diisopropyl azodicarboxylate were added and the reaction mixture was stirred for 2 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90/10, isocratic). This gave 11 mg (9% of theory) of the target compound.
- Example 45A A solution of 200 mg (about 0.328 mmol) of Example 45A, 15 mg (0.459 mmol) of methanol and 120 mg (0.459 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. 96 mg (0.487 mmol) of diisopropyl azodicarboxylate were added and the reaction mixture was stirred for 2 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 5 mg (5% of theory) of the target compound.
- Example 50A were taken up in 20 ml benzene and treated with 227 mg (1.002 mmol) DDQ. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and the residue was taken up in about 2 ml of acetonitrile. The precipitate was filtered off and the remaining solution was then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 190 mg (70% of theory) of the target compound.
- Example 234 mg (0.561 mmol) of example 52A were taken up in 20 ml of benzene and 140 mg (0.617 mmol) of DDQ were added. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and the residue was taken up in about 2 ml of acetonitrile. The precipitate was filtered off and the remaining solution then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). 111 mg (66% of theory) of the target compound were thus obtained.
- Example (87 mg, 0.235 mmol) of Example 54A was taken up in 20 ml of benzene and 59 mg (0.259 mmol) of DDQ were added. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and the residue was taken up in about 2 ml of acetonitrile. The precipitate was filtered off and the remaining solution was then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 50 mg (64% of theory) of the target compound.
- Example 56A 149 mg (0.837 mmol) of N-bromosuccinimide, 20.3 mg (0.084 mmol) of dibenzoyl peroxide and 173 g (1.26 mmol) of potassium carbonate were taken up under an argon atmosphere in 10 ml of dioxane and stirred overnight at reflux temperature. After cooling, the mixture was concentrated, the residue taken up in water and then extracted with ethyl acetate (2x). The combined organic phases were dried with magnesium sulfate and the volatile components were separated on a rotary evaporator. The mixture was then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 184 mg (66% of theory) of the target compound.
- preparative HPLC eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10
- Example 57A 90 mg (0.271 mmol) of Example 57A, 22 ⁇ l (0.284 mmol) of 2-propanol and 74.5 mg (0.284 mmol) of triphenylphosphine were taken up in 5 ml of THF and stirred for 20 minutes at room temperature. Subsequently, 55 ⁇ l (0.459 mmol) of diisopropyl azodicarboxylate were added and the
- Reaction mixture reacted for 2 h at room temperature.
- the volatile components were removed by distillation under reduced pressure and the crude product was purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 66 mg (65% of theory) of the target compound.
- Example 57A 90 mg (0.271 mmol) of Example 57A, 26 ⁇ l (0.284 mmol) of cyclopentanol and 74.5 mg (0.284 mmol) of triphenylphosphine were taken up in 5 ml of THF and stirred at room temperature for 20 minutes. Subsequently, 55 ⁇ l (0.459 mmol) of diisopropyl azodicarboxylate were added and the
- Reaction mixture reacted for 2 h at room temperature.
- the volatile components were removed by distillation under reduced pressure and the crude product was purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). This gave 75 mg (69% of theory) of the target compound.
- Example 45A A mixture of 200 mg (about 0.328 mmol) of Example 45A, 34 mg (0.459 mmol) of n-propanol and 120 mg (0.459 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature touched. Then, 93 mg (0.459 mmol) of diisopropyl azodicarboxylate was added and the reaction mixture was further stirred for 2 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 19 mg (17% of theory) of the target compound.
- Example 45A A mixture of 200 mg (about 0.328 mmol) of Example 45A, 34 mg (0.459 mmol) of 2-methyl-1-propanol and 120 mg (0.459 mmol) of triphenylphosphine in 2 ml of THF was stirred for 20 minutes at room temperature. Then, 93 mg (0.459 mmol) of diisopropyl azodicarboxylate was added and the reaction mixture was further stirred for 2 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 15 mg (13% of theory) of the target compound.
- aqueous phase was extracted with dichloromethane and the organic phase was washed with water and a saturated aqueous sodium chloride solution, dried over sodium sulfate and concentrated.
- the crude product was mixed with acetonitrile and the resulting crystals were filtered off and dried under high vacuum to constant weight. This gave 1.47 g (40% of theory) of the target compound in 88% purity (LC-MS), which was reacted without further purification steps.
- Example 63 A A solution of 80 mg (about 0.219 mmol) of Example 63 A, 14 mg (0.307 mmol) of ethanol and 80 mg (0.307 mmol) of triphenylphosphine in 1.5 ml of THF was stirred for 20 minutes at room temperature. 62 mg (0.307 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for a further 2.5 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 40 mg (52% of theory) of the target compound.
- Example 63A A solution of 80 mg (about 0.219 mmol) of Example 63A, 18 mg (0.307 mmol) of 2-propanol and 80 mg (0.307 mmol) of triphenylphosphine in 1.5 ml of THF was stirred for 20 minutes at room temperature. 62 mg (0.307 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for a further 2.5 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 47 mg (59% of theory) of the target compound.
- Example 63A A solution of 80 mg (about 0.219 mmol) of Example 63A, 22 mg (0.307 mmol) of 2-methylpropanol and 80 mg (0.307 mmol) of triphenylphosphine in 1.5 ml of THF was stirred for 20 minutes at room temperature. 62 mg (0.307 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for a further 2.5 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 47 mg (57% of theory) of the target compound.
- Example 63 A A solution of 80 mg (about 0.219 mmol) of Example 63 A, 22 mg (0.307 mmol) of cyclopropylmethanol and 80 mg (0.307 mmol) of triphenylphosphine in 1.5 ml of THF was stirred for 20 minutes at room temperature. 62 mg (0.307 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was further stirred at room temperature for 2.5 hours. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 42 mg (51% of theory) of the target compound.
- Example 63 A A solution of 80 mg (about 0.219 mmol) of Example 63 A, 23 mg (0.307 mmol) of 2-methoxyethanol and 80 mg (0.307 mmol) of triphenylphosphine in 1.5 ml of THF was stirred for 20 minutes at room temperature. 62 mg (0.307 mmol) of diisopropyl azodicarboxylate were added and then the reaction mixture was stirred for a further 2.5 h at room temperature. The mixture was concentrated by rotary evaporation and purified by preparative HPLC without elution (eluent: acetonitrile / water 90:10, isocratic). This gave 44 mg (53% of theory) of the target compound.
- Example 2A 100 mg (0.300 mmol) Example 2A were taken up in 5 ml of ethanol and admixed with 1.50 ml (3.00 mmol) of a 2M aqueous potassium hydroxide solution. It was stirred at reflux temperature. After cooling, the reaction was concentrated, taken up in water and acidified with 1N hydrochloric acid. It was then extracted with ethyl acetate. After drying with magnesium sulfate, the solvent was removed by distillation under reduced pressure and the crude material purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 -> 90:10). This gave 73 mg (76% of theory) of the target compound.
- preparative HPLC eluent: acetonitrile / water, gradient 10:90 -> 90:10
- Example 5 A To a solution of 41 mg (0.113 mmol) of Example 5 A in 2 ml of ethanol was added 1.13 ml (2.260 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. overnight. For workup, the ethanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 1 with 1 N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum. 44 mg (64% of theory) of the target compound were obtained.
- Example 6A To a solution of 40 mg (0.105 mmol) of Example 6A in 2 ml of ethanol was added 1.05 ml (2.111 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. overnight. For workup, the ethanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum. 25 mg (67% of theory) of the target compound were obtained.
- Example 7A To a solution of 42 mg (0.125 mmol) of Example 7A in 2 ml of ethanol was added 1.25 ml (2.509 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. overnight. For workup, the ethanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum. 20 mg (52% of theory) of the target compound were obtained.
- Example 8A To a solution of 14 mg (0.031 mmol) of Example 8A in 2 ml of ethanol was added 0.316 ml (0.632 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 4 h. For workup, the ethanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum. 9 mg (69% of theory) of the target compound were obtained.
- Example I IA To a solution of 26 mg (0.077 mmol) of Example I IA in 2 ml of ethanol was added 0.78 ml (1.553 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. overnight. For workup, the ethanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum. 20 mg (84% of theory) of the target compound were obtained.
- Example 12A To a solution of 68 mg (0.196 mmol) of Example 12A in 2 ml of ethanol was added 1.96 ml (3.921 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the ethanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum. 53 mg (85% of theory) of the target compound were obtained.
- Example 16A 690 mg (1.99 mmol) Example 16A were taken up in 25 ml dioxane and treated with 558 mg (9.95 mmol) potassium hydroxide. The reaction was stirred for 2 hours at reflux temperature. After cooling, the reaction mixture was concentrated on a rotary evaporator. Then the residue was taken up in water, acidified with 1N hydrochloric acid and washed with
- Example 17A To a solution of 43 mg (0.134 mmol) of Example 17A in 2 ml of ethanol was added 1.34 ml (2.687 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. Thirty mg (74% of theory) of the target compound were obtained.
- Example 18A To a solution of 50 mg (0.049 mmol) of Example 18A in 2 ml of ethanol was added 1.49 ml (2.987 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and in
- Example 19A To a solution of 76 mg (0.218 mmol) of Example 19A in 3 ml of ethanol was added 2.18 ml (4.357 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and in
- Example 2OA To a solution of 51 mg (0.141 mmol) of Example 2OA in 2 ml of ethanol was added 1.40 ml (2,810 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 0 C for 3h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 40 mg (86% of theory) of the target compound were obtained.
- Example 21A To a solution of 36 mg (0.100 mmol) of Example 21A in 1.5 ml of ethanol was added 1.00 ml (2,006 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C for 3h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 17 mg (52% of theory) of the target compound were obtained.
- Example 22A To a solution of 50 mg (0.134 mmol) of Example 22A in 2 ml of ethanol was added 1.34 ml (2.673 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 22 mg (48% of theory) of the target compound were obtained.
- Example 26A To a solution of 33 mg (0.095 mmol) of Example 26A in 1.5 ml of ethanol was added 0.95 ml (1.903 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 8O 0 C for 3h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid posed. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 29 mg (93% of theory) of the target compound were obtained.
- Example 27A To a solution of 33 mg (0.095 mmol) of Example 27A in 2 ml of ethanol was added 1.23 ml (2.469 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 34 mg (81% of theory) of the target compound were obtained.
- Example 28A To a solution of 40 mg (0.106 mmol) of Example 28A in 2 mL of ethanol was added 1.06 mL (2.123 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 32 mg (86% of theory) of the target compound were obtained.
- Example 29A To a solution of 41 mg (0.105 mmol) of Example 29A in 2 ml of ethanol was added 1.05 ml (2.093 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and in Drying oven dried overnight at 40 0 C. 31 mg (82% of theory) of the target compound were obtained.
- Example 30A To a solution of 68 mg (0.175 mmol) of Example 30A in 3 ml of ethanol was added 1.75 ml (3.497 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C for 3h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and in
- Example 31A To a solution of 68 mg (0.175 mmol) of Example 31A in 2 ml of ethanol was added 1.15 ml (2.293 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 6 h. For workup, the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid and the volatile components were distilled off in vacuo. The resulting crystals were filtered off with suction and dried under high vacuum. 23 mg (97% of theory) of the target compound were obtained.
- example 35A 270 mg (0.78 mmol) of example 35A were taken up in 14 ml of pyridine and 521 mg (3.89 mmol) of lithium iodide were added. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 -> 90:10). This gave 121 mg (47% of theory) of the target compound.
- Example (70 mg, 0.203 mmol) of Example 37A was taken up in 5 ml of pyridine, and 136 mg (1.02 mmol) of lithium iodide were added. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 -> 90:10). This gave 42 mg (59% of theory) of the target compound.
- Example 38A To a solution of 60 mg (0.159 mmol) of Example 38A in 2.8 ml of ethanol was added 1.59 ml (3.189 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 6 h. For workup, the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid and the volatile components were distilled off in vacuo. The resulting crystals were filtered off, dried under high vacuum and then purified by preparative HPLC (eluent: acetonitrile / water, gradient 50:50). This gave 15 mg (28% of theory) of the target compound.
- preparative HPLC eluent: acetonitrile / water, gradient 50:50
- Example 39A To a solution of 20 mg (0.060 mmol) of Example 39A in 2 ml of ethanol was added 0.60 ml (1.202 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 10 h and allowed to stand at room temperature over the weekend. For workup, the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The volatile components were then distilled off in vacuo. The resulting crystals were filtered off with suction and dried under high vacuum. This gave 12 mg (64% of theory) of the target compound
- Example 4OA To a solution of 9 mg (0.026 mmol) of Example 4OA in 1 ml of ethanol was added 0.26 ml (0.523 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 8O 0 C 10h and over the weekend at room temperature, allowed to stand. For workup, the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The volatile components were then distilled off in vacuo. The resulting crystals were filtered off with suction and dried under high vacuum. This gave 5 mg (59% of theory) of the target compound.
- Example 41A To a solution of 50 mg (0.129 mmol) of Example 41A in 2.4 ml of ethanol was added 1.28 ml (2.571 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 6 h. For workup, the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The volatile components were then distilled off in vacuo. The resulting crystals were filtered off with suction and dried under high vacuum. This gave 29 mg (60% of theory) of the target compound.
- Example 42A To a solution of 25 mg (0.067 mmol) of Example 42A in 1.2 ml of ethanol was added 0.67 ml (1334 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 6 h. For workup, the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The volatile components were then distilled off in vacuo. The resulting crystals were filtered off with suction and dried under high vacuum. This gave 23 mg (94% of theory) of the target compound.
- Example 43A To a solution of 40 mg (0.110 mmol) of Example 43A in 2.2 ml of ethanol was added 1.1 ml (2,205 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 6 h. For workup, the reaction mixture was adjusted to pH 1 with 1N hydrochloric acid. The volatile components were then distilled off in vacuo. The resulting crystals were filtered off with suction and dried under high vacuum. This gave 15 mg (38% of theory) of the target compound.
- Example 46A To a solution of 10 mg (0.029 mmol) of Example 46A in 0.6 ml of ethanol was added 0.29 ml (0.582 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. This gave 7.3 mg (75% of theory) of the target compound.
- Example 47A To a solution of 4.4 mg (0.014 mmol) of Example 47A in 0.3 ml of ethanol was added 0.14 ml (0.276 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 3 mg (69% of theory) of the target compound were obtained.
- Example 48A To a solution of 10 mg (0.030 mmol) of Example 48A in 0.64 ml of ethanol was added 0.30 ml (0.595 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the volatile components were distilled off in vacuo. The residue was taken up in water and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and in
- Example (184 mg, 0.617 mmol) of Example 51A was taken up in 5 ml of pyridine and 413 mg (3.08 mmol) of lithium iodide were added. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90-> 90:10). After drying under high vacuum, 59 mg (34% of theory) of the target compound were obtained.
- Example 47 mg (0.140 mmol) Example 55A were taken up in 5 ml pyridine and treated with 93 mg (0.70 mmol) lithium iodide. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 ⁇ 90:10). After drying under high vacuum, 21 mg (47% of theory) of the target compound were obtained.
- example 58A 65 mg (0.173 mmol) of example 58A were taken up in 4 ml of pyridine and admixed with 116 mg (0.867 mmol) of lithium iodide. The reaction was stirred overnight at reflux temperature. After cooling, the batch was concentrated on a rotary evaporator and then purified by preparative HPLC (eluent: acetonitrile / water, gradient 10:90 -> 90:10). After drying under high vacuum, 46 mg (74% of theory) of the target compound were obtained.
- preparative HPLC eluent: acetonitrile / water, gradient 10:90 -> 90:10
- Example 6OA To a solution of 19.2 mg (0.055 mmol) of Example 6OA in 1.1 ml of ethanol was added 0.55 ml (1.107 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the reaction mixture was distilled off in vacuo and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 15 mg (80% of theory) of the target compound were obtained.
- Example 61A To a solution of 15.4 mg (0.043 mmol) of Example 61A in 0.86 ml of ethanol was added 0.42 ml (0.854 mmol) of a 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the reaction mixture was distilled off in vacuo and adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried in a drying oven at 40 0 C overnight. 11.5 mg (77% of theory) of the target compound were obtained.
- Example 65A To a solution of 45 mg (0.125 mmol) of Example 65A in 2.3 ml of methanol was added 1.25 ml (2.491 mmol) of 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the methanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum overnight. 38 mg (87% of theory) of the target compound were obtained.
- Example 66A To a solution of 43 mg (0.114 mmol) of Example 66A in 2 ml of methanol was added 1.14 ml (2.277 mmol) of 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the methanol was removed on a rotary evaporator and the reaction mixture was adjusted to pH 2 with 1N hydrochloric acid. The resulting crystals were filtered off, washed with water and dried under high vacuum overnight. 20 mg (98% of theory) of the target compound were obtained.
- Example 67A To a solution of 40 mg (0.106 mmol) of Example 67A in 2 mL of methanol was added 1.06 mL (2.123 mmol) of 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the methanol was removed on a rotary evaporator, the reaction mixture was adjusted to pH 2 with 1N hydrochloric acid and extracted with dichloromethane. The organic phase was dried over magnesium sulfate and concentrated. This gave 29 mg (73% of theory) of the target compound.
- Example 68A To a solution of 42 mg (0.111 mmol) of Example 68A in 2 ml of methanol was added 1.11 ml (2.212 mmol) of 2M aqueous sodium hydroxide solution. The reaction mixture was stirred at 80 ° C. for 3 h. For workup, the methanol was removed on a rotary evaporator, the reaction mixture was adjusted to pH 2 with 1N hydrochloric acid and extracted with dichloromethane. The organic phase was dried over magnesium sulfate and concentrated. This gave 34 mg (85% of theory) of the target compound.
- a cellular assay is used to identify activators of the peroxisome proliferator-activated receptor alpha (PPAR-alpha).
- the GAL4-PPAR ⁇ expression construct contains the ligand binding domain of PPARa (amino acids 167-468), which is PCR amplified and cloned into the vector pcDNA3.1. This vector already contains the GAL4 DNA binding domain (amino acids 1-147) of the vector pFC2-dbd (Stratagene).
- the reporter construct containing five copies of the GAL4 binding site upstream of a thymidine kinase promoter, expresses the firefly luciferase (Photinus pyralis) upon activation and binding of GAL4-PPAR ⁇ .
- CHO (Chinese hamster ovary) cells stably expressing the above-described GAL4-PPAR ⁇ chimera and the luciferase reporter gene construct are in the medium (Optimem, GIBCO), 2% activated charcoal-purified fetal calf serum (Hyclone) the day before the test. , 1.35 mM sodium pyruvate (GIBCO), 0.2% sodium bicarbonate (GIBCO) with 1 x 10 3 cells plated in 96-well microtiter plates and maintained in a cell incubator (96% humidity, 5% v / v CO 2 , 37 ° C).
- the substances to be tested are taken up in the above-mentioned medium, but without the addition of calf serum, and added to the cells.
- the luciferase activity is measured using a video camera.
- the measured relative light units give as a function of the substance concentration a sigmoidal stimulation curve.
- the EC 50 values are calculated using the computer program GraphPad PRISM (version 3.02).
- B-3 Test description for the discovery of pharmacologically active substances which increase or inhibit the apoprotein Al (ApoAl) and HDL-cholesterol (HDL-C) in the serum of transgenic mice transfected with the human ApoAl gene (hApoAl) .
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Die vorliegende Anmeldung betrifft neue substituierte 2-Phenylpyrimidin-5-carbonsäure-Derivate, Verfahren zu ihrer Herstellung, ihre Verwendung zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, vorzugsweise zur Behandlung und/oder Prophylaxe kardiovaskulärer Erkrankungen, insbesondere von Dyslipidämien, Arteriosklerose und Herzinsuffizienz.
Description
Substituierte 2-Phenylpyrimidin-5-carbonsäuren und ihre Verwendung
Die vorliegende Anmeldung betrifft neue substituierte 2-Phenylpyrimidin-5-carbonsäure-Derivate, Verfahren zu ihrer Herstellung, ihre Verwendung zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, vorzugsweise zur Behandlung und/oder Prophylaxe kardiovaskulärer Erkrankungen, insbesondere von Dyslipidämien, Arteriosklerose und Herzinsuffizienz.
Trotz vielfacher Therapieerfolge bleiben kardiovaskuläre Erkrankungen ein ernstes Problem der öffentlichen Gesundheit. Während die Behandlung mit Statinen durch Hemmung der HMG-CoA- Reduktase sehr erfolgreich sowohl die Plasmakonzentrationen von LDL-Cholesterin (LDL-C) als auch die Mortalität von Risikopatienten senken, so fehlen heute überzeugende Behandlungsstrategien zur Therapie von Patienten mit ungünstigem HDL-C/LDL-C-Verhältnis oder der Hyper- triglyceridämie.
Fibrate stellen neben Niacin bisher die einzige Therapieoption für Patienten dieser Risikogruppen dar. Sie senken erhöhte Triglyceride um 20-50%, erniedrigen LDL-C um 10-15%, verändern die LDL-Partikelgröße von atherogenem LDL geringer Dichte zu normal dichtem und weniger atherogenem LDL und erhöhen die HDL-Konzentration um 10-15%.
Fibrate wirken als schwache Agonisten des Peroxisom-Proliferator-aktivierten Rezeptors (PPAR)- alpha (Nature 1990, 347, 645-50). PPAR-alpha ist ein nuklearer Rezeptor, der die Expression von Zielgenen durch Bindung an DNA-Sequenzen im Promoter-Bereich dieser Gene [auch PPAR Response-Elemente (PPRE) genannt] reguliert. PPREs sind in einer Reihe von Genen identifiziert worden, welche für Proteine kodieren, die den Lipid-Metabolismus regulieren. PPAR-alpha ist hoch in der Leber exprimiert und seine Aktivierung führt unter anderem zu einer gesenkten VLDL- ProduktionASekretion sowie zu einer reduzierten Apolipoprotein CHI (ApoCIII)-Synthese. Im Gegensatz dazu wird die Synthese von Apolipoprotein Al (ApoAl) gesteigert.
Ein Nachteil von bisher zugelassenen Fibraten ist ihre nur schwache Interaktion mit dem Rezeptor (EC50 im μM-Bereich), was wiederum zu den oben beschriebenen relativ geringen pharmakologischen Effekten führt.
WO 99/41253 offenbart substituierte Pyrimidine zur Behandlung viraler Infektionen. In WO 2004/111014 werden substituierte Pyrimidine zur Behandlung von cystischer Fibrose beansprucht. WO 2005/049573 und WO 2005/049606 beschreiben substituierte Pyrimidincarbonsäureester als Syntheseintermediate ohne biologische Wirkung. WO 2005/110416 offenbart 4,5-disubstituierte 2- Arylpyrimidine als C5a-Rezeptorliganden zur Behandlung von inflammatorischen, immunologischen und kardiovaskulären Erkrankungen. In WO 2006/124874 werden unter
anderem substituierte Pyrimidine zur Behandlung von Krebs beschrieben. In WO 2006/097220 werden 4-Phenoxy-2-phenylpyrimidincarbonsäuren und in WO 2008/031500 und WO 2008/031501 4-Phenoxy- bzw. 2-Phenoxynikotinsäuren als PPAR-alpha-Modulatoren zur Behandlung von Dyslipidämien und Arteriosklerose beansprucht.
Aufgabe der vorliegenden Erfindung war die Bereitstellung neuer Verbindungen, die als PPAR- alpha-Modulatoren zur Behandlung und/oder Prophylaxe insbesondere kardiovaskulärer Erkrankungen eingesetzt werden können und eine verbesserte metabolische Stabilität gegenüber Verbindungen aus dem Stand der Technik aufweisen.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)

in welcher
R1 für (C3-Cio)-Alkyl, (C3-C7)-Cycloalkyl oder -ORA steht,
wobei (C3-CiO)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, (Ci-C4)-Alkoxy, Trifluormethoxy und (C3-C7)-Cycloalkyl substituiert sein kann,
worin (C3-C7)-Cycloalkyl seinerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (CrC4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (CrC4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei (C3-C7)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (Ci-C4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (Ci-C4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei
RA für (CrC10)-Alkyl oder (C3-C7)-Cycloalkyl steht,
wobei (Ci-Cio)-Alkyl mit einem Substituenten ausgewählt aus der Gruppe Hydroxy, (Ci-C4)-Alkoxy und (C3-C7)-Cycloalkyl substituiert sein kann,
worin (C3-C7)-Cycloalkyl seinerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (Ci-C4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (CrC4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei (C3-C7)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (Ci-C4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (Ci-C4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei in allen genannten Cycloalkyl-Gruppen eine CH2-Einheit gegen Sauerstoff ausgetauscht sein kann,
R2 für (C i -C4)-Alkyl oder Cyclopropyl steht,
wobei (Ci-C4)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (d-C4)-Alkoxy substituiert sein kann,
R3 für Wasserstoff oder Fluor steht,
R4 für Wasserstoff, Fluor, Chlor, Methyl oder Trifluormethyl steht,
R5 für Wasserstoff, Halogen, Nitro, Cyano, Trifluormethyl, Methyl, Ethyl, Trifluormethoxy oder Methoxy steht,
R6 für Wasserstoff, Fluor, Chlor, Methyl oder Trifluormethyl steht,
wobei mindestens einer der Reste R3, R4, R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze,
mit Ausnahme der Verbindung 4-Cyclopropyl-6-(methoxymethyl)-2-[4-(trifluormethyl)phenyl]- pyrimidin-5-carbonsäure.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereo- isomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung umfasst deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Malein- säure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
AIs Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" umfaßt Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Insbesondere umfasst die vorliegende Erfindung auch hydrolysierbare Ester-Derivate der Carbonsäuren der Formel (I). Hierunter werden Ester verstanden, die in physiologischen Medien und insbesondere in vivo auf enzymatischem oder chemischem Wege zu den freien Carbonsäuren hydro- lysiert werden können. Als solche Ester werden geradkettige oder verzweigte (Ci-C6)-Alkylester, in denen die Alkylgruppe mit Hydroxy, (Ci-C4)-Alkoxy, Amino, MOnO-(C1 -C4)-alkylamino und/ oder Di-(C i-C4)-alkylamino substituiert sein kann, bevorzugt. Besonders bevorzugt sind die Methyl- oder Ethylester der Verbindungen der Formel (I).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Alkyl steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkylrest mit der jeweils angegebenen Anzahl an Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, iso-Butyl, 1-Methylpropyl, tert.-Butyl, n-Pentyl, iso-Pentyl, 1- Ethylpropyl, 1-Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, n-Hexyl, 1-Methylpentyl, 2- Methylpentyl, 3-Methylpentyl, 4-Methylpentyl, 3,3-Dimethylbutyl, 1-Ethylbutyl, 2-Ethylbutyl, 1 ,4-Dimethylpentyl, 4,4-Dimethylpentyl und 1,4,4-Trimethylpentyl.
Cycloalkyl steht in Rahmen der Erfindung für einen monocyclischen, gesättigten Alkylrest mit 3 bis 7 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
Alkoxy steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy, 1-Methylpropoxy, n-Butoxy, iso-Butoxy und tert.-Butoxy.
Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und Iod ein. Bevorzugt sind Chlor oder Fluor.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevor- zugt. Ganz besonders bevorzugt ist die Substitution mit einem Substituenten.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 für (C3-C8)-Alkyl, Cyclopropyl, Cyclopentyl, Cyclohexyl oder -ORA steht,
wobei (C3-C8)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, Methoxy, Ethoxy und Trifiuormethoxy substituiert sein kann,
und
wobei Cyclopropyl, Cyclopentyl und Cyclohexyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, Methyl, Ethyl, Trifluormethyl und Methoxy substitutiert sein können,
und
wobei
RA für Methyl, Ethyl, (C3-C8)-Alkyl, Cyclopropyl, Cyclopentyl oder Cyclohexyl steht,
wobei Methyl und Ethyl mit einem Substituenten ausgewählt aus der Gruppe Cyclopropyl, Cyclopentyl und Cyclohexyl substituiert sind,
worin Cyclopropyl, Cyclopentyl und Cyclohexyl ihrerseits mit 1 oder 2
Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, Methyl, Ethyl, Trifluormethyl und Methoxy substitutiert sein können,
wobei (C3-C8)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxy, Methoxy und Ethoxy substituiert sein kann,
und
wobei Cyclopropyl, Cyclopentyl und Cyclohexyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, Methyl, Ethyl, Trifluormethyl und Methoxy substitutiert sein können,
und
wobei in allen genannten Cyclopentyl- und Cyclohexyl-Gruppen eine CH2-Einheit gegen Sauerstoff ausgetauscht sein kann,
R2 für (Ci-C4)-Alkyl, Trifluormethyl oder Cyclopropyl steht,
R3 für Wasserstoff steht,
R4 für Wasserstoff oder Fluor steht,
R5 für Wasserstoff, Fluor, Chlor, Trifluormethyl oder Methyl steht,
R6 für Wasserstoff oder Methyl steht,
wobei mindestens einer der Reste R4, R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze.
Besonders bevorzugt sind Verbindungen der Formel (I), in welcher
R1 für (C3-C6)-Alkyl, Cyclopentyl, Cyclohexyl, Tetrahydrofuranyl oder -ORA steht,
wobei
RA für Methyl, (C3-C6)-Alkyl, Cyclopropyl, Cyclopentyl oder Cyclohexyl steht,
worin Methyl mit einem Substituenten ausgewählt aus der Gruppe Cyclopropyl, Cyclopentyl und Cyclohexyl substituiert ist,
worin Cyclopropyl, Cyclopentyl und Cyclohexyl ihrerseits mit 1 oder 2
Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Trifluormethyl substitutiert sein können,
worin (C3-Co)-Al]CyI mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxy und Methoxy substituiert sein kann,
und
worin Cyclopropyl, Cyclopentyl und Cyclohexyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Trifluormethyl substitutiert sein können,
R2 für Methyl, Ethyl, n-Propyl, Isopropyl, Trifluormethyl oder Cyclopropyl steht,
R3 für Wasserstoff steht,
R4 für Wasserstoff oder Fluor steht,
R5 für Wasserstoff, Chlor oder Methyl steht,
R6 für Wasserstoff oder Methyl steht,
wobei mindestens einer der Reste R4, R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze.
Ganz besonders bevorzugt sind Verbindungen der Formel (I), in welcher
R1 für Isopropyl, iso-Butyl, 1-Methylpropyl, iso-Pentyl, 1 -Methylbutyl oder-ORA steht,
wobei
RA für Isopropyl, iso-Butyl, 1-Methylpropyl, 1 -Methylbutyl oder 3-Methylbutyl steht,
R2 für Methyl, Ethyl oder Trifluormethyl steht,
R3 für Wasserstoff steht,
R4 für Wasserstoff steht,
R5 für Wasserstoff, Chlor oder Methyl steht,
R6 für Wasserstoff oder Methyl steht,
wobei mindestens einer der Reste R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R1 für Isopropyl, iso-Butyl, 1- Methylpropyl, iso-Pentyl oder 1 -Methylbutyl steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R1 für -ORA steht, worin RA für Isopropyl, iso-Butyl, 1-Methylpropyl, 1-Methylbutyl oder 3-Methylbutyl steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R2 für Methyl oder Ethyl steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R2 für Trifluormethyl steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R3, R4 und R6 für Wasserstoff stehen, und R5 für Chlor oder Methyl steht.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verbindung 4-Cyclopropyl-6- (methoxymethyl)-2-[4-(trifluormethyl)phenyl]-pyrimidin-5-carbonsäure zur Prophylaxe und/oder Behandlung von kardiovaskulären Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der Verbindung 4- Cyclopropyl-6-(methoxymethyl)-2-[4-(trifluormethyl)phenyl]-pyrimidin-5-carbonsäure zur
Herstellung von Arzneimitteln oder pharmazeutischen Zusammensetzungen zur Prophylaxe und/oder Behandlung von kardiovaskulären Erkrankungen.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Ver- bindungen der Formel (I), dadurch gekennzeichnet, dass man eine Verbindung der Formel (II)

in welcher R2, R3, R4, R5 und R6 jeweils die oben angegebenen Bedeutungen haben,
und
R7 für (C,-C4)-Alkyl steht,
entweder
[A] in einem inerten Lösungsmittel unter Mitsunobu-Bedingungen mit einer Verbindung der Formel (EI-A)
R1 — H (m-A),
in welcher
R'A für -ORA steht,
worin RΛ die oben angegebene Bedeutung hat,
zu Verbindungen der Formel (IV-A)

in welcher R1A, R2, R3, R4, R5, R6 und R7 jeweils die oben angegebenen Bedeutungen haben,
umsetzt und diese durch basische oder saure Hydrolyse in die Carbonsäuren der Formel (I-A)

in welcher R , 1A , τ R>2 , τ R> 3 , r R>4 , r R> 5 und j τ R>6 jeweils die oben angegebenen Bedeutungen haben,
überführt,
oder
[B] mit Hilfe eines geeigneten Chlorierungsmittels, wie beispielsweise Phosphoroxychlorid, in eine Verbindung der Formel (V)

in welcher R2, R3, R4, R5, R6 und R7 jeweils die oben angegebenen Bedeutungen haben, überführt,
und diese anschliessend in einem inerten Lösungsmittel in Gegenwart einer Base und eines geeigneten Palladium-Katalysators mit einer Verbindung der Formel (IH-B)
R1B— X1 (m-B),
in welcher
R IB für (C3-Cio)-Alkyl oder (C3-C7)-Cycloalkyl steht,
wobei (C3-Cio)-Alkyl mit einem oder zwei Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, (CrQ)-AIkOXy, Trifluormethoxy und (C3-C7)-Cycloalkyl substituiert sein kann,
worin (C3-C7)-Cycloalkyl seinerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (Ci-C4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (CrC4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei die genannten (C3-C7)-Cycloalkyl-Gruppen mit einem oder zwei Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy,
Oxo, (CrC4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (CrC4)-Alkoxy und Trifluormethoxy substitutiert sein können,
und
X1 für eine Gruppe der Formel -B(OR8)2 oder -ZnHaI steht,
worin
HaI für Halogen, insbesondere Chlor, Brom oder Iod steht,
und
R8 für Wasserstoff oder (CrC4)-Alkyl steht
oder
beide Reste R8 zusammen eine -C(CH3)2-C(CH3)2-Brücke bilden,
zu Verbindungen der Formel (IV-B)

in welcher R1B, R2, R3, R4, R5, R6 und R7 jeweils die oben angegebenen Bedeutungen haben,
umsetzt und diese durch basische oder saure Hydrolyse in die Carbonsäuren der Formel
(I-B)

in welcher R , R , R , R , R und R jeweils die oben angegebenen Bedeutungen haben,
überfuhrt,
und die Verbindungen der Formeln (I-A) und (I-B) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Basen oder Säuren zu ihren Solvaten, Salzen und/oder Solvaten der Salze umsetzt,
Die Verbindungen der Formeln (III-A) und (III-B) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaturbekannten Verfahren hergestellt werden.
Die Verbindungen der Formel (II) können hergestellt werden, indem man Verbindungen der For- mel (VI)

in welcher R2 und R7 die oben angegebenen Bedeutungen haben,
zunächst in einem inerten Lösungsmittel in Gegenwart einer geeigneten Base mit einer Verbindung der Formel (VII)

in welcher R > 3 , τ R> 4 , τ R} 5 und j D R6 die oben angegebenen Bedeutungen haben,
zu einer Verbindung der Formel (VIII)

in welcher R2, R3, R4, R5, R6 und R7 jeweils die oben angegebenen Bedeutungen haben,
umsetzt und diese anschliessend in einem inerten Lösungsmittel in Gegenwart einer Base und einer Bromquelle, wie beispielsweise N-Bromsuccinimid, sowie eines geeigneten Radikalstarters, wie beispielsweise Dibenzoylperoxid, zu einer Verbindung der Formel (IX)

in welcher R2, R3, R4, R5, R6 und R7 jeweils die oben angegebenen Bedeutungen haben,
oxidiert.
Die Verbindungen der Formeln (VI) und (VII) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaturbekannten Verfahren hergestellt werden.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfϊndungsgemäßen Verbindungen der Formel (I), in welcher R1 für R1B steht, worin R1B die oben genannten Bedeutungen hat, dadurch gekennzeichnet, dass man eine Verbindung der Formel (X)

in einem inerten Lösungsmittel in Gegenwart einer geeigneten Base oder Säure mit einer Verbindung der Formel (XI)

in welcher R1B die oben angegebene Bedeutung hat,
zu einer Verbindung der Formel (XII)

in welcher R1B, R2 und R7 jeweils die oben angegebenen Bedeutungen haben,
umsetzt, diese anschliessend mit einer Verbindung der Formel (VE) in einem inerten Lösungsmittel in Gegenwart einer geeigneten Base zu Verbindungen der Formel (XHI)

in welcher R1B, R2, R3, R4, R5, R6 und R7 jeweils die oben angegebenen Bedeutungen haben,
umsetzt, diese in einem inerten Lösungsmittel mit einem geeigneten Oxidationsmittel wie beispielsweise 2,3-Dichlor-5,6-dicyano-l,4-benzochinon zu Verbindungen der Formel (XIV)

in welcher R1B, R2, R3, R4, R5, R6 und R7 jeweils die oben angegebenen Bedeutungen haben,
oxidiert und diese durch basische oder saure Hydrolyse in die Carbonsäuren der Formel (I-B)

und die Verbindungen der Formel (I-B) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Basen oder Säuren zu ihren Solvaten, Salzen und/oder Solvaten der Salze umsetzt.
Die Verbindungen der Formeln (X) und (XI) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaturbekannten Verfahren hergestellt werden.
Die Verbindung 4-Cyclopropyl-6-(methoxymethyl)-2-[4-(trifluormethyl)phenyl]-pyrimidin-5- carbonsäure kann in Analogie zu oben beschriebenen Verfahren [B] oder wie in WO 2005/049573 beschrieben dargestellt werden.
Die Umsetzung (EI) — > (HI) erfolgt ohne Lösungsmittel oder gegebenenfalls in einem unter den Reaktionsbedingungen geeigneten inerten Lösungsmittel wie beispielsweise Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid, N-Methylpyrrolidon (ΝMP) oder Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt erfolgt die Reaktion ohne Lösungsmittel.
Die Umsetzung (ET) — > (IH) erfolgt im Allgemeinen in einem Temperaturbereich von 00C bis +1600C, bevorzugt bei +200C bis +1200C, gegebenenfalls in einer Mikrowelle. Die Reaktion kann
bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Übergangsmetall-Katalysatoren, Katalysatorliganden und Hilfsbasen für die Kupplungsreaktionen (V) + (III-B) → (IV-B) sind literaturbekannt [vgl. z.B. J. Hassan et al., Chem. Rev. JO2, 1359-1469 (2002)] und kommerziell erhältlich. Bevorzugt werden Palladium- oder Nickel-Katalysatoren verwendet.
Im Falle der Boronsäure-Kupplungen (V) + (HI-B) [X1 = -B(OR8)2] → (IV-B) erfolgt die Umsetzung in Gegenwart einer Hilfsbase und gegebenenfalls eines zusätzlichen Katalysatorliganden. Bevorzugt wird hierbei Bis-(triphenylphosphin)-palladium(II)chlorid als Katalysator, Tris-(o-tolyl)-phosphin als weiterer Ligand und wässrige Kaliumcarbonat-Lösung als Hilfsbase verwendet. Im Falle von Zink-organischen Verbindungen [X2 = -ZnHaI in (IH-A) und X4 = -ZnHaI in (VI)] wird bevorzugt Tetrakis-(triphenylphosphin)-palladium(0) als Katalysator eingesetzt.
Inerte Lösungsmittel für die Boronsäure-Kupplungen (V) + (IE-B) [X1 = -B(OR8)2] → (IV-B) sind beispielsweise Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder tert. -Sutanol, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder
Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid, NN'-
Dimethylpropylenharnstoff (DMPU), N-Methylpyrrolidon (ΝMP), Pyridin, Acetonitril oder auch Wasser. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt wird Dimethylformamid oder Dioxan verwendet.
Die Kupplungsreaktionen (V) + (IH-B) [X1 = -B(OR8)2] → (IV-B) erfolgen im Allgemeinen in einem Temperaturbereich von -200C bis +1500C, bevorzugt bei O0C bis +800C, gegebenenfalls in einer Mikrowelle. Die Umsetzungen können bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar).
Die Mitsunobu-Reaktion (H) + (HI-A) → (IV-A) [siehe: a) Hughes, D. L. "The Mitsunobu Reaction," Organic Reactions; John Wiley & Sons, Ltd, 1992, vol. 42, p. 335. b) Hughes, D. L. Org. Prep. Proceed. Int. 1996, 28, 127.] erfolgt unter Verwendung von Triphenylphosphin, oder Tri-n-butylphosphin, 1 ,2-Bis(diphenylphosphino)ethan (DPPE), Diphenyl(2-pyridyl)phosphin (Ph2P-Py), (p-Dimethylaminophenyl)diphenylphosphin (DAP-DP), tris(4-Dimethylaminophenyl)- phosphin (tris-DAP) und eines geeigneten Dialkylazodicarboxylats, wie beispielsweise Diethylazodicarboxylat (DEAD), Diisopropylazodicarboxylat (DIAD), Di-tert-butyl- azodicarboxylat, NNN'N'-Tetramethylazodicarboxamid (TMAD), l,l '-(Azodicarbonyl)-
dipiperidin (ADDP) oder 4,7-Dimethyl-3,5,7-hexahydro-l,2,4,7-tetrazocin-3,8-dion (DHTD). Bervorzugt werden Triphenylphosphin und Diisopropylazodicarboxylat (DIAD) verwendet.
Inerte Lösungsmittel für die Mitsunobu-Reaktion (ET) + (IQ-A) → (IV-A) sind beispielsweise Ether wie Tetrahydrofuran, Diethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Halogenkohlenwasserstoffe wie Dichlormethan, Dichlorethan oder andere Lösungsmittel wie Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt wird THF verwendet.
Die Mitsunobu-Reaktion (U) + (IH-A) — >• (IV-A) erfolgt im Allgemeinen in einem Temperaturbereich von -78°C bis +1800C, bevorzugt bei 00C bis +500C, gegebenenfalls in einer Mikrowelle. Die Umsetzungen können bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar).
Die Hydrolyse der Carbonsäureester in den Verfahrensschritten (IV-A) -» (I-A) und (IV-B) — > (I-B) erfolgt nach üblichen Methoden, gegebenenfalls in einer Mikrowelle, indem man die Ester in inerten Lösungsmitteln mit Säuren oder Basen behandelt, wobei die bei letzterem zunächst ent- stehenden Salze durch nachfolgendes Behandeln mit Säure in die freien Carbonsäuren überführt werden. Im Falle der tert.-Butylester erfolgt die Esterspaltung bevorzugt mit Säuren.
Als inerte Lösungsmittel eignen sich für die Hydrolyse der Carbonsäureester Wasser oder die für eine Esterspaltung üblichen organischen Lösungsmittel. Hierzu gehören insbesondere Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder tert. -Butanol, Ether wie Diethyl- ether, Tetrahydrofuran, Dioxan oder Glykoldimethylether, oder andere Lösungsmittel wie Aceton, Acetonitril, Dichlormethan, Dimethylformamid oder Dimethylsulfoxid. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Im Falle einer basischen Ester-Hydrolyse werden bevorzugt Gemische von Wasser mit Dioxan, Tetrahydrofuran, Methanol und/oder Ethanol eingesetzt. Im Falle der Umsetzung mit Trifluoressigsäure wird bevorzugt Dichlormethan und im Falle der Umsetzung mit Chlorwasserstoff bevorzugt Tetrahydrofuran, Diethylether, Dioxan oder Wasser verwendet.
Als Basen eignen sich für die Ester-Hydrolyse die üblichen anorganischen Basen. Hierzu gehören insbesondere Alkali- oder Erdalkalihydroxide wie beispielsweise Natrium-, Lithium-, Kaliumoder Bariumhydroxid, oder Alkali- oder Erdalkalicarbonate wie Natrium-, Kalium- oder Calcium- carbonat. Bevorzugt werden Natriumhydroxid oder Kaliumhydroxid eingesetzt.
Als Säuren eignen sich für die Esterspaltung im Allgemeinen Schwefelsäure, Chlorwasserstoff/ Salzsäure, Bromwasserstoff/Bromwasserstoffsäure, Phosphorsäure, Essigsäure, Trifluoressigsäure,
Toluolsulfonsäure, Methansulfonsäure oder Trifluormethansulfonsäure oder deren Gemische gegebenenfalls unter Zusatz von Wasser. Bevorzugt sind Chlorwasserstoff oder Trifluoressigsäure im Falle der tert.-Butylester und Salzsäure im Falle der Methylester.
Die Esterspaltung erfolgt im Allgemeinen in einem Temperaturbereich von 00C bis +1000C, bevor- zugt bei 00C bis +500C. Die Umsetzung kann bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar).
Die Herstellung der erfindungsgemäßen Verbindungen kann durch die folgenden Syntheseschemata veranschaulicht werden:
Schema 1

b) c)

[a): POCl3, Rückflußtemperatur; b) R -B(OH)2 oder R1B-ZnCl, K2CO3, Pd(PPh3),,, DMF, RT;
c): NaOH, Ethanol/Wasser, Rückflußtemperatur oder Mikrowelle, 140 0C d): R1A-H, Triphenylphosphin, DIAD, Raumtemperatur].
Schema 2


[a): Natriumethanolat, Ethanol, Rückflußtemperatur; b): N-Bromsuccinimid, K2CO3, kat. Dibenzoylperoxid, Rückflußtemperatur]
Schema 3


[a): katalytisch Piperidin oder Essigsäure, Dichlormethan, Rückflußtemperatur am Wasserabscheider; b): Triethylamin, 1-Butanol, Rückflußtemperatur; c): DDQ, Benzol, 700C; d): KOH, Ethanol/Wasser, Rückflußtemperatur].
Die erfmdungsgemäßen Verbindungen besitzen wertvolle pharmakologische Eigenschaften und können zur Vorbeugung und Behandlung von Erkrankungen bei Menschen und Tieren verwendet werden.
Die erfindungsgemäßen Verbindungen sind hochwirksame PPAR-alpha-Modulatoren und weisen zudem eine erhöhte metabolische Stabilität auf. Sie eignen sich insbesondere zur primären und/oder sekundären Prävention sowie Behandlung von kardiovaskulären Erkrankungen, die durch Störungen im Fettsäure- und Glukose-Metabolismus hervorgerufen werden. Solche Erkrankungen umfassen Dyslipidämien (Hypercholesterolämie, Hypertriglyceridämie, erhöhte Konzentrationen
der postprandialen Plasma-Triglyceride, Hypoalphalipoproteinämie, kombinierte Hyperlipidämien), Arteriosklerose sowie metabolische Erkrankungen (Metabolisches Syndrom, Hyperglykämie, Insulin-abhängiger Diabetes, Nicht-Insulin-abhängiger Diabetes, Gestationsdiabetes, Hyperinsulinämie, Insulinresistenz, Glukose-Intoleranz, Fettsucht (Adipositas) und diabetische Spätfolgen wie Retinopathie, Nephropathie und Neuropathie).
Als hochwirksame PPAR-alpha-Modulatoren eignen sich die erfindungsgemäßen Verbindungen insbesondere auch zur primären und/oder sekundären Prävention sowie Behandlung der Herzinsuffizienz.
Im Sinne der vorliegenden Erfindung umfasst der Begriff Herzinsuffizienz auch spezifischere oder verwandte Krankheitsformen wie Rechtsherzinsuffϊzienz, Linksherzinsuffizienz, Globalinsuffϊ- zienz, durch Hypertonie induzierte Herzinsuffizienz, ischämische Kardiomyopathie, dilatative Kardiomyopathie, angeborene Herzfehler, Herzklappenfehler, Herzinsuffizienz bei Herzklappenfehlern, Mitralklappenstenose, Mitralklappeninsuffizienz, Aortenklappenstenose, Aortenklappeninsuffϊzienz, Trikuspidalstenose, Trikuspidalinsuffizienz, Pulmonalklappenstenose, Pulmonalklappeninsuffizienz, kombinierte Herzklappenfehler, Herzmuskelentzündung (Myokarditis), chronische Myokarditis, akute Myokarditis, virale Myokarditis, diabetische Herzinsuffizienz, alkoholtoxische Kardiomyopathie, kardiale Speichererkrankungen, diastolische Herzinsuffizienz sowie systolische Herzinsuffizienz.
Weitere unabhängige Risikofaktoren für kardiovaskuläre Erkrankungen, welche sich durch die erfindungsgemäßen Verbindungen behandeln lassen, sind Bluthochdruck, Ischämie, Myokardinfarkt, Angina pectoris, Herzmuskelschwäche, Restenose, pulmonale Hypertonie, erhöhte Spiegel von Fibrinogen und von LDL geringer Dichte sowie erhöhte Konzentrationen von Plasminogen- aktivator-Inhibitor 1 (PAI-I).
Darüber hinaus können die erfindungsgemäßen Verbindungen auch zur Behandlung und/oder Prä- vention von Krebserkrankungen wie beispielsweise Hautkrebs, Brustkrebs, Hirntumoren, Kopf- Hals-Tumoren, Liposarcomen, Karzinomen des Auges, des Magen-Darm-Traktes, der Schilddrüse, der Leber, der Bauchspeicheldrüse, der Atemwegsorgane, der Niere, der Harnleiter, der Prostata, des Genitaltraktes und deren Fernmetastasen sowie Lyphome, Sarkome und Leukämien eingesetzt werden.
Darüber hinaus können die erfindungsgemäßen Verbindungen auch zur Behandlung und/oder Prävention von mikro- und makrovaskulären Schädigungen (Vasculitis), Reperfusionsschäden, arteriellen sowie venösen Thrombosen, Ödemen, von Erkrankungen des Zentralen Nervensystems und neurodegenerativen Störungen (Schlaganfall, Alzheimer'sche Krankheit, Parkinson'sche Krankheit,
Demenz, Epilepsie, Depressionen, Multiple Sklerose), von Entzündungserkrankungen, Immunerkrankungen (Morbus Crohn, Colitis ulcerosa, Lupus erythematodes, rheumatoide Arthritis, Asthma), chronisch-obstruktiven Atemwegserkrankungen (chronische Bronchitis, COPD), Nierenerkrankungen (Glomerulonephritis), Schilddrüsenerkrankungen (Hyperthyreose), Erkrankungen der Bauchspeicheldrüse (Pankreatitis), Leberfibrose, Hauterkrankungen (Psoriasis, Akne, Ekzeme, Neurodermitis, Dermatitis, Keratitis, Narbenbildung, Warzenbildung, Frostbeulen), Sepsis, viralen Erkrankungen (HPV, HCMV, HIV), Kachexie, Osteoporose, Gicht, Inkontinenz sowie zur Wundheilung, Angiogenese und zum Anti-Aging eingesetzt werden.
Die Wirksamkeit der erfindungsgemäßen Verbindungen lässt sich z.B. in vitro durch den im Beispielteil beschriebenen Transaktivierungsassay prüfen.
Die Wirksamkeit der erfindungsgemäßen Verbindungen in vivo lässt sich z.B. durch die im Beispielteil beschriebenen Untersuchungen prüfen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor ge- nannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prä- vention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Weiterer Gegenstand der vorliegenden Erfindung sind die erfindungsgemäßen Verbindungen zur Verwendung in einem Verfahren zur Behandlung und/oder Prophylaxe von Dyslipidämien, Arteriosklerose und Herzinsuffizienz.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prävention der zuvor genannten Erkrankungen.
Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt: den Fettstoffwechsel verändernde Wirkstoffe, Antidiabetika, Blutdruck-Senker, durchblutungsfördernd und/
oder antithrombotisch wirkende Mittel sowie Antioxidantien, Chemokin-Rezeptor-Antagonisten, p38-Kinase-Inhibitoren, NPY-Agonisten, Orexin-Agonisten, Anorektika, PAF-AH-Inhibitoren, Antiphlogistika (COX-Inhibitoren, LTB4-Rezeptor-Antagonisten), Analgetika (Aspirin), Antidepressiva und andere Psychopharmaka.
Gegenstand der vorliegenden Erfindung sind insbesondere Kombinationen mindestens einer der erfindungsgemäßen Verbindungen mit mindestens einem den Fettstoffwechsel verändernden Wirkstoff, einem Antidiabetikum, einem blutdrucksenkenden Wirkstoff und/oder einem antithrombotisch wirkenden Mittel.
Die erfϊndungsgemäßen Verbindungen können vorzugsweise mit einem oder mehreren
• den Fettstoffwechsel verändernden Wirkstoffen, beispielhaft und vorzugsweise aus der Gruppe der HMG-CoA-Reduktase-Inhibitoren, Inhibitoren der HMG-CoA-Reduktase-Expres- sion, Squalensynthese-Inhibitoren, ACAT-Inhibitoren, LDL-Rezeptor-Induktoren, Choleste- rin-Absorptionshemmer, polymeren Gallensäureadsorber, Gallensäure-Reabsorptionshemmer, MTP-Inhibitoren, Lipase-Inhibitoren, LpL-Aktivatoren, Fibrate, Niacin, CETP-Inhibitoren, PPAR-γ- und/oder PPAR-δ-Agonisten, RXR-Modulatoren, FXR-Modulatoren, LXR-Modula- toren, Thyroidhormone und/oder Thyroidmimetika, ATP-Citrat-Lyase-Inhibitoren, Lp(a)- Antagonisten, Cannabinoid-Rezeptor 1 -Antagonisten, Leptin-Rezeptor-Agonisten, Bombesin- Rezeptor-Agonisten, Histamin-Rezeptor-Agonisten sowie der Antioxidantien / Radikalfänger;
• Antidiabetika, die in der Roten Liste 2004/Η, Kapitel 12 genannt sind, sowie beispielhaft und vorzugsweise jenen aus der Gruppe der Sulphonylharnstoffe, Biguanide, Meglitinid-Derivate,
Glukosidase-Inhibitoren, Oxadiazolidinone, Thiazolidindione, GLP 1 -Rezeptor- Agonisten, Glukagon- Antagonisten, Insulin-Sensitizer, CCK 1 -Rezeptor- Agonisten, Leptin-Rezeptor- Agonisten, Inhibitoren von Leberenzymen, die an der Stimulation der Glukoneogenese und/ oder Glykogenolyse beteiligt sind, Modulatoren der Glukoseaufhahme sowie der Kalium- kanalöffher, wie z.B. denjenigen, die in WO 97/26265 und WO 99/03861 offenbart sind;
• den Blutdruck senkenden Wirkstoffen, beispielhaft und vorzugsweise aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Inhibitoren, beta-Rezeptoren- Blocker, alpha-Rezeptoren-Blocker, ECE-Inhibitoren und der Vasopeptidase-Inhibitoren;
• antithrombotisch wirkenden Mitteln, beispielhaft und vorzugsweise aus der Gruppe der Thrombozytenaggregationshemmer oder der Antikoagulantien;
• Diuretika;
• Aldosteron- und Mineralokorticoid-Rezeptor-Antagonisten;
• Vasopressin-Rezeptor-Antagonisten;
• organischen Nitraten und NO-Donatoren;
• positiv-inotrop wirksamen Verbindungen;
• Verbindungen, die den Abbau von cyclischem Guanosinmonophosphat (cGMP) und/oder cyclischem Adenosinmonophosphat (cAMP) inhibieren, wie beispielsweise Inhibitoren der Phosphodiesterasen (PDE) 1, 2, 3, 4 und/oder 5, insbesondere PDE 5-Inhibitoren wie Sildenafil, Vardenafil und Tadalafil sowie PDE 3-Inhibitoren wie Milrinone;
• natriuretischen Peptiden, wie z.B. "atrial natriuretic peptide" (ANP, Anaritide), "B-type natriuretic peptide" oder "brain natriuretic peptide" (BNP, Nesiritide), "C-type natriuretic peptide" (CNP) sowie Urodilatin;
• Calcium-Sensitizern, wie beispielhaft und vorzugsweise Levosimendan;
• Kalium-Supplements;
• NO-unabhängigen, jedoch Häm-abhängigen Stimulatoren der Guanylatcyclase, wie insbeson- dere den in WO 00/06568, WO 00/06569, WO 02/42301 und WO 03/095451 beschriebenen
Verbindungen;
• NO- und Häm-unabhängigen Aktivatoren der Guanylatcyclase, wie insbesondere den in WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 und WO 02/070510 beschriebenen Verbindungen;
• Inhibitoren der humanen neutrophilen Elastase (HNE), wie beispielsweise Sivelestat und DX- 890 (Reltran);
• die Signaltransduktionskaskade inhibierenden Verbindungen, wie beispielsweise Tyrosin- kinase-Inhibitoren, insbesondere Sorafenib, Imatinib, Gefitinib und Erlotinib; und/oder
• den Energiestoffwechsel des Herzens beeinflussenden Verbindungen, wie beispielweise Eto- moxir, Dichloracetat, Ranolazine und Trimetazidine
kombiniert werden.
Unter den Fettstoffwechsel verändernden Wirkstoffen werden vorzugsweise Verbindungen aus der Gruppe der HMG-CoA-Reduktase-Inhibitoren, Squalensynthese-Inhibitoren, ACAT-Inhibitoren, Cholesterin-Absorptionshemmer, MTP-Inhibitoren, Lipase-Inhibitoren, Thyroidhormone und/oder Thyroidmimetika, Niacin-Rezeptor-Agonisten, CETP-Inhibitoren, PPAR-gamma-Agonisten, PPAR-delta-Agonisten, polymeren Gallensäureadsorber, Gallensäure-Reabsoφtionshemmer, Anti- oxidantien / Radikalfänger sowie der Cannabinoid-Rezeptor 1 -Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem HMG-CoA-Reduktase-mhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosu- vastatin, Cerivastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK-475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindun- gen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterin-Absorptionshernmer, wie beispielhaft und vorzugsweise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide oder JTT-130, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verab- reicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidhormon und/oder Thyroidmimetikum, wie beispielhaft und vorzugsweise D-Thyroxin oder 3,5,3'-Triiodothyronin (T3), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindun- gen in Kombination mit einem Agonisten des Niacin-Rezeptors, wie beispielhaft und vorzugsweise Niacin, Acipimox, Acifran oder Radecol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP-Inhibitor, wie beispielhaft und vorzugsweise Torcetrapib, JTT-705 oder CETP Vaccine (Avant), verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindun- gen in Kombination mit einem PPAR-gamma-Agonisten, wie beispielhaft und vorzugsweise Pio- glitazone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-delta-Agonisten, wie beispielhaft und vorzugsweise GW- 501516, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimide, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugs- weise ASBT (= IBAT)-Inhibitoren wie z.B. AZD-7806, S-8921, AK-105, BARI-1741, SC-435 oder SC-635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Antioxidans / Radikalfänger, wie beispielhaft und vorzugsweise Probucol, AGI-1067, BO-653 oder AEOL-10150, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cannabinoid-Rezeptor 1 -Antagonisten, wie beispielhaft und vorzugsweise Rimonabant oder SR-147778, verabreicht.
Unter Antidiabetika werden vorzugsweise Insulin und Insulinderivate sowie oral wirksame hypo- glykämische Wirkstoffe verstanden. Insulin und Insulinderivate umfasst hierbei sowohl Insuline tierischen, menschlichen oder biotechnologischen Ursprungs als auch Gemische hieraus. Die oral wirksamen hypoglykämischen Wirkstoffe umfassen vorzugsweise Sulphonylharnstoffe, Biguanide, Meglitinid-Derivate, Glukosidase-Inhibitoren und PPAR-gamma-Agonisten.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Insulin verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Sulphonylharnstoff, wie beispielhaft und vorzugsweise Tolbutamid, Glibenclamid, Glimepirid, Glipizid oder Gliclazid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindun- gen in Kombination mit einem Biguanid, wie beispielhaft und vorzugsweise Metformin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Meglitinid-Derivat, wie beispielhaft und vorzugsweise Repaglinid oder Nateglinid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Glukosidase-Inhibitor, wie beispielhaft und vorzugsweise Miglitol oder Acarbose, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-gamma-Agonisten beispielsweise aus der Klasse der Thia- zolidindione, wie beispielhaft und vorzugsweise Pioglitazone oder Rosiglitazone, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Inhibitoren, beta-Rezeptoren-Blocker, alpha-Rezeptoren-Blocker sowie der Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindun- gen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise einem Schleifen- diuretikum wie Furosemid, Bumetanid oder Torsemid, oder einem Thiazid- oder Thiazid-ähnlichen Diuretikum wie Chlorthiazid oder Hydrochlorthiazid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Aldosteron- oder Mineralokortikoid-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Spironolacton oder Eplerenon, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vasopressin-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Conivaptan, Tolvaptan, Lixivaptan oder SR-121463, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindun- gen in Kombination mit einem organischen Nitrat oder NO-Donator, wie beispielhaft und Vorzugs-
weise Natriumnitroprussid, Nitroglycerin, Isosorbidmononitrat, Isosorbiddinitrat, Molsidomin oder SESf-I, oder in Kombination mit inhalativem NO verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einer positiv-inotrop wirksamen Verbindung, wie beispielhaft und vor- zugsweise Herzglycosiden (Digoxin), beta-adrenergen und dopaminergen Agonisten wie Isopro- terenol, Adrenalin, Noradrenalin, Dopamin oder Dobutamin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Calcium-Antagonisten, wie beispielhaft und vorzugsweise Nifedipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin AQ-Antagonisten, wie beispielhaft und vorzugsweise Losartan, Valsartan, Candesartan, Embusartan oder Telmisartan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Inhibitor, wie beispielhaft und vorzugsweise Enalapril, Capto- pril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem beta-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Metipra- nolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol, Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucin- dolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem alpha-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Antisympathotonikum, wie beispielhaft und vorzugsweise Reser- pin, Clonidin oder alpha-Methyl-Dopa, oder in Kombination mit einem Kaliumkanal-Agonisten, wie beispielhaft und vorzugsweise Minoxidil, Diazoxid, Dihydralazin oder Hydralazin, verabreicht.
Unter antithrombotisch wirkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer oder der Antikoagulantien verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindun- gen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximelaga- tran, Melagatran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem GPÜb/IHa-Antagonisten, wie beispielhaft und vorzugsweise Tiro- fiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Rivaroxa- ban (BAY 59-7939), DU-176b, Apixaban, Otamixaban, Fidexaban, Razaxaban, Fondaparinux, Idraparinux, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Kombinationen enthaltend mindestens eine der erfindungsgemäßen Verbindungen sowie einen oder mehrere weitere Wirkstoffe ausgewählt aus der Gruppe bestehend aus HMG-CoA-Reduktase-Inhibitoren (Statine), Diuretika, beta-Rezeptoren-Blocker, organische Nitrate und NO-Donatoren, ACE-Inhibitoren, Angiotensin Aü-Antagonisten, Aldosteron- und Mineralokortikoid-Rezeptor-Antagonisten, Vasopressin-Rezep- tor-Antagonisten, Thrombozytenaggregationshemmer und Antikoagulantien, sowie deren Verwendung zur Behandlung und/oder Prävention der zuvor genannten Erkrankungen.
Die erfindungsgemäßen Verbindungen können zur Behandlung und/oder Prophylaxe von Krebserkrankungen allein oder bei Bedarf in Kombination mit anderen Anti-Tumor Wirkstoffen eingesetzt werden. Gegenstand der vorliegenden Erfindung sind besonders Kombinationen mindestens einer der erfindungsgemäßen Verbindungen mit mindestens einem anderen AntiTumor Wirkstoff ausgewählt aus der Gruppe bestehend aus Alkylanzien, Antimetaboliten, von
Pflanzen abgeleitete Anti-Tumor Wirkstoffe, Wirkstoffe der Hormontherapie, Topoisomerase Inhibitoren, Camptothecin-Derivate, Kinase Inhibitoren, zielgerichtete Medikamente, Antikörper, Immunokonjugate, Interferon und/oder Immunmodulatoren, Antiangiogen-wirksame Verbindungen, Antisense-RNA und RNA- Interferenz, und weitere Anti-Tumor Medikamente.
Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
• Alkylanzien wie beispielsweise Chlormethin-N-oxid, Zyklophosphamid, Ifosfamid, Thiotepa, Ranimustin, Νimustin, Temozolomid, Altretamin, Apaziquon, Brostallicin, Bendamustin, Carmustin, Estramustin, Fotemustin, Glufosfamid, Mafosfamid und Mitolactol; Platinkoordinierte Alkylanzien wie beispielhaft Cisplatin, Carboplatin, Eptaplatin, Lobaplatin, Νedaplatin, Oxaliplatin und Satraplatin;
• Antimetabolite wie beispielsweise Methotrexat, 6-Mercaptopurin ribosid, Mercaptopurin, 5- Fluoruracil allein oder in Kombination mit Leucovorin, Tegafur, Doxifluridin, Carmofur, Cytarabin, Cytarabin Ocfosfat, Enocitabin, Gemcitabin, Fludarabin, 5-Azacitidin, Capecitabin, Cladribin, Clofarabin, Decitabin, Eflornithin, Ethynylcytidin, Cytosin- Arabinosid, Hydroxyharnstoff, Melphalan, Νelarabin, Νolatrexed, Ocfosfit, Dinatrium
Premetrexed, Pentostatin, Pelitrexol, Raltitrexed, Triapin, Trimetrexat, Vidarabin, Vincristin, und Vinorelbin;
• Wirkstoffe der Hormontherapie wie beispielsweise Exemestan, Lupron, Anastrozol, Doxercalciferol, Fadrozol, Formestan, 11-beta Hydroxysteroid Dehydrogenase- 1 Inhibitoren, 17-alpha Hydroxylase/17,20 Lyase Inhibitoren wie Abirateron Acetat, 5-alpha-Reduktase
Inhibitoren wie beispielsweise Finasterid und Epristerid, Anti-Östrogene wie Tamoxifen- Zitrat und Fulvestrant, Trelstar, Toremifen, Raloxifen, Lasofoxifen, Letrozol, Anti-Androgene wie Bicalutamid, Flutamid, Mifepriston, Νilutamid, Casodex sowie Anti-Progesterone, und Kombinationen davon;
• von Pflanzen abgeleitete Anti-Tumor Wirkstoffe wie beispielsweise Mitosehemmer wie Epothilone (Sagopilon, Ixabepilon und Epothilon B), Vinblastin, Vinflunin, Docetaxel, und Paclitaxel;
• Zytotoxische Topoisomerase Inhibitoren wie beispielsweise Aclarubicin, Doxorubicin, Amonafid, Belotecan, Camptothecin, 10-Hydroxycamptothecin, 9-Aminocamptothecin, Diflomotecan, Irinotecan, Topotecan, Edotecarin, Epimbicin, Etoposide, Exatecan, Gima- tecan, Lurtotecan, Mitoxantron, Pirambicin, Pixantron, Rubitecan, Sobuzoxan, Tafluposid, und Kombinationen davon;
• Immunologische Wirkstoffe beispielhaft und vorzugsweise aus der Gruppe der Interferone wie z. B. Interferon alpha, Interferon alpha-2a, Interferon alpha-2b, Interferon beta, Interferon gamma- Ia und Interferon gamma-nl, und andere Immunstimulanzien wie z. B. L19-IL2 und andere IL2 Derivative, Filgrastim, Lentinan, Sizofilan, TheraCys, Ubenimex, Aldesleukin, Alemtuzumab, BAM-002, Dacarbazin, Daclizumab, Denileukin, Gemtuzumab, Ozogamicin,
Ibritumomab, Imiquimod, Lenograstim, Lentinan, Melanom-Impfstoff (Corixa), Molgramostim, Sargramostim, Tasonermin, Tecleukin, Thymalasin, Tositumomab, Vimlizin, Epratuzumab, Mitumomab, Oregovomab, Pemtumomab und Proveng;
• Immunmodulatoren wie beispielsweise Krestin, Lentinan, Sizofiran, Picibanil, ProMun und Ubenimex;
• Antiangiogen-wirksame Verbindungen wie beispielsweise Acitretin, Aflibercept, Angiostatin, Aplidin, Asentar, Axitinib, Recentin, Bevacizumab, Brivanib-Alaninat, Cilengtid, Combreta- statin, DAST, Endostatin, Fenretinid, Halofuginon, Pazopanib, Ranibizumab, Rebimastat, Removab, Revlimid, Sorafenib, Vatalanib, Squalamin, Sunitinib, Telatinib, Thalidomid, Ukrain und Vitaxin;
• VEGF Inhibitoren wie beispielsweise Sorafenib, DAST, Bevacizumab, Sunitinib, Recentin, Axitinib, Aflibercept, Telatinib, Brivanib alaninat, Vatalanib, Pazopanib und Ranibizumab;
• Antikörper wie beispielsweise Trastuzumab, Cetuximab, Bevacizumab, Rituximab, Ticilimumab, Ipilimumab, Lumiliximab, Catumaxomab, Atacicept, Oregovomab und Alemtu- zumab;
• EGFR (HERl) Inhibitoren wie beispielsweise Cetuximab, Panitumumab, Vectibix, Gefitinib, Erlotinib und Zactima;
• HER2 Inhibitoren wie beispielsweise Lapatinib, Tratuzumab und Pertuzumab;
• mTOR Inhibitoren wie beispielsweise Temsirolimus, Sirolimus/Rapamycin und Everolimus;
• cMet Inhibitoren;
• PI3K und AKT Inhibitoren;
• CDK Inhibitoren wie beispielsweise Roscovitin und Flavopiridol;
• Spindle assembly Checkpoint Inhibitoren und zielgerichtete Mitosehemmer wie PLK Inhibitoren, Aurora Inhibitoren (z.B. Hesperadin), Checkpoint-Kinase Inhibitoren und KSP Inhibitoren;
• HDAC Inhibitoren wie beispielsweise Panobinostat, Vorinostat, MS275, Belinostat und LBH589;
• Inhibitoren der Histon-Methyltransferasen wie beispielsweise Vidaza;
• HSP90 und HSP70 Inhibitoren;
• Proteasom Inhibitoren wie Bortezomib und Carfϊlzomib;
• Serin-/Threonin-Kinase Inhibitoren wie beispielsweise MEK Inhibitoren und Raf Inhibitoren wie Sorafenib;
• Farnesyl Transferase Inhibitoren wie beispielsweise Tipifarnib;
• Tyrosin-Kinase Inhibitoren wie beispielsweise Dasatinib, Nilotibib, DAST, Bosutinib, Sorafenib, Bevacizumab, Sunitinib, AZD2171, Axitinib, Aflibercept, Telatinib, Imatinib Mesylat, Brivanib Alaninat, Pazopanib, Ranibizumab, Vatalanib, Cetuximab, Panitumumab, Vectibix, Gefitinib, Erlotinib, Lapatinib, Tratuzumab, Pertuzumab und c-Kit Inhibitoren;
• Vitamin D Rezeptor Agonisten;
• Kortikoide, z.B. Dexamethason;
• Thalidomid oder Thalidolid Analoga, z.B. Lenalidomid
• Bcl-2 Protein Inhibitoren wie beispielsweise Obatoclax, Oblimersen Natrium und Gossypol;
• CD20 Rezeptor Antagonisten wie beispielsweise Rituximab;
• Ribonukleotid Reduktase Inhibitoren wie Gemcitabin;
• Tumor Nekrose Apoptose einleitende Ligand-Rezeptor 1 Agonisten wie beispielsweise Mapatumumab;
• 5-Hydroxytryptamin Rezeptor Antagonisten wie beispielsweise rEV598, Xaliprod, Palonosetron-Hydrochlorid, Granisetron, Zindol und AB-1001 ;
• Integrin Inhibitoren einschliesslich Alpha5-betal Integrin Inhibitoren z. B. E7820, JSM 6425, Volociximab und Endostatin;
• Androgen Rezeptor Antagonisten einschliesslich z.B. Nandrolon Decanoat, Fluoxymesteron, Android, Prost-aid, Andromustin, Bicalutamid, Flutamid, Apo-cyproteron, Apo-flutamid, Chlormadinon Acetat, Androcur, Tabi, Cyproteron Acetat und Nilutamid;
• Aromatase Inhibitoren wie z. B. Anastrozol, Letrozol, Testolacton, Exemestan, Amino- glutethimid und Formestan;
• Matrix Metalloproteinase Inhibitoren;
• Weitere zur Krebstherapie eingesetzte Wirkstoffe einschliesslich z. B. Alitretinoin, Ampligen, Atrasentan Bexaroten, Bortezomib, Bosentan, Calcitriol, Exisulind, Fotemustin, Brondonat,
Miltefosin, Mitoxantron, I-Asparaginase, Procarbazin, Dacarbazin, Hydroxycarbamid, Pegaspargase, Pentostatin, Tazaroten, Velcad, Gallium-Nitrat, Canfosfamid, Darinaparsin und Tretinoin.
Die erfindungsgemäßen Verbindungen können auch zur Behandlung von Krebserkrankungen in Verbindung mit Strahlentherapie und/ oder chirurgischen Eingriffen eingesetzt werden.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfmdungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfmdungsgemäßen Verbindungen in geeigneten Appli- kationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfmdungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Frei-
setzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augenpräparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streu- puder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale und die intravenöse Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharma- zeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige PoIy- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispiels- weise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindest- menge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
Abkürzungen und Akronyme:
abs. absolut
Ac2O Acetanhydrid
AcOH Essigsäure aq. wässrig d Tage
DC Dünnschichtchromatographie
DCI direkte chemische Ionisation (bei MS) dd Dublett von Dublett (bei NMR)
DDQ 2,3-Dichlor-5,6-dicyano-l,4-benzochinon
DIAD Diisopropylazodicarboxylat
DMF Dimethylformamid
DMSO Dimethylsulfoxid dt Dublett von Triplett (bei NMR) d. Th. der Theorie (bei Ausbeute) eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS) h Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
LHMDS Lithium-N,N-bistrimethylsilylamid
LC-MS Flüssigchromatographie-gekoppelte Massenspektrometrie min Minute(n)
MPLC Mitteldruckchromatographie
MS Massenspektrometrie mz Multiple«, zentriert (bei NMR) n-Bu n-Butyl
NMR Kernresonanzspektrometrie o-Tol ortho-Tolyl
Ph Phenyl
PvP reverse phase (bei HPLC)
RT Raumtemperatur
R. Retentionszeit (bei HPLC) sbr Singulett, breit (bei NMR) sept Septett (bei NMR) t-Bu tert.-Butyl
THF Tetrahydrofuran tt Triplett von Triplett (bei NMR)
UV Ultraviolett-Spektrometrie v/v Volumen-zu- Volumen-Verhältnis (einer Mischung)
LC-MS- und HPLC-Methoden:
Methode 1 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 2 (LC-MSV Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2.5 μ MAX-RP 100A Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90%A -» 0.1 min 90%A -» 3.0 min 5%A -» 4.0 min 5%A -» 4.01 min 90%A; Fluss: 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 3 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith SpeedROD RP-18e 100 x 4.6 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure; Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 10% B-> 7.0 min 95% B-» 9.0 min 95% B; Ofen: 35°C; Fluss: 0.0 min 1.0 ml/min -> 7.0 min 2.0 ml/min-> 9.0 min 2.0 ml/min; UV-Detektion: 210 nm
Methode 4 (LC-MS): Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2.5 μ MAX-RP 100A Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90% A -> 0.1 min 90% A ^ 3.0 min 5% A ■» 4.0 min 5% A -» 4.1 min 90% A; Fluss: 2 ml/min; Ofen: 500C; UV-Detektion: 208- 400 nm.
Methode 5 (LC-MS): Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Thermo Hypersil GOLD 3μ 20 x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 100% A -> 0.2 min 100% A -» 2.9 min 30% A -> 3.1 min 10% A -»5.5 min 10% A; Ofen:50°C; Fluss: 0.8 ml/min; UV-Detektion: 210 nm.
Methode 6 (LC-MS): Instrument: Micromass QuattroPremier mit Waters UPLC Acquity; Säule: Thermo Hypersil GOLD l,9μ 50 x lmm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure , Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90% A -> 0.1 min 90% A ^ 1.5 min 10% A -» 2.2 min 10% A; Ofen:50°C; Fluss: 0.33 ml/min; UV-Detektion: 210 nm.
Methode 7 (LC-MS): Instrument: Micromass Quattro Micro MS mit HPLC Agilent Serie 1100; Säule: Thermo Hypersil GOLD 3μ 20 x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure , Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 100% A^ 3.0 min 10% A -> 4.0 min 10% A^ 4.01 min 100% A (Fluss 2.5ml)-> 5.00 min 100% A Ofen: 500C; Fluss:2 ml/min; UV-Detektion: 210 nm
Ausgangsverbindungen und Intermediate:
Beispiel IA
2-(4-Chloφhenyl)-6-ethyl-4-(2-methylpropyl)-l,4-dihydropyrimidin-5-carbonsäuremethylester

1.04 g (7.97 mmol) 3-Oxopentansäuremethylester, 686 mg (7.97 mmol) 3-Methylbutanal, 79 μl (0.797 mmol) Piperidin und 46 μl (0.797 mmol) Essigsäure wurden in 20 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so 1.56 g Methyl-5- methyl-2-propanoylhex-2-enoat, welches ohne weitere Reinigungsoperationen umgesetzt wurde. 470 mg (ca. 2.37 mmol) des so gewonnenen Rohmaterials wurden mit 453 mg (2.37 mmol) 4-Chlorbenzamidin-Hydrochlorid versetzt. Anschließend wurde in 10 ml 1-Butanol aufgenommen, mit 0.397 ml (2.85 mmol) Triethylamin versetzt und 4 h bei Rückflußtemperatur umgesetzt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und der Rückstand in Wasser aufgenommen. Nach dreimaliger Extraktion mit Essigsäureethylester wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet, das Lösungsmittel am Rotationsverdampfer abgetrennt und anschließend durch präparative HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 -> 90:10) aufgereinigt. Man erhielt so 154 mg (19% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 0.93 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.89 (d, 3H), 0.95 (d, 3H), 1.14 (t, 3H), 1.54 (mz, IH), 1.82 (mz, IH), 2.69 (mz, IH), 2.81 (mz, IH), 3.68 (s, 3H), 4.54 (dd, IH), 7.65 (d, 2H), 7.88 (d, 2H).
Beispiel 2A
2-(4-Chlorphenyl)-4-ethyl-6-(2-methylpropyl)pyrimidin-5-carbonsäuremethylester

150 mg (0.448 mmol) Beipiel IA wurden in 10 ml Benzol aufgenommen und mit 112 mg (0.493 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, in Wasser aufgenommen und anschließend mit Essigsäureethylester extrahiert. Es wurde mit Magnesiumsulfat getrocknet und das Lösemittel unter vermindertem Druck destillativ abgetrennt. Anschließend wurde mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 107 mg (72% d. Th.) der Zielverbindung.
LC-MS (Methode 1): Rt = 1.82 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.92 (d, 6H), 1.29 (t, 3H), 2.23 (sept, IH), 2.65 (d, 2H), 2.79 (q, 2H), 3.94 (s, 3H), 7.62 (d, 2H), 8.42 (d, 2H).
Beispiel 3A
2-(4-Chloφhenyl)-4-methyl-6-oxo-l,4,5,6-tetrahydropyrimidin-5-carbonsäureethylester

Zu einer Lösung aus 3.66 g (53.703 mmol) Natriumethylat und 5.00 g (29.537 mmol) 4-Chlorbenzamidin-Hydrochlorid in 50 ml Ethanol wurden unter Argonatmosphäre 5.00 g (26.851 mmol) Ethylidenmalonsäurediethylester zugegeben. Das Reaktionsgemisch wurde 1.5 h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, in Dichlormethan aufgenommen und anschließend mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Man erhielt so 6.86 g (75% d. Th.) Rohprodukt in 86%iger Reinheit (LC-MS), welches ohne weitere Reinigungsoperation umgesetzt wurde.
LC-MS (Methode 1): R, = 0.87 min; MS (ESIpos): m/z = 295 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.20 (t, 3H), 1.28 (d, 3H), 3.40 (d, IH), 3.97-4.03 (m, IH), 4.16 (q, 2H), 7.52 (d, 2H), 7.85 (d, 2H), 9.53 (sbr, IH).
Beispiel 4A
2-(4-Chloφhenyl)-4-methyl-6-oxo-l,6-dihydropyrimidin-5-carbonsäureethylester

Eine Lösung aus 3.00 g (ca. 8.753 mmol) Beispiel 3A, 1.56g (8.753 mmol) N-Bromsuccinimid, 212 mg (0.875 mmol) Dibenzoylperoxid und 1.81g (13.130 mmol) im Mörser gemahlenes Kaliumcarbonat in 150 ml Dioxan wurde Ih bei Rückflußtemperatur unter Argonatmosphäre gerührt. Nach dem Abkühlen wurde der Ansatz mit Wasser versetzt und anschließend mit Dichlormethan und Essigsäureethylester extrahiert. Die organische Phase wurde mit einer gesättigten wässrigen Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Man erhielt so 2.5g (66% d. Th.) Rohprodukt in 71%iger Reinheit (LC-MS), welches ohne weitere Reinigungsoperation umgesetzt wurde.
LC-MS (Methode 1): R, = 1.04 min; MS (ESIpos): m/z = 293 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.28 (t, 3H), 2.32 (s, 3H), 4.28 (q, 2H), 7.62 (d, 2H), 8.13 (d, 2H), 13.11 (sbr, IH).
Beispiel 5A
2-(4-Chlorphenyl)-4-methyl-6-(3-methylbutoxy)pyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.342 mmol) Beispiel 4A, 42 mg (0.478 mmol) 3 -Methyl- 1-butanol und 125 mg (0.478 mmol) Triphenylphosphin in 4 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 96 mg (0.478 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 3h bei Raumtemperatur weitergerührt. Die Mischung wurde ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser,: 90:10, isokratisch). Man erhielt so 41 mg (46 % d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.84 min; MS (ESIpos): m/z = 363 [M+H]+.
Beispiel 6A
2-(4-Chlorphenyl)-4-(3-methoxy- 1 -methylpropoxy)-6-methylpyrimidin-5-carbonsäureethylester
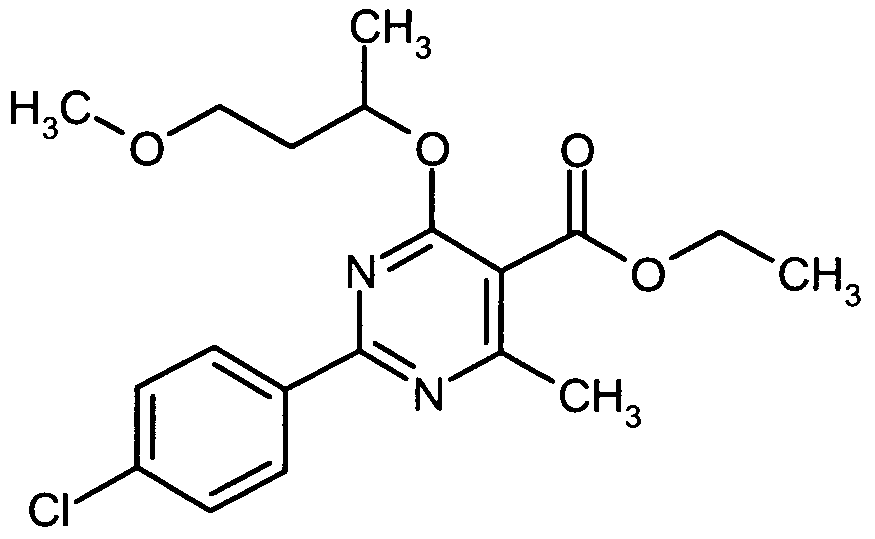
Eine Lösung aus 100 mg (ca. 0.342 mmol) Beispiel 4A, 50 mg (0.478 mmol) 4-Methoxybutan-2-ol und 125 mg (0.478 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 96 mg (0.478 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 3h bei Raumtemperatur weitergerührt. Die Mischung wurde ohne weitere Aufarbeitung mittels präparativer HPLC auf gereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 40 mg (43 % d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.75 min; MS (ESIpos): m/z = 379 [M+H]+.
Beispiel 7A
2-(4-Chlθφhenyl)-4-methyl-6-(l-methylethoxy)pyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.342 mmol) Beispiel 4A, 28 mg (0.478 mmol) Isopropanol und 125 mg (0.478 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 96 mg (0.478 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 3h bei Raumtemperatur weitergerührt. Die Mischung wurde ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 42 mg (51 % d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.69 min; MS (ESIpos): m/z = 335 [M+H]+.
Beispiel 8A
2-(4-Chloφhenyl)-4-methyl-6-{[3-(trifluormethyl)cyclohexyl]oxy}pyrimidin-5- carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.342 mmol) Beispiel 4A, 28 mg (0.478 mmol) 3-(Trifluormethyl)cyclohexanol und 125 mg (0.478 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 96 mg (0.478 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 3h bei
Raumtemperatur weitergerührt. Die Mischung wurde ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 14 mg (13% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.09 min; MS (ESIpos): m/z = 443 [M+H]+.
Beispiel 9A
2-(4-Chlθφhenyl)-4-(l-methylethyl)-6-oxo-l,4,5,6-tetrahydropyrimidin-5-carbonsäureethylester

Zu einer Lösung aus 1.59 g (23.336 mmol) Natriumethylat und 2.45 g (12.835 mmol) 4-Chlorbenzamidin-Hydrochlorid in 10 ml Ethanol unter Argonatmosphäre wurden 2.5 g (11.668 mmol) (2-Methylpropyliden)malonsäuredieethylester zugegeben. Das Reaktionsgemisch wurde 6.5h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, in Dichlormethan aufgenommen und anschließend mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Man erhielt so 3.03 g (60% d. Th.) Rohprodukt (75 %ige Reinheit (LC-MS)), welches ohne weitere Reinigungsoperation umgesetzt wurde.
LC-MS (Methode 3): R, = 2.23 min; MS (ESIpos): m/z = 323 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.87 (d, 3H), 1.07 (d, 3H), 1.20 (t, 3H), 1.70-1.80 (m, IH), 3.50 (d, IH), 3.76 (dd, IH), 4.16 (q, 2H), 7.53 (d, 2H), 7.88 (d, 2H), 11.01 (sbr, IH).
Beispiel IQA
2-(4-Chlθφhenyl)-4-(l-methylethyl)-6-oxo-l,6-dihydropyrimidin-5-carbonsäureethylester

LC-MS (Methode 3): R, = 2.50 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.18 (d, 6H), 1.26 (t, 3H), 4.23 (q, 2H), 7.54 (d, 2H), 8.20 (d, 2H).
Beispiel IIA
2-(4-Chloφhenyl)-4-methoxy-6-(l-methylethyl)pyrimidin-5-carbonsäureethylester

Eine Lösung aus 92 mg (ca. 0.200 mmol) Beispiel 10A, 9 mg (0.201 mmol) Methanol und 74 mg (0.281 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Dann 57 mg (0.281 mmol) Diisopropylazodicarboxylat wurden zugegeben, und das Reaktionsgemisch wurde 1.5h bei Raumtemperatur weitergerührt. Die Mischung wurde ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 26 mg (39% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.75 min; MS (ESIpos): m/z = 335 [M+H]+.
Beispiel 12A
2-(4-Chlorphenyl)-4-(cyclopropylmethoxy)-6-methylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.342 mmol) Beispiel 4A, 34 mg (0.478 mmol) Cyclopropylmethanol und 125 mg (0.478 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 96 mg (0.478 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 3h bei Raumtemperatur weitergerührt. Die Mischung wurde ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90: 10, isokratisch). Man erhielt so 68 mg (81% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.31 min; MS (ESIpos): m/z = 347 [M+H]+.
Beispiel 13A
2-(4-Chlorphenyl)-6-ethyl-4-( 1 -methylethyl)- 1 ,4-dihydropyrimidin-5 -carbonsäuremethylester

2.15 g (16.5 mmol) 3-Oxopentansäuremethylester, 1.19 g (16.5 mmol) 2-Methylpropionaldehyd, 163 μl (1.65 mmol) Piperidin und 95 μl (1.65 mmol) Essigsäure wurden in 40 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so ca. 3 g 4- Methyl-2-propanoylpent-2-ensäuremethylester, der ohne weitere Reinigungsoperationen umgesetzt wurde. 1.52 g (ca. 8.25 mmol) des so gewonnenen Rohmaterials wurden mit 1.58 g (8.25 mmol) 4-Chlorbenzamidin-Hydrochlorid versetzt. Anschließend wurde in 20 ml 1-Butanol aufgenommen, mit 1.38 ml (9.90 mmol) Triethylamin versetzt und 4 h bei Rückflußtemperatur gerührt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und der Rückstand in Wasser aufgenommen. Nach dreimaliger Extraktion mit Essigsäureethylester wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet, das Lösungsmittel am Rotationsverdampfer
abgetrennt und anschließend durch präparative HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — » 90: 10) aufgereinigt. Man erhielt so 1.10 g (41% d. Th., bezogen auf 4-Chlorbenzamidin- Hydrochlorid) der Zielverbindung.
LC-MS (Methode 1): R, = 0.85 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.83 (d, 3H), 0.90 (d, 3H), 1.14 (t, 3H), 1.72 (mz, IH), 2.73 (mz, 2H), 3.66 (s, 3H), 4.37 (d, IH), 7.62 (d, 2H), 7.89 (d, 2H).
Beispiel 14A
2-(4-Chlorphenyl)-4-ethyl-6-( 1 -methylethyl)pyrimidin-5 -carbonsäuremethylester

940 mg (2.93 mmol) Beipiel 13A wurden in 50 ml Benzol aufgenommen und mit 731 mg (3.22 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — > 90:10) aufgereinigt. Man erhielt so 390 mg (41% d. Th.) der Zielverbindung und 190 mg (21% d. Th.) Beispiel 7 direkt durch partielle Verseifung.
LC-MS (Methode 1): R, = 1.77 min; MS (ESIpos): m/z = 319 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.26-1.32 (9H), 2.79 (q, 2H), 3.07 (sept, IH), 3.95 (s, 3H), 7.63 (d, 2H), 8.45 (d, 2H).
Beispiel 15A
2-(4-Chlorphenyl)-6-ethyl-4-( 1 -methylbutyl)- 1 ,4-dihydropyrimidincarbonsäuremethylester

1.56 g (12.0 mmol) 3-Oxopentansäuremethylester, 1.20 g (12.0 mmol) 2-Methylpentanal, 118 μl (1.20 mmol) Piperidin und 68 μl (1.20 mmol) Essigsäure wurden in 30 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so 2.67 g 4-Methyl-2- propanoylhept-2-ensäuremethylester, welcher ohne weitere Reinigungsoperationen umgesetzt wurde. 2.53 g (11.9 mmol) des so gewonnenen Rohmaterials wurden mit 2.28 g (11.9 mmol) 4-Chlorbenzamidin-Hydrochlorid versetzt. Anschließend wurde in 40 ml 1-Butanol aufgenommen, mit 2.00 ml (14.3 mmol) Triethylamin versetzt und über Nacht bei Rückflußtemperatur gerührt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und der Rückstand in Wasser aufgenommen. Nach dreimaliger Extraktion mit Essigsäureethylester wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet und das Lösungsmittel am Rotationsverdampfer abgetrennt. Anschließend wurde mittels präparativer MPLC (Biotage 4OM Kartusche; Laufmittel: Isohexan/ Essigsäureethylester 4/1) aufgereinigt. Man erhielt so 1.37 g (26% d. Th., bezogen auf 4-Chlorbenzamidin-Hydrochlorid) der Zielverbindung.
LC-MS (Methode 2): R, = 1.29 min; MS (ESIpos): m/z = 349 [M+H]+.
Beispiel 16A
2-(4-Chloφhenyl)-4-ethyl-6-(l-methylbutyl)pyrimidin-5-carbonsäuremethylester

1.37 g (3.14 mmol) Beipiel 15A wurden in 40 ml Benzol aufgenommen und mit 784 mg (3.46 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels
präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90: 10) aufgereinigt. Man erhielt so 690 mg (58% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.91 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.82 (t, 3H), 1.12 (mz, IH), 1.22 (mz, IH), 1.12 (d, 3H), 1.29 (t, 3H), 1.56 (mz, IH), 1.84 (mz, IH), 2.77 (q, 2H), 2.90 (mz, IH), 3.95 (s, 3H), 7.62 (d, 2H), 8.43 (d, 2H).
Beispiel 17A
2-(4-Chlθφhenyl)-4-ethoxy-6-methylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.304 mmol) Beispiel 4A, 19 mg (0.426 mmol) Ethanol und 111 mg (0.426 mmol) Triphenylphosphin in 1.8 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 86 mg (0.426 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90: 10, isokratisch). Man erhielt so 45 mg (58% d. Th.) der Zielverbindung
LC-MS (Methode 3): Rt = 3.13 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.31 (t, 3H), 1.36 (t, 3H), 4.35 (q, 2H), 4.57 (q, 2H), 7.61 (d, 2H), 8.38 (d, 2H).
Beispiel 18A
2-(4-Chlθφhenyl)-4-methyl-6-propoxypyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.304 mmol) Beispiel 4A, 25 mg (0.426 mmol) n-Propanol und 111 mg (0.426 mmol) Triphenylphosphin in 1.8 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 86 mg (0.426 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90: 10, isokratisch). Man erhielt so 52 mg (51% d. Th.) der Zielverbindung
LC-MS (Methode 3, MHZ-Z2-GEM-1): R, = 3.26 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSOKI6): δ [ppm] = 0.98 (t, 3H), 1.31 (t, 3H), 1.71-1.80 (m, 2H), 4.36 (q, 2H), 4.48 (t, 2H), 7.60 (d, 2H), 8.38 (d, 2H).
Beispiel 19A
2-(4-Chloφhenyl)-4-methyl-6-(2-methylpropoxy)pyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.304 mmol) Beispiel 4A, 31 mg (0.426 mmol) 2-Methyl-l-propanol und 111 mg (0.426 mmol) Triphenylphosphin in 1.8 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 86 mg (0.426 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90: 10, isokratisch). Man erhielt so 78 mg (73% d. Th.) der Zielverbindung
LC-MS (Methode 3): R, = 3.36 min; MS (ESIpos): m/z = 349 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.99 (d, 6H), 1.32 (t, 3H), 2.01-2.11 (m, IH), 4.31 (d, 2H), 4.35 (q, 2H), 7.60 (d, 2H), 8.38 (d, 2H).
Beispiel 2OA
2-(4-Chloφhenyl)-4-methyl-6-[(l-methylcyclopropyl)methoxy]pyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.304 mmol) Beispiel 4A, 36 mg (0.426 mmol) (l-Methylcyclopropyl)methanol und 111 mg (0.426 mmol) Triphenylphosphin in 1.8 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 86 mg (0.426 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90: 10, isokratisch). Man erhielt so 53 mg (48% d. Th.) der Zielverbindung
LC-MS (Methode 3): R, = 3.34 min; MS (ESIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSO-Cl6): δ [ppm] = 0.39 (sbr, 2H), 0.58 (sbr, 2H), 1.15 (s, 3H), 1.32 (t, 3H), 4.34-4.39 (m, 4H), 7.59 (d, 2H), 8.36 (d, 2H).
Beispiel 21 A
2-(4-Chloφhenyl)-4-(cyclopentyloxy)-6-methylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.304 mmol) Beispiel 4A, 37 mg (0.426 mmol) Cyclopenthanol und 111 mg (0.426 mmol) Triphenylphosphin in 1.8 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 86 mg (0.426 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser = 50/50). Man erhielt so 38 mg (34% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.37 min; MS (ESIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.30 (t, 3H), 1.61-1.79 (m, 6H), 1.99-2.03 (m, 2H), 2.47 (s, 3H), 4.34 (q, 2H), 5.63-5.67 (m, IH), 7.61(d, 2H), 8.38 (d, 2H).
Beispiel 22A
2-(4-Chlorphenyl)-4-(cyclopentylmethoxy)-6-methylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.304 mmol) Beispiel 4A, 42 mg (0.426 mmol) Cyclopentylmethanol und 111 mg (0.426 mmol) Triphenylphosphin in 1.8 ml THF wurde 20 Minuten bei
Raumtemperatur gerührt. Es wurden 86 mg (0.426 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur gerührt. Die Mischung wurde
einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser: 90:10, isokratisch). Man erhielt so 52 mg (46% d. Th.) der Zielverbindung
LC-MS (Methode 3): R, = 3.47 min; MS (ESIpos): m/z = 375[M+H]+.
Beispiel 23A
4-Chlor-2-(4-Chloφhenyl)-6-methylpyrimidin-5-carbonsäureethylester

1.5 g (5.124 mmol) Beispiel 4A in 19.1 ml (204.971 mmol) Phosphoroxychlorid wurden 2h bei Rückflußtemperatur gerührt. Das Reaktionsgemisch wurde einrotiert. Der Rückstand wurde mit einer 25%igen wässrigen Ammoniumhydroxid-Lösung versetzt, mit IN Salzsäure auf pH 7 gestellt und anschließend mit Dichlormethan extrahiert. Die organische Phase wurde über Natriumsulfat getrocknet und eingeengt. Man erhielt so 1.38 g (87% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.10 min; MS (ESIpos): m/z = 311[M+H]+.
Beispiel 24A
2-(4-Chlorphenyl)-6-oxo-4-propyl-l,4,5,6-tetrahydropyrimidin-5-carbonsäureethylester

Zu einer Lösung aus 0.76 g (11.201 mmol) Natriumethylat und 1.17 g (6.161 mmol) 4-Chlorbenzamidin-Hydrochlorid in 12 ml Ethanol wurden unter Argonatmosphäre 1.2 g (5.600 mmol) n-Butylidenmalonsäurediethylester zugegeben. Das Reaktionsgemisch wurde 2.5h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, in Dichlormethan aufgenommen und anschließend mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über
Natriumsulfat getrocknet und eingeengt. Man erhielt so 1.25 g (56% d. Th.) Rohprodukt in 82%iger Reinheit (LC-MS), welches ohne weitere Reinigungsoperation umgesetzt wurde.
LC-MS (Methode 2): R, = 1.75 min; MS (ESIpos): m/z = 323 [M+H]+.
Beispiel 25A
2-(4-Chlorphenyl)-6-oxo-4-propyl- 1 ,6-dihydropyrimidin-5 -carbonsäureethylester

Eine Lösung aus 1.25g (ca. 3.175 mmol) Beispiel 24A, 0.57 g (3.175 mmol) N-Bromsuccinimid, 565 mg (0.318 mmol) Dibenzoylperoxid und 6.58 g (1.703 mmol) im Mörser gemahlenes Kaliumcarbonat in 58 ml Dioxan wurde unter Argonatmosphäre Ih bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz mit Wasser versetzt und anschließend mit Dichlormethan und Essigsäureethylester extrahiert. Die organische Phase wurde mit einer gesättigten wässrigen Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Das Rohprodukt wurde mit Ether versetzt und der ausgefallene Feststoff wurde abfiltriert und im Hochvakuum getrocknet. Man erhielt 335 mg (33% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.05 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.92 (t, 3H), 1.28 (t, 3H), 1.66-1.75 (m, 2H), 4.28 (q, 2H), 7.62 (d, 2H), 8.14 (d, 2H), 13.10 (sbr, IH).
Beispiel 26A
2-(4-Chloφhenyl)-4-ethoxy-6-propylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 60 mg (0.187 mmol) Beispiel 25 A, 12 mg (0.262 mmol) Ethanol und 69 mg (0.262 mmol) Triphenylphosphin in 1.2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 53 mg (0.262 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2.5h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser: 90/10, isokratisch). Man erhielt so 35 mg (53% Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.07 min; MS (ESIpos): m/z = 349 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.93 (t, 3H), 1.31 (t, 3H), 1.36 (t, 3H), 1.70-1.79 (m, 2H), 2.69 (t, 2H), 4.36 (q, 2H), 4.56 (q, 2H), 7.61 (d, 2H), 8.38 (d, 2H).
Beispiel 27A
2-(4-Chlorphenyl)-4-propoxy-6-propylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 60 mg (0.187 mmol) Beispiel 25A, 16 mg (0.262 mmol) n-Propanol und 69 mg (0.262 mmol) Triphenylphosphin in 1.2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 53 mg (0.262 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2.5h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser: 90: 10, isokratisch). Man erhielt so 46 mg (68% Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.19 min; MS (ESIpos): m/z = 363 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.95 (t, 3H), 0.99 (t, 3H), 1.29 (t, 3H), 1.70-1.80 (m, 4H), 2.69 (t, 2H), 4.35 (q, 2H), 4.48 (t, 2H), 7.61 (d, 2H), 8.38 (d, 2H).
Beispiel 28A
2-(4-Chloφhenyl)-4-(2-methylpropoxy)-6-propylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 60 mg (0.187 mmol) Beispiel 25A, 19 mg (0.262 mmol) 2-Methyl-l-propanol und 69 mg (0.262 mmol) Triphenylphosphin in 1.2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 53 mg (0.262 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2.5 h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser = 90/10, isokratisch). Man erhielt so 47 mg (60% Th.) der Zielverbindung.
LC-MS (Methode 2): Rt = 3.28 min; MS (ESIpos): m/z = 377 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.83 (t, 3H), 0.98 (d, 6H), 1.31 (t, 3H), 1.70-1.79 (m, 2H), 2.00-2.11 (m, IH), 2.70 (t, 2H), 4.30 (d, 2H), 4.36 (q, 2H), 7.61 (d, 2H), 8.38 (d, 2H).
Beispiel 29A
2-(4-Chlorphenyl)-4-[(l-methylcyclopropyl)methoxy]-6-propylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 60 mg (0.187 mmol) Beispiel 25 A, 22 mg (0.262 mmol) 1- Methylcyclopropanmethanol und 69 mg (0.262 mmol) Triphenylphosphin in 1.2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 53 mg (0.262 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2.5 h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser = 90/10, isokratisch). Man erhielt so 42 mg (58% Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.28 min; MS (ESIpos): m/z = 389 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.39 (dd, 2H), 0.58 (dd, 2H), 0.95 (t, 3H), 1.23 (s, 3H), 1.33 (t, 3H), 1.70-1.80 (m, 2H), 2.70 (t, 2H), 4.35 (s, 2H), 4.37 (q, 2H), 7.60 (d, 2H), 8.37 (d, 2H).
Beispiel 3OA
2-(4-Chloφhenyl)-4-(cyclopentyloxy)-6-propylpyrimidin-5-carbonsäureethylester

Eine Lösung aus 90 mg (0.281 mmol) Beispiel 25A, 34 mg (0.393 mmol) Cyclopentanol und 103 mg (0.393 mmol) Triphenylphosphin in 1.8 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 80 mg (0.393 mmol) Diisopropylazodicarboxylat zugegeben und das
Reaktionsgemisch 2.5 h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne
weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser: 90: 10, isokratisch). Man erhielt so 70 mg (64 % Th.) der Zielverbindung.
LC-MS (Methode 2): Rt = 3.31 min; MS (ESIpos): m/z = 389 [M+H]+.
1H-NMR (400 MHz, DMSO-de): δ [ppm] = 0.93 (t, 3H), 1.30 (t, 3H), 1.60-1.79 (m, 8H), 1.99-2.03 (m, 2H), 2.68 (t, 2H), 4.34 (q, 2H), 5.63-5.67 (m, IH), 7.61 (d, 2H), 8.38 (d, 2H).
Beispiel 31 A
2-(4-Chloφhenyl)-4-ethoxy-6-(l-methylethyl)pyrimidin-5-carbonsäureethylester

Eine Lösung aus 60 mg (ca. 0.187 mmol) Beispiel 10A, 12 mg (0.262 mmol) Ethanol und 69 mg (0.262 mmol) Triphenylphosphin in 1.2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 53 mg (0.262 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2.5 h bei Raumtemperatur gerührt. Die Mischung wurde eingeengt und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser = 90/10, isokratisch). Man erhielt so 40 mg (61% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.81 min; MS (ESIpos): m/z = 349 [M+H]+.
Beispiel 32A
2-(4-Chloφhenyl)-6-ethyl-4-(l-ethylpropyl)-l,4-dihydropyrimidin-5-carbonsäuremethylester

LC-MS (Methode 1): R, = 1.00 min; MS (ESIpos): m/z = 349 [M+H]+.
Beispiel 33A
2-(4-Chloφhenyl)-4-ethyl-6-(l-ethylpropyl)pyrimidin-5-carbonsäuremethylester

595 mg (1.71 mmol) Beipiel 32A wurden in 40 ml Benzol aufgenommen und mit 426 mg (1.88 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 249 mg (43% d. Th.) der Zielverbindung.
LC-MS (Methode 1): Rt = 1.88 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.72 (t, 6H), 1.29 (t, 3H), 1.68 (mz, 2H), 1.75-1.88 (m, 2H), 2.59 (mz, IH), 2.77 (q, 2H), 3.94 (s, 3H), 7.62 (d, 2H), 8.43 (d, 2H).
Beispiel 34A
2-(4-Chlθφhenyl)-6-ethyl-4-(tetrahydrofuran-3-yl)-l,4-dihydropyrimidin-5-carbonsäure- methylester

1.30 g (10.0 mmol) 3-Oxopentansäuremethylester, 1.00 g (10.0 mmol) Tetrahydrofuran-3- carbaldehyd, 99 μl (1.00 mmol) Piperidin und 57 μl (1.00 mmol) Essigsäure wurden in 45 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so ca. 2.2 g 3-Oxo-2-(tetrahydrofuran-3-ylmethylidene)pentansäuremethylester, welcher ohne weitere Reingungsschritte umgesetzt wurde. 2.12 g des so gewonnenen Rohmaterials wurden mit 1.91 g (10.0 mmol) 4-Chlorbenzamidin-Hydrochlorid versetzt. Anschließend wurde in 30 ml 1- Butanol aufgenommen, mit 1.74 ml (12.5 mmol) Triethylamin versetzt und über Nacht bei Rückflußtemperatur gerührt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und der Rückstand in Wasser aufgenommen. Nach dreimaliger Extraktion mit Essigsäureethylester wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet, das Lösungsmittel am Rotationsverdampfer abgetrennt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90: 10) aufgereinigt. Man erhielt so 512 mg (11% d. Th., bezogen auf 4-Chlorbenzamidin-Hydrochlorid) der Zielverbindung.
LC-MS (Methode 3): R, = 0.84 min; MS (ESIpos): m/z = 349 [M+H]+.
Beispiel 35A
2-(4-Chloφhenyl)-4-ethyl-6-(tetrahydrofuran-3-yl)pyrimidin-5-carbonsäuremethylester

500 mg (1.43 mmol) Beipiel 34A wurden in 45 ml Benzol aufgenommen und mit 358 mg (1.58 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und der Rückstand in ca. 6 ml Acetonitril aufgenommen. Das Präzipitat wurde abfϊltriert und die verbleibende Lösung anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — > 90: 10) aufgereinigt. Man erhielt so 290 mg (55% d. Th.) der Zielverbindung.
LC-MS (Methode 3): Rt = 3.05 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.29 (t, 3H), 2.18-2.35 (m, 2H), 2.80 (q, 2H), 3.60 (quint, IH), 3.81-3.90 (m, 2H), 3.96 (s, 3H), 4.01 (mz, IH), 4.07 (t, IH), 7.63 (d, 2H), 8.43 (d, 2H).
Beispiel 36A
2-(4-Chloφhenyl)-4-cyclopentyl-6-ethyl-l,4-dihydropyrimidin-5-carbonsäuremethylester

1.30 g (10.0 mmol) 3-Oxopentansäuremethylester, 0.98 g (10.0 mmol) Cyclopentancarbaldehyd, 99 μl (1.00 mmol) Piperidin und 57 μl (1.00 mmol) Essigsäure wurden in 45 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so ca. 2.20 g 2-(Cyclopentylmethylidene)-3-oxopentansäuremethylester, welcher ohne weitere
Reingungsschritte umgesetzt wurde. 2.10 g des so gewonnenen Rohmaterials wurden mit 1.91 g
(10.0 mmol) 4-Chlorbenzamidin-Hydrochlorid versetzt. Anschließend wurde in 30 ml 1-Butanol aufgenommen, mit 1.74 ml (12.5 mmol) Triethylamin versetzt und über Nacht bei Rückflußtemperatur gerührt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und der Rückstand in Wasser aufgenommen. Nach dreimaliger Extraktion mit Essigsäureethylester wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet, das Lösungsmittel am Rotationsverdampfer abgetrennt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 245 mg (7% d. Th., bezogen auf 4-Chlorbenzamidin-Hydrochlorid) der Zielverbindung.
LC-MS (Methode 1): R, = 0.93 min; MS (ESIpos): m/z = 347 [M+H]+.
Beispiel 37A
2-(4-Chloφhenyl)-4-cyclopentyl-6-ethylpyrimidin-5-carbonsäuremethylester

245 mg (0.706 mmol) Beipiel 36A wurden in 40 ml Benzol aufgenommen und mit 176 mg (0.777 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückfiußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und der Rückstand in ca. 5 ml Acetonitril aufgenommen. Das Präzipitat wurde abfiltriert und die verbleibende Lösung anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — > 90:10) aufgereinigt. Man erhielt so 70 mg (26% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.86 min; MS (ESIpos): m/z = 345 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.28 (t, 3H), 1.61-1.76 (m, 2H), 1.79-2.03 (m, 6H), 2.76 (q, 2H), 3.19 (quint, IH), 3.94 (s, 3H), 7.61 (d, 2H), 8.41 (d, 2H).
Beispiel 38A
2-(4-Chlorphenyl)-4-(l-methylethyl)-6-propoxypyrimidin-5-carbonsäureethylester

Eine Lösung aus 60 mg (0.187 mmol) Beispiel 1OA, 15 mg (0.262 mmol) n-Propanol und 69 mg (0.262 mmol) Triphenylphosphin in 1.2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Dann 53 mg (0.262 mmol) Diisopropylazodicarboxylat wurden zugegeben, und das Reaktionsgemisch wurde 2.5 h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Acetonitril/Wasser = 50/50). Man erhielt so 60 mg (88 % Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.89 min; MS (ESIpos): m/z = 363 [M+H]+.
Beispiel 39A
4-Butyl-2-(4-chlorphenyl)-6-methylpyrimidin-5 -carbonsäureethylester

100 mg (0.321 mmol) Beispiel 23A wurden in 2.5 ml DMF gelöst. Anschließend wurden 1.28 ml (0.643 mmol) 1 -Butylzinkbromid-Lösung (0.5M in THF) und 18 mg (0.016 mmol) Tetrakis(triphenylphosphin)palladium(0) addiert. Das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt und über das Wochenende bei Raumtemperatur stehen gelassen. Das Reaktionsgemisch wurde ohne weitere Aufarbeitung mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 90:10, isokratisch) gereinigt. Man erhielt 20 mg (19% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.81 min; MS (ESIpos): m/z = 333 [M+H]+.
Beispiel 4OA
2-(4-Chlorphenyl)-4-methyl-6-(l-methylpropyl)pyrimidin-5-carbonsäureethylester

100 mg (0.321 mmol) Beispiel 23 A wurden in 2.5 ml DMF gelöst. Anschließend wurden 1.28 ml (0.643 mmol) 2-Butylzinkbromid-Lösung (0.5M in THF) und 18 mg (0.016 mmol) Tetrakis(triphenylphosphin)palladium(0) addiert. Das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt und über das Wochenende bei Raumtemperatur stehen gelassen. Das Reaktionsgemisch wurde ohne weitere Aufarbeitung mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 90:10, isokratisch) gereinigt. Man erhielt 8 mg (8% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.80 min; MS (ESIpos): m/z = 333 [M+H]+.
Beispiel 41 A
2-(4-Chlorphenyl)-4-(cyclopentyloxy)-6-(l-methylethyl)pyrimidin-5-carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.143 mmol) Beispiel 10A, 17 mg (0.201 mmol) Cyclopentanol und 53 mg (0.201 mmol) Triphenylphosphin in 1.4 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 41 mg (0.201 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch über Nacht bei Raumtemperatur gerührt. Die Mischung wurde ohne weitere
Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser: 90:10, isokratisch ). Man erhielt so 54 mg (85 % Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.97 min; MS (ESIpos): m/z = 389 [M+H]+.
Beispiel 42A
2-(4-Chlorphenyl)-4-(cyclopropylmethoxy)-6-( 1 -methylethyl)pyrimidin-5 -carbonsäureethylester

Eine Lösung aus 100 mg (ca. 0.143 mmol) Beispiel 10A, 157 mg (0.201 mmol) Cyclopropylmethanol und 53 mg (0.201mmol) Triphenylphosphin in 1.4 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 53 mg (0.262 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch über Nacht bei Raumtemperatur gerührt. Die Mischung wurde ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser: 90/10, isokratisch ). Man erhielt so 28 mg (52 % Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.88 min; MS (ESIpos): m/z = 375 [M+H]+.
Beispiel 43A
2-(4-Chlorphenyl)-4-( 1 -methylethoxy)-6-( 1 -methylethyl)pyrimidin-5 -carbonsäureethylester

LC-MS (Methode 1): R, = 1.88 min; MS (ESIpos): m/z = 363 [M+H]+.
Beispiel 44A
2-(4-Chlθφhenyl)-4-cyclopropyl-6-oxo-l,4,5,6-tetrahydropyrimidin-5-carbonsäuremethylester

3.0 g (22.707 mmol) Malonsäuredimethylester, 1.59 g (22.707 mmol) Cyclopropancarboxaldehyd, 0.22 ml (2.271 mmol) Piperidin und 0.13 ml (2.271 mmol) Essigsäure wurden in 50 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so (Cyclopropylmethyliden)malonsäuredimethylester, der ohne weitere Reinigungsoperationen umgesetzt wurde. Zu einer Lösung aus 3.09 g (45.414 mmol) Natriumethylat und 4.77 g (24.978 mmol) 4-Chlorbenzamidin-Hydrochlorid in 50 ml Ethanol wurde unter Argonatmosphäre 4.1 g (ca. 22.707 mmol) des so gewonnenen Rohmaterials addiert. Das Reaktionsgemisch wurde 3.5 h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, in Essigsäureethylester aufgenommen und mit gesättigter wässriger Natriumchlorid-Lösung gewaschen. Dann wurde mit Natriumsulfat getrocknet und eingeengt. Man erhielt so 4.57 g (43% d. Th.) Rohprodukt in 66%-iger Reinheit (LC-MS), welches ohne weitere Reinigungsoperation eingesetzt wurde.
LC-MS (Methode 2): R, = 1.35 min; MS (ESIpos): m/z = 307 [M+H]+.
Beispiel 45A
2-(4-Chloφhenyl)-4-cyclopropyl-6-oxo-l,6-dihydropyrimidin-5-carbonsäuremethylester

Eine Lösung aus 4.57 g (ca. 8.929 mmol) Beispiel 44A, 1.58g (8.829 mmol) N-Bromsuccinimid, 433 mg (0.1.786 mmol) Dibenzoylperoxid und 1.85 g (13.394 mmol) im Mörser gemahlenes Kaliumcarbonat in 60 ml Dioxan wurde unter Argonatmosphäre 1.5h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz mit einer gesättigten wässrigen Natriumthiosulfat- Lösung versetzt und anschließend wurden die flüchtigen Komponenten am Rotationsverdampfer abgetrennt. Das Rohmaterial wurde mit Essigsäureethylester aufgenommen, mit Wasser und einer gesättigten wässrigen Natriumchlorid-Lösung gewaschen. Die organische Phase wurde mit Natriumsulfat getrocknet und eingeengt. Man erhielt so 3.3 g (60% d. Th.) Rohprodukt in 50%- iger Reinheit (LC-MS). Dieses wurde ohne weitere Reinigungsoperation umgesetzt.
LC-MS (Methode 3): Rt = 2.23 min; MS (ESIpos): m/z = 305 [M+H]+.
Beispiel 46A
2-(4-Chlorphenyl)-4-cyclopropyl-6-( 1 -methylethoxy)pyrimidin-5 -carbonsäuremethylester

Eine Lösung aus 200 mg (ca. 0.341 mmol) Beispiel 45A, 28 mg (0.478 mmol) Isopropanol und 125 mg (0.478 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 96 mg (0.487 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90/10, isokratisch). Man erhielt so 11 mg (9% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.34 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ρpm]= 1.08-1.10 (m, 2H), 1.20-1.23 (m, 2H), 1.34 (d, 6H), 2.04-2.07 (m, IH), 3.88 (s, 3H), 5.49-5.54 (m, IH), 7.58 (d, 2H), 8.30 (d, 2H).
Beispiel 47A
2-(4-Chloφhenyl)-4-cyclopropyl-6-methoxypyrimidin-5-carbonsäureπiethylester

Eine Lösung aus 200 mg (ca. 0.328 mmol) Beispiel 45 A, 15 mg (0.459 mmol) Methanol und 120 mg (0.459 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 96 mg (0.487 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 5 mg (5% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.12 min; MS (ESIpos): m/z = 319 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm]= 1.09-1.13 (m, 2H), 1.22-1.24 (m, 2H), 2.07-2.12 (m, IH), 3.89 (s, 3H), 4.06 (s, 3H), 7.59 (d, 2H), 8.34 (d, 2H).
Beispiel 48A
2-(4-Chloφhenyl)-4-cyclopropyl-6-ethoxypyrimidin-5-carbonsäuremethylester

Eine Lösung aus 200 mg (ca. 0.328 mmol) Beispiel 45 A, 21 mg (0.459 mmol) Ethanol und 120 mg (0.459 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es
wurden 96 mg (0.487 mmol) Diisopropylazodicarboxylat zugegeben und das Reaktionsgemisch 2h bei Raumtemperatur gerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser = 90/10, isokratisch). Man erhielt so 11 mg (11% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.26 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm]= 1.08-1.12 (m, 2H), 1.21-1.23 (m, 2H), 1.35 (t, 3H), 2.06-2.11 (m, IH), 3.89 (s, 3H), 4.56 (q, 2H), 7.58 (d, 2H), 8.31 (d, 2H).
Beispiel 49A
4-Methyl-2-propanoylpent-2-ensäuremethylester (E/Z-Isomerengemisch)

33.18 g (255 mmol) 3-Oxopentansäuremethylester, 18.39 g (255 mmol) 2-Methylpropionaldehyd, 2.52 ml (25.5 mmol) Piperidin und 1.46 ml (25.5 mmol) Essigsäure wurden in 250 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so ca. 50g der Zielverbindung in 88%-iger Reinheit (GC-MS) als E/Z-Isomerengemisch. Das so erhaltene Rohprodukt wurde ohne weitere Reinigungsoperationen eingesetzt.
GC (Methode 5): R, = 3.49 & 3.59 min (E/Z-Isomerengemisch)
MS (DCI): m/z = 185 [M+H]+.
Beispiel 5OA
6-Ethyl-4-(l -methylethyl)-2-(3-methylphenyl)-l ,4-dihydropyrimidin-5-carbonsäuremethylester

0.54 g (ca. 2.93 mmol) Beipiel 49A wurden mit 0.50 g (2.93 mmol) 3-Methylbenzamidin-
Hydrochlorid versetzt. Anschließend wurde in 15 ml 1-Butanol aufgenommen, mit 0.51 ml (3.66 mmol) Triethylamin versetzt und über Nacht bei Rückflußtemperatur gerührt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und der Rückstand in Wasser aufgenommen.
Nach dreimaliger Extraktion mit Essigsäureethylester wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet, das Lösungsmittel am Rotationsverdampfer abgetrennt und anschließend durch präparative HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — > 90:10) aufgereinigt. Man erhielt so 380 mg (31% d. Th., bezogen auf 4-Chlorbenzamidin-Hydrochlorid) der Zielverbindung in 71 %-iger Reinheit (LC-MS).
LC-MS (Methode 1): R, = 0.84 min; MS (ESIpos): m/z = 301 [M+H]+.
Beispiel 51A
4-Ethyl-6-(l-methylethyl)-2-(3-methylphenyl)pyrimidin-5-carbonsäuremethylester

380 mg (0.911 mmol) Beipiel 50A wurden in 20 ml Benzol aufgenommen und mit 227 mg (1.002 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und der Rückstand in ca. 2 ml Acetonitril aufgenommen. Das Präzipitat wurde abfiltriert und die verbleibende Lösung anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 190 mg (70% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.71 min; MS (ESIpos): m/z = 299 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.25-1.32 (m, 9H), 2.42 (s, 3H), 2.75 (q, 2H), 3.05 (sept, IH), 3.93 (s, 3H), 7.37 (d, IH), 7.43 (t, IH), 8.20-8.27 (m, 2H).
Beispiel 52A
6-Ethyl-4-( 1 -methylethyl)-2-(4-methylphenyl)- 1 ,4-dihydropyrimidin-5 -carbonsäuremethylester

0.54 g (ca. 2.93 mmol) Beipiel 49A wurden mit 0.50 g (2.93 mmol) 4-Methylbenzamidin- Hydrochlorid versetzt. Anschließend wurde in 15 ml 1-Butanol aufgenommen, mit 0.511 ml (3.66 mmol) Triethylamin versetzt und über Nacht bei Rückflußtemperatur gerührt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und der Rückstand in Wasser aufgenommen. Nach dreimaliger Extraktion mit Essigsäureethylester wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet, das Lösungsmittel am Rotationsverdampfer abgetrennt und anschließend durch präparative HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — » 90: 10) aufgereinigt. Man erhielt so 237 mg (19% d. Th., bezogen auf 4-Chlorbenzamidin-Hydrochlorid) der Zielverbindung in 72 %-iger Reinheit (LC-MS).
LC-MS (Methode 1): Rt = 0.85 min; MS (ESIpos): m/z = 301 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.84 (d, 3H), 0.91 (d, 3H), 1.16 (t, 3H), 1.76 (m, IH), 2.40 (s, 3H), 2.76 (q, 2H), 3.68 (s, 3H), 4.37 (d, IH), 7.39 (d, 2H).
Beispiel 53A
4-Ethyl-6-(l-methylethyl)-2-(4-methylphenyl)pyrimidin-5-carbonsäuremethylester

234 mg (0.561 mmol) Beipiel 52A wurden in 20 ml Benzol aufgenommen und mit 140 mg (0.617 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und der Rückstand in ca. 2 ml Acetonitril aufgenommen. Das Präzipitat wurde abfϊltriert und die verbleibende Lösung anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 111 mg (66% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.72 min; MS (ESIpos): m/z = 299 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.24-1.31 (m, 9H), 2.39 (s, 3H), 2.75 (q, 2H), 3.05 (sept, IH), 3.94 (s, 3H), 7.36 (d, 2H), 8.34 (d, 2H).
Beispiel 54A
2-(4-Chloφhenyl)-6-(methoxymethyl)-4-(l-methylethyl)-l,4-dihydropyrimidin-5-carbonsäure- methylester

1.51 g (10.0 mmol) 4-Methoxy-3-oxo-butansäuremethylester, 721 mg (10.0 mmol) 2-Methylpropionaldehyd, 99 μl (1.00 mmol) Piperidin und 57 μl (1.00 mmol) Essigsäure wurden in 45 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so ca. 2.30g 2-(Methoxyacetyl)-4-methylpent-2-ensäuremethylester, der ohne weitere Reinigungs-operationen umgesetzt wurde. 1.91 g (ca. 10.00 mmol) des so gewonnenen Rohmaterials wurden mit 2.00 g (10.00 mmol) 4-Chlorbenzamidin-Hydrochlorid
versetzt. Anschließend wurde in 30 ml 1-Butanol aufgenommen, mit 1.74 ml (12.50 mmol) Triethylamin versetzt und über Nacht bei Rückflußtemperatur gerührt. Das Lösemittel wurde bei vermindertem Druck destillativ abgetrennt und das Rohprodukt anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 87 mg (3% d. Th., bezogen auf 4-Chlorbenzamidin-Hydrochlorid) der Zielverbindung.
LC-MS (Methode 2): R1 = 0.86 min; MS (ESIpos): m/z = 337 [M+H]+.
Beispiel 55A
2-(4-Chlorphenyl)-4-(methoxymethyl)-6-(l-methylethyl)pyrimidin-5-carbonsäuremethylester

87 mg (0.235 mmol) Beipiel 54A wurden in 20 ml Benzol aufgenommen und mit 59 mg (0.259 mmol) DDQ versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und der Rückstand in ca. 2 ml Acetonitril aufgenommen. Das Präzipitat wurde abfiltriert und die verbleibende Lösung anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 50 mg (64% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.75 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.29 (d, 6H), 3.16 (sept, IH), 3.88 (s, 3H), 4.63 (s, 2H), 7.63 (d, 2H), 8.43 (d, 2H).
Beispiel 56A
2-(4-Chlorphenyl)-6-oxo-4-(trifluormethyl)-l,4,5,6-tetrahydropyrimidin-5-carbonsäuremethylester (einzelnes Diastereomer)

7.17 g (54.3 mmol) Malonsäuredimethylester, 8.69 g (54.3 mmol) Trifluoracetaldehyd- Ethylhemiacetal, 1.39 ml (16.3 mmol) Piperidin und 0.93 ml (16.3 mmol) Essigsäure wurden in 100 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so ca. 15 g (2,2,2-Trifluorethyliden)malonsäuredimethylester, welcher ohne weitere Reinigungsoperationen direkt umgesetzt wurde. 1.01 g (ca. 4.76 mmol) der so gewonnenen Rohsubstanz und 1.00 g (5.23 mmol) 4-Chlorbenzamidin-Hydrochlorid wurden in 25 ml Methanol aufgenommen und unter Argonatmosphäre mit 1.98 ml (9.52 mmol, 27.5 Gew%-ig in Methanol) Natriummethanolat-Lösung versetzt. Das Reaktionsgemisch wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, in Wasser aufgenommen und anschließend mit Essigsäureethylester extrahiert (2x). Die vereinigten organischen Phasen wurden mit Magnesiumsulfat getrocknet und dann wurden am Rotationsverdampfer die flüchtigen Komponenten abgetrennt. Der Rückstand wurde in wenig Acetonitril aufgenommen und anschließend wurde mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10) aufgereinigt. Man erhielt so 0.28 g (15% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.19 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 3.74 (s, 3H), 4.00 (d, IH), 4.92 (mz, IH), 7.57 (d, 2H), 7.89 (d, 2H), 11.60 (s, IH).
Beispiel 57A
2-(4-Chlθφhenyl)-6-oxo-4-(trifluormethyl)-l,6-dihydropyrimidin-5-carbonsäuremethylester

LC-MS (Methode 1): R, = 1.23 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSO-de): δ [ppm] = 3.86 (s, 3H), 7.67 (d, 2H), 8.15 (d, 2H).
Beispiel 58A
2-(4-Chlθφhenyl)-4-(l-methylethoxy)-6-(trifluormethyl)pyrimidm-5-carbonsäuremethylester

90 mg (0.271 mmol) Beispiel 57A, 22 μl (0.284 mmol) 2-Propanol und 74.5 mg (0.284 mmol) Triphenylphosphin wurden in 5 ml THF aufgenommen und 20 Minuten bei Raumtemperatur gerührt. Anschließend wurden 55 μl (0.459 mmol) Diisopropylazodicarboxylat zugegeben und das
Reaktionsgemisch 2h bei Raumtemperatur umgesetzt. Die flüchtigen Komponenten wurden unter vermindertem Druck destillativ abgetrennt und das Rohprodukt mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90: 10). Man erhielt so 66 mg (65% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.24 min; MS (ESIpos): m/z = 375 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ρpm]= 1.40 (d, 6H), 3.91 (s, 3H), 5.64 (sept, IH), 7.67 (d, 2H), 8.38 (d, 2H).
Beispiel 59A
2-(4-Chlθφhenyl)-4-(cyclopentyloxy)-6-(trifluormethyl)pyrimidin-5-carbonsäuremethylester

90 mg (0.271 mmol) Beispiel 57A, 26 μl (0.284 mmol) Cyclopentanol und 74.5 mg (0.284 mmol) Triphenylphosphin wurden in 5 ml THF aufgenommen und 20 Minuten bei Raumtemperatur gerührt. Anschließend wurden 55 μl (0.459 mmol) Diisopropylazodicarboxylat zugegeben und das
Reaktionsgemisch 2h bei Raumtemperatur umgesetzt. Die flüchtigen Komponenten wurden unter vermindertem Druck destillativ abgetrennt und das Rohprodukt mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90:10). Man erhielt so 75 mg (69% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.37 min; MS (ESIpos): m/z = 401 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm]= 1.59-1.76 (m, 4H), 1.76-1.87 (m, 2H), 1.98-2.11 (m, 2H), 3.90 (s, 3H), 5.74 (mz, IH), 7.67 (d, 2H), 8.39 (d, 2H).
Beispiel 6QA
2-(4-Chlorphenyl)-4-cyclopropyl-6-propoxypyrimidin-5 -carbonsäuremethylester

Eine Mischung von 200 mg (ca. 0.328 mmol) Beispiel 45A, 34 mg (0.459 mmol) n-Propanol und 120 mg (0.459 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur
gerührt. Dann 93 mg (0.459 mmol) Diisopropylazodicarboxylat wurden zugegeben, und das Reaktionsgemisch wurde 2h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 19 mg (17% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.36 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm]= 0.97 (t, 3H), 1.08-1.13 (m, 2H), 1.20-1.24 (m, 2H), 1.71-1.80 (m, 2H), 2.06-2.12 (m, IH), 3.89 (s, 3H), 4.47 (t, 2H), 7.59 (d, 2H), 8.31 (d, 2H).
Beispiel 61A
2-(4-Chloφhenyl)-4-cyclopropyl-6-(2-methylpropoxy)pyrimidin-5-carbonsäuremethylester

Eine Mischung von 200 mg (ca. 0.328 mmol) Beispiel 45A, 34 mg (0.459 mmol) 2-Methyl-l- propanol und 120 mg (0.459 mmol) Triphenylphosphin in 2 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Dann 93 mg (0.459 mmol) Diisopropylazodicarboxylat wurden zugegeben, und das Reaktionsgemisch wurde 2h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 15 mg (13% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.43 min; MS (ESIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm]= 0.97 (dt, 3H), 1.08-1.13 (m, 2H), 1.21-1.24 (m, 2H), 2.01-2.13 (m, 2H), 3.89 (s, 3H), 4.29 (d, 2H), 7.59 (d, 2H), 8.31 (d, 2H).
Beispiel 62A
2-(4-Chlorophenyl)-4-(2-methylpropyl)-6-oxo- 1 ,4,5 ,6-tetrahydropyrimidin-5- carbonsäuremethylester

3.0 g (22.707 mmol) Malonsäuredimethylester, 2.15 g (24.978 mmol) 3-Methylbutanal, 0.22 ml (2.271 mmol) Piperidin und 0.13 ml (2.271 mmol) Essigsäure wurden in 40 ml Dichlormethan gelöst und über Nacht am inversen Wasserabscheider bei Rückflußtemperatur umgesetzt. Die flüchtigen Komponenten wurden am Rotationsverdampfer abgetrennt. Man erhielt so Dimethyl-(3- methylbutyliden)propandioat, welches ohne weitere Reingungsoperationen umgesetzt wurde. Zu einer Mischung von 3.52 g (51.777 mmol) Natriumethylat und 5.44 g (24.477 mmol) 4- Chlorbenzolcarboximidamid-Hydrochlorid in 50 ml Methanol unter Argonatmosphäre wurden 5.18g (ca. 25.888 mmol) des so gewonnenen Rohmaterials zugegeben. Das Reaktionsgemisch wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, mit Wasser versetzt, und die wässrige Phase wurde mit Essigsäureethylester und Dichloromethan extrahiert. Anschließend wurde die organische Phase mit Wasser und gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Man erhielt so 5.02 g (40% d. Th.) Rohprodukt in 66% -iger Reinheit (LC-MS), welches ohne weitere Reinigungsschritte umgesetzt wurde.
LC-MS (Methode 2): R, = 1.83 min; MS (EIpos): m/z = 323[M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.93 (dd, 6H), 1.15-1.23 (dd, IH), 1.46-1.53 (dd, IH), 2.06 (sbr, IH), 3.44 (d, IH), 3.69 (s, 3H), 3.93-4.00 (dd, IH), 7.52 (d, IH), 7.86 (d, IH), 11.15 (sbr, IH).
Beispiel 63A
2-(4-Chloφhenyl)-4-(2-methylpropyl)-6-oxo-l,6-dihydropyrimidin-5-carbonsäuremethylester

Eine Mischung von 5.02 g (ca. 10.268 mmol) Beispiel 62A, 1.83 g (10.268 mmol) N- Bromsuccinimid, 497 mg (2.054 mmol) Dibenzoylperoxid und 2.13 g (15.402 mmol) im Mörser gemahlenes Kaliumcarbonat in 120 ml Dioxan unter Argonatmosphäre wurde 3 h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz mit einer gesättigten wässrigen Natriumthiosulfat-Lösung versetzt und anschließend wurde das Dioxan eingeengt. Die wässrige Phase wurde mit Dichlormethan extrahiert, und die organische Phase wurde mit Wasser und einer gesättigten wässrigen Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Das Rohprodukt wurde mit Acetonitril versetzt und die entstandenen Kristalle wurden abgesaugt und im Hochvakuum bis zur Gewichtskonstanz getrocknet. Man erhielt 1.47 g (40% d. Th.) der Zielverbindung in 88%-iger Reinheit (LC-MS), welches ohne weitere Reinigungsschritte umgesetzt wurde.
LC-MS (Methode 2): R, = 2.02 min; MS (EIpos): m/z = 321[M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.92 (d, 6H), 2.11-2.21 (m, IH), 2.41 (d, 2H), 3.80 (s, 3H), 7.61 (d, 2H), 8.14 (d, 2H), 13.13 (sbr, IH).
Beispiel 64A
2-(4-Chlorphenyl)-4-ethoxy-6-(2-methylpropyl)pyrimidin-5- carbonsäuremethylester

Eine Lösung aus 80 mg (ca. 0.219 mmol) Beispiel 63 A, 14 mg (0.307 mmol) Ethanol und 80 mg (0.307 mmol) Triphenylphosphin in 1.5 ml THF wurde 20 Minuten bei Raumtemperatur gerührt.
Es wurden 62 mg (0.307 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 40 mg (52 % d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.01 min; MS (EIpos): m/z = 349 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.90 (d, 6H), 1.36 (t, 3H), 2.16-2.26 (m, IH), 2.59 (d, 2H), 3.92 (s, 3H), 4.58 (q, 2H) 7.61 (d, 2H), 8.36 (d, 2H).
Beispiel 65A
2-(4-Chlorphenyl)-4-( 1 -methylethoxy)-6-(2-methylpropyl)pyrimidin-5- carbonsäuremethylester

Eine Lösung aus 80 mg (ca. 0.219 mmol) Beispiel 63A, 18 mg (0.307 mmol) 2-Propanol und 80 mg (0.307 mmol) Triphenylphosphin in 1.5 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 62 mg (0.307 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 47 mg (59 % d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.12 min; MS (EIpos): m/z = 363[M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.90 (d, 6H), 1.20 (d, 6H), 2.12-2.26 (m, IH), 2.57 (d, 2H), 3.87 (s, 3H), 5.51-5.57 (m, IH), 7.61 (d, 2H), 8.36 (d, 2H).
Beispiel 66A
2-(4-Chlθφhenyl)-4-(2-methylpropoxy)-6-(2-methylpropyl)pyrimidin-5- carbonsäuremethylester

Eine Lösung aus 80 mg (ca. 0.219 mmol) Beispiel 63A, 22 mg (0.307 mmol) 2-Methylpropanol und 80 mg (0.307 mmol) Triphenylphosphin in 1.5 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 62 mg (0.307 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 47 mg (57 % d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.25 min; MS (EIpos): m/z = 377 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.91 (d, 6H), 0.97 (d, 6H), 1.99-2.10 (m, IH), 2.14- 2.23 (m, IH), 2.60 (d, 2H), 3.88 (s, 3H), 4.31 (d, 2H), 7.61 (d, 2H), 8.38 (d, 2H).
Beispiel 67A
2-(4-Chloφhenyl)-4-(cyclopropylmethoxy)-6-(2-methylpropyl)pyrimidm-5- carbonsäuremethylester
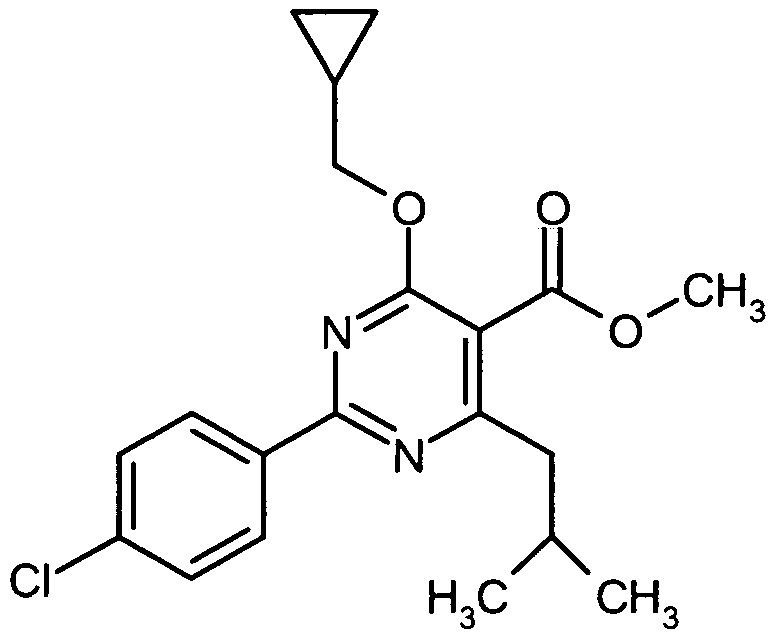
Eine Lösung aus 80 mg (ca. 0.219 mmol) Beispiel 63 A, 22 mg (0.307 mmol) Cyclopropylmethanol und 80 mg (0.307 mmol) Triphenylphosphin in 1.5 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 62 mg (0.307 mmol) Diisopropylazodicarboxylat zugegeben
und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 42 mg (51 % d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 3.10 min; MS (EIpos): m/z = 375 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.37-0.40 (m, 2H), 0.55-0.59 (m, 2H), 0.91 (d, 6H), 1.26-1.31 (m, IH), 2.16-2.26 (m, IH), 2.59 (d, 2H), 3.88 (s, 3H), 4.39 (d, 2H), 7.61 (d, 2H), 8.38 (d, 2H).
Beispiel 68A
2-(4-Chloφhenyl)-4-(2-methoxyethoxy)-6-(2-methylpropyl)pyrimidin-5- carbonsäuremethylester

Eine Lösung aus 80 mg (ca. 0.219 mmol) Beispiel 63 A, 23 mg (0.307 mmol) 2-Methoxyethanol und 80 mg (0.307 mmol) Triphenylphosphin in 1.5 ml THF wurde 20 Minuten bei Raumtemperatur gerührt. Es wurden 62 mg (0.307 mmol) Diisopropylazodicarboxylat zugegeben und dann wurde das Reaktionsgemisch 2.5 h bei Raumtemperatur weitergerührt. Die Mischung wurde einrotiert und ohne weitere Aufarbeitung mittels präparativer HPLC aufgereinigt (Eluent: Acetonitril/Wasser 90:10, isokratisch). Man erhielt so 44 mg (53 % d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.84 min; MS (EIpos): m/z = 379 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.91 (d, 6H), 2.14-2.26 (m, IH), 2.60 (d, 2H), 3.30 (s, 3H), 3.69-3.71 (m, 2H), 3.87 (s, 3H), 4.60-4.67 (2H), 7.61 (d, 2H), 8.38 (d, 2H).
Ausführungsbeispiele:
Beispiel 1
2-(4-Chlorphenyl)-4-ethyl-6-(2-methylpropyl)pyrimidin-5-carbonsäure
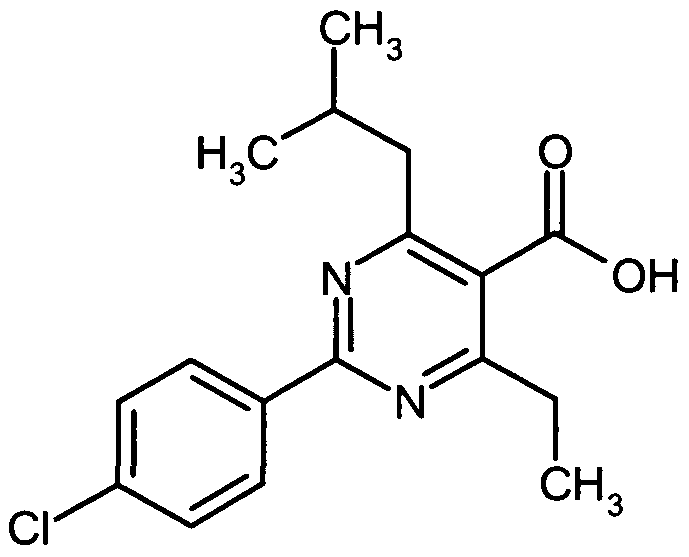
100 mg (0.300 mmol) Beipiel 2A wurden in 5 ml Ethanol aufgenommen und mit 1.50 ml (3.00 mmol) einer 2M wässrigen Kaliumhydroxid-Lösung versetzt. Es wurde Ih bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz eingeengt, in Wasser aufgenommen und mit IN Salzsäure angesäuert. Anschließend wurde mit Essigsäureethylester extrahiert. Nach dem Trocknen mit Magnesiumsulfat wurde das Lösemittel unter vermindertem Druck destillativ abgetrennt und das Rohmaterial durch präparative HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — > 90:10) aufgereinigt. Man erhielt so 73 mg (76% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.55 min; MS (ESIpos): m/z = 319 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.93 (d, 6H), 1.30 (t, 3H), 2.26 (sept, IH), 2.70 (d, 2H), 2.83 (q, 2H), 7.61 (d, 2H), 8.42 (d, 2H), 13.91 (sbr, IH).
Beispiel 2
2-(4-Chlorphenyl)-4-methyl-6-(3-methylbutoxy)pyrimidin-5-carbonsäure

LC-MS (Methode 1): R, = 1.53 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm]= 0.93 (d, 6H), 1.64 (dd, 2H), 1.72-1.80 (m, IH), 2.48 (s. 3H), 4.56 (t, 2H), 7.60 (d, 2H), 7.38 (d, 2H), 13.52 (sbr, IH).
Beispiel 3
2-(4-Chloφhenyl)-4-(3-methoxy-l-methylpropoxy)-6-methylpyrimidin-5-carbonsäure

Zu einer Lösung aus 40 mg (0.105 mmol) Beispiel 6A in 2 ml Ethanol wurden 1.05 ml (2.111 mmol) einer 2M wässrigen Natriumhydroxid-Lösung addiert. Das Reaktionsgemisch wurde über Nacht bei 800C gerührt. Zur Aufarbeitung wurde das Ethanol am Rotationsverdampfer abgetrennt und das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Hochvakuum getrocknet. Man erhielt 25 mg (67% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 2.68 min; MS (ESIpos): m/z = 351 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.38 (d, 3H), 1.80-1.91 (m, 2H), 2.48 (s, 3H), 3.19 (s, 3H), 3.43 (t, 2H), 5.49-5.54 (m, IH), 7.60 (d, 2H), 8.36 (d, 2H), 13.48 (sbr, IH).
Beispiel 4
2-(4-Chlorphenyl)-4-methyl-6-(l-methylethoxy)pyrimidin-5-carbonsäure

Zu einer Lösung aus 42 mg (0.125 mmol) Beispiel 7A in 2 ml Ethanol wurden 1.25 ml (2.509 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde über Nacht bei 800C gerührt. Zur Aufarbeitung wurde das Ethanol am Rotationsverdampfer abgetrennt und das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Hochvakuum getrocknet. Man erhielt 20 mg (52% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.34 min; MS (ESIpos): m/z = 307 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.36 (d, 6H), 2.47 (s, 3H), 5.50-5.57 (m, IH), 7.60 (d, 2H), 8.36 (d, 2H), 13.46 (sbr, IH).
Beispiel 5
2-(4-Chloφhenyl)-4-methyl-6-{[3-(trifluormethyl)cyclohexyl]oxy}pyrimidin-5-carbonsäure

Zu einer Lösung aus 14 mg (0.031 mmol) Beispiel 8A in 2 ml Ethanol wurden 0.316 ml (0.632 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 4h bei 800C gerührt. Zur Aufarbeitung wurde das Ethanol am Rotationsverdampfer abgetrennt und das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Hochvakuum getrocknet. Man erhielt 9 mg (69% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.56 min; MS (ESIpos): m/z = 415 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.37-2.18 (m, 9H), 5.74 (m, IH), 7.60 (d, 2H), 8.36 (d, 2H), 13.55 (sbr, IH).
Beispiel 6
2-(4-Chlorphenyl)-4-methoxy-6-(l-methylethyl)pyrimidin-5-carbonsäure

Zu einer Lösung aus 26 mg (0.077 mmol) Beispiel I IA in 2 ml Ethanol wurden 0.78 ml (1.553 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde über Nacht bei 800C gerührt. Zur Aufarbeitung wurde das Ethanol am Rotationsverdampfer abgetrennt und das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Hochvakuum getrocknet. Man erhielt 20 mg (84% d. Th.) der Zielverbindung.
LC-MS (Methode 4): R, = 2.66 min; MS (ESIpos): m/z = 307 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.26 (d, 6H), 3.09-3.16 (m, IH), 4.07 (s, 3H), 7.62 (d, 2H), 8.42 (d, 2H), 13.62 (sbr, IH).
Beispiel 7
2-(4-Chloφhenyl)-4-(cyclopropylmethoxy)-6-methylpyrimidin-5-carbonsäure
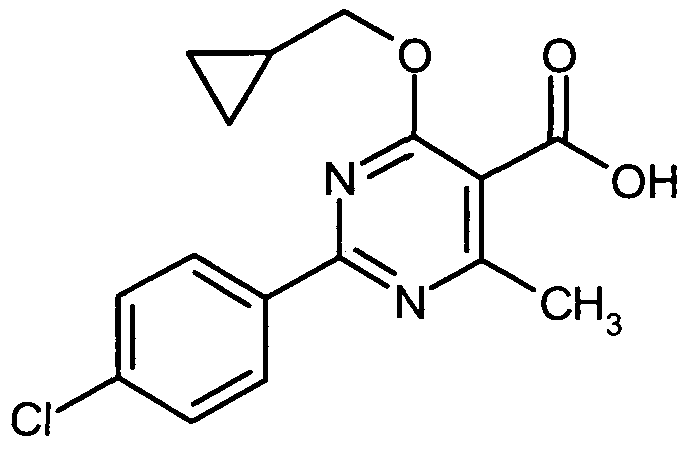
LC-MS (Methode 2): R, = 2.23 min; MS (ESIpos): m/z = 319 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.37-0.41 (m, 2H), 0.55-0.59 (m, 2H), 1.22-1.31 (m, IH), 4.38 (d, 2H), 7.60 (d, 2H), 8.35 (d, 2H), 13.52 (sbr, IH).
Beispiel 8
2-(4-Chlorphenyl)-4-ethyl-6-( 1 -methylethyl)pyrimidin-5-carbonsäure

390 mg (1.22 mmol) Beipiel 14A wurden in 10 ml Dioxan aufgenommen und mit 343 mg (6.17 mmol) Kaliumhydroxid versetzt. Der Ansatz wurde 2h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde das Reaktionsgemisch am Rotationsverdampfer eingeengt. Anschließend wurde der Rückstand in Wasser aufgenommen, mit IN Salzsäure angesäuert und mit Essigsäureethylester extrahiert (2x). Dann wurden die vereinigten organischen Phasen mit Magnesiumsulfat getrocknet und das Lösemittel am Rotationsverdampfer abgetrennt. Das Rohprodukt wurde abschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90: 10) aufgereinigt. Man erhielt so 256 mg (69% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.08 min; MS (ESIpos): m/z = 305 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.26-1.34 (m, 9H), 2.81 (q, 2H), 3.18 (sept, IH), 7.62 (d, 2H), 8.44 (d, 2H), 13.95 (sbr, IH).
Beispiel 9
2-(4-Chlorphenyl)-4-ethyl-6-( 1 -methylbutyl)pyrimidin-5-carbonsäure

690 mg (1.99 mmol) Beipiel 16A wurden in 25 ml Dioxan aufgenommen und mit 558 mg (9.95 mmol) Kaliumhydroxid versetzt. Der Ansatz wurde 2h bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde das Reaktionsgemisch am Rotationsverdampfer eingeengt. Anschließend wurde der Rückstand in Wasser aufgenommen, mit IN Salzsäure angesäuert und mit
Essigsäureethylester extrahiert (2x). Dann wurden die vereinigten organischen Phasen mit
Magnesiumsulfat getrocknet und das Lösemittel am Rotationsverdampfer abgetrennt. Das Rohprodukt wurde abschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient
10:90 → 90:10) aufgereinigt. Man erhielt so 382 mg (58% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.23 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.83 (t, 3H), 1.13 (mz, IH), 1.24 (mz, IH), 1.26 (d, 3H), 1.30 (t, 3H), 1.57 (mz, IH), 1.86 (mz, IH), 2.82 (q, 2H), 3.03 (mz, IH), 7.62 (d, 2H), 8.43 (d, 2H), 13.92 (sbr, IH).
Beispiel 10
2-(4-Chlθφhenyl)-4-ethoxy-6-methylpyrimidin-5-carbonsäure
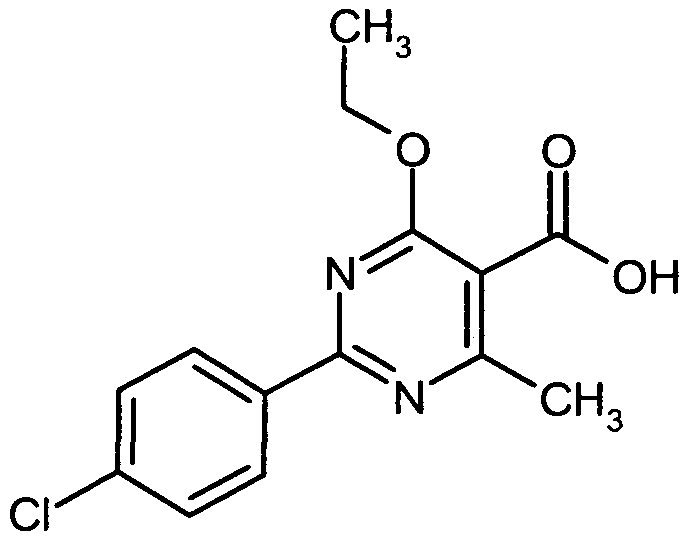
LC-MS (Methode 3): R, = 2.53 min; MS (ESIpos): m/z = 293 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] =1.37 (t, 3H), 2.48 (s, 3H), 4.57 (q, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 13.53 (sbr, IH).
Beispiel 11
2-(4-Chloφhenyl)-4-methyl-6-propoxypyrimidin-5-carbonsäure

Zu einer Lösung aus 50 mg (0.049 mmol) Beispiel 18A in 2 ml Ethanol wurden 1.49 ml (2.987 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im
Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 42 mg (90% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 2.69 min; MS (ESIpos): m/z = 307 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] =0.98 (t, 3H), 1.72-1.80 (m,2H), 2.48 (s, 3H), 4.48 (t, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 13.51 (sbr, IH).
Beispiel 12
2-(4-Chloφhenyl)-4-methyl-6-(2-methylpropoxy)pyrimidin-5-carbonsäure

Zu einer Lösung aus 76 mg (0.218 mmol) Beispiel 19A in 3 ml Ethanol wurden 2.18 ml (4.357 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im
Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 36 mg (52% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 2.82 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSO-df,): δ [ppm] =0.98 (d, 6H), 2.01-2.11 (m, IH), 2.48 (s, 3H), 4.30 (d, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 13.52 (sbr, IH).
Beispiel 13
2-(4-Chlorphenyl)-4-methyl-6-[( 1 -methylcyclopropyl)methoxy]pyrimidin-5-carbonsäure

Zu einer Lösung aus 51 mg (0.141 mmol) Beispiel 2OA in 2 ml Ethanol wurden 1.40 ml (2.810 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch
wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 40 mg (86% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 2.84 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] =0.39 (dd, 2H), 0.59 (dd, 2H), 1.15 (s, 3H), 2.48 (s, 2H), 4.35 (s, 2H), 7.59 (d, 2H), 8.36 (d, 2H), 13.52 (sbr, IH).
Beispiel 14
2-(4-Chloφhenyl)-4-(cyclopentyloxy)-6-methylpyrimidin-5-carbonsäure

Zu einer Lösung aus 36 mg (0.100 mmol) Beispiel 21A in 1.5 ml Ethanol wurden 1.00 ml (2.006 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 80°C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 17 mg (52% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.41 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] =1.62-1.77 (m, 6H), 2.00-2.09 (m, 2H), 2.48 (s, 3H), 5.62-5.65 (m, IH), 7.60 (d, 2H), 8.36 (d, 2H), 13.45 (sbr, IH).
Beispiel 15
2-(4-Chloφhenyl)-4-(cyclopentylmethoxy)-6-methylpyrimidin-5-carbonsäure

Zu einer Lösung aus 50 mg (0.134 mmol) Beispiel 22A in 2 ml Ethanol wurden 1.34 ml (2.673 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 22 mg (48% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.57 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSOd5): δ [ppm] =1.32-1.79 (m, 8H), 2.30-2.37 (m, IH), 2.48 (s, 3H), 4.41 (d, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 13.50 (sbr, IH).
Beispiel 16
2-(4-Chlorphenyl)-4-ethoxy-6-propylpyrimidin-5-carbonsäure

Zu einer Lösung aus 33 mg (0.095 mmol) Beispiel 26A in 1.5 ml Ethanol wurden 0.95 ml (1.903 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 8O0C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2
gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 29 mg (93% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.47 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] =0.93 (t, 3H), 1.37 (t, 3H), 1.71-1.80 (m, 2H), 2.71 (t, 2H), 4.57 (q, 2H), 7.61 (d, 2H), 8.38 (d, 2H), 13.53 (sbr, IH).
Beispiel 17
2-(4-Chloφhenyl)-4-propoxy-6-propylpyrimidin-5-carbonsäure
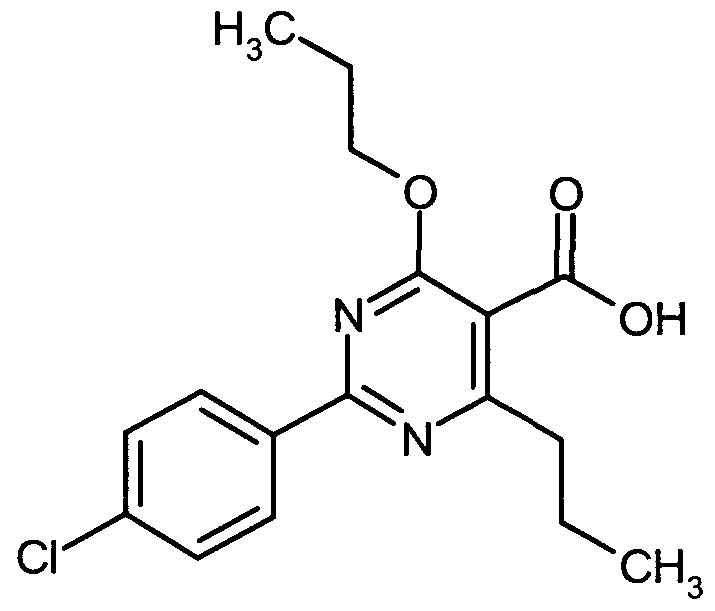
Zu einer Lösung aus 33 mg (0.095 mmol) Beispiel 27A in 2 ml Ethanol wurden 1.23 ml (2.469 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 34 mg (81% d. Th.) der Zielverbindung.
LC-MS (Methode 1): Rt = 1.55 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] =0.93 (t, 3H), 0.98 (t, 3H), 1.72-1.81 (m, 4H), 2.72 (t, 2H), 4.48(t, 2H), 7.60 (d, 2H), 8.38 (d, 2H), 13.52 (sbr, IH).
Beispiel 18
2-(4-Chloφhenyl)-4-(2-methylpropoxy)-6-propylpyrimidin-5-carbonsäure

Zu einer Lösung aus 40 mg (0.106 mmol) Beispiel 28 A in 2 ml Ethanol wurden 1.06 ml (2.123 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 32 mg (86% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.62 min; MS (ESIpos): m/z = 349 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] =0.94 (t, 3H), 0.98 (d, 6H), 1.74-1.79 (m, 2H), 2.01-2.12 (m, IH), 2.72 (t, 2H), 4.30 (d, IH), 7.60 (d, 2H), 8.38 (d, 2H), 13.54 (sbr, IH).
Beispiel 19
2-(4-Chloφhenyl)-4-[(l-methylcyclopropyl)methoxy]-6-propylpyrimidin-5-carbonsäure

Zu einer Lösung aus 41 mg (0.105 mmol) Beispiel 29A in 2 ml Ethanol wurden 1.05 ml (2.093 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im
Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 31 mg (82% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.62 min; MS (ESIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] =0.39 (dd, 2H), 0.59 (dd, 2H), 0.94 (t, 3H), 1.16 (s, 3H), 1.72-1.80 (m, 2H), 2.74 (t, 2H), 4.30 (s, IH), 7.59 (d, 2H), 8.37 (d, 2H), 13.55 (sbr, IH).
Beispiel 20
2-(4-Chloφhenyl)-4-(cyclopentyloxy)-6-propylpyrimidin-5-carbonsäure

Zu einer Lösung aus 68 mg (0.175 mmol) Beispiel 30A in 3 ml Ethanol wurden 1.75 ml (3.497 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 80°C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im
Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 58 mg (91% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.63 min; MS (ESIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] =0.93 (t, 3H), 1.60-1.80 (m, 8H), 2.01-2.10 (m, 2H), 2.70 (t, 2H), 5.61-5.66 (m, IH), 7.60 (d, 2H), 8.38 (d, 2H), 13.47 (sbr, IH).
Beispiel 21
2-(4-Chlorphenyl)-4-ethoxy-6-( 1 -methylethyl)pyrimidin-5-carbonsäure

Zu einer Lösung aus 68 mg (0.175 mmol) Beispiel 31A in 2 ml Ethanol wurden 1.15 ml (2.293 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 6h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt und die flüchtigen Komponenten im Vakuum abdestilliert. Die entstandenen Kristalle wurden abgesaugt und im Hochvakuum getrocknet. Man erhielt 23 mg (97% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R4 = 1.51 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] =1.27 (d, 6H), 1.37 (t, 3H), 3.08-3.15 (m, IH), 4.57 (q, 2H), 7.61 (d, 2H), 8.38(d, 2H), 13.56 (sbr, IH).
Beispiel 22
2-(4-Chlorphenyl)-4-ethyl-6-( 1 -ethylpropyl)pyrimidin-5-carbonsäure

245 mg (0.76 mmol) Beipiel 33 A wurden in 10 ml Pyridin aufgenommen und mit 473 mg (3.53 mmol) Lithiumiodid versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — > 90: 10) aufgereinigt. Man erhielt so 171 mg (73% d. Th.) der Zielverbindung.
LC-MS (Methode 2): Rt = 2.76 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSO-(I6): δ [ppm] = 0.74 (t, 6H), 1.31 (t, 3H), 1.69 (mz, 2H), 1.75-1.89 (m, 2H), 2.749 (mz, IH), 2.83 (q, 2H), 7.61 (d, 2H), 8.42 (d, 2H), 13.86 (sbr, IH).
Beispiel 23
2-(4-Chloφhenyl)-4-ethyl-6-(tetrahydrofuran-3-yl)pyrimidin-5-carbonsäure

270 mg (0.78 mmol) Beipiel 35A wurden in 14 ml Pyridin aufgenommen und mit 521 mg (3.89 mmol) Lithiumiodid versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — » 90: 10) aufgereinigt. Man erhielt so 121 mg (47% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.29 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.30 (t, 3H), 2.19-2.37 (m, 2H), 2.84 (q, 2H), 3.66 (quint, IH), 3.80-3.91 (m, 2H), 4.01 (mz, IH), 4.08 (t, IH), 7.62 (d, 2H), 8.42 (d, 2H), 14.08 (sbr, IH).
Beispiel 24
2-(4-Chlorphenyl)-4-cyclopentyl-6-ethylpyrimidin-5-carbonsäure

LC-MS (Methode 3): R, = 3.12 min; MS (ESIpos): m/z = 331 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.30 (t, 3H), 1.63-1.72 (m, 2H), 1.80-2.03 (m, 6H), 2.81 (q, 2H), 7.61 (d, 2H), 8.41 (d, 2H), 13.86 (sbr, IH).
Beispiel 25
2-(4-Chlorphenyl)-4-( 1 -methylethyl)-6-propoxypyrimidin-5-carbonsäure

Zu einer Lösung aus 60 mg (0.159 mmol) Beispiel 38A in 2.8 ml Ethanol wurden 1.59 ml (3.189 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 6h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt und die flüchtigen Komponenten im Vakuum abdestilliert. Die entstandenen Kristalle wurden abgesaugt, im Hochvakuum getrocknet und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 50:50) aufgereinigt. Man erhielt so 15 mg (28% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.58 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.97 (t, 3H), 1.20 (d, 6H), 1.70-1.77 (m, 2H), 4.34 (t, IH), 7.54 (d, 2H), 8.34 (d, 2H).
Beispiel 26
4-Butyl-2-(4-chlθφhenyl)-6-methylpyrimidin-5-carbonsäure

Zu einer Lösung aus 20 mg (0.060 mmol) Beispiel 39A in 2 ml Ethanol wurden 0.60 ml (1.202 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 10h bei 800C gerührt und über das Wochenende bei Raumtemperatur stehen gelassen. Zur Aufarbeitung wurde das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die flüchtigen Komponenten wurden dann im Vakuum abdestilliert. Die entstandenen Kristalle wurden abgesaugt und im Hochvakuum getrocknet. Man erhielt so 12 mg (64% d. Th.) der Zielverbindung
LC-MS (Methode 3): Rt = 2.96 min; MS (ESIpos): m/z = 305 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.91 (t, 3H), 1.36 (q, 2H), 1.69-1.77 (m, 2H), 7.60 (d, 2H), 8.40 (d, 2H), 13.89 (sbr, IH).
Beispiel 27
2-(4-Chlθφhenyl)-4-methyl-6-(l-methylpropyl)pyrimidin-5-carbonsäure

Zu einer Lösung aus 9 mg (0.026 mmol) Beispiel 4OA in 1 ml Ethanol wurden 0.26 ml (0.523 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 10h bei 8O0C gerührt und über das Wochenende bei Raumtemperatur stehen gelassen. Zur Aufarbeitung wurde das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die flüchtigen Komponenten wurden dann im Vakuum abdestilliert. Die entstandenen Kristalle wurden abgesaugt und im Hochvakuum getrocknet. Man erhielt so 5 mg (59% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 2.98 min; MS (ESIpos): m/z = 305 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.78 (t, 3H), 1.26 (d, 3H), 1.58-1.67 (m, IH), 1.83-1.90 (m, IH), 2.91-2.97 (m, IH), 7.60 (d, 2H), 8.40 (d, 2H), 13.45 (sbr, IH).
Beispiel 28
2-(4-Chlorphenyl)-4-(cyclopentyloxy)-6-( 1 -methylethyl)pyrimidin-5-carbonsäure

Zu einer Lösung aus 50 mg (0.129 mmol) Beispiel 41A in 2.4 ml Ethanol wurden 1.28 ml (2.571 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 6h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die flüchtigen Komponenten wurden dann im Vakuum abdestilliert. Die entstandenen Kristalle wurden abgesaugt und im Hochvakuum getrocknet. Man erhielt so 29 mg (60% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.77 min; MS (ESIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.25 (d, 6H), 1.60-1.76 (6H), 2.01-2.03 (2H), 6H), 3.07-3.13 (m, IH), 5.62-5.64 (m, IH), 7.60 (d, 2H), 8.40 (d, 2H), 13.45 (sbr, IH).
Beispiel 29
2-(4-Chlorphenyl)-4-(cyclopropylmethoxy)-6-( 1 -methylethyl)pyrimidin-5 -carbonsäure

Zu einer Lösung aus 25 mg (0.067 mmol) Beispiel 42A in 1.2 ml Ethanol wurden 0.67 ml (1.334 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 6h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die flüchtigen Komponenten wurden dann im Vakuum abdestilliert. Die entstandenen Kristalle wurden abgesaugt und im Hochvakuum getrocknet. Man erhielt so 23 mg (94% d. Th.) der Zielverbindung.
LC-MS (Methode 2): Rt = 2.63 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSO-dβ): δ [ppm] = 0.38 (dd, 2H), 0.57 (dd, 2H), 1.24-1.32 (m, 7H), 3.09- 3.15 (m, IH), 4.37 (d, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 15.53 (sbr, IH).
Beispiel 30
2-(4-Chlorphenyl)-4-( 1 -methylethoxy)-6-( 1 -methylethyl)pyrimidin-5-carbonsäure

Zu einer Lösung aus 40 mg (0.110 mmol) Beispiel 43A in 2.2 ml Ethanol wurden 1.1 ml (2.205 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 6h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch mit IN Salzsäure auf pH 1 gestellt. Die flüchtigen Komponenten wurden dann im Vakuum abdestilliert. Die entstandenen Kristalle wurden abgesaugt und im Hochvakuum getrocknet. Man erhielt so 15 mg (38% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.05 min; MS (ESIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.26 (d, 6H), 1.36 (d, 6H), 3.06-3.12 (m, IH), 5.49- 5.56 (m, IH), 7.61 (d, 2H), 8.38 (d, 2H), 13.51 (sbr, IH).
Beispiel 31
2-(4-Chlorphenyl)-4-cyclopropyl-6-( 1 -methylethoxy)pyrimidin-5 -carbonsäure

Zu einer Lösung aus 10 mg (0.029 mmol) Beispiel 46A in 0.6 ml Ethanol wurden 0.29 ml (0.582mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 7.3 mg (75% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.44 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm]= 1.08-1.11 (2H), 1.18-1.21 (2H), 1.35 (d, 6H), 2.08-2.14 (m, IH), 5.48-5.55 (m, IH), 7.58 (d, 2H), 8.29 (d, 2H), 13.49 (sbr, IH).
Beispiel 32
2-(4-Chloφhenyl)-4-cyclopropyl-6-methoxypyrimidin-5-carbonsäure

LC-MS (Methode 1): R1 = 1.39 min; MS (ESIpos): m/z = 305 [M+H]+.
1H-NMR (400 MHz, DMSO-Cl6): δ [ppm]= 1.08-1.13 (m, 2H), 1.17-1.23 (m, 2H), 2.13-2.18 (m, IH), 4.06 (s, 3H), 7.58 (d, 2H), 8.33 (d, 2H), 13.62 (sbr, IH).
Beispiel 33
2-(4-Chlorphenyl)-4-cyclopropyl-6-ethoxypyrimidin-5-carbonsäure

Zu einer Lösung aus 10 mg (0.030 mmol) Beispiel 48A in 0.64 ml Ethanol wurden 0.30 ml (0.595 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurden die flüchtigen Komponenten im Vakuum abdestilliert. Der Rückstand wurde mit Wasser aufgenommen und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im
Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 8.8 mg (93% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.46 min; MS (ESIpos): m/z = 319 [M+H]+.
1H-NMR (400 MHz, DMSO-(I6): δ [ppm]= 1.08-1.13 (m, 2H), 1.18-1.26 (m, 2H), 1.36 (t, 3H), 2.13-2.18 (m, IH), 4.55 (q, 2H), 7.58 (d, 2H), 8.31 (d, 2H).
Beispiel 34
4-Ethyl-6-(l-methylethyl)-2-(3-methylphenyl)pyrimidin-5-carbonsäure

184 mg (0.617 mmol) Beipiel 51A wurden in 5 ml Pyridin aufgenommen und mit 413 mg (3.08 mmol) Lithiumiodid versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — »• 90:10) aufgereinigt. Nach der Trocknung am Hochvakuum erhielt man 59 mg (34% d. Th.) der Zielverbindung.
LC-MS (Methode 1): Rt = 1.42 min; MS (ESIpos): m/z = 285 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 1.25-1.35 (m, 9H), 2.42 (s, 3H), 2.81 (q, 2H), 3.17 (sept, IH), 7.36 (d, IH), 7.43 (t, IH), 8.21-8.27 (m, 2H), 13.88 (sbr, IH).
Beispiel 35
4-Ethyl-6-(l-methylethyl)-2-(4-methylphenyl)pyrimidin-5-carbonsäure

111 mg (0.372 mmol) Beipiel 53A wurden in 5 ml Pyridin aufgenommen und mit 249 mg (1.86 mmol) Lithiumiodid versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt. Es wurde in Wasser aufgenommen, mit IN Salzsäure sauer eingestellt und dann mit Essigsäureethylester extrahiert. Die organische Phase wurde mit Wasser und mit gesättigter wässriger Natriumchlorid-Lösung gewaschen. Anschließend wurde mit Magnesiumsulfat getrocknet, das Lösemittel abgetrennt und
dann mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 → 90: 10) aufgereinigt. Nach der Trocknung am Hochvakuum erhielt man 81 mg (77% d. Th.) der Zielverbindung.
LC-MS (Methode 1): Rt = 1.43 min; MS (ESIpos): m/z = 285 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.26-1.32 (m, 9H), 2.39 (s, 3H), 2.80 (q, 2H), 3.16 (sept, IH), 7.35 (d, 2H), 8.33 (d, 2H), 13.84 (sbr, IH).
Beispiel 36
2-(4-Chloφhenyl)-4-(methoxymethyl)-6-(l-methylethyl)pyrimidin-5-carbonsäure

47 mg (0.140 mmol) Beipiel 55A wurden in 5 ml Pyridin aufgenommen und mit 93 mg (0.70 mmol) Lithiumiodid versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: AcetonitrilAVasser, Gradient 10:90 — > 90:10) aufgereinigt. Nach der Trocknung am Hochvakuum erhielt man 21 mg (47% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.38 min; MS (ESIpos): m/z = 321 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.30 (d, 6H), 3.28 (sept, IH), 3.32 (s, 3H), 4.63 (s, 2H), 7.63 (d, 2H), 8.43 (d, 2H).
Beispiel 37
2-(4-Chlθφhenyl)-4-(l-methylethoxy)-6-(trifluormethyl)pyrimidin-5-carbonsäure

65 mg (0.173 mmol) Beipiel 58A wurden in 4 ml Pyridin aufgenommen und mit 116 mg (0.867 mmol) Lithiumiodid versetzt. Der Ansatz wurde über Nacht bei Rückfiußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 — > 90:10) aufgereinigt. Nach der Trocknung am Hochvakuum erhielt man 46 mg (74% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.12 min; MS (ESIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSO-O6): δ [ρpm]= 1.40 (d, 6H), 5.63 (sept, IH), 7.66 (d, 2H), 8.36 (d, 2H), 14.21 (sbr, IH).
Beispiel 38
2-(4-Chloφhenyl)-4-(cyclopentyloxy)-6-(trifluormethyl)pyrimidin-5-carbonsäure

90 mg (0.173 mmol) Beipiel 59A wurden in 5 ml Pyridin aufgenommen und mit 124 mg (0.923 mmol) Lithiumiodid versetzt. Der Ansatz wurde über Nacht bei Rückflußtemperatur gerührt. Nach dem Abkühlen wurde der Ansatz am Rotationsverdampfer eingeengt und anschließend mittels präparativer HPLC (Eluent: Acetonitril/Wasser, Gradient 10:90 -» 90:10) aufgereinigt. Nach der Trocknung am Hochvakuum erhielt man 47 mg (66% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.30 min; MS (ESIpos): m/z = 387 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm]= 1.60-1.76 (m, 4H), 1.77-1.86 (m, 2H), 2.01-2.12 (m, 2H), 5.74 (sept, IH), 7.66 (d, 2H), 8.38 (d, 2H), 14.17 (sbr, IH).
Beispiel 39
2-(4-Chlorophenyl)-4-cyclopropyl-6-propoxypyrimidin-5-carbonsäure

Zu einer Lösung von 19.2 mg (0.055 mmol) Beispiel 6OA in 1.1 ml Ethanol wurden 0.55 ml (1.107 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch im Vakuum abdestilliert und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 15 mg (80% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R1 = 1.54 min; MS (ESIpos): m/z = 333 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm]= 0.98 (t, 3H), 1.09-1.11 (m, 2H), 1.19-1.21 (m, 2H), 1.72-1.81 (m, 2H), 2.11-2.17 (m, IH), 4.46 (t, 2H), 7.58 (d, 2H), 8.31 (d, 2H), 13.55 (sbr, IH).
Beispiel 40
2-(4-Chlorophenyl)-4-cyclopropyl-6-(2-methylpropoxy)pyrimidin-5-carbonsäure

Zu einer Lösung von 15.4 mg (0.043 mmol) Beispiel 61A in 0.86 ml Ethanol wurden 0.42 ml (0.854 mmol) einer 2M wässrigen Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch im Vakuum abdestilliert und mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Trockenschrank über Nacht bei 400C getrocknet. Man erhielt 11.5 mg (77% d. Th.) der Zielverbindung.
LC-MS (Methode 1): R, = 1.61 min; MS (ESIpos): m/z = 347 [M+H]+.
1H-NMR (400 MHz, DMSO-Cl6): δ [ppm]= 0.98 (d, 6H), 1.09-1.14 (m, 2H), 1.16-1.22 (m, 2H), 2.01-2.09 (m, IH), 2.18 (m, IH), 4.28 (d, 2H), 7.58 (d, 2H), 8.31 (d, 2H), 13.56 (sbr, IH).
Beispiel 41
2-(4-Chlθφhenyl)-4-ethoxy-6-(2-methylpropyl)pyrimidin-5- carbonsäure

Zu einer Lösung von 38 mg (0.109 mmol) Beispiel 64A in 2 ml Methanol wurden 1.09 ml (2.173 mmol) 2M wässriger Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurde das Reaktionsgemisch im Vakuum abdestilliert, mit IN Salzsäure auf pH 2 gestellt und mit Dichloromethan extrahiert. Die organische Phase wurde über Natriumsulfat getrocknet und eingeengt. Man erhielt so 29 mg (97% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.54 min; MS (EIpos): m/z = 335 [M+H]+.
1H-NMR (400 MHz, DMSO-Cl6): δ [ppm] = 0.91 (d, 6H), 1.37 (t, 3H), 2.18-2.30 (m, IH), 2.62 (d, 2H), 4.57 (q, 2H), 7.60 (d, 2H), 8.38 (d, 2H), 13.51 (sbr, IH).
Beispiel 42
2-(4-Chlθφhenyl)-4-(l-methylethoxy)-6-(2-methylpropyl)pyrimidin-5- carbonsäure

Zu einer Lösung aus 45 mg (0.125 mmol) Beispiel 65 A in 2.3 ml Methanol wurden 1.25 ml (2.491 mmol) 2M wässriger Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurde das Methanol am Rotationsverdampfer abgetrennt und das Reaktionsgemisch mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Hochvakuum über Nacht getrocknet. Man erhielt 38 mg (87% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.65 min; MS (EIpos): m/z = 349 [M+H]+.
1H-NMR (400 MHz, DMSO-(I6): δ [ppm] = 0.91 (d, 6H), 1.37 (d, 6H), 2.18-2.32 (m, IH), 2.61 (d, 2H), 5.48-5.58 (m,lH), 7.60 (d, 2H), 8.36 (d, 2H), 13.45 (sbr, IH).
Beispiel 43
2-(4-Chlθφhenyl)-4-(2-methylpropoxy)-6-(2-methylpropyl)pyrimidin-5- carbonsäure

Zu einer Lösung aus 43 mg (0.114 mmol) Beispiel 66A in 2 ml Methanol wurden 1.14 ml (2.277 mmol) 2M wässriger Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurde das Methanol am Rotationsverdampfer abgetrennt und das Reaktionsgemisch mit IN Salzsäure auf pH 2 gestellt. Die entstandenen Kristalle wurden abgesaugt, mit Wasser gewaschen und im Hochvakuum über Nacht getrocknet. Man erhielt 20 mg (98% d. Th.) der Zielverbindung.
LC-MS (Methode 3): R, = 3.24 min; MS (EIpos): m/z = 363 [M+H]+.
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.91 (d, 6H), 0.98 (d, 6H), 2.01-2.11 (m, IH), 2.19- 2.29 (m, IH), 2.63 (d, 2H), 4.30 (d, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 13.51 (sbr, IH).
Beispiel 44
2-(4-Chlorphenyl)-4-(cyclopropylmethoxy)-6-(2-methylpropyl)pyrimidin-5- carbonsäure
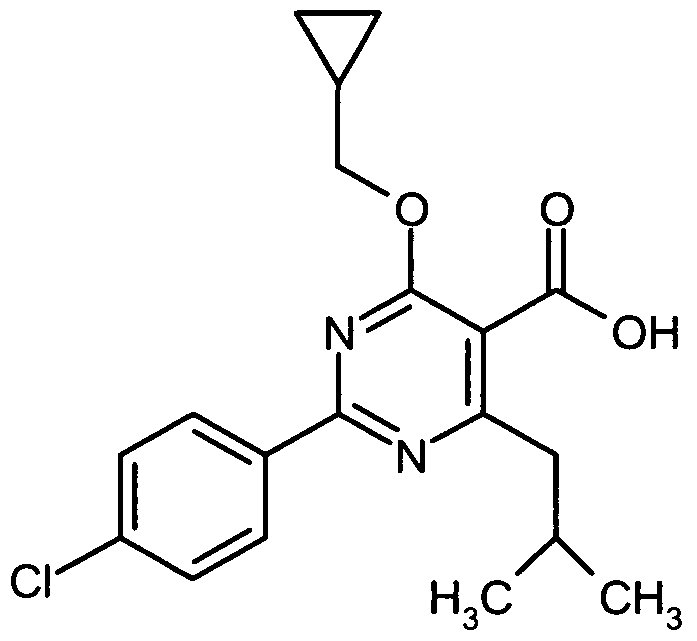
Zu einer Lösung aus 40 mg (0.106 mmol) Beispiel 67A in 2 ml Methanol wurden 1.06 ml (2.123 mmol) 2M wässriger Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurde das Methanol am Rotationsverdampfer abgetrennt, das Reaktionsgemisch mit IN Salzsäure auf pH 2 gestellt und mit Dichloromethan extrahiert. Die
organische Phase wurde über Magnesiumsulfat getrocknet und eingeengt. Man erhielt so 29 mg (73% d. Th.) der Zielverbindung.
LC-MS (Methode 2): R, = 2.67 min; MS (EIpos): m/z = 361 [M+H]+.
1H-NMR (400 MHz, DMSO-Cl6): δ [ppm] = 0.37-0.41 (2H), 0.55-0.60 (2H), 0.92 (d, 6H), 1.23-1.35 (m, IH), 2.19-2.29 (m, IH), 2.63 (d, 2H), 4.37 (d, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 13.52 (sbr, IH).
Beispiel 45
2-(4-Chloφhenyl)-4-(2-methoxyethoxy)-6-(2-methylpropyl)pyrimidin-5- carbonsäure

Zu einer Lösung aus 42 mg (0.111 mmol) Beispiel 68A in 2 ml Methanol wurden 1.11 ml (2.212 mmol) 2M wässriger Natriumhydroxid-Lösung gegeben. Das Reaktionsgemisch wurde 3h bei 800C gerührt. Zur Aufarbeitung wurde das Methanol am Rotationsverdampfer abgetrennt, das Reaktionsgemisch mit IN Salzsäure auf pH 2 gestellt und mit Dichloromethan extrahiert. Die organische Phase wurde über Magnesiumsulfat getrocknet und eingeengt. Man erhielt so 34 mg (85% d. Th.) der Zielverbindung.
LC-MS (Methode 4) R, = 2.65 min; MS (EIpos): m/z = 365 [M+H]+.
1H-NMR (400 MHz, DMSOd6): δ [ppm] = 0.91 (d, 6H), 2.20-2.26 (m, IH), 2.63 (d, 2H), 3.31 (s, 3H), 3.71 (dd, 2H), 4.66 (dd, 2H), 7.60 (d, 2H), 8.37 (d, 2H), 13.55 (sbr, IH).
B. Bewertung der pharmakologischen Wirksamkeit
Die pharmakologische Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
B-I: Zellulärer Transaktivierungs-Assay:
a) Testprinzip:
Ein zellulärer Assay wird eingesetzt zur Identifizierung von Aktivatoren des Peroxisom- Proliferator-aktivierten Rezeptors alpha (PPAR-alpha).
Da Säugetierzellen verschiedene endogene nukleare Rezeptoren enthalten, die eine eindeutige Interpretation der Ergebnisse komplizieren könnten, wird ein etabliertes Chimärensystem ein- gesetzt, in dem die Liganden-Bindungsdomäne des humanen PPARα-Rezeptors an die DNA- Bindungsdomäne des Hefe-Transkriptionsfaktors GAL4 fusioniert wird. Die so entstehende GAL4-PPARα-Chimäre wird in CHO-Zellen mit einem Reporterkonstrukt co-transfϊziert und stabil exprimiert.
b) Klonierung:
Das GAL4-PPARα-Expressions-Konstrukt enthält die Ligandenbindungsdomäne von PP ARa (Aminosäuren 167-468), welche PCR-amplifiziert wird und in den Vektor pcDNA3.1 hinein- kloniert wird. Dieser Vektor enthält bereits die GAL4-DNA-Bindungsdomäne (Aminosäuren 1- 147) des Vektors pFC2-dbd (Stratagene). Das Reporterkonstrukt, welches fünf Kopien der GAL4- Bindestelle vorgeschaltet vor einem Thymidinkinase-Promoter enthält, führt zur Expression der Firefly-Luciferase (Photinus pyralis) nach Aktivierung und Bindung von GAL4-PPARα.
c) Testablauf:
CHO (chinese hamster ovary)-Zellen, die die oben beschriebene GAL4-PPARα-Chimäre und das Luciferase-Reportergenkonstrukt stabil exprimieren, werden am Tag vor dem Test in Medium (Optimem, GIBCO), 2% Aktivkohle-gereinigtes fötales Kälberserum (Hyclone), 1.35 mM Natriumpyruvat (GIBCO), 0.2% Natriumbicarbonat (GIBCO) mit 1 x 103 Zellen in 96-Loch- Mikrotiterplatten ausplattiert und in einem Zellinkubator (96% Luftfeuchtigkeit, 5% v/v CO2, 37°C) gehalten. Am Testtag werden die zu prüfenden Substanzen in oben genanntem Medium, allerdings ohne Zusatz von Kälberserum, aufgenommen und zu den Zellen hinzugegeben. Nach einer Stimulationszeit von 6 h wird die Luciferaseaktivität mit Hilfe einer Videokamera gemessen. Die gemessenen relativen Lichteinheiten ergeben in Abhängigkeit von der Substanzkonzentration
eine sigmoide Stimulationskurve. Die Berechnung der EC50- Werte erfolgt mit Hilfe des Computerprogramms GraphPad PRISM (Version 3.02).
In der folgenden Tabelle sind die EC50- Werte repräsentativer Beispielverbindungen aufgeführt:
Tabelle

Zur Bestimmung der Wirkung auf die Plasma-Fibrinogen-Konzentration werden männliche Wistar-Ratten oder NMRI-Mäuse für einen Zeitraum von 4-9 Tagen per Schlundsonden-Applika- tion oder über Futterbeimischung mit der zu untersuchenden Substanz behandelt. Anschließend wird in Terminalnarkose Citratblut durch Herzpunktion gewonnen. Die Plasma-Fibrinogen-Spiegel werden nach der Clauss-Methode [A. Clauss, Acta Haematol. 17, 237-46 (1957)] durch Messung der Thrombinzeit mit humanem Fibrinogen als Standard bestimmt.
B-3: Testbeschreibung zur Auffindung von pharmakologisch wirksamen Substanzen, die das Apoprotein Al (ApoAl) und das HDL-Cholesterin (HDL-C) im Serum von transgenen Mäusen, die mit dem humanen ApoAl -Gen (hApoAl) transfiziert sind, erhöhen bzw. die
Serumtriglvzeride (TG) senken:
Die Substanzen, die auf ihre HDL-C erhöhende Wirkung in vivo untersucht werden sollen, werden männlichen transgenen hApoAl -Mäusen oral verabreicht. Die Tiere werden einen Tag vor Versuchsbeginn randomisiert Gruppen mit gleicher Tierzahl, in der Regel n = 7-10, zugeordnet.
Während des gesamten Versuches steht den Tieren Trinkwasser und Futter ad libitum zur Verfügung. Die Substanzen werden einmal täglich 7 Tage lang oral verabreicht. Zu diesem Zweck werden die Testsubstanzen in einer Lösung aus Solutol HS 15 + Ethanol + Kochsalzlösung (0.9%) im Verhältnis 1+1+8 oder in einer Lösung aus Solutol HS 15 + Kochsalzlösung (0.9%) im Verhält- nis 2+8 gelöst. Die Applikation der gelösten Substanzen erfolgt in einem Volumen von 10 ml/kg Körpergewicht mit einer Schlundsonde. Als Kontrollgruppe dienen Tiere, die genauso behandelt werden, aber nur das Lösungsmittel (10 ml/kg Körpergewicht) ohne Testsubstanz erhalten.
Vor der ersten Substanzapplikation wird jeder Maus zur Bestimmung von ApoAl, Serumcholesterin, HDL-C und Serumtriglyzeriden (TG) Blut durch Punktion des retroorbitalen Venen- plexus entnommen (Vorwert). Anschließend wird den Tieren mit einer Schlundsonde die Testsubstanz zum ersten Mal verabreicht. 24 Stunden nach der letzten Substanzapplikation (am 8. Tag nach Behandlungsbeginn) wird jedem Tier zur Bestimmung der gleichen Parameter erneut Blut durch Punktion des retroorbitalen Venenplexus entnommen. Die Blutproben werden zentrifugiert und nach Gewinnung des Serums werden TG, Cholesterin, HDL-C und humanes ApoAl mit einem Cobas Integra 400 plus-Gerät (Cobas Integra, Fa. Roche Diagnostics GmbH, Mannheim) unter Verwendung der jeweiligen Kassetten (TRIGL, CHOL2, HDL-C und APOAT) bestimmt. HDL-C wird durch Gelfiltration und Nachsäulenderivatisierung mit MEGA Cholesterol-Reagens (Fa. Merck KGaA) analog zur Methode von Garber et al. [J. Lipid Res. 4J., 1020-1026 (2000)] bestimmt.
Die Wirkung der Testsubstanzen auf die HDL-C-, hApoAl- bzw. TG-Konzentrationen wird durch Subtraktion des Messwertes der 1. Blutentnahme (Vorwert) von dem Messwert der 2. Blutentnahme (nach Behandlung) bestimmt. Es werden die Differenzen aller HDL-C-, hApoAl - bzw. TG- Werte einer Gruppe gemittelt und mit dem Mittelwert der Differenzen der Kontrollgruppe verglichen. Die statistische Auswertung erfolgt mit Student 's t-Test nach vorheriger Überprüfung der Varianzen auf Homogenität.
Substanzen, die das HDL-C der behandelten Tiere, verglichen mit dem der Kontrollgruppe, statistisch signifikant (p<0.05) um mindestens 20% erhöhen oder die TG statistisch signifikant (p<0.05) um mindestens 25% senken, werden als pharmakologisch wirksam angesehen.
B-4: DOCA/Salz-Modell:
Die Verabreichung von Desoxycorticosteronacetat (DOCA) in Kombination mit einer Hochsalzdiät und einseitiger Nierenentfernung induziert bei der Ratte einen Hypertonus, der durch relativ niedrige Reninspiegel charakterisiert ist. Als Folge dieser endokrinen Hypertonie (DOCA ist eine direkte Vorstufe von Aldosteron) kommt es in Abhängigkeit von der gewählten DOCA-Konzen-
tration zu einer Hypertrophie des Herzens und weiteren Endorgan-Schäden, z.B. der Niere, die u.a. durch Proteinurie und Glomerulosklerose charakterisiert sind. In diesem Rattenmodell lassen sich somit Testsubstanzen auf vorhandene antihypertrophe und Endorgan-schützende Wirkung hin untersuchen.
Etwa 8 Wochen alte (Körpergewicht zwischen 250 und 300 Gramm), männliche Sprague Dawley (SD)-Ratten werden linksseitig uninephrektomiert. Dazu werden die Ratten mit 1.5-2%-igem Iso- fiuran in einer Mischung aus 66% N2O und 33% O2 anästhesiert und die Niere über einen Flankenschnitt entfernt. Als spätere Kontrolltiere dienen sogenannte sham-operierte Tiere, denen keine Niere entfernt wird.
Uninephrektomierte SD-Ratten erhalten 1% Natriumchlorid im Trinkwasser und einmal wöchentlich eine subkutane Injektion von Desoxycorticosteronacetat (gelöst in Sesamöl; Fa. Sigma) zwischen die Schulterblätter gespritzt (Hochdosis: 100 mg/kg/Woche s.c; Normaldosis: 30 mg/kg/Woche s.c).
Die Substanzen, die auf ihre protektive Wirkung in vivo untersucht werden sollen, werden per Gavage oder über das Futter (Fa. Ssniff) oder Trinkwasser verabreicht. Die Tiere werden einen
Tag vor Versuchsbeginn randomisiert und Gruppen mit gleicher Tierzahl, in der Regel n = 10, zugeordnet. Während des gesamten Versuchs steht den Tieren Trinkwasser und Futter ad libitum zur Verfügung. Die Substanzen werden einmal täglich 4-6 Wochen lang per Gavage, Futter oder
Trinkwasser verabreicht. Als Plazebogruppe dienen Tiere, die genauso behandelt werden, aber ent- weder nur das Lösungsmittel oder das Futter bzw. Trinkwasser ohne Testsubstanz erhalten.
Die Wirkung der Testsubstanzen wird durch Messung hämodynamischer Parameter [Blutdruck, Herzfrequenz, Inotropie (dp/dt), Relaxationszeit (tau), maximaler linksventrikulärer Druck, links- ventrikulärer enddiastolischer Druck (LVEDP)], Gewichtsbestimmung von Herz, Niere und Lunge, Messung der Proteinausscheidung sowie durch Messung der Genexpression von Bio- markern (z.B. ANP, Atrial Natriuretic Peptide, und BNP, Brain Natriuretic Peptide) mittels RT/TaqMan-PCR nach RNA-Isolation aus kardialem Gewebe bestimmt.
Die statistische Auswertung erfolgt mit Srudent's t-Test nach vorheriger Überprüfung der Varianzen auf Homogenität.
B-5: Bestimmung der metabolischen Stabilität
Zur Bestimmung der metabolischen Stabilität von Testverbindungen werden diese in vitro mit Lebermikrosomen oder bevorzugt mit primären frischen Hepatozyten verschiedener Tierspezies (z.B. von Ratte und Hund) als auch humanen Ursprungs inkubiert, um Metabolitenprofile eines
möglichst kompletten hepatischen Phase I- und Phase E-Metabolismus zu erhalten und zu vergleichen.
Die Testverbindungen werden mit einer Konzentration von 10-20 μM inkubiert. Dazu werden Stammlösungen der Substanzen mit einer Konzentration von 1-2 mM in Acetonitril hergestellt und dann mit einer l: 100-Verdünnung in den Inkubationsansatz pipettiert. Die Lebermikrosomen werden in 50 mM Kaliumphosphat-Puffer (pH 7.4) mit und ohne NADPH-generierendem System, bestehend aus 1 mM NADP+, 10 mM Glucose-6-phosphat und 1 Einheit Glucose-6-phosphat- Dehydrogenase, bei 37°C inkubiert. Primäre Hepatozyten werden in Suspension in Williams E- Medium ebenfalls bei 37°C inkubiert. Nach einer Inkubationszeit von 0-4 Stunden werden die Inkubationsansätze mit Acetonitril abgestoppt (Endkonzentration ca. 30%) und das Protein bei ca. 15000 x g abzentrifugiert. Die so abgestoppten Proben werden entweder direkt analysiert oder bis zur Analyse bei -200C gelagert.
Die Analyse erfolgt mittels Hochleistungsflüssigkeits-Chromatographie mit Ultraviolett- und massenspektrometrischer Detektion (HPLC-UV-MS/MS). Dazu werden die Überstände der Inku- bationsproben mit geeigneten C18-reversed-phase-Säulen und variablen Eluenten-Gemischen aus Acetonitril und 10 mM wässriger Ammoniumformiat-Lösung chromatographiert. Die UV-Chroma- togramme in Verbindung mit massenspektrometrischen MS/MS-Daten dienen zur Identifizierung und Strukturaufklärung der Metabolite.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überfuhrt werden:
Tablette:
Zusammensetzung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.
Claims
Patentansprüche
1. Verbindung der Formel (I)

in welcher
R1 für (C3-C10)-Alkyl, (C3-C7)-Cycloalkyl oder -ORA steht,
wobei (C3-Cio)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, (Ci-C4)-Alkoxy, Trifluormethoxy und (C3-C7)-Cycloalkyl substituiert sein kann,
worin (C3-C7)-Cycloalkyl seinerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (CrO-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (CrC4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei (C3-C7)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (C]-C4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (Ci-C4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei
RA für (d-Cio)-Alkyl oder (C3-C7)-Cycloalkyl steht,
wobei (Ci-Cio)-Alkyl mit einem Substituenten ausgewählt aus der Gruppe Hydroxy, (Ci-C4)-Alkoxy und (C3-C7)-Cycloalkyl substituiert sein kann,
worin (C3-C7)-Cycloalkyl seinerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (C,-C4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (Ci-C4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei (C3-C7)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (Ci-C4)- Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (C,-C4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei in allen genannten Cycloalkyl-Gruppen eine Ctt-Einheit gegen Sauerstoff ausgetauscht sein kann,
R2 für (CrC4)-Alkyl oder Cyclopropyl steht,
wobei (Ci-C4)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C4)-Alkoxy substituiert sein kann,
R3 für Wasserstoff oder Fluor steht,
R4 für Wasserstoff, Fluor, Chlor, Methyl oder Trifluormethyl steht,
R5 für Wasserstoff, Halogen, Nitro, Cyano, Trifluormethyl, Methyl, Ethyl, Trifluormethoxy oder Methoxy steht,
R6 für Wasserstoff, Fluor, Chlor, Methyl oder Trifluormethyl steht,
wobei mindestens einer der Reste R3, R4, R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze,
mit Ausnahme der Verbindung 4-Cyclopropyl-6-(methoxymethyl)-2-[4- (trifluormethyl)phenyl]-pyrimidin-5-carbonsäure.
Verbindung der Formel (I) nach Anspruch 1, in welcher
R1 für (C3-C8)-Alkyl, Cyclopropyl, Cyclopentyl, Cyclohexyl oder -ORA steht,
wobei (C3-C8)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, Methoxy, Ethoxy und Trifluormethoxy substituiert sein kann,
und
wobei Cyclopropyl, Cyclopentyl und Cyclohexyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, Methyl, Ethyl, Trifluormethyl und Methoxy substitutiert sein können,
und
wobei
RA für Methyl, Ethyl, (C3-C8)-Alkyl, Cyclopropyl, Cyclopentyl oder
Cyclohexyl steht,
wobei Methyl und Ethyl mit einem Substituenten ausgewählt aus der Gruppe Cyclopropyl, Cyclopentyl und Cyclohexyl substituiert sind,
worin Cyclopropyl, Cyclopentyl und Cyclohexyl ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der
Gruppe Fluor, Hydroxy, Oxo, Methyl, Ethyl, Trifluormethyl und Methoxy substitutiert sein können,
wobei (C3-C8)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxy, Methoxy und Ethoxy substituiert sein kann,
und
wobei Cyclopropyl, Cyclopentyl und Cyclohexyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, Methyl, Ethyl, Trifluormethyl und Methoxy substitutiert sein können,
und
wobei in allen genannten Cyclopentyl- und Cyclohexyl-Gruppen eine CH2-Einheit gegen Sauerstoff ausgetauscht sein kann,
R2 für (CrC4)-Alkyl, Trifluormethyl oder Cyclopropyl steht,
R3 für Wasserstoff steht,
R4 für Wasserstoff oder Fluor steht,
R5 für Wasserstoff, Fluor, Chlor, Trifluormethyl oder Methyl steht,
R6 für Wasserstoff oder Methyl steht,
wobei mindestens einer der Reste R4, R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze.
3. Verbindung der Formel (I) nach Anspruch 1 oder 2, in welcher
R1 für (C3-C6)-Alkyl, Cyclopentyl, Cyclohexyl, Tetrahydrofuranyl oder -ORA steht,
wobei
RA für Methyl, (C3-C6)-Alkyl, Cyclopropyl, Cyclopentyl oder Cyclohexyl steht,
worin Methyl mit einem Substituenten ausgewählt aus der Gruppe Cyclopropyl, Cyclopentyl und Cyclohexyl substituiert ist,
worin Cyclopropyl, Cyclopentyl und Cyclohexyl ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Trifluormethyl substitutiert sein können,
worin (C3-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxy und Methoxy substituiert sein kann,
und
worin Cyclopropyl, Cyclopentyl und Cyclohexyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Trifluormethyl substitutiert sein können,
R2 für Methyl, Ethyl, n-Propyl, Isopropyl, Trifluormethyl oder Cyclopropyl steht,
R3 für Wasserstoff steht,
R4 für Wasserstoff oder Fluor steht,
R5 für Wasserstoff, Chlor oder Methyl steht,
R6 für Wasserstoff oder Methyl steht,
wobei mindestens einer der Reste R4, R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze.
4. Verbindung der Formel (I) nach Anspruch 1 , 2 oder 3, in welcher
R1 für Isopropyl, iso-Butyl, 1-Methylpropyl, iso-Pentyl, 1-Methylbutyl oder -ORA steht,
wobei
RΛ für Isopropyl, iso-Butyl, 1-Methylpropyl, 1-Methylbutyl oder 3-
Methylbutyl steht,
R2 für Methyl, Ethyl oder Trifluormethyl steht,
R3 für Wasserstoff steht,
R4 für Wasserstoff steht,
R5 für Wasserstoff, Chlor oder Methyl steht,
R6 für Wasserstoff oder Methyl steht,
wobei mindestens einer der Reste R5 und R6 von Wasserstoff verschieden ist,
sowie ihre Salze, Solvate und Solvate der Salze.
5. Verfahren zur Herstellung von Verbindungen der Formel (I), wie in den Ansprüchen 1 bis 4 definiert, dadurch gekennzeichnet, dass man eine Verbindung der Formel (H)

in welcher R2, R3, R4, R5 und R6 jeweils die in den Ansprüchen 1 bis 4 angegebenen Bedeutungen haben,
und
R7 für (Ci-C4)-AIkyl steht,
entweder
[A] in einem inerten Lösungsmittel unter Mitsunobu-Bedingungen mit einer Verbindung der Formel (DI-A)
R1 — H (m-A),
in welcher
R1 für -ORA steht,
worin RΛ die in den Ansprüchen 1 bis 4 angegebene Bedeutung hat,
zu Verbindungen der Formel (IV-A)

in welcher R1Λ, R2, R3, R4, R5, R6 und R7 jeweils die zuvor angegebenen Bedeutungen haben,
umsetzt und diese durch basische oder saure Hydrolyse in die Carbonsäuren der Formel (I- A)

in welcher R1A, R2, R3, R4, R5 und R6 jeweils die zuvor angegebenen Bedeutungen haben,
überführt,
oder
[B] mit Hilfe eines geeigneten Chlorierungsmittels, wie beispielsweise Phosphoroxychlorid, in eine Verbindung der Formel (V)

in welcher R2, R3, R4, R5, R6 und R7 jeweils die zuvor angegebenen Bedeutungen haben, überführt,
und diese anschliessend in einem inerten Lösungsmittel in Gegenwart einer Base und eines geeigneten Palladium-Katalysators mit einer Verbindung der Formel
(m-B)
R1B— x1 (m-B),
in welcher
R1B für (C3-Cio)-Alkyl oder (C3-C7)-Cycloalkyl steht,
wobei (C3-Cio)-Alkyl mit einem oder zwei Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, (Ci-C4)-Alkoxy, Trifluormethoxy und (C3-C7)-Cycloalkyl substituiert sein kann,
worin (C3-C7)-Cycloalkyl seinerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (Ci-C4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (Ci-C4)-Alkoxy und Trifluormethoxy substitutiert sein kann,
und
wobei die genannten (C3-C7)-Cycloalkyl-Gruppen mit einem oder zwei Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Hydroxy, Oxo, (CrC4)-Alkyl, Trifluormethyl, 2,2,2-Trifluorethyl, (CrC4)- Alkoxy und Trifluormethoxy substitutiert sein können,
und
X1 für eine Gruppe der Formel -B(OR8)2 oder -ZnHaI steht,
worin
HaI für Halogen, insbesondere Chlor, Brom oder Iod steht,
und
R8 für Wasserstoff oder (CrC4)-Alkyl steht
oder
beide Reste R8 zusammen eine -C(CH3)2-C(CH3)2-Brücke bilden,
zu Verbindungen der Formel (IV-B)

in welcher R1B, R2, R3, R4, R5, R6 und R7 jeweils die zuvor angegebenen Bedeutungen haben,
umsetzt und diese durch basische oder saure Hydrolyse in die Carbonsäuren der Formel (I-B)

in welcher R1B, R2, R3, R4, R5 und R6 jeweils die zuvor angegebenen Bedeutungen haben,
überfuhrt,
und die Verbindungen der Formeln (I-A) und (I-B) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Basen oder Säuren zu ihren Solvaten, Salzen und/oder Solvaten der Salze umsetzt.
Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 4 definiert, zur Behandlung und/oder Prophylaxe von Krankheiten.
Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 4 definiert, zur Verwendung in einem Verfahren zur Behandlung und/oder Prophylaxe von Dyslipidämien, Arteriosklerose und Herzinsuffizienz.
8. Verwendung einer Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 4 definiert, zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Dys- lipidämien, Arteriosklerose und Herzinsuffizienz.
9. Arzneimittel enthaltend eine Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 4 definiert, in Kombination mit einem inerten, nicht-toxischen, pharmazeutisch geeigneten
Hilfsstoff.
10. Arzneimittel enthaltend eine Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 4 definiert, in Kombination mit einem oder mehreren weiteren Wirkstoffen ausgewählt aus der Gruppe bestehend aus HMG-CoA-Reduktase-Inhibitoren, Diuretika, beta-Rezeptoren- Blocker, organische Nitrate und NO-Donatoren, ACE-Inhibitoren, Angiotensin An¬
Antagonisten, Aldosteron- und Mineralokortikoid-Rezeptor-Antagonisten, Vasopressin- Rezeptor- Antagonisten, Thrombozytenaggregationshemmer sowie Antikoagulantien.
11. Arzneimittel nach Anspruch 9 oder 10 zur Behandlung und/oder Prophylaxe von Dyslipid- ämien, Arteriosklerose und Herzinsuffizienz.
12. Verfahren zur Behandlung und/oder Prophylaxe von Dyslipidämien, Arteriosklerose und Herzinsuffizienz in Menschen und Tieren unter Verwendung einer wirksamen Menge mindestens einer Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 4 definiert, oder eines Arzneimittels, wie in einem der Ansprüche 9 bis 11 definiert.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102007061757.9 | 2007-12-20 | ||
| DE102007061757A DE102007061757A1 (de) | 2007-12-20 | 2007-12-20 | Substituierte 2-Phenylpyrimidin-5-carbonsäuren und ihre Verwendung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009080248A1 true WO2009080248A1 (de) | 2009-07-02 |
Family
ID=40344489
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/010698 Ceased WO2009080248A1 (de) | 2007-12-20 | 2008-12-16 | Substituierte 2-phenylpyrimidin-5-carbonsäuren und ihre verwendung |
Country Status (2)
| Country | Link |
|---|---|
| DE (1) | DE102007061757A1 (de) |
| WO (1) | WO2009080248A1 (de) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011037223A1 (ja) | 2009-09-28 | 2011-03-31 | 興和株式会社 | 内臓脂肪重量の低下剤 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014050134A1 (ja) | 2012-09-27 | 2014-04-03 | 興和株式会社 | 脂質異常症治療剤 |
| WO2015030033A1 (ja) | 2013-08-28 | 2015-03-05 | 興和株式会社 | 脂質異常症治療剤 |
| US12569481B2 (en) | 2020-06-12 | 2026-03-10 | Vanderbilt University | Methods of treatment for gastrointestinal motility disorders |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050096337A1 (en) * | 2003-11-05 | 2005-05-05 | Jean Ackermann | Phenyl derivatives, their manufacture and use as pharmaceutical agents |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU727775B2 (en) | 1996-01-17 | 2000-12-21 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| PL338013A1 (en) | 1997-07-16 | 2000-09-25 | Novo Nordisk As | Derivatives of condensed 1,2,4-thiadiazine, their production and application |
| BR9908004A (pt) | 1998-02-17 | 2001-12-18 | Tularik Inc | Composto, composição e método para prevençãoou supressão de uma infecção viral |
| DE19834044A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Neue substituierte Pyrazolderivate |
| DE19834047A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Substituierte Pyrazolderivate |
| DE19943634A1 (de) | 1999-09-13 | 2001-04-12 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943636A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943635A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943639A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Dicarbonsäurederivate mit neuartigen pharmazeutischen Eigenschaften |
| AR031176A1 (es) | 2000-11-22 | 2003-09-10 | Bayer Ag | Nuevos derivados de pirazolpiridina sustituidos con piridina |
| DE10110749A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Substituierte Aminodicarbonsäurederivate |
| DE10110750A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE10220570A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Ag | Carbamat-substituierte Pyrazolopyridine |
| DE602004022819D1 (de) | 2003-06-06 | 2009-10-08 | Vertex Pharma | Von atp-bindende kassette transportern |
| RU2367659C2 (ru) | 2003-11-05 | 2009-09-20 | Ф.Хоффманн-Ля Рош Аг | Гетероарильные производные в качестве активаторов рецепторов, активируемых пролифераторами пероксисом (ppar) |
| JP4906715B2 (ja) | 2004-05-08 | 2012-03-28 | ニューロジェン・コーポレーション | 4,5−ジ置換−2−アリールピリミジン類 |
| DE102005027150A1 (de) | 2005-03-12 | 2006-09-28 | Bayer Healthcare Ag | Pyrimidincarbonsäure-Derivate und ihre Verwendung |
| WO2006124874A2 (en) | 2005-05-12 | 2006-11-23 | Kalypsys, Inc. | Inhibitors of b-raf kinase |
-
2007
- 2007-12-20 DE DE102007061757A patent/DE102007061757A1/de not_active Withdrawn
-
2008
- 2008-12-16 WO PCT/EP2008/010698 patent/WO2009080248A1/de not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050096337A1 (en) * | 2003-11-05 | 2005-05-05 | Jean Ackermann | Phenyl derivatives, their manufacture and use as pharmaceutical agents |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011037223A1 (ja) | 2009-09-28 | 2011-03-31 | 興和株式会社 | 内臓脂肪重量の低下剤 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| KR20150063035A (ko) | 2012-09-27 | 2015-06-08 | 교와 가부시키가이샤 | 지질 이상증 치료제 |
| WO2014050134A1 (ja) | 2012-09-27 | 2014-04-03 | 興和株式会社 | 脂質異常症治療剤 |
| US9572798B2 (en) | 2012-09-27 | 2017-02-21 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US9931321B2 (en) | 2012-09-27 | 2018-04-03 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US10258609B2 (en) | 2012-09-27 | 2019-04-16 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US11013722B2 (en) | 2012-09-27 | 2021-05-25 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| WO2015030033A1 (ja) | 2013-08-28 | 2015-03-05 | 興和株式会社 | 脂質異常症治療剤 |
| KR20210107168A (ko) | 2013-08-28 | 2021-08-31 | 교와 가부시키가이샤 | 지질 이상증 치료제 |
| EP3939586A1 (de) | 2013-08-28 | 2022-01-19 | Kowa Company, Ltd. | Kombination von pemafibrate und omega-3 fettsäuren bei dyslipedmie |
| KR20230050472A (ko) | 2013-08-28 | 2023-04-14 | 교와 가부시키가이샤 | 지질 이상증 치료제 |
| KR20240152981A (ko) | 2013-08-28 | 2024-10-22 | 교와 가부시키가이샤 | 지질 이상증 치료제 |
| US12569481B2 (en) | 2020-06-12 | 2026-03-10 | Vanderbilt University | Methods of treatment for gastrointestinal motility disorders |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102007061757A1 (de) | 2009-06-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2197846A1 (de) | Substituierte 6-phenylnikotinsäuren und ihre verwendung | |
| WO2009080248A1 (de) | Substituierte 2-phenylpyrimidin-5-carbonsäuren und ihre verwendung | |
| WO2009080242A1 (de) | Substituierte 4-aminopyrimidin-5-carbonsäuren und ihre verwendung | |
| EP2635576B1 (de) | Benzyl-substituierte carbamate und ihre verwendung | |
| EP2235011B1 (de) | Substituierte pyrrolo[2, 3-b]- und pyrazolo[3, 4-b]pyridine als adenosin rezeptor liganden | |
| EP3660015A1 (de) | Verfahren zur herstellung von (4s)- 4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl- 1,4-dihydro-1,6-naphthyridin-3-carbox-amid und dessen aufreinigung für die verwendung als pharmazeutischer wirkstoff | |
| EP2066637A1 (de) | Neue substituierte bipyridin-derivate und ihre verwendung als adenosin rezeptor liganden | |
| WO2007128454A1 (de) | 3-tetrazolylindazole und 3-tetrazolylpyrazolopyridine sowie ihre verwendung | |
| EP2066634A1 (de) | 4-phenoxy-nikotinsäure-derivate und ihre verwendung als ppar-modulatoren | |
| EP3191452A1 (de) | Substituierte n,2-diarylchinolin-4-carboxamide und ihre anti-inflammatorische verwendung | |
| AU2017217931A1 (en) | Halo-substituted piperidines as orexin receptor modulators | |
| WO2008031501A2 (de) | 2-phenoxynikotinsäure-derivate und ihre verwendung | |
| EP2556856B1 (de) | 6-Cyano-substituierte Pyrido[2,3-d]pyrimidine als Adenosin Rezeptor Liganden zur Behandlung von Kardiovaskulären Erkrankungen | |
| EP1866289A1 (de) | Pyrimidincarbonsäure-derivate und ihre verwendung | |
| EP1797045A1 (de) | Neue pyrimidin-derivate und ihre verwendung als ppar-alpha-modulatoren | |
| WO2020165031A1 (de) | Substituierte isochinolin-piperidinylmethanon-derivate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08864639 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08864639 Country of ref document: EP Kind code of ref document: A1 |