WO2009110010A2 - Stable oral dosage form - Google Patents
Stable oral dosage form Download PDFInfo
- Publication number
- WO2009110010A2 WO2009110010A2 PCT/IN2009/000163 IN2009000163W WO2009110010A2 WO 2009110010 A2 WO2009110010 A2 WO 2009110010A2 IN 2009000163 W IN2009000163 W IN 2009000163W WO 2009110010 A2 WO2009110010 A2 WO 2009110010A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dosage form
- solid dosage
- stable
- amlodipine
- oral
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2095—Tabletting processes
Definitions
- the present invention relates to a stable, oral dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and olmesartan or its pharmaceutically acceptable salt
- calcium channel blockers and angiotensin II receptor antagonists are widely used clinically as medicaments for the treatment and prophylaxis of hypertension.
- United States patent application number US200602S2805 (herein after referred to as '805) relates to a pharmaceutical composition comprising pharmaceutically effective amounts of the following active ingredients:
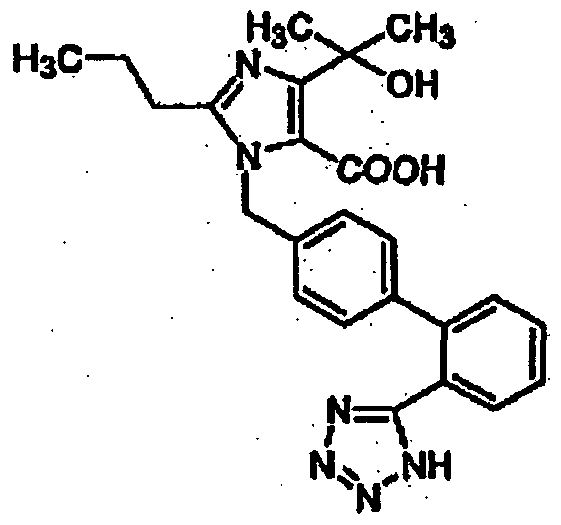
- an angiotensin II receptor antagonist which is (5-methyl-2-oxo-l,3-dioxolen-4-yl) methyl $-(l-hydroxy-l-methylethyl)-2-propyl-[[2'-(lh-tetrazol-5-yl)biphenyl-4-yl)methyl]imidadol-5- carboxylate and
- (B) a calcium channel blocker is amlodipine.
- an angiotensin II receptor antagonist selected from the group consisting of a compound having a formula (I), a pharmacologically acceptable ester thereof and a pharmacologically acceptable salt thereof;
- a calcium channel blocker selected from the group consisting of 1,4-dihydropyridine compound and a pharmacologically acceptable salt thereof.
- PCT patent publication, WO200700106S (hereinafter referred to as publication '06S) disclosed that prior art compositions made by direct compression had poor dissolution characteristics and claims a method comprising a step of preparing by wet granulation, a composition comprising an angiotensin II receptor antagonist and a calcium channel blocker to achieve improved dissolution of angiotensin II receptor antagonist.
- PCT patent publication, WO2007001066 (hereinafter referred to as publication '066) claims a pharmaceutical composition comprising an angiotensin II receptor antagonist, a calcium channel blocker and at least one substance selected from a hydrophilic polymer, an acidic substance and a fluidizing agent.
- composition in the form of tablets may be made by any method known in the art including direct compression:
- PCT patent publication, WO2007001067 claims a solid dosage form with improved dissolution wherein the angiotensin II receptor antagonist and the calcium channel blocker are not intimately mixed rather each of the active ingredient is mixed separately with pharmaceutical excipients and isolated from each other by granulation or compression of at least one or both the mixtures.
- the compositions can be made in the form of tablets by any of the methods known in the art including direct compression.
- the publication '107 also claims a solid dosage form comprising olmesartan medoxomil and amlodipine or its pharmacologically acceptable salts thereof, having less than 0.4 % concentration (w/w) of 3-ethyl-S- methyl-2-[ (2-aminoethoxy)methyl]-4- (2-chloroophenyl)-6-methylpyridine-3,5-dicarobxylate (Impurity D).
- the publication further claims a solid dosage form comprising olmesartan medoxomil and amlodipine or its pharmacologically acceptable salts thereof, having less than S.I % concentration (w/w) of total impurities.
- the solid dosage form of the invention comprises olmesartan medoxomil and amlodipine or a pharmaceutically acceptable salt thereof, which are characterized by having less than 2.5 % concentration (w/w) of RNH-6270, less than 0.4 % concentration (w/w) of impurity D and less than 5.1 % concentration (w/w) of total impurities and by being substantially free of reducing sugar.
- the present invention provides a stable, oral, solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants, the solid dosage form being free of hydrophilic or hydrophobic binders.
- the present invention also provides a stable, oral, solid dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt, one or more antioxidants and pharmaceutically acceptable disintegrants, diluents and lubricants.
- the present invention further provides a stable, oral solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants and wherein the solid dosage form is prepared by slugging.
- the present invention provides a stable, oral, solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and one or more pharmaceutically acceptable diluents, disintegrants and lubricants, the said solid dosage form being free of hydrophilic or hydrophobic binders.
- the present invention also provides a stable, oral, solid dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt, one or more antioxidants and pharmaceutically acceptable disintegrants, diluents and lubricants.
- the present invention further provides a stable, oral solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants and wherein the solid dosage form is prepared by slugging.
- the present invention provides dosage forms having adequate rates of dissolution and chemical stability of both amlodipine besylate and olmesartan medoxomil.
- the term 'adequate rate of dissolution' as used herein means that the dissolution of each active ingredient is more than 80 % in about 45 minutes.
- the term 'without appreciable degradation* as used herein generally, means that the total impurities of active ingredients and known impurities do not exceed the acceptable limits of the labeled amount of active ingredient during the shelf life of the product or when kept at accelerated stability conditions for example, at 40 ° C ⁇ 2 ° C and 75 % ⁇ 5 % relative humidity for a period of three months in closed containers, with or without desiccants.
- Amlodipine is known to be very hygroscopic and known to degrade via a catalytic oxidative route to form Impurity D (II). It was found that the composition of the present invention showed less than 0.2 % of Impurity D (II) during the shelf life of the product
- Amlodipine used in the solid dosage form of the present invention can be used as a pharmaceutically acceptable salt such as a besylate, maleate, fumarate, camsylate, hydrochloride, hydrobromide, lactate, tartarate, citrate, mesylate, nicotinate, gluconate and the like, as well as in the form of a free base.
- amlodipine besylate is used.
- the amount of amlodipine besylate used in the dosage form of the present invention ranges from about 1 mg to about 100 mg by weight the dosage form.
- the amount of olmesartan medoxomil used in the dosage form of the present invention ranges from about 1 mg to about 100 mg.
- the amount of amlodipine besylate ranges from about 5 mg to about 20 mg and the amount of olmesartan medoxomil ranges from about 10 mg to about SO mg per unit dosage form.
- amlodipine or its pharmaceutically acceptable salt are 'intimately mixed' together.
- 'intimately mixed' means that the two drugs are mixed and blended and are capable of coming in contact with each other.
- the oral, solid, dosage form of the present invention is free of hydrophilic or hydrophobic binders.
- the term 'binder' used herein is known in the art and connotes an excipient that promotes binding of particles to form strong aggregates such as granules, pellets, slugs, pills or tablets.
- hydrophilic binders include, but are not limited to, cellulose derivatives such as low molecular weight grades of hydroxypropyl methyl cellulose, methyl cellulose, hydroxypropyl cellulose and sodium carboxymethyl cellulose; synthetic polymers such as polyvinylpyrrolidone, aminoalkyl methacrylate copolymer, carboxyvinyl polymer, polyvinyl alcohol and polyethylene glycol); gum Arabic, agar, gelatin and sodium alginate and mixtures thereof.
- cellulose derivatives such as low molecular weight grades of hydroxypropyl methyl cellulose, methyl cellulose, hydroxypropyl cellulose and sodium carboxymethyl cellulose
- synthetic polymers such as polyvinylpyrrolidone, aminoalkyl methacrylate copolymer, carboxyvinyl polymer, polyvinyl alcohol and polyethylene glycol
- gum Arabic agar, gelatin and sodium alginate and mixtures thereof.
- hydrophilic or hydrophobic binder does not include diluents that have the ability to consolidate upon compression, for example, directly compressible diluents such as directly compressible grades of microcrystalline cellulose, spray dried lactose, mannitol and the like.
- the amount of diluent that may be used may range from about S % to about 95 % by weight of the oral dosage form.
- microcrystalline cellulose is used as the diluent.
- Microcrystalline cellulose is a purified, partially deloplymerized cellulose, that occurs as a white, odorless, tasteless and crystalline powder composed of porous particles. It is commercially available in different particle sizes and moisture grades which have different properties and specifications. It is available commercially under the brand names of Avicel * by FMC Corporation, Embocel* by Edward Mendell Co. Inc and Vicacel ® by J.E Rettenmaier and S ⁇ hne GmbH. These commercially available brands are available with varying mean particle size, and moisture content.
- Avicel ® by FMC Corporation is available as Avicel PH-101 with a nominal mean particle size of 50 microns, Avicel PH- 102 with a nominal mean particle size of 100 microns, Avicel PH-103 with a nominal mean particle size of 50 microns and moisture content of less than 3.0 %, Avicel PH- 105 with a nominal mean particle size of 20 microns and moisture content of less than 5.0 %, Avicel PH- 112 with a nominal mean particle size of 100 microns and moisture content of less than 1.5 %, Avicel PH-113 with a nominal mean particle size of 50 microns and moisture content of less than 1.5 %, Avicel PH-200 with a nominal mean particle size of 180 microns and moisture content of less than 5.0 %, Avicel PH-301 with a nominal mean particle size of 50 microns and moisture content of less than 5.0 %, Avicel PH-302 with a nominal mean particle size of 100 microns and moisture content of less than 5.0 %
- the Vivacel brand is available as Vivacel 101, 102, 12 and 20 with the nominal mean particle size of 50, 100, 180 and 20 microns respectively, with a moisture content of less than 5 %.
- the microcrystalline cellulose having mean particle in the range of 150 ⁇ ms to about 200 ⁇ ms, preferably a grade having mean particle size of about 180 ⁇ ms is used.
- the solid dosage form comprises substantial amount of a reducing agent
- a reducing sugar is a type of sugar with an aldehyde group, which allows a sugar to act as a reducing agent, for example, in a Maillard reaction or a Benedicts reaction.
- reducing sugars include, but are not limited to, lactose, glucose, fructose, glyceraldehyde, arabinose, mannose, galactose, maltose, xylose, cellobiose, mellibiose, maltotriose, and the like and mixtures thereof.
- substantially amount' means amount of the reducing agent in more than 10 % by weight of the solid dosage form, preferably more than 20 % by weight of the solid dosage form.
- disintegrants used in the oral, solid dosage form of the present invention include, but are not limited to, microcrystalline cellulose, croscarmellose sodium, cross linked polyvinyl pyrrolidone, sodium starch glycolate, starch and its derivatives such as partially gelatinized starch, low substituted hydroxyl propyl cellulose, ion exchange resins like polacrillin potassium, effervescent couple such as sodium bicarbonate and citric acid and the like and mixtures thereof.
- One preferred embodiment of the present invention uses croscarmellose sodium as the disintegrants.
- the amount of disintegrants that may be used ranges from about 0.1 % to about 50 % by weight of the dosage form, preferably from about 1 % to about 30 % by weight of the oral dosage form.
- lubricants that are used in the solid dosage form of the present invention, include, but are not limited to, the group comprising of sodium lauryl sulfate, talc, magnesium stearate, sodium stearyl fumarate, stearic acid, glyceryl behenate, hydrogenated vegetable oil, zinc stearate and the like and mixtures thereof.
- the amount of lubricants used may range from about 0.1 % to about 2 % by weight of the dosage form.
- the oral, solid, dosage form comprises one or more antioxidants.
- the antioxidants may be selected from the group comprising butylated hydroxy anisole, butylated hydroxy toluene, alpha tocopherol and the like and mixtures thereof.
- the amount of butylated hydroxyl toluene that may be used in the oral solid dosage form ranges from about 0.01 % to about 2 % by weight of the unit dosage form, preferably 0.05 % to about 0.S % by weight of the unit dosage form.
- the oral, solid dosage form of the present invention may be in the form of powders, pills, pellets, tablets, powder filled into capsules, sachets, granules, slugs, compacts.
- the solid dosage form is in the form of compressed tablets.
- the present invention provides dosage forms that require few steps for manufacture into tablet dosage form.
- the present invention provides a solid oral dosage form that does not necessarily require wet granulation as a means for improving dissolution. Instead, a dry granulation or direct compression which avoids the energy intensive steps of drying and also minimizing the moisture content in the final dosage form may be utilized.
- oral dosage form of the present invention may be prepared by any suitable method such as wet granulation, dry granulation or direct compression; dry granulation processes such as slugging and roller compaction are preferred.
- the oral dosage form is prepared by roller compaction.
- roller compaction the blend of powders is fed to two counter-rotating rolls which draw the powder between the rolls due to friction and compact the powder.
- the compacts formed by roller compaction are usually referred to as 'ribbons'.
- the ribbons are further milled into smaller granules and then may be filled into capsules, or converted into tablets by compressing the milled compacts.
- the oral dosage form may be prepared by the process of slugging.
- the term 'slugging* as used herein means that the intimate mixture of the two active ingredients is converted into larger particles without the use of any solvent.
- the oral dosage form is prepared by mixing together the two drugs and the pharmaceutically acceptable excipients and converting the powder blend into slugs of sizes of, for example, about 10 mm to about 20 mm round diameter, preferably about IS mm in diameter using round punches. The slugs are further milled into smaller granules and then may be filled into capsules, or converted into tablets by compressing the milled slugs.
- the amlodipine besylate is mixed with an alcoholic solution of butylated hydroxyl toluene (BHT).
- BHT butylated hydroxyl toluene
- the alcohol is evaporated to get a dry amlodipine besylate with BHT.
- This mixture of amlodipine besylate and BHT is further mixed with olmesartan or its pharmaceutically acceptable salt to get an 'intimate mixture' of the said two drugs.
- This intimate mixture may be further mixed with other excipients such as disintegrants, lubricants and then converted into compacts or slugs.
- the compacts or slugs may be further milled and converted into granules. These granules may be filled into capsules, or may be compressed into tablets.
- an inert excipient such as diluent, disintegrants or lubricant or mixtures thereof, is mixed with an alcoholic solution of butylated hydroxyl toluene.
- the alcohol is evaporated to get a dry excipient with the antioxidant
- This mixture of the excipient and antioxidant is further mixed with the blend of amlodipine or its pharmaceutically acceptable salt and olmesartan or its pharmaceutically acceptable salt to get an 'intimate mixture' of the said two drugs.
- This intimate mixture may be further mixed with other excipients and converted into compacts or slugs.
- the compacts or slugs may be further milled and converted into granules. These granules may be filled into capsules, or may be compressed into tablets.
- Butylated hydroxyl toluene was dissolved in isopropyl alcohol. Croscarmellose sodium (half portion) was wetted with the alcoholic solution of butylated hydroxyl toluene. The wet mass was dried. The specified amounts of olmesartan medoxomil, lactose anhydrous, colloidal silicon dioxide (half amount) and microcrystalline cellulose was sifted individually through ASTM # 40 mesh and mixed and then blended for about 20 minutes. This blend was mixed with the specified amount of amlodipine besylate. The mixture was further mixed in a blender for 10 minutes. Magnesium stearate was added to the above blend and blended for further S minutes.
- This lubricated mixture was slugged using 16 mm round plain punches at a target weight of 1400 mg.
- the slugs were milled through clit mill fitted with 2.00 mm screen at slow speed with knives forward.
- the remaining half portion of both croscarmellose sodium and colloidal silicon dioxide were sifted through ASTM # 40 mesh.
- Magnesium stearate was sifted through # 80 ASTM mesh.
- the slugs were mixed with the blend of croscarmellose sodium, colloidal silicon dioxide and lubricated with magnesium stearate and compressed into tablets using suitable punches.
- the core tablets were coated with aqueous dispersion of Opadry II white in purified water.
- the tablets prepared according to example 1 were subjected to in vitro dissolution in USP type 2 apparatus (paddle) in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in Table 2a.
- the tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and silica gel dessicant at different temperature and humidity conditions.
- the stability data given below indicates that the dosage form prepared according to example 1 was stable upon storage.
- Butylated hydroxyl toluene was dissolved in isopropyl alcohol. Croscarmellose sodium (half portion) was wetted with the alcoholic solution of butylated hydroxyl toluene. The wet mass was dried and mixed with the specified amounts of olmesartan medoxomil, lactose anhydrous, colloidal silicon dioxide (half amount) and microcrystalline cellulose. The material was mixed in a blender for 10 minutes. Magnesium stearate was added to the above blend and blended for further 5 minutes. This material was slugged using 16 mm round plain punches at a target weight of 1400 mg. The slugs were milled through cut mill fitted with 2.00 mm screen at slow speed with knives forward.
- the remaining half portion of croscarmellose sodium, colloidal silicon dioxide and amlodipine besylate were sifted through ASTM # 40 mesh.
- the remaining amount of magnesium stearate was sifted through # 80 ASTM mesh.
- the slugs were mixed with the blend of croscarmellose sodium, colloidal silicon dioxide and amlodipine besylate an lubricated with magnesium stearate and compressed into tablets using suitable punches.
- the core tablets were coated with aqueous dispersion of Opadry II white in purified water.
- the tablets prepared according to example 2 were subjected to in vitro dissolution in USP type 2 apparatus in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in Table 4a.
- magnesium stearate All the listed ingredients except magnesium stearate were sifted through 40 mesh.
- Olmesartan medoxomil was mixed with part of following excipients: lactose anhydrous, microcrystalline cellulose, low substituted hydroxypropyl cellulose and colloidal silicon dioxide in a blender for 20 minutes. This blend was lubricated with part of sifted magnesium stearate and slugged. The slugs were milled.
- Sifted amlodipine besylate part of the following ingredients namely microcrystalline cellulose, lactose anhydrous, colloidal silicon dioxide were mixed with the milled slugs. This blend was compressed into tablets. The cores were coated with HPMC based Opadry film coating.
- the tablets prepared according to example 3 were subjected to in vitro dissolution in USP type 2 apparatus in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in table 6.
- the tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and silica gel dessicant at different temperature and humidity conditions.
- the tablets of example 4 were found to retain 94.1 % of amlodipine besylate and 95.0 % of olmesartan medoxomil upon storage at 40 0 C and 75 % relative humidity for three months.
- the stability data given below indicates that the dosage form prepared according to example 3 was stable upon storage.
- magnesium stearate All the listed ingredients except magnesium stearate were sifted through 40 mesh.
- Olmesartan medoxomil was mixed with the specified amounts of following excipients: lactose anhydrous, microcrystalline cellulose and a portion of the colloidal silicon dioxide and portion of croscarmellose sodium in a blender for 20 minutes. This blend was lubricated with part of sifted magnesium stearate and slugged. The slugs were milled. Butylated hydroxy! toluene was dissolved in isopropyl alcohol. Amlodipine besylate was wetted with this solution.
- the tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and silica gel dessicant at different temperature and humidity conditions.
- the tablets of example 4 were found to retain 94.4 % of amlodipine besylate and 93.0 % of olmesartan medoxomil upon storage at 40 0 C and 75 % relative humidity for three months.
- the stability data given below indicates that the dosage form prepared according to example 1 was stable upon storage.
- Table 8 b Result of the stability study
- magnesium stearate was sifted through 40 mesh.
- Magnesium stearate was sifted through ASTM # 60 mesh.
- Olmesartan medoxomil was mixed with the specified amounts of following excipients: lactose anhydrous, microcrystalline cellulose and a portion of the colloidal silicon dioxide and portion of croscarmellose sodium in a blender for 20 minutes. This blend was lubricated with part of sifted magnesium stearate and slugged. The slugs were milled. Butylated hydroxy! toluene was dissolved in isopropyl alcohol. Amlodipine besylate was wetted with this solution.
- the tablets prepared according to example 5 were subjected to in vitro dissolution in USP type 2 apparatus in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in table 10a.
- the tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and SiIiCa 1 gel dessicant at different temperature and humidity conditions.
- the tablets of example 5 were found to retain 97.09 % of amlodipine besylate and 95.01 % of olmesartan medoxomil upon storage at 40 0 C and 75 % relative humidity for three months.
- the stability data given below indicates that the dosage form prepared according to example 5 was stable upon storage.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
The present invention relates to a stable, oral dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and olmesartan or its pharmaceutically acceptable salt.
Description
STABLE ORAL DOSAGE FORM
The present invention relates to a stable, oral dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and olmesartan or its pharmaceutically acceptable salt
BACKGROUND OF THE INVENTION
Currently, calcium channel blockers and angiotensin II receptor antagonists are widely used clinically as medicaments for the treatment and prophylaxis of hypertension.
United States patent application number US200602S2805 (herein after referred to as '805) relates to a pharmaceutical composition comprising pharmaceutically effective amounts of the following active ingredients:
(A) an angiotensin II receptor antagonist which is (5-methyl-2-oxo-l,3-dioxolen-4-yl) methyl $-(l-hydroxy-l-methylethyl)-2-propyl-[[2'-(lh-tetrazol-5-yl)biphenyl-4-yl)methyl]imidadol-5- carboxylate and
(B) a calcium channel blocker is amlodipine.
United States patent application number US20060009S02 (herein after referred to as '302) claims a pharmaceutical composition comprising pharmaceutically effective amounts of the following active ingredients:
(A) an angiotensin II receptor antagonist selected from the group consisting of a compound having a formula (I), a pharmacologically acceptable ester thereof and a pharmacologically acceptable salt thereof;

PCT patent publication, WO200700106S (hereinafter referred to as publication '06S) disclosed that prior art compositions made by direct compression had poor dissolution characteristics and claims a method comprising a step of preparing by wet granulation, a composition comprising an angiotensin II receptor antagonist and a calcium channel blocker to achieve improved dissolution of angiotensin II receptor antagonist.
PCT patent publication, WO2007001066 (hereinafter referred to as publication '066) claims a pharmaceutical composition comprising an angiotensin II receptor antagonist, a calcium channel blocker and at least one substance selected from a hydrophilic polymer, an acidic substance and a fluidizing agent. The patent application states that a combination of the angiotensin II receptor antagonist and a calcium channel blocker with additional hydrophilic polymer, acidic substance or fluidizing agent affords improved dissolution properties. The composition in the form of tablets may be made by any method known in the art including direct compression:
PCT patent publication, WO2007001067 (hereinafter referred to as publication '067) claims a solid dosage form with improved dissolution wherein the angiotensin II receptor antagonist and the calcium channel blocker are not intimately mixed rather each of the active ingredient is mixed separately with pharmaceutical excipients and isolated from each other by granulation or compression of at least one or both the mixtures. The compositions can be made in the form of tablets by any of the methods known in the art including direct compression.
PCT patent application, WO2008032107 (hereinafter referred to as publication '107) claims a solid dosage form comprising olmesartan medoxomil and amlodipine or its pharmacologically acceptable salts thereof, having less than 2.5 % concentration (w/w) of 4-(l-hydroxy-lmethylethyl)-2-propyl-l-[[2'- (1 H-tetrazol-5-yl) biphenyl-4-yl] methyl]-lH-imidazol-5-carboxylic acid (RNH-6270). The publication '107 also claims a solid dosage form comprising olmesartan medoxomil and amlodipine or its pharmacologically acceptable salts thereof, having less than 0.4 % concentration (w/w) of 3-ethyl-S- methyl-2-[ (2-aminoethoxy)methyl]-4- (2-chloroophenyl)-6-methylpyridine-3,5-dicarobxylate (Impurity D). The publication further claims a solid dosage form comprising olmesartan medoxomil and amlodipine or its pharmacologically acceptable salts thereof, having less than S.I % concentration (w/w) of total impurities. The '107 publication describes that there is a degradation of amlodipine because of presence of lactose due to the Maillard reaction in a Olmetec® based formulation. The publication further states that the solid dosage form of the invention comprises olmesartan medoxomil and amlodipine or a pharmaceutically acceptable salt thereof, which are characterized by having less than 2.5 % concentration (w/w) of RNH-6270, less than 0.4 % concentration (w/w) of impurity D and less than 5.1 % concentration (w/w) of total impurities and by being substantially free of reducing sugar.
None of the above mentioned prior arts disclose whether the compositions retain both amlodipine besylate or olmesartan medoxomil without appreciable degradation wherein the composition contains substantial amount of lactose. The free base amlodipine and olmesartan are drugs with ester function and are susceptible to hydrolysis. Yeung et al in the journal publication entitled Liquid chromatography assay for amlodipine: chemical stability and pharmacokinetics in rabbits J. Pharm. Biomed. Anal. vol.No.9 (7). paye No. 565-71 «1991 provides a liquid chromatography method for studying the stability of amlodipine besylate. Celebier et al, in the journal publication titled 'Determination of olmesartan medoxomil in tablets by UV- Vis spectrophotometry, Pharmazie vol. 62(6): page No. 419-422 (2007) discusses the analytical method for determination of stability of olmesartan medoxomil in the
pharmaceutical dosage forms. Both olmesartan medoxomil and amlodipine are chemical compounds that seem to have some stability problems. Therefore, there lies a need for a combination for a combination of these two drugs which also has acceptable drug stability. We surprisingly found that the solid dosage form comprising olmesartan medoxomil and amlodipine besylate in spite of having substantial amounts of reducing sugars provided a composition that was stable over a period of time.
OBEJCTS OF THE INVENTION
It was the object of the present invention to provide dosage forms having adequate rates of dissolution and chemical stability of both amlodipine besylate and olmesartan medoxomil.
It was also the object of the present invention to provide dosage forms by processes that do not necessarily require separation of the amlodipine besylate and olmesartan medoxomil.
It was the object of the present invention to provide dosage forms that require few steps for manufacture into tablet dosage form.
It was further an object of the present invention to provide dosage forms that do not necessarily require wet granulation as a means for improving dissolution but instead can be made by dry granulation or direct compression thus avoiding the energy intensive steps of drying and also minimizing the moisture content in the final dosage form.
SUMMARY OF THE INVENTION
The present invention provides a stable, oral, solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants, the solid dosage form being free of hydrophilic or hydrophobic binders.
The present invention also provides a stable, oral, solid dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt, one or more antioxidants and pharmaceutically acceptable disintegrants, diluents and lubricants.
The present invention further provides a stable, oral solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants and wherein the solid dosage form is prepared by slugging.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides a stable, oral, solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and one or more pharmaceutically acceptable diluents, disintegrants and lubricants, the said solid dosage form being free of hydrophilic or hydrophobic binders.
The present invention also provides a stable, oral, solid dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt, one or more antioxidants and pharmaceutically acceptable disintegrants, diluents and lubricants.
The present invention further provides a stable, oral solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants and wherein the solid dosage form is prepared by slugging.
The present invention provides dosage forms having adequate rates of dissolution and chemical stability of both amlodipine besylate and olmesartan medoxomil. The term 'adequate rate of dissolution' as used herein, means that the dissolution of each active ingredient is more than 80 % in about 45 minutes. The term 'without appreciable degradation* as used herein generally, means that the total impurities of active ingredients and known impurities do not exceed the acceptable limits of the labeled amount of active ingredient during the shelf life of the product or when kept at accelerated stability conditions for example, at 40 ° C ± 2 ° C and 75 % ± 5 % relative humidity for a period of three months in closed containers, with or without desiccants.
Amlodipine is known to be very hygroscopic and known to degrade via a catalytic oxidative route to form Impurity D (II). It was found that the composition of the present invention showed less than 0.2 % of Impurity D (II) during the shelf life of the product

(II: Impurity D)
Olmesartan medoxomil is known to form the acid impurity III under basic or acidic conditions and in presence of water because of hydrolysis of the ester bond. It was found that the composition of the present invention showed less than 2 % of Impurity A (III) during the shelf life of the product. Another impurity E (IV) arising from route was found to be less than 0.2 % by weight of the composition during the storage of the solid dosage form over a period of time. It was found that the composition of the present invention showed less than 3 % of the total impurities when stored during the shelf life of the product
Amlodipine used in the solid dosage form of the present invention can be used as a pharmaceutically acceptable salt such as a besylate, maleate, fumarate, camsylate, hydrochloride, hydrobromide, lactate, tartarate, citrate, mesylate, nicotinate, gluconate and the like, as well as in the form of a free base. In one preferred embodiment, amlodipine besylate is used. The amount of amlodipine besylate used in the dosage form of the present invention ranges from about 1 mg to about 100 mg by weight the dosage form. The amount of olmesartan medoxomil used in the dosage form of the present invention ranges from about 1 mg to about 100 mg. In the preferred embodiment, the amount of amlodipine besylate ranges from about 5 mg to about 20 mg and the amount of olmesartan medoxomil ranges from about 10 mg to about SO mg per unit dosage form.
According to the present invention, the amlodipine or its pharmaceutically acceptable salt are 'intimately mixed' together. The term 'intimately mixed' as used herein means that the two drugs are mixed and blended and are capable of coming in contact with each other.
The oral, solid, dosage form of the present invention is free of hydrophilic or hydrophobic binders. The term 'binder' used herein is known in the art and connotes an excipient that promotes binding of particles to form strong aggregates such as granules, pellets, slugs, pills or tablets. The Examples of the hydrophilic binders include, but are not limited to, cellulose derivatives such as low molecular weight grades of hydroxypropyl methyl cellulose, methyl cellulose, hydroxypropyl cellulose and sodium carboxymethyl cellulose; synthetic polymers such as polyvinylpyrrolidone, aminoalkyl methacrylate copolymer, carboxyvinyl polymer, polyvinyl alcohol and polyethylene glycol); gum Arabic, agar, gelatin and sodium alginate and mixtures thereof. The term hydrophilic or hydrophobic binder does not include
diluents that have the ability to consolidate upon compression, for example, directly compressible diluents such as directly compressible grades of microcrystalline cellulose, spray dried lactose, mannitol and the like.
Examples of diluents that may be used in the oral dosage form of the present invention includes, but are not limited to, microcrystalline cellulose, starch, sugars and the like and mixture thereof. The amount of diluent that may be used may range from about S % to about 95 % by weight of the oral dosage form.
In one preferred embodiment, microcrystalline cellulose is used as the diluent. Microcrystalline cellulose is a purified, partially deloplymerized cellulose, that occurs as a white, odorless, tasteless and crystalline powder composed of porous particles. It is commercially available in different particle sizes and moisture grades which have different properties and specifications. It is available commercially under the brand names of Avicel * by FMC Corporation, Embocel* by Edward Mendell Co. Inc and Vicacel® by J.E Rettenmaier and Sβhne GmbH. These commercially available brands are available with varying mean particle size, and moisture content. For example, Avicel ® by FMC Corporation, is available as Avicel PH-101 with a nominal mean particle size of 50 microns, Avicel PH- 102 with a nominal mean particle size of 100 microns, Avicel PH-103 with a nominal mean particle size of 50 microns and moisture content of less than 3.0 %, Avicel PH- 105 with a nominal mean particle size of 20 microns and moisture content of less than 5.0 %, Avicel PH- 112 with a nominal mean particle size of 100 microns and moisture content of less than 1.5 %, Avicel PH-113 with a nominal mean particle size of 50 microns and moisture content of less than 1.5 %, Avicel PH-200 with a nominal mean particle size of 180 microns and moisture content of less than 5.0 %, Avicel PH-301 with a nominal mean particle size of 50 microns and moisture content of less than 5.0 %, Avicel PH-302 with a nominal mean particle size of 100 microns and moisture content of less than 5.0 %, Embocel 50M with a nominal mean particle size of 51 microns and moisture content of less than 5.0 %, Embocel 9OM with a nominal mean particle size of 91 microns and moisture content of less than 5.0 %,. The Vivacel brand is available as Vivacel 101, 102, 12 and 20 with the nominal mean particle size of 50, 100, 180 and 20 microns respectively, with a moisture content of less than 5 %. In one preferred embodiment, where the oral dosage form is prepared by slugging, the microcrystalline cellulose having mean particle in the range of 150 μms to about 200 μms, preferably a grade having mean particle size of about 180 μms is used.
In one embodiment of the present invention, the solid dosage form comprises substantial amount of a reducing agent A reducing sugar is a type of sugar with an aldehyde group, which allows a sugar to act as a reducing agent, for example, in a Maillard reaction or a Benedicts reaction. Examples of "reducing sugars" include, but are not limited to, lactose, glucose, fructose, glyceraldehyde, arabinose, mannose, galactose, maltose, xylose, cellobiose, mellibiose, maltotriose, and the like and mixtures thereof. The term 'substantial amount' as used herein means amount of the reducing agent in more than 10 % by weight of the solid dosage form, preferably more than 20 % by weight of the solid dosage form.
Examples of disintegrants used in the oral, solid dosage form of the present invention include, but are not limited to, microcrystalline cellulose, croscarmellose sodium, cross linked polyvinyl pyrrolidone, sodium starch glycolate, starch and its derivatives such as partially gelatinized starch, low substituted hydroxyl propyl cellulose, ion exchange resins like polacrillin potassium, effervescent couple such as sodium bicarbonate and citric acid and the like and mixtures thereof. One preferred embodiment of the present invention uses croscarmellose sodium as the disintegrants. The amount of disintegrants that may be used ranges from about 0.1 % to about 50 % by weight of the dosage form, preferably from about 1 % to about 30 % by weight of the oral dosage form.
Examples of lubricants, that are used in the solid dosage form of the present invention, include, but are not limited to, the group comprising of sodium lauryl sulfate, talc, magnesium stearate, sodium stearyl fumarate, stearic acid, glyceryl behenate, hydrogenated vegetable oil, zinc stearate and the like and mixtures thereof. The amount of lubricants used may range from about 0.1 % to about 2 % by weight of the dosage form.
According to one preferred embodiment of the present invention, the oral, solid, dosage form comprises one or more antioxidants. The antioxidants may be selected from the group comprising butylated hydroxy anisole, butylated hydroxy toluene, alpha tocopherol and the like and mixtures thereof. In one preferred embodiment, the amount of butylated hydroxyl toluene that may be used in the oral solid dosage form ranges from about 0.01 % to about 2 % by weight of the unit dosage form, preferably 0.05 % to about 0.S % by weight of the unit dosage form.
The oral, solid dosage form of the present invention may be in the form of powders, pills, pellets, tablets, powder filled into capsules, sachets, granules, slugs, compacts. In one preferred embodiment of the present invention, the solid dosage form is in the form of compressed tablets.
The present invention provides dosage forms that require few steps for manufacture into tablet dosage form. In one preferred embodiment, the present invention provides a solid oral dosage form that does not necessarily require wet granulation as a means for improving dissolution. Instead, a dry granulation or direct compression which avoids the energy intensive steps of drying and also minimizing the moisture content in the final dosage form may be utilized.
Although the oral dosage form of the present invention may be prepared by any suitable method such as wet granulation, dry granulation or direct compression; dry granulation processes such as slugging and roller compaction are preferred.
In one embodiment of the present invention, the oral dosage form is prepared by roller compaction. Generally, in roller compaction, the blend of powders is fed to two counter-rotating rolls which draw the powder between the rolls due to friction and compact the powder. The compacts formed by roller
compaction are usually referred to as 'ribbons'. The ribbons are further milled into smaller granules and then may be filled into capsules, or converted into tablets by compressing the milled compacts.
In an alternative embodiment of the present invention, the oral dosage form may be prepared by the process of slugging. The term 'slugging* as used herein means that the intimate mixture of the two active ingredients is converted into larger particles without the use of any solvent. In a preferred embodiment of the present invention, the oral dosage form is prepared by mixing together the two drugs and the pharmaceutically acceptable excipients and converting the powder blend into slugs of sizes of, for example, about 10 mm to about 20 mm round diameter, preferably about IS mm in diameter using round punches. The slugs are further milled into smaller granules and then may be filled into capsules, or converted into tablets by compressing the milled slugs.
In one embodiment of the present invention, the amlodipine besylate is mixed with an alcoholic solution of butylated hydroxyl toluene (BHT). The alcohol is evaporated to get a dry amlodipine besylate with BHT. This mixture of amlodipine besylate and BHT is further mixed with olmesartan or its pharmaceutically acceptable salt to get an 'intimate mixture' of the said two drugs. This intimate mixture may be further mixed with other excipients such as disintegrants, lubricants and then converted into compacts or slugs. The compacts or slugs may be further milled and converted into granules. These granules may be filled into capsules, or may be compressed into tablets.
In an another embodiment of the present invention, an inert excipient such as diluent, disintegrants or lubricant or mixtures thereof, is mixed with an alcoholic solution of butylated hydroxyl toluene. The alcohol is evaporated to get a dry excipient with the antioxidant This mixture of the excipient and antioxidant is further mixed with the blend of amlodipine or its pharmaceutically acceptable salt and olmesartan or its pharmaceutically acceptable salt to get an 'intimate mixture' of the said two drugs. This intimate mixture may be further mixed with other excipients and converted into compacts or slugs. The compacts or slugs may be further milled and converted into granules. These granules may be filled into capsules, or may be compressed into tablets.
The examples that follow do not limit the scope of the invention and are merely used as illustrations.
EXAMPLE 1
Table 1

The tablets prepared according to example 1 were subjected to in vitro dissolution in USP type 2 apparatus (paddle) in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in Table 2a.
Table 2a: in vitro dissolution in peak vessel dissolution of tablets of Example 1

The tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and silica gel dessicant at different temperature and humidity conditions. The stability data given below indicates that the dosage form prepared according to example 1 was stable upon storage.
Table 2 b: Result of the stabilit stud

Table 3

Butylated hydroxyl toluene was dissolved in isopropyl alcohol. Croscarmellose sodium (half portion) was wetted with the alcoholic solution of butylated hydroxyl toluene. The wet mass was dried and mixed with the specified amounts of olmesartan medoxomil, lactose anhydrous, colloidal silicon dioxide (half amount) and microcrystalline cellulose. The material was mixed in a blender for 10 minutes. Magnesium stearate was added to the above blend and blended for further 5 minutes. This material was slugged using 16 mm round plain punches at a target weight of 1400 mg. The slugs were milled through cut mill fitted with 2.00 mm screen at slow speed with knives forward. The remaining half portion of croscarmellose sodium, colloidal silicon dioxide and amlodipine besylate were sifted through ASTM # 40 mesh. The remaining amount of magnesium stearate was sifted through # 80 ASTM mesh. The slugs were mixed with the blend of croscarmellose sodium, colloidal silicon dioxide and amlodipine besylate an lubricated with magnesium stearate and compressed into tablets using suitable punches. The core tablets were coated with aqueous dispersion of Opadry II white in purified water.
The tablets prepared according to example 2 were subjected to in vitro dissolution in USP type 2 apparatus in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in Table 4a.
Table 4a: in vitro dissolution in peak vessel dissolution of tablets of example 2

The tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and silica gel dessicant at different temperature and humidity conditions. The stability data given below indicates that the dosage form prepared according to example 2 was stable upon storage. Table 4 b: Result of the stability study

Table 5

All the listed ingredients except magnesium stearate were sifted through 40 mesh. Magnesium stearate was sifted through ASTM # 60 mesh. Olmesartan medoxomil was mixed with part of following excipients: lactose anhydrous, microcrystalline cellulose, low substituted hydroxypropyl cellulose and colloidal silicon dioxide in a blender for 20 minutes. This blend was lubricated with part of sifted magnesium stearate and slugged. The slugs were milled. Sifted amlodipine besylate, part of the following ingredients namely microcrystalline cellulose, lactose anhydrous, colloidal silicon dioxide were mixed with the milled slugs. This blend was compressed into tablets. The cores were coated with HPMC based Opadry film coating.
The tablets prepared according to example 3 were subjected to in vitro dissolution in USP type 2 apparatus in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in table 6.
Table 6a: in vitro dissolution in peak vessel dissolution of tablets of Example 3

The tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and silica gel dessicant at different temperature and humidity conditions. The tablets of example 4 were found to retain 94.1 % of amlodipine besylate and 95.0 % of olmesartan medoxomil upon storage at 40 0C and 75 % relative humidity for three months. The stability data given below indicates that the dosage form prepared according to example 3 was stable upon storage.
Table 4 b: Result of the stabilit stud

Table 7

All the listed ingredients except magnesium stearate were sifted through 40 mesh. Magnesium stearate was sifted through ASTM # 60 mesh. Olmesartan medoxomil was mixed with the specified amounts of following excipients: lactose anhydrous, microcrystalline cellulose and a portion of the colloidal silicon dioxide and portion of croscarmellose sodium in a blender for 20 minutes. This blend was lubricated with part of sifted magnesium stearate and slugged. The slugs were milled. Butylated hydroxy! toluene was dissolved in isopropyl alcohol. Amlodipine besylate was wetted with this solution. The wet mass was dried in tray drier till the isopropyl alcohol was evaporated completely. Amlodipine besylate treated with BHT was mixed with part of the following ingredients namely, colloidal silicon dioxide and croscarmellose sodium and mixed with the milled slugs. Magnesium stearate was added to this mixture and blended for 5-10 minutes. This blend was compressed into tablets using suitable punches. The cores were coated with Opadry film coating. The tablets prepared according to example 4 were subjected to in vitro dissolution in USP type 2 apparatus in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in table 8a.
Table 8a: in vitro dissolution in peak vessel dissolution of tablets of Example 4

The tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and silica gel dessicant at different temperature and humidity conditions. The tablets of example 4 were found to retain 94.4 % of amlodipine besylate and 93.0 % of olmesartan medoxomil upon storage at 40 0C and 75 % relative humidity for three months. The stability data given below indicates that the dosage form prepared according to example 1 was stable upon storage. Table 8 b: Result of the stability study

EXAMPLE 5
Table 9

AH the listed ingredients except magnesium stearate were sifted through 40 mesh. Magnesium stearate was sifted through ASTM # 60 mesh. Olmesartan medoxomil was mixed with the specified amounts of following excipients: lactose anhydrous, microcrystalline cellulose and a portion of the colloidal silicon dioxide and portion of croscarmellose sodium in a blender for 20 minutes. This blend was lubricated with part of sifted magnesium stearate and slugged. The slugs were milled. Butylated hydroxy! toluene was dissolved in isopropyl alcohol. Amlodipine besylate was wetted with this solution. The wet mass was dried in tray drier till the isopropyl alcohol was evaporated completely. Amlodipine besylate treated with BHT was mixed with part of the following ingredients namely, colloidal silicon dioxide and croscarmellose sodium and mixed with the milled slugs. Magnesium stearate was added to this mixture and blended for S-10 minutes. This blend was compressed into tablets using suitable punches. The cores were coated with Opadry film coating.
The tablets prepared according to example 5 were subjected to in vitro dissolution in USP type 2 apparatus in 900 ml of pH 6.8 phosphate buffer rotating at 50 rpm. The dissolution was tested in peak dissolution vessel. The results of dissolution are tabulated in table 10a.
Table 10a: in vitro dissolution in peak vessel dissolution of tablets of Example 5
The tablets were stored in high density polyethylene bottles stoppered with CRC caps containing charcoal and SiIiCa1 gel dessicant at different temperature and humidity conditions. The tablets of example 5 were found to retain 97.09 % of amlodipine besylate and 95.01 % of olmesartan medoxomil upon storage at 40 0C and 75 % relative humidity for three months. The stability data given below indicates that the dosage form prepared according to example 5 was stable upon storage.
Table IQ b: Result of the stabilit stud

Claims
Claims:
1. A stable, oral, solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants, the solid dosage form being free of hydrophilic or hydrophobic binders.
2. A stable oral, solid dosage form as claimed in claim 1 wherein the diluent is a reducing sugar present in substantial amount
3. A stable, oral, solid dosage form as claimed in claim 1 wherein the amlodipine is in the form of besylate salt and the olmesartan is in the form of medoxomil salt.
4. A stable, oral solid dosage form as claimed in claim 1 wherein the composition is prepared by slugging.
5. A stable, oral, solid dosage form comprising therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt, one or more antioxidants and pharmaceutically acceptable disintegrants, diluents and lubricants.
6. A stable, oral solid dosage form as claimed in claim S wherein the diluent is a reducing sugar present in substantial amount.
7. A stable, oral, solid dosage form as claimed in claim S wherein the amlodipine is in the form of besylate salt and the olmesartan is in the form of medoxomil salt
8. A stable, oral solid dosage form as claimed in claim S wherein the antioxidant is selected from the group consisting of butylated hydroxytoluene, butylated hydroxyl anisole, alpha tocopherol and mixtures thereof.
9. A stable, oral solid dosage form consisting essentially of an intimate mixture of therapeutically effective amounts of amlodipine or its pharmaceutically acceptable salt and therapeutically effective amounts of olmesartan or its pharmaceutically acceptable salt and pharmaceutically acceptable disintegrants, diluents and lubricants, wherein the solid dosage form is prepared by slugging.
10. A stable, oral solid dosage form as claimed in claim 8 wherein the diluent comprises reducing sugar present in substantial amounts. 11. A stable, oral, solid dosage form as claimed in claim 9 wherein the amlodipine is in the form of besylate salt and the olmesartan is in the form of medoxomil salt.
12. A stable, oral solid dosage form as claimed in claim 3 wherein the impurity D of amlodipine is less than 0.20 % by weight and the acid impurity A of olmesartan medoxomil is less than 2.0 % by weight, when the said solid dosage form is stored in closed containers at 40 ° C ± 2 ° C and 75 % ± 5 % relative humidity for a period of three months in closed containers, with or without desiccants.
13. A stable, oral solid dosage form as claimed in claim 7 wherein the impurity D of amlodipine is less than 0.20 % by weight and the acid impurity A of olmesartan medoxomil is less than 2.0 % by weight, when the said solid dosage form is stored in closed containers at 40 ° C ± 2 ° C and 75 % ± 5 % relative humidity for a period of three months in closed containers, with or without desiccants.
14. A stable, oral solid dosage form as claimed in claim 11 wherein the impurity D of amlodipine is less than 0.2 % by weight and the acid impurity A of olmesartan medoxomil is less than 2.0 % by weight, when the said solid dosage form is stored in closed containers at 40 ° C ± 2 ° C and 75 % ± 5 % relative humidity for a period of three months in closed containers, with or without desiccants. S IS. A stable, oral solid dosage form as claimed in claim 2 wherein the impurity D of amlodipine is less than 0.2 % by weight and the acid impurity A of olmesartan medoxomil is less than 2.0 % by weight, when the said solid dosage form is stored in closed containers at 40 ° C ± 2 ° C and 75 % ± 5 % relative humidity for a period of three months in closed containers, with or without desiccants.
16. A stable, oral solid dosage form as claimed in claim 6 wherein the impurity D of amlodipine is less0 than 0.2 % by weight and the acid impurity A of olmesartan medoxomil is less than 2.0 % by weight, when the said solid dosage form is stored in closed containers at 40 ° C ± 2 ° C and 75 % ± 5 % relative humidity for a period of three months in closed containers, with or without desiccants.
17. A stable, oral solid dosage form as claimed in claim 10 wherein the impurity D of amlodipine is less than 0.2 % by weight and the acid impurity A of olmesartan medoxomil is less than 2.0 % by weight, S when the said solid dosage form is stored in closed containers at 40 ° C ± 2 ° C and 75 % ± 5 % relative humidity for a period of three months in closed containers, with or without desiccants.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN478MU2008 | 2008-03-07 | ||
| IN478/MUM/2008 | 2008-03-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009110010A2 true WO2009110010A2 (en) | 2009-09-11 |
| WO2009110010A3 WO2009110010A3 (en) | 2010-01-07 |
Family
ID=41056433
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2009/000163 Ceased WO2009110010A2 (en) | 2008-03-07 | 2009-03-09 | Stable oral dosage form |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009110010A2 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012020368A1 (en) * | 2010-08-08 | 2012-02-16 | Abdi Ibrahim Ilac Sanayi Ve Ticaret Anonim Sirketi | Olmesartan formulations |
| WO2016155815A1 (en) * | 2015-04-01 | 2016-10-06 | Ceva Sante Animale | Oral solid dosage form of amlodipine and veterinary uses thereof |
| WO2017068532A1 (en) * | 2015-10-23 | 2017-04-27 | Ftf Pharma Private Limited | Oral solution of dihydropyridine derivatives |
| CN119868296A (en) * | 2025-02-14 | 2025-04-25 | 宁波大红鹰药业股份有限公司 | Olmesartan medoxomil amlodipine tablet and preparation method thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050187262A1 (en) * | 2004-01-12 | 2005-08-25 | Grogan Donna R. | Compositions comprising (S)-amlodipine and an angiotensin receptor blocker and methods of their use |
| CA2613417C (en) * | 2005-06-27 | 2011-11-29 | Daiichi Sankyo Company, Limited | Pharmaceutical preparation containing an angiotensin ii receptor antagonist and a calcium channel blocker |
-
2009
- 2009-03-09 WO PCT/IN2009/000163 patent/WO2009110010A2/en not_active Ceased
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012020368A1 (en) * | 2010-08-08 | 2012-02-16 | Abdi Ibrahim Ilac Sanayi Ve Ticaret Anonim Sirketi | Olmesartan formulations |
| EP2425859A1 (en) * | 2010-08-08 | 2012-03-07 | Abdi Ibrahim Ilac Sanayi ve Ticaret Anonim Sirketi | Olmesartan formulations |
| WO2016155815A1 (en) * | 2015-04-01 | 2016-10-06 | Ceva Sante Animale | Oral solid dosage form of amlodipine and veterinary uses thereof |
| WO2016156550A1 (en) * | 2015-04-01 | 2016-10-06 | Ceva Sante Animale | Oral solid dosage form of amlodipine and veterinary uses thereof |
| AU2016239689B2 (en) * | 2015-04-01 | 2021-06-24 | Ceva Sante Animale | Oral solid dosage form of amlodipine and veterinary uses thereof |
| US11147804B2 (en) | 2015-04-01 | 2021-10-19 | Ceva Sante Animale | Oral solid dosage form of amlodipine and veterinary uses thereof |
| WO2017068532A1 (en) * | 2015-10-23 | 2017-04-27 | Ftf Pharma Private Limited | Oral solution of dihydropyridine derivatives |
| CN119868296A (en) * | 2025-02-14 | 2025-04-25 | 宁波大红鹰药业股份有限公司 | Olmesartan medoxomil amlodipine tablet and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009110010A3 (en) | 2010-01-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2010213594B2 (en) | Delayed release, oral dosage compositions that contain amorphous CDDO-Me | |
| RU2423975C2 (en) | Solid medication of olmesartan medoxomil and amlodipine | |
| EP2229938B9 (en) | Ezetimibe compositions | |
| EP1849459A1 (en) | Ezetimibe compositions | |
| US20110142930A1 (en) | Pharmaceutical Compositions of Atorvastatin | |
| WO2009034541A9 (en) | Controlled release pharmaceutical dosage forms of trimetazidine | |
| WO2018199282A1 (en) | Orally administrable enzalutamide-containing pharmaceutical composition | |
| JP2012503613A (en) | Compact cinacalc set | |
| EP4074308A1 (en) | Elagolix formulation | |
| US20080305158A1 (en) | Methods For the Preparation of Stable Pharmaceutical Solid Dosage Forms of Atorvastatin and Amlodipine | |
| AU2013365715B2 (en) | A pharmaceutical composition containing candesartan cilexetil and amlodipine | |
| CN101804029A (en) | Atorvastatin liposome and preparation method thereof, and medicine composition containing atorvastatin | |
| CA2465693A1 (en) | Pharmaceutical compositions of atorvastatin | |
| WO2009110010A2 (en) | Stable oral dosage form | |
| AU2003240164A1 (en) | Extended release formulation of divalproex sodium | |
| WO2012136839A1 (en) | Dry formulation and pharmaceutical composition comprising fesoterodine or a salt or a solvate thereof | |
| US20080038332A1 (en) | Stable pharmaceutical formulation comprising atorvastatin calcium | |
| US20080268049A1 (en) | Stable Solid Dosage Forms of Amlodipine and Benazepril | |
| WO2006059217A1 (en) | Stable solid dosage forms of amlodipine besylate and processes for their preparation | |
| TWI414310B (en) | Elution-improved pharmaceutical preparation | |
| CA2450001A1 (en) | Stable pharmaceutical compositions containing pravastatin | |
| CN118477074A (en) | Olmesartan medoxomil amlodipine pharmaceutical composition and preparation method thereof | |
| EP2839829A1 (en) | Sustained release tablet containing levodropropizine and method for preparing same | |
| WO2004075825A2 (en) | Dosage forms of amlodipine and processes for their preparation | |
| CA2609026A1 (en) | Formulations containing losartan and/or its salts |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09718495 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09718495 Country of ref document: EP Kind code of ref document: A2 |