WO2009127922A2 - 심혈관계 질환 치료용 약제학적 제제 - Google Patents
심혈관계 질환 치료용 약제학적 제제 Download PDFInfo
- Publication number
- WO2009127922A2 WO2009127922A2 PCT/IB2009/000331 IB2009000331W WO2009127922A2 WO 2009127922 A2 WO2009127922 A2 WO 2009127922A2 IB 2009000331 W IB2009000331 W IB 2009000331W WO 2009127922 A2 WO2009127922 A2 WO 2009127922A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- release
- cellulose
- amlodipine
- pharmaceutical formulation
- copolymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the present invention is designed to control and release each drug at a specific rate by applying the so-called Chronotherapy principle and XenoMotics, which are administered by staggering the time of expression of pharmacological action in each body
- the present invention relates to a pharmaceutical formulation comprising, as an active ingredient, a controlled release dihydropyridine calcium channel blocker and a statin lipid lowering agent.
- Hyperbaric pressure often coexists with coronary artery disease and is a major cause of heart disease. Hypertension is closely related to atherosclerosis due to hyperlipidemia. In other words, if the blood pressure rises, the arterial sclerosis worsens, and if the arteriosclerosis worsens, the blood pressure rises further to exacerbate each other. Such symptoms are recognized as a serious risk factor for developing cardiovascular disease. Hyperlipidemia in the "one type of gokol Leste 3 ⁇ 4 increase is the my elevated serum low-density lipoprotein (LDL), and call test Te serum chongkol less hedge the increase of the specific.
- LDL my elevated serum low-density lipoprotein
- lowering the level of serum lipids, especially LDL cholesterol may lower the likelihood of developing cardiovascular disease, delay the progression of atherosclerosis, or induce regression of sinus sclerosis.
- Hypercholesterolemia is involved in the early development of atherosclerosis, characterized by an uneven distribution of lipid deposits in the veins, including the coronary, jugular and peripheral arteries. This irregular distribution of atherosclerosis is characteristic of coronary artery injury and cardiovascular disease (Am J Cardiol 1987 59 (14): 91G).
- Atherosclerosis and hypertension are cyclically exacerbating symptoms. Therefore, both hyperlipidemia patients and hypertensive patients should be treated for isochronism and hypertension isochronously and prevent exacerbations [Hypertens Res 2001; 24: 3-ll, Hyper tensRes2003; 6: 1-36, Hyper tensRes2003; 26: 979-990].
- Clinical studies have shown that synergistic effects of co-administered statin-based lipid-lowering drugs are synergistic. Kramsch et al., Journal of Human Hyperteasion (1995) (Sup l.
- Dihydropyridine calcium channel blockers are not only high-pressure drugs, but they are also known to be angina pectoris, and statin drugs are not only lipid lowering agents but also known for their anti-inflammatory effects in various conditions.
- Both drug groups can be developed in a dosage form that is administered once a day, which is a common feature that taking at dinner is the pharmacologically ideal dosage time.
- the drug is prescribed in combination of two yakmulgun may include simvastatin of the dihydropyridine calcium channel blocker of the 'amlodipine and statin lipid-lowering drugs.
- amlodipine is synergistic with lipid lowering agents as well as its inherent anti-pressing action. .
- Simvastatin to increase the lipid-lowering effect of simvastatin Simvastatin also has a function to increase the blood pressure-lowering effect of the drug unique through synergistic action with Amlodi 3 ⁇ 4 as well as intrinsic lipid-lowering action There is an advantage.
- eNOs N0 production system
- amlodipine has a function of promoting NO release.
- atorvastatin has a phenoxy group, and this functional group catalyzes antioxidant activity due to proton donation and electron stabilization.
- Statin-based lipid lowering agents are ⁇ ( ⁇ ⁇ reductase inhibitors) and have the following general information, where statin-based lipid lowering agents and HMG-CoA reductase inhibitors are used interchangeably.
- Statin-based hypolipidemia mainly consists of sylvastatin, atorvastate
- Atherosclerotic or diabetic patients have abnormal N0 production (eNOs) in the vessel wall. This decreases N0 production and increases blood pressure.
- Statin-based lipid lowering agents including simvastatin, increase the normal levels of eNOS in these 3 ⁇ 4 tube walls. This is also one of the complex prescription effects in which lipid lowering action helps anti-pressure [Am J Physiol Renal Physiol Vol 281 Issue 5: F802-F809, 2001].
- Metabolic enzymes that primarily activate statin-based lipid lowering agents are Cytokrum P450 3A4 And 2C9, metabolites are excreted from the liver through the biliary tract while acting in the liver
- statin-based lipid lowering agents inhibit cytokine P450 enzymes that activate statins
- statins and intermediate metabolites leak into the blood. The concentration will increase.
- statin-based body metabolism and active metabolites liberated by hyperlipidemia can cause serious side effects of increased muscle lysis [Cl in Pharmacol Thar 1998; 63: 332-341, CI in Pharraacol Therl998; 64: 177-182, Physici ansDeskRef erence 2006 (Zocor), JPharmacolExpTher 1997; 282: 294-300, PharmacolExpTher 1999; 290: 1116-1125, L if eSc # 200; 76: 281-292 .: Drug Metab Dispos 1991; 19: 740-
- the dihydropyridine-based chamomile antagonist when a simple combination of two drugs, the dihydropyridine-based chamomile antagonist first reaches the liver and induces * inhibition of cytokine P450 enzymes, and a large part of the starin-based lipid lowering agents that subsequently enter the liver or enter simultaneously Since it is not metabolized by cytokine P450 enzymes, a large amount of blood leaks out, delays excretion, or accumulates, and is not metabolized by the cytokine P450 enzyme.
- the hydroxy acid migrates to hypertension, causing blood levels to be higher than necessary, which can cause muscle disorders such as muscle solubility.
- Simvastatin itself is a deactivated lactone-based compound and is primarily active in metabolites S, such as ⁇ -hydroxy acid of simvasta3 ⁇ 4, an active form that has a hypolipidemic activity that enters the liver. Simvastatin is metabolized in several stages by the cytokine P450 3A4 in the liver, and activated metabolites exert potent lipid-lowering effects. It is well known that simvapatin has been shown to reduce the incidence of coronary heart disease 1 and reduce mortality in large clinical trials [Lancet 199; 34: 1383-1389.
- simvastatin strongly inhibits HMG-CoA reductase, which plays a key role in the synthesis of cholesterol in the liver, and at the same time acts as an inhibitor of inflammation-inducing factors ["Scandinavian Simvastat in Survival Study "published in the Lancet, 1994, 344, 1383-89], Since pitavasta 3 ⁇ 4 is metabolized by cytochrome P450 2C9 and acts in the liver, it is excreted, and when administered with a drug that inhibits cytokine P450 enzymes, phytavastatin inhibits intrahepatic metabolism, thereby increasing blood concentration, This can cause side effects such as muscle lysis.
- Staggered channel blockers are one of the most commonly used anticancer drugs in combination with statin-based lipid-lowering agents. There are dihydropyridine-based staggered channel blockers in calcium channel blockers, and dihydropyridine-based staggered channel blockers, such as amlodipine and lercanidipine.
- amlodipine is most commonly prescribed as an antihypertensive and angina treatment agent worldwide [Cardiologyl 992; 80 (StiE »pll): S31—S36, JCardi ovascPharmaco 11988; 12 (Suppl7): S110-S113, Lancet 2000; 356: 359 -365, Hyper tens Res 2002; 25: 717-725, Hyper tens Res 2002; 25: 329-333.
- the dihydropyridine-based chasm channel blocker is an antihypertensive drug that lowers blood pressure by blocking permeate in 3 ⁇ 4 tubular smooth muscle and inducing peripheral artery dilation, and has common characteristics effective for angina due to convulsive vasoconstriction. It is also known that calcium channel blockers may have beneficial effects in the treatment of early cervical lesions [Lancet, 335, p. 1109-1139, 1990; and Circulation, 82, p.19401953, 1990].
- an increase in blood pressure during the morning is a 3 ⁇ 4 pressure increase due to 3 ⁇ 4 wall wall spasm caused by stress stimulation, and dihydropyridine calcium channel blockers are primarily responsible for relaxing these spasms. Because of the action, especially in the morning after waking up in the morning, the blood pressure-lowering effect appears strongly. Therefore, if the dihydropyridine kill: sump channel blocker in the evening hours is reacted in the evening, it can reach the maximum concentration of 3 ⁇ 4 before waking up, and the strongest blood pressure drop during the waking routine. Can be.
- Amlodi 3 ⁇ 4 a representative example of a dihydropyridine-based scab channel blocker, is an antihypertensive drug that lowers blood pressure by blocking peripheral vascular smooth muscle influx and inducing peripheral veins, which is effective in angina due to convulsive vasoconstriction.
- the chemical name is 3– to 3 ⁇ 4-5-methyl ⁇ 2 ⁇ (2-aminoespecialime 3 ⁇ 4) -4- (2-chlorophenyl) -1, 4-dihydro-6-methyl ⁇ 3, 5 ⁇ pyridinedica
- the half-life is very consistent with 30 to 50 hours. It is a very useful chest channel blocker that shows activity over a long period of time [European Patent Publication No.
- amlodipine is a 24 hour daily lasting medicine, and taking it in the evening hours is the most powerful effect of lowering blood pressure from the morning of the next morning to midday.
- Nitte dipin is when oral ruyeo a single drug mostly varies in the form of a metabolite of the inert type, 60-80% of the dose excreted in urine and 3 ⁇ 4 the kidney, it is "arranged in byeoneu part via the bile duct.
- the metabolism of nifedipine is involved in the cytokine P450 enzyme system and has a potent inhibitory effect on cytokine P450 3A4.
- Nifedipine's excretion half-life is known to be about two hours.
- the nisinidipine formulations provided are provided in a slow release form throughout the day. In terms of pathophysiology ' blown, especially morning weather.
- the increase in 3 ⁇ 4 pressure in the morning is the increase in blood pressure caused by vascular wall spasm caused by stress stimulation, nifedipine is the main action to relax this mild vascular contraction. Especially after morning waking, the blood pressure of morning sickness is effectively lowered.
- cytokine P450 3A4 enzymes are mainly metabolized by cytokine P450 3A4 enzymes and also by cytokine P450 3A4 enzymes such as cytokine P450 2C9 [Arc lnternMed. 2002Feb25; 162 (4): 405-. 12., Dru ⁇ et abDi spos .2000Feb; 28 (2) : 125-30.] Due to these characteristics, a combination of statin-based lipid lowering agents and dihydropyridine-based drugs requiring cytokine P450 appeal is required. Should be administered with a time delay to avoid interaction other drugs [MedCheml991; 34: 1838-1844,
- Korean Registered Patent No. 582347 discloses ammodyne, a dihydropyridine calcium channel blocker, which is rapidly released and the mandrel is eluted, and the statin is gradually eluted over 24 hours, thereby preventing the statin drug from being eluted and absorbed at one time. Disclosed is a composite agent.
- statins in the present invention it is considered as an irrational agent that reduces the efficacy of statins in the present invention as a complex system of concepts that are different from the concept of eluting statins first and metabolizing them in the liver first. If the amlodipine is introduced into the thrower liver, the production of cytochrome ⁇ 3 ⁇ 450 3A4 is inhibited in the liver, so that a small amount of statins, which are subsequently introduced into the liver, is not metabolized by the liver and is released into the blood.
- the patent only increases the side effects such as muscle lysis due to the release of statins, which should act in the liver, into 3 ⁇ 4.
- Korean Patent No. 742432 is a patent for a pharmaceutical preparation including amlodicec 3 ⁇ 4 sillate and simvastatin, and a method for preparing the same, but Cytok, which is required by simvastatin because both components are eluted simultaneously and absorbed into the liver
- comb P450 3A4 is inhibited by amlodipine
- simvastatin is released in excess of 30% into the blood along with the activator, which is not sufficiently activated in the liver, resulting in decreased lipid lowering and increased side effects.
- the present invention is a simple compounding concept, and this simple North compound is being removed due to the lack of progress.
- Pfizer's Korean Patent Publication No. 200,7002144 was also rejected by the Korean Intellectual Property Office because amlodipine and ato a vastatin are simple combinations.
- the present inventors have completed the present invention to develop a pharmaceutical preparation effective for the prevention and treatment of hypertension and hyperlipidemia and thereby cardiovascular disease or metabolic syndrome.
- the present invention controls the release of an agent comprising a dihydropyridine-based chamomile channel blocker containing several star 3 ⁇ 4 lipid-lowering agents within about 2 hours of dissolution and absorption time in the gastrointestinal tract.
- an agent comprising a dihydropyridine-based chamomile channel blocker containing several star 3 ⁇ 4 lipid-lowering agents within about 2 hours of dissolution and absorption time in the gastrointestinal tract.
- the present invention also provides a preparation comprising a dihydropyridine-based chamois blocker and a statin-based lipid lowering agent, by optimizing the drug delivery time of the drug to prevent metabolic syndrome and insulin-resistant patients, and patients suspected of diabetes mellitus or diabetes mellitus.
- Maximize the effect of calcium channel blocker which is the prevention or treatment of cardiovascular disease, cardiopulmonary disease, lung disease or kidney disease, and the effect of HMG-CoA reductase inhibitor, which is the prevention or treatment of heart disease caused by coronary atherosclerosis such as angina pectoris or myocardial infarction, It is intended to provide a pharmaceutical formulation that can avoid the interaction between the two drugs and the side effects thereby.
- the present invention is a preparation comprising a dihydropyridine-based chamomile channel blocker and a statin-based lipid lowering agent, which is added to convenience by taking one tablet once daily between evening hours and preferably 5 to 11 pm.
- a pharmaceutical preparation that can further increase the patient's medication woowoonggi cold.
- the present invention relates to a pharmaceutical formulation designed to control and release each drug at a specific rate by applying the so-called Chronotherapy principle, which is administered at a time difference in the pharmacological action expression time of each complex component.
- the pharmaceutical formulation of the present invention applies the principle of GhronotheraDy and Xenobiotics of the drug to the expression of pharmacological action of each of the complex components, and is controlled at the specific rate and absorbed into the body. It is a specially designed drug delivery system that can produce an ideal effect.
- the present invention provides a pre-release compartment comprising a statin-based lipid lowering agent, an isomer thereof, or a pharmaceutically acceptable salt thereof, and a dihydropyridine calcium channel blocker, an isomer thereof, as a pharmacologically active ingredient.
- a delayed-release compartment comprising a pharmaceutically acceptable salt thereof.
- the present invention is a dihydropyridine-based chamomile antagonist as an active ingredient, a delayed-release compartment made of granules, pellets or tablets, and a statin-based lipid lowering agent as an active ingredient.
- a pharmaceutical formulation with controlled release properties even with timed dissolution which is a capsul formulation comprising an exudative compartment.
- the dihydropyridine-based scabin antagonist is lowered in starlin-based lipids
- the release can be adjusted to be absorbed by the liver 2 to 4 hours later.
- the dihydropyridine-based scab antagonist is amlodipine, lercanidipine, lassipinepine, felodipine, vanidipine, benidipine, silnidipine, isradinine, mandidi 3 ⁇ 4, nicardidipine, nifedipine, nimodipine.
- Nilvadipine, nisulodidi3 ⁇ 4, nirenedipine, azanidipine may be selected from pharmaceutically acceptable salts thereof and isomerism thereof, and may be included in the range of 1 to 400 rag in the formulation.
- the delayed-release compartment may include at least one release controlling substance selected from an enteric polymer, a water-insoluble polymer, a hydrophobic compound, a hydrophilic polymer, an osmotic membrane coating base, and an osmotic pressure-controlling agent.
- a release controlling substance selected from an enteric polymer, a water-insoluble polymer, a hydrophobic compound, a hydrophilic polymer, an osmotic membrane coating base, and an osmotic pressure-controlling agent.
- Semipermeable membrane coating base Coating base is polyvinyl acetate, a full methacrylate copolymer as a poly (ethyl acrylate- Meth 3 ⁇ 4 methacrylate copolymer, 3 ⁇ 4 (ethyl arc 3 ⁇ 4rate-meth 3 ⁇ 4 methacrylate-trimethyl meth) No 3 ⁇ 4 decacrylate) copolymer, ethyl shellulose, saltose ester, 3 ⁇ 4 chloro ether, cellulose acylate, cellulose dicylate, cellulose triacylate, cellulose One or two or more mixtures selected from acetate, salose acetate and cellulose triacetate;
- the third pressure regulating agent is magnesium sulfate, magnesium chloride, sodium chloride, lithium chloride, calcom sulfate, sodium sulfate, lithium sulfate and sodium sulfate selected one or two or more mixtures;
- the statin-based lipid lowering agent is one or two selected from simvastatin, lovastat3 ⁇ 4, atorvastatin, pitavastatin, rosuvastated, flu; vastatin, pravasta 3 ⁇ 4, pharmaceutically acceptable salts thereof and isomers thereof. It may be a combination of the above.
- the present invention also relates to (a) a delayed-release compartment comprising a dihydropyridine base antagonist, a pharmaceutically acceptable salt thereof or an isomer thereof, a pharmaceutically acceptable carrier or diluent as an active ingredient (star as W active ingredient)
- a layer comprising a preservative-release compartment prepared using a base lipid lowering agent, a pharmaceutically acceptable salt thereof, or an isomer thereof, a pharmaceutically acceptable carrier or diluent, and
- the first agent and the second agent Provided is a kit comprising container means for thickening.
- the present invention also provides a dihydropyridine-based calcium antagonist active ingredient, a delayed-release compartment prepared as a tablet, and a statin-based lipid lowering agent as an active ingredient.
- the present invention provides a pharmaceutical formulation with controlled release, capable of dissolution of time, which is a coated tablet comprising a pre-release compartment coated on the surface of a single agent.
- the present invention also provides a delayed-release compartment comprising the dihydropyridine calcium antagonist as an active ingredient and constituting the inner core to release the drug by osmotic pressure, and a statin-based lipid lowering agent as the active ingredient,
- the present invention relates to a pre-release compartment comprising a statin-based lipid lowering agent, an isomer thereof, or a pharmaceutically acceptable salt thereof, and a dihydropyridine calcium channel blocker, an isomer thereof, as a pharmacologically active ingredient.
- it provides a pharmaceutical formulation comprising a delayed-release compartment comprising a pharmaceutically acceptable salt thereof.
- the formulation according to the present invention is physically separated or partitioned to control the release property between the two active ingredients to obtain different release rates between the two ingredients, thereby improving the problem of co-administration or isoadministration of the existing single agent. It provides a more relaxing therapeutic effect.
- the agent of the present invention is a pre-release compartment and a dihydropyridine calcium antagonist that elutes statin-based lipid lowering agents to be absorbed in the small intestine so that they can be absorbed in the liver 2 to 4 hours later than statin-based lipid lowering agents. It is a pharmaceutical formulation comprising a delayed-release compartment.
- the present invention provides a delayed release compartment comprising simvastatin, an isomer thereof, or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and an amlodipine, other isomers or a pharmaceutically acceptable salt thereof, as a pharmacologically active ingredient.
- Pharmaceutical formulations comprising a release compartment (hereinafter referred to as simvastared / amlodipine formulations) are provided.
- the present invention provides a prior-release compartment comprising atorvastatin, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a pharmacological activity
- a prior-release compartment comprising atorvastatin, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and a pharmacological activity
- Pharmaceutical formulations comprising amlodipine, isomers thereof or pharmaceutically acceptable salts thereof as active ingredients provide a pharmaceutical formulation comprising a delayed-release compartment (hereinafter referred to as atorvastatin / amlodipine formulation).
- the present invention also provides a prior-release compartment comprising atorvastatin, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and nifedipine, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient. It provides a pharmaceutical formulation (hereinafter referred to as ' 1 atorvastatin / nifedipine formulation') comprising a delayed-release compartment comprising.
- the present invention also provides a prior-release compartment comprising pitavastatin, an isomer thereof or a pharmaceutically acceptable salt thereof as an pharmacologically active ingredient, and amlodipine, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient.
- a pharmaceutical formulation comprising a delayed-release compartment containing the following (hereinafter referred to as a 'pitavastatin / amlodipine formulation') is provided.
- the present invention provides a prior-release compartment comprising Roschvastatin, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient, and amlodipine, an isomer thereof or a pharmaceutically acceptable salt thereof as a pharmacologically active ingredient. It provides a pharmaceutical formulation ( hereinafter referred to as "roschvastatin / amlodipine formulation") comprising a delayed-release compartment comprising a.
- the present invention provides that the pharmacologically active component of the lipophilic compartment and the pharmacologically active component of the prior-release compartment are bendipine several pitavastatin, nimodipine and pravastatin, nivaldi 3 ⁇ 4 and pitavastatin, nisoldipine and lovastatin ni.
- the present invention also preferably provides a statin-based lipid lowering agent release of statin-based lipid lowering agent, which is initiated after about 1 hour of statin lowering agent release * and is completed before about 8 hours. Providing a pharmaceutical agent which is then commenced and completed about 6 hours prior,
- Nifedipine in the dihydropyridine-based calcium channel blocker as an active ingredient
- the preparation is delayed until the release of nifedipine is sufficiently delayed and the elution is initiated after about 1 to 8 hours.
- atorvastatin elutes at least 90% of the drug within 1 hour, and nifedipine is sufficiently released. It is delayed and adjusted to begin dissolution about 2-6 hours after oral administration.
- the release of simvastatin / amlodipine formulations, or pitavastatin / amlodipine formulations, amlodipine of the present invention is initiated about 1 hour after each statin-based lipid lowering agent and is completed before about 8 hours, or is preferred. Preferably about 2 hours later.
- Amtodipine release is initiated about 1 hour after atorvastatin release and is completed before about 8 hours, or preferably after about 1 hour Rochevastatin / Amlodipine Formulation
- Completed before about 6 hours and amlodipine release can be initiated after about 1 hour of roschvastatin release, preferably after about 2 hours.
- the pharmaceutical formulation of the present invention has a time for which release of less than about 403 ⁇ 4 of the total amount of dihydropyridine calcium channel blockers is maintained within about 2 hours, preferably within about 3 hours, more preferably after the start of release of the statin-based lipid lowering agent.
- the drug is provided within about 4 hours, so that the effect of the dihydropyridine calcium channel blocker can be effectively generated after a certain delay time.
- the release of about 403 ⁇ 4 or less of the total amount of amlodipine is maintained within about 2 hours, preferably within about 3 hours. More preferably within about 4 hours and phytavastatin / amlodipine formulations or Roschvastarin / amlodipine formulations can dissolve up to 4 hours of amlodipine 1: 40% of this total, preferably not more than 303 ⁇ 4 .
- statin-based lipid lowering agent is first released and 8M, preferably 90% or more of the elution is completed within 1 hour, and the dihydropyridine-based calm channel blocker starts to dissolve after about 2 hours. And less than 40%, preferably less than about 30%, of the total amount within about 4 hours.
- each statin-based lipid lowering agent is at least 803 ⁇ 4 within 1 hour, preferably at least 90% This elutes and the amlodipine or nifedi 3 ⁇ 4 starts to elute after 2 hours, and the dissolution rate up to 4 hours is 40%, preferably not more than 303 ⁇ 4. It is adjusted to be released to the level.
- Rochevastatin is released more than 80% within 30 minutes, preferably 15 minutes, and amlodipine release is delayed more than 403 ⁇ 4 within 4 hours after oral administration. And preferably a pharmaceutical formulation released below 3 ".
- atorvastatin / nifedi tube preparation atorvastatin elutes at least 80% within 1 hour, and nifedipine is S-cutted so that elution is started after about 1 to 8 hours due to the delayed release. Atorvastatin elutes 90% or more within 1 hour, and nifedipine can be controlled to initiate elution approximately 2-6 hours after oral administration due to a significant delay in release.
- the pharmaceutical composition of the present invention exhibits the most effective effect when taken between 5 and 11 o'clock in the evening.
- Pre-release compartment refers to the compartment that is released earlier than the delayed-release compartment in the pharmaceutical formulation of the present invention.
- the pharmacologically active ingredient of the prior release compartment comprises a statin-based lipid lowering agent or a pharmaceutically acceptable salt thereof, and may further comprise a pharmaceutically acceptable additive as necessary.
- Statin-based lipid lowering agents of the present invention include simvastatin, lovastat3 ⁇ 4, atorvastatin, pitavastar 3 ⁇ 4, rosuvastatin fluvastatin, pravasta 3 ⁇ 4, and the like, including its optical isomers, racemates It includes all of them.
- isomers of atorvastatin include (RJ isomers, a, s) isomers, (s, s) isomers, or
- the (S,) isomers and their racemates include, but are not limited to, the ( ⁇ , ⁇ ) isomer (R, S) isomer, the (S, R) isomer, or the (S, S) isomer. Isomers are mentioned.
- the active ingredient in the pre-release compartment may comprise about 1 to about 160 mg, preferably about 2 to about 80 mg, as a unit dosage star 3 ⁇ 4 lipid lowering agent.
- simvastatin is about 1 ⁇ about 160 mg, preferably about 2 to about 80 rag
- atorvastatin is about 1 to 160 mg, preferably 5 to 160 mg, more preferably 10 to 80 mg
- the tin may comprise about 1 to 2 (g Roschvastatin 1 to 160 mg, preferably 5 to 80 rag.
- the prerelease compartmentalizing statin-based lipid lowering agent is at least about 803 ⁇ 4 of the total amount of the statin-based lipid lowering agent in the unit formulation within 1 hour, preferably within 30 minutes, more preferably within 15 minutes of the start of release. More than% is released, which can quickly indicate the efficacy.
- simvastatin is at least about 80% of the total amount within 1 hour after initiation of release, preferably at least about go% and atorvastatin is at least about 80%, preferably at least 90% within 1 hour of onset of release.
- Pitavastatin is within 1 hour after initiation of release, preferably within 30 minutes, more preferably within about 15 % or more of the total amount Rochevastatin within 1 hour after initiation of release, preferably within 30 minutes, More preferably, at least about 80% of the total amount can be released within 15 minutes.
- the formulations of the present invention may also be formulated using additives such as pharmaceutically acceptable diluents, binders, disintegrants, lubricants, stabilizers, pH adjusting agents, dissolution aids, colorants, fragrances, etc. without departing from the effect of the present invention.
- additives such as pharmaceutically acceptable diluents, binders, disintegrants, lubricants, stabilizers, pH adjusting agents, dissolution aids, colorants, fragrances, etc.
- Its content is preferably 100 to 30,000 parts by weight based on 100 parts by weight of the statin-based lipid lowering agent, more preferably 100 to 20,000 parts by weight.
- simvastatin / amlodipine formulation 100 to 100 parts by weight of simvastatin is added.
- atorvastatin / amlodipine formulation preferably 100-20,000 parts by weight, in the atorvastatin / amlodipine formulation, pitavastatin / amlodipine formulation, or atorvastatin / nisudipine formulation, 1 to 100 parts by weight for each .
- statin lipid lowering agent Preferably it is 1-30 parts by weight and the Roschvastatin / Amlodi 3 ⁇ 4 preparation may contain 0.01-100 parts by weight per 1 part of Roschvastatin.
- the diluent is starch, microcrystalline 3 ⁇ 4, lactose , Glucose, manny, alginate, alkaline earth metal salts, cleats, polyethylene glycols, dicalcium phosphate, or mixtures thereof. .
- the binder is starch, microcrystalline cellulose, highly dispersible silica, manny, sucrose, lactose, pulley ethylene glycol, pulley vinylpyridone, hydroxypropyl methyl salose, hydroxy propyl cellulose, Natural gums, synthetic gums, copovidones, l, or combinations thereof may be used.
- the disintegrant is a starch or modified starch, such as sodium starch glycolate, corn starch, potato starch or starch gelatinized starch, bentonite, monmo 3 ⁇ 4 lonite, or clay microcrystalline cell such as vegum, Cells such as oxypropylcellose or carboxymethyl 3 ⁇ 4rose such as sodium alginate or alginic acid.
- Crosslinked cells such as alginate croscarmel lose sodium Crosslinked polymers such as guarose gum guar gum, xanthan gum and crosslinked polymers such as xanthan gum Polivinyl pyridone (crospovidone) Effervescent agents such as sodium bicarbonate and citric acid Or a combination thereof.
- the lubricant is talc, stearic acid, magnesium stearate, calcium stearate, etc., lauryl sulfate sodium, hydrogenated vegetable oil, natzyl benzoate, sodium stearyl fumarate, glyceryl behanate, glycerol monomonate, glyceryl mono Stearate, glycer palmitostearate, polyethylene glycol and the like can be used.
- the stabilizer may be a salt of an alkali metal, a salt of an alkaline earth metal, or an alkalizing agent thereof, and preferably, calcium carbonate, sodium carbonate, sodium bicarbonate, magnesium oxide, magnesium carbonate, sodium citrate, and the like.
- Ascorbic acid, citric acid, butylated hydroxy anisole, butylated hydroxy toluene, tocope may be used as the derivative.
- the pH adjusting agent may be an acidifying agent such as acetic acid, adipic acid, ascorbic acid, malic acid, succinic acid, tartaric acid, fumaric acid, citric acid and a basicizing agent such as precipitated calcium carbonate, ammonia water, meglumine.
- an acidifying agent such as acetic acid, adipic acid, ascorbic acid, malic acid, succinic acid, tartaric acid, fumaric acid, citric acid and a basicizing agent such as precipitated calcium carbonate, ammonia water, meglumine.
- the dissolution aid may be a lauryl sulphate, polysorbate, such as pulley oxyethylene sorbitan fatty acid esters, docuate sodium, and the like.
- formulation of the present invention may be formulated using a pharmaceutically acceptable additive selected from various additives selected from colorants and fragrances.
- a delayed-release compartment means a compartment in which the active ingredient is released after a certain time of release of the active release compartment active ingredient.
- the delayed-release compartment comprises (1) the pharmacologically active amlodi 3 ⁇ 4, an isomer thereof or a pharmaceutically acceptable salt thereof, and (2-1) a release controlling substance and / or (2-2) an osmotic regulator and a semipermeable membrane.
- a coating base, and, if necessary, (3) may further include a pharmaceutically acceptable additive.
- the delayed-release compartment according to the present invention comprises a commercially available statin-based lipid lowering agent. It can also be taken at the same time.
- the pharmacologically active ingredient of the delayed-release compartment includes dihydropyridine-based chamomile channel blockers, isomers thereof and pharmaceutically acceptable salts.
- Dihydropyridine calcium channel blockers can be used to select components that are inhibited by Cytokrum P450 enzymes, for example, amlodipine, lercanidipine, lassidipine, felodipine, vanidipine, benidipine, silinidi. 3 ⁇ 4, device radio 3 ⁇ 4, Mani dipin, you carboxylic dipin, nifedipine, Nemo dipin, carbonyl body pin, nisul Lodi pin nitrendipine, O 3 ⁇ 4 you dipin but include ", nor not they are limited in kind between, as set forth above It can be selected and used in dihydropyridine type antagonism which is inhibited by the torque P450 type enzyme.
- Cytokrum P450 enzymes for example, amlodipine, lercanidipine, lassidipine, felodipine, vanidipine, benidipine, silinidi. 3 ⁇ 4, device radio 3 ⁇ 4, Mani dipin, you carboxylic dipin
- amlodipine, nifedipine and pharmaceutically acceptable salts thereof can be used, and pharmaceutically acceptable salts of amlodipine include, specifically, maleic acid salts of amlodipine and besylate salts of amlodipine.
- the active ingredient in the delayed-release compartment is a unit-depleted dihydropyridine-based scab channel blocker, which is based on a daily adult (65-75 kg adult male), about 1 to about 400 mg of unit dosage, and preferably about 2 "-about 120 mg may be included.
- the increase in amlodapine is about 1 to about 40 mg, preferably about 2 to about 20 mg, nifedipine may contain about 1 to about 90 mg per day.
- Dihydropyridine-based Calcium channel blockers have a release time of about 40% or less of the total amount of unit dosages reached release within about 2 hours, preferably within about 3 hours, more preferably about 4 hours after initiation of the star 3 ⁇ 4 lipid lowering agent.
- simvastatin / amlodipine formulations or atorvastatin / nifedipine formulations which require less than about 40% of the total amount of amlodipine or nifedipine to reach release.
- the atorvastatin / amlodipine formulation has a time of less than 203 ⁇ 4 of the total amount of amlodi 3 ⁇ 4 to reach release within about 2 hours, preferably Is within about 3 hours, more preferably about 4 hours, and the pitavastatin / amlodipine formulation is released up to 4 hours at 40% or less of the total amount of amplified amlodipine, preferably at 30% or less, and Rochevastatin / amlodipine.
- the formulations may release up to 3 hours of up to 303 ⁇ 4 of the total amount of amlodipine, preferably up to 20%.
- the delayed-release compartment in the pharmaceutical formulations of the present invention comprises at least one release agent selected from an elutable polymer, a water insoluble polymer, a hydrophobic compound, a hydrophilic polymer, and a mixture thereof.
- the release control material may be used in an amount of 10 to 3,000 parts by weight with respect to 100 parts by weight of dihydropyridine-based chamomile 3 ⁇ 4 agent, the amount of which is used in the above range. If less than sufficient delayed release is not obtained, if the dose exceeds the above range drug release is excessively delayed to obtain a significant clinical effect.
- the release-controlling material in the simvastatin / amlodipine formulation is preferably an enteric polymer comprising "and the common compounds of a hydrophilic polymer, or comprises a subeul soluble polymer, relative to 1 part amlodipine increase, 0.1 - comprises 100 parts by weight .
- the release controlling substance may include 0.05-100 parts by weight, preferably 0.05-30 parts by weight, with respect to 1 part by weight of amlodipine, and preferably may be a mixture of an enteric polymer and a hydrophilic polymer.
- the enteric polymer and the hydrophilic polymer mixture may be included in 0.5 to 10 parts by weight and 0.5 to 20 parts by weight, respectively, based on 1 part by weight of amlodipine.
- the release controlling substance in the pitavastatin / amlodipine formulation is available at 0.05-100 parts by weight with respect to 1 part of amlodipine, and preferably includes water-insoluble polymers and polymers and hydrophilic polymers in the roschvastatin / amlodipine formulation.
- the release control material is available in an amount of 0.05 to 100 parts by weight based on 1 part by weight of amlodipine, preferably comprising at least one release control material selected from a water-insoluble polymer and a hydrophilic polymer, and in the atorvastatin / nifedipine formulation.
- the release control material may be used in an amount of 5 to 300 parts by weight with respect to 100 parts by weight of nifedipine, and may preferably include an enteric polymer and a hydrophilic polymer.
- an enteric polymer is one that is soluble or stable under acidic conditions of less than pH 5, and refers to a polymer that is dissolved or decomposed under conditions of pH 5 or higher, such as an enteric cellulose derivative, an enteric acrylic acid system, and the like. It is selected from the group consisting of a co-polymer, an enteric maleic acid copolymer, an enteric polyvinyl derivative, and a combination thereof.
- the enteric cellulose derivatives include hydroxypropyl methyl cellulose acetate succinate, hydroxypropyl decyl queryl phthalate, hydroxyspecific methyl ethyl cellulose phthalate, cellulose acetate phthalate, and cellulose.
- enteric acrylic acid co-polymers selected from phthalate, methyl hydroxyethyl cellulose and their mixtures include styrene-arc 3 ⁇ 4 acid copolymers, methyl acrylate-acrylic acid copolymers, and methyl methacrylate acrylate.
- Acid Copolymers Arc Butyl-Styrene-Acrylate Acid Copolymers, Methac- Acid-Methac Methyl acrylate copolymer (Example 3 ⁇ 4 units, Eudragit L 100, Eudragit S, Degussa, Germany), Methacrylic acid, Ethyl acrylate copolymer (Example 3 ⁇ 4 units, Eudragit L 100-55, Degussa Germany ), At least one of the above-mentioned enteric maleic acid-based co-polymers selected from methyl acrylate-methacrylic acid-acetic acid octyl co-polymer and their mixtures is selected from the group consisting of vinyl acetate-maleic anhydride co-polymer styrene-maleic anhydride copolymer, styrene Monoester maleic acid copolymer, vinyl methyl ether-maleic anhydride copolymer, ethylene-maleic anhydride co
- enteric polymers for simvastatin / amlodipine formulations include polyvinylacetate phthalate, hydroxypropyltetylcelose phthalate, 3 ⁇ 4 talc, cellulose acetate phthalate, cellulose propionate phthalate, poly (methacrylate, methyl Methacrate) Copolymer and pulley (methacrylate, Ethacrylate) Copolymer may be at least one selected from 0.1 to 20 parts by weight, preferably 0.5-10 parts by weight relative to 1 part by weight of amlodipine. Can be included 3 ⁇ 4.
- Preferred enteric polymers in atorvastatin / amlodipine formulations are hydroxypropylmethyl.
- Cell 3 ⁇ 4 low phthalate, arc 3 ⁇ 4 methyl-acrylic acid co-polymer, methacrylic acid-methyl methacrylate co-polymer, polyvinyl alcohol phthalate, polyvinylacetacetal phthalate are used, and more preferably hydroxypropyl methyl May be at least one selected from cellulose phthalate S, methacrylate acrylic acid-methacrylic acid copolymer, methacrylic acid-methacrylic acid 3 ⁇ 4 acid tetramethyl co-polymerization, 0.1 to 20 parts by weight relative to 1 part by weight of amlodipine, preferably Can be included in 0.5 ⁇ : 10 increments.
- enteric polymers in the pitavastatin / amlodipine formulations include arc 3 ⁇ 4 acid copolymers, preferably methyl methacrylate acrylic acid copolymers (product name, acryl-is). It may be included in an amount of 0.1 to 20 parts by weight, preferably 0.5 to 10 parts by weight.
- Preferred enteric polymers in the Rochevastatin / Amlodipine formulation are at least one selected from methylmethacrylic acid co-polymer and hydroxypropylmethylcell of rosophthalate, and are included in an amount of 0.1-20 parts by weight, preferably 0.5-10 parts by weight, relative to amlodipine. If less than 0.1 parts by weight, there is a problem that is easily dissolved at pH ' below 5, and if more than 20 parts by weight there is a problem that the total weight of the formulation is unnecessarily increased or excessively delayed dissolution.
- enteric cellulose derivatives In the atorvastatin / nifedipine preparations, enteric cellulose derivatives, enteric acrylic acid co-polymers, and enteric pulley vinyl derivatives are preferably used, and more preferably hydroxypropylme 3 ⁇ 43 ⁇ 4rollosephthalate and hydroxypropylmerolo.
- Osacetate succinate, methacrylic acid-methacrylic acid 3 ⁇ 4 copolymer may be used and may be included in an amount of 5 to 150 parts by weight, preferably 10 to 50 parts by weight, based on 100 parts by weight of nipadidi pin.
- a water-insoluble polymer is a polymer which is insoluble in pharmaceutically acceptable water to control the release of the drug.
- the water-soluble polymer is preferably a polyvinylacetate; a polymethacrylate co-polymer; a pulley (ethyl acrylate-methyl methacrylate) co-polymer; a poly (ethyl arc ⁇ late-methyl methacrylate 3 ⁇ 4late ⁇ trimethylaminoethyl methacrylate) copolymer, ethyl cellulose, cellulose acetate, more preferably polyvinyl acetate, poly (ethyl arc methacrylate—meth methacrylate Late trimethylamino 3 ⁇ 4 methacrylate) copolymer, polybutak 3 ⁇ 4 tate co-polymer, poly (3 ⁇ 4 acrylate-methyl methacrylate) copolymer, and the like can be used. —30 parts by weight, preferably from 0.5 to 20 parts by weight.
- the water-insoluble polymer is preferably polyvinyl acetate, Eudragit RS P0, ethyl shellose, sal-rose acetate, and more preferably polyvinylacetate, Eudragit RS. P0, ethyl cellulose may be used, and may be included in an amount of about 0.1-30 parts by weight, preferably 0.5-20 parts by weight, based on 1 part by weight of the active ingredient amlodipine.
- the water-insoluble polymer may preferably be an acrylic copolymer roll, more preferably Eudragit RS30D, and 0.1-30 parts by weight, preferably 0.5-, based on 1 part by weight of amlodipine. It may be included in 20 parts by weight.
- the soluble polymer is composed of 3 ⁇ 4ribinal acetate (Collicott S 30D), saloose acetate and ply (ethyl arc 3 ⁇ 4rate-methyl methacrylate-trimethylaminoethylmethac 3 ⁇ 4). It is preferable that at least one selected from the rate) co-polymer (Eudragit RS30D), it can be included in the amount of 0.1-30 parts by weight, preferably 0.5-20 parts by weight relative to amlodipine.
- the water-soluble polymer preferably uses an acrylic acid copolymer, more preferably a poly (3 ⁇ 4H-acrylate-methyl methacrylate-trimetha3 ⁇ 4aminoethylmethacrylate) copolymer ( Eudragit RS30D and Eudragit LR30D) and may be included in an amount of 5 to 200 parts by weight, preferably 10 to 100 parts by weight, based on 100 parts by weight of nifedipine.
- an acrylic acid copolymer more preferably a poly (3 ⁇ 4H-acrylate-methyl methacrylate-trimetha3 ⁇ 4aminoethylmethacrylate) copolymer ( Eudragit RS30D and Eudragit LR30D) and may be included in an amount of 5 to 200 parts by weight, preferably 10 to 100 parts by weight, based on 100 parts by weight of nifedipine.
- hydrophobic compounds refer to substances which are insoluble in the pharmaceutically acceptable water controlling the release of the drug, such as fatty acids and fatty acid esters, fatty acid alcohols, waxes, inorganic substances, and And at least one selected from the group consisting of these mixtures.
- the exciple fatty acids and fatty acid esters are at least one selected from glyceryl palmitostearate, glycerol 3 ⁇ 4 stearate, glyceryl bihenate, cetyl palmitate, glyceryl monooleate, stearic acid, and combinations thereof;
- Fatty acid alcohols include one or more selected from cetostearyl alcohol, cetyl alcohol, stearyl alcohol, and mixtures thereof;
- the waxes may be at least one selected from carnauba wax, beeswax, microcrystalline wax, and mixtures thereof.
- Inorganic materials may be used at least one selected from talc, precipitated carbonated carbon, hydrogen hydrogen phosphate, zinc oxide, titanium oxide, carbonine, bentonite, ponmo 3 ⁇ 4 ronite, non-gum and their common substances.
- the hydrophobic compounds preferably contain fatty acid esters, glyceryl palmitostearate, glyceryl stearate, glyceryl nonarate, more preferably gloxe 3 ⁇ 4 stearate, glycerol bihe Nate may be used and may be included in an amount of 0, 1-20, and preferably 0,5-10, relative to 1 part of amlodipine.
- the hydrophobic compound preferably uses fatty acid esters, fatty acid alcohols, waxes, inorganic materials, more preferably fatty acid esters, fatty acid alcohols, and amlodipine. 0.1-20 parts by weight, preferably 0, 5-10 parts by weight based on 1 part by weight.
- the hydrophobic compound is preferably fatty acid esterolol, more preferably glycerol 3 ⁇ 4 stearate, and 0.1 to 20 parts by weight, preferably 0.5 to 1 part by weight of amlodipine. It can be included in 40 increments.
- the hydrophobic compound may be included in an amount of 0.1-20 parts by weight, preferably 0.5-10 parts by weight, relative to amlodipine.
- the hydrophobic compounds are preferably in fatty acids. More preferably, glycerol 3 ⁇ 4 stearate may be used, and may be included in an amount of 5 to 300 parts by weight, preferably 15 to 100 parts by weight, based on 100 parts by weight of nife dipine.
- glycerol 3 ⁇ 4 stearate may be used, and may be included in an amount of 5 to 300 parts by weight, preferably 15 to 100 parts by weight, based on 100 parts by weight of nife dipine.
- the hydrophilic polymer is a pharmaceutically acceptable water-soluble polymer that controls the release of the drug, sugars, 3 ⁇ 4 roll derivatives, gums, proteins, poly bi3 ⁇ 4 derivatives, polymethacrylate copolymers, 3 ⁇ 43 ⁇ 4 derivatives and carboxyvinylated polyethers can be selected and used as the saccharides, specifically dextrins, 3 ⁇ 4 liddextrins, dextrans, peck3 ⁇ 4 and peck derivatives, alginates, pulligaltaxuronic acids, xylans, arabinoxylans, Arabinogalactan, starch, hydrated propyl starch, amylose, amylopected, etc.
- cellulose derivatives can be selected, and as cellulose derivatives, hydroxypropylme 3 ⁇ 4cellose, hydroxypropylpropyl ** loose , Hydroxymethylcellose, hydroxy to 3 ⁇ 4 ⁇ rose, methylsalorose, carboxymethylshell loose, romium, hydroxypropyl methylcell Osacetate succinate, hydroxyethylmethylsulloose, etc.
- gums guar gum, locust bean gum, tragacanta, carrageenan, acacia gum, gum arabic, gellan gum, xanthan gum, etc.
- gelatin, casein, zein and the like can be used as a protein
- polyvinyl alcohol, polyvinyl pyrrolidone and polyvinyl acetal diethylamino acetate and the like can be selected and used as a polyvinyl derivative
- poly (3 ⁇ 4-acrylate to methacrylic acid) copolymers can be selected and used as polyethylene derivatives, such as pulley terylene glycol, Lithium oxide and the like can be selected and used, and carbomer can be used as the carboxyvinyl pulley.
- the hydrophilic polymer is preferably starch, hydroxypropylmethylshell, hydroxypropylshell, carboxymethiolose, sodium nitrate, xanthan gum, polyvinyl alcohol or Carbomer may be used and may be included as 0.05-30 parts by weight, preferably 0.5-20 parts by weight, relative to 1 part by weight of Amrodda 3 ⁇ 4.
- the hydrophilic polymer is preferably a cell using a loose derivative, a polyvinyl derivative, a carbocyvinyl polymer, a pulley ethylene derivative, more preferably hydroxypropyl ⁇ loose.
- Hydroxypropyl methyl cell Hydroxypropyl methyl cell, cellulose acetate succinate, polyvinylpyridone, carbomer, pulley ethylene oxide can be used and 0.05 to 30 parts by weight, preferably 0.5 to 20 parts by weight relative to 1 part by weight of amlodipine. have.
- the hydrophilic polymer may preferably be carboxyvinyl 3 ⁇ 4 mer, more preferably carbomer, 0.05 to 30 parts by weight, preferably 1 part by weight of amlodipine, preferably It may be included in an amount of 0.5 to 20 parts by weight.
- the hydrophilic polymer preferably comprises a hydroxypropylcelose and / or a polymethathirate copolymer, more preferably a pulley (methacrylic acid methylmethylmethacrylate).
- S may be used and may be included in an amount of 0.05 to 30 parts by weight, preferably 0.5 to 20 parts by weight, for 1 part by weight of amlodipine.
- the sing-group hydrophilic polymer preferably contains a cell as a loose derivative, a carboxyvinyl plymer, more preferably 3 ⁇ 4 salicylate propylme loose, Carbomers may be used and may be included in an amount of from 5 to 200 parts by weight, preferably 10 to 100 parts by weight, based on the weight of nizadipine loo.
- the preferred release control agents are polyvinylacetate, fullime 1-tacrylate co-concentration ⁇ poly (ethyl arc 3 ⁇ 4rate, decyl methacrylate 3 ⁇ 4tate, trimeth 3 ⁇ 4 amino to 3 ⁇ 4 methacrylate S ), Copolymer ⁇ , Carboxy vinyl hornmer, 3 ⁇ 4 Cellulose, "Loose Acetate, Carboxyme 3 ⁇ 4 Cellulose Sodium, Polyethylene Oxide Hydroxypropyl Methyl Chloride, Hydroxylpropyl Cellulose, And at least one selected from the group consisting of hydroxypropyl methylcells and phthalates, or selected from the group consisting of carboxyvinal polymers
- Preferred release control substances for atorbasin / tin / amlodipine formulations include polyvinylacetate, polymethacrylate co-polymer, carboxyvinyl polymer, hydroxypropylmetholose, hydroxypropyl3 ⁇ 4 At least one selected from the group consisting of cellulose, 3 ⁇ 4 roll acetate, ethyl cellulose, polyethylene oxide, and hydroxypropyl methyl pentose phthalate carboxyvinyl plymer, polymeth 3 ⁇ 4late ⁇ polymer and And a mixture of carboxyvinyl plymer and hydroxypropylme 3 ⁇ 4cell selected from the group consisting of hydroxypropylmethylcellose.
- the preferred release control substances are hydroxypropylcellose, hydroxypropylmethylsalose, hydroxypropylmethylcellosephthalate, shell ⁇ rose acetate, polyvinyl At least one selected from the group consisting of acetates, polymethacrylic acid copolymers, ethylcel ' loose, li (methacrylate methylmethacrylate) copolymers and methyl methacrylate methacrylic acid copolymers, or One is selected from the group consisting of hydroxypropylcelose, cellulose acetate, ethylcellose, poly (meth3 ⁇ 4late methylmethacrylate) copolymer.
- the preferred release controlling substances are hydroxyspecificpropylsalose, hydroxyethoxypropylmerose, hydroxypropylmethyl 3 ⁇ 4rollosephthalate, pylooseaceti 1, selected from the group consisting of eth, polyvinylacetate, fully methacrylate copolymer, ethyl 3 ⁇ 4 loose, fully (methacrylate, methyl methacrylate) copolymer, and methacrylic acid 3 ⁇ 4 terpacrylic acid copolymer Or the one or more selected from the group consisting of hydroxy propyl shellulose, salulose acetate, ethyl gelose, poly t decacrylate, and meth methacrylate).
- preferred release control substances are hydroxypropyl methyl 3 ⁇ 4 cell cellulose phthalate, hydroxy propyl methyl cell cellulose acetate succinate, methac 3 ⁇ 4 acid-methacrylate methyl copolymer, poly (ethylacryl Suntec from the group consisting of latex-methyl methacrylate-tritetyladinoethylmethacrylate), a glycerol 3 ⁇ 4 stearate, hydroxy propylmethylshell, hydroxypropyl3 ⁇ 4, and carbomer
- the release controlling substance is hydroxypropylmethylsal, loose phthalate, hydroxypropylmethylcell, loose acetate.
- enteric polymers selected from succinate or methacrylic acid methacrylic acid methacrylic acid copolymers and 3 ⁇ 41 hydroxypropylmethelose, hydrophilic polymers selected from cellulose, hydroxypropyl3 ⁇ 4ose, or carbomer .
- enteric polymers selected from succinate or methacrylic acid methacrylic acid methacrylic acid copolymers and 3 ⁇ 41 hydroxypropylmethelose, hydrophilic polymers selected from cellulose, hydroxypropyl3 ⁇ 4ose, or carbomer .
- the delayed-release compartment of the present invention includes an osmotic pressure control agent and may be a compartment coated with a semipermeable membrane coating base.
- osmotic seclective refers to components used to control the release rate of drugs using the principle of osmotic pressure, such as' magnesium sulfate, magnesium chloride, sodium chloride, calcium chloride, sodium phosphate, And at least one member selected from the group consisting of calcium phosphate, ammonium acetate, lithium chloride, sodium sulfate, sodium sulfate, sodium sulfate, and combinations thereof.
- sodium chloride, potassium chloride, sodium phosphate *, calcium phosphate can be used.
- the osmotic pressure regulator is 0.01 to 10 parts by weight, preferably 0.05 to 5 parts by weight, based on 1 part by weight of amlodipine, atorvastatin / amlodipine formulations, pitavastatin / amlodipine formulations or roschvastatin /
- 0.05 to 30 parts by weight, preferably 0.1-20 parts by weight, relative to 1 part by weight of amlodipine, in the atorvastatin / nifedipine formulation, in the amount of 5 to 150 ⁇ , preferably 10 to 100 parts by weight may be included.
- the semipermeable membrane coating base is a pharmaceutically usable coating base, which is used in the coating layer of the pharmaceutical formulation to form a membrane which allows some components to pass but does not pass other components.
- the above-mentioned water insoluble polymer may also be used.
- the semi-permeable membrane skipping agent is, for example, polyvinyl acetate, plymethac 3 ⁇ 4 lay ⁇ copolymer, ply (ethyl arc 3 ⁇ 4 rate, meth hexa decacrylate) copolymer, pulley (ethyl arc 3 ⁇ 4 acetate, detyl meth 3 ⁇ 4). , Trimeth 3 ⁇ 4 aminoethyl meth 3 ⁇ 4 rate) copolymer, ethyl cellulose, cellulose.
- the semipermeable membrane coating agent is preferably cellulose acetate, polyvinyl acetate, cellulose acetate, ethyl cellulose, polymethacrylate co-polymer, 1 part by weight of amlodipine. 0.01 to 10 parts by weight, preferably 0.05 parts by weight to 1.25 parts by weight.
- the semipermeable membrane coating agent is preferably a poly (ethyl ac 3 ⁇ 4, methyl methacrylate: trimethylaminoethyl methacrylate) co-polymer, cellulose ester, ethyl Salose, cellulose acetate may be used, and may be included in an amount of 0.5 to 2Q, preferably 1 to 10 parts by weight, relative to 1 part by weight of amlodipine.
- the semipermeable membrane coating base may preferably be cellulose acetate and may be included in an amount of 0.1 to 20 parts by weight, preferably 1 to 10 parts by weight, based on 1 part by weight.
- the semipermeable membrane coating agent may be included in an amount of 0.05 parts by weight, 30 parts by weight, preferably 0.1 parts by weight to 20 parts by weight, based on 1 part by weight of amlodipine.
- the semipermeable membrane coating agent may preferably use 3 ⁇ 4 rhorose acetate and may be included in an amount of 5 to 2000, and preferably 10 to 500, based on 100 parts by weight of nifedi 3 ⁇ 4.
- the formulations of the present invention are referred to as pharmaceutically acceptable substances of (2-1) or (2 ⁇ 2) without diminishing the effects of the present invention, diluents, binders, disintegrants, lubricants, pH thereof.
- Commonly used additives such as crude agents, antifoams, and solubilizers can be formulated by further use without departing from the nature of delayed release.
- starch, microcrystalline shellulose, lactose, glucose, mannitol, alginate, alkaline earth metal salts, clay, polyethylene 3 ⁇ 4 recall, decalum phosphate, or a combination thereof can be used as a diluent.
- starch As a binder, starch, microcrystalline salose, highly disperse silica, manny, sucrose, lactose, polyethylene glycol, polyvinylpyridin, hydroxypropylmethylcellose, hydroxypropylcellose, natural gum, synthetic Gum, copovidone, povidone, gelaline, or a combination thereof may be used,
- starch or modified 3 ⁇ 4-minute starch such as sodium starch glycolate, corn starch, potato starch or starch gelled starch, bentonite > montmorillonite, or vegeum, etc.
- Cells such as salose or carboxymethyl gelose Crosslinked polyvinyl chloride such as sodium alginate or alginic acid Alginate croscarmel lose sodium such as alginic acid 3 ⁇ 4 Gurose gum such as guar gum and xanthan gum
- Crosslinked polymers such as pyridone (crospovidone), effervescent agents such as sodium bicarbonate, citric acid, or a mixture thereof can be used.
- Lubricant destearic acid, stearic acid magnesite, stearic acid kum, lauryl sulfur sulphate fl ", hydrogenated vegetable oil, natzoate, colloidal silicon dioxide, sodium stearyl fumarey S, glycerol 3 ⁇ 4 behenate, glycerol Peel mono-rays, ⁇ riceryl monostearate, glyceryl gulmitostearate and polyethylene glycol may be used.
- the £ control agent is an acidifying agent such as acetic acid, adipic acid, ascorbic acid, malic acid, succinic acid, tartaric acid, fumaric acid, citric acid, and a basic agent such as precipitated calcium carbonate, ammonia water, and meglumine. Etc. can be used.
- the antifoaming agent may use dimethicone, oleyl alcohol, propylene glycol alginate, simethicone such as simethicone emulsion, and the like.
- the dissolution aid can be used pleoxyethylene sorbitan fatty acid esters such as sodium lauryl sulfate, polysorbate, docuate or lithium.
- additives selected from colorants, perfumes may be selected to formulate the formulations of the present invention.
- the range of additives that can be used in the present invention is not limited to the use of such additives, and one of the additives may be formulated to contain a range of dosages by selection,
- purified water, ethane, methylene chloride, and the like may be used as the solvent of the binder and the delayed-release additive, but preferably, purified water or ethane may be used.
- the available 3 ⁇ 4 exclusion range is not limited to the use of such additives, and the above-mentioned additives may be formulated to contain a range of dosages by choice.
- the delayed-release compartment of the present invention consists of particles or granules obtained by mixing, granulating or coating a dihydropyridine-based calcium antagonist, a time difference controlling substance and a conventional additive used in pharmaceuticals.
- the pre-release compartment of the present invention is made of granulated black granules through a conventional process for producing oral solids such as mixing, coalescing, drying and granulation, together with a statin-based lipid lowering agent, together with pharmaceutically acceptable additives. If the fluidity of the statin-based lipid-lowering agent mixture can be directly tableted, the composition can be obtained by mixing. If the fluidity is not *, the composition can be obtained by compression, granulation, and granulation. Thus it can be configured as a pre-release compartment.
- the present invention provides a single tablet such as a biphasic matrix tablet, a multilayer tablet, a nucleated tablet containing a delayed-release dihydropyridine-based antagonist discontinuous phase in a continuous-release statin-based lipid lowering agent continuous phase as described below. It can be carried out by the production method and administration method, but is not limited thereto.
- the granules constituting the delayed-release compartment and the prior-release compartment are mixed with pharmaceutical additives, and the tablets are compressed into double tablets or three tablets in parallel, using multiple tableting machines. Possible tablets for oral administration can be obtained.
- the granules constituting the delayed-release compartment may be mixed with a pharmaceutical additive and tableted to form a nucleus tablet, or the pharmaceutical coating may be applied to exhibit delayed-release properties. After mixing with a conventional additive and tableting as an outer layer, a tablet for oral administration can be obtained in the form of delayed release to the inner nucleus and a form in which the inner layer is surrounded by the inner layer.
- the granules constituting the delayed-release compartment may be mixed and compressed with pharmaceutical additives or the drug layer constituting the prior-release compartment may be water-soluble after applying a special pharmaceutical coating even if it exhibits delayed release. After dissolving and dispersing in a film coating solution Tablets for oral administration can be obtained by coating the layers of the delayed-release tablet layer.
- the osmotic material is contained in the tablet, and then compressed into tablets. Then, the surface of the tablet is coated with a semi-permeable polymer to form a nucleus tablet.
- a pharmaceutical additive and tableting as an outer layer to obtain a tablet for oral administration in the form of a delayed-release compartment into the inner core and the immediate release layer on the surface of the inner core.
- the present invention relates to a delayed-release compartment and a prior-release compartment comprising two-phase granules, radial granules or pellets and delayed-release tablets, delayed-release granules or pallets and pre-release tablets, and delayed-release tablets and pre-release tablets. It can be carried out by the production method and administration method of the layered camsul agent, but is not limited thereto.
- the granules constituting the delayed-release second agent and immediate-release first agent according to the present invention are mixed with pharmaceutical additives as needed to layer the capsules in a controlled release. Possible capping agent can be obtained.
- the granules constituting the delayed-release compartment are coated as they are or tableted to show delayed release, and the tablets are compressed or coated after the tablets are granulated or coated.
- the tablets may be layered on the capsule to obtain a capsular agent capable of time release.
- the granules constituting the open-release compartment are coated as they are or tableted to show delayed release, and the powder, granules or pellets constituting the pre-release compartment are laminated on the capsule to enable time lag release. I can get sage,
- the granules constituting the pre-release compartment is compressed into tablets and, if necessary, prepared into tablets
- the granules and 3 ⁇ 43 ⁇ 4 constituting the delayed-release compartment may be layered on the capsule to obtain a capsulant capable of time release.
- the granules or pellets constituting the delayed-release compartment and the sacking or pellets constituting the prior-release compartment may be laminated on the capsule to obtain a capsulant capable of timed release.
- the osmotic material is contained in the tablet, and then compressed into tablets. Then, the surface of the tablet is coated with a semipermeable polymer to form a nucleus tablet. Water can be mixed with pharmaceutical additives and then laminated to the capsule to obtain a timed release agent *.
- the present invention separately prepares granule pellets or pre-release tablet layers constituting the prior release compartment, and calls for granules or delayed-release tablet layers constituting the delayed release compartment. It can be manufactured as a kit that can be layered together at work, blister, bottle, etc.
- the pharmaceutical preparations of the present invention can be formulated in a variety of formulations and can be formulated, for example, as viscous tablets, coated tablets, multilayer tablets, or nucleated tablets, powders, granules, or capsulants.
- the pharmaceutical formulations of the present invention may be in the form of a two-phase matrix tablet consisting of delayed-release compartments—pre-releasing compartments surrounding them.
- the matrix of the delayed-release compartment and the prior-release compartment may be prepared as a preparation for oral administration by post-mixing or compressing the pharmaceutically acceptable additives in the material constituting the delayed-release compartment and the pre-release compartment.
- Two phase S present formulations The pharmaceutical formulation of the present invention may be in the form of a two-phase matrix tablet obtained by slicing after uniformly mixing the delayed-release compartment and the prior-release compartment, and preferably, the delayed-release compartment is prepared in a granular form. It is.
- the pharmaceutical formulation of the present invention may be in the form of a film skipping tablet consisting of a slow-release compartment and a film-coating layer consisting of a pre-release compartment surrounding the denture of the tablet, the film coating layer as the film coating layer is dissolved Simvastatin will be eluted.
- the pharmaceutical formulation of the present invention is a delayed-release compartment obtained by mixing the pharmaceutical additives in the fruits and forests constituting the delayed-release compartment and the prior-release compartment, and tableting in two tablets or three tablets using a multiple tableting machine.
- the pre-release compartment may be multi-layered form forming a multi-layer structure.
- This formulation is a tablet for oral administration specially formulated for pre-release and delayed release in layers.
- the pharmaceutical formulation of the present invention may be a nucleated tablet consisting of the inner layer consisting of a delayed-release compartment and a layer consisting of a prior-release compartment enclosing the surface of the inner core
- the nucleated tablet may be an osmotic nucleated tablet.
- the osmotic nucleus tablet is a tablet containing the osmotic pressure-controlling agent inside the tablet for delayed release and then tableted, and then the surface of the tablet is coated with a semi-permeable film coating base to form the inner core, and the granules constitute a pre-release compartment.
- the pharmaceutical formulations of the present invention may comprise particles, granules, pal3 ⁇ 4, or It may be a capsular formulation comprising particles, granules, pellets, or tablets consisting of tablets and prerelease compartments.
- the formulation of the present invention may further form a coating layer on the outside of the delayed-release compartment and / or the prior-release compartment. That is, the surface of the particles, granules, pellets, or tablets composed of delayed-release compartments and / or pre-release compartments may be coated for the purpose of release control or formulation stability.
- the pharmaceutical formulation of the present invention may be in the form of a kit comprising a delayed-release compartment, and a prior-release compartment.
- the present invention prepares particles, granules, 3 ⁇ 43 ⁇ 4, or tablets constituting the pre-release compartment, separately prepares fruiting, 3 ⁇ 43 ⁇ 4 or tablets constituting the delayed-release compartment, foils, blisters, bottles, etc. It may be in the form of a kit prepared in a form that can be filled together and taken simultaneously.
- the formulation according to the present invention is also provided in a state such as uncoated tablets without additional coating, but may be a formulation in the form of a coated tablet further comprising a coating layer by forming a coating layer on the outside of the formulation, if necessary.
- a coating layer By forming the coating layer, it is possible to provide a formulation that can further ensure the stability of the active ingredient.
- the method of forming the coating layer can be appropriately selected by the choice of a person skilled in the art in the method of forming the coating layer outside the film on the surface of the tablet layer, a method such as a fluidized bed coating method, a fan coating method, can be applied, preferably For example, 3 ⁇ 4 coating can be applied.
- the coating layer may be formed by using a coating agent, an auxiliary coating agent, or a combination thereof.
- the coating agent may be a 3 ⁇ 4 loose derivative such as hydroxypropyl methyl salose, hydroxy propyl propyl cellulose, a sugar derivative, or a 3 ⁇ 4 lye.
- Vinyl derivatives, macrophages, fatty acids, gelatin, or mixtures thereof, and the like, and coating aids include polyethylene glycol, ethyl salose, 3 ⁇ 4 riselides, titanium oxide, talc, diethyl phthalate, or mixtures thereof. A mixture or the like can be used.
- the coating layer may be included in the range of 0.5 to 15% by weight (% w / w) relative to the total tablet weight increase.
- the present invention also provides a pharmaceutical formulation according to the present invention for evening administration.
- lipid synthesis in the liver becomes vigorous after early dinner, and the general population including people with hypertension Extravasation "falls between night and dawn, and blood pressure rises in the morning after waking, peaking during the day (active).
- the formulation of the present invention is used in the evening
- the pre-release simvastated liver enzyme is administered at the time of activating the enzyme, which shows more lipid lowering effect
- the delayed-release amlodipine effectively lowers the 3 ⁇ 4 pressure after dawn, so that blood pressure can be equalized from morning to morning. It is possible to maintain a competitive mutual antagonism of the drug and to maximize the effect of each active ingredient
- the present invention is a method for treating cardiovascular diseases comprising administering a pharmaceutical formulation of the present invention to a mammal To provide.
- cardiovascular disease is applied to hypertension, or hypertension and complications of those with metabolic syndrome, such as diabetes mellitus, obesity, high altitudes, and coronary artery disease.
- Pharmaceutical formulations of the invention may be used in any suitable manner in the art, for example
- Chrontherpeut ics 2003, Peter Redfern, PhP
- it can be preferably formulated according to each disease or component, and specifically can be prepared by the method comprising the following steps. .
- amalodipine is mixed with an enteric polymer, a water-insoluble polymer, a hydrophobic compound, or one or two selected release controlling substances selected from hydrophilic polymers, and a conventional 3 ⁇ 4 gauze used in pharmacy. Delayed-release granules or tablets are obtained by granulation or coating, and tableting, or by mixing, combining, drying, granulating, or tableting amlodipine with osmotic pressure ⁇ ablation and the usual additives used in pharmaceutical preparations. Coating to obtain delayed-release granules or tablets.
- the second step consists in administering simvastatin and conventionally acceptable pharmaceutically acceptable additives, followed by conventionally released granules through conventional processes for producing oral solids via mixing, association, drying, granulation, black coating, and tableting, or Obtaining tablets.
- the granules or tablets obtained in the first step and the second step are mixed with a pharmaceutical excipient, tableted or filled to obtain a preparation for oral administration.
- the first step and the second step may be reversed or executed simultaneously.
- the pharmaceutical formulation of the present invention may be prepared by the above process, and the formulation method of the third step will be described in more detail as follows, but is not limited thereto. [A] Preparation of two-phase matrix tablet
- the tablets are obtained by mixing with the granules prepared in the second step and compressing them with a certain amount of weight.
- the resulting tablets may be film coated as needed for the purpose of improving stability or properties.
- the after-coated tablets or add the granules to a release-controlling material coating and appointed other by a predetermined amount and dried as black obtained in step 1 is manufactured to obtain the coating in addition, after the simvastatin to the film coating solution of the water-soluble corresponding 'dissolution
- an oral dosage form film-coated tablet containing the active ingredient in the film coating may be prepared.
- the granules obtained in the first step can be prepared as is or in a double tablet using a tableting machine.
- Coated multilayer tablets can be prepared by formulating or coating triple or more multi-layered tablets by adding a release complement S layer as needed, or by formulation design.
- the coated tablets or granules obtained in the first step are additionally coated as they are or with a release control material, dried, and then compressed into a predetermined amount, which is then further coated with black as an inner core, followed by a nucleated tableting machine together with the granules obtained in the second step.
- the coated nucleated tablet may be prepared by coating or preparing a nucleated tablet in the form of a pre-release layer submerged on the surface of the first-stage tablet by tableting.
- the granules obtained in the first step are additionally coated as is or with a release control material, and the dried granules or tablets and the granules or systems obtained in the second step are placed in a capsular bed electric machine and layered by an effective amount of each active ingredient in 3 ⁇ 4 sul of a certain size. Capsules can be prepared.
- 3 ⁇ 4 slabs can be prepared by layering the capsules with a capsul layered device.
- the amlodipine-containing preparation obtained in the first step and the simvastatin-containing preparation obtained in the second step can be prepared as a kit that can be layered together in a foil, blister, bottle, or the like.
- the complex drug system of the present invention as described above is formulated into a complex formulation including a dihydropyridine-based Calm channel blocker and a statin-lipid lowering agent as an active ingredient to separately formulate each active ingredient by administering only one hour in the evening It is easier to guide the medication than to take it at the time of moxibustion, and it can reduce the side effects due to the metabolic interference action between drugs, and the shale control and lipid control effects of each drug are their own. Eggplant appears to be better than the effect.
- statin lipid lowering agent represented by simvastatin
- dihydropyridine type antagonist represented by amlodipine
- the present invention is a so-called time difference dosing treatment (Chronotherapeutics) to maximize the therapeutic effect on the basis of heterogeneous pharmacokinetics Ofenobiotics to improve the side effects that can occur from the combination of different drugs from the pharmacokinetic point of view
- Chronotherapeutics a so-called time difference dosing treatment
- It is formulated as an effective active ingredient of dihydropyridine-calcin antagonist and statin-based lipid lowering agent, which affects or receives the same enzyme cytochrome P 450 system, and they are eluted from the body.
- according to the present invention can be configured differently the release rate of the drug to prevent the antagonism and side effects between the drugs and at the same time can take the synergism of the drug.
- the complex preparation can be taken at a time, the medication guide for the patient and the patient's medication are easy.
- compositions comprising a pre-release compartment containing a statin-based lipid lowering agent of the present invention and a delayed-release compartment containing a dihydropyridine-based calcium channel blocker may be used for patients with metabolic syndrome and insulin resistance and diabetes or diabetes mellitus.
- a statin-based lipid lowering agent of the present invention may be used for patients with metabolic syndrome and insulin resistance and diabetes or diabetes mellitus.
- a delayed-release compartment containing a dihydropyridine-based calcium channel blocker may be used for patients with metabolic syndrome and insulin resistance and diabetes or diabetes mellitus.
- Example 1 is a graph showing the comparative elution curves of amlodipine / simvastatin nucleated tablets prepared in Example 1-1 and a control drug (Curve?: Simvastatin monotherapy, Novask: amlodipine monotherapy).
- Fig. 2 is a graph showing the curves of comparative coagulation between the combination preparation of amlodipine / simvastatin prepared in accordance with Examples 1-4 and 10 and the reference drug (Zoko: Simvastad single agent, Novask: Amlodipine single agent).
- Fig. 4 is a graph showing the comparative dissolution curves of the complex preparation of amlodipine / atorvastatin prepared in accordance with Examples 1-13 and the reference drug (lipitor: atorvastatin monotherapy, Novask: amlodipine monotherapy).
- FIG. 5 is a graph showing the comparative elution curves of the lercanidi 3 ⁇ 4 / simvastatin complex preparation prepared according to Examples 1-16 and the control agent (Zoko: simvasta3 ⁇ 4 monotherapy, zanidib: lercanidipine monotherapy).
- FIG. 6 is a graph showing the comparative elution curves of the laxidipine / simvastatin complex preparation prepared according to Examples 1-18 and the control drug (Zoko: simvapatin monoclonal, Dr.r: lacidipine monolith).
- Fig. 7 is a graph showing the curves of all the elution of amlodipine besylate / simvastarin nucleated tablets prepared according to Examples 1-20 and the comparative drug (Zoko: simvastatin monotherapy, MSD, Novasque: amlodipine monotherapy, Pfizer).
- FIG. 8 is a comparative elution of a capsul agent containing an amlodipine pulsate / simvastatin combination tablet and a control agent (Zoko: simvastatin monotherapy, MSD, Novask: amlodipine monotherapy, Pfizer) prepared according to Example 1-22.
- ⁇ This is a graph showing the curve.
- Figure 9 is a combination preparation of amlodipine besylate / atorvastatin calcium prepared in accordance with Examples 1-30 and the control drug (lipitor: atorvasta 3 ⁇ 4 single agent, Pfizer, Novask: amlodi 3 ⁇ 4 single agent, Pfizer) A graph showing the comparative dissolution curves of.
- FIG. 10 is a biphasic preparation in the form of pelodipine / atorvasta 3 ⁇ 4 capsulant prepared in accordance with Examples 1-36 and the control drug (lipitor: atorvastatin monotherapy, Pfizer, non-novol: felodipine monotherapy, hand poison drug)
- lipitor atorvastatin monotherapy, Pfizer, non-novol: felodipine monotherapy, hand poison drug
- FIG. 11 is a biphasic preparation in the form of isradipine / lluvastatin capsules prepared according to Example 1 ′ 4 0 and a control drug (Lescol: Polubasta 3 ⁇ 4 single agent, Novartis, Dynazek: Sradipine Monolithic drug, Daewoong Pharmaceuticals).
- Figure 1 is a graph showing the dissolution test results * (S)-amlodipine / simvastatin complex formulation prepared according to Examples 1-51, 55.
- Figure 13 is a graph showing the dissolution test results of the (S) ⁇ amlodipine / simvastatin composite formulation prepared according to Examples 1-52, 53, 54.
- £ 14 is the clinical trial result according to Experimental Example 1-14 above. It is a graph comparing the blood concentration of hydroxy acid.
- FIG. 15 is a graph comparing blood concentrations of simvastatin ⁇ -hydroxy acid and simvastatin between experimental groups as the clinical test results according to Experimental Examples 1-14.
- Figure 16 is a graph comparing the concentration of amlodipine in the time difference between the administration group and the co-administration group as a clinical test results except the experimental example ⁇ - ⁇ .
- Fig. 17 is a graph showing the comparative elution curves of amlodipine / simvastatin nucleated tablets prepared according to Example II-1 and the reference drug (2: Ko: simvastatin monotherapy, MSD, Novask: amlodipine monotherapy, Pfizer).
- FIG. 13 is a graph showing the comparative elution curves of the pharmaceutical preparation of amlodipine / simvastatin and the control agent (Zoko: simvastatin monotherapy, Novasque: amlodipine monotherapy) prepared according to Examples II-4 and 10.
- FIG. 13 is a graph showing the comparative elution curves of the pharmaceutical preparation of amlodipine / simvastatin and the control agent (Zoko: simvastatin monotherapy, Novasque: amlodipine monotherapy) prepared according to Examples II-4 and 10.
- FIG. 13 is a graph showing the comparative elution curves of the pharmaceutical preparation of amlodipine / simvastatin and the control agent (Zoko: simvastatin monotherapy, Novasque: amlodipine monotherapy) prepared according to Examples II-4 and 10.
- FIG. 13 is a graph showing the comparative elution curves of the pharmaceutical preparation of amlodipine / simvastatin and the control agent
- FIG. 19 is a graph showing the comparative elution curves of amlodapine besylate / simvastatin nucleated tablets prepared according to Example II-11 and a control drug (Zoko: simvastatin monotherapy, Novask: amlodipine monotherapy).
- FIG. 20 is a graphidi showing a comparative dissolution curve of a capsule containing an amlodi 3 ⁇ 4 besylate / simvastatin complex tablet prepared according to Example 11-13 and a control agent (Zoko: simvastatin monotherapy, Novask: amlodipine monotherapy) .
- FIG. 21 is a graph showing the comparative elution of amlodipine / atorvastatin nucleated tablets prepared according to Example III-1 and a control agent (Ripto: atorvastatin monotherapy, Novask: Amlodi 3 ⁇ 4 monotherapy).
- FIG. 22 is a graph showing the dissolution patterns of amlodipine of Examples III-1, 2 and 3, and
- FIG. 23 is a graph showing the dissolution profiles of Examples III-6 and 7.
- FIG. 24 is a graph showing the Atorvasta 3 ⁇ 4 3/4 concentration-time profile of Example III-1.
- Example III-1 is a graph showing the amlodi 3 ⁇ 4 hyperemia concentration ⁇ time profile of Example III-1.
- FIG. 27 is a graph showing the amlodipine elution aspect of Examples 2, 3.
- 28 is a graph showing the amlodipine elution pattern of Examples IV-17 and 18.
- FIG. 29 is a graph showing the comparative elution of Roschvastatin / Amlodi 3 ⁇ 4 nucleated tablets prepared versus Example (Cresto: Roschvastatin Monotherapy, Novask: Amlodipine Monotherapy) prepared according to Example V-4.
- FIG. 31 is a graph showing the comparative elution of atorvastatin / nifedipon nucleated tablets prepared according to Example VI-1 and a control agent (lipitor: atorvastatin monotherapy, Procadia XL: nifedipine monotherapy).
- Example 32 is a graph showing the elution aspect of Example VI-6.
- Example 34 is a graph showing the dissolution profiles of Example "VI-10.
- nucleated tablet tableting machine (RUD-1: Ki lian) as the inner core of the amlodipine core tablet and the composition containing simvastatin at a rate of 30 revolutions per minute, hardness 7 ⁇ 9kp, thickness 6.0 ⁇ , a 'compressed into tablets with a diameter 9.5 ⁇ then using a Hi-coater (SFC- 30N, Sejong machinery Co., Ltd. Korea) to form a film-coated layer, thereby preparing a press-coated tablet.
- Example 1-2 Definition of amlodipine-simvastatin nucleated tablet :
- Amodi 3 ⁇ 4 malate and microcrystalline cell were apologized as No. 35 and mixed with a double cone mixer, and then added to a fluidized bed granulator (GPCG 1: Glatt) as shown in Table 1-2.
- Hydroxylpropyl 3 ⁇ 4cell was sprayed to form a granule by spraying the binder solution prepared by dissolving cellulose in water and drying. Again, the granules were coated by spraying a hydroxypropylmethylcell solution containing a phthalate phthalate dissolved in 1: 1 shaker of ethane and methylene chloride.
- Magnesium stearate was added to this mixture and mixed with a final double cone mixer. The final mixture was then rotated at a speed of 30 revolutions per minute using a rotary tableting machine (MRC-33: Sejong), hardness 7 9 kp, thickness 3.0 kPa, diameter 5.5 The tablet was compressed into a nuclear tablet.
- Amlodipine-simvastatin nucleated tablets were prepared in the same manner as in Example 1-1, 3) Tableting and Coating.
- Example 1-3 Preparation of Amlodipon-Simvastatin Nucleated Tablets
- amlodipine malate and microcrystalline cellulose were used as No. 35 Sieve apologies, mix with a double cone mixer, feed into a high-speed mixer, add Colicoat SR30D, combine, and granulate with a sieve No. 20 using an oscillator. It was sifted. Magnesium stearate was added to the final mixture in a double cone mixer, and the final mixture was used at a speed of 30 revolutions per minute using a rotary tablet press (M C-33: Sejong), hardness 7-9 kPa, thickness 3.0 kPa, diameter. A tablet of 5.5 mm was used to give a two-nuclear tablet.
- Example 1-4 Preparation of Amlodipine-simvasta multilayered tablets
- the binding solution made by amplifying amlodipine maleate and microcrystalline shell loose with No. 35 sieve and mixing with a double cone mixer and dissolving hydroxypropyl methyl shellose in water separately The particles were sprayed to form granules and dried.
- the granules were dissolved in 1: 1 shaker of ethane and methylene chloride, the granules were coated by spraying 3 ⁇ 4 hydroxypropylmethylcell with a phthalate phthalate solution. Magnesium stearate was added to this and mixed with the final double conmixer.
- Amlodipine-simvastatin multi-layered tablets were prepared in the same manner as in Example 1-4, 3) Tableting and Coating.
- Example VII-6 Preparation of Amlodipine-simvastared Two-Phase Matrix Tablets
- amlodipine malate and microcrystalline cells were mixed with apple No. 35 with apples, mixed with them, and colloidal SR30D was added to the high speed mixer. After the association, granulation was carried out using an oscillator with No. 20 sieve, and it was dried at 60 I;
- apples simvastatin, microcrystalline 3 ⁇ 4, and Manny were No. 35, and were mixed with a high-speed ⁇ agitator.
- hydroxypropyl shellulose and citric acid were dissolved in the tulle to prepare a binding solution, and the mixture was added to a high speed shaker with the mixture of the main ingredients, and then granulated using an oscillator. It was dried at ° C, and then sieved to No. 20 sieve, and butylated hydrated cyanuric sol was added thereto and mixed.
- Each of the final S compounds prepared above was mixed with a double cone mixer, sodium starch glyconate, After colloidal silicon dioxide was mixed, stearic acid magnesium was added and mixed with a double cone.
- Example 1-7 Preparation of Amlodiated-simvastatin Two-Phase Matrix Tablets
- the amlodipine malate and the microcrystalline cell rhodes were apologized as No. 35 and mixed in a Dubucon mixer as shown in the following Tables 1-2.
- the mixture was introduced into a fluidized bed granulator (GPCG 1: Glatt), and a binder solution, which was prepared by dissolving hydroxypropylmethyl ⁇ loose in water, was sprayed to form granules and dried.
- the granules were sprayed again by spraying Eudragit RS P0 solution dissolved in ethanol and 1: 1 shake solution of methylten chloride.
- Amlodipine-simvastarin biphasic matrix tablets were prepared in the same manner as in Example 1-6, 3) Postmixing, Tableting and Coating.
- Example I Amlodipine-simvastatin biphasic matrix tablets were prepared according to post mixing, tableting and coating methods. Examples 1-9: Amlodipine® simvastared two-phase capsular formulation
- amlodipine maleate and microcrystalline gelose were apologized with No. 35 and mixed in a double cone mixer, and then put into a layered granulator (GPCG 1: Glatt) and colicoat SR30D. Sprayed to form granules and dried.
- lovastatin, microcrystalline cellulose and manny were appled into No. 35 sieve and mixed with a high speed mixer. Separately, dissolve hydroxypropyl 3 ⁇ 4 and citric acid in water to gradate the binder solution, and then combine it with a main component mixture in a high-speed mixer, combine it, and granulate it with a No. 20 sieve using an oscillator. After drying at ° C it was again established as No. 20 sieve. In addition, butanated hydrated cyanosol, starch glycoic acid ' natrop, colloidal silicon dioxide were mixed, and stearic acid magnesium was added to the final mixture in a double cone mixer.
- the tablet was compressed using a multi-layer tablet press (MRC-37T: Sejong). That is, the composition containing the lovastarin is placed in the primary powder feeder, and the composition containing amlodipine is placed in the secondary powder feeder at a speed of 30 revolutions per minute at a speed of 30 revolutions per minute to minimize the infiltration of the layers. It was compressed into 9 kp, 6.0 mm thick, 9.5 mm diameter and formed a film coating layer as a high coater to prepare a multi-layer point.
- MRC-37T Sejong
- Example 1-11 Svastatin pre-release compartment of Example 1-11 and the ingredients and contents shown in Table 1-3 It was prepared according to the preparation method.
- amlodipine-lovastatin multi-layered tablet was prepared in the same manner as in Example 1-11, 3) Tableting and coating method.
- Example 1-13 Preparation of amlodipine-atorvastad multilayered tablets
- atorvastatin, microcrystalline ⁇ loose, and Manni were apologized as No. 35 and mixed with a high speed mixer.
- hydroxypropyl3 ⁇ 4loose and citric acid are dissolved in water to prepare a binding solution, which is mixed with a main component: in a high-speed mixer, and fed together, and then granulated using a No. 20 ceto oscillator. After drying, it was established as No. 20 sieve again.
- Butyl hydroxyanisole, sodium starch glyconate, and colloidal silicon dioxide were mixed therein, and magnesium stearate was added to the final mixture in a double cone mixer.
- Example 1-15 Preparation of Amlodipine-atorvastatin Nucleated Tablets
- Press-coated tablet tableting machine at the rate of the inner core of amlodipine tablet core using (RUD-1 Ki Han) and atorvastatin on the '30 turns per minute to the composition in an outer layer comprising eu, hardness 7 ⁇ 9 kp, thickness of 6.0 ⁇ After tableting to a diameter of 9.5 kPa, a coating layer was formed using a high coater roll to prepare a nucleated tablet.
- Example 1-16 Preparation of Lercanidipine-simvastatin Multi-Layered Tablet
- Apples with lercanidipine and microcrystalline salolosol No. 35 ⁇ as shown in the following Tables 1 to 3 were mixed with a double cone mixer, and the above mixture was added to a granular granulator (GPCG 1: Glatt).
- GPCG 1 Glatt
- hydroxypropylmethylshellulose was dissolved in water and sprayed to form a binder solution, which was then dried to form granules.
- the granules were sprayed with a solution of hydrated propylpropyl ⁇ 3 ⁇ 4cell dissolved in 1: 1 shaker of ethanol and tertylene chloride, and then the granules were coated.
- Magnesium stearate was added to this and mixed with the final double conmixer.
- the tablet was compressed using a multi-layer tablet press (MRC-37T: Sejong). That is The composition containing simvastatin is placed in the primary powder feeder, and the composition containing lercanidipine is placed in the secondary powder feeder at a speed of 30 revolutions per minute, at a speed of 7 to 9 kp, to minimize the incidence of filling.
- the film was hit with a thickness of 6,0 mm 3 and a diameter of 9.5 mm, and a film coating layer was formed as a high coater to prepare a multilayer tablet.
- Example IVII 17 Preparation of Lerke "Nidi 3 ⁇ 4-Simvasta3 ⁇ 4 Multi-Layered Tablets
- lercanidipine and the microcrystalline cell were apologized with a Rooseol No. 35 sieve, mixed with a debulcon mixer, fed into a high-speed mixer, and then added with Colicoat SR30D.
- Granulation was carried out using an oscillator, which was dried at 60 ° C. using a silver water dryer, and then re-formed as No. 20.
- Magnesium stearic acid was added thereto and finally mixed with a double cone mixer.
- Lercanidipine-simvastatin multi-layered tablets were prepared in the same manner as in Example 1-16, 3) Tableting and Coating Method.
- Example 1-18 Manufacture of Lacidipine-simvastatin Multi-Layered Tablets
- the acedipine and the microcrystalline cellulose were apologized with a No. 35 sieve and mixed with a double cone mixer, and the above mixing tool was placed on a homogeneous layered granulator (GPCG 1: Glatt).
- GPCG 1 Glatt
- the mixture was added and sprayed separately, hydroxypropylmethylcell was dissolved in cellulose roll water to form granules and dried.
- the granules were sprayed with a hydroxypropylmeth 3 ⁇ 4cell, which was dissolved in a 1: 1 mixture of ethane and methylene chloride, and sprayed with a phthalate phthalate solution to coat the granules.
- Magnesium stearate was added to this and mixed with the final double cone mixer.
- Lacidipine-simvastatin multi-layered tablets were prepared in the same manner as in Example 1-18, 3) Tableting and Coating.
- Example I-20 Preparation of Amrodafine-Simvastatin Nucleated Tablets
- amlodipine besylate and microcrystalline cells of aloe and decalom phosphate were apologized with No. 35 sieve, mixed with a double cone filter, and added to a fluidized bed granulator (GPCG 1: Glatt).
- GPCG 1 Glatt
- the binding solution made by dissolving hydroxypropylmethylshellose in water was sprayed to form granules and dried.
- Carbomer 71G was added to the granules in a powder form, and then stearic acid was added to the granules, and the resultant mixture was mixed with a final double cone mixer, and the final mixture was mixed with Bundang (MRC-33: Sejong).
- simvastatin, microcrystalline cellulose, corn starch, and lactose were apples in No. 35 as shown in Table 1-4 and mixed with a high speed mixer.
- prepare a binding solution by dissolving hydroxyaluminum salulose and citric acid in water, and incorporating it into a high-speed mixer with the main component mixture, and then coalescing it using an oscillator with a No. 20 sieve. It was dried at 60 ° C. and then sieved to No. 20 sieve again.
- Butylated hydroxyanisole, starch glyconate natate here. s colloidal silicon dioxide was mixed, and stearic acid magnesium was added and finally mixed in a double cone mixer.
- nucleated tablet tablet machine (RUD-1: Ki Han) as the inner core of the amlodipine core tablet and simvastatin-containing composition at a rate of 30 revolutions per minute, hardness 7 to 9 kp, thickness 6.0 mm, diameter 9.5 Nucleated tablets were prepared by tableting with a pan and then forming a film nose layer as a high coater.
- the aloe 3 ⁇ 4 besylate and microcrystalline cell were apologized with S-ose and decalcium phosphate as No. 35 and mixed with a double cone mixer, and hydroxypropylmethylcellose was separately added to water.
- the binder solution was dissolved and sprayed to form granules.
- the granules were coated by spraying a coating solution consisting of 3 ⁇ 4 hydroxypropylmethylshell loose phthalate and acetylated monoglycerides when the granules were dissolved in 1: 1 shaker of ethane and salt methylene. Stearic acid magnesite was added thereto and mixed in the final double cone mixer.
- Example 1-22 Amlodipine tablets-3/4 tablet containing simvastatin tablets
- amlodipine besylate and microcrystalline 3 ⁇ 4 rose were apologized as No. 35, mixed with a double cone mixer, and then added to a granular granulator (GPCG 1: Giatt), and then separately.
- Granules were formed and dried by spraying a binder solution made by dissolving hydroxypropylmethylcell 3 ⁇ 4 rose in water.
- Carbomer 71G was added to the granules in powder form, and then stearic acid and magnesium were added to the final double cone mixer, and the final mixture was mixed with a rotary tablet press (MRC-33: Sejong).
- tableting was carried out with a hardness of 7-9 kp, a thickness of 3.0 mm and a diameter of 5.0 mm.
- hydroxypropyl methylcell (5 weight 3 ⁇ 4) and acetylated monoglyceride solution dissolved in 1: 1 shaker of ethane and methylene chloride and acetylated monoglyceride solution were coated with a high coater. To form a tablet.
- simvastatin, the crystalline salose, and Manny were apples with No. 35 and mixed with a high speed mixer. Separately, hydroxypropyl3 ⁇ 4loose and citric acid are dissolved in water to prepare a binding solution, which is then added to a high-speed mixer combined with the main component mixture, which is then granulated using an oscillator with a No. 20 sieve. After drying at ° C it was again established as No. 20 sieve. Add the mixed hydroxyanisole, sodium starch glyconate, and colloidal silicon dioxide, and add the stearic acid magnesium to the double-mixer for final mixing.
- amlodipine tablets of step 1) and the simvastatin tablets of step 2) were layered onto the hard gelatin capsulization of No. 3 using a capsule layer electric machine.
- simvasta3 ⁇ 4, microcrystalline cellulose, lactose and starch starch were appled with No. 35 sieve and mixed with a high speed mixer.
- hydroxypropylcellolose and citric acid are dissolved in water to prepare a binding solution, which is added to a high-speed mixer with the main component mixture, and then combined.Then, it is granulated using an oscillator with a No. 20 sieve. After drying at 1C it was again established as No. 20 sieve.
- butylated hydroxyanisole, sodium starch glyconate and colloidal silicon dioxide were mixed, and a stearic acid magnesium was added to the final mixture in a double cone mixer.
- amlodipine tablet of step 1) and the simvastatin granules of step 2) were layered into hydroxypropylmethylcell No. 2 in the hard capsule of cellulose using a 3 ⁇ 4 capsule layer electric machine.
- Example 1-24 Amlodipine-Simvastar 3 ⁇ 4 Coated Tablets
- simvastatin As shown in Table 1-4, simvastatin, butyl tated hydroxyanisole, hydrated propylmethylsalose, colloidal silicon oxide, polyethylene glycol 6000, titanium oxide, talc and ethane A pre-release simvastatin coating solution was prepared by dissolving and dispersing in methylene chloride shake solution.
- amlodipine tablets prepared above were administered to the high coater and then first coated with a simvastared coating solution.
- Example I-2 6 Amlodipine-simvastatin blister packaging kit
- Example 1-22 amlodipine-atorvastatin nucleated tablets
- Example 1-28 Amlodipine-atorvastatin multilayer tablet
- Example 1-22 amlodipine-atorvastatin capsule was prepared.
- Example 1-30 Amlodipine-Atorvasta 3 ⁇ 4 capsule
- nucleated tablet tableting machine (RUD—l: KHian) as the inner nucleus ⁇ and the composition containing ato 5vastatin as the inner core ⁇ , at a rate of 30 revolutions per minute, hardness 7 ⁇ 9 kp, thickness
- the tablets were compressed into 6,0 mm 3 and 9.5 mm diameter, and then a film coating layer was formed as a high coater to prepare nucleated tablets.
- Realization ⁇ 1-33 Amlodipine ⁇ Atorvasta 3 ⁇ 4 Blister Packaging Kit
- Example 1-34 Nifedipine-simvastatin Nucleated Tablets
- tablets were made with a hardness of 7-9 kp, 3.0 mm thick, and 5.5 mm diameter.
- hydroxypropylmethyl3 ⁇ 4-loose phthalate solution and acetylated monoglyceride solution dissolved in a 1: 1 mixture of ethane and methylene chloride were formed using a high coater to form a film coating layer.
- Table 1-5 shows the ingredients and contents and the examples. 1-20, 2) "simvastatin pre-release compartments were prepared according to the preparation method.
- nucleated tablet tableting machine (RUD-1: Ki l ian) as the inner core of the nifedipine core tablet and the composition containing simvastatin as the outer layer at a speed of 30 revolutions per minute, hardness 7 — 9 3, thickness 6.0 ⁇ , Girum Nucleated tablets were prepared by tableting at 9.5 mm and then forming a film coating layer as a high coater.
- Example 1-34 Except for using atorvastatin instead of simvastatin, according to the ingredients and contents of Table 1-5, according to the preparation method of Example 1-34 was prepared nipydipine- atorvastatin nucleated tablets.
- apples pelped and microcrystalline cellulose were mixed with a sieve No. 35, mixed in a double cone mixer, and put into a fluidized bed granulator (GPCG. 1: Glatt), and hydroxy separately.
- a binder solution made by dissolving propyl cellulose in water was sprayed to form granules and dried.
- the granules were then coated with hydroxypropylmethylsalloosephthalate solution dissolved in 1: 1 shaker of ethane and methylene chloride to coat the granules.
- the granules were mixed with 71G rolls of carbomer, and then mixed with a double cone mixer.
- the final mixture was used at a speed of 30 revolutions per minute using a rotary tableting machine (MRC-33: Sejong). , Tablets with a hardness of 7 to 9 kp, a thickness of 3.0 mm and a diameter of 5.0 mm were tableted.
- Example 1-22 the ingredients and contents shown in Table 1-7 and 2) the atorvastatin pre-release compartment of Example 1-22 were prepared according to the method S.
- vanidipine hydrochloride and microcrystalline cells were replaced with 35 Apples and common combined in a double cone mixer, open inflicting a then Kollicoat SR30D commitment to high-speed common stapler Union and then a No. 20 sieve and granulated using an oscillator, this hermit dryer this use at 60 X is dried again 20 Issue It was sifted.
- Magnesium stearate was added to this mixture and finally mixed with a double cone mixer. The final mixture was used at a speed of 30 revolutions per minute using a rotary milling machine (MRC # 33: Sejong), hardness 7-9kp, thickness 3.0mm, diameter Tablets of 5.5 mm were compressed.
- lovastatin, microcrystalline cellulose, and Manny were apologized to No. 35 and mixed with a high speed mixer.
- hydroxypropyl salulose and citric acid were dissolved in water to prepare a binding solution, which was added to a high-speed mixer with the main component mixture, and then united. After drying at C, it was established as No. 20 sieve again.
- Butylrate hydroxyanisole, starch glyconate sodium, colloidal silicon dioxide was mixed, and the final mixture was mixed with a double cone mixer after stearic acid magnesite was added to the rotary tableting machine (MRC-33 : Tablets of hardness 7-9 kp, thickness 5.0 kPa, and diameter 5.5 kPa were hit at a speed of 30 revolutions per minute using Sejong.
- Example 1-36 Lodiphan delayed-release compartment of Example 1-36 was prepared according to S.
- Benidin tablets of step 1) and the final composition of 2) were encapsulated in capsule layer electrophoresis # 1 gelatin It was stratified in hard cap surgery.
- silinidipine instead of vanidied, as prepared, according to the ingredient content shown in Table 1-6, was prepared according to the method of preparing a delayed-release layer of Example 1-37 1) vanidipine.
- Pravastatin sodium was used in place of simvastatin, according to the preparation method of Example 1-11, 2) simvastatin pre-release compartment, as shown in Table 1-7.
- Isradipine and microcrystalline cells were apples as a No. 35 sieve as shown in Table 1-6, and the mixtures were prepared by mixing in a double cone mixer. Separately, hydroxypropyl cellulose was dissolved in water to prepare a binder solution. While spraying the defect solution to the crystalline sucrose seeds in the CF granulator, the mixed powder was dispersed in the crystalline sucrose seeds to prepare pellets. The obtained 3 ⁇ 4 was dried at 50 ° C. until the water content was 2% or less, thereby preparing isoldipine-containing core 3 ⁇ 4lets. Again, Pelhat was commented by spraying a solution of hydroxypropylmethyl 3 ⁇ 4 rose phthalate dissolved in 3 ⁇ 43 ⁇ 4 of ethane and acetone in 1: 1 shake solution.
- simvastatin pre-release compartment of Example 1-1 was prepared according to the preparation method of 2) simvastatin pre-release compartment of Example 1-1 as shown in Table 1-7.
- Mandihydrochloride 3 ⁇ 4 and microcrystalline cell were apologized with No. 35 sieve and mixed with a double cone mixer, and then poured into a fluidized bed granulator (GPCG 1: Glatt), and separately The combined solution prepared by dissolving oxypropylmethylcell in water was sprayed to form granules and dried. Again, the granules were coated by spraying a hydroxypropylmexylsaloseophthalate solution dissolved in ethanol and 1: 1 shake solution of methylene chloride. Magnesium squalene was added to the mixture and the final mixture was mixed using a double cone mixer.
- lovastatin, microcrystalline cellulose and manny were apologized as No. 35 and mixed with a high speed mixer.
- Separately hydroxypropyl to prepare a binding solution by dissolving trehalose with citric acid in water saelreul and put in a high-speed common stapler with the main component mixture is granulated using an oscillator with a union and then sieve No. 20 and this, using a hermit dryer 60 ° C It was dried at and then reconstituted with No. 20 sieve.
- Butylated hydric cyanosol, sodium starch glycolate, colloidal silicon dioxide, and magnesium stearate were added thereto and finally mixed with a double cone mixer.
- the tablet was tableted with a warning of 7 to 9 kp, a thickness of 3.0 mm, and a diameter of 5.5 mm at a speed of 30 revolutions per minute using the Rotary Mixer (MRC-33; Sejong).
- MRC-33 Rotary Mixer
- Nicardipine hydrochloride was used in place of the felodisome, which was prepared according to the ingredients and contents shown in the following Tables 1-6 and 1) 3 ⁇ 4 rhodipine delayed-release compartment of Example I ′ 36.
- rosuvastatin kite instead of simvastatin is shown in Table 1-7 below. It was prepared according to the preparation method of 2) simvastatin pre-release compartment of Example 1-1 as well as egg component and content.
- nucleated tablet press (RUD-1: Ki l iaii) as the inner core of the nicardipine nucleus and the composition containing rosuvastatin as an outer layer at a speed of 30 revolutions per minute, hardness 7 to 9 kp, thickness 6.0 kPa
- a nucleus tablet was prepared by forming a film coating layer as a high coater.
- nifedipine instead of felodipine, it was prepared according to the ingredients and contents shown in the following Table 1-8 and 1) 3 ⁇ 4 rhodipine delayed-release compartment of Examples 1-36.
- nucleated tablet tablet machine (RUD-1: KHian) as the inner core of nifedipine core tablets and simvastatin-containing composition at a rate of 30 revolutions per minute, hardness 7 ⁇ 9 kp, thickness 6.0 kPa, diameter 9.5 mm Nucleated tablets were prepared by forming a film coating layer as a high coater after tableting.
- ROD-1 KHian
- Example 1-44 Preparation of Amlodipine-Pitavastatin Nucleated Tablets
- Amlodipine-rosuvastatin nucleated tablets were prepared according to the preparation method of Examples 1-20, except that rosuvastatin carsum was used instead of simvastatin.
- Example 1-46 Preparation of Nimodipine-pravastatin Coated Tablets
- Nimodibyun and microcrystalline cell were apologized with No. 35 sieve and common with a double cone mixer, which was then poured into a fluidized bed granulator (GPCG 1: Glatt), and hydroxypropyl separately.
- the binding solution made by dissolving methylcellose in water was sprayed to form granules and dried.
- the granules were coated by spraying a hydroxypropylmethyl gel solution containing phthalate and ethanol in a 1: 1 mixture of ethylene chloride and methylene chloride.
- the carbomer 71G was added to the granules in powder form, and then stear 3 ⁇ 4 acid magnesium was added and mixed with a final double cone mixer.
- Nimodipine tablet roll using a high coater Dissolves and disperses hydroxytitanium ⁇ titanium oxide and talc in ethanol and constant water, and dissolves pravastatin sodium salt as shown in the following Tables 1-9.
- a pre-release coating solution containing pullulvastatin nat salt was prepared, and the coating solution was prepared by forming a coating layer on the outer layer of the nimodipine delayed-release compartment with the coating solution.
- nibaldimun and microcrystalline cells were apologized with No. 35 sieve and mixed with a double cone mixer.
- hydroxypropylmethylcell was dissolved in water to spray a binder solution.
- the granules were coated by spraying a solution of hydroxyspitalymethylmethylcell dissolved in a 1: 1 shake solution of ol and methylene chloride.
- the granules were poured into carbomer 71G roll powder, and then stearic acid magnesium was added to the final double cone mixer.
- tableting was carried out using a multi-layer tablet press (ffiC-37T: Sejong). That is, the composition containing the pitavasta 3 ⁇ 4 in the primary powder feeder, and the composition containing nibaldipine in the secondary powder feeder at a speed of 30 revolutions per minute to minimize the infiltration of the layers, hardness 7 It was tableted to ⁇ 9 kp, 6.0 ram thick, 9,5 mm in diameter, and a film coating layer was formed as a high coater to prepare a tablet in a multi-layered form.
- Example 1-48 Nisoldipine-Lovastatin Multi-Layered Tablet S
- Granules were formed and dried. Again, the granules were coated by spraying the Eudragit RS P0 solution dissolved in 1: 1 shaker of ethane and methylene chloride. Stearic acid magnesium was added to this and mixed with the final double cone mixer.
- lovastatin in place of simvastatin was prepared according to the method of preparation of 2) simvastatin pre-release compartment of Example 1-1 with the components and contents shown in the following Table 1-9.
- pravastad nat "salt, microcrystalline shellulose, manniolol apologies with 35 No. 35 and mixed with a high-speed mixer Separately hydroxypropyl cellulose and citric acid in water It was prepared by dissolving in a binder solution, and putting it in a high speed mixer together with the main component mixture, and then granulating it with an oscillator using No. 20 sieve and drying it at 60 ° C. using a silver water dryer, and then reconstituting it with No. 20 sieve.
- Butylated hydroxyanisole, sodium starch glyconate, and colloidal silicon dioxide were mixed, and finally mixed with a stearic acid magnesite with a double cone filter, and the final mixture was rotated by a tablet press (MRC-33: Sejong). Using a tablet at a rate of 30 revolutions per minute, a viscosity of 7 to 9 kp, 4.0 mm thick, and 8.5 mm in diameter was tableted.
- (S) -Amlodipine core tablets were prepared by compressing the final mixture with a hardness of 6 to 10 kp, a thickness of 3.0 mm, and a diameter of 5.5 mm at a rate of 30 revolutions per minute using a rotary tablet press (MRC-33: tax). .
- simvastatin, microcrystalline salulose and manni were sieved through a No. 20 sieve, and then mixed in a high-speed mixer for 10 minutes. Separately, hydrophilic prophyllose and citric acid were purified. It was dissolved in to prepare a binding solution. While adding the binding solution to the above mixed moles, the granulation was completed, the granulation was completed, and granulation was completed. Butylated hydroxyanisole, starch sodium glycolate, and colloidal silicon dioxide, which were previously sieved through a No. 35 sieve, were administered to a double cone mixer together with the above granules, and then mixed for about 10 minutes. Was passed through a sieve No. 35 and administered to a double cone mixer, followed by final mixing for about 4 minutes to complete the preparation of simvasta3 ⁇ 4 layer granules.
- nucleated tablet press (RUD-1: Ki lian) as the inner core of the Amlodi 3 ⁇ 4 nuclear tablet and the composition containing simvastatin as the outer layer, at a speed of 30 revolutions per minute, hardness 7-13 kp, thickness 6.0 kPa
- the inner core was prepared by tableting with a diameter of 9.5 mm 3.
- the film coating layer composition of Table 1-8 was dissolved in a solvent to prepare a film coating solution.
- the inner core prepared above was administered to a high coater and then coated with a film coating solution to complete nucleated tablet manufacturing.
- Example 1-51 Preparation of (S) -amlodipine-simvastatin nucleated tablets
- Example 1-50 It was prepared in the same manner.
- Example 1-52 Preparation of (S) -amlodipine-simvastatin capsulant
- (S) -amlodipine besylate and microcrystalline cell loose were mixed with a No. 35 sieve and mixed with a double cone mixer, and then put into a fluidized bed granulator (GPCG 1: Glati).
- GPCG 1 fluidized bed granulator
- the binding solution prepared by dissolving parohydroxypropylcellose in purified water to form granules
- the granules were dried, and the resultant granules were infused with carbomer 71G in powder form for 10 minutes in a double cone mixer.
- stearic acid magnesium which was sieved through a No. 35 sieve was added and mixed for 4 minutes.
- (S) -Amlodipine tablets prepared by tableting the final mixture with a hardness of 6 ⁇ 10 kp, a thickness of 3.0 ⁇ and a diameter of 5.5 ⁇ at a speed of 30 revolutions per minute using a rotary tablet press (M C-33: Sejong).
- M C-33: Sejong a rotary tablet press
- simvasta layered granules 35 sieve and administering to a double cone mixer, the final mixture was finally mixed for about 4 minutes to complete the preparation of the simvasta layered granules.
- Simvastatin tablets were prepared by tableting the granules at a speed of 30 revolutions per minute using a rotary tableting machine (MRC-33: Sejong, Korea) at a hardness of 6 to 10 kp, a thickness of 3.0 mm and a diameter of 5.5 mm. Coating with a film layer component of 8 completed simvastatin tablet preparation.
- (S) —Amlodipine besylate and microcrystalline shell rollose were apples in No. 35, mixed in a double cone mixer, and then put into a fluidized bed granulator (GPCG 1: Glatt).
- GPCG 1 Glatt
- the binding solution made by dissolving hydroxypropylcellose in purified water was sprayed to form granules, and then dried.
- Magnesium Stearate which was sieved through No. 35, was added, followed by final mixing for 4 minutes.
- (S) amlodipine tablets prepared by tableting the final mixture with a hardness of 6 to 10 kp, a thickness of 3.0 mm and a diameter of 5.5 at a rate of 30 revolutions per minute using a rotary tableting machine (M C-33: Sejong).
- the preparation of the (S) -amlodi 3 ⁇ 4 tablet was completed by co-coating with li (methacrylate, 3 ⁇ 4 meth 3 ⁇ 4 tate) copolymer.
- simvastatin, microcrystalline salose, and Manni were sieved through a No. 20 sieve and mixed for 10 minutes in a high speed mixer. Separately, the combined solution was prepared by dissolving hydrocyclopropyl cellulose and citric acid in purified water. While adding the binding solution to the mixture, granulation was completed by granulation, drying, and sizing to complete granulation. Butylated hydride cyanosol, starch glyconate sodium, and colloidal silicon dioxide, which were previously sieved through a No. 35 sieve, were administered to a Dubolcon mixer together with the above granules, and then mixed for about 10 minutes. The sieve was sifted through No. 35 sieve and then administered to the Dulon method Final mixing was completed for minutes to complete the preparation of the simvastatin layer granules.
- Capsule preparation was completed by simultaneously laminating the above-mentioned (S) -amlodipine tablets and simvastatin granules in the hydrophilic cipropylmethyl 3 ⁇ 4 rollose hard cap capsule of No. 1 using a capsular layer electrolysis.
- Example 1-54 (S) —Amlodipine Tablet + Simvastar3 ⁇ 4 Blister Packaging Kit
- Example 1-55 Preparation of (S) -amlodiated-simvastatin coated tablets
- (S) -Amlodipine tablets prepared by tableting the final mixture with a hardness of 6 ⁇ 10 kp, a thickness of 4.0 ma, and a diameter of 8.0 ⁇ at a speed of 30 revolutions per minute using a rotary tablet press (MRC-33: Sejong). It was coated with a pulley (methacrylate, methyl methacrylate) copolymer below Table 1-10 to complete the preparation of (S)-amlodipine tablets.
- simvastatin, butylated hydroxyanisole, hydroxypropyl decylose, and colloidal silicon oxide were dissolved in ethane and ethylene chloride shaker to prepare a simvastatin coating solution.
- the second step Han (S) —Amlodipine tablets were administered to the high coater, followed by a first skip with a simvastatin coating solution.
- Example 1-56 Preparation of (S) -amlodipine-atorvastatin capsulant:
- Atorvastatin calum, butylated hydroxy anisole and microcrystalline salose were apologized as No. 35 as shown in Table 1-11, and mixed in a double cone mixer to prepare a mixture.
- hydroxypropyl 3 ⁇ 4 was dissolved in water to prepare a binder solution.
- a pellet was prepared by spraying the mixed powder on the crystalline sucrose seed while spraying the binder solution on the crystalline sucrose seed in the CF granulator.
- the pel3 ⁇ 4 obtained was dried at 50 ° C until the water content was 23 ⁇ 4> or less to prepare 3 ⁇ 43 ⁇ 4 containing atorvastatin sting.
- (S) -amlodi 3 ⁇ 4 besylate and microcrystalline cellulose were apologized as No. 35, and mixed in a double cone mixer to prepare a mixed powder.
- the binder solution was prepared by dissolving hydroxyipropyl cellulose in water. While spraying the binder solution to the crystalline sucrose seed in the CF granulator ' , the mixed powder was dispersed in the crystalline sucrose: seed to prepare pel3 ⁇ 4. The pellets were dried under arc until the water content was below 2% to remove (s) -amlodipine besylate containing core 3 ⁇ 4.
- atorvastatin scabbard, microcrystalline cellulose and mannyul were sieved through No. 20 sieve, and then mixed in a high speed mixer for 10 minutes.
- the binder solution was prepared by dissolving hydroxyipropylpropylcellulose and citric acid in purified water. Combination of the above While adding the binder solution to the water, the association was completed, then granulated, dried, and granulated to complete the granulation roll. Butylated hydroxyanisole, sodium starch guliconate, and colloidal silicon dioxide, which were previously sieved through a No.
- Capsule preparation was completed by simultaneously laminating the above-mentioned (S) -amlodipine besylate felhett and atorvastatin Calm tablets on hydroxypropylmethol 3 Cellulose hard capsing using capsule layer electrolysis.
- Example 1-58 Preparation of (S) -amlodipine-atorvastatin capsule.
- Example 1-56 It was prepared according to the preparation method of Example 1-56 2) atorvastatin calcium pre-release compartment as shown in Table 1-11.
- Capsule preparation was completed by simultaneously laminating the above-mentioned (S) -amlodipine granules and atorvastatin 3 ⁇ 4 on No. 1 hydrospecylmethylcellose hard capsule using a capsular layer electrolysis.
- Example 1-60 (S) Preparation of amlodipine-atorvastatin 3 ⁇ 4 sludge
- atorvastatin calcium, microcrystalline cellulose and mannrol were sieved through No. 20 sieve, and then mixed in a high speed mixer for 10 minutes. Separately, the combined solution was prepared by dissolving hydroxypropylcellose and citric acid in purified water. Adding the binder solution to the mixture above, after completing the association, granulation, drying, and granulation were completed to complete the granulation. Butane-rated hydroxyanisole, sodium starch glycolate, and roidoid silicon dioxide, which were previously sieved through a No. 35 sieve, were administered to a double cone mixer together with the above granules, and then mixed for about 10 minutes. The sieved sieve was sieved through a No. 35 sieve and administered to a double cone mixer, followed by final mixing for about 4 minutes to complete the atorvasta layered granule production roll.
- Capsule preparation was completed by simultaneously filling the above (S) -amlodipine 3 ⁇ 43 ⁇ 4 and atorvastatin granules with No. 1 hydroxypropylmethylcell in a hard capsule capsule.
- nucleated tableting machine (RUD-1: Kilian) as a layer of amlodipine inner core as a composition containing simvastatin, and then using a high coater (SFC-30N, Sejong Machinery, Korea), the components described in Table II-2 and Nucleated tablets were prepared by forming a film coating layer by content.
- SFC-30N Sejong Machinery, Korea
- Amlodipine-Simvasta3 ⁇ 4 nucleated tablets were prepared according to 3) Tableting and coating method of Example II-1 with the ingredients and contents shown in Table II-2. 9 000331
- Amlodipine-simvastatin nucleated tablets were prepared according to 3) tableting and coating method of Example II-1 with the mechanical components and contents of Table II-1.
- the binding solution prepared by amplifying amlodipine malate and microcrystalline salulose with No. 35 according to the ingredients and contents shown in Table II ⁇ 2 and mixing with a double cone mixer and dissolving hydroxypropylmethylcellose in purified water (1 » wAv) was sprayed to form granules and dried. Again, the granules were coated by spraying Eudragit RS P0 solution (20% wAv) dissolved in a 1: 1 mixture of ethane and methylene chloride. Magnesium stearate was added thereto and mixed in a final double cone mixer.
- Amlodipine-simvastatin multi-layered tablets were prepared according to the method of Example II-4, 3) Tableting and Coating according to Table II-2.
- Each of the final compositions prepared above was mixed with a double cone mixer, starch glyconate natto, 3 ⁇ 4-loid silicon dioxide, and mixed with stearic acid magnesium.
- Amlodipine—simvasta3 ⁇ 4 two-phase matrix tablets were prepared according to 3) postmixing, tableting and coating methods of Example ⁇ -6 with the ingredients and contents of Table ⁇ -2.
- amlodipine wheat rate and microcrystalline shell
- Example II-2 Amlodipine-simvastatin biphasic matrix tablets were prepared according to the method of Example II-6 in 3) Post-mixing Tableting and Coating.
- Example II-9 Amlodipine-Simvastar 3 ⁇ 4 Biphasic Capsule Formulation
- Example II-8 several contents of the ingredients shown in Table 11-2 and 1) of Example II-8 were prepared according to the preparation method of the amlodipine delayed-release compartment.
- Example II-ll Preparation of amlodi / 4-simvastatin nucleated tablets
- hydroxypropyl merlophthalate and acethiated monoglycerides were introduced into a 1: 1 mixture of ethane and chlorene chlorene chloride, and then dissolved as a coating solution (20fa / w).
- the inner core was prepared by forming a film coating layer using a coater (SFC-30N, Sejong Machinery, South Korea).
- simvastatin, microcrystalline selreul as agarose, corn starch and lactose as ingredients and contents shown in heunhap was 35 navale S apples and 'high-speed common stapler.
- hydroxypropylcellulose and citric acid were dissolved in purified water to prepare a binder solution (10% w / w), which was added to a high-speed mixer combined with the main component mixture, and then granulated using an oscillator. It was dried at 60 ° C using a silver water dryer and then re-established as No. 20.
- Butylated hydration cyanisole, sodium starch glyconate, and colloidal silicon dioxide were mixed therein, and magnesium stearate was added thereto, followed by ⁇ condensation in a double cone mixer.
- Example I 13 Amtodipine Tablets—Preparation of Capsulfonate Containing Simvastatin Tablets
- a solution of hydroxypropyl 3 ⁇ 4 cells in a 1: 1 shaker solution of ethanol and methylene chloride was added to a solution of chloropropylphthalate and acetylated monoterminicete as a coating solution (20% w / w).
- a high coater SFC-30N, Sejong Machinery, Korea.
- Butylated hydroxyanisole, sodium starch glyconate and colloidal silicon dioxide were mixed, and finally mixed with magnesium stearate in a double cone mixer, and the final mixture was mixed with a rotary tablet press (MRC-30: Sejong, Korea). ) was compressed into tablets.
- the tablet is a high coater (SFC-30N, Sejong machine, Korea) by fusing a coating solution (201 ⁇ 4 / w) prepared by dissolving and dispersing hydroxy propylose 2910, polyethylene glycol 6000, titanium oxide and talc in 803 ⁇ 4 ethane. ),
- amlodipine tablets of step 1> and the simvastatin tablets of step 2) were layered onto No. 3 hard 3 ⁇ 4 latin capsules using 3 ⁇ 4 slabs.
- Example 11-14 Preparation of Amlodipine-Simvastatin Capsulant
- simvastatin, microcrystalline cells, apples, lactose, and corn starch as No. 35 sieve were mixed with a high speed mixer. Separately, hydrated special propyl sal was dissolved in purified water, and a binder solution (103 ⁇ 4 w / w) was prepared, and the mixture was thrown into a high-speed mixer with a main ingredient mixture, and then granulated using an oscillator. After drying at 60 using was again established as No. 20 sieve. To this was added butylated hydroxyanisole, starch glyconate Na: trium, and colloidal silicon dioxide, and magnesium stearate was added for final mixing in a double cone mixer.
- amlodipine tablet of step 1) and the simvastatin granules of step 2) were layered onto the hydroxy hard capsules of No. 2 hydroxypropylme 3 ⁇ 4 cells using a capsular bed electric machine.
- Example 11-15 Amlodipine—simvastatin coated tablet S
- amlodipine tablets prepared above were administered to a high coater (SFC-30N, Sejong Machinery, Korea), and then first coated with a simvastatin coating solution.
- Example 11-16 Preparation S of Simvastar3 ⁇ 4 Rapid-Amlodipine Osmotic Nucleated Tablets
- Example II—11) 3) Simvastatin immediate-amlodipine-invasive nucleated tablets were prepared according to the method of tableting and skipping.
- Example 11-3 Tablets Amlodithi of Example A— Example II—13 And simvastatin tablets were prepared in the same manner as in Example 11-13, except that the blister packaging was packaged for isochronization instead of simultaneously filling in the capsule, Example 11-18: (S) -Amlodi 3 ⁇ 4 ⁇ Preparation of Vastatin Nucleated Tablets
- Nucleating tablets were prepared by tableting at a rate of 30 revolutions per minute using a nucleated tableting machine (RUD-1: Kil iai *) as the outer layer of a composition containing amlodipine inner cores.
- the coating layer composition was dissolved in a solvent to prepare a film coating solution.
- the nucleated tablet prepared above was administered to a high coater (SFC-30N, Sejong Machinery, Korea) and coated with a film coating solution to complete nucleated tablet preparation. 19 : (S) -Amlodipine-Zombatin Nucleated Tablets
- Example 11-20 Preparation of (S) -Amlodipine-Simvastatin Capsulant
- (S) -Ambro dipine tablets prepared by tableting at the speed of 30 revolutions per minute using a rotary tablet press (MRC-30: Sejong) rolls were prepared using a poly (methacrylate, methacrylate) copolymer. Coating with a solution (20taAr) dispersed in purified water to complete the preparation of (S)-amlodipine tablets.
- Capsule preparation was completed by layering the above (S) -amlodipine tablets and simvastatin tablets on the gelatine hard cap capsules of No. 3 using a capsular layer electrophoresis.
- Example 11-21 Preparation of (S) -Amlodipine-Simvasta 3 ⁇ 4 capsulant 1) (S) —the ⁇ (tablet) of the amlodied delayed-release compartment
- sivastatin, microcrystalline cellulose, and manni were sieved through a No. 20 sieve with the ingredients and contents shown in Table ⁇ -4, followed by mixing for 10 minutes in a high speed mixer.
- the hydroxydemic propyl sal was dissolved in purified water of loose and citric acid to prepare a binding solution (103 ⁇ 4 w / w).
- the binder was added to the above mixture, granulation, drying, and granulation were completed to complete granulation rolls. It was then administered to a double cone mixer with granules on buhalated hydride cyanosol, starch glycolate, and colloidal silicon dioxide, which were previously sieved through No. 35, and then mixed for about 10 minutes.
- Magnesium acid was sieved through a No. 35 sieve and poured into a double cone mixer, followed by final mixing for about 4 minutes to complete the preparation of simvastatin layer granules.
- Example II-23 (S) —Preparation of Amlodipine-Simvastar 3 ⁇ 4 Coated Tablet
- (S) -Amlodipine and the microcrystalline cell were anointed with No. 35 sieve, mixed with a double cone mixer, and then put into a fluidized bed granulator (GPCG 1: Glatt).
- GPCG 1 Glatt
- a combined solution 103 ⁇ 4 of hydroxypropylcellose was dissolved in purified water, sprayed to form a grove, and dried.
- Carbomer 71G was added to the granules in a powdered state: 10 minutes in a double cone mixer. After mixing, the stearic acid was passed through a No.
- simvastar3 ⁇ 4 butylated hydrated cyanuric sol, hydroxypropylmethylshellose 2910, and colloidal silicon oxide were added to a shake solution of ethane and methylene chloride 1: 1. Melt to prepare a simvastatin coating solution,
- the (S) -amlodipine tablets prepared above were administered to a high coater (SFC-30N, Sejong Machinery, South Korea), followed by primary codontification with a simvastar3 ⁇ 4 coating solution.
- amlodipine besylate and microcrystalline cell were apologized as No. 35 sieve and mixed with a double cone mixer, and then put into a fluidized bed granulator (GPCG 1: Glatt), and hydroxypropyl separately.
- Granules were formed by spraying the binding solution prepared by dissolving methyl salose in purified water and drying the granules.
- Carbomer 71G »powder was added to the granules and stearic acid was added to the final double cone mixer.
- the final mixture was compressed into tablets using a rotary tablet press (MRC-33: Sejong) and used as the inner core.
- atorvastatin chamomile trihydrate, carbohydrate carbonate, microcrystalline cellulose, lactose, pregelatinized starch and lauryl sulfate; "(sodium lauryl sulfate) was appointed in No. 35 and mixed with a high-speed mixer. Loose was dissolved in purified water to prepare a binding solution, which was added together with the main ingredient mixture in a high speed mixer, and then granulated using an oscillator as a No. 20 sieve, dried at 60 X using a silver water dryer, and then re-established as No. 20 sieve. Croscarmellose sodium was added to the mixture, and magnesium stearate was added to the final mixture in a double cone mixer.
- nucleated tablet tableting machine (RUD ⁇ 1: Ki lian)
- the composition containing amlodipine inner core and atorvastatin was used as an external insect, followed by rubbing using a high coater (SFC-30N, Sejong Machinery, Korea).
- a nucleated tablet was prepared by forming a film coating layer with an excess content.
- Example III-2 Preparation of Amlodiphan-Atorvastated Nucleated Tablets
- nucleated tablet tableting machine (RUD-1: Ki liai)
- the composition containing the amlodipine inner core and atorvastatin as an outer layer was compressed into tablets, and then used a high coater (SFC-30N, Sejong Machinery, Korea).
- Nucleated tablets were prepared by forming a film coat layer with an excess content.
- Example III-3 Preparation of Amlodipine-Atorvastated Nucleated Tablets
- nucleated tablet tableting machine (RUD-1: Ki H an)
- the composition containing amlodipine inner core and atorvastatin as an outer layer was compressed into tablets, and then used a high coater (SFC-30N, Sejong Machinery, Korea). to form a film coating layer components and the content was prepared with a press-coated tablet of example ⁇ -4: amlodipine-atorvastatin multi-layered manufacturing
- the atorvastared stale anhydrous, calcium carbonate, microcrystalline cellulose, lactose and sodium lauryl sulfate were appled with a No. 35 sieve and mixed with a high speed mixer. .
- prepare a defect solution by dissolving hydroxypropyl ⁇ loose in purified water add it to a high-speed mixer mixed with a main ingredient mixture, combine it, and granulate it with a No. 20 sieve using an oscillator, and dry it at 60 3 ⁇ 4 using a silver water dryer. Again it was determined as No. 20 body. Croscarmellose sodium was added to this mixture, and stearic acid magnesium was added to the final mixture using a double cone mixer. 3) tableting and coating
- the tablet was compressed using a multi-layer tablet press (M4C-37T: Sejong). That is, the composition containing atorvastatin was placed in a primary powder feeder, and the composition containing amlodipine was placed in a secondary powder feeder. The tablets were compressed to minimize the infiltration between the layers, and a multi-layered tablet was prepared by forming a film coating layer using the ingredients and contents of Table III-1 as a high coater (SFC-30M, Sejong Machinery, Korea).
- Example II-5 Preparation of amlodipine-atorvastatin multilayer tablet S
- atorvastatin calcium strontium pentahydrate, calcium carbonate, microcrystalline 3 ⁇ 4-lose, lactose and sodium lauryl sul fate as apple No. 35 were used as apples.
- a special liquid was prepared by dissolving the loose-special propyl shell in purified water, putting it in a high-speed mixer with a main component mixture, and then combining it with J1 and then granulating it using an oscillator in a No. 20 sieve. It was dried at and then reconstituted with No. 20 sieve. Croscarmellose sodium was added to the mixture, and magnesium stearate was added thereto, followed by final mixing with a double cone mixer.
- the tablet was compressed using a multi-layer tablet press (MRC-37T: Sejong). That is, the composition containing Atorvasta 3 ⁇ 4 was placed in a primary powder feeder, and the composition containing amlodipine was placed in a secondary powder feeder. The tablets were compressed to minimize the infiltration between the layers, and a multi-layered tablet was prepared by forming a fill # coating layer using the ingredients and contents of Table II 1-1 as a high coater (SFC-30N, Sejong Machinery, Korea).
- Example 6-6 Preparation of Amlodipine-Atorvastatin Biphasic Matrix Tablets
- amlodipine vasylate and microcrystalline shell loose were mixed with apple No. 35 and mixed, and then fed into a high-speed mixer, and co-coated SR30D was added.
- association was completed, granulated with 20 housings using an oscillator, dried at 60 using a silver water dryer, and then re-established as 20 :.
- atorvastatin calcium trihydrate, calcium carbonate, microcrystalline salose, lactose, mannose, and crospovidone were appled with No. 35 and mixed with a high-speed mixer.
- Loose and plysorbate 80 were dissolved in purified water to prepare a binding solution, and the mixture was introduced into a high-speed mixer with the common substance of the above-mentioned ingredients. Then, it was granulated using an oscillator. Dried at I; and back to 20.
- Each of the final compositions prepared above was mixed with a double cone mixer, and sodium starch glycolate was mixed, and then a magnesium stearate was added thereto, followed by final mixing with a double cone mixer.
- amlodipine besylate and microcrystalline shell loose were apologized as No. 35 and mixed with a double cone mixer.
- the above mixture was introduced into a fluidized bed granulator (GPCG 1: Glatt).
- granules were formed by spraying a binder solution made by dissolving 3 ⁇ 4 cell cellulose in hydrated propyl-metholose in purified water, and drying the granules by spraying 3 ⁇ 4 Eudragit RS P0 solution upon dissolving in granules.
- GPCG 1 Glatt
- Example III ⁇ 6-2 It was prepared by the crime prevention of Example III ⁇ 6-2) with the ingredients and contents shown in the following Table III-1
- Example ⁇ -6-2 In the same manner as in Example ⁇ -6-2), except that atorvastatin calcium anhydride was used instead of atorvastatin chame trihydrate with the ingredients and contents shown in the following Table 111-1.
- Example III-9 (S) -Amlodipine-Atorvastatin Phase 2 Capsule Preparation
- Apples of atorvastatin calcium trihydrate, calcium carbonate, microcrystalline cellulose, lactose, pregelatinized starch, and sodium lauryl sulfate were identified as No. 35 as shown in Table III-1.
- Faro hydroxypropylcellose was dissolved in purified water to prepare a binder, which was added to a high-speed mixer with a mixture of the main ingredients, and then granulated using an oscillator roll. It dried at 60X: using the dryer roll, and it was made up again with No. 20 sieve.
- atorvastatin strontium pentahydrate, calcium 3 ⁇ 4 acid, microcrystalline 3 ⁇ 4 rate loose, lactose, manni and crospovidone were apologized by No. 35 and mixed with a high speed mixer. Dissolve the propylcellulose and pulley sorbate 80 in purified water to remove the defect solution, add it to the high-speed mixer together with the mixture of the main component, and then unite with the No. 20 sieve. It was dried at 60 ° C. and then sieved to No. 20 sieve again.
- Steps 1) and 2) were mixed with a Dubucon mixer, sodium starch glycolate was added thereto, followed by mixing with a double cone mixer, and magnesium stearate was added for final mixing.
- the final mixed mixture was put into a powder feeder and layered into No. 1 3 ⁇ 4 liquor using a 3 ⁇ 4 sul charger.
- Example II-11 Preparation of Amlodipine-Atorvasta 3 ⁇ 4 Nucleated Tablets
- a hydroxypropyl methyl salulose phthalate solution and acetylated monoglycerides dissolved in 1: 1 shaker of ethane and methylene chloride were added at a ratio of 100: 1 to a high coater (SFC-30N, Sejong Machinery, Korea) was used to form a film coating layer to remove the inner core.
- Atorvastatin 3 ⁇ 4 calcium trihydrate, stearic acid, chestnut, microcrystalline gellose, oak starch and lactose were apologized as No. 35 and mixed in a high-speed mixer as shown in the following table II- 2. Dissolve propyl-l-loose and plysorbate 80 in purified water to remove the binding solution, add it to a high-speed mixer with the main component mixture, combine it, and granulate it with the No. 20 sieve using an oscillator. The mixture was dried at 60'C, and then sieved to No. 20. Here, sodium starch glyconate was mixed, and magnesium stearate was added and finally mixed with a double cone mixer.
- Example II 1-11-2 It was prepared by the method of Example II 1-11-2) with the ingredients and contents shown in Table II 1-2.
- the tablets were compressed using a multi-layer tablet press (MRC) 37T: Sejong). That is, the composition containing the atorvastatin was placed in a primary powder feeder, and the composition containing amlodisome was transferred to a secondary powder feeder. Tablets were put in a condition that minimizes the intrusion between layers, and a multi-layered tablet was prepared by forming a film coating layer using the ingredients and contents of Table ⁇ -2 as a high coater (SFC-30N, Sejong Machinery, Korea).
- MRC multi-layer tablet press
- atorvastatin chamomile trihydrate, carbonate chamomile, and microcrystalline salose were apologized as No. 35 and mixed with a high speed mixer.
- Dissolve the sorbate 80 in purified water to make a binding solution add it to a high speed mixer with the main component mixture and combine it, and granulate it using the oscillator with No. 20 sieve.
- Nathium starch glyconate is mixed, and the final mixture is mixed with a stearic acid magnesium bosom with a double cone mixer, and the final mixture is placed into a rotary tablet press (MRC- 33: King Sejong).
- the tablet is a coating liquid prepared by dissolving and dispersing hydrated propyl cellulose, polystyrene glycol 6000, titanium oxide and talc ethane in. It was coated with a high coater (SFC-30N, Sejong Machinery, Korea).
- amlodipine tablet of step 1) and the atorvastar 3 ⁇ 4 tablet of step 2) were layered into capsule 3 using a 3 ⁇ 4 sulfoner.
- Example 111-14 Preparation of Amlodi 3 ⁇ 4 ⁇ atorvastatin 3 ⁇ 4
- the atorvastatin calcium trihydrate, calcium carbonate, microcrystalline cellulose, lactose, corn starch and lau 3 ⁇ 4 sodium sulfate as apples No. 35 were apples and mixed with a high speed mixer. Separately, hydroxypropyl 3 ⁇ 4 loose was added to purified water to prepare a binding solution, and the mixture was added to a high-speed mixer mixed with the main ingredient mixture. Then, the mixture was granulated using an oscillator with No. 20 sieve and dried at 60 13 using a silver water dryer. Again, No. 20 sieve was added. Here, sodium starch glyconate was mixed, and magnesium stearate was added and finally mixed with a double cone mixer.
- amlodipine tablet of step 1) and the atorvastatin granules of step 2) were layered onto hydroxypropylmethylcellol.rose hard 3 ⁇ 4 liquor of No. 2 using a capsular bed electric machine.
- Example 111-15 Preparation of Amlodi 3 ⁇ 4-Atorvasta Coated Tablets
- Decalum Phosphate 3 ⁇ 4 35 as shown in Table ⁇ -2, mixed with a double cone mixer, and put into a fluidized bed granulator (GPCG 1: Glatt).
- GPCG 1 Glatt
- hydroxypropylme 3 ⁇ 4 ⁇ loose was dissolved in purified water and sprayed with a defect solution to form granules and dried.
- the carbomer 71G was added to the above-mentioned vinegar in powder form, and then magnesium stearate was added thereto.
- the atorvastatin calcium trihydrate, hydrated propylpropylcellulose was dissolved and dispersed in 803 ⁇ 4 ethane to prepare a pre-release atorvastatin coating solution.
- amlodipine tablets prepared above were administered to a high coater (SFC-30N, Sejong Machinery, Korea), followed by primary treatment with an atorvastatin coating solution.
- Example III—16 Preparation of Amlodipine-Atorvastated Seborrheic Nucleated Tablets
- Example 111-14-2 It was prepared by the method of Example 111-14-2) with the ingredients and contents shown in Table III-3 below.
- amlodipine delayed-release compartment (tablet) and atorvastatin pre-release compartment (tablet) by the method of Examples III-13-1) and 13-2) as shown in the following components III and III. It was manufactured by packaging to allow simultaneous use in the bolister packaging.
- the nucleation point was subtracted by the following method with the ingredients and contents listed in the table.
- Amlodipine besylate, microcrystalline 3 ⁇ 4 rose, anhydrous calcium hydrogen phosphate, pregelatinized starch (Starch 1500G, Colorcon, USA) were apples in No. 35 and mixed for 5 minutes with a double cone mixer to prepare a mixture. Separately, hydroxypropylcell was dissolved in purified water roll to form a binding solution, which was then combined, granulated, and dried. After drying, it is established as No. 18 again. The sieved material is placed in a fluidized bed corrugator, and separately 3 ⁇ 4 cellulose acetate: ⁇ 320S (32% acetal group) and saloseacetate S 398-10NF (acetal group 39.8%) are added to ethane and methylene chloride.
- the melted solution was prepared and the above granules were placed in a fluid bed granulation coater (GPC &-1; Glatt, Germany) and nosed. Coating completed. After that, magnesium stearate was added and mixed for 4 minutes, and the tablets were tableted with a rotary tablet press (MRC-37, Sejong Machinery, Korea) equipped with a 5 mm diameter bias.
- a fluid bed granulation coater GPC &-1; Glatt, Germany
- a nucleated tableting machine (RUTK1: i lian, Germany) equipped with 11 mm bias was used as phytavastatin calcium pre-release granules of 1) as the outer layer, and the amlodipine besylate delayed-release tablet of 2) was used as a nuclear tablet. Tableting.
- Amlodipine besylate, microcrystalline cellulose, anhydrous hydrogen phosphate, and pregelatinized starch were appointed as No. 35 and mixed in a double cone mixer for 5 minutes to prepare a mixture.
- hydroxypropylcellulose was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried. After drying, it is established as No. 18 again.
- the granules were placed in a fluidized bed coater, and a solution obtained by dissolving Eudragit RS30D (ETOiiik Degussa, Germany) and triethyl citrate (Vert lus, England) in methylene chloride was prepared. GPCG-1; Glatt, Germany) and coated. After the coating was completed, the stearic acid was added thereto, mixed for 4 minutes, and tableted with a rotary tablet press (MRC-37: Sejong) equipped with a 5 mm diameter bias to prepare a nuclear tablet.
- MRC-37 Sej
- Example IV-3 Preparation of Amlodipine-Pitavata 3 ⁇ 4 nucleated tablets Nucleated tablets were prepared by the following method using the ingredients and contents shown in Table IV-1.
- Amlodipine tesylate, microcrystalline shellulose, and di-mannee were apples with No. 35 sieve, and mixed with each other for 5 minutes to prepare a mixture.
- pulley vinylphyllolidon (Koll idon 30, BASF, Germany) was dissolved in purified water and combined, granulated, and dried as a binder. After drying, it is established as No. 18 again.
- the formulations are placed in a fluidized bed coater and separately placed in 3 ⁇ 4 shell (HERCULES, USA) and poly (methachite, methyl methrate) copolymer (Evonik degussa, USA) with ethane and methylene chloride.
- Example IV-4 Preparation of Amlodipine-Pipavatatin Nucleated Tablets
- Nucleated tablets were prepared by the following method, using ingredients and contents set forth in Table IV-1.
- Amlodichet malate, microcrystalline shellose, anhydrous hydrogen phosphate, and di-mannee were appled in No. 35 and mixed for 5 minutes with a double cone mixer to prepare a mixture. Separately, hydroxypropyl cells were dissolved in purified water to form a binding solution, which was then combined, granulated, and dried. After drying, it is settled again with No. 18 sieve. Stearic acid magnesium sieved through No. 35 sieve was added to the sieved material, followed by mixing for 4 minutes to prepare amlodipine delayed-release layer granules.
- Nuclear tablets were prepared by tableting the amlodipine fed granules with a rotary tablet press (MRC—37: Sejong) equipped with a 5 mm diameter punch. Separately, dissolve and disperse arc 3 ⁇ 4-ises (methac3 ⁇ 4 acid copolymer type C, talc, PEG, colloidal silicon dihexaside, sodium bicarbonate, SLS, Colorcon, USA) in purified water to prepare a 3 ⁇ 4 coating solution. Lodi 3 ⁇ 4 tablet was formed as a coater as a high coater (SFC—30N, Sejong machine, Korea) to complete the amlodipine tablet manufacturing roll.
- a rotary tablet press MRC—37: Sejong
- arc 3 ⁇ 4-ises metalhac3 ⁇ 4 acid copolymer type C, talc, PEG, colloidal silicon dihexaside, sodium bicarbonate, SLS, Colorcon, USA
- Lodi 3 ⁇ 4 tablet was formed as a coater as a high
- Amalodipine malate and microcrystalline mullose were apples in No. 35 and mixed for 5 minutes with a double cone mixer to prepare a mixture.
- the mixture was added to a double cone mixer and colicoat SR30D (30% suspension of main component pulley vinyl acetate, manufactured by BASF, Germany) was added and granulated using an oscillator. After drying at 60 1C it was again established as No. 18.
- Magnesium stearate which was sieved through No. 35 sieve, was added to the sieved material, mixed for 4 minutes, and tableted with a rotary tableting machine (M C-37: Sejong) equipped with a 5 mm diameter bias to prepare a nuclear tablet.
- a coating solution was prepared by dissolving and dispersing hydroxypropylmethelloose phthalate (Shin— etsu, Japan) in ethane and methylene chloride.
- the above Amlodi 3 ⁇ 4 tablets were coated on a high coater (SFC-30N, Sejong Machinery, Korea). ) To form a coating layer to complete the amlodipine oak preparation.
- Example IV-6 Preparation of Amlodipine-Pitabatatin Nucleated Tablets
- Amlodipine besylate, pregelatinized starch, corn starch were apples with No. 35 sieve and mixed for 5 minutes with a sticking mixer to prepare a mixture.
- hydroxypropylmethyl ⁇ loose was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried. After construction, it is established as No. 18 body again.
- the granules are placed in a fluidized bed cobalt, and separately hydrated propylmethylsalose phthalate is added to ethane and methalene chloride, and the above granules are added to a soft layer granulation coater (GPCG-1; Glatt, Germany ) And coated. After the coating was completed, magnesium stearate was added thereto, mixed for 4 minutes, and tableted with a Totari tableting machine (MRC-37: Sejong) equipped with a 5 mm diameter punch to prepare a core tablet.
- MRC-37 Sejong
- Example IV-7 Amlodipine-Pitavapatin Two-Phase Matrix Tablet Preparation
- hydroxypropylsal was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried.
- the dried product was placed in a fluidized bed coater, and a solution obtained by dissolving Dalo Cell Acetate 320S (acetal group 32%) and Cellulose Acetate 398-10NF10 (acetal group 39.8%) in ethane and methylene chloride was prepared.
- the above granules were put into a coating of granular layer coating machine (GPCG-1: Glatt, Geramny) and coated.
- Rix tablets were prepared by the following method, with the components and contents listed in Table IV-1.
- Amalodipine malate, microcrystalline 3 ⁇ 4 rose, pregelatinized starch were apples in No. 35 sieve and double cone mixed with each other for 5 minutes to prepare a mixture.
- polyvinylpyridone was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried.
- the dry granules were placed in a fluidized bed coater and separately prepared by dissolving in Eudragit RS30D and triethylcitrate methylene chloride, and the above granules were placed in a fluidized bed granular coronate (GPCG-1; Glatt, Germany) and coated.
- Example IV-9 Amlodipine-Pitabatated Biphasic Matrix Tablet ⁇
- a mixture of S-amlodipine tesylate, microcrystalline salose, and di-manny apples with No. 35 was mixed for 5 minutes with a double cone mixer.
- the mixture was added to a high-speed mixer, fed with Colicoat SR30D, and then granulated using No. 20 sieve using an oscillator.
- the roll was dried at 60 1 C using a dry water dryer and re-established into No. 18 sieve.
- Example IV-10 Amlodi 3 ⁇ 4-Pitabatatin Biphasic Matrix Tablet Preparation
- Amalodifun besylate, microcrystalline salose, anhydrous calcium hydrogen phosphate, pregelatinized starch were appled in a No. 35 sieve and mixed for 5 minutes with a double cone mixer to prepare a mixture.
- polyvinylpyridone was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried.
- Example IV-11 Preparation of Amlodipine-Pitabata Multi-Layered Tablets
- a multilayer tablet was prepared by the following method.
- a mixture of pitavasta calder, meta metasilicate aluminate, microcrystalline cellulose, lactose hydrate and apple No. 35 was ap- plied and shaken in a double cone mixer for 5 minutes. Separately, hydroxypropylcellose was dissolved in purified water to prepare a binding solution, which was associated with the main ingredient mixture. When the coalition is received, the granules are granulated using the oscillator 20 and dried at 60 ° C using a hot water dryer. After drying, it was reestablished as No. 18. The starch sodium starch glycolate was mixed, magnesium stearate was added, and the final mixture was mixed with a double cone mixer.
- Amalodipine besylate, microcrystalline shell loose, 3 ⁇ 4 starch, corn starch were apples with No. 35 sieve and mixed for 5 minutes with a double cone filter to prepare a mixture.
- pulley vinylpy * ridone was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried. After drying, the mixture was again sieved to No. 18 sieve.
- GPCG fluidized bed granulator
- the above tablet was prepared by preparing a coating solution in which hydroxypropylmethylcellulose 2910, 3 ⁇ 4 Li 3 ⁇ 4lenglycol 6,000, Tal 3, and titanium oxide were dissolved and dispersed in ethane and purified water (SFC-30N: Sejong Machinery, Korea) to form a film coating layer to prepare a tablet in the form of a multi-layered tablet.
- SFC-30N Sejong Machinery, Korea
- a multilayer tablet was prepared by the following method.
- Example IV 13 Preparation of Amrodafine-Pitabata 3 ⁇ 4 Polyplyum Tablets
- a multilayer tablet was prepared by the following method.
- S-amlodipine besylate, microcrystalline cellulose, anhydrous calcium hydrogen phosphate, pregelatinized starch were appled with a No. 35 sieve, and mixed with a double cone mixer for 5 minutes to prepare a mixture.
- the mixture was added to a high-speed mixer, fed with a coat coat SR30D, and then granulated with a No. 20 sieve using an oscillator, which was dried at 60 'C using a silver water dryer, and then back to No. 18 sieve. After adding 3 ⁇ 4 magnesium stearate, the mixture was mixed for 4 minutes.
- Example IV-11 In the same manner as 3) of Example IV-11, multimodal tablets were prepared by post-mixing, tableting, and coring.
- Example IV-14 Amlodiphan-Pitabata Multilayer Tablet Preparation
- Amlodipine besylate, anhydrous calcium hydrogen phosphate, and di-mannney were appointed as No. 35 and mixed with each other for 5 minutes to prepare a mixture.
- hydroxypropylcell was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried. After drying, it was established as No. 18 body again.
- the sieved material was placed in a gas-coated coater and separately prepared by dissolving Eudragit RS30D and triethyl citrate in methylene chloride. . After completion of the coating, magnesium stearate was thrown up and mixed for 4 minutes.
- Example IV The same method as 3) of 11) was followed by post-mixing, tableting, and coating to prepare tablets in the form of a multilayer tablet.
- Example IV-15 Amlodipine-Pitabatatin Capsule Preparation (Pallet-Granule)
- a capsulant was prepared by the following method.
- the sugar spheres were poured into a fluid bed granulator (GPCG1: Glatt), and then separately dissolved in hydrated propyl salulose and pitavastatin kalmos in water, sprayed with 3 ⁇ 4 binding solution to form a pitavastatin-containing pellet. Formed and dried.
- GPCG1 fluid bed granulator
- step 1) and 2) was filled in capsul (Seheung capsal, Korea) using a capsul layered electric to complete the preparation of the timed release formulation in the form of capsule.
- Example IV-17 Preparation of Amlodipine-Pitabatatin Capsul (Pel Osmotic Tablets)
- capsules were prepared by the following method: 1) Preparation of a prerelease compartment (pitavastatin immediate release pellets)
- Amlodipine besylate, microcrystalline cellulose, anhydrous hydrogen phosphate, corn starch were apples with No. 35 sieve, and the mixture was prepared for 5 minutes in a double cone mixer. Separately, the hydrophilic cyclofilel was dissolved in purified water to form a binding solution, and then combined, granulated, and dried. Sodium chloride and magnesium stearate were added to the sieved material, followed by mixing for 4 minutes. The mixture was compressed into tablets using a rotary tableting machine (MRC-37: Sejong) equipped with a 5 mm diameter bias.
- MRC-37 Sejong
- a solution obtained by dissolving cellulose acetate 320S (acetal group 32%) and chlorocellulose acetate 398-10 F (acetal group 39.8%) in ethane and methylene chloride was prepared and used as a high coater (SFC-30N). : Sejong Machinery, Korea) to form a film coating layer to prepare an osmotic tablet of amlodipine.
- Amalodipine besylate, microcrystalline cellulose and pregelatinized starch were apples in No. 35 and mixed for 5 minutes using a double cone mixer. Separately, hydroxypropyl ⁇ was dissolved in purified water and combined to form a binding solution. Granulated and dried. When the case is over, clean it again with No. 18 sieve. Magnesium stearate, which was sieved through No. 35 sieve, was added to the sieved material, mixed for 4 minutes, and the mixture was compressed into tablets using a rotary tablet press (MRC-37: Sejong) equipped with a 5 mm diameter bias.
- MRC-37 Sejong
- amlodipine tablets were prepared as a high coater (SFC-30N, Sejong Machinery, Korea) by preparing a nose 3 ⁇ 4 solution in which 3 ⁇ 4-ise was dissolved and dispersed in purified water. A coating layer was formed to complete the preparation of amlodipine oak,
- the capsule was prepared in the following manner.
- Example IV-20 Amlodipine-Pitabatatin Capsule Manufacture (tablet-tablet)
- a capsulant was prepared by the following method.
- the coating solution was prepared by dissolving and dispersing latex, meth methacrylate copolymer (Coiorcon, USA) in purified water to form a coating layer as a high coater (SPC-30N, Sejong Machinery Co., Ltd. Korea). Article was completed.
- Example IV-21 Preparation of Amlodipine-Pitabatatin Capsule (Granules-Granules)
- Amlodipine besylate, microcrystalline cellulose, and di-mannee were apples with No. 35 sieve and mixed with each other for 5 minutes to prepare a mixture.
- Collicoat SR30D was added and associated with the main ingredient mixing mole. After the association, granulation was carried out using an oscillator in No. 20 and dried using a silver water dryer. After drying, it was established as No. 18 body again.
- a capsul was prepared by the following method.
- 3 ⁇ 4 knee was prepared by the following method.
- Amlodipine besylate, anhydrous calcium phosphate and pregelatinized starch were apples in No. 35 sieve and mixed for 5 minutes using a double cone mixer to prepare a mixture. Separately, hydroxypropylsal was dissolved in purified water to form a binding solution, which was then combined, granulated, and dried. After drying, it is settled again with No. 18 sieve. Magnesium stearate, which was sieved through No. 35 sieve, was put into the tablets, mixed for 4 minutes, and the mixture was compressed into tablets using a rotary tablet press (MRC-37: Sejong) equipped with a 5 mm diameter bias.
- MRC-37 Sejong
- Capsuljeol was prepared by the following method with the ingredients and contents shown in Table IV-3.
- Amalodipine besylate, microcrystalline salose, anhydrous hydrogen phosphate, and hydroxypropylmethyl cellulose were apples in No. 35 and mixed for 5 minutes with a double cone filter to prepare a mixture. Purified water was added and combined with the main ingredient mixture. After the association, granulation was carried out using an oscillator in No. 20 sieve, and it was dried using a dry roll of silver. After drying, it was again erected with No. 18 sieve. Magnesium stearate was added to the tablets for final mixing, and amlodipine delayed-release granules were prepared.
- Example IV-25 Preparation of Pitavastar 3 ⁇ 4-Amlodipine Tesylate Blister Packaging Kit A pitavastatin-amlodi 3 ⁇ 4 besylate blister packaging kit was prepared by the following security.
- Example IV-7 and 1) and the amlodipine besylate delayed-release granules prepared in Example IV—7 2) were prepared using a rotary tablet press (MRC—33: Sejong Machinery, Korea After each tablet is purified using a blister packaging machine (Minister A, Heunga Engineer 3 ⁇ 4), the blister packaging container (silver foil, the same good grade) is used. PVDC, Jeonmin industry) to prepare a blister packaging kit by packaging each tablet for simultaneous use.
- Nucleated tablets were prepared by the following method, using ingredients and contents set forth in Table V-1.
- Roschvasta Calcium (MSN, INDIA), Tribasic Chest Phosphate, Microcrystalline Salose (Avicel PHlOl, FMC Biopolymer, USA), Lactose Hydrate (DMV, Germany), Pregelatinized Starch (Starch 1500G, Colorcon, USA) Sieve apologies, and mixed with a double cone mixer (Dasan and Martech, Korea) for 5 minutes in real, prepared a fire.
- hydroxypropyl salulose HPC-L, Nippon Soda, Japan
- HPC-L hydroxypropyl salulose
- Amlodipine besylate, crystalline cellulose, anhydrous calcium hydrogen phosphate, pregelatinized starch (Starch 1500G, Colorcon, USA) were apples in No. 35, and mixed with a double cone mixer for 5 minutes in a real environment to prepare a mixture.
- hydroxypropylcellose was dissolved in purified water to prepare a conjugated solution, which was associated with the main component mixture. After the coalition, it was granulated with an oscillator using No. 18 sieve, which was dried at using a silver water dryer (HW-C, Samgong, Japan) and erected with No. 20 sieve.
- 3 ⁇ 4 loose acetate 320S (acetal group 32%) and shellloose acetate 398-10 F (acetal group 39.8%) were dissolved in a mixture of ethane and methylene chloride to prepare a fluidized bed granulation coater with the above formulation. (GPCG-1; Glatt, Germany) and coated. After completion of coating, magnesium stearate was added, mixed for 4 minutes, and equipped with a rotary tablet press (MRC-30, Sejong Pharmatech, Korea) equipped with a 5 mm diameter punch. Tableting gave a nuclear tablet, the titled delayed-release compartment. (3) tableting and coating
- the tablets were compressed into nuclear tablets. Separately hydroxypropylme 3 ⁇ 43 ⁇ 4rose 2910 (S in-ets, Japan), pulley 3 ⁇ 4 lenglycol 6,000 (BASF, Germany), talc (Luzenac, France), titanium oxide (Tioside Americas, USA) and ethane.
- Example V-2 Preparation of Amlodipine-Roschvastatin Nucleated Tablets
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
본 발명은 약리학적 활성성분으로 스타틴계 지질저하제, 그의 이성질체, 또는 그의 약학적으로 허용가능한 염을 포함하는 선방출성 구획, 및 약리학적 활성성분으로 디히드로피리딘계 칼슘 채널 차단제, 그의 이성질체, 또는 그의 약제학적으로 허용가능한 염을 포함하는 지연방출성 구획을 포함하는 약제학적 제제를 제공한다. 본 발명에 의한 스타틴계 지질저하제를 함유하는 선방출성 구획, 및 디히드로피리딘계 칼슘 채널 차단제를 함유하는 지연방출성 구획을 포함하는 약제학적 제제는 대사 증후군 및 인슐린 저항성 환자 및 당뇨병 혹은 당뇨병 전증으로 의심되는 환자들의 심혈관 질환, 심폐 질환, 폐질환 또는 신장 질환의 예방 또는 치료에서 약리학적, 임상학적, 과학적 및 경제적으로 각각의 약물의 단일 제제 및 단순 복합 제제보다 매우 유용하다. 본 발명에 의한 제제는 스타틴계 지질저하제와 디히드로피리딘계 칼슘채널 차단제의 용출속도를 달리 구성하여 약물 상호간의 길항 작용 및 부작용을 방지하고, 2종의 약물을 단회 투여함으로써 환자의 복용을 용이하게 하는 효과를 나타낸다.
Description
【명세서】
【발명의 명칭】 심혈관계 질환 치료용 약제학적 제제
【기슬분야】
본 발명숀 복합성분 각각의 체내 약리 작용 발현 시간에 시차를 두어 투약하 는 이른바 시간차 투약 (Chronotherapy) 원리와 약물의 체내 대사 원리 (XenoMotics)를 적용하여 특정 속도로 각 약물을 제어 방출할 수 있도록 설계한 방출성이 제어된 디히드로피리딘계 칼슘 채널 차단제 및 스타틴계 지질저하제를 활 성성분으로 포함하는 약제학적 제제에 관한 것이다.
【배경기술】
고¾압은 관상 동맥 질환과 공존하는 경우가 많으며 , 심장 질환을 초래하는 주요 원인이다. 고혈압은 고지혈증에 기인한 동맥 경화와 서로 밀접한 관련이 있 다. 즉, 혈압이 상승하면 등맥경화가 악화되고 동맥경화가 악화되면 혈압이 더욱 상승하여 서로를 악화시키는 작용을 나타낸다. 이 같은 증상은 심혈관 질환을 발병 시키는 심각한 위험인자로서 인식되고 있다. 고지혈증의' 한가지 유형인 고콜레스테 를¾증은 상승된 혈청내 저밀도지단백 (LDL)콜테스테를 및 혈청내 총콜레스헤를의 상승을 특정으로 한다. 따라서, 혈청내 지질 , 특히 LDL콜레스테를 수치를 낮추면 심혈관 질환의 발병 가능성을 낮추고, 동맥경화증의 진행을 지연시키거나 또는 동 떽경화증의 퇴행을 유도할 수 있다 [참조: American diabetes associat ion, Diabetic care 2000; 23: S57] .
고콜레스테를혈증은, 관상 동맥, 경등맥 및 말초 동맥을 포함하는 등맥 내에 지질 침적튤이 블균등하게 분포하는 것을 특징으로 하는 죽상동멕경화증의 초기 발 병에 관여한다. 이러한 불규칙한 죽종 분포는 관상 동맥 손상, 심혈관 질환의 특징 이 된다 [참조: Am J Cardiol 1987 59(14) : 91G] .
동맥경화와 고혈압은 익 "순환적으토 증상을 상호 악화시키는. 증상이며, 따라 서 고지혈증 환자나 고혈압 환자는 모두가 동덱경화와 고혈압을 등시에 치료해야 하며 악화를 방지해야 한다 [HypertensRes2001;24:3-ll, Hyper tensRes2003; 6: 1-36 , Hyper tensRes2003; 26: 979-990] . 이에 , ¾압강하 효과를 나타내는 칼슴채널차단제와 콜레스테를 합성 억제 효
과를 니:타내는 스타틴계 지질저하제를 병용 투여하면 상승효과를 나타낸다는 임상 에 대한 연구는 많이 발표되어 왔다. 크람시 (Kramsch)등의 문헌 [Journal of Human Hyperteasion(1995) (Sup l . l) , 53-59]에 의하면 죽상동맥경화증을 예방 치료하기 위해 암로디핀과 지질저하제를 배합하여 더욱 좋은 치료 효과를 나타내고 있다. 주크마 (Jukema) 등의 문헌 [Arteriosclerosis, Thr ombos i sandVascu 1 arBi o 1 ogy , voI .16No ,3, 1996,425-43()]에서도 칼슴채널차단제가 지질저하제와 배합함으로써 상 호 상송적 효과를 얻을 수 있음을 입증하고 있다. 또한 오레코브 (Orekhov) 등의 문 헌 [Int J Cardiol , 62, p.67-77, 199기에는 등맥경화증의 치료를 위해 지질저하제 로바스타틴과 배합한 암로디핀의 용도가 알려진 바 있다. 디히드로피리딘계 칼슘 채널 차단제는 고¾압 치료제일 뿐 아니라 협심증 치 제임은 주지의 사실이고, 또한 스타틴계 약물은 지질저하제일 뿐 아니라 여러 병 태에 대한 항염 작용을 나타내는 것으로도 잘 알려진 약이며, 그 증 심바스타틴은 아피 영 ^에서는 없이 약국에ᅵ서 구입이 가능할 정도로 안정성을 인정 받는 약 이다 [Cardiologyl992;80(Suppll) :S31-S36, JCardiovascPharmacol 1988; 12(Suppl7): S110-S113, Lancet2000; 356 :359-365, Hypertens Res 2002; 25: 717-725, HypertensRes2002;25:329-333,: Am J Manag Care. 2004 Oct ; 10(11 Suppl) :S332- 8. :Curr Control Trials Cardiovasc Med 2000, 1: 161165] . 이 두 약물군은 모두가 1일 1희 투여하는 제형으로 개발할 수 있어 , 저녁 식사시 복용하는 것이 약리학적 으로 가장 이상적인 투약 시간대임이 공통된 특성이다. 두 약물군 중 병용 처방되는 사례가 많은 약물로는 디히드로피리딘계 칼슘 채널 차단제 중' 암로디핀과 스타틴계 지질저하제 중 심바스타틴을 들 수 있다. 이 러한 두 약물의 병용시 암로디핀은 고유의 항압작용 뿐 아니라 지질저하제와 상승. . , 작용을 통해 심바스타틴의 지질저하 작용을 높여주는 기능을 발휘하고, 심바스타틴 역시 고유의 지질저하작용뿐만 아니리- 암로디 ¾과의 상승작용을 통해 약물고유의 혈압강하 작용을 증가시키는 기능을 발휘하게 되는 장점이 있다.
또다른 빈용 처방례인 암로디핀과 아토르바스타틴을 병용할 경우에는 다음과 같은 장첨이 있다.
동맥내에 지질 침적물이 블균등하게 분포함을 특징으로 하는 죽상등맥경화증 을 보인 환자는 혈관벽 내의 N0 생성계 (eNOs)가 비정상적이며 , 이 때문에 생성
이 감소하여 혈압을 높이게 된다. 일반적인 HMG~CoA 환원효소 억제제는 NO 방출 효 과가 거의 없지만 암로디핀은 NO방출을 촉진하는 기능을 갖는다. 그러나 암로디된 과 아토르바^타틴의 븍합투여는 NO에 대한 상승효과가 ¾씬 더 발휘된다. 아토르 바스타틴이 타 스타틴계 약물과 달리 페녹시기 (phenoxy group)가 있으며 이 작용기 에 의해 양자 공여 및 전자 안정화로 인하여 항산화 작용을 촉매함을 알 수 있다. 이는 02'-는 혈관 수축 작용을 NO'-는 혈관 확장 작용을 하는 것으로 즉, 암로디핀 과 아토르바스타틴의 복합투여는 이러한 혈관벽 내 eNOS 에 대한 상승효과로 인해 지질저하제인 아토르바스타틴이 칼슴채널 길항제인 암로디핀의 효과를 도와주는 복 합처방인 것이다 [The American Journal of Medicine, 118, 54-61 , 2005] 스타틴계 지질저하제 및 디히드리피리딘계 칼슴 채널 차단제에 대해 구체적 으로 살피본다.
스타틴 (Statin)계 지질저하제는 ΗΜ(Η Α 환원효소 억제제로 다음과 같은 일 반 정보를 가지며, 본원에서 스타틴계 지질저하제 및 HMG-CoA 환원효소 억제제는 상호 교환적으로 사용된다. 스타틴계 지질저하제는 협심증이나 심근경색 등, 관상 동맥경화증으로 인한 심장병 예방과 치료에 일차 선택 약이다 [Lancetl995;346:750- 753, AmJCar dio 11998; 82: 57T-59T, AmJCardiol 1995; 76: 107C-112C , Hyper tensRes 2003; 26: 699-704 , HypertensRes2003; 26: 273-280 , BrMedBul 12001; 59: 3-16, AmJMedl998; 104(Suppl 1) :6S-8S, CI inPharmacoki net 2002 ;41 :343-370. ] . 스타틴계 지 질저하제 증에는 실바스타틴, 아토르바스타된, 플루바스타린, 로바스타된 , 피타바 스타틴 , 프라바스타틴 , 로수바스타틴및 그의 약학적으로 허용되는 염과 이성질체가 주로 사용되며 그 증 심바스타틴이 가장 대표적으로 처방된다.
스타틴계 지질저하제는 간 내에서의 지질 합성은 초저녁 식사 이후 왕성해지 므로 스타틴계 약물은 초저녁에 복용토특 권장되어 왔다 [Arterioscler Thromb 11: 816-826, Am J Cardiol . 2006 Jan 1; 97(1) :44-7. Epub 2005 Nov 8] .
죽상동맥경화 환자나 당뇨병 환자는 혈관벽 내의 N0 생성계 (eNOs)가 비정상 적이다. 때문에 N0 생성이 감소하여 혈압을 증가시키게 된다. 심바스타틴을 포함한 스타틴계 지질저하제는 바로, 이러한 ¾관벽 내 eNOS를 정상 수준으로 증가시켜 놓 는다. 이것 또한 지질저하 작용이 항압작용을 도와주는 복합 처방 효과 중 하나다 [Am J Physiol Renal Physiol Vol 281 Issue 5: F802-F809, 2001] .
스타린계 지질저하제를 주로 활성화시키는 대사효소는 사이토크룸 P450 3A4
및 2C9이며, 대사물은 간 내에서 작용하면서 간에서부터 담도를 통해 배설된다
[DrugMetabDisposl990l 18: 138-145, DrugMetabDisposl990; 18:476-483 DrugMetabDisposl997; 25: 1191-1199 1997;53 :828-847 C I inPharraacoki net 1996; 5: 348-371 , Ar chB i ochemB i op ysl991 ; 290 : 355-361
Drug etabDisposl997; 25:321-331: DrugMetabDispos .1999Mar; 27(3): 410-6. ] .
따라서 , 스타틴계 지질저하제와 병용하는 약물이 스타틴계를 활성화 시키는 사이토크름 P450 효소를 억제하는 경우에는 스타틴계 지질저하제의 간내 대사가 억 제되어 스타틴계 물질과 중간 대사물질이 혈중으로 유출되어 ¾증농도가 증가하게 된다. 이와 같이 혈증으로 유리된 스타틴계 본체물질 및 활성대사물질은 근육융해 증갈은 심각한 부작용을 유발활 수 있다 [Cl inPharmacolThar 1998 ;63: 332-341, CI inPharraacolTherl998; 64: 177-182, Physici ansDeskRef erence 2006(Zocor ) , JPharmacolExpTher 1997;282:294-300, PharmacolExpTher 1999 ;290: 1116-1125, L if eSc Ϊ200 ; 76: 281-292.: Drug Metab Dispos 1991 ;19: 740-
748:EurJCl inP armaco 11996; 50 :209-215] .
즉 , 두 약물의 단순 병용시 디히드로피리딘계 칼슴채널 길항제가 먼저 간에 도달하고 사이토크름 P450 효소의 유도 * 억제하게 되며, 뒤따라 간으로 들어오거 나 동시에 들어오게 되는 스타린계 지질저하제의 상당 부분은 사이토크름 P450 효 소의 대사 작용을 받지 못하므로 혈증으로 많은 양이 유출되거나 배설이 지연되거 나 축적 작용을 일으키게 되며, 상기 사이토크름 P450 효소에 의해 대사 되지 않은 스타 ¾계 지질저하제나 그의 β -히드록시산이 혈증으로 이행하여 혈중 농도가 필요 이상 높아져 근육 용해증과 같은 근 장애를 유발시킬 수 있는 것이다.
심바스타틴은 그 자체가 블활성 락론 (Lactone)계 화합물이며 일차적으로 간 으로 들어가서 지 질저하작용을 지닌 활성형인 심바스타¾의 |3ᅳ히드특시산 ( β— hydroxy acid) 등 활성 대사물 S 변한다. 심바스타틴은 간 내의 사이토크름 P450 3A4 에 의해 여러 단계로 대사되고 , 활성화된 대사물질들은 강력한 지 질 저하 작용 을 발휘한다. 심바스파틴이 관상동맥성 심장질환 발병 1:을 감소시키고 사망를을 감 소시키는 효과가 대단위 임상시험을 통해 입증되어은 것도 잘 알려진 사실인 데 [Lancet 199 ; 34 : 1383-1389. ] , 이는 간 내에서의 콜레스테를을 합성하는 과정에서 핵심 역할을 하는 HMG-CoA 환원 효소를 심바스타틴이 강력하게 억제하기 때문이며 동시에 염증 유발 인자를 억제하는 작용을 발휘하기 때문이다 [ "Scandinavian Simvastat in Survival Study" published in the Lancet , 1994 , 344, 1383-89] ,
피타바스타 ¾은 사이토크롬 P450 2C9에 의해 대사되고 간 내에서 작용하면서 배설되므로, 사이토크름 P450효소를 억제하는 약물과 병 투여시 피타바스타틴의 간 내 대사가 억제되어 혈중농도가 증가하게 되고 , 이로 인해 근육용해증과 같은 부작용이 발생할 수 있다 .
칼슴 채널 차단제는 스타틴계 지질저하제와 가장 많이 병용 처방 되는 항암 제의 하나로서 칼슘 채널 차단제 증에서 대표적으로 디히드로피리딘계 칼슴 채널 차단제가 있으며 디히드로피리딘계 칼슴 채널 차단제로서, 암로디핀 , 레르카니디 핀 , 라시디핀, 펠로디핀 , 바니디핀 , 베니디핀 , 실니디핀, 이스라디핀, 마니디핀 , 니카르디핀, 니페디핀, 니모디핀 , 닐바디된 , 니술로디핀 니트렌디핀 , 아¾니디핀 및 그의 염 및 이성질체가 주로 사용되며 그 중에서도 암로디핀이 가장 대표적으로 처방된다. 특히 암로디핀은 항압제 및 협심증 치료제로서 전 세계적으로 가장 많이 처방 되고 있다 [Cardiologyl992 ;80(StiE»pll) :S31— S36 , JCardi ovascPharmaco 11988; 12 (Suppl7) :S110-S113, Lancet 2000; 356: 359-365 , Hyper tensRes2002 ;25: 717-725 , Hyper tensRes2002; 25: 329-333 · ],
디히드로피리딘계 칼슴 채널 차단제는 ¾관 평활근 내 칼슴 유입을 차단하여 말초 동맥 확장을 유도함으로써 혈압을 강하하는 항고혈압 약물로서 경련성 혈관수 축으로 인한 협심증에 유효한 공통의 특성을 갖는다. 또한, 칼슘채널 차단제가 초 기 등맥경화성 병소의 치료에 유리한 효과를 가져을 수 있음이 알려져 있다 [Lancet , 335, p.1109-1139, 1990 ; 및 Circulation, 82, p.19401953, 1990] .
병태생리학적인 견지에서 볼 때 , 특히 아침 기상 후 오전 중의 혈압 상승은 스트레스 자극에 기인하는 ¾관벽 경련에 의한 ¾압 상승인데 , 디히드로피리딘계 칼슘 채널 차단제는 이러한 경련성 혈관 수축을 이완시키는 작용이 주작용이므로 특히 아침 기상 후 오전 중의 혈압 강하 작용이 강하게 나타난다. 따라서 , 저녁시 간 대에 루여한 만약 디히드로피리던계 킬:슴 채널 차단제를 저녁시간에 복응하게 되면 기상전에 최고 ¾증농도에 도달하게 되고 , 기상 후 일과 중에 가장 강력하게 혈압 강하 작용을 나타나게 할 수 있다.
디히드로피리딘계 칼슴 채널 차단제의 대표적인 예인 암로디 ¾은 혈관 평활 근 내 칼슴 유입을 차단하여 말초 등맥 확장을 유도함으로써 혈압을 강하하는 항고 혈압 약물로서 경련성 혈관수축으로 인한 협심증에 유효한 특성을 가지며 , 그 화학 명이 3—에 ¾-5-메틸ᅳ 2ᅳ(2-아미노에특시메 ¾ )-4-(2-클로로페닐 )-1 , 4-디하이드로 -6- 메틸ᅳ 3, 5ᅳ피리딘디카르복실레이트로서 반감기가 30 ~ 50 시간으로 매우 지속적이
고 장기간에 걸쳐 활성을 나타내는 매우 유용한 칼슴채널차단제이다 [유럽특허공개 제 89, 167호 및 미국 특허 제 4, 572, 909호] . 암로디핀을 단일제로 경구 투여하면 소 장에서 :수되어 간에서 40% 이상 분해되고 , 나머지 & 정도만이 혈증으로 나가게 되며 층분한 혈압 강하 작융을 발휘한다. 또한 암로디핀은 1일 24 시간 지속형 약 물이며 , 저녁 시간대에 복용하는 것이 익일 아침 시간부터 한낮 동안에 혈압 저하 작용을 가장 강력하게 발휘한다.
니떼디핀은 단일제로 경구 루여하면 대부분이 불활성형의 대사체의 형태로 변하며 , 투여 ¾의 60~80%가 신장을 통해 뇨로 배설되고 , 일부가 담도를 거쳐 변으 로' 배설된다. 니페디핀의 대사는 사이토크름 P450 효소계에 관여하며 , 사이토크름 P450 3A4 에 강력한 억제 작용을 나타낸다. 니폐디핀의 배설 반감기는 약 2시간 남 짓으로 알려져 있다. 투여시 하루 종일 혈압강하 작용을 ¾기 위해서 , 시관되는 니 꽤디핀 제제는 하루동안 서서히 방출되는 형태로 제공되고 있다. 병태생리학'적인 견지에서 블 때 , 특히 아침 기상. 이후 오전 중의 ¾압 상승은 스트레스 자극에 기 인하는 혈관벽 경련에 의한 혈압 상승인데 , 니폐디핀은 이러한 경 ¾성 혈관 수축을 이완시키는 작용이 주작용이다. 특히 아침 기상 후 오전 증의 혈압을 효과적으로 강하시킨다. 파라서, 니페디핀을 저녁시간 (17~23시)에 복용하게 되면 기상전에 최 고 혈중농도에 도달하게 되고, 기상 후 일과 중에 가장 강력하게 ¾입" 강하 작용을 나타나게 할 수 있다. ' 상기 살핀 바와 같이 , 사이토크름 P450 효소가 이미 존계할 때는 암로디핀과 같은 디히드로피리딘계 약물은 이 효소에 의해 일부가 억제될 수 있으나, 곧 사이 토크롬 P 450 3A4 효소의 생성을 억제하는 작용을 발휘하게 된다. 디히드로피리던계 약물은 대부분 사이토크름 P450 3A4 효소에 의해 주로 대사되며 사이토크름 P450 2C9 등 사이토크름 P450 3A4 이의의 효소에 의해서도 대사된다 [Arc lnternMed .2002Feb25; 162(4): 405-12. , Dru^et abDi spos .2000Feb; 28(2): 125- 30. ] 이러한 특성 때문에 사이토크름 P450 호소를 ' 필요로 하는 약물인 스타틴계 지질저하제와 디히드로피리딘계 약물의 병용시에는 약물외 상호작용을 막기 위해 시간차를 두고 투여해야 한다 [MedCheml991;34: 1838-1844,
EurJCl inPharraacol2000; 55: 843-852. ] .
따라서 , 스타틴계를 활성화시키는 사이토크름 P450 효소의 ' 생성을 억제하는 디히드로피리딘계 칼습 채널 차단제와 같은 약물과 병용 투여시에는 특별한 투여법
이 필요하다> 그러나 지금까지 디히드로괴리딘계 칼슘 채널 차단제와 스타틴계 지 질저하제를 시간차를 두고 작용할 수 있도록 한 약제학적 제제는 발명된 바 없고 허가된 바도 없다. 또한 처방의 들은 이 두 약물을 병용처방 하면서도 환자에게 이 두 약물을 시간차를 두고 복용하라는 등의 적절한 복약지도를 해주지 못하고 있다. 상기 두 약물을 등시 북용할 경우 스타된계 지질저하제의 혈중 농도가 상숑 하여 부작용을 초래할 수 있으며 , 두 약물을 병용함으로써 얻을 수 있는 보다 효과 적인 혈압 강하 작용 및 효과적인 지질 저하 작용도 얻기 어렵다. 이에 대하여 신이치로 니시오 (Shinidiiro Nishio) 등의 [Hypertens Res , 2005, Vol .28, No.3] 연구는 고지혈증을 지닌 고혈암 환자에게 디히드로피리딘계 칼슘 채널 차단제인 암로디핀과 스타틴계 지질저하제인 심바스타틴의 단일제를 동 시에 투여할 경우와 심바스타틴 단일제만을 투여한 경우를 비교 실험한 경우를 발 표하고 있다.
아래 표 1의 실험 결과에 의하면, 심바스타틴과 암로디편을 동시에 투약하면 암로디핀으로 인해 사이토크름 P450 3A4 효소가 억제되어 심바스타린과 활성 대사체 의 혈증 농도가 활성 역가 측정법으로 측정해 보았을 때 3» 이상 증가했음이 밝혀 졌으며 , 이로 인한 부작용 발생 위험 가능성을 발표하고 있다 [Hypertension Research Vol . 28 (2005) , No.3 March 223-227] .
[표 1]

상기 표 1에 나타낸 바와 같이, 지질저하 성분의 혈중 농도가 심바스타¾만 을 단일 투여했을 때 보다 복합 처방시의 혈중 농도가 활성 역가 측정치로 블 때
30% 정도 더 높게 나타났다. 그럼에도 불구하고, 지질 저하 작용은 상승하지 않았 다. 간에서 작용해야 할 심바스타틴 활성 대사체의 ¾중 농도가 필요이상 높아지면 오히려 콜레스테를의 생합성 저해 효과는 감소되고, 근육 융해증과 같은 중대한 부 작용이 나타날 확률이 높아질 뿐인 것이다.
한국등록 특허 제 582347호는 디히드로피리딘계 칼슘 채널 차단제인 암로디된 이 속방출되어 맨 던저 용출되게 하고 스타틴계는 24시간을 통해 서서히 용출되게 하여 스타린계 약물이 일시에 용출되어 흡수되는 것을 억제하는 복합제에 관하여 개시하고 있다. 이는 본 발명에서 스타틴계롤 먼저 용출하여 간에서 먼저 대사되게 하는 개념과는 전혀 다론 개념의 복합계로서, 는리적으로나 약리학적으로 스타틴계 의 약효를 감소시키는 비합리적 제재로 사료된다. 홰냐하면 암로디핀이 던저 간에 유입되면 사이토크롬 ί¾50 3A4의 생성이 간에서 억제되므로 곧 이어 간으로 유입되 는 소량의 스타틴계는 간에서 전혁 대사 받지 못하고 혈증으로 유출되고 만다. 상 기 특허는 간에서 작용해야 할 스타틴이 ¾중으로 유출되어 근육 융해 등의 부작용 만 증가시킬 뿐이다.
또한, 한국등록 특허 제 742432호는 암로디편 ¾실레이트 및 심바스타틴을 포 함하는 약제학적 제제 및 이의 제조방법에 관한 특허이나, 두 성분이 동시에 용출 되어 간으로 흡수되므로 심바스타틴이 필요로 하는 사이토크콤 P450 3A4가 암로디 핀에 의해 억제띄기 때문에 , 심바스타틴은 간에서 층분히 활성화되지 못한 채 증간 활성체와 함께 혈중으로 30% 이상이 방출되어 지질저하작용은 감소되고 부작용만 증가시킬 수 있다 (본 발명의 임상 실험을 통해서도 입증되어 있음) . 따라서 , 상기 발명은 단순 복합제 개념이며 이러한 단순 북합제는 진보성 부족으로 인해 거철되 고 있다. 또한, 화이자의 한국공개특허 제 200으 7002144호 역시 암로디핀과 아토 a 바스타틴이 단순 복합제라 하여 한국 특허청으로부터 거절된 바 있다.
즉, 상기한 결과 등으로 볼 때 그 동안 단일 제제를 단순히 복합 처방하거 나, 디히드로피리딘계 칼슴 채널 차단제를 스타틴계 지질저하제 보다 던저 복용하 는 것으로부터 나타나기 쉬운 문제점인 약물의 상호 길항 작용을 예방할 수 있는 투약법 혹은 약제학적인 제제의 개발의 필요성이 더욱 커지게 되었다.
이에 , 본 발명자들은 고혈압과 고지혈증 및 그로 인한 심혈관계 질환 또는 대사증후군의 예방 및 치료에 효과적인 약제학적 제제를 개발하기 위해 연구한 결 과 본 발명을 완성하였다.
【발명의 상세한 설명 I
【기술적 과제】
본 발명은 디히드로피리딘계 칼슴 채널 차단제 몇 스타 ¾계 지질저하제를 포 함하는 제제의 방출을 제어하여 위장관내 용출 및 흡수 시간에 있어서 약 2시간 내
지 약 4시간 이상의 차이를 나타내는 약제학적 제제를 제공하고자 한다.
본 발명은 또한 디히드로피리딘계 칼슴 채녈 차단제 및 스타틴계 지질저하제 를 포함하는 제제로서 , 약물의 약물전달시간을 최적화하여 대사 증후군 및 인술린 저항성 환자, 및 당뇨병 흑은 당뇨병 ¾증으로 의심되는 환자들의 심혈관 질환, 심 폐질환, 폐질환 또는 신장 질환의 예방 또는 치료라는 칼슘 채널 차단제의 효과와 협심증이나 심근경색 등 관상동맥경화증으로 인한 심장병 예방 또는 치료라는 HMG- CoA 환원 효소 억제제의 효과를 극대화하고, 두 약물간 상호작용 및 이로 인한 부 작용을 피할 수 있는 약제학적 제제를 제공하고자 한다.
나아가 본 발명은 디히드로피리딘계 칼슴 채널 차단제 및 스타틴계 지질저하 제를 포함하는 제제로서 , 저녁시간대, 바람직하게는 저녁 5시 내지 11시 사이에 1 일 1회 1정을 복용함으로써 편리성까지 부가하여 환자의 복약 순웅도를 더욱 높여 춥 수 있는 약제학적 제제를 제공하고자 한다.
【기술적 해결방법】
본 발명은 복합성분 각각의 체내 약리 작용 발현 시간에 시차를 두어 투약하 는 이른바 시간차予약 이론 (Chronotherapy) 원리롤 적용하여 특정 속도로 각 약물 을 제어 방출할 수 있도록 설계된 약제학적 제제에 관한 것이다, 즉, 본 발명의 약 제학적 제제는 복합 성분 각각의 체내 약리 작용 발현에 시 차투약 (GhronotheraDy)원리와 약물의 채내대사 (Xenobiotics)원리를 적용하여 특정 속도로 체내에서 제어 방출하여 체내 흡수시 가장 이상적인 효과를 나타낼 수 있도특 설계 된 약물 송달 시스템이다.
본 발명은 약리학적 활성성분으로 스타틴계 지질저하제 , 그의 이성질쳬 , 또 는 그의 약학적으로 허용가능한 염을 포함하는 선방출성 구획, 및 약리학적 활성성 분으로 디히드로피리딘계 칼슘 채널 차단제, 그의 이성질체 , 또는 그의 약제학적으 로 허용가능한 염을 포함하는 지연방출성 구획을 포함하는 약제학적 제제를 제공한 다. 본 발명은 디히드로피리딘계 칼슴 ¾항제를 활성성분으로 하며 , 과립, 펠렛 또는 정제로 제조된 지연방출성 구획과, 스타틴계 지질저하제를 활성성분으로 하 며., 과립 , 또는 정제로 제조된 선방출성 구획을 포함하여 이루어진 캡술제인 시간차 용출이 가능하도특 방출성이 조절된 약제학적 제제를 제공한다.
본 발명의 제제 증 상기 디히드로피리딘계 칼슴 길항제는 스타린계 지질저하
제 보다 2시간내지 4시간늦게 간에서 흡수되도록 방출성이 조절될수있다.
본 발명에 있어서 디히드로피리딘계 칼슴 길항제는 암로디핀, 레르카니디 핀, 라시디핀, 펠로디핀, 바니디핀, 베니디핀, 실니디핀, 이스라디핀, 마니디 ¾, 니카르디핀, 니페디핀, 니모디핀, 닐바디핀, 니술로디¾, 니트렌디핀, 아¾니디핀 이들의 약학적으로 허용 가능한 염 및 이들의 이성질체 증에서 선택된 것일 수 있 으며 , 제제중 1 ~ 400 rag범위로포함될수 있다.
본발명에 있어서 상기 지연방출성 구획은장용증합체, 수불용성 중합체, 소 수성 화합물, 친수성 고분자, 삼투성 반투막코팅기제 및 삼투압조절제 중에서 선 택된 1종 이상의 방출제어 물질을포함할수 있으며 상기 장용증합체는폴리비닐아 세테이트프탈테이트, 히드록시프로필메 ¾셸롤로오스프탈레이트, 쉘락, 셀를로오 ώ 아세테이트프탈례이트, 샐를로오스프로피오네이트프탈테이트, 풀리 (메타크릴레이 트, 메틸메타크릴테이트)공중합체 및 플리 (메타크릴레이트, 에틸아크릴레이트)공중 합체 중에서 선택된 1종또는 2종 이상의 흔합물 반투과성막코팅기제 코팅기제 는 폴리비닐 아세테이트, 풀리메타크릴레이트 공중합체로서 플리 (에틸아크릴레이트 -메 ¾ 메타크릴레이트) 공중합체, ¾리 (에틸아크 ¾레이트-메 ¾메타크릴레이트 -트리 메틸아더노에¾데타크릴레이트)공중합체, 에틸셸를로오스, 샐를토오스 에스테르, ¾를로오스 에.테르, 쎌를로오스 아실레이트, 셀를로오스 디아실레이트, 셀를로오스 트리아실레이트, 셀를로오스 아세테이트, 샐를로오스 아세테이트 및 셀를로오스 트리아세테이트에서 선택된 1종또는 2종 이상의 흔합물인 것 ; 상기 삼루압조절제 는 황산마그네슘, 염화마그네슴, 염화나트륨, 염화리튬, 황산칼콤, 황산나트륨, 황 산리튬및 황산나트콤선택된 1종또는 2종 이상의 흔합물인 것;
상기 스타틴계 지질저하제는 심바스타틴, 로바스타¾, 아토르바스타틴, 피타바스타 틴, 로수바스타된, 플루;바스타틴, 프라바스타 ¾, 이들의 약제학적으로 허용가능한 염 및 이들의 이성질체 중에서 선택된 1종또는 2종 이상의 흔합물인 것일 수 있 다. 본 발명은 또한 (a) 활성성분으로서 디히드로피리딘계 칼슴길항제, 이의 약 학적으로 허용가능한 염 또는 이의 이성질체꽈, 약학적으로 허용가능한 담체 또는 희석제가 포함된 지연방출성 구획 (W 활성성분으로서 스타된계 지질저하제, 이의 약학적으로 허용가능한 염 또는 이의 이성질체와, 약학적으로 헉용가능한 담체 또 는 희석제를 이용하여 제조한선방출성 구획 및 (c) 상기 제 1제와제 2제를함께 층
진하기 위한 용기 수단을 포함하는 키트를 제공한다.
본 발명은 또한, 디히드로피리딘계 칼슘 길항제롤 활성성분으로 하며 , 정제 로 제조된 지연방출성 구획과, 스타틴계 지질저하제를 활성성분으로 하며 , 상기 제
1제의 표면에 코팅되는 선방출성 구획을 포함하여 이루어진 코팅정제인 시간차 용 출이 가능하도톡 방출성이 조절된 약제학적 제제를 제공한다.
본 발명은 또한 디히드로피리딘계 칼슘 길항제를 활성성분으로 하며 삼투압 에 의해 약물을 방출하는 내핵을 구성하는 지연방출성 구획과, 스타틴계 지질저하 제 * 활성성분으로 하며, 상기 내핵의 외층을 구정하는 선방촐성 구획을 포함하여 이루어진 이증층의 단일정인 시간차 용출이 가능하도록 방출성이 조절된 약제학적 제제를 제공한다. 본 발명은 약리학적 활성성분으로 스타린계 지질저하제, 그의 이성질체, 또 는 그의 약학적으로 허용가능한 염을 포함하는 선방출성 구획 , 및 약리학적 활성성 분으로 디히드로피리딘계 칼슘 채널 차단제 , 그의 이성질체 , 또는 그의 약제학적으 로 허용가능한 염을 포함하는 지연방출성 구획을 포함하는 약제학적 제제를 제공한 다.
본 발명에 의한 제제는 두 활성성분간의 방출성을 제어하는 물리적으로 분리 되거나 구획되어 두 성분간의 상이한 방출속도를 얻을 수 있도특함으로써 , 기존 단 일제제의 병용투여 또는 등시투여의 문제점을 개선하여 보다 유웅한 치료효과를 제 공한다.
본 발명의 제제는 스타틴계 지질저하제를 일차로 용출시켜 소장에서 던저 흡 수되도록 하는 선방출성 구획 및 디히드로피리딘계 칼슘길항제는 스타틴계 지질저 하제 보다 2시간 내지 4시간 늦게 간에서 흡수될 수 있도록 하는 지연방출성 구획 을 포함하는 약제학적 제제이다.
본 발명은 약리학적 활성성분으로 심바스타틴 , 이의 이성질체 또는 이의 약 제학적으로 허용 가능한 염을 포함하는 선방출성 구획 , 및 약리학적 활성성분으로 암로디핀 , 이외 이성질체 또는 그의 약제학적으로 허용 가능한 염을 포함하는 지연 방출성 구획을 포함하는 약제학적 제제 (이하 '심바스타된 /암로디핀 제제 '라 함)를 제공한다.
또한, 본 발명은 약리학적 활성성분으로 아토르바스타틴, 이의 이성질체 또 는 이의 약제학적으로 허용 가능한 염을 포함하는 선방출성 구획, 및 약리학적 활
성성분으로 암로디핀, 이의 이성질체 또는 그의 약제학적으로 허용 가능한 염을 포 함학는 지연방출성 구획을 포함하는 약제학적 제제 (이하 '아토르바스타틴 /암로디핀 제제 '라 함)롤 제공한다 .
또한, 본 발명은 약리학적 활성성분으로 아토르바스타틴, 이의 이성질체 또 는 이의 약제학적으로 허용 가능한 염을 포함하는 선방출성 구획 , 및 약리학적 활 성성분으로 니폐디핀, 이의 이성질체 또는 그의 약제학적으로 허용 가능한 염을 포 함하는 지연방출성 구획을 포함하는 약제학적 제제 (이하 1아토르바스타틴 /니페디핀 제제 '라 함)를 제공한다.
또한, 본 발명은 약리학적 활성성분으로 피타바스타틴 , 이의 이성질체 또는 이의 약제학적으로 허용 가능한 염을 포함하는 선방출성 구획 , 및 약리학적 활성성 분으로 암로디핀, 이의 이성질체 또는 그의 약제학적으로 허용 가능한 염을 포함하 는 지연방출성 구획을 포함하는 약제학적 제제 (이하 . '피타바스타틴 /암로디핀 제제 ' 라 함)를 제공한다.
또한, 본 발명은 약리학적 활성성분으로 로슈바스타틴 , 이의 이성질체 또는 이의 약제학적으로 허용 가능한 염을 포함하는 선방출성 구획 , 및 약리학적 활성성 분으로 암로디핀 , 이의 이성질체 또는 그의 약제학적으로 허용 가능한 염을 포함하 는 지연방출성 구획을 포함하는 약제학적 제제 (:이하 '로슈바스타틴 /암로디핀 제제 ' 라 함)를 제공한다.
또한, 본 발명은 지방출성 구획의 약리학적 활성성분 및 선방출성 구획의 약 리학적 활성성분이 각각 베니디핀 몇 피타바스타틴 , 니모디핀 및 프라바스타틴, 니 발디 ¾ 및 피타바스타틴 , 니솔디핀 및 로바스타틴 니카르디핀 및 로수바스타틴, 니트렌디핀 및 프라바스타틴 , 니페디핀 및 풀루바스타틴 , 라시디핀 및 심바스타틴, 레르카니디핀 및 심바스타틴 , 마니디핀 및 로바스타¾, 바니디핀 및 로바스타¾, 실니디핀 및 프라바스타틴, 암로디핀 및 로바스타된, 이스라디핀 및 4루바스타틴 , 아졀니디핀 및 심바스타틴 또는 펠로디핀 및 아토르바스타틴인 약제학적 제제를 제 공한다.
본 발명은 또한 계제 중 디히드로피리딘계 칼슴 채널 차단제의 방출이 , 스타 틴계 지질저하제 방 * 약 1시간 이후에 개시되고 약 8시간 이전에 완료되는, 바람 직하게는 스타틴계 지질저하제 방출 약 2시간 이후에 개시되고 약 6시간 이전에 완 료되는 약제학적 계제를 제공한다,
상기 디히드로피리딘계 칼슘 채널 차단제 중 니페디핀을 활성성분으로 포함
하는 제제는 니폐디핀의 방출이 충분히 지연되어 약 1~8시간이 지난 후 용출이 개 시되도콕 절되고, 바람직하게는 아토르바스타틴은 1시간 이내에 약물의 90%이상 용출되고, 니페디핀은 방출이 층분히 지연되어 경구 투여 후 약 2~6 시간이 지난 후 용출이 개시되도록 조절된다.
예를 들어, 본 발명의 심바스타틴 /암로디핀 제제 , 또는 피타바스타틴 /암로디 핀 제제 증 암로디핀의 방출은, 각 스타틴계 지질저하제 방촐 약 1시간 이후에 개 시되고 약 8시간 이전에 완료되거나, 바람직하게는 약 2시간 이후에 기)시되고 약. 6 시갚 이전에 완료되며 본 발명의 아토르바스타 ¾/암로디핀 제제 증 암토디핀 방출 은, 아토르바스타틴 방출 약 1시간 이후에 개시되고 약 8시간 이전에 완료되거나, 바람직하게는 약 1시간 이후에 개시되고 약 6시간 이전에 완료되고 로슈바스타틴 / 암로디핀 제제 증 암로디핀 방출은 로슈바스타틴 방출 약 1시간 이후에, 바람직하게 는 약 2시간 이후에 개시될 수 있다,
본 발명의 약제학적 제제는 스타틴계 지질저하제의 방출 개시 후, 디히드로 피리딘계 칼슘 채널 차단제 총량의 약 40¾ 이하 방출이 유지되는 시간이 약 2시간 이 내 , 바람직하게는 약 3시간 이내 , 더욱 바람직하게는 약 4시간 이내인 제제를 제공하 여, 디히드로피리딘계 칼슘 채널 차단제의 약효를 일정 지연시간 경과 후 효과적으 로 발생할 수 있게 한다. 예를 들어 심바스타틴 /암로디핀 제제 또는 아토르바스타 틴 /암로디핀 제제 , 심바스타틴 또는 아토르바스타틴의 방출 개시 후, 암로디핀 총 량의 약 40¾ 이하 방출이 유지되는 시간이 약 2시간 이내 , 바람직하게는 약 3시간 이 내 , 더욱 바람직하게는 약 4시간 이내이며 피타바스타틴 /암로디핀 제제 또는 로슈바 스타린 /암로디핀 제제는 암로디핀의 4시간까지의 용출 1:이 총량의 40%를, 바람직하 게는 30¾를 넘지 않을 수 있다. 본 발명의 보다 바람직한 약제학적 제제는, 스타틴계 지질저하제가 먼저 방 출되어 1시간 이내에 8M, 바람직하게는 90% 이상이 용출 완료되고 디히드로피리딘 계 칼슴 채널 차단제는 약 2시간 이후에 용출이 개시되어 , 약 4시간 이내에 총량의 40%, 바람직하게는 약 30%이내로 방출되는 제제이다. 예를 들어, 심바스타틴 /암로 디핀 제제 , 아토르바스타틴 /암로디핀 제제, 아토르바스타린 /니페디핀 제제, 또는 피타바스타틴 /암로디핀 제제에서 각 스타틴계 지질저하제는 1시간 이내 80¾ 이상 이, 바람직하게는 90% 이상이 용출되고 암로디핀 또는 니페디 ¾은 은 2시간 이후에 용출이 개시 되며 , 4시간까지의 용출률이 40%를, 바람직하게는 30¾를 넘지 않는 수
준으로 방출되도록 조절된다. 또한 로슈바스타틴 /암로디휜 제제에서 로슈바스타틴 은 30분 이내 , 바탐직하게는 15분 이내에 80% 이상이 방출되고, 암로디핀은 방출이 층분히 지연되어 경구 투여 후 총 4시간 이내에 40¾이하로, 바람직하게는 3» 이하 로 방출된 약제학적 제제를 제공한다. 또한, 아토르바스타틴 /니페디관 제제에 있어 서는 아토르바스타틴이 1시간 이내 80% 이상이 용출되고, 니페디핀은 방출이 층분 히 지연되어 약 1~8시간이 지난 후 용출이 개시되도록 S절되고, 바람직하게는 아 토르바스타틴은 1시간 이내에 90%이상 용출되고 , 니페디핀은 방출이 층분히 지연되 어 경구 투여 후 약 2~6 시간이 지난 후 용출이 개시되도록 조절될 수 있다. 본 발명의 약제학적 조성물은 1일 1희 ᅳ 저녁 5시 내지 11시 사이에 복용되는 경우, 가장 유효한 효과를 발휘한다.
또한 상기 제시된 선방출성 구획과 제어방출성 구획은 다양한 제형으로 구현 가능하다.
본 발명의 약제학적 제제의 각 구성성분을 보다 상세히 설명하면 다^과 같 다.
1. 선 (先)방출성 구획
선방출성 구획은 본 발명의 약제학적 제제에 있어서 지연방출성 구획에 비해 먼저 방출되는 구획을 의미한다.
(1) 약리학적 활성성분
선방출성 구획의 약리학적 활성성분은 스타틴계 지질저하제 또는 그의 약제학적으 로 허용가능한 염을 포함하며 , 필요에 따라 약제학적으로 허응 가능한 첨가제를 추 가로 포함할 수 있다.
본 발명의 스타틴계 지질저하제는심바스타린 , 로바스타¾, 아토르바스타틴 , 피타바 스타 ¾, 로수바스타틴ᅤ 플루바스타틴, 프라바스타 ¾ 등을 들 수 있으며 , 그의 광학 이성질체를 포함하는 이성질체, 라세미쳬 등을 모두 포함한다. 예를 들어 , 아토르 바스타틴의 이성질체에는 (RJ 이성질체 , a,s) 이성질체 , (s,s) 이성질체 또는
(S, )이성질체와 이들의 라세미체를, 로슈바스타틴의 이성질체에는 (Κ,β)이성질 체 (R,S)이성질체 , (S,R)이성질체 또는 (S,S)이성질체와 같은 광학 이성질체를 들 수 있다.
선방출성 구획 중 활성성분은 단위제제 증 스타 ¾계 지질저하제로 약 1 ~ 약 160 mg, 바람직하기로는 약 2 ~ 약 80 mg 포함될 수 있다. 예를 들어, 심바스타틴은 약 1 ^ 약 160 mg, 바람직하기로는 약 2 ~ 약 80 rag이 아토르바스타틴은 약 1 ~ 160 mg, 바람직하기로는 5 ~ 160 mg, 보다 바람직하기로는 10 ~ 80 mg이 피타바스타 틴은 약 1 ~ 2( g이 로슈바스타틴은 l~160mg, 바람직하기로는 5 ~ 80 rag이 포함될 수 있다.
선방출성 구획 증 스타틴계 지질저하제는 방출개시 후 1시간 이내에 , 바람직하게는 30분 이내에 , 보다 바람직하게는 15분 이내에 단위제제 중 스타틴계 지질저하제 총 량의 약 80¾ 이상이, 바람직하게는 약 90 % 이상이 방출되어 , 약효를 신속하게 나 타낼 수 있다. 예를 들어 , 심바스타틴은 방출개시 후 1시간 이내에 총량의 약 80% 이상이, 바람직하게는 약 go % 이상이 아토르바스타틴은 방출개시 후 1시간 이내에 총량의 약 80% 이상, 바람직하게는 90% 이상이 피타바스타틴은 방출개시 후 1시간 이내 , 바람직하게는 30분 이내, 보다 바람직하게는 15 분 이내에 총량의 약 80 % 이상이 로슈바스타틴은 방출개시 후 1시간 이내, 바람직하게는 30분 이내, 보다 바 람직하게는 15분 이내에 총량의 약 80% 이상이 방출될 수 있다.
(2) 약제학적으로 히용가능한 첨가제
본 발명의 제제는 또한 본 발명의 효과를 해치지 않는 범위 안에서 약제학적으로 허용 가능한 희석제, 결합제, 붕해제, 윤활제 , 안정화제 , pH 조절제, 용해보조제 , 착색제 , 향료 등의 첨가제를 사용하여 제제화할 수 있다. 이의 함량은스타틴계 지 질저하제 100 증량부에 대하여 100 ~ 30,000 중량부가 바람직하며, 더욱 바탐직하게 는 100~ 20,000 중량부이다.예를 들어 , 심바스타틴 /암로디핀 제제에서는심바스타틴 100 증량부에 대하여 100 ~ 30,000 중량부, 바람직하게는 100- 20,000 중량부이며 아토르바스타틴 /암로디핀 제제 , 피타바스타틴 /암로디핀 제제 , 또는 아토르바스타틴 /니꿰디핀 제제에서는 각 .스타틴계 지질저하제 1중량부에 대하여 1~100중량부 , 바 람직하게는 1~30증량부이며 로슈바스타틴 /암로디 ¾ 제제에서는 로슈바스타틴 1중 량부에 대하여 0.01-100 증량부를 포함할 수 있다, 상기 희석제는 전분, 미세결정성 ¾를로오스, 유당, 포도당, 만니를, 알기네이트, 알칼리토금속류염 , 클테이 , 폴리에틸렌글리콜, 디칼슘포스페이트, 또는 이들의 혼 합물 등을 사용할 수 있다.
상기 결합제는 전분, 미세결정성 셀를로오스, 고분산성 실리카, 만니를, 자당, 유 당, 풀리에틸렌글리콜, 풀리비닐피를리돈, 히드록시프로필메틸샐를로오스, 히드톡 시프로필셀를로오스 , 천연검 , 합성검, 코포비돈, 라된 , 또는 이들의 흔합물 등을 사용할 수 있다.
상기 붕해제는 전분글리콘산나트륨, 옥수수전분, 감자전분 또는 전젤라틴화전분 등 의 전분 또는 변성전분 벤토나이트 , 몬모 ¾로나이트, 또는 비검 (veegum) 등의 클레 이 미세결정성셀를로오스, 히드록시프로필셀를로오스 또는 카르복시메틸 ¾를로오스 등의 셀를로오스류 알긴산나트륨 또는 알긴산' . 등의 알긴류 크로스카멜로스 (croscarmel lose)나트륨 등의 가교 셀를로오스류 구아검 , 잔탄검 등의 검류 가교 풀리비닐피를리돈 (crospovidone) 등의 가교 증합체 증탄산나트륨, 시트르산 등의 비등성 제제 , 또는 이들의 흔합물을 사용할 수 있다.
상기 윤활제는 탈크, 스테아린산, 스테아르산 마그네슴, 스테아린산 칼슘 등, 라우 릴설페이트나트륨, 수소화식물성오일, 나트꼼벤조에이트, 나트륨스테아릴푸마레이 트, 글리세릴 베해네이트, 글리세 ¾ 모노레이트 , 글리세릴모노스테아레이트, 글리 세¾ 팔미토스테아레이트 및 폴리에틸렌글리콜 등을 사용할 수 있다.
상기 안정화제는 알칼리금속의 염, 알칼리토금속의 염, 또는 이들의 흔함물인 알칼 리화제를 사용할 수 있으며 , 바탐직하게는 탄산칼슴, 탄산나트륨, 탄산수소나트륨, 산화마그네슘, 탄산마그네슴, 구연산나트륨 등을 사용할 수 있으며 , 아스코르빈산ᅳ 구연산, 부틸레이티드히드록시 아니솔, 부틸레이티드히드톡시 를루엔, 토코페를 유 도체를 사용할 수도 있다.
상기 pH 조절제는 초산, 아디프산, 아스코르브산, 사과산, 숙신산, 주석산, 푸마르 산, 구연산과 같은 산성화제와 침강 탄산 칼슘, 암모니아수, 메글루민와 같은 염기 성화제 등을 사용할 수 있다.
상기 용해보조제는 라우 ¾황산나트륨, 폴리소르베이트 등의 풀리옥시에틸렌 소르비 탄 지방산 에스테류, 도큐세이트 나트륨 등을 사용할 수 있다.
이외에도 착색제 , 향료 중에서 선택된 다양한 첨가제로서 약제학적으로 허용 가능 한 첨가제를 선택 사용하여 본 발명의 제제를 제제화할 수 있다.
본 발명에서 사용가능한 첨가제의 범위가 상기 첨가제를 사용하는 것으로 한정되는 것은 아니며, 상기한 첨가제는 선택에 의하여 통상 범위의 용량을 함유하여 제제화 할 수 있다.
2. 지연방출성 구획 본 발명에서 지연방출성 구획은 선방출성 구획 활성성분의 방출 일정 시간 후에 그 활성성분이 방출되는 구획을 의미한다. 지연방출성 구획은 (1)약리학적 활성성분인 암로디 ¾, 이의 이성질체 또는 그의 약제학적으로 허용되는 염 , 및 (2-1)방출제어 물질 및 /또는 (2-2)삼투압 조절제 및 반투과성막 코팅기제를 포함하며, 필요에 따 라, (3) 약제학적으로 허용 가능한 첨가제를 추가로 포함할 수 있다ᅳ 본 발명에 의 한 지연방출성 구획은 상업적으로 판매되는 스타틴계 지질저하제를 포함하는 제제 와 동시에 복용될 수도 있다.
( 1) 약리학적 활성성분
지연방출성 구획의 약리학적 활성성분은 디히드로피리딘계 칼슴 채널 차단제 , 그의 이성질체 및 약학적으로 허용 가능한 염을 포함한다.
디히드로피리딘계 칼슘 채널 차단제는 사이토크룸 P450계 효소의 저해를 받는 성분 을 션택 사용할 수 있으며 , 예를 들면 , 암로디핀, 레르카니디핀 , 라시디핀 , 펠로디 핀, 바니디핀 , 베니디핀 , 실니디 ¾, 이스라디¾, 마니디핀 , 니카르디핀, 니페디핀, 니모디핀, 닐바디핀 , 니술로디핀 니트렌디핀 , 아¾니디핀 '등이 있으나, 이들이 종 류에 한정하지 아니하고 상기 제시된 바와 같이 사이토크름 P450 계 효소의 저해를 받는 디히드로피리딘계 칼슴길항제 증에서 선택 사용가능하다. 바람직하기로는 암 로디핀 , 니페디핀 및 이의 약학적으로 허용 가능한 염을 사용할 수 있으며 , 암로디 핀의 약제학적으로 허용가능한 염은 구체적으로 암로디핀의 말레인산 염 및 암로디 핀의 베실산염을 들 수 있다.
지연방출성 구획 중 활성성분은 단위제제 증 디히드로피리딘계 칼슴 채널 차단제로 1일 성인 (체중 65 ~ 75 kg의 성 인 남자) 기준, 단위제제 증 약 1 ~ 약 400 mg, 바 람직하기로는 약 2 "- 약 120 mg 포함될 수 있다. 그 증 암로다핀은 약 1 약 40mg, 바람직하게는 약 2〜약 20mg이 , 니페디핀은 1일 약 1 ~ 약 90mg을 포함할 수 있다. 디히드로피리딘계 칼슘 채널 차단제는 스타 ¾계 지질저하제의 방촐 개시 후, 단위 제제 증 총량의 약 40% 이하가 방출에 도달하는 시간이 약 2시간 이내 , 바람직하게 는 약 3시간 이내, 더욱 바람직하게는 약 4시간 이내이며, 그 결과 약효 발생시간 을 지연시킨다. 예를 들어 , 심바스타틴 /암로디핀 제제 또는 아토르바스타틴 /니페디 핀 제제는 암로디핀 또는 니페디핀 총량의 약 40% 이하가 방출에 도달하는 시간이
약 2시간 이내 , 바람직하게는 약 3시간 이내 , 더욱 바람직하게는 약 4시간 이내 아 토르바스타틴 /암로디핀 제제는 암로디 ¾ 총량의 20¾ 이하가 방출에 S달하는 시간 이 약 2시간 이내 , 바람직하게는 약 3시간 이내 , 더욱 바람직하게는 약 4시간이며 피 타바스타틴 /암로디핀 제제는 4시간까지 제제 증 암로디핀 총량의 40% 이하로, 바람 직하게는 30% 이하로 방출되며 , 로슈바스타틴 /암로디핀 제제는 3시간 까지 암로디 핀 총량의 30¾ 이하가, 바람직하게는 20%이하가 방출될 수 있다.
(2-1) 방출제어물질
본 발명의 약제학적 제제 중 지연방출성 구획은 장융성 고분자, 수불용성 중합체, 소수성 화합물, 친수성 고분자 및 이들의 흔합물 증에서 선택된 1종 이상의 방출제 어물질을 포함한다. 상기 방출제어 물질은 디히드로피리딘계 칼슴 ¾항제 100 증량 부에 대하여 10 ~ 3,000 증량부 사용 가능한데 , 사용량이 상기 범위. 미만이면 충 분한 지연방출성을 얻을 수 없고, 시용량이 상기 범위를 초과하면 약물방출이 지나 치게 지연되어 유의성 있는 임상적 효과를 얻을 수 없다.
심바스타틴 /암로디핀 제제에서 상기 방출제어물질은 바람직하게는 장용성 고분자 '및 친수성 고분자의 흔합물을' 포함하거나, 수블용성 중합체를 포함하며 , 암로디핀 1증량부에 대하여 , 0,1 - 100 중량부를 포함한다.
아토르바스타틴 /암로디핀 제제에서 상기 방출제어물질은 암로디핀 1증량부에 대하 여, 0.05—100 증량부, 바람직하게는 0.05 - 30 증량부를 포함될 수 있으며 바람직 하게는 장용성 고분자 및 친수성 고분자의 흔합물일 수 있고, 상기 장용성 고분자 및 친수성 고분자 흔합물은 각각 암로디핀 1중량부에 대하여 각각 0.5~10증량부 및 0.5~20증량부로 포함될 수 있다
피타바스타린 /암로디핀 제제에서 상기 방출제어물질은 암로디핀 1 증량부에 대하여 0.05-100 중량부로 사용가능하고, 바탐직하게는 수불용성 중합체 및 중합체 및 친 수성 고분자를 포함하며 로슈바스타틴 /암로디핀 제제에서 상기 방출제어물질은 암 로디핀 1 중량부에 대하여 0.05~100 증량부 사용가능하고, 바람직하게는 수불용성 증합체 및 친수성 고분자 중에서 선택된 1종 이상의 방출제어물질을 포함하며 아토 르바스타틴 /니페디핀 제제에서 상기 방출제어물질은 니페디핀 100 증량부에 대하여 5 내지 300 증량부로 사용가능하고, 바람직하게는 장용성 고분자 및 친수성 고분자 를 포함할 수 있다.
본 발명의 지연방출성 구획에서 , 장용성 고분자는 pH 5 미만의 산성 조건하에서 블 용성이거나 또는 안정한 것으로, pH 5 이상의 조건하에서 용해되거나 또는분해되는 고분자를 말하며 , 예컨대 장용성 셀를로오스 유도체 , 장용성 아크릴산계 공증합체, 장용성 말레인산계 공중합체 , 장용성 폴리비닐 유도체 , 및 이들의 흔합물로 이루어 진 군에서 선택된 것이다. 여기서, 장용성 셀를로오스 유도체는 히드록시프로필메 틸셀를로오스아세테이트숙시네이트, 히드록시프로필데틸쎌롤로오스프탈레이트, 히 드특시메틸에틸셀롤로오스프탈레이트, 셀를로오스아세테이트프탈테이트, 샐를로오 스아세테이트숙시네이트, 샐를로오스아세테이트말레이트, 샐롤로오스 ¾조에이트프 탈레이트 ¾를로오스프로피오네이트프탈레이트, 메틸셀를로오스프탈레이트, 카르 복시메틸에틸셀를로오스, 에틸히드록시에틸셀를로오스프탈레이트 , 메틸히드록시에 틸셀를로오스 및 이들의 흔합물 증에서 선택된 1종 이상 상기 장용성 아크릴산계 공증합체는 스티 렌 -아크 ¾산 공중합체, 아크릴산메틸ᅳ아크릴산 공중합체, 아크 ¾산 메틸메타크릴산 공증합체 , 아크 ¾산부틸—스티렌-아크¾산 공중합체, 메타크 ¾산-메 타크릴산메틸 공중합체 (예 ¾대 , 유드라짓 L 100, 유드라짓 S, 데구사, 독일), 메타 크릴산,아크릴산에틸공증합체 (예 ¾대, 유드라짓 L 100-55, 데구사 독일) , 아크 릴산메틸-메타크 ¾산-아크¾산옥틸공증합체 및 이들의 흔합물 증에서 선택된 1종 이상 상기 장용성 말레인산계 공증합체는 아세트산비닐-말레인산 무수물 공증합체 스티렌—말레인산 무수물 공중합체, 스티렌-말레인산모노에스테롤 공중합체 , 비닐메 틸에테르-말레인산 무수물 공중합체, 에틸렌-말레인산 무수물 공증합체, 비닐부틸 에테르-말테인산 무수물 공증합체 , 아크 ¾로니트릴-크 ¾산메틸 ' 말레인산 무수물 공증합체 , 아크 ¾산부틸—스티렌-말레인산 무수물 공증합체 및 이들의 혼합물 중에 서 선택된 1종 이상 장용성 플리비닐 유도체는 폴리비닐알콜프탈레이트, 플리비닐 아세탈프탈례이트, 플리비닐부뫼레이트프탈레이트, 폴리비닐아세트아세탈프탈레이 트 및 이들의 흔합물 증에서 선택된 1종 이상일 수 있다.
심바스타틴 /암로디핀 제제에서 바람직한 장용성 고분자는 폴리비닐아세테이트프탈 레이트, 히드록시프로필떼틸셀를로오스프탈레이트, ¾탁, 셀를로오스아세테이트프 탈레이트, 셀를로오스프로피오네이트프탈레이트, 폴리 (메타크릴레이트, 메틸메타크 ¾레이트)공증합체 및 풀리 (메타크¾레이트, 에¾아크릴테이트)공증합체 증에서 선 택된 1 종 이상일 수 있으며 암로디핀 1 중량부 대비 0.1~20 중량부, 바람직하게는 0.5-10 중량부로 포함 ¾ 수 있다.
아토르바스타틴 /암로디핀 제제에서 바람직한 장용성 고분자는 히드록시프로필메틸
셀 ¾로오스프탈레이트, 아크 ¾산메틸-아크릴산 공증합체 , 메타크릴산-메타크릴산메 틸 공증합체, 폴리비닐알콜프탈테이트, 플리비닐아세트아세탈프탈레이트를 사용하 며 , 보다 바람직하게는 히드록시프로필메틸셀를로오스프탈레이 S , 아크릴산메 ¾ -아 크¾산 공증합체 , 메타크릴산-메타크 ¾산떼틸 공증합체 증에서 선택된 1종 이상일 수 있으며 암로디핀 1중량부 대비 0.1 20 중량부, 바람직하게는 0.5ᅳ: 10 증량부로 포함될 수 있다.
피타바스타틴 /암로디핀 제제에서 바람직한 장용성 고분자는 아크 ¾산계 공중합체를 들 수있으며 바람직하게는 아크릴산메틸메타크될산 공중합체 (제품명 , 아크릴-이 즈)를 사용할 수 있고, 활성성분인 암로디핀 1 중량부에 대해 0.1~20 중량부, 바람 직하게는 0.5~10 증량부로 포함될 수 있다.
로슈바스타틴 /암로디핀 제제에서 바람직한 장용성 고분자는 아크릴산메틸메타크릴 산 공증합체 및 히드톡시프로필메틸셀를로소스프탈레이트 중에서 선택된 1종 이상 이며 암로디핀 대비 0.1~20 중량부 , 바람직하게는 0.5-10 중량부로 포함될 수 있으 며 , 0.1 중량부 미만인 경우에는 pH' 5 미만에서 쉽게 용해되는 문제점이 있고, 20 중량부 초과인 경우에는 불필요하게 제제 총중량이 커지거나 과도하게 용출이 지연 되는 문제점이 있다 .
아토르바스타틴 /니페디핀 제제에서 바람직하게는 장용성 셀를로오스 유도체 , 장용 성 아크릴산계 공증합체 , 장용성 풀리비닐 유도체가 사용가능하며 , 보다 바람직하 게는 히드록시프로필메 ¾¾롤로오스프탈레이트, 히드톡시프로필메 를로오스아세 테이트숙시네이트, 메타크릴산-메타크릴산메 ¾공중합체를 사용할 수 있으며 니패디 핀 100증량부에 대해 5 내지 150 중량부, 바람직하게는 10 내지 50 중량부로 포함 될 수 있다.
상기 제제들에서 장용성 고분자가 각 하한의 미만으로 포함되는 경우에는 pH 5 미 만에서 쉽게 용해되는 문제점이 있고, 각 상한 초과로 포함되는 경우에는 불필요하 게 제제 총중량이 커지거나 과도하게 용출이 지연되는 문제점이 있다. 본 발명의 지연방출성 구획에서 , 수불용성 증합체는 약물의 방출을 제어하는 약제 학적으로 허용가능한 물에 용해되지 않는 증합체를 말하며 예¾대, 폴리비닐 아세 테이트, 플리메타크필레이트 공중합체 , 플리 (에틸아크릴테이트—메필 메타크릴레이 트〉 공중합쳬 , 폴리 (에될아크 ¾레이트 -녜틸 메타크 ¾레이트-트리메틸아미노에¾메 타크릴테이트)공증합체 , 에틸셀를로오스, 쉘를로오스 에스테르, 셀롤로오스 에테
르, ¾를로오스 아실레이트, 셀를로오스 디아실레이트 샐롤로오스 트리아실레이 트, ¾를로오스 아세테이트, 로오스 디아세테이트, ¾롤로오스 트리아세테이트 및 이들의 혼합물로 이루어진 군에서 선택된 1종 이상을 들 수 있다.
심바스타틴 /암로디 ¾ 제제에 있어서 수블용성 중합체는 바람직하게는 플리비닐 아 세테이;트, 플리메타크릴레이트 공증합체, 풀리 (에틸아크렬레이트 -메틸 메타크릴레 이트) 공증합체 , 폴리 (에틸아크 ¾레이트 -메틸 메타크 ¾레이트ᅳ트리메틸아미노에틸 메타크릴레이트)공중합체 , 에틸셀를로오스, 셀를로오스 아세테이트를, 보다 바탐직 하게는 폴리비닐 아세테이트, 폴리 (에틸아크 ¾레이트—메¾ 메타크릴레이트-트리메 틸아미노에 ¾메타크릴레이트)공중합체 , 폴리쩨타크 ¾테이트 공증합체, 폴리 (에 ¾아 크¾레이트—메틸 메타크릴레이트) 공중합체등을 사용할 수 있으며 암로디핀 1증량 부 대비 0.1—30 중량부 바람직하게는 0.5~20 증량부로 포함될 수 있다.
아토르바스타틴 /암로디핀 제제에 있어서 수불용성 증합체는 바람직하게는 폴리비닐 아세테이트, 유드라짓 RS P0, 에틸셸를로오스, 샐 ·로오스아세테이트를 사용하며 , 보다 바람직하게는 폴리비닐아세테이트, 유드라짓 RS P0, 에틸셀롤로오스를 사용할 수 있고, 활성성분인 암로디핀 1 중량부 대비 약 0.1-30 중량부, 바람직하게는 0.5-20 중량부로 포함될 수 있다.
피타바스타틴 /암로디핀 제제에 있어서 수불용성 중합체는 바람직하게는 아크릴산계 공중합체롤, 보다 바람직하게는 유드라짓 RS30D를 사용할 수 있으며 암로디핀 1중 량부에 대해 0.1-30 중량부, 바람직하게는 0.5-20 중량부로 포함될 수 있다.
로슈바스타틴 /암로디핀 제제에 있어서, 수블용성 증합체는 ¾리비날 아세테이트 (콜 리코트 S 30D) , 샐를로오스아세테이트 및 플리 (에틸아크 ¾레이트 -메틸 메타크릴레 이트-트리메틸아미노에틸메타크 ¾레이트)공증합체 (유드라짓 RS30D) 증에서 선택된 1종 이상인 것이 바람직하며, 암로디핀 대비 0.1-30 증량부, 바람직하게는 0.5-20 중량부로 포함 ¾ 수 있다.
아토르바스타틴 /니꽤디핀 제제에 있어서 수블용성 중합체는 바람직하게는 아크릴산 공중합체를 사용하며, 보다 바람직하게는 폴리 (에 ¾아크릴레이트 -메틸 메타크릴레 이트-트리메 ¾아미노에틸메타크릴레이트)공중합체 (유드라짓 RS30D 및 유드라짓 LR30D와 흔합물)을 사용하며 니폐디핀 100 증량부에 대해 5 내지 200 중량부, 바람 직하게는 10 내지 100증량부로 포함될 수 있다. 상기 플리 (에틸아크 ¾레이트 -메틸 메타크 ¾레이트 -트리메¾아미노에틸메타크 ¾레이트)공중합체는 니페디핀 100중량부 에 대해 20 내지 100 증량부를 사용하는 것이 바람직하다.
상기 제제들에서 수블용성 증합채가 각 하한의 미만으로 포함되는 경우에는 약물의 방출이제어되지 않는 문제점이 있고 , 각 상한 초과로 포함되는 경우에는 불필요하 게 과도하게 용출이 지연되는 문제점이 있다. 본 발명의 지연방출성 구획에서 , 소수성 화합물은 약물의 방출을 제어하는 약제학 적으로 허용가능한 물에 용해되지 않는 물질을 말하며 , 예컨대 지방산 및 지방산 에스테르류, 지방산 알코을류, 왁스류, 무기물질, 및 이들의 흔합물로 이루어진 군 에서 선택된 1종 이상을 들 수 있다. 여기세 지방산 및 지방산 에스테르류는 글리 세릴 팔미토스테아레이트, 글리세 ¾ 스테아레이트, 글리세릴 비헤네이트, 세틸 팔 미테이트, 글리세릴 모노 올레이트, 스테아린산 및 이들의 흔합물 증에서 선택된 1 종 이상; 지방산 알코을류는 세토스테아릴 알코올, 세틸알코을 , 스테아릴알코을 및 이들의 흔합물 증에서 선택된 1종 이상; 왁스류는 카르나우바왁스 , 밀납, 미결정왁 스, 및 이¾의 혼합물 증에서 선택된 1종 이상;' 무기물질은 탈크, 침강탄산칼슴, 인산일수소칼슴, 산화아연 , 산화티탄, 카을린, 벤토나이트, 폰모 ¾로나이트, 비검 및 이들의 흔함물 중에서 선택된 1종 이상을 사용할 수 있다.
심바스타틴 /암로디 ¾ 제제에 있어서, 소수성 화합물은 바람직하게는 지방산 에스테 르류, 글리세릴 팔미토스테아레이트, 글리세릴 스테아레이트, 글리세릴 비해네이트 를, 보다 바람직하게는 글푀세 ¾ 스테아레이트, 글리세 ¾ 비헤네이트를 사용할 수 있으며 암로디핀 1증량부 대비 0, 1—20 증량부, 바람직하게는 0,5~10 증량부로 포함 될 수 있다,
아토르바스타틴 /암로디핀 제제에 있어서, 소수성 화합물은 바람직하게는 지방산 에 스테르류, 지방산 알코을류, 왁스류, 무기물질을 사용하며, 보다 바람직하게는 지 방산 에스테르류, 지방산 알코올류를 사용할 수 있으며, 암로디핀 1중량부 대비 0.1-20 중량부, 바람직하게는 0, 5-10 증량부로 포함될 수 있다.
피타바스타틴 /암로디핀 제제에 있어서 , 소수성 화합물은 바람직하게는 지방산 에스 테르류롤 사용하며 , 보다 바람직하게는 글리세 ¾ 스테아레이트를 사용할 수 있으며 암로디핀 1 중량부에 대해 0.1 20 증량부 , 바람직하게는 0.5~40 증량부로 포함될 수 있다.
로수바스타틴 /암로디핀 제제에 있어서 , 소수성 화합물은 암로디핀 대비 0.1-20 중 량부, 바람직하게는 0.5~10 중량부 포함 ¾ 수 있다.
아토르바스타틴 /니페디핀 제제에 있어서 , 소수성 화함물은 바람직하게는 지방산 에
스테르류를, 보다 바람직하게는 글리세 ¾ 스테아레이트를 사용할 수 있으며, 니페 디핀 100 증량부에 대해 5 내지 300 증량부, 바람직하게는 15 내지 100 중량부로 포함될 수 있다. 상기 제제들에서 소수성 화합물이 각 하한의 미만으로 포함되는 경우에는 약물의 방출이제어되지 않는 문제점이 있고, 각 상한 초과로 포함되는 경우에는 불필요하 게 과도하게 용출이 지연되는 문제점이 있다. 본 발명에 있어서 친수성 고분자는 약물의 방출을 제어하는 약제학적으로 허용가능 한 물에 용해되는 고분자 물질을 당류, ¾롤로오스 유도체, 검류, 단백질류, 폴리 비¾ 유도체, 폴리메타크릴레이트 공중합체 , 폴리에 ¾¾ 유도체 및 카르복시비닐증 합체 둥을 선택 사용할 수 있으며 , 구체적으로 당류로서 덱스트린 , ¾리덱스트린, 덱스트란, 펙¾ 및 펙된 유도체 , 알긴산염 , 풀리갈탁투론산, 자일란, 아라비노자일 란, 아라비노갈락탄, 전분, 히드특시프로필스타치 , 아밀로오스, 아밀로펙된등을 선 택 사용할 수 있고, 셀를로오스 유도체로서 히드록시프로필메 ¾셀를로오스, 히드톡 시프로필¾*로오스, 히드록시메틸셀를로오스, 히드록시에 ¾쎌를로오스, 메틸샐롤 로오스, 카르복시메틸셸를로오스 나트 ·롬, 히드록시프로필 메틸셀 ·로오스 아세테이 트 숙시네이트, 히드록시에틸메틸쎌를로오스 등을 선택하여 사용할 수 있으며, 검 류로서 구아검 , 로커스트 콩 검, 트라가칸타, 카라기난, 아카시아검 , 아라비아검 , 젤란검 , 잔탄검 등을 선택 사용할 수 있으며, 단백질류로서 젤라틴, 카제인 , 및 제 인 등을 선택 사용할 수 있고, 플리비닐 유도체로서 폴리비닐 알코을, 폴리비닐 피 롤리돈 및 폴리비닐아세탈디에틸아미노아세테이트 등을 선택 사용할 수 있으며 , Φ 리메타크¾레이트 공중합체로서 폴리 (부틸 메타크 ¾레이트 , (2-디메털아미노에 ¾)메 타크 ¾레이트-메틸메타크릴레이트) 공중합체, 폴리 (메타크¾산-메 ¾메타크 ¾테이 B) 공증합체 , 폴리 (메타크릴산ᅳ에 ¾아크릴레이트) 공중합체 등을 선택하여 사용할 수 있으며 , 폴리에틸렌 유도체로서 풀리에털렌글리콜, *리에틸렌 옥사이드 등을 선택 사용할 수 있으며 , 카르복시비닐풀리머로서 카보머 * 사용할 수 있다.
심바스타틴 /암로디 ¾ 제제에 있어서 , 상기 친수성 고분자는 바람직하게는 전분, 히 드록시프로필메틸셸를로오스, 히드록시프로필셸를로오스, 카르복시메 ¾¾를로오스 나트晉, 잔탄검 , 플리비닐 알코을 또는 카보머를 사용할 수 있으며 암로다 ¾ 1중량 부 대비 0.05-30 중량부, 바람직하게는 0.5~20중량부로 포함 ¾ 수 있다.
아토르바스타틴 /암로디 ¾ 제제에 있어서 , 상기 친수성 고분자는 바람직하게는 셀를 로오스 유도체 , 폴리비닐유도체 , 카르북시비닐폴리머 , 풀리에틸렌 유도체를 사용하 며 , 보다 바람직하게는 히드록시프로필쩨 를로오스, 히드톡시프로필메틸셀를로 오스아세테이트 숙시네이트, 폴리비닐피를리돈, 카보머 , 풀리에틸렌 옥사이드를 사 용할 수 있으며 암로디핀 1증량부 대비 0.05~30 증량부, 바람직하게는 0.5~20중량 부로 포함될 수 있다.
피타바스타틴 /암로디핀 제제에 있어서, 상기 친수성 고분자는 바람직하게는 카르복 시비닐 ¾ 머를, 보다 바람직하게는 카보머를 사용할 수 있으며 , 암로디핀 1 증량 부에 대해 0.05~30 증량부, 바람직하게는 0.5~20증량부로 포함될 수 있다.
로슈바스타틴 /암로디핀 제제에 있어서 , 상기 친수성 고분자는 바람직하게는 히드록 시프로필셀를로오스 및 /또는 폴리메타크 ¾레이트 공중합체를, 보다 바람직하게는 풀리 (메타크¾산ᅳ메틸메타크릴례이 S)를 사용할 수 있으며 암로디핀 1증량부에 대 하여 0.05~30 증량부, 바람직하게는 0.5 20증량부로 포.함될 수 있다.
아토르바스타틴 /니폐디핀 제제에 있어서 , 싱-기 친수성 고분자는 바람직하게는 셀를 로오스 유도체, 카르복시비닐플리머를, 보다 바람직하게는 히드특시프로필메 ¾샐를 로오스, 히드특시프로필 를로오스 , 카보머를 사용할 수 있으며 니꽤디핀 loo 중량 부에 대해 5 내지 200 증량부, 바람직하게는 10 내지 100 증량부로 포함될 수 있 다.
상기 제제들에서 친수성 고분자가 각 하한의 미만으로 포함되는 경우에는 약물의 방출속도가≤절되지 않는 문제점이 있고, 각 상한 초과로 포함되는 경우에는 과도 하게 용출이 지연되는 문제점이 있다. 심바스타틴 /암로디핀 제제에 있어서 , 바람직한 방출제어물질은 풀리비닐아세테이 트, 풀리메 1타크렬레이트 공증합쳬 ᅳ 폴리 (에틸아크 ¾레이트, 데틸 메타크 ¾테이트, 트리메 ¾아미노에 ¾메타크릴레이 S)공중합쳬, 카르복시비닐 뿔리머 , 에¾셀를로오 스, " 를로오스 아세테이트, 카르복시메 ¾셀롤로오스 나트륨, 플리에틸렌 옥사이 드 히드록시프로필메틸셸를로오스 ᅳ 히드특시프로필셀를로오스, 및 히드톡시프로필 메틸셀를로오스프탈레이트로 구성된 군으로부터 선택된 1종 이상의 것 ; 또는 카르 복시비날폴리머 , 히드록시프로필메틸셀를로오스, 몇 히드록시프로필.메¾셀를로오스 프탈레이트로 구성된 군으로부터 선택된 1종 이상의 것 ; 또는 카르복시비닐풀리머 및 히드록시프로필메 로오스를 포함하는 것일 수 있다.
아토르바스 }·틴 /암로디핀 제제에 있어서 , 바람직한 방출제어물질은 폴리비닐아세테 이트, 플리메타크릴레이트 공증합체 , 카르복시비닐 폴리머 , 히드록시프로필메 ¾샐 를로오스, 히드록시프로필¾를로오스, ¾롤로오스 아세테이트, 에틸셀를로오스, 플 리에틸렌 옥사이드 및 히드록시프로필메틸쩔를로오스프탈레이트로 구성된 군으로부 터 선택된 1종 이상의 것 카르복시비닐플리머, 폴리메타크 ¾레이트 ^증합체 및 히 드톡시프로필메틸셀를로오스로 구성된 군으로부터 선택된 1종 이 ^의 것 또는 카르 복시비닐플리머 및 히드록시프로필메 ¾셀를로오스의 흔합물을 들 수 있다.
피타바스타틴 /암로디핀 제제에 있어서, 바람직한 방출제어물질은 히드록시프로필셀 를로오스, 히드록시프로필 메틸샐를로오스, 히드톡시프로필메틸셀를로오스프탈레이 트, 셸 ■로오스아세테이트, 폴리비닐아세테이트, 폴리메타크 ¾레이트공중합체 , 에 틸셀'를로오스, *리 (메타크¾레이트 메틸메타크릴레이트)공증합체 및 아크릴산메틸 메타크 ¾산 공중합체로 이루어진 군으로부터 선택된 하나 이상의 것 , 또는 히드록 시프로필셀를로오스, 셀를로오스아세테이트, 에틸셀를로오스, 폴리 (메타크¾레이트 메틸메타크 ¾레이트)공증합체로 이루어진 군으로부터 선택된 하나 이싱;이다.
로슈바스타틴 /암로디편 제제에 있어서 , 바람직한 방출제어물질은 히드특시프로필샐 를로오스, 히드톡시프로필메 를로오스, 히드록시프로필메틸 ¾롤로오스프탈레이 트, 쩔를로오스아세티에트, 풀리비닐아세테이트, 풀리메타크릴레이트공증합체, 에 틸 ¾를로오스, 풀리 (메타크릴레이트, 메틸메타크 ¾레이트)공증합체, 및 아크릴산메 ¾떼파크릴산 공중합체로 이루어진 군으로부터 선택된 1 이상의 것 , 또는 히드록시 프로필셸를로오스, 샐를로오스아세테이트, 에틸젤를로오스, 폴리 t데타크릴레이트, 메¾메타크리레이트)공증합체로 이루어진 군으로부터 선택된 1 이상의 것이다. 아토르바스타틴 /니폐디핀 제제에 있어서 , 바람직한 방출제어물질은 히드톡시프로필 메 ¾셀를로오스프탈레이트 , 히드록시프로필메틸셀를로오스아세테이트숙시네이트 , 메타크 ¾산-메타크릴산메틸공증합체 , 플리 (에틸아크릴레이트 -메틸 메타크 ¾레이트- 트리떼틸아디노에틸메타크 ¾레이트)공증합체 , 글리세 ¾ 스테아레이트, 히드톡시프 로필메틸셸를로오스, 히드록시프로필¾를로오스, 및 카보머로 이루어진 군으로부터 선텍된 하나 이상이며, 특히 바람직하게는 방출제어물질은 히드록시프로필메틸샐를 로오스프탈레이트, 히드록시프로필메틸셀를로오스아세테이트. 숙시네이트 또는 메타 크 ¾산 메타크릴산메 ¾공증합체로부터 선택되는 하나 이상의 장용성 고분자 및 ¾1 드록시프로필메¾셀를로오스, 히드록시프로필¾를로오스, 또는 카보머로부터 선택 되는 친수성 고분자를 포함한다.
(2-2) 삼투압 조절제 및 반투과성막 코팅기제
본 발명의 지연방출성 구획은 삼투압 조절제를 포함하며 , 반투과성막 코팅기제로 코팅된 구획일 수 있다. 본 발명의 지연방출성 구획에서 , 삼투압쵸절제는 삼투압의 원리를 이용하여 약물의 방출속도를 조절하는데 사용되는 성분을 말하몌 예컨대' 황산마그네슴, 염화마그네 슴, 염화나트륨, 염화칼쁌, 인산나트륨, 인산칼晉, 초산암모늄, 염화리꼼, 황산칼 , 황산나트 황산리튬, 황산나트륨 및 이들의 흔합물로 이루어진 군에서 선택 된 1종 이상을 들 수 있다. 바람직하게는 염화나트륨, 염화칼튬, 인산나트 *, 인산 칼튬을 사용할 수 있다.
심바스타틴 /암로디핀 제제에 있어서 , 삼투압 조절제는 암로디핀 1 중량부에 대해서 0.01 증량부 ~ 10 중량부, 바람직하게는 0.05 중량부- 5 중량부로 아토르바스타틴 / 암로디핀 제제 , 피타바스타틴 /암로디핀 제제 또는 로슈바스타틴 /암로디핀 제제에 있어서는 암로디핀 1중량부 대비 0.05 30 증량부, 바람직하게는 0.1-20중량부로 아 토르바스타틴 /니폐디핀 제제에 있어서는 니페디핀 100증량부에 대해 5 내지 150 중 량^, 바람직하게는 10 내지 100증량부로 포함될 수 있다.
상기 제제들에서 삼투압 조절제가 각 하한의 미만으로 프함되는 경우에는 삼투압 발생 효과가 미약한 문제점이 있고, 각 상한 초과로 포함되는 경우에는 블필요하게 제제 총중량을 증가시키거나 적합한 약물 방출속도를 구현할 수 없는 문제점이 있 다. 본 할명의 지연방출성 구획에서 , 반투과성막 코팅기제는 약학적으로 사용가능한 코 팅기제로서, 약학적 제제의 코팅층에 배합하여 일부 성분은 통과시키지만, 다른 성 분은 통과시키지 않는 막을 ¾성하는데 사용하는 물질을 말하며, 상기 언급된 수불 용성 증합체를 사용할 수 도 있다. 본 발명에서 반투과성막 코뜀기제는 예컨대 폴 리비닐 아세테이트, 플리메타크 ¾레이≤ 공중합체 , 플리 (에틸아크 ¾레이트, 메¾ 데타크릴레이트) 공중합체, 풀리 (에틸아크 ¾테이트, 데틸 메타크 ¾레이트, 트리메 ¾아미노에틸메타크 ¾레이트)공증합체 , 에틸셀롤로오스, 셀를로오스. 에스테르 , 샐 를로오스 에테르, ¾를로오스 아실테이트, ^를로오스 디아실테이트, 셀를로오스 트리아실레이트, 셀를로오스 아세테이트 , 셸를로오스 디아세테이트 , 셸를로오스 트
리아세테이트, 및 이들의 흔합물로 이루어진 군에서 선택된 1종 이상을 들 수 있 다.
심바스타틴 /암로디핀 제제에 있어서, 반투과성막 코팅기제는 바람직하게는 셀를로 오스 아세뒈이트, 플리비닐 아세테이트 , 셀를로오스 아세테이트, 에틸셀를로오스, 폴리메타크릴레이트 공증합체를 사용할 수 있으며 암로디핀 1 중량부에 대해서 0.01 중량부 ~ 10 중량부, 바람직하게는 0.05 중량부 ― 1.25 중량부로 포함될 수 ¾다.
아토르바스타 ¾/암로디핀 제제에 있어서 , 반투과성막 코팅기제는 바람직하게는 플 리 (에틸아크 ¾레이트, 메틸 메타크릴레이:트, 트리메틸아미노에틸메타크릴레이트)공 증합체 , 셀를로오스 에스테르, 에틸샐를로오스, 셀를로오스 아세테이트를 사용할 수 있으며 암로디핀 1증량부 대비 0.5~2Q 증량부, 바람직하게는 1~10증량부로 포함 될 수 있다.
피타바스타틴 /암로디핀 제제에 있어서 , 반투과성막 코팅기제는 바람직하게는 셀롤 로오스 아세테이트를 사용할 수 있으며 1증량부에 대해 0.1 내지 20 증량부, 바람 직하게는 1 내지 10증량부로 포함될 수 있다.
로슈바스타틴 /암로디핀 제제에 있어서 , 반투과성막 코팅기제는 암로디핀 1 증량부 에 대해서 0.05 증량부 30 중량부, 바람직하게는 0.1 중량부 ~ 20 중량부로 포함 될 수 있다.
아토르바스타틴 /니페디핀 제제에 있어서 , 반투과성막 코팅기제는 바람직하게는 ¾ 를로오스 아세테이트를 사용할 수 있으며 니페디 ¾ 100중량부에 대해 5 내지 2000 증량부, 바람직하게는 10 내지 500증량부로 포함될 수 있다.
상기 제제들에서 반투과성막 코팅기제가 각 하한의 미만으로 포함되는 경우에는 층 분한 지연시간을 갖기 어려운 등 방출속도가 조절되지 않는 문제점이 있고, 각 상 한 초과로 포함되는 경우에는 약불의 방출이 일어나지 않거나 과도하게 (예 : 9시간 이상 등) 용출이 지연되는 문제점이 있다.
(3) 약제학적으로 허용가능한 첨가제
본 발명의 제제는 본 발명의 효과를 해치지 않는 범위 안에서 약학적으로 허용 가 능한 (2-1) 또는 (2ᅳ 2)의 물질로 언급한 것 이의의 희석제 , 결합제 , 붕해제, 윤활 제 , pH 조¾제 , 소포제 , 용해보 제 등의 통상적으로 사용되는 첨가제를 지연방출 성의 성격을 벗어나지 않는 범위내에서 추가로 사용하여 제제 "할 수 있다.
예를 들어, 희석제로 전분, 미결정셸를로오스, 유당, 포도당, 만니틀, 알기네이트 , 알칼리토금속류염 , 클레이 , 플리에틸렌 ¾리콜, 디칼슴포스떼이트, 또는 이들의 흔 합물 등을 사용할 수 있으며 결합제로 전분, 미결정샐를로오스, 고분산성 실리카, 만니를, 자당, 유당, 폴리에틸렌글리콜, 폴리비닐피를리든, 히드톡시프로필메틸셀 를로오스, 히드록시프로필셀를로오스, 천연검 , 합성검 , 코포비돈, 포비돈, 젤라린 , 또는 이들의 흔합물 등을 사용할 수 있다,
붕해제로는 전분글리콘산나트륨, 옥수수전분, 감자전분 또는 전젤라된화전분 등의 전분 또는 변성 ¾분 벤토나이트 > 몬모릴로나이트, 또는 비검 (veegum) 등의 클테이 미결정젤를로오스, 히드특시프로필샐를로오스 또는 카르복시메틸젤를로오스 등의 셀를로오스류 알긴산나트륨 또는 알긴산 등의 알긴류 크로스카멜로스 (croscarmel lose)나트륨 등의 가교 ¾를로오스류 구아검 , 잔탄검 등의 검류 가교 폴리비닐피를리돈 (crospovidone) 등의 가교 증합체 중탄산나트륨, 시트르산 등의 비등성 제제, 또는 이들의 흔합물을 사용할 수 있다.
윤활제로 탈 스테아린산, 스테아 ¾산 마그네슴, 스테아린산 칼슴 등, 라우릴황 산나트 fl", 수소화식물성오일 , 나트 조에이트, 콜로이드성 이산화규소, 나트륨스 테아릴푸마레이 S, 글리세 ¾ 베혜네이트, 글리세필 모노레이:트, ^리세릴모노스테 아레이트, 글리세릴 괄미토스테아레이트 및 플리에틸렌글리콜 등을 사용할 수 있 다.
본 발명의 약제학적으로 허용 가능한 첨가제로서 £ 조절제는 초산, 아디프산, 아 스코르브산, 사과산, 숙신산, 주석산, 푸마르산, 구연산과 같은 산성화제와 침강 탄산 칼슘, 암모니아수, 메글루민와 같은 염기성화제 등을 사용할 수 있다.
본 발명의 약제학적 허용 가능한 첨가제로서 소포제는 디메시콘, 을레일 알코을 , 프로필렌글리클 알지네이트, 시메티콘 에멀견과 같은 시메티콘류 등을 사용할 수 있다.
본 발명의 약제학적으로 허용가능한 첨가제에서 , 용해보조제는, 라우릴황산나트륨, 폴리소르베이트 등의 플리옥시에틸렌 소르비탄 지방산 에스테르류, 도큐세이트 나 트튬등을 사용할 수 있다.
이외에도 착색제 , 향료 증에서 선택된 다양한 첨가제로서 약학적으로 허용 가능한 첨가제를 선택 사용하여 본 발명의 제제를 제제화할 수 있다. 본 발명에서 사용가 능한 첨가제의 범위가 상기 첨가제를 사용하는 것으로 한정되는 것은 아니며 , 상기 한 첨가제는 선택에 의하여 통상 범위의 용량을 함유하여 제제화할 수 있다,
또한 본 발명의 지연방출제제에서 , 결합용매와 지연 방출성 첨가채의 용매로 정제 수, 에탄을, 염화메틸렌등을 사용할 수 있으몌 바람직하게는 정제수 또는 에탄을 을 사용할 수 있다.
본 발명의 약제학적으로 허용가능한 첨가제에서, 사용가능한 ¾가제외 범위가 상기 첨가제를 사용하는 것으로 한정되는 것은 아니며, 상기한 첨가제는 선택에 의하여 통상 범위의 용량을 함유하여 제제화할 수 있다. 본 발명의 지연방출성 구획은 디히드로피리딘계 칼슘길항제와, 시간차 제어 물질 및 약제학적으로 사용되는 통상의 첨가제의 흔합, 제립 혹은 코팅 방법에 의해 얻 어진 입자 또는 과립으로 이루어진다.
본 발명의 선방출성 구획은 스타틴계 지질저하제를 약제학적으로 혀용되는 첨가제 와 함께 흔합, 연합, 건조 및 제림과 같은 경구 고형제를 생산하기 위한 통상의 과 정을 통하여 ¾어진 입자 흑은 과립으로 제조할 수 있다, 스타틴계 지질.저하제 흔 합물의 유동성이 직접 타정이 가능할 경우 흔합하여 조성물을 얻을 수 있고, 유동 성이 *지 않을 경우 압착, 제립 및 정립하여 과립화하여 조성물을 얻을 수 있으 며 , 이렇게 선방출성 구획으로 구성할 수 있다.
본 발명은 다음과 같이 단일 정제로 타정하여 선방출형 스타틴계 지질저하제 연속 상 내에 지연방출성 디히드로피리딘계 칼슴길항제 불연속상이 위치하는 2상 매트릭 스 정제 , 다층정제 , 유핵정제와 같은 단일 정제의 제조방법 및 투여 방법으로 실시 가 가능하나, 이에 한정되는 것은 아니다.
또한, 지연방출성 구획과 선방출성 구획을 구성하는 과립물을 약제학적인 첨가제와 흔합하여 다중 타정기를 사용하여 각층이 평행한 2중정 흑은 3증정으로 타정하여 각 층에 따른 선방출과 지연 방출이 가능한 경구 투여용 정제를 얻을 수 있다. 또한, 지연방출성 구획을 구성하는 과립물을 약제학적인 첨가제와 흔합 및 타정하 여 이를 핵정으로 하거나, 지연방출성을 나타내도록 약제학적인 코팅을 시행한 후, 선방출성 구획을 구성하는 과립물을 약제학적인 첨가제와 흔합한 뒤 외층으로 하여 타정함으로써 내핵으로 지연방출성을 갖고 상기 내핵의 표면을 속방층이 둘러싼 형 태의 경구 투여용 정제를 얻을 수 있다.
또한, 지연방출성 구획을 구성하는 과립물을 약제학적인 첨가제와 흔합 및 타정하 거나, 지연방출성을 나타내도특 약제학적인 코팅을 시행한 후, 선방출성 구획을 구 성하는 약물층을 수용성의 특징을 나타내는 필름코팅용액에 용해 및 분산시킨 후
지연방출성 정제층의 의층에 코팅함으로써 경구투여용 정제를 얻을 수 있다.
또한, 디히드로피리딘계의 방출 제어를 위해 삼투성 물질을 정제의 내부에 함유하 게 하여 타정한 후, 반투과성 고분자로 정제의 표면을 코팅하여 이를 핵정으로 하 고, 선방출성 구획을 구성하는 과립물을 약제학적 인 첨가제와 흔합한 뒤 외층으로 하여 타정함으로써 내핵으로 지연방출성 구획을 갖고 상기 내핵의 표면을 속방층이 들러싼 형태의 경구 투여용 정제를 얻을 수 있다.
본 발명은 지연방출성 구획과 선방출성 구획 이 2상의 과립 , 선방 성 과립 또는 펠 뻣과 지연방출성 정제 , 지연방출성 과립 또는 팰¾과 선방출성 정제 , 및 지연방출 성 정제와 선방출성 정제로 층전된 캠술제의 제조방법 및 투여 방법으로 실시가 가 능하나, 이에 한정되는 것은 아니다
이하, 본 발명을 구체적으로 설명하면 , 본 발명으로 상기 지연방출성 제 2제와 속방 성 제 1제를 구성하는 과립물을 필요에 따라 약제학적인 첨가제와 흔합하여 캡슐에 층전하여 2상의 제어 방출이 가능한 캡슬제를 얻을 수 있다.
또한, 지연방출성 구획을 구성하는 과립물올 그대로 또는 정제로 타정한 후 지연방 출성을 나타내도록 코팅을 하고, 선방출성 구획을 구성하는 과립물을 타정하거나 코팅한 후, 지연방출성 정제와 선방출성 정제를 캡술에 층전하여 시간차 방출이 가 능한 캡슬제를 얻을 수 있다.
또한, ᅵ연방출성 구획을 구성하는 과립물을 그대로 또는 정제로 타정한 후 지연방 출성을 나타내도록 코팅을 하고, 선방출성 구획을 구성하는 분말, 과립 또는 펠렛 을 캡슐에 층전하여 시간차 방출이 가능한 캡슬제를 얻을 수 있다,
또한, 선방출성 구획을 구성하는 과립물을 타정 및 필요시 코팅하여 정제로 제조 후, 지연방출성 구획을 구성하는 과립 및 ¾¾을 캡슬에 층전하여 시간차 방출이 가능한 캡술제를 얻을 수 있다.
또한, 지연방출성 구획을 구성하는 과립 또는 펠¾과, 선방출성 구획을 구성하는 과림 또는 펠¾을 캡술에 층전하여 시간차 방출이 가능한 캡술제를 얻을 수 있다. 또한, 디히드로피리딘계 칼슴길항제의 방출 제어를 위해 삼투성 물질을 정제의 내 부에 함유하게 하여 타정한 후, 반투과성 고분자로 정제의 표면을 코팅하여 이를 핵정으로 하고, 선방출성 구획을 구성하는 과립물을 약제학적인 첨가제와 흔합한 뒤 캡술에 층전하여 시간차 방출이 가능한 캠술제 * 얻을 수 있다.
본 발명은 선방출성 구획을 구성하는 과립물 펠¾ 또는 선방출성 정제층을 별도로 제조하고, 지연방출성 구획을 구성하는 과립물, 또는 지연방출성 정제층을 호
일 , 블리스터 , 병 등에 같이 층전하여 동시에 복용이 가능한 키트로 제조가 가능하 다. 본 발명의 약제학적 제제는 다양한 제형으로 제 s할 수 있으며 , 예를 들어 나정 , 코팅정 , 다층정 , 또는 유핵정 등의 점제 , 분말제 , 과립제, 또는 캡술제 등으로 제 형화할 수 있다 .
본 발명의 약제학적 제제는 지연방출성 구획괴-, 이를 둘러싸는 선방출성 구획으로 이루어진 2상의 매트릭스 정제 형 태일 수 있다 . 상기 제조된 지연방출성 구획과 선 방출성 구획을 이루는 성물에 약제학적으로 허용가능한 첨가제들을 후흔합하여 타정하거나 혹은 캡술에 층전하여 경구 투여용 제제로 제조하여 지연방출성 구획과 선방출성 구획의 매트릭스가 2상으 S 존재하는 제제일 수 있다. 본 발명의 약제학 적 제제는 지연방출성 구획 및 선방출성 구획이 균일하게 흔합된 후 타점하여 얻어 지는 2상의 매트릭스 정제 형태일 수 있으며, 바람직하게는 상기 지연방출성 구획 은 입 (粒)상으로 제조된 것이다.
또한, 본 발명의 약제학적 제제는 지연방출성 구획으로 이루어진 점제와 상기 정제 의 의부를 둘러싸는 선방출성 구획으로 이루어진 필름코팅층으로 구성된 필름코뜀 정 형태일 수 있으며 , 필름코팅층이 용해됨 에 따라 필름코팅층의 심바스타틴이 던 저 용출되게 된다.
또한, 본 발명의 약제학적 제제는 지연방출성 구획과 선방출성 구획을 구성하는 과 림물에 약제학적인 첨가제를 흔합하고, 다중 타정기를 사용하여 2중정 흑은 3증정 으로 타정하여 얻어진 , 지연방출성 구획과 선방출성 구획 이 다층구조를 이루는 다 층정 형태일 수 있다. 이 제제는 층별로 선방출과 지연방출이 가능하도특 제제화된 경구 투여용 정제이다.
또한, 본 발명의 약제학적제제는 지연방출성 구획으로 이루어진 내핵과 상기 내핵 의 의면을 들러싸고 있는 선방출성 구획으로 이루어진 의층으로 구성 된 유핵정 형 태일 수 었다, 상기 유핵정은 삼투성 유핵정일 수 있으며 , 상기 삼투성 유핵정은 지연방출을 위해 삼투압조절제를 정제의 내부에 함유하게 하여 타정한 후, 반투과 성막 코팅기제로 정제의 표면을 코팅하여 이를 내핵으로 하고, 선방출성 구획을 구 성하는 과립물을 약제학적인 첨가제와 흔합한 뒤 외층으로 하여 타정함으로써 지연 방출성의 내핵을 갖고 상기 내핵의 표면을 선방출층이 둘러싼 형태의 제형이다. 본 발명의 약제학적 제제는 지연방출성 구획으로 이루어진 입자, 과림, 팰¾, 또는
정제와 선방출성 구획으로 이루어진 입자, 과립, 펠렛 , 또는 정제를 포함하는 캡술 제 형테일 수 있다.
본 발명의 제제는 지연방출성 구획 및 /또는 선방출성 구획의 외부에 코팅층을 추가 로 형성할 수 있다. 즉 지연방출성 구획 및 /또는 선방출성 구획으로 이루어진 입 자, 과립, 펠¾, 또는 정채 등의 표면에 방출제어 또는 제제 안정을 위한 목적으로 코팅을 할 수 있다.
또한 본 발명의 약제학적 제제는 지연방출성 구획 , 및 선방출성 구획을 포함하는 키트 형태일 수 있으며 . 구체적으로 본 발명은 선방출성 구획을 구성하는 입자, 과 립물, ¾¾, 또는 정제를 제조하고, 지연방출성 구획을 구성하는 과림물, ¾¾ 또 는 정제를 별도로 제조하여, 호일 , 블리스터 , 병 등에 같이 충전하여 동시에 복용 이 가능한 형 태로 제조한 키트 형태일 수 있다.
본 발명에 따른 제제는, 추가의 코팅이 없는 나정 등의 상태로도 제공되지만, 필요 에 따라 상기 제제의 외부에 코팅층을 형성시켜, 코팅층을 추가로 포함하는 코팅정 형태의 제제일 수 있다. 코팅층을 형성함으로써, 활성성분의 안정성을 더욱 확보할 수 있는 제제를 제공할 수 있다.
코팅층을 형성하는 방법은 정제층의 표면에 필름상외 코팅층을 형성할 수 있는 방 법 증에서 당업자의 선택에 의하여 적절히 선택할 수 있으며 , 유동층 코팅법 , 팬 코팅법 등의 방법을 적용할 수 있으며 , 바람직하게는 ¾ 코팅법을 적용할 수 있다. 코팅층은 피막제, 피막 보조제 또는 이들의 흔합불을 사용하여 형성할 수 있으며, 구체적으로 피막제는 히드록시프로필메틸샐를로오스 히드톡시프로필셸를로오스 등 과 같은 ¾를로오스 유도체 , 당 유도체 , ¾리비닐 유도체 , 확스류, 지방류, 젤라 틴 , 또는 이들의 혼합물 등을 사용할 수 있고, 피막 보조제는 플리에틸렌글리콜, 에틸샐를로오스, ¾리세라이드류, 산화티탄, 탈크, 디에틸프탈레이트, 또는 이들의 흔합물 등을 사용할 수 있다. 코팅층은 정제 총 증량에 대하여 0.5 15 증량퍼센트 (% w/w) 범위로 포함될 수 있다. 본 발명은 또한 저녁투여용인 본 발명에 의한 약제학적 제제를 제공한다.
본 발명의 제제는 하루애 한 번 특히 저녁시간 (17 - 23시 )에 복용함으로써, 각각의 유효 성분의 효과는 최대화하고 부작용은 최소화할 수 있다. 일반적으로 고혈압, 고지혈증 환자의 치료에서 고려해야할 증요한 요소는 생체리듬이다. 일례로 간 내 에서의 지질 합성은 초저녁 식사 이후 왕성해지고 , 고혈압 질환자를 포함한 일반인
외 혈입"은 밤과 새벽 사이에 하강하고 기상 후 오전 증에 혈압이 상승하기시작하여 일과시간 중 (활등 중)에 최고조에 이르게 된다. 이러한 사실을 감안할 때 , 본 발명 의 제제를 저녁시간에 북용할 경우 선방출되는 심바스타된이 간 효소가 활성화 되 는 시간대에 투여 되어 더 큰 지질 저하 효과를 보이고 지연방출되는 암로디핀에 의해 새벽 이후의 ¾압이 효과적으로 강하되어 새벽부터 오전 시간에 혈압을 균등 하게 유지시켜 줄 수 있으므로 약물의 경쟁적 상호 길항 작용을 피하며 각각의 유 효 성분의 효과를 최대화할 수 있다. 본 발명은 본 발명의 약제학적 제제를 포유류에게 투여하는 단계를 포함하는 심혈 관계 질환 치료방법을 제공한다 .
상기 심 ¾관계 질환은 고혈압, 또는 당뇨병 , 비만증, 고지 ¾중, 관상 동맥 질환 등 이 복합적으로 나타나는 이른바 대사성 증후군을 지닌 자들의 고혈압과 합병증등을 적용한다. 본 발명의 약제학적 제제는 당 분야의 적절한 방법으로, 일 예로
Chrontherpeut ics (2003, Peter Redfern, PhP) 에 개시되어 있는 시간차 투약 원리 를 이용하여 각 질환에 따라 또는 성분에 따라 바람직하게 제제화 할 수 있으며 , 구체적으로 이하의 단계롤 포함하는 방법에 의해 제조될 수 있다 .
제 1 단계는 암로디핀을 장용성 고분자, 수불용성 증합체 , 소수성 화합물, 친수성 고분자 증에서 선텍된 방출제어물질 1종 또는 2종과 약제학적으로 사용되는 통상의 ¾가제를 투여하여 흔합, 연합, 건조, 제립 또는 코팅, 및 타정을 통해 지연방출성 과립 또는 정제를 얻거나, 암로디핀을 삼투압≤절제와 약제학적으로 사용되는 통상 의 첨가제를 투여하여 흔합, 연합, 건조, 제립 또는 타정한 후 반투과성막 코팅기 제로 코팅 하여 지연방출성 과립 또는 정제를 얻는 단계이다 . 제 2 단계는 심바스타틴과 약제학적으로 허용되는 통상의 첨가제를 투여하여 흔합, 연합, 건조 , 제립 흑은 코팅 , 및 파정을 통해 경구 고형제를 생산하기 위한 통상의 과정을 통하여 엄어진 선방출성 과립 또는 정제를 얻는 단계이다.
제 3 단계는 상기 제 1 단계 및 제 2 단계에서 얻어진 각각의 과립 흑은 정제를 약 제학적인 부형제와 흔합하여 타정 또는 충전하여 경구 투여용 제제를 얻는 단계이 다.
상기 제 1 단계와 상기 제 2 단계는 순서를 바꾸거나, 동시에 실시할 수 있다. 상기 과정에 의하여 본 발명의 약제학적 제제가 제조될 수 있으며 , 제 3단계의 제제 화 방법을 보다 상세하게 설명하면 다음과 같으나, 이에 한정되는 것은 아니다. [가] 2상의 매트릭스 정제의 제조
제 1 단계에서 얻어진 입자 또는 과립을 그대로 또는 방출제어물질로 추가 코팅한 후, 제 2 단계에서 제조한 과립과 흔합하여 일정량의 무게로 타정하여 정제를 제≤ 한다. ¾어진 정제를 안정성 또는 성상 개선의 목적으로 필요에 따라 필름 코팅을 할 수 있다.
[나] 활성성분을 함유한 필름코팅정의 제조
제 1 단계에서 얻어진 코팅정 또는 과립을 그대로 또는 방출제어물질로 추가 코팅 하고 건조한 후 일정량으로 타정하여 그대로 흑은 추가로 코팅하여 정제를 제조한 후, 별도로 심바스타틴을 수용성의 필름코팅용액에 ' 용해 후 분산시켜제 1 단계에서 얻은 정제 의층에 코팅함으로써 필름코팅에 활성성분을 함유한 경구투여형 필름코 팅정제를 제조할 수 있다.
[다] 다층정의 제조
제 1 단계에서 얻어진 과립을 그대로 또는 방출제어.물질로 추가 코¾하고 건조하여 얻은 과립과 제 2 단계에서 얻어진 과립을 타정기를 이용하여 2중정으로 제조할 수 있다. 제형설계 또는 필요에 따라 방출 보 S층을 추가하여 3중 또는 그 이상의 다 층정을 제 S하거나 코팅하여 코팅 다층정을 제조할 수 있다.
[라] 유핵정의 제조
제 1 단계에서 얻어진 코팅정 또는 과립을 그대로 또는 방출제어물질로 추가 코팅 하고 건조한 후 일정량으로 타정하여 그대로 흑은 추가로 코팅을 하여 내핵으로 한 후, 제 2 단계예서 얻은 과립과 함께 유핵정타정기로 타정하여 1단계 정제의 표면 을 선방출층이 물러싼 형태의 유핵정을 제조하거나 코팅하여 코팅 유핵정을 제조할 수 있다.
[마 Ϊ 캡슐제 (과립 또는 정제 함유)의 제조 '
제 1 단계에서 얻어진 과립을 그대로 또는 방출제어물질로 추가 코팅하고 건조한 과립 또는 정제와, 제 2 단계에서 얻은 과립 또는 정계를 캡슬층전기에 넣고 일정 크기의 ¾술에 각 주성분 유효량 해당 량만큼 층전하여 캡슐제를 제조할 수 있다.
[바] ¾슬제 (펠햇)의 제조
(1) 암로디핀과 방출제어물질 , 필요에 따라 약제학적으로 허용 가능한 첨가제를 물 , 유기용매, 또는 혼합용매에 용해시키거나 현탁시켜 설탕 구형과립에 코팅 , 건조 후 필요에 따라 방출제어물질 단독 또는 2종 이상을 사용하여 물, 유기용데 , 또는 흔합용메에 용해시킨 후 코팅, 건조한 후 제 2 단계에서 얻은 과립 또는 제 3 단계 에서 얻은 정제와 흔합 후 캡슬충진기로 캡슬에 층전하여 캡술제를 제조할 수 있 다 .
(2) 심바스타틴과 제제학적으로 허용 가능한 첨가제를 물, 유기용메 , 또는 흔합용 매에 용해시키거나 현탁시켜 설탕 구형과립에 코팅 , 건조 후 상기 (1)의 암로디핀을 함유한 방출제어 ¾과 흔합 후 캡술층진기로 캡슬에 층전하여 ¾슬제를 제조할 수 있다.
[사] 키트의 제조
제 1 단계에서 얻어진 암로디핀 함유 제제와, 제 2 단계에서 얻은 심바스타틴 함유 제제를 호일 , 블리스터 , 병 등에 같이 층전하여 동시에 복용이 가능한 키트로 제조 할 수 있다. 상기와 같은 본 발명의 복합 약물 시스템은 디히드로피 리딘계 칼슴 채널 차 단제와 스타틴계 지질저하제를 활성성분으로 포함하여 복합 제제화되어 저녁 시간 대에 단 1희씩 투여케 함으로써 각 활성성분을 별도로 제제화하여 이를 뜸시에 복 용케 하는 경우보다 복약지도가 쉽고, 또한 약물 상호간의 대사 간섭 작용이 일어 나지 않아 이에 따른 부작용을 감소시킬 수 있으며, 각 약물이 가지는 혈암조절과 지질조절효과가 이들의 자체가 단독으로 가지는 효과보다 향상되어 나타난다. 【유리한 효과】
심바스타틴으로 대표되는 스타틴계 지질저하제와, 암로디핀으로 대표되는 디 히드로피리딘계 칼슴길항제의 위장관내 용출 및 흠수 시간에 각각 차이를 두도톡
할 경우 스타틴계 지질저하제의 필요 이상의 혈증 농도 상승을 막고 축적 작용을 막아 그 부작용을 예방할 수 있을 것으로 판단하여 이들을 혈증 농도 상승을 막고 그 부작용을 예방할 수 있을 것임을 확인하였다.
상슬한 바와 같이, 본 발명은 이종약물의 병용시 발생할 수 있는 부작용을 약물동태학적 관점에서 개선하고자 하는 이종 약물 대사학 Ofenobiotics)을 근거로 하여 치료 효과를 극대화시키는 이른바 시간 차 투약 치료볕 (Chronotherapeutics) 을 제제화시킨 것으로서, 등일한 효소인 사이토크롬 P 450계 호소에 영향을 주거나 또는 받는 성분인 디히드로피리딘계 칼슴길항제와 스타틴계 지질 저하제롤 유효 활 성성분으로 복합하여 사용하면서 , 이들이 체내에서 용출되는 시간을 제어할 수 있 도록 지연방출성 물질을 차별성 있게 사용함으로써 활성성분을 특정 속도로 시간차 를 두고 송달 가능하므로, 상기 약풀성분을 따로따로 동시에 복용하는 복합 처방의 경우 보다 만성 순환기 질환의 예방과 치료에서 약리학적 , 임상학적 , 과학적 및 경 제적으로 보다 유용한 약제학적 제제를 실현케 할 수 있다.
또한, 본 발명에 의하면 상기 약물의 방출속도를 달리 구성하여 약물 상호간 의 길항 작용 및 부작용을 방지함과 동시에 약흐의 상승 작용을 엄을 수 있다.
또한ᅳ 본 발명에 의하면 복합 제제를 한번에 복용할 수 있으므로 환자에 대 한 복약 지도 및 환자의 복약이 용이하다.
또한, 본 발명의 스타틴계 지질저하제를 함유하는 선방출성 구획 , 및 디히드 로피리딘계 칼슘 채널 차단제를 함유하는 지연방출성 구획을 포함하는 약제학적 제 제는 대사 증후군 및 인슐린 저항성 환자 및 당뇨병 혹은 당뇨병 전증으로 의심되 는 환자들의 심혈관 질환, 심폐 질환, 폐질환 또는 신장 질환의 예방 또는 치료에 서 약리학적, 임상학적 과학적 및 경제적으로 각각의 약물의 단일 제제 및 단순 복합 제제보다 매우 유용하다.
【도면의 간단한 '설명】
도 1은 실시예 1-1에 따라 제초된 암로디핀 /심바스타틴 유핵정제와 대조약 ( 조≤ : 심바스타틴 단일제 , 노바스크 : 암로디핀 단일제)의 비교 용출의 곡선을 나 타낸 그래프이다,
도 2는 실시예 1-4, 10에 따라 제조된 암로디핀 /심바스타틴의 복합 제제와 대조약 (조코 : 심바스타된 단일제 , 노바스크 : 암로디핀 단일제)의 비교 응출의 곡 선을 나타낸 그래프이다.
도 3은 실시예 1-11에 따라 제 S된 암로디핀 /로바스타틴의 복합 제제와 대조
약 (메바코 : 로바스타틴 단일제, 노바스크 : 암로디핀 단일제)의 비교 용출의 곡선 을 나타낸 그래프이다.
도 4는 실시예 1—13에 따라 제조된 암로디핀 /아토르바스타틴의 복함 제제와 대조약 (리피토 : 아토르바스타틴 단일제 , 노바스크 : 암로디핀 단일제)의 비교 용 출의 곡선을 나타낸 그래프이다,
도 5는 실시예 1-16에 따라 제조된 레르카니디 ¾/심바스타틴의 복합 제제와 대조약 (조코 : 심바스타¾ 단일제, 자니딥 : 레르카니디핀 단일제)의 비교 용출의 곡선을 나타낸 그래프이다.
도 6은 실시예 1-18에 따라 제조된 라시디핀 /심바스타틴의 복함 제제와 대조 약 (조코 : 심바스파틴 단일제 , 박사르 : 라시디핀 단일체)의 비교 용출의 곡선을 나타낸 그래프이다.
도 7은 실시예 1-20에 따라 제조된 암로디핀 베실레이트 /심바스타린 유핵정 제와 대조약 (조코:심바스타틴 단일제 , MSD, 노바스크 : 암로디핀 단일제 , 화이자) 의 비교 용출의 곡선올 나파낸 그래프이다.
도 8은 상기 실시예 1-22에 따라 ᅳ제조된 암로디핀 폐실레이트 /심바스타틴 복 합정제를 함유한 캡술제와 대조약 (조코:심바스타틴 단일제, MSD, 노바스크:암로디 핀 단일제, 화이자)의 비교 용출곡선을 나타낸 그래프이다ᅵ.
도 9는 상기 실시예 1-30에 따라 제조된 암로디핀 베실레이트 /아토르바스타 틴 칼슘의 복합 제제와 대조약 (리피토 : 아토르바스타 ¾ 단일제, 화이자, 노바스 크:암로디 ¾ 단일제 , 화이자)의 비교 용출곡선을 나타낸 그래프이다.
도 10은 상기 실시예 1-36에 따라 제조된 펠로디핀 /아토르바스타 ¾ 캡술제 형태의 2상 제제와 대조약 (리피토:아토르바스타틴 단일제 , 화이자, 무노발:펠로디 핀 단일제 , 한독약품)의 비교 용출곡선을 나타낸 그래프이다.
도 11은 상기 실시예 1ᅳ40에 따라 제조된 이스라디핀 /를루바스타틴 캡슬제 형태의 2상 제제와 대조약 (레스콜:폴루바스타 ¾ 단일제, 노바티스, 다이나쩌크:이 스라디핀 단일제, 대웅제약)의 비교 용출곡선을 나타낸 그래프이다.
도 I2은 상기 실시예 1-51, 55에 따라 제조된 (S)-암로디핀 /심바스타틴 복합 제제의 용출시험 결과 * 나타낸 그래프이다.
도 13은 상기 실시예 1-52, 53, 54에 따라 제조된 (S)ᅳ암로디핀 /심바스타틴 복합제제의 용출시험 결과를 나타낸 그래프이다.
£ 14는 상기 실험 예 1-14에 의한 임상시험결과로서 실험군간 심바스타¾ β
-히드록시산의 혈중 농도를 비교한 그래프이다.
도 15는 상기 실험예 1-14에 의한 임상시험결과로서 실험군간 심바스타틴 β -히드록시산 및 심바스타틴의 혈중 농도를 비교한 그래프이다.
도 16은 상기 실험예 Ι-Μ에 외한 임상시험결과로서 시간차 투여군과 동시투 여군 간의 암로디핀의 혈증 농도를 비교한 그래프이다.
도 17은 실시 예 II— 1에 따라 제조된 암로디핀 /심바스타틴 유핵정제와 대조약 (2:코 : 심바스타틴 단일제 , MSD,노바스크 : 암로디핀 단일제 , 화이자)의 비교 용 출의 곡선을 나타낸 그래프이다.
도 13은 실시예 Π-4, 10에 따라 제조된 암로디핀 /심바스타틴의 약제학적 제 제와 대조약 (조코 : 심바스타틴 단일제 , 노바스크 : 암로디핀 단일제)의 비교 용출 의 곡선을 나타낸 그래프이다.
도 19는 실시예 IIᅳ 11에 따라 제조된 암로다핀 베실레이트 /심바스타틴 유핵 정제와 대조약 (조코:심바스타틴 단일제 , 노바스크 : 암로디핀 단일제)의 비교 용출 의 곡선을 나타낸 그래프이다.
도 20은 상기 설시예 11-13에 따라 제조된 암로디 ¾ 베실레이트 /심바스타틴 복합정제를 함유한 캡슬제와 대조약 (조코:심바스타틴 단일제 , 노바스크 :암로디핀 단일제)의 비교 용출곡선을 나타낸 그래프이디-.
도 21은 실시예 III-1에 따라 제조된 암로디핀 /아토르바스타틴 유핵정제와 대조약 (리퍼토: 아토르바스타틴 단일제 , 노바스크 : 암로디 ¾ 단일제)의 비교 용출 의 곡선을 나타낸 그래프이다. - 도 22는 실시예 III-1, 2, 3의 암로디핀 용출 양상을 나타낸 그래프이다, 도 23은 실시예 ΙΠ-6, 7의 용출 양상을 나타낸 그래프이다.
도 24는 실시예 III-1의 아토르바스타 ¾ ¾증 농도 -시간 프로필을 나타낸 그 래프이다.
도 25는 실시 예 III-1의 암로디 ¾ 혈증 농도ᅳ시간 프로필을 나타낸 그래프이 다.
도 26은 실시예 IV-2에 따라. 제조된 피타바스타틴핀 /암로디핀 유핵정제와 대 조약 (리바로 : 피타바스타틴 단일제, 노바스크 : 암로디핀 단일제 )의 비교 용출의 곡선을 나타낸 그래프이다.
도 27은 실시예 2, 3의 암로디핀 용출 양상을 나타낸 그래프이다. 도 28은 실시예 IV-17, 18의 암로디핀 용출 양상을 나타낸 그래프이다.
도 29는 실시예 V-4에 따라 제조된 로슈바스타틴된 /암로디 ¾ 유핵정제와 대 조약 (크레스토: 로슈바스타틴 단일제, 노바스크: 암로디핀 단일제)의 비교 용출의 곡선을 나타낸 그래프이다.
도 30은 실시예 및 실시예 V-18 에 따라 제조된 제제 내의 로슈바스타 틴 /암로디핀의 용출 양상을 나타낸 그래프이다,
도 31은 실시예 VI-1에 따라 제조된 아토르바스타틴핀 /니페디꾄 유핵정제와 대조약 (리피토: 아토르바스타틴 단일제, 프로카디아 XL: 니페디핀 단일제)의 비교 용출의 곡선을 나타낸 그래프이다.
도 32는 실시예 VI-6의 용출 양상을 나타낸 그래프이다.
도 33은 실시예 의 용출 양상을 나타낸 그래프이다.
도 34는 실시예 "VI-10의 용출 양상을 나타낸 그래프이다.
【발명의 실시를 위한 형태】
본 발명의 이점 및 특징 , 그리고 그것들을 달성하는 방법은 상세하게 후술되 어 있는 실시예들을 참조하면 명확해질 것이다. 그러나 본 발명은 이하에서 개시되 는 실시예들에 한정되는 것이 아니라 서로 다른 다양한 형태로 구현될 것이며 , 단 지 본 실시예들은 본 발명의 개시가 완.전하도록 하고, 본 발명이 속하는 기슬 분야 에서 통상의 지식을 가진 자에게 발명의 범주를 완전하게 알려주기 위해 제공되는 것이며 , 본 발명은 청구항의 범주에 의해 정의될 뿐이다. 실시예 1-1: 암로디핀―심바스타틴 유핵정의 제조
1) 암로디핀 지연방출성 구획의 제조
다음 표 1-2에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정 셀를로오 스를 35호체로 사과하고 더블콘믹서토 흔합한 후 유동층과립기 (GPCG 1: Glatt)에 투입하고, 따로 히드륙시프로필메틸셀를로오스를 물에 녹여 만든 결합액을 분무하 여 과립을 형성, 건조하였다. 다시 상기의 과림에 카보머 71G를 분말상태로 투입한 후, 스테아린산 마그네슘을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 흔함물을 로타리 타정기 (MRC-33: 세종)를 사용하여 분당 30희전의 속도로 경도 7~9kp, 두께 3.0隨, 지름 5.5腿으로 타정하여 이를 핵정으로 하였다,
2) 심바스타¾ 선방출성 구획의 제조
다음 표 r-2에 나타난 성분 및 함량과 같이 심바스타틴 , 미결정샐를로오스, 만니를 을 35호체로 사과하고 고뇩흔합기로 흔합하였다. 따로 히드특시프로필셀롤로오스와
구연산을 물에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 부틸레이티드 히드록시아니솔, 전분 글리콘산 나트 ", 콜로이드성 이산화규소를 흔합하고, 스테 아린산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
3) 타정 및 코팅
유핵정 타정기 (RUD-1: Ki l ian)를 사용하여 암로디핀 핵정을 내핵으로 하고 심바스 타틴을 포함하는 조성물을 의층으로 하여 분당 30회전의 속도로, 경도 7~— 9kp, 두 께 6.0聽, 지름 9.5腿으로' 타정한 다음 하이코터 (SFC— 30N, 세종 기계 한국)를 사 용하여 필름 코팅 층을 형성하여 유핵정을 제조하였다. 실시예 1-2 : 암로디핀 - 심바스타틴 유핵정의 쩨 :
1) 암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 1—2에 나타난 성분 및 함량과 같이 암로디 ¾ 말레이트와 미결정 셀를로오 스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과립기 (GPCG 1: Glatt)에 투입하고, 따로 히드록시프로필메 ¾셀를로오스를 물에 녹여 만든 결합액을 분무하 여 과립을 형성, 건조하였다. 다시 상기의 과립에 에탄을과 염화메틸렌의 1:1흔액 에 용해시킨 히드록시프로필메틸셀를로오스프탈레이트 용액을 분무하여 과립을 코 팅하였다. 여기에 스테아린산 마그네슴을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC-33 : 세종)를 사용하여 분당 30회전의 속도로, 경도 7 9 kp, 두께 3.0 腿, 지름 5.5 腿으로 타정하여 이를 핵정으로 하였다 .
2) 심바스타틴 선방출성 구획의 제조
다음 표 1-2에 나타난 성분 및 함량과 실시예 1-1의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
실시예 1-1의 3) 타정 및 코팅 방법에 따라 조작하여 암로디핀 -심바스타틴 유핵정 을 제조하였다. 실시예 1—3 : 암로디괸 - 심바스타틴 유핵정의 제조
1) 암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 1-2에 나타난 함량과 같이 암로디핀 말레이트와 미결정셀를로오스를 35호
체로 사과하고 더블콘믹서로 흔합하고, 고속흔합기에 투입한 후 콜리코트 SR30D를 가하여 연합한 다음 20호체로 오실레이터를 이용하여 제립하고, 이를 은수 건조기 를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 스테아린산 마그네슘을 넣어 더블콘믹서로 최종 혼합하고, 상기 최종 흔합물을 로타리 타정기 (M C-33 : 세종)를 사용하여 분당 30회전의 속도로, 경도 7~9kp, 두깨 3.0圃, 지 름 5.5mm으로 타정하여 이¾ 핵정으로 하였다.
2) 심바스타¾ 선방출성 구획의 제조
다음 표 1—2에 나타난 성분 및 함량과 셜시예 1-1의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
실시예 1-1의 3) 타정 및 코팅 방법에 따라 조작하여 암로디된—심바스타틴 유핵정 을 제조하였다. 실시예 1-4 : 암로디핀 - 심바스타된 다층정의 제조
1) 암로디핀 지 연방출성 구획외 제조 (내핵)
다음 표 1-2에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정 셸를로오 스를 35호체로 사과하고 더블콘믹서로 흔합하고, 따로 히드록시프로필메틸셸를로오 스를 물에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과 립에 에탄을과 염화메틸렌의 1:1흔액에 용해시 ¾ 히드록시프로필메틸셀를로오스프 탈레이트 용액을 분무하여 과립을 코팅하였다. 여기에 스테아린산 마그네숨을 넣어 최종 더블 콘믹서로 흔합하였다.
2) 심바스타틴 선방출성 구획의 제
다음 표 1-2에 나타난 성분 및 함량과 실시예 1-1의 2) 심바스타틴 선방출성 구획 의 제 S 방법에 따라 제조하였다.
3 타정 및 코¾
본 공정에서는 다층정 타정기 (MRC-37T : 세종)를 사용하여 타정하였다. 즉, 상기 심바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 암로디핀을 포함하는 조 성불을 2차 분말 공급기에 넣어 층간외 혼입을 최소화할 수 있는 조건으로 분당 30 희전의 속도로, 경도 7 ~ 9 1φ, 두께 6.0 mm, 지름 9.5顏으로 타정하고, 하이코 터로서 필름 코팅층을 형성하여 다층정 을 제 a하였다,
실시예 1-5 : 암로디 ¾ - 심바스타틴 다층정의 제조
1) 암로디핀 지연방출성 구획의 제조
다음 표 1-2에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정 셀를로오 스를 35호체로 사과하고 더블콘믹서로 흔합하고, 따토 히드록시프로필메 ¾¾€ -로오 스를 물에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조하였다. 다시 .상기의 과 립에 에탄을과 염화데틸렌의 1: 1흔액에 용해시킨 유드라짓 RS P0 용액을 분무하여 과립을 코팅하였다. 여기에 스테아린산 마그네슴을 넣어 최종 더블 콘 ¾서로 흔합 하였다.
2) 심바스타틴 선방출성 구획의 제 S
다음 표 1—2에 나타난 성분 및 함량과 실시예 1-1의 2) 심바스타틴 선방 *성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코¾
실시예 1-4의 3) 타정 및 코팅 방법에 따라 조작하여 암로디핀 -심바스타틴 다층정 을 제조하였다. 실시예 Ϊ-6 : 암로디핀 - 심바스타된 2상 매트릭스 정제의 제조
1) 암로디핀 지연방출성 구획의 제 S '
다음 표 1-2,에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정셀를로오스 를 35호체로 사과하고 혼합하고, 이를 고속 흔합기에 투업하여 콜리코트 SR30D를 첨가한 후 연합하였다 . 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고, 이를 은수 건조기를 이용하여 60 I;에서 건 S한 다음 다시 20 호로 정립하였다,
2) 심바스타틴 선방출성 구획의 제
다음 표 1—2에 나타낸 성분 및 함량과 같이 심바스타틴, 미결정 ¾를로오스, 만니를 을 35호체로 사과하고 고속 Φ합기로 흔합하였다. 따로 히드록시프로필셸를로오스와 구연산을 튤에 녹여 결합액을 제조하고 이를 상기 주성분의 혼합물과 함께 고속흔 합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하고, 여기에 부틸테 이티드히드특시아니솔ᅳ을 넣고 흔합하였다.
3) 후혼합, 타정 및 코뜀
상기 제조된 각각의 최종 S성물을 더블콘믹서로 흔합하고, 전분 글리콘산 나트륨,
콜로이드성 이산화규소를 흔합한 후 스테아린산 마그네슴을 넣어 더블콘먹서로 최 중 흔합하였다.
상기 최종 흔합물을 로타리 타정기 (MRC-33 : 세종)롤 사용하여 분당 30희전의 속도 로, 경도 7 ~ 9 kp, 두께 6,0 ram, 지름 9.5顯으로 타정한 다음 하이코터로서 필 름 코팅 층을 형성하여 2상 매트릭스 정제를 제조하였다. 실시예 1-7 : 암로디된 - 심바스타틴 2 상 매트릭스 정제의 제
1) 암로디핀 지연방출성 과립의 제조
다음 표 1—2에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정 셀를로오 스를 35호체로 사과하고 더불콘믹서로 흔합하였다. 상기의 흔합물을 유동층과립기 (GPCG 1: Glatt)에 투입하고 따로 히드록시프로필메틸 ^를로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과립에 에탄을과 염화메 틸텐의 1: 1흔액에 용해시킨 유드라짓 RS P0 용액을 분무하여 과립을 코¾하였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1-2에 나타난 성분 및 함량과 실시 예 1-6의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
실시예 1-6의 3) 후흔합, 타정 및 코팅 방법에 따라 조작하여 암로디핀 -심바스타린 2상 매트릭스 정제를 제조하였다. 실시 예 1-8 : 암로디핀 - 심바스타띈 2 상 매트릭스 정제의 제조
1) 암로디핀 지연방출성 구획의 제조
다음 표 1-2에 나타난 성분 및 함량과 같이 . 암로디핀 말레이트와 미결정 셸를로오 스롤 35호체로 사과하고 더블콘믹서로 흔합하였다. 상기의 흔합물을 유등층과립기 (GPCG 1: Glatt ) 에 투입하고 따로 히드특시^로필메틸셀를로오스를 물에 녹여 만 든 결합액을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄을과 염 화메틸렌의 1: 1흔액에 용해시킨 히드콕시프로필메틸샐를로오스 H탈레이트 용액을 분무하여 과립을 코팅하였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1-2에 나타난 성분 및 함량과 실시예 1-6의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
실시 예 I ^의 3) 후흔합, 타정 및 코팅 방법에 따라 조작하여 암로디핀 -심바스타틴 2상 매트릭스 정제를 제조하였다. 실시예 1~9 : 암로디핀 ᅳ 심바스타된 2상 캡술 제제
1) 암로디핀 지연방출성 구획의 제조 (과림)
다음 표 Iᅳ 2에 나타난 성분 및 함량과 실시예 1-8의 1) 암로디핀 지연방출성 구획 의 제조 방법에 따라 제조하였다.
2) 심바스타틴 선방출성 구획의 제조 (과립 )
다음 표 1—2에 나타난 성분 및 함량과 실시예 1-6의 2) 심바스타된 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 흔합 및 캡슬층전
공정 1)과 2)의 최종 조성물을 더블콘믹서로 흔합하고, 여기에 전분글리콘산나트륨 을 투입한 후 더불콘믹서로 흔합하고, 콜로이드성 이산화규소를 흔합하고, 스테아 린산 마그네슘을 넣어 최종 흔합하였다. 최종 혼합된 흔합물을 분말 공급기에 투입 하고 캡술층전기를 이용하여 1호 ¾라틴 경질캡슐에 층전하였다. 실시예 1-10 : 암로디핀 ᅳ 심바스타틴 2상 캡술 제제
1) 암로디핀 지연방출성 구획의 제조 (과립 )
다음 표 1-2에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정젤를로오스 를 35호체로 사과하고 더블콘믹서로 흔합한 다음 이를 유등층과립기 (GPCG 1 : Glatt)에 투입하고 콜리코트 SR30D 를 분무하여 과립을 형성 , 건조시켰다.
2) 심바스타틴 선방출성 구획의 제조 (과립)
다음 표 1-2에 나타난 성분 및 함량과 실시예 1-6의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 혼합 및 캡슬 층전
실시예 1-9의 3) 후흔합, 타정 및 코팅 방법에 따라 조작하여 암로디괸 -심바스타틴 2상 ¾슐제제를 제조하였다. 실시예 1-11 : 암로디 ¾ - 로바스타틴 다층정의 제조
1) 암로디핀 지연방출성 구획의 제조
다음 표 I— 3에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정 셀를로오 스를 35호체로 사과하고 더블콘믹서로 흔합하고 , 상기의 흔합물을 유등층과립기 (GPCG 1 : Glatt)에 투입하고 따로 히드록시프로필메틸셀를로오스를 물에 녹여 만 든 결합액을 분무하여 과립을 형성, 건 S하였다. 다시 상기의 과립에 에탄을과 염 화메틸렌의 1: 1혼액에 용해시 ¾ 히드록시프로필메털셀를로오스프탈레이트 용액을 분무하여 과립을 코팅하였다. 여기에 스테아린산 마그네슴을 넣어 최종 더블 콘먹 서로 흔합하였다.
2) 로바스타린 선방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 같이 로바스타틴, 미결정셀를로오스, 만니를 을 35호체로 사과하고 고속흔합기로 혼합하였다. 따로 히드톡시프로필¾를로오스와 구연산을 물에 녹여 결합액을 계조하고 이를 주성분 흔합물과 함께 고속흔합기에 투업하고 연합한 다음 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기 를 이용하여 60 °C에서 건조한 후 다시 20호체로 정립하였다. 여기에 부¾테이티드 히드특시아니솔, 전분 글리콘산' 나트롭, 콜로이드성 이산화규소를 흔합하고, 스테 아린산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
3) 타정 및 코팅
본 공정에서는 다층정 타정기 (MRC— 37T : 세종)를 사용하여 타정하였다. 즉, 상기 로바스타린을 포함하는 조성물을 1차 분말공급기에 넣고, 암로디핀을 포함하는 조 성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 분당 30 회전외 속도로, 경도 7 ~ 9 kp, 두께 6.0 腿, 지름 9.5 隱으로 타정하고 하이코 터로서 필름 코팅층을 형성하여 다층점을 제조하였다. 실시예 1-12 : 암로디핀 - 로바스타틴 다층정의 제조
1) 암로디핀 지연방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정 ¾를로오스 를 35 호체로 사과하고 더불콘믹서로 흔합한 후 고속흔합기에 투입하고 콜리코트 S 30D 를 가하여 연합한 다음 20호체로 오실레이터를 이용하여 제립하고, 이를 은 수 건조기를 이용하여 60 에서 건조한 후 다시 20호체로 정 립하였다. 여기에 스 테아린산 마그네슘을 넣어 더블콘믹서로 최종 흔합하였다.
2) 로바스타¾ 선방출성 구획의 제조
다흠 표 1-3에 나타난 성분 및 함량과 실시예 1-11의 2) S바스타틴 선방출성 구획
의 제조 방법에 따라 제조하였다.
3) 타정 및 코뜀
실시예 1-11의 3) 타정 및 코팅 방법에 따라 조작하여 암로디핀 -로바스타틴 다층정 을 제조하였다. 실시예 1-13 : 암로디핀 - 아토르바스타된 다층정의 제조
1) 암로디핀 지연방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 실시예 1-11의 1) 암로디핀 지연방출성 구획 의 제조 방법에 따라 제조하였다.
2) 아토르바스타틴 선방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 같이 아토르바스타틴 , 미결정 ^를로오스, 만 니를을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드록시프로필¾를로오 스와 구연산을 물에 녹여 결합액을 제조하고 이를 주성분 흔합 :과 함께 고속흔합 기에 투입하고 연합한 다음 20호체토 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 에서 건조한 후 다시 20호체로 정 립하였다. 여기에 부틸레 이티드히드록시아니솔, 전분 글리콘산 나트륨, 콜로이드성 이산화규소를 흔합하고, 스테아린산 파그네슘을 넣어 더블콘믹서로 최종 흔합하였다.
3) 타정 및 코팅
본 공정에서는 다층정 타정기 (MRC-37T : 세종)를 사용하여 타정하였다. 즉, 상기 아토르바스타틴을 포함하는 S성물을 1차 분말공급기에 넣고, 암로디핀을 포함하는 조성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 분당 30희전의 속도로 경도 7 ~ 9 >, 두께 6.0 纖, 지름 9.5腿으로 타정하고, 하이 코터로서 필름 코팅층을 형성하여 다층정 형태의 지연방출성 다층정을 제조하였다. 실시예 1-14 : 암로디핀 - 아토르바스타틴 다층정의 제조
1) 암로디핀 지연방출성 구획의 제조
다음 표 ί~3에 나타난 성분 및 함량과 실시예 1-12의 1) 암로디핀 지연방출성 구획 의 제조 방법에 따라 제조하였다.
2) 아토르바스타틴 선방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 실시예 1-13의 2) 아토르바스타틴 선방출성 구획의 제 방법에 따라 제조하였다.
3) 타정 및 코팅
실시예 1-13의 3) 타정 및 코팅 방법에 따라 조작하여 암로디핀ᅳ아토 s바스타틴 다 층정을 제조하였다. 실시예 1-15 : 암로디핀 - 아토르바스타틴 유핵정의 제조
1) 암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 1-3에 나타난 성분 및 함량과 실시예 1-1의 1) 암로디핀 지연방출성 구획 의 제조 방법에 따라 제조하였다.
2) 아토르바스타 ¾ 선방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 실시예 1-13의 2) 아토르바스타 ¾ 선방출성 구획의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
유핵정 타정기 (RUD-1 : Ki Han)를 사용하여 암로디핀 핵정을 내핵으로 하고 아토르 바스타틴을 ᅳ포함하는' 조성물을 외층으로 하여 분당 30회전의 속도로, 경도 7 〜 9 kp, 두께 6.0删, 지름 9.5赚으로 타정한 다음 하이코터롤 사용하여 필름 코팅 층 을 형성하여 유핵정을 제조하였다. 질시예 1-16 : 레르카니디핀 - 심바스타틴 다층정의 제조
1) 레르카니디핀 지연방출성 구획의 제조
다음 표 1—3에 나타난 성분 및 함량과 같이 레르카니디핀과 미결정 샐롤로오스롤 35호쳬로 사과하고 더블콘믹서로 흔합하고, 상기의 흔합물을 유등층과립기 (GPCG 1 : Glatt)에 투입하고 따로 히드록시프로필메틸셸를로오스를 물에 녹여 만든 결합액 을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄올과 염화떼틸렌의 1: 1흔액에 용해시킨 히드특시프로필쨰 ¾셀를로오스프탈레이트 용액을 분무하여 과 립을 코팅하였다. 여기에 스테아린산 마그네슴을 넣어 최종 더블 콘믹서로 흔합하 였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1—3에 나타난 성분 및 함량과 실시예 1-1의 2) 심바스타¾ 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
본 공정에서는 다층정 타정기 (MRC-37T : 세종)롤 사용하여 타정하였다. 즉, 상기
심바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 레르카니디핀을 포함하는 조성물을 2차 분말 공급기에 넣어 충간의 흔입을 최소화할 수 있는 조건으로 분당 30회전의 속도로, 경도 7 ~- 9 kp, 두께 6,0讓, 지름 9.5 mm으로 타점하고, 하이 코터로서 필름 코팅층을 형성하여 다층정을 제조하였다. 실시예 Iᅳ 17 : 레르키 "니디 ¾ - 심바스타¾ 다층정의 제조
1) 레르카니디핀 지연방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 같이 레르카니디핀과 미결정셀를로오스롤 35 호체로 사과하고 더불콘믹서로 혼합한 후 고속흔합기에 투입하고 콜리코트 SR30D를 가하여 연합한 다음 20호체로 오실레이터를 이용하여 제림하고, 이를 은수 건조기 를 이용하여 60 °C에서 건조한 후 다시 20호체로 정립하였다. 여기에 스테아린산 마그네슴을 넣어 더블콘믹서로 최종 혼합하였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 실시예 1-1의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코뜀
실시예 1-16의 3) 타정 및 코팅 방법에 따라 조작하여 레르카니디핀 -심바스타틴 다 층정을 제조하였다. 실시예 1-18 : 라시디핀 - 심바스타틴 다층정의 제조
1) 라시디핀 지연방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 같이 라시디핀과 미결정 쎌를로오스를 35호 체로 사과하고 더블콘믹서로 흔합하고 , , 상기의 흔합툴을 유등층과림기 (GPCG 1 : Glatt)에 투입하고 따로 히드록시프로필메틸셀를로오스롤 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과립에 에탄을과 염화메틸렌의 1: 1흔액에 용해시킨 히드록시프로필메 ¾셀를로오스프탈래이트 용액을 분무하여 과 립을 코팅하였다. 여기에 스테아린산 마그네슴을 넣어 최종 더블 콘먹서로 혼합하 였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 실시예 1—11의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코렁
본 공정에서는 다층정 타정기 (MRC-37T : 세종)를 사용하여 타정하였다. 즉, 상기 심바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 라시디핀을 포함하는 조 성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 분당 30 회전의 속도로, 경도 7 9 kp, 두께 6.0 賺, 지름 9.5 咖으로 타정하고, 하이코 터로서 필름 코팅층을 형성하여 다층정을 제조하였다. 실시예 1-19 : 라시디 ¾ - 심바스타된 다층정의 제조
1) 라시디핀 지연방출성 구획의 제조
다음 표 Ϊ-3에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정셀를로오스 를 35 호체로 사과하고 더블콘믹서로 흔합한 후 고속혼합기에 투입하고 콜리코트 SR30D 를 가하여 연합한 다음 20호체로 오실레이터를 이용하여 제립하고, 이를 은 수 건조기를 이용하여 60 X:에서 건조한 후 다시 20호체로 정립하였다. 여기에 스 테아린산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 Iᅳ 3에 나타난 성분 및 함량과 실시예 1-1의 2) 심바스타린 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
실시예 1-18의 3) 타정 및 코팅 방법에 따라 조작하여 라시디핀—심바스타틴 다층정 을 제조하였다. 실시예 Iᅳ 20 : 암로다핀 -심바스타틴 유핵정의 제조
1) 암로디핀 지연방출성 구획의 제조 〔내핵)
다음 표 1-4에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 셀를로 오스, 디칼슴 포스페이트를 35호체로 사과하고 더블콘먹서로 흔합한 후 유동층과립 기 (GPCG 1: Glatt)에 투입하고, 따로 히드록시프로필메틸셸를로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과립에 카보머 71G 를 분말상태로 투입한 후, 여기에 스테아린산 마그네슴을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 흔합물을 로타리 타점기 (MRC-33 : 세종)를 사용하여 분당 30 희전의 속도로, 경도 7 ~ 9 kp, 두깨 3.0 麵, 지름 5.5 咖으로. 타정하였다. 다시 상기의 정제에 에탄을과 염화메틸렌의 1: 1흔액에 용해시킨 히드톡시프로필메 ¾¾를
로오스프탈레이트 용액과 아세틸레이티드모노글리세리드 용액을 하이코터롤 사용하 여 필름 코팅 층을 형성하여 핵정을 제조하였다.
2) 심바스타틴 선방출성 구획의 제조
다을 표 1-4에 나타난 성분 및 함량과 같이 심바스타틴 , 미결정셀를로오스, 옥수수 전분 및 유당을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드특시프로필 샐를로오스와 구연산을 물에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제림하고 이를 은수 건 S기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 부틸레이티드히드록시아니솔, 전분 글리콘산 나트. s 콜로이드성 이산화규소를 흔 합하고, 스테아린산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
3) 타정 및 코팅
유핵정 타정기 (RUD-1: Ki Han)를 사용하여 암로디핀 핵정을 내핵으로 하고 심바스 타틴을 포함하는 조성물을 의층으로 하여 분당 30회전의 속도로, 경도 7 〜 9 kp , 두께 6.0 mm, 지름 9.5 讓으로 타정한 다음 하이코터로서 필름 코 ¾ 층을 형성하여 유핵정을 제조하였다. 실시예 1—21 : 암로디핀 -심바스타된 다층정
1) 암로디핀 지연방출성 구획의 제조
다음 표 1-4에 나타난 성분 및 함량과 같이 솬로디 ¾ 베실레이트와 미결정 셀를 S 오스 및 디칼슴포스페이트를 35호체로 사과하고 더블콘믹서로 흔합하고, 따로 히드 록시프로필메틸셀를로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성 건조 하였다. 다시 상기의 과립에 에탄을과 염확메틸렌의 1:1흔액에 용해시 ¾ 히드 록시프로필메틸셸를로오스프탈레이트과 아세틸레이티드모노글리세리드로 구성된 코 팅액을 분무하여 과립을 코팅하였다. 여기에 스테아린산 마그네습을 넣어 최종 더 블콘믹서로 흔합하였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1-3에 나타난 성분 및 함량과 실시예 1-1의 2) 심바스타¾ 선방출의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
본 공점에서는 다층정 타정기 (MRC-37T : 세종)롤 사용하여 타정하였다. 즉 , 상기 심바스타틴을 포함하는 조성불을 1차 분밀:공급기에 넣고, 암로디핀을 포함하는 조
성물을 2차 분말 공급기에 넣어 층¾의 흔입을 최소화할 수 있는 조건으로 분당 30 회전의 속도로, 경도 7 ~ 9 kp, 두께 6.0 mm, 지름 9,5議으로 타정하고 , 하이코 터로서 필름 코팅층을 형성하여 다층정을 제조하였다, 실시예 1-22 : 암로디핀 정제 -심바스타틴 정제를 함유한 ¾술제
1) 암로디핀 지연방출성 구획의 제조 (정제)
다음 표 1-4에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 ¾를로 오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유등층과립기 (GPCG 1: Giatt)에 투입하고, 따로 히드록시프로필메틸셀 ¾로오스를 물에 녹여 만든 결합액을 분무하 여 과립을 형성 , 건조하였다 . 다시 상기의 과립에 카보머 71G를 분말상태로 투입한 후, 여기에 스테아린산 마그네숨을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC-33 : 세종)를 사용하여 분당 30회전의 속도로, 경도 7 - 9 kp, 두께 3.0 mm, 지름 5.0 mm으로 타정하였다. 다시 상기의 정제에 에탄을과 염화메틸렌의 1:1흔액에 용해시킨 히드톡시프로필메틸셀를로오스프탈테이트 용액 (5 중량 ¾)과 아세틸레이티드모노글리세리드 용액을 하이코터를 사용하여 필름 코팅 을 형성하여 정제를 제조하였다.
2) 심바스타틴 선방출성 구획의 제조 (정제)
다음 표 1-4에 나타난 성분 및 함량과 같이 심바스타틴 , 더결정샐를로오스, 만니를 을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드록시프로필¾를로오스와 구연산을 물에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함깨 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제림하고 이를 은수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 부¾레이티드 히드록시아니솔, 전분 글리콘산 나트륨, 콜로이드성 이산화규소를 흔합하고, 스테 아린산 마그네슴을 넣어 더블콘믹서로 최종 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC-33: 세종)를 사용하여 분당 30회전의 속도로, 경도 7 ~ 9 kp, 두께 4.0 腿, 지름 5.0 賺의 정제를 타정하였다. 상기 정제는 히드록시프로필샐를로오 스, 플리에 ¾렌글리콜 6000, 산화티탄 및 탈크를 80% 에탄올에 용해 및 분산시켜 제조한 코팅액을 이용하여 하이코터로 코팅하였다.
3) ¾슬충전
공정 1)의 암로디핀 정제와 공정 2)의 심바스타틴 정제를 캡슐층전기를 이용하여 3 호의 경질 젤라틴 캡술에 층전하였다.
실시예 1-23 : 암로디핀 -심바스타틴 ¾술제
1) 암로디핀 지연방출성 구획의 제조 (정제)
다음 표 1-4에 나타난 성분 및 함량과 실시예 1-22의 1) 암로디핀 지연방출성 구획 의 제 방법에 따라 제조하였다.
2) 심바스타틴 선방출성 구획의 제조' (과립 )
다음 표 1-4에 나타난 성분 및 함량과 같이 심바스타¾ , 미결정셸롤로오스, 유당, 욱수수전분을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드록시프로필셀 롤로오스와 구연산을 물에 녹여 결합액을 제 a하고 이를 주성분 흔합물과 함께 고 속흔합기에 투입하고 연합한 다음 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 1C에서 건조한 후 다시 20호체로 정립하였다. 여기에 부¾레이티드히드록시아니솔, 전분 글리콘산 나트륨, 콜로이드성 이산화규소를 흔 합하고, 스테아린산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
3) 술층전
공정 1)의 암로디핀 정제와 공정 2)의 심바스타틴 과립을 ¾슐층전기를 이용하여 2 호의 히드록시프로필메틸셀를로오스 경질캡슐에 층전하였다. 실시예 1-24 : 암로디핀-심바스타 ¾ 코팅정
1) 암로디된 지연방출성 구획의 제조 (정제)
다음 표 1—4에 나타난 성분 및 함량과 실시예 1-20의 1) 암로디핀 지연방출성 구획 의 제조 방법에, 따라 제조하였다.
2) 심바스타틴 선방출 ^성 코팅액의 제초
다음 표 1-4에 나타난 성분 및 함량과 같이 심바스타틴, 부틸테이티드히드록시아니 솔, 히드특시프로필메틸샐를로오스, 콜토이드성이산화규소, 폴리에틸렌클리콜 6000, 산화티탄, 탈크를 에탄을과 염화메틸렌 흔액에 용해 및 분산시켜 선방출성 심바스타틴 코팅액을 제조하였다.
3) 1차 코팅
위에서 제조한 암로디핀정을 하이코터에 투여한 후 심바스타된 코팅액으로 1차 코 팅하였다.
4) 2차 코팅
표 1-4의 필름코팅층 조성불을 용매에 녹여 필름코팅액을 제조한 후 1차 코팅완료
된 정제에 2차 코팅을 하여 필름코팅정을 제조하였다. 실시 예 1-25 : 심바스타¾ 속방-암로디 ¾ 삼투성 유핵정의 제조
1) 암로디핀 지연방출성 구획의 제조 (정제 )
다음 표 1—4에 나타난 성분 및 함량과 ¾—이 암로디핀 베실레이트와 미결정 샐를로 오스 몇 염화나≤륨을 35호체로 사과하고 더블콘믹서로 흔합한 후 여기에 스테아린 산 마그네슘을 넣어 최종 더블콘믹서로 흔합하고 상기 최종 흔합물을 로타리 타정 기 (MRC-33 : 세종)를 사용하여 분당 30희 ¾의 속도로, 경도 7 ~ 9 kp, 두께 3.0 腿, 지름 5.5舰으로 타정하였다. 타정 후 삼투성 기제로서 콜리코드 Si? 30D와 트 리에틸시트레이트를 정제수에 분산시킨 후 하이코터를 이용하여 내핵에 코팅하여 삼투성 핵정을 제조하였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1—4에 나타난 성분 및 함량과 실시예 1-20의 2) 심바스타틴 선방출성 구획 의 제조 방법에 파라 제조하였다.
3) 타정 및 코팅
실시 예 1—20의 3) 타정 및 코팅 방법에 따라 조작하여 심바스타틴 속방-암로디핀 삼투성 유핵정제를 제조하였다. 실시예 I-26 : 암로디핀 -심바스돠틴 블리스터 포장 키트
다음 표 1—4에 나타난 성분 및 함량과 같이 제조하되, 실시예 Iᅳ 22의 암로디핀 정 과 심바스타틴 정제를 ¾슬에 동시 충전하는 것 대신 블리스터 포장용기에 동시복 용 가능하도록 포장하는 것을 제외하고는 실시예 1-22와 같이 제조하였다. 실시예 1—27 : 암로디핀-아토르바스타틴 유핵정
심바스타틴 대신에 아토르바스타틴을 사용한 것 이외에는 표 1-4의 성분과 함량에 의해 , 실시예 1-20의 제조방법에 따라 암로디핀-아토르바스타틴 유핵정을 제 하였 다. 실시예 1-28 : 암로디핀-아토르바스타틴 다층정
심바스타틴 대신에 아토르바스타된을 사용한 것 이의에는 표 1-4의 성분과 함량에 의해, 실시예 1-21의 제 S방법에 따라 암로디핀-아토르바스타틴 다층정을 제조하였
다. 실시예 1-29 : 암로디핀-아토르바스타틴 캡술제
심바스타틴 대샨에 아토르바스타틴을 사용한 것 이외에는 표 1-4의 성분과 함량에 의해 , 실시예 1-22의 제조방법에 따라 암로디핀-아토르바스타틴 캡술을 제 S하였 다. 실시예 1-30 : 암로디핀-아토르바스타 ¾ 캡슬제
1) 암로디핀 지연방출성 구획의 제조 (정제)
다음 표 1-4에 나타난 성분 및 함량과 실시예 1-22의 1) 암로디핀 지연방출성 구획 의 제조 방법에 따라 제조하였다.
2) 아토르바스파틴 선방출성 구획의 제조 (과립 )
다음 표 1—4에 나타난 성분 및 함량과 실시예 1-23의 2) 아토르바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
3) 캡슬층전
공정 1)의 암로디된 정제와 공정 2)의 아토르바스타틴 과립을 캡술층전기롤 이용하 여 히드특시프로필메 ¾¾를로오스 경질캡술에 층전하였다. 실시예 1-31 : 암로디된-아토르바스타된 코팅정제의 제조
선방출성 구획 제조시 심바스타틴 대신에 아토르바스타틴을 사용한 것과 콜로이드 성 이산화규소를 사용하지 않은 것 이외에는 표 1-5의 성분과 함량에 의해 , 실시예 1-24의 제조방법에 따라 암로디핀-아토르바스타된 코팅정제를 제조하였다. 실시예 1-32 : 아토르바스타틴-암토디핀 삼투성 유핵정의 제조
1) 암로디핀 지연방출성 구획의 제조 (내핵)
다훔 표 1-5에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 셀^로 오스 및 염화나트륨을 35호체로 사과하고 더블콘믹서로 흔합한 후 여기에 스 τ세아린 산 마그네슴을 넣어 최종 더블콘믹서로 흔합하고 상기 최종 혼합몰을 로타^ 파정 기 (MRC-33 : 세종)를 사용하여 분당 30회전외 속도로, 경도 7 ~ 9 kp, ^께 3.0 賺, 지름 5.5 麵으로 타정하였다. 타정 후 삼투성 기계로서 유드라짓 RS 30D와 트 리에틸시트레이트를 정제수에 분산시킨 후 하이코터를 이용하여 내 ^에 코팅하여
삼투성 핵정을 제조하였다.
2) 아토르바스타 ¾ 선방출성 구획의 제조
다음 표 1-5에 나타난 성분 및 함량과 실시예 1-25의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
유핵정 타정기 (RUD—l: KHian)를 사용하여 암로디된 삼투성 핵정을 내핵^로 하고 아토 5바스타틴을 포함하는 조성물을 의층으로 하여 분당 30회전의 속도로, 경도 7 ~ 9 kp, 두께 6,0賺, 지름 9.5腦으로 타정한 다음 하이코터로서 필름 코팅 층을 형성하여 유핵정을 제조하였다. 실시여 ΐ 1-33 : 암로디핀ᅳ아토르바스타 ¾ 블리스터 포장 키트
다음 표 1-5에 나타난 성분 및 함량과 같이 제조하되 , 실시예 1-29의 암로디핀 정 과 아토르바스타틴 정제를 캡슬에 등시 층전하는 것 대신 블리스터 포장용기에 동 시 복용 가능하도특 포장하는 것을 제외하고?는 실시예 1-29와 같이 제 S하였다. 실시예 1-34 : 니페디핀 -심바스타틴 유핵정
1) 니페디핀 지연방출성 구획의 제 s (내핵 )
다음 표 1—5에 나타난 성분 및 함량과 같이 니페디핀을 Ϊ리에 ¾렌글리콜 6000과 혼협1한 후 용융시켜 고체분산체를 제조하였다. 제조된 고체분산체를 35호체로 사과 하고, 35호체로 사과한 미결정셀를로오스 및 라우 ¾ 황산나트륨과 함께 더블콘믹서 로 흔합하였다 . 다시 상기의 과립에 카보머 71G를 분말상태로 투입한 후, 여기에 스테아린산 마그네슘을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 흔합물을 로 타리 타정기 (MRC— 33 : 세종)를 사용하여 분당 30희전의 속도로, 경도 7 ~ 9 kp, 두깨 3.0 mm, 지름 5.5 mm으로 타정하였다. 다시 상기의 정제에 에탄을과 염화메틸 렌의 1: 1혼액에 용해시킨 히드록시프로필메틸 ¾를로오스프탈레이트 용액과 아세틸 레이티드모노글리세리드 용액을 하이코터를 사용하여 필름 코팅 층을 형성하여 핵 정을 제조하였다.
2) 실바스타¾ 선방출성 구획의 제조
다음 표 1-5에 나타난 성분 및 함량과 실시예. 1-20의 2) "심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3) 타정 및 코¾
유핵정 타정기 (RUD-1: Ki l ian)를 사용하여 니페디핀 핵정을 내핵으로 하고 심바스 타틴을 포함하는 조성물을 외층으로 하여 분당 30회전의 속도로, 경도 7 — 9 3 , 두깨 6.0 画, 지룸 9.5 mm으토 타정한 다음 하이코터로서 필름 코팅 층을 형성하여 유핵정을 제조하였다.
"실시예 1-35 : 니페디핀-아토르바스타된 유핵정
심바스타틴 대신 아토르바스타틴을 사용한 것 이외에는 표 1-5의 성분과 함량에 의 해 , 실시예 1-34의 제조방법에 따라 니뙈디핀-아토르바스타틴 유핵정을 제조하였 다. 실시예 1-36 : ¾로디핀―아토르바스파된 2상 ¾슐제제
1) 펠로디핀 지연방출성 구획의 제조 (정제)
다음 표 1-6에 나타난 성분 및 함량과 같이 펠로디된과 미결정 셸롤로오스를 35호 체로 사과하고 더블콘믹서로 혼합한 후 유동층과립기 (GPCG. 1: Glatt)에 투입하고, 따로 히드록시프 S필메틸셀를로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과립에 에탄을과 염화메틸렌의 1:1흔액에 용해시킨 히드록시프로필메틸샐를로오스프탈레이트 용액을 분투하여 과립을 코팅하였다. 여 기에 과립에 카보머 71G롤 ^ ¾상꿰 ^^한 ? , 스 의^ 종 더블콘믹서로 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC-33 : 세종)롤 사용하여 분당 30회전의 속도로, 경도 7 ~ 9 kp, 두께 3.0 醒, 지름 5.0 mm의 정 제를 타정하였다.
2) 아토르바스타 ¾ 선방출성 구획의 제조 (정제)
다음 표 1-7에 나타난 성분 및 함량과 실시예 1-22의 2) 아토르바스타틴 선방출성 구획의 제 S 방법에 따라 제조하였다.
3) ¾슐층전
공정 1)의 ¾로디핀 정제와 공점 2)의 아토르바스타된 정제를 캡술층전기 1: 이용하 여 2호 ¾라틴 경질캡슐에 층전하였다. 실시예 1-37 : 바니디핀 - 로바스타틴 2상 캡술 제제
1) 바니디 ¾ 지연방출성 구획의 제조 (정제 )
다음 표 1-6에 나타난 함량과 같이 염산바니디핀과 미결정셀를로오스를 35호체로
사과하고 더블콘믹서로 흔합하고 , 고속흔합기에 투입한 후 콜리코트 SR30D 를 가하 여 연합한 다음 20호체로 오실레이터를 이용하여 제립하고, 이를 은수 건조기를 이 용하여 60 X 에서 건조한 다음 다시 20호체로 정립하였다. 여기에 스테아린산 마그 네슴을 넣어 더블콘믹서로 최종 흔합하고, 상기 최종 흔합물을 로타리 파정기 (MRCᅳ 33 : 세종)를 사용하여 분당 30회전의 속도로, 경도 7~9kp, 두께 3.0mm, 지름 5.5 mm의 정제를 타정하였다.
2) 로바스타틴 선방출성 구획의 제조 (정제)
다음 표 1-7에 나타난 성분 및 함량과 같이 로바스타틴 , 미결정쨀를로오스, 만니를 을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드록시프로필샐를로오스와 구연산을 물에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 'C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 부틸레이티르 히드록시아니솔, 전분 글리콘산 나트튬, 콜로이드성 이산화규소를 흔합하고, 스테 아린산 마그네습을 넣어 더블콘먹서로 최종 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC-33 : 세종)를 사용하여 분당 30회전의 속도로, 경도 7 ~ 9 kp, 두께 5.0瞧, 지름 5.5匪의 정제를 타점하였다.
3) 캡술층전
공정 1)의 바니디핀 정제와 공정 2)의 로바스타틴 정제를 ¾술층전기를 이용하여 2 호의 히드톡시프로필메틸셀를로오스 경질캡술에 층전하였다. 실시예 1-38 : 베니디 ¾ - 피타바스타틴 2상 ¾슬제제
1) 떼니디핀 지연방출성 구획의 제조 (정제)
로디핀 대신에 염산패니디핀을 사용한 것 이의에, 다음 표 1ᅳ6에 나파난 성분 및 함량과 실시예 1-36의 1) 로디판 지연방출성 구획의 제조 방법에 따라 제 S하였 다.
2) 피타바스타틴 선방출성 구획의 제조 (과립 )
심바스타틴 대신에 피타바스타틴 칼슴을 사용한 것 이외에 , 다음 표 1—7에 나타난 성분 및 함량과 같이 실시예 1-1의 2) 심바스타틴 선방출성 구획의 제조방법에 따 라 제조하였다.
3) 캡슬층전
공정 1)의 베니디핀 정제와 2)의 최종 조성물을 캡술층전기를 이융하여 1호 젤라틴
경질캡술에 층전하였다. 실시예 1-39 : 실니디핀 - 프라바스타 ¾ 2상 캡술제제
1) 실니디핀 지연방출성 구획의 제조 (점제)
바니디된 대신에 실니디핀을 사용한 것 이의에 , 다음 표 1-6에 나타난 성분 함량과 같이 실시예 1-37의 1) 바니디핀의 지연방출성층의 제조방법에 따라 제조하였다.
2) 프라바스타틴 선방출성 구획의 제조 (과립)
심바스타틴 대신에 프라바스타틴 나트륨을 사용한 것을 이의에, 다음 표 1-7에 나 타난 성분 및 함량과 같이 실시예 1-11의 2) 심바스타틴 선방출성 구획의 제조 방 법에 따라 제 :하였다.
3) 캡술층전
공정 1)의 실니디핀 정제와 2)의 최종 S성물 ¾술층전기를 이용하여 1호의 히드록 시프로필메틸셀를로오스 경질캡슬에 층전하였다. 실시예 1-40 : 이스라디핀 ᅳ플루바스타틴 2상 ¾술제제
1) 이스라디¾ 지연방출형 펠햇의 제조
표 1-6에 나타난 성분 및 함량과 같이 이스라디핀 및 미결정셀를로오스를 35호체로 사과하고, 더블콘믹서에서 흔합하여 흔합분을 제조하였다. 따로 , 히드록시프로필 셀롤로오즈를 물에 용해시켜 결합제용액을 제조하였다. CF과립기 내에서의 결정성 슈크로오즈 시드에 결함제 용액을 분무하면서 , 흔합분말을 결정성 슈크로오즈 시드 에 산포하여 펠렛을 제조하였다. 얻어진 ¾을 수분함량이 2%이하로 될 때까지 50 C에서 건조하여 이스라디핀 함유 코어 ¾렛올 제조하였다. 다시 , 상기의 ¾¾에 에 탄을과 아세톤의 1: 1흔액에 용해시킨 히드록시프로필메틸 ¾를로오스 프탈레이트용 액을 분무하여 펠햇을 코평하였다
2) 플루바스타틴 선방출성 구획의 제조 (과림 )
심바스타틴 대신에 플루바스타틴 나트 "을 사용한 것을 이외에, 다음 표 1-7에 나 타난 성분 및 함량과 같이 실시예 1-1의 2) 심바스타틴 선방출성 구획의 제조 방법 에 따라 제조하였다.
3) 캡슬층전
공정 1)과 2)의 최종 조성물을 캡술층전기를 이용하여 1호 ¾라¾ 경질캡술에 층전 하였다.
실시예 1-41 : 마니디핀 - 로바스타틴 2상 ¾슐제제
1) 마니디핀 지연방출성 구획의 제조 (과립 )
다음 표 1-6에 나타난 성분 및 함량과 같이 염산마니디 ¾과 미결정셀를로오스를 35 호체로 사과하고 더블콘믹서로 흔합한 후 유동층 과립기 (GPCG 1 : Glatt)에 투입하 고, 따로 히드록시프로필메틸셀를로오스롤 물에 녹여 만든 결합액을 분무하여 과립 을 형성 , 건조하였다. 다시 상기의 과림에 에탄을과 염화메틸렌의 1: 1흔액에 용해 시킨 히드록시프로필메필샐를로오스프탈레이트 용액을 분무하여 과립을 코팅하였 다. 여기에 스쩨아린산 마그네슘을 넣고 더블콘믹서를 이용하여 최종 흔합을 하였 다 -
2) 로바스타틴 선방출성 구획의 제 (정제)
다음 표 1—7에 나타난 성분 및 함량과 같이 로바스타틴, 미결정쎌를로오스, 만니를 을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드록시프로필샐를로오스와 구연산을 물에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 부틸레이티드 히드특시아니솔, 전분글리콘산 나트륨, 콜로이드성 이산화규소 및 스테아린산 마그 네슴을 넣고 더블콘믹서로 최종 흔합하였다.
상기 최 흔합물올 로타리 타점기 (MRC— 33; 세종)를 이용하여 분당 30회전의 속도 로, 경고 7~9kp, 두께 3.0顧, 지름 5.5蘭의 정제를 타정하였다.
3) 흔합 및 ¾술층전
공정 1)과 2)의 최종 조성물을 캡술층전기롤 이용하여 2호 젤라틴 경질캡슬에 층전 하였다. 실시예 1-42 : 니카르디핀 - 로수바스타된 유핵정의 제조
1) 니카르디핀 지연방출성 구획의 제조 (내핵)
펠로디편 대신에 염산니카르디핀을 사용한 것 이의에 , 다음 표 1-6에 나타난 성분 및 함량과 실시예 Iᅳ 36의 1) ¾로디핀 지연방출성 구획의 제 S 방법에 따라 제조하 였다.
2) 로수바스타틴 선방출성 구획의 제조
심바스타틴 대신에 로수바스타틴 칼슴을 사용한 것을 이의에 , 다음 표 1-7에 나타
난 성분 및 함량과 같이 실시예 1-1의 2) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
유핵정 타정기 (RUD-1: Ki l iaii)를 사용하여 니카르디핀 핵정을 내핵으로 하고 로수 바스타틴을 포함하는 조성물을 외층으로 하여 분당 30회전의 속도로, 경도 7 ~ 9 kp, 두께 6.0 麵, 지름 9.5 ram으로 타정한 다음 하이코터로서 필름 코팅 층을 형성 하여 유핵정을 제조하였다. 실시예 1-43 : 니페디핀 - 플루바스타된 유핵정의 제조
1) 니페디핀 지연방출성 구획의 제조 (내핵 )
펠로디핀 대신에 니페디핀을 사용한 것 이외에, 다음 표 1-8에 나타난 성분 및 함 량과 실시 예 1-36의 1) ¾로디핀 지연방출성 구획의 제조 방법에 따라 제조하였다.
2) 플루바스타틴 선방출성 구획의 제 S
심바스타틴 대신에 플루바스타틴 나트륨을 사용한 것을 이외에 , 다음 표 1-9에 나 타난 성분 및 함량과 같이 실시예 1-1의 2) 심바스타틴 선방출성 구획의 제조 방법 에 따라 제조하였다.
3) 타정 및 코팅
유핵정 타정기 (RUD-1: KHian)를 사응하여 니페디핀 핵정을 내핵으로 하고 심바스 타틴을 포함하는 조성물을 의층으로 하여 분당 30회전의 속도로, 경도 7 ~ 9 kp, 두께 6.0 瞧, 지름 9.5 mm으로 타정한 다음 하이코터로서 필름 코팅 층을 형 여 유핵정을 제조하였다. 질시예 1-44 : 암로디핀-피타바스타틴 유핵정의 제조
심바스타틴 대신에 피타바스타 ¾ 칼슴을 사용한 것 이의에는 표 1-8 및 9의 성분과 함량에 의해 , 실시예 1-20의 제 S방법에 따라 암로디된-피타바스타틴 유핵정을 제 조하였다. 실시예 1-45 : 암로디핀-로수바스타틴 유핵정의 제조
심바스타틴 대신에 로수바스타틴 칼숨을 사용한 것 이외에는 표 1—8 및 9의 성분과 함량에 의해 , 실시 예 1-20의 제조방법에 따라 암로디핀-로수바스타틴 유핵정을 제 조하였다.
시예 1-46: 니모디핀 - 프라바스타틴 코팅정의 제조
1) 니모디핀 지연방출성 구획의 제조 (정제)
다음 표 1-8에 나타난 성분 및 함량과 같이 니모디휜과 미결정 셀를로오스를 35호 체로 사과하고 더블콘믹서로 흔함한 후 유동층과립기 (GPCG 1: Glatt)에 투¾하고, 따로 히드록시프로필메틸셀를로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄을과 염화메 ¾렌의 1: 1흔액에 용해시킨 히드록시프로필메틸젤를로오스프탈레이트 용액을 분무하여 과립을 코팅하였다. 여 기에 과립에 카보머 71G를 분말상태로 투입한 후, 스테아 ¾산 마그네슴을 넣어 최 종 더블콘믹서로 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC 33 : 세종) ¾ 사용하여 분당 30희전의 속도로 , 경도 7 ~ 9 kp, 두께 3.0醒, 지름 7.0腿의 정 제를 타정하였다.
2) 프라바스타틴 선방출성부의 코팅
하이코터를 사용하여 니모디핀 정제롤 다음 표 1—9에 나타난 성분 및 함량과 같이 에탄올 및 정계수에 히드특시프로필셀를로오스 τ 산화티탄 및 탈크를 용해 , 분산시 키고 프라바스타틴 나트륨염을 용해시켜 풀루바스타틴 나트晉염을 포함한 선방출성 코팅용액을 제조하고, 이 코팅 액으로 니모디핀 지연방출성 구획의 외층에 코팅층을 형성하여 코팅정을 겨ᅵ조하였다.
3) 코팅
상기 2) 프라바스타틴 선방출성부의 코팅부 위에 하이코터 (SFC-30N)를 사용하여 표 1-7에 나타난 성분 및 함량과 같이 코팅층을 형성하여 필름코팅정제를 완성하였다. 실시예 1-47 : 니발디핀 - 피타바스타틴 다층정의 제조
1) 니발디핀 지연방출성 구획 과¾의 제조
다음 표 1-8에 나타난 성분 및 함량과 같이 니발디뮌과 미결정 셀를로오스를 35호 체로 사과하고 더블콘믹서로 흔합하고, 따로 히드록시프로필메틸셀를로오스를 물에 녹여 만든 결합액을 분무하여 과림을 형성 , 건조하였다. 다시 상기의 과립에 에된: 올과 염화메틸렌의 1: 1흔액에 용해시킨 히드특시프로필메틸셀를로오스프탈레이트 용액을 분무하여 과립을 코팅하였다. 여기에 과립에 카보머 71G롤 분말상태로 루입 한 후, 스테아린산 마그네슴을 넣어 최종 더블 콘믹서로 흔함하였다.
2) 피타바스타틴 선방출성 구획의 제조
심바스타틴 대신에 피타바스타틴 칼슴을 사용한 것을 이외에 , 다음 표 1-9에 나타 난 성분 및 함량과 같이 실시예 1-1의 2) 심바스타틴 선방출성 구획의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
본 공정에서는 다층정 타정기 (ffiC-37T : 세종)를 사용하여 타정하였다. 즉, 상기 피타바스타 ¾을 포함하는 조성물을 1차 분말공급기에 넣고 , 니발디핀을 포함하는 조성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 분당 30회전의 속도로 , 경도 7 ~ 9 kp, 두께 6.0 ram, 지름 9,5麵으로 타정하고, 하이 코터로서 필름 코팅층을 형성하여 다층정 형 태의 정제를 제조하였다. 실시예 1-48 : 니솔디핀 - 로바스타틴 다층정의 제 S
1) 니솔디핀 지연방출성 구획 과립의 제조
다음 표 1—8에 나타난 성분 및 함량과 같이 니솔디핀과 미결정 셀를로오스를 35호 체로 사과하고 더블콘믹서로 흔합하고, 따로 히드특시프로필메틸셀를로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄 을과 염화메 ¾렌의 1:1흔액에 용해시킨 유드라짓 RS P0 용액을 분무하여 과립을 코 팅하였다. 여기에 스테아린산 마그네슴을 넣어 최종 더블 콘믹서로 흔합하였다.
2) 로바스타틴 선방출성 구획의 제조
심바스타틴 대신에 로바스타틴을 사용한 것을 이의에 , 다음 표 1-9에 나타난 성분 및 함량과 같이 실시예 1-1의 2) 심바스타틴 선방출성 구획의 제조 방법에 따라 제 조하였다.
3) 타정 및 코팅
본 공정에서는 다층정 타정기 (MRC-37T : 세종)를 사용하여 타정하였다. 즉 상기 로바스타틴을 포함하는 S성물을 1차 분말공급기에 넣고, 니솔디핀을 포함하는 조 성불을 2차 분말 공급기에 넣어 간의 혼입을 최소화할 수 있는 조건으로 분당 30 회전의 속도로, 경도 7 ~ 9 kp, 두께 6.0 mm, 지름 9.5瞧으로 타정하고 , 하이코 터로서 필름 코팅층을 형성하여 다층정 형태의 정제를 제조하였다. 실시예 Iᅳ 49 : 니트렌디핀 - 프라바스타틴 블리스터 포장 키트
1) 니트렌디핀 지연방출성 구획의 제조
펠로디핀 대신에 니트렌디핀을 사용한 것 이외에 , 다음 표 1—8에 나타난 성분 및
함량과 실시예 I— 36의 1) 펠로디핀 지연방출성 구획의 제조 방법에 따라 제조하였 다.
2) 프라바스타틴 선방출성 구획의 제조
다음 표 1-9에 나타난 성분 및 함량과 같이 프라바스타된 나트 "염, 미결정셸를로 오스, 만니를올 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드록시프로필 셀를로오스와 구연산을 물에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 부틸레이티드히드록시아니솔, 전분 글리콘산 나트륨, 콜로이드성 이산화규소를 흔 합하고, 스테아린산 마그네슴을 넣어 더블콘먹서로 최종 흔합한 후 상기 최종 흔합 물을 로타리 타정기 (MRC-33 : 세종)를 사용하여 분당 30희전의 속도로, 경도 7 ~ 9 kp, 두깨 4.0 纖, 지름 8.5 mm의 점제를 타정하였다.
3) 포장
공정 1)의 니트렌디핀 정제와 공정 2)의 프라바스타틴 정제를 하나의 블리스터에 포장하며 동시 복용하도록 명시하였다. 실시예 1-50: (S)-암로디핀 - 심바스타틴 유핵정의 제조
1) (S 암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 1-10에 나타난 성분 및 함량과 같이 (s)-암로디핀 베실레이트와 미결정 셀 를로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유등층과립기 (GPCG 1: Glatt)에 투입하고, 따로 히드록시프로필셀를로오스를 정제수에 녹여 만든 결합액 을 분무하여 과립을 형성시킨 후, 건조하였다. 상기의 생성된 과립에 카보머 71G를 분말상태로 투입하여 더블콘믹서에서 10분간 혼합한 후, 35호체로 체과한 스테아린 산 마그네슴을 넣고 4분간 최종 흔합하였다. 상기의 최종 흔합물을 로타리 타정기 (MRC-33 : 세증)를 사용하여 분당 30희전의 속도로, 경도 6 ~ 10 kp, 두께 3.0 腿, 지름 5.5 讓로 타정하여 (S)-암로디핀 핵정을 제조하였다.
2) 심바스타틴 선방출성 구획의 제조
다음 표 1-10에 나타난 성분 및 함량과 같이 심바스타틴, 미결정샐롤로오스 , 만니 를을 20호체로 체과한 후, 고속흔합기에서 10분간 흔합하였다, 따로 히드특시프로 필셀롤로오스와 구연산을 정제수에 녹여 결합액을 제조하였다. 위의 흔합몰에 결합 액을 가하면서, 연합을 완료한 후 제립 건 , 정립하여 과립제조를 완료하였다.
여기에 미리 35호 체로 체과한 부틸레이티드히드록시아니솔, 전분글리콘산나트튬, 콜로이드성 이산화규소를 위의 과립과 함께 더블콘믹서에 투여한 후, 약 10분간 흔합하고 , 스테아린산 마그네슴을 35호 체로 체과하여 더블콘믹서에 투여한 후 약 4분간 최종 흔합하여 심바스타¾층 과립 제조를 완료하였다,
3) 타정 및 코팅
유핵정 타정기 (RUD-1: Ki lian)를 사용하여 암로디 ¾ 핵정을 내핵으로 하고 심바스 타틴을 포함하는 조성물을 외층으로 하여 분당 30회전의 속도로, 경도 7 - 13 kp, 두께 6.0 薩, 지름 9.5 腿으로 타정하여 내핵을 제조하였다. 따로 표 1—8의 필름코 팅층 조성물을 용매에 녹여 필름코팅 액을 제조하였다. 위에서 제조한 내핵을 하이 코터에 투여한 후 필름코팅 액으로 코팅하여 유핵정 제조를 완료하였다. 실시예 1-51 : (S)-암로디핀 - 심바스타틴 유핵정의 제조
다음 표 1—10에 나타난 성분 및 함량과 같이 제조하되 , 실시예 1-50의 핵정 제조 후 플리 (메타크릴레이트, 메틸메타크릴레이트)공증합체로 장용 코팅한 것을 제외하 고는 실시예 1-50과 동일하게 제조하였다. 실시예 1-52 : (S)-암로디핀―심바스타틴 캡술제의 제조
1) (s)-암로디관 지연방출성 구획의 제조 (정제)
다음 표 1-10에 나타난 성분 및 함량과 같이 (S)-암로디핀 베실테이트와 미결정 셀 를로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과립기 (GPCG 1: Glati)에 투입하고, 파로 히드특시프로필셀를로오스를 정제수에 녹여 만든 결합액 을 분무하여 과립을 형성시킨 후, 건조하였다, 상기의 생성된 과립에 카보머 71G를 분말상태로 투입하여 더블콘믹서에서 10분간 흔합한 후, 35호체로 체과한 스테아린 산 마그네슴을 넣고 4분간 휙종 흔합하였다. 상기의 최종 혼함물을 로타리 타정기 (M C-33 : 세종)를 사용하여 분당 30회전의 속도로 , 경도 6 ᅳ 10 kp, 두께 3.0 誦, 지름 5.5 画으로 타정하여 제조한 (S)-암로디핀 정제를 폴리 (메타크릴레이트 , 메틸메타크릴레이트)공중합체로 코팅하여 (S)-암로디핀 정제 제조를 완료하였다.
2) 심바스타틴 선방출성 구획의 제조 (정제)
다음 표 1-10에 나타난 성분 및 함량과 같이 심바스타된 , 미결정 ¾를로오스, 만니 를을 20호체로 체과한 후, 고속흔합기에서 10분간 흔합하였다. 따로 히드록시프로 필샐를로오스와 구연산을 정제수에 녹여 결합액을 제 하였다. 위의 혼합물에 결합
액을 가하면서, 연합을 완료한 후 제립 , 건조, 정립하여 과립제조롤 완료하였다. 여기에 미리 35호 체로 체과한 부틸레이티드히드록시아니솔, 전분글리콘산나트튬, 콜로이드성 이산화규소를 위의 과립과 함께 더블콘믹서에 투여한 후, 약 10분간 흔합하고, 스테아린산 마그네슘을 35호 체로 체과하여 더블콘믹서에 투여한 후 약 4분간 최종 흔합하여 심바스타된층 과립 제조를 완료하였다. 상기의 과립을 로타리 타정.기 (MRC-33 : 세종)를 사용하여 분당 30회전의 속도로, 경도 6 〜 : 10 kp, 두께 3.0 醒, 지름 5.5 醒으로 타정하여 제조한 심바스타틴 정제를 표 1—8의 필름층 성 분으로 코팅하여 심바스타틴 정제 제조를 완료하였다.
3) 캡슬층전
캡술층전기를 사용하여 위의 (S)ᅳ암로디괸 정제와 심바스타틴 정제를 3호의 라틴 경질캡술에 등시 층전하여 ¾술제 제조를 완료하였다. 실시예 1-53 : (S)-암로디핀 - 심바스타틴 ¾술제의 제조
1) (S)-암로디핀 지연방출성 구획의 제≤ (정제 )
다음 표 1-10에 나타난 성분 및 함량과 같이 (S)—암로디핀 베실레이트와 미결정 셸 롤로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과림기 (GPCG 1: Glatt)에 투입하고, 따로 히드록시프로필셀를로오스를 정제수에 녹여 만든 결합액 을 분무하여 과립을 형성시 ¾ 후, 건조하였다. 상기의 생성된 과립에 카보머 71G를 분말상태로 투입하여 더블콘믹서에서 10분? } 흔합한 후, 35호체로 체과한 스테아린 산 마그네슘을 넣고 4분간 최종 흔합하였다. 상기의 최종 흔합물을 로타리 타정기 (M C-33 : 세종)를 사용하여 분당 30희전의 속도로, 경도 6 ~ 10 kp, 두께 3.0 腿, 지름 5.5 으로 타정하여 제조한 (S) 암로디핀 정제를 ¾리 (메타크릴레이트, 메¾메타크 ¾테이트)공중합체로 코텅하여 (S)-암로디 ¾ 정제 제조를 완료하였다.
2) 심바스타된층 선방출성 구획의 제 s (과립)
다음 표 1-10에 나타난 성분 및 함량과 같이 심바스타틴, 미결정샐를로오스, 만니 를을 20호체로 체과한 후, 고속흔합기에서 10분간 혼합하였다. 따로 히드특시프로 필셀롤로오스와 구연산을 정제수에 녹여 결합액을 제조하였다. 위의 혼합물에 결합 액을 가하면서 , 연합을 완료한 후 제립, 건조, 정립하여 과립제조률 완료하였다. 여기에 미리 35호 체로 체과한 부틸레이티드히드특시아니솔, 전분글리콘산나트튬, 콜로이드성 이산화규소를 위의 과립과 함께 더볼콘믹서에 투여한 후, 약 10분간 흔 합하고, 스테아린산 마그네슴을 35호 체로 체과하여 더블론먹서에 투여한 후 약 4
분간 최종 흔합하여 심바스타틴층 과립 제조를 완료하였다.
3) 캡술층전
캡슬층전기를 사용하여 위의 (S)-암로디핀 정제와 심바스타틴 과립을 1호의 히드특 시프로필메틸 ¾롤로오스 경질캡술에 동시 층전하여 캡술제 제조를 완료하였다. 실시예 1-54 : (S)—암로디핀정 + 심바스타¾ 블리스터 포장 키트
다음 표 1-10에 나타난 성분 및 함량과 같이 제조하되 , 실시예 1-50의 (S)-암로디핀 정과 심바스타틴 정제를 캡술에 등시 층전하는 것 대신 PTP 포장용기에 등시복용 가능하도록 포장하는 것을 제외하고는 실시예 1-52와 같이 제조하였다. 실시예 1-55 : (S)-암로디된 - 심바스타틴 코팅정제의 제조
1) (S)-암로디핀 지연방출성 구획의 제조 (정제)
다음 표 1-10에 나타난 성분 및 함량과 같이 , (S)-암로디핀과 미결정 셀를로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과림기 (GPCG 1: Glatt)에 투입하 고, 따로 히드록시프로필셸를로오스를 정제수에 녹여 만든 결합액을 분무하여 과립 을 형성시 ¾ 후, 건조하였다. 상기의 ' 생성된 과립에 카보머 71G롤 분말상태로 투입 하여 더블콘믹서에서 10분간 흔합한 후, 35호체로 체과한 스테아린산 마그네슴을 넣고 4분간 최종 흔합하였다. 상기의 최종 흔합물을 로타리 타정기 (MRC-33 : 세종) 를 사용하여 분당 30회전의 속도로, 경도 6 ᅳ 10 kp, 두께 4.0 ma, 지름 8.0廳으 로 타정하여 제조한 (S)-암로디핀 정제를 아래 표 1-10외 풀리 (메타크릴레이트, 메 틸메타크릴레이트)공중합체로 코팅하여 (S)-암로디핀 정제 제조를 완료하였다.
2) 심바스타틴코팅액의 제
다음 표 1-10에 나타난 성분 및 함량과 같이 심바스타틴, 부틸레이티드히드록시아 니솔, 히드록시프로필데틸샐를로오스, 콜로이드성이산화규소를 에탄을 , 염화데틸렌 흔액에 녹여 심바스타틴 코팅액을 제조한다.
3) 1차 코팅
위에서 제 2:한 (S)—암로디핀정을 하이코터에 투여한 후 심바스타틴 코팅액으로 1차 코뜀하였다.
4) 2차 코팅
표 1—10의 필름코팅층 조성물을 용매에 녹여 필름코팅액을 제조한 후 1차 코팅완료 된 정제에 2차 코팅을 하여 필름코팅정을 제조하였다.
실시 예 1-56 : (S)-암로디핀 - 아토르바스타틴 캡술제의 제:
1) (S)-암로디핀 지연방출성 구획의 제조 (정제)
다음 표 1-11에 나타난 성분 및 함량과 같이 실시예 1-53의 1) (S)-암로디핀 지연 방출성 구획의 제조방법에 따라 제조하였다.
2) 아토르바스타틴층 선방출성 구획의 제 2: (펠헷)
표 1-11에 나타난 성분 및 함량과 같이 아토르바스타틴 칼슴, 부틸레이티드히드록 시 아니솔 및 미결정샐를로오스를 35호체로 사과하고, 더블콘믹서에서 흔합하여 흔 합분을 제조하였다 . 따로 히드록시프로필 ¾를로오스를 물에 용해시켜 결합제 용 액을 제조하였다. CF과립기 내에서의 결정성 슈크로오스 시드에 결합제 용액을 분 무하면서 , 흔합분말을 결정성 슈크로오즈 시드에 산포하여 펠렛을 제조하였다. 얻 어진 펠¾을 수분함량이 2¾>이하로 될 때까지 50 °C에서 건조하여 아토르바스타틴 칼슴 함유 ¾¾을 제조하였다.
3) 캡슬 층전
캡슐층전기를 사용하여 위의 (S)-암로디핀 정제와 아토 5바스타틴 칼슴 펠렛을 2호 의 히드록시프로필메틸셀를로오스 경질캡술에 등시 층전하여 캡술제 제조를 완료하 였다. 실시예 1-57 : (S)-암로디핀 - 아토르바스타틴 캡슐제의 제조
1) (S)-암로디핀 지연방출성 구획의 제조 (펠?
표 1-11에 나타난 성분 및 함량과 같이 (S)-암로디 ¾ 베실레이트 및 미결정셀를로 오스를 35호체로 사과하고, 더블콘믹서에서 흔합하여 흔합분을 제조하였다. 따로 , 히드특시프로필 셀를로오즈를 물에 용해시켜 결합제 용액을 제조하였다. CF과립기' 내에서의 결정성 슈크로오즈 시드에 결합제 용액을 분무하면서, 혼합분말을 결정성 슈크로오 : 시드에 산포하여 펠¾을 제조하였다. 얻어진 펠렛을 수분함량이 2%이하 로 될 때까지 arc에서 건조하여 (s)-암로디핀 베실레이트 함유 코어 ¾을 제 하였다.
2) 아토르바스타틴 선방출성 구획의 제조 (정제)
다음 표 1-11에 나타난 성분 및 함량과 같이 아토르바스타틴 칼슴, 미결정셀를로오 스, 만니를을 20호체로 체과한 후, 고속혼합기에서 10분간 흔합하였다. 따로 히드 특시프로필쎌를로오스와 구연산을 정제수에 녹여 결합액을 제조하였다. 위의 흔합
물에 결합액을 가하면서, 연합을 완료한 후 제립 , 건조, 정립하여 과립제조롤 완료 하였다. 여기에 미리 35호 체로 체과한 부틸레이티드히드록시아니솔, 전분굴리콘산 나트륨, 콜로이드성 이산화규소를 위의 과림과 함깨 더블콘믹서에 투여한 후, 약 10분간 흔합하고, 스테아린산 마그네슘을 35호 체로 체과하여 더블콘믹서에 투여한 후 약 4분간 최종 혼합하여 아토르파스타틴 과립 제조를 완료하였다. 상기의 과립 을 로타리 타정기 (MRC— 33 : 세종)를 사용하여 분당 30희전의 속도로, 경도 6 ~ 10 kp, 두께 3.0 ram, 지름 5.5 !舰으로 타정하여 제 S한 아토르바스타련 칼슘 정제를 표 1-11의 코팅층 성분으로 코팅하여 아토르바스타된 칼슴 정제 제조를 완료하였 다.
3) 캡술층전
캡술층전기를 사용하여 위의 (S)-암로디핀 베실레이트 펠헷과 아토르바스타틴 칼슴 정제를 1호의 히드록시프로필메 ¾셀를로오스 경질캡술에 동시 층전하여 캡술제 제 조를 완료하였다. 실시예 1-58 : (S)-암로디핀 - 아토르바스타틴 캡슬제의 제조
1) (S)-암로디핀 지연방출성 구획의 제조 (팰 ¾)
표 1—11에 나타난 성분 및 함량과 같이 실시예 1-57의 1) (S)ᅳ암로디핀 지연방출성 구획의 제조방법에 따라 제조하였다.
2) 아토르바스타틴 선방출성 구획의 제조 (펠 ¾)
다음 표 1-11에 나타난 성분 및 함량과 같이 실시예 1-56의 2) 아토르바스타틴 칼 슘 선방출성 구획의 제조방법에 따라 제조하였다.
3) 캡술층전
캡슬층전기를 사용하여 위의 (S)-암로디편 ¾¾와 아토르바스타린 펠렛을 2호의 히 드록시프로필메틸셀를로오스 경질캡슐에 등시 층전하여 ¾슬제 제조를 완료하였다. 실시예 1-59 : (S)-암로디핀 아토르바스타련 캡술제의 제조
1) (S)—암로디핀 지연방출성 구획의 제 (과립 )
표 i-ii에 나타난 성분 및 함량과 같이 (s)-암로디핀 떼실레이트와 미결정 ¾를로오 스를 35호체로 사과하고 더블콘믹서로 흔합한 다음 이를 유동층과림기 (GPCG 1 : Glatt)에 투입하고 콜리코트 SR30D 를 분무하여 과립을 형성, 건조시켰다.
2) 아토르바스타 ¾ 선방출성 구획의 제조 (펠벳)
다음 표 1-11에 나타난 성분 및 함량과 같이 실시예 1-56의 2) 아토르바스타틴 선 방출성 구획의 제조방법에 따라 제 하였다.
3) 캡술층전
캡슬층전기를 사용하여 위의 (S)-암로디핀 과립과 아토르바스타틴 ¾을 1호의 히 드특시프로필메틸셀를로오스 경질캡술에 동시 층전하여 캡슬제 제조를 완료하였다. 실시예 1—60 : (S) 암로디핀 - 아토르바스타틴 ¾슬제의 제조
1) (S)-암로디 ¾ 지연방출성 구획의 제조 (¾¾)
표 1 11에 나타난 성분 및 함량과 같이 실시예 1—57의 1) (S)-암로디된 지연방출성 구획의 제조방법에 따라 제조하였다.
2) 아토르바스타틴 선방출성 구획의 제 (과립 )
다음 표 1-11에 나타난 성분 및 함량과 같이 아토르바스타틴 칼슘, 미결정셀를로오 스 , 만니롤을 20호체로 체과한 후, 고속혼합기에서 10분간 흔합하였다. 따로 히드 톡시프로필셀를로오스와 구연산을 정제수에 녹여 결합액을 제조하였다. 위의 혼합 물에 결합액을 가하면서 , 연합을 완료한 후 제립 , 건조 , 정립하여 과립계조를 완료 하였다. 여기에 미리 35호 체로 체과한 부¾레이티드히드록시아니솔, 전분글리콘산 나트륨, 를로이드성 이산화규소를 위의 과립과 함께 더블콘믹서에 투여한 후, 약 10분간 흔합하고, 스테아린산 파그네슴을 35호 체로 체과하여 더블콘믹서에 투여한 후 약 4분간 최종 흔합하여 아토르바스타된층 과립 제조롤 완료하였다.
3) ¾슬층전
캡술층전기를 사용하여 위의 (S)-암로디핀 ¾¾과 아토르바스타틴 과립을 1호의 히 드록시프로필메틸셀를로오스 경질캡슬에 동시 충전하여 캡술제 제조를 완료하였다.
[표; [-2]

-3]

/:/ΗΙ><ί ϊεεοοο600Ζ

[표 1-5]

[표 1-6]

IB2009/000331
75
[표 1—7]



[6-IS]
LL
iccooo/60ozai/i3d zz6Li mz OAV
[표 1-10]

-11]

1)암로디핀 지연방출성 구획의 제조
다음 표 Π-2에 나타난 성분 및 함량으로 암로디핀 말레이트와 미결정 셀를로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과립기 (GPCG 1 Glatt)에 투입하 따로 히드록시프로필메틸 ·€를로오스롤 정제수에 녹여 만든 결합액 (10¾ w/w)을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과립에 카보머 71G를 분말상태로 투입한 후, 스테아르산 마그네슘을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 혼함물을 로타리 타정기 (MRC-30: 세종)를 사웅하여 타정하고 이를 내핵으로 하였
다.
2)심바스타틴 선방출성 구획의 제조
다음 표 Π-2에 나타난 성분 및 함량으로 심바스타틴, 미결정셀를로오스 , 만니를을 35호체로 사과하고 고속혼합기로 흔합하였다. 따로 히드록시프로필셀를로오스와 구 연산을 정제수에 녹여 결합액 (1W w/w)을 제조하고 이를 주성분 흔합물과 함께 고 속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은 수 건조기를 이용하여 60 °C에서 건 2:한 다음 다시 20호체로 정립하였다. 여기에 부틸레이티드히드특시아니솔, ¾분 글리콘산 나트륨, 콜로이드성 이산화규소를 흔 합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 혼합하였다.
3)타정 및 코팅
유핵정 타정기 (RUD-1: Kilian)를 사용하여 암로디핀 내핵을 심바스타틴을 포함하는 조성물을 의층으로 하여 타정한 다음 하이코터 (SFC-30N, 세종 기계 , 한국)를 사용 하여 표 II-2 기재 성분 및 함량으로 필름 코팅 층을 형성하여 유핵정을 제조하였 다. . 실시예 Π-2 : 암로디핀 -심바스타틴 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (내핵 )
다음 표 Π— 2에 나타난 성분 및 함량으로 암로디핀 말테이트와 미결정 셀를로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유등출과립기 (GPCG 1: Glatt)에 투입하 고, 따로 히드록시프로필메틸셀를로오스를 정제수에 녹여 만든 결합액 (10% w/w)을 분무하여 과립을 형성, 건조하였다, 다시 상기의 과림에 에탄을과 염화메틸렌의 1:1흔액에 용해시킨 히드특시프로필메 ¾샐를로오스프탈레이트 용액 (20 /w)을 분무 하여 과립을 코팅하였다. 여기에 스테아르산 마그네슴을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC— 30: 세종)를 사용하여 타정하고 이를 내핵으로 하였다.
2)심바스타틴 선방출성 구획의 제 S
다음 표 Π-2에 나타난 성분 및 함량으로 실시예 II-1의 2) 심바스타된 선방출성 구 획의 제조 방법에 따라 제조하였다.
3)타정 및 코팅
표 II-2 기재 성분 및 함량으로 실시예 II-1의 3) 타정 및 코팅 방법에 따라 암로 디핀-심바스타¾ 유핵정을 제조하였다.
9 000331
81
실시예 11-3 : 암로디핀 -심바스타틴 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 Π-2에 나타난 함량으로 암로디핀 말레이트와 미결정샐를로오스를 35호체로 사과하고 더블콘믹서로 흔합하고, 고속흔합기에 투입한 후 콜리코트 SR30D롤 가하 여 연합한 다음 20호체로 오실레이터를 이용하여 제립하고 , 이를 은수 건조기를 이 용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 스테아르산 마그 네슴을 넣어 더블콘믹서로 최종 흔합하고, 상기 최종 혼합물을 로타리 타정기 (MRC- 30: 세종)를 사용하여 타정하고 이를 내핵으로 하였다.
2)심바스타틴 선방출성 구획의 제조
다음 표 Π-2에 나타난 성분 및 함량으로 실시예 II— 1외 2) 심바스타틴 선방출성 구 획의 제조 방법에 따라 제조하였다.
3)타정 및 코팅
표 IIᅳ 2 기계 성분 및 함량으로 실시예 II-1의 3) 타정 및 코팅 방법에 따라 암로 디핀—심바스타틴 유핵정을 제 a하였다. 실시예 II-4 : 암로디핀 -심바스타틴 다층정의 제 3:
1)암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 II— 2에 나타난 성분 및 함량으로 암로디핀 말테이트와 더결정 셀를로오스롤 35호체로 사과하고 더붙콘믹서로 흔합하고, 따로 히드특시프로필메틸샐를로오스를 정제수에 녹여 만든 결합액 (10% w/w)을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과림에 에탄올과 염화메틸렌의 1:1흔액에 용해시킨 히드록시프로필메틸셀롤 로오스프탈레이트 용액을 분무하여 과립을 코팅하였다. 여기에 스테아르산 마그네 숨을 넣어 최종 더블 콘믹서로 흔합하였다.
2)심바스타틴 선방출성 구획의 제조
다음 표 II-2에 나타난 성분 및 함량과 실시예 Π-1의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3)타정 및 코^
본 공정에서는 다층정 타정기 (MRC-37T : 세종)를 사용하여 타정하였다. 즉, 상기 심바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 암로디핀을 포함하는 조 성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 타정하
고, 하이코터 (SFC-30N, 세종 기계 , 한국)로서 필름 코팅층을 형성하여 다층정을 제 조하였다. 실시예 II-5 : 암로디핀 -심바스타틴 다층정외 제조
1)암로디핀 지연방출성 구획의 제조
다음 표 IIᅳ 2에 나타난 성분 및 함량으로 암로디핀 말레이트와 미결정 샐를로오스를 35호체로 사과하고 더블콘믹서로 흔합하고, 따로 히드록시프로필메틸셀를로오스를 정제수에 녹여 만든 결합액 (1» wAv)을 분무하여 과립을 형성, 건조하였다. 다시 상기의 과립에 에탄을과 염화메틸렌의 1: 1혼액에 용해시킨 유드라짓 RS P0 용액 (20%wAv)을 분무하여 과립을 코팅하였다. 여기에 스테아르산 마그네슘을 넣어 최종 더블 콘믹서로 흔합하였다.
2)심바스타틴 선방출성 구획의 제조
다음 표 II-2에 나타난 성분 및 함량으로 실시예 II-1의 2) 심바스타틴 선방출성 구 획의 제조 방법에 따라 제조하였다.
3)타점 및 코팅
표 II-2 기재 성분 및 함량으로 실시예 II— 4의 3) 타정 및 코팅 방법에 따라 암로 디핀 -심바스타틴 다층정을 제조하였다. 실시예 II— 6 : 암로디 ¾ -심바스타된 2상 매트릭스 정제의 제조
1)암로디핀 지연방출성 구획의 제조
다음 표 IIᅳ 2에 나타난 성분 및 함량으로 암로디 ¾ 말레이트와 미결정셀를로오스를 35호체로 사과하고 흔합하고 , 이를 고속 흔합기에 투입하여 콜리코트 SR30D를 첨가 한 후 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고, 이를 은수 건조기롤 이용하여 60 에서 건조한 다음 다시 20 호로 정립하였다.
2)심바스타¾ 선방출성 구획의 제조
다음 표 II-2에 나타낸 성분 및 함량으로 심바스타틴, 미결정셀를로오스, 만니를을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드록시프로필셀를로오스와 구 연산을 정제수에 녹여 결합액 (10% wAv)을 제조하고 이를 상기 주성분의 흔합물과 함께 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제림하고 이를 은수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 점립하고, 여 기에 부틸레이티드히드록시아니솔을 넣고 혼합하였다.
3)후혼합, 타정 및 코팅
상기 제조된 각각의 최종 조성물을 더블콘믹서로 흔합하고 전분 글리콘산 나트晉, ¾로이드성 이산화규소를 흔합한 후 스테아르산 마그네슴을 넣어 더블콘믹서로 최 종 흔함하였다.
상기 최종 흔합물을 로타리 타정기 (MKC-30: 세종)를 사용하여 타정한 다음 하이코 터 (SFC-30N, 세종 기계 , 한국)로서 필름 코팅 층을 형성하여 2상 매트릭스 정제를 제조하였다. 실시예 II-7 : 암로디 ¾ -심바스타틴 2 상 매트릭스 정제의 제조
1)암로디핀 지연방출성 구획인 과립의 제조
다음 표 11-2에 나타난 성분 및 함량으로 암로디핀 말레이트와 미결정 셀를로오스를 35호체로 사과하고 더블콘믹서로 흔함하였다. 상기의 흔합물을 유동층과립기 (GPCG 1: Glatt)에 투입하고 따로 히드특시프로필데틸셀를로오스를 정제수에 녹여 만든 결합액 (10¾ w/w)을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄을 과 염화메틸렌의 1:1흔액에 용해시 ¾ 유드라짓 RS P0 용액 (20¾ΛΪ)을 분무하여 과 립을 코팅하였다.
2)심바스타틴 선방출성 구획의 제조
다음 표 Π-2에 나타난 성분 및 함량과 실시예 Π-6의 2) 심바스타틴 선방출성 구획 의 제 ; 방법에 따라 제조하였다.
3)타정 및 코팅
표 ΙΙ-2 기재 성분 및 함량으로 실시예 Π-6의 3) 후흔합, 타정 및 코팅 방법에 따 라 암로디핀—심바스타¾ 2상 매트릭스 정제를 제조하였다. 실시예 Π-8 : 암로디핀 -심바스타틴 2 상 매트헉스 정제의 제
υ암로디핀 지연방출성 구획의 제조
다음 표 11-2에 나타난 성분 및 함량으로 암로디핀 밀:레이트와 미결정 셸를로오스를
35호체로 사과하고 더블콘믹서로 흔합하였다. 상기의 흔합물을 유동층과립기 (GPCG 1: Glatt) 에 투입하고 따로 히드록시프로필메필셀를로오스를 정제수에 녹여 만든 결합액 (10 w/w)을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄을 과 염화메틸렌의 1:1흔액에 용해시킨 히드록시프로필메틸셀를로오스프탈레이트 용 액 (2 w/w)을 분무하여 과립을 코팅하였다.
2)심바스타틴 선방촐성 구획의 제조
다음 표 II-2에 나타난 성분 및 함량과 실시예 II-6의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3)타정 및 코팅
표 II-2 기재 성분 및 함량으로 실시예 II— 6의 3) 후흔합 타정 및 코팅 방법에 따 랴 암로디핀 -심바스타틴 2상 메트릭스 정제를 제조하였다 . 실시예 II— 9 : 암로디핀-심바스타 ¾ 2상 캡술 제제
1)암로디핀 지연방출성 구획의 제조 (과립)
다음 표 11-2에 나타난 성분 몇 함량과 실시예 II— 8의 1) 암로디핀 지연방출성 구획 의 제조 방법에 따라 제조하였다.
2)심바스타틴 선방출성 구획의 제조 (과림 )
다음 표 IIᅳ 2에 나타난 성분 및 함량과 실시예 Ι Γ-6의 2) 심바스타¾ 선방출성 구획 의 제조 방법에 따라 제조하였다.
3)흔합 및 캡술층전
공정 1)과 2)의 최종 조성물을 더블콘믹서로 흔합하고, 여기에 전분 ¾리콘산나트륨 을 투입한 후 더블콘믹서로 흔합하고, 콜로이드성 이산화규소를 흔합하고, 스테아 르산 마그네슴을 넣어 최종 흔합하였다 . 최종 흔합된 혼합물을 분말 공급기에 투입 하고 캡술층전기를 이용하여 1호 ¾라된 경질캡술에 층전하였다. 실시예 11-10 : 암로디 ¾ -심바스타 ¾ 2상 캡술 제제
1)암로디핀 지연방출성 구획의 제조 (과립)
다음 표 11-2에 나타난 성분 및 함량으로 암로디핀 말레이트와 미결정셀를로오스 » 35호체로 사과하고 더블콘믹서로 흔합한 다음 이를 유동층과립기 (GPCG 1 : Glatt) 에 투입하고 콜리코트 SR30D 를 분무하여 과립을 형성 , 건 :시켰다.
2)심바스타틴 선방출성 구획의 제조 (과립 )
다음 표 II-2에 나타난 성분 및 함량과 실시예 Π-6의 2) 심바스타틴 선방출성 구획 의 제조 방법에 따라 제조하였다.
3)흔합 및 캡슬 층전
표 II-2 기재 성분 및 함량으로 실시예 II-9의 3) 흔합 및 ¾슬층전 방법에 따라 암토디핀 -심바스타된 2상 캡슬제제를 제조하였다,
실시예 II-ll : 암로디 ¾ -심바스타틴 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (내핵 )
다음 표 11-3에 나타난 성분 및 함량으로 암로디핀 베실레이트와 미결정 ¾를로오 스, 디칼슘 포스폐이트를 35호체로 사과하고 더블콘믹서로 흔합한 후 유등충과림기 (GPCG 1: Glatt)에 투입하고, 따로 히드록시프로필메틸 ¾를로오스를 정제수에 녹여 만든 결합액 (1« w/w)을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 카보머 71G를 분말상태로 투입한 후, 여기에 스테아르산 마그네슘을 넣어 최종 더 볼콘믹서로 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC-30: 세종)를 사용하 여 타정하였다.
다시 상기의 정제에 에탄을과 염화떼 ¾렌의 1 :1흔액에 히드록시프로필메 를로오 스프탈레이트와 아세 ¾레이티드모노글리세리드를 루입하여 용해한 액을 코팅 액 (20fa/w)으로 하여 하이코터 (SFC-30N, 세종 기계 , 한국)를 사용하여 필름 코팅 층 을 형성하여 내핵을 제조하였다.
2)심바스타틴 선방출성 구획의 제조
다음 표 11-3에 나타난 성분 및 함량으로 심바스타틴 , 미결정셀를로오스 , 옥수수전 분 및 유당을 35호체 S 사과하고' 고속흔합기로 흔합하였다. 따로 히드록시프로필셀 를로오스와 구연산을 정제수에 녹여 결합액 (10% w/w)을 제조하고 이를 주성분 흔합 물과 함깨 고속흔합기에 투입하고 연합한 후 20호체로 오실테이터를 이용하여 제립 하고 이를 은수 건조기를 이용하여 60 Ό에서 건조한 다음 다시 20호체로 정립하였 다. 여기에 부틸레이티드히드특시아니솔, 전분 글리콘산 나트륨, 콜로이드성 이산 화규소를 혼합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 ^종 혼합하였다.
3)타정 및 코팅
유핵정 타정기 (RUD-l: Ki lian)를 사용하여 암로디핀 내핵을 심바스타틴을 Ϊ함하는 S성물을 의층으로 하여 타정한 다음 하이코터 (SFC-30N,' 세종 기계 , 한국)로서 표 3 기재 성분 및 함량으로 필름 코팅 층을 형성하여 유핵정을 제조하였다. 실시예 Π-12 : 암로디 ¾ -심바스타된 다층정의 제조
1)암로디핀 지연방출성 구획의 제≤:
다음 표 II— 3에 나타난 성분 및 함량으로 암로디핀 베실레이트와 미결정 셸를로오스 및 디칼슴포스페이트를 35호체로 사과하고 더볼콘믹서로 혼함하고, 따로 히드록시
프로필데¾셀롤로오스를 정제수에 녹여 만든 결합액 (10% w/w)을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄을과 염화메틸렌의 1:1흔액에 용해시킨 히드특시프로필쩨틸셀를로오스프탈레이트과 아세틸레이티드모노 ¾리세리드로 구성 된 코팅액 (20¾ V)을 분무하여 과립을 코팅하였다. 여기에 스테아르산 마그네슴을 넣어 최종 더블콘믹서로 흔합하였다.
2)심바스타틴 선방출성 구획의 제조
다음 표 11-3에 나타난 성분 및 함량과 실시예 I I-1의 2) 심바스타¾ 선방출 성 구 획의 제조 방법에 따라 제조하였다.
3)타정 및 코팅
본 공점에서는 다층정 타정기 (MRC-37T : 세종)를 사용하여 타정하였다. 즉, 상기 심바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 암로디핀을 포함하는 조 성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 타정하 고, 하이코터 (SFC-30N, 세종 기계 , 한국)로서 표 3 기재 성분 및 함량으로 필름 코 팅층을 형성하여 다층정을 제조하였다. 실시예 I 13 : 암토디핀 정제 -심바스타틴 정제를 함유한 캡술제의 제조
1)암로디핀 지연방출성 구획의 제조 (정제)
다음. 표 II-3에 나타난 성분 및 함량으로 암로디핀 베실레이트와 머결정 ¾롤로오스 를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과립기 (GPCG 1: Glatt)에 투 입하고 따로 히드특시프로필떼틸셀를토오스를 정제수에 늑여 만든 결합액 (10% w/w)을 분무하여 과립을 형성, 건 하였다. 다시 상기의 과림에 카보머 71G를 분말 상태로 투입한 후, 여기에 스테아르산 마그네슘을 넣어 최종 더블콘믹서로 흔함한 후 상기 최종 흔합물을 로타리 타정기 (MRC-30: 세종)를 사용하여 타정하였다. 다시 상기의 점제에 에탄올과 염화메틸렌의 1:1 흔액에 히드록시프로필데 ¾셀를로오스프 탈레이트와 아세틸레이티드모노끝리세리드를 투입하여 용해한 액을 코팅 액 (20%w/w) 으로 하여 하이코터 (SFC-30N, 세종 기계, 한국)를 사용하여 필름 코팅 층을 형성하 여 정제를 제조하였다.
2)심바스타틴 선방출성 구획의 제조 (정제)
다음 표 Π-3에 나타난 성분 및 함량으로 심바스타된, 미결정쎌를로오스 , 만니를을 35호체로 사과하고 고속혼합기로 혼합하였다. 따로 히드록시프로필셀롤로오스와 구 연산을 정제수에 녹여 결합액 (10¾ w/w)을 제조하고 이를 주성분 흔합물과 함깨 고
속흔합기에 투입하고 연함한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은 수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정 립하였다. 여기에 부틸레이티드히드록시아니솔, 전분 글리콘산 나트륨, 콜로이드성 이산화규소를 흔 합하고, 스테아르산 마그네슘을 넣어 더블콘믹서로 최종 혼합한 후 상기 최종 흔합 물을 로타리 타정기 (MRC-30: 세종)를 사용하여 타정하였다. 상기 정제는 히드록시 프로필 로오스 2910, 폴리에틸렌글리콜 6000, 산화티탄 및 탈크를 80¾ 에탄을에 용해 및 분산시켜 제조한 코팅액 (20¼/w)을 이융하여 하이코터 (SFC-30N, 세종 기 계, 한국)로 코팅하였다,
3)캡술층전
공정 1〉의 암로디핀 정제와 공정 2)의 심바스타틴 정제를 ¾슬충 ·전기를 이용하여 3 호의 경질 ¾라틴 캡술에 층전하였다. 실시예 11-14 : 암로디핀 -심바스타틴 캡술제의 제조
1)암로디핀 지연방출성 구획의 제 S (정제)
다음 표 11-3에 나타난 성분 및 함량과 실시예 Π-13의 1) 암로디핀 지연방출성 구 획의 제조 방볍에 따라 제조하였다.
2)심바스타틴 선방출성 구획의 제조 (과립)
다음 표 11-3에 나타난 성분 및 함량으로 심바스타틴, 미결정셀를토오스, 유당, 옥 수수전분을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드특시프로필샐를 로오스와 구연산을 정제수에 녹여 결합액 (10¾ w/w)을 제조하고 이를 주성분 혼합물 과 함께 고속흔합기에 투업하고 연합한 다음 20호체로 오실레이터를 이용하여 제립 하고 이를 온수 건조기를 이용하여 60 에서 건조한 후 다시 20호체로 정립하였 다. 여기에 부¾레이티드히드톡시아니솔, 전분 글리콘산 나:트륨, 콜로이드성 이산 화규소를 흔합하고, 스테아르산 마그네슘을 넣어 더블콘믹서로 최종 흔합하였다.
3) ¾술충전
공정 1)의 암로디핀 정제와 공정 2)의 심바스타틴 과립을 캡슬층전기를 이용하여 2 호의 히드록시프로필메 ¾셀를로오스 경질캡슬에 층전하였다. 질시예 11-15 : 암로디핀—심바스타틴 코팅정의 제 S
1)암로디핀 지연방출성 구획의 제조 (정제)
다음 표 11-3에 나타난 성분 및 함량과 실시예 11-11의 1) 암로디핀 지연방출성 구
획의 제조 방법에 따라 제조하였다.
2)심바스타틴 선방출성 코팅 액의 제조
다음 표 11-3에 나타난 성분 및 함량으로 심바스타틴, 부틸레이티드히드특시아니솔, 히드록시프로필메틸셀를로오스 2910, 콜로이드성이산화규소, 풀리에 ¾렌클리콜 6000, 산화티탄, 탈크를 80% 에탄을에 용해 및 분산시켜 선방출성 심바스타틴 코팅 액 (2W w)을 제조하였다.
3) 1차 코팅
위에서 제조한 암로디핀정을 하이코터 (SFC-30N, 세종 기계, 한국)에 투여한 후 심 바스타린 코팅액으로 1차 코팅하였다.
4) 2차 코팅
표 II-3의 필름코팅층 조성물을 80% 에.탄을 및 정제수 흔액에 녹여 필름코팅 액을 제조한 후 1차 코팅완료된 정제에 2차 코팅을 하여 필름코팅정을 제조하였다. 실시예 11-16 : 심바스타¾ 속방-암로디핀 삼투성 유핵정의 제 S
1)암로디핀 지연방출성 구획의 제조 (정제)
다음 표 3에 나타난 성분 및 함량으로 암로디 ¾ 베실레이트와 미결정 셀를로오스 및 염화나트륨을 35호체로 사과하고 더블콘믹서로 흔합한 후 여기에 스테아르산 마그 네슘을 넣어 더블콘믹서로 최종 흔합하고 상기 최종 흔합물을 로타리 타정기 (MRC- 30: 세종)를 사용하여 타정하였다. 타정 후 반투과성막 코팅기제로서 콜리코트 S 30D와 가소제로써 트리에틸시트레이트를 정제수에 분산시킨 후 (20%w/w) 하이코터 (SFC-30N, 세종 기계, 한국)를 이용하여 내핵에 코¾하여 삼투성 내핵을 제조하였 다.
2)심바스타틴 선방출성 구획의 제조
다음 표 11-3에 나타난 성분 및 함량과 실시예 11-11의 2) 심바스타틴 선방출성 구 획의 제조 방법에 따라 제조하였다.
3) 타정 및 코팅
실시예 II— 11의 3) 타정 및 코뜀 방법에 따라 조작하여 심바스타틴 속방-암로디핀 살투성 유핵정제를 제조하였다. 실시 예 II一 17 : 암로디핀 -심바스타틴 블리스터 포장 키트
다음 표 11-3에 나타난 성분 및 함량으로 제조하되, 실 Aᅵ예 II— 13의 암로디휜 정제
과 심바스타틴 정제를 캡슬에 동시 충전하는 것 대신 블리스터 포장용기에 등시복 용 가능하도록 포장하는 것을 계외하고는 실시예 11-13과 같이 제조하였다, 실시예 11-18: (S)-암로디 ¾~ 바스타틴 유핵정의 제조
1) (S)-암로디핀 지연방출성 구획의 제조 (내핵 )
다음 표 Π-4에 나타난 성분 및 함량으로 (S)-암로디핀 베실레이트와 미결정 샐를 로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유등층과립기 (GPCG 1: Glatt) 에 루입하고, 따로 히드록시프로필셀를로오스롤 정제수에 녹여 만든 결합액 (10% w 을 분무하여 과립을 형성시킨 후, 건조하였다. 상기의 생성된 과립에 카보머 71G를 분말상태로 투입하여 더블콘믹서에서 10분간 흔합한 후, 35호체로 체과한 스 테아르산 마그네슘을 넣고 4분간 최종 혼합하였다. 상기의 최종 흔합물을 로타리 타정기 (MRC-30: 세종)를 사용하여 분당 30희전의 속도로 타정하여 (S)-암로디핀 내 핵올 제조하였다.
2)심바스타틴 선방출성 구획의 제조
다음 표 Π-4에 나타난 성분 및 함량으로 심바스타틴 , 미결정셀를로오스, 만니를을 20호체로 체과한 후 , 고속흔합기에서 10분간 흔합하였다. 따로 히드톡시프로필^를 로오스와 구연산을 정제수에 녹여 결합액 (10% w/w)을 제조하였다. 위의 흔합물에 결합액을 가하면서 , 연합을 완료한 후 제립, 건조, 정립하여 과립제조를 완료하였 다. 여기에 미리 35호 체로 체과한 부¾레이티드히드록시아니솔, 전분글리콘산나트 륨을 위의 과립과 함께 더블콘믹서에 투여한 후, 약 10분간 흔합하고, 스테아르산 마그네슴을 35호 체로 체과하여 더블콘믹서에 투여한 후 약 4분간 최종 흔합하여 심바스타틴층 과립 제조를 환료하였다.
3)타정 및 코팅.
유핵점 타정기 (RUD-1: Kil iai * 사용하여 암로디핀 내핵을 심바스타된을 포함하는 조성물을 외층으로 하여 분당 30회전의 속도로 타정하여 유핵정을 제조하였다. 따 로 표 Π-4의 필 *코팅층 조성물을 용매에 녹여 필름코팅액을 제조하였다. 위에서 제조한 유핵정을 하이코터 (SFC-30N, 세종 기계 , 한국)에 투여한 후 필름코팅액으로 코팅하여 유핵정 제조를 완료하였다. 실시예 11—19 : (S)-암로디핀 -싶바스타틴 유핵정의 제쵸
다음 표 Π-4에 나타난 성분 및 함량으로 제조하되 ᅳ 실시예 Π-18의 핵정 제조 후
폴리 (데타크릴레이트, 메틸메파크 ¾레이트)공증합체를 정제수 분산시킨 용액
(20¼/w)으로추가적으로 코팅한 것을 제의하고는 실시예 IIᅳ 18과 등일하게 제조하 였다, 실시예 11-20 : (S)-암로디핀 -심바스타틴 캡술제의 제조
1) (s)-암로디핀 지연방출성 구획의 제조 (정제)
다음 표 Π-4에 나타난 성분 및 함량으로 (S)—암로디핀 베실레이트와 미결정 셀를 로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동충과립기 (GPCG 1: Glatt) 에 투입하고, 따로 히드록시프로필셸를로오스를 정제수에 녹여 만든 결합액 (10% w/iv)을 분무하여 과립을 형성시킨 후, 건 S하였다, 상기의 생성된 과립에 카보머 71G를 분말상태로 투입하여 더블콘믹서에서 W분간 흔합한 후, 35호체로 체과한 스 테아르산 마그네슴을 넣고 4분간 최종 흔합하였다. 상기의 최종 혼합물을 로타리 타정기 (MRC-30: 세종)롤 사용하여 분당 30회전의 속도로 타정하여 제조한 (S)-암로 디핀 정제를 폴리 (메타크릴레이트, 메¾메타크릴레이트)공증합체를 정제수에 분산 시킨 용액 (20taAr)으로 코팅하여 (S)-암로디핀 정제 제조를 완료하였다.
2)심바스타틴 선방출성 구획의 제조 (정제)
다음 표 Π—4에 나타난 성분 및 함량으로 심바스타틴 , 미결정셀롤로오스, 만니를을 20호체로 체과한 후, 고속흔합기에서 10분간 흔합하였다, 따로 히드록시프로필셀를 로오스와 구연산을 정제수에 녹여 결합액 (10¾ w/w)을 제 2:하였다. 위의 흔함물에 . 결합액을 가하면서 , 연합을 완료한 후 제림 , 건조, 정 립하여 과립제조를 완료하였 다. 여기에 미리 35호 체로 체과한 부틸레이티드히드록시아니솔, 전분글리콘산나트 콤, 콜로이드성 이산화규소를 위의 과립과 함께더블콘믹서에 투여한 후, 약 10분간 흔합하고, 스테아르산 마그네슘을 35호 체로 체과하여 더블콘믹서에 투여한 후 약 4분간 최종 혼합하여 심바스타¾충 과립 제조를 완료하였다. 상기의 과립을 로타리 타정기 (MRC-30: 세종)를 사용하여 분당 30희전의 속도로 타정하여 제조한 심바스타 ¾ 정제를 표 8의 필름층 성분으로 코팅하여 심바스타틴 정제 제조를 완료하였다.
3)캡술층전
캡슬층전기를 사용하여 위의 (S)-암로디핀 정제와 심바스타틴 정제를 3호의 젤라틴 경질캡술에 등시 층전하여 캡술제 제조를 완료하였다. 실시예 11-21 : (S)-암로디핀—심바스타¾ 캡술제의 제조
1) (S)—암로디된 지연방출성 구획의 제≥ (정제 )
다을 표 II-4애 나타난 성분 및 함량으로 실시예 11-20-1)의 방법에 의해 (S)-암로 디핀 정제 제조를 완료하였다.
2)심바스타틴층 선방출성 구획의 제조 (과립 )
다음 표 Π-4에 나타난 성분 및 함량으로 심바스타틴 , 미결정셀를로오스, 만니를을 20호체로 체과한 후, 고속흔합기에서 10분간 혼합하였다. 따로 히드특시프로필샐를 로오스와 구연산을 정제수에 녹여 결합액 (10¾ w/w)을 제조하였다. 위의 흔합물에 결합액을 가하면서 , 연합을 완료한 후 제립 , 건조, 정립하여 과립제조롤 완료하였 다. 여기에 미리 35호 체로 체과한 부될레이티드히드특시아니솔, 전분글리콘산나트 륨, 콜로이드성 이산화규소롤 위의 과립과 함께 더블콘믹서에 투여한 후, 약 10분 간 흔합하고, 스테아르산 마그네슘을 35호 체로 체과하여 더블콘믹서에 루여한 후 약 4분간 최종 혼합하여 심바스타틴층 과립 제조를 완료하였다.
3)캡슐층전
¾술층전기를 사용하여 위의. (S)-암로디핀 정제와 심바스파린 과립을 1호의 히드록 시프로필메틸셀를로오스 경질캡술에 동시 층전하여 캡술제 제조를 완료하였다. 실시예 11-22 : (S)-암로디편정 + 심바스타된 블리스터 포장 키트
다음 표 Π-4에 나타난 성분 및 함량으로 제조하되, 실시예 IIᅳ 20의 (S)-암로디핀 정제와 심바스타틴 정제롤 캡술에 동시 층전하는 것 대신 PTP 포장용기에 동시복용 가능하도톡 포장하는 것을 제의하고는 실시예 11-20과 같이 제조하였다. 실시예 IIᅳ 23 : (S)—암로디핀-심바스타 ¾ 코팅정제의 제조
i)(s)-암로디핀 지연방출성 구획의 제 s (정제)
다음 표 II-4에 나타난 성분 및 함량으로, (S)-암로디핀과 미결정 셀를로오스를 35 호체로 사과하고 더블콘믹서로 흔합한 후 유동층과립기 (GPCG 1: Glatt)에 투입하 고 , 따로 히드록시프로필셀를로오스를 정제수 에 녹여 만든 결합액 (10¾ 을 분 무하여 과림을 형성시킨 후, 건조하였다. 상기의 생성된 과립에 카보머 71G를 분밀: 상태로 투입하여 더블콘믹서에서 10분간 혼합한 후, 35호체로 체과한 스테아르산 마그네슴을 넣고 4분간 최종 흔함하였다. 상기의 최종 흔합물을 로타리 타정기 (M C-30: 세종)를 사용하여 분당 30희전의 속도로 타정하여 제조한 (S)-암로디핀 정제를 아래 표 Π-4의 플리 (메타크¾레이트, 메틸메타크 ¾테이트)공증합체를 정제
숙에 분산시 ¾ 용액 (20¾v/w)으로 코팅하여 (S)-암로디핀 정제 제조를 완료하였다 .
2)심바스타틴 코팅 액의 제조
다음 표 II-4에 나타난 성분 및 함량으로 심바스타¾, 부틸레이티드히드특시아니 솔, 히드록시프로필메틸셸를로오스 2910, 콜로이드성이산화규소를 에탄을과 염화메 틸렌 1:1의 흔액에 녹여 심바스타틴 코팅액을 제조하였다,
3) 1차 코팅
위에서 제조한 (S)-암로디핀정을 하이코터 (SFC-30N, 세종 기계 , 한국)에 투여한 후 심바스타¾ 코팅 액으로 1차 코텅하였다.
4) 2차 코팅
표 II-4의 필름코팅층 조성물을 용매에 녹여 필름코팅 액을 제조한 후 1차 코팅 완료 된 정제에 2차 코팅을 하여 필름코팅정을 제조하였다.
-2]


C6 ιεεοοο/6θοζ9ΐχ3ί
[표 11-3]


실시예 II I-l: 암로디핀-아토르바스타틴 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (내핵 )
다음 표 III-1에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 셀를로 오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과립기 (GPCG 1 : Glatt)에 투입하고, 따로 히드록시프로필메틸샐를로오스를 정제수에 녹여 만든 결합액을 분 무하여 과립을 형성, 건조하였다.다시 상기의 과립에 카보머 71G» 분말상태로 투 입한 후, 스테아르산 마그네슴을 넣어 최종 더블콘믹서로 혼합한 후 상기 최종 흔 합물을 로타리 타정기 (MRC-33: 세종)를 사용하여 타정하고 이를 내핵으로 하였다.
2)아토르바스타틴 선방출성 구획의 제 S
다음 표 II 1-1에 나타난 성분 및 함량과 같이 아토르바스타틴 칼슴 삼수화물, 탄산 칼슴, 미결정셀를로오스, 유당, 전호화전분 및 라우릴황산나; "(sodium lauryl sulfate)을 35호체로 사과하고 고속흔합기로 흔합하였다.따로 히드특시프로필셀를
로오스을 정제수에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 고속혼합기 에 투입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조 기를 이용하여 60 X 에서 건조한 다음 다시 20호체로 정립하였다. 여기에 크로스카 르델로오스 나트륨을 흔합하고, 스테아르산 마그네슘을 넣어 더블콘믹서로 최종 흔 합하였다.
3)타정 및 코팅
유핵정 타정기 (RUDᅳ 1: Ki lian)를 사용하여 암로디핀 내핵과 아토르바스타틴을 포함 하는 조성물을 외충으로 하여 타점한 다음 하이코터 (SFC-30N, 세종 기계 , 한국)를 사용하여 표 III-1기재 성분과 함량으로 필름 코팅층을 형성하여 유핵정을 제조하였 다. 실시예 III-2: 암로디판-아토르바스타된 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 III-1에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결점 셀를로 오스를 35호체로 사과하고 더블콘 ^서로 흔합한 후 유동층과립기 (GPCG 1: Glatt)에 투¾하고, 따로 히드록시프로필떼 ¾젤를로오스를 정제수에 녹여 만든 결합액을 분 무하여 과립을 형성, 건 S하였다. 다시 상기의 과립에 에탄을과 염화메틸렌의 1:1 흔액에 용해시킨 히드록시프로필데틸셀를로오스프탈레이트 용액을 분무하여 과립을 코팅하였다.여기에 스테아르산 마그네슴을 넣어 최종 더블콘믹서로 혼합한 후 상기 최종 혼함물을 로타리 타정기 (MRC-33: 세종)를 사용하여 타정하고 이를 내핵으로 하였다.
2)아토르바스타틴 선방출성 구획의 제조
다음 표 ΙΠ-1에 나타난 성분 및 함량으로 실시예 III— 1의 방법에 따라 제조하였다.
3)타정 및 코팅
유핵정 타정기 (RUD-1: Ki liai)를 사용하여 암로디핀 내핵과 아토르바스타틴을 포함 하는 조성물을 외층으로 하여 타정한 다음 하이코터 (SFC-30N, 세종 기계 , 한국)를 사용하여 표 ΙΠ-1기재 성분과 함량으로 필름 코¾ 층을 형성하여 유핵정을 제조하 였다. 실시예 ΙΠ-3: 암로디핀-아토르바스타된 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 III— 1에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 ^를로 오스를 35호체로 사과하고 더블콘믹서로 흔합하였다.상기의 흔합물을 유동층과¾기 (GPCG 1: Giatt)에 투입하고 따로 히드록시프로필메틸셀를로오스를 정제수에 늑여 만든 결합액을 분무하여 과립을 형성, 건조하였다.다시 상기의 과립에 염화메틸렌 에 용해시킨 유드라짓 RS P0 용액올 분무하여 과립을 코팅하였다. 여기에 스테아르 산 마그네슘을 넣어 더블콘믹서로 최종 흔합하고 , 상기 최종 흔합물을 로타리 타정 기 (MRC— 33: 세종)를 사용하여 타정하고 이를 핵정으로 하였다.
2)아토르바스타틴 선방출성 구획의 제조
다음 표 III-1에 나타난 성분 및 함량으로 실시예 ΙΠ-1-2)의 방법에 따라 제조하였 다.
3)타정 및 코팅
유핵정 타정기 (RUD-1: Ki H an)를 사용하여 암로디핀 내핵과 아토르바스타틴을 포함 하는 조성물을 외층으로 하여 타정한 다음 하이코터 (SFC-30N, 세종 기계, 한국)를 사용하여 표 IIIᅳ 1기재 성분과 함량으로 필름 코팅 층을 형성하여 유핵정을 제조하 였다, 실시예 ΙΠ-4: 암로디핀-아토르바스타틴 다층정의 제조
1)암로디핀 지연방출성 구획의 제조
다음 표 III-1에 나타난 성분. 및 함량과 같이 암로디편 말레이트와 미결정셀를로오 스롤 35호체로 사과하고 흔합하고, 이를 고속 흔합기에 투입하여 콜리코트 SR30D를 첨가한 후 연합하였다.연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고, 이를 은수 건 기를 이용하여 60 1C에서 건조한 다음 다시 20 호로 정팁하였다. 여 기에 스테아르산 마그네슘을 넣어 더블콘믹서로 최종 흔합하였다.
2)아토르바스타틴 선방출성 구획의 제조
다음 표 III-1에 나타난 성분 및 함량과 같이 아토르바스타된 칼슴 무수물, 탄산칼 슘, 미결정셀를로오스, 유당 및 라우 ¾황산나트륨 (sodium lauryl sulfate)을 35호 체로 사과하고 고속흔합기로 흔합하였다. 따로 히드톡시프로필^를로오스을 정제수 에 녹여 결함액을 제조하고 이를 주성분 흔합물과 함깨 고속혼합기에 투입하고 연 합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 ¾에서 건조한 다음 다시 20호체로 정 ¾하였다. 여기에 크로스카르멜로오스 나 트륨을 흔합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
3)타정 및 코팅
본 공정에서는 다층정 타정기 (M4C-37T: 세종)를 사용하여 타정하였다.즉, 상기 아 토르바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 암로디핀을 포함하는 조성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 타정 하고, 하이코터 (SFC-30M, 세종 기계, 한국)로서 표 III-1기재 성분과 함량으로 필 름 코팅층을 형성하여 다층정을 제조하였다. 실시예 ΠΙ-5: 암로디핀-아토르바스타틴 다층정의 제 S
1)암로디핀 지연방출성 구획의 채조
다음 표 III 1에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정 ¾를로오 스를 35호체로 사과하고 더블콘믹서로 흔합하고, 따로 히드록시프로필메 ¾¾를로오 스를 정제수에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 다시 상기 의 과립에 에탄을과 염화메틸렌의 1:1흔액에 용해시킨 히드록시프로필메틸셀를로오 스프탈레이트 용액을 분무하여 과팀을 코텅하였다.여기에 스테아르산 마그네슘을 넣어 최종 더블 콘믹서로 흔합하였다.
2)아토르바스타틴 선방출성 구획의 제조
다음 표 III-1에 나타난 성분 및 함량과 같이 아토르바스타틴 칼슘 스트론륨 오수화 물, 탄산칼슘, 미결정 ¾를로오스, 유당 및 라우 ¾황산나트륨 (sodium lauryl sul fate)을 35호체로 사과하고 고속흔합기로 흔합하였다. 따로 히드특시프로필셸를 로오스을 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함깨 고속흔합기 에 투입하 J1 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조 기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정립하였다. 여기에 크로스카 르멜로오스 나트륨을 흔합하고, 스테아 3산 마그네슘을 넣어 더블콘믹서로 최종 흔 합하였다.
3)타정 및 코팅
본 공정에서는 다층정 타정기 (MRC-37T: 세종)를 사용하여 타정하였다.즉, 상기 아 토르바스타 ¾을 포함하는 조성물을 1차 분말공급기에 넣고, 암로디핀을 포함하는 조성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 타정 하고, 하이코터 (SFC-30N, 세종 기계 , 한국)로서 표 II 1-1기재 성분과 함량으로 필 # 코팅층을 형성하여 다층정을 제조하였다.
실시예 ίΠ-6: 암로디핀―아토르바스타틴 2상 매트릭스 정제의 제조
1)암로디핀 지연방출성 구획의 제조
다음 표 Π 1-1에 나타난 성분 및 함량과 같이 암로디핀 배실레이트와 미결정셸를로 오스를 35호체로 사과하고 흔합하고, 이를 고속 흔합기에 투입하여 콜리코;트 SR30D 를 첨가한 후 연합하였다. 연합이 끝나면 20호채로 오실레이터를 이용하여 제립하 고 , 이를 은수 건조기를 이용하여 60 에서 건조한 다음 다시 20 :로 정립하였 다.
2)아토르바스타 ¾ 선방출성 구획의 제조
다음 표 ΠΙ-1에 나타낸 성분 및 함량과 같이 아토르바스타틴 칼슘 삼수화물, 탄산 칼슘, 미결정샐를로오스, 유당, 만니를 및 크로스포비돈을 35호체로 사과하고 고속 흔합기로 흔합하였다.따로 히드톡시프로필셸를로오스과 풀리소르베이트 80을 정제 수에 녹여 결합액을 제조하고 이를 상기 주성분의 흔함물과 함께 고속흔합기에 투 입하고 연합한 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 I;에서 건조한 다음 다시 20호체로 정림하였다.
3)후흔합, 타정 및 코팅
상기 제조된 각각의 최종 조성물을 더블콘믹서로 흔합하고, 전분 글리콜산 나트튬 을 혼합한 후 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
상기 최종 흔합물을 로타리 타정기 (MRC— 33: 세종)를 사용하여 타점한 다음 하이코 터 (SFC-30N, 세종 기계 , 한국)로서 표 II 1-1기재 성분과 함량으로 필름 코팅 층을 형성하여 2상 매트릭스 정제를 제조하였다. 실시예 III-7: 암로디뭔-아토르바스타틴 2 상 매트릭스 정제의 제조
1)암로디편 지연방출성 구획의 제조
다음 표 III-1에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 셸를로 오스를 35호체로 사과하고 더블콘믹서로 흔합하였다.상기의 흔합물을 유동층과림기 (GPCG 1: Glatt)에 루입하고 따로 히드특시프로필메 ¾셀를로오스를 정제수에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조하였다.다시 상기의 과립에 염화메 ¾렌 에 용해시 ¾ 유드라짓 RS P0 용액을 분무하여 과립을 코팅하였다.
2)아토르바스타틴 선방출성 구획의 제조
다음 표 III-1에 나타낸 성분 및 함량으로 실시예 IIIᅳ 6-2)의 방범으로 제조하였다
3)돠정 및 코팅
상기 제≥된 각각의 최종 조성물을 더블콘믹서로 흔합하고, 전분 글리콜산 나트륨 을 흔합한 후 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다. 상기 최 종 흔합물을 로타리 타정기 (MRC-33: 세종)를 사용하여 타정한 다음 하이코터 (SFC- 30N, 세종 기계 , 한국)로서 표 III-1기재 성분과 함량으로 필름 코팅 층을 형성하 여 2상 매트릭스 정제를 제 하였다. 실시예 III-8: (S)-암로디핀-아토르바스타틴 2 상 매트릭스 정제의 제조
1)암로디핀 지연방출성 구획의 제조
다음 표 III— 1에 나타낸 성분 및 함량으로 암로디핀 베실테이트 대신 S-암로디핀 베 실레이트를 사용한 것을 제의하고 실시예 III-6-1)의 방법과 동일하게 제 :하였다.
2)아토르바스타틴 선방출성 구획외 제조
다음 표 111ᅳ1에 나타낸 성분 및 함량으로 아토르바스타틴 칼슴 삼수화물 대신 아토르바스타틴 칼슘 무수물을 사용한 것을 제외하고 실시예 ΠΙ-6-2)의 방법 과 동일하게 제 S하였다.
3)타정 및 코팅
다음 표 III— 1에 나타낸 성분 및 함량으로 실시예 III-6-3)의 방법과 등일 하게 제조하였다. 실시예 III-9: (S)-암로디핀-아토르바스타텬 2상 캡술 제제
1)암로디핀 지연방출성 구획의 제조 (과립)
다음 표 II 1-1에 나타난 성분 및 함량과 같이 S-암로디핀 베실테이트와 미결정 셀를 로오스를 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과¾기 1 (1 1: Glatt) 에 투입하고, 따로 히드톡시프로필데 ¾셀를로오스를 정제수에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조하였다. 다시 상기의 과립에 에탄을과 염화께틸렌의 1:1흔액에 용해시킨 히드록시프로필메틸샐를로오스프탈레이트 용액을 분무하여 과 립을 코팅하였다.
2)아토르바스타 ¾ 선방출성 구획의 제 (과립)
다음 표 III-1에 나타낸 성분 및 함량과 같이 아토르바스타틴 칼슘 삼수화물, 탄산 칼슘, 미결정셀를로오스, 유당, 전호화전분 및 라우릴황산나트륨을 35호체로 사과
하고 고속흔합기로 흔합하였다.파로 히드록시프로필셀를로오스을 정제수에 녹여 결 합액을 제조하고 이를 상기 주성분의 혼합물과 함께 고속흔합기에 투입하고 연합한 후 20호쳬로 오실레이터롤 이용하여 제립하고 이를 은수 건조기롤 이용하여 60 X: 에서 건조한 다음 다시 20호체로 정립하였다.
3)흔합 및 캡슬층전
공정 1)과 2)의 최종 조성물을 더블콘믹서로 흔합하고, 여기에 전분글리클산나트 " 을 투업한 후 더블콘믹서로 흔합하고, 스뒈아르산 마그네슘을 넣어 최종 혼합하였 다. 최종 흔합된 혼합몰을 분말 공급기에 투입하고 캡슬층전기를 이용하여 1호 캡 술에 층전하였다. 실시예 111-10: 암로디핀-아토르바스타틴 2상 술 제제의 제조
1)암로디핀 지연방출성 구획의 제조 (과립)
다음 표 ΙΠ-1에 나타난 성분 및 함량과 같이 암로디핀 말레이트와 미결정셀를로오 스를 35호체로 사과하고 더불콘믹서로 흔합한 다음 이를 유동층과립기 (GPCG 1: Glatt )에 투입하고 콜리코트 SR30D 를 분무하여 과립을 형성, 건조시켰다.
2)아토르바스타된 선방출성 구획의 제조 (과립)
다윰 표 II 1-1에 나타낸 성분 및 함량과 같이 아토르바스타틴 스트론튬 오수화읗, ¾산칼슘, 미결정 ¾률로오스, 유당, 만니를 및 크로스포비돈을 35호체로 사과하고 고속흔합기로 흔합하였다.따로 히드록시프로필셀롤로오스과 풀리소르베이트 80을 정제수에 녹여 결함액을 제 하고 이를 상기 주성분의 흔합물과 함께 고속흔합기애 투입하고 연합한 후 20호체로 오실테이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 °C에서 건조한 다음 다시 20호체로 정림하였다.
3)흔합 및 캡슬 층전
공정 1)과 2)의 최종 조성물을 더불콘믹서로 흔합하고, 여기에 전분글리콜산나트륨 을 투입한 후 더블콘믹서로 흔합하고, 스테아르산 마그네슘을 넣어 최종 흔합하였 다. 최종 흔합된 흔합물을 분말 공급기에 투입하고 ¾술충전기를 이용하여 1호 ¾ 술에 층전하였다. 실시예 ΠΙ-11: 암로디핀-아토르바스타 ¾ 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (내핵)
다음 표 ΙΠ-2에 나타난 성분 및 함량과 같이 암로디핀 베실테이트와 미결정 쉘를로
오스, 디칼슘 포스페이트를 35호체로 사과하고 더블콘믹서로 혼합한 후 유등층과립 7) (GPCG 1: Glatt)에 투입하고 , 따로 히드록시프로필메틸젤률로오스를 정제수에 녹 여 만든 결합액을 분무하여 과립을 형성 , 건조하였다.다시 상기의 과립에 카보머 71G를 분말상태로 투입한 후, 여기에 스테아르산 마그네슴을 넣어 최종 더블콘믹서 로 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC— 33: 세종)를 사용하여 타정하 였다 >
다시 상기의 정제에 에탄을과 염화메틸렌의 1:1흔액에 용해시킨 히드록시프로필메 틸샐를로오스프탈레이트 용액과 아세틸레이티드모노글리세리드를 100:1로 *합한 용엑을 하이코터 (SFC-30N, 세종 기계 , 한국)를 사용하여 필름 코팅 층을 형성하여 내핵을 제 하였다.
2)아토르바스타틴 선방출성 구획의 제조
다음 표 ΙΠ— 2에 나타난 성분 및 함량과 같이 아토르바스타틴 ¾:슘 삼수화물, 스테 아르산 칼슴, 미결정젤를로오스, 욱수수전분 및 유당을 35호체로 사과하고 고속혼 합기로 흔합하였다.따로 히드록시프로필 ·€를로오스와 플리소르베이트 80을 정제수 에 녹여 결합액을 제 하고 이를 주성분 흔합물과 함께 고속혼합기에 투입하고 연 합한 후 20호체로 오실례이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 'C에서 건조한 다음 다시 20호체로 정립하였다.여기에 전분 글리콘산 나트륨를 흔합하고, 스테아르산 마그네슘을 넣어 더블콘믹서로 최종 흔합하였다.
3)타정 및 코팅
유핵정 타정기 (Rl -1: Ki l ian)를 사용하여 암로디핀 내핵과 아토르바스타틴을 포함 하는 조성물을 의층으로 하여 타정한 다음 하이코터 (SFC-30N, 세종 기계, 한국)로 서 표 III-2기재 성분과 함량으로 필름 코팅 층을 형성하여 유핵정을 제조하였다. 실시예 ΙΠ-12: (S)—암로디핀-아토르바스타된 다층정의 제조
1)암로디핀 지연방출성 구획의 제
다음 표 II 1—2에 나타난 성분 및 함량과 같이 S-암로디핀 베실레이트와 미결정 셀를 로오스 및 디칼슘포스페이트를 35호체로 사과하고 더블콘먹서로 흔합하 따로 히 드록시프로필메틸셀를로오스를 정제수에 녹여 만든 결합액을 분무하여 과립을 형 성, 건조하였다.다시 상기의 과립에 에탄을과 염화메틸렌의 1: 1흔액에 용해시킨 히 드록시프로필메¾¾를로오스프탈레이트 용액과 아세틸레이티드모노글리세리드를 100:1로 흔합한 용액을 분무하여 과립을 코팅하였다, 여기에 스테아르산 마그네슴
을 넣어 최종 더블콘믹서로 흔합하였다.
2)아토르바스타틴 선방출성 구획의 제조
다음 표 II 1-2에 나타난 성분 및 함량으로 실시예 II 1-11-2)의 방법으로 제조하였 다.
3)타정 및 코팅
본 공정에서는 다층정 타정기 (MRCᅳ 37T: 세종)를 사용하여 타정하였다.즉 , 상기 아 토르바스타틴을 포함하는 조성물을 1차 분말공급기에 넣고, 암로디편을 포함하는 조성물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 타정 하고, 하이코터 (SFC-30N, 세종 기계 , 한국)로서 표 ΠΙ-2기재 성분과 함량으로 필 름 코팅층을 형성하여 다층정을 제조하였다. 질시예 II 1-13: 암로디핀-아토르바스타된 캡슬제의 제조
1)암로디핀 지연방출성 구획의 제조 (정제)
다음 표 II 1-2에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 샐를로 오스를 35호체로 사과하고 더불콘믹서로 흔합한 후 유등층과립기 (GPCG 1: Glatt)에 투입하고, 따로 히드록시프로필떼틸셸를로오스를 정제수에 녹여 만든 결합액을 분 무하여 과립을 형성 , 건조하였다.다시 상기의 과립에 카보머 71G를 분팔상태로 투 입한 후, 여기에 스테아르산 마그네슘을 넣어 최종 더블콘믹서로 흔합한 후 상기 최종 혼합물을 로타리 타정기 (MRC-33: 세종)를 사용하여 타정하였다.다시 상기의 정제에 에탄을과 염화메틸렌의 1:1흔액에 용훼시킨 히드록시프로필메틸셀를로오스 프탈레이트 용액과 아세틸레이티드모노글리세리드를 100:1로 흔합한 용액을 하이코 터 (SFC-30N, 세종 기계 , 한국)를 사용하여 필름 코팅 층을 형성하여 정제를 제조하 였다.
2)아토르바스타틴 선방출성 구획의 제조 (정제)
다음 표 II 1-2에 나파난 성분 및 함량과 같이 아토르바스타틴 칼슴 삼수화물 , 탄산 칼슴 및 미결정샐를로오스를 35호체로 사과하고 고속흔합기로 흔합하였다.따로 히 드록시프로필셀롤로오스와 ΐ리소르베이트 80을 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합불과 함께 고속흔합기에 투입하고 연합한 후 20호체로 오실레이터 를 이용하여 제립하고 이를 은수 건조기롤 이용하여 60 t에서 건조한 다음 다시 20호체토 정립하였다.여기에 전분 글리콘산 나트튬을 흔합하고 , 스테아르산 마그네 슴을 넣어 더블콘믹서로 최종 흔합한 후 상기 최종 흔합불을 로타리 타정기 (MRC-
33: 세종)를 사용하여 타정하였다. 상기 정제는 히드특시프로필셀를로오스, 플리에 틸렌글리콜 6000, 산화티탄 및 탈크를 瞧 에탄을에 용해 및 분산시켜 제조한 코팅 액을. 이용하여 하이코터 (SFC-30N, 세종 기계 , 한국)로 코팅하였다.
3)¾술층전
공정 1)의 암로디핀 정제와 공정 2)의 아토르바스타 ¾ 정제를 ¾술충전기를 이용하 여 3호 캡술에 층전하였다. 실시예 111-14: 암로디 ¾ᅳ아토르바스타틴 ¾술제의 제조
1)암로디핀 지연방출성 구획의 제조 (정제)
다음 표 III— 2에 나타난 성분 및 함량으로 실시예 III-13-1)의 방법에 의해 제조하 였다.
2〉아토르바스파틴 선방출성 구획의 제 (과립 )
다음 표 II 1—2에 나파난 성분 및 함량과 같이 아토르바스타틴 칼슘 삼수화물, 탄산 칼슘, 미결정셀를로오스, 유당, 옥수수전분 및 라우 ¾황산나트튬을 35호체로 사과 하고 고속혼합기로 흔합하였다. 따로 히드록시프로필 ¾를로오스을 정제수에 늑여 결합액을 제조하고 이를 주성분 혼합물과 함깨 고속흔합기에 투입하고 연합한 다음 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60 13에서 건조한 후 다시 20호체로 정 ¾하였다.여기에 전분 글리콘산 나트륨를 흔합하고, 스 테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
3)¾슬충전
공정 1)의 암로디핀 정제와 공정 2)의 아토르바스타틴 과립을 캡슬층전기를 이용하 여 2호의 히드록시프로필메틸셀롤.로오스 경질 ¾술에 층전하였다. 실시예 111-15: 암로디 ¾ -아토르바스타된 코팅정의 제조
1)암로디핀 지연방출성 구획의 제조 (정제)
다음 표 ΠΙ-2에 나타난 성분 및 함량과 같이 암로디핀 베실레이트와 미결정 ¾를로 오스 , 디칼슴 포스페이트 ¾ 35호체로 사과하고 더블콘믹서로 흔합한 후 유동층과립 기 (GPCG 1: Glatt)에 투입하고, 따로 히드록시프로필메 ¾· 를로오스를 정제수에 녹 여 만든 결함액을 분무하여 과립을 형성 , 건조하였다.다시 상기의 과림에 카보머 71G를 분말상태로 투입한 후, 여기에 스테아르산 마그네슘을 넣어 최종 더불콘먹서 로 흔함한 후 상기 최종 흔합불을 로타리 타정기 (M C-33: 세종)를 사용하여 타정하
였다.다시 상기의 정제에 에탄을과 염화데 ¾렌의 1:1흔액에 용해시킨 히드록시프로 필메틸셸를로오스프탈레이트 용액과 아세틸레이티드모노글리세리드를 100:1혼합한 용액을 하이코터 (SFC-30N, 세종 기계 , 한국)를 사용하여 필름 코팅 층을 형성하여 정제를 제조하였다.
2)아토르바스타틴 선방출성 코팅액의 제 S
다음 표 II 1-2에 나타난 성분 및 함량과 같이 아토르바스타틴 칼슘 삼수화물, 히드 특시프로필셀를로오스를 80¾ 에탄을에 용해 및 분산시켜 선방출성 아토르바스타틴 코팅액을 제조하였다.
3)차 코팅
위에서 제조한 암로디핀정을 하이코터 (SFC-30N, 세종 기계, 한국)에 투여한 후 아 토르바스타틴 코팅액으로 1차 코뒹하였다.
4)차 코팅
표 ΠΙ— 2의 필름코뜀층 조성물을 용매에 녹여 필름코팅액을 제조한 후 1차 코팅완 료된 정제에 2차 S팅을 하여 필름코팅정을 제조하였다. 실시예 III— 16: 암로디핀―아토르바스타된 삼루성 유핵정의 제조
1)암로디핀 지연방출성 구획의 제조 (삼투성 정제)
다음 표 III-2에 나타난 성분 및 함량과 같이 암로디핀 펴ᅵ실레이 트와 미결정 셀를로 오스 및 염화나트晉을 35호체로 사과하고 더블콘믹서로 흔합한 후 여기에 스테아르 산 마그네슘을 넣어 최종 더블콘믹서로 혼합하고 상기 최종 혼합물을 로타리 타정 기 (MRC-33: 세종)를 사용하여 타정하였다.타정 후 트리에틸시 S레이트와 삼투성 기 제로서 콜리코트 S 30D를 정제수에 분산시킨 후 하이코터 (SFC-30N, 세종 기계, 한 국 )를 이용하여 내핵에 코팅하여 삼투성 내핵을 제조하였다.
2)아토르바스타틴 선방출성 구획의 제조 (과립)
다음 표 IIIᅳ 2에 나타난 성분 및 함량으로 실시예 111-14— 2)의 방법으로 제조하였 다.
3)타정 및 코팅
유핵정 타정기 (RUD— 1: Kil ian)를 사용하여 암로디핀 내핵과 아토르바스타틴을 포함 하는 조성물을 외충으로 하여 타점한 다음 하이코터 (SFC-30N, 세종 기계 , 한국)로 서 필름 코¾ 층을 형성하여 삼투성 유핵정제를 제조하였다.
실시예 I IIᅳ 17: 암로디핀-아토르바스타 ¾ 블리스터 포장 키트의 제조
다음 표 IIIᅳ 2에 나타난 성분 및 함량과 같이 실시예 III-13-1) 및 13-2)의 방법으 로 암로디핀 지연방출성 구획 (정제) 및 아토르바스타틴 선방출성 구획 (정제)을 제 조하고, 볼리스터 포장용기에 동시복용 가능하도록 포장하여 제조하였다 .
-1]


[표 II 1-2]

2)g¾a트 sreoD - 추성 gsi m cwa¾s 3o¾ ¾학액, - at스 s
실시예 IVᅳ 1: 암로디핀-피타바타 ¾ 유핵정 제 2:
표 에 기채된 성분과 함량으로, 이하의 방법에 의해 유핵점을 제 하였다.
1) 선방출성 구획 (피타바스타틴 선방출 과립 )제조
피타바스타틴 칼슘, 메타규산알루민산마그네슴, ᅵ결정셀를로오스 (AvkelPHlOl, F C Biopolymer , USA) , 유당수화물 (DMV, Germany) , 전호화전분 (Starch 1500G, Colorcon, USA)을 달아 35호체토 사과하고, 더블콘믹서에서 5분간 흔합하여 흔합물 을 제조하였다. 따로 히드록시프로필셀를로오스 OFC-L, Nippon Soda, Japan)* 정 제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함깨 연합하였다. 연합이 끝 나면 18호체로 오실례이터 R402, ERWEKA, Gennany)* 이용하여 제립하고 이를 은 수 건조기 (H-W-C, 삼공사, Japan)를 이용하여 60°C에서 건조한다, 건조가 끝나면 다시 20호체로 정립하였다. 정립물에 저치환도히드록시프로필샐를로오스을 흔합하 고, 스테아르산 마그네슘 (Nitika Chemical , India)을 넣어 더블콘믹서로 최종 흔합 하였다.
2) 지연방출성 구획 (암로디핀 지연방출 정제)제조
암로디핀 떼실레이트, 미결정 ¾를로오스, 무수인산수소칼슘, 전호화전분 (Starch 1500G, Colorcon, USA)을 35호체로 사과하고 더블콘믹서로 5분간 혼합하여 혼합물 을 제조하였다. 따로 히드록시프로필셀를로오스롤 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18호체로 정립한다. 정립물을 유동 층 코령기에 넣고, 따로 ¾를로오스아세테이:巨 320S (아세탈기 32 %), 샐를로오스아 세테이 S 398-10NF (아세탈기 39.8%)를 에탄을과 염화메틸렌에 녹인 액을 조제하여 위의 조립물을 유동층 과립 코팅기 (GPC&— 1 ; Glatt , Germany)에 넣고 코 ¾하였다, 코팅 완료. 후, 스테아르산 마그네슘을 투입하여 4 분간 흔합하고, 직경 5議 편치 가 장착된 로타리 타정기 (MRC-37 ,세종기계 , 한국)로 타정을 하여 핵정을 제 2:하였 다.
3) 하정 코팅
11 mm 편치가 장착된 유핵정타정기 (RUTK1 : i lian, 독일)로 상기 1)의 피타바스타 틴 칼슘 선방출 과립을 외층으로 사용하고, 상기 2)의 암로디핀 베실레이트 지연방 출 정제를 핵정으로 하여 타정하였다. 따로 히드톡시프로필메틸쎌를로오스
2910(Shin-etsu, Japan) , 풀리에 ¾렌글리콜 6,000(BASF, Germany) , ¾크 2^3(:, France) , 산화티탄 (Tioside Americas, USA)을 에탄을과 정제수에 용해 및 분산시킨 코팅 액을 조제하여 상기 정제를 하이코터 (SFC-30N, 세종 기계, 한국)로서 필름 코 팅층을 형성하여 유핵정 형태의 정제롤 제조하였다. 실시예 IV-2 : 암로디핀ᅳ피타바타틴 유핵정 제조
표 IV— 1에 기재된 성분과 함량으로 이하의 방법에 의해 유핵정을 제조하였다.
1) 선방출성 구획 (피타바스타 ¾ 선방출 과립)제조
피타바스타틴 칼슴, 메타규산알루민산마그네슴, 피결정셸를로오스, 유당수화물, 전 호화전분을 달아 35호체로 사과하고, 더블콘믹서에서 5분간 흔함하여 흔합물을 제 조하였다. 따로 히드록시프로필¾를로오스를 정제수에 녹여 결합액을 제조하고 이 를 주성분 혼합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용 하여 제립하고 이를 온수 건조기를 이용하여 60¾에서 건 한다. 건조가 끝나면 다 시 20호체로 정 립하였다. 정립물에 저치환도히드록시프로필셀롤로오스를 흔합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
2) 지연방출성 구획 (암로디핀 지연방출 정제)제조
암로디핀 베실레이트, 미결정셀를로오스, 무수인산수소칼슴, 전호화전분을 35호체 로 사과하고 더블콘믹서로 5분간 흔합하여 흔합물을 제조하였다. 따로 히드록시프 로필셀를로오스를 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조 가 끝나면 다시 18호체로 정립한다. 정립물을 유동층 코팅기에 넣고, 1다로 유드라 짓 RS30D(ETOiiik Degussa, Germany) 및 :트리에틸시트레이트 (Vert lus, England)를 염화메틸렌에 녹인 액을 조제하여 위의 조립물을 유동층 과립 코팅기 (GPCG—1 ; Glatt , Germany)에 넣고 코팅하였다. 코팅 완료 후, 스테아르산 마그네슴을 투입하 여 4 분간 흔합하고, 직경 5 麵 편치가 장착된 로타리 타정기 (MRC-37 : 세종)로 타 정을 하여 핵정을 제조하였다.
3) 타정 및 코팅
실시예 IV-1외 3)과 동일 방법으로 타정 및 코팅하여 유핵정 형태의 정제를 제조하 였다, 실시예 IV— 3 : 암로디핀-피타바타 ¾ 유핵정 제조
표 IV— 1에 기재된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다,
1) 선방출성 구획 (피타바스타틴 선방출 과립〉제조
피타바스타린 칼습, 메타규산알루민산마그네슘, 미결정샐를로오스, 유당수화물, 전 호화전분을 달아 35호체로 사과하고, 더블콘믹서에서 5분간 흔합하여 흔합물을 제 조하였다. 따로 히드록시프로필샐를로오스롤 정제수에 녹여 결함액을 제조하고 이 롤 주성분 흔합물과 함깨 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용 하여 제립하고 이를 은수 건 :기를 이용하여 6(TC에서 건 :한다. 건조가 끝나면 다 시 20호체로 정립하였다. 정 립물에 저치환도히드록시프로필셸를로오스를 흔합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
2) 지연방출성 구획 (암로디핀 지연방출 정제)제조
암로디핀 떼실레이트, 미결정셸를로오스, 디-만니를을 35호체로 사과하고 더블콘믹 서로 5분간 흔합하여 흔합물을 제조하였다. 따로 풀리비닐필로리돈 (Koll idon 30, BASF, Germany)을 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조 가 끝나면 다시 18호체로 정립한다. 정립물을 유동층 코팅기에 넣고, 따로 따로 에 ¾셸를로오스 (HERCULES, USA) , 폴리 (메타크¾붸이트, 메틸메타크 ¾레이트)공중합체 (Evonik degussa, USA)를 에탄을과 염화메털렌에 녹인 액을 조제하여 위의 조립물 을 유동층 과립 코팅기 (GPCGᅳ 1 ; Glatt , Germany)에 넣고 코팅하였다. 코팅 완료 후, 스테아르산 마그네슘을 투입하여 4 분간 혼합하고, 직경 5瞧 펀치가 장착 ¾ 로타리 타정기 (MRC-37 : 세종)로 티 -점을 하여 핵정을 제 하였다.
3) 타정 및 코팅
실시예 IV-1의 3)과 등일 방법으로 타정 및 코팅하여 유핵정 형태의 정제 » 제조하였다, 실시예 IV-4 : 암로디핀 -피파바타틴 유핵정 제조
표 IV-1에 기채된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다.
1) 선방출성 구획 (피타바스타틴 선방출 과립 )외 제 s
피타바스타 ¾ 칼슴, 메타규산알루민산마그네슴, 미결정 ¾를로오스, 유당수화물, 전 호화전분을 달아 35호체로 사과하고, 더블콘믹서에서 5분간 흔합하여 혼합물을' 제 조하였다. 따로 히드톡시프로필셀롤로오스를 정제수에 녹여 결합액을 제 a하고 이
를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용 하여 제립하고 이를 은수 건조기를 이용하여 6(rc에서 건 s한다. 건 s가 끝나면 다 시 20호체로 정립하였다. 정 립물에 전분글리를산나트륨 (DMV, Germany) , 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 혼합하였다. '
2) 지연방출성 구획 (암로디핀 지연방출 점제)의 제조
암로디괸 말레이트, 미결정셸를로오스, 무수인산수소칼슴, 디-만니를을 35호체로 사과하고 더블콘믹서로 5분간 흔합하여 혼합물을 제조하였다. 따로 히드록시프로필 셀를로오스를 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18호체로 정림한다. 정립물에 35호체로 체과한 스테아르산 마그네슴을 투입 후, 4 분간 흔합하여 암로디핀 지연 방출층 과립을 제조하였다. 암로디핀 지 연방출 과립을 직경 5 咖 펀치가 장착된 로타리 타정기 (MRC— 37 : 세종)로 타정하여 핵정을 제 S하였다. 따로 아크 ¾ -이즈 (메타크¾산 공중합체 type C, 탈크, PEG, 콜 로이달실리콘다이육사이드, 중탄산나트륨, SLS, Colorcon, USA)를 정제수에 용해 및 분산시 ¾ 코팅액을 조제하여 위외 암로디 ¾ 정제를 하이코터 (SFC— 30N, 세종 기 계 , 한국)로서 코팅층을 형성하여 암로디핀 정제 제조롤 완료하였다.
3) 타정 및 코팅
11 醒 편치가 장착된 유핵정타정기 (励-1, ilian, 독일)에서 피타바스파틴 속방출 과¾을 외층으로 사용하고, 상기 2)의 암로디핀 지연방출 정제롤 핵정으로 하여 타 정하였다. 따로 히드록시프로필메틸셀를로오스 2910, 플리에틸렌글리콜 6,000, 탈 크, 산화티탄을 에탄을과 정제수에 용해 및 분산시 ¾ 코팅 액을 조제하여 위의 핵정 을 하이코터 (SFC-30N, 세종 기계 , 한국)로서 필름코팅층을 형성하여 유핵정 형태의 정제를 제조하였다. ' 실시예 IV-5 : 암로디핀-피타바타 ¾ 유핵정 제조
표 에 기채된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다. 1) 선방출성 구획 (피타바스타틴 속방출 과립)의 제조
피타바스타틴 ¾슘, 메타규산알루민산마그네슴, 미결정셸를로오스, 유당수화물, 옥 수수전분 (DMV, Germany)을 달아 35호체로 사과하고, 더블콘믹서에서 5분간 흔함하 여 혼합물을 제 하였다. 따로 히드특시프로필셀를로오스를 정제수에 녹여 결합액
을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실 레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60t:에서 건 :한다. 건 조가 끝나면 다시 20호체로 정 립하였다. 정립물에 저치환도히드록시프로필셸를로오 스를 흔합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하였다.
2) 지연방출성 구획 (암로디편 지연방출 정제)의 제 S
암로디핀 말레이트, 미결정쎌를로오스을 35호체로 사과하고 더블콘믹서로 5분간 흔 합하여 흔합물을 제조하였다. Φ합물을 더블콘믹서에 투입하고 콜리코트 SR30D (주 성분 풀리비닐아세테이트 30% 현탁액 , BASF사 제조, Germany)를 가하여 연합한 다 음 20호채로 오실레이터를 이용하여 제립하고, 이를 은수 건조기 * 이용하여 60 1C 에서 건조한 후 다시 18호쳬로 정립하였다. 정립물에 35호체로 체과한 스테아르산 마그네슘을 투입하여, 4 분간 흔합한 후, 직경 5讀 편치가 장착된 로타리 타정기 (M C-37 : 세종)로 타정을 하여 핵정을 제조하였다. 따로 히드록시프로필메털셀를 로오스프탈레이트 (Shin— etsu, Japan)를 에탄을과 염화메틸렌에 용해 및 분산시킨 코팅 액을 조제하여 위의 암로디 ¾ 정제를 하이코터 (SFC-30N, 세종 기계 , 한국)로서 코팅층을 형성하여 암로디핀 점제 제조를 완료하였다.
3) 타정 및 코팅
실시예 IV-1의 3)과 동일 방법으로 타정 및 코팅하여 유핵정 형태의 정제롤 제조하 였다. 실시예 IV-6 : 암로디핀 -피타바타틴 유핵정 제조
표 IV-1에 기재된 성분과 함량으로 , 이하의 방뜁에 의해 유핵정을 제조하였다. 1) 선방출성 구획 (피타바스타틴 속방출 과립)의 제조
피타바스타틴 칼슘, 메타규산알루민산마그네슘, 더결정셀롤로오스, 유당수화물, 옥 수수전분을 달아 35호체로 사과하고, 더블콘믹서에서 5분간 흔합하여 흔함물을 제 조하였다. 따로 히드특시프로필셀를로오스를 정제수에 녹여 결합액을 제 S하고 이 를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용 하여 제립하고 이를 은수 건조기를 이용하여 arc에서 건 s한다. 건조가 끝나면 다 시 20호체로 정립하였다. 정립물에 전분글리콜산나트륨 (DMV, Germany) , 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합 하였다.
2) 지연방출성 구획 (암로디핀 지연방출 정제 )의 제조
암로디핀 베실레이트, 전호화전분, 옥수수전분을 35호체로 사과하고 더붙콘믹서로 5분간 흔합하여 흔합물을 제조하였다. 따로 히드록시프로필메틸 ^를로오스를 정제 수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건 가 끝나면 다시 18호체 로 정립한다. 정립물을 유동층 코¾기에 넣고 , 따로 히드특시프로필메틸샐를로오스 프탈레이트를 에탄을과 염화메될렌에 늑인 액을 조제하여 위의 립물을 유등층 과 립 코팅기 (GPCG-1 ; Glatt, Germany)에 넣고 코팅하였다. 코팅 완료 후, 스테아르 산 마그네슘을 투입하여, 4 분간 흔합한 후 직경 5墮펀치가 장착된 토타리 타정 기 (MRC-37 : 세종)로 타정을 하여 핵정을 제조하였다.
3) 타정 및 코팅
실시예 IV-1의 3)과 동일 방법으 S 타정 및 코팅하여 유핵정 형태의 정제롤 제조하 였다. 실시예 IV-7 : 암로디핀ᅳ피타바파틴 2상 매트릭스 정제 제조
표 IV-1에 기재된 성분과 함량으로, 이하의 방범예 의훼 2상 메트릭스 정제를 제조 하였다.
1) 선방출성 구획 (피타바스타 ¾ 속방출 과립)의 제조
피타바스타틴 칼슴, 메타규산알루민산마그네슴, 유당수화물, 전호화전분을 달아 35 호체로 사과하고 더블콘믹서에서 5분간 흔합하여 Φ합물을 제조하였다. 따로 히드 록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함깨 연합하였다. 연합이 끝나면 20호체 오실레이터를 이용하여 제¾하고 이를 온 수 건조기를 이용하여 601C에서 건조 후 다시 18호체로 정립하였다.
2) 지연방출성 구획 (암로디¾ 지연성 속방층 과립)의 제조
암로디핀 베실테이트, 미결정셀를로오스, 무소인산수소칼슘을 35호체로 사과하고 더블콘먹서로 5분간 흔합하여 혼합물을 제조하였다. 따로 히드록시프로필샐를로오 스를 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조물을 유동층 코팅기에 넣고, 다로 셀를로오스아세테이트 320S (아세탈기 32 %) , 셀를로오스아세 테이트 398-10NF10 (아세탈기 39.8%)를 에탄을과 염화메틸렌에 녹인 액을 조제하여
위의 조립물을 유등층 과립 코팅기 (GPCG-1: Glatt , Geramny)에 넣고 코팅하였다.
3) 후흔합, 타정 및 코¾
상기 1)과 2)의 산물을 더블콘믹서에 넣고 흔합하였다. 이 흔합물에 전분 글리콜산 나트튬, 및 스테아르산 마그네슘을 넣어 더블콘믹서로 최종 흔합하였다. 최종 흔합 물을 로타리 타점기 (MRC-33 : 세종 기계 , 한국 )를 사용하여 타정하였다. 따로 히드 록시프로필메틸■€를로오스 2910, 폴리에틸렌글리¾ 6,000, 탈크 , 및 ·산화티탄을 에 탄올과 정제수에 용해 및 분산시킨 코팅액을 조제하여 , 위의 정제에 하이코터 (SFC- 30N:세종 기계, 한국)를 이용하여 필름코팅층을 형성하여 2상 매트릭스 정제를 제 조하였다. 실시예 IV-8 : 암로디핀 -피타바타틴 2상 메트릭스 정제 제조
표 IV-1에 기계된 성분과 함량으로, 이하의 방법에 의해 2상 매: 릭스 정제를 제조 하였다.
1) 선방출성 구획 (피타바스타 ¾ 속방출 과립)의 제조
피타바스타틴 칼슴', 메타규산알루민산마그네슴, 미결정셀롤로오스를 달아 35호체로 사과하고 더블콘믹서에서 5분간 흔함하여 흔함물을 제조하였다. 따로 히드록시프로 필쎌를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합 하였다. 연함이 끝나면 20호체 오실테이터를 이용하여 제립하고 이를 은수 건조기 를 이용하여 6( C에서 건조 후 다시 18호체로 정립하였다.
2) 지연방출성 구획 (암로디핀 지연성 속방층 과립)의 제조
암로디핀 말레이트, 미결정 ¾를로오스, 전호화전분을 35호체로 사과하고 더블콘믹 서로 5분간 흔합하여 흔합물을 제조하였다. 따로 폴리비닐피를리돈을 정제수에 녹 여 결합액으로 하여 연합, 제립 및 건조하였다. 건조블을 유동층 코팅기에 넣고, 따로 유드라짓 RS30D 및 트리에틸시트레이트롤 염화메틸렌에 녹인 액을 조제하여 위의 조립물을 유동층 과립 코텅기 (GPCG— 1 ; Glatt , Germany)에 넣고 코팅하였다.
3) 후흔합, 타정 및 코팅
질시예 IV-7의 3)과 동일 방법으로 후흔합, 타정 및 코팅하여 2상 매트릭스 형태의 정제를 제 하였다.
질시예 IVᅳ 9 : 암로디핀ᅳ피타바타된 2상 매트릭스 정제 제≤
표 IV-2에 기재된 성분과 함량으로, 이하외 방법에 외해 2상 메트힉스 정제를 제조 하였다.
1) 선방출성 구획 (피타바스타틴 속방출 과립 )의 제조
피타바스타틴 칼슘, 메타규산알루민산마그네슘, 미결정셀를로오스, 유당수화물을 달아 35호체로 사과하고 더블콘믹서에서 5분간 흔합하여 흔합물을 제조하였다. 따 로 히드록시프로필셸 ·로오스를 정제수에 녹여 결합액을 제조하고 이롤 주성분 흔 합물과 함께 연합하였다. 연합이 끝나면 20호체 오실레이터를 이용하여 제림하고 이를 은수 건조기를 이용하여 60°C에서 건조 후 다시 18호체로 정립하였다.
2) 지연방출성 구획 (암로디핀 지연성 속방층 과립)외 제조
S-암로디핀 떼실레이트, 미결정샐를로오스, 디-만니를을 35호체로 사과하고 더블콘 믹서로 5분간 흔합하여 혼합물을 제조하였다. 흔합물을 고속흔합기에 투입하고 콜 리코트 SR30D를 가하여 연합한 다음 20호체로 오실레이터를 이용하여 제립하고, 이 롤 은수 건조기를 이용하여 60 1C에서 건조한 후 다시 18호체로 정 립하였다.
3) 후흔합, 타정 및 코뜀
실시예 IV-7의 3)과 동일 방법으로 후흔합, 타정 및 코팅하여 2상 매트릭스 형태의 정제를 제조하였다. 실시예 IV-10 : 암로디 ¾ -피타바타틴 2상 매트릭스 정제 제조
표 IV-2에 기재된 성분과 함량으로, 이하의 방법에 의해 2상 매트릭스 정제를 제조 하였다.
1) 선방출성 구획 (피타바스타틴 속방출 과립 )의 제조
피타바스타틴 칼슘 , 메타규산알루민산마그네슘, 육수수전분, 전호화전분을 달아 35 호체로 사과하고 더블콘믹서에서 5분간 흔합하여 흔합물을 제효하였다. 따로 히드 록시프로필셀롤로오스를 정제수에 녹여 결합액을 제 S하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체 오실레이터를 이용하여 제립하고 이를 은 수 건조기를 이용하여 60 에서 건조 후 다시 18호체로 정림하였다.
2) 지연방출성 구획 (암로디핀 지연성 속방층 과립 )의 제조
암로디펀 베실레이트 , 미결정샐를로오스, 무수인산수소칼슘, 전호화전분을 35호체 로 사과하고 더블콘믹서로 5분간 흔합하여 혼합물을 제조하였다. 따로 폴리비닐피 를리돈을 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건 ¾물을 유 동층 코팅기에 넣고, 따로 따로 에틸샐를로오스 (HERCULES, USA), 폴리 (메타크릴레 이트, 메틸메타크 ¾테이트)공증합체를 에탄을과 염화메틸렌에 녹인 액을 조제하여 위의 조림물을 유등층 과립 코¾기 ½« -1 ; Glatt , Germany)에 넣고 코팅하였다.
3) 후흔합, 타정 및 코팅
실시예 IV-7의 3)과 등일 방법으로 후혼합, 타정 및 코팅하여 2상 매트릭스 형태의 정제를 제조하였다. 실시예 IV-11: 암로디핀 -피타바타된 다층정 제조
표 IV-2에 기쳬된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다.
1) 선방출성 구획 (피타바스타된 속방출 과립)의 제조
피타바스타된 칼슴, 메타규산알루민산마그네슘, 미결정셀를로오스, 유당수화물을 달아 35호체로 사과하고 더블콘믹서에서 5분간 흔함하여 혼합물을 제조하였다. 따 로 히드록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔 합물과 함께 연합하였다. 연합이 ^나면 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 60°C에서 건조한다. 건조가 끝나면 다시 18호체로 정 립하였다. 정립물에 전분 글리콜산 나트륨을 흔합하고, 스테아르산 마그네슘을 넣 어 더블콘믹서로 최종 흔합하였다.
2) 지연방출성 구획 (암로디핀 지연성 속방층 과립 )의 제조
암로디핀 베실테이트, 미결정쉘를로오스, ¾호화전분, 옥수수전분을 35호체로 사과 하고 더블콘먹서로 5분간 흔합하여 흔합물을 제조하였다. 따로 풀리비닐피 *리돈을 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18 호체로 점림하였다. 정림물을 유동층 코팅기에 넣고, 따로 에 ¾셀를로오스, 폴리 ( 메타크릴레이트, 데¾데타크릴레이트)공중합체를 에탄을과 염화메틸렌에 녹인 액을 조제하여 위의 팁물을 유동층 과립 코팅기 (GPCG-1 ; Glatt , Germany)에 넣고 코 팅하였다. 코팅 완료 후, 스테아르산 마그네슴을 투입 후, 4 분간 흔합하였다.
3) 후혼합, 파정 및 코팅
다층정 타정기 (MRC-37T: 세종)를 사용하여 타정하였다. 상기 1)의 최종 흔합물을 1 차 분말공급기에 넣고, 상기 2)의 최종 흔합물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으토 타정하였다. 따로, 히드록시프로필메틸셀를로오 스 2910, ¾리에 ¾렌글리콜 6,000, 탈 3, 및 산화티탄을 에탄을과 정제수에 용해 및 분산시킨 코팅액을 조제하여 위의 정제를 하이코터 (SFC-30N: 세종 기계, 한국) 로서 필름코팅층을 형성하여 다층정 형태의 정제를 제조하였다. 실시예 IV-12 : 암로디핀 -피타바타틴 다층정 제조
표 IV-2에 기재된 성분과 함량으로, 이하외 방법에 의해 다층정을 제조하였다.
1) 선방출성 구획 (피타바스타틴 속방출 과립 )의 제조
피타바스타틴 칼슴, 메타규산알루민산마그네슴, 미결정 ¾를로오스 , 옥수수전분을 달아 35호체로 사과하고 더블콘믹서에서 5분간 흔합하여 흔합물을 제조하였다. 따 로 히드특시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔 합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 60'C에서 건조한다. 건조가 끝나면 다시 18호체로 정 립하였다. 정립물에 전분 글리를산 나트륨을 흔합하고, 스테아르산 마그네슴을 넣 어 더블콘믹서로 최종 흔합하였다.
2) 지연방출성 구획 (암로디핀 지연성 속방층 과립)의 제조
암로디핀 말레이트, 미결정 ^¾로오스, 전호화전분, 디-만니를을 35호체로 사과하 고 더블콘믹서로 5분간 흔합하여 흔합물을 제조하였다. 따로 풀리비닐피를리돈을 정제수에 늑여 결합액으로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18 호체로 정립하였다. 정립물을 유동층 코팅기애 넣고, 따로 셀#로오스아세테이트 320S (아세탈기 32 %) , ¾를로오스아세테이트 398-10NF (아세탈기 39.8¾)* 에탄을과 염화메틸렌에 ^인 액을 조제하여 워의 조팁물을 유동층 과립 코팅기 (GPCG-1: Glatt , Gerarany)에 넣고 코팅하였다. 코팅 완료 후, 스테아르산 마그네슘을 투입 후 , 4 분간 혼합하였다.
3) 후흔함, 타정 및 코팅
실시예 IV-11의 3)과 등일 방범으로 후흔합, 타정 및 코팅하여 다층점 형태의 정제 를 제조하였다. 실시예 IV— 13 : 암로다핀-피타바타 ¾ 다총정 제조
표 IV-2에 기재된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다.
1) 선방출성 구획 (피타바스타틴 속방출 과립 )의 제조
피타바스타틴 칼슘, 메타규산알루민산마그네슘, 머결정셀를로오스 , 유당수화물, 옥 수수전분을 달아 35호체로 사과하고 더블콘믹서에서 5분간 혼합하여 흔합물을 제조 하였다. 따로 히드특시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연함이 끝나면 20호체로 오실레이터를 이용하여 제립하: a 이를 온수 건조기를 이용하여 60°C에서 건조한다. 건조가 끝나면 다시 18 호체로 정립하였다. 정립물에 저치환도히드록시프로필 ¾를로오스를 혼함하고, 스테 아르산 마그네슘을 넣어 더블콘믹서로 최종 흔합하였다.
2) 지연방출성 구획 (암로디핀 지연성 속방총 과립 )의 제조
S-암로디핀 베실레이트, 미결정셀를로오스, 무수인산수소칼슘 , 전호화전분을 35호 체로 사과하고 더블콘믹서로 5분간 흔합하여 흔합물을 제조하였다. 흔합물을 고속 흔합기에 투입하고 誉리코트 SR30D를 가하여 연합한 다음 20호체로 오실레이터를 이용하여 제립하고, 이를 은수 건조기를 이용하여 60 'C에서 건조한 후 다시 18호 체로 정림하였다. 정 ¾물 에스테아르산 마그네슘을 투입 후, 4 분간 흔합하였다.
3) 후혼합, 타정 및 코팅
실시예 IV-11의 3)과 등일 방법으로 후흔합, 타정 및 코¾하여 다층정 형태의 정제 를 제조하였다. 실시예 IV-14 : 암로디판 -피타바타된 다층정 제조
표 IV-2에 기재된 성분과 함량으로, 이하의 방범에 의해 다층정을 제조하였다. 1) 선방출성 구획 (피타바스타린 속방출 과 ¾)의 제조
피타바스타틴 칼슘, 메타규산알루민산마그네슴, 미결정셀를로오스, 전호화전분을 달아 35호체로 사과하고 더블콘믹서에서 5분간 혼합하여 흔합물을 제조하였다. 따 S. 히프록시프로필셸를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔
합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60t에서 건조한다. 건조가 끝나면 다시 18호체로 정 립하였다. 정립물에 저치환도히드톡시프로필셀를로오스를 흔합하고, 스테아르산 마 그네슘을 넣어 더블콘믹서로 최종 흔합하였다.
2) 지연방출성 구획 (암로디핀 지연성 속방층 과립〉의 제조
암로디핀 베실레이트, 무수인산수소칼슘, 디-만니를을 35호체로 사과하고 더블콘믹 서로 5분간 흔합하여 흔합물을 제조하였다. 따로 히드톡시프로필셀를로오스를 정제 수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18호체 로 정 립하였다. 정립물을 유뜸층 코팅기에 넣고, 따로 유드라짓 RS30D 및 트리에틸 시트레이트를 염화메틸렌에 녹인 액을 조제하여 위의 S립물을 유등층 과립 코팅기 (GPCG— 1; Glatt , Germany)에 넣고 코팅하였다. 코팅 완료 후, 스테아르산 마그네슘 을 투업 후, 4 분간 혼합하였다.
3) 후혼합, 타정 및 코팅
실시예 IV— 11의 3)과 등일 방법으로 후흔합, 타정 및 코팅하여 다층정 형태의 정제 쁠 제조하였다. 실시예 IV— 15 : 암로디핀 -피타바타틴 캡술제 제조 (팰렛-과립 )
표 IV-2에 기재된 성분과 함량으로, 이하의 방법에 의해 ¾술제를 제조하였다.
1) 선방출성구획 (피타바스타틴 ¾¾)의 제조
슈가 시드 (Sugar sphere) (NP Pharmaceutical , France)를 유동층 과립기 (GPCG, Glatt , Germany)에 투입한 뒤 , 따로 정제수에 히드록시프로필셀를로오스와 피타바 스타 ¾ 칼슘을 용해시킨 결합액을 분무하여 펠¾을 형성 , 건조하였다.
2) 지연방출성 구획 (암로디¾ 지연방출성 과립 )의 제조
암로디핀 떼실레이트, 미결정셀롤로오스, 무수인산수소칼슴을 35호채로 사과하 JZ 더블콘믹서로 5분간 흔합하여 흔합물을 제조하였다. 콜리코트 SR30D 넣어 주성분 흔합물과 함깨 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하 고 이를 온수 건조기를 이용하여 건조하였다. 건조가 끝나면 다시 18호체 S 정립하 였다.
3) 후혼합, 타정 및 코팅
상기 1)과 2)의 최종 산물을 더블콘믹서로 흔합하였다. 흔합물에 스테아르산 마그 네슴을 넣어 최종 흔합하였다. 최종 흔합된 흔합물을 분말공급기에 투입하고 캡슐 충전기 (SF 40N, 세종기계 , 한국)를 이용하여 캡슬 (서홍갑샐 , 한국)에 층전하여 캡 술형태의 시간차 방출 제제의 제조를 완료하였다. 실시예 ιν-16 : 암로디핀 -피타바타틴 캡술제 제조
표 I -2에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다.
1) 선방출성 구획 (피타바스타틴 속방 * ¾¾)의 제조
슈가 시드 (Sugar sphere)를 유동층 과립기 (GPCG1 : Glatt)에 투¾한 뒤 따로 물에 히드특시프로필샐를로오스와 피타바스타틴 칼습을 용해시 ¾ 결합액을 분무하여 피 타바스타틴 함유 펠펫을 형성, 건조하였다.
2) 지연방출성 구획 (암로디핀 지연방출 펠렛)의 제조
슈가 시드 (Sugar sphere)를 유동층 과립기 (GPCG1 : Glatt)에 투입한 뒤 , 따로 물에 히드록시프로필씰를로오스, 암로디핀 베실레이트를 용해시킨 결합액을 분무하여 암 로디핀 함유 펠햇을 형성 , 건조하였다. 다시 상기워 ¾헷에 히드톡시프로필메틸샐 를로오스프탈레이트 (Shin-etsu, Japan)를 에탄을과 염화메틸렌에 녹인 액을 분무하 여 암로디핀 지연성 펠¾을 제조하였다,
3) 후흔합, 타정 및 코팅
공정 1)과 2)의 최종 산물을 캡술층전기를 이용하여 캡술 (서흥캅샐, 한국)에 충전 하여 캡술형태의 시간차 방출 제제의 제조를 완료하였다. 실시예 IV-17 암로디핀 -피타바타틴 캡술제 제조 (펠 삼투성 정제)
표 IV-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡슐제를 제조하였다, 1) 선방출성 구획 (피타바스타틴 속방출 펠렛)의 제조
슈가 시드 (Sugar sphere)를 유동층 과립기 (GPCG1 : Glatt)에 투입한 뒤 따로 물에 히드록시프로필셀를로오스와 피타바스타틴 칼슴을 용해시킨 결합액을 분무하여 피 타바스타틴 함유 ¾¾을 형성, 건조하였다.
2) 지연방출성 구획 (암로디핀 삼투성 정제 )의 제조
암로디핀 베실레이트, 미결정셀를로오스, 무수인산수소칼슴 , 옥수수전분을 35호체 로 사과하고 더블콘믹서로 5분간 흔함하여 흔합물을 제조하였다. 따로 히드특시프 로필셀를로오스를 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다, 건조 가 끝나면 다시 18호체로 정립하였다. 정립물에 염화나트륨 및 스테아르산 마그네 슴을 넣고 4분간 흔합하였다. 상기 흔합물을 직경 5 mm 편치가 장착된 로타리 타정 기 (MRC-37 : 세종)로 타정을 하여 정제를 제조하였다. 따로 샐를.로오스아세테이트 320S (아세탈기 32 %) , 셸롤로오스아세테이트 398-10 F (아세탈기 39.8%)를 에탄을과 염화메틸렌에 녹인 액을 조제하여 , 상기 정제에 하이코터 (SFC-30N : 세종기계 , 한 국)로 필름코팅층을 형성하여 암로디핀의 삼투성 정제를 제조하였다.
3) 후흔합, 타정 및 코팅
상기 1)과 2)의 최종 산물을 캡슐층전기 (EXC 40F, 서흥캅셀, 한국)롤 이용하여 ¾ 술 (서홍캅셀 , 한국)에 층전하여 캡슬형태의 지연방출제제의 제조를 완료하였다. 실시예 IV-18 : 암로디핀 -피타바타틴 캡술제 제조 (¾¾ -정제)
표 IV-3에 기계된 성분과 함량으로, 이하의 방법에 의해 슐제를 제조하였다.
1) 선방출성 구획 (피타바스타틴 속방출 펠 ¾)의 제조
슈가 시드 (Sugar sphere)를 유동층 과립기 (GPCG1 : Watt)에 투입한 뒤 따로 물에 히드록시프로필셸를로오스와 피타바스타틴 칼슘을 용해시킨 결합액을 분무하여 피 타바스타틴 함유 ¾¾을 형성, 건조하였다 .
2) 지연방출성 구획 (암로디핀 지연방출 정제 )의 제 S
암로디핀 베실레이트, 미결정셀를로오스, 전호화전분을 35호체로 사과하고 더블콘 믹서로 5분간 흔합하여 흔합물을 제조하였다, 따로 히드록시프로필쎌를로오스롤 정 제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건 :가 끝나면 다시 18호 체로 정 림한다. 정립물에 35호체로 체과한 스테아르산 마그네슘을 투입하여 , 4 분 간 흔합한 후, 혼합물을 직경 5讓편치가 장착된 로타리 타정기 (MRC— 37 : 세종)로 타정하여 정제를 제조하였다. 따로 아크 ¾-이즈를 정제수에 용해 및 분산시킨 코¾ 액을 조제하여 위의 암로디핀 정제를 하이코터 (SFC-30N, 세종 기계 , 한국)로서 코
팅층을 형성하여 암로디핀 점제 제조를 완료하였다,
3) 후흔합, 타정 및 코팅
상기 1)과 2)의 최종 산물을 캡술층전기를 이용하여 캡슐 (서흥칼셀 , 한국)에 층전 하여 캡슬형태의 시간차 방출 제제의 제조를 완료하였다. 실시예 IV-19 : 암로디핀 -피타바타틴 캡슬제 제조 (정제- ¾¾)
표 IVᅳ 3에 기재된 성분과 함량으로, 이하의 방법에 외해 캡술제를 제조하였다.
1) 선방출성 구획 (괴타바스타틴 속방출 정제)의 제조
피타바스타틴 칼습, 메타규산알루민산마그네슴, 미결정셀를로오스, 옥수수전분을 35호체로 사과하고 더불콘믹서로 5분간 흔합하여 흔함물을 제조하였다. 따로 히드 특시프로필셀를로오스를 정제수에 녹여 결할액으로 하여 연합 , 제립 및 건조하였 다, 건조가 끝나면 다시 18호체로 정립한다. 정림물에 전분 글리콜산 나트륨 및 스 테아르산 마그네슘을 투입하여, 4분간 흔합한 후, 직경 5薩편치가 장착된 로타리 타정기 (MSC-37, 세종)로 타정을 하여 정제를 제조하였다.
2) 지연방출성 구획 (HMG~CoA 환원 효소 억제제외 지연성 속방층 ¾¾)의 제조 슈가 시드 (Sugarsphere)를 유동층 과립기 (GPCG1 : Glatt)에 투입한 뒤 , 따로 물에 풀리비닐피를리돈, S-암로디핀 베실레이트를 용해시킨 결합액을 분무하여 암로디핀 함유 ¾¾을 형성 , 건조하였다. 다시 상기의 펠 95에 유드라짓 RS30D(Evonik Degussa, Germany) 및 트리에털시트레이트 (Vertel lus , England)를 염화메 ¾렌에 늑 인 액을 분무하여 암로디핀 지연성 펠렛을 제 S하였다.
3) 후흔합, 타정 및 코팅
상기 1)과 2)의 산물을 ¾슬층전기를 이용하여 캡술 (서흥캅셀, 한국)에 층전하여 캡술형태의 시간차 방출 제제의 제조를 완료하였다, 실시예 IV-20 : 암로디핀 -피타바타틴 캡슬제 제조 (정제-정제)
표 IV-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다.
1) 선방출성 구획 (피타바스타틴 속방출 정제)의 제조
피타바스타된 칼슴, 메타규산알루민산마그네슴, 옥수수전분을 달아 35호체로 사과
하고 더블콘믹서에서 5분간 흔합하여 흔합물을 제조하였다, 따로 히드특시프로필셀 를로오스를 정제수에 녹여 결합액을 제조하고 이롤 주성분 흔합물과 함께 연합하였 다. 연함이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60°C에서 건조한다. 건조가 끝나면 다시 18호체로 정립하였다. 정립물에 35호체로 체과한 전분 글리콜산 나트 Ϋ과 스테아르산 마그네슴을 투입하여, 4 분간 흔합한 후 직경 5 ram 펀치가 장착된 로타리 타정기 (M¾>37, 세종기계, 한국)로 타 정하여, 피다바스타틴 정제 제조를 완료하였다.
2) 지연방출성 구획 (암로디핀 지연성 정제)의 제 3:
S-암로디핀 떼실레이트, 미결정샐롤로오스, 무수인산칼슴을 35호체로 사과하고 더 블콘믹서로 5분간 흔합하여 흔합물을 제 2:하였다, 따로 콜리코트 SR30D을 결합액으 로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18호체로 정 립한다. 정 립 물에 35호체로 체과한 스테아르산 마그네슴을 투입하여 , 4 분간 흔합한 후 직경 5 mm 편치가 장착된 로타리 타정기 (M 37 : 세종)로 타정하여 정제를 제조하였다ᅳ 따로 폴리 (메타크릴레이트, 메¾메타크릴레이트)공중합체 (Coiorcon, USA)를 정제수 에 용해 및 분산시킨 코팅액을 조제하여 위의 암로디핀 정제를 하이코터 (SPC-30N, 세종 기계 ᅳ 한국)로서 코팅층을 형성하여 암로디핀 정제 제 ;를 완료하였다.
3) 후흔합, 타정 및 코텅
상기 1)과 2)의 산물을 캡술층전기를 이용하여 캡술 (서홍갑¾, 한국)에 층전하여 캡슬형태의 제어 방출 제제의 제조를 완료하였다. 실시예 IV-21 : 암로디핀 -피타바타틴 캡슬제 제조 (과립—과립 )
표 IV-3에 기채된 성분과 함량으로, 이하의 방뜁에 의해 캡술제를 제조하였다. 1) 선방출성 구획 (피타바스타틴 속방층 과립)의 제조
피타바스타틴 칼슘, 메타규산알루민산마그네슴, 유당수화물을 달아 35호체로 사과 하고, 더블콘믹서에서 5분간 흔합하여 흔합물을 제조하였다. 따로 히드록시프로필 샐롤로오스를 정제수에 녹여 결합액을 제 2:하고 이를 주성분 흔합물과 함께. 연합하 였다. 연합이 끝나면 20호체로 오실테이터를 이용하여 제립하고 이를 은수 건조기 를 이용하여 6(TC에서 건조한다, 건조가 끝나면 다시 18호체로 정립하였다.
2) 지연방출성 구획 (암로디핀 지연성 과립)의 제조
암로디핀 베실레이트 , 미결정셀를로오스, 디-만니를을 35호체로 사과하고 더볼콘믹 서로 5분간 흔합하여 혼합물을 제조하였다. 콜리코트 SR30D 넣어 주성분 혼합몰과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 건조하였다. 건조가 끝나면 다시 18호체로 정립하였다.
3) 후흔합, 타정 및 코팅
상기 1)과 2)의 산물을 더블콘믹서로 흔합하였다. 흔합물에 전분 ¾리콜산 나트륨 을 투입하고 더블콘믹서로 흔합하였다. 다시 흔합물에 스테아르산 마그네슴을 넣어 최종 흔합하였다. 최종 흔합된 혼합물을 분말공급기에 투입하고 캡슬충전기를 이용 하여 ¾술 (서흥캅셀, 한국)에 충전하여 캡슐형태의 시간차 방출제제의 제 를 완료 하였다. 실시예 IV-22 : 암로디판 -피타바타틴 캡술제 제 (과립—펠 ¾)
표 IV-3에 기계된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다 .
1) 선방출성 구획 (피타바스타틴 속방층 과립 )의 제조
피타바스타린 칼슴, 메타규산알루민산마그네슘, 미결정셀를로오스, 전호화전분을 달아 35호체로 사과하고 더블콘믹서에서 5분간 흔합하여 흔합물을 제조하였다. 따 로 히드록시프로필셸 *로오스 * 정제수에 녹여 결합액을 제조하고 이롤 주성분 혼 합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터 * 이용하여 제립하고 이를 은수 건조기를 이용하여 6(rc에서 건조한다. 건조가 끝나면 다시 18호체로 정 림하였다 .
2) 지연방출성 구획 (암로디¾ 지연성 펠렛)의 제조
슈가 시드 (Sugar sphere)를 유동층 과립기 (GPCG1 : Glatt)에 루입한 뒤 , 따로 정제 수에 폴리비닐피를리돈 및 암로디핀 베실레이트를 용해시킨 결함액을 분무하여 암 로디핀 함유 ¾¾을 형성 , 건조하였다. 다시 상기의 펠렛에 유드라짓 RS30D와 트리 에틸시트레이트를 염화쩨 ¾렌에 녹인 액을 분무하여 암토디핀 지연성 펠렛을 제조 하였다.
3) 후흔합, 타정 및 코팅
상기 1)과 2)의 산물을 더블콘믹서로 흔합하였다. 흔합물에 전분글리콜산나트륨을 투입하고 더블콘믹서로 혼합하였다. 다시 흔합물에 스테아르산 마그네슴을 넣어 최 종 흔합하였다. 최종 흔합된 흔합물을 분말공급기애 루입하고 캡술층전기를 이용하 여 캡술 (서홍캅셀, 한국)에 층전하여 캡술형태의 시간차 방출 제제의 제조를 완료 하였다. 실시예 IV-23 : 암로디핀 -피타바타틴 캡술제 제조 (과립-정제)
표 IV-3에 기쳬된 성분과 함량으로 , 이하의 방법에 의해 ¾슬제를 제조하였다.
1) 선방출성 구획 (피타바스타틴 속방출 과립)의 제조
피타바스타틴 칼슘, 메타규산알루민산마그네슴, 유당수화물, 옥수수전분을 달아 35 호체로 사과하고, 더블콘믹서에서 5분간 흔합하여 흔합물을 제조하였다. 흔합물에 정제수를 넣어 연합하였다. 연합이 끝나면 18호체로 오실레이터롤 이용하여 제림하 고 이를 은수 건조기를 이용하여 60°C에서 건조한다. 건조가 끝나면 다시 20호체로 정 립하였다ᅵ. 정립물에 전분 글리콜산나트튬을 투업하고 더불콘믹서로 흔합하였다. 다시 혼합물에 스테아르산 마그네슴을 넣어 최종 흔합하였다.
2) 지연방출성 구획 (암로디핀 지연방출 정제)의 제조
암로디핀 베실레이트, 무수인산칼슘, 전호화전분을 35호체로 사과하고 더블콘믹서 로 5분간 흔합하여 흔합물을 제조하였다. 따로 히드록시프로필샐를로오스를 정제수 에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18호체로 정림한다. 정 립물에 35호체로 체과한 스테아르산 마그네슴을 투입하여 , 4 분간 흔 합한 후 흔합물을 직경 5 讓 편치가 장착된 로타리 타정기 (MRC-37 : 세종)로 타정 하여 정제를 제조하였다. 따로 아크 ¾-이즈를 정제수에 용해 및 분산시 ¾ 코팅 액을 조제하여 위의 암로디 ¾ 정제를 하이코터 (SFC-30N, 세종 기계, 한국)로서 코팅층을 형성하여 암로디핀 정제 제조를 완료하였다
3) 후혼합, 타정 및 코¾
상기 1)과 2)의 산물을 캡술층전기 » 이용하여 캡슐 (서홍캅샐, 한국)에 층전하여슬형태의 시간차 방출 제제의 제조를 완료하였다 실시예 IV-24 : 암로디핀-피타바타 ¾ 캡술제 제 S (정제-과립)
표 IV-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제롤 제조하였다.
1) 선방출성 구획 (피타바스타틴 속방출 정제)의 제조
피타바스타틴 칼슴, 메타규산알루민산마그네슘, 미결정셸를로오스, 전호화전분을 달아 35호체로 사과하고 더블콘믹서에서 5분간 흔합하여 흔합물을 제조하였다. 따 로 히드록시프로필셀를로오스을 정제수에 녹여 결합액을 제조하고 이롤 주성분 흔 합물과 함께 연합하였다. 연합이 끝나면 20호훼로 오실레이터를 이용하여 제립하고 이를 은수 건조기롤 이용하여 60°C에서 건조한다. 건조가 끝나면 다시 18호체로 정 립하였다. 정립물에 35호체로 체과한 저치환도히드특시프로필 ¾를로오스와 스테아 르산 마그네슘을 투입하여 , 4 분간 흔합한 후, 흔합물을 직경 5画편치가 장착된 로타리 타정기 (MRC— 37, 세종, 한국)로 타정하여 피타바스타틴 정제 제조를 완료하 였다.
2) 지연방출성 구획 (암로디핀 지연방출성 과립 )의 제조
암로디핀 베실테이트, 미결정샐를로오스, 무수인산수소칼슴 및 히드록시프로필메틸 셀를로오스를 35호체로 사과하고 더블콘먹서로 5분간 흔합하여 흔합물을 제조하였 다. 정제수를 넣어 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체로 오 실레이터를 이용하여 제립하고, 이를 은수 건조기롤 이용하여 건조하였다. 건조가 끝나면 다시 18호체로 점 립하였다. 정 립물에 스테아르산 마그네슴을 넣어 최종흔합 하였으며, 암로디핀 지연방출성 과립을 제조하였다.
3) 후혼합, 타정 및 코팅
상기 1)과 2)의 산물을 캡슬층전기를 이용하여 캡슬 (서흥캅셀, 한국)에 층전하여 캡슬형태의 시간차 방출 제제의 제조를 완료하였다. 실시예 IV-25 : 피타바스타 ¾—암로디핀 떼실레이트 블리스터 포장키트 제조 이하의 방범에 의해피타바스타틴- 암로디 ¾ 베실레이트 블리스터 포장키트를 제조 하였다.
실시 예 IV-7의 1)에서 제조한 피타바스타틴 칼슴 선방출 과립과 실시예 IV— 7 의 2) 에서 제조한 암로디핀 베실레이트 지연 방출 과립 각각을 로타리 타정기 (MRC— 33:세 종기계 , 한국)를 사용하여 타정하여 각각의 정제를 제 :한 후, 블리스터 포장기 (Minister A, 흥아엔지니어 ¾ )를 이용하여 블리스터 포장용기 (은박, 동일양행 ,
PVDC, 전민산업 )에 각각의 정제를 동시복용 가능하도록 포장하여 블리스터 포장키 트를 제조하였다.
[표 IV-l]


[표 IV-2]
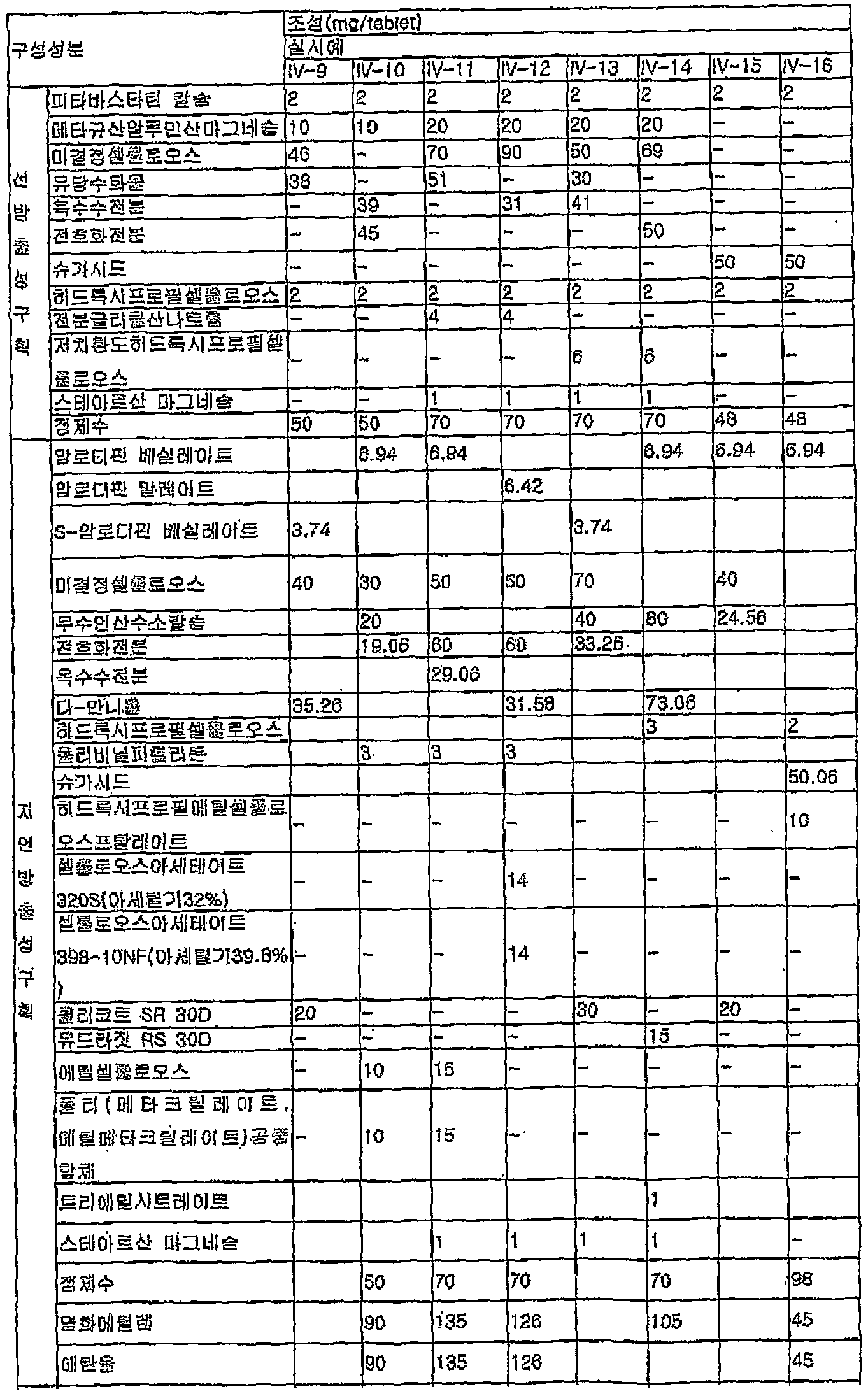

[표 IV-33


표 V-1에 기채된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출 과립)제조
로슈바스타된 칼슘 (MSN, INDIA),삼염기칼슴인산염 , 미결정샐를로오스 (AvicelPHlOl, FMC Biopolymer, USA) , 유당수화물 (DMV, Germany) , 전호화전분 (Starch 1500G, Colorcon, USA)을 35호체로 사과하고, 더블콘믹서 (다산과마텍 , 한국)을 이용하여 실은에서 5분간 흔합하여 혼합불을 제조하였다, 별도로 히드록시프로필샐를로오스 (HPC-L, Nippon Soda, Japan)를 정제수에 녹여 결합액을 제 S하고 이를 주성분 흔 합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기 (IHf— C, 삼공사, Japan)를 이용하여 6013에서 건조 시킨 후 20호 체로 정 립하였다. 정립물에 크로스포비돈을 흔합하고, 스테아르산 마그네슴 (Nitika Chemical , India)을 넣어 더블콘믹서로 최종 흔합하여 표제의 선방출성 구획을 제 ≥하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 정제)제조
암로디핀 베실레이트, 머결정셀롤로오스, 무수인산수소칼슘, 전호화전분 (Starch 1500G, Colorcon, USA)을 35호체로 사과하고 더블콘믹서로 5분간 실은에서 흔합하 여 흔합물을 제조하였다. 별도로 히드록시프로필셀를로오스를 정제수에 녹여 결 합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제림하고 이를 은수 건조기 (H-W—C, 삼공사, Japan)를 이용 하여 에서 건조 시킨 후 20호체로 점립하였다. 별도로 ¾를로오스아세테이트 320S (아세탈기 32 %) , 셸를로오스아세테이트 398-10 F (아세탈기 39.8%)를 에탄을과 염화메틸렌 혼액에 녹인 액을 조제하여 위의 정립물과 함께 유동층 과립 코팅기 (GPCG-1 ; Glatt , Germany)에 넣고 코팅하였다, 코팅 완료 후, 스테아르산 마그네 슘을 투입하여 4 분간 흔합하고, 직경 5舰펀치가 장착된 로타리 타정기 (MRC-30, 세종파마텍 , 한국)로 타정을 하여 표제의 지연방출성 구획인 핵정을 제조하였다.
(3) 타정 및 코팅
11 mm 편치가 장착된 유핵정타정기 (RUD-1: Ri l ian, 독일)로 상기 (1)의 로슈바스타 ¾ 칼슴 선방출 과립을 외층으로 사용하고, 상기 (2)의 암로디핀 폐실레이트 지연 방출성 정제를 핵정으로하여 타정하였다. 별도로 히드록시프로필메 ¾¾를로오스 2910(S in-ets , Japan) , 풀리에 ¾렌글리콜 6,000(BASF, Germany) , 탈크 (Luzenac, France), 산화티탄 (Tioside Americas , USA)을 에탄을과 정제수에 용해 및 분산시 ¾ 코팅액을 조제하여 상기 정제를 하이코터 (SFC-30N, 세종 기계 , 한국)로서 필晉 코 팅층을 형성하여 유핵정 형태의 정제를 제조하였다. 실시예 V— 2 : 암로디핀-로슈바스타틴 유핵정 제조
표 V-1에 기재된 성분과 함량으로, 이하의 방뜁에 의해 유핵정을 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출 과립)제조
로슈바스타틴 칼슴, 삼염기칼슘인산염, 미결정 ¾를로오스, 유당수화물, 전호화전분 을 달아 35호체로 사과하고, 더블콘믹서에서 실은에서 5분간 흔합하여 흔합물을 제 조하였다. 별도로 히드록시프로필셸롤로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터롤 이 용하여 제림하고 이 » 온수 건조기를 이용하여 6C C에서 건조시킨 후 20호체로 정 립하였다. 정 립물에 크로스포비돈을 흔합하고, 스테아르산 마그네슘을 넣어 더블콘 먹서로 최종 흔합하여 표제의 선방출성 구획을 제 하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 정제)제조
암로디핀 베실레이트, 미결정쎌를로오스, 무수인산수소칼습 , 전호화전분을 35호체 로 사과하고 더블콘믹서로 실은에서 5분간 흔합하여 흔합물을 제조하였다. 별도로 히드록시프로필셀롤로오스를 정제수에 녹여 결합액으로 하여 연합 및 제립하고 이 를 은수 건조기를 이용하여 에서 건조시 ¾ 후 20호체로 정 림하였다. 별도로 유 드라짓 RS30D(Evomk Degussa, Germany) 및 트리에 ¾시트레이 CVertellus, England)를 염화메털렌에 녹인 액을 조제하고 위의 정립물을 유동층 과림 코팅기에 넣고 코팅하였다. 코¾ 완료 후, 스테아르산 마그네슘을 투입하여 4 분간 흔합하 고, 직경 5舰 편치가 장착된 토타리 타정기 (MRCᅳ 30, 세종파마텍 , 한국)로 타정을 하여 표제의 지연방출성 구획인 핵정을 제≤하였다,
(3) 타정 및 코팅
실시 예 V-1의 (3)과 동일 방법으로 타정 및 코팅하여 유핵정 형태의 정제를 제조하 였다. 실시예 V— 3: 암로디 ¾ -로슈바스타 ¾ 유핵정 제조
표 V-1에 기재된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다,
(1) 선방출성 구획 (로슈바스타 ¾ 선방출 과립)제조
로슈바스타틴 칼슘, 삼염기칼슘인산염, 미결정셀를로오스, 유당수화물 , 전호화전분 을 35호체로 사과하고 더블콘믹서에서 실은에서 5분간 혼합하여 흔합물을 제조하 였다. 별도로 히드톡시프로필¾를로오스를 정제수에 녹여 결합액을 제조하고 이롤 주성분 흔합물과 함깨 연합하였다. 연합이 끝나면 18호체로 오실테이터를 이용하여 제립하고 이를 은수 건조기 * 이용하여 60 2에서 건조시켰다. 건조가 끝나면 다시 20호체로 정 립한 후 정 립물에 크로스포비돈을 흔합하고 , 스테아르산 마그네슴을 넣 어 더블콘믹서로 최종 흔합하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 정제 )제조
암로디핀 베실테이 S , 미결정셀를로오스 , 디-만니톨 (Roquette, USA)을 35호체로 사 과하고 더블콘믹서로 5분간 혼합하여 흔합물을 제조하였다. 별도로 풀리비닐필로리 돈 (Kol l idcm 30, BASF, Germany)을 정제수에 녹여 결합액으로 하여 연합 및 제립하 고 이를 은수 건조기를 이용하여 에서 건조시킨 후 20호체로 정립하였다. 별도 로 에 ¾셀를로오스 (HERCULES, USA) , *리 (메타크¾레이트 , 메틸메타크릴레이트)공 증합체 (Evonik degussa, USA)를 에탄을과 염화메틸렌 흔액에 녹인 액을 조제하여 위의 정 립물을 유동층 과립 코팅기에 넣고 코팅하였다. 코팅 완료 후 , 스테아르산 마그네슴을 투입하여 4 분간 흔합하고, 직경 5 mm 펀치가 장착된 로타리 타정기로 타정을 하여 표제의 지연방출성 구획을 제조하였다,
(3) 타정 및 코팅
실시 예 V-1의 (3)과 동일 방법으로 타정 및 코팅하여 유핵정 형태의 정제를 제조하 였다.
실시예 V-4: 암로디핀-로슈바스타틴 유핵정 제조
표 V-1에 기재된 성분과 ᅳ함량으로, 이하의 방법에 의해 유핵정을 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 과립 )의 제조
로슈바스타틴 칼슘, 삼염기칼슴인산염 , 미결정쎌 ·로오스, 유당수화물, 전호화전분 을 35호체로 사과하고, 더블콘믹서에서 5분간 실은에서 흔합하여 흔합물을 제조하 였다. 별도로 히드록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 상기 주성분 흔합물과 함께 실은에서 연합하였다. 연합이 끝나면 18호체로 오실레 이터를 이용하여 제립하고 이롤 은수 건조기를 이용하여 60 ^에서 건조시킨 후 20 호체로 정립하였다. 정립물에 전분글리콜산나트륨 (DMV, Germany) , 스떼아르산 마그 네슴을 넣고 더블콘믹서로 실온에서' 최종 흔합하여 표제의 선방출성 구획을 제조하 였다.
(2) 지연방출성 구획 (암로디편 지연방출성 정제)의 제조
암로디핀 말레이트 (대희화학, 한국) 미결정셀를로오스, 무수인산수소칼슘, 디 -만니 를을 35호체로 사과하고 더블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였 다. 별도로 히드록시프로필샐를로오스를 정제수에 녹여 결합액으로 제조한 후, 이 에 주성분의 흔합물과 연합 및 제립하고 이를 은수 건조기를 이용하여 60 C에서 건 조시킨 후 20호체로 정립하였다. 정립물에 35호체로 체과한 스테아르산 마그네슘을 투입 후 4 분간 흔합하여 HMG-CoA 환원호소억제제 지연용출층 과립을 제조하였다. HMG-CoA 환원호소 억제제 지연성 속용출 과립을 직경 5 mm 편치가 장착된 로타리 타정기로 타정하여 핵정을 제 하였다. 별도로 아크릴 -이즈 (메타크¾산 공중합체 type C, 탈크, PEG, 콜로이달실리콘다이옥사이드, 중탄산나트晉, SLS, Colorcon, USA)를 정제수에 용해 몇 분산시 ¾ 코팅 액을 제하여 위의 암로디핀 정제를 하이 코터로서 코팅층을 형성하여 표제의 지연방출성 구획인 암로디핀 정제를 제조하였 다. '-
(3) 타정 및 코팅
질시예 V-1의 (3)과 등일 방법으로 타정 및 코팅하여 유핵정 형태의 정제를 제조하 였다. 실시예 V-5: 암로디 ¾-로슈바스타린 유핵정 제조
표 V-l에 기재된 성분과 함량으로, 이하의 방범에 의해 유핵정을 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성과립 )의 제조
로슈바스타틴 칼슴, 삼염기칼슴인산염, 미결정셀를로오스, 유당수화물, 옥수수전분
(Colorcon, USA)을 달아 35호체로 사과하고 , 더블콘믹서에서 5분간 실은에서 흔합 하여 흔합물을 제조하였다, 별도로 히드록시프로필셀를로오스를 정제수에 녹여 결 합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합 후 18호체로 오실레 이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 6( C에서 건조시켰다. 건 조가 끝나면 다시 20호체로 정 립하였다. 정림물에 크로스포비돈을 혼합하고, 스테 아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하여 표제의 선방출성 구획을 제조 하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 ^정제)의 제조
암로디편 말레이트, 미결정셸롤로오스을 35호체로 사과하고 더블콘믹서로 5분간 실 은에서 혼합하여 흔합물을 제조하였다. 흔합물을 더블콘믹서에 투입하고 콜리코트 SR30D(BASF, Germany)를 가하여 연합한 다음 18호체로 오실레이터롤 이용하여 제립 하고, 이를 은수 건조기를 이용하여 60 °C에서 건조한 후 20호체로 정림하였다. ¾ 립물에 35호체로 체과한 스테아 ¾산 마그네슴을 투입하고, 4 분간 흔합한 후, 직경 5 腿 편치가 장착된 로타리 타정기로 타정하여 핵정 ¾ 제조하였다. 별도로 히드특 시프로필데필셀를로오스프탈레이트 (Shin-etsu, Japan)를 에탄을과 염화메틸렌 흔액 에 용해 및 분산시켜 코팅 액을 조제하였다. 이 코팀액을 이용하여 암로디핀 핵정에 하이코터로 코팅층을 형성시켜 표제의 지연방출성 구획인 암로디핀 정제를 제조하 였다.
(3) 타정 및 코팅
실시예 V— 1외 (3)과 등일 방법으로 타정 및 코팅하여 유핵정 형태의 정제를 제조하 였다. 실시예 V-6: 암로디핀—로슈바스타틴 유핵정 제조
표 V-1에 기재된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다. (1) 선방출성 구획 (로슈바스타 ¾ 선방출성 과립 )의 제조
로슈바스타틴 칼슴, 삼염기칼숨인산염 , 미결정샐를로오스, 유당수화물, 옥수수전분
을 달아 35호체로 사과하고, 더블콘먹서를 이용하여 실은에서 5분간 흔합하여 흔합 물을 제조하였다. 별도로 히드록시프로필샐를로오스를 정제수에 녹여 결합액을 제 조하고 이를 주성분 흔합물과 함깨 연합한 후 18호체로 오실레이터를 이용하여 제 립하고 이를 온수 건조기를 이용하여 6( C에서 건조시켰다. 건조가 끝나면 다시 20 호체로 정립하였다, 정 립툴에 전분 글리툴산 나트쿰 (DMV, Germany) , 스테아르산 마 그네슴을 넣어 더블콘믹서로 최종 혼합하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 정제)의 제조
암로디핀 베실레이트, 전호화전분, 옥수수전분을 35호체로 사과하고 더불콘믹서로 실은에서 5분간 흔합하여 흔합물을 제조하였다. 별도로 히드록시프로필메틸셀를로 오스를 정제수에 녹여 결함액을 제조하고 이에 주성분 흔합물을 넣고 연합 및 제립 하고 이를 은수 건조기를 이용하여 60°C에서 건조시킨 후 20호체로 정 립하였다. 별 도로 히드록시프로필데틸 ¾를로오스프탈레이트를 에탄을과 염화메틸렌 흔액에 녹여 코팅액을 조제하고 이와 정립물을 유등층 과림 코팅기 (GPCG-1 ; Glatt , Germany)에 넣고 상기 코팅액으로 코령하였다. 코팅 완료 후, 스테아린산 마그네슘을 투입하 여 , 4 분간 혼합한 후 직경 5 mm 편치가 장착된 로타리 타정기로 타정을 하여 표제 의 지연방출성 구획인 핵정을 제조하였다.
(3) 타정 및 코팅
실시예 v-i의 (3)과 등일 방법으로 타정 및 코팅하여 유핵정 형태의 정제를 제 2:하였다. 실시예 V-7: 암로디핀-로슈바스타틴 2상 매트릭스 정제 제조
표 V-1에 기재된 성분과 함량으로, 이하의 방법에 의해 2상 매트릭스 점제를 제조 하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 과립)의 제조
로슈바스타틴 칼슴ᅳ' 삼염기칼슴인산염 , 유당수화물, 전호화전분을 달아 35호체로 사과하고 더블콘믹서로 실은에서 5분간 흔합하여 혼합물을 제조하였다. 별도로 히 드록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함깨 연합하였다. 연합이 끝나면 20호체 오실레이터를 이용하여 제립하고 이를 온 수 건조기를 이용하여 60°C에서 건조 후 다시 18호체로 정립하여 표제의 선방출 구 획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출 과립)의 제조
암로디핀 베실레이트 , 머결정쎌롤로오스, 무소인산수소칼슴을 35호체로 사과하고 더블콘믹서로 실은에서 5분간 혼합하여 혼합물을 제조하였다. 별도로 히드록시프로 필¾를로오스를 정제수에 녹여 결합액으로하고 상기 주성분 흔합물과 연합 및 제립 하고 이를 은수 건조기를 이용하여 6C C에서 건 S시킨 후 20호체로 정 립하였다. 별 도로 셀를로오스아세테이트 320S (아세탈기 32 %) 및 셀를로오스아세테이트 398- 10NF (아세탈기 39.8%)를 에탄을과 염화메틸렌 흔액에 녹여 코팅 액을 제조한 후 이 롤 이용하여 위 정 림물을 유동층 과립 코팅 기 (GPCG-1: Glatt , Geratnny)에 넣고 코 팅하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 산물을 더블콘믹서에 넣고 실은에서 흔합하였다. 이 흔합물에 전 분 글리콜산 나트륨,, 및 스테아르산 마그네슴을 넣고 더블콘믹서로 최종 흔합하였 다. 최종 흔합물을 직경 10麵 핀치가 장착된 로타리 타정기를 사용하여 파정하였 다. 별도로 히드록시프로필메 ¾셀를로오스 2910, 풀리에틸렌글리콜 6,000, 탈크 , 및 산화티탄을 에탄을과 정제수에 용해 및 분산시켜 코팅 액을 조제하고, 위의 정제 에 하이코터 (SFC— 30N:세종 기계 , 한국)를 이용하여 필름코팅층을 형성하여 2상 메 : 릭스 정제를 제조하였다. 실시예 V-8: 암로디핀-로슈바스타된 2상 매트릭스 정제 제조
표 V-1에 기계된 성분과 함량으로, 이하의 방법에 의해 2상 메트릭스 정제를 제 : 하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 과립 )의 제조
로슈바스타틴 칼슘, 삼염기칼슘인산염, 미결정 ·¾*로오스을 35호체로 사과하고 더 블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도로 히드록시프로필 샐를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하 였다 . 연합이 끝나면 20호체 오실레이터를 이용하여 제림하고 이를 은수 건조기를 이용하여 6(TC에서 건조 후 다시 18호체로 정립하여 표제의 선방출성 구획을 제조 하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 과립 )의 제조
암로디핀 말레이트, 미결정셀를로오스, 전호화전분을 35호체로 사과하고 더블콘믹 서로 5분간 실온에서 흔합하여 흔합물을 제조하였다. 별도로 폴리비닐피를리돈을 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조물을 유동층 코팅 기에 넣고, 별도로 유드라짓 RS30D 및 트리에틸시트레이트를 염화메틸렌에 녹인 액 을 조제하여 위의 조립물을 유동층 과림 코팅기에 넣고 코팅하여 표제의 지연방출 성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅 、
실시예 V-7의 (3)과 등일 방법으로 후혼합, 타정 및 코팅하여 2상 매트릭스 형태의 정제를 제조하였다. 실시예 V-9: (S)-암로디핀—로슈바스타틴 2상 매트릭스 정제 제조
표 V-2에 기재된 성분과 함량으로, 이하의 방법에 의해 2상 매트릭스 정제를 제조 하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출 과립)의 제조
로슈바스타틴 칼슘, 삼염기칼슘인산염 , 더결정셀 ¾로오스, 유당수화물을 35호체로 사과하고 더블콘믹서로 5분간 실온에서 흔합하여 흔합물을 제조하였다. 별도로 히 드록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체 오실레이터롤 이용하여 제립하고 이를 은 수 건조기를 이용하여 6CTC에서 건조 후 다시 18호체로 정립하여 표제의 선방출성 구획을 제조하였다,
(2) 지연방출성 구획 (암로디핀 지연방출성 과립〉의 제조
S-암로디핀 베실레이트 (Glochem, INDIA) , 미결점셀를로오스, 디-만니를을 35호체로 사과하고 더블콘믹서로 5분간 실은에서 혼합하여 흔합물을 제조하였다, 혼합물을 고속흔합기에 투업하고 콜리코트 SR30D를 가하여 연합한 다음 20호체로 오실레이터 를 이용하여 제립하고, 이를 은수 건조기를 이용하여 60 °C에서 건조한 후 다시 18 호체로 정립하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
실시 예 V-7의 (3)과 동일 방법으로 후흔합, 타정 및 코팅하여 2상 매트릭스 형태의 정제를 제조하였다. ' 실시예 V-10: 암로디핀-로슈바스타틴 2상 매트릭스 정제 제조
표 V-2에 기재된 성분과 함량으로, 이하의 방법에 의해 2상 매트릭스 정제를 제조 하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 과립 )의 제조
로슈바스타틴 칼슘, 삼염기칼슴인산염 , 옥수수전분, 전호화전분을 달아 35호체로 사과하고 더블콘믹서로 5분간 실온에서 혼합하여 흔합물을 제조하였다. 별도로 히 드록시프로필셀를로오스를 정제수에 늑여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체 오실레이터를 이용하여 제립하고 이를 은 수 건조기를 이용하여 6(TC에서 건조 투 다시 18호체로 정 립하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 과립)의 제조
암로디핀 베실레이트, 미결정 ¾를로오스, 무수인산수소칼슘, 전호화전분을 35호체 로 사과하고 더블콘믹서로 5분간 실은에서 흔합하여 흔함물을 제조하였다. 별도로 폴리비닐피를리돈을 정제수에 녹여 결합액을 제조하고, 주성분 흔합물과 함께 합 및 제립하고 이를 은수 건조기를 이용하여 60¾에서 건 시킨 후 20호체로 정립하 였다. 정립물을 유등충 코팅기에 넣고, 별도로 에틸셀를로오스 (HERCULES, USA) , 폴 리 (메타크릴레이트 , 메틸메타크릴레이트)공증합체를 에탄을과 염화메틸렌흔액에 녹 인 코팅 액을 조제하고, 이를 코팅액으로 사용하여 위의 조립물을 유동층 파립 코팅 기 (GPCG-1 ; Glatt , Germany)로 코팅하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
실시예 V-7의 (3)과 동일 방법으로 후흔합, 타정 및 코팅하여 2상 매트릭스 형 태의 정제를 제조하였다. 질시예 V-11: 암로디핀-로슈바스타틴 다충정 제조
표 V-2에 기재된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다. (1) 선방출성 구획 (로슈바스타틴 선방출성 과립 )의 제조
로슈바스타틴 칼슴, 삼염기칼슘인산염 , 더결정셀를로오스 , 유당수화불을 달아 35호 체로 사과하고 더블콘믹서로 5분간 실온에서 흔합하여 흔합물을 제조하였다. 별도 로 히드록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔 합물과 함께 연합하였다. 연합이 끝나면 20호쳬로 오실레이터롤 이용하여 제립하고 이를 은수 건조기를 이용하여 60t;에서 건조시킨 후 18호체로 정 림하였다. 이 후 정립물과 전분 글리콘산 나트륨을 흔합하고 , 스테아르산 마그네슴을 넣어 더블콘믹 서로 최종 흔합하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 과립)의 제조
암로디 ¾ 베실레이트, 미결정쉘를로오스, 전호화전분, 육수수전분을 35호체로 사과 하고 더블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도로 플리비 닐피를리돈을 정제수에 녹여 결합액을 제조하고 연합 및 제립하고 이를 은수 건조 기를 이용하여 60¾에서 건조시킨 후 20호체로 정립하였다. 정팀물을 유동층 코팅 기에 넣고, 별도로 에틸썰를로오스, 폴리 (메타크¾레이트, 메틸메타크 ¾레이트〕공 증합체를 에탄을과 염화메틸렌 흔액에 녹여 코팅 액을 조제하고 , 이 코팅액을 사용 하여 위의 조립물을 유동층 과립 코팅기로 코팅하였다. 코팅 완료 후, 스테아르산 마그네슴을 투입 후 , 4 분간 혼합하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)의 최종 결과물을 다층정 타정기 (MRC-37T: 세종)의 1차 분말공급기에 넣 고 상기 (2)의 최종 결과물을 2차 분말 공급기에 넣어 층간의 흔입을 최소화할 수 있는 조건으로 타정하였다. 별도로, 히드특시프로필메틸셀를로오스 2910, 풀리에릴 렌글리콜 6,000, 탈크 , 및 산화티탄을 에탄을과 정제수에 용해 및 분산시켜 코팅액 을 조제하고 이를 이용하여 위의 정제를 하이코터로 필름코팅층을 형성시켜 표제의 다층정 형태의 정제를 제조하였다. 실시예 V-12: 암로디핀-토슈바스타틴 다층정 제조
표 V-2에 기재된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다. (1) 선방출성 구획 (로슈바스파틴 선방출성 과립)의 제 S
로슈바스타틴 칼슴, 삼염기칼슘인샅염, 미결정셀를로오스, 옥수수전분을 35호체로 사과하고 더블콘믹서로 5분간 실은에서 흔합하여 혼합물을 제조하였다. 별도로 히
드특시프로필 ¾를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용하여 6( C에서 건조한 후 18호체로 정립하였다. 정립물에 전분 글리클산 나트튬을 흔합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합 하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 과림)의' 제조
암로디핀 말레이트, 미결정셀를로오스, 전호화전분, 디ᅳ만니를을 35호체로 사과하 고 더블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도로 폴리비닐 피를리돈을 정제수에 녹여 결합액을 제조한 후 이와 주성분 흔함물을 연합 및 제립 하고 이를 온수 건조기를 이용하여 60'C에서 건조시킨 후 20호체로 정립하였다. 정 립물을 유동층 코팅기에 넣고, 별도로 셸를로오스아세테이트 320S (아세탈기 32 %) , 셀를로오스아세테이트 398-10NF (아세탈기 39.8%)를 에탄을과 염화메틸렌 1: 1 혼액에 녹인 액을 조제하여 위의 조립물을 유동층 과립 코팅기에 넣고 코팅하였다. 코팅 완료 후. 스테아르산 마그네슘을 투입 후, 4 분¾ 흔합하여 표제의 지연방출성 구 획을 제조하였다.
(3) 후흔합, 타정 및 코¾
실시예 V-11의 (3)과 동일 방법으로 후흔함, 타정 및 코팅하여 다층정 형태의 정제 를 제조하였다. 실시예 V-13: (S)-암로디핀-로슈바스타된 다층정 제조
표 V— 2에 기재된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 속방출 과립)의 제조
로슈바스타틴 칼슘, 삼염기칼슘인산염, 미결정셀를로오스, 유당수화물, 육수수전분 을 달아 35호체로 사과하고 더블콘믹서에서 5분간 흔합하여 혼합물을 제조하였다. 별도 S 히드록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하 고 이를 은수 건조기를 이용하여 6013에서 건조시 ¾ 후, 18호체로 정 립하였다. 정 립물에 크로스포비돈을 흔합하고, 스테아르산 마그네슘을 넣어 더블콘믹서로 최종 흔합하여 표제의 선방출성 구획흩 제조하였다.
C2) 지연방출성 구획 (암로디핀 지연방출성 과립)의 제조
S-암로디핀 베실레이트, 미결정셀를로오스, 무수인산수소칼숨, 전호화전분을 35호 체로 사과하고 더블콘믹서로 5분간 실은에서 혼합하여 흔합물을 제조하였다. 흔합 물을 고속흔합기에 루입하고 콜리코트 SR30D를 가하여 연함한 다음 20호체로 오실 레이터를 이용하여 제립하고, 이를 은수 건조기를 이용하여 60 에서 건조한 후 다시 18호체로 정립하였다. 정립물에스테아린산 마그네슴을 투입 후, 4 분간 흔합 하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
실시예 의 (3)과 동일 방범으로 후흔합, 타정 및 코팅하여 다층정 형태의 정제 롤 제조하였다. 실시예 V-14: 암로디핀-로슈바스타틴 다층정 제조
표 V-2에 기계된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다.
(1) 선방출성 구획 (로슈바 ώ타틴 선방출성 과립)의 제조
로슈바스타틴 칼슘, 삼염기칼슴인산염 , 미결정셀를로오스, 전호화전분을 35호체로 사과하고 더블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제 S하였다 .· 별도로 히 드록시프로필젤를로오스를 정제수에 늑여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다ᅳ 이 후 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조 기를 이용하여 60°C에서 시킨 후 18호체로 정립하였다. 정립물에 크로스포비돈을 흔 합하고, 스테아르산 마그네슴을 넣어 더블콘믹서로 최종 흔합하여 , 표제의 선방출 성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 과립)의 제 S
암로디핀 베실레이트, 무수인산수소칼슴, 디-만니를을 35호체로 사과하고 더블콘먹 서로 5분간 실은에서 흔합하여 흔합물을 제 2:하였다. 별도로 히드록시프로필¾를로 오스를 정제수에 녹여 결합액으로 하여 연합, 제립 및 건조하였다. 건조가 끝나면 다시 18호체로 정 립하였다. 정 립물을 유동층 코팅기에 넣고, 별도로 유드라짓 RS30D 및 트리에 ¾시트레이트를 염화메틸렌에 녹인 액을 조제하여 위의 조립물을 유동층 과립 코팅기에 넣고 코팅하였다.' 코팅 완료 후, 스테아르산 마그네슴을 투 입 후, 4 분간 흔합하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
실시예 V— 11의 (3)과 등일 방법으로 후흔합, 타정 및 코팅하여 다층정 형태의 정제 를 제조하였다. 질시예 V-15: 암로디핀—로슈바스타틴 캡술제 제조 (펠렛-과립)
표 V— 2에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제 a하였다.
(1) 선방출성 구획 (로슈바스타틴 ¾¾)의 제조
슈가 시드 (Sugar sphere) (NP Pharmaceutical , France)를 유동층 과립기 (GPCG, Glatt , Germany)에 투입한 뒤, 별도로 정제수에 히드록시프로필셀를로오스와 로슈 바스타틴 칼슴을 응해시 ¾ 결합액을 분무하여 ¾¾을 형성, 건조하여 표제의 선방 출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 과립)의 제조
암로디핀 볘실레이트, 미결정 ¾를로오스, 무수인산수소칼슘을 35호체로 사과하고 더붙콘믹서로 5분간 실온에서 흔합하여 ¾합물을 제조하였다. 콜리코트 SR30D 넣어 주성분 혼합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 건조하한 후 다시 18호체로 정립하여 표제 의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2〉의 최종 결과물을 더블콘믹서로 실은에서 흔합하고 흔합물에 스테아 르산 마그네슴을 넣어 최종 흔합하였다. 최종 흔합된 흔합물을 분말공급기에 투입 하고 캡슬충전기 (SF 40N, 세종기계 , 한국)를 이용하여 캡술 (서흥칼셀, 한국)에 층 전하여 표제의 캡술제를 제조하였다. 실시예 V— 16: 암로디핀-로슈바스타틴 캡술제 제조
표 V-2에 기제된 성분과 함량으로, 이하의 방뜁에 의해 캡술제를 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 펠 ¾)의 제조
슈가 시드를 유동층 과립기에 투입한 뒤 별도로 물에 히드록시프로필 ·€를로오스와 로슈바스타틴 칼슴을 용해시 ¾ 결합액을 분무하고 건조시켜 표제의 선방출성 구획 인 로슈바스타틴 함유 ¾¾을 형성하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 펠렛)의 제조
슈가 시드롤 유동층 과립기에 투업한 뒤 , 별도로 정제수에 히드록시'프로필셀를로오 스 및 암로디편 베실레이트를 용해시킨 결합액을 분무하여 암로디괸 함유 펠¾을
형성 , 건조하였다. 다시 상기의 펠¾에 히드록시프로필메 를로오스프탈레이트
(Shin-etsu, Japan)를 에탄을과 염화메틸렌 흔액에 녹인 액을 분무하여 표제의 지 연방출성 구획인 암로디핀 지연방출 펠렛을 제조하였다.
(3) 후흔합, 타정 및 코팅
공정 (1)과 (2〕의 최종 결과물을 캡술층전기를 이용하여 슬 (서흥캅 ·€, 한국)에 층전하여 ¾술형태의 약제학적 제제의 제조를 완료하였다. 실시예 V-17: 암로디핀-로슈바스타틴 캡술제 제조 (펠탯- 삼투성 정제)
표 V-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡슬제를 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 ¾¾)의 제조
슈가 시드를 유동층 과립기에 투입한 뒤 별도로 정제수에 히드록시프로필셀를로오 스와 로슈바스타틴 칼슴을 용해시킨 결합액을 분무하고 건조시켜 , 표제의 선방출성 구획인 로슈바스타린 함유 펠¾을 제조하였다.
(2) 지연방출성 구획 (암로디핀 삼투성 정제 )의 제조
암로디핀 베실레이트, 미결정 ¾를로오스, 무수인산수소칼슴, 옥수수전분을 35호체 로 사과하고 더블콘믹서로 5분간 실은에서 흔합하여 혼합물을 제조하였다. 별도로 히드록시프로필샐를로오스를 정제수에 녹여 결합액으로 제조하고 , 이를 주성분 흔 합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기롤 이용하여 60¾에서 건조시킨 후 18호체로 정립하였다. 그 정립 물에 염화나트튬 및 스테아르산 마그네슴을 넣고 4분간 흔합하였다. 상기 흔합물을 직경 5 mm. 편치가 장착된 로타리 타정기로 타정을 하여 정제를 제조하였다. 별도로 셀를로오스아세테이트 320S (아세탈기 32 ¾) , 셀를로오스아세테이트 398-10NF (아세 탈기 39.8%)를 에탄을과 염화메틸렌 흔액에 녹여 코팅액을 조제하였다. 이를 이용 하여 상기 정제에 하이코터로 필름코팅층을 형성하여 표제의 지연방출성 구획인 암 로디핀의 삼투성 정제 * 제조하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 최종 결과물을 캡술층전기 (EXC 40F, 서흥캅셸, 한국)를 이용하여 캡술 (서흥캅셀, 한국)에 층전하여 표제의 캡술형태의 지연용출제제를 제조하였다. 실시여 i V-18: 암로디핀-로슈바스타틴 캡술제 제조 (펠 ¾-정제)
표 V-3에 기쟤된 성분과 함량으로, 이하의 방법에 의해 캡슐제를 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 펠렛)의 제조
슈가 시드를 유등층 과립기에 투입한 뒤 별도로 물에 히드록시프로필셀를로오스와 로슈바스타된 칼슘을 용해시킨 결합액을 분무하고 건조시켜 표제의 선방출성 로슈 바스타틴 함유 펠렛을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지 연방출성 점제)의 제조 .
암로디휜 베실레이트, 미결정젤를로오스, 전호화전분을 35호체로 사과하고 더불콘 믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도로 히드록시프로필셸를 로오스를 정제수에 녹여 결합액을 제조한 후 주성분 흔합물과 함께 연합하였다. 연 합이 끝나면 20호체로 오실레이터를 이용하여 제 립하고 이를 온수 건 기를 이용하 여 6CTC에서 건조시킨 후 18호훼로 정 립하였다. 정 립물에 35호체로 체과한 스테아 르산 마그네슘을 투입하여 , 4 분간 흔합한 후, 혼합물; i: 직경 5 随 편치가 장착된 로타리 타정기토 타정하여 정제를 제조하였다. 별도로 아크 ¾ -이 S를 정제수에 용 해 및 분산시킨 코팅 액을 조제하고 이를 이용하여 상기 정제를 하이코터로 코팅층 을 형성시켜 표제의 지연방출성 암로디핀 정제 제조하였다 .
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 최종 결과물을 캡술층전기를 이용하여 캡슐 (서흥캅셀, 한국)에 층전하여 표제의 ¾술제롤 제조하였다. 실시 예 V-19: (S)—암로디 ¾-로슈바스타틴 캡슬제 제조 (정제- ¾¾)
표 V-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다.
(1) 선방출성 구획 (로슈바스타된 선방출 정제)의 제조
토슈바스타틴 칼슘, 삼염기칼슴인산염, 미결정 셀를로오스, 옥수수전분을 35호체로 사과하고 더블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도로 히 드록시프로필쉘를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60°C에서 건조시킨 후 18호체로 정 립하였다. 점립물에 전 분 글리콘산 나트륨 및 스테아르산 마그네슘을 투입하여 , 4분간 흔합한 후, 직경 5 画 편치가 장착된 로타리 타정기로 타정하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀의 지연방출성 ¾¾)의 제조
슈가 시드 (Sugarsphere)를 유동층 과립기에 투입한 뒤 별도로정제수에 플리비닐피
를리돈, Sᅳ암로디핀 베실레이트를 용해시킨 결합액을 분무하여 암로디핀 함유 ¾ 을 형성 , 건조하였다. 다시 상기의 펠¾에 유드라짓 RS30D(Evonik: Degussa, Germany) 및 트리에틸시트레이트 (Vertellus, England)를 염화메 ¾¾에 녹인 액을 분무하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 최종 결과물을 캡술층전기를 이용하여 캡술 (서흥캅¾, 한국)에 층전하여 표제의 캡슐제를 제조하였다. 실시예 V-20: (S)-암로디핀-로슈바스타된 캡슐제 제조 (정제-정제)
표 V-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출성 정제)의 제조
로슈바스타틴 칼슘, 삼염기칼슴인산염 , 옥수수전분을 35호체로 사과하고 더불콘믹 서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도로 히드록시프로필¾를로 오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건조기를 이용 하여 601C에서 건조시킨 후 18호체로 정립하였다. 정립물에 35호체로 체과한 전분 글리콜산 나트튬과 스테아르산 마그네슴을 투입하여 , 4 분간 흔함한 후 직경 5 舰 펀치가 장착된 로타리 타정기로 타정하여, 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연성 정제 )외 제조
S-암로디핀 베실레이트 , 미결정셀를로오스, 무수인산칼슘을 35호체로 사과하고 더 블콘믹서로 5분간 실은에서흔합하여 흔합물을 제조하였다. 별도로 콜리코트 SR30D 을 결합액으로 하여 주성분 흔합물과 함께 연합하였다. 연합이 끝나던 20호체로 오 실레이터를 이용하여 제림하고 이를 온수 건조기를 이용하여 에서 건 ¾시 ¾ 후 18호체로 정립하였다. 정립물에 35호체로 체과한 스테아르산 마그네슘을 투입하여 , 4 분간 혼합한 후 직경 5腿 편치가 장착된 로타리 타정기로 타정하여 정제를 제조 하였다. 별도로 폴리 (메타크릴레이트, 메틸메타크 ¾레이트)공중합체 (Colorcon, USA)를 정제수에 용해 및 분산시킨 코팅 액을 S제하여 위의 암로디 ¾ 정제를 하이 코터로서 코¾층을 형성하여 표제의 지연방출성 구획을 제조하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 최종 결과물을 캡술층전기를 이용하여 캡술 (서흥캅셸, 한국)에 층전하여 표제의 캡술제를 제조하였다, 실시예 V-21: 암로디 ¾-로슈바스타틴 캡슬제 제조 (과립-과립)
표 V-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡슬제를 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 속방층 과립)의 제조
로슈바스타틴 칼슴, 삼염기칼슘인산염 , 유당수화물을 달아 35호체로 사과하고, 더 블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도로 히드록시프로필 셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하 였다. 연합이 끝나면 20호체로 오실레이터롤 이용하여 제립하고 이를 은수 건조기 를 이용하여 에서 건조시 ¾ 후 18호체로 정 립하여 표제의 선방출성 구획을 제 조하였다.
(2) 지연방출성 구획 (암로디핀 지연성 과립〉의 제조
암로디핀 베실레이트, 미결정셀롤로오스, 디-만니를을 35호체로 사과하고 더블콘믹 서로 5분간 실은에서 혼합하여 흔합물을 제조하였다. 콜리코트 SR30D 넣어 주성분 흔합물과 함께 연합하고 20호체로 오실레이터롤 이용하여 제 ¾하고 이를 은수 건조 기를 이용하여 60°C에서 건조하였다, 건 3:후 18호체로 정립하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 최종 결과물을 더블콘믹서로 흔합하였다. 흔합물에 전분 글리콜 산 나트륨을 투입하고 더블콘믹서로 흔합하였다. 다시 흔합물에 스테아르산 마그네 슴을' 넣어 최종 혼합하였다. 최종 혼합된 흔합물을 분말공급기에 투입하고 캡술충 전기를 이용하여 캡술 (서흥 "셸, 한국)에 층전하여 표제의 캡술제를 제조하였다 실시예 V-22: 암로디핀ᅳ로슈바스타틴 캡슬제 제조 (과립-펠 ¾)
표 V-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 선방출 과립)의 제 :
로슈바스타틴 칼슴, 삼염기칼슘인산염, 미결정샐를로오스, ¾호화전분을 35호체로 사과하고 더블콘먹서에서 5분간 실은에서 흔합하여 혼함물을 제조하였다. 별도로 히드특시프로필셀를로오스를 정제수에 녹여 결합액을 제조하 i 이를 주성분 흔합물 과 함께 연합하였다. 연합이 끝나면 20호체로 오실테이터를 이용하여 제립하고 이
를 은수 건 S기롤 이용하여 601:에서 건조한다. 건조가 끝나면 다시 18호체로 정립 하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디¾ 지연방출성 ¾¾)의 제조
슈가 시드를 유동층 과립기에 투입한 뒤 , 별도로 ᅵ정제수에 풀리비닐피를리돈 및 암 로디핀 베실레이트를 용해시킨 결합액을 분무하여 암로디핀 함유 ¾렛을 형성 , 건 조하였다. 다시 상기의 ¾¾에 유드라짓 RS30D와 트리에틸시트레이트를 염화메틸렌 에 녹인 액을 분무하여 표제의 지연방출성 구획을 제조하였다
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 최종 결과물을 더블콘믹서로 흔합하였다. 흔합물에 전분글리콘산 나트룸을 투입하고 더블콘믹서로 흔합하였다. 다시 혼합물에 스테아르산 마그네슴 을 넣어 최종 흔합하였다. 최종 혼합된 흔합물을 분말공급기에 투입하고 캡술층전 기 * 이용하여 캡술 (서흥캅셀, 한국)에 층전하여 표제의 캡술제를 제조하였다. 실시예 V-23: 암로디핀-로슈바스타틴 캡술제 제 : (과립ᅳ정제)
표 V— 3에 기계된 성분과 함량으로, 이하의 방법에 의해 캡슐제를 제조하였다.
(1) 선방출성 구획 (로슈바스타틴 속방출성 과립)의 제조
로쥬바스타틴 칼 , 삼염기칼슘인산염, 유당수화물, 옥수수전분을 달아 35호체로 사과하고, 더블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 흔합물에 정제수를 넣어 연 "한 후 18호체로 오실레이터를 이용하여 제립하고 이를 은수 건 조기를 이용하여 60t에서 건조시켰다. 이 후 20호체로 정립하고 정립물에 전분 글 리콜산 나트륨을 투입하고 더불콘믹서로 흔합하였다. 다시 흔합물에 스테아르산 마 그네슴을 넣고 최종 흔합하여 S제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 정제 )의 제조
암로디핀 베실레이트, 무수인산수소칼슴, 전호화전분을 35호체로 사과하고 더블콘 믹서로 5분간 실은에서 흔합하여 혼합물을 제조하였다. 별도로 히드록시프로필셀를 로오스를 정제수에 녹여 결합액으로 하여 연합, 제림 및 건조하였다. 건조가 끝나 면 다시 18호체로 정립한다, 정립물에 35호체로 채과한 스테아르산 마그네슘을 투 입하여 , 4 분간 흔합한 후 흔합물을 직경 5 瞧 펀치가 장착된 로타리 타점기로 타 정하여 정제를 제조하였다. 별도로 아크릴-이즈를 정제수에 용해 및 분산시킨 코팅 액을 조제하고 이를 이용하여 위의 암로디 ¾ 정채 * 하이코터로 코팅층을 '형성시켜
표제의 지연방출성 구획을 제조하였다.
(3) 후혼합, 타정 및 코팅
상기 (1)과 (2)의 산물을 술층전기를 이용하여 캠술 (서흥캅샐, 한국)에 층전하여 표제의 캡슬제를 제 S하였다. 실시예 V-24: 암로디핀-로슈바스타틴 ¾슬제 제조 (정제-과립 )
표 V-3에 기채된 성분과 함량으로, 이하의 방법에 의해 ¾슬제를 제 하였다.
(1) 선방출성 구획 (로슈바스타틴 속방출 정제)의 제조
로슈바스타틴 칼슘 , 삼염기칼슘인산염, 미결정셀를로오스, 전호화전분을 달아 35호 체로 사과하고 더블콘믹서로 5분간 실은에서 흔합하여 흔합물을 제조하였다. 별도 로 히드록시프로필셀를로오스을 정제수에 녹여 결합액을 제조하고 이를 주성분 흔 합물과 함께 연합하였다. 연합 후 20호체로 오실레이터를 이용하여 제립하고 이를 온수 건 S기를 이용하여 60¾에서 건조시 ¾ 후 18호체로 정립하였다. 정림물에 35 호쳬로 체과한 크로스포비돈과 스테아르산 마그네슴을 투입하여 , 4 분간 흔합한 후 , 흔합물을 직경 5 m 펀치가 장착된 로타리 타정기로 타정하여 표제의 선방출성 구획을 제조하였다.
(2) 지연방출성 구획 (암로디핀 지연방출성 과립)의 제조
암로디핀 베실레이트, 미결정샐롤로오스, 무수인산수소칼슴 및 히드록시프로필메 ¾ 셀를로오스를 35호체로 사과하고 더블콘믹서로 5분간 흔합하여 흔합물을 제조하였 다. 정제수를 넣어 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체로 오 실레이터를 이용하여 제립하고., 이를 은수 건≥기를 이용하여 60'C에서 건조시 ¾ 후 18호체로 정립하였다. 정립물에 스테아린산 마그네슴을 넣어 최종흔합하여 표제 의 지연방출성 구획을 제≥하였다.
(3) 후흔합, 타정 및 코팅
상기 (1)과 (2)의 최종 결과물을 캡술층전기를 이용하여 캡슐 (서흥갑¾, 한국)에 층전하여 표제의 캡술제를 제조하였다. 실시예 V-25: 로슈바스타 ¾ -암로디¾ 떼실레이트 블리스터 포장키트 제조 이하의 방법에 의해로슈바스타틴- 암로디핀 베실레이트 블리스터 포장키트를 제 S
하였다.
실시예 V— 7 의 (1)에서 제조한 로슈바스타틴 칼슴 선용출 과립과 실시예 V— 7 의 (2)에서 제조한 암로디핀 베실례이트 지연 용출 과립 각각을 7ι 편치가 장착된 로 타리 타정기를 사용하여 타정하여 각각의 정제를 제조한 후, 블리스터 포장기 (Minister A, 흥아엔지니어링 )를 이용하여 블리스터 포장용기 (은박, 동일양행 , 전민산업 )에 각각의 정제롤 동시복용 가능하도록 포장하여 블리스터 포장키 트롤 제조하였다
[^V-l]


[표 V-2]


[표 Vᅳ 3]
조성 (mg/tablet)
구성성분 실시에
V-17 V-18 V-19 V-20 V-21 V-22 V-23 V-24 로슈바스라틴 ¾습 10,4 10.4 10.4 10.4 10.4 10.4 10.4 10.4 샴영기 ¾승인산영 - - 20 20 30 £0 30 20 口 1결정셀 s로오스 ᅳ - 50 - - 40 - 41 선 유당수화¾ - - ― - 22.6 - 20 ᅳ 방 옥수수전분 - - 15.6 61.6 - - 15.6 -
Sg화전분 - - - - - 20.6 - 30.6
50 50
성
: c:록시 로 ¾ ϋΐ ^로오
2 2 1 1 1 1 2 전분 ¾¾¾^나트9 - - 2 6 - ᅳ 2
크로스포비돈 - - - ᅳ 4 스 ειιοι·르산 Diᅳ그 ui승 - ᅳ 1 1 1 1 정 1수 48 48 50 50 20 50 30 50 암≥ E! Hil실레이트 6.94 6.94 6.94 6.94 6.94 6.94 s-앙로 as Ηΐι실 eii이트 3.74 3.74
0'|결정셀롭로오스 30 50 - 50 30 25 무수인산수소 ¾슘 10 - 25.26 23.06 22.06 s호화전분 - 25.66 - - - 40
18'.6
옥 "4»전분 - - -
6
디 -만니§ - - 13.06
.지 si.드록人 i 로 a셀 s로오
2 2 ᅳ
연 - - 2 -
1.26 1.06
•§l드록人 I프로 galls셀를
- - - - - 20 로 -오스
성
슈 드 50 50
구
아크 a-이즈 ᅳ 9 - - 획 - - 9. -
^률로오스 oAiigi이트
12 ᅳ - - ― - - -
320S{0U-1IS^|32%)
셀룰로오스 ·ί)ᅡ세 ΕΙΙΟί트
398-10IF(0 i)ia기 39.8 12 - - - - - - - %)
S리 S트 SR 30D - - - 10 20 - - -
^드 BIS RS 30D - 12 ᅳ 12 - ᅳ
S¾(D|Eᅡ크릴레 0!트 ,
oiiSDiiEᅡ크 a ioj트)공중 - - ᅳ 10 - - - 합 ι
트 5.1에 S I트러!이트 - 3 3 ~ 영화 나트 g 10 一 - 스 E)l아르산 마그너|송 1 1 - 1 - - 1 1 정제수 30 86 49 45 76 30 염 SKHIS ! 108 84 105
에탄을 108 45
후 전분 g¾§ iL}eg ᅳ ― - ᅳ 3 3 - - 스 Eli아르산아그 WI승 ᅳ - - 2
합 2 - -
¾슬 63- 63 63 63 63 63 63 ' 63 if계 220 220 233 263 202 233 224 247 실시예 VI-1 : 니페디핀-아토르바스타틴 유핵정 제조
표 vi-i에 기재된 성분과 함량으로 , 이하의 방법에 의해 유핵정을 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과립) 제조
아토르바스타틴 칼슘 무수물, 탄산 칼슘 (Precipitated calcium carbonate, Nitto Funka, 일본), 미결정셀를로오스 (Avicel PH101, FMC Biopolymer, 미국), 유당수화 물 (DMV, 독일), 전호화전분 (Starch 1500G, Colorcon, 미국), 라우 ¾황산나트륨 (Texapon 12P, Cognis Corp, 미국)롤 달아 35호체로 사과하고, 더불콘 흔합기 (다 산파마텍, 한국)에서 5분간 흔합하여 혼합물을 제조하였다. 따로 히드록시프로필¾ 를로오스 (HPC-L, Nippon Soda, 일본)를 정제수에 녹여 결합액을 제조하고 이를 주 성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터 (AR402, Erweka, 독일 )를 이용하여 제립하고 이를 은수 건 기 (H- C, 삼공사, 일본)를 이 용하여 60°C에서 건 한다. 건조가 끝나면 다시 20호체로 정 립하였다. 정립물에 크 로스카르멜로오스나트쁌 (Primellose, D V, 독일)을 흔합하고, 스테아르산 마그네슴 CNitika Chemical , 인도)을 넣어 더블콘 흔합기로 최종 흔합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 정제) 제조
니페디핀, 미결정셀를로오스, 라우 ¾황산나트튬을 35호체로 사과하고 더블콘 흔합 기로 5분간 흔합한 후 유등층 과립기 (GPCG 1, Glatt , 독일)에 투입하고, 따로 히드 록시프로필메틸셀를로오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조
하였다. 여기에 콜리돈 SR(Basf , 득일)을 흔합하고 , 스테아르산 마그네슴을 넣어 최종 흔합한 후 상기 최종 흔합물을 로타리 타정기 (MRC-30, 세종 기계 , 한국)로 타 정하여 이를 핵정으로 하였다.
상기 제조한 핵정에 코팅기 (SFC— 30F, 세종 기계 , 한국)를 사용하여 , 80% 에탄을에 용해시킨 유드라짓 L 100 용액을 분무하여 코팅 핵정으로 제조하였다.
3) 타정 및 코팅
유핵정 타정기 (RUE L, 킬리안, 득일)로 상기 1)의 아토르바스타틴 칼슘 선방출 과 립을 외층으로 사용하고, 상기 2)의 니페디핀 지연방출성 서방 정제를 핵정으로 하 여 타정하였다. 따로 80% 에탄을에 히드록시프로필메틸 ¾를로오스 2910(Stiin-etsu, 일본), 폴리에틸렌글리튤 6000 (Basf , 독일)을 용해시키고, 탈크 (Luz ac , 프랑스) , 산화티탄 (Tioside Americas, 미국)을 분산시켜 코팅 액을 제조한 후 상기 유핵정에 코팅기로서 코팅층을 형성하여 필름코팅된 유핵정 형태의 정제를 제조하였다. 실시예 VI-2 : 니페디핀-아토르바스타된 유핵 ¾ 제조
표 에 기채된 성분과 함량으로, 이하외 방법에 의해 유핵정을 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과림)제조
아토르바스타틴 칼슴 삼수화물, 탄산 칼슘, 미결정셀를로오스, 유당수화물 , 전호화 전분 , 라우 ¾황산나트콤를 달아 35호체로 사과하: a, 더블콘 흔합기에서 5분간 흔합 하여 흔합물을 제조하였다. 따로 히드록시프로필¾를로오스를 정제수에 녹여 결함 액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연함이 끝나면 18호체로 오 실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60t:에서 건조한다. 건조가 끝나면 다시 20호체로 정립하였다. 정립물애 크로스카르델로오스나트 "을 흔합하고, 스테아르산 마그네슴을 넣어 더블콘 흔합기로 최종 흔합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 정제)제조
니페디핀, 미결정셀를로오스, 라우릴황산나트튬을 35호체로 사과하고 더블콘 흔합 기로 5분간 혼합한 후 유등층 과립기에 투입하고, 따로 히드록시프로필메틸셀를로 오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 여기에 히드 특시프로필메털젤를로오스 (Benecel , Hercules, 미국) 및 히드록시프로필셀를로오스 (Klucel , Hercules , 미국)를 흔합하고, 스테아르산 마그네슴을 넣어 최종 흔합한
후 상기 최종 흔합물을 로타리 타정기로 타정하여 이를 핵정으로 하였다.
상기 제 2:한 핵정에 코팅기를 사용하여, 정제수에 유드라짓 RS 30D (에보닉 , . 독일) 및 유드라짓 RL 30D(에보닉 , 독일)를 분산시켜 제조한 코팅액을 분무하여 코¾ 핵 정으로 제조하였다.
3) 타정 및 코팅
실시예 VI— 1의 3)과 동일 방법으로 타정 및 코팅하여 필름코팅된 유핵정 형태의 정 제를 제조하였다. 실시예 VI-3 : 니페디핀-아토르바스타 ¾ 유핵정 제조
표 에 기재된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과립)제조
아토르바스타틴 스트른튬 오수화물, 탄산 칼슴, 미결정샐를로오스, 유당수화물, 전 호화전분, 라우릴황산나트륨를 달아 35호체로 사과하고, 더블콘 혼합기에서 5분간 흔합하여 흔합불을 제조하였다. 따로 히드특시프로필샐를로오스롤 정제수에 녹여 결함액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체 로 오실레이터를 이용하여 제립하고 이롤 은수 건조기를 이용하여 에서 건조한 다. 건조가 끝나면 다시 20호체로 정립하였다. 정 립물에 크로스카르멜로오스나트륨 을 혼합하고, 스테아르산 마그네슴을 넣어 더블콘 흔합기로 최종 흔합하였다.
2) 지연방출성 구획 (니폐디핀 지연방출성 서방 정제)제조
니페디핀 , 미결정셀를로오스, 라우릴황산나트륨을 35호체로 사과하고 더블콘 흔합 기로 5분간 흔합한 후 유동층 과립기에 투입하고, 따로 히드특시프로필메털셀를로 오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성 , 건조하였다. 여기에 유드 라짓 RS P0 (에보닉 , 독일) , 유드라짓 RL P0 (에보닉 , 독일 ) 및 염화나트튬을 흔합하 고, 스테아르산 마그네슴을 넣어 최종 흔합한 후 상기 최종 혼합물을 로타리 타정 기로 타정하여 이를 핵정으로 하였다.
상기 제조한 핵정에 코팀기를 사용하여 , 정제수에 셀를로오스아세테이트프탈레이:트 (Aquacoat cPD, FMC, 머국), 플리에틸렌글리콜 6000(Basf , 득일) 및 트리에틸시트 레이트 (Vertel lus, 영국)를 회석시켜 제조한 코팅액을 분무하여 코팅 핵점으로 제 조하였다.
3) 타정 및 코팅
실시예 VI-1의 3)과 동일 방법으로 타정 및 코팅하여 필름코팅된 유핵정 형태의 정 제를 제조하였다. ᅳ 실시 예 VI-4 : 니페디판ᅳ아토르바스타틴 유핵정 제조
표 VI-1에 기재된 성분과 함량으로, 이하의 방법에 의해 유핵정을 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과립)의 제조
아토르바스타틴 칼슘 삼수화물, ¾산 칼슴 , 미결점셀를로오스, 유당수화물, 전호화 전분을 달아 35호체로 사과하고, 더블콘 흔합기에서 5분간 혼합하여 흔합물을 제조 하였다. 따로 히드록시프로필셀를로오스와 T een 80(AkzoNobel , 네덜란드)를 정제수 에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이롤 은수 건조기를 이용하여 60°C에서 건조한다. 건조가 끝나면 다시 20호체로 정 립하였다. 정립물에 전분글리콜산나트륨 ζ丽, 득일) , 스테아르산 마그네슴을 넣어 더블콘 흔합기로 최종 혼합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 정제)의 제쵸
니페디된, 미결정샐를로오스, 라우릴황산나트晉을 35호체로 사과하고 더블콘 흔합 기로 5분간 흔합한 후 유등층 과립기에 투입하고, 따로 히드록시프로필메틸셀를로 오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 여기에 다시 정제수에 에틸셀를로오스 (Aquacoat ECD, FMC, 미국)를 희석하고 플리에털렌글리콜 6000을 용해시켜 제조한 용액을 분무하여 과립을 코팅 , 건조하였다. 상기 제조한 과립에 스테아르산 마그네슴을 넣어 최종 흔합한 후 로타리 타정기로 타정하여 이 핵정으로 하였다,
상기 제조한 핵정에 코팅기를 사용하여, 정제수에 히드특시프로필메틸셀롤로오스아 세테이트숙시네이트 (Shinᅳ etsu, 일본) 및 트리에틸시트레이트를 녹여 제 ;한 코팅 액을 분무하여 코팅 핵정으로 제조하였다.
3) 타정 및 코팅
실시예 VI-1의 3)과 동일 방법으로 타정 및 코팅하여 필름코팅된 유핵정 형태의 정 제를 제조하였다.
실시예 VI-5 : 니페디핀ᅳ아토르바스타띈 유핵정 제조
표 VI-1에 기재된 성분과 함량으로, 이하의 방법에 의훼 유핵정을 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과립 )의 제조
아토르바스타틴 칼습 삼수화물 , 탄산 칼슴, 미결정셀를로오스 유당수화물, 옥수수 전분 (DMV, 독일) , 라우릴황산나트륨를 달아 35호체로 사과하고 더불콘 흔합기에서 5분간 흔합하여 흔합물을 제조하였다. 따로 히드록시프로필 " 를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 18 호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60°C에서 건 조한다. 건조가 끝나면 다시 20호체로 정립하였다. 점립물에 크로스키"르멜로오스나 트륨을 흔합하고, 스테아르산 마그네슴을 넣어 더블콘 흔합기로 최종 흔합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 정제)의 제조
니페디핀 미결정셀를로오스, 라우릴황산나트륨을 35호체로 사과하고 더블콘 혼합 기로 5분간 흔합한 후 유동층 과립기에 투입하고, 따로 히드록시프로필메틸 ^를로 오스프탈레이트 (Shin-etsu, 일본)롤 물에 녹여 만든 결합액을 분무하여 과림을 형 성, 건조하였다. 상기 제조한 과림에 카보머 71G를 흔합하고, 스테아르산 마그네슴 을 넣어 최종 흔합한 후 로타리 타정기로 타정하여 이를 핵정으로 하였다.
3) 타정 및 코팅
실시예 VI-1의 3)과 등일 방법으로 타정 및 코팅하여 유핵정 형태의 정제를 제조하 였다. 실시예 VI-6 : 니페디핀-아토르바스타틴 2상 매트릭스 정제 제 2:
표 VI-1에 기재된 성분과 함량으로, 이하의 방법에 의해 2상 매트릭스 정제를 제조 하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과립 )의 제조
아토르바스타틴 칼슘 무수물, 탄산칼슘, 유당수화물 , 전호화전분을 달아 35호체로 사과하고 더블콘 흔합기에서 5분간 흔함하여 흔합물을 제조하였다. 따로 히드록시 프로필 ¾를로오스과 Tween 80을 정제수에 녹여 결합액을 제조하고 이를 주성분 흔 합물과 함께 연합하였다. 연합이 끝나면 20호체 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용하여 60t에서 건조 후 다시 18호체로 정립하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 과립 )의 제
니페디핀 , 미결정셀를로오스, 라우릴황산나트톰을 35호체로 사과하고 더블콘 흔합 기로 5분간 흔합한 후 유등층 과립기에 투입하고, 따로 히드록시프로필메틸셀롤로 오스를 물에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 여기에 다시 정제수에 에틸쎌를로오스 (Aquacoat ECD, FMC, 미국)를 희석하고 를리에틸렌글리콜 6000을 용해시켜 제조한 용액을 분무하여 과립을 코뜀 , 건조하였다. 상기 제조한 과립에 별도로 정제수에 히드록시프로필메틸셸를로오스아세테이 S숙시네이트 (Shin- etsu, 일본) 및 트리에틸시트레이트를 녹여 제조한 코팅액을 분무하여 과립을 코¾ 하였다.
3) 후흔합, 타정 및 코팅
상기 1)과 2)의 산물을 더블콘 흔합기에 넣고 흔합하였다. 이 흔합불에 전분 글리 콘산 나트륨을 흔합하고, 스테아르산 마그네슴을 넣어 최종 흔합하였다. 최종 흔합 물을 로타리 타정기로 타정하여 2상 매트헉스 정제를 제조하였다. 따로 에탄을과 정제수에 히드톡시프로필메 ¾¾를로오스 2910, 플리에 ¾렌글리콜 6, 000을 용해시키 고, 탈크 , 산화티탄을 분산시킨 코팅 액을 조제하고, 코팅기를 이용하여 상기 제조 한 2상 매트릭스 정제에 필름코팅층을 형성하였다. 실시예 VI-7 : 니페디핀-아토르바스타틴 2상 매트릭스 정제 제조
표 VI-1에 기재된 성분과 함량으로, 이하의 방법에 의해 2상 매트릭스 정제를 제 하였다. -
1) 선방출성 구획 (아토르바스타틴 선방출 과립)의 제조
아토르바스타틴 칼슘 삼수화물, 탄산칼슴, 미결정셀률로오스 , 라우릴황산나트륨을 달아 35호체로 사과하고 더블콘 흔합기에서 5분간 흔합하여 흔합물을 제조하였다. 따로 히드록시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체 오실레이터를 이용하여 제림하고 이를 은수 건조기를 이용하여 6(VC에서 건조 후 다시 18호체로 정 립하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 과립)의 제조
니페디핀을 ¾리에틸렌글리콜 6000과 흔합한 후 용융시켜 고체분산체를 제조하였 다. 제조된 고체분산체를 35호체로 사과하고 35호체로 사과한 머결정샐를로오스, 라우릴황산나트륨 및 카보머 71G와 함께 더블콘 흔합기로 흔합하였다. 상기의 흔합 물에 히드록시프로필메틸쩔를로오스를 불에 녹여 만든 결합액을 분무하여 과립을 형성, 건조하였다. 다시 별도로 에탄을과 정제수에 용해시킨 히드록시프로필메틸셀
를로오스프탈레이트 용액과 아세 ¾레이티드모노글리세리드 (Riken, 일본) 용액흘 분 무하여 과립을 코팅하였다.
3) 후흔합, 타정 및 코팅
실시예 6의 3)과 동일 방법으로 후흔합, 타정 및 코팅하여 2상 매트릭스 형태의 정 제를 체조하였다. 실시예 VI-8 : 니페디핀-아토르바스타틴 다층정 제조
표 VI-2에 기재된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과립)의 제조
아토르바스타틴 칼슴 무수물ᅳ 탄산킬:슴, 미결정샐를로오스, 유당수화물, 라우릴황 산나트륨을 달아 35호체로 사과하고 더블콘 흔합기에서 5분간 흔합하여 흔합물을 제조하였다. 따로 히드록시프로필셀롤로오스를 정제수에 녹여 결합액을 제 S하고 이를 주성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이 용하여 제립하고 이를 은수 건조기를 이용하여 6CTC에서 건조한다. 건조가 끝나면 다시 18호체로 정립하였다, 정립물에 전분 글리콘산 나트꼼을 흔합하고 , 스테아르 산 마그네슘을 넣어 더블콘 흔합기로 최종 흔합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 과림)의 제조
실시예 VI-6의 2)과 동일한 방법으로 니페디핀 지연방출성 서방 과립을 저 1조하였 .
3) 후흔합, 타정 및 코팅
다층정 타정기 (MRC—37T, 세종파마텍 , 한국)를 사용하여 타정하였다. 상기 1)의 최 종 흔합물을 1차 분말공급기에 넣고, 상기 2)의 최종 흔합물을 2차 분말 공급기에 넣어 층간의 혼입을 최소화할 수 있는 조건으로 타정하였다. 따로, 히드특시프로필 메틸셀를로오스 2910, 폴리에틸렌글리콜 6,000, 탈크, 및 산화티탄을 에탄을과 정 제수에 용해 및 분산시킨 코팅 액을 조제하여 위의 정제를 코팅기로서 필름코팅층을 형성하여 다층정 형태의 정제를 제조하였다. -' 실시예 VI-9 : 니패디 ¾ -아토르바스파틴 다층정 제조
표 VI-2에 기재된 성분과 함량으로, 이하의 방법에 의해 다층정을 제조하였다. 1) 선방출성 구획 (아토르바스타틴 선방출 과립 )의 제조
아토르바스타된 칼슴 삼수화물, 탄산칼슴, 미결정■§를로오스, 옥수수전분을 달아
35호체로 사과하고 더블콘 혼합기에서 5분간 흔합하여 혼합물을 제조하였다. 따로 히드록시프로필셸를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물 과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이 를 은수 건조기를 이용하여 60 C에서 건조한다. 건조가 끝나면 다시 18호체로 정립 하였다. 정립물에 전분 글리콘산 나트륨을 흔합하고, 스테아르산 마그네슘을 넣어 더블콘 흔합기로 최종 흔합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 과립 )의 제조
실시예 VI-7의 2)과 동일한 방법으로 니패디핀 지연방출성 서방 과립을 제조하였 다.
3) 후흔합, 타정 및 코팅
실시예 VI-8의 3)과 동일 방법으로 후흔합, 타정 및 코팅하여 다층정 형태의 정제 를 제조하였다. 실시예 VI-10 : 니페 핀-아토르바스타틴 캡술제 제조 (펠렛-과립)
표 VI-2에 기재된 성분과 함량으로, 이하의 방범에 의해 캡슐제를 제조하였다.
1) 선방출성구획 (아토르바스타틴 ¾¾)의 제조
슈가 시드 (Sugar sphere, MP Pharmaceutical , 프랑스)를 35 호체로 체과하고 유동 층 과립기에 루입한 뒤 . 따로 정제수에 히드록시프로필셀를로오스와 아토르바스타 틴 칼슴 삼수화물을 용해시킨 결합액을 분무하여 펠 을 형성, 건조하였다.
2) 지연방출성 구획 (니꽤디핀 지연방출성 서방 과립 )의 제조
실시예 VI-7의 2)과 동일한 방법으로 니페디핀 지연방출성 서방 과립을 제조하였 다.
3) 후흔합 및 캡슬층진
상기 1)과 2)의 최종 산물을 더블콘 흔합기로 흔합하였다. 흔합물에 스테아르산 마 그네슘을 넣어 최종 흔합하였다. 상기 최종 흔합물을 분말공급기에 투입하고 ¾술 충진기 (SF 40N, 세종파마웩 , 한국) 이용하여 ¾술 (서홍갑셀, 한국)에 층전하여 캡술형태의 지연방출성 서방 제제의 제조를 완료하였다. 실시 예 VI-11 : 니페디핀-아토르바스타틴 캡술제 제조 (펠 ¾-¾렛)
표 VI-2에 기재된 성분과 함량으로, 이하의 방법에 의해 술제를 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 ¾¾)의 제조
슈가 시드를 35 호체로 쳬과하고 유동층 과립기에 투입한 뒤 따로 물에 히드록시프 로필 ¾를로오스와 아토르바스타틴 칼슴 삼수화물을 용해시킨 결합액을 분무하여 아 토르바스타틴 함유 ¾을 형성 , 건조하였다.
2) 지연방출성 구획 (니페디핀 지연성 선방 ¾¾)외 제조
슈가 시드를 35호체로 체과하고 유등층 과림기에 투입한 뒤 , 따로 물에 히드록시프 로필메틸셀를로오스, 폴리에틸렌글리콜 6000, 라우 ¾황산나트륨, 및 니페디핀을 용 해시킨 결합액을 분무하여 니페디핀 함유 펠¾을 형성, 건조하였다. 다시 상기의 ¾¾에 *리코트 30D(Basf , 독일)를 에탄을과 염화메될렌에 녹인 액을 분무하여 니페디핀 지연성 ¾¾을 제조하였다. 다시 상기의 펠¾에 아크 ¾ -이즈 (Colorcon, 미국)를 에탄올과 정제수에 녹인 액을 분무하여 니페디핀 지연방출성 서방 ¾을 제조하였다.
3) 캡슬층진 - 공정 1)과 2)의 최종 산물을 캡슐층진기를 이용하여 캡슬에 층전하여 ¾슬형태의 제제의 제조를 완료하였다. 실시예 VI-12 : 니페디 ¾ -아토르바스타틴 캡술제 제 : (펠 5-삼투성 정제) 표 VIᅳ 2에 기채된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 펠헷)의 제조
슈가 시드 (Sugar sphere)를 35 호체로 체과하고 유동층 과립기에 투업한 뒤 따로 물에 히드톡시프로필샐를로오스와 아토르바스타틴 ¾슴 삼수화물을 용해시킨 결합 액을 분무하여 아토르바스타틴 함유 펠¾을 형성, 건조하였다.
2) 지연방출성 구획 (니페디핀 삼투성 정제)의 제 S
실시예 VI— 3의 2)과 동일한 방법으로 니페디핀 지연방출성 서방 정제을 제조하였 다.
3) ¾슐층진
상기 1)과 2)의 최종 산물을 캡슬층진기를 이용하여 캡슐에 층전하였다. 실시예 VI-13 : 니페디 ¾—아토르바스타된 ¾슬제 제조 (펠 ¾-정제〉
표 에 기재된 성분과 함량으로, 이하의 방법에 의해 캡슬제를 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 ¾¾)의 제조
슈가 시드를 35 호체로 체과하고 유등층 과립기에 투입한 뒤 따로 물에 히드록시프
로필샐를로오스와 아토르바스타틴 칼슘 삼수화물을ᅵ 용해시킨 결합액을 분무하여 아 토르바스타틴 함유 했을 형성■, 건조하였다.
2) 지연방출성 구획 (니페디된 지연방출성 서방 정제.)의 제 S
실시예 VI-1의 2)과 동일한 방법으로 니페디핀 지연방출성 서방 정제을 제조하였 다.
3) 캡술층진
상기 1)과 2)의 최종 산물을 캠술층전기를 이용하.여 캡슬에 층전하였다. 실시예 VI-14 : 니페디 ¾ -아 S르바스타틴 캡술제 제조 (정제-펠렛)
표 VI-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡술제를 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 정제)의 제 S
아토르바스타린 칼슴 삼수화물, 탄산칼슘, 미결정 ¾를로오스, 옥수수전분 , 라우릴 황산나트 "을 35호체로 사과하고 더블콘 흔합기로 5분간 흔합하여 흔합물을 제조하 였다. 따로 히드록시프로필셀를로오스를 점제수에 녹여 결합액으로 하여 연합, 제 립 및 건조하였다. 건조가 끝나면 다시 18호체로 정립한다. 정 립물에 전분 글리콘 산 나트륨 및 스테아르산 마그네슘을 투입하여 , 4분간 흔합한 후, 직경 5咖 펀치 가 장착된 로타리 타정기 (MRC-37, 세종)로 타정을 하여 정제를 제조하였다.
2) 지연방출성 구획 (니페디핀의 지연방출성 서방 팰 ¾)의 제조
실시예 VI— 11의 2)과 동일한 방법으로 니페디핀 지였.방출성 서방 펠렛을 제조하였 다.
3) 캡술층진
상기 1)과 2)의 산물을 캡슬층전기롤 이용하여 캡술에 층전하였다. 실시 예 VI -15 : 니페디휜-아토르바스타틴 캡술제 제조 (정제-정제)
표 VI-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캠술제를 제조하였다. 1) 선방출성 구획 (아토르바스타틴 선방출 정제)의 제조
아토르바스타틴 칼슴 삼수화물, 탄산칼슴, 옥수수전분, 라우 ¾황산나트륨을 달아 35호체로 사과하고 더블콘 흔함기에서 5분간 흔합하여 흔합물을 제조하였다, 따로 히드특시프로필셀를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물 과 함깨 연합하였다. 연합이 끝나면 20호체로 오실레이터를 이용하여 제립하고 이 롤 온수 건조기를 이용하여 60t!에서 건조한다. 건 a가 끝나면 다시 18호체로 정림
하였다. 정립물에 35호체로 체과한 전분글리콘산나트륨과 스테아르산 마그네슴을 투입하여 , 4 분간 혼합한 후 직경 5腿 편치가 장착된 로타리 타정기로 타정하여 , 아토르바스타 ¾ 점제. 제 롤 완료하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 정제)의 제≥
실시예 의 2)과 동일한 방법으로 니페디핀 지연방출성 서방 정제를 제조하였 다.
3) 캡술층진 .
싱-기 1)과 2)의 산물올 ¾슬층전기를 이용하여 캡술에 층전하였다. 실시예 VI-16 :니페디핀-아토르바스타틴 캡술제 제조 (과림-과림)
표 VI -3에 기쟤된 성분과 함량으로, 이하의 방법에 외해 ¾슐제를 제조하였다.
1) 선방출성 구획 (아토르바스타¾ 선방출충 과립 )의 계조
아토르바스타틴 칼슴 삼수화물, 탄산칼슘, 유당수화물, 라우릴황산나트틈을 달아 35호체로 사과하고, 더블콘 흔합기에서 5분간 흔합하여 흔합물을 제조하였다. 흔합 물에 크로스카르멜로오스나트륨을 넣어 최종흔합하였다.
2) 지연방출성 구획 (니폐디핀 지연방출성 서방 과립)의 제조
실시 예 VI-6의 2)과 동일한 방¾으로 니페디핀 지연방출성 서방 과립을 제조하였 다.
3) 캡슬층진
상기 1)과 2)의 산물을 더블콘 혼합기로 흔합하였다. 흔합물에 전분, 글리콘산나트 륨을 투입하고 더블콘 흔합기로 흔합하였다. 다시 흔합물에 스테아르산 마그네슘을 넣어 최종 흔합하였다. 최종 흔합된 흔합물을 분말공급기에 투입하고 캡슬층전기롤 이용하여 ¾술에 층전하였다. 실시예 VI-17 : 니페디 ¾ -아토르바스타췬 캡술제 제조 (과립- ¾)
표 VI-3에 기재된 성분과 함량으로, 이하의 방법에 의해 캡슬제롤 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출층 과립〉의 제조
아토르바스타틴 칼슘 삼수화물, 탄산칼슘, 미결정셀를로오스 , 전호화전분, 라우 ¾ 황산나트륨을 달아 35호체로 사과하고 더블콘 흔합기에서 5분간 흔합하여 흔합물을 제조하였다. 따로 히드륙시프로필 ¾를로오스를 정제수에 녹여 결합액을 제조하고 이를 주성분 흔합물과 함께 연함하였다. 연합이 끝나면 20호체로 오실레이터를 이
용하여 제립하고 이를 은수 건조기를 이용하여 6(rc에서 건조한다. 건조가 끝나면 다시 18호체로 점 ¾하고, 여기에 크로스카르멜로오스나트 "을 흔합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 ¾¾)의 제조
실시예 의 2)과 동일한 방법으로 니피ᅵ디핀 지연방출성 서방 펠¾을 제조하였 다.
3) 캡술충진
상기 1)과 2)의 산물을 더블콘 흔합기로 흔합하였다. 흔합물에 전분글리콘산나트륨 을 투입하고 더블콘 혼합기로 흔합하였다. 다시 흔합물에 스테아르산 마그네슴을 넣어 최종 흔합하였다. 최종 흔합된 흔합물을 분말공급기에 투입하고 캡술층전기를 이용하여 ¾술에 층전하였다. ' 실시 예 VI-18 : 니페디핀-아토르바스타 ¾ 캡술제 제조 (과립—정제)
표 VI-3예 기재된 성분과 함량으로, 이하의 방법에 의해 캡슬제를 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 과립 )의 제조
아토르바스타틴 칼슴 삼수화물, 탄산칼슴, 유당수화물, 옥수수전분을 달아 35호체 로 사과하고, 더블콘 흔합기에서 5분간 흔합하여 흔합물을 제조하였다. 따로 Tween 80 을 정제수에 녹여 결합액을 제조하고 이를 주성분 혼합물과 함께 연합하였다. 연합이 끝나면 18호체로 오실레이터를 이용하여 제립하고 이를 은수 건조기를 이용 하여 60 에서 건조한다. 건조가 끝나면 다시 20호체로 정립하였다. 정립물에 전분 글리콘산나트륨을 투입하고 더블콘 흔합기로 혼합하였다. 다시 흔합물에 스테아르 산 마그네슴을 넣어 최종 흔합하였다.
2) 지연방출성 구획 (니페디핀 지연방출성 서방 정제)의 제조
실시 예 VI-1의 2)과 동일한 방법으로 니페디핀 지연방출성 서방 정제를 제조하였 다.
3) ¾슬층진
상기 1)과 2)의 산물을 캡술층전기를 이용하여 캡술에 층전하였다. 실시예 VI-19 : 니폐디핀-아토르바스타된 캡슬제 제조 (정제-과립 )
표 VI-3에 기재된 성분과 함량으로, 이하의 방법에 의해 ¾술제를 제조하였다.
1) 선방출성 구획 (아토르바스타틴 선방출 정제)의 제 2:
아토르바스타틴 칼숨 삼수화물, 탄산칼슴 미결정샐를로오스, 전호화전분을 달아
35호체로 사과하고 더블콘 흔합기에서 5분간 흔합하여 흔합물을 제조하였다. 따로 히드톡시프로필젤를로오스와 Tween 80을 정계수에 녹여 결합액을 제 S하고 이를 주 성분 흔합물과 함께 연합하였다. 연합이 끝나면 20호체로 오실레이터롤 이용하여 제립하고 이를 은수 건조기를 이용하여 6(TC에서 건조한다. 건조가 끝나면 다시 18 호체로 정립하였다. 정립물에 35호체로 체과한 크로스카르델로오스 나트튬과 스테 아르산 마그네슴을 투입하여 , 4 분간 흔합 ¾ 후, 흔합물을 직경 5 mm 편치가 장착 된 로타리 타정기 (MRC-37, 세종, 한국)로 타정하여 아토르바스타틴 정제 제조를 완 료하였다,
2) 지연방출성 구획 (니페디핀 지연방출성 과립 )의 제 a
실시예 VIᅳ 7의 2)과 동일한 방법으로 니페디핀 지연방출성 서방 과립을 제조하였 다.
3) 캡술층진
상기 1)과 2)의 산물을 ¾슬층전기를 이용하여 캡술에 층전하였다. 실시예 VI-20 : 아토르바스타틴-니폐디핀 블리스터 포장키트 제조
이하의 방법에 의해 아토르바스타틴-니페디핀 블리스터 포장키트를 제조하였다. 실시예 VI-7의 1)에서 제조한 아토르바스타틴 칼숨 삼수화물 선방출 과립과 실시예 VI-7 외 2)에서 제조한 니페디 ¾ 지연방출성 서방 과립 각각을 로타리 타정기로 타 정하여 각각의 정제를 제조한 후, 블리스터 포장기 (Minister A, 흥아엔지니어링 )를 이용하여 블리스터 포장용기 (은박, 동일양행 , PTOC, 전민산업, 이상 한국)에 각각 의 정제를 동시북용 가능하도특 포장하여 블리스터 포짱키트를 제조하였다.
[표



[표 VI一 2]
S¾(mg/tablet)
?성성분
VI-1 V!-1 Vl-I VI-1
Vl-8 VI-9
0 1 2 3
10,3
아토르바스 Efg 말승무수 g
5
10,8 10.8 10.8 10.8 10.8
01·토르 iᅡ스라¾ ¾슘삼수화물
5 S 5 5 5
&산말 s 60 60
01곁정셀 BS 스 27 36.5
유당수 ae 40
!방 *성구획
욱^수전 C 30
아시드 50 50 50 50 라무릴황^나트 g 3 3
SI드^ AI프로§셀 g로오스 2.65 2.65 3.15 2,15 3,15 S.15 전분 §리 S산 i S^ 6 6
스日 1아 Ξ산 아그 Ul¾ 1 1
uffli ia 30 30 30 30 30 30
*가시드 50· αι§정셀 B로2스 65 39 39 28 27 라우¾¾산^트§ 3 3 3 3 3
SI드록 Λ|Ξ£§0ΙΙ§¾§Ε£스 2 3 3 2 2 2 히드 '특 AIS로 S0«뭡설!醫 £2'스프 g¾이트 12 12
히£¾시2£2에§¾를¾오스아세日|0|트숙시네이
ίθ
트
SR 58 지연방 β성구
볼리코 SR 30D 12
10 10
유드라 S UOO 15 유£라5! RS- PO 15 유드 ¾ RL PO 25
0|-3¾-012; 10 로 £스 서 iE|K)|g프 ¾21ί이트 12
OiiSS를로오스 15
탠 §리촐 6000 3 30 30 30 3 트리에 s시트 all이트 2 2 asm=g 15 owi @ B)I이曰뜨 α≤노§리세 ¾드 3 3
스레아르산 마그네숨 1 1 후 s¾ 스 HI아 s산 οᅡ그 ui¾ 1
히드쪽 Λ|≥로 SWI S !를로오스 2910 3.2 3.2
플리에 a !gai書 ΘΟΟΟ 3.2 3.2
3 S #
산& 탄 2 2
1.6 1.6
¾술 63 63 63 63 합계 890 290 258 263 263 263
[^VI-3]

상기 실시예 1-1에 따라 제조된 암로디관 말레이 S/심바스타틴 유핵정제와 대조약 ( 조코:심바스타틴 단일제 , MSD, 노바스크 :암로디핀 단일제, 화이자)의 비교 용출시 험을 실시하였다. 암로디핀 성분 용출시험의 경우 2시간을 기점으로 용출액을 인공 위 액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 각 성분별 용출시험 방 법은 아래와 같으며, 그 결과를 첨부 도면 도 1과 같이 나타내었다.
도 1에 의하면 본 발명의 유핵정제는 하기 조건에서 용출 시험 시 심바스타틴 성분 은 대조 제제인 조코와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되 었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용출 속 도를 확인할 수 있다. 본 발명의 암로디핀 말레이트 /심바스타띈의 유핵정제는 1시 간까지의 암로디핀 성분의 용출률이 모두 50% 이내로서 대 S 제제의 용출를 (약 99%) 보다 ¾씬 느리다.
이처 ¾ 본 발명의 암로디핀 말레이트 /심바스타틴의 유핵정제는 대조약인 암로디핀 단일제와 달리 암로디핀의 초기 방출이 심바스타틴 보다 매우 느리기 때문에 심바 스타틴 보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다. 실험예 1-2 :암로디핀 -심바스타린 다층정의 비교 용출 시험 (comparative dissolution prof i le test)
상기 실시 예 1-4, 10에 따라 제조된 암로디핀 말례이트 /심바스타틴의 복합 제제와 대조약 (조코:심바스타틴 단일제 , 노바스크:암로디핀 단일제)의 비교 용출시험을 실 시하였다. 심바스타틴과 암로디핀 성분의 각각의 용출 특성을 하기의 방법으로 측 정하였으며 , 암로디핀 성분 용출시험의 경우 2시간을 기점으로 용출액을 인공 위액 에서 인공 장액으로 변경하여 용츌시험을 진행하였다. 그 결과롤 첨부도면 도 2와 같이 나타내었다.
도 2에 의하면 본 발명의 실시예 1-4, 10은 하기 2:건에서 용출 시험시 심바스타' 성분은 대조 제제인 조코와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확 인되었으나 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용출 속도를 확인할 수 있다, 본 발명의 암로디핀 말레이트 /심바스타틴의 다층 정제는 1 시간까지의 암로디핀 성분의 용출를이 모두 50% 이내로서 대조 제제의 용출률 (약
99¾)보다 훨씬 느리다.
이처럼 본 발명의 암로디핀 말레이트 /심바스타틴의 다층 정제는 대조약인 암로디핀 단일제와 달리 암로디핀의 초기 방출이 심바스타틴 보다 매우 느리기 ' 때문에 심바 스파틴 보다 던저 간에서 대사를 받을 확를이 낮아지게 된다,
실험예 1-3 : 암로디핀ᅳ로바스타틴 다층정의 비교 용출 시험 (comparat ive dissolution prof i le test)
상기 실시예 1-11에 따라 제조된 암로디핀 말레이트 /로바스타틴의 복합 제제와 대 조약 (메바코 : 로바스타틴 단일제 , 노바스크: 암로디 ¾ 단일제, 화이자)의 비교 용 출시험을 실시하였다. 암로디핀 성분 용출시험을 경우 2시간을 기점으로 용출액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 각 성분별 용출시 험 방법은 아래와 같으며 , 그 결과를 첨부도면 도 3과 같이 나타내었다.
도 3에 의하면 본 발명의 실시예 1-11은 하기 조건에서 용출 시험시 로바스타틴 성 분은 대조 제제인 메바코와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확 인되었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용출 속도를 확인할 수 있다. 본 발명의 암로디핀 말레이트 /로바스타틴의 다층 정제는 1 시간까지의 암로디핀 성분의 용출률이 모두 5 이내로서 대조 제제의 용출률 (약 99%)보다 ¾찐 느리다.
이처럼 본 발명의 암로디편 말테이트 /로바스타틴의 다층 정제는 대조약인 암 S디핀 단일제와 달리 암로디핀의 초기 방출이 로바스타틴보다 매우 느리기 때문에 로바스 타틴보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다. 질험예 1-4: 암로디핀 -아토르바스타틴의 비교 용출 시험 (comparative dissolution profi le test)
상기 실시예 1-13에 따라 제조된 암로디핀 말레이트 /아토르바스타틴의 복합 제제와 대조약 (리피토 : 아토르바스타틴 단일제 , 화이자, 노바스크:암로디핀 단일제, 화이 자)의 비교 용출시험을 실시하였다. 암로디핀 성분 용출시험을 경우 2시간을 기점 으로 용 *액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 그 결과 « 첨부 도면 도 4와 같이 나타내었다,
도 4에 외하면 본 발명의 실시예 1-13은 하기 조건에서 용출 시험시 아토르바스타
틴 성분은 대조 제제인 리피토와 비교하여 거의 동등한 용출특성을 나타내는 것으 로 확인되었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용출 속도를 확인할 수 있다. 본 발명의 암로디핀 말레이트 /아토르바스타틴의 다층 정제는 1시간까지의 암로디핀 성분의 용출률이 모두 50¾ 이내로서 대조 제제의 용 출를 (약 99%) 보다 훨씬 느리다.
이처럼 본 발명의 암로디핀 말레이트 /아토르바스타린의 다층 정제는 대조약인 암로 디핀 단일제와 달리 암로디핀의 초기 방출이 아토르바스타틴보다 매우 느리기 때문 에 아토르바스타 ¾보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다. 실험예 1-5 : 레르카니디핀-심바스타 ¾ 다층정의 비교 용출시험 (comparat ive dissolution profi le test)
상기 실시예 1-16에 따라 제조된 레르카니디핀 /심바스타¾의 복합 제제와 대조약 ( 조코:심바스타틴 단일제, MSD, 자니딥 :레르카니디핀 단일제 , 엘지생명과학)의 비교 용출시험을 실시하였다. 레르카니디핀 성분 용출시험을 경우 2시간을 기점으로 용 출액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 그 결과를 첨부 도면 도 5와 같이 나타내었다.
도 5에 의하면 본 발명의 실시예 1-16은 하기 조건에서 용출 시험 시 심바스타틴 성분은 대조 제제인 조코와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확 인되었으나, 레르카르디핀 성분은 대조 제제인 자니딥과 비교할 때 매우 늦어진 용 출 속도를 확인할 수 있다. 본 발명의 레르카르디핀 /심바스타틴의 다층 정제는 1시 간까지의 레르카르디핀 성분의 용출를이 모두 50¾ 이내로서 대조 제제의 용촐를 (약 99%) 보다 ¾씬 느리다.
이처럼 본 발명의 레르카르디핀 /심바스타틴의 다층 정제는 대조약인 레르카르디핀 단일제와 달리 레르카르디핀의 초기 방출이 심바스타틴 보다 매우 느리기 때문에 심바스타된 보다 던저 간에서 대사를 받을 확를이 낮아지게 된다. 실험예 1—6 : 라시디핀 -심바스타틴 다층정의 비교 용출시험 (comparat ive dissolution profile test)
상기 실시예 1-18에 따라 제조된 라시디핀 /심바스타틴의 복합 제제와 대조약 ( 코: 심바스타틴 단일제, MSD, 박사르:라시디핀 단일제 , 글락소 스미스클라인)의 비교 용출시험을 실시하였다. 라시디핀 성분 용출시험을 경우 2시간을 기점으로 용출액
을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 그 결과를 첨부 도면 도 6과 같이 나타내었다.
도 6에 의하면 본 발명의 실시예 1-18은 하기 조건에서 용출 시험시 심바스타틴 성 분은 대조 제제인 초코와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인 되었으나, 라시디 ¾ 성분은 대조 제제인 박사르와 비교할 때 매우 늦어진 용출 속 도를 확인할 수 있다. 본 발명의 라시디핀 /심바스타틴의 다층 정제는 1시간까지의 라시디핀 성분의 용출를이 모두 50% 이내로서 대조 제제의 용출를 (약 99%)보다 ¾ 씬 느리다.
이처럼 본 발명외 라시디핀 /심바스타틴의 다층 정제는 대조약인 라시디핀 단일제와 달리 라시디핀의 초기 방출이 심바스타틴 보다 메우 느리기 때문에 심바스타틴 보 다 먼저 간에서 대사를 받을 확를이 낮아지게 된다, 실험예 1-7 : 암로디편—심바스타틴 유핵정의 비교 용출시험 (comparative dissolution profi le test)
상기 실시예 1-20에 따라 제조된 암로디핀 베실레이트 /심바스타틴 유핵정제와 대조 약 (조코:심바스타틴 단일제, MSD, 노바스크 : 암로디핀 단일제 , 화이자)의 비교 용출시험을 실시하였다. 암로디핀 성분 용출시험의 경우 2시간을 기점으로 용출액 을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 각 성분별 용출 시험 방법은 아래와 같으며 , 그 결과를 첨부 도면 도 7과 같이 나타내었다.
도 7에 의하면 본 발명의 유핵정제는 하기 조건에서 용출 시험 시 심바스타틴 성분 은 대조 제제인 조코와 비교하여 거의 등등한 용출특성을 나타내는 것으로 확인되 었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용출 속 도를 확인할 수 있다. 본 발명의 암로디핀 베실테이트 /심바스타틴의 유핵정제는 1 시간까지의 암로디핀 성분의 용출를이 모두 50% 이내로서 대조 제제의 용출를보다 훨씬 느리다.
이처럼 본 발명의 암로디핀 베실레이트 /심바스타틴의 유핵정제는 대조약인 암로디 핀 단일제와 달리 암로디핀의 초기 방출이 심바스타틴 보다 매우 느리기 때문에 심 바스타틴 보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다. 실험예 1-8 : 암로디핀 -심바스타틴 2상 캡슬제제의 비교 용출시험 (comparative dissolution profi le test)
상기 실시예 1-22에 따라 제조된 암로디핀 베실레이트 /심바스타틴 복합정제를 함유 한 캡술제와 대조약 (조코:심바스타틴 단일제, MSD, 노바스크:암로디핀 단일제 , 화 이자)의 비교 용출시험을 실시하였다. 암로디편 성분 용 #시험의 경우 2시간을 기 점으로 용출액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 각 성분별 용출시험 방법은 아래와 같으며, 그 결과를 첨부 도면 도 8과 같이 나타 내었다.
도 8에 의하면 본 발명의 2상 캡술제제는하기 조건에서 용출 시험 시 심바스타틴 성분은 대조 제제인 조코와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확 인되었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용출 속도를 확인할 수 있다. 본 발명의 암로디 ¾ 베실레이트 /심바스타틴 복합정제를 함 유한 캡슬제는 1시간까지의 암로디핀 성분의 용출률이 모두 50¾ 이내로서 대조 제 제의 용출률 보다 휠씬 느리다.
이처럼 본 발명의 암로디핀 베실레이트 /심바스타틴 복합정제를 함유한 캡술제는 대 조약인 암로디핀 단일제와 달리 맘로디핀의 초기 방출이 심바스타틴보다 데우 느리 기 때분에 심바스타틴 보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다. 실험예 1-9 : 암로디핀-아토르바스타틴 2상 캡슬제제의 비교 용출 시험 (comparative dissolution profi le test)
상기 실시예 I— 30에 따라 제조된 암로디핀 베실레이트 /아토르바스타틴 칼슴의 복합 제제와 대조약 (리피토 : 아토르바스타틴 단일제 , 화이자, 노바스크:암로디핀 단일 제 , 화이자)의 비교 용출시험을 실시하였다. 암로디핀 성분 용출시험을 경우 2시간 을 기점으로 용출액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였 다. 그 결과를 첨부 도면 도 9와 같이 나타내었다.
도 9에 의하면 본 발명의 실시예 1-30은 하기 조건에서 용출 시험시 아토르바스타 틴 성분은 대조 제제인 리피토와 비교하여 거의 동등한 용출특성을 나타내는 것으 로 확인되었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용출 속도를 확인할 수 있다. 본 발명의 암로디핀 베실레이트 /아토르바스타틴 칼슴 의 복합 제제는 1시간까지의 암로디핀 성분의 용출률이 모두 50% 이내로서 대조 제 제의 용출률보다 휠씬 느리다.
이처럼 본 발명의 암로디핀 베실레이트 /아토르바스타틴 칼슘의 복합 제제는 대조약 인 암로디핀 단일제와 달리 암로디핀의 초기 방출이 아토르바스타틴보다 매우 느리
기 때문에 아토르바스타틴보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다, 실험예 1-10 : 펠로디핀-아토르바스타틴 2상 캡슬제제의 비교 용출시험 (comparative dissolution profi le test)
상기 실시예 1-36에 따라 제조된 '펠로디핀 /아토르바스타틴 2상 캡슬체제와 대조약 ( 리피토:아토르바스타틴 단일제, 화이자, 무노발: ¾로디핀 단일제 , 한독약품)의 비 교 용출시험을 실시하였다. 펠로디핀 성분 용출시험의 경우 2시간을 기점으로 응출 액을 계면활성제로서 라우릴 황산 나트튬을 함유한 인공 위액에서 계면활성제로서 라우 ¾ 황산 나트툼을 함유한 인공 장액으로 ¾경하여 용출시험을 진행하였다. 각 성분별 용출시험 방법은 아래와 같으며 , 그 결과를 첨부 도면 도 10과 같이 나타내 었다.
도 10에 의하면 본 발명의 2상 캡슬제제는 하기 조건에서 용출 시험 시 아토르바스 타틴 성분은 대조 제제인 리피토와 비교하여 거의 등등한 용출특성을 나타내는 것 으로 확인되었으나, ¾로디핀 성분은 대조 제제인 무노발과 용출결과를 비교할 때 초기 약물방출이 지연이 된 것을 확인할 수 있다.
이처럼 본 발명의 ¾로디된 /아토르바스타틴의 2상 캡술제제는 대조약인 펠로디핀 단일제와 달리 ¾로디핀의 초기 방출이 아토르바스타 ¾ 보다 매우 느리기 때문에 아토르바스타틴 보다 던저 간에서 대사를 받을 확를이 낮아지게 된다. 실험예 1-11 : 이스라디핀—풀루바스타¾ 2상 캡술제제의 비교 용출시험 (comparative dissolution profile test)
상기 실시 예 1-40에 따라 제조된 이스라디핀 /플루바스타틴 2상 캡슬제제와 대조약 (레스콜:플루바스타틴 단일제, 노바티스, 다이나써크:이스라디핀 단일제 , 대웅제 약)의 비교 용출시험을 실시하였다. 이스라디핀 성분 응출시험의 경우 2시간을 기 점으로 용출액을 계면활성제로서 라우 ¾ 황산 나트륨을 함유한 인공 위액에서 계면 활성제로서 라우 ¾ 황산 나트콤을 함유한 인공 장액으로 변경하여 용출시험을 진행 하였다. 각 성분별 용출시험 방법은 아래와 같으며 , 그 결과를 첨부 도면 도 11과 같이 나타내었다.
도 11에 의하면 본 발명의 2상 캡슬제제는 하기 조건에서 용출 시험 시 플루바스타 틴 성분은 대조 제제인 레스콜과 비교하여 거의 동등한 용출특성을 나타내는 것으 로 확인되었으나, 이스라디핀 성분은 대조 제제인 다이나써크와 비교할 때 매우 늦
어진 용출 속도를 확인할 수 있다. 본 발명의 이스라디핀 성분은 대조 제제인 다이 나써크와 용출결과를 비교할 때 초기 약물방출이 지연이 된 것을 확인할 수 있다. 이처럼 본 발명의 이스라디핀 /*루바스타틴의 2상 캡술제제는 대조약인 이스라디핀 단일제와 달리 이스라디¾의 초기 방출이 플루바스타틴 보다 매우 느리기 때문에 플루바스타틴 보다 먼저 간에서 대사를 받을 확률이 낮아지게 된다. 실험예 1ᅳ12 : (S)ᅳ암로디핀 -심바스타틴 제제의 붕해시험 (Disintegration test) 상기 실시예 1-51, 52, 54에서 얻은 제제를 대한약전 8개정 일반시험뜁 중 붕해시 험법에 따라 진행하되 시험액 등의 시험 조건은 다음과 같다.
상세한 시험방법은 시험기를 수축에 달고 비커 속에 넣어 1 분간 29 ~ 32 회 왕복, 진폭 53 ~ 57 mm로 부드럽게 상하운동을 하도록 조절하였다, 시험기가 가장 아래로 내려갔을 때 아래의 망 면이 비커의 바닥으로부터 25 廳가 되도특 하고 비커에 넣 는 시험액의 양은 시험기가 가장 아래로 내려갔을 때 시험기의 윗면이 액의 표면과 일치하도록 하였다. 액의 온도는 37.2 °C로 유지하였다. 시험액으로는 붕해시험법 1액 (pH 약 1.2), 붕해시험법 2액 (PH 6.8)을 사용하였다.
각 실시 예의 검체를 각각 6 개를 취하여 시험기의 유리관에 1 개씩 넣고 시험기를 미리 은도 및 액량을 조절한 비커 중의 시험액에 담그고 일정시간 상하운등을 한 다음 시험기를 가만히 시험액에서 꺼내고 유리관내의 검체상태롤 관찰하였다. 붕해시간 등의 결과는 표 1-12와 같이 나타났다.
표 1-12외 결과에서 확인하는 바와 같이 본 발명에 따른 상기 실시예의 심바스타틴 층 정제 또는 과립은 모든 조건에서 빠른 붕해를 나타내었다. 반면 , (S)-암로디핀 층의 정제는 퉁해시험 결과 pH 1.2 붕해시험액에서는 2시간등안 붕해가 되지 않았 고 pH 6.8에서는 일정한 지연시간 후 붕해가 개시띔을 확인할 수 있었다. 위의 실 험결과를 통해 , 본 발명외 약제학적 제제를 환자가 복용하였을 때 심바스타틴 성분 이 먼저 붕해 , 용출되어 흡수가 이루어지고, 일정시간이 지난 후 암로디핀 성분이 용출 흡수될 것이라는 것을 보여주었다. 이는 본 발명이 개발하고자 하는 약제학적 제제의 목적과 부합되는 것이다.
[표 1-12]
 실험예 1-13 : (S)-암로디핀 /심바스타된 약제학적 제제의 용출시험 (Dissolution profile test)
실험예 1-13 : (S)-암로디핀 /심바스타된 약제학적 제제의 용출시험 (Dissolution profile test)
상기 실시예 1-51, 52, 53, 54, 55에 따라 제조된 (S)-암로디핀 /심바스타틴 약제학 적 제제를 용출 시험하였다. 용출시험은 염산용액 (산성환경 )에서 2시간등안 용출을 진행한 후, pH 6.8(인공장액)완층액에서 추가로 용출시험을 진행하였다. 단 , 심바스타틴 성분의 경우에는 일반 용출 조건에서는 용해도가 낮아 검출하기가 힘 들므로, 별도의 조건으로 용출시험을 진행하였으며 각 성분별 용출시험 방법은 아 래와 같으며, 그 결과를 첨부 도면 도 12, 도 13과 같이 나타내었다.
도 12와 13에 의하면 본 발명의 (S)-암로디핀 /심바스타틴 약제학적 제제는 본 발명 이 의도한 것처럼 심바스타¾은 빨리 용출되어 흡수되고, (S)-암로디핀은 심바스타 된아 흡수된 후에 용출되어 흡수될 수 있도특 제제화 되었다. 또한, 환자의 복용이 편할 수 있도록 다양한 제형으로 제조가 가능한 반면, 용출 그래프에서 확인할 수 있듯이 제형 차이에 의한 각 .성분의 용출률 차이는 거의 발생하지 않았다. 실험예 1-14 : 임상시험 (clinical study)
본 실험은 시판증인 '조코정 1 (싶바스타틴 20 rag, MSD) 및 '노바스크정 ' (떼실 산 암로디핀 6.944 mg, 화이자) 동시투여군과 실험군으로서 'S코정 ' 및 '노바스크 정 '을 본 발명의 실시예에 의하여 제공되는 조성물에서와 같은 방출시간을 갖도록
시간차 투여함으로써 본 발명에 의한 조성물로서의 효과를 갖도특 하여 표 1-13과 같이 임상실험을 실시하였다.
[표 1-13]


본 실험은 발명의 효과를 뒷받침하는 실험으로서 시판을 목적으로 의약품의 허가를 위해 진행되는 국내의 임상시험기준과 실험군수를 줄여 실시되었으나, 시험기간 증 피험자 관리 및 기타 임상시험으로써의 기준은 엄수하여 실시되었다.
비교 임상실험의 상세 결과는 표 1-14 및 도 14 ~ 16과 같다.
[표 1-14]
^ 비교 임상 ¾과 밝혀진 임상결과의 약동학 /약력학

1. 혈압 강하면에서 시간차 투여군은 수축기 혈압과 확장기 혈압에서 41일째 의 혈암 측정에서 동시 루여군보다 낮은 혈압 수치를 나타내었다
2. 총콜레스테롤, 저밀도지질단백콜레스테를의 저하면에서 시간차 투여군은 동시 투여군보다 41일째 지질 상태가 가장 낮은 수치를 보였다
3. 고지혈증의 가장 중요한 지질인 중성 지질 저하상태는 시간차 투여군이 우수하였고 , 동시 투여군은 증성 지질 저하 작용이 전혀 나타나지 않았다. 스 타틴계는 콜레스테를 합성 저해제이지만 콜레스테를' 합성 반웅 자체가 지질 단백
합성 반웅의 율속 조절작용을 하므로 증성 지질저하 작용도 8¾-2 범위로 알려져 은 약물이다. 이 수치와 비교해 볼 때 본 임상에서 나타난 중성 지질저하 작용 은 시간차 투여군이 20%로 나타난 반면, 동시 투여군은 거의 작용이 나타나지 않았 다.
이는 등시 루여군은 간 내 효소에 의해 상호 길항 작용을 일으켜 심바스타틴 이 혈증으로 유출띔으로써 저하작용이 중성 지질 저하까지는 미치지 돗한 것으로 해석할 수 있다.
4. 또한, 지질개선은 HDL 수치가 증가해야 함은 주지의 사실인바 시간차 투 여군의 경우 HDL 증가를 관찰할 수 있었다.
5. 약물요법의 안전성을 평가하기 위한 투약 41일째의 혈액 내 Biomark 변화 를 보면
1) S-GPT, S-GOT, CPK, y -GPT, 알칼라인 포스파타제 (Alkal ine Phosphatase) 수치에서 시간차 투여 법이 보다 안전한 Biomarks 수치를 나타내었다.
2) 이는 동시 투여군이 시간차 투여 군보다 보다 많은 심바스타틴이 혈증으 로 유출되므로 이로 인한 심바스타틴의 블필요한 염증 유발 반웅 때문으로 유추할 수 있다.
6. 임상적 부작용 관찰에 의하면 치료 대상자 중 단 2명에서 가벼운 부작용 3건 관찰되었다. 시간차 투여군에서는 설사가 1예 나타났고 등시 투여군에서는 1 명에서 피로증상과 기침이 각 1예씩 나타났다.
본 비교 임상을 통해 나타난 약물의 혈증농도변화는 아리)와 같다.
1) 심바스타틴 β -히드록시산외 혈^ 농도 : 혈중에서 낮은 수치를 보여야 할 심바스되 "틴 β-히드록시산의 혈증농도에 있어서 시간차 투여법이 다른 동시요법이 나 심바스타틴 단독 요법보다 40%나 감소되어 있음을 볼 수가 있다 (표 Iᅳ 15 및 도 14) .
이는 시간차 투여법이 지질 저하작용은 보다 우수하고 부작용은 더욱 낮출 수 있다는 약등학적 증명이 된다.
[표 1-15]
심바스타틴 P-히드록시산의 혈증농도 증가비을

2) 심바스타틴과 심바스타틴 β -히드특시산 합계 : 표 1-16 및 도 15에서 보 는 바와 같이 심바스타틴과 심바스타틴 -히드록시산의 ¾중농도 합계는 간 내에 서의 작용하는 심바스타틴외 농도가 시간차 투여법의 경우가 압도적으로 높음을 볼 수 있으며 , 이는 시간차 투여법 이 동시 투여법에 비해 지질 저하작용이 우수하고 부작용 발현도가 낮을 수 있음을 입증한다.
[표 1—16]
심바스꽈¾ 및 심바스타린 ρ-히도륙시 의 합^ 혈중 ¾:도 증가비을

3) 암로디핀의 ¾증농도 : 암로디핀 혈중 농도는 혈압 강하 작용과 직결되고 지질 저하작용도 증가시키는 부가적 작용을 가지고 있으므로 혈증 농도가 높을수특 혈압 강하 작용이 우수하다.
[표 1-17]
암로디핀의 혈증농도

도 14 ~ 15에 드러나는 바와 같이 , 싶바스타틴으로 대표되는 스타틴계 지질저하제 와, 암로디핀으로 대표되는 디히드로피리딘계 칼슴길항제의 위장관내 훔수 시간에 각각 차이를 두도록 할 경우 스타틴계 지질저하제 (심바스타틴) 및 활성형인 변화체 (심바스타된 βᅳ히드록시산)의 혈증 농도가 대조군에 비하여 현저히 저하띔으로써 이의 축적에 의한 근융해증외 부작용을 예방할 수 있음이 확인되었으며 , 또한 이는 본 발명에 의한 조성물이, 시간차 투여에 의하여 스타틴계 지질저하제가 먼저 흡수 되어 간에서 활성형으로 전환되게 하고 비활성형 대사꼴은 사이토크톰 Ρ450 3Α4 호 소에 의해 대사되어 배설되고, 층분한 홉수 시차를 두고 뒤이어 투여된 디히드로피 리딘계 칼슘길항제 즉, 암로디핀이 위장관에서 흡수띔으로써 암로디핀의 사이토크 름 Ρ450 3Α4에 대한 억제작용이 심바스타틴에 대해 아무런 영향이 미치지 않음을 보여주고 있다.
또한, 도 16에 드러나는 바와 같이 , 시간차 투여군은 암로디핀으로 대표되는 디히 드로피리딘계 칼슴 ¾항제의 Tmax(약물의 최대 혈중농도 도달시간)가 약 4 내지 5시 간 지연뜀으로써 시험군 전반에 걸쳐 저녁 7시에 동시 복용되었으나, 저녁 12시에 투여된 것과 동일한 지연된 혈압강하효과를 나타냄을 알 수 있다. 이로서 본 발명에 의한 조성물이 통상적인 동시투여군과는 달리, 평균혈압이 최고조에 이르는 투여 익 일 아침부터 한낮에 이르^ 시간동안의 최적외 혈압강하효과를 가지게 됨을 알 수 있다.
이로서 , 본 발명에 의한 디히드로피리딘계 칼슴길항제와 스타틴계 지질저하제의 약 제학적 제제와 같이 , 시간차 투여한 경우가 디히드로피리딘계 칼슴길항제와 스타뛰 계 지질저하제의 단일 제제 각각을 등시에 투여할 때의 경우 보다 스타틴계 지질저 하제의 부작용을 현저히 경감시킬 뿐만 아니라, 혈압 강하의 목적으로 투여된 디히 드로괴리딘계 칼슘길항제의 임상적인 최적의 효과가 발현되게 함을 알 수 있다,
표 I— 18은 암로디핀과 심바스타틴의 동시투여군 및 본 발명에 따르는 시간차 투여 군간의 지질을 측정한 결과이다 . 시간차 투여군은 '노바스크정ᅳ (베실산 암로디 핀 6.944 rag)과 '조코정 ' (심바스타틴 20 mg)의 저녁시간대 등시 투여군에 비하여, 지질저하제로서 투여된 심바스타틴의 LDL-콜테스테를 및 총콜레스테를의 ¾증농도 수치를 현저히 저하시킴을 알 수 확인할 수 있었다. 이는 본 발명에 의한 조성 물이 동시 투여된 디히드로피리딘계 칼슘길항제인 암로디핀에 의한 사이토크틈 P450 3A4 효소가 저해띔에 따른 영향을 받지 않고 , 투여된 약물 전량이 간에서 활 성형으로 전환되어 혈중 콜레스테를 수치를 증가시키는 HMG-CoA 환원효소를 저해함 에 따른 것이다.
[표 1-18]
사간차 투여 ¾ 퐁시투여의 誉레스테률, HDL, LDL 및 증성지질 버교 걸과 < 0.05, p < 0.01 vs 스크리닝 ;)

한편, 표 1-19 및 표 1-20은 암로디 ¾과 심바스타틴의 동시투여군 및 본 발명 에 따르는 시간차 투여군 간의 혈압을 측정한 결과이다. 디히드로피리딘계 칼 슘길항제인 암로디핀에 의한 혈압강하작용이 본 발명에 의해 제공되는 시험군에서 평균 좌위 수축기 혈압과 평균 좌뷔 이완기 ¾압이 유의적으로 저하됨이 드러나고 있으며 이는 도 16에서 보여지고 있는 시간차 투여군의 암로디핀 혈증농도의 상승 이 환자군의 실제 혈압강하호과로 드러남을 보여주고 있다. 이로서 본 발명에 의해 의도된 바와 같이 약 4시간에 걸친 시간차를 두고 혈압 강하의 목적으로 투여 된 암로디핀의 지연된 약물방출을 통하여 시간차 투여군이 동시 투여군에 비하여 우수한 혈압 강하효과롤 가짐이 증명되었다.
1
195
[표 1-19]
시긴'차 루약과 동시 투이의 혈압 비교
(* ρ < 0.05, ** p < 0.01 va Scr)

[표 1-20]
시 ¾차 투^파 ·시 무여의 맥압 및 맥박수 버 a
(* p < Q.D5, ** p < O.Oi vs 스3'리닝')

결론적으로, 상기 임상시험을 통하여 본 발명에 의한 디히드로피리딘계 칼슘 길항제와 스타틴계 지질저하제의 약제학적 제제를 시간차 투여한 경우가 디히드로 피리딘계 칼슴길항제와 스타틴계 지질저하제의 단일 제제 각각을 동시에 투여할 때 의 경우보다 동일한 투여용량에서도 스타틴계 지질저하제의 항고지혈증 효과가 더 욱 우수하게 발휘될 뿐만 아니라 혈압 강하의 목적으로 투여된 디히드로피리딘계 칼슘길항제의 상승된 혈압강하 효과가 증명되었으며, 방출시간 연장에 의한 최적효 과 발현을 예축됨을 알 수 있다.
실험예 Π-l : 유핵정의 비교 용출시험 (comparative dissolution profile test) 상기 실시예 11ᅳ1의 암로디핀 말레이트 /심바스타틴 유핵정제와 대조약 (조코 :심바스 타틴 단일제, MSD,노바스크 :암로디핀 단일제 , 화이자)의 비교 용출시험을 실시하였 다, 암로디핀 성분 용출시험의 경우 2시간을 기점으로 용출액을 인공 위액에서 인 공 장액으로 변경하여 용출시험을 진행하였다. 각 성분별 용출시험 방법은 아래와 같으며,그 결과를 도 17과 같이 나타내었다.
도 17에 의하면 본 발명의 유핵정제 중 심바스타틴 성분은 대 제제인 2:코와 비 교하여 거의 동등한 용출특성을 나타낸 반면 , 암로디핀 성분은 3시간까지의 용출를 이 30¾ 이내로서 대조 제제가 1시간 대에 이미 거의 99% 용출된 것과 비교해 볼 때 현저히 지연되어 용출띔을 확인할 수 있었다.
이처럼 본 발명의 암로디핀 말레이트 /심바스타틴의 유핵정제는 대조약인 암로디핀 단일제와 달리 암로디핀의 초기 방출이 심바스타틴 보다 매우 느리기 때문에 심바 스타린 보다 던저 ¾에서 대사를 받을 확률이 현저히 낮아지게 ¾을 알 수 있다 실험예 II-2 : 다층정의 비교 용출 시험 (comparat ive dissolution profi le test) 상기 실시예 Π— 4, 5외 암로디핀 말례이트 /심바스타틴의 약제학적 제제와대 S약 (조 코:심바스타틴 단일제, 노바스크 :암로디핀 단일제 )의 비교 용출시험을 실시하였다. 심바스타틴과 암로디핀 성분의 각각의 용출 특성을 하기의 방법으로 측정하였으며 , 암로디핀 성분 용출시험을 경우 2시간을 기점으로 용출액을 인공 위 액에서 인공 장 액으로 변경하여 용출시험을 진행하였다. 그 결과를 첨부도면 도 18와 갈이 나타내 었다.
도 18에 의하면 본 발명의 다층정 증 심바스타틴 성분은 대조 제제인 조코와 비교 하여 거의 동등한 용출특성을 나타낸 반면 , 암로디핀 성분은 3시간까지의 용출률이 모두 40% 이내로서 대조 제제가 1시간 대에 . 이미 거의 99% 용출된 것과 비교해 블 때 현저히 지연되어 용출됨을 확인할 수 있었다.
이처럼 본 발명의 암로디핀 말레이트 /심바스타틴의 다층 정제는 대조약인 암로디핀 단일제와 달리 암로디편의 초기 방출이 심바스타틴 보다 메우 느리기 때문에 심바 스타틴 보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다.
실험예 II-3 : 유핵정의 비교 용출시험 (comparative dissolution prof i le test) 상기 실시 예 ii-ii의 암로디 ¾ 베실레이트 /심바스타¾ 유핵정 제와 대조약 (조코:심 바스타틴 단일제, MSD,노바스크 : 암로디핀 단일제 , 화이자)의 비교 용출시험을 실 시하였다. 암로디핀 성분 용출시험의 경우 2시간을 기점으로 용출액을 인공 위액에 서 인공 장액으로 변경하여 용출시험을 진행하였다. 각 성분별 용출시험 방법은 아 래와 같으며 , 그 결과롤 첨부 도면 도 19와 같이 나타내었다 .
도 19에 의하면 본 발명의 유핵정 중 심바스타틴 성분은 대조 제제인 조코와 비교 하여 거의 동등한 용출특성을 나타낸 반면 , 암로디 ¾ 성분은 3시간까지의 용출를이 모두 40% 이내로서 대조 제제가 1시간 대에 이미 거외 99% 용출된 것과 비교해 블 때 현저히 지연되어 용출띔을 확인할 수 있었다.
이처럼 본 발명의 암로디핀 테실레이트 /심바스타틴의 유핵정제는 대조약인 암로디 핀 단일제와 달리 암로디핀의 초기 방출이 심바스타틴 보다 매우 느리기 때문에 심 바스타틴 보다 먼저 간에서 대사를 받을 획 ·*이 낮아지게 된다.
실험예 II-4 : 2상 캡술제제의 비교 용출시험 (comparative dissolution profi le test)
상기 실시 예 Π-13의 암로디핀 베실레이트 /심바스타틴 복합정제를 함유한 캡술제와 대조약 (조코:심바스타틴 단일제, MSD,노바스크 :암로디핀 단일제 , 화이자)의 비교 용출시험을 실시하였다. 암로디핀 성분 용출시험의 경우 2시간을 기점으로 용출액 을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다. 각 성분별 용출 시험 방법은 아래와 갈으며 , 그 결과 첨부 도면 도 20과 갈이 나타내었다.
도 20에 의하면 본 발명의 2상 캡술제제 중 심바스타틴 성분은 대조 제제인 조코와 비교하여 거의 동등한 용출특성을 나타낸 반면 , 암로디핀 성분은 3시간까지의 용출 *이 30¾ 이내로서 대조 제제가 1시간 대에 이미 거의 99% 용출된 것과 비교해 블 때 현저히 지연되어 용출됨을 확인할 수 있었다.
이처럼 본 발명의 암로디핀 테실레이트 /싶바스타¾ 복합정제를 함유한 랩슬제는 대 조약인 암로디 ¾ 단일제와 달리 암로디핀외 초기 방출이 심바스타틴보다 매우 느리 기 때문에 심바스타틴 보다 먼저 간예서 대사를 받을 확를이 낮아지게 된다.
실험예 III-l : 암로디핀-아토르바스타뷘 유핵정의 비교 용출시험 (comparative dissolution profile test)
상기 실시예 III— 1에 따라 제조된 암로디핀 /아토르바스타틴 유핵정제와 대조약 (리 피토: 아토르바스타틴 단일제, 화이자,노바스크: 암로디핀 단일제 , 화이자)의 비교 용출시험을 실시하였다.암로디핀 성분 용출시험의 경우 2시간을 기점으토 용출액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다.각 성분별 용출시험 방법은 아래와 같으며 , 그 결과를 도 21과 같이 나타내었다.
도 21에 의하면 본 발명의 유핵정제는 하기 조건에서 용출 시험 시 아토르바스타틴 성분은 대조 제제인 리피토와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 때 매우 늦어진 용 출 속도롤 확인할 수 있다. 본 발명의 암로디 ¾/아토르바스타틴의 유핵정제는 4시 간까지의 암로디핀 성분의 용출률이 2 이내로서 1시간 대에 이미 약 99%에 이른 대조 제제의 용출를보다 훨씬 낮아 뛰어난 지연방출 효과를 나타낼 뿐만 아니라, 이후 다시 약 2시간 이내에 80¾ 수준의 용출에 이르는 급격한 방출을 나타냄을 확 인할 수 있었다.
이처럼 본 발명의 암로디핀 /아토르바스타틴의 유핵정제는 대조약인 암로디핀 단일 제와 달리 암로디핀의 초기 방출이 아토르바스타틴 보다 현저히 지연되어 본 발명 의 유핵 정제 복용시 제제 중 암로디핀이 제제 증 아토르바스타틴보다 먼저 간에서 대사를 받을 확률이 현저히 낮아지게 띔을 알 수 있다. 실험예 II 1-2: 아토르바스타 ¾-암로디 ¾ 유핵정의 비교 용출 시험 (comparative dissolution profile test)
상기 실시예 ΠΙ-1, 2, 3의 제제에 대하여 암로디핀의 비교 용출시험을 실시하였 다. 암로디핀의 용출시험 방법은 실시예 III-1과 동일하였으며, 그 결과를 도 22에 나타내었다.
도 22에 의하면 실시예 III-1의 조건에서 용출 시험 시 본 발명의 제어 방출 유핵 정은 친수성 고분자를 타정 전 후혼합한 것 (실시 예 ΠΙ-1)보다, 타정 후 장용성 고 분자로 코팅을 하였을 때 (실시예 I II— 2) 및 타정 후 수불용성 중합체로 코팅을 하 였을 때 (실시예 IIIᅳ 3) , 암로디핀 성분이 의도한 시간까지 지연시간을 가진 후 비 교적 더 급격하게 방출되는 것을 확인할 수 있었다. 암로디핀 성분의 용출를은 총 480분까지 90% 이내이며 , 방출제어 ¾질 및 방출제어 코팅의 종류에 따라 암로디핀
베실레이트 성분이 급격하게 방출되었다.
이처럼 본 발명의 아토르바스타틴 /암로디핀의 유핵정은 방출제어물질을 사용함으로 써 의도한 시간까지 지연 시간을 가진 후 급격하게 암로디핀를 방출시킬 수 있다. 또한, 본 발명의 아토르바스타틴 /암로디핀의 유핵정은 대조약인 아토르바스타틴 단 일제와 암로디편 단일제를. 등시 복용하였을 경우의 용출 양상과는 달리 암로디핀의 초기 방출이 아토르바스타린보다 매우 느리기 때문에 아토르바스타틴이 던저 간에 서 대사롤 받은 후 대사 관련 효소인 사이토크룸 P450이 재생될 시간을 층분히 확 보할 수 있어 , 부작용 등 발생 가능성을 감소시킬 수 있다. 실험예 ΙΠ-3 : 아토르바스타틴-암로디판 2상 매트릭스정의 비교 용출 시험 (comparative dissolut ion profi le test)
상기 실시 예 111-6, 7의 제제에 대하여 비교 용출시험을 실시하였다. 각 성분볕 용 출시험 방법은 실시예 III— 1과 같으며, 그 결과를 도 23에 나타내었다 .
도 23에 의하면 실시예 ΠΙ-l의 조건에서 용출 시험 시 본 발명의 제어 방출 2상 매트릭스정제는 결합제로 수불용성 중합체를 사용한 것 (실시예 ΙΠ-6)과 과립 제조 후 수블용성 증합체로 코팅을 하였을 때 (실시예 ΠΙ-7) , 암로디핀 베실레이트 성분 이 의도한 시간까지 지연시간을 가진 후 비교적 급격하게 방출되는 것을 확인할 수 있었다. 암로디핀 베실테이트 성분의 용출를은 총 480분까지 9 이내이며, 방출제 어물질 및 방출제어 코팅의 종류에 따라 암로디핀 베실레이트 성분이 급격하게 방 출되었다.
이처럼 본 발명의 아토르바스타틴 칼슴 /암로디핀 베실레이트의 2상 매트릭스 정제 는 방출제어물질을 사용함으로써 의도한 시간까지 지연 시간을 가진 후 급격하게 암로디핀 베실레이트를 방출시킬 수 있다.
또한, 본 발명의 아토르바스타틴 칼슘 /암로디핀 베실레이트의 유핵정은 대 S약인 아토르바스타틴 칼슘 단일제와 암로디핀 베실레이 S 단일제를 동시 복용하였을 경 우의 용출 양상과는 달리 암로디핀 베실레이트의 초기 방출이 아토르바스타틴보다 매우 느리기 때문에 아토르바스타틴 칼슴이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크름 Ρ450 이 재생될 시간을 층분히 확보할 수 있어 , 부작용 등 발생 가능성을 감소시킬 수 있다. 실험예 II 1-4: 혈중농도시험
본 실험은 대조군으로서 시관증인 '리피토정' (아토르바스타틴 칼슴 10.85mg, 화 이자)과 '노바스크정, (베실산 암로디핀 6.9 ng, 화이자)을 단득 투여 군으로 하 고, 실험군으로 실시예 III-1에서 제조된 제제를 투여하여 본 발명에 의한 방출 지 연효과를 확인하는 혈중농도시험을 실시하였다.
한편 , 대조군 1, 대조군 2, 시험군에 대하여 각 시험군당 피험자는 6명 , 총 18명으로 시험을 실시하였으며 , 자세한 내용은 표 ΙΠ-3과 같다.
[표 III— 3]

대조군 1과 시험군에 의한 아토르바스타틴의 혈중농도 시험결과는 표 III-4( 아토르바스타틴의 혈종농도) 및 도 24에 나타내었다.
[표 ΠΙ-4]

시험군과 대조군 1의 평균 Tmax 값의 차이는 없었으며 , Cn x 값과 AUC(0-∞) 값에서도 유의적인 차이가 없었다.
대조군 2와 시험군에 의한 암로디핀의 혈증농도 시험결과는 표 III-5(암로디 핀의 혈중농도) 및 도 25에 나타내었다.
[표 πι-5]

대조군 2에 의한 암로디핀의 3 는6.5시간임을 알 수 있었으며 , 시험군의 암로디핀의 Tmax가 대조군 2에 의한 암로디핀의 Tma 에 비해서 약 3시간 이상 지연 됨이 확인되었다.
한편 , 암로디핀의 Qnax 및 AUC 값은 시험군과 대조군 두 군에서 유의적인 차 이가 없었다.
이로서 본 발명에 의한 약제학적 제제 (실시예 III-1)로 1일 1희 저녁에 투약 함으로써 , 병용처방에 따론 약물 상호작용을 배제할 수 있음을 확인하였다. 실험예 IV-1 : 암로디 ¾-피타바스타틴 유핵정의 비교 용 *실험 (comparative dissolution profile test)
상기 실시예 IV—1에 따라 제조된 피타바스타틴 /암로디 ¾ 베실레이트 유핵정과 대조 약 (리바로 :피타바스타틴 단일제 , 중의제약, 노바스크:암로디핀 단일제 , 화이자)의 비교 용출실험을 실시하였다.암로디핀 성분 용출시험의 경우 2시간을 기점으로 용 출액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다.각 성분별 용출시험 방법은 아래와 같으며 , 그 ¾과를 도 26에나타내었다.
도 26에 의하면 본 발명의 유핵정제는 하기 건에서 용출 시험 시 아토르바스타된 성분은 대조약인 리바로와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인 되었으나, 암로디핀 성분은 대조약인 노바스크와 비교할 때 매우 늦어진 용출 속도 를 확인할 수 있다.본 발명의 암로디핀 베실레이트 /피타바스티"틴의 유핵정은 1시간 까지의 암로디 ¾ 성분외 용출를이 모두 50% 이내로서 대조약의 용출를 (약 99%) 보다 훨씬 느리다.
이처럼 본 발명의 암로디핀 베실례이트 /피타바스타 ¾의 유핵정은 대조약인 암로디 핀 단일제와 달리 암로디핀의 초기 방출이 피타바스타틴 보다 매우 느리기 때문에 피타바스타틴보다 먼저 간에서 대사를 받을 확물이 낮아지게 된다.
실험예 IV-2:피타바스타 ¾ -암로디핀 유핵정의 비교 용출 실험 (comparative dissolution profi le test)
상기 실시예 IV-1 , 2, 3의 제제에 대하여 비교 용출실험을 실시하였다. 각 성분별 용출시험 방법은 실험예 IV-1과 같으며 , 그 결과를 도 27에 나타내었다.
도 27에 의하면 실험예 IV-1의 조건에서 용출 시험 시 본 발명의 방출성이 제어된 유핵정은 정제 타정후 장용성 코팅을 제조한 것 (실시예 IV— 1, 2)과 장용성 기제로 과립 제조하였을 때 (실시예 IV-3) 암로디핀 성분이 의도한 시간까지 지연시간을 가 진 후 비교적 급격하게 방출되는 것을 확인할 수 있었다. 암로디핀 성분의 용출를 은 총 480분까지 9W 이내이며 , 장용성 기제 및 장용성 코팅의 종류에 따라 암로디 핀 성분이 급격하게 방출되었다.
이처럼 본 발명의 피타바스타틴 칼슘 /암로디핀 베실레이트의 유핵정은 장용코팅을 함으로써 의도한 시간까지 지연 시간을 가진 후 급격하게 암로디핀을 방출시 ¾ 수 있다.
또한, 본 발명의 피타바스타틴 칼슴 /암로디핀 베실레이트외 유핵정은 대조약인 피 타바스타틴 칼슴 단일제와 암로디핀 단일제를 등시 복용하였을 경우의 용출 양상과 는 달리 암로디핀 베실레이트외 초기 방출이 피타바스타틴 칼슘보다 매우 느리기 때분에 피타바스타틴 칼슴이 먼저 간에서 대사롤 받은 후 대사 관련 효소인 사이토 크름 P450 이 재생될 시간을 층분히 확보할 수 있어, 부작용 등 발생 가능성을 감 소시킬 수 있다. 실험예 IV-3 :피타바스타틴-암로디핀 캡술제의 비교 용출 실험 (comparative dissolution profi le test)
상기 실시예 IV-17, 18 의 제제에 대하여 비교 용출실험을 실시하였다. 각 성분별 용출시험 방법은 실시예 IV-1과 같으며, 그 결과를 도 28에 나타내었다.
도 28에 의하면 실험예 IV-1의 조건에서 용출 시험 시 본 발명의 제어 방출 정제는 삼투성 정제 (실시예 IV-17)와 정제 타정 후 장용성 코팅을 하였을 때 (실시예 IV- 18) , 암로디 ¾ 성분이 의도한 시간까지 지연시간을 가진 후 비교적 급격하게 방출 되는 것을 확인할 수 있었다. 암로디핀 성분의 용출률은 총 480분까지 90% 이내이 며, 장용성 기제 및 장용성 코팅의 종류에 따라 암로디핀 성분이 급격하게 방출되 었다.
이처럼 본 발명의 피타바스타틴 칼슴 /암로디핀 베실레이트의 다층정숀 장용코팅을
함으로써 의도한 시간까지 지연 시간을 가진 후 급격하게 암로디핀 칼슴을 방출시 킬 수 있다.
또한, 본 발명의 피타바스타틴 칼슴 /암로디핀 베실레이트의 다층정은 대조약인 피 타바스타틴 칼슴 단일제와 암로디핀 베실레이트 단일제를 동시 북용하였을 경우의 용출 양상과는 달리 암로디 ¾ 베실테이트의 초기 방출이 피타바스타틴보다 매우 느 리기 때문에 피타바스타틴 칼슘이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크룸 P450 이 재생될 시간을 층분히 확보할 수 있어, 부작용 등 발생 가능성 을 감소시킬 수 있다, 실험예 V-1: 암로디핀-로슈바스타틴 유핵정의 비교 용출시험 (comparative dissolution profile test)
상기 실시예 V-4에 따라 제조된 로슈바스타된 /암로디된 베실레이트 유핵정과 대조 약 (크레스토:로슈바스타 ¾ 단일제 , 아스트라제네캬,노사스크:암로디핀 단일제, 화 이자)의 비교 용출시험을 실시하였다 .암로디핀 성분 용출시험의 경우 2시간을 기점 으로 용출액을 인공 위액에서 인공 장액으로 변경하여 용출시험을 진행하였다.각 성분별 용출시험 방법은 아래와 같다.
그 결과를 첨부 도면 도 29과 갈이 나타내었다.
도 29에 의하면 본 발명의 유핵정의 로슈바스타틴 성분은 대조 제제인 크레스토와 비교하여 거의 등등한 용출특성을 나타내는 것으로 확인되었으나, 암로디핀 성분은 대조 제제인 노바스크와 비교할 패 매우 늦어진 용출 속도를 확인할 수 있다. 구체 적으로 본 발명의 암로디핀 /로슈바스타틴의 유핵정은 용출시험 시작 후 1시간까지 의 암로디편 성분이 거의 용출되지 않아, 대조 제제의 1시간의 용출를 (약 99%)보다 ¾씬 느린 것을 확인 할 수 있었다.
이처럼 본 발명의 암로디핀 /로슈바스타틴의 유해정은 대조약인 암로디핀 단일제와 달리 암로디핀의 초기 용출이 로슈바스타된보다 매우 느리기 패문에 로슈바스타틴 보다 먼저 간에서 대사를 받을 확를이 낮아지게 된다. 실험예 V-2: 로슈바스타틴-암로디핀 캠술의 비교 용출 시험 (comparative dissolution profi le test)
상기 실시예 V-17, 18 제제에 대하여 실험예 V~l과 같은 방법으로 용출 시험을 실 시하였으며 , 각 제제에서의 암로디핀의 용출률을 측정하여 도 30에 나타내었다.
도 30에 의하면 실시 예 V-17(정제 타정 후 반투과성막 코팅을 한 경우) 및 질시예 V-18(정제 타정 후 장용성 코팅을 한 경우)에서 암로디핀은 로슈바스타틴 용출 개 시후 1시간 30분 이후에야 비로소 급격하게 용출되는 것을 확인할 수 있었다. 또 한, 암로디핀 성분의 용출률은 총 480분이내 90% 이상 용출되었다.
이처럼 본 발명의 로슈바스타틴 /암로디핀 캡슬은 반투과성막코팅 및 장용코팅을 함 으로써 의도한 시간까지 지연 시간을 가진 후 급격하게 암로디핀 베실레이트를 ' 용 출시킨다.
또한, 본 발명의 로슈바스타틴 칼슘 /암로디 ¾ 베실레이트의 ¾슬제는 대조약인 로 슈바스타틴 칼슘 단일제와 암로디핀 베실레이트 단일제를 동시 복용하였을 경우의 용출 양상과는 달리 암로디핀 베질레이트의 초기 용출이 로슈바스타틴보다 매우 느 리기 때문에 로슈바스타틴 칼슘이 먼저 간에서 대사를 받은 후 대사 관련 효소인 사이토크兽 P450 이 재생될 시간을 층분히 확보할 수 있어, 부작용 등 발생 가능선 을 감소시킬 수 있다. 실험예 : 니페디편-아토르바스타틴 유핵정의 비교 용출실험 (comparative dissolution profile test)
상기 실시예 VI-1에 따라 제조된 아토르바스타틴 /니페디핀 유핵정과 대조약 (리피 토: 아토르바스타틴 단일제 프로카디아 XL: 니페디핀 단일제 , 이상 화이자)의 비교 용출시험을 실시하였다. 각 성분별 용출시험 방법은 아래와 같으며 , 그 결과를 첨 부 도 31과 같이 나타내었다.
도 31에 의하면 하기 조건에서 용출 실험시 본 발명의 유핵정 내 아토르바스타틴 성분은 대 제제인 리피토와 비교하여 거의 동등한 용출특성을 나타내는 것으로 확인되었으나, 니폐디핀 성분은 대조 제제인 프로카디아 XL과 비교할 때 방출 시간 이 지연된 것을 확인할 수 있다.
이처럼 본 발명의 아토르바스타틴 /니페디핀의 유핵정은 대조약인 아토르바스타틴 단일제와 니페디핀 단일제롤 등시 복용하였을 경우의 용출 양상과는 달리 니페디핀 의 초기 방출이 아토르바스타틴과 층분한 시간차를 두고 재시되기 때문에 간으로 두 약물이 등시에 유입되어 대사적 상호작용이 발생하는 것을 차단할 수 있다. 실험예 VI-2 : 니페디핀-아토르바스타틴 2상 매트릭스정의 비교 용출실험 (comparative dissolution profi le test)
상기 실시예 에 따라 제조된 아토르바스타틴 /니페디핀 2상 매트릭스정제의 용 출시험을 실시하였다. 니떼디핀의 용출시험 방법은 아래와 같으며 , 그 결과를 첨부 도 32와 같이 나타내었다.
도 32에 의하면 본 발명의 2상 메트릭스정제는 하기 조건에서 용출 시험 시 니페디 핀 성분은 대조 제제인 프로카디아 XL (도 31의 방출 모습 참조)과 비교할 때 방출 지연시간이 늦어진 것을 확인할 수 있다.
이처럼 본 발명의 아토르바스타틴 /니꽤디핀의 2상 매트릭스정은 니페디핀의 초기 방출이 아토르바스타틴보다 층분한 시간차를 두고 개시되기 때문에 간으로 두 약물 이 등시에 유입되어 대사적 상호작용이 발생하는 것을 차단할 수 있다. 실험예 VI-3 : 니페디관-아토르바스타틴 다층정의 비교 용출시험 (comparative dissolution profile test)
상기 실시예 VI— 8에 따라 제조된 아토르바스타틴 /니쩨디편 다층정외 용출시험을 실 시하였다, 니페디핀의 용 *시험 방법은 아래의" 같으뎌 . 그 결과흩 첨부 도 33과 같 이 나타내었다.
도 33에 의하면 본 발명의 다층정제는 하기 조건에서 용출 시험 시 니페디 ¾ 성분 은 대조 제제인 프로카디아 XL (도 31의 방출 모습 참조)과 비교할 때 방출 지연시간 이 늦어진 것을 확인할 수 있다.
이처 ¾ 본 발명의 아토르바스타틴 /니페디핀의 다층정은 니페디핀의 초기 방 ¾이 아 토르바스타틴보다 층분한 시간차를 두고 개시되기 때문에 간으로 두 약물이 동시에 유입되어 대사적 상호작용이 발생하는 것을 차단할 수 있다. 실험예 VI-4 : 니페디핀-아토르바스타틴 캡술제의 비교 용출시험 (comparative dissolut ion profi le test)
상기 실시예 VI-10에 따라 제조된 아토르바스타틴 /니페디괸 캡술제의 용출시험을 실시하였다. 니페디핀의 용출시험 방볍은 아래와 같으며, 그 결과를 ¾부 도면 도
34와 같이 나타내었다.
도 34에 의하면 본 발명의 캡술제는 하기 조건에서 용출 시험 시 니페디핀 성분은 대조 제제인 프로外디아 XL (도 31의 방출 모습 참조)과 비교할 때 방출 지연시간이 늦어진 것을 확인할 수 있다.
이처럼 본 발명의 아토르바스타 ¾/니페디핀의 캡술제는 니 디핀의 초기- 방출이 아
토르바스타틴보다 층분한 시간차를 두고 개시되기 때문에 간으로 두 약불이 동시에 유입되어 대사적 상호작용이 발생하는 것을 차단할 수 있다.
[각 활성성분의 용출시험 방법 ]
[암로디핀 시험방법 ]
용출시험 근거 : 대한약전 제 8 개정 증 일반시험법의 용출시험 ¾
시험 방범 : 패들법 (Paddle method) , 75희전 /분
시험 액 : 0.01M 염산용액 , 750mL (0 2시간)
H 6.8 인공장액, l,OO0mL (2시간 이후)
분석방법 : 자의가시부흡광광도법 ( 검 * 파장 = 최대 240nm )
[레르카니디핀 시험방법 ]
용출시험 근거 : 대한약전 제 8 7)정 중 일반시험법의 용출시험법
시험 방법 : 패들법 (Paddle method) , 75회전 /분
시험 액 : 0.01M 염산용액, 750inL (0~2시간)
pH 6.8 인공장액, l,000mL (2시간 이후)
분석방법 : 고속액체크로마토그래프법
검출 파장 : 356 nra
이등상 : 아세토니] ¾ : 0.0IM 인산염 완층액 = 45 : 55(pH=4.0)
¾럼 : 안지름 4.6腿, 길이 250幽의 스테인테스관에 옥타데실실릴화한
유속 : 1.0 mL/분
[라시디핀 시험방법]
용출시험 근거 : 대한약전 제 8 개정 증 일반시험법의 용출시험법
시험 방법 : 패들법 (Paddle method) , 75회전 /분
시험액 : 0.01M 염산용액 , 750mL (0~2시간)
PH6.8 인공장액 , l,000mL (2시간 이후)
분석방법 : 고속액체크로마토그래프법
검출 파장 : 282iM
이동상 : 아세토니트릴 : 0.05M 암모늄 아세테이트 완충액 = 80 : 20
컬럼 : 안지름 4.6績, 길이 250誦의 스테인레스관에 옥타데실실릴화한 ¾
유속 : l.OmL/분
[펠로디핀 시험방법 ]
용출시험 근거 : 대한약전 제 8 개정 증 일반시험법의 용출시험법
시험 방법 : 패들법 (Paddle method), 50회전 /분
시험액 : 0.01M염산용액, 750mL-라우릴 황산 나트륨, 2¾ (0~2시간)
pH 6.8 인공장액 , l,000nL-라우릴 황산 나트륨, 2% (2시간 이후) 분석방법 : 자외가시부흡광광도법 ( 검출 파장 = 최대 254nm )
[이스라디핀 시험방뜁]
용출시험 근거 : 대한약전 제 8개정 중 일반시험법의 용출시험법
시험 방법 : 패돌범 (Paddle method), 50희전 /분
시험액 : 0.011 염산용액, 750mL-라우릴 황산 나트쁌, 2¾ (0—2시간)
pH 6,8 인공장액, l,000aiL-라우릴 황산 나트륨, 2% (2시간 이후) 분석방법 : 자외가시부홉광광도법 ( 검출 파장 = 최대 328nm )
[니폐디¾ 시험방법]
용출시험 근거 : 대한약전 제 8 개정 증 일반시험법의 응출시험법
시험 방법 : 패들법 (Paddle method), 100회전 /분
시험액 : 1%라우 ¾황산나트 ", pH 6.8 인산완층액, 900mL (0~24시간)
분석방법 : 고속액체크로마토그래프법 (검출 파장 : 350 nm )
[심바스타틴 시험방법]
용촐시험 근거 : 미국약전 (USP 29)중의 'Simvastatin tablet1항
시험 방¾ : 패읗법 (Paddle method), 50희전 /분
시험액 : pH=7.0 완층액 ( 조성 = 계면활성제로서 라우릴 황산 나트륨 0.5% 증량 /증 량을함유하는 0.0M 인산이수소나트륨용액 ), 900mL
분석방법 : 자의가시부흡광광도법 ( 검출 파장 =최대 247, 최소 257nm )
[로바스타틴 시험방법 ]
용출시험 근거 : 미국약전 JSF 29)중의 'Lovastatin tablet'항
시험 방볍 : 패들법 (Paddle method) , 50회전 /분
시험액 : pH=7.0 완층액 ( 조성 = 계면활성제로서 라우릴 황산 나트륨 약 증량 / 증량을 함유하는 0.01M 인산이수소나트륨 용액 ) , 900mL
분석방법 : 고속액체크로마토그래프법
검출 파장 : 230nra
이등상 : 아세토니트 ¾ : 0.02M 인산이수소나트륨 완층액 (pH-4.0) : 메탄을 = 5 : 3 : 1
¾럼 : 안지름 4.6麵, 길이 250 mm의 스테인레스관에 옥타데실실 ¾화한 ¾ 유속 : 1.5raL/분
[아토르바스타틴 시험방법;!
용출시험 근거 : 대한약전 제 8 개정 증 일반시험법의 용출시험법
시험 방법 : 패들법 (Paddle method) , 50회전 /분
시험액 : PH=7.0 완층액 ( 조성 = 계면활성제로서 라우릴 황산 나트륨 Ά 증량 /증량 을 함유하는 0.01M 인산이수소나트륨 용액 ) , 900mL
분석방법 : 고속액체크로마토그래프법
검출 파장 : 247
이등상 : 메탄을 :0.025M 인산이수소나트튬 완층액 (pH-4.0) = 67:33 (pH=4.0) 컬럼 : 안지름 4·6ΙΜ, 길이 250匪의 스테인레스관에 옥타데실실 ¾화한 ¾ 유속 : 1.5mL/분
[플루바스타틴 시험방법]
용출시험 근거 : 디국약전 (USP 30)중의 'Fluvastat in capsules '항
시험 방법 : 패들법 (Paddle method) , 50회전 /분
시험 액 : 정제수, 900mL
분석방법 : 고속액체크로마토그래프법
검출 파장 : 235nm
이등상 : 메탄을 /아세토니트릴 (3:2)혼액 : 0.016M 인산이수소암모늄 완층액 (pH 3.5) = 7:3
컬럼 : 안지름 4.6瞧, 길이 100 mm의 스테인레스관에 옥타데실실 ¾화한 겔 유속 : 2.0mLJ분
[피타바스타린 시험방법 ]
용출시험 근거 : 대한약전 제 9 개정 증 일반시험법의 용출시험법
시험 방법 : 패들법 (Paddle method) , 50희전 /분
시험액 : 0.0 염산용액 , 750mL (0—2시간)
pH 6.8 인공장액, l,000mL (2시간 이후)
분석방법 : 자외가시부흡광광도법 ( 검출 파장 - 254nra )
[로슈바스타틴 시험방 ¾]
용출시험 근거 : 대한약전 제 9 개정 증 일반시험법의 용출시험법
시험 방법 : 패들법 (Paddle method) , 50회전 /분
시험액 : 0.01M 염산용액 , 750mL (0~2시간)
6.8 인공장액, l.OOOmL (2시간 이후)
분석방법 : 자의가시부흡광광도법 ( 검출 과장 = 220nm )
【¾업상 이용가능성】
상술한 바와 같이 , 본 발명은 이종약물의 병용시 발생할 수 있는 부작용을 약물등태학적 관점에서 개선하고자 하는 이종 약물 대사학 (Xenobiotics)을 근거로 하여 치료 효과를 극대화시키는 이른바 시간 차 투약 치료법 (Chronotherapeutics) 을 제제화시킨 것으로서 , 동일한 효소인 사이토크름 P 450계 효소에 영향을 주거나 또는 효소활성을 저해하는 성분인 디히드로피리딘계 칼슴길항제와 스타틴계 지질 저하제를 유효 활성성분으로 복합하여 사응하면서, 이들이 체내에서 용출되는 시간 을 제어할 수 있도록 시간차 방출성 물질 및 속방성 물질을 차별성 있게 사용함으 로써 활성성분을 특정 속도로 시간차를 두고 송달 가능하므로, 상기 약물성분을 따 로따로 동시에 복용하는 복합 처방의 경우 보다 만성 순환기 질환의 예방과 치료에 서 약리학적, 임상학적 , 과학적 및 경제적으로 보다 유용한 기능성 시간차 루여 약 제학적 제제를 실현케 할 수 있다.
. 또한, 본 발명에 의하면 상기 약물의 방출속도를 달리 구성하여 약불 상호간 의 ¾항 작용 및 부작용을 방지함과 등시에 약효의 상승 작용을 얻을 수 있다. 또한, 본 발명에 의하면 복합 제제를 한 번에 복용할 수 있으므로 환자에 대 한 복약 지도 및 환자의 복약이 용이하다.
Claims
【청구의 범위】
【청구항 1】
디히드로피리딘계 칼슴 채¾ 차단제, 그의 이성질쳬, 또는 그의 약제학적으' 로 허용가능한 염을 활성성분으로 하며, 과립으로 제조된 지연방출성 구획 및 스타 틴계 지질저하제를 활성성분으로 하며, 펠¾ 또는 정제로 제 S된 선방출성 구획을 포함하는 캡술제인 약제학적 제제.
【청구항 2】 ·
디히드로피리딘계 칼슴 채널 차단제, 그의 이성질체, 또는그의 약제학적으 로 허용가능한 염을 활성성분으로하며, 또는 정제로제조된 지연방출성 구획 및 스타 ¾계 지질저하제롤 활성성분으로하며 , 과립, ¾렛, 또는정제로 제조된 선 방출성 구획을포함하여 이루어진 캡술제인 약제학적 제제.
【청구항 3】
(a) 활성성분으로서 디히드로피리딘계 칼슘길항제, 그의 이성질체, 또는 그 의 약제학적으로 허용가능한 염 및 약제학적으로허용가능한 담체를 포함하는 지연 방출성 구획
(b) 활성성분으로서 스타틴계 지질저하제, 그의 이성질체, 또는 그의 약제학적으로 허용가능한 염 및 약제학적으로 허용가능한 담체를 포함하는 선방출성 구획 및 (c) 상기 지연방출성 구획과 선방출성 구획을 함께 층진하기 위한 용기 수단을 포함하 는 키트인 약제학적 제제.
【청구항 4】
디히드로피리딘계 칼슴 채¾ 차단제를 활성성분으로하며, 정제로 제 된 지 연방출성 구획 및 스타틴계 지질저하제를 활성성분으로 하며, 상기 지연방출성 구 획의 표면에 코팅되는 선방출성 구획을포함하여 이루어진 코팅정제인 약제학적 제 제.
【청구항 5】
디히드 a피리딘계 칼슘 채널 차단제를 활성성분으로 하며 , 삼투압 조절제를 포함하여 삼투암에 의해 꺅물을 방출하는 내핵을 구성하는 지연방출성 구획 및 스 타된계 지질저하제를 활성성분으로 하며, 상기 내핵의 외층을 구성하는 선방출성 구획을포함하여 이루어진 삼투성 유핵정인 약제학적 제제.
【청구항 6]
제 1 항 내지 제 5항 증 어느 한 항에 있어서, 상기 디히드로피리딘계 칼슴
채널 차단제가스타틴계 지질저하제 보다 2시간 내지 4시간 늦게 간에서 흡수되도 록방출성이 조절된 약제학적 제제.
【청구항 7】
제 1항내지 제 5항증어느 한항에 있어서, 상기 디히드로피리딘계 칼슴채 ¾ 차단제는 암로디핀, 레르카니디핀, 라시디핀, ¾로디핀, 바니디핀, 베니디핀, 실니디핀, 이스라디핀, 마니디핀, 니카르디핀, 니페디핀, 니모디핀, 닐바디핀, 니 술로디핀, 니트렌디핀 아¾니디 ¾ 그의 이성질체, 및 약학적으로 허용 가능한중 에서 선택된 1종이상인 약제학적 제제.
【청구항 8]
제 7항에 있어서, 상기 디히드로피리딘계 칼슴채널 차단제는 제제 증 1 ~ 400 mg범위로포함되는 약제학적 제제.
【청구항 9 ½
제 1항내지 제 5항중 어느한항에 있어서, 상기 스타 ¾계 지질저하제는 심 바스타틴, 로바스타틴, 아토르바스타된, 피타바스타틴, 로수바스타된, 플투바스타 틴, 프라바스타틴, 그의 이성질체, 및 그의 약제학적으로 허용 가능한 염 중에서 선택된 1종 이상인흔합물인 약제학적 제제.
【청구항 10】
약리학적 활성성분으로 심바스타틴, 그의 이성질체 또는 그의 약제학적으로 허용가능한 염을포함하는선방출성 구획 및 약리학적 활성성분으로 암로디핀, 그 의 이성질체 또는 그의 약제학적으로 허용 가능한 염을 포함하는지연방출성 구획 을포함하는 약제학적 제제.
【청구항 11】
약리학적 활성성분으로 아토르바스타틴, 또는 이의 약학적으로 허용가능한 염을 포함하는 선방출성 구획 및 약리학적 활성성분으로 암로디핀 또는 그의 약계 학적으로허용가능한염을포함하는지연방출성 구획을포함하는약제학적 제제 .
【청구항 12】
약리학적 활성성분으로 피타바스타틴 또는 그의 약제학적으로 허용 가능한 염을 포함하는 선방출성 구획 및 약리학적 활성성분으로 암로디핀 또는 그의 약제 학적으로허용가능한염을포함하는지연방출성 구획을포함하는약제학적 제제. 【청구항 13】
약리학적 활성성분으로 로슈바스타틴 , 그의 이성질체 또는 그의 약학적으로 허용 가능한 염을 포함하는 선방출성 구획 및 약리학적 활성성분으로 암로디핀, 그 의 이성질체 또는 그의 약학적으로 허웅 가능한 염을 포함하는 지연방출성 구획을 포함하는 약학적인 제제 .
【청구항 14】
약리학적 활성성분으로 아토르바스타틴 또는 그의 약제학적으로 허용 가능한 염을 포함하는 선방출성 구획 및 약리학적 활성성분으로 니페디핀 또는 그의 약제 학적으로 허용 가능한 염을 포함하는 지연방출성 구획올 포함하는 약제학적 제제 .
【청구항 15]
제 1 항 내지 제 5항 및 제 10항 내지 제 14항 증 어느 한 항에 있어서, 상기 지연방출성 구획은 장용성 고분자, 수블용성 중합체 , 소수성 화합물, 친수성 고분 자 및 이들의 혼합물 증에서 선택된 방출제어물질을 1종 이상 포함하는 약제학적 제제 .
【청구항 16]
제 15항에 있어서 , 상기 장용성 고분차는 장용성 셀를로오스 유도채 , 장용성 아크릴산계 공중합체 , 장용성 말레인산계 공중합체 , 장용성 풀리비닐 유도체, 및 이들의 흔합물로 이루어진 군에서 선택된 1종 이상인 약제학적 제제 .
【청구항 17]
제 16 항에 있어서, 상기 장용성 샐를로오스 유도체는 히드록시프로필메틸젤 를로오스아세테이트숙시네이트, 히드톡시프로필메털샐를로오스프탈레이트, 히드특 시메틸에틸셸를로오스프탈레이트, 쎌를로오스아세테이트프탈레이트, ^를로오스아 세테이트숙시네이트, 샐를로오스아세테이트말레이트, 샐를로오스벤조에이트프탈레 이트, 샐를로오스프로피오네이트프탈레이트, 메틸셀를로오스프탈레이트, 카르복시 메¾에틸셀를로오스, 에틸히드록시에¾¾를로오 ώ프탈레이트, 메틸히드록시에¾¾ 를로오스 및 이들의 흔합물 증에서 선택된 1종 이상이고 ; 상기 장용성 아크릴산계 공증합체는 스티¾ -아크릴산 공중합체, 아크 ¾산메 ¾ -아크릴산 공중합체 , 아크 ¾산 메틸메타크 ¾산 공증합체 , 아크릴산부틸 -스티렌―아크릴산 공증합체 , 데타크 ¾산―메 타크릴산메 ¾ 공중합체, 메타크 ¾산 ' 아크릴산에틸공중합체, 아크릴산메틸-메타크 ¾산-아크¾산옥틸공중합체 및 이들의 흔합물 증에서 선택된 1종 이상이며 ; 상기
장용성 말레인산계 공증합쳬는 아세트산비닐-말레인산 무수물 공중합체 , 스티렌-말 레인산 무수몰 공증합체 , 스티렌-말레인산모노에스테르 공중합체 , 비닐메틸에테르一 말레인산 무수툴 공중합체 , 에틸렌-말레인산 무수물 공증합체 , 비닐부틸에테르―말 레인산 무수물 공중합체 , 아크릴로니트릴-크릴산메틸 · 말레인산 무수물 공중합체, 아크릴산부 ¾-스티'렌-말레인산 무수물 공중합체 및 이들의 혼합물 증에서 선택된 1 종 이상이고; 상기 장용성 플리비닐 유도체는플리비날알콜프탈레이트, 플리비닐아 세탈프탈레이트, 폴리비닐부티레이트프탈레이트,및 플리비닐아세:트아세탈프탈레이 트 및 이들외 흔합물 중에서 선택된 1종 이상인 약제학적 제제 .
【청구항 18】
제 15 항에 있어서 , 상기 수불용성 중합체는 ¾리비닐 아세테이트 , 풀리메타 크¾레이트 공증합체, *리 (에틸아크릴레이≤-메틸 메타크릴레이트) 공증합체 , 풀 리 (에틸아크 ¾레이트-메틸 쩨타크 ¾레이트 -트리메틸아미노에틸메타크릴레이트)공증 합체 , 에 ¾셀를로오스, 셀를로오스 에스테르, ¾를로오스 에테르, 셀를로오스 아실 레이트, ¾를로오스 디아실레이트, 첼를로오스 트리아실레이트, 셀를로오스 아세테 이트> 샐를로오스 디아세테이트 , 셀를로오스 트리아세테이트 및 이들의 흔합물 중 에서 선택된 1 종 이상인 약제학적 제제 .
【청구항 19】
제 15 항에 있어서 , 상기 소수성 화합물은 지방산 및 지방산 에스테르류, 지 방산 알코올류, 왁스류, 무기물질 및 이들의 흔함물 증에서 선택된 1종 이상인 약 제학적 제제 .
【청구항 20]
제 19 항에 있어서 , 상기 지방산 및 지방산 에스테르류로는 글리세릴 괄미토 스테아레이트, 글리세 ¾ 스테아레이트, 글리세릴 비해네이트, 세틸 괄미테이트 , 글 리세 ¾ 모노 을레이트, 스테아린산 및 이들의 흔합물 중에서 선택된 1종 이상 이 고; 상기 지방산 알코을류는 세토스테아 ¾ 알코을, 세 ¾알코을, 스테아릴알코을 및 이들의 흔합물 증에서 선택된 1종 이상 이며 ; 상기 왁스류는 카르나우바왁스, 밀 납, 미결정왁스 및 이들의 흔합물중에서 선택된 1종 이상 이고 : 상기 무기물질은 탈크, 침강탄산칼슘, 인산일수소칼슴, 산화아연 , 산화티탄, 카을린 , 벤토나이트, 몬모틸로나이트, 비검 및 이들의 흔합물 중에서 선택된 1종 이상인 약제학적 제제 , 【청구항 211
제 15항에 있어서, 상기 친수성 고분자는 당류, 셀를로오스 유도체, 검류,
단백질류, 풀리비닐 유도체, 플리메타크 ¾레이트 공증합체, 폴리에틸¾ 유도체. 카 르복시비닐공중합체 및 이들의 혼합물 중에서 선택된 1종 이상인 약제학적 제제. 【청구항 22】
제 21 항에 있어서, 상기 당류는 덱스트린, 플리덱스트린, 덱스트란, 펙틴 및 펙틴 유도체, 알긴산염, 폴리갈락투론산, 자일란, 아라비노자일란, 아라비노갈 락탄, 전분, 히드록시프로필스타치, 아밀로오스, 아밀로펙틴 및 이들의 흔합물 중 에서 선택된 1종 이상이고; 상기 셀를로오스유도체는 히드록시프로필메틸셀를로오 스, 히드록시프로필셀를로오스, 히드록시메털셀를로오스, 히드록시에털셀를로오스, 메틸셸를로오스, 카르복시메틸셸를로오스 나트 히드록시프로필 메 ¾¾를로오스 아세테이트 숙시네이트, 히드록시에틸메틸셀를로오스 및 이들의 혼합물 중에서 선 택된 1종 이상이며; 상기 검류는 구아검 , 로커스트 콩검 , 트라가칸타, 카라기난, 아카시아검 , 아라비아검, 젤란검, 잔탄검 및 이들외 혼합물 증에서 선택된 1종 이 상이고; 상기 단백질류는 ¾라된, 카제인, 제인 및 이들의 흔합물 증에서 선택 1종 이상이며; 상기 폴리비닐 유도체는 플리비닐 알코올, 플리비닐 피를리돈, 폴리비닐 아세탈디에 ¾아미노아세테이트 및 이들의 흔합물 증에서 선택된 1종 이상이며; 상 기 폴리메타크릴테이트 공중합체는 플리 (부틸 메타크 ¾테이트, (2-디메틸아미노에 틸)메타크릴레이트-메틸떼타크릴테이트) 공증합체, 플리 (메타크릴산-메 ¾메타크릴 레이트) 공증합체 및 풀리 (메타크릴산-에틸아크릴레이트) 공증합체, 및 이읗의 흔 합물 중에서 선택된 1종 이상이고; 상기 를리에틸렌 유도체는 볼리에틸렌글리콜, 폴리에틸렌 옥사이드 및 이들외 흔합물 증에서 선택된 1종 이상이며; 상기 카르복 시비닐 증합체는 카보머인 것인 약제학적 제제.
【청구항 23】
제 10항 내지 제 14항 충 어느 한 항에 있어서, 상기 제제는 지연방출성 구획 및 선방출성 구획이 균일하게 흔합된 후 타정하여 얻어지는 2상의 매트릭스 정제 형태인 약제학적 제제.
【청구항 241
제 10항 내지 제 14항 증 어느한 항에 있어서 , 상기 약제학적 제제는 지연방 출성 구획으로 이루어진 정제와 상기 정제의 의부를 둘러싸는 선방출성 구획으로 이루어진 필름코팅층으로구성된 필 *코팅정 형태인 약제학적 제제.
【청구항 25】
제 10항내지 제 14항중 어느 한항에 있어서, 상기 약제학적 제제는상기 지
연방출성 구획과상기 선방출성 구획이 층을 이루는 다층정 형태인 약제학적 제제. 【청구항 26]
제 ισ항 내지 제 14항 증 어느 한 항에 있어서, 상기 제제는 지연방출성 구획 으로 이루어진 내핵정과, 상기 내핵의 외면을 들러싸고 있는 선방출성 구획으로 이 투어진 외층으로구성된 유핵정 형태인 약제학적 제제.
【청구항 27】
제 26항에 있어서, 상기 유핵정은 삼투성 유핵정인 약제학적 제제.
【청구항 281
제 10항 내지 제 14항 중 어느 한 항에 있어서, 상기 약제학적 쩨제는 지연방 출성 구획으로 이루어진 입자, 과립, ¾¾, 또는 정제와, 선방출성 구획으로 이투 어진 입자, 과립, ¾, 또는정제를포함하는 캡술제 형태인 약제학적 제제. 【청구항 29】
제 10항 내지 제 14항 증 어느 한 항에 있어서, 상기 지연방출성 구획, 또는 상기 선방출성 구획 증 하나 이상의 외부에 코팅층을추가로 포함하는 약제학적 제 제.
【청구항 30】
제 10항 내지 제 14항 증 어느 한 항에 있어서, 상기 지연방출성 구획은 삼투 압조절제를포함하며 반투과성막코팅기제로코팅된 것인 약제학적 제재 .
【청구항 31]
제 30항에 있어서, 상기 삼투압 조절제는 황산마그네슘, 염화마그네 , 염 화나트륨, 염화칼륨, 인산나트륨, 인산칼晉, 초산암모늄, 염화리튬, 황산칼륨, 황 산나트콤, 황산리튬, 황산나트품, 및 이돌의 흔합물로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제.
【청구항 32】
제 30항에 있어서, 상기 반투과성막코팅기제는 *리비닐 아세테이트, 플리 메타크릴례이트 공증합체, 폴리 (에틸아크 ¾레이트, 메¾ 메타크 ¾레이트) 공증합 체, 폴리 (에틸아크릴레이트, 메¾ 메타크 ¾테이트, 트리메틸아미노에틸폐타크릴레 이트)공증함체, 에될샐를로오스, 셀 1"로오스 에스테르, 셸를로오스 에테르, ·를로 오스 아실레이트, ■€를로오스 디아실테이트, 셀를로오스 트리아실레이트, 셀를로오 스 아세테이트, 쌜를로오스 디아세테이트, 셀를로오스 트리아세테이트, 및 이들의 흔합 "로 이루어진 군에서 선택된 1종 이상인 것인 약제학적 제제 .
【청구항 33】
제 10항 내지 제 14항 중 어느 한 항에 있어서, 외부에 코팅층을 추가로 포함 하는코팅정 형태인 약제학적 제제.
【청구항 34]
제 10항내지 제 14항증 어느 한항에 있어서 , 상기 제제는지연방출성 구획 , 및 선방출성 구획을포함하는키트 형태인 약제학적 제제.
【청구항 35]
제 10항 내지 제 14항 중 어느 한 항에 있어서, 상기 제제는 저녁투여용인 약 제학적 제제.
【청구항 36]
제 1항 내지 제 5항 및 제 10 항 내지 제 14항 중 어느 한 항의 약제학적 제 제를포유류에게 투여하는 단계를포함하는 심혈관계 질환치료방법.
【청구항 37】
제 10 항에 있어서, 심바스타틴은그 방출개시 후 1시간 이내에 제제 중 심바 스타틴총량의 80¾ 이상이 방출되는 것인 약제학적 제제.
【청구항 38]
제 10항에 있어서, 암로디핀은 심바스타틴 방출개시 후 1시간 이후에 방출이 개시되고, 8시간 내에 방출이 완료되는 것인 약제학적 제제.
【청구항 39]
제 38 항에 있어서, 암로디핀은 심바스타틴 방출 개시 후 3시간 이내에 단위 제제 중총량의 40% 이내가방출되는 것인 약제학적 제제.
ί청구항 40】
제 10항에 있어서, 상기 심바스타틴은 약제학적 제제 중 1 내지 160 mg 범위 로 포함되는 약제학적 제제. - 【청구항 41]
제 10항에 있어서, 상기 암로디핀은 약제학적 제제 증 1 내지 40 mg 범위로 포함되는 약제학적 제제.
【청구항 42]
제 10 항에 있어서, 상기 지연방출성 구획은 장용성 고분자 및 친수성 고분 자를방출제어물질로포함하는 것인 약제학적 제제.
【청구항 43]
제 15 항에 있어서 , 상기 제 10항의 장용성 고분자는 풀리비닐아세테이트프탈 레이트, 히드록시프로필메틸셀를로오스프탈레이트, 쉘락, ¾를로오스아세테이트프 탈테이트, ¾롤로오스프로피오네이트프탈레이트 플리 (메타크릴레이트, 메틸메타크 릴레이트)공중합체 및 폴리 (데타크¾레이트 , 에틸아크릴테이트)공중합체 및 이돌의 흔합물 중에서 선텍된 1 종 이상인 약제학적 제제 .
【청구항 44]
제 42항에 있어서 , 상기 장용성 고분자는 플리비닐아세테이트프탈레이트, 히 드록시프로필메틸셀를로오스프탈레이트, «락, 셀 *로오스아세테이:트프탈레이트 , 샐를로오스프로피오네이트프탈레이트, *리 (메타크릴레이트, 메틸메타크필레이트) 공중합체 및 플리 (매타크릴레이트, 에틸아크릴레이트)공중합체 및 이들의 흔할물 증에서 선택된 1 종 이상인 약제학적 제제 .
【청구항 45】
겨 1 15 항에 있어서 , 상기 제 10항의 수불용성 증합체는 폴리비닐 아세테이트, 플리메타크릴레이트 공증합체 , 풀리 (에털아크릴레이트—메틸 데타크릴레이트) 공중 합체 , 풀리 (에틸아크 ¾테이트, 메¾ 메타크 ¾레이트, 트리메틸아미노에 ¾메타크 ¾ 레이트)공증합체 , 에틸셀를로오스, 셀를로오스 아세테이트, 및 이들의 흔합물 중에 서 선택된 1종 이상인 약제학적 제제 .
I청구항 46]
제 15 항에 있어서 , 상기 제 10항의 친수성 고분자는 전분, 히드록시프로필메 틸셀를로오스, 히드특시프로필셀를로오스, 카르복시메 ¾·¾를로오스 나트륨, 잔탄 검 , 폴리비닐 알코을, 카르복시비닐폴리머 및 이들의 흔합물로 이루어진 군에서 선 택된 것인 약제학적 제제 .
【청구항 471
제 42 항에 있어서, 상기 친수성 고분자는 전분 히드록시프로필메틸셀를로 오스, 히드륙시프로필셀를로오스, 카르복시메틸셸를로오스 나트콤, 잔탄검 , 폴리비 닐 알코올, 카르복시비닐폴리머 및 이들의 혼합물로 이루어진 군에서 선택된 것인 약제학적 제제 .
【청구항 48)
제 15항에 있어서 , 상기 제 10항의 방출제어물질이 암로디핀 1증량부에 대하여 0.1 내지 100 중량부로 함유된 약제학적 제제 .
【청구항 49】
제 15항에 있어서, 상기 제 10항의 방출제어물질은 ¾리비닐아세테이트, 폴리 메타크 ¾레이트 공중합체, 폴리 (에틸아크릴레이트, 메틸 메타크 ¾레이트, 트리메틸 아미노에틸메타크 ¾레이트)공중합체, 카르북시비닐 플리머, 에틸셀를로오스, 셀를 로오스 아세테이트, 카르툭시메틸셀를로오스 나트륨, 폴리에틸렌 옥사이드, 히드특 시프로필메틸셀 ■로오스ᅳ 히드톡시프로필^를로오스, 및 히드록시프로필메틸셸를로 오스프탈테이트로구성된 군으로부터 선택된 1종 이상의 것인 약제학적 제제. 【청구항 50]
제 49항에 있어서, 상기 방출제어물질은 카르복시비닐플리머, 히드록시프로필 메틸쎌를로오스, 및 히드록시프로필메틸샐를로오스프탈레이트로 구성된 군으로부터 선택된 1종 이상의 것인 약제학적 제제.
【청구항 51】
제 42항 또는 제 49항에 있어서, 상기 방출제어물질은 카르복시비닐폴리머 및 히드록시프로필메틸셀를로오스의 흔합불인 약^학적 제제.
【청구항 52]
제 11항에 있어서, 상기 아토르바스타틴은그 방출개시 후 1시간 이내에 단위 제제 중 아토르바스타틴 총량의 90% 이상이 방출되는 것인 약제학적 제제.
【청구항 53]
제 11항에 있어서, 상기 암로디핀은 아토르바스타틴 방출 1시간 이후에 방출 개시되고 8시간 이전에 방출 완료되는 것인 약제학적 제제 .
【청구항 54]
제 53항에 있어서, 상기 암로디 ¾은 아토르바스타틴 방출 1시간 이후에 방출 개시되고 6시간 이전에 방출 완료되는 것인 약제학적 제제.
【청구항 55】
제 53항에 있어서, 상기 약제학적 제제는 아토르바스타틴 방출 개시 후 4시간 이내에 단위 제제 중 암로디핀 총량의 20%이내가방출되는 것인 약제학적 제제. 【청구항 56)
제 11항에 있어서, 상기 암로디핀은 그의 (S)이성질체, (R) 이성질체, 및 라 세미체로 이루어진 군에서 선택된 것인 약제학적 제제.
【청구항 57】
제 11항에 있어서, 상기 아토르바스타틴은그의 (RI?)이성질체, (RS) 이성질체 (SR)이성질체, (SS) 이성질체, 및 라세미체로 이루어진 군에서 선택된 것인 약제학
적 제제 .
【청구항 58]
제 n항에 있어서 , 상기 지연방출성 구획은 장용성 고분자 및 친수성 고분자 의 혼합물인 방출제어물질을 포함하는 것인 약제학적 제제 .
【청구항 59]'
제 58항에 있어서 , 상기 장용성 고분자 및 친수성 고분자는 각각 암로디핀 1 증량부 대비 0.5 내지 10 중량부 및 0.5 내지 20 중량부로 포함되는 약제학적 제 제 .
【청구항 60]
제 58항 또는 제 59항에 있어세 상기 장용성 고분자는 장용성 셀를로오스 유 도체로서 히드록시프로필메 ¾셀를로오스아세테이트숙시네이트, 히드록시프로필메틸 쉘를로오스프탈레이트, 히드록시메 ¾에틸셀를로오스프탈테이트, 롤로오스아세테 이트프탈레이트, ¾롤로오스아세테이트숙시네이트, 셀를로오스아세꿰이트말레이트, ¾를로오스벤조에이트프탈레이트, 셀를로오스프로괴오네이트프탈레이트, 메틸셀를 로오스프탈레이트, 카르복시메틸에틸 ¾를로오스, 에¾히드특시에틸샐를로오스프탈 테이트, 및 메틸히드특시에털샐를로오스 증에서 선택된 하나 이상; 장용성 아크릴 산계 공중합체로서 스티렌 -아크 ¾산 공중합체, 아크릴산메틸 -아크 ¾산 공중합체, 아크릴산메틸메타크릴산 공중합체, 아크 ¾산부틸 -스티렌-아크릴산 공중합체, 메타 크¾산-메타크 ¾산메 ¾ 공증합채 , 펴]타크릴산■아크릴산에틸공중합체, 및 아크릴산 메틸-메타크릴산-아크릴산옥틸공중합체 중에서 선택된 하나 이상; 장용성 말레인산 계 공중합체로서 아세트산비닐-말레인산 무수물 공증합체 , 스티렌-말테인산 무수물 공증합체, 스티렌-말레인산모노에스테르 공중합체, 비닐메필에테르-말레인산 무수 물 공증합체,' 에 ¾렌-말레인산 무수물 공증합체 , 비닢부 ¾에테르-말레인산 무수물 공증합체 , 아크릴로니트 ¾ -크릴산메틸 · 말레인산 무수물 공중합체 , 및 아크 ¾산부 ¾ -스티렌-말레인산 무수물 공중합체 증에서 선택된 하나 이상; 장용성 ¾리비닐 유도체로서 «■리비닐알콜프탈레이트, 폴리비닐아세탈프탈레이트, 플리비닐부티레이 트프탈레이트 및 폴리비닐아세트아세탈프탈레이트 중에서 선택된 하나 이상 및 그 흔합물 증에서 선택된 하나 이상이고,
상기 친수성 고분자는 셀를로오스 유도체로서 히드록시프로필메틸셀를로오스, 히드 록시프로필셀를로오스, 히드록시메틸젤를로오스, 히드특시에틸셀를로오스, 메틸셀 를로오스, 카르복시메될셀를로오스 4트 1", 히드특시프로필 떼틸셀롤로오스 아세테
이: 숙시네이트 , 및 히드록시에틸데¾¾를로오스 중에서 선텍된 하나 이상; 검류 로서 구아검, 로커스트 콩 검 , 트라가칸타, 카라기난, 아카시아검 , 아라비아검 , 젤 란검 , 및 잔탄검 증에서 선택된 하나 이상; 단백질류로서 ¾라틴 , 카제인 , 및 제인 증에서 선택된 하나 이상 ; 플리비닐 유도체로서 플리비닐 알코을, 폴리비닐 피를리 돈, 및 폴리비닐아세탈디에 ¾아미노아세테이트 증에서 선택된 하나 이상; 폴리메타 크릴레이트 공증함체로서 풀리 (부 ¾ 메타크릴레이트— (2-디메틸아미노에틸)메타크 ¾ 레이트ᅳ데틸데타크릴레이트) 공중합체, 풀리 (메타크릴산— 메틸메타크 ¾레이트) 공 증합체 , 및 플리 (데타크¾산-에틸아크 ¾레이트) 공증합체 중에서 선택된 하나 이 상; ¾리에틸렌 유도체로서 플리에틸렌 글리콜 , 및 폴리에틸렌 옥사이드 중에서 선 택된 하나 이상 카르복시비닐폴리머로서 카보머 및 그 흔합물 중에서 선택된 하나 이상인 약제학적 제제 .
【청구항 61】
제 11 항에 있어서, 상기 암로디 ¾은 단위제제 증 1 내지 20iiig 포함되는 것 인 약제학적 제제 .
【청구항 62】
제 11항에 있어서 , 상기 아토르바스타틴은 단위제제 중 5 내지 160 mg 포함되 는 것인 약제학적 제제 .
【청구항 63】
제 15항에 있어서 , 상기 제 11항의 방출제어물질은 플리비닐아세테이트, 폴리 메타크 ¾레이트 공중합체, 카르복시비닐 폴리머 , 히드톡시프로필메털샐를로오스, 히드특시프로필 ·€를로오스, 쎌를로오스 아세테이트, 에틸쌜를로오스, 폴리에틸렌 옥사이드 및 히드록시프로필메틸셀를로오스프탈레이트로 구성된 군은로부터 '선택된 1종 이상의 것인 약제학적 제제 .
【청구항 64】
제 63항에 있어서 r 상기 방출제어물질은 카르복시비닐풀리머, 폴리메타크 ¾레 이트 공중합체 및 히드록시프로필메 ¾셀를로오스로 구성된 군으로부터 선택된 1종 이상의 것인 약제학적 제제 .
【청구항 65】
제 58항 또는 제 63항에 있어서 , 상기 방출제어물질은 카르복시비닐폴리머 및 히드톡시프로필메 ¾셀를로오스인 약제학적 제제 .
【청구항 66]
제 12항에 있어서 , 상기 선방출성 구획의 피타바스타틴은 방출개시 후 15분 이내에 피타바스타틴 총량의 80¾ 이상이 방출되는 약제학적 제제 .
【청구항 67]
제 12항에 있어서 , 상기 암로디 ¾은 피타바스타틴 방출 개시 후 2시간 내지 4 시간 이후에 .방출되는 약제학적 제제 .
【청구항 68]
제 12항에 있어서, 상기 피타바스타틴은 제제 중 1 내지 20mg으로 포함되는 약제학적 제제 .
【청구항 69]
제 12항에 있어서 , 싱'기 피타바스타 ¾은 그의 이성질체이고, 상기 암로디핀은 그의 (S)이성질체, (R)이성질체 , 및 라세피체로 이루어진 군에서 선택되는 것인 약 제학적 제제 .
【청구항 701
제 12항에 있어서 , 상기 암로디된은 제제 증 1 내지 20mg으로 포함되는 약제 학적 제제 .
【청구항 71】
제 12항에 있어서, 상기 암로디 ¾은 피타바스타틴 방출 개시 후 4시간 경과시 까지 단위 제제 증 총량의 40¾ 이하 방출되는 약제학적 제제 .
【청구항 72]
제 12항에 있어서 , 상기 지연방출성 구획은 수블용성 중합체 및 친수성 고분 자의 흔합물인 방출제어물걸을 포함한 약제학적 제제 .
【청구항 73]
제 15항에 있어서 , 상기 제 12항의 방출제어물질이 암로디핀 1증량부에 대하여 0.05 내지 100 중량부로 함유된 약제학적 제제 .
【청구항 ¾】
제 72항에 있어서, 상기 수불용성 증합체는 폴리비닐 아세테이트, 풀리메타크 ¾레이트 공중합체, 폴리 (에틸아크릴레이트 -메틸 메타크 ¾레이트) 공중합체 , ¾리 ( 에 ¾아크릴레이트-메틸 메타크릴레이트-트리메틸아미노에 ¾메타크 ¾레이트)공중합 체, 에틸셀를로오스, 셀를로오스 에스테르 , ¾를로오스 에테르, 로오스 아실레 이트, 셀를로오스 디아실레이트, 셀를로오스 트리아실레이트, 셸를로오스아세테이 트, 셀를로오스 디아세테이트, ¾를로오스 트리아세테이트 및 이들의 혼합물로 이
루어진 군에서 선택된 것이고,
상기 친수성 고분자는 샐를로오스 유도체로서 히드록시프로필메틸셀를로오스, 히드 특시프로필샐를로오스, 히드록시메틸셀를로오스, 히드특시에틸셀를로오스, 메틸셀 를로오스, 카르복시메틸셸를로오스 나트륨, ■&ᅵ드록시프로필 메틸 " 를로오스 아세테 이트 숙시네이트, 및 히드톡시에 ¾메¾셀를로오스 증에서 선택된 하나 이상; 검류 로서 구아검 , 로커스트 콩 검 , 트라가칸타, 카라기난, 아카시아검 , 아라비아검, 젤 란검 , 및 잔탄검 증에서 선택된 하나 이상; 단백질류로서 젤라틴 , 카제인 , 및 제인 증에서 선택된 하나 이상; 폴리비닐 유도체로서 폴리비닐 알코을, 풀리비닐 피를리 돈, 및 플리비닐아세탈디에틸아미노아세테이트 증에서 선택된 하나 이상; 폴리메타 크릴레이트 공중합체로서 플리 (부틸 메타크 ¾레이트 -(2-디메틸아미노에틸)메타크 ¾ 레이트-메틸메타크릴레이트) 공증합체, 풀리 (메타크릴산— 메틸메타크 ¾레이트) 공 증합훼, 및 폴리 (폐타크¾산-에틸아크 ¾레이트) ^증함쾌 증세서 선택된 하나 이 상; 폴리에틸렌 유도체로서 플리에 ¾렌 글리클, 몇 폴리에틸렌 옥사이드 증에서 선 택된 하나 이상; 카르복시비닐폴리머로서 카보머 및 그 흔합물 중에서 선택된 하나 이상인 약제학적 제제 .
【청구항 75】
제 15항에 있어서 , 상기 제 12항의 방출제어물질은 히드특시프로필셀를로오스, 히드록시 프로필메틸 ¾를로오스, 히드특시프로필메틸셀를로오스프탈레이트, 셀를로 오스 아세테이트, 폴리비닐아세테이트, 폴리비닐피를리돈, ¾리에틸렌옥사이드, 폴 리메돠크¾레이트공중합체, 에틸셸를로오스, 풀리 (메타크¾레이트 메틸메타크릴레 이트)공중함체 및 아크릴산메틸 메타크릴산 공중합체로 이루어진 군으로부터 선택 된 하나 이상의 것인 약제학적 제제 .
【청구항 76】
제 72 항 또는 제 75 항에 있어서 , 상기 제어방출물질은 히드특시프로필¾를 로오스, 셀를로오스아세테이트 에 ¾셀를로오스 및 폴리 (메타크릴레이트메틸메타크 ¾레이트) 공중합체로 이루어진 군으로부터 선택된 하나 이상인 것인 약제학적 제 제 .
【청구항 77】
제 13항에 있어서 , 상기 로슈바스타틴이 방출 개시 후 15분 이내에 로슈바스 타틴 총량의 80¾ 이상이 방출되는 약제학적 제제 .
【청구항 78】
제 13항에 있어서 , 상기 암로디핀이 로슈바스타틴 방촐개시 후 2시간 이후에 방출되는 약제학적 제제 .
【청구항 791
제 13항에 있어서, 상기 암로디핀이 로슈바스타틴 용출개시 후 3시간 이내에 단위제제 중 암로디 ¾ 총량의 20¾이하로 방출되는 약제학적 제제 .
【청구항 80J
제 13항에 있어서 , 상기 암로디 ¾이 8시간 이내에 방출 완 S되는 약제학적 제 제 .
【청구항 81】
제 13항에 있어서 , 상기 로슈바스타틴은 제제 중 5 내지 80mg인 약제학적 제 제 .
【청구항 82]
제 13항에 있어서, 상기 암로디핀은 제제 중 1 내지 20m 인 약제학적 제제 . 【청구항 83】
제 13항에 있어서 , 상기 지연방출성 구획은 장용성 고분자 및 수블용성 중합 체 증에서 선텍된 1종 이상인 약제학적 제제 .
ί청구항 84】
제 15항에 있어서 , 상기 제 13항의 방출제어물질이 암로디 ¾ 1중량부에 대하여 0-05증량부 내지 100증량부로 포함되는 ¾인 약제학적 체체 .
【청구항 85]
제 15항에 있어서 , 상기 제 13항의 장용성 고분자가 아크 ¾산메틸메타크 ¾산 공중합체 및 히드록시프로필메틸 ¾를로소스프탈테이트 증에서 선택된 1종 이상인 약제학적 제제 .
【청구항 86】
제 15항에 있어서 , 상기 제 13항의 장용성 고분자가 암로디핀 1 중량부에 대해 서 0.1 중량부 내지 20 중량부인 약제학적 제제 .
【청구항 87】
제 15항에 있어서 , 상기 제 13항의 수불용성 증합체는 플리비닐아세테이트, 셀 를로오스아세테이트 및 풀리 (에틸아크 ¾레이트-메¾메타크 ¾레이트-트리메틸아머노 에킬메타크 ¾레이트) 공중합체 중에서 선택된 1종 이상인 약제학적 제제 .
【청구항 88]
제 15항에 있어서, 상기 제 13항의 수볼용성 증합체는 암로디핀 1 중량부에 대 해서 0.1 증량부 내지 30 증량부로 포함되는 약제학적 제제 .
【청구항 891
제 15항에 있어서 , 상기 제 13항의 소수성 화합물이 암로디핀 1 증량부에 대해 서 으 1 증량부 내지 20 증량부인 약제학적 제제 .
【청구항 901
제 15 항에 있어서 , 상기 제 13항의 친수성 고분자가 암로디 ¾ 1 중량부에 대 해서 0.05 증량부 내지 30 증량부인 약제학적 제제 .
【청구항 911
제 15항에 있어서, 상기 제 13항의 방출제어물질이 히드특시프로필셸를로오스, 히드록시프로필메틸셀를로오스, 히드록시프로필메틸셀를로오스프탈레이트 , 샐를로 오스아세테이트 , 폴리비닐아세테이트 , 폴리메타크릴레이트 공중합체, 에틸썰를로오 스, 폴리 (데타크필레이트 -메틸메타크 ¾테이트) 공중합체 , 및 아크 ¾산메틸메타크 ¾ 산 공증합체로 이루어진 군으로부터 선택된 1 종 이상인 것인 약제학적 제제 .
【청구항 92】
제 14항에 있어서, 상기 선방출성 구획의 아토르바스타틴은 방출개시 후 1시 간 이내에 아토르바스타틴 총량의 8(» 이상이 방출되는 약제학적 제제 .
【청구항 93]
제 14항에 있어서 , 상기 니패디핀은. 아토르바스타틴 방출개시 후 2시간 내지 6시간 이후에 방출되는 약제학적 제제 .
【청구항 94】
겨 114항에 있어서 , 아토르바스타 ¾은 제제 증 1 내지 160mg으로 S함되는 약 제학적 제제 .
【청구항 95]
제 14항에 있어서 , 상기 아토바스타틴은 그의 이성질체인 약제학적 제제 . 【청구항 96]
제 14항에 있어서, 니페디핀은 제제 증 1 내지 90mg으로 포함되는 약제학적 제제 .
【청구항 97】
제 14 항에 있어서 , 상기 지연방출성 구획은 장용성 고분자 및 친수성 고분
자를 포함하는 약제학적 제제 .
【청구항 98]
제 15항에 있어서 , 상기 제 14항의 방출제어물질이 니페디핀 100 증량부에 대 하여 5 내지 300 증량부로 함유된 약제학적 제제 .
【청구항 99]
제 97항에 있어서, 상기 제 14항의 방출제어물질이 니페디핀 100 증량부에 대 하여 5 내지 300 증량부로 함유된 약제학적 제제 .
【청구항 100】
제 97항 또는 제 98항에 있어서, 상기 장용성 고분자는 장용성 샐를로오스 유 도체로서 히드록시프로필메 ¾셀를로오스아세테이트숙시네이트, 히드록시프로필메틸 샐를로오스프탈레이트, 히드록시메 ¾에¾샐를로오스프탈레이트 , 셀를로오스아세테 이트프탈테이트, 썰를로오스아세테이트숙시네이트 , 셸를로오스아세테이트말레이트, 샐를로오스벤조에이트프탈레이트, ¾를로오스프로피오네이트프탈레이 S, 메틸셀를 토오스프탈레이트, 카르복시메틸에틸셀를로오스, 에틸히드록시에¾셀를로오스프탈 레이트, 및 메¾히드록시에틸셀를로오스 증에서 선택된 하나 이상; 장용성 아크 ¾ 산계 공증합체토서 스티렌 -아크 ¾산 공중합체 , 아크릴산메 ¾ᅳ아크 ¾산 공중합체 , 아크릴산매틸메타크릴산 공중합체 , 아크 ¾산부틸-스티렌—아크릴산 공중합체 , 메타 크릴산-메타크릴산메틸 공중합체 , 메타크 ¾산 * 아크릴산에틸공증합체 , 및 아크릴산 메틸-메타크 ¾산ᅳ아크¾산옥틸공증합체 증에서 선택된 하나 이상; 장용성 말테인산 계 공중합체로서 아세트산비닐-말레인산 무수물 공중합체 , 스티렌-말테인산 무수불 공증합체, 스티렌-말레인산모노에스테르 공증합체, 비닐메될에테르-말레인산 무수 물 공중합체 . 에¾렌ᅳ말레인산 무수 " 공중합체 , 비닐부 ¾에테르-말레인산 무수불 공중합체, 아크릴로니트릴-크필산메틸 · 말레인산 무수물 공증합체 , 및 아크릴산부 털 -스티렌-말레인산 무수물 공중합체 중에서 선택된 하나 이상; 또는 장용성 폴리 비닐 유도체로서 리비날알콜프탈레이트, 폴리비닐아세탈프탈레이트, 폴리비닐부티 테이트프탈레이트 및 플리비닐아세트아세탈프탈레이트 중에서 선택된 하나 이상이 고,
상기 친수성 고분자는 셀롤로오스 유도체 S서 히드록시프로필메 ¾셀를로오스, 히 드록시프로필 ¾를로오스, 히드륵시메틸셀를로오스, 히드록시에틸셸를로오스, 메¾ 셀롤로오스, 카르복시메틸 ¾를로오스 나트튬, 히드록시프로필 메틸셀를로오스 아세 테이트 숙시네이트, 및 히드록시에 ¾메¾셀를로오스 증에서 선택된 하나 이상; 검
류로서 구아검 , 로커스트 콩 검, 트라가칸타, 카라기난, 아카시아검 , 아라비아검 , 젤란검, 및 잔탄검 중에서 선택된 하나 이상 단백질류로서 젤라틴 , 카제인 , 및 제 인 증에서 선택된 하나 이상 플리비닐 유도체로서 폴리비닐 알코을, 폴리비닐 피를 리돈, 및 풀리비닐아세탈디에틸아미노아세테이트 중에서 선택된 하나 이상; 플리메 타크릴레이트 공중합체로서 폴리 (부틸 메타크릴레이트— (2-디메틸아미노에틸)메타크 릴레이트-메틸메타크릴레이트) 공중합체 , 플리 (메타크릴산- 메틸쩨타크릴레'이트) 공증합체 , 및 플리 (메타크릴산-에틸아크릴레이트) 공증합체 증에서 선택된 하나 이 상; 풀리에 ΐ톈 유도체로서 풀리에 ¾렌 글리콜, 및 폴리에틸렌 옥사이드 증에서 선 택된 하나 이상 카르복시비닐폴리머로서 카보머 및 그 흔합물 중에서 선택된 하나 이상인 약제학적 제제 .
【청구항 101]
제 15항에 있어서, 상기 제 14항의 방출제어물질은 히드특시프로필메틸셀를로 오스프탈레이트 , 히드록시프로필메필셀를로오스아세테이트숙시네이트, 메타크릴산— 메타크릴산메 ¾공중합체, 글리세 ¾ 스테아레이트, 히드록시프로필메 ¾셀를로오스 , 히드록시프로필쎌를로오스 , 카보머 , 및 이들의 흔합물로 이루어진 군으로부터 선택 되는 약제학적 제제 .
【청구항 102】
제 97항 또는 제 101항에 있어서 , 방출제어물질은 히드록시프로필메틸셀를로오 스프탈레이트, 히드록시프로필메틸셸를로오스아세테이트 숙시네이트 , 메타크릴산- 메타크릴산메틸공증합체 , 히드록시프로필메틸셀롤로오스, 히드특시프로필셀를로오 스, 카보머 , 및 이들의 흔합물로 이루어진 군으로부터 선택되는 약제학적 제제 .
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2008-0016516 | 2008-02-22 | ||
| KR20080016516 | 2008-02-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009127922A2 true WO2009127922A2 (ko) | 2009-10-22 |
| WO2009127922A3 WO2009127922A3 (ko) | 2009-12-10 |
Family
ID=41199515
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2009/000331 Ceased WO2009127922A2 (ko) | 2008-02-22 | 2009-02-23 | 심혈관계 질환 치료용 약제학적 제제 |
| PCT/IB2009/006277 Ceased WO2009127974A2 (ko) | 2008-02-22 | 2009-02-23 | 심혈관계 질환 치료용 약제학적 제제 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2009/006277 Ceased WO2009127974A2 (ko) | 2008-02-22 | 2009-02-23 | 심혈관계 질환 치료용 약제학적 제제 |
Country Status (2)
| Country | Link |
|---|---|
| KR (5) | KR20090091084A (ko) |
| WO (2) | WO2009127922A2 (ko) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103442699A (zh) * | 2011-03-23 | 2013-12-11 | 韩美药品株式会社 | 包含ω-3脂肪酸酯及HMG-CoA还原酶抑制剂的口服复合组成物 |
| RU2715771C2 (ru) * | 2015-02-27 | 2020-03-03 | ЭКОЛАБ ЮЭсЭй ИНК. | Композиции для улучшения нефтеотдачи |
| US10808165B2 (en) | 2016-05-13 | 2020-10-20 | Championx Usa Inc. | Corrosion inhibitor compositions and methods of using same |
| US11203709B2 (en) | 2016-06-28 | 2021-12-21 | Championx Usa Inc. | Compositions for enhanced oil recovery |
| US12031128B2 (en) | 2021-04-07 | 2024-07-09 | Battelle Memorial Institute | Rapid design, build, test, and learn technologies for identifying and using non-viral carriers |
| US12109223B2 (en) | 2020-12-03 | 2024-10-08 | Battelle Memorial Institute | Polymer nanoparticle and DNA nanostructure compositions and methods for non-viral delivery |
| US12441996B2 (en) | 2023-12-08 | 2025-10-14 | Battelle Memorial Institute | Use of DNA origami nanostructures for molecular information based data storage systems |
| US12458606B2 (en) | 2023-09-29 | 2025-11-04 | Battelle Memorial Institute | Polymer nanoparticle compositions for in vivo expression of polypeptides |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101143559B1 (ko) | 2009-09-25 | 2012-05-24 | 기아자동차주식회사 | 오일유로 통합형 엔진브레이크 장치 |
| KR101466617B1 (ko) * | 2011-11-17 | 2014-11-28 | 한미약품 주식회사 | 오메가-3 지방산 및 HMG-CoA 환원효소 억제제를 포함하는 안정성이 증가된 경구용 복합 제제 |
| KR101378973B1 (ko) * | 2012-04-13 | 2014-03-28 | 한미약품 주식회사 | 구형에 가까운 형태의 다중 투여 단위 정제를 포함하는 경질 캡슐 복합 제형 및 이의 제조방법 |
| JP2015503555A (ja) * | 2012-05-11 | 2015-02-02 | ハナル・バイオファーマ・カンパニー・リミテッド | ボセンタン制御放出性経口製剤 |
| KR20140001357A (ko) * | 2012-06-26 | 2014-01-07 | 현대약품 주식회사 | 아세브로필린을 포함하는 속효성과 지속성을 동시에 갖는 약제학적 조성물 |
| CN104161960A (zh) * | 2014-09-02 | 2014-11-26 | 济南超舜中药科技有限公司 | 一种治疗肺心病的醋饮及其制备方法 |
| PL3320903T3 (pl) * | 2015-07-08 | 2021-12-06 | Hk Inno.N Corporation | Kompozycja farmaceutyczna zawierająca amlodypinę, walsartan i rosuwastatynę |
| US10413543B2 (en) | 2015-09-01 | 2019-09-17 | Sun Pharma Advanced Research Company Ltd. | Stable multiparticulate pharmaceutical composition of rosuvastatin |
| KR20190043076A (ko) * | 2017-10-17 | 2019-04-25 | 한미약품 주식회사 | 암로디핀, 로자탄 및 로수바스타틴을 포함하는 당뇨병을 동반한 심혈관계 질환의 예방 또는 치료용 약학 조성물 및 이를 포함하는 복합제제 |
| KR102265977B1 (ko) * | 2018-07-16 | 2021-06-16 | 주식회사 코피텍 | 방습성이 개선된 필름 코팅용 조성물 및 이를 코팅한 정제 |
| KR102730602B1 (ko) * | 2021-06-24 | 2024-11-18 | 주식회사 종근당 | 칸데사르탄, 암로디핀 및 아토르바스타틴을 포함하는 약제학적 복합제제 |
| KR102608853B1 (ko) * | 2021-11-15 | 2023-12-01 | 재단법인 아산사회복지재단 | 대사질환 개선용 생체발광 캡슐 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AP1207A (en) * | 1997-08-29 | 2003-09-20 | Pfizer Prod Inc | Combination therapy. |
| IL156585A0 (en) * | 2001-01-26 | 2004-01-04 | Schering Corp | Combinations of sterol absorption inhibitor(s) with cardiovascular agent(s) for the treatment of vascular conditions |
| KR20050016452A (ko) * | 2002-05-29 | 2005-02-21 | 노파르티스 아게 | Dpp ⅳ 억제제와 심혈관계 화합물의 조합물 |
| KR100582347B1 (ko) * | 2004-12-30 | 2006-05-22 | 한미약품 주식회사 | 3-하이드록시-3-메틸글루타릴 조효소 a 환원효소 억제제및 고혈압 치료제의 복합제제 및 그의 제조방법 |
| KR100762847B1 (ko) * | 2006-01-27 | 2007-10-04 | 씨제이 주식회사 | 멀티플 유닛 타입 서방성 경구 제제 및 그 제조방법 |
| WO2008023869A1 (en) * | 2006-08-24 | 2008-02-28 | Hanall Pharmaceutical Co., Ltd. | Combined pharmeceutical formulation with controlled-release comprising dihydropyridine calcium channel blockers and hmg-coa reductase inhibitors |
-
2009
- 2009-02-23 WO PCT/IB2009/000331 patent/WO2009127922A2/ko not_active Ceased
- 2009-02-23 KR KR1020090015058A patent/KR20090091084A/ko not_active Withdrawn
- 2009-02-23 KR KR1020090014652A patent/KR101207618B1/ko not_active Expired - Fee Related
- 2009-02-23 KR KR1020090015061A patent/KR20090091085A/ko not_active Withdrawn
- 2009-02-23 KR KR1020090014851A patent/KR101181172B1/ko not_active Expired - Fee Related
- 2009-02-23 KR KR1020090015023A patent/KR20090091083A/ko not_active Withdrawn
- 2009-02-23 WO PCT/IB2009/006277 patent/WO2009127974A2/ko not_active Ceased
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103442699A (zh) * | 2011-03-23 | 2013-12-11 | 韩美药品株式会社 | 包含ω-3脂肪酸酯及HMG-CoA还原酶抑制剂的口服复合组成物 |
| CN103442699B (zh) * | 2011-03-23 | 2015-07-01 | 韩美药品株式会社 | 包含ω-3脂肪酸酯及HMG-CoA还原酶抑制剂的口服复合组成物 |
| RU2715771C2 (ru) * | 2015-02-27 | 2020-03-03 | ЭКОЛАБ ЮЭсЭй ИНК. | Композиции для улучшения нефтеотдачи |
| US10767104B2 (en) | 2015-02-27 | 2020-09-08 | Ecolab Usa Inc. | Compositions for enhanced oil recovery |
| US10808165B2 (en) | 2016-05-13 | 2020-10-20 | Championx Usa Inc. | Corrosion inhibitor compositions and methods of using same |
| US11203709B2 (en) | 2016-06-28 | 2021-12-21 | Championx Usa Inc. | Compositions for enhanced oil recovery |
| US11912925B2 (en) | 2016-06-28 | 2024-02-27 | Championx Usa Inc. | Compositions for enhanced oil recovery |
| US12109223B2 (en) | 2020-12-03 | 2024-10-08 | Battelle Memorial Institute | Polymer nanoparticle and DNA nanostructure compositions and methods for non-viral delivery |
| US12433910B2 (en) | 2020-12-03 | 2025-10-07 | Battelle Memorial Institute | Polymer nanoparticle and DNA nanostructure compositions and methods for non-viral delivery |
| US12031128B2 (en) | 2021-04-07 | 2024-07-09 | Battelle Memorial Institute | Rapid design, build, test, and learn technologies for identifying and using non-viral carriers |
| US12458606B2 (en) | 2023-09-29 | 2025-11-04 | Battelle Memorial Institute | Polymer nanoparticle compositions for in vivo expression of polypeptides |
| US12441996B2 (en) | 2023-12-08 | 2025-10-14 | Battelle Memorial Institute | Use of DNA origami nanostructures for molecular information based data storage systems |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20090091083A (ko) | 2009-08-26 |
| WO2009127974A2 (ko) | 2009-10-22 |
| KR20090091080A (ko) | 2009-08-26 |
| KR20090091077A (ko) | 2009-08-26 |
| KR101181172B1 (ko) | 2012-09-18 |
| WO2009127922A3 (ko) | 2009-12-10 |
| KR20090091085A (ko) | 2009-08-26 |
| KR20090091084A (ko) | 2009-08-26 |
| WO2009127974A3 (ko) | 2010-03-25 |
| KR101207618B1 (ko) | 2012-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009127922A2 (ko) | 심혈관계 질환 치료용 약제학적 제제 | |
| CA2664893C (en) | Controlled release complex composition comprising angiotensin-ii-receptor blockers and hmg-coa reductase inhibitors | |
| KR101205633B1 (ko) | 심혈관계 질환 치료용 약제학적 제제 | |
| CN101505737B (zh) | 包含二氢吡啶类钙通道阻断剂和HMG-CoA还原酶抑制剂的控释组合药物制剂 | |
| EP2275093A2 (en) | Pharmaceutical formulation | |
| EP2255796A2 (en) | Pharmaceutical preparation | |
| KR20090114190A (ko) | 방출성이 제어된 HMG―CoA 환원 효소 억제제와안지오텐신―Ⅱ―수용체 차단제의 복합 조성물 | |
| KR101869406B1 (ko) | 방출제어형 경질캡슐 제제 | |
| CN105431140A (zh) | 含有缓释二甲双胍和速释HMG-CoA还原酶抑制剂的复合制剂 | |
| CN102316856A (zh) | 药物制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09733176 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09733176 Country of ref document: EP Kind code of ref document: A2 |