WO2009135977A1 - Procedimiento para la preparación de ácido oregánico - Google Patents
Procedimiento para la preparación de ácido oregánico Download PDFInfo
- Publication number
- WO2009135977A1 WO2009135977A1 PCT/ES2009/070138 ES2009070138W WO2009135977A1 WO 2009135977 A1 WO2009135977 A1 WO 2009135977A1 ES 2009070138 W ES2009070138 W ES 2009070138W WO 2009135977 A1 WO2009135977 A1 WO 2009135977A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- stereoisomers
- mixtures
- integer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 COC(CC(C(OC)=O)=C(CCCNC*)C(OC)=O)=O Chemical compound COC(CC(C(OC)=O)=C(CCCNC*)C(OC)=O)=O 0.000 description 8
- FWXRAMAZAJHBPG-GMYFYABVSA-N CC(C/C(/C(OC)=O)=C(\CCCC[O]=C)/C([U]C)=O)=N Chemical compound CC(C/C(/C(OC)=O)=C(\CCCC[O]=C)/C([U]C)=O)=N FWXRAMAZAJHBPG-GMYFYABVSA-N 0.000 description 1
- JCFGUWUPLSBGRK-FQSKSLCWSA-N CC/C=C(\C(\CC(NC)=C)=C(\CC)/C(OC)=O)/OC Chemical compound CC/C=C(\C(\CC(NC)=C)=C(\CC)/C(OC)=O)/OC JCFGUWUPLSBGRK-FQSKSLCWSA-N 0.000 description 1
- LBDBGTPPCRSRNT-VQHVLOKHSA-N CN(CO)/C(/C(OC)=O)=C(\CC(OC)=O)/C(OC)=O Chemical compound CN(CO)/C(/C(OC)=O)=C(\CC(OC)=O)/C(OC)=O LBDBGTPPCRSRNT-VQHVLOKHSA-N 0.000 description 1
- JWUJQDFVADABEY-RXMQYKEDSA-N C[C@H]1OCCC1 Chemical compound C[C@H]1OCCC1 JWUJQDFVADABEY-RXMQYKEDSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/02—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms with only carbon-to-carbon double bonds as unsaturation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/24—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfuric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/28—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reaction of hydroxy compounds with sulfonic acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C305/00—Esters of sulfuric acids
- C07C305/02—Esters of sulfuric acids having oxygen atoms of sulfate groups bound to acyclic carbon atoms of a carbon skeleton
- C07C305/14—Esters of sulfuric acids having oxygen atoms of sulfate groups bound to acyclic carbon atoms of a carbon skeleton being acyclic and unsaturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/72—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/73—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton to carbon atoms of non-condensed six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/42—Unsaturated compounds containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/03—Preparation of carboxylic acid esters by reacting an ester group with a hydroxy group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/307—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/31—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by introduction of functional groups containing oxygen only in singly bound form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/313—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by introduction of doubly bound oxygen containing functional groups, e.g. carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/62—Halogen-containing esters
- C07C69/65—Halogen-containing esters of unsaturated acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/732—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids of unsaturated hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/734—Ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/738—Esters of keto-carboxylic acids or aldehydo-carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/09—Geometrical isomers
Definitions
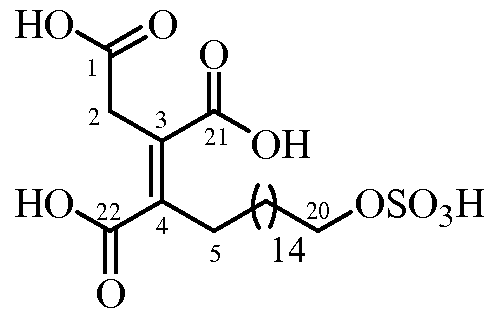
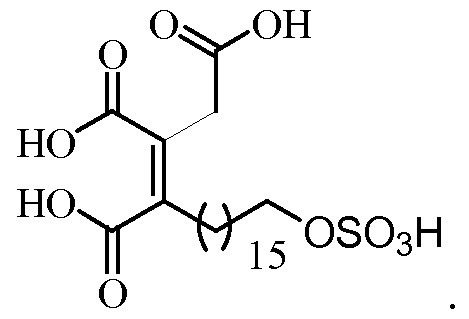
- the present invention relates to a new process for the synthesis of Oregon acid and derivatives thereof, to the intermediate compounds of the synthesis and to the use of these compounds in the preparation of Oregon acid and its derivatives.
- Ras proteins It is a natural product that belongs to the family of alkyl citrates and that has Ras FTase inhibitory activity.
- the inhibition of the farnesylation process of Ras proteins is considered as an effective mechanism to control the growth of tumors promoted by mutated Ras proteins, since such inhibition prevents mutated Ras proteins from entering the cell membrane and being activated, being unable to carry out its biological action.
- For a study of the biological activity of Oregon acid see: Silverman, K. C; Jayasuriya, H .; Cáscales, C; Vilella, D .; Bills, G. F .; Jenkins, R. G .; Singh, S. B .; Lingham, R. B. Biochem. Biophis Res. Commun. 1997, 252, 478-481.
- Oregon acid was isolated by Dr. Jayasuriya's group in 1996 from an unidentified endophytic fungus (MF 6046), isolated from live leaves of Berber ⁇ s oregana (Berberidaceae) in Lord Ellis Summit, Humboldt Co. California, USA.
- MF 6046 unidentified endophytic fungus
- Berber ⁇ s oregana Berber ⁇ s oregana
- Dr. Gibbs' group represented Oregon acid with a Z (correct) geometry in the double-substituted tetrasubstituted bond between positions C3-C4
- the authors of the invention have prepared the first total synthesis described to date of the Oregon acid and derivatives thereof.
- This synthesis uses simple starting substrates, implies a reasonable number of stages, requires few protective groups and does not include complex transformations. Therefore, it allows to obtain the Oregon acid with good yield, and following conditions that would make possible the synthesis on a large scale and in a short time.
- a first aspect of the invention relates to a process for obtaining a compound of formula (VIII), intermediate of the oranic acid and its derivatives, which already comprises the final skeleton of the molecule, which comprises subjecting a reaction of hydrogenation a compound of formula (VI) or (VII); or deprotect the trialkylsilyl group of a compound of formula (XVI).
- a further aspect of the present invention relates to a process for obtaining compounds of formula (I), their stereoisomers, or mixtures thereof, from compounds of formula (VI), (VII) or (XVI), their stereoisomers, or mixtures thereof through compounds of formula (VIII), their stereoisomers, or mixtures thereof.
- Additional aspects of the invention relate to compounds of formula (I), (IV), (V), (VI), (VII), (VIII), (IX), (XV), (XVI) and (XVIII) defined in the present document.
- Another aspect of the invention corresponds to the use of at least one compound selected from the compounds of formula (II), (III), (IV), (V), (VI),
- Alkyl refers to a hydrocarbon chain radical consisting of carbon and hydrogen atoms, which does not contain unsaturation and is attached to the rest of the molecule by a single bond. In each case the number of carbon atoms of the alkyl group is specified. For example, when “C 1 -C 4 alkyl” is indicated it refers to an alkyl group of one, two, three or four carbon atoms, that is, methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl or tert-butyl.
- Aryl refers to an aromatic substituent, preferably C 6 -CiS, more preferably C 6 -Ci 4 , more preferably C 6 -CiO, which comprises a single aromatic ring or multiple fused rings in which at least one of them is aromatic.
- Preferred aryl groups are phenyl or naphthyl, preferably phenyl.
- Halogen means -F, -Cl, -Br or -I.
- Trialkylsilyl means a radical of the formula -Si (R ') (R ") R'", wherein each of R ', R “and R'” is independently selected from a phenyl group and an alkyl group Ci-C 6 .
- Non-limiting examples of trialkylsilyl groups can be trimethylsilyl, triethylsilyl, tri- ⁇ -propylsilyl; dimethylisopropylsilyl, diethylisopropylsilyl, dimethylthexylsilyl, tert-butyldimethylsilyl, tert-butyldiphenylsilyl.
- Bn means benzyl (- (CH 2 ) -phenyl).
- Leaving group is an atom or group of atoms that activates the carbon to which they are attached to a nucleophilic reagent, such that said nucleophile, or part of said nucleophile, is attached to the molecule and the leaving group emerges from the same.
- Outgoing groups are known to the person skilled in the art, for example, OTs
- alkanoyl such as a Ci-C 6 alkanoyl group, such as acyl and the like
- carboxamido (- (C O) NH 2 ); trialkylsilyl.
- substituted alkyl includes groups such as cyanoethyl, acetylmethyl, carboxamidomethyl (-CH 2 CONH 2 ), 2-trimethylsilylethyl.
- the carbon-linked chain of position 4 will be called the "side chain".
- all the compounds of the present invention that contain a double bond between the C3-C4 positions can have said double bond with Z configuration, with E configuration or they can be found as mixtures of Z / E isomers.
- the present invention encompasses said stereoisomers or mixtures thereof, all useful in the synthesis of compounds of formula (I).
- the compounds of the invention also refers to including compounds that differ only in the presence of one or more isotopically enriched atoms.
- the compounds having the present structures except for the substitution of a hydrogen for a deuterium or for tritium, or the substitution of a carbon for a carbon enriched in 13 C or 14 C, are within the scope of this invention.
- the reaction products obtained can be purified, if desired, by conventional methods, such as crystallization, chromatography and crushing.
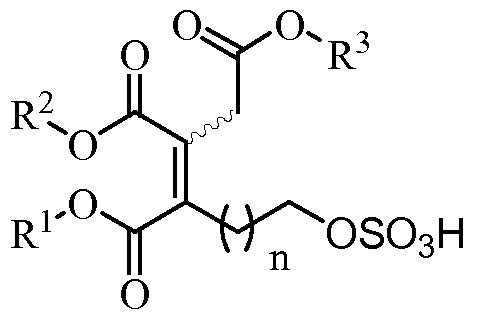
- the present invention relates to a process for obtaining a compound of formula (VIII), its stereoisomers or mixtures thereof where
- R 1 , R 2 and R 3 are independently selected from C 1 -C 20 alkyl, substituted or unsubstituted; and n is an integer between 2 and 20; characterized in that it comprises at least one of the following steps (a), (b) or (c) (a) subjecting a compound of formula (VI), its stereoisomers or mixtures thereof, to a hydrogenation reaction
- R 1 , R 2 and R 3 are as defined above; mi is an integer that is selected from 1, 2, 3, 4 and 5; and m2 is an integer between 0 and 13;
- n, R 1 , R 2 and R 3 are as defined above; or (c) removing the trialkylsilyl group of a compound of formula (XVI), its stereoisomers or mixtures thereof
- a metal catalyst is added on a solution of a tri-ester of formula (VI) or (VII) in a suitable solvent, such as 1,4-dioxane.
- a suitable solvent such as 1,4-dioxane.
- the solvent is degassed by passing a stream of an inert gas.
- the suspension thus prepared is stirred under hydrogen atmosphere for a time of between 10 and 210 minutes, preferably between 20 and 150 minutes, more preferably between 25 and 80 minutes.
- heterogeneous hydrogenation reactions are performed are known to the person skilled in the art.
- Various metal catalysts are used, in which the metal is selected from platinum, palladium, rhodium or ruthenium.
- Some commercial hydrogenation reaction catalysts useful for the purposes of the present invention are, for example, Ni (Ra), Pd / C, Rh (Al 2 Os). Among these, the preferred catalyst is Pd / C.
- the reaction medium 0.02-1.5M
- Pd / C of a Concentration between 3 and 7%, preferably 5%
- favors obtaining directly an alcohol of formula (VIII) as the only product favors obtaining directly an alcohol of formula (VIII) as the only product.
- Preferred embodiments for the direct obtaining of compounds of formula (VIII) are also those wherein between 0.01-0.1 equivalents, preferably 0.04-0.06 equivalents, of Pd / C are added.
- the solvent is 1,4-dioxane.
- the reaction time is between 50 and 70 minutes.
- Preferred conditions for obtaining a compound of formula (VII) from compounds of formula (VI) are those in which the concentration of the compound of formula (VI) in the reaction medium and the concentration of Pd / C is slightly higher.
- the concentration of the compound of formula (VI) in the reaction medium is greater than 0.15M, preferably 0.16-0.25M), more preferably approximately 0.2M, and Pd / C of a concentration between 8 and 12%, preferably 10%, is used.
- Preferred embodiments for obtaining compounds of formula (VII) are also those in which 0.01-0.1 equivalents, preferably 0.04-0.06 equivalents, of Pd / C are added.
- the solvent is 1,4-dioxane.
- option (c) comprises the removal of the trialkylsilyl group, that is, the transformation of R 4 into a hydrogen, of a compound of formula (XVI).
- Such transformation is usually understood as a deprotection and can be performed under different conditions (see for example, Kocienski, PJ Protecting Groups; Thi eme: Stuttgart, 2000. pp.: 187-230).
- the trialkylsilyl group is removed from a compound of formula (XVI) to give a compound of formula (VIII) in dilute acid medium, such as, for example, 1% HCl.
- the invention relates to compounds of (VI), (VII), or (XVI) as defined above, immediate precursors of the compounds of formula (VIII).
- n is an integer between 8 and 20, preferably between 12 and 17, more preferably between 13 and 16, more preferably n is 15.
- mi is an integer that is selected from 2, 3 or 4, preferably mi is 3.
- m2 is an integer that is selected from 5-12, preferably 8-12, more preferably m2 is 10.
- R 1 , R 2 and R 3 groups act as protecting groups for the acid groups of the compounds of formula (I) and, as seen below, are eliminated in the last stages of the synthesis. Therefore, any substituted or unsubstituted C1-C20 alkyl protecting group, orthogonal to the other protecting groups used in the synthesis (for example, benzyl and trialkylsilyl) is useful for the purposes of the present invention.
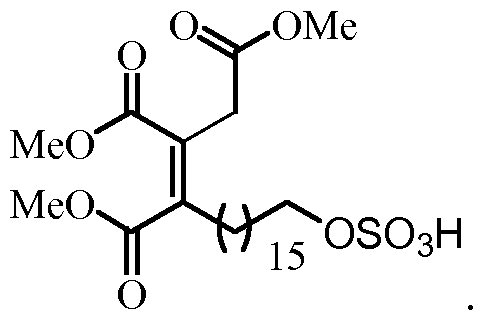
- each of R 1 , R 2 and R 3 is independently selected from a C1-C4 alkyl group, preferably C1-3, more preferably, R 1 , R 2 and R 3 are methyl groups.
- Another aspect of the present invention relates to a process for the synthesis of a compound of formula (VI).
- Said compound of formula (VI), its stereoisomers or mixtures thereof, can be prepared by reacting a compound of formula (V) with a phosphonate of formula (XIII) in the presence of a base:
- R 1 is selected from a substituted or unsubstituted C1-C20 alkyl; and R is selected from C1-C4 alkyl substituted or unsubstituted by one or more halogen, benzyl, phenyl and tolyl atoms.
- the phosphonate is in excess with respect to the compound of formula (V), more preferably in a ratio between 1.5 to 3 equivalents, even more preferable in a proportion of 2.2 equivalents.
- this transformation also requires the presence of a base, such as lithium bases, such as n-BuLi, t-BuLi, 5-BuLi, LDA, LiHMDS; sodium bases, such as NaH, NaHMDS; or potassium bases, such as KHMDS, ⁇ -BuOK.
- the base is NaH.
- the amount of base depends on the amount of phosphonate used.
- the base is in excess with respect to the compound of formula (V), more preferably in a proportion between 1.5-3.
- the reaction is performed using THF solvent at room temperature.
- the reaction leads to mixtures of the C3-C4 geometric isomers of the compound (VI) in different proportions. Both isomers are within the scope of the present invention, and allow the synthesis of oreganic acid and its derivatives, as discussed below.
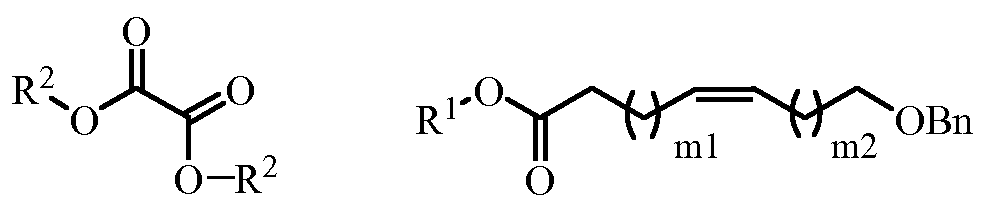
- a compound of formula (V) can be prepared by reacting a compound of formula (IV) with an oxalate of formula (XII) in the presence of a base:
- R 1 and R 2 are independently selected from a substituted or unsubstituted C1-C20 alkyl; mi is an integer that is selected from 0, 1, 2, 3, 4 and 5; and m2 is an integer between 0 and 13.
- this transformation also requires the presence of a base, such as lithium bases, such as n-BuLi, ⁇ -BuLi, 5-BuLi, LDA, LiHMDS; sodium bases, such as NaH, NaHMDS; or potassium bases, such as KHMDS, ⁇ -BuOK
- a base such as lithium bases, such as n-BuLi, ⁇ -BuLi, 5-BuLi, LDA, LiHMDS
- sodium bases such as NaH, NaHMDS
- potassium bases such as KHMDS, ⁇ -BuOK
- the base is NaH.
- reaction is conveniently carried out in a mixture of MeOH / THF, using a temperature of about 70 0 C.
- a compound of formula (IV) can be prepared by reacting a compound (III) with a compound of formula (XI ):
- R 1 is selected from a substituted or unsubstituted C1-C20 alkyl, mi is an integer that is selected from 0, 1, 2, 3, 4 and 5; m2 is an integer between 0 and 13; X is a halogen; and each of the groups Ar is independently selected from among the aryl groups C 6 -Ci 0 .
- the base used is n-BuLi
- the aryl is phenyl
- the halogen is Br. Therefore, it is possible to prepare a compound of formula (VI) by the following synthetic sequence:
- the compound of formula (XXI) is 1,12-dodecanediol.
- deprotonation of the latter with a base, such as NaH followed by treatment with a benzylating agent, such as BnBr, leads to 12-benzyloxy-l-dodecanol (6).
- the alcohol is replaced by a halogen.
- the halogen is bromine. This bromination reaction can be carried out in the presence of PPh 3 and CBr 4 .
- the halogenated compound of formula (X) is transformed into a compound of formula (XI) by treatment with triphenylphosphma.
- the compounds of formula (III) can be prepared by the oxidation of alcohols of formula (lia) (lia) where
- R 1 is selected from a substituted or unsubstituted C1-C20 alkyl; and mi is an integer that is selected from 0, 1, 2, 3, 4 and 5;
- Oxidizing agents for the transformation of alcohols into aldehydes are known to those skilled in the art (Larock, RC Comprehensive Organic transformations; John Wiley & Sons: New York, 1999, pp .: 1234-1249). Agree with a preferred embodiment, said oxidizing agent is selected from the group consisting of PCC (pyridinium chlorochromate), MnO 2 , and DMSO / (COCl) 2 / Et 3 N, preferably PCC.
- the treatment of ⁇ -caprolactone (compound of formula (XXIVa) in done mi is 3) in acidic medium using as an solvent an alcohol of formula R 1 OH, preferably wherein R 1 is a C 1 alkyl -
- the oxidizing reagent is PCC.
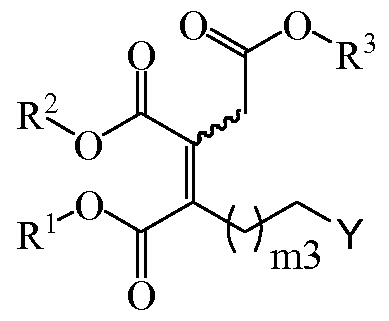
- the side chain of a compound of formula (VIII) can be elongated to provide a compound of formula (VIII) with a longer chain.
- the compound of formula (VIII), its stereoisomers or mixtures thereof is a compound of formula (Villa), its stereoisomers or mixtures thereof,
- R 1 , R 2 and R 3 are independently selected from C 1 -C 20 alkyl, substituted or unsubstituted; and m3 is an integer between 2 and 18; additionally includes the following stages
- n, R 1 , R 2 and R 3 are as defined above, and
- preferred leaving groups are those active against organometallic reagents (nuecleophilic groups) employed in the formation of carbon-carbon bonds, preferably Grinard reagents transmetalated or not with Zn, Al, or Cu.
- Preferred leaving groups are -OTs, -I, -Br, Cl, -SO3H.
- the leaving group is selected from Br and OTs.
- the hydroxyl group is activated as the corresponding tosylate (OTs) in the presence of a base, such as pyridine.
- treatment of a compound of formula (VIII) with CBr 4 and PPI1 3 results in the compound of formula (XVIII) in which Y is Br.
- the compounds of formula (XVI) can be prepared by reacting a compound of formula (XV) with a phosphonate of formula (XIII) in the presence of a base
- R is selected from C1-C4 alkyl substituted or unsubstituted by one or more halogen, benzyl, phenyl and tolyl atoms; and R is selected from a substituted or unsubstituted C1-C20 alkyl; and n, R 1 , R 2 and R 4 are as defined above.
- the reactions of the compounds of formula (V) or (XV) with the phosphonates of formula (XIII) can give rise to mixtures of Z (cis) / E (trans) isomers that can be separated, subjected to the next step as a mixture or enrich in isomer Z (cis) according to the procedure described below.
- the compounds of formula (XV) can be obtained by reacting an oxalate of formula (XII) with a compound of formula (XIV)
- R 2 is selected from a substituted or unsubstituted C1-C20 alkyl; and n, R 1 and R 4 are as defined above.
- a compound of formula (XIV) is prepared by treating an alcohol of formula (II) with a base and a silylating agent
- R 1 and n are as defined above.
- Non-limiting examples of conditions under which this protection transformation of the hydroxyl group can be carried out can be found in, for example, Dalla, V .; Catteau, JP Tetrahedron 1999, 55, 6497-6510, and trialkylsilyl groups that can be used in this reaction, as well as reagents suitable for introduction and removal, are known to the person skilled in the art (eg see Greene, TW; Wuts, PGM Greene 's Protective Groups in Organic Synthesis; John Wiley & Sons: Hoboken, 2007.
- the base used is imidazole and the silylating agent is TBDMSCl.
- the compounds of formula (II) can be prepared by reacting a compound of formula (XXIV) in the presence of an alcohol of formula R 1 OH, wherein R 1 and n are as defined above.
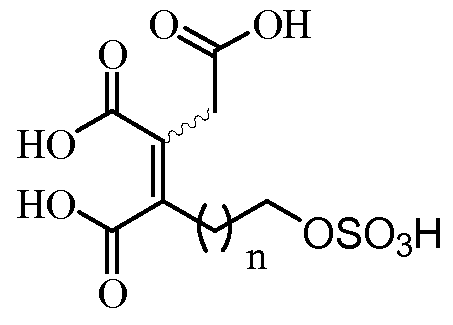
- a further aspect of the present invention relates to a process for the synthesis of a compound of formula (I),
- n is an integer between 2 and 20; its stereoisomers or mixtures thereof, characterized in that it comprises at least one of the following steps (ai), (aii) or (aiii)
- n, R 1 , R 2 and R 3 are as defined above; and additionally understands
- sulphating agents are known to the person skilled in the art. The most common are SO3 and HSO3CI. Preferably the sulphating agent is the S ⁇ 3 -pyridine complex.
- the process of the invention comprises obtaining a compound of formula cis- (I), cis- (VI), cis- (VII), cis- (VIII), cis- (VIIIa ), cis- (IX), cis- (XVI), cis- (XVIa) or cis- (XVIII) or enrichment in the cis- isomer of a mixture of cis and trans isomers of a compound of formula (I), (VI), (VII), (VIII), (Villa), (IX), (XVI), (XVIa) or (XVIII), by exposure to a basic medium of a compound of formula trans- (I), trans - (VI), trans - (VII), trans - (VIII), trans - (VIIIa), trans - (IX), trans - (XVI), trans - (XVIa) or trans - (XVIII), or a mixture of cis and trans iso
- the process of the invention comprises transforming a compound of formula trans - (VI), trans - (VII), trans - (IX) and / or trans - (XVI) into the corresponding compound of formula trans - (VI), trans - (VII), trans - (IX) and / or trans - (XVI), or in mixtures of the corresponding cis / trans isomers.
- said basic medium comprises S ⁇ 3-pyridine.
- Another additional aspect is a compound of formula (I), its stereoisomers or mixtures thereof, as defined above, except
- Another additional aspect is a compound of formula (IX), its stereoisomers or mixtures thereof, as defined above, except
- a further aspect of the present invention is directed to a process for the preparation of a compound of formula (VIIIb), which comprises reacting a compound of formula (XXX) with a hydride, preferably NaBH 4 (see Burke, SD; Danheiser, RL Handbook of Reagents for Organic Synthesis: Oxidizing and Reducing Agents; John Wiley & Sons: Chichester, 1999. pp .: 394-400).
- a hydride preferably NaBH 4
- R 1 , R 2 and R 3 are independently selected from C1-C20 alkyl, substituted or unsubstituted; and neither is an integer between 1 and 16, preferably between 3 and 12, more preferably between 5 and 10, more preferably 8;
- said compound of formula (XXX) is prepared by reacting a compound of formula (XXXI) with a compound of formula (XXXII) in the presence of a compound of formula PAr 3 , preferably triphenylphosphma, wherein each of the groups Ar is independently selected from a C 6 -CiO aryl group, and where n, R 1 , R 2 and R 3 are as defined above.
- a compound of formula PAr 3 preferably triphenylphosphma, wherein each of the groups Ar is independently selected from a C 6 -CiO aryl group, and where n, R 1 , R 2 and R 3 are as defined above.
- said compound of formula (XXXI) is prepared by reacting a compound of formula (XXXIII) with an oxalate of formula (XII) in the presence of a base, where neither and R 2 are as is They have defined above.
- the compounds of formula (XXXI) can be obtained under the conditions described in Seki, K .; Isegawa, J .; Fukuda, M .; Ohki, M. Chem. Pharm. Buil 1984, 32, 1568-1577; or Ashton, W. T .; Doss, G. A. J. Heterocycl Chem. 1993, 30, 307-311, which are incorporated by reference in their entirety.
- the dioxane was degassed by passing a stream of argon before being used.
- the general procedure used to create the H 2 atmosphere necessary to carry out the hydrogenation reactions consisted of: after emptying the reaction flask, a stream of H 2 was connected. The empty / H 2 process was repeated twice, and finally two balloons filled with H 2 were connected. The reactions under high pressure conditions were carried out in glass vials of the Kimble brand.
- IR infrared
- LRMS Low resolution mass spectra
- the elemental analyzes were performed with the Perkin-Elmer 240C and Heraus CHN-O-Rapid analyzers. The calculated and observed data are expressed in percentages.
- the product was purified by chromatographic column (hexane / CH 2 Cl 2 , 2: 1), obtaining (2.45 g, rto. 86%) 12-benzyloxy-1-bromo do decane (7), as a clear oil.
- the product was purified by trituration with MeOH, obtaining (0.126 g, quantitative ortho) (Z) -3,4- bis (methoxycarbonyl) -8-sulfooxy-3-octenoate methyl (31-Z), as a white solid .
- the product was purified by chromatographic column (hexane / AcOEt, 10: 1), obtaining (0.754 g, rto. 50%) (3Z, 8Z) -20-benzyloxy-3,4-bis (methoxycarbonyl) -3,8- methyl icosadienoate (23-2) and (0.261 g, rto. 18%) (£ 3, 8Z) -20-benzyloxy-3,4-bis (methoxycarbonyl) -3,8-methyl icosadienonate (23-E) , both as a transparent oil.
- He product was purified by trituration with MeOH, obtaining (0.119 g, quantitative rto.) a mixture of (£) -3,4-bis (methoxycarbonyl) -20-sulfooxy-3-methyl icosenoate (37-E) and (Z ) -3,4-bis (methoxycarbonyl) -20-methyl sulfooxy-3-icosenoate (37-Z) in a 2: 3 ratio, respectively, as a white solid.
- the product was purified by graphic chromate column (hexane / AcOEt, 6: 1), obtaining e (0.240 g, rto. 64%) (2Z, 4Z) -3,4-bis (methoxycarbonyl) -6-oxo-2, 4- methyl pentadecadienoate (86), as a colorless oil.
- LRMS m / z 382 (M +, 1), 351 (4), 323 (90), 291 (11), 265 (3), 255 (10), 227 (32), 196 (100).
- LRMS m / z 386 (M +, 0), 353 (0), 320 (2), 295 (1), 265 (0), 235 (1), 198 (91), 166 (100), 139 (28), 111 (10).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description




















































Claims

























Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011507949A JP2011519903A (ja) | 2008-05-06 | 2009-05-05 | オレガン酸の調製方法 |
| EP09742183A EP2287146A4 (en) | 2008-05-06 | 2009-05-05 | PROCESS FOR THE PREPARATION OF OREGANIC ACID |
| CN2009801239430A CN102066309A (zh) | 2008-05-06 | 2009-05-05 | 制备奥瑞盖尼酸的方法 |
| US12/991,520 US20110301370A1 (en) | 2008-05-06 | 2009-05-05 | Method for preparing oreganic acid |
| AU2009245667A AU2009245667A1 (en) | 2008-05-06 | 2009-05-05 | Process for preparing oreganic acid |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES200801304A ES2328893B1 (es) | 2008-05-06 | 2008-05-06 | Procedimiento para la preparacion de acido oreganico. |
| ESP200801304 | 2008-05-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009135977A1 true WO2009135977A1 (es) | 2009-11-12 |
Family
ID=41258529
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/ES2009/070138 Ceased WO2009135977A1 (es) | 2008-05-06 | 2009-05-05 | Procedimiento para la preparación de ácido oregánico |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20110301370A1 (es) |
| EP (1) | EP2287146A4 (es) |
| JP (1) | JP2011519903A (es) |
| CN (1) | CN102066309A (es) |
| AU (1) | AU2009245667A1 (es) |
| ES (1) | ES2328893B1 (es) |
| WO (1) | WO2009135977A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103936635B (zh) * | 2011-06-17 | 2016-10-12 | 广东东阳光药业有限公司 | 鱼腥草衍生物及其在药物中的应用 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AUPQ480399A0 (en) * | 1999-12-22 | 2000-02-03 | Commonwealth Scientific And Industrial Research Organisation | Unsaturated fatty acids and their uses in therapy |
-
2008
- 2008-05-06 ES ES200801304A patent/ES2328893B1/es not_active Expired - Fee Related
-
2009
- 2009-05-05 CN CN2009801239430A patent/CN102066309A/zh active Pending
- 2009-05-05 US US12/991,520 patent/US20110301370A1/en not_active Abandoned
- 2009-05-05 WO PCT/ES2009/070138 patent/WO2009135977A1/es not_active Ceased
- 2009-05-05 AU AU2009245667A patent/AU2009245667A1/en not_active Abandoned
- 2009-05-05 JP JP2011507949A patent/JP2011519903A/ja not_active Withdrawn
- 2009-05-05 EP EP09742183A patent/EP2287146A4/en not_active Withdrawn
Non-Patent Citations (20)
| Title |
|---|
| ARMAREGO, W. L.; PERRIN, D. D.: "Purification of Laboratory Chemicals", 1996, BUTTERWORTH-HEINEMANN |
| ASHTON, W. T.; DOSS, G. A., J. HETEROCYCL. CHEM., vol. 30, 1993, pages 307 - 311 |
| BOSONE, E.; FARINA, P.; GUAZZI, G.; INNOCENTI, S.; MAROTTA, V., SYNTHESIS, 1983, pages 942 - 944 |
| BURKE, S.D.; DANHEISER, R.L.: "Handbook of Reagents for Organic Synthesis: Oxidizing and Reducing Agents", 1999, JOHN WILEY & SONS, pages: 394 - 400 |
| DALLA, V.; CATTEAU, J. P., TETRAHEDRON, vol. 55, 1999, pages 6497 - 6510 |
| GARCIA, J.; LOPEZ, M.; ROMEO, J., TETRAHEDRON: ASYMMETRY, vol. 10, 1999, pages 2617 - 2626 |
| GIBBS, R. A.; ZAHN, T. J.; SEBOLT-LEOPOLD, J. S, CURR. MED. CHEM., vol. 8, 2001, pages 1437 - 1465 |
| GREENE, T. W.; WUTS, P. G. M.: "Greene's Protective Groups in Organic Synthesis", 2007, JOHN WILEY & SONS |
| JAYASURIYA, H.; BILLS, G. F.; CASCALES, C.; ZINK, D. L.; GOETZ, M. A.; JENKINS, R. G.; SILVERMAN, K. C.; LINGHAM, R. B.; SINGH, S., BIOORG. MED. CHEM. LETT., vol. 6, 1996, pages 2081 - 2084 |
| KOCIENSKI, P. J., PROTECTING GROUPS, 2000, pages 187 - 230 |
| KOCIENSKI, P. J., PROTECTING GROUPS, 2000, pages 393 - 425 |
| LAROCK, R. C.: "Comprehensive Organic transformations", 1999, JOHN WILEY & SONS, pages: 1234 - 1249 |
| MARYANOFF B. ET AL.: "The Wittig olefination reaction and modifications involving phosphoryl- stabilized carbanions", CHEMICAL REVIEWS, vol. 89, 1989, pages 863 - 927, XP008146334 * |
| MARYANOFF, B. E.; REIZT, A. B., CHEM. REV., vol. 89, 1989, pages 863 - 927 |
| NISHIMURA, S.: "Heterogenous Catalytic Hydrogenation for Organic Synthesis", 2001, JOHN WILEY & SONS |
| See also references of EP2287146A4 |
| SEKI, K.; ISEGAWA, J.; FUKUDA, M.; OHKI, M., CHEM. PHARM. BULL., vol. 32, 1984, pages 1568 - 1577 |
| SILVERMAN K. ET AL.: "Oreganic acid, a potent inhibitor of Ras famesyl-protein transferase", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, vol. 232, 1997, pages 478 - 481, XP008146353 * |
| SILVERMAN, K. C.; JAYASURIYA, H.; CASCALES, C.; VILELLA, D.; BILLS, G. F.; JENKINS, R. G.; SINGH, S. B.; LINGHAM, R. B., BIOCHEM. BIOPHIS. RES. COMMUN., vol. 232, 1997, pages 478 - 481 |
| YAVARI, I.; SAMZADEH-KERMANI, A. R., TETRAHEDRON LETT., vol. 39, 1998, pages 6343 - 6344 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102066309A (zh) | 2011-05-18 |
| EP2287146A1 (en) | 2011-02-23 |
| JP2011519903A (ja) | 2011-07-14 |
| AU2009245667A1 (en) | 2009-11-12 |
| EP2287146A4 (en) | 2012-06-27 |
| ES2328893A1 (es) | 2009-11-18 |
| ES2328893B1 (es) | 2010-09-22 |
| US20110301370A1 (en) | 2011-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105315247B (zh) | 贝前列素的生产方法 | |
| JP6272888B2 (ja) | 胆汁酸誘導体を調製するためのプロセス | |
| ES2355586T3 (es) | Procedimiento de preparación de drospirenona. | |
| ES2699151T3 (es) | Nuevo procedimiento de fabricación del (E,Z)-7,9-dodecadienil-1-acetato | |
| WO2009135977A1 (es) | Procedimiento para la preparación de ácido oregánico | |
| CN104781231A (zh) | 制备曲伏前列素的方法 | |
| WO2006026274A2 (en) | Process for preparing chloromethyl di-tert-butylphosphate | |
| ES2316555T3 (es) | Reacciones de adicion sin catalizar. | |
| KR102436114B1 (ko) | 신규한 이노토디올의 제조방법 | |
| US20080221340A1 (en) | Process for the Production of Nebivolol | |
| CN103387587B (zh) | 恩替卡韦中间体及其制备方法 | |
| WO2003031401A1 (en) | Process for preparation of 19-norvitamin d derivatives | |
| PL163316B1 (pl) | Sposób wytwarzania pochodnych kwasu glutarowego PL PL PL PL PL | |
| JP2011074061A (ja) | スルホニウム塩の製造方法およびそれによって製造されたスルホニウム塩 | |
| Satoh et al. | The first example of an amide-carbonyl stabilized oxiranyl anion: Generation from epoxysilane, its properties, and trapping with electrophiles | |
| ES2293574T3 (es) | Derivados de heptino quirales para la preparacion de epotilonas y procesos para su preparacion. | |
| CN101130529A (zh) | 光学活性2,3-二羟基丁内半缩醛衍生物的制备方法 | |
| Okazaki et al. | Total Synthesis and Absolute Configuration Revision of Biofloranate F | |
| JP2022022148A (ja) | ラタノプロステン・ブノド及びその中間体の製造方法、並びにそれらを含む組成物 | |
| KR19980065189A (ko) | 무스콘의 신규합성중간체 및 그의 제조방법 | |
| WO2009137691A2 (en) | 1,3-diol synthesis via controlled, radical-mediated c-h functionalization | |
| JPS6254303B2 (es) | ||
| JP2002322114A (ja) | カルビノール誘導体 | |
| CA3137236A1 (en) | Process and intermediates for the preparation of eldecalcitol | |
| JP2000186098A (ja) | A−76202及びその類縁化合物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980123943.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09742183 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011507949 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009742183 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009245667 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2009245667 Country of ref document: AU Date of ref document: 20090505 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12991520 Country of ref document: US |