WO2009146152A2 - Non-myelosuppressive compounds, pharmaceutical compositions thereof, and methods of treatment - Google Patents
Non-myelosuppressive compounds, pharmaceutical compositions thereof, and methods of treatment Download PDFInfo
- Publication number
- WO2009146152A2 WO2009146152A2 PCT/US2009/039679 US2009039679W WO2009146152A2 WO 2009146152 A2 WO2009146152 A2 WO 2009146152A2 US 2009039679 W US2009039679 W US 2009039679W WO 2009146152 A2 WO2009146152 A2 WO 2009146152A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- cancer
- compound
- alkyl
- alkenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC[C@](C)C([C@@](C)C([C@](C)(CC=C)C(O*)=CN*CC1*=C1)=O)=* Chemical compound CC[C@](C)C([C@@](C)C([C@](C)(CC=C)C(O*)=CN*CC1*=C1)=O)=* 0.000 description 10
- XITNRWBAAWMPAZ-MLWJPKLSSA-N CC(C)[C@@H]1SPCC(C)N1 Chemical compound CC(C)[C@@H]1SPCC(C)N1 XITNRWBAAWMPAZ-MLWJPKLSSA-N 0.000 description 1
- WLHCBQAPPJAULW-UHFFFAOYSA-N Cc(cc1)ccc1S Chemical compound Cc(cc1)ccc1S WLHCBQAPPJAULW-UHFFFAOYSA-N 0.000 description 1
- IVSZLXZYQVIEFR-UHFFFAOYSA-N Cc1cccc(C)c1 Chemical compound Cc1cccc(C)c1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 1
- UFWYSUDGLSPWIA-UHFFFAOYSA-N NC1=CC2=PP=PC2=C1 Chemical compound NC1=CC2=PP=PC2=C1 UFWYSUDGLSPWIA-UHFFFAOYSA-N 0.000 description 1
- BSJGHOQJDCMQMT-UHFFFAOYSA-N c1ccc2P=P[P]c2c1 Chemical compound c1ccc2P=P[P]c2c1 BSJGHOQJDCMQMT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/14—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
Definitions
- 2-Methoxyestradiol also called 2ME2 or PANZEM ®
- 2ME2 microtubule-targeting drug
- PANZEM ® microtubule-targeting drug
- Data from those clinical trials has shown clinical efficacy, including cases of stable disease in patients with advanced breast cancer and multiple myeloma and a durable partial response in a patient with ovarian carcinoma who had failed three prior chemotherapy regimens.
- the safety data from those clinical trials have demonstrated that 2ME2 is very well tolerated with moderate side effects. This is in marked contrast with other approved MTDs widely used in cancer chemotherapy such as the taxanes and Vinca alkaloids.
- Clinical administration of these drugs is known to induce severe adverse effects, mostly associated with peripheral neuropathy and myelosuppression, which limit the clinical applicability of MTDs and diminish their efficacy.
- R 2 is selected from the group consisting of aryl, alkaryl, alkenaryl, alkynaryl, heteroaryl, heteroalkenyl, alkyl, alkenyl, cyclo-alkyl, aryloxy, alkoxy, or alkenoxy;
- FIG. 11 illustrates the specificity of H ⁇ l tubulin antibody tested by Western blot analysis of total cell extracts from hematopoietic tissue or the epithelial cancer cell lines PC3 (prostate), HT-29 (colorectal) and 1A9 (ovarian) and 1A9 cells transiently transfected with an H ⁇ l-encoding plasmid; evidence for the presence of tubulin in all samples is shown by the positive staining for total ⁇ -tubulin (DM 1 ⁇ );
- FIG. 12 provides confocal images of 1A9 transiently transfected with a full length H ⁇ l plasmid; cells were treated overnight with the indicated drug concentrations; cells were co-stained with ⁇ -tubulin (clone 2.1) and H ⁇ l-specific antibodies; DNA was counterstained with DAPI;
- compound as used herein means a chemical entity, whether in the solid, liquid, or gaseous phase, and whether in a crude mixture or purified and isolated.
- alkyl refers to methyl, trifluoromethyl, ethyl, propyl, isopropyl, cyclopropyl, butyl, isobutyl, t-butyl, pentyl, cyclopentyl, isopentyl, neopentyl, hexyl, isohexyl, cyclohexyl, cyclohexylmethyl, 3-methylpentyl, 2,2-dimethybutyl, and 2,3- dimethylbutyl.
- Substituted alkenyl refers to alkenyl substituted with one or more moieties selected from the group consisting of halo (e.g., Cl, F, Br, and I); halogenated alkyl (e.g., CF 3 , 2-Br-ethyl, CH 2 F, CH 2 Cl, CH 2 CF 3 , or CF 2 CF 3 ); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryloxy; nitro; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate.
- heteroalkenyl as used herein means an alkenyl group as described above having one or more carbon atoms replace with a heteroatom, such as N, O, or S.
- alkynyl as used herein means alkyl moieties wherein at least one saturated C-C bond is replaced by a triple bond.
- alkynyl refers to groups comprising 1 to 10 carbon atoms ("C 1-10 alkynyl”).
- alkynyl refers to groups comprising 1 to 8 carbon atoms (“C 1-8 alkynyl”), 1 to 6 carbon atoms (“Ci_ 6 alkynyl”), or 1 to 4 carbon atoms (“C 1-4 alkynyl”).
- alkynyl can be ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1- pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1- hexynyl, 2-hexynyl, 3-hexynyl, A- hexynyl, or 5 -hexynyl.
- aryl as used herein means a stable monocyclic, bicyclic, or tricyclic carbon ring of up to 8 members in each ring, wherein at least one ring is aromatic as defined by the Huckel 4n+2 rule. Multiple aryl rings may be fused, and aryl rings may be fused or unfused with one or more cyclic hydrocarbon, heteroaryl, or heterocyclic rings. Exemplary aryl groups according to the invention include phenyl, naphthyl, tetrahydronaphthyl, and biphenyl.
- alkaryl and "alkylaryl” as used herein means an alkyl group as defined above linked to the molecule through an aryl group as defined above.
- alkylene as used herein means an alkyl group having two free valencies (i.e., a divalent alkyl radical);
- amide as used herein means a compound having the general formula
- antiproliferative agent means a compound that decreases the hyperproliferation of cells.
- abnormal cell proliferation means a disease or condition characterized by the inappropriate growth or multiplication of one or more cell types relative to the growth of that cell type or types in an individual not suffering from that disease or condition.
- the present invention provides analogues of docetaxel wherein the tert-butyl group on the C3'-NHC0 is replaced with an analogue-forming group. In one embodiment, the present invention provides analogues of paclitaxel wherein the C3' phenyl group is replaced with an analogue-forming group.
- FIG. 1 shows paclitaxel in the bioactive T-Taxol conformation at the ⁇ -tubulin taxane binding site in the class I ⁇ -tubulin isotype.
- the phenyl ring on the C3'-NHCO is clamped between residues 23 and 231 (valine and alanine for this isotype) in the class I (HM40) ⁇ -tubulin isotype.
- FIG. 2 shows paclitaxel in the bioactive T-Taxol conformation at the ⁇ -tubulin taxane binding site in the class VI ⁇ -tubulin isotype.
- alkyl, alkenyl, and alkynyl groups include but are not restricted to straight chain, branched chain, cyclic, and polycyclic (e.g., norbornyl) groups, as well as aryl or alkyl substituted groups.
- Aryl groups can include but are not limited to benzene, naphthalene, and other condensed aromatic hydrocarbons optionally substituted with alkyl, halogen, amino, ester, azido, nitro, and similar polar and nonpolar substituents.
- Heteroalkyl and heteroalkenyl groups include but are not limited to the fully saturated and di- and tetrahydro variations of the heteroaryl systems mentioned above including tetrahydrofuran, piperidine, morpholine, and alkylated and esterified variations, as well as condensed polycyclic systems, such as tropane, quinuclidine, l,4-diazabicyclo[2.2.2.0]-octane and similar systems substituted by alkyl and ester functionalities.
- the invention encompasses compounds according to Formula (3), wherein Ri can be selected from the group consisting of bridged polycyclo compounds, fused polycyclo compounds, and substituted aromatic compounds.
- bridged polycyclo compounds that may be used as Ri include compounds having from 6 to 15 carbon atoms, from 6 to 14 carbon atoms, from 6 to 13 carbon atoms, from 6 to 12 carbon atoms, or from 6 to 11 carbon atoms.
- Non-limiting examples of fused polycyclo compounds that may be used as Ri include compounds having from 6 to 15 carbon atoms, from 6 to 14 carbon atoms, from 6 to 13 carbon atoms, from 6 to 12 carbon atoms, or from 6 to 11 carbon atoms.
- the compounds may be two fused rings wherein one ring has 6 atoms and the other ring has 4 atoms, 5 atoms, 6 atoms, 7 atoms, or 8 atoms.
- the compounds also may be two fused rings wherein one ring has 5 atoms and the other ring has 4 atoms, 5 atoms, 7 atoms, or 8 atoms.
- Non-limiting examples of substituted aromatic compounds that may be used as Ri include compounds having from 7 to 20 carbon atoms, from 8 to 20 carbon atoms, from 8 to 18 carbon atoms, from 8 to 16 carbon atoms, or from 8 to 14 carbon atoms.
- the compounds may be phenyl rings that are one or more of ortho-, meta-, or para- substituted.
- substituents may include alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, C(O)O-alkyl, C(O)O-alkenyl, or C(O)O-alkynyl, all of which may be linear or branched.
- Specific non-limiting examples of substituted aromatic compounds that may be encompassed by the present invention as an Ri group, according to Formula (3) include the following (wherein W can be a substituent, as described in the preceding paragraph).
- substituted aromatic compounds that may be encompassed by the present invention as an R 2 group, according to Formula (3), include the following.
- Non-limiting examples of cycloalkanes or cycloalkenes that may be used as R3 include C3-C7 compounds, particularly C3-C5 compounds.
- Specific non-limiting examples of cycloalkanes or cycloalkenes that may be encompassed by the present invention as an R 3 group, according to Formula (3), include the following.
- X5 is selected from the group consisting of
- the known fragment (I) was prepared according to a known procedure (Koch G., et al., 2004, Synlett 4:693-697 and Nicolaou KC, et al., 1997, J. Am. Chem. Soc. 119:79747991). Fragment (II) was prepared as shown below.
- the H ⁇ l selective MTC does not bind H ⁇ l but does bind other ⁇ -tubulin isotypes, the H ⁇ l selective MTC would be expected to be effective to treat the cancer of the patient in a non-myelosuppressive fashion, as otherwise disclosed herein.
- the foregoing is provided for exemplary purposes and is not intended to limit the scope of the invention. Rather, the same testing procedure could be used to evaluate whether a patient would respond to compounds selective for other ⁇ -tubulin isotypes.
- the testing methods of the invention may include testing of various cell components to determine the appropriate method of treatment.
- the method can comprises isolating and testing nucleic acids from the cancer cells.
- the method may also comprise isolating and testing proteins from the cancer cells.
- the present invention has identified that compounds that selectively do not inhibit H ⁇ l -tubulin are useful as myelo-sparing MTCs. Accordingly, in certain embodiments, the invention provides a method for identifying myelo-sparing microtubule targeting compounds, the method comprising the following steps: i) screening a library of compounds against H ⁇ l -tubulin to determine whether the compounds individually inhibit the H ⁇ l -tubulin; ii) discarding agents that inhibit the H ⁇ l -tubulin; and iii) retaining agents that do not inhibit the H ⁇ l -tubulin.
- an initial screen can be performed in cells (e.g., epithelial cancer cells) transfected with an H ⁇ l tubulin encoding mammalian expression plasmid, and the cells then can be exposed to varying concentrations of each compound to be tested and compared to a control.
- the effect of each compound on H ⁇ l -specific tubulin can be assessed by a cell-based tubulin polymerization assay as described below in relation to FIG. 4 in conjunction with immunostaining with the H ⁇ l -specific antibody to assess the effects of each of the compounds on H ⁇ l microtubule polymer mass.
- the method comprises the following steps: i) screening a library of compounds against a ⁇ -tubulin with an amino acid other than valine at ⁇ 236, tyrosine at ⁇ 200, or valine at ⁇ 316; ii) discarding compounds that inhibit the ⁇ -tubulin; and iii) retaining compounds that do not inhibit the ⁇ -tubulin.
- optical isomers of the compounds according to the present invention include the following: i) physical separation of crystals whereby macroscopic crystals of the individual enantiomers are manually separated.
- This technique may particularly be used when crystals of the separate enantiomers exist (i.e., the material is a conglomerate), and the crystals are visually distinct; ii) simultaneous crystallization whereby the individual enantiomers are separately crystallized from a solution of the racemate, possible only if the latter is a conglomerate in the solid state; iii) enzymatic resolutions whereby partial or complete separation of a racemate by virtue of differing rates of reaction for the enantiomers with an enzyme; iv) enzymatic asymmetric synthesis, a synthetic technique whereby at least one step of the synthesis uses an enzymatic reaction to obtain an enantiomeric ally pure or enriched synthetic precursor of the desired enantiomer; v) chemical asymmetric synthesis whereby the desired enantiomer is synthesized from an achiral precursor under conditions that produce asymmetry (i.e., chirality) in the product, which may be achieved using chiral catalysts or chiral aux
- the desired enantiomer is then released from the diastereomers; viii) kinetic resolutions comprising partial or complete resolution of a racemate (or of a further resolution of a partially resolved compound) by virtue of unequal reaction rates of the enantiomers with a chiral, non-racemic reagent or catalyst under kinetic conditions; ix) enantiospecific synthesis from non-racemic precursors whereby the desired enantiomer is obtained from non-chiral starting materials and where the stereochemical integrity is not or is only minimally compromised over the course of the synthesis; x) chiral liquid chromatography whereby the enantiomers of a racemate are separated in a liquid mobile phase by virtue of their differing interactions with a stationary phase.
- the compounds of the invention can comprise an MTC having an enantiomeric purity for a single enantiomer of at least about 75%.
- the compounds of the invention have an enantiomeric purity of at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or at least about 99.5%.
- the compounds of the invention may have such enantiomeric purity for the (S) isomer.
- the compounds of the invention may have such enantiomeric purity for the (R) isomer.
- compositions include propionic acid, glycolic acid, oxalic acid, malic acid, malonic acid, benzoic acid, cinnamic acid, mandelic acid, salicylic acid, and the like.
- pharmaceutically acceptable salts include, but are not limited to, sulfates, pyrosulfates, bisulfates, sulfites, bisulf ⁇ tes, phosphates, monohydrogenphosphates, dihydrogenphosphates, metaphosphates, pyrophosphates, chlorides, bromides, iodides, acetates, propionates, decanoates, caprylates, acrylates, formates, isobutyrates, caproates, heptanoates, propiolates, oxalates, malonates, succinates, suberates, sebacates, fumarates, maleates, butyne-l,4-dioates, hexyne-1,6- dioates,
- prodrug ligands are known.
- alkylation, acylation, or other lipophilic modification of one or more heteroatoms of the compound, such as a free amine or carboxylic acid residue reduces polarity and allows passage into cells.
- the compounds of the invention can be prepared and delivered together with one or more pharmaceutically acceptable carriers therefore, and optionally, other therapeutic ingredients.
- Carriers should be acceptable in that they are compatible with any other ingredients of the composition and not harmful to the recipient thereof.
- a carrier may also reduce any undesirable side effects of the agent.
- Such carriers are known in the art. See, Wang et al. (1980) J. Parent. Drug Assn. 34(6):452-462, herein incorporated by reference in its entirety.
- Hard capsules containing the compound may be made using a physiologically degradable composition, such as gelatin.
- Such hard capsules comprise the compound, and may further comprise additional ingredients including, for example, an inert solid diluent such as calcium carbonate, calcium phosphate, or kaolin.
- Soft gelatin capsules containing the compound may be made using a physiologically degradable composition, such as gelatin.
- Such soft capsules comprise the compound, which may be mixed with water or an oil medium such as peanut oil, liquid paraffin, or olive oil.
- Sublingual tablets are designed to dissolve very rapidly.
- compositions include ergotamine tartrate, isosorbide dinitrate, and isoproterenol HCL.
- the compositions of these tablets contain, in addition to the drug, various soluble excipients, such as lactose, powdered sucrose, dextrose, and mannitol.
- Transdermal drug delivery patches may also be comprised of a reservoir underlying the backing layer that is separated from the skin of the recipient by a semi-permeable membrane and adhesive layer. Transdermal drug delivery may occur through passive diffusion or may be facilitated using electrotransport or iontophoresis.
- the MTCs according to the present invention are useful in a variety of methods of treatment.
- the inventive MTCs may be useful in any methods of treatment where conventional taxanes and/or epothilones may find use.
- the MTC of the invention provide such treatment with designed specificity to avoid the undesirable side effects of conventional taxanes and/or epothilones, particularly myelosuppression.
- the MTCs of the invention may be useful in a method of treating cancer.
- the MTCs of the invention may be useful in method of treating and/or preventing abnormal cell proliferation.
- a hypotonic lysis buffer that recovers less than 10% of microtubule polymer in taxane-treated samples and their respective untreated control was used.
- the percentage of polymerized tubulin was determined by dividing the densitometric value of polymerized tubulin (P) by the total tubulin content (P+S).
- a similar assay was performed using human platelets obtained from peripheral blood from a healthy donor following centrifugation of platelet-rich plasma at 300xg for 10 minutes.
- Class VI ⁇ -tubulin isotype mRNA amounts were analyzed by quantitative real-time PCR using probe 14 of the Universal Human Probe Library detection system (Roche) in conjunction with the following specific primers for class VI ⁇ -tubulin isotype (H ⁇ l/TUBBl gene, accession # NM_030773): Forward 5 '-GGATGCGTGAAATTGTCCAT-S ' and reverse 5 '-AGTCGATCCCGTGTTCCTC-S'. Primers were used at a final concentration of 200 nM each.
- Umbilical cord blood cells were obtained from the public cord blood bank of the New York- Presbyterian Hospital Weill Cornell Medical Center.
- Peripheral blood mononuclear cells PBMCs
- PBMCs Peripheral blood mononuclear cells
- CD34 positive cells were sorted using a monoclonal Ab (StemCell Technologies, Inc.).
- Statistical analysis was performed using a single factor ANOVA test (p ⁇ 0.05 was considered statistically significant).
- a 2ME2-resistance cell model was generated consisting of the parental 1A9 human ovarian carcinoma cells and its drug-resistant counterpart 1A9/2MRC cells (2MRC).
- the 2MRC cells were more than 80-fold resistant to 2ME2 and did not exhibit significant cross resistance to any of the other classes of microtubule inhibitors, including the colchicine - site binding agents (combretastatin, ENMD MKC-I and colchicine) as well as the 2ME2 analog ENMDl 198, also undergoing clinical development (See Table 2 below). These results indicate that the 2MRC cells display a 2ME2-specif ⁇ c resistant phenotype.
- the second genetic alteration is a missense mutation resulting in a change in amino acid 236 from Valine to Isoleucine ( ⁇ V236I).
- Subcloning of the cDNA PCR products followed by direct sequencing further showed that the two genetic alterations were mutually exclusive since 100% of the clones analyzed had either the frame-shift mutation (L187fsX193) or the missense mutation (1236), indicating that the two alterations occurred in different alleles. This result suggests that the 2MRC cells functionally express only the HM40 ⁇ 236 mutant tubulin only responsible for the 2ME2 resistance.
- H ⁇ l model predicts that while PTX encounters no difficulty binding to the class VI ⁇ -tubulin isotype, 2ME2 is hindered from occupying the colchicine pocket primarily by the 1236 side chain. Therefore, H ⁇ l tubulin is predicted to display resistance to the effects of 2ME2 while retaining its sensitivity to PTX.
- tubulin immunofluorescence with a ⁇ -tubulin antibody recognizing all ⁇ -isotypes but H ⁇ l showed overlap between the two staining (see merge).
- Treatment with 2ME2 had no effect on the microtubule network of H ⁇ l -expressing cells, even at the dose of 100 ⁇ M, as shown by the intact, well organized microtubule cytoskeleton in the transfected cells.
- 2ME2 treatment caused extensive microtubule depolymerization with short microtubule fragments scattered throughout the cytoplasm and an increase in the number of atypical mitotic figures, consistent with the drug-sensitive phenotype of the non-transfected 1A9 cells (FIG. 5).
- mice deficient in ⁇ 1 -tubulin develop moderate thrombocytopenia as a result of reduced proplatelet formation and their spherocytic platelets carry a structurally defective marginal band and reduced microtubule content.
- the role of ⁇ l -tubulin is limited to a few clinical studies showing that a Q43P polymorphism in H ⁇ l is present in approximately 11% of the general population and in approximately 24% of a small cohort of patients with macrothombocytopenia.
- Heterozygous carriers show defects in platelet aggregation that protects against thrombotic disorders and appears to reduce the risk of cardiovascular disease in men, but increases the risk of intracerebral hemorrhage.
- a solution of compound (j) (908 mg, 0.735 mmol) in THF (7.5 mL) was added a stock solution of hydrogen fluoride in pyridine (HF.Py) (this stock solution was prepared by addition of 1.25 mL HF.Py to 3.5 mL pyridine in 6.125 mL THF) at 0 0 C.
- the resulting reaction mixture was warmed to 25 0 C by removing the ice-bath and allowed to stir at that temperature for 2 h.
- Tje epothilone analog of formula (19) was prepared from the diene of compound (t), using a similar procedure to that described for the conversion of compound (q) to the compound of Formula (18a) as a colorless oil in 44% yield (55% starting material was recovered).
- Iodide - Compound of Formula (44) To a solution of primary alcohol of Formula (43) (19 mg, 0.02497 mmol) in THF (2.5 mL) was added PPh 3 (13 mg, 0.0499 mmol, 2 eq), followed by imidazole (5.1 mg, 0.07491 mmol, 3 eq) and iodine (25 mg, 0.09988 mmol, 4 eq). The resulting reaction mixture was allowed to stir at 25 0 C for 30 min prior to being quenched by the addition of saturated Na 2 S 2 O 3 solution (10 mL).
- Epothilone D analog - Compound of Formula (45) Epothilone D analog - Compound of Formula (45). Iodide of Formula (44) (19.3 mg, 0.022 mmol) and sodiumcyanoborohydride (14 mg, 0.22 mmol, 10 eq) were dissolved in HMPA (0.2 mL) and the resulting mixture was heated at 45 0 C for 22 h. After cooling to 25 0 C, the reaction mixture was diluted with 0.2 mL of EtOAc and then directly loaded onto preparative thin layer chromatography, eluting with 15% EtOAc in hexanes, to yield pure epothilone D analog of Formula (45) (10 mg, 60.6%) as a colorless oil.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides microtubule targeting compounds that are myelo-sparing. Particularly, the MTCs of the invention are structurally designed to exhibit specificity for inhibiting β-tubulin isotypes that are not related to the myelosuppression associated with other known MTCs. The invention further provides pharmaceutical compositions incorporating the inventive MTCs and methods of treating cancer using the MTCs. The invention still further provides various assays for identifying myelo-sparing compounds and determining whether such compounds are indicated for use with specific patients.
Description
NON-MYELOSUPPRESSIVE COMPOUNDS, PHARMACEUTICAL COMPOSITIONS THEREOF, AND METHODS OF TREATMENT
BACKGROUND
Microtuble-targeting drugs (MTDs), such as taxanes, are highly active, cytotoxic, anti-tumor agents which work by disrupting the function of a cell's microtubules. Accordingly, MTDs represent one of the most effective classes of cancer chemotherapeutics. Working essentially as mitotic inhibitors, MTDs stabilize GDP- tubulin in microtubules, thus locking cells in a permanent state of mitosis, eventually leading to cell death.
The utility of known MTDs (e.g., therapeutic benefits in both hematopoietic and solid tumors), however, is limited because of a wide array of negative side effects, the most detrimental being drug-induced myelosuppression, a condition in which bone marrow activity is decreased, resulting in reduced production of red blood cells, white blood cells, and platelets. Myelosuppression is a frequent side-effect of typical cancer treatments, such as those utilizing paclitaxel, docetaxel, and other compounds in the taxane family of drugs, as well as other MTDs.
Microtubules are polymers of α- and β-tubulin dimers that polymerize end-to-end in protofilaments that then bundle in hollow cylindrical filaments. Typically, the proto filaments arrange themselves in an imperfect helix with one turn of the helix containing 13 tubulin dimers, each from a different proto filament. Microtubules are nucleated and organized by the microtubule organizing centers (MTOCs), such as centrioles and basal bodies. They are part of a structural network (the cytoskeleton) within the cell's cytoplasm, but, in addition to structural support, microtubules take part in many other processes, as well. They are capable of growing and shrinking in order to generate force, and there are also motor proteins that allow organelles to move along the microtubule. A notable structure involving microtubules is the mitotic spindle used by eukaryotic cells to segregate their chromosomes correctly during cell division. Microtubules are also part of the cilia and flagella of eukaryotic cells. Current MTDs bind to microtubules, and/or to their constituent tubulin heterodimers (or specifically to the β- tubulin subunit), and affect microtubule polymerization and dynamics, as described above. In recent years, extensive research has been done to find a way to mitigate the side effects of taxanes by altering the administration thereof. For example, DHA -paclitaxel,
PG-paclitaxel, and tumor-activated Taxol prodrugs are have been proposed. One line of research has linked paclitaxel to docosahexaenoic acid (DHA), a fatty acid that is easily taken up by tumor cells, in an attempt to limit the cytotoxicity of the taxane until the bond with DHA is cleaved within the cell. Other research has bound paclitaxel to a polyglutamate polymer since tumor cells tend to exhibit greater porosity to polyglutamate polymers than do normal cells. Still further research is directed toward tumor-activated prodrugs (TAP), which are designed for accurate targeting by the action of a monoclonal antibody which is very specific to certain cells. Such research merely masks or delays the underlying toxicity of the MTD, however. A more desirable solution is the introduction of MTDs that exhibit cancer treatment activity without the undesirable effects. The present invention provides such compounds.
SUMMARY OF THE INVENTION 2-Methoxyestradiol (also called 2ME2 or PANZEM®) is a microtubule-targeting drug (MTD) with potent antiangiogenic activity and is currently in phase II oncology clinical trials. Data from those clinical trials has shown clinical efficacy, including cases of stable disease in patients with advanced breast cancer and multiple myeloma and a durable partial response in a patient with ovarian carcinoma who had failed three prior chemotherapy regimens. In addition, the safety data from those clinical trials have demonstrated that 2ME2 is very well tolerated with moderate side effects. This is in marked contrast with other approved MTDs widely used in cancer chemotherapy such as the taxanes and Vinca alkaloids. Clinical administration of these drugs is known to induce severe adverse effects, mostly associated with peripheral neuropathy and myelosuppression, which limit the clinical applicability of MTDs and diminish their efficacy.
Previously, it has not been known why 2ME2 does not induce myelosuppression and consequently, there has been no molecular ground that would enable the design of a cell-type specific microtubule inhibitor devoid of dose-limiting and life-threatening toxicities. The present invention, however, has overcome this limitation in the art and provides families of compounds that are tubulin inhibitors (and thus provide potent chemotherapeutic effects) while being at least partially, preferably completely, devoid of any undesirable myelosuppressive effects. The ability to provide such compounds with minimal side effects significantly augments the chances of clinical success by allowing the
use of a truly therapeutic dose rather than the maximally tolerated dose. In specific embodiments, the families of compounds are analogues of taxane compounds and/or analogues of epothilone compounds, wherein the analogues have specific side chain alterations that reduce myelosuppression, and specifically white blood cell suppression, while allowing the compounds themselves to retain the chemotherapeutic effects.
The present invention identifies an important molecular mechanism responsible for the drug-induced myelosuppression associated with the clinical use of tubulin inhibitors, such as the taxanes and Vinca alkaloids, in cancer chemotherapy. Despite the clinical success of these drugs, their toxicities to normal tissues (primarily myelosuppression) represent serious obstacles for the treatment of cancer patients. Previously, there has been no molecular ground to render this class of agents specific for the cytoskeleton of tumor cells while sparing bone marrow from dose-limiting, life -threatening toxicities. The present invention, however, has identified the mechanism by which 2ME2 achieves specific tumor targeting without inducing myelosuppression in cancer patients and has applied that mechanism in theory to arrive at new and separate families of compounds that are specific to the dominant tubulin isotypes in tumor cells without affecting the tubulin isotypes in normal human cell, particularly bone marrow cells. Specifically, the present invention shows that an acquired tubulin mutation that confers 2ME2 resistance in vitro is naturally expressed by the hematopoietic-specific Hβl tubulin isotype, which in turn protects the bone marrow from drug-induced myelosuppression.
In the era of targeted molecular therapies, an important aspect of clinical efficacy, namely drug-induced toxicities, is invariably overlooked. However, the ability to design a small molecule with minimal side-effects significantly augments the chances of clinical success by allowing the use of a truly therapeutic dose rather than the maximally tolerated dose. Herein are described the first tubulin isotype-targeted chemotherapeutics, setting a new paradigm for the entire class of antimitotics, and providing a model that is applicable to the design of new tubulin inhibitors devoid of myelosuppressive side effects.
In one aspect, the present invention provides microtubule targeting compounds. In some embodiments, the MTCs are derivatives or analogues of taxane compounds. In other embodiments, the MTCs are epothilone compounds. In one embodiment, the invention is directed to a MTC according to the following formula that does not inhibit β-tubulin isotype Hβl,

wherein:
Ri is selected from the group consisting of aryl, alkaryl, alkenaryl, alkynaryl, heteroaryl, heteroalkenyl, alkyl, alkenyl, cyclo-alkyl, aryloxy, alkoxy, and alkenoxy;
R2 is selected from the group consisting of aryl, alkaryl, alkenaryl, alkynaryl, heteroaryl, heteroalkenyl, alkyl, alkenyl, cyclo-alkyl, aryloxy, alkoxy, or alkenoxy; and
R3 is selected from the group consisting of alkyl, alkenyl, alkynyl, alkaryl, alkenaryl, alkynaryl, acyl, and any of the foregoing groups wherein one or more carbon atoms is replaced with one or more atoms selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof; and wherein the compound excludes paclitaxel and docetaxel. Groups R1, R2, and R3 further may be defined in relation to other embodiments.
For example, Ri may be selected from the group consisting of bridged polycyclics, fused polycyclics, and substituted aromatics; R2 may be selected from the group consisting of substituted aromatics, cycloalkanes, cycloalkenes, fused polycyclics, and branched alkyl or alkenyl; and R3 may be selected from the group consisting of cycloalkanes, cycloalkenes, and linear or branched alkyl, alkenyl, or alkynyl where one or more carbon atoms is optionally replaced with an atom selected from the group consisting of O, S, and N. In other embodiments, R1, R2, and R3 may be described by specific compounds or groups.
The present invention also provides pharmaceutical compositions comprising MTCs. In certain embodiments, the pharmaceutical compositions comprising taxane-type MTCs, as described herein. In other embodiments, the pharmaceutical compositions comprising an epothilone-type MTCs, as described herein. In another aspect, the invention also provides methods of treating cancer in a patient through administration of one or more MTCs according to the invention. In one embodiment, the method comprises administering to the patient a taxane-type MTCs, as described herein. In another embodiment, the method comprises administering to the patient an epothilone-type MTCs, as described herein. In specific embodiments, the cancer being treated can be selected from the group consisting of non-small-cell lung cancer, ovarian cancer, breast cancer, prostate cancer, colo-rectal cancer, renal cancer, gastric cancer, gall bladder cancer, liver cancer, pancreatic cancer, small intestine cancer, testicular cancer, head cancer, neck cancer, melanoma, hepatocellular carcinoma, fallopian tube cancer, endometrial cancer, peritoneal cancer, solid tumors, non-Hodgkin's lymphoma, chronic lymphocytic leukemia, gliomas, and Kaposi's sarcoma. In other embodiments, the cancer being treated is a type wherein a majority of the cancer cells express a β-tubulin isotype that does not substantially include Hβl. In still further embodiments, the cancer is a type wherein the cells do not express Hβl -tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316. The invention also provides a method of reducing myelosuppression in cancer treatment associated with administration of a microtubule targeting compound, hi specific embodiments, the method can comprise administering an MTC according to the invention.
In another aspect, the invention provides various assays around the inventive MTCs. In one embodiment, the invention is directed to a method of determining whether a patient with cancer will exhibit a favorable response to treatment with a microtubule targeting compound that does not inhibit β-tubulin isotype Hβl. Specifically, the method can comprise: i) testing cancer cells from said patient to determine whether the cells overexpress Hβl, wherein a positive result in the test means the cells overexpress Hβland a negative result means the cells do not overexpress Hβl ; ii) correlating a positive result in the test to treatment with a compound other than an MTC that does not inhibit β-tubulin isotype Hβl ; and
iii) correlating a negative result in the test to treatment with an MTC that does not inhibit β-tubulin isotype Hβl.
In another embodiment, the invention is directed to a method to determine whether a patient with cancer will exhibit a favorable response to treatment with a microtubule targeting compound that does not inhibit β-tubulin isotype Hβl, wherein the method can comprise: i) testing cancer cells from the patient to determine whether the cells express β- tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316, wherein a positive result in the test means the cells express Hβl -tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316, and a negative result in the test means the cells do not express Hβl -tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316; ii) correlating a positive result in the test with treatment with an MTC that does not inhibit β-tubulin isotype Hβl; and iii) correlating a negative result in the test with treatment with a compound other than an MTC that does not inhibit β-tubulin isotype Hβl.
In another embodiment, the invention provides a method of identifying a myelo- sparing microtubule targeting compound. Particularly, the method can comprise: i) screening a library of compounds in an assay that determines the ability of the compounds to inhibit Hβl -tubulin; ii) discarding compounds that inhibit said Hβl -tubulin; and iii) retaining compounds that do not inhibit said Hβl -tubulin.
In still another embodiment, the invention provides a method for identifying a myelo-sparing microtubule targeting compound, wherein the method can comprise: i) screening a library of compounds in an assay that determines the ability of the compounds to inhibit β-tubulin with an amino acid other than valine at β236, tyrosine at β200, or valine at β316; ii) discarding compounds that inhibit said β-tubulin; and iii) retaining compounds that do not inhibit said β-tubulin.
BRIEF DESCRIPTION OF THE DRAWINGS
Having thus described the invention in general terms, reference will now be made to the accompanying drawings, which is not necessarily drawn to scale, and wherein:
FIG. 1 is an illustration of the compound paclitaxel in the bioactive T-Taxol conformation at the β-tubulin taxane binding site in the class I β-tubulin isotype;
FIG. 2 is an illustration of the compound paclitaxel in the bioactive T-Taxol conformation at the β-tubulin taxane binding site in the class VI β-tubulin isotype;
FIG. 3 is provides confocal images of 1A9 and 2MRC cells treated overnight with the specified concentrations of 2ME2 or PTX, wherein cells were fixed and immunostained with an antibody against a-tubulin (clone YL1/2);
FIG. 4 shows 1A9 (upper panel) and 2MRC cells (lower panel) that were treated with 2ME2 or TXT for 6 hours, wherein control and 2ME2 treated samples were lysed with a microtubulestabilizing buffer (MTSB) while PTX treated samples and their corresponding control were lysed using a hypotonic buffer (LSB) for 10 min at 37°C; the polymerized (P) and the soluble (S) tubulin fractions were separated by centrifugation, loaded on adjacent lanes in SDS-PAGE, and immunoblotted with an antibody against a-tubulin (clone YL1/2) or acetylated a-tubulin; the percentage of polymerized tubulin (%P) was determined by dividing the densitometric value of polymerized tubulin (P) by the total tubulin content (P+S); the %P values for total and acetylated tubulin represent the median values of 3 different experiments;
FIG. 5 illustrates the impaired ability of 2ME2 to induce cell cycle arrest and apoptosis in 2MRC cells, wherein 1A9 and 2MRC cells were treated with 2ME2 and PTX for 24 hours, and measurement of cellular DNA content and analysis of the cell cycle distribution was performed by flow cytometry; FIG. 6 shows a Western blot analysis for the cleaved forms of caspace-3 and PARP p85 in 1A9 and 2MRC cells treated with 2ME2 and PTX for 48 hours, wherein immunostaining for β-actin was used as a loading control;
FIG. 7 illustrates how an acquired β-tubulin mutation confers 2ME2 -resistance in 2MRC cells where total RNA from 1A9 and 2MRC cells was subjected to RT-PCR amplification using primers specific for class I β-tubulin (HM40/TUBB, RNA accession # NM 178014); the PCR products were cloned into pCR4-TOPO vector followed by direct sequencing; a minimum of 20 clones for each PCR reaction were analyzed; two genetic
alterations were identified in HM40 in expressed in the 2MRC cells; arrows depict the location of the modified nucleotides;
FIG. 8 provides a model of 2ME2 in the colchicine binding site with V236 (left panel) or the acquired mutation 1236 (right panel) highlighted to demonstrate proximity; atomic coordinates were obtained from PDB 1 SAO; 2ME2 replaces colchicine (insert: ROCS best fit of colchicine and 2ME2);
FIG. 9 human β-tubulin isotypes amino acid sequences aligned using Clustal W method; only residues 221-240 are shown; HM40 sequence is used as the reference; non- conserved residues are highlighted; FIG. 10 provides a class VI β-tubulin model (Hβl, left panel) or class I β-tubulin
(HM40, right panel) with bound 2ME2; important binding-site residues are depicted as a ball model. 1236, F200 and 1316 occupy the 2ME2 binding site in HA forcing close contacts that disrupt ligand binding (left panel);
FIG. 11 illustrates the specificity of Hβl tubulin antibody tested by Western blot analysis of total cell extracts from hematopoietic tissue or the epithelial cancer cell lines PC3 (prostate), HT-29 (colorectal) and 1A9 (ovarian) and 1A9 cells transiently transfected with an Hβl-encoding plasmid; evidence for the presence of tubulin in all samples is shown by the positive staining for total α-tubulin (DM 1 α);
FIG. 12 provides confocal images of 1A9 transiently transfected with a full length Hβl plasmid; cells were treated overnight with the indicated drug concentrations; cells were co-stained with β-tubulin (clone 2.1) and Hβl-specific antibodies; DNA was counterstained with DAPI;
FIG. 13 illustrates test results wherein human platelets were obtained from peripheral blood, plated, and treated overnight with the indicated drug concentrations; control and 2ME2 treated platelets were lysed with a microtubule-stabilizing buffer
(MTSB) while PTX treated platelets and their corresponding control were lysed using a hypotonic buffer (LSB) for 10 min at 37°C. %P = P/(P+S)*100;
FIG. 14 illustrates mRNA expression of Hβl measured by means of quantitative RT- PCR in 20 human tissues; the amount of the Hβl mRNA was normalized with the GUS mRNA content in each sample; the tissues are ordered according to their Hβl content;
FIG. 15 illustrates that anticancer doses of 2ME2 do not affect microtubule cytoskeleton in hematopoietic tissue where eight weeks old male C57BL6 mice were treated with a single dose at the indicated drug concentration; animals were sacrificed 24
hours after dosing and bone marrow aspirates were collected, cytospun onto slides, fixed, and immunostained with an antibody against α-tubulin; DNA was counterstained with DAPI; solid and dashed arrows point at cells with normal and aberrant mitotic spindles, respectively; arrowheads point at depolymerized microtubules in the VCR- treated mice or to microtubules bundles in the PTX-treated group;
FIG. 16 provides quantification of percentage of normal and aberrant mitotic figures as a fraction of total mitosis from mice bone marrow aspirates for each animal group; a minimum of 600 cells were counted for each treatment;
FIG. 17 illustrates human bone marrow aspirates obtained from a patient with a pre-malignant BM disorder (MGUS) and following ficoll separation, cells were plated onto coverslips and allowed to attach overnight before treating the cells ex vivo with the indicated concentrations of drugs for 24 hours; cells were fixed and immunostained with an α-tubulin antibody; DNA was counterstained with DAPI;
FIG. 18 illustrates stem/progenitor cells isolated from umbilical cord blood using a CD34 positive selection and treated and immunostained;
FIG. 19 illustrates expression of Hβl in CD34+ cells, wherein stem/progenitor cells were isolated from umbilical cord blood using a CD34 positive selection; cells were plated onto coverslips and allowed to attach before treating the cells ex vivo with the indicated concentrations of drugs for 24 hours; cells were fixed and co-stained with antibodies against Hβl -tubulin and α-tubulin antibody (clone YL 1/2); DNA was counterstained with DAPI; the effects of the drug treatments were evaluated in interphase microtubules (first panel) and mitotic cells (second panel).
DETAILED DESCRIPTION OF THE INVENTION The present invention now will be described more fully hereinafter through reference to various embodiments. These embodiments are provided so that this disclosure will be thorough and complete, and will fully convey the scope of the invention to those skilled in the art. Indeed, the invention may be embodied in many different forms and should not be construed as limited to the embodiments set forth herein; rather, these embodiments are provided so that this disclosure will satisfy applicable legal requirements. As used in the specification, and in the appended claims, the singular forms "a", "an", "the", include plural referents unless the context clearly dictates otherwise.
I. Definitions
The term "compound" as used herein means a chemical entity, whether in the solid, liquid, or gaseous phase, and whether in a crude mixture or purified and isolated.
The terms "microtubule -targeting drug (MTD)," "microtubule -targeting compound (MTC)," and "microtubule-targeting agent (MTA)" as used herein are used interchangeably and mean any compound wherein at least one mechanism of action of the compound relates to disruption of a cell's microtubules.
The term "alkyl" as used herein means saturated straight, branched, or cyclic hydrocarbon groups (including fused rings and polycyclic systems). In particular embodiments, alkyl refers to groups comprising 1 to 10 carbon atoms ("C1-10 alkyl"). In further embodiments, alkyl refers to groups comprising 1 to 8 carbon atoms ("C1-8 alkyl"), 1 to 6 carbon atoms ("Ci_6 alkyl"), or 1 to 4 carbon atoms ("C1-4 alkyl"). In specific embodiments, alkyl refers to methyl, trifluoromethyl, ethyl, propyl, isopropyl, cyclopropyl, butyl, isobutyl, t-butyl, pentyl, cyclopentyl, isopentyl, neopentyl, hexyl, isohexyl, cyclohexyl, cyclohexylmethyl, 3-methylpentyl, 2,2-dimethybutyl, and 2,3- dimethylbutyl. Substituted alkyl refers to alkyl substituted with one or more non- interfering substituents, such as but not limited to halo (e.g., Cl, F, Br, and I); halogenated alkyl (e.g., CF3, 2-Br-ethyl, CH2F, CH2Cl, CH2CF3, or CF2CF3); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryl; aryloxy; nitro; cycloalkyl; acetylene; alkanoyloxy; ketone; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate.
The term "alkenyl" as used herein means alkyl moieties wherein at least one saturated C-C bond is replaced by a double bond. In particular embodiments, alkenyl refers to groups comprising 1 to 10 carbon atoms ("C1-10 alkenyl"). In further embodiments, alkenyl refers to groups comprising 1 to 8 carbon atoms ("C1-8 alkenyl"), 1 to 6 carbon atoms ("Ci_6 alkenyl"), or 1 to 4 carbon atoms ("C1-4 alkenyl"). In specific embodiments, alkenyl can be vinyl, allyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl, 3- butenyl, 1-pentenyl, 2-pentenyl, 3-pentenyl, 4-pentenyl, 1-hexenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, or 5-hexenyl. Substituted alkenyl refers to alkenyl substituted with one or more moieties selected from the group consisting of halo (e.g., Cl, F, Br, and I); halogenated alkyl (e.g., CF3, 2-Br-ethyl, CH2F, CH2Cl, CH2CF3, or CF2CF3); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryloxy; nitro; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate.
The term "heteroalkenyl" as used herein means an alkenyl group as described above having one or more carbon atoms replace with a heteroatom, such as N, O, or S.
The term "alkynyl" as used herein means alkyl moieties wherein at least one saturated C-C bond is replaced by a triple bond. In particular embodiments, alkynyl refers to groups comprising 1 to 10 carbon atoms ("C1-10 alkynyl"). In further embodiments, alkynyl refers to groups comprising 1 to 8 carbon atoms ("C1-8 alkynyl"), 1 to 6 carbon atoms ("Ci_6 alkynyl"), or 1 to 4 carbon atoms ("C1-4 alkynyl"). In specific embodiments, alkynyl can be ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1- pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1- hexynyl, 2-hexynyl, 3-hexynyl, A- hexynyl, or 5 -hexynyl. Substituted alkynyl refers to alkynyl substituted with one or more moieties selected from the group consisting of halo (e.g., Cl, F, Br, and I); halogenated alkyl (e.g., CF3, 2-Br-ethyl, CH2F, CH2Cl, CH2CF3, or CF2CF3); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryloxy; nitro; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate. The term "alkoxy" as used herein means straight or branched chain alkyl groups linked by an oxygen atom (i.e., -O-alkyl or -alkyl-O-alkyl), wherein alkyl is as described above. In particular embodiments, alkoxy refers to oxygen-linked groups comprising 1 to 10 carbon atoms ("C1-10 alkoxy"). In further embodiments, alkoxy refers to oxygen-linked groups comprising 1 to 8 carbon atoms ("C1-8 alkoxy"), 1 to 6 carbon atoms ("Ci_6 alkoxy"), or 1 to 4 carbon atoms ("C1-4 alkoxy"). Substituted alkoxy refers to alkoxy substituted with one or more moieties selected from the group consisting of halo (e.g., Cl, F, Br, and I); halogenated alkyl (e.g., CF3, 2-Br-ethyl, CH2F, CH2Cl, CH2CF3, or CF2CF3); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryloxy; nitro; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate.
The term "alkenoxy" as used herein means straight or branched chain alkenyl groups linked by an oxygen atom (i.e., -O-alkenyl, -alkenyl-O-alkyl, or -alkyl-O- alkenyl), wherein alkyl and alkenyl are as described above. In particular embodiments, alkenoxy refers to oxygen-linked groups comprising 1 to 10 carbon atoms ("C1-10 alkenoxy"). In further embodiments, alkenoxy refers to oxygen-linked groups comprising 1 to 8 carbon atoms ("C1-8 alkenoxy"), 1 to 6 carbon atoms ("Ci_6 alkenoxy"), or 1 to 4 carbon atoms ("C1-4 alkenoxy"). Substituted alkenoxy refers to alkenoxy substituted with one or more moieties selected from the group consisting of halo (e.g., Cl, F, Br, and I);
halogenated alkyl (e.g., CF3, 2-Br-ethyl, CH2F, CH2Cl, CH2CF3, or CF2CF3); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryloxy; nitro; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate.
The term "halo" or "halogen" as used herein means fluorine, chlorine, bromine, or iodine.
The term "heterocycle" or "heterocyclic" as used herein means one or more rings of at least 5 atoms, preferably 5, 6, 7, 8, 9, 10, or 11 atoms, with or without unsaturation or aromatic character and having at least one ring atom which is not carbon. Preferred heteroatoms include sulfur, oxygen, and nitrogen. Multiple rings may be fused, as in quinoline or benzofuran. "Substituted heterocycle" is heterocycle having one or more side chains formed from non-interfering substituents.
The term "aryl" as used herein means a stable monocyclic, bicyclic, or tricyclic carbon ring of up to 8 members in each ring, wherein at least one ring is aromatic as defined by the Huckel 4n+2 rule. Multiple aryl rings may be fused, and aryl rings may be fused or unfused with one or more cyclic hydrocarbon, heteroaryl, or heterocyclic rings. Exemplary aryl groups according to the invention include phenyl, naphthyl, tetrahydronaphthyl, and biphenyl. The aryl group can be substituted with one or more non-interfering substituents, such as, for example, hydroxyl, amino, alkylamino, arylamino, alkoxy, aryloxy, nitro, cyano, sulfonic acid, sulfate, phosphonic acid, phosphate, or phosphonate.
The term "heteroaryl" as used herein means an aryl group containing from one or more (particularly one to four) non-carbon atom(s) (particularly N, O, or S) or a combination thereof, which heteroaryl group is optionally substituted at one or more carbon or nitrogen atom(s) with alkyl, -CF3, phenyl, benzyl, or thienyl, or a carbon atom in the heteroaryl group together with an oxygen atom form a carbonyl group, or which heteroaryl group is optionally fused with a phenyl ring. Heteroaryl rings may also be fused with one or more cyclic hydrocarbon, heterocyclic, aryl, or heteroaryl rings. Heteroaryl includes, but is not limited to, 5-membered heteroaryls having one hetero atom (e.g., thiophenes, pyrroles, furans); 5 membered heteroaryls having two heteroatoms in 1,2 or 1,3 positions (e.g., oxazoles, pyrazoles, imidazoles, thiazoles, purines); 5-membered heteroaryls having three heteroatoms (e.g., triazoles, thiadiazoles); 5-membered heteroaryls having 3 heteroatoms; 6-membered heteroaryls with one heteroatom (e.g., pyridine, quinoline, isoquinoline, phenanthrine, 5,6-cycloheptenopyridine); 6-membered
heteroaryls with two heteroatoms (e.g., pyridazines, cinno lines, phthalazines, pyrazines, pyrimidines, quinazolines); 6-membered heretoaryls with three heteroatoms (e.g., 1,3,5- triazine); and 6-membered heteroaryls with four heteroatoms. Substituted heteroaryl is heteroaryl having one or more non-interfering groups as substituents. The term "aryloxy" as used herein means aryl groups linked by an oxygen atom
(i.e., -O-aryl, or -aryl-O-aryl), wherein aryl as described above. Substituted aryloxy refers to aryloxy substituted with one or more moieties selected from the group consisting of halo (e.g., Cl, F, Br, and I); halogenated alkyl (e.g., CF3, 2-Br-ethyl, CH2F, CH2Cl, CH2CF3, or CF2CF3); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryloxy; nitro; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate.
The terms "aralkyl" and "arylalkyl" as used herein mean an aryl group as defined above linked to the molecule through an alkyl group as defined above.
The terms "alkaryl" and "alkylaryl" as used herein means an alkyl group as defined above linked to the molecule through an aryl group as defined above.
The term "alkenaryl" as used herein means an alkenyl group as defined above linked to the molecule through an aryl group as defined above.
The term "alkynaryl" as used herein means an alkynyl group as defined above linked to the molecule through an aryl group as defined above The term "acyl" as used herein means a carboxylic acid ester in which the non- carbonyl moiety of the ester group is selected from straight, branched, or cyclic alkyl or lower alkyl; alkoxyalkyl including methoxymethyl; aralkyl including benzyl; aryloxyalkyl such as phenoxymethyl; aryl including phenyl optionally substituted with one or more non-interfering substituents, such as halogen, C1-Ce alkyl or C1-Ce alkoxy; sulfonate esters such as alkyl or aralkyl sulphonyl including methanesulfonyl; mono-, di-, or triphosphate ester; trityl or monomethoxytrityl; substituted benzyl; trialkylsilyl such as dimethyl-t- butylsilyl or diphenylmethylsilyl. Aryl groups in the esters optimally comprise a phenyl group. Substituted acyl refers to alkoxy substituted with one or more moieties selected from the group consisting of halo (e.g., Cl, F, Br, and I); halogenated alkyl (e.g., CF3, 2- Br-ethyl, CH2F, CH2Cl, CH2CF3, or CF2CF3); hydroxyl; amino; carboxylate; carboxamido; alkylamino; arylamino; alkoxy; aryloxy; nitro; azido; cyano; thio; sulfonic acid; sulfate; phosphonic acid; phosphate; and phosphonate.
The term "amino" as used herein means a moiety represented by the structure NR2, and includes primary amines, and secondary and tertiary amines substituted by alkyl (i.e., alkylamino). Thus, R2 may represent two hydrogen atoms, two alkyl moieties, or one hydrogen atom and one alkyl moiety. The terms "alkylamino" and "arylamino" as used herein mean an amino group that has one or two alkyl or aryl substituents, respectively.
The term "nitro" as used herein means a group having the structure NO2;
The term "alkylene" as used herein means an alkyl group having two free valencies (i.e., a divalent alkyl radical); The term "amide" as used herein means a compound having the general formula
Ri(CO)NR2R3, wherein any of Ri, R2, and R3 can be hydrogen or hydrocarbon;
The term "non-interfering substituents" as used herein means any groups that yield stable compounds. Suitable non-interfering substituents or radicals include, but are not limited to, halo, C1-C10 alkyl, C2-C10 alkenyl, C2-C10 alkynyl, Ci-Ci0 alkoxy, C7-C12 aralkyl, C7-C12 alkaryl, C3-C10 cycloalkyl, C3-C10 cycloalkenyl, phenyl, substituted phenyl, toluoyl, xylenyl, biphenyl, C2-C12 alkoxyalkyl, C7-C12 alkoxyaryl, C7-C12 aryloxyalkyl, C6-Ci2 oxyaryl, Ci-C6 alkylsulfinyl, Ci-Ci0 alkylsulfonyl, -(CH2)Jn-O-(C1-C10 alkyl) wherein m is from 1 to 8, aryl, substituted aryl, substituted alkoxy, fluoroalkyl, heterocyclic radical, substituted heterocyclic radical, nitroalkyl, -NO2, -CN, -NRC(O)-(C1- Ci0 alkyl), -C(O)-(Ci-Ci0 alkyl), C2-Ci0 thioalkyl, -C(O)O-(Ci-Ci0 alkyl), -OH, -SO2, =S, - COOH, -NR2, carbonyl, -C(O)-(Ci-Ci0 alkyl)-CF3, -C(O)-CF3, -C(O)NR2, -(Ci-Ci0 alkyl)- S-(C6-Ci2 aryl), -C(O)-(C6-Ci2 aryl), -(CH2)m-O-(CH2)m-O-(Ci-Ci0 alkyl) wherein each m is from 1 to 8, -C(O)NR2, -C(S)NR2, -SO2NR2, -NRC(O)NR2, -NRC(S)NR2, salts thereof, and the like. Each R as used herein is H, alkyl or substituted alkyl, aryl or substituted aryl, aralkyl, or alkaryl.
The term "analogue" as used herein means a compound in which one or more individual atoms or functional groups have been replaced, either with a different atom or a different functional, generally giving rise to a compound with similar properties.
An "analogue-forming group" as used herein means any group that replaces an identified group on an identified compound to form an analogue of the identified compound.
The term "derivative" as used herein means a compound that is formed from a similar, beginning compound by attaching another molecule or atom to the beginning
compound. Further, derivatives, according to the invention, encompass one or more compounds formed from a precursor compound through addition of one or more atoms or molecules or through combining two or more precursor compounds.
The term "prodrug" as used herein means any compound which, when administered to a mammal, is converted in whole or in part to a compound of the invention.
The term "active metabolite" as used herein means a physiologically active compound which results from the metabolism of a compound of the invention, or a prodrug thereof, when such compound or prodrug is administered to a mammal. The term "intermittent administration" as used herein means administration of a therapeutically effective dose of a composition according to the invention, followed by a time period of discontinuance, which is then followed by another administration of a therapeutically effective dose, and so forth.
"Pharmaceutically acceptable excipient" or "pharmaceutically acceptable carrier" refers to an excipient that can be included in the compositions of the invention and that causes no significant adverse toxicological effects to the patient.
"Pharmacologically effective amount," "physiologically effective amount," "therapeutically effective amount", and "therapeutically effective dose" are used interchangeably herein to mean the amount of a conjugate of the invention present in a pharmaceutical preparation that is needed to provide a desired level of active agent and/or conjugate in the bloodstream or in the target tissue. The precise amount will depend upon numerous factors, e.g., the particular active agent, the components and physical characteristics of the pharmaceutical preparation, intended patient population, patient considerations, and the like, and can readily be determined by one skilled in the art, based upon the information provided herein and available in the relevant literature.
The term "antiproliferative agent" as used herein means a compound that decreases the hyperproliferation of cells.
The term "abnormal cell proliferation" as used herein means a disease or condition characterized by the inappropriate growth or multiplication of one or more cell types relative to the growth of that cell type or types in an individual not suffering from that disease or condition.
The term "cancer" as used herein means a disease or condition characterized by uncontrolled, abnormal growth of cells, which can spread locally or through the
bloodstream and lymphatic system to other parts of the body. The term includes both tumor- forming or non-tumor forming cancers, and includes various types of cancers, such as primary tumors and tumor metastasis.
The term "tumor" as used herein means an abnormal mass of cells within a multicellular organism that results from excessive cell division that is uncontrolled and progressive, also called a neoplasm. A tumor may either be benign or malignant.
Compounds or groups illustrated with a bond terminated by the following symbol
» is intended to indicate an available point of attachment for adding the group as a substituent to another group or compound.
II. Compounds
Seven β-tubulin isotypes have been identified (classes I, II, III, IVa, IVb, V, and VI). Class VI β-tubulin (βl -tubulin or Hβl) is dominant in human bone marrow and CD34-positive stem/progenitor cells. Class V β-tubulin is the dominant tubulin isotype in tumor cells. The present invention has identified that residue Vβ236 is highly conserved across all human β-tubulin isotypes with the exception of Hβl, which shows a Vβ236I mutation that is believed to be the source of myelosuppression by known MTCs, such as taxanes or Vinca alkaloids. Through analysis of the three-dimensional structure of β- tubulin and the binding of paclitaxel based on an electron-crystallographic structure of the paclitaxel-tubulin binding complex, we have been able to identify families of compounds that can block the dominant tubulin isotypes in tumor cells but fail to do so in bone marrow cells containing the class VI isotype. As more fully described herein, the compounds of the invention are useful to ablate cancers and their progression in tumors while avoiding the serious myelosuppressive side effect.
A. Taxanes
In certain embodiments, the compounds of the present invention are taxane compounds having appropriate substituents to impart β-tubulin isotype specificity. In some embodiments, the taxane compounds of the invention are analogues of paclitaxel, the structure of which is shown below in Formula (1). In other embodiments, the taxane analogues of the invention are analogues of docetaxel, the structure of which is shown below in Formula (2). In other embodiments, the taxane compounds can be described independent of any known taxane compounds.
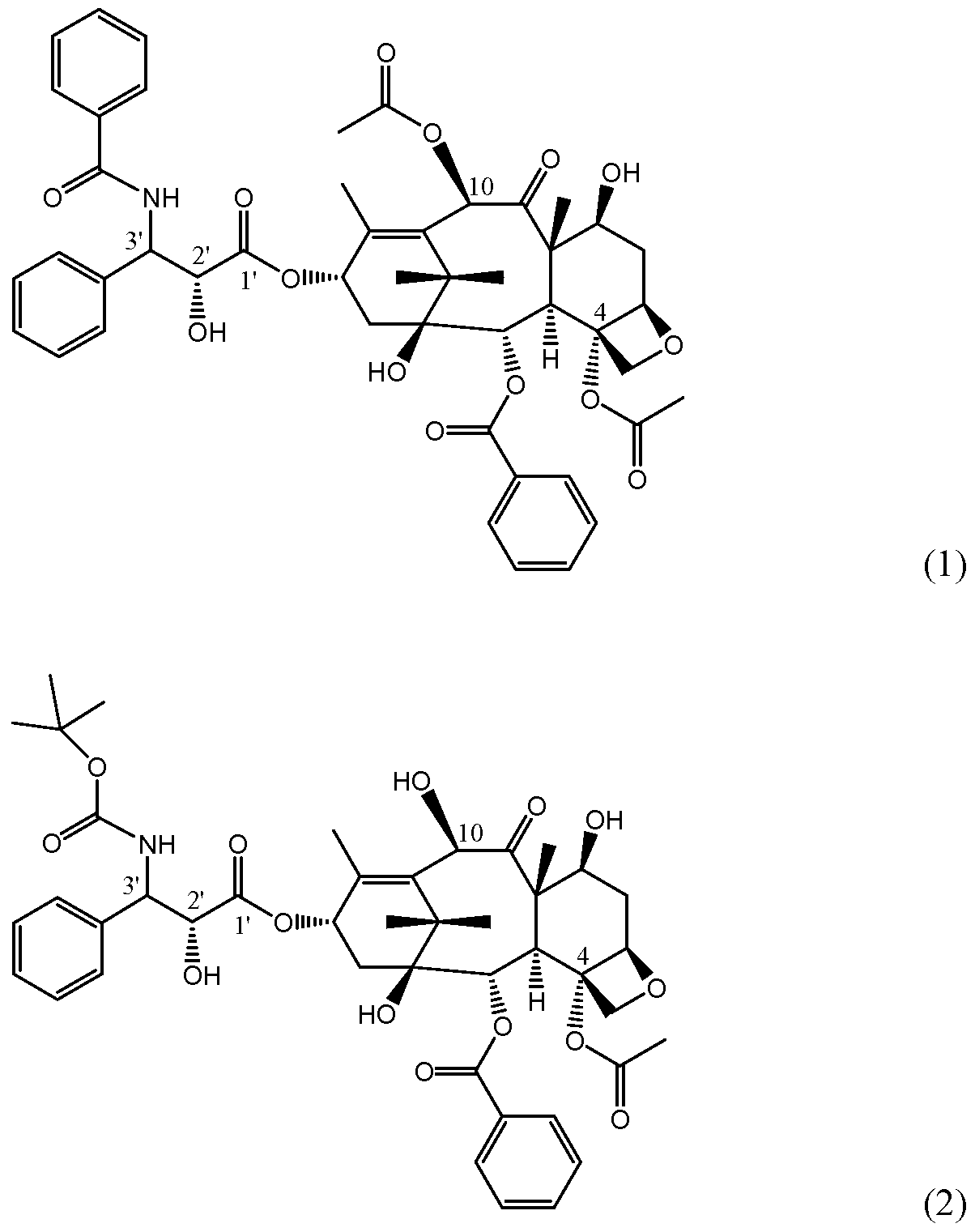
In one embodiment, the present invention provides analogues of paclitaxel wherein the phenyl group on the C3'-NHC0 is replaced with an analogue-forming group.
In one embodiment, the present invention provides analogues of docetaxel wherein the tert-butyl group on the C3'-NHC0 is replaced with an analogue-forming group. In one embodiment, the present invention provides analogues of paclitaxel wherein the C3' phenyl group is replaced with an analogue-forming group.
In one embodiment, the present invention provides analogues of docetaxel wherein the C3' phenyl group is replaced with an analogue-forming group.
In one embodiment, the present invention provides analogues of paclitaxel wherein the methyl group on the C4 O-acetyl is replaced with an analogue-forming group.
In one embodiment, the present invention provides analogues of docetaxel wherein the methyl group on the C4 O-acetyl is replaced with an analogue-forming group.
In specific embodiments of the invention, an analogue-forming group is a group that sterically hinders binding of the formed compound with a β-tubulin isotype. In one embodiment, the analogue-forming group is a group that sterically hinders binding of the formed compound with the class VI β-tubulin isotype. Although not wishing to be bound by theory, it is believed that MTCs are bioactive toward β-tubulin via interaction of the compound with a specific binding site. FIG. 1 shows paclitaxel in the bioactive T-Taxol conformation at the β-tubulin taxane binding site in the class I β-tubulin isotype. As illustrated therein, the phenyl ring on the C3'-NHCO is clamped between residues 23 and 231 (valine and alanine for this isotype) in the class I (HM40) β-tubulin isotype. FIG. 2 shows paclitaxel in the bioactive T-Taxol conformation at the β-tubulin taxane binding site in the class VI β-tubulin isotype. As illustrated therein, the phenyl ring on the C3'-NHCO again is clamped between residues 23 and 231 (methionine and leucine for this isotype) in the class VI (HM40) β-tubulin isotype. The present invention has recognized that appropriate substitution on the phenyl ring to form a more bulky substituent (or complete replacement of the phenyl ring with a more bulky group) can result in a steric clash with the larger methionine and leucine class VI residues but still bind with other isotypes, such as the class I isotype. This manipulation based on steric hindrance can provide MTCs exhibiting β-tubulin isotype selectivity.
In certain embodiments, an analogue-forming group useful to replace the phenyl group on the C3 '-NHCO in paclitaxel or to replace the tert-butyl group on the C3 '-NHCO in docetaxel can include aryl, alkaryl, alkenaryl, alkynaryl, heteroaryl, heteroalkenyl, alkyl, alkenyl, cyclo-alkyl (including fused ring structures and polycyclic ring systems), aryloxy, alkoxy, or alkenoxy.
In certain embodiments, an analogue-forming group useful to replace the C3' phenyl group in paclitaxel or docetaxel can include aryl, alkaryl, alkenaryl, alkynaryl, heteroaryl, heteroalkenyl, alkyl, alkenyl, cyclo-alkyl (including fused ring structures and polycyclic ring systems), aryloxy, alkoxy, or alkenoxy.
In certain embodiments, an analogue-forming group useful to replace the methyl group on the C4 O-acetyl in paclitaxel or docetaxel can include alkyl, alkenyl, alkynyl, alkaryl, alkenaryl, alkynaryl, acyl, and any of the foregoing groups wherein one or more carbon atoms is replaced with one or more atoms selected from the group consisting of O, S, and N.
The foregoing also can apply to analogues of paclitaxel or docetaxel at other carbon atoms. Thus, the invention can encompass any analogue of paclitaxel or docetaxel that is sterically hindered from binding with a specific β-tubulin isotype, preferably class VI β-tubulin.
In some embodiments, the present invention provides MTCs according to the structure provided below in Formula (3),

wherein:
Ri is selected from the group consisting of aryl, alkaryl, alkenaryl, alkynaryl, heteroaryl, heteroalkenyl, alkyl, alkenyl, cyclo-alkyl, aryloxy, alkoxy, and alkenoxy; R-2 is selected from the group consisting of aryl, alkaryl, alkenaryl, alkynaryl, heteroaryl, heteroalkenyl, alkyl, alkenyl, cyclo-alkyl, aryloxy, alkoxy, or alkenoxy; and R3 is selected from the group consisting of alkyl, alkenyl, alkynyl, alkaryl, alkenaryl, alkynaryl, acyl, and any of the foregoing groups wherein one or more carbon atoms is replaced with one or more atoms selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
It is understood that the foregoing compounds exclude the known taxane compounds paclitaxel and docetaxel.
In specific embodiments, alkyl, alkenyl, and alkynyl groups include but are not restricted to straight chain, branched chain, cyclic, and polycyclic (e.g., norbornyl) groups, as well as aryl or alkyl substituted groups. Aryl groups can include but are not limited to benzene, naphthalene, and other condensed aromatic hydrocarbons optionally substituted
with alkyl, halogen, amino, ester, azido, nitro, and similar polar and nonpolar substituents. Heteroaryl includes but in not limited to furan, thiophene, pyrazole, diazole, triazole, oxazole, thiazole, oxadiazole, thiadiazoles, isoxazole, isothiazoles, pyridine, 1,2-diazine, 1,3-diazine, 1,4-diazine, triazine, benzofuran, benzothiophenes, quinoline, acridine, indole, phenazine, phenothiazines, purines, pyrimidines, and the corresponding keto analogues (i.e., amides and lactones). Heteroalkyl and heteroalkenyl groups include but are not limited to the fully saturated and di- and tetrahydro variations of the heteroaryl systems mentioned above including tetrahydrofuran, piperidine, morpholine, and alkylated and esterified variations, as well as condensed polycyclic systems, such as tropane, quinuclidine, l,4-diazabicyclo[2.2.2.0]-octane and similar systems substituted by alkyl and ester functionalities.
In certain embodiments, the invention encompasses compounds according to Formula (3), wherein Ri can be selected from the group consisting of bridged polycyclo compounds, fused polycyclo compounds, and substituted aromatic compounds. Non-limiting examples of bridged polycyclo compounds that may be used as Ri include compounds having from 6 to 15 carbon atoms, from 6 to 14 carbon atoms, from 6 to 13 carbon atoms, from 6 to 12 carbon atoms, or from 6 to 11 carbon atoms. In particular, the compounds may be bicyclo[2.1.1]-alkanes or -alkenes, bicyclo[2.1.2]- alkanes or -alkenes, bicyclo[2.2.2]-alkanes or -alkenes, bicyclo[3.1.1]-alkanes or alkenes, bicyclo[2.1.3]-alkanes or -alkenes, or bicyclo[3.1.3]-alkanes or -alkenes. Of course, the invention would encompass other bicyclo systems, in addition to those described above, as well as tricyclo systems (e.g., adamantane).
Specific non-limiting examples of bridged polycyclo compounds that may be encompassed by the present invention as an Ri group, according to Formula (3), include the following. If the point of attachment is not specified, the group may be attached to taxane compound at any available ring carbon.

Specific non-limiting examples of fused polycyclo compounds that may be encompassed by the present invention as an Ri group, according to Formula (3), include the following (wherein A can be an atom selected from C, O, S, and N).

Non-limiting examples of substituted aromatic compounds that may be used as Ri include compounds having from 7 to 20 carbon atoms, from 8 to 20 carbon atoms, from 8 to 18 carbon atoms, from 8 to 16 carbon atoms, or from 8 to 14 carbon atoms. In particular, the compounds may be phenyl rings that are one or more of ortho-, meta-, or para- substituted. Such substituents may include alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, C(O)O-alkyl, C(O)O-alkenyl, or C(O)O-alkynyl, all of which may be linear or branched. Specific non-limiting examples of substituted aromatic compounds that may be encompassed by the present invention as an Ri group, according to Formula (3), include the following (wherein W can be a substituent, as described in the preceding paragraph).

In certain embodiments, the invention encompasses compounds according to Formula (3), wherein R2 can be selected from the group consisting of substituted aromatic compounds, cycloalkanes or cycloalkenes, fused polycyclo compounds, and branched alkyl or alkenyl compounds.
Non-limiting examples of substituted aromatic compounds that may be used as R2 include meta- and/or para-substituted phenyl compounds, as well as 7-membered and 8- membered substituted aromatic compounds. Non-limiting examples of suitable substituents may include alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, C(O)O- alkyl, C(O)O-alkenyl, or C(O)O -alkynyl, all of which may be linear or branched.
Specific non-limiting examples of substituted aromatic compounds that may be encompassed by the present invention as an R2 group, according to Formula (3), include the following.
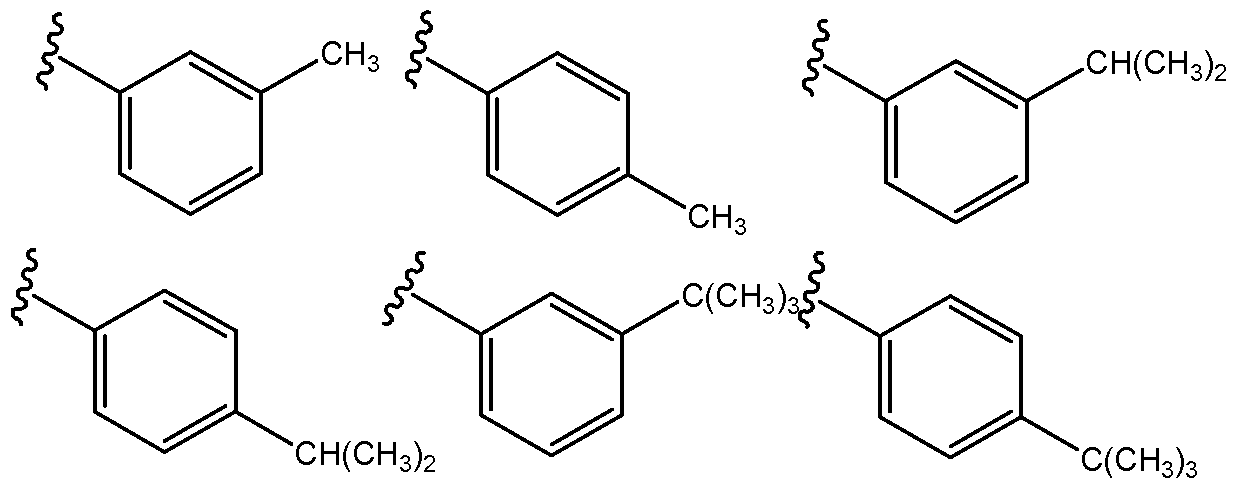
Non-limiting examples of cycloalkanes or cycloalkenes that may be used as R2 include C4-Cs cycloalkyl or cycloalkenyl, which may be substituted or unsubstituted. Non-limiting examples of suitable substituents may include alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, C(O)O-alkyl, C(O)O-alkenyl, or C(O)O -alkynyl, all of which may be linear or branched. Preferred substituents are Ci-C6, Ci-C5, or Ci-C4. A specific non-limiting example of a cycloalkane that may be encompassed by the present invention as an R2 group, according to Formula (3), include the following.

Non-limiting examples of fused polycyclo compounds that may be used as R2 include compounds having from 6 to 15 carbon atoms, from 6 to 14 carbon atoms, from 6 to 13 carbon atoms, from 6 to 12 carbon atoms, or from 6 to 11 carbon atoms. In particular, the compounds may be two fused rings wherein one ring has 6 atoms and the other ring has 4 atoms, 5 atoms, 6 atoms, 7 atoms, or 8 atoms. The compounds also may be two fused rings wherein one ring has 5 atoms and the other ring has 4 atoms, 5 atoms, 7 atoms, or 8 atoms. The ring structures may include one or more double bonds in the ring system, may be aromatic, may have one or more carbon atoms replaced with an atom selected from the group consisting of O, S, and N, and may be optionally substituted, such as by alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, alkynoxy, or acyl. Such substituents preferably comprise from 1 to 10 carbon atoms. Specific non-limiting examples of fused polycyclo compounds that may be encompassed by the present invention as an R2 group, according to Formula (3), include the following (wherein A can be an atom selected from C, O, S, and N).

Non-limiting examples of branched alkyl or alkenyl compounds that may be used as R2 include alkyl and alkenyl compounds having a total of 2 to 10 carbon atoms, one or more of which may be replaced by one or more atoms selected from the group consisting of O, S, and N. Preferred compounds are C3-C6, C3-C5, or C3-C4.
Specific non-limiting examples of branched alkyl or alkenyl compounds that may be encompassed by the present invention as an R2 group, according to Formula (3), include the following.

In certain embodiments, the invention encompasses compounds according to Formula (3), wherein R3 is a group that is relatively long and thin and can thus populate a narrow space that forms a specific binding site and also interfere with another residue. Such groups can be selected from the group consisting of cycloalkanes or cycloalkenes and linear or branched alkyl compounds, alkenyl compound, and alkynyl compounds, wherein one or more carbon atoms in the alkylene, alkenylene, or alkynylene may be replaced with an atom selected from the group consisting of O, S, and N.
Non-limiting examples of cycloalkanes or cycloalkenes that may be used as R3 include C3-C7 compounds, particularly C3-C5 compounds. Specific non-limiting examples of cycloalkanes or cycloalkenes that may be encompassed by the present invention as an R3 group, according to Formula (3), include the following.

Non-limiting examples of linear or branched alkyl compounds, alkenyl compound, and alkynyl compounds that may be used as R3 include C3-C12 compounds, particularly
C3-C10 compounds. In embodiments incorporating a heteroatom, it is preferable for the compound to be a linear or branched alkane. Specific non-limiting examples of linear or branched alkyl compounds, alkenyl compound, and alkynyl compounds that may be encompassed by the present invention as an R3 group, according to Formula (3), include the following.

Examples of specific taxane compounds useful as MTCs according to the present invention are provided below by the structures of Formula (4), Formula (5), Formula (6), and Formula (7). Of course, it is understood that the following are only examples of some of the compounds encompassed by the invention and are not intended to limit the scope of the invention.
 B. Epothilones
B. Epothilones
Epothilone A (EpoA) and Epothilone B (EpoB), shown below in Formula (8) and Formula (9), respectively, were originally isolated as antifungal agents from the soil- derived mycobacterium Sorangium cellulosum. The discovery of their taxol-like tubulin- polymerization activity and the elucidation of their absolute configuration led to great scientific interest in the development of epothilones as potential anticancer agents. The chemistry, biology, and structure activity relationships (SAR) of the epothilones have been extensively reviewed, and epothilone-based drug discovery research so far has delivered seven compounds which have entered clinical development. The most advanced compounds at this time are epothilone B (also known as Epo 906 or patupilone) and the lactam analogue of epothilone B (known as ixabepilone or IXEMPRA™), which has been approved for clinical use to treat certain forms of breast cancer. Ixabepilone is shown in Formula (10). Epothilone D (known as deoxyepothilone B or KOS-862), shown in Formula (11) and ZK-EPO, shown in Formula (12), are other known epothilones.


The present invention provides a number of new epothilone compounds that are useful as MTCs according to the invention. In certain embodiments, epothilone compounds according to the invention can include those having the structure shown below according to Formula (13),

Y is N or O;
Xi is selected from the group consisting of alkyl, alkenyl, alkynyl, C(=O)alkyl, C(=O) alkenyl, C(=O)alkynyl, C(C=O)aiyl, C(=O)alkaryl, C(=O)alkenaryl, C(=O)alkynaryl, C(=O)Oalkyl, C(=O)Oalkenyl, C(=O)Oalkynyl, C(=O)Oarylalkyl, C(=O)Oarylalkenyl, and C(=O)Oarylalkynyl; X2 is selected from the group consisting of H, alkyl, alkenyl, alkynyl, and cycloalkyl;
X3 and X4 independently are selected from the group consisting of alkyl, NX6, O, and S;
X4- is OH, or X4 and X4- may be combined to form a bicyclic ring system, wherein the formed ring has 5-7 ring members, optionally including one or more heteroatoms selected from N and O, and optionally being substituted with NX6, O, OH, halogen, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, or alkynoxy;
X5 is selected from the group consisting of

Xe is selected from the group consisting of H and alkyl; X7 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, S- alkyl, S-acyl CH2-S-alkyl, CH2-S-acyl, and NX6; and
Xs is selected from the group consisting of H, alkyl, alkenyl, alkynyl, and cycloalkyl; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof. It is understood that the foregoing compounds exclude the known epothilone compounds described in Formulas (8) - (12). Preferably, the epothilone compounds according to the invention have the same stereochemistry as EpoA and EpoB. Of course, the invention does not exclude other stereoisomers.
In other embodiments, epothilone compounds according to the invention can include those having the structure shown above according to Formula (13), wherein X4 is OH, X2 and X4 may be combined to form a bicyclic ring system, wherein the formed ring has 5-7
ring members, optionally including one or more heteroatoms selected from N and O, and optionally being substituted with NX6, O, OH, halogen, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, or alkynoxy, and wherein the compound is otherwise as described previously.
In further embodiments, epothilone compounds according to the invention can include those having the structure shown below according to Formula (14),

wherein: Xi is selected from the group consisting of alkyl, alkenyl, alkynyl, C(=O)alkyl,
C(=0) alkenyl, C(=O)alkynyl, C(C=O)aryl, C(=O)alkaryl, C(=O)alkenaryl, C(=O)alkynaryl, C(=O)Oalkyl, C(=O)Oalkenyl, C(=O)Oalkynyl, C(=O)Oarylalkyl, C(=O)Oarylalkenyl, and C(=O)Oarylalkynyl;
X2 and Xs independently are selected from the group consisting of H, alkyl, alkenyl, alkynyl, and cycloalkyl; and
X7 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, S- alkyl, S-acyl CH2-S-alkyl, CH2-S-acyl, and NX6; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof. Examples of specific epothilone compounds useful as MTCs according to the present invention are provided below in Formula (15) through Formula (22). Of course, it is understood that the following are only examples of some of the compounds encompassed by the invention and are not intended to limit the scope of the invention.




One or more of the compounds described herein may exist in more than one diasteriomeric form. For example, as shown in Example 2 below, the compound of Formula (18) can be provided as the compound of Formula (18a) or (18b).
In further embodiments, epothilone compounds according to the invention can include those having the structure shown below according to Formula (35),

wherein: each X6 is independently H or alkyl; each X9 is independently H, NX6, or OX6; and
Zi is a heterocycle group; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
In further embodiments, epothilone compounds according to the invention can include those having the structures shown below according to Formula (36) through Formula (39),


Xio is selected from the group consisting of alkyl, alkenyl, and aryl; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof. In still other embodiments, epothilone compounds according to the invention can include those having the structures shown below according to Formula (40) through
Formula (46).


Epothilone compounds useful as MTCs according to the invention may be prepared according to a variety of methods. For example, bridged epothilones, such as those shown in Formula (15), Formula (16), and Formula (30), may be prepared with a ring closing metathesis (RCM) reaction as the penultimate step. Specifically, performing an RCM on compounds A and B, or protected versions thereof, in the reaction scheme shown below would yield the desired products. The reaction scheme shown below is particularly a retrosynthetic analysis for the bridged epothilones of Formula (15) and Formula (16).

X=12,13-olefm, or X=O A - X=12,13-olefm B - X=O
The known fragment (I) was prepared according to a known procedure (Koch G., et al., 2004, Synlett 4:693-697 and Nicolaou KC, et al., 1997, J. Am. Chem. Soc. 119:79747991). Fragment (II) was prepared as shown below.

Treatment of methyl acetoacetate with benzyl chloromethyl ether provided the starting diketone, which was subjected to catalytic asymmetric reduction to provide the hydroxyester. The benzyl group on the hydroxyester was deprotected and the resulting diol was selectively protected with tert-butyldimethylsilyl chloride to give ester in 95% overall yield. The ester was treated with lithium diisopropyl amide (LDA) and allyl iodide to provide an allyl derivative in excellent yield and diastereoselectivity, which was subjected to LDA and methyl iodide to provide compound the next intermediate as the major diastereomer in 53% yield (d.r. 3: 1). The above transformation presumably proceeds as formulated through the bracketed structure. The configuration of the allyl derivative was initially assigned based on literature precedent (Eggen M, et al., 2000, J. Org. Chem. 65:7792-7799). The assignment was subsequently established by converting it to the ketal. Selective irradiation of the C4-methyl group resulted in signal enhancement of the
protons H3-H, on the adjacent carbons. Irradiation of the proton H3 of the (S) chiral center resulted in signal enhancement of the C4-methyl group and the Hb and protons, but not of the CH2 protons of the allyl group. These results confirm the (R) configuration of the newly introduced center at the C4 carbon. The remaining transformations proceeded in 52% overall yield for the five steps.
The compound of Formula (17) can be prepared through Aldol coupling of fragments (I) and (II) using LDA as the base, as shown in the reaction scheme below. The first formed alcohol is converted to an acid or ester intermediate and thence to the intermediate lactone and the final lactone by methods similar to those described in the literature (Nicolaou, KC et al., 1997, J. Am. Chem. Soc. 119:7974-7991 and Nicolaou, KC et al., 2000, Chem. Eur. J. 6:2783-2800).



Furthermore, the H-3 signal is downshifted from 4.5 ppm to 5.18 ppm relative to the starting material, indicating that the hydroxyl group at C-3 is esterified. The structure of the compound of Formula (18) was thus established as indicated. Since the acryloyl double bond did not participate in the anomalous metathesis reaction, the reaction was carried out on further epothilone derivatives, each lacking this double bond. As expected, reaction occurred as before to give the internal lactones of 26-hydroxy epothilone D, Formula (20), epothilone B, Formula (21), and epothilone D Formula (22). These schemes are shown below.

50% formic /- R1 = TBS, R2 = OTr acid, 79% VR1 = TBS, R2 = OH \ I2, Imadazole,
NaCNBH3, ( Ri = TBS, R2 = I V PPh3, 89% 60.6% VR1 = TBS, R2 = H 76% R1 = R2 = H "J HF, Py,
The mechanism of this unusual "metathesis" reaction, as shown below, is probably analogous to those proposed for the ruthenium-catalyzed transformations of amino alcohols to lactams (Naota, T., et al., 1991, Synlett, 693-694) or of amines and alcohols to amides (Gunanathan C, 2007, Science 317:790-792). It is proposed that the ruthenium catalyst reacts initially in the normal way with the allyl double bond of an epothilone derivative such as the starting material in the reaction scheme shown below to give the first shown intermediate. This compound could then lose styrene to give the shown complex, which is presumably too sterically encumbered to react with the acryloyl double bond. The complex is thus trapped by the internal C3 hydroxyl group to give the next intermediate, which undergoes oxidation by loss of a hydride to give the next intermediate, and then hydrolysis.

C. Modes of Action The compounds of the present invention may be described, in some embodiments, in light of the specific mode of action. For example, as described above, certain taxane compounds of the invention are believed to exhibit specific β-tubulin selectivity.
2-Methoxyestradiol (2ME2) is a naturally occurring metabolite of 17β-estradiol with antitumor and antiangiogenic activities in a wide variety of human cell lines, including those that are resistant to different chemotherapeutic agents such as taxanes (microtubule-stabilizing drug) and methotrexate (DNA synthesis interfering agents) and tamoxifen (anti-hormonal therapy). Importantly, 2ME2 anticancer properties are independent of estrogen receptors α and β for which 2ME2 has a very low affinity.
Mechanistically, 2ME2 binds at the colchicine-binding site in both the α/β-tubulin heterodimers and to the microtubule polymer resulting in depolymerization of microtubule cytoskeleton. Work according to the present invention has provided a unifying mechanism of action for the antitubulin and antiangiogenic properties of 2ME2 showing that 2ME2 inhibits tumor angiogenesis by effectively inhibiting the hypoxia
inducible factor- lα (HIF- lα) levels and its transcriptional activity, including transcription of VEGF in vitro and in vivo. Notably, inhibition of HIF-I α occurred downstream of disruption of the microtubule cytoskeleton, providing a functional relationship between the antitubulin and antiangiogenic effects of 2ME2. We further revealed that 2ME2 shares this mechanism of action with other widely used microtubule- targeting agents (MTAs), such as the taxanes and Vinca alkaloids, suggesting a common mechanism of antiangiogenic activity for all MTAs. These results identified the microtubule cytoskeleton as an important component of the angiogenic process and provided a molecular insight into the mechanism underlying the reported antiangiogenic activity of MTAs.
The present invention has identified that the acquired HM40/TUBB Vβ236I tubulin mutation in the human ovarian carcinoma 2ME2 -resistant cells (2MRC) confers drug resistance by impairing the ability of 2ME2 to interact with tubulin. More importantly, the present invention has identified that the Ileβ236 residue, naturally encoded by the hematopoietic -specific Hβl tubulin isotype, protects the bone marrow (BM) from potential 2ME2 -induced myelosuppressive effects. The present invention has expanded on the knowledge that the effects of 2ME2 are tubulin isotype-specifϊc and has been applied towards the design of new MTAs devoid of myelosuppressive side effects. Such compounds have been described herein. In specific embodiments, the invention provides compounds that selectively inhibit one or more specific β-tubulin isotypes. For example, the MTCs of the invention may be selective in that they only bind to one or more β-tubulins selected from class I β-tubulin, class II β-tubulin, class III β-tubulin, class IVa β-tubulin, class IVb β-tubulin, or class V β- tubulin. In other embodiments, the MTCs of the invention may be selective in that they will not bind to one or more specific β-tubulin. For example the MTCs of the invention specifically may not bind to class VI β-tubulin (Hβl). Thus, one or more compounds according to the invention may be described as a MTC that does not inhibit β-tubulin isotype Hβl. It is understood that the MTC according to such embodiments would specifically exclude 2-Methoxyestradiol. The compounds of the invention may further be described in terms of the negative effects that are typically associated with MTCs but are not exhibited by the MTCs of the invention. For example, the present invention provides MTCs that are not
myelosuppressive (i.e., "non-myelosuppressive microtubule targeting compounds"). In still further embodiments, the invention encompasses MTCs that do not suppress healthy bone marrow.
The provision of MTCs having β-tubulin isotype specificity according to the present invention can extend into various analytical methods to assess the likelihood that a patient with cancer will respond to treatment using one or more of the compounds of the present invention or any further compounds that exhibit β-tubulin isotype specificity.
As described herein, certain types of cancer may be characterized by an overexpression of one or more β-tubulin isotypes. Treatment by administration of a MTC that selective does not bind the β-tubulin isotype being overexpressed in the cancer patient would not be expected to be effective.
In specific embodiments, the present invention provides MTCs that selectively do not inhibit β-tubulin isotype Hβl yet do inhibit one or more further β-tubulin isotypes. The invention thus further provides methods to determine whether a patient with cancer will respond to treatment with the MTC that selectively does not inhibit β-tubulin isotype Hβl (i.e., an Hβl selective MTC) but may inhibit one or more further β-tubulin isotypes. In one embodiment, the method comprises the following steps: i) testing cancer cells from the patient to determine whether the cells overexpress Hβl, wherein a positive result in the test means the cells overexpress Hβland a negative result means the cells do not overexpress Hβl ; ii) correlating a positive result in the test with treatment with a compound other than the Hβl selective MTC; and iii) correlating a negative result in the test with treatment with the Hβl selective MTC. If the cancel cells of the patient do not overexpress Hβl, then the cells would be expected to express a majority of β-tubulin that is not Hβl. Since the Hβl selective MTC does not bind Hβl but does bind other β-tubulin isotypes, the Hβl selective MTC would be expected to be effective to treat the cancer of the patient in a non-myelosuppressive fashion, as otherwise disclosed herein. Of course, it understood that the foregoing is provided for exemplary purposes and is not intended to limit the scope of the invention. Rather, the same testing procedure could be used to evaluate whether a patient would respond to compounds selective for other β-tubulin isotypes.
As described herein, the effectiveness of the compounds of the present invention to provide β-tubulin isotype selectivity at least partially arises from the steric hindrance of the inventive compounds with prevents binding in isotypes with certain amino acid arrangements while not preventing binding in isotypes with certain other amino acid arrangements. Thus, identifying such specific amino acid sequences can allow for making a determination of whether a patient with cancer will respond to treatment with an Hβl - specific compound according to the invention. Correlating amino acid structure to selectivity for other β-tubulin isotypes could be similarly achieved according to the invention. In specific embodiments, the invention thus provides methods to determine whether a patient with cancer will respond to treatment with an MTC according to the invention that selectively does not inhibit β-tubulin isotype Hβ 1 but may inhibit one or more further β- tubulin isotypes. In one embodiment, the method comprises the following steps: i) testing the patient's cancer cells to determine whether they express β-tubulin isotypes encoding valine at β236, tyrosine at β200, or valine at β316, wherein a positive result in the test means the cells express Hβl-tubulin isotypes encoding valine at β236, tyrosine at β200, or valine at β316, and a negative result in the test means the cells do not express Hβl-tubulin isotypes encoding valine at β236, tyrosine at β200, or valine at β316; ii) correlating a positive result in the test with treatment with an MTC according to the invention that selectively does not inhibit β-tubulin isotype Hβl but may inhibit one or more further β-tubulin isotypes; and iii) correlating a negative result in the test with treatment with a compound other than the Hβl -selective compound of the invention.
The testing methods of the invention may include testing of various cell components to determine the appropriate method of treatment. For example, the method can comprises isolating and testing nucleic acids from the cancer cells. The method may also comprise isolating and testing proteins from the cancer cells.
For example, a real time quantitative RT-PCR has been performed with primers specific for each of the 7 beta-tubulin isotypes, and the results were normalized with amplification of an endogenous gene (e.g., beta-glucorunidase). This method can be applied to test the relative beta-tubulin isotype expression in various samples. One non- limiting example includes frozen surgical tumor samples (and from matched surrounding normal tissue) from NSCLC patients that have received taxane-based chemotherapy
before surgery. The desired outcome from such testing can be to determine the contribution of the relative beta-tubulin isotype expression to tumorigenesis (normal versus tumor) and to response to therapy (responders versus non-responders).
In light of the understanding around β-tubulin isotype specificity obtained according to the present invention, it is also possible to screen compounds (including the compounds of the invention as well as compounds that may not be encompassed by the presently described compounds) to determine whether the compounds would be effective for use as MTCs that are nonmyelosuppressive (i.e., are myelo-sparing).
For example, the present invention has identified that compounds that selectively do not inhibit Hβl -tubulin are useful as myelo-sparing MTCs. Accordingly, in certain embodiments, the invention provides a method for identifying myelo-sparing microtubule targeting compounds, the method comprising the following steps: i) screening a library of compounds against Hβl -tubulin to determine whether the compounds individually inhibit the Hβl -tubulin; ii) discarding agents that inhibit the Hβl -tubulin; and iii) retaining agents that do not inhibit the Hβl -tubulin.
Of course, as used herein, it is understood that the term "library" can refer to any collection of compounds and can encompass a single compound, thousands of compounds, or any number of compounds falling therein. Moreover, the library of compounds can include compounds that are categorized according to any methodology used in the art, including a random sampling of compounds. In certain embodiments, the screens described herein can be based on high-throughput screening of a large library of small organic molecules. In other embodiments, the screen can be based on utilization of a library of small molecules tailored to the properties of microtubule targeting agents. For example, in one method for compound screening, a highly specific antibody was developed that recognizes Hβl tubulin only. In one embodiment, an initial screen can be performed in cells (e.g., epithelial cancer cells) transfected with an Hβl tubulin encoding mammalian expression plasmid, and the cells then can be exposed to varying concentrations of each compound to be tested and compared to a control. The effect of each compound on Hβl -specific tubulin can be assessed by a cell-based tubulin polymerization assay as described below in relation to FIG. 4 in conjunction with immunostaining with the Hβl -specific antibody to assess the effects of each of the compounds on Hβl microtubule polymer mass. Co-staining with a beta-tubulin antibody
(which is commercially available) that recognizes all β-tubulin isotypes but Hβl also can be performed to assess the effects of each compound on the other constitutively expressed beta-tubulin isotypes.
In further embodiments, a secondary screening assay can be performed according to a method similar to that described above that assess the effects of each compound on Hβl -containing microtubules or total β-tubulin by immunofluorescence followed by confocal microscopy as described below in relation to FIG. 12.
Screening of compounds similar to that described above also can be carried out using the knowledge of amino acid effect on compound binding with various β-tubulin isotypes, as described previously. In other words, the invention provides a method to identify myelo-sparing MTCs based on the known amino acid structures that lead to steric hindrance, as described herein, in specific relation to Hβ 1 -tubulin. In one embodiment, the method comprises the following steps: i) screening a library of compounds against a β-tubulin with an amino acid other than valine at β236, tyrosine at β200, or valine at β316; ii) discarding compounds that inhibit the β-tubulin; and iii) retaining compounds that do not inhibit the β-tubulin.
In specific embodiments, the screening based on amino acid encoding can include the use of β-tubulin that positively encodes specific amino acids. For example, the amino acid at β236 can be isoleucine. In other embodiments, the amino acid at β200 can be phenylalanine. In still other embodiments, the amino acid at β316 can be isoleucine.
The screens can also be carried out in a variety of manners. For example, in certain embodiments, the screen is in silico. In other embodiments, the screen it protein- based. In still further embodiments, the screen is cell-based. In specific embodiments, the cells used in the screen can be specific cell types, such as 2MRC cells or bone marrow cells.
Myelo-sparing MTCs identified according to the inventive screening methods particularly may include those compounds that bind β-tubulin isotypes with valine at β236, tyrosine at β200, or valine at β316 and that do not bind to β-tubulin isotypes with an amino acid other than isoleucine at β236, phenylalanine at β200, or isoleucine at β316. In specific embodiments, the identified compounds do not bind β-tubulin isotypes that comprise an isoleucine at β236, phenylalanine at β200, or isoleucine at β316. Preferably,
the identified compounds do not bind β-tubulin isotype Hβl. In other embodiments, the identified compounds do bind β-tubulin isotypes with valine at β236 and do not bind β- tubulin isotypes with isoleucine at β236.
In specific embodiments, the present invention can provide specific antibodies. For example, in one embodiment, the invention provides an antibody specific to human Hβl -tubulin. In certain embodiments, the antibody binds to residues 443-451 of the C- terminus of human Hβl-tubulin.
III. Biologically Active Variants Biologically active variants of the compounds set forth above are particularly also encompassed by the invention. Such variants should retain the general biological activity of the original compounds; however, the presence of additional activities would not necessarily limit the use thereof in the present invention. Such activity may be evaluated using standard testing methods and bioassays recognizable by the skilled artisan in the field as generally being useful for identifying such activity.
According to one embodiment of the invention, suitable biologically active variants comprise one or more analogues or derivatives of the compounds described above. Indeed, compounds such as those described above may give rise to an entire family of analogues or derivatives having similar activity and, therefore, usefulness according to the present invention. Likewise, a single compound, such as those described above, may represent a single family member of a greater class of compounds useful according to the present invention. Accordingly, the present invention fully encompasses not only the compounds described above, but analogues and derivatives of such compounds, particularly those identifiable by methods commonly known in the art and recognizable to the skilled artisan.
The compounds disclosed herein may contain chiral centers, which may be either of the (R) or (S) configuration, or may comprise a mixture thereof. Accordingly, the present invention also includes stereoisomers of the compounds described herein, where applicable, either individually or admixed in any proportions. Stereoisomers may include, but are not limited to, enantiomers, diastereomers, racemic mixtures, and combinations thereof. Such stereoisomers can be prepared and separated using conventional techniques, either by reacting enantiomeric starting materials, or by separating isomers of compounds of the present invention. Isomers may include geometric isomers. Examples of geometric
isomers include, but are not limited to, cis isomers or trans isomers. Other isomers are contemplated among the compounds of the present invention. The isomers may be used either in pure form or in admixture with other isomers of the compounds described herein.
Various methods are known in the art for preparing optically active forms and determining activity. Such methods include standard tests described herein other similar tests which are well known in the art. Examples of methods that can be used to obtain optical isomers of the compounds according to the present invention include the following: i) physical separation of crystals whereby macroscopic crystals of the individual enantiomers are manually separated. This technique may particularly be used when crystals of the separate enantiomers exist (i.e., the material is a conglomerate), and the crystals are visually distinct; ii) simultaneous crystallization whereby the individual enantiomers are separately crystallized from a solution of the racemate, possible only if the latter is a conglomerate in the solid state; iii) enzymatic resolutions whereby partial or complete separation of a racemate by virtue of differing rates of reaction for the enantiomers with an enzyme; iv) enzymatic asymmetric synthesis, a synthetic technique whereby at least one step of the synthesis uses an enzymatic reaction to obtain an enantiomeric ally pure or enriched synthetic precursor of the desired enantiomer; v) chemical asymmetric synthesis whereby the desired enantiomer is synthesized from an achiral precursor under conditions that produce asymmetry (i.e., chirality) in the product, which may be achieved using chiral catalysts or chiral auxiliaries; vi) diastereomer separations whereby a racemic compound is reacted with an enantiomerically pure reagent (the chiral auxiliary) that converts the individual enantiomers to diastereomers. The resulting diastereomers are then separated by chromatography or crystallization by virtue of their now more distinct structural differences and the chiral auxiliary later removed to obtain the desired enantiomer; vii) first- and second-order asymmetric transformations whereby diastereomers from the racemate equilibrate to yield a preponderance in solution of the diastereomer from the desired enantiomer or where preferential crystallization of the diastereomer from the desired enantiomer perturbs the equilibrium such that eventually in principle all the
material is converted to the crystalline diastereomer from the desired enantiomer. The desired enantiomer is then released from the diastereomers; viii) kinetic resolutions comprising partial or complete resolution of a racemate (or of a further resolution of a partially resolved compound) by virtue of unequal reaction rates of the enantiomers with a chiral, non-racemic reagent or catalyst under kinetic conditions; ix) enantiospecific synthesis from non-racemic precursors whereby the desired enantiomer is obtained from non-chiral starting materials and where the stereochemical integrity is not or is only minimally compromised over the course of the synthesis; x) chiral liquid chromatography whereby the enantiomers of a racemate are separated in a liquid mobile phase by virtue of their differing interactions with a stationary phase. The stationary phase can be made of chiral material or the mobile phase can contain an additional chiral material to provoke the differing interactions; xi) chiral gas chromatography whereby the racemate is volatilized and enantiomers are separated by virtue of their differing interactions in the gaseous mobile phase with a column containing a fixed non-racemic chiral adsorbent phase; xii) extraction with chiral solvents whereby the enantiomers are separated by virtue of preferential dissolution of one enantiomer into a particular chiral solvent; and xiii) transport across chiral membranes whereby a racemate is placed in contact with a thin membrane barrier. The barrier typically separates two miscible fluids, one containing the racemate, and a driving force such as concentration or pressure differential causes preferential transport across the membrane barrier. Separation occurs as a result of the non-racemic chiral nature of the membrane which allows only one enantiomer of the racemate to pass through. The compounds of the invention may be provided in an enantiomerically enriched form, such as a mixture of enantiomers in which one enantiomer is present in excess (given as a mole fraction or a weight fraction). Enantiomeric excess is understood to exist where a chemical substance comprises two enantiomers of the same compound and one enantiomer is present in a greater amount than the other enantiomer. Unlike racemic mixtures, these mixtures will show a net optical rotation. With knowledge of the specific rotation of the mixture and the specific rotation of the pure enantiomer, the enantiomeric excess (abbreviated "ee") can be determined by known methods. Direct determination of
the quantities of each enantiomer present in the mixture is possible with NMR spectroscopy and chiral column chromatography.
In specific embodiments, the compounds of the invention can comprise an MTC having an enantiomeric purity for a single enantiomer of at least about 75%. In further embodiments, the compounds of the invention have an enantiomeric purity of at least about 80%, at least about 85%, at least about 90%, at least about 95%, at least about 96%, at least about 97%, at least about 98%, at least about 99%, or at least about 99.5%. In one embodiment, the compounds of the invention may have such enantiomeric purity for the (S) isomer. In another embodiment, the compounds of the invention may have such enantiomeric purity for the (R) isomer.
The compounds described herein can also be in the form of an ester, amide, salt, solvate, prodrug, or metabolite provided they maintain pharmacological activity according to the present invention. Esters, amides, salts, solvates, prodrugs, and other derivatives of the compounds of the present invention may be prepared according to methods generally known in the art, such as, for example, those methods described by J. March, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 4th Ed. (New York: Wiley- Interscience, 1992), which is incorporated herein by reference.
Examples of pharmaceutically acceptable salts of the compounds useful according to the invention include acid addition salts. Salts of non-pharmaceutically acceptable acids, however, may be useful, for example, in the preparation and purification of the compounds. Suitable acid addition salts according to the present invention include organic and inorganic acids. Preferred salts include those formed from hydrochloric, hydrobromic, sulfuric, phosphoric, citric, tartaric, lactic, pyruvic, acetic, succinic, fumaric, maleic, oxaloacetic, methanesulfonic, ethanesulfonic, p-toluenesulfonic, benzesulfonic, and isethionic acids. Other useful acid addition salts include propionic acid, glycolic acid, oxalic acid, malic acid, malonic acid, benzoic acid, cinnamic acid, mandelic acid, salicylic acid, and the like. Particular example of pharmaceutically acceptable salts include, but are not limited to, sulfates, pyrosulfates, bisulfates, sulfites, bisulfϊtes, phosphates, monohydrogenphosphates, dihydrogenphosphates, metaphosphates, pyrophosphates, chlorides, bromides, iodides, acetates, propionates, decanoates, caprylates, acrylates, formates, isobutyrates, caproates, heptanoates, propiolates, oxalates, malonates, succinates, suberates, sebacates, fumarates, maleates, butyne-l,4-dioates, hexyne-1,6- dioates, benzoates, chlorobenzoates, methylbenzoates, dinitrobenzoates,
hydroxybenzoates, methoxyenzoates, phthalates, sulfonates, xylenesulfonates, phenylacetates, phenylpropionates, phenylbutyrates, citrates, lactates, γ-hydroxybutyrates, glycolates, tartrates, methanesulfonates, propanesulfonates, naphthalene- 1 -sulfonates, naphthalene -2-sulfonates, and mandelates. An acid addition salt may be reconverted to the free base by treatment with a suitable base. Preparation of basic salts of acid moieties which may be present on a compound useful according to the present invention may be prepared in a similar manner using a pharmaceutically acceptable base, such as sodium hydroxide, potassium hydroxide, ammonium hydroxide, calcium hydroxide, triethylamine, or the like. Esters of the compounds according to the present invention may be prepared through functionalization of hydroxyl and/or carboxyl groups that may be present within the molecular structure of the compound. Amides and prodrugs may also be prepared using techniques known to those skilled in the art. For example, amides may be prepared from esters, using suitable amine reactants, or they may be prepared from anhydride or an acid chloride by reaction with ammonia or a lower alkyl amine. Moreover, esters and amides of compounds of the invention can be made by reaction with a carbonylating agent (e.g., ethyl formate, acetic anhydride, methoxyacetyl chloride, benzoyl chloride, methyl isocyanate, ethyl chloroformate, methanesulfonyl chloride) and a suitable base (e.g., A- dimethylaminopyridine, pyridine, triethylamine, potassium carbonate) in a suitable organic solvent (e.g., tetrahydrofuran, acetone, methanol, pyridine, N,N-dimethylformamide) at a temperature of 0 0C to 60 0C. Prodrugs are typically prepared by covalent attachment of a moiety, which results in a compound that is therapeutically inactive until modified by an individual's metabolic system. Examples of pharmaceutically acceptable solvates include, but are not limited to, compounds according to the invention in combination with water, isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid, or ethanolamine.
In the case of solid compositions, it is understood that the compounds used in the compositions of the invention may exist in different forms. For example, the compounds may exist in stable and metastable crystalline forms and isotropic and amorphous forms, all of which are intended to be within the scope of the present invention. If a compound useful according to the invention is a base, the desired salt may be prepared by any suitable method known to the art, including treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, maleic acid,
succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, pyranosidyl acids such as glucuronic acid and galacturonic acid, alpha-hydroxy acids such as citric acid and tartaric acid, amino acids such as aspartic acid and glutamic acid, aromatic acids such as benzoic acid and cinnamic acid, sulfonic acids such a p-toluenesulfonic acid or ethanesulfonic acid, or the like.
If a compound of the invention is an acid, the desired salt may be prepared by any suitable method known to the art, including treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary), an alkali metal or alkaline earth metal hydroxide or the like. Illustrative examples of suitable salts include organic salts derived from amino acids such as glycine and arginine, ammonia, primary, secondary and tertiary amines, and cyclic amines such as piperidine, morpholine and piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
The present invention further includes prodrugs and active metabolites of the compounds of the invention. Any of the compounds described herein can be administered as a prodrug to increase the activity, bioavailability, or stability of the compound or to otherwise alter the properties of the compound. Typical examples of prodrugs include compounds that have biologically labile protecting groups on a functional moiety of the active compound. Prodrugs include compounds that can be oxidized, reduced, aminated, deaminated, hydroxylated, dehydroxylated, hydrolyzed, dehydrolyzed, alkylated, dealkylated, acylated, deacylated, phosphorylated, and/or dephosphorylated to produce the active compound.
A number of prodrug ligands are known. In general, alkylation, acylation, or other lipophilic modification of one or more heteroatoms of the compound, such as a free amine or carboxylic acid residue, reduces polarity and allows passage into cells. Examples of substituent groups that can replace one or more hydrogen atoms on a free amine and/or carboxylic acid moiety include, but are not limited to, the following: aryl; steroids; carbohydrates (including sugars); 1,2-diacylglycerol; alcohols; acyl (including lower acyl); alkyl (including lower alkyl); sulfonate ester (including alkyl or arylalkyl sulfonyl, such as methanesulfonyl and benzyl, wherein the phenyl group is optionally substituted with one or more substituents as provided in the definition of an aryl given herein); optionally substituted arylsulfonyl; lipids (including phospholipids); phosphotidylcholine; phosphocholine; amino acid residues or derivatives; amino acid acyl residues or
derivatives; peptides; cholesterols; or other pharmaceutically acceptable leaving groups which, when administered in vivo, provide the free amine and/or carboxylic acid moiety. Any of these can be used in combination with the disclosed compounds to achieve a desired effect.
IV. Pharmaceutical Compositions
While it is possible for individual compounds according to the present invention to be administered in the raw chemical form, it is preferred for the compounds to be delivered as a pharmaceutical composition. Accordingly, there are provided by the present invention pharmaceutical compositions comprising one or more compounds as described herein. As such, the compositions of the present invention comprise the pharmaceutically active compounds, as described above, or pharmaceutically acceptable esters, amides, salts, solvates, analogs, derivatives, or prodrugs thereof. Further, the inventive compositions can be prepared and delivered in a variety of combinations. For example, the composition can comprise a single composition containing all of the active ingredients. Alternately, the composition can comprise multiple compositions comprising separate active ingredients but intended to be administered simultaneously, in succession, or in otherwise close proximity of time.
The compounds of the invention can be prepared and delivered together with one or more pharmaceutically acceptable carriers therefore, and optionally, other therapeutic ingredients. Carriers should be acceptable in that they are compatible with any other ingredients of the composition and not harmful to the recipient thereof. A carrier may also reduce any undesirable side effects of the agent. Such carriers are known in the art. See, Wang et al. (1980) J. Parent. Drug Assn. 34(6):452-462, herein incorporated by reference in its entirety.
Compositions of the present invention may include short-term, rapid-onset, rapid- offset, controlled release, sustained release, delayed release, and pulsatile release compositions, providing the compositions achieve administration of a compound as described herein. See Remington 's Pharmaceutical Sciences (18th ed.; Mack Publishing Company, Eaton, Pennsylvania, 1990), herein incorporated by reference in its entirety. Pharmaceutical compositions according to the present invention are suitable for various modes of delivery, including oral, parenteral (including intravenous, intramuscular, subcutaneous, intradermal, intra-articular, intra-synovial, intrathecal, intra-arterial,
intracardiac, subcutaneous, intraorbital, intracapsular, intraspinal, intrastemal, and transdermal), topical (including dermal, buccal, and sublingual), vaginal, urethral, and rectal administration. Administration can also be via nasal spray, surgical implant, internal surgical paint, infusion pump, or via catheter, stent, balloon or other delivery device. The most useful and/or beneficial mode of administration can vary, especially depending upon the condition of the recipient and the disorder being treated.
The pharmaceutical compositions may be conveniently made available in a unit dosage form, whereby such compositions may be prepared by any of the methods generally known in the pharmaceutical arts. Generally speaking, such methods of preparation comprise combining (by various methods) the active compounds of the invention with a suitable carrier or other adjuvant, which may consist of one or more ingredients. The combination of the active ingredients with the one or more adjuvants is then physically treated to present the composition in a suitable form for delivery (e.g. , shaping into a tablet or forming an aqueous suspension). Pharmaceutical compositions according to the present invention suitable for oral dosage may take various forms, such as tablets, capsules, caplets, and wafers (including rapidly dissolving or effervescing), each containing a predetermined amount of the active agent. The compositions may also be in the form of a powder or granules, a solution or suspension in an aqueous or non-aqueous liquid, and as a liquid emulsion (oil-in-water and water-in-oil). The active agents may also be delivered as a bolus, electuary, or paste. It is generally understood that methods of preparations of the above dosage forms are generally known in the art, and any such method would be suitable for the preparation of the respective dosage forms for use in delivery of the compositions according to the present invention. In one embodiment, compound may be administered orally in combination with a pharmaceutically acceptable vehicle such as an inert diluent or an edible carrier. Oral compositions may be enclosed in hard or soft shell gelatin capsules, may be compressed into tablets or may be incorporated directly with the food of the patient's diet. The percentage of the composition and preparations may be varied; however, the amount of substance in such therapeutically useful compositions is preferably such that an effective dosage level will be obtained.
Hard capsules containing the compound may be made using a physiologically degradable composition, such as gelatin. Such hard capsules comprise the compound, and
may further comprise additional ingredients including, for example, an inert solid diluent such as calcium carbonate, calcium phosphate, or kaolin. Soft gelatin capsules containing the compound may be made using a physiologically degradable composition, such as gelatin. Such soft capsules comprise the compound, which may be mixed with water or an oil medium such as peanut oil, liquid paraffin, or olive oil.
Sublingual tablets are designed to dissolve very rapidly. Examples of such compositions include ergotamine tartrate, isosorbide dinitrate, and isoproterenol HCL. The compositions of these tablets contain, in addition to the drug, various soluble excipients, such as lactose, powdered sucrose, dextrose, and mannitol. The solid dosage forms of the present invention may optionally be coated, and examples of suitable coating materials include, but are not limited to, cellulose polymers (such as cellulose acetate phthalate, hydroxypropyl cellulose, hydroxypropyl methylcellulose, hydroxypropyl methylcellulose phthalate, and hydroxypropyl methylcellulose acetate succinate), polyvinyl acetate phthalate, acrylic acid polymers and copolymers, and methacrylic resins (such as those commercially available under the trade name EUDRAGIT®), zein, shellac, and polysaccharides.
Powdered and granular compositions of a pharmaceutical preparation of the invention may be prepared using known methods. Such compositions may be administered directly to a patient or used in the preparation of further dosage forms, such as to form tablets, fill capsules, or prepare an aqueous or oily suspension or solution by addition of an aqueous or oily vehicle thereto. Each of these compositions may further comprise one or more additives, such as dispersing or wetting agents, suspending agents, and preservatives. Additional excipients (e.g., fillers, sweeteners, flavoring, or coloring agents) may also be included in these compositions. Liquid compositions of the pharmaceutical composition of the invention which are suitable for oral administration may be prepared, packaged, and sold either in liquid form or in the form of a dry product intended for reconstitution with water or another suitable vehicle prior to use.
A tablet containing one or more compounds according to the present invention may be manufactured by any standard process readily known to one of skill in the art, such as, for example, by compression or molding, optionally with one or more adjuvant or accessory ingredient. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active agents.
Adjuvants or accessory ingredients for use in the compositions of the present invention can include any pharmaceutical ingredient commonly deemed acceptable in the art, such as binders, fillers, lubricants, disintegrants, diluents, surfactants, stabilizers, preservatives, flavoring and coloring agents, and the like. Binders are generally used to facilitate cohesiveness of the tablet and ensure the tablet remains intact after compression. Suitable binders include, but are not limited to: starch, polysaccharides, gelatin, polyethylene glycol, propylene glycol, waxes, and natural and synthetic gums. Acceptable fillers include silicon dioxide, titanium dioxide, alumina, talc, kaolin, powdered cellulose, and microcrystalline cellulose, as well as soluble materials, such as mannitol, urea, sucrose, lactose, dextrose, sodium chloride, and sorbitol. Lubricants are useful for facilitating tablet manufacture and include vegetable oils, glycerin, magnesium stearate, calcium stearate, and stearic acid. Disintegrants, which are useful for facilitating disintegration of the tablet, generally include starches, clays, celluloses, algins, gums, and crosslinked polymers. Diluents, which are generally included to provide bulk to the tablet, may include dicalcium phosphate, calcium sulfate, lactose, cellulose, kaolin, mannitol, sodium chloride, dry starch, and powdered sugar. Surfactants suitable for use in the composition according to the present invention may be anionic, cationic, amphoteric, or nonionic surface active agents. Stabilizers may be included in the compositions to inhibit or lessen reactions leading to decomposition of the active agents, such as oxidative reactions.
Solid dosage forms may be formulated so as to provide a delayed release of the active agents, such as by application of a coating. Delayed release coatings are known in the art, and dosage forms containing such may be prepared by any known suitable method. Such methods generally include that, after preparation of the solid dosage form (e.g., a tablet or caplet), a delayed release coating composition is applied. Application can be by methods, such as airless spraying, fluidized bed coating, use of a coating pan, or the like. Materials for use as a delayed release coating can be polymeric in nature, such as cellulosic material (e.g., cellulose butyrate phthalate, hydroxypropyl methylcellulose phthalate, and carboxymethyl ethylcellulose), and polymers and copolymers of acrylic acid, methacrylic acid, and esters thereof.
Solid dosage forms according to the present invention may also be sustained release (i.e., releasing the active agents over a prolonged period of time), and may or may not also be delayed release. Sustained release compositions are known in the art and are
generally prepared by dispersing a drug within a matrix of a gradually degradable or hydrolyzable material, such as an insoluble plastic, a hydrophilic polymer, or a fatty compound. Alternatively, a solid dosage form may be coated with such a material.
Compositions for parenteral administration include aqueous and non-aqueous sterile injection solutions, which may further contain additional agents, such as antioxidants, buffers, bacteriostats, and solutes, which render the compositions isotonic with the blood of the intended recipient. The compositions may include aqueous and nonaqueous sterile suspensions, which contain suspending agents and thickening agents. Such compositions for parenteral administration may be presented in unit-dose or multi-dose containers, such as, for example, sealed ampoules and vials, and may be stores in a freeze- dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example, water (for injection), immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules, and tablets of the kind previously described. The compositions according to the present invention may also be administered transdermally, wherein the active agents are incorporated into a laminated structure (generally referred to as a "patch") that is adapted to remain in intimate contact with the epidermis of the recipient for a prolonged period of time. Typically, such patches are available as single layer "drug-in-adhesive" patches or as multi-layer patches where the active agents are contained in a layer separate from the adhesive layer. Both types of patches also generally contain a backing layer and a liner that is removed prior to attachment to the skin of the recipient. Transdermal drug delivery patches may also be comprised of a reservoir underlying the backing layer that is separated from the skin of the recipient by a semi-permeable membrane and adhesive layer. Transdermal drug delivery may occur through passive diffusion or may be facilitated using electrotransport or iontophoresis.
Compositions for rectal delivery of the compositions of the present invention include rectal suppositories, creams, ointments, and liquids. Suppositories may be presented as the active agents in combination with a carrier generally known in the art, such as polyethylene glycol. Such dosage forms may be designed to disintegrate rapidly or over an extended period of time, and the time to complete disintegration can range from a short time, such as about 10 minutes, to an extended period of time, such as about 6 hours.
Topical compositions may be in any form suitable and readily known in the art for delivery of active agents to the body surface, including dermally, buccally, and sublingually. Typical examples of topical compositions include ointments, creams, gels, pastes, and solutions. Compositions for topical administration in the mouth also include lozenges.
In certain embodiments, the compounds and compositions disclosed herein can be delivered via a medical device. Such delivery can generally be via any insertable or implantable medical device, including, but not limited to stents, catheters, balloon catheters, shunts, or coils. In one embodiment, the present invention provides medical devices, such as stents, the surface of which is coated with a compound or composition as described herein. The medical device of this invention can be used, for example, in any application for treating, preventing, or otherwise affecting the course of a disease or condition, such as those disclosed herein.
In another embodiment of the invention, the pharmaceutical composition comprising one or more compounds described herein is administered intermittently. Administration of the therapeutically effective dose may be achieved in a continuous manner, as for example with a sustained-release composition, or it may be achieved according to a desired daily dosage regimen, as for example with one, two, three, or more administrations per day. By "time period of discontinuance" is intended a discontinuing of the continuous sustained-released or daily administration of the composition. The time period of discontinuance may be longer or shorter than the period of continuous sustained- release or daily administration. During the time period of discontinuance, the level of the components of the composition in the relevant tissue is substantially below the maximum level obtained during the treatment. The preferred length of the discontinuance period depends on the concentration of the effective dose and the form of composition used. The discontinuance period can be at least 2 days, at least 4 days or at least 1 week. In other embodiments, the period of discontinuance is at least 1 month, 2 months, 3 months, 4 months or greater. When a sustained-release composition is used, the discontinuance period must be extended to account for the greater residence time of the composition in the body. Alternatively, the frequency of administration of the effective dose of the sustained- release composition can be decreased accordingly. An intermittent schedule of administration of a composition of the invention can continue until the desired therapeutic effect, and ultimately treatment of the disease or disorder, is achieved.
Administration of the composition according to the invention comprises administering a single pharmaceutically active compound as described herein; administering a pharmaceutically active compound as described herein with one or more further pharmaceutically active compounds described herein; or administering one or more pharmaceutically active compounds described herein in combination with one or more further pharmaceutically active compounds (i.e., co-administration). Accordingly, it is recognized that the pharmaceutically active compounds in the compositions of the invention can be administered in a fixed combination (i.e., a single pharmaceutical composition that contains both active materials). Alternatively, the pharmaceutically active compounds may be administered simultaneously (i.e., separate compositions administered at the same time). In another embodiment, the pharmaceutically active compounds are administered sequentially (i.e., administration of one or more pharmaceutically active compounds followed by separate administration or one or more pharmaceutically active compounds). One of skill in the art will recognized that the most preferred method of administration will allow the desired therapeutic effect.
Delivery of a therapeutically effective amount of a composition according to the invention may be obtained via administration of a therapeutically effective dose of the composition. Accordingly, in one embodiment, a therapeutically effective amount is an amount effective to treat abnormal cell proliferation. In another embodiment, a therapeutically effective amount is an amount effective to treat inflammation. In yet another embodiment, a therapeutically effective amount is an amount effective to treat arthritis. In still another embodiment, a therapeutically effective amount is an amount effective to treat asthma.
The active compound is included in the pharmaceutical composition in an amount sufficient to deliver to a patient a therapeutic amount of a compound of the invention in vivo in the absence of serious toxic effects. The concentration of active compound in the drug composition will depend on absorption, inactivation, and excretion rates of the drug as well as other factors known to those of skill in the art. It is to be noted that dosage values will also vary with the severity of the condition to be alleviated. It is to be further understood that for any particular subject, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, and that the dosage ranges set forth herein are exemplary only and are not intended to limit the scope or
practice of the claimed composition. The active ingredient may be administered at once, or may be divided into a number of smaller doses to be administered at varying intervals of time.
A therapeutically effective amount according to the invention can be determined based on the body weight of the recipient. For example, in one embodiment, a therapeutically effective amount of one or more compounds of the invention is in the range of about 0.1 μg/kg of body weight to about 5 mg/kg of body weight per day. Alternatively, a therapeutically effective amount can be described in terms of a fixed dose. The effective dosage range of pharmaceutically acceptable salts and prodrugs can be calculated based on the weight of the parent nucleoside to be delivered. If a salt or prodrug exhibits activity in itself, the effective dosage can be estimated as above using the weight of the salt or prodrug, or by other means known to those skilled in the art.
It is contemplated that the compositions of the invention comprising one or more compounds described herein will be administered in therapeutically effective amounts to a mammal, preferably a human. An effective dose of a compound or composition for treatment of any of the conditions or diseases described herein can be readily determined by the use of conventional techniques and by observing results obtained under analogous circumstances. The effective amount of the compositions would be expected to vary according to the weight, sex, age, and medical history of the subject. Of course, other factors could also influence the effective amount of the composition to be delivered, including, but not limited to, the specific disease involved, the degree of involvement or the severity of the disease, the response of the individual patient, the particular compound administered, the mode of administration, the bioavailability characteristics of the preparation administered, the dose regimen selected, and the use of concomitant medication. The compound is preferentially administered for a sufficient time period to alleviate the undesired symptoms and the clinical signs associated with the condition being treated. Methods to determine efficacy and dosage are known to those skilled in the art. See, for example, Isselbacher et al. (1996) Harrison 's Principles of Internal Medicine 13 ed., 1814-1882, herein incorporated by reference.
V. Articles of Manufacture
The present invention also includes an article of manufacture providing a composition comprising one or more compounds described herein. The article of manufacture may contain one or more of the compounds described herein in combination with one or more further therapeutic agents. The article of manufacture can include a vial or other container that contains a composition suitable for use according to the present invention together with any carrier, either dried or in liquid form. In particular, the article of manufacture can comprise a kit including a container with a composition according to the invention. In such a kit, the composition can be delivered in a variety of combinations. For example, the composition can comprise a single dosage comprising all of the active ingredients. Alternately, where more than one active ingredient is provided, the composition can comprise multiple dosages, each comprising one or more active ingredients, the dosages being intended for administration in combination, in succession, or in other close proximity of time. For example, the dosages could be solid forms (e.g., tablets, caplets, capsules, or the like) or liquid forms (e.g., vials), each comprising a single active ingredient, but being provided in blister packs, bags, or the like, for administration in combination.
The article of manufacture further includes instructions for carrying out the method of the invention. Such instructions may be in various forms, such as a label on the container, an insert included in a box in which the container is packaged, or a variety of computer readable formats. The instructions can also be printed on the box in which the vial is packaged. The instructions contain information such as sufficient dosage and administration information so as to allow the subject or a worker in the field to administer the pharmaceutical composition. It is anticipated that a worker in the field encompasses any doctor, nurse, technician, spouse, or other caregiver that might administer the composition. The pharmaceutical composition can also be self-administered by the subject.
VI. Methods of Treatment
The MTCs according to the present invention are useful in a variety of methods of treatment. In certain embodiments, the inventive MTCs may be useful in any methods of treatment where conventional taxanes and/or epothilones may find use. As described herein, though, the MTC of the invention provide such treatment with designed specificity to avoid the undesirable side effects of conventional taxanes and/or epothilones, particularly myelosuppression.
In specific embodiments, the MTCs of the invention may be useful in a method of treating cancer. In other embodiments, the MTCs of the invention may be useful in method of treating and/or preventing abnormal cell proliferation.
In some embodiments, the MTCs of the invention can be used to treat a variety of cancer types, including but not limited to non-small-cell lung cancer, ovarian cancer (e.g., germ cell ovary cancer), breast cancer (including brain and CNS tumors caused by breast cancer), prostate cancer (e.g., hormone refractory prostate cancer), colo-rectal cancer, renal cancer, gastric cancer, gall bladder cancer, liver cancer, pancreatic cancer, small intestine cancer, testicular cancer, head cancer, neck cancer, melanoma, hepatocellular carcinoma, fallopian tube cancer, endometrial cancer, peritoneal cancer, solid tumors, non- Hodgkin's lymphoma, chronic lymphocytic leukemia, gliomas, and Kaposi's sarcoma.
The epothilone compounds described herein particularly have shown significant antiproliferative activity against A2780 ovarian cancer and the PC3 prostate cancer cell lines, with activities up to tenfold greater than that of epothilone D (Table 1). As seen in Table 1, the formation of the internal lactone further increased the potency of the compounds in the A2780 assay, with increases ranging from fivefold to twofold.
Table 1
Cmpd, IC5O (oM) EC50 Tb Ka (XlO7M"1) Hill Slope
A2780 assembly μM
Epo. D 40 0.44 15.2 2.9
Formula (17) 1480 1.240 0.72 1.01
Formula (19) 34 0.465 2.21 1.4
Formula (20) 250 0.605 1.26 0.91
Formula (41) 8.9 0.204 6.84 1.92
Formula (21) 4.2 0.141 8.50 2.45
Formula (46) 39 0.467 6.99 1.73
Formula (22) 7.9 0.181 8.98 2.48
VII. Experimental
The present invention will now be described with specific reference to various examples. The following examples are not intended to be limiting of the invention and are rather provided as exemplary embodiments.
EXAMPLE 1 Identifying Non-Myelosuppressive Microtuble Targeting Compounds
The 2-Methoxyestradiol (2ME2), ENMD-1198, and ENMD- MKC-I used in the following testing were provided by EntreMed Inc. Combretastatin A4 was obtained from Tocris. Taxotere (TXT) provided by Aventis. Vincristine (VCR), vinblastine (VBL), epothilone B (EpoB), and colchicine were obtained from Calbiochem. Paclitaxel (PTX) was obtained from Sigma. The following primary antibodies were used: rat anti-α- tubulin (clone YL1/2; Chemicon International; 1 : 1000 dilution); mouse anti acetylated a- tubulin (Sigma; 1 :2000 dilution); caspase-3 cleavage; PARP p85 cleavage; rabbit polyclonal antibody to human actin (Sigma; 1 :5000 dilution); polyclonal rabbit Hβl (1 : 15000 dilution); mouse α-tubulin (clone DMIa, Sigma; 1 :2000 dilution); and mouse β-tubulin (clone 2.1, Sigma; 1 :2000 dilution). For immunofluorescence, specific secondary antibodies conjugated to Alexa Fluor goat 488 and 568 were used (Molecular Probes; 1 :500 dilution). For western blotting, specific secondary antibodies were Alexa Fluor 680 (Molecular Probes; 1 : 15000 dilution), IRdye 800 (Rockland Immunochemicals; 1: 15000 dilution), or horseradish peroxidase conjugated (Amersham Pharmacia; 1 :2000 dilution). DNA was counterstained with DAPI (Sigma; lug/ml).
A peptide corresponding to residues 443-451 of the C-terminus of the human class VI β-tubulin (Hβl) was designed. For immunization purposes, the Hβl peptide was conjugated to KLH. For ELISA analysis, Hβl peptide was conjugated to BSA. For affinity purification columns, the unconjugated peptide was used. Rabbit polyclonal Hβl antibody was produced by Pocono Rabbit Farm & Laboratory, Inc. according to their protocol. The peptides used in this project were synthesized by Sigma Genomics. All cells were cultured in RPMI 1640 (Cellgro). All media were supplemented with 10% FBS (CellGro) and antibiotics (Cellgro). Cells were cultured at 37°C in a humidified atmosphere and 5% CO2. 2ME2 resistant cell lines were obtained by selecting the human ovarian carcinoma cell line 1A9 (a subclone of A2780 cells) in the presence of increasing concentrations of 2ME2 for a period of time superior to two years. The 1A9 cells were initially cultivated in the presence of twice the IC50 value for 2ME2 (0.6 μM), and upon acquisition of drug resistance the concentrations of 2ME2 were progressively increased up to 50 μMin order to obtain late-stage resistant cells (2MRC). The stability of the resistant phenotype was assessed in 2MRC cells by incubating those cells in the
presence of drug- free media for more than 6 months, with no reduction in the relative resistant to 2ME2. Cell survival was assessed by using the SRB assay as described by Skehan, P., et al, 1990, J. Natl. Cancer Inst., 82: 1107-1112.
Immunofluorescence microscopy was performed as described by Mabjeesh, N.J., et al., 2003, Cancer Cell. 3:363-375, and Escuin, D., et al.,2005, Cancer Res. 65:9021-9028. Total cell extracts from different epithelial cancer cell lines or from hematopoietic tissue were immunoblotted with the indicated antibodies. Blotted proteins were detected using the Odyssey infrared imaging system (LI-COR).
Quantitative drug-induced tubulin polymerization was done as described by Giannakakou, P., et al., 1997, J. Biol. Chem. 272: 17118-17125, and Giannakakou, P., et al., 1998, Int. J. Cancer 75:57-63. Untreated and 2ME2 treated cells were lysed in a glycerol-containing microtubule-stabilizing buffer (MTSB) allowing almost 50% of endogenous microtubule polymers to be recovered in the pellet fraction (P) after centrifugation, thereby maximizing our ability to detect the microtubule depolymerizing effects of 2ME2. Conversely, to maximize the sensitivity for detecting drug-induced tubulin polymerization, a hypotonic lysis buffer (LSB) that recovers less than 10% of microtubule polymer in taxane-treated samples and their respective untreated control was used. Thus, the use of these distinct buffers accounts for the differences observed in the % P in the two controls samples. The percentage of polymerized tubulin (%P) was determined by dividing the densitometric value of polymerized tubulin (P) by the total tubulin content (P+S). A similar assay was performed using human platelets obtained from peripheral blood from a healthy donor following centrifugation of platelet-rich plasma at 300xg for 10 minutes.
Cells were imaged using a Zeiss LSM 5 LIVE confocal microscope using a 4Ox/ 1.3 EC Plan Neofluar objective, a 63x/l .4 Plan APOCHROMAT objective and lOOx/1.4 Plan APOCHROMAT objectives. All images were acquired and analyzed using Zeiss LSM 5 LIVE software.
Cells were plated on 6-well plates at density of 0.5xl06 cells and incubated overnight. Any floating cells were removed by replacing fresh medium. Compounds were added and incubated for 24 hours. All floating and adherent cells were harvested.
After washing with PBS containing 0.1% BSA, cells were fixed with 75% ethanol (in 0.1% BSA-PBS) and incubated at -200C overnight. The fixed cells were washed with PBS thrice
and stained with propidium iodide (PI) solution (50 μg/ml PI and 10 us RNase in PBS) overnight. The stained cells were analyzed by a BD FACSCalibur flow cytometer.
Total RNA was isolated using Qiagen RNeasy kit (Qiagen Inc) and RT-PCR was performed using Protoscript First Strand cDNA synthesis kit (New England Biolabs). PCR-amplifϊed cDNA of class I β-tubulin (HM40/TUBB gene, RNA accession # NM_178014) was performed using the following primers :
5' - CTTGCCCCATACATACCTT - 3';
5 ' - GTAAGACGGCTAAGGGAACTG - 3'. PCR products were direct sequenced using the following primers: 5 ' - TCTGGGGCAGGTAACAACT - 3 ';
5'- AGTTGTTACCTGCCCCAGA - 3 ';
5' - CTCCGCAAGTTGGCAGTCAAC - 3 ';
5 ' - TGGCCTCCAGATGGCAGTC - 3';
5' - GGGGATCCATTCCACAAAGTA - 3'; 5' - GGACCATGTTGACTGCCAAC - 3';
5' - GACTGCCATCTTGAGGCCAC - 3'.
Subcloning of the cDNA PCR-amplified products was performed using the TOPO TA cloning system (Invitrogen), followed by direct sequencing as described above. A minimum of 20 clones were analyzed for each sample. All PCR reactions were performed in 50 μl reactions. PCR reactions were carried out in an iCycler Thermocycler (BioRad) using a touchdown temperature program that include an initial denaturation at 95°C for 5 min followed by 15 cycles of denaturation at 95°C for 30 seconds, annealing at 65°C (the annealing temperature was decrease by 0.50C per cycle) for 30 seconds, extension at 68° for 1 min 30sec. PCR parameters for the proceeding 20 cycles were: 95°C for 30 sec, 57°C for 30 sec, 68°C for 1 min and 30sec, and one final cycle of extension at 72°C for 5 minutes. DNA sequencing was performed using Big Dye Terminator chemistry and AmpliTaq-FS DNA Polymerase in an Applied Biosystems Automated 3730 DNA Analyzer. Multiple sequence alignment of β-tubulin isotypes was performed using the Clustal W method (Thompson, J.D., et al., 1994, Nucleic Acids Res. 22:4673-4680) using MegAlign program (Lasergene, DNASTAR).
The structure of colchicine-depleted tubulin in complex with stathmin (PDB code: 1 SAO) was employed for the analysis. A 3-D molecular representation of 2ME2 was constructed with the Maestro v7.5 model builder and geometry optimized with the
MMFFs force field coupled to a GBSA/H20 solvation model. Conformational searches for both 2ME2 and colchicine were performed with Omega with default parameters, resulting in a single conformation for 2ME2 and twenty-three for colchicine. The latter conformer dataset was scanned against the 2ME2 structure with ROCS (Rapid Overlay of Chemical Structures) and ranked using a combination Shape Tanimoto and Scaled Color Score (ComboScore). The best overlap, with a score of 0.7, was then manually docked into the protein. The A-ring of 2ME2 overlaps with the C-ring of colchicine, the methoxy groups are positioned similarly and the hydroxyl and carbonyl groups are aligned. The bovine brain tubulin model was converted to Hβl in Maestro by mutating all the appropriate residues after removing 2ME2 from the pocket. Optimization of the new protein model with 100 ps of molecular dynamics (MD) at 300K using MMFFs, the GBSA/H2O solvation model and a time step of 1.5 fs followed. To determine if the PTX binding site is significantly reshaped by this treatment, the optimized PTX -bound tubulin devoid of stathmin complexation was utilized for comparison. Twenty different cDNAs from human non-tumoral tissues were obtained from the human multiple tissue cDNA (MTC) panels I and II and the human immune system MTC panel (Clontech). All samples correspond to adult tissues except for a sample from fetal liver that corresponded to a 18-24 week fetuses. Class VI β-tubulin isotype mRNA amounts were analyzed by quantitative real-time PCR using probe 14 of the Universal Human Probe Library detection system (Roche) in conjunction with the following specific primers for class VI β-tubulin isotype (Hβl/TUBBl gene, accession # NM_030773): Forward 5 '-GGATGCGTGAAATTGTCCAT-S ' and reverse 5 '-AGTCGATCCCGTGTTCCTC-S'. Primers were used at a final concentration of 200 nM each. The PCR amplification reaction was performed in 12 μl final volume, using the Universal Master Mix (PE Applied Biosystems) with an initial step at 95°C for 10 min, followed by 55 cycles of 95°C for 15 seconds, and 6O0C for 1 minute in a Sequence Detection System 7900HT (Applied Biosystems). Quantification was done using a standard curve constructed with serial 1/10 dilutions of the peripheral blood leukocyte cDNA sample. Normalization was carried out with the internal standard (3 -glucuronidase (GUS). PCR reactions were performed in triplicates.
Eight weeks old male C57BL6 mice (2 animals per group) were treated with the indicated drug concentrations for 24 h prior to sacrifice by CO2 inhalation. Mice were treated with 2ME2 p.o. at 600 mpk or i.v. at 200 mpk. Control mice were untreated or
treated with VCR 0.5 mpk i.p. or PTX 40 mpk ip. BM was flushed from femurs with Hanks Balanced salt solution and cells were cytospun onto slides and fixed in PHEMO for 10 minutes at room temperature. Human BM samples were obtained from a consented patient with monoclonal gammopathy of undetermined significance. Umbilical cord blood cells (UCBCs) were obtained from the public cord blood bank of the New York- Presbyterian Hospital Weill Cornell Medical Center. Peripheral blood mononuclear cells (PBMCs) from both BM and UCBC were isolated using Ficoll-Paque PLUS (GE Healthcare) and CD34 positive cells were sorted using a monoclonal Ab (StemCell Technologies, Inc.). Experiments presented in the figures are representative of three or more different repetitions. Statistical analysis was performed using a single factor ANOVA test (p < 0.05 was considered statistically significant).
A 2ME2-resistance cell model was generated consisting of the parental 1A9 human ovarian carcinoma cells and its drug-resistant counterpart 1A9/2MRC cells (2MRC). The 2MRC cells were more than 80-fold resistant to 2ME2 and did not exhibit significant cross resistance to any of the other classes of microtubule inhibitors, including the colchicine - site binding agents (combretastatin, ENMD MKC-I and colchicine) as well as the 2ME2 analog ENMDl 198, also undergoing clinical development (See Table 2 below). These results indicate that the 2MRC cells display a 2ME2-specifϊc resistant phenotype.
Table 2
1A9 2MRC R.R,
2ME2 (μM) 0.6 52 83.0
EN MD 1198 (μM) 0.26 2.15 8.3
EN MD MKC-I (μM) 0.17 0.53 3.1
Colchicine (nM) 11.2 12.1 1.08
Combretastatin (nM) 3.2 3.6 1.13
Vincristine (nM) 2.2 17.1 7.77
Vinblastine (nM) 1.4 9.5 6.78
Paclitaxel (nM) 2.1 1.9 0.91
Taxotere (nM) 0.3 0.2 0.67
Epothilone B (nM) 0.3 0.2 0.67
Table 2 shows the cytotoxicity profile of 2MRC cells to MTAs. The parental 1A9 and drug-resistant 2MRC cells were treated with various MTAs and their IC50 values were obtained following for 72 hour treatment. Relative resistance (R.R.) is calculated as the ratio of the IC50 of each drug against the resistant 2MRC cells divided by that obtained against the parental 1A9 cells.
To investigate the effects of 2ME2 on its target, tubulin, the parental and 2ME2- resistant cells were treated with increasing concentrations of 2ME2, and the relative ability of the drug to depolymerize microtubules was examined (See FIG. 3 and FIG. 4). Confocal microscopy analysis revealed that 2ME2 treatment of the parental 1A9 cells resulted in a dose-dependent depolymerization of interphase microtubules together with an increase of aberrant mitotic arrest evidenced by the presence of multiple asters. In contrast, the microtubule cytoskeleton of 2MRC cells was well organized and remained largely unaffected by 2ME2, even at the highest dose of 100 μM where microtubules are only slightly affected (FIG. 3). On the other hand, paclitaxel (PTX) treatment was equally effective at stabilizing microtubules in both parental and resistant cells as seen by the induction of microtubule bundles and aberrant mitotic figures in both 2ME2-sensitive and - resistant cell lines.
To assess the effects of 2ME2 on microtubule polymer mass in parental and drug- resistant cells, a cell-based tubulin polymerization assay was performed (FIG. 4). This quantitative assay was based on the principle that tubulin exists in a dynamic equilibrium between soluble tubulin dimers and microtubule polymers that remain detergent-insoluble following centrifugation. Microtubule-stabilizing drugs shift this equilibrium towards the microtubule polymer recovered in the pellet fraction (P); while drugs that depolymerize microtubules shift the equilibrium towards the pool of soluble tubulin recovered in the supernatant (S). Treatment of the parental 1A9 cells with 2ME2 resulted in a dose- dependent microtubule depolymerization manner, as evidenced by the decrease in tubulin polymer mass from 47% in untreated cells to 18% in cells treated with 100 μM of 2ME2. In contrast, 2ME2 treatment of 2MRC cells had no effect on microtubule polymer mass, in agreement with the results obtained from FIG. 3. Treatment with TXT resulted in a robust increase in tubulin polymerization in both cell lines, consistent with the similar IC50 values of TXT in both 1A9 and 2MRC cells (FIG. 4).
To further characterize the effects of 2ME2 on microtubule stability in the parental 1 A9 and drug-resistant 2MRC cells, the same blot was re -probed with an
antibody against acetylated α-tubulin. Tubulin acetylation is a post-translational modification that occurs at the conserved lysine 40 residue of α-tubulin and has been associated with microtubule stability. Drugs that hyperstabilize microtubules such as the taxanes, enhance tubulin acetylation while the opposite is observed with drugs that depolymerize microtubules. As shown in FIG. 4, 2ME2 treatment decreased the levels of acetylated tubulin in the parental drug-sensitive cells while it had no effect on tubulin acetylation in the drug-resistant 2MRC cells. As expected, TXT treatment resulted in a robust increase of tubulin acetylation in both cell lines.
The effect of 2ME2 treatment on cell cycle distribution and apoptosis in parental and resistant cells was also investigated. Flow cytometry analysis of DNA content in asynchronous 1A9 cells treated with 2ME2 revealed a dose-dependent increase in the percentage of cells that accumulate in G2/M (FIG. 5). In contrast, 2ME2 had no effect on the 2MRC cells that remained largely unaffected with the exception of a partial G2/M arrest observed when the highest concentration was used (100 gM). Treatment with PTX, on the other hand, was equally effective at inducing G2/M arrest in 2ME2-sensitive and -resistant cells. In agreement with the cell cycle results, the ability of 2ME2 to induce apoptosis in the 2MRC cells was equally compromised, as shown by the drug effects on caspase-3 and PARP p85 cleavage (FIG. 6).
Taken together, the data show that the ability of 2ME2 to induce microtubule- depolymerization, cell cycle arrest, and apoptotic cell death is severely impaired in the 2MRC cells, likely due to impaired drug-target engagement.
Testing further illustrated that an acquired β-tubulin mutation confers 2ME2- resistance in 2MRC cells. Drug resistance to cancer chemotherapy is often attributed to alterations in the drug's cellular target(s) that protect the cells from the toxic effects of the drug allowing them to continue proliferating, hi the case of MTAs, acquired mutations in the β-tubulin gene confer resistance to different microtubule disrupting agents including PTX and the epothilones A and B. In order to investigate whether β-tubulin mutations account for the resistant phenotype in the 2MRC cells, the gene status of the class I β- tubulin isotype (gene HM40/TUBB) was analyzed since it is the predominantly expressed β-tubulin isotype in most epithelial cancer cell types including the 1A9 cells. Direct sequencing of the HM40 β-tubulin cDNA revealed two acquired gene alterations in the 2MRC cells (FIG. 7). The first alteration consists of a single nucleotide deletion that causes a frame-shift mutation at residue 187 leading to a premature termination codon
(PTC) at residue 193 (L187fsX193). The second genetic alteration is a missense mutation resulting in a change in amino acid 236 from Valine to Isoleucine (βV236I). Subcloning of the cDNA PCR products followed by direct sequencing further showed that the two genetic alterations were mutually exclusive since 100% of the clones analyzed had either the frame-shift mutation (L187fsX193) or the missense mutation (1236), indicating that the two alterations occurred in different alleles. This result suggests that the 2MRC cells functionally express only the HM40 β236 mutant tubulin only responsible for the 2ME2 resistance.
To assess the role of V236I mutation on the protein-ligand interaction, the crystal structure of tubulin complexed with stathmin and colchicine was used, and 2ME2 was docked into the colchicine binding pocket at the α/β-tubulin interface. Both ligands were assumed to occupy a common site in agreement with their competitive binding (FIG. 8 insert). The five-membered D-ring of 2ME2 resides near V236, with certain hydrogens of the ligand making van der Waals contacts at 2.2-2.5 A (FIG. 8, left panel). The Vβ236I exchange introduces a methyl group into the site, reduces the size of the binding pocket severely, and prevents 2ME2 from occupying it (FIG. 8, right panel). Thus, the present model suggests that the 2ME2 binding site is reorganized by the V236I mutation in the HM40 β-tubulin isotype.
Our testing further illustrated that Hβl tubulin is resistant to the depolymerizing effects of 2ME2. Data indicated that the single point mutation at β236 likely mediates the 2ME2 -specific drug-resistance. As mentioned earlier, this mutation is located in the HM40 β-tubulin isotype, which is the predominantly expressed isotype in these cells, accounting for more than 85% of total β-tubulin mRNA expression. There are at least seven well characterized classes of human β-tubulin isotypes, many of which are ubiquitously expressed while some are tissue-specific; however, their precise function and tissue specificity are not well understood. To investigate whether residue V236 is conserved across the family of β-tubulin isotypes, protein sequence alignment was performed (FIG. 9). This analysis revealed that residue V236 is highly conserved across all human β-tubulin isotypes with the only exception of the hematopoietic -specific class VI β-tubulin (βl -tubulin or Hβl). Strikingly, Hβl tubulin encodes an He at residue β236, which is the same as the acquired mutation found in the drug-resistant 2MRC cells. This result together with the impact of Iβ236 on 2ME2 -tubulin interaction led to the hypothesis that the ability of 2ME2 to interact with Hβl tubulin will be similarly
compromised. A 3D model of the latter tubulin was constructed by replacing residues in the X-ray structure of bovine β-tubulin/colchicine (FIG. 10, right panel) with the corresponding Hβl residues (FIG. 10, left panel). As described for the Vβ236I mutant, the 2ME2/colchicine binding site is markedly congested by residue 1236, as well as 1316 and F200; a valine and a tyrosine present in the original X-ray structure, respectively (FIG. 10). The 2ME2 binding pocket in the Hβl model is more hydrophobic than in the X- ray structure, with several of the polar residues replaced by hydrophobic counterparts. As expected, the crowded and volume-truncated center prevents 2ME2 from occupying the site. Residue S239 is another example of a non-conserved residue that encodes a cysteine in the tubulin isotype used in the X-ray structure. However, the change of β239 from cysteine to serine in Hβl does not appear to affect 2ME2 binding directly, as the side chains of S239 point away from 2ME2 even though this residue sits along the 2ME2 binding site. For comparison, the taxane site in the Hβl model was also examined to determine if site-related residues in this isotype-specific model might influence PTX binding (data not shown). A number of residue replacements, including a pair on the critical M- loop, do not interact directly with the bound ligand, but a pair of changes does have the potential for interfering with PTX binding. In an optimized model of the taxane binding site, the benzamide phenyl ring of PTX is packed tightly between V23 and A231. In Hβ 1 , these residues are a Met and Leu, respectively, bulkier residues that accommodate themselves with additional space in the subsite provided by an S374A mutation.
Extensive molecular dynamics on the tubulin-PTX complex illustrates that this region of the binding site is expansive, highly flexible, and able to accommodate changes in structure. Thus, the Hβ l model predicts that while PTX encounters no difficulty binding to the class VI β-tubulin isotype, 2ME2 is hindered from occupying the colchicine pocket primarily by the 1236 side chain. Therefore, Hβl tubulin is predicted to display resistance to the effects of 2ME2 while retaining its sensitivity to PTX.
To test these predictions experimentally, 1A9 drug-sensitive cells were transiently transfected with a plasmid encoding Hβ l, and the effects of 2ME2 treatment on Hβl tubulin were assessed. Currently, there is no available antibody specific for Hβl tubulin; thus, a rabbit polyclonal Hβ 1 antibody was generated, which is highly specific for Hβ 1 as shown by the immunoblot in FIG. 11. The results in FIG. 12 show that the Hβl tubulin isotype was fully incorporated into interphase microtubules as evidenced by the extensive, fine and intricate Hβl microtubule network depicted in untreated 1A9 cells. In addition,
tubulin immunofluorescence with a β-tubulin antibody recognizing all β-isotypes but Hβl showed overlap between the two staining (see merge). Treatment with 2ME2 had no effect on the microtubule network of Hβl -expressing cells, even at the dose of 100 μM, as shown by the intact, well organized microtubule cytoskeleton in the transfected cells. In contrast, in the surrounding non-transfected cells, 2ME2 treatment caused extensive microtubule depolymerization with short microtubule fragments scattered throughout the cytoplasm and an increase in the number of atypical mitotic figures, consistent with the drug-sensitive phenotype of the non-transfected 1A9 cells (FIG. 5). On the other hand, treatment with either PTX or vincristine (VCR) disrupted the microtubule cytoskeleton in both Hβl transfected and non-transfected cells resulting in microtubule bundling with an increase in the density of cellular microtubules and inhibition of microtubule assembly, respectively. These results are similar to those obtained with the 2MRC cells following treatment with 2ME2 (FIG. 3) lending support to the hypothesis that Hβl tubulin remains insensitive to 2ME2. To rule out potential artifacts due to both the overexpression of Hβl as well as the presence of other β-tubulin isotypes, the 2ME2-resistance properties of Hβl in human platelets were investigated, where the Hβl isotype accounts for approximately 90% of total β- tubulin. Similar to the experiment shown in FIG. 4, a tubulin polymerization assay was performed in isolated human platelets treated with 2ME2 or PTX. As shown in FIG. 13, 2ME2 treatment of platelets had no effect on microtubule polymer mass as seen by the drug's inability to shift the dynamic Hβl tubulin equilibrium towards the soluble pool (S) of tubulin, indicative of depolymerization. In contrast, PTX treatment clearly induced microtubule polymerization shown by the increase in the percentage of polymerized tubulin (%P) from 24% in untreated cells to 59% in cells treated with 0.1 μM PTX (FIG. 13). Taken together, the data show that the binding of 2ME2 to Hβl is impaired and thus Hβl, and hence the tissues where it is expressed, would be naturally resistant to the effects of 2ME2.
The testing further illustrated that Hβl is specifically expressed in hematopoietic tissues and protects them against the effects of 2ME2. It has been reported that βl -tubulin is expressed specifically in hematopoietic tissue, such as megakaryocytes where its expression is critically important for platelet biogenesis. Most of the research reported for βl -tubulin has been focusing on the role of this isotype in mice; while there is very little expression and functional data of Hβl tubulin in humans. Thus, the expression
pattern of Hβl in 20 human normal tissues including brain, heart, lung, colon, breast, ovary, kidney, and prostate was investigated. The results by quantitative RT-PCR (FIG. 14) clearly showed that Hβl expression is restricted to hematopoietic tissues with the highest expression levels found in peripheral blood (PB) and BM followed by the spleen, small intestine and placenta. In addition, high levels of Hβ 1 expression were found in human fetal liver which is a major site of hematopoiesis in embryonic development. Collectively, our data show that Hβ 1 expression is restricted to organs involved in hematopoiesis in humans, similar to the reported expression for this β-tubulin isotype in mice.
These results together with previous data showing insensitivity of the Hβl isotype to the depolymerizing effects of 2ME2 indicated that the BM of patients receiving 2ME2 as anticancer therapy possibly would remain largely unaffected by the toxic effects of this MTA. hi accordance with this prediction, the safety data from recent oncology clinical trials have demonstrated that 2ME2 at clinically active doses is well tolerated with minimal if any side effects on myelosuppression. To directly test the hypothesis that Hβl expression in the BM confers natural resistance to 2ME2, the in vivo effects of this agent in hematopoietic tissue in mice was investigated. Importantly, the drug-resistance-conferring 1236 Hβl tubulin residue is conserved across different mammalian species, including mouse, rat, and dog. To that end, non-tumor bearing, 8-week old C57BL6 mice were treated with 2ME2 at 600 mpk, po, or 200 mpk, iv. The oral 2ME2 dose was chosen to significantly exceed the daily dose (150 mpk, po) previously reported to inhibit tumor growth and angiogenesis in vivo. The 200 mpk iv dose was chosen as the maximum injectable iv dose expected to be 100% bioavailable. Mice were also treated with effective doses of PTX and VCR serving as controls for other MTDs with profound myelosuppressive dose-limiting toxicity. Importantly, the drug-resistance-conferring 1236 Hβl tubulin residue is conserved across different mammalian species, including mouse, rat, and dog.
Confocal microscopy of BM aspirates (FIG. 15) revealed that 2ME2 did not induce any significant effect on interphase microtubules as compared to the untreated animals. In addition, the percentage of normal mitotic figures (%NM) in untreated animals (92% of total mitosis) exhibiting ring-form nuclei with condensed chromatin and commonly found in most normal mature murine leukocytes remained unchanged in the 2ME2- treated animals (94% NM for 200 mpk iv group and 85% in 600 mpk po group) (FIG. 16). In contrast, both PTX and VCR treatments resulted in significant disruption of the
microtubule cytoskeleton, shown by the distinct bundling and depolymerization, respectively. In addition, both PTX and VCR treatments resulted in a decrease in the percentage of normal mitotic figures with a concomitant significant increase (p<0.05) in the percentage of aberrant mitosis (69% and 73% respectively, as compared to 8% in untreated animals).
To investigate the effects of 2ME2 in human hematological tissue, BM aspirates were obtained from a patient with a pre -malignant disorder in the BM (MGUS: monoclonal gammopathy of undetermined significance) with a life-long risk of progression to multiple myeloma, and the BM cells were treated ex vivo with 2ME2 or PTX. Similar to the results obtained with the animal BM, treatment with 2ME2, even at concentrations 200- 1000-fold higher than those achieved clinically had no effect on the microtubule cytoskeleton; whereas PTX treatment, at doses well within the range of the drug's plasma levels in patients, resulted in near-complete microtubule disruption as seen by the accumulation of distinct perinuclear microtubule bundles (FIG. 17). Taken together these results clearly show the lack of 2ME2 activity in mouse as well as human BM, recapitulating the minor effects of 2ME2 in BM suppression in patients.
Chemotherapy-induced myelosuppression is the result of apoptosis induction in the rapidly proliferating CD34+ hematopoietic progenitor cells (HPCs) that reside in the BM and to a lesser extent in the relatively quiescent hematopoietic stem cells (HSCs). In order to extend our observations to a cell population more closely related to the stem/progenitor BM cells, cord blood was used as a rich source of primitive, undifferentiated CD34+ stem/progenitor cells. CD34+ cells were isolated from UCBCs and treated ex vivo with 2ME2 or PTX (FIG. 18). Similar to the results obtained in the BM, 2ME2 failed to induce any microtubule depolymerization in CD34+ cells, whereas PTX treatment resulted in the formation of distinct microtubule bundles, indicative of microtubule-stabilization. In addition, immunofluorescence staining with our Mi-specific antibody showed that CD34+ cells stained positive for Hβl (FIG. 19), and similar to the results in FIG. 18, 2ME2 did not induce microtubule-depolymerization on either interphase or mitotic microtubules. Collectively, the results show that in humans, Hβl is selectively expressed in hematopoietic tissues, confers natural protection against the depolymerizing effects of 2ME2, and provide a molecular explanation for the lack of myelosuppression associated with 2ME2 treatment in cancer patients.
The described testing thus identified that the β-tubulin residue Valβ236 is important for 2ME2 binding, and its mutation to He leads to more than 80-fold resistance to 2ME2. Molecular modeling of 2ME2 binding to tubulin revealed that the β236 residue is within van der Waals contact of the C-and D-rings of 2ME2 and that its mutation to He decreases available ligand space in an already tight volume at the plus end of Helix 7 in the protein thus precluding 2ME2 from binding. Therefore, cells that functionally express only this mutant tubulin, like the 2MRC cells, will remain insensitive to 2ME2 due to impaired drug-tubulin interaction. Molecular modeling predicts that the 2ME2 analog, ENMDl 198, is tolerated by the Iβ236 mutation since its unique A-ring functionality relative to 2ME2 can make productive interactions with GTP and its binding site, avoiding, at the same time, a steric clash with 1236. In agreement with the modeling data, the cytotoxicity analysis shows that the 2MRC cells harboring the Iβ236 mutation remain sensitive to the effects of ENMDl 198, suggesting that this residue does not impair this drug's binding to tubulin, in agreement with the ENMDl 198-induced myelosuppression and neutropenia seen clinically
Importantly, the Iβ236 2ME2 -resistance conferring residue is naturally encoded by the class VI β-tubulin, or Hβl isotype, in humans. Hβl-tubulin has, in addition to the 1236, two other residues that are different from the tubulin in the original X-ray structure as well as from the ubiquitous HM40 tubulin, namely Y200F and V3161. These additional amino acid changes not present in the HM40 tubulin in the drug-resistant 2MRC cells render the 2ME2 -binding pocket in the Hβl more hydrophobic, and further prevent 2ME2 from occupying the site.
These results suggest that the presence of all three binding site modifications (F200, 1236, and 1316) in Hβl, would greatly impact the binding of 2ME2 rendering this hematopoietic-specific isotype insensitive to the effects of the drug.
Not much is actually known about the role of the human Hβl tubulin, as this isotype has been primarily studied in mice (Mβl) where its expression is hematopoietic specific, restricted to the marginal band of platelets and to mature megakaryocytes as well as fetal erythroblast. Functionally, Mβl plays an important role in the biogenesis of platelets from terminally differentiated megakaryocytes a function regulated by the transcription factor NF -E2, as well as other transcription factors. In addition, Mβl is required to reorganize platelet marginal bands correctly during platelet activation. Moreover, mice deficient in β 1 -tubulin develop moderate thrombocytopenia as a result of
reduced proplatelet formation and their spherocytic platelets carry a structurally defective marginal band and reduced microtubule content. In humans, the role of βl -tubulin is limited to a few clinical studies showing that a Q43P polymorphism in Hβl is present in approximately 11% of the general population and in approximately 24% of a small cohort of patients with macrothombocytopenia. Heterozygous carriers show defects in platelet aggregation that protects against thrombotic disorders and appears to reduce the risk of cardiovascular disease in men, but increases the risk of intracerebral hemorrhage. Hβl expression is restricted to hematopoietic tissues, with the highest levels found in peripheral blood leukocytes followed by the BM and to a lesser extend the spleen and the placenta, all of them well known hematopoietic tissues. In agreement with the mRNA expression pattern of Hβl, the testing described herein showed that Hβl protein is present in the marginal band of platelets, as well as in the BM and the CD34+ isolated UCBC cells, using a specifically generated Hβl -specific antibody (FIG. 11, FIG. 12, and FIG. 19). The functional data in FIG. 12 and FIG. 13 further show that the 1236 harboring Hβl tubulin, whether naturally present in platelets or exogenously expressed in 1A9 cells, is not sensitive to the depolymerizing effects of 2ME2, lending further support to the drug- binding-impairing role of Iβ236.
To further confirm that the lack of drug-induced myelosuppression and neutropenia associated with 2ME2 clinical use stems from the inability of this drug to affect Hβl tubulin in the BM of patients and especially in the CD34+ HSP cell niche, the BMs of mice and humans following systemic or ex vivo 2ME2 administration, respectively, were analyzed, hi both cases, no cytoskeletal effect was detected by the 2ME2 treatment, in contrast to the robust microtubuledisruption observed with other MTAs such as PTX and VCR. Hβl showed negligible expression in all of the different tumor types analyzed including lung, prostate, ovary, breast, and larynx. These results likely indicate that
2ME2, although not causing neutropenia, would still be expected to be effective in these tumors. Further, CD34+ UCBC cells were isolated, which similarly to the treated BM cells did not show any microtubule-perturbation following ex vivo treatment with 2ME2. PTX treatment on the other hand had profound effects on microtubule-stabilization, a result consistent with the essentially unaltered taxane binding site in Hβ 1. Molecular modeling predicts that ENMDl 198 is tolerated by the Iβ236 mutation since its unique A-ring functionality relative to 2ME2 can make productive interactions with GTP and its binding site, avoiding, at the same time, a steric clash with 1236. In agreement with the modeling
data, the cytotoxicity analysis showed that the 2MRC cells harboring the Iβ236 mutation remain sensitive to the effects of ENMDl 198, suggesting that this residue does not impair this drug's binding to tubulin, a finding consistent with the ENMDl 198-induced myelosuppression and neutropenia seen clinically. The knowledge gained herein provides a new molecular insight into mechanisms of drug-induced myelosuppression that has in part led to the new MTCs described herein that are devoid of myelosuppressive side effects, but retaining anticancer activity.
EXAMPLE 2 Preparation of Epothilone Compounds
Preparation of (S)-BINAP-RuBr2 catalyst. A degassed solution of acetone (8 mL) was added to equimolar amounts of (5)2,2'-bis(diphenylphosphino)-l, r-binaphthyl [(S)- BINAP] (32 mg) and bis-(methylallyl)-l,5-cyclooctadiene ruthenium (II) (16 mg). To the resulting suspension, a solution of HBr in methanol (0.29 M, 0.44 mL, 2.5 eq) was added and the resulting reaction mixture was stirred for 1 h. The volatiles were concentrated in vacuum to provide the catalyst as a light brown solid, which was used directly for the reduction.

(a) (b)
Methyl (3S)-5-benzyloxy-3-hydroxypentanoate (14). (S)-BINAP-RuBr2 catalyst was added to a solution of the ketoester (a) (3.38 g, 14 mmol) in CH3OH (30 mL) and the resulting mixture was hydrogenated (Parr hydrogenator) at 50 psi for 2 days. The insoluble material was removed by filtration through a Celite pad, and the filtrate was concentrated under reduced pressure. The crude material obtained was subjected to column chromatography over silica gel (10-25% EtOAc in Hexanes) to provide compound (b) (2.08 g, 95% yield based on unrecovered starting material). [α]D + 9.6 (c 5.4, CHCl3).
Reported 1I a]D + 11 (CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.32-7.21 (m, 5H), 4.50 (s, 2H), 4.25-4.22 (m, 2H), 3.73-3.62 (m, IH), 3.69 (s, 3H), 2.90 (br.s, IH), 2.51 (d, J = 8.8 Hz, 2H), 1.85-1.70 (m, 2H).

(b) (C)
Methyl (3S)-3,5-dihydroxypentanoate (15). Pd-C (10%, 0.6 g) was added to the solution of the benzyl ether (b) (3.4 g, 14.2 mmol) in CH3OH (20 mL), and the resulting mixture was hydrogenated in a sealed tube at 35 psi for 4 h. The insoluble material was removed by filtration through a silica gel pad, and the filtrate was concentrated in vacuo to give residue, which was subjected to silica gel chromatography with 50% ethyl acetate in hexanes to provide the diol of compound (c) (2.1 g, 100%). [α]D + 19.13 (c 1.8, CHCl3). 1H NMR (400 MHz, CDCl3) δ 4.20 (m, IH), 3.90 (br.s, IH), 3.75 (m, 2H), 3.65 (s, 3H), 3.40 (br.s, IH), 2.46 (dd, J= 5.8, 1.2 Hz, 2H), 1.65 (q, J = 5.6 Hz, 2H). 13CNMR (100 MHz, CDCl3) 8 173.2, 67.4, 60.5, 51.9, 41.7, 38.1.
To the solution of the diol (1.9 g, 6.75 mmol) obtained from the above reaction, in CH2CI2 (15 mL) was sequentially added imidazole (0.69 g, 10 mmol, 1.5 eq) and TBSCl (1 g, 6.75 mmol, 1 eq), and the reaction was allowed to proceed at 25 0C with stirring for 30 min. A saturated solution of aqueous NaHCO3 was added to quench the reaction and the mixture was extracted with EtOAc (20 mL x 3). The combined organic extracts were dried (Na2SO4), and the solvent was removed in vacuo. The crude product obtained was subjected to silica gel column chromatography eluting with 20-25% EtOAc in hexanes to afford compound (c) (1.7 g, 95% yield) as a colorless oil. [α]D + 9.6 (c 0.74, CHCl3). 1H NMR (400 MHz, CDCl3) 8 4.20 (m, IH), 3.80 (m, 211), 3.62 (s, 311), 3.60 (d, J = 1.2 Hz, IH), 2.44 (m, 2H), 1.63 (m, 211), 0.82 (s, 9H), 0.01 (s, 6H). 13C NMR (100 MHz, CDCl3) 8 172.8, 67.7, 61.6, 51.7, 41.8, 38.3, 26.0, 18.3, -5.35, -5.37. HRFABMS: calcd for Cj2H27O4Si (M+H) 263.1679, found 263.1669.

Methyl (S)-24(S)-l-hydroxy-3-(tert-butyl-dimethylsilyloxy)propyl)pen-4-enoate. A solution of LDA (2 M, 3.96 mmol, 2.6 eq) in tetrahydrofuran (THF) was added to a solution of compound (c) (0.4 g, 1.52 mmol) in THF (15 mL) at -78 0C, and the resulting solution was allowed to warm to -20 0C and stir for 30 min at -20 0C. AUyI. iodide (0.2 mL, 2.28 mmol, 1.5 eq) in hexamethylphosphoramide (HMPA) (0.76 mL, 4.27 mmol, 1.08 eq to LDA) was added to the above reaction mixture that was recooled to -78 0C. The subsequent reaction mixture was warmed to -20 0C, and the reaction proceeded at -20 0C with stirring for 1 h prior to being quenched by the addition of saturated NH4Cl solution (50 mL). The two layers were separated and aqueous phase was extracted with ether (20 x 3). The combined organic extracts were washed with water and brine, the organic fraction was dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure to give the crude mass. Purification of the product by silica gel column chromatography eluting with 10% ethyl acetate in hexanes yielded methyl -2-allyI-3-hydroxy-5-O- 'butyldimethylsilyloxy pentanoate (0.336 g, 72%). [α]D + 5.4 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) 8 5.70 (m, IH), 5.04 (dd, J = 17.2, 1.6 Hz, IH), 5.96 (dt, J- 10.0, 0.8 Hz, IH), 3.92 (m, IH), 3.82 (m, IH), 3.75 (m, IH), 3.64 (s, 311), 3.40 (d, J= 4.4 Hz, IH), 2.51 (m, IH), 2.32 (m, 211), 1.66 (m, 2H), 0.83 (s, 9H), 0.01 (s, 6H). 13CNMR (100 MHz, CDCl3) 8 174.9,
135.2, 1 17.1, 71.6, 61.9, 51.7, 51.4, 36.6, 33.2, 26.0, 18.3, -5.3. HRFABMS: calcd for Ci5H31O4Si (M+H) 303.1992, found 303.2002.
Methyl (R)-2((S)-l-hydroxy-3-(terr-butyl-dimethylsilyloxy)propyl)-2- methylpen-4-enoate - compound (d). To a freshly prepared solution of LDA (0.6 M, 15.73 mmol, 2.6 eq) in THF (24 mL), a solution of methyl-2-allyl-3- hydroxy-5-O- 'butyldimethylsilyloxy pentanoate, obtained from the above reaction, (1.83 g, 6 mmol) in THF (15 mL) was added at -78 0C, and the resulting solution was warmed to -20 0C and stirred for 4 h at that temperature. Then a solution of methyl iodide (0.6 mL, 9.69 mmol, 1.6 eq) in HMPA (17 mmol, 1.08 eq to LDA) was added to the above reaction
mixture that was re -cooled to -78 0C. The subsequent reaction mixture was re -warmed to - 20 0C and allowed to stir at that temperature for 2 h. The reaction was quenched by the addition of saturated ammonium chloride (50 mL). The two layers were separated and aqueous phase was re-extracted with ether (20 mL x 3). T he combined organic phases were washed with water and brine, the organic fraction was dried (TS^SO4), and the solvents were removed under reduced pressure. The crude oil obtained was subjected to silica gel column chromatography using 5% ether in hexane as eluent to provide compound (d). (major) (1.09 g, 57% yield) (minor) (0.35 g, 18% yield). Compound (d): [α]D + 17.4 (c 1.3, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.70 (m, IH), 5.04 (dd, /= 17.2, 1.6 Hz, IH), 3.98 (dd, J= 10.8, 0.8 Hz, IH), 3.93 (br.d, J= 10.4, IH), 3.85-3.71 (m, 2H), 3.60 (s, 3H), 3.43 (d, J= 2.4 Hz, 1 11), 2.47 (dd, J = 13.6, 7.2 Hz,, IH), 2.26 (dd, J= 13.6, 7.2 Hz, IH), 1.66-1.54 (in, IH), 1.45(dt, J = 12.8, 1.0 Hz, IH), 1.19 (s, 3H), 1.08 (s, 9H), 0.8 (s, 6H). 13CNMR (IOO MHz, CDCl3) 8δ 176.2, 134.4, 118.0, 75.7, 63.0, 51.7, 51.3, 40.8, 34.3.2, 26.0, 18.3, 16.2, -5.3. HRFABMS: calcd for Ci6H33O4Si (M+H) 317.2148, found 317.2166.

Methyl (R)-2-((S)-l,3-bis(terr-butyldimethylsilyloxy)propyl)-2-methylpen-4- enoate. To a solution of compound (d) (1.97 g, 6.23 mmol) in dichloromethane (25 mL) was added 2,6-lutidine (1.15 mL, 9.96 mmol, 1.6 eq) and tert-butyldimethylsilyl triflate (TBSOTf) (2.24 mL, 9.35 mmol, 1.5 eq) at -78 0C, and the resulting reaction mixture was allowed to stir at -78 0C over 6 h. Saturated ammonium chloride (50 mL) was added to quench the reaction. The organic layer was separated and the aqueous phase was re- extracted with dichloromethane (20 mL x 3). The combined dichloromethane phase was dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. The crude product obtained was subjected to silica gel chromatography eluting with 5% ether in hexanes to furnish bis-TBS ether (2.22 g, 83%). [α]D + 5Λ (c 1, CHCl3). 1FINMR (400 MHz, CDCl3)δ 5.62 (m, IH), 4.99-4.95 (m, 2H), 3.98 (dd, J= 8.0, 3.2 Hz, IH), 3.58 (s, 3H), 3.61-3.51 (m, 2H), 2.38 (dd, J= 13.6, 7.2 Hz, IH), 2.22 (dd, T 13.6,
7.2 Hz, IH), 1.62-1.44 (m, 2H), 1.04 (s, 3H), 0.85 (s, 911), 0.84 (s, 9H), 0.06 (s, 311), 0.04 (s, 3H), -0.009 (s, 3H), -0.01 (s, 3H). 13CNMR (100 MHz, CDCl3) δ 176.0, 134.5, 117.9, 73.6, 60.0, 52.8, 51.6, 42.3, 37.7, 26.2, 26.0, 18.5, 18.4, 14.8, -3.6, -3.8, -5.1.
(S)-2((S)-l,3-bis(terf-butyldimethylsilyloxy)propyl)-2-methylpen-4-en-l-ol - compound (e). A solution of diisobutylaluminum hydride (DIBAL-H) (IM, 17 mmol, 3.5 eq) was added dropwise to a solution of bis-TBS ether (2.08 g, 4.83 mmol), obtained from the above reaction, in dichloromethane (45 mL) at -78 0C. The reaction was allowed to proceed at -78 0C with stirring for 45 min. Methanol (5 mL) was added and the solution was allowed to warm to 25 0C. Then a saturated solution of sodium/potassium tartrates (50 mL) was added to the mixture, which was stirred at 25 0C until the two layers were clearly separated. Organic layer was separated and the aqueous phase was re-extracted with dichloromethane (30 mL x 3). The combined organic extracts were dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. Purification of the crude product obtained by silica gel column chromatography with 3 % EtOAc in hexanes as eluent to yield 17 (1.83 g, 94%). [α]D -16.9 (c 0.87, CHCl3). 'H NMR (400 MHz, CDCl3) δ 5.80 (m, IH), 5.05-5.01 (m, 21-1), 3.77-3.63 (m, 4H), 3.32 (dd, J = 11.2, 6.8 Hz,, IH), 2.97 (t, J= 4.4 Hz, IH), 2.25-1.91 (m, 311), 1.67-1.58 (m, IH), 0.98 (s, 3H), 0.90 (s, 18H), 0.099 (s, 31-1), 0.092 (s, 3H), 0.06 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 134.6, 117.8, 76.1, 68.5, 60.8, 42.2, 39.7, 36.2, 26.3, 26.1, 19.4, 18.4, -3.7, -3.9, -5.0, -5.1.

(R)-2((S)-l,3-bis(tert-butyldimethylsilyloxy)propyl)-2-methylpent-4-enal. To a solution of compound (e) (1.8 g, 4.5 mmol) in a 1 : 1 mixture of CH2Cl2 and dimethyl sulfoxide (DMSO) (40 mL), was added triethylamine (3.16 mL, 22.7 mmol, 5eq) followed by the sulfur trioxide-pyridine complex (SO3.Py) (3.16 g, 22.78 mmol, 5 eq) at 0 0C and the resulting reaction mixture was stirred for 30 min. The reaction was quenched by the
addition of saturated NH4Cl solution (50 mL). Organic layer was separated and the aqueous phase was extracted with dichloromethane (20 mL x 3). The combined organic phases were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified via silica gel column chromatography eluting with 5 % ether in hexanes to yield (R)-2-((5)-l,3-bis(tertbutyldimethylsilyloxy)propy l)-2- methylpent-4-enal (1.75 g, 97%). [α]D -3.8 (c 1.1 , CHCl3). 1H NMR (400 MHz, CDCl3) δ 9.60 (s, IH), 5.65 (m, IH), 5.08-5.03 (m, 2H), 4.00 (dd, J = 7.6, 3.2 Hz, IH), 3.68-3.56 (m, 2H), 2.50 (dd, J= 14.2, 6.8 Hz, IH), 2.25 (dd, J= 14.2, 6.8 Hz, IH), 1.76- 1.66 (m, IH), 1.65-1.56 (m, IH), 1.00 (s, 3H), 0.885 (s, 9H), 0.880 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 206.7, 133.7, 118.4, 72.9, 59.7, 54.3, 37.2, 37.0, 26.2, 26.1, 18.5, 18.4, 15.7, -3.6, -4.0, -5.11, -5.13.
(4S)-4-((S)-l,3-bis(tert-butyldimethylsilyloxy)propyl)-4-methylhept-6-en-3-ol. To a solution of (R)-2((S)-l,3-bis(tert-butyldimethylsilyloxy)propyl)-2-methylpent-4-enal (1.5 g, 3.75 mmol), obtained from the above reaction, in THF (20 mL) was added ethyl magnesium bromide (1 M, 6.56 mL, 6.56 mmol, 1.7 eq) at 0 0C and the resulting reaction mixture was allowed to stir at 0 0C for 1 h. Saturated NH4Cl solution (50 mL) was added to quench the reaction. Organic layer was separated and the aqueous phase was re- extracted with dichloromethane (20 mL x 3). The organic extracts were combined and dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure. The crude product obtained was purified via silica gel column chromatography eluting with 2.5 % ether in hexanes to produce the product as a diastereomeric mixture (9: 1) (68%) and the alcohol of compound (e), as an undesired byproduct (31%). The diastereomeric mixture of (4S)-((S)-l,3-bis(tertbutyldimethylsilyloxy)propyl)-4-methylhept-6-en-3-ol was subjected to the next reaction without further purification. HRFABMS : calcd for C23H5i03Si2 (M+H) 431.3371, found 431.3372.
(R)-4((S)-l,3-bis(tert-butyldimethylsilyloxy)propyl)^t-methylhept-6-en-3-one - compound (f). To a solution of (4S)-4- ((S)-1, 3-bis(tert-butyldimethylsilyloxy)propyl)- 4-methylhept-6-en-3-ol (1.545 g, 3.59 mmol) in a 1 : 1 mixture Of CH2Cl2 and DMSO (36 mL), was added triethylamine (2.50 mL, 17.97 mmol, 5 eq) followed by SO3. Py complex (2.82 g, 17.96 mmol, 5 eq) at 0 0C. The reaction was allowed to proceed at 0 0C with stirring for 2 h prior to being quenched by the addition of saturated NH4Cl solution (50 mL). Organic layer was separated and the aqueous phase was extracted with
dichloromethane (20 mL x 3). The combined organic extracts were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure to give crude product, which was purified via silica gel chromatography eluting with 3% ether in hexanes to give compound (f) (1.16 g, 75%). [α]D -0.0 (c 1.8, CHCl3). Η NMR (400 MHZ, CDCl3) δ 5.60 (m, IH), 5.00-4.97 (m, 2H), 4.05 (dd, J = 8.0, 2.8 Hz, IH), 3.58 (dd, J = 8.0, 4.8 Hz, 2H), 2.55-2.35 (m, 3H), 2.22 (dd, J = 14.2, 6.8 Hz, IH), 1.52-1.35 (m, 2H), 1.09 (s, 31-1), 0.98 (t, J = 7.2 Hz, 3H), 0.90 (s, 9H), 0.87 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H), 0.02 (s, 3H), 0.01 (s, 3H). 13CNMR (100 MHz, CDCl3) 5 214.6, 134.3, 117.9, 73.3, 59.8, 57.4, 42.3, 37.8, 32.8, 26.3, 26.0, 18.6, 18.4, 14.9, 7.6, -3.5, -3.6, -5.13, -5.15. HRFABMS: calcd for C23H49O3Si2 (M+H) 429.3220, found 429.3208.

(f) (h)
Aldol product (h). A solution of ketone compound (f) ( 1070 mg, 2.50 mmol, 2.3 eq to aldehyde compound (g)) in THF (6 mL) was added to a solution of freshly prepared LDA 0.6 M, 2.875 mmol, 1.15 eq to compound (f)) in THF at -78 0C. After stirring for 1 h at -78 0C, the solution was allowed to warm up to -40 0C and stir at that temperature for 30 min. The reaction mixture was then recooled to -78 0C and a cold (-78 0C) solution of aldehyde (g) (753 mg, 1.086 mmol) in THF (6 mL) was rapidly introduced to the above reaction mixture. Upon completion of the addition, stirring was continued for a further 5 min before the reaction was quenched by the rapid injection of AcOH (0.31 mL, 5.21 mmol, 4.8 eq) as a solution in THF (1 mL). The whole reaction mixture was warmed to 25 0C and partitioned between ether and saturated aqueous NH4Cl solution. The aqueous phase was re-extracted with ether (20 mL x 3), and the combined organic extracts were dried (Na2SO4), and the solvents were removed under reduced pressure. Flash column
chromatography (silica gel, 3 to 20% ether in hexanes) of crude mass obtained provided compound (h) (884 mg, 74%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J= 8.8, Hz, 6H), 7.24-7.15 (m, 9H), 6.86 (s, IH), 6.46 (s, IH), 5.61 (m, IH), 5.56 (t, J= 7.2 Hz, IH), 4.98-4.95 (m, 2H), 4.14 (t, J= 6.8 Hz, IH), 3.83 (dd, J= 7.6, 2.4 Hz, IH), 3.65- 3.50 (m, 2H), 3.39 (br.s, 2H), 3.27 (br.s, IH), 3.19-3.12 (m, 2H), 2.67 (s, 3H), 2.44-2.24 (m, 3H), 2.06-1.99 (m, 2H), 2.01 (s, 3H), 1.70-1.57 (m, 311), 1.39-1.34 (m, 2H), 1.31-1.11 (m, 2H), 1.09-1.00 (m, 2H), 0.94 (d, J= 6.8 Hz, 3H), 0.89 (s, 3H), 0.87 (s, 9H), 0.86 (s, 9H), 0.85 (s, 9H), 0.65 (d, J= 6.8 Hz, 3H), 0.08 (s, 3H), 0.07 (s, 3H), 0.04 (s, 3H), 0.004 (s, 6H), 0.01 (s, 3H). 13C NMR (IOO MHZ, CDCl3) 8 222.4, 165.0, 153.9, 145.1, 143.2, 138.9, 134.8, 129.5, 128.4, 127.5, 123.2, 119.5, 119.1, 115.8, 87.2, 79.5, 75.0, 74.4, 68.1, 61.2, 61.0, 58,2, 43.2, 42.1, 38.5, 36.1, 35.8, 33.8, 30.5, 29.8, 26.9, 26.7, 26.6, 26.3, 21.8, 19.9, 19.2, 19.0, 17.9, 15.9, 15.0, 14.8, 9.6, -2.8, -3.2, -3.8, -4.0, -4.47, -4.49. HRFABMS: calcd for C66H103NO6SSi3 (M+Na) 1144.6712, found 1144.6646.

(h) G)
Tetrakis-[tert-butyldimethylsilyl] -ether - compound Q). To a solution of compound (h) (864 mg, 0.77 mmol) in dichloromethane (8 mL) was added 2,6-lutidine (0.38 mL, 3.3 mmol, 4.3 eq) followed by TBSOTf (0.57 mL, 2.46 mmol, 3.2 eq) at 0 0C, and the reaction was allowed to proceed with stirring for 2 h at 0 0C. Saturated NaHCO3 solution was added to quench the reaction. Two layers were separated and the aqueous layer was re-extracted with ether (20 mL x 3). The combined organic extracts were dried (Na2SO4), and the solvents were removed under reduced pressure. The crude mass obtained was subjected to column chromatography over silica gel, eluting with 5% ether in hexanes, to yield the tetrakis-TBS ether of compound (i) (908 mg, 95%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ7.39 (d, J= 6.8 Hz, 611), 7.22-7.16 (m, 9H), 6.85 (s, IH), 6.46 (s, IH), 5.59-5.50 (m, 211), 4.92 (d, J= 3.2 Hz, IH), 4.89 (d, J= 11.6 Hz, IH), 4.14 (t, J= 6.0 Hz, IH), 3.73 (dd, J= 7.6, 2.4 Hz, 111), 3.63-3.50 (m, 311), 3.453.38 (m, 2H), 3.01 (m, IH), 2.66
(s, 3H), 2.39-2.23 (m, 31-1), 2.02-1.88 (m, 2H), 2.01 (s, 314), 1.70-1.60 (m, IH), 1.40-1.28 (m, 2H), 1.30-1.15 (m, 4H), 1.17 (s, 314), 1.01-0.96 (m, IH), 0.97 (d, J= 6.8 Hz, 31-1), 0.86 (s, 91-1), 0.855 (s, 9H), 0.85 (s, 911), 0.84 (s, 9H), 0.75 (d, J= 6.8 Hz, 3H), 0.05 (s, 3H), 0.04 (s, 3H), 0.03 (s, 311), 0.01 (s, 3H), -0.010 (s, 3H), -0.015 (s, 6H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3) 5 217.2, 164.4, 153.4, 144.6, 142.5, 138.3, 134.6, 128.9, 127.9, 127.0, 122.9, 1 19.0, 118.2, 115.3, 86.7, 78.9, 76.5, 74.7, 67.6, 60.6, 56.9, 45.5, 41.2, 40.0, 37.8, 35.3, 31.8, 29.5, 26.8, 26.47, 26.42, 26.2, 26.1 19.4, 18.7, 18.69, 18.51, 18.48, 17.2, 14.3, 14.2, -3.3, -3.4, -3.6, -3.7, -4.3, -4.5, -4.9. HRFABMS: calcd for C72 H118NO6SSi4 (M+H) 1236.7757, found 1236.7673.

Tris-[tert-butyldimethylsilyl] -ether - compound (k). To a solution of compound (j) (908 mg, 0.735 mmol) in THF (7.5 mL) was added a stock solution of hydrogen fluoride in pyridine (HF.Py) (this stock solution was prepared by addition of 1.25 mL HF.Py to 3.5 mL pyridine in 6.125 mL THF) at 0 0C. The resulting reaction mixture was warmed to 25 0C by removing the ice-bath and allowed to stir at that temperature for 2 h. Saturated NaHCO3 solution (50 mL) was added to quench the reaction, and this mixture was extracted with ethyl acetate (30 mL x 3). The combined organic extracts were dried over anhydrous Na2SO4, the solvents were removed under reduced pressure. The crude product obtained was subjected to column chromatography over silica gel employing 14% EtOAc in hexanes as eluent to yield the desired primary alcohol of compound (k) (793 mg, 96%) as a colorless syrup. IH NMR (400 MHz, CDCl3) δ 7.40 (d, J= 8.0 Hz, 6H), 7.23-7.15 (m, 9H), 6.86 (s, IH), 6.47 (s, IH), 5.58-5.48 (in, 2H), 4.93 (d, J= 14.0 Hz, 3H), 4.98 (d, J= 17.2 Hz, 3H), 4.14 (t, J= 6.8 Hz, IH), 3.94 (dd, J= 6.8, 3.2 Hz, IH), 3.63-3.59 (m, 3H), 3.50-3.38 (m, 2H), 3.02-2.96 (m, IH), 2.66 (s, 311), 2.42-2.23 (m, 311), 2.05-1.94 (m, IH), 2.00 (s, 3H), 1.65-1.47 (m, 3H), 1.311.16 (m, IH), 1.18 (s 31-1), 1.06-0.94 (m, 2H), 0.99 (d, J= 6.8 Hz, 31-1), 0.865 (s, 911), 0.859 (s, 9H), 0.84 (s, 9H),
0.75 (d, J= 6.4 Hz, 3H), 0.06 (s, 31-1), 0.049 (s, 311), 0.048 (s, 3H), 0.024 (s, 311), -0.008 (s, 3H), -0.02 (s, 311). 13CNMR (IOO MHz, CDCl3) 5218.2, 164.5, 153.4, 144.5, 142.5, 138.3, 133.9, 128.9, 127.9, 127.0, 122.9, 119.0; 1 18.3, 115.3, 86.7, 78.9, 76.1, 73.2, 67.6,
60.3, 57.2, 45.2, 41.0, 40.0, 38.1, 35.3, 31.7, 29.5, 26.7, 26.4, 26.3, 26.1 19.4, 18.7, 18.6,
18.4, 17.5, 17.05, 14.5, 14.2, -3.5, -3.6, -3.8, -4.3, -4.6. HRFABMS: calcd for C66H104NO6SSi3 (M+l-1) 1122.6892, found 1122.6902.

Oxidation of compound (k) to aldehyde compound (1). To a solution of primary alcohol obtained in the preceding step (777 mg, 0.693 mmol) in 1:1 mixture of dichloromethane and DMSO (7 mL), was added triethylamine (0.48 mL, 3.465 mmol, 5 eq) followed by SO3. Py (551 mg, 3.465 mmol, 5 eq) at 0 0C and the reaction was allowed to proceed at 0 0C with stirring for 1 h. Saturated NH4Cl solution (30 mL) was added to quench the reaction, and the mixture was extracted with ether (20 mL x 3). The combined organic extracts were dried over anhydrous Na2SO4, and the organic solvents were removed under reduced pressure. The crude product obtained was purified via silica gel column chromatography employing 15 % ether in hexanes as eluent to give compound (1) (722 mg, 93%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 9.70 (s, 1 H), 7.40 (d, J= 7.2 Hz, 6H), 7.23-7.15 (m, 9H), 6.86 (s, 1 H), 6.47 (s, IH), 5.56-5.46 (m, 2H), 4.96 (d, J= 9.2 Hz, IH), 4.91 (d, J= 18.4 Hz, IH), 4.34 (t, J= 4.4 Hz, 111), 4.14 (t, J= 6.0 Hz, IH), 3.59 (dd, J= 6.8, 3.2 Hz, IH), 3.43 (d, J= 12.0 Hz, IH), 3.39 (d, J= 12.0 Hz, IH), 3.02-2.97 (m, IH), 2.66 (s, 3H), 2.60 (dd, J= 18.4, 4.8 Hz, IH), 2.40-2.25 (m, 4H), 2.01-1.83 (m, 3H), 2.00 (s, 3H), 1.58-1.33 (m, 2H), 1.20-0.92 (m, 21-1), 0.97 (d, J= 7.2 Hz, 3H), 0.865 (s, 9H), 0.835 (s, 9H), 0.829 (s, 9H), 0.75 (d, J= 6.8 Hz, 3H), 0.06 (s, 3H), 0.05 (s, 3H), 0.01 (s, 3H), -0.008 (s, 3H), -0.02 (s, 6H). 13CNMR (100 MHz, CDCl3) δ 217.5, 200.9, 164.5, 153.4, 144.5, 142.5, 138.3, 133.4, 128.9, 127.9, 127.0, 122.9, 119.0, 127.9, 127.0, 122.9, 119.0, 49.3, 45.4, 40.6, 39.9, 35.3, 31.7, 29.4, 26.7, 26.4, 26.3, 26.1, 19.4, 18.7,
18.4, 18.3, 18.2, 17.1, 14.2, -3.5, -3.7, -3.9, -4.2, -4.3, -4.6.

Oxidation of compound (1) to carboxylic acid compound (m). To a solution of compound (1) (722 mg, 0.645 mmol) in t-BuOH:H20 (4.5: 1, 25.3 mL) was added sequentially, 2-methyl-2-butene (5.11 mL, 48.9 mmol, 75 eq) in THF (15 mL), NaH2PO4 (271 mg, 2.26 mmol, 3.5 eq), and NaClO2 (511 mg, 4.5 mmol, 7 eq). The reaction was allowed to proceed with stirring at 25 0C for 1 h, then volatiles were removed under reduced pressure, and the residue was partitioned between ethyl acetate and brine solution. Two layers were separated and aqueous layer was re-extracted with ethyl acetate (20 mL x 3). The combined organic extracts were dried over anhydrous Na2SO4, and the organic solvents were removed in vacua pressure to furnish the crude carboxylic acid of compound (m) (836 mg) as oil. This crude product was used directly for the next reaction without further purification.

Hydroxy acid - compound (n). A solution of the carboxylic acid of compound (m) (836 mg, ca. 0.645 mmol) in THF (6.5 mL) at O 0C was treated with tetra-w- butylammonium fluoride (TBAF) (3.87 mL, 1 M in THF, 3.87 mmol, 6 eq) and then the mixture was allowed to warm up to 25 0C. The reaction was quenched after being stirred for 21 h at 25 0C by the addition of saturated aqueous NH4Cl solution (30 mL), and the
mixture was extracted with EtOAc (20 mL x 3). The combined organic extracts were dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure to give crude product. Column chromatography of this crude product over silica gel, eluting with 50% ethyl acetate in hexanes, yielded hydroxyacid (n) (581 mg, 88% for two steps) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J= 7.6, Hz, 6H),
7.24-7.15 (m, 9H), 6.86 (s, IH), 6.46 (s, IH), 5.61-5.51 (m, 2H), 5.01-4.93 (m, 2H), 4.38 (t, J = 4.4 Hz, IH), 4.18 (t, J = 6.4 Hz, IH), 3.66 (dd, J = 6.8, 3.2 Hz, IH), 3.56 (d, J = 12.0 Hz, IH), 3.52 (d, J= 12.0 Hz, IH), 3.12-104 (m, IH), 2.98 (s, 3H), 2.54-2.37 (m, 4H), 2.35-2.25 (m, IH), 2.17-2.05 (m, 2H), 2.07 (s, 3H), 2.05-1.95 (m, IH), 1.92-1.84 (m, 3.1-1), 1.72-1.41 (m, 6H), 1.13 (s, 3H), 1.03 (d, J = 7.2 Hz, 3H), 0.85 (s, 9H), 0.83 (s, 9H), 0.79 (d, J= 6.8 Hz, 3H), 0.09 (s, 3H), 0.08 (s, 3H), 0.02 (s, 3H), -0.01 (s, 3H). 13C NMR (IOO MHZ, CDCl3) 5 217.1, 165.5, 152.8, 144.6, 142.3, 140.8, 134.1, 129.0, 128.0, 127.2, 121.8, 119.1, 118.7, 115.7, 92.6, 87.0, 76.8, 73.5, 67.7, 57.4, 45.2, 39.9, 37.0, 34.2, 31.9, 29.5, 27.0, 26.5, 26.3, 26.0, 23.7, 19.1, 18.8, 18.6, 17.7, 17.3, 15.2, 15.1, -3.4, -3.6, -4.2. HRFABMS: calcd for C60H88NO7SSi2 (M+H) 1022.5820, found 1022.5796.

(n) (o)
Macrolactone - compound (o). To a solution of hydroxyacid compound (n) (581 mg, 0.569 mmol) in THF (6 mL) was added triethylamine (0.48 mL, 3.45 mmol, 6 eq), followed by 2,4,6-trichlorobenzoyl chloride (0.21 mL, 1.36 mmol, 2.4 eq) at 0 0C and the resulting reaction mixture was stirred for 1 h at that temperature. The mixture was then added to a solution of 4-(dimethylamino)pyridine (DMAP) (153 mg, 1.25 mmol, 2.2 eq) in toluene (115 ml, 0.005 M based on 2Oe) at 75 0C over 2 h, via syringe pump. After addition was complete, the reaction mixture was stirred for an additional 2 h. Toluene was removed under reduced pressure and the residue was filtered through a short plug of silica gel, eluting with 50% ether in hexanes, to give a crude product. Purification of this product by
column chromatography over silica gel, eluting with 3 to 20% ether in hexanes, furnished lactone compound (o) (312 mg, 54.6%) as a colorless syrup. 1H NMR (400 MHz, CDCl3) δ 7.45-7.40 (m, 6H), 7.26-7.19 (m, 9H), 6.96 (s, IH), 6.56 (s, IH), 5.58 (m, 21-1), 5.03-4.99 (m, 2H), 3.85 (d, J= 8.4 Hz, IH), 3.50-3.44 (m, 3H), 3.01 (m, 111), 2.82-2.58 (m, 2H), 2.71 (s, 3H), 2.40-2.15 (m, 3H), 2.13 (s, 3H), 1.45 (m, 3H), 1.30 (m, 2H), 1.10 (d, J= 6.8 Hz, 3H), 1.07 (s, 3H), 0.94 (s, 9 H), 0.84 (d, J = 6.4 Hz, 3H), 0.71 (s, 9H), 0.09 (s, 3H), 0.06 (s, 3H), 0.04 (s, 3H), -0.3 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 214.5, 171.3, 164.9, 152.6, 144.4, 141.9, 138.8, 133.1, 128.9, 127.9, 127.1, 120.6, 119.3, 118.9, 116.2, 86.6, 79.6, 74.6, 66.9, 66.0, 56.9, 48.2, 41.0, 39.1, 37.7, 32.7, 31.4, 28.8, 27.2, 26.6, 26.4, 19.4, 18.9, 18.7, 15.6, 15.5, -2.9, -3.3, -3.4, -5.4. HRFABMS: calcd for C60H86NO6SSi2 (M+H) 1004.5714, found 1004.5724.

(4R)-4-Allyl-4-demethyl-26-hydroxyepothilone D - Formula (17). To a solution of the macrolactone of compound (o) (1 12 mg, 0.1 12 mmol) in THF (15 mL) was added HF. Py (4.3 mL) at 0 0C. The resulting reaction mixture was allowed to warm up to 25 0C, and the reaction was allowed to proceed with stirring at 25 0C for 62 h prior to being quenched by careful, portionwise addition into saturated aqueous NaHCO3 solution (10 mL) with further addition of sufficient solid NaHCO3 to ensure complete neutralization. The mixture was then extracted with EtOAc (10 mL x 3), the combined organic extracts were dried (Na2SO4), and the solvent was removed under reduced pressure. Purification of the crude product obtained by preparative thin layer chromatography over silica gel eluting with 70% ethyl acetate in hexanes gave the compound of Formula (17) (47 mg, 79%) as a colorless oil. [α]D -79 (c 0.6, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.94 (s, IH), 6.62 (s, IH), 5.63 (m, IH), 5.44 (dd, J = 9.8, 4.8 Hz, IH), 5.30 (d, J = 8.4 Hz, IH), 5.13 (dd, J = 17.2, 1.6 Hz, IH), 5.10 (dd, J = 10.4, 1.6 Hz, IH), 4.45 (d, J = 11.2 Hz, IH), 4.06 (d, J = 13.2 Hz, IH), 4.00 (d, J = 13.2 Hz, IH), 3.90 (bs, IH), 3.66 (d, J =
6.0 Hz, IH), 3.22 (qt, J = 6.8, 1.2 Hz, IH), 3.13 (bs, IH), 2.67 (s, 3H), 2.60 (m, 3H), 2.50 (m, IH), 2.38-2.30 (m, 3H), 2.05 (s, 3H), 1.96 (m, 3H), 1.80 (m, IH), 1.62 (m, 2H), 1.45 (m, 2H), 1.30 (m, 2H), 1.17 (d, J = 6.8 Hz, 3H), 1.02 (s, 3H), 1.0 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ6 220.0, 170.5, 165.3, 152.0, 142.1, 139.2, 133.5, 122.2, 119.2, 119.1, 115.8, 78.5, 73.3, 71.6, 66.5, 58.0, 41.5, 40.2, 39.8, 37.4, 31.6, 28.2, 27.0, 24.7, 17.7, 15.7, 14.5, 13.8. HRFABMS: calcd for C29H44NO6S (M+H) 534.2889, found 534.2925.

12,13-a-Epoxide - compound (p). To a solution of allylic alcohol of Formula (17) (26 mg, 0.049 mmol) and 4 A molecular sieves (60 mg) in CH2Cl2 (0.5 mL) at -20 0C was added diethyl-D-tartrate (12 mg, 0.0588 mmol, 1.2 eq) in CH2Cl2 (0.1 mL) and titanium isopropoxide (14 mg, 0.049 mmol, 1.0 eq) in CH2Cl2 (0.1 mL). After stirring at that temperature for 1 h, t-butyl hydroperoxide (30 μL, 5 M in decane, 0.0147 mmol, 3.0 eq) was added and the resulting reaction mixture was stirred at -20 0C for an additional 2 h. The reaction mixture was then filtered through celite into saturated aqueous Na2SO4 solution (10 mL), rinsing with EtOAc (10 mL). The subsequent biphasic mixture was stirred for 1 h and two layers were separated. The aqueous phase was re-extracted with EtOAc (10 mL x 3), and the combined organic extracts were dried over anhydrous Na2SO4, and the solvents were removed in vacua to give a crude oil. Purification of this oil by preparation thin layer chromatography over silica gel using 80% EtOAc in hexanes as eluent furnished a mixture of α-epoxide triol of compound (p) and β-epoxide triol of compound (r) (22.6 mg, 84%, containing around 20% β-eposide diastereoisomer, which cannot be separated via chromatography over silica gel). This mixture was used directly for the next step without further purification.

(P) (q)
26-Acryloyloxy-α-epoxide - compound (q). To a solution of the mixture of triol compounds (p) and (r) (22.6 mg, 0.041 mmol) in dichloromethane (1.5 mL) was added sequentially Et3N (29 μL, 0.205 mmol, 5 eq), acryloyl chloride (6 μL, 0.0738 mmol, 1.8 eq) and DMAP (1 mg) at 0 0C. The resulting reaction mixture was allowed to stir for 30 min prior to being quenched by the addition of saturated NaHCO3. The mixture was extracted with ethyl acetate (10 mL x 3), the combined organic extracts were dried over anhydrous Na2SO4, and the solvents were removed in vacuo. The crude product obtained was subjected to preparative thin layer chromatography over silica gel, eluting with 30% EtOAc in hexances for three times, to provide pure 26-acryloyloxy-α- epoxide compound (q) (15.7 mg, 79%) as a colorless syrup. 1H NMR (400 MHz, CDCl3) δ 6.93 (s, IH), 6.58 (s, IH), 6.39 (d, J= 17.6 Hz, IH), 6.08 (dd,J= 17.6, 10.4 Hz, IH), 5.84 (d, J= 10.8 Hz, IH), 5.66 (m, IH), 5.50 (d,J= 10.8 Hz, IH), 5.12 (d, J= 16.4 Hz, IH), 5.09 (m, IH), 4.50 (m, IH), 4.18 (d, J= 12.8 Hz, IH), 4.06 (d, J= 12.4 Hz, IH), 3.91 (m, IH), 3.28 (m, IH), 3.12 (dd, J= 7.6, 5.2 Hz, IH), 2.66 (s, 3H), 2.68-2.61 (m, 3H), 2.56- 2.44 (m, 2H), 2.12-2.01 (m, 2H), 2.07 (s, 3H), 1.88-1.71 (m, 3H), 1.49-1.27 (m, 4H), 1.08-0.99 (m, IH), 1.03 (s, 3H), 1.00 (d, 6.8 Hz, 3H), 0.91 (d, J= 7.2 Hz, 3H). 13C NMR (IOO MHz, CDCl3) 5 216.5, 170.2, 165.7, 164.9, 152.0, 137.6, 133.8, 131.7, 127.7, 119.4, 118.5, 116.3, 76.8, 72.2, 71.0, 66.9, 59.6, 59.1, 57.2, 43.2, 40.2, 39.6, 35.5, 31.3, 30.4, 25.8, 19.1, 19.0, 15.7, 15.2, 10.1. HRFABMS: calcd for C32H46NO8S (M+H) 604.2944, found 604.2981.

Internal lactone Epothilone - compound of Formula (18a). To a solution of diene compound (q) (12 mg, 0.0199 mmol) in dichloromethane (6 mL) was added second- generation Grubbs catalyst (6 mg, 0.006965 mmol, 0.35 eq) in dichloromethane (3.5 mL) for a period of 3 h via syringe pump under N2. The resulting reaction mixture was allowed to stir for 20 h at 25 0C, and then the dichloromethane was removed under reduced pressure. The residue obtained was subjected to preparative thin layer chromatography over silica gel, eluting with 50% EtOAc in hexanes, to furnish the compound of Formula (18a) (2.84 mg, 23.7%) and starting material of compound (q) (8.18 mg, 68.2%). The compound of Formula (18a) was obtained as a colorless oil. IR(fϊlm) cm"1: 3506 (OH), 1786 (5-membered lactone), 1733, 1729 (CO, COO), 1697(unsaturated ester). 1H NMR (400 MHz, CD3CN) and 13CNMR (100 MHz, CD3CN) see Table 3 (wherein δ is in ppm and J - Hz). HRFABMS: calcd for C3iH42NO9S (M+H) 604.2580, found 604.2538.
Table 3
Atom S(C)(HSQC) S(H) 1H-1H COSY HMBC(H~*C)
C(I) 167.7(CO) - - -
CH2(2) 35.3(CH2) 2.92 (d, J = 7.2) 2.62 H-3, H-2(2.62) N/A
(d, J = 12) H-3, H-2(2.92) C-I, C-3, C-4
CH(3) 77.9(CH) 5.13 (t, J= 7.6) H2-2 C- l,C-2,C-5,C-23(4-CH3)
C(4) 55.0(C) - - -
C(5) 212.1(CO) - - -
CH(6) 44.7(CH) 3.37 (m) H-7,H3-24(6-CH3) C-5,C-7,C-24(6-CH3)
CH(7) 76.3(CH) 3.70 (m) H-6 C-6, C-9
CH(8) 35.7(C) 1.38 (m) H3-25(8-CH3) C-25
CH2(9) 30.0(CH2) 1.58 (m) H2- IO C-12
CH2(IO) 22.1(CH2) 1.61 (m) H2-9 C-9, C-12
CH2(I l) 29.0(CH2) 2.04 (m), 1.55 (m) - C-12, C-13
C(12) 61.9(C) - - -
CH(13) 57.1(CH) 3.04 (dd, J = 7.6, 5.2) H2- 14 C-I l, C-14
CH2(14) 29.2(CH2) 2.10 (m) H- 13, H-15 C-12, C-13, C-15
1.74 (m) H- 13, H-15 C-12, C-13
CH(15) 76.5(CH) 5.39 (m) H-14 N/A
C(16) 135.9(C) - - -
CH(17) 118.4(CH) 6.51 (s) - C-15,C- 16,C-18,C-19,C-27
C(18) 158.0(C) - - -
CH(19) 118.1(CH) 7.25 (s) - C-18, C-20
C(20) 165.2(C) - - -
CH3(21) 18.6(CH3) 2.64 (s) H2-21 C-18, C-19, C-20
CH2(22) 41.0(CH2) 3.52 (d, J = 16.4) 2.53 H-22(2.53) C-l",C-4,C-5,C-23(4-CH3)
(d, J = 16.4) H-22(3.52) C-l",C-3,C-4,C-23(4-CH3)
CH3(23) 16.7(4-CH3) 1.15 (s) - C-3, C-4, C-5, C-22
CH3(24) 15.0(6-CH3) 1.10 (d, J = 7.2) H-6 C-5, C-6, C-7
CH3(25) 17.3(8-CH3) 0.94 (d, J = 6.4) H-8 C-7, C-8, C-9
CH2(26) 65.4(CH2) 4.39 (d, J= 12.8) H-26(4.12) C-12, C- 13, C- I '
4.12 (d, J= 12.4) H-26(4.39) C-12, C- 13, C- I '
CH3(27) 15.3(16-CH3) 2.17 (s) - C- 15,C- 16,C-17,C- 18


Formula (17) (r)
12,13-β-Epoxide - compound (r). The β-epoxide triol of compound (r) was prepared as a pure compound using diethyl -L-tartrate instead of diethyl -D-tartrate following the procedure described previously for the synthesis of α-epoxide triol of compound (p). Compound (r) was obtained as a colorless oil in 83% yield. 1H NMR (400 MHz, CDCl3) δ 6.97 (s, IH), 6.59 (s, IH), 5.66 (m, IH), 5.42 (d, J= 7.2 Hz, IH), 5.13 (d, J= 16.4 Hz, IH), 5.10 (d, J= 10.8 Hz, 11-1), 4.37 (d, J= 11.2 Hz, IH), 4.29 (s, IH), 3.73 (d, J= 12.8 Hz, 111), 3.69 (dd, J= 6.0, 2.0 Hz, IH), 3.59 (d, J= 12.0 Hz, IH), 3.41 (m, IH), 3.12 (dd, J= 8.0, 3.6 Hz, IH), 2.85 (br.s, IH), 2.69 (s, 3H), 2.64-2.53 (m, 4H), 2.34 (m, IH), 2.18 (m, IH), 2.06 (s, 3H), 2.11-1.88 (m, 3H), 1.78-1.63 (m, 2H), 1.55-1.23 (m, 3H), 1.10 (d, J= 6.8 Hz, 311), 1.04 (s, 3H), 0.98 (d, J= 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 219.7, 170.8, 165.8, 152.6, 138.3, 133.6, 119.2, 119.1, 116.2, 77.1, 72.9, 72.89, 71.6, 64.4, 64.0, 57.9, 42.3, 39.8, 39.7, 35.9, 31.6, 31.3, 29.0, 20.7, 16.7, 16.1, 15.5, 12.0. HRFABMS: calced for C29H44NO7S (M+H) 550.2838, found 550.2836.

(r) (S)
(4R)-4-AUyl-4-demethyl-26-(acryloyloxy)epothilone B - compound (s). 26-
Acryloyloxy-β-epoxide of compound (s) was synthesized from β-epoxide triol of compound (r) employing the same procedure as that described for 26-acryloyloxy-α-
epoxide of compound (q). Compound (s) was obtained as a colorless syrup in 80% yield. 1H NMR (400 MHz, CDCl3) δ 6.93 (s, 1-H), 6.55 (s, IH), 6.37 (d, J= 16.4 Hz, 11-1), 6.08 (dd, J= 16.4, 10.8 Hz, IH), 5.83 (d, J= 10.8 Hz, IH), 5.40 (dd, J= 8.8, 1.6 Hz, 111), 5.12 (d, J= 16.8 Hz, IH), 5.08 (m, IH), 4.38 (m, IH), 4.28 (d, J= 12.4 Hz, 11-1), 4.03 (d, J= 12.4 Hz, IH), 3.67 (m, IH), 3.36 (m, IH), 2.95 (dd, J= 8.4, 2.8 Hz, IH), 2.78 (br.s, IH), 2.67- 2.64 (m, IH), 2.65 (s, 3H), 2.59 (d, J= 7.6 Hz, 2H), 2.56 (dd, J= 14.0, 2.8 Hz, IH), 2.33 (d, J= 14.0, 1.6 Hz, 111), 2.17 (dt, J= 18.8, 3.2 Hz, IH), 2.09-2.02 (m, IH), 2.04 (s, 3H), 1.98-1.87 (m, 2H), 1.78-1.70 (m, 2H), 1.58-1.48 (m, 2H), 1.43-1.36 (m, 2H), 1.30-1.24 (m, IH), 1.06 (d, J= 6.8 Hz, 3H), 1.02 (s, 3H), 0.95 (d, J= 6.4 Hz, 3H). 13C NMR (IOO MHz, CDCl3) 5 219.6, 170.8, 165.9, 165.6, 151.8, 138.1, 133.6, 131.8, 128.0, 119.8, 119.1, 116.3, 77.1, 72.6, 71.7, 66.4, 62.2, 58.9, 57.9, 42.1, 39.8, 35.8, 31.6, 31.5, 29.3, 20.6, 19.2, 16.7, 16.2, 16.0, 15.4, 11.7. HRFABMS: calcd for C32H46NO8S (M+H) 604.2944, found 604.2976.


Internal lactone epothilone - Formula (18b). The epothilone analog of Formula (18b) was prepared from the diene of compound (s) according to a similar procedure to that described for the conversion of compound (q) to the compound of Formula (18a). The compound of Formula (18b) was obtained as a colorless oil in 24.8% yield (69.7% starting material was recovered). IR(film) cm"1 : 3506 (OH), 1786 (5-membered lactone), 1733, 1730 (CO, COO), 1697(unsaturated ester).1!! NMR (400 MHz, CDCl3) δ 7.13 (s, IH), 6.62 (s, IH), 6.37 (d, J= 17.6 Hz, IH), 6.05 (dd, J= 17.6, 10.4 Hz, IH), 5.84 (d, J= 10.8 Hz, IH), 5.37 (m, IH), 4.98 (dd, J= 10.8, 2.8 Hz, IH), 4.17 (d, J= 12.8 Hz, IH), 4.07 (d, J= 12.4 Hz, IH), 3.78 (m, IH), 3.27 (d, J= 16.4 Hz, IH), 3.21 (m, IH), 3.01 (dd, J= 7.6, 5.2 Hz, IH), 2.67 (s, 3H), 2.51 (d, J= 16.4 Hz, IH), 2.73-2.50 (m, 3H), 2.20-2.14 (m, IH), 2.16 (s, 311), 1.92-
1.83 (m, 2H), 1.74-1.64 (m, 3H), 1.47-1.38 (m, 311), 1.33-1.13 (m, 21 1), 1.19 (d, J= 7.2 Hz, 3H), 1.18 (s, 311), 1.00 (d, J= 6.4 Hz, 3H). 13C NMR (100 MHz, CD3CN) δ 211.9, 174.1, 168.0, 165.6, 165.3, 152.9, 137.2, 131.3, 128.2, 119.8, 118.1, 77.7, 77.6, 76.9, 66.5, 61.6, 60.1, 55.0, 44.8, 41.2, 35.4, 34.9, 32.5, 28.8, 28.1, 21.9, 18.6, 17.8, 16.7, 15.4, 14.2. HRFABMS: calcd for C3IH42NO9S (M+H) 604.2580, found 604.2574.

(4R)-4-Allyl-4-demethyl-26-(acryloyloxy)epothilone D - Compound (t). 26-
Acryloyloxy-macrolactone of compound (t) was prepared directly from the triol of Formula (17) following the procedure described previously for the preparation of compound (q). Compound (t) was obtained as a colorless oil in 50% yield. 1H NMR (400 MHz, CDCl3) δ 6.92 (s, IH), 6.55 (s, IH), 6.36 (dd, J= 17.2, 1.6 Hz, IH), 6.09 (dd, J= 17.2, 10.4 Hz, IH), 5.84 (dd, J= 10.6, 1.2 Hz, IH), 5.60 (m, IH), 5.45 (dd, 10.2, 4.8 Hz, IH), 5.27 (d, J= 8.0 Hz, IH), 5.11 (dd, J= 17.2, 1.6 Hz, IH), 5.07 (dd, J= 10.4, 1.6 Hz, IH), 4.58 (d, J= 12.8 Hz, IH), 4.49 (d, J= 12.8 Hz, IH), 4.41 (dd, J= 11.0, 3.2 Hz, IH), 3.64 (d, J= 5.6 Hz, IH), 3.49 (bs, IH ), 3.19 (qd, J= 5.6, 1.2 Hz, IH), 3.08 (d, J= 1.6 Hz, IH), 2.66 (m, IH), 2.65 (s, 3H), 2.60-2.42 (m, 3H), 2.38-2.26 (m, 3H), 2.04 (s, 3H), 2.03 (m, IH), 1.77 (m, 2H), 1.63 (m, IH), 1.40 (m, IH), 1.28 (m, IH), 1.11 (d, J= 7.2 Hz, 3H), 1.0 (s, 3H), 0.99 (d, J= 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 219.6, 170.2, 165.9, 151.7, 138.9, 137.0, 133.4, 131.0, 128.4, 125.3, 1 19.2, 118.9, 115.8, 78.2, 73.6, 73.3, 71.6, 67.6, 57.7, 41.4, 40.1, 39.6, 37.6, 32.4, 31.5, 28.3, 24.6, 19.0, 16.0, 15.5, 14.6, 12.1. HRFABMS: calcd for C32H46NO7S (M+H) 588.2995, found 588.3007.

(t) Formula (19)
Internal lactone Epothilone - Compound of Formula (19). Tje epothilone analog of formula (19) was prepared from the diene of compound (t), using a similar procedure to that described for the conversion of compound (q) to the compound of Formula (18a) as a colorless oil in 44% yield (55% starting material was recovered). 1H NMR (400 MHz, CDCl3) δ 7.25 (s, IH), 6.50 (s, IH), 6.27 (dd, J= 17.6, 1.2 Hz, IH), 6.01 (dd, J= 17.6, 10.4 Hz, IH), 5.61 (dd, J= 10.4, 1.2 Hz, IH), 5.35 (m, IH), 5.32 (t, J= 4.4, IH), 5.00 (dd, J= 12.0, 2.8 Hz, IH), 4.86 (d, J= 14.8 Hz, IH), 4.23 (d, J= 14.0 Hz, IH), 3.80 (m, IH), 3.66 (d, J= 16.4 Hz, 111), 3.56 (m, IH), 2.68 (s, 3H), 2.60-2.54 (m, 2H), 2.46 (d, J= 16.0 Hz, IH), 2.42-2.32 (m, 3H), 2.21 (s, 3H), 1.78- 1.64 (m, 4H), 1.34-1.22 (m, 3H), 1.20 (d, J= 6.8 Hz, 3H), 1.17 (s, 3H), 1.03 (d, J= 7.2 Hz, 311). 13CNMR (100 MHz, CDCl3) δ 211.8, 174.2, 167.4, 166.1, 164.7, 153.2, 136.3, 134.8, 131.5, 128.2, 118.7, 1 18.5, 67.2, (three carbon's signals were hidden among solvent signal), 55.3, 43.8, 41.6, 36.7, 35.4, 29.5, 28.8, 27.4, 19.6, 17.9, 16.7, 16.4, 15.9. HRFABMS: calcd for C3XH42NO8S (M+l-1) 588.2626, found 588.2611.

Formula (17) Formula (20)
Internal lactone Epothilone - compound of Formula (20). The epothilone analog of Formula (20) was prepared directly from the triol of Formula (17) by the procedure described previously for the synthesis of the compound of Formula (18a) as a colorless oil in 17.7%
yield (71.7% starting material was recovered). 1H NMR (500 MHz, CDCl3) δ 7.15 (s, IH), 6.62 (s, IH), 5.48 (dd, J= 8.2, 7.4 Hz, IH), 5.31 (dd, J= 6.0, 3.6 Hz, IH), 5.05 (dd, J= 10.4, 2.7 Hz, IH), 4.08 (d, J= 12.4 Hz, IH), 4.01 (d, J= 12.9 Hz, IH), 3.76 (m, IH), 3.40 (br.d, IH), 3.31 (d, J= 17.0 Hz, IH), 3.28 (m, IH), 2.70 (s, 3H), 2.70-2.68 (m, 3H), 2.46 (d, J= 17.0 Hz, IH), 2.51-2.32 (m, 3H), 2.11 (s, 3H), 1.78-1.64 (m, 3H), 1.34- 1.22 (m, 3H), 1.22 (d, J= 6.6 Hz, 3H), 1.19 (s, 3H), 1.03 (d, J= 6.9 Hz, 3H). 13CNMR (100 MHz, CD3CN) 5 212.2, 174.1, 168.2, 164.7, 152.1, 143.5, 134.7, 119.9, 119.1, 118.7, 77.9, 77.0, 70.3, 65.4, 55.7, 44.1, 41.3, 36.7, 35.8, 29.9, 28.3, 27.8, 26.6, 17.8, 17.7, 16.1, 15.9, 15.0. HRFABMS: calcd for C28H40NO7S (M+H) 534.2525, found 534.2513.

(r) Formula (41)
(4R)-4-Allyl-4-demethyl-epothilone B- Formula (41). To a stirred solution of 12,13-β-epoxy-triol of compound (r) (14.1 mg, 0.02568 mmol) in dichloromethane (0.5 mL) at 0 0C was added Et3N (1 1 ttL, 0.077 mmol, 3 eq) followed by tosyl chloride (7.3 mg, 0.0385 mmol, 1.5 eq) and 4-DMAP (3.1 mg, 0.02568 mmol, 1.0 eq). The resulting reaction mixture was warmed to 25 0C and stirred for 1 h before saturated aqueous NH4Cl solution (10 mL) was added. The mixture was extracted with EtOAc (10 mL x 3), the combined organic extracts were dried (Na2SO4), and the solvents were removed in vacuo. The residue obtained was then dissolved in acetone (1 mL) and treated with NaI (12 mg, 0.07704 mmol, 3 eq). After stirring at 25 0C for 30 h, the solvent was removed under reduced pressure and the residue was purified by preparative thin layer chromatography (40% EtOAc in hexanes) to provide iodide (12.5 mg, 74% for two steps) as a colorless syrup. 1H NMR (400 MHz, CDCl3) δ 6.97 (s, IH), 6.58 (s, IH), 5.67 (m, IH), 5.41 (d, J= 7.6 Hz, IH), 5.14 (d, J= 16.8 Hz, IH), 5.11 (d, J= 10.0 Hz, IH), 4.41 (m, IH), 4.09 (br.s, IH), 3.69 (m, IH), 3.38 (m, 11-1), .31 (d, J= 10.4 Hz, IH), 3.09 (d,J= 10.4 Hz, IH), 3.01 (dd, J= 8.0, 3.2 Hz, IH), 2.86 (br.s, IH), 2.69 (s, 3H), 2.72-2.53 (m, 3H), 2.42-2.34 (m, 2H),
2.07 (s, 3H), 2.181.92 (m, 2H), 1.86-1.69 (m, 3H), 1.56-1.46 (m, 3H), 1.38-1.30 (m, IH), 1.11 (d, J= 6.8 Hz, 3H), 1.05 (s, 3H), 0.99 (d, J= 6.4 Hz, 3H). 13CNMR (IOO MHZ, CDCl3) δ 219.9, 170.8, 165.6, 151.7, 138.0, 133.5, 119.9, 119.2, 116.4, 77.0, 73.0, 71.8,
64.7, 63.1, 57.8, 42.3, 39.8, 39.7, 35.9, 32.8, 31.0, 30.5, 21.1, 19.2, 16.7, 16.0, 15.5, 12.6, 12.0. HRFABMS: calcd for C29H43NO6SI (M+H) 660.1856, found 660.1819.
Iodide prepared in the preceding step (12.5 mg, 0.018968 mmol) and sodiumcyanoborohydride (12 mg, 0.18968 mmol, 10 eq) were dissolved in HMPA and the resulting mixture was heated at 45 0C for 43 h. After cooling to 25 0C, brine (10 mL) was added and the mixture was extracted with EtOAc (10 mL x 5). The combined organic extracts were dried (Na2SO4), the solvents were removed under reduced pressure, and the residue was passed through a short plug of silica gel to remove traces of HMPA (40% EtOAc in hexanes). The solvent was evaporated, and the residue was purified by preparative thin layer chromatography using 20% EtOAc in hexanes as eluent to provide pure epothilone B analog of Formula (41) (5 mg, 49.5%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.96 (s, IH), 6.58 (s, IH), 5.67 (m, IH), 5.41 (d, J= 8.8 Hz, IH), 5.14 (d, J= 17.2 Hz, IH), 5.11 (d, J = 9.6 Hz, IH), 4.41 (br.d, J= 9.2 Hz, IH), 4.25 (br.s, IH), 3.69 (br.s, IH), 3.38 (m, IH), 2.88 (br.s, IH), 2.79 (dd,J= 6.8, 5.6 Hz, IH), 2.69 (s, 3H), 2.66- 2.55 (m, 3H), 2.42-2.33 (m, IH), 2.18-2.142 (m, IH), 2.07 (s, 3H), 1.94-1.86 (m, IH), 1.82-1.69 (m, 2H), 1.69-1.46 (m, 2H), 1.42-1.34 (in, 3H), 1.28 (s, 3H), 1.11 (d, J= 6.8 Hz, 3H), 1.06 (s, 3H), 099 (d, J= 6.4 Hz, 3H). 13CNMR (100 MHz, CDCIl3) δ 219.9, 170.9, 165.6, 151.8, 138.5, 133.6, 1 19.57, 1 19.51, 116.2, 77.4, 72.9, 71.7, 63.1, 62.1,
57.8, 42.1, 39.8, 39.7, 36.0, 33.4, 32.3, 31.7, 22.8, 21.1, 19.2, 16.7, 16.0, 15.4, 11.9. HRFABMS: calcd for C29H44NO6S (M+H) 534.2889, found 534.2853.



Primary Alcohol - Compound of Formula (43). To a solution of macrolactone compound (o) (70 mg, 0.0698 mmol) in ether (1.5 mL) at -5 0C was added formic acid (1.5 mL) and the reaction was allowed to proceed with stirring for 5 h at -5 0C. Water (10 mL) and then solid NaHCO3 were sequently added until cessation of effervescence. The mixture was extracted with ether (10 mL x 3), the combined organic extracts were dried (Na2SO4), and the solvents were removed in vacuo. Purification of the crude product via preparative thin layer chromatography eluting with 50% ether in hexanes to yield primary alcohol of Formula (43) (42 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 6.93 (s, IH), 6.51 (s, IH), 5.58 (m, IH), 5.43 (m, IH), 5.00-4.97 (m, 3H), 3.94 (dd, J = 13.2, 4.4 Hz, IH), 4.10-4.05 (m, 21-1), 3.86 (dd, J = 8.4, 4.8 Hz, IH), 3.04 (m, IH), 2.76-2.53 (m, 3H), 2.67 (s, 3H), 2.46-2.20 (m, 3H), 2.18-1.92 (m, 3H), 2.08 (s, 3H), 1.75-1.55 (m, 4H), 1.06 (s, 3H), 0.89 (s, 9 H), 1.20-0.74 (m, 7 H), 0.77 (s, 9H), 0.03 (s, 9H), -0.19 (s, 3H). 13C NMR (IOO MHz, CDCl3) δ 214.4, 171.0, 164.8, 152.5, 143.9, 138.5, 133.0, 120.7, 119.6,
118.9, 116.2, 79.5, 74.6, 66.7, 60.7, 57.0, 48.2, 41.0, 39.2, 37.8, 32.6, 31.5, 28.3, 27.4, 26.6, 26.4, 19.6, 19.4, 18.9, 15.5, 14.4, -3.0, -3.25, -3.4, -5.3. HRFABMS: calcd for C4IH72NO6SSi2 (M+H) 762.4619, found 762.4654.

Formula (43) Formula (44)
Iodide - Compound of Formula (44). To a solution of primary alcohol of Formula (43) (19 mg, 0.02497 mmol) in THF (2.5 mL) was added PPh3 (13 mg, 0.0499 mmol, 2 eq), followed by imidazole (5.1 mg, 0.07491 mmol, 3 eq) and iodine (25 mg, 0.09988 mmol, 4 eq). The resulting reaction mixture was allowed to stir at 25 0C for 30 min prior to being quenched by the addition of saturated Na2S2O3 solution (10 mL). The subsequent mixture was extracted with ether (10 mL x 3), the combined organic extracts were dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure. The residue obtained was purified by preparative thin layer chromatography eluting with 5% EtOAc in hexanes to furnish iodide of Formula (44) (19.3 mg, 89%) as a colorless oil. 1H NMR (SOO MHz, CDCl3) δ 6.96 (s, IH), 6.56 (s, IH), 5.61 (m, IH), 5.48 (m, IH), 5.04-5.01 (m, 3H), 4.12 (m, IH), 4.00 (d, J= 9.0 Hz, IH), 3.89 (d, J= 8.6 Hz, 2H), 3.05 (m, IH), 2.81 (br.s 111), 2.71 (s, 3H), 2.65 (m, 2H), 2.44-2.22 (m, 3H), 2.10 (s, 3H), 1.72- 1.47 (m, 411), 1.26-1.07 (m, 2H), 1.11 (d, J= 7.2 Hz, 31-1), 1.10 (s, 3H), 0.98 (d, J= 6.6 Hz, 3H), 0.92 (s, 9 H), 0.82 (s, 91-1), 0.11 (s, 3H), 0.07 (s, 3H), -0.009 (s, 3H), -0.17 (s, 3H). 13C NMR (125 MHz, CDCl3) 5214.3, 170.7, 164.8, 152.4, 143.9, 138.3, 132.9, 124.0, 1 19.7, 118.9, 1 16.3, 80.7, 74.5, 60.5, 56.8, 48.2, 40.9, 33.2, 31.2, 28.6, 27.0, 26.4, 26.2, 19.3, 18.7, 18.6, 15.4, 14.3, 13.1, -3.1, -3.35, -3.6, -5.3. HRFABMS: calcd for C4IH71NO5SSi2I (M+H) 872.3636, found 872.3594.

Formula (44) Formula (45)
Epothilone D analog - Compound of Formula (45). Iodide of Formula (44) (19.3 mg, 0.022 mmol) and sodiumcyanoborohydride (14 mg, 0.22 mmol, 10 eq) were dissolved in HMPA (0.2 mL) and the resulting mixture was heated at 45 0C for 22 h. After cooling to 25 0C, the reaction mixture was diluted with 0.2 mL of EtOAc and then directly loaded onto preparative thin layer chromatography, eluting with 15% EtOAc in hexanes, to yield pure epothilone D analog of Formula (45) (10 mg, 60.6%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 6.95 (s, IH), 6.54 (s, IH), 5.61 (m, IH), 5.16 (t, J= 7.6 Hz, IH), 5.05-4.97 (m, 3H), 4.12 (in, IH), 3.90 (d, J= 8.6 Hz, IH), 3.07 (m, IH), 2.83 (br.s IH), 2.70 (s, 311), 2.68 (m, 2H), 2.45-2.33 (m, 3H), 2.10 (s, 3H), 1.73 (m, IH), 1.66 (s, 31-1), 1.12-0.82 (m, 2H), 1.11 (d, J= 7.2 Hz, 3H), 1.10 (s, 311), 0.98 (d, J= 6.6 Hz, 311), 0.94 (s, 9 H), 0.83 (s, 911), 0.10 (s, 3H), 0.07 (s, 3H), -0.007 (s, 3H), -0.13 (s, 3H). 13CNMR (125 MHz, CDCl3) δ 214.4, 171.0, 164.6, 152.6, 147.0, 138.9, 133.0, 119.4, 119.1, 118.8, 115.9, 79.8, 74.7, 60.5, 56.8, 40.9, 32.7, 31.9, 31.4, 26.5, 26.3, 23.1, 21.1, 19.3, 18.8, 18.7, 14.3, 3.1, -3..5, -3.6, -5.3. HRFABMS: calcd for C4iH72NO5SSi2 (M+H) 746.4670, found 746.4688.

(4R)-4-Allyl-4-demethyl-epothilone D - Compound of Formula (46). To a solution of the compound of Formula (45) (13.3 mg, 0.0134 mmol) in THF (6 mL) was added HF.Py (6 mL) at 0 0C. The resulting reaction mixture was allowed to warm up to 25
0C and the reaction was allowed to proceed for 24 h at 25 0C. The reaction was quenched by careful, portionwise addition into saturated aqueous NaHCθ3 solution (10 mL) with further addition of sufficient solid NaHCθ3 to ensure complete neutralization. The mixture was then extracted with EtOAc (10 mL x 3), the combined organic extracts were dried (Na2SO4), and the solvents were removed under reduced pressure to afford crude oil. Preparative thin layer chromatography of this oil over silica gel, eluting with 40% ethyl acetate in hexanes, gave the compound of Formula (46) (7 mg, 76%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.91 (s, IH), 6.54 (s, IH), 5.60 (m, IH), 5.23 (d, J= 8.8 Hz, IH), 5.11-5.06 (m, 3H), 4.39 (br.d, J= 11.6 Hz, IH), 3.66 (d, J= 5.2 Hz, IH), 3.47 (d, J= 5.2 Hz, IH), 3.17 (m, 2H), 2.66 (s, 3H), 2.65-2.44 (in, 4H), 2.31- 2.20 (m, 311), 2.03 (s, 3H), 1.87-1.74 (m, 3H), 1.62 (s, 3H), 1.32-1.20 (m, 2H), 1.12 (d, J= 6.8 Hz, 3H), 1.00 (s, 3H), 0.99 (d, J= 6.8 Hz, 3H). 13CNMR (100 MHz, CDCl3) δ 212.4, 170.3, 165.0, 151.9, 139.3, 138.2, 133.2, 121.1, 1 19.0, 118.9, 1 15.5, 78.7, 73.7, 71.6, 57.5, 41.2, 39.8, 39.5, 38.1, 32.6, 3 1.5, 31.4, 24.6, 22.9, 19.0, 15.9, 15.4, 14.6, 12.4. HRFABMS: calcd for C29H44NO5S (M+H) 518.2940, found 518.2897.

Formula (46) Formula (22)
Internal lactone epothilone D - Compound of Formula (22). The epothilone D analog of Formula (22) was obtained from (4R)-4-allyl-4-demethyl epothilone D (compound of formula (46)), according to the procedure described previously for the synthesis of the compound of Formula (18a), as a colorless oil in 35.6% yield (55.7% starting material was recovered). 1H NMR (500 MHz, CDCl3) δ 7.14 (s, IH), 6.60 (s, 114), 5.27 (d, J= 8.8 Hz, IH), 5.16 (dd, J= 8.8, 5.2 Hz, IH), 5.05 (dd, J= 10.4, 3.3 Hz, IH), 3.75 (m, IH), 3.30 (d, J= 17.0 Hz, 1 11), 3.27 (m, IH), 2.73-2.62 (m, 2H), 2.70 (s, 3H), 2.51 (dd, J= 10.1, 3.5 Hz, IH), 2.46 (d, J= 17.0 Hz, IH), 2.18 (s, 3H), 1.90-1.84 (m, IH), 1.72-1.63(m, IH), 1.67 (s, 3H), 1.51 -1.49 (m, IH), 1.39-1.30 (m, IH), 1.22 (d, J = 6.8 Hz, 3H), 1.17 (s, 3H), 1.03 (d, J= 6.8 Hz, 3H). 13CNMR (125 MHz, CDCl3) δ 212.1,
172.9, 167.5, 164.8, 152.7, 138.5, 137.1, 120.8, 119.6, 117.3, 80.6, 78.1, 76.3, 55.3, 43.1, 41.0, 37.1, 35.9, 32.3, 32.0, 30.7, 26.8, 23.3, 19.4, 17.2, 16.8, 15.4, 15.3,12.4. HRFABMS: calcd for C29H40NO6S (M+H) 518.2576, found 518.2549.
Many modifications and other embodiments of the inventions set forth herein will come to mind to one skilled in the art to which these inventions pertain having the benefit of the teachings presented in the foregoing descriptions. Therefore, it is to be understood that the inventions are not to be limited to the specific embodiments disclosed and that modifications and other embodiments are intended to be included within the scope of the appended claims. Although specific terms are employed herein, they are used in a generic and descriptive sense only and not for purposes of limitation.
Claims
1. A microtubule targeting compound according to the following formula that does not inhibit β-tubulin isotype Hβl,

Ri is selected from the group consisting of bridged polycyclics, fused polycyclics, and substituted aromatics;
R-2 is selected from the group consisting of substituted aromatics, cycloalkanes, cycloalkenes, fused polycyclics, and branched alkyl or alkenyl; and
R3 is selected from the group consisting of cycloalkanes, cycloalkenes, and linear or branched alkyl, alkenyl, or alkynyl where one or more carbon atoms is optionally replaced with an atom selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
2. The microtubule targeting compound of claim 1, wherein the compound is the compound of Formula (4)

3. The microtubule targeting compound of claim 1, wherein the compound is the compound of Formula (5)

4. The microtubule targeting compound of claim 1, wherein the compound is the compound of Formula (6)

5. The microtubule targeting compound of claim 1, wherein the compound is the compound of Formula (7)  or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
6. The microtubule targeting compound of claim 1, wherein Ri is selected from the group consisting of

7. The microtubule targeting compound of claim 1, wherein R2 is selected from the group consisting of 
wherein A is C, O, S, or N.
8. The microtubule targeting compound of claim 1, wherein R3 is selected from the group consisting of


9. The microtubule targeting compound of claim 1, wherein: Ri is selected from the group consisting of



R2 is selected from the group consisting of


and
R3 is selected from the group consisting of


10. A pharmaceutical composition comprising a pharmaceutically acceptable carrier and a microtubule targeting compound according to the following formula that does not inhibit β-tubulin isotype Hβl,
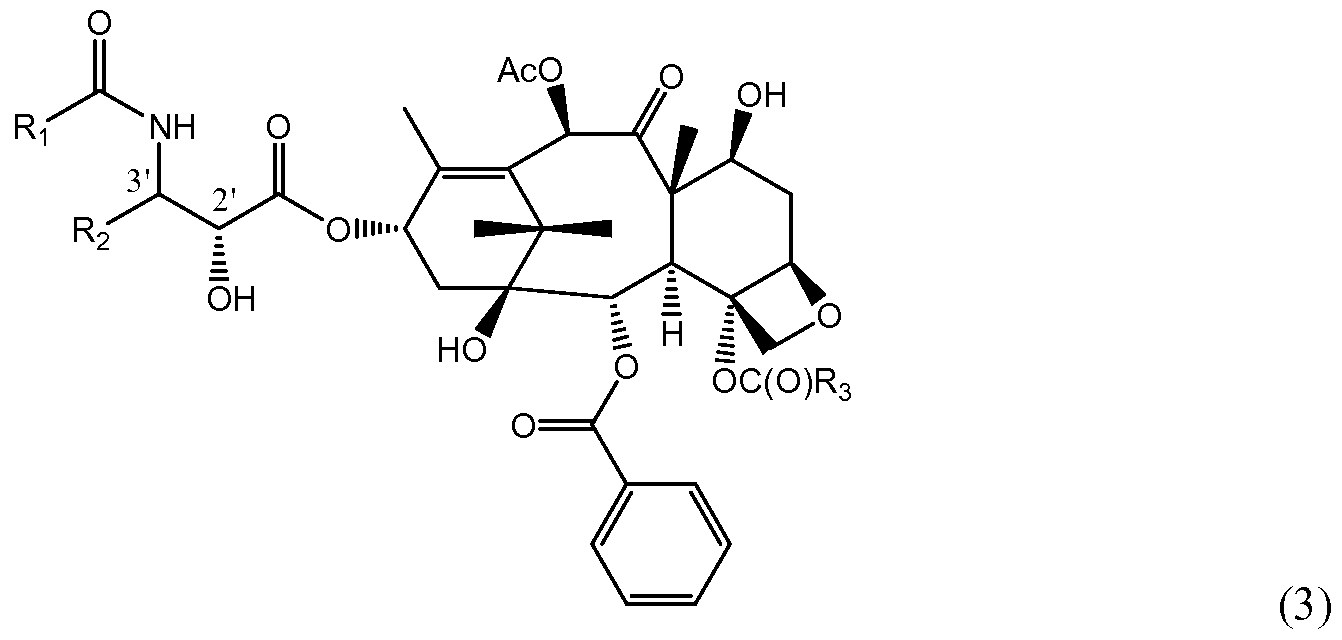
Ri is selected from the group consisting of bridged polycyclics, fused polycyclics, and substituted aromatics;
R-2 is selected from the group consisting of substituted aromatics, cycloalkanes, cycloalkenes, fused polycyclics, and branched alkyl or alkenyl; and
R3 is selected from the group consisting of cycloalkanes, cycloalkenes, and linear or branched alkyl, alkenyl, or alkynyl where one or more carbon atoms is optionally replaced with an atom selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
11. The pharmaceutical composition of claim 10, wherein the microtubule targeting compound is the compound of Formula (4)

12. The pharmaceutical composition of claim 10, wherein the microtubule targeting compound is the compound of Formula (5)

13. The pharmaceutical composition of claim 10, wherein the microtubule targeting compound is the compound of Formula (6)

14. The pharmaceutical composition of claim 10, wherein the microtubule targeting compound is the compound of Formula (7)

15. The pharmaceutical composition of claim 10, wherein Ri is selected from the group consisting of

16. The pharmaceutical composition of claim 10, wherein R2 is selected from the group consisting of


wherein A is C, O, S, or N.
17. The pharmaceutical composition of claim 10, wherein R3 is selected from the group consisting of
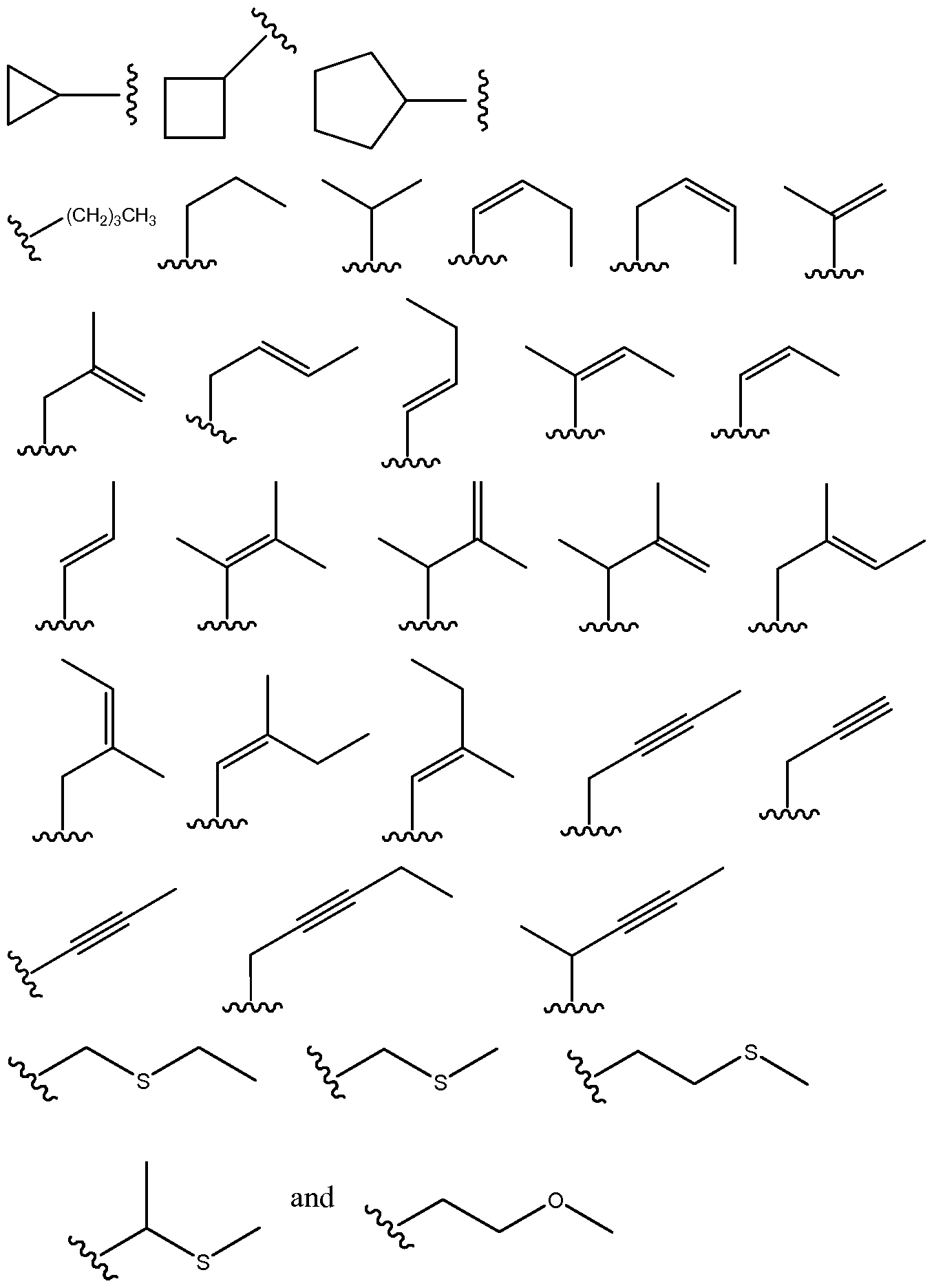
18. The pharmaceutical composition of claim 10, wherein: Ri is selected from the group consisting of 
R-2 is selected from the group consisting of 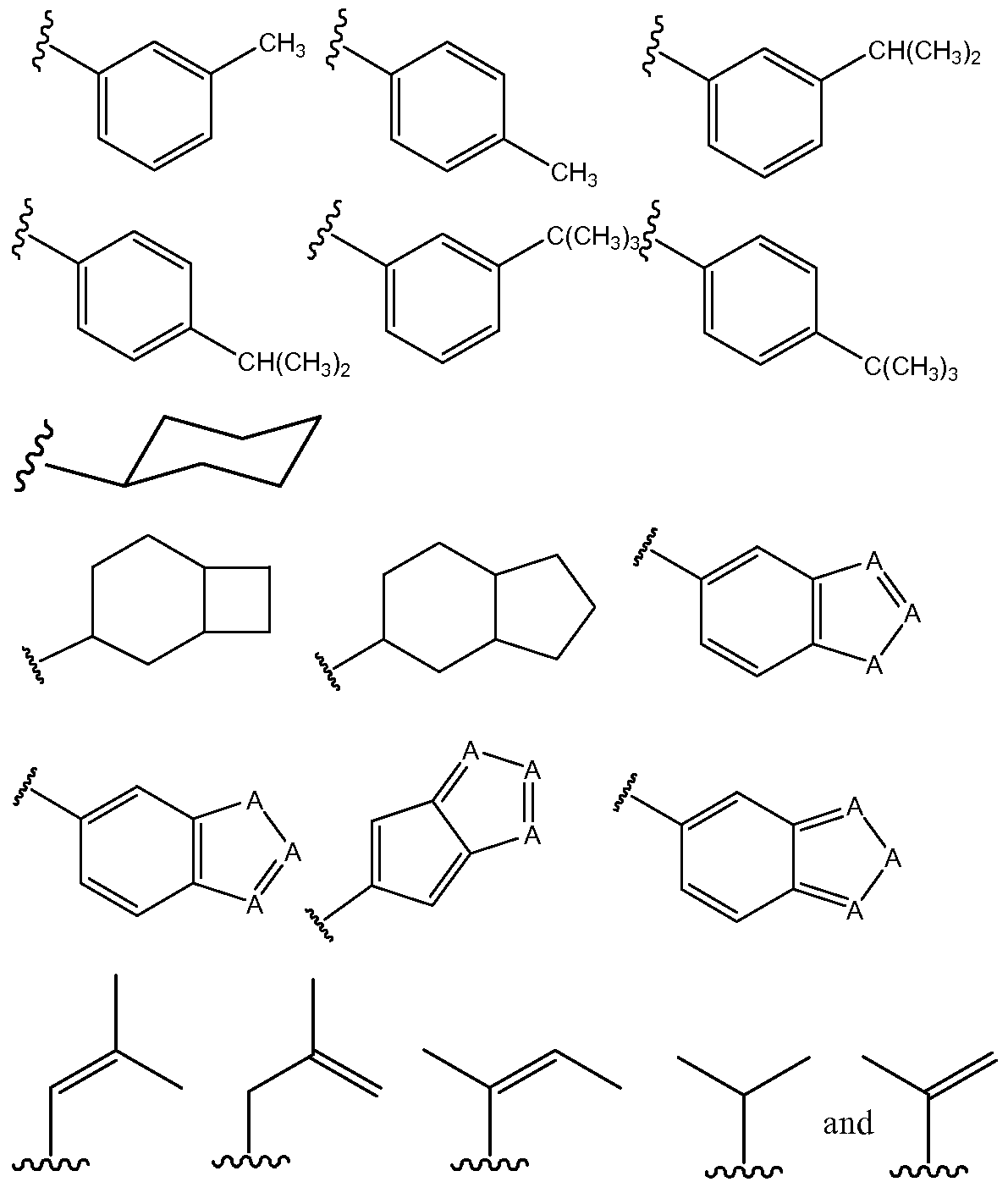 and
and
R3 is selected from the group consisting of

19. A method of treating cancer in a patient, the method comprising administering to the patient a microtubule targeting compound according to the following formula that does not inhibit β-tubulin isotype Hβl  wherein:
wherein:
Ri is selected from the group consisting of bridged polycyclics, fused polycyclics, and substituted aromatics;
R-2 is selected from the group consisting of substituted aromatics, cycloalkanes, cycloalkenes, fused polycyclics, and branched alkyl or alkenyl; and
R3 is selected from the group consisting of cycloalkanes, cycloalkenes, and linear or branched alkyl, alkenyl, or alkynyl where one or more carbon atoms is optionally replaced with an atom selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
20. The method of claim 19, wherein the microtubule targeting compound is the compound of Formula (4)

21. The method of claim 19, wherein the microtubule targeting compound is the compound of Formula (5)

22. The method of claim 19, wherein the microtubule targeting compound is the compound of Formula (6)

23. The method of claim 19, wherein the microtubule targeting compound is the compound of Formula (7)  or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
24. The method of claim 19, wherein Ri is selected from the group consisting of

25. The method of claim 19, wherein R2 is selected from the group consisting of 
wherein A is C, O, S, or N.
26. The method of claim 19, wherein R3 is selected from the group consisting of


27. The method of claim 19, wherein: Ri is selected from the group consisting of

R-2 is selected from the group consisting of


and
R3 is selected from the group consisting of


28. The method of claim 19, wherein the cancer is selected from the group consisting of non-small-cell lung cancer, ovarian cancer, breast cancer, prostate cancer, colo-rectal cancer, renal cancer, gastric cancer, gall bladder cancer, liver cancer, pancreatic cancer, small intestine cancer, testicular cancer, head cancer, neck cancer, melanoma, hepatocellular carcinoma, fallopian tube cancer, endometrial cancer, peritoneal cancer, solid tumors, non-Hodgkin's lymphoma, chronic lymphocytic leukemia, gliomas, and Kaposi's sarcoma.
29. The method of claim 19, wherein the cancer is a type wherein a majority of the cancer cells express a β-tubulin isotype that does not substantially include Hβl.
30. The method of claim 19, wherein the cancer is a type wherein the cells do not express Hβl -tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316.
31. A method of reducing myelosuppression in cancer treatment associated with administration of a microtubule targeting compound, the method comprising administering a microtubule targeting compound according to the following formula that does not inhibit β-tubulin isotype Hβl  wherein:
wherein:
Ri is selected from the group consisting of bridged polycyclics, fused polycyclics, and substituted aromatics;
R-2 is selected from the group consisting of substituted aromatics, cycloalkanes, cycloalkenes, fused polycyclics, and branched alkyl or alkenyl; and
R3 is selected from the group consisting of cycloalkanes, cycloalkenes, and linear or branched alkyl, alkenyl, or alkynyl where one or more carbon atoms is optionally replaced with an atom selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
32. The method of claim 31 , wherein the microtubule targeting compound is the compound of Formula (4)

33. The method of claim 31 , wherein the microtubule targeting compound is the compound of Formula (5)

34. The method of claim 31 , wherein the microtubule targeting compound is the compound of Formula (6)

35. The method of claim 31, wherein the microtubule targeting compound is the compound of Formula (7)  or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
36. The method of claim 31, wherein Ri is selected from the group consisting of

37. The method of claim 31, wherein R2 is selected from the group consisting of 
wherein A is C, O, S, or N.
38. The method of claim 31, wherein R3 is selected from the group consisting of


39. The method of claim 31, wherein: Ri is selected from the group consisting of



R2 is selected from the group consisting of
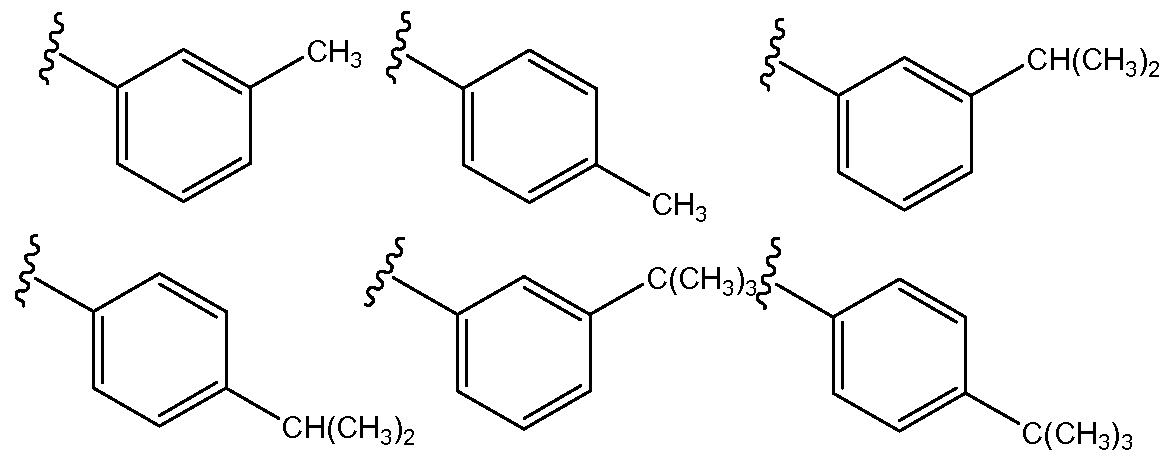

and
R3 is selected from the group consisting of


40. A method of determining whether a patient with cancer will exhibit a favorable response to treatment with a microtubule targeting compound that does not inhibit β-tubulin isotype Hβl, the method comprising: i) testing cancer cells from said patient to determine whether said cells overexpress Hβl, wherein a positive result in the test means the cells overexpress Hβland a negative result means the cells do not overexpress Hβl; ii) correlating a positive result in the test to treatment with a compound other than a microtubule targeting compound that does not inhibit β-tubulin isotype Hβl; and iii) correlating a negative result in the test to treatment with a microtubule targeting compound that does not inhibit β-tubulin isotype Hβl.
41. The method of claim 40, wherein the microtubule targeting compound that does not inhibit β-tubulin isotype Hβl is a compound of the following formula

Ri is selected from the group consisting of bridged polycyclics, fused polycyclics, and substituted aromatics; R-2 is selected from the group consisting of substituted aromatics, cycloalkanes, cycloalkenes, fused polycyclics, and branched alkyl or alkenyl; and
R3 is selected from the group consisting of cycloalkanes, cycloalkenes, and linear or branched alkyl, alkenyl, or alkynyl where one or more carbon atoms is optionally replaced with an atom selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
42. The method of claim 41, wherein the microtubule targeting compound is the compound of Formula (4)

43. The method of claim 41, wherein the microtubule targeting compound is the compound of Formula (5)

44. The method of claim 41 , wherein the microtubule targeting compound is the compound of Formula (6)

45. The method of claim 41, wherein the microtubule targeting compound is the compound of Formula (7)

46. The method of claim 41, wherein Ri is selected from the group consisting of

47. The method of claim 41, wherein R2 is selected from the group consisting of


wherein A is C, O, S, or N.
48. The method of claim 41, wherein R3 is selected from the group consisting of

49. The method of claim 41, wherein: Ri is selected from the group consisting of 
R-2 is selected from the group consisting of  and
and
R3 is selected from the group consisting of

50. The method of claim 40, wherein said step of testing cancer cells comprises testing nucleic acids.
51. The method of claim 40, wherein said step of testing cancer cells comprises testing one or more proteins.
52. The method of claim 40, wherein the cancer is selected from the group consisting of non-small-cell lung cancer, ovarian cancer, breast cancer, prostate cancer, colo-rectal cancer, renal cancer, gastric cancer, gall bladder cancer, liver cancer, pancreatic cancer, small intestine cancer, testicular cancer, head cancer, neck cancer, melanoma, hepatocellular carcinoma, fallopian tube cancer, endometrial cancer, peritoneal cancer, solid tumors, non-Hodgkin's lymphoma, chronic lymphocytic leukemia, gliomas, and Kaposi's sarcoma.
53. A method to determine whether a patient with cancer will exhibit a favorable response to treatment with a microtubule targeting compound that does not inhibit β-tubulin isotype Hβl, the method comprising: i) testing cancer cells from said patient to determine whether the cells express β- tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316, wherein a positive result in the test means the cells express Hβ 1 -tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316, and a negative result in the test means the cells do not express Hβl -tubulin isotypes encoding one or more of valine at β236, tyrosine at β200, or valine at β316; ii) correlating a positive result in the test with treatment with microtubule targeting compound that does not inhibit β-tubulin isotype Hβl; and iii) correlating a negative result in the test with treatment with a compound other than a microtubule targeting compound that does not inhibit β-tubulin isotype Hβl.
54. The method of claim 53, wherein the microtubule targeting compound that does not inhibit β-tubulin isotype Hβl is a compound of the following formula

Ri is selected from the group consisting of bridged polycyclics, fused polycyclics, and substituted aromatics;
R-2 is selected from the group consisting of substituted aromatics, cycloalkanes, cycloalkenes, fused polycyclics, and branched alkyl or alkenyl; and
R3 is selected from the group consisting of cycloalkanes, cycloalkenes, and linear or branched alkyl, alkenyl, or alkynyl where one or more carbon atoms is optionally replaced with an atom selected from the group consisting of O, S, and N; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
55. The method of claim 54, wherein the microtubule targeting compound is the compound of Formula (4)
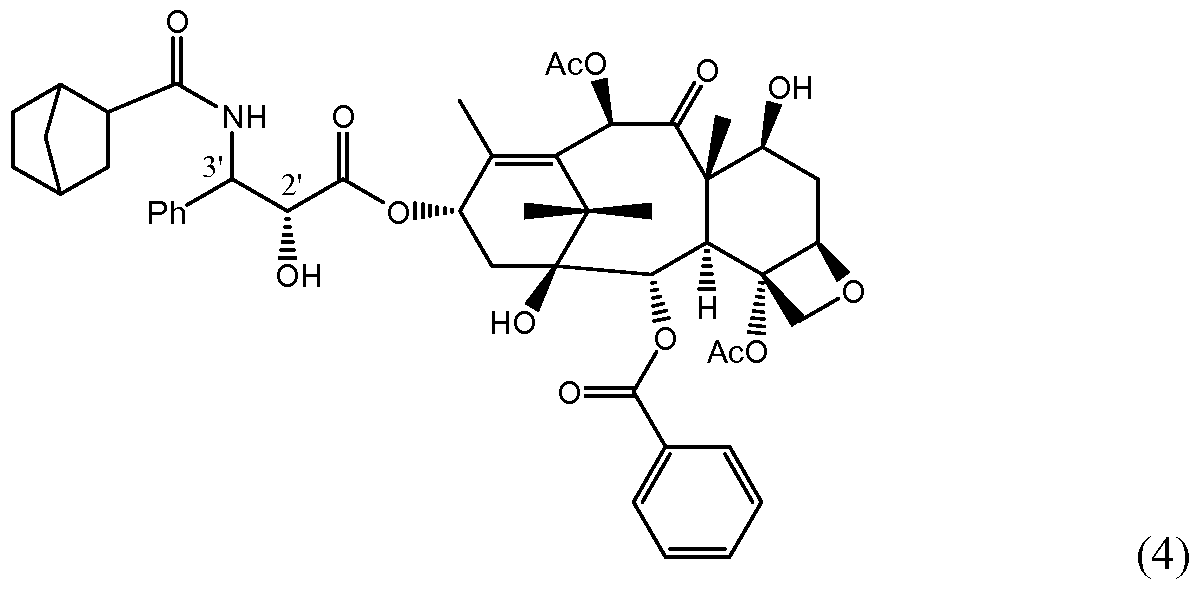
56. The method of claim 54, wherein the microtubule targeting compound is the compound of Formula (5)

57. The method of claim 54, wherein the microtubule targeting compound is the compound of Formula (6)

58. The method of claim 54, wherein the microtubule targeting compound is the compound of Formula (7)  or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
59. The method of claim 54, wherein Ri is selected from the group consisting of

60. The method of claim 54, wherein R2 is selected from the group consisting of 
wherein A is C, O, S, or N.
61. The method of claim 54, wherein R3 is selected from the group consisting of


62. The method of claim 54, wherein: Ri is selected from the group consisting of

R-2 is selected from the group consisting of


and
R3 is selected from the group consisting of


63. The method of claim 53, wherein said step of testing cancer cells comprises testing nucleic acids.
64. The method of claim 53, wherein said step of testing cancer cells comprises testing one or more proteins.
65. The method of claim 53, wherein the cancer is selected from the group consisting of non-small-cell lung cancer, ovarian cancer, breast cancer, prostate cancer, colo-rectal cancer, renal cancer, gastric cancer, gall bladder cancer, liver cancer, pancreatic cancer, small intestine cancer, testicular cancer, head cancer, neck cancer, melanoma, hepatocellular carcinoma, fallopian tube cancer, endometrial cancer, peritoneal cancer, solid tumors, non-Hodgkin's lymphoma, chronic lymphocytic leukemia, gliomas, and Kaposi's sarcoma.
66. A method of identifying a myelo-sparing microtubule targeting compound, the method comprising: i) screening a library of compounds in an assay that determines the ability of the compounds to inhibit Hβl -tubulin; ii) discarding compounds that inhibit said Hβl -tubulin; and iii) retaining compounds that do not inhibit said Hβl -tubulin.
67. A method for identifying a myelo-sparing microtubule targeting compound, the method comprising: i) screening a library of compounds in an assay that determines the ability of the compounds to inhibit β-tubulin with an amino acid other than valine at β236, tyrosine at β200, or valine at β316; ii) discarding compounds that inhibit said β-tubulin; and iii) retaining compounds that do not inhibit said β-tubulin.
68. The method of claim 67, wherein the amino acid at β236 is isoleucine, the amino acid at β200 is phenylalanine, or the amino acid at β316 is isoleucine.
69. The method of claim 68, wherein the amino acid at β236 is isoleucine.
70. The method of claim 67, wherein the screen is in silico.
71. The method of claim 67, wherein the screen is protein-based.
72. The method of claim 67, wherein the screen is cell-based.
73. The method of claim 72, wherein the cells used in the screen are 2MRC cells.
74. The method of claim 72, wherein the cells are bone marrow cells.
75. The method of claim 67, wherein the screen is based on high-throughput screening of a large library of small organic molecules.
76. The method of claim 67, wherein said library comprises small molecules believed to exhibit the properties of microtubule targeting agents.
77. A microtubule targeting compound according to the following formula 
wherein:
Y is N or O;
Xi is selected from the group consisting of alkyl, alkenyl, alkynyl, C(=O)alkyl, C(=O) alkenyl, C(=O)alkynyl, C(C=O)aryl, C(=O)alkaryl, C(=O)alkenaryl, C(=O)alkynaryl, C(=O)Oalkyl, C(=O)Oalkenyl, C(=O)Oalkynyl, C(=O)Oarylalkyl, C(=O)Oarylalkenyl, and C(=O)Oarylalkynyl;
X2 is selected from the group consisting of H, alkyl, alkenyl, alkynyl, and cycloalkyl;
X3 and X4 independently are selected from the group consisting of alkyl, NX6, O, and S;
Xr is OH, or X4 and X4- may be combined to form a bicyclic ring system, wherein the formed ring has 5-7 ring members, optionally including one or more heteroatoms selected from N and O, and optionally being substituted with NX6, O, OH, halogen, alkyl, alkenyl, alkynyl, alkoxy, alkenoxy, or alkynoxy;
X5 is selected from the group consisting of

X6 is selected from the group consisting of H and alkyl;
X7 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, S- alkyl, S-acyl CH2-S-alkyl, CH2-S-acyl, and NX6; and Xs is selected from the group consisting of H, alkyl, alkenyl, alkynyl, and cycloalkyl; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
78. A microtubule targeting compound according to the following formula

wherein: Xi is selected from the group consisting of alkyl, alkenyl, alkynyl, C(=O)alkyl,
C(=O) alkenyl, C(=O)alkynyl, C(C=O)aryl, C(=O)alkaryl, C(=O)alkenaryl, C(=O)alkynaryl, C(=O)Oalkyl, C(=O)Oalkenyl, C(=O)Oalkynyl, C(=O)Oarylalkyl, C(=O)Oarylalkenyl, and C(=O)Oarylalkynyl;
X2 and Xs independently are selected from the group consisting of H, alkyl, alkenyl, alkynyl, and cycloalkyl; and
X7 is selected from the group consisting of alkyl, alkenyl, alkynyl, cycloalkyl, S- alkyl, S-acyl CH2-S-alkyl, CH2-S-acyl, and NX6; or a pharmaceutically acceptable ester, amide, salt, solvate, prodrug, or enantiomer thereof.
79. A microtubule targeting compound according to the following formula
80. A microtubule targeting compound according to any of the following formulas:



Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12306708P | 2008-04-04 | 2008-04-04 | |
| US61/123,067 | 2008-04-04 | ||
| US8166108P | 2008-07-17 | 2008-07-17 | |
| US61/081,661 | 2008-07-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009146152A2 true WO2009146152A2 (en) | 2009-12-03 |
| WO2009146152A3 WO2009146152A3 (en) | 2010-01-21 |
Family
ID=41377876
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/039679 Ceased WO2009146152A2 (en) | 2008-04-04 | 2009-04-06 | Non-myelosuppressive compounds, pharmaceutical compositions thereof, and methods of treatment |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009146152A2 (en) |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6399638B1 (en) * | 1998-04-21 | 2002-06-04 | Bristol-Myers Squibb Company | 12,13-modified epothilone derivatives |
| MXPA03010909A (en) * | 2001-06-01 | 2004-02-17 | Bristol Myers Squibb Co | Epothilone derivatives. |
-
2009
- 2009-04-06 WO PCT/US2009/039679 patent/WO2009146152A2/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009146152A3 (en) | 2010-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10017500B2 (en) | 1,3-benzodioxole derivative | |
| EP2814490B1 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP1506203B1 (en) | Synthesis of epothilones, intermediates thereto, analogues and uses thereof | |
| JP2021513534A (en) | Tetrahydroquinazoline derivative useful as an anticancer agent | |
| JP2020532547A (en) | Spiro ring compound and its preparation and usage | |
| RS51023B (en) | 6-ALKENYL, 6-ALKINYL AND 6-EPOXY-EPOTYLON DERIVATIVES, PROCEDURE FOR THEIR PRODUCTION AND THEIR USE IN PHARMACEUTICAL PREPARATIONS | |
| EP3452483B1 (en) | Enhancer of zeste homolog 2 inhibitors | |
| CA2914723A1 (en) | Processes of making and crystalline forms of a mdm2 inhibitor | |
| EP3686196A1 (en) | Polycyclic compound acting as ido inhibitor and/or ido-hdac dual inhibitor | |
| US20210292309A1 (en) | Bryostatin compounds and methods of preparing the same | |
| AU2010205201A1 (en) | Imidazothiazole derivative having proline ring structure | |
| EP2757103B1 (en) | Pyrrolidine-3-ylacetic acid derivative | |
| US20250197416A1 (en) | Parthenolide derivatives and use thereof | |
| EP3035929B1 (en) | 7a-amide substituted-6,6-difluoro bicyclic himbacine derivatives | |
| WO2023097020A1 (en) | Heterobifunctional compounds as hpk1 degraders | |
| EP3035930A1 (en) | 7a-heterocycle substituted- 6, 6-difluoro bicyclic himbacine derivatives | |
| EP1722791A2 (en) | Synthesis of epothilones, intermediates thereto, analogues and uses thereof | |
| WO2009146152A2 (en) | Non-myelosuppressive compounds, pharmaceutical compositions thereof, and methods of treatment | |
| NL2031175B1 (en) | Dhcr24 inhibitory compounds | |
| FR2916443A1 (en) | NOVEL ANALOGUE DERIVATIVES OF HERBIMYCIN A, COMPOSITIONS CONTAINING SAME AND USE THEREOF. | |
| JP2018514526A (en) | Epoxyazulene derivatives useful for the treatment of cancer and diabetes | |
| UA73118C2 (en) | Derivatives of 6-alkenyl, 6-alkinyl and 6-epoxyepothilone, method for obtaining thereof and use in pharmaceutic preparates | |
| Jiang | Design, Synthesis and Biological Evaluation of C4-C9 Bridged Epothilone Analogs | |
| HK40032870B (en) | Spirocycle compounds and methods of making and using same | |
| HK40032870A (en) | Spirocycle compounds and methods of making and using same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09755514 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09755514 Country of ref document: EP Kind code of ref document: A2 |