WO2010024430A1 - 2,4-ジアミノピリミジン化合物 - Google Patents
2,4-ジアミノピリミジン化合物 Download PDFInfo
- Publication number
- WO2010024430A1 WO2010024430A1 PCT/JP2009/065149 JP2009065149W WO2010024430A1 WO 2010024430 A1 WO2010024430 A1 WO 2010024430A1 JP 2009065149 W JP2009065149 W JP 2009065149W WO 2010024430 A1 WO2010024430 A1 WO 2010024430A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- amino
- alkyl
- added
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a 2,4-diaminopyrimidine compound useful as an active ingredient of a pharmaceutical composition, particularly a pharmaceutical composition for suppressing acute rejection in transplantation.
- PLC Protein kinase C
- PKC activated by calcium and diacylglycerol is classical PKC (cPKC), activated by DAG, but PKC that does not require calcium in its activation is novel PKC (nPKC), active
- cPKC calcium and diacylglycerol
- nPKC novel PKC
- aPKC atypical PKC
- each subfamily consists of a plurality of isozymes, and cPKC is classified as PKC ⁇ , PKC ⁇ , PKC ⁇ , nPKC is classified as PKC ⁇ , PKC ⁇ , PKC ⁇ , PKC ⁇ , and aPKC is classified as PKC ⁇ and PKC ⁇ .
- calcineurin which is a target molecule of FK506 and cyclosporin A widely used in current transplantation medicine
- PKC ⁇ are in a complementary relationship, so calcineurin inhibitor and PKC ⁇
- Patent Document 1 reports that the compound represented by the formula (A) inhibits PKC ⁇ and is useful as an immunosuppressant. Although a compound having a pyrimidine structure is disclosed as a specific compound, there is no specific disclosure of the compound of the present invention. (R2 in the formula is Etc. For other symbols, see the publication. )
- Patent Document 2 reports that the compound represented by the formula (B) inhibits PKC ⁇ and is useful as an immunosuppressant. Although a compound having a pyrimidine structure is disclosed as a specific compound, there is no specific disclosure of the compound of the present invention. (R3 in the formula is Represents. For other symbols, see the publication. )
- Patent Document 3 it is reported that the compound represented by the formula (C) inhibits PKC ⁇ and is useful as an immunosuppressant.
- a compound having a pyrimidine structure is disclosed as a specific compound, there is no specific disclosure of the compound of the present invention.
- R1 in the formula is Represents. For other symbols, see the publication. )
- the compound represented by the formula (D) has an inhibitory activity against cyclin-dependent kinase (CDK), Aurora B kinase, etc., and is used for treatment and prevention of diseases characterized by excessive or abnormal cell proliferation. It has been reported to be useful. Although a compound having a pyrimidine structure is disclosed as a specific compound and is described as being useful for immunosuppression in organ transplantation, there is no specific disclosure of the compound of the present invention. (See the official gazette for symbols in the formula.)
- Patent Document 5 the compound represented by formula (E) inhibits polo-like kinase (PLK) and is useful for the prevention and / or treatment of tumors, neurodegenerative diseases, and diseases related to immune system activation. It has been reported. Although a compound having a pyrimidine structure is disclosed as a specific compound, there is no specific disclosure of the compound of the present invention, and there is no description regarding PKC ⁇ inhibitory activity or description of being useful for suppression of acute rejection in transplantation. . (See the official gazette for symbols in the formula.)
- Patent Document 6 reports that the compound represented by the formula (F) inhibits G protein-coupled receptor protein 88 (GPR88) and is useful for the prevention and / or treatment of central diseases.
- GPR88 G protein-coupled receptor protein 88
- a compound having a pyrimidine structure is disclosed as a specific compound, there is no specific disclosure of the compound of the present invention, and there is no description regarding PKC ⁇ inhibitory activity or description of being useful for suppression of acute rejection in transplantation. .
- R1 represents hydrogen
- A represents an optionally substituted heterocyclic group, an optionally substituted heterocyclic alkyl, an optionally substituted C 3-8 cycloalkyl, etc.
- An object of the present invention is to provide a 2,4-diaminopyrimidine compound useful as an active ingredient of a drug having PKC ⁇ inhibitory activity, particularly a pharmaceutical composition for suppressing acute rejection in transplantation.
- the present inventors have a structure such as aralkyl on the 2-position amino group of 2,4-diaminopyrimidine and a structure such as adamantylalkyl on the 4-position amino group.
- the present invention was completed by discovering that a compound or a salt thereof characterized by having an excellent PKC ⁇ inhibitory activity. That is, the present invention relates to a compound of formula (I) or a pharmaceutically acceptable salt thereof, as well as a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable salt.
- the present invention relates to a pharmaceutical composition containing a form. (The symbols in the formula have the following meanings.
- R 1 is Any one group selected from the group consisting of: R 4 represents —OH, an optionally substituted amine, or —CH 2 NH 2 ; n1 represents 0 or 1; R 5 represents —OH, (C 1-6 alkyl optionally substituted with —OH or —NH 2 ) or —CN; R 6 represents C 1-6 alkyl optionally substituted with —H or aryl; p represents 0 or 1; q represents 1, 2, 3 or 4; R 13 represents —H or C 1-6 alkyl; R 2 represents —CN, —CF 3 , —NO 2 or halogen; A represents a single bond or C 1-6 alkylene; R 3 is Any one group selected from the group consisting of: R 9 is the same or different from each other, halogen, optionally substituted C 1-6 alkyl, —OH, —CN, cycloalkyl, —Q— (optionally substituted C 1-6 alkyl) or Represents an optionally substituted aryl
- the present invention also relates to a pharmaceutical composition for suppressing acute rejection in transplantation containing a compound of formula (I) or a pharmaceutically acceptable salt thereof, that is, a compound of formula (I) or a pharmaceutical thereof
- a pharmaceutical composition for suppressing acute rejection in transplantation containing a compound of formula (I) or a pharmaceutically acceptable salt thereof, that is, a compound of formula (I) or a pharmaceutical thereof
- the present invention relates to an agent for suppressing acute rejection in transplantation containing an acceptable salt.
- the present invention relates to the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of an agent for suppressing acute rejection in transplantation, as well as a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the present invention relates to a method for suppressing acute rejection in transplantation, which comprises administering an effective amount of a salt acceptable to a patient to a patient.
- the compound of formula (I) or a pharmaceutically acceptable salt thereof has a PKC ⁇ inhibitory action and can be used as an inhibitor of acute rejection in transplantation.
- R 1 is Any one group selected from the group consisting of: R 4 represents —OH, an optionally substituted amine, or —CH 2 NH 2 ; n1 represents 0 or 1; R 5 represents —OH, (C 1-6 alkyl optionally substituted with —OH or —NH 2 ) or —CN; R 6 represents C 1-6 alkyl optionally substituted with —H or aryl; p represents 0 or 1; q represents 1, 2, 3 or 4; R 13 represents —H or C 1-6 alkyl; R 2 represents —CN, —CF 3 , —NO 2 or halogen; A represents a single bond or C 1-6 alkylene; R 3 is Any one group selected from the group consisting of: R 9 is the same or different from each other, halogen, optionally substituted C 1-6 alkyl, —OH, —OH,
- R 4 is —OH, —NR 7 R 8 or —CH 2 NH 2 ; R 7 and R 8 are the same or different from each other, (a) -H; (b) C 1-6 alkyl.
- the C 1-6 alkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 12).
- the cycloalkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 6). 1) -OH 2) -NHR 11 3) Halogen 4) Oxo 5) C 1-6 alkyl optionally substituted with -OH, and 6) heterocycloalkyl optionally substituted by (halogen, —OH, —CH 2 OH or —COCH 3 ); (d) Heterocycloalkyl.
- the heterocycloalkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 11).
- R 1 is And R 3 is The compound according to [2] or a pharmaceutically acceptable salt thereof.
- R 4 is -NR 7 R 8 R 7 and R 8 are the same or different from each other, (b) C 1-6 alkyl.
- the C 1-6 alkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 12).
- the cycloalkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 6). 1) -OH 2) -NHR 11 3) -F 4) Oxo 5) C 1-6 alkyl optionally substituted with -OH, and 6) (halogen, —OH, —CH 2 OH or —COCH 3 ) optionally substituted (azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl or morpholinyl); R 11 is -H; n1 is 1; R 2 is —CN, —CF 3 , —NO 2 or —F; A is C 1-6 alkylene; R 9 is (i) -F, -Cl or -Br (j) C 1-6 alkyl optionally substituted with -OH or halogen, (k) -OH, (l) -CN, (m) cyclopropyl, (n) -Q- (C 1-6 alkyl optional
- R 7 and R 8 are the same or different from each other, (b) C 1-6 alkyl.
- the C 1-6 alkyl may be substituted with the following groups. 9) -OH, -CH 3, cyclohexyl substituted with one or more groups selected from the group consisting of -CH 2 OH and, 10) piperidinyl optionally substituted with —OH or (C 1-6 alkyl optionally substituted with —OH, —OCH 3 , —CN or —F); Or (c) Cycloalkyl.
- the cycloalkyl may be substituted with one or more groups selected from the group consisting of the following 1), 2) and 5).
- a pharmaceutical composition comprising the compound according to [1] or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable excipient.
- a PKC ⁇ inhibitor comprising the compound according to [1] or a pharmaceutically acceptable salt thereof.
- a pharmaceutical composition for suppressing acute rejection in transplantation comprising the compound according to [1] or a pharmaceutically acceptable salt thereof.
- a method for suppressing acute rejection in transplantation comprising administering an effective amount of the compound or salt thereof according to [1] to a patient.
- C 1-6 alkyl means linear or branched alkyl having 1 to 6 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec -Butyl, tert-butyl, n-pentyl, n-hexyl group and the like.
- C 1-6 alkylene means linear or branched C 1-6 alkylene such as methylene, ethylene, trimethylene, tetramethylene, pentamethylene, hexamethylene, propylene, methylmethylene. Ethylethylene, 1,2-dimethylethylene, 1,1,2,2-tetramethylethylene, and the like. Another embodiment is C 1-4 alkylene, yet another embodiment is C 1-2 alkylene, and yet another embodiment is methylene or ethylene.
- halogen means F, Cl, Br and I.
- cycloalkyl is a C 3-10 saturated hydrocarbon ring group, which may have a bridge.
- cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl and the like Another embodiment is C 3-8 cycloalkyl, yet another embodiment is C 3-6 cycloalkyl, and yet another embodiment is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl.
- aryl is a C 6-14 monocyclic to tricyclic aromatic hydrocarbon ring group, such as phenyl or naphthyl, and in another embodiment, phenyl.
- the “heterocycle” means i) a 3- to 8-membered, or in another embodiment, a 5- to 7-membered single atom containing 1 to 4 heteroatoms selected from oxygen, sulfur and nitrogen.
- a ring heterocycle, and ii) the monocyclic heterocycle is selected from the group consisting of a monocyclic heterocycle, a benzene ring, a C 5-8 cycloalkane and a C 5-8 cycloalkene And a ring group selected from bi to tricyclic heterocycles containing 1 to 5 heteroatoms selected from oxygen, sulfur and nitrogen. Ring atoms such as sulfur or nitrogen may be oxidized to form oxides or dioxides.
- heterocycle examples include the following embodiments.
- monocyclic saturated heterocycle (a) containing 1 to 4 nitrogen atoms, such as azepanyl, diazepanyl, aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, piperidyl, pyrazolidinyl, piperazinyl, azocanyl, etc .; (B) those containing 1 to 3 nitrogen atoms and 1 to 2 sulfur atoms and / or 1 to 2 oxygen atoms, such as thiomorpholinyl, thiazolidinyl, isothiazolidinyl, oxazolidinyl, morpholinyl and the like; (C) those containing 1 to 2 sulfur atoms, such as tetrahydro-2H-thiopyranyl; (D) those containing 1 to 2 sulfur atoms and 1 to 2 oxygen atoms, such as oxathiolanyl; (a) containing
- (1) monocyclic unsaturated heterocyclic groups (a) those containing 1 to 4 nitrogen atoms, such as pyrrolyl, imidazolyl, pyrazolyl, pyridyl, dihydropyridyl, tetrahydropyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, Triazolyl, tetrazolyl, triazinyl, dihydrotriazinyl, azepinyl and the like; (B) those containing 1 to 3 nitrogen atoms and 1 to 2 sulfur atoms and / or 1 to 2 oxygen atoms, for example thiazolyl, isothiazolyl, thiadiazolyl, dihydrothiazinyl, oxazolyl, isoxazolyl, oxadiazolyl, Oxazinyl and the like; (C) those containing 1 to 2 sulfur atoms, such as thienyl, thie
- condensed polycyclic saturated heterocyclic group (a) containing 1 to 5 nitrogen atoms, such as quinuclidinyl, 7-azabicyclo [2.2.1] heptyl, 3-azabicyclo [3.2.2] nonanyl ; (B) those containing 1 to 4 nitrogen atoms, and 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, such as trithiadiazaindenyl, dioxoleumidazolidinyl and the like; (C) those containing 1 to 3 sulfur atoms and / or 1 to 3 oxygen atoms, such as 2,6-dioxabicyclo [3.2.2] oct-7-yl;
- condensed polycyclic unsaturated heterocyclic group (a) containing 1 to 5 nitrogen atoms for example, indolyl, isoindolyl, indolinyl, indolizinyl, benzimidazolyl, dihydrobenzimidazolyl, tetrahyzolobenzimidazolyl, quinolyl, tetrahydro Quinolyl, isoquinolyl, tetrahydroisoquinolyl, indazolyl, imidazopyridyl, benzotriazolyl, tetrazolopyridazinyl, carbazolyl, acridinyl, quinoxalinyl, dihydroquinoxalinyl, tetrahydroquinoxalinyl, phthalazinyl, dihydroindazo Ril, benzopyrimidinyl, naphthyridinyl, quinazolinyl, cinnolinyl, etc .
- heterocycloalkyl refers to the monocyclic saturated heterocyclic group described in (1) and the condensed polycyclic saturated group described in (3) among the above “heterocycle”. It is a tero ring group, and ring atoms such as sulfur or nitrogen may be oxidized to form an oxide or a dioxide.
- Another embodiment is the monocyclic saturated heterocycle described in (1) in which sulfur or nitrogen as a ring atom may be oxidized to form an oxide or a dioxide, and in another embodiment, azetidinyl Pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, tetrahydro-2H-thiopyranyl, tetrahydrothiopyranyl dioxide, tetrahydro-2H-pyranyl.
- the “nitrogen-containing heterocycloalkyl” is a monocyclic ring containing at least one nitrogen atom described in (1) (a), (b) among the above “heterocycle”.
- Another embodiment is a monocyclic saturated heterocycle containing at least one nitrogen atom described in (1) (a), (b), and in another embodiment, azetidinyl, pyrrolidinyl, Piperidyl, piperazinyl, morpholinyl.
- heteroaryl means an aromatic group among (2) monocyclic unsaturated heterocyclic group and (4) condensed polycyclic unsaturated heterocyclic group of the above “heterocycle”. It is a heterocyclic group having a property.
- Another embodiment is (2) a heterocyclic group (monocyclic heteroaryl) having aromaticity among monocyclic unsaturated heterocyclic groups, and further another is pyridyl.
- optionally substituted means unsubstituted or having 1 to 5 substituents, and in another embodiment, unsubstituted or substituted It means having 1-3. “Substituted” means having 1 to 5 substituents, and in another aspect, having 1 to 3 substituents. In addition, when it has a some substituent, those substituents may be the same, or may mutually differ.
- “Protected —OH” means that the OH group is protected with a protecting group generally used for protecting a hydroxyl group. In another embodiment, it means that it is protected by an acyl group, an ether group, a silyl ether group or an acetal group. In still another embodiment, when it has a methyl group or two OH groups adjacent to each other, It means being protected with a dimethylmethylene group or a benzylidene group.
- R 1 is A compound that is (2) The compound according to (1), wherein R 4 is —OH, —NR 7 R 8 or —CH 2 NH 2 , and in another embodiment, is —NR 7 R 8 .
- R 7 and R 8 are the same or different from each other, (a) -H; (b) C 1-6 alkyl.
- the C 1-6 alkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 12).
- the cycloalkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 6). 1) -OH 2) -NHR 11 3) Halogen, in another embodiment, -F 4) Oxo 5) C 1-6 alkyl optionally substituted with —OH, in another embodiment, —CH 3 or —CH 2 OH, and 6) Heterocycloalkyl optionally substituted with (halogen, —OH, —CH 2 OH or —COCH 3 ), another embodiment is (—F, —OH, —CH 2 OH or —COCH 3 ) Optionally substituted with (azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl or morpholinyl); (d) heterocycloalkyl, in another embodiment azetidinyl, piperidinyl, tetrahydro-2H-pyranyl or tetrahydro-2H-thiopyranyl.
- the heterocycloalkyl may be substituted with one or more groups selected from the group consisting of the following 1) to 11).
- C 1-6 alkyl which may be substituted with (—OH, —OCH 3 , —CN, halogen or —CONH 2 )
- another embodiment includes (—OH, —OCH 3 , —CN, — F or -CONH 2 ) optionally substituted C 1-6 alkyl
- Aryl another embodiment is phenyl
- aryl in another embodiment phenyl
- R 14 and R 15 are the same as or different from each other, and are —H, C 1-6 alkyl or heterocycloalkyl, and in another embodiment, are —H, methyl, or tetrahydro-2H-pyranyl.
- (6) The compound according to (1), wherein n1 is 1.
- R 5 is —OH, —CH 2 OH, —CH 2 NH 2 or —CN.
- R 6 is substituted with -H or aryl are also C 1-6 alkyl, in another embodiment, it is a C 1-6 alkyl optionally substituted with -H or phenyl Compound.
- the compound wherein R 2 is —CN.
- R 3 is A compound that is (12) R 9 is the same or different from each other, (i) halogen, in another embodiment, -F, -Cl or -Br; (j) optionally substituted C 1-6 alkyl, in another embodiment, -OH or halogen optionally substituted C 1-6 alkyl, in a further aspect, -OH or -F C 1-6 alkyl optionally substituted by (k) -OH; (l) -CN; (m) cycloalkyl, in another embodiment cyclopropyl; (n) -Q- (optionally substituted C 1-6 alkyl), another embodiment is -Q- (substituted with halogen, -OH, -OCH 3 , -CN or -CONH 2 May be C 1-6 alkyl); Or (o) aryl which may
- R 10 is —Cl, —CH 3 , —OCH 3 , —OCH 2 CH 3 , —OCH (CH 3 ) 2 , —SCH 3 , —SCH 2 CH 3 , —SCH (CH 3 ) 2 , -SOCH 3 , -SO 2 CH 3 , -S- (cyclopentane) or -OCF 3 , and in another embodiment, (11) which is -Cl, -CH 3 , -OCH 3 or -SCH 3 The described compound.
- R 12 is —H or —Cl.
- tautomers and geometric isomers may exist depending on the type of substituent.
- the compound of the formula (I) may be described in only one form of an isomer, but the present invention also includes other isomers, separated isomers, or those And mixtures thereof.
- the compound of formula (I) may have an asymmetric carbon atom or axial asymmetry, and optical isomers based on this may exist.
- the present invention also includes separated optical isomers of the compound of formula (I) or a mixture thereof.
- the present invention includes a pharmaceutically acceptable prodrug of the compound represented by the formula (I).
- Pharmaceutically acceptable prodrugs are compounds that are converted to the compounds of the present invention by solvolysis or under physiological conditions. Examples of groups that form prodrugs include those described in Prog. Med., 5, 2157-2161 (1985) and “Development of pharmaceuticals” (Yodogawa Shoten, 1990), Volume 7, Molecular Design 163-198. Is mentioned.
- the salt of the compound of the formula (I) is a pharmaceutically acceptable salt of the compound of the formula (I), and may form an acid addition salt or a salt with a base depending on the type of substituent. is there.
- inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid Acid addition with organic acids such as lactic acid, malic acid, mandelic acid, tartaric acid, dibenzoyl tartaric acid, ditoluoyl tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, aspartic acid, glutamic acid Salts, salts with inorganic bases such as sodium, potassium, magnesium, calcium and
- the present invention also includes various hydrates and solvates of the compound of the formula (I) and pharmaceutically acceptable salts thereof, and crystalline polymorphic substances.
- the present invention also includes compounds labeled with various radioactive or non-radioactive isotopes.
- the compound of the formula (I) and pharmaceutically acceptable salts thereof can be produced by applying various known synthetic methods utilizing characteristics based on the basic structure or the type of substituent. At that time, depending on the type of functional group, it is effective in terms of production technology to replace the functional group with an appropriate protective group (a group that can be easily converted into the functional group) at the stage from the raw material to the intermediate. There is a case.
- protecting groups include protecting groups described in “Greene's Protective Groups in Organic Synthesis (4th edition, 2006)” by PGM Wuts and TW Greene. These may be appropriately selected according to the reaction conditions. In such a method, after carrying out the reaction by introducing the protective group, the desired compound can be obtained by removing the protective group as necessary.
- the prodrug of the compound of formula (I) introduces a specific group at the stage from the raw material to the intermediate, or reacts further using the obtained compound of formula (I), as in the case of the protecting group.
- the reaction can be carried out by applying a method known to those skilled in the art, such as ordinary esterification, amidation, dehydration and the like.
- typical production methods of the compound of the formula (I) will be described. Each manufacturing method can also be performed with reference to the reference attached to the said description.
- the manufacturing method of this invention is not limited to the example shown below.
- Manufacturing method 1 (In the formula, Lv 1 and Lv 2 represent a leaving group. The same applies hereinafter.)
- a compound (3) is produced by a nucleophilic substitution reaction between the compound (1) and the amine compound (2).
- This is a method for producing the invention compound (I).
- the leaving group include halogen, methanesulfonyloxy, methylsulfinyl, methylsulfonyl, p-toluenesulfonyloxy group and the like.
- the compound (1) and the compound (2), or the compound (3) and the compound (4) are used in an equivalent amount or in excess, and the mixture thereof is used in a solvent inert to the reaction or in the absence of solvent
- the mixture is stirred under cooling to heating under reflux, preferably at 0 ° C. to 80 ° C., usually for 0.1 hour to 5 days.
- the solvent used here include, but are not limited to, aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as diethyl ether, tetrahydrofuran, dioxane and dimethoxyethane, dichloromethane and 1,2-dichloroethane.
- halogenated hydrocarbons such as chloroform, N, N-dimethylformamide, N-methylpyrrolidone, 1,3-dimethyl-2-imidazolidinone, dimethyl sulfoxide, ethyl acetate, acetonitrile and mixtures thereof.
- the reaction is carried out in the presence of an organic base such as triethylamine, N, N-diisopropylethylamine or N-methylmorpholine, or an inorganic base such as potassium carbonate, sodium carbonate or potassium hydroxide. May be advantageous.
- an organic base such as triethylamine, N, N-diisopropylethylamine or N-methylmorpholine
- an inorganic base such as potassium carbonate, sodium carbonate or potassium hydroxide.
- Production Method 2 Other Production Methods Further, some compounds represented by the formula (I) can be obtained from the compounds of the present invention obtained as described above by known amidation, alkylation, reductive amination, hydroxyl group of carbonyl group. It can also be produced by arbitrarily combining processes that can be usually employed by those skilled in the art, such as reduction of the above. For example, it can be produced by applying the following reaction, the method described in Examples below, a method obvious to those skilled in the art, or a modification thereof.
- An amide compound can be obtained by amidating a carboxylic acid compound and an amine compound.
- the carboxylic acid compound and the amine compound are used in the same amount or in excess, and the mixture is used in the presence of a condensing agent, in a solvent inert to the reaction, from cooling to heating, preferably -20.
- the mixture is usually stirred for 0.1 hour to 5 days at -60 ° C.
- the solvent used here are not particularly limited, but are aromatic hydrocarbons such as benzene, toluene or xylene, halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane or chloroform, diethyl ether, tetrahydrofuran.
- Ethers such as dioxane and dimethoxyethane, N, N-dimethylformamide, dimethyl sulfoxide, ethyl acetate, acetonitrile or water, and mixtures thereof.
- condensing agents include, but are not limited to, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide, dicyclohexylcarbodiimide, 1,1′-carbonyldiimidazole, diphenyl phosphate azide, and phosphorus oxychloride. Is not to be done. It may be preferred for the reaction to use an additive (eg 1-hydroxybenzotriazole).
- an organic base such as triethylamine, N, N-diisopropylethylamine, or N-methylmorpholine
- an inorganic base such as potassium carbonate, sodium carbonate, or potassium hydroxide
- the method of making it react with an amine compound after converting carboxylic acid into a reactive derivative can also be used.
- reactive derivatives of carboxylic acids include acid halides obtained by reacting with halogenating agents such as phosphorus oxychloride and thionyl chloride, mixed acid anhydrides obtained by reacting with isobutyl chloroformate, 1-hydroxy
- reaction between these reactive derivatives and amine compounds is carried out in a solvent inert to the reaction of halogenated hydrocarbons, aromatic hydrocarbons, ethers, etc., under cooling to heating, preferably from ⁇ 20 ° C. Can be performed at 60 ° C.
- Alkylation An alkylamine compound can be obtained by alkylating an amine compound by reacting with a compound having a leaving group. Alkylation can be carried out in the same manner as in Production Method 1.
- An amine compound can be alkylated by reducing an imine compound prepared from a carbonyl compound and a primary or secondary amine compound. In this reaction, an equivalent amount or an excess amount of a carbonyl compound and a primary or secondary amine compound are used in an excess amount, and these mixtures are heated in a solvent inert to the reaction in the presence of a reducing agent at ⁇ 45 ° C. to heating under reflux. The mixture is preferably stirred at 0 ° C. to room temperature, usually for 0.1 hour to 5 days.
- Examples of the solvent used here are not particularly limited, but alcohols such as methanol and ethanol, halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane and chloroform, diethyl ether, tetrahydrofuran, dioxane, dimethoxyethane and the like. And ethers thereof, and mixtures thereof.
- Examples of the reducing agent include sodium cyanoborohydride, sodium triacetoxyborohydride, sodium borohydride and the like. It may be preferable to carry out the reaction in the presence of a dehydrating agent such as molecular sieves or an acid such as acetic acid, hydrochloric acid, titanium (IV) isopropoxide complex.
- an imine may be generated by condensation between a carbonyl compound and a primary or secondary amine compound, and may be isolated as a stable intermediate.
- the target product can be obtained by a reduction reaction after isolating the imine intermediate.
- a reduction catalyst for example, palladium carbon, Raney nickel, etc.
- a solvent such as methanol, ethanol, ethyl acetate, in the presence or absence of an acid such as acetic acid or hydrochloric acid.
- An alcohol compound can be obtained by reducing a carbonyl compound.
- the carbonyl compound is treated with an equal amount or an excess amount of a reducing agent, usually for 0.1 hour to 3 days, in a solvent inert to the reaction under cooling to heating, preferably at -20 ° C to 80 ° C.
- a solvent inert to the reaction under cooling to heating, preferably at -20 ° C to 80 ° C.
- the solvent used here are not particularly limited, but ethers such as diethyl ether, tetrahydrofuran, dioxane and dimethoxyethane, alcohols such as methanol, ethanol and 2-propanol, and aromatics such as benzene, toluene and xylene.
- Examples include hydrocarbons, N, N-dimethylformamide, dimethyl sulfoxide, ethyl acetate, and mixtures thereof.
- reducing agent hydride reducing agents such as sodium borohydride or diisobutylaluminum hydride, metal reducing agents such as sodium, zinc and iron, and the reducing agents in the following documents are preferably used.
- the raw materials used in the production of the compound of the present invention are, for example, the methods described in the following production examples, known methods described in “Production method 2: Other production methods” It can be produced from known compounds available by applying methods obvious to those skilled in the art, or modifications thereof.
- the compounds of formula (I) are isolated and purified as free compounds, pharmaceutically acceptable salts, hydrates, solvates or crystalline polymorphic substances thereof.
- the pharmaceutically acceptable salt of the compound of formula (I) can also be produced by subjecting it to a conventional salt formation reaction. Isolation and purification are performed by applying ordinary chemical operations such as extraction, fractional crystallization, and various fractional chromatography.
- Various isomers can be produced by selecting an appropriate raw material compound, or can be separated by utilizing a difference in physicochemical properties between isomers.
- optical isomers can be obtained by general optical resolution of racemates (for example, fractional crystallization leading to diastereomeric salts with optically active bases or acids, chromatography using chiral columns, etc.). Further, it can also be produced from a suitable optically active raw material compound.
- Test Method 1 Measurement of Human PKC ⁇ Enzyme Inhibitory Activity The test was performed using the HTRF R KinEASE TM S1 kit (CIS bio). Place 4 ⁇ L of the drug solution in a 384-well plate (CORNING), 3 ⁇ L of STK Substrate 1-biotin (final 250 nM) and Full-length human PKC ⁇ (Carna Biosciences, final 31 ng / mL) mixture and let stand at room temperature for 30 minutes, then ATP solution 3 ⁇ L (final 30 ⁇ M) was dispensed, and the enzyme reaction was performed at room temperature for 1 hour.
- Test method 2 Measurement of human IL-2 production inhibitory activity i) Preparation of plasmid A DNA fragment (445 bp) of the Human IL-2 promoter region corresponding to the DNA nucleotide sequence described in the database was cloned, inserted into pGL3 basic, a vector for reporter gene assay, and pGL3-IL2-pro-43 was inserted. I got it. ii) Maintenance and passage of Jurkat cells Jurkat, Clone E6-1 (ATCC No. TIB-152), a human T cell line cultured cell, was used as a medium with 10% FBS RPMI 1640 (Sigma) at 37 ° C, 5% CO 2.
- the cells were cultured under saturated humidity conditions, and subcultured when they reached about 90% confluent state.
- iii) Transfection and seeding After measuring the number of cells using a hemocytometer, prepare a cell suspension using 10% FBS RPMI 1640 (Sigma) so that the cell concentration is 2.5 x 10 7 cells / mL. Then, 10 ⁇ g of pGL3-IL2-pro-43 was mixed. Then, mix by adding 400 ⁇ L of Jurkat cells prepared in each plasmid mixture was adjusted to 2.5 ⁇ 10 7 cells / mL, it was the total amount added to the Gene Pulsor R Cuvette (BIO-RAD ).
- a mixed solution diluted 250-fold was added at a rate of 25 ⁇ L / well. This was cultured under conditions of 37 ° C., 5% CO 2 and saturated humidity for about 14 hours. The assay was performed in duplicate.
- the substrate solution attached to the Bright-Glo TM Luciferase Assay System (Promega) was added in an amount of 100 ⁇ L / well and gently mixed.
- the multi-label counter (ARVO SX, WALLAC) was set to reaction temperature: 25 ° C., Shaking Duration: 1 sec, Measurement time: 1 sec, and the measurement well of each 96 wells plate was set to measure Firefly luciferase activity.
- Test method 3 measurement of cytochrome P450 (CYP3A4) enzyme inhibitory activity i) Inhibition test I (calculation of residual rate I) Using a 96-well plate, substrate 2 ⁇ M (midazolam), test compound 5 ⁇ M, and human liver microsome (0.1 mg protein / mL) in 100 mM phosphate buffer (pH 7.4) containing 0.1 mM EDTA, 1 mM NADPH, total volume 150 ⁇ L And incubated at 37 ° C for 20 minutes. Thereafter, 130 ⁇ L of an aqueous solution containing 80% acetonitrile was added to stop the reaction.
- CYP3A4 enzyme inhibitory activity
- Residual rate II (%) Ai, II / Ao, II / (Ai, I / Ao, I) x 100
- Ai, II Metabolite production after reaction in the presence of test compound in inhibition test II
- the compound of formula (I) has a PKC ⁇ inhibitory action and has reduced CYP inhibition, so it is clear that it is useful as an inhibitor of acute rejection in transplantation and the like. .
- a pharmaceutical composition containing one or more of the compounds of formula (I) or a pharmaceutically acceptable salt thereof as an active ingredient is a pharmaceutical excipient or drug that is usually used in the art. It can be prepared by a commonly used method using a carrier or the like. Administration is orally by tablets, pills, capsules, granules, powders, solutions, etc., or injections such as intra-articular, intravenous, intramuscular, suppositories, eye drops, ophthalmic ointments, transdermal solutions, Any form of parenteral administration such as an ointment, a transdermal patch, a transmucosal liquid, a transmucosal patch, and an inhalant may be used.
- a solid composition for oral administration tablets, powders, granules and the like are used.
- one or more active ingredients are combined with at least one inert excipient such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, polyvinylpyrrolidone. And / or mixed with magnesium aluminate metasilicate.
- the composition may contain an inert additive, for example, a lubricant such as magnesium stearate, a disintegrant such as sodium carboxymethyl starch, a stabilizer, or a solubilizing agent according to a conventional method. .
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups or elixirs and the like, and commonly used inert diluents such as purified water. Or ethanol.
- the liquid composition may contain solubilizers, wetting agents, auxiliaries such as suspending agents, sweeteners, flavors, fragrances, and preservatives.
- the injection for parenteral administration contains a sterile aqueous or non-aqueous solution, suspension or emulsion.
- aqueous solvent include distilled water for injection or physiological saline.
- non-aqueous solvents include propylene glycol, polyethylene glycol or vegetable oil such as olive oil, alcohols such as ethanol, or polysorbate 80 (a pharmacopeia name).
- Such compositions may further contain isotonic agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers, or solubilizing agents. These are sterilized by, for example, filtration through a bacteria-retaining filter, blending with a bactericide or irradiation. These can also be used by producing a sterile solid composition and dissolving or suspending it in sterile water or a sterile solvent for injection before use.
- External preparations include ointments, plasters, creams, jellies, poultices, sprays, lotions, eye drops, eye ointments and the like.
- ointment bases include commonly used ointment bases, lotion bases, aqueous or non-aqueous solutions, suspensions, emulsions, and the like.
- ointments or lotion bases include polyethylene glycol, propylene glycol, white petrolatum, white beeswax, polyoxyethylene hydrogenated castor oil, glyceryl monostearate, stearyl alcohol, cetyl alcohol, lauromacrogol, sorbitan sesquioleate, etc. Can be mentioned.
- a transmucosal agent such as an inhalant or a nasal agent is used in a solid, liquid, or semi-solid state, and can be produced according to a conventionally known method.
- known excipients, and further pH adjusters, preservatives, surfactants, lubricants, stabilizers, thickeners and the like may be appropriately added.
- an appropriate device for inhalation or insufflation can be used.
- a known device such as a metered dose inhalation device or a nebulizer
- the compound is administered alone or as a powder in a formulated mixture or as a solution or suspension in combination with a pharmaceutically acceptable carrier. be able to.
- the dry powder inhaler or the like may be for single or multiple administration, and a dry powder or a powder-containing capsule can be used. Alternatively, it may be in the form of a pressurized aerosol spray using a suitable propellant, for example, a suitable gas such as chlorofluoroalkane, hydrofluoroalkane or carbon dioxide.
- a suitable propellant for example, a suitable gas such as chlorofluoroalkane, hydrofluoroalkane or carbon dioxide.
- the daily dose is about 0.0001 to 100 mg / kg per body weight, which should be administered once or divided into 2 to 4 times.
- the appropriate daily dose is about 0.0001 to 10 mg / kg per body weight, and is administered once to several times a day.
- about 0.0001 to 1 mg / kg per body weight is administered once to several times a day.
- the dose is appropriately determined according to individual cases in consideration of symptoms, age, sex, and the like.
- the compound of the formula (I) can be used in combination with various therapeutic agents or preventive agents for diseases for which the compound of the formula (I) is considered to be effective.
- the combination may be administered simultaneously, separately separately, or at desired time intervals.
- the simultaneous administration preparation may be a compounding agent or may be separately formulated.
- the manufacturing method of the compound of Formula (I) is demonstrated in detail.
- this invention is not limited to the compound as described in the following Example.
- the manufacturing method of a raw material compound is shown in a manufacture example.
- the production method of the compound of the formula (I) is not limited to the production methods of the specific examples shown below, and the compound of the formula (I) may be a combination of these production methods or a person skilled in the art. It can also be produced by methods that are self-evident.
- HPLC analysis conditions are as follows. (Analysis conditions) Column: YMC-Pack ODS-AM (S-5 ⁇ m, 12nm) (150x4.6mm ID), column temperature: 40 ° C, detection method: UV (254nm), flow rate: 1mL / min, eluent A: acetonitrile, eluent B: pH3 buffer (0.05M NaH 2 PO 4 adjusted phosphoric acid to pH3 by adding to an aqueous solution) Time program: Time (minutes) 0 20 30 Liquid A (%) 10 60 60 B liquid (%) 90 40 40 40
- the reaction mixture was diluted with EtOAc, and the organic layer was washed successively with water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure. The resulting residue was purified by silica gel flash column chromatography (hexane-EtOAc) to give benzyl tert-butyl [(1S, 3R, 5S) -tricyclo [3.3.1.1 3,7 ] decane-1,4- 113 mg of diylbis (methylene)] bisrel-carbamate were obtained.
- N- (5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) glycine tert-butyl ester (616 mg) in dichloromethane (6.16 ml) in trifluoroacetic acid (3.4 ml ) was added and stirred at room temperature. After completion of the reaction, diisopropyl ether was added to the reaction mixture. The precipitated solid was collected by filtration, washed with diisopropyl ether, and dried under reduced pressure to give N- (5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) glycine tris. 484 mg of fluoroacetate was obtained.
- the reaction mixture was extracted with EtOAc, and the organic layer was washed 3 times with water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure. MeOH was added to the resulting residue to precipitate a solid, which was collected by filtration. The filtrate was concentrated under reduced pressure, and a solid was precipitated again with MeOH and collected by filtration. The filtrate was concentrated under reduced pressure, and a solid was precipitated again with MeOH and collected by filtration. The obtained solid was dried under reduced pressure to obtain benzyl rel-[(1R, 2R, 3S, 5S) -5-carbamoyladamantan-2-yl] carbamate (2.9 g).
- the obtained crude product was further purified by silica gel flash column chromatography (chloroform-MeOH) to obtain benzyl rel-[(1R, 2S, 3S, 5S) -5-( ⁇ [2- ⁇ [ 2- (Trifluoromethoxy) benzyl] amino ⁇ -5- (trifluoromethyl) pyrimidin-4-yl] amino ⁇ methyl) adamantan-2-yl] carbamate (100 mg) and benzyl rel-[(1R, 2S, 3S , 5S) -5-( ⁇ [4- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ -5- (trifluoromethyl) pyrimidin-2-yl] amino ⁇ methyl) adamantan-2-yl] carbamate ( 50 mg) was obtained.
- the configuration of the obtained product is a compound (benzyl rel-( ⁇ (1R, 3S, 4R, 5R) -4-[(trans-4- ⁇ [tert- Butyl- (dimethyl) silyl] oxy ⁇ cyclohexyl) amino] adamantan-1-yl ⁇ methyl) carbamate) as a starting material, rel-trans-4- ⁇ [(1R, 2S, 3S, 5S)- 5- (aminomethyl) adamantan-2-yl] amino ⁇ cyclohexanol was subjected to Example 45, and the HPLC retention time (15.1 min) of the resulting product was consistent with Example 42 (transalcohol). Were determined.
- the catalyst was filtered off through celite, washed with MeOH, the filtrate was concentrated under reduced pressure, and rel-trans-4- ⁇ [(1R, 2S, 3S, 5S) -5- (aminomethyl) adamantan-2-yl] 263.8 mg of amino ⁇ cyclohexanol was obtained.
- the obtained residue was purified by amino silica gel flash column chromatography (chloroform-MeOH) and then silica gel flash column chromatography (chloroform-MeOH) to give 1- [1- (tetrahydro-2H-pyran-2-yl 0.42 g of a mixture of) -1H-benzimidazol-5-yl] methanamine and 1- [1- (tetrahydro-2H-pyran-2-yl) -1H-benzimidazol-6-yl] methanamine was obtained.
- N-bromosuccinimide (19.7 g) was added to a solution of tert-butyl (2-methoxybenzyl) carbamate (25.0 g) in MeCN (200 ml), and the mixture was stirred at room temperature overnight.
- N-bromosuccinimide (10.0 g) was added to the reaction mixture, and the mixture was further stirred at room temperature for 8 hours.
- the reaction mixture was concentrated under reduced pressure, and the residue was diluted with EtOAc, washed successively with water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel flash column chromatography (toluene) to obtain 9.55 g of tert-butyl (5-bromo-2-methoxybenzyl) carbamate.
- tert-butyl rel- (cis-4- ⁇ [(1R, 2S , 3S, 5S) -5- ⁇ [(5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] methyl ⁇ adamantan-2-yl] amino ⁇ cyclohexyl) Carbamate (86.6 mg) is eluted first, followed by tert-butyl rel- (trans-4- ⁇ [(1R, 2S, 3S, 5S) -5- ⁇ [(5-cyano-2- ⁇ [2- ( Trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] methyl ⁇ adamantan-2-yl] amino ⁇ cyclohexyl) carbamate (
- the reaction mixture was diluted with EtOAc, washed successively with water, saturated aqueous ammonium chloride solution, water, saturated aqueous sodium hydrogen carbonate, water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure.
- the organic layer was dried over anhydrous sodium sulfate, and after removing the desiccant, the solvent was distilled off under reduced pressure.
- the obtained residue was purified by silica gel flash column chromatography (hexane-EtOAc) to obtain 400 mg of tert-butyl ⁇ [2- (cyclopentylsulfonyl) pyridin-3-yl] methyl ⁇ carbamate.
- each production example compound was produced using the corresponding raw material.
- the structure of each production example compound, production method and physicochemical data are shown in the following table.
- Example 1 rel-1-[(1R, 3S, 5S) -5- ⁇ [(5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] methyl ⁇ adamantane-2
- EtOH EtOH
- Pd / C 50% water-containing product
- Example 3 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-
- iodobenzene 14.2 ⁇ l
- cesium acetate 50.8 mg
- copper (I) iodide 20.2 mg
- microwave irradiation was performed at 90 ° C. for 24 hours. Stir for hours.
- the reaction mixture was diluted with EtOAc, and the organic layer was washed successively with water and saturated brine, and dried over anhydrous sodium sulfate.
- Example 5 rel- (4R) -1-[(1R, 3S, 5S) -5- ⁇ [(5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] methyl ⁇
- adamantane-2-yl] -4-hydroxy-D-proline 23 mg
- ammonium chloride 6.3 mg
- HOBt 15.9 mg
- WSC 18.3 mg
- Example 6 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5- Nicotinic acid (14.3 mg), HOBt (15.7 mg), and WSC (18.1 mg) were sequentially added to a solution of carbonitrile (50 mg) in DMF (1.0 ml), and the mixture was stirred overnight at room temperature. The reaction mixture was diluted with EtOAc, and the organic layer was washed successively with water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure.
- Example 7 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-
- Sodium carbonate (44.8 mg) and 1,3-dibromopropane (43.2 ⁇ l) were added to a solution of carbonitrile (50 mg) in N, N-dimethylacetamide (2.5 ml), and microwave irradiation was performed at 100 ° C. for 30 minutes. Stir.
- the reaction mixture was diluted with EtOAc, and the organic layer was washed successively with water and saturated brine, and dried over anhydrous sodium sulfate.
- Example 16 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5- DIPEA (22.1 ⁇ l) and 2-bromoethanol (4.5 ⁇ l) were added to a solution of carbonitrile (30 mg) in DMF (0.6 ml), and the mixture was heated and stirred at 60 ° C. The reaction mixture was diluted with EtOAc, and the organic layer was washed successively with water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure.
- Example 32 4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile (50 mg) in DMF (1 ml) was added (2,2-dimethyl-1,3-dioxane-5-yl) methyl 4-methylbenzenesulfonate (47.7 mg) and potassium carbonate (29.2 mg), For 12 hours. Water was added to the reaction mixture and extracted with EtOAc. The organic layer was dried over magnesium sulfate, and after removing the desiccant, the solvent was distilled off under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (hexane-EtOAc), and 4-( ⁇ [(1S, 3R, 4S, 5S) -4- ⁇ [(2,2-dimethyl-1,3-dioxane- 21 mg of 5-yl) methyl] amino ⁇ adamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile were obtained.
- Example 36 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-
- oxiran-2-ylmethanol 8.2 ⁇ l
- Oxilan-2-ylmethanol 82.1 ⁇ l was added, and the mixture was further stirred at room temperature for 48 hours.
- reaction mixture was purified by amino silica gel flash column chromatography (chloroform-MeOH) to give rel-4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(2,3-dihydroxypropyl) 25 mg of amino] adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile were obtained.
- Example 39 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-
- DIPEA 36.9 ⁇ l
- 2,2,2-trifluoroethyl trifluoromethanesulfonate (18.3 ⁇ l)
- reaction mixture was directly purified by silica gel flash column chromatography (chloroform-MeOH) to obtain rel-4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(2,2 55 mg of 2,2-trifluoroethyl) amino] adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile was obtained.
- Example 40 4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile
- 1,3-dimethylimidazolidin-2-one 0.5 ml
- 2,2-bis (bromomethyl) propane-1,3-diol 221.8 mg
- potassium carbonate 146.3 mg
- reaction mixture was directly purified by silica gel column chromatography (chloroform-MeOH), and [( ⁇ (1S, 3R, 4S, 5S) -4- [3,3-bis (hydroxymethyl) azetidin-1-yl-adamantane- 1 mg of 1-yl ⁇ methyl) amino] -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile was obtained.
- Examples 41 and 42 rel-4-[( ⁇ (1R, 3S, 5R) -4-[(trans-4-hydroxycyclohexyl) amino] adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (trifluoromethoxy) Benzyl] amino ⁇ pyrimidine-5-carbonitrile stereoisomer mixture (39 mg) was separated and purified by reversed-phase liquid chromatography (eluent: 0.2% formic acid / MeOH / water mixture).
- Example 43 rel-trans-4- ⁇ [(1R, 2S, 3S, 5S) -5- (aminomethyl) adamantan-2-yl] amino ⁇ cyclohexanol (80 mg) of 1,3-dimethylimidazolidin-2-one ( To the solution, DIPEA (0.20 ml) and 4-chloro-2- ⁇ [2- (methylsulfanyl) benzyl] amino ⁇ pyrimidine-5-carbonitrile (108.6 mg) were added and stirred overnight at room temperature.
- reaction mixture was purified by amino silica gel flash column chromatography (chloroform-MeOH) to give rel-4-[( ⁇ (1R, 3S, 4R, 5R) -4-[(trans-4-hydroxycyclohexyl) 73.4 mg of amino] adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (methylsulfanyl) benzyl] amino ⁇ pyrimidine-5-carbonitrile was obtained.
- Example 63 rel-N-[(1R, 2S, 3S, 5S) -5- ⁇ [(5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] methyl ⁇ adamantane
- 2-yl] glycine ethyl ester 24 mg
- 4M aqueous lithium hydroxide solution 0.1 ml
- 1M hydrochloric acid 0.4 ml was added to the reaction mixture and stirred, and then the reaction mixture was concentrated under reduced pressure.
- Example 64 rel-4-( ⁇ [(1S, 3R, 5S) -4-oxoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile
- dichloromethane 0.8 ml
- trans-4-aminocyclohexanol 14.7 mg
- sodium triacetoxyborohydride 53.9 mg
- Saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, and the mixture was extracted with EtOAc. The organic layer was washed with saturated brine, and dried over anhydrous sodium sulfate.
- Example 68 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (methylsulfanyl) benzyl] amino ⁇ pyrimidine-5-carbo 4-Hydroxycyclohexanone (39.4 mg) and sodium triacetoxyborohydride (146.3 mg) were added to a solution of nitrile (50 mg) in dichloromethane (3.5 ml), and the mixture was stirred at room temperature for 2 hours.
- Example 105 rel-4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(4-hydroxycyclohexyl) amino] adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (trifluoromethoxy)
- MeOH MeOH
- 37% aqueous formalin solution 28.4 ⁇ l
- sodium cyanoborohydride 8.3 mg
- Example 117 rel-4-( ⁇ [(1S, 3R, 5S) -4-oxoadamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile
- sodium borohydride (13.5 mg) and stirred at room temperature.
- the reaction mixture was diluted with EtOAc, and the organic layer was washed successively with water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure.
- Example 120 4-[(1-Azabicyclo [2.2.2] oct-3-ylmethyl) amino] -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile (40 mg) in dichloromethane (1 ml) To the solution, 77% MCPBA (water-containing product, 21 mg) was added under ice cooling, and the mixture was stirred at room temperature for 3 hours. Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with EtOAc. The organic layer was dried over anhydrous sodium sulfate, and after removing the desiccant, the solvent was distilled off under reduced pressure.
- MCPBA water-containing product, 21 mg
- Example 121 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4- (tetrahydro-2H-thiopyran-4-ylamino) adamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoro
- Example 122 4- ⁇ [(3-endo) -8-benzyl-8-azabicyclo [3.2.1] oct-3-yl] amino ⁇ -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-
- ammonium formate 100 mg
- Pd / C 50% water-containing product
- Example 123 tert-butyl rel- ⁇ (1R, 2S, 3S, 5S) -5-[( ⁇ 2-[(2-chlorobenzyl) amino] -5-cyanopyrimidin-4-yl ⁇ amino) methyl] adamantane-2- Ir ⁇ carbamate (70 mg) in dichloromethane (1 ml) was added trifluoroacetic acid (0.135 ml) and stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure, an aqueous potassium carbonate solution was added to the resulting residue, and the mixture was stirred at room temperature for 3 hours.
- Example 170 tert-butyl 3- ⁇ [(5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] methyl ⁇ -8-azabicyclo [3.2.1] octane-8-
- 4M hydrogen chloride dioxane solution (0.28 ml)
- the reaction mixture was concentrated under reduced pressure and 4-[(8-azabicyclo [3.2.1] oct-3-ylmethyl) amino] -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile. 56 mg of dihydrochloride was obtained.
- Example 175 4-( ⁇ [1- (Tetrahydro-2H-pyran-2-yl) -1H-benzimidazol-5-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-
- 1M hydrochloric acid (1 ml) was added to the reaction mixture, and the mixture was further stirred at 60 ° C. for 24 hours.
- the reaction solution was concentrated under reduced pressure, and EtOAc and EtOH were added to the resulting residue.
- Example 176 Benzyl rel-[(1R, 2S, 3S, 5S) -5- ⁇ [(5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] methyl ⁇ adamantane- 10% Pd / C (50% water-containing product, 15 mg) was added to a solution of 2-yl] carbamate (55 mg) in MeOH (3 ml), and the mixture was stirred at room temperature for 6 hours under a hydrogen atmosphere at normal pressure. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure.
- the obtained residue was purified by preparative alumina thin layer chromatography (chloroform-MeOH) to obtain rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-aminoadamantan-1-yl 20.0 mg of] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile was obtained.
- Example 186 Benzyl rel- ⁇ (1R, 2S, 3S, 5S) -5-[( ⁇ 5-cyano-2-[(2,5-dichlorobenzyl) amino] pyrimidin-4-yl ⁇ amino) methyl] adamantane-2- Yl ⁇ carbamate (45 mg) in acetic acid (1.5 ml) was added 48% hydrobromic acid (1.5 ml) and stirred at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure, the residue was dissolved in EtOAc, washed successively with aqueous potassium carbonate solution, water and saturated brine, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure.
- Example 188 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4- ⁇ [(2-phenyl-1,3-dioxan-4-yl) methyl] amino ⁇ adamantan-1-yl] methyl ⁇ amino)
- THF 1.5 ml
- 1M hydrochloric acid 1.5 ml
- the solid obtained by filtering the precipitate and drying under reduced pressure was purified by amino silica gel column chromatography (chloroform-MeOH) to obtain rel-4-[( ⁇ (1S, 3R, 4S, 5S ) -4-[(2,4-dihydroxybutyl) amino] adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile 41.2 mg Obtained.
- Example 191 rel-4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(2,2-dimethyl-1,3-dioxan-5-yl) amino] adamantan-1-yl ⁇ methyl) amino]-
- 2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile 55 mg
- 1M hydrochloric acid 1.0 ml
- Example 207 rel-2-[(2-Chlorobenzyl) amino] -4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(2-chloroethyl) amino] adamantan-1-yl ⁇ methyl) amino] pyrimidine
- Potassium iodide (13.8 mg) and DIPEA (0.02 ml) were added to a DMF (0.27 ml) solution of -5-carbonitrile (27.0 mg) and (2S) -pyrrolidin-2-ylmethanol (7.9 mg) at 75 ° C.
- (2S) -pyrrolidin-2-ylmethanol (1.1 mg) was added, and the mixture was further stirred at the same temperature for 2 hr.
- Example 220 rel-4-[( ⁇ (1R, 3S, 4R, 5R) -4-[(cis-4-aminocyclohexyl) amino] adamantan-1-yl ⁇ methyl) amino] -2-[(2-chlorobenzyl) DIPEA (80.4 ⁇ l) and 2-bromoethanol (8.2 ⁇ l) were added to a DMI (1.0 ml) solution of amino] pyrimidine-5-carbonitrile (30 mg), and the mixture was heated and stirred at 120 ° C.
- DMI 1.0 ml
- reaction mixture was diluted with chloroform and purified by amino silica gel flash column chromatography (chloroform-MeOH) to obtain rel-2-[(2-chlorobenzyl) amino] -4-( ⁇ [ 17.6 mg of (1R, 3S, 4R, 5R) -4-( ⁇ cis-4-[(2-hydroxyethyl) amino] cyclohexyl ⁇ amino) adamantan-1-yl] methyl ⁇ amino) pyrimidine-5-carbonitrile Obtained.
- Example 235 rel-2-[(2-chlorobenzyl) amino] -4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(4-oxocyclohexyl) amino] adamantan-1-yl ⁇ methyl) amino]
- pyrimidine-5-carbonitrile 50 mg
- dichloromethane 3.0 ml
- 4,4-difluoropiperidine hydrochloride (30.4 mg)
- sodium triacetoxyborohydride (61.2 mg) were sequentially added, Stir at room temperature. After completion of the reaction, saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, and the mixture was extracted with chloroform.
- Example 281 rel-4-[( ⁇ (1R, 3S, 4R, 5R) -4-[(cis-4-aminocyclohexyl) amino] adamantan-1-yl ⁇ methyl) amino] -2-[(2-chlorobenzyl) 1-Acetylpiperidin-4-one (21.7 mg) and sodium triacetoxyborohydride (48.9 mg) were sequentially added to a solution of amino] pyrimidine-5-carbonitrile (40 mg) in dichloromethane (2.0 ml) and stirred at room temperature. did. After completion of the reaction, saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, and the mixture was extracted with chloroform. The solvent was evaporated under reduced pressure.
- the obtained residue was purified by amino silica gel flash column chromatography (chloroform-MeOH) to obtain rel-4-( ⁇ [(1R, 3S, 4R, 5R) -4-( ⁇ cis-4-[( 54 mg of 1-acetylpiperidin-4-yl) amino] cyclohexyl ⁇ amino) adamantan-1-yl] methyl ⁇ amino) -2-[(2-chlorobenzyl) amino] pyrimidine-5-carbonitrile were obtained.
- Example 338 rel-4-( ⁇ [(1S, 3R, 4S, 5S) -4-( ⁇ [trans-4-( ⁇ [tert-butyl (dimethyl) silyl] oxy ⁇ methyl) cyclohexyl] methyl ⁇ amino) adamantane-1 1M hydrochloric acid (0.6 ml) was added to a suspension of -yl] methyl ⁇ amino) -2-[(2-cyanobenzyl) amino] pyrimidine-5-carbonitrile (68 mg) in MeOH (1.4 ml) at room temperature. Stir for hours. After evaporating the solvent under reduced pressure, the residue was diluted with chloroform, and saturated aqueous sodium hydrogen carbonate was added under ice-cooling.
- Example 351 4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(3- ⁇ [tert-butyldimethyl) silyl] oxypropyl) (methyl) amino] adamantan-1-yl ⁇ methyl) under ice cooling Amino] -2- ⁇ [2- (methylsulfanyl) benzyl] amino ⁇ pyrimidine-5-carbonitrile (55 mg) in THF (2 mL) was added 1M tetrabutylammonium fluoride in THF (0.20 mL) at room temperature. Stir for 1 hour.
- the solvent was distilled off under reduced pressure and purified by amino silica gel flash column chromatography (chloroform-MeOH) as it was, and 4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(3-hydroxypropyl) (methyl 25 mg of) amino] adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (methylsulfanyl) benzyl] amino ⁇ pyrimidine-5-carbonitrile was obtained.
- Example 353 rel-4-[( ⁇ (1S, 3R, 4S, 5S) -4- [3- (1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl) azetidin-1-yl] Adamantan-1-yl ⁇ methyl) amino] -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile (64 mg) in EtOH (1.28 ml) and THF (1.28 ml) To the mixture was added hydrazine monohydrate (18.9 ⁇ l), and the mixture was heated to reflux. The insoluble material was removed by filtration, and the filtrate was concentrated under reduced pressure.
- Chloroform was added to the resulting residue, washed twice with water, and dried over anhydrous sodium sulfate. After removing the desiccant, the solvent was distilled off under reduced pressure, and the residue was purified by amino silica gel flash column chromatography (chloroform-MeOH) to obtain rel-4-( ⁇ [(1S, 3R, 4S, 5S) 14 mg of 4- (3-aminoazetidin-1-yl) adamantan-1-yl] methyl ⁇ amino) -2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidine-5-carbonitrile was obtained. .
- Example 356 rel-2-[(2-chlorobenzyl) amino] -4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(piperidin-4-ylmethyl) amino] adamantan-1-yl ⁇ methyl) amino 1-Fluoro-3-iodopropane (18 mg) and DIPEA (0.017 ml) were added to a DMI solution (1 ml) of pyrimidine-5-carbonitrile (40 mg), and microwave irradiation was performed at 100 ° C. for 1 hour.
- reaction mixture was diluted with chloroform and purified by amino silica gel flash column chromatography (hexane-EtOAc) to obtain rel-2-[(2-chlorobenzyl) amino] -4-( ⁇ [(1S, 3R , 4S, 5S) -4-( ⁇ [1- (3-Fluoropropyl) piperidin-4-ylmethyl] methyl ⁇ amino) adamantan-1-yl ⁇ methyl ⁇ amino) pyrimidine-5-carbonitrile was obtained in an amount of 20 mg.
- Example 376 Under ice cooling, tert-butyl 4- ⁇ 4-[(5-cyano-2- ⁇ [2- (trifluoromethoxy) benzyl] amino ⁇ pyrimidin-4-yl) amino] butanoyl ⁇ piperazine-1-carboxylate ( 205 mg) in dichloromethane (4.1 ml) was added TFA (0.75 ml) and stirred at room temperature for 18 hours. The reaction mixture was concentrated, an aqueous potassium carbonate solution was added under ice cooling, and the mixture was stirred at room temperature. The precipitated solid was collected by filtration, dried, and purified by amino silica gel flash column chromatography (chloroform-MeOH).
- Example 412 rel-4-[( ⁇ (1S, 3R, 4S, 5S) -4-[(1,4-Dioxaspiro [4.5] dec-8-ylmethyl) amino] adamantan-1-yl ⁇ methyl) amino] -2- 1M hydrochloric acid (2 ml) was added to a THF solution (3 ml) of ( ⁇ [2- (methylsulfanyl) pyridin-3-yl] methyl ⁇ amino) pyrimidine-5-carbonitrile (140 mg), and the mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure, saturated aqueous sodium hydrogen carbonate was added, and the mixture was extracted with EtOAc.
- each Example compound was produced using the corresponding raw material.
- the structure of each example compound, production method and physicochemical data are shown in the following table.
- the compound of formula (I) or a pharmaceutically acceptable salt thereof has a PKC ⁇ inhibitory action and can be used as an inhibitor of acute rejection in transplantation.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Transplantation (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
各isozymeの発現分布は比較的広範にわたっているが、nPKCのひとつであるPKCθの発現は、Tリンパ球と骨格筋に限局している。また、PKCθのノックアウトマウスの表現型が、T細胞シグナル伝達阻害やT cell anergyの誘導であり、また、骨格筋の異常はみとめられていないことから、PKCθが副作用の少ない免疫抑制剤のターゲットとして有望である。









即ち、本発明は、式(I)の化合物又はその製薬学的に許容される塩、並びに、式(I)の化合物又はその製薬学的に許容される塩及び製薬学的に許容される賦形剤を含有する医薬組成物に関する。

R1は、

R4は、-OH、置換されていてもよいアミン又は-CH2NH2を示し;
n1は、0又は1を示し;
R5は、-OH、(-OH又は-NH2で置換されていてもよいC1-6アルキル)又は-CNを示し;
R6は、-H又はアリールで置換されていてもよいC1-6アルキルを示し;
pは、0又は1を示し;
qは、1、2、3又は4を示し;
R13は、-H又はC1-6アルキルを示し;
R2は、-CN、-CF3、-NO2又はハロゲンを示し;
Aは、単結合又はC1-6アルキレンを示し;
R3は、

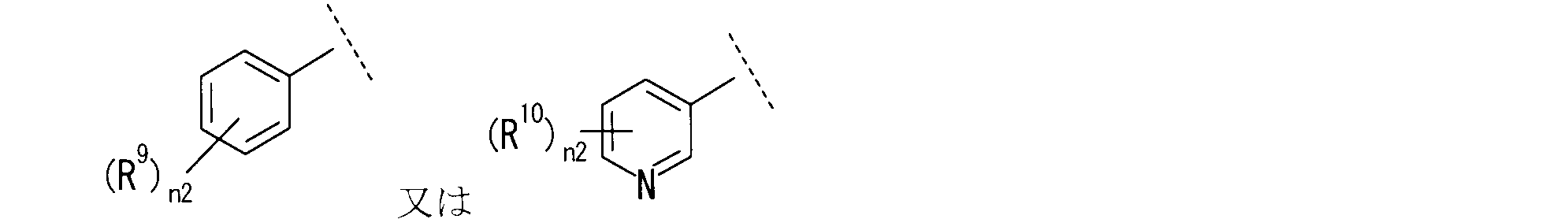
R9は、同一又は互いに異なって、ハロゲン、置換されていてもよいC1-6アルキル、-OH、-CN、シクロアルキル、-Q-(置換されていてもよいC1-6アルキル)又は置換されていてもよいアリールを示し;
Qは、-O-、-S-、-SO-、-SO2-又は-NHSO2-を示し;
n2は、0、1、2又は3を示し;
R10は、ハロゲン、C1-6アルキル、-CN、-O-(C1-6アルキル)、-S-(C1-6アルキル)、-SO-(C1-6アルキル)、-SO2-(C1-6アルキル)、-S-(シクロアルキル)又は-OCF3を示し;
R12は、-H又はハロゲンを示す。)
なお、特に記載がない限り、本明細書中のある化学式中の記号が他の化学式においても用いられる場合、同一の記号は同一の意味を示す。
さらに、本発明は、移植における急性拒絶反応抑制剤の製造のための式(I)の化合物又はその製薬学的に許容される塩の使用、並びに、式(I)の化合物又はその製薬学的に許容される塩の有効量を患者に投与することからなる移植における急性拒絶反応の抑制方法に関する。
[1]
式(I)の化合物又はその製薬学的に許容される塩。
R1は、

R4は、-OH、置換されていてもよいアミン又は-CH2NH2を示し;
n1は、0又は1を示し;
R5は、-OH、(-OH又は-NH2で置換されていてもよいC1-6アルキル)又は-CNを示し;
R6は、-H又はアリールで置換されていてもよいC1-6アルキルを示し;
pは、0又は1を示し;
qは、1、2、3又は4を示し;
R13は、-H又はC1-6アルキルを示し;
R2は、-CN、-CF3、-NO2又はハロゲンを示し;
Aは、単結合又はC1-6アルキレンを示し;
R3は、

R9は、同一又は互いに異なって、ハロゲン、置換されていてもよいC1-6アルキル、-OH、-CN、シクロアルキル、-Q-(置換されていてもよいC1-6アルキル)又は置換されていてもよいアリールを示し;
Qは、-O-、-S-、-SO-、-SO2-又は-NHSO2-を示し;
n2は、0、1、2又は3を示し;
R10は、ハロゲン、C1-6アルキル、-CN、-O-(C1-6アルキル)、-S-(C1-6アルキル)、-SO-(C1-6アルキル)、-SO2-(C1-6アルキル)、-S-(シクロアルキル)又は-OCF3を示し;
R12は、-H又はハロゲンを示す。)
[2]
R4が-OH、-NR7R8又は-CH2NH2であり;
R7、R8が、互いに同一または異なって、
(a)-H;
(b)C1-6アルキル。ここで該C1-6アルキルは、以下の1)~12)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)保護された-OH
3)ハロゲン
4)-COOH
5)-CONH2
6)オキソ
7)アリール
8)ヘテロアリール
9)-OH、保護された-OH、(-OHで置換されていてもよいC1-6アルキル)、ハロゲン、-CN、-NR14R15、-CONR14R15、-SO2NR14R15、(-OHで置換されていてもよいC1-6アルキル)-O-、及びオキソからなる群から選択される1つ以上の基で置換されていてもよいシクロアルキル
10)-OH又は(-OH、-OCH3、-CN又はハロゲンで置換されていてもよいC1-6アルキル)で置換されていてもよいヘテロシクロアルキル
11)(-OH又は-NH2で置換されていてもよいヘテロシクロアルキル)-CO-、及び、
12)(ヘテロシクロアルキル)-NH-CO-;
(c)シクロアルキル。ここで該シクロアルキルは、以下の1)~6)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)-NHR11
3)ハロゲン
4)オキソ
5)-OHで置換されていてもよいC1-6アルキル、及び、
6)(ハロゲン、-OH、-CH2OH又は-COCH3)で置換されていてもよいヘテロシクロアルキル;
(d)ヘテロシクロアルキル。ここで、該ヘテロシクロアルキルは以下の1)~11)からなる群から選択される1つ以上の基で置換されてもよい。
1)(-OH、-OCH3、-CN、ハロゲン又は-CONH2)で置換されていてもよいC1-6アルキル
2)シクロアルキル
3)アリール
4)ヘテロシクロアルキル
5)ヘテロシクロアルキル-CO-
6)-COCH3
7)-CONH2
8)-COCH2OH
9)-COOCH2CH3
10)-SO2CH3
11)オキソ、及び
12)ハロゲン;
(e)アリール;
(f)ニコチノイル;
(g)-SO2CH3;
又は
(h)R7とR8がそれらが結合する窒素原子と一体となって、(-OH、-NH2、-COOH、-COCH3、-CONH2および-CH2OH)からなる群から選択される1つ以上の基で置換されていてもよい含窒素へテロシクロアルキルであり;
R11が、-H、(ハロゲン又は-OH)で置換されていてもよいC1-6アルキル、ハロゲンで置換されていてもよいシクロアルキル、-COCH3で置換されていてもよいヘテロシクロアルキル又は-COCH3であり;
R14、R15が、互いに同一又は異なって、-H、C1-6アルキル又はヘテロシクロアルキルである、[1]に記載の化合物又はその製薬学的に許容される塩。
[3]
R1が

R3が、
[4]
R4が-NR7R8であり、
R7、R8が、互いに同一または異なって、
(b)C1-6アルキル。ここで該C1-6アルキルは、以下の1)~12)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)メチル基で保護された-OH又は互いに近接する二つのOH基を有する場合はジメチルメチレン基又はベンジリデン基で保護された-OH
3)-F
4)-COOH
5)-CONH2
6)オキソ
7)フェニル
8)ピリジル
9)-OH及び(-OHで置換されていてもよいC1-6アルキル)からなる群から選択される1つ以上の基で置換されていてもよいシクロヘキシル
10)-OH又は(-OH、-OCH3、-CN又は-Fで置換されていてもよいC1-6アルキル)で置換されていてもよい(ピペリジニル又はピロリジニル)
11)(ピペラジニル)-CO-又は(-OH又は-NH2で置換されていてもよいピペリジニル)-CO-、及び、
12)(ピペリジニル)-NH-CO-;
又は
(c)シクロアルキル。ここで該シクロアルキルは、以下の1)~6)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)-NHR11
3)-F
4)オキソ
5)-OHで置換されていてもよいC1-6アルキル、及び、
6)(ハロゲン、-OH、-CH2OH又は-COCH3)で置換されていてもよい(アゼチジニル、ピロリジニル、ピペリジニル、ピペラジニル又はモルホリニル)であり;
R11が、-Hであり;
n1が1であり;
R2が、-CN、-CF3、-NO2又は-Fであり;
Aが、C1-6アルキレンであり;
R9が、
(i)-F、-Cl又は-Br
(j)-OH又はハロゲンで置換されていてもよいC1-6アルキル、
(k)-OH、
(l)-CN、
(m)シクロプロピル、
(n)-Q-(ハロゲン、-OH、-OCH3、-CN又は-CONH2で置換されていてもよいC1-6アルキル)
又は
(o)-CH2NH2で置換されていてもよいフェニルであり;
n2が1である、[3]に記載の化合物又はその製薬学的に許容される塩。
[5]
R7、R8が、互いに同一または異なって、
(b)C1-6アルキル。ここで該C1-6アルキルは、以下の基で置換されていてもよい。
9)-OH、-CH3、-CH2OHからなる群から選択される1つ以上の基で置換されたシクロヘキシル、及び、
10)-OH又は(-OH、-OCH3、-CN又は-Fで置換されていてもよいC1-6アルキル)で置換されていてもよいピペリジニル;
又は
(c)シクロアルキル。ここで該シクロアルキルは、以下の1)、2)、5)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)-NHR11、及び、
5)-OHで置換されていてもよいC1-6アルキルであり;
R11が、-Hであり;
R2が、-CNであり;
Aが、メチレンであり;
R9が、
(i)-F、-Cl又は-Br
(j)-OH又は-Fで置換されていてもよいC1-6アルキル
(k)-OH、
(l)-CN、
(m)シクロプロピル、
(n)-Q-(ハロゲン、-OH、-OCH3、-CN又は-CONH2で置換されていてもよいC1-6アルキル)
又は
(o)-CH2NH2で置換されていてもよいフェニルであり;
R10が、-Cl、-CH3、-OCH3、-OCH2CH3、-OCH(CH3)2、-SCH3、-SCH2CH3、-SCH(CH3)2、-SOCH3、-SO2CH3、-S-(シクロペンタン)又は-OCF3である、[4]に記載の化合物又はその製薬学的に許容される塩。
[6]
[1]に記載の化合物又はその製薬学的に許容される塩、及び製薬学的に許容される賦形剤を含有する医薬組成物。
[7]
[1]に記載の化合物又はその製薬学的に許容される塩を含有する、PKCθ阻害剤。
[8]
[1]に記載の化合物又はその製薬学的に許容される塩を含有する、移植における急性拒絶反応の抑制用医薬組成物。
[9]
移植における急性拒絶反応抑制剤の製造のための[1]に記載の化合物又はその製薬学的に許容される塩の使用。
[10]
[1]に記載の化合物又はその塩の有効量を患者に投与することからなる移植における急性拒絶反応の抑制方法。
本明細書中において、「C1-6アルキル」とは、直鎖又は分枝状の炭素数が1から6のアルキル、例えばメチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチル、n-ヘキシル基等である。
(1)単環式飽和へテロ環
(a)1~4個の窒素原子を含むもの、例えば、アゼパニル、ジアゼパニル、アジリジニル、アゼチジニル、ピロリジニル、イミダゾリジニル、ピペリジル、ピラゾリジニル、ピペラジニル、アゾカニル等;
(b)1~3個の窒素原子、ならびに1~2個の硫黄原子および/または1~2個の酸素原子を含むもの、例えば、チオモルホリニル、チアゾリジニル、イソチアゾリジニル、オキサゾリジニル、モルホリニル等;
(c)1~2個の硫黄原子を含むもの、例えば、テトラヒドロ-2H-チオピラニル等;
(d)1~2個の硫黄原子および1~2個の酸素原子を含むもの、例えば、オキサチオラニル等;
(e)1~2個の酸素原子を含むもの、例えば、オキシラニル、オキセタニル、ジオキソラニル、テトラヒドロフラニル、テトラヒドロ-2H-ピラニル、1,4-ジオキサニル等;
(a)1~4個の窒素原子を含むもの、例えば、ピロリル、イミダゾリル、ピラゾリル、ピリジル、ジヒドロピリジル、テトラヒドロピリジニル、ピリミジニル、ピラジニル、ピリダジニル、トリアゾリル、テトラゾリル、トリアジニル、ジヒドロトリアジニル、アゼピニル等;
(b)1~3個の窒素原子、ならびに1~2個の硫黄原子および/または1~2個の酸素原子を含むもの、例えば、チアゾリル、イソチアゾリル、チアジアゾリル、ジヒドロチアジニル、オキサゾリル、イソオキサゾリル、オキサジアゾリル、オキサジニル等;
(c)1~2個の硫黄原子を含むもの、例えば、チエニル、チエピニル、ジヒドロジチオピラニル、ジヒドロジチオニル等;
(d)1~2個の硫黄原子および1~2個の酸素原子を含むもの、具体的には、ジヒドロオキサチオピラニル等;
(e)1~2個の酸素原子を含むもの、例えば、フリル、ピラニル、オキセピニル、ジオキソリル等;
(a)1~5個の窒素原子を含むもの、例えば、キヌクリジニル、7-アザビシクロ[2.2.1]ヘプチル、3-アザビシクロ[3.2.2]ノナニル等;
(b)1~4個の窒素原子、ならびに1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、トリチアジアザインデニル、ジオキソロイミダゾリジニル等;
(c)1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、2,6-ジオキサビシクロ[3.2.2]オクト-7-イル等;
(a)1~5個の窒素原子を含むもの、例えば、インドリル、イソインドリル、インドリニル、インドリジニル、ベンゾイミダゾリル、ジヒドロベンゾイミダゾリル、テトラヒゾロベンゾイミダゾリル、キノリル、テトラヒドロキノリル、イソキノリル、テトラヒドロイソキノリル、インダゾリル、イミダゾピリジル、ベンゾトリアゾリル、テトラゾロピリダジニル、カルバゾリル、アクリジニル、キノキサリニル、ジヒドロキノキサリニル、テトラヒドロキノキサリニル、フタラジニル、ジヒドロインダゾリル、ベンゾピリミジニル、ナフチリジニル、キナゾリニル、シンノリニル等;
(b)1~4個の窒素原子、ならびに1~3個の硫黄原子および/または1~3個の酸素原子を含むもの、例えば、ベンゾチアゾリル、ジヒドロベンゾチアゾリル、ベンゾチアジアゾリル、イミダゾチアゾリル、イミダゾチアジアゾリル、ベンゾオキサゾリル、ジヒドロベンゾオキサゾリル、ジヒドロベンゾオキサジニル、ベンゾオキサジアゾリル、ベンゾイソチアゾリル、ベンゾイソオキサゾリル等;
(c)1~3個の硫黄原子を含むもの、例えば、ベンゾチエニル、ベンゾジチオピラニル、ジベンゾ[b,d]チエニル等;
(d)1~3個の硫黄原子および1~3個の酸素原子を含むもの、例えば、ベンゾオキサチオピラニル、フェノキサジニル等;
(e)1~3個の酸素原子を含むもの、例えば、ベンゾジオキソリル、ベンゾフラニル、ジヒドロベンゾフラニル、イソベンゾフラニル、クロマニル、クロメニル、ジベンゾ[b,d]フラニル、メチレンジオキシフェニル、エチレンジオキシフェニル等;
など。
(1)R1が

(2)R4が、-OH、-NR7R8又は-CH2NH2であり、別の態様としては、-NR7R8である(1)に記載の化合物。
(3)R7、R8が、互いに同一または異なって、
(a)-H;
(b)C1-6アルキル。ここで該C1-6アルキルは、以下の1)~12)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)保護された-OH、別の態様としては、メチル基で保護された-OH又は互いに近接する二つのOH基を有する場合はジメチルメチレン基又はベンジリデン基で保護された-OH
3)ハロゲン、別の態様としては、-F
4)-COOH
5)-CONH2
6)オキソ
7)アリール、別の態様としては、フェニル
8)ヘテロアリール、別の態様としては、ピリジル
9)-OH、保護された-OH、(-OHで置換されていてもよいC1-6アルキル)、ハロゲン、-CN、-NR14R15、-CONR14R15、-SO2NR14R15、(-OHで置換されていてもよいC1-6アルキル)-O-及びオキソからなる群から選択される1つ以上の基で置換されていてもよいシクロアルキル、別の態様としては、-OH及び(-OHで置換されていてもよいC1-6アルキル)からなる群から選択される1つ以上の基で置換されていてもよいシクロヘキシル、さらに別の態様としては、-OH、-CH3、-CH2OHからなる群から選択される1つ以上の基で置換されたシクロヘキシル
10)-OH又は(-OH、-OCH3、-CN又はハロゲンで置換されていてもよいC1-6アルキル)で置換されていてもよいヘテロシクロアルキル、別の態様としては、-OH又は(-OH、-OCH3、-CN又はFで置換されていてもよいC1-6アルキル)で置換されていてもよい(ピペリジニル又はピロリジニル)
11)(-OH又は-NH2で置換されていてもよいヘテロシクロアルキル)-CO-、別の態様としては、(ピペラジニル)-CO-又は(-OH又は-NH2で置換されていてもよいピペリジニル)-CO-、及び、
12)(ヘテロシクロアルキル)-NH-CO-、別の態様としては、(ピペリジン)-NH-CO-;
(c)シクロアルキル、別の態様としてはシクロブチル又はシクロヘキシル。ここで、該シクロアルキルは、以下の1)~6)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)-NHR11
3)ハロゲン、別の態様としては、-F
4)オキソ
5)-OHで置換されていてもよいC1-6アルキル、別の態様としては、-CH3又は-CH2OH、及び、
6)(ハロゲン、-OH、-CH2OH又は-COCH3)で置換されていてもよいヘテロシクロアルキル、別の態様としては、(-F、-OH、-CH2OH又は-COCH3)で置換されていてもよい(アゼチジニル、ピロリジニル、ピペリジニル、ピペラジニル又はモルホリニル);
(d)ヘテロシクロアルキル、別の態様としては、アゼチジニル、ピペリジニル、テトラヒドロ-2H-ピラニル又はテトラヒドロ-2H-チオピラニル。ここで、該ヘテロシクロアルキルは以下の1)~11)からなる群から選択される1つ以上の基で置換されてもよい。
1)(-OH、-OCH3、-CN、ハロゲン又は-CONH2)で置換されていてもよいC1-6アルキル、別の態様としては、(-OH、-OCH3、-CN、-F又は-CONH2)で置換されていてもよいC1-6アルキル
2)シクロアルキル、別の態様としてはシクロプロピル
3)アリール、別の態様としてはフェニル
4)ヘテロシクロアルキル、別の態様としてはテトラヒドロ-2H-ピラニル
5)ヘテロシクロアルキル-CO-、別の態様としては、モルホリニル-CO-
6)-COCH3
7)-CONH2
8)-COCH2OH
9)-COOCH2CH3
10)-SO2CH3
11)オキソ、及び、
12)ハロゲン;
(e)アリール、別の態様としてはフェニル;
(f)ニコチノイル;
(g)-SO2CH3;
又は
(h)R7とR8がそれらが結合する窒素原子と一体となって、(-OH、-NH2、-COOH、-COCH3、-CONH2および-CH2OH)からなる群から選択される1つ以上の基で置換されていてもよい含窒素へテロシクロアルキル、別の態様としては(-OH、-NH2、-COOH、-COCH3、-CONH2および-CH2OH)からなる群から選択される1つ以上の基で置換されていてもよい(アゼチジニル、ピロリジニル、ピペリジニル又はピペラジニル)
である(2)に記載の化合物。
(4)R11が、-H、(ハロゲン又は-OH)で置換されていてもよいC1-6アルキル、ハロゲンで置換されていてもよいシクロアルキル、-COCH3で置換されていてもよいヘテロシクロアルキル又は-COCH3であり、別の態様としては、-H、(-F又は-OH)で置換されていてもよいC1-6アルキル、-Fで置換されていてもよいシクロアルキル、-COCH3で置換されていてもよいヘテロシクロアルキル又は-COCH3であり、さらに別の態様としては、(-F又は-OH)で置換されていてもよいC1-6アルキル、-Fで置換されていてもよいシクロヘキシル、テトラヒドロ-2H-ピラニル、-COCH3で置換されたピペリジニル又は-COCH3である(3)に記載の化合物。
(5)R14、R15が、互いに同一又は異なって、-H、C1-6アルキル又はヘテロシクロアルキルであり、別の態様としては、-H、メチル、又はテトラヒドロ-2H-ピラニルである(3)に記載の化合物。
(6)n1が、1である(1)に記載の化合物。
(7)R5が、-OH、-CH2OH、-CH2NH2又は-CNである化合物。
(8)R6が、-H又はアリールで置換されていてもよいC1-6アルキルであり、別の態様としては、-H又はフェニルで置換されていてもよいC1-6アルキルである化合物。
(9)R2が、-CNである化合物。
(10)Aが、C1-6アルキレンであり、別の態様としては、メチレン又はエチレンであり、さらに別の態様としては、メチレンである化合物。
(11)R3が、

(12)R9が、同一又は互いに異なって、
(i)ハロゲン、別の態様としては、-F、-Cl又は-Br;
(j)置換されていてもよいC1-6アルキル、別の態様としては、-OH又はハロゲンで置換されていてもよいC1-6アルキル、さらに別の態様としては、-OH又は-Fで置換されていてもよいC1-6アルキル;
(k)-OH;
(l)-CN;
(m)シクロアルキル、別の態様としてはシクロプロピル;
(n)-Q-(置換されていてもよいC1-6アルキル)、別の態様としては、-Q-(ハロゲン、-OH、-OCH3、-CN又は-CONH2で置換されていてもよいC1-6アルキル);
又は
(o)置換されていてもよいアリール、別の態様としては-CH2NH2で置換されていてもよいアリール、さらに別の態様としては、-CH2NH2で置換されていてもよいフェニル
である化合物であり、さらに別の態様としては、-Cl、-O-CF3、-O-CHF2又は-SCH3ある(11)に記載の化合物。
(13)n2が、1である(11)に記載の化合物。
(14)R10が、-Cl、-CH3、-OCH3、-OCH2CH3、-OCH(CH3)2、-SCH3、-SCH2CH3、-SCH(CH3)2、-SOCH3、-SO2CH3、-S-(シクロペンタン)又は-OCF3であり、別の態様としては、-Cl、-CH3、-OCH3又は-SCH3である(11)に記載の化合物。
(15)R12が、-H又は-Clである化合物。
(16)上記(1)~(15)に記載の基のうち二以上の組み合わせである化合物。
また、式(I)の化合物には、不斉炭素原子や軸不斉を有する場合があり、これに基づく光学異性体が存在しうる。本発明は、式(I)の化合物の光学異性体の分離されたもの、あるいはそれらの混合物も包含する。
式(I)の化合物及びその製薬学的に許容される塩は、その基本構造あるいは置換基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することができる。その際、官能基の種類によっては、当該官能基を原料から中間体へ至る段階で適当な保護基(容易に当該官能基に転化可能な基)に置き換えておくことが製造技術上効果的な場合がある。このような保護基としては、例えば、ウッツ(P. G. M. Wuts)及びグリーン(T. W. Greene)著、「Greene's Protective Groups in Organic Synthesis(第4版、2006年)」に記載の保護基等を挙げることができ、これらの反応条件に応じて適宜選択して用いればよい。このような方法では、当該保護基を導入して反応を行なったあと、必要に応じて保護基を除去することにより、所望の化合物を得ることができる。
また、式(I)の化合物のプロドラッグは、上記保護基と同様、原料から中間体へ至る段階で特定の基を導入、あるいは得られた式(I)の化合物を用いてさらに反応を行なうことで製造できる。反応は通常のエステル化、アミド化、脱水等、当業者に公知の方法を適用することにより行うことができる。
以下、式(I)の化合物の代表的な製造法を説明する。各製法は、当該説明に付した参考文献を参照して行うこともできる。なお、本発明の製造法は以下に示した例には限定されない。

本製法は、化合物(1)とアミン化合物(2)の求核置換反応により化合物(3)を製造し、さらに得られた化合物(3)とアミン化合物(4)の求核置換反応により、本発明化合物(I)を製造する方法である。ここで、脱離基の例には、ハロゲン、メタンスルホニルオキシ、メチルスルフィニル、メチルスルホニル、p-トルエンスルホニルオキシ基等が含まれる。
この反応では、化合物(1)と化合物(2)、もしくは化合物(3)と化合物(4)を等量若しくは一方を過剰量用い、これらの混合物を、反応に不活性な溶媒中、又は無溶媒下、冷却下から加熱還流下、好ましくは0℃~80℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、ジメトキシエタン等のエーテル類、ジクロロメタン、1,2-ジクロロエタン、クロロホルム等のハロゲン化炭化水素類、N,N-ジメチルホルムアミド、N-メチルピロリドン、1,3-ジメチル-2-イミダゾリジノン、ジメチルスルホキシド、酢酸エチル、アセトニトリル及びこれらの混合物が挙げられる。トリエチルアミン、N,N-ジイソプロピルエチルアミン若しくはN-メチルモルホリン等の有機塩基、又は炭酸カリウム、炭酸ナトリウム若しくは水酸化カリウム等の無機塩基の存在下で反応を行うのが、反応を円滑に進行させる上で有利な場合がある。なお、先に化合物(1)と(4)を反応させた後、化合物(2)を反応させても良い。
〔文献〕
S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善)
さらに、式(I)で示されるいくつかの化合物は、以上のように得られた本発明化合物から公知のアミド化、アルキル化、還元的アミノ化、カルボニル基の水酸基への還元等、当業者が通常採用しうる工程を任意に組み合わせることにより製造することもできる。例えば、以下の反応、後述の実施例に記載の方法、当業者にとって自明な方法、あるいはそれらの変法を適用することによって製造することができる。
カルボン酸化合物とアミン化合物をアミド化することにより、アミド化合物を得ることができる。
この反応では、カルボン酸化合物とアミン化合物とを等量若しくは一方を過剰量用い、これらの混合物を、縮合剤の存在下、反応に不活性な溶媒中、冷却下から加熱下、好ましくは-20℃~60℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、ベンゼン、トルエン若しくはキシレン等の芳香族炭化水素類、ジクロロメタン、1,2-ジクロロエタン若しくはクロロホルム等のハロゲン化炭化水素類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、ジメチルスルホキシド、酢酸エチル、アセトニトリル又は水、及びこれらの混合物が挙げられる。縮合剤の例としては、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド、ジシクロヘキシルカルボジイミド、1,1’-カルボニルジイミダゾール、ジフェニルリン酸アジド、オキシ塩化リンが挙げられるが、これらに限定されるものではない。添加剤(例えば1-ヒドロキシベンゾトリアゾール)を用いることが反応に好ましい場合がある。トリエチルアミン、N,N-ジイソプロピルエチルアミン若しくはN-メチルモルホリン等の有機塩基、又は炭酸カリウム、炭酸ナトリウム若しくは水酸化カリウム等の無機塩基の存在下で反応を行うことが、反応を円滑に進行させる上で有利な場合がある。
また、カルボン酸を反応性誘導体へ変換した後にアミン化合物と反応させる方法も用いることができる。カルボン酸の反応性誘導体の例としては、オキシ塩化リン、塩化チオニル等のハロゲン化剤と反応して得られる酸ハロゲン化物、クロロギ酸イソブチル等と反応して得られる混合酸無水物、1-ヒドロキシベンゾトリアゾール等と縮合して得られる活性エステル等が挙げられる。これらの反応性誘導体とアミン化合物との反応は、ハロゲン化炭化水素類、芳香族炭化水素類、エーテル類等の反応に不活性な溶媒中、冷却下~加熱下、好ましくは、-20℃~60℃で行うことができる。
〔文献〕
S. R. Sandler及びW. Karo著、「Organic Functional Group Preparations」、第2版、第1巻、Academic Press Inc.、1991年
日本化学会編「実験化学講座(第5版)」16巻(2005年)(丸善)
アミン化合物を脱離基を有する化合物と反応させてアルキル化することにより、アルキルアミン化合物を得ることができる。
アルキル化は製法1と同様の方法で行うことができる。
カルボニル化合物と一級又は二級アミン化合物より調製されるイミン化合物を還元することにより、アミン化合物をアルキル化することができる。
この反応では、カルボニル化合物と一級又は二級アミン化合物とを等量若しくは一方を過剰量用い、これらの混合物を、還元剤の存在下、反応に不活性な溶媒中、-45℃~加熱還流下、好ましくは0℃~室温において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定されないが、メタノール、エタノール等のアルコール類、ジクロロメタン、1,2-ジクロロエタン若しくはクロロホルム等のハロゲン化炭化水素類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、ジメトキシエタン等のエーテル類、及びこれらの混合物が挙げられる。還元剤としては、シアン化水素化ホウ素ナトリウム、トリアセトキシ水素化ホウ素ナトリウム、水素化ホウ素ナトリウム等が挙げられる。モレキュラーシーブス等の脱水剤、又は酢酸、塩酸、チタニウム(IV)イソプロポキシド錯体等の酸存在下で反応を行うことが好ましい場合がある。反応によっては、カルボニル化合物と一級又は二級アミン化合物との縮合によりイミンが生成し、安定な中間体として単離できる場合がある。そのような場合には、イミン中間体を単離した後、還元反応により目的物を得ることができる。また、前記還元剤での処理の代わりに、メタノール、エタノール、酢酸エチル等の溶媒中、酢酸、塩酸等の酸の存在下又は非存在下で、還元触媒(例えば、パラジウム炭素、ラネーニッケル等)を用いて反応を行うこともできる。この場合、反応を常圧から50気圧の水素雰囲気下で、冷却下から加熱下で行うことが好ましい。
〔文献〕
A. R. Katritzky及びR. J. K. Taylor著、「Comprehensive Organic Functional Group Transformations II」、第2巻、Elsevier Pergamon、2005年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善)
カルボニル化合物を還元することによりアルコール化合物を得ることができる。
この反応では、反応に不活性な溶媒中、冷却下から加熱下、好ましくは-20℃から80℃で、カルボニル化合物を等量若しくは過剰量の還元剤で、通常0.1時間~3日間処理する。ここで用いられる溶媒の例としては、特に限定されないが、ジエチルエーテル、テトラヒドロフラン、ジオキサン、ジメトキシエタン等のエーテル類、メタノール、エタノール、2-プロパノール等のアルコール類、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、ジメチルスルホキシド、酢酸エチル及びこれらの混合物が挙げられる。還元剤としては、水素化ホウ素ナトリウム若しくはジイソブチルアルミウムヒドリド等のヒドリド還元剤、ナトリウム、亜鉛、鉄等の金属還元剤、その他、下記文献中の還元剤が好適に用いられる。
〔文献〕
M. Hudlicky著、「Reductions in Organic Chemistry, 2nd ed (ACS Monograph :188)」、ACS、1996年
R. C. Larock著、「Comprehensive Organic Transformations」、第2版、VCH Publishers, Inc.、1999年
T. J. Donohoe著、「Oxidation and Reduction in Organic Synthesis (Oxford Chemistry Primers 6)」、Oxford Science Publications、2000年
日本化学会編「実験化学講座(第5版)」14巻(2005年)(丸善)
本発明化合物の製造に使用する原料、つまりアミン化合物(2)、アミン化合物(4)は、例えば、後述の製造例に記載の方法、「製法2:その他の製法」に記載した公知の方法または当業者にとって自明な方法、あるいはそれらの変法等を適用することによって、入手可能な公知化合物から製造することができる。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー等、通常の化学操作を適用して行なわれる。
各種の異性体は、適当な原料化合物を選択することにより製造でき、あるいは異性体間の物理化学的性質の差を利用して分離することができる。例えば、光学異性体は、ラセミ体の一般的な光学分割法(例えば、光学活性な塩基又は酸とのジアステレオマー塩に導く分別結晶化や、キラルカラム等を用いたクロマトグラフィー等)により得られ、また、適当な光学活性な原料化合物から製造することもできる。
HTRFR KinEASETM S1キット(CIS bio)を使用して試験を実施した。384穴プレート(CORNING)へ薬液4μL、STK Substrate 1-biotin(final 250nM)およびFull-length human PKCθ(Carna Biosciences、final 31ng/mL)混合液3μLを入れて室温で30分静置後、ATP液(final 30μM)3μLを分注し、酵素反応を室温で1時間行った。その後、Sa-XL665(final 31.25nM)および抗体STK-Antibody-Cryptate(final 800倍希釈)を含む反応停止液10μLを分注し、室温で1時間放置した。Discovery(PACKARD)にて620nm(Cryptate)および665nm(XL665)の蛍光強度を測定し、Vehicleを0%、Blankを100%抑制として、抑制率およびIC50値を算出した。
試験結果を、表1に示す。Exは後記実施例化合物番号を示す。
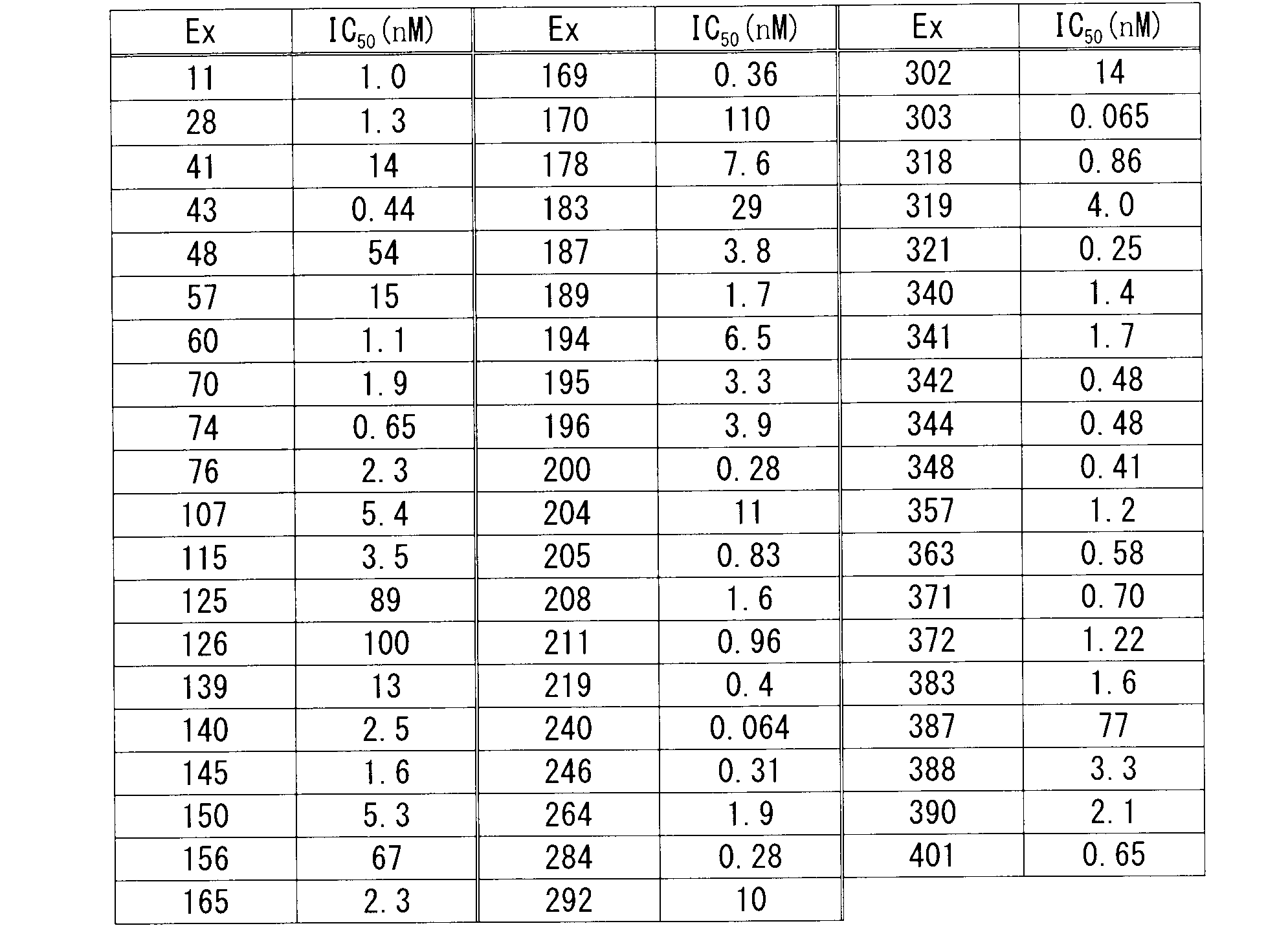
i)プラスミドの調製
データベース記載のDNA塩基配列に対応するHuman IL-2 promoter領域のDNA断片(445bp)をクローニングし、レポータージーンアッセイ用VectorであるpGL3 basicに挿入し、pGL3-IL2-pro-43を取得した。
ii)Jurkat細胞の維持・継代
ヒトT細胞系培養細胞であるJurkat, Clone E6-1(ATCC No.TIB-152)を10%FBS RPMI 1640(シグマ)を培地として、37℃、5% CO2、飽和湿度条件下にて培養し、confluentの約90%状態になった時点で、継代を行った。
iii)トランスフェクションおよび播種
血球計数板を用いて細胞数を計測後、細胞濃度が2.5 x 107cells/mLになるように、10%FBS RPMI 1640(シグマ)を用いて細胞懸濁液を調製し、pGL3-IL2-pro-43 10μgを混合した。次いで、調製した各plasmid混合液に2.5x107cells/mLに調製したJurkat細胞を400μL加えて混ぜ、Gene PulsorR Cuvette(BIO-RAD)に全量添加した。Gene PulsorRII(BIO-RAD)により300V, 975μFにてplasmidを導入し、plasmid導入済みJurkat細胞全量を、10%FBS RPMI 1640 2.5mLに軽く懸濁した後、96 well plate(Corning Coster)に50μL/wellにて播種し、37℃、5% CO2、飽和湿度条件下で、約10時間培養した。
iv)ヒトIL-2産生抑制活性の測定
薬物溶液を25μL/wellずつ添加し、さらに抗CD3抗体、抗CD28抗体(Pharmingen)(ともに終濃度1μg/mLの1000倍液)を10%FBS RPMI1640で250倍希釈した混合液を25μL/wellずつ添加した。これを、37℃、5% CO2、飽和湿度条件下で、約14時間培養した。アッセイはduplicateにて実施した。
Bright-GloTM Luciferase Assay System(Promega)付属の基質溶液を100μL/wellずつ加え、穏やかに混和した。マルチラベルカウンター(ARVO SX、WALLAC)を反応温度:25℃、Shaking Duration:1sec、Measurement time:1secに設定し、各96 wells plateの測定wellを設定して、Firefly luciferase活性を測定した。
i)阻害試験I (残存率Iの算出)
96穴プレートを用いて、基質2μM(ミダゾラム)、試験化合物5μM及びヒト肝ミクロソーム(0.1mg protein/mL)を0.1mM EDTA、1mM NADPHを含む100mMリン酸緩衝液(pH7.4)、総量150μL中で37℃で20分間インキュベーションした。その後アセトニトリル80%含有水溶液130μLを加えて反応を停止した。その後サンプルをLC/MS/MSで分析し、下記の数式1を用いて残存率Iを算出した。
(数式1)
残存率 I (%) = Ai,I/ Ao,I x 100
Ai,I=阻害試験Iで試験化合物存在下における反応後の代謝物の生成量
Ao,I=阻害試験Iで試験化合物非存在下における反応後の代謝物の生成量
ii)阻害試験II(残存率IIの算出)
96穴プレートを用いて、試験化合物5μM及びヒト肝ミクロソーム(0.1mg protein/mL)を0.1mM EDTA、1mM NADPHを含む100mMリン酸緩衝液(pH7.4)総量145μL中で、37℃で30分間インキュベーションした。その後、基質であるミダゾラム2μMを添加して総量を150μLにし、37℃で20分間インキュベーションした。インキュベーション後、アセトニトリル80%含有水溶液130μLを加えて反応を停止し、サンプルをLC/MS/MSで分析し、下記の式2を用いて残存率IIを算出した。
(数式2)
残存率 II(%) = Ai,II/Ao,II /(Ai,I/Ao,I)x 100
Ai,II=阻害試験IIで試験化合物存在下における反応後の代謝物の生成量
Ao,II=阻害試験IIで試験化合物非存在下における反応後の代謝物の生成量
試験結果を、表2に示す。Exは後記実施例化合物番号を示す。
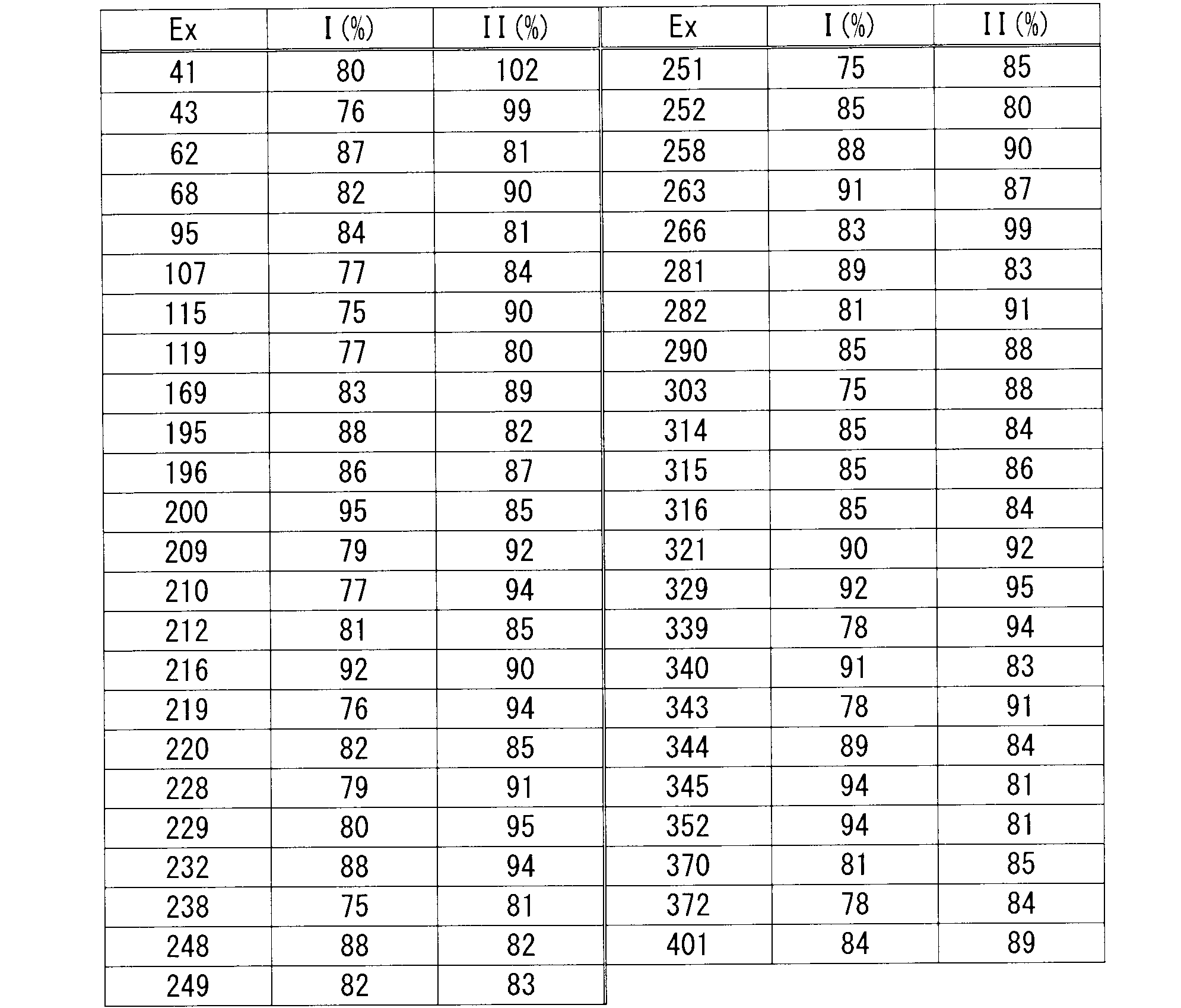
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、又は、関節内、静脈内、筋肉内等の注射剤、坐剤、点眼剤、眼軟膏、経皮用液剤、軟膏剤、経皮用貼付剤、経粘膜液剤、経粘膜貼付剤、吸入剤等による非経口投与のいずれの形態であってもよい。
経口投与のための液体組成物は、薬剤的に許容される乳濁剤、溶液剤、懸濁剤、シロップ剤又はエリキシル剤等を含み、一般的に用いられる不活性な希釈剤、例えば精製水又はエタノールを含む。当該液体組成物は不活性な希釈剤以外に可溶化剤、湿潤剤、懸濁剤のような補助剤、甘味剤、風味剤、芳香剤、防腐剤を含有していてもよい。
PEx:製造例番号、Ex:実施例番号、Str:構造式(構造式中に、例えばHClの記載がある場合は、その化合物が塩酸塩であることを意味し、2HClの記載がある場合は、その化合物が2塩酸塩であることを意味する。)、rel:相対配置(PEx又はExの番号の下にrelの記載がある場合は、Strの欄に記載した構造式におけるアダマンタン骨格部分の立体表記が相対配置を表していることを意味する。)、Syn:製造法(数字のみの場合は同様に製造した実施例番号を、数字の前にPがある場合は同様に製造した製造例番号をそれぞれ示す。)、Dat:物理化学的データ、NMR1:DMSO-d6中の1H NMRにおけるδ(ppm)、NMR2:CDCl3中の1H NMRにおけるδ(ppm)、NMR3:D2O中の1H NMRにおけるδ(ppm)、FAB+:FAB-MS (陽イオン)、ESI+:ESI-MS (陽イオン)、ESI-:ESI-MS (陰イオン)、TEA:トリエチルアミン、TFA:トリフルオロ酢酸、THF:テトラヒドロフラン、DMF:N,N-ジメチルホルムアミド、DME:ジメトキシエタン、DMI:1,3-ジメチル-2-イミダゾリジノン、MeOH:メタノール、EtOH:エタノール、EtOAc:酢酸エチル、MeCN:アセトニトリル、HOBt:1-ヒドロキシベンゾトリアゾール、WSC:3-エチル-1-(3-ジメチルアミノプロピル)カルボジイミド、DEAD:ジエチルアゾジカルボキシレート、DIPEA:ジイソプロピルエチルアミン、MCPBA:m-クロロ過安息香酸、LAH:水素化アルミニウムリチウム、Pd/C:パラジウム炭素、TLC1:TLC分析(条件:展開溶媒;MeOH/クロロホルム=1/9、シリカゲルプレート(シリカゲル60 F254, Merck))、TLC2:TLC分析(条件:展開溶媒;ヘキサン/EtOAc=1/1、アミノシリカゲルプレート(TLCプレート(NH), FUJI SILYSIA))、TLC3:TLC分析(条件:展開溶媒;EtOAc、アミノシリカゲルプレート(TLCプレート(NH), FUJI SILYSIA))、HPLC:HPLC分析、rt:保持時間。
(分析条件)
カラム:YMC-Pack ODS-AM (S-5μm, 12nm) (150x4.6mm I.D.)、カラム温度:40℃、検出方法:UV(254nm)、流速:1mL/min、溶出液A:アセトニトリル、溶出液B:pH3緩衝液(0.05M NaH2PO4水溶液にリン酸を加えてpH3に調整)
タイムプログラム:
時間(分) 0 20 30
A液(%) 10 60 60
B液(%) 90 40 40
rel-[(1R,3S,5S)-5-({[(ベンジルオキシ)カルボニル]アミノ}メチル)アダマンタン-2-イル]酢酸(250mg)のトルエン(3ml)溶液にTEA(127μl)とジフェニルリン酸アジド(196μl)を順次加え、80℃にて1時間撹拌した。室温まで放冷後、反応混合液にヨウ化銅(I)(69mg)とtert-ブタノール(3ml)を順次加え、80℃にて1時間撹拌した。反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、ベンジル tert-ブチル [(1S,3R,5S)-トリシクロ[3.3.1.13,7]デカン-1,4-ジイルビス(メチレン)]ビスrel-カルバマートを113mg得た。
氷冷下、60%水素化ナトリウム(オイル懸濁品、25.5mg)のTHF(1ml)懸濁液にホスホノ酢酸トリエチル(0.128ml)を滴下し、10分間撹拌した。反応混合液に同温度にてベンジル rel-{[(1S,3R,5S)-4-オキソアダマンタン-1-イル]メチル}カルバマート(100mg)を分割添加し、反応混合液を室温にて1時間撹拌した。反応混合液にEtOAcと水を加え、有機層を分取した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、rel-(2E)-[(1R,3S,5R)-5-({[(ベンジルオキシ)カルボニル]アミノ}メチル)トリシクロ[3.3.1.13,7]デク-2-イリデン]酢酸エチルを120mg得た。
tert-ブチル rel-{(1R,2S,3S,5S)-5-[({2-[(3-ブロモベンジル)アミノ]-5-シアノピリミジン-4-イル}アミノ)メチル]アダマンタン-2-イル}カルバマート(136mg)のDME(2.7ml)溶液に(3-アミノメチルフェニル)ボロン酸 塩酸塩(89.8mg)、テトラキス(トリフェニルホスフィン)パラジウム(0)(41.5mg)、炭酸ナトリウム(101.6mg)、水(0.34ml)を加え、窒素気流下、140℃にて6時間撹拌した。反応混合液に(3-アミノメチルフェニル)ボロン酸 塩酸塩(89.8mg)、2M炭酸ナトリウム水溶液(0.479ml)を加え、140℃にてさらに4時間撹拌した。反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、tert-ブチル rel-[(1R,2S,3S,5S)-5-({[2-({[3'-(アミノメチル)ビフェニル-3-イル]メチル}アミノ)-5-シアノピリミジン-4-イル]アミノ}メチル)アダマンタン-2-イル]カルバマートを126.6mg得た。
N-(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)グリシン tert-ブチルエステル(616mg)のジクロロメタン(6.16ml)溶液にトリフルオロ酢酸(3.4ml)を加え、室温にて撹拌した。反応終了後、反応混合液にジイソプロピルエーテルを加えた。析出した固体を濾取し、ジイソプロピルエーテルで洗浄後、減圧下乾燥することでN-(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)グリシン トリフルオロ酢酸塩を484mg得た。
1H-ベンズイミダゾール-5-カルボン酸メチルエステル(8.5g)のTHF(85ml)溶液に3,4-ジヒドロ-2H-ピラン(5.3ml)と(1S)-(+)-10-カンファースルホン酸(1.1g)を加え、24時間加熱還流した。反応混合液に3,4-ジヒドロ-2H-ピラン(4.4ml)と(1S)-(+)-10-カンファースルホン酸(10.1g)を追加し、さらに12時間加熱還流した。反応混合液をEtOAcと水の混合液に注入し、有機層を分取した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-カルボン酸メチルエステルと1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-6-カルボン酸メチルエステルの混合物(7.46g)を得た。
氷冷下、tert-ブチル rel-[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]カルバマート(200mg)とTEA(0.12ml)のジクロロメタン(4ml)溶液にクロロ蟻酸ベンジル(0.11ml)を加え、室温で4時間撹拌した。反応液をEtOAcで希釈し、0.1M塩酸、水、飽和重曹水、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除き、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、ベンジル rel-({(1S,3R,4S,5S)-4-[(tert-ブトキシカルボニル)アミノ]アダマンタン-1-イル}メチル)カルバマートを295.0mg得た。
氷冷下、rel-1-[(1'R,3'S,5'S)-5'H-スピロ[1,3-ジオキソラン-2,2'-トリシクロ[3.3.1.13,7]デカン]-5'-イル]メタンアミン(2.15g)のTHF(21.5ml)懸濁液にクロロ蟻酸ベンジル(1.92ml)と1M水酸化ナトリウム水溶液(13.5ml)を順次滴下した。反応混合液を室温まで昇温し、室温にて3時間撹拌した。反応混合液をEtOAcで希釈後、硫酸水素ナトリウム水溶液にてpH3にし、有機層を分取した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することにより、ベンジル rel-[(1'R,3'S,5'S)-5'H-スピロ[1,3-ジオキソラン-2,2'-トリシクロ[3.3.1.13,7]デカン]-5'-イルメチル]カルバマートを2.66g得た。
メタンスルホン酸{トランス-3-[(tert-ブトキシカルボニル)アミノ]シクロブチル}メチル(80.7mg)とアジ化ナトリウム(93.9mg)のDMF(0.81ml)と水(0.081ml)の懸濁液を120℃で40分間撹拌した。反応液を冷却後、EtOAcで希釈し、水、飽和食塩水の順で洗い、無水硫酸ナトリウムで乾燥した。乾燥剤を除き、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル [トランス-3-(アジドメチル)シクロブチル]カルバマートを63.1mg得た。
tert-ブチル [トランス-3-(アジドメチル)シクロブチル]カルバマート(270mg)のMeOH(13.5ml)溶液に10%Pd/C(50%含水品、81mg)を加え、水素雰囲気下、常圧にて室温で40分間撹拌した。触媒をセライトで濾別し、MeOHで洗浄後、濾液を減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルム-MeOH-濃アンモニア水)にて精製することにより、tert-ブチル [トランス-3-(アミノメチル)シクロブチル]カルバマートを120.5mg得た。
N-(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)グリシン トリフルオロ酢酸塩(30mg)のDMF(0.9ml)溶液にtert-ブチル (2-アミノエチル)カルバマート(25.0mg)、HOBt(9.3mg)、WSC(24.2mg)を順次加え、室温にて撹拌した。反応終了後、反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、tert-ブチル (2-{[N-(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)グリシル]アミノ}エチル)カルバマートを10mg得た。
氷冷下、ベンジル rel-[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]カルバマートとピリジン(1.4ml)のジクロロメタン(50ml)溶液に無水トリフルオロ酢酸(2.5ml)のジクロロメタン(20ml)溶液を加え、同温度で30分間撹拌した。ピリジン(0.128ml)、トリフルオロ酢酸無水物(0.225ml)をさらに加え、氷冷下30分間撹拌した。氷冷下、反応混合液に水を加えて撹拌後、EtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することでベンジル rel-[(1R,2S,3S,5S)-5-{[(トリフルオロアセチル)アミノ]メチル}アダマンタン-2-イル]カルバマートを7.0g得た。
rel-(1R,3S,5R,7S)-4-{[(ベンジルオキシ)カルボニル]アミノ}アダマンタン-1-カルボン酸(12g)のジクロロメタン(120ml)溶液に塩化オキサリル(4.8ml)を加え、室温にて撹拌した。反応終了後、反応混合物を減圧下濃縮し、トルエンを加え、さらに減圧下濃縮した。得られた残渣を1,4-ジオキサン(12ml)に溶解し、氷冷下、28%アンモニア水(110g)に滴下した。反応混合液をEtOAcにて抽出し、有機層を水で3回、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣にMeOHを加えて固体を析出させ、濾取した。濾液を減圧下濃縮し、再度MeOHで固体を析出させ、濾取した。濾液を減圧下濃縮し、再々度MeOHで固体を析出させ、濾取した。得られた固体を減圧下乾燥することでベンジル rel-[(1R,2R,3S,5S)-5-カルバモイルアダマンタン-2-イル]カルバマート(2.9g)を得た。濾液を減圧下濃縮し、得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、ベンジル rel-[(1R,2S,3S,5S)-5-カルバモイルアダマンタン-2-イル]カルバマートを1.9g得た。
tert-ブチル rel-[(1R,2S,3S,5S)-5-{[(トリフルオロアセチル)アミノ]メチル}アダマンタン-2-イル]カルバマート(4.6g)のMeOH(46ml)と水(23ml)の混合溶液に炭酸カリウム(16.9g)を加え、室温にて撹拌した。反応終了後、反応混合液をEtOAcで希釈し、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することでtert-ブチル rel-[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]カルバマートを3.78g得た。
氷冷下、窒素気流下でベンジル rel-[(1R,2S,3S,5S)-5-カルバモイルアダマンタン-2-イル]カルバマート(500mg)のTHF(5.0ml)溶液に1.17M ボラン-テトラヒドロフラン錯体のTHF溶液(3.9ml)を滴下し、3時間加熱還流した。反応混合液を氷冷後、注意深く水を滴下し、ジクロロメタン-炭酸カリウム水溶液に撹拌下注入した。有機層を分取し、さらにジクロロメタンで2回抽出した。得られた有機層を合わせ、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することでベンジル rel-[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]カルバマートを530mg得た。
氷冷下、イミノジカルボン酸ジ-tert-ブチル(1.88g)のDMF(28ml)溶液にカリウム tert-ブトキシド(970mg)を少量ずつ加え、室温にて1時間撹拌した。反応混合物に酢酸 3-(ブロモメチル)-4-クロロフェニル(1.90g)のDMF(10ml)溶液を氷冷下滴下し、室温にて2時間撹拌した。反応混合物に水を加え、EtOAcで抽出した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、酢酸 3-{[ビス(tert-ブトキシカルボニル)アミノ]メチル}-4-クロロフェニルを2.79g得た。
アルゴン雰囲気下、[2-(ベンジルオキシ)フェニル]メタノール(20.2g)のクロロホルム(160ml)溶液に室温にて塩化チオニル(13.8ml)のクロロホルム(40ml)溶液をゆっくりと加えた。室温にて90分間撹拌した後、減圧下揮発性物質を留去することにより1-(ベンジルオキシ)-2-(クロロメチル)ベンゼンを得た。次に、アルゴン雰囲気下、イミノジカルボン酸ジ-tert-ブチル(41.0g)のDMF(500ml)溶液に室温にてカリウム tert-ブトキシド(21.2g)を加えた。同温にて70分間撹拌した後、1-(ベンジルオキシ)-2-(クロロメチル)ベンゼンのDMF(60ml)溶液を加えた。同温にて15時間撹拌した後、水を加えてさらに90分間撹拌した。析出物を濾取して水で洗浄した後、減圧下乾燥した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(クロロホルム)で精製することにより、[2-(ベンジルオキシ)ベンジル]イミド二炭酸ジ-tert-ブチルを34.5g得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(30mg)のDMF(0.6ml)溶液に、DIPEA(22μl)、ブロモ酢酸エチル(5.8μl)を加え、60℃にて撹拌した。反応終了後、反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-N-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]グリシン エチルエステル(26.1mg)を得た。
(2-ヒドロキシベンジル)イミド二炭酸ジ-tert-ブチル(500mg)のDMF(5.0ml)溶液に2-ブロモアセトアミド(320mg)、炭酸カリウム(641mg)およびヨウ化カリウム(385mg)を加え、80℃にて3時間撹拌した。室温まで放冷後、水を加えて析出する生成物を濾取することにより、[2-(2-アミノ-2-オキソエトキシ)ベンジル]イミド二炭酸ジ-tert-ブチルを546mg得た。
tert-ブチル (5-ホルミル-2-メトキシベンジル)カルバマート(1.0g)のTHF(3.0ml)とEtOH(6.0ml)の混合溶液に水素化ホウ素ナトリウム(192.5mg)を加え、室温にて撹拌した。反応終了後、反応混合液に水を加えてEtOAcで抽出し、有機層を無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル [5-(ヒドロキシメチル)-2-メトキシベンジル]カルバマートを1.09g得た。
4-クロロ-2-(メチルスルファニル)ピリミジン-5-カルボニトリル(2.2g)の1,3-ジメチルイミダゾリジン-2-オン溶液にDIPEA(4.13ml)、tert-ブチル rel-[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]カルバマート(3.99g)を加え、室温にて撹拌した。反応終了後、反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル rel-[(1R,2S,3S,5S)-5-({[5-シアノ-2-(メチルスルファニル)ピリミジン-4-イル]アミノ}メチル)アダマンタン-2-イル]カルバマートを4.76g得た。
氷冷下、2,4-ジクロロピリミジン-5-カルボニトリル(1.00g)のDMF(15ml)溶液に2-(メチルチオ)ベンジルアミン(881mg)のDMF(5ml)溶液およびDIPEA(1.2ml)を滴下し、同温度で1時間撹拌した。2-(メチルチオ)ベンジルアミン(44mg)のDMF(2ml)溶液を追加し、室温にてさらに1時間撹拌した。反応混合物にEtOAcと水を加え、分液した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルム)で精製することにより、4-クロロ-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリルを709mg得た。
氷-食塩水浴で冷却した2,4-ジクロロ-5-(トリフルオロメチル)ピリミジン(300mg)のDMF(6.0ml)溶液にDIPEA(252.9μl)と1-[2-(トリフルオロメトキシ)フェニル]メタンアミン(277.5mg)を加え-20℃で撹拌した。反応終了後、反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することで、4-クロロ-N-[2-(トリフルオロメトキシ)ベンジル]-5-(トリフルオロメチル)ピリミジン-2-アミンと2-クロロ-N-[2-(トリフルオロメトキシ)ベンジル]-5-(トリフルオロメチル)ピリミジン-4-アミンの混合物を507mg得た。
4-クロロ-N-[2-(トリフルオロメトキシ)ベンジル]-5-(トリフルオロメチル)ピリミジン-2-アミンと2-クロロ-N-[2-(トリフルオロメトキシ)ベンジル]-5-(トリフルオロメチル)ピリミジン-4-アミンの混合物(90mg)のDMF(1.0ml)溶液にDIPEA(84.3μl)、ベンジル rel-[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]カルバマート(79.9mg)を加え、室温にて撹拌した。反応終了後、反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、ベンジル rel-[(1R,2S,3S,5S)-5-({[2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}-5-(トリフルオロメチル)ピリミジン-4-イル]アミノ}メチル)アダマンタン-2-イル]カルバマートの粗精製物とベンジル rel-[(1R,2S,3S,5S)-5-({[4-{[2-(トリフルオロメトキシ)ベンジル]アミノ}-5-(トリフルオロメチル)ピリミジン-2-イル]アミノ}メチル)アダマンタン-2-イル]カルバマートの粗精製物を得た。得られたそれぞれの粗精製物をさらにシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、ベンジル rel-[(1R,2S,3S,5S)-5-({[2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}-5-(トリフルオロメチル)ピリミジン-4-イル]アミノ}メチル)アダマンタン-2-イル]カルバマート(100mg)とベンジル rel-[(1R,2S,3S,5S)-5-({[4-{[2-(トリフルオロメトキシ)ベンジル]アミノ}-5-(トリフルオロメチル)ピリミジン-2-イル]アミノ}メチル)アダマンタン-2-イル]カルバマート(50mg)を得た。
ベンジル rel-[(1R,2S,3S,5S)-5-({[5-シアノ-2-(メチルスルフィニル)ピリミジン-4-イル]アミノ}メチル)アダマンタン-2-イル]カルバマート(50mg)の1,3-ジメチルイミダゾリジン-2-オン(1.0ml)溶液に、3-ブロモアニリン(0.114ml)、4M塩化水素ジオキサン溶液(2.6μl)を加え、100℃にて3時間撹拌した。室温まで放冷後、反応混合液に水を加えて析出した固体を濾取し、水とヘキサンで洗浄後、減圧下乾燥することで、ベンジル rel-{(1R,2S,3S,5S)-5-[({2-[(3-ブロモフェニル)アミノ]-5-シアノピリミジン-4-イル}アミノ)メチル]アダマンタン-2-イル}カルバマートを46mg得た。
rel-[(1R,3S,5S)-5-({[(ベンジルオキシ)カルボニル]アミノ}メチル)アダマンタン-2-イル]酢酸エチル(300mg)のMeOH(6.0ml)溶液に、4M水酸化リチウム水溶液(1.2ml)を加え、60℃にて3時間撹拌した。反応混合液をEtOAcで希釈後、硫酸水素カリウム水溶液を加えてpH2とし、有機層を分取した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することでrel-[(1R,3S,5S)-5-({[(ベンジルオキシ)カルボニル]アミノ}メチル)アダマンタン-2-イル]酢酸を264.6mg得た。
rel-(1R,3S,5R,7S)-4-{[(ベンジルオキシ)カルボニル]アミノ}アダマンタン-1-カルボン酸メチル(15g)の1,4-ジオキサン(75ml)とMeOH(75ml)の混合溶液に1M水酸化ナトリウム水溶液(87.4ml)を加え、60℃にて4時間撹拌した。反応混合液を室温まで放冷後、10%硫酸水素カリウム水溶液にてpH4とし、EtOAcにて抽出した。得られた有機層を飽和食塩水で1回洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することでrel-(1R,3S,5R,7S)-4-{[(ベンジルオキシ)カルボニル]アミノ}アダマンタン-1-カルボン酸を12g得た。
窒素雰囲気下、水素化リチウムアルミニウム(1.2g)のTHF(100ml)懸濁液に1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-カルボン酸メチルエステルと1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-6-カルボン酸メチルエステルの混合物(7.0g)のTHF(100ml)溶液を-10℃以下で滴下し、氷冷下1時間撹拌した。水素化リチウムアルミニウム(0.8g)を分割して追加し、氷冷下でさらに30分間撹拌した。同温度で水(6.0ml)、15%水酸化ナトリウム水溶液(6.0ml)、水(3.0ml)を順次加え、室温にて30分間撹拌した。不溶物をセライトで濾去し、濾液を減圧下濃縮することで[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-イル]メタノールと[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-6-イル]メタノールの混合物を5.48g得た。
氷冷下、rel-(2E)-[(1R,3S,5R)-5-({[(ベンジルオキシ)カルボニル]アミノ}メチル)トリシクロ[3.3.1.13,7]デク-2-イリデン]酢酸エチル(350mg)のMeOH(6.0ml)溶液に窒素雰囲気下、塩化ニッケル(II)(23.7mg)を加え、水素化ホウ素ナトリウムを分割添加後、同温度にて1時間、室温にて3時間撹拌した。反応混合液に水を加え、EtOAcにて抽出した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することでrel-[(1R,3S,5S)-5-({[(ベンジルオキシ)カルボニル]アミノ}メチル)アダマンタン-2-イル]酢酸エチルを310mg得た。
rel-(1R,3S,5R,7S)-4-オキソアダマンタン-1-カルボン酸メチル(500mg)のジクロロメタン(7.5ml)溶液にベンジルアミン(0.262ml)とトリアセトキシ水素化ホウ素ナトリウム(763mg)を順次加え、室温にて2時間撹拌した。反応混合液に飽和重曹水を加え撹拌の後、ジクロロメタンで抽出し有機層を分取した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-(1R,3S,5R,7S)-4-(ベンジルアミノ)アダマンタン-1-カルボン酸メチルを757mg得た。
ベンジル rel-{[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}カルバマート(760mg)のジクロロメタン(22.8ml)溶液に、4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキサノン(1.22ml)とトリアセトキシ水素化ホウ素ナトリウム(1.02g)を加え、室温にて4時間撹拌した。反応混合液に飽和重曹水を加え、EtOAcで抽出した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去し、減圧下溶媒を留去して得られた残渣をアミノシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、先ずベンジル rel-({(1R,3S,4R,5R)-4-[(シス-4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキシル)アミノ]アダマンタン-1-イル}メチル)カルバマートを674.8mg溶出し、続いてベンジル rel-({(1R,3S,4R,5R)-4-[(トランス-4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキシル)アミノ]アダマンタン-1-イル}メチル)カルバマートを435.8mg溶出した。
得られた生成物の立体配置は、アミノシリカゲルカラムクロマトグラフィーにおいて後で溶出される化合物(ベンジル rel-({(1R,3S,4R,5R)-4-[(トランス-4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキシル)アミノ]アダマンタン-1-イル}メチル)カルバマート)を原料として製造例134で得られたrel-トランス-4-{[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]アミノ}シクロヘキサノールを実施例45に供し、得られた生成物のHPLC保持時間(15.1 min)が実施例42(トランスアルコール体)と一致したことにより決定した。
氷冷下、tert-ブチル rel-[(1R,2S,3S,5S)-5-({[5-シアノ-2-(メチルスルファニル)ピリミジン-4-イル]アミノ}メチル)アダマンタン-2-イル]カルバマート(4.7g)のジクロロメタン(50ml)溶液に75%MCPBA(含水品)(2.77g)を加え、同温度で撹拌した。反応終了後、反応混合液をEtOAcで希釈し、有機層を飽和重曹水、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、tert-ブチル rel-[(1R,2S,3S,5S)-5-({[5-シアノ-2-(メチルスルフィニル)ピリミジン-4-イル]アミノ}メチル)アダマンタン-2-イル]カルバマートを5.02g得た。
氷冷下、tert-ブチル rel-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]カルバマート(147mg)のジクロロメタン(5.0ml)溶液に75%MCPBA(含水品、69.6mg)を加え、同温度で1時間撹拌した。反応混合液に飽和重曹水を加えてEtOAcで抽出した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAcで開始し、途中でクロロホルム-MeOHに変更)にて精製することにより、tert-ブチル rel-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(メチルスルフィニル)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]カルバマート(123.9mg)とtert-ブチル rel-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(メチルスルホニル)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]カルバマート(28.1mg)を得た。
氷冷下、LAH(88mg)のTHF(20ml)懸濁液にtert-ブチル 3-シアノ-8-アザビシクロ[3.2.1]オクタン-8-カルボキシレート(550mg)を加え、室温で4時間撹拌した。氷冷下、水を加えてEtOAcで抽出し、有機層を無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下、溶媒留去することによりtert-ブチル3-(アミノメチル)-8-アザビシクロ[3.2.1]オクタン-8-カルボキシレートを600mg得た。
{2-[2-(メトキシメトキシ)エトキシ]ベンジル}イミド二炭酸ジ-tert-ブチル(288mg)のメタノール(1.4ml)溶液に4M塩化水素ジオキサン溶液(3.5ml)を加え、室温にて2時間撹拌した。減圧下溶媒を留去することにより、2-[2-(アミノメチル)フェノキシ]エタノール 塩酸塩を140mg得た。
氷冷下、ベンジル rel-({(1S,3R,4S,5S)-4-[(tert-ブトキシカルボニル)アミノ]アダマンタン-1-イル}メチル)カルバマート(295.0mg)のジクロロメタン(3.54ml)溶液にトリフルオロ酢酸(3.54ml)を加え、室温にて2時間撹拌した。反応液を減圧下濃縮後、残渣に炭酸カリウム水溶液を加えてアルカリ性にし、EtOAcで抽出した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除き、減圧下溶媒を留去してベンジル rel-{[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}カルバマートを242.8mg得た。
[2-(2-メトキシエトキシ)ベンジル]イミド二炭酸ジ-tert-ブチル(341mg)の1,4-ジオキサン(1.7ml)溶液に室温にて4M塩化水素ジオキサン溶液(3.5ml)を加え、2時間撹拌した。減圧下溶媒を留去することにより、1-[2-(2-メトキシエトキシ)フェニル]メタンアミン 塩酸塩を155mg得た。
tert-ブチル (2-{4-[(メチルスルホニル)アミノ]フェニル}エチル)カルバマート(900mg)のジクロロメタン(18ml)溶液にトリフルオロ酢酸(2.89ml)を加え、室温にて終夜撹拌した。反応混合液を減圧下濃縮後、残渣にトルエンを加え再度減圧下濃縮した。得られた残渣にジエチルエーテルを加えて析出した固体を濾取し、ジエチルエーテルで洗浄後、減圧下乾燥した。得られた固体をEtOHに懸濁し、1M水酸化ナトリウム水溶液を加えてアルカリ性とし、次いで1M塩酸でpH7に調整後、クロロホルムで抽出した。有機層をあわせて無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することでN-[4-(2-アミノエチル)フェニル]メタンスルホンアミドを95mg得た。
氷冷下、ベンジル rel-[(1R,2S,3S,5S)-5-{[(トリフルオロアセチル)アミノ]メチル}アダマンタン-2-イル]カルバマート(7.0g)のEtOH(175ml)溶液にジ-tert-ブチル ジカーボネート(5.58g)、10%Pd/C(50%含水品、7.0g)、シクロヘキサ-1,4-ジエン(15.9ml)を順次加え、室温にて1時間撹拌した。触媒を濾過除去後、濾液を減圧下濃縮した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル rel-[(1R,2S,3S,5S)-5-{[(トリフルオロアセチル)アミノ]メチル}アダマンタン-2-イル]カルバマートを4.67g得た。
ベンジル rel-({(1R,3S,4R,5R)-4-[(トランス-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)カルバマート(380mg)のMeOH(11.4ml)溶液に10%Pd/C(50%含水品、76mg)を加え、水素雰囲気下、常圧にて35℃で2.5時間撹拌した。触媒をセライトで濾別し、MeOHで洗浄後、濾液を減圧下濃縮し、rel-トランス-4-{[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]アミノ}シクロヘキサノールを263.8mg得た。
酢酸 3-{[ビス(tert-ブトキシカルボニル)アミノ]メチル}-4-クロロフェニル(2.79g)のメタノール(56ml)溶液に炭酸カリウム(1.45g)を加え、室温にて1時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、析出物を濾取することにより、(2-クロロ-5-ヒドロキシベンジル)イミド二炭酸ジ-tert-ブチルを2.15g得た。
rel-4-{[(1'R,3'S,5'S)-5'H-スピロ[1,3-ジオキソラン-2,2'-トリシクロ[3.3.1.13,7]デカン]-5'-イルメチル]アミノ}-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(385mg)のTHF(23.1ml)と水(30.8ml)の混合溶液にトシル酸一水和物(1.42g)を加え、室温にて終夜撹拌した。反応混合液を減圧下濃縮し、残渣に飽和重曹水を加えアルカリ性とした後、EtOAcで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、rel-4-({[(1S,3R,5S)-4-オキソアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを300mg得た。
ベンジル rel-[(1'R,3'S,5'S)-5'H-スピロ[1,3-ジオキソラン-2,2'-トリシクロ[3.3.1.13,7]デカン]-5'-イルメチル]カルバマート(2.5g)のTHF(25ml)と水(25ml)の混合溶液にトシル酸一水和物(6.65g)を加え、室温にて終夜撹拌した。反応混合液を減圧下濃縮し、残渣に飽和重曹水を加えてアルカリ性とした後、EtOAcで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去することで、ベンジル rel-{[(1S,3R,5S)-4-オキソアダマンタン-1-イル]メチル}カルバマートを2.26g得た。
ベンジル rel-({(1R,3S,4R,5R)-4-[(トランス-4-{[tert-ブチル(ジメチル)シリル]オキシ}シクロヘキシル)アミノ]アダマンタン-1-イル}メチル)カルバマート(446mg)のTHF(8.92ml)溶液に1M テトラブチルアンモニウム フルオリドのTHF溶液(2.54ml)を加え、70℃で5.5時間撹拌した。減圧下溶媒を留去し、残渣に水を加えてクロロホルムで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した後、乾燥剤を除いて減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルム-MeOHで開始し、途中でクロロホルム-MeOH-濃アンモニア水に変更)により精製し、ベンジル rel-({(1R,3S,4R,5R)-4-[(トランス-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)カルバマートを385.1mg得た。
2-{[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-イル]メチル}-1H-イソインドール-1,3(2H)-ジオンと2-{[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-6-イル]メチル}-1H-イソインドール-1,3(2H)-ジオンの混合物(3.99g)のEtOH(79.8ml)とTHF(79.8ml)の混合溶液にヒドラジン一水和物(2.14ml)を加え、加熱還流した。反応終了後、不溶物を濾去し、濾液を減圧下濃縮した。得られた残渣をジクロロメタンで希釈し、1M水酸化ナトリウム水溶液で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)、次いでシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、1-[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-イル]メタンアミンと1-[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-6-イル]メタンアミンの混合物を0.42g得た。
rel-(1R,3S,5R,7S)-4-(ベンジルアミノ)アダマンタン-1-カルボン酸メチル(720mg)のEtOH(7.2ml)と水(0.72ml)の混合溶液に10%Pd/C(50%含水品、144mg)と蟻酸アンモニウム(455mg)を加え、30分間加熱還流した。反応混合液を室温まで放冷後、セライトにて触媒を濾別し、濾液を減圧下濃縮することでrel-(1R,3S,5R,7S)-4-アミノアダマンタン-1-カルボン酸メチルを482mg得た。
[2-(ベンジルオキシ)ベンジル]イミド二炭酸ジ-tert-ブチル(34.5g)のメタノール(170ml)およびTHF(170ml)溶液に10%Pd/C(3.5g)を加え、水素雰囲気下、常圧にて14時間撹拌した。反応混合物を濾過し、濾液を減圧下濃縮することにより、(2-ヒドロキシベンジル)イミド二炭酸ジ-tert-ブチルを27.0g得た。
酢酸 4-クロロ-3-メチルフェニル(2.06g)の四塩化炭素(20.6ml)溶液にN-ブロモスクシンイミド(1.99g)および2,2'-アゾビス(イソブチロニトリル)(366mg)を加え、1時間加熱還流した。反応液に水を加え、EtOAcで抽出し、有機層を飽和塩化ナトリウム水溶液で洗浄後、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)で精製することにより、酢酸 3-(ブロモメチル)-4-クロロフェニルを1.87g得た。
tert-ブチル (2-メトキシベンジル)カルバマート(25.0g)のMeCN(200ml)溶液にN-ブロモコハク酸イミド(19.7g)を加え、室温にて終夜撹拌した。反応混合液にN-ブロモコハク酸イミド(10.0g)を追加し、さらに室温にて8時間撹拌した。反応混合液を減圧下濃縮後、残渣をEtOAcで希釈し、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(トルエン)にて精製することにより、tert-ブチル (5-ブロモ-2-メトキシベンジル)カルバマートを9.55g得た。
tert-ブチル (5-ブロモ-2-メトキシベンジル)カルバマート(5.0g)のDMF(50ml)溶液にビス(トリフェニルホスフィン)パラジウム(II) ジクロリド(222mg)、トリフェニルホスフィン(83mg)、炭酸水素ナトリウム(1.61g)を加え、一酸化炭素雰囲気下、常圧にて110℃で加熱撹拌した。反応混合液をEtOAcで希釈し、水、炭酸ナトリウム水溶液、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル (5-ホルミル-2-メトキシベンジル)カルバマートを1.69g得た。
氷冷下、[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-イル]メタノールと[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-6-イル]メタノールの混合物(2.0g)のトルエン(40ml)溶液にコハク酸イミド(1.52g)、トリフェニルホスフィン(2.71g)、DEAD(1.62ml)を加え、同温度で3時間撹拌した。反応混合液をEtOAcで希釈し、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、2-{[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-イル]メチル}-1H-イソインドール-1,3(2H)-ジオンと2-{[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-6-イル]メチル}-1H-イソインドール-1,3(2H)-ジオンの混合物を4.12g得た。
氷冷下、tert-ブチル [トランス-3-(ヒドロキシメチル)シクロブチル]カルバマート(60mg)とTEA(0.066ml)のジクロロメタン(4ml)溶液にメタンスルホニル クロリド(0.035ml)を加え、同温で30分間撹拌した。反応液をEtOAcで希釈し、水、飽和食塩水の順で洗い、無水硫酸ナトリウムで乾燥した。乾燥剤を除き、減圧下溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、メタンスルホン酸{トランス-3-[(tert-ブトキシカルボニル)アミノ]シクロブチル}メチルを83.3mg得た。
氷冷下、tert-ブチル [2-(4-アミノフェニル)エチル]カルバマート(1.5g)のクロロホルム(15ml)溶液に、TEA(0.973ml)とメタンスルホニル クロリド(0.540ml)を順次加え、室温にて3時間撹拌した。TEA(1.326ml)とメシルクロリド(0.737ml)を順次追加し、室温にてさらに3時間撹拌した。反応混合液を減圧下濃縮し、得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル (2-{4-[(メチルスルホニル)アミノ]フェニル}エチル)カルバマートを1.03g得た。
氷冷下、tert-ブチル ピペリジン-4-イルカルバマート(400mg)とDIPEA(0.30ml)のジクロロメタン(6ml)溶液にクロロアセチル クロリド(0.175ml)を加え、同温度で30分間、室温で2.5時間撹拌した。反応混合物にEtOAcと0.5M塩酸を加えて分液後、有機層を水、飽和重曹水、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することによりtert-ブチル [1-(クロロアセチル)ピペリジン-4-イル]カルバマートを545mg得た。
6-クロロニコチノニトリル(400mg)とtert-ブチル ピペリジン-4-イルカルバマート(693mg)のDMF(4.8ml)溶液に炭酸カリウム(598mg)を加え、120℃で2時間撹拌した。反応混合物にEtOAcと水を加えて分液後、有機層を水(3回)、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、析出する粉末を濾取した。EtOAcにて洗浄後、乾燥し、tert-ブチル [1-(5-シアノピリジン-2-イル)ピペリジン-4-イル]カルバマートを504mg得た。
氷冷下、4-[({(1S,3R,4S,5S)-4-[(3-{[tert-ブチルジメチル)シリル]オキシプロピル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリル(65mg)のMeOH(1.3mL)溶液に35%ホルマリン水溶液(34mg)および水素化シアノホウ素ナトリウム(27mg)を加え、室温で5時間撹拌した。反応混合物に飽和重曹水(5mL)を加え、析出した白色固体を濾取した。得られた固体をクロロホルムに溶解し、アミノシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製し、4-[({(1S,3R,4S,5S)-4-[(3-{[tert-ブチルジメチル)シリル]オキシプロピル)(メチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリルを60mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(100mg)のEtOAc(1.0ml)溶液にtert-ブチル (4-オキソシクロヘキシル)カルバマート(67.7mg)とチタニウム(IV)イソプロポキシド(250μl)を加え、室温にて20分間撹拌した。次いで、反応混合液に酸化白金(12mg)を加え、水素雰囲気下、室温にて220分間撹拌した。反応混合液に水、EtOAcを順次加えて撹拌し、不溶物をセライトで濾去した。濾液を減圧下濃縮し、残渣をEtOAcで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル rel-(cis-4-{[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]アミノ}シクロヘキシル)カルバマート(86.6mg)を先ず溶出し、続いてtert-ブチル rel-(trans-4-{[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]アミノ}シクロヘキシル)カルバマート(28.4mg)を溶出した。
氷冷下、(4-メトキシピリジン-3-イル)メタノール(58mg)のクロロホルム(0.6ml)溶液に塩化チオニル(0.046ml)を加え、室温で1時間撹拌した。反応液を減圧濃縮し、残渣に飽和重曹水を加え、EtOAcで抽出した。有機層を水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、3-(クロロメチル)-4-メトキシピリジンを65mg得た。
氷冷下、tert-ブチル 5-シアノ-4,5,6,7-テトラヒドロ-1H-ベンズイミダゾール-1-カルボキシラート(100mg)のMeOH(3mL)溶液に塩化コバルト(II)6水和物(192mg)と水素化ホウ素ナトリウム(61mg)をゆっくり加え、室温で1時間撹拌した。反応液に1M塩酸(1mL)を加え、不溶物をセライトで濾去した。濾液をクロロホルム(10mL)で洗浄し、水層に1M塩酸(4mL)を加えて減圧濃縮し、1-(4,5,6,7-テトラヒドロ-1H-ベンズイミダゾール-5-イル)メチルアミン 2塩酸塩を72mg得た。
tert-ブチル (2-ビニルベンジル)カルバマート(30mg)のアセトン(0.15ml)-水(0.075ml)溶液に4%四酸化オスミウム水溶液(41mg)および4-メチルモルホリン N-オキシド(23mg)を室温で加えた。同温で2時間撹拌した後、氷冷下で10%亜硫酸ナトリウム水溶液を加えた。混合物をEtOAcで抽出し、有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル [2-(1,2-ジヒドロキシエチル)ベンジル]カルバマートを24mg得た。
氷冷下、tert-ブチル 5-カルバモイル-4,5,6,7-テトラヒドロ-1H-ベンズイミダゾール-1-カルボキシラート(500mg)のTHF(5mL)溶液にトリフルオロ酢酸無水物(0.32mL)とピリジン(0.32mL)を加え、室温で1時間撹拌した。反応液に飽和重曹水(10mL)を加え、EtOAc(40mL)で抽出した。有機層を水(10mL)および飽和食塩水(10mL)で洗浄後、無水硫酸ナトリウムで乾燥した後、乾燥剤を除いて減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製し、tert-ブチル 5-シアノ-4,5,6,7-テトラヒドロ-1H-ベンズイミダゾール-1-カルボキシラートを380mg得た。
氷冷下、2-(メチルスルファニル)ニコチノニトリル(382mg)のMeOH(6ml)溶液に塩化コバルト(II)6水和物(1.69g)を加え、水素化ほう素ナトリウム(346mg)を同温度で少量ずつ加えた。室温で3時間撹拌した後、析出物をセライトにより濾去した。濾液を減圧下濃縮し、残渣に1M塩酸を加え、クロロホルムで洗浄した。水層に28%アンモニア水を加えて塩基性にした後、クロロホルムで抽出し、有機層を無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去し、1-[2-(メチルスルファニル)ピリジン-3-イル]メチルアミンを237mg得た。
氷冷下、rel-4-({[(1S,3R,4S,5S)-4-(3-ヒドロキシアゼチジン-1-イル)アダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のTHF(2ml)とトルエン(3ml)の混合溶液にコハク酸イミド(19.8mg)、トリフェニルホスフィン(29.8mg)、DEAD(17.8μl)を加え、室温にて撹拌した。コハク酸イミド(33.4mg)、トリフェニルホスフィン(74.4mg)、DEAD(44.5μl)、THF(2ml)、トルエン(1ml)を追加し、室温にてさらに撹拌した。反応終了後、反応混合液をEtOAcで希釈し、有機層を水(3回)、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[3-(1,3-ジオキソ-1,3-ジヒドロ-2H-イソインドール-2-イル)アゼチジン-1-イル]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを70.7mg得た。
氷冷下、rel-(1S,3R,4S,5S)-4-アミノアダマンタン-1-カルボン酸(100mg)の1,4-ジオキサン(0.7ml)懸濁液に1M水酸化ナトリウム水溶液(0.62ml)を加え、同温度で10分間撹拌して溶解した。氷冷下、ジ-tert-ブチル ジカーボネート(115mg)の1,4-ジオキサン(0.1ml)溶液を滴下し、室温で4時間撹拌した。氷冷下、1M塩酸(0.74ml)を加えてEtOAcで抽出し、水(2回)、次いで飽和食塩水で洗浄した。有機層を無水硫酸ナトリウムで乾燥し、濾過後減圧濃縮すると結晶が析出したので、乾固する前にヘキサン(3ml)で懸濁し、濾取してrel-(1S,3R,4S,5S)-4-[(tert-ブトキシカルボニル)アミノ]アダマンタン-1-カルボン酸を111.1mg得た。
ドライアイス-アセトン浴で冷却した(メトキシメチル)(トリフェニル)ホスホニウム クロリド(164.57g)のTHF(500ml)懸濁液に、窒素気流下、-55℃以下でn-ブチルリチウムのヘキサン溶液(濃度1.65M、281.3ml)を滴下した。滴下後、反応混合液を昇温させ、室温にて1時間撹拌した。撹拌後、反応混合液を氷冷し、4-ヒドロキシ-4-メチルシクロヘキサノン(20.51g)のTHF(205ml)溶液を滴下した。滴下後、反応混合液を室温まで昇温し、15時間撹拌した。反応混合液に水およびEtOAcを順次加えて撹拌した後、有機層を分取した。水層をさらにEtOAcで抽出し、有機層を合わせて飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、4-(メトキシメチレン)-1-メチルシクロヘキサノール(21.37g)を得た。
tert-ブチル rel-[(1R,2S,3S,5S)-5-(アジドメチル)アダマンタン-2-イル]カルバマート(300mg)のTHF(3ml)溶液にトリフェニルホスフィン(300mg)を加え、室温で4時間撹拌した。反応混合物に水(1.8ml)を加え、室温で2時間撹拌した後、減圧下で溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(先ずEtOAcのみで副生成物を溶出した後、溶出溶媒をMeOH/クロロホルム/28%NH3aq.(1/9/0.1)に変更)にて精製することによりtert-ブチル rel-[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]カルバマートを270mg得た。
tert-ブチル rel-[(1R,2S,3S,5S)-5-{[(2-クロロ-5-フルオロピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]カルバマート(100mg)と2-(トリフルオロメトキシ)ベンジルアミン(280mg)のDMI(0.8ml)溶液にDIPEA(0.127ml)を加え、165℃でマイクロウェーブを4時間照射した。反応液をEtOAcで希釈し、水、飽和塩化アンモニウム水溶液、水、飽和重曹水、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル rel-[(1R,2S,3S,5S)-5-{[(5-フルオロ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]カルバマートを78.3mg得た。
氷冷下、tert-ブチル [(2-クロロピリジン-3-イル)メチル]カルバマート(450mg)のDMF溶液(2ml)にシクロペンタンチオール(0.65ml)および水素化ナトリウム(ミネラルオイル約40%添加、220mg)を加えた。室温で4時間撹拌した。次いで反応液を氷冷し、シクロペンタンチオール(0.40ml)と水素化ナトリウム(ミネラルオイル約40%添加、140mg)を追加し、室温で2時間撹拌した。再び反応液を氷冷し、飽和塩化アンモニウム水溶液を加え、クロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥し、乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、tert-ブチル {[2-(シクロペンチルスルホニル)ピリジン-3-イル]メチル}カルバマートを400mg得た。
室温で、rel-(1S,3R,4S,5S)-4-[(tert-ブトキシカルボニル)アミノ]アダマンタン-1-カルボン酸(99.0mg)のDME(0.99ml)懸濁液にN-メチルモルホリン(0.044ml)を加えて溶解した。氷冷下、クロロ炭酸イソブチル(0.052ml)を滴下し、同温度で40分間撹拌した。析出した白色不溶物を濾去し、DME(0.5ml)で洗浄した。濾液を氷冷し、水素化ホウ素ナトリウム(25.3mg)を加え、次いでMeOH(0.495ml)をゆっくりと滴下した。室温で1時間撹拌した後、反応液を氷冷し、EtOAcで希釈した。1M塩酸(1.0ml)を加えて酸性にし、有機層を分取した。有機層を水(2回)、飽和重曹水、水、次いで飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製した。目的物を含むフラクションを減圧濃縮後、残渣をEtOAcに溶解し、0.1ml程度まで減圧濃縮した。ヘキサン(1.5ml)を少しずつ加えて結晶化し、結晶を濾取してtert-ブチル rel-[(1R,2S,3S,5S)-5-(ヒドロキシメチル)アダマンタン-2-イル]カルバマートを77.3mg得た。
rel-(1R,3S,5R,7S)-4-オキソアダマンタン-1-カルボン酸(1.0g)をアンモニアのMeOH溶液(濃度8M、20ml)に溶解し、10%Pd/C (50%含水品、100mg)を加え、3気圧の水素雰囲気下、25℃で10時間撹拌した。大量に析出した生成物を水(20ml)で溶解し、触媒をセライトで濾去した。MeOHを減圧留去した残渣にアセトニトリル(30ml)を滴下して室温で1時間撹拌した。析出物を濾取し、MeCN(10ml)で洗浄後、45℃で減圧乾燥してrel-(1S,3R,5S)-4-アミノアダマンタン-1-カルボン酸をトランス体とシス体の比率が3.5 : 1の混合物として982mg得た。
rel-(1S,3R,5S)-4-アミノアダマンタン-1-カルボン酸(トランス体とシス体の比率が3.5 : 1の混合物、100mg)を水(4ml)に懸濁し、75℃で30分間撹拌した。撹拌しながら室温に戻し、MeCN(4ml)をゆっくり滴下した後、同温度で30分間撹拌した。析出物を濾取し、アセトニトリル(1ml)で洗浄後、45℃で減圧乾燥してrel-(1S,3R,4S,5S)-4-アミノアダマンタン-1-カルボン酸を50.0mg得た。
4-(メトキシメチレン)-1-メチルシクロヘキサノール(5.0g)のMeCN(50ml)溶液に、水(8.6ml)とTFA(3.6ml)を順次加え、室温にて4時間攪拌した。反応混合液を飽和重曹水にて中性に調節した後、EtOAcで4回抽出した。有機層を合わせて飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製し、まずシス-4-ヒドロキシ-4-メチルシクロヘキサンカルバルデヒド(2.37g)を溶出し、次いでトランス-4-ヒドロキシ-4-メチルシクロヘキサンカルバルデヒド(2.7g)を溶出した。






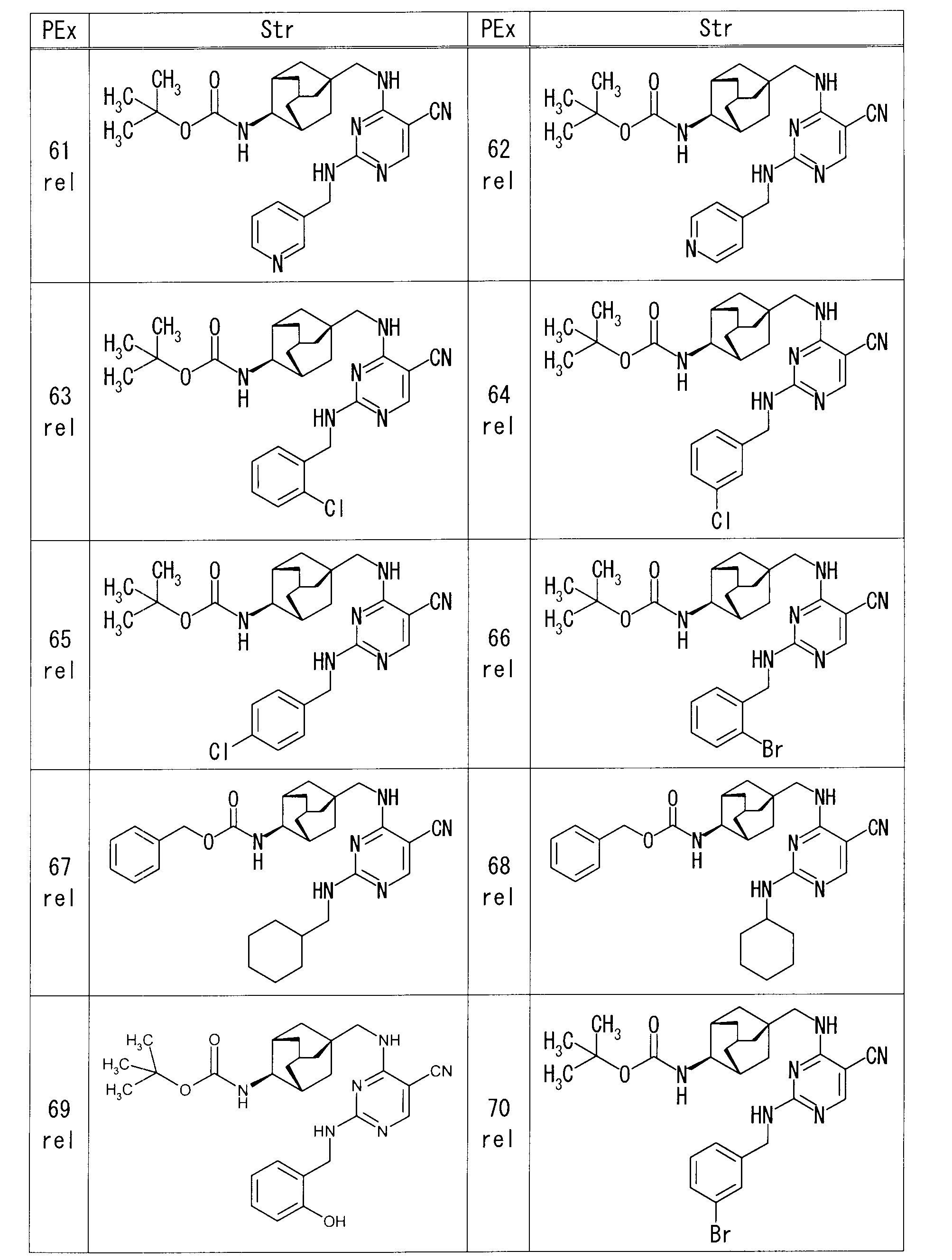







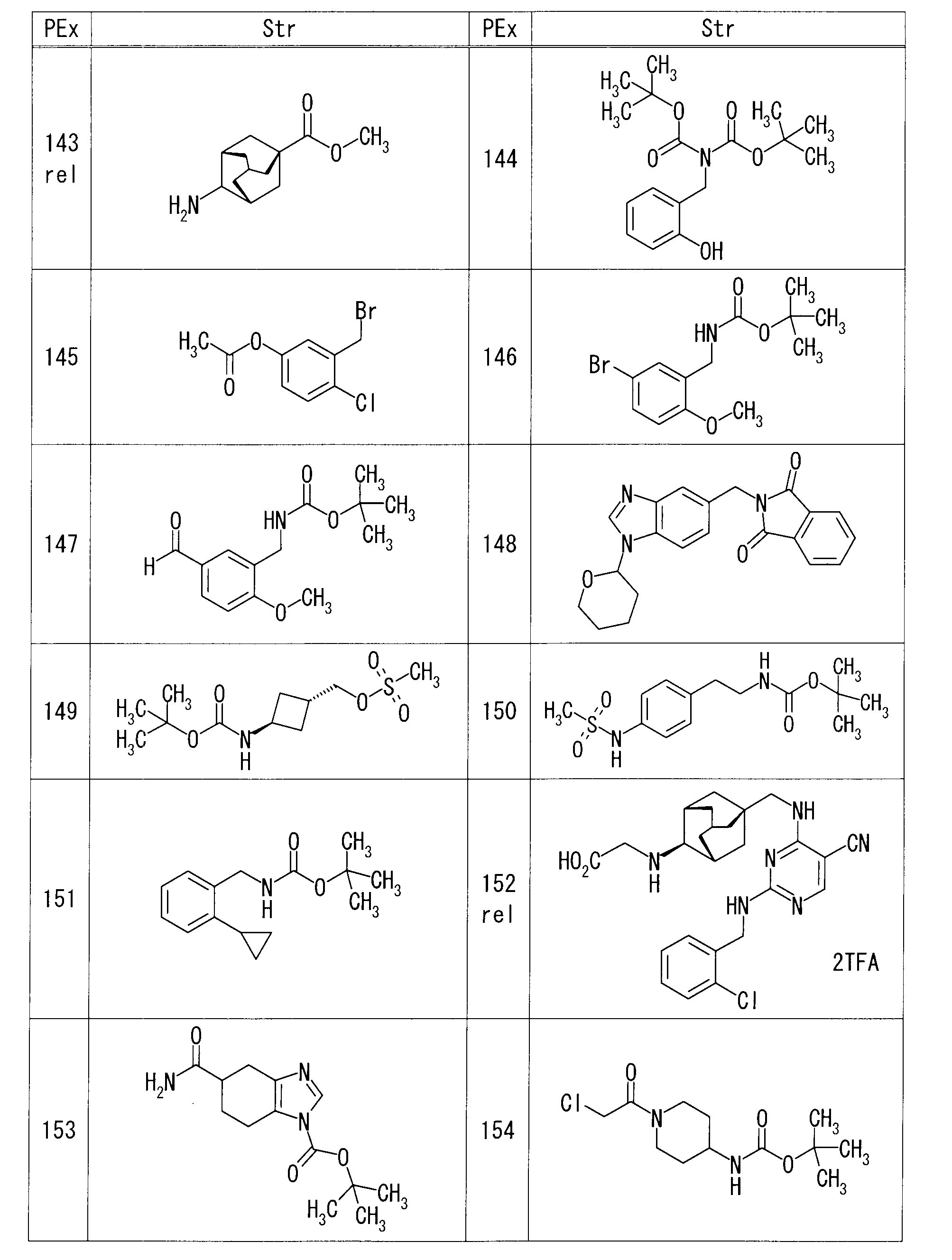


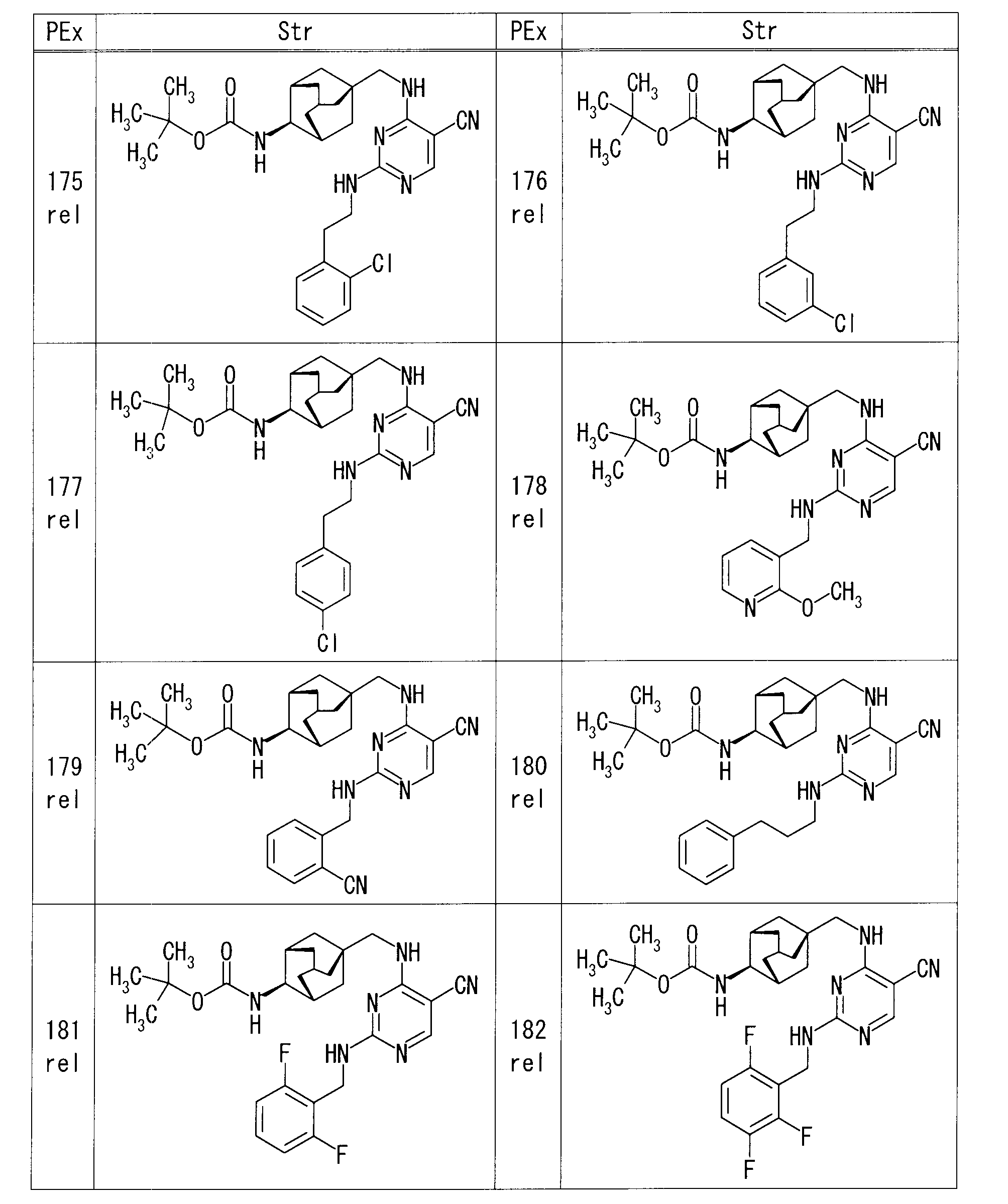


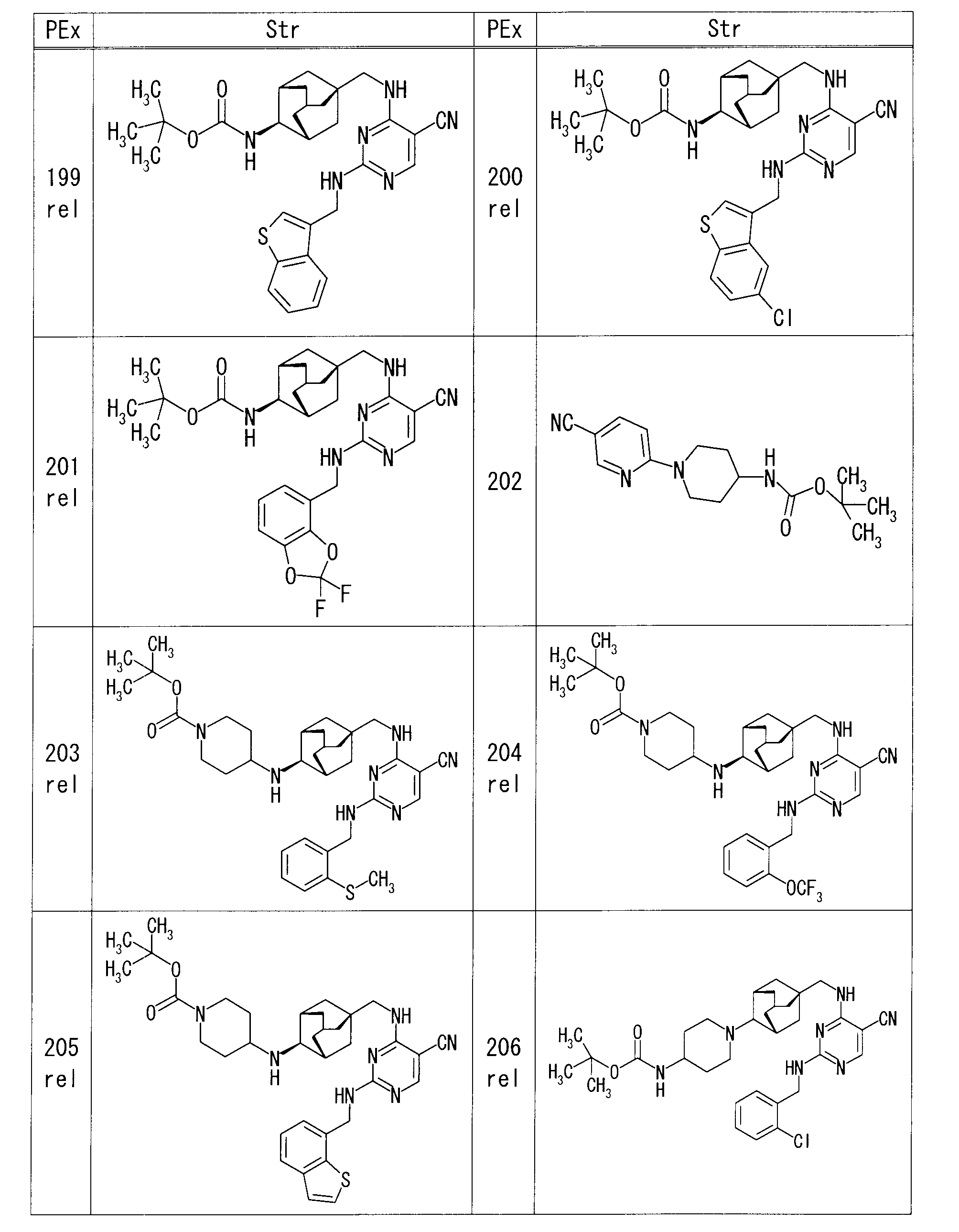



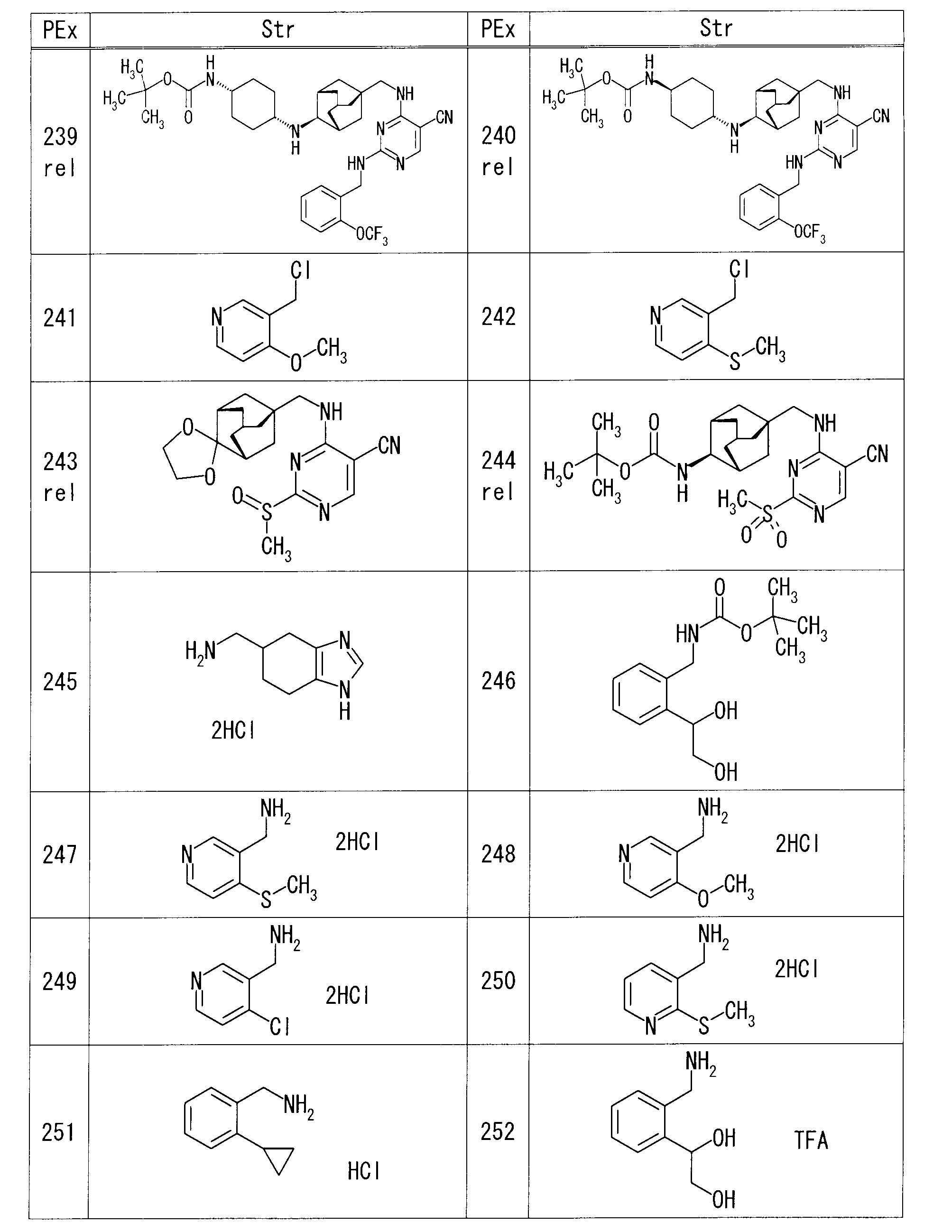







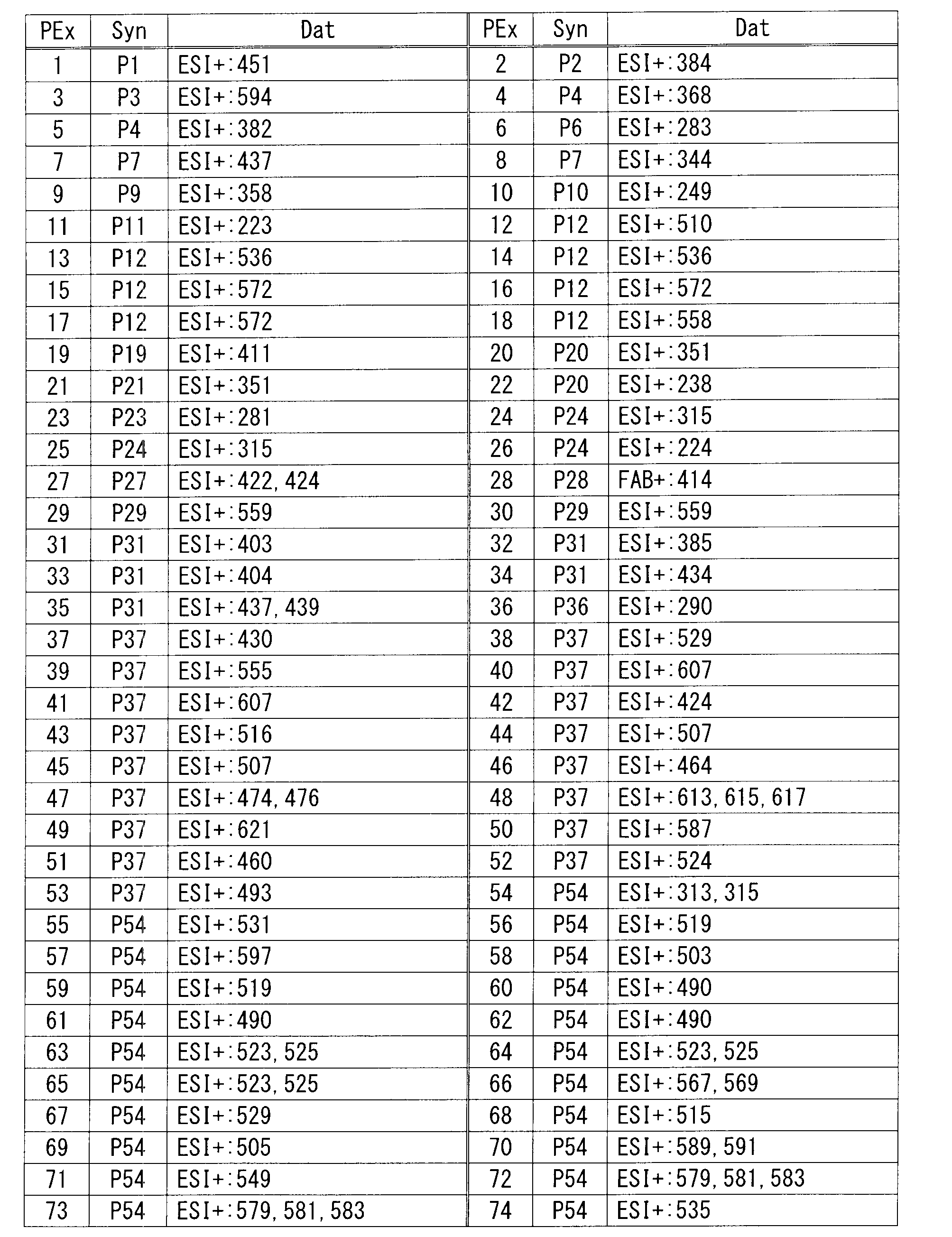
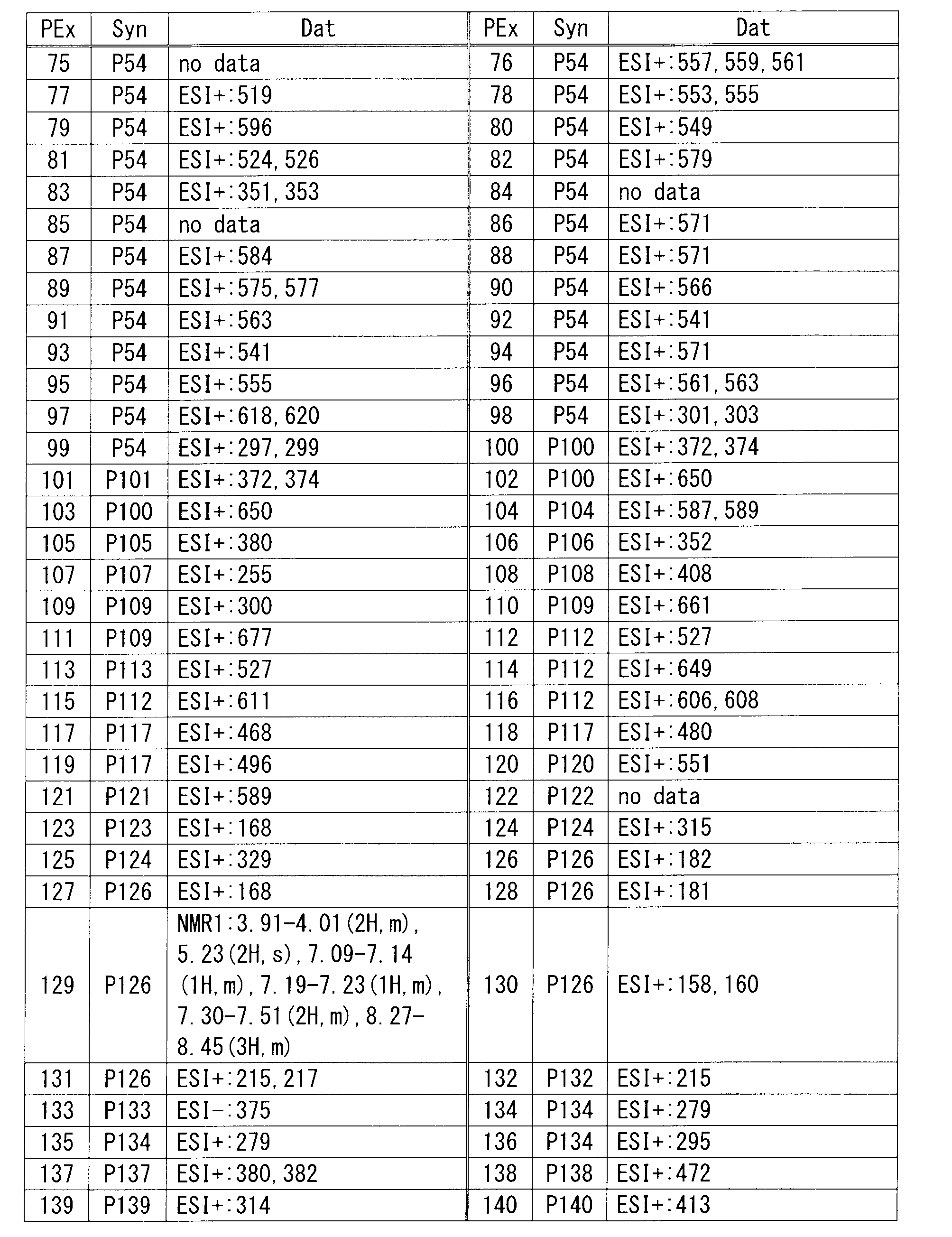




rel-1-[(1R,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]-D-プロリン ベンジルエステル(100mg)のEtOH(5ml)溶液に10%Pd/C(50%含水品、20mg)を加え、水素雰囲気下、常圧にて室温で撹拌した。反応終了後、セライト濾過にて触媒を濾別し、濾液を減圧下濃縮した。得られた残渣を分取用アルミナ薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-1-[(1R,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]-D-プロリンを74.4mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のDMF(0.2ml)溶液にヨードベンゼン(14.2μl)、酢酸セシウム(50.8mg)、ヨウ化銅(I)(20.2mg)を加え、マイクロウェーブ照射下、90℃にて24時間撹拌した。反応混合液をEtOAcで希釈後、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-({[(1S,3R,4S,5S)-4-アニリノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを5.1mg得た。
氷冷下、rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のDMF(1.0ml)溶液にDIPEA(27.7μl)、塩化アセチル(10.3μl)を順次加え、同温度で1時間撹拌した。反応混合物をEtOAcで希釈後、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-N-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]アセトアミドを46mg得た。
rel-(4R)-1-[(1R,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]-4-ヒドロキシ-D-プロリン(23mg)のDMF(0.5ml)溶液に、塩化アンモニウム(6.3mg)、HOBt(15.9mg)、WSC(18.3mg)を順次加え、室温にて終夜撹拌した。反応混合物をEtOAcで希釈後、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(THF)、次いでアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-(4R)-1-[(1R,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]-4-ヒドロキシ-D-プロリンアミドを10mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のDMF(1.0ml)溶液に、ニコチン酸(14.3mg)、HOBt(15.7mg)、WSC(18.1mg)を順次加え、室温にて終夜撹拌した。反応混合物をEtOAcで希釈後、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、rel-N-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]ニコチンアミドを58mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のN,N-ジメチルアセトアミド(2.5ml)溶液に、炭酸ナトリウム(44.8mg)、1,3-ジブロモプロパン(43.2μl)を加え、マイクロウェーブ照射下、100℃にて30分間撹拌した。反応混合物をEtOAcで希釈後、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-({[(1S,3R,4S,5S)-4-アゼチジン-1-イルアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを45mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(30mg)のDMF(0.6ml)溶液に、DIPEA(22.1μl)、2-ブロモエタノール(4.5μl)を加え、60℃で加熱撹拌した。反応混合物をEtOAcで希釈後、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)、次いで分取用アルミナ薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[(2-ヒドロキシエチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを25mg得た。
4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のDMF(1ml)溶液に(2,2-ジメチル-1,3-ジオキサン-5-イル)メチル 4-メチルベンゼンスルホナート(47.7mg)および炭酸カリウム(29.2mg)を加え、70℃で12時間撹拌した。反応混合物に水を加え、EtOAcで抽出した。有機層を硫酸マグネシウムで乾燥し、乾燥剤を除去後、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン-EtOAc)で精製し、4-({[(1S,3R,4S,5S)-4-{[(2,2-ジメチ-1,3-ジオキサン-5-イル)メチル]アミノ}アダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを21mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のEtOH(1.0ml)とTHF(0.5ml)混合溶液にオキシラン-2-イルメタノール(8.2μl)を加え、室温にて24時間撹拌した。オキシラン-2-イルメタノール(82.1μl)を追加し、室温にてさらに48時間撹拌した。反応混合液をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[(2,3-ジヒドロキシプロピル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを25mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)の1,3-ジメチルイミダゾリジン-2-オン(1.0ml)溶液に、DIPEA(36.9μl)、2,2,2-トリフルオロエチルトリフルオロメタンスルホネート(18.3μl)を加え、室温にて撹拌した。反応終了後、反応混合液をそのままシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[(2,2,2-トリフルオロエチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを55mg得た。
4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)の1,3-ジメチルイミダゾリジン-2-オン(0.5ml)溶液に2,2-ビス(ブロモメチル)プロパン-1,3-ジオール(221.8mg)および炭酸カリウム(146.3mg)を加え、室温で4日間撹拌した。反応混合物をそのままシリカゲルカラムクロマトグラフィー(クロロホルム-MeOH)により精製し、[({(1S,3R,4S,5S)-4-[3,3-ビス(ヒドロキシメチル)アゼチジン-1-イル-アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを5mg得た。
rel-4-[({(1R,3S,5R)-4-[(trans-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルの立体異性体混合物(39mg)を逆相液体クロマトグラフィー(溶出液:0.2%蟻酸/MeOHと水の混合液)で分離分取精製することにより、rel-4-[({(1R,3S,4R,5R)-4-[(trans-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルの蟻酸塩とrel-4-[({(1R,3S,4S,5R)-4-[(trans-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルの蟻酸塩をそれぞれ単一異性体として得た。次いで、それらの蟻酸塩をそれぞれアミノシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、rel-4-[({(1R,3S,4R,5R)-4-[(trans-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを19.1mg、rel-4-[({(1R,3S,4S,5R)-4-[(trans-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを10.4mg得た。
rel-trans-4-{[(1R,2S,3S,5S)-5-(アミノメチル)アダマンタン-2-イル]アミノ}シクロヘキサノール(80mg)の1,3-ジメチルイミダゾリジン-2-オン(1.0ml)溶液に、DIPEA(0.20ml)、4-クロロ-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリル(108.6mg)を加え、室温にて終夜撹拌した。反応混合液をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1R,3S,4R,5R)-4-[(trans-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリルを73.4mg得た。
rel-N-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]グリシン エチルエステル(24mg)のTHF(2.4ml)溶液に、4M水酸化リチウム水溶液(0.1ml)を加え、室温にて攪拌した。反応終了後、反応混合液に1M塩酸(0.4ml)を加えて撹拌後、反応混合液を減圧下濃縮した。得られた残渣に水を加えて固体を濾取後、減圧下乾燥した。得られた固体にEtOAcとジイソプロピルエーテルを加えて懸濁後、固体を濾取し、減圧下乾燥することによりrel-N-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]グリシンを11.0mg得た。
rel-4-({[(1S,3R,5S)-4-オキソアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(40mg)のジクロロメタン(0.8ml)溶液に、trans-4-アミノシクロヘキサノール(14.7mg)とトリアセトキシ水素化ホウ素ナトリウム(53.9mg)を加え、室温にて終夜撹拌した。反応混合液に飽和重曹水を加え、EtOAcで抽出後、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1R,3S,5R)-4-[(trans-4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを18mg得た。
rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のジクロロメタン(3.5ml)溶液に4-ヒドロキシシクロヘキサノン(39.4mg)とトリアセトキシ水素化ホウ素ナトリウム(146.3mg)を加え、室温にて2時間撹拌した。反応混合液に飽和重曹水を加え、EtOAcで抽出後、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[(4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリルを60.6mg得た。
rel-4-[({(1S,3R,4S,5S)-4-[(4-ヒドロキシシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のMeOH(2ml)溶液に37%ホルマリン水溶液(28.4μl)と水素化シアノホウ素ナトリウム(8.3mg)を加え、室温にて5時間撹拌した。反応混合液に飽和重曹水を加え、EtOAcで抽出後、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[(4-ヒドロキシシクロヘキシル)(メチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを37.6mg得た。
rel-4-({[(1S,3R,5S)-4-オキソアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(30mg)のMeOH(3ml)溶液に水素化ホウ素ナトリウム(13.5mg)を加え、室温にて撹拌した。反応終了後、反応混合物をEtOAcで希釈後、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-({[(1S,3R,5S)-4-ヒドロキシアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを25.6mg得た。
氷冷下、rel-2-[(2-メトキシベンジル)アミノ]-4-[({(1S,3R,4S,5S)-4-[(4-オキソシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]ピリミジン-5-カルボニトリル(40mg)のTHF(2ml)溶液に、窒素雰囲気下で0.97M臭化メチルマグネシウムのTHF溶液(0.40ml)を滴下し、同温度で3時間撹拌した。反応混合液に水を加えてEtOAcで抽出後、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH-28%アンモニア水)にて精製することにより、先ずrel-4-[({(1S,3R,4S,5S)-4-[(4-ヒドロキシ-4-メチルシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-[(2-メトキシベンジル)アミノ]ピリミジン-5-カルボニトリル(isomer A)を4.3mg、次いでrel-4-[({(1S,3R,4S,5S)-4-[(4-ヒドロキシ-4-メチルシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-[(2-メトキシベンジル)アミノ]ピリミジン-5-カルボニトリル(isomer B)を5.3mg得た。
4-[(1-アザビシクロ[2.2.2]オクタ-3-イルメチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(40mg)のジクロロメタン(1ml)溶液に、氷冷下77%MCPBA(含水品、21mg)を加え、室温で3時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、EtOAcで抽出した。有機層を無水硫酸ナトリウムで乾燥し、乾燥剤を除去後、溶媒を減圧下留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)で精製し、4-[(1-オキシド-アザビシクロ[2.2.2]オクタ-3-イルメチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを19mg得た。
rel-4-({[(1S,3R,4S,5S)-4-(テトラヒドロ-2H-チオピラン-4-イルアミノ)アダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(88mg)のジクロロメタン(2ml)溶液に75%MCPBA(含水品、106.1mg)を加え、室温にて6時間撹拌した。反応混合液に飽和重曹水を加えてEtOAcで抽出し、有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)、次いで分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[(1,1-ジオキシドテトラヒドロ-2H-チオピラン-4-イル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを8.8mg得た。
4-{[(3-endo)-8-ベンジル-8-アザビシクロ[3.2.1]オクト-3-イル]アミノ}-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(16mg)のMeOH(1ml)溶液に蟻酸アンモニウム(100mg)と触媒量の10%Pd/C(50%含水品)を加え、6時間加熱還流した。反応混合液を室温まで放冷した後、触媒を除去し、濾液を減圧下濃縮した。得られた残渣をEtOAcに溶解し、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、4-[(3-endo)-8-アザビシクロ[3.2.1]オクト-3-イルアミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを8.0mg得た。
tert-ブチル rel-{(1R,2S,3S,5S)-5-[({2-[(2-クロロベンジル)アミノ]-5-シアノピリミジン-4-イル}アミノ)メチル]アダマンタン-2-イル}カルバマート(70mg)のジクロロメタン(1ml)溶液にトリフルオロ酢酸(0.135ml)を加え室温にて終夜撹拌した。反応混合液を減圧下濃縮し、得られた残渣に炭酸カリウム水溶液を加え、室温にて3時間撹拌した。析出した固体を濾取して水洗後、減圧下乾燥することでrel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-[(2-クロロベンジル)アミノ]ピリミジン-5-カルボニトリルを55mg得た。
tert-ブチル 3-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}-8-アザビシクロ[3.2.1]オクタン-8-カルボキシレートのジオキサン(0.6ml)溶液に4M塩化水素ジオキサン溶液(0.28ml)を加え、室温で10時間撹拌した。反応混合物を減圧下濃縮し、4-[(8-アザビシクロ[3.2.1]オクタ-3-イルメチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル 2塩酸塩を56mg得た。
4-({[1-(テトラヒドロ-2H-ピラン-2-イル)-1H-ベンズイミダゾール-5-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のEtOH(3ml)溶液に1M塩酸(1ml)を加え、60℃にて24時間撹拌した。反応混合液に1M塩酸(1ml)を追加し、60℃にてさらに24時間撹拌した。反応液を減圧下濃縮し、得られた残渣にEtOAcとEtOHを加えた。析出した固体を濾取し、EtOAcで洗浄後、減圧下乾燥することで4-[(1H-ベンズイミダゾール-5-イルメチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル 2塩酸塩を53.1mg得た。
ベンジル rel-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]カルバマート(55mg)のMeOH(3ml)溶液に10%Pd/C(50%含水品、15mg)を加え、水素雰囲気下、常圧にて室温で6時間撹拌した。触媒を濾去し、濾液を減圧下濃縮した。得られた残渣を分取用アルミナ薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを20.0mg得た。
ベンジル rel-{(1R,2S,3S,5S)-5-[({5-シアノ-2-[(2,5-ジクロロベンジル)アミノ]ピリミジン-4-イル}アミノ)メチル]アダマンタン-2-イル}カルバマート(45mg)の酢酸(1.5ml)溶液に48%臭化水素酸(1.5ml)を加え、室温で24時間撹拌した。反応液を減圧下濃縮後、残渣をEtOAcで溶解し、炭酸カリウム水溶液、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-[(2,5-ジクロロベンジル)アミノ]ピリミジン-5-カルボニトリルを18mg得た。
rel-4-({[(1S,3R,4S,5S)-4-{[(2-フェニル-1,3-ジオキサン-4-イル)メチル]アミノ}アダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}-5-カルボニトリル(54mg)のTHF(1.5ml)溶液に1M塩酸(1.5ml)を加え、室温で4時間撹拌した。反応混合物を氷冷し、飽和重曹水を加えた。析出物を濾取し、減圧下乾燥することによって得られた固体をアミノシリカゲルカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-[({(1S,3R,4S,5S)-4-[(2,4-ジヒドロキシブチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを41.2mg得た。
rel-4-[({(1S,3R,4S,5S)-4-[(2,2-ジメチル-1,3-ジオキサン-5-イル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(55mg)のTHF(1.0ml)溶液に1M塩酸(1.0ml)を加え、室温にて攪拌した。反応終了後、反応混合液を減圧下濃縮後、残渣にジクロロメタンを加え減圧下濃縮した。得られた残渣にジエチルエーテルを加え、析出した固体を濾取し、ジエチルエーテルで洗浄後、減圧下乾燥することで、rel-4-({[(1S,3R,4S,5S)-4-{[2-ヒドロキシ-1-(ヒドロキシメチル)エチル]アミノ}アダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル 2塩酸塩を50.5mg得た。
氷冷下、rel-4-({[(1S,3R,4S,5S)-4-アミノアダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(50mg)のDMF(1.0ml)溶液にDIPEA(27.7μl)とメタンスルホニルクロリド(8.6μL)を順次加え、同温度で1時間撹拌した。反応混合液をEtOAcで希釈し、有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去した。得られた残渣を分取用シリカゲル薄層クロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-N-[(1R,2S,3S,5S)-5-{[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]メチル}アダマンタン-2-イル]メタンスルホンアミドを31mg得た。
氷冷下、rel-2-[(2-クロロベンジル)アミノ]-4-({[(1S,3R,5S)-4-ピペラジン-1-イルアダマンタン-1-イル]メチル}アミノ)ピリミジン-5-カルボニトリル(18mg)のDMF(360μl)溶液に、トリエチルアミン(3.6μl)、無水酢酸(7.6μl)を順次加え室温にて攪拌した。反応終了後、反応混合液をEtOAcで希釈し、水(3回)、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、rel-4-({[(1S,3R,5S)-4-(4-アセチルピペラジン-1-イル)アダマンタン-1-イル]メチル}アミノ)-2-[(2-クロロベンジル)アミノ]ピリミジン-5-カルボニトリルを16.2mg得た。
rel-2-[(2-クロロベンジル)アミノ]-4-[({(1S,3R,4S,5S)-4-[(2-クロロエチル)アミノ]アダマンタン-1-イル}メチル)アミノ]ピリミジン-5-カルボニトリル(27.0mg)と(2S)-ピロリジン-2-イルメタノール(7.9mg)のDMF(0.27ml)溶液にヨウ化カリウム(13.8mg)とDIPEA(0.02ml)を加え、75℃で2時間撹拌後、(2S)-ピロリジン-2-イルメタノール(1.1mg)を追加して同温度でさらに2時間撹拌した。減圧下溶媒を留去し、得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、2-[(2-クロロベンジル)アミノ]-4-({[(1R,3R,4S,5S)-4-({2-[(2S)-2-(ヒドロキシメチルピロリジン-1-イル]エチル}アミノ)アダマンタン-1-イル]メチル}アミノ)ピリミジン-5-カルボニトリルを25.4mg得た。
rel-4-[({(1R,3S,4R,5R)-4-[(cis-4-アミノシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-[(2-クロロベンジル)アミノ]ピリミジン-5-カルボニトリル(30mg)のDMI(1.0ml)溶液に、DIPEA(80.4μl)と2-ブロモエタノール(8.2μl)を加え、120度にて加熱撹拌した。反応終了後、反応混合物をクロロホルムで希釈し、そのままアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-2-[(2-クロロベンジル)アミノ]-4-({[(1R,3S,4R,5R)-4-({cis-4-[(2-ヒドロキシエチル)アミノ]シクロヘキシル}アミノ)アダマンタン-1-イル]メチル}アミノ)ピリミジン-5-カルボニトリルを17.6mg得た。
rel-2-[(2-クロロベンジル)アミノ]-4-[({(1S,3R,4S,5S)-4-[(4-オキソシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]ピリミジン-5-カルボニトリル(50mg)のジクロロメタン(3.0ml)溶液に4,4-ジフルオロピペリジン塩酸塩(30.4mg)、トリエチルアミン(26.7μl)、トリアセトキシ水素化ホウ素ナトリウム(61.2mg)を順次加え、室温にて撹拌した。反応終了後、反応混合液に飽和重曹水を加えてクロロホルムで抽出し、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-2-[(2-クロロベンジル)アミノ]-4-({[(1S,3R,4S,5S)-4-{[4-(4,4-ジフルオロピペリジン-1-イル)シクロヘキシル]アミノ}アダマンタン-1-イル]メチル}アミノ)ピリミジン-5-カルボニトリルを13.1mg得た。
rel-4-[({(1R,3S,4R,5R)-4-[(cis-4-アミノシクロヘキシル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-[(2-クロロベンジル)アミノ]ピリミジン-5-カルボニトリル(40mg)のジクロロメタン(2.0ml)溶液に1-アセチルピペリジン-4-オン (21.7mg)とトリアセトキシ水素化ホウ素ナトリウム(48.9mg)を順次加え、室温にて撹拌した。反応終了後、反応混合液に飽和重曹水を加えてクロロホルムで抽出し、減圧下溶媒を留去した。得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-({[(1R,3S,4R,5R)-4-({cis-4-[(1-アセチルピペリジン-4-イル)アミノ]シクロヘキシル}アミノ)アダマンタン-1-イル]メチル}アミノ)-2-[(2-クロロベンジル)アミノ]ピリミジン-5-カルボニトリルを54mg得た。
rel-4-({[(1S,3R,4S,5S)-4-({[trans-4-({[tert-ブチル(ジメチル)シリル]オキシ}メチル)シクロヘキシル]メチル}アミノ)アダマンタン-1-イル]メチル}アミノ)-2-[(2-シアノベンジル)アミノ]ピリミジン-5-カルボニトリル(68mg)のMeOH(1.4ml)懸濁液に1M塩酸(0.6ml)を加え、室温で1時間撹拌した。減圧下溶媒を留去した後、残渣をクロロホルムで希釈し、氷冷下飽和重曹水を加えた。混合物をクロロホルム-MeOH (10:1)混合溶媒で抽出し、有機層を水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、得られた残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製し、rel-2-[(2-シアノベンジル)アミノ]-4-({[(1S,3R,4S,5S)-4-({[trans-4-(ヒドロキシメチル)シクロヘキシル]メチル}アミノ)アダマンタン-1-イル]メチル}アミノ)ピリミジン-5-カルボニトリルを49.4mg得た。
氷冷下、4-[({(1S,3R,4S,5S)-4-[(3-{[tert-ブチルジメチル)シリル]オキシプロピル)(メチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリル(55mg)のTHF(2mL)溶液に1M テトラブチルアンモニウム フルオリドのTHF溶液(0.20mL)を加え、室温で1時間撹拌した。溶媒を減圧留去し、そのままアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製し、4-[({(1S,3R,4S,5S)-4-[(3-ヒドロキシプロピル)(メチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(メチルスルファニル)ベンジル]アミノ}ピリミジン-5-カルボニトリルを25mg得た。
rel-4-[({(1S,3R,4S,5S)-4-[3-(1,3-ジオキソ-1,3-ジヒドロ-2H-イソインドール-2-イル)アゼチジン-1-イル]アダマンタン-1-イル}メチル)アミノ]-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル(64mg)のEtOH(1.28ml)とTHF(1.28ml)の混合溶液にヒドラジン一水和物(18.9μl)を加え、加熱還流した。不溶物を濾過除去後、濾液を減圧下濃縮した。得られた残渣にクロロホルムを加え、水で2回洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-4-({[(1S,3R,4S,5S)-4-(3-アミノアゼチジン-1-イル)アダマンタン-1-イル]メチル}アミノ)-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリルを14mg得た。
rel-2-[(2-クロロベンジル)アミノ]-4-[({(1S,3R,4S,5S)-4-[(ピペリジン-4-イルメチル)アミノ]アダマンタン-1-イル}メチル)アミノ]ピリミジン-5-カルボニトリル(40mg)のDMI溶液(1ml)に1-フルオロ-3-ヨードプロパン(18mg)とDIPEA(0.017ml)を加え、100℃で1時間マイクロウェーブを照射した。反応混合物をクロロホルムで希釈し、そのままアミノシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン-EtOAc)にて精製することにより、rel-2-[(2-クロロベンジル)アミノ]-4-({[(1S,3R,4S,5S)-4-({[1-(3-フルオロプロピル)ピペリジン-4-イルメチル]メチル}アミノ)アダマンタン-1-イル}メチル}アミノ)ピリミジン-5-カルボニトリルを20mg得た。
氷冷下、tert-ブチル 4-{4-[(5-シアノ-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-4-イル)アミノ]ブタノイル}ピペラジン-1-カルボキシラート(205mg)のジクロロメタン(4.1ml)溶液にTFA(0.75ml)を加え、室温にて18時間撹拌した。反応混合液を濃縮し、氷冷下、炭酸カリウム水溶液を加え、室温で撹拌した。析出した固体をろ取し、乾燥後、アミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製した。所望の化合物を含むフラクションを濃縮し、残渣に4M塩化水素 酢酸エチル溶液を加えた後、濃縮乾固することにより4-{[4-オキソ-4-(ピペラジン-1-イル)ブチル]アミノ}-2-{[2-(トリフルオロメトキシ)ベンジル]アミノ}ピリミジン-5-カルボニトリル 2塩酸塩を65.6mg得た。
rel-4-[({(1S,3R,4S,5S)-4-[(1,4-ジオキサスピロ[4.5]デカ-8-イルメチル)アミノ]アダマンタン-1-イル}メチル)アミノ]-2-({[2-(メチルスルファニル)ピリジン-3-イル]メチル}アミノ)ピリミジン-5-カルボニトリル(140mg)のTHF溶液(3ml)に1M塩酸(2ml)を加え、室温で2時間撹拌した。反応混合物を減圧下濃縮後、飽和重曹水を加えてEtOAcで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。乾燥剤を除去後、減圧下溶媒を留去し、残渣をアミノシリカゲルフラッシュカラムクロマトグラフィー(クロロホルム-MeOH)にて精製することにより、rel-2-({[2-(メチルスルファニル)ピリジン-3-イル]メチル}アミノ)-4-({[(1S,3R,4S,5S)-4-{[(4-オキソシクロヘキシル)メチル]アミノ}アダマンタン-1-イル]メチル}アミノ)ピリミジン-5-カルボニトリルを100mg得た。



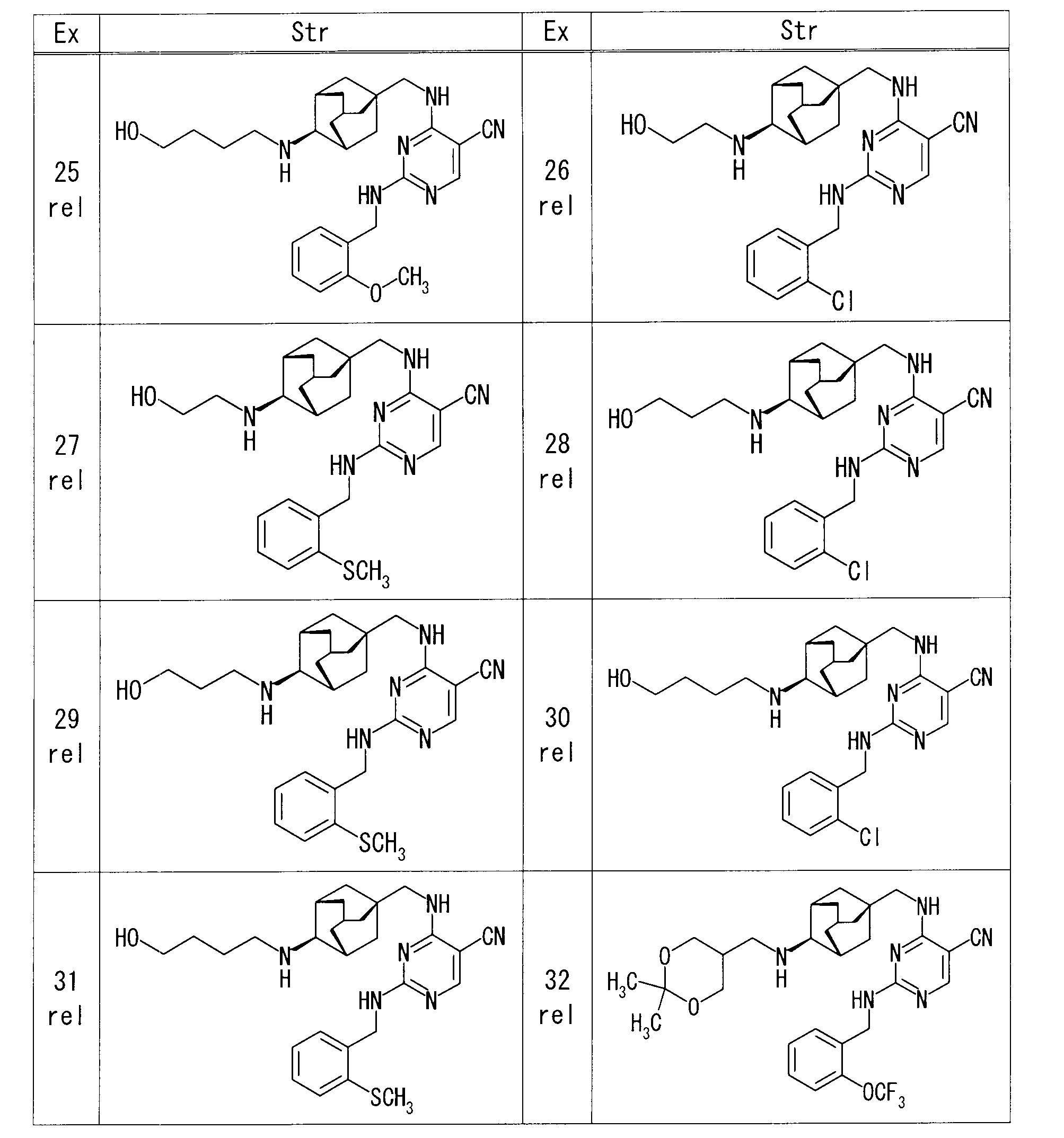


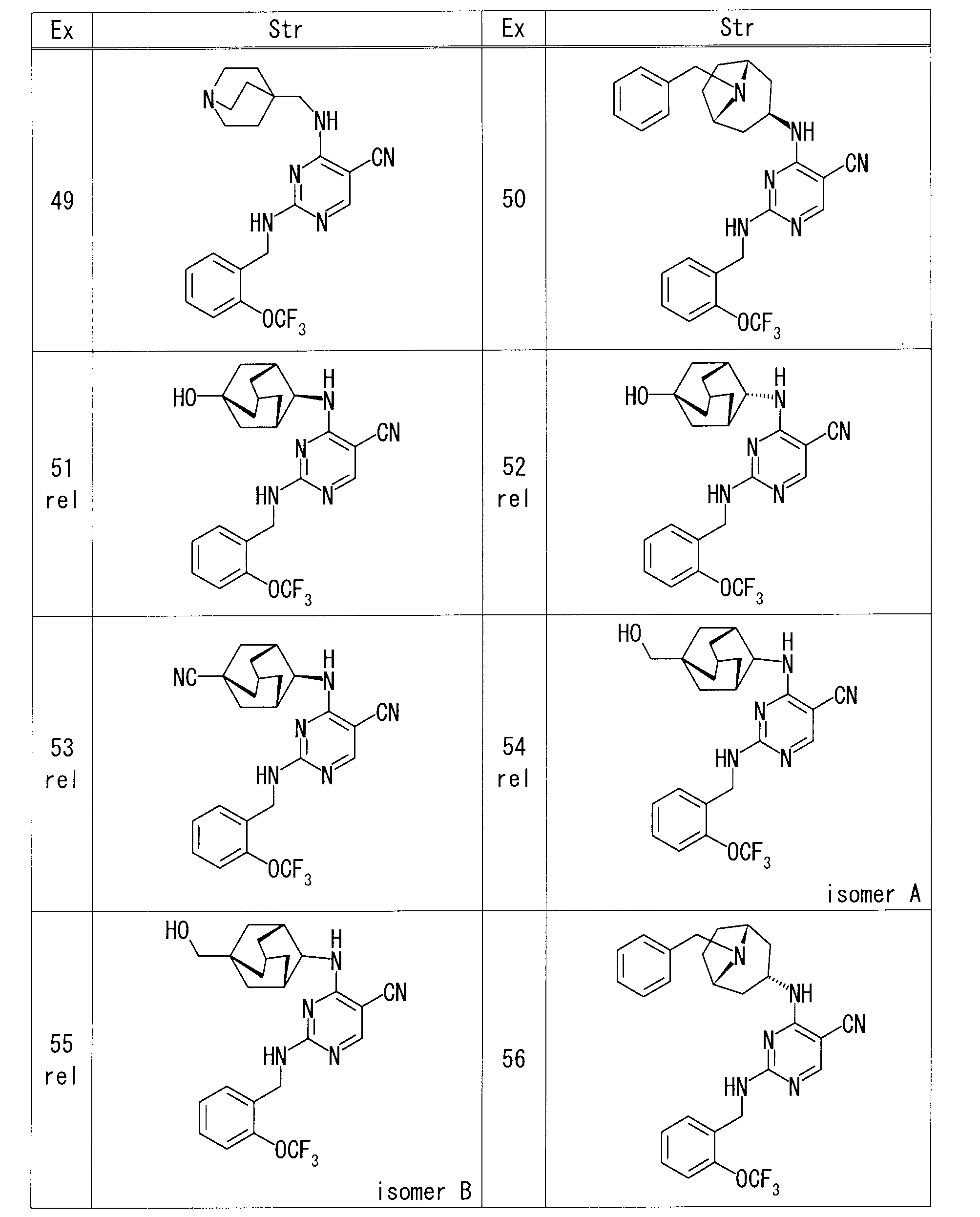
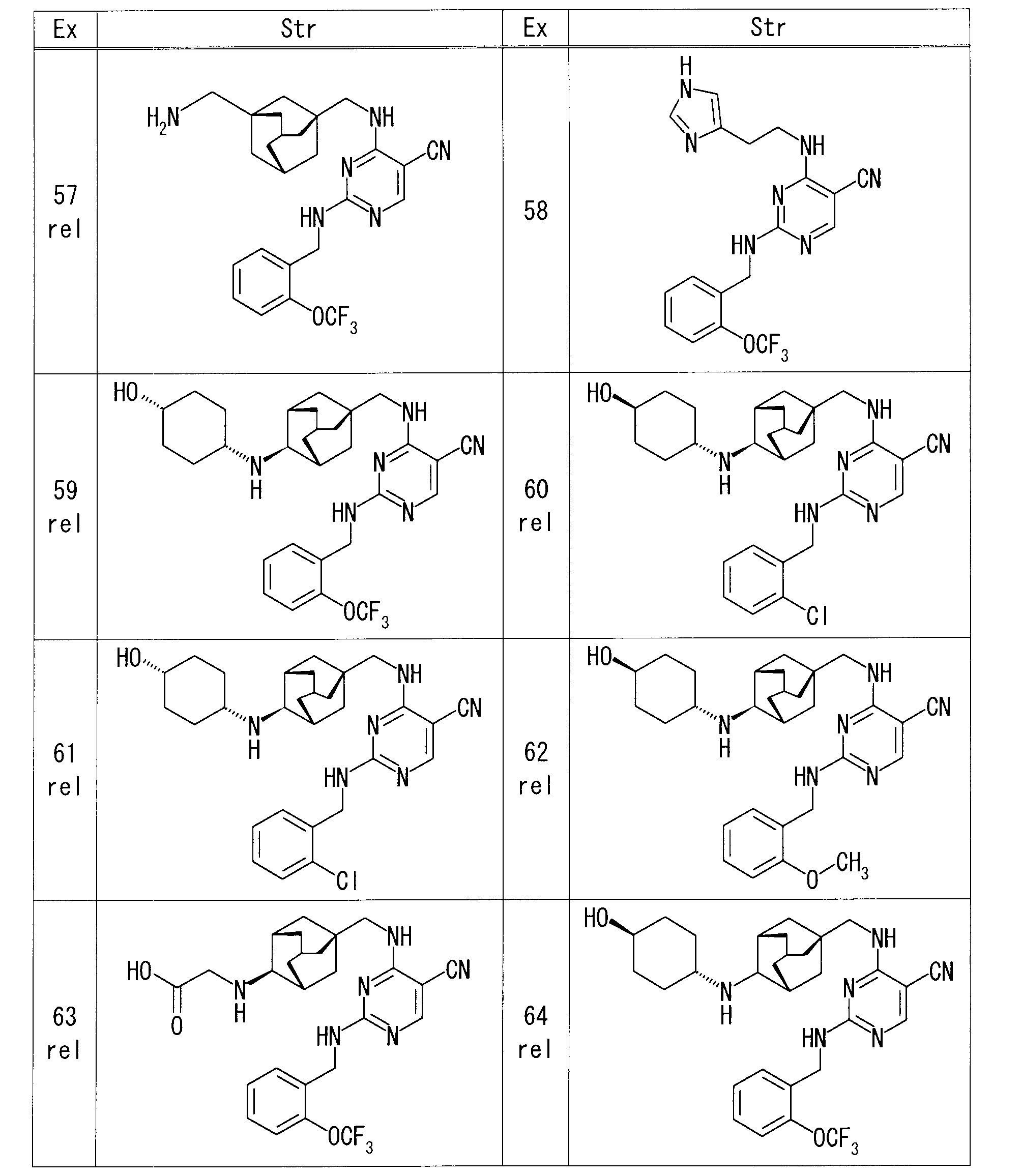

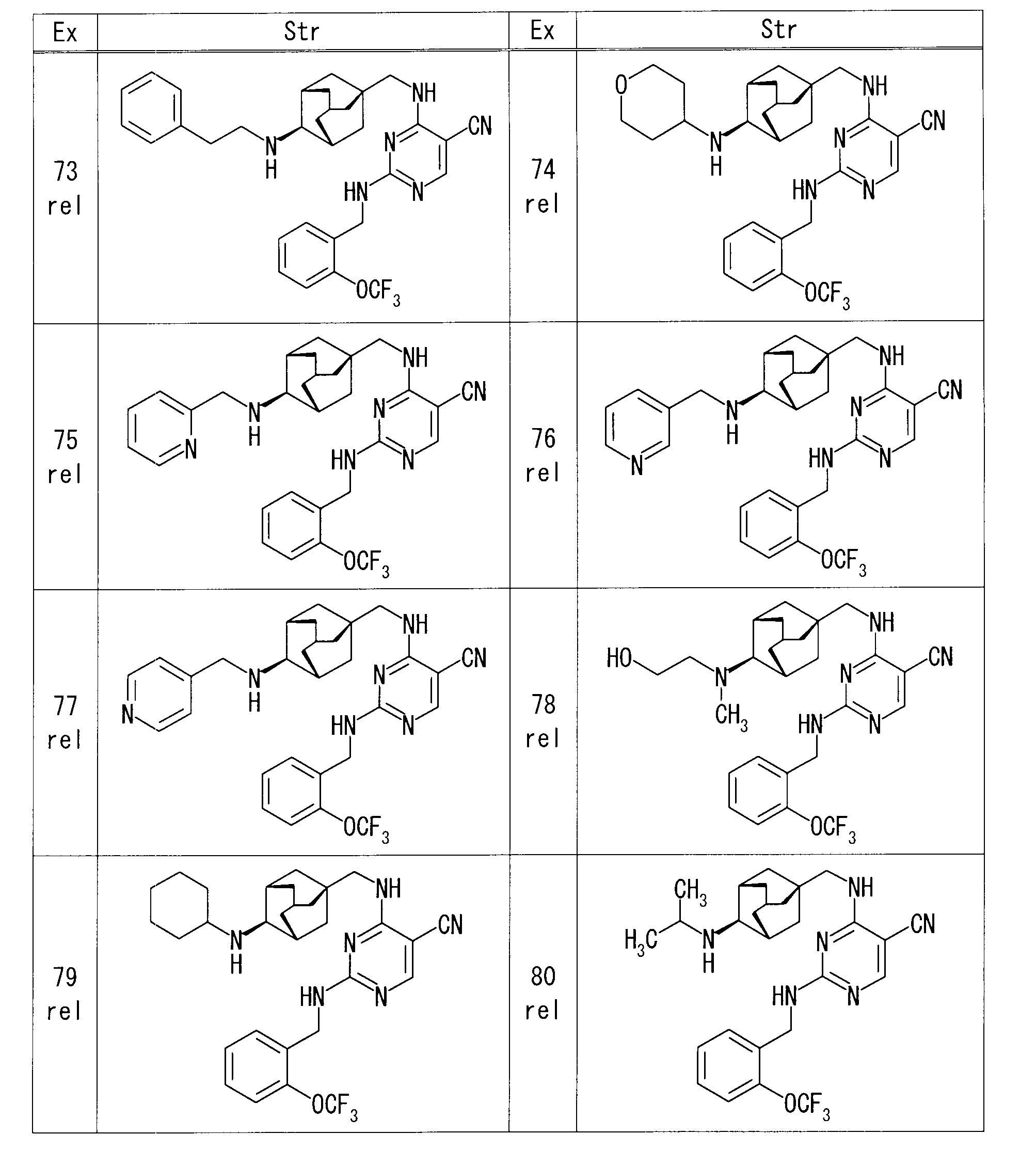


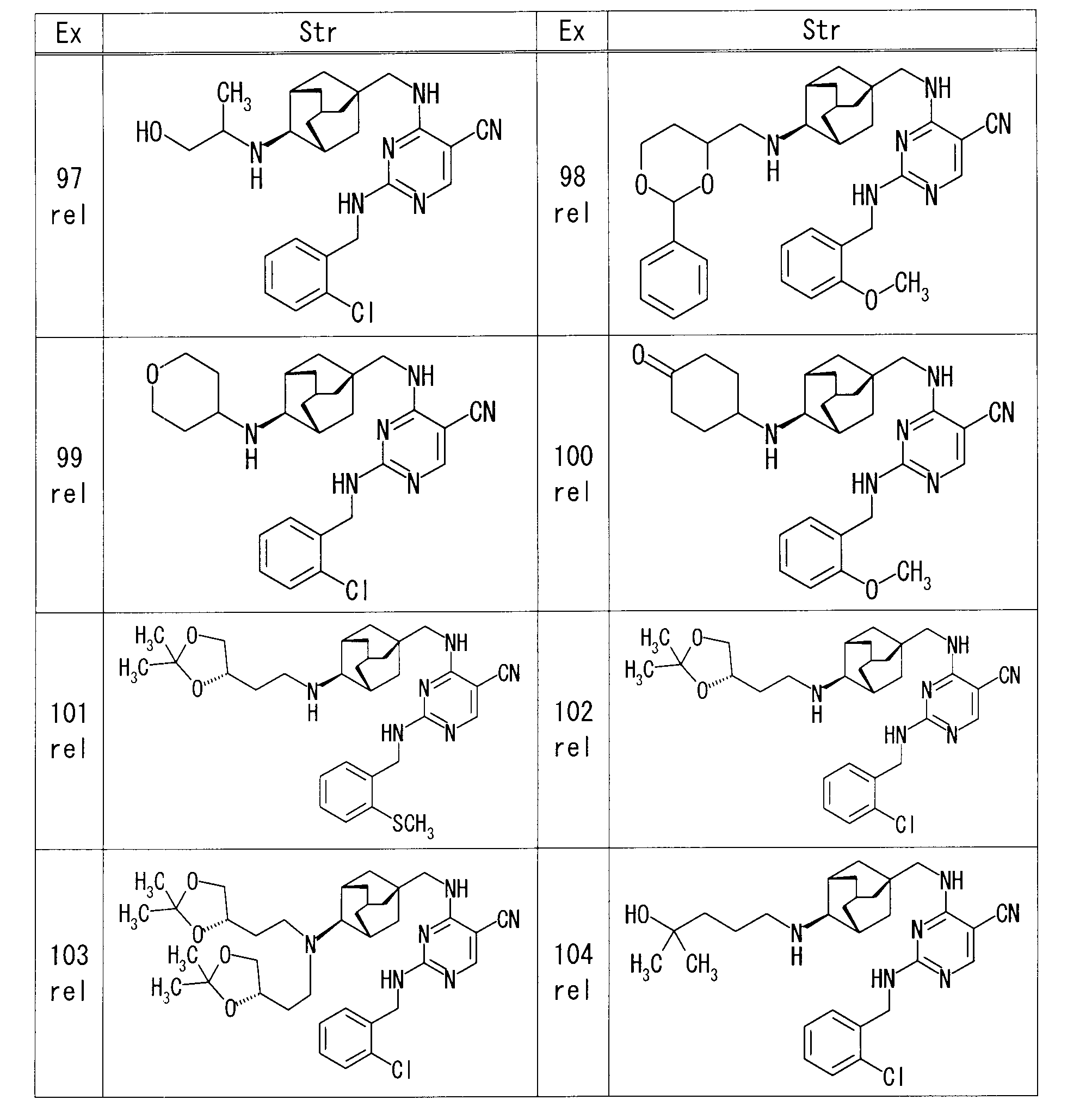





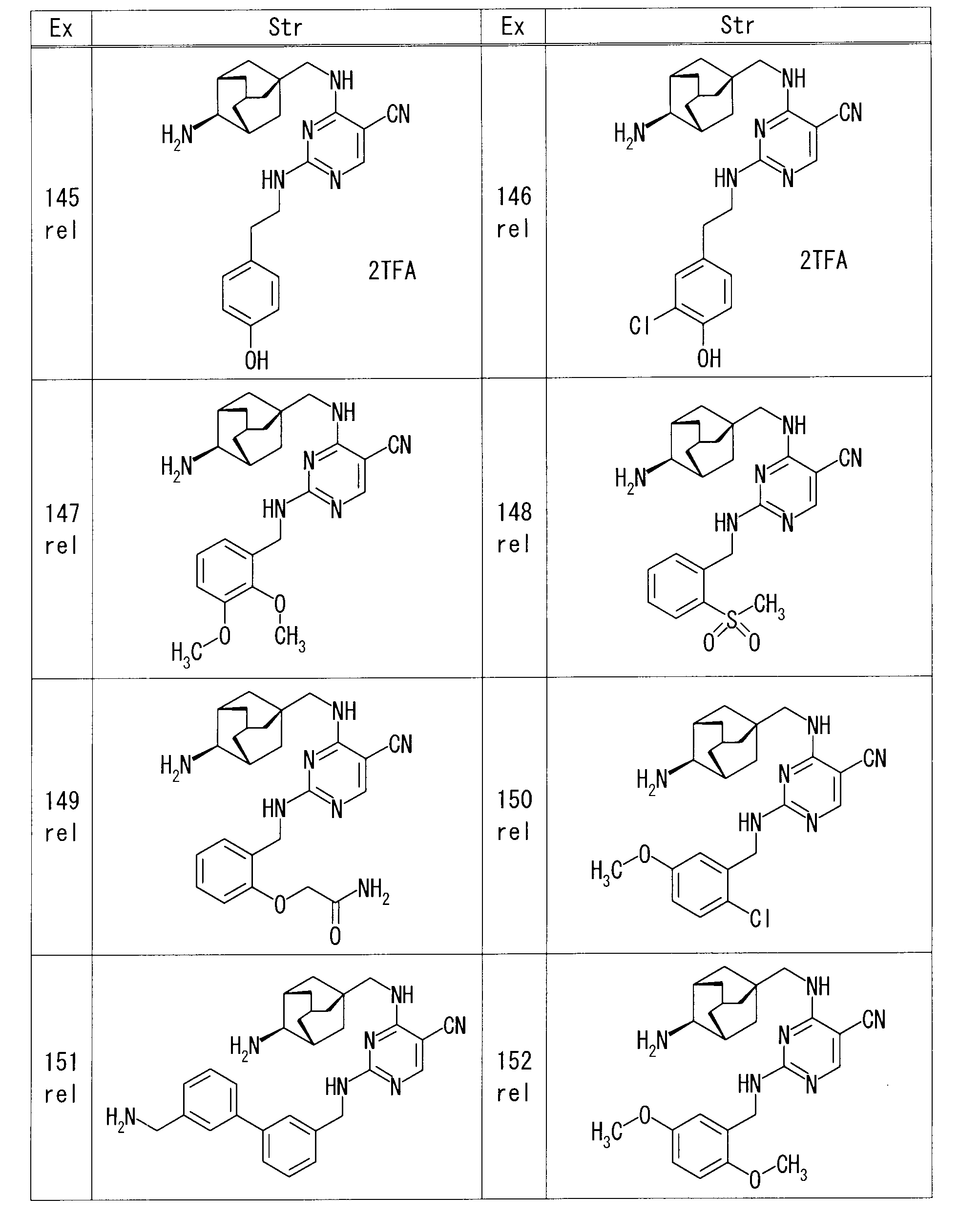









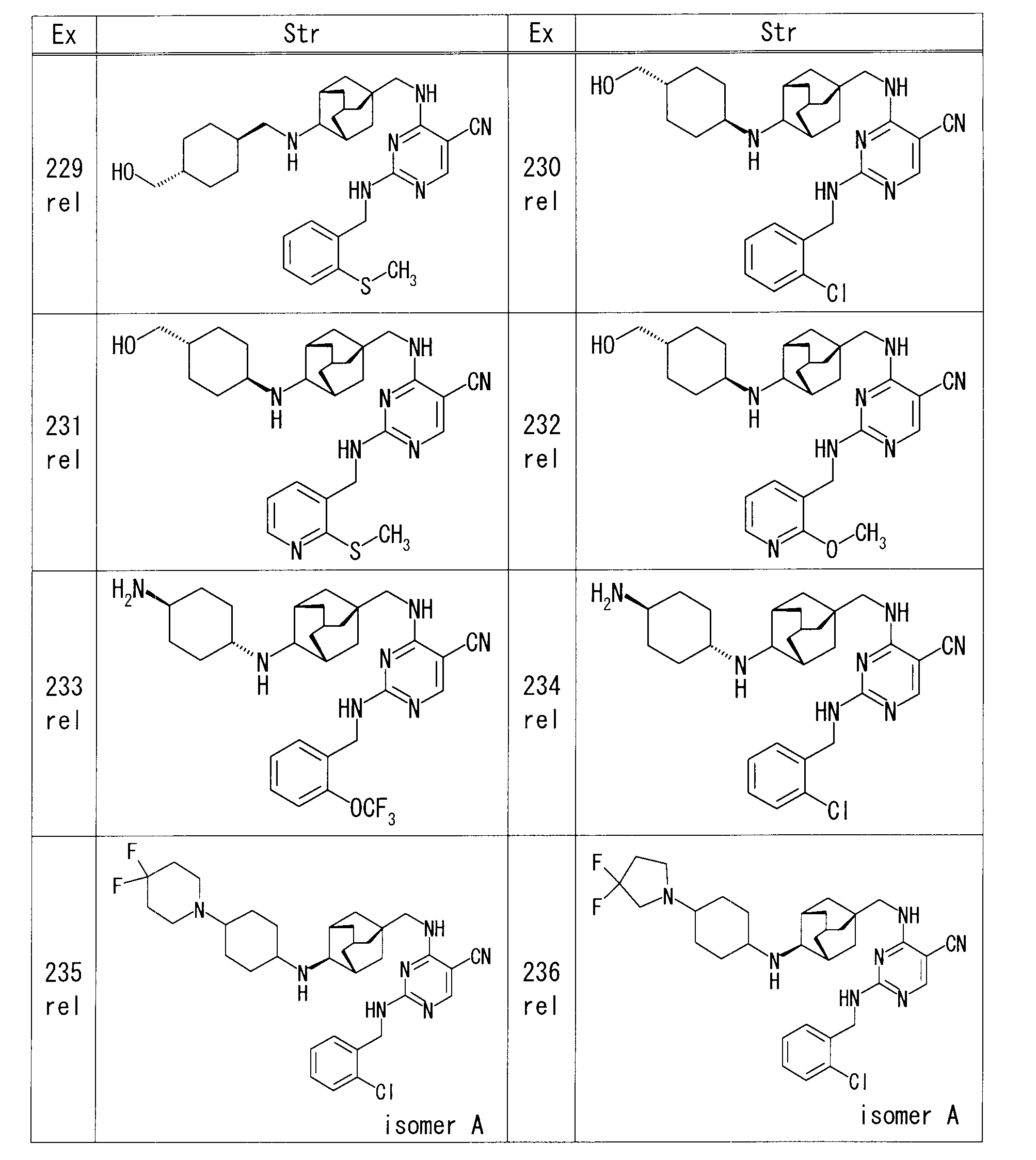
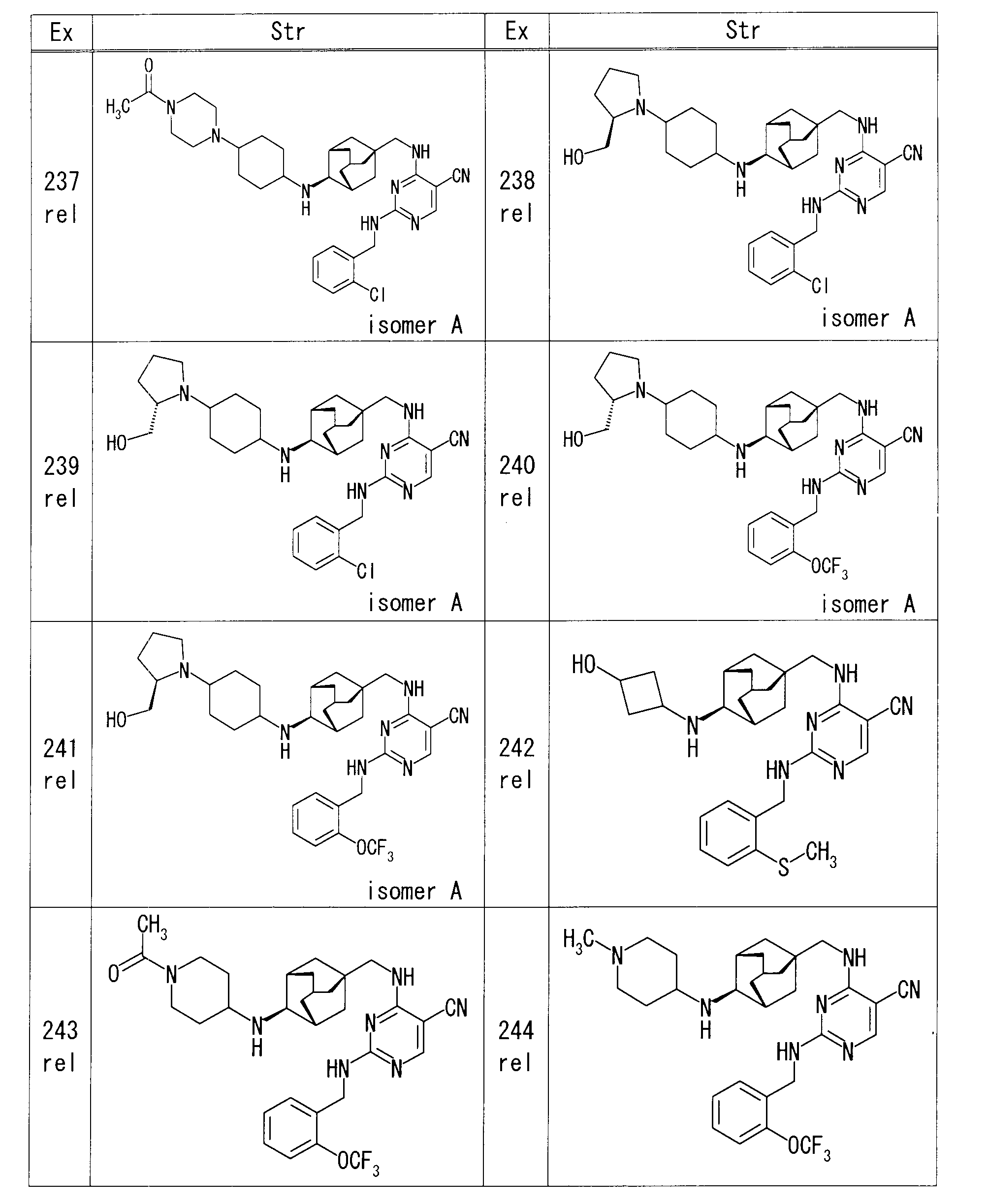
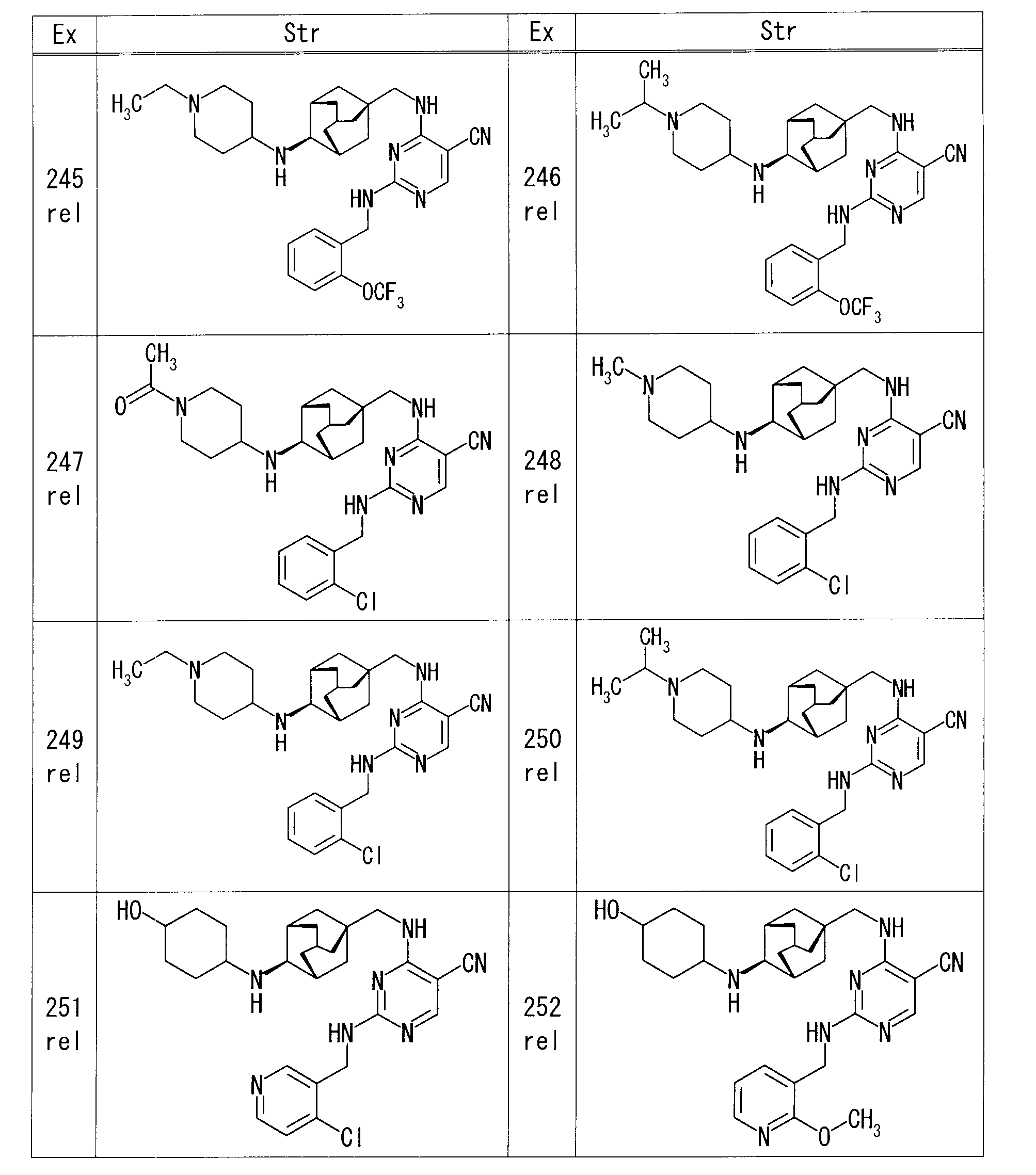



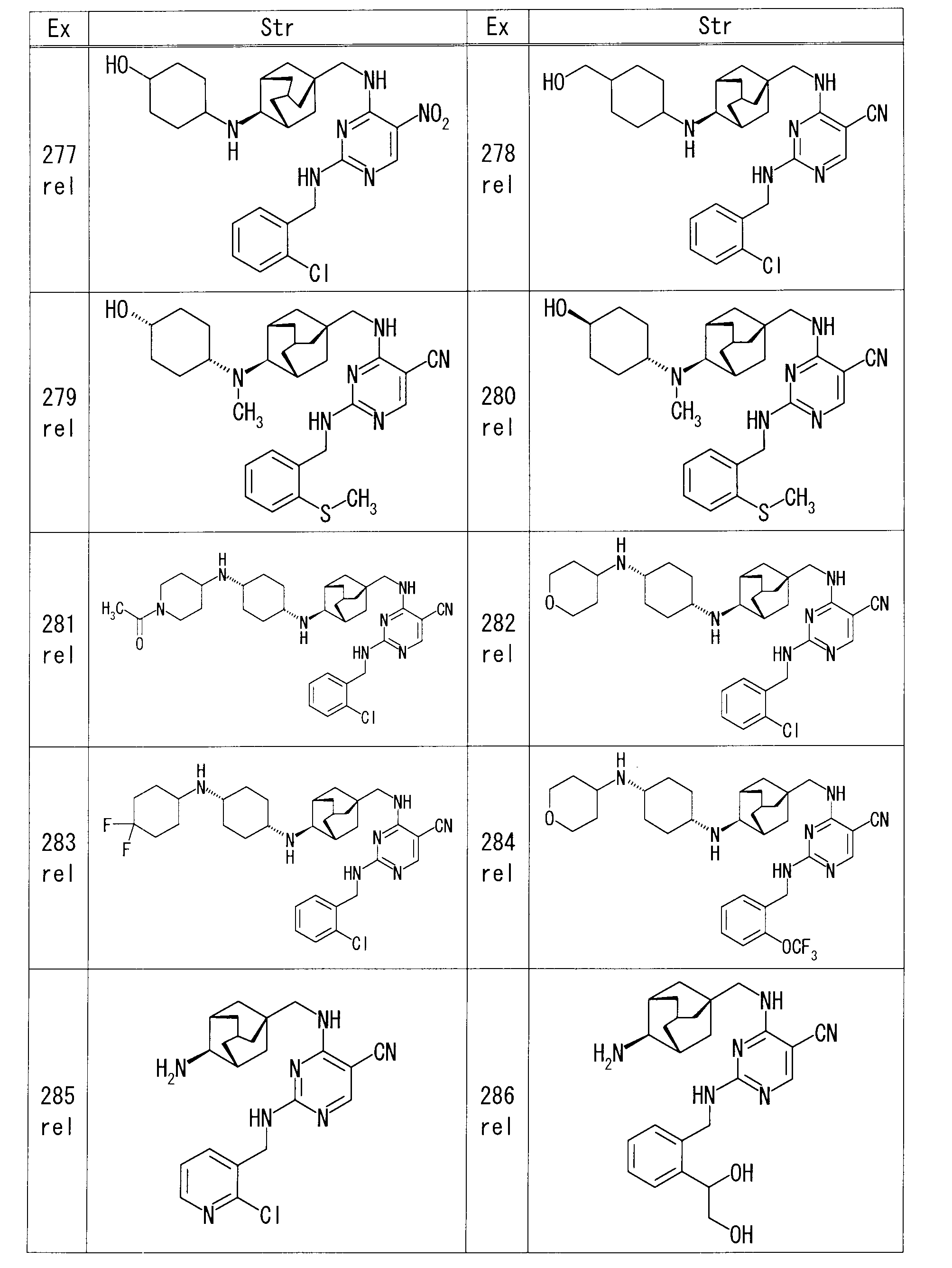


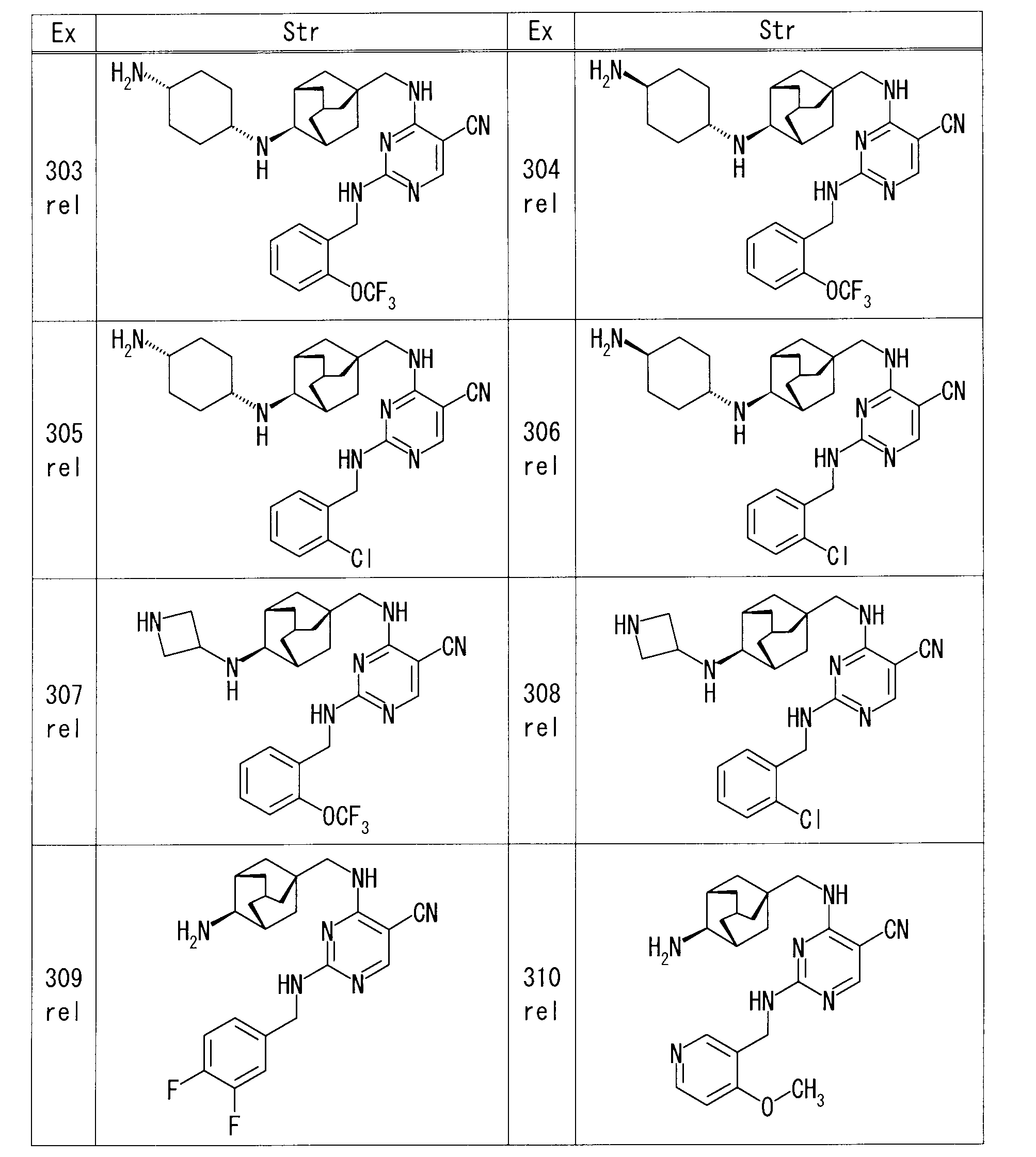


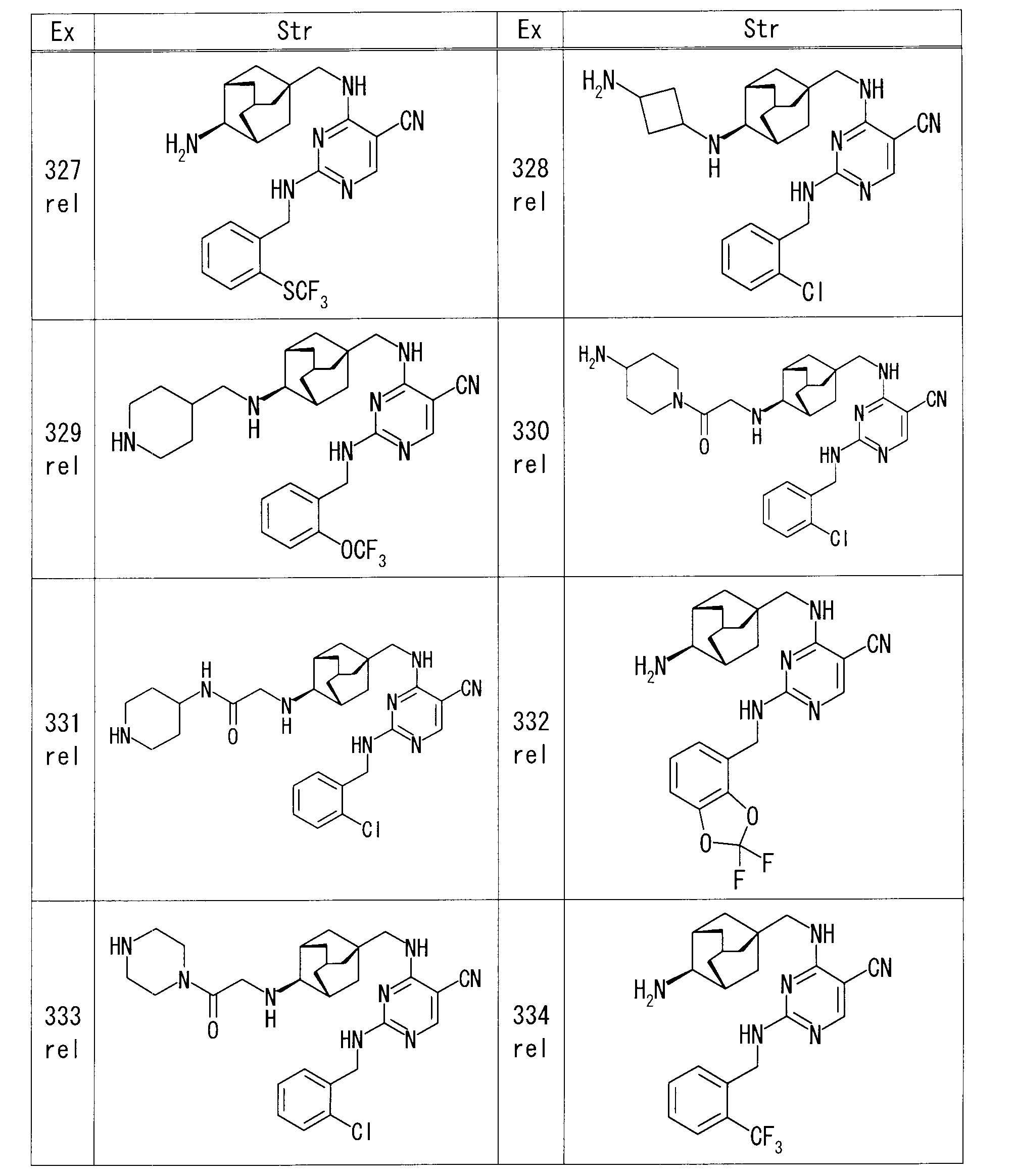



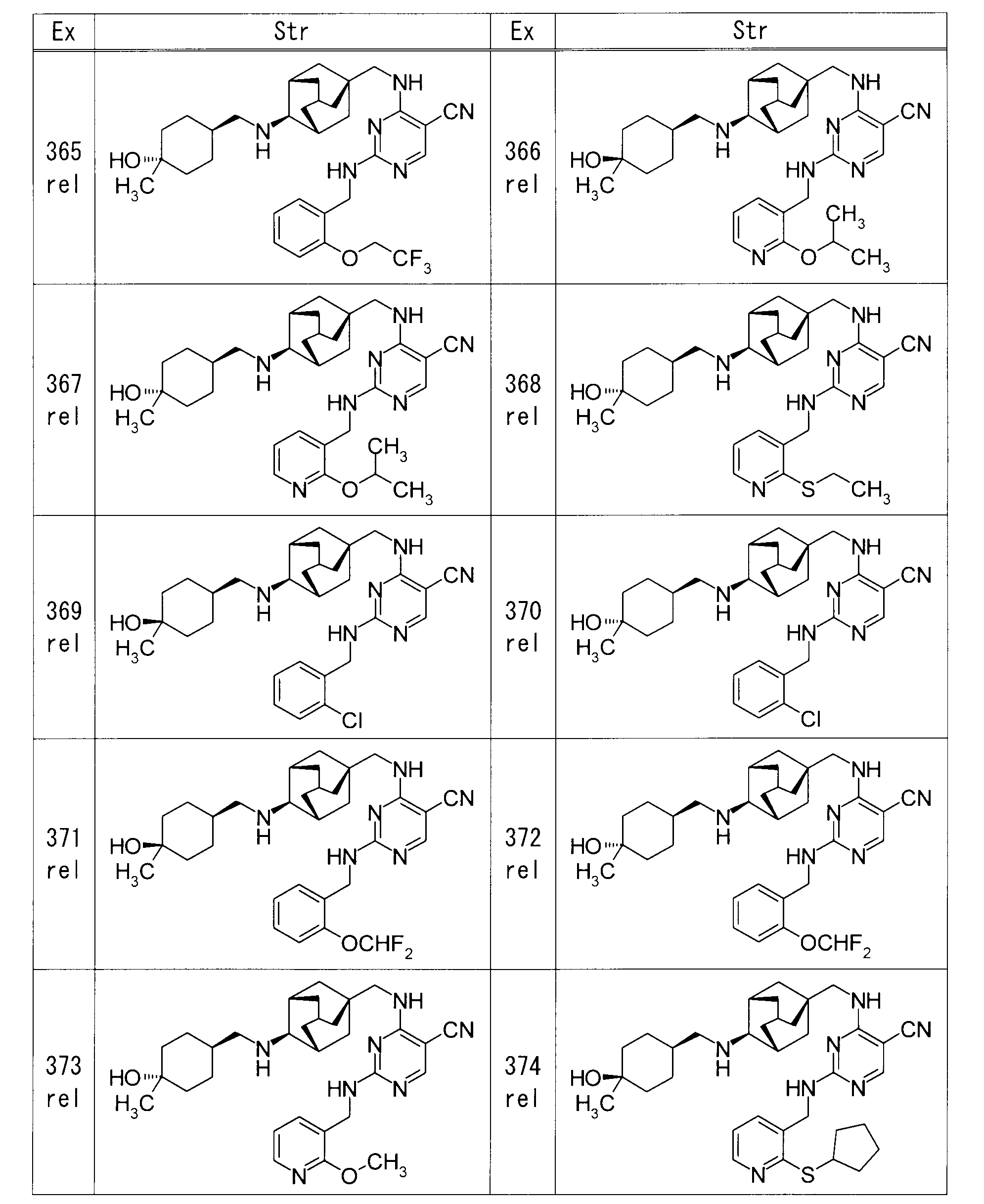




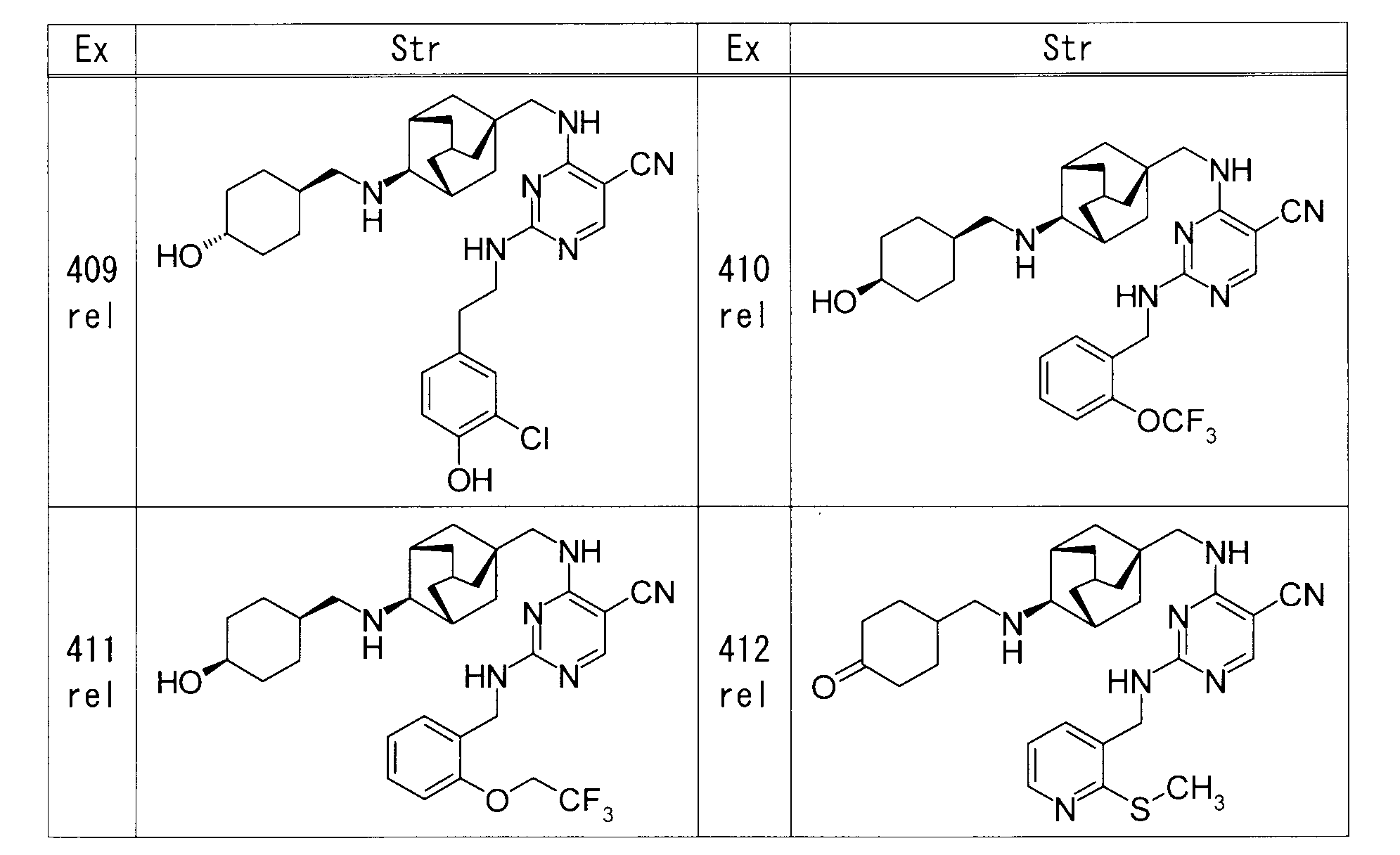

























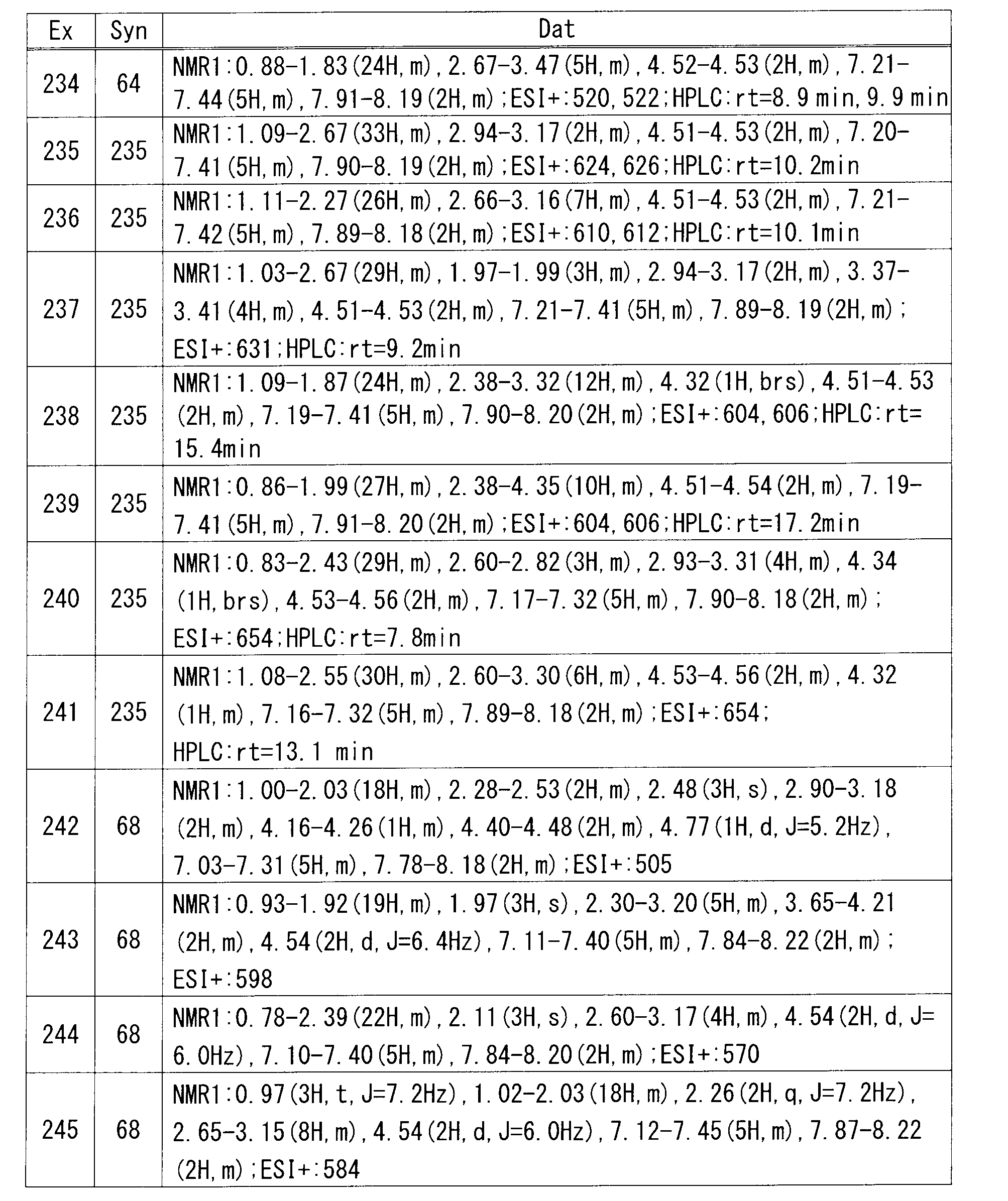













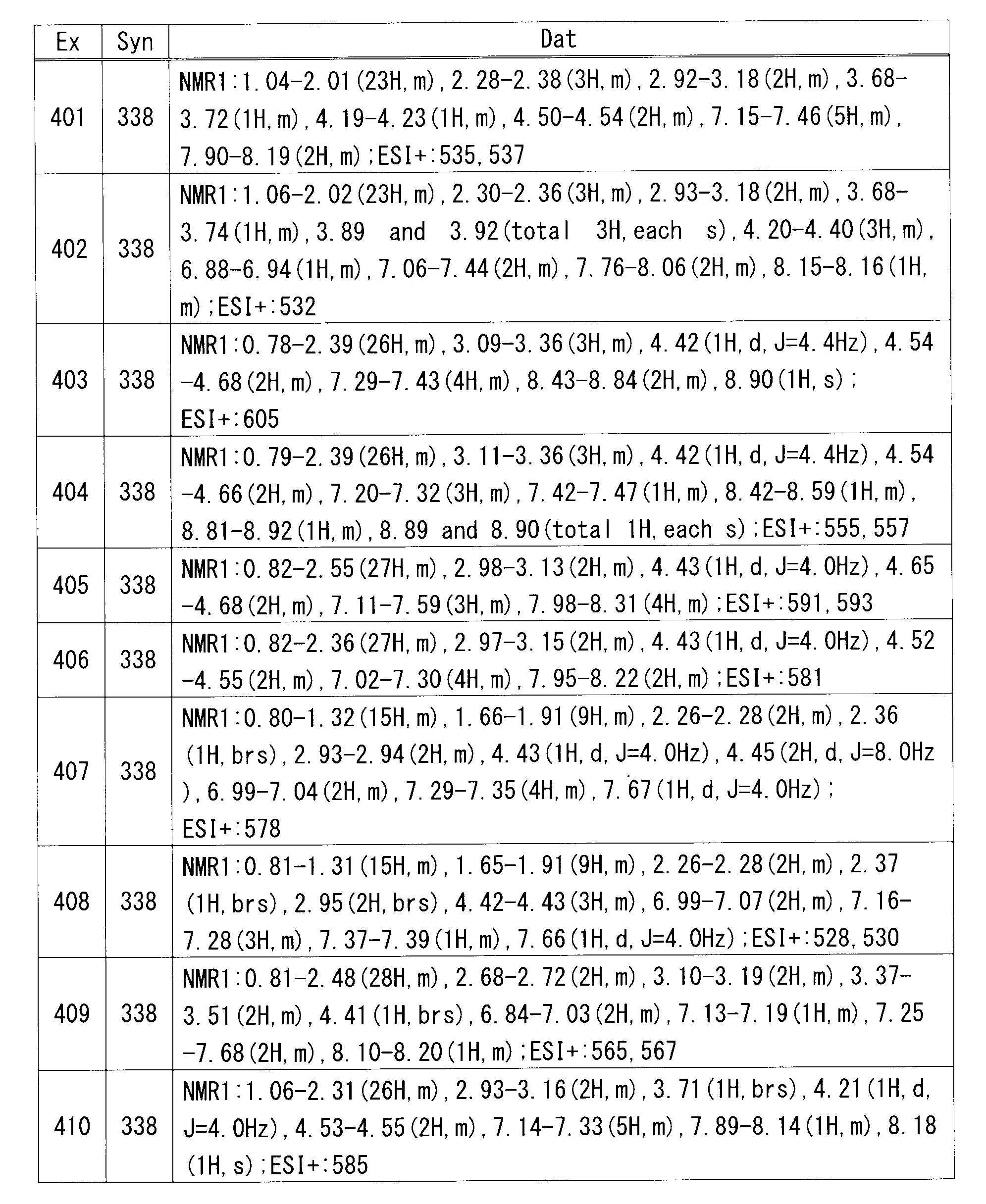
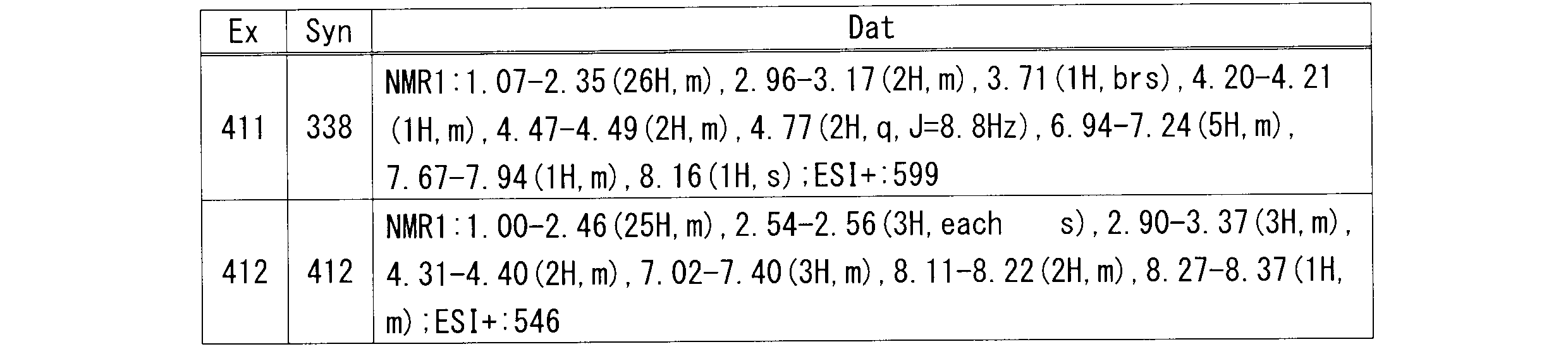





Claims (10)
- 式(I)の化合物又はその製薬学的に許容される塩。

R1は、

R4は、-OH、置換されていてもよいアミン又は-CH2NH2を示し;
n1は、0又は1を示し;
R5は、-OH、(-OH又は-NH2で置換されていてもよいC1-6アルキル)又は-CNを示し;
R6は、-H又はアリールで置換されていてもよいC1-6アルキルを示し;
pは、0又は1を示し;
qは、1、2、3又は4を示し;
R13は、-H又はC1-6アルキルを示し;
R2は、-CN、-CF3、-NO2又はハロゲンを示し;
Aは、単結合又はC1-6アルキレンを示し;
R3は、
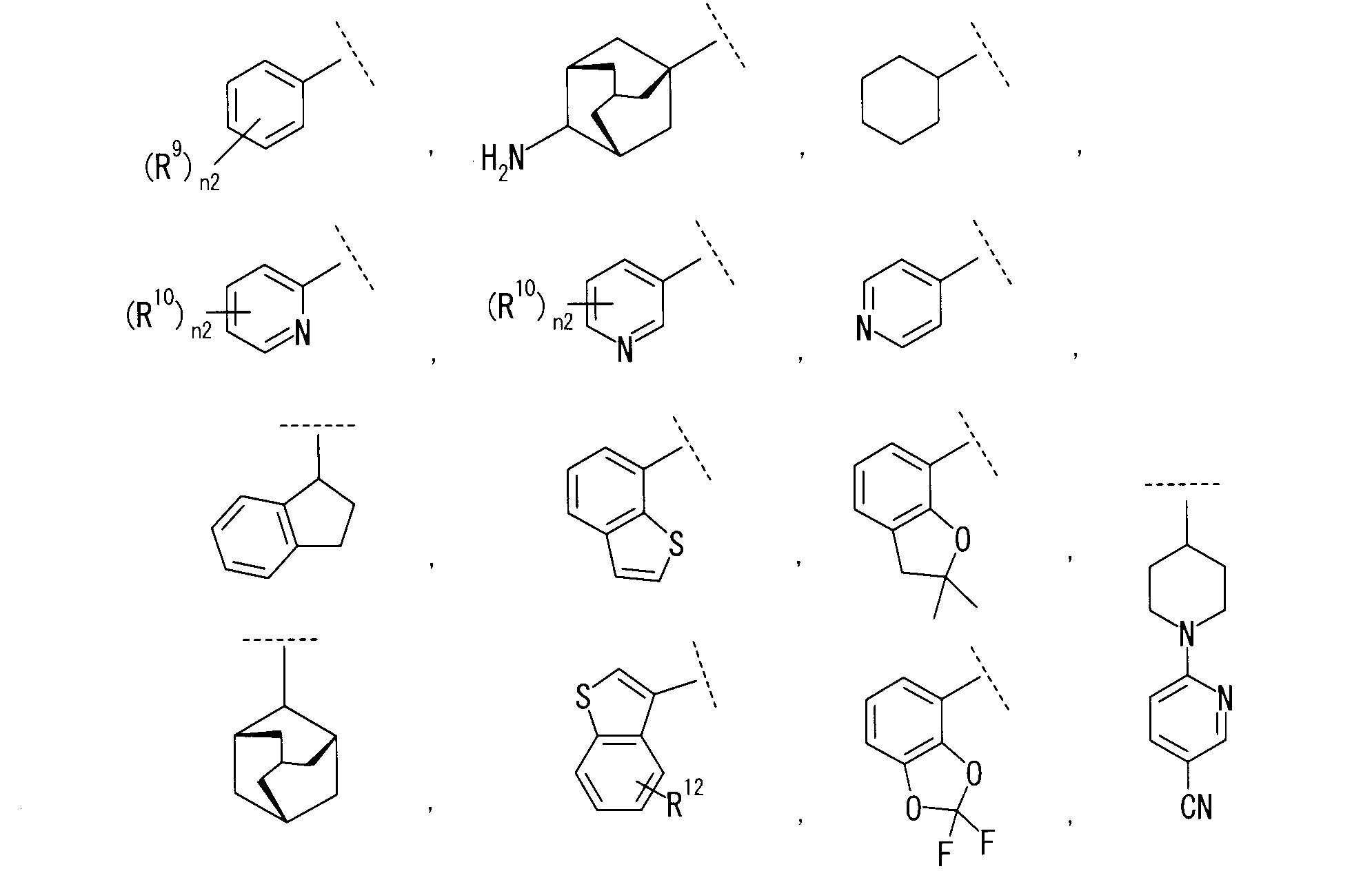
R9は、同一又は互いに異なって、ハロゲン、置換されていてもよいC1-6アルキル、-OH、-CN、シクロアルキル、-Q-(置換されていてもよいC1-6アルキル)又は置換されていてもよいアリールを示し;
Qは、-O-、-S-、-SO-、-SO2-又は-NHSO2-を示し;
n2は、0、1、2又は3を示し;
R10は、ハロゲン、C1-6アルキル、-CN、-O-(C1-6アルキル)、-S-(C1-6アルキル)、-SO-(C1-6アルキル)、-SO2-(C1-6アルキル)、-S-(シクロアルキル)又は-OCF3を示し;
R12は、-H又はハロゲンを示す。) - R4が-OH、-NR7R8又は-CH2NH2であり;
R7、R8が、互いに同一または異なって、
(a)-H;
(b)C1-6アルキル。ここで該C1-6アルキルは、以下の1)~12)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)保護された-OH
3)ハロゲン
4)-COOH
5)-CONH2
6)オキソ
7)アリール
8)ヘテロアリール
9)-OH、保護された-OH、(-OHで置換されていてもよいC1-6アルキル)、ハロゲン、-CN、-NR14R15、-CONR14R15、-SO2NR14R15、(-OHで置換されていてもよいC1-6アルキル)-O-、及びオキソからなる群から選択される1つ以上の基で置換されていてもよいシクロアルキル
10)-OH又は(-OH、-OCH3、-CN又はハロゲンで置換されていてもよいC1-6アルキル)で置換されていてもよいヘテロシクロアルキル
11)(-OH又は-NH2で置換されていてもよいヘテロシクロアルキル)-CO-、及び、
12)(ヘテロシクロアルキル)-NH-CO-;
(c)シクロアルキル。ここで該シクロアルキルは、以下の1)~6)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)-NHR11
3)ハロゲン
4)オキソ
5)-OHで置換されていてもよいC1-6アルキル、及び、
6)(ハロゲン、-OH、-CH2OH又は-COCH3)で置換されていてもよいヘテロシクロアルキル;
(d)ヘテロシクロアルキル。ここで、該ヘテロシクロアルキルは以下の1)~11)からなる群から選択される1つ以上の基で置換されてもよい。
1)(-OH、-OCH3、-CN、ハロゲン又は-CONH2)で置換されていてもよいC1-6アルキル
2)シクロアルキル
3)アリール
4)ヘテロシクロアルキル
5)ヘテロシクロアルキル-CO-
6)-COCH3
7)-CONH2
8)-COCH2OH
9)-COOCH2CH3
10)-SO2CH3
11)オキソ、及び
12)ハロゲン;
(e)アリール;
(f)ニコチノイル;
(g)-SO2CH3;
又は
(h)R7とR8がそれらが結合する窒素原子と一体となって、(-OH、-NH2、-COOH、-COCH3、-CONH2および-CH2OH)からなる群から選択される1つ以上の基で置換されていてもよい含窒素へテロシクロアルキルであり;
R11が、-H、(ハロゲン又は-OH)で置換されていてもよいC1-6アルキル、ハロゲンで置換されていてもよいシクロアルキル、-COCH3で置換されていてもよいヘテロシクロアルキル又は-COCH3であり;
R14、R15が、互いに同一又は異なって、-H、C1-6アルキル又はヘテロシクロアルキルである請求項1に記載の化合物又はその製薬学的に許容される塩。 - R1が

R3が、

- R4が-NR7R8であり;
R7、R8が、互いに同一または異なって、
(b)C1-6アルキル。ここで該C1-6アルキルは、以下の1)~12)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)メチル基で保護された-OH又は互いに近接する二つのOH基を有する場合はジメチルメチレン基又はベンジリデン基で保護された-OH
3)-F
4)-COOH
5)-CONH2
6)オキソ
7)フェニル
8)ピリジル
9)-OH及び(-OHで置換されていてもよいC1-6アルキル)からなる群から選択される1つ以上の基で置換されていてもよいシクロヘキシル
10)-OH又は(-OH、-OCH3、-CN又は-Fで置換されていてもよいC1-6アルキル)で置換されていてもよい(ピペリジニル又はピロリジニル)
11)(ピペラジニル)-CO-又は(-OH又は-NH2で置換されていてもよいピペリジニル)-CO-、及び、
12)(ピペリジニル)-NH-CO-;
又は
(c)シクロアルキル。ここで該シクロアルキルは、以下の1)~6)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)-NHR11
3)-F
4)オキソ
5)-OHで置換されていてもよいC1-6アルキル、及び、
6)(ハロゲン、-OH、-CH2OH又は-COCH3)で置換されていてもよい(アゼチジニル、ピロリジニル、ピペリジニル、ピペラジニル又はモルホリニル)であり;
R11が、-Hであり;
n1が1であり;
R2が、-CN、-CF3、-NO2又は-Fであり;
Aが、C1-6アルキレンであり;
R9が、
(i)-F、-Cl又は-Br
(j)-OH又はハロゲンで置換されていてもよいC1-6アルキル、
(k)-OH、
(l)-CN、
(m)シクロプロピル、
(n)-Q-(ハロゲン、-OH、-OCH3、-CN又は-CONH2で置換されていてもよいC1-6アルキル)
又は
(o)-CH2NH2で置換されていてもよいフェニルであり;
n2が1である請求項3に記載の化合物又はその製薬学的に許容される塩。 - R7、R8が、互いに同一または異なって、
(b)C1-6アルキル。ここで該C1-6アルキルは、以下の基で置換されていてもよい。
9)-OH、-CH3、-CH2OHからなる群から選択される1つ以上の基で置換されたシクロヘキシル、及び、
10)-OH又は(-OH、-OCH3、-CN又は-Fで置換されていてもよいC1-6アルキル)で置換されていてもよいピペリジニル;
又は
(c)シクロアルキル。ここで該シクロアルキルは、以下の1)、2)、5)からなる群から選択される1つ以上の基で置換されていてもよい。
1)-OH
2)-NHR11、及び、
5)-OHで置換されていてもよいC1-6アルキルであり;
R11が、-Hであり;
R2が、-CNであり;
Aが、メチレンであり;
R9が、
(i)-F、-Cl又は-Br
(j)-OH又は-Fで置換されていてもよいC1-6アルキル
(k)-OH、
(l)-CN、
(m)シクロプロピル
(n)-Q-(ハロゲン、-OH、-OCH3、-CN又は-CONH2で置換されていてもよいC1-6アルキル)
又は
(o)-CH2NH2で置換されていてもよいフェニルであり;
R10が、-Cl、-CH3、-OCH3、-OCH2CH3、-OCH(CH3)2、-SCH3、-SCH2CH3、-SCH(CH3)2、-SOCH3、-SO2CH3、-S-(シクロペンタン)又は-OCF3である請求項4に記載の化合物又はその製薬学的に許容される塩。 - 請求項1に記載の化合物又はその製薬学的に許容される塩、及び製薬学的に許容される賦形剤を含有する医薬組成物。
- 請求項1に記載の化合物又はその製薬学的に許容される塩を含有する、PKCθ阻害剤。
- 請求項1に記載の化合物又はその製薬学的に許容される塩を含有する、移植における急性拒絶反応の抑制用医薬組成物。
- 移植における急性拒絶反応抑制剤の製造のための請求項1に記載の化合物又はその製薬学的に許容される塩の使用。
- 請求項1に記載の化合物又はその塩の有効量を患者に投与することからなる移植における急性拒絶反応の抑制方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2735730A CA2735730A1 (en) | 2008-09-01 | 2009-08-31 | 2,4-diaminopyrimidine compound |
| EP09810074A EP2325175A4 (en) | 2008-09-01 | 2009-08-31 | 2,4-DIAMINOPYRIMIDINVERBINDUNG |
| BRPI0918472A BRPI0918472A2 (pt) | 2008-09-01 | 2009-08-31 | composto 2,4-diaminopirimidina |
| MX2011002306A MX2011002306A (es) | 2008-09-01 | 2009-08-31 | Compuesto de 2,4-diaminopirimidina. |
| JP2010526805A JP5644499B2 (ja) | 2008-09-01 | 2009-08-31 | 2,4−ジアミノピリミジン化合物 |
| US13/061,311 US20110159019A1 (en) | 2008-09-01 | 2009-08-31 | 2,4-diaminopyrimidine compound |
| CN2009801341763A CN102137848A (zh) | 2008-09-01 | 2009-08-31 | 2,4-二氨基嘧啶化合物 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-223323 | 2008-09-01 | ||
| JP2008223323 | 2008-09-01 | ||
| JP2009-121482 | 2009-05-20 | ||
| JP2009121482 | 2009-05-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010024430A1 true WO2010024430A1 (ja) | 2010-03-04 |
Family
ID=41721589
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/065149 Ceased WO2010024430A1 (ja) | 2008-09-01 | 2009-08-31 | 2,4-ジアミノピリミジン化合物 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20110159019A1 (ja) |
| EP (1) | EP2325175A4 (ja) |
| JP (1) | JP5644499B2 (ja) |
| KR (1) | KR20110049902A (ja) |
| CN (1) | CN102137848A (ja) |
| BR (1) | BRPI0918472A2 (ja) |
| CA (1) | CA2735730A1 (ja) |
| MX (1) | MX2011002306A (ja) |
| TW (1) | TW201014832A (ja) |
| WO (1) | WO2010024430A1 (ja) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010134533A1 (ja) * | 2009-05-20 | 2010-11-25 | アステラス製薬株式会社 | アミノシクロヘキシルアルキル基を有する2,4-ジアミノピリミジン化合物 |
| WO2011078101A1 (ja) * | 2009-12-22 | 2011-06-30 | 塩野義製薬株式会社 | アダマンタンアミン誘導体 |
| JP2012067033A (ja) * | 2010-09-22 | 2012-04-05 | Kuraray Co Ltd | アミノメチルピリジン誘導体の製造方法 |
| WO2012059932A1 (en) * | 2010-11-01 | 2012-05-10 | Aurigene Discovery Technologies Limited | 2, 4 -diaminopyrimidine derivatives as protein kinase inhibitors |
| US20130029987A1 (en) * | 2011-04-22 | 2013-01-31 | Bennett Brydon L | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9156798B2 (en) | 2013-12-20 | 2015-10-13 | Signal Pharmaceuticals, Llc | Substituted diaminopyrimidyl compounds, compositions thereof, and methods of treatment therewith |
| US9181221B2 (en) | 2010-05-21 | 2015-11-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9365524B2 (en) | 2014-01-30 | 2016-06-14 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9513297B2 (en) | 2014-12-16 | 2016-12-06 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-Jun N-terminal kinase in skin |
| WO2017072039A1 (de) | 2015-10-26 | 2017-05-04 | Bayer Cropscience Aktiengesellschaft | Kondensierte bicyclische heterocyclen-derivate als schädlingsbekämpfungsmittel |
| US9796685B2 (en) | 2014-12-16 | 2017-10-24 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-Methylcyclohexylamino)-pyrimidine-5-carboxamide |
| AU2016244228B2 (en) * | 2011-04-22 | 2019-02-21 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US10252981B2 (en) | 2015-07-24 | 2019-04-09 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US10689351B2 (en) | 2015-01-29 | 2020-06-23 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| WO2020173861A1 (de) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Kondensierte bicyclische heterocyclen-derivate als schädlingsbekämpfungsmittel |
| WO2020173860A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2024146945A1 (en) | 2023-01-07 | 2024-07-11 | Syngenta Crop Protection Ag | Novel carboxamide and sulfonamide pesticidal compounds |
| WO2025109114A1 (en) | 2023-11-24 | 2025-05-30 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
| WO2025149629A1 (en) | 2024-01-12 | 2025-07-17 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
| WO2025149637A1 (en) | 2024-01-12 | 2025-07-17 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
| WO2026008750A1 (en) | 2024-07-05 | 2026-01-08 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2577829T3 (es) | 2010-06-04 | 2016-07-19 | F. Hoffmann-La Roche Ag | Derivados de aminopirimidina como moduladores de la LRRK2 |
| EP2604591A4 (en) | 2010-08-09 | 2015-07-22 | Shionogi & Co | PROCESS FOR THE PREPARATION OF AMINOADAMANTYL CARBAMATE DERIVATIVES |
| NO2638031T3 (ja) | 2010-11-10 | 2018-03-10 | ||
| EP2760843B1 (en) * | 2011-09-26 | 2016-03-02 | Bristol-Myers Squibb Company | Selective nr2b antagonists |
| EP2760856B1 (en) | 2011-09-30 | 2016-09-14 | Bristol-Myers Squibb Company | Selective nr2b antagonists |
| WO2025136129A1 (en) * | 2023-12-22 | 2025-06-26 | Captor Therapeutics S.A. | Targeted protein degradation using bifunctional compounds that bind ubiquitin ligase and target protein kinase c |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003032997A1 (de) | 2001-10-17 | 2003-04-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Pyrimidinderivate, arzneimittel enthaltend diese verbindungen, deren verwendung und verfahren zu ihrer herstellung |
| WO2004043936A1 (ja) | 2002-11-14 | 2004-05-27 | Kyowa Hakko Kogyo Co., Ltd. | Plk阻害剤 |
| WO2004048343A1 (en) * | 2002-11-28 | 2004-06-10 | Schering Aktiengesellschaft | Chk-, pdk- and akt-inhibitory pyrimidines, their production and use as pharmaceutical agents |
| WO2004054617A1 (ja) | 2002-12-13 | 2004-07-01 | Kyowa Hakko Kogyo Co., Ltd. | 中枢疾患の予防および/または治療剤 |
| WO2004067516A1 (en) | 2003-01-30 | 2004-08-12 | Boehringer Ingelheim Pharmaceuticals, Inc. | 2,4-diaminopyrimidine derivatives useful as inhibitors of pkc-theta |
| WO2005118544A2 (en) * | 2004-05-18 | 2005-12-15 | Rigel Pharmaceuticals, Inc. | Cycloalkyl substituted pyrimidinediamine compounds and their uses |
| WO2006014482A1 (en) | 2004-07-08 | 2006-02-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Pyrimidine derivatives useful as inhibitors of pkc-theta |
| WO2007076247A1 (en) | 2005-12-21 | 2007-07-05 | Boehringer Ingelheim International Gmbh | Pyrimidine derivatives useful as inhibitors of pkc-theta |
| WO2009012421A1 (en) * | 2007-07-17 | 2009-01-22 | Rigel Pharmaceuticals, Inc. | Cyclic amine substituted pyrimidinediamines as pkc inhibitors |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0016877D0 (en) * | 2000-07-11 | 2000-08-30 | Astrazeneca Ab | Chemical compounds |
| BR0209774A (pt) * | 2001-05-29 | 2004-06-01 | Schering Ag | Pirimidinas inibidoras de cdk, sua preparação e aplicação como medicamento |
| CN102317265B (zh) * | 2009-01-14 | 2015-03-25 | 里格尔药品股份有限公司 | 用于治疗炎性疾病的2,4-嘧啶二胺化合物 |
-
2009
- 2009-08-31 JP JP2010526805A patent/JP5644499B2/ja not_active Expired - Fee Related
- 2009-08-31 CA CA2735730A patent/CA2735730A1/en not_active Abandoned
- 2009-08-31 US US13/061,311 patent/US20110159019A1/en not_active Abandoned
- 2009-08-31 BR BRPI0918472A patent/BRPI0918472A2/pt not_active IP Right Cessation
- 2009-08-31 EP EP09810074A patent/EP2325175A4/en not_active Withdrawn
- 2009-08-31 CN CN2009801341763A patent/CN102137848A/zh active Pending
- 2009-08-31 MX MX2011002306A patent/MX2011002306A/es not_active Application Discontinuation
- 2009-08-31 WO PCT/JP2009/065149 patent/WO2010024430A1/ja not_active Ceased
- 2009-08-31 KR KR1020117007428A patent/KR20110049902A/ko not_active Withdrawn
- 2009-09-01 TW TW098129376A patent/TW201014832A/zh unknown
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003032997A1 (de) | 2001-10-17 | 2003-04-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Pyrimidinderivate, arzneimittel enthaltend diese verbindungen, deren verwendung und verfahren zu ihrer herstellung |
| WO2004043936A1 (ja) | 2002-11-14 | 2004-05-27 | Kyowa Hakko Kogyo Co., Ltd. | Plk阻害剤 |
| WO2004048343A1 (en) * | 2002-11-28 | 2004-06-10 | Schering Aktiengesellschaft | Chk-, pdk- and akt-inhibitory pyrimidines, their production and use as pharmaceutical agents |
| WO2004054617A1 (ja) | 2002-12-13 | 2004-07-01 | Kyowa Hakko Kogyo Co., Ltd. | 中枢疾患の予防および/または治療剤 |
| WO2004067516A1 (en) | 2003-01-30 | 2004-08-12 | Boehringer Ingelheim Pharmaceuticals, Inc. | 2,4-diaminopyrimidine derivatives useful as inhibitors of pkc-theta |
| WO2005118544A2 (en) * | 2004-05-18 | 2005-12-15 | Rigel Pharmaceuticals, Inc. | Cycloalkyl substituted pyrimidinediamine compounds and their uses |
| WO2006014482A1 (en) | 2004-07-08 | 2006-02-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Pyrimidine derivatives useful as inhibitors of pkc-theta |
| WO2007076247A1 (en) | 2005-12-21 | 2007-07-05 | Boehringer Ingelheim International Gmbh | Pyrimidine derivatives useful as inhibitors of pkc-theta |
| WO2009012421A1 (en) * | 2007-07-17 | 2009-01-22 | Rigel Pharmaceuticals, Inc. | Cyclic amine substituted pyrimidinediamines as pkc inhibitors |
Non-Patent Citations (13)
| Title |
|---|
| "Bunshi Sekkei", vol. 7, 1990, HIROKAWA SHOTEN, article "lyakuhin no Kaihatsu", pages: 163 - 198 |
| "Comprehensive Organic Functional Group Transformations II", vol. 2, 2005, ELSEVIER PERGAMON |
| "Comprehensive Organic Transformations", 1999, VCH PUBLISHERS, INC. |
| "Green's Protective Groups in Organic Synthesis", 2006 |
| "Jikken Kagaku Koza", vol. 14, 2005, MARUZEN |
| "Jikken Kagaku Koza", vol. 16, 2005, MARUZEN |
| "Organic Functional Group Preparations", vol. 1, 1991, ACADEMIC PRESS INC. |
| "Oxidation and Reduction in Organic Synthesis", 2000, OXFORD SCIENCE PUBLICATIONS |
| "Reductions in Organic Chemistry", 1996, ACS |
| CYWIN, C.L. ET AL.: "Discovery of potent and selective PKC-0 inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 17, no. L, 2007, pages 225 - 230, XP002592849 * |
| PROG. MED., vol. 5, 1985, pages 2157 - 2161 |
| See also references of EP2325175A4 * |
| VERMA, S. ET AL.: "Substituted aminobenzimidazole pyrimidines as cyclin-dependent kinase inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 15, no. 8, 2005, pages 1973 - 1977, XP025314402 * |
Cited By (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010134533A1 (ja) * | 2009-05-20 | 2010-11-25 | アステラス製薬株式会社 | アミノシクロヘキシルアルキル基を有する2,4-ジアミノピリミジン化合物 |
| JP5686413B2 (ja) * | 2009-12-22 | 2015-03-18 | 塩野義製薬株式会社 | アダマンタンアミン誘導体 |
| WO2011078101A1 (ja) * | 2009-12-22 | 2011-06-30 | 塩野義製薬株式会社 | アダマンタンアミン誘導体 |
| EP2518051A4 (en) * | 2009-12-22 | 2013-10-16 | Shionogi & Co | ADAMANTANAMINE DERIVATIVE |
| US9738644B2 (en) | 2010-05-21 | 2017-08-22 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9181221B2 (en) | 2010-05-21 | 2015-11-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| JP2012067033A (ja) * | 2010-09-22 | 2012-04-05 | Kuraray Co Ltd | アミノメチルピリジン誘導体の製造方法 |
| WO2012059932A1 (en) * | 2010-11-01 | 2012-05-10 | Aurigene Discovery Technologies Limited | 2, 4 -diaminopyrimidine derivatives as protein kinase inhibitors |
| CN105001165A (zh) * | 2011-04-22 | 2015-10-28 | 西格诺药品有限公司 | 取代的二氨基嘧啶其组合物,和用其治疗的方法 |
| JP2023179797A (ja) * | 2011-04-22 | 2023-12-19 | シグナル ファーマシューティカルズ,エルエルシー | 置換されたジアミノカルボキサミドおよびジアミノカルボニトリルピリミジン、その組成物、ならびに、それを用いた治療方法 |
| US9139534B2 (en) * | 2011-04-22 | 2015-09-22 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| TWI681952B (zh) * | 2011-04-22 | 2020-01-11 | 美商標誌製藥公司 | 經取代之二胺基甲醯胺及二胺基甲腈嘧啶、其組合物、及以該等治療之方法 |
| JP2014514322A (ja) * | 2011-04-22 | 2014-06-19 | シグナル ファーマシューティカルズ,エルエルシー | 置換されたジアミノカルボキサミドおよびジアミノカルボニトリルピリミジン、その組成物、ならびに、それを用いた治療方法 |
| CN103492370A (zh) * | 2011-04-22 | 2014-01-01 | 西格诺药品有限公司 | 取代的二氨基甲酰胺和二氨基甲腈嘧啶,其组合物,和用其治疗的方法 |
| US12129237B2 (en) | 2011-04-22 | 2024-10-29 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| CN103492370B (zh) * | 2011-04-22 | 2016-10-26 | 西格诺药品有限公司 | 取代的二氨基甲酰胺和二氨基甲腈嘧啶,其组合物,和用其治疗的方法 |
| CN106946795B (zh) * | 2011-04-22 | 2020-06-02 | 西格诺药品有限公司 | 取代的二氨基甲酰胺和二氨基甲腈嘧啶,其组合物,和用其治疗的方法 |
| US10266500B2 (en) | 2011-04-22 | 2019-04-23 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| AU2016244228B2 (en) * | 2011-04-22 | 2019-02-21 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US10919865B2 (en) | 2011-04-22 | 2021-02-16 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| JP2021138755A (ja) * | 2011-04-22 | 2021-09-16 | シグナル ファーマシューティカルズ,エルエルシー | 置換されたジアミノカルボキサミドおよびジアミノカルボニトリルピリミジン、その組成物、ならびに、それを用いた治療方法 |
| JP7374955B2 (ja) | 2011-04-22 | 2023-11-07 | シグナル ファーマシューティカルズ,エルエルシー | 置換されたジアミノカルボキサミドおよびジアミノカルボニトリルピリミジン、その組成物、ならびに、それを用いた治療方法 |
| US9701643B2 (en) | 2011-04-22 | 2017-07-11 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| CN106946795A (zh) * | 2011-04-22 | 2017-07-14 | 西格诺药品有限公司 | 取代的二氨基甲酰胺和二氨基甲腈嘧啶,其组合物,和用其治疗的方法 |
| US20130029987A1 (en) * | 2011-04-22 | 2013-01-31 | Bennett Brydon L | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US10040770B2 (en) | 2011-04-22 | 2018-08-07 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US11325890B2 (en) | 2011-04-22 | 2022-05-10 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9546180B2 (en) | 2011-08-29 | 2017-01-17 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9783505B2 (en) | 2013-12-20 | 2017-10-10 | Signal Pharmaceuticals, Llc | Substituted diaminopyrimidyl compounds, compositions thereof, and methods of treatment therewith |
| US9556126B2 (en) | 2013-12-20 | 2017-01-31 | Signal Pharmaceuticals, Llc | Substituted diaminopyrimidyl compounds, compositions thereof, and methods of treatment therewith |
| JP2017500325A (ja) * | 2013-12-20 | 2017-01-05 | シグナル ファーマシューティカルズ,エルエルシー | 置換されたジアミノピリミジル化合物、それらの組成物、及びそれらによる治療方法 |
| US9156798B2 (en) | 2013-12-20 | 2015-10-13 | Signal Pharmaceuticals, Llc | Substituted diaminopyrimidyl compounds, compositions thereof, and methods of treatment therewith |
| US9814713B2 (en) | 2014-01-30 | 2017-11-14 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US9365524B2 (en) | 2014-01-30 | 2016-06-14 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US11241430B2 (en) | 2014-01-30 | 2022-02-08 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US10517873B2 (en) | 2014-01-30 | 2019-12-31 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US10226461B2 (en) | 2014-01-30 | 2019-03-12 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US10197579B2 (en) | 2014-12-16 | 2019-02-05 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-Jun N-terminal kinase in skin |
| US10590089B2 (en) | 2014-12-16 | 2020-03-17 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US9513297B2 (en) | 2014-12-16 | 2016-12-06 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-Jun N-terminal kinase in skin |
| US10131639B2 (en) | 2014-12-16 | 2018-11-20 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US9796685B2 (en) | 2014-12-16 | 2017-10-24 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-Methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10689351B2 (en) | 2015-01-29 | 2020-06-23 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10975039B2 (en) | 2015-01-29 | 2021-04-13 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US11492332B2 (en) | 2015-01-29 | 2022-11-08 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10774033B2 (en) | 2015-07-24 | 2020-09-15 | Celgene Corporation | Methods of synthesis of (1R, 2R, 5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US11192847B2 (en) | 2015-07-24 | 2021-12-07 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US10252981B2 (en) | 2015-07-24 | 2019-04-09 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US11780801B2 (en) | 2015-07-24 | 2023-10-10 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methyl-cyclohexanol hydrochloride and intermediates useful therein |
| WO2017072039A1 (de) | 2015-10-26 | 2017-05-04 | Bayer Cropscience Aktiengesellschaft | Kondensierte bicyclische heterocyclen-derivate als schädlingsbekämpfungsmittel |
| WO2020173860A1 (en) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
| WO2020173861A1 (de) | 2019-02-26 | 2020-09-03 | Bayer Aktiengesellschaft | Kondensierte bicyclische heterocyclen-derivate als schädlingsbekämpfungsmittel |
| WO2024146945A1 (en) | 2023-01-07 | 2024-07-11 | Syngenta Crop Protection Ag | Novel carboxamide and sulfonamide pesticidal compounds |
| WO2025109114A1 (en) | 2023-11-24 | 2025-05-30 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
| WO2025149629A1 (en) | 2024-01-12 | 2025-07-17 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
| WO2025149637A1 (en) | 2024-01-12 | 2025-07-17 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
| WO2026008750A1 (en) | 2024-07-05 | 2026-01-08 | Syngenta Crop Protection Ag | Novel carboxamide compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2735730A1 (en) | 2010-03-04 |
| EP2325175A1 (en) | 2011-05-25 |
| KR20110049902A (ko) | 2011-05-12 |
| JP5644499B2 (ja) | 2014-12-24 |
| CN102137848A (zh) | 2011-07-27 |
| US20110159019A1 (en) | 2011-06-30 |
| TW201014832A (en) | 2010-04-16 |
| MX2011002306A (es) | 2011-05-25 |
| EP2325175A4 (en) | 2012-03-21 |
| JPWO2010024430A1 (ja) | 2012-01-26 |
| BRPI0918472A2 (pt) | 2015-12-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5644499B2 (ja) | 2,4−ジアミノピリミジン化合物 | |
| US20130150364A1 (en) | Heterocyclic compound | |
| AU2014356069B2 (en) | Novel amino pyrimidine derivatives | |
| JPWO2010092962A1 (ja) | へテロ環誘導体 | |
| JP5532366B2 (ja) | ピラジンカルボキサミド化合物 | |
| JP6422986B2 (ja) | 化合物 | |
| JP2014005206A (ja) | アリールアミノヘテロ環カルボキサミド化合物 | |
| WO2010058846A1 (ja) | 4,6-ジアミノニコチンアミド化合物 | |
| CN110891954A (zh) | 富含亮氨酸的重复激酶2的抑制剂 | |
| EP3601296B1 (en) | 2-oxo-thiazole derivatives as a2a inhibitors and compounds for use in the treatment of cancers | |
| KR20120002581A (ko) | 피리미딘 화합물 | |
| CA2950564C (en) | 2-acylaminothiazole derivative or salt thereof | |
| TW201522330A (zh) | 2-醯胺噻唑衍生物或其鹽 | |
| JP2007055940A (ja) | ピラゾロピリミジン誘導体 | |
| JPWO2015056771A1 (ja) | 含硫黄二環式化合物 | |
| JPWO2007111227A1 (ja) | アシルアミノピペリジン化合物 | |
| WO2010134533A1 (ja) | アミノシクロヘキシルアルキル基を有する2,4-ジアミノピリミジン化合物 | |
| JP2011173853A (ja) | 置換アミノアダマンチルアルキル基を有する2,4−ジアミノピリミジン化合物 | |
| JP2011173854A (ja) | ビシクロ環置換アルキル基を有する2,4−ジアミノピリミジン化合物 | |
| WO2021187605A1 (ja) | 含窒素複素環αシアノカルボニル化合物 | |
| WO2003078441A1 (en) | Aminomethyl-substituted thiazolobenzimidazole derivative | |
| HK40019115B (en) | 2-oxo-thiazole derivatives as a2a inhibitors and compounds for use in the treatment of cancers | |
| HK40019115A (en) | 2-oxo-thiazole derivatives as a2a inhibitors and compounds for use in the treatment of cancers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980134176.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09810074 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010526805 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2735730 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1433/CHENP/2011 Country of ref document: IN Ref document number: 2009810074 Country of ref document: EP Ref document number: MX/A/2011/002306 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20117007428 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0918472 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110301 |