WO2010035747A1 - α-トリフルオロメチル-α,β-不飽和エステル類の製造方法 - Google Patents
α-トリフルオロメチル-α,β-不飽和エステル類の製造方法 Download PDFInfo
- Publication number
- WO2010035747A1 WO2010035747A1 PCT/JP2009/066527 JP2009066527W WO2010035747A1 WO 2010035747 A1 WO2010035747 A1 WO 2010035747A1 JP 2009066527 W JP2009066527 W JP 2009066527W WO 2010035747 A1 WO2010035747 A1 WO 2010035747A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- substituted
- trifluoromethyl
- alkyl group
- organic base
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(*)C(C(F)(F)F)(C(O*)=O)O Chemical compound *C(*)C(C(F)(F)F)(C(O*)=O)O 0.000 description 4
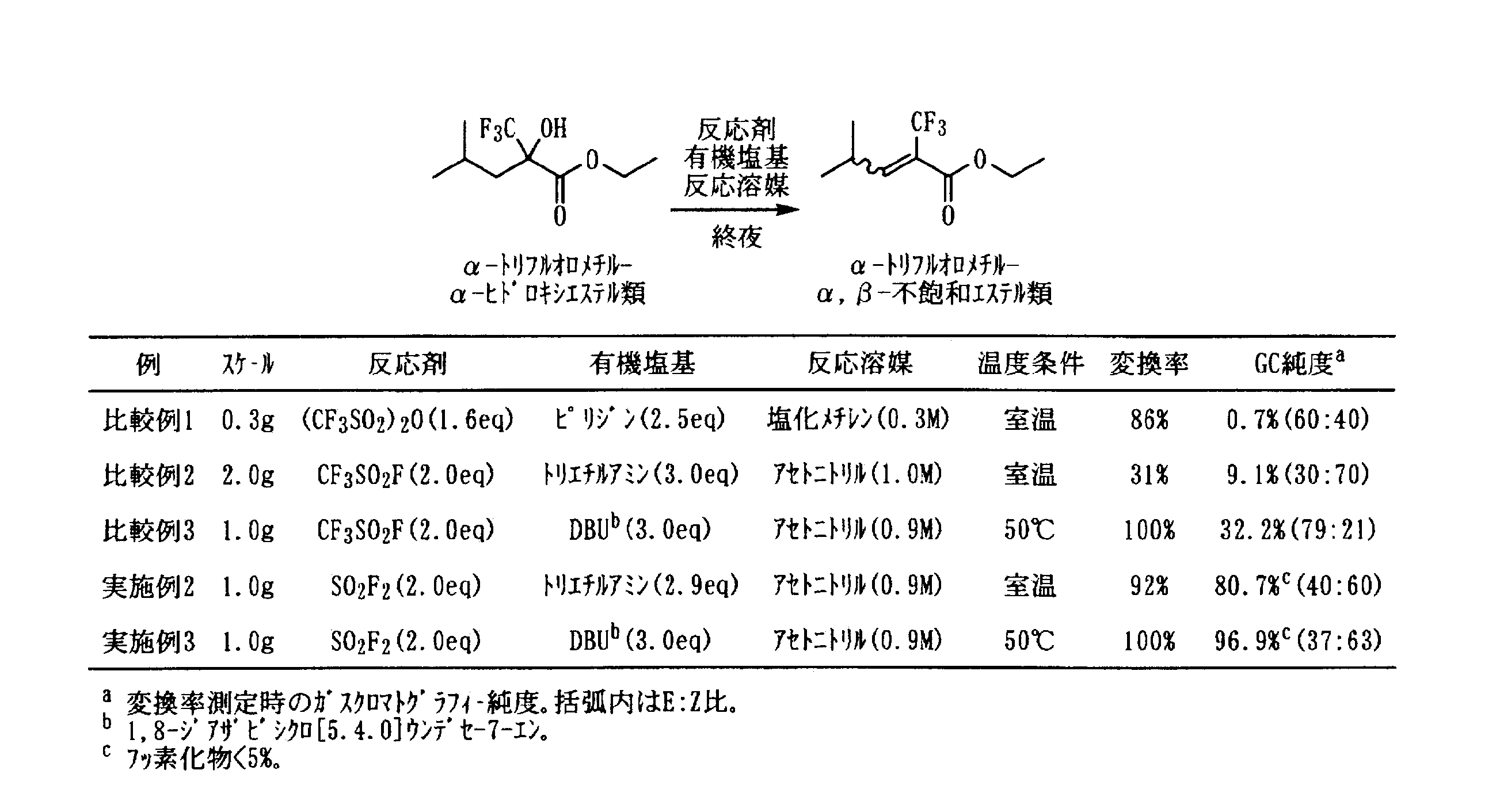
- AQIMLRWXURJTES-ALCCZGGFSA-N CCOC(/C(/C(F)(F)F)=C/C(C)C)=O Chemical compound CCOC(/C(/C(F)(F)F)=C/C(C)C)=O AQIMLRWXURJTES-ALCCZGGFSA-N 0.000 description 1
- IFRHWQSRTKCGRL-CSKARUKUSA-N CCOC(/C(/C(F)(F)F)=C\c1ccccc1)=O Chemical compound CCOC(/C(/C(F)(F)F)=C\c1ccccc1)=O IFRHWQSRTKCGRL-CSKARUKUSA-N 0.000 description 1
- OUGSSAWTQCAUKE-UHFFFAOYSA-N CCOC(C(CC(C)C)(C(F)(F)F)O)=O Chemical compound CCOC(C(CC(C)C)(C(F)(F)F)O)=O OUGSSAWTQCAUKE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/317—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/317—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
- C07C67/327—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups by elimination of functional groups containing oxygen only in singly bound form
Definitions
- the present invention relates to a process for producing ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters important as intermediates for medicines and agricultural chemicals.
- ⁇ -Trifluoromethyl- ⁇ , ⁇ -unsaturated esters are important as pharmaceutical and agrochemical intermediates.
- Production techniques related to the present invention include thionyl chloride (SOCl 2 ), diphosphorus pentoxide (P 2 O 5 ), acetic anhydride [(CH 3 CO) 2 O] or trifluoromethanesulfonic anhydride [( Examples using CF 3 SO 2 ) 2 O] have been reported (Non-Patent Documents 1 to 6, Patent Document 1).
- the method using trifluoromethanesulfonic anhydride is considered the most excellent method because it can be applied to a raw material substrate in which the acidity of the ⁇ -position proton is low (desired reaction does not proceed easily).
- the present applicant has disclosed a dehydroxyfluorination reaction of alcohols by a combination of sulfuryl fluoride (SO 2 F 2 ) and an organic base (Patent Document 2).
- An object of the present invention is to provide a practical method for producing ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters. For that purpose, it is necessary to solve the problems of the prior art.
- Non-Patent Documents 1 to 6 a raw material substrate having high ⁇ -position proton acidity due to an adjacent electron-withdrawing group and an intermediate leaving group (derived from the hydroxyl group of the raw material substrate) are conjugated. It is limited to the case where it can be easily detached by electron extrusion, and the substrate application range is very narrow.
- trifluoromethanesulfonic anhydride has two trifluoromethanesulfonyl (CF 3 SO 2 ) groups, but only one of them is used to derive the intermediate leaving group, and the atom economy From this point of view, it is not a preferable dehydrating agent.
- trifluoromethanesulfonic acid CF 3 SO 3 H
- CF 3 SO 3 H trifluoromethanesulfonic acid
- one of the ⁇ -position substituents is a hydrogen atom
- the other is an alkyl group, a substituted alkyl group, an alkenyl group, a substituted alkenyl group, an aromatic ring group or a substituted aromatic ring group, and an ester group
- the alkyl ester is more preferable.
- the raw material substrate is easily available, the desired reaction proceeds well, and the ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters obtained are particularly important as intermediates for medicines and agrochemicals.
- the organic base 1,5-diazabicyclo [4.3.0] non-5-ene (DBN) or 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) is more preferable. It was also revealed that it was preferable. By using the organic base, the desired reaction proceeds even better.
- the production conditions of the present invention are similar to the dehydroxyfluorination reaction conditions disclosed in Patent Document 2 and actually produce a fluorinated product in which the ⁇ -position hydroxyl group is substituted with a fluorine atom [Scheme-1 ( See Example 1)].
- Scheme-1 See Example 1
- ⁇ -trifluoromethyl- ⁇ -hydroxyesters which are raw material substrates of the present invention are used, ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters which are dehydrates can be selectively obtained. I found it.
- the present inventors have found a very useful method for producing ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters, and have reached the present invention.
- the present invention includes [Invention 1] to [Invention 3] and provides a practical method for producing ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters.
- the organic base is 1,5-diazabicyclo [4.3.0] non-5-ene (DBN) or 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU).
- DBN 1,5-diazabicyclo [4.3.0] non-5-ene
- DBU 1,8-diazabicyclo [5.4.0] undec-7-ene
- the production method of the present invention has a wide substrate application range and can obtain the target product with high productivity and high yield. Furthermore, since hardly separated impurities are hardly produced as a by-product, the target product can be obtained with high chemical purity.
- the sulfuryl fluoride used in the present invention has a high atom economy, and can be easily treated with inorganic salts such as fluorite (CaF 2 ) and calcium sulfate (CaSO 4 ) that are not particularly problematic in waste treatment.
- sulfuryl fluoride is widely used as a fumigant, and in a larger amount than a dehydrating agent such as trifluoromethanesulfonic anhydride or fluorosulfuric anhydride [(FSO 2 ) 2 O] disclosed in Patent Document 1. And it can be obtained at a low cost.
- the present invention solves all the problems of the prior art and is a manufacturing method that can be implemented industrially.
- no report has been made of the use of sulfuryl fluoride, a fumigant, as a dehydrating agent in organic synthesis.
- ⁇ -trifluoromethyl- ⁇ -hydroxyesters represented by the general formula [1] are reacted with sulfuryl fluoride in the presence of an organic base to produce ⁇ -trimethyl represented by the general formula [2].
- This is a method for producing fluoromethyl- ⁇ , ⁇ -unsaturated esters.
- R 1 and R 2 of the ⁇ -trifluoromethyl- ⁇ -hydroxyester represented by the general formula [1] are each independently a hydrogen atom, an alkyl group, a substituted alkyl group, an alkenyl group, a substituted alkenyl group, an alkynyl group, It represents a substituted alkynyl group, aromatic ring group, substituted aromatic ring group, alkylcarbonyl group, substituted alkylcarbonyl group, alkoxycarbonyl group, substituted alkoxycarbonyl group, arylcarbonyl group, substituted arylcarbonyl group, cyano group or nitro group.
- a hydrogen atom, an alkyl group, a substituted alkyl group, an alkenyl group, a substituted alkenyl group, an aromatic ring group, and a substituted aromatic ring group are preferable, particularly one is a hydrogen atom, and the other is an alkyl group, a substituted alkyl group, An alkenyl group, a substituted alkenyl group, an aromatic ring group, and a substituted aromatic ring group are more preferable.
- the alkyl group can have a straight chain or branched chain structure having 1 to 18 carbon atoms, or a cyclic structure (when the number of carbon atoms is 3 or more).
- a single bond of any two adjacent carbon atoms in the above alkyl group is replaced with a double bond in any number, and the stereochemistry of the double bond is E-form, Z-form, or E-form. And a Z-form mixture can be taken.
- a single bond of any two adjacent carbon atoms in the above alkyl group can be replaced with a triple bond in any number.
- the aromatic ring group is an aromatic hydrocarbon group having 1 to 18 carbon atoms, such as a phenyl group, a naphthyl group, an anthryl group, or a pyrrolyl group, a furyl group, a thienyl group, an indolyl group, a benzofuryl group, a benzothienyl group, etc.
- An aromatic heterocyclic group containing a hetero atom such as a nitrogen atom, an oxygen atom or a sulfur atom can be employed.
- the alkyl group (R) of the alkylcarbonyl group (—COR) is the same as the above alkyl group.
- the alkyl group (R) of the alkoxycarbonyl group (—CO 2 R) is the same as the above alkyl group.
- the aryl group (Ar) of the arylcarbonyl group (—COAr) is the same as the above aromatic ring group.
- alkyl group, alkenyl group, alkynyl group, aromatic ring group, alkylcarbonyl group, alkoxycarbonyl group and arylcarbonyl group may have a substituent on any carbon atom in any number and in any combination.
- a substituted alkyl group, a substituted alkenyl group, a substituted alkynyl group, a substituted aromatic ring group, a substituted alkylcarbonyl group, a substituted alkoxycarbonyl group and a substituted arylcarbonyl group respectively.
- substituents include fluorine, chlorine, bromine, iodine halogen atoms, azide groups, nitro groups, methyl groups, ethyl groups, propyl groups and other lower alkyl groups, fluoromethyl groups, chloromethyl groups, bromomethyl groups and other lower groups.
- Lower alkyl groups such as haloalkyl groups, methoxy groups, ethoxy groups, propoxy groups, etc., lower haloalkoxy groups such as fluoromethoxy groups, chloromethoxy groups, bromomethoxy groups, dimethylamino groups, diethylamino groups, dipropylamino groups, etc.
- Lower alkylthio groups such as amino group, methylthio group, ethylthio group and propylthio group, lower alkoxycarbonyl groups such as cyano group, methoxycarbonyl group, ethoxycarbonyl group and propoxycarbonyl group, aminocarbonyl group (CONH 2 ), dimethylaminocarbonyl group , Diethyl Lower aminocarbonyl group such as minocarbonyl group, dipropylaminocarbonyl group, unsaturated group such as alkenyl group, alkynyl group, aromatic ring group such as phenyl group, naphthyl group, pyrrolyl group, furyl group, thienyl group, phenoxy group, Aromatic ring oxy groups such as naphthoxy group, pyrrolyloxy group, furyloxy group, thienyloxy group, aliphatic heterocyclic groups such as piperidyl group, piperidino group, morpholinyl group, hydroxyl
- “Lower” means a linear or branched chain or cyclic group having 1 to 6 carbon atoms (when the number of carbon atoms is 3 or more).
- “unsaturated group” is a double bond (alkenyl group)
- both E-form and Z-form geometric isomerism can be adopted.
- ProtectiveiGroups In Organic Synthesis, Third Edition, 1999, John Wiley & Sons, Inc. Can be used (two or more functional groups can be protected with one protecting group).
- “Unsaturated group”, “aromatic ring group”, “aromatic ring oxy group” and “aliphatic heterocyclic group” include halogen atom, azide group, nitro group, lower alkyl group, lower haloalkyl group, lower alkoxy group.
- Group, lower haloalkoxy group, lower alkylamino group, lower alkylthio group, cyano group, lower alkoxycarbonyl group, aminocarbonyl group, lower aminocarbonyl group, hydroxyl group, hydroxyl group protector, amino group, amino group protector Thiol group, thiol group protector, aldehyde group, aldehyde group protector, carboxyl group, carboxyl group protector and the like can be substituted.
- R 3 in the ⁇ -trifluoromethyl- ⁇ -hydroxyester represented by the general formula [1] represents an alkyl group or a substituted alkyl group. Of these, an alkyl group is preferable, and a lower alkyl group is more preferable.
- the alkyl group and the substituted alkyl group are the same as the alkyl group and the substituted alkyl group of R 1 and R 2 described above.
- the amount of sulfuryl fluoride used may be 0.7 mol or more with respect to 1 mol of ⁇ -trifluoromethyl- ⁇ -hydroxyester represented by the general formula [1], and usually 0.8 to 10 mol. In particular, 0.9 to 5 mol is more preferable.
- organic base examples include triethylamine, diisopropylethylamine, tri-n-propylamine, tri-n-butylamine, tri-n-pentylamine, pyridine, 2,3-lutidine, 2,4-lutidine, 2,6-lutidine, 3,4 -Lutidine, 3,5-lutidine, 2,4,6-collidine, 3,5,6-collidine, 4-dimethylaminopyridine, 1,5-diazabicyclo [4.3.0] non-5-ene, 1 , 8-diazabicyclo [5.4.0] undec-7-ene, N, N, N ′, N ′, N ′′ -pentamethylguanidine, 1,5,7-triazabicyclo [4.4.0 And phosphazene bases such as dece-5-ene, BEMP and t-Bu-P4.
- triethylamine, diisopropylethylamine, tri-n-butylamine, pyridine, 2,6-lutidine, 2,4,6-collidine, 4-dimethylaminopyridine, 1,5-diazabicyclo [4.3.0] non-5 Ene and 1,8-diazabicyclo [5.4.0] undec-7-ene are preferred, especially triethylamine, diisopropylethylamine, 1,5-diazabicyclo [4.3.0] non-5-ene and 1,8- Diazabicyclo [5.4.0] undec-7-ene is more preferred.
- organic bases can be used alone or in combination.
- the organic base may be used in an amount of 0.7 mol or more with respect to 1 mol of ⁇ -trifluoromethyl- ⁇ -hydroxyester represented by the general formula [1], and usually 0.8 to 10 mol. Particularly preferred is 0.9 to 5 moles. In the case of using a combination of organic bases, it means the total amount used, and the more basic one can be used catalytically (see Examples 5 to 7).
- Reaction solvents include aliphatic hydrocarbons such as n-hexane, cyclohexane and n-heptane, aromatic hydrocarbons such as benzene, toluene and xylene, and halogenated carbons such as methylene chloride, chloroform and 1,2-dichloroethane.
- Hydrogen type diethyl ether, tetrahydrofuran, diisopropyl ether, ether type such as tert-butyl methyl ether, ester type such as ethyl acetate and n-butyl acetate, nitrile type such as acetonitrile and propionitrile, N, N-dimethylformamide, Examples thereof include amides such as N, N-dimethylacetamide and 1,3-dimethyl-2-imidazolidinone, and dimethyl sulfoxide.
- n-hexane, n-heptane, toluene, xylene, methylene chloride, tetrahydrofuran, diisopropyl ether, tert-butyl methyl ether, ethyl acetate, acetonitrile, propionitrile, N, N-dimethylformamide and dimethyl sulfoxide are particularly preferable.
- n-Heptane, toluene, methylene chloride, tetrahydrofuran, tert-butyl methyl ether, ethyl acetate, acetonitrile and N, N-dimethylformamide are more preferred.
- These reaction solvents can be used alone or in combination.
- the reaction of the present invention can be carried out without a solvent.
- the reaction solvent may be used in an amount of 0.01 L (liter) or more with respect to 1 mol of ⁇ -trifluoromethyl- ⁇ -hydroxyester represented by the general formula [1], and usually 0.03 to 30 L. In particular, 0.05 to 20 L is more preferable.
- the temperature condition may be in the range of ⁇ 30 to + 150 ° C., usually ⁇ 20 to + 140 ° C. is preferable, and ⁇ 10 to + 130 ° C. is more preferable.
- the reaction time may be in the range of 24 hours or less, and varies depending on the raw material substrate and reaction conditions. Therefore, the progress of the reaction is traced by analysis means such as gas chromatography, liquid chromatography, nuclear magnetic resonance, etc.
- the end point is preferably the point at which almost disappeared.
- the reaction completion solution (concentrate the reaction solvent if necessary) is used as an organic solvent (for example, n-hexane, n-heptane, toluene, xylene, methylene chloride, diisopropyl ether, tert-butyl methyl ether, ethyl acetate).
- an organic solvent for example, n-hexane, n-heptane, toluene, xylene, methylene chloride, diisopropyl ether, tert-butyl methyl ether, ethyl acetate.
- the ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated ester represented by the general formula [2] can be obtained as a crude product by concentrating the recovered organic layer.
- a crude product can be obtained by distillation under reduced pressure directly from the reaction end solution, and the post-treatment operation can be simplified.
- the crude product can be purified to a high chemical purity by activated carbon treatment, distillation, recrystallization, column chromatography or the like, if necessary.
- the wavy line of ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters represented by the general formula [2] indicates that the stereochemistry of the double bond is E-form, Z-form, or a mixture of E-form and Z-form
- the stereochemistry of the resulting product varies depending on the raw material substrate and reaction conditions.
- ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated esters are produced by reacting ⁇ -trifluoromethyl- ⁇ -hydroxyesters with sulfuryl fluoride in the presence of an organic base. (Aspect 1).
- one of the ⁇ -position substituents is a hydrogen atom
- the other is an alkyl group, a substituted alkyl group, an alkenyl group, a substituted alkenyl group, an aromatic ring group or a substituted aromatic ring group
- the ester group is an alkyl ester (Aspect 2).
- the organic base is more preferably 1,5-diazabicyclo [4.3.0] non-5-ene or 1,8-diazabicyclo [5.4.0] undec-7-ene. Preferred (Aspect 3). By using the organic base of this embodiment, the desired reaction proceeds even better.
- Example 1 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula ⁇ -trifluoromethyl- ⁇ -hydroxyesters represented by 2.00 g (10.75 mmol, 1.00 eq), 5.4 mL (1.99 M) of acetonitrile and 2.17 g (21.44 mmol, 1.99 eq) of triethylamine And was immersed in a refrigerant bath at ⁇ 78 ° C., 2.19 g (21.46 mmol, 2.00 eq) of sulfuryl fluoride (SO 2 F 2 ) was blown from the bomb and stirred at room temperature overnight.
- SUS stainless steel
- the conversion rate was 86% from 19 F-NMR of the reaction completed liquid. From 19 F-NMR when measuring the conversion rate, An ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated ester represented by the formula: The production ratio of the fluorinated product represented by is 70:30. No post-treatment was performed on the reaction end solution. 1 H- and 19 F-NMR are shown below.
- Example 2 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula ⁇ -trifluoromethyl- ⁇ -hydroxyesters represented by 1.00 g (4.38 mmol, 1.00 eq), acetonitrile 5.0 mL (0.88 M) and triethylamine 1.30 g (12.85 mmol, 2.93 eq) was added, and immersed in a refrigerant bath at ⁇ 78 ° C., 0.89 g (8.72 mmol, 1.99 eq) of sulfuryl fluoride (SO 2 F 2 ) was blown from the bomb and stirred at room temperature overnight.
- SUS stainless steel
- the conversion rate was 92% from the gas chromatography of the reaction completion liquid.
- the gas chromatography purity at the time of measuring the conversion rate was 80.7% ⁇ intermediate [LG; leaving group (OSO 2 F)] 10.1%, fluorinated compound ⁇ 5% ⁇ , and the E: Z ratio was 40: 60.
- the reaction-terminated liquid is diluted with 30 mL of ethyl acetate, washed with 30 mL of saturated aqueous potassium carbonate solution, washed with 30 mL of water, dried over anhydrous magnesium sulfate, and the collected organic layer is concentrated under reduced pressure and subjected to short column chromatography (silica gel, acetic acid).
- Example 3 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula ⁇ -trifluoromethyl- ⁇ -hydroxyesters represented by the formula: 1.00 g (4.38 mmol, 1.00 eq), 5.0 mL (0.88 M) of acetonitrile and 1,8-diazabicyclo [5.4.0] undece 1.97 g (12.94 mmol, 2.95 eq) of ⁇ 7-ene (DBU) was added, immersed in a refrigerant bath at ⁇ 78 ° C., and 0.90 g (8.82 mmol, 2.82 mmol) of sulfuryl fluoride (SO 2 F 2 ). 01 eq) was blown from a cylinder and stirred at 50 ° C. overnight.
- SUS stainless steel
- the conversion rate was 100% from the gas chromatography of the reaction completed liquid.
- Gas chromatographic purity (corrected excluding DBU peak) at the time of measuring the conversion rate was 96.9% ⁇ intermediate [LG; leaving group (OSO 2 F)] 1.2%, fluoride ⁇ 5% ⁇ .
- the E: Z ratio was 37:63.
- the reaction-terminated liquid is diluted with 30 mL of ethyl acetate, washed with 30 mL of saturated aqueous potassium carbonate solution, washed with 30 mL of water, dried over anhydrous magnesium sulfate, and the collected organic layer is concentrated under reduced pressure and subjected to short column chromatography (silica gel, acetic acid).
- Example 4 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula 2.00 g (7.63 mmol, 1.00 eq), 10.0 mL (0.76 M) of acetonitrile and 1,8-diazabicyclo [5.4.0] undece 3.48 g (22.86 mmol, 3.00 eq) of ⁇ 7-ene (DBU) was added and immersed in a ⁇ 78 ° C. refrigerant bath to give 1.56 g (15.29 mmol, 2.29 mmol, sulfuryl fluoride (SO 2 F 2 )). 00eq) was blown from a cylinder and stirred at 50 ° C. overnight.
- SUS stainless steel
- the conversion was 95% by gas chromatography of the reaction completed liquid.
- the gas chromatographic purity (corrected excluding the peaks of DBU and raw material substrate-derived impurities) at the time of conversion rate measurement was 90.6% (fluoride ⁇ 5%), and the E: Z ratio was 90:10.
- the reaction-terminated liquid is diluted with 30 mL of ethyl acetate, washed with 30 mL of saturated aqueous potassium carbonate solution, washed with 30 mL of water, dried over anhydrous magnesium sulfate, and the collected organic layer is concentrated under reduced pressure and subjected to short column chromatography (silica gel, acetic acid).
- Example 5 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula ⁇ -trifluoromethyl- ⁇ -hydroxyesters represented by the formula: 1.00 g (3.81 mmol, 1.00 eq), acetonitrile 5.0 mL (0.76 M), 1,8-diazabicyclo [5.4.0] undece Add 0.29 g (1.90 mmol, 0.50 eq) of ⁇ 7-ene (DBU) and 0.77 g (7.61 mmol, 2.00 eq) of triethylamine, soak in a refrigerant bath at ⁇ 78 ° C., and add sulfuryl fluoride (SO 2). 2 F 2 ) 0.78 g (7.64 mmol, 2.01 eq) was blown from the bomb and stirred at 50 ° C. overnight.
- SUS stainless steel
- the conversion was 95% by gas chromatography of the reaction completed liquid.
- the gas chromatographic purity (corrected excluding the peaks of DBU and impurities derived from the raw material substrate) at the time of measuring the conversion rate was 80.2% (fluoride ⁇ 5%), and the E: Z ratio was 90:10.
- the reaction-terminated liquid is diluted with 20 mL of ethyl acetate, washed with 20 mL of saturated aqueous potassium carbonate solution, washed with 20 mL of water, dried over anhydrous sodium sulfate, and the collected organic layer is concentrated under reduced pressure and dried under vacuum to obtain the following formula.
- Example 6 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula ⁇ -trifluoromethyl- ⁇ -hydroxyesters represented by the formula: 3.00 g (13.26 mmol, 1.00 eq), acetonitrile 10.0 mL (1.33 M), 1,8-diazabicyclo [5.4.0] undece Add 1.00 g (6.57 mmol, 0.50 eq) of ⁇ 7-ene (DBU) and 2.68 g (26.48 mmol, 2.00 eq) of triethylamine, and immerse in a refrigerant bath at ⁇ 78 ° C. to obtain sulfuryl fluoride (SO 2 F 2 ) (4.06 g, 39.78 mmol, 3.00 eq) was blown from the bomb and stirred at 50 ° C. overnight.
- SUS stainless steel
- the conversion rate was 100% from the gas chromatography of the reaction completed liquid.
- the gas chromatography purity at the time of measuring the conversion rate was 96.1% (fluorinated product ⁇ 5%), and the E: Z ratio was 92: 8.
- the reaction-terminated liquid was diluted with 30 mL of ethyl acetate, washed with 30 mL of saturated aqueous potassium carbonate solution, washed twice with 30 mL of water, dried over anhydrous magnesium sulfate, and the collected organic layer was concentrated under reduced pressure and dried under vacuum.
- Example 7 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula 50.00 g (249.80 mmol, 1.00 eq), 83.0 mL (3.01 M) of acetonitrile, 1,8-diazabicyclo [5.4.0] undece 19.00 g (124.80 mmol, 0.50 eq) of ⁇ 7-ene (DBU) and 63.20 g (624.57 mmol, 2.50 eq) of triethylamine were added and immersed in a refrigerant bath at ⁇ 78 ° C. to obtain sulfuryl fluoride (SO 2 F 2 ) 51.00 g (499.71 mmol, 2.00 eq) was blown from the bomb and stirred at room temperature overnight.
- SUS stainless steel
- the conversion was 99% by gas chromatography of the reaction completed liquid.
- the gas chromatography purity at the time of measuring the conversion rate was 82.1% (fluoride 9.0%), and the E: Z ratio was 78:22.
- the reaction-terminated liquid is directly distilled under reduced pressure (boiling point 52-58 ° C./vacuum degree 5000 Pa) to obtain the following formula: 17.21 g of a crude product of ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated ester represented by the formula (1) was obtained.
- the yield was 38%.
- the gas chromatography purity was 82.8% (fluoride 10.5%), and the E: Z ratio was 79:21. 1 H- and 19 F-NMR are shown below.
- the conversion was 86% by gas chromatography of the reaction completed liquid. From gas chromatography at the time of conversion rate measurement, The purity of the ⁇ -trifluoromethyl- ⁇ , ⁇ -unsaturated ester represented by the formula is 0.7% ⁇ intermediate [LG; leaving group (OSO 2 CF 3 )] 83.8% ⁇ , and E: The Z ratio was 60:40. No post-treatment was performed on the reaction end solution.
- Comparative Example 2 was carried out in the same manner by changing the reactants with reference to Example 2.
- Comparative Example 3 was carried out in the same manner by changing the reactants with reference to Example 3.
- Comparative Example 4 was carried out in the same manner with reference to Comparative Example 1 except that the raw material substrate was changed (see the reaction formulas in Tables 1 and 2).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description



一般式[1]


[式中、R1およびR2はそれぞれ独立に水素原子、アルキル基、置換アルキル基、アルケニル基、置換アルケニル基、アルキニル基、置換アルキニル基、芳香環基、置換芳香環基、アルキルカルボニル基、置換アルキルカルボニル基、アルコキシカルボニル基、置換アルコキシカルボニル基、アリールカルボニル基、置換アリールカルボニル基、シアノ基またはニトロ基を表し、R3はアルキル基または置換アルキル基を表す。一般式[2]の波線は、二重結合の立体化学がE体、Z体、またはE体とZ体の混合物であることを表す]
一般式[3]


[式中、R4はアルキル基、置換アルキル基、アルケニル基、置換アルケニル基、芳香環基または置換芳香環基を表し、R5はアルキル基を表す。一般式[4]の波線は、二重結合の立体化学がE体、Z体、またはE体とZ体の混合物であることを表す]
発明1または発明2において、有機塩基が1,5-ジアザビシクロ[4.3.0]ノン-5-エン(DBN)または1,8-ジアザビシクロ[5.4.0]ウンデセ-7-エン(DBU)であることを特徴とする、発明1または発明2に記載のα-トリフルオロメチル-α,β-不飽和エステル類の製造方法。
実施例により本発明の実施の形態を具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
ステンレス鋼(SUS)製耐圧反応容器に、下記式



19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;α-トリフルオロメチル-α,β-不飽和エステル類/96.06(s、3F)、フッ素化物/31.88(s、1F)、83.73(s、3F)。
ステンレス鋼(SUS)製耐圧反応容器に、下記式

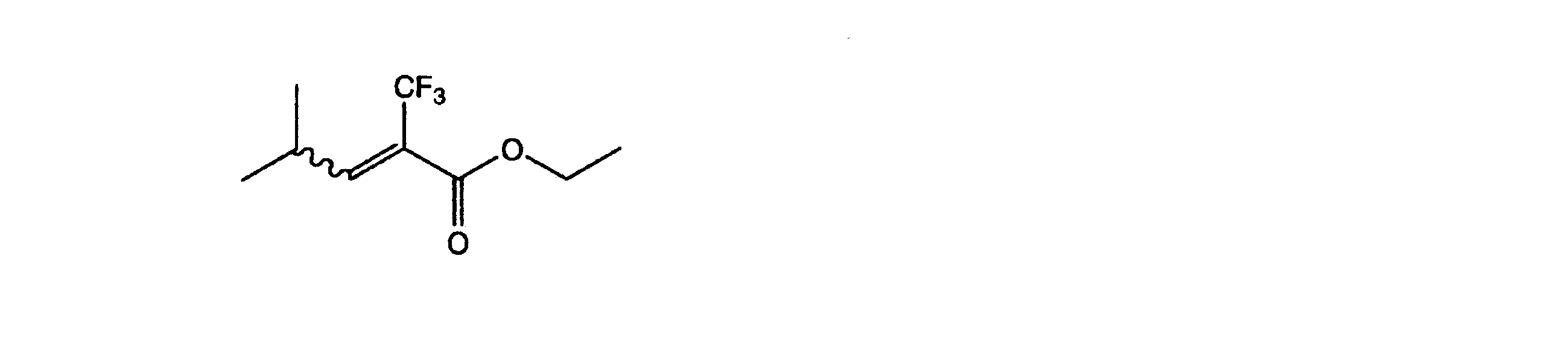
19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;E体/97.80(s、3F)、Z体/103.05(s、3F)。
ステンレス鋼(SUS)製耐圧反応容器に、下記式


ステンレス鋼(SUS)製耐圧反応容器に、下記式
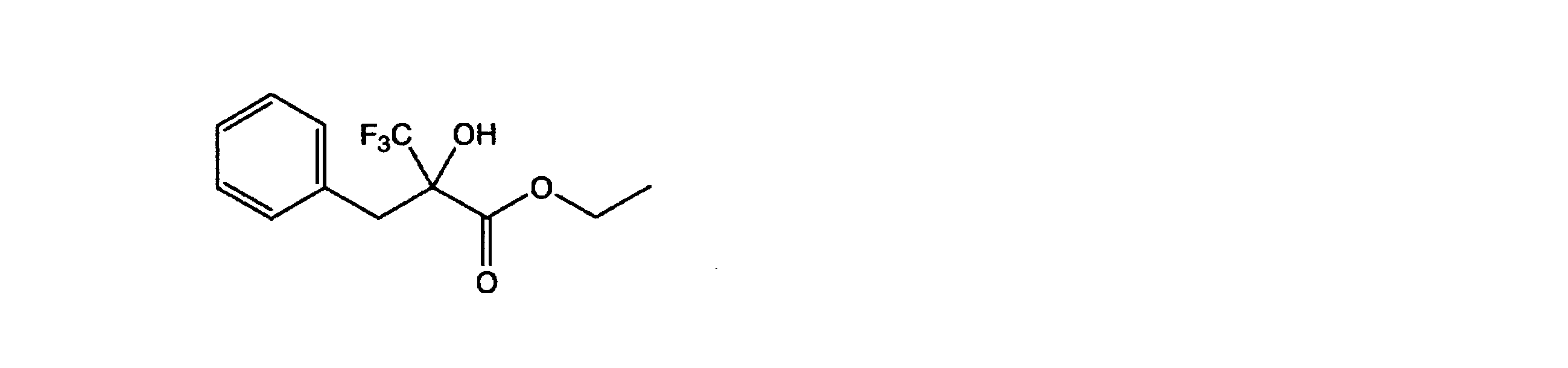

19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;E体/97.85(s、3F)、Z体/103.81(s、3F)。
ステンレス鋼(SUS)製耐圧反応容器に、下記式


ステンレス鋼(SUS)製耐圧反応容器に、下記式


19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;E体/98.05(s、3F)、Z体/103.85(s、3F)。
ステンレス鋼(SUS)製耐圧反応容器に、下記式


19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;E体/97.64(s、3F)、Z体/103.00(s、3F)。

19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;30.08(s、1F)、80.39(s、3F)。
下記式


Claims (3)
- 一般式[1]


[式中、R1およびR2はそれぞれ独立に水素原子、アルキル基、置換アルキル基、アルケニル基、置換アルケニル基、アルキニル基、置換アルキニル基、芳香環基、置換芳香環基、アルキルカルボニル基、置換アルキルカルボニル基、アルコキシカルボニル基、置換アルコキシカルボニル基、アリールカルボニル基、置換アリールカルボニル基、シアノ基またはニトロ基を表し、R3はアルキル基または置換アルキル基を表す。一般式[2]の波線は、二重結合の立体化学がE体、Z体、またはE体とZ体の混合物であることを表す] - 一般式[3]
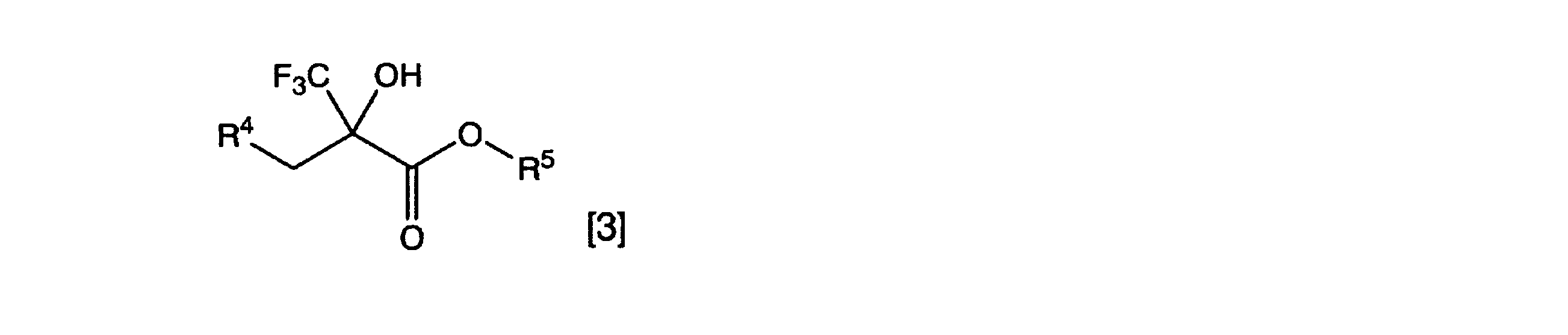

[式中、R4はアルキル基、置換アルキル基、アルケニル基、置換アルケニル基、芳香環基または置換芳香環基を表し、R5はアルキル基を表す。一般式[4]の波線は、二重結合の立体化学がE体、Z体、またはE体とZ体の混合物であることを表す] - 請求項1または請求項2において、有機塩基が1,5-ジアザビシクロ[4.3.0]ノン-5-エン(DBN)または1,8-ジアザビシクロ[5.4.0]ウンデセ-7-エン(DBU)であることを特徴とする、請求項1または請求項2に記載のα-トリフルオロメチル-α,β-不飽和エステル類の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09816162.3A EP2332902B1 (en) | 2008-09-26 | 2009-09-24 | Process for producing alpha-trifluoromethyl-alpha,beta-unsaturated ester |
| US13/060,565 US8653295B2 (en) | 2008-09-26 | 2009-09-24 | Process for producing α-trifluoromethyl-α,β-unsaturated ester |
| CN200980137940.2A CN102164883B (zh) | 2008-09-26 | 2009-09-24 | α-三氟甲基-α,β-不饱和酯类的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-247053 | 2008-09-26 | ||
| JP2008247053A JP5277837B2 (ja) | 2008-09-26 | 2008-09-26 | α−トリフルオロメチル−α,β−不飽和エステル類の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010035747A1 true WO2010035747A1 (ja) | 2010-04-01 |
Family
ID=42059746
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/066527 Ceased WO2010035747A1 (ja) | 2008-09-26 | 2009-09-24 | α-トリフルオロメチル-α,β-不飽和エステル類の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8653295B2 (ja) |
| EP (1) | EP2332902B1 (ja) |
| JP (1) | JP5277837B2 (ja) |
| CN (1) | CN102164883B (ja) |
| WO (1) | WO2010035747A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111233666A (zh) * | 2020-01-16 | 2020-06-05 | 山东师范大学 | 一种高效合成三氟甲基化合物的方法、三氟甲基化合物及应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6078941A (ja) * | 1983-10-07 | 1985-05-04 | Daikin Ind Ltd | α−フルオロアクリル酸エステルの製造法 |
| JP2003342211A (ja) * | 2002-05-29 | 2003-12-03 | Ihara Chem Ind Co Ltd | 4,4−ジフルオロ−3−ブテン−1−オール誘導体の製造法 |
| US20060004195A1 (en) | 2004-06-30 | 2006-01-05 | Xiaohu Deng | Alpha,beta-unsaturated esters and acids by stereoselective dehydration |
| JP2006290870A (ja) | 2005-03-18 | 2006-10-26 | Central Glass Co Ltd | スルフリルフルオリドを用いるフッ素化反応 |
| JP2008013519A (ja) * | 2006-07-07 | 2008-01-24 | Central Glass Co Ltd | 光学活性2−フルオロアルコール誘導体の製造方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60260434A (ja) * | 1984-06-04 | 1985-12-23 | Shin Etsu Chem Co Ltd | 光伝送用無水ガラス素材の製造方法 |
| AU703560B2 (en) * | 1995-10-10 | 1999-03-25 | Dow Agrosciences Llc | A method for making alpha,beta-unsaturated-beta- trifluoromethyl-carboxylates and related compounds |
-
2008
- 2008-09-26 JP JP2008247053A patent/JP5277837B2/ja not_active Expired - Fee Related
-
2009
- 2009-09-24 EP EP09816162.3A patent/EP2332902B1/en not_active Not-in-force
- 2009-09-24 WO PCT/JP2009/066527 patent/WO2010035747A1/ja not_active Ceased
- 2009-09-24 CN CN200980137940.2A patent/CN102164883B/zh not_active Expired - Fee Related
- 2009-09-24 US US13/060,565 patent/US8653295B2/en not_active Expired - Fee Related
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6078941A (ja) * | 1983-10-07 | 1985-05-04 | Daikin Ind Ltd | α−フルオロアクリル酸エステルの製造法 |
| JP2003342211A (ja) * | 2002-05-29 | 2003-12-03 | Ihara Chem Ind Co Ltd | 4,4−ジフルオロ−3−ブテン−1−オール誘導体の製造法 |
| US20060004195A1 (en) | 2004-06-30 | 2006-01-05 | Xiaohu Deng | Alpha,beta-unsaturated esters and acids by stereoselective dehydration |
| JP2008505092A (ja) * | 2004-06-30 | 2008-02-21 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | 立体選択的脱水によるα,β−不飽和エステル類および酸類 |
| JP2006290870A (ja) | 2005-03-18 | 2006-10-26 | Central Glass Co Ltd | スルフリルフルオリドを用いるフッ素化反応 |
| JP2008013519A (ja) * | 2006-07-07 | 2008-01-24 | Central Glass Co Ltd | 光学活性2−フルオロアルコール誘導体の製造方法 |
Non-Patent Citations (9)
| Title |
|---|
| "Seriya Khimicheskaya (Russia)", 1992, IZVESTIYA AKADEMI NAUK, pages: 2617 - 2623 |
| JOURNAL OF FLUORINE CHEMISTRY (NETHERLANDS), vol. 21, 1982, pages 377 - 384 |
| JOURNAL OF FLUORINE CHEMISTRY (NETHERLANDS), vol. 51, 1991, pages 323 - 334 |
| JOURNAL OF THE CHEMICAL SOCIETY (U.K.), 1961, pages 4519 - 4521 |
| MENDELEEV COMMUNICATIONS (RUSSIA), 2006, pages 175 - 177 |
| See also references of EP2332902A4 |
| TETRAHEDRON (U.K.), vol. 58, 2002, pages 8565 - 8571 |
| TETRAHEDRON LETTERS (U.K.), vol. 45, 2004, pages 183 - 185 |
| ZHURNAL ORGANICHESKOI KHIMII (RUSSIA), vol. 25, 1989, pages 2523 - 2527 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2332902A4 (en) | 2013-09-04 |
| EP2332902B1 (en) | 2014-07-23 |
| US8653295B2 (en) | 2014-02-18 |
| EP2332902A1 (en) | 2011-06-15 |
| CN102164883B (zh) | 2014-03-19 |
| US20110160477A1 (en) | 2011-06-30 |
| CN102164883A (zh) | 2011-08-24 |
| JP5277837B2 (ja) | 2013-08-28 |
| JP2010077069A (ja) | 2010-04-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5326510B2 (ja) | α−置換エステル類の製造方法 | |
| JP5338138B2 (ja) | ハロゲン化α−フルオロエーテル類の製造方法 | |
| JP5359252B2 (ja) | フルオロ硫酸エステル類の製造方法 | |
| CN101878192B (zh) | 4-全氟异丙基苯胺类的制造方法 | |
| JP4940790B2 (ja) | 脱ヒドロキシフッ素化剤 | |
| JP5793996B2 (ja) | フルオロ硫酸芳香環エステル類の製造方法 | |
| JP5369853B2 (ja) | α−フルオロ−β−アミノ酸類の製造方法 | |
| JP5240078B2 (ja) | 2−フルオロアクリル酸エステルの製造方法 | |
| JP5277837B2 (ja) | α−トリフルオロメチル−α,β−不飽和エステル類の製造方法 | |
| JP5476895B2 (ja) | ヒドロキシル基置換生成物の製造方法 | |
| JP5374895B2 (ja) | 光学活性フルオロアミン類の製造方法 | |
| JP2009155248A (ja) | 酸弗化物類の製造方法 | |
| JP4610252B2 (ja) | 4−フルオロプロリン誘導体の製造方法 | |
| JP2013180976A (ja) | α,α−ジフルオロ芳香族化合物の製造方法 | |
| JP2008174552A (ja) | 4−パーフルオロイソプロピルアニリン類の製造方法 | |
| JP5600994B2 (ja) | 2−フルオロイソ酪酸エステルの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980137940.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09816162 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13060565 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 841/KOLNP/2011 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009816162 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |