WO2010090318A1 - セレンテラミド類縁体 - Google Patents
セレンテラミド類縁体 Download PDFInfo
- Publication number
- WO2010090318A1 WO2010090318A1 PCT/JP2010/051806 JP2010051806W WO2010090318A1 WO 2010090318 A1 WO2010090318 A1 WO 2010090318A1 JP 2010051806 W JP2010051806 W JP 2010051806W WO 2010090318 A1 WO2010090318 A1 WO 2010090318A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- calcium
- coelenterazine
- benzyl
- ethyl acetate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*N(*)c1ncc(-c2ccc(*)cc2)nc1Cc1ccccc1 Chemical compound C*N(*)c1ncc(-c2ccc(*)cc2)nc1Cc1ccccc1 0.000 description 6
- RFVFNOZQFFCGKP-UHFFFAOYSA-N CC(Oc(cc1)ccc1-c(nc1Cc2ccccc2)cnc1NS(Cc1ccccc1)(=O)=O)=O Chemical compound CC(Oc(cc1)ccc1-c(nc1Cc2ccccc2)cnc1NS(Cc1ccccc1)(=O)=O)=O RFVFNOZQFFCGKP-UHFFFAOYSA-N 0.000 description 1
- VWSUFHSKGFWHEH-UHFFFAOYSA-N O=S(Cc1ccccc1)(Nc1ncc(-c2ccccc2)nc1Cc1ccccc1)=O Chemical compound O=S(Cc1ccccc1)(Nc1ncc(-c2ccccc2)nc1Cc1ccccc1)=O VWSUFHSKGFWHEH-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D241/20—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/43504—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from invertebrates
- C07K14/43595—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from invertebrates from coelenteratae, e.g. medusae
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
Definitions
- the present invention relates to a coelenteramide analog and a fluorescent protein containing the coelenteramide analog.
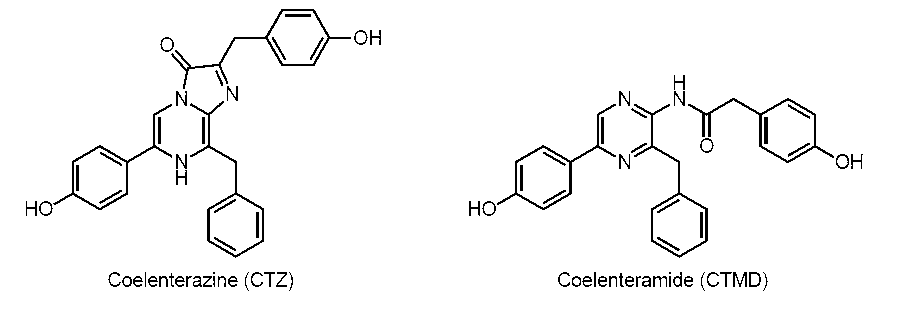
- Coelenteramide is an oxidation product of coelenterazine. It is produced during the luminescence process of calcium-binding photoproteins derived from coelenterate and the luminescence process by coelenterazine (CTZ) luciferases such as Renilla and Gaussia.
- CTZ coelenterazine
- the calcium-binding photoprotein is a photoprotein that specifically binds to Ca 2+ and emits light instantaneously, and is composed of a complex of coelenterazine peroxide and apoprotein (Non-patent Document 1. Head, JF et. al. (2000) Nature 405: 372-376).
- a typical photoprotein is aequorin derived from luminescent aquatic jellyfish.
- aequorin derived from luminescent aquatic jellyfish.
- mytrocomin, criticin-I, and criticin-II are known.
- the light emission mechanism is considered to be basically the same.
- BFP is a fluorescent protein having heat stability and has also been proved to have luciferase activity using CTZ as a substrate.
- a green-like fluorescent protein (gFP) was produced by adding a chelating agent and removing Ca 2+ from apoaequorin-Ca 2+ .
- the regeneration method to aequorin with luminous ability by adding CTZ to this gFP was also established.
- a protein having fluorescence ability and having luciferase activity is only BFP and can be regenerated to aequorin. Therefore, it may be used as a reporter protein in the field of cell biology, and has attracted attention.
- Non-Patent Documents 4-21 disclose the following fluorescent coelenteramide-related compounds.
- Non-Patent Documents 4-21 disclose the structures of the above-mentioned coelenteramide-related compounds and their fluorescence characteristics.
- the present inventors have found that specific thioamide-type coelenteramide analogs and specific sulfonamide-type coelenteramide analogs exhibit fluorescence characteristics different from conventional ones. As a result, the present invention has been completed. That is, the present invention provides the following coelenteramide analogs, fluorescent proteins, and the like.
- R 1 is substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, linear or branched alkyl optionally substituted by an aliphatic cyclic group, aliphatic cyclic group, or heterocyclic Group
- R 2 is hydrogen or — (SO 2 ) R 4
- R 3 is hydrogen, hydroxyl group, methoxy, or acetoxy
- R 4 is substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, or linear or branched alkyl optionally substituted by an aliphatic cyclic group
- X 1 is —C ( ⁇ S) — or —SO 2 —.
- R 1 is phenyl, p-methylphenyl, p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, p-nitrophenyl, benzyl, ⁇ -hydroxybenzyl, 4-methylbenzyl, 4-hydroxybenzyl, 4- Methoxybenzyl, 4-acetoxybenzyl, 4-nitrobenzyl, phenylethyl, methyl, ethyl, propyl, 2-methylpropyl, 2-methylpropanyl, cyclohexylmethyl, cyclohexylethyl, adamantylmethyl, cyclopentylmethyl, cyclohexyl, or thiophene Being 2-yl,
- R 2 is hydrogen, benzenesulfonyl, p-toluenesulfonyl, 4-hydroxyphenylsulfonyl, 4-hydroxyphenylsulfonyl, 4-
- Blue fluorescent protein (7)
- a method for producing a blue fluorescent protein which comprises reacting with.
- a green fluorescent protein comprising the compound according to any one of (1) to (5a) above and an apoprotein of a calcium-binding photoprotein.
- Production of a green fluorescent protein comprising treating the blue fluorescent protein according to (6) with a chelating agent for removing calcium ions or divalent or trivalent ions that can be substituted for calcium ions Method.
- a method for producing a calcium-binding photoprotein comprising reacting coelenterazine or an analog thereof with the green fluorescent protein according to (9). (12) The method according to (11) above, wherein the reaction between the fluorescent protein and coelenterazine or an analog thereof is performed in the presence of a reducing agent.
- the coelenteramide analogs of some aspects of the present invention exhibit fluorescence properties that are different from conventional ones.
- the coelenteramide analog of a preferred embodiment of the present invention exhibits a relatively high fluorescence ability in an aqueous solvent.
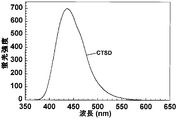
- FIG. 7 is a graph showing a fluorescence spectrum of c-38 compound (CTSD) having a final concentration of 18 ⁇ M in 50 mM Tris-HCl (pH 7.6) containing 10 mM CaCl 2 (Example 3).
- CTSD fluorescence spectrum of c-38 compound
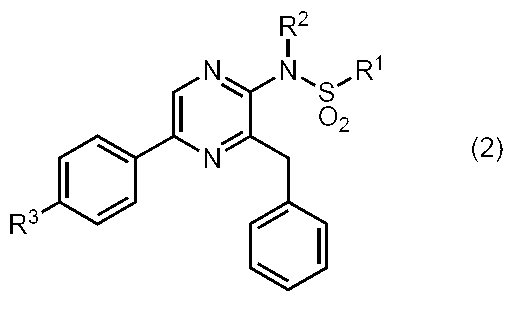
- Coelenteramide analog The present invention provides a compound represented by the following general formula (1) (coelenteramide analog of the present invention).
- R 1 is substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, linear or branched alkyl optionally substituted by an aliphatic cyclic group, aliphatic cyclic group, or heterocyclic Group
- R 2 is hydrogen or — (SO 2 ) R 4
- R 3 is hydrogen, hydroxyl group, methoxy, or acetoxy
- R 4 is substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, or linear or branched alkyl optionally substituted by an aliphatic cyclic group
- X 1 is —C ( ⁇ S) — or —SO 2 —.
- “Substituted or unsubstituted aryl” for R 1 is, for example, aryl having 1 to 5 substituents, or unsubstituted aryl.
- the substituent include halogen (fluorine, chlorine, bromine, iodine, etc.), hydroxyl group, alkyl having 1 to 6 carbon atoms, alkoxyl having 1 to 6 carbon atoms, amino, and 1 to 6 carbon atoms. And at least one selected from the group consisting of dialkylamino and the like.
- the substituent is a hydroxyl group.
- “Substituted or unsubstituted aryl” specifically includes phenyl, p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, p-nitrophenyl, p-aminophenyl, p-dimethylaminophenyl, and the like. And preferred are phenyl, p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, p-nitrophenyl and the like.
- substituted or unsubstituted aryl is substituted aryl, such as p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, or p-nitrophenyl.
- “Substituted or unsubstituted arylalkyl” for R 1 is, for example, arylalkyl having 1 to 5 substituents, or unsubstituted arylalkyl.
- substituents include halogen (fluorine, chlorine, bromine, iodine, etc.), hydroxyl group, alkyl having 1 to 6 carbon atoms, alkoxyl having 1 to 6 carbon atoms, amino, or 1 to 6 carbon atoms. And dialkylamino.
- “Substituted or unsubstituted arylalkyl” means, for example, benzyl, ⁇ -hydroxybenzyl, 4-methylbenzyl, 4-hydroxybenzyl, 4-methoxybenzyl, 4-acetoxybenzyl, 4-nitrobenzyl, phenylethyl 4-hydroxy Benzyl, 4-dimethylaminobenzyl, etc., preferably benzyl, ⁇ -hydroxybenzyl, 4-methylbenzyl, 4-hydroxybenzyl, 4-methoxybenzyl, 4-acetoxybenzyl, 4-nitrobenzyl, or phenylethyl Etc.
- substituted or unsubstituted arylalkyl is arylalkyl substituted with a hydroxyl group, such as ⁇ -hydroxybenzyl or 4-hydroxybenzyl.
- substituted or unsubstituted arylalkyl is unsubstituted arylalkyl, such as benzyl or phenylethyl.
- Linear or branched alkyl optionally substituted by an aliphatic cyclic group for R 1 is unsubstituted linear or branched alkyl or, for example, 1 to 10 aliphatic cyclic Straight or branched chain alkyl substituted by a group.
- the aliphatic cyclic group include cyclohexyl, cyclopentyl, adamantyl, cyclobutyl, and cyclopropyl.
- the aliphatic cyclic group is cyclohexyl, cyclopentyl, adamantyl or the like.
- “Straight or branched alkyl optionally substituted by an aliphatic cyclic group” includes, for example, methyl, ethyl, propyl, 2-methylpropyl, 2-methylpropanyl, cyclohexylmethyl, cyclohexylethyl, adamantyl Methyl, cyclopentylmethyl, cyclobutylmethyl, cyclopropylmethyl, etc., preferably methyl, ethyl, propyl, 2-methylpropyl, 2-methylpropanyl, cyclohexylmethyl, cyclohexylethyl, adamantylmethyl, cyclopentylmethyl, etc. It is.
- straight chain or branched alkyl optionally substituted by an aliphatic cyclic group is a straight chain alkyl optionally substituted by an aliphatic cyclic group.
- Examples of the “aliphatic cyclic group” for R 1 include cyclohexyl, cyclopentyl, adamantyl, cyclobutyl, and cyclopropyl.
- the aliphatic cyclic group is cyclohexyl or the like.
- the “heterocyclic group” of R 1 is, for example, a 5- to 7-membered ring containing 1 to 3 atoms selected from the group consisting of N, O, and S in addition to carbon as atoms constituting the ring.
- “Heterocyclic group” includes, for example, thiophen-2-yl, 2-furanyl, 4-pyridyl and the like.
- a “heterocyclic group” is a sulfur containing heterocyclic group, such as thiophen-2-yl.
- R 1 is phenyl, p-methylphenyl, p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, p-nitrophenyl, benzyl, ⁇ -hydroxybenzyl, 4-methyl.
- R 1 is p-methylphenyl, p-nitrophenyl, benzyl, methyl or the like.
- substituents include halogen (fluorine, chlorine, bromine, iodine, etc.), hydroxyl group, alkyl having 1 to 6 carbon atoms, alkoxyl having 1 to 6 carbon atoms, amino, and 1 to 6 carbon atoms. And at least one selected from the group consisting of dialkylamino and the like.
- the substituent is a hydroxyl group.
- “Substituted or unsubstituted aryl” specifically includes phenyl, p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, p-nitrophenyl, p-hydroxybenzyl, p-dimethylaminobenzyl and the like. And preferred are phenyl, p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, p-nitrophenyl and the like.
- substituted or unsubstituted aryl is substituted aryl, such as p-hydroxyphenyl, p-methoxyphenyl, p-acetoxyphenyl, or p-nitrophenyl.
- “Substituted or unsubstituted arylalkyl” for R 4 is, for example, arylalkyl having 1 to 6 substituents, or unsubstituted arylalkyl.
- substituents include halogen (fluorine, chlorine, bromine, iodine, etc.), hydroxyl group, alkyl having 1 to 6 carbon atoms, alkoxyl having 1 to 6 carbon atoms, amino, and 1 to 6 carbon atoms. And dialkylamino.
- “Substituted or unsubstituted arylalkyl” means, for example, benzyl, ⁇ -hydroxybenzyl, 4-methylbenzyl, 4-hydroxybenzyl, 4-methoxybenzyl, 4-acetoxybenzyl, 4-nitrobenzyl, phenylethyl, 4-ethyl Hydroxybenzyl, 4-dimethylaminobenzyl, etc., preferably benzyl, ⁇ -hydroxybenzyl, 4-methylbenzyl, 4-hydroxybenzyl, 4-methoxybenzyl, 4-acetoxybenzyl, 4-nitrobenzyl, or phenyl Such as ethyl.
- substituted or unsubstituted arylalkyl is arylalkyl substituted with a hydroxyl group, such as ⁇ -hydroxybenzyl or 4-hydroxybenzyl.
- substituted or unsubstituted arylalkyl is unsubstituted arylalkyl, such as benzyl or phenylethyl.
- Linear or branched alkyl optionally substituted by an aliphatic cyclic group for R 4 is unsubstituted linear or branched alkyl, or, for example, 1 to 10 aliphatic cyclic Straight or branched chain alkyl substituted by a group.
- the aliphatic cyclic group include cyclohexyl, cyclopentyl, adamantyl, cyclobutyl, and cyclopropyl.
- the aliphatic cyclic group is cyclohexyl, cyclopentyl, adamantyl or the like.
- “Straight or branched alkyl optionally substituted by an aliphatic cyclic group” includes, for example, methyl, ethyl, propyl, 2-methylpropyl, 2-methylpropanyl, cyclohexylmethyl, cyclohexylethyl, adamantyl Methyl, cyclopentylmethyl, cyclobutylmethyl, cyclopropylmethyl, etc., preferably methyl, ethyl, propyl, 2-methylpropyl, 2-methylpropanyl, cyclohexylmethyl, cyclohexylethyl, adamantylmethyl, cyclopentylmethyl, etc. It is.
- straight chain or branched alkyl optionally substituted by an aliphatic cyclic group is a straight chain alkyl optionally substituted by an aliphatic cyclic group.
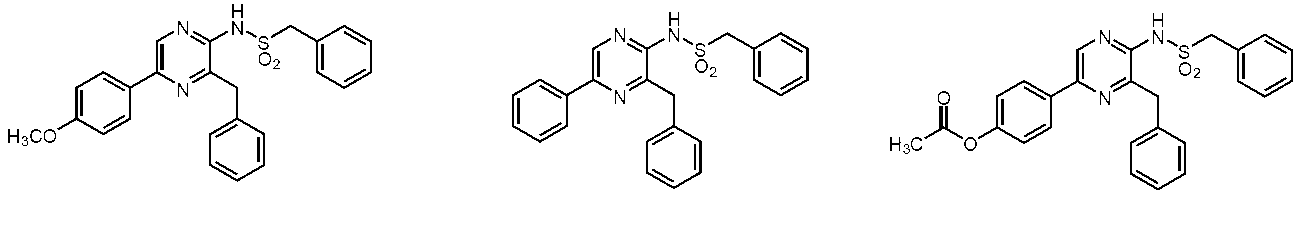
- R 2 is hydrogen, benzenesulfonyl, p-toluenesulfonyl, 4-hydroxyphenylsulfonyl, 4-methoxyphenylsulfonyl, 4-acetoxyphenylsulfonyl, 4-nitrophenylsulfonyl, benzylsulfonyl, ⁇ -hydroxybenzylsulfonyl, 4-methylbenzylsulfonyl, 4-hydroxybenzylsulfonyl, 4-methoxybenzylsulfonyl, 4-acetoxybenzylsulfonyl, 4-nitrobenzylsulfonyl, phenylethylsulfonyl, methanesulfonyl, ethylsulfonyl, propylsulfonyl, 2 -Methylpropylsulfonyl, 2-methylpropanylsulfon
- the compound represented by the general formula (1) is a compound represented by the following general formula (2) (the sulfonamide coelenteramide of the present invention).
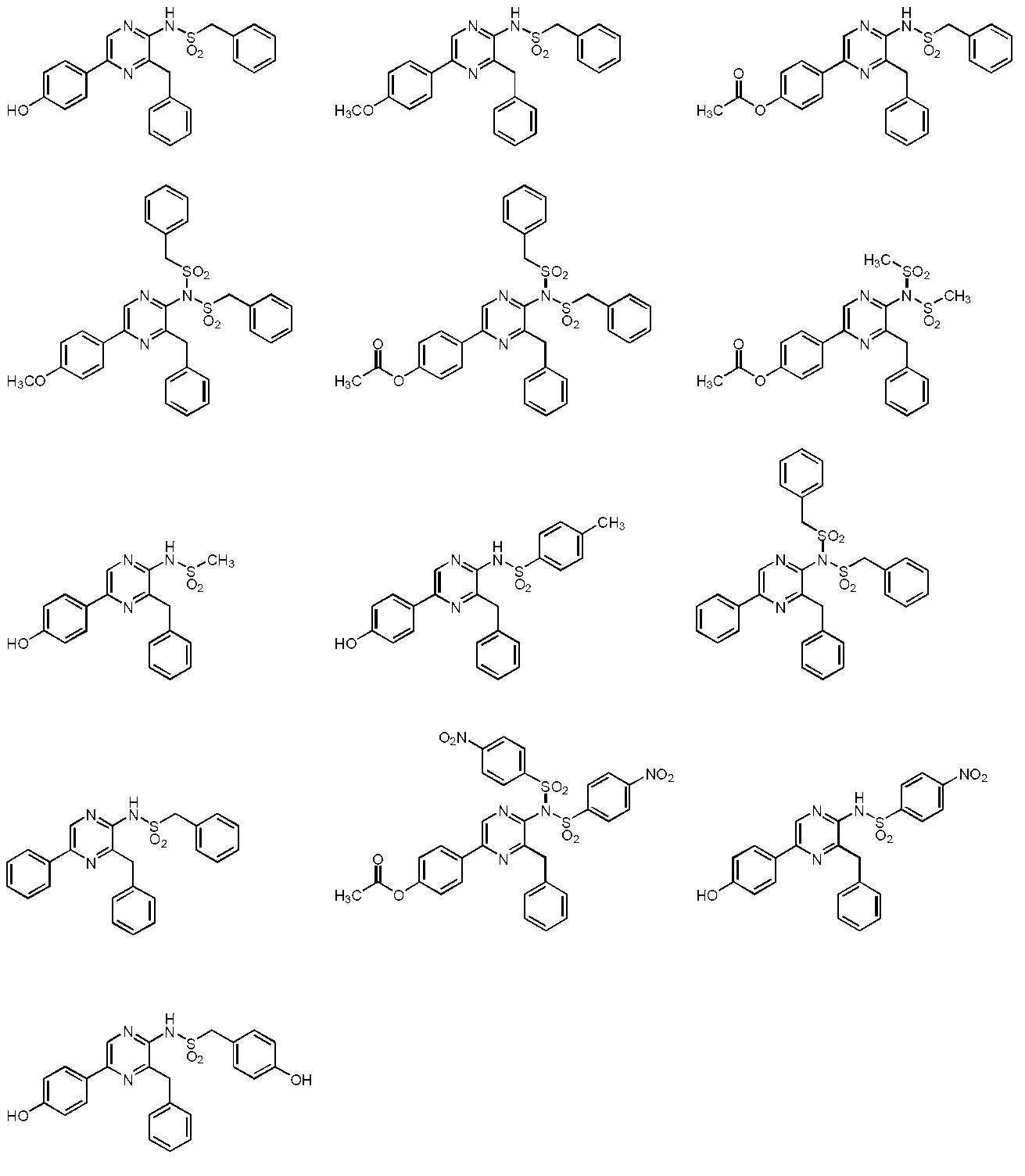
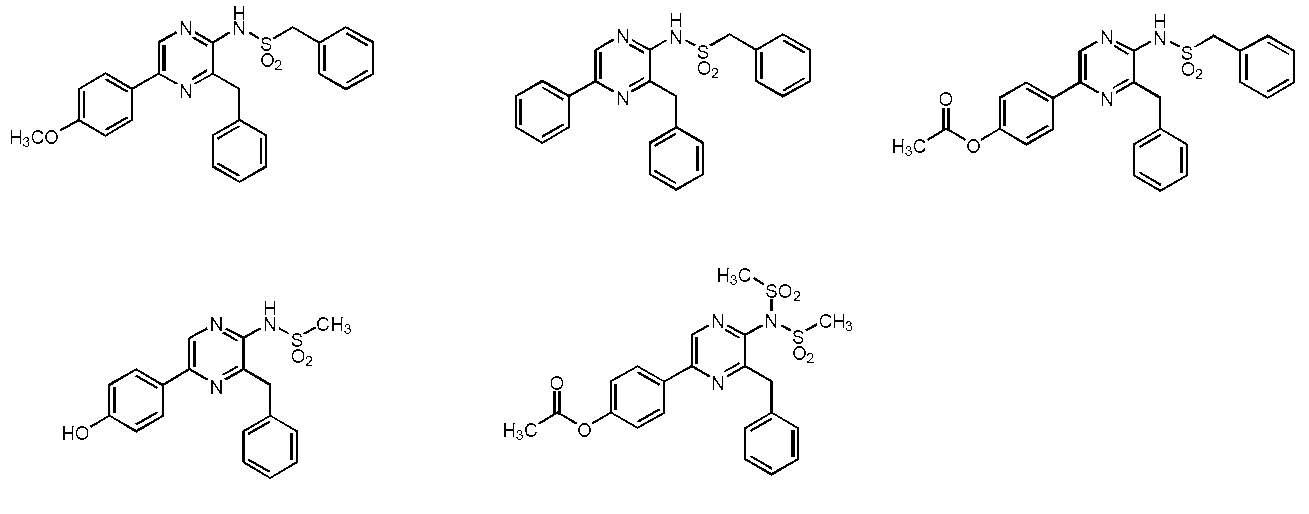
- the compound represented by the general formula (2) is a compound selected from the group consisting of the following compounds.
- the compound represented by the general formula (2) is a compound selected from the group consisting of the following compounds.
- the compound represented by the general formula (2) is a compound selected from the group consisting of the following compounds.
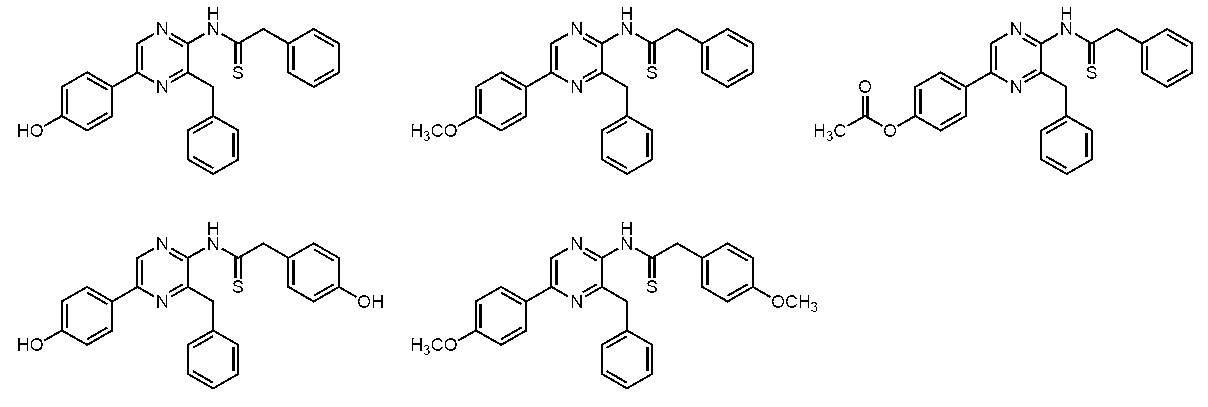
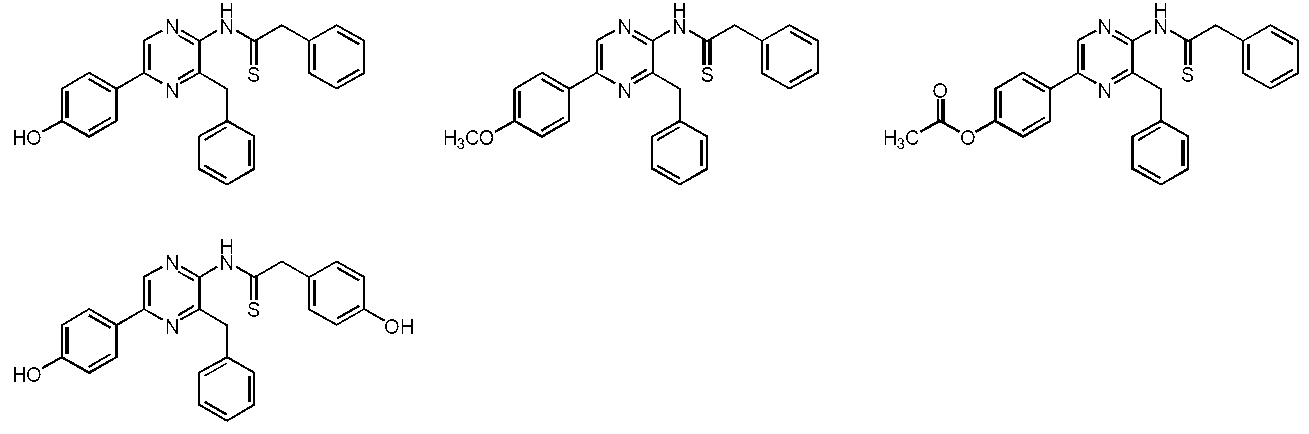
- the compound represented by the general formula (1) is a compound represented by the following general formula (3) (the thioamide coelenteramide of the present invention).
- the compound represented by the general formula (3) is a compound selected from the group consisting of compounds represented by the following formula.
- the compound represented by the general formula (3) is a compound selected from the group consisting of compounds represented by the following formula.
- the sulfonamide coelenteramide represented by the general formula (2) is, for example, General formula (4) (Wherein R 3 is as defined above), General formula (5) Can be produced by reacting a compound represented by the formula (wherein R 1 is as defined above).
- the compound represented by the general formula (4) can be produced by a known production method.
- the compound represented by the general formula (4) is Kishi, Y. et al., Tetrahedron Lett., 13, 2747-2748 (1972), or Adamczyk, M. et al., Org. Prep. Proced. Int. ., 33, 477-485 (2001) or a method analogous thereto.
- the compound represented by the general formula (4) can be produced as follows.
- a cyclization reaction between a substituted phenylglyoxal aldoxime and a glycinonitrile derivative is performed using a Lewis acid catalyst such as titanium tetrachloride to form pyrazine oxide, and then catalytic hydrogen using Raney Ni or the like as a catalyst. It can be produced by reduction, or by carrying out a Suzuki-Miyaura coupling reaction between a 2-amino-5-bromopyrazine derivative and a substituted phenylboric acid or a substituted phenylboric acid pinacol ester.
- the compound represented by the general formula (5) can also be produced by a known production method, or a commercially available product can be obtained. Specifically, for example, 1) excess thionyl chloride is allowed to act on the corresponding substituted benzyl sulfonic acid or a salt thereof and heated under reflux and then concentrated under reduced pressure, or 2) the corresponding substituted benzyl sulfonic acid or The salt is subjected to oxalyl dichloride for the corresponding carboxylic acid in the presence of a catalytic amount of N, N-dimethylformamide (DMF) in a solvent such as dichloromethane, and then concentrated under reduced pressure, or 3)
- the substituted benzyl Grignard reagent can be produced by reacting sulfuryl chloride with any method or a method analogous thereto.
- Benzylsulfonyl chloride can be purchased from Tokyo Chemical Industry Co., Ltd., Wako Pure Chemical Industries, Ltd., Kanto Chemical Co., Ltd.
- the solvent used in the method for producing the compound represented by the general formula (2) is not particularly limited as long as it is other than aqueous or alcohols, and various solvents can be used.
- pyridine, dichloromethane, chloroform, acetonitrile, tetrahydrofuran, ethyl acetate, acetone, toluene, dioxane, ether, and the like can be used alone or as a mixture.
- the reaction temperature and the reaction time are not particularly limited, but are, for example, ⁇ 20 ° C. to 200 ° C., 0.25 hour to 72 hours, preferably ⁇ At 20 ° C. to 100 ° C., 0.5 hour to 36 hours, more preferably at 0 ° C. to 50 ° C., for 1 hour to 24 hours.
- the thioamide type coelenteramide represented by the general formula (3) is, for example, General formula (6) (Wherein R 1 , R 2 , and R 3 are as defined above) are reacted with Lawesson's reagent or diphosphorus pentasulfide (phosphorus tetrasulfide). be able to.
- the compound represented by the general formula (6) can be produced by a known production method. Specifically, for example, the compound represented by the general formula (4) and the acid halide represented by the general formula (7) or an analog thereof in an organic solvent, in the presence of a base, or in a basic organic solvent. It can be produced by a reaction method or a method analogous thereto.
- R 1 is as defined above, and X is halogen (eg, fluorine, chlorine, bromine, or iodine), or R 1 C ( ⁇ O) —).
- halogen eg, fluorine, chlorine, bromine, or iodine
- the solvent used in the method for producing the compound represented by the general formula (3) is not particularly limited as long as it is other than water-based or alcohols, ketones, and esters, and various solvents can be used.
- toluene, benzene, dioxane, tetrahydrofuran, ether, dichloromethane, chloroform, pyridine and the like can be used alone or in combination.
- the reaction temperature and the reaction time are not particularly limited.
- the reaction temperature is 0 to 200 ° C., 0.5 to 72 hours, preferably room temperature to At 200 ° C. for 1 hour to 48 hours, more preferably at 60 ° C. to 150 ° C. for 2 hours to 24 hours.
- BFP blue fluorescent protein
- gFP green fluorescent protein
- the blue fluorescent protein (BFP) of the present invention is a complex in which the coelenteramide analog of the present invention is coordinated to the apoprotein of the calcium-binding photoprotein. That is, the BFP of the present invention contains the coelenteramide analog of the present invention, the apoprotein of the calcium-binding photoprotein, and divalent or trivalent ions that can be substituted for calcium ions or calcium ions. BFP can generate fluorescence by being excited by light, and can also emit light by contacting BFP with coelenterazine or an analog thereof.
- BFP is produced from the coelenteramide analog of the present invention as follows. That is, in the presence of calcium ions or divalent or trivalent ions that can be substituted for calcium ions, the coelenteramide analog of the present invention (for example, a compound represented by the general formula (1)), an apoprotein of a calcium-binding photoprotein BFP is produced by reacting with.
- the coelenteramide analog of the present invention for example, a compound represented by the general formula (1)
- an apoprotein of a calcium-binding photoprotein BFP is produced by reacting with.
- the coelenteramide analog of the present invention used to produce BFP in the present invention is as described above.
- Examples of the coelenteramide analog of the present invention include compounds produced by the above production method.
- the divalent or trivalent ions that can be substituted for the calcium ions used to produce BFP are divalent or trivalent ions that cause a luminescent reaction when reacted with a calcium-binding photoprotein instead of calcium ions. It is a trivalent ion. That is, it acts on calcium-binding photoproteins in the same manner as calcium ions.
- Examples of calcium ions or divalent or trivalent ions that can be substituted for calcium ions include calcium ions (Ca 2+ ), magnesium ions (Mg 2+ ), strontium ions (Sr 2+ ), barium ions (Ba 2+ ), lead Ion (Pb 2+ ), Cobalt ion (Co 2+ ), Nickel ion (Ni 2+ ), Cadmium ion (Cd 2+ ), Yttrium ion (Y 3+ ), Lanthanum ion (La 3+ ), Samarium ion (Sm 3+ ), Europium ion (Eu 3+ ), dysprosium ion (Dy 3+ ), thulium ion (Tm 3+ ), yttrium ion (Yb 3+ ), and the like can be given.
- divalent metal ions are preferred. More preferred are divalent metal ions other than transition metals, such as Ca 2+ , Sr 2+ , or Pb
- the apoprotein of the calcium-binding photoprotein used to produce BFP in the present invention is, for example, apoaequorin, apocritin-I, apocritin-II, apooberin, apomytrocomin, apomineopsin, or apovelvoin is there.
- the apoprotein is apoaequorin, apocrytin-I, apocritin-II, apomytrocomin, etc., for example, apoaequorin.
- the apoprotein may be collected from nature or may be produced by genetic engineering. Furthermore, as long as the apoprotein can form BFP, its amino acid sequence may be mutated from a natural one by a gene recombination technique.
- the base sequence and amino acid sequence of an apoprotein (natural apoprotein) of a photoprotein collected from nature are as follows. That is, the base sequence of natural apoaequorin is shown in SEQ ID NO: 1, and the amino acid sequence is shown in SEQ ID NO: 2.
- the nucleotide sequence of natural apocritin-I is shown in SEQ ID NO: 3, and the amino acid sequence is shown in SEQ ID NO: 4.
- the nucleotide sequence of natural apocritin-II is shown in SEQ ID NO: 5, and the amino acid sequence is shown in SEQ ID NO: 6.
- the base sequence of natural apomitrocomine is shown in SEQ ID NO: 7, and the amino acid sequence is shown in SEQ ID NO: 8.
- the base sequence of natural apooverin is shown in SEQ ID NO: 9, and the amino acid sequence is shown in SEQ ID NO: 10.
- the base sequence of natural apovelvoin is shown in SEQ ID NO: 11, and the amino acid sequence is shown in SEQ ID NO: 12.
- the apoprotein mutated by the recombinant technique is, for example, a protein selected from the group consisting of the following (a) to (c).
- A a protein comprising an amino acid sequence in which one to a plurality of amino acids are deleted, substituted, inserted and / or added in the amino acid sequence of a natural apoprotein, and having an apoprotein activity or function of a calcium-binding photoprotein
- B a protein comprising an amino acid sequence having 90% or more identity to the amino acid sequence of a natural apoprotein and having an apoprotein activity or function of a calcium-binding photoprotein
- a natural apoprotein It consists of an amino acid sequence encoded by a polynucleotide that hybridizes under stringent conditions with a polynucleotide comprising a base sequence complementary to the protein base sequence, and has an apoprotein activity or function of a calcium-binding photoprotein protein.
- the above-mentioned “natural type apoprotein” is, for example, apoaequorin, apocrytin-I, apocrytin-II, apoovelin, apomytrocomin, apomineopsin, or apovelvoin.
- the apoprotein is apoaequorin, apocrytin-I, apocritin-II, apomaitrocomin, or the like, and preferably apoaequorin.
- the amino acid sequences or base sequences of these natural apoproteins are as described above.
- apoprotein activity or function of calcium-binding photoprotein means, for example, the activity or function of forming a calcium-binding photoprotein by binding a protein with a peroxide of coelenterazine or a peroxide of coelenterazine analog.
- a protein binds to a peroxide of coelenterazine or a peroxide of coelenterazine to form a calcium-binding photoprotein” specifically means that (1) the protein binds to a peroxide of coelenterazine or a peroxide of coelenterazine analog.
- a photoprotein containing a protein and a peroxide of coelenterazine or a coelenterazine analog by contacting the coelenterazine or its derivative in the presence of oxygen. It also means forming a (complex).
- contact means that a protein and coelenterazine or an analog thereof are present in the same reaction system, for example, adding a protein to a container containing coelenterazine or an analog thereof, For example, adding coelenterazine or an analog thereof to the container accommodated, or mixing the protein with coelenterazine or an analog thereof.
- coelenterazine analog refers to a compound that can constitute a calcium-binding photoprotein such as aequorin as an apoprotein, like coelenterazine.
- Coelenterazine or its analogs include, for example, coelenterazine, h-coelenterazine, f-coelenterazine, cl-coelenterazine, n-coelenterazine, cp-coelenterazine, ch-coelenterazine, hch-coelenterazine, fch-coelenterazine, ef-coelenterazine Ech-coelenterazine or hcp-coelenterazine. Methods for obtaining these coelenterazines or their analogs will be described later.
- amino acid sequence in which one or more amino acids are deleted, substituted, inserted and / or added is, for example, 1 to 20, 1 to 15, 1 to 10 1 to 9, 1 to 8, 1 to 7, 1 to 6 (1 to several), 1 to 5, 1 to 4, 1 to 3, 1 to 2, 1 is there.
- the smaller the number of amino acids deleted, substituted, inserted or added the better.
- Two or more of the amino acid residue deletions, substitutions, insertions and additions may occur simultaneously.
- Such areas include “Sambrook J. et al., Molecular Cloning: A Laboratory Manual, Third Edition, Cold Spring Harbor Laboratory Press (2001) '',“ Ausbel F. M.
- the range of “90% or more” in the “amino acid sequence having 90% or more identity” is, for example, 90% or more, 91% or more, 92% or more, 93% or more, 94% or more, 95% or more. 96% or more, 97% or more, 98% or more, 99% or more, 99.1% or more, 99.2% or more, 99.3% or more, 99.4% or more, 99.5% or more, 99.6 % Or more, 99.7% or more, 99.8% or more, or 99.9% or more.
- identity of amino acid sequences and base sequences can be determined using an analysis program such as BLAST (for example, see Altzshul S. F. et al., J. Mol. Biol. 215, 403 (1990)). . When using BLAST, the default parameters of each program are used.
- polynucleotide hybridizing under stringent conditions refers to a polynucleotide consisting of a base sequence complementary to the base sequence of the natural apoprotein or a polynucleotide encoding the amino acid sequence of the natural apoprotein.
- a polynucleotide obtained by using a colony hybridization method, a plaque hybridization method, a Southern hybridization method, or the like using all or part of the probe as a probe. Specifically, hybridization was performed at 65 ° C.
- the “stringent conditions” referred to here may be any of low stringent conditions, medium stringent conditions, and high stringent conditions.
- Low stringent conditions are, for example, conditions of 5 ⁇ SSC, 5 ⁇ Denhardt's solution, 0.5% (w / v) SDS, 50% (v / v) formamide, 32 ° C.
- the “medium stringent conditions” are, for example, conditions of 5 ⁇ SSC, 5 ⁇ Denhardt's solution, 0.5% (w / v) SDS, 50% (v / v) formamide, 42 ° C.
- “High stringent conditions” are, for example, conditions of 5 ⁇ SSC, 5 ⁇ Denhardt's solution, 0.5 (w / v)% SDS, 50% (v / v) formamide, 50 ° C.
- a polynucleotide eg, DNA
- multiple factors such as temperature, probe concentration, probe length, ionic strength, time, and salt concentration can be considered as factors that affect hybridization stringency. Those skilled in the art will select these factors as appropriate. It is possible to achieve similar stringency.
- Alkphos Direct Labeling Reagents manufactured by Amersham Pharmacia
- Alkphos Direct Labeling Reagents can be used, for example.
- follow the protocol attached to the kit incubate with the labeled probe overnight, and then wash the membrane with primary wash buffer containing 0.1% (w / v) SDS at 55 ° C. , Hybridized DNA can be detected.
- the polynucleotide encoding the amino acid sequence of the apoprotein is about 60% or more, 65% or more, 70% or more, 75% or more, 80% or more, 85% or more, 88% or more, 90% or more, 92% or more, 95% or more, 97% or more, 98% or more, 99% or more, 99.3% or more, 99.5%
- DNA having 99.7% or more, 99.8% or more, or 99.9% or more of identity can be mentioned.
- the identity of amino acid sequences and base sequences can be determined using the method described above.
- recombinant apoprotein that can be used in the present invention, for example, recombinant aequorin described in Shimomura, O. and Inouye, S.Protein Express. Purif. (1999) 16: 91-95, Inouye, S. and Sahara, Y. Protein Express. Purif. (2007) 53: ⁇ ⁇ ⁇ Recombinant Critin I described in 384-389, or Inouye, S. J. Biochem. (2008) 143: 711-717 Tin-II can be mentioned.
- the apoprotein has all cysteine residues replaced with serine residues.
- BFP loses chemiluminescence activity when the free SH group of a cysteine residue in an apoprotein is oxidized to produce an SS bond. Therefore, an apoprotein in which a cysteine residue is substituted with a serine residue and cannot generate an SS bond hardly loses chemiluminescence activity and does not generate an SS bond, so that the activity continues.
- the amount of the coelenteramide analog of the present invention used for the production of BFP is not particularly limited, but for example, 1 mol to 5 mol, preferably 1 mol to 2 mol, more preferably 1 mol to 1.2 mol with respect to 1 mol of apoprotein. It is.
- the reaction of the coelenteramide analog of the present invention with the apoprotein and calcium ions or divalent or trivalent ions that can be substituted for calcium ions is preferably carried out in the presence of a reducing agent.
- a reducing agent examples include dithiothreitol (DTT) and mercaptoethanol.
- DTT dithiothreitol
- mercaptoethanol examples include dithiothreitol (DTT) and mercaptoethanol.
- DTT dithiothreitol
- mercaptoethanol mercaptoethanol
- the amount of reducing agent used for the production of BFP is not particularly limited as long as it does not affect the regeneration of BFP, but apoaequorin has an S—S bond formation due to the presence of three cysteine residues.
- the concentration to prevent is preferred.
- the final concentration is 1 mM dithiothreitol or 0.1% (v / v) mercaptoethanol.
- the reaction temperature and reaction time in the production of BFP are not particularly limited.
- the reaction temperature is 0 ° C. to 42 ° C. for 0.1 hour to 2 hours, 4 ° C. to 37 ° C. for 0.1 hour to 2 hours, or 4 ° C. to 15 ° C. 0.1 to 24 hours.
- the BFP thus obtained may be further subjected to purification.
- Purification of BFP can be performed according to a normal separation / purification method. Separation / purification methods include, for example, ammonium sulfate precipitation, gel filtration chromatography, ion exchange chromatography, affinity chromatography, reverse phase high performance liquid chromatography, dialysis, ultrafiltration, etc., alone or in appropriate combination. be able to.
- BFP blue fluorescent protein
- the BFP of the present invention acts on a luminescent substrate to cause it to emit light, and thus can be utilized as a luminescent catalyst. Accordingly, the present invention provides a light emitting method including bringing coelenterazine or an analog thereof into contact with the BFP of the present invention.
- contact means that BFP and coelenterazine or an analogue thereof are present in the same reaction system, for example, adding BFP to a container containing coelenterazine or an analogue thereof, For example, adding coelenterazine or an analog thereof to the container accommodated, or mixing BFP and coelenterazine or an analog thereof.
- the luminescent substrate used in the luminescent method of the present invention is, for example, coelenterazine or an analog thereof.
- coelenterazine analog refers to a compound that can constitute a calcium-binding photoprotein such as aequorin as an apoprotein, like coelenterazine.
- Coelenterazine or its analog used as a luminescent substrate is, for example, coelenterazine, h-coelenterazine, f-coelenterazine, cl-coelenterazine, n-coelenterazine, cp-coelenterazine, ch-coelenterazine, hch-coelenterazine, fch-coelenterazine, e-coelenterazine , Ef-coelenterazine, ech-coelenterazine, or hcp-coelenterazine, and preferably coelenterazine, h-coelenterazine, or e-coelenterazine.
- Light emission occurs.
- the normal light emission time is 0.5 to 3 hours, but the light emission time can be made longer or the light emission time can be made shorter depending on the selection of conditions.
- the BFP of the present invention can also be used as a reporter protein for measuring transcriptional activity of a promoter or the like.
- a vector is constructed in which a polynucleotide encoding an apoprotein is fused to a target promoter or other expression control sequence (for example, an enhancer).
- the vector is introduced into a host cell, and the coelenteramide analog of the present invention and calcium ion or divalent or trivalent ion replaceable with calcium ion are brought into contact therewith, and fluorescence derived from the fluorescent protein of the present invention By detecting this, the activity of the target promoter or other expression control sequence can be measured.
- contact means that a host cell, a coelenteramide analog, and calcium ions or divalent or trivalent ions that can be substituted for calcium ions are present in the same culture system / reaction system. Adding a coelenteramide analog and calcium ions or divalent or trivalent ions that can be replaced with calcium ions to a host cell culture vessel; host cell or coelenteramide analog and divalent or replacing calcium ions or calcium ions Alternatively, mixing with trivalent ions, culturing host cells in the presence of coelenteramide analogs and calcium ions or divalent or trivalent ions that can replace calcium ions, and the like are included.
- the BFP of the present invention can be used as a detection marker by fluorescence.
- the detection marker of the present invention can be used for detecting a target substance in, for example, an immunoassay or a hybridization assay.
- the BFP of the present invention can be used by binding to a target substance (protein or nucleic acid) by a commonly used method such as a chemical modification method. A detection method using such a detection marker can be performed by a normal method.
- the detection marker of the present invention is expressed, for example, as a fusion protein of an apoprotein and a target substance, introduced into a cell by a technique such as a microinjection method, and the coelenteramide analog and calcium ion of the present invention. Alternatively, it can also be used to measure the distribution of the target substance by contacting divalent or trivalent ions that can be replaced with calcium ions.
- contact means that cells, coelenteramide analogs and calcium ions or divalent or trivalent ions that can be substituted for calcium ions are present in the same culture system / reaction system, for example, Adding a coelenteramide analog and calcium ions or divalent or trivalent ions replaceable with calcium ions to a cell culture vessel; divalent or trivalent replaceable cells, coelenteramide analog and calcium ions or calcium ions; And culturing host cells in the presence of coelenteramide analogs and calcium ions or divalent or trivalent ions that can be substituted for calcium ions.
- Such distribution of the target substance can also be measured using a detection method such as fluorescence imaging.
- the apoprotein can be expressed in the cell and used in addition to being introduced into the cell by a technique such as a microinjection method.
- the BFP of the present invention generates fluorescence upon receiving light excitation. Therefore, the BFP of the present invention can be suitably used as a fluorescent base material for amusement goods. Examples of amusement goods include fluorescent soap bubbles, fluorescent ice, fluorescent glaze, fluorescent paints, and the like.
- the amusement product of the present invention can be manufactured by a usual method.
- the green fluorescent protein (gFP) of the present invention is a complex in which the coelenteramide analog of the present invention is coordinated to the apoprotein of the calcium-binding photoprotein. That is, the gFP of the present invention includes the coelenteramide analog of the present invention and the apoprotein of the calcium-binding photoprotein. gFP can generate fluorescence upon excitation of light.
- the gFP of the present invention can be obtained by removing calcium ions or divalent or trivalent ions replaceable with calcium ions from the aforementioned BFP.
- BFP is treated with a chelating agent for removing calcium ions or divalent or trivalent ions capable of substituting with calcium ions. Can be removed from BFP.
- the chelating agent used to produce gFP in the present invention is not particularly limited as long as it is strongly bound to calcium ions or divalent or trivalent ions that can be substituted for calcium ions.
- chelating agents include ethylenediaminetetraacetic acid (EDTA), ethylene glycol bis ( ⁇ -aminoethyl ether) N, N, N ′, N′-tetraacetic acid (EGTA), trans-1,2-diaminocyclohexane N, N , N ′, N′-tetraacetic acid (CyDTA), N- (2-hydroxyethyl) iminodiacetic acid (HIDA), and the like.
- the divalent or trivalent ions that can be substituted for calcium ions are as described above.
- the amount of chelating agent used for the production of gFP is not particularly limited as long as it does not affect gFP regeneration. Since it is shown that 3 mol of calcium ions bind to 1 mol of ionic apoaequorin, for example, 3 mol% or more is preferable.
- the reaction temperature and reaction time in the production of gFP are not particularly limited.
- the reaction temperature is 0 ° C. to 42 ° C. for 0.1 hour to 2 hours, 4 ° C. to 37 ° C. for 0.1 hour to 2 hours, or 4 ° C. to 15 ° C. 0.1 to 24 hours.
- the gFP thus obtained may be further subjected to purification.
- the purification of gFP can be performed according to a normal separation / purification method. Separation / purification methods include, for example, ammonium sulfate precipitation, gel filtration chromatography, ion exchange chromatography, affinity chromatography, reverse phase high performance liquid chromatography, dialysis, ultrafiltration, etc., alone or in appropriate combination. be able to.
- the gFP of the present invention can also be used as a reporter protein for measurement of transcriptional activity of a promoter or the like.
- a vector is constructed in which a polynucleotide encoding an apoprotein is fused to a target promoter or other expression control sequence (for example, an enhancer).
- the vector is introduced into a host cell, and the coelenteramide analog of the present invention is brought into contact therewith to produce BFP, and then calcium ions or divalent or trivalent ions that can be substituted with calcium ions are removed.
- the activity of the target promoter or other expression control sequence can be measured by generating gFP by contacting with a chelating agent for the purpose and detecting the fluorescence derived from the gFP of the present invention.
- the gFP of the present invention can be used as a detection marker by fluorescence.
- the detection marker of the present invention can be used for detecting a target substance in, for example, an immunoassay or a hybridization assay.
- the gFP of the present invention can be used by binding to a target substance (protein or nucleic acid) by a commonly used method such as a chemical modification method. A detection method using such a detection marker can be performed by a normal method.
- the detection marker of the present invention is expressed, for example, as a fusion protein of an apoprotein and a target substance, introduced into a cell by a technique such as a microinjection method, and further contacted with the coelenteramide analog of the present invention.
- Distribution of the target substance by, for example, generating BFP by contacting with a chelating agent for removing calcium ions or divalent or trivalent ions that can be substituted for calcium ions, etc. Can also be used to measure. Such measurement of the distribution of the target substance can also be performed using a detection method such as fluorescence imaging.
- the apoprotein can be expressed in the cell and used in addition to being introduced into the cell by a technique such as a microinjection method.
- amusement article material The gFP of the present invention can be suitably used as a fluorescent material for amusement article materials.
- Examples of amusement goods include fluorescent soap bubbles, fluorescent ice, fluorescent glaze, fluorescent paints, and the like.
- the amusement product of the present invention can be manufactured by a usual method.
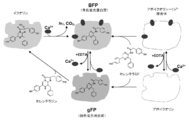
- Calcium-binding photoprotein As shown in FIG. 1, calcium-binding photoproteins such as aequorin are present in the presence of a chelating agent for removing calcium ions such as EDTA or divalent or trivalent ions that can be substituted for calcium ions. Below, it can manufacture by making coelenterazine or its analog react with gFP.
- the calcium-binding photoprotein of the present invention can be produced from the gFP of the present invention. That is, the calcium-binding photoprotein of the present invention can be obtained by reacting gFP with coelenterazine, which is a luminescent substrate, or an analog thereof.
- the reaction between gFP and coelenterazine or its analog is performed by bringing gFP into contact with coelenterazine or its analog.
- Contact means that the gFP of the present invention and coelenterazine or an analog thereof are present in the same reaction system.
- the gFP of the present invention is added to a container containing coelenterazine or an analog thereof.
- Coelenterazine or its analogs used in the production of the calcium-binding photoprotein of the present invention include, for example, coelenterazine, h-coelenterazine, f-coelenterazine, cl-coelenterazine, n-coelenterazine, cp-coelenterazine, ch-coelenterazine, hch-coelenterazine , Fch-coelenterazine, e-coelenterazine, ef-coelenterazine, ech-coelenterazine, hcp-coelenterazine, etc., preferably coelenterazine, h-coelenterazine, or e-coelenterazine.
- the method for obtaining these coelenterazines or their analogs is as described above.
- the amount of coelenterazine or its analog used for the production of calcium-binding photoprotein is not particularly limited, but may be, for example, 1.2 mol or more with respect to 1 mol of gFP.
- the reaction temperature and reaction time in the production of the calcium-binding photoprotein are not particularly limited.
- the reaction temperature is 0 to 42 ° C. for 0.1 to 2 hours, 4 to 37 ° C. for 0.1 to 2 hours, or 4 to 4 ° C. It is 0.1 to 24 hours at 15 ° C.
- the reaction between the fluorescent protein and coelenterazine or its analog is preferably carried out in the presence of calcium ions or a chelating agent for removing divalent or trivalent ions that can be substituted for calcium ions.
- the chelating agent used for producing gFP in the present invention is the same as described above.
- the reaction between the fluorescent protein and coelenterazine or an analog thereof is performed in the presence of a reducing agent.
- a reducing agent used at this time is, for example, dithiothreitol (DTT) or mercaptoethanol.
- DTT dithiothreitol
- the amount of the reducing agent used for the production of the calcium-binding photoprotein is not particularly limited as long as it does not affect the regeneration. However, since apoaequorin has three cysteine residues, it has an SS bond. A concentration that prevents formation is preferred. For example, the final concentration is 1 mM dithiothreitol or 0.1% (v / v) mercaptoethanol.
- the calcium-binding photoprotein of the present invention is a photoprotein (holoprotein) that emits light by the action of calcium ions. Therefore, the photoprotein of the present invention can be used for detection or quantification of calcium ions.
- a photoprotein composed of apoprotein and a peroxide of coelenterazine analog is used.
- the photoprotein can be produced according to the method described above. Detection or quantification of calcium ions can be performed by directly adding the sample solution to the photoprotein solution and measuring the generated luminescence. Alternatively, calcium ions can be detected or quantified by adding a photoprotein solution to the sample solution and measuring the generated luminescence.
- the detection or quantification of calcium ions can be performed by measuring the luminescence of the photoprotein of the present invention by calcium ions using a luminescence measuring device.
- a luminescence measuring device a commercially available device such as Centro® LB® 960 (manufactured by Bertrand) can be used. Quantification of the calcium ion concentration can be measured by creating a luminescence standard curve for a known calcium ion concentration using a photoprotein.
- BRET Bioluminescence Resonance Energy Transfer
- the photoprotein of the present invention when used as a donor protein and an organic compound or a fluorescent protein is used as an acceptor, the interaction between the proteins is detected by causing bioluminescence resonance energy transfer (BRET) between them. be able to.
- the organic compound used as an acceptor is Hoechist3342, Indo-1 or DAP1.
- the fluorescent protein used as an acceptor is green fluorescent protein (GFP), blue fluorescent protein (BFP), mutant GFP fluorescent protein, phycobilin, or the like.
- the physiological function to be analyzed is orphan receptor (especially G protein-coupled receptor), apoptosis, or transcriptional regulation by gene expression.
- the enzyme to be analyzed is a protease, esterase or phosphorylase.
- Analysis of physiological functions by the BRET method can be performed by a known method, for example, the method described in Biochem. J. 2005, 385, 625-637, or Expert ⁇ Opin. Ther Tarets, 2007 11: 541-556 It can be performed according to.
- the enzyme activity can also be measured by a known method, for example, according to the method described in Nat Methods 2006, 3: 165-174, or Biotechnol J. 2008, 3: 311-324. Can do.
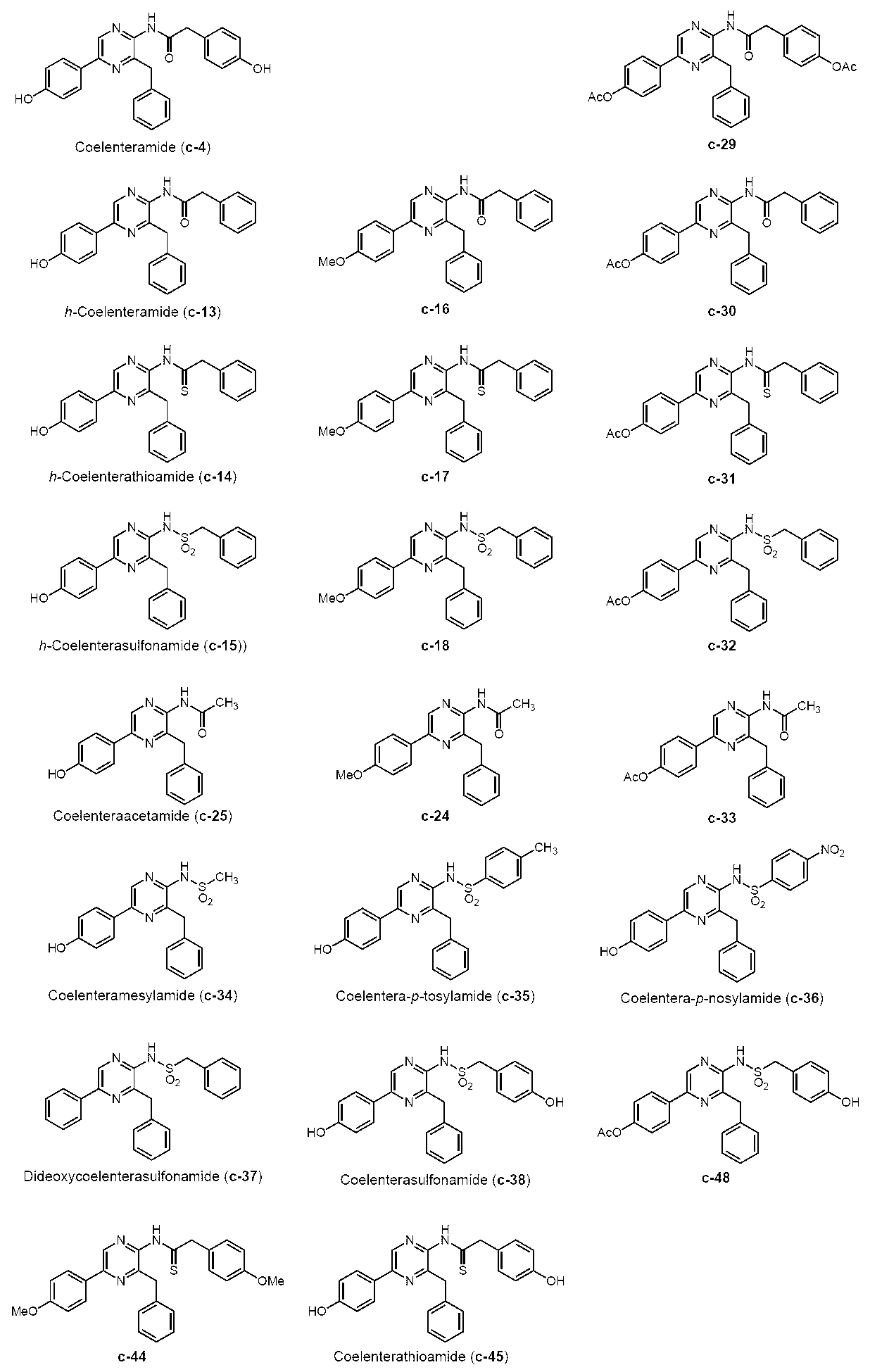
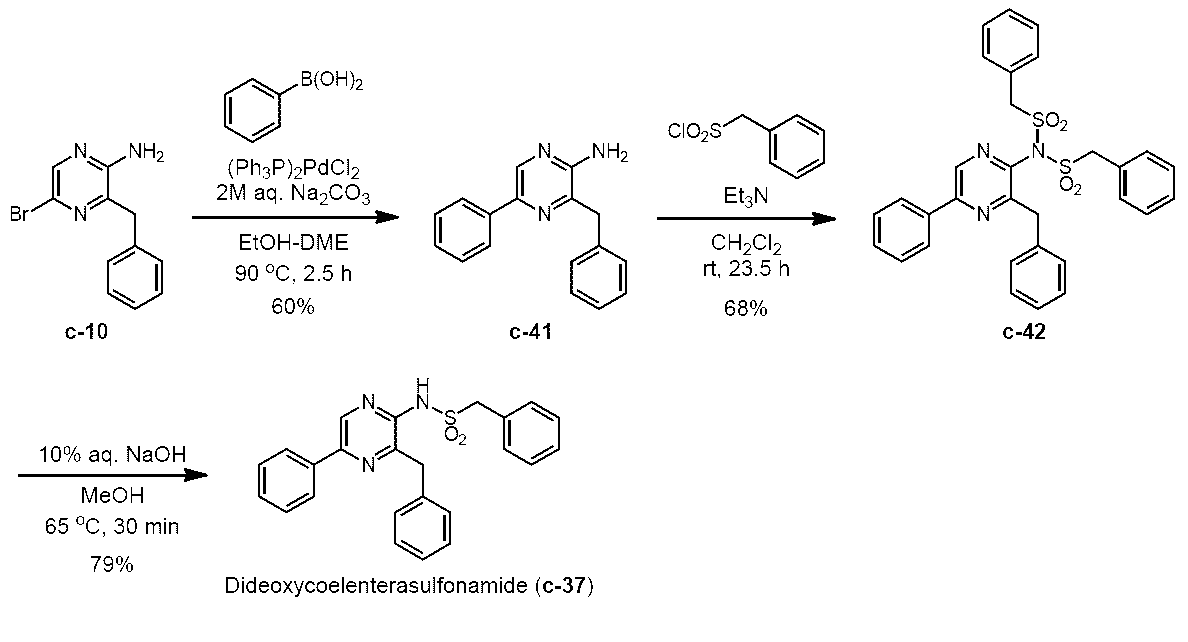
- CTMD derivatives were synthesized by the following synthesis method based on h-coelenteratiothioamide, h-coelentersulfonamide, and h-coelenteramide.
- the fluorescence ability of the synthetic compound was evaluated by determining the solubility in an aqueous solvent and the fluorescence quantum yield in an aqueous solvent or an organic solvent.
- Phenylacetyl chloride (Wako), 4- (dimethylamino) pyridine (Wako), 1.0 M boron tribromide / dichloromethane solution (Aldrich), Lawson's reagent (Aldrich), benzylsulfonyl chloride (Wako), acetic anhydride (Wako) , Triethylamine (Wako), sodium hydroxide (Wako), dehydrated pyridine (Wako), acetyl chloride (Wako), methanesulfonyl chloride (Wako), p-toluenesulfonyl chloride (Kanto), p-nitrobenzenesulfonyl chloride (Aldrich), Phenylboronic acid (Acros organics), sodium carbonate (Wako), and dichlorobis (triphenylphosphine) palladium (II) (Aldrich).
- IR infrared spectrometer spectrum
- HRMS high resolution mass spectrometry
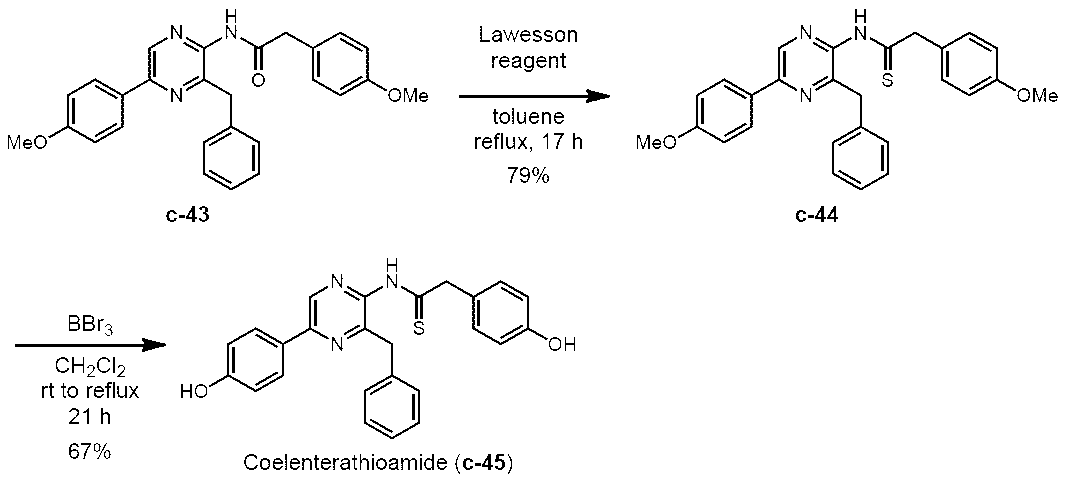
- 3-benzyl-5- (4-methoxyphenyl) -2- (phenylacetylamino) pyrazine (c-16) (539 mg, 1.32 mmol) was suspended in dehydrated toluene (50 mL) at room temperature. Lawesson reagent (320 mg, 790 ⁇ mol) was added to this while stirring, and the mixture was heated to reflux for 17 hours. After refluxing, the mixture was allowed to cool to room temperature and concentrated under reduced pressure using a rotary evaporator.
- a saturated aqueous sodium hydrogen carbonate solution and ethyl acetate were added thereto to stop the reaction, and the aqueous layer and the organic layer were separated, followed by extraction with ethyl acetate three times.
- the organic layer was washed 3 times with water and once with saturated brine, and then dried over anhydrous sodium sulfate. After anhydrous sodium sulfate was removed by filtration, the filtrate was concentrated under reduced pressure using a rotary evaporator.
- IR (KBr, cm -1 ) 513, 596, 640, 654, 710, 746, 851, 910, 1016, 1136, 1167, 1198, 1217, 1373, 1423, 1452, 1466, 1493, 1508, 1535, 1630, 1746, 3148, 3289.
- triethylamine (145 ⁇ L, 1.04 mmol), dehydrated dichloromethane (1.0 mL), and benzylsulfonyl chloride (131 mg, 687 ⁇ mol) were sequentially added thereto, followed by stirring for 1 hour.
- triethylamine (145 ⁇ L, 1.04 mmol), dehydrated dichloromethane (1.0 mL), and benzylsulfonyl chloride (131 mg, 687 ⁇ mol) were sequentially added thereto, followed by stirring for 2 hours.
- a saturated aqueous sodium hydrogen carbonate solution was added thereto to stop the reaction, and the mixture was extracted twice with dichloromethane.
- the organic layer was washed once with water and once with a saturated aqueous solution of sodium sulfate, and then dried using anhydrous sodium sulfate.
- Anhydrous sodium sulfate was removed by filtration, and the filtrate was concentrated under reduced pressure using a rotary evaporator.
- 2-amino-3-benzyl-5-phenylpyrazine (c-41) (852 mg, 3.26 mmol) was dissolved in dehydrated dichloromethane (25 mL), and triethylamine (1.38 mL, 9.87 mmol) was added thereto. In addition, it was cooled to 0 ° C. Benzylsulfonyl chloride (1.87 g, 9.80 mmol) was added thereto, and the mixture was warmed to room temperature and stirred for 23.5 hours.
- Example 1 The fluorescence quantum yield of each synthesized CTMD analog was determined by the following procedure. The fluorescence spectrum was measured at 25 ° C. using an FP-6500 fluorescence spectrophotometer from Jasco (JASCO Corporation, Tokyo). Specifically, a quartz cell (optical path length 10 mm) is used, and the CTMD analog is dissolved in methanol (MeOH) or 67 mM phosphate buffer solution (PB) (pH 7.4) so that the final concentration is 750 nM.
- MeOH methanol
- PB 67 mM phosphate buffer solution
- Measurement was performed three times at a wavelength of 330 nm, emission / excitation bandwidth: 3 nm, response: 0.5 seconds, scan speed: 100 nm / min, and the average spectrum was defined as a compound fluorescence spectrum.
- the fluorescence quantum yield was determined after spectral correction.
- Quinine sulfate was used as a standard control solution for fluorescence quantum yield. After quinine sulfate (Wako Pure Chemical Industries) was dissolved in a 0.1 N aqueous sulfuric acid solution, the excitation light was measured at 366 nm, under the above-described fluorescence measurement conditions. The relative fluorescence quantum yield (fluorescence intensity) of the compound was calculated by setting the quantum yield of quinine sulfate to 0.55.
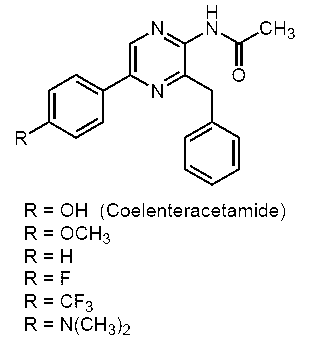
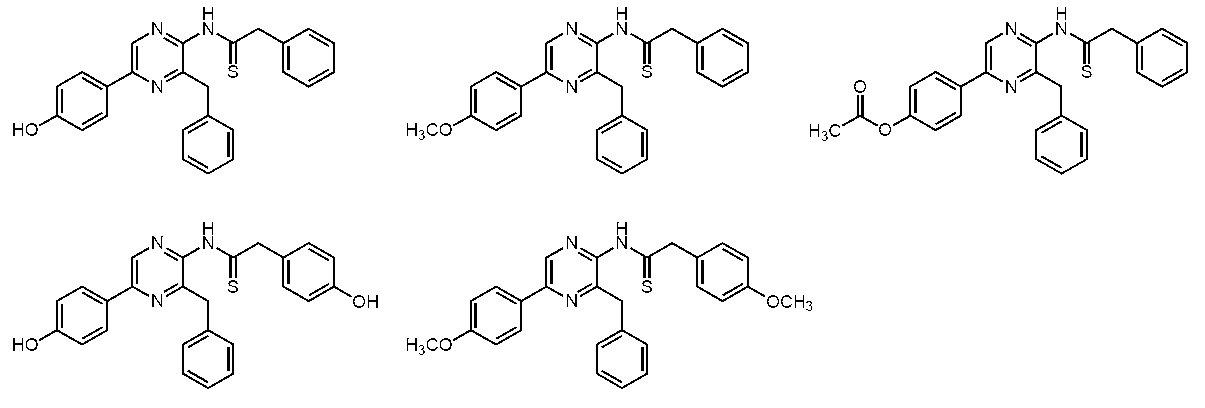
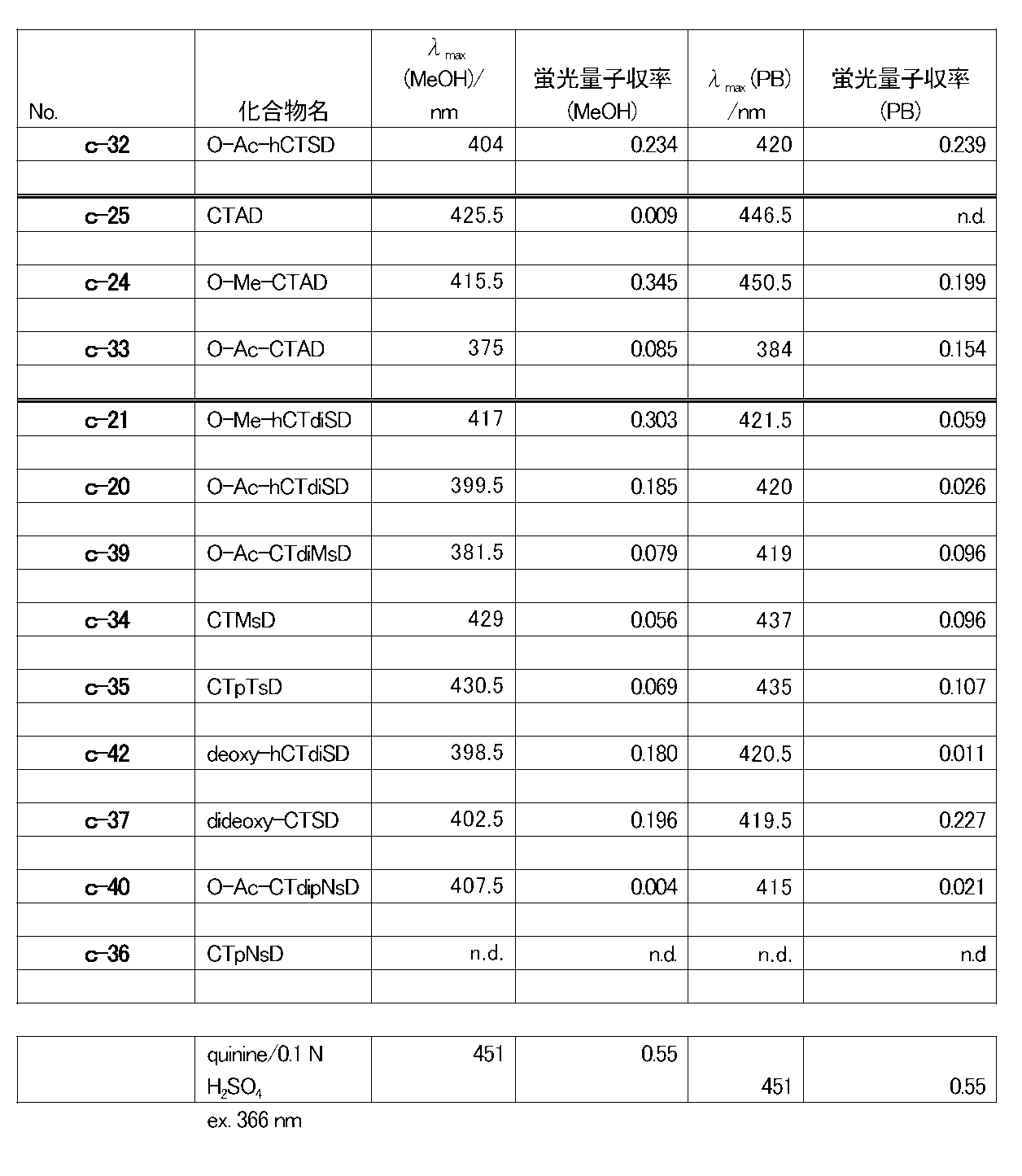
- the coelenteramide analogs of the present invention (c-14, c-17, c-31, c-15, c-18, c-32, c-21, c-20, c-39, c- 34, c-35, c-42, c-37, c-40, or c-36) have different fluorescence characteristics from known CTMD (c-4), h-CTMD (c-13), etc. I understood.
- c-15, c-18, c-32, c-21, c-20, c-39, c-34, c-35, c-42, or c- No. 37 has a fluorescence quantum yield of 0.090 or more in an organic solvent or an aqueous solvent, and was found to have strong fluorescence ability.
- c-15, c-18, c-32, c-21, c-20, c-35, c-42, or c-37 has a fluorescence quantum yield of 0.100 in an organic or aqueous solvent. This is the above, and it has been found that it has particularly strong fluorescence ability.
- fluorescent compounds are known to have a significantly reduced fluorescence ability in an aqueous solution even if they have a strong fluorescence ability in an organic solvent.
- c-18, c-32, or c- As for 37 it was found that the fluorescence quantum yield was 0.100 or more in an organic solvent or an aqueous solvent, and particularly strong fluorescence ability was maintained.
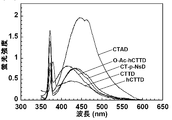
- Example 2 The fluorescence quantum yield obtained in Example 1 is a numerical value calculated by measuring the fluorescence spectrum with the final concentration of the coelenteramide analog being 750 nM, but when the final concentration is 750 nM as in Example 1, Among the coelenteramide analogs of the present invention, c-14, c-31, and c-36 cannot detect fluorescence spectra in methanol and phosphate buffer, and c-25 is in phosphate buffer. The fluorescence spectrum could not be detected with (nd: not detectable).
- FIG. 3 also shows the fluorescence spectrum of c-45 (CTTD) in 50 mM Tris-HCl (pH 7.6) containing 10 mM CaCl 2 .
- the fluorescence spectrum data of c-4, c-14, c-31, c-25 and c-36 are summarized in Table 2.
- the coelenteramide analog of the present invention containing c-14, c-31, and c-36 has fluorescence ability in an organic solvent and an aqueous solution. Further, it was confirmed that c-45 has fluorescence ability at least in an aqueous solution.
- c-44 CTTD
- c-17 hCTTD is considered to have fluorescence ability similar to c-17 because c-17 (hCTTD) has fluorescence ability.
- the coelenteramide analog of the preferred embodiment of the present invention has fluorescence ability in both an organic solvent and an aqueous solution, and thus can be applied to a wide range of applications such as bioassay and in vivo molecular imaging. It is.
- Example 3 For c-38, the fluorescence spectrum was measured at 25 ° C. using an FP-6500 fluorescence spectrophotometer from Jasco (JASCO Corporation, Tokyo). Specifically, using a quartz cell (optical path length 10 mm), c-38 was dissolved in 50 mM Tris-HCl (pH 7.6) containing 10 mM CaCl 2 so that the final concentration was 18 ⁇ M, and the excitation wavelength was 330 The measurement was carried out three times at nm, emission / excitation bandwidth: 3 nm, response: 0.5 seconds, scan speed: 100 nm / min, and the average spectrum was taken as the fluorescence spectrum.
- FIG. 4 shows the fluorescence spectrum of c-38 (CTSD) with 50 mM Tris-HCl (pH 7.6) containing 10 mM CaCl 2 . From FIG. 4, it was found that c-38 has fluorescence ability.
- [Sequence Listing Free Text] [SEQ ID NO: 1] This is the base sequence of natural apoaequorin.
- SEQ ID NO: 2] is the amino acid sequence of natural apoaequorin.
- SEQ ID NO: 3 This is the base sequence of natural apocrytin-I.
- SEQ ID NO: 4] is an amino acid sequence of natural apocrytin-I.
- SEQ ID NO: 5 This is the base sequence of natural apocrytin-II.
- SEQ ID NO: 6] is the amino acid sequence of natural apocrytin-II.
- SEQ ID NO: 7] is the base sequence of natural apomytrocomin.
- [SEQ ID NO: 8] is an amino acid sequence of natural apomytrocomin.
- [SEQ ID NO: 9] This is the base sequence of natural apooverin.
- [SEQ ID NO: 10] is the amino acid sequence of natural apooverin.
- [SEQ ID NO: 11] This is the base sequence of natural apovelvoin.
- [SEQ ID NO: 12] is an amino acid sequence of natural apovelvoin.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Peptides Or Proteins (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description









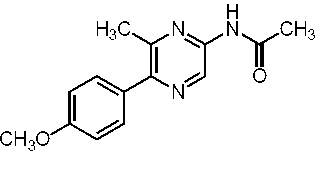
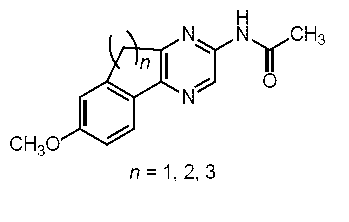
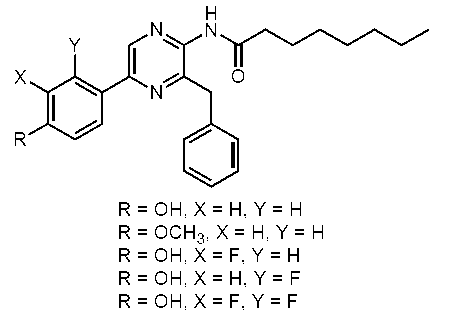


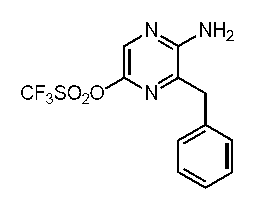

すなわち、本発明は、以下に示す、セレンテラミド類縁体、及び蛍光蛋白質などを提供する。

で表わされる化合物
(式中、
R1は、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、脂肪族環式基によって置換されていてもよい直鎖若しくは分枝鎖のアルキル、脂肪族環式基、又は複素環式基であり、
R2は、水素、又は-(SO2)R4であり、
R3は、水素、水酸基、メトキシ、又はアセトキシであり、
R4は、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、又は脂肪族環式基によって置換されていてもよい直鎖若しくは分枝鎖のアルキルであり、
X1は、-C(=S)-、又は-SO2-である。)。
(2)一般式(1)において、
R1は、フェニル、p-メチルフェニル、p-ヒドロキシフェニル、p-メトキシフェニル、p-アセトキシフェニル、p-ニトロフェニル、ベンジル、α-ヒドロキシベンジル、4-メチルベンジル、4-ヒドロキシベンジル、4-メトキシベンジル、4-アセトキシベンジル、4-ニトロベンジル、フェニルエチル、メチル、エチル、プロピル、2-メチルプロピル、2-メチルプロパニル、シクロヘキシルメチル、シクロヘキシルエチル、アダマンチルメチル、シクロペンチルメチル、シクロヘキシル、又はチオフェン-2-イルであること、
を特徴とする、上記(1)に記載の化合物。
(3)一般式(1)において、
R2は、水素、ベンゼンスルホニル、p-トルエンスルホニル、4-ヒドロキシフェニルスルホニル、4-メトキシフェニルスルホニル、4-アセトキシフェニルスルホニル、4-ニトロフェニルスルホニル、ベンジルスルホニル、α-ヒドロキシベンジルスルホニル、4-メチルベンジルスルホニル、4-ヒドロキシベンジルスルホニル、4-メトキシベンジルスルホニル、4-アセトキシベンジルスルホニル、4-ニトロベンジルスルホニル、フェニルエチルスルホニル、メタンスルホニル、エチルスルホニル、プロピルスルホニル、2-メチルプロピルスルホニル、2-メチルプロパニルスルホニル、シクロヘキシルメチルスルホニル、シクロヘキシルエチルスルホニル、アダマンチルメチルスルホニル、又はシクロペンチルメチルスルホニルであること、
を特徴とする、上記(1)又は(2)に記載の化合物。
(4)下記化合物

からなる群から選択される、上記(1)~(3)のいずれか1項に記載の化合物。
(5)下記化合物
からなる群から選択される、上記(1)~(3)のいずれか1項に記載の化合物。
(5a)下記化合物

からなる群から選択される、上記(1)~(3)のいずれか1項に記載の化合物。
(6)上記(1)~(5a)のいずれか1項に記載の化合物、カルシウム結合型発光蛋白質のアポ蛋白質、及びカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを含む、青色蛍光蛋白質。
(7)カルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンの存在下、上記(1)~(5a)のいずれか1項に記載の化合物と、カルシウム結合型発光蛋白質のアポ蛋白質とを反応させることを含む、青色蛍光蛋白質の製造方法。
(8)前記反応を、還元剤の存在下において行う、上記(7)に記載の方法。
(9)上記(1)~(5a)のいずれか1項に記載の化合物、及びカルシウム結合型発光蛋白質のアポ蛋白質を含む、緑色蛍光蛋白質。
(10)上記(6)に記載の青色蛍光蛋白質を、カルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを除去するためのキレート剤で処理することを含む、緑色蛍光蛋白質の製造方法。
(11)上記(9)に記載の緑色蛍光蛋白質に、セレンテラジン又はその類縁体を反応させることを含む、カルシウム結合型発光蛋白質の製造方法。
(12)蛍光蛋白質とセレンテラジン又はその類縁体との反応を、還元剤の存在下において行う、上記(11)に記載の方法。
1.セレンテラミド類縁体
本発明は、下記一般式(1)で表わされる化合物(本発明のセレンテラミド類縁体)を提供する。

(式中、
R1は、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、脂肪族環式基によって置換されていてもよい直鎖若しくは分枝鎖のアルキル、脂肪族環式基、又は複素環式基であり、
R2は、水素、又は-(SO2)R4であり、
R3は、水素、水酸基、メトキシ、又はアセトキシであり、
R4は、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、又は脂肪族環式基によって置換されていてもよい直鎖若しくは分枝鎖のアルキルであり、
X1は、-C(=S)-、又は-SO2-である。)。
(式中、R1、R2、及びR3は、前記の通りである。)
(式中、R1、R2、及びR3は、前記の通りである。)
2.1.スルホンアミド系セレンテラミド類縁体の製造方法
本発明のセレンテラミド類縁体のうち、下記一般式(2)で表わされるスルホンアミド系セレンテラミドの製造方法について説明する。

(式中、R1、R2、及びR3は、前記の通りである。)
一般式(4)

で表わされる化合物(式中、R3は、前記の通りである。)と、
一般式(5)

で表わされる化合物(式中、R1は、前記の通りである。)を反応させることにより、製造することができる。
本発明のセレンテラミド類縁体のうち、下記一般式(3)で表わされるスルホンアミド系セレンテラミドの製造方法について説明する。
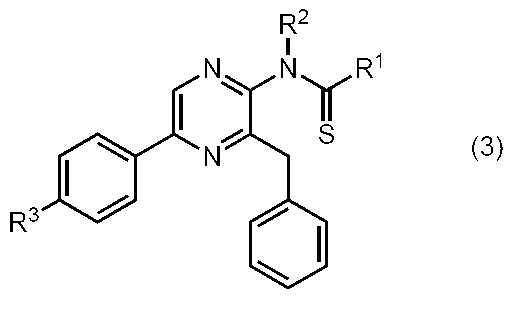
(式中、R1、R2、及びR3は、前記の通りである。)
一般式(6)

で表わされる化合物(式中、R1、R2、及びR3は、前記の通りである。)に、ローソン試薬、又は五硫化二リン(十硫化四リン)を反応させることにより、製造することができる。
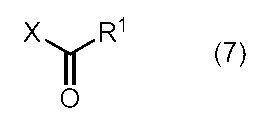
(式中、R1は前記の通りであり、Xはハロゲン(例えば、フッ素、塩素、臭素、又はヨウ素)、又はR1C(=O)-である。)
図1に示すように、青色蛍光蛋白質(BFP)は、セレンテラミド又はその類縁体に、アポイクオリン等のアポ蛋白質を反応させることにより製造することができる。一方、緑色蛍光蛋白質(gFP)は、BFPを、EDTA等のカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを除去するためのキレート剤で処理することにより製造することができる。
3.1.1.青色蛍光蛋白質(BFP)の製造方法
本発明の青色蛍光蛋白質(BFP)は、カルシウム結合型発光蛋白質のアポ蛋白質に、本発明のセレンテラミド類縁体が配位した複合体である。すなわち、本発明のBFPは、本発明のセレンテラミド類縁体、カルシウム結合型発光蛋白質のアポ蛋白質、及びカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを含む。BFPは、光の励起を受けて蛍光を発生することができるとともに、BFPとセレンテラジン又はその類縁体と接触させることで、発光を生じさせることもできる。
(a)天然型アポ蛋白質のアミノ酸配列において1~複数個のアミノ酸が欠失、置換、挿入及び/又は付加したアミノ酸配列からなり、かつ、カルシウム結合型発光蛋白質のアポ蛋白質活性若しくは機能を有する蛋白質、
(b)天然型アポ蛋白質のアミノ酸配列に対して90%以上の同一性を有するアミノ酸配列からなり、かつ、カルシウム結合型発光蛋白質のアポ蛋白質活性若しくは機能を有する蛋白質、及び
(c)天然型アポ蛋白質の塩基配列に相補的な塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズするポリヌクレオチドによってコードされるアミノ酸配列からなり、かつ、カルシウム結合型発光蛋白質のアポ蛋白質活性若しくは機能を有する蛋白質。
ハイブリダイゼーションは、Sambrook J. et al., Molecular Cloning: A Laboratory Manual, Third Edition, Cold Spring Harbor Laboratory Press (2001)、Ausbel F. M. et al., Current Protocols in Molecular Biology, Supplement 1~38, John Wiley and Sons (1987-1997)、又はGlover D. M. and Hames B. D., DNA Cloning 1: Core Techniques, A practical Approach, Second Edition, Oxford University Press (1995)等の実験書に記載されている方法に準じて行うことができる。
(1)発光触媒としての利用
本発明のBFPは、発光基質に作用しそれを発光させるので、発光触媒として利用できる。そこで、本発明は、本発明のBFPに、セレンテラジン又はその類縁体を接触させることを含む、発光方法を提供する。ここで、「接触」とは、BFPとセレンテラジン又はその類縁体とを同一の反応系に存在させることを意味し、例えば、セレンテラジン又はその類縁体を収容した容器にBFPを添加すること、BFPを収容した容器にセレンテラジン又はその類縁体を添加すること、又はBFPとセレンテラジン又はその類縁体とを混合すること、などが含まれる。
本発明のBFPは、レポーター蛋白質としてプロモーターなどの転写活性の測定に利用することもできる。アポ蛋白質をコードするポリヌクレオチドを、目的のプロモーター又は他の発現制御配列(例えば、エンハンサーなど)に融合したベクターを構築する。前記ベクターを宿主細胞に導入し、さらに、これに、本発明のセレンテラミド類縁体及びカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを接触させ、本発明の蛍光蛋白質に由来する蛍光を検出することにより、目的のプロモーター又は他の発現制御配列の活性を測定することができる。ここで、「接触」とは、宿主細胞とセレンテラミド類縁体とカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンとを同一の培養系・反応系に存在させることを意味し、例えば、宿主細胞の培養容器にセレンテラミド類縁体及びカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを添加すること、宿主細胞とセレンテラミド類縁体とカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンとを混合すること、宿主細胞をセレンテラミド類縁体及びカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンの存在下で培養することなどが含まれる。
本発明のBFPは、蛍光による検出マーカとして利用することができる。本発明の検出マーカは、例えば、イムノアッセイ又はハイブリダイゼーションアッセイなどにおける目的物質の検出に利用することができる。本発明のBFPを化学修飾法など通常用いられる方法により目的物質(蛋白質或いは核酸など)と結合させて使用することができる。このような検出マーカを用いた検出方法は、通常の方法によって行うことができる。
本発明のBFPは、光の励起をうけて蛍光を生じる。よって、本発明のBFPは、アミューズメント用品の材料の蛍光基材として好適に使用することができる。アミューズメント用品としては、たとえば、蛍光シャボン玉、蛍光アイス、蛍光飴、蛍光絵の具等があげられる。本発明のアミューズメント用品は、通常の方法によって製造することができる。
3.2.1.緑色蛍光蛋白質(gFP)の製造
本発明の緑色蛍光蛋白質(gFP)は、カルシウム結合型発光蛋白質のアポ蛋白質に、本発明のセレンテラミド類縁体が配位した複合体である。すなわち、本発明のgFPは、本発明のセレンテラミド類縁体、及びカルシウム結合型発光蛋白質のアポ蛋白質を含む。gFPは、光の励起を受けて蛍光を発生することができる。
(1)レポーター蛋白質としての利用
本発明のgFPは、レポーター蛋白質としてプロモーターなどの転写活性の測定に利用することもできる。アポ蛋白質をコードするポリヌクレオチドを、目的のプロモーター又は他の発現制御配列(例えば、エンハンサーなど)に融合したベクターを構築する。前記ベクターを宿主細胞に導入し、さらに、これに、本発明のセレンテラミド類縁体を接触させてBFPを生成させた後、カルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを除去するためのキレート剤を接触させることでgFPを生成させ、本発明のgFPに由来する蛍光を検出することにより、目的のプロモーター又は他の発現制御配列の活性を測定することができる。
本発明のgFPは、蛍光による検出マーカとして利用することができる。本発明の検出マーカは、例えば、イムノアッセイ又はハイブリダイゼーションアッセイなどにおける目的物質の検出に利用することができる。本発明のgFPを化学修飾法など通常用いられる方法により目的物質(蛋白質或いは核酸など)と結合させて使用することができる。このような検出マーカを用いた検出方法は、通常の方法によって行うことができる。また、本発明の検出マーカは、例えば、アポ蛋白質と目的物質との融合蛋白質として発現させ、マイクロインジェクション法などの手法により細胞内に導入し、さらに、これに、本発明のセレンテラミド類縁体を接触させてBFPを生成させた後、カルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを除去するためのキレート剤を接触させることでgFPを生成させること等によって、前記目的物質の分布を測定するために利用することもできる。このような目的物質などの分布の測定は、蛍光イメージング等の検出法などを利用して行うこともできる。なお、アポ蛋白質は、マイクロインジェクション法などの手法により細胞内に導入する以外に、細胞内で発現させて用いることもできる。
本発明のgFPは、アミューズメント用品の材料の蛍光材として好適に使用することができる。アミューズメント用品としては、たとえば、蛍光シャボン玉、蛍光アイス、蛍光飴、蛍光絵の具等があげられる。本発明のアミューズメント用品は、通常の方法によって製造することができる。
図1に示すように、イクオリン等のカルシウム結合型発光蛋白質は、EDTA等のカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを除去するためのキレート剤の存在下、gFPに、セレンテラジン又はその類縁体を反応させることで、製造することが出来る。
本発明のカルシウム結合型発光蛋白質は、本発明のgFPから製造することができる。すなわち、本発明のカルシウム結合型発光蛋白質は、gFPに、発光基質であるセレンテラジン又はその類縁体を反応させることによって、得ることができる。
蛍光蛋白質とセレンテラジン又はその類縁体との反応を、カルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを除去するためのキレート剤の存在下において行うのが好ましい。本発明においてgFPを製造するのに用いるキレート剤は、前記と同様である。
(1)カルシウムイオンの検出又は定量
本発明のカルシウム結合型発光蛋白質は、カルシウムイオンの作用によって発光する発光蛋白質(ホロ蛋白質)である。よって、本発明の発光蛋白質は、カルシウムイオンの検出又は定量に使用することができる。
本発明のカルシウム結合型発光蛋白質は、生物発光共鳴エネルギー移動(BRET)法による分子間相互作用の原理を利用した生理機能の解析や酵素活性の測定等の分析方法に利用することができる。
CTMDの基本骨格をベースとして、h-セレンテラチオアミドとh-セレンテラスルホンアミドを、下記合成スキームにより合成した。また、比較として、h-セレンテラミドも合成した。


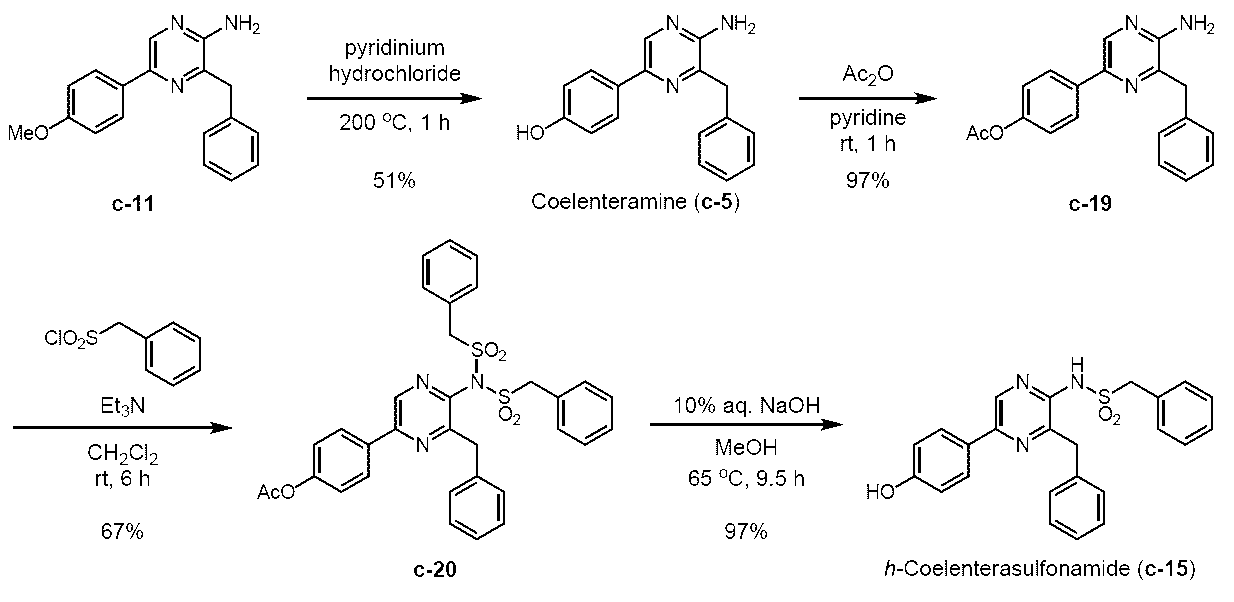





[材料及び方法]
(1)化学薬品
全ての化学薬品は特に記さない限り、市販のものをそのまま使用した。
反応、抽出、及びクロマトグラフィー用の溶媒には、酢酸エチル、n-ヘキサン、ジクロロメタン、脱水ジクロロメタン、クロロホルム、メタノール、エタノール、ジエチルエーテル、アセトン、トルエン、及び1,2-ジメトキシエタンの市販品(Wako)をそのまま用いた。
反応試薬は以下に挙げるものを使用した。
フェニルアセチルクロリド (Wako)、4-(ジメチルアミノ)ピリジン (Wako)、1.0 M 三臭化ホウ素/ジクロロメタン溶液 (Aldrich)、ローソン試薬 (Aldrich)、ベンジルスホニルクロリド (Wako)、無水酢酸 (Wako)、トリエチルアミン (Wako)、水酸化ナトリウム (Wako)、脱水ピリジン (Wako)、アセチルクロリド (Wako)、メタンスルホニルクロリド (Wako)、p-トルエンスルホニルクロリド (Kanto)、p-ニトロベンゼンスルホニルクロリド (Aldrich)、フェニルボロン酸 (Acros organics)、炭酸ナトリウム (Wako)、及びジクロロビス(トリフェニルホスフィン)パラジウム(II) (Aldrich)。
分析用薄層クロマトグラフィー(TLC)には、MERCK社製、Silicagel 60 F254(Cat. No. 1.05715.)を用いた。スポットの検出は、紫外線ランプ(254 nm又は365 nm)による方法、ヨウ素吸着、及びリンモリブデン酸の酸性水溶液に浸した後にホットプレートで焼くことで行った。分取用フラッシュカラムクロマトグラフィーには、順相シリカゲルとして関東化学社製、Silicagel 60N Cat. No. 37563-85、メッシュ40-50 μm、又は関東化学社製、Silicagel 60N Cat. No. 37565-84、メッシュ63-200 μmを使用した。なお、本実施例においてクロマトグラフィー用の混合溶媒の比率は、特に説明が無い限りv/v である。
融点(Mp.)は、YANACO社製、微量融点測定装置MP-J3を用いて測定した。
紫外吸収スペクトル(UV)及びOD値(330 nm)の測定には、日本分光社製、V-560を用いて25 ℃で測定した。各サンプルの30 μMメタノール溶液及びpH.7.4リン酸緩衝水溶液(PB)を調製し、石英セル(光路長10 mm)中で測定した。難溶性の化合物は、はじめに少量のDMSOに溶解し、それを各溶媒で希釈することでサンプル調製を行った。DMSOを用いた場合には、その含有割合を記載した。全ての測定は、バンド幅0.5 nm、レスポンスMedium、スキャン速度200 nm/minの条件で測定した。
1H(400 MHz)核磁気共鳴スペクトル(NMRスペクトル)は、Varian社製、Unity Plus 400を用いてDMSO-d6中で測定した。1H NMRの化学シフトの基準には、測定溶媒であるDMSO-d6中の残存非重水素化ジメチルスルホキシドのピークをδ2.49とした。
13C(75.5 MHz)核磁気共鳴スペクトル(NMRスペクトル)は、Varian社製、MERCURY 300を用いてDMSO-d6中で測定した。13C NMRの化学シフトの基準には、測定溶媒であるDMSO-d6のピークをδ 39.5とし、ピークが重複した化合物についてはピークを分離するために重メタノールを加え、その旨を記載した。
赤外分光計スペクトル(IR)は、DRS-8000を装着したSHIMADZU社製、IRPrestige-21 spectrophotometerを用いて、拡散反射法により測定した。
高分解能質量分析スペクトル(HRMS)は、JEOL社製JMS-700を用いて、電子衝撃イオン化(EI)法、又は高速原子衝突(FAB+)法、又はBrucker社製MicrOTOFを用いてエレクトロスプレーイオン化法(ESI+)により測定した。
元素分析は、YANACO社製CHN CORDER MT-5を用いて行った。
3-ベンジル-5-(4-メトキシフェニル)-2-(フェニルアセチルアミノ)ピラジン(c-16)
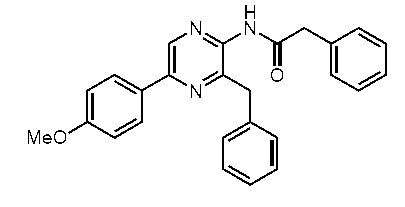
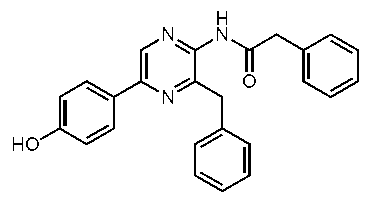
3-ベンジル-5-(4-ヒドロキシフェニル)-2-(フェニルアセチルアミノ)ピラジン(h-セレンテラミド) (c-13)
3-ベンジル-5-(4-メトキシフェニル) -2-(フェニルチオアセチルアミノ)ピラジン(c-17)

3-ベンジル-5-(4-ヒドロキシフェニル)-2-(フェニルチオアセチルアミノ)ピラジン(h-セレンテラチオアミド) (c-14)
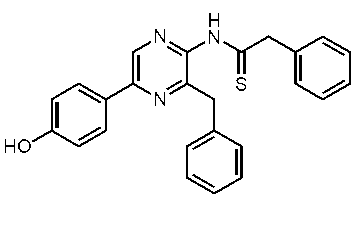
3-ベンジル-2-ベンジルスルホニルアミノ-5-(4-メトキシフェニル)ピラジン (c-18)
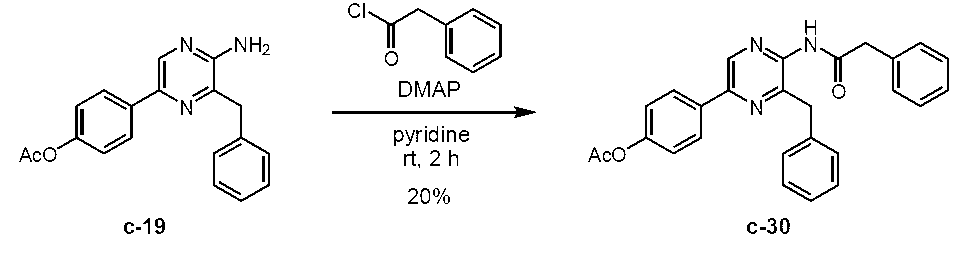
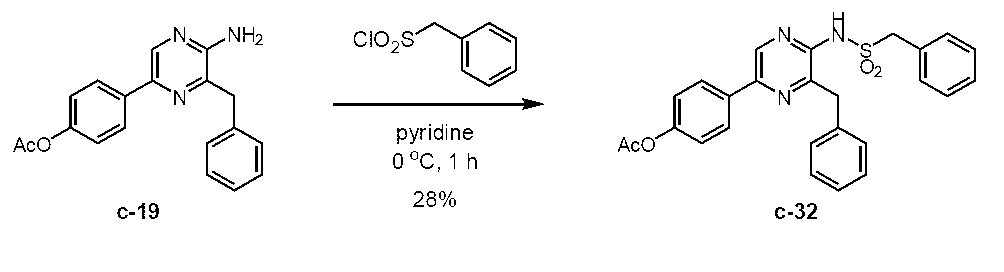
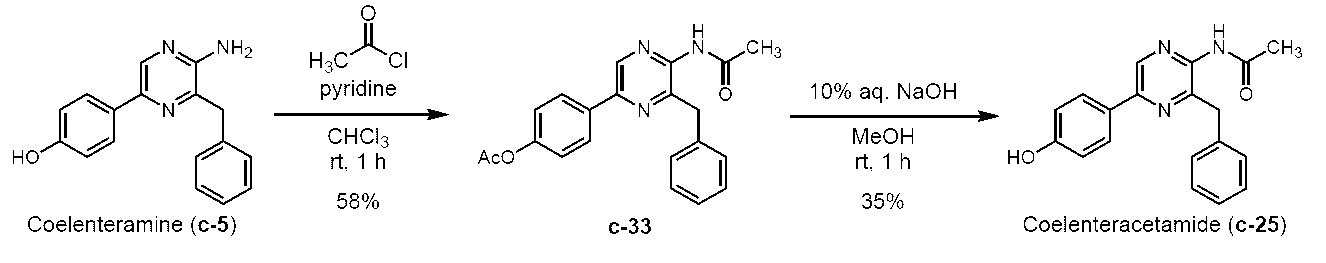
5-(4-アセトキシフェニル)-2-アミノ-3-ベンジルピラジン (c-19)

アルゴン雰囲気下、2-アミノ-3-ベンジル-5-(4-ヒドロキシフェニル)ピラジン (セレンテラミン) (c-5) (Adamczyk, M. et al., Org. Prep. Proced. Int., 33, 477-485 (2001)に記載の方法によって製造) (303 mg, 1.09 mmol)をピリジン (2 mL)に溶解し、0 ℃に冷却した。これに、無水酢酸 (133 μL, 1.40 mmol)を加え、室温に昇温後1時間撹拌した。これに、飽和炭酸水素ナトリウム水溶液と酢酸エチルを加え反応を停止し、水層と有機層を分離した後、酢酸エチルで3回抽出した。有機層を水で3回、飽和食塩水で1回洗浄した後、無水硫酸ナトリウムを用いて乾燥した。無水硫酸ナトリウムをろ過により除去した後、ろ液をロータリーエバポレーターで減圧濃縮した。残渣をカラムクロマトグラフィー (50 g, n-ヘキサン/酢酸エチル = 1/1)で精製し、5-(4-アセトキシフェニル)-2-アミノ-3-ベンジルピラジン(c-19)を淡黄色固体として得た (337 mg, 96.7%)。Rf = 0.31 (n-ヘキサン/酢酸エチル = 3/2)。Mp. 183.5-185.5 ℃。1H NMR (400 MHz, DMSO-d6) δ2.27 (s, 3H), 4.07 (s, 2H), 6.42 (s, 2H), 7.12-7.17 (AA’BB’, 2H), 7.19 (d, J = 7.2 Hz, 1H), 7.27 (t, J = 7.5 Hz, 2H), 7.33 (d, J = 7.0 Hz, 2H), 7.90-7.95 (AA’BB’, 2H), 8.41 (s, 1H)。IR (KBr, cm-1) 513, 596, 640, 654, 710, 746, 851, 910, 1016, 1136, 1167, 1198, 1217, 1373, 1423, 1452, 1466, 1493, 1508, 1535, 1630, 1746, 3148, 3289。
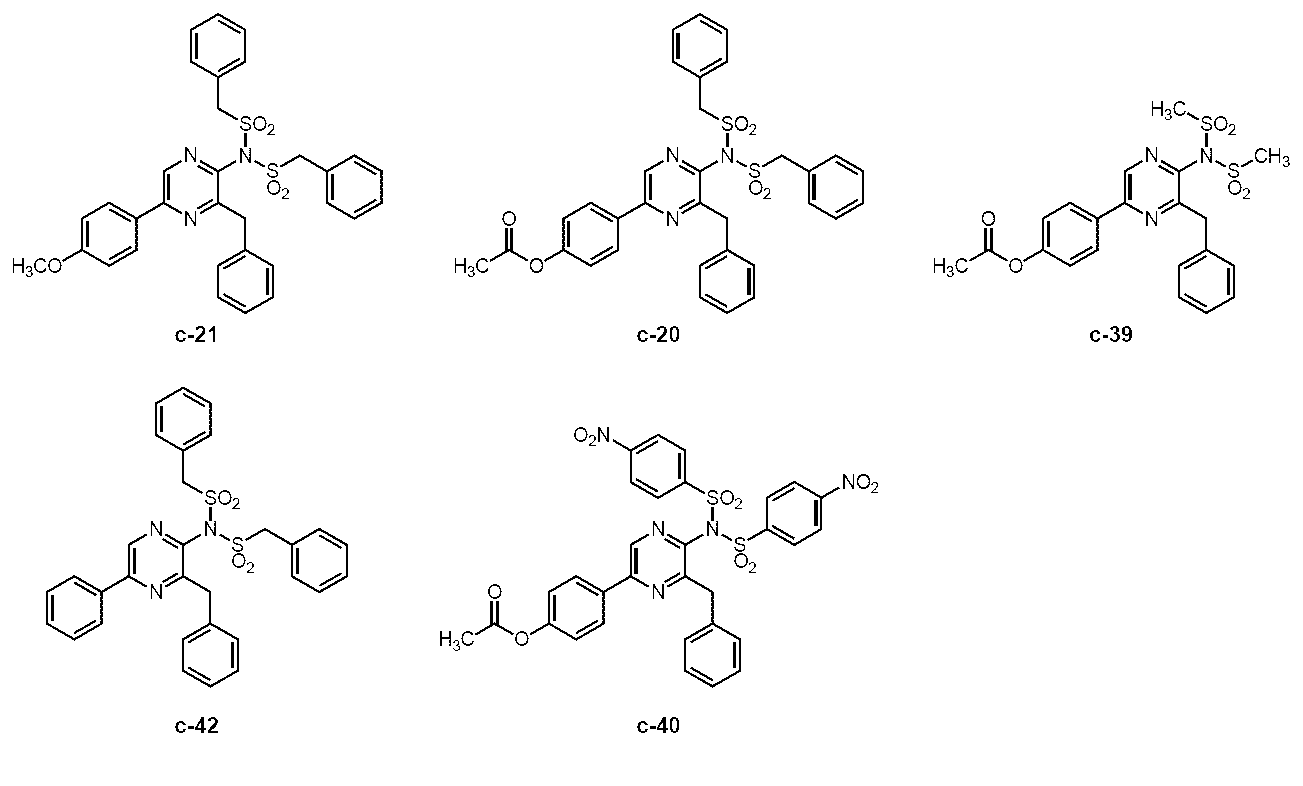
5-(4-アセトキシフェニル)-3-ベンジル-2-ビス(ベンジルスルホニル)アミノピラジン (c-20)

3-ベンジル-2-ベンジルスルホニルアミノ-5-(4-ヒドロキシフェニル)ピラジン (h-セレンテラスルホンアミド) (c-15)

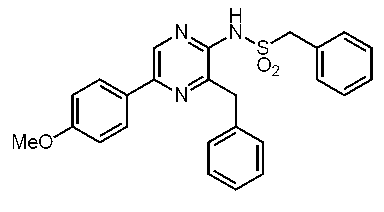
3-ベンジル-2-ビス(ベンジルスルホニル)アミノ-5-(4-メトキシフェニル)ピラジン (c-21)

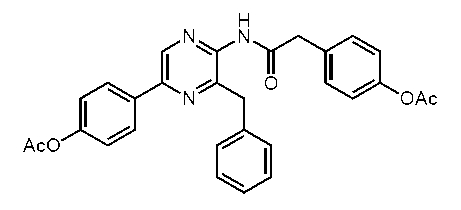
5-(4-アセトキシフェニル)-2-(4-アセトキシフェニル)アセチルアミノ-3-ベンジルピラジン (c-29)
5-(4-アセトキシフェニル)-3-ベンジル-2-(フェニルアセチルアミノ)ピラジン (c-30)
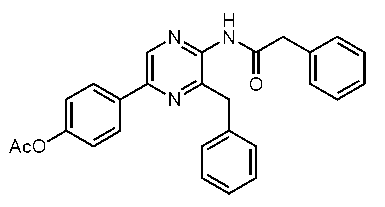
5-(4-アセトキシフェニル)-3-ベンジル-2-(フェニルチオアセチルアミノ)ピラジン (c-31)

5-(4-アセトキシフェニル)-3-ベンジル-2-(ベンジルスルホニルアミノ)ピラジン (c-32)
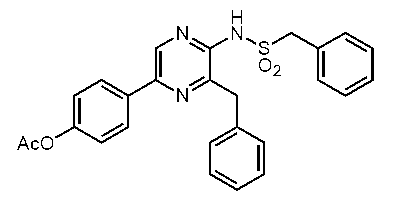
5-(4-アセトキシフェニル)-2-アセチルアミノ-3-ベンジルピラジン (c-33)
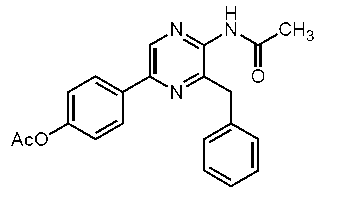
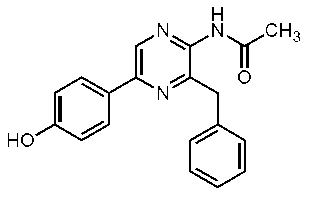
2-アセチルアミノ-3-ベンジル-5-(4-ヒドロキシフェニル)ピラジン(セレンテラアセトアミド) (c-25)
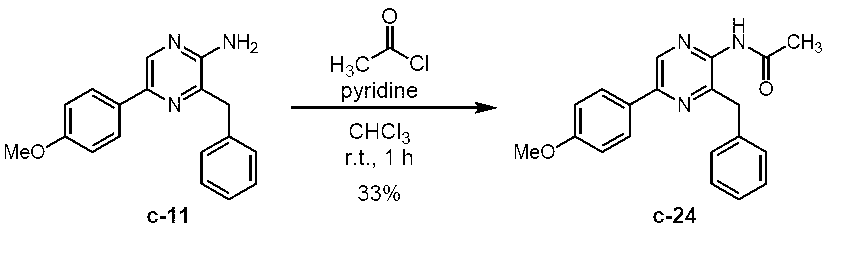
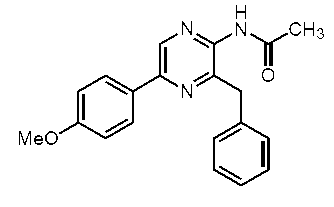
2-アセチルアミノ-3-ベンジル-5-(4-メトキシフェニル)ピラジン (c-24)
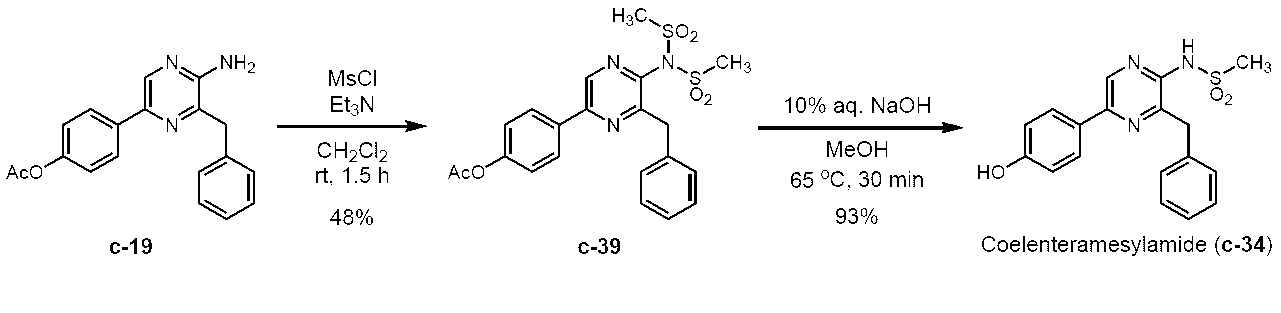
5-(4-アセトキシフェニル)-3-ベンジル-2-ビス(メタンスルホニル)アミノピラジン (c-39)

3-ベンジル-5-(4-ヒドロキシフェニル)2-(メタンスルホニルアミノ)ピラジン(セレンテラメシルアミド) (c-34)
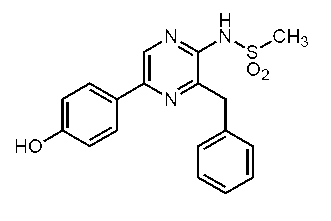
3-ベンジル-5-(4-ヒドロキシフェニル)-2-[(4-メチルフェニル)スルホニルアミノ]ピラジン(セレンテラ-p-トシルアミド) (c-35)
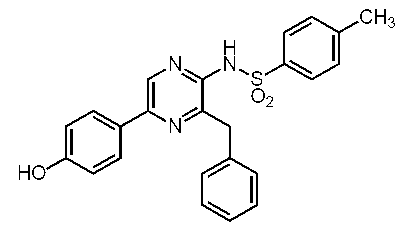
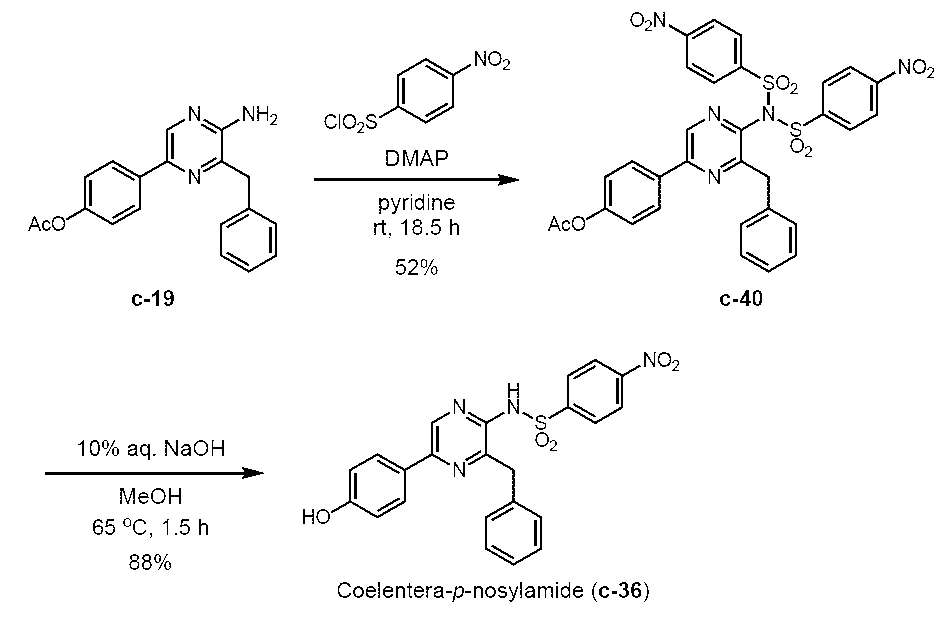
5-(4-アセトキシフェニル)-3-ベンジル-2-ビス(4-ニトロフェニルスルホニル)アミノピラジン(c-40)

3-ベンジル-5-(4-ヒドロキシフェニル)-2-[(4-ニトロフェニル)スルホニルアミノ]ピラジン(セレンテラ-p-ノシルアミド) (c-36)

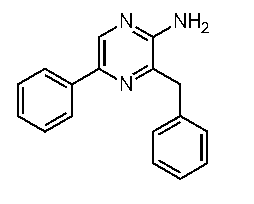
2-アミノ-3-ベンジル-5-フェニルピラジン (c-41)
アルゴン雰囲気下、2-アミノ-3-ベンジル-5-ブロモピラジン (c-10) (1.66 g, 6.27 mmol)を1,2-ジメトキシエタン(17 mL)、エタノール (13 mL)に溶解し、これに室温で撹拌しながら2 M 炭酸ナトリウム水溶液 (31.4 mL, 62.8 mmol)、ジクロロビス(トリフェニルホスフィン)パラジウム(II) (221 mg, 315 μmol)、フェニルボロン酸 (996 mg, 8.16 mmol)を順次加え、90 ℃で2.5時間撹拌した。室温まで放冷後、飽和食塩水と酢酸エチルを加え反応を停止し、有機層を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥した。無水硫酸ナトリウムをろ過することで除去し、ろ液をロータリーエバポレーターで減圧濃縮した。残渣をカラムクロマトグラフィー (50 g, n-ヘキサン/酢酸エチル = 3/2 → 1/1)で精製した後、得られた固体を酢酸エチルを用いて2回再結晶し、2-アミノ-3-ベンジル-5-フェニルピラジン (c-41)を黄色固体として得た(982 mg, 59.9%)。Rf = 0.30 (n-ヘキサン/酢酸エチル = 3/2)。1H NMR (400 MHz, DMSO-d6) δ 4.07 (s, 2H), 6.41 (s, 2H), 7.18 (t, J = 7.5 Hz, 1H), 7.24-7.31 (m, 3H), 7.34 (d, J = 7.3 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.90 (d, J = 7.8 Hz, 2H), 8.41 (s, 1H)。IR (KBr, cm-1) 596, 664, 694, 716, 733, 754, 773, 908, 937, 1070, 1152, 1217, 1233, 1396, 1427, 1450, 1462, 1493, 1543, 1636, 3024, 3125, 3291, 3487。HRMS (EI+) m/z 261.1269 (M, C17H15N3requires 261.1266)。
3-ベンジル-2-ビス(ベンジルスルホニル)アミノ-5-フェニルピラジン (c-42)

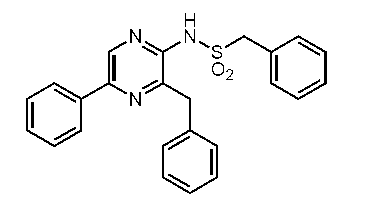
3-ベンジル-2-ベンジルスルホニルアミノ-5-フェニルピラジン(ジデオキシセレンテラスルホンアミド) (c-37)
3-ベンジル-5-(4-メトキシフェニル)-2-[(4-メトキシフェニル)チオアセチルアミノ]ピラジン (c-44)

3-ベンジル-5-(4-ヒドロキシフェニル)-2-[(4-ヒドロキシフェニル)チオアセチルアミノ]ピラジン(セレンテラチオアミド) (c-45)

5-(4-アセトキシフェニル)-3-ベンジル-2-ビス(4-ヨードベンジルスルホニル)アミノピラジン (c-46)
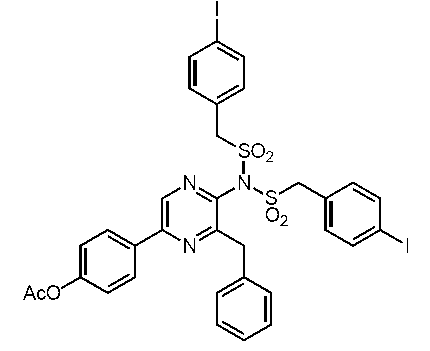
5-(4-アセトキシフェニル)-3-ベンジル-2-[(4-ヒドロキシベンジル)スルホニルアミノ]ピラジン (c-48)

上記で得られた混合物をアセトン(10 mL)に溶解し、これに室温にてoxone (247 mg, 401 μmol)を水(3 mL)に溶解したものを加え、そのまま室温で15分間攪拌した。これに飽和炭酸水素ナトリウム水溶液を加え反応を停止し、ジクロロメタンで3回抽出した。有機層を水で1回、飽和食塩水で1回洗浄した後、無水硫酸ナトリウムを用いて乾燥した。無水硫酸ナトリウムをろ過することで除去し、ろ液をロータリーエバポレーターで減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(50 g, n-ヘキサン/酢酸エチル = 2/1、および50 g, n-ヘキサン/酢酸エチル = 3/2)にて精製し、得られたを無色固体を(68.5 mg)を酢酸エチルに溶解し、n-ヘキサンを加え化合物を析出させた。ろ過により析出した固体を回収し、減圧下で乾燥することにより、5-(4-アセトキシフェニル)-3-ベンジル-2-[(4-ヒドロキシベンジル)スルホニルアミノ]ピラジン (c-48) を無色固体として得た (44.8 mg, 20.0%, 2 steps)。Rf = 0.45 (n-ヘキサン/酢酸エチル = 1/1)。1H NMR (400 MHz, DMSO-d6) δ2.30 (s, 3H), 4.26 (s, 2H), 4.81 (s, 2H), 6.68-6.79 (AA’BB’, 2H), 7.08-7.32 (m, 9H), 8.02-8.14 (AA’BB’, 2H), 8.94 (s, 1H), 9.55 (s, 1H), 10.45 (s, 1H)。13C NMR (67.8 MHz, DMSO-d6) δ20.9, 37.9, 59.1, 115.3 (2C), 119.8, 122.4 (2C), 126.3, 127.2 (2C), 128.3 (2C), 128.9 (2C), 132.0 (2C), 133.3, 136.8, 137.9, 145.0, 145.4, 145.5, 151.3, 157.7, 169.1。IR (KBr, cm-1) 536, 598, 702, 841, 891, 920, 1015, 1153, 1204, 1233, 1327, 1373, 1422, 1454, 1514, 1599, 1746, 3269。
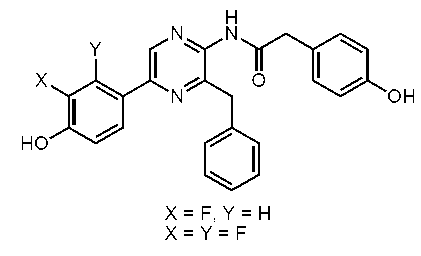
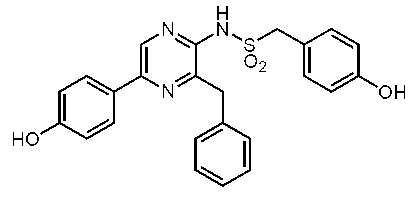
3-ベンジル-2-(4-ヒドロキシベンジル)スルホニルアミノ-5-(4-ヒドロキシフェニル)ピラジン (セレンテラスルホンアミド) (c-38)
[実施例1]
合成した各CTMD類縁体の蛍光量子収率決定を以下の手順で行った。蛍光スペクトルは、Jasco(日本分光株式会社、東京)のFP-6500蛍光分光光度計を用いて25℃で測定した。具体的には、石英セル(光路長10mm)を用い、最終濃度が750 nMになるようにCTMD類縁体をメタノール (MeOH)又は67mMリン酸緩衝液(PB)(pH 7.4)に溶解し、励起波長:330 nm、 発光/励起帯域幅: 3 nm、レスポンス:0.5 秒、スキャン速度:100 nm/分で3回測定を行い、その平均スペクトルを、化合物蛍光スペクトルとした。蛍光量子収率決定には、スペクトル補正を行った後に使用した。
実施例1で求めた蛍光量子収率は、セレンテラミド類縁体の最終濃度を750 nMとして、蛍光スペクトルを測定し算出した数値であるが、実施例1のように最終濃度を750 nMとした場合、本発明のセレンテラミド類縁体のうち、c-14、c-31、及びc-36については、メタノール中およびリン酸緩衝液中とも蛍光スペクトルを検出できず、c-25は、リン酸緩衝液中で蛍光スペクトルを検出できなかった(n.d.:検出不可)。そこで、蛍光スペクトルを検出できなかったc-14、c-31、c-25 及びc-36について、これらのセレンテラミド類縁体の最終濃度を30 μMとしたことを除いて実施例1と同じ条件で蛍光スペクトルを測定した。また、同様に、c-4についても、最終濃度を30 μMとして蛍光スペクトルを測定した。さらに、c-45についても、最終濃度を 18μMとしたことを除いて実施例1と同じ条件で10 mM CaCl2を含む50 mM Tris-HCl (pH 7.6)中で蛍光スペクトルを測定した。
c-14 (hCTTD)、c-31 (O-Ac-h-CTTD) 及び c-36 (CT-p-NsD) のメタノール中での蛍光スペクトルを図2示し、また、リン酸緩衝液中での、c-14 (hCTTD)、c-31 (O-Ac-h-CTTD)、 C-25 (CTAD) 及び c-36 (CT-p-NsD)の蛍光スペクトルを図3に示す。また、10 mM CaCl2を含む50 mM Tris-HCl (pH 7.6)中での、c-45 (CTTD)の蛍光スペクトも図3に示す。
また、c-4、c-14、c-31、c-25 及び c-36の蛍光スペクトルデータを表2にまとめた。
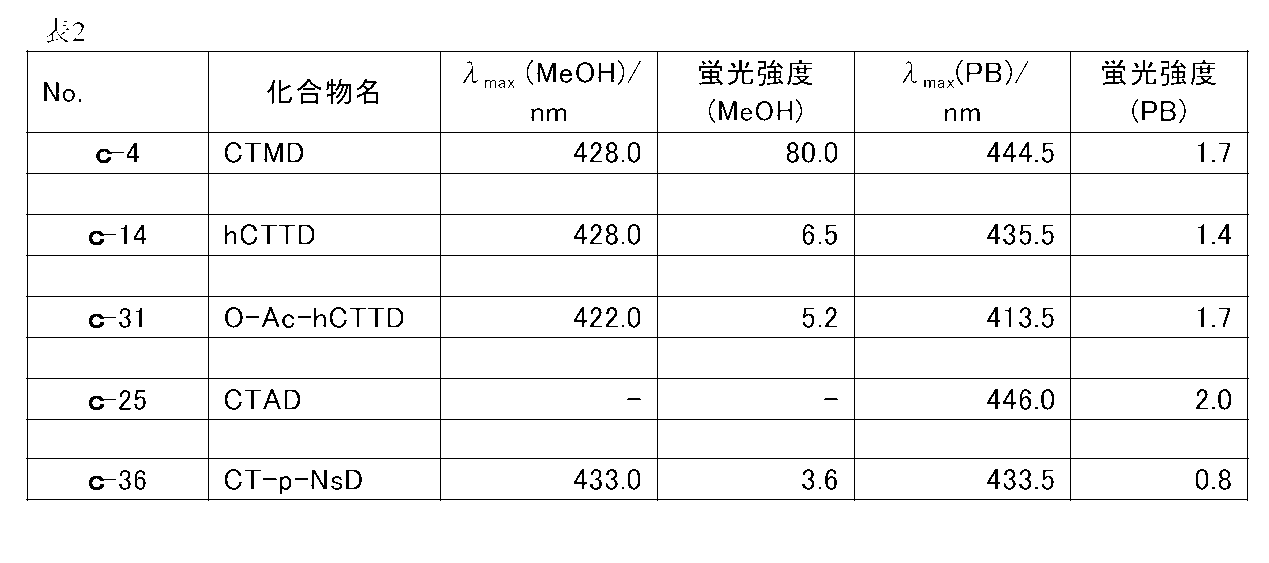
尚、c-44(CTTD)については、c-17(hCTTD)に蛍光能があることから、c-17と同様、蛍光能を有すると考えられる。
c-38について、蛍光スペクトルを、Jasco(日本分光株式会社、東京)のFP-6500蛍光分光光度計を用いて25℃で測定した。具体的には、石英セル(光路長10mm)を用い、最終濃度が18μMになるようにc-38を10 mM CaCl2を含む50 mM Tris-HCl (pH 7.6)に溶解し、励起波長:330 nm、 発光/励起帯域幅: 3 nm、レスポンス:0.5 秒、スキャン速度:100 nm/分で3回測定を行い、その平均スペクトルを、蛍光スペクトルとした。
c-38(CTSD)の10 mM CaCl2を含む50 mM Tris-HCl (pH 7.6)での蛍光スペクトルを図4に示す。
図4から、c-38は蛍光能を有することが分かった。
〔配列番号:1〕天然型アポイクオリンの塩基配列である。
〔配列番号:2〕天然型アポイクオリンのアミノ酸配列である。
〔配列番号:3〕天然型アポクライティン-Iの塩基配列である。
〔配列番号:4〕天然型アポクライティン-Iのアミノ酸配列である。
〔配列番号:5〕天然型アポクライティン-IIの塩基配列である。
〔配列番号:6〕天然型アポクライティン-IIのアミノ酸配列である。
〔配列番号:7〕天然型アポマイトロコミンの塩基配列である。
〔配列番号:8〕天然型アポマイトロコミンのアミノ酸配列である。
〔配列番号:9〕天然型アポオベリンの塩基配列である。
〔配列番号:10〕天然型アポオベリンのアミノ酸配列である。
〔配列番号:11〕天然型アポベルボインの塩基配列である。
〔配列番号:12〕天然型アポベルボインのアミノ酸配列である。
Claims (12)
- 下記一般式(1)
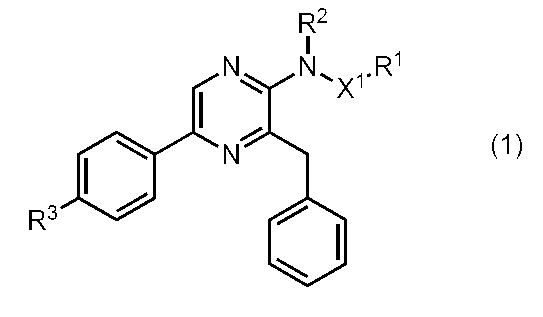
で表わされる化合物
(式中、
R1は、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、脂肪族環式基によって置換されていてもよい直鎖若しくは分枝鎖のアルキル、脂肪族環式基、又は複素環式基であり、
R2は、水素、又は-(SO2)R4であり、
R3は、水素、水酸基、メトキシ、又はアセトキシであり、
R4は、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、又は脂肪族環式基によって置換されていてもよい直鎖若しくは分枝鎖のアルキルであり、
X1は、-C(=S)-、又は-SO2-である。)。 - 一般式(1)において、
R1は、フェニル、p-メチルフェニル、p-ヒドロキシフェニル、p-メトキシフェニル、p-アセトキシフェニル、p-ニトロフェニル、ベンジル、α-ヒドロキシベンジル、4-メチルベンジル、4-ヒドロキシベンジル、4-メトキシベンジル、4-アセトキシベンジル、4-ニトロベンジル、フェニルエチル、メチル、エチル、プロピル、2-メチルプロピル、2-メチルプロパニル、シクロヘキシルメチル、シクロヘキシルエチル、アダマンチルメチル、シクロペンチルメチル、シクロヘキシル、又はチオフェン-2-イルであること、
を特徴とする、請求項1に記載の化合物。 - 一般式(1)において、
R2は、水素、ベンゼンスルホニル、p-トルエンスルホニル、4-ヒドロキシフェニルスルホニル、4-メトキシフェニルスルホニル、4-アセトキシフェニルスルホニル、4-ニトロフェニルスルホニル、ベンジルスルホニル、α-ヒドロキシベンジルスルホニル、4-メチルベンジルスルホニル、4-ヒドロキシベンジルスルホニル、4-メトキシベンジルスルホニル、4-アセトキシベンジルスルホニル、4-ニトロベンジルスルホニル、フェニルエチルスルホニル、メタンスルホニル、エチルスルホニル、プロピルスルホニル、2-メチルプロピルスルホニル、2-メチルプロパニルスルホニル、シクロヘキシルメチルスルホニル、シクロヘキシルエチルスルホニル、アダマンチルメチルスルホニル、又はシクロペンチルメチルスルホニルであること、
を特徴とする、請求項1又は2に記載の化合物。 - 下記化合物
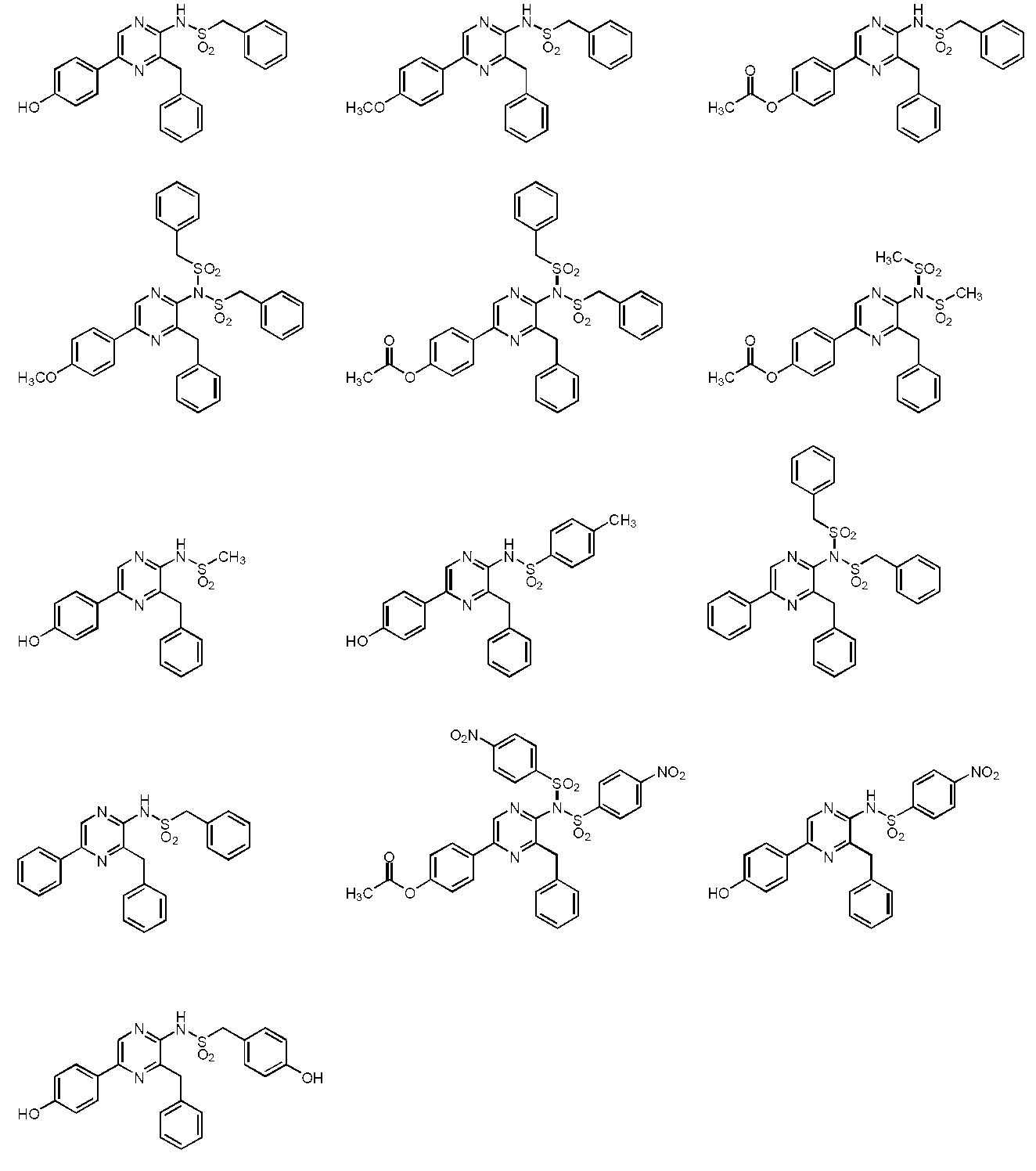
からなる群から選択される、請求項1~3のいずれか1項に記載の化合物。 - 下記化合物
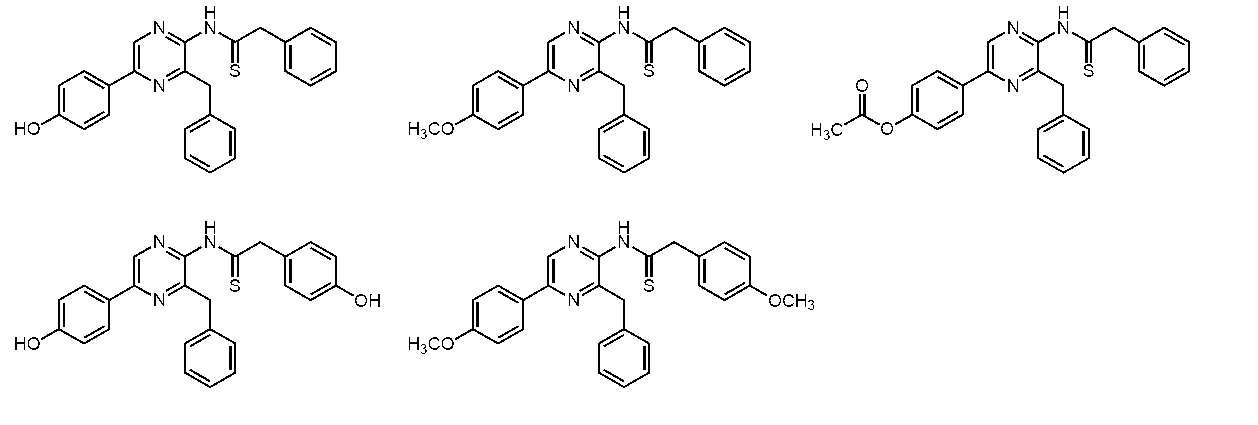
からなる群から選択される、請求項1~3のいずれか1項に記載の化合物。 - 請求項1~5のいずれか1項に記載の化合物、カルシウム結合型発光蛋白質のアポ蛋白質、及びカルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを含む、青色蛍光蛋白質。
- カルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンの存在下、請求項1~5のいずれか1項に記載の化合物と、カルシウム結合型発光蛋白質のアポ蛋白質とを反応させることを含む、青色蛍光蛋白質の製造方法。
- 前記反応を、還元剤の存在下において行う、請求項7に記載の方法。
- 請求項1~5のいずれか1項に記載の化合物、及びカルシウム結合型発光蛋白質のアポ蛋白質を含む、緑色蛍光蛋白質。
- 請求項6に記載の青色蛍光蛋白質を、カルシウムイオン又はカルシウムイオンと置換可能な2価若しくは3価のイオンを除去するためのキレート剤で処理することを含む、緑色蛍光蛋白質の製造方法。
- 請求項9に記載の緑色蛍光蛋白質に、セレンテラジン又はその類縁体を反応させることを含む、カルシウム結合型発光蛋白質の製造方法。
- 蛍光蛋白質とセレンテラジン又はその類縁体との反応を、還元剤の存在下において行う、請求項11に記載の方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/146,710 US8431525B2 (en) | 2009-02-09 | 2010-02-08 | Coelenteramide analogs |
| JP2010549535A JP5534346B2 (ja) | 2009-02-09 | 2010-02-08 | セレンテラミド類縁体 |
| GB1114142.1A GB2479692B (en) | 2009-02-09 | 2010-02-08 | Coelenteramide analogs |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009027904 | 2009-02-09 | ||
| JP2009-027904 | 2009-02-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010090318A1 true WO2010090318A1 (ja) | 2010-08-12 |
Family
ID=42542204
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/051806 Ceased WO2010090318A1 (ja) | 2009-02-09 | 2010-02-08 | セレンテラミド類縁体 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8431525B2 (ja) |
| JP (1) | JP5534346B2 (ja) |
| GB (1) | GB2479692B (ja) |
| WO (1) | WO2010090318A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102432614A (zh) * | 2011-11-16 | 2012-05-02 | 泰州凯美迪生物医药技术有限公司 | 腔肠素的合成方法 |
| US8883432B2 (en) | 2010-04-06 | 2014-11-11 | Jnc Corporation | Coelenterazine analogues and coelenteramide analogues |
| US10854747B2 (en) | 2017-07-10 | 2020-12-01 | Micron Technology, Inc. | NAND memory arrays, devices comprising semiconductor channel material and nitrogen, and methods of forming NAND memory arrays |
| EP4206184A4 (en) * | 2020-09-07 | 2024-11-20 | Sumitomo Pharma Co., Ltd. | PHENOL DERIVATIVE |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3416968B1 (en) | 2016-02-15 | 2022-07-27 | Promega Corporation | Coelenterazine analogues |
| JP7212612B2 (ja) | 2016-07-28 | 2023-01-25 | プロメガ コーポレイション | セレンテラジン類縁体 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005014633A1 (ja) * | 2003-08-12 | 2005-02-17 | Chisso Corporation | 蛍光蛋白質 |
| JP2006271327A (ja) * | 2005-03-30 | 2006-10-12 | Chisso Corp | 蛍光活性を有するルシフェラーゼの活性増強方法 |
-
2010
- 2010-02-08 WO PCT/JP2010/051806 patent/WO2010090318A1/ja not_active Ceased
- 2010-02-08 GB GB1114142.1A patent/GB2479692B/en not_active Expired - Fee Related
- 2010-02-08 JP JP2010549535A patent/JP5534346B2/ja not_active Expired - Fee Related
- 2010-02-08 US US13/146,710 patent/US8431525B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005014633A1 (ja) * | 2003-08-12 | 2005-02-17 | Chisso Corporation | 蛍光蛋白質 |
| JP2006271327A (ja) * | 2005-03-30 | 2006-10-12 | Chisso Corp | 蛍光活性を有するルシフェラーゼの活性増強方法 |
Non-Patent Citations (3)
| Title |
|---|
| KOTARO MORI ET AL.: "Real light emitter in the bioluminescence of the calcium-activated photoproteins aequorin and obelin: light emission from the singlet-excited state of coelenteramide phenolate anion in a contact ion pair", TETRAHEDRON, vol. 62, no. 26, 2006, pages 6272 - 6288 * |
| MASAKI KUSE ET AL.: "Novel synthetic route of aryl-aminopyrazine", TETRAHEDRON, vol. 60, no. 4, 2004, pages 835 - 840 * |
| OSAMU SHIMOMURA ET AL.: "Light-emitters involved in the luminescence of coelenterazine", LUMINESCENCE, vol. 15, no. 1, 2000, pages 51 - 58 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8883432B2 (en) | 2010-04-06 | 2014-11-11 | Jnc Corporation | Coelenterazine analogues and coelenteramide analogues |
| US9056840B2 (en) | 2010-04-06 | 2015-06-16 | Jnc Corporation | Coelenterazine analogues and coelenteramide analogues |
| US9075058B2 (en) | 2010-04-06 | 2015-07-07 | Jnc Corporation | Coelenterazine analogues and coelenteramide analogues |
| CN102432614A (zh) * | 2011-11-16 | 2012-05-02 | 泰州凯美迪生物医药技术有限公司 | 腔肠素的合成方法 |
| US10854747B2 (en) | 2017-07-10 | 2020-12-01 | Micron Technology, Inc. | NAND memory arrays, devices comprising semiconductor channel material and nitrogen, and methods of forming NAND memory arrays |
| EP4206184A4 (en) * | 2020-09-07 | 2024-11-20 | Sumitomo Pharma Co., Ltd. | PHENOL DERIVATIVE |
| US12590063B2 (en) | 2020-09-07 | 2026-03-31 | Sumitomo Pharma Co., Ltd. | Phenol derivative |
Also Published As
| Publication number | Publication date |
|---|---|
| US20110288280A1 (en) | 2011-11-24 |
| US8431525B2 (en) | 2013-04-30 |
| JPWO2010090318A1 (ja) | 2012-08-09 |
| GB2479692B (en) | 2013-10-30 |
| GB201114142D0 (en) | 2011-10-05 |
| JP5534346B2 (ja) | 2014-06-25 |
| GB2479692A (en) | 2011-10-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5534346B2 (ja) | セレンテラミド類縁体 | |
| JP5582505B2 (ja) | セレンテラジン類縁体及びその製造方法 | |
| JP5605727B2 (ja) | セレンテラジン類縁体及びセレンテラミド類縁体 | |
| EP2454261B1 (en) | Imidazo[1,2-alpha]pyrazin-3(7h)-one derivatives bearing a new electron-rich structure | |
| JP5353279B2 (ja) | セレンテラミド又はその類縁体の製造方法 | |
| JP5772892B2 (ja) | セレンテラミド又はその類縁体の製造方法 | |
| JP5549113B2 (ja) | カルシウム結合発光蛋白質、それをコードする遺伝子およびその用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10738646 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13146710 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010549535 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 1114142 Country of ref document: GB Kind code of ref document: A Free format text: PCT FILING DATE = 20100208 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1114142.1 Country of ref document: GB |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10738646 Country of ref document: EP Kind code of ref document: A1 |