WO2010098251A1 - ロキソプロフェン誘導体及びそれを含有する医薬 - Google Patents
ロキソプロフェン誘導体及びそれを含有する医薬 Download PDFInfo
- Publication number
- WO2010098251A1 WO2010098251A1 PCT/JP2010/052463 JP2010052463W WO2010098251A1 WO 2010098251 A1 WO2010098251 A1 WO 2010098251A1 JP 2010052463 W JP2010052463 W JP 2010052463W WO 2010098251 A1 WO2010098251 A1 WO 2010098251A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- loxoprofen
- atom
- substituted
- acceptable salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C(O)=O)c1ccc(CC(CCC2)C2=O)cc1*=C Chemical compound CC(C(O)=O)c1ccc(CC(CCC2)C2=O)cc1*=C 0.000 description 2
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/49—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups
- C07C205/56—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups having nitro groups bound to carbon atoms of six-membered aromatic rings and carboxyl groups bound to acyclic carbon atoms of the carbon skeleton
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/40—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino groups bound to carbon atoms of at least one six-membered aromatic ring and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C229/42—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino groups bound to carbon atoms of at least one six-membered aromatic ring and carboxyl groups bound to acyclic carbon atoms of the same carbon skeleton with carboxyl groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by saturated carbon chains
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/62—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/42—Unsaturated compounds containing hydroxy or O-metal groups
- C07C59/54—Unsaturated compounds containing hydroxy or O-metal groups containing six-membered aromatic rings and other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/42—Unsaturated compounds containing hydroxy or O-metal groups
- C07C59/56—Unsaturated compounds containing hydroxy or O-metal groups containing halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/76—Unsaturated compounds containing keto groups
- C07C59/88—Unsaturated compounds containing keto groups containing halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/76—Unsaturated compounds containing keto groups
- C07C59/90—Unsaturated compounds containing keto groups containing singly bound oxygen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C65/00—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C65/32—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing keto groups
- C07C65/34—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing keto groups polycyclic
- C07C65/36—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing keto groups polycyclic containing rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/74—Esters of carboxylic acids having an esterified carboxyl group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C69/757—Esters of carboxylic acids having an esterified carboxyl group bound to a carbon atom of a ring other than a six-membered aromatic ring having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/76—Esters of carboxylic acids having a carboxyl group bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Definitions
- the present invention relates to a novel loxoprofen derivative having an excellent anti-inflammatory effect and high safety. Specifically, the present invention relates to a loxoprofen derivative useful as a pharmaceutical agent that does not exhibit side effects such as gastrointestinal disorders.
- Loxoprofen is widely used clinically as a pharmaceutical product having an excellent analgesic, anti-inflammatory and antipyretic action as a hydrate of its sodium salt (generic name: loxoprofen sodium hydrate).
- Various proposals have been made so far for various derivatives of loxoprofen that maintain excellent analgesic, anti-inflammatory and antipyretic actions.
- derivatives represented by the following formulas A, B and C are known (patents): References 1-3).
- Patent Document 1 discloses a compound represented by the above formula (A), which is reported to have anti-inflammatory, analgesic and antipyretic effects.
- Patent Document 2 discloses a wide range of derivatives represented by the above formula (B), but they are different from loxoprofen derivatives provided by the present invention.
- Patent Document 3 discloses a wide range of derivatives represented by the above formula (C), which are more than existing nonsteroidal acidic anti-inflammatory agents (acidic NSAIDs) represented by aspirin or indomethacin. Furthermore, it has been reported that it has a strong anti-inflammatory action and analgesic action, and has extremely few side effects such as gastrointestinal tract disorders.
- acidic NSAIDs nonsteroidal acidic anti-inflammatory agents
- an object of the present invention is to provide a novel loxoprofen derivative having an excellent anti-inflammatory / analgesic action while avoiding side effects such as gastrointestinal disorders.
- the present invention for solving such a problem has the following formula (I) or (II):
- R 1 and R 2 represent a halogen atom or a substituted or unsubstituted phenyl group
- a pharmacologically acceptable salt thereof
- the present invention relates to a loxoprofen derivative in which the halogen atoms of R 1 and R 2 are selected from a chlorine atom, a bromine atom, a fluorine atom, and an iodine atom in the above formulas (I) and (II) Its pharmacologically acceptable salt.
- the substituent in the substituted phenyl group represented by R 1 is a halogen atom, a hydroxyl group, a substituted or unsubstituted lower alkyl group, a lower alkylthio group, a lower alkoxy group, a nitro group.
- a loxoprofen derivative which is a group, amino group or carboxyl group or a pharmacologically acceptable salt thereof, and the substituent in the substituted phenyl group of R 2 in the above formula (II) is a halogen atom or a substituted lower alkyl group Is a loxoprofen derivative or a pharmacologically acceptable salt thereof.
- the present invention is a medicament containing, as an active ingredient, a loxoprofen derivative represented by the above formula (I) or (II) or a pharmacologically acceptable salt thereof.
- a particularly preferred present invention is a loxoprofen derivative in which the substituent R 1 in the above formula (I) is a fluorine atom, a bromine atom, a p-hydroxyphenyl group or a p-aminophenyl group, or a pharmacologically acceptable salt thereof. And a pharmaceutical containing these compounds as active ingredients.
- the loxoprofen derivative provided by the present invention is a novel compound that has not been known so far, and has no side effects such as gastrointestinal disorders observed in conventional acidic NSAIDs, and moreover than loxoprofen that is used clinically. Strong anti-inflammatory and analgesic action. Therefore, since the safety range is large, it can be said that it is extremely effective in that it can be used safely for humans.
- FIG. 9 shows changes in the amount of gastric mucosa PGE 2 when a compound corresponding to 40 mg / kg of loxoprofen is administered in Test Example 3. It is the figure which showed the carrageenan edema (%) 6 hours after administering the test compound of loxoprofen equivalent to 10 mg / kg in Test Example 3.
- R 1 and R 2 represent a halogen atom or a substituted or unsubstituted phenyl group
- a pharmacologically acceptable salt thereof
- the halogen atom in the substituent “R 1 ” or “R 2 ” refers to a halogen atom selected from a chlorine atom, a bromine atom, a fluorine atom or an iodine atom.
- the lower alkyl group as a substituent in the substituted phenyl group represented by the substituent “R 1 ” or “R 2 ” refers to a substituted or unsubstituted alkyl group having about 1 to 6 carbon atoms. , Methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group, hexyl group and the like.
- the substituents for these lower alkyl groups mean a hydroxyl group, an amino group, a nitro group and the like.
- the lower alkoxy group is a lower alkyloxy group having about 1 to 6 carbon atoms, and specifically includes a methoxy group, an ethoxy group, a propoxy group, a butoxy group, an isobutoxy group, a sec-butyloxy group, and a tert-butyloxy group.
- Pentyloxy group isopentyloxy group, hexyloxy group and the like.
- the position of the substituent in the substituted phenyl group and the number thereof are not particularly limited, but it is preferably a mono-substituted phenyl group and the substitution position is preferably a meta position or a para position.
- the methyl group of the phenylpropionic acid moiety can be in the ⁇ -position or ⁇ -position, but the present invention
- the coordination of the methyl group may be both or a mixture thereof.
- the coordination of the hydroxyl group (position 1) and the phenyl group (position 2) of the cyclopentane ring in the formula (II-a) can take a cis- or trans-coordination.
- -It may be a cis isomer, a 1,2-trans isomer, or a mixture of diastereomers thereof.
- novel loxoprofen derivative provided by the present invention can be specifically produced as follows.
- the manufacturing method demonstrated below is one specific manufacturing method, It is not limited to this, It cannot be overemphasized that it can manufacture with reference to a general chemical textbook.
- the loxoprofen derivative in which the substituent “R 1 ” is a halogen atom can be synthesized, for example, according to the production scheme 1 shown in the following chemical reaction formula. . [Production Scheme 1]
- arithmetic numbers represent specific compound numbers, and Roman numerals represent process numbers in Production Scheme 1 (the same applies in the following production schemes).
- reaction conditions reaction time, reaction temperature, etc.
- reaction reagents, solvents, catalysts, etc. in each production process based on the following production schemes are exemplified as preferred production examples, and are not limited thereto. Absent.
- the amino group terminal was converted to an aldehyde compound (2a-d) by a formylation reaction.
- Such conversion can be performed, for example, by a diazotization reaction with hydrochloric acid / sodium nitrate / copper sulfate / sodium sulfite / sodium acetate as the i step and then with a hydroxyamine hydrochloride in the presence of paraformaldehyde as the ii step.
- the target aldehyde compound can be obtained by decomposition with hydrochloric acid in step iii.
- the resulting aldehyde compound (2a-d) is then subjected to, for example, a Wittig reaction (step iv) with MeOCH 2 P (Ph 3 ) Cl, C 6 H 18 KNSi 2 in toluene, and further in acetone, for example.
- a phenyl aldehyde compound (3a-d) having an extended carbon chain by acid treatment with hydrochloric acid or the like (step v) is converted into a phenylacetic acid compound (step vi) by oxidation with periodic acid (step vi) in the presence of 2 mol% of PFC. 4a-d), an esterification reaction with an acid-alcohol (step vii) is carried out to convert it into a phenylacetic acid ester compound (5a-d).
- phenylacetic acid ester compound (5a-d) was subjected to an ⁇ -methylation reaction (step viii) with methyl iodide in the presence of about 2 mol of lithium diisopropylamide (LDA) in dry tetrahydrofuran. Then, it was sequentially converted into a propionic acid ester compound (6a-d).
- the reaction is preferably carried out at a low temperature, for example, it is preferably carried out at a temperature of about ⁇ 78 ° C. to about ⁇ 40 ° C.
- a compound (7a-d) in which a methyl group terminal is converted to bromomethylene step ix) by refluxing with N-bromosuccinimide (NBS) and azoisobutyronitrile (AIBN) in carbon tetrachloride.
- N-bromosuccinimide N-bromosuccinimide
- AIBN azoisobutyronitrile
- step x methyl 2-oxocyclopentanecarboxylate
- potassium carbonate eg dry acetone
- the loxoprofen derivative in the case where the substituent “R 1 ” is a substituted or unsubstituted phenyl group includes, for example, production scheme 2 and It can be synthesized according to Production Scheme 3. [Production Scheme 2]
- the loxoprofen derivative (19-25, 31-35) in which the substituent “R 1 ” is a substituted or unsubstituted phenyl group in the loxoprofen derivative of the present invention is synthesized.
- the process is shown. First, the carboxylic acid terminal of the loxoprofen derivative (9c) in which the aromatic ring obtained in Production Scheme 1 was substituted with bromine was mixed with methanol in 1,2-dichloroethane in the presence of 4-dimethylaminopyridine (4-DMAP).
- step i) compounds (10c, 11c) in which the carboxyl group is esterified with benzyl alcohol (BnOH) (step i) are synthesized, and Suzuki-Miyaura cross-coupling reaction with various boron compounds using these as substrates ( By carrying out step ii), an ester-protected biphenyl type loxoprofen derivative (12-17) was obtained.
- BnOH benzyl alcohol
- the Suzuki - Miyaura cross-coupling reaction for example, boron compounds having a substituent shown as R 2 above using the [R 2 -PhB (OH) 2 ], the presence of about 2 moles of sodium carbonate, aqueous tetrahydrofuran It can be performed by refluxing with Pd (PPh 3 ) 4 .
- the compound (17) in which the biphenyl end is substituted with a nitro group is converted to an amino group by a reduction reaction using palladium-carbon (compound 18), and finally these compounds (12-18)
- the target biphenyl type loxoprofen derivative (19-25) was obtained by catalytic reduction or hydrolysis of the ester group terminal.
- Catalytic reduction is performed by, for example, using 10% palladium-carbon as a catalyst and absorbing hydrogen gas in an alcohol solvent such as methanol or ethanol.
- Hydrolysis is performed by ordinary alkali hydrolysis (for example, alkali metal in an alcohol solvent). (Hydrolysis with hydroxide).
- the Suzuki-Miyaura cross-coupling reaction with a boronic acid compound (step i) and the hydrolysis / decarboxylation reaction (step ii) are the same reactions as performed in the above reaction scheme 2 and reaction scheme 1. Can be implemented.
- 4-DMAP 4-dimethylaminopyridine
- step iii the target reduced loxoprofen derivative (40a, c; 41a, c; 42; 43) was obtained by catalytic reduction or hydrolysis (step iii or iv) of the terminal end of the ester group.
- the catalytic reduction or hydrolysis reaction employs the same means as in the method of step iv or v in production scheme 2.
- the loxoprofen derivative which is the target compound of the present invention can be obtained by the production method described above.
- the physicochemical properties of the obtained compounds are summarized in Tables 1 to 3 below.
- the loxoprofen derivative provided by the present invention can be used as a free carboxylic acid or as a pharmacologically acceptable salt thereof.
- the pharmacologically acceptable salt include alkali metal salts such as sodium salt and potassium salt, or ammonium salt.
- the active ingredient which is the derivative or a pharmaceutically acceptable salt thereof is used alone or in a conventional manner. It can be administered orally or parenterally as appropriate dosage forms such as capsules, tablets, injections and the like together with the form.
- a capsule can be prepared by mixing a loxoprofen derivative or a salt thereof with an excipient such as lactose, starch or a derivative thereof, or a cellulose derivative and filling it into a gelatin capsule.
- the tablets are kneaded by adding water and a binder such as sodium carboxymethyl cellulose, alginic acid, gum arabic, etc., and if necessary, granulated, and further lubricants such as talc and stearic acid.
- a binder such as sodium carboxymethyl cellulose, alginic acid, gum arabic, etc.
- lubricants such as talc and stearic acid.
- loxoprofen derivative or a salt thereof is dissolved in a sterilized distilled water or a sterilized physiological saline together with a solubilizing agent, and enclosed in an ampoule to prepare an injectable preparation. If necessary, stabilizers, buffer substances and the like may be contained. These parenteral preparations can be administered intravenously or intravenously.
- the dosage of the loxoprofen derivative provided by the present invention cannot be unconditionally limited by various factors such as the symptoms, severity, age, presence or absence of complications of the patient to be treated.
- the active ingredient is usually 0.1 to 1000 mg / day / human, preferably 1 to In the case of parenteral administration, it can be appropriately selected and administered within the range of about 1/100 to 1/2 of the dose in the case of parenteral administration.
- these dosages can be appropriately increased or decreased depending on the age, symptoms, etc. of the patient.
- Test Example 1 Human whole blood assay (in vitro) The test was performed according to the method described in Inflamm. Res., 45: 68-74 (1996).
- B In vitro COX-2 assay. Blood draw subjects selected those who had not taken NSAIDs for at least one week and were healthy on the day of collection. Blood is collected in a test tube (Venojectll blood collection tubes, manufactured by Terumo Corporation) treated with heparin, and lipopolysaccharide (LPS), which is an inflammatory stimulant [Sigma-Aldrich Japan Inc, # L2880 from E. coli055: B5, diluted with phosphate buffered saline (PBS) to a final concentration of 100 ⁇ g / mL].
- test tube Vehicle
- LPS lipopolysaccharide
- Test Example 2 Renal injury assay (in vivo) The test was performed according to the method described in Biochem. Biophysical. Res. Com., 323; 1032-39 (2004).
- A Liposome membrane preparation method Egg yolk phosphatidylcholine (10 ⁇ M, 7.7 mg: manufactured by Kanto Chemical Co., Inc.) was dissolved in chloroform / methanol (1: 2, v / v), dried, and then dissolved in 1.5 mL of diethyl ether.
- Test Example 3 Animal assay (in vivo test) A: The gastric ulcer formation test by the loxoprofen derivative followed the method described in Biochem. Pharmacol., 67; 575-85 (2004). Wister male rats (body weight: 180-200 g) were fasted for 18 hours, and the test compound was orally administered (the dose was adjusted to be equal to that of loxoprofen). After 8 hours, the stomach was removed, and the area of the ulcer formed in the stomach was measured. The area of all ulcers was summed to obtain the ulcer coefficient. Further, the amount of PGE 2 in the gastric mucosa was measured by ELISA. The protocol followed the protocol attached to the kit.
- FIG. 1 shows the results when a test compound corresponding to 40 mg / kg of loxoprofen is administered
- FIG. 2 shows the results when a test compound corresponding to 50 mg / kg of loxoprofen is administered
- FIG. 3 shows gastric mucosa PGE.
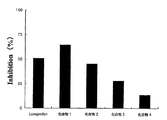
- the loxoprofen derivative of the present invention (compounds 1 to 4) has a significantly lower ulcer coefficient than that of loxoprofen, and almost no ulcer was formed in compounds 3 and 4. It was not.
- the amount of PGE 2 in the gastric mucosa was higher in compounds 2, 3 and 4 than in loxoprofen.
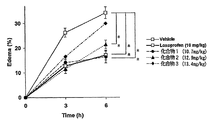
- FIG. 4 shows carrageenin edema (%) 6 hours after administration of a test compound corresponding to loxoprofen 10 mg / kg
- FIG. 5 shows edema inhibition rate (%).
- the edema suppression rate is based on the following calculation.
- Inhibition rate (%) 100 ⁇ (edema volume at the time of compound administration / edema volume at the time of vehicle administration) ⁇ 100
- FIG. 6 shows changes in foot volume 3 hours and 6 hours after administration.
- the loxoprofen derivative (compounds 1 to 4) of the present invention exhibits a good anti-inflammatory action and does not cause ulceration, which is a side effect. Is found to be a well-separated compound.
- Formulation Example 1 Tablet compound 1 50 mg Lactose 100mg Hydroxypropylcellulose 150mg Magnesium stearate 50mg Based on the above formulation, the granules were prepared and then tableted to prepare tablets having a weight of 350 mg by a conventional method.
- Formulation Example 2 Granule compound 2 50 mg Lactose 100mg Corn starch 150mg Based on the above formulation, granules containing 50 mg of active ingredient in 200 mg granules were prepared by a conventional method.
- the loxoprofen derivative provided by the present invention is a novel compound that has not been known so far, and has no side effects such as gastrointestinal disorders observed in conventional NSAIDs, and is clinically used. Anti-inflammatory and analgesic action is stronger than loxoprofen. Therefore, since its safety range is large, it is extremely effective in that it can be used safely for humans, and its industrial contribution is great.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pain & Pain Management (AREA)
- Pharmacology & Pharmacy (AREA)
- Rheumatology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
これまでに、ロキソプロフェンの優れた鎮痛、抗炎症、解熱作用を維持した各種誘導体の提案が種々行われてきており、例えば、次式A、B及びCで示される誘導体が知られている(特許文献1~3)。

さらに、特許文献3には上記式(C)で形式的に示される誘導体が広範囲に開示されており、これらはアスピリン或いはインドメタシンに代表される既存の非ステロイド性酸性消炎剤(酸性NSAIDs)よりも更に強力な抗炎症作用及び鎮痛作用を有し、胃腸管障害等の副作用が極めて少ないものであると報告されている。
したがって、これまでに、抗炎症・鎮痛作用を示す化合物について、その薬理作用と副作用の分離が種々検討されてきているが、これといった成果が上げられていないのが現状である。
本発明者は、かかる現状下、ロキソプロフェン誘導体について検討を行った結果、胃腸障害等の副作用を回避し、優れた抗炎症、鎮痛効果を有する化合物を合成することに成功し、本願発明を完成させるに至った。

で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
したがって、その安全域が大きいことから、ヒトに対して安全に使用できる点で、極めて有効なものといえる。

で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
これらの低級アルキル基の置換基としては水酸基、アミノ基、ニトロ基等を意味する。

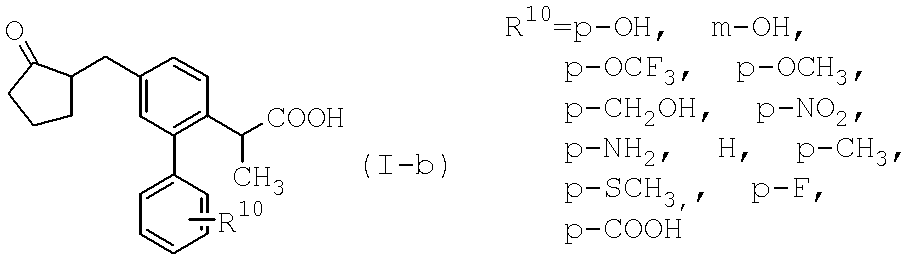

更に、式(II-a)におけるシクロペンタン環の水酸基(1位)とフェニル基(2位)の配位は、シス-及びトランス-配位を取り得るが、本発明においては、1,2-シス体であっても、または1,2-トランス体であっても、或いはそのジアステレオマーの混合物であってもよい。
なお、以下に説明する製造方法は、具体的な一製造方法であり、これに限定されるものでなく、一般的な化学教科書を参照し、製造し得ることはいうまでもない。
[製造スキーム1]
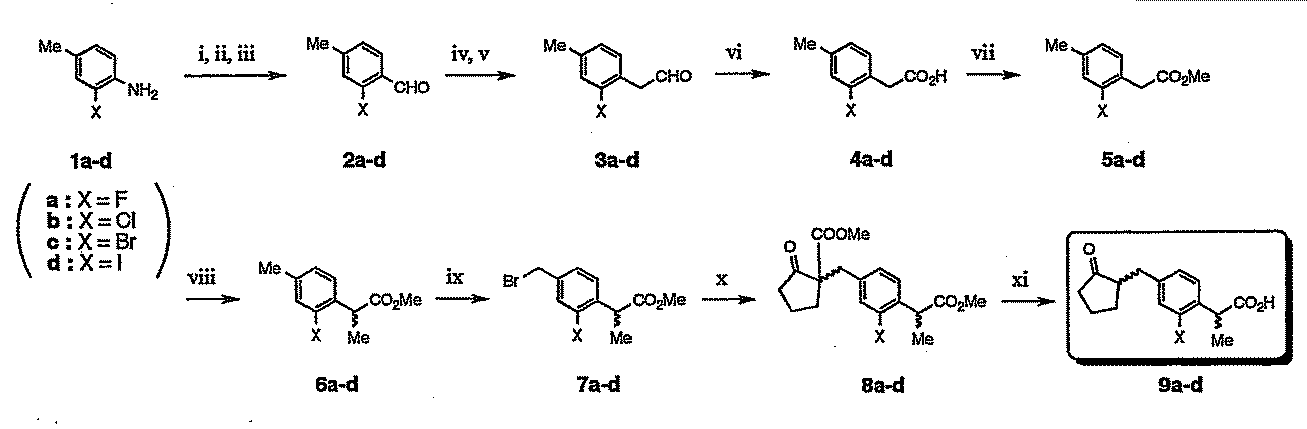
また、以下の各製造スキームに基づく各製造工程における反応条件(反応時間、反応温度等)、反応試薬、溶媒、触媒等は、好ましい製造例として例示するものであり、これらに限定されるものではない。
かかる変換は、例えば、第i工程として、塩酸/硝酸ナトリウム/硫酸銅/亜硫酸ナトリウム/酢酸ナトリウムによるジアゾ化反応の後、第ii工程としてパラホルムアルデヒドの存在下、ヒドロキシムアミン塩酸塩との処理の後、第iii工程として塩酸による分解で目的とするアルデヒド化合物を得ることができる。
最後に、これらの中間体化合物(8a-d)を、例えば、酢酸/塩酸による酸加水分解・脱炭酸することによって、目的とする本発明の式(I)で示されるロキソプロフェン誘導体のなかで、置換基「R1」がハロゲン原子である場合のロキソプロフェン誘導体(9a-d)が製造される。
[製造スキーム2]

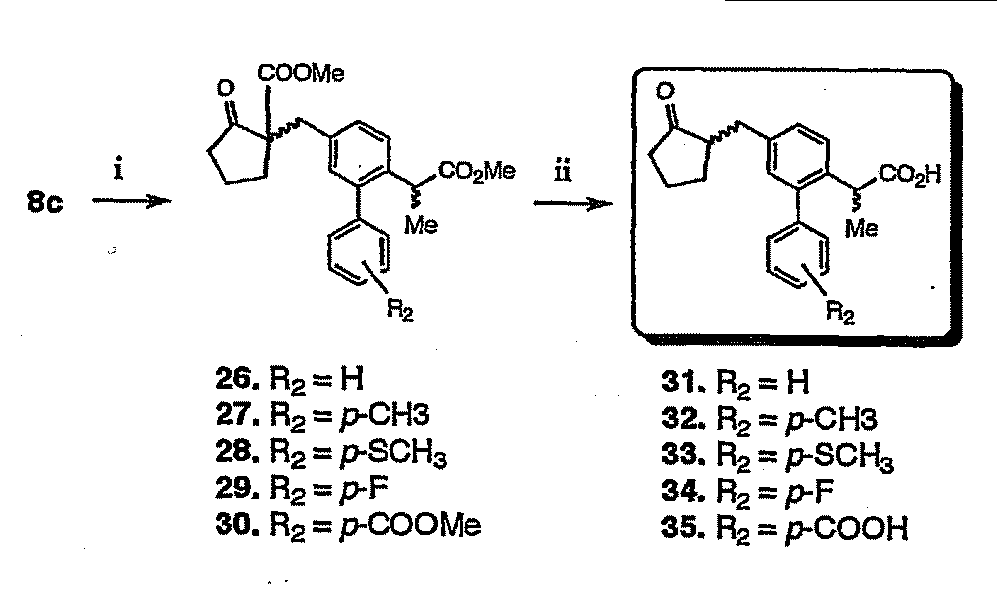
まず、製造スキーム1で得られた芳香環が臭素で置換されたロキソプロフェン誘導体(9c)のカルボン酸末端を、1,2-ジクロルエタン中、4-ジメチルアミノピリジン(4-DMAP)の存在下、メタノールまたはベンジルアルコール(BnOH)によりカルボキシル基をエステル化(第i工程)でエステル保護した化合物(10c、11c)を合成し、これらを基質とした種々のボロン化合物との鈴木-宮浦クロスカップリング反応(第ii工程)を行うことにより、エステルで保護されたビフェニル型のロキソプロフェン誘導体(12-17)を得た。
接触還元は、例えば、10%パラジウム-炭素を触媒とし、メタノール或いはエタノール等のアルコール溶媒中水素ガスを吸収させることにより、また加水分解は、通常のアルカリ加水分解(例えば、アルコール溶媒中、アルカリ金属水酸化物による加水分解)で実施することができる。
[製造スキーム4]
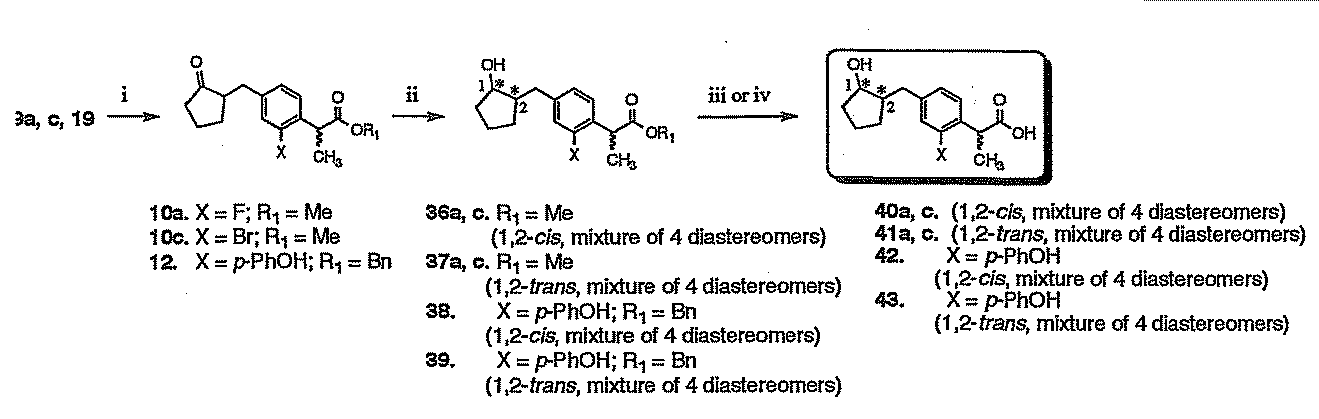
この場合の接触還元または加水分解反応は、製造スキーム2における第iv又はv工程の方法と同様の手段が採用される。



薬理学的に許容される塩としては、ナトリウム塩、カリウム塩などのアルカリ金属塩或いはアンモニウム塩を挙げることができる。
また、投与経路、剤形、投与回数等によっても異なるものであるが、一般的には、経口投与の場合は、有効成分として、通常、0.1~1000mg/日/ヒト、好ましくは1~500mg/日/ヒトの範囲内、また、非経口投与の場合は、経口投与の場合における投与量の約1/100~1/2量程度の範囲内で適宜選別し、投与することができる。なお、これらの投与量は、患者の年齢、症状等により適宜増減することが可能であることは勿論である。


試験は、Inflamm. Res., 45: 68-74 (1996) に記載される方法に準じて行った。
A:インビトロにおけるCOX-1アッセイ
採血対象者は、少なくとも1週間以上にNSAIDsを服用しておらず、採取日に健康である人を選択した。
血液は、血液凝固抑制剤無添加で採取し、直後にアッセイに用いた。採取した血液を500μLずつチューブ(Protein Lobingdin tube, Eppenduf Co. LTD., Tokyo, Japan)に分注し、適切な溶媒(DMSO又はMilliQ water)に溶解させた試験化合物2μL(最終濃度:0.1μM-1000μM)を添加し、37℃/24時間血液凝固が認められるまでインキュベーションした。
インキュベーション後、サンプルを12,000×g/5分間の遠心分離を行い、血清を分離した。血中タンパクを除外するため、得られた血清100μLをエタノール400μLに添加し、再び12,000×g/5分間の遠心分離を行った。上清中のTXB2を酵素免疫測定法(EIA)kit[Cayman (Ann, Arbor, MI,USA)#519031]を用いて定量した。プロトコルは、付属のプロトコルに従った。
採血対象者は、少なくとも1週間以上にNSAIDsを服用しておらず、採取日に健康である人を選択した。
血液は、ヘパリン処理が施された試験管(Venojectll blood collection tubes、テルモ社製)に採取し、炎症性刺激物質であるリポポリサッカロイド(LPS)[Sigma-Aldrich Japan Inc,#L2880 from E. coli055:B5、最終濃度が100μg/mLとなるように燐酸緩衝生理食塩水(PBS)で希釈した]を添加した。500μLずつチューブに分注し、適切な溶媒(DMSO又はMilliQ water)に溶解させた試験化合物2μL(最終濃度:0.1μM-1000μM)を添加し、COX-2を誘導するために37℃/24時間インキュベーションした。
インキュベーション後、サンプルを12,000×g/5分間の遠心分離を行い、血清を分離した。血中タンパクを除外するため、得られた血清100μLをエタノール400μLに添加し、再び12,000×g/5分間の遠心分離を行った。上清中のPGE2を酵素免疫測定法(EIA)kit[Cayman (Ann, Arbor, MI,USA)#514040]を用いて定量した。プロトコルは、付属のプロトコルに従った。
試験は、Biochem. Biophysical. Res. Com., 323; 1032-39 (2004) に記載の方法に従った。
A:リポソーム膜調製法
卵黄ホスファチジルコリン(10μM、7.7mg:関東化学社製)をクロロホルム/メタノール(1:2、v/v)に溶解し、乾燥後、1.5mLのジエチルエーテルに溶解した。100mM calcein-NaOH(pH7.4)を1mL加え、1分間超音波処理した後、ジエチルエーテルをconventional rotary evaporator(25℃)で除去し、リン酸緩衝液に懸濁したものをリポソーム膜溶液とした(reversed-phase evaporation法)。
(1)水を溶剤として用いる測定方法
30μLのリポソーム膜溶液を、5mLのリン酸緩衝液に懸濁させ、遠心(10,500×g/20分間)し、リン酸緩衝液で2回洗浄した。ペレットを1mLのリン酸緩衝液に懸濁し、その懸濁液を膜傷害実験に用いた。
1.5mLチューブに6μLずつ分注し、そのリポソーム懸濁液に、濃度が異なるテストサンプルのリン酸緩衝液6μLを加え、30℃/10分間インキュベーションした。氷上で冷却後、384穴wellに混合溶液を10μL分注した後、漏出したcalceinの蛍光強度を測定した(励起波長:490nm)。
リポソーム懸濁液を遠心(10,500×g/20分間)し、リン酸緩衝液で2回洗浄した。ペレットを5mLのリン酸緩衝液に懸濁し、その懸濁液の0.75mLを50mLのリン酸緩衝液で希釈したものを膜傷害実験に用いた。
1.5mLチューブに400μLずつ分注し、そのリポソーム懸濁液に、濃度が異なるテストサンプルのDMSO溶液を加え、30℃/10分間インキュベーションした。氷上で冷却後、96穴wellに混合溶液を200μL分注した後に、漏出したcalceinの蛍光強度を測定した(励起波長:490nm)。
100%膜傷害のコントロールとして、triton X-100の25%溶液を用い、各テストサンプルの蛍光強度を測定後、100%膜傷害のコントロールの値に対する割合を百分率で表したものを膜傷害(calcein release)とした。

A:ロキソプロフェン誘導体による胃潰瘍の形成
試験は、Biochem. Pharmacol., 67; 575-85 (2004) に記載の方法に従った。
Wister系雄性ラット(体重:180-200g)を18時間絶食させ、試験化合物を経口投与した(投与量は、ロキソプロフェンと等しい物質量となるようにした)。8時間後、胃を摘出し、胃内部に生じた潰瘍の面積を測定した。全ての潰瘍の面積を合計し、潰瘍係数とした。
また、胃粘膜のPGE2量は、ELISA法により測定した。
なお、プロトコルは、キット付属のプロトコルに従った。
図1にロキソプロフェン40mg/kg量に相当する試験化合物を投与した場合の結果を示し、図2にロキソプロフェン50mg/kg量に相当する試験化合物を投与した場合の結果を示し、図3に胃粘膜PGE2量の変化を示した。
図中に示した結果からも判明するように、本発明のロキソプロフェン誘導体(化合物1~4)は、ロキソプロフェンに比較して顕著に潰瘍係数が低く、化合物3及び4では殆ど潰瘍の形成が認められないものであった。
また、胃粘膜のPGE2量は、化合物2、3及び4において、ロキソプロフェンより高いものであった。
試験は、Br. J. Pharmacol., 151; 285-91 (2007) に記載の方法に従った。
Wister系雄性ラット(体重:180-200g)を18時間絶食させ、試験化合物を経口投与した(投与量は、ロキソプロフェンと等しい物質量となるようにした)。1時間後、左足蹠皮下に1%カラゲニン(生理食塩水に溶解)100μLを注射し、浮腫を惹起させた。
カラゲニン投与前、投与後3時間及び6時間後の足容積を、Plethysmometerを用いて測定した。
また、足浮腫PGE2量は、ELISA法により測定した。なお、プロトコルは、キット付属のプロトコルに従った。
図4にロキソプロフェン10mg/kg量に相当する試験化合物を投与した6時間後におけるカラゲニン浮腫(%)を示し、図5に浮腫抑制率(%)を示した。
なお、浮腫抑制率は、以下の計算による。
抑制率(%)=100-(化合物投与時の浮腫容積/Vehicle投与時の浮腫容積)×100
また、図6に、投与後3時間及び6時間後の足容積の変化を示した。
化合物1 50mg
乳糖 100mg
ヒドロキシプロピルセルロース 150mg
ステアリン酸マグネシウム 50mg
上記処方を基本とし、顆粒を調製後、打錠し重量350mgの錠剤を、常法により調製した。
化合物2 50mg
乳糖 100mg
トウモロコシデンプン 150mg
上記処方を基本とし、200mg顆粒中有効成分50mg含有の顆粒を常法により調製した。
したがって、その安全域が大きいことから、ヒトに対して安全に使用できる点で、極めて有効なものであり、産業上の貢献度は多大なものである。
Claims (6)
- 次式(I)又は(II):

で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩。 - 式(I)及び(II)中、R1及びR2のハロゲン原子が塩素原子、臭素原子、フッ素原子、ヨウ素原子から選択されるものである請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
- 式(I)中、R1の置換フェニル基における置換基が、ハロゲン原子、水酸基、置換若しくは非置換低級アルキル基、低級アルキルチオ基、低級アルコキシ基、ニトロ基、アミノ基又はカルボキシル基である請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
- 式(II)中、R2の置換フェニル基における置換基が、ハロゲン原子又は置換低級アルキル基である請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
- 請求項1に記載の式(I)又は(II)で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬。
- 請求項2~4のいずれかに記載のロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201080009343.4A CN102333754B (zh) | 2009-02-26 | 2010-02-18 | 洛索洛芬衍生物和含有其的药品 |
| EP10746132.9A EP2402305B1 (en) | 2009-02-26 | 2010-02-18 | Loxoprofen derivative and pharmaceutical containing same |
| KR1020117022258A KR101708073B1 (ko) | 2009-02-26 | 2010-02-18 | 록소프로펜 유도체 및 그것을 함유하는 의약 |
| US13/148,566 US8431614B2 (en) | 2009-02-26 | 2010-02-18 | Loxoprofen derivative and pharmaceutical preparation containing the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-043801 | 2009-02-26 | ||
| JP2009043801A JP5390883B2 (ja) | 2009-02-26 | 2009-02-26 | ロキソプロフェン誘導体及びそれを含有する医薬 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010098251A1 true WO2010098251A1 (ja) | 2010-09-02 |
Family
ID=42665458
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/052463 Ceased WO2010098251A1 (ja) | 2009-02-26 | 2010-02-18 | ロキソプロフェン誘導体及びそれを含有する医薬 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8431614B2 (ja) |
| EP (1) | EP2402305B1 (ja) |
| JP (1) | JP5390883B2 (ja) |
| KR (1) | KR101708073B1 (ja) |
| CN (1) | CN102333754B (ja) |
| WO (1) | WO2010098251A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013062028A1 (ja) * | 2011-10-25 | 2013-05-02 | 塩野義製薬株式会社 | Hiv複製阻害剤 |
| WO2013099553A1 (ja) * | 2011-12-27 | 2013-07-04 | 株式会社Lttバイオファーマ | 2-フルオロフェニルプロピオン酸誘導体 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101465025B1 (ko) * | 2013-02-27 | 2014-11-26 | 한미정밀화학주식회사 | 입체선택적인 록소프로펜 (2s, 1''r, 2''s) 트랜스-알코올의 제조방법 |
| WO2014167509A2 (en) * | 2013-04-10 | 2014-10-16 | Shasun Pharmaceuticals Limited | Loxoprofen polymorphs and process for preparation of the same |
| CN109134261A (zh) * | 2017-06-15 | 2019-01-04 | 北京蓝丹医药科技有限公司 | 一种洛索洛芬衍生物 |
| CN109134262A (zh) * | 2017-06-15 | 2019-01-04 | 北京蓝丹医药科技有限公司 | 一种洛索洛芬衍生物 |
| CN111635315B (zh) * | 2019-03-01 | 2024-03-12 | 华创合成制药股份有限公司 | 一种解热镇痛药物及其制备方法和用途 |
| CN111635309B (zh) * | 2019-03-01 | 2024-03-12 | 华创合成制药股份有限公司 | 一种新型解热镇痛药物及其制备方法和用途 |
| CN113121331B (zh) * | 2019-12-31 | 2024-01-05 | 华创合成制药股份有限公司 | 具有环丙基的苯氧芳酸类化合物及其药学上可接受的盐,以及它们的制备方法和应用 |
| WO2022052936A1 (zh) * | 2020-09-09 | 2022-03-17 | 南京海融医药科技股份有限公司 | 一种芳基丙酸衍生物、药物组合物及其制备方法和应用 |
| CN115557839B (zh) * | 2021-12-23 | 2024-02-13 | 南京海融医药科技股份有限公司 | 一种包含芳基丙酸衍生物的脂肪乳剂及其制备方法 |
| WO2023115511A1 (zh) * | 2021-12-24 | 2023-06-29 | 南京海融医药科技股份有限公司 | 一种芳基丙酸衍生物及其乳状制剂 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS53135958A (en) * | 1977-04-05 | 1978-11-28 | Sankyo Co Ltd | Substituted phenylacetic acid derivatives and their preparation |
| JPS54103852A (en) | 1978-01-27 | 1979-08-15 | Schering Ag | Novel phenyl acetate derivative*its manufacture and inflammation therapeutic medicine containing said derivative |
| WO1993002999A1 (fr) | 1991-08-10 | 1993-02-18 | Hisamitsu Pharmaceutical Co., Inc. | Derive de l'acide phenylalcanoique, production de ce compose et separation d'isomeres optiques dudit derive |
| JPH0584699A (ja) | 1991-09-24 | 1993-04-06 | Shima Seiki Mfg Ltd | 裁断装置 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4161538A (en) | 1977-04-05 | 1979-07-17 | Sankyo Company Limited | Substituted phenylacetic acid derivatives and process for the preparation thereof |
| JPS6033718B2 (ja) | 1981-06-27 | 1985-08-05 | 石川島播磨重工業株式会社 | 船舶の操舵装置 |
-
2009
- 2009-02-26 JP JP2009043801A patent/JP5390883B2/ja not_active Expired - Fee Related
-
2010
- 2010-02-18 EP EP10746132.9A patent/EP2402305B1/en not_active Not-in-force
- 2010-02-18 US US13/148,566 patent/US8431614B2/en not_active Expired - Fee Related
- 2010-02-18 CN CN201080009343.4A patent/CN102333754B/zh not_active Expired - Fee Related
- 2010-02-18 WO PCT/JP2010/052463 patent/WO2010098251A1/ja not_active Ceased
- 2010-02-18 KR KR1020117022258A patent/KR101708073B1/ko not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS53135958A (en) * | 1977-04-05 | 1978-11-28 | Sankyo Co Ltd | Substituted phenylacetic acid derivatives and their preparation |
| JPS54103852A (en) | 1978-01-27 | 1979-08-15 | Schering Ag | Novel phenyl acetate derivative*its manufacture and inflammation therapeutic medicine containing said derivative |
| WO1993002999A1 (fr) | 1991-08-10 | 1993-02-18 | Hisamitsu Pharmaceutical Co., Inc. | Derive de l'acide phenylalcanoique, production de ce compose et separation d'isomeres optiques dudit derive |
| JPH0584699A (ja) | 1991-09-24 | 1993-04-06 | Shima Seiki Mfg Ltd | 裁断装置 |
Non-Patent Citations (5)
| Title |
|---|
| BIOCHEM. BIOPHYSICAL. RES. COM., vol. 323, 2004, pages 1032 - 39 |
| BIOCHEM. PHARMACOL., vol. 67, 2004, pages 575 - 85 |
| BR. J. PHARMACOL., vol. 151, 2007, pages 285 - 91 |
| INFLAMM. RES., vol. 45, 1996, pages 68 - 74 |
| See also references of EP2402305A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013062028A1 (ja) * | 2011-10-25 | 2013-05-02 | 塩野義製薬株式会社 | Hiv複製阻害剤 |
| WO2013099553A1 (ja) * | 2011-12-27 | 2013-07-04 | 株式会社Lttバイオファーマ | 2-フルオロフェニルプロピオン酸誘導体 |
| JP2013133323A (ja) * | 2011-12-27 | 2013-07-08 | Ltt Bio-Pharma Co Ltd | 2−フルオロフェニルプロピオン酸誘導体 |
| CN103998412A (zh) * | 2011-12-27 | 2014-08-20 | 日本株式会社Ltt生物医药 | 2-氟苯基丙酸衍生物 |
| US9221786B2 (en) | 2011-12-27 | 2015-12-29 | Ltt Bio-Pharma Co., Ltd. | 2-fluorophenyl propionic acid derivatives |
| CN103998412B (zh) * | 2011-12-27 | 2017-05-31 | 日本株式会社 Ltt 生物医药 | 2‑氟苯基丙酸衍生物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2402305B1 (en) | 2013-10-02 |
| EP2402305A4 (en) | 2012-07-18 |
| KR20110137321A (ko) | 2011-12-22 |
| KR101708073B1 (ko) | 2017-02-17 |
| CN102333754B (zh) | 2014-04-16 |
| JP5390883B2 (ja) | 2014-01-15 |
| US8431614B2 (en) | 2013-04-30 |
| JP2010195727A (ja) | 2010-09-09 |
| CN102333754A (zh) | 2012-01-25 |
| EP2402305A1 (en) | 2012-01-04 |
| US20120016158A1 (en) | 2012-01-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5390883B2 (ja) | ロキソプロフェン誘導体及びそれを含有する医薬 | |
| JP3880067B2 (ja) | 新規化合物並びに抗炎症及び抗血栓活性を有する組成物 | |
| JP4043046B2 (ja) | 消炎、鎮痛および抗血栓活性を有するニトロ化合物およびそれらの組成物 | |
| Bandgar et al. | Synthesis and biological evaluation of orally active prodrugs of indomethacin | |
| CN102844290B (zh) | 用于治疗转甲状腺素蛋白淀粉样变性的1-(2-氟联苯-4-基)-烷基羧酸衍生物 | |
| JPS58162554A (ja) | 2−(p−イソブチルフエニル)プロピオン酸誘導体、その製造法およびそれを有効成分とする医薬 | |
| PT86811B (pt) | Processo para a preparacao de amidas de acidos gordos saturados inibidoras do acil-coenzima a: colesterol aciltransferase e de composicoes farmaceuticas que as contem | |
| WO2010139482A1 (en) | Glomerulonephritis treatment | |
| ITMI20010985A1 (it) | Farmaci per il morbo di alzheimer | |
| JPH02268178A (ja) | 3―ホルミルアミノ―7―メチルスルホニルアミノ―6―フェノキシ―4h―1―ベンゾピラン―4―オンまたはその塩を含有する抗炎症製剤 | |
| JPH0215048A (ja) | ネオペンチルエステル誘導体、その製造法およびその医薬としての使用 | |
| Venu et al. | Synthesis and crystallographic analysis of benzophenone derivatives—The potential anti-inflammatory agents | |
| CN100545162C (zh) | 一种非甾体解热镇痛抗关节炎的新化合物及其药物组合物 | |
| JPS5973516A (ja) | 抗炎症剤 | |
| CN104418818A (zh) | 帕瑞昔布钠无水化合物 | |
| JPH0247463B2 (ja) | ||
| WO2018133765A1 (zh) | 2-乳酰氨基苯甲酸的制备方法与应用 | |
| CN103998412B (zh) | 2‑氟苯基丙酸衍生物 | |
| CN109535126B (zh) | 一种用于抗炎的cox/lox抑制剂及其制备方法和应用 | |
| RU2542100C1 (ru) | Сокристаллическая форма теофиллина с дифлунисалом или диклофенаком | |
| JPS5936614B2 (ja) | 安息香酸誘導体およびそれらの塩の製造方法 | |
| US4529737A (en) | Analgesic and anti-inflammatory arylalkanoic acid phthalidyl esters and pharmaceutical compositions thereof | |
| CN102260276A (zh) | 普拉格雷枸橼酸盐及其制备方法 | |
| WO2022121222A1 (zh) | 吡啶巯乙酸类化合物及其制备方法、药学衍生物或配剂以及应用 | |
| WO2023274242A1 (zh) | 苯基[a]吲哚[2,3-g]并喹嗪类化合物的晶型、其盐和盐的晶型及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080009343.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10746132 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010746132 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20117022258 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13148566 Country of ref document: US |