WO2011148586A1 - 末端不飽和ポリオレフィン、及びその製造方法 - Google Patents
末端不飽和ポリオレフィン、及びその製造方法 Download PDFInfo
- Publication number
- WO2011148586A1 WO2011148586A1 PCT/JP2011/002728 JP2011002728W WO2011148586A1 WO 2011148586 A1 WO2011148586 A1 WO 2011148586A1 JP 2011002728 W JP2011002728 W JP 2011002728W WO 2011148586 A1 WO2011148586 A1 WO 2011148586A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- polyolefin
- propylene
- butene
- copolymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F10/04—Monomers containing three or four carbon atoms
- C08F10/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F290/00—Macromolecular compounds obtained by polymerising monomers on to polymers modified by introduction of aliphatic unsaturated end or side groups
- C08F290/02—Macromolecular compounds obtained by polymerising monomers on to polymers modified by introduction of aliphatic unsaturated end or side groups on to polymers modified by introduction of unsaturated end groups
- C08F290/04—Polymers provided for in subclasses C08C or C08F
- C08F290/042—Polymers of hydrocarbons as defined in group C08F10/00
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/50—Partial depolymerisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/10—Homopolymers or copolymers of propene
- C08L23/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/18—Homopolymers or copolymers of hydrocarbons having four or more carbon atoms
- C08L23/20—Homopolymers or copolymers of hydrocarbons having four or more carbon atoms having four to nine carbon atoms
- C08L23/22—Copolymers of isobutene; Butyl rubber; Homopolymers or copolymers of other iso-olefins
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D155/00—Coating compositions based on homopolymers or copolymers, obtained by polymerisation reactions only involving carbon-to-carbon unsaturated bonds, not provided for in groups C09D123/00 - C09D153/00
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J123/00—Adhesives based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Adhesives based on derivatives of such polymers
- C09J123/02—Adhesives based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Adhesives based on derivatives of such polymers not modified by chemical after-treatment
- C09J123/10—Homopolymers or copolymers of propene
- C09J123/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J123/00—Adhesives based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Adhesives based on derivatives of such polymers
- C09J123/02—Adhesives based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Adhesives based on derivatives of such polymers not modified by chemical after-treatment
- C09J123/10—Homopolymers or copolymers of propene
- C09J123/14—Copolymers of propene
- C09J123/142—Copolymers of propene at least partially crystalline copolymers of propene with other olefins
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J123/00—Adhesives based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Adhesives based on derivatives of such polymers
- C09J123/02—Adhesives based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Adhesives based on derivatives of such polymers not modified by chemical after-treatment
- C09J123/18—Homopolymers or copolymers of hydrocarbons having four or more carbon atoms
- C09J123/20—Homopolymers or copolymers of hydrocarbons having four or more carbon atoms having four to nine carbon atoms
- C09J123/22—Copolymers of isobutene; Butyl rubber ; Homo- or copolymers of other iso-olefines
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J155/00—Adhesives based on homopolymers or copolymers, obtained by polymerisation reactions only involving carbon-to-carbon unsaturated bonds, not provided for in groups C09J123/00 - C09J153/00
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K3/00—Materials not provided for elsewhere
- C09K3/10—Materials in mouldable or extrudable form for sealing or packing joints or covers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F110/00—Homopolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F110/04—Monomers containing three or four carbon atoms
- C08F110/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/04—Monomers containing three or four carbon atoms
- C08F210/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2810/00—Chemical modification of a polymer
- C08F2810/40—Chemical modification of a polymer taking place solely at one end or both ends of the polymer backbone, i.e. not in the side or lateral chains
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/10—Homopolymers or copolymers of propene
- C08L23/14—Copolymers of propene
- C08L23/142—Copolymers of propene at least partially crystalline copolymers of propene with other olefins
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2200/00—Chemical nature of materials in mouldable or extrudable form for sealing or packing joints or covers
- C09K2200/06—Macromolecular organic compounds, e.g. prepolymers
- C09K2200/0615—Macromolecular organic compounds, e.g. prepolymers obtained by reactions only involving carbon-to-carbon unsaturated bonds
- C09K2200/0617—Polyalkenes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2200/00—Chemical nature of materials in mouldable or extrudable form for sealing or packing joints or covers
- C09K2200/06—Macromolecular organic compounds, e.g. prepolymers
- C09K2200/0642—Copolymers containing at least three different monomers
Definitions
- the present invention relates to a terminal unsaturated polyolefin and a method for producing the same. More specifically, the present invention relates to a terminal unsaturated polyolefin having a large number of unsaturated groups at both ends and having a narrow molecular weight distribution, and an efficient method for producing a terminal unsaturated polyolefin capable of reducing by-products.
- High molecular weight polyolefins are widely used as industrial parts because they have high chemical stability, excellent mechanical properties, and are inexpensive. On the other hand, low molecular weight polyolefins are limited to use as waxes, but higher functionality is expected.
- Patent Document 1 discloses polypropylene obtained by pyrolyzing isotactic polypropylene at 370 ° C. (for example, the number of vinylidene groups per molecule is 1.8), and Patent Document 2 is pyrolysis obtained by pyrolyzing polybutene at 370 ° C. Polybutenes (for example, 1.53 to 1.75 vinylidene groups per molecule) are disclosed.
- the vinylidene number is increased by high-decomposition of the high molecular weight product, but there is a problem that the yield decreases due to the generation of a large amount of by-products.
- Non-Patent Document 1 discloses pyrolytic polypropylene (for example, the number of vinylidene groups per molecule is 1.66 to 1.80). From the relationship between the molecular weight and the number of vinylidenes, the number of vinylidene groups per molecule is 1 It is disclosed that it is difficult to make it .8 or more, especially 2.0 or more. In addition, the method of Non-Patent Document 1 has a problem in yield because it can be obtained only as a mixture of saturated at both ends, saturated at one end, and unsaturated at both ends.
- the polyolefin obtained is a polyolefin having high stereoregularity [mmmm] or no stereoregularity.
- Patent Documents 3 to 6 In addition to the method using thermal decomposition, methods for controlling terminal unsaturated groups using a catalyst (for example, a metallocene catalyst) are disclosed (Patent Documents 3 to 6).
- a catalyst for example, a metallocene catalyst
- Patent Document 3 atactic polypropylene having a terminal vinylidene group is produced by polymerization with a zirconocene dichloride catalyst.
- Patent Document 4 discloses an example of producing isotactic polypropylene having a terminal vinylidene group using polypropylene having stereoregularity [mm] of 88.8 and 94%.
- Patent Document 5 discloses a polyolefin having a terminal vinylidene group with high purity and high selectivity using a low stereoregular polyolefin having a stereoregularity [mmmm] of 30 to 80%.
- the maximum number of terminal unsaturated groups is 1.0 per molecule, and a high degree of unsaturation cannot be achieved.
- Patent Document 6 discloses an unsaturated group introduction method by copolymerization. Specifically, a method for introducing a large amount of diolefin residues per molecule by ethylene / propylene / diolefin copolymerization is disclosed. However, in the case of copolymerization with a diolefin, it is difficult to introduce an unsaturated group at the terminal, and there is a problem that a side reaction such as crosslinking proceeds during the polymerization reaction.
- the polyolefin exhibiting stereoregularity is mainly highly crystalline isotactic polypropylene.
- flexibility, hardness control, and liquid and solid state control are required, and the molecular weight is in a low molecular weight to medium molecular weight region, and the number of terminal unsaturated groups and There has been a demand for a polyolefin base material with controlled stereoregularity.
- JP 2003-40921 A JP 2003-137927 A JP-A-6-65110 JP-A-4-226506 WO2008 / 047860 JP 2002-38129 A
- the following terminal unsaturated polyolefin and the like are provided.
- Propylene homopolymer a propylene copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and the content of ethylene and the ⁇ -olefin having 4 to 10 carbon atoms is 10 mol% or less
- a butene-1 homopolymer or a butene-1 copolymer comprising butene-1, ethylene, propylene and one or more ⁇ -olefins having 5 to 10 carbon atoms, ethylene
- a terminal unsaturated polyolefin comprising a butene-1 copolymer having a content of propylene and the ⁇ -olefin having 5 to 10 carbon atoms of 10 mol% or less, A terminal unsaturated polyolefin satisfying the following (1) to (4).
- the mesopentad fraction [mmmm] of the propylene chain part or butene-1 chain part is 20 to 80 mol%.
- the number of terminal vinylidene groups per molecule is 1.3 to 2.5.
- the weight average molecular weight Mw is 500 to 100,000.
- the molecular weight distribution Mw / Mn is 1.1 to 2.6.
- Propylene homopolymer a propylene copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and the content of ethylene and the ⁇ -olefin having 4 to 10 carbon atoms is 10 mol% or less
- a butene-1 homopolymer and a butene-1 copolymer comprising butene-1, ethylene, propylene and one or more ⁇ -olefins having 5 to 10 carbon atoms, ethylene
- Propylene homopolymer a propylene copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and the content of ethylene and the ⁇ -olefin having 4 to 10 carbon atoms is 10 mol% or less A butene-1 homopolymer; and a butene-1 copolymer comprising butene-1, ethylene, propylene and one or more ⁇ -olefins having 5 to 10 carbon atoms, ethylene, A terminal unsaturated polyolefin obtained by decomposing a raw material polyolefin selected from propylene and a butene-1 copolymer having a content of 10 to 10 mol% of propylene and an ⁇ -olefin having 5 to 10 carbon atoms, A terminal unsaturated polyolefin satisfying the following (7) to (10).
- the number of terminal vinylidene groups per molecule is 1.3 to 2.5 (8) fv ⁇ ⁇ 2 (Mp / Mm) +2 (where Mp is the number of terminal unsaturated polyolefins) (Mm is the number average molecular weight of the raw material polyolefin, where Mp / Mm is 0.05 to 0.8) (9) The weight average molecular weight Mw is 500 to 100,000. (10) The molecular weight distribution Mw / Mn is 1.1 to 2.6. A terminal unsaturated polyolefin obtained by decomposing the propylene homopolymer or the propylene-based copolymer, 8.
- Propylene homopolymer a propylene copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and the content of ethylene and the ⁇ -olefin having 4 to 10 carbon atoms is 10 mol% or less
- a butene-1 homopolymer and a butene-1 copolymer comprising butene-1, ethylene, propylene, and one or more ⁇ -olefins having 5 to 10 carbon atoms.
- a method for producing a graft copolymer comprising contacting a body (a) with a copolymerizable monomer (b), or a thermoplastic resin composition comprising the graft copolymer. 18.
- Propylene homopolymer a propylene copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and the content of ethylene and the ⁇ -olefins having 4 to 10 carbon atoms is 10 mol% A butene-1 homopolymer; and a butene-1 copolymer comprising butene-1, ethylene, propylene and one or more ⁇ -olefins having 5 to 10 carbon atoms; At both ends of a polyolefin selected from ethylene, propylene, and a butene-1 copolymer having a content of 10 to 10% by mole of the ⁇ -olefin having 5 to 10 carbon atoms, the following [I], [II], [ Polyolefin chain derived from at least one selected from III] and [IV], or poly derived from at least one selected from Group A and at least one selected from Group B A graft copolymer having olefin chain
- a thermoplastic resin composition comprising the graft copolymer according to 21.18 and a polyolefin. 22.
- the use which uses the graft copolymer of 24.18, or the thermoplastic resin composition of 21 as an adhesive agent, a resin compatibilizing agent, a dispersion, and a coating material Use in which the functionalized polyolefin according to 25.11 or 12 is used as an adhesive, a resin compatibilizing agent, a dispersion, or a coating material. Use in which the functionalized polyolefin described in 26.11 or 12 is used as a reactive hot melt adhesive, a sealing material, or a potting material.
- a low and medium stereoregular terminal unsaturated polyolefin having a high degree of unsaturation can be provided.
- ADVANTAGE OF THE INVENTION According to this invention, the manufacturing method of terminal unsaturated polyolefin with little generation amount of a by-product can be provided.
- the first terminal unsaturated polyolefin of the present invention is a propylene homopolymer; a propylene-based copolymer comprising propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and ethylene and carbon atoms having 4 to 10 carbon atoms.
- the first terminal unsaturated polyolefin of the present invention is a low stereoregular to medium stereoregular terminal unsaturated polyolefin and has flexibility, solvent solubility, low temperature fluidity and low crystallinity, It further has contradictory properties of heat resistance and solvent resistance due to the reaction of unsaturated groups.
- the first terminal unsaturated polyolefin of the present invention is a polyolefin comprising any of the following polymers (i) to (iv), preferably (i) a propylene homopolymer.
- a propylene homopolymer ii) a propylene-based copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, wherein the content of ethylene and ⁇ -olefins having 4 to 10 carbon atoms is A propylene copolymer of 10 mol% or less
- iii) a butene-1 homopolymer iv) a butene-1 copolymer comprising butene-1, ethylene, propylene and one or more ⁇ -olefins having 5 to 10 carbon atoms
- Examples of the ⁇ -olefin of the copolymers (ii) and (iv) include butene-1, pentene-1, heptene-1, hexene-1, heptene-1, octene-1, decene-1, 4-methylpentene. -1,3-methylbutene-1 and the like.
- the content of ethylene and ⁇ -olefin, or the content of ethylene, propylene and ⁇ -olefin is preferably more than 0 mol% and 5 mol% or less.
- the first terminal unsaturated polyolefin of the present invention has a propylene chain part or a butene-1 chain part mesopentad fraction [mmmm] of 20 to 80 mol%, preferably 25 to 70 mol%, more preferably Is 30 to 60 mol%.
- the mesopentad fraction of the propylene chain part or butene-1 chain part is less than 20 mol%, the crystallinity is not exhibited, so the strength is poor, and sufficient adhesive strength to crystalline polypropylene when used as an adhesive. May not be expressed.
- it exceeds 80 mol% the crystallinity becomes high, so that the low-temperature meltability becomes poor and the workability such as coating property may be lowered.
- the mesopentad fraction [mmmm] is based on the methyl group signal of the 13 C-NMR spectrum in accordance with the method proposed by A. Zambelli et al. In “Macromolecules, 6,925 (1973)”. The mesoprobability in pentad units in the measured polypropylene molecule or polybutene chain. As the mesopentad fraction [mmmm] increases, the stereoregularity increases.
- the 13 C-NMR spectrum can be measured according to the following equipment and conditions according to the assignment of peaks proposed by “Macromolecules, 8, 687 (1975)” by A. Zambelli et al. it can.
- Apparatus JNM-EX400 type 13 C-NMR apparatus manufactured by JEOL Ltd.
- Method Proton complete decoupling method Concentration: 220 mg / mL
- Solvent 90:10 (volume ratio) of 1,2,4-trichlorobenzene and heavy benzene Mixed solvent temperature: 130 ° C
- Pulse width 45 °
- Pulse repetition time 4 seconds Integration: 10,000 times
- the number of terminal vinylidene groups per molecule is 1.3 to 2.5, preferably 1.35 to 2.5, and more preferably 1.40 to 2.5. 2.0.
- the number of terminal vinylidene groups per molecule is less than 1.3, heat resistance may be insufficiently imparted by a reaction starting from the terminal vinylidene group.
- polyolefin contains a lot of branched structures, and the branched structure is different from the linear structure in the melt fluidity, so that the behavior such as coating may change. is there.
- the number of terminal vinylidene groups can also be determined using 13 C-NMR.
- all terminal group species are determined, and their abundance is measured.
- the number of terminal vinylidene groups per molecule is calculated from the ratio of terminal vinylidene groups to the total unsaturated groups.
- the selectivity of the terminal vinylidene group can be determined. The measurement of the number of terminal vinylidene groups using 1 H-NMR and 13 C-NMR will be described below using a propylene polymer as an example.
- the peak intensity of the amount of terminal vinyl groups in 13 C-NMR is calculated as follows using (A) and (B) obtained by 1 H-NMR spectrum.
- 13 C-NMR terminal vinyl group content peak intensity (B) / (A) ⁇ ⁇ 7>
- T the total concentration (T) of the end groups is expressed as follows.
- T (B) / (A) ⁇ ⁇ 7> + ⁇ 4> + ⁇ 5> + ⁇ 6> + ⁇ 7> Therefore, the ratio of each terminal is as follows.
- Terminal vinylidene group ⁇ 7> / T ⁇ 100 Unit: mol%
- Terminal vinyl group (B) / (A) ⁇ ⁇ 7> ⁇ 100
- E) n-propyl terminal ⁇ 5> / T ⁇ 100
- F) n-butyl group terminal ⁇ 6> / T ⁇ 100
- G) iso-butyl end ⁇ 4> / T ⁇ 100
- the number of terminal vinylidene groups per molecule is 2 ⁇ (C) / 100 units: pieces / molecule.
- the first terminal unsaturated polyolefin of the present invention has a weight average molecular weight Mw of 500 to 100,000, preferably 700 to 90,000, more preferably 800 to 80,000.
- Mw weight average molecular weight
- the weight average molecular weight is less than 500, the flexibility of the polyolefin may be hindered when heat resistance is imparted to the polyolefin by a reaction based on the terminal vinylidene group.
- the weight average molecular weight exceeds 100,000, the melt viscosity becomes large and workability such as coating property may be deteriorated.
- the weight average molecular weight can be measured by gel permeation chromatography (GPC) method.
- the first terminal unsaturated polyolefin of the present invention has a molecular weight distribution Mw / Mn of 1.1 to 2.6, preferably 1.1 to 2.55, more preferably 1.1 to 2.5. It is. It is technically difficult to make the molecular weight distribution less than 1.1. On the other hand, when the molecular weight distribution is more than 2.6, it corresponds to an increase in low molecular weight polyolefin, which may cause a decrease in mechanical properties.
- the molecular weight distribution Mw / Mn can be determined by measuring the weight average molecular weight (Mw) and the number average molecular weight (Mn) by the GPC method.
- the weight average molecular weight (Mw) and the number average molecular weight (Mn) are determined by the Universal Calibration method using the constants K and a of the Mark-Houwink-Sakurada formula in order to convert the polystyrene-equivalent molecular weight into the molecular weight of the corresponding polymer.
- the GPC measurement apparatus and measurement conditions are, for example, as follows.
- Detector RI detector for liquid chromatography
- Waters 150C Column: TOSO GMHHR-H (S) HT
- Solvent 1,2,4-trichlorobenzene Measurement temperature: 145 ° C
- Flow rate 1.0 mL / min
- Sample concentration 0.3% by mass
- the first terminal unsaturated polyolefin of the present invention is a propylene homopolymer; a propylene-based copolymer comprising propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and ethylene and carbon atoms having 4 to 10 carbon atoms.
- terminal unsaturated polyolefin can reduce the amount of a liquid or gaseous by-product, purity can be improved. Moreover, terminal unsaturated polyolefin can be manufactured at low cost. Since the stereoregularity [mmmm] is 20 to 80 mol%, the raw material polyolefin can be easily melted at a low temperature or has good solubility in a solvent. It is wide and can be decomposed at relatively low temperatures. Thereby, it has the merit which can control a side reaction. In addition, in the decomposition using the organic peroxide in combination, the decomposition can be efficiently performed with a milder and shorter reaction, and the above-mentioned merit of the raw material polyolefin can be increased. In particular, when the raw material polyolefin has a terminal unsaturated group in advance, the above merits can be maximized.
- the propylene-based polymer (propylene homopolymer and propylene-based copolymer) and butene-based polymer (butene-1 homopolymer and butene-1 copolymer) which are raw material polyolefins of the first terminal unsaturated polyolefin Is the same as the propylene polymer and butene polymer of the first terminal unsaturated polyolefin.
- the stereoregularity [mmmm] of the propylene chain part or butene-1 chain part of the raw material polyolefin is the same as that of the first terminal unsaturated polyolefin.
- the weight average molecular weight Mw of the raw material polyolefin is 4000 to 1,000,000, preferably 5000 to 900,000, more preferably 6000 to 800,000.
- the decomposition rate may not be set high.
- the weight average molecular weight of the raw material polyolefin exceeds 1,000,000, the decomposition rate can be set high, but the viscosity at the time of decomposition is high, and there is a possibility that constraints such as stirring power and stirring uniformity in the process may occur. .
- the raw material polyolefin preferably has 0.4 to 1.0 terminal unsaturated groups per molecule, more preferably 0.45 to 1.0, still more preferably 0.5 to 1.0, most preferably Preferably 0.55 to 1.0.
- the number of terminal unsaturated groups per molecule is less than 0.4, the number of terminal vinylidene groups due to decomposition does not increase so much, and in order to increase the terminal vinylidene groups, it is necessary to decompose the high molecular weight body. There is a fear.
- the number of terminal unsaturated groups per molecule exceeds 1.0, it is difficult to produce by the technique using a polymerization catalyst described later.
- terminal unsaturated group examples include a vinyl group, a vinylidene group, and a trans (vinylene) group, and a vinylidene group is preferable.
- Vinylidene groups are radically polymerizable and have a wide range of applications for various reactions, and can meet various requirements.
- Raw material polyolefin can be manufactured by using a metallocene catalyst which consists of a combination of the following component (A), (B) and (C), for example, and using hydrogen as a molecular weight regulator. Specifically, it can be produced by the method disclosed in WO2008 / 047860.
- A Transition metal compound containing a metal element belonging to Groups 3 to 10 of the periodic table having a cyclopentadienyl group, a substituted cyclopentadienyl group, an indenyl group, or a substituted indenyl group.
- B Reacts with a transition metal compound. That can form an ionic complex
- C Organoaluminum compound
- the transition metal compound containing a metal element belonging to Groups 3 to 10 of the periodic table having a cyclopentadienyl group, a substituted cyclopentadienyl group, an indenyl group or a substituted indenyl group as the component (A) can be represented by the following general formula (I ) Is represented.
- M represents a metal element of Groups 3 to 10 of the periodic table, and specific examples include titanium, zirconium, hafnium, yttrium, vanadium, chromium, manganese, nickel, cobalt, palladium, and lanthanoid series. Metal etc. are mentioned. Among these, titanium, zirconium and hafnium are preferable from the viewpoint of olefin polymerization activity and the like, and zirconium is most preferable from the viewpoint of yield of terminal vinylidene group and catalytic activity.
- E 1 and E 2 are substituted cyclopentadienyl group, indenyl group, substituted indenyl group, heterocyclopentadienyl group, substituted heterocyclopentadienyl group, amide group (—N ⁇ ), phosphine group (—P ⁇ ), Hydrocarbon group [>CR-,> C ⁇ ] and silicon-containing group [>SiR-,> Si ⁇ ] (where R is hydrogen, a hydrocarbon group having 1 to 20 carbon atoms, or a heteroatom-containing group)
- a ligand selected from among (A) is shown, and a crosslinked structure is formed through A 1 and A 2 .
- E 1 and E 2 may be the same as or different from each other.
- a cyclopentadienyl group, a substituted cyclopentadienyl group, an indenyl group and a substituted indenyl group are preferable, and at least one of E 1 and E 2 is a cyclopentadienyl group, A substituted cyclopentadienyl group, an indenyl group or a substituted indenyl group;
- X represents a ⁇ -bonding ligand, and when there are a plurality of Xs, the plurality of Xs may be the same or different, and may be cross-linked with other X, E 1 , E 2 or Y.
- X include a halogen atom, a hydrocarbon group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, an aryloxy group having 6 to 20 carbon atoms, an amide group having 1 to 20 carbon atoms, carbon Examples thereof include silicon-containing groups having 1 to 20 carbon atoms, phosphide groups having 1 to 20 carbon atoms, sulfide groups having 1 to 20 carbon atoms, acyl groups having 1 to 20 carbon atoms, and the like.
- the halogen atom include a chlorine atom, a fluorine atom, a bromine atom, and an iodine atom.
- hydrocarbon group having 1 to 20 carbon atoms include alkyl groups such as methyl group, ethyl group, propyl group, butyl group, hexyl group, cyclohexyl group, and octyl group; vinyl group, propenyl group, cyclohexenyl group and the like
- An arylalkyl group such as benzyl group, phenylethyl group, phenylpropyl group; phenyl group, tolyl group, dimethylphenyl group, trimethylphenyl group, ethylphenyl group, propylphenyl group, biphenyl group, naphthyl group, methylnaphthyl group Group, anthracenyl group, aryl group such as phenanthonyl group, and the like.
- alkyl groups such as methyl group, ethyl group, and propyl group
- aryl groups such as phenyl
- Examples of the alkoxy group having 1 to 20 carbon atoms include alkoxy groups such as methoxy group, ethoxy group, propoxy group, and butoxy group, phenylmethoxy group, and phenylethoxy group.
- Examples of the aryloxy group having 6 to 20 carbon atoms include phenoxy group, methylphenoxy group, and dimethylphenoxy group.
- Examples of the amide group having 1 to 20 carbon atoms include a dimethylamide group, a diethylamide group, a dipropylamide group, a dibutylamide group, a dicyclohexylamide group, and a methylethylamide group, a divinylamide group, and a dipropenylamide group.
- Alkenylamide groups such as dicyclohexenylamide group; arylalkylamide groups such as dibenzylamide group, phenylethylamide group and phenylpropylamide group; arylamide groups such as diphenylamide group and dinaphthylamide group.
- Examples of the silicon-containing group having 1 to 20 carbon atoms include monohydrocarbon-substituted silyl groups such as methylsilyl group and phenylsilyl group; dihydrocarbon-substituted silyl groups such as dimethylsilyl group and diphenylsilyl group; trimethylsilyl group, triethylsilyl group, Trihydrocarbon-substituted silyl groups such as tripropylsilyl group, tricyclohexylsilyl group, triphenylsilyl group, dimethylphenylsilyl group, methyldiphenylsilyl group, tolylsilylsilyl group and trinaphthylsilyl group; hydrocarbons such as trimethylsilyl ether group A substituted silyl ether group; a silicon-substituted alkyl group such as a trimethylsilylmethyl group; a silicon-substituted aryl group such as a tri
- Examples of the phosphide group having 1 to 20 carbon atoms include methyl sulfide groups, ethyl sulfide groups, propyl sulfide groups, butyl sulfide groups, hexyl sulfide groups, cyclohexyl sulfide groups, octyl sulfide groups and the like; vinyl sulfide groups, propenyl sulfides Group, alkenyl sulfide group such as cyclohexenyl sulfide group; arylalkyl sulfide group such as benzyl sulfide group, phenylethyl sulfide group, phenylpropyl sulfide group; phenyl sulfide group, tolyl sulfide group, dimethylphenyl sulfide group, trimethylphenyl sulfide group, Ethyl phenyl
- Examples of the sulfide group having 1 to 20 carbon atoms include alkyl sulfide groups such as methyl sulfide group, ethyl sulfide group, propyl sulfide group, butyl sulfide group, hexyl sulfide group, cyclohexyl sulfide group, octyl sulfide group; vinyl sulfide group, propenyl sulfide Group, alkenyl sulfide group such as cyclohexenyl sulfide group; arylalkyl sulfide group such as benzyl sulfide group, phenylethyl sulfide group, phenylpropyl sulfide group; phenyl sulfide group, tolyl sulfide group, dimethylphenyl sulfide group, trimethylphenyl sulfide group, E
- acyl group having 1 to 20 carbon atoms examples include formyl group, acetyl group, propionyl group, butyryl group, valeryl group, palmitoyl group, thearoyl group, oleoyl group and other alkyl acyl groups, benzoyl group, toluoyl group, salicyloyl group, Examples thereof include arylacyl groups such as cinnamoyl group, naphthoyl group and phthaloyl group, and oxalyl group, malonyl group and succinyl group respectively derived from dicarboxylic acid such as oxalic acid, malonic acid and succinic acid.
- Y represents a Lewis base, and when there are a plurality of Y, the plurality of Y may be the same or different, and may be cross-linked with other Y, E 1 , E 2 or X.
- Specific examples of the Lewis base of Y include amines, ethers, phosphines, thioethers and the like.
- Examples of the amine include amines having 1 to 20 carbon atoms, and specifically include methylamine, ethylamine, propylamine, butylamine, cyclohexylamine, methylethylamine, dimethylamine, diethylamine, dipropylamine, dibutylamine, dicyclohexylamine.
- Alkylamines such as methylethylamine; alkenylamines such as vinylamine, propenylamine, cyclohexenylamine, divinylamine, dipropenylamine, dicyclohexenylamine; arylalkylamines such as phenylamine, phenylethylamine, phenylpropylamine; Examples include arylamines such as naphthylamine.
- ethers include aliphatic single ether compounds such as methyl ether, ethyl ether, propyl ether, isopropyl ether, butyl ether, isobutyl ether, n-amyl ether, and isoamyl ether; methyl ethyl ether, methyl propyl ether, methyl isopropyl ether, Aliphatic hybrid ether compounds such as methyl-n-amyl ether, methyl isoamyl ether, ethyl propyl ether, ethyl isopropyl ether, ethyl butyl ether, ethyl isobutyl ether, ethyl n-amyl ether, ethyl isoamyl ether; vinyl ether, allyl ether, methyl Aliphatic unsaturated ether compounds such as vinyl ether, methyl allyl ether, ethyl vinyl ether, ethy
- phosphines include phosphines having 1 to 20 carbon atoms. Specific examples include monohydrocarbon-substituted phosphines such as methylphosphine, ethylphosphine, propylphosphine, butylphosphine, hexylphosphine, cyclohexylphosphine, and octylphosphine; dimethylphosphine, diethylphosphine, dipropylphosphine, dibutylphosphine, dihexylphosphine, dicyclohexyl Dihydrocarbon-substituted phosphines such as phosphine and dioctylphosphine; alkyl phosphines such as trihydrocarbon-substituted phosphines such as trimethylphosphine, triethylphosphine, tripropylphosphine, tribu
- a 1 and A 2 are divalent bridging groups for bonding two ligands, which are a hydrocarbon group having 1 to 20 carbon atoms, a halogen-containing hydrocarbon group having 1 to 20 carbon atoms, and a silicon-containing group.
- R 1 represents a hydrogen atom, a halogen atom, a hydrocarbon group having 1 to 20 carbon atoms or a halogen-containing hydrocarbon group having 1 to 20 carbon atoms, and they may be the same as each other May be different.
- q is an integer of 1 to 5 and represents [(valence of M) -2], and r represents an integer of 0 to 3.
- At least one is preferably a crosslinking group composed of a hydrocarbon group having 1 or more carbon atoms.
- an ethylene group, an isopropylidene group, and a dimethylsilylene group are preferable.
- R 2 and R 3 are each a hydrogen atom or a hydrocarbon group having 1 to 20 carbon atoms, and they are the same as each other. However, they may be different from each other, and may be bonded to each other to form a ring structure, and e represents an integer of 1 to 4.
- transition metal compound represented by the general formula (I) include specific examples described in WO2008 / 066168.
- analogous compound of the metal element of another group may be sufficient.
- a transition metal compound belonging to Group 4 of the periodic table is preferred, and a zirconium compound is particularly preferred.
- the compound represented by the general formula (II) is preferable.
- M represents a metal element belonging to Groups 3 to 10 of the periodic table
- a 1a and A 2a each represent a bridging group represented by the general formula (a) in the general formula (I).
- CH 2 , CH 2 CH 2 , (CH 3 ) 2 C, (CH 3 ) 2 C (CH 3 ) 2 C, (CH 3 ) 2 Si and (C 6 H 5) 2 Si are preferred.
- a 1a and A 2a may be the same as or different from each other.
- R 4 to R 13 each represent a hydrogen atom, a halogen atom, a hydrocarbon group having 1 to 20 carbon atoms, a halogen-containing hydrocarbon group having 1 to 20 carbon atoms, a silicon-containing group, or a heteroatom-containing group.
- halogen atom the hydrocarbon group having 1 to 20 carbon atoms, and the silicon-containing group are the same as those described in the general formula (I).
- halogen-containing hydrocarbon group having 1 to 20 carbon atoms examples include p-fluorophenyl group, 3,5-difluorophenyl group, 3,4,5-trifluorophenyl group, pentafluorophenyl group, 3,5-bis ( (Trifluoro) phenyl group, fluorobutyl group and the like.
- heteroatom-containing group examples include C1-C20 heteroatom-containing groups.
- nitrogen-containing groups such as dimethylamino group, diethylamino group, and diphenylamino group; phenylsulfide group, methylsulfide group, and the like
- Sulfur-containing groups of: Phosphorus-containing groups such as dimethylphosphino group and diphenylphosphino group; Oxygen-containing groups such as methoxy group, ethoxy group and phenoxy group.
- R 4 and R 5 a group containing a hetero atom such as a halogen atom, oxygen, or silicon is preferable because of high polymerization activity.
- R 6 to R 13 are preferably a hydrogen atom or a hydrocarbon group having 1 to 20 carbon atoms.
- X and Y are the same as in general formula (I).
- q is an integer of 1 to 5 and represents [(valence of M) -2], and r represents an integer of 0 to 3.
- transition metal compounds represented by the above general formula (II) when both indenyl groups are the same, examples of the transition metal compounds belonging to Group 4 of the periodic table include specific examples described in WO2008 / 066168. . Further, it may be a compound similar to a metal element other than Group 4. A transition metal compound belonging to Group 4 of the periodic table is preferred, and a zirconium compound is particularly preferred.
- transition metal compounds represented by the general formula (II) when R 5 is a hydrogen atom and R 4 is not a hydrogen atom, transition metal compounds belonging to Group 4 of the periodic table are disclosed in WO2008 / 066168. Specific examples of the description are given. Further, it may be a compound similar to a metal element other than Group 4. A transition metal compound belonging to Group 4 of the periodic table is preferred, and a zirconium compound is particularly preferred.
- a high purity terminal unsaturated olefin polymer having a relatively low molecular weight can be obtained, and A borate compound is preferable in terms of high catalyst activity.
- Specific examples of the borate compound include those described in WO2008 / 066168. These can be used individually by 1 type or in combination of 2 or more types.
- dimethylanilinium tetrakis (pentafluorophenyl) borate, triphenylcarbenium tetrakis (pentafluorophenyl) borate, and tetrakis (Perfluorophenyl) methylanilinium borate and the like are preferable.
- the catalyst used in the production method of the present invention may be a combination of the component (A) and the component (B), and an organoaluminum compound is used as the component (C) in addition to the component (A) and the component (B). Also good.
- organoaluminum compound (C) trimethylaluminum, triethylaluminum, triisopropylaluminum, triisobutylaluminum, trinormalhexylaluminum, trinormaloctylaluminum, dimethylaluminum chloride, diethylaluminum chloride, methylaluminum dichloride, ethylaluminum dichloride.
- Dimethylaluminum fluoride, diisobutylaluminum hydride, diethylaluminum hydride and ethylaluminum sesquichloride These organoaluminum compounds may be used alone or in combination of two or more.
- trialkylaluminum such as trimethylaluminum, triethylaluminum, triisopropylaluminum, triisobutylaluminum, trinormalhexylaluminum and trinormaloctylaluminum is preferable, and triisobutylaluminum, trinormalhexylaluminum and trimethylaluminum Normal octyl aluminum is more preferred.
- the amount of component (A) used is usually 0.1 ⁇ 10 ⁇ 6 to 1.5 ⁇ 10 ⁇ 5 mol / L, preferably 0.15 ⁇ 10 ⁇ 6 to 1.3 ⁇ 10 ⁇ 5 mol / L, More preferably, it is 0.2 ⁇ 10 ⁇ 6 to 1.2 ⁇ 10 ⁇ 5 mol / L, and particularly preferably 0.3 ⁇ 10 ⁇ 6 to 1.0 ⁇ 10 ⁇ 5 mol / L.
- the amount of component (A) used is 0.1 ⁇ 10 ⁇ 6 mol / L or more, the catalytic activity is sufficiently expressed, and when it is 1.5 ⁇ 10 ⁇ 5 mol / L or less, the heat of polymerization is easy. Can be removed.
- the use ratio (A) / (B) of the component (A) and the component (B) is preferably 10/1 to 1/100, more preferably 2/1 to 1/10 in terms of molar ratio.
- (A) / (B) is in the range of 10/1 to 1/100, an effect as a catalyst can be obtained and the catalyst cost per unit mass polymer can be suppressed.
- the use ratio (A) / (C) of the component (A) and the component (C) is preferably 1/1 to 1/10000, more preferably 1/5 to 1/2000, and even more preferably 1 in terms of molar ratio. / 10 to 1/1000.
- the preliminary contact may be performed using the above-described component (A) and component (B), or component (A), component (B) and component (C).
- the preliminary contact can be performed by bringing the component (A) into contact with, for example, the component (B).
- the method is not particularly limited, and a known method can be used. Such preliminary contact is effective in reducing the catalyst cost, such as improving the catalyst activity and reducing the proportion of the (B) component used as the promoter.
- the first terminal unsaturated polyolefin is produced by decomposing the raw material polyolefin in an inert gas atmosphere, and the decomposition is preferably a thermal decomposition reaction or a radical decomposition reaction.
- a radical decomposition reaction is particularly preferred because the reaction proceeds under relatively mild conditions.
- the thermal decomposition reaction is performed by heat-treating the raw material polyolefin.
- the heating temperature can be adjusted by setting a target molecular weight and taking into consideration the results of experiments conducted in advance, and is preferably 300 to 400 ° C, more preferably 310 to 390 ° C. If the heating temperature is less than 300 ° C, the thermal decomposition reaction may not proceed. On the other hand, when heating temperature exceeds 400 degreeC, there exists a possibility that the terminal unsaturated polyolefin obtained may deteriorate.
- the thermal decomposition time (heat treatment time) is preferably 30 minutes to 10 hours, and more preferably 60 to 240 minutes. When the thermal decomposition time is less than 30 minutes, the amount of terminal unsaturated polyolefin obtained may be small. On the other hand, when the thermal decomposition time exceeds 10 hours, the terminal unsaturated polyolefin obtained may be deteriorated.
- the thermal decomposition reaction is performed by, for example, using a stainless steel reaction vessel equipped with a stirrer as the thermal decomposition device, filling this vessel with an inert gas such as nitrogen or argon, and placing the raw material polyolefin into heat melting.
- the molten polymer phase can be bubbled with an inert gas and heated at a predetermined temperature for a predetermined time while extracting a volatile product.
- the radical decomposition reaction can be carried out at a temperature of 160 to 300 ° C. by adding 0.05 to 2.0 mass% of an organic peroxide with respect to the raw material polyolefin.
- the decomposition temperature is preferably 170 to 290 ° C, more preferably 180 to 280 ° C. When the decomposition temperature is less than 160 ° C., the decomposition reaction may not proceed. On the other hand, when the decomposition temperature is higher than 300 ° C., the decomposition proceeds vigorously, and the decomposition may be completed before the organic peroxide is sufficiently diffused uniformly into the molten polymer by stirring, which may reduce the yield.
- the organic peroxide to be added is preferably an organic peroxide having a 1-minute half-life temperature of 140 to 270 ° C.
- specific examples of the organic peroxide include the following compounds: diisobutyryl peroxide, Cumylperoxyneodecanoate, di-n-propylperoxydicarbonate, diisopropylperoxydicarbonate, di-sec-butylperoxydicarbonate, 1,1,3,3-tetramethylbutylperoxyneodecano Eight, di (4-t-butylcyclohexyl) peroxydicarbonate, di (2-ethylhexyl) peroxydicarbonate, t-hexylperoxyneodecanoate, t-butylperoxyneoheptanoate, t-hexyl Peroxypivalate, t-butyl peroxypivalate, di (3 , 5-trimethylhexanoyl) peroxide,
- the amount of the organic peroxide added is preferably 0.1 to 1.8% by mass, more preferably 0.2 to 1.7% by mass with respect to the raw material polyolefin.
- the addition amount is less than 0.05% by mass, the decomposition reaction rate may be slowed and the production efficiency may be deteriorated.
- the addition amount exceeds 2.0 mass%, the odor resulting from the decomposition of the organic peroxide may cause a problem.
- the decomposition time of the decomposition reaction is, for example, 30 seconds to 10 hours, preferably 1 minute to 1 hour.
- the decomposition time is less than 30 seconds, not only does the decomposition reaction not proceed sufficiently, but a large amount of undecomposed organic peroxide may remain.
- the decomposition time exceeds 10 hours, there is a concern that the cross-linking reaction, which is a side reaction, may progress, and the resulting polyolefin may be yellowed.
- the radical decomposition reaction can be carried out by using, for example, either a batch method or a melt continuous method.
- a radical thermal decomposition reaction can be carried out by dropping an organic oxide and heating at a predetermined temperature for a predetermined time.
- the organic peroxide may be dropped within the range of the decomposition time, and the dropping may be either continuous dropping or divided dropping. Further, the reaction time from the dropping end time is preferably within the range of the reaction time.
- the organic peroxide may be dissolved in a solvent and dropped as a solution.
- the solvent is preferably a hydrocarbon solvent, and specific examples include aliphatic hydrocarbons such as heptane, octane, decane, dodecane, tetradecane, hexadecane, and nanodecane; methylcyclopentane, cyclohexane, methylcyclohexane, cyclooctane, And alicyclic hydrocarbons such as cyclododecane; and aromatic hydrocarbons such as benzene, toluene, xylene, ethylbenzene, and trimethylbenzene.
- a solvent having a boiling point of 100 ° C. or higher is preferable.
- the raw material polyolefin may be dissolved in a solvent during decomposition.
- the decomposition temperature is usually in the range of 100 to 250 ° C., preferably in the range of 120 to 200 ° C.
- the reaction time as viewed from the average residence time is, for example, 20 seconds to 10 minutes.
- the melt continuous method can improve the mixing state and shorten the reaction time compared to the batch method.
- a method of impregnating the raw material polyolefin with the organic peroxide using the above-mentioned apparatus, or a method of individually supplying and mixing the raw material polyolefin and the organic peroxide can be applied.
- the impregnation of the raw material polyolefin with the organic peroxide is performed by adding a predetermined amount of the organic peroxide to the raw material polyolefin in the presence of an inert gas such as nitrogen, and stirring the mixture at room temperature to 40 ° C.
- the raw material pellets can be uniformly absorbed and impregnated.
- the obtained raw material polyolefin (impregnated pellet) impregnated with the organic peroxide is decomposed by melt extrusion, or the impregnated pellet is added to the raw material polyolefin as a master batch and decomposed to obtain a terminal unsaturated polyolefin.
- the raw material polyolefin is absorbed and impregnated as a solution in which the organic peroxide is previously dissolved in a hydrocarbon solvent. It is good to let them.
- the raw material polyolefin and the organic peroxide are supplied to the extruder hopper at a constant flow rate, or the organic peroxide is supplied at a constant flow rate in the middle of the barrel. Can be implemented.
- the second terminal unsaturated polyolefin of the present invention is a propylene homopolymer; a propylene-based copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms.
- the number of terminal vinylidene groups per molecule is 1.3 to 2.5 (8) fv ⁇ ⁇ 2 (Mp / Mm) +2 (where Mp is the number of terminal unsaturated polyolefins) (Mm is the number average molecular weight of the raw material polyolefin, where Mp / Mm is 0.05 to 0.8) (9) The weight average molecular weight Mw is 500 to 100,000. (10) The molecular weight distribution Mw / Mn is 1.1 to 2.6.
- the propylene polymer (propylene homopolymer and propylene copolymer) and butene polymer (butene-1 homopolymer and butene-1 copolymer) which are the raw material polyolefins of the second terminal unsaturated polyolefin of the present invention.
- the polymer is the same as the propylene polymer and butene polymer of the first terminal unsaturated polyolefin.
- the weight average molecular weight of the raw material polyolefin is preferably 4000 to 1,000,000, and more preferably 10,000 to 800,000.
- the weight average molecular weight of the raw material polyolefin is less than 4000, the number of terminal vinylidene groups of the terminal unsaturated polyolefin is not sufficiently increased, and the volatile components of by-products may increase.
- the weight average molecular weight of the raw material polyolefin exceeds 1,000,000, the melt viscosity is high, and stirring may be hindered.
- the raw material polyolefin is preferably a polyolefin having a terminal unsaturated group, more preferably a polyolefin having a terminal vinylidene group.
- the number of the terminal vinylidene groups per molecule is preferably 0.4 to 1.0, and more preferably 0.5 to 1.0. More preferably, the number is 0.6 to 1.0.
- Mp / Mm decomposition rate
- the manufacturing method of raw material polyolefin is the same as that of raw material polyolefin used for manufacture of 1st terminal unsaturated polyolefin. That is, it can be produced by using a metallocene catalyst comprising a combination of the following components (A), (B) and (C) disclosed in WO2008 / 047860 and using hydrogen as a molecular weight regulator.
- A Transition metal compound containing a metal element belonging to Groups 3 to 10 of the periodic table having cyclopentadienyl group substitution, cyclopentadienyl group, indenyl group, and substituted indenyl group.
- B Reacting with transition metal compound
- C Organoaluminum compound that can form an ionic complex
- the raw material polyolefin used for the production of the second terminal unsaturated polyolefin when the stereoregularity [mmmm] of the raw material polyolefin used for the production of the second terminal unsaturated polyolefin is 20 to 80 mol%, the raw material polyolefin uses a borate compound as the component (B) and hydrogen is present. Otherwise, it can be produced by polymerizing using a trace amount of hydrogen, or using methylaluminoxane as the component (B) and polymerizing without using hydrogen.
- the catalyst used for the production of the raw material polyolefin is not limited to the metallocene catalyst disclosed in WO2008 / 047860, and the following catalysts can be used.
- Catalyst represented by the following formula (A-2), (A-3) or (A-4) CpM 1 R 5 e R 6 f R 7 g (A-2) Cp 2 M 1 R 5 h R 6 i (A-3) (Cp-A-Cp) M 1 R 5 h R 6 i (A-4)
- M 1 is a transition metal of Group 4 of the periodic table
- Cp is a cyclopentadienyl group, a substituted cyclopentadienyl group, an indenyl group, a substituted indenyl group, a tetrahydroindenyl group, a substituted tetrahydroindenyl group , A fluorenyl group, a substituted fluorenyl group, an octahydrofluorenyl group, a substituted octahydrofluorenyl group and an azulenyl group.
- R 5 , R 6 and R 7 each independently represent a ligand, and A represents a covalent bridge.
- e, f and g each represents an integer of 0 to 3
- h and i each represents an integer of 0 to 2.
- e, f, and g are not simultaneously 0, and h and i are not simultaneously 0.
- Two or more of R 5 , R 6 and R 7 may be bonded to each other to form a ring.
- two Cp may be the same or different from each other. ]
- the catalyst represented by the formula (A-2), (A-3) or (A-4) include bis (cyclopentadienyl) zirconium dichloride, bis (cyclopentadienyl) zirconium dimethyl, bis (Cyclopentadienyl) zirconium dihalide, bis (methylcyclopentadienyl) zirconium dichloride, bis (pentamethylcyclopentadienyl) zirconium dichloride, isopropylidene (cyclopentadienyl) (fluorenyl) zirconium dichloride, ethylene-bis (Indenyl) zirconium dichloride, dimethylsilyl-bis (indenyl) zirconium dichloride, and the like.
- Catalyst represented by the following formula (A-5) [Wherein M 2 is a titanium atom, a zirconium atom or a hafnium atom.
- Cp is a cyclic unsaturated hydrocarbon group such as cyclopentadienyl group, substituted cyclopentadienyl group, indenyl group, substituted indenyl group, tetrahydroindenyl group, substituted tetrahydroindenyl group, fluorenyl group or substituted fluorenyl group, or a chain An unsaturated hydrocarbon group.
- X 2 represents a hydrogen atom, a halogen atom, an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, an alkylaryl group or an arylalkyl group, or an alkoxy group having 1 to 20 carbon atoms.
- R 9 is a group selected from a hydrogen atom or an alkyl, aryl, silyl, halogenated alkyl, halogenated aryl group and combinations thereof having up to 20 non-hydrogen atoms, and R 10 has 1 to 10 carbon atoms. Or an aryl group having 6 to 10 carbon atoms, or may form a bonded ring system of one or more R9 and up to 30 non-hydrogen atoms. s represents 1 or 2. ]
- catalyst represented by the formula (A-5) include (tertiary ptylamide) (tetramethyl- ⁇ 5-cyclopentadienyl) 1,2 ethanediylzirconium dichloride: (tertiary ptylamide) (tetra Methyl- ⁇ 5-cyclopentadienyl) -1,2-ethanediyltitanium dichloride; (methylamide) (tetramethyl- ⁇ 5-cyclopentadienyl) -1,2-ethanediylzirconium dichloride: (methylamide) (tetra Methyl- ⁇ 5-cyclopentadie) -1,2-ethanediyltitanium dichloride; (ethylamide) (tetramethyl- ⁇ 5-cyclopentadienyl) methylenetitanium dichloride: (tertiary butylamide) dimethyl (tetramethyl- ⁇ 5- Cyclo
- the number of terminal vinylidene groups per molecule and the weight average molecular weight of the second terminal unsaturated polyolefin are the same as those of the first terminal unsaturated polyolefin.
- the second terminal unsaturated polyolefin satisfies the following formula. (8) fv ⁇ ⁇ 2 (Mp / Mm) +2 [Wherein, Mp is the number average molecular weight of the terminal unsaturated polyolefin, and Mm is the number average molecular weight of the raw material polyolefin. However, Mp / Mm is 0.05 to 0.8]
- the above formula (8) is a polyolefin having a high number of terminal unsaturated groups by using a raw material polyolefin having terminal unsaturated groups in advance, and having the same decomposition rate and a high number of terminal unsaturated groups even when compared with the same molecular weight. Is generated.
- Mp / Mm is an index corresponding to the decomposition rate.
- Mp / Mm is, for example, 0.05 to 0.8, and preferably 0.06 to 0.7.
- Mp / Mm is less than 0.05, the decomposition rate is high, and there is a possibility that by-products increase.
- Mp / Mm exceeds 0.8, the number of terminal vinylidene groups is small, and when polyolefin is used as an adhesive or the like, sufficient heat resistance may not be obtained.
- the second terminal unsaturated polyolefin has a molecular weight distribution Mw / Mn of 1.1 to 2.6, preferably 1.1 to 2.55, and more preferably 1.1 to 2.5.
- Mw / Mn molecular weight distribution of 1.1 to 2.6, preferably 1.1 to 2.55, and more preferably 1.1 to 2.5.
- the production of terminally unsaturated polyolefins with a molecular weight distribution of less than 1.1 is technically difficult.
- the molecular weight distribution is more than 2.6, it means that the low molecular weight polyolefin is increased, and the mechanical properties may be lowered.
- the second terminal unsaturated polyolefin is preferably a polyolefin obtained by decomposing a propylene homopolymer or a propylene copolymer, more preferably a propylene homopolymer, and these propylene homopolymer and propylene copolymer.
- the union has an atactic structure, a syndiotactic structure or an isotactic structure.
- the syndioctatic structure means that [rrrr] of the polyolefin is 30 to 95 mol%, and the isotactic structure means that [mmmm] is 30 to 95 mol%.
- the decomposition of the raw material polyolefin is the same as the decomposition of the raw material polyolefin in the method for producing the first terminal unsaturated polyolefin, and preferably a thermal decomposition method or a radical decomposition reaction.
- the decomposition reaction is performed by a radical decomposition reaction by a continuous melt process, and the raw material polyolefin is impregnated with the organic peroxide, the raw material polyolefin is highly ordered (mmmm ⁇ 90 mol% or rrrr ⁇ 90 mol). %), The impregnation property may be lowered.
- the first and second terminal unsaturated polyolefins of the present invention are preferably functionalized polyolefins in which 5 mol% or more of the terminal vinylidene groups have functional groups, and more preferably 10 mol% or more of the terminal vinylidene groups are functional groups.
- the functional group is preferably one or more functional groups selected from a hydroxyl group, an epoxy group, an alkoxysilicon group, an alkylsilicon group, a carboxyl group, an amino group, and an isocyanate group.
- the first and second terminal unsaturated polyolefins preferably have an acid anhydride structure.
- An acid anhydride structure is a structure in which one molecule of water is lost from two carboxyl groups of a carboxylic acid, and two acyl groups share one oxygen atom.
- R 1 COOCOR 2 For example, maleic anhydride, succinic anhydride, phthalic anhydride and the like can be mentioned.
- terminal unsaturated polyolefin When the terminal unsaturated polyolefin has a functional group, compatibility with a polar compound and dispersibility can be improved, and it becomes easy to obtain a composition with various polymers. Moreover, since terminal unsaturated polyolefin has a functional group, the solubility and dispersibility to polar solvents, such as water, can be improved, and it can be used as an emulsion adhesive or a coating material. For application to polyolefin materials, adhesion and paintability can be imparted, and the surface condition of organic inorganic pigments can be improved, making it possible to produce polyolefin master batches. Can be granted.
- Examples of methods for introducing functional groups include maleic anhydride ene addition reaction; introduction of hydroxyl groups with formic acid / hydrogen peroxide; epoxidation with peracetic acid; trimethoxysilane, triethoxysilane, triisopropoxysilane, methyldimethoxysilane, Introduction of alkoxysilicon groups by reaction with alkoxysilanes such as ethyldiethoxysilane, phenyldimethoxysilane, phenyldiethoxysilane; introduction of alkylsilicon groups by reaction with alkylsilanes such as tri-hexylsilane, tri-normal octylsilane; Carboxylation with copper bromide / tertiary butyl peroxyacetate; introduction of amino group by reaction of anhydrous maleate and diamine compound; introduction of isocyanate group by reaction of anhydrous maleate and diisocyanate compound It is.
- the functional group is introduced by hydroboration with BH 3 .THF; boronation with 9-boranebicyclo [3,3,1] nonane; metalation with isobutylaluminum hydride or the like; halogen with dibromo or hydrogen bromide Hydroformylation with formic acid / cobalt catalyst; aldehyde formation with carbon monoxide / dicobalt octacarbonyl catalyst; sulfonation with acetic anhydride / sulfuric acid
- the modified polymer obtained by reacting the functionalized polyolefin of the present invention with a polyfunctional compound having two or more functions can be used for applications such as a sealing material, a potting material, a reactive hot melt adhesive, and a paint.
- a polyfunctional compound having two or more functions examples include water, isocyanate compounds such as TDI and MDI, amine compounds such as hexamethylenediamine, polyethylene glycol and polypropylene glycols containing terminal hydroxyl groups, and terminal epoxy groups.
- Polybutadiene-modified products; polyacrylic acid and acrylic acid copolymers can be mentioned.
- the reaction between the functionalized polyolefin and the polyfunctional compound having two or more functions can be carried out in accordance with a generally used method, for example, in accordance with a method described in JP2009-185171A.
- the functional group of the functionalized polyolefin is a hydroxyl group
- the bifunctional or higher polyfunctional compound that can be suitably used is a compound having an isocyanate group, a carboxylic acid group, and an epoxy group.
- an epoxy group is a compound having a hydroxyl group, an amino group, or an isocyanate group. In the case of alkoxy silicon, it has a hydroxyl group such as water.
- a carboxyl group it is a compound having a hydroxyl group, an epoxy group, or an amino group.
- an amino group it is an epoxy group, an isocyanate group, or a carboxyl group.
- an isocyanate group it is a compound having an active hydrogen such as a hydroxyl group or an amino group.
- an acid anhydride structure it is a hydroxyl group, an amino group, an epoxy group, or an isocyanate group.
- the crosslinked product obtained by moisture curing the functionalized polyolefin of the present invention containing an alkoxysilicon group as a functional group can be used for a sealing material, a potting material, a reactive hot melt adhesive, and the like.
- Moisture curing of a functionalized polyolefin containing an alkoxysilicon group as a functional group can usually be carried out by a treatment in contact with moisture or moisture.
- a silanol condensation catalyst may be used as the curing catalyst at this time. Examples of the silanol condensation catalyst include organometallic catalysts and tertiary amines.
- organic metals examples include dibutyltin dilaurate, dibutyltin diacetate, dibutyltin dioctate, tin octenoate, lead octenoate, lead naphthenate and the like.
- Tertiary amines include N-triethylamine, N-methylmorpholine bis (2-dimethylaminoethyl) ether, N, N, N ′, N ′′, N ′′, N ′′ -pentamethyldiethylenetriamine, N, N, N '-Trimethylaminoethyl-ethanolamine, bis (2-dimethylaminoethyl) ether, N-methyl-N'-dimethylaminoethylpiperazine, imidazole compounds in which the secondary amine functional group of the imidazole ring is substituted with a cyanoethyl group, etc.
- These catalysts may be used alone or in combination of two or more.
- dibutyltin dilaurate, dibutyltin diacetate, and dibutyltin dioctate are particularly preferable.
- the amount of these added is usually 0.005 to 2.0% by mass, preferably 0.01 to 0.5% by mass, based on the graft copolymer.
- the curing reaction is also affected by temperature, and is slower as the temperature is lower, and requires a curing period of about one week at room temperature.
- the terminal-modified polyolefin obtained by reacting the first and second terminal unsaturated polyolefins of the present invention with an organohydrogenpolysiloxane having two or more SiH groups per molecule has high solvent solubility and heat resistance. Have. Therefore, the terminal-modified polyolefin can surface-treat the filler not only in a molten state but also in a solution or emulsion state.
- the residue of the terminal-modified polyolefin produced by reacting the first and second terminal unsaturated polyolefins of the present invention with an organohydrogenpolysiloxane having two or more SiH groups per molecule is preferably the following ( Polysiloxane residues that satisfy a), (b) and (c): (A) having a siloxane terminal (A unit) represented by formula (A) or a siloxane main chain (B unit) represented by formula (B), or a structure of both (b) a polysiloxane molecular main chain; (C) having a siloxane repeating unit (C unit) represented by the formula (C): (c) the number of A units is 0 to 2 / molecule, the number of B units is 0 to 10 / molecule, Unit and B unit cannot be 0 at the same time.
- the total of A unit, B unit and C unit is 5 to 1500 per molecule, preferably 5 to 200, more preferably 10 to 150, and the polysiloxane terminal other than the bonding site with polyolefin is R 7 or OR 8 (R 7 and R 8 each independently represents an unsubstituted or substituted monovalent hydrocarbon group having 1 to 12 carbon atoms).
- R 7 and R 8 each independently represents an unsubstituted or substituted monovalent hydrocarbon group having 1 to 12 carbon atoms).
- R 2 to R 6 each represents an unsubstituted or substituted monovalent hydrocarbon group having 1 to 12 carbon atoms.
- * -Si represents a bonding site with a polyolefin.
- Examples of the unsubstituted monovalent hydrocarbon group having 1 to 12 carbon atoms represented by R 2 to R 8 include a methyl group, an ethyl group, an isopropyl group, and a phenyl group.
- Examples of the substituted monovalent hydrocarbon group having 1 to 12 carbon atoms include a hydrocarbon group in which the above hydrocarbon group is substituted with a hydrogen atom, an alkoxy group, an amino group, or the like.
- the A unit is a group corresponding to the reacted siloxane molecule end, and the unit B is a group present in the main chain of the reacted siloxane molecule.
- the number of A units, B units, and C units is an integer. However, when the polysiloxane residue satisfying the above (a) to (c) has a molecular weight distribution, the number of the A unit, B unit and C unit is a positive number because it is expressed as an average value.
- organohydrogenpolysiloxane having two or more SiH groups per molecule examples include hydridopolydimethylsiloxane having one terminal, trimethylsiloxy group-blocking methylhydrogenpolysiloxane having both molecular chains, and trimethylsiloxy group-blocking dimethylsiloxane having both molecular chains.
- Methylhydrogensiloxane copolymer, molecular chain both ends silanol group blocked methylhydrogenpolysiloxane, molecular chain both ends silanol group blocked dimethylsiloxane ⁇ Methylhydrogensiloxane copolymer, molecular chain both ends dimethylhydrogensiloxy group blocked Dimethylpolysiloxane, dimethylhydrogensiloxy group-capped methylhydrogenpolysiloxane with molecular chain terminals, and dimethylhydrogensiloxy-capped dimethylsiloxa group with molecular chain terminals ⁇ Meth
- the polysiloxane residue is appropriately selected according to the intended use of the terminal-modified polyolefin.
- the terminal-modified poly ⁇ -olefin is used for the purpose of imparting lubricity or wear resistance to the resin, a single-terminal hydridopolydimethylsiloxane residue is preferred.
- the resin is further provided with melt properties, mechanical properties such as flexibility and impact resistance, and gas permeability, a polysiloxane residue having 2 to 10 hydride bond residues is preferable.
- the polysiloxane residue containing an alkoxy group is preferable.
- the graft copolymer of the present invention obtained by graft polymerization using the first and second terminal unsaturated polyolefins of the present invention contains a side chain polyolefin chain and a specific monomer that do not contain an irregular structure such as a crosslinked structure.
- the graft copolymer of the present invention contains 20 to 100% by mass of the first or second terminal unsaturated polyolefin of the present invention and polyolefins other than the terminal unsaturated polyolefin. 100 parts by mass of a combination of ⁇ 80% by mass, one or more monomers selected from the following [I] to [IV], 0.2 to 300 parts by mass, or one or more selected from the following group A and B It is obtained by graft polymerization of 0.2 to 300 parts by mass of one or more monomer mixtures selected from the group at 40 to 230 ° C. in the presence of 0.001 to 10 parts by mass of a radical initiator (graft copolymer).
- graft copolymer graft copolymer
- Method for producing coalescence I when the above components are polymerized, only a graft copolymer is obtained, and in addition to the graft copolymer, a thermoplastic resin formed by polymerizing a monomer that did not contribute to the graft polymerization is included. A product (mixture) may be obtained.
- polyolefins other than terminal unsaturated polyolefins include polyolefins that are substantially free of terminal unsaturated groups and that are obtained using Ziegler catalysts, metallocene catalysts, and the like.
- the polyolefins (1) to (3) may be mentioned, and preferred is a polyolefin composed of the same olefin species as the first or second terminal unsaturated polyolefin.
- Polyethylene resins such as high density polyethylene (HDPE), low density (LDPE), L-LDPE (2) Isotactic polypropylene, syndiotactic polypropylene, atactic polypropylene, propylene and ethylene, 4 to 12 carbon atoms Copolymer composed of one or more ⁇ -olefins, polypropylene resin such as block polypropylene (3) Polymer composed of one or more olefins having 6 to 28 carbon atoms
- the combination of the first or second terminal unsaturated polyolefin of the present invention and a polyolefin other than these includes a melt mixture, a powder mixture, pellets, etc. of the first or second terminal unsaturated polyolefin and the other polyolefin.
- a melt mixture a powder mixture, pellets, etc. of the first or second terminal unsaturated polyolefin and the other polyolefin.
- examples thereof include a dry blend mixture, a solution dissolved in a hydrocarbon solvent, or a mixture in a suspended state generated by cooling or reprecipitation after dissolution.
- the mixing ratio of the first or second terminal unsaturated polyolefin of the present invention and the other polyolefin is 20 to 100% by mass of the first or second terminal unsaturated polyolefin and 0 to 80% by mass of the other polyolefin. Preferably 30 to 100% by mass of the first or second terminal unsaturated polyolefin and 0 to 70% by mass of the other polyolefin, more preferably 40 to 100% by mass of the first or second terminal unsaturated polyolefin and the other.
- the polyolefin is 0 to 60% by mass, more preferably 50 to 100% by mass of the first or second terminal unsaturated polyolefin and 0 to 50% by mass of other polyolefins.
- the amount of the graft copolymer produced after the graft polymerization reaction is preferably increased.
- the graft copolymer of the present invention or a thermoplastic resin composition (thermoplastic resin) containing the copolymer of the present invention in the same type of thermoplastic resin as the polyolefin other than the first or second terminal unsaturated polyolefin is added and used, if the graft copolymer is present in a sufficient amount in the polyolefin other than the first or second terminal unsaturated polyolefin and is in a good dispersion state, the resulting composition is obtained.
- Thermoplastic resin composition II is preferable because the graft copolymer is easily dispersed throughout. Therefore, when the amount of polyolefin other than the first or second terminal unsaturated polyolefin is 80% by mass or less, the polyolefin becomes an appropriate amount, which is preferable from the viewpoint of dispersibility.
- [I] to [IV] examples include the following compounds.
- [I] Acrylic acid and its derivatives (1) Acrylic acid (2) Acrylic acid esters such as methyl acrylate, ethyl acrylate, butyl acrylate, normal octyl acrylate, 2-ethylhexyl acrylate; polyethylene glycol monoacrylate, Polyethylene glycol polypropylene glycol acrylate, poly (ethylene glycol-n-tetramethylene glycol) monoacrylate, propylene glycol polybutylene glycol monoacrylate, polypropylene glycol monoacrylate and the like long-chain polyalkylene glycols having a molecular weight of 30,000 or less (3) Acrylic acid Acrylic acid metal salt consisting of acrylic acid and typical metal elements such as sodium, potassium acrylate, magnesium acrylate, calcium acrylate, etc.
- Acrylic acid esters containing an oxygen, nitrogen, sulfur or silicon atom in the ester residue such as glycidyl acrylate, 2-hydroxyethyl acrylate, 2-hydroxypropyl acrylate, 2-hydroxy-3-phenoxypropyl
- Acrylates having functional groups such as acrylate, 4-hydroxybutyl acrylate, acryloyloxyethyl isocyanate, methacryloyloxyethyl isocyanate, 3-acryloxypropyltrimethoxysilane, 3-acryloxypropyltriethoxysilane; polyethylene glycol monoacrylate , Polyethylene glycol polypropylene glycol acrylate, poly (ethylene glycol-n-tetramethylene glycol) monoacrylate, propylene glycol polybuty Long-chain polyalkylene glycols having a molecular weight of 30,000 or less, such as polyglycol monoacrylate and polypropylene glycol monoacrylate (5) Acrylamide (6) N-substituted acrylamide
- Vinyl esters and derivatives thereof or alkoxy vinyl silanes Vinyl esters and derivatives thereof such as vinyl acetate, vinyl propionate, vinyl isolacrate, vinyl pivalate, vinyl undecanoate, vinyl palmitate; trimethoxyvinyl silane, triethoxyvinyl silane, etc. Alkoxy vinyl silane
- Preferred monomers and preferred monomer combinations include the following.
- acrylic acid and derivatives thereof the above-mentioned compounds are all preferable, and in particular, all compounds except the acrylic acid metal salt are more preferable.
- Graft polymerization is possible only with methacrylic acids and their derivatives, but [II] methacrylic acids and their derivatives by combining [I] acrylic acid and its derivatives / [II] methacrylic acids and their derivatives The amount of graft polymerization of the derivative is preferable. In particular, combinations of acrylic acid and acrylic acid esters with methacrylic acid and methacrylic acid esters are preferable.
- a preferable molar ratio of acrylic acid and its derivative / [II] methacrylic acid and its derivative is [I] / [II] (molar ratio) of 0.1-2, preferably 0.2-1. 5, more preferably 0.3 to 1.2, and still more preferably 0.5 to 1.0.
- [I] / [II] (molar ratio) is 0.1 or more, the amount of graft polymerization of [II] methacrylic acid and derivatives thereof increases, and when it is 2 or less, it does not participate in graft polymerization [I].
- Acrylic acid and its derivatives / [II] methacrylic acid and its copolymers are preferred because they do not by-produce.
- Graft polymerization is possible with styrene and its derivatives alone, but the combination of [I] acrylic acid and its derivatives / [VI] styrene and its derivatives increases the amount of graft polymerization of styrene and its derivatives. .
- combinations of acrylic acid, acrylic acid esters, styrene, and derivatives thereof are preferable.
- a preferred molar ratio of [I] acrylic acid and its derivative / [VI] styrene and its derivative is [I] / [VI] (molar ratio) of 0.1 to 2, preferably 0.2 to 1.5, More preferably, it is in the range of 0.3 to 1.2, and still more preferably in the range of 0.5 to 1.0.
- the monomers consisting of Group A and Group B are the following compounds, and the compounds represented by the formulas [I], [II], [III] and [IV] are as described above.
- Group A [V] Maleic anhydride and its substitute [VI] Maleic acid and its ester [VII] Maleimide and its substitute B group: [I] Acrylic acid and its derivative [II] Methacrylic acid and its derivative [III] Vinyl ester and its derivative or alkoxy vinyl silane [IV] Styrene and its derivative [VIII] ⁇ -olefin
- Examples of the monomers represented by [V] to [VIII] include the following compounds.
- Maleic anhydride such as maleic anhydride, methyl maleic anhydride, dimethyl maleic anhydride, phenyl maleic anhydride, diphenyl maleic anhydride and its substitutes
- Maleic acid methyl maleic acid, dimethyl maleate, maleate Diethyl acid, dibutyl maleate, monomethyl maleate, maleic acid and esters thereof
- the monomer one or more selected from the group A compounds and the group B compounds are used.
- the monomer of group A is a monomer in which the same kind of monomers are difficult to be polymerized because the electron density of double bonds is small. Therefore, in the present invention, the content of the monomer of the A group is improved by polymerization in combination with the monomer of the B group. Moreover, in this invention, since the reactivity of 1st and 2nd terminal unsaturated polyolefin can be improved by using the monomer of A group, the effect that a graft copolymer can be manufactured efficiently. Can also be obtained.
- the ⁇ -olefin having 2 to 28 carbon atoms is selected in consideration of the relationship between the graft polymerization temperature and its boiling point. In melt graft polymerization, the higher the temperature, the easier the handling in terms of reaction operation to use a higher boiling ⁇ -olefin. Further, in the graft polymerization using a solvent, it is possible to use from a gaseous state to an ⁇ -olefin having a high boiling point.
- the combination of the group A compound and the group B compound is such that the group A compound / group B compound (molar ratio) is usually about 0.1 to 2, preferably 0.5 to 1.5, more preferably The range is 0.8 to 1.2, more preferably 0.9 to 1.1.
- the molar ratio is 0.1 or more, the amount of graft polymerization of the group A compound increases, and when it is 2 or less, a copolymer of the group A compound / group B compound not involved in the graft polymerization is by-produced. Not preferred.
- the combination of the compounds of Group A and Group B is preferably a combination of [V] maleic anhydride of Group A and its substituent and a compound of Group B, and [V] maleic anhydride of Group A and [I] of Group B are preferred.
- a combination of acrylic acid and derivatives thereof, [III] vinyl ester and derivatives thereof or alkoxyvinylsilane, and [VIII] ⁇ -olefin is more preferable.
- the radical initiator used in the graft polymerization of the present invention is not particularly limited, and can be appropriately selected from conventionally known radical initiators such as various organic peroxides and azo compounds, Both compounds are suitable radical initiators.
- the organic peroxide include dibenzoyl peroxide, di-8,5,5-trimethylhexanoyl peroxide, dilauroyl peroxide, didecanoyl peroxide, and di (2,4-dichlorobenzoyl) peroxide.
- Diacyl peroxides such as 2,5-dimethylhexane-2,5-dihydroperoxide, di-t-butyl Peroxide, dicumyl peroxide, 2,5-dimethyl-2,5-di (t-butylperoxy) hexane, 2,5-dimethyl-2,5-di (t-butylperoxy) hexyne-3, Dialkyl pars such as ⁇ , ⁇ 'bis (t-butylperoxy) diisopropylbenzene Peroxyketals such as xoxides, 1,1-bis-t-butylperoxy-3,3,5-trimethylcyclohexane, 2,2-bis (t-butylperoxy) butane, t-butylperoxyoct Alkyl peresters such as ate,
- the radical initiator is in the range of 0.001 to 10 parts by mass, preferably 0.005 to 5 parts by mass with respect to 100 parts by mass of the combination of the first or second terminal unsaturated polyolefin and the polyolefin other than these polyolefins. Used.
- the amount used is preferably in the range of 1 to 250 parts by mass, more preferably 5 to 200 parts by mass, and still more preferably 10 to 180 parts by mass.
- the amount used is 0.2 parts by mass or more, the amount of monomers to be copolymerized in the graft polymer increases, and functions such as compatibilization are likely to be exhibited.
- the coalescence is preferable because no by-product is formed.
- the graft polymerization method is not particularly limited.
- a graft copolymer or a thermoplastic resin composition containing the copolymer can be produced by melt-kneading and reacting using the above.
- Reaction conditions include a temperature of 40 to 230 ° C. and 0.01 to 0.5 hours.
- the graft polymerization reaction may be carried out in the presence of a Lewis acid.
- the temperature is preferably 40 to 140 ° C.
- hydrocarbon solvents such as butane, pentane, hexane, cyclohexane and toluene, halogenated hydrocarbon solvents such as chlorobenzene, dichlorobenzene and trichlorobenzene, and appropriate organic solvents such as liquefied ⁇ -olefins or solvent-free
- a graft copolymer or a thermoplastic resin composition containing the copolymer can also be produced.
- the reaction conditions include a temperature of 20 to 230 ° C., preferably a temperature of 40 to 140 ° C. and 0.1 to 10 hours in the presence of Lewis acid.
- Lewis acid used in the graft polymerization of the present invention examples include the following compounds.
- Group 2-4 group element halides chlorine, bromine, fluorine, iodine
- alkylates hydrocarbon groups having 1 to 20 carbon atoms
- alkyl halides (2) aluminum, boron, Lewis acid consisting of zinc, tin, magnesium and calcium atoms
- Lewis acids include magnesium chloride, calcium chloride, zinc chloride, boron trichloride, aluminum trichloride, gallium trichloride, silicon tetrachloride, silicon tetrachloride, and compounds in which chlorine atoms are converted to bromine and fluorine atoms.
- the amount of Lewis acid used in the graft polymerization reaction is such that Lewis acid / monomer (mol / mol) is 0.01 to 1, preferably 0.05 to 1, more preferably 0.1 to 0.5.
- the monomer means [I] acrylic acid and derivatives thereof, [II] methacrylic acids and derivatives thereof, [III] vinyl esters and derivatives thereof, or alkoxyvinylsilane, [IV] Monomers selected from styrene and derivatives thereof, or group A; [V] maleic anhydride and substituted products thereof, [VI] maleic acid and esters thereof, [VII] maleimide and substituted products thereof, group B; [I] Means a monomer selected from acrylic acid and derivatives thereof, [II] methacrylic acids and derivatives thereof, [III] vinyl esters and derivatives thereof or alkoxyvinylsilanes, [IV] styrene and derivatives thereof, and [VIII] ⁇ -olefins. ing.
- the Lewis acid is added before the radical initiator is added, and the graft polymerization reaction is performed, or the graft polymerization reaction is performed by using the monomer [I] to [VIII] in contact with the Lewis acid in advance. .
- the graft copolymer or the thermoplastic resin composition containing the graft copolymer is preferably subjected to graft polymerization in the presence of a chain transfer agent.
- graft polymerization in the presence of a chain transfer agent, the melt fluidity of the graft copolymer can be improved.
- melt fluidity of the graft copolymer can be improved without lowering the heat resistance.
- it has the advantage that the by-product of a gel or a puff can be prevented.
- chain transfer agent A reagent that causes a chain transfer reaction is called a chain transfer agent.
- the chain transfer agent receives radicals from the growing polymer chain and stops the elongation of the polymer, but the chain transfer agent that receives the radicals is a reaction agent that can attack the monomer and start polymerization again. Used mainly to adjust the molecular weight of the polymer.
- a kind and addition amount can be selected suitably.
- the efficiency of the chain transfer agent can be evaluated by the chain transfer constant.
- the chain transfer constant of the chain transfer agent for each monomer can be referred to, for example, Polymer Handbook 3rd Edition (J. BRANDRUP and EH IMMERGUT edition, published by JOHN WILEY & SON).
- the chain transfer constant can also be obtained experimentally with reference to Takayuki Otsu and Masato Kinoshita "Experimental Methods for Polymer Synthesis” Kagaku Dojin, published in 1972.
- mercaptans are suitable. Specific examples include the following. Alkyl mercaptans such as isopropyl mercaptan, n-butyl mercaptan, t-butyl mercaptan, n-octyl mercaptan, n-dodecyl mercaptan, t-dodecyl mercaptan, n-pentyl mercaptan, n-lauryl mercaptan.
- Alkyl mercaptans such as isopropyl mercaptan, n-butyl mercaptan, t-butyl mercaptan, n-octyl mercaptan, n-dodecyl mercaptan, t-dodecyl mercaptan, n-pentyl mercaptan, n-lauryl mercaptan.
- Thiophenols such as thiophenolthio- ⁇ -naphthol, m-bromothiophenol, p-bromothiophenol, m-toluenethiol, p-toluenethiol; Mercaptan having an ester group such as 2-ethylhexyl mercaptoacetate, methoxybutyl mercaptoacetate, mercaptans having alkoxysilicon such as 3-mercaptopropyltrimethoxysilane, 3-mercaptopropyltriethoxysilane, etc. From the viewpoint of odor, long-chain alkyl alkyl mercaptans or mercaptans having a large molecular weight are preferred.
- n-octyl mercaptan examples include n-octyl mercaptan, n-dodecyl mercaptan, t-dodecyl mercaptan, n-pentyl mercaptan, n-lauryl mercaptan, 2-ethylhexyl mercaptoacetate, and in applications requiring heat resistance, Examples include 3-mercaptopropyltrimethoxysilane and 3-mercaptopropyltriethoxysilane.
- the use amount of the chain transfer agent is 0.001 to 50% by mass, preferably 0.01 to 30% by mass, more preferably 0.1 to 15% by mass with respect to the monomer. % Range.
- the amount is 0.001% by mass, the control of the molecular weight is insufficient, and a gel-like product is easily generated due to an increase in the molecular weight.
- it exceeds 50% by mass the molecular weight is greatly decreased, and the main chain chain composed of the monomer is shorter than the side chain chain, so that the adhesive strength and the compatibilizing performance are decreased.
- the graft copolymer of the present invention or the thermoplastic resin composition containing the graft copolymer is substantially free of terminal unsaturated polyolefin (thermally decomposed or radically decomposed low-order polyolefin) and homoradical polymerizability. Obtained by bringing the monomer (a) copolymerizable with the monomer (a) into contact with a mixture of the monomer (a) not present in the monomer (a method for producing a graft copolymer) II). According to the said manufacturing method, the reaction rate of monomer (a) and (b) improves, and the isolation
- Monomer (a) (i) Definition of Monomer (a) Monomer (a) refers to a monomer that lacks single radical polymerizability and has substantially no single radical polymerizability. More specifically, it refers to the monomer specifically shown below. (ii) Specific Examples of Monomer (a) Maleic anhydride and substituted products thereof are the same as [V] of Group A in the above-mentioned graft copolymer production method I.
- maleic anhydride And maleic anhydrides such as acid, methylmaleic anhydride, dimethylmaleic anhydride, phenylmaleic anhydride, diphenylmaleic anhydride, and substituted products thereof.
- -Maleic acid and its ester are the same as [VI] of group A in the above-mentioned graft copolymer production method I.
- maleic acid, methyl maleic acid, dimethyl maleate, diethyl maleate, maleate Mention may be made of dibutyl acid, monomethyl maleate, maleic acid and esters thereof.
- ⁇ -olefin is substantially the same as [VIII] of Group B in the above-mentioned graft copolymer production method I, specifically, ethylene, propylene, 1-butene, 1-hexene, 1-heptene, Carbons such as 1-octene, 1-decene, 1-undecene, 1-dodecene, 1-tridecene, 1-tetradecene, 1-pentadecene, 1-hexadecene, 1-heptadecene, 1-octadecene, 1-nonadecene, 1-eicosene Examples thereof include branched olefins such as ⁇ -olefins having a number of 2 to 28 and isobutene. -Vinyl silane and its substitutes. Specific examples include vinyltrimethoxysilane and vinyltriethoxysilane.
- Monomer (b) is a monomer copolymerizable with the monomer (a) or two monomers selected from the monomers (a). A monomer copolymerizable with the above.
- Specific Example (a) When the monomer (a) is maleic anhydride and its substituted product, and maleic acid and its ester. Vinyl ester and its derivatives. Except for vinyl alkoxysilane, production of the graft copolymer The same as [III] in Method I, specifically, vinyl esters such as vinyl acetate, vinyl propionate, vinyl isolacrate, vinyl pivalate, vinyl undecanoate, vinyl palmitate, and derivatives thereof.
- ⁇ -olefins The same as the ⁇ -olefins in the monomer (a) can be exemplified.
- Styrene and its derivatives The same as [IV] in the above-mentioned graft copolymer production method I, specifically, ⁇ -methylstyrene, p-methylstyrene, p-ethylstyrene, p-propylstyrene, p- Isopropyl styrene, p-butyl styrene, p-tert-butyl styrene, p-phenyl styrene, o-methyl styrene, o-ethyl styrene, o-propyl styrene, o-isopropyl styrene, m-methyl styrene, m-ethyl styrene, alkyl styrenes such as m-isopropyl styrene, m-butyl styrene
- Acrylic acid (2) Acrylic acid esters such as methyl acrylate, ethyl acrylate, butyl acrylate, normal octyl acrylate, 2-ethylhexyl acrylate; polyethylene glycol monoacrylate, polyethylene glycol polypropylene glycol acrylate, poly ( Ethylene glycol-n-tetramethylene glycol) monoacrylate, propylene glycol polybutylene glycol monoacrylate, polypropylene glycol monoacrylate, etc., long-chain polyalkylene glycols having a molecular weight of 30,000 or less (3) sodium acrylate, potassium acrylate, acrylic acid Acrylic acid metal salt consisting of acrylic acid and typical metal elements such as magnesium and calcium acrylate (4) Oxygen, nitrogen, Acrylic esters containing yellow and silicon atoms, such as glycidy
- methacrylic acid -Methacrylic acid and ⁇ -alkyl substituted acrylic acid (hereinafter sometimes collectively referred to as "methacrylic acid”) and derivatives thereof [II] in the above-mentioned graft copolymer production method I
- methacrylic acid ⁇ -alkyl substituted acrylic acid
- derivatives thereof [II] in the above-mentioned graft copolymer production method I Specific examples thereof include monomers having an alkyl group such as a methyl group (preferably an alkyl group having 6 or less carbon atoms) at the ⁇ -position of the monomers exemplified as acrylic acid and derivatives thereof. It is done.
- Preferable combinations of the monomers (a) and (b) include the following.
- Maleic anhydride and its substitutes / ⁇ -olefins more preferably maleic anhydride or maleic acid / ⁇ -olefins having 4 to 28 carbon atoms, more preferably maleic anhydride / ⁇ -olefins having 6 to 18 carbon atoms
- Maleic anhydride and substituted products thereof / acrylic acid and derivatives thereof more preferably maleic anhydride or maleic acid / acrylic acid, acrylic esters, more preferably maleic anhydride / acrylic acid, acrylic esters
- Maleic anhydride and its substituted / vinyl ester and derivatives thereof more preferably maleic anhydride or maleic acid / vinyl acetate, more preferably maleic anhydride / vinyl acetate
- solvent 1 Specific examples of the solvent for dissolving the thermal decomposition product or radical decomposition product and the monomers (a) and (b) include aromatic hydrocarbons such as benzene, toluene, xylene and mixed xylene.
- Aliphatic hydrocarbons such as hexane, heptane, octane and decane; alicyclic hydrocarbons such as cyclohexane and methylcyclohexane; carboxylic acid esters such as methyl acetate, ethyl acetate, propyl acetate and butyl acetate; acetone and methyl ethyl ketone And ketones such as tetrahydrofuran, cyclic ethers such as tetrahydrofuran and tetrahydropyran; and halogenated hydrocarbons such as chlorobenzene.
- the single solvent system aromatic hydrocarbons such as toluene and xylene; and cyclic ethers such as tetrahydrofuran are preferable.
- the mixed solvent system it is necessary that the monomer (a), the monomer (b), and the thermal decomposition product or radical decomposition product are uniformly dissolved. At that time, it is used after confirming that each of the three components is dissolved in a mixed solvent having the same solvent type and composition.
- one or more selected from aliphatic hydrocarbons and alicyclic hydrocarbons, and one or more mixed solvents selected from carboxylic acid esters and ketones include hexane / ethyl acetate, hexane / butyl acetate, hexane / methyl ethyl ketone, and combinations in which hexane is replaced with heptane, decane, cyclohexane or methylcyclohexane.
- the mixed composition is preferably in the range of 25/75 to 75/25% by mass, more preferably in the range of 30/70 to 70/30% by mass, and in the range of 35/65 to 65/35% by mass.
- the mixed solvent system which consists of the combination of the solvent used for a single solvent system, and the combination of the solvent used for a single solvent system, and carboxylic acid ester or ketones can also be used.
- the mixed composition is preferably in the range of 30/70 to 70/30% by mass.
- (A) In case of melting reaction The reaction is carried out in the same manner as in the case of using a solvent, except that the reaction is carried out in the molten state of the thermal decomposition product or radical decomposition product without using the solvent. It is preferable that the monomer (a) is dissolved in the molten thermal decomposition product or radical decomposition product. Specifically, this is the case where the monomer (a) is an ⁇ -olefin, vinyl silane, or a substituted product thereof.
- the graft copolymer of the present invention is a propylene homopolymer; a propylene copolymer composed of propylene, ethylene and one or more ⁇ -olefins having 4 to 10 carbon atoms, and ethylene and ⁇ -olefins having 4 to 10 carbon atoms.
- a graft copolymer having a polyolefin chain derived satisfy the following (c).
- the graft ratio of the graft copolymer is preferably 1 to 150% by mass, more preferably 2 to 130% by mass, and further preferably 5 to 100% by mass.
- the graft ratio is 1% by mass or more, the number of side chains is appropriate, and functions such as compatibilization and adhesion can be sufficiently exerted.
- the graft rate is 150% by mass or less, the reactive polyolefin component is appropriate and functions sufficiently. Can demonstrate.
- the graft ratio can be measured, for example, as follows.
- the solvent and the soluble polymer component that were not involved in the graft reaction were dissolved and removed by the solvent, and the mass of the insoluble graft copolymer component (W2) and the mass of the reactive polyolefin used as the raw material (W1) were as follows: Calculate as follows.
- Graft ratio (mass%) (W2-W1) / W1 ⁇ 100
- the proportion of propylene chain or butene-1 chain mesopentad [mmmm] of the graft copolymer is preferably 20 to 80 mol%, more preferably 30 to 70 mol%.
- the polyolefin selected from the propylene homopolymer, propylene copolymer, butene-1 homopolymer and butene-1 copolymer used in the production of the graft copolymer of the present invention has an atactic structure or syndiotactic It preferably has a tic structure.
- the graft copolymer of the present invention preferably has an intrinsic viscosity [ ⁇ ] measured at 135 ° C. in decalin of 0.01 to 2.5 dl / g, more preferably 0.02 to 2.2, still more preferably. Is 0.05 to 2.0.
- the intrinsic viscosity [ ⁇ ] is 0.01 dl / g or more, functions such as resin compatibilization are improved, and when it is 2.5 dl / g or less, dispersibility in the resin is improved.
- the weight average molecular weight of the graft copolymer of the present invention is usually 500 to 400,000, preferably 700 to 350,000, more preferably 1000 to 300,000, and most preferably 1500 to 250,000.
- the molecular weight distribution (Mw / Mn) of the graft copolymer of the present invention is 1.6 to 6, preferably 1.8 to 5, and more preferably 1.8 to 4.
- control the reactivity of unsaturated groups, controlling the number of terminal unsaturated groups per molecule, etc. it is possible to suppress side reactions other than graft polymerization and further reduce the molecular weight distribution. is there.
- evaluation of the weight average molecular weight and molecular weight distribution of a graft copolymer can use the above-mentioned GPC method.
- the graft copolymer of the present invention preferably has a gel fraction of 5% by mass or less, and more preferably contains substantially no gel component.
- the gel fraction can be evaluated by, for example, the following method.
- the graft is made of a stainless steel 400 mesh (opening 0.034 mm) net in a glass separable flask equipped with a stirrer. Add 50 mg of copolymer and fix to a stirring blade. A solvent containing 0.1% by mass of an antioxidant (BHT) is added and dissolved while stirring at a temperature below the boiling point for 4 hours. After dissolution, the collected soot is sufficiently dried in vacuum, and the insoluble part is determined by weighing.
- the gel component (gel fraction) defined as an insoluble part is calculated by the following formula.
- composition of the present invention and use thereof
- the graft copolymer of the present invention, and the thermoplastic resin composition containing the graft copolymer of the present invention and polyolefin are preferably further fillers. And / or pigments.
- the filler includes an inorganic filler and an organic filler.
- the inorganic filler include talc, white carbon, silica, mica, bentonite, aluminum compound, magnesium compound, barium compound, diatomaceous earth, glass beads or glass fibers, metal powder, or metal fibers.
- the organic filler include starch (for example, powdered starch), fibrous leather (for example, natural organic fibers composed of cellulose such as cotton and hemp), and fibers composed of synthetic polymers such as nylon, polyester and polyolefin.
- the pigment examples include inorganic pigments and organic pigments (for example, azo pigments and polycyclic pigments).
- Inorganic pigments include oxides (titanium dioxide, zinc white (zinc oxide), iron oxide, chromium oxide, iron black, cobalt blue, etc., hydroxides: alumina white, iron oxide yellow, viridian, etc.), sulfides ( Zinc sulfide, lithopone, cadmium yellow, vermilion, cadmium red, etc.), chromate (yellow lead, molybdate orange, zinc chromate, strontium chromate, etc.), silicate (white carbon, clay, talc, ultramarine blue, etc.), sulfate (Precipitated barium sulfate, barite powder, etc., carbonates: calcium carbonate, lead white, etc.), ferrocyanide (bituminous), phosphate (manganese violet), carbon (carbon black), etc.
- oxides titanium dioxide,
- azo pigments that are organic pigments include soluble azo (Kermin 6B, Lakelet C, etc.), insoluble azo (Disazo Yellow, Lakelet 4R, etc.), condensed azo (chromophthalo Yellow 3G, chromophthalscarlet RN, etc.), azo complex salts (nickel) Azo yellow, etc.) and pendidalone azo (permanent orange HL, etc.).
- Polycyclic pigments that are organic pigments include isoindolinone, isoindoline, quinophthalone, pyrazolone, flavatron, anthraquinone, diketo-pyrrolo-pyrrole, pyrrole, pyrazolone, pyranthrone, perinone, perylene, quinacridone, indigoid, oxazine, imidazolone , Xanthene, carbonium, violanthrone, phthalocyanine, nitroso and the like.
- (a) / (b) is less than 0.005
- improvement of the wettability and adhesiveness of the filler or pigment surface is insufficient, and in the masterbatch or resin dispersion composition, the dispersibility of the filler or pigment and the interfacial adhesiveness. May be insufficient.
- component (b) not involved in the surface treatment exists, which is not preferable because the production cost increases.
- composition of the present invention can be suitably used as an adhesive, a resin compatibilizer, a dispersion, or a coating material.
- the composition of the present invention can be used as a hot melt adhesive substrate.
- additives such as oils, tackifiers, antioxidants and the like are used within a normal range.
- the composition of the present invention can be dissolved in a solvent and used as a solvent-type adhesive, and can be applied and sprayed to form a film on the surface of an adhesive substrate and adhere to an adherend.
- the composition of the present invention can also be used as an adhesive by dispersing it in a polar solvent such as water or making it into an emulsion.
- the composition of the present invention can be formed into a sheet shape or a film shape, sandwiched between adhesive substrates, heated to a temperature higher than the temperature at which the adhesive flows, and bonded by cooling and solidification.
- composition of the present invention can be used as a resin compatibilizer by adding, for example, 0.005 to 15% by mass to the resin composition containing polyolefin as an essential component.
- the composition of the present invention can be made into a dispersion in which the graft copolymer is finely dispersed by dissolving in a solvent at room temperature or by heating.
- concentration of the graft copolymer is in the range of 5 to 30% by mass.
- the solvent include aliphatic hydrocarbon compounds such as hexane, heptane and decane; aromatic hydrocarbon compounds such as benzene, toluene and xylene; alicyclic hydrocarbon compounds such as cyclohexane and methylcyclohexane; halogens such as chlorobenzene Hydrocarbons; ether compounds such as tetrahydrofuran and tetrahydropyran.
- An emulsion can be obtained by using a polar solvent as the solvent.
- the polar solvent include water; alcohols such as methanol, ethanol and butanol; and carboxylic acid esters such as methyl acetate, ethyl acetate and butyl acetate.
- Etc. can be illustrated.
- a method for producing by adding a 20 to 30 mass% solution of tetrahydrofuran to water at 20 to 50 ° C. little by little, then removing tetrahydrofuran under reduced pressure, and adjusting the amount of water to a desired concentration Etc. can be illustrated.
- Other methods include known methods such as a method of directly dispersing in a polar solvent with high-speed stirring or a high shear field. If necessary, an anionic, cationic, or nonionic surfactant or a water-soluble polymer compound may be used as an additive.
- the molten functionalized polyolefin and the graft copolymer of the present invention can be uniformly coated on a substrate and coated by a cooling and solidifying step.
- the functionalized polyolefin of the present invention can be used as an adhesive, a resin compatibilizing agent, a dispersion, and a coating material as described above, and can also be used as a reactive hot melt adhesive, a sealing material, and a potting material. .
- the reactive hot melt adhesive is mainly composed of a functionalized polyolefin containing alkoxysilicon and the graft copolymer of the present invention, and optionally contains an oil, a tackifier, an inorganic filler, and a silanol condensation catalyst.
- oils such as naphthenic oil, paraffinic oil, and aroma oil, oils obtained by mixing these, and liquid rubbers such as liquid polybutene and liquid isopolybutylene. You may use these individually by 1 type or in mixture of 2 or more types.
- tackifiers examples include rosin and derivatives thereof, terpene resins and hydrogenated resins thereof, styrene resins, coumarone-indene resins, dicyclopentadiene (DCPD) resins and hydrogenated resins thereof.
- One of these tackifier resins may be used alone, or two or more thereof may be used as a mixture.
- Preferred tackifying resins include terpene resins and their hydrogenated resins, styrene resins, dicyclopentadiene (DCPD) resins from the viewpoint of the balance between removability and adhesion to curved and uneven surfaces. And its hydrogenated resin, aliphatic (C5) petroleum resin and its hydrogenated resin, aromatic (C9) petroleum resin and its hydrogenated resin, and C5 -C9 copolymer petroleum resin It is preferable to use one kind of resin or a mixture of two or more kinds selected from the group of hydrogenated resins.
- DCPD dicyclopentadiene
- Inorganic fillers include silica, alumina, zinc oxide, titanium oxide, calcium oxide, magnesium oxide, iron oxide, tin oxide, antimony oxide, ferrites, calcium hydroxide, magnesium hydroxide, aluminum hydroxide, basic magnesium carbonate, Calcium carbonate, zinc carbonate, barium carbonate, dosonite, hydrotalcite, calcium sulfate, barium sulfate, calcium silicate, talc, clay, mica, montmorillonite, bentonite, sepiolite, imogolite, serisalite, glass fiber, glass beads, silica balun , Aluminum nitride, boron nitride, silicon nitride, carbon black, graphite, carbon fiber, carbon balun, zinc borate, and various magnetic powders.
- An inorganic filler may be used in place of the inorganic filler, and surface treatment may be performed with various coupling agents such as silane and titanate.
- this treatment method a method of directly treating an inorganic filler with various coupling agents such as a dry method, a slurry method or a spray method, an integral blend method such as a direct method or a masterbatch method, or a dry concentrate method, etc. The method is mentioned.
- the silanol condensation catalyst is preferably used in admixture.
- the silanol condensation catalyst include organic tin metal compounds such as dibutyltin dilaurate, dibutyltin diacetate, and dibutyltin dioctate; organic bases, organic acids such as ethylamine acid, fatty acids, and the like, with dibutyltin dilaurate and dibutyltin being particularly preferable. Diacetate, dibutyltin dioctate. The amount of these added is 0.005 to 2.0% by mass, preferably 0.01 to 0.5% by mass, based on the ⁇ -olefin polymer modified product.
- curing can be carried out by bringing it into contact with moisture or moisture in the presence or absence of a silanol condensation catalyst and curing it at room temperature.
- a reactive hot melt adhesive may be left in the air, or immersion or steam may be introduced into a water tank.
- the temperature may be room temperature, but a high temperature is preferable because crosslinking is performed in a short time.
- the functionalized polyolefin can be used as a sealing material and a potting material, and can be prepared in the same manner as the reactive hot melt adhesive when it requires a crosslinking performance.
- a functionalized polyolefin and a melt mainly composed of the graft copolymer of the present invention are used for sealing or potting and fixed by cooling and solidification.
- the polymerization temperature was set to 83 ° C., and propylene and hydrogen were continuously fed so that the hydrogen concentration in the gas phase part of the reactor was 0.86% and the total pressure in the reactor was maintained at 0.7 MPa ⁇ G. A polymerization reaction was performed.
- Irganox 1010 (manufactured by Ciba Specialty Chemicals), which is a stabilizer, is added to the resulting polymerization solution so that its content is 500 ppm by mass, and n-heptane, which is a solvent, is removed to reduce the content.
- Crystalline polypropylene was obtained. This low crystalline polypropylene was made into resin pellets by underwater cutting. The resulting low crystalline polypropylene has a stereoregularity [mmmm] of 45 mol%, a weight average molecular weight (Mw) of 45,600, and a number of terminal unsaturated groups (here, number of terminal vinylidene groups) of 0.95 / It was a molecule.
- Production Example 2 [Production of low crystalline polypropylene]
- the polymerization temperature was set to 67 ° C.
- the hydrogen concentration in the gas phase of the reactor was 0.74 mol%
- the total pressure in the reactor was maintained at 0.75 MPa ⁇ G.
- Resin pellets of low crystalline polypropylene were obtained in the same manner as in Production Example 1 except that propylene and hydrogen were continuously supplied.
- the obtained low crystalline polypropylene had a stereoregularity [mmmm] of 46 mol%, a weight average molecular weight (Mw) of 129,000, and a terminal unsaturated group number (terminal vinylidene group number) of 0.97 / molecule. It was.
- the polymerization temperature is set to 70 ° C., and propylene and hydrogen are continuously supplied so that the hydrogen concentration in the gas phase part of the reactor is 0.23 mol% and the total pressure in the reactor is maintained at 0.46 MPa ⁇ G. A polymerization reaction was performed.
- Irganox 1010 (manufactured by Ciba Specialty Chemicals), which is a stabilizer, is added to the resulting polymerization solution so that its content is 500 ppm by mass, and n-heptane, which is a solvent, is removed to reduce the content.
- Crystalline polypropylene was obtained. This low crystalline polypropylene was made into resin pellets by underwater cutting. The obtained low crystalline polypropylene had a stereoregularity [mmmm] of 59 mol%, a weight average molecular weight (Mw) of 45,000, and a terminal unsaturated group number (terminal vinylidene group number) of 0.96 per molecule.
- Example 1 [Production of high-end unsaturated polypropylene and its radical decomposition] Using the polypropylene of Production Example 1 as the raw material polyolefin, radical-decomposed polypropylene was produced under the conditions shown in Table 1. Specifically, 40 g of the polypropylene produced in Production Example 1 was charged into a stainless steel reactor (with an internal volume of 500 mL) equipped with a stirrer. It stirred for 30 minutes under nitrogen stream. Stirring was stopped and the resin temperature was raised to 120 ° C. using a mantle heater. Stirring was resumed after confirming that it was in a molten state, and the mantle heater was controlled so that the resin temperature was constant at 270 ° C.
- Table 2 shows the results of evaluating the weight-average molecular weight Mw, number-average molecular weight Mn, molecular weight distribution Mw / Mn, vinylidene group content, and number of terminal vinylidene groups per molecule of the radically decomposed polypropylene obtained.
- the by-product generation rate (by-product generation amount [g] / charged raw material [g]) was 0.01135.
- Examples 2 to 9 A radically decomposed polypropylene was produced and evaluated in the same manner as in Example 1 except that the conditions in Table 1 were used. The results are shown in Table 2.
- the organic peroxide was added in two portions, but this addition was performed by adding the first organic peroxide in the same addition time and reaction time as in Example 1. After that, the second organic peroxide was added and reacted again at the same addition time and reaction time as the first time, and the by-product generation rate in Example 2 (by-product generation amount [g] / The charged raw material [g]) was 0.00304, and the generation rate of by-products in Example 4 was 0.00844.
- tertiary butyl cumyl peroxide in Examples 3 and 5 to 7 is Perbutyl C (manufactured by NOF Corporation), and the trade name of diisopropylbenzene hydroperoxide in Example 8 is PARK Mill P (NOF Corporation). Made by Co., Ltd.).
- Comparative Example 1 [Production of terminally unsaturated polypropylene from high molecular weight polypropylene] A radically decomposed polypropylene was produced and evaluated in the same manner as in Example 1 except that a commercially available polypropylene J300SP (manufactured by Prime Polymer Co., Ltd.) was used as the raw material polyolefin.
- J300SP commercially available polypropylene J300SP
- the obtained decomposed polypropylene had a weight average molecular weight of 371,000, and the number of terminal unsaturated groups (number of terminal vinylidene groups) was 0.08 per molecule.
- the stereoregularity [mmmm] was 96%.
- J3000GV commercially available polypropylene J3000GV
- the resulting decomposed polypropylene had a weight average molecular weight of 155000 and a terminal unsaturated group number (terminal vinylidene group number) of 0.12 per molecule.
- the stereoregularity [mmmm] was 96 mol%.
- Example 10 Manufacture of highly terminal unsaturated polypropylene (manufacture of continuous radical decomposition polypropylene by melt extruder)]
- composition comprising polypropylene and organic peroxide
- Compositions comprising raw material polypropylene and organic peroxide were produced under the conditions shown in Table 3. Specifically, 1000 g of the polypropylene obtained in Production Example 2 was put into a universal mixing stirrer (model: 5DM-L-03-rr, Dalton mixer) and stirred at 126 rpm for 30 minutes under a nitrogen stream.
- Examples 11 and 12 A decomposed polypropylene was produced and evaluated in the same manner as in Example 10 except that the melt radical decomposition extrusion was performed under the conditions shown in Table 4. The results are shown in Table 5.
- Comparative Example 3 A decomposed polypropylene was produced and evaluated in the same manner as in Example 10 except that the composition comprising polypropylene and an organic peroxide produced under the conditions shown in Table 3 was used and melt radical decomposition extrusion was carried out under the conditions shown in Table 4. The results are shown in Table 5.
- the decomposed polypropylene of Comparative Example 3 had a stronger odor than the decomposed polypropylenes of Examples 10-12.
- Reference examples 1 to 3 A composition comprising polypropylene and an organic peroxide produced under the conditions shown in Table 3 was evaluated in the same manner as in Example 10. The results are shown in Table 3.
- Example 13 [Production of high-end unsaturated polypropylene by pyrolysis] Using the polypropylene of Production Example 1 as the raw material polyolefin, pyrolyzed polypropylene was produced under the conditions shown in Table 6. Specifically, 100 g of the polypropylene produced in Production Example 1 was put into a stainless steel reactor (with an internal volume of 500 mL) equipped with a stirrer. The mixture was stirred for 30 minutes under a nitrogen stream. Stirring was stopped, and the resin temperature was raised to 150 ° C. using a mantle heater.
- Examples 14 and 15 A decomposed polypropylene was produced and evaluated in the same manner as in Example 13 except that the thermal decomposition was performed under the conditions shown in Table 6. The results are shown in Table 7.
- Comparative Example 4 Except for using a commercially available polypropylene J300SP (manufactured by Prime Polymer Co., Ltd.) as a raw material polyolefin, an attempt was made to produce pyrolytic polypropylene in the same manner as in Example 13. However, at 300 ° C., J300SP is highly viscous and difficult to stir uniformly. The temperature of the molten resin could not be controlled to a predetermined temperature, and it was impossible to produce pyrolytic polypropylene.
- J300SP manufactured by Prime Polymer Co., Ltd.
- Example 16 [Production of copolymer with polar monomer] (1) Production of radically decomposed low-order polypropylene Radical-decomposed low-order polypropylene was produced in the same manner as in Example 8 except that the reaction scale was increased to 5 times, and recovered in a molten state at a temperature of 140 ° C. After cooling and solidifying, it was cut in advance to form a plate of about 5 ⁇ 5 ⁇ 1 cm. This was cooled in a dry ice ethanol bath, pulverized with a pulverizer to form pulverized pellets, air-dried, and dried in a vacuum dryer at 40 ° C. for 8 hours.
- Examples 17-21 [Production of copolymer with polar monomer] A graft copolymer was produced and evaluated in the same manner as in Example 16 except that the copolymer was produced under the conditions shown in Table 8. The results are shown in Table 9.
- the trade name of 1,1-di (tertiary hexyl proxy) cyclohexane is Perhexa HC (manufactured by NOF Corporation).
- Example 22 [Production of copolymer with polar monomer] A graft copolymer was produced under the conditions shown in Table 10 using the radically decomposed polypropylene produced in Example 16 (1) (the polypropylene of Example 8 having a reaction scale of 5 times). Specifically, after a 200-ml reaction vessel equipped with a stirrer was sufficiently purged with nitrogen, 30 g of polypropylene produced in Example 16 (1), 1.0 g of maleic anhydride, and 100 ml of dehydrated toluene were added and stirred. Started. At this time, the mixture was dissolved with stirring while bubbling with nitrogen. After dissolution, nitrogen bubbling was stopped and the temperature was kept constant at 110 ° C. while maintaining the nitrogen atmosphere.
- Examples 23-26 [Production of copolymer with polar monomer] A graft copolymer was produced and evaluated in the same manner as in Example 22 except that the copolymer was produced under the conditions shown in Table 10. The results are shown in Table 11.
- Example 27 Manufacture of cured products
- 20 grams of the copolymer of Example 16 was dissolved by heating at 140 ° C., and about 40 milligrams of dibutyltin dilaurate was added and stirred until uniform.
- This composition was formed into a sheet having a thickness of 1 mm using a hot press and allowed to stand in an atmosphere of 23 ° C. and 50% humidity for 168 hours to produce a cured product.
- the obtained cured product was heated on a hot plate controlled at 120 ° C. for 10 minutes, and when the shape change was observed, the shape was retained.
- the results are shown in Table 12.
- cured material obtained on the hotplate showed rubber elasticity, and returned
- Example 28 Manufacture of cured products
- a cured product was produced and evaluated in the same manner as in Example 27 except that the copolymer of Example 17 was used instead of the copolymer of Example 16. The results are shown in Table 12.
- Example 29 Manufacture of cured products
- 5 g of the copolymer of Example 17 was dissolved in 50 ml of hexane under a nitrogen atmosphere and dissolved uniformly.
- Ten milligrams of dibutyltin dilaurate was added thereto and stirred until uniform, then poured into a 5 ⁇ 10 cm container and allowed to stand to evaporate hexane.
- the obtained composition was dried under reduced pressure at 40 ° C. for 8 hours to prepare a sheet.
- the sheet was left in an atmosphere of 23 ° C. and 50% humidity for 168 hours to produce a cured product.
- the obtained cured product was evaluated in the same manner as in Example 27. The results are shown in Table 12.
- Comparative Example 5 A copolymer was produced in the same manner as in Example 17 except that the low-order polypropylene of Production Example 1 was used instead of the polypropylene of Example 16 (1). Instead of using the copolymer of Example 16, a sheet was prepared in the same manner as in Example 27 except that 20 grams of this copolymer was used, and the sheet was left in an atmosphere of 23 ° C. and 50% humidity for 168 hours. A cured product was produced. The obtained cured product was evaluated in the same manner as in Example 27. The results are shown in Table 12.
- Example 30 Manufacture of cured products 100 parts of the copolymer of Example 19 and 3 parts of polypropylene glycol (average molecular weight 250) and 3 parts (NOF Co., Ltd., Uniol D-250) were uniformly mixed at 140 ° C. to form a sheet of 1 mm thickness, and then 140 parts. Treated at 0 ° C. for 8 hours. The sheet (cured product) once cooled was heated on a hot plate controlled at 120 ° C. for 10 minutes, and the shape change was observed. The results are shown in Table 12.
- Comparative Example 6 A copolymer was produced in the same manner as in Example 19 except that the low-order polypropylene of Production Example 1 was used instead of the polypropylene of Example 16 (1). A sheet was prepared and evaluated in the same manner as in Example 30 except that 20 grams of this copolymer was used instead of using the copolymer of Example 19. The results are shown in Table 12.
- the reaction mixture was added to a large amount of methanol, and polypropylene was recovered by filtration. After air drying, vacuum drying was performed at a temperature of 80 ° C. to obtain 210 g of polypropylene.
- the molecular weight of the obtained polypropylene was 35000 in terms of weight average molecular weight (Mw), and the molecular weight distribution (Mw / Mn) was 1.85.
- the number of terminal vinylidene groups was 0.9 / molecule, and the stereoregularity (mmmm) was 78.9 mol%.
- Production Example 5 [Production of terminally unsaturated atactic PP] Polymerization was carried out in the same manner as in Production Example 4 except that 10 ⁇ mol of biscyclopentadienyl zirconium was used instead of ethylene bisindenylzirconium dichloride, the propylene pressure was 0.5 MPa, and polymerization was performed at a temperature of 20 ° C. for 90 minutes. After completion of the polymerization, the mixture was poured into 1 liter of methanol containing 2 mL of 12N hydrochloric acid, washed with water after stirring, and liquid polypropylene was obtained. The obtained liquid polypropylene was dried under reduced pressure at 80 ° C. to obtain 104 g of polypropylene.
- the molecular weight of the obtained polypropylene was 7340 in terms of weight average molecular weight (Mw), and the molecular weight distribution (Mw / Mn) was 1.77.
- the number of terminal vinylidene groups was 0.95 / molecule, and the stereoregularity (mmmm) was 9.0 mol%.
- Example 31 [Production of high-end unsaturated polypropylene] 40 g of the polypropylene produced in Production Example 4 was charged into a stainless steel reactor (with an internal volume of 500 mL) equipped with a stirrer. The mixture was stirred for 30 minutes under a nitrogen stream. Stirring was stopped and the resin temperature was raised to 160 ° C. using a mantle heater. After confirming that it was in a molten state, stirring was resumed. The mantle heater was controlled so that the resin temperature was constant at 270 ° C. To this, 0.4 ml of cumene hydroperoxide was added dropwise over 4 minutes. After completion of the dropwise addition, the reaction was performed for 4 minutes, and then cooled to 110 ° C. Paraxylene was added thereto and heated to obtain a uniform solution, which was then recovered, the solvent was removed, and then dried under reduced pressure at 150 ° C. for 10 hours to obtain radically decomposed polypropylene.
- the yield of the obtained decomposed polypropylene was 98.87% by mass with respect to the charged polypropylene, and the amount of by-products was very small.
- the weight average molecular weight (Mw) of the radically decomposed polypropylene was 13200, and the molecular weight distribution (Mw / Mn) was 1.62.
- the number of terminal vinylidene groups was 1.85 / molecule.
- Example 32 [Production of high-end unsaturated polypropylene] 40 g of atactic polypropylene produced in Production Example 5 was charged into a stainless steel reactor (with an internal volume of 500 mL) equipped with a stirrer. The mixture was stirred for 30 minutes under a nitrogen stream. Stirring was stopped, and the resin temperature was raised to 160 ° C. using a mantle heater. After confirming that it was in a molten state, stirring was resumed. The mantle heater was controlled so that the resin temperature was kept constant at 270 ° C. To this, 0.4 ml of cumene hydroperoxide was added dropwise over 4 minutes. After completion of the dropwise addition, the reaction was performed for 4 minutes, and then cooled to 110 ° C.
- n-heptane was added at 40 ° C. to obtain a homogeneous solution, which was collected, and after removing the solvent, drying under reduced pressure at 100 ° C. for 10 hours was performed to obtain radically decomposed polypropylene.
- the yield of the obtained decomposed polypropylene was 98.25% by mass with respect to the charged polypropylene, and the amount of by-product was very small.
- the weight average molecular weight (Mw) of the radically decomposed polypropylene was 2900, and the molecular weight distribution (Mw / Mn) was 1.76.
- the number of terminal vinylidene groups was 1.9 / molecule.
- Example 33 [Production of hydrosilylated polypropylene] A platinum-divinyltetramethyldisiloxane complex (SIP6831.0, manufactured by Azumax Co., Ltd.) 130 ppm was added as a catalyst to 5 g of the radically decomposed polypropylene and 0.2 g of diethoxymethylsilane produced in Example 1, and reacted at 100 ° C. for 5 hours. I let you. In the obtained polypropylene, the unsaturated bond in the vicinity of 1640 cm ⁇ 1 disappeared from the measurement of the infrared absorption spectrum, so that it was confirmed that the hydrosilylation proceeded, and it was confirmed that the hydrosilylated polypropylene was produced.
- SIP6831.0 platinum-divinyltetramethyldisiloxane complex
- Example 34 [Production of hydrosilylated polypropylene] Instead of the radically decomposed polypropylene of Example 1, the polypropylene of Example 32 was used, The reaction was conducted in the same manner as in Example 33 except that 0.5 g of diethoxymethylsilane was used. As for the obtained polypropylene, the unsaturated bond near 1640 cm ⁇ 1 disappeared from the measurement of infrared absorption spectrum, so that it was confirmed that the hydrosilylation proceeded, and it was confirmed that the hydrosilylated polypropylene was produced.
- Example 35 Manufacture of cured products. It was confirmed that 4 g of the hydrosilylated polypropylene of Example 33 was dissolved in 50 ml of hexane under a nitrogen atmosphere and dissolved uniformly. Ten milligrams of dibutyltin dilaurate was added thereto and stirred until uniform, then poured into a 5 ⁇ 10 cm container and allowed to stand to evaporate hexane. The obtained composition was dried under reduced pressure at 40 ° C. for 8 hours to prepare a sheet. The sheet was left in an atmosphere of 23 ° C. and 50% humidity for 168 hours to produce a cured product. The obtained cured product was heated on a hot plate controlled at 120 ° C. for 10 minutes, and its shape change was observed. As a result, it was confirmed that it was a cured product that retained its shape in a heated state and exhibited rubber elasticity.
- Example 36 [Manufacture of cured products] 1.2 g of polymethyl H siloxane (HMS-991, Azmax Co., Ltd.) is mixed with 10 g (6.07 mmol) of the radically decomposed polypropylene obtained in Example 32 which is liquid at room temperature, and further a platinum-divinyltetramethyldisiloxane complex. 10 mg (Azmax Corp. SIP6831.0) was added and mixed, then poured into a 5 ⁇ 10 cm container and stored at room temperature for 10 days to obtain a cured product. The cured product was heated for 10 minutes on a hot plate controlled at 120 ° C., and the shape change was observed. As a result, it was confirmed that it was a cured product that retained its shape in a heated state and exhibited rubber elasticity.
- polymethyl H siloxane HMS-991, Azmax Co., Ltd.
- Hydrogen was introduced at a pressure of 0.1 MPa and the temperature was controlled at 60 ° C. Further, propylene was continuously introduced at a gauge pressure of 0.6 MPa, and polymerization was performed for 40 minutes.
- the obtained polypropylene was 156 grams, the weight average molecular weight was 93,000, the number average molecular weight was 48,300, and the number of terminal vinylidene groups was 0.051 per molecule.
- the stereoregularity [mmmm] was 46 mol%.
- Example 38 [Production of high-end unsaturated polypropylene] 40 g of the polypropylene produced in Production Example 6 was charged into a stainless steel reactor (with an internal volume of 500 mL) equipped with a stirrer. The mixture was stirred for 30 minutes under a nitrogen stream. Stirring was stopped and the resin temperature was raised to 120 ° C. using a mantle heater. After confirming that it was in a molten state, stirring was resumed. The mantle heater was controlled so that the resin temperature was constant at 270 ° C. To this, 0.8 ml of cumene hydroperoxide (trade name: Park Mill H, manufactured by NOF Corporation) was dropped over 20 minutes.
- cumene hydroperoxide trade name: Park Mill H, manufactured by NOF Corporation
- the resulting radically decomposed polypropylene had a weight average molecular weight (Mw) of 15,800, a molecular weight distribution (Mw / Mn) of 1.45, and the number of terminal vinylidene groups per molecule (fv) of 1.60. . Moreover, the amount of by-products [by-product (g) / charged raw material (g)] was 0.015.
- Example 39 Manufacture of highly terminal unsaturated polypropylene (manufacture of continuous radical decomposition polypropylene by melt extruder)]
- (1) Production of Composition Composed of Polypropylene and Organic Peroxide 2,700 g of the polypropylene obtained in Production Example 2 was put into a universal mixing stirrer (type: 5DM-L-03-rr, Dalton mixer) and nitrogen stream The mixture was stirred at 126 rpm for 30 minutes. While a nitrogen stream was stopped and a nitrogen atmosphere state was secured, a homogeneous solution of 20 ml of hexane and 24 grams of diisopropylbenzene hydroperoxide (trade name: Park Mill P, manufactured by NOF Corporation) was stirred for 20 minutes. It was dripped. It stirred for 20 minutes after completion
- the bonded test pieces were cured for 7 days in an atmosphere of 23 ° C. and 50% RH and moisture-cured.
- the measurement of the heat resistant creep temperature was carried out according to JIS K6833. Specifically, the lower part of the test piece was cut off so that the length of the bonded portion was 25 mm, and a 500 g weight was attached to the lower end of the test piece suspended in the thermostat. The temperature of the thermostatic bath was maintained at 38 ° C. for 15 minutes, and then the temperature was increased at a rate of 2 ° C. for 5 minutes to obtain a heat resistant creep temperature when the weight dropped. As a result, the heat resistant creep temperature was 81 ° C.
- Example 40 A copolymer was produced in the same manner as in Example 39 except that the amount of 3- (triethoxysilyl) propyl methacrylate was 21.4 g in Example 39 (3). As a result, the B viscosity was 6,900 mPaS and the heat-resistant creep temperature was 71 ° C.
- Example 41 In Example 40, 6.0 g of 2-ethylhexyl mercaptoacetate was used as a chain transfer agent in a homogeneous solution of 2-ethylhexyl acrylate, 3- (triethoxysilyl) propyl methacrylate, and tertiarybutyl cumyl peroxide. A copolymer was produced in the same manner except that this mixed solution was added dropwise. As a result, the B viscosity was 4,600 mPaS and the heat-resistant creep temperature was 69 ° C. Compared to Example 40, the use of a chain transfer agent was able to significantly reduce the melt viscosity without impairing the heat-resistant creep characteristics.
- Example 42 [Production of copolymer with polar monomer] A glass 3 liter separable flask having a stirrer and an inlet was sufficiently dried and then placed in a nitrogen atmosphere. To this, 500.0 g of the decomposed polypropylene produced in Example 39 (2) and 5.65 g of maleic anhydride were added. To this was added 492 g of dehydrated ethyl acetate, and stirring was started at room temperature. To this was added 484 g of heptane. It heated so that it might be in a boiling state (internal temperature 80.6 degreeC) using the oil bath.
- a uniform solution consisting of 54.2 g of acrylic acid, 10.0 g of azobisisobutyronitrile, and 88.5 g of dehydrated ethyl acetate was charged at a constant rate over 115 minutes. The reaction was continued for an additional 180 minutes after the end of the addition. After completion of the reaction, a part of the reaction mixture was sampled, a large amount of ethyl acetate was added to separate it into a liquid part and a solid, and unreacted monomers present in the liquid part were quantified by gas chromatogram method. In addition, the graft copolymer was recovered by heating and reducing the reaction mixture and further reducing the pressure at 130 ° C. for 8 hours.
- the unreacted monomer remained at a rate of 0.18% by mass with respect to the produced graft copolymer.
- the graft copolymer dried under heating under reduced pressure was white. Further, when the infrared absorption spectrum of the graft copolymer was measured, no absorption based on the vinylidene group present at 888 cm ⁇ 1 was observed, so it was confirmed that the graft reaction proceeded efficiently. Further, the B viscosity of the graft copolymer was 12,500 mPaS.
- Example 42 Comparative Example 7 In Example 42, the same procedure as in Example 42 was performed, except that acrylic acid and maleic anhydride were previously charged in the reaction vessel and a dehydrated ethyl acetate solution of azobisisobutyronitrile was added. As a result, the unreacted monomer remained at a ratio of 1.02% by mass with respect to the produced graft copolymer. Further, the graft copolymer dried under heating under reduced pressure turned yellow. When the infrared absorption spectrum was measured, it was confirmed that the efficiency of the graft reaction was low because of the presence of absorption based on the vinylidene group.
- Example 43 A graft copolymerization reaction was carried out in the same manner as in Example 42 except that 6 g of n-dodecyl mercaptan was added dropwise to a homogeneous solution of acrylic acid, azobisisobutyronitrile, and dehydrated ethyl acetate. After completion of the reaction, a part was extracted, poured into a large amount of tetrahydrofuran, dissolved, and filtered through a 400 mesh stainless steel sieve. As a result, no residue remained on the mesh. Further, the B viscosity of the recovered graft copolymer was 4500 mPaS.
- Example 44 Production of Low Stereoregularity Polybutene A pressure-resistant autoclave made of stainless steel with an internal capacity of 1.4 liters was heated and depressurized to 80 ° C. and sufficiently dried to form a nitrogen atmosphere. To this, 400 ml of dehydrated heptane and 1 ml of a heptane solution containing 0.5 mmol of triisobutylaluminum were added and stirred at room temperature for 5 minutes.
- the weight average molecular weight (Mw) of this product was 9,800, the molecular weight distribution (Mw / Mn) was 2.2, and the number of terminal vinylidene groups per molecule was 0.7 / molecule.
- the stereoregularity (mmmm) was 61.6 mol%.
- the resulting radically decomposed low-order polybutene has a weight average molecular weight Mw of 2,200, a number average molecular weight Mn of 1,300, a molecular weight distribution Mw / Mn of 1.7, Mp / Mm of 0.224, and a terminal vinylidene group.
- the number per molecule was 1.71 / molecule.
- the terminal unsaturated polyolefin of the present invention uses a terminal vinylidene group as a reaction point, thereby imparting adhesiveness, paintability and coating properties to a chemically inert polyolefin material, and an alloy material with a resin other than polyolefin.
- a terminal vinylidene group as a reaction point, thereby imparting adhesiveness, paintability and coating properties to a chemically inert polyolefin material, and an alloy material with a resin other than polyolefin.
- it can be widely used for reactive hot melt adhesives, sealing materials, potting materials and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Macromonomer-Based Addition Polymer (AREA)
Abstract
Description
特許文献1及び特許文献2では、高分子量体を高分解することでビニリデン数を増加しているが、大量の副生成物の発生により、収率が低下する課題がある。
特許文献3では、ジルコノセンジクロリド触媒による重合で末端ビニリデン基を有するアタクチックポリプロピレンを製造している。特許文献4では、立体規則性[mm]が88.8及び94%のポリプロピレンを用いて、末端ビニリデン基を有するアイソタクチックポリプロピレンの製造例を開示している。特許文献5では、立体規則性[mmmm]が30~80%の低立体規則性ポリオレフィンを用いて、高純度及び高選択性の末端ビニリデン基を有するポリオレフィンを開示している。
しかし当該方法では、末端不飽和基は、1分子当たり最大1.0個であり、高い不飽和度を達成することはできない。
しかし、ジオレフィンとの共重合による場合は、末端に不飽和基を導入することは困難であり、重合反応の際に架橋等の副反応が進行する問題があった。
本発明の他の目的は、副生成物の発生量が少ない末端不飽和ポリオレフィンの製造方法を提供することである。
1.プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;又はブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体からなる末端不飽和ポリオレフィンであって、
下記(1)~(4)を満たす末端不飽和ポリオレフィン。
(1)プロピレン連鎖部又はブテン-1連鎖部のメソペンタッド分率[mmmm]が20~80モル%である
(2)末端ビニリデン基の1分子当り数が1.3~2.5である
(3)重量平均分子量Mwが500~100,000である
(4)分子量分布Mw/Mnが1.1~2.6である
2.前記プロピレン単独重合体からなる1に記載の末端不飽和ポリオレフィン。
3.プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンであって、下記(5)及び(6)を満たす原料ポリオレフィンを不活性気体存在下で分解する1又は2に記載の末端不飽和ポリオレフィンの製造方法。
(5)プロピレン連鎖部又はブテン-1連鎖部の立体規則性[mmmm]が20~80モル%である
(6)重量平均分子量Mwが4000~1,000,000である
4.前記分解が、前記原料ポリオレフィンを300~400℃の温度で30分~10時間加熱処理する熱分解反応である3に記載の末端不飽和ポリオレフィンの製造方法。
5.前記分解が、温度160~300℃で、有機過酸化物を前記原料ポリオレフィンに対して0.05~2質量%添加するラジカル分解反応である3に記載の末端不飽和ポリオレフィンの製造方法。
6.前記有機過酸化物の1分間半減期温度が、140~270℃である5に記載の末端不飽和ポリオレフィンの製造方法。
7.プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンを分解して得られる末端不飽和ポリオレフィンであって、
下記(7)~(10)を満たす末端不飽和ポリオレフィン。
(7)末端ビニリデン基の1分子当りの数(fv)が1.3~2.5である
(8)fv≧-2(Mp/Mm)+2(式中、Mpは末端不飽和ポリオレフィンの数
平均分子量であり、Mmは原料ポリオレフィンの数平均分子量である。但し、Mp/Mmは0.05~0.8である)
(9)重量平均分子量Mwが500~100,000である
(10)分子量分布Mw/Mnが1.1~2.6である
8.前記プロピレン単独重合体又は前記プロピレン系共重合体を分解して得られる末端不飽和ポリオレフィンであって、
前記プロピレン単独重合体又は前記プロピレン系共重合体のプロピレン連鎖部が、アタクチック構造、シンジオタクチック構造又はアイソタクチック構造を有する7に記載の末端不飽和ポリオレフィン。
9.前記原料ポリオレフィンがプロピレン単独重合体である7又は8に記載の末端不飽和ポリオレフィン。
10.プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1と、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンであって、重量平均分子量が4000~1,000,000である原料ポリオレフィンを不活性気体存在下で分解する7~9のいずれかに記載の末端不飽和ポリオレフィンの製造方法。
11.1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィンの末端ビニリデン基の5モル%以上が官能基を有する官能化ポリオレフィン。
12.前記官能基が、水酸基、エポキシ基、イソシアナート基、アルコキシ珪素基、アルキル珪素基、カルボキシル基、アミノ基、及び酸無水物構造からなる群から選択される1種以上である11に記載の官能化ポリオレフィン。
13.前記官能基として、少なくともアルコキシ珪素基を含む11又は12に記載の官能化ポリオレフィンを湿気硬化してなる架橋体。
14.1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィンとSiH基を1分子当たり2個以上有するオルガノハイドロジェンポリシロキサンとを反応させて得られる反応物。
15.1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィン20~100質量%、及び前記末端不飽和ポリオレフィン以外のポリオレフィン0~80質量%との組合わせ100質量部と、下記[I]~[IV]から選ばれる単量体1種以上0.2~300質量部、又は下記A群から選ばれる1種以上とB群から選ばれる1種以上の単量体混合物0.2~300質量部を、ラジカル開始剤0.001~10質量部の存在下、40~230℃でグラフト重合させるグラフト共重合体、又は前記グラフト共重合体を含む熱可塑性樹脂組成物の製造方法。
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
A群:
[V]無水マレイン酸及びその置換体
[VI]マレイン酸及びそのエステル
[VII]マレイミド及びその置換体
B群:
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
[VIII]α-オレフィン
16.連鎖移動剤の存在下にグラフト重合を行う、15に記載のグラフト共重合体、又は前記グラフト共重合体を含む熱可塑性樹脂組成物の製造方法。
17.1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィンと、単独ラジカル重合性を実質的に持たない単量体(a)とを共存させた混合物に、前記単量体(a)と共重合可能な単量体(b)を接触させることからなるグラフト共重合体、又は前記グラフト共重合体を含む熱可塑性樹脂組成物の製造方法。
18.プロピレン単独重合体;プロピレンと、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1と、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択されるポリオレフィンの両末端に、下記[I]、[II]、[III]及び[IV]から選ばれる1種以上から誘導されるポリオレフィン連鎖、又は下記A群から選ばれる1種以上及びB群から選ばれる1種以上から誘導されるポリオレフィン連鎖を有するグラフト共重合体であって、
下記(c)を満たすグラフト共重合体。
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
A群:
[V]無水マレイン酸及びその置換体
[VI]マレイン酸及びそのエステル
[VII]マレイミド及びその置換体
B群:
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
[VIII]α-オレフィン
(c)分子量分布(Mw/Mn)が1.6~6
19.前記ポリオレフィン連鎖のプロピレン連鎖又はブテン-1連鎖のメソペンタッド分率[mmmm]が20~80モル%である18記載のグラフト共重合体。
20.前記ポリオレフィンが、アタクチック構造又はシンジオタクチック構造であるプロピレン単独重合体である18記載のグラフト共重合体。
21.18に記載のグラフト共重合体及びポリオレフィンを含有する熱可塑性樹脂組成物。
22.18に記載のグラフト共重合体、又は21に記載の熱可塑性樹脂組成物がさらに熱可塑性樹脂を含む組成物。
23.18に記載のグラフト共重合体、又は21に記載の熱可塑性樹脂組成物がさらにフィラー及び/又は顔料を含む組成物。
24.18に記載のグラフト共重合体、又は21に記載の熱可塑性樹脂組成物を接着剤、樹脂相溶化剤、分散体、コーティング材として用いる用途。
25.11又は12に記載の官能化ポリオレフィンを接着剤、樹脂相溶化剤、分散体、コーティング材として用いる用途。
26.11又は12に記載の官能化ポリオレフィンを反応性ホットメルト接着剤、シール材、ポッティング材として用いる用途。
本発明によれば、副生成物の発生量が少ない末端不飽和ポリオレフィンの製造方法が提供できる。
本発明の第1の末端不飽和ポリオレフィンは、プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;又はブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体からなる末端不飽和ポリオレフィンであって、下記(1)~(4)を満たす:
(1)プロピレン連鎖部、又はブテン-1連鎖部のメソペンタッド分率[mmmm]が20~80モル%である
(2)末端ビニリデン基の1分子当り数が1.3~2.5である
(3)重量平均分子量Mwが500~100,000である
(4)分子量分布が1.1~2.6である
(i)プロピレン単独重合体
(ii)プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体
(iii)ブテン-1単独重合体
(iv)ブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体
また、上記(ii)及び(iv)の共重合体における、エチレン及びαオレフィンの含有量、又はエチレン、プロピレン及びαオレフィンの含有量は、好ましくは0モル%超5モル%以下である。エチレン、及びαオレフィンの含有量、又はエチレン、プロピレン及びαオレフィンの含有量が10モル%超の場合、αオレフィンのポリプロピレン及びポリブテンとの親和性が低下し、ポリオレフィンの接着強度等が低下するおそれがある。
プロピレン連鎖部、又はブテン-1連鎖部のメソペンタッド分率20モル%未満では、結晶性を示さないため、強度に乏しく、接着剤として用いた場合等に、結晶性ポリプロピレンに対して十分な接着強度を発現できないおそれがある。一方、80モル%を越える場合、結晶性が高くなるため、低温溶融性が乏しくなり、塗布性等の作業性が低下するおそれがある。
尚、13C-NMRスペクトルの測定は、エイ・ザンベリ(A.Zambelli)等により「Macromolecules,8,687(1975)」で提案されたピークの帰属に従い、下記の装置及び条件にて行うことができる。
方法:プロトン完全デカップリング法
濃度:220mg/mL
溶媒:1,2,4-トリクロロベンゼンと重ベンゼンの90:10(容量比)混合溶媒
温度:130℃
パルス幅:45°
パルス繰り返し時間:4秒
積算:10000回
末端ビニリデン基の1分子当り数が1.3未満の場合、末端ビニリデン基を起点とした反応による耐熱性の付与が不十分となるおそれがある。一方、末端ビニリデン基の1分子当り数が2.5超の場合、ポリオレフィンは分岐構造を多く含み、分岐構造は、直鎖状構造と溶融流動性が異なるため塗布等の挙動が変化するおそれがある。
末端ビニリデン基の1分子当り数(個)=(Mn/M)×(C/100)
全末端基量に対する末端ビニリデン基の存在割合から1分子当りの末端ビニリデン基数(末端不飽和基がビニリデン基である場合の末端不飽和基数)を、全不飽和基に対する末端ビニリデン基の存在割合から末端ビニリデン基の選択性を決定することができる。
1H-NMR及び13C-NMRを利用した末端ビニリデン基の個数の測定を、プロピレン重合体を例にして以下説明する。
プロピレン重合体では、<2>末端ビニリデン基のメチレン基(4.8~4.6ppm)、<1>末端ビニル基のメチレン基(5.10~4.90ppm)が観測され、全プロピレンに対する割合は次式で計算できる。尚、<3>は、プロピレン連鎖(0.6~2.3ppm)のメチン、メチレン、メチル基に相当するピーク強度に対応する。
末端ビニリデン基量(A)=(<2>/2)/[(<3>+4×<1>/2+3×<2>/2)/6]×100 単位:mol%
末端ビニル基量(B)=(<1>/2)/[(<3>+4×<1>/2+3×<2>/2)/6]×100 単位:mol%
プロピレン重合体では、<5>n-プロピル末端の末端メチル基(14.5ppm付近)、<6>n-ブチル基末端の末端メチル基(14.0ppm付近)、<4>iso-ブチル末端のメチン基(25.9ppm付近)、及び<7>末端ビニリデン基のメチレン基(111.7ppm付近)が観察される。
13C-NMRの末端ビニル基量ピーク強度=(B)/(A)×<7>
ここで末端基の全濃度(T)は以下のように表わされる。
T=(B)/(A)×<7>+<4>+<5>+<6>+<7>
従って、各末端の割合は、以下となる。
(C)末端ビニリデン基=<7>/T×100 単位:mol%
(D)末端ビニル基=(B)/(A)×<7>×100
(E)n-プロピル末端=<5>/T×100
(F)n-ブチル基末端=<6>/T×100
(G)iso-ブチル末端=<4>/T×100
1分子当たりの末端ビニリデン基の個数は2×(C)/100 単位:個/分子となる。
重量平均分子量が500未満である場合、末端ビニリデン基を基点とした反応によってポリオレフィンに耐熱性を付与する場合に、ポリオレフィンの柔軟性が阻害されるおそれがある。一方、重量平均分子量が100,000を越える場合、溶融粘度が大きくなって、塗布性等の作業性が低下するおそれがある。
尚、上記重量平均分子量は、ゲルパーミエイションクロマトグラフィー(GPC)法により測定できる。
分子量分布が1.1未満とすることは技術的に困難である。一方、分子量分布が2.6超の場合、低分子量ポリオレフィンが増加することに対応するため、力学物性の低下をもたらすおそれがある。
重量平均分子量(Mw)及び数平均分子量(Mn)は、ポリスチレン換算分子量を対応するポリマーの分子量に換算するため、Mark-Houwink-桜田の式の定数K及びaを用いてUniversal Calibration法により求めることができる。
具体的には「「サイズ排除クロマトグラフィー」」森定雄著、P67~69、1992年、共立出版」に記載の方法によって決定できる。尚、K及びαは、「「Polymer
Handbook」 John Wiley&Sons,Inc.」に記載されている。また、新たに算出する絶対分子量に対する極限粘度の関係から定法によって決定することもできる。
検出器:液体クロマトグラフィー用RI検出器 ウオーターズ 150C
カラム:TOSO GMHHR-H(S)HT
溶媒:1,2,4-トリクロロベンゼン
測定温度:145℃
流速:1.0mL/分
試料濃度:0.3質量%
本発明の第1の末端不飽和ポリオレフィンは、プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンであって、下記(5)及び(6)を満たす原料ポリオレフィンを不活性気体存在下で分解することにより製造できる。
(5)プロピレン連鎖部又はブテン-1連鎖部の立体規則性[mmmm]が20~80モル%である
(6)重量平均分子量Mwが4000~1,000,000である
立体規則性[mmmm]が20~80モル%であるため、原料ポリオレフィンは低温で容易に溶融できる、又は溶媒への良好な溶解性を有するため、分解反応場の選定範囲及び分解設定温度範囲が広く、比較的低温で分解することが可能である。これにより、副反応を制御できるメリットを有している。加えて、有機過酸化物を併用する分解では、より温和でかつ短時間の反応で効率よく分解でき、原料ポリオレフィンの上記メリットを大きくすることができる。特に原料ポリオレフィンが末端不飽和基を予め有している場合では、上記メリットを最大化できる。
また、原料ポリオレフィンのプロピレン連鎖部又はブテン-1連鎖部の立体規則性[mmmm]についても、第1の末端不飽和ポリオレフィンと同様である。
原料ポリオレフィンの重量平均分子量が4000未満の場合、分解率を高く設定することができないおそれがある。一方、原料ポリオレフィンの重量平均分子量が1,000,000超の場合、分解率を高く設定できるが、分解時の粘度が高く、プロセス上の攪拌動力、攪拌均一性等の制約が生じるおそれがある。
1分子当りの末端不飽和基の数が0.4個未満の場合、分解による末端ビニリデン基の数がさほど増加せず、末端ビニリデン基を増加させるために、高分子量体を分解する必要が生じるおそれがある。一方、1分子当りの末端不飽和基の数が1.0個超は、後述する重合触媒による技術では製造が困難である。
尚、上記末端不飽和基は、ビニル基、ビニリデン基、トランス(ビニレン)基等が挙げられ、好ましくはビニリデン基である。ビニリデン基はラジカル重合性、各種反応の適用範囲が広く、多様な要求に対応できる。
(A)シクロペンタジエニル基、置換シクロペンタジエニル基、インデニル基、置換インデニル基を有する周期律表第3族~10族の金属元素を含む遷移金属化合物
(B)遷移金属化合物と反応してイオン性の錯体を形成しうる化合物
(C)有機アルミニウム化合物

E1及びE2はそれぞれ、置換シクロペンタジエニル基,インデニル基,置換インデニル基,ヘテロシクロペンタジエニル基,置換ヘテロシクロペンタジエニル基,アミド基(-N<),ホスフィン基(-P<),炭化水素基〔>CR-,>C<〕及びケイ素含有基〔>SiR-,>Si<〕(但し、Rは水素又は炭素数1~20の炭化水素基あるいはヘテロ原子含有基である)の中から選ばれた配位子を示し、A1及びA2を介して架橋構造を形成している。E1及びE2は互いに同一でも異なっていてもよい。このE1及びE2としては、シクロペンタジエニル基、置換シクロペンタジエニル基,インデニル基及び置換インデニル基が好ましく、E1及びE2のうちの少なくとも一つは、シクロペンタジエニル基、置換シクロペンタジエニル基、インデニル基又は置換インデニル基である。
Xはσ結合性の配位子を示し、Xが複数ある場合、複数のXは同じでも異なっていてもよく、他のX,E1,E2又はYと架橋していてもよい。このXの具体例としては、ハロゲン原子,炭素数1~20の炭化水素基,炭素数1~20のアルコキシ基,炭素数6~20のアリールオキシ基,炭素数1~20のアミド基,炭素数1~20のケイ素含有基,炭素数1~20のホスフィド基,炭素数1~20のスルフィド基,炭素数1~20のアシル基等が挙げられる。
ハロゲン原子としては、塩素原子、フッ素原子、臭素原子、ヨウ素原子が挙げられる。炭素数1~20の炭化水素基として具体的には、メチル基、エチル基、プロピル基、ブチル基、ヘキシル基、シクロヘキシル基、オクチル基等のアルキル基;ビニル基、プロペニル基、シクロヘキセニル基等のアルケニル基;ベンジル基、フェニルエチル基、フェニルプロピル基等のアリールアルキル基;フェニル基、トリル基、ジメチルフェニル基、トリメチルフェニル基、エチルフェニル基、プロピルフェニル基、ビフェニル基、ナフチル基、メチルナフチル基、アントラセニル基、フェナントニル基等のアリール基等が挙げられる。なかでもメチル基、エチル基、プロピル基等のアルキル基やフェニル基等のアリール基が好ましい。
炭素数1~20のケイ素含有基としては、メチルシリル基、フェニルシリル基等のモノ炭化水素置換シリル基;ジメチルシリル基、ジフェニルシリル基等のジ炭化水素置換シリル基;トリメチルシリル基、トリエチルシリル基、トリプロピルシリル基、トリシクロヘキシルシリル基、トリフェニルシリル基、ジメチルフェニルシリル基、メチルジフェニルシリル基、トリトリルシリル基、トリナフチルシリル基等のトリ炭化水素置換シリル基;トリメチルシリルエーテル基等の炭化水素置換シリルエーテル基;トリメチルシリルメチル基等のケイ素置換アルキル基;トリメチルシリルフェニル基等のケイ素置換アリール基等が挙げられる。なかでもトリメチルシリルメチル基、フェニルジメチルシリルエチル基等が好ましい。
炭素数1~20のアシル基としては、ホルミル基、アセチル基、プロピオニル基、ブチリル基、バレリル基、パルミトイル基、テアロイル基、オレオイル基等のアルキルアシル基、ベンゾイル基、トルオイル基、サリチロイル基、シンナモイル基、ナフトイル基、フタロイル基等のアリールアシル基、シュウ酸、マロン酸、コハク酸等のジカルボン酸からそれぞれ誘導されるオキサリル基、マロニル基、スクシニル基等が挙げられる。
このような架橋基のうち、少なくとも一つは炭素数1以上の炭化水素基からなる架橋基であることが好ましい。このような架橋基としては、例えば一般式(a)で表されるものが挙げられ、その具体例としては、メチレン基,エチレン基,エチリデン基,プロピリデン基,イソプロピリデン基,シクロヘキシリデン基,1,2-シクロヘキシレン基,ビニリデン基(CH2=C=),ジメチルシリレン基,ジフェニルシリレン基,メチルフェニルシリレン基,ジメチルゲルミレン基,ジメチルスタニレン基,テトラメチルジシリレン基,ジフェニルジシリレン基等を挙げることができる。これらの中で、エチレン基,イソプロピリデン基及びジメチルシリレン基が好適である。

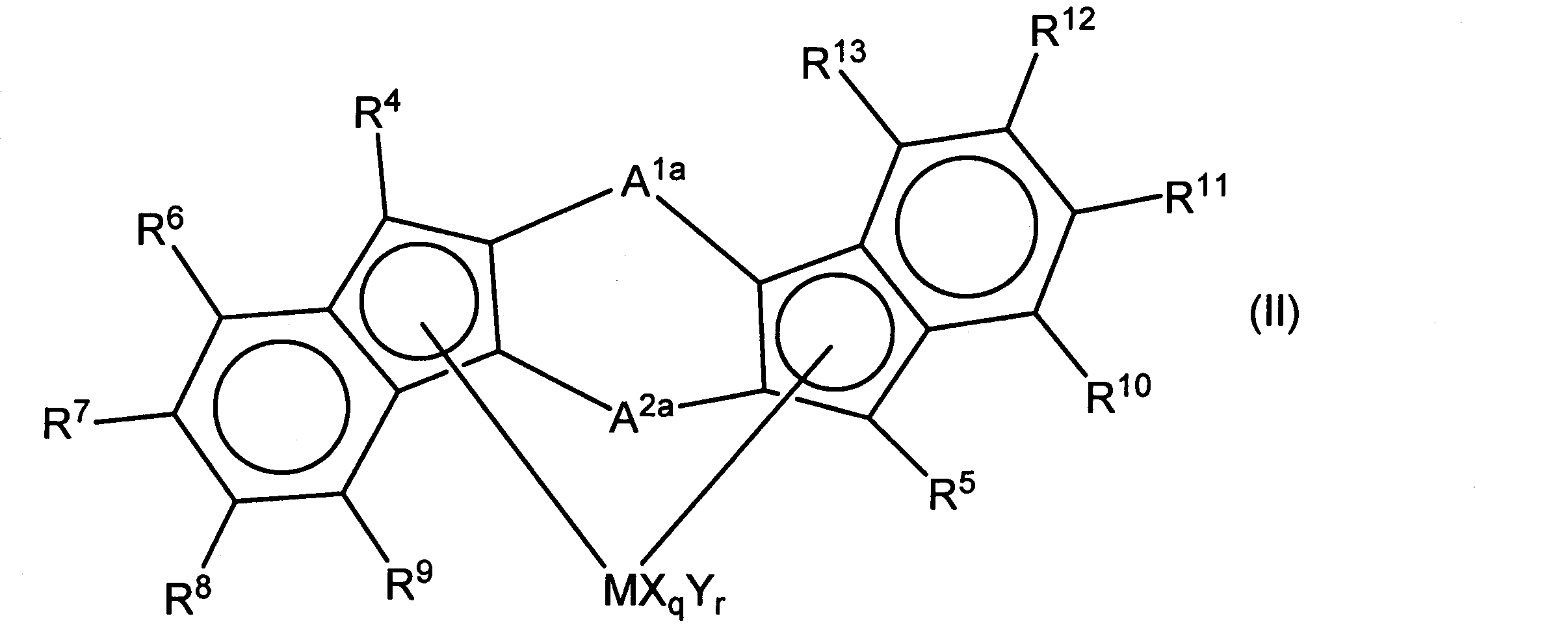
(C)成分の有機アルミニウム化合物としては、トリメチルアルミニウム、トリエチルアルミニウム、トリイソプロピルアルミニウム、トリイソブチルアルミニウム、トリノルマルへキシルアルミニウム、トリノルマルオクチルアルミニウム、ジメチルアルミニウムクロリド、ジエチルアルミニウムクロリド、メチルアルミニウムジクロリド、エチルアルミニウムジクロリド、ジメチルアルミニウムフルオリド、ジイソブチルアルミニウムヒドリド、ジエチルアルミニウムヒドリド及びエチルアルミニウムセスキクロリド等が挙げられる。これらの有機アルミニウム化合物は一種用いてもよく、二種以上を組み合わせて用いてもよい。
これらのうち、本発明においては、トリメチルアルミニウム、トリエチルアルミニウム、トリイソプロピルアルミニウム、トリイソブチルアルミニウム、トリノルマルへキシルアルミニウム及びトリノルマルオクチルアルミニウム等のトリアルキルアルミニウムが好ましく、トリイソブチルアルミニウム、トリノルマルへキシルアルミニウム及びトリノルマルオクチルアルミニウムがより好ましい。
(A)成分と(B)成分との使用割合(A)/(B)は、モル比で好ましくは10/1~1/100、より好ましくは2/1~1/10である。(A)/(B)が10/1~1/100の範囲にあると、触媒としての効果が得られると共に、単位質量ポリマー当たりの触媒コストを抑えることができる。また、目的とする末端不飽和オレフィン系重合体中にホウ素が多量に存在するおそれがない。
(A)成分と(C)成分との使用割合(A)/(C)は、モル比で好ましくは1/1~1/10000、より好ましくは1/5~1/2000、さらに好ましくは1/10~1/1000である。(C)成分を用いることにより、遷移金属当たりの重合活性を向上させることができる。(A)/(C)が1/1~1/10000の範囲にあると、(C)成分の添加効果と経済性のバランスが良好であり、また、目的とする末端不飽和オレフィン系重合体中にアルミニウムが多量に存在するおそれがない。
本発明の製造方法においては、上述した(A)成分及び(B)成分、あるいは(A)成分、(B)成分及び(C)成分を用いて予備接触を行うこともできる。予備接触は、(A)成分に、例えば(B)成分を接触させることにより行うことができるが、その方法に特に制限はなく、公知の方法を用いることができる。このような予備接触により触媒活性の向上や、助触媒である(B)成分の使用割合の低減等、触媒コストの低減に効果的である。
加熱温度は、目標とする分子量を設定し、予め実施した実験結果を勘案して調整することができ、好ましくは300~400℃であり、より好ましくは310~390℃である。加熱温度が300℃未満の場合、熱分解反応が進まないおそれがある。一方、加熱温度が400℃超の場合、得られる末端不飽和ポリオレフィンが劣化するおそれがある。
また、熱分解時間(加熱処理時間)は、好ましくは30分~10時間であり、より好ましくは60~240分である。熱分解時間が30分未満の場合、得られる末端不飽和ポリオレフィンの生成量が少なくおそれがある。一方、熱分解時間が10時間超の場合、得られる末端不飽和ポリオレフィンが劣化するおそれがある。
上記分解温度は、好ましくは170~290℃であり、より好ましくは180~280℃である。分解温度が160℃未満の場合、分解反応が進まないおそれがある。一方、分解温度が300℃超の場合、分解が激しく進行し、攪拌により十分に有機過酸化物が溶融ポリマーに均一拡散する前に分解が終了してしまい、収率が低下するおそれがある。
上記有機過酸化物の滴下は、上記分解時間の範囲内で滴下するとよく、当該滴下は連続的な滴下及び分割した滴下のいずれでもよい。また、滴下終了時間からの反応時間は、上記反応時間の範囲内とするとよい。
上記溶媒は、好ましくは炭化水素系溶媒であり、具体例としてはヘプタン、オクタン、デカン、ドデカン、テトラデカン、ヘキサデカン、ナノデカン等の脂肪族炭化水素;メチルシクロペンタン、シクロヘキサン、メチルシクロへキサン、シクロオクタン、シクロドデカン等の脂環式炭化水素;及びベンゼン、トルエン、キシレン、エチルベンゼン、トリメチルベンゼン等の芳香族炭化水素が挙げられる。これら溶媒のなかでも、沸点が100℃以上の溶媒が好ましい。
装置は、単軸又は二軸の溶融押出機を用いることができ、好ましくはバレル途中に注入口を有し、減圧脱気が可能な押出機であって、L/D=10以上である押出機である。
尚、有機過酸化物が固体である、又は有機過酸化物が原料ポリオレフィンに対して溶解性が低い場合は、有機過酸化物を予め炭化水素溶媒に溶解させた溶液として、原料ポリオレフィンに吸収含浸させるとよい。
本発明の第2の末端不飽和ポリオレフィンは、プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンを分解して得られる末端不飽和ポリオレフィンであって、下記(7)~(10)を満たす。
(7)末端ビニリデン基の1分子当りの数(fv)が1.3~2.5である
(8)fv≧-2(Mp/Mm)+2(式中、Mpは末端不飽和ポリオレフィンの数平均分子量であり、Mmは原料ポリオレフィンの数平均分子量である。但し、Mp/Mmは0.05~0.8である)
(9)重量平均分子量Mwが500~100,000である
(10)分子量分布Mw/Mnが1.1~2.6である
原料ポリオレフィンの重量平均分子量が4000未満の場合、末端不飽和ポリオレフィンの末端ビニリデン基数が十分に上がらず、副生物の揮発成分が増加するおそれがある。一方、原料ポリオレフィンの重量平均分子量が1,000,000超の場合、溶融粘度が高く攪拌に支障が生じるおそれがある。
原料ポリオレフィンが末端ビニリデン基を有するポリオレフィンである場合、1分子当りの当該末端ビニリデン基の数は、好ましくは0.4個~1.0個であり、より好ましくは0.5個~1.0個であり、さらに好ましくは0.6個~1.0個である。
原料ポリオレフィンの1分子当りの末端ビニリデン基の数が0.4個未満の場合、得られる第2の末端不飽和ポリオレフィンの末端ビニリデン数を増加するためには分解率(Mp/Mm)を高める必要があり、副生成物量が増加するおそれがあるうえ、特に高分子量であってかつ所望の末端ビニリデン基数を有する末端不飽和ポリオレフィンの製造が困難となるおそれがある。
(A)シクロペンタジエニル基置換、シクロペンタジエニル基、インデニル基、置換インデニル基を有する周期律表第3族~10族の金属元素を含む遷移金属化合物
(B)遷移金属化合物と反応してイオン性の錯体を形成しうる化合物
(C)有機アルミニウム化合物
CpM1R5 eR6 fR7 g (A-2)
Cp2M1R5 hR6 i (A-3)
(Cp-A-Cp)M1R5 hR6 i (A-4)
[式中、M1は、周期律表第4族遷移金属である
Cpはシクロペンタジエニル基,置換シクロペンタジエニル基,インデニル基,置換インデニル基,テトラヒドロインデニル基,置換テトラヒドロインデニル基,フルオレニル基,置換フルオレニル基,オクタヒドロフルオレニル基,置換オクタヒドロフルオレニル基及びアズレニル基から選ばれる基である。
R5,R6及びR7は、それぞれ独立に配位子を示し、Aは共有結合による架橋を示す。
e,f及びgはそれぞれ0~3の整数を、h及びiはそれぞれ0~2の整数を示す。但しe、f及びgは同時に0にならず、h及びiも同時に0にならない。
R5,R6及びR7は、その2以上が互いに結合して環を形成していてもよい。また、式(A-3)式及び式(A-4)において、2つのCpは同一のものであってもよく、互いに異なるものであってもよい。]

Cpはシクロペンタジエニル基、置換シクロペンタジエニル基、インデニル基、置換インデニル基、テトラヒドロインデニル基、置換テトラヒドロインデニル基、フルオレニル基又は置換フルオレニル基等の環状不飽和炭化水素基又は鎖状不飽和炭化水素基を示す。
X2は、水素原子、ハロゲン原子、炭素数1~20のアルキル基、炭素数6~20のアリール基、アルキルアリール基若しくはアリールアルキル基又は炭素数1~20のアルコキシ基を示す。
Zは、SiR9 2,CR9 2,SiR9 2SiR9 2,CR9 2CR9 2,CR9 2CR9 2CR9 2,CR9=CR9,CR9 2SiR9 2又はGeR9 2を示し、Y2は-N(R10)-,-O-,-S-又は-P(R10)-を示す。
上記R9は、水素原子又は20個までの非水素原子をもつアルキル,アリール,シリル,ハロゲン化アルキル,ハロゲン化アリール基及びそれらの組合せから選ばれる基であり、R10は炭素数1~10のアルキル若しくは炭素数6~10のアリール基であるか、又は1個若しくはそれ以上のR9と30個までの非水素原子の結合環系を形成してもよい。
sは1又は2を示す。]
M2がチタン原子である触媒が好ましい。
尚、触媒活性が低い場合は、極微量の水素を重合反応場に添加することで末端ビニリデン基の生成量を大きく減少させずに、活性を高めることができる。
(8)fv≧-2(Mp/Mm)+2
[式中、Mpは末端不飽和ポリオレフィンの数平均分子量であり、Mmは原料ポリオレフィンの数平均分子量である。但し、Mp/Mmは0.05~0.8である]
上記式(8)は、予め末端不飽和基を有する原料ポリオレフィンを用いることで、同一分解率では、末端不飽和基数が高く、また、同一分子量で比較した場合でも、末端不飽和基数が高いポリオレフィンが生成されることを示す。
Mp/Mmは、例えば0.05~0.8であり、好ましくは0.06~0.7である。Mp/Mmが0.05未満の場合、分解率が高く、副生成が増加するおそれがある。一方、Mp/Mmが0.8超の場合、末端ビニリデン基の数が少なく、ポリオレフィンを接着剤等として用いる場合に、十分に耐熱性が得られないおそれがある。
分子量分布が1.1未満の末端不飽和ポリオレフィンの製造は、技術的に困難である。一方、分子量分布が2.6超である場合、低分子量のポリオレフィンが増加していることを意味し、力学物性が低下するおそれがある。
尚、シンジオクタチック構造は、ポリオレフィンの[rrrr]が30~95モル%であることを意味し、アイソタクチック構造とは[mmmm]が30~95モル%を意味する。
但し、分解反応を溶融連続法によるラジカル分解反応で実施する場合であって、有機過酸化物を原料ポリオレフィンに含浸させる場合は、原料ポリオレフィンが高規則性(mmmm≧90モル%又はrrrr≧90モル%)では、含浸性が低下するおそれがある。
当該官能基は、好ましくは水酸基、エポキシ基、アルコキシ珪素基、アルキル珪素基、カルボキシル基、アミノ基及びイソシアナート基から選択される1以上の官能基である。
また、第1及び第2の末端不飽和ポリオレフィンは、好ましくは酸無水物構造を有する。酸無水物構造とは、カルボン酸のカルボキシル基2個から1分子の水が失われ、2つのアシル基が1個の酸素原子を共有する構造である。一般に、R1COOCOR2で示される。例えば、無水マレイン酸、無水コハク酸、無水フタル酸等が挙げられる。
上記2官能以上の多官能性化合物としては、例えば水、;TDI,MDI等のイソシアネート化合物;ヘキサメチレンジアミン等のアミン化合物;末端水酸基を含有するポリエチレングリコール、ポリプロピレングリコール類;末端エポキシ基を含有するポリブタジエン変性物;ポリアクリル酸及びアクリル酸共重合体類等を挙げることができる。
具体的には、官能化ポリオレフィンの官能基が水酸基の場合、好適に用いることができる2官能以上の多官能性化合物としてはイソシアナート基、カルボン酸基、エポキシ基を有する化合物である。同様に、エポキシ基の場合は、水酸基、アミノ基、イソシアナート基を有する化合物である。アルコキシ珪素の場合は水等の水酸基を有するものである。カルボキシル基の場合は水酸基、エポキシ基、アミノ基を有する化合物である。アミノ基の場合はエポキシ基、イソシアナート基、カルボキシル基である。イソシアナート基の場合は水酸基、アミノ基等の活性水素を有する化合物である。酸無水物構造の場合は水酸基、アミノ基、エポキシ基、イソシアナート基である。
官能基としてアルコキシ珪素基を含む官能化ポリオレフィンの湿気硬化は、通常は水分又は湿気と接触させる処理により実施できる。この際の硬化触媒として、シラノール縮合触媒を使用してもよい。
シラノール縮合触媒としては、例えば有機金属触媒類、3級アミン類等を挙げることができる。有機金属類としては、例えばジブチル錫ジラウレート、ジブチル錫ジアセテート、ジブチル錫ジオクテート、オクテン酸錫、オクテン酸鉛、ナフテン酸鉛等を挙げることができる。3級アミン類としては、N-トリエチルアミン、N-メチルモルホリンビス(2-ジメチルアミノエチル)エーテル、N,N,N’,N”,N”,N”-ペンタメチルジエチレントリアミン、N,N,N’-トリメチルアミノエチル-エタノールアミン、ビス(2-ジメチルアミノエチル)エーテル、N-メチル-N’-ジメチルアミノエチルピペラジン、イミダゾール環の第2級アミン官能基をシアノエチル基で置換したイミダゾール化合物等を挙げることができる。これら触媒は、1種のみを単独で用いてもよく、2種以上を混合して用いてもよい。
上記触媒の中で特に好ましいのはジブチル錫ジラウレート、ジブチル錫ジアセテート、ジブチル錫ジオクテートである。これらの添加量はグラフト共重合体に対して、通常0.005~2.0質量%であり、好ましくは0.01~0.5質量%である。硬化反応は温度によっても影響を受け、低温ほど遅く、室温では1週間程度の養生期間が必要となる。
(a)式(A)で表されるシロキサン末端(Aユニット)又は式(B)で表される
シロキサン主鎖(Bユニット)、あるいは両者の構造を有する
(b)ポリシロキサン分子主鎖に、式(C)で表されるシロキサンの繰り返し単位(Cユニット)を有する
(c)Aユニットの数が0~2個/分子であり、Bユニットの数が0~10個/分子であり、AユニットとBユニットは同時に0とはならない。Aユニット、Bユニット及びCユニットの合計が1分子当り5~1500個、好ましくは5~200個、より好ましくは10~150個であり、ポリオレフィンとの結合部位以外のポリシロキサン末端はR7又はOR8(R7及びR8は、それぞれ独立に非置換又は置換の炭素数1~12の1価の炭化水素基を示す)である。

本発明の第1及び第2の末端不飽和ポリオレフィンを用いてグラフト重合してなる本発明のグラフト共重合体は、架橋構造等の不規則構造を含まない、側鎖ポリオレフィン連鎖と特定単量体から誘導される主鎖からなる高い構造制御性を有するグラフト共重合体であり、異種材料間の分散性向上剤、相溶化剤として有用である。
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はアルコキシビニルシラン
[IV]スチレン及びその誘導体
A群:
[V]無水マレイン酸及びその置換体
[VI]マレイン酸及びそのエステル
[VII]マレイミド及びその置換体
B群:
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はアルコキシビニルシラン
[IV]スチレン及びその誘導体
[VIII]α-オレフィン
例えば、不飽和基を含有する反応性ポリオレフィンを水添処理した不飽和基を含まないポリオレフィン、ポリエン成分を用いないで製造した不飽和基を含まないポリオレフィンであり、より具体的には、以下の(1)~(3)のポリオレフィンが挙げられ、好ましくは第1又は第2の末端不飽和ポリオレフィンと同一のオレフィン種で構成されるポリオレフィンである。
(1)高密度ポリエチレン(HDPE)、低密度(LDPE)、L-LDPE等のポリエチレン樹脂
(2)アイソタクチックポリプロピレン、シンジオタクチックポリプロピレン、アタクチックポリプロピレン、プロピレンとエチレン、炭素数4~12のα-オレフィンの一種以上からなる共重合体、ブロックポリプロピレン等のポリプロピレン樹脂
(3)炭素数6~28のオレフィン一種以上からなる重合体
第1又は第2の末端不飽和ポリオレフィンの含有量が20質量%以上であると、グラフト重合反応後のグラフト共重合体の生成量が上昇し好ましい。
また、他の観点から、第1又は第2の末端不飽和ポリオレフィン以外のポリオレフィンと同種の熱可塑性樹脂に、本発明のグラフト共重合体又は該共重合体を含む熱可塑性樹脂組成物(熱可塑性樹脂組成物I)を添加して用いる場合、グラフト共重合体が第1又は第2の末端不飽和ポリオレフィン以外のポリオレフィン中に充分な量存在し、良好な分散状態であれば、得られる組成物(熱可塑性樹脂組成物II)全体へ容易にグラフト共重合体が分散するので好ましい。従って、第1又は第2の末端不飽和ポリオレフィン以外のポリオレフィンが80質量%以下であると、当該ポリオレフィンが適正量となり分散性の観点から好ましい。
[I]アクリル酸及びその誘導体
(1)アクリル酸
(2)アクリル酸メチル,アクリル酸エチル,アクリル酸ブチル,アクリル酸ノルマルオクチル、アクリル酸2-エチルヘキシル等のアクリル酸エステル類;ポリエチレングリコールモノアクリレート、ポリエチレングリコールポリプロピレングリコールアクリレート、ポリ(エチレングリコール-n-テトラメチレングリコール)モノアクリレート、プロピレングリコールポリブチレングリコールモノアクリレート、ポリプロピレングリコールモノアクリレート等の分子量30000以下の長鎖ポリアルキレン型グリコール類
(3)アクリル酸ナトリウム、アクリル酸カリウム、アクリル酸マグネシウム、アクリル酸カルシウム等のアクリル酸と典型金属元素からなるアクリル酸金属塩
(4)エステル残基に酸素、窒素、硫黄、珪素原子を含むアクリル酸エステル類、例えば、アクリル酸グリシジル、アクリル酸2-ヒドロキシエチル、アクリル酸2-ヒドロキシプロピル、2-ヒドロキシ-3-フェノキシプロピルアクリレート、4-ヒドロキシブチルアクリレート、アクリロイルオキシエチルイソシアネート、メタクリロイルオキシエチルイソシアネート、3-アクリロキシプロピルトリメトキシシラン、3-アクリロキシプロピルトリエトキシシラン等の官能基を有するアクリル酸エステル類;ポリエチレングリコールモノアクリレート、ポリエチレングリコールポリプロピレングリコールアクリレート、ポリ(エチレングリコール-n-テトラメチレングリコール)モノアクリレート、プロピレングリコールポリブチレングリコールモノアクリレート、ポリプロピレングリコールモノアクリレート等の水酸基を有する分子量30000以下の長鎖ポリアルキレングリコール類
(5)アクリルアミド
(6)置換基に酸素、窒素、硫黄、珪素原子を含むN-置換アクリルアミド、例えば、N-メチルアクリルアミド、N-エチルアクリルアミド、N-イソプロピルアクリルアミド、N-シクロへキシルアクリルアミド、N,N-ジメチルアクリルアミド、N,N-ジエチルアクリルアミド、N,N-ジブチルアクリルアミド、N,N-ジシクロへキシルアクリルアミド、N-(2-ヒドロキシエチル)-アクリルアミド、N-(2-ヒドロキシプロピル)-アクリルアミド、N,N-ジメチルアミノエチルアクリアミド、N-メチロールアクリルアミド等のN-置換アクリルアミド
(7)アクリロニトリル
上記[I]の単量体のα位にメチル基等のアルキル基(好ましくは、炭素数6以下のアルキル基)を有する単量体
酢酸ビニル、プロピオン酸ビニル、イソラク酸ビニル、ピバリン酸ビニル、ウンデカン酸ビニル、パルミチン酸ビニル等のビニルエステル及びその誘導体;トリメトキシビニルシランやトリエトキシビニルシラン等のアルコキシビニルシラン
α-メチルスチレン、p-メチルスチレン、p-エチルスチレン、p-プロピルスチレン、p-イソプロピルスチレン、p-ブチルスチレン、p-tert-ブチルスチレン、p-フェニルスチレン、o-メチルスチレン、o-エチルスチレン、o-プロピルスチレン、o-イソプロピルスチレン、m-メチルスチレン、m-エチルスチレン、m-イソプロピルスチレン、m-ブチルスチレン、メシチルスチレン、2,4-ジメチルスチレン、2,5-ジメチルスチレン、3,5-ジメチルスチレン等のアルキルスチレン類;p-メトキシスチレン、o-メトキシスチレン、m-メトキシスチレン等のアルコキシスチレン類;p-クロロスチレン、m-クロロスチレン、o-ブロモスチレン、p-フルオロスチレン、m-フルオロスチレン、o-フルオロスチレン、o-メチル-p-フルオロスチレン等のハロゲン化スチレン類;トリメチルシリルスチレン、ビニル安息香酸等のスチレン及びその誘導体
[I]アクリル酸及びその誘導体としては、上記の化合物が全て好ましく、特に、アクリル酸金属塩を除く全ての化合物がより好ましい。
[II]メタアクリル酸類及びその誘導体のみでもグラフト重合は可能であるが、[I]アクリル酸及びその誘導体/[II]メタアクリル酸類及びその誘導体を組合せることにより、[II]メタアクリル酸類及びその誘導体のグラフト重合量が上昇し好ましい。特に、アクリル酸、アクリル酸エステル類とメタアクリル酸、メタアクリル酸エステル類の組合せが好ましい。
[I]アクリル酸及びその誘導体/[II]メタアクリル酸類及びその誘導体の好ましいモル比は、[I]/[II](モル比)が0.1~2、好ましくは0.2~1.5、より好ましくは0.3~1.2、さらに好ましくは0.5~1.0の範囲の範囲である。
[I]/[II](モル比)が0.1以上であると、[II]メタアクリル酸類及びその誘導体のグラフト重合量が上昇し、2以下であるとグラフト重合に関与しない[I]アクリル酸及びその誘導体/[II]メタアクリル酸類及びその誘導体からなる共重合体が副生しないため好ましい。
[I]アクリル酸及びその誘導体/[VI]スチレン及びその誘導体の好ましいモル比は、[I]/[VI](モル比)が0.1~2、好ましくは0.2~1.5、より好ましくは0.3~1.2、さらに好ましくは0.5~1.0の範囲の範囲である。
[I]/[VI](モル比)が0.1以上であると[VI]スチレン及びその誘導体のグラフト重合量が上昇し、2以下であるとグラフト重合に関与しない[I]アクリル酸誘導体及びその/[VI]スチレン及びその誘導体からなる共重合体が副生しないため好ましい。
A群:
[V]無水マレイン酸及びその置換体
[VI]マレイン酸及びそのエステル
[VII]マレイミド及びその置換体
B群:
[I]アクリル酸及びその誘導体
[II]メタアクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はアルコキシビニルシラン
[IV]スチレン及びその誘導体
[VIII]α-オレフィン
[V]無水マレイン酸、メチル無水マレイン酸、ジメチル無水マレイン酸、フェニル無水マレイン酸、ジフェニル無水マレイン酸等の無水マレイン酸及びその置換体
[VI]マレイン酸、メチルマレイン酸、マレイン酸ジメチル、マレイン酸ジエチル、マレイン酸ジブチル、マレイン酸モノメチル、等マレイン酸及びそのエステル
[VII]マレイミド、N-アルキル置換マレイミド、N-メチルマレイミド、N-エチ
ルマレイミド、N-フェニルマレイミド等のマレイミド及びその置換体
[VIII]エチレン、プロピレン、1-ブテン、1-へキセン、1-ヘプテン、1-オクテン、1-デセン、1-ウンデセン、1-ドデセン、1-トリデセン、1-テトラデセン、1-ペンタデセン、1-ヘキサデセン、1-ヘプタデセン、1-オクタデセン、1-ノナデセン、1-エイコセン等の炭素数2~28のα-オレフィン
上記A群の単量体は、二重結合の電子密度が小さいため、同種の単量体同士が重合しにくい単量体である。従って、本発明においてはB群の単量体と併用して重合することで、A群の単量体の含有量を向上させる。また、本発明においては、A群の単量体を使用することで、第1及び第2の末端不飽和ポリオレフィンの反応性を高めることができるため、効率よくグラフト共重合体を製造できるという効果も得られる。
また、[VIII]炭素数2~28のα-オレフィンは、グラフト重合温度とその沸点の関係を考慮して選択される。溶融グラフト重合では、温度が高いほど高沸点のα-オレフィンを用いるほうが反応操作上、取り扱いが容易である。また、溶媒を用いるグラフト重合では、ガス状から高沸点のα-オレフィンまで使用可能である。
モル比が0.1以上であると、A群の化合物のグラフト重合量が上昇し、2以下であるとグラフト重合に関与しないA群の化合物/B群の化合物からなる共重合体が副生せず、好ましい。
、[VIII]α-オレフィンとの組合せがより好ましい。
上記有機過酸化物としては、例えばジベンゾイルパーオキシド,ジ-8,5,5-トリメチルヘキサノイルパーオキシド,ジラウロイルパーオキシド,ジデカノイルパーオキシド,ジ(2,4-ジクロロベンゾイル)パーオキシド等のジアシルパーオキシド類、t-ブチルヒドロパーオキシド,キュメンヒドロパーオキシド,ジイソプロピルベンゼンヒドロパーオキシド,2,5-ジメチルヘキサン-2,5-ジヒドロパーオキシド等のヒドロパーオキシド類、ジ-t-ブチルパーオキシド,ジクミルパーオキシド,2,5-ジメチル-2,5-ジ(t-ブチルパーオキシ)ヘキサン,2,5-ジメチル-2,5-ジ(t-ブチルパーオキシ)ヘキシン-3、α,α’ビス(t-ブチルパーオキシ)ジイソプロピルベンゼン等のジアルキルパーオキシド類、1,1-ビス-t-ブチルパーオキシ-3,3,5-トリメチルシクロヘキサン,2,2-ビス(t-ブチルパーオキシ)ブタン等のパーオキシケタール類、t-ブチルパーオキシオクトエート,t-ブチルパーオキシピバレート,t-ブチルパーオキシネオデカノエート,t-ブチルパーオキシベンゾエート等のアルキルパーエステル類、ジ-2-エチルヘキシルパーオキシジカーボネート,ジイソプロピルパーオキシジカーボネート,ジ-sec-ブチルパーオキシジカーボネート,t-ブチルパーオキシイソプロピルカーボネート等のパーオキシカーボネート類等が挙げられ、これらの中で、ジアルキルパーオキシド類が好ましい。アゾ系化合物としては、アゾビスイソブチロニトリル、アゾビスイソバレロニトリル等が挙げられる。
ラジカル開始剤は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。
ラジカル開始剤は、第1又は第2の末端不飽和ポリオレフィンとこれらポリオレフィン以外のポリオレフィンの組合わせ100質量部に対し、0.001~10質量部、好ましくは0.005~5質量部の範囲で用いられる。
その使用量は、好ましくは1~250質量部、より好ましくは5~200質量部、さらに好ましくは10~180質量部の範囲である。
使用量が0.2質量部以上であると、グラフト重合体における共重合する単量体量が上昇し、相溶化等の機能を発現し易く、300質量以下であるとグラフト反応に関与しない重合体が副生せず好ましい。
反応条件としては、ルイス酸共存下で行う場合は、20~230℃の温度で、好ましくは40~140℃の温度で0.1~10時間が挙げられる。
通常用いられる高温条件でグラフト重合を行った場合、第1又は第2の末端不飽和ポリオレフィンの分解による分子量や粘度の低下や、架橋反応等によるゲルの発生が生じ易い。しかしながら、本発明の条件では、比較的低温でグラフト重合を行うため、分子量や粘度の低下がなく、架橋反応等の副反応も進行しない。
(1)周期律表2族~4族元素のハロゲン化物(塩素、臭素、フッ素、ヨウ素)、アルキル化物(炭素数1~20の炭化水素基)、ハロゲン化アルキル物
(2)アルミニウム、硼素、亜鉛、スズ、マグネシウム、カルシウム原子からなるルイス酸
ここで、単量体とは、[I]アクリル酸及びその誘導体、[II]メタクリル酸類及びその誘導体、[III]ビニルエステル及びその誘導体又はアルコキシビニルシラン、[IV]
スチレン及びその誘導体から選ばれる単量体、又はA群;[V]無水マレイン酸及びその
置換体、[VI]マレイン酸及びそのエステル、[VII]マレイミド及びその置換体、B群
;[I]アクリル酸及びその誘導体、[II]メタクリル酸類及びその誘導体、[III]ビ
ニルエステル及びその誘導体又はアルコキシビニルシラン、[IV]スチレン及びその誘導体、[VIII]α-オレフィンから選ばれる単量体を意味している。
ルイス酸/単量体(モル/モル)が0.01以上であると、グラフト率が高く、1以下であると脱灰によるルイス酸残渣の除去が不必要なため、着色がない等好ましい。
連鎖移動剤の存在下にグラフト重合を行うことにより、グラフト共重合体の溶融流動性が向上させることができる。特に、耐熱用途では耐熱性を低下することなくグラフト共重合体の溶融流動性が向上させることができる。また、ゲルやブツの副生を防止できるという利点を有する。
(1)連鎖移動剤の定義
連鎖移動反応を起こす試薬を連鎖移動剤という。連鎖移動剤は成長ポリマー鎖からラジカルを受け取り、ポリマーの伸長を止めるが、ラジカルを受け取った連鎖移動剤はモノマーを攻撃して再び重合を開始させることができる反応試剤である。主に重合体の分子量を調整するために用いられる。前記連鎖移動剤については、併用する重合性モノマーの種類に応じて、適宜、種類及び添加量を選択することができる。連鎖移動剤の効率は連鎖移動定数により評価することができる。各モノマーに対する連鎖移動剤の連鎖移動定数は、例えば、ポリマーハンドブック第3版(J.BRANDRUP及びE.H.IMMERGUT編、JOHN WILEY&SON発行)を参照することができる。また、該連鎖移動定数は大津隆行、木下雅悦共著「高分子合成の実験法」化学同人、昭和47年刊を参考にして、実験によっても求めることができる。
連鎖移動剤としては、メルカプタン類が好適である。具体的には、下記のものが挙げられる。
・イソプロピルメルカプタン、n-ブチルメルカプタン、t-ブチルメルカプタン、n-オクチルメルカプタン、n-ドデシルメルカプタン、t-ドデシルメルカプタン、n-ペンチルメルカプタン、n-ラウリルメルカプタン等のアルキルメルカプタン。
・チオフェノールチオ-β-ナフトール、m-ブロモチオフェノール、p-ブロモチオフェノール、m-トルエンチオール、p-トルエンチオール等のチオフェノール類。
・メルカプト酢酸2-エチルヘキシル、メルカプト酢酸メトキシブチル等のエステル基を有するメルカプタン
・3-メルカプトプロピルトリメトキシシラン、3-メルカプトプロピルトリエトキシシラン等のアルコキシ珪素を有するメルカプタン類等
上記のうち好ましいものとしては、長鎖アルキルのアルキルメルカプタン又は、分子量の大きなメルカプタン類が臭気の点で好ましい。具体的には、n-オクチルメルカプタン、n-ドデシルメルカプタン、t-ドデシルメルカプタン、n-ペンチルメルカプタン、n-ラウリルメルカプタン、メルカプト酢酸2-エチルヘキシルが挙げられ、また、耐熱性を要求する用途においては、3-メルカプトプロピルトリメトキシシラン、3-メルカプトプロピルトリエトキシシランが挙げられる。
連鎖移動剤の使用量は、単量体に対して0.001-50質量%、好ましくは0.01-30質量%、より好ましくは0.1-15質量%の範囲である。0.001質量%では分子量の制御が不十分で、分子量増大によるゲル状物が発生し易くなる。50質量%を超えると分子量の低下が大きくなり、単量体からなる主鎖連鎖が側鎖連鎖に比べて短くなるため、接着強度や相溶化性能が低下する。
上記製造方法によれば、単量体(a)及び(b)の反応率が向上し、未反応単量体の分離工程を省略できる。また、残留単量体に起因するグラフト共重合体の黄変を回避できる。
(1)単量体(a)
(i)単量体(a)の定義
単量体(a)は、単独ラジカル重合性に乏しく、単独ラジカル重合性を実質的に持たない単量体をいう。より詳細には、以下に具体的に示す単量体を指す。
(ii)単量体(a)の具体例
・無水マレイン酸及びその置換体は、前記グラフト共重合体の製造方法IにおけるA群の[V]と同じであり、具体的には、無水マレイン酸、メチル無水マレイン酸、ジメチル無水マレイン酸、フェニル無水マレイン酸、ジフェニル無水マレイン酸等の無水マレイン酸及びその置換体が挙げられる。
・マレイン酸及びそのエステルは、前記グラフト共重合体の製造方法IにおけるA群の[VI]と同じであり、具体的には、マレイン酸、メチルマレイン酸、マレイン酸ジメチル、マレイン酸ジエチル、マレイン酸ジブチル、マレイン酸モノメチル、等マレイン酸及びそのエステルが挙げられる。
・α-オレフィンは、前記グラフト共重合体の製造方法IにおけるB群の[VIII]とほぼ同じであり、具体的には、エチレン、プロピレン、1-ブテン、1-へキセン、1-ヘプテン、1-オクテン、1-デセン、1-ウンデセン、1-ドデセン、1-トリデセン、1-テトラデセン、1-ペンタデセン、1-ヘキサデセン、1-ヘプタデセン、1-オクタデセン、1-ノナデセン、1-エイコセン等の炭素数2~28のα-オレフィン、イソブテン等の分岐オレフイン類が挙げられる。
・ビニルシラン及びその置換体。具体例としては、ビニルトリメトキシシラン、ビニルトリエトキシシラン等が挙げられる。
(i)単量体(b)の定義
単量体(b)は、上記単量体(a)とラジカル共重合可能な単量体又は上記単量体(a)の中から選ばれる2種以上と共重合可能な単量体をいう。
(ii)具体例
(イ)単量体(a)が無水マレイン酸及びその置換体、及びマレイン酸及びそのエステルの場合
・ビニルエステル及びその誘導体
ビニルアルコキシシランを除き、前記グラフト共重合体の製造方法Iにおける[III]と同じであり、具体的には、酢酸ビニル、プロピオン酸ビニル、イソラク酸ビニル、ピバリン酸ビニル、ウンデカン酸ビニル、パルミチン酸ビニル等のビニルエステル及びその誘導体が挙げられる。
・α-オレフィン
上記単量体(a)におけるα-オレフィンと同じものが例示できる。
前記グラフト共重合体の製造方法Iにおける[IV]と同じであり、具体的には、α-メチルスチレン、p-メチルスチレン、p-エチルスチレン、p-プロピルスチレン、p-イソプロピルスチレン、p-ブチルスチレン、p-tert-ブチルスチレン、p-フェニルスチレン、o-メチルスチレン、o-エチルスチレン、o-プロピルスチレン、o-イソプロピルスチレン、m-メチルスチレン、m-エチルスチレン、m-イソプロピルスチレン、m-ブチルスチレン、メシチルスチレン、2,4-ジメチルスチレン、2,5-ジメチルスチレン、3,5-ジメチルスチレン等のアルキルスチレン類;p-メトキシスチレン、o-メトキシスチレン、m-メトキシスチレン等のアルコキシスチレン類;p-クロロスチレン、m-クロロスチレン、o-ブロモスチレン、p-フルオロスチレン、m-フルオロスチレン、o-フルオロスチレン、o-メチル-p-フルオロスチレン等のハロゲン化スチレン類;トリメチルシリルスチレン、ビニル安息香酸等のスチレン及びその誘導体が挙げられる。
前記グラフト共重合体の製造方法Iにおける[I]と同じであり、具体的には下記のものが挙げられる。
(1)アクリル酸
(2)アクリル酸メチル,アクリル酸エチル,アクリル酸ブチル,アクリル酸ノルマルオクチル、アクリル酸2-エチルヘキシル等のアクリル酸エステル類;ポリエチレングリコールモノアクリレート、ポリエチレングリコールポリプロピレングリコールアクリレート、ポリ(エチレングリコール-n-テトラメチレングリコール)モノアクリレート、プロピレングリコールポリブチレングリコールモノアクリレート、ポリプロピレングリコールモノアクリレート等の分子量30000以下の長鎖ポリアルキレン型グリコール類
(3)アクリル酸ナトリウム、アクリル酸カリウム、アクリル酸マグネシウム、アクリル酸カルシウム等のアクリル酸と典型金属元素からなるアクリル酸金属塩
(4)エステル残基に酸素、窒素、硫黄、珪素原子を含むアクリル酸エステル類、例えば、アクリル酸グリシジル、アクリル酸2-ヒドロキシエチル、アクリル酸2-ヒドロキシプロピル、2-ヒドロキシ-3-フェノキシプロピルアクリレート、4-ヒドロキシブチルアクリレート、アクリロイルオキシエチルイソシアネート、メタクリロイルオキシエチルイソシアネート、3-アクリロキシプロピルトリメトキシシラン、3-アクリロキシプロピルトリエトキシシラン等の官能基を有するアクリル酸エステル類;ポリエチレングリコールモノアクリレート、ポリエチレングリコールポリプロピレングリコールアクリレート、ポリ(エチレングリコール-n-テトラメチレングリコール)モノアクリレート、プロピレングリコールポリブチレングリコールモノアクリレート、ポリプロピレングリコールモノアクリレート等の水酸基を有する分子量30000以下の長鎖ポリアルキレングリコール類
(5)アクリルアミド
(6)置換基に酸素、窒素、硫黄、珪素原子を含むN-置換アクリルアミド、例えば、N-メチルアクリルアミド、N-エチルアクリルアミド、N-イソプロピルアクリルアミド、N-シクロへキシルアクリルアミド、N,N-ジメチルアクリルアミド、N,N-ジエチルアクリルアミド、N,N-ジブチルアクリルアミド、N,N-ジシクロへキシルアクリルアミド、N-(2-ヒドロキシエチル)-アクリルアミド、N-(2-ヒドロキシプロピル)-アクリルアミド、N,N-ジメチルアミノエチルアクリアミド、N-メチロールアクリルアミド等のN-置換アクリルアミド
(7)アクリロニトリル
前記グラフト共重合体の製造方法Iにおける[II]と同じであり、具体的には、上記アクリル酸及びその誘導体として例示した単量体のα位にメチル基等のアルキル基(好ましくは、炭素数6以下のアルキル基)を有する単量体が挙げられる。
・上記(イ)で述べた組合せと逆になり、無水マレイン酸及びその置換体及びマレイン酸及びそのエステルが挙げられる。
・アクリル酸及びその誘導体
前記グラフト共重合体の製造方法Iにおける[I]と同じである。
・スチレン及びその誘導体
前記グラフト共重合体の製造方法Iにおける[IV]と同じである。
(i)無水マレイン酸及びその置換体/α-オレフィン
・より好ましくは無水マレイン酸又はマレイン酸/炭素数4~28のαオレフィン
・さらに好ましくは無水マレイン酸/炭素数6~18のαオレフィン
(ii)無水マレイン酸及びその置換体/アクリル酸及びその誘導体
・より好ましくは無水マレイン酸又はマレイン酸/アクリル酸、アクリル酸エステル類
・さらに好ましくは無水マレイン酸/アクリル酸、アクリル酸エステル類
(iii)無水マレイン酸及びその置換体/ビニルエステル及びその誘導体
・より好ましくは無水マレイン酸又はマレイン酸/酢酸ビニル
・さらに好ましくは無水マレイン酸/酢酸ビニル
(iv)ビニルシラン及びその置換体/アクリル酸及びその誘導体
・より好ましくはビニルトリメトキシシラン、ビニルトリエトキシシラン/アクリル酸エステル類、アクリロニトリル
単量体(a)/単量体(b)=0.01~0.5、好ましくは0.02~0.4、より好ましくは0.03~0.35である。
0.01未満ではグラフト共重合体中の単量体(a)の含有量が低下し、接着等の性能が低下する。0.5を超えると単量体(a)が未反応物として残存し、着色等の不具合や製造コストの増加を招く。
(i)基本となる製造方法
単量体(a)の全量を、前記本発明の末端不飽和ポリオレフィンの熱分解物又はラジカル分解物(以下、熱分解物又はラジカル分解物という)と共存させた混合物(#)と、単量体(b)とを接触させてグラフト共重合を実施する。好ましくは、単量体(b)を混合物(#)に添加し、その添加時間が20分以上、最長でラジカル開始剤の少なくとも98%が分解する時間(通常は48時間以内)とすることが好ましい。
(ア)溶媒を用いる場合
1)分解物又はラジカル分解物と単量体(a)を溶媒に溶解した溶液に、単量体(b)とラジカル開始剤の各々又は混合溶液を滴下する方法。
2)熱分解物又はラジカル分解物と単量体(a)及びラジカル開始剤を溶媒に溶解した溶液に、単量体(b)を滴下する方法。
1)熱分解物又はラジカル分解物と単量体(a)、及び(b)を溶解する溶媒の具体例としては、ベンゼン、トルエン、キシレン、混合キシレン等の芳香族炭化水素類;ヘキサン、ヘプタン、オクタン、デカン等の脂肪族炭化水素類;シクロヘキサン、メチルシクロヘキサン等の脂環式炭化水素類;酢酸メチル、酢酸エチル、酢酸プロピル、酢酸ブチル等のカルボン酸エステル類;アセトン、メチルエチルケトン等のケトン類、テトラヒドロフラン、テトラヒドロピラン等の環状エーテル類;及びクロルベンゼン等のハロゲン化炭化水素類が挙げられる。
単独溶媒系としては、トルエン、キシレン等の芳香族炭化水素類;及びテトラヒドロフラン等の環状エーテル類が好ましい。
混合溶媒系は、単量体(a)、単量体(b)及び熱分解物又はラジカル分解物が均一に溶解することが必要であり、溶媒種を適宜選定して用いる。その際、予め上記3成分の各々が溶媒種及び組成が同一の混合溶媒に溶解することを確認して用いる。
具体的には、脂肪族炭化水素類及び脂環式炭化水素類から選ばれた一種以上と、カルボン酸エステル類及びケトン類から選ばれた一種以上の混合溶媒であり、より具体的な組合せとしては、ヘキサン/酢酸エチル、ヘキサン/酢酸ブチル、ヘキサン/メチルエチルケトン、及びヘキサンをヘプタン、デカン、シクロヘキサン又はメチルシクロヘキサンに置き換えた組み合わせが挙げられる。その混合組成は25/75~75/25質量%の範囲であることが好ましく、30/70~70/30質量%の範囲であることがより好ましく、35/65~65/35質量%の範囲であることがさらに好ましい。一方の溶媒種が25質量%未満、又は75質量%を超えると不溶成分が生じ易く好ましくない。また、単独溶媒系に用いられる溶媒同士の組合せ及び単独溶媒系に用いられる溶媒とカルボン酸エステル類又はケトン類との組合せから成る混合溶媒系も使用できる。その混合組成は30/70~70/30質量%の範囲であることが好ましい。
溶媒を用いず、熱分解物又はラジカル分解物の溶融状態で反応を行うこと以外は溶媒を用いる場合と同様に実施する。
溶融した熱分解物又はラジカル分解物に単量体(a)が溶解する場合が好ましい。具体的には、単量体(a)がαオレフィン類、ビニルシラン及びその置換体の場合である。
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はアルコキシビニルシラン
[IV]スチレン及びその誘導体
A群:
[V]無水マレイン酸及びその置換体
[VI]マレイン酸及びそのエステル
[VII]マレイミド及びその置換体
B群:
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はアルコキシビニルシラン
[IV]スチレン及びその誘導体
[VIII]α-オレフィン
(c)分子量分布(Mw/Mn)が1.6~6
グラフト率が1質量%以上であると、側鎖数が適正で、相溶化、接着等の機能を十分に発揮でき、150質量%以下であると、反応性ポリオレフィン成分が適正で、機能を充分発揮できる。
溶媒によりグラフト反応に関与しなかった単量体及び可溶性の重合体成分を溶解除去し、不溶のグラフト共重合体成分の質量(W2)と原料として用いた反応性ポリオレフィンの質量(W1)から以下のようにして算出する。
グラフト率(質量%)=(W2-W1)/W1×100
また、上記方法に限定されず、不溶のグラフト共重合体成分のNMR測定から定法により決定できる。
極限粘度〔η〕が0.01dl/g以上であると、樹脂相溶化等の機能が上昇し、2.5dl/g以下であると樹脂への分散性が向上し好ましい。
尚、グラフト共重合体の重量平均分子量及び分子量分布の評価は、上述のGPC法を用いることができる。
ゲル分率は、例えば以下の方法で評価できる。
酸化防止剤(BHT)0.1質量%を含む溶媒を投入し、沸点以下の温度で4時間攪拌しながら溶解する。溶解後、回収した籠を十分真空乾燥し、秤量により不溶部を求める。
不溶部として定義するゲル成分(ゲル分率)は以下の式で算出する。
[メッシュ内残量(g)/仕込試料量(g)]×100(単位:%)
溶媒としては、パラキシレン、トルエン等が挙げられる。
通常、上記式において、ゲル成分が0~1.5質量%の範囲であれば、ゲル成分を含まないと規定する。
本発明のグラフト共重合体、並びに本発明のグラフト共重合体及びポリオレフィンを含有する熱可塑性樹脂組成物(以下、これらをまとめて単に本発明の組成物という場合がある)は、好ましくはさらにフィラー及び/又は顔料を含む。
無機フィラーとしては、タルク、ホワイトカーボン、シリカ、マイカ、ベントナイト、アルミニウム化合物、マグネシウム化合物、バリウム化合物、珪藻土、ガラスビーズ又はガラス繊維、金属粉又は金属繊維等が挙げられる。
有機フィラーとしては、でんぷん(例えば粉末状でんぷん)、繊維状皮革(例えば綿、麻等のセルロースからなる天然有機繊維)、及びナイロン、ポリエステル、ポリオレフィン等の合成高分子からなる繊維等が挙げられる。
無機顔料としては、酸化物(二酸化チタン、亜鉛華(酸化亜鉛)、酸化鉄、酸化クロム、鉄黒、コバルトブルー等、水酸化物として:アルミナ白、酸化鉄黄、ビリジアン等)、硫化物(硫化亜鉛、リトポン、カドミウムエロー、朱、カドミウムレッド等)、クロム酸塩(黄鉛、モリブデートオレンジ、ジンククロメート、ストロンチウムクロメート等)、珪酸塩(ホワイトカーボン、クレー、タルク、群青等)、硫酸塩(沈降性硫酸バリウム、バライト粉等、炭酸塩として:炭酸カルシウム、鉛白等)が挙げられ、これらのほかフェロシアン化物(紺青)、燐酸塩(マンガンバイオレット)、炭素(カーボンブラック)等も用いることができる。
有機顔料であるアゾ系顔料としては、溶性アゾ(カーミン6B、レーキレットC等)、不溶性アゾ(ジスアゾエロー、レーキレット4R等)、縮合アゾ(クロモフタルエロー3G、クロモフタルスカーレットRN等)、アゾ錯塩(ニッケルアゾエロー等)、ペンズイダゾロンアゾ(パーマネントオレンジHL等)が挙げられる。有機顔料である多環式系顔料としては、イソインドリノン、イソインドリン、キノフタロン、ピラゾロン、フラバトロン、アントラキノン、ジケト-ピロロ-ピロール、ピロール、ピラゾロン、ピランスロン、ペリノン、ペリレン、キナクリドン、インジゴイド、オキサジン、イミダゾロン、キサンテン、カルボニウム、ビオランスロン、フタロシアニン、ニトロソ等が挙げられる。
(a)/(b)が0.005未満ではフィラー又は顔料表面の濡れ性、接着性の改良が不十分で、マスターバッチ又は樹脂分散組成物において、フィラー又は顔料の分散性及び、界面接着性が不足するおそれがある。一方、(a)/(b)が20を超えると、表面処理に関与しない(b)成分が存在し、製造コストが上昇して好ましくない。
本発明の組成物は、溶媒に溶解して溶媒型接着剤として用いることができ、塗布、噴霧して接着基材表面に皮膜を形成し被着体と接着することができる。また、本発明の組成物を、水等の極性溶媒に分散させる又はエマルジョンとすることでも接着剤として用いることができる。そのほか、本発明の組成物をシート状又はフィルム状に成形し、接着基材間に挟み込み、接着剤が流動する温度以上に加熱して接着し、冷却固化により接着することができる。
上記溶媒としては、ヘキサン、ヘプタン、デカン等の脂肪族炭化水素系化合物;ベンゼン、トルエン、キシレン等の芳香族炭化水素化合物;シクロヘキサン、メチルシクロヘキサン等の脂環式炭化水素化合物;クロルベンゼン等のハロゲン化炭化水素;テトラヒドロフラン、テトラヒドロピラン等のエーテル化合物等が挙げられる。
溶媒に極性溶媒を用いることでエマルジョンとすることができる。極性溶媒としては水;メタノール、エタノール、ブタノール等のアルコール類、酢酸メチル、酢酸エチル、酢酸ブチル等のカルボン酸エステル類等が挙げられる。
具体的には、テトラヒドロフランの20~30質量%の溶液を20~50℃の水に少量ずつ添加した後、減圧状態でテトラヒドロフランを除去し、水の量を所望の濃度に調整して製造する方法等を例示できる。他の方法としては、高速攪拌又は高せん断場で極性溶媒に直接分散する方法等、公知の方法を挙げることができる。必要に応じて、アニオン、カチオン、ノニオンタイプの界面活性剤や水溶性高分子化合物を添加剤として用いてもよい。
好ましい粘着性付与樹脂としては、再剥離性と、曲面及び凹凸面への接着性とのバランスの観点から、テルペン系樹脂及びその水素添加型樹脂、スチレン系樹脂、ジシクロペンタジエン(DCPD)系樹脂及びその水素添加型樹脂、脂肪族系(C5系)石油樹脂及びその水素添加型樹脂、芳香族系(C9系)石油樹脂及びその水素添加型樹脂、並びにC5系-C9系の共重合石油樹脂及びその水素添加型樹脂の群から選ばれる1種の樹脂又は2種以上の混合物を用いることが好ましい。
シラノール縮合触媒としては、ジブチル錫ジラウレート、ジブチル錫ジアセテート、ジブチル錫ジオクテート等の有機錫金属化合物;有機塩基、エチルアミン酸等の有機酸、脂肪酸等であり、特に好ましいのはジブチル錫ジラウレート、ジブチル錫ジアセテート、ジブチル錫ジオクテートである。これらの添加量はα-オレフィン重合体変性物に対して、0.005~2.0質量%であり、好ましくは0.01~0.5質量%である。
水分又は湿気を接触させるには、例えば反応性ホットメルト接着剤を空気中に放置してもよいし、水槽に浸漬、スチームを導入してもよい。また温度は常温でもよいが、高温にすると短時間で架橋させるので好ましい。
架橋性能を必要としない場合は官能化ポリオレフィン、本発明のグラフト共重合体を主成分とした溶融物をシール又はポッティングに用い冷却固化により固定化する。
[低結晶性ポリプロピレンの製造]
攪拌機付きの内容積20Lのステンレス製反応器に、n-ヘプタンを24L/h、トリイソブチルアルミニウムを15mmol/h、及びジメチルアニリニウムテトラキスペンタフルオロフェニルボレートと(1,2’-ジメチルシリレン)(2,1’-ジメチルシリレン)-ビス(3-トリメチルシリルメチルインデニル)ジルコニウムジクロライドとトリイソブチルアルミニウムとプロピレンとを質量比1:2:20で接触させて得られた触媒成分を、ジルコニウム換算で6μmol/hで連続供給した。
重合温度を83℃に設定し、反応器の気相部の水素濃度を0.86ル%、反応器内の全圧を0.7MPa・Gに保つように、プロピレン及び水素を連続供給し、重合反応を行った。
得られた低結晶性ポリプロピレンの立体規則性[mmmm]は45モル%であり、重量平均分子量(Mw)は45,600、末端不飽和基数(ここでは、末端ビニリデン基数)は0.95個/分子であった。
[低結晶性ポリプロピレンの製造]
製造例1の重合反応において、重合温度を67℃に設定し、反応器の気相部の水素濃度を0.74モル%、反応器内の全圧が0.75MPa・Gに保つように、プロピレン及び水素を連続供給した以外は製造例1と同様にして低結晶性ポリプロピレンの樹脂ペレットを得た。
得られた低結晶性ポリプロピレンの立体規則性[mmmm]は46モル%であり、重量平均分子量(Mw)は129,000、末端不飽和基数(末端ビニリデン基数)は0.97個/分子であった。
[低結晶性ポリプロピレンの製造]
攪拌機付きの内容積20Lのステンレス製反応器に、n-ヘプタンを12L/h、トリイソブチルアルミニウムを15mmol/h、及びジメチルアニリニウムテトラキスペンタフルオロフェニルボレートと(1,2’-ジメチルシリレン)(2,1’-ジメチルシリレン)-(3-トリメチルシリルメチルインデニル)(インデニル)ジルコニウムジクロライドとトリイソブチルアルミニウムとプロピレンとを質量比1:2:20で接触させて得られた触媒成分を、ジルコニウム換算で24μmol/hで連続供給した。
重合温度を70℃に設定し、反応器の気相部の水素濃度を0.23モル%、反応器内の全圧を0.46MPa・Gに保つように、プロピレン及び水素を連続供給し、重合反応を行った。
得られた低結晶性ポリプロピレンの立体規則性[mmmm]は59モル%であり、重量平均分子量(Mw)は45000、末端不飽和基数(末端ビニリデン基数)は0.96個/分子であった。
[高末端不飽和ポリプロピレンの製造及びそのラジカル分解]
原料ポリオレフィンとして製造例1のポリプロピレンを用い、表1の条件でラジカル分解ポリプロピレンを製造した。
具体的には、攪拌装置付きステンレス製反応器(内容量500mL)に製造例1で製造したポリプロピレン40gを投入した。窒素気流下に30分間攪拌した。
攪拌を停止し、マントルヒーターを用いて樹脂温度を120℃に昇温した。溶融状態になったことを確認して攪拌を再開し、マントルヒーターを樹脂温度が270℃で一定になるように制御した。この溶融樹脂に、キュメンハイドロパーオキサイド(商品名:パークミルH、日油株式会社製)0.4ミリリットル(0.36g)を4分にわたり滴下した。滴下終了後、4分間反応させ、空冷して110℃まで冷却した。110℃で温度を保ったまま、トルエン200ミリリットルを投入し、均一溶液を調製した。
このトルエン溶液をテフロン(登録商標)コート製のバットに回収し、トルエンを除去して、100℃で8時間減圧乾燥することでラジカル分解ポリプロピレンを得た。
尚、実施例1において、副生成物の発生率(副生成物発生量[g]/仕込み原料[g])は、0.01135であった。
表1の条件とした他は実施例1と同様にしてラジカル分解ポリプロピレンを製造し、評価した。結果を表2に示す。実施例5、6及び7では有機過酸化物を2回に分けて添加しているが、当該添加は、実施例1と同様の添加時間及び反応時間で第1回目の有機過酸化物を添加し反応させ、その後再び1回目と同様の添加時間及び反応時間で2回目の有機過酸化物を添加し反応させた
実施例2における副生成物の発生率(副生成物発生量[g]/仕込み原料[g])は、0.00304であり、実施例4における副生成物の発生率は、0.00844であった。
尚、実施例3及び5~7のターシャリブチルクミルパーオキサイドの商品名はパーブチルC(日油株式会社製)であり、実施例8のジイソプロピルベンゼンハイドロパーオキサイドの商品名はパークミルP(日油株式会社製)である。


[高分子量ポリプロピレンからの末端不飽和ポリプロピレンの製造]
原料ポリオレフィンとして市販のポリプロピレンJ300SP(プライムポリマー株式会社製)を用いた他は実施例1と同様にしてラジカル分解ポリプロピレンを製造し、評価した。
J300SP(立体規則性[mmmm]=96モル%、重量平均分子量(Mw)=440000)の190℃における溶融粘度は2000Pasを大きく越えるため、攪拌が困難であり、多大な攪拌動力が必要であった。その結果、有機化酸化物の攪拌混合が十分できず、有機過酸化物が液滴として溶融ポリプロピレン表面に長く滞留した。
得られた分解ポリプロピレンの重量平均分子量は371000であり、末端不飽和基数(末端ビニリデン基数)は0.08個/分子であった。また、立体規則性[mmmm]は96%であった。
[比較的低分子量のポリプロピレンを用いた末端不飽和ポリプロピレンの製造]
原料ポリオレフィンとして市販のポリプロピレンJ3000GV(プライムポリマー株式会社製、立体規則性[mmmm]=97モル%、重量平均分子量(Mw)=230000)を用いた他は実施例1と同様にしてラジカル分解ポリプロピレンを製造し、評価した。
J3000GVの190℃における溶融粘度は2000Pasを越えるが、攪拌は可能であった。
得られた分解ポリプロピレンの重量平均分子量は155000であり、末端不飽和基数(末端ビニリデン基数)は0.12個/分子であった。また、立体規則性[mmmm]は96モル%であった。
一方で、低分子量のポリプロピレンを原料とした場合は、攪拌の問題は回避可能であるが、不飽和度の高いポリプロピレンの製造には限界があることが比較例1及び2からわかる。
[高末端不飽和ポリプロピレンの製造(溶融押出機による連続ラジカル分解ポリプロピレンの製造)]
(1)ポリプロピレン及び有機過酸化物からなる組成物の製造
表3に示す条件で原料ポリプロピレン及び有機過酸化物からなる組成物を製造した。具体的には、製造例2で得たポリプロピレン1000gを万能混合攪拌器(形式:5DM-L-03-rr、ダルトンミキサー)に投入し、窒素気流下126rpmで30分間攪拌した。窒素気流を停止し、窒素雰囲気状態を確保しながら、これにヘキサン20ミリリットル、ジイソプロピルベンゼンハイドロパーオキサイド(商品名:パークミルP、日油株式会社製)4.3グラムの均一溶液を20分間にわたり攪拌しながら滴下した。滴下終了後20分間攪拌した。
攪拌終了後、ペレットを目視により観察したところペレットは乾燥状態であり、均一に有機過酸化物が吸収されていることを確認した。
東洋精機製二軸ラボプラストミル(20mmφ、L/D=27)を用い、バレル温度230℃、ダイス温度200℃、回転数100rpm、吐出量2800グラム/時間で押出し、吐出物を空冷し固まり状の分解ポリプロピレンを製造した。この際のホッパー部及び定量フィーダー部の汚れ及び付着を目視で確認した。結果を表4に示す。
得られた分解ポリプロピレンについて、実施例1と同様にして評価した。結果を表5に示す。
溶融ラジカル分解押出を表4の条件で実施した他は実施例10と同様にして分解ポリプロピレンを製造し、評価した。結果を表5に示す。
表3の条件で製造したポリプロピレン及び有機過酸化物からなる組成物を用い、溶融ラジカル分解押出を表4の条件で実施した他は実施例10と同様にして分解ポリプロピレンを製造し、評価した。結果を表5に示す。
比較例3の分解ポリプロピレンは、実施例10~12の分解ポリプロピレンと比較して臭気が強かった。
表3の条件で製造したポリプロピレン及び有機過酸化物からなる組成物を実施例10と同様にして評価した。結果を表3に示す。



[熱分解による高末端不飽和ポリプロピレンの製造]
原料ポリオレフィンとして製造例1のポリプロピレンを用い、表6の条件で熱分解ポリプロピレンを製造した。
具体的には、攪拌装置付きステンレス製反応器(内容量500mL)に製造例1で製造したポリプロピレン100gを投入した。窒素気流下に30分間攪拌した。
攪拌を停止し、マントルヒーターを用い樹脂温度を150℃に上昇した。溶融状態になったことを確認して攪拌を再開し、マントルヒーターを樹脂温度が320℃と一定になるように制御し、240分間熱分解を実施した。反応終了後、100℃に冷却し、トルエン200ミリリットルを投入して均一溶液を溶液した。
このトルエン溶液をテフロン(登録商標)コート製のバットに回収し、トルエンを除去して、100℃で8時間減圧乾燥すること熱分解ポリプロピレンを得た。得られた熱分解ポリプロピレンを実施例1と同様にして評価した。結果を表7に示す。
熱分解を表6の条件で実施した他は実施例13と同様にして分解ポリプロピレンを製造し、評価した。結果を表7に示す。

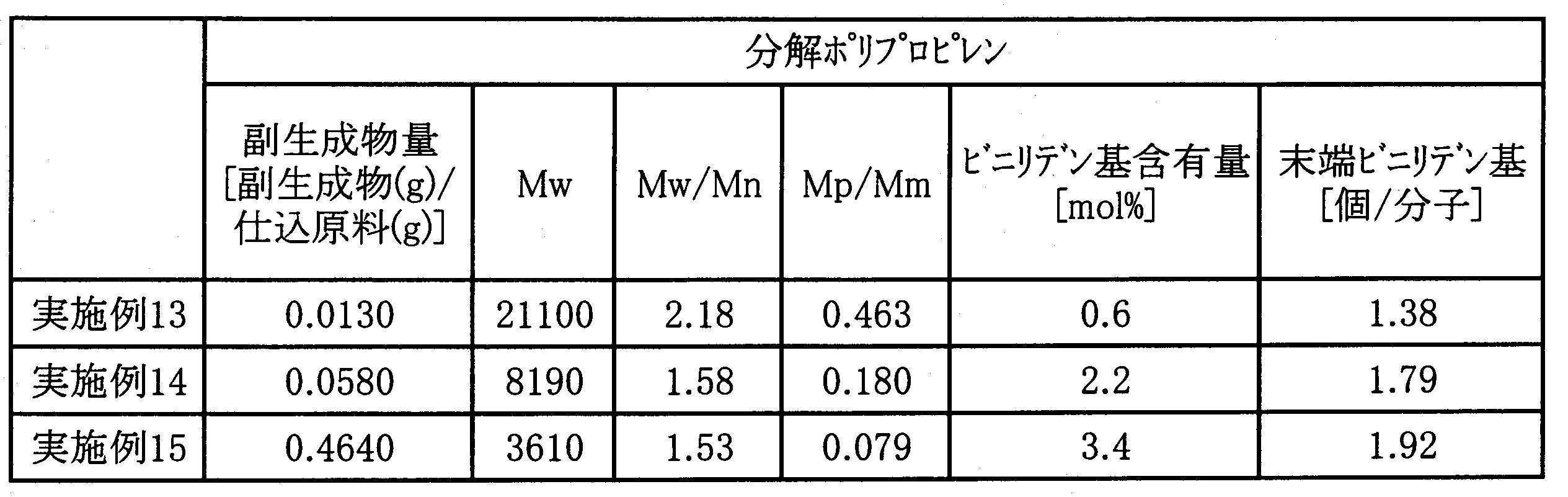
原料ポリオレフィンとして市販のポリプロピレンJ300SP(プライムポリマー株式会社製)を用いた他は実施例13と同様にして熱分解ポリプロピレンの製造を試みたが、320℃ではJ300SPは高粘度で均一攪拌が困難なため、溶融樹脂の温度を所定温度に制御ことができず熱分解ポリプロピレンを製造することはできなかった。
一方で実施例13~15から、予め末端不飽和基を有するものを原料ポリプロピレンとした場合、低分子量ポリプロピレンを用いて末端不飽和基数のより高い不飽和ポリプロピレンを製造できるため、攪拌が容易であり、製造スケールの制限なく末端不飽和基数の高いポリプロピレンを効率よく製造できることが分かる。
[極性モノマーとの共重合体の製造]
(1)ラジカル分解低規則性ポリプロピレンの製造
反応スケールを5倍とした他は実施例8と同様にしてラジカル分解低規則性ポリプロピレンを製造し、温度140℃で溶融状態のまま回収した。冷却固化後、予め切断し約5×5×1cm程度のプレート状にした。これをドライアイスエタノールバスで冷却した後、粉砕機で粉砕して粉砕ペレットとし、風乾後、40℃の減圧乾燥器で8時間乾燥した。
(1)で製造したラジカル分解ポリプロピレンを用い、表8の条件でグラフト共重合体を製造した。
具体的には、攪拌装置付き200ミリリットルの反応容器を十分に窒素置換した後、(1)で製造したポリプロピレンの粉砕ペレット30gを投入し、10分間窒素気流下で攪拌した。これを温度140℃で溶融した。これにアクリル酸2-エチルヘキシル7.7ミリリットル(6.70g)、3-(トリエトキシシリル)プロピル=メタクリレート1.1ミリリットル(1.15g)、及び有機化酸化物であるターシャリブチルクミルパーオキサイド(パーブチルC、日本油脂株式会社製)0.2ミリリットル(0.18g)の混合溶液を30分にわたり攪拌しながら添加した。添加終了後、さらに10分間反応させ、140℃を保ったまま減圧状態で20分間保持した。得られた共重合体を窒素雰囲気下で回収し、窒素雰囲気下に保存した。
溶媒:アセトン:100mL
試料:5g
温度:30℃
抽出時間:8時間
[固液分離方法]
静置分離後、上澄みをろ過し、ろ液を濃縮後乾燥により可溶部重合体を回収。可溶部重合体のNMR測定を実施。
[極性モノマーとの共重合体の製造]
共重合体の製造を表8の条件で実施した他は実施例16と同様にしてグラフト共重合体を製造し、評価した。結果を表9に示す。
尚、1,1-ジ(ターシャリヘキシルプロキシ)シクロヘキサンの商品名はパーヘキサHC(日本油脂株式会社製)である


[極性モノマーとの共重合体の製造]
実施例16(1)で製造したラジカル分解ポリプロピレン(反応スケールを5倍にした実施例8のポリプロピレン)を用い、表10の条件でグラフト共重合体を製造した。
具体的には、攪拌装置付き200ミリリットルの反応容器を十分に窒素置換した後、実施例16(1)で製造したポリプロピレン30g、無水マレイン酸1.0グラム、及び脱水トルエン100ミリリットルを投入し攪拌を開始した。この際、窒素バブリングしながら攪拌溶解した。溶解後、窒素バブリングを停止し窒素雰囲気を保ったまま、温度110℃で一定に制御した。この溶液に脱水トルエン10ミリリットルに溶解したAIBN(アゾビスイソブチロニトリル)0.25グラム及びアクリル酸5ミリリットルを別々の投入口から同時に滴下を開始した。滴下時間はAIBNが150分、アクリル酸は120分であった。AIBN滴下終了後60分間反応させた。反応終了後、トルエンを200ミリリットル追加して希釈した後に回収し、溶媒を除去及び乾燥して共重合体を得た。
得られた共重合体を実施例1と同様にして評価した。結果を表11に示す。
[極性モノマーとの共重合体の製造]
共重合体の製造を表10の条件で実施した他は実施例22と同様にしてグラフト共重合体分を製造し、評価した。結果を表11に示す。


[硬化物の製造]
実施例16の共重合体20グラムを140℃で加熱溶解し、ジブチル錫ジラウリレートを約40ミリグラム添加して均一になるまで攪拌した。この組成物を熱プレスを用いて厚さ1mmのシートに成形し、23℃、湿度50%の雰囲気下に168時間放置し、硬化物を製造した。
得られた硬化物を120℃に制御したホットプレート上に10分間加熱し、その形状変化を観察したところ、形状は保持していた。結果を表12に示す。また、ホットプレート上で得られた硬化物はゴム弾性を示し、伸長した後に開放することで元に戻った。
[硬化物の製造]
実施例16の共重合体を用いる代わりに、実施例17の共重合体を用いた他は実施例27と同様にして硬化物を製造し、評価した。結果を表12に示す。
[硬化物の製造]
実施例17の共重合体5gをヘキサン50ミリリットルに窒素雰囲気下で溶解し、均一に溶解することを確認した。これにジブチル錫ジラウリレートを10ミリグラム添加し、均一になるまで攪拌した後、5×10cmの容器に注ぎ込み静置してヘキサンを蒸発させた。得られた組成物を40℃で8時間減圧乾燥してシートを作成した。このシートを23℃、湿度50%の雰囲気下に168時間放置して硬化物を製造した。
得られた硬化物について、実施例27と同様に評価した。結果を表12に示す。
実施例16(1)のポリプロピレンを用いる代わりに製造例1の低規則性ポリプロピレンを用いた他は実施例17と同様にして共重合体を製造した。
実施例16の共重合体を用いる代わりに、この共重合体20グラムを用いた他は実施例27と同様にしてシートを作成し、23℃、湿度50%の雰囲気下に168時間放置して硬化物を製造した。
得られた硬化物について、実施例27と同様に評価した。結果を表12に示す。
[硬化物の製造]
実施例19の共重合体100部とポリプロピレングリコール(平均分子量250)、(日油株式会社、ユニオールD-250)3部を140℃で均一混合し、1mm厚のシート状にした後、引続き140℃で8時間処理した。
一旦冷却したシート(硬化物)を120℃に制御したホットプレート上で10分間加熱し、その形状変化を観察した。結果を表12に示す。
実施例16(1)のポリプロピレンを用いる代わりに製造例1の低規則性ポリプロピレンを用いた他は実施例19と同様にして共重合体を製造した。
実施例19の共重合体を用いる代わりに、この共重合体20グラム用いた他は実施例30と同様にしてシートを作製し、評価した。結果を表12に示す。
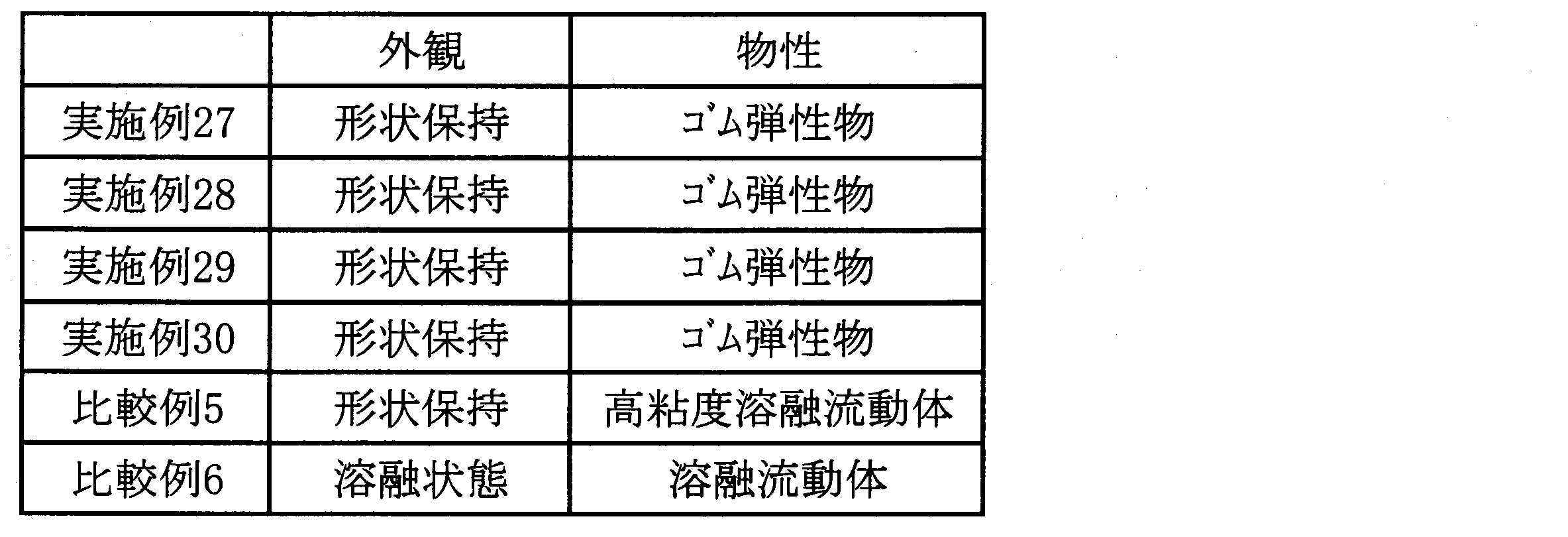
[末端不飽和高立体規則性IPP(アイソタクチックポリプロピレン)の製造]
攪拌機付きの内容積1.6Lのステンレス製反応器に、窒素雰囲気下、脱水トルエンを400mL、メチルアルミノキサン2mmolのトルエン溶液2mLを投入し、攪拌した。これにエチレンビスインデニルジルコニウムジクロリドを1.5μモル投入し、温度を40℃に昇温した後、プロピレンを0.7MPa張込み全圧を一定にして5時間重合した。
重合終了後、大量のメタノールに反応混合物を投入し、ろ過によりポリプロピレンを回収した。風乾後、温度80℃で減圧乾燥を実施し、210gのポリプロピレンを得た。
得られたポリプロピレンの分子量は重量平均分子量(Mw)で35000であり、分子量分布(Mw/Mn)は1.85であった。また、末端ビニリデン基数は0.9個/分子であり、立体規則性(mmmm)は78.9モル%であった。
[末端不飽和アタクチックPPの製造]
エチレンビスインデニルジルコニウムジクロリドの代わりにビスシクロペンタジエニルジルコニウム10μモルを用い、プロピレン圧を0.5MPaとし、温度20℃で90分間重合した他は製造例4と同様にして重合を実施した。
重合終了後、12N塩酸を2mL含む1リットルのメタノールに投入し、攪拌後に水洗し、液状のポリプロピレンを得た。得られた液状ポリプロピレンを80℃で減圧乾燥を実施し、104gのポリプロピレンを得た。
得られたポリプロピレンの分子量は重量平均分子量(Mw)で7340であり、分子量分布(Mw/Mn)は1.77であった。また、末端ビニリデン基数は0.95個/分子であり、立体規則性(mmmm)は9.0モル%であった。
[高末端不飽和ポリプロピレンの製造]
攪拌装置付きステンレス製反応器(内容量500mL)に製造例4で製造したポリプロピレン40gを投入した。窒素気流下に30分間攪拌した。
攪拌を停止し、マントルヒーターを用い樹脂温度を160℃に上昇させた。溶融状態になったことを確認して、攪拌を再開した。マントルヒーターを樹脂温度が270℃で一定になるように制御した。これに、キュメンハイドロパーオキサイド0.4ミリリットルを4分にわたり滴下した。滴下終了後、4分間反応し、その後110℃まで冷却した。これにパラキシレンを投入し加温して均一溶液とした後、これを回収し、溶媒を除去した後、150℃で減圧乾燥を10時間行ってラジカル分解ポリプロピレンを得た。
[高末端不飽和ポリプロピレンの製造]
攪拌装置付きステンレス製反応器(内容量500mL)に製造例5で製造したアタクチックポリプロピレン40gを投入した。窒素気流下に30分間攪拌した。
攪拌を停止し、マントルヒーターを用い樹脂温度を160℃に上昇した。溶融状態になったことを確認して、攪拌を再開した。マントルヒーターを樹脂温度が270℃と一定になるように制御した。これに、キュメンハイドロパーオキサイド0.4ミリリットルを4分にわたり滴下した。滴下終了後、4分間反応し、その後110℃まで冷却した。
反応終了後、40℃でn-ヘプタンを投入し、均一溶液とした後、これを回収し、溶媒を除去した後、100℃で減圧乾燥を10時間行いラジカル分解ポリプロピレンを得た。
[ハイドロシリル化ポリプロピレンの製造]
実施例1で製造したラジカル分解ポリプロピレン5g及びジエトキシメチルシラン0.2gに、触媒として白金-ジビニルテトラメチルジシロキサン錯体(アズマックス株式会社製 SIP6831.0)を130ppm添加し、100℃で5時間反応させた。
得られたポリプロピレンについて、赤外線吸収スペクトルの測定から1640cm-1付近の不飽和結合が消失したことから、ハイドロシリレーションが進行したことが確認でき、ハイドロシリル化ポリプロピレンが製造されたことを確認した。
[ハイドロシリル化ポリプロピレンの製造]
実施例1のラジカル分解ポリプロピレンの代わりに実施例32のポリプロピレンを用い、
ジエトキシメチルシラン0.5gを用いた他は実施例33と同様にして反応を実施した。
得られたポリプロピレンについて、赤外線吸収スペクトルの測定から1640cm-1付近の不飽和結合が消失したことから、ハイドロシリレーションが進行したことが確認でき、ハイドロシリル化ポリプロピレンが製造されたことを確認した
[硬化物の製造]
実施例33のハイドロシリル化ポリプロピレン4gをヘキサン50ミリリットルに窒素雰囲気下で溶解し、均一に溶解することを確認した。これにジブチル錫ジラウリレートを10ミリグラム添加し、均一になるまで攪拌した後、5×10cmの容器に注ぎ込み静置してヘキサンを蒸発させた。得られた組成物を40℃で8時間減圧乾燥してシートを作成した。このシートを23℃、湿度50%の雰囲気下に168時間放置して硬化物を製造した。
得られた硬化物を120℃に制御したホットプレート上で10分間加熱し、その形状変化を観察した。その結果、加熱状態で形状保持し、ゴム弾性を示す硬化物であることを確認した。
[硬化物の製造]
室温で液状である実施例32で得られたラジカル分解ポリプロピレン10g(6.07mmol)にポリメチルHシロキサン(HMS-991、アズマックス株式会社)1.2gを混合し、さらに白金-ジビニルテトラメチルジシロキサン錯体(アズマックス(株)製
SIP6831.0)を10mg添加し混合した後、5×10cmの容器に注ぎ込み、室温状態で10日保存して硬化物を得た。
120℃に制御したホットプレート上で硬化物を10分間加熱し、その形状変化を観察した。その結果、加熱状態で形状を保持し、ゴム弾性を示す硬化物であることを確認した。
(1)ラジカル分解アタクチックポリプロピレン/アクリル酸2-エチルヘキシル/3-(トリエトキシシリル)プロピル=メタクリレート共重合体の製造
攪拌装置付き50ミリリットルの反応容器を十分に窒素置換した後、実施例32で製造したアタクチックポリプロピレン10gを投入し、10分間窒素気流下で攪拌した。これを温度140℃で溶融した。これにアクリル酸2-エチルヘキシル2.5ミリリットル(2.2g)、3-(トリエトキシシリル)プロピル=メタクリレート0.3ミリリットル(0.31g)、有機過酸化物(パーブチルC、日本油脂株式会社製)0.1ミリリットル(0.09g)を混合溶解し、この混合液を10分にわたり攪拌しながら添加した。添加終了後、さらに10分間反応させた。140℃を保ったまま、減圧状態で20分間保持し、得られた共重合体を窒素雰囲気下に保存した。得られたグラフト共重合体の分子量分布は2.3であった。
(1)で得られた共重合体を100℃に加熱し、ジブチル錫ジラウリレートを10ミリグラム添加し、均一になるまで攪拌した後、5×10cmの容器に注ぎ込み静置した。これを23℃、湿度50%の雰囲気下に168時間放置して硬化物を製造した。
得られた硬化物を120℃に制御したホットプレート上で10分間加熱し、その形状変化を観察した。その結果、加熱状態で形状保持し、ゴム弾性を示す硬化物であることを確認した。
[低結晶性ポリプロピレンの製造]
内容量1.4リットルのステンレス製耐圧オートクレーブに窒素雰囲気下、n-ヘプタン400ミリリットル、トリイソブチルアルミニウム1ミリモル、メチルアルミノキサン(アルベマール社製、トルエン溶液)2ミリモルを添加し5分間攪拌した。
攪拌を停止し、(1,2’-ジメチルシリレン)(2,1’-ジメチルシリレン)-ビス(3-トリメチルシリルメチルインデニル)ジルコニウムジクロライド1マイクロモルを含むn-ヘプタン1ミリリットルを添加した。水素をゲージ圧で0.1MPa導入し、温度60℃に制御した。さらにプロピレンをゲージ圧で0.6MPaの圧力になるように導入し続け、40分間重合した。
得られたポリプロピレンは156グラムであり、重量平均分子量は93,000、数平均分子量は48,300、末端ビニリデン基数は0.051個/分子であった。立体規則性[mmmm]は46モル%であった。
[高末端不飽和ポリプロピレンの製造]
攪拌装置付きステンレス製反応器(内容量500mL)に製造例6で製造したポリプロピレン40gを投入した。窒素気流下に30分間攪拌した。
攪拌を停止し、マントルヒーターを用い樹脂温度を120℃に上昇させた。溶融状態になったことを確認して、攪拌を再開した。マントルヒーターを樹脂温度が270℃で一定になるように制御した。これに、キュメンハイドロパーオキサイド(商品名:パークミルH、日油株式会社製)0.8ミリリットルを20分にわたり滴下した。滴下終了後、4分間反応し、空冷して110℃まで冷却した。この温度を保ったまま、トルエン200ミリリットルを投入し、均一溶液を調製した。
このトルエン溶液を回収しテフロン(登録商標)コート製のバットに回収し、トルエンを除去し、100℃で8時間減圧乾燥することでラジカル分解ポリプロピレンを得た。
[高末端不飽和ポリプロピレンの製造(溶融押出機による連続ラジカル分解ポリプロピレンの製造)]
(1)ポリプロピレン及び有機過酸化物からなる組成物の製造
製造例2で得たポリプロピレン2,700gを万能混合攪拌器(形式:5DM-L-03-rr、ダルトンミキサー)に投入し、窒素気流下126rpmで30分間攪拌した。窒素気流を停止し、窒素雰囲気状態を確保しながら、これにヘキサン20ミリリットル、ジイソプロピルベンゼンハイドロパーオキサイド(商品名:パークミルP、日油株式会社製)24グラムの均一溶液を20分間にわたり攪拌しながら滴下した。滴下終了後20分間攪拌した。
攪拌終了後、ペレットを目視により観察したところペレットは乾燥状態であり、均一に有機過酸化物が吸収されていることを確認した。
東洋精機製二軸ラボプラストミル(20mmφ、L/D=27)を用い、バレル温度280℃、ダイス温度200℃、回転数120rpm、吐出量2,100グラム/時間で押出し、吐出物を空冷し固まり状の分解ポリプロピレンを製造した。ホッパー部及び定量フィーダー部の汚れ及び付着を目視で確認した結果、ホッパー部及びホッパー下部、及びスクリューに付着等の汚れは認められなかった。分解ポリプロピレンの重量平均分子量は48,700、Mp/Mmは0.378、末端不飽和基数(末端ビニリデン基数)は1.62個/分子であった。
攪拌装置付き3リットルの反応容器を十分に窒素置換した後、上記(2)で製造した分解ポリプロピレン500gを投入し、窒素気流下、温度160℃とし融解を確認した。その後30分間攪拌した。内温を160℃に制御しながら、アクリル酸2-エチルヘキシル63.1g、3-(トリエトキシシリル)プロピル=メタクリレート32.2g、及び有機化酸化物であるターシャリブチルクミルパーオキサイド(パーブチルC、日本油脂株式会社製)1.5ミリリットルの混合溶液を35分にわたり攪拌しながら添加した。添加終了後、さらに15分間反応させた。得られたグラフト共重合体を窒素雰囲気下で回収し、窒素雰囲気下に保存した。
(i)B粘度の測定
JISK-6862に準拠し、190℃で溶融した上記共重合体の粘度をブルックフィールド型粘度計で測定した。上記粘度計には、東機産業(株)製TVB-10型粘度計及びM2ローター(No21)を用い、H-2型少量サンプルアダプタを使用して測定した。
その結果、B粘度は5,840mPaSであった。
上記(3)で製造したグラフト共重合体35gを180℃で加熱溶融し、硬化触媒としてジラウリン酸ジブチル錫(IV)0.07gを添加し、十分に攪拌した。この溶融樹脂0.5gを、幅25mm、長さ200mmの綿帆布(JIS10号)の端部から80mmにステンレス製のヘラで均一に塗布して、幅25mm、長さ100mm、厚さ2mmのポリプロピレン板に重ね合わせた。次いで接着部に2kgの錘を載せて120℃のオーブンで10分間熱融着し、そのまま取り出して室温まで冷却した。貼り合わせた試験片は、23℃、50%RH雰囲気中で7日間養生して湿気硬化させた。
耐熱クリープ温度の測定はJIS K6833に準拠して実施した。具体的には、接着部分の長さが25mmとなるように試験片下部を切り捨て、恒温槽内に吊り下げた試験片の下端に500gの錘を取り付けた。恒温槽の温度を、38℃で15分間保持してから、5分間に2℃の割合で温度を上昇させて、錘が落下したときの耐熱クリープ温度とした。その結果、耐熱クリープ温度は81℃であった。
実施例39(3)において3-(トリエトキシシリル)プロピル=メタクリレートの仕込み量を21.4gとすること以外は実施例39と同様にして共重合体を製造した。その結果、B粘度は6,900mPaSであり、耐熱クリープ温度は71℃であった。
実施例40に於いて、アクリル酸2-エチルヘキシル、3-(トリエトキシシリル)プロピル=メタクリレート、ターシャリブチルクミルパーオキサイドの均一溶液の中に、連鎖移動剤としてメルカプト酢酸2-エチルヘキシル6.0gを添加しこの混合溶液を滴下すること以外は同様にして共重合体を製造した。その結果、B粘度は4,600mPaSであり、耐熱クリープ温度は69℃であった。
実施例40と比較すると連鎖移動剤の使用により、耐熱クリープ特性を損なうことなく、溶融粘度を大幅に低下することができた。
[極性モノマーとの共重合体の製造]
攪拌装置及び投入口を有するガラス製3リットルのセパラブルフラスコを十分乾燥した後、窒素雰囲気下とした。これに実施例39(2)で製造した分解ポリプロピレン500.0g、無水マレイン酸5.65gを投入した。これに脱水処理した酢酸エチル492gを投入し室温で攪拌を開始した。これにヘプタン484gを投入した。オイルバスを用いて沸騰状態(内温80.6℃)になる様に加熱した。これにアクリル酸54.2g、アゾビスイソブチロニトリル10.0g、脱水酢酸エチル88.5gから成る均一溶液を115分間かけ一定の速度で投入した。投入終了後さらに180分間反応を続行した。反応終了後、反応混合物を一部サンプリングし、多量の酢酸エチルを添加して液部と固体に分け、液部に存在する未反応単量体をガスクロマトグラム法で定量した。また、グラフト共重合体は反応混合液を加温減圧により液体を回収し、さらに、130℃で8時間減圧した。
その結果、未反応単量体は生成したグラフト共重合体に対して0.18質量%の割合で残存した。
また、加熱減圧乾燥したグラフト共重合体は白色であった。
また、グラフト共重合体の赤外線吸収スペクトルを測定したところ、888cm-1に存在するビニリデン基に基づく吸収が認められないことから、グラフト反応が効率的に進行したことを確認した。
また、グラフト共重合体のB粘度は12,500mPaSであった。
実施例42に於いて、アクリル酸及び無水マレイン酸を予め反応槽に仕込み、アゾビスイソブチロニトリルの脱水酢酸エチル溶液を投入すること以外は実施例42と同様に実施した。
その結果、未反応単量体は生成したグラフト共重合体に対して1.02質量%の割合で残存した。
また、加熱減圧乾燥したグラフト共重合体は黄色に変色した。
赤外線吸収スペクトルを測定したところ、ビニリデン基に基づく吸収が存在したことからグラフト反応の効率が低いことを確認した。
実施例42に於いて、アクリル酸、アゾビスイソブチロニトリル、脱水酢酸エチルの均一溶液にn-ドデシルメルカプタン6gを加えたものを滴下すること以外は同様にしてグラフト共重合反応を実施した。反応終了後、一部を抜出し、大量のテトラヒドロフランに投入し、溶解させた後、400メッシュのステンレス製篩でろ過した。その結果、メッシュ上に残存するものは認められなかった。
また、回収したグラフト共重合体のB粘度は4500mPaSであった。
(1)低立体規則性ポリブテンの製造
内容量1.4リットルの攪拌装置ステンレス製耐圧オートクレーブを80℃に加熱減圧して十分に乾燥し、窒素雰囲気とした。これに脱水ヘプタン400ミリリットル、トリイソブチルアルミニウム0.5ミリモルを含むヘプタン溶液1ミリリットルを投入し、室温下で5分間攪拌した。その後、ジメチルアニリニウムテトラキスペンタフルオロフェニルボレート3.2マイクロモルを含むヘプタンスラリー2.5ミリリットル、(1,2’-ジメチルシリレン)(2,1’-ジメチルシリレン)-ビス(3-トリメチルシリルメチルインデニル)ジルコニウムジクロライド1.6マイクロモルを含むヘプタン溶液3.0ミリリットルを投入し水素25ミリリットルを注入し、攪拌を開始した。その後、耐圧容器に1-ブテン200ミリリットルをバランスラインを介して投入し、温度90℃に制御して90分間重合を実施した。重合終了後、冷却脱圧し、反応混合物を回収し、乾燥によりポリ1-ブテン89グラムを製造した。
このものの重量平均分子量(Mw)は9,800、分子量分布(Mw/Mn)は2.2であり一分子当たりの末端ビニリデン基数は0.7個/分子であった。立体規則性(mmmm)は61.6モル%であった。
攪拌装置付きステンレス製反応器(内容量500mL)に上記(1)製造したポリブテン80gを投入した。窒素気流下に30分間攪拌した。
攪拌を停止し、マントルヒーターを用いて樹脂温度を120℃に昇温した。溶融状態になったことを確認して攪拌を再開し、マントルヒーターを樹脂温度が270℃で一定になるように制御した。この溶融樹脂に、キュメンハイドロパーオキサイド(商品名:パークミルH、日油株式会社製)1.6ミリリットル(1.42g)を60分にわたり滴下した。滴下終了後、4分間反応させ、空冷して110℃まで冷却した。110℃で温度を保ったまま、トルエン200ミリリットルを投入し、均一溶液を調製した。
このトルエン溶液をテフロン(登録商標)コート製のバットに回収し、トルエンを除去して、100℃で8時間減圧乾燥することでラジカル分解低規則性ポリブテンを得た。
得られたラジカル分解低規則性ポリブテンの重量平均分子量Mwは2,200、数平均分子量Mnは1,300、分子量分布Mw/Mnは1.7、Mp/Mmは0.224、末端ビニリデン基の1分子当りの数(末端ビニリデン基数)は1.71個/分子であった。
この明細書に記載の文献の内容を全てここに援用する。
Claims (26)
- プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;又はブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体からなる末端不飽和ポリオレフィンであって、
下記(1)~(4)を満たす末端不飽和ポリオレフィン。
(1)プロピレン連鎖部又はブテン-1連鎖部のメソペンタッド分率[mmmm]が20~80モル%である
(2)末端ビニリデン基の1分子当り数が1.3~2.5である
(3)重量平均分子量Mwが500~100,000である
(4)分子量分布Mw/Mnが1.1~2.6である - 前記プロピレン単独重合体からなる請求項1に記載の末端不飽和ポリオレフィン。
- プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンであって、下記(5)及び(6)を満たす原料ポリオレフィンを不活性気体存在下で分解する請求項1又は2に記載の末端不飽和ポリオレフィンの製造方法。
(5)プロピレン連鎖部又はブテン-1連鎖部の立体規則性[mmmm]が20~80モル%である
(6)重量平均分子量Mwが4000~1,000,000である - 前記分解が、前記原料ポリオレフィンを300~400℃の温度で30分~10時間加熱処理する熱分解反応である請求項3に記載の末端不飽和ポリオレフィンの製造方法。
- 前記分解が、温度160~300℃で、有機過酸化物を前記原料ポリオレフィンに対して0.05~2質量%添加するラジカル分解反応である請求項3に記載の末端不飽和ポリオレフィンの製造方法。
- 前記有機過酸化物の1分間半減期温度が、140~270℃である請求項5に記載の末端不飽和ポリオレフィンの製造方法。
- プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンを分解して得られる末端不飽和ポリオレフィンであって、
下記(7)~(10)を満たす末端不飽和ポリオレフィン。
(7)末端ビニリデン基の1分子当りの数(fv)が1.3~2.5である
(8)fv≧-2(Mp/Mm)+2(式中、Mpは末端不飽和ポリオレフィンの数
平均分子量であり、Mmは原料ポリオレフィンの数平均分子量である。但し、Mp/Mmは0.05~0.8である)
(9)重量平均分子量Mwが500~100,000である
(10)分子量分布Mw/Mnが1.1~2.6である - 前記プロピレン単独重合体又は前記プロピレン系共重合体を分解して得られる末端不飽和ポリオレフィンであって、
前記プロピレン単独重合体又は前記プロピレン系共重合体のプロピレン連鎖部が、アタクチック構造、シンジオタクチック構造又はアイソタクチック構造を有する請求項7に記載の末端不飽和ポリオレフィン。 - 前記原料ポリオレフィンがプロピレン単独重合体である請求項7又は8に記載の末端不飽和ポリオレフィン。
- プロピレン単独重合体;プロピレン、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1と、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択される原料ポリオレフィンであって、重量平均分子量が4000~1,000,000である原料ポリオレフィンを不活性気体存在下で分解する請求項7~9のいずれかに記載の末端不飽和ポリオレフィンの製造方法。
- 請求項1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィンの末端ビニリデン基の5モル%以上が官能基を有する官能化ポリオレフィン。
- 前記官能基が、水酸基、エポキシ基、イソシアナート基、アルコキシ珪素基、アルキル珪素基、カルボキシル基、アミノ基、及び酸無水物構造からなる群から選択される1種以上である請求項11に記載の官能化ポリオレフィン。
- 前記官能基として、少なくともアルコキシ珪素基を含む請求項11又は12に記載の官能化ポリオレフィンを湿気硬化してなる架橋体。
- 請求項1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィンとSiH基を1分子当たり2個以上有するオルガノハイドロジェンポリシロキサンとを反応させて得られる反応物。
- 請求項1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィン20~100質量%、及び前記末端不飽和ポリオレフィン以外のポリオレフィン0~80質量%との組合わせ100質量部と、下記[I]~[IV]から選ばれる単量体1種以上0.2~300質量部、又は下記A群から選ばれる1種以上とB群から選ばれる1種以上の単量体混合物0.2~300質量部を、ラジカル開始剤0.001~10質量部の存在下、40~230℃でグラフト重合させるグラフト共重合体、又は前記グラフト共重合体を含む熱可塑性樹脂組成物の製造方法。
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
A群:
[V]無水マレイン酸及びその置換体
[VI]マレイン酸及びそのエステル
[VII]マレイミド及びその置換体
B群:
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
[VIII]α-オレフィン - 連鎖移動剤の存在下にグラフト重合を行う、請求項15に記載のグラフト共重合体、又は前記グラフト共重合体を含む熱可塑性樹脂組成物の製造方法。
- 請求項1、2、7、8及び9のいずれかに記載の末端不飽和ポリオレフィンと、単独ラジカル重合性を実質的に持たない単量体(a)とを共存させた混合物に、前記単量体(a)と共重合可能な単量体(b)を接触させることからなるグラフト共重合体、又は前記グラフト共重合体を含む熱可塑性樹脂組成物の製造方法。
- プロピレン単独重合体;プロピレンと、エチレン及び1種以上の炭素数4~10のαオレフィンからなるプロピレン系共重合体であり、エチレン及び前記炭素数4~10のαオレフィンの含有量が10モル%以下であるプロピレン系共重合体;ブテン-1単独重合体;及びブテン-1と、エチレン、プロピレン及び1種以上の炭素数5~10のαオレフィンからなるブテン-1系共重合体であり、エチレン、プロピレン及び前記炭素数5~10のαオレフィンの含有量が10モル%以下であるブテン-1系共重合体から選択されるポリオレフィンの両末端に、下記[I]、[II]、[III]及び[IV]から選ばれる1種以上から誘導されるポリオレフィン連鎖、又は下記A群から選ばれる1種以上及びB群から選ばれる1種以上から誘導されるポリオレフィン連鎖を有するグラフト共重合体であって、
下記(c)を満たすグラフト共重合体。
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
A群:
[V]無水マレイン酸及びその置換体
[VI]マレイン酸及びそのエステル
[VII]マレイミド及びその置換体
B群:
[I]アクリル酸及びその誘導体
[II]メタクリル酸類及びその誘導体
[III]ビニルエステル及びその誘導体又はビニルアルコキシシラン
[IV]スチレン及びその誘導体
[VIII]α-オレフィン
(c)分子量分布(Mw/Mn)が1.6~6 - 前記ポリオレフィン連鎖のプロピレン連鎖又はブテン-1連鎖のメソペンタッド分率[mmmm]が20~80モル%である請求項18記載のグラフト共重合体。
- 前記ポリオレフィンが、アタクチック構造又はシンジオタクチック構造であるプロピレン単独重合体である請求項18記載のグラフト共重合体。
- 請求項18に記載のグラフト共重合体及びポリオレフィンを含有する熱可塑性樹脂組成物。
- 請求項18に記載のグラフト共重合体、又は請求項21に記載の熱可塑性樹脂組成物がさらに熱可塑性樹脂を含む組成物。
- 請求項18に記載のグラフト共重合体、又は請求項21に記載の熱可塑性樹脂組成物がさらにフィラー及び/又は顔料を含む組成物。
- 請求項18に記載のグラフト共重合体、又は請求項21に記載の熱可塑性樹脂組成物を接着剤、樹脂相溶化剤、分散体、コーティング材として用いる用途。
- 請求項11又は12に記載の官能化ポリオレフィンを接着剤、樹脂相溶化剤、分散体、コーティング材として用いる用途。
- 請求項11又は12に記載の官能化ポリオレフィンを反応性ホットメルト接着剤、シール材、ポッティング材として用いる用途。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012517118A JP5890774B2 (ja) | 2010-05-26 | 2011-05-17 | 末端不飽和ポリオレフィン、及びその製造方法 |
| CN2011800260126A CN102906125A (zh) | 2010-05-26 | 2011-05-17 | 末端不饱和聚烯烃、及其制造方法 |
| EP11786290.4A EP2578607A4 (en) | 2010-05-26 | 2011-05-17 | TERMINAL UNSATURATED POLYOLEFIN AND MANUFACTURING METHOD THEREFOR |
| US13/700,008 US8871855B2 (en) | 2010-05-26 | 2011-05-17 | Terminally unsaturated polyolefin and method for producing the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010120768 | 2010-05-26 | ||
| JP2010-120768 | 2010-05-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011148586A1 true WO2011148586A1 (ja) | 2011-12-01 |
Family
ID=45003587
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/002728 Ceased WO2011148586A1 (ja) | 2010-05-26 | 2011-05-17 | 末端不飽和ポリオレフィン、及びその製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8871855B2 (ja) |
| EP (1) | EP2578607A4 (ja) |
| JP (1) | JP5890774B2 (ja) |
| CN (1) | CN102906125A (ja) |
| WO (1) | WO2011148586A1 (ja) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012124345A1 (ja) * | 2011-03-16 | 2012-09-20 | 出光興産株式会社 | 反応性ポリオレフィン、その製造方法、及びそれを含む組成物 |
| WO2013118841A1 (ja) * | 2012-02-08 | 2013-08-15 | 出光興産株式会社 | 末端不飽和α-オレフィン重合体及びその製造方法 |
| WO2014046134A1 (ja) * | 2012-09-18 | 2014-03-27 | 出光興産株式会社 | 官能化α-オレフィン重合体、それを用いた硬化性組成物及び硬化物 |
| JP2014098072A (ja) * | 2012-11-13 | 2014-05-29 | Idemitsu Kosan Co Ltd | 官能化α−オレフィン重合体、それを用いた硬化性組成物及び硬化物 |
| JP2014223752A (ja) * | 2013-05-16 | 2014-12-04 | 三井化学株式会社 | 包装材用の積層フィルムおよび包装材 |
| WO2018131670A1 (ja) * | 2017-01-13 | 2018-07-19 | 株式会社日本触媒 | 共重合体および樹脂組成物 |
| WO2020027222A1 (ja) * | 2018-08-02 | 2020-02-06 | 出光興産株式会社 | ポリプロピレン系接着剤及びその製造方法 |
| KR20210121043A (ko) * | 2018-12-28 | 2021-10-07 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| KR20210121029A (ko) * | 2018-12-28 | 2021-10-07 | 다우 글로벌 테크놀로지스 엘엘씨 | 텔레켈릭 폴리올레핀을 포함하는 경화성 조성물 |
| KR20210121027A (ko) * | 2018-12-28 | 2021-10-07 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| JP2021181558A (ja) * | 2020-05-19 | 2021-11-25 | 三洋化成工業株式会社 | 熱硬化性樹脂組成物 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5861147A (en) | 1997-06-09 | 1999-01-19 | The Procter & Gamble Company | Methods for controlling environmental odors on the body using compositions comprising uncomplexed cyclodextrins and perfume |
| WO2014069606A1 (ja) * | 2012-11-02 | 2014-05-08 | 出光興産株式会社 | ポリオレフィン、これを含む粘接着剤組成物及びこれを用いた粘着テープ |
| CN107868283A (zh) * | 2017-11-30 | 2018-04-03 | 安徽坤大化学锚固有限公司 | 一种高抗低温型树脂锚固剂及其制备工艺 |
| CN113454130B (zh) | 2018-12-28 | 2024-12-13 | 陶氏环球技术有限责任公司 | 遥爪聚烯烃和用于制备遥爪聚烯烃的方法 |
| US12098160B2 (en) | 2018-12-28 | 2024-09-24 | Dow Global Technologies Llc | Organometallic chain transfer agents |
| WO2020212807A1 (en) * | 2019-04-19 | 2020-10-22 | 3M Innovative Properties Company | Polyolefin composition comprising polydioorganosiloxane block copolymer having c16-c70 alkyl block suitable for release coating and pressure sensitive adhesive article |
| CN111808552A (zh) * | 2020-07-08 | 2020-10-23 | 陈坚 | 一种多用途反应型热熔胶及其制造方法 |
| CN117447789B (zh) * | 2021-08-04 | 2026-02-06 | 中国石油化工股份有限公司 | 一种含酸酐改性聚丙烯复合材料及其制备方法与应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0641235A (ja) * | 1992-04-01 | 1994-02-15 | Mitsui Toatsu Chem Inc | シンジオタクチックポリプロピレンワックス、その製造方法及び該ワックスを使用する熱ロール定着用トナー組成物 |
| JP2003535193A (ja) * | 2000-05-30 | 2003-11-25 | バーゼル、ポリオレフィン、ゲゼルシャフト、ミット、ベシュレンクテル、ハフツング | ファイバーグレードのプロピレンポリマーの製造方法 |
| WO2008047860A1 (en) * | 2006-10-20 | 2008-04-24 | Idemitsu Kosan Co., Ltd. | Highly pure, terminal-unsaturated olefin polymer and process for production thereof |
| WO2008066168A1 (en) * | 2006-12-01 | 2008-06-05 | Idemitsu Kosan Co., Ltd. | Graft copolymer, thermoplastic resin composition comprising the graft copolymer, and those production method |
| JP2008133320A (ja) * | 2006-11-27 | 2008-06-12 | Idemitsu Kosan Co Ltd | 末端変性ポリα−オレフィン、その製造方法及びそれを含む組成物 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04226506A (ja) | 1990-07-11 | 1992-08-17 | Mitsubishi Petrochem Co Ltd | 水酸基含有重合体 |
| DE4205932A1 (de) | 1992-02-27 | 1993-09-02 | Basf Ag | Verfahren zur herstellung von propenoligomeren |
| EP0563834B1 (en) | 1992-04-01 | 1995-10-11 | MITSUI TOATSU CHEMICALS, Inc. | Syndiotactic polypropylene wax, production process thereof, and heating roll fixing-type toner composition making use of the wax |
| EP0667359B1 (en) * | 1992-10-28 | 2004-03-31 | Idemitsu Kosan Company Limited | Olefin copolymers and process for producing the same |
| US6096721A (en) * | 1995-04-13 | 2000-08-01 | Milkhaus Laboratory, Inc. | Method for treating mucositis by sublingual administration of DNA |
| US6100224A (en) * | 1997-10-01 | 2000-08-08 | Exxon Chemical Patents Inc | Copolymers of ethylene α-olefin macromers and dicarboxylic monomers and derivatives thereof, useful as additives in lubricating oils and in fuels |
| US7176269B2 (en) | 2000-07-25 | 2007-02-13 | Mitsui Chemicals, Inc. | Curable composition and its use |
| JP4120139B2 (ja) | 2000-07-25 | 2008-07-16 | 三井化学株式会社 | 複層ガラス用シーリング材 |
| KR100734496B1 (ko) | 2000-07-25 | 2007-07-03 | 미쓰이 가가쿠 가부시키가이샤 | 경화성 조성물 및 그 용도 |
| JP4125884B2 (ja) | 2001-11-06 | 2008-07-30 | 孝志 澤口 | 末端ビニリデン基を有するオリゴ(1−ブテン) |
| EP1364973B1 (en) | 2000-11-24 | 2010-11-17 | San-Ei Kougyou Corporation | Functional substances derived from oligoolefins having functional groups at the ends |
| JP4866988B2 (ja) | 2001-07-30 | 2012-02-01 | Jnc株式会社 | アイソタクチック−アタクチック−ブロックポリプロピレン |
| JP2006077163A (ja) | 2004-09-10 | 2006-03-23 | Mitsui Chemicals Inc | 末端に極性基を有するポリオレフィンの製造方法 |
| JP5356698B2 (ja) | 2008-02-29 | 2013-12-04 | 学校法人日本大学 | 生分解性重合体ブロックを有するトリブロック共重合体およびその製造方法 |
| JP5555410B2 (ja) * | 2008-03-14 | 2014-07-23 | 出光興産株式会社 | ポリオレフィン系グラフト共重合体および接着剤組成物 |
-
2011
- 2011-05-17 EP EP11786290.4A patent/EP2578607A4/en not_active Withdrawn
- 2011-05-17 CN CN2011800260126A patent/CN102906125A/zh active Pending
- 2011-05-17 WO PCT/JP2011/002728 patent/WO2011148586A1/ja not_active Ceased
- 2011-05-17 US US13/700,008 patent/US8871855B2/en active Active
- 2011-05-17 JP JP2012517118A patent/JP5890774B2/ja not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0641235A (ja) * | 1992-04-01 | 1994-02-15 | Mitsui Toatsu Chem Inc | シンジオタクチックポリプロピレンワックス、その製造方法及び該ワックスを使用する熱ロール定着用トナー組成物 |
| JP2003535193A (ja) * | 2000-05-30 | 2003-11-25 | バーゼル、ポリオレフィン、ゲゼルシャフト、ミット、ベシュレンクテル、ハフツング | ファイバーグレードのプロピレンポリマーの製造方法 |
| WO2008047860A1 (en) * | 2006-10-20 | 2008-04-24 | Idemitsu Kosan Co., Ltd. | Highly pure, terminal-unsaturated olefin polymer and process for production thereof |
| JP2008133320A (ja) * | 2006-11-27 | 2008-06-12 | Idemitsu Kosan Co Ltd | 末端変性ポリα−オレフィン、その製造方法及びそれを含む組成物 |
| WO2008066168A1 (en) * | 2006-12-01 | 2008-06-05 | Idemitsu Kosan Co., Ltd. | Graft copolymer, thermoplastic resin composition comprising the graft copolymer, and those production method |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2578607A4 * |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012124345A1 (ja) * | 2011-03-16 | 2012-09-20 | 出光興産株式会社 | 反応性ポリオレフィン、その製造方法、及びそれを含む組成物 |
| WO2013118841A1 (ja) * | 2012-02-08 | 2013-08-15 | 出光興産株式会社 | 末端不飽和α-オレフィン重合体及びその製造方法 |
| JPWO2013118841A1 (ja) * | 2012-02-08 | 2015-05-11 | 出光興産株式会社 | 末端不飽和α−オレフィン重合体及びその製造方法 |
| WO2014046134A1 (ja) * | 2012-09-18 | 2014-03-27 | 出光興産株式会社 | 官能化α-オレフィン重合体、それを用いた硬化性組成物及び硬化物 |
| JPWO2014046134A1 (ja) * | 2012-09-18 | 2016-08-18 | 出光興産株式会社 | 官能化α−オレフィン重合体、それを用いた硬化性組成物及び硬化物 |
| JP2014098072A (ja) * | 2012-11-13 | 2014-05-29 | Idemitsu Kosan Co Ltd | 官能化α−オレフィン重合体、それを用いた硬化性組成物及び硬化物 |
| JP2014223752A (ja) * | 2013-05-16 | 2014-12-04 | 三井化学株式会社 | 包装材用の積層フィルムおよび包装材 |
| KR20190055838A (ko) * | 2017-01-13 | 2019-05-23 | 가부시키가이샤 닛폰 쇼쿠바이 | 공중합체 및 수지 조성물 |
| WO2018131670A1 (ja) * | 2017-01-13 | 2018-07-19 | 株式会社日本触媒 | 共重合体および樹脂組成物 |
| JPWO2018131670A1 (ja) * | 2017-01-13 | 2019-08-08 | 株式会社日本触媒 | 共重合体および樹脂組成物 |
| CN110167981A (zh) * | 2017-01-13 | 2019-08-23 | 株式会社日本触媒 | 共聚物及树脂组合物 |
| KR102190057B1 (ko) * | 2017-01-13 | 2020-12-14 | 가부시키가이샤 닛폰 쇼쿠바이 | 공중합체 및 수지 조성물 |
| WO2020027222A1 (ja) * | 2018-08-02 | 2020-02-06 | 出光興産株式会社 | ポリプロピレン系接着剤及びその製造方法 |
| JPWO2020027222A1 (ja) * | 2018-08-02 | 2021-08-12 | 出光興産株式会社 | ポリプロピレン系接着剤及びその製造方法 |
| US12110424B2 (en) | 2018-08-02 | 2024-10-08 | Idemitsu Kosan Co., Ltd. | Polypropylene-based adhesive and method for producing same |
| JP7356425B2 (ja) | 2018-08-02 | 2023-10-04 | 出光興産株式会社 | ポリプロピレン系接着剤及びその製造方法 |
| KR20210121029A (ko) * | 2018-12-28 | 2021-10-07 | 다우 글로벌 테크놀로지스 엘엘씨 | 텔레켈릭 폴리올레핀을 포함하는 경화성 조성물 |
| KR20210121043A (ko) * | 2018-12-28 | 2021-10-07 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| KR102879248B1 (ko) * | 2018-12-28 | 2025-11-04 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| JP2022515522A (ja) * | 2018-12-28 | 2022-02-18 | ダウ グローバル テクノロジーズ エルエルシー | 不飽和ポリオレフィンを含む硬化性組成物 |
| JP2022516119A (ja) * | 2018-12-28 | 2022-02-24 | ダウ グローバル テクノロジーズ エルエルシー | テレケリックポリオレフィンを含む硬化性組成物 |
| JP2022516122A (ja) * | 2018-12-28 | 2022-02-24 | ダウ グローバル テクノロジーズ エルエルシー | 不飽和ポリオレフィンを含む硬化性組成物 |
| JP2022516118A (ja) * | 2018-12-28 | 2022-02-24 | ダウ グローバル テクノロジーズ エルエルシー | 不飽和ポリオレフィンを含む硬化性組成物 |
| KR20210121030A (ko) * | 2018-12-28 | 2021-10-07 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| JP7539387B2 (ja) | 2018-12-28 | 2024-08-23 | ダウ グローバル テクノロジーズ エルエルシー | 不飽和ポリオレフィンを含む硬化性組成物 |
| KR20210121027A (ko) * | 2018-12-28 | 2021-10-07 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| JP7592597B2 (ja) | 2018-12-28 | 2024-12-02 | ダウ グローバル テクノロジーズ エルエルシー | 不飽和ポリオレフィンを含む硬化性組成物 |
| JP7609784B2 (ja) | 2018-12-28 | 2025-01-07 | ダウ グローバル テクノロジーズ エルエルシー | テレケリックポリオレフィンを含む硬化性組成物 |
| JP7609783B2 (ja) | 2018-12-28 | 2025-01-07 | ダウ グローバル テクノロジーズ エルエルシー | 不飽和ポリオレフィンを含む硬化性組成物 |
| KR102880619B1 (ko) * | 2018-12-28 | 2025-11-04 | 다우 글로벌 테크놀로지스 엘엘씨 | 텔레켈릭 폴리올레핀을 포함하는 경화성 조성물 |
| KR102873284B1 (ko) * | 2018-12-28 | 2025-10-21 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| KR102879257B1 (ko) | 2018-12-28 | 2025-10-31 | 다우 글로벌 테크놀로지스 엘엘씨 | 불포화 폴리올레핀을 포함하는 경화성 조성물 |
| JP7627164B2 (ja) | 2020-05-19 | 2025-02-05 | 三洋化成工業株式会社 | 熱硬化性樹脂組成物 |
| JP2021181558A (ja) * | 2020-05-19 | 2021-11-25 | 三洋化成工業株式会社 | 熱硬化性樹脂組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| US8871855B2 (en) | 2014-10-28 |
| CN102906125A (zh) | 2013-01-30 |
| JPWO2011148586A1 (ja) | 2013-07-25 |
| US20130066007A1 (en) | 2013-03-14 |
| EP2578607A1 (en) | 2013-04-10 |
| EP2578607A4 (en) | 2014-01-22 |
| JP5890774B2 (ja) | 2016-03-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5890774B2 (ja) | 末端不飽和ポリオレフィン、及びその製造方法 | |
| JP5512973B2 (ja) | グラフト共重合体又は該共重合体を含有する熱可塑性樹脂組成物及びそれらの製造方法 | |
| JP5555410B2 (ja) | ポリオレフィン系グラフト共重合体および接着剤組成物 | |
| JPWO2009096375A1 (ja) | ポリオレフィン系グラフト共重合体を含有する水系分散体 | |
| JP5241294B2 (ja) | ポリオレフィン系グラフト共重合体と付加重合ポリマーを配合してなる樹脂組成物 | |
| JP5374051B2 (ja) | 変性グラフト共重合体 | |
| JP5903000B2 (ja) | α−オレフィン重合体 | |
| WO2009090949A1 (ja) | グラフト共重合体を含有するエンジニアリングプラスチック系樹脂組成物 | |
| JP6002230B2 (ja) | 官能化α−オレフィン重合体、それを用いた硬化性組成物及び硬化物 | |
| WO2012124345A1 (ja) | 反応性ポリオレフィン、その製造方法、及びそれを含む組成物 | |
| JP2012201821A (ja) | グラフト共重合体の製造方法、及び当該製造方法により得られるグラフト共重合体を含む熱可塑性樹脂組成物 | |
| JP2014098072A (ja) | 官能化α−オレフィン重合体、それを用いた硬化性組成物及び硬化物 | |
| JP2014040526A (ja) | 官能化α−オレフィン重合体、それを用いた硬化性組成物及び硬化物 | |
| JP2014040516A (ja) | 硬化性粘接着組成物 | |
| WO2013183611A1 (ja) | α-オレフィン重合体 | |
| JP2009138137A (ja) | ポリオレフィン系樹脂組成物 | |
| JP5074013B2 (ja) | ブロック共重合体又は該共重合体を含有する熱可塑性樹脂組成物及びそれらの製造方法 | |
| JP5957472B2 (ja) | 末端不飽和α−オレフィン重合体及びその製造方法 | |
| JP2012201822A (ja) | グラフト共重合体の製造方法、及び当該製造方法により得られるグラフト共重合体を含む熱可塑性樹脂組成物 | |
| JP2016164233A (ja) | 官能化α−オレフィン重合体、及びそれを含む樹脂組成物 | |
| JP5466501B2 (ja) | 酸化変性プロピレン系重合体及びその製造方法 | |
| CN118119647A (zh) | 固化性树脂 | |
| JP2014047220A (ja) | 官能化α−オレフィン重合体、それを用いた硬化性組成物及び硬化物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180026012.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11786290 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012517118 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13700008 Country of ref document: US Ref document number: 2011786290 Country of ref document: EP |