WO2012013606A1 - Verfahren zur herstellung von dialkylcarbonaten, kupfer-haltiger katalysator und verwendung eines kupfer-haltigen katalysators - Google Patents
Verfahren zur herstellung von dialkylcarbonaten, kupfer-haltiger katalysator und verwendung eines kupfer-haltigen katalysators Download PDFInfo
- Publication number
- WO2012013606A1 WO2012013606A1 PCT/EP2011/062679 EP2011062679W WO2012013606A1 WO 2012013606 A1 WO2012013606 A1 WO 2012013606A1 EP 2011062679 W EP2011062679 W EP 2011062679W WO 2012013606 A1 WO2012013606 A1 WO 2012013606A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- copper
- cucl
- compound
- dmim
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0277—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides comprising ionic liquids, as components in catalyst systems or catalysts per se, the ionic liquid compounds being used in the molten state at the respective reaction temperature
- B01J31/0292—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides comprising ionic liquids, as components in catalyst systems or catalysts per se, the ionic liquid compounds being used in the molten state at the respective reaction temperature immobilised on a substrate
- B01J31/0294—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides comprising ionic liquids, as components in catalyst systems or catalysts per se, the ionic liquid compounds being used in the molten state at the respective reaction temperature immobilised on a substrate by polar or ionic interaction with the substrate, e.g. glass
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C68/00—Preparation of esters of carbonic or haloformic acids
- C07C68/01—Preparation of esters of carbonic or haloformic acids from carbon monoxide and oxygen
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/70—Oxidation reactions, e.g. epoxidation, (di)hydroxylation, dehydrogenation and analogues
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/10—Complexes comprising metals of Group I (IA or IB) as the central metal
- B01J2531/16—Copper
Definitions
- the invention relates to a process for the preparation of dialkyl carbonates, a copper-containing catalyst and the use of a copper-containing catalyst according to the invention.
- Dialkyl carbonates are industrially important compounds which find application in various fields. Some members of this class of compounds, such as dimethyl carbonate, are low toxicity intermediates that can replace toxic intermediates, such as phosgene or dimethyl sulfate, in many reactions. Another advantage is that they are not corrosive and In addition, no environmentally harmful by-products arise when using them. Because of these properties, dialkyl carbonates are of great importance in the chemical industry for a variety of syntheses. Dimethyl carbonate can be used to improve the octane rating of gasoline and thus replace environmentally problematic lead compounds. In addition, it can be used as a non-toxic and biodegradable solvent.
- MeO ' OMe A further disadvantage is that the catalysts lead to corrosion of the reactor and that the catalyst is deactivated by water, which is formed as a by-product in the reaction.
- ENICHEM process of oxidative methanol carbonylation are summarized in a review article [N. Keller, G. Rebmann, V. Keller, J. Mol. Cat. A 2010, 317, 1].
- the BAYER company describes a process in which a hydrophilic electrolytic inorganic salt melt of KCl and CuCl is used as a catalytic solvent [Z. Kricsfalussy, H. Waldmann, H.-J. Traenckner (BAYER), EP 0 636 601 A1, 1995 and Z. Kricsfalussy, H. Waldmann, H.-J. Traenckner, Ind. Eng. Chem. Res. 1998, 37, 865.].
- the catalytic solvent (KCl / CuCl melt) is placed in a reactor vessel.
- the educts are fed via a gassing stirrer as gas.
- the actual reaction product dimethyl carbonate forms an azeotropic mixture with the by-produced water, which is distilled out of the reactor vessel. In this way, the water can be at least partially separated and a deactivation of the catalyst can be limited by water.
- the method has the disadvantage that the reaction water can not be completely separated by the distillation as an azeotrope with DMC and therefore accumulates with increasing reaction time in the hydrophilic salt melt.
- the water content in the molten salt is brought to a considered advantageous water content of less than 10 wt.%.
- this is only possible at high temperatures of 120 - 300 ° C.
- these conditions lead to the hydrolysis of DMC to CO2 and thus to a poor CO selectivity or to a bad one
- a particular characteristic of the said process is the implementation of a heterogeneous gas-solid reaction in which the starting materials and products are in the gas phase, while the catalyst is present in the solid phase.
- the advantage of heterogeneous process management lies in the fact that the Catalyst and by-produced water are not in the same phase. By this configuration, the deactivation of the catalyst can be limited by the water.
- Another advantage of using a two-phase system is the easier product separation.
- a serious disadvantage is that a diffusion of the reactants to each other and in particular to active centers within the solid catalyst phase due to physical laws is very limited. In addition, only a limited solubility of the educts in the solid catalyst phase. Of the total, in the solid catalyst phase, active catalyst sites active only those are efficiently used, which are arranged on the catalyst surface. This reduces sales.
- a further disadvantage is that in the process mentioned a creeping deactivation of the solid catalyst with a reactivation of the catalytic centers must be countered by organohalogen compounds. This complicates the process and leads to an increase in costs.
- this method has the disadvantage that it is carried out homogeneous catalysis in a methanol solution.
- the water accumulation in the hydrophilic catalyst phase leads to the above with increasing reaction time to deteriorating CO and product selectivity and thus to a low yield.
- the separation of product and catalyst in homogeneous processes is extremely complicated and costly. Due to the toxicity and combustibility of methanol, the process is associated with a relatively high risk potential and therefore requires special safety measures. The use of the solvent is therefore associated with a cost.
- the object of the invention is therefore to overcome these and other disadvantages in the prior art and to provide a catalytic process for the preparation of dialkyl carbonates available, which overcomes the disadvantages of known methods and provides the highest possible dialkyl carbonate yield.
- the method should have the lowest possible risk and also allow easy and rapid separation of the reaction product from the catalyst.
- the process should be simple to perform and inexpensive to use.
- the catalysts should also allow high conversion rates in the absence of a solvent.
- Catalysts supported on solid phases are particularly easy to separate from further constituents of a reaction mixture. Due to their easy separability, additional purification steps, such as extraction or chromatography, are eliminated, especially during product recovery. Consequently, with the aid of the support, both the expenditure of time and the amount of material required in the method can be significantly reduced.
- the application of the copper-containing catalyst to a porous support significantly increases the surface area of the catalyst phase and thus the exchange surface between the catalyst phase and the educts and products which is available during the reaction. In this way, in the process with a simple means very high reaction rates and thus high conversions can be achieved.
- the supported catalyst can be introduced into a fixed bed reactor and used, for example, in a continuous process.
- the known advantages of a continuous reaction such as improved reaction control, smaller reaction volume, small space requirement of the reactor, etc. can be used.
- Suitable support materials are particularly porous inorganic and organic substances.
- Suitable inorganic carriers are SiO 2 , Al 2 O 3 , zeolites and carbon.
- Suitable organic support materials are polymers such. B. polystyrene.
- Vankelecom P.A. Jacobs, Adv. Synth. Catal. 2006, 348,, 1413; N.E. Leadbeater, M. Marco, Chem. Rev. 2002, 102,, 3217.).
- the liquid catalyst phase With the aid of the liquid catalyst phase, it is possible in an advantageous manner to provide a means in multiphase systems which additionally promotes the discharge of the water from the catalyst phase into an educt / product phase.
- the polarity of the catalyst phase is made such that a hydrophobic character results, due to which the discharge of the water is forced.
- the copper-containing catalyst, or an additive contained in the liquid catalyst phase have a hydrophobic or lipophilic character.
- the liquid catalyst phase can consequently be used to ensure, with a comparatively simple means, that the process according to the invention gives a high conversion even in the case of long transit times. Due to the polarity, the transverse solubility can also be influenced in multiphase continuous systems.
- the process uses a copper-containing catalyst which is present in a liquid catalyst phase also provides an important prerequisite for achieving high diffusion rates between the catalyst phase and the reactants and products in the process. you can. In this way, the reactants can easily diffuse into the liquid catalyst phase and be reacted there quickly with the aid of the catalyst, so that high conversion rates can be achieved in the process.
- the process according to the invention can also be carried out in a 3-phase system with a gaseous educt / product phase. It is also particularly advantageous because with a suitable choice of process conditions, the by-produced water is gaseous.
- the catalyst comprises a copper-containing coordination compound.
- a coordination compound in the context of the invention includes both neutral complexes and the salts of cationic and anionic complexes. Copper-containing coordination compounds have proven to be very efficient catalysts in the preparation of dialkyl carbonates and.
- the copper-containing coordination compound has a melting point of less than 120 ° C, preferably less than 15 ° C, more preferably less than 100 ° C and most preferably less than 80 ° C. This is particularly advantageous because the process can thereby be carried out even at relatively low temperatures, without the use of a solvent for providing the catalyst phase liquid under process conditions is necessary. This property thus provides an important prerequisite for the entire process to be carried out in the absence of a solvent.
- Solvents are often toxic and / or flammable, so their use is usually associated with an increase in the hazard potential of a process. Furthermore, the use of solvents has a negative impact on the atom economy of a production process and is therefore associated with increased process costs. In a solvent-free implementation of the method, this can be avoided. The possibility of such a procedure is therefore particularly attractive.
- the at least one copper-containing coordination compound forms the liquid catalyst phase.
- the copper-containing coordination compound contains at least one nitrogen (III) compound as AADonorligand and / or a quaternized nitrogen (III) compound as an organic cation, wherein the nitrogen (III ) - compound is selected from the group of A / -Alkylimidazolen, N, N-dialkylaminopyridines, AAAlkylpyrazolen or AAAlkyl-pentaorganoguanidinen.
- This embodiment is particularly advantageous since the said copper-containing coordination compounds provide particularly good results in processes for the preparation of dialkyl carbonates by catalytic oxidative carbonylation of an alcohol.
- the Lewis acidity of the copper-containing catalyst can be reduced. Reducing the Lewis acidity limits the ability of the catalyst to also catalyze the undesired hydrolysis of the dialkyl carbonate. By this configuration, therefore, the selectivity of the catalytic reaction can be increased and thus the efficiency of the process can be increased.
- the copper-containing co-ordination compound contains at least one A / -alkylimidazole as AADonorligand and / or a quaternized A / -Alkylimidazol, i. an A / alkylimidazolium ion, as an organic cation.
- the copper-containing coordination compound has at least one lipophilic substituent. It is advantageous, in particular, that a lipophilic substituent, such as e.g. a long alkyl substituent can be provided very easily and because of its polarity is suitable for promoting the discharge of the water from the catalyst phase. In this way, the deactivation of the catalyst can be counteracted.
- the lipophilic substituent is preferably an aliphatic substituent having a number from 1 to 25 carbon atoms, and most preferably an alkyl substituent having from 1 to 25 carbon atoms.
- the lipophilic substituent may have substituents such as e.g. Have alkyl chains.
- an advantage of this embodiment is in particular that the lipophilic substituent is located on a component of the catalyst, thereby directly counteracting deactivation of the catalyst by water.
- a particularly important embodiment provides that the lipophilic substituent is provided on the nitrogen (III) compound. It is furthermore preferred that the lipophilic substituent is arranged on the nitrogen of the nitrogen (III) compound. With this embodiment, particularly high yields could be achieved in processes for the preparation of dialkyl carbonates.
- a further advantage is that the melting point of the coordination compounds can often be lowered by the introduction of the lipophilic substituent, especially in the case of copper-containing coordination compounds which have at least one A / -alkylimidazole.
- a copper-containing coordination compound can be readily provided which is liquid under process conditions.
- a preferred embodiment of a method according to the invention also provides that gaseous starting materials are brought into contact with the catalyst phase, and that the products are removed via the gas phase.
- a product is water.
- the catalyst is an ionic liquid, wherein the copper-containing coordination compound is contained in the anion and / or in the cation of the ionic liquid. This is particularly suitable since particularly good dialkyl carbonate yields could be achieved with processes designed in this way.
- Ionic liquids are salts that already melt at a temperature of ⁇ 100 ° C. They are usually composed of an organic cation and an organic or inorganic anion. They are characterized by a number of advantageous properties. For example, they are not inflammable, have a very low vapor pressure and also have very good dissolution properties. In this way it can be ensured that the educts used in the process are soluble in the catalyst phase. A further advantage is that ionic liquids have a low melting point and often also a relatively high thermal stability. Thus, the method according to the invention can be carried out in a wide temperature range.
- dialkyl carbonate yields can also be achieved in that the anion of the ionic liquid is either a copper-containing coordination compound or from [CuCl 2 ] - , [CuBr 2 ] - , [Cul 2 ] - , [BF 4 ] " and [PF 6 ] "is selected.
- the copper-containing coordination compound is selected from the ionic liquids [Cu (Im 12 ) 4 ] [PF 6 ], [Cu (Im 12 ) 2 ] [CuCl 2 ], [Cu (Im 12 ) 2 ] [CuBr 2 ], [Cu (lm 6 ) 2 ] [CuCl 2 ], [Cu (lm 6 ) 2 ] [CuBr 2 ], [DMIM] [CuCl 2 ], [DMIM] [CuCl 2 ] [DMIM] 2 [CuCl 3 ],
- An important embodiment of the process provides that branched and unbranched alkanols having 1 to 4 carbon atoms are reacted. Very particular preference is given to the catalytic oxidative carbonylation of methanol to dimethyl carbonate.
- An important embodiment provides that the process in a temperature range of 60 ° C to 200 ° C, preferably from 70 ° C to 140 ° C, more preferably from 75 ° C to 120 ° C and most preferably from 80 ° C to 1 15 ° C is performed. Especially at temperatures below
- the process is carried out as a continuous process. Continuous synthesis runs are known.
- a method according to the invention can be based on all of the known execution protocols.
- the catalyst can be placed in a fixed bed reactor which is separate from the remainder of the reaction mixture, i. is continuously flowed through by the reacted alcohol, the carbon monoxide and the oxygen.
- a particularly advantageous development provides that the continuous process is carried out in the gas phase.
- an additive is contained in the liquid catalyst phase.
- the additive can advantageously be used to optimally match the physical properties such as the viscosity of the catalyst phase or the starting material solubility (in particular the solubility of the alkanol) to the process conditions.
- the additive is preferably a low-volatility compound such as, for example, a silicone oil or a perfluorinated polyether oil.
- a particularly preferred process for the preparation of dialkyl carbonates provides that the catalyst is applied to the solid phase support by non-covalent interaction. It is advantageous here that in the preparation of the catalyst used in the process, the attachment of this to the solid phase carrier is eliminated.
- the support of the catalyst is thus comparatively simple. In contrast to a covalent support, in the case of a non-covalently supported catalyst, no special linker for the application of the catalyst to the solid phase support has to be introduced. This reduces the cost of producing the catalyst and thus the cost of the entire process.
- the solid phase support is formed by SiO 2 .
- SiO 2 is a low-cost carrier material that is available in many different designs in terms of its shape, its particle size and its surface finishes.
- the support material can also be used in a wide temperature range.
- the solid-phase support of SiO 2 is formed with a particle size between 0.05 mm and 4 mm and a BET surface area of 250 to 1000 m 2 / g.
- a solid phase support of SiO 2 which has a particle size between 0.063 mm and 0.2 mm and a BET surface area of 300 - 400m 2 / g.
- the process uses a copper-containing catalyst supported on a solid phase support, in which the mass ratio of the copper-containing catalyst to the solid phase support is between 0.075 and 1, 350, more preferably between 0.150 and 0.750, and most particularly preferably between 0.300 and 0.590.
- the invention further provides a copper-containing catalyst which is supported on a solid phase support and which is present in a catalyst phase which has a melting point of less than 120 ° C., and which contains at least one copper phase.
- the copper-containing coordination compound contains at least one nitrogen (III) compound as AADonorligand and / or a quaternized nitrogen (III) compound as an organic cation, wherein the nitrogen (III) compound is selected from the group of A / alkylimidazoles, A /, A / dialkylaminopyridines, AAalkylpyrazoles or AAalkylpentaorganoguanidines.
- the copper-containing catalyst is supported on a solid phase support and is present in a catalyst phase containing a
- a catalyst can be provided, which can be particularly easily separated from further components of the reaction mixture when using the catalyst in a catalytic process.
- the application of the copper-containing catalyst to a porous support significantly increases the surface area of the catalyst phase and thus the exchange surface between the catalyst phase and the educts and products which is available during the reaction. In this way, when using a catalyst according to the invention in a catalytic process very high reaction rates and thus high conversions can be achieved.
- a further advantage is that a liquid catalyst phase is provided in a catalyst according to the invention. This can provide an important prerequisite for achieving high rates of diffusion between the catalyst phase and the starting materials and products. Infol- The educts can readily be diffused into the liquid catalyst phase and reacted there quickly with the aid of the catalyst, so that high conversion rates can be achieved when using a catalyst according to the invention in a catalytic reaction.
- the fact that the copper-containing catalyst is present in a catalyst phase having a melting point of less than 120 ° C also provides an important prerequisite for the catalyst to be used in a process in which the catalytic reaction is already takes place at relatively low temperatures even in the absence of a solvent in a liquid phase.
- Solvents are often toxic and / or flammable, so that their use is usually associated with an increase in the hazard potential of a process. Furthermore, the use of solvents has a negative impact on the atom economy of a process and is therefore associated with increased costs. In a solvent-free reaction, this can be avoided. A catalyst that makes this possible is therefore particularly attractive.
- the catalyst phase has a melting point of less than 120 ° C, preferably less than 15 ° C, more preferably less than 100 ° C and most preferably less than 80 ° C.
- the melting point of the liquid phase the lower the temperatures, the catalyst can be used without the addition of a solvent in the liquid phase.
- the copper-containing coordination compound has a melting point of less than 120 ° C, preferably less than 15 ° C, more preferably less than 100 ° C and most preferably less than 80 ° C.
- a catalyst phase can be particularly easily se, which has a melting point less than 120 ° C, or less than 15 ° C, less than 100 ° C or less than 80 ° C.
- the catalyst has at least one lipophilic substituent. This is particularly advantageous since the introduction of the lipophilic substituent can influence the melting point of the catalyst.
- a further advantage is that the polarity of the catalyst can be adapted to its intended use by the lipophilic substituent.
- the lipophilic substituent can reduce the transverse solubility in the polar phase. In this way, detachment of the catalyst from the solid phase support or removal of the catalyst from the catalyst phase can be prevented.
- the lipophilic substituent is preferably an aliphatic substituent having a number from 1 to 25 carbon atoms, and most preferably an alkyl substituent having from 1 to 25 carbon atoms.
- the lipophilic substituent may itself also have substituents such as e.g. Have alkyl chains.
- a particularly important embodiment provides that the lipophilic substituent on the nitrogen (III) compound is provided. This is particularly advantageous because catalysts constructed in this way often have a melting point of less than 120.degree.
- the catalyst is an ionic liquid, wherein the copper-containing coordination compound is contained in the anion and / or in the cation of the ionic liquid.
- Ionic liquids in which the copper-containing coordination compound is contained in the anion and / or in the cation of the ionic liquid and in particular those in which the copper-containing coordination compound at least a least one A / -Alkylimidazol as AADonorligand and / or a quaternized N-alkylimidazole as an organic cation, generally show a high thermal stability. This is particularly advantageous, since even with long reaction times, no decomposition of the catalyst takes place, so that the process yields constant conversions.
- a particularly high thermal stability could be achieved also if the anion of the ionic liquid represents either a copper-containing coordination compound or from [CuCl 2] ⁇ , [CuBr 2] ⁇ [Cul 2] ⁇ , [BF 4] "and [ PF 6 ] " is selected.
- catalysts according to the invention it is also possible to influence the properties (for example reactant solubility, flow properties) of the catalyst phase by adding further, low-volatility components.
- these components such as Silicone oils and perfluorinated polyether oils are mixed with the catalytically active liquid prior to application of the liquid film to the support and then applied to the support material, if appropriate with the aid of a solvent.
- the catalyst is applied by noncovalent interaction to a solid phase support.
- the solid phase support of the catalyst is formed by SiO 2 .
- SiO 2 is a low-cost carrier material that is available in many different particle sizes and surface finishes. The support material can also be used in a wide temperature range, this has an advantageous effect on the applications of the catalyst.
- the solid phase support is preferably formed by SiO 2 having a particle size between 0.05 and 4 mm and a BET surface area of 250 to 1000 m 2 / g. Particularly preferred is a support of SiO 2 having a particle size between 0.063 - 0.2 mm and a BET surface area of 300 - 400m 2 / g.
- the mass ratio of the copper-containing catalyst to the solid phase support is between 0.075-1.350 and more preferably between 0.150-0.750 and very particularly preferably between 0.300-0.590.
- the copper-containing coordination compound is selected from the ionic liquids
- the invention additionally provides for a use of a catalyst according to the invention in an oxidative carbonylation. This is particularly advantageous since the catalysts according to the invention have led to particularly high conversions when used in such reactions.
- the catalysts according to the invention are particularly suitable for such an application since they have, on the one hand, a copper complex and, on the other hand, high thermal stability. Oxidative carbonylations can be catalyzed by copper complexes. In addition, these reactions are also often carried out at elevated temperatures.
- a catalyst according to the invention in an oxidative carbonylation of alkanols in the presence of oxygen is particularly preferred. and carbon monoxide to dialkyl carbonates.
- very particular preference is given to the use of a catalyst according to the invention in an oxidative carbonylation of branched and unbranched alcohols having 1 to 4 carbon atoms to form dialkyl carbonates.
- good results could be achieved with the ionic liquids according to the invention in the oxidative carbonylation of methanol to dimethyl carbonate.
- a particularly simple process for the preparation of the catalyst according to the invention provides that either the copper-containing coordination compound itself, or in an in situ preparation of these, the components required for the preparation of the copper-containing coordination compound under inert gas and optionally at elevated Temperature and / or be applied with shaking and / or the action of ultrasound on the solid phase support.
- a solvent which forms a solution with the corresponding component (s) of the catalyst. Solvents such as acetonitrile, methanol, tetrahydrofuran or dichloromethane are particularly well suited for this purpose.
- the solid phase carrier is treated with an excess of said solution. After the mass transfer between the solid phase support and the solution, the excess solution is removed.
- Another method is the spraying of the solid phase carrier with such a solution.
- an added solvent is removed during or after the spraying process.
- the catalyst supported on a solid phase support is preferably prepared by displacing the solid phase support with a solution of the catalyst or, in the case of an in situ preparation with a solution of the corresponding components, in a solvent and then evaporating off the solvent.
- Fig. 1 is a schematic representation of a continuous gas phase reactor.
- FIG. 1 shows a schematic representation of a continuous gas-phase reactor.
- a continuous gas-phase reactor Such a reactor was used to carry out the continuous processes described in the working examples in Table 3.
- the metering of the gases takes place via mass flow controller 1.
- the system pressure is set at the pressure control valve 7.
- the liquid educt (in a reaction according to Table 3, methanol) is conveyed via an HPLC piston pump 2 and brought in a heated evaporator 3 in the gaseous state.
- the fixed bed reactor 4 is a stainless steel pipe. If required, the volumetric flow of the reaction gases can be passed through a second, identically constructed reactor 5 or via a bypass 6 past the reactor. All pipelines of the experimental apparatus are electrically heatable.
- the products are detected by an online gas chromatograph 8 with Restek RTX-5 column and flame ionization detector.
- Crystals of I suitable for single crystal structure analysis were obtained from MeOH at room temperature, taken up in inert oil, and transferred to the cold N 2 cooling gas stream of the diffractometer. 1 .2 [Cu (lm 12 ) 2 ] [CuCl 2 ] (II)
- IR (substance) ⁇ ' 3122, 3051 w, 2949, 2916vs, 2847s, 1690w, 161v, 1533, 1520, 1466, 1442, 1399w, 1375, 1357, 1288, 1255w, 1240, 1 1 10s, 1053, 1039, 1026, 1006w, 962, 890w, 845, 833, 757vs, 730, 721, 654s, 630, 506w, 444w and 428w.
- IR (Substance) v - / cm -3 ' 3122, 31 12, 3049w, 2949, 2916vs, 2847s, 1685w, 1608w, 1533, 1520, 1465, 1442, 1397w, 1376w, 1341w, 1288, 1255w, 1239, 1 1 10, 1 100, 1053w, 1039w, 1026w, 960w, 889w, 843, 832, 755s, 728, 722, 654s, 630, 505w, 444w and 427w.
- Crystals of III suitable for single crystal structure analysis were prepared from CH 3 N / Et 2 O at -20 ° C., taken up in inert oil and transferred to the N 2 cooling gas stream of the diffractometer 1 .4 [Cu (Im 6 ) 2 ] [CuCl 2 ] (IV)
- 1-hexylimidazole (Im 6 ) (4.84 g, 31.8 mmol) was added to CuCl (2.97 g, 30.00 mmol).
- the reaction mixture was mixed with CH 3 CN (5 ml) for homogenization and sonicated for about 10 minutes.
- the solvent was removed in vacuo and the viscous liquid was digested with ether. After phase separation, the ether phase was decanted.
- Ci 8 H 32 CuN 4 calculates 367.1 91 7); m / z (ESI, negative mode, CH 3 CN)
- Ci 8 H 32 CuN 4 calculates 367.1 91 7); m / z (ESI, negative mode, CH 3 CN)
- Schirenk tube was added to CuCl (200 mg, 2.02 mmol) a solution of [DMIM] Cl (869 mg, 3.03 mmol) in CH 3 CN (5 mL). The reaction mixture was sonicated at room temperature for about 10 minutes. After removal of the solvent in vacuo and freeze drying of the greyish
- CD 3 CN 14.4 (CH 3 (17)), 23.3, 26.6, 29.6, 30.0, 30.2, 30.3, 30.6, 32.6 (CH 2 ( 7-16)), 37.0 (CH 3 (18)), 50.5 (CH 2 (6)), 123.2 (CH (5)), 124.6 (CH (4)) and 136, 8 (CH (2)); m / z (ESI; CH 3 CN) 251, 2480 (. [C 6 H 31 N 2] ⁇ 100% C 6 H 31 N 2 calculates 251, 2482); m / z (ESI, negative mode, CH 3 CN) 222.7651 ([CuBr 2 ] ⁇ 23%, CuBr 2 calculates 220.7648), 264 (35) and 314 (23).
- an ultrasound head is immersed in the water bath and operated at a frequency of 2 s -1 After 15 minutes, a vacuum pump is connected to the flask and the pressure is slowly reduced from 10 5 Pa to 5 * 10 2 Pa. After about 45 minutes a turquoise powder in front, which turns dark green when suddenly in contact with air.
- the suspension is heated in a water bath with stirring to 50 ° C.
- an ultrasound head is immersed in the water bath and operated at a frequency of 2 s -1
- a vacuum pump is connected to the flask and the pressure is slowly reduced from 10 5 Pa to 5 * 10 2 Pa. After about 45 minutes a blue powder before, which discolored suddenly dark green in contact with air.
- an ultrasound head is immersed in the water bath and operated at a frequency of 2 s -1 After 15 minutes, a vacuum pump is connected to the flask and the pressure is slowly reduced from 10 5 Pa to 5 * 10 2 Pa. After about 45 minutes a bluish powder, which suddenly turns dirty green on contact with air.
- the weighing of the unsupported copper (II) compound takes place outside of an inert gas box.
- 6.34 g of [CuBr 2 (Im 12 ) 4 ] are weighed into a 500 ml Schlenk flask.
- 90 ml of acetonitrile are added and the mixture is stirred for 15 minutes on a magnetic stirrer.
- 10.52 g of silica gel (particle size 0.063-0.200 mm, BET surface area 335 m 2 / g, pore volume 0.966 ml / g) are added and the suspension is stirred for a further 15 minutes.
- the suspension is heated in a water bath with stirring to 50 ° C.
- an ultrasound head is immersed in the water bath and operated at a frequency of 2 s -1 After 15 minutes, a vacuum pump is connected to the flask and the pressure is slowly reduced from 10 5 Pa to 5 * 10 2 Pa. After about 45 minutes C.
- the reaction apparatus (without reactor) was preheated to 100.degree.
- the copper-containing catalyst was placed in a fixed bed reactor and the fixed bed reactor was screwed into a plant as shown in FIG.
- the heating mantle was placed around the fixed bed reactor and the fixed bed reactor was heated to 100 ° C.
- the volume flow of the reaction gases CO and air was adjusted via two separate mass flow controllers and passed through a bypass (100 ° C) past the fixed bed reactor.
- the volumetric flows were 72 ml / min CO and 51.4 ml / min air.
- the system pressure was set to 5 bar at the pressure control valve.
- Entry 3 of Table 3 shows that increasing the reactor temperature to 120 ° C results in a further increase in the conversion and activity of the catalyst.
- Entry 4 to 7 show that with a further increase in the residence time by increasing the amount of catalyst and increasing the system pressure very significant increases in sales can be achieved.
- the invention is not limited to any of the prescribed embodiments, but can be modified in many ways.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Ein Verfahren zur Herstellung eines Dialkylcarbonats, bei welchem ein Alkanol mit Kohlenmonoxid und Sauerstoff in Anwesenheit eines Kupfer-haltigen Katalysators, der auf einem Festphasenträger geträgert ist, umgesetzt wird, sieht erfindungsgemäß vor, dass der Katalysator auf dem Festphasenträger unter Verfahrensbedingungen in einer flüssigen Katalysatorphase vorliegt, und dass der Katalysator wenigstens eine Kupfer-haltige Koordinationsverbindung umfasst. Zudem ist der Kupfer-haltige Katalysator, auf einem Festphasenträger geträgert, und liegt in einer Katalysatorphase vor, die einen Schmelzpunkt kleiner 120°C aufweist. Eine zum Katalysator gehörende Kupfer-haltige Koordinationsverbindung umfasst mindestens eine Stickstoff(lll)-Verbindung als AADonorligand und/oder eine quarternisierte Stickstoff(lll)-Verbindung als organisches Kation. Dabei ist die Stickstoff(lll)-Verbindung ausgewählt aus der Gruppe von A/-Alkylimidazolen, A/,A/-Dialkylaminopyridinen, N-Alkylpyrazolen oder AAAlkyl-penta-organoguanidinen. Die Verwendung eines Katalysators bei einer oxidativen Carbonylierung ist zudem Gegenstand der Erfindung.
Description
zur Herstellung von Dialkylcarbonaten, Kupfer-haltiger Katalysator und Verwendung eines Kupfer-haltigen Katalysators
Die Erfindung betrifft ein Verfahren zur Herstellung von Dialkylcarbonaten, einen Kupfer-haltigen Katalysator und die Verwendung eines erfindungsgemäßen Kupfer-haltigen Katalysators.
Dialkylcarbonate sind großtechnisch wichtige Verbindungen, die in verschiede- nen Bereichen Anwendung finden. Einige Vertreter dieser Verbindungsklasse, wie z.B. Dimethylcarbonat, sind Zwischenprodukte mit geringer Toxizität, die giftige Zwischenprodukte, wie Phosgen oder Dimethylsulfat, bei vielen Reaktionen ersetzen können. Vorteilhaft ist ebenfalls, dass sie nicht korrosiv sind und
bei ihrer Verwendung zudem keine umweltschädlichen Nebenprodukte entstehen. Aufgrund dieser Eigenschaften sind Dialkylcarbonate in der chemischen Industrie für eine Vielzahl von Synthesen von großer Bedeutung. Dimethylcar- bonat kann zur Verbesserung der Oktanzahl von Benzin eingesetzt werden und somit umweltproblematische Bleiverbindungen ersetzen. Zudem kann es als ungiftiges und biologisch abbaubares Lösungsmittel verwendet werden.
Aufgrund der vielfältigen Anwendbarkeit der Dialkylcarbonate besteht ein großes Interesse an effizienten Herstellungsverfahren für diese Verbindungen.
Im Stand der Technik sind Verfahren zur Herstellung von Dialkylcarbonaten durch Umsetzung der entsprechenden Alkohole mit Kohlenmonoxid und Sauerstoff in der Gegenwart von Cu-Salzen nach folgender Umsatzgleichung bekannt:
2 ROH + CO + 0.5 O2 > RO-C(O)-OR + H2O
Problematisch bei derartigen oxidativen Carbonylierungsreaktionen, die üblicherweise CuCI2 und basische Kupfer(ll)-halogenide verwenden, ist jedoch zum einen, dass die Cu-Salze aufgrund ihrer Eigenschaft als Lewis-Säure, wie nachfolgend am Beispiel der Umsetzung von Methanol zu Dimethylcarbonat (DMC) dargestellt, nicht nur die Bildung (1 ) des Dialkylcarbonats katalysieren, sondern auch dessen Hydrolyse (2) zu dem entsprechenden Alkohol und Kohlendioxid. Die Hydrolyse kann hierbei auch auf der Stufe eines katalytischen Intermedia- tes der Kupferkomplexe stattfinden.
2 MeOH + CO + 0.5 02 + H20 (1 )
O Cu-Kat.
+ H20 ■*> 2 MeOH + C02
MeO' OMe
Nachteilig ist weiterhin, dass die Katalysatoren zur Korrosion des Reaktors führen und dass der Katalysator durch Wasser, das bei der Reaktion als Nebenprodukt entsteht, deaktiviert wird. Die ursprünglich unter dem Begriff ENICHEM-Verfahren zusammengefassten Methoden der oxidativen Methanol-Carbonylierung sind in einem Übersichtsartikel zusammengefasst [N. Keller, G. Rebmann, V. Keller, J. Mol. Cat. A 2010, 317, 1 ].
Die Firma BAYER beschreibt ein Verfahren, bei dem eine hydrophile elektische anorganische Salzschmelze aus KCl und CuCI als katalytisches Lösungsmittel eingesetzt wird [Z. Kricsfalussy, H. Waldmann, H.-J. Traenckner (BAYER), EP 0 636 601 A1 , 1995 sowie Z. Kricsfalussy, H. Waldmann, H.-J. Traenckner, Ind. Eng. Chem. Res. 1998, 37, 865.]. Bei dem Verfahren wird das katalytische Lösungsmittel (KCI/CuCI-Schmelze) in einem Reaktorbehälter vorgelegt. Die Edukte werden über einen Begasungsrührer als Gas zugeführt. Das eigentliche Reaktionsprodukt Dimethylcarbonat bildet mit dem als Nebenprodukt entstehenden Wasser ein azeotropes Gemisch, welches aus dem Reaktorbehälter herausdestilliert wird. Auf diese Weise kann das Wasser zumindest teilweise abgetrennt und eine Deaktivierung des Katalysators durch Wasser eingeschränkt werden.
Das Verfahren hat jedoch den Nachteil, dass das Reaktionswasser durch die Destillation als Azeotrop mit DMC nicht vollständig abgetrennt werden kann und sich daher mit zunehmender Reaktionsdauer in der hydrophilen Salzschmelze anreichert. Um dem entgegenzuwirken, wird der Wassergehalt in der Salzschmelze auf einen als vorteilhaft angesehenen Wassergehalt von weniger als 10 Gew.% gebracht. Dies ist jedoch erst bei hohen Temperaturen von 120 - 300 °C möglich. Diese Bedingungen führen jedoch zur Hydrolyse von DMC zu CO2 und damit zu einer schlechten CO-Selektivität bzw. zu einer schlechten
Ausbeute an DMC.
Weiterhin wird bei den hohen Reaktionstemperaturen die Bildung von Nebenprodukten wie Methylchlorid und Dimethoxymethan beobachtet. Die Bildung dieser Nebenprodukte wirkt sich negativ auf die Atomökonomie des Verfahrens aus und führt zudem zu einem erhöhten Gefahrenpotential. Beispielsweise ist Methylchlorid hochentzündlich und gesundheitsschädlich. Es steht zudem im Verdacht, krebserregendes Potential zu besitzen. Die hohe Reaktionstemperatur, die bei diesem Verfahren notwendig ist, ist daher äußerst unvorteilhaft.
Desweiteren muss aufgrund der Verfahrensbedingungen ein hochtemperatur- beständig geschmierter, druckfest gelagerter und abgedichteter Autoklaven- Begasungsrührer verwendet werden. Die Technologie eines solchen Autokla- ven-Begasungsrührers ist teuer und störungsanfällig. Nachteilig bei der Verwendung eines solchen Autoklaven-Begasungsrührers sind zudem die hohen Energieverluste durch den mechanischen Energieeintrag und die Reibungsver- luste.
US 005387708 A beschreibt einen typisch heterogenen Prozess der oxidativen Carbonylierung von Methanol in der Gasphase [D. C. Molzahn, M. E. Jones, G. E. Hartwell, J. Puga, (DOW CHEMICAL), 1995]. Die Katalysatoren bestehen aus (a) einem Kupferhalogenid, Kupferoxyhalogenid oder Kupfercarboxylat- halogenid, (b) einem quaternären Ammoniumsalz und (c) einem festen Trägermaterial SiO2, AI2O3 oder Zeolithe Y. Die Trägerung der Salze findet in verschiedenen Stufen teilweise bei Temperaturen zwischen 400 - 700 °C statt. Es wird davon ausgegangen, dass salzartige Verbindungen des Typs
[NR1 R2R3R4]X [CuyXz] auf der Oberfläche des Trägers als Feststoff deponiert sind. Aufgrund der hohen Temperatur ist die Herstellung derartiger Katalysatoren verhältnismäßig kostenintensiv.
Besonderes Kennzeichen des genannten Verfahrens ist die Durchführung einer heterogenen Gas-Feststoff-Reaktion, bei der sich die Edukte und Produkte in der Gasphase befinden, während der Katalysator in der Festphase vorliegt. Der Vorteil der heterogenen Prozessführung liegt insbesondere darin, dass sich der
Katalysator und das als Nebenprodukt gebildete Wasser nicht in der gleichen Phase befinden. Durch diese Ausgestaltung kann die Deaktivierung des Katalysators durch das Wasser eingeschränkt werden. Vorteilhaft bei Verwendung eines Zwei-Phasen-Systems ist zudem die erleichterte Produktabtrennung.
Ein gravierender Nachteil ist jedoch, dass eine Diffusion der Reaktionspartner zueinander und insbesondere zu aktiven Zentren innerhalb der festen Katalysatorphase aufgrund physikalischer Gesetzmäßigkeiten stark begrenzt ist. Auch besteht nur eine begrenzte Löslichkeit der Edukte in der festen Katalysatorpha- se. Von den insgesamt, in der festen Katalysatorphase vorhandenen, aktiven Reaktionsstellen des Katalysators sind folglich nur diejenigen effizient nutzbar, die auf der Katalysator-Oberfläche angeordnet sind. Dies schmälert den Umsatz. Nachteilig ist weiterhin, dass bei dem genannten Verfahren einer schleichenden Deaktivierung des festen Katalysators mit einer Reaktivierung der katalytischen Zentren durch Organohalogenverbindungen begegnet werden muss. Dies verkompliziert den Prozess und führt zu einer Erhöhung der Kosten. Eine Publikation über Ligandeneffekte auf die Selektivität der Kupferkatalysierten aeroben oxidativen Carbonylierung von Methanol zu Dimethylcar- bonat (DMC) [V. Raab, M. Merz, J. Sundermeyer, J. Mol. Catal. A 2001 , 175, 51 .] beschreibt die Wirkung von AADonorliganden auf die Selektivität neutraler Komplexverbindungen. So ist bekannt, dass 1 -Methylimidazol-, 4- Dimethylaminopyridin- und Pentaorganoguanidin-Liganden die Sauerstoffaufnahme des Kupfers und die Selektivität und Produktivität neutraler Kupferkomplex-Katalysatoren vorteilhaft beeinflussen.
Dieses Verfahren hat jedoch den Nachteil, dass es homogenkatalytisch in einer Methanol-Lösung durchgeführt wird. Es kommt zu einer unvorteilhaften Anreicherung des Nebenproduktes Wasser in der Katalysatorphase, die, wie auch die Produktphase, eine methanolhaltige hydrophile Phase ist.
Die Wasseranreicherung in der hydrophilen Katalysatorphase führt nach dem obengenannten mit zunehmender Reaktionszeit zu schlechter werdender CO- und Produkt-Selektivität und damit zu einer geringen Ausbeute. Hinzu kommt, dass die Abtrennung von Produkt und Katalysator bei homogenen Verfahren äußerst aufwendig und kostenintensiv ist. Aufgrund der Toxizität und der Brennbarkeit von Methanol ist das Verfahren mit einem relativ hohen Gefahrenpotential verbunden und bedarf daher besonderer Sicherheitsmaßnahmen. Der Einsatz des Lösungsmittels ist deshalb auch mit einem Kostenaufwand verbunden.
Aufgabe der Erfindung ist es daher, diese und weitere Nachteile im Stand der Technik zu überwinden und ein katalytisches Verfahren zur Herstellung von Dialkylcarbonaten zur Verfügung zu stellen, welches die genannten Nachteile bekannter Verfahren überwindet und eine möglichst hohe Dialkylcarbonat- Ausbeute liefert. Das Verfahren soll ein möglichst geringes Gefahrenpotential aufweisen und zudem eine einfache und rasche Abtrennung des Reaktionsprodukts vom Katalysator ermöglichen. Darüber hinaus soll das Verfahren einfach durchzuführen und kostengünstig in der Anwendung sein. Es ist weiterhin Aufgabe der Erfindung einen Kupfer-haltigen Katalysator zur Verfügung zu stellen, der mit einfachen Mitteln kostengünstig herstellbar und in einem Reaktionsgemisch möglichst leicht von anderen Bestandteilen abtrennbar ist. Die Katalysatoren sollen zudem auch in Abwesenheit eines Lösungsmittels hohe Umsatzraten ermöglichen.
Hauptmerkmale der Erfindung sind jeweils im kennzeichnenden Teil von Anspruch 1 , sowie in Anspruch 12 und 17 angegeben. Ausgestaltungen sind Gegenstand der jeweiligen Unteransprüche.
Bei einem Verfahren zur Herstellung eines Dialkylcarbonats, bei welchem ein Alkanol mit Kohlenmonoxid und Sauerstoff in Anwesenheit eines Kupfer- haltigen Katalysators, der auf einem Festphasenträger geträgert ist, umgesetzt wird, sieht die Erfindung vor, dass der Katalysator auf dem Festphasenträger unter Verfahrensbedingungen in einer flüssigen Katalysatorphase vorliegt, und dass der Katalysator wenigstens eine Kupfer-haltige Koordinationsverbindung umfasst.
Auf Festphasen geträgerte Katalysatoren können besonders leicht von weiteren Bestandteilen eines Reaktionsgemisches abgetrennt werden. Durch die leichte Abtrennbarkeit entfallen insbesondere bei der Produktgewinnung zusätzliche Reinigungsschritte wie Extraktion oder Chromatographie. Folglich können mit Hilfe der Trägerung sowohl der Zeitaufwand als auch der bei dem Verfahren erforderliche Materialeinsatz deutlich reduziert werden.
Durch das Aufbringen des Kupfer-haltigen Katalysators auf einen porösen Träger wird die Oberfläche der Katalysatorphase und damit die bei der Reaktion zur Verfügung stehende Austauschfläche zwischen der Katalysatorphase und den Edukten und Produkten signifikant erhöht. Auf diese Weise können bei dem Verfahren mit einem einfachen Mittel sehr hohe Reaktionsgeschwindigkeiten und somit hohe Umsätze erzielt werden.
Bei der Verwendung von Katalysatoren, die keine derart große Oberfläche aufweisen müssen für gewöhnlich Reaktoren verwendet werden, die kostenauf- wendige und störanfällige Rühreinrichtungen besitzen. Dies kann durch das erfindungsgemäße Verfahren vermieden werden.
Des Weiteren kann der geträgerte Katalysator in einen Festbettreaktor eingebracht und beispielsweise in einem kontinuierlichen Verfahren eingesetzt wer- den. Auf diese Weise können bei dem Verfahren die bekannten Vorteile einer kontinuierlichen Reaktionsführung wie z.B. verbesserte Reaktionskontrolle,
kleineres Reaktionsvolumen, geringer Platzbedarf des Reaktors usw. genutzt werden.
Durch das verhältnismäßige geringe Volumen des auf dem Festphasenträger geträgerten Katalysators können auch die Korrosionsprobleme verringert werden, was sich ebenfalls vorteilhaft auf die Kosten des Verfahrens auswirkt.
Als Trägermaterialien eignen sich besonders poröse anorganische und organische Substanzen. Als anorganische Träger kommen SiO2, AI2O3, Zeolithe und Kohlenstoff in Frage. Als organische Trägermaterialien eignen sich Polymere, wie z. B. Polystyrol. Eine Übersicht über Trägermaterialien und deren Anwendungen bieten Dioos et at, und Leadbeater et at. (B. M. L. Dioos, I. F. L.
Vankelecom, P. A. Jacobs, Adv. Synth. Catal. 2006, 348, , 1413; N. E. Leadbeater, M. Marco, Chem. Rev. 2002, 102, , 3217.).
Mit Hilfe der flüssigen Katalysatorphase kann in mehrphasigen Systemen in vorteilhafter Weise ein Mittel bereitgestellt werden, welches das Austragen des Wassers von der Katalysatorphase in eine Edukt/Produkt-Phase zusätzlich fördert. Dazu kann z.B. die Polarität der Katalysatorphase derart gestaltet werden, dass ein hydrophober Charakter resultiert, aufgrund dessen das Austragen des Wassers forciert wird. So können beispielsweise der Kupfer-haltige Katalysator, oder ein in der flüssigen Katalysatorphase enthaltenes Additiv, einen hydrophoben bzw. lipophilen Charakter aufweisen. Durch die flüssige Katalysatorphase kann folglich mit einem vergleichsweise einfachen Mittel sichergestellt werden, dass das erfindungsgemäße Verfahren auch bei langen Laufzeiten einen hohen Umsatz liefert. Durch die Polarität kann in mehrphasigen kontinuierlichen Systemen zudem die Querlöslichkeit beeinflusst werden.
Die Tatsache, dass bei dem Verfahren ein Kupfer-haltiger Katalysator verwen- det wird, der in einer flüssigen Katalysatorphase vorliegt, schafft zudem eine wichtige Vorraussetzung dafür, dass bei dem Verfahren hohe Diffusionsraten zwischen der Katalysatorphase und den Edukten und Produkten erreicht wer-
den können. Auf diese Weise können die Edukte leicht in die flüssige Katalysatorphase diffundieren und dort rasch mit Hilfe des Katalysators umgesetzt werden, so dass bei dem Verfahren hohe Umsatzraten erreicht werden können. Durch das Vorhandensein einer flüssigen Katalysatorphase auf einem Festphasenträger kann zudem ermöglicht werden, dass das erfindungsgemäße Verfahren auch in einem 3-Phasensystem mit einer gasförmigen Edukt/Produkt-Phase durchgeführt werden kann. Es ist auch deshalb besonders vorteilhaft, da bei einer geeigneten Wahl der Verfahrensbedingungen auch das als Nebenprodukt entstehende Wasser gasförmig ist. Auf diese Weise kann es leicht aus der Katalysatorphase ausgetragen werden, so dass eine Deaktivierung des Katalysators und eine damit verbundene Verringerung des Umsatzes wirksam vermieden werden kann. Von großer Bedeutung bei dem erfindungsgemäßen Verfahren ist zudem, dass der Katalysator eine Kupfer-haltige Koordinationsverbindung umfasst. Eine Koordinationsverbindung im Sinne der Erfindung schließt sowohl Neutralkomplexe als auch die Salze kationischer und anionischer Komplexe ein. Kupfer-haltige Koordinationsverbindungen haben sich als z.T. sehr effiziente Katalysatoren bei der Herstellung von Dialkylcarbonaten und erwiesen.
Übersichtsartikel zu den Vorteilen der Verwendung von Katalysatoren, die in einer flüssigen Katalysatorphase vorliegen, welche auf einem Festphasenträger geträgert ist, bei der Durchführung katalytischer Reaktionen finden sich bei- spielsweise bei Reek et al. (J. N. H. Reek, P. W. N. M. van Leeuwen, A. G. J. van der Harn, A. D. de Haan, Supported Catalysts, in Catalyst Separation, Recovery and Recycling (Ed.: D. J. Cole-Hamilton, R. P. Tooze), Springer, 2006), bei Wight und Davies (A. P. Wight, M. E. Davies, Design and Preparation of Organic-Inorganic Hydrid Catalysts, Chem. Rev. 2002, 102, 3589.) und bei Cornils et at. (B. Cornils et at. (Ed.) Multiphase homogeneous catalysis, Wiley- VCH, 2005.
Eine besonders wichtige Ausführungsform sieht vor, dass die Kupfer-haltige Koordinationsverbindung einen Schmelzpunkt kleiner 120 °C, bevorzugt kleiner 1 15 °C, besonders bevorzugt kleiner 100 °C und ganz besonders bevorzugt kleiner 80 °C aufweist. Dies ist besonders vorteilhaft, da das Verfahren dadurch auch bei verhältnismäßig geringen Temperaturen durchgeführt werden kann, ohne dass der Einsatz eines Lösungsmittels für das Bereitstellen der unter Verfahrensbedingungen flüssigen Katalysatorphase notwendig ist. Diese Eigenschaft schafft somit eine wichtige Voraussetzung dafür, dass das gesamte Verfahren in Abwesenheit eines Lösungsmittels durchgeführt werden kann.
Lösungsmittel sind häufig toxisch und/oder brennbar, so dass ihr Einsatz üblicherweise mit einer Erhöhung des Gefahrenpotentials eines Verfahrens verbunden ist. Des Weiteren hat die Verwendung von Lösungsmitteln einen negativen Einfluss auf die Atomökonomie eines Produktionsprozesses und ist des- halb mit erhöhten Verfahrenskosten verbunden. Bei einer lösungsmittelfreien Durchführung des Verfahrens kann dies vermieden werden. Die Möglichkeit einer derartigen Verfahrensführung ist daher besonders attraktiv.
In einer besonders vorteilhaften und einfachen Ausgestaltung bildet daher die wenigstens eine Kupfer-haltige Koordinationsverbindung die flüssige Katalysatorphase.
Eine bevorzugte Ausführungsform des erfindungsgemäßen Verfahrens sieht vor, dass die Kupfer-haltige Koordinationsverbindung mindestens eine Stick- stoff(lll)-Verbindung als AADonorligand und/oder eine quarternisierte Stick- stoff(lll)-Verbindung als organisches Kation enthält, wobei die Stickstoff(lll)- Verbindung ausgewählt ist aus der Gruppe von A/-Alkylimidazolen, N,N- Dialkylaminopyridinen, AAAlkylpyrazolen oder AAAlkyl-pentaorganoguanidinen. Diese Ausgestaltung ist besonders vorteilhaft, da die genannten Kupfer-haltigen Koordinationsverbindungen in Verfahren zur Herstellung von Dialkylcarbonaten durch katalytische oxidative Carbonylierung eines Alkohols besonders gute Ergebnisse liefern.
Durch die Verwendung einer Stickstoff(lll)-Verbindung als AADonorligand kann die Lewis-Acidität des Kupfer-haltigen Katalysators verringert werden. Durch die Verringerung der Lewis-Acidität wird die Fähigkeit des Katalysators, auch die unerwünschte Hydrolyse des Dialkylcarbonats zu katalysieren, eingeschränkt. Durch diese Ausgestaltung kann daher die Selektivität der katalytischen Reaktion erhöht und somit die Effizienz des Verfahrens gesteigert werden.
In einer besonders vorteilhaften Ausführungsform enthält die Kupfer-haltige Ko- ordinationsverbindung mindestens ein A/-Alkylimidazol als AADonorligand und/oder eine quarternisiertes A/-Alkylimidazol, d.h. ein A/-Alkylimidazolium-lon, als organisches Kation. Auf diese Weise konnten bei einem Verfahren zur Herstellung von Dialkylcarbonaten besonders gute Umsätze erzielt werden. Des Weiteren ist bei einem erfindungsgemäßen Verfahren bevorzugt, dass die Kupfer-haltige Koordinationsverbindung wenigstens einen lipophilen Substituen- ten aufweist. Vorteilhaft dabei ist insbesondere, dass ein lipophiler Substituent wie z.B. ein langer Alkyl-Substituent sehr leicht zur Verfügung gestellt werden kann und aufgrund seiner Polarität dazu geeignet ist, das Austragen des Was- sers aus der Katalysatorphase zu fördern. Auf diese Weise kann der Deaktivie- rung des Katalysators entgegengewirkt werden.
Der lipophile Substituent ist bevorzugt ein aliphatischer Substituent mit einer Anzahl von 1 bis 25 Kohlenstoffatomen und ganz besonders bevorzugt ein Al- kyl-Substituent mit 1 bis 25 Kohlenstoffatomen. Der lipophile Substituent kann zudem seinerseits Substituenten wie z.B. Alkylketten aufweisen.
Vorteilhaft bei dieser Ausgestaltung ist insbesondere, dass der lipophile Substituent an einem Bestandteil des Katalysators lokalisiert ist, dadurch wird einer Deaktivierung des Katalysators durch Wasser unmittelbar entgegengewirkt.
Eine besonders wichtige Ausführungsform sieht vor, dass der lipophile Substi- tuent an der Stickstoff(lll)-Verbindung vorgesehen ist. Bevorzugt ist weiterhin, dass der lipophile Substituent am Stickstoff der Stickstoff(lll)-Verbindung angeordnet ist. Durch diese Ausgestaltung konnten bei Verfahren zur Herstellung von Dialkylcarbonaten besonders hohe Ausbeuten erzielt werden.
Vorteilhaft ist weiterhin, dass durch die Einführung des lipophilen Substituenten insbesondere bei Kupfer-haltigen Koordinationsverbindungen, die mindestens ein A/-Alkylimidazol aufweisen, häufig der Schmelzpunkt der Koordinationsver- bindungen gesenkt werden kann. So kann eine Kupfer-haltige Koordinationsverbindung einfach zur Verfügung gestellt werden, die unter Verfahrensbedingungen flüssig ist.
Eine bevorzugte Ausgestaltung eines erfindungsgemäßen Verfahrens sieht zu- dem vor, dass gasförmige Edukte mit der Katalysatorphase in Kontakt gebracht werden, und dass die Produkte über die Gasphase abgeführt werden. Ein Produkt ist Wasser. Vorteilhaft bei dieser Ausführungsform ist, dass das Wasser im gasförmigen Zustand leicht aus der Katalysatorphase ausgetragen werden kann. Durch die genannte Ausgestaltung kann daher verhindert werden, dass der Katalysator durch das als Nebenprodukt entstehende Wasser deaktiviert und dadurch der Umsatz verringert wird.
In einer besonders wichtigen Ausführungsform eines erfindungsgemäßen Verfahrens ist der Katalysator eine ionische Flüssigkeit, wobei die Kupfer-haltige Koordinationsverbindung im Anion und/oder im Kation der ionischen Flüssigkeit enthalten ist. Dies ist besonders geeignet, da bei derart ausgestalteten Verfahren besonders gute Dialkylcarbonat-Ausbeuten erreicht werden konnten.
Ionische Flüssigkeiten sind Salze, die bereits bei einer Temperatur von <100 °C schmelzen. Sie setzen sich üblicherweise aus einem organischen Kation und einem organischen oder anorganischen Anion zusammen. Sie zeichnen sich durch eine Reihe vorteilhafter Eigenschaften aus. Beispielsweise sind sie nicht
entzündlich, besitzen einen sehr geringen Dampfdruck und verfügen zudem über sehr gute Lösungseigenschaften. Auf diese Weise kann sichergestellt werden, dass die beim Verfahren verwendeten Edukte in der Katalysatorphase löslich sind. Vorteilhaft ist weiterhin, dass ionische Flüssigkeiten einen niedrigen Schmelzpunkt und häufig auch eine relativ hohe thermische Stabilität aufweisen. Somit kann das Verfahren erfindungsgemäß in einem breiten Temperaturbereich durchgeführt werden.
Ionische Flüssigkeiten, bei denen die Kupfer-haltige Koordinationsverbindung im Anion und/oder im Kation der ionischen Flüssigkeit enthalten ist, wobei die Kupfer-haltige Koordinationsverbindung mindestens ein A/-Alkylimidazol als N- Donorligand und/oder ein quarternisiertes A/-Alkylimidazol als organisches Kation umfasst, zeigen im Allgemeinen eine hohe thermische Stabilität. Dies ist besonders vorteilhaft, da dadurch auch bei langen Reaktionszeiten keine Zerset- zung des Katalysators erfolgt, so dass das Verfahren konstante Umsätze liefert.
Besonders gute Dialkylcarbonat-Ausbeuten können zudem dadurch erreicht werden, dass das Anion der ionischen Flüssigkeit entweder eine Kupfer-haltige Koordinationsverbindung darstellt oder aus [CuCI2]~, [CuBr2]~, [Cul2]~, [BF4]" und [PF6]" ausgewählt ist.
In einer ganz besonders bevorzugten Ausführungsform des Verfahrens ist die Kupfer-haltige Koordinationsverbindung ausgewählt aus den ionischen Flüssigkeiten [Cu(lm12)4][PF6], [Cu(lm12)2][CuCI2], [Cu(lm12)2][CuBr2], [Cu(lm6)2][CuCI2], [Cu(lm6)2][CuBr2], [DMIM][CuCI2], [DMIM][CuCI2] · [DMIM]2[CuCI3],
[DMIM][CuBr2], [DMIM]2[CuCI4], [DMIM]2[CuBr4] oder den neutralen Komplexen [Cu(lm12)l]6, [CuCI2(lm10)2], [CuBr2(lm12)4], [Cu4OCI6(lm11)4)].
Eine wichtige Ausgestaltung des Verfahrens sieht vor, dass verzweigte und un- verzweigte Alkanole mit 1 bis 4 Kohlenstoffatomen umgesetzt werden. Ganz besonders bevorzugt ist dabei die katalytische oxidative Carbonylierung von Methanol zu Dimethylcarbonat.
Eine wichtige Ausführungsform sieht vor, dass das Verfahren in einem Temperaturbereich von 60 °C bis 200 °C, bevorzugt von 70 °C bis 140 °C, besonders bevorzugt von 75 °C bis 120 °C und ganz besonders bevorzugt von 80 °C bis 1 15 °C durchgeführt wird. Insbesondere bei Temperaturen unterhalb von
120 °C kann die Bildung von unerwünschten und aufgrund ihrer speziellen physikalischen und toxischen Eigenschaften problematischen Nebenprodukten wie Methylchlorid und Methyldimetoxymethan vermieden werden. Eine weitere wichtige Ausführungsform sieht vor, dass das Verfahren als kontinuierliches Verfahren durchgeführt wird. Kontinuierliche Syntheseführungen sind bekannt. Einem erfindungsgemäßen Verfahren können sämtliche der bekannten Ausführungsprotokolle zu Grunde gelegt werden. Der Katalysator kann beispielsweise in einem Festbettreaktor angeordnet werden, der von dem übri- gen Reaktionsgemisch, d.h. von dem umzusetzenden Alkohol, dem Kohlenmo- noxid und dem Sauerstoff, kontinuierlich durchströmt wird.
Eine besonders vorteilhafte Weiterbildung sieht dabei vor, dass das kontinuierliche Verfahren in der Gasphase durchgeführt wird. Im Allgemeinen wird bei der Durchführung des Verfahrens als Gasphasenreaktion bevorzugt ein Gesamtdruckbereich von 1 bar bis 100 bar und besonders bevorzugt ein Gesamtdruckbereich von 5 bis 50 bar verwendet.
Eine weitere Ausführungsform des Verfahrens sieht vor, dass in der flüssigen Katalysatorphase ein Additiv enthalten ist. Das Additiv kann in vorteilhafter Weise dazu verwendet werden, um insbesondere die physikalischen Eigenschaften wie die Viskosität der Katalysatorphase oder die Eduktloslichkeit (insbesondere die Löslichkeit des Alkanols) optimal auf die Verfahrensbedingungen abzustimmen. Bei dem Additiv handelt es sich bevorzugt um eine schwerflüchtige Ver- bindung wie z.B. ein Silikonöl oder ein perfluoriertes Polyetheröl.
Ein besonders bevorzugtes Verfahren zur Herstellung von Dialkylcarbonaten sieht vor, dass der Katalysator durch nicht-kovalente Wechselwirkung auf den Festphasenträger aufgebracht ist. Vorteilhaft ist hierbei, dass bei der Herstellung des bei dem Verfahren verwendeten Katalysators die Anknüpfung dessen an den Festphasenträger entfällt. Die Trägerung des Katalysators ist somit vergleichsweise einfach. Im Gegensatz zu einer kovalenten Trägerung muss bei einem nicht-kovalent geträgerten Katalysator kein spezieller Linker für das Aufbringen des Katalysators auf den Festphasenträger eingeführt werden. Dies reduziert die Kosten für die Herstellung des Katalysators und damit auch die Kosten des gesamten Verfahrens.
In einer wichtigen Ausführungsform des Verfahrens wird der Festphasenträger von SiO2 gebildet. SiO2 ist ein kostengünstiges Trägermaterial, das in vielen verschiedenen Ausführungen bezüglich seiner Form, seiner Partikelgröße und seiner Oberflächenbeschaffenheiten verfügbar ist. Das Trägermaterial kann ferner in einem weiten Temperaturbereich eingesetzt werden.
In einer wichtigen Ausgestaltung des Verfahrens wird der Festphasenträger von SiO2 mit einer Partikelgröße zwischen 0,05 mm und 4 mm und einer BET- Oberfläche von 250 - 1000m2/g gebildet. Besonders bevorzugt ist die Verwendung eines Festphasenträgers aus SiO2, das eine Partikelgröße zwischen 0,063 mm und 0,2 mm und eine BET-Oberfläche von 300 - 400m2/g aufweist.
Es ist weiterhin vorgesehen, dass bei dem Verfahren ein auf einem Festpha- senträger geträgerter Kupfer-haltiger Katalysator verwendet wird, bei welchem das Massenverhältnis des Kupfer-haltigen Katalysators zum Festphasenträger zwischen 0,075 und 1 ,350, besonders bevorzugt zwischen 0,150 und 0,750 und ganz besonders bevorzugt zwischen 0,300 und 0,590 liegt. Die Erfindung sieht zudem einen Kupfer-haltigen Katalysator vor, der auf einem Festphasenträger geträgert ist, und der in einer Katalysatorphase vorliegt, die einen Schmelzpunkt kleiner 120°C aufweist, und der wenigstens eine Kupfer-
haltige Koordinationsverbindung umfasst, wobei die Kupfer-haltige Koordinationsverbindung mindestens eine Stickstoff(lll)-Verbindung als AADonorligand und/oder eine quarternisierte Stickstoff(lll)-Verbindung als organisches Kation enthält, wobei die Stickstoff(lll)-Verbindung ausgewählt ist aus der Gruppe von A/-Alkylimidazolen, A/,A/-Dialkylaminopyridinen, AAAlkylpyrazolen oder AAAlkyl- pentaorganoguanidinen.
Aufgrund der Tatsache, das der Kupfer-haltige Katalysator auf einem Festphasenträger geträgert ist und in einer Katalysatorphase vorliegt, die einen
Schmelzpunkt kleiner 120 °C aufweist, kann ein Katalysator zur Verfügung gestellt werden, der bei der Verwendung des Katalysators in einem katalytischen Verfahren besonders leicht von weiteren Bestandteilen des Reaktionsgemischs abgetrennt werden kann.
Durch die leichte Abtrennbarkeit können insbesondere bei der Produktisolation zusätzliche Reinigungsschritte wie Extraktion oder Chromatographie entfallen. Auf diese Weise können durch den Einsatz eines erfindungsgemäßen Katalysators in einem katalytischen Verfahren, sowohl der Zeitaufwand, als auch der erforderliche Materialeinsatz, deutlich reduziert werden.
Durch das Aufbringen des Kupfer-haltigen Katalysators auf einen porösen Träger wird die Oberfläche der Katalysatorphase und damit die bei der Reaktion zur Verfügung stehende Austauschfläche zwischen der Katalysatorphase und den Edukten und Produkten signifikant erhöht. Auf diese Weise können bei der Verwendung eines erfindungsgemäßen Katalysators in einem katalytischen Verfahren sehr hohe Reaktionsgeschwindigkeiten und somit hohe Umsätze erzielt werden.
Vorteilhaft ist weiterhin, dass bei einem erfindungsgemäßen Katalysator eine flüssige Katalysatorphase vorgesehen ist. Dadurch kann eine wichtige Voraussetzung dafür geschaffen werden, dass hohe Diffusionsraten zwischen der Katalysatorphase und den Edukten und Produkten erreicht werden können. Infol-
gedessen können die Edukte leicht in die flüssige Katalysatorphase diffundieren und dort rasch mit Hilfe des Katalysators umgesetzt werden, so dass bei der Verwendung eines erfindungsgemäßen Katalysators in einer katalytischen Reaktion hohe Umsatzraten erreicht werden können.
Des Weiteren wird durch die Tatsache, dass der Kupfer-haltige Katalysator in einer Katalysatorphase vorliegt, die einen Schmelzpunkt kleiner 120°C aufweist, auch eine wichtige Voraussetzung dafür geschaffen, dass der Katalysator in einem Verfahren eingesetzt werden kann, bei dem die katalytische Reaktion bereits bei verhältnismäßig geringen Temperaturen auch in Abwesenheit eines Lösungsmittels in einer flüssigen Phase stattfindet.
Lösungsmittel sind häufig toxisch und/oder brennbar, so dass ihr Einsatz üblicherweise mit einer Erhöhung des Gefahrenpotentials eines Verfahrens ver- bunden ist. Des Weiteren hat die Verwendung von Lösungsmitteln einen negativen Einfluss auf die Atomökonomie eines Verfahrens und ist deshalb mit erhöhten Kosten verbunden. Bei einer lösungsmittelfreien Reaktionsführung kann dies vermieden werden. Ein Katalysator, der dies ermöglicht, ist daher besonders attraktiv.
Eine weitere besonders wichtige Ausführungsform sieht vor, dass die Katalysatorphase einen Schmelzpunkt kleiner 120°C, bevorzugt kleiner 1 15°C, besonders bevorzugt kleiner 100°C und ganz besonders bevorzugt kleiner 80°C aufweist. Je niedriger der Schmelzpunkt der flüssigen Phase, bei desto geringeren Temperaturen kann der Katalysator auch ohne den Zusatz eines Lösungsmittels in flüssiger Phase eingesetzt werden.
Eine besonders vorteilhafte Weiterbildung sieht vor, dass die Kupfer-haltige Koordinationsverbindung einen Schmelzpunkt kleiner 120°C, bevorzugt kleiner 1 15°C, besonders bevorzugt kleiner 100 °C und ganz besonders bevorzugt kleiner 80 °C aufweist. Auf diese Weise kann besonders leicht eine Katalysatorpha-
se zur Verfügung gestellt werden, die einen Schmelzpunkt kleiner 120°C, bzw. kleiner 1 15°C, kleiner 100°C oder kleiner 80 °C aufweist.
Des Weiteren ist bevorzugt, dass der Katalysator wenigstens einen lipophilen Substituenten aufweist. Dies ist besonders vorteilhaft, da durch die Einführung des lipophilen Substituenten der Schmelzpunkt des Katalysators beeinflusst werden kann.
Vorteilhaft ist weiterhin, dass durch den lipophilen Substituenten die Polarität des Katalysators auf dessen Verwendungszweck abgestimmt werden kann. Bei einer Verwendung des Katalysators in einem heterogenen System mit einer polaren Edukt- und/oder Produkt-Phase kann durch den lipophilen Substituenten die Querlöslichkeit in der polaren Phase verringert werden. Auf diese Weise kann ein Ablösen des Katalysators von dem Festphasenträger bzw. ein Austra- gen des Katalysators aus der Katalysatorphase verhindert werden.
Der lipophile Substituent ist bevorzugt ein aliphatischer Substituent mit einer Anzahl von 1 bis 25 Kohlenstoffatomen und ganz besonders bevorzugt ein Al- kyl-Substituent mit 1 bis 25 Kohlenstoffatomen. Der lipophile Substituent kann unter Umständen seinerseits ebenfalls Substituenten wie z.B. Alkylketten aufweisen. Eine besonders wichtige Ausführungsform sieht vor, dass der lipophile Substituent an der Stickstoff(lll)-Verbindung vorgesehen ist. Dies ist besonders vorteilhaft, da Katalysatoren, die derart aufgebaut sind, häufig einen Schmelzpunkt kleiner 120°C aufweisen.
Eine besonders wichtige Ausführungsform sieht vor, dass der Katalysator eine ionische Flüssigkeit ist, wobei die Kupfer-haltige Koordinationsverbindung im Anion und/oder im Kation der ionischen Flüssigkeit enthalten ist. Ionische Flüssigkeiten, bei denen die Kupfer-haltige Koordinationsverbindung im Anion und/oder im Kation der ionischen Flüssigkeit enthalten ist, und insbesondere solche, bei denen die Kupfer-haltige Koordinationsverbindung mindes-
tens ein A/-Alkylimidazol als AADonorligand und/oder ein quarternisiertes N- Alkylimidazol als organisches Kation umfasst, zeigen im Allgemeinen eine hohe thermische Stabilität. Dies ist besonders vorteilhaft, da dadurch auch bei langen Reaktionszeiten keine Zersetzung des Katalysators erfolgt, so dass das Verfah- ren konstante Umsätze liefert.
Eine besonders hohe thermische Stabilität konnte zudem erreicht werden, wenn das Anion der ionischen Flüssigkeit entweder eine Kupfer-haltige Koordinationsverbindung darstellt oder aus [CuCI2]~, [CuBr2]~, [Cul2]~, [BF4]" und [PF6]" aus- gewählt ist.
Bei erfindungsgemäßen Katalysatoren besteht zudem die Möglichkeit, die Eigenschaften (z.B. Eduktlöslichkeit, Fließeigenschaften) der Katalysatorphase durch Zugabe weiterer, schwerflüchtiger Komponenten zu beeinflussen. Diese Komponenten, wie z.B. Silikonöle und perfluorierte Polyetheröle, werden vor der Aufbringung des Flüssigkeitsfilms auf den Träger mit der katalytisch aktiven Flüssigkeit vermischt und anschließend ggf. unter Zuhilfenahme eines Lösungsmittels auf das Trägermaterial aufgebracht. In einer besonders bevorzugten Ausführungsform ist der Katalysator durch nicht-kovalente Wechselwirkung auf einen Festphasenträger aufgebracht ist. Vorteilhaft hierbei ist insbesondere, dass keine speziellen Maßnahmen für das Aufbringen notwendig sind. So muss beispielsweise kein spezieller Substituent für das Aufbringen der Katalysators eingeführt werden. Dies reduziert die Kos- ten für die Herstellung des Katalysators.
In einer weiteren wichtigen Ausführungsform wird der Festphasenträger des Katalysators von SiO2 gebildet. SiO2 ist ein kostengünstiges Trägermaterial, das in vielen verschiedenen Partikelgrößen und Oberflächenbeschaffenheiten ver- fügbar ist. Das Trägermaterial ist zudem ebenfalls in einem weiten Temperaturbereich einsetzbar, dies wirkt sich vorteilhaft auf die Einsatzmöglichkeiten des Katalysators aus.
In einer bevorzugten Ausgestaltung des erfindungsgemäßen Katalysators wird der Festphasenträger bevorzugt von SiO2 mit einer Partikelgröße zwischen 0,05 und 4 mm und einer BET-Oberfläche von 250 - 1000m2/g gebildet. Besonders bevorzugt ist ein Träger aus SiO2 mit einer Partikelgröße zwischen 0,063 - 0,2 mm und einer BET-Oberfläche von 300 - 400m2/g.
Es ist weiterhin bevorzugt, dass das Massenverhältnis des Kupfer-haltigen Katalysators zum Festphasenträger zwischen 0,075 - 1 ,350 und besonders be- vorzugt zwischen 0,150 - 0,750 und ganz besonders bevorzugt zwischen 0,300 - 0,590 liegt.
In einer ganz besonders bevorzugten Ausführungsform ist die Kupfer-haltige Koordinationsverbindung ausgewählt aus den ionischen Flüssigkeiten
[Cu(lm12)4][PF6], [Cu(lm12)2][CuCI2], [Cu(lm12)2][CuBr2], [Cu(lm6)2][CuCI2],
[Cu(lm6)2][CuBr2], [DMIM][CuCI2], [DMIM][CuCI2] · [DMIM]2[CuCI3],
[DMIM][CuBr2], [DMIM]2[CuCI4], [DMIM]2[CuBr4] oder den neutralen Komplexen [Cu(lm12)l]6, [CuCI2(lm10)2], [CuBr2(lm12)4], [Cu4OCI6(lm11)4)]. Die Erfindung sieht zudem eine Verwendung eines erfindungsgemäßen Katalysators bei einer oxidativen Carbonylierung vor. Die ist besonders vorteilhaft, da die erfindungsgemäßen Katalysatoren bei der Verwendung in derartigen Reaktionen zu besonders hohen Umsätzen geführt haben. Die erfindungsgemäßen Katalysatoren sind für eine derartige Anwendung besonders geeignet, da sie zum einen einen Kupfer-Komplex und zum anderen eine hohe thermische Stabilität aufweisen. Oxidative Carbonylierungen können von Kupfer-Komplexen katalysiert werden. Hinzu kommt, dass diese Reaktionen ebenfalls häufig bei erhöhten Temperaturen durchgeführt werden.
Besonders bevorzugt ist die Verwendung eines erfindungsgemäßen Katalysators bei einer oxidativen Carbonylierung von Alkanolen in Gegenwart von Sau-
erstoff und Kohlenmonoxid zu Dialkylcarbonaten. Ganz besonders bevorzugt ist die Verwendung eines erfindungsgemäßen Katalysators bei einer oxidativen Carbonylierung von verzweigten und unverzweigten Alkoholen mit 1 bis 4 Kohlenstoffatomen zu Dialkylcarbonaten. Insbesondere konnten mit den erfin- dungsgemäßen ionischen Flüssigkeiten gute Ergebnisse bei der oxidativen Carbonylierung von Methanol zu Dimethylcarbonat erzielt werden.
Ein besonders einfaches Verfahren zur Herstellung des erfindungsgemäßen Katalysators sieht vor, dass entweder die Kupfer-haltige Koordinationsverbin- dung selbst, oder bei einer in situ Herstellung dieser, die Komponenten die zur Herstellung der Kupfer-haltige Koordinationsverbindung erforderlich sind, unter Schutzgas und gegebenenfalls bei erhöhter Temperatur und/oder unter Schütteln und/oder Einwirkung von Ultraschall auf den Festphasenträger aufgebracht werden. Unter Umständen kann zudem ein Lösemittel verwendet werden, wel- ches mit der bzw. den entsprechenden Komponente/n des Katalysators eine Lösung bildet. Lösungsmittel wie Acetonitril, Methanol, Tetrahydrofuran oder Dichlormethan sind dafür besonders gut geeignet. Der Festphasenträger wird mit einem Überschuss der genannten Lösung versetzt. Nach dem Stoffaustausch zwischen dem Festphasenträger und der Lösung wird die überschüssige Lösung entfernt. Eine andere Methode ist die Besprühung des Festphasenträgers mit einer solchen Lösung. Gegebenenfalls wird ein zugesetztes Lösemittel während oder nach dem Sprühvorgang entfernt. Bevorzugt wird der auf einem Festphasenträger geträgerte Katalysator durch Versetzen des Festphasenträgers mit einer Lösung des Katalysators, bzw. bei einer in situ Herstellung mit einer Lösung der entsprechenden Komponenten, in einem Lösemittel und anschließendem Verdampfen des Lösemittels hergestellt.
Weitere Merkmale, Einzelheiten und Vorteile der Erfindung ergeben sich aus dem Wortlaut der Ansprüche sowie aus den folgenden Beschreibungen von Ausführungsbeispielen anhand der Abbildungen. Darin zeigt:
Fig. 1 eine schematische Darstellung eines kontinuierlichen Gasphasenreaktors.
Figur 1 zeigt eine schematische Darstellung eines kontinuierlichen Gasphasen- reaktors. Ein derartiger Reaktor wurde zur Durchführung der in den Ausführungsbeispielen in Tabelle 3 beschriebenen kontinuierlichen Verfahren verwendet. Die Dosierung der Gase (bei einer Reaktion gemäß Tabelle 3, Kohlenmo- noxid und synthetische Luft) erfolgt über Massendurchflussregler 1 . Der Anlagendruck wird am Druckregelventil 7 eingestellt. Das flüssige Edukt (bei einer Reaktion gemäß Tabelle 3, Methanol) wird über eine HPLC-Kolbenpumpe 2 gefördert und in einem beheizten Verdampfer 3 in den gasförmigen Zustand gebracht. Bei dem Festbettreaktor 4 handelt es sich um ein Edelstahlrohr. Bei Bedarf kann der Volumenstrom der Reaktionsgase durch einen zweiten, baugleichen Reaktor 5 oder über einen Bypass 6 am Reaktor vorbei geleitet wer- den. Alle Rohrleitungen der Versuchsapparatur sind elektrisch beheizbar. Die Detektion der Produkte erfolgt durch einen online-Gaschromatographen 8 mit Restek RTX-5-Säule und Flammenionisationsdetektor.
Ausführungsbeispiele:
Aus Gründen der Übersichtlichkeit wird 1 -Dodecylimidazol nachfolgend mit „Im12", 1 -Undecylimidazol mit„Im11", 1 -Decylimidazol mit„Im10", 1 -Hexylimidazol mit„Im6" und das 1 -Dodecyl-3-methylimidazolium-Kation als„DMIM" abgekürzt. A. Darstellung der Kupfer-haltigen Koordinationsverbindungen
1 . Ionische Flüssigkeiten
1 .1 [Cu(lm12)4][PF6] (I)
In einem Schlenkrohr wurde zu [Cu(CH3CN)4][PF6] (186 mg, 0,50 mmol) 1 - Dodecylimidazol (Im12) (473 mg, 2,00 mmol) gegeben. Die Reaktionsmischung
wurde bei Raumtemperatur für ca. 10 min mit Ultraschall behandelt. Aus dem Cu(l)-Salz freigewordenes CH3CN wurde im Vakuum entfernt und der farblose viskose Rückstand wurde gefriergetrocknet. [Cu(lm12)4][PF6] (I) (574 mg, 99%) wurde als farbloser Feststoff erhalten (gefunden: C, 62,15; H, 9,74; N, 10,13. C60H112CuF6N8P berechnet C, 62,44; H, 9,78; N, 9,71 %); Smp. 27,0 °C (aus MeOH), Zers. 234,0 °C; 4(300,1 MHz; CD3CN) 0,88 (3 H, t, CH3), 1 ,26 (18 H, br s, CH2(8-16)), 1 ,68-1 ,84 (2 H, m, CH2(7)), 3,98 (2 H, t, CH2(6)), 6,75-7,40 (2 H, m, CH(4-5)) und 7,67 (1 H, br s, CH(2)); 4(75,5 MHz; CD3CN) 14,4 (CH3), 23,4, 27,1 , 29,7, 30,1 , 30,2, 30,3, 30,4, 31 ,5, 32,7 (CH2(7-16)) und 48,1
(CH2(6)); m/z (ESI; CH3CN) 237,2325 ([Ci5H28N2 +H]\ 47%. Ci5H29N2 berechnet 237,2325), 340 (56) und 535,3793 ([C30H56CuN4]+, 100. C30H56CuN4 berechnet 535,3796); m/z (ESI, negativer Modus; CH3CN) 144,9645 ([PF6]~, 100%. F6P berechnet 144,9647) und 313 ([2 PF6 + Na]", 43). IR (Substanz) v— /cm "' = 3138w, 2922vs, 2852vs, 1684w, 1516s, 1465s, 1403, 1376, 1284, 1236s, 1 108s, 1088s, 1032, 836vs, 732s, 661 s, 627 und 557s. Für die Einkristallstrukturuntersuchung geeignete Kristalle von I wurden aus MeOH bei Raumtemperatur erhalten, in inertem Öl aufgenommen und in den kalten N2- Kühlgasstrom des Diffraktometers überführt. 1 .2 [Cu(lm12)2][CuCI2] (II)
In einem Schlenkrohr wurde zu CuCI (1 ,21 g, 12,22 mmol) eine Lösung von Im12 (3,06 g, 12,95 mmol) in CH3CN (5 mL) gegeben. Das Reaktionsgemisch wurde ca. 30 min lang mit Ultraschall behandelt. Das Lösungsmittel wurde im Vakuum entfernt, der erhaltene Feststoff mit Et2O gewaschen und anschließend im Vakuum getrocknet. [Cu(lm12)2][CuCI2] (II) (4,06 g, 99%) wurde als feinkristalliner farbloser Feststoff erhalten (gefunden: C, 53,87; H, 8,58; N, 8,45.
C30H56CI2Cu2N4 berechnet: C, 53,72; H, 8,41 ; N, 8,35%); Smp. 73,0 °C (aus CH3CN), Zers. 281 ,4 °C; 4(300,1 MHz; CD3CN) 0,88 (3 H, t, CH3), 1 ,26 (18 H, br s, CH2(8-16)), 1 ,69-1 ,85 (2 H, m, CH2(7)), 4,01 (2 H, t, CH2(6)) und 7,1 1 -8,68 (3 H, m, Im-H); 4(75,5 MHz; CD3CN) 14,4 (CH3), 23,4, 27,0, 29,7, 30,0, 30,1 , 30,2, 30,3, 30,8, 31 ,2, 32,6 (CH2(7-16)) und 48,9 (CH2(6)); m/z (ESI; CH3CN)
237 ([C15H28N2 + H]\ 83%), 340 (80) und 535,3793 ([C30H56CuN4]+, 100.
C30H56CuN4 berechnet 535,3796); m/z (ESI, negativer Modus; CH3CN)
132,8681 ([CuCI2]\ 33%. CI2Cu berechnet 132,8679), 179 (100), 223 (74), 233
(23), 277 (22) und 323 (19). IR (Substanz) ^^' = 3122, 3051 w, 2949, 2916vs, 2847s, 1690w, 161 Ow, 1533, 1520, 1466, 1442, 1399w, 1375, 1357, 1288, 1255w, 1240, 1 1 10s, 1053, 1039, 1026, 1006w, 962, 890w, 845, 833, 757vs, 730, 721 , 654s, 630, 506w, 444w und 428w.
1 .3 [Cu(lm12)2][CuBr2] (III)
Nach derselben Vorgehensweise wie im Falle von II, jedoch unter Verwendung von CuBr■ SMe2 oder CuBr an Stelle von CuCI, wurde [Cu(lm12)2][CuBr2] (III) in quantitativer Ausbeute als farbloser Feststoff erhalten (gefunden: C, 47,44; H, 7,53; N, 7,32. C3oH56Br2Cu2N4 berechnet C, 47,43; H, 7,43; N, 7,38%); Smp. 66,0 °C (aus CH3CN), Zers. 278,3 °C; *(300,1 MHz; CD3CN) 0,88 (3 H, t,
CH3), 1 ,26 (18 H, br s, CH2(8-16)), 1 ,76 (2 H, quin, 3J 7,1 , CH2(7)), 3,99 (2 H, t, 3J 7,2, CH2(6)), 6,78-7,48 (2 H, m, CH(4-5)) und 7,67-8,1 1 (1 H, m, CH(2)); (75,5 MHz; CD3CN) 14.4 (CH3), 23,4, 27,0, 29,7, 30,0, 30,1 , 30,2, 30,3, 30,9, 31 ,3, 32,6 (CH2(7-16)) und 48,8 (CH2(6)); m/z (ESI; CH3CN) 237 ([Ci5H28N2 + H]\ 45%), 340 (24) und 535,3786 ([C30H56CuN4]+, 100. C30H56CuN4 berechnet 535,3796); m/z (ESI, negativer Modus; CH3CN) 222,7651 ([CuBr2]\ 25%. Br2Cu berechnet 222,7648), 264 (57) und 313 (88). IR (Substanz) v-/cm "' = 3122, 31 12, 3049w, 2949, 2916vs, 2847s, 1685w, 1608w, 1533, 1520, 1465, 1442, 1397w, 1376w, 1341 w, 1288, 1255w, 1239, 1 1 10, 1 100, 1053w, 1039w, 1026w, 960w, 889w, 843, 832, 755s, 728, 722, 654s, 630, 505w, 444w und 427w. Für die Einkristallstrukturuntersuchung geeignete Kristalle von III wurden aus CH3N/Et2O bei -20 °C erhalten, in inertem Öl aufgenommen und in den N2- Kühlgasstrom des Diffraktometers überführt. 1 .4 [Cu(lm6)2][CuCI2] (IV)
In einem Schlenkrohr wurde zu CuCI (2,97 g, 30,00 mmol) 1 -Hexylimidazol (Im6) (4,84 g, 31 ,8 mmol) gegeben. Das Reaktionsgemisch wurde zwecks Homogenisierung mit CH3CN (5 ml_) versetzt und ca. 1 0 min lang mit Ultraschall behandelt. Das Lösungsmittel wurde im Vakuum entfernt, die viskose Flüssig- keit mit Ether digeriert. Nach Phasenseparation wurde die Etherphase dekantiert. [Cu(lm6)2][CuCI2] (IV) (7,46 g, 99%) wurde nach Trocknen im Vakuum als farblose, viskose Flüssigkeit erhalten. Glaspunkt -60,3 °C (aus CH3CN);
<5H(300,1 MHZ; CD3CN) 0,87 (3 H, t, CH3), 1 ,28 (6 H, br s, CH2(8-1 0)), 1 ,76 (2
H, quin, 3J 7,1 , CH2(7)), 4,00 (2 H, t, 3J 7,2, CH2(6)), 6,83-7,41 (2 H, m, CH(4- 5)) und 7,86 (1 H, s, CH(2)); (75,5 MHz; CD3CN) 14,2 (CH3), 23,2, 26,7, 31 ,2,
31 ,9 (CH2(7-1 0)), 48,5 (CH2(6)) und 1 29,6 (Im-CH); m/z (ESI; CH3CN) 256 ([C9H16N2 + CH3CN + Cu]\ 21 %) und 367,1 912 ([C18H32CuN4]\ 1 00.
Ci8H32CuN4 berechnet 367,1 91 7); m/z (ESI, negativer Modus; CH3CN)
132,8676 ([CuCI2]\ 84%. CI2Cu berechnet 132,8679), 224 (100), 233 (92), und 333 (16).
I .5 [Cu(lm6)2][CuBr2] (V)
Nach derselben Vorgehensweise wie im Falle von IV, jedoch unter Verwendung von CuBr anstelle von CuCI, wurde [Cu(lm6)2][CuBr2] (V) in 98%iger Ausbeute als gelbe, viskose Flüssigkeit erhalten. Glaspunkt -53,9 °C (aus CH3CN);
^(300,1 MHz; CD3CN) 0,87 (3 H, t, CH3), 1 ,28 (6 H, br s, CH2(8-1 0)), 1 ,76 (2 H, quin, 3J 7,1 , CH2(7)), 3,99 (2 H, t, 3J 7,2, CH2(6)), 6,63-7,50 (2 H, m, CH(4- 5)) und 7,89 (1 H, s, CH(2)); (75,5 MHz; CD3CN) 14,2 (CH3), 23,1 , 26,7, 31 ,3, 31 ,8 (CH2(7-1 0)), 48,5 (CH2(6)) und 1 29,8 (Im-CH); m/z (ESI; CH3CN) 256 ([C9H16N2 + CH3CN + Cu]\ 1 8%) und 367,1 91 1 ([Ci8H32CuN4]+, 1 00.
Ci8H32CuN4 berechnet 367,1 91 7); m/z (ESI, negativer Modus; CH3CN)
222,7648 ([CuBr2]\ 99%. Br2Cu berechnet 222,7648), 31 2 (1 00) und 367 (36). 1 .6 [DMIM][CuCI2] (VI)
In einem Schlenkrohr wurde zu CuCI (200 mg, 2,02 mmol) eine Lösung von 1 - Dodecyl-3-methylimidazoliumchlorid ([DMIM]CI) (580 mg, 2,02 mmol) in CH3CN (5 mL) gegeben. Die Reaktionsmischung wurde bei Raumtemperatur für ca. 1 0 min mit Ultraschall behandelt. Das Lösungsmittel wurde im Vakuum entfernt, hellgelbe hochviskose Rückstand gefriergetrocknet. [DMIM][CuCI2] (VI) (780 mg, quantitative Ausbeute) wurde als hellgelbes Öl erhalten (gefunden: C, 49,64; H, 7,83; N, 7,32. Ci6H3iCI2CuN2 berechnet C, 49,80; H, 8,1 0; N, 7,26%); Smp. -1 6,2 °C (aus CH3CN), Zers. 293,0 °C; <5H(300,1 MHZ; CD3CN) 0,80-0,92 (3 H, m, CH2CH3), 1 ,26 (1 8 H, br s, CH2(8-1 6)), 1 ,72-1 ,88 (2 H, m, CH2(7)), 3,78-3,86 (3 H, m, CH3), 4,06-4,1 7 (2 H, m, CH2(6)), 7,30-7,42 (2 H, m, CH(4- 5)) und 8,45-8,52 (1 H, m, CH(2)); (75,5 MHz; CD3CN) 14,3 (CH3(1 7)), 23,3, 26,6, 29,5, 30,0, 30,2, 30,3, 30,6, 32,6 (CH2(7-1 6)), 36,9 (CH3(1 8)), 50,5 (CH2(6)), 1 23,2 (CH(5)), 1 24,6 (CH(4)) und 136,8 (CH(2)); m/z (ESI; CH3CN) 251 ,2479 ([Ci6H31 N2]\ 1 00%. Ci6H31 N2 berechnet 251 ,2482); m/z (ESI, negati- ver Modus; CH3CN) 1 32,8680 ([CuCI2]\ 90%. CI2Cu berechnet 1 32,8679), 1 70
(20), 233 (5) [Cu2CI3]" und 321 (1 0). IR (Substanz) ^ /cm ' = 3140w, 31 00, 2921 vs, 2852s, 1569, 1464, 1 377w, 1 338w, 1 1 65s, 1 090w, 1 022w, 836, 743, 650, 620s und 404. Für die Einkristallstrukturanalyse geeignete Kristalle wurden aus der Schmelze bei Raumtemperatur nach mehreren Monaten erhalten, in inertem Öl aufgenommen und in den N2-Kühlgasstrom des Diffraktometers ü- berführt, wobei sich zeigte, dass es sich um die Verbindung [DMIM][CuCI2]■ [DMIM]2[CuCI3] (VII) handelte.
1 .7 [DMIM][CuCI2] · [DMIM]2[CuCI3] (VII)
Die gezielte Synthese von VII wurde wie folgt durchgeführt: In einem
Schirenkrohr wurde zu CuCI (200 mg, 2,02 mmol) eine Lösung von [DMIM]CI (869 mg, 3,03 mmol) in CH3CN (5 mL) gegeben. Das Reaktionsgemisch wurde bei Raumtemperatur ca. 1 0 min lang mit Ultraschall behandelt. Nach Entfernen des Lösungsmittels im Vakuum und Gefriertrocknung des gräulichen
Rückstandes wurde [DMIM][CuCI2]■ [DMIM]2[CuCI3] (VII) erhalten (1 069 mg, quantitative Ausbeute) (gefunden: C, 53,86; H, 8,65; N, 8,24. C48H93CI5Cu2N6
berechnet C, 54,46; H, 8,85; N, 7,94%); Smp. 26,2 °C (aus CH3CN), Zers.
243,7 °C; <5H(300,1 MHz; CD3CN) 0,87 (3 H, t, CH2CH3), 1 ,26 (18 H, br s, CH2(8-16)), 1 ,82 (2 H, quin, 3J 7,0, CH2(7)), 3,85 (3 H, s, CH3), 4,15 (2 H, t, 3J 7,4, CH2(6)), 7,37-7,40 (1 H, m, CH(5)), 7,40-7,43 (1 H, m, CH(4)) und 8,91 (1 H, s, CH(2)); (75,5 MHz; CD3CN) 14,4 (CH3(17)), 23,4, 26,7, 29,6, 30,0, 30,1 , 30,2, 30,3, 30,6, 32,6 (CH2(7-16)), 36,9 (CH3(18)), 50,5 (CH2(6)), 123,2 (CH(5)), 124,5 (CH(4)) und 137,3 (CH(2)); m/z (ESI; CH3CN) 251 ,2480 ([Ci6H31 N2]+, 100%. Ci6H31N2 berechnet 251 ,2482); IR (Substanz) ,»,/™ -· = 3138w, 3086, 2950, 2920vs, 2850s, 1639w, 1568, 1464, 1378, 1336, 1288w, 1221 w, 1 163vs, 1 104w, 1020w, 880w, 829, 819s, 763, 723, 652, 642, 618vs, 601 und 41 1 s.
1 .8 [DMIM][CuBr2] (VIII)
Analog zu der Vorgehensweise bei der Herstellung von VI, jedoch unter Ver- wendung der entsprechenden Bromide, wurde [DMIM][CuBr2] (VIII) in quantitativer Ausbeute in Form eines nahezu farblosen Feststoffes erhalten (gefunden: C, 40,31 ; H, 6,69; N, 5,86. Ci6H31 Br2CuN2 berechnet C, 40,48; H, 6,58; N, 5,90%); Smp. 26,7 °C (aus CH3CN), Zers. 322,3 °C; *(300,1 MHz; CD3CN) 0,87 (3 H, t, CH2CH3), 1 ,27 (18 H, br s, CH2(8-16)), 1 ,82 (2 H, quin, 3J 7,2, CH2(7)), 3,83 (3 H, s, CH3), 4,13 (2 H, t, 3J 7,2, CH2(6)), 7.35 (1 H, t, 4J 1 ,8, CH(5)), 7,38 (1 H, t, 4J 1 ,8, CH(4)) und 8,50 (1 H, s, CH(2)); (75,5 MHz;
CD3CN) 14,4 (CH3(17)), 23,3, 26,6, 29,6, 30,0, 30,2, 30,3, 30,6, 32,6 (CH2(7- 16)), 37,0 (CH3(18)), 50,5 (CH2(6)), 123,2 (CH(5)), 124,6 (CH(4)) und 136,8 (CH(2)); m/z (ESI; CH3CN) 251 ,2480 ([Ci6H31 N2]\ 100%. Ci6H31N2 berechnet 251 ,2482); m/z (ESI, negativer Modus; CH3CN) 222,7651 ([CuBr2]\ 23%. CuBr2 berechnet 220.7648), 264 (35) und 314 (23). IR (Substanz) ^/cm ' = 3139w, 3099, 3062, 2915vs, 2849vs, 1619w, 1570, 1466, 1429, 1377, 1337, 1284w, 1 165vs, 1088w, 1019w, 842, 754, 721 , 657, 623, 615 und 412w. Für die Einkristallstrukturanalyse geeignete Kristalle von VIII wurden aus CH3CN/EtOAc bei 4 °C erhalten, in inertem Öl aufgenommen und in den N2-Kühlgasstrom des Diffraktometers überführt, dabei konnte gezeigt werden, dass es sich um die oben genannte Verbindung handelte.
1 .9 [DMIM]2[CuCI4] (IX)
In einem Schlenkrohr wurde zu einer Schmelze von wasserfreiem CuCI2 (67 mg, 0,50 mmol) und [DMIM]CI (287 mg, 1 ,00 mmol) CH3CN (20 ml_) zwecks Homogenisierung gegeben. Das Reaktionsgemisch wurde bei Raumtemperatur für ca. 10 min mit Ultraschall behandelt. Das Lösungsmittel wurde im Vakuum entfernt und der gelbe, hochviskose Rückstand gefriergetrocknet.
[DMIM]2[CuCI4] (IX) (354 mg, quantitative Ausbeute) wurde als gelber Feststoff erhalten (gefunden: C, 53,91 ; H, 8,74; N, 7,65. C32H62CI4CuN4 berechnet C, 54,27; H, 8,82; N, 7,91 %); Smp. 27,3 °C (aus CH3CN), Zers. 237,2 °C; <5H(300,1 MHz; CD3CN) 0,80-0,94 (3 H, m, CH2CH3), 1 ,18-1 ,59 (18 H, m, CH2(8-16)),
2.09 (2 H, br, CH2(7)), 4,12 (3 H, br, CH3), 4,32 (2 H, br, CH2(6)), 7,41 (2 H, br, CH(4-5)) und 9,33 (1 H, br, CH(2)); (75,5 MHz; CD3CN) 13,9 (CH3(17)), 22,8, 27,9, 29,6, 29,8, 29,9, 30,0, 30,1 , 30,2, 32,1 (CH2(7-16)), 41 ,7 (CH3(18)), 54,0 (CH2(6)), 123,3 (CH(5)), 125,0 (CH(4)) und 130,5 (CH(2)); m/z (ESI; CH3CN) 251 ,2480 ([Ci6H31 N2]\ 100%. Ci6H31 N2 berechnet 251 ,2482); IR (Substanz) vmax / cm ' = 3143Wj 3-100, 2916vs, 2849vs, 1633w, 1571 , 1558, 1468, 1431 , 1375w, 1340w, 1287w, 1 166vs, 1094w, 1022w, 859, 838, 765, 720, 662, 653, 619vs und 412w.
1 .10 [DMIM]2[CuBr4] (X)
Unter Verwendung derselben Prozedur wie im Falle von IX wurde
[DMIM]2[CuBr4] (X) in quantitativer Ausbeute als dunkelvioletter Feststoff erhalten, (gefunden: C, 43,81 ; H, 7,00; N, 6,49. C32H62Br4CuN4 berechnet C, 43,38; H, 7,05; N, 6,32%); Smp. 27,3 °C (aus CH3CN), Zers. 224,2 °C; ^(300,1 MHz; CD3CN) 0,88 (3 H, t, CH2CH3), 1 ,20-1 ,62 (18 H, m, CH2(8-16)), 2,12 (2 H, br, CH2(7)), 4,17 (3 H, br, CH3), 4,41 (2 H, br, CH2(6)), 7,50 (2 H, br, CH(4-5)) und 9,52 (1 H, br, CH(2)); (75,5 MHz; CD3CN) 13,8 (CH3(17)), 22,7, 27,9, 29,4, 29,8, 29,9, 30,0, 30,1 , 32,0 (CH2(7-16)), 41 ,3 (CH3(18)), 53,2 (CH2(6)), 123,5 (CH(5)), 125,2 (CH(4)) und 133,2 (CH(2)); m/z (ESI; CH3CN) 251 ,2480
([Ci6H3iN2]+, 100%. Ci6H3i N2 berechnet 251 ,2482); IR (Substanz) ^'m"' = 3133w, 3095, 3056, 2953, 2919vs, 2851 vs, 1717w, 1569, 1466, 1428, 1376, 1335w, 1 162vs, 1089w, 1020w, 860, 842, 746s, 720, 660, 649 und 621 s. 2. Neutralkomplexe
2.1 [Cu(lm12)l]6 (XI)
In einem Schlenkrohr wurde zu Cul (952 mg, 5,00 mmol) eine Lösung von Im12 (1 ,27 g, 5,35 mmol) in CH3CN (50 mL) gegeben. Nachdem für 2 h bei Raumtemperatur gerührt worden war, wurde das Lösungsmittel im Vakuum entfernt. Der farblose Feststoff wurde mit Et2O gewaschen und im Vakuum getrocknet. [Cu(lm12)l]6 (XI) (691 mg, 32%) wurde als farbloser Feststoff erhalten (gefunden: C, 41 ,95; H, 6,93; N, 6,62. C9oH168Cu6l6Ni2 berechnet C, 42,21 ; H, 6,61 ; N, 6,56%); Smp. 76,7 °C (aus CH3CN), Zers. 253,5 °C; <5H(300,1 MHZ; CD3CN) 0,88 (3 H, t, CH3), 1 ,27 (18 H, br s, CH2(8-16)), 1 ,68-1 ,86 (2 H, m, CH2(7)), 4,01 (2 H, t, CH2(6)) und 7,17-9,41 (3 H, m, Im-H); (75,5 MHz; CD3CN) 14,4 (CH3) und 23,4, 27,1 , 29,7, 30,0, 30,1 , 30,2, 30,3, 31 ,3, 32,7 (CH2(7-16)); m/z (ESI; CH3CN) 237 ([Ci5H28N2 + H]\ 25%), 340 (12) und 535 ([C30H56CuN4]+, 100). IR (Nujol) raax / cm ' = 3129w, 31 1 1 , 1601 w, 1524, 1516, 1285, 1235, 1 109s, 1037, 1024, 937, 827, 814, 745, 731 , 654, 627 und 619. Der Einsatz eines Cul : lm12- Verhältnisses von 1 : 2 an Stelle von 1 : 1 führte zu demselben Ergebnis. Für die Einkristallstrukturanalyse geeignete Kristalle von XI wurden durch Umkrisal- lisation aus CH3CN bei Raumtemperatur erhalten, in inertem Öl aufgenommen und in den N2-Kühlgasstrom des Diffraktometers überführt.
2.2 [CuCI2(lm10)2] (XII)
In einem Schlenkrohr wurden zu CuCI2■ 2 H2O (528 mg, 3,10 mmol) 1 - Decylimidazol (Im10) (1 ,33 g, 6,38 mmol) und CH3CN (10 mL) gegeben.
Nachdem das dunkelgrüne Reaktionsgemisch für ca. 5 min bei
Raumtemperatur mit Ultraschall behandelt worden war, wurde das
Lösungsmittel im Vakuum entfernt. Der blaue Feststoff wurde mit Et2O digeriert und im Vakuum getrocknet. [CuCl2(lm10)2] (XII) wurde als blauer, feinkristalliner Feststoff erhalten (gefunden: C, 56,42; H, 8,76; N, 10,08. C9oH168Cu6l6Ni2 berechnet C, 56,66; H, 8,78; N, 10,17%); Smp. 66 °C (aus CH3CN). Für die Einkristallstrukturanalyse von XII geeignete Kristalle wurden durch
Umkristallisation aus CH3CN bei 4 °C erhalten, in inertem Öl aufgenommen und in den N2-Kühlgasstrom des Diffraktometers überführt.
2.3 [CuBr2(lm12)4] (XIII)
Zu einer Suspension von CuBr2 (246 mg, 1 ,10 mmol) in 15 mL CH3CN wurde bei Raumtemperatur eine Lösung von Im12 (1 ,06 g, 4,48 mmol) in 10 mL CH3CN gegeben. Aus der grünen Lösung fiel sofort ein blau-violetter Feststoff aus, der abfiltriert und im Vakuum getrocknet wurde. [CuBr2(lm12)4] (XIII) (528 mg, 41 %) wurde als blaue-violetter, feinkristalliner Feststoff erhalten (gefunden: C, 61 ,35; H, 9,63; N, 9,58. C6oHn2Br2CuN8 berechnet C, 61 ,65; H, 9,66; N, 9,59%); Smp. 1 12 °C (aus CH3CN). Für die Einkristallstrukturanalyse von XIII geeignete Kristalle wurden aus CH3CN bei Raumtemperatur erhalten, in inertem Öl aufgenommen und in den N2-Kühlgasstrom des Diffraktometers überführt, dabei konnte gezeigt werden, dass es sich um die oben genannte Verbindung handelt.
2.4 [Cu4OCI6(/7-BuOH)4)] (XIV) Wasserfreies CuCI2 (21 ,41 g 159,20 mmol) wurde mit CuO (7,60 g, 95,50 mmol) in 120 mL n-BuOH suspendiert. Die dunkelbraune Suspension wurde für 24 h unter Rückfluss erhitzt. Anschließend wurde heiß filtriert, um überschüssiges CuO abzutrennen. Das gelbe Filtrat wurde im Vakuum bis zur Trockene eingeengt und der Rückstand mit Pentan digeriert. Der so erhaltene goldfarbe- ne, feinkristalline Feststoff wurde im Vakuum getrocknet. Für die Einkristallstrukturanalyse von XIV geeignete Kristalle wurden aus Pentan bei 4 °C erhal-
ten, in inertem Öl aufgenommen und in den N2-Kühlgasstrom des Diffraktome- ters überführt.
Durch Umsetzung von XIV mit z. B. vier Äquivalenten 1 -Undecylimidazol (Im11) in einem Et2O/Aceton-Gemisch wird nach Umkristallisation aus CH3CN die Neutralverbindung [Cu4OCI6(lm11)4)] erhalten.
B. Herstellung eines erfindungsgemäßen Katalysators 1 .1 Trägerung der Kupfer-haltigen Koordinationsverbindung
[Cu(lm12)2][CuBr2]
Das Abwiegen der ungeträgerten Cu(l)-enthaltenden ionischen Flüssigkeit erfolgt in der Inertgasbox oder mittels Schlenktechnik. Zur Herstellung einer Kata- lysatorcharge für einen kontinuierlichen Versuch werden 6,17 g
[Cu(lm12)2][CuBr2] in einen 500 ml Schlenkkolben eingewogen. Es werden 90 ml Acetonitril zugegeben und für 15 Minuten gerührt. Anschließend werden 10,50 g Kieselgel (Partikelgröße 0,063 - 0,200 mm, BET-Oberfläche 335 m2/g, Porenvolumen 0,966 ml/g) hinzugefügt und die Suspension weitere 15 Minuten gerührt. Dann wird die Suspension in einem Wasserbad unter Rühren auf 50 °C aufgeheizt. Gleichzeitig wird ein Ultraschallkopf in das Wasserbad getaucht und mit einer Frequenz von 2 s"1 betrieben. Nach 15 Minuten wird an den Kolben eine Vakuumpumpe angeschlossen und der Druck langsam von 105 Pa auf 5*102 Pa reduziert. Nach etwa 45 Minuten liegt ein türkises Pulver vor, welches sich bei Luftkontakt schlagartig dunkelgrün verfärbt.
1 .2 Trägerung der Kupfer-haltigen Koordinationsverbindung
[Cu(lm6)2][CuBr2] Das Abwiegen der ungeträgerten Cu(l)-enthaltenden ionischen Flüssigkeit erfolgt in der Inertgasbox oder mittels Schlenktechnik. Zur Herstellung einer Katalysatorcharge für einen kontinuierlichen Versuch werden 2,87 g
[Cu(lm6)2][CuBr2] in einen 500 ml Schlenkkolben eingewogen. Es werden 40 ml Acetonitril zugegeben und für 15 Minuten gerührt. Anschließend werden 10,01 g Kieselgel (Partikelgröße 0,063 - 0,200 mm, BET-Oberfläche 335 m2/g, Porenvolumen 0,966 ml/g) hinzugefügt und die Suspension weitere 15 Minuten gerührt. Dann wird die Suspension in einem Wasserbad unter Rühren auf 50 °C aufgeheizt. Gleichzeitig wird ein Ultraschallkopf in das Wasserbad getaucht und mit einer Frequenz von 2 s"1 betrieben. Nach 15 Minuten wird an den Kolben eine Vakuumpumpe angeschlossen und der Druck langsam von 105 Pa auf 5*102 Pa reduziert. Nach etwa 45 Minuten liegt ein blaues Pulver vor, welches sich bei Luftkontakt schlagartig intensiv dunkelgrün verfärbt.
1 .3 Trägerung der Kupfer-haltigen Koordinationsverbindung
[Cu(lm12)2][CuCI2] Das Abwiegen der ungeträgerten Cu(l)-enthaltenden ionischen Flüssigkeit erfolgt in der Inertgasbox oder mittels Schlenktechnik. Zur Herstellung einer Katalysatorcharge für einen kontinuierlichen Versuch werden 2,80 g
[Cu(lm12)2][CuCI2] in einen 500 ml Schlenkkolben eingewogen. Es werden 1 10 ml Acetonitril zugegeben und für 15 Minuten gerührt. Anschließend werden 10,01 g Kieselgel (Partikelgröße 0,063 - 0,200 mm, BET-Oberfläche 335 m2/g, Porenvolumen 0,966 ml/g) hinzugefügt und die Suspension weitere 15 Minuten gerührt. Dann wird die Suspension in einem Wasserbad unter Rühren auf 50 °C aufgeheizt. Gleichzeitig wird ein Ultraschallkopf in das Wasserbad getaucht und mit einer Frequenz von 2 s"1 betrieben. Nach 15 Minuten wird an den Kolben eine Vakuumpumpe angeschlossen und der Druck langsam von 105 Pa auf 5*102 Pa reduziert. Nach etwa 45 Minuten liegt ein bläuliches Pulver vor, welches sich bei Luftkontakt schlagartig schmutzig grün verfärbt.
Trägerung der Kupfer-haltigen Koordinationsverbindung
[Cu(lm6)2][CuCI2]
Das Abwiegen der ungeträgerten Cu(l)-enthaltenden ionischen Flüssigkeit erfolgt in der Inertgasbox oder mittels Schlenktechnik. Zur Herstellung einer Katalysatorcharge für einen kontinuierlichen Versuch werden 2,80 g
[Cu(lm6)2][CuCl2] in einen 500 ml Schlenkkolben eingewogen. Es werden 40 ml Acetonitril zugegeben und für 15 Minuten gerührt. Anschließend werden 10,00 g Kieselgel (Partikelgröße 0,063 - 0,200 mm, BET-Oberfläche 335 m2/g, Porenvolumen 0,966 ml/g) hinzugefügt und die Suspension weitere 15 Minuten gerührt. Dann wird die Suspension in einem Wasserbad unter Rühren auf 50 °C aufgeheizt. Gleichzeitig wird ein Ultraschallkopf in das Wasserbad getaucht und mit einer Frequenz von 2 s"1 betrieben. Nach 15 Minuten wird an den Kolben eine Vakuumpumpe angeschlossen und der Druck langsam von 105 Pa auf 5*102 Pa reduziert. Nach etwa 45 Minuten liegt ein bläuliches Pulver vor, welches sich bei Luftkontakt schlagartig intensiv grün verfärbt. 1 .5 Trägerung der Kupfer-haltigen Koordinationsverbindung [CuBr2(lm12)4]
Das Abwiegen der ungeträgerten Kupfer(ll)-Verbindung erfolgt außerhalb einer Inertgasbox. Zur Herstellung einer Katalysatorcharge für einen kontinuierlichen Versuch werden 6,34 g [CuBr2(lm12)4] in einen 500 ml Schlenkkolben eingewo- gen. Es werden 90 ml Acetonitril zugegeben und für 15 Minuten auf einem Magnetrührer gerührt. Anschließend werden 10,52 g Kieselgel (Partikelgröße 0,063 - 0,200 mm, BET-Oberfläche 335 m2/g, Porenvolumen 0,966 ml/g) hinzugefügt und die Suspension weitere 15 Minuten gerührt. Dann wird die Suspension in einem Wasserbad unter Rühren auf 50 °C aufgeheizt. Gleichzeitig wird ein Ultraschallkopf in das Wasserbad getaucht und mit einer Frequenz von 2 s"1 betrieben. Nach 15 Minuten wird an den Kolben eine Vakuumpumpe angeschlossen und der Druck langsam von 105 Pa auf 5*102 Pa reduziert. Nach etwa 45 Minuten liegt ein blau-violettes Pulver vor. 1 .6 Trägerung der Kupfer-haltigen Koordinationsverbindung [CuBr2(lm12)4] gelöst in einem schwerflüchtigen Silikonöl
Das Abwiegen der ungetragerten Kupfer(ll)-Verbindung erfolgt außerhalb einer Inertgasbox. Zur Herstellung einer Katalysatorcharge werden 6.1 1 g
[CuBr2(lm12)4] in einen 500 ml Schlenkkolben eingewogen. Es werden 90 ml Tetrahydrofuran zugegeben und für 15 Minuten gerührt. Anschließend werden zunächst 3,12 g Silikonöl AS 4 (Fluka, Viskosität 6 mPa sec ) eingetragen und nach weiteren 2 Minuten 16,21 g Kieselgel (Partikelgröße 0,063 - 0,200 mm, BET-Oberfläche 335 m2/g, Porenvolumen 0,966 ml/g) hinzugefügt und die Suspension weitere 15 Minuten gerührt. Dann wird die Suspension in einem Wasserbad unter Rühren auf 50 °C aufgeheizt. Gleichzeitig wird ein Ultraschallkopf in das Wasserbad getaucht und mit einer Frequenz von 2 s"1 betrieben. Nach 15 Minuten wird an den Kolben eine Vakuumpumpe angeschlossen und der Druck langsam von 105 Pa auf 5*102 Pa reduziert. Nach etwa 45 Minuten liegt ein blaues Pulver vor. C. Katalytische Aktivität von ungeträgerten Kupfer-haltigen Koordinationsverbindungen in der oxidativen Carbonylierung von Methanol in Anwesenheit von CO und O2
Tabelle 1 : Katalysatoraktivitäten der ungeträgerten ionischen Flüssigkeiten in der Dimethylcarbonat-Synthese3
Katalysator Umsatz/% Selektivität/%
[Cu(lrr 2][CuCI2] (II) 60 83
[Cu(lm12)2][CuBr2] (III) 62 89 a Reaktionsbedingungen : 5 mol% Cu (Gesamtmenge) in Bezug auf MeOH (30 mmol), 3 bar 02, 50 bar CO, 4 h, 120 °C. Tabelle 2: Variation der Katalysatorkonzentration der ungeträgerten ionischen Flüssigkeiten in der Dimethylcarbonat-Synthese"
Katalysator/mol% 0,5 1 ,0 2,0 5,0
Umsatz/% 32 37 40 44
Selektivität/% 88 79 85 88
a Reaktionsbedingungen : Katalysator III ( mol% Cu in Bezug auf MeOH), 1 50 mmol
MeOH, 5 bar 02, 35 bar CO, 4 h, 120 °C.
D. Ergebnisse eines erfindungsgemäßen Verfahrens
Zur Durchführung eines kontinuierlichen Versuches des erfindungsgemäßen Verfahrens wurde wie folgt vorgegangen: Die Reaktionsapparatur (ohne Reak- tor) wurde auf 100 °C vorgeheizt. Der Kupfer-haltige Katalysator wurde in einen Festbettreaktor gegeben und der Festbettreaktor wurde in eine Anlage entspr. Fig. 1 geschraubt. Der Heizmantel wurde um den Festbettreaktor gelegt und der Festbettreaktor auf 100°C geheizt. Der Volumenstrom der Reaktionsgase CO und Luft wurde über zwei separate Massendurchflussregler eingestellt und über einen Bypass (100°C) am Festbettreaktor vorbei geleitet. In einem üblichen Versuch betrugen die Volumenströme 72 ml/min CO und 51 ,4 ml/min Luft. Der Anlagendruck wurde am Druckregelventil auf 5 bar eingestellt. Nun wurde über eine Mikroliterkolbenpumpe 0,069 ml/min Methanol^ dosiert. Sobald der online-Gaschromatograph ein stabiles Methanolsignal innerhalb der Detekti- onsgrenzen anzeigte, wurde von Bypass auf Reaktor umgestellt. Die GC- Analyse erfolgte automatisch in regelmäßigen Zeitabständen.
Als Edukte kamen Methanol, Kohlenstoffmonoxid und synthetische Luft zum Einsatz. Die Versuche wurden mit den in den Ausführungsbeispielen unter A. und B. hergestellten geträgerten Katalysatoren durchgeführt.
Tabelle 3: Versuchsergebnisse
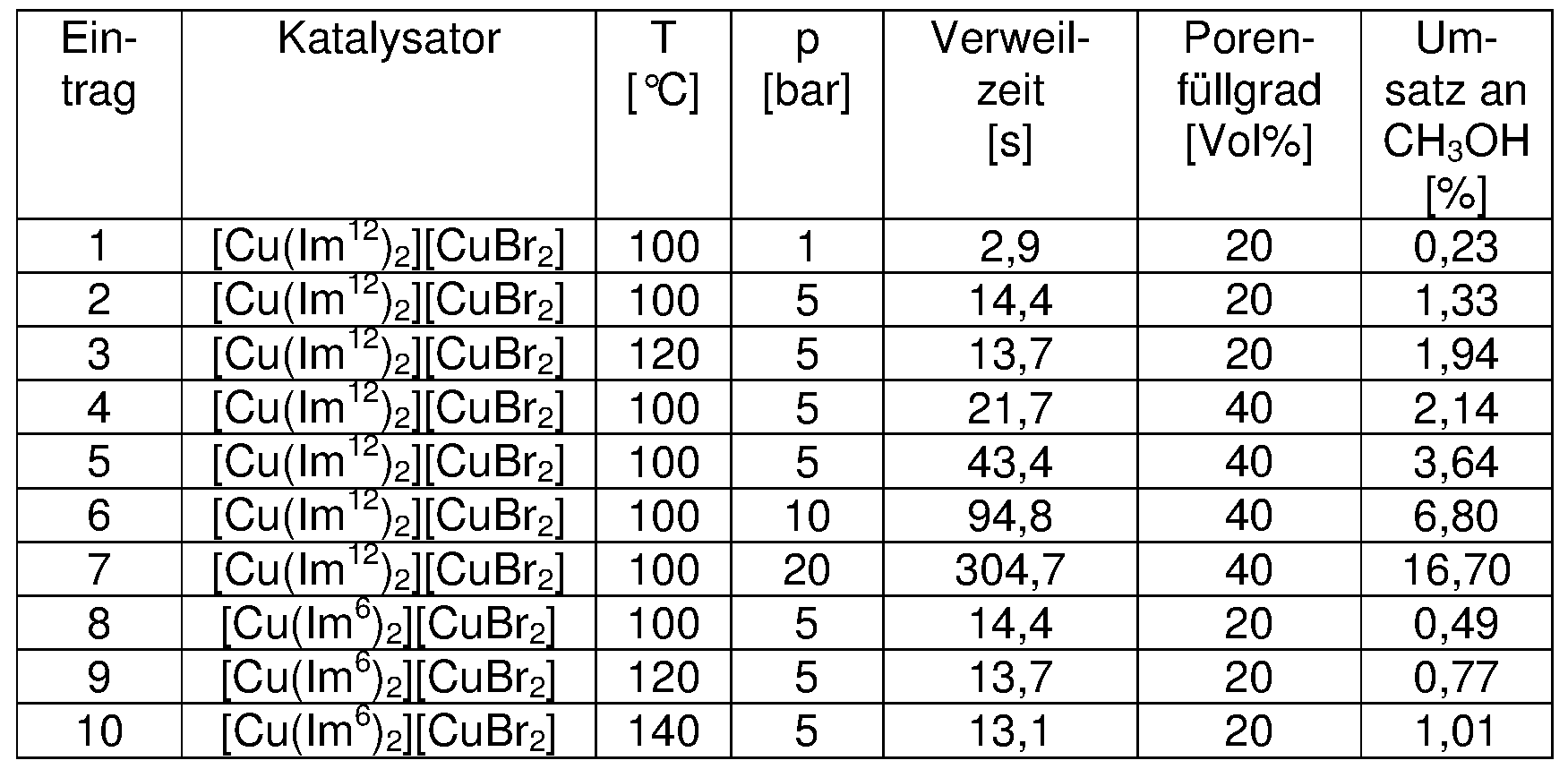

Kieselgel: Merck KGaA Typ 10184 (Partikelgröße 0,063 - 0,200 mm, BET- Oberfläche 335 m2/g, Porenvolumen 0,966 ml/g), T = Temperatur, p = Druck. Eintrag 1 aus Tabelle 3 zeigt, dass mit dem erfindungsgemäßen Katalysatorsystem bei einer extrem kurzen Verweilzeit von 2,9 Sekunden und einem sehr niedrigen Druck von 1 bar schon ein konstanter Umsatz von 0,23 % erreicht werden kann. Eintrag 2 aus Tabelle 3 zeigt, dass die Erhöhung des Anlagendrucks und damit die Erhöhung der Verweilzeit bei sonst gleichen Parametern zu einer deutlichen Steigerung des Umsatzes auf 1 ,33 % führt.
Eintrag 3 aus Tabelle 3 zeigt, dass eine Erhöhung der Reaktortemperatur auf 120 °C eine weitere Zunahme des Umsatzes und der Aktivität des Katalysators zur Folge hat. Eintrag 4 bis 7 zeigen, dass mit einer weiteren Steigerung der Verweilzeit durch Erhöhung der Katalysatormenge und Steigerung des Anlagendrucks sehr deutliche Umsatzzunahmen erzielt werden können. Die Erfindung ist nicht auf eine der vorgeschriebenen Ausführungsformen beschränkt, sondern in vielfältiger Weise abwandelbar.
Sämtliche aus den Ansprüchen, der Beschreibung, den Figuren und den Ausführungsbeispielen hervorgehenden Merkmale, Vorteile und Verfahrensschritte, können sowohl für sich als auch in den verschiedensten Kombinationen erfindungswesentlich sein.
Bezugszeichen
1 Massendurchflussregler
2 Kolbenpumpe
3 Verdampfer
4,5 Festbettreaktor
6 Bypass
7 Druckventil
8 Online-Gaschromatograph
Claims
Patentansprüche
Verfahren zur Herstellung eines Dialkylcarbonats, bei welchem ein Alkanol mit Kohlenmonoxid und Sauerstoff in Anwesenheit eines Kupfer-haltigen Katalysators, der auf einem Festphasenträger geträgert ist, umgesetzt wird, dadurch gekennzeichnet,
■ dass der Katalysator auf dem Festphasenträger unter Verfahrensbedingungen in einer flüssigen Katalysatorphase vorliegt, und
■ dass der Katalysator wenigstens eine Kupfer-haltige Koordinationsverbindung umfasst.
Verfahren nach Anspruch 1 , dadurch gekennzeichnet, dass die Kupfer- haltige Koordinationsverbindung einen Schmelzpunkt kleiner 120°C aufweist.
Verfahren nach Anspruch 1 oder 2, dadurch gekennzeichnet, dass die Kupfer-haltige Koordinationsverbindung mindestens eine Stickstoff (NO- Verbindung als AADonorligand und/oder eine quarternisierte Stickstoff (NO- Verbindung als organisches Kation enthält, wobei die Stickstoff (III)- Verbindung ausgewählt ist aus der Gruppe von A/-Alkylimidazolen, N,N- Dialkylaminopyridinen, AAAlkylpyrazolen oder AAAlkyl-pentaorgano- guanidinen.
Verfahren nach einem der Ansprüche 1 bis 3, dadurch gekennzeichnet, dass die Kupfer-haltige Koordinationsverbindung wenigstens einen li- pophilen Substituenten aufweist.
Verfahren nach Anspruch 4, dadurch gekennzeichnet, dass der lipophile Substituent an der Stickstoff(lll)-Verbindung vorgesehen ist.
6. Verfahren nach einem der Ansprüche 1 bis 5, dadurch gekennzeichnet, dass gasförmige Edukte mit der Katalysatorphase in Kontakt gebracht werden, und dass die Produkte über die Gasphase abgeführt werden.
Verfahren nach einem der Ansprüche 1 bis 6, dadurch gekennzeichnet, dass der Katalysator eine ionische Flüssigkeit ist, wobei die Kupfer-haltige Koordinationsverbindung im Anion und/oder im Kation der ionischen Flüssigkeit enthalten ist.
Verfahren nach einem der vorhergehenden Ansprüche, dadurch gekennzeichnet, dass die Kupfer-haltige Koordinationsverbindung aus den ionischen Flüssigkeiten [Cu(lm12)4][PF6], [Cu(lm12)2][CuCI2],
[Cu(lm12)2][CuBr2], [Cu(lm6)2][CuCI2], [Cu(lm6)2][CuBr2], [DMIM][CuCI2], [DMIM][CuCI2] · [DMIM]2[CuCI3], [DMIM][CuBr2], [DMIM]2[CuCI4],
[DMIM]2[CuBr4] oder den neutralen Komplexen [Cu(lm12)l]6, [CuCI2(lm10)2], [CuBr2(lm12)4], [Cu4OCI6(lm11)4)] ausgewählt ist.
Verfahren nach einem der vorhergehenden Ansprüche, dadurch gekennzeichnet, dass verzweigte und unverzweigte Alkanole mit 1 bis 4 Kohlenstoffatomen umgesetzt werden.
Verfahren nach einem der vorhergehenden Ansprüche, dadurch gekennzeichnet, dass das Verfahren in einem Temperaturbereich von 60 °C bis 200 °C, bevorzugt von 70 °C bis 140°C, besonders bevorzugt von 75 °C bis 120°C und ganz besonders bevorzugt von 80 °C bis 1 15°C durchgeführt wird. 1 Verfahren nach einem der vorhergehenden Ansprüche, dadurch gekennzeichnet, dass das Verfahren als kontinuierliches Verfahren durchgeführt wird.
12. Kupfer-haltiger Katalysator, der auf einem Festphasenträger geträgert ist, und der in einer Katalysatorphase vorliegt, die einen Schmelzpunkt kleiner 120°C aufweist, und der wenigstens eine Kupfer-haltige Koordinationsverbindung umfasst, wobei die Kupfer-haltige Koordinationsverbindung mindestens eine Stickstoff(lll)-Verbindung als AADonorligand und/oder eine quarternisierte Stickstoff(lll)-Verbindung als organisches Kation enthält, wobei die Stickstoff(lll)-Verbindung ausgewählt ist aus der Gruppe von N- Alkylimidazolen, A/,A/-Dialkylaminopyridinen, AAAlkylpyrazolen oder N- Alkyl-pentaorganoguanidinen.
13. Kupfer-haltiger Katalysator nach Anspruch 12, dadurch gekennzeichnet, dass, der Katalysator wenigstens einen lipophilen Substituenten aufweist.
14. Kupfer-haltiger Katalysator nach Anspruch 13, dadurch gekennzeichnet, dass der lipophile Substituent an der Stickstoff(lll)-Verbindung vorgesehen ist.
15. Kupfer-haltiger Katalysator nach einem der Ansprüche 12 bis 14, dadurch gekennzeichnet, dass der Katalysator eine ionische Flüssigkeit ist, wobei die Kupfer-haltige Koordinationsverbindung im Anion und/oder im Kation der ionischen Flüssigkeit enthalten ist.
16. Kupfer-haltiger Katalysator nach einem der Ansprüche 12 bis 15, dadurch gekennzeichnet, dass die Kupfer-haltige Koordinationsverbindung aus den ionischen Flüssigkeiten [Cu(lm12)4][PF6], [Cu(lm12)2][CuCI2],
[Cu(lm12)2][CuBr2], [Cu(lm6)2][CuCI2], [Cu(lm6)2][CuBr2], [DMIM][CuCI2], [DMIM][CuCI2] · [DMIM]2[CuCI3], [DMIM][CuBr2], [DMIM]2[CuCI4],
[DMIM]2[CuBr4] oder den neutralen Komplexen [Cu(lm12)l]6, [CuCI2(lm10)2], [CuBr2(lm12)4], [Cu4OCI6(lm11)4)] ausgewählt ist.
17. Verwendung eines Katalysators nach einem der Ansprüche 12 bis 16 als Katalysator bei einer oxidativen Carbonylierung.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102010036631.5A DE102010036631B4 (de) | 2010-07-26 | 2010-07-26 | Verfahren zur Herstellung von Dialkylcarbonaten, Kupferhaltiger Katalysator und Verwendung eines Kupfer-haltigen Katalysators |
| DE102010036631.5 | 2010-07-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012013606A1 true WO2012013606A1 (de) | 2012-02-02 |
Family
ID=44583822
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/062679 Ceased WO2012013606A1 (de) | 2010-07-26 | 2011-07-22 | Verfahren zur herstellung von dialkylcarbonaten, kupfer-haltiger katalysator und verwendung eines kupfer-haltigen katalysators |
Country Status (2)
| Country | Link |
|---|---|
| DE (1) | DE102010036631B4 (de) |
| WO (1) | WO2012013606A1 (de) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106916303A (zh) * | 2017-02-28 | 2017-07-04 | 华南理工大学 | 一种十二烷基咪唑配体一价铜催化剂催化制备聚三唑的方法 |
| CN109387596A (zh) * | 2017-08-02 | 2019-02-26 | 中国石油化工股份有限公司 | 耐硫变换催化剂小型评价装置及其使用方法 |
| CN109884240A (zh) * | 2019-01-24 | 2019-06-14 | 铜仁学院 | 一种带急激装置的固定床催化剂评价装置 |
| CN113145175A (zh) * | 2021-04-29 | 2021-07-23 | 沈阳化工大学 | 一种以吡啶氯化铜为活性组分催化剂制备方法及其应用 |
| CN113387810A (zh) * | 2021-06-11 | 2021-09-14 | 重庆微而易科技有限公司 | 一种一步合成碳酸甲乙酯的方法 |
| CN115463675A (zh) * | 2022-10-26 | 2022-12-13 | 高化学(江苏)化工新材料有限责任公司 | 一种新型合成碳酸二甲酯的催化剂及其制备方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102012202621A1 (de) * | 2012-02-21 | 2013-08-22 | Wacker Chemie Ag | Verfahren zur Befüllung eines Reaktors mit einem Katalysator |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0636601A1 (de) | 1993-07-30 | 1995-02-01 | Bayer Ag | Verfahren zur Herstellung von Dialkylcarbonaten |
| US5387708A (en) | 1993-12-10 | 1995-02-07 | The Dow Chemical Company | Production of dialkyl carbonates using copper catalysts |
| US20050054868A1 (en) * | 2003-09-05 | 2005-03-10 | Stibrany Robert T. | Use of a ionic halide free copper catalyst for the production of dialkyl carbonates |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4747298B2 (ja) * | 2005-10-07 | 2011-08-17 | 国立大学法人 千葉大学 | ビスイミダゾリン配位子及びそれを用いた触媒 |
-
2010
- 2010-07-26 DE DE102010036631.5A patent/DE102010036631B4/de not_active Expired - Fee Related
-
2011
- 2011-07-22 WO PCT/EP2011/062679 patent/WO2012013606A1/de not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0636601A1 (de) | 1993-07-30 | 1995-02-01 | Bayer Ag | Verfahren zur Herstellung von Dialkylcarbonaten |
| US5387708A (en) | 1993-12-10 | 1995-02-07 | The Dow Chemical Company | Production of dialkyl carbonates using copper catalysts |
| US20050054868A1 (en) * | 2003-09-05 | 2005-03-10 | Stibrany Robert T. | Use of a ionic halide free copper catalyst for the production of dialkyl carbonates |
Non-Patent Citations (12)
| Title |
|---|
| "Multiphase homogeneous catalysis", 2005, WILEY-VCH |
| A. P. WIGHT, M. E. DAVIES: "Design and Preparation of Organic-Inorganic Hydrid Catalysts", CHEM. REV., vol. 102, 2002, pages 3589 |
| B. M. L. DIOOS, F. L. VANKELECOM, P. A. JACOBS, ADV. SYNTH. CATAL., vol. 348, 2006, pages 1413 |
| D. C. MOLZAHN, M. E. JONES, G. E. HARTWELL, J. PUGA, DOW CHEMICAL, 1995 |
| J. N. H. REEK, P. W. N. M. VAN LEEUWEN, A. G. J. VAN DER HAM, A. D. DE HAAN: "Supported Catalysts, in Catalyst Separation, Recovery and Recycling", 2006, SPRINGER |
| N. E. LEADBEATER, M. MARCO, CHEM. REV., vol. 102, 2002, pages 3217 |
| N. KELLER, G. REBMANN, V. KELLER, J. MOL. CAT. A, vol. 317, 2010, pages 1 |
| RAAB, V. ET AL: "Ligand effects in the copper catalyzed aerobic oxidative carbonylation of methanol to dimethyl carbonate (DMC)", JOURNAL OF MOLECULAR CATALYSIS A: CHEMICAL , 175(1-2), 51-63 CODEN: JMCCF2; ISSN: 1381-1169, 2001, XP002659818 * |
| SASAKI T ET AL: "IMMOBILIZED METAL ION-CONTAINING IONIC LIQUIDS: PREPARATION, STRUCTURE AND CATALYTIC PERFORMANCE IN KHARASCH ADDITION REACTION", CHEMICAL COMMUNICATIONS - CHEMCOM; [6015D], ROYAL SOCIETY OF CHEMISTRY, GB, no. 19, 21 May 2005 (2005-05-21), pages 2506 - 2508, XP009079807, ISSN: 1359-7345, DOI: 10.1039/B500349K * |
| SASAKI, TAKEHIKO ET AL: "Immobilized metal ion-containing ionic liquids : Preparation, structure and catalytic performances in Kharasch addition reaction and Suzuki cross-coupling reactions", JOURNAL OF MOLECULAR CATALYSIS A: CHEMICAL , 279(2), 200-209 CODEN: JMCCF2; ISSN: 1381-1169, 2008, XP002659817 * |
| V. RAAB, M. MERZ, J. SUNDERMEYER, J. MOL. CATAL. A, vol. 175, 2001, pages 51 |
| Z. KRICSFALUSSY, H. WALDMANN, H.-J. TRAENCKNER, IND. ENG. CHEM. RES., vol. 37, 1998, pages 865 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106916303A (zh) * | 2017-02-28 | 2017-07-04 | 华南理工大学 | 一种十二烷基咪唑配体一价铜催化剂催化制备聚三唑的方法 |
| CN106916303B (zh) * | 2017-02-28 | 2019-04-09 | 华南理工大学 | 一种十二烷基咪唑配体一价铜催化剂催化制备聚三唑的方法 |
| CN109387596A (zh) * | 2017-08-02 | 2019-02-26 | 中国石油化工股份有限公司 | 耐硫变换催化剂小型评价装置及其使用方法 |
| CN109387596B (zh) * | 2017-08-02 | 2022-04-08 | 中国石油化工股份有限公司 | 耐硫变换催化剂小型评价装置及其使用方法 |
| CN109884240A (zh) * | 2019-01-24 | 2019-06-14 | 铜仁学院 | 一种带急激装置的固定床催化剂评价装置 |
| CN113145175A (zh) * | 2021-04-29 | 2021-07-23 | 沈阳化工大学 | 一种以吡啶氯化铜为活性组分催化剂制备方法及其应用 |
| CN113145175B (zh) * | 2021-04-29 | 2023-09-05 | 沈阳化工大学 | 一种以吡啶氯化铜为活性组分催化剂制备方法及其应用 |
| CN113387810A (zh) * | 2021-06-11 | 2021-09-14 | 重庆微而易科技有限公司 | 一种一步合成碳酸甲乙酯的方法 |
| CN114163333A (zh) * | 2021-06-11 | 2022-03-11 | 重庆微而易科技有限公司 | 一种一步合成碳酸甲乙酯的方法 |
| CN114163333B (zh) * | 2021-06-11 | 2023-09-19 | 重庆微而易科技有限公司 | 一种一步合成碳酸甲乙酯的方法 |
| CN115463675A (zh) * | 2022-10-26 | 2022-12-13 | 高化学(江苏)化工新材料有限责任公司 | 一种新型合成碳酸二甲酯的催化剂及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102010036631A1 (de) | 2012-01-26 |
| DE102010036631B4 (de) | 2021-12-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012013606A1 (de) | Verfahren zur herstellung von dialkylcarbonaten, kupfer-haltiger katalysator und verwendung eines kupfer-haltigen katalysators | |
| EP1480956B1 (de) | Halogenfreie ionische flüssigkeiten | |
| EP1425268B2 (de) | Organische sulfate als ionische flüssigkeiten | |
| EP0807619B1 (de) | Verfahren zur Herstellung von Diarylcarbonaten | |
| DE60304618T2 (de) | Verfahren zur Carbonylierung von Alkoholen, mittels eines Katalysators auf Rhodium- oder Iridiumbasis in einem nicht-wässrigen ionischen Lösungsmittel, mit effizienter Wiederverwendung des Katalysators | |
| Raab et al. | Ligand effects in the copper catalyzed aerobic oxidative carbonylation of methanol to dimethyl carbonate (DMC) | |
| EP0749955B1 (de) | Verfahren zur Herstellung von Diarylcarbonaten | |
| EP0801051A1 (de) | Verfahren zur kontinuierlichen Herstellung von Diarylcarbonaten | |
| DE112007003012T5 (de) | Alkylierungsverfahren unter Verwendung eines mit Alkyhalogenid arbeitenden Ionenflüssigkeitskatalysators | |
| WO2012084691A1 (de) | Verfahren zur herstellung von ameisensäure durch umsetzung von kohlendioxid mit wasserstoff | |
| WO2004067492A1 (de) | Acetoacetylierung von alkoholen, thiolen und aminen im mikroreaktor | |
| DE4403075A1 (de) | Verfahren zur kontinuierlichen Herstellung von Diarylcarbonaten | |
| EP0413215B1 (de) | Verfahren zur Herstellung von Dialkylcarbonaten | |
| EP2588438A1 (de) | Verfahren zur herstellung von ameisensäure durch umsetzung von kohlendioxid mit wasserstoff | |
| EP2588439A1 (de) | Verfahren zur herstellung von ameisensäure durch umsetzung von kohlendioxid mit wasserstoff | |
| EP2791125B1 (de) | HERSTELLUNG VON 5-HYDROXYMETHYLFURFURAL (HMF) AUS SACCHARIDLÖSUNGEN IN GEGENWART EINES LÖSEMITTELS MIT EINEM SIEDEPUNKT GRÖßER 60°C UND KLEINER 200°C (BEI NORMALDRUCK, KURZ LEICHTSIEDER GENANNT) | |
| DE19943844A1 (de) | Verfahren zur Herstellung von Carbonsäurechloriden | |
| EP2729438A1 (de) | Verfahren zur herstellung von ameisensäure durch umsetzung von kohlendioxid mit wasserstoff | |
| EP1140779B1 (de) | Verfahren zur herstellung von diarylcarbonaten | |
| DE69507414T2 (de) | Verfahren zur Herstellung von Octadienen | |
| DE102005040752A1 (de) | Eisen-katalysierte allylische Alkylierung | |
| DE2018054C3 (de) | Verfahren zur Herstellung von Octadienol | |
| EP0931046A2 (de) | VERFAHREN ZUR HERSTELLUNG VON n-BUTYLALKYLETHERN | |
| EP3816170B1 (de) | Verfahren zur herstellung von phosphorsäurearylesterdiamiden | |
| EP4273119A1 (de) | Verfahren zur herstellung von c5-aldehyden |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11741167 Country of ref document: EP Kind code of ref document: A1 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11741167 Country of ref document: EP Kind code of ref document: A1 |