WO2012164741A1 - 消泡剤 - Google Patents
消泡剤 Download PDFInfo
- Publication number
- WO2012164741A1 WO2012164741A1 PCT/JP2011/062796 JP2011062796W WO2012164741A1 WO 2012164741 A1 WO2012164741 A1 WO 2012164741A1 JP 2011062796 W JP2011062796 W JP 2011062796W WO 2012164741 A1 WO2012164741 A1 WO 2012164741A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrophobic
- silica
- carbon atoms
- antifoaming agent
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D19/00—Degasification of liquids
- B01D19/02—Foam dispersion or prevention
- B01D19/04—Foam dispersion or prevention by addition of chemical substances
- B01D19/0404—Foam dispersion or prevention by addition of chemical substances characterised by the nature of the chemical substance
- B01D19/0409—Foam dispersion or prevention by addition of chemical substances characterised by the nature of the chemical substance compounds containing Si-atoms
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D19/00—Degasification of liquids
- B01D19/02—Foam dispersion or prevention
- B01D19/04—Foam dispersion or prevention by addition of chemical substances
- B01D19/0404—Foam dispersion or prevention by addition of chemical substances characterised by the nature of the chemical substance
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D19/00—Degasification of liquids
- B01D19/02—Foam dispersion or prevention
- B01D19/04—Foam dispersion or prevention by addition of chemical substances
- B01D19/0404—Foam dispersion or prevention by addition of chemical substances characterised by the nature of the chemical substance
- B01D19/0413—Foam dispersion or prevention by addition of chemical substances characterised by the nature of the chemical substance compounds containing N-atoms
Definitions
- the present invention relates to an antifoaming agent. More specifically, defoaming suitable for the fields of paint industry (water-based paint, paper coating paint), chemical industry, food industry, petroleum industry, civil engineering and construction industry, textile industry, paper pulp industry, pharmaceutical industry or wastewater treatment process, etc. It relates to the agent.
- Patent Documents 1 and 2 using an alkylene oxide derivative as an emulsifier is known.
- JP 2008-188480 A Special table 2011-506086 gazette (WO2009 / 080428 pamphlet)
- the emulsion type antifoaming agent described in Patent Document 1 has a problem that the product stability is insufficient, and the emulsifier becomes a foaming component, which is sufficient in a wide temperature range (5 to 70 ° C.), particularly in a high temperature range. There is a problem that the defoaming property cannot be obtained.
- the antifoaming agent described in Patent Document 2 also has the same problem because an emulsifier is essential for emulsification as in the emulsion type antifoaming agent of Patent Document 1. That is, an object of the present invention is to provide an antifoaming agent that is excellent in antifoaming properties over a wide temperature range and excellent in product stability.
- the antifoaming agent of the present invention is characterized by hydrophobic dry silica (S) obtained by hydrophobizing silica by a dry method and having a primary particle size of 5 to 100 nm, water, and general formula (1).
- the gist is that the hydrophobic compound (Q) containing 1 to 25% by weight of the represented ester compound (E) is an essential component.
- R 1 is an alkyl group having 1 to 21 carbon atoms or an alkenyl group having 2 to 21 carbon atoms
- D is involved in an ester bond from a compound (D ′) containing 2 to 6 carbon atoms and 1 to 6 hydroxyl groups
- D ′ is involved in an ester bond from a compound (D ′) containing 2 to 6 carbon atoms and 1 to 6 hydroxyl groups
- a feature of the production method of the present invention is a method for producing the antifoaming agent, which includes a step of adding and mixing hydrophobic dry silica (S) to a mixed liquid composed of the hydrophobic liquid (Q) and water. Is the gist.
- the antifoaming agent of the present invention is excellent in antifoaming property in a wide temperature range (5 to 70 ° C.) and excellent in product stability. Does not cause aggregation or scum.
- the antifoaming agent can be easily produced.
- Hydrophobic dry silica can be used without limitation as long as it is a silica obtained by hydrophobizing silica by a dry method having a primary particle diameter of 5 to 100 nm.
- the dry silica having a primary particle diameter of 5 to 100 nm includes dry synthetic silica (SN) among amorphous synthetic silica (SN). That is, the amorphous synthetic silica (SN) includes dry method (pyrolysis method, melting method) silica (SD) and wet method (gel method, precipitation method) silica (SW). Silica by a dry method having a primary particle diameter of 5 to 100 nm can be used as the dry method silica.
- amorphous synthetic silica examples include the following. Of these, wet method (gel method, precipitation method) silica (SW) is used in hydrophobic wet silica (B3) described later.
- Pyrolytic silica It is obtained by burning a silicon compound such as silicon tetrachloride in an oxyhydrogen flame and is likely to exist as primary particles (fine particles).
- Fused silica It is obtained by melting natural silica powder or the like in a flame and tends to exist as primary particles (fine particles).
- Gel silica It is obtained by neutralizing sodium silicate with an acid in an acidic environment and filtering and drying the resulting precipitate, and has an aggregate structure.
- Precipitated silica It is obtained by neutralizing sodium silicate with an acid in an alkaline environment and filtering and drying the resulting precipitate. The pore volume is large and the specific surface area is large.
- these amorphous synthetic silicas have hydroxysilyl groups (silanol groups) on the surface of the silica particles, they show hydrophilicity.
- pyrolytic silica is preferable from the viewpoints of defoaming properties and product stability.
- Amorphous synthetic silica can be easily obtained from the market, and trade names are exemplified below.
- Admafine series ⁇ Admatechs "Admafine” is a registered trademark of Toyota Motor Corporation. ⁇ , Fuselex series ⁇ Tatsumori Co., Ltd. ⁇ , Denka fused silica series ⁇ Electrochemical Industry Co., Ltd. ⁇ , etc.
- Hydrophobization of silica ⁇ amorphous synthetic silica (SN), etc. ⁇ by a dry method having a primary particle size of 5 to 100 nm can be performed by a known method such as amorphous synthetic silica (SN) and lipophilicity.
- a dry treatment method and a solvent ⁇ organic solvent, mineral oil, animal and vegetable oil, etc. ⁇ in which a lipophilic compound ⁇ a compound described later such as halosilane and alkoxysilane ⁇ is reacted with the surface of silica particles to be hydrophobized while stirring the mixture with the compound
- a wet treatment method in which a lipophilic compound is adsorbed or reacted on the surface of silica particles to be hydrophobized.
- condensation of a functional group on the surface of silica ⁇ amorphous synthetic silica (SN) etc.) and a functional group possessed by a lipophilic compound by a dry method having a primary particle size of 5 to 100 nm Reactions can be utilized, and wet processing methods include (2) physical adsorption to pores of silica ⁇ amorphous synthetic silica (SN) etc. ⁇ by a dry method having a primary particle diameter of 5 to 100 nm, and (3 ) Utilization of surface adsorption of silica ⁇ amorphous synthetic silica (SN), etc. ⁇ by a dry method having a primary particle size of 5 to 100 nm and ionic functional groups of lipophilic compounds can be used. .
- the method using the condensation reaction (1) is preferable.
- lipophilic compounds examples include halosilanes and alkoxysilanes.
- halosilane examples include alkylhalosilanes having an alkyl group having 1 to 12 carbon atoms and arylhalosilanes having an aryl group having 6 to 12 carbon atoms, such as methyltrichlorosilane, dimethyldichlorosilane, trimethylchlorosilane, trimethylbromosilane, Examples thereof include ethyltrichlorosilane, phenyltrichlorosilane, diphenyldichlorosilane, and t-butyldimethylchlorosilane.
- alkoxysilane examples include those having an alkyl group or an aryl group having 1 to 12 carbon atoms and an alkoxy group having 1 to 2 carbon atoms, such as methyltrimethoxysilane, dimethyldimethoxysilane, phenyltrimethoxysilane, Diphenyldimethoxysilane, o-methylphenyltrimethoxysilane, p-methylphenyltrimethoxysilane, n-butyltrimethoxysilane, i-butyltrimethoxysilane, hexyltrimethoxysilane, octyltrimethoxysilane, decyltrimethoxysilane, dodecyl Trimethoxysilane, tetraethoxysilane, methyltriethoxysilane, dimethyldiethoxysilane, phenyltriethoxysilane, diphenyldiethoxysilane,
- the lipophilic compound in addition to the above, known coupling agents (silane coupling agents other than those described above, titanate coupling agents, zircoaluminate coupling agents, etc.) and the like can also be used.
- alkylhalosilanes and alkoxysilanes are preferable from the viewpoints of defoaming properties and product stability, and alkoxysilanes are more preferable.
- the M value of hydrophobic dry silica (S) is preferably 30 to 80, more preferably 30 to 75, particularly preferably 35 to 75, and most preferably 40 to 70.
- the M value is a concept representing the degree of hydrophobicity, and the higher the M value, the lower the hydrophilicity.
- S hydrophobic dry silica
- ⁇ M value calculation method 0.2 g of sample ⁇ hydrophobic dry silica (S) ⁇ is added to 50 mL of water in a 250 mL beaker, followed by dropwise addition of methanol from the burette until the entire sample is suspended. At this time, the solution in the beaker is constantly stirred with a magnetic stirrer, and the time when the entire amount of the sample is uniformly suspended in the solution is taken as the end point, and the volume percentage of methanol in the liquid mixture in the beaker at the end point becomes the M value.
- Hydrophobic dry silica (S) can be easily obtained from the market.
- trade names include the following.
- metal fine particles (aluminum, titanium, etc.) having a primary particle diameter of 5 to 100 nm can also be used.
- the primary particle diameter (nm) of hydrophobic dry silica (S) is 5 to 100, preferably 5 to 80, more preferably 10 to 60, and particularly preferably 10 to 40. Within this range, antifoaming properties and product stability are further improved. In general, there is no change in the primary particle size before and after hydrophobization.
- the primary particle size of the hydrophobic dry silica (S) is JIS Z8901-2006 “Test powder and test particles” 5.44 Particle size distribution (c) A sample prepared by a sprinkling method is used. JIS Z8827-1: 2008 (corresponding international standard; ISO133322-1; the present disclosure by reference to the disclosure content disclosed in this document) Equivalent to a circle calculated using computer software for image processing ⁇ for example, WinRoof manufactured by Mitani Corporation) according to “Particle size—Image analysis method—Part 1: Static image analysis method” It is the number average value of diameters.
- ester compound (E) any compound represented by the general formula (1) can be used without limitation.
- the alkyl group having 1 to 21 carbon atoms may be a linear alkyl group or a branched alkyl group. Groups etc. can be used.
- linear alkyl group examples include methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl and icosyl. .
- branched alkyl groups include isopropyl, isobutyl, t-butyl, isopentyl, neopentyl, isohexyl, 2-ethylhexyl, isotridecyl, isotetradecyl, isooctadecyl, 2-propylheptyl, 2-butyloctyl, 2-hexyldecyl, 2 -Octyldodecyl, 2-dodecylhexyl, 3,5,5-trimethylhexyl, 3,7,11-trimethyldodecyl and the like.
- linear alkenyl groups include vinyl, allyl, propenyl, butenyl, pentenyl, heptenyl, octenyl, nonenyl, decenyl, undecenyl, dodecenyl, tridecenyl, tetradecenyl, pentadecenyl, hexadecenyl, heptadecenyl, octadecenyl, nonadecenyl, and the like.
- Examples of the branched alkenyl group include isobutenyl, isopentenyl, neopentenyl, isohexenyl, isotridecenyl and isooctadecenyl.
- alkyl groups are preferable, more preferably straight chain alkyl groups, and particularly preferably straight chain alkyl groups having 8 to 20 carbon atoms, Most preferred are dodecyl and octadecyl.
- examples of the compound (D ′) containing 2 to 6 carbon atoms and 1 to 6 hydroxyl groups include monoalcohol having an alkyl group having 2 to 6 carbon atoms and 2 to 6 carbon atoms. Of 2 to 6 hydric alcohols.
- Examples of the monoalcohol having an alkyl group having 2 to 6 carbon atoms include ethyl alcohol, n-propyl alcohol, n-butyl alcohol, n-pentyl alcohol and n-hexyl alcohol. Of these, n-butyl alcohol and n-hexyl alcohol are preferred, and n-butyl alcohol is more preferred.
- divalent to hexavalent alcohol having 2 to 6 carbon atoms examples include ethylene glycol, propylene glycol, diethylene glycol, dipropylene glycol, tetramethylene glycol, hexamethylene glycol, glycerin, trimethylolpropane, cyclohexylene glycol, pentaerythritol, sorbitan and sorbitol. Etc. Of these, diethylene glycol, dipropylene glycol and trimethylolpropane are preferred.
- Examples of the adduct obtained by adding an alkylene oxide having 2 to 4 carbon atoms to the compound (D ′) containing 2 to 6 carbon atoms and 1 to 6 hydroxyl groups in the general formula (1) include the above compounds. Any alkylene oxide of (D ′) can be used without limitation.
- alkylene oxide having 2 to 4 carbon atoms examples include ethylene oxide, propylene oxide and butylene oxide.
- the order of reaction (block shape, random shape and combinations thereof) and the use ratio are not limited, but the block shape Or it is preferable that the combination of block shape and random shape is included.
- These chemical reactions may be carried out in any form such as anionic polymerization, cationic polymerization or coordination anionic polymerization. These polymerization forms may be used alone or in combination according to the degree of polymerization.
- p is the number of hydroxyl groups of the compound (D ′) or the number of hydroxyl groups of the adduct obtained by adding alkylene oxide to the compound (D ′) in the case of a full ester of a monovalent to trivalent alcohol.
- a partial ester it corresponds to a number smaller than the number of these hydroxyl groups (the number of hydroxyl groups involved in the ester bond among all hydroxyl groups).
- ester compound (E) examples include ethylene glycol formate diester, glycerin acetic acid triester, trimethylolpropane butyric acid triester, tetramethylene glycol capric acid diester, polyethylene glycol acrylic acid diester, glycerin polypropylene oxide adduct lauric acid diester, ethylene glycol Stearic acid diester, propylene glycol behenic acid diester, glycerin crotonic acid triester, trimethylolpropane oleic acid triester, tetramethylene glycol linoleic acid diester, ethylene glycol alagidonic acid diester, glycerine erucic acid triester, monococonut oil fatty acid sorbitan ester , Sorbitan monostearate, sorbitan monooleate Ether, trioleate sorbitan esters and polyoxyethylene coconut oil fatty acid sorbitan esters and the like.
- the HLB value of the ester compound (E) is preferably 2 to 7, more preferably 3 to 7, particularly preferably 4 to 7, and most preferably 4 to 6. Within this range, product stability and antifoaming properties at high temperatures are further improved.
- HLB is a concept that represents the balance between hydrophilic and hydrophobic groups in a molecule, and the value is "Surfactant properties and applications" (author Takao Karime, publisher Koyukibo, Ltd., September 1980) (Issued on the 1st of January), page 89 to page 90, and “calculation method of HLB by emulsification test”.
- the ester compound can be calculated by the following test method.
- the ester compound (X) whose HLB value is unknown and the emulsifier (A) whose HLB value is known are mixed at different ratios to emulsify the oil agent whose HLB value is known.
- the HLB value of the ester compound (X) is calculated from the mixing ratio when the thickness of the emulsified layer becomes maximum using the following formula.
- HLB value of oil agent ⁇ (W A ⁇ HLB A ) + (W X ⁇ HLB X ) ⁇ ⁇ (W A + W X )
- W A is the weight fraction of the emulsifier (A) based on the total weight of the ester compound (X) and the emulsifier (A)
- W X is the ester compound (X) based on the total weight of the ester compound (X) and the emulsifier (A).
- HLB A is the HLB value of the emulsifier (A)
- HLB X is the HLB value of the ester compound (X).
- the content (% by weight) of the ester compound (E) is preferably 1 to 25, more preferably 3 to 25, particularly preferably 5 to 25, and most preferably 5 based on the weight of the hydrophobic liquid (Q). ⁇ 20. Within this range, the product stability and antifoaming properties at high temperatures are further improved.
- the ester compound (E) can be easily obtained from the market, and examples thereof include the following products. Ionette S-60C (manufactured by Sanyo Chemical Industries, Ltd., sorbitan monostearate, HLB value: 4.7, “Ionette” is a registered trademark of the company). same as below.
- Ionette S-20 (manufactured by Sanyo Chemical Industries, sorbitan monolaurate, HLB value: 8.6), Ionette S-85 (sorbitan trioleate, HLB value: 1.8), Ionet MO-400 (polyoxy) Ethylene monooleate, HLB value: 11.8), Rheodor SP-L10 (manufactured by Kao Corporation, sorbitan monolaurate, HLB value: 8.6), Emazole L-10 (F) (manufactured by Kao Corporation) Sorbitan monolaurate, HLB value: 8.6), Rhedol TW-L120 (polyoxyethylene sorbitan monolaurate, HLB value: 16.7), Nonion L-2 (manufactured by NOF Corporation, polyoxyethylene) Monolaurate, HLB value: 9.9), Nonion CP-08R (Sorbitan monocaprylate, HLB value: 9.6), Unigri K-207 ⁇ manufactured by NOF
- the hydrophobic liquid (Q) contains a hydrophobic substance in addition to the ester compound (E).
- a hydrophobic substance it does not dissolve easily in water and can be used without limitation as long as it is uniformly mixed with the ester compound (E) and is liquid (25 ° C.).
- Hydrocarbon oil (A1), organopolysiloxane (A2) Polyether compound (A3), fatty acid metal salt (B1), fatty acid amide (B2), and hydrophobic wet silica (B3) obtained by hydrophobizing silica by a wet method having a volume average particle diameter of 1 to 15 ⁇ m.
- the hydrophobic liquid (Q) includes at least one selected from the group consisting of an ester compound (E), a hydrocarbon oil (A1), an organopolysiloxane (A2), and a polyether compound (A3), and a fatty acid metal salt. (B1), fatty acid amide (B2) and at least one selected from the group consisting of hydrophobic wet silica (B3) obtained by hydrophobizing silica by a wet method having a volume average particle diameter of 1 to 15 ⁇ m. .
- Hydrocarbon oil (A1) includes mineral oil and synthetic lubricating oil.
- the mineral oil includes mineral oil having a kinematic viscosity at 40 ° C. of 5 to 40 mm 2 / s, and examples thereof include spindle oil, machine oil, and refrigeration oil.
- spindle oil As a trade name of mineral oil ⁇ Numerical values in parentheses are pour points (° C).
- Synthetic lubricating oils include polyolefin oil ( ⁇ -olefin oil), polybutene oil, alkylbenzene oil (alkylate oil) and isoparaffin oil.
- Isoparaffin oil includes isoparaffin oil having a kinematic viscosity at 25 ° C. of 1 to 20 mm 2 / s.
- linearene ⁇ Idemitsu Kosan Co., Ltd., “Linearene” is a registered trademark of the company.
- Dialen ⁇ Mitsubishi Chemical Corporation “Dialen” is a registered trademark of the same company.
- NAS-5H ⁇ NOF Corporation ⁇ .
- the pour point of the hydrocarbon oil (A1) is preferably ⁇ 50 to 2 ° C.
- the pour point is measured according to JIS K2269-1987 (3. Pour point test method).
- the organopolysiloxane (A2) includes a polymer having a siloxane bond as a main chain and a side chain having an alkyl group, an aryl group, an alkoxyl group, a polyoxyalkylene group, etc., and a polyalkylsiloxane ⁇ dimethylpolysiloxane, methyl Ethylpolysiloxane, diethylpolysiloxane, hydrogenmethylpolysiloxane, methylphenylpolysiloxane and dimethylsiloxane-alkoxy (4 to 12 carbon atoms) methylsiloxane copolymer ⁇ and polyalkylethylene at the side chain or terminal of the polyalkylsiloxane And / or modified silicone to which a polyoxypropylene chain or the like is added.
- the kinematic viscosity at 25 ° C. of the organopolysiloxane (A2) is preferably from 50 to 100,000 mm 2 / s.
- the kinematic viscosity at 25 ° C. is measured according to JIS K2283: 2000 (corresponding international standards; ISO2909: 1981 and ISO3104: 1994; the disclosure disclosed in this document is incorporated herein by reference).
- Examples of the polyether compound (A3) include monoalcohols having an alkyl group having 2 to 6 carbon atoms, and compounds obtained by adding an alkylene oxide having 2 to 4 carbon atoms to a polyhydric alcohol having 2 to 6 carbon atoms. .
- Examples of the monoalcohol having an alkyl group having 2 to 6 carbon atoms include ethyl alcohol, n-propyl alcohol, n-butyl alcohol, n-pentyl alcohol and n-hexyl alcohol. Of these, n-butyl alcohol and n-hexyl alcohol are preferred, and n-butyl alcohol is more preferred.
- polyhydric alcohol having 2 to 6 carbon atoms examples include ethylene glycol, propylene glycol, diethylene glycol, dipropylene glycol, tetramethylene glycol, hexamethylene glycol, glycerin, diglycerin, trimethylolpropane, cyclohexylene glycol, pentaerythritol, sorbitan and Examples include sorbitol. Of these, diethylene glycol, dipropylene glycol and trimethylolpropane are preferred.
- alkylene oxide having 2 to 4 carbon atoms examples include ethylene oxide, propylene oxide and butylene oxide.
- the order of reaction (block, random, and combinations thereof) and the use ratio are not limited, but the block or block It is preferable to include a combination with a random state.
- These chemical reactions may be carried out in any form such as anionic polymerization, cationic polymerization or coordination anionic polymerization. These polymerization forms may be used alone or in combination according to the degree of polymerization.
- the number average molecular weight of the polyether compound (A3) is preferably 500 to 5,000.
- the fatty acid metal salt (B1) includes a compound represented by the general formula (2).
- R 1 represents an alkyl group having 1 to 21 carbon atoms or an alkenyl group having 2 to 21 carbon atoms
- M represents a 1 to 3 valent metal atom
- q represents an integer of 1 to 3.
- alkyl group having 1 to 21 carbon atoms or the alkenyl group having 2 to 21 carbon atoms (R 1 ) is the same as that described in the general formula (1).
- alkyl groups straight chain alkyl groups and straight chain alkenyl groups
- more preferably straight chain alkyl groups are preferable, more preferably straight chain alkyl groups, and particularly preferably straight chain alkyl groups having 8 to 20 carbon atoms, Most preferred are dodecyl and octadecyl.
- Examples of the monovalent to trivalent metal atom (M) include alkali metals (lithium, sodium, potassium, etc.), alkaline earth metals (barium, calcium, magnesium, etc.), transition metals (zinc, nickel, iron, copper, manganese, Cobalt, silver, gold, platinum, palladium, titanium, zirconium, cadmium, etc.), Periodic Table Group 13 metals (aluminum, etc.), Group 14 metals (tin, lead, etc.) or lanthanoid metals (lanthanum, cerium, etc.) And the like.
- a bivalent to trivalent metal atom is preferable, a zinc atom, a magnesium atom and an aluminum atom are more preferable, and an aluminum atom is particularly preferable.
- the fatty acid metal salt (B1) may be composed of one residue obtained by removing a hydrogen atom from a fatty acid and one metal atom, or one metal atom and plural (2 to 3). (Preferably) fatty acid residues.
- the compound represented by the general formula (2) may be a single compound having one kind of q or a plurality of mixtures having different q. Moreover, the mixture from which the kind of alkyl group or alkenyl group differs may be sufficient.
- the fatty acid metal salt (B1) may be composed of one kind of metal atom and plural kinds of fatty acids, or may be composed of plural kinds of metal atoms and one kind of fatty acid.
- the meaning of both a salt and a complex is contained in a fatty acid metal salt.
- Preferred examples of the fatty acid metal salt (B1) include zinc laurate, zinc behenate, calcium stearate, zinc stearate, aluminum stearate and magnesium stearate. Such a preferable fatty acid metal salt (B1) can be easily obtained from the market, and examples thereof include the following products.
- the fatty acid amide (B2) includes a compound represented by the general formula (3).
- R 2 represents an alkyl group having 9 to 21 carbon atoms or an alkenyl group having 10 to 21 carbon atoms, and t represents an integer of 1 to 3.
- the alkyl group having 9 to 21 carbon atoms or the alkenyl group having 10 to 21 carbon atoms (R 2 ), the alkyl group having 1 to 21 carbon atoms or the alkenyl group having 2 to 21 carbon atoms (R 1 ) of the general formula (1) ) are the same as those applicable to carbon number.
- Examples of the fatty acid amide (B2) include ethylene biscetylamide, ethylene bisstearylamide, ethylene bispalmitylamide, ethylene bismyristylamide, ethylene bislaurylamide, ethylene bisoleylamide, ethylene bisoctylamide, propylene bisstearylamide, Examples include propylene bispalmitylamide, propylene bismyristylamide, propylene bislaurylamide, propylene bisoleylamide, butylene bisstearylamide, butylene bispalmitylamide, butylene bismyristylamide, butylene bislaurylamide, and butylene bisoleylamide. .
- any hydrophobized silica obtained by a wet method having a volume average particle diameter of 1 to 15 ⁇ m can be used without limitation.
- Silica by a wet method having a volume average particle diameter of 1 to 15 ⁇ m includes silica by a wet method among amorphous synthetic silica (SN). That is, as described above, amorphous synthetic silica (SN) includes dry method (pyrolysis method, melting method) silica (SD) and wet method (gel method, precipitation method) silica (SW). Among these, silica by a wet method having a volume average particle diameter of 1 to 15 ⁇ m can be used as the wet method silica.
- Hydrophobic wet silica is a hydrophobized silica obtained by hydrophobizing wet process silica (SW) with a lipophilic compound and having a volume average particle size of 1 to 15 ⁇ m (distinguishable from the previously described hydrophobic dry silica). ) Is included.
- Hydrophobization of wet silica (SW) with a lipophilic compound can be performed by a known method.
- wet silica (SW) is treated with a solvent (organic solvent (toluene, xylene, biphenyl, dimethyl sulfoxide, etc.) and kinematic viscosity 5 to
- a solvent organic solvent (toluene, xylene, biphenyl, dimethyl sulfoxide, etc.)
- kinematic viscosity 5 to A wet treatment method in which a lipophilic compound is adsorbed or reacted on the surface of silica particles in a 30 mm 2 / s (40 ° C.) paraffin oil, mineral oil, or the like ⁇ .
- adsorbing or reacting a lipophilic compound on the surface of wet silica (SW) by a wet treatment method (1) physical adsorption to the pores of wet silica (SW), and (2) wet silica (SW) Electroadsorption between the surface charge of the lyophilic compound and the ionic functional group of the lipophilic compound can be used.
- the method (1) based on physical adsorption is preferable.
- silicone oil and modified silicone oil can be used.
- silicone oil examples include dimethylsiloxane and cyclotetradimethylsiloxane having a kinematic viscosity of 10 to 3000 (mm 2 / s, 25 ° C.).
- a part of the methyl group of the dimethylsiloxane is an alkyl group having 2 to 6 carbon atoms, an alkoxyl group having 2 to 4 carbon atoms, a phenyl group, a hydrogen atom, a halogen (such as chlorine and bromine) atom, and / or Alternatively, those substituted with an aminoalkyl group having 2 to 6 carbon atoms are included.
- the amount (% by weight) of the lipophilic compound used is preferably 5 to 70, more preferably 7 to 50, and particularly preferably 10 to 30 based on the weight of wet silica (SW). Within this range, the defoaming property is further improved.
- the heating temperature is preferably 100 to 400, more preferably 120 to 300, and particularly preferably 140 to 250.
- the volume average particle diameter ( ⁇ m) of the hydrophobic wet silica (B3) is preferably 1 to 15, more preferably 1.5 to 14, and particularly preferably 2 to 13. Within this range, the defoaming property is further improved. In general, there is no change in the volume average particle diameter before and after hydrophobization.
- the volume average particle size of the hydrophobic wet silica is JIS Z8825-1: 2001 (corresponding international standard; ISO13320-1: 1999 Particle-size analysis-Laser-diffraction-methods-Part-1: General-principles; see the disclosure disclosed in this document Incorporated into the present application by a laser diffraction particle size analyzer (for example, Microtrac Model No. manufactured by Lees & Northrup).
- the refractive index of methanol is 1.329
- the refractive index of the measurement sample is a literature value (“A GUIDE It is calculated as a 50% cumulative volume average particle diameter using FOR ENTERING MICROTRAC "RUN INFORMATION" (F3) DATA, created by Lees & Northrup).
- Hydrophobic wet silica (B3) can be easily obtained from the market, and examples thereof include the following products.
- the hydrophobic liquid (Q) can contain a wax.
- a wax As waxes, plant waxes (carnauba wax, rice wax, etc.), animal waxes (beeswax, etc.), mineral waxes (montan wax, etc.), synthetic waxes (polyethylene, polypropylene, polyethylene oxide, polypropylene oxide, etc.), ethylene-propylene copolymer And ethylene-butene copolymer, ethylene- ⁇ -olefin copolymer, ethylene-vinyl acetate copolymer, ethylene-acrylic acid copolymer and ethylene-propylene-maleic anhydride copolymer). .
- the content (% by weight) is preferably 20 to 95, more preferably 25 to 25, based on the weight of the hydrophobic liquid (Q). 95, particularly preferably 30 to 90, most preferably 30 to 80. Within this range, the antifoaming property is further improved.
- the content is preferably 5 to 95, more preferably 5 to 5 based on the weight of the hydrophobic liquid (Q). 90, particularly preferably 10 to 85, most preferably 10 to 80. Within this range, the antifoaming property is further improved.
- the content (% by weight) is preferably from 5 to 95, more preferably from 5 to 95, based on the weight of the hydrophobic liquid (Q). 90, particularly preferably 10 to 85, most preferably 10 to 80. Within this range, the antifoaming property is further improved.
- the content (% by weight) is preferably 0.5 to 6, more preferably based on the weight of the hydrophobic liquid (Q). It is 0.8 to 6, particularly preferably 1 to 6, and most preferably 1 to 5. Within this range, the antifoaming property is further improved.
- the content (% by weight) is preferably 0.5 to 6, more preferably 0, based on the weight of the hydrophobic liquid (Q). 8 to 6, particularly preferably 1 to 6, and most preferably 1 to 5. Within this range, the antifoaming property is further improved.
- the content (% by weight) is preferably 0.5 to 5, more preferably based on the weight of the hydrophobic liquid (Q). Is 0.5 to 4, particularly preferably 1 to 4, and most preferably 1 to 3. Within this range, the antifoaming property is further improved.
- the content (% by weight) is preferably 0.5 to 10, more preferably 0.5 to 8, based on the weight of the hydrophobic liquid (Q). Particularly preferred is 1 to 7, and most preferred is 1 to 5. Within this range, the antifoaming property is further improved.
- tap water As water, tap water, industrial water, distilled water, ion exchange water, ground water, etc. can be used. Of these, tap water, industrial water, distilled water and ion exchange water are preferred.
- the antifoaming agent of the present invention may contain a surfactant or the like in addition to the hydrophobic dry silica (S) having a primary particle size of 5 to 100 nm, water and the hydrophobic liquid (Q).
- S hydrophobic dry silica
- Q hydrophobic liquid
- surfactant known nonionic, cationic, anionic or amphoteric surfactants can be used.
- Nonionic surfactants include higher alkylamine alkylene oxide adducts, higher fatty acid amide alkylene oxide adducts, acetylene glycol alkylene oxide adducts, polyoxyalkylene-modified silicones (polyether-modified silicones), and polyglycerin fatty acid esters. Etc. However, the above-mentioned polyether compound (A3) is not included in the nonionic surfactant.
- Cationic surfactants include higher alkylamine salts, higher alkylamine alkylene oxide adducts, solomin A type cationic surfactants, sapamin A type cationic surfactants, Arcobel A type cationic surfactants, and imidazoline type cationic surfactants. Agents, higher alkyltrimethylammonium salts, higher alkyldimethylbenzylammonium salts, sapamine-type quaternary ammonium salts and pyridinium salts.
- Anionic surfactants include fatty acid alkali metal salts, fatty acid ammonium salts, fatty acid amine salts, ⁇ -olefin sulfonates, alkylbenzene sulfonic acids and their salts, alkyl sulfate esters, alkyl ether sulfate esters, N-acyl alkyls. Examples include taurine salts and alkylsulfosuccinates. However, the aforementioned fatty acid metal salt (B1) is not included in the anionic surfactant.
- amphoteric surfactants include higher alkylaminopropionates and higher alkyldimethylbetaines.
- the content (% by weight) of the hydrophobic dry silica (S) having a primary particle diameter of 5 to 100 nm is determined according to the defoaming agent (hydrophobic dry silica (S) having a primary particle diameter of 5 to 100 nm, water, hydrophobic 0.02 to 2, preferably 0.05 to 1.8, particularly preferably 0.05 to 2, based on the weight of the liquid (Q) and, if necessary, the antifoaming agent of the present invention comprising a surfactant (hereinafter the same). Is 0.07 to 1.5, most preferably 0.1 to 1. Within this range, antifoaming properties and product stability are further improved.
- the water content (% by weight) is preferably 8 to 70, more preferably 14 to 69, particularly preferably 19 to 64, and most preferably 24 to 64, based on the weight of the antifoaming agent. Within this range, the antifoaming property is further improved.
- the content (% by weight) of the hydrophobic liquid (Q) is preferably 28 to 90, more preferably 30 to 85, particularly preferably 35 to 80, and most preferably 35 to 75, based on the weight of the antifoaming agent. It is. Within this range, the antifoaming property is further improved.
- the content is preferably 0.3 to 4, more preferably 0.6 to 3, particularly preferably 0.9 to 4, based on the weight of the antifoaming agent. 2, most preferably 1-2. In this range, the product stability may be further improved.
- the viscosity (mPa ⁇ s / 25 ° C.) of the antifoaming agent of the present invention is preferably 300 to 3000, more preferably 300 to 2500, particularly preferably 500 to 2500, and most preferably 500 to 2000. Within this range, the product stability and antifoaming properties are further improved.
- the viscosity is JIS K7233-1986 4.2 single cylinder rotational viscometer method (corresponding international standard: ISO2555Resins25inRethe liquid state or as emulsions or dispersions-Determination of Brookfield RV viscosity, ISO3104Petroleum products-Transparent and-opaw Detemination of kinematic viscosity and calculation of dynamic viscosity, ISO3105 Glass capillary kinematic viscometer-Specifucation and opreting instructions; the disclosure disclosed in this document is incorporated herein by reference).
- the antifoaming agent of the present invention can be produced by a known method or the like. For example, the following method or the like can be applied.
- ⁇ Manufacturing method 1> A method comprising the step (1) of adding and mixing hydrophobic dry silica (S) to a mixed liquid comprising a hydrophobic liquid (Q) and water.
- a method comprising the step (2) of adding a hydrophobic dry silica (S) to a mixed liquid composed of a hydrophobic liquid (Q) and a part of water, and further adding and mixing the remaining water.
- a method comprising a step (3) of adding and mixing water to a mixed liquid comprising a hydrophobic liquid (Q) and a hydrophobic dry silica (S).
- the temperature for addition and mixing is about 10 to 70 ° C.
- these may be added all at once, may be added successively little by little, or divided (for example, divided into 2 to 10 times). And may be added.
- the production methods (1) to (3) they may be added and mixed (emulsified) with a homogenizer, a disper mill or the like.
- the antifoaming agent of the present invention contains a surfactant, the surfactant can be added and mixed in an arbitrary stage.
- the hydrophobic liquid (Q) may include a mixing step (4) in which the ester compound (E) and a hydrophobic substance other than the ester compound (E) are simply mixed uniformly. Step (5) may be included.
- the mixing step (4) may be performed before or after the above production methods (1) to (3) ⁇ steps (1) to (3) ⁇ .
- the hydrophobic liquid (Q) contains the fatty acid metal salt (B1) and / or the fatty acid amide (B2) (including the case of containing a wax)
- the heating and mixing step (5) may be performed before or after the above production methods 1 to 3 ⁇ steps (1) to (3) ⁇ , but is preferably performed before.
- the heating temperature is preferably 80 to 200, more preferably 90 to 190, particularly preferably 100 to 180, and most preferably 110 to 170.
- the hydrophobic liquid (Q) contains the fatty acid amide (B2) and includes the heating and mixing step (5)
- the fatty acid amide (B2) is dissolved (or melted) and then cooled with stirring.
- a cooling step (6) followed by a heat treatment step (7) (preferably about 3 hours) in which heat treatment is performed with stirring at 60 to 80 ° C. preferable.
- the hydrophobic liquid (Q) does not contain the fatty acid amide (B2), it is preferable not to include the cooling step (6) and the heat treatment step (7).
- the hydrophobic liquid (Q) is prepared through a cooling step (8) for cooling to 40 ° C. or lower to obtain the hydrophobic liquid (Q).
- each of the cooling step (6), the heat treatment step (7), and the cooling step (8) is performed after the heating and mixing step (5), the above production methods 1 to 3 ⁇ steps (1) to (3) ⁇ It may be performed before or after, but is preferably performed before.
- the hydrophobic liquid (Q) includes the hydrocarbon oil (A1) and the fatty acid amide (B2)
- a part of the hydrocarbon oil (A1) is used in the heating and mixing step (5), and the remaining hydrocarbon oil (A1) may be added in the subsequent cooling step (6), heat treatment step (7) and / or cooling step (8).
- the fatty acid metal salt (B1), fatty acid amide (B2) and / or ester may be obtained with a ball mill, disper mill, homogenizer or gorin homogenizer.
- Compound (E) may be refined.
- the antifoaming agent of the present invention is effective for aqueous foaming liquid. Therefore, it can be used as an antifoaming agent for paints (water-based paints, etc.) and an antifoaming agent for various production processes (paper making process, fermentation process, wastewater treatment process, monomer stripping process, polymer polymerization process, etc.).
- examples of the binder contained in the emulsion paint include vinyl acetate resin, acrylic resin, styrene resin, halogenated olefin resin, urethane resin, silicone resin, or fluorine atom-containing silicone resin. is there.
- a method for adding the antifoaming agent of the present invention when applied to a paint, (1) a method of adding at the time of pigment dispersion and / or (2) after preparation of the paint, etc. may be mentioned. Moreover, when applying to various manufacturing processes, any of (1) Supply of raw materials, (2) Before heating and / or pressure reduction treatment, and / or (3) Final finishing process etc. may be used.
- the amount (% by weight) of the antifoaming agent of the present invention is preferably 0.1 to 5, more preferably 0.3 to 4, particularly preferably 0. 5 to 4, most preferably 0.5 to 3.
- the addition amount (% by weight) of the antifoaming agent of the present invention is preferably 0.005 to 1, more preferably 0.006 to 08, based on the weight of the aqueous liquid. Particularly preferred is 0.008 to 0.6, and most preferred is 0.01 to 0.5.
- part means “part by weight”
- % means “% by weight”.
- ⁇ Production Example 1> In a stainless steel container, 5 parts of an ester compound (e1) ⁇ IONET S-80, sorbitan monooleate, HLB: 4.3, manufactured by Sanyo Chemical Industries, Ltd. ⁇ and a hydrocarbon oil (a11) ⁇ Cosmo Pure Spin RB, manufactured by Cosmol Bricantz Co., Ltd., pour point -12.5 ° C., “Pure Spin” is a registered trademark of the same company. ⁇ 80 parts, polyether compound (a31) ⁇ New Pole LB-1715, polyoxypropylene (degree of polymerization: 40) butyl ether, manufactured by Sanyo Chemical Industries, Ltd., "New Pole” is a registered trademark of the same company.
- ester compound (e3) ⁇ Leodol MS-60, glycerin monostearate, HLB: 3.5, manufactured by Kao Corporation ⁇ and hydrocarbon oil (a13) ⁇ Cosmo Pure Spin G, Cosmolbricans Co., Ltd., 74 parts pour point -7.5 ° C., organopolysiloxane (a21) ⁇ KF96-1,000cs, dimethyl silicone oil, Shin-Etsu Chemical Co., Ltd., 1,000 mm 2 / s / 25 ° C ⁇ 5 parts, hydrophobic wet silica (b31) ⁇ Nipsil SS-50, manufactured by Tosoh Silica Co., Ltd., volume average particle diameter 1 ⁇ m, M value 65, “Nipsil” is a registered trademark of the same company.
- the mixture was heated to 180 ° C. while stirring at 3000 rpm with a homogenizer, and further heated and stirred at this temperature for 3 hours. Then, stirring the obtained mixture, it cooled by air cooling to 30 degreeC, and obtained the hydrophobic liquid (q4).
- ⁇ Production Example 10> In a stainless steel container, 5 parts of an ester compound (e5) ⁇ ionet S-85 ⁇ , 40 parts of an organopolysiloxane (a24) ⁇ KF96-50cs ⁇ , an organopolysiloxane (a25) ⁇ KF96-100,000 cs, dimethyl silicone 10 parts of oil, manufactured by Shin-Etsu Chemical Co., Ltd., 100,000 mm 2 / s / 25 ° C., and 5 parts of fatty acid amide (b22) ⁇ Alflow AD-281F, ethylenebisoleylamide, manufactured by NOF Corporation ⁇ Then, the temperature was raised to 120 ° C.
- an ester compound (e5) ⁇ ionet S-85 ⁇ 40 parts of an organopolysiloxane (a24) ⁇ KF96-50cs ⁇ , an organopolysiloxane (a25) ⁇ KF96-100,000 cs, dimethyl silicone 10 parts
- Example 1 In a stainless steel container, 90 parts of the hydrophobic liquid (q1) obtained in Production Example 1 and 8.2 parts of ion-exchanged water (hereinafter simply referred to as water) were stirred and mixed to obtain a mixture, Hydrophobic dry silica (s1) ⁇ Aerosil RY200, manufactured by Nippon Aerosil Co., Ltd., primary particle size: 12 nm, M value while stirring the mixture at 3000 rpm with a homogenizer at room temperature (15-20 ° C., the same applies hereinafter) : 75 ⁇ 1.8 parts was added and mixed in 30 minutes, and stirring was further continued for 30 minutes to obtain an antifoaming agent (1) of the present invention.
- s1 Hydrophobic dry silica
- s1 Hydrophobic dry silica (s1) ⁇ Aerosil RY200, manufactured by Nippon Aerosil Co., Ltd., primary particle size: 12 nm
- M value while stirring the mixture at 3000 rpm with a homogen
- Example 2 In a stainless steel container, 28 parts of the hydrophobic liquid (q2) obtained in Production Example 2 and 12 parts of water and a surfactant (1) ⁇ Naroacty CL-40, manufactured by Sanyo Chemical Industries, Ltd., nonionic surfactant , "Naroacty” is a registered trademark of the same company ⁇ 2 parts are mixed with stirring to obtain a liquid mixture, and then the hydrophobic dry silica (s2) while stirring the liquid mixture at 3000 rpm with a homogenizer at room temperature ⁇ Aerosil R104, manufactured by Nippon Aerosil Co., Ltd., primary particle size: 16 nm, M value: 40 ⁇ 0.02 part was added and mixed in 60 minutes, and stirring was continued for another 30 minutes, followed by stirring at 3000 rpm. 57.98 parts was added and mixed in 60 minutes to obtain an antifoaming agent (2) of the present invention.
- a surfactant (1) ⁇ Naroacty CL-40, manufactured by Sanyo Chemical Industries, Ltd., noni
- Example 3 In a stainless steel container, 80 parts of the hydrophobic liquid (q3) obtained in Production Example 3 and 4 parts of water and a surfactant (2) ⁇ SY Glister CRS-75, manufactured by Sakamoto Pharmaceutical Co., Ltd., nonionic surfactant , Polyglycerin-condensed ricinoleic acid ester ⁇ 0.95 part with stirring and mixing to obtain a mixed solution, and then the hydrophobic dry silica (s3) ⁇ Aerosil RX200 while stirring the mixed solution at 3000 rpm with a homogenizer at room temperature.
- a surfactant (2) ⁇ SY Glister CRS-75, manufactured by Sakamoto Pharmaceutical Co., Ltd., nonionic surfactant , Polyglycerin-condensed ricinoleic acid ester ⁇ 0.95 part with stirring and mixing to obtain a mixed solution, and then the hydrophobic dry silica (s3) ⁇ Aerosil RX200 while stirring the mixed solution at 3000 rpm with a
- the antifoaming agent (3) of the present invention was obtained by adding and mixing for 30 minutes.
- Example 4 In a stainless steel container, 70 parts of the hydrophobic liquid (q4) obtained in Production Example 4 and 7 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. However, 0.1 part of hydrophobic dry silica (s4) ⁇ Aerosil R972, manufactured by Nippon Aerosil Co., Ltd., primary particle size: 16 nm, M value: 50 ⁇ was added and mixed in 30 minutes, and stirring was further continued for 30 minutes. Then, while stirring at 3000 rpm, 22.9 parts of water was added and mixed in 30 minutes to obtain an antifoaming agent (4) of the present invention.
- s4 hydrophobic dry silica
- Example 5 In a stainless steel container, 60 parts of the hydrophobic liquid (q5) obtained in Production Example 5 and 12 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. However, 0.2 part of hydrophobic dry silica (s5) ⁇ Aerosil R974, manufactured by Nippon Aerosil Co., Ltd., primary particle size: 12 nm, M value: 45 ⁇ was added and mixed in 30 minutes, and stirring was further continued for 30 minutes. Then, while stirring at 3000 rpm, 27.8 parts of water was added and mixed in 30 minutes to obtain an antifoaming agent (5) of the present invention.
- hydrophobic dry silica ⁇ Aerosil R974, manufactured by Nippon Aerosil Co., Ltd., primary particle size: 12 nm, M value: 45 ⁇ was added and mixed in 30 minutes, and stirring was further continued for 30 minutes. Then, while stirring at 3000 rpm, 27.
- Example 6 In a stainless steel container, 50 parts of the hydrophobic liquid (q6) obtained in Production Example 6 and 10 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. While adding 0.5 parts of hydrophobic dry silica (s3) ⁇ Aerosil RX200 ⁇ over 60 minutes, stirring was continued for another 30 minutes, and then 39.5 parts of water was added over 30 minutes while stirring at 3000 rpm. By mixing, an antifoaming agent (6) of the present invention was obtained.
- Example 7 In a stainless steel container, 45 parts of the hydrophobic liquid (q7) obtained in Production Example 7 and 15 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. While adding 0.3 parts of hydrophobic dry silica (s1) ⁇ Aerosil RY200 ⁇ over 30 minutes, stirring was continued for another 30 minutes, and then 39.7 parts of water was added over 45 minutes while stirring at 3000 rpm. By mixing, the antifoaming agent (7) of the present invention was obtained.
- Example 8> In a stainless steel container, 40 parts of the hydrophobic liquid (q8) obtained in Production Example 8 and 10 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. Then, 1 part of hydrophobic dry silica (s2) ⁇ Aerosil R104 ⁇ was added and mixed in 45 minutes, stirring was continued for another 30 minutes, and then 49 parts of water was added and mixed in 45 minutes while stirring at 3000 rpm. An antifoaming agent (8) of the present invention was obtained.
- s2 hydrophobic dry silica
- Example 9 In a stainless steel container, 85 parts of the hydrophobic liquid (q9) obtained in Production Example 9 and 13.5 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was mixed at 3000 rpm with a homogenizer at room temperature. While stirring, 1.5 parts of hydrophobic dry silica (s4) ⁇ Aerosil R972 ⁇ was added and mixed in 60 minutes, and stirring was further continued for 30 minutes to obtain an antifoaming agent (9) of the present invention.
- s4 hydrophobic dry silica
- Example 10 In a stainless steel container, 35 parts of the hydrophobic liquid (q10) obtained in Production Example 10, 25 parts of water, and 0.93 part of surfactant (1) ⁇ Naroacty CL-40 ⁇ are mixed with stirring. Then, 0.07 parts of hydrophobic dry silica (s1) ⁇ Aerosil RY200 ⁇ was added and mixed for 15 minutes while stirring the mixture at 3000 rpm with a homogenizer at room temperature, and stirring was continued for another 30 minutes. Then, while stirring at 3000 rpm, 39 parts of water was added and mixed in 45 minutes to obtain an antifoaming agent (10) of the present invention.
- Example 11 In a stainless steel container, 30 parts of the hydrophobic liquid (q11) obtained in Production Example 11 and surfactant (3) ⁇ Naroacty CL-70, manufactured by Sanyo Chemical Industries, Ltd., nonionic surfactant ⁇ Surfactant (4) ⁇ SN Wet 984, manufactured by San Nopco Co., Ltd., Nonionic Surfactant ⁇ 2.6 parts were mixed with stirring to obtain a mixed solution, and then the mixed solution was mixed at 3000 rpm with a homogenizer at room temperature.
- hydrophobic dry silica (s5) ⁇ Aerosil R974 ⁇ was added and mixed in 30 minutes, and stirring was continued for another 30 minutes, and then 66 parts of water was added in 90 minutes while stirring at 3000 rpm.
- an antifoaming agent (11) of the present invention was obtained.
- Example 12 In a stainless steel container, 75 parts of the hydrophobic liquid (q12) obtained in Production Example 12, 24 parts of water, and 0.4 part of surfactant (1) ⁇ Naroacty CL-40 ⁇ are mixed with stirring. After stirring, the mixture was stirred at 3000 rpm with a homogenizer at room temperature, 0.6 parts of hydrophobic dry silica (s3) ⁇ Aerosil RX200 ⁇ was added and mixed for 45 minutes, and stirring was continued for another 30 minutes. Thus, an antifoaming agent (12) of the present invention was obtained.
- Example 13 In a stainless steel container, 55 parts of the hydrophobic liquid (q13) obtained in Production Example 13 and 20 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. Then, 2 parts of hydrophobic dry silica (s4) ⁇ Aerosil R972 ⁇ was added and mixed in 75 minutes, and stirring was further continued for 30 minutes. Then, 23 parts of water was added and mixed in 30 minutes while stirring at 3000 rpm. An antifoaming agent (13) of the present invention was obtained.
- s4 hydrophobic dry silica
- Example 14 In a stainless steel container, 35 parts of the hydrophobic liquid (q9) obtained in Production Example 9, 35 parts of the hydrophobic liquid (q13) obtained in Production Example 13, and surfactant (5) ⁇ SN wet 980, San Nopco 3 parts of Nonionic Surfactant, manufactured by Co., Ltd., were stirred and mixed to obtain a mixed solution, and then the hydrophobic dry silica (s5) ⁇ Aerosil R974) was stirred at 3000 rpm with a homogenizer at room temperature. ⁇ Add 0.2 parts in 30 minutes, continue stirring for another 30 minutes, and then add and mix 26.8 parts of water in 60 minutes while stirring at 3000 rpm. )
- ⁇ Comparative Example 1> In a stainless steel container, 55 parts of the hydrophobic liquid (q5) obtained in Production Example 5 and 20 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. While adding 0.5 parts of hydrophobic wet silica (b32) ⁇ Sipernat D10 ⁇ over 30 minutes, stirring was continued for another 30 minutes, and then 24.5 parts of water was added over 30 minutes while stirring at 3000 rpm. By mixing, a defoaming agent (U1) for comparison was obtained.
- ⁇ Comparative Example 2> In a stainless steel container, 60 parts of the hydrophobic liquid (q12) obtained in Production Example 12 and 10 parts of water were stirred and mixed to obtain a mixed liquid, and then the mixed liquid was stirred at 3000 rpm with a homogenizer at room temperature. While adding 0.5 parts of silica ⁇ non-hydrophobized silica, Aerosil 200CF, Nippon Aerosil Co., Ltd., primary particle size: 12 nm ⁇ over 30 minutes, stirring was further continued for 30 minutes, followed by stirring at 3000 rpm. Then, 29.5 parts of water was added and mixed in 60 minutes to obtain a comparative antifoaming agent (U2).
- U2 comparative antifoaming agent
- polyoxyethylene lauryl ether sulfate sodium salt Carribbon (registered trademark) EN-200, Sanyo Chemical Industries Co., Ltd.] 2 parts and 96 parts of ion-exchanged water were added, followed by uniform stirring and mixing for 10 minutes to obtain an effervescent test solution.
- Defoaming test method 750 ml of a foaming test solution was put into a glass cylindrical foam tube having a capacity of about 2000 ml, the temperature was adjusted to 5 ° C or 70 ° C, and the liquid level at this time was read as a reference height. Next, using a circulation pump, the foamability test solution is dropped at 3,000 ml / min from the bottom of the foam pipe from the top of the foam pipe (height 150 mm from the reference height) into the foam pipe. The foaming test solution was continuously circulated.
- the evaluation sample ⁇ the concentration obtained by removing water from the antifoaming agent becomes the foaming test solution with a micropipette.
- the foam height (the height of the uppermost end of the foam—the reference height: mm) is changed to 30 seconds after the start of the test. After, measurements were taken after 1 minute and after 5 minutes. These smaller bubble heights mean higher defoaming properties.
- ⁇ Defoaming property evaluation 2> (1) Preparation of emulsion base paint Grinding and letdown were carried out using an Excel auto homogenizer (model ED, manufactured by Nippon Seiki Co., Ltd.) equipped with impeller blades with the raw material composition shown in Table 2. To paint. The obtained paint was diluted with water so as to be 80 KU (25 ° C.) with a stoma viscometer (JIS K5600-2-2: 1999) to obtain an emulsion base paint.
- Emulsion paints (1) to (18) were obtained by stirring and mixing in an auto homogenizer at 25 ° C., 5000 rpm for 5 minutes.
- the defoamers of Examples 1 to 14 were used for the emulsion paints (1) to (14), and the defoamers of Comparative Examples 1 to 4 were used for the emulsion paints (15) to (18).
- the emulsion coating material (19) was obtained like the above except not adding an antifoamer for blanks.
- Emulsion paints (1) to (19) whose temperature was adjusted to 5 ° C. or 40 ° C. were stirred and mixed at 4000 rpm for 3 minutes in an Excel auto homogenizer equipped with impeller blades to entrain the foam. After 15 seconds, the specific gravity of the emulsion paints (1) to (19) including bubbles was measured with a 50 ml specific gravity cup. The larger the specific gravity value, the less the foam bites and the better the defoaming property.
- Horizontal defoaming tester ⁇ newspaper white water in a cuboid container with open top surface is pumped up from the bottom of one end of the cuboid container with a circulation pump (3000ml / min) 20cm above the level of newspaper paper white water (cuboid) It is a circulation type defoaming tester in which newspaper paper white water is foamed by dropping from the top of the other end of the container, while newspaper paper white water flows 20 cm from the other end to one end and then pumped up by a pump. And a tester capable of measuring the length of bubbles from the other end on the liquid surface in the rectangular parallelepiped container. ; See Japanese Patent No.
- the evaluation sample ⁇ concentration excluding the water from the antifoaming agent] Is the amount of foam in a rectangular parallelepiped container after adding using a micropipette (measured in 5 minutes after the start of the test, in mm, the smaller the value, the more defoaming occurs)
- the defoaming property was evaluated on the basis of the fact that the property is good.
- a paper coating paint for evaluation (with an evaluation sample) is applied to one side of a commercially available medium quality paper (basis weight 63 g / m 2 ) so that the coating amount is 15 g / m 2.
- the coated paper was obtained by blade coating (temperature 50 ° C.) at a coating speed of 50 m / min.
- this coated paper was subjected to a super calender process (conditions: temperature 40 ° C., linear pressure 60 kg / cm, twice) to obtain an evaluation paper.
- the number (number) of bubble marks in the paper surface of the evaluation paper (area: 20 cm ⁇ 20 cm) was measured visually. The smaller the number, the better the defoaming property.
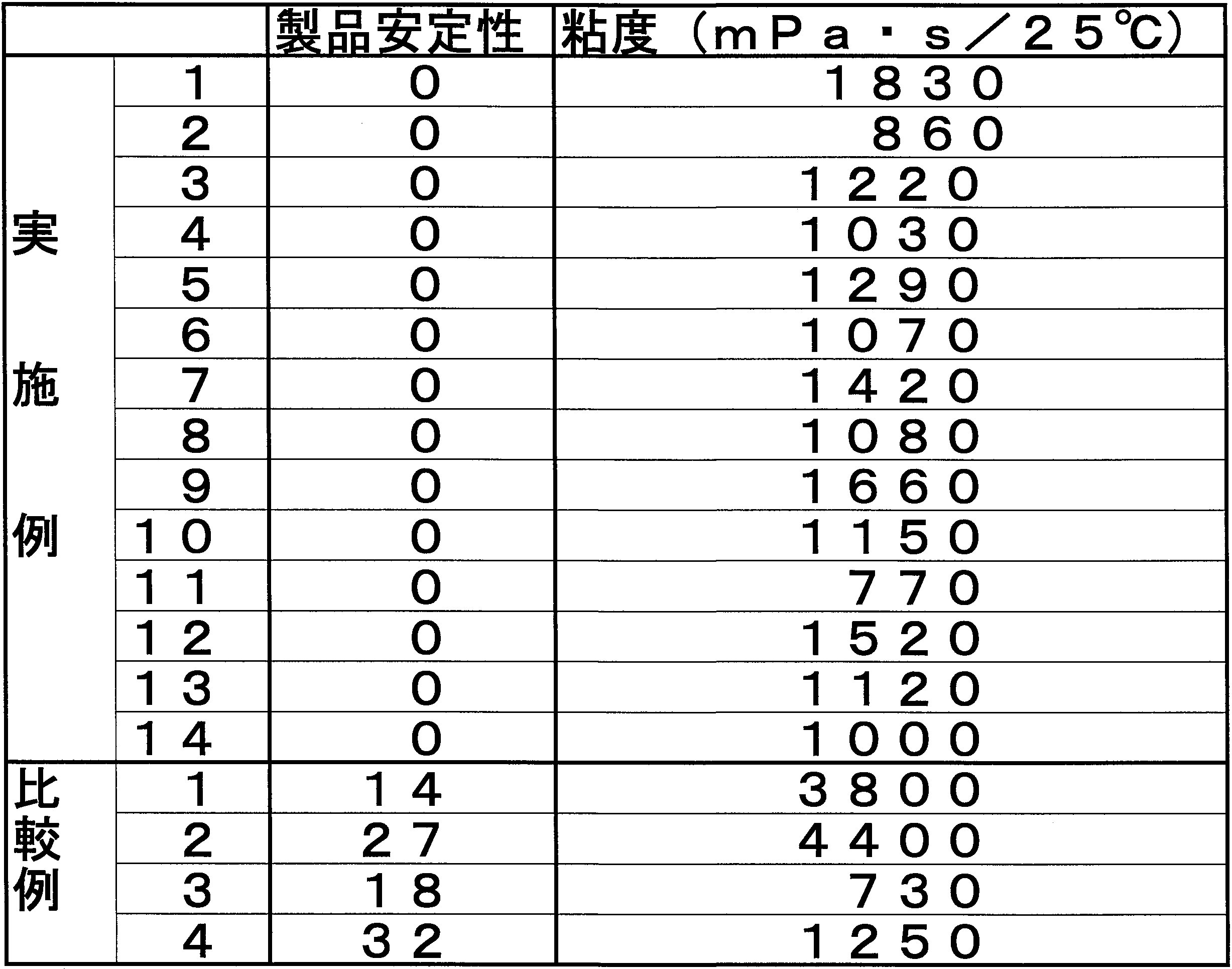
- the antifoaming agent of the present invention exhibits excellent antifoaming properties in a wide temperature range (5 to 70 ° C.) and excellent product stability as compared with a comparative antifoaming agent. I was able to confirm that.
- the antifoaming agent of the present invention can be used for any application, but is effective for aqueous foaming liquids, for example, paint industry (water-based paint, paper coating paint), chemical industry, food industry, petroleum industry. It is suitable in fields such as civil engineering and construction industry, textile industry, pulp and paper industry, pharmaceutical industry or wastewater treatment process.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Toxicology (AREA)
- Dispersion Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Degasification And Air Bubble Elimination (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
また、特許文献2に記載の消泡剤においても、エマルション化には特許文献1のエマルション型消泡剤と同じように乳化剤を必須とすることから、同じ問題を抱えている。
すなわち、本発明の目的は、広い温度範囲で消泡性に優れ、且つ製品安定性に優れる消泡剤を提供することである。
(R1-COO)p-D (1)
(2)溶融法シリカ:天然のシリカ粉末等を火炎中で溶融して得られ、一次粒子(微粒子)として存在し易い。
(3)ゲル法シリカ:酸性環境下にて珪酸ソーダを酸で中和し、生じた析出物をろ過、乾燥することによって得られ、凝集構造を有する。
(4)沈降法シリカ:アルカリ性環境下にて珪酸ソーダを酸で中和し、生じた析出物をろ過、乾燥することによって得られ、細孔容積が大きく、比表面積が大きい。
Aerosilシリーズ{日本アエロジル株式会社及びエボニック デグサ社、「Aerosil」はエボニック デグサ ゲーエムベーハーの登録商標である。}、Reolosilシリーズ{株式会社トクヤマ、「Reorosil」は株式会社トクヤマの登録商標である。}、Cab-O-Silシリーズ{キャボット社、「Cab-O-Sil」はキャボットコーポレーションの登録商標である。}等。
Admafineシリーズ{アドマテックス社、「Admafine」はトヨタ自動車株式会社の登録商標である。}、Fuselexシリーズ{株式会社龍森}、デンカ溶融シリカシリーズ{電気化学工業株式会社}等。
Nipsilシリーズ{東ソー・シリカ株式会社、「Nipsil」は東ソー・シリカ株式会社の登録商標である。}、Sipernatシリーズ{エボニック デグサ ジャパン株式会社、「Sipernat」はエボニック デグサ ゲーエムベーハーの登録商標である。}、Carplexシリーズ{DSL.ジャパン株式会社、「Carplex」はDSL.ジャパン株式会社の登録商標である。}、FINESILシリーズ{株式会社トクヤマ、「FINESIL」は株式会社トクヤマの登録商標である。}、TOKUSIL{株式会社トクヤマ、「TOKUSIL」は株式会社トクヤマの登録商標である。}、Zeosil{ローディア社、「Zeosil」はロディア シミ の登録商標である。}、MIZUKASILシリーズ{水澤化学工業株式会社、「MIZUKASIL」は水沢化学工業株式会社の登録商標である。}等。
Carplexシリーズ、SYLYSIAシリーズ{富士シリシア株式会社、「SYLYSIA」は有限会社ワイ・ケイ・エフ の登録商標である。}、Nipgelシリーズ{東ソー・シリカ株式会社、「Nipgel」は東ソー・シリカ株式会社の登録商標である。}、MIZUKASILシリーズ{水澤化学工業株式会社、「MIZUKASIL」は水沢化学工業株式会社の登録商標である。}等。
これらのうち、消泡性及び製品安定性等の観点の他に凝集を防ぎ一次粒子径を保ちやすいという観点から、(1)の縮合反応を利用する方法が好ましい。
試料{疎水性乾式シリカ(S)}0.2gを容量250mLのビーカー中の50mLの水に添加し、続いてメタノールをビュレットから試料の全量が懸濁するまで滴下する。この際ビーカー内の溶液をマグネティックスターラーで常時攪拌し、試料の全量が溶液中に均一懸濁された時点を終点とし、終点におけるビーカー内の液体混合物のメタノールの容量百分率がM値となる。
Aerosil シリーズ(R972、RX200、RY200、R202、R805及びR812等){日本アエロジル株式会社及びエボニック デグサ社}、Reolosil MT及びDMシリーズ(MT-10、DM-10及びDM-20等){株式会社トクヤマ}、(TS-530TS-610TS-720等){キャボットカーボン社}等。
HLB値が未知のエステル化合物(X)とHLB値が既知の乳化剤(A)を異なった比率で混合し、HLB値が既知の油剤の乳化を行う。乳化層の厚みが最大となったときの混合比率から下記式を用いてエステル化合物(X)のHLB値を算出する。
(油剤のHLB値)={(WA×HLBA)+(WX×HLBX)}÷(WA+WX)
イオネットS-60C{三洋化成工業(株)製、ソルビタンモノステアレート、HLB値:4.7、「イオネット」は同社の登録商標である。以下同じ。}、イオネットS-80(ソルビタンモノオレエート、HLB値:4.3) 、イオネットS-80C(ソルビタンモノオレエート、HLB値:4.3) イオネットDO-20(ポリオキシエチレンジオレエート、HLB値:5.3)、レオドールSP-P10{花王(株)製、ソルビタンモノパルミテート、HLB値:6.7、「レオドール」は同社の登録商標である。以下同じ。}、レオドールSP-S10(ソルビタンモノステアレート、HLB値:4.7)、レオドールSP-S30(ソルビタントリステアレート、HLB値:2.1)、レオドールSP-O10(ソルビタンモノオレエート、HLB値:4.3)、レオドールMS-5O(グリセロールモノステアレート、HLB値:2.8)、レオドールMO-6O(グリセロールモノオレエート、HLB値:2.8)、エマゾールMO-50{花王(株)製、グリセロールモノオレエート、HLB値:2.8、「エマゾ-ル」は同社の登録商標である。以下同じ。}、モノグリD{日油(株)製、グリセリン脂肪酸モノエステル、HLB値:3.8}、モノグリMB(グリセロールモノステアレート、HLB値:5.5)、ノニオンPP-40Rペレット{日油(株)製、ソルビタンモノパルミテート、HLB値:6.7}及びノニオンBP-70R(ソルビタンモノベヘネート、HLB値:3.9)等。
疎水性物質としては、水に容易に溶解せず、エステル化合物(E)と均一混合されて液体(25℃)であれば制限なく使用でき、炭化水素油(A1)、オルガノポリシロキサン(A2)、ポリエーテル化合物(A3)、脂肪酸金属塩(B1)、脂肪酸アミド(B2)及び体積平均粒子径が1~15μmである湿式法によるシリカを疎水化した疎水性湿式シリカ(B3)が含まれる。
鉱物油としては、40℃における動粘度が5~40mm2/sの鉱物油が含まれ、スピンドル油、マシン油及び冷凍機油等が挙げられる。鉱物油の商品名として{括弧内の数値は流動点(℃)である。}、コスモピュアスピンG(-10)、コスモピュアスピンE(0)、コスモSP-10(-12.5)、コスモSP-32(-40)及びコスモSC22(-15)(以上、コスモ石油株式会社、「コスモ」及び「ピュアスピン」は同社の登録商標である。)、並びにスタノール35(-15)、スタノール43N(-15)(エクソンモービルコーポレーション)等が挙げられる。
イソパラフィン油としては、25℃における動粘度が1~20mm2/sのイソパラフィン油が含まれ、商品名として、リニアレン{出光興産(株)、「リニアレン」は同社の登録商標である。}、ダイアレン{三菱化学(株)、「ダイアレン」は同社の登録商標である。}及びNAS-5H{日油(株)}等が挙げられる。
炭化水素油(A1)の流動点は、-50~2℃が好ましい。
流動点は、JIS K2269-1987(3.流動点試験法)に準拠して測定される。
25℃における動粘度は、JIS K2283:2000(対応国際規格;ISO2909:1981及びISO3104:1994;この文献に開示された開示内容を参照により本出願に取り込む。)に準拠して測定される。
(R1-COO)q-M (2)
このような好ましい脂肪酸金属塩(B1)は市場からも容易に入手でき、たとえば以下の商品等が挙げられる。
R2-CONH-(CH2)t-NHCO-R2 (3)
体積平均粒子径が1~15μmである湿式法によるシリカとしては、非晶質合成シリカ(SN)のうち、湿式法によるシリカが含まれる。すなわち、非晶質合成シリカ(SN)としては、上記の通り、乾式法(熱分解法、溶融法)シリカ(SD)及び湿式法(ゲル法、沈降法)シリカ(SW)が含まれるが、これらのうち、体積平均粒子径が1~15μmである湿式法によるシリカを湿式法シリカとして使用できる。
Nipsil SSシリーズ(SS-10、SS-40、SS-50及びSS-115等){東ソー・シリカ株式会社、「Nipsil」は東ソー・シリカ株式会社 の登録商標である。)}、Sipernat D及びCシリーズ(D10、D17、C600及びC630等){デグサジャパン株式会社}、並びにSYLOPHOBICシリーズ(100、702、505及び603等){富士シリシア化学株式会社、「SYLOPHOBIC」は富士シリシア化学株式会社の登録商標である。}等。
ワックスとしては、植物ワックス(カルナウバワックス及びライスワックス等)、動物ワックス(蜜蝋等)、鉱物ワックス(モンタンワックス等)、合成ワックス(ポリエチレン、ポリプロピレン、酸化ポリエチレン、酸化ポリプロピレン等、エチレン-プロピレン共重合体、エチレン-ブテン共重合体、エチレン-α-オレフィン共重合体、エチレン-酢酸ビニル共重合体、エチレン-アクリル酸共重合体及びエチレン-プロピレン-無水マレイン酸共重合体等)等が挙げられる。
<製造方法1>
疎水性液体(Q)及び水からなる混合液に疎水性乾式シリカ(S)を添加混合する工程(1)を含む方法。
疎水性液体(Q)及び水の一部からなる混合液に疎水性乾式シリカ(S)を添加混合し、さらに残りの水を添加混合する工程(2)を含む方法。
疎水性液体(Q)及び疎水性乾式シリカ(S)からなる混合液に、水を添加混合する工程(3)を含む方法。
本発明の消泡剤に界面活性剤を含有する場合、界面活性剤は任意段階で添加混合できる。
また、工程(1)~(4)、(7)及び/又は(8)の後に、ボールミル、ディスパーミル、ホモジナイザー又はゴーリンホモジナイザー等で脂肪酸金属塩(B1)、脂肪酸アミド(B2)及び/又はエステル化合物(E)を微細化処理してもよい。
ステンレス製容器内に、エステル化合物(e1){イオネットS-80、ソルビタンモノオレエート、HLB:4.3、三洋化成工業(株)製}5部と、炭化水素油(a11){コスモピュアスピンRB、コスモルブリカンツ(株)製、流動点-12.5℃、「ピュアスピン」は同社の登録商標である。}80部と、ポリエーテル化合物(a31){ニューポールLB-1715、ポリオキシプロピレン(重合度:40)ブチルエーテル、三洋化成工業(株)製、「ニューポール」は同社の登録商標である。}10部と、脂肪酸金属塩(b11){アルミニウムステアレート900、トリステアリン酸アルミニウム、日油(株)製}5部とを投入した後、ホモジナイザー{ハイフレックスディスパーサーHG-92G、タイテック(株)製、以下同じ}にて3000rpmで攪拌しつつ150℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q1)を得た。
ステンレス製容器内に、エステル化合物(e2){イオネットDL-200、ポリオキシエチレン(重合度:4)グリコールジラウリン酸エステル、HLB:6.6、三洋化成工業(株)製}25部と、炭化水素油(a12){NCL22、谷口石油(株)製、流動点-47.5℃}40部と、脂肪酸アミド(b21){アルフロー H-50S、エチレンビスステアリルアミド、日油(株)製、「アルフロー」は同社の登録商標である。}5部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ130℃まで昇温し、この温度にてさらに1時間加熱攪拌した。次いで炭化水素油(a12)(NCL22)を30部加え、70℃にて3時間攪拌した。その後得られた混合物を攪拌しながら30℃まで空冷にて冷却し、疎水性液体(q2)を得た。
ステンレス製容器内に、エステル化合物(e3){レオドールMS-60、グリセリンモノステアレート、HLB:3.5、花王(株)製}20部と、炭化水素油(a13){コスモピュアスピンG、コスモルブリカンツ(株)製、流動点-7.5℃}74部と、オルガノポリシロキサン(a21){KF96-1,000cs、ジメチルシリコーンオイル、信越化学工業(株)製、1,000mm2/s/25℃}5部と、疎水性湿式シリカ(b31){Nipsil SS-50、東ソー・シリカ(株)製、体積平均粒子径1μm、M値65、「Nipsil」は同社の登録商標である。}1部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ180℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q3)を得た。
ステンレス製容器内に、エステル化合物(e4){レオドールMO-60、グリセリンモノオレエート、HLB:2.8、花王(株)製}3部と、オルガノポリシロキサン(a22){KF96-3,000cs、ジメチルシリコーンオイル、信越化学工業(株)製、3,000mm2/s/25℃}94部と、疎水性湿式シリカ(b32){Sipernat D10、デグサジャパン(株)製、体積平均粒子径5μm、M値72、「SIPERNAT」はエボニック デグサ ゲーエムベーハーの登録商標である。}3部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ180℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q4)を得た。
ステンレス製容器内に、エステル化合物(e5){イオネットS-85、ソルビタントリオレエート、HLB:1.8、三洋化成工業(株)製}2.5部と、オルガノポリシロキサン(a23){KF96-5,000cs、ジメチルシリコーンオイル、信越化学工業(株)製、5,000mm2/s/25℃}80部と、ポリエーテル化合物(a32){ニューポールPP-950、ポリオキシプロピレン(重合度:16)グリコール、三洋化成工業(株)製}17部と、疎水性湿式シリカ(b32){Sipernat D10}0.5部投入した後、ホモジナイザーにて3000rpmで攪拌しつつ180℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q5)を得た。
ステンレス製容器内に、エステル化合物(e1){イオネットS-80}10部と、炭化水素油(a13){コスモピュアスピンG}84部と、ポリエーテル化合物(a33){ニューポール50HB-260、ポリオキシプロピレン(重合度:7)-ポリオキシエチレン(重合度:10)ブチルエーテル、三洋化成工業(株)製}5部と、脂肪酸金属塩(b12){アルミニウムステアレート600、ジステアリン酸アルミニウム、日油(株)製}1部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ150℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q6)を得た。
ステンレス製容器内に、エステル化合物(e1){イオネットS-80}14部と、炭化水素油(a12){NCL22}70部と、ポリエーテル化合物(a33){ニューポール50HB-260}10部と、脂肪酸金属塩(b11){アルミニウムステアレート900}6部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ150℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q7)を得た。
ステンレス製容器内に、エステル化合物(e3){レオドールMS-60}1部と、炭化水素油(a11){コスモピュアスピンRB}88部と、オルガノポリシロキサン(a24){KF96-50cs、ジメチルシリコーンオイル、信越化学工業(株)製、50mm2/s/25℃}10部と、脂肪酸金属塩(b11){アルミニウムステアレート900}0.5部及び脂肪酸アミド(b21){アルフローH-50S}0.5部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ130℃まで昇温し、さらに1時間攪拌した。その後得られた混合物を攪拌しながら5時間かけて30℃まで空冷にて冷却し、疎水性液体(q8)を得た。
ステンレス製容器内に、エステル化合物(e3){レオドールMS-60}17部と、ポリエーテル化合物(a31){ニューポールLB-1715}80部と、脂肪酸金属塩(b13){オーラブライトMA-76、ジステアリン酸マグネシウム、日油(株)製、「オーラブライト」は同社の登録商標である。}3部とを投入した後、にて3000rpmで攪拌しつつ150℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q9)を得た。
ステンレス製容器内に、エステル化合物(e5){イオネットS-85}5部と、オルガノポリシロキサン(a24){KF96-50cs}40部、オルガノポリシロキサン(a25){KF96-10万cs、ジメチルシリコーンオイル、信越化学工業(株)製、10万mm2/s/25℃}10部と、脂肪酸アミド(b22){アルフロー AD-281F、エチレンビスオレイルアミド、日油(株)製}5部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ120℃まで昇温し、この温度にてさらに1時間加熱攪拌した。次いでオルガノポリシロキサン(a24){KF96-50cs}40部を加え、65℃にて3時間攪拌した。その後得られた混合物を攪拌しながら30℃まで空冷にて冷却し、疎水性液体(q10)を得た。
ステンレス製容器内に、エステル化合物(e4){レオドールMO-60}11部と、オルガノポリシロキサン(a21){KF96-1,000cs}85部と、疎水性湿式シリカ(b32){Sipernat D10}4部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ180℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q11)を得た。
ステンレス製容器内に、エステル化合物(e2){イオネットDL-200}5.5部と、ポリエーテル化合物(a31){ニューポールLB-1715}90部と、疎水性湿式シリカ(b31){Nipsil SS-50}4.5部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ180℃まで昇温し、この温度にてさらに3時間加熱攪拌した。その後、得られた混合物を攪拌しながら、30℃まで空冷にて冷却し、疎水性液体(q12)を得た。
ステンレス製容器内に、エステル化合物(e1){イオネットS-80}25部と、炭化水素油(a12){NCL22}30部と、ポリエーテル化合物(a31){ニューポールLB-1715}40.5部と、疎水性湿式シリカ(b31){Nipsil SS-50}3部と、ワックス{エポレンE-10Jワックス、酸化ポリエチレンワックス、イーストマンケミカルジャパン(株)製、「EPOLENE」はウエストレイク ロングビュー コーポレーションの登録商標である。}1.5部とを投入した後、ホモジナイザーにて3000rpmで攪拌しつつ180℃まで昇温し、さらに同温度で3時間攪拌した。その後得られた混合物を攪拌しながら30℃まで空冷にて冷却し、疎水性液体(q13)を得た。
ステンレス製容器内で、製造例1で得た疎水性液体(q1)90部とイオン交換水(以下、単に水と略称す)8.2部とを攪拌混合して混合液を得た後、室温(15~20℃、以下同じ)下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s1){Aerosil RY200、日本アエロジル(株)製、一次粒子径:12nm、M値:75}1.8部を30分間で添加混合して、さらに30分攪拌を続けて、本発明の消泡剤(1)を得た。
ステンレス製容器内で、製造例2で得た疎水性液体(q2)28部と水12部及び界面活性剤(1){ナロアクティー CL-40、三洋化成工業(株)製、ノニオン界面活性剤、「ナロアクティー」は同社の登録商標である}2部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s2){Aerosil R104、日本アエロジル(株)製、一次粒子径:16nm、M値:40}0.02部を60分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水57.98部を60分間で添加混合して、本発明の消泡剤(2)を得た。
ステンレス製容器内で、製造例3で得た疎水性液体(q3)80部と水4部及び界面活性剤(2){SYグリスター CRS-75、阪本薬品工業(株)製、ノニオン界面活性剤、ポリグリセリン縮合リシノール酸エステル}0.95部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s3){Aerosil RX200、日本アエロジル(株)製、一次粒子径:12nm、M値:70}0.05部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水15部を30分間で添加混合して、本発明の消泡剤(3)を得た。
ステンレス製容器内で、製造例4で得た疎水性液体(q4)70部と水7部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s4){Aerosil R972、日本アエロジル(株)製、一次粒子径:16nm、M値:50}0.1部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水22.9部を30分間で添加混合して、本発明の消泡剤(4)を得た。
ステンレス製容器内で、製造例5で得た疎水性液体(q5)60部と水12部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s5){Aerosil R974、日本アエロジル(株)製、一次粒子径:12nm、M値:45}0.2部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水27.8部を30分間で添加混合して、本発明の消泡剤(5)を得た。
ステンレス製容器内で、製造例6で得た疎水性液体(q6)50部と水10部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s3){Aerosil RX200}0.5部を60分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水39.5部を30分間で添加混合して、本発明の消泡剤(6)を得た。
ステンレス製容器内で、製造例7で得た疎水性液体(q7)45部と水15部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s1){Aerosil RY200}0.3部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水39.7部を45分間で添加混合して、本発明の消泡剤(7)を得た。
ステンレス製容器内で、製造例8で得た疎水性液体(q8)40部と水10部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s2){Aerosil R104}1部を45分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水49部を45分間で添加混合して、本発明の消泡剤(8)を得た。
ステンレス製容器内で、製造例9で得た疎水性液体(q9)85部と水13.5部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s4){Aerosil R972}1.5部を60分間で添加混合して、さらに30分攪拌を続けて、本発明の消泡剤(9)を得た。
ステンレス製容器内で、製造例10で得た疎水性液体(q10)35部と水25部及び界面活性剤(1){ナロアクティー CL-40}0.93部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s1){Aerosil RY200}0.07部を15分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水39部を45分間で添加混合して、本発明の消泡剤(10)を得た。
ステンレス製容器内で、製造例11で得た疎水性液体(q11)30部と界面活性剤(3){ナロアクティー CL-70、三洋化成工業(株)製、ノニオン界面活性剤}1部及び界面活性剤(4){SNウエット984、サンノプコ(株)製、ノニオン界面活性剤}2.6部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s5){Aerosil R974}0.4部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水66部を90分間で添加混合して、本発明の消泡剤(11)を得た。
ステンレス製容器内で、製造例12で得た疎水性液体(q12)75部と水24部及び界面活性剤(1){ナロアクティー CL-40}0.4部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s3){Aerosil RX200}0.6部を45分間で添加混合して、さらに30分攪拌を続けて、本発明の消泡剤(12)を得た。
ステンレス製容器内で、製造例13で得た疎水性液体(q13)55部と水20部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性乾式シリカ(s4){Aerosil R972}2部を75分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水23部を30分間で添加混合して、本発明の消泡剤(13)を得た。
ステンレス製容器内で、製造例9で得た疎水性液体(q9)35部と、製造例13で得た疎水性液体(q13)35部と、界面活性剤(5){SNウエット980、サンノプコ(株)製、ノニオン界面活性剤}3部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて混合液を3000rpmで攪拌しながら、疎水性乾式シリカ(s5){Aerosil R974}0.2部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水26.8部を60分間で添加混合して、本発明の消泡剤(14)を得た。
ステンレス製容器内で、製造例5で得た疎水性液体(q5)55部と水20部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、疎水性湿式シリカ(b32){Sipernat D10}0.5部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水24.5部を30分間で添加混合して、比較用の消泡剤(U1)を得た。
ステンレス製容器内で、製造例12で得た疎水性液体(q12)60部と水10部とを攪拌混合して混合液を得た後、室温下、ホモジナイザーにて3000rpmで混合液を攪拌しながら、シリカ{非疎水化シリカ、Aerosil 200CF、日本アエロジル(株)製、一次粒子径:12nm}0.5部を30分間で添加混合して、さらに30分攪拌を続け、次いで3000rpmで攪拌しながら、水29.5部を60分間で添加混合して、比較用の消泡剤(U2)を得た。
ステンレス製容器内で、製造例7で得た疎水性液体(q7)65部と、界面活性剤(6){ノニオンTA-405、日油(株)製、ポリオキシエチレン-ポリオキシプロピレン-アルキルエーテル、HLB:5}2部と、界面活性剤(7){ノニオンOT-221、日油(株)製、ポリオキシエチレンソルビタン-モノオレート、HLB:15.7}3部と、界面活性剤(8){ナイミーンS-204、日油(株)製、ポリオキシエチレン-ステアリルアミン、HLB:8、「ナイミーン」は同社の登録商標である。}2部とを攪拌混合してから40℃まで加熱攪拌て混合液を得た後、混合液を室温まで放冷しつつホモジナイザーにて3000rpmで攪拌しながら水28部を60分間で添加混合し、ホモジナイザーにて3000rpmで30分攪拌を続けて、比較用の消泡剤(U3)を得た。
ステンレス製容器内で、α-オレフィン{出光興産(株)製、リニアレン2024、「リニアレン」は同社の登録商標である。}7部と、高級アルコール{カルコール220-80、花王(株)製、「カルコール」は同社の登録商標である。}15部と、ステアリルステアレート{エキセバールSS、花王(株)製、「エキセバール」は同社の登録商標である。}10部と、界面活性剤(9){ソフタノール30{日本触媒(株)製、炭素数12~14の直鎖型第2級アルコール1モル当たり、プロピレンオキシド3モルを付加した後、エチレンオキシド5モルを付加したもの}3部とを攪拌混合してから、85℃まで加熱攪拌して混合液を得た後、ホモジナイザーにて3000rpmで撹拌しつつ、90℃の温水65部を45分掛けて添加混合し、その後3000rpmで撹拌しつつ室温まで冷却して、比較用の消泡剤(U4)を得た。
(1)発泡性試験液の調製
ステンレスビーカーに、ポリオキシエチレンアルキルエーテル[ナローアクティーCL140、HLB=14.7、三洋化成工業(株)製]2部、ポリオキシエチレンラウリルエーテル硫酸エステルナトリウム塩[キャリボン(登録商標)EN-200、三洋化成工業(株)製]2部及びイオン交換水96部を投入後、10分間均一撹拌混合を行い、発泡性試験液を得た。
容量約2000mlのガラス製円柱型発泡管に発泡性試験液を750ml入れ、5℃又は70℃に温度調節し、このときの液面を基準高さとして読み取った。次いで、循環ポンプを用いて、発泡性試験液を3,000ml/分で、発泡管の底部から抜きながら、発泡管の上部(基準高さから150mmの高さ)から発泡管内に落下させることにより、発泡性試験液を循環させ続けた。この循環により発泡性試験液が泡立って、泡高さが基準高さより100mmの位置に達した時点で、マイクロピペットにて、評価試料{消泡剤から水を除いた濃度が発泡性試験液に対して300ppmとなる量}を添加した後、発泡性試験液の循環を継続して変化する泡高さ(泡の最上端高さ-基準高さ:mm)を試験開始5秒後、30秒後、1分後及び5分後に測定を行った。これらの泡高さの小さい方が消泡性が高いことを意味している。

(1)エマルションベース塗料の調製
表2に示した原料組成にて、インペラー型羽根を装着したエクセルオートホモジナイザー(日本精器株式会社製、モデルED)を用いて、グラインディング及びレットダウンを実施して塗料化した。得られた塗料を、ストマー粘度計(JIS K5600-2-2:1999)で80KU(25℃)になるように水で希釈してエマルションベース塗料を得た。

注2:サンノプコ(株)製増粘剤
注3:竹原化学工業(株)製炭酸カルシウム
注4:石原産業(株)製二酸化チタン
注5:BASF社製アクリルエマルション、「ACRONAL」は、ビ-エ-エスエフ アクチエンゲゼルシヤフトの登録商標である。
注6:サンノプコ(株)製防腐剤
注7:イーストマンケミカル社製造膜調整剤、「テキサノール」は吉村化学株式会社の登録商標である。
注8:サンノプコ(株)製増粘剤
エマルションベース塗料に、評価試料{消泡剤から水を除いた濃度がエマルションベース塗料に対して0.5%となる量}を加えて、インペラー型羽根を装着したエクセルオートホモジナイザーにて25℃、5000rpm、5分間攪拌混合してエマルション塗料(1)~(18)を得た。エマルション塗料(1)~(14)には実施例1~14の消泡剤を用い、エマルション塗料(15)~(18)には比較例1~4の消泡剤を用いた。また、ブランク用として消泡剤を加えないこと以外、上記と同様にして、エマルション塗料(19)を得た。
5℃又は40℃に温度調整したエマルション塗料(1)~(19)をインペラー型羽根を装着したエクセルオートホモジナイザーにて4000rpm、3分間攪拌混合して泡を巻き込ませてから15秒後に、泡を巻き込んだエマルション塗料(1)~(19)の比重を50mlの比重カップにて測定した。比重値が大きいほど泡の噛み込みが少ないことを表し、消泡性に優れている。
約250mlのエマルション塗料(1)~(19)を250mlサンプル瓶に入れて、密閉状態で40℃にて1ケ月間静置保管した後、『(3)初期消泡性の評価』と同様にして、比重を測定し、これを持続消泡性とした。

ホリゾンタル消泡試験機{上面が解放された直方体容器に入れた新聞抄紙白水を直方体容器の一端部の底部から循環ポンプにて汲み上げて(3000ml/分)新聞抄紙白水の液面から20cm上部(直方体容器の他端部の上部)から落下させることにより新聞抄紙白水を発泡させる一方、新聞抄紙白水が他端部から一端部へ20cm流れた後、ポンプにて汲み上げられる循環式消泡試験機であって、直方体容器内の液面上における他端部からの泡の長さを測定できる試験機。;特許第3799393号公報参照}に某製紙工場から採取した新聞紙抄紙白水500mlを入れ、30℃(又は70℃)に温度調整した。次いで新聞抄紙白水を循環(3000ml/分)しながら、新聞抄紙白水の落下地点からの泡長さが100mmに達したとき、落下する新聞抄紙白水に評価試料{消泡剤から水を除いた濃度が新聞抄紙白水に対して5ppmとなる量}をマイクロピペットを用いて添加した後の直方体容器内の泡長さ(試験開始2分後、5分後に測定、mm表示、値が小さいほど消泡性が良いことを表す)で消泡性を評価した。

(1)紙塗工塗料の調製
以下の原料組成にて、インペラー型羽根を装着したエクセルオートホモジナイザーを用いて塗料化して、評価用の紙塗工塗料を得た(評価試料入り)。また、評価試料を使用しないこと以外、上記と同様にして、消泡剤なしの紙塗工塗料を得た。

注2:エンゲルハード(株)製
注3:竹原化学工業(株)製
注4:竹原化学工業(株)製
注5:サンノプコ(株)製分散剤
注6:ジェイエスアール(株)製JSR0629
注7:王子コーンスターチ(株)製
注8:サンノプコ(株)製湿潤剤
注9:消泡剤から水を除いた量として1.0部となる量
評価用の紙塗工塗料(評価試料入り)を、市販の中質紙(坪量63g/m2)の片面に塗工量が15g/m2になるように塗工速度50m/minでブレード塗工(温度50℃)して塗工紙を得た。次いでこの塗工紙をスーパーカレンダー処理(条件;温度40℃、線圧60Kg/cm、2回通し)を行い評価用紙とした。評価用紙の紙面中(面積:20cm×20cm)の泡痕の数(個)を目視にて計測した。数字が小さいほど消泡性が良好である。

実施例、比較例で得た消泡剤をガラス容器(内寸;直径20mm、内容液高さ70mm)に充填して密栓した後、60℃にて1週間静置して、分離する水層(下層)の厚さ(単位:mm)を評価した。
消泡剤を作成した直後に、JIS K7233-1986の4.2単一円筒回転粘度計法に準拠して、回転粘度計(VISCOMETER TV-20、TOKI SANGYO CO,LTD.製、ローターNo.3、60又は30rpm、液温25℃)で測定した。
Claims (10)
- 乾式法によるシリカを疎水化して得られ、一次粒子径が5~100nmである疎水性乾式シリカ(S)と、
水と、
一般式(1)で表されるエステル化合物(E)を1~25重量%含有する疎水性液体(Q)とを
必須成分とすることを特徴とする消泡剤。
(R1-COO)p-D (1)
R1は炭素数1~21のアルキル基又は炭素数2~21のアルケニル基、Dは2~6個の炭素原子及び1~6個の水酸基を含有する化合物(D’)からエステル結合に関与した水酸基を除いた残基又はこの化合物(D’)に炭素数2~4のアルキレンオキシドを付加させた付加体からエステル結合に関与した水酸基を除いた残基、pは1~3の整数を表す。 - 一次粒子径が5~100nmである疎水性乾式シリカ(S)のM値が30~80である請求項1に記載の消泡剤。
- 消泡剤の重量に基づいて、疎水性乾式シリカ(S)の含有量が0.02~2重量%、水の含有量が8~70重量%、疎水性液体(Q)の含有量が28~90重量%である請求項1又は2に記載の消泡剤。
- 疎水性液体(Q)が、
一般式(1)で表されるエステル化合物(E)と、
炭化水素油(A1)、オルガノポリシロキサン(A2)及びポリエーテル化合物(A3)からなる群より選ばれる少なくとも1種と、
脂肪酸金属塩(B1)、脂肪酸アミド(B2)及び体積平均粒子径が1~15μmである湿式法によるシリカを疎水化した疎水性湿式シリカ(B3)からなる群より選ばれる少なくとも1種とからなる請求項1~3のいずれかに記載の消泡剤。 - 疎水性液体(Q)に炭化水素油(A1)を含み、炭化水素油(A1)の流動点が-50~2℃である請求項1~4のいずれかに記載の消泡剤。
- 疎水性液体(Q)にオルガノポリシロキサン(A2)を含み、オルガノポリシロキサン(A2)の25℃における動粘度が50~10万mm2/sである請求項1~5のいずれかに記載の消泡剤。
- 疎水性液体(Q)にポリエーテル化合物(A3)を含み、ポリエーテル化合物(A3)が炭素数2~4のアルキレンオキシドの付加体からなり、数平均分子量が500~5,000である請求項1~6のいずれかに記載の消泡剤。
- 疎水性液体(Q)に脂肪酸金属塩(B1)を含み、脂肪酸金属塩(B1)が一般式(2)で表される化合物である請求項1~7のいずれかに記載の消泡剤。
(R1-COO)q-M (2)
R1は炭素数1~21のアルキル基又は炭素数2~21のアルケニル基、Mは1~3価の金属原子、qは1~3の整数を表す。 - 疎水性液体(Q)に脂肪酸アミド(B2)を含み、脂肪酸アミド(B2)が一般式(3)で表される化合物である請求項1~8のいずれかに記載の消泡剤。
R2-CONH-(CH2)t-NHCO-R2 (3)
R2は炭素数9~21のアルキル基又は炭素数10~21のアルケニル基、tは1~3の整数を表す。 - 請求項1~9のいずれかに記載された消泡剤を製造する方法であって、疎水性液体(Q)及び水からなる混合液に疎水性乾式シリカ(S)を添加混合する工程を含むことを特徴とする製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2011/062796 WO2012164741A1 (ja) | 2011-06-03 | 2011-06-03 | 消泡剤 |
| KR1020137033936A KR101740095B1 (ko) | 2011-06-03 | 2011-06-03 | 소포제 |
| CN201180071397.8A CN103596655B (zh) | 2011-06-03 | 2011-06-03 | 消泡剂 |
| US14/113,464 US9345991B2 (en) | 2011-06-03 | 2011-06-03 | Defoaming agent |
| JP2013517792A JP5934857B2 (ja) | 2011-06-03 | 2011-06-03 | 消泡剤 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2011/062796 WO2012164741A1 (ja) | 2011-06-03 | 2011-06-03 | 消泡剤 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012164741A1 true WO2012164741A1 (ja) | 2012-12-06 |
Family
ID=47258626
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/062796 Ceased WO2012164741A1 (ja) | 2011-06-03 | 2011-06-03 | 消泡剤 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9345991B2 (ja) |
| JP (1) | JP5934857B2 (ja) |
| KR (1) | KR101740095B1 (ja) |
| CN (1) | CN103596655B (ja) |
| WO (1) | WO2012164741A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016140078A1 (ja) * | 2015-03-02 | 2016-09-09 | サンノプコ株式会社 | 消泡剤 |
| JP2017196571A (ja) * | 2016-04-27 | 2017-11-02 | サンノプコ株式会社 | 消泡剤 |
| JP2025161523A (ja) * | 2024-04-12 | 2025-10-24 | サンノプコ株式会社 | 消泡剤 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106102856A (zh) * | 2014-06-27 | 2016-11-09 | 圣诺普科有限公司 | 消泡剂 |
| JP6393177B2 (ja) * | 2014-12-10 | 2018-09-19 | 東ソー・シリカ株式会社 | 疎水性シリカ及びその製造方法 |
| CN105457342A (zh) * | 2015-11-24 | 2016-04-06 | 宜兴市丰烨化学有限公司 | 一种聚醚硅油乙二醇消泡剂及其制备方法 |
| JP6665307B2 (ja) * | 2016-02-02 | 2020-03-13 | ザ プロクター アンド ギャンブル カンパニーThe Procter & Gamble Company | 組成物 |
| CN110575786B (zh) * | 2018-06-11 | 2021-09-14 | 江苏四新科技应用研究所股份有限公司 | 一种新型疏水组合物 |
| JP7242287B2 (ja) * | 2018-12-21 | 2023-03-20 | 日華化学株式会社 | 皮革用材の製造方法 |
| CN116783252A (zh) * | 2020-12-25 | 2023-09-19 | Agc硅素技术株式会社 | 疏水性二氧化硅粒子及其用途、以及疏水性二氧化硅粒子的制造方法 |
| CN113185875A (zh) * | 2021-05-06 | 2021-07-30 | 湖北巴乐福化工科技有限公司 | 一种防火涂料及其制备方法 |
| CN114307260B (zh) * | 2021-11-06 | 2023-10-27 | 南京瑞思化学技术有限公司 | 一种聚醚组合物的制备方法 |
| US20240390820A1 (en) * | 2023-05-25 | 2024-11-28 | Solenis Technologies, L.P. | Defoaming agent and compositions containing the defoaming agent |
| CN117599472B (zh) * | 2023-11-23 | 2026-04-07 | 深圳市海沃维斯科技有限公司 | 一种用于涤纶染色的耐高温消泡剂、制备方法及其应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5013282A (ja) * | 1973-06-08 | 1975-02-12 | ||
| JPH06508548A (ja) * | 1991-06-26 | 1994-09-29 | ヘンケル・コマンディットゲゼルシャフト・アウフ・アクチェン | 消泡剤 |
| JPH10286404A (ja) * | 1997-04-11 | 1998-10-27 | Hakuto Co Ltd | 消泡剤組成物 |
| JP2007222812A (ja) * | 2006-02-24 | 2007-09-06 | Shin Etsu Chem Co Ltd | 消泡剤組成物 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0312195D0 (en) | 2003-05-28 | 2003-07-02 | Syngenta Ltd | Formulation |
| JP4916901B2 (ja) | 2007-01-31 | 2012-04-18 | 東邦化学工業株式会社 | 消泡剤組成物 |
| DE102007047211A1 (de) * | 2007-10-02 | 2009-04-09 | Wacker Chemie Ag | Entschäumerzusammensetzungen |
| DE102007061455A1 (de) | 2007-12-20 | 2009-06-25 | Evonik Degussa Gmbh | Entschäumerformulierung |
| JP4899004B2 (ja) | 2009-01-13 | 2012-03-21 | サンノプコ株式会社 | 消泡剤 |
| JP4898994B2 (ja) * | 2009-02-02 | 2012-03-21 | サンノプコ株式会社 | 消泡剤 |
| JP5211279B2 (ja) | 2009-10-09 | 2013-06-12 | サンノプコ株式会社 | 消泡剤 |
-
2011
- 2011-06-03 WO PCT/JP2011/062796 patent/WO2012164741A1/ja not_active Ceased
- 2011-06-03 JP JP2013517792A patent/JP5934857B2/ja active Active
- 2011-06-03 US US14/113,464 patent/US9345991B2/en active Active
- 2011-06-03 CN CN201180071397.8A patent/CN103596655B/zh active Active
- 2011-06-03 KR KR1020137033936A patent/KR101740095B1/ko not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5013282A (ja) * | 1973-06-08 | 1975-02-12 | ||
| JPH06508548A (ja) * | 1991-06-26 | 1994-09-29 | ヘンケル・コマンディットゲゼルシャフト・アウフ・アクチェン | 消泡剤 |
| JPH10286404A (ja) * | 1997-04-11 | 1998-10-27 | Hakuto Co Ltd | 消泡剤組成物 |
| JP2007222812A (ja) * | 2006-02-24 | 2007-09-06 | Shin Etsu Chem Co Ltd | 消泡剤組成物 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016140078A1 (ja) * | 2015-03-02 | 2016-09-09 | サンノプコ株式会社 | 消泡剤 |
| JPWO2016140078A1 (ja) * | 2015-03-02 | 2017-12-07 | サンノプコ株式会社 | 消泡剤 |
| JP2017196571A (ja) * | 2016-04-27 | 2017-11-02 | サンノプコ株式会社 | 消泡剤 |
| JP2025161523A (ja) * | 2024-04-12 | 2025-10-24 | サンノプコ株式会社 | 消泡剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2012164741A1 (ja) | 2014-07-31 |
| JP5934857B2 (ja) | 2016-06-15 |
| KR20140033449A (ko) | 2014-03-18 |
| CN103596655A (zh) | 2014-02-19 |
| US9345991B2 (en) | 2016-05-24 |
| US20140100293A1 (en) | 2014-04-10 |
| KR101740095B1 (ko) | 2017-05-25 |
| CN103596655B (zh) | 2015-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5934857B2 (ja) | 消泡剤 | |
| JP5771795B2 (ja) | 消泡剤 | |
| JP6442660B2 (ja) | 疎水性微粒湿式シリカ、その製造方法及び消泡剤 | |
| CN101903074B (zh) | 消泡剂配制品 | |
| JP2006199962A (ja) | 消泡剤組成物 | |
| JP2005535452A (ja) | シリコーン泡制御組成物 | |
| JP5540171B2 (ja) | 消泡剤組成物 | |
| CN103814072A (zh) | 有机硅泡沫控制组合物及其制备方法 | |
| CN103804978A (zh) | 本体型涂料消泡剂 | |
| JP2013103204A (ja) | 粉末消泡剤及びその製造方法 | |
| JP6627040B2 (ja) | 消泡剤 | |
| CN106999802A (zh) | 消泡剂 | |
| JP6298954B2 (ja) | 消泡剤 | |
| CN100509106C (zh) | 消泡剂 | |
| JP4057508B2 (ja) | 消泡剤組成物 | |
| CN100518918C (zh) | 表面活性剂 | |
| WO2022145345A1 (ja) | シリコーン系消泡剤用オイルコンパウンド及びその製造方法、並びにシリコーン系消泡剤用オイルコンパウンドを含む消泡剤 | |
| JP2014214286A (ja) | 作動液 | |
| JP7751888B2 (ja) | 消泡剤 | |
| JP3944690B2 (ja) | 消泡剤組成物 | |
| JP3100302B2 (ja) | 消泡剤用オイルコンパウンドの製造方法及びそれを含有する消泡剤組成物 | |
| ES1263125U (es) | Composicion antiespumante | |
| WO2026018581A1 (ja) | 分散状消泡剤 | |
| JP2001269503A (ja) | 表面処理された粉体及び消泡剤組成物 | |
| JP2020025928A (ja) | 消泡剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11866618 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013517792 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14113464 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20137033936 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11866618 Country of ref document: EP Kind code of ref document: A1 |