WO2012172839A1 - リン酸鉄リチウムの製造方法 - Google Patents
リン酸鉄リチウムの製造方法 Download PDFInfo
- Publication number
- WO2012172839A1 WO2012172839A1 PCT/JP2012/055487 JP2012055487W WO2012172839A1 WO 2012172839 A1 WO2012172839 A1 WO 2012172839A1 JP 2012055487 W JP2012055487 W JP 2012055487W WO 2012172839 A1 WO2012172839 A1 WO 2012172839A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- iron phosphate
- lithium iron
- acid
- aqueous solution
- carbon
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/45—Phosphates containing plural metal, or metal and ammonium
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/5825—Oxygenated metallic salts or polyanionic structures, e.g. borates, phosphates, silicates, olivines
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to a method for producing lithium iron phosphate (method for producing lithium iron phosphate).
- a small lithium ion battery which is a secondary battery widely spread mainly in portable devices, has been improved in performance by improving a negative active material, an electrolytic solution, and the like.
- the cathode active material no major technological innovation has been observed since the commercialization of small lithium ion batteries until now, and lithium cobalt oxide (LiCoO 2 ) containing an expensive rare metal is the main. Has been used.
- This lithium cobaltate is expensive and does not have sufficient thermal and chemical stability, and at a high temperature of about 180 ° C, there is a risk of releasing oxygen and igniting the organic electrolyte. The problem remains. Therefore, for the development of lithium-ion battery applications that include large equipment, it is indispensable to develop positive active materials that are cheaper than conventional lithium cobaltate, are thermally and chemically stable, and have high safety. It is.
- lithium iron phosphate olivine type lithium iron phosphate (LiFePO 4 , Hereinafter, it is simply referred to as “lithium iron phosphate”.
- This lithium iron phosphate does not release oxygen up to about 400 ° C. due to the strong P—O bond in the crystal structure, so it is a highly safe active material, and also has good long-term stability and quick charge characteristics. Active material.
- lithium iron phosphate In applying lithium iron phosphate to the positive electrode active material, in order to ensure high-speed charge / discharge characteristics (high-speed charge / discharge charge characteristic), which is a characteristic required for the positive electrode active material, electronic conduction of lithium iron phosphate is required. It is required to improve the property and shorten the diffusion distance of lithium ions. As a solution to this problem, it is effective to coat the surface of lithium iron phosphate particles with a conductive material and to refine the lithium iron phosphate particles to a particle size of about 100 nm to increase the reaction surface area. ing. There is also a report that doping with other elements is effective in improving electron conductivity and stabilizing the crystal structure.
- Patent Document 1 a metal iron and a compound that liberates phosphate ions are first reacted in an aqueous solution, and then lithium carbonate or lithium hydroxide is added to prepare a precursor of lithium iron phosphate (precursor).
- precursor of lithium iron phosphate precursor of lithium iron phosphate (precursor).
- the dried product is subjected to primary firing in the temperature range of 300 to 450 ° C., followed by adding a substance that generates conductive carbon by thermal decomposition and firing at 500 to 800 ° C.
- Patent Document 2 discloses that a precursor is prepared by dissolving iron powder, a lithium salt, and a phosphoric acid group compound in an aqueous citric acid solution, and then the precursor is spray-dried and then fired at a temperature of 500 ° C. or higher. How to do is described.
- Patent Document 3 first, iron powder is reacted in an aqueous solution containing phosphoric acid and citric acid, then lithium hydroxide is added, and further converted into a conductive oxide by metal oxide or baking. A method is described in which a precursor is prepared by adding a metal salt and dried, and finally a dried product of the precursor is calcined.
- Patent Document 1 since iron metal having a high purity of 99.9% or more is reacted with phosphoric acid when the precursor is prepared, iron phosphate which is a poorly soluble divalent iron compound. Aggregated particles of (Fe 3 (PO 4 ) 2 ⁇ 8H 2 O) are generated and grown, and the solution becomes a creamy high-viscosity substance exhibiting white to light blue. As a result, stirring of the solution becomes insufficient, and unreacted metallic iron tends to remain, and the raw materials are not mixed uniformly. Patent Document 1 also discloses that an acid such as hydrochloric acid or oxalic acid is added in order to promote the reaction of unreacted metallic iron.
- an acid such as hydrochloric acid or oxalic acid is added in order to promote the reaction of unreacted metallic iron.
- Patent Document 2 is a method in which dissolution of iron powder is performed in a citric acid aqueous solution in which a lithium salt and a phosphate group compound coexist, so that it takes a long time to dissolve, and is not economical. .
- Patent Document 2 describes that effective divalent iron is produced by oxidizing iron with an organic acid or mixed organic acid, but divalent iron is stably present only by mixing raw materials. There is a problem that it is difficult.
- the lithium salt is lithium nitrate
- nitrate ions act as an oxidizing agent at the time of firing, and when lithium salt is lithium acetate, the raw material is expensive. It is inappropriate for cost reduction.
- the present invention has been made in view of such circumstances.
- a precursor of lithium iron phosphate uniformly mixed at the atomic level is prepared, and is inexpensive and has excellent discharge capacity.
- An object is to provide a method for producing lithium iron oxide.
- the present inventors have intensively studied to achieve the above object.
- hydroxycarboxylic acid is used when reacting the iron particles with phosphoric acid. It was found that it is effective to continue supplying oxygen chemically bound to the surface of the iron particles by coexistence and reaction in an oxidizing atmosphere.
- a chelate of iron phosphate that is uniformly dispersed in an aqueous solution is obtained, and then, by adding a lithium source, a precursor of lithium iron phosphate in which raw materials are uniformly mixed at an atomic level is obtained.
- lithium iron phosphate having a high discharge capacity can be produced by adding a carbon source to the above solution of lithium iron phosphate precursor, drying and firing in a non-oxidizing atmosphere. Further, when improving various properties of lithium iron phosphate by substituting with other elements, the metal or compound of the substituting element is added to the aqueous solution containing phosphoric acid and hydroxycarboxylic acid, thereby uniformly substituted phosphoric acid. It was found that iron lithium was obtained. That is, the present invention provides the following (1) to (11).
- an aqueous solution preparation step of preparing an aqueous solution containing phosphoric acid and hydroxycarboxylic acid; To the aqueous solution, iron particles containing 0.1 to 2% by mass of oxygen are added, and the phosphoric acid and the hydroxycarboxylic acid in the aqueous solution are reacted with the iron particles in an oxidizing atmosphere to perform the first reaction.
- a first production process for producing a liquid A second production step of producing a second reaction liquid by adding a lithium source to the first reaction liquid; A third production step of producing a third reaction solution by adding a carbon source to the second reaction solution; A precursor generating step of drying the third reaction solution to generate a lithium iron phosphate precursor; A firing step of firing the lithium iron phosphate precursor in a non-oxidizing atmosphere to obtain lithium iron phosphate; A method for producing lithium iron phosphate.
- the third reaction solution is prepared by adding a carbon source to the second reaction solution so that the amount of carbon contained in the fired lithium iron phosphate is 1 to 5% by mass.
- the manufacturing method of lithium iron phosphate as described in (1) which consists of producing.
- the carbon source is at least one selected from the group consisting of carbon black, acetylene black, ketjen black, vapor grown carbon fiber (VGCF), carbon nanofiber, graphite and fullerene.
- VGCF vapor grown carbon fiber
- the method for producing lithium iron phosphate of the present invention includes an aqueous solution preparation step of preparing an aqueous solution containing phosphoric acid and hydroxycarboxylic acid, 1st preparation which adds the iron particle containing 2 mass% oxygen, and makes the said phosphoric acid and the said hydroxycarboxylic acid, and the said iron particle in the said aqueous solution react in the oxidizing atmosphere, and produces the 1st reaction liquid.
- Manufacturing step a precursor generation step of drying the third reaction solution to generate a lithium iron phosphate precursor, and baking the lithium iron phosphate precursor in a non-oxidizing atmosphere to lithium iron phosphate
- a baking step to obtain iron phosphate This is a method for producing lithium.
- the production method of the present invention will be described in detail.
- the aqueous solution preparation step is a step of preparing an aqueous solution containing phosphoric acid and hydroxycarboxylic acid. Details of phosphoric acid and hydroxycarboxylic acid contained in the aqueous solution prepared in the aqueous solution preparation step will be described later. In addition, it does not specifically limit as water contained in the said aqueous solution, For example, ion-exchange water and distilled water are used suitably.
- first manufacturing step iron particles containing 0.1 to 2% by mass of oxygen are added to the aqueous solution prepared in the aqueous solution preparation step, and phosphoric acid and the phosphoric acid in the aqueous solution are added in an oxidizing atmosphere.
- This is a step of producing a first reaction liquid by reacting hydroxycarboxylic acid with iron particles.
- iron particles containing 0.1 to 2% by mass of oxygen are added to an aqueous solution containing phosphoric acid and hydroxycarboxylic acid, and reacted in an oxidizing atmosphere, thereby allowing iron phosphate to react. A chelate is formed.
- iron powder can be used as iron particles.
- iron powder include reduced iron powder obtained by reducing mill scale (iron oxide) with coke; atomized iron obtained by pulverizing and cooling molten steel with high-pressure water. Powder; electrolytic iron powder obtained by electrolyzing an iron salt aqueous solution and deposited on the cathode; and the like.
- the average particle size of the iron powder is preferably 100 ⁇ m or less because it is advantageous for promoting the subsequent reaction with phosphoric acid and hydroxycarboxylic acid to form a chelate of iron phosphate. More preferably, it is 80 ⁇ m.
- the average particle size was determined according to JIS M 8706.
- the average particle size of ordinary general industrial iron powder is 70 to 80 ⁇ m, but since particles with a maximum particle size of 150 to 180 ⁇ m are also included, coarse particles can be removed by sieving as necessary, or by mechanical pulverization. It is better to use a larger reaction area, for example, by refining coarse particles.
- iron particles used in the present invention may be appropriately referred to as “iron”, “metallic iron”, “iron powder”, or the like.
- oxygen contained in iron particles means oxygen chemically bonded to iron.
- the oxygen content of the iron particles By setting the oxygen content of the iron particles to 0.1 to 2% by mass, preferably 0.6 to 2% by mass, formation of an iron phosphate chelate at the initial stage of the reaction is promoted.
- the oxygen content of the iron particles is less than 0.1% by mass, the direct reaction between metallic iron and phosphoric acid gives priority to iron phosphate (Fe) which is a hardly soluble divalent iron compound. 3 (PO 4 ) 2 ⁇ 8H 2 O) aggregated particles are easily generated and grown. For this reason, the aqueous solution may become a creamy high-viscosity substance exhibiting white to light blue.
- the oxygen content of iron particles is, for example, 0 in the case of reduced iron powder obtained by reducing mill scale (iron oxide) with coke, or atomized iron powder obtained by pulverizing molten steel with high-pressure water and drying after cooling. .5 to 1.5% by mass. Further, when the iron particles are reduced with hydrogen, the oxygen content is 0.1 to 0.4 mass%.
- the iron particles used in the present invention do not particularly require expensive hydrogen-reduced iron particles.
- the oxygen content of the iron particles is quantified using TC436 manufactured by LECO, based on the JIS Z 2613 (1992) vacuum melting infrared absorption method.
- the atmosphere in which the iron particles are added to the aqueous solution containing phosphoric acid and hydroxycarboxylic acid for the reaction needs to be an oxidizing atmosphere.
- the reaction that forms a chelate (hereinafter also referred to as “chelation reaction”) proceeds and oxygen on the surface of the iron particles is consumed, the chelation reaction cannot be sustained, and the direct reaction between metallic iron and phosphate ions takes precedence. As a result, aggregated particles of poorly soluble iron phosphate are generated and grow. Therefore, in the present invention, the atmosphere during the reaction is an oxidizing atmosphere, whereby the surface of the iron particles is appropriately oxidized to replenish oxygen chemically bound to the iron particles, and the chelate reaction is continued.
- the oxidizing atmosphere in the present invention is a state in which the surface of the iron particles in the aqueous solution can be appropriately oxidized.
- This state is, for example, a method in which the interface of the aqueous solution is brought into contact with the oxygen-containing gas; Examples of specific operations in these methods include stirring in an air atmosphere; supply of oxygen by bubbling air; and the like.
- the phosphoric acid is preferably an aqueous solution of orthophosphoric acid (H 3 PO 4 ), but an aqueous solution of higher-order condensed phosphoric acid (H n + 2 P n O 3n + 1 ) can also be used.
- Orthophosphoric acid is usually available as a 75 to 85% by weight aqueous solution which is an industrial product.
- 1 mol is a stoichiometric equivalent with respect to 1 mol of iron, but about 0.1 mol may be added in excess.
- Hydroxycarboxylic acid is a carboxylic acid having a hydroxyl group and a carboxy group, and functions as a chelating agent when forming a chelate of iron phosphate.
- the hydroxycarboxylic acid used in the present invention include lactic acid, tartaric acid, malic acid, citric acid and the like, which have a strong chelating power against iron. Among them, a chelating body having a strong chelating power and hardly oxidizing is formed. Citric acid is preferred.
- Hydroxycarboxylic acid functions as a reducing agent because it remains as charcoal during firing.
- the residual carbon ratio of the hydroxycarboxylic acid is preferably 3% by mass or more. This is because if the residual carbon ratio is less than 3% by mass, the obtained lithium iron phosphate precursor may be oxidized by a small amount of oxygen in the atmosphere. Moreover, since the residual carbon amount after baking will become excess when a residual carbon rate exceeds 20 mass%, it is preferable that a residual carbon rate shall be 20 mass% or less.
- the residual carbon ratio of the hydroxycarboxylic acid is lactic acid: 7% by mass, tartaric acid: 7% by mass, malic acid: 12% by mass, and citric acid monohydrate: 7% by mass. Moreover, since the oxalic acid dihydrate of carboxylic acid, acetic acid, etc. are less than 1 mass%, the reducing action at the time of baking is small.
- the “remaining carbon ratio” in the present invention is the amount of carbon remaining after firing in accordance with JIS G 1211 (1995) high-frequency induction furnace combustion-infrared absorption method and divided by the original amount of hydroxycarboxylic acid. This is the value obtained.
- the amount of hydroxycarboxylic acid added is preferably 0.1 to 0.5 mol, more preferably 0.15 to 0.3 mol, relative to 1 mol of iron.
- the amount added is less than 0.1 mol, the effect of chelation with hydroxycarboxylic acid is reduced, so that iron iron and phosphate ions react directly to produce and grow poorly soluble iron phosphate agglomerated particles.
- the aqueous solution becomes a creamy high-viscosity material exhibiting white to light blue, resulting in insufficient stirring of the aqueous solution, unreacted metallic iron tends to remain, raw materials are not uniformly mixed, etc. May cause problems.
- the iron phosphate chelate formed is uniformly dispersed in the aqueous solution (the raw materials are uniformly mixed). As a result, the apparent discharge capacity of the finally obtained lithium iron phosphate may be reduced. On the other hand, if the addition amount is within the above range, unreacted metallic iron hardly remains, the raw materials are uniformly mixed, and the apparent discharge capacity of the obtained lithium iron phosphate is good. Become.
- the aqueous solution temperature is preferably 10 to 40 ° C, more preferably 20 to 30 ° C.
- the temperature of the aqueous solution is controlled within the range of 10 to 40 ° C.
- oxygen on the iron particle surface is consumed by the chelate reaction, and then the newly appearing iron particle surface comes into contact with dissolved oxygen or air bubbles in the aqueous solution.
- the aqueous solution temperature is less than 10 ° C., the chelation reaction of the iron particles becomes slow, and it may take a long time to complete the reaction.
- oxidation for supplementing oxygen cannot catch up with the surface of the iron particles where oxygen is consumed, and therefore, direct reaction between metallic iron and phosphoric acid is prioritized and hardly soluble. Agglomerated particles of iron phosphate may be generated and grow, and the aqueous solution may become a creamy high-viscosity substance exhibiting white to light blue.
- hydroxycarboxylic acid is chelated with iron via oxygen or hydroxyl groups present on the surface of iron particles.
- the phosphoric acid oxidizes and binds iron to form an iron phosphate chelate represented by the following formula (1), and a first reaction liquid in which this chelate is uniformly dispersed is obtained. Guessed.
- the second production process is a process for producing a second reaction liquid by adding a lithium source to the first reaction liquid produced in the first production process.
- the second preparation step lithium iron phosphate in which the raw materials are uniformly mixed at the atomic level by adding a lithium source to the first reaction liquid in which the chelate represented by the above formula (1) is uniformly dispersed The precursor of is obtained.
- the lithium source added to the first reaction solution is not particularly limited as long as it is a water-soluble lithium salt, but lithium hydroxide and lithium carbonate are preferred because no harmful gas is generated during firing.
- reaction solution When a lithium source is added to the first reaction solution, the reaction solution turns dark green, and a second reaction solution having a pH of 6 to 7 is obtained. Further, when an X-ray diffraction analysis is performed on the dried product obtained by drying the second reaction solution, a crystalline compound is not detected, and an amorphous phase caused by a chelate mixed uniformly at the atomic level is confirmed.
- This chelate of lithium iron phosphate is present in a dispersed state in the second reaction solution, but a part of the chelate may be present as aggregated particles and may become a precipitate. In such a case, it is desirable that the aggregated particles are finely pulverized by wet mechanical pulverization in order to make the precursor solution uniform before drying performed in a later step.
- wet pulverization method include ultrasonic irradiation, a wet jet mill, and a bead mill.
- the third production process is a process for producing a third reaction liquid by adding a carbon source to the second reaction liquid produced in the second production process.
- a substance that thermally decomposes during firing to generate carbon, or conductive carbon is preferable.
- Substances that thermally decompose during firing to produce carbon include those that melt during wetting and wet the surface of lithium iron phosphate particles, or produce gaseous substances that deposit carbon on the surface of lithium iron phosphate particles.
- sugars such as glucose, fructose, maltose, sucrose, ascorbic acid and erythorbic acid
- water-soluble polymers such as polyvinyl alcohol (PVA) and polyethylene glycol
- PVA polyvinyl alcohol
- the conductive carbon include carbon black, acetylene black, ketjen black, vapor grown carbon fiber (VGCF), carbon nanofiber, graphite, fullerene and the like. These may be used alone or in combination of two or more.
- the amount of carbon source added to the second reaction liquid is preferably such that the amount of carbon contained in the lithium iron phosphate after calcination is 1 to 5% by mass, more preferably 1.5 to 3% by mass. preferable.
- the amount of carbon When the amount of carbon is less than 1% by mass, the conductivity of lithium iron phosphate may be insufficient, and the performance of lithium iron phosphate particles as a positive electrode active material may not be sufficiently extracted. There is. On the other hand, when the amount of carbon is more than 5% by mass, the apparent discharge capacity may be reduced. However, if the amount of carbon source added is within the above range, the conductivity of lithium iron phosphate is sufficient, and the apparent discharge capacity is also good.
- generation process is a process of drying the 3rd reaction liquid produced at the 3rd production process, and producing
- the lithium iron phosphate precursor which is a dried product of the third reaction solution, is in powder form, and the particle size is not particularly limited, but is preferably 50 to 200 ⁇ m from the viewpoint of ease of handling.
- the method for drying the third reaction liquid is not particularly limited, but it is preferable to employ a spray drying method because of the good drying efficiency.
- the spray drying method is a method in which a sample solution is sprayed and dried in high-temperature heated air, so that a powder having a uniform shape can be obtained.
- the inlet temperature (heated air temperature) of the spray drying apparatus is preferably set to 100 to 250 ° C.
- the inlet temperature is set to 150 to 250 ° C.
- the ultimate temperature of the dried product to be generated is 100 to 150 ° C., depending on the balance with the amount of liquid to be fed.
- the firing step is a step of obtaining lithium iron phosphate by firing the lithium iron phosphate precursor produced in the precursor production step in a non-oxidizing atmosphere.
- hydroxyl groups and organic substances contained in the lithium iron phosphate precursor are thermally decomposed to H 2 O, CO 2 , H 2 and hydrocarbons are removed, and the dried product having an amorphous phase is crystallized to be converted into a crystal of lithium iron phosphate having an olivine structure, and pyrolytic carbon is deposited on the surface of lithium iron phosphate particles.
- the non-oxidizing atmosphere means, for example, an inert gas atmosphere such as nitrogen or argon having an oxygen concentration of 1000 ppm or less; a reducing gas atmosphere containing a reducing gas such as hydrogen or carbon monoxide; Say.
- the reason why baking is performed in a non-oxidizing atmosphere is to prevent oxidation.
- the firing temperature (firing temperature) in the firing step is preferably 300 ° C. or higher, more preferably 500 ° C. or higher, and even more preferably 600 to 800 ° C.
- the calcination temperature is less than 300 ° C.
- the volatile components H 2 O, CO 2 , H 2 and hydrocarbons may not be thermally decomposed and removed, and crystallization may not occur.
- the firing temperature exceeds 800 ° C., the resulting crystal particles are coarsened and by-products such as Fe 2 P may be generated.
- the firing temperature is within the above range, the thermal decomposition removal and crystallization of volatile components proceed sufficiently, and the coarsening of crystal particles and the generation of by-products such as Fe 2 P are also suppressed.
- the primary particle size of lithium iron phosphate obtained by firing a lithium iron phosphate precursor in a non-oxidizing atmosphere is preferably 200 nm or less, and more preferably 50 to 150 nm, because the lithium ion diffusion distance is shortened. preferable.
- the primary particle size of lithium iron phosphate is determined by using the Scherrer equation by X-ray diffraction analysis performed using Ultimate IV (X-Ray: Cu-K ⁇ 1) manufactured by Rigaku. .
- the primary particle diameter is a crystallite diameter and is a minimum unit of crystals.
- lithium iron phosphate precursor second reaction solution
- a carbon source is added to this solution.
- lithium iron phosphate having a high discharge capacity can be produced by firing in a non-oxidizing atmosphere.
- the positive electrode active material is required to have high-speed charge / discharge characteristics
- lithium iron phosphate obtained by the production method of the present invention is suitably used as a positive electrode active material for a secondary battery such as a lithium ion battery.
- inexpensive iron particles are used, cost reduction is also realized.
- lithium iron phosphate obtained by substituting iron with other elements can also be obtained.
- the discharge characteristics may be more excellent.
- the metal or compound of the element that replaces iron may be dissolved in advance in an aqueous solution containing phosphoric acid and hydroxycarboxylic acid. Thereby, the element to substitute can be mixed uniformly.
- Examples of the metal or compound of the element that replaces iron include Ti (OH) 4 , TiOSO 4 .H 2 O in the case of titanium; FeV, V 2 O 5 , VOSO 4 .2H 2 O in the case of vanadium; In the case of Mg, Mg, MgO, Mg (OH) 2 ; in the case of zirconium, ZrO (CH 3 COO) 2 ; in the case of manganese, MnCO 3 .nH 2 O, Mn (CH 3 COO) 2 ;
- the amount of substitution depends on the kind of element, from the viewpoint of easy expression of the effect of substitution, substitution of 0.01 mol% or more of iron element is preferable, and 0.05 mol% or more is more preferable.
- the upper limit of the substitution amount varies greatly depending on factors such as the ionic radius, valence, and coordination number of the substitution element, and thus cannot be determined unconditionally, but when the substitution amount exceeds the threshold value, There is a tendency that characteristics are deteriorated due to localization of electrons due to generation or change in band structure.
- Example 1 Distilled water: 2000 g of phosphoric acid: 10 mol and citric acid monohydrate: 2 mol were dissolved in this mixture, and iron powder (manufactured by JFE Steel, oxygen content: 0.68% by mass, average) Particle size: 80 ⁇ m): 10 mol was added, and the mixture was reacted for 1 day with stirring under an air atmosphere at a liquid temperature of 25 ° C. Next, lithium carbonate: 5 mol was added to form lithium iron phosphate, and finally, ascorbic acid: 80 g as a carbon source was added to prepare a precursor solution. This precursor was dried at an inlet temperature of 200 ° C.
- Example 2 Lithium iron phosphate was prepared in the same manner as in Example 1 except that lactic acid: 2 mol was used instead of citric acid monohydrate.
- Example 3 Lithium iron phosphate was prepared in the same manner as in Example 1 except that 2 mol of malic acid was used instead of citric acid monohydrate.
- Example 4 Lithium iron phosphate was prepared in the same manner as in Example 1 except that tartaric acid: 2 mol was used instead of citric acid monohydrate.
- Example 5 Lithium iron phosphate was prepared in the same manner as in Example 1 except that iron powder (manufactured by JFE Steel Corporation, oxygen content: 0.41% by mass, average particle size: 80 ⁇ m) was used.
- Example 6 To a mixed solution of phosphoric acid and citric acid monohydrate, zirconium oxyacetate: 0.1 mol of zirconium source (1 mol% substitution of iron element) is added and dissolved, and the same iron powder as in Example 1 is added to this mixed solution: Lithium iron phosphate was prepared in the same manner as in Example 1 except that 9.9 mol was added.
- Example 7 To the mixed solution of phosphoric acid and citric acid monohydrate, manganese carbonate (MnCO 3 .nH 2 O): 7 mol of manganese source (70 mol% substitution of iron element) was added, and this mixed solution was the same as in Example 1.
- Iron powder Lithium iron phosphate was prepared in the same manner as in Example 1 except that 3 mol was added.
- Example 8 Lithium iron phosphate was prepared in the same manner as in Example 1 except that 80 g of sucrose was used instead of 80 g of ascorbic acid as the carbon source.
- Example 9 Ascorbic acid as a carbon source: Ketjen black paste (L-W W-376R, concentration 20% by mass): 80 g was used instead of 80 g, and lithium iron phosphate was prepared in the same manner as in Example 1. did.
- Example 10 Lithium iron phosphate was prepared in the same manner as in Example 1 except that ascorbic acid: 95 g was used instead of ascorbic acid: 80 g as the carbon source.
- Example 11 To the mixed solution of phosphoric acid and citric acid monohydrate, vanadium oxide (V 2 O 5 ): 0.05 mol (substitution of 1 mol% of iron element) as a vanadium source was added, and this mixed solution was the same as in Example 1.
- Iron powder Lithium iron phosphate was prepared in the same manner as in Example 1 except that 9.9 mol was added.
- Lithium iron phosphate was prepared in the same manner as in Example 1 except that the stirring after adding the iron powder was performed in a nitrogen atmosphere.
- Comparative Example 2 Lithium iron phosphate was prepared in the same manner as in Example 1 except that iron powder (FEE04PB manufactured by High Purity Chemical Laboratory Co., Ltd., oxygen content: less than 0.1% by mass, particle size: 53 ⁇ m or less) was used. did.
- iron powder FEE04PB manufactured by High Purity Chemical Laboratory Co., Ltd., oxygen content: less than 0.1% by mass, particle size: 53 ⁇ m or less
- Lithium iron phosphate was prepared in the same manner as in Example 1 except that 2 mol of oxalic acid dihydrate was used instead of citric acid monohydrate.
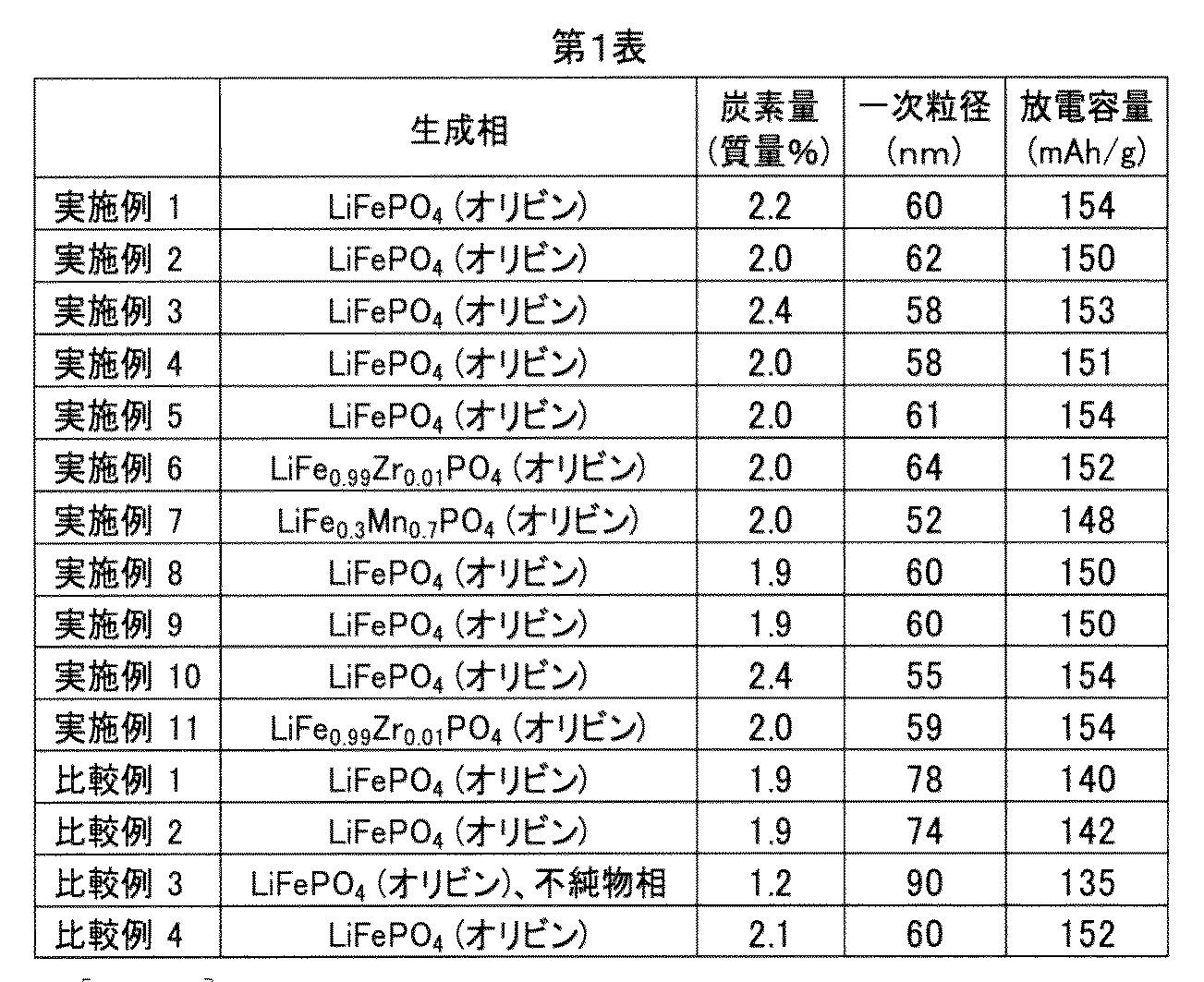
- Each lithium iron phosphate prepared in Examples 1 to 11 and Comparative Examples 1 to 4 was subjected to identification analysis (identification of a generated phase) by X-ray diffraction analysis and carbon quantification. Moreover, the primary particle size was measured about each lithium iron phosphate. X-ray diffraction analysis was performed using Ultimate IV (X-Ray: Cu-K ⁇ 1) manufactured by Rigaku. The carbon content of the lithium iron phosphate was quantified using EMIA620 manufactured by HORIBA. The primary particle size was determined from the X-ray diffraction analysis using the Scherrer equation. The identification analysis, the carbon content and the primary particle size measurement results are shown in Table 1 below.
- each lithium iron phosphate prepared in Examples 1 to 11 and Comparative Examples 1 to 4 was measured by the following method.
- As the negative electrode a half cell (made by Hosen) was assembled using metallic lithium.
- the carbon content is 1.9 to 2.4% by mass
- the primary particle size is 52 to 64 nm
- the discharge capacity is High olivine-type lithium iron phosphate was obtained.
- Comparative Examples 1 to 3 lithium iron phosphate having a sufficient discharge capacity was not obtained. In Comparative Examples 1 to 3, it is presumed that phosphorus, iron, and lithium are not uniformly mixed at the atomic level. In addition, when Comparative Example 4 having many steps up to secondary firing is compared with Examples 1 to 11 in which the steps are simpler, the discharge capacity of the obtained lithium iron phosphate is compared with Examples 1 to 11. It was found to be equivalent to Example 4.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Battery Electrode And Active Subsutance (AREA)
Abstract
Description
そのため、大型機器を睨んだリチウムイオン電池の用途展開のためには、従前のコバルト酸リチウムよりも安価であり、かつ、熱的、化学的に安定で安全性の高い正極活物質の開発が不可欠である。
このリン酸鉄リチウムは、結晶構造中の強固なP−O結合により約400℃まで酸素を放出しないことから、安全性の高い活物質であり、さらには、長期安定性や急速充電特性も良好な活物質である。
かかる問題の解決策としては、リン酸鉄リチウム粒子の表面に導電性物質を被覆し、かつ、リン酸鉄リチウム粒子を約100nmの粒径に微細化して反応表面積を増大させることが有効とされている。また、他元素をドープすることが、電子伝導性の改善や結晶構造の安定化に有効であるという報告もある。
例えば、特許文献1には、まず金属鉄とリン酸イオンを遊離する化合物とを水溶液中で反応させ、その後、炭酸リチウムまたは水酸化リチウムを加えてリン酸鉄リチウムの前駆体(precursor)を調製して乾燥し、この乾燥物を300~450℃の温度範囲で一次焼成、ついで熱分解により導電性炭素を生成する物質を加えて500~800℃で焼成する方法が記載されている。
なお、特許文献1には、未反応金属鉄の反応を促進するために、塩酸、シュウ酸等の酸を添加することも開示されている。しかし、塩酸を添加する場合には生成物が酸化されやすく、またシュウ酸を添加する場合には安定なシュウ酸鉄が単体で沈殿物として生成する等、均一な前駆体を調製することが難しい。
そして、特許文献1に記載された方法において導電性炭素として添加するカーボンブラック等は、前駆体中に原子レベルで均一に混合することが難しいため、前駆体が酸化されやすい一次焼成の温度範囲において還元剤としての効果は小さい。
さらに、特許文献1に記載された方法では、焼成を2回行うため工程数が多い。
さらに、特許文献2には、有機酸または混合有機酸で鉄を酸化して有効な2価鉄を生成させると記載されているが、単に原料を混合するだけでは2価鉄を安定して存在させることは難しいという問題がある。
そして、特許文献2に記載された方法において、リチウム塩が硝酸リチウムである場合には硝酸イオンが焼成時に酸化剤として作用し、また、酢酸リチウムである場合には原料が高価であるため、低コスト化を図る上では不適当である。
なお、特許文献3には、導電性酸化物として酸化バナジウム(V2O5)を加える例が記載されているが、酸化バナジウムをリン酸鉄リチウムの前駆体が生成した後に加えているため、バナジウムはドープされずにリン酸鉄リチウム粒子の周囲に付着する。
すなわち、本発明は、以下の(1)~(11)を提供する。
前記水溶液に、0.1~2質量%の酸素を含有する鉄粒子を添加し、酸化雰囲気下で当該水溶液中の前記リン酸および前記ヒドロキシカルボン酸と前記鉄粒子とを反応させて第1反応液を作製する第1の作製工程と、
前記第1反応液にリチウム源を添加して第2反応液を作製する第2の作製工程と、
前記第2反応液に炭素源を添加して第3反応液を作製する第3の作製工程と、
前記第3反応液を乾燥させてリン酸鉄リチウム前駆体を生成させる前駆体生成工程と、
前記リン酸鉄リチウム前駆体を非酸化性雰囲気下で焼成してリン酸鉄リチウムを得る焼成工程と、
を備えるリン酸鉄リチウムの製造方法。
以下、本発明の製造方法について、詳細に説明する。
水溶液準備工程は、リン酸およびヒドロキシカルボン酸を含む水溶液を準備する工程である。水溶液準備工程で準備される水溶液に含まれるリン酸およびヒドロキシカルボン酸の詳細は後述する。なお、当該水溶液に含まれる水としては、特に限定されず、例えば、イオン交換水、蒸留水が好適に用いられる。
第1の作製工程は、水溶液準備工程で準備された水溶液に、0.1~2質量%の酸素を含有する鉄粒子(iron particles)を添加し、酸化雰囲気下で当該水溶液中のリン酸およびヒドロキシカルボン酸と鉄粒子とを反応させて第1反応液を作製する工程である。
第1の作製工程においては、0.1~2質量%の酸素を含有する鉄粒子を、リン酸およびヒドロキシカルボン酸を含む水溶液に添加し、酸化雰囲気中で反応させることにより、リン酸鉄のキレート体が形成される。
通常の一般工業用鉄粉の平均粒径は70~80μmであるが、最大粒径が150~180μmの粒子も含まれるため、必要に応じて篩いにより粗粒を除去したり、機械式粉砕で粗粒を微細化する等、反応面積を大きくして使用したほうがよい。
これに対して、鉄粒子の酸素含有量が0.1質量%未満である場合は、金属鉄とリン酸との直接反応が優先して難溶性の2価鉄化合物であるリン酸鉄(Fe3(PO4)2・8H2O)の凝集粒子が生成・成長しやすくなる。そのため、水溶液が、白色~淡青色を呈するクリーム状の高粘度物質となる場合がある。その結果、水溶液の撹拌が不十分となり、未反応の金属鉄が残存し易い、原料が均一に混合されない等の支障をきたす。
また、鉄粒子の酸素含有量が2質量%を超えると、鉄粉表面に酸化鉄のスケールが偏析するため、リン酸とヒドロキシカルボン酸の水溶液との反応が妨げられる。
鉄粒子の酸素含有量は、例えば、ミルスケール(酸化鉄)をコークスで還元しただけの還元鉄粉や、溶鋼を高圧水で粉化、冷却後の乾燥しただけのアトマイズ鉄粉の場合、0.5~1.5質量%である。また、この鉄粒子を水素還元した場合の酸素含有量は0.1~0.4質量%となる。
本発明に使用する鉄粒子は、高価な水素還元処理した鉄粒子を特に必要としない。
なお、鉄粒子の酸素含有量は、JIS Z 2613(1992年)真空融解赤外線吸収法に準拠し、LECO社製TC436を用いて定量したものである。
キレート体が形成される反応(以下「キレート反応」ともいう)が進んで鉄粒子表面の酸素が消費されると、キレート反応は持続できず、金属鉄とリン酸イオンとの直接反応が優先して難溶性のリン酸鉄の凝集粒子が生成・成長してしまう。
そこで、本発明では、上記反応時の雰囲気を酸化雰囲気とすることにより、鉄粒子表面を適度に酸化して鉄粒子に化学結合した酸素を補給し、キレート反応を持続させる。
リン酸の添加量は、鉄1molに対して1molが化学量論的当量であるが、0.1mol程度過剰に添加してもよい。
本発明において用いられるヒドロキシカルボン酸としては、例えば、鉄に対するキレート力の強い乳酸、酒石酸、リンゴ酸、クエン酸等が挙げられ、なかでも、キレート力が強く、かつ、酸化されにくいキレート体を形成するクエン酸が好ましい。
ヒドロキシカルボン酸の残炭率としては、乳酸:7質量%、酒石酸:7質量%、リンゴ酸:12質量%、クエン酸一水和物:7質量%である。また、カルボン酸のシュウ酸二水和物、酢酸などは1質量%未満であるため、焼成時の還元作用が小さい。
上記添加量が0.1mol未満の場合は、ヒドロキシカルボン酸によるキレート化の効果が小さくなるため、金属鉄とリン酸イオンとが直接反応して難溶性のリン酸鉄の凝集粒子が生成・成長し、水溶液が白色~淡青色を呈するクリーム状態の高粘度物質となってしまい、その結果、水溶液の撹拌が不十分となり、未反応の金属鉄が残存しやすい、原料が均一に混合されない等の支障をきたす場合がある。
また、上記添加量が0.5molを超える場合には、形成されるリン酸鉄のキレート体が水溶液中に均一に分散する(原料が均一に混合される)が、焼成後の残炭量が過剰となり、その結果、最終的に得られるリン酸鉄リチウムの見かけ上の放電容量が低下する場合がある。
これに対し、上記添加量が上記範囲内であれば、未反応の金属鉄が残存しにくくなり、原料が均一に混合され、また、得られるリン酸鉄リチウムの見かけ上の放電容量が良好になる。
水溶液温度を10~40℃の範囲に制御すると、上記キレート反応により鉄粒子表面の酸素が消費され、続いて新たに現れた鉄粒子表面が水溶液中の溶存酸素や空気バブル等と接触することにより適度に酸化されることで、連続的にリン酸鉄のキレート体を形成させることが可能となる。
これに対して、水溶液温度が10℃未満である場合には、鉄粒子のキレート反応が遅くなり、完全に反応が終了するまでに長時間を要する場合がある。
また、水溶液温度が40℃を超える場合には、酸素が消費された鉄粒子表面に酸素を補うための酸化が追いつかず、そのため、金属鉄とリン酸との直接反応が優先して難溶性のリン酸鉄の凝集粒子が生成・成長し、水溶液が白色~淡青色を呈するクリーム状の高粘度物質となってしまう場合がある。

第2の作製工程は、第1の作製工程で作製された第1反応液にリチウム源を添加して第2反応液を作製する工程である。
第2の作製工程では、上記式(1)で表されるキレート体が均一に分散した第1反応液にリチウム源を添加することにより、原料が原子レベルで均一に混合されたリン酸鉄リチウムの前駆体が得られる。
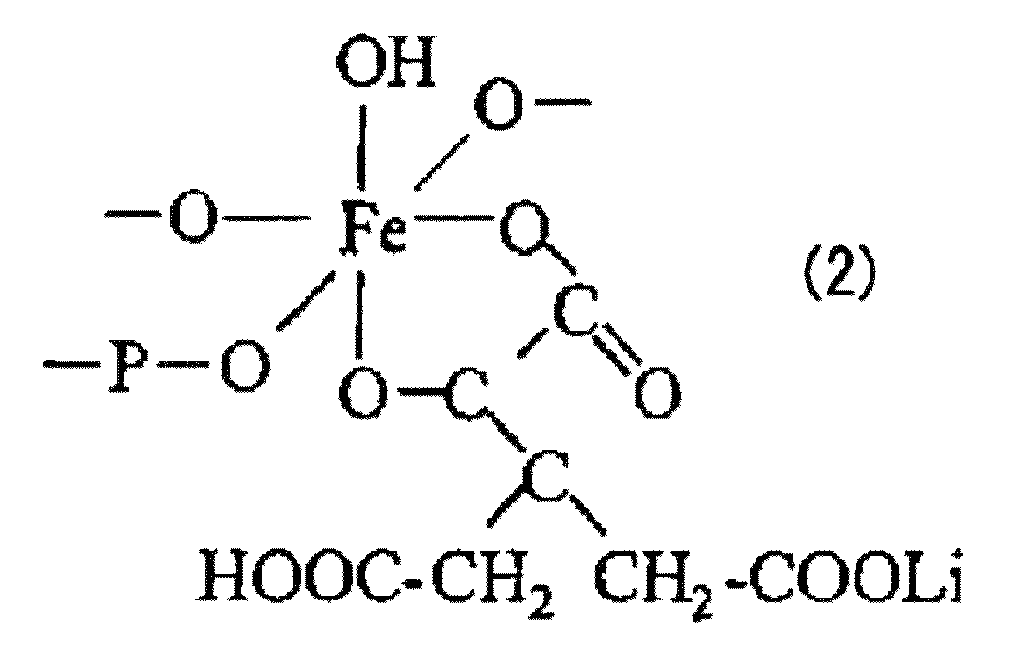
このような場合には、後の工程で行われる乾燥の前に、前駆体溶液の均一化を図るため、凝集粒子を湿式で機械粉砕して微細化することが望ましい。なお、湿式粉砕方法としては、例えば、超音波照射、湿式ジェットミル、ビーズミル等による方法が挙げられる。
第3の作製工程は、第2作製工程で作製された第2反応液に炭素源を添加して第3反応液を作製する工程である。
焼成時に熱分解して炭素を生成する物質としては、焼成時に溶融してリン酸鉄リチウム粒子の表面を濡らすもの、または、ガス状物質を生成しリン酸鉄リチウム粒子の表面に炭素を析出するものが好ましく、具体的には、例えば、グルコース、フルクトース、マルトース、スクロース、アスコルビン酸、エリソルビン酸などの糖;ポリビニルアルコール(PVA)、ポリエチレングリコールなどの水溶性高分子;等を用いることができる。
導電性炭素としては、例えば、カーボンブラック、アセチレンブラック、ケッチェンブラック、気相成長炭素繊維(VGCF)、カーボンナノファイバー、グラファイト、フラーレン等を用いることができる。
これらは、1種単独で用いてもよく、2種以上を併用してもよい。
しかし、炭素源の添加量が上記範囲内であれば、リン酸鉄リチウムの導電性が十分となり、見かけ上の放電容量も良好となる。
前駆体生成工程は、第3の作製工程で作製された第3反応液を乾燥させてリン酸鉄リチウム前駆体を生成させる工程である。
第3反応液の乾燥物であるリン酸鉄リチウム前駆体は、粉末状であり、その粒径は特に限定されないが、取り扱いのしやすさの観点から、50~200μmが好ましい。
スプレードライ法を採用する場合には、スプレードライ装置の入口温度(加熱空気温度)を100~250℃とすることが好ましい。入口温度を150~250℃にすれば、生成する乾燥物の到達温度は、送液量とのバランスにも依存するが、100~150℃となる。
焼成工程は、前駆体生成工程で生成したリン酸鉄リチウム前駆体を非酸化性雰囲気下で焼成してリン酸鉄リチウムを得る工程である。
前駆体生成工程で生成したリン酸鉄リチウム前駆体を、非酸化性雰囲気下で焼成することにより、リン酸鉄リチウム前駆体に含まれる水酸基および有機物が熱分解によりH2O,CO2,H2および炭化水素として除去され、アモルファス相を有する乾燥物が結晶化しオリビン構造であるリン酸鉄リチウムの結晶体に変化するとともに、熱分解炭素がリン酸鉄リチウムの粒子表面に析出する。
焼成温度が300℃未満の場合には、揮発成分であるH2O,CO2,H2および炭化水素の熱分解除去が不十分となる場合があるうえ、結晶化が生じない場合もある。一方、焼成温度が800℃を超えると、得られる結晶粒子の粗大化が進行し、Fe2Pなどの副生成物が生成する場合がある。
しかし、焼成温度が上記範囲内であれば、揮発成分の熱分解除去および結晶化が十分に進行し、また、結晶粒子の粗大化やFe2Pなどの副生成物の生成も抑制される。
なお、本発明において、リン酸鉄リチウムの一次粒径は、Rigaku社製UltimaIV(X−Ray:Cu−Kα1)を使用して行ったX線回折分析によりScherrer式を用いて求めたものである。一次粒径とは、結晶子径のことで結晶の最小単位である。
正極活物質には高速充放電特性が要求されることから、本発明の製造方法により得られるリン酸鉄リチウムは、リチウムイオン電池等の二次電池用正極活物質として好適に用いられる。
なお、本発明においては、安価な鉄粒子が使用されているため、低コスト化も実現されている。
この場合、例えば、上述した水溶液準備工程において、リン酸およびヒドロキシカルボン酸を含む水溶液に、鉄と置換する元素の金属または化合物を予め溶解させればよい。これにより、置換する元素を均一に混合することができる。
一方、置換量の上限値については、置換元素のイオン半径、価数、配位数などの要因により大きく変化するため、一概には決められないが、置換量が閾値を超えると、不純物相の生成やバンド構造の変化による電子の局在化などにより特性が悪くなる傾向がある。
蒸留水:2000gに、85質量%のリン酸:10molおよびクエン酸一水和物:2molを溶解し、この混合溶液に鉄粉(JFEスチール社製、酸素含有量:0.68質量%、平均粒径:80μm):10molを添加して、液温:25℃、空気雰囲気下で撹拌しながら1日間反応させた。次いで、炭酸リチウム:5molを加えてリン酸鉄リチウムとし、最後に炭素源のアスコルビン酸:80gを加えて前駆体溶液を調製した。この前駆体を、スプレードライヤ(大川原化工機社製FOC16)を用いて、入口温度:200℃で乾燥し、SEM観察による平均粒径が約50μmの乾燥粉末を得た。この乾燥粉末を窒素気流中にて、700℃×5hの焼成を施し、最後に目開き63μmで篩い、リン酸鉄リチウムを調製した。
なお、鉄粉の酸素含有量は、LECO社製TC436を用いて定量した。
クエン酸一水和物に代えて乳酸:2molを使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
クエン酸一水和物に代えてリンゴ酸:2molを使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
クエン酸一水和物に代えて酒石酸:2molを使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
鉄粉(JFEスチール社製、酸素含有量:0.41質量%、平均粒径:80μm)を使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
リン酸およびクエン酸一水和物の混合溶液に、ジルコニウム源のオキシ酢酸ジルコニウム:0.1mol(鉄元素の1mol%置換)を加えて溶解させ、この混合溶液に実施例1と同じ鉄粉:9.9molを添加したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
リン酸およびクエン酸一水和物の混合溶液に、マンガン源の炭酸マンガン(MnCO3・nH2O):7mol(鉄元素の70mol%置換)を加えて、この混合溶液に実施例1と同じ鉄粉:3molを添加したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
炭素源のアスコルビン酸:80gに代えてスクロース:80gを使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
炭素源のアスコルビン酸:80gに代えてケッチェンブラックペースト(ライオン社製W−376R、濃度20質量%):80gを使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
炭素源のアスコルビン酸:80gに代えてアスコルビン酸:95gを使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
リン酸およびクエン酸一水和物の混合溶液に、バナジウム源の酸化バナジウム(V2O5):0.05mol(鉄元素の1mol%置換)を加えて、この混合溶液に実施例1と同じ鉄粉:9.9molを添加したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
鉄粉を添加した後の撹拌を窒素雰囲気下で行ったこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
鉄粉(高純度化学研究所社製FEE04PB、酸素含有量:0.1質量%未満、粒径:53μm以下)を使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
クエン酸一水和物に代えてシュウ酸二水和物:2molを使用したこと以外は、実施例1と同様にして、リン酸鉄リチウムを調製した。
蒸留水:2000gに、85質量%のリン酸:10molおよびクエン酸一水和物:2molを溶解し、この混合溶液に鉄粉(JFEスチール社製、酸素含有量:0.68質量%、平均粒径:80μm):10molを添加して、液温:25℃、空気雰囲気下で撹拌しながら1日間反応させた。次いで、炭酸リチウム:5molを加えてリン酸鉄リチウムの前駆体溶液を調製した。この前駆体溶液を、スプレードライヤ(大川原化工機社製FOC16)を用いて、入口温度:200℃で乾燥し、SEM観察による平均粒径が約50μmの乾燥粉末を得た。この乾燥粉末を窒素気流中にて、400℃×5hの一次焼成を施し、次いでこの焼成粉に炭素源のアスコルビン酸:80gを加えて湿式ボールミルにて粉砕・混合を行った。得られた混合物を乾燥後、窒素気流中にて700℃×10hの二次焼成を施し、最後に目開き63μmで篩い、リン酸鉄リチウムを調製した。
X線回折分析は、Rigaku社製UltimaIV(X−Ray:Cu−Kα1)を使用して行った。炭素の定量は、HORIBA社製EMIA620を使用し、リン酸鉄リチウムの炭素含有量を定量した。一次粒径は、X線回折分析よりScherrer式を用いて求めた。
上記同定分析、炭素量および一次粒径の測定結果を、下記第1表に示す。
アルミ箔の集電体に、リン酸鉄リチウム:VGCF:ポリフッ化ビニリデン(クレハ社製KFL#1320)=86:4:10(質量比)のペーストを10mg/cm2塗布して、正極を作製した。負極は金属リチウムを用いてハーフセル(宝泉製)を組み立てた。電解液は、1M−LiPF6/EC(エチレンカーボネート):EMC(エチルメチルカーボネート)=3:7(質量比)を使用した。測定条件は、定電流充電を0.2mA/cm2で4.0Vまで行った後、定電流放電を0.2mA/cm2で2.5Vまで行い、放電容量を求めた。ただし、実施例7のみ定電流充電を4.5Vまで行った。求めた放電容量を下記第1表に示す。
なお、二次焼成まで行う工程の多い比較例4と、工程がより簡略な実施例1~11とを対比すると、得られたリン酸鉄リチウムの放電容量については、実施例1~11は比較例4と同等であることが分かった。
Claims (11)
- リン酸およびヒドロキシカルボン酸を含む水溶液を準備する水溶液準備工程と、
前記水溶液に、0.1~2質量%の酸素を含有する鉄粒子を添加し、酸化雰囲気下で当該水溶液中の前記リン酸および前記ヒドロキシカルボン酸と前記鉄粒子とを反応させて第1反応液を作製する第1の作製工程と、
前記第1反応液にリチウム源を添加して第2反応液を作製する第2の作製工程と、
前記第2反応液に炭素源を添加して第3反応液を作製する第3の作製工程と、
前記第3反応液を乾燥させてリン酸鉄リチウム前駆体を生成させる前駆体生成工程と、
前記リン酸鉄リチウム前駆体を非酸化性雰囲気下で焼成してリン酸鉄リチウムを得る焼成工程と、
を備えるリン酸鉄リチウムの製造方法。 - 前記水溶液準備工程の水溶液に、鉄と置換する元素の金属または化合物を溶解させる溶解工程を有する、請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記鉄と置換する元素が、チタン、バナジウム、ジルコニウムとマンガンからなるグループから選択された一つである、請求項2に記載のリン酸鉄リチウムの製造方法。
- 前記ヒドロキシカルボン酸が3質量%以上の残炭率を有する請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記ヒドロキシカルボン酸が、乳酸、酒石酸、リンゴ酸とクエン酸からなるグループから選択された少なくとも一つである請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記ヒドロキシカルボン酸が、前記鉄粒子中の鉄1molに対して0.1~0.5molの添加量を有する請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記第3の作製工程は、焼成後のリン酸鉄リチウムに含まれる炭素量が1~5質量%になるように前記第2反応液に炭素源を添加して第3反応液を作製することからなる、請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記炭素源は、焼成時に熱分解して炭素を精製する物質、または、導電性炭素である、請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記炭素源が、グルコース、フルクトース、マルトース、スクロース、アスコルビン酸とエリソルビン酸からなるグループから選択された少なくとも一つである請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記炭素源が、カーボンブランク、アセチレンブラック、ケッチェンブラック、気相成長炭素繊維(VGCF)、カーボンナノファイバー、グラファイトとフラーレンからなるグループから選択された少なくとも一つである請求項1に記載のリン酸鉄リチウムの製造方法。
- 前記リン酸鉄リチウムが、二次電池用正極活物質である、請求項1に記載のリン酸鉄リチウムの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12801341.4A EP2698347A4 (en) | 2011-06-17 | 2012-02-27 | METHOD FOR PRODUCING LITHIUM STEEL PHOSPHATE |
| US14/122,354 US20140127111A1 (en) | 2011-06-17 | 2012-02-27 | Method for producing lithium iron phosphate |
| KR1020137033512A KR20140024923A (ko) | 2011-06-17 | 2012-02-27 | 인산철리튬의 제조방법 |
| CN201280029356.7A CN103596877A (zh) | 2011-06-17 | 2012-02-27 | 磷酸铁锂的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-135055 | 2011-06-17 | ||
| JP2011135055A JP2013001605A (ja) | 2011-06-17 | 2011-06-17 | リン酸鉄リチウムの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012172839A1 true WO2012172839A1 (ja) | 2012-12-20 |
Family
ID=47356838
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/055487 Ceased WO2012172839A1 (ja) | 2011-06-17 | 2012-02-27 | リン酸鉄リチウムの製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20140127111A1 (ja) |
| EP (1) | EP2698347A4 (ja) |
| JP (1) | JP2013001605A (ja) |
| KR (1) | KR20140024923A (ja) |
| CN (1) | CN103596877A (ja) |
| TW (1) | TW201300315A (ja) |
| WO (1) | WO2012172839A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2746220A3 (de) * | 2012-12-21 | 2014-07-30 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Mit einem Interkalationsmaterial beschichtete Partikeln mit großer Oberfläche, Verfahren zu deren Herstellung sowie Verwendung der Partikeln in Hybridelektroden und hochkapazitativen Doppelschichtkondensatoren und schnellen Batterien |
| CN105409033A (zh) * | 2013-05-08 | 2016-03-16 | 台湾立凯电能科技股份有限公司 | 电池复合材料及其前驱物的制备方法 |
| CN113195412A (zh) * | 2018-12-21 | 2021-07-30 | 霍加纳斯股份有限公司 | 含纯铁化合物 |
| CN113929070A (zh) * | 2021-10-09 | 2022-01-14 | 湖北万润新能源科技股份有限公司 | 一种高倍率磷酸铁锂正极材料的制备方法 |
| CN115506006A (zh) * | 2022-08-23 | 2022-12-23 | 合肥国轩高科动力能源有限公司 | 一种电池级单晶磷酸铁的制备方法 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014201459A (ja) * | 2013-04-02 | 2014-10-27 | Jfeケミカル株式会社 | リン酸鉄リチウムの製造方法 |
| TWI606633B (zh) * | 2016-12-29 | 2017-11-21 | 尚志精密化學股份有限公司 | 電池複合材料的前驅漿料及電池複合材料的製備方法 |
| CN109205586B (zh) * | 2018-09-07 | 2019-12-10 | 高延敏 | 一种工业化的磷酸铁锂制造方法及其制备的磷酸铁锂复合材料 |
| CN113683071A (zh) * | 2021-07-20 | 2021-11-23 | 广东邦普循环科技有限公司 | 一种高倍率磷酸铁锂的制备方法 |
| CN113830747B (zh) * | 2021-09-17 | 2023-05-05 | 湖北亿纬动力有限公司 | 一种低温启动型磷酸铁锂正极材料及其制备方法 |
| CN114188508B (zh) * | 2021-10-28 | 2023-02-14 | 厦门理工学院 | 一种磷酸铁锂正极材料、制备方法及应用 |
| CN114314548B (zh) | 2021-12-29 | 2022-11-25 | 湖北万润新能源科技股份有限公司 | 钛、锆共掺杂碳包覆磷酸铁锂材料及其制备方法与应用 |
| CN114506833B (zh) * | 2022-01-25 | 2023-08-18 | 佛山市德方纳米科技有限公司 | 一种锂电池正极材料及其制备方法 |
| KR102746787B1 (ko) | 2022-05-25 | 2024-12-26 | 후베이 알티 어드밴스드 머터리얼스 그룹 컴퍼니 리미티드 | 인산망간철리튬의 제조 방법 |
| TWI838175B (zh) * | 2023-03-21 | 2024-04-01 | 台灣立凱電能科技股份有限公司 | 碳包覆磷酸鋰鐵材料的製造方法 |
| WO2025076720A1 (zh) * | 2023-10-11 | 2025-04-17 | 万华化学(烟台)电池材料科技有限公司 | 一种磷酸锰铁锂正极材料及其制备方法和锂离子电池 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005116393A (ja) * | 2003-10-09 | 2005-04-28 | Sumitomo Osaka Cement Co Ltd | 電極材料粉体の製造方法と電極材料粉体及び電極並びにリチウム電池 |
| JP2006131485A (ja) * | 2004-11-03 | 2006-05-25 | Tatung Co | オリビン型リン酸鉄リチウム正極材料の製造方法 |
| JP2007305585A (ja) * | 2006-05-11 | 2007-11-22 | Aquire Energy Co Ltd | 充電式バッテリ製造用の陰極材料 |
| JP2011042553A (ja) * | 2009-03-13 | 2011-03-03 | Jfe Chemical Corp | リン酸鉄リチウムの製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101442117B (zh) * | 2008-12-22 | 2012-03-21 | 上海电力学院 | 碳包覆磷酸铁锂的制备方法 |
| CN102034962B (zh) * | 2009-09-30 | 2013-11-06 | 清华大学 | 一种锂离子电池正极物质的制备方法 |
| CN101764205A (zh) * | 2009-10-14 | 2010-06-30 | 孙琦 | 一种碳包覆磷酸铁锂的制备方法 |
-
2011
- 2011-06-17 JP JP2011135055A patent/JP2013001605A/ja active Pending
-
2012
- 2012-02-27 EP EP12801341.4A patent/EP2698347A4/en not_active Withdrawn
- 2012-02-27 KR KR1020137033512A patent/KR20140024923A/ko not_active Ceased
- 2012-02-27 CN CN201280029356.7A patent/CN103596877A/zh active Pending
- 2012-02-27 US US14/122,354 patent/US20140127111A1/en not_active Abandoned
- 2012-02-27 WO PCT/JP2012/055487 patent/WO2012172839A1/ja not_active Ceased
- 2012-03-27 TW TW101110529A patent/TW201300315A/zh unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005116393A (ja) * | 2003-10-09 | 2005-04-28 | Sumitomo Osaka Cement Co Ltd | 電極材料粉体の製造方法と電極材料粉体及び電極並びにリチウム電池 |
| JP2006131485A (ja) * | 2004-11-03 | 2006-05-25 | Tatung Co | オリビン型リン酸鉄リチウム正極材料の製造方法 |
| JP2007305585A (ja) * | 2006-05-11 | 2007-11-22 | Aquire Energy Co Ltd | 充電式バッテリ製造用の陰極材料 |
| JP2011042553A (ja) * | 2009-03-13 | 2011-03-03 | Jfe Chemical Corp | リン酸鉄リチウムの製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| JIS G 1211, 1995 |
| JIS Z 2613, 1992 |
| See also references of EP2698347A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2746220A3 (de) * | 2012-12-21 | 2014-07-30 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Mit einem Interkalationsmaterial beschichtete Partikeln mit großer Oberfläche, Verfahren zu deren Herstellung sowie Verwendung der Partikeln in Hybridelektroden und hochkapazitativen Doppelschichtkondensatoren und schnellen Batterien |
| CN105409033A (zh) * | 2013-05-08 | 2016-03-16 | 台湾立凯电能科技股份有限公司 | 电池复合材料及其前驱物的制备方法 |
| CN105409033B (zh) * | 2013-05-08 | 2018-07-17 | 台湾立凯电能科技股份有限公司 | 电池复合材料及其前驱物的制备方法 |
| CN113195412A (zh) * | 2018-12-21 | 2021-07-30 | 霍加纳斯股份有限公司 | 含纯铁化合物 |
| US12291450B2 (en) | 2018-12-21 | 2025-05-06 | Höganäs Ab (Publ) | Pure iron containing compound |
| CN113929070A (zh) * | 2021-10-09 | 2022-01-14 | 湖北万润新能源科技股份有限公司 | 一种高倍率磷酸铁锂正极材料的制备方法 |
| CN115506006A (zh) * | 2022-08-23 | 2022-12-23 | 合肥国轩高科动力能源有限公司 | 一种电池级单晶磷酸铁的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013001605A (ja) | 2013-01-07 |
| EP2698347A1 (en) | 2014-02-19 |
| US20140127111A1 (en) | 2014-05-08 |
| EP2698347A4 (en) | 2014-10-29 |
| KR20140024923A (ko) | 2014-03-03 |
| CN103596877A (zh) | 2014-02-19 |
| TW201300315A (zh) | 2013-01-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5388822B2 (ja) | リン酸鉄リチウムの製造方法 | |
| JP5581065B2 (ja) | リン酸鉄リチウムの製造方法 | |
| WO2012172839A1 (ja) | リン酸鉄リチウムの製造方法 | |
| JP5995726B2 (ja) | 微粒子混合物、正極活物質材料、正極、2次電池及びこれらの製造方法 | |
| CN102123943B (zh) | 制备包含锂、钒和磷酸根的多孔材料的方法 | |
| JP5004413B2 (ja) | 燐酸アンモニウム鉄及びリチウムイオン二次電池用正極材料の製造方法、並びにリチウムイオン二次電池 | |
| JP7131911B2 (ja) | 高出力アプリケーション用のナノスケールポア構造のカソードおよび材料合成方法 | |
| WO2014163124A1 (ja) | リン酸鉄リチウムの製造方法 | |
| CN103636035A (zh) | 锂离子二次电池用正极活性物质的制造方法 | |
| JP4252331B2 (ja) | リチウムイオン電池用正極活物質の製造方法 | |
| JP2005190882A (ja) | リチウム電池用正極活物質の製造方法とリチウム電池用正極活物質及びリチウム電池用電極並びにリチウム電池 | |
| JP5750086B2 (ja) | リチウムイオン二次電池正極材用リン酸鉄リチウム粒子およびその製造方法、ならびにこれを用いたリチウムイオン二次電池正極およびリチウムイオン二次電池 | |
| JP5648732B2 (ja) | 正極活物質及びリチウムイオン二次電池 | |
| JP2013251227A (ja) | リチウムイオン二次電池正極材用リン酸鉄リチウム粒子およびその製造方法、ならびに上記リン酸鉄リチウム粒子を用いたリチウムイオン二次電池正極およびその正極を備えるリチウムイオン二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12801341 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012801341 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14122354 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20137033512 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |