WO2012173217A1 - ピロロキノリンキノンジナトリウム塩の結晶およびその製造方法 - Google Patents
ピロロキノリンキノンジナトリウム塩の結晶およびその製造方法 Download PDFInfo
- Publication number
- WO2012173217A1 WO2012173217A1 PCT/JP2012/065303 JP2012065303W WO2012173217A1 WO 2012173217 A1 WO2012173217 A1 WO 2012173217A1 JP 2012065303 W JP2012065303 W JP 2012065303W WO 2012173217 A1 WO2012173217 A1 WO 2012173217A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal
- crystals
- pqq
- precursor
- disodium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/66—Preparation of oxygen-containing organic compounds containing the quinoid structure
Definitions
- the present invention relates to crystals of pyrroloquinoline quinone disodium salt and a method for producing the same.
- PQQ pyrroloquinoline quinone
- Non-Patent Document 1 pyrroloquinoline quinone
- PQQ may be a new vitamin (for example, Non-Patent Document 1).
- PQQ is present not only in bacteria but also in eukaryotic molds and yeasts, and plays an important role as a coenzyme.
- PQQ promotes cell growth, anti-cataract, liver disease prevention and treatment, wounds
- Many physiological activities such as healing action, anti-allergic action, reverse transcriptase inhibitory action and glyoxalase I inhibitory action-anticancer action have been revealed. Therefore, PQQ attracts attention as a useful substance in the pharmaceutical, food and cosmetic fields.
- PQQ is expected to be applied as a therapeutic agent for heart (Patent Document 1), skin (Patent Document 2), nerve (Patent Document 3) and the like.
- PQQ is expected to have an anti-aging effect
- PQQ is expected to have an anti-aging effect
- its application is expected as a substance having a beautifying effect.
- PQQ can be produced by an organic chemical synthesis method, a fermentation method, or the like, and can be provided as an alkali metal salt, particularly a disodium salt crystal (hereinafter sometimes referred to as “disodium crystal”). Many.
- This PQQ disodium crystal is a pentahydrate crystal and has been subjected to structural analysis by single crystal analysis (Non-patent Document 1).
- Non-Patent Document 4 a method of crystallizing by adjusting the pH of the solution
- Patent Document 5 a precipitation method using a water-soluble organic solvent
- the obtained solid has low crystallinity or is a method using an edible solvent.
- the storage stability of the obtained crystals is not necessarily high.
- PQQ In applying PQQ to pharmaceuticals and cosmetics, it is desired to have high crystallinity, few impurities, stable solubility in solvents or high dispersibility in solvents, and high storage stability. In addition, it is further desired that it is harmless to the human body when applied to food and medicine. Further, when applied to dermatological drugs and cosmetics, high permeability to the skin is desired.
- An object of the present invention is to provide a new PQQ disodium crystal that is excellent in dispersibility to a solvent and permeability to skin and is useful in the pharmaceutical field, cosmetic field or food field, and an efficient production method thereof.
- the present inventors have found that new crystal forms (crystal form 1 and crystal form 2) of PQQ disodium crystals can be obtained by preparing PQQ disodium crystals under specific conditions.
- PQQ disodium crystals have thermal stability in edible oil.
- good crystalline form 1 and crystalline form 2 can be obtained in a short time. It was found that the following crystals were obtained.
- crystal form 1 and crystal form 2 PQQ disodium crystals are highly dispersible in oil and highly permeable to the skin. The present invention is based on such knowledge.
- the pyrroloquinoline quinone disodium crystal according to the above [1] or [2] which exhibits a 2 ⁇ angle peak at 0.4, 28.5 ⁇ 0.4 ° (crystal form 1).
- a functional food or nutrient comprising the pyrroloquinoline quinone disodium crystal according to any one of [1] to [4] above.
- a method for producing a pyrroloquinoline quinone disodium crystal according to any one of [1] to [4] A method for producing a pyrroloquinoline quinone disodium crystal, comprising a step of obtaining a crystal (precursor crystal) which is a precursor of the pyrroloquinoline quinone disodium crystal and then drying the precursor crystal.
- the precursor crystal is crystallized by adjusting the pH of an aqueous solution of ethanol or isopropanol having a concentration of 10 to 90% in which a sodium salt of pyrroloquinoline quinone is dissolved to a range of 2 to 5.
- the precursor crystals were 9.1 ⁇ 0.4, 10.3 ⁇ 0.4, 13.8 ⁇ 0.4, 17.7 ⁇ 0. 0 by powder X-ray diffraction using Cu K ⁇ radiation.
- the PQQ disodium crystal of the present invention has not only high purity but also improved dispersibility in solution and permeability to skin, and is useful as a component of cosmetics, pharmaceuticals or functional foods.
- FIG. 1 is a graph showing the results of powder X-ray diffraction of PQQ disodium crystals (crystal form 1).
- FIG. 2 is a diagram showing the results of powder X-ray diffraction when the temperature of the drying process is changed.
- FIG. 3 is a diagram showing the results of powder X-ray diffraction of PQQ disodium crystals prepared by an ethanol precipitation method.
- the PQQ disodium crystal of the present invention is a crystal having a low water content (hereinafter sometimes referred to as “low water content crystal”). Specifically, the PQQ disodium crystal of the present invention is a PQQ disodium crystal having a water content of 7% or less.
- the moisture content in the crystal is more preferably 4.6% or less, and further preferably 3% or less, from the viewpoint of crystal stability.
- the PQQ disodium crystal of the present invention is, for example, the PQQ disodium crystal of crystal form 1 or crystal form 2 described herein, while the PQQ disodium crystal of the present invention is crystal form 1, crystal form 2 And a mixture of one or more crystals selected from crystals of other crystal forms, and the PQQ disodium crystal of crystal form 1 or crystal form 2 of the present invention is, for example, PQQ disodium of the present invention
- a crystal serving as a crystal precursor hereinafter, sometimes simply referred to as “precursor crystal”.
- the PQQ disodium crystal of the present invention is preferably 11.4 ⁇ 0.4, 13.5 ⁇ 0.4, 18.0 ⁇ 0.4, 18.18 by powder X-ray diffraction using Cu K ⁇ radiation.
- This is a PQQ disodium crystal (crystal form 1) showing 2 ⁇ angle peaks at 7 ⁇ 0.4, 26.0 ⁇ 0.4, 28.5 ⁇ 0.4 °.
- This peak can be observed with a general powder X-ray diffractometer equipped with a monochromator. Since the measurement data includes measurement errors, the crystal form defined in the present invention is a crystal form having a reasonable identity with respect to the peak angle.
- the PQQ disodium crystal of the present invention is preferably 8.7 ⁇ 0.4, 11.5 ⁇ 0.4, 12.0 ⁇ 0.4, 17.17 by powder X-ray diffraction using Cu K ⁇ radiation. It is a PQQ disodium crystal (crystal form 2) showing a peak of 2 ⁇ angle at 4 ⁇ 0.4, 18.7 ⁇ 0.4 °.
- the crystal form of crystal form 1 can be observed as a low moisture content crystal at ordinary temperature (normal temperature), and crystal form 2 cannot be observed in a normal humidity environment and is 180 ° C. or higher. It is necessary to measure in a high temperature environment or a dry atmosphere.
- the PQQ disodium crystal of the present invention is produced by a method comprising a step of obtaining a precursor crystal of the PQQ disodium crystal of the present invention and drying the precursor crystal.
- the precursor crystal of PQQ disodium crystal refers to a PQQ disodium crystal having a water content exceeding 4.6%, preferably exceeding 7%.
- the water content of the precursor crystal of PQQ disodium crystal is preferably 14% or less.
- PQQ sodium salt that is, PQQ monosodium salt, PQQ disodium salt and PQQ trisodium salt
- PQQ sodium salt can be used, and preferably PQQ trisodium is used.
- the PQQ sodium salt may contain sodium chloride or the like as impurities that may be included in the production process. In this case, the impurities can be appropriately removed during the production process of the present invention to increase the purity of the product. is there.
- a precursor crystal of PQQ disodium crystal is obtained under the condition that the solid of PQQ sodium salt is not completely dissolved in an aqueous solution having an ethanol or isopropanol concentration of 10 to 90% by weight (that is, a condition in which the PQQ sodium salt is saturated).
- Crystallization can be achieved by adding a sodium salt (eg, a solid of PQQ sodium salt) and then adding an acid to adjust the pH of the aqueous solution to a range of 2-5.
- the concentration of ethanol or isopropanol in the aqueous solution is preferably 35 to 65% by weight.
- the range in which PQQ sodium salt does not completely dissolve that is, the condition in which PQQ is saturated
- the range in which PQQ sodium salt does not completely dissolve varies depending on the alcohol concentration used.
- PQQ trisodium is used as the PQQ sodium salt.
- the concentration is 0.5 to 800 g / L, more preferably 0.5 to 100 g / L, and further preferably 5 to 60 g / L.
- the PQQ concentration can be used.
- the aqueous solution is a 50% ethanol aqueous solution
- the usable PQQ trisodium concentration is 1 to 200 g / L.
- the pH of the aqueous solution can be adjusted to a range of 2.5 to 3.5 more preferably.
- the precursor crystal can be obtained by reacting for 0.1 to 96 hours after setting the pH of the aqueous solution to a predetermined value. More preferably, the reaction time can be 6 to 72 hours in order to obtain large crystals.
- the reaction temperature can be 0 to 90 ° C, more preferably 10 to 60 ° C.
- the crystallization conditions can be freely selected in consideration of the presence or absence of stirring and the strength of the strength of the resulting crystal.
- the obtained precursor crystal can be obtained by filtration, centrifugation, and decantation. Furthermore, it is possible to provide this as a precursor crystal by washing with alcohol or the like, but it is also possible to provide the precipitated crystal as a precursor crystal as it is without performing such a separation operation. .
- the precursor crystals thus obtained are preferably 9.1 ⁇ 0.4, 10.3 ⁇ 0.4, 13.8 ⁇ 0.4 by powder X-ray diffraction using Cu K ⁇ radiation, PQQ disodium showing 2 ⁇ angle peaks at 17.7 ⁇ 0.4, 18.3 ⁇ 0.4, 24.0 ⁇ 0.4, 27.4 ⁇ 0.4, 39.5 ⁇ 0.4 ° It is a crystal, and preferably contains 12.7% of water as crystal water.
- the precursor crystal is dried.
- the PQQ disodium crystal of the present invention can be obtained.
- the precursor crystals can be dried by freeze drying, atmospheric pressure drying, or vacuum drying.
- the drying conditions such as drying time and drying temperature
- the drying temperature varies depending on the method, but can be, for example, ⁇ 80 to 250 ° C., preferably ⁇ 60 to 250 ° C.
- the lower limit of the drying temperature is a starting temperature at the time of freeze-drying
- the upper limit is a temperature at which decomposition of PQQ disodium does not occur.
- the drying temperature can be ⁇ 80 to 0 ° C., preferably ⁇ 60 to 0 ° C. for freeze drying, 40 to 250 ° C. for atmospheric drying, and 0 for vacuum drying. It can be up to 250 ° C. More specifically, in order to obtain PQQ disodium crystals of crystal form 1, drying under reduced pressure is not particularly limited. For example, it can be dried at 50 ° C. for 20 hours or more, preferably 70 hours or more, In the normal pressure drying, heating can be performed at 120 ° C. or higher and lower than 180 ° C.
- crystal form 2 of PQQ disodium can also be obtained by first preparing crystal form 1 and then phase-changing the crystal.
- the drying step can be terminated using the water content in the crystal as an index.
- the drying can be terminated by using as an index that the moisture content of the crystals is 7% by weight or less, preferably 4.6% by weight or less, more preferably 3% by weight or less.
- drying is preferably performed in a nitrogen gas atmosphere rather than in normal air. Below, it is easy to raise drying temperature and to shorten drying time.
- the precursor crystals can be dried by a method in which the precursor is heated in oil (for example, fried in oil).
- the heating temperature is preferably 120 to 200 ° C. under normal pressure from the viewpoint of proceeding the conversion to crystal form 1 or crystal form 2.
- the oil temperature is not particularly limited in order to obtain crystal form 1 PQQ disodium crystal, but can be, for example, 120 ° C. or higher and less than 180 ° C., and to obtain crystal form 2 PQQ disodium crystal. , A temperature higher than that, that is, 180 to 200 ° C.
- the heating temperature may exceed 200 ° C.
- the heating time is preferably 30 minutes or longer.
- the oil is not particularly limited as long as it is edible, and edible oils such as soybean oil, medium chain fatty acid oil, corn oil, fish oil, olive oil, rapeseed oil, rice oil, fish oil and coconut oil can be used.
- the mixture of oil and PQQ disodium crystals obtained in this step can be used as a slurry of PQQ disodium crystals with a low water content, for example, for soft capsules, or the oil can be removed with hexane or the like. May be used.
- PQQ has been known for its high reactivity and was not known for its thermal stability in edible oil, but according to the present invention, PQQ disodium crystals have high temperature stability. Moreover, surprisingly, it is converted into a new crystal form 1 or crystal form 2 by heating.
- the method of heating precursor crystals in oil is advantageous in that PQQ disodium crystals of crystal form 1 or crystal form 2 can be obtained in a short time.
- the crystal form 1 and crystal form 2 PQQ disodium crystals of the present invention have high dispersibility in edible oil and high permeability to the skin.
- the PQQ disodium crystals of crystal form 1 and crystal form 2 of the present invention are low moisture content crystals and can have high storage stability.
- the PQQ disodium crystal obtained in the present invention has an advantage of high purity because it is a crystal. Since the PQQ disodium crystal of crystal form 2 is a crystal that is relatively easy to absorb moisture in a wet atmosphere, it is preferably stored in a dry atmosphere.
- the PQQ disodium crystal of the present invention can be suitably used as foods, functional foods, nutrients, cosmetics, pharmaceuticals or quasi drugs for humans or animals.
- Functional food here means foods taken for the purpose of maintaining nutrition or supplementing nutrition, such as health foods, nutritional supplements, functional nutritional foods, nutritional health foods, and foods for specified health use.
- Specific forms of foods, functional foods, nutrients, cosmetics, pharmaceuticals or quasi drugs include capsules (eg, gelatin capsules, soft capsules), tablets, chewable tablets, drinks, etc. It is not limited to these. Since the PQQ disodium crystals of crystal form 1 and crystal form 2 of the present invention have a low water content, they are advantageous for tablet molding and storage.
- the crystal form 1 and crystal form 2 PQQ disodium crystals of the present invention are also excellent in dispersibility in fats and oils and permeability to the skin as described above. Accordingly, the present invention provides a pharmaceutical composition, a cosmetic composition, a functional food, and a nutrient containing the PQQ disodium crystal of the present invention.
- the pharmaceutical composition of the present invention can be made into a pharmaceutical composition for transdermal administration.
- the crystal of the present invention is excellent in dispersibility in fats and oils, it is suitable for prescription into an oil dispersion preparation.
- the pharmaceutical composition or cosmetic composition of the present invention is preferably in the form of a dispersion preparation such as an emulsion or suspension, a semi-solid preparation such as an ointment or cream, or a molded preparation such as a soft capsule. Can be provided.
- additives include sweeteners, coloring agents, preservatives, thickening stabilizers, antioxidants, color formers, bleaching agents, antibacterial agents. Antifungal agents, gum bases, bittering agents, enzymes, brighteners, acidulants, seasonings, emulsifiers, strengthening agents, manufacturing agents, fragrances, spice extracts and the like can be used.
- the PQQ disodium crystal of the present invention is generally used for ordinary foods such as miso, soy sauce, instant miso soup, ramen, fried noodles, curry, corn soup, merboard tofu, marvo eggplant, pasta sauce, pudding, cake It can be added to bread and the like.
- the pharmaceutical composition of the present invention may contain the PQQ disodium crystal of the present invention and at least one pharmaceutical additive.
- the cosmetic composition of the present invention may contain the PQQ disodium crystal of the present invention and at least one cosmetic additive.
- a pharmaceutical additive or cosmetic additive can be appropriately selected by those skilled in the art according to the formulation of the pharmaceutical composition or cosmetic composition.
- Powder X-ray diffraction uses M18XCE apparatus made by Mac Science Co., Ltd. or RINT2500 made by Rigaku Co., Ltd.
- X-ray Cu / tube voltage 40 kV / tube current 100 mA
- the water content (% by weight) of the crystal was measured by the Karl Fischer method.
- Reference Example 1 A raw material PQQ trisodium was obtained by a culture method in accordance with the description in Precursor Crystal Patent No. 2751183. 60 g of a hydrated PQQ trisodium salt containing 20 g of PQQ was added to 1 L of an aqueous solution having an ethanol concentration of 50% by weight and saturated. At this time, the solid was not completely melted. Hydrochloric acid was added thereto at room temperature to adjust the pH to 3.5. Hydrochloric acid was added slowly dropwise over about 2 hours. After addition of hydrochloric acid, the mixture was stirred for 2 days, and then the obtained aqueous ethanol solution was filtered to obtain hydrous PQQ disodium crystals in a yield of 99 mol%.
- the obtained crystals were 9.1 ⁇ 0.4, 10.3 ⁇ 0.4, 13.8 ⁇ 0.4, 17.7 ⁇ 0.4, 18.3. Peaks of 2 ⁇ angles were shown at ⁇ 0.4, 24.0 ⁇ 0.4, 27.4 ⁇ 0.4, and 39.5 ⁇ 0.4 °.
- the obtained crystals had a moisture content of 12.7% and a purity of 87.3%. there were.
- Example 1 Crystalline form 1
- the precursor crystal obtained in Reference Example 1 is placed in a vacuum dryer (product name: FS-420, manufactured by Advantech Co., Ltd.) with a diaphragm type vacuum pump (DRYFAST ULTRA, product number: 2032 manufactured by A Gardner Denver Product). The pressure was reduced to the apparatus limit (2 torr) and then dried at 50 ° C. for 77 hours.
- the obtained crystals were 11.4 ⁇ 0.4, 13.5 ⁇ 0.4, 18.0 ⁇ 0.4, 18.7 ⁇ 0.4, 26.0. It was found to be a PQQ disodium crystal (crystal form 1) showing peaks of 2 ⁇ angles at ⁇ 0.4 and 28.5 ⁇ 0.4 ° (FIG. 1).
- the PQQ disodium crystal obtained in this example was a crystal having a new crystal form (crystal form 1) different from the crystal form of the precursor crystal having a high water content.
- the water content of the obtained crystal was 0.7%, and the water content of the crystal was significantly reduced compared to the precursor crystal.
- the purity of the obtained crystal was 99.3%.
- Example 2 Crystalline form 1
- the precursor crystal obtained in Reference Example 1 was placed in a vacuum dryer (manufactured by Advantech, product number: FS-420) and dried at 120 ° C. for 1 day under atmospheric pressure.
- a vacuum dryer manufactured by Advantech, product number: FS-420
- the obtained crystal was the same as the crystal form 1 obtained in Example 1.
- the water content of the obtained crystal was 2.2% and the purity was 97.8%.
- Example 3 Crystalline form 1 (in oil, 0.5 hours at 150 ° C.) 9.6 g of the precursor crystal obtained in Reference Example 1 was added to 65 g of Nisshin Oilio cooking oil ODO and heated at 150 ° C. for 30 minutes (fried). As a result of powder X-ray diffraction, it was found that the obtained crystal was the same as the crystal form obtained in Example 1. Although the heating time was short as compared with Examples 1 and 2, Crystal Form 1 was obtained in a short time by heating the precursor crystals in oil.
- the amount of impurities contained in the crystal did not increase and the obtained crystal had a purity of 99% or more.
- the water content of the obtained crystals was 1.9%.
- the heating time was shorter than that of Examples 1 and 2, the water content of the obtained crystals was greatly reduced compared to the precursor crystals, which was equivalent to the water content of the crystal form of Example 1 or 2. It was.
- Example 4 Crystal mixture having a water content of 7%
- the precursor crystal obtained in Reference Example 1 was placed in a vacuum dryer, and the apparatus was set using a diaphragm-type vacuum pump (DRYFAST ULTRA manufactured by A Gardner Denver Product, product number: 2032). The pressure was reduced to the limit (2 torr) and then dried at 50 ° C. for 22 hours.
- DFRAST ULTRA diaphragm-type vacuum pump
- the obtained crystals were 11.4 ⁇ 0.4, 13.5 ⁇ 0.4, 18.0 ⁇ 0.4, 18.7 ⁇ 0.4, 26.0. It was found to be a mixture of crystal form 1 PQQ disodium crystal showing 2 ⁇ angle peaks at ⁇ 0.4, 28.5 ⁇ 0.4 ° and the precursor crystal of Reference Example 1. As a result of measuring the water content, the water content of the obtained crystal was 7% and the purity was 93%. The resulting crystal mixture could then be sequentially converted to crystalline Form 1 crystals by reducing the water content (see, eg, Example 1 dried for 77 hours under the same conditions).
- Example 5 Relationship between heating temperature and obtained crystal form
- the relationship between the heating temperature in the drying step and the obtained crystal form was examined.
- the precursor crystal obtained in Reference Example 1 was heated at a temperature change rate of 20 ° C./min, and measurement was started 15 minutes after reaching each set temperature.
- thermogravimetric analysis of the crystal of crystal form 2 revealed that the water content of the crystal of crystal form 2 was approximately 0%. However, this crystal was unstable, and when it was allowed to stand at 30 ° C. for a while (for example, half a day or more), it absorbed moisture and became a crystal having the same crystal form as the precursor crystal (FIG. 2).
- Comparative Example 1 PQQ disodium crystal different from Reference Example 1
- the precursor crystal of Reference Example 1 was dissolved in water, and ethanol was added to precipitate red PQQ disodium crystal.
- the water content of the obtained crystals was 14.5%.
- the obtained crystals were dried under reduced pressure for 96 hours in the same manner as in Example 1, and powder X-ray analysis was performed.
- the crystal obtained by this method had a structure with low crystallinity showing a peak of 2 ⁇ angle at 9.4, 22.4 ° (FIG. 3).
- the water content of the obtained crystals was 9.6%, and the purity was 90.4%.
- Example 6 Dispersibility test of crystal form 1 and crystal form 2 PQQ disodium crystals
- the dispersibility of crystal form 1 and crystal form 2 PQQ disodium crystals and conventional crystals in edible oils and fats was examined. Compared.
- crystal form 1 and crystal form 2 PQQ disodium crystals were prepared as follows.
- the crystal of crystal form 1 is obtained by adding 3 g of the PQQ disodium salt precursor of Reference Example 1 to a glass container, and using a diaphragm vacuum pump (AFGardner Denver Product DRYFAST ULTRA, product number: 2032) to limit the equipment ( The pressure was reduced to 2 torr) and then dried under reduced pressure at 150 ° C. for 4 hours to obtain a dark brown solid. After drying, the inside of the container was returned to atmospheric pressure, and the glass container lid was immediately closed.
- a diaphragm vacuum pump AFGardner Denver Product DRYFAST ULTRA, product number: 2032
- the crystal of crystal form 2 was prepared by adding 3 g of the PQQ disodium salt precursor crystal of Reference Example 1 to a 200 mL eggplant flask and heating at 200 ° C. for 4 hours while refluxing nitrogen gas at a flow rate of 100 mL / min. Obtained as a brown solid. After drying, the temperature in the flask was lowered to room temperature while refluxing nitrogen. In this example, the PQQ disodium crystal (precursor crystal) of Reference Example 1 was used as a control.
- the precursor crystals all precipitated to the bottom of the vessel in 0.5 minutes, while crystals of Form 1 and Form 2 were 30 minutes and 45 minutes, respectively, before settling. It took a minute. From this, it was suggested that the crystals of crystal form 1 and crystal form 2 have improved dispersibility in edible fats and oils than the precursor crystals. Table 1 suggests that the dispersibility of the crystals of crystal form 1 and crystal form 2 in the edible fat is improved by 50 times or more as compared with the precursor crystal.
- PQQ disodium crystals of the present invention are more dispersible than conventional crystals, and are considered suitable for, for example, the production of soft capsule formulations.
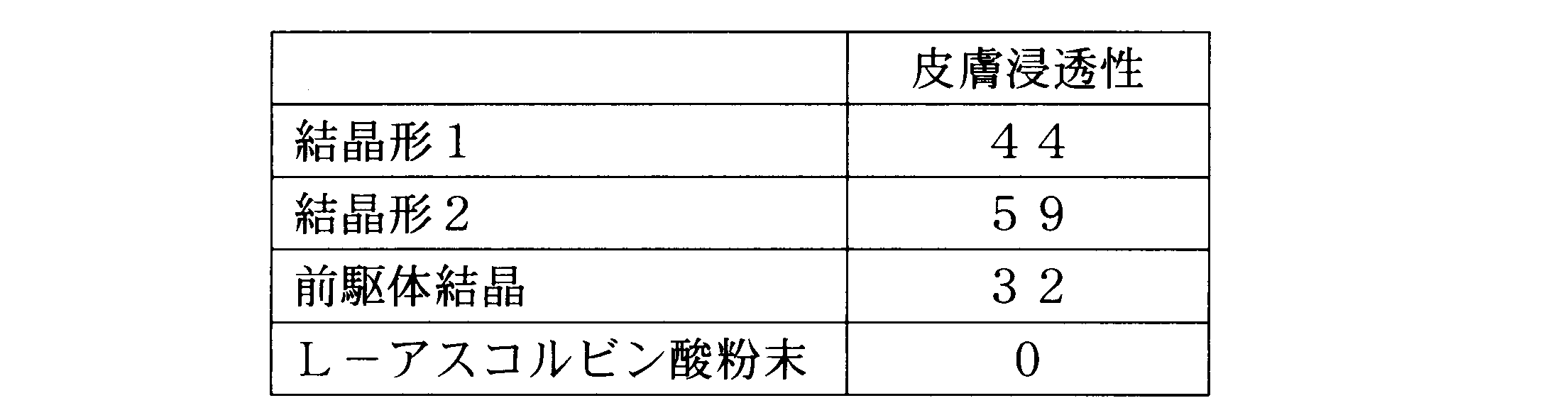
- Example 7 Penetration test of crystal form 1 and crystal form 2 PQQ disodium crystals into skin
- the crystal form 1 and crystal form 2 PQQ disodium crystals prepared in Example 6 were used to The permeability was compared.
- Example 6 Each of the PQQ disodium crystals of crystal form 1 and crystal form 2 prepared in Example 6 and the precursor crystal of Reference Example 1 were mixed with L-ascorbic acid powder (made by Wako Co., Ltd., special grade) in a weight ratio of 1: 4. To prepare a test composition, which was used for the penetration test. As a control, L-ascorbic acid powder was used.
- the penetration test was performed using porcine skin.
- the pig's skin was washed with tap water to completely wipe off the water.
- Each 5 mg of the test composition was brought into contact with the pig skin, and then fixed to the skin with tape. After 80 minutes, the tape was peeled off and the skin was washed with tap water to remove the test composition attached to the skin surface.
- the high permeability of PQQ crystals to the skin was evaluated by the degree of skin coloring.
- a photograph of the skin after treatment is taken into a computer, and changes in skin brightness are measured using image processing software (product name: Paint (software attached to Windows XP), manufactured by Microsoft Corporation). Evaluation was made by measuring. At this time, the change in skin brightness is expressed by the following formula: Calculated by
- the crystals of crystal form 1 and crystal form 2 showed higher permeability to the skin than the precursor crystals. This suggests that the PQQ disodium crystals of crystal form 1 and crystal form 2 are more suitable for use in cosmetics and pharmaceuticals for transdermal administration.
- Formulation Example 1 Gelatin capsule preparation Coenzyme Q10 (manufactured by Mitsubishi Gas Chemical Co., Inc.) 8 g, soybean protein hydrolyzate (Nisshin Oilio Co., Ltd.) 2 g, and PQQ disodium crystal (crystal form 1) 2 g of Example 1 in a plastic bag And shaken well to prepare a composition for oral consumption. The obtained composition was sealed in a gelatin capsule in an amount of 10 mg to obtain a gelatin capsule preparation.
- Formulation example 2 Soft capsule formulation of oil dispersion 2700 g of safflower salad oil (Nisshin Oillio Co., Ltd.), 300 g of glycerin fatty acid ester (manufactured by Riken Vitamin Co., Ltd.) and 300 g of beeswax (Yokohama Yushi Kogyo Co., Ltd.) are stirred at 70 ° C. After that, the PQQ disodium crystals of Example 1 (Crystal Form 1) were added and mixed with a mixer. The obtained mixture was enclosed in a soft capsule to obtain a soft capsule preparation.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Genetics & Genomics (AREA)
- Ophthalmology & Optometry (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Coloring Foods And Improving Nutritive Qualities (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Cosmetics (AREA)
Abstract
Description
〔1〕水分含量が7重量%以下であるピロロキノリンキノンジナトリウム結晶。
〔2〕水分含量が4.6重量%以下である、上記〔1〕に記載のピロロキノリンキノンジナトリウム結晶。
〔3〕Cu Kα放射線を用いた粉末X線回折で11.4±0.4,13.5±0.4,18.0±0.4,18.7±0.4,26.0±0.4,28.5±0.4°に2θ角度のピークを示す、上記〔1〕または〔2〕に記載のピロロキノリンキノンジナトリウム結晶(結晶形1)。
〔4〕Cu Kα放射線を用いた粉末X線回折で8.7±0.4,11.5±0.4,12.0±0.4,17.4±0.4,18.7±0.4°に2θ角度のピークを示す、上記〔1〕または〔2〕に記載のピロロキノリンキノンジナトリウム結晶(結晶形2)。
〔5〕上記〔1〕~〔4〕のいずれかに記載のピロロキノリンキノンジナトリウム結晶を含んでなる、医薬組成物。
〔6〕上記〔1〕~〔4〕のいずれかに記載のピロロキノリンキノンジナトリウム結晶を含んでなる、化粧組成物。
〔7〕上記〔1〕~〔4〕のいずれかに記載のピロロキノリンキノンジナトリウム結晶を含んでなる、機能性食品または栄養剤。
〔8〕上記〔1〕~〔4〕のいずれかに記載のピロロキノリンキノンジナトリウム結晶を製造する方法であって、
前記ピロロキノリンキノンジナトリウム結晶の前駆体となる結晶(前駆体結晶)を得、次いで、前駆体結晶を乾燥させる工程を含んでなる、ピロロキノリンキノンジナトリウム結晶の製造方法。
〔9〕前記前駆体結晶が、ピロロキノリンキノンのナトリウム塩を溶解したエタノール又はイソプロパノール濃度10~90%水溶液のpHを2~5の範囲に調整することにより結晶化される、上記〔8〕に記載のピロロキノリンキノンジナトリウム結晶の製造方法。
〔10〕前記前駆体結晶が、Cu Kα放射線を用いた粉末X線回折で9.1±0.4,10.3±0.4,13.8±0.4,17.7±0.4,18.3±0.4,24.0±0.4,27.4±0.4,39.5±0.4°に2θ角度のピークを示すピロロキノリンキノンジナトリウム結晶である、上記〔8〕または〔9〕に記載のピロロキノリンキノンジナトリウム結晶の製造方法。
〔11〕乾燥が、結晶を油中で加熱することにより行われる、上記〔8〕~〔10〕のいずれかに記載のピロロキノリンキノンジナトリウム結晶の製造方法。
X線:Cu/管電圧40kV/管電流100mA
スキャンスピード:4.000°/min
サンプリング幅:0.020°
で行った。
特許第2751183号公報の記載に従って、原料のPQQトリナトリウムを培養法で得た。PQQ20gを含む含水PQQトリナトリウム塩の固体60gをエタノール濃度50重量%の水溶液1Lに加え、飽和させた。このとき、固体は溶け切っていなかった。ここに室温下で塩酸を加え、pHを3.5に調整した。塩酸の添加は、約2時間かけてゆっくり滴下して行った。塩酸添加後は、2日間攪拌し、その後、得られたエタノール水溶液を濾過して含水PQQジナトリウム結晶を収率99mol%で得た。粉末X線回折を行ったところ、得られた結晶は、9.1±0.4,10.3±0.4,13.8±0.4,17.7±0.4,18.3±0.4,24.0±0.4,27.4±0.4,39.5±0.4°に2θ角度のピークを示した。温度40℃、相対湿度(RH)75%の環境に得られた結晶を置き、水分含量を測定した結果、得られた結晶の水分含量は12.7%であり、純度は87.3%であった。
参考例1で得られた前駆体結晶を減圧乾燥器(アドバンテック社製、製品名:FS-420)に入れて、ダイヤフラム型真空ポンプ(A Gardner Denver Product社製DRYFAST ULTRA、製品番号:2032)で装置限界(2トル)まで減圧してから、50℃で77時間乾燥させた。
参考例1で得られた前駆体結晶を減圧乾燥器(アドバンテック社製、製品番号:FS-420)に入れ、大気圧下120℃で1日乾燥させた。粉末X線回折を行ったところ、得られた結晶は、実施例1で得られた結晶形1と同一であることが分かった。水分含量を測定した結果、得られた結晶の水分含量は2.2%であり、純度は97.8%であった。
参考例1で得られた前駆体結晶9.6gを日清オイリオ製食用油ODO65gに加えて、150℃で30分間加熱した(揚げた)。粉末X線回折を行ったところ、得られた結晶は、実施例1で得られた結晶形と同一であることが分かった。加熱時間は実施例1や2と比較して短かったが、前駆体結晶を油中で加熱することにより、短時間で結晶形1が得られた。
参考例1で得られた前駆体結晶を減圧乾燥器に入れて、ダイヤフラム型真空ポンプ(A Gardner Denver Product社製DRYFAST ULTRA、製品番号:2032)で装置限界(2トル)まで減圧してから、50℃で22時間乾燥させた。
本実施例では、乾燥工程における加熱温度と得られる結晶形との関係を調べた。
30℃では前駆体結晶と同様の結晶、
120℃では実施例4の水分含量7%の結晶混合物と同様の前駆体結晶と結晶形1が混合した結晶混合物、
150℃では結晶形1の結晶、
180℃では、8.7±0.4,11.5±0.4,12.0±0.4,17.4±0.4,18.7±0.4°に2θ角度のピークを示す新たな結晶形のPQQジナトリウム結晶(結晶形2)となっていた。それぞれの温度で処理した結晶の粉末X線回折の結果を図2に示す。結晶形2の結晶の熱重量分析を行ったところ、結晶形2の結晶の水分含量はほぼ0%であることが明らかとなった。しかし、この結晶は不安定であり、30℃にしばらく(例えば、半日以上)静置すると吸湿して前駆体結晶と同様の結晶形の結晶になっていた(図2)。
参考例1の前駆体結晶を水に溶解し、エタノールを加えることで赤色のPQQジナトリウム結晶を析出させた。水分含量を測定した結果、得られた結晶の水分含量は14.5%であった。その後さらに、得られた結晶を実施例1と同様に減圧下で96時間乾燥させ、粉末X線解析を行った。しかしながら、この方法により得られた結晶は、9.4,22.4°に2θ角度のピークを示す結晶性の低い構造を有することが分かった(図3)。また、水分含量を測定した結果、得られた結晶の水分含量は9.6%であり、純度は90.4%であった。
本実施例では、結晶形1および結晶形2のPQQジナトリウム結晶と従来の結晶の食用油脂中での分散性を比較した。

本実施例では、実施例6で調製した結晶形1および結晶形2のPQQジナトリウム結晶を用いて、皮膚に対する浸透性を比較した。

コエンザイムQ10(三菱瓦斯化学社製)8g、大豆タンパク加水分解物(日清オイリオ社製)2g、および実施例1のPQQジナトリウム結晶(結晶形1)2gをポリ袋に入れ、良く振って混合して経口摂取用組成物を調製した。得られた組成物をゼラチンカプセルに10mgずつ封入し、ゼラチンカプセル製剤を得た。
サフラワーサラダ油(日清オイリオ社製)2700g、グリセリン脂肪酸エステル(理研ビタミン社製)300g、および蜜蝋(横浜油脂工業社製)300gを合わせて70℃で攪拌した後、実施例1のPQQジナトリウム結晶(結晶形1)を加えてミキサーで混合した。得られた混合物をソフトカプセルに封入して、ソフトカプセル製剤を得た。
Claims (11)
- 水分含量が7重量%以下であるピロロキノリンキノンジナトリウム結晶。
- 水分含量が4.6重量%以下である、請求項1に記載のピロロキノリンキノンジナトリウム結晶。
- Cu Kα放射線を用いた粉末X線回折で11.4±0.4,13.5±0.4,18.0±0.4,18.7±0.4,26.0±0.4,28.5±0.4°に2θ角度のピークを示す、請求項1または2に記載のピロロキノリンキノンジナトリウム結晶(結晶形1)。
- Cu Kα放射線を用いた粉末X線回折で8.7±0.4,11.5±0.4,12.0±0.4,17.4±0.4,18.7±0.4°に2θ角度のピークを示す、請求項1または2に記載のピロロキノリンキノンジナトリウム結晶(結晶形2)。
- 請求項1~4のいずれか一項に記載のピロロキノリンキノンジナトリウム結晶を含んでなる、医薬組成物。
- 請求項1~4のいずれか一項に記載のピロロキノリンキノンジナトリウム結晶を含んでなる、化粧組成物。
- 請求項1~4のいずれか一項に記載のピロロキノリンキノンジナトリウム結晶を含んでなる、機能性食品または栄養剤。
- 請求項1~4のいずれかに記載のピロロキノリンキノンジナトリウム結晶を製造する方法であって、
前記ピロロキノリンキノンジナトリウム結晶の前駆体となる結晶(前駆体結晶)を得、次いで、前駆体結晶を乾燥させる工程を含んでなる、ピロロキノリンキノンジナトリウム結晶の製造方法。 - 前記前駆体結晶が、ピロロキノリンキノンのナトリウム塩を溶解したエタノール又はイソプロパノール濃度10~90%水溶液のpHを2~5の範囲に調整することにより結晶化される、請求項8に記載のピロロキノリンキノンジナトリウム結晶の製造方法。
- 前記前駆体結晶が、Cu Kα放射線を用いた粉末X線回折で9.1±0.4,10.3±0.4,13.8±0.4,17.7±0.4,18.3±0.4,24.0±0.4,27.4±0.4,39.5±0.4°に2θ角度のピークを示すピロロキノリンキノンジナトリウム結晶である、請求項8または9に記載のピロロキノリンキノンジナトリウム結晶の製造方法。
- 乾燥が、結晶を油中で加熱することにより行われる、請求項8~10のいずれか一項に記載のピロロキノリンキノンジナトリウム結晶の製造方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NO12800143A NO2722331T3 (ja) | 2011-06-16 | 2012-06-15 | |
| EP12800143.5A EP2722331B1 (en) | 2011-06-16 | 2012-06-15 | Crystal of pyrroloquinolinequinone disodium salt, and method for producing same |
| PL12800143T PL2722331T3 (pl) | 2011-06-16 | 2012-06-15 | Kryształ soli disodowej pirolochinolinochinonu oraz sposób jego wytwarzania |
| ES12800143.5T ES2651238T3 (es) | 2011-06-16 | 2012-06-15 | Cristal de sal disódica de pirroloquinolina quinona y procedimiento para su fabricación |
| US14/125,788 US9174983B2 (en) | 2011-06-16 | 2012-06-15 | Pyrroloquinoline quinone disodium salt crystal and method for producing the same |
| CN201280028888.9A CN103619842B (zh) | 2011-06-16 | 2012-06-15 | 吡咯并喹啉醌二钠盐的结晶及其制造方法 |
| DK12800143.5T DK2722331T3 (da) | 2011-06-16 | 2012-06-15 | Pyrroloquinolinquinondinatriumsaltkrystal og fremgangsmåde til fremstilling af samme |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011134279 | 2011-06-16 | ||
| JP2011-134279 | 2011-06-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012173217A1 true WO2012173217A1 (ja) | 2012-12-20 |
Family
ID=47357196
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/065303 Ceased WO2012173217A1 (ja) | 2011-06-16 | 2012-06-15 | ピロロキノリンキノンジナトリウム塩の結晶およびその製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9174983B2 (ja) |
| EP (1) | EP2722331B1 (ja) |
| JP (1) | JPWO2012173217A1 (ja) |
| CN (1) | CN103619842B (ja) |
| DK (1) | DK2722331T3 (ja) |
| ES (1) | ES2651238T3 (ja) |
| NO (1) | NO2722331T3 (ja) |
| PL (1) | PL2722331T3 (ja) |
| WO (1) | WO2012173217A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014071772A1 (zh) * | 2012-11-09 | 2014-05-15 | 诸城市浩天药业有限公司 | 吡咯并喹啉醌的二钠盐结晶 |
| JP2017100979A (ja) * | 2015-11-30 | 2017-06-08 | 株式会社協和 | 胎盤抽出物の製造方法 |
| JPWO2018003531A1 (ja) * | 2016-06-29 | 2019-04-18 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンモノナトリウム及びその製造方法、並びにそれを含む組成物 |
| KR102120670B1 (ko) * | 2019-01-03 | 2020-06-10 | 주식회사 누베파마 | 돌연변이 균주를 이용한 항산화 물질 생산 방법 |
| CN112125899A (zh) * | 2019-06-24 | 2020-12-25 | 浙江医药股份有限公司 | 吡咯并喹啉醌二钠盐结晶、其制备方法及包含其的组合物 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104744462B (zh) | 2009-07-16 | 2016-09-21 | 三菱瓦斯化学株式会社 | 吡咯并喹啉醌的钠盐结晶、其制法、功能性食品和医药 |
| MA39715A (fr) * | 2014-04-16 | 2015-10-22 | Anthem Biosciences Private Ltd | Formes polymorphes de l'acide 4,5-dihydro-1h-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylique et de son sel de disodium, procédé de préparation desdites formes polymorphes et leur utilisation |
| US10131661B2 (en) * | 2015-01-19 | 2018-11-20 | Mitsubishi Gas Chemical Company, Inc. | High-solubility acetone-added pyrroloquinoline quinone salt |
| US10364244B2 (en) | 2015-09-25 | 2019-07-30 | Zhejiang Hisun Pharmaceutical Co., Ltd. | Crystal form of pyrroloquinoline quinone sodium salt and preparation method and use thereof |
| CN115716825A (zh) * | 2021-08-24 | 2023-02-28 | 浙江医药股份有限公司新昌制药厂 | 吡咯并喹啉醌二钠盐结晶、其制备方法及包含其的组合物 |
| CN116850085A (zh) * | 2023-08-18 | 2023-10-10 | 水羊化妆品制造有限公司 | 吡咯并喹啉醌二钠盐在修护皮肤屏障中的应用 |
| CN117327069B (zh) * | 2023-09-27 | 2024-04-30 | 山东原力泰医药科技有限公司 | 吡咯并喹啉醌二钠盐晶型及其制备方法和应用 |
| CN117777147A (zh) * | 2024-02-23 | 2024-03-29 | 内蒙古金达威药业有限公司 | 一种吡咯并喹啉醌二钠盐的晶型及其制备方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62246575A (ja) * | 1986-04-17 | 1987-10-27 | Mitsubishi Gas Chem Co Inc | ピロロキノリンキノンの精製方法 |
| JPH0267284A (ja) * | 1988-09-01 | 1990-03-07 | Mitsubishi Gas Chem Co Inc | ピロロキノリンキノン類の回収・精製方法 |
| JPH07113024A (ja) | 1993-08-27 | 1995-05-02 | Dainippon Ink & Chem Inc | 共重合体樹脂エマルジョン、その製造方法及び防湿加工用組成物 |
| JP2751183B2 (ja) | 1988-02-26 | 1998-05-18 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンの製造方法 |
| JP2005530786A (ja) | 2002-05-15 | 2005-10-13 | シーエルエフ メディカル テクノロジー アクセラレーション プログラム インコーポレイテッド | 心損傷の処置のためのピロロキノリンキノンおよびその使用方法 |
| JP2007269769A (ja) | 2006-03-10 | 2007-10-18 | Ultizyme International Ltd | 神経変性疾患関連蛋白質凝集線維化抑制剤 |
| WO2011007633A1 (ja) | 2009-07-16 | 2011-01-20 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンのナトリウム塩結晶 |
| WO2011055796A1 (ja) * | 2009-11-06 | 2011-05-12 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンのフリー体 |
| JP2011246442A (ja) | 2010-04-28 | 2011-12-08 | Rohto Pharmaceutical Co Ltd | 光老化抑制剤および皮膚菲薄化抑制剤 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1248869A2 (en) * | 2000-01-07 | 2002-10-16 | Transform Pharmaceuticals, Inc. | High-throughput formation, identification, and analysis of diverse solid-forms |
| JP2011098911A (ja) | 2009-11-06 | 2011-05-19 | Mitsubishi Gas Chemical Co Inc | ピロロキノリンキノン粉体 |
-
2012
- 2012-06-15 PL PL12800143T patent/PL2722331T3/pl unknown
- 2012-06-15 EP EP12800143.5A patent/EP2722331B1/en active Active
- 2012-06-15 DK DK12800143.5T patent/DK2722331T3/da active
- 2012-06-15 CN CN201280028888.9A patent/CN103619842B/zh active Active
- 2012-06-15 US US14/125,788 patent/US9174983B2/en active Active
- 2012-06-15 ES ES12800143.5T patent/ES2651238T3/es active Active
- 2012-06-15 WO PCT/JP2012/065303 patent/WO2012173217A1/ja not_active Ceased
- 2012-06-15 JP JP2013520591A patent/JPWO2012173217A1/ja active Pending
- 2012-06-15 NO NO12800143A patent/NO2722331T3/no unknown
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62246575A (ja) * | 1986-04-17 | 1987-10-27 | Mitsubishi Gas Chem Co Inc | ピロロキノリンキノンの精製方法 |
| JP2751183B2 (ja) | 1988-02-26 | 1998-05-18 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンの製造方法 |
| JPH0267284A (ja) * | 1988-09-01 | 1990-03-07 | Mitsubishi Gas Chem Co Inc | ピロロキノリンキノン類の回収・精製方法 |
| JPH07113024A (ja) | 1993-08-27 | 1995-05-02 | Dainippon Ink & Chem Inc | 共重合体樹脂エマルジョン、その製造方法及び防湿加工用組成物 |
| JP2005530786A (ja) | 2002-05-15 | 2005-10-13 | シーエルエフ メディカル テクノロジー アクセラレーション プログラム インコーポレイテッド | 心損傷の処置のためのピロロキノリンキノンおよびその使用方法 |
| JP2007269769A (ja) | 2006-03-10 | 2007-10-18 | Ultizyme International Ltd | 神経変性疾患関連蛋白質凝集線維化抑制剤 |
| WO2011007633A1 (ja) | 2009-07-16 | 2011-01-20 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンのナトリウム塩結晶 |
| WO2011055796A1 (ja) * | 2009-11-06 | 2011-05-12 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンのフリー体 |
| JP2011246442A (ja) | 2010-04-28 | 2011-12-08 | Rohto Pharmaceutical Co Ltd | 光老化抑制剤および皮膚菲薄化抑制剤 |
Non-Patent Citations (2)
| Title |
|---|
| ISHIDA, T. ET AL.: "Molecular and Crystal Structure of PQQ (Methoxatin), a Novel Coenzyme of Quinoproteins: Extensive Stacking Character and Metal Ion Interaction", J. AM. CHEM. SOC., vol. 111, 1989, pages 6822 - 6828, XP008149475 * |
| ISHIDA, T. ET AL.: "Molecular and crystal structure of PQQ(methoxatin), a novel coenzyme of quinoproteins: extensive stacking character and metal ion interaction", JOURNAL OF AMERICAN CHEMICAL SOCIETY, vol. 111, 1989, pages 6822 - 6828, XP008149475, DOI: doi:10.1021/ja00199a050 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014071772A1 (zh) * | 2012-11-09 | 2014-05-15 | 诸城市浩天药业有限公司 | 吡咯并喹啉醌的二钠盐结晶 |
| JP2017100979A (ja) * | 2015-11-30 | 2017-06-08 | 株式会社協和 | 胎盤抽出物の製造方法 |
| JPWO2018003531A1 (ja) * | 2016-06-29 | 2019-04-18 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンモノナトリウム及びその製造方法、並びにそれを含む組成物 |
| US11021476B2 (en) | 2016-06-29 | 2021-06-01 | Mitsubishi Gas Chemical Company, Inc. | Pyrroloquinoline quinone monosodium and method for producing the same, and composition comprising the same |
| JP7335070B2 (ja) | 2016-06-29 | 2023-08-29 | 三菱瓦斯化学株式会社 | ピロロキノリンキノンモノナトリウム及びその製造方法、並びにそれを含む組成物 |
| KR102120670B1 (ko) * | 2019-01-03 | 2020-06-10 | 주식회사 누베파마 | 돌연변이 균주를 이용한 항산화 물질 생산 방법 |
| CN112125899A (zh) * | 2019-06-24 | 2020-12-25 | 浙江医药股份有限公司 | 吡咯并喹啉醌二钠盐结晶、其制备方法及包含其的组合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2722331B1 (en) | 2017-09-06 |
| DK2722331T3 (da) | 2017-11-20 |
| JPWO2012173217A1 (ja) | 2015-02-23 |
| CN103619842A (zh) | 2014-03-05 |
| PL2722331T3 (pl) | 2018-02-28 |
| EP2722331A1 (en) | 2014-04-23 |
| NO2722331T3 (ja) | 2018-02-03 |
| ES2651238T3 (es) | 2018-01-25 |
| EP2722331A4 (en) | 2014-11-05 |
| US20140128609A1 (en) | 2014-05-08 |
| CN103619842B (zh) | 2016-06-22 |
| US9174983B2 (en) | 2015-11-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012173217A1 (ja) | ピロロキノリンキノンジナトリウム塩の結晶およびその製造方法 | |
| CN103857676B (zh) | 吡咯喹啉醌醇加成物 | |
| JP4859006B2 (ja) | 安定なアスタキサンチン−シクロデキストリン包接化合物及びその製造方法、並びに該包接化合物を含有する液剤、飲食物、飼料、医薬品及び化粧品 | |
| JP6299984B2 (ja) | 黄色系還元型ピロロキノリンキノン結晶及びその製造方法、並びに、食品、医薬品、ゲル、組成物及び組成物の製造方法 | |
| TWI445536B (zh) | 大豆皂醇組成物 | |
| JP2023099033A (ja) | ピロロキノリンキノンモノナトリウム及びその製造方法、並びにそれを含む組成物 | |
| EP2995605B1 (en) | Method for preparing a reduced coenzyme q10 powder. | |
| JP2013112677A (ja) | ピロロキノリンキノンジナトリウム結晶 | |
| JP6902329B2 (ja) | 皮脂腺細胞の活性化の抑制剤 | |
| CN101835782B (zh) | 利用过氧化氢或过酸由鼠尾草酸生产鼠尾草酚的方法 | |
| JP2013053115A (ja) | α環状ジペプチドの製造方法 | |
| US11912875B2 (en) | Method for preparing a colour-stable preparation of a magnesium chlorophyllin alkali metal salt or alkali earth metal salt from natural sources of chlorophyll | |
| JP2015145350A (ja) | セロビオースリピッドの塩及びその用途 | |
| CN1933824A (zh) | 血中持续性辅酶q组合物 | |
| JP5891970B2 (ja) | 新規ケルセチン誘導体 | |
| JP2013245211A (ja) | 新規ケルセチン誘導体 | |
| JP2013192514A (ja) | 牡蠣エキスの製造方法 | |
| JP6188422B2 (ja) | 抗炎症剤、ヒアルロニダーゼ阻害剤、抗アレルギー剤 | |
| JP5564906B2 (ja) | 生体吸収性に優れたコエンザイムq10とピロロキノリンキノンを共に含む経口摂取用組成物 | |
| JP2024160658A (ja) | ニコチンアミドアデニンジヌクレオチド増加促進剤、及びその製造方法 | |
| JP2024160656A (ja) | 組成物、添加剤、及び組成物の製造方法 | |
| TW201408302A (zh) | 大豆皂醇組成物 | |
| JP2011195506A (ja) | 大豆タンパクを含むピロロキノリンキノンとシクロデキストリン包接コエンザイムq10を共に含む経口摂取用組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12800143 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013520591 Country of ref document: JP Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012800143 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14125788 Country of ref document: US |