WO2013024763A1 - 核酸アプタマーの作製方法 - Google Patents
核酸アプタマーの作製方法 Download PDFInfo
- Publication number
- WO2013024763A1 WO2013024763A1 PCT/JP2012/070188 JP2012070188W WO2013024763A1 WO 2013024763 A1 WO2013024763 A1 WO 2013024763A1 JP 2012070188 W JP2012070188 W JP 2012070188W WO 2013024763 A1 WO2013024763 A1 WO 2013024763A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nucleic acid
- stranded
- target substance
- stranded nucleic
- acid molecule
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images










Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/543—Immunoassay; Biospecific binding assay; Materials therefor with an insoluble carrier for immobilising immunochemicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1048—SELEX
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/115—Aptamers, i.e. nucleic acids binding a target molecule specifically and with high affinity without hybridising therewith ; Nucleic acids binding to non-nucleic acids, e.g. aptamers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6834—Enzymatic or biochemical coupling of nucleic acids to a solid phase
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/5308—Immunoassay; Biospecific binding assay; Materials therefor for analytes not provided for elsewhere, e.g. nucleic acids, uric acid, worms, mites
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/16—Aptamers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/10—Applications; Uses in screening processes
- C12N2320/13—Applications; Uses in screening processes in a process of directed evolution, e.g. SELEX, acquiring a new function
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/30—Special therapeutic applications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2330/00—Production
- C12N2330/30—Production chemically synthesised
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/46—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans from vertebrates
- G01N2333/47—Assays involving proteins of known structure or function as defined in the subgroups
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/46—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans from vertebrates
- G01N2333/47—Assays involving proteins of known structure or function as defined in the subgroups
- G01N2333/4701—Details
- G01N2333/4703—Regulators; Modulating activity
Definitions
- the present invention relates to an efficient method for producing a nucleic acid aptamer, particularly a DNA aptamer.
- nucleic acid aptamers have attracted attention as other active nucleic acids such as siRNA as a new active ingredient for drugs or diagnostic agents that replace low molecular weight compounds, and various research and development have been promoted in various countries around the world. ing.
- a nucleic acid aptamer is a functional nucleic acid that binds firmly and specifically to a target substance such as a protein by its three-dimensional structure and can inhibit or suppress its function.
- Nucleic acid aptamers are usually used as nucleic acid molecules that bind to a target substance from a nucleic acid library containing a random base sequence by an in vitro selection method (in vitro selection method) called the SELEX (systematic evolution of ligands by exponential enrichment) method.
- in vitro selection method in vitro selection method
- SELEX systematic evolution of ligands by exponential enrichment
- RNA aptamers composed of RNA have been the mainstream of nucleic acid aptamers.
- RNA is unstable and high in production cost, so in recent years it is stable in vivo and can be produced at low cost.
- Patent Document 4 Non-Patent Documents 5 to 8
- it has been difficult to efficiently produce DNA aptamers as compared to RNA aptamers.
- SELEX method as a method for isolating a complex formed by binding a target substance and a nucleic acid aptamer, (1) a complex is obtained by trapping a protein on a nitrocellulose filter using hydrophobic interaction.
- a method of recovering a complex by shifting the gel mobility during gel electrophoresis or (3) A target substance is labeled in advance, and the target substance is affinityd based on the label.
- a method of immobilizing it on a carrier and mixing it with a DNA library is employed.
- An object of the present invention is to develop and provide a method for efficiently and simply producing a nucleic acid aptamer having high specificity and binding property to a target substance, particularly a DNA aptamer.
- Another object of the present invention is to provide a nucleic acid molecule that contains non-Watson-Crick base pairing in a double-stranded region and can specifically and firmly bind to a target substance.
- an object of the present invention is to provide a target substance function inhibitor containing the nucleic acid molecule as an active ingredient and a pharmaceutical composition containing the same.
- the present inventors have developed a method for producing a new nucleic acid aptamer that can suppress non-specific adsorption of DNA or the like and reduce the background by modifying the SELEX method.
- Successful in the conventional SELEX method, a target substance is first bound to a solid phase carrier, and a single-stranded nucleic acid library is added thereto, whereby a nucleic acid aptamer bound to the target substance on the solid phase carrier is recovered.
- the target substance and the single-stranded nucleic acid library were first mixed to form a complex of the single-stranded nucleic acid and the target substance, and then adsorbed to the target substance and / or the solid phase carrier.
- a method for producing a nucleic acid aptamer wherein a single-stranded nucleic acid library and a target substance are mixed in a solution to form a complex of a single-stranded nucleic acid and a target substance.
- the solution after the formation step and the solid phase carrier are mixed, and the complex is immobilized on the solid phase carrier via the binding substance adsorbed on the target substance and / or the solid phase carrier, and immobilized on the solid phase carrier.
- a recovery step of recovering the conjugated complex from the solution an amplification step of amplifying the single-stranded nucleic acid by a nucleic acid amplification method after recovering the single-stranded nucleic acid in the complex, and a double strand obtained in the amplification step
- the said manufacturing method including the single-stranded nucleic acid preparation process of forming a three-dimensional structure within a molecule
- the secondary structure contains one or more double-stranded regions in which 5 to 20 consecutive bases base pair with each other, and
- a nucleic acid molecule that binds to a target substance comprising at least one double-stranded region in which consecutive 5 to 20 bases base pair with each other, and 1 to 10 base pairs in the double-stranded region
- the nucleic acid molecule comprising a Watson-Crick base pair.
- nucleic acid molecule according to (11), wherein the nucleic acid molecule is a single-stranded nucleic acid or a double-stranded nucleic acid.
- nucleic acid molecule according to any one of (11) to (13), wherein the target substance is a peptide.
- nucleic acid molecule according to (14), wherein the peptide is a transcriptional regulatory factor, a signal transduction factor, a protein ligand, or a receptor protein.
- nucleic acid molecule according to (17) comprising a double-stranded region consisting of the base sequence represented by SEQ ID NOs: 3 and 4, SEQ ID NOs: 5 and 6, or SEQ ID NOs: 7 and 8.
- a target substance function inhibitor comprising the nucleic acid molecule according to any one of (11) to (19) as an active ingredient.
- (22) A method for detecting a target substance to which a nucleic acid molecule present in a sample binds using the nucleic acid molecule according to any one of (11) to (15).
- a nucleic acid aptamer having high specificity and binding property to a target substance, particularly a DNA aptamer can be produced efficiently and simply.
- nucleic acid molecule containing the non-Watson-Crick base pairing in the double-stranded region of the present invention it is possible to provide a nucleic acid molecule that can specifically and firmly bind to the target substance.
- consensus sequence (a) found in the double-stranded region of the nucleic acid molecule of the present invention targeting NF- ⁇ B p50 and the consensus sequence found in the DNA aptamer obtained by the production method of the present invention The base sequence ((b) to (d)), the sequence number of each chain, and their base pairing are shown.
- the base sequence of the consensus sequence (a) the base pair described as “W-W” indicates “a-t” or “t-a”.
- ” between the bases of the double-stranded region is a Watson-Crick base pair, “ ⁇ ” or “ ⁇ ” is “ag” or “ga”, or “gt” or “tg”, respectively.
- the top is the sequence of the single-stranded nucleic acid library used in the production method.
- a sequence similar to the natural consensus DNA sequence (SEQ ID NO: 29) to which NF- ⁇ B binds is surrounded by a black frame.
- the underline indicates a region presumed to form a double-stranded region (stem structure) by base pairing in the molecule.
- Bold letters indicate bases that are mutated in similar clone sequences compared to 5R01 or 5R14 or 5R05 sequences.
- a hyphen (-) indicates a single nucleotide deletion mutation.
- suction of the single stranded DNA to the various magnetic beads as a solid-phase carrier by real-time PCR is shown.
- the top is a known NF- ⁇ B p50 binding consensus sequence.
- the white box indicates the 5'-end primer binding region
- the black box indicates the 3'-end primer binding region.
- SPR Surface plasmon resonance
- FIG. 9 shows an SPR sensorgram in which the interaction between the three clones shown in FIG. 8 and the control Cont-68 and NF- ⁇ B p50 was detected.
- nucleic acid or “nucleic acid molecule” refers to a biopolymer composed of nucleotides as structural units in principle and linked by phosphodiester bonds.
- a DNA in which only deoxyribonucleotides having any base of adenine, guanine, cytosine and thymine are linked
- an RNA in which only ribonucleotides having any base of adenine, guanine, cytosine and uracil are linked, or those Natural nucleic acids that exist in nature such as combinations are applicable.
- the nucleic acid of the present invention can include a non-natural nucleotide or a non-natural nucleic acid in part or in whole.
- non-natural nucleotide refers to an artificially constructed or artificially chemically modified nucleotide having similar properties and / or structure to the natural nucleotide, or a natural nucleotide.
- a nucleotide that does not exist in nature including a nucleoside or base having properties and / or structures similar to those of the nucleoside or base constituting the type nucleotide. Examples include abasic nucleosides, arabino nucleosides, 2'-deoxyuridines, ⁇ -deoxyribonucleosides, ⁇ -L-deoxyribonucleosides, and other nucleosides with sugar modifications.
- nucleoside having the above-mentioned sugar modification includes substituted pentasaccharide (2′-O-methylribose, 2′-deoxy-2′-fluororibose, 3′-O-methylribose, 1 ′, 2′-deoxy. Ribosides), arabinose, substituted arabinose sugars, substituted hexamonosaccharides and alpha-anomeric sugar modifications.
- the non-natural nucleotide of the present invention may be an artificially constructed base analog or an artificially chemically modified base (modified base).
- Base analog includes, for example, 2-oxo (1H) -pyridin-3-yl group, 5-substituted-2-oxo (1H) -pyridin-3-yl group, 2-amino-6- (2 -Thiazolyl) purin-9-yl group, 2-amino-6- (2-thiazolyl) purin-9-yl group, 2-amino-6- (2-oxazolyl) purin-9-yl group and the like.
- Modified bases include, for example, modified pyrimidines (eg, 5-hydroxycytosine, 5-fluorouracil, 4-thiouracil), modified purines (eg, 6-methyladenine, 6-thioguanosine) and other heterocyclic rings. Examples include bases.
- non-natural nucleic acid refers to an artificially constructed nucleic acid analog having a structure and / or property similar to that of a natural nucleic acid.
- examples include peptide nucleic acids (PNA: Peptide Nucleic Acid), peptide nucleic acids having a phosphate group (PHONA), cross-linked nucleic acids (BNA / LNA: Bridged Nucleic Acid / Locked Nucleic Acid), morpholino nucleic acids, and the like.
- nucleic acids and nucleic acid analogs such as methylphosphonate DNA / RNA, phosphorothioate DNA / RNA, phosphoramidate DNA / RNA, 2′-O-methyl DNA / RNA can also be included.
- modified nucleic acids hereinafter, for convenience, the above-mentioned non-natural nucleotides or non-natural nucleic acids are collectively referred to as “modified nucleic acids”.
- nucleic acid aptamer is an aptamer composed of nucleic acids, and is targeted by a three-dimensional structure formed based on the secondary structure and tertiary structure of a single-stranded nucleic acid via hydrogen bonding or the like.
- a ligand molecule that binds strongly and specifically to a substance and has the ability to specifically inhibit or suppress a function such as physiological activity of a target substance.
- an RNA aptamer composed only of RNA and a DNA aptamer composed only of DNA are generally known, but the nucleic acid constituting the nucleic acid aptamer in the present specification is not particularly limited.
- a DNA aptamer for example, a DNA aptamer, an RNA aptamer, an aptamer composed of a combination of DNA and RNA, an aptamer including a modified nucleic acid as a part thereof, an aptamer composed only of a modified nucleic acid, and the like are included.
- it is a DNA aptamer.
- target substance refers to a substance that can be bound by a nucleic acid molecule, particularly a nucleic acid aptamer.
- the type of target substance is not particularly limited as long as it is a biological substance to which a nucleic acid molecule can bind. Examples include peptides (oligopeptides or polypeptides), nucleic acids, lipids, sugars (including sugar chains), or low molecular compounds. Preferably it is a peptide, more preferably a polypeptide, ie a protein.
- the target substance can be appropriately selected according to the purpose. Usually, it is selected for the purpose of inhibiting, suppressing or enhancing the specific biological function of the biological material.
- Specific biological functions include, for example, catalytic function or gene expression control function (including control of transcription, translation, transport, etc.), apoptosis control function, and protein-protein interaction that is widely responsible for cell information transmission. Examples include interactions between biological materials.
- the target substance to be used may be any naturally derived substance, chemically synthesized substance, genetically modified substance or the like. It is preferable to use a purified single substance free from impurities. Further, when the target substance is a polypeptide, it may be a fusion polypeptide in which a tag sequence is fused. Examples of the tag sequence include hexahistidine (His), FLAG, HA, myc, and GFP.
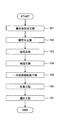
- a first embodiment of the present invention is a method for producing a nucleic acid aptamer. According to the present invention, a background due to nonspecific adsorption of a single-stranded nucleic acid can be reduced, and a nucleic acid aptamer having high specificity for a target substance, particularly a DNA aptamer, can be produced efficiently and simply. .
- the production method of the present invention comprises a complex formation step (101), an immobilization step (102), a recovery step (103), an amplification step (104), and a single-stranded nucleic acid preparation step (105).
- the manufacturing method of this invention can include the repetition process (106) and / or the selection process (107) as arbitrary processes as needed.
- the selection step (107) can be performed between the amplification step (104) and the single-stranded nucleic acid preparation step (105) and / or after the repetition step (106).
- each process will be specifically described.
- Complex formation step 101) is a step of mixing a single-stranded nucleic acid library and a target substance in a solution to form a complex of the single-stranded nucleic acid and the target substance. It is.
- the “single-stranded nucleic acid library” refers to a pool composed of a plurality of identical and / or different single-stranded nucleic acids including candidate nucleic acid aptamer molecules.
- a double-stranded nucleic acid formed by pairing all or part of the bases of the single-stranded nucleic acid may be included in a part thereof. Since a single-stranded nucleic acid library is a library containing nucleic acid aptamer candidates as described above, each single-stranded nucleic acid constituting the library forms a higher-order structure by self-folding in principle. .
- the primary structure of the single-stranded nucleic acid constituting the library has primer binding regions (201, 203) to which a primer binds on the 5 ′ end side and the 3 ′ end side, and is located between them.
- a central region (202) The base length of each primer region is 15 to 40 bases.
- the base length of the central region is 20 to 80 bases. Therefore, the base length of the single-stranded nucleic acid constituting the single-stranded nucleic acid library is in the range of 50 to 160 bases.
- the primer binding regions (201, 203) on the 5 ′ end side and 3 ′ end side have a base sequence corresponding to the forward primer (204) and a base sequence complementary to the reverse primer (205), respectively.
- the base sequence of each primer is a sequence in which the primer does not form a secondary structure in the molecule and / or a sequence in which the forward primer and the reverse primer do not form a continuous double-stranded region, and the Tm value is 50 to 80 °C, 55-75 °C, or within the range of 60-70 °C, Tm value of both primers should not be significantly different, and the GC content of each primer is 40-60% or 45-55% Is preferred.
- the base sequence of the central region (202) of each single-stranded nucleic acid constituting the single-stranded nucleic acid library is composed of a random or specific base sequence.
- the central region is desirably random in principle.
- the specific base sequence refers to a base sequence in a single-stranded nucleic acid subjected to a predetermined selection pressure.
- “single-stranded nucleic acid subjected to a predetermined selection pressure” is used after the second round (second round), for example, when the production method of the present invention includes an iterative process (106) described later. This constitutes a single-stranded nucleic acid constituting a single-stranded nucleic acid library.
- the single-stranded nucleic acid library may be appropriately prepared according to a method known in the art.
- the method of the present invention aims to produce an unknown nucleic acid aptamer that can bind to a target substance
- the single-stranded nucleic acid library used for the first time is composed of a large number of different single-stranded nucleic acid populations. It is preferable that Therefore, in this case, for example, a single-stranded nucleic acid library may be prepared by chemical synthesis using a nucleic acid synthesizer.
- a target library can be obtained by inputting the designed base sequence into a synthesis program using a DNA synthesizer.
- the base sequence may be outsourced to each manufacturer to produce a desired single-stranded nucleic acid library.
- the production method of the present invention includes an iterative process (106) described later, a single-stranded nucleic acid library used in the second and subsequent rounds is obtained in the round immediately before the iterative process (106). It may be prepared based on the single-stranded nucleic acid.
- the single-stranded nucleic acid library used for the first time is preliminarily subjected to the intramolecular higher-order structure formation treatment of the single-stranded nucleic acid described in the single-stranded nucleic acid preparation step (105) described later. It is preferable to go.
- the single-stranded nucleic acid library is preliminarily stored in a solid-phase carrier (two or more kinds). When using these solid phase carriers, it is preferable to remove single-stranded nucleic acids that bind nonspecifically to each solid phase carrier). This process involves adding and mixing an appropriate amount of the solid phase carrier used in the solid phase immobilization step in the solution containing the single stranded nucleic acid library, and then recovering and removing the solid phase carrier. Use as a rally is enough.
- complex refers to a single-stranded nucleic acid constituting a single-stranded nucleic acid library, specifically, a nucleic acid aptamer candidate molecule formed by a single-stranded nucleic acid and a target substance.
- the type and properties of the solution used in this step are not particularly limited as long as the solution can form a complex between the nucleic acid and the target substance.
- it is water or aqueous solution.
- the pH may be in the range of 5.0 to 9.0, preferably 6.0 to 8.0, more preferably 6.5 to 7.6.
- the salt concentration may be in the range of 20 to 500 mM, preferably 50 to 300 mM, more preferably 90 to 180 mM in the final concentration.
- a preferred aqueous solution is a buffer.
- an appropriate salt eg, NaCl, CH 3 COOK
- a pH buffer solution eg, phosphate buffer, citrate-phosphate buffer, Tris-HCl buffer or HEPES buffer
- PBS buffer 1.1 mM KH 2 PO 4 , 155 mM NaCl, 3 mM Na 2 HPO 4 , pH 7.4
- the composition of the pH buffer is based on the known composition described in, for example, Sambrook, J. et.al., (2001) Molecular Cloning: A Laboratory Manual Third Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. Fine adjustments may be made as necessary.
- the above solution may further contain a reducing agent or a surfactant as required.
- the reducing agent examples include DTT (dithiothreitol) and 2-mercaptoethanol.
- the final concentration of the reducing agent in the solution may be in the range of 0.5 to 10 mm, preferably 1 to 5 mm.
- the surfactant is preferably a nonionic surfactant.
- a nonionic surfactant for example, Nonidet P40 (NO-40), Triton X-100, Triton X-114, Brij-35, Brij-58, Tween-20, Tween-40, Tween-60, Tween-80, n-octyl- ⁇ - Examples include glucoside, MEGA-8, MEGA-9, and MEGA-10.
- the final concentration of the surfactant in the solution may be in the range of 0.005% to 0.1%, preferably 0.01% to 0.08% by volume / volume (V / V).
- the solution used in this step may contain a competitive substance.
- “competitive substance” refers to a substance that competes with the target substance for binding with the single-stranded nucleic acid.
- the type of the competitive substance is not particularly limited as long as it can compete with the single-stranded nucleic acid for the target substance.
- a nucleic acid, peptide, lipid, sugar, or low molecular weight compound can be mentioned.
- a substance having the same properties as the single-stranded nucleic acid that is the target nucleic acid aptamer for example, a substance having the same binding site on the target substance is preferable.
- Such a substance corresponds to a nucleic acid (single-stranded nucleic acid and / or double-stranded nucleic acid) having a base sequence similar to the target single-stranded nucleic acid.
- the target substance is a transcriptional regulatory factor
- the base sequence on the genome sequence to which the transcriptional regulatory factor originally binds corresponds.
- the competitor is the nucleic acid, even if the competitor forms a complex with the target substance, it is designed and prepared so that it does not have a primer binding region (201, 203) like a single-stranded nucleic acid By doing so, it can be removed without being amplified in the amplification step (104) described later.
- By containing the competitive substance in the solution it becomes possible to produce a nucleic acid aptamer that binds to the target substance more firmly.
- a single-stranded nucleic acid library and a target substance are mixed at 9: 1 to 1: 9, preferably 5: 5 (volume: volume), and 4 to 40 ° C., preferably Incubation may be performed within a range of 15 to 37 ° C. for 5 minutes to 30 minutes or more, for example, about 10 minutes to 1 hour, preferably about 20 minutes to 40 minutes.
- the formed complex may be washed before the next immobilization step (102).
- the washing of the complex may be performed using a method known in the art based on the type of the target substance, the molecular size and characteristics of the complex. For example, when the target substance is a protein, the complex can be separated from the free single-stranded nucleic acid using an ultrafiltration membrane through which only the nucleic acid passes depending on the molecular size.
- the composition of the washing buffer may be the same composition as the buffer used for forming the complex.
- the cleaning buffer may contain a reducing agent or a surfactant.
- concentration and composition of the reducing agent and surfactant may be the same as the buffer used for complex formation.
- the remaining free single-stranded nucleic acid can be removed by washing operation in the subsequent immobilization step (102) and recovery step (103). Strand nucleic acids can be removed. Therefore, washing may be performed as necessary.
- the “immobilization step” (102) is a step of mixing the solution after the previous step and the solid phase carrier to immobilize the complex on the solid phase carrier.
- the “solid phase carrier” is a solid state carrier, for example, a magnetic bead, a polymeric polysaccharide support (eg, Sepharose, Sephadex, agarose), silica, glass, metal (eg, gold, Platinum, silver), plastics, ceramics, resins (natural or synthetic resins), or combinations thereof.
- a polymeric polysaccharide support eg, Sepharose, Sephadex, agarose
- silica glass
- metal eg, gold, Platinum, silver
- plastics ceramics
- resins natural or synthetic resins
- the solid phase carrier itself may be hydrophilic, or the solid phase carrier may be hydrophobic and the surface thereof may be subjected to a hydrophilic coating treatment.
- the shape of the carrier is not particularly limited. For example, a spherical shape, a substantially spherical shape, a flat plate shape, a substantially flat plate shape, a fiber shape, and the like can be given.
- a substantially spherical particle such as a bead is particularly preferable as the shape of the solid phase carrier in this step because of its large binding surface area and high operability.
- immobilization refers to linking the complex to a solid phase carrier.
- the complex is immobilized on the solid phase carrier through a target substance and / or a binder adsorbed on the solid phase carrier.
- the “binding agent” refers to a molecule that mediates the connection between a target substance and a solid phase carrier.
- the binder can include not only a single molecule but also two or more different molecules to be linked, as long as it can intervene the linkage between the target substance and the solid phase carrier.
- Specific examples of the connector include, for example, a low molecular weight compound, an amino acid or peptide, a nucleic acid or a constituent thereof (including nucleoside or nucleotide), or a combination thereof.
- the low molecular compound herein is a natural product or a chemical synthesis product having a molecular weight of about several hundred to several thousand.
- vitamins including biotin
- terpenoids eg carotenoids, heme, chlorophyll
- polyphenols eg flavonoids, catechins, tannins
- Peptides include proteins (including recombinant antibodies such as humanized antibodies or multivalent antibodies) or proteins including enzymes or functional fragments thereof.
- Nucleic acids include DNA, RNA, nucleic acid analogues such as LNA (Locked Nucleic Acid: LNA is a registered trademark) or PNA (Peptide Nucleic Acid), or fragments thereof.
- Preferred conjugates in the present invention include, for example, a conjugate consisting of biotin and avidin or streptavidin, a lectin-biotin and avidin in which lectin is further bound to the biotin, a conjugate consisting of streptavidin or neutravidin, and one or more antibodies Or a binder consisting of an antibody and protein A, G or L.
- the binder is adsorbed on the target substance, the solid support or both.
- adsorption refers to immobilizing a binder on a target substance or solid phase carrier by chemical adsorption, physical adsorption and / or affinity.
- chemical adsorption includes chemical bonds such as covalent bonds, ionic bonds, and hydrogen bonds
- physical adsorption includes Coulomb forces, van der Waals forces, and hydrophobic interactions.
- the binder When the binder is adsorbed only on one of the target substance or the solid phase carrier, the binder can specifically recognize and bind to the partner substance on which the binder is not adsorbed. For example, when a binder is adsorbed on the solid phase carrier, the binder specifically recognizes and binds to the target substance. More specifically, for example, when protein A or protein A bound to the antibody is adsorbed on a solid phase carrier as a binder, the antibody specifically recognizes and binds to the target substance. Therefore, by mixing the target substance and the solid phase carrier in the solution, the target substance and the solid phase carrier are linked via the connector.
- Part or all of the target substance after the complex formation step (101) forms a complex with a single-stranded nucleic acid that is a candidate for a nucleic acid aptamer. Therefore, the complex can be immobilized on the solid phase carrier by this step.
- the binder When the binder is adsorbed on each of the target substance and the solid phase carrier, the binder (for convenience, the binder adsorbed on the target substance is hereinafter referred to as “first binder” and adsorbed on the solid phase carrier.
- the combined binders are referred to as “second binders” and can bind specifically to each other.
- biotin which is the first binder
- avidin, streptavidin, or neutravidin which is the second binder
- the target substance and the solid phase carrier are linked through the binding of biotin and avidin, streptavidin, or neutravidin.
- the target substance when the target substance is adsorbed with the anti-target substance mouse monoclonal IgG antibody as the first binder and the rabbit anti-mouse IgG antibody or protein A as the second binder with the solid phase carrier, the target substance and By mixing the solid phase carrier in the solution, the anti-target substance mouse monoclonal IgG antibody and the rabbit anti-mouse IgG antibody or protein A specifically bind to each other.
- the target substance and the solid phase carrier are linked via an antibody-antibody or antibody-protein A bond.
- the complex can be immobilized on a solid phase carrier.
- this step it is the target substance in the complex that directly contributes to the immobilization of the complex to the solid phase carrier via the binder. Therefore, in this step, not only the complex but also a free target substance that does not form the complex can be simultaneously immobilized on the solid phase carrier. However, even if a target substance in a free state that does not carry a single-stranded nucleic acid is immobilized on a solid phase carrier, the effect on the present invention is aimed at reducing nonspecific adsorption of single-stranded nucleic acid. Or very minor and does not constitute a particular impediment to achieving the present invention.
- the adsorption method of the binder to the target substance and the solid phase carrier varies depending on the type of the target substance, the solid phase carrier and / or the binder. Therefore, what is necessary is just to adsorb
- the binder and / or the adsorption method does not inhibit or dissociate the binding between the single-stranded nucleic acid in the complex and the target substance.
- the target substance or the solid phase carrier has a functional group
- a functional group for example, an active functional group capable of covalent bonding with the functional group (for example, an aldehyde group, a carboxyl group, a sulfo group, an amino group, A thiol group, a cyano group, a nitro group), or a bond into which such an active functional group is introduced, a nucleophilic addition reaction, a nucleophilic substitution reaction or an electrophilic substitution between the two functional groups.
- the binder can be adsorbed to the target substance or the solid phase carrier through a covalent bond by a chemical reaction such as a reaction.
- Such a combination of functional groups capable of covalent bonding includes, for example, an amino group and an aldehyde group, an amino group and an ester group, a thiol group and a maleimide group, an azide group and an acetylene group, an azide group and an amino group, a hydrazine group and a ketone.
- a method of covalently bonding these functional groups by chemical reaction is a technique well known in the art.
- the target is a protein and the protein is adsorbed using biotin as a binder
- an active ester group is introduced into biotin using N-hydroxysuccinimide ester (NHS), and then the amino group of the protein and the ester By forming an amide bond with the group, the protein can be adsorbed with biotin.
- biotinylation reagents are commercially available from manufacturers, and they may be used.
- the first binder is an antibody that specifically recognizes and binds to the epitope of the antigen
- the first substance is brought into contact with the first in an appropriate solution by affinity binding.
- the binder can be adsorbed to the target substance.
- the target substance is a polypeptide and an antibody capable of specifically recognizing the polypeptide is present
- the antibody can be adsorbed to the polypeptide as a first binder.
- the tag sequence can be specifically recognized if it is possible to synthesize a fusion polypeptide of the polypeptide and the tag sequence.
- the resulting antibody can be adsorbed to the polypeptide as the first binder.
- the adsorption time of the binder is not limited, but when the binder is adsorbed to the target substance, it is preferably after the complex formation step (101) and before this step. This is because, when the binder is adsorbed to the target substance before the complex formation step (101), the binding between the target substance and the single-stranded nucleic acid may be suppressed or inhibited by the adsorption of the binder. Therefore, when the binder is adsorbed to the target substance, after the complex formation step (101), an appropriate time before this step, for example, a solution containing the complex obtained after the complex formation step (101).
- the binder may be adsorbed to the target substance by any of the adsorption methods described above.
- the binder When the binder is adsorbed on the solid phase carrier, it is preferably adsorbed before mixing the solution containing the complex and the solid phase carrier at least in this step. This is because the use of the solid phase carrier on which the binder is adsorbed makes it possible to more reliably fix the target substance to the solid phase carrier. Therefore, when the binder is adsorbed to the solid phase carrier, the binder can be adsorbed to the solid phase carrier by any of the above adsorption methods before mixing the solution containing the complex and the solid phase carrier at least in this step. That's fine.
- the biotin adsorption to the protein is
- the solution containing the complex obtained after the complex formation step (101) may be adsorbed according to the attached protocol using, for example, a commercially available biotinylation reagent. Thereafter, unadsorbed biotin is preferably washed and removed by a method known in the art such as ultrafiltration.
- the adsorption of streptavidin to the magnetic beads may be performed by previously adsorbing the streptavidin to the magnetic beads using a known method, independently of the complex formation step (101).
- a magnetic bead when a magnetic bead has a Tosyl group or an epoxy group, it can be adsorbed as it is with a primary amino group of streptavidin as it is by simply mixing with streptavidin. Further, when the magnetic beads have a carboxyl group, they can be adsorbed by a covalent bond with the primary amino group of streptavidin by activation with carbodiimide. These are methods well known in the art. Further, commercially available magnetic beads on which streptavidin is adsorbed in advance may be purchased and used.
- binders used for immobilizing the target substance on the solid phase carrier.
- this is a case where a plurality of different first binders are adsorbed on the target substance.
- a case where biotin and an anti-target substance antibody are adsorbed to one target substance as independent first binders can be mentioned.
- the immobilization step (102) and the recovery step (103) described later are performed using different solid phase carriers on which the second binders corresponding to the respective first binders are adsorbed.
- the background of the single-stranded nucleic acid mixed by nonspecific adsorption can be further reduced.
- a target substance in which the aforementioned biotin and anti-target substance antibody are adsorbed as a first binder using magnetic beads that are solid phase carriers in which streptavidin is adsorbed as a second binder, After performing the immobilization step (102) and the recovery step (103), again using the Sepharose beads that are solid phase carriers on which protein G is adsorbed as the second binder, the immobilization step (102) and the recovery step ( 103).
- the target substance in a free state that does not form a complex with the single-stranded nucleic acid together with the complex is also immobilized on the solid phase carrier.
- the target substance that does not form such a complex is also removed at the same time. It will not be.
- Recovery Step 103) is a step of recovering the complex immobilized on the solid phase carrier from the solution.
- the complex is immobilized on the solid phase carrier after the immobilization step (102) via a connector.
- This step is characterized in that the complex-solid phase carrier is separated from the solution and recovered based on the characteristics of the solid phase carrier.
- the characteristics of the solid phase carrier refer to properties specific to the solid phase carrier. For example, magnetic force, specific gravity, fluorescence, luminescence, affinity, etc. are included.
- the complex-solid phase carrier in the solution after the immobilization step (102) is recovered using a magnet (magnetic magnet), and then the buffer is stored. It can be recovered by washing and removing the single-stranded nucleic acid non-specifically adsorbed to the target substance or solid phase carrier.
- the solid support is a polymer polysaccharide support, silica, metal (including magnetic beads) or glass, the complex-solid support is precipitated by centrifugation, the supernatant is removed, and then a buffer is used. By washing, the complex-solid phase carrier can be similarly recovered.
- the solid phase carrier is a polymer polysaccharide support or the like carrying a fluorescent substance, it can be recovered by a fluorescence detector such as FACS.
- FACS fluorescence detector
- a buffer having the same composition as the buffer used in the complex formation step (101) can be used.
- the buffer may contain a reducing agent or a surfactant as necessary.
- the reducing agent to be used include DTT (dithiothreitol) and 2-mercaptoethanol.
- the final concentration of the reducing agent in the solution may be in the range of 0.5 to 10 ⁇ mM and 1 to 5 ⁇ mM.
- the surfactant is preferably a nonionic surfactant.
- Nonidet P40 (NO-40), Triton X-100, Triton X-114, Brij-35, Brij-58, Tween-20, Tween-40, Tween-60, Tween-80, n-octyl- ⁇ - Glucoside, MEGA-8, MEGA-9 and MEGA-10 are preferred.
- the final concentration of the surfactant in the solution may be in the range of 0.005% to 0.1%, 0.01% to 0.08% by volume / volume (V / V).
- Washing may be performed once to several times using the above buffer. Two to three times are preferable.
- the washing temperature and washing time are not particularly limited, but may be 15 to 50 ° C. or 20 to 40 ° C. for 10 minutes to 1 hour.
- the “amplification step” (104) is a step of recovering the single-stranded nucleic acid in the complex and amplifying it by a nucleic acid amplification method.
- the complex is eluted from the complex-solid phase carrier recovered in the recovery step (103).
- the elution method varies depending on the type of the binder.
- the binder is an antibody
- the complex-solid phase carrier is dissociated by acid treatment or the like, and then alkali is added as necessary to neutralize the complex-solid phase carrier.
- the complex can be eluted.
- the binder is biotin and avidin, streptavidin or neutravidin
- the biotin and avidin or streptoid can be treated by heating in a solution containing 7 M or more urea and / or 2 M or more ⁇ -mercaptoethanol.
- the binding of avidin or neutravidin can be dissociated and the complex can be eluted. Furthermore, when the target substance is a sugar chain-modified substance and the binder is a lectin, the complex can be eluted by adding a sugar such as glucose. These methods may be appropriately performed according to methods known in the art.
- the method for recovering the complex single-stranded nucleic acid differs depending on the type of target substance forming the complex. Therefore, it may be carried out based on a method known in the art for recovering nucleic acid from a complex consisting of the target substance and nucleic acid.
- the target substance is a peptide such as a protein
- the target single-stranded nucleic acid can be recovered by coagulating and removing the protein by a protein denaturation method such as an alkali method or a phenol / chloroform method.
- the target substance is a lipid or low molecular weight compound
- heat treatment is performed to destroy the double-stranded structure of the nucleic acid
- a chelating agent is added to the elution buffer, or elution One that has been dissociated by the alcohol precipitation method or the like after dissociating the binding between the target substance and nucleic acid by destroying the double-stranded structure of the nucleic acid by heat treatment with the buffer pH changed from the binding buffer Strand nucleic acids can be recovered.
- a linker capable of being cleaved with light irradiation or a reducing agent is added to the binder, so that the solid phase carrier is combined with the nucleic acid to which the target substance is bound.
- the nucleic acid molecule can also be recovered by the alcohol precipitation method or the like by the above-described operation.
- the target substance that is immobilized on the solid phase carrier together with the complex in the immobilization step (102) and that does not form the complex can also be removed in this step.
- nucleic acid amplification method refers to a method of amplifying a specific region of a nucleic acid serving as a template using an enzyme such as a primer and a polymerase.
- an enzyme such as a primer and a polymerase.
- any method known in the art may be used.
- PCR polymerase chain reaction
- ICAN isothermal gene amplification
- the polymerase used in the reaction is appropriately determined depending on the nucleic acid amplification method to be used, but usually a DNA polymerase, particularly a heat resistant DNA polymerase is used.
- a DNA polymerase particularly a heat resistant DNA polymerase is used.
- thermostable nucleic acid polymerases are commercially available from manufacturers such as TaKaRa, New England Biolab, Roche, Life Technology, Roche, and Promega. You can also.
- a polymerase having high fidelity is preferred as a polymerase used in the nucleic acid amplification method, but the polymerase used in this step does not necessarily need to have high fidelity, and often like Taq polymerase. It may be a polymerase into which an error can be introduced.
- the reaction conditions for the nucleic acid amplification method take into account the length of the base sequence to be amplified, the amount of the template nucleic acid, that is, the amount of recovered single-stranded nucleic acid, the Tm value of the primer, the optimum reaction temperature and the optimum pH of the polymerase to be used, etc. And decide.
- the sequence corresponding to the 5'-end primer binding region (201) constituting each single-stranded nucleic acid of the single-stranded nucleic acid library is used as the forward primer (204), and the 3'-end side.
- a sequence complementary to the primer binding region (203) is used for the reverse primer (205).
- the double-stranded nucleic acid as the amplification product is selectively separated and purified from the reaction solution after the amplification reaction based on the label,
- the other single-stranded nucleic acid complementary to the target single-stranded nucleic acid among the double-stranded nucleic acids can be conveniently separated and removed based on the label.
- the temperature and reaction time for each step are, for example, a heat denaturation step at 90 ° C to 98 ° C for 30 seconds to 1 minute, and an annealing step at 50 ° C to 60 ° C.
- the extension step may be performed at 70 ° C. to 75 ° C. for 40 seconds to 2 minutes for 30 seconds to 1 minute.
- the number of cycles is usually 10 to 40 cycles. Preferably it is 15 to 20 cycles.
- the obtained amplified nucleic acid may be purified as necessary before the single-stranded preparation step. Unreacted deoxynucleotides and primers, polymerase, etc. can be removed by purification.
- the purification method may be any method known in the art. For example, an ethanol precipitation method or a purification method using a spin gel filtration column can be used. The latter is preferable because the nucleic acid can be purified quickly and easily. Such columns are commercially available from companies in the bio-related business, and they can also be used.
- the “single-stranded nucleic acid preparation step” (105) is a step of making the double-stranded nucleic acid obtained in the amplification step (104) into a single strand.
- the nucleic acid after the amplification step (104) is usually not a nucleic acid aptamer composed of a single-stranded nucleic acid that specifically binds to a target substance, but two single-stranded nucleic acids having a base sequence complementary thereto. It exists in the state of a strand nucleic acid. Therefore, in this step, after preparing the double-stranded nucleic acid into a single strand, an intramolecular higher-order structure is formed in the target single-stranded nucleic acid to prepare a nucleic acid aptamer.
- Single-stranded double-stranded nucleic acid is generally performed by heat denaturation.
- Thermal denaturation may be performed in the range of 60 to 90 ° C.
- the solution used at the time of denaturation may contain 1 to 7 M urea.
- electrophoresis can be performed using a denaturing gel, and a double-stranded single strand can be purified and purified by a method known in the art, such as purification by eluting a band of the desired size from the gel. .
- the single-stranded nucleic acid prepared in this step includes a mixture of a nucleic acid capable of forming a target nucleic acid aptamer and a pair having a base sequence complementary to the target single-stranded nucleic acid.
- the single-stranded nucleic acid having such a complementary base sequence is removed to form a target nucleic acid aptamer. Only the nucleic acid may be separated. Separation of only one single-stranded nucleic acid having such a desired base sequence can be achieved, for example, by labeling the reverse primer with a label as described above.
- the purified double-stranded nucleic acid is denatured, single-stranded, and fractionated by the difference in mobility of both strands by denaturing gel electrophoresis, and only the desired single-stranded nucleic acid is separated from the gel and purified. can do.
- the single-stranded nucleic acid may be heated and cooled.
- a single-stranded nucleic acid is dissolved in a buffer (for example, PBS buffer) used in the complex formation step (101) and dissolved at 80 to 98 ° C., preferably 85 to 95 ° C. for 30 seconds to 5 minutes, preferably May be heat-denatured for 30 seconds to 3 minutes, and then allowed to stand at room temperature or the like, and then slowly cooled or stepwise cooled to form an intramolecular higher-order structure.
- the stepwise cooling may be carried out by once cooling at 50 to 70 ° C. for about 1 to 20 minutes, and then lowering the temperature to 15 to 35 ° C. for cooling.
- nucleic acid aptamer that specifically binds to the target substance can be produced.
- the nucleotide sequence of the obtained nucleic acid aptamer can be identified using a normal nucleic acid cloning technique known in the art.
- the nucleic acid aptamer obtained can be denatured linearly and inserted into a suitable cloning vector, and then the base sequence can be determined by cycle sequencing reaction or the like.
- cycle sequencing reaction or the like are known methods and can be carried out using a sequencer using a commercially available kit such as Big ⁇ Dye Terminator Cycle Sequencing Kit from Life Technologies.
- Step “Repeat Step” (106) means from complex formation step (101) to single-stranded nucleic acid preparation step (105) (hereinafter, a series of these steps is referred to as “round”. This is a step of repeating a plurality of times.
- This step is an optional step as described above. However, in order to narrow down nucleic acid aptamers having higher specificity with the target substance after the single-stranded nucleic acid preparation step (105), it is preferable to repeat this step two or more rounds. Specifically, for example, 2 to 15 rounds, 2 to 8 rounds, or 2 to 5 rounds.
- the single-stranded nucleic acid library used in the complex formation step (101) is basically a single-stranded nucleic acid obtained in the immediately preceding round of single-stranded nucleic acid preparation step (105) except for the first round.
- the first round is preferably composed of a large number of different single-stranded nucleic acid populations as described above, it is generally desirable to use a single-stranded nucleic acid library prepared by chemical synthesis.
- the conditions in each round, that is, the complex formation step (101) to the single-stranded nucleic acid preparation step (105) are the same as necessary.
- round conditions include, for example, changing the composition of the solution and buffer used in each round. Specifically, in the first half of the buffer, wash conditions should be mild to acquire more nucleic acid aptamer candidates, and in the second half of the buffer, about 3 M urea should be mixed to make the washing conditions stricter. Thus, it is possible to separate only single-stranded nucleic acids that bind more strongly to the target substance. Furthermore, the concentration of the target substance and the single-stranded nucleic acid library in the complex formation step (101) can be changed in each round.
- the concentration of the target substance and single-stranded nucleic acid library is lowered each time the rounds are repeated, and the complex formation conditions are made stricter, so that only single-stranded nucleic acids that bind tightly to the target substance are separated. Can do.
- the “selection step” (107) is a step of selecting a single-stranded nucleic acid molecule having a predetermined structure from the single-stranded nucleic acid obtained after the repeating step (106). This step is an optional step, and can be selectively performed when the purpose is to produce a nucleic acid aptamer that binds more strongly to the target substance, as will be described later.
- the predetermined structure here means that when a single-stranded nucleic acid molecule forms a secondary structure as a nucleic acid aptamer, the predicted secondary structure includes one or more double-stranded regions, and at least one of the two This refers to a structure containing non-Watson-Crick base pairs in the chain region.
- the present inventors have a non-Watson-Crick base pair in a double-stranded region predicted to be a binding region with a target substance. It has been found that it binds very firmly. This process is based on the knowledge.
- This step is for the single-stranded nucleic acid preparation step (105) described above for each single-stranded nucleic acid obtained after the repeating step (106), that is, after the single-stranded nucleic acid preparation step (105) that is the final step of the round.
- the secondary structure includes one or more double-stranded regions in which 5 to 20 bases, 7 to 18 bases, 8 to 17 bases, or 10 to 15 bases base pair with each other, that is, a stem region.
- a single-stranded nucleic acid in which at least one of the double-stranded regions comprises 1 to 5 base pairs, 1 to 7 base pairs, 1 to 8 base pairs, or 1 to 10 base pairs consisting of non-Watson-Crick base pairs Select an array.
- non-Watson-Crick base pair means guanine and cytosine (g), guanine (g), adenine (a), cytosine (c), thymine (t) and uracil (u).
- gc A base pair other than adenine and thymine (at) or uracil (au). Also included are base pairs in a hydrogen bonding mode different from the base pairs formed with normal double-stranded DNA (Nagaswamy U., et al., Nucl. Acid Res. 2000, 28: 375-376).
- This step can be performed after each round and before the start of the next round, in addition to the end of the final round.
- the single-stranded nucleic acid having a predetermined structure obtained in this step as a single-stranded nucleic acid library in the next round, by tightening the conditions of each step in one round, It is possible to produce a nucleic acid aptamer that binds to a target substance even more firmly.
- a nucleic acid aptamer having high specificity and having a binding ability 100 to 1000 times stronger than that of a conventional method for producing a nucleic acid aptamer, particularly a DNA aptamer, is efficiently produced. can do.
- Nucleic acid molecule The second embodiment of the present invention is a nucleic acid molecule that binds to a target substance.
- the nucleic acid molecule of the present invention contains one or more double-stranded regions in the molecule, and at least one of the double-stranded regions contains non-Watson-Crick base pairs.
- the nucleic acid molecule of the present invention is, in principle, a natural nucleic acid such as DNA, RNA, or a combination thereof, as described in the section “1. Definition” above. Moreover, the nucleic acid of the present invention may partially or entirely contain non-natural nucleotides or non-natural nucleic acids.
- a preferred nucleic acid form of the invention is DNA.
- Double-stranded region refers to a region in which base pairs are continuously formed between nucleotide strands constituting a nucleic acid molecule.
- the length of consecutive base pairs is 5 to 20 bases, 7 to 18 bases, 8 to 17 bases, or 10 to 15 bases.
- each double-stranded region is composed of the same or different base pairs.
- the length of each double-stranded region may be the same or different.
- each double-stranded region may be separated by a region that does not form base pairs with each other (for example, including a mismatch site, a gap, a bulge structure, or an internal loop structure), or may be continuous Good.
- non-Watson-Crick base pair refers to a base pair other than guanine and cytosine, adenine and thymine, or uracil in each base of guanine, adenine, cytosine, thymine, and uracil.
- a base pair consisting of guanine and adenine or thymine, or guanine and guanine (g-g), adenine and adenine (a-a), or the like is preferable.
- the non-Watson-Crick base pair may be contained in at least one of the double-stranded regions present in the nucleic acid molecule of the present invention.
- the non-Watson-Crick base pairs contained in one double-stranded nucleic acid region are 1 to 5 base pairs, 1 to 7 base pairs, 1 to 8 base pairs, or 1 to 10 base pairs.
- the position of the non-Watson-Crick base pair in one double-stranded nucleic acid region is not particularly limited.
- the double-stranded region containing non-Watson-Crick base pairs is a region directly involved in binding to the target substance. Therefore, the base sequence differs depending on the target substance. Based on the base sequence of a double-stranded region already known to bind to the target substance, any base sequence may be used as long as a non-Watson-Crick base pair is introduced.
- the nucleic acid molecule of the present invention that binds to a specific target substance has a base sequence of a double-stranded region that is expected to be a target substance binding site in a known decoy DNA, RNA aptamer, or DNA aptamer that binds to the target substance. Any non-Watson-Crick base pair may be introduced into both strands.
- the nucleic acid molecule of the present invention binds to a target substance.
- the “target substance” refers to a biological substance that can be a binding target of a nucleic acid molecule, as described in the section “1. Definition” above.
- the type of the target substance is not particularly limited, and examples thereof include peptides, nucleic acids, lipids, sugars, and low molecular compounds. Preferably it is a peptide, more preferably a polypeptide, ie a protein.
- a transcriptional regulatory factor a signal transducing factor, a protein ligand (including cytokines and chemokines), or a receptor protein that binds to a nucleic acid having a specific base sequence is preferable as a target substance of the nucleic acid molecule of the present invention.
- transcription regulatory factors include NF- ⁇ B, SP1, E2F, AP-1, and STAT-1.
- signal transducing factor examples include Raf, Cytohesin 1, Phospholipase A 2 and HER3.
- protein ligands include, for example, VEGF, EGF, NGF, HGF, KGF, bFGF, PDGF, IL-2, -3, -6, -8, -10, -20, IFN- ⁇ , - ⁇ , - ⁇ , TGF- ⁇ , BMP, Activin, TNF- ⁇ , Wnt, RANKL.
- the nucleic acid molecule of the present invention can inhibit, suppress or enhance the specific biological function of the target substance by binding to the target substance. Usually, it has a function inhibiting or suppressing action.
- the nucleic acid molecule of the present invention consists of a double-stranded nucleic acid and / or a single-stranded nucleic acid.
- each of the double-stranded nucleic acid and the single-stranded nucleic acid fragment will be specifically described.
- each nucleotide chain is not limited, but is preferably in the range of 5 to 50-mer, 7 to 40-mer, or 10 to 35-mer, for example.
- the length of each strand that is base paired need not be the same.
- one nucleotide chain may be 7-mer or longer than the other nucleotide chain.
- the long-side nucleotide chain may form a hairpin structure by intramolecular annealing in a single-stranded region that does not correspond to the other nucleotide chain.
- the stem region formed in the hairpin structure is also included in the double-stranded region of the present invention.
- each nucleotide chain constituting the double-stranded nucleic acid can include a single-stranded region that does not form base pair with each other on the 5 ′ side and / or 3 ′ side of the double-stranded region.
- the double-stranded nucleic acid also includes a dumbbell-shaped closed circular nucleic acid in which these single-stranded regions form a loop structure like a linker nucleic acid, and both nucleotide strands of the double-stranded nucleic acid are linked.
- dumbbell-type nucleic acid has a degradation resistance against a nucleolytic enzyme such as a nuclease compared to a linear double-stranded nucleic acid, and is preferable as the nucleic acid molecule of the present invention.
- one or both of the nucleotide strands constituting the double-stranded nucleic acid is the hairpin DNA described in International Application No. PCT / JP2011 / 059619 on the 5 ′ side and / or 3 ′ side, preferably 3 ′ side. It can also be included.
- this hairpin type DNA the three DNA nucleic acid regions of the first nucleic acid region, the second nucleic acid region, and the third nucleic acid region are linked in order from the 5 ′ end side to the 3 ′ end side, respectively. It has a structure.
- the “first nucleic acid region” is a nucleic acid region consisting of an arbitrary nucleotide of 2 to 5-mer.
- the base of this nucleic acid region may be guanine, adenine, cytosine or thymine, but is preferably guanine or cytosine. This is because when the stem structure is formed with the third nucleic acid region described later, the Tm value increases as the gc content increases, and the stem structure can be stably maintained. Therefore, it is most preferable that the entire base sequence of the first nucleic acid region is composed of g and / or c.
- the “second nucleic acid region” is a nucleic acid region consisting of a 5′-gna-3 ′ or 5′-gnna-3 ′ base sequence. Each n in the sequence independently consists of either a natural base (g, a, t, or c), a base analog, or a modified base.
- the “third nucleic acid region” is a nucleic acid region having a base sequence complementary to the first nucleic acid region. Therefore, the base sequence of the third nucleic acid region is determined by the base sequence of the first nucleic acid region, and the first nucleic acid region and the third nucleic acid region form a base pair in the molecule. As a result, the first nucleic acid region and the third nucleic acid region have a stem structure that is completely base-paired with each other, and the second nucleic acid region that exists between the first nucleic acid region and the third nucleic acid region has a loop structure. As a whole, for example, a hairpin DNA consisting of 7 to 14-mer nucleotides having the nucleotide sequence of SEQ ID NO: 37 or 38 is formed.
- the degradation resistance of the double-stranded nucleic acid to the nucleolytic enzyme is improved and stable in vivo. It becomes possible to improve the nature.
- the single-stranded nucleic acid constituting the nucleic acid molecule of the present invention forms a secondary structure by intramolecular annealing, and has one or more stem structures and one or more loop structures in the molecule.
- the double-stranded region is contained within this stem structure.
- the stem structure may include one or more mismatch sites and / or one or more bulge structures.
- the length of the nucleotide chain of the single-stranded nucleic acid is not particularly limited as long as it has a length that can include at least one double-stranded region in the molecule. For example, a range of 15 to 100-mer, 20 to 90-mer, or 30 to 80-mer is preferable.
- the single-stranded nucleic acid can also contain the above-mentioned hairpin DNA on the 5 'side and / or 3' side, preferably 3 'side.
- a suitable form includes a nucleic acid aptamer.
- DNA aptamers are particularly preferred from the standpoint of nucleic acid stability.
- DNA encoding double-stranded RNA or single-stranded RNA consists of double-stranded RNA or single-stranded RNA (for example, RNA aptamer).
- DNA encoding them can also be used.
- the base sequence of such DNA is, for example, a base sequence in which uracil (U) in the base sequence constituting double-stranded RNA or single-stranded RNA is replaced with thymine (T).
- RNA aptamer is used as a template, and all or part of the DNA is complementary to the 3 'terminal nucleotide sequence of the aptamer.
- It can be prepared by performing a reverse transcription reaction using a plumer.
- a technique known in the art may be used.
- it can be carried out according to the method described in Molecular Cloning described above.
- the DNA of the present invention can also be produced by chemical synthesis methods known in the art based on the base sequence information of double-stranded RNA and single-stranded RNA.
- the DNA encoding the double-stranded RNA or single-stranded RNA may be inserted into an expression vector in a state where it can be expressed.
- “Expressable state” means that DNA encoding a double-stranded RNA or a single-stranded RNA is linked downstream of a promoter in an expression vector so that the corresponding RNA can be expressed.
- a plasmid or virus capable of autonomously growing in a host can be used. Examples of such plasmids include pET, pGEX6p, pMAL, pREST, etc. when the host is Escherichia coli, pUB110, pTP5, etc.
- the host when the host is Bacillus subtilis, and the host is yeast. In this case, YEp13, YEp24, YCp50, etc., and when the host is a plant, pBI, pRI, or pGW binary vectors can be mentioned.
- the virus when the host is E. coli, ⁇ phage ( ⁇ gt11, ⁇ ZAP, etc.), when the host is a mammal, retrovirus, adenovirus, adeno-associated virus, vaccinia virus, etc., when the host is an insect, When the host is a plant such as baculovirus, cauliflower mosaic virus (CaMV), kidney bean golden mosaic virus (BGMV), tobacco mosaic virus (TMV) and the like can be mentioned.
- CaMV cauliflower mosaic virus
- BGMV kidney bean golden mosaic virus
- TMV tobacco mosaic virus
- nucleic Acid Molecules with NF- ⁇ B as Target Substances The nucleic acid molecules of the present invention with NF- ⁇ B as a target substance are described below with specific examples.
- NF- ⁇ B is one of the transcription factors that play a major role in the immune response, and is involved in inflammatory reactions, cell proliferation, apoptosis, etc., so it has attracted attention as a drug discovery target and can inhibit its function.
- Molecular development is taking place around the world. For example, decoy DNA containing a double-stranded DNA fragment having a common sequence to which NF- ⁇ B binds (WO / 1996/035430; Miyake T., et al., Mol. Ther., 2001, 19: 181-187; Kim KH, et al., Exp. Mol. Pathol., 2008, 86: 114-120; Isomura I., A. Morita, Microbiol.
- RNA aptamers Lebruska LL and Maher III LJ, Biochemistry, 1999, 38: 3168-3174; NJ Reiter, LJ Maher III, SE Butcher, Nucleic Acids Res., 2008, 36: 1227-1236; Chan R., et al., Nucleic Acids Res., 2006, 34, e36; SE Wurster, LJ Maher III, RNA, 2008, 14: 1037-1047).
- both of these nucleic acid molecules had a binding ability (dissociation constant, Kd) to NF- ⁇ B of about several nM.
- the nucleic acid molecule of the present invention has a binding ability to NF- ⁇ B on the order of pM, and has a binding ability 100 times higher than that of known nucleic acid molecules.
- the nucleic acid molecule of the present invention that targets p50 of NF- ⁇ B as a target substance includes a double-stranded region consisting of the base sequence represented by SEQ ID NOs: 1 and 2 shown in FIG. 3 (a) as a consensus sequence. .
- the base pair described as “W-W” indicates “a-t” or “t-a”.
- ” between the bases of the double-stranded region is Watson-Crick base pairing, “ ⁇ ” or “ ⁇ ” is “ag” or “ga”, or “gt” or “tg”, respectively.
- the nucleic acid molecule of the present invention targeting p50 of NF- ⁇ B contains one or more double-stranded regions comprising the above consensus sequence
- double-stranded nucleic acid and / or single-stranded nucleic acid Either form may be sufficient.
- the hairpin DNA may be contained on the 5 ′ side and / or 3 ′ side, preferably 3 ′ side of the consensus sequence.
- a DNA aptamer is a suitable example.
- these DNA aptamers have a binding ability 100 to 1000 times or more stronger than conventionally known RNA aptamers and DNA aptamers using NF- ⁇ B p50 as a target substance.
- at least one double-stranded region contained in the nucleic acid molecule has a non-Watson-Crick base pair, so that the binding ability of a conventional nucleic acid aptamer to a target substance can be increased. It can be improved dramatically.
- Target substance function inhibitor The third embodiment of the present invention is a target substance function inhibitor.
- the target substance function inhibitor of the present invention contains the nucleic acid molecule of Embodiment 2 as an active ingredient.
- target substance function inhibition refers to biological functions such as catalytic function or gene expression control function (including control of transcription, translation, transport, etc.) and apoptosis control function possessed by the target substance. Inhibiting or suppressing by binding of a nucleic acid molecule as an active ingredient.
- the content of the nucleic acid molecule in the target substance function inhibitor of the present invention may be a pharmaceutically effective amount.
- the “pharmaceutically effective amount” means a dose necessary for the nucleic acid molecule of the second embodiment, which is an active ingredient of a target substance function inhibitor, to exhibit target substance function inhibition, which is its medicinal effect, In addition, it refers to a dose that has little or no harmful side effects on the living body or the like to be administered.
- the specific amount depends on the type of the target substance, the inhibitory activity of the nucleic acid molecule, the dosage form to be used, and, for the purpose of administration to the living body, information on the living body that is the subject and the administration route. Different.
- a pharmaceutically effective amount range and a suitable route of administration are generally formulated based on data obtained from cell culture assays and animal experiments.
- the final dose is determined and adjusted according to the judgment of the doctor according to the individual subject.
- the information of the subject to be considered includes the degree or severity of the disease, the general health condition, age, weight, sex, diet, drug sensitivity, resistance to treatment, and the like.
- the content of the nucleic acid molecule of the present invention per dosage unit of the target substance function inhibitor is, for example, the above-described NF- ⁇ B p50 for a human adult male (body weight 60 kg) that does not require the use of other drugs.
- the nucleic acid molecule of the present invention having a target substance as a target substance is administered by injection, about 0.01% (w / v) to about 20% (w / v), preferably about 0.1% ( W / v) to about 10% (w / v) may be included.
- the nucleic acid can be divided into several times to reduce the burden on the subject.
- composition A fourth embodiment of the present invention is a pharmaceutical composition.
- the pharmaceutical composition of the present invention contains at least one target substance function inhibitor described in the third embodiment.
- the pharmaceutical composition of the present invention can also contain a pharmaceutically acceptable carrier.
- “Pharmaceutically acceptable carrier” refers to its action in order to facilitate the formulation of a pharmaceutical composition ordinarily used in the field of pharmaceutical technology and application to a living body, and to maintain the effect of the target substance function inhibitor. A substance added within a range not inhibiting or suppressing.
- Carriers include, for example, excipients, binders, disintegrants, fillers, emulsifiers, flow control agents, lubricants or surfactants.
- excipient examples include sugars such as monosaccharides, disaccharides, cyclodextrins and polysaccharides (specifically, but not limited to, glucose, sucrose, lactose, raffinose, mannitol, sorbitol, inositol, Including dextrin, maltodextrin, starch and cellulose), metal salts (eg, sodium phosphate or calcium phosphate, calcium sulfate, magnesium sulfate), citric acid, tartaric acid, glycine, low, medium, high molecular weight polyethylene glycol (PEG), Pluronics, or combinations thereof.
- sugars such as monosaccharides, disaccharides, cyclodextrins and polysaccharides (specifically, but not limited to, glucose, sucrose, lactose, raffinose, mannitol, sorbitol, inositol, Including dextrin, mal
- binder examples include starch paste, corn, wheat, rice, or potato starch, gelatin, tragacanth, methylcellulose, hydroxypropylmethylcellulose, sodium carboxymethylcellulose, and / or polyvinylpyrrolidone.
- disintegrant examples include the starch, carboxymethyl starch, cross-linked polyvinyl pyrrolidone, agar, alginic acid, sodium alginate or salts thereof.
- filler examples include the sugar and / or calcium phosphate (for example, tricalcium phosphate or calcium hydrogen phosphate).
- emulsifier examples include sorbitan fatty acid ester, glycerin fatty acid ester, sucrose fatty acid ester, and propylene glycol fatty acid ester.
- Examples of the “flow additive modifier” and “lubricant” include silicate, talc, stearate or polyethylene glycol.
- the drug composition of the present invention comprises a flavoring agent, a solubilizing agent (solubilizing agent), a suspending agent, a diluent, a surfactant, a stabilizer, an absorption accelerator (if necessary)
- a flavoring agent for example, quaternary ammonium salts, sodium lauryl sulfate, etc., extenders, moisturizers, humectants (eg, glycerin, starch, etc.), adsorbents (eg, starch, lactose, kaolin, bentonite, colloidal silicic acid, etc.
- Disintegration inhibitors eg, sucrose, stearin, cocoa butter, hydrogenated oil, etc.
- coating agents colorants, preservatives, antioxidants, fragrances, flavoring agents, sweetening agents, buffering agents, and the like.
- surfactant examples include lignosulfonic acid, naphthalene sulfonic acid, phenol sulfonic acid, alkali metal salt, alkaline earth metal salt and ammonium salt of dibutyl naphthalene sulfonic acid, alkyl aryl sulfonate, alkyl sulfate, alkyl sulfonate.
- the pharmaceutical composition of the present embodiment can include one or more of the above carriers.
- the pharmaceutical composition of the present invention can also contain other drugs as long as the pharmacological effect of the nucleic acid of the present invention is not lost.
- a predetermined amount of antibiotics may be contained.
- the dosage form of the pharmaceutical composition of the present invention is not particularly limited as long as it is a form that does not inactivate the active ingredient and can exhibit its pharmacological effect in vivo after administration. Usually, it varies depending on the administration method and / or prescription conditions.
- dosage forms suitable for oral administration include solid preparations (including tablets, pills, sublinguals, capsules, drops, troches), granules, powders, powders, liquids, and the like.
- the solid preparation can be made into a dosage form known in the art, for example, a sugar-coated tablet, a gelatin-encapsulated tablet, an enteric tablet, a film-coated tablet, a double tablet, or a multilayer tablet, if necessary. .
- Parenteral administration is subdivided into systemic administration and local administration, and local administration is further subdivided into intra-tissue administration, transepidermal administration, transmucosal administration, and rectal administration.
- the dosage form can be made suitable for.
- dosage forms suitable for systemic or intra-tissue administration include injections that are liquids.
- Suitable dosage forms for transepidermal or transmucosal administration include, for example, liquids (including coating agents, eye drops, nasal drops, and inhalants), suspensions (including emulsions and creams), and powders (points). Nasal and suction agents), pastes, gels, ointments, plasters and the like.
- Examples of dosage forms suitable for rectal administration include suppositories.
- the dosage form of the pharmaceutical composition may be liquid, solid (including semi-solid), or a combination thereof. Solutions, oily dispersions, emulsions, suspensions, powders, powders, pastes, gels, pellets, tablets and granules can be used.
- each dosage form are not particularly limited as long as the dosage form is within the range of dosage forms known in the art for each dosage form.
- the nucleic acid molecule of the second embodiment is dissolved in a pharmaceutically acceptable solvent, a pharmaceutically acceptable carrier is added as necessary, and a method commonly used in the art is used. Can be manufactured.
- Examples of the “pharmaceutically acceptable solvent” include water, ethanol, propylene glycol, ethoxylated isostearyl alcohol, polyoxylated isostearyl alcohol, polyoxyethylene sorbitan fatty acid esters and the like. These are preferably sterilized and preferably adjusted to be isotonic with blood as necessary.
- the pharmaceutical composition of the present embodiment can be administered to a living body in a pharmaceutically effective amount for treatment or prevention of a target disease or the like.
- the living body to be administered is a vertebrate, preferably a mammal, more preferably a human.
- the pharmaceutical composition of the present invention may be either systemic administration or local administration. It can select suitably according to the kind of disease, onset location, or a progression degree. If the onset site is a local disease, local administration directly administered to and around the onset site by injection or the like is preferable. This is because a sufficient amount of the nucleic acid molecule of the present invention can be administered to a site (tissue or organ) to be treated, and other tissues are hardly affected. On the other hand, when the treatment site cannot be specified or the onset is a systemic disease, systemic administration by intravenous injection or the like is preferable, although there is no limitation. This is because the nucleic acid molecule of the present invention is distributed throughout the body through the bloodstream, so that it can be administered even to a lesion that cannot be found by diagnosis.
- the pharmaceutical composition of the present invention can be administered by any appropriate method that does not deactivate the active ingredient.
- it may be parenteral (for example, injection, aerosol, application, eye drop, nose drop) or oral.
- parenteral for example, injection, aerosol, application, eye drop, nose drop
- oral Preferably, it is an injection.
- the injection site is not particularly limited. Any site may be used as long as the nucleic acid molecule as an active ingredient can bind to the target substance and suppress its function.
- Intravenous injection such as intravenous injection or intraarterial injection is preferable. This is because the pharmaceutical composition of the present invention can be distributed throughout the body through the blood stream as described above, and is less invasive.
- Target Substance Detection Method is a method for detecting a target substance using the nucleic acid molecule described in the second embodiment.
- the nucleic acid molecule described in the second embodiment can bind to the target substance of the nucleic acid molecule very firmly and specifically, the target substance present in the sample is detected using the property of the nucleic acid molecule. can do.
- a known detection method may be used as the detection method itself.
- SPR method quartz crystal microbalance method, turbidimetric method, colorimetric method, or fluorescent method can be used.
- SPR surface plasmon resonance
- the SPR method is a measurement method that utilizes this phenomenon, and can measure the adsorbate on the surface of the metal thin film that is the sensor portion with high sensitivity.
- the nucleic acid molecule of the second embodiment is immobilized on the surface of the metal thin film in advance, and the sample passes through the surface of the metal thin film, and the sample passes through the binding between the nucleic acid molecule and the target substance.
- the target substance in the sample can be detected by detecting the difference between adsorbates on the front and back metal surfaces.
- a substitution method, an indirect competition method, and the like are known, and any of them may be used.
- the QCM (Quartz Crystal Microbalance) method uses a phenomenon in which when a substance is adsorbed on the surface of an electrode attached to a crystal resonator, the resonance frequency of the crystal resonator decreases according to its mass. .
- a QCM sensor using this method can quantitatively capture an extremely small amount of adsorbate based on the amount of change in the water resonance frequency.
- the amount of change in the water resonance frequency caused by the binding between the nucleic acid molecule and the target substance by immobilizing the nucleic acid molecule in advance on the electrode surface in the same manner as in the SPR method and bringing the sample into contact with the electrode surface.
- the target substance in the sample can be detected quantitatively.
- This technique is well known in the art. For example, see Christopher J., et al. (2005) Self-Assembled Monolayers of a Form of Nanotechnology, Chemical Review, 105: 1103-1169.
- the turbidimetric method irradiates a solution with light and optically measures the attenuation of scattered light scattered by a substance suspended in the solution or transmitted light that has passed through the solution using a colorimeter or the like. It is a method of measuring the amount of substance in it.
- the target substance in the sample can be quantitatively detected by measuring the absorbance before and after adding the nucleic acid molecule of the second embodiment to the sample.
- the target substance can be detected by using it together with an antibody against the target substance.
- a method using an ELISA sandwich method may be used. In this method, first, the nucleic acid molecule of the second embodiment is immobilized on a solid phase carrier, and then a sample is added to bind the target substance present in the sample and the nucleic acid molecule. Subsequently, after washing the sample, an anti-target substance antibody is added to bind to the target substance. After washing, the target substance in the sample can be detected by detecting the anti-target substance antibody using a secondary antibody with an appropriate label.
- beads made of materials such as polystyrene, polycarbonate, polyvinyltoluene, polypropylene, polyethylene, polyvinyl chloride, nylon, polymethacrylate, latex, gelatin, agarose, cellulose, sepharose, glass, metal, ceramics or magnetic materials
- Insoluble carriers in the form of microplates, test tubes, sticks or test pieces can be used.
- the nucleic acid molecule is represented by SEQ ID NOS: 1 and 2 shown in FIG. 3A as the consensus sequence described in the second embodiment.
- a nucleic acid molecule comprising a double-stranded region consisting of a base sequence.
- the base sequences shown by SEQ ID NOs: 3 and 4 shown in FIG. 3 (b) the base sequences shown by SEQ ID NOs: 5 and 6 shown in FIG. 3 (c), and SEQ ID NOs: 7 and 7 shown in FIG.
- the hairpin described in the second embodiment is more specifically, for example, on the 5 ′ side and / or 3 ′ side, preferably 3 ′ side of the consensus sequence. 4 or a DNA aptamer containing the central region represented by SEQ ID NOs: 9 to 21 in FIG. 4, more specifically, for example, clones 5R01, 5R09, 5R43, 5R14, 5R13, 5R34, 5R10, 5R26, 5R27, 5R11, 5R05, 5R28 and 5R19.
- the sample may be used after treating the sample with a nuclease inhibitor before measurement.
- NF- ⁇ B p50 detection kit The sixth embodiment of the present invention is a kit for detecting NF- ⁇ B p50 containing one or more NF- ⁇ B p50-binding nucleic acid molecules of the second embodiment. This kit can detect NF- ⁇ B p50 in a sample using the NF- ⁇ B p50-binding nucleic acid according to the above embodiment.
- this kit is used for diluting and washing a labeled secondary antibody, a substrate necessary for detection of a label, a positive control and a negative control, or a sample as necessary.
- the buffer solution to be used can be included.
- instructions for using the kit can be included.
- Example 1 Development of highly efficient production method of nucleic acid aptamer> I. Examination of conditions for solid phase carrier In the method for producing the nucleic acid aptamer of the present invention, the optimum conditions for the solid phase carrier were examined in order to reduce non-specific adsorption of the single-stranded nucleic acid to the solid phase carrier used.
- Magnetic beads A (NewEngland Biolabs, Hydrophilic Streptavidin Magentic Beads) have a hydrophilic bead surface
- magnetic beads B (NewEngland Biolabs, Streptavidin Magnetic Beads) have a hydrophobic bead surface.
- These magnetic beads were used to verify nonspecific adsorption of single-stranded DNA on the magnetic beads.
- the amount of DNA adsorbed non-specifically to the magnetic beads was detected by real-time PCR.
- a reverse primer represented by SEQ ID NO: 23 and a forward primer represented by SEQ ID NO: 24 were used.
- the reverse primer complementary to the single-stranded DNA constituting the library is biotinylated at the 5 'end thymine (t). Therefore, the PCR amplified product was mixed with streptavidin and then subjected to denaturing gel electrophoresis, whereby the unreacted reverse primer and the complementary sequence of the single-stranded DNA shown in sequence 22 in which the 5 'end was biotinylated Since it binds to streptavidin and has a low mobility, the target single-stranded DNA amplification product can be selectively prepared by the gel shift method.
- PCR reaction conditions were as follows: in the presence of SYBR® Green® I (Lonza®) and ROX® Dye (Life Technologies), 95 ° C for 30 seconds, 50 ° C for 30 seconds, and 65 ° C for 3 minutes in 2 steps.
- the amplification process was detected by Mx3005P (Agilent Technologies).
- the target substance is NF- ⁇ B p50, which is a transcription factor, and a DNA aptamer that binds strongly thereto is prepared. It was.
- magnetic beads A Hydrophilic Streptavidin Beads, NewngEngland Biolabs
- streptavidin magnetic beads Hydrophilic Streptavidin Beads, New England Biolabs. Incubate at room temperature for 30 minutes, remove the magnetic beads by a magnetic stand and centrifugation, and collect the supernatant.
- the target protein recombinant human NF- ⁇ B p50 (rhNF- ⁇ B, Promega) was mixed at the ratio shown in Table 1, and incubated at 25 ° C. for 30 minutes, whereby single-stranded DNA and NF- ⁇ B p50 were mixed.
- the complex of was formed.
- the single-stranded nucleic acid library for complex formation, the concentration ratio of NF- ⁇ B p50, the volume of the reaction solution used in this step, and the like were adjusted for each round. Details are described in the repetitive step (106) described later.
- Biotin and streptavidin were used as binders.
- Biotin is the first binder adsorbed on the target substance, and streptavidin corresponds to the second binder adsorbed on the magnetic beads.
- NF- ⁇ B p50 Since the used NF- ⁇ B p50 is not biotinylated, first, 0.09 volume of 10 mM biotinylation reagent (EZ-link Sulfo-NHS-LC-Biotin, Thermo Scientific) was added and incubated at 25 ° C. for 15 minutes. As a result, NF- ⁇ B50p50 contained in the complex in the solution and free NF- ⁇ B p50 not formed of the complex were biotinylated.
- biotinylation reagent EZ-link Sulfo-NHS-LC-Biotin, Thermo Scientific
- the unreacted biotinylation reagent was removed by a washing operation by ultrafiltration using Microcon 50 (Merck).
- the washed solution containing the biotinylated complex is mixed with streptavidin magnetic beads (Hydrophilic Streptavidin Beads, New England Biolabs) (6.8 to 8 ⁇ g per pmol in terms of p50 monomer) and incubated at room temperature for 10 minutes, and biotin-strept
- streptavidin magnetic beads Hydrophilic Streptavidin Beads, New England Biolabs
- the magnetic beads are washed with 40 ⁇ mL of PBS buffer containing 0.05% Nonidet P-40 and 2.5 mM DTT (hereinafter referred to as ⁇ Buffer A ”) and incubated at 37 ° C. with stirring for 30 minutes. This washing operation was repeated once more.
- PCR a reverse primer represented by SEQ ID NO: 26 and a forward primer represented by SEQ ID NO: 27 were used.
- the reverse primer is biotinylated at the 5 'end thymine (t).
- PCR was performed using ExTaq DNA polymerase (Takara Bio) at a final concentration of 0.025 U / ⁇ L and an attached buffer.
- Double-stranded DNA was recovered from the PCR solution containing the amplified product after the amplification step (104) by ethanol precipitation, and then single-stranded DNA was prepared by gel shift method using streptavidin. Specifically, the recovered double-stranded DNA (0.4 mL of PCR solution) is suspended in 10 ⁇ L of SA buffer (10 mM Tris-HCl pH 7.6, 50 mM NaCl, 1 mM EDTA) at 75 ° C. After heating for 3 minutes, 10 ⁇ L of streptavidin solution (5 mg / mL, dissolved in SA buffer) was added and incubated at 25 ° C. for 30 minutes.
- SA buffer 10 mM Tris-HCl pH 7.6, 50 mM NaCl, 1 mM EDTA
- the obtained single-stranded DNA was dissolved in PBS buffer solution (1.1 mM KH 2 PO 4 , 155 mM NaCl, 3 mM Na 2 HPO 4 , pH 7.4), heated at 90 ° C. for 3 minutes, and then at 60 ° C. for 3 minutes. The mixture was cooled and then heated and cooled at 25 ° C. to form a higher order structure in the single-stranded DNA molecule. Thereafter, this solution was mixed with PBS buffer containing 0.1% Nonidet P-40 and 5 mM DTT in an equal amount, and used as a new single-stranded nucleic acid library in the next round.
- PBS buffer solution 1.1 mM KH 2 PO 4 , 155 mM NaCl, 3 mM Na 2 HPO 4 , pH 7.4
- a single-stranded nucleic acid library consisting of single-stranded DNA prepared by chemical synthesis and gel purification as described above was directly used.
- the total number of molecular species of single-stranded DNA in this round was 333 pmol. This corresponds to about 2 ⁇ 10 13 molecules of single-stranded DNA.
- the concentrations of the single-stranded nucleic acid library and NF- ⁇ B p50 were gradually lowered in order to tighten the conditions for complex formation of single-stranded DNA-NF- ⁇ B p50.
- NF- ⁇ B mini46 46-mer
- SEQ ID NO: 28 An amount was added.
- 40 mL of buffer A was used, and washing was performed twice at 37 ° C. for 30 minutes.
- washing was further performed by inverting and mixing for 15 minutes at room temperature with 1 mL of buffer with 3 M urea added to buffer A. As a result, washing conditions were tightened, and single-stranded DNA fragments that strongly bind to NF- ⁇ B p50 were selected.
- FIG. 4 shows the nucleotide sequence, clone name, clone number, and SEQ ID NO of the obtained NF- ⁇ B p50-binding DNA aptamer.
- the base region of the central region is roughly divided into three sequence groups, that is, a sequence group having a base sequence represented by SEQ ID NOs: 9 to 11, a sequence group having a base sequence represented by SEQ ID NOs: 12 to 18, And it was classified into a sequence group having the base sequence shown in SEQ ID NO: 19-21.
- Each sequence had a sequence similar to the natural consensus DNA sequence to which NF- ⁇ B binds as shown in SEQ ID NO: 29. Sequences similar to those consensus were found to be located in the stem portion of the hairpin structure, as shown in FIG. Interestingly, it was revealed that the sequence of the stem portion contained non-Watson-Crick base pairs of GA and GT, unlike the natural DNA sequence.
- Example 2 Binding ability of DNA aptamer binding to NF- ⁇ B> Among the NF- ⁇ B p50-binding DNA aptamer clones obtained in Example 1, 5R01, 5R14, and 5R05 (see FIG. 4), which had a large number of clones in each sequence group, were used to bind to NF- ⁇ B p50. Was analyzed.
- Example 1 Single-stranded DNA (full length 95-mer) of each clone was PCR-amplified using the clone plasmid obtained in Example 1 as a template and the biotinylated primers of Sequence 26 and Sequence 27, and then the Example 1 was prepared by the gel shift method using streptavidin in the same procedure as the single-stranded nucleic acid preparation step shown in 1.
- the binding ability of each NF- ⁇ Bp50 binding DNA aptamer clone to NF- ⁇ Bp50 was measured by the surface plasmon resonance (SPR) method using BIACORE3000 (GE Healthcare).
- SPR surface plasmon resonance
- buffer A was used as a running buffer, and measurement was performed at a set temperature of 25 ° C.
- the sensor chip (SA chip) coated with streptavidin has a base sequence complementary to the 3 ′ end region of each clone, and the base at the 5 ′ end is represented by SEQ ID NO: 30
- Detection of binding / dissociation between immobilized clonal DNA and NF- ⁇ B50p50 is achieved by injecting 2.5 nM or 5 nM NF- ⁇ B p50 solution (diluted with buffer A and converted to dimer) using Kinetic Injection mode. Monitored.
- the measurement conditions were a flow rate of 20 ⁇ L / min, a protein injection of 6 minutes, and a protein dissociation measurement after completion of the injection of 6 minutes.
- Regeneration of the chip was performed by injecting 5 ⁇ L (corresponding to 15 seconds) of 2 ⁇ M NaCl solution.
- the DNA immobilized by hybridization can be removed by injection of 0.05 ⁇ M NaOH solution at a flow rate of 25 ⁇ L / min by 5 ⁇ L (corresponding to 12 seconds), and the same chip is used for binding analysis with different DNA sequences. I was able to use it.
- FIG. This figure is a sensorgram in which the interaction between the above three clones and NF- ⁇ B p50 is detected.
- both clones bind very strongly to NF- ⁇ B p50, and from the pattern of the sensorgram after completion of NF- ⁇ B p50 injection, dissociation of the bound clone from NF- ⁇ B p50 was found to be extremely slow.
- Example 3 Binding ability of DNA aptamer mutant binding to NF- ⁇ B p50> NF- ⁇ B p50 binding DNA aptamer clones 5R01, 5R14, and 5R05 that were verified in Example 2 were analyzed for their ability to bind to NF- ⁇ B p50.
- the immobilization of various single-stranded DNAs on the SA chip by hybridization to the probe was performed at 20 ⁇ L / min at a flow rate of 20 ⁇ L / min with various DNA fragment (68-mer) solutions diluted to 250 nM with PBS. It was performed by injecting ⁇ L (equivalent to 1 minute).
- FIG. 9 shows the result. This figure is a sensorgram of SPR in which the interaction between Cont-68 and NF- ⁇ B p50 for the above three clones and control was detected.
- Example 4 Binding ability of NF- ⁇ B p50-binding DNA aptamer 5R01-68 mutant to NF- ⁇ B p50>
- the common structure of each clone of the NF- ⁇ B p50-binding DNA aptamer obtained in Example 1 includes non-Watson-Crick base pairs that are different from ordinary double-stranded DNA (FIG. 4). .
- FIG. This figure shows SPR sensorgrams when mutant 5R01mut-68 (A), control 5R01-68 (B) and Cont-68 (C) were detected for interaction with NF- ⁇ B p50. It is. Mutant 5R01mut-68 was more than 20 times more potent than Cont-68, but the dissociation rate constant was 10 times greater than 5R01-68, and 5R01-mut with non-Watson-Crick base pairs. It was found that it was easier to dissociate from NF- ⁇ B p50 than 68.
- Example 5 Binding specificity of NF- ⁇ B p50-binding DNA aptamer to NF- ⁇ B p50> The NF- ⁇ B p50 binding selectivity of the NF- ⁇ B p50 binding DNA aptamer obtained in Example 1 was examined.
- a 59-mer DNA fragment Taq-59 (SEQ ID NO: 36) incorporating a known anti-Taq DNA aptamer was prepared by chemical synthesis and applied to various proteins in the same manner as 5R01-68 and 5R01mut-68. The binding was verified by SPR method.
- FIG. This figure shows NF- ⁇ B p50 ( ⁇ ), Taq DNA polymerase ( ⁇ ), and AP-1 ( ⁇ ) for 5R01-68 (A), 5R01mut-68 (B), and Taq-59 for control. It is a sensorgram of SPR when the interaction with is detected. From this figure, the binding ability of 5R01-68 and 5R01mut-68 to AP-1 and Taq DNA polymerase is very weak, and the DNA aptamer obtained by the production method of the present invention is specific to the target substance NF- ⁇ B p50. It became clear that they could be combined.
- Table 2 summarizes the dissociation constants (Kd values) for NF- ⁇ B p50 calculated from the results of the SPR method for DNA aptamers and the like verified in the above examples.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Medicinal Chemistry (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Analytical Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Tropical Medicine & Parasitology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
Abstract
Description
本明細書で使用する一般的な用語の定義について以下で説明する。
2-1.概要
本発明の第1の実施形態は、核酸アプタマーの製造方法である。本発明によれば、一本鎖核酸の非特異的吸着によるバックグラウンドを低減し、標的物質に対して特異性の高い核酸アプタマー、特にDNAアプタマーを効率的に、かつ簡便に製造することができる。
本発明の工程フローを図1に示す。この図が示すように、本発明の製造方法は、複合体形成工程(101)、固定化工程(102)、回収工程(103)、増幅工程(104)及び一本鎖核酸調製工程(105)を必須の工程として含む。また、本発明の製造方法は、必要に応じて反復工程(106)及び/又は選択工程(107)を任意の工程として含むことができる。このうち、選択工程(107)は、増幅工程(104)と一本鎖核酸調製工程(105)間、及び/又は反復工程(106)後に行うことができる。以下、それぞれの工程について、具体的に説明をする。
「複合体形成工程」(101)とは、一本鎖核酸ライブラリーと標的物質とを溶液中で混合し、一本鎖核酸と標的物質の複合体を形成させる工程である。
「固定化工程」(102)とは、前工程後の溶液と固相担体を混合して、複合体を固相担体に固定化する工程である。
「回収工程」(103)とは、前記固相担体に固定化された複合体を溶液から回収する工程である。
「増幅工程」(104)とは、複合体中の一本鎖核酸を回収後、核酸増幅法によって増幅させる工程である。
「一本鎖核酸調製工程」(105)とは、増幅工程(104)で得られた二本鎖核酸を一本鎖化する工程である。
「反復工程」(106)とは、複合体形成工程(101)から一本鎖核酸調製工程(105)まで(本明細書では、以降、これらの一連の工程を「ラウンド」と称する)を複数回繰り返す工程である。
「選択工程」(107)とは、反復工程(106)後に得られる一本鎖核酸から、所定の構造を有する一本鎖核酸分子を選択する工程である。本工程は、任意の工程であり、後述するように、標的物質とより強固に結合する核酸アプタマーを製造することを目的とする場合に選択的に行うことができる。
本発明の製造方法によれば、従来法の核酸アプタマー、特にDNAアプタマーの製造方法と比較して、特異性が高く、かつ100~1000倍以上強い結合能を有する核酸アプタマーを効率的に製造することができる。
本発明の第2の実施形態は、標的物質に結合する核酸分子である。
本発明の核酸分子は、その分子内に、二本鎖領域を一以上含み、かつ前記二本鎖領域の少なくとも一つが非ワトソン・クリック型塩基対を含むことを特徴とする。
二本鎖核酸からなる場合、各ヌクレオチド鎖の長さは、限定はしないが、例えば、5~50-mer、7~40-mer、又は10~35-merの範囲が好ましい。塩基対合する各鎖の長さは、同じである必要はない。例えば、一方のヌクレオチド鎖が他方のヌクレオチド鎖に対して7-mer以上長い場合が挙げられる。この場合、長い側のヌクレオチド鎖は、他方のヌクレオチド鎖と対応しない一本鎖領域内で分子内アニールによってヘアピン構造を形成してもよい。このヘアピン構造内に形成されたステム領域も本発明の二本鎖領域に包含される。
本発明の核酸分子を構成する一本鎖核酸は、分子内アニールによって二次構造を形成し、分子内に一以上のステム構造及び一以上のループ構造を有する。前記二本鎖領域は、このステム構造内に含まれる。さらに、ステム構造は、一以上のミスマッチ部位及び/又は一以上のバルジ構造を含んでいてもよい。
本発明の核酸分子が二本鎖RNA又は一本鎖RNA(例えば、RNAアプタマー)からなる場合、それらをコードするDNAも利用することができる。そのようなDNAの塩基配列は、例えば、二本鎖RNA又は一本鎖RNAを構成する塩基配列中のウラシル(U)をチミン(T)に置換した塩基配列となる。
以下にNF-κBを標的物質とする本発明の核酸分子を具体例を挙げて説明する。
これらのDNAアプタマーは、下記実施例で示すように、従来知られていたNF-κB p50を標的物質とするRNAアプタマーやDNAアプタマーよりも100~1000倍以上強い結合能を有する。このように、本発明の核酸分子によれば、核酸分子内に含まれる少なくとも一つの二本鎖領域が非ワトソン・クリック型塩基対を有することで、従来の核酸アプタマーの標的物質に対する結合能を飛躍的に向上させることができる。
本発明の第3の実施形態は、標的物質の機能阻害剤である。
本発明の標的物質機能阻害剤は、前記実施形態2の核酸分子を有効成分とする。
本発明の第4の実施形態は、医薬組成物である。
本発明の医薬組成物は、前記実施形態3に記載の標的物質機能抑制剤を少なくとも一つ含有する。また、本発明の医薬組成物は、製薬上許容可能な担体を含むことができる。「製薬上許容可能な担体」とは、製剤技術分野において通常使用する医薬組成物の製剤化や生体への適用を容易にし、前記標的物質機能抑制剤の効果を維持するために、その作用を阻害又は抑制しない範囲で添加される物質をいう。担体には、例えば、賦形剤、結合剤、崩壊剤、充填剤、乳化剤、流動添加調節剤、潤滑沢剤又は界面活性剤が挙げられる。
本発明の医薬組成物を製造するには、原則として当該分野で公知の製剤化方法を応用すればよい。例えば、Remington’s Pharmaceutical Sciences(Merck Publishing Co.,Easton,Pa.)に記載の方法を参照することができる。
本実施形態の医薬組成物は、目的とする疾患等の治療又は予防のために製薬上有効な量を生体に投与することができる。投与する対象となる生体は、脊椎動物、好ましくは哺乳動物、より好ましくはヒトである。
本発明の第5の実施形態は、前記第2実施形態に記載の核酸分子を用いた標的物質の検出方法である。
第2実施形態に記載の核酸分子は、その核酸分子の標的物質と極めて強固に、かつ特異的に結合し得るため、核酸分子のその性質を利用して試料中に存在する標的物質を検出することができる。
本発明の検出方法によれば、第2実施形態の核酸分子の標的物質への強固かつ特異的な結合を利用して、試料中の微量な標的物質を高感度に検出することが可能となる。
本発明の第6の実施形態は、第2実施形態のNF-κB p50結合性核酸分子を一以上含むNF-κB p50を検出するためのキットである。本キットは、前記態様であるNF-κB p50結合性核酸を使用して試料中のNF-κB p50を検出することができる。
I.固相担体の条件検討
本発明の核酸アプタマーの製造方法において、使用する固相担体への一本鎖核酸の非特異的吸着を低減させるために、固相担体の最適条件を検討した。
固相担体には、メーカー市販のストレプトアビジンでコートされた2種類の磁気ビーズを用いた。磁気ビーズA(NewEnglandBiolabs社、Hydrophilic Streptavidin Magentic Beads)は、ビーズ表面が親水性であり、磁気ビーズB(NewEnglandBiolabs社、Streptavidin Magnetic Beads)は、ビーズ表面が疎水性である。
結果を図5に示す。この図が示すように、疎水性表面を有する磁気ビーズBは、親水性表面を有する磁気ビーズAよりもより多くの増幅産物が得られた。この結果は、親水性表面を有する固相担体の方が疎水性表面を有する固相担体よりも一本鎖核酸の非特異的吸着が少なく、本発明の製造方法における固相担体に適していることが示された。
次に、本発明の核酸アプタマー製造方法を用いて、標的物質を転写因子であるNF-κB p50とし、それに強結合するDNAアプタマーの作製を行った。
化学合成した配列番号25で示す、43塩基からなるランダムな塩基配列の中央領域(Nで示す)を含む一本鎖DNAからなる一本鎖核酸ライブラリー(全長95-mer)を用いて、複合体形成工程(101)から一本鎖核酸調製工程(105)を1ラウンドとして、後述の反復工程(106)を行った。
前記一本鎖核酸ライブラリーは、PBS緩衝液(1.1 mM KH2PO4、155 mM NaCl、3 mM Na2HPO4、pH 7.4)に溶解した。一本鎖DNA分子内で高次構造を形成させるため、90℃で3分間加熱後、60℃で3分間冷却し、さらにその後25℃に置く加熱・冷却処理を行った。その後、この溶液と等量の0.1 % Nonidet P-40及び5 mM DTTを含むPBS緩衝液と混合した。

本実施例では、結合子としてビオチン及びストレプトアビジンを用いた。ビオチンは、標的物質に吸着する第1結合子であり、ストレプトアビジンは、前記磁気ビーズに吸着された第2結合子に相当する。
固定化工程(102)後の磁気ビーズに400 μLの溶出液(100 mMクエン酸ナトリウム pH 5.0、7 M 尿素、3 mM EDTA)を加えて、90℃で5分間加熱して、溶出液を回収した。その後、この回収した溶出液にフェノール・クロロホルム処理を行い、イソプロピルアルコールによる沈殿操作により、NF-κB p50に結合していた一本鎖DNAを回収した。
PCRには、配列番号26で示すリバースプライマーと配列番号27で示すフォワードプライマーを用いた。リバースプライマーは、5′末端のチミン(t)をビオチン化修飾している。PCRは、最終濃度0.025 U/μLのExTaq DNA ポリメラーゼ(タカラバイオ社)と添付バッファを用いて行った。反応組成は、最終濃度が2 mM MgCl2、0.2 mM dNTP (N=A,G,C,T)となるように調製し、サイクル条件は、94℃を30秒、50℃を30秒、そして72℃を1分の3ステップで、DNA増幅量に応じて、15サイクル又は20サイクルで行った。
増幅工程(104)後の増幅産物を含むPCR溶液からエタノール沈殿により二本鎖DNAを回収した後、ストレプトアビジンを利用したゲルシフト法により一本鎖DNAを調製した。具体的には、回収された二本鎖DNA(PCR溶液0.4 mL)を、10 μLのSA緩衝液(10 mM Tris-HCl pH 7.6、50 mM NaCl、1 mM EDTA)に懸濁し、75℃で3分間加熱後、10 μLのストレプトアビジン溶液(5 mg/mL、SA緩衝液に溶解)を加えて、25℃で30分間インキュベーションした。増幅工程(104)で使用されたリバースプライマーの5′末端の塩基は、ビオチン化されていることから、この操作によって、ビオチン化二本鎖DNAとアビジンの複合体が形成される。この複合体を含む溶液に、同容量の変性ゲル用ローディングバッファ(10 M尿素、1×TBE溶液)を加えて、75℃で3分間加熱して、一本鎖化した後、7 M 尿素を含む6 % ポリアルアミド変性ゲルで電気泳動し、増幅された目的の一本鎖DNAをゲルから回収することによって、不要なビオチン化された一本鎖DNAを除去した。得られた一本鎖DNAをPBS緩衝液(1.1 mM KH2PO4、155 mM NaCl、3 mM Na2HPO4、pH 7.4)に溶解し、90℃で3分間加熱後、60℃で3分間冷却し、さらにその後25℃に置く加熱・冷却処理を行って一本鎖DNA分子内で高次構造を形成させた。その後、この溶液と等量の0.1 % Nonidet P-40及び5 mM DTTを含むPBS緩衝液と混合し、新たな一本鎖核酸ライブラリーとして次のラウンドに使用した。
本実施例では、本工程を5ラウンド行った。各ラウンドにおける一本鎖核酸ライブラリー及び標的物質であるNF-κB p50の濃度等の条件については、前記表1に示す通りである。
5ラウンド終了後に得られた各一本鎖DNAにおける中央領域の塩基配列を決定し、得られたNF-κB p50結合性DNAアプタマーの一次構造、及びその塩基配列から推定される二次構造を同定した。
得られたNF-κB p50結合性DNAアプタマーの塩基配列、クローン名、クローン数及び配列番号を図4に示す。この図で示すように、中央領域の塩基配列は、大きく3つの配列群、すなわち、配列番号9~11で示す塩基配列を有する配列群、配列番号12~18で示す塩基配列を有する配列群、そして配列番号19~21で示す塩基配列を有する配列群に分類された。それぞれの配列には、配列番号29で示すNF-κBの結合する天然型のコンセンサスDNA配列と類似した配列が存在していた。それらのコンセンサスに類似する配列は、図5で示すように、ヘアピン構造のステム部分に位置することがわかった。興味深いことに、このステム部分の配列には、天然型のDNA配列と異なり、非ワトソン・クリック型のG-AとG-Tの塩基対が含まれていることが明らかとなった。
実施例1で得られたNF-κB p50結合性DNAアプタマークローンのうち、各配列群においてクローン数の多かった5R01、5R14、5R05(図4参照)を用いて、NF-κB p50への結合能について解析した。
各クローンの一本鎖DNA(全長95-mer)は、実施例1で得られたクローンのプラスミドを鋳型として、ビオチン化された配列26及び配列27のプライマーを用いてPCR増幅した後、実施例1で示した一本鎖核酸調製工程と同様の手順で、ストレプトアビジンを利用したゲルシフト法により調製した。
図7に結果を示す。この図は、上記3つのクローンとNF-κB p50との相互作用を検出したセンサグラムである。この図が示すように、いずれのクローンもNF-κB p50に非常に強く結合すること、そして、NF-κB p50インジェクション終了後のセンサグラムのパターンから、結合したクローンのNF-κB p50からの解離が極めて遅いことが明らかとなった。BIACORE3000付属の解析ソフトを用いて、反応モデル1:1 binding with mass transferでのフィッティングにより解離定数 Kdを算出した結果、Kd=2.1~3.5 pMで、解離速度定数も10-5(1/s)のオーダーであった。
実施例2で検証したNF-κB p50結合性DNAアプタマークローン5R01、5R14、5R05を切り詰めた変異体におけるNF-κB p50への結合能について解析した。
各NF-κB p50結合性DNAアプタマークローンから5′末端側のプライマー配列領域を含む配列を取り除いた一本鎖DNA変異体(68-mer、配列番号31、32、33)の結合能について、実施例2と同様にSPR法により解析した。各種DNA断片の配列を図8に示す。これら結合解析に使用した一本鎖DNA変異体は、化学合成とゲル精製により調製した。なお、図中、Cont-68(配列番号34)は、既知のNF-κB p50結合コンセンサス配列を含む二本鎖DNAをアデニンのテトラループ(AAAA)で連結した一本鎖DNAに相当する。また、SPR解析において、各種一本鎖DNAのプローブへのハイブリダイゼーションによるSAチップへの固定化は、流速20 μL/minで、PBSにより250nMに希釈した各種DNA断片(68-mer)溶液を20 μL(1分間相当)インジェクションすることで行った。
図9に結果を示す。この図は、上記3つのクローン及び対照用のCont-68とNF-κB p50との相互作用を検出したSPRのセンサグラムである。
実施例1で得られたNF-κB p50結合性DNAアプタマーの各クローンの共通構造は、前述のように、通常の二本鎖DNAとは異なる非ワトソン・クリック型塩基対を含む(図4)。そこで、NF-κB p50結合性DNAアプタマー5R01の変異体であって、実施例3で調製した5R01-68の二本鎖領域中に存在する5箇所の非ワトソン・クリック型塩基対、すなわち、2箇所のG-T塩基対及び3箇所のG-A塩基対を、全てワトソン・クリック型塩基対であるG-C塩基対に変えた配列番号35で示される変異体5R01mut-68について、NF-κB p50への結合能をSPR法により測定した。
変異体5R01mut-68は、配列番号35で示される塩基配列に基づき、化学合成により調製した。実施例2と同様の方法によりNF-κB p50に対する結合能を検証した。同時に、非ワトソン・クリック型塩基対を含む5R01-68とCont-68を対照用として用い、同様に結合能を測定した。
図10に結果を示す。この図は、変異体5R01mut-68(A)、対照用の5R01-68(B)及びCont-68(C)のそれぞれについて、NF-κB p50との相互作用を検出したときのSPRのセンサグラムである。変異体5R01mut-68は、Cont-68よりも結合能が20倍以上強かったが、5R01-68と比較すると解離速度定数が10倍大きくなっており、非ワトソン・クリック型塩基対を有する5R01-68よりもNF-κB p50から解離しやすくなっていることがわかった。この結果から、本発明の製造方法で得られたNF-κB p50結合性DNAアプタマーの各クローンにおける共通構造中の非ワトソン・クリック型塩基対は、NF-κB p50との結合に寄与していることが明らかとなった。
実施例1で得られたNF-κB p50結合性DNAアプタマーのNF-κB p50結合選択性を調べた。
実施例3で調製した非ワトソン・クリック型塩基対を有する5R01-68及び、実施例4で調製した非ワトソン・クリック型塩基対を含まない5R01mut-68を固定化したセンサーチップを用いて、NF-κB p50以外の転写因子であるAP-1タンパク質(プロメガ社)、及びTaq DNAポリメラーゼ (Roche社、1U= 0.05 pmol換算)への結合をSPR法によって検証した。
図11に結果を示す。この図は、5R01-68(A)、5R01mut-68(B)、対照用のTaq-59のそれぞれについて、NF-κB p50(●)、Taq DNAポリメラーゼ(○)、及びAP-1(△)との相互作用を検出したときのSPRのセンサグラムである。この図から、5R01-68及び5R01mut-68のAP-1やTaq DNAポリメラーゼへの結合能は非常に弱く、本発明の製造方法で得られたDNAアプタマーが標的物質であるNF-κB p50に特異的に結合できることが明らかとなった。

Claims (25)
- 核酸アプタマーの製造方法であって、
一本鎖核酸ライブラリーと標的物質とを溶液中で混合して、一本鎖核酸と標的物質の複合体を形成させる複合体形成工程、
複合体形成工程後の溶液と固相担体を混合し、標的物質及び/又は固相担体に吸着させた結合子を介して、複合体を固相担体に固定化する固定化工程、
固相担体に固定化された複合体を溶液から回収する回収工程、
複合体中の一本鎖核酸を回収後、該一本鎖核酸を核酸増幅法によって増幅させる増幅工程、及び
増幅工程で得られた二本鎖核酸を一本鎖化した後、分子内立体構造を形成させる一本鎖核酸調製工程を含む前記製造方法。 - 一本鎖核酸調製工程で得られる一本鎖核酸を新たな一本鎖核酸ライブラリーに用いて、複合体形成工程から一本鎖核酸調製工程までを複数回繰り返す反復工程をさらに含む、請求項1に記載の製造方法。
- 反復工程において複合体形成工程から一本鎖核酸調製工程までを2~15回繰り返す、請求項2に記載の製造方法。
- 反復工程後に得られる一本鎖核酸の中から、連続する5~20塩基が互いに塩基対合する二本鎖領域を二次構造中に一以上含み、かつ前記二本鎖領域内の1~10塩基対が非ワトソン・クリック型塩基対からなる一本鎖核酸分子を選択する選択工程をさらに含む、請求項2又は3に記載の製造方法。
- 複合体形成工程において、前記溶液が前記一本鎖核酸との間で標的物質との結合を競合し合う競合物質を含む、請求項1~4のいずれか一項に記載の製造方法。
- 前記核酸がDNAである、請求項1~5のいずれか一項に記載の製造方法。
- 前記標的物質がペプチドである、請求項1~6のいずれか一項に記載の製造方法。
- 前記結合子がビオチン及びアビジン、ストレプトアビジン又はニュートラアビジンである、請求項1~7のいずれか一項に記載の製造方法。
- 前記固相担体が親水性である、請求項1~8のいずれか一項に記載の製造方法。
- 複合体形成工程及び/又は回収工程で使用する溶液又はバッファが界面活性剤を含む、請求項1~9のいずれか一項に記載の製造方法。
- 標的物質に結合する核酸分子であって、
連続する5~20塩基が互いに塩基対合する二本鎖領域を一以上含み、かつ
前記二本鎖領域内の1~10塩基対が非ワトソン・クリック型塩基対からなる前記核酸分子。 - 前記核酸分子が一本鎖核酸又は二本鎖核酸からなる、請求項11に記載の核酸分子。
- 前記核酸分子がDNAである、請求項12に記載の核酸分子。
- 前記標的物質がペプチドである、請求項11~13のいずれか一項に記載の核酸分子。
- 前記ペプチドが転写調節因子、シグナル伝達因子、タンパク質リガンド、又は受容体タンパク質である、請求項14に記載の核酸分子。
- 前記転写調節因子がNF-κBである、請求項15に記載の核酸分子。
- 前記NF-κBがp50であって、配列番号1及び2で示される塩基配列からなる二本鎖領域を含む、請求項16に記載の核酸分子。
- 配列番号3及び4、配列番号5及び6、又は配列番号7及び8で示される塩基配列からなる二本鎖領域を含む、請求項17に記載の核酸分子。
- 配列番号9~21で示される塩基配列を含む、請求項18に記載の核酸分子。
- 請求項11~19のいずれか一項に記載の核酸分子を有効成分とする標的物質の機能阻害剤。
- 請求項20に記載の標的物質機能阻害剤を含む医薬組成物。
- 請求項11~15のいずれか一項に記載の核酸分子を用いて試料中に存在するその核酸分子が結合する標的物質を検出する方法。
- 請求項16~19のいずれか一項に記載の核酸分子を用いて試料中のNF-κB p50を検出する方法。
- 表面プラズモン共鳴測定法、水晶振動子マイクロバランス測定法、比濁法、比色法又は蛍光法を用いて検出する、請求項22又は23に記載の方法。
- 請求項16~19のいずれか一項に記載の核酸分子を一以上含むNF-κB p50検出用キット。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2012295913A AU2012295913A1 (en) | 2011-08-12 | 2012-08-08 | Production method for nucleic acid aptamer |
| CN201280039329.8A CN103732748A (zh) | 2011-08-12 | 2012-08-08 | 核酸适配体的制造方法 |
| CA2844750A CA2844750A1 (en) | 2011-08-12 | 2012-08-08 | Method for preparing nucleic acid aptamer |
| EP12824560.2A EP2743349A4 (en) | 2011-08-12 | 2012-08-08 | PROCESS FOR PREPARING A NUCLEIC ACID TAPER |
| US14/237,645 US20140194320A1 (en) | 2011-08-12 | 2012-08-08 | Method for preparing nucleic acid aptamer |
| US15/188,307 US20160299131A1 (en) | 2011-08-12 | 2016-06-21 | Method for preparing nucleic acid aptamer |
| US15/188,297 US9857362B2 (en) | 2011-08-12 | 2016-06-21 | Method for preparing nucleic acid aptamer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011177112 | 2011-08-12 | ||
| JP2011-177112 | 2011-08-12 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/237,645 A-371-Of-International US20140194320A1 (en) | 2011-08-12 | 2012-08-08 | Method for preparing nucleic acid aptamer |
| US15/188,307 Division US20160299131A1 (en) | 2011-08-12 | 2016-06-21 | Method for preparing nucleic acid aptamer |
| US15/188,297 Division US9857362B2 (en) | 2011-08-12 | 2016-06-21 | Method for preparing nucleic acid aptamer |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013024763A1 true WO2013024763A1 (ja) | 2013-02-21 |
Family
ID=47715083
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/070188 Ceased WO2013024763A1 (ja) | 2011-08-12 | 2012-08-08 | 核酸アプタマーの作製方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (3) | US20140194320A1 (ja) |
| EP (2) | EP3222727A1 (ja) |
| JP (2) | JPWO2013024763A1 (ja) |
| CN (1) | CN103732748A (ja) |
| AU (1) | AU2012295913A1 (ja) |
| CA (1) | CA2844750A1 (ja) |
| WO (1) | WO2013024763A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103898119A (zh) * | 2014-03-24 | 2014-07-02 | 湖南大学 | 一种多西紫杉醇的核酸适配体、核酸适配体衍生物及其用途 |
| CN104004763A (zh) * | 2014-05-22 | 2014-08-27 | 中国科学院化学研究所 | 一种亲和丙型病毒性肝炎核心蛋白的核酸适体及其应用 |
| WO2017047750A1 (ja) * | 2015-09-18 | 2017-03-23 | 公立大学法人大阪市立大学 | 細胞内移行性dna及びそれを用いた目的分子の細胞内導入方法 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101719285B1 (ko) * | 2014-11-04 | 2017-03-23 | 한국과학기술원 | 표적 물질에 의해 조절되는 핵산 중합효소 활성을 이용한 생체물질의 검출 및 정량 방법 |
| EP3266871B1 (en) * | 2015-03-06 | 2021-04-28 | TAGCyx Biotechnologies Inc. | Method for stabilizing dna aptamers |
| WO2016158851A1 (ja) | 2015-03-30 | 2016-10-06 | 日産化学工業株式会社 | 血管内皮増殖因子受容体に結合する核酸アプタマー |
| US20180371460A1 (en) * | 2015-06-26 | 2018-12-27 | University Of Utah Research Foundation | Depletion of Abundant Serum Proteins to Facilitate Biomarker Discovery |
| TWI731028B (zh) * | 2016-01-22 | 2021-06-21 | 國立大學法人東京大學 | 用以篩選核酸適體之方法 |
| MX2021012471A (es) * | 2019-04-11 | 2022-01-04 | Univ Arkansas | Aptameros de adn especificos de cd40 como adyuvantes de vacunas. |
| GB201917630D0 (en) * | 2019-12-03 | 2020-01-15 | Ge Healthcare Bio Sciences Ab | Method for determination of aggregates |
| CN114853869B (zh) * | 2019-12-10 | 2023-12-26 | 湖南赛奥维生物技术有限公司 | 一种碱性成纤维细胞生长因子替代物及其组合物和应用 |
| CN111499620A (zh) * | 2020-04-28 | 2020-08-07 | 辽宁科技学院 | 一种用葛根素核酸适配体提取废水中葛根素的方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991019813A1 (en) | 1990-06-11 | 1991-12-26 | The University Of Colorado Foundation, Inc. | Nucleic acid ligands |
| WO1992014843A1 (en) | 1991-02-21 | 1992-09-03 | Gilead Sciences, Inc. | Aptamer specific for biomolecules and method of making |
| WO1994008050A1 (en) | 1992-09-29 | 1994-04-14 | Nexagen, Inc. | Nucleic acid ligands and methods for producing the same |
| WO1996035430A1 (en) | 1995-05-12 | 1996-11-14 | Fujisawa Pharmaceutical Co., Ltd. | REMEDY AND PREVENTIVE FOR DISEASES CAUSED BY NF-λB |
| WO1996040159A1 (en) | 1995-06-07 | 1996-12-19 | Merck & Co., Inc. | Capped synthetic rna, analogs, and aptamers |
| JP2003128559A (ja) * | 1995-05-12 | 2003-05-08 | Anges Mg Inc | NF−κBに起因する疾患の治療および予防剤 |
| WO2007004748A1 (ja) * | 2005-07-05 | 2007-01-11 | Ribomic Inc. | 免疫グロブリンgに結合する核酸とその利用法 |
| JP2011177112A (ja) | 2010-03-01 | 2011-09-15 | Nippon Steel Engineering Co Ltd | 芝生育成促進材及び芝生育成方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK164963C (da) | 1990-03-27 | 1993-02-15 | Polysheet As | Fastgoerelsesindretning til fastgoerelse af en beskyttelses- eller afskaermningsbeklaedning til et stillads eller lignende understoetningskonstruktion samt vaerktoej til brug ved montering af fastgoerelsesindretningen |
| US20030054360A1 (en) * | 1999-01-19 | 2003-03-20 | Larry Gold | Method and apparatus for the automated generation of nucleic acid ligands |
| DE60026641T2 (de) * | 1999-07-15 | 2007-01-25 | Qiagen Gmbh | Verfahren zur Trennung von teilchenförmigen Substraten aus einer Lösung indem man den Teilchenverlust minimiert |
| US7601497B2 (en) * | 2000-06-15 | 2009-10-13 | Qiagen Gaithersburg, Inc. | Detection of nucleic acids by target-specific hybrid capture method |
| US7374882B2 (en) * | 2000-11-28 | 2008-05-20 | Riken | Method for base sequencing and biologically active nucleic acids |
| WO2006125124A2 (en) * | 2005-05-18 | 2006-11-23 | Nanosphere, Inc. | Substrate functionalization method for high sensitivity applications |
| EP2260103A4 (en) * | 2008-03-12 | 2012-06-13 | Univ Syracuse | DIRECT SELECTION OF STRUCTURAL DEFINED APTAMERS |
| US8440801B2 (en) | 2008-07-14 | 2013-05-14 | The University Of Tokyo | Aptamer against IL-17 and use thereof |
| WO2010019847A2 (en) * | 2008-08-15 | 2010-02-18 | Georgia State University Research Foundation, Inc. | Aptamer inhibition of thrombus formation |
| US9540650B2 (en) * | 2011-11-18 | 2017-01-10 | Tagcyx Biotechnologies | Nucleic acid fragment binding to target protein |
-
2012
- 2012-08-08 CA CA2844750A patent/CA2844750A1/en not_active Abandoned
- 2012-08-08 WO PCT/JP2012/070188 patent/WO2013024763A1/ja not_active Ceased
- 2012-08-08 JP JP2013528982A patent/JPWO2013024763A1/ja active Pending
- 2012-08-08 US US14/237,645 patent/US20140194320A1/en not_active Abandoned
- 2012-08-08 AU AU2012295913A patent/AU2012295913A1/en not_active Abandoned
- 2012-08-08 EP EP17167130.8A patent/EP3222727A1/en not_active Withdrawn
- 2012-08-08 EP EP12824560.2A patent/EP2743349A4/en not_active Withdrawn
- 2012-08-08 CN CN201280039329.8A patent/CN103732748A/zh active Pending
-
2016
- 2016-06-21 US US15/188,307 patent/US20160299131A1/en not_active Abandoned
- 2016-06-21 US US15/188,297 patent/US9857362B2/en not_active Expired - Fee Related
-
2017
- 2017-08-09 JP JP2017154747A patent/JP2018019697A/ja not_active Withdrawn
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991019813A1 (en) | 1990-06-11 | 1991-12-26 | The University Of Colorado Foundation, Inc. | Nucleic acid ligands |
| WO1992014843A1 (en) | 1991-02-21 | 1992-09-03 | Gilead Sciences, Inc. | Aptamer specific for biomolecules and method of making |
| WO1994008050A1 (en) | 1992-09-29 | 1994-04-14 | Nexagen, Inc. | Nucleic acid ligands and methods for producing the same |
| WO1996035430A1 (en) | 1995-05-12 | 1996-11-14 | Fujisawa Pharmaceutical Co., Ltd. | REMEDY AND PREVENTIVE FOR DISEASES CAUSED BY NF-λB |
| JP2003128559A (ja) * | 1995-05-12 | 2003-05-08 | Anges Mg Inc | NF−κBに起因する疾患の治療および予防剤 |
| WO1996040159A1 (en) | 1995-06-07 | 1996-12-19 | Merck & Co., Inc. | Capped synthetic rna, analogs, and aptamers |
| WO2007004748A1 (ja) * | 2005-07-05 | 2007-01-11 | Ribomic Inc. | 免疫グロブリンgに結合する核酸とその利用法 |
| JP2011177112A (ja) | 2010-03-01 | 2011-09-15 | Nippon Steel Engineering Co Ltd | 芝生育成促進材及び芝生育成方法 |
Non-Patent Citations (25)
| Title |
|---|
| BOCK L.C. ET AL., NATURE, vol. 355, 1992, pages 564 - 566 |
| CHAN R. ET AL., NUCLEIC ACIDS RES., vol. 34, 2006, pages E36 |
| CHO M. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 107, 2010, pages 15373 - 15378 |
| CHRISTOPHER J. ET AL.: "Self-Assembled Monolayers of a Form of Nanotechnology", CHEMICAL REVIEW, vol. 105, 2005, pages 1103 - 1169 |
| CITARTAN, M. ET AL.: "Conditions optimized for the preparation of single-stranded DNA (ssDNA) employing lambda exonuclease digestion in generating DNA aptamer", WORLD J. MICROBIOL. BIOTECHNOL., vol. 27, May 2011 (2011-05-01), pages 1167 - 1173, XP055143874 * |
| FAN X. ET AL., J. T. LIS, vol. 101, 2004, pages 6934 - 6939 |
| HAMULA C.L. ET AL., ANAL. CHEM., vol. 80, 2008, pages 7812 - 7819 |
| ISOMURA I.; A. MORITA, MICROBIOL. IMMUNOL., vol. 50, 2006, pages 559 - 563 |
| JEONG S. ET AL., OLIGONUCLEOTIDES, vol. 20, 2010, pages 155 - 161 |
| KIM K.H. ET AL., EXP. MOL. PATHOL., vol. 86, 2008, pages 114 - 120 |
| KIM, K. H. ET AL.: "Transcriptional regulation of NF-KB by ring type decoy oligodeoxynucleotide in an animal model of nephropathy", EXPERIMENTAL AND MOLECULAR PATHOLOGY, vol. 86, 2009, pages 114 - 120, XP026079977 * |
| LAUHON C.T.; SZOSTAK J.W., J. AM. CHEM. SOC., vol. 117, 1995, pages 1246 - 1257 |
| LEBRUSKA L.L.; MAHER III L.J., BIOCHEMISTRY, vol. 38, 1999, pages 3168 - 3174 |
| MAIRAL, T. ET AL.: "Aptamers: molecular tools for analytical applications", ANAL. BIOANAL. CHEM., vol. 390, 2008, pages 989 - 1007, XP019584708 * |
| MANN M.J., INVEST., vol. 106, 2000, pages 1071 - 1075 |
| MIYAKE T. ET AL., MOL. THER., vol. 19, 2001, pages 181 - 187 |
| MIYAKE, T. ET AL.: "Systemic Administration of Ribbon-type Decoy Oligodeoxynucleotide Against Nuclear Factor KB and Ets Prevents Abdominal Aortic Aneurysm in Rat Model", MOLECULAR THERAPY, vol. 19, no. 1, January 2011 (2011-01-01), pages 181 - 187, XP055143876 * |
| N.J. REITER; L.J. MAHER III; S.E. BUTCHER, NUCLEIC ACIDS RES., vol. 36, 2008, pages 1227 - 1236 |
| NAGASWAMY U. ET AL., NUCL. ACID RES., vol. 28, 2000, pages 375 - 376 |
| S.E. WURSTER; L.J. MAHER III, RNA, vol. 14, 2008, pages 1037 - 1047 |
| SAMBROOK, J. ET AL.: "Molecular Cloning: A Laboratory Manual Third Ed.,", 2001, COLD SPRING HARBOR LABORATORY PRESS |
| See also references of EP2743349A4 |
| TOK J. ET AL., ELECTROPHORESIS, vol. 31, 2010, pages 2055 - 2062 |
| WOCHNER, A. ET AL.: "Semi-automated selection of DNA aptamers using magnetic particle handling", BIOTECHNIQUES, vol. 43, no. 3, 2007, pages 344,346,348,350,352 - 353, XP055143871 * |
| ZHAO X. ET AL., NUCLEIC ACIDS RES., vol. 34, 2006, pages 3755 - 3761 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103898119A (zh) * | 2014-03-24 | 2014-07-02 | 湖南大学 | 一种多西紫杉醇的核酸适配体、核酸适配体衍生物及其用途 |
| CN104004763A (zh) * | 2014-05-22 | 2014-08-27 | 中国科学院化学研究所 | 一种亲和丙型病毒性肝炎核心蛋白的核酸适体及其应用 |
| CN104004763B (zh) * | 2014-05-22 | 2016-03-16 | 中国科学院化学研究所 | 一种亲和丙型病毒性肝炎核心蛋白的核酸适体及其应用 |
| WO2017047750A1 (ja) * | 2015-09-18 | 2017-03-23 | 公立大学法人大阪市立大学 | 細胞内移行性dna及びそれを用いた目的分子の細胞内導入方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2013024763A1 (ja) | 2015-03-05 |
| AU2012295913A8 (en) | 2014-04-03 |
| JP2018019697A (ja) | 2018-02-08 |
| US20160289680A1 (en) | 2016-10-06 |
| EP3222727A1 (en) | 2017-09-27 |
| CA2844750A1 (en) | 2013-02-21 |
| US20140194320A1 (en) | 2014-07-10 |
| AU2012295913A1 (en) | 2014-03-13 |
| EP2743349A1 (en) | 2014-06-18 |
| US20160299131A1 (en) | 2016-10-13 |
| US9857362B2 (en) | 2018-01-02 |
| EP2743349A4 (en) | 2015-03-25 |
| CN103732748A (zh) | 2014-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2018019697A (ja) | 核酸アプタマーの作製方法 | |
| AU777043B2 (en) | Nucleic acid ligands to CD40ligand | |
| JP6307675B2 (ja) | 標的タンパク質に結合する核酸断片 | |
| JP6407195B2 (ja) | 改善されたオフ速度(off−rate)を持つアプタマーを生成するための方法 | |
| US11104904B2 (en) | Method for stabilizing DNA aptamers | |
| JP6721189B2 (ja) | vWFに結合するDNAアプタマー | |
| Jain et al. | Nucleic acid therapeutics: a focus on the development of aptamers | |
| WO2017127762A1 (en) | Methods for improved aptamer selection | |
| WO2016158851A1 (ja) | 血管内皮増殖因子受容体に結合する核酸アプタマー | |
| WO2015147017A1 (ja) | Fgf2に対するアプタマー及びその使用 | |
| WO2020163974A1 (zh) | 针对骨硬化蛋白的适体的诊断用途 | |
| WO2020204151A1 (ja) | Fgf9に対するアプタマー及びその使用 | |
| CA3003576C (en) | Dna aptamer that binds to vwf | |
| JP2025152676A (ja) | フラビウイルスに対するアプタマー及びその使用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12824560 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013528982 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14237645 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2844750 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2012295913 Country of ref document: AU Date of ref document: 20120808 Kind code of ref document: A |