WO2013035270A1 - 樹脂担持触媒および樹脂担持触媒の製造方法 - Google Patents
樹脂担持触媒および樹脂担持触媒の製造方法 Download PDFInfo
- Publication number
- WO2013035270A1 WO2013035270A1 PCT/JP2012/005388 JP2012005388W WO2013035270A1 WO 2013035270 A1 WO2013035270 A1 WO 2013035270A1 JP 2012005388 W JP2012005388 W JP 2012005388W WO 2013035270 A1 WO2013035270 A1 WO 2013035270A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- catalyst
- supported catalyst
- fine particles
- supported
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/26—Catalysts comprising hydrides, coordination complexes or organic compounds containing in addition, inorganic metal compounds not provided for in groups B01J31/02 - B01J31/24
- B01J31/28—Catalysts comprising hydrides, coordination complexes or organic compounds containing in addition, inorganic metal compounds not provided for in groups B01J31/02 - B01J31/24 of the platinum group metals, iron group metals or copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- B01J21/063—Titanium; Oxides or hydroxides thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the alkali- or alkaline earth metals or beryllium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/14—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of germanium, tin or lead
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/20—Vanadium, niobium or tantalum
- B01J23/22—Vanadium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/26—Chromium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/32—Manganese, technetium or rhenium
- B01J23/34—Manganese
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/32—Manganese, technetium or rhenium
- B01J23/36—Rhenium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/42—Platinum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/44—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/46—Ruthenium, rhodium, osmium or iridium
- B01J23/462—Ruthenium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/46—Ruthenium, rhodium, osmium or iridium
- B01J23/464—Rhodium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/48—Silver or gold
- B01J23/50—Silver
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/745—Iron
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/75—Cobalt
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/755—Nickel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/06—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing polymers
- B01J31/08—Ion-exchange resins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/22—Organic complexes
- B01J31/2204—Organic complexes the ligands containing oxygen or sulfur as complexing atoms
- B01J31/2208—Oxygen, e.g. acetylacetonates
- B01J31/2226—Anionic ligands, i.e. the overall ligand carries at least one formal negative charge
- B01J31/223—At least two oxygen atoms present in one at least bidentate or bridging ligand
- B01J31/2239—Bridging ligands, e.g. OAc in Cr2(OAc)4, Pt4(OAc)8 or dicarboxylate ligands
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/391—Physical properties of the active metal ingredient
- B01J35/393—Metal or metal oxide crystallite size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/613—10-100 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/615—100-500 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/16—Reducing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/16—Reducing
- B01J37/18—Reducing with gases containing free hydrogen
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/40—Substitution reactions at carbon centres, e.g. C-C or C-X, i.e. carbon-hetero atom, cross-coupling, C-H activation or ring-opening reactions
- B01J2231/42—Catalytic cross-coupling, i.e. connection of previously not connected C-atoms or C- and X-atoms without rearrangement
- B01J2231/4205—C-C cross-coupling, e.g. metal catalyzed or Friedel-Crafts type
- B01J2231/4261—Heck-type, i.e. RY + C=C, in which R is aryl
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2235/00—Indexing scheme associated with group B01J35/00, related to the analysis techniques used to determine the catalysts form or properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/80—Complexes comprising metals of Group VIII as the central metal
- B01J2531/82—Metals of the platinum group
- B01J2531/824—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/165—Polymer immobilised coordination complexes, e.g. organometallic complexes
- B01J31/1658—Polymer immobilised coordination complexes, e.g. organometallic complexes immobilised by covalent linkages, i.e. pendant complexes with optional linking groups, e.g. on Wang or Merrifield resins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/22—Organic complexes
- B01J31/2204—Organic complexes the ligands containing oxygen or sulfur as complexing atoms
- B01J31/2208—Oxygen, e.g. acetylacetonates
- B01J31/2226—Anionic ligands, i.e. the overall ligand carries at least one formal negative charge
Definitions
- the present invention relates to a resin-supported catalyst and a method for producing a resin-supported catalyst.
- a catalyst supported on a carrier (hereinafter referred to as a supported catalyst) can be easily recovered and reused, and is an effective means particularly when an expensive noble metal is used as the catalyst (for example, Patent Document 1 and 2).
- the binding force between the catalyst and the carrier is not always strong.
- the catalyst may be detached from the carrier during use, and the detached catalysts may aggregate with each other. In that case, the catalyst activity gradually decreases by repeatedly using the supported catalyst.
- Patent Document 1 As a method for suppressing the deterioration of the activity of the supported catalyst due to repeated use, for example, there is a method described in Patent Document 1.
- a mixture of a thermosetting resin and a catalyst composed of a metal or a metal compound or a precursor thereof is heated to 500 ° C. or more in a non-oxidizing atmosphere to carbonize the thermosetting resin, so that the metal or A metal compound is supported on a carbide.
- the cured body of the thermosetting resin is carbonized, it is carbonized while maintaining its shape although it is reduced.
- many catalysts made of metals or metal compounds are stable under the condition that the thermosetting resin is carbonized. For this reason, after mixing a catalyst etc. with a thermosetting resin, the thermosetting resin is hardened and carbonized, and the carbon material which fixed the catalyst substance firmly is obtained.
- Patent Document 2 fine particles having catalytic activity are supported on the surface of spherical resin particles, and a coating layer having a specific thickness made of a thermosetting resin is formed so as to cover the spherical resin particles. Is described. By providing a coating layer made of a thermosetting resin in this way, the detachment of fine particles having catalytic activity during use is suppressed, and separation and recovery from reaction products after use is facilitated. .
- the carriers may aggregate.
- the reaction product is less likely to come into contact with the catalyst, resulting in a reduction in reaction efficiency. That is, the activity per carrier is reduced.
- the present invention has been made in view of the above circumstances, and provides a resin-supported catalyst having excellent repeatability and utilization efficiency.
- thermosetting resin having a phenolic hydroxyl group As a result of intensive research on improving the repetitive characteristics and utilization efficiency of a catalyst on a resin-supported catalyst using a cured product of a thermosetting resin as a catalyst carrier, the present inventors have found that a thermosetting resin having a phenolic hydroxyl group. When the cured product of the above was used as a carrier for supporting a catalyst, it was possible to suppress the release of catalyst fine particles from the carrier, so that a supported catalyst with excellent catalyst utilization efficiency was obtained, and the present invention was completed. .
- thermosetting resin Fine particles having catalytic activity carried on the surface of the cured body; Including A resin-supported catalyst in which the thermosetting resin has a phenolic hydroxyl group is provided.
- a curing step of curing a thermosetting resin having a phenolic hydroxyl group A supporting step of supporting fine particles having catalytic activity on the surface of the cured body obtained in the curing step;
- a method for producing a resin-supported catalyst comprising:
- the resin-supported catalyst according to the present embodiment includes a cured product of a thermosetting resin having a phenolic hydroxyl group, and fine particles having catalytic activity supported on the surface of the cured product of the thermosetting resin (hereinafter referred to as “catalyst particles”). It is also called).
- the resin-supported catalyst in the present embodiment since the catalyst fine particles are supported on the surface of the cured body of the thermosetting resin, the reaction product easily reaches the catalyst fine particles. Therefore, the utilization efficiency of the catalyst is excellent. Further, the resin-supported catalyst in the present embodiment is excellent in the catalyst repeatability. The reason is not necessarily clear, but is considered as follows.
- a cured body of a thermosetting resin having a phenolic hydroxyl group on the surface is used as a carrier. And it is thought that it is carry
- the catalyst fine particles are supported on the phenolic hydroxyl group that remains in the cured body of the thermosetting resin that is the catalyst carrier.
- the catalyst fine particles are supported on the phenolic hydroxyl group, so that it is considered that the catalyst fine particles are uniformly dispersed on the catalyst support as compared with the conventional supported catalyst. For this reason, it is considered that the aggregation of the catalyst fine particles is less likely to occur at the time of use, and the catalyst repetitive characteristics and the use efficiency are improved.
- thermosetting resin is used as a catalyst carrier in this embodiment.
- the cured body of the thermosetting resin may be formed in, for example, a particle shape, a film shape, or a uniform layer shape.
- the thermosetting resin before the curing treatment is not particularly limited as long as it is a thermosetting resin having a phenolic hydroxyl group, but preferably contains a phenol resin or a derivative thereof.
- the phenolic hydroxyl group equivalent of the thermosetting resin is preferably 30 g / eq or more, and more preferably 35 g / eq or more.
- the phenolic hydroxyl group equivalent of the thermosetting resin is not less than the above lower limit, a cured product having a hydroxyl group on the surface can be obtained, and the catalyst can be prevented from being detached during use.
- the phenolic hydroxyl group equivalent of the thermosetting resin is 500 g / eq or less, preferably 400 g / eq or less, more preferably 350 g / eq or less. is there.
- a resin-supported catalyst having excellent catalytic activity can be provided.
- the phenolic hydroxyl group equivalent of a thermosetting resin is below the said upper limit, the phenolic hydroxyl group on the surface of a hardening body increases, and the retention of a catalyst can be maintained without weakening.
- the phenolic hydroxyl group equivalent can be quantified by a known method such as an acetylation method.
- the phenol resin in this embodiment is obtained by reacting phenols and aldehydes in the presence of an alkaline or acidic catalyst, and has at least one phenolic hydroxyl group in the molecule. Yes.
- examples thereof include a phenol resin, a cresol resin, a resorcin resin, a xylenol resin, a naphthol resin, a bisphenol A resin, an aralkyl phenol resin, a biphenyl aralkyl phenol resin, and a modified phenol resin using cashew nut oil having a phenolic hydroxyl group.
- xylene-modified phenol resins containing substances having phenolic hydroxyl groups and various modified phenol resins such as phenol-modified rosin, oil-modified phenol resins modified with terpene oil, rubber-modified phenol resins modified with rubber, etc. can do.
- phenols used for obtaining the phenol resin those having a phenolic hydroxyl group in the molecule are preferable, and further, a substituent other than the phenolic hydroxyl group may be contained.
- cresols such as phenol, o-cresol, m-cresol, p-cresol, mixed cresol mixed with these, 2,3-xylenol, 2,4-xylenol, 2,5-xylenol, 2,6-xylenol, Xylenol such as 3,4-xylenol and 3,5-xylenol, ethylphenol such as o-ethylphenol, m-ethylphenol and p-ethylphenol, butylphenol such as isopropylphenol, butylphenol and p-tert-butylphenol, p- Halogenated phenols such as alkylphenols such as tert-amylphenol, p-oct
- aldehydes used for obtaining the phenol resin include formaldehyde, paraformaldehyde, trioxane, acetaldehyde, propionaldehyde, polyoxymethylene, chloral, hexamethylenetetramine, furfural, glyoxal, n-butyraldehyde, capro
- the method for reacting the phenols and aldehydes is not particularly limited, and a known method can be adopted.
- the catalyst for obtaining the phenol resin is not particularly limited, and examples thereof include an acid catalyst, a base catalyst, and a transition metal salt catalyst.
- the acid catalyst that can be used include inorganic acids such as hydrochloric acid, sulfuric acid, and phosphoric acids, and organic acids such as oxalic acid, p-toluenesulfonic acid, and organic phosphonic acid.
- the base catalyst include alkali metal hydroxides such as sodium hydroxide, potassium hydroxide and lithium hydroxide, alkaline earth metal hydroxides such as calcium hydroxide and barium hydroxide, ammonia, alkylamine and the like. These amines can be used.
- the transition metal salt catalyst include zinc oxalate and zinc acetate.
- the specific surface area by the BET method of the resin-supported catalyst in the present embodiment is preferably 300 m 2 / g or less, particularly preferably 200 m 2 / g or less.
- the specific surface area is not more than the above upper limit value, the amount of catalyst fine particles taken up to the inside of the cured body of the thermosetting resin can be suppressed. Therefore, the utilization efficiency of the catalyst can be further improved.
- thermosetting resin in the present embodiment is not particularly limited. Solid, powder form, spherical form, etc. are mentioned, Any form can be used.
- thermosetting resin may be mixed with an organic or inorganic filler and cured after molding, or may be cured after impregnating another substrate.
- the catalyst fine particles supported on the surface of the cured body of the thermosetting resin in the present embodiment may be any metal, metal oxide, and metal compound as long as they have catalytic activity, and are particularly limited. It is not a thing.
- metals such as titanium, chromium, cobalt, nickel, copper, ruthenium, rhodium, palladium, rhenium, osmium, platinum, iron, zinc, manganese, magnesium, calcium, silver, vanadium, tin, oxides thereof, and other organic titanium And at least one selected from the group consisting of metal compounds and complexes.
- complex containing at least 2 or more types of these can also be used. Among these, palladium or platinum is particularly preferably used.
- the average particle diameter of the catalyst fine particles is preferably 1 ⁇ m or less. By doing so, it is possible to provide a resin-supported catalyst having even better catalytic activity. Further, nano-sized metal fine particles having an average particle diameter of 1 nm to 100 nm can also be used.
- the production method according to the present embodiment includes a step of producing a cured body that becomes a catalyst carrier by curing a thermosetting resin having a phenolic hydroxyl group, and catalyst fine particles are supported on the surface of the cured body that is a catalyst carrier. And a process.
- thermosetting resin a method for curing the thermosetting resin will be described. Although it does not specifically limit as a hardening processing method of the thermosetting resin in this embodiment, A well-known method is employable.
- thermosetting resin When a resol type phenol resin is used as the thermosetting resin, it can be cured by heating. Alternatively, a method of mixing acids such as p-toluenesulfonic acid and phenolsulfonic acid and curing at normal temperature or by heating can be used.
- a curing agent such as hexamethylenetetramine is mixed with an additive compound and cured by heating, a thermosetting resin such as an epoxy resin, a polyisocyanate, or a melamine resin. And a method of mixing with an additive compound and curing by heating.
- the curing temperature of the thermosetting resin in this embodiment is not particularly limited, it is preferably 250 ° C. or lower. When the curing temperature is not more than the above upper limit, an economical curing rate can be obtained, and decomposition of the main chain of the phenol resin can be suppressed.
- the support of the catalyst fine particles on the cured body of the thermosetting resin is not particularly limited as this embodiment, but a known method can be adopted.
- the resin-supported catalyst obtained by the present embodiment may be formed on the surface of the substrate.
- a catalyst in which a resin-supported catalyst is formed on a substrate surface will be described as a “substrate-supported catalyst”.
- the substrate-supported catalyst according to this embodiment is in the form of a sheet or a plate, it can be deformed into various shapes according to the shape of the reaction apparatus.
- the shape of the catalyst sheet can be changed into various shapes according to the shape of the reaction tube in the reaction apparatus, such as a folded state or a rolled state.
- the communication hole is provided in the sheet-like resin-supported catalyst, it can be used as a catalyst filter.
- the shape of the substrate is not particularly limited.
- a particle shape, a sheet shape, or a plate shape is used.
- a sheet shape or a plate shape is preferable.
- the base material is preferably a porous body or a mesh structure, and more preferably a mesh structure.
- the base material is preferably a porous body or a mesh structure, and more preferably a mesh structure.
- the porous body may have a plurality of irregularities or may be provided with a plurality of holes (hereinafter referred to as communication holes) communicating from the front surface to the back surface.
- the base material according to the present embodiment is more preferably a plate having a mesh structure. By doing so, it is possible to significantly improve the contact efficiency between the catalyst fine particles and the reactant. That is, the reaction activity can be improved, and a substrate-supported catalyst having a further excellent catalytic activity can be provided.
- the communication holes may form a honeycomb structure.
- the mechanical strength of the substrate itself can be improved. It is also possible to disperse the catalyst fine particles uniformly and at a high density.
- the substrate according to the present embodiment may be a compound or polymer containing a polar functional group such as a carbonyl group, an imide group and a hydroxy group in the chemical structure, or a compound or polymer containing no polar functional group. It may be.
- a polar functional group such as a carbonyl group, an imide group and a hydroxy group in the chemical structure
- a compound or polymer containing no polar functional group may be.
- the compound or polymer containing a polar functional group include cellulose, polyurethane, polyamide, and polyester.
- compounds and polymers that do not contain polar functional groups include fluorine resins such as polyethylene, polypropylene, polymethylpentene, polybutene, polybutadiene, polystyrene, polyisobutylene, polytetrafluoroethylene, natural rubber, styrene butadiene rubber, and butyl rubber. Is used.
- the method for forming the cured body of the thermosetting resin on the surface of the substrate can be appropriately selected depending on the shape of the substrate.
- a method of impregnating and curing a solid or powder resin solution or liquid resin, a method of heating and melting a solid or powder resin and impregnating and curing the substrate, etc. Used.
- a method of coating a cured body of a thermosetting resin is used.
- the present invention relates to a specific method for producing catalyst fine particles used in the first embodiment.
- Non-patent Documents 1 and 2 a method of reducing catalyst fine particles in advance using a reducing agent has been used (Non-patent Documents 1 and 2).
- the reducing agent used at this time include a reducing agent selected from the group consisting of hydrogen, carbon monoxide, aldehydes, carboxylic acids, amines, metal hydrides, and hydrazine.
- the method of reducing the catalyst fine particles in advance using the reducing agent is insufficient from the viewpoint of aggregation of the catalyst fine particles.
- the catalyst fine particles are reduced not by a reducing agent but by a phenolic hydroxyl group, and a resin-supported catalyst having excellent repeatability and utilization efficiency can be manufactured.
- aggregation of catalyst fine particles can be prevented by reducing the catalyst fine particles using a phenolic hydroxyl group. That is, when the catalyst fine particles are reduced by the phenolic hydroxyl group, the catalyst fine particles can be uniformly supported on the surface of the cured body of the thermosetting resin that is the catalyst carrier. For this reason, it leads to the improvement of reaction efficiency, and the resin carrying catalyst excellent in the repetition characteristic and utilization efficiency can be manufactured.
- the reducing agent when reducing the catalyst fine particles with the phenolic hydroxyl group, does not exhibit a reducing action on the material forming the catalyst fine particles and does not inhibit the reduction reaction of the catalyst fine particles with the phenolic hydroxyl group.
- It may be contained in the catalyst-supported reaction solution.
- the reducing agent may be contained in the catalyst-supported reaction solution as long as it is 0 mol or more and 10 mol or less with respect to 1 mol of the material forming the catalyst fine particles, and more preferably 0 mol or more and 5 mol or less. preferable.
- the catalyst fine particles can be reduced on the surface of the thermosetting resin having a phenolic hydroxyl group. Therefore, the catalyst fine particles can be uniformly and finely supported on the surface of the cured body of the thermosetting resin. Therefore, it is possible to obtain a resin-supported catalyst having excellent repeatability and utilization efficiency.
- the reducing agent is most preferably not contained in the catalyst-supported reaction solution. By doing so, aggregation of the supported catalyst fine particles is less likely to occur compared to the resin-supported catalyst obtained by the conventional method. For this reason, the resin-supported catalyst obtained by the method according to the present embodiment is remarkably superior in the repeatability and utilization efficiency of the resin-supported catalyst compared to the resin-supported catalyst obtained by the conventional method.
- the reducing agent molecules that inhibit the dispersion of the catalyst fine particles are contained in the catalyst-supported reaction solution when reducing the catalyst fine particles. Therefore, it is considered that the catalyst fine particles are uniformly and finely dispersed on the surface of the cured body of the thermosetting resin. For this reason, it is considered that the resin-supported catalyst according to the present embodiment supports the catalyst fine particles more firmly on the surface of the cured body of the thermosetting resin than the conventional resin-supported catalyst.
- the catalyst fine particles are reduced in advance using a reducing agent.
- reducing agent used herein include aldehydes such as hydrogen, carbon monoxide and formaldehyde, carboxylic acids such as formic acid and oxalic acid, amines such as triethylamine, sodium borohydride and lithium aluminum hydride. Metal hydrides and hydrazine are mentioned.
- the catalyst fine particles are reduced using a reducing agent as in the conventional method for producing a resin-supported catalyst
- the catalyst fine particles are supported on the catalyst carrier in the procedure described below.
- the catalyst fine particles are reduced in advance using a reducing agent.
- the catalyst fine particles reduced and precipitated by the reducing agent are supported on the catalyst carrier. That is, in the conventional method for producing a resin-supported catalyst, catalyst fine particles are reduced, and the precipitated catalyst fine particles are dispersed in a solution, and then the catalyst fine particles are supported on the catalyst carrier.
- catalyst fine particles are reduced by a phenolic hydroxyl group. Specifically, the catalyst fine particles are reduced on the surface of the thermosetting resin having a phenolic hydroxyl group. That is, in the method for producing a resin-supported catalyst according to the present embodiment, the catalyst fine particles are brought into contact with a cured body of a thermosetting resin that is a catalyst carrier, and then the catalyst fine particles are reduced and precipitated. According to this method for producing a resin-supported catalyst, the catalyst fine particles can be uniformly and finely supported on the catalyst carrier. By doing so, it is possible to produce a resin-supported catalyst having excellent repeatability and utilization efficiency.
- the reason for this is not necessarily clear, but it is thought that the precipitation of the reduced catalyst fine particles and the loading of the precipitated catalyst fine particles on the catalyst carrier occur almost simultaneously. That is, compared with the conventional method for producing a resin-supported catalyst, the catalyst fine particles can be supported on the catalyst carrier without dispersing the precipitated catalyst fine particles in the solvent, and therefore the aggregation of the catalyst fine particles can be highly suppressed. Conceivable.
- the catalyst fine particles are reduced with phenolic hydroxyl groups in the liquid phase and supported on a catalyst carrier having phenolic hydroxyl groups.
- a catalyst carrier having phenolic hydroxyl groups When reducing the catalyst fine particles with the phenolic hydroxyl group, it is preferable not to add the reducing agent. By doing so, the catalyst fine particles can be reduced on the surface of the thermosetting resin having a phenolic hydroxyl group. Therefore, the catalyst fine particles can be uniformly and finely supported on the surface of the cured body of the thermosetting resin.
- the first implementation is performed as a method for forming a cured body of a thermosetting resin, catalyst fine particles, a substrate-supported catalyst, a substrate, and a cured body of a thermosetting resin on the surface of the substrate.
- the first implementation is performed as a method for forming a cured body of a thermosetting resin. The same thing as what is described in a form can be used.
- Example A1 (Production of cured phenol resin)
- Example A1 Production of cured phenol resin
- 1300 parts of phenol, 1600 parts of 43% formaldehyde aqueous solution, 800 parts of water and 30 parts of triethylamine and polyvinyl alcohol (Kuraray Poval PVA117) as a suspending agent , Saponification degree 98%, polymerization degree 1700) 30 parts were added and heated at 100 ° C. for 3 hours to synthesize a spherical phenol resin cured product.
- the reaction product was collected after 1 hour of reaction, freeze-dried, and the phenolic hydroxyl group equivalent measured by the acetylation method was 115 g / eq.
- the synthesized spherical phenol resin cured product was subjected to solid-liquid separation and dried at 150 ° C. to obtain a spherical phenol resin cured product having an average particle size of 100 ⁇ m.
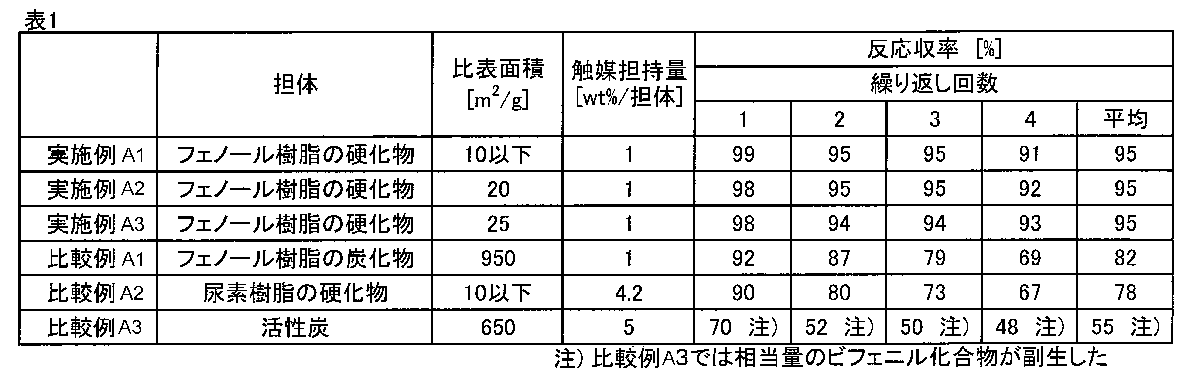
- the amount of palladium catalyst supported in the resin-supported catalyst was 1 wt%. Moreover, the specific surface area of this resin supported catalyst was 10 m ⁇ 2 > / g or less.
- the supported amount of palladium catalyst was measured using a commercially available atomic absorption spectrophotometer, and the specific surface area was measured by nitrogen gas and BET three-point method.
- Example A2 50 parts of methyl ethyl ketone was mixed with 50 parts of a commercially available resol type phenolic resin (Sumitomo Bakelite, Sumilite Resin (r) PR-50087, phenolic hydroxyl group equivalent 130 g / eq), and a filter paper (ADVANTEC (r) No. 590). And then dried at 180 ° C. for 1 hour to obtain a cured phenolic resin (phenol resin content 50 wt%). Further, a resin-supported catalyst was obtained in the same manner as in Example A1. The supported amount of palladium catalyst of this resin-supported catalyst was 1 wt%, and the specific surface area was 20 m 2 / g.
- Example A3 Example A2 except that 30 parts of a commercially available novolak-type phenol resin (Sumitomo Bakelite, Sumilite Resin (r) PR-310, phenolic hydroxyl group equivalent 105 g / eq) was dissolved in 70 parts of methyl ethyl ketone. Thus, a resin-supported catalyst (phenol resin content 50 wt% in the catalyst support) was obtained. The amount of palladium catalyst supported on this resin-supported catalyst was 1 wt%, and the specific surface area was 25 m 2 / g.
- a commercially available novolak-type phenol resin Suditomo Bakelite, Sumilite Resin (r) PR-310, phenolic hydroxyl group equivalent 105 g / eq
- Example A1 The spherical phenol resin cured product obtained in Example A1 was carbonized and activated at 900 ° C. under a nitrogen stream and an air stream in a commercially available carbonization furnace. Further, a resin activated carbon-supported catalyst was obtained in the same manner as in Example A1. The supported amount of the palladium catalyst of this resin activated carbon supported catalyst was 1 wt%, and the specific surface area was 950 m 2 / g.
- Comparative Example A2 A commercially available palladium-urea resin (Pd Encat 30 manufactured by Wako Pure Chemical Industries, Ltd.) using a cured product of a thermosetting resin not containing a phenolic hydroxyl group as a catalyst carrier was used as Comparative Example A2.
- the amount of palladium catalyst supported on the palladium-urea resin was 4.2 wt%, and the specific surface area was 10 m 2 / g or less.
- Comparative Example A3 A commercially available palladium-activated carbon (Palladium-activated carbon, manufactured by Wako Pure Chemical Industries, Ltd.) was used as Comparative Example A3.
- the supported amount of palladium catalyst of palladium-activated carbon was 5 wt%, and the specific surface area was 650 m 2 / g.
- the catalytic activity of the supported catalyst was evaluated by the reaction yield of trans-methyl cinnamate obtained by Heck reaction between iodobenzene and methyl acrylate.
- methyl acrylate 23 ⁇ L (0.25 mmol) were dissolved in acetonitrile (2 mL).
- 50 mg of the supported catalyst was added to the obtained solution.
- the mixture was heated using an oil bath and stirred at 120 ° C. for 12 hours. After completion of the reaction, the supported catalyst was filtered off.
- reaction yield is defined by (C 0 -C f ) / C 0 ⁇ 100 (%), where C 0 is the number of moles of iodobenzene before the reaction, and C f is the number of moles of iodobenzene after the reaction. It is.
- Example B1 (Production of cured phenol resin)
- a sheet-like polypropylene nonwoven fabric (manufactured by Japan Vilene Co., Ltd.) is immersed for 1 minute at room temperature in a phenol resin solution in which liquid phenolic resin (Sumitomo Bakelite Co., Ltd., Sumilite Resin PR-50087) and methanol are mixed at a weight ratio of 1: 1. Air-dried at room temperature for 30 minutes. After drying, it was heated at 90 ° C. for 30 minutes, and further heated at 150 ° C. for 30 minutes to obtain a sheet-like phenol resin carrier containing 30% by weight of phenol resin.
- liquid phenolic resin Suditomo Bakelite Co., Ltd., Sumilite Resin PR-50087
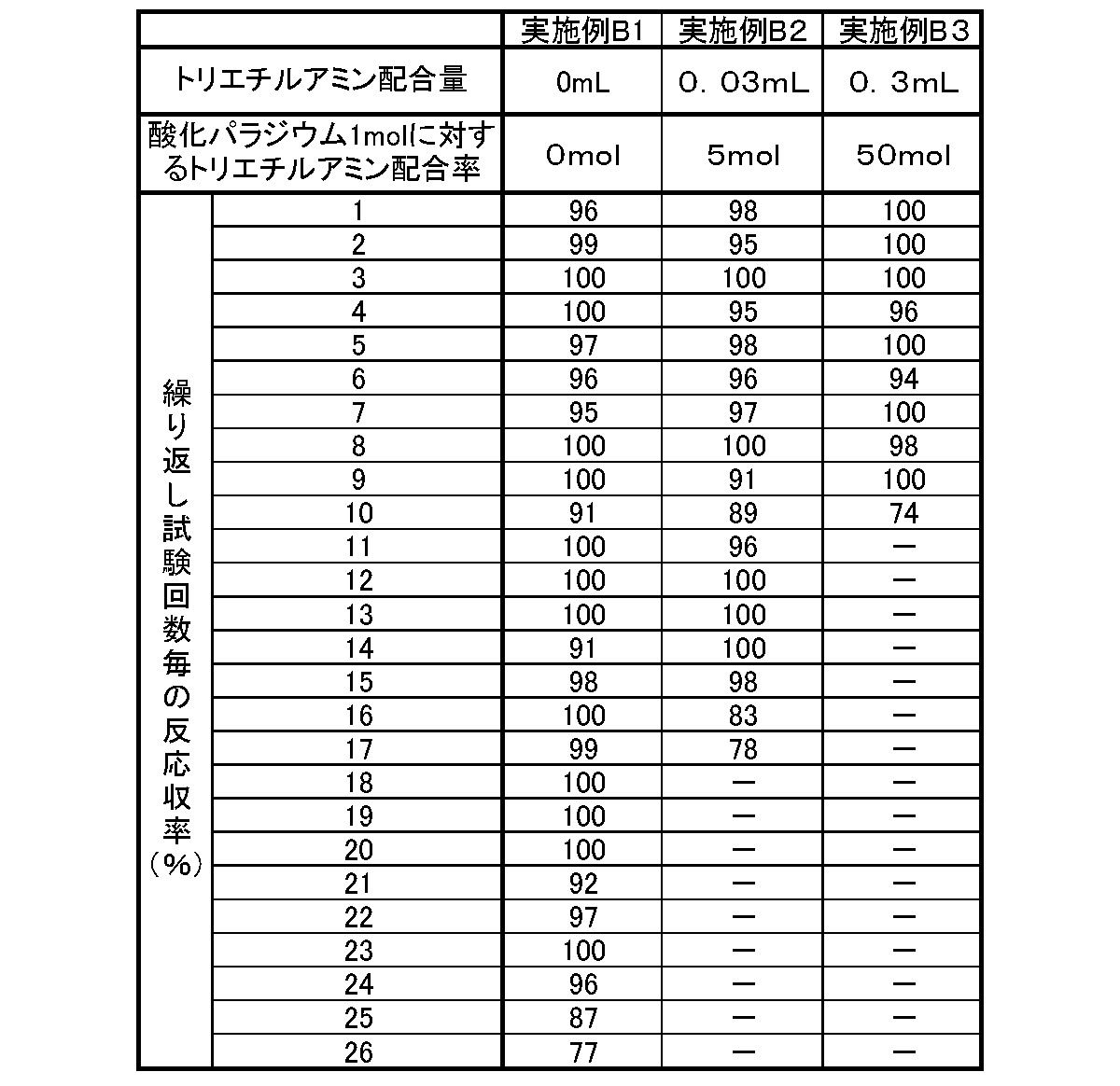
- Example B2 10 mg of palladium acetate (manufactured by Wako Pure Chemical Industries, Ltd.), 1 g of a phenol resin carrier produced in the same manner as Example B1, 0.03 mL of triethylamine (manufactured by Wako Pure Chemical Industries, Ltd., 5 mol relative to palladium acetate), acetonitrile ( 10 mL) (manufactured by Wako Pure Chemical Industries, Ltd.) was added and heated in a sealed tube at 100 ° C. for 12 hours. Subsequently, the phenol resin carrier was taken out with tweezers, then washed and dried to obtain a phenol resin-supported catalyst on which palladium particles were supported. The amount of palladium catalyst supported in the phenol resin supported catalyst was 1% by weight.
- Example B3 10 mg of palladium acetate (manufactured by Wako Pure Chemical Industries, Ltd.), 1 g of a phenol resin carrier prepared in the same manner as Example B1, 0.3 mL of triethylamine (50 mol relative to palladium acetate, manufactured by Wako Pure Chemical Industries, Ltd.), acetonitrile ( 10 mL) (manufactured by Wako Pure Chemical Industries, Ltd.) was added and heated in a sealed tube at 100 ° C. for 12 hours. Subsequently, the phenol resin carrier was taken out with tweezers, washed and dried to obtain a phenol resin-supported catalyst on which palladium particles were supported. The amount of palladium catalyst supported in the phenol resin supported catalyst was 1% by weight.
- the catalytic activity of the supported catalyst was evaluated by the reaction yield of trans-methyl cinnamate obtained by Heck reaction between iodobenzene and methyl acrylate.
- 230 ⁇ L (2.0 mmol) of iodobenzene, 230 ⁇ L (2.5 mmol) of methyl acrylate, and 350 ⁇ L (2.5 mmol) of triethylamine were dissolved in acetonitrile (20 mL).
- 500 mg of the supported catalyst was added to the obtained solution.
- the mixture was heated using an oil bath and stirred at 120 ° C. for 12 hours. After completion of the reaction, the supported catalyst was removed by tweezers or collected by filtration.
- the reaction yield was calculated from the area ratio of the chromatogram before and after the reaction.
- reaction yield is defined by (C 0 -C f ) / C 0 ⁇ 100 (%), where C 0 is the number of moles of iodobenzene before the reaction, and C f is the number of moles of iodobenzene after the reaction. It is.
- the phenol resin-supported catalysts of the examples are excellent in terms of repeated use of the catalyst as the amount of reducing agent used in the production process is reduced. That is, when the resin-supported catalyst described in the examples is used, excellent catalytic activity can be maintained for a long time without exchanging the catalyst.
- the present invention includes the following aspects.
- [1-1] A cured product of a thermosetting resin having a phenolic hydroxyl group;
- a resin-supported catalyst comprising fine particles having catalytic activity supported on the surface of a cured product of the thermosetting resin.
- [1-2] In the resin-supported catalyst according to [1-1], The resin-carrying catalyst whose phenolic hydroxyl group equivalent of the said thermosetting resin is 30 g / eq or more and 500 g / eq or less.
- thermosetting resin is a resin-supported catalyst obtained by curing the thermosetting resin at 250 ° C. or lower.
- a resin-supported catalyst, wherein the fine particles having catalytic activity include one or more of a metal, a metal oxide, and a metal compound.
- a method for producing a resin-supported catalyst, wherein a hydroxyl equivalent of the thermosetting resin is 30 g / eq or more and 500 g / eq or less.
- thermosetting resin contains a phenol resin.
- the said hardening process is a manufacturing method of the resin supported catalyst including the process of hardening
- a reducing agent selected from the group consisting of hydrogen, carbon monoxide, aldehydes, carboxylic acids, amines, metal hydrides, and hydrazine is used in an amount of 0 mol or more with respect to 1 mol of the material.
- the method for producing a resin-supported catalyst according to [2-1] which is contained in an amount of 10 mol or less.
- any reducing agent selected from the group consisting of hydrogen, carbon monoxide, aldehydes, carboxylic acids, amines, metal hydrides, and hydrazine is not added.
- a metal in which the fine particles are titanium, chromium, cobalt, nickel, copper, ruthenium, rhodium, palladium, rhenium, osmium, platinum, iron, zinc, manganese, magnesium, calcium, silver, vanadium, tin, and
- the resin according to any one of [2-1] to [2-3], which is formed from a material containing any one or more of oxides, other metal compounds and complexes of organic titanium A method for producing a supported catalyst.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inorganic Chemistry (AREA)
- Catalysts (AREA)
- Crystallography & Structural Chemistry (AREA)
Abstract
Description
熱硬化性樹脂の硬化体と、
前記硬化体の表面に担持された触媒活性を有する微粒子と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する樹脂担持触媒が提供される。
前記硬化工程において得られた硬化体の表面に触媒活性を有する微粒子を担持させる担持工程と、
を含む樹脂担持触媒の製造方法が提供される。
本実施形態における樹脂担持触媒は、触媒微粒子が熱硬化性樹脂の硬化体の表面に担持されているため、反応物が触媒微粒子に到達しやすい。そのため、触媒の利用効率が優れている。
また、本実施形態における樹脂担持触媒は触媒の繰り返し特性に優れている。その理由は必ずしも明らかではないが、以下のように考えられる。本実施形態における樹脂担持触媒では、担体として表面にフェノール性水酸基を有する熱硬化性樹脂の硬化体が用いられている。そして、触媒担体に存在するフェノール性水酸基により担持されているものと考えられる。こうすることにより、触媒微粒子は、従来の担持触媒と比較して、強固に担持されているものと考えられる。したがって、触媒担体である熱硬化性樹脂の硬化体から、触媒微粒子が脱離しにくくなり、触媒の繰り返し特性と利用効率が向上したものと考えられる。
熱硬化性樹脂の硬化体は、本実施形態において触媒担体として用いられる。熱硬化性樹脂の硬化体は、例えば、粒子状、膜状、均一な層状に形成していてもよい。硬化処理前の熱硬化性樹脂としては、フェノール性水酸基を有する熱硬化性樹脂であればとくに限定されないが、フェノール樹脂またはその誘導体を含むことが好ましい。
例えば、フェノール樹脂、クレゾール樹脂、レゾルシン樹脂、キシレノール樹脂、ナフトール樹脂、ビスフェノールA樹脂、アラルキルフェノール樹脂、ビフェニルアラルキルフェノール樹脂、およびフェノール性水酸基を有するカシューナッツ油などによる変性フェノール樹脂などが挙げられる。また、フェノール性水酸基を有する物質を含む、キシレン変性フェノール樹脂、およびフェノール類とロジン、テルペン油などで変性した油変性フェノール樹脂、ゴムで変性したゴム変性フェノール樹脂などの各種変性フェノール樹脂なども使用することができる。
本実施形態における熱硬化性樹脂の硬化体の表面に担持される触媒微粒子としては、触媒活性を有するものであれば金属、金属酸化物および金属化合物のいずれであってもよく、とくに限定されるものではない。例えばチタン、クロム、コバルト、ニッケル、銅、ルテニウム、ロジウム、パラジウム、レニウム、オスミニウム、白金、鉄、亜鉛、マンガン、マグネシウム、カルシウム、銀、バナジウム、スズなどの金属ならびにその酸化物、その他の有機チタンなどの金属化合物および錯体などの中から選ばれる少なくとも1種からなるものが挙げられる。また、これらのうちの少なくとも二種類以上を含む複合体も使用することもできる。これらの中でも、とくにパラジウムまたは白金が好適に用いられる。
本実施形態における熱硬化性樹脂の硬化処理方法としてはとくに限定されないが、公知の方法を採用することができる。
本実施形態における熱硬化性樹脂の硬化体への上記触媒微粒子の担持方法について詳細に説明する。本実施形態においては、熱硬化性樹脂の硬化体を作製後、その硬化体の表面に触媒微粒子を担持させることが好ましい。こうすることで、触媒微粒子が硬化体の内部に取り込まれるのを抑制することができる。
本実施形態に係る基材担持触媒は、シート状あるいは板状であることで、反応装置の形状に合わせて、種々の形状に変形させることが可能である。触媒シートの形状は、例えば、折りたたまれた状態、丸めた状態など反応装置における反応管の形状に合わせて種々の形状に変形させることができる。シート状の樹脂担持触媒において連通孔が設けられている場合、触媒フィルターとして用いることも可能である。
次に、基材の形状は、特に限定されるものではないが、例えば、粒子状、シート状あるいは板状が用いられる。これらの中でも、シート状あるいは板状であることが好ましい。
本実施形態において熱硬化性樹脂の硬化体を基材の表面に形成する方法は、基材の形状によって、適宜方法を選択することができる。例えば、基材がメッシュ状である場合、固形や粉末の樹脂の溶液や液状樹脂を基材に含浸硬化する方法、固形や粉末の樹脂を加熱溶融して基材に含浸硬化する方法、等が用いられる。これに対し、基材が粒子状である場合、熱硬化性樹脂の硬化体をコーティングする方法、等が用いられる。こうすることで、基材に均一に熱硬化性樹脂の硬化体を形成することが可能である。
第1の実施形態において用いられている触媒微粒子の具体的な製造方法に関する。
しかしながら、還元剤を用いて事前に触媒微粒子を還元する方法は、触媒微粒子の凝集という観点では、不十分であった。
上述したように従来の樹脂担持触媒の製造方法では、還元剤を用いて事前に触媒微粒子を還元している。ここで用いている還元剤としては、例えば、水素、一酸化炭素、ホルムアルデヒドなどのアルデヒド類、ギ酸やシュウ酸などのカルボン酸類、トリエチルアミンなどのアミン類、水素化ホウ素ナトリウムや水素化アルミニウムリチウムなどの金属ヒドリド、およびヒドラジンが挙げられる。
(実施例A1)
(フェノール樹脂の硬化体の作製)
撹拌装置、還流冷却器および温度計を備えた5Lの円筒型セパラブルフラスコ中にフェノール1300部、43%ホルムアルデヒド水溶液1600部、水800部およびトリエチルアミン30部および懸濁剤としてポリビニルアルコール(クラレポバールPVA117、けん化度98%、重合度1700)30部を入れ100℃で3時間加熱し球状フェノール樹脂硬化体を合成した。なお、反応1時間経過時に反応物を採取し、凍結乾燥しアセチル化法でのフェノール性水酸基当量を測定したところ、フェノール性水酸基当量は115g/eqだった。合成した球状フェノール樹脂硬化体を固液分離し、150℃で乾燥し平均粒径100μmの球状フェノール樹脂硬化体を得た。
酢酸パラジウム(和光純薬工業社製)5mgと、触媒担体である上記の球状フェノール樹脂硬化体500mgと、トリエチルアミン(和光純薬工業社製)0.15mLと、アセトニトリル(和光純薬工業社製)5mLを配合し、封管中、100℃で12時間加熱した。つづいて、分散液をろ過した後、ろ過物を洗浄し、乾燥することで、パラジウム粒子が表面に担持された樹脂担持触媒を得た。樹脂担持触媒中におけるパラジウム触媒の担持量は1wt%であった。また、この樹脂担持触媒の比表面積は、10m2/g以下であった。なお、パラジウム触媒の担持量は、市販の原子吸光分光光度計を用いて測定し、比表面積は窒素ガス、BET3点法で測定した。
市販のレゾール型フェノール樹脂(住友ベークライト社製、スミライトレジン(r)PR-50087、フェノール性水酸基当量130g/eq)50部にメチルエチルケトン50部を混合し、濾紙(ADVANTEC(r)No.590)に含浸して180℃で1時間乾燥しフェノール樹脂硬化体(フェノール樹脂分50wt%)を得た。さらに、実施例A1と同様の方法で樹脂担持触媒を得た。この樹脂担持触媒のパラジウム触媒の担持量は1wt%、比表面積は、20m2/gであった。
フェノール樹脂を市販のノボラック型フェノール樹脂(住友ベークライト社製、スミライトレジン(r)PR-310、フェノール性水酸基当量105g/eq)30部をメチルエチルケトン70部に溶解した以外は、実施例A2と同様の方法で樹脂担持触媒(触媒担体中のフェノール樹脂分50wt%)を得た。この樹脂担持触媒のパラジウム触媒の担持量は1wt%、比表面積は、25m2/gであった。
実施例A1で得られた球状フェノール樹脂硬化体を市販の炭化賦活炉で、窒素気流下および空気気流下で900℃で炭化賦活した。さらに、実施例A1と同様の方法で樹脂活性炭担持触媒を得た。この樹脂活性炭担持触媒のパラジウム触媒の担持量は1wt%、比表面積は、950m2/gであった。
フェノール性水酸基を含有しない熱硬化性樹脂の硬化体を触媒担体とした、市販のパラジウム‐尿素樹脂(和光純薬工業社製、Pdエンキャット30)を比較例A2とした。このパラジウム‐尿素樹脂のパラジウム触媒の担持量は4.2wt%、比表面積は10m2/g以下であった。
市販のパラジウム‐活性炭(和光純薬工業社製、パラジウム‐活性炭素)を比較例A3とした。このパラジウム‐活性炭素のパラジウム触媒の担持量は5wt%、比表面積は650m2/gであった。
担持触媒の触媒活性は、ヨードベンゼンと、アクリル酸メチルとのヘック反応で得られるトランス-桂皮酸メチルの反応収率により評価した。
ヨードベンゼン23μL(0.20mmol)、アクリル酸メチル23μL(0.25mmol)、トリエチルアミン35μL(0.25mmol)をアセトニトリル(2mL)に溶解した。得られた溶液に担持触媒を50mg加えた。混合物をオイルバスを用いて加熱し、120℃で12時間攪拌した。反応終了後、担持触媒をろ別した。
ろ液を減圧濃縮し、残渣をシリカゲルクロマトグラフィー(n-ヘキサン:酢酸エチル=5:1)で精製し、トランス-桂皮酸メチルを得た。
なお、必要に応じて、トランス-桂皮酸メチルを取り出すことなく、反応液を一定量取り出し、液体クロマトグラフィーを用いて分析した。以下、とくに断らない限り、反応前後のクロマトグラムの面積比から反応収率を算出した。ここで、反応収率は、(C0-Cf)/C0×100(%)で定義され、C0は反応前のヨードベンゼンのモル数、Cfは反応後のヨードベンゼンのモル数である。
反応終了後、反応液から担持触媒を一旦取り出し、洗浄した。その後、再度担持触媒として上記のヘック反応をおこなった。これらのヘック反応の一連の操作を4回繰り返した。
(実施例B1)
(フェノール樹脂の硬化体の作製)
液状フェノール樹脂(住友ベークライト社製、スミライトレジンPR-50087)とメタノールを重量比1対1で混合したフェノール樹脂溶液に、シート状のポリプロピレン不織布(日本バイリーン社製)を室温で1分間浸し、常温で30分間自然乾燥した。乾燥後、90℃で30分間加熱し、さらに150℃で30分間加熱してフェノール樹脂を30重量%含むシート状のフェノール樹脂担体を得た。
酢酸パラジウム(和光純薬工業社製)10mgと、触媒担体である上記フェノール樹脂触媒担体1gと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は1重量%であった。パラジウム触媒の担持量は、市販の原子吸光分光光度計を用いて測定した。
酢酸パラジウム(和光純薬工業社製)10mgと、実施例B1と同様に作製したフェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製,酢酸パラジウムに対して5mol)0.03mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することによって、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は1重量%であった。
酢酸パラジウム(和光純薬工業社製)10mgと、実施例B1と同様に作製したフェノール樹脂担体1gと、トリエチルアミン(和光純薬工業社製,酢酸パラジウムに対して50mol)0.3mLと、アセトニトリル(和光純薬工業社製)10mLを配合し、封管中、100℃で12時間加熱した。つづいて、フェノール樹脂担体をピンセットで取り出した後、洗浄し乾燥することで、パラジウム粒子が担持されたフェノール樹脂担持触媒を得た。フェノール樹脂担持触媒中におけるパラジウム触媒の担持量は1重量%であった。
担持触媒の触媒活性は、ヨードベンゼンと、アクリル酸メチルとのヘック反応で得られるトランス-桂皮酸メチルの反応収率により評価した。
ヨードベンゼン230μL(2.0mmol)、アクリル酸メチル230μL(2.5mmol)、トリエチルアミン350μL(2.5mmol)をアセトニトリル(20mL)に溶解した。得られた溶液に担持触媒を500mg加えた。混合物をオイルバスを用いて加熱し、120℃で12時間攪拌した。反応終了後、担持触媒をピンセットによって取り出すか、あるいは濾別することによって回収した。
反応終了後、反応液から担持触媒を回収し、洗浄した。その後、回収した担持触媒を用いて上記のヘック反応をおこなった。これらのヘック反応の一連の操作を、トランス-桂皮酸メチルの反応収率が80%を下回るまで繰り返した。その結果を下記表2に示す。
[1-1]フェノール性水酸基を有する熱硬化性樹脂の硬化物と、
前記熱硬化性樹脂の硬化物の表面に担持された触媒活性を有する微粒子と
を含む、樹脂担持触媒。
[1-2][1-1]に記載の樹脂担持触媒において、
前記熱硬化性樹脂のフェノール性水酸基当量が30g/eq以上500g/eq以下である、樹脂担持触媒。
[1-3][1-1]または[1-2]に記載の樹脂担持触媒において、
前記熱硬化性樹脂が、フェノール樹脂を含む、樹脂担持触媒。
[1-4][1-1]乃至[1-3]いずれか一つに記載の樹脂担持触媒において、
BET法による比表面積が、300m2/g以下である、樹脂担持触媒。
[1-5][1-1]乃至[1-4]いずれか一つに記載の樹脂担持触媒において、
前記熱硬化性樹脂の硬化物は、前記熱硬化性樹脂を250℃以下で硬化することにより得られる、樹脂担持触媒。
[1-6][1-1]乃至[1-5]いずれか一つに記載の樹脂担持触媒において、
前記熱硬化性樹脂の硬化後に、前記触媒活性を有する微粒子を前記硬化物の表面に担持させた、樹脂担持触媒。
[1-7][1-1]乃至[1-6]いずれか一項に記載の樹脂担持触媒において、
前記触媒活性を有する微粒子が、金属、金属酸化物および金属化合物のいずれか1種以上を含む、樹脂担持触媒。
[1-8]フェノール性水酸基を有する熱硬化性樹脂を硬化処理する硬化工程と、
前記熱硬化性樹脂の硬化物の表面に触媒活性を有する微粒子を担持させる担持工程と
を含む、樹脂担持触媒の製造方法。
[1-9][1-8]に記載の樹脂担持触媒の製造方法において、
前記熱硬化性樹脂の水酸基当量が30g/eq以上500g/eq以下である、樹脂担持触媒の製造方法。
[1-10][1-8]または[1-9]に記載の樹脂担持触媒の製造方法において、
前記熱硬化性樹脂が、フェノール樹脂を含む、樹脂担持触媒の製造方法。
[1-11][1-8]乃至[1-10]いずれか一つに記載の樹脂担持触媒の製造方法において、
前記硬化工程は前記熱硬化性樹脂を250℃以下で硬化する工程を含む、樹脂担持触媒の製造方法。
前記硬化工程において得られた硬化体の表面に触媒活性を有する微粒子を担持させる担持工程と、
を有し、
前記微粒子は、金属、金属酸化物および金属化合物のいずれか1種以上からなる材料により形成されたものであって、
前記担持工程は、前記フェノール性水酸基によって前記微粒子を還元する還元工程を含む樹脂担持触媒の製造方法。
[2-2]前記還元工程は、水素、一酸化炭素、アルデヒド類、カルボン酸類、アミン類、金属ヒドリド、およびヒドラジンから成る群より選択される還元剤を、前記材料1molに対して、0mol以上10mol以下含有することを特徴とする[2-1]に記載の樹脂担持触媒の製造方法。
[2-3]前記還元工程において、水素、一酸化炭素、アルデヒド類、カルボン酸類、アミン類、金属ヒドリド、およびヒドラジンからなる群より選択される還元剤は、いずれも加えないことを特徴とする[2-1]に記載の樹脂担持触媒の製造方法。
[2-4]前記微粒子が、チタン、クロム、コバルト、ニッケル、銅、ルテニウム、ロジウム、パラジウム、レニウム、オスミニウム、白金、鉄、亜鉛、マンガン、マグネシウム、カルシウム、銀、バナジウム、スズからなる金属ならびにその酸化物、その他の有機チタンからなる金属化合物および錯体のいずれか1種以上を含む材料により形成されたものである[2-1]乃至[2-3]のいずれか一つに記載の樹脂担持触媒の製造方法。
[2-5]前記熱硬化性樹脂がフェノール樹脂である[2-1]乃至[2-4]のいずれか一つに記載の樹脂担持触媒の製造方法。
[2-6]前記硬化工程は前記熱硬化性樹脂を250℃以下で硬化する工程である[2-1]乃至[2-5]のいずれか一つに記載の樹脂担持触媒の製造方法。
[2-7]基材を準備する工程をさらに含んでおり、
前記担持工程が、前記基材の表面に熱硬化性樹脂の前記硬化体を形成するとともに、前記熱硬化性樹脂の表面に触媒活性を有する微粒子を担持させる工程である[2-1]乃至[2-6]のいずれか一つに記載の樹脂担持触媒の製造方法。
[2-8]前記熱硬化性樹脂におけるフェノール性水酸基当量が500g/eq以下である[2-1]乃至[2-7]のいずれか一つに記載の樹脂担持触媒の製造方法。
Claims (16)
- 熱硬化性樹脂の硬化体と、
前記硬化体の表面に担持された触媒活性を有する微粒子と、
を含み、
前記熱硬化性樹脂がフェノール性水酸基を有する樹脂担持触媒。 - 前記熱硬化性樹脂がフェノール樹脂である請求項1に記載の樹脂担持触媒。
- 前記熱硬化性樹脂におけるフェノール性水酸基当量が500g/eq以下である請求項1または2に記載の樹脂担持触媒。
- BET法により測定された比表面積が300m2/g以下である請求項1乃至3のいずれか一項に記載の樹脂担持触媒。
- 前記微粒子は、金属、金属酸化物および金属化合物のいずれか1種以上からなる材料により形成されたものである請求項1乃至4のいずれか一項に記載の樹脂担持触媒。
- 前記微粒子は、チタン、クロム、コバルト、ニッケル、銅、ルテニウム、ロジウム、パラジウム、レニウム、オスミニウム、白金、鉄、亜鉛、マンガン、マグネシウム、カルシウム、銀、バナジウム、スズからなる金属ならびにその酸化物、その他の有機チタンからなる金属化合物および錯体のいずれか1種以上を含む材料により形成されたものである請求項1乃至5のいずれか一項に記載の樹脂担持触媒。
- フェノール性水酸基を有する熱硬化性樹脂を硬化させる硬化工程と、
前記硬化工程において得られた硬化体の表面に触媒活性を有する微粒子を担持させる担持工程と、
を含む樹脂担持触媒の製造方法。 - 前記微粒子は、金属、金属酸化物および金属化合物のいずれか1種以上からなる材料により形成されたものである請求項7に記載の樹脂担持触媒の製造方法。
- 前記担持工程は、前記フェノール性水酸基によって前記微粒子を還元する還元工程を含む請求項8に記載の樹脂担持触媒の製造方法。
- 前記還元工程は、水素、一酸化炭素、アルデヒド類、カルボン酸類、アミン類、金属ヒドリド、およびヒドラジンからなる群より選択される還元剤を、前記材料1molに対して、0mol以上10mol以下含有する請求項9に記載の樹脂担持触媒の製造方法。
- 前記還元工程は、前記還元剤を、用いない工程である請求項10に記載の樹脂担持触媒の製造方法。
- 前記微粒子が、チタン、クロム、コバルト、ニッケル、銅、ルテニウム、ロジウム、パラジウム、レニウム、オスミニウム、白金、鉄、亜鉛、マンガン、マグネシウム、カルシウム、銀、バナジウム、スズからなる金属ならびにその酸化物、その他の有機チタンからなる金属化合物および錯体のいずれか1種以上を含む材料により形成されたものである請求項7乃至11のいずれか一項に記載の樹脂担持触媒の製造方法。
- 前記熱硬化性樹脂がフェノール樹脂である請求項7乃至12のいずれか一項に記載の樹脂担持触媒の製造方法。
- 前記硬化工程は前記熱硬化性樹脂を250℃以下で硬化する工程である請求項7乃至13のいずれか一項に記載の樹脂担持触媒の製造方法。
- 前記熱硬化性樹脂におけるフェノール性水酸基当量が500g/eq以下である請求項7乃至14のいずれか一項に記載の樹脂担持触媒の製造方法。
- 前記微粒子は、前記熱硬化性樹脂を硬化させた後に、前記硬化体の表面に担持される請求項7乃至15のいずれか一項に記載の樹脂担持触媒の製造方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/342,315 US9314783B2 (en) | 2011-09-06 | 2012-08-28 | Resin-supported catalyst and method for preparing resin-supported catalyst |
| KR1020147008844A KR20140059277A (ko) | 2011-09-06 | 2012-08-28 | 수지 담지 촉매 및 수지 담지 촉매의 제조 방법 |
| RU2014113353/04A RU2014113353A (ru) | 2011-09-06 | 2012-08-28 | Катализатор, нанесенный на смолу, и способ получения катализатора, нанесенного на смолу |
| CA2848167A CA2848167A1 (en) | 2011-09-06 | 2012-08-28 | Resin-supported catalyst and method for preparing resin-supported catalyst |
| EP12829809.8A EP2754490A4 (en) | 2011-09-06 | 2012-08-28 | ART RESIN-BASED CATALYST AND METHOD FOR PRODUCING THE ARTIFICIAL RESIN-BASED CATALYST |
| CN201280042953.3A CN103764283A (zh) | 2011-09-06 | 2012-08-28 | 树脂担载催化剂和树脂担载催化剂的制造方法 |
| IN1623DEN2014 IN2014DN01623A (ja) | 2011-09-06 | 2012-08-28 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011193647A JP5885969B2 (ja) | 2011-09-06 | 2011-09-06 | 樹脂担持触媒および樹脂担持触媒の製造方法 |
| JP2011-193647 | 2011-09-06 | ||
| JP2012-174359 | 2012-08-06 | ||
| JP2012174359A JP5931641B2 (ja) | 2012-08-06 | 2012-08-06 | 樹脂担持触媒の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013035270A1 true WO2013035270A1 (ja) | 2013-03-14 |
Family
ID=47831746
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/005388 Ceased WO2013035270A1 (ja) | 2011-09-06 | 2012-08-28 | 樹脂担持触媒および樹脂担持触媒の製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9314783B2 (ja) |
| EP (1) | EP2754490A4 (ja) |
| KR (1) | KR20140059277A (ja) |
| CN (1) | CN103764283A (ja) |
| CA (1) | CA2848167A1 (ja) |
| IN (1) | IN2014DN01623A (ja) |
| RU (1) | RU2014113353A (ja) |
| TW (1) | TW201323086A (ja) |
| WO (1) | WO2013035270A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103483851A (zh) * | 2013-09-22 | 2014-01-01 | 苏州市湘园特种精细化工有限公司 | 一种热固性废弃塑料的再加工方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104368390B (zh) * | 2014-10-09 | 2016-06-29 | 浙江工业大学 | 一种表面接枝多点桥连负载型tempo催化剂及其制备 |
| CN107778141B (zh) * | 2016-08-30 | 2021-08-06 | 中国石油化工股份有限公司 | 一种1,4-丁二醇的纯化方法 |
| CN106423155A (zh) * | 2016-11-03 | 2017-02-22 | 中国检验检疫科学研究院 | 一种制备棒状Au@TiO2 复合纳米光催化材料的方法 |
| CN108404983B (zh) * | 2018-03-23 | 2020-08-04 | 华东师范大学 | 一种有序介孔酚醛树脂聚合物负载银催化剂的制备及其应用 |
| CN113318793A (zh) * | 2021-04-30 | 2021-08-31 | 浙江工商大学 | 一种产双氧水和去除污染物的新材料及其制备方法和应用 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02307976A (ja) * | 1989-05-19 | 1990-12-21 | Unitika Ltd | 繊維用抗菌剤およびその製造方法 |
| JPH07238183A (ja) * | 1994-02-28 | 1995-09-12 | Catalysts & Chem Ind Co Ltd | 抗菌性樹脂成形品およびその製造法 |
| JP2000140643A (ja) | 1998-11-13 | 2000-05-23 | Dainippon Ink & Chem Inc | 金属系触媒の固定方法 |
| JP2002201284A (ja) * | 2001-01-09 | 2002-07-19 | Nippon Paint Co Ltd | 金属微粒子担持樹脂粒子およびその製造方法 |
| JP2007237065A (ja) * | 2006-03-08 | 2007-09-20 | Miyoshi Oil & Fat Co Ltd | 光触媒活性を有する抗菌剤及び抗菌処理方法 |
| JP2010022980A (ja) | 2008-07-23 | 2010-02-04 | Kyocera Chemical Corp | 球状触媒粒子とその製造方法 |
| JP2010207777A (ja) * | 2009-03-12 | 2010-09-24 | Osaka Municipal Technical Research Institute | カラムリアクター及びその製造方法 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5104575A (en) * | 1987-09-30 | 1992-04-14 | Union Carbide Chemicals & Plastics Technology Corporation | Alkoxylation using heterogeneous calcium catalysts and products therefrom |
| US5488023A (en) * | 1994-08-12 | 1996-01-30 | Corning Incorporated | Method of making activated carbon having dispersed catalyst |
| WO1999017874A1 (en) * | 1997-10-08 | 1999-04-15 | Corning Incorporated | Method of making activated carbon-supported catalysts |
| JPH11140277A (ja) * | 1997-11-10 | 1999-05-25 | Sumitomo Bakelite Co Ltd | エポキシ樹脂組成物及びこれを用いた半導体装置 |
| JP3460820B2 (ja) * | 1999-12-08 | 2003-10-27 | 日本電気株式会社 | 難燃性エポキシ樹脂組成物 |
| JP2002241590A (ja) * | 2001-02-20 | 2002-08-28 | Ajinomoto Co Inc | 難燃性エポキシ樹脂組成物 |
| JP2003147052A (ja) * | 2001-11-12 | 2003-05-21 | Nec Corp | 難燃性エポキシ樹脂組成物 |
| US20070060720A1 (en) * | 2003-07-03 | 2007-03-15 | Yukihiro Kiuchi | Epoxy resin composition |
| JP5234726B2 (ja) * | 2007-09-20 | 2013-07-10 | 花王株式会社 | 3級アミンの製造法 |
| US8314045B1 (en) * | 2009-10-27 | 2012-11-20 | Entreprises Sinoncelli S.A.R.L. | Solid acid catalyst |
| JP5413127B2 (ja) * | 2009-10-29 | 2014-02-12 | 住友ベークライト株式会社 | 半導体封止用樹脂組成物、これを用いる半導体装置及び半導体封止用樹脂組成物の製造方法 |
| IN2014DN07208A (ja) * | 2012-03-21 | 2015-04-24 | Sumitomo Bakelite Co |
-
2012
- 2012-08-28 CN CN201280042953.3A patent/CN103764283A/zh active Pending
- 2012-08-28 CA CA2848167A patent/CA2848167A1/en not_active Abandoned
- 2012-08-28 EP EP12829809.8A patent/EP2754490A4/en not_active Withdrawn
- 2012-08-28 KR KR1020147008844A patent/KR20140059277A/ko not_active Withdrawn
- 2012-08-28 WO PCT/JP2012/005388 patent/WO2013035270A1/ja not_active Ceased
- 2012-08-28 RU RU2014113353/04A patent/RU2014113353A/ru unknown
- 2012-08-28 IN IN1623DEN2014 patent/IN2014DN01623A/en unknown
- 2012-08-28 US US14/342,315 patent/US9314783B2/en not_active Expired - Fee Related
- 2012-08-30 TW TW101131499A patent/TW201323086A/zh unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02307976A (ja) * | 1989-05-19 | 1990-12-21 | Unitika Ltd | 繊維用抗菌剤およびその製造方法 |
| JPH07238183A (ja) * | 1994-02-28 | 1995-09-12 | Catalysts & Chem Ind Co Ltd | 抗菌性樹脂成形品およびその製造法 |
| JP2000140643A (ja) | 1998-11-13 | 2000-05-23 | Dainippon Ink & Chem Inc | 金属系触媒の固定方法 |
| JP2002201284A (ja) * | 2001-01-09 | 2002-07-19 | Nippon Paint Co Ltd | 金属微粒子担持樹脂粒子およびその製造方法 |
| JP2007237065A (ja) * | 2006-03-08 | 2007-09-20 | Miyoshi Oil & Fat Co Ltd | 光触媒活性を有する抗菌剤及び抗菌処理方法 |
| JP2010022980A (ja) | 2008-07-23 | 2010-02-04 | Kyocera Chemical Corp | 球状触媒粒子とその製造方法 |
| JP2010207777A (ja) * | 2009-03-12 | 2010-09-24 | Osaka Municipal Technical Research Institute | カラムリアクター及びその製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| "Catalyst Handbook", 10 December 2008, KODANSHA LTD., pages: 309 - 311 |
| MUROI TAKAGI: "Industrial noble metal catalysts -practice and reaction of practical metal catalyst", 26 May 2003, JETTY CO., LTD., pages: 6 - 7 |
| See also references of EP2754490A4 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103483851A (zh) * | 2013-09-22 | 2014-01-01 | 苏州市湘园特种精细化工有限公司 | 一种热固性废弃塑料的再加工方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2848167A1 (en) | 2013-03-14 |
| US9314783B2 (en) | 2016-04-19 |
| US20140213434A1 (en) | 2014-07-31 |
| IN2014DN01623A (ja) | 2015-05-15 |
| EP2754490A4 (en) | 2014-12-10 |
| CN103764283A (zh) | 2014-04-30 |
| EP2754490A1 (en) | 2014-07-16 |
| TW201323086A (zh) | 2013-06-16 |
| KR20140059277A (ko) | 2014-05-15 |
| RU2014113353A (ru) | 2015-10-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9314783B2 (en) | Resin-supported catalyst and method for preparing resin-supported catalyst | |
| Zhao et al. | Ultrafine and highly dispersed platinum nanoparticles confined in a triazinyl-containing porous organic polymer for catalytic applications | |
| Yang et al. | Highly dispersed ultrafine palladium nanoparticles encapsulated in a triazinyl functionalized porous organic polymer as a highly efficient catalyst for transfer hydrogenation of aldehydes | |
| CN109999820B (zh) | 一种用于氢化石油树脂制备的镍基催化剂及其制备方法与应用 | |
| US9527067B2 (en) | Inorganic/polymeric hybrid catalytic materials containing metal nano-particles therein | |
| JP2010070738A (ja) | フェノール樹脂球形粒子の硬化物の製造方法 | |
| KR101578071B1 (ko) | 귀금속 담지 촉매의 고분산 제조 방법 | |
| CN107876047B (zh) | 一种α,β-不饱和醛/酮加氢用Pd/C催化剂的制备方法 | |
| Cheong et al. | Au-Pd core-shell nanoparticles as alcohol oxidation catalysts: Effect of shape and composition | |
| Shen et al. | Hierarchical Pd@ PC-COFs as efficient catalysts for phenol hydrogenation | |
| CN108380182A (zh) | 酚醛树脂基活性炭及其制备方法和应用 | |
| Villa et al. | Material science for the support design: a powerful challenge for catalysis | |
| CN115007184A (zh) | 一种氮掺杂碳负载的钌-铁双金属催化剂及其制备方法和应用 | |
| CN110327975B (zh) | 氢甲酰化催化剂及其制备方法和应用 | |
| EP2952252A1 (en) | Selective hydrogenation catalyst, production method for selective hydrogenation catalyst, and selective hydrogenation method | |
| JP5931641B2 (ja) | 樹脂担持触媒の製造方法 | |
| JP5886145B2 (ja) | 基材担持触媒および基材担持触媒の製造方法 | |
| WO2013140705A1 (ja) | 基材担持触媒および基材担持触媒の製造方法 | |
| JP5885969B2 (ja) | 樹脂担持触媒および樹脂担持触媒の製造方法 | |
| JP5905753B2 (ja) | 基材担持触媒および基材担持触媒の製造方法 | |
| JP2014169236A (ja) | 飽和炭化水素又は不飽和炭化水素の合成方法、合成物及び触媒複合体 | |
| JP2014148485A (ja) | 求電子部位を有する化合物または不飽和炭化水素の水素化方法、水素化物および触媒複合体 | |
| JPH0147433B2 (ja) | ||
| CN115945201B (zh) | 一种金属掺杂活性炭微球的制备方法和应用 | |
| CN108144654B (zh) | 一种酚醛交联法制备三维石墨烯负载纳米Pd催化剂及其在硝基苯加氢中的应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12829809 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14342315 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2848167 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012829809 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012829809 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20147008844 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2014113353 Country of ref document: RU Kind code of ref document: A |