WO2013100132A1 - ペプチド化合物の環化方法 - Google Patents
ペプチド化合物の環化方法 Download PDFInfo
- Publication number
- WO2013100132A1 WO2013100132A1 PCT/JP2012/084103 JP2012084103W WO2013100132A1 WO 2013100132 A1 WO2013100132 A1 WO 2013100132A1 JP 2012084103 W JP2012084103 W JP 2012084103W WO 2013100132 A1 WO2013100132 A1 WO 2013100132A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- amino acid
- compound
- residue
- acid analog
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*(NC)=C)=O Chemical compound CC(*(NC)=C)=O 0.000 description 15
- ANOKIZYHJHWXPE-HNNXBMFYSA-N CC(C)(C)OC(N[C@@H](CSSC(C)(C)C)C(NCc1ccccc1)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CSSC(C)(C)C)C(NCc1ccccc1)=O)=O ANOKIZYHJHWXPE-HNNXBMFYSA-N 0.000 description 2
- QJQDQUCENWAMAS-UHFFFAOYSA-N CCN(C(C)C)C(C)CC(C#N)Br Chemical compound CCN(C(C)C)C(C)CC(C#N)Br QJQDQUCENWAMAS-UHFFFAOYSA-N 0.000 description 2
- JTTARFOFOAZVQT-ZETCQYMHSA-N C[C@@H](COC(C)(C)C)C(NCC(N)=O)=O Chemical compound C[C@@H](COC(C)(C)C)C(NCC(N)=O)=O JTTARFOFOAZVQT-ZETCQYMHSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N C1CCNCC1 Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- QWMMBQYDEVMKTK-VIFPVBQESA-N CC(C)(C)OC(N(C)[C@@H](CSSC(C)(C)C)C(O)=O)=O Chemical compound CC(C)(C)OC(N(C)[C@@H](CSSC(C)(C)C)C(O)=O)=O QWMMBQYDEVMKTK-VIFPVBQESA-N 0.000 description 1
- LFKDJXLFVYVEFG-UHFFFAOYSA-N CC(C)(C)OC(N)=O Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 1
- FFTXXCRVJUBYOP-KIYNQFGBSA-N CC(C)(C)OC(N[C@@H](CC(CN=N)SSC(C)(C)C)C(OCC#N)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CC(CN=N)SSC(C)(C)C)C(OCC#N)=O)=O FFTXXCRVJUBYOP-KIYNQFGBSA-N 0.000 description 1
- JJEICSIUYZNFAJ-FQEVSTJZSA-N CC(C)(C)OC(N[C@@H](CC(NCC(OCc1ccccc1)=O)=O)C(OCc1ccccc1)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CC(NCC(OCc1ccccc1)=O)=O)C(OCc1ccccc1)=O)=O JJEICSIUYZNFAJ-FQEVSTJZSA-N 0.000 description 1
- PPQRALRLGBAWHD-QMMMGPOBSA-N CC(C)(C)OC(N[C@@H](CSSC(C)(C)C)C(O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CSSC(C)(C)C)C(O)=O)=O PPQRALRLGBAWHD-QMMMGPOBSA-N 0.000 description 1
- PINZAMFDEPERMX-QFIPXVFZSA-N CC(C)(C)OC([C@H](CCC(N(C)C)=O)NC(OCC1c(cccc2)c2-c2ccccc12)=O)=O Chemical compound CC(C)(C)OC([C@H](CCC(N(C)C)=O)NC(OCC1c(cccc2)c2-c2ccccc12)=O)=O PINZAMFDEPERMX-QFIPXVFZSA-N 0.000 description 1
- FXVOKJIKEGWHLZ-OJSPPFRZSA-N CC(C)(C)SSCC[C@@H](C(O[C@H]([C@@H](COP(O)(O[C@@H](C1)C(COP(O)(O)=O)O[C@H]1N(C=CC(N)=N1)C1=O)=O)OC1[n]2c3ncnc(N)c3nc2)[C@H]1O)=O)N Chemical compound CC(C)(C)SSCC[C@@H](C(O[C@H]([C@@H](COP(O)(O[C@@H](C1)C(COP(O)(O)=O)O[C@H]1N(C=CC(N)=N1)C1=O)=O)OC1[n]2c3ncnc(N)c3nc2)[C@H]1O)=O)N FXVOKJIKEGWHLZ-OJSPPFRZSA-N 0.000 description 1
- BBMJELDHCZJMQA-LBPRGKRZSA-N CC(C)(C)SSC[C@@H](C(NCc1ccccc1)=O)N Chemical compound CC(C)(C)SSC[C@@H](C(NCc1ccccc1)=O)N BBMJELDHCZJMQA-LBPRGKRZSA-N 0.000 description 1
- AQCYZSJTMQORHF-INIZCTEOSA-N CC(C)(C)SSC[C@@H](C(NCc1ccccc1)=O)NSc(nccc1)c1N[O-] Chemical compound CC(C)(C)SSC[C@@H](C(NCc1ccccc1)=O)NSc(nccc1)c1N[O-] AQCYZSJTMQORHF-INIZCTEOSA-N 0.000 description 1
- WQBCZTUSIWRNDW-YFKPBYRVSA-N CC(C)[C@H](C)C(N)=O Chemical compound CC(C)[C@H](C)C(N)=O WQBCZTUSIWRNDW-YFKPBYRVSA-N 0.000 description 1
- RZUGYIVFTYWGQW-HECZYDGTSA-N CCC(N(C)C(Cc1ccccc1)C([N](C)(C)CC(N[C@@H](CC(C)N1[C@@H](C)C(N)=O)C1=O)=O)=O)=O Chemical compound CCC(N(C)C(Cc1ccccc1)C([N](C)(C)CC(N[C@@H](CC(C)N1[C@@H](C)C(N)=O)C1=O)=O)=O)=O RZUGYIVFTYWGQW-HECZYDGTSA-N 0.000 description 1
- NKWDQTKTGYQWLT-UHFFFAOYSA-N CCCNCC(OCC#N)=O Chemical compound CCCNCC(OCC#N)=O NKWDQTKTGYQWLT-UHFFFAOYSA-N 0.000 description 1
- BLCATMOIZIKIBU-WNKKXLCOSA-N CCSSC[C@@H](C(O[C@H]([C@@H](COP(O)(O[C@@H](C1)[C@@H](COP(O)(O)=O)O[C@H]1N(C=CC(N)=N1)C1=O)=O)OC1[n]2c3ncnc(N)c3nc2)[C@H]1O)=O)N(C)C(OCc(c([N+]([O-])=O)c1)cc(OC)c1OC)=O Chemical compound CCSSC[C@@H](C(O[C@H]([C@@H](COP(O)(O[C@@H](C1)[C@@H](COP(O)(O)=O)O[C@H]1N(C=CC(N)=N1)C1=O)=O)OC1[n]2c3ncnc(N)c3nc2)[C@H]1O)=O)N(C)C(OCc(c([N+]([O-])=O)c1)cc(OC)c1OC)=O BLCATMOIZIKIBU-WNKKXLCOSA-N 0.000 description 1
- QEXYOHMTAOCTDV-TUSQITKMSA-N C[C@@H](C(N(C)[C@@H](Cc1ccccc1)C([N](C)(C)CC(N)=O)=O)=O)NC([C@H](Cc1ccccc1)NC(OC(C)(C)C)=O)=O Chemical compound C[C@@H](C(N(C)[C@@H](Cc1ccccc1)C([N](C)(C)CC(N)=O)=O)=O)NC([C@H](Cc1ccccc1)NC(OC(C)(C)C)=O)=O QEXYOHMTAOCTDV-TUSQITKMSA-N 0.000 description 1
- MQQOAUOXXYAZOQ-LOJRBXKRSA-N C[C@@H](C(N)=O)N(C(C)C[C@@H]1C)C1=O Chemical compound C[C@@H](C(N)=O)N(C(C)C[C@@H]1C)C1=O MQQOAUOXXYAZOQ-LOJRBXKRSA-N 0.000 description 1
- YIVCZMHNRHERGG-XHNBAOFPSA-N C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C([C@H](C1)NC(OCC2c(cccc3)c3-c3c2cccc3)=O)=O)[O]=C1OCc1ccccc1)=O Chemical compound C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C([C@H](C1)NC(OCC2c(cccc3)c3-c3c2cccc3)=O)=O)[O]=C1OCc1ccccc1)=O YIVCZMHNRHERGG-XHNBAOFPSA-N 0.000 description 1
- IPBNVVDEDQVEQX-ANZMCEIWSA-N C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C([C@H](C1)NC(OCC2c3ccccc3-c3c2cccc3)=O)=O)[O]=C1O)=O Chemical compound C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C([C@H](C1)NC(OCC2c3ccccc3-c3c2cccc3)=O)=O)[O]=C1O)=O IPBNVVDEDQVEQX-ANZMCEIWSA-N 0.000 description 1
- LGMFOSIGAPINCO-ZNZJXIMRSA-N C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C1=O)c2ccccc2C[C@@H]1N(C)C([C@@](C1)([C@@H]1C(O)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O Chemical compound C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C1=O)c2ccccc2C[C@@H]1N(C)C([C@@](C1)([C@@H]1C(O)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O LGMFOSIGAPINCO-ZNZJXIMRSA-N 0.000 description 1
- MLVBDSXLHGQJCA-AGZMXDBFSA-N C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C1=O)c2ccccc2C[C@@H]1N(C)C([C@@](C1)([C@@H]1C(OCc1ccccc1)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O Chemical compound C[C@@H](C(N1CCCCC1)=O)NC([C@H](Cc1ccccc1)N(C1=O)c2ccccc2C[C@@H]1N(C)C([C@@](C1)([C@@H]1C(OCc1ccccc1)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O MLVBDSXLHGQJCA-AGZMXDBFSA-N 0.000 description 1
- YDIRYGJEZROABP-RYUDHWBXSA-N C[C@@H](CC(OCc1ccccc1)=O)C(N(C)[C@@H](C)C(N)=O)=O Chemical compound C[C@@H](CC(OCc1ccccc1)=O)C(N(C)[C@@H](C)C(N)=O)=O YDIRYGJEZROABP-RYUDHWBXSA-N 0.000 description 1
- ODTUTIKYAWGREV-VIFPVBQESA-N C[C@@H](Cc1ccccc1)C(N=C)=O Chemical compound C[C@@H](Cc1ccccc1)C(N=C)=O ODTUTIKYAWGREV-VIFPVBQESA-N 0.000 description 1
- SNHMUERNLJLMHN-UHFFFAOYSA-N Ic1ccccc1 Chemical compound Ic1ccccc1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 description 1
- UXGRDCMGDKKPFT-UHFFFAOYSA-N NC(C1)(C1c1ccc(C(F)(F)F)cc1)C(OCC#N)=O Chemical compound NC(C1)(C1c1ccc(C(F)(F)F)cc1)C(OCC#N)=O UXGRDCMGDKKPFT-UHFFFAOYSA-N 0.000 description 1
- WVHTZZOXKNLOAG-DIBZTSHWSA-N NC(C=CN1C[C@@H](C2)O[C@H](COP(O)(O)=O)[C@H]2OP(O)(OC[C@H]([C@H]2O)O[C@@H](C[n]3c4ncnc(N)c4nc3)[C@@H]2O)=O)=NC1=O Chemical compound NC(C=CN1C[C@@H](C2)O[C@H](COP(O)(O)=O)[C@H]2OP(O)(OC[C@H]([C@H]2O)O[C@@H](C[n]3c4ncnc(N)c4nc3)[C@@H]2O)=O)=NC1=O WVHTZZOXKNLOAG-DIBZTSHWSA-N 0.000 description 1
- KBUYLLSQUSBBQT-UHFFFAOYSA-N NC(SSC(N)=O)=O Chemical compound NC(SSC(N)=O)=O KBUYLLSQUSBBQT-UHFFFAOYSA-N 0.000 description 1
- HMMRSEKWXWQVIW-UHFFFAOYSA-N NS(c(ccc([N+]([O-])=O)c1)c1[N+]([O-])=O)(=O)=O Chemical compound NS(c(ccc([N+]([O-])=O)c1)c1[N+]([O-])=O)(=O)=O HMMRSEKWXWQVIW-UHFFFAOYSA-N 0.000 description 1
- GNDKYAWHEKZHPJ-UHFFFAOYSA-N NS(c(cccc1)c1[N+]([O-])=O)(=O)=O Chemical compound NS(c(cccc1)c1[N+]([O-])=O)(=O)=O GNDKYAWHEKZHPJ-UHFFFAOYSA-N 0.000 description 1
- LOVVSIULYJABJF-UHFFFAOYSA-N NSc(cccc1)c1[N+]([O-])=O Chemical compound NSc(cccc1)c1[N+]([O-])=O LOVVSIULYJABJF-UHFFFAOYSA-N 0.000 description 1
- BDEZGPKAMAVGBE-UHFFFAOYSA-N NSc(nccc1)c1[N+]([O-])=O Chemical compound NSc(nccc1)c1[N+]([O-])=O BDEZGPKAMAVGBE-UHFFFAOYSA-N 0.000 description 1
- GJADKSXFFUHKBR-HKBQPEDESA-N OC(CCCCc1cccc(C[C@@H](C(N2CCCCC2)=O)NC(OCC2c3ccccc3-c3ccccc23)=O)c1)=O Chemical compound OC(CCCCc1cccc(C[C@@H](C(N2CCCCC2)=O)NC(OCC2c3ccccc3-c3ccccc23)=O)c1)=O GJADKSXFFUHKBR-HKBQPEDESA-N 0.000 description 1
- NSERYVHZQRDIQX-IBGZPJMESA-N OC(COC([C@H](CCC1)N1C(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O Chemical compound OC(COC([C@H](CCC1)N1C(OCC1c2ccccc2-c2c1cccc2)=O)=O)=O NSERYVHZQRDIQX-IBGZPJMESA-N 0.000 description 1
- DXKLSDIIIUXGOJ-NRFANRHFSA-N OC(C[C@@H](C(N1CCCCC1)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O)=O Chemical compound OC(C[C@@H](C(N1CCCCC1)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O)=O DXKLSDIIIUXGOJ-NRFANRHFSA-N 0.000 description 1
Images






















































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/64—Cyclic peptides containing only normal peptide links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/107—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides
- C07K1/113—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides without change of the primary structure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K11/00—Depsipeptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K11/00—Depsipeptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K11/02—Depsipeptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof cyclic, e.g. valinomycins ; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K19/00—Hybrid peptides, i.e. peptides covalently bound to nucleic acids, or non-covalently bound protein-protein complexes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/1013—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing O or S as heteroatoms, e.g. Cys, Ser
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/08—Linear peptides containing only normal peptide links having 12 to 20 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/50—Cyclic peptides containing at least one abnormal peptide link
- C07K7/54—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring
- C07K7/56—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring the cyclisation not occurring through 2,4-diamino-butanoic acid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/06—Libraries containing nucleotides or polynucleotides, or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/10—Libraries containing peptides or polypeptides, or derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to a novel cyclization method for peptide compounds, a novel peptide compound and a library containing them.
- Non-Patent Document 1 It has been considered that effective inhibition of tough targets, which have been difficult to find drugs other than antibodies, is possible even with compounds having a molecular weight of 500 to 2000 (Non-patent Document 2).
- cases that are outside the range of rule of 5 proposed by Lipinski can be blocked by oral agents and intracellular targets, mainly natural products.
- Display Library display library
- Non-Patent Document 4 Non-Patent Document 4
- Cyclosporin A is a representative example of a peptide produced by an 11-residue microorganism that inhibits an orally administrable intracellular target (cyclophilin).
- Peptides generally have low metabolic stability and membrane permeability, but it has also been clarified that they can be improved by non-naturalization such as peptide cyclization or N-methylation (Non-patent Document 5). ).
- Non-Patent Document 6 A successful example of cyclization of an integrin-inhibiting peptide and clinical trials as an oral agent by non-naturalization has been reported (Non-Patent Document 6), but such drug development is brought about at the end of a long research period. There are few examples that have been made. For example, oral development of high-value pharmaceutical injections such as insulin, GLP-1 (glucagon-like peptide-1), PTH (parathyroid hormone), calcitonin has not been successful.
- Non-Patent Document 22 peptides containing non-natural amino acids and hydroxycarboxylic acid derivatives
- Non-Patent Document 22 peptides containing non-natural amino acids and hydroxycarboxylic acid derivatives
- Display® Library containing non-natural amino acids has become a reality.
- peptides containing N-methyl amino acids can be synthesized in ribosomes by utilizing tRNA that binds a non-natural amino acid and a cell-free translation system such as PureSystem (registered trademark) (Non-Patent Documents 7, 8, 9, 10, 11).
- PureSystem registered trademark
- An example of a challenge to Display® library containing one unnatural amino acid was also reported (Non-patent Documents 12 and 13).
- an example has been reported in which a display of a peptide containing N-methylamino acid is produced (Non-patent Document 23).
- Non-patent Document 19 a method of cyclizing two Cys via mesitylene has been reported.
- This approach results in a more stable thioether-mediated cyclization, but the effect is partial and leaves room for improvement.
- thioethers are susceptible to oxidative metabolism. It has been reported that it is decomposed into RSCH 2 R ′ ⁇ RSH + R′CHO by cytochrome P450 and metabolized to sulfoxide by flavin-containing monooxygenase (Non-patent Document 20). When the former is generated, it becomes a reactive metabolite, which leads to toxicity.
- Non-Patent Document 21 Non-Patent Document 25, Non-Patent Document 26, Non-Patent Document.
- Reference 27 all generate a structure in which the active species obtained by cleaving the main chain amide bond and the amino acid main chain amino group are chemically reacted. It cannot be applied as a method.
- These techniques are useful as a technique for condensation cyclization of many natural products such as cyclosporin A with a main chain carboxylic acid and a main chain amino group.
- the Display library since the main chain carboxylic acid end needs to be bound to mRNA, it is not possible to use a method for generating an active species by decomposing the main chain amide bond.
- Non-Patent Document 12 by cross-linking the amino group of methionine at the N-terminal and the amino group of lysine arranged downstream (C-terminal side) with disuccinimidyl glutarate (DSG)
- a cyclization method (Non-patent Document 11, Patent Document 1) by introducing an amino acid derivative having a chloroacetyl group as a translation initiation amino acid and forming a thioether by intramolecular cyclization reaction by arranging Cys downstream
- the improvement of the above points was insufficient, and development of a new cyclization method instead of these was demanded.
- Non-patent Document 24 For example, in the SS cyclization method using two cysteines, it is necessary to make two amino acids specific (cysteine), but in the cyclization method by cross-linking using DSG, there are three sites including lysine. As a result, the diversity of the structure is reduced in a peptide library having a certain number of residues. Further, it has been reported that when the structure of the cyclization site is changed, the activity (strength of drug efficacy) of the peptide having the cyclization site is greatly reduced (Non-patent Document 24).
- Kessler, H. et. Al. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anti-cancer Agents in Medicinal Chemistry 2010, 10, 753 Roberts, R. W., et al. Encodamers: Unnatural peptide oligomers encoded in RNA. Chem. Bio. 2003, 10, 1043. Forster, A. C. et. Al., Specificity of translation for N-alkyl amino acids. J. Am. Chem. Soc.
- peptides with membrane permeability and metabolic stability are designed and those compound groups are displayed. Realization of Display library technology that selects peptides with medicinal effects is considered effective. Hit compounds that have membrane permeability and metabolic stability to some extent can be easily synthesized and chemically modified unlike natural products, so hit compounds are the same as conventional low molecular drug discovery within rule of 5. It is possible to carry out structural optimization while keeping the structural change of the compound small, and it can be expected that a clinically developed compound can be created relatively easily.
- druglikeness of medium molecular peptides that are outside the scope of rule of 5 (drug-likeness: In this specification, preferably, both membrane permeability and metabolic stability are achieved. I thought that it was essential to understand the conditions that satisfy the conditions) and to construct a display library containing unnatural amino acids consisting of molecular groups that satisfy the conditions. On the other hand, in order to effectively use the limited scale of medium molecule peptide display (which means that there is a limitation on the peptide chain length in consideration of drug likeness), peptides with completely different structures (non-natural type) It is important to include a large amount of peptides containing amino acids.
- the present inventors have clarified for the first time the conditions required for a drug-like cyclic peptide.
- the present inventors have found a method for synthesizing a display library composed of peptide compounds having highly diverse cyclized portions that satisfy the above conditions.
- the present invention includes the following.
- a method for producing a peptide compound having a cyclic part 1) An amino acid residue and / or an amino acid analog residue, or an acyclic peptide compound composed of an amino acid residue and / or an amino acid analog residue and an N-terminal carboxylic acid analog is encoded with the peptide compound A process of translating and synthesizing from the nucleic acid
- the acyclic peptide compound comprises an amino acid residue or amino acid analog residue having a reactive site on one of the side chains on the C-terminal side, and an amino acid residue or amino acid analog having another reactive site on the N-terminal side.
- a body residue or an N-terminal carboxylic acid analog 1) Combine the reaction point of the amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog on the N-terminal side with the reaction point of the amino acid residue or amino acid analog residue in the side chain on the C-terminal side And forming an amide bond or a carbon-carbon bond.
- a method for producing a peptide compound having a cyclic part 1) An amino acid residue and / or an amino acid analog residue, or an acyclic peptide compound composed of an amino acid residue and / or an amino acid analog residue and an N-terminal carboxylic acid analog is encoded with the peptide compound A process of translating and synthesizing from the nucleic acid
- the acyclic peptide compound comprises an amino acid residue or amino acid analog residue having an active ester group in the side chain, and an amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog having a reaction auxiliary group near the amine.
- the amino acid, amino acid analog or N-terminal carboxylic acid analog having a reaction auxiliary group in the vicinity of the amine is a compound N-1 or a compound N-2 represented by the following general formula: Any of the methods; Wherein R1 represents a hydrogen atom, S—R23 (R23 represents an optionally substituted alkyl group, aryl group or aralkyl group), or a protective group for the HS group; R2 and R3 each independently represent a hydrogen atom or an optionally substituted alkyl group, alkenyl group, alkynyl group, aryl group, heteroaryl group, aralkyl group or cycloalkyl group; Or R2 and R3 represent a substituent that forms a ring, or R2 or R3 represents a substituent that forms a ring with R4; R4 represents an alkylene group which may have a substituent, an arylene group which may have a substituent or a divalent aralkyl group which may have a
- a method for producing a peptide compound having a cyclic part 1) An amino acid residue and / or an amino acid analog residue, or an acyclic peptide compound composed of an amino acid residue and / or an amino acid analog residue and an N-terminal carboxylic acid analog is encoded with the peptide compound A process of translating and synthesizing from the nucleic acid
- the acyclic peptide compound is an amino acid residue or amino acid analog residue having an active ester group in the side chain and an amino acid residue having an N-terminal main chain amino group, or an amino group in the main chain or side chain.
- Comprising an amino acid analog residue having an N-terminal carboxylic acid analog comprising: [11]
- the active ester group is an alkylthioester group or an aralkylthioester group, and includes a step of converting the active ester group into a more active active ester group by adding an activator after the translational synthesis in step 1).
- the translational synthesis of the N-terminal site in step 1) is introduced using a translation initiation tRNA acylated with a translatable amino acid other than formylmethionine, a translatable amino acid analog or a translatable N-terminal carboxylic acid analog.
- the start codon is skipped, and a translatable amino acid other than Met, a translatable amino acid analog or a translatable N-terminal carboxylic acid analog is introduced into the N-terminal
- the translational synthesis of the N-terminal site in step 1) is performed using a method of cleaving the N-terminal amino acid, amino acid analog or carboxylic acid analog with aminopeptidase. The method described.
- N-terminal formyl Met is removed by treatment with methionine aminopeptidase, and other translatable amino acids, translatable amino acid analogs or translatable N-terminal carboxylic acids at the N-terminal
- the method according to [17] wherein the method is performed using a method for introducing an acid analog.
- the non-cyclic peptide compound includes an ⁇ -hydroxycarboxylic acid and an amino acid or amino acid analog having an amino group which may be protected in the side chain, 3) including a step of chemically reacting an ⁇ -hydroxycarboxylic acid moiety and an amino acid or amino acid analog moiety having an amino group optionally protected by a side chain to form a branched moiety [1] to [21]
- the method in any one of.
- a method for producing a peptide compound having a cyclic part and a linear part 1) A non-cyclic peptide compound composed of an amino acid residue and / or an amino acid analog residue, or an amino acid residue and / or an amino acid analog residue, an N-terminal carboxylic acid analog, and an ⁇ -hydroxycarboxylic acid
- a step of translating and synthesizing from the nucleic acid encoding the peptide compound wherein the acyclic peptide compound is i) an amino acid residue (or amino acid analog residue) having a reactive site in one of the side chains on the C-terminal side, and an amino acid residue or amino acid analog residue having another reactive site on the N-terminal side Or an N-terminal carboxylic acid analog, ii) an ⁇ -hydroxycarboxylic acid having Rf5 at the ⁇ -position between the two reaction points described in i) above (Rf5 is a hydrogen atom, an optionally substituted alkyl group, an aralkyl group,
- the number of amino acid residues and / or amino acid analog residues contained between the ⁇ -hydroxycarboxylic acid described in ii) of step 1) and the amino acid residue or amino acid analog residue having an amino group in the side chain is The method according to [23], which is 7 or less. [25] The method according to [23] or [24], wherein the ⁇ -hydroxycarboxylic acid described in ii) of step 1) is contained in the acyclic peptide compound as Cys-Pro- ⁇ -hydroxycarboxylic acid. [26] From [23], the amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog having another reactive site on the N-terminal side described in i) of step 1) has a reaction auxiliary group.
- step 1 The amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog having another reactive site on the N-terminal side described in i) of step 1) does not have a reaction auxiliary group, and described in ii)
- the amino group of the amino acid residue or amino acid analog residue having an amino group in the side chain has a protecting group, and by adding an activator, the cyclization reaction of step 2) is carried out.
- the method includes the step of removing the amino group-protecting group of the amino acid residue or amino acid analog residue having the amino group described in ii) in the side chain, [23] The method according to any one of [26].
- a method for producing a peptide compound having a cyclic part 1) An amino acid residue and / or an amino acid analog residue, or an acyclic peptide compound composed of an amino acid residue and / or an amino acid analog residue and an N-terminal carboxylic acid analog is encoded with the peptide compound A process of translating and synthesizing from the nucleic acid
- the acyclic peptide compound comprises an amino acid residue or amino acid analog residue having a reactive site in one of the side chains, and an amino acid, amino acid analog residue or N-terminal carboxylic acid having another reactive site at the N-terminus.
- the peptide compound-nucleic acid complex is synthesized using a nucleic acid in which puromycin is bound to the 3 ′ end of a nucleic acid encoding an acyclic peptide compound used for translational synthesis via a linker, [33] the method of. [35] The method according to [33] or [34], wherein the spacer is a peptide, RNA, DNA, or a polymer of hexaethylene glycol, or a combination thereof. [36] The method according to any one of [1] to [35], wherein the peptide compound is produced by translating a nucleic acid library comprising a plurality of nucleic acids having different sequences.
- [37] A peptide compound or a peptide compound-nucleic acid complex produced by the production method according to any one of [1] to [36]. [38] A library comprising a plurality of peptide compounds according to [37] or peptide compounds-nucleic acid complexes having different structures.
- a peptide compound having a cyclic part which has the following (i) to (iv): (I) the total number of amino acids and amino acid analogs is 9 to 13 including a cyclic part consisting of 5 to 12 total amino acid and amino acid analog residues; (Ii) at least two N-substituted amino acids and at least one amino acid that is not N-substituted, (Iii) The ClogP value is 6 or more, (Iv) The bond of the amino acid or amino acid analog forming the cyclic part is at least a bond comprising an active ester group in the side chain of the amino acid or amino acid analog and the amine group of another amino acid or amino acid analog Have one.
- Amino acids and amino acid analogs contained in peptide compounds are amino acids or amino acid analogs capable of translational synthesis, or amino acids or amino acids obtained by chemically modifying the translatable amino acids or side chains or N-substitution sites of amino acid analogs
- the peptide compound according to [39] which is an amino acid or an amino acid analog selected from derivatives.
- the peptide compound according to [39] or [40] further comprising at least one linear moiety consisting of 1 to 8 in total of amino acid and amino acid analog residues.
- R54 is a peptide composed of 0-8 amino acid residues
- R55 is a C1-C6 alkyl group which may have a substituent, a C5-C10 aryl group, an aralkyl group, an ester group, an amide group which may have a substituent, * Indicates a binding site in the annular portion. ).
- Forming a non-cyclic peptide compound-nucleic acid complex (Ii) The complex acyclic peptide compound translated and synthesized in step (i) is cyclized by an amide bond or a carbon-carbon bond, and the total number of amino acids in the cyclic portion and amino acid analogs is 5-12.
- Forming a cyclic compound comprising: and (Iii) A step of contacting the peptide compound-nucleic acid complex library having a cyclic moiety obtained in step (ii) with a biological molecule and selecting a complex having binding activity to the biological molecule.
- the manufacturing method as described in [46] including the following processes; (Iv) obtaining sequence information of the peptide compound from the nucleic acid sequence of the complex selected in the step (iii), and (V) A step of chemically synthesizing the peptide compound based on the sequence information obtained in the step (iv).
- step (ii) After the step of forming the cyclic compound of step (ii), wherein the acyclic peptide compound comprises an ⁇ -hydroxycarboxylic acid and an amino acid or amino acid analog having an amino group in the side chain which may be protected;
- the method according to [46] or [47] comprising a step of chemically reacting an ⁇ -hydroxycarboxylic acid moiety with an amino acid having an amino group in the side chain or an amino acid analog moiety to form a branched moiety.
- the amino acid or amino acid analog on the N-terminal side that undergoes a cyclization reaction is an amino acid or amino acid analog selected from the compound represented by Compound N-1 or Compound N-2, and the amino acid or amino acid analog on the C-terminal side Is an amino acid or an amino acid analog selected from the compound represented by the compound C-1, and includes a step of removing the reaction auxiliary group after the step of obtaining the cyclic compound of step (ii). 50].
- the nucleic acid sequence has a spacer at the 3 ′ end, and the C-terminus of the peptide compound to be translated and synthesized forms a complex with the nucleic acid sequence via the spacer, according to any of [46] to [51] The manufacturing method as described.
- the peptide compound-nucleic acid complex is synthesized using a nucleic acid in which puromycin is bound to the 3 ′ end of a nucleic acid encoding an acyclic peptide compound used for translational synthesis via a linker, according to [52]. the method of. [54] [52] The production method according to [53], wherein the spacer is a peptide, RNA, DNA, or a polymer of hexaethylene glycol.
- a drug compound having a drug-like (particularly membrane permeability and metabolic stability) cyclic part or a peptide compound having a cyclic part and a linear part which can be translationally synthesized, and A display library is provided.
- the display library of the present invention is a library rich in diversity, there is a high probability that a hit compound for a desired target molecule can be obtained.
- the hit compound obtained from the display library of the present invention already has excellent membrane permeability and metabolic stability, the conventional low molecular weight compound does not undergo a large structural change. It is possible to efficiently carry out optimization as a pharmaceutical product in the same way as the above idea.
- the present invention provides Scaffold for new drugs different from low molecular weight compounds and antibodies that have been known as drugs, and provides a new drug discovery system for efficiently creating drugs. is there.
- FIG. 2 is a diagram showing a general method for synthesizing aminoacylated pdCpA compounds in which side chain carboxylic acids are active esterified. It is the figure which showed the mass-spectrum analysis of the translation product of mRNA which codes the peptide sequence P-1 containing Glu (SBn). It is the figure which showed having analyzed the mass spectrum of the translation product of mRNA which codes the peptide sequence P-2 containing Asp (SMe). In the translation of Asp (SMe), a compound de-MeSHed from the translated full-length peptide was detected as the main product (translated peptide P-3).
- FIG. 2 is a diagram showing a general method for synthesizing aminoacylated pdCpA compounds in which side chain carboxylic acids are active esterified. It is the figure which showed the mass-spectrum analysis of the translation product of mRNA which codes the peptide sequence P-1 containing Glu (SBn). It is the figure which showed having analyzed the mass
- FIG. 6 is a diagram showing the production of peptide P-4 by hydrolysis of translated peptide P-6. It is the figure which showed translation of the peptide sequence which does not contain Tyr but contains thioester. It is the figure which showed the translational synthesis of the thioester containing peptide which does not have an N terminal amino group.
- FIG. 3 is a diagram showing translational synthesis of a model peptide in which an amino acid N-alkylated into a C-terminal amino acid following an amino acid whose side chain is thioesterified is introduced. It is the figure which showed the comparison of the chemical reactivity of a thioester, a glycine derivative, or a cysteine derivative.
- FIG. 3 is a diagram showing translational synthesis of a model peptide in which an amino acid N-alkylated into a C-terminal amino acid following an amino acid whose side chain is thioesterified is introduced. It is the figure which showed the comparison of the chemical
- FIG. 8 is a diagram showing a continuation of FIG. 8-1.
- FIG. 3 shows the synthesis of amide 5d-1 by reaction of thioester 5b-1 with a glycine derivative under the imidazole addition condition.
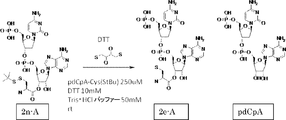
- FIG. 2 shows a general method for synthesizing aminoacylated pdCpA of a cysteine derivative.
- FIG. 2 is a diagram showing a synthesis example of aminoacylated pdCpA of a cysteine derivative. It is the figure which showed the peptide translation synthesis which made Cys (StBu) N terminal.
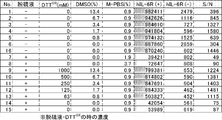
- FIG. 3 is a graph showing the stability of compound 2n-A and compound 2e-A in a translation simulation solution.
- FIG. 32 is a diagram showing a continuation of FIG. 30-1. It is the figure which showed the radical desulfurization reaction using a model substrate. It is the figure which showed the conditions examination of the radical desulfurization reaction using a model substrate.
- FIG. 5 is a diagram showing desulfurization reaction conditions for translated peptide P-50 and a mass spectrum of the obtained peptide P-51. It is the figure which showed the influence evaluation to the protein of desulfurization reaction.
- FIG. 1 It is a figure which shows the analysis result by the mass spectrum of the translation reaction material obtained by the translational synthesis of the peptide containing the benzylthioesterified aspartic acid derivative. It is a figure which shows the analysis result by the mass spectrum of the product obtained by the amide cyclization experiment of the peptide using the thioester on translation peptide P141, and N terminal alpha-amino group. 2 is a graph showing a comparison of synthesis conditions of Compound 10.
- FIG. It is a figure which shows the result of having analyzed the production
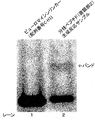
- FIG. It is a figure which shows the analysis result by electrophoresis of the reaction product of a peptide cyclization reaction.
- FIG. It is a figure which shows the result of having analyzed the production
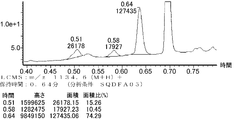
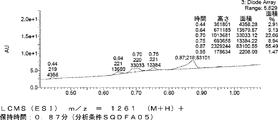
- FIG. 61 is a mass chromatograph of LCMS 0.3 to 0.6 minutes.
- FIG. 62 shows peptide P-E1 (peak I) containing an N-terminal formylmethionine, thioester cyclized with a side chain thiol group and a carboxylic acid, and the N-terminal formylmethionine removed, resulting in an exposed N-terminal amino acid.
- FIG. 7 is a diagram showing the result of MALDI-MS analysis of compound P-E2 (peak II) amide-cyclized with a group nitrogen atom and an Asp side chain carboxylic acid. It is a figure which shows the result of having analyzed the production
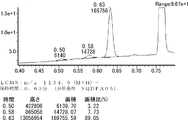
- FIG. 65 is a mass chromatograph of LCMS 0.3 to 0.6 minutes. The peptide components are all eluted from 0.3 minutes to 0.6 minutes under the present analysis conditions. Therefore, the mass chromatographs from 0.3 to 0.6 minutes are integrated and averaged for the purpose of evaluating the selectivity of the reaction.
- LCMS is a mass chromatograph averaged over the entire range.
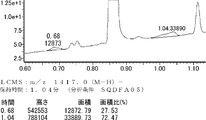
- LCMS is a mass chromatograph averaged over the entire range.
- LCMS is a mass chromatograph averaged over the entire range. It is a figure which shows the result of having analyzed the production
- the peptide compound having a cyclic part of the present invention is a compound formed by amide bond or ester bond of an amino acid or an amino acid analog, and the cyclic part forms an amide bond or a carbon-carbon bond.
- a compound obtained by further chemically modifying the compound is also included in the peptide compound of the present invention.
- the peptide compound of the present invention may have a linear portion, and can be described as, for example, Scheme A (Schemes A-1 and A-2).
- the peptide compound having a cyclic part may further have a linear part.
- the number of amide bonds or ester bonds (the number and length of amino acids or amino acid analogs) is not particularly limited, but when it has a linear part, it is preferably within 30 residues including the cyclic part and the linear part.
- the total number of amino acids including the cyclization site and the linear site is more preferably 13 residues or less.
- the total number of amino acids is more preferably 9 or more.
- the number of amino acids and amino acid analogs constituting the cyclic part is preferably 5-12.
- the number of amino acids and amino acid analogs constituting the cyclic part is more preferably 5 to 11, and further preferably 7 to 11 residues. 9 to 11 residues are particularly preferred.
- the number of amino acids and amino acid analogs (number of units) in the linear portion is preferably 0-8. Furthermore, 0 to 3 are preferable.
- amino acids may include amino acid analogs.
- druglikeness or “druglike” means that a peptide compound targets at least an oral agent, or an intracellular region of an intracellular protein, nucleic acid, membrane protein or a transmembrane domain of a membrane protein. In this case, it means having membrane permeability and metabolic stability that can be used as a medicine.
- the cyclic part of the peptide compound having a cyclic part of the present invention is not particularly limited as long as it is a peptide that forms a ring, but the post-translational cyclization part has a functional group capable of satisfying both membrane permeability and metabolic stability (Druglikeness). It must be the cyclization unit that forms. If it is such a cyclization method, it will not specifically limit. Examples thereof include an amide bond formed from a carboxylic acid and an amine, and a carbon-carbon bond reaction using a transition metal such as a Suzuki reaction, a Heck reaction (Heck reaction), and a Sonogashira reaction as a catalyst. Accordingly, the peptide compound of the present invention contains at least one set of functional groups capable of these coupling reactions. In particular, from the viewpoint of metabolic stability, a functional group that forms an amide bond by a binding reaction is preferably included.
- cyclic part of the peptide compound of the present invention for example, a cyclic part formed by cyclization by a chemical reaction after translation synthesis as shown in Scheme A is preferable. Furthermore, a circular part that can be formed under reaction conditions that do not affect nucleic acids such as RNA and DNA after translation is preferred.
- the formation of the annular portion is preferably a drug-like cyclization.
- Drag-like cyclization means a bond in which the bond produced is drug-like.
- the bond includes a heteroatom that can be easily oxidized and does not include a bond that hinders metabolic stability.
- Examples of the bond generated by cyclization include an amide bond formed by a bond between an active ester and an amine, and a bond generated by a Heck reaction product formed by a carbon-carbon double bond and an aryl halide. Since these require a reactive functional group in the triangular unit (cyclized N-terminal unit) or crossing unit described in Scheme A, amino acids suitable for drug-like are not necessarily selected for the triangular unit or crossing unit. However, it is converted into a compound having a drug-like functional group after post-translational modification. In the present invention, such a bond is also included in the drug-like cyclized bond.
- the curve part of Scheme A is a site that is cyclized after translation (post-translational cyclization part), and this part is a variety of post-translational modification chemical reactions represented by carbon-carbon bond formation reactions such as amide bond or Heck reaction. To form an annular portion.
- “translational synthesis” means that a peptide compound is translated and synthesized from a nucleic acid (eg, DNA, RNA) encoding the peptide compound. Translation is a process of obtaining a linear peptide by repeating amide bond or ester bond reaction using mRNA as a template by the action of ribosome.
- Post-translational modification refers to a chemical reaction that occurs after translation automatically or by adding another reagent other than the action of ribosome, and includes, for example, a cyclization reaction and a deprotection reaction.
- Post-translational cyclization involves a ring-forming reaction among post-translational modifications.
- Scheme A A scheme for explaining the peptide compound of the present invention.
- the white circle unit, the black circle unit, the triangular unit, and the square unit each mean an amino acid or an amino acid analog.
- a triangular unit may be an N-terminal carboxylic acid analog, for example, each of the eight black circle units may be a different type of amino acid or an amino acid analog, some of which Alternatively, the amino acid or amino acid analog may be chemically or skeleton-converted to a compound having another skeleton by chemical modification that can be performed after translation. 1 unit is the amino acid or amino acid analog at the end of post-translational modification. However, this also includes those in which an amino acid or amino acid analog translated by one tRNA is chemically or skeleton-converted to a compound having another skeleton by post-translational modification.
- amino acids include amino acid analogs, and in this specification, post-translational cyclization may be simply referred to as cyclization. is there.
- the cyclic part is one triangular ( ⁇ ) unit (residue) (cyclized N-terminal unit), eight black circle ( ⁇ ) units (cyclic part main chain unit) and one It is a part composed of white circle ( ⁇ ) units (intersection units), and the straight chain part is a part composed of six square ( ⁇ ) units (straight chain main chain units).
- the annular part is a part composed of one triangular unit, eight black circle units and one crossing unit, and the straight chain part is a part composed of four squares and three square units.
- the crossing unit refers to a functional group possessed by the amino acid or amino acid analog of the triangular unit in the peptide compound on the amino acid side chain in the peptide compound before cyclization (non-cyclized peptide compound) formed after translation.
- the white circle unit in Scheme A corresponds to this.
- the crossing unit is preferably selected from the above amino acids or amino acid analogs and capable of translation from a nucleic acid.
- the derivative itself is translated when the derivative is translated.
- Asp (SBn) a compound in which the side chain methylene chain of Asp is freely substituted is allowed as an intersection unit (for example, R28 and R29 of compound C-3 are not translated). May be).
- the crossover unit the amino group and carboxyl group of the main chain are used for covalent bond formation in translation synthesis, and the third functional group is necessary for post-translational cyclization, so that it has a total of three or more functional groups.
- the posttranslational cyclization site is cyclized by the functional group of the side chain site of the crossing unit.
- the amino acid or amino acid analog having a functional group that cyclizes with the crossing unit, or the N-terminal carboxylic acid analog is not particularly limited as long as it has a functional group that cyclizes with the crossing unit.
- the triangular unit is arranged at the N-terminal.
- the main chain amino group can be used as the cyclized functional group.
- post-translational cyclization can be performed by the main chain amino group of the triangular unit and an amide bond.
- the side chain of the triangular unit may not have a reactive functional group.
- Reaction auxiliary groups such as SH groups (thiol groups) can also be introduced into the side chain.
- an amino acid analog is used as the triangular unit
- the hydroxyl group of the main chain may be used as a reactive functional group, or a reactive functional group arranged in a side chain may be used.
- an N-terminal carboxylic acid analog is used, an amino group or a hydroxyl group may be used as a reactive functional group as described above, but as a free reactive unit having no amino group or hydroxyl group.
- Various functional groups can be introduced.
- the triangular unit is preferably selected from the above amino acids, amino acid analogs, or N-terminal carboxylic acid analogs and translated. In addition, even if it is difficult to translate itself, it is not essential that the derivative is translated as in the case of the intersection unit.
- the crossing unit and the triangular unit can be incorporated at a desired position in the peptide compound before cyclization as long as they can be cyclized, but cyclization or post-translational modification after cyclization is applied. It is preferable that the amino acid, amino acid analog or N-terminal carboxylic acid analog in the subsequent cyclic site is incorporated at a position where the total number is 5 to 12. Furthermore, the amino acid or amino acid analog or N-terminal carboxylic acid analog of the cyclic part after cyclization or post-translational modification after cyclization is incorporated at a position where the total number is 5 to 11. It is preferable.
- the triangular unit is arranged at the N-terminal in Scheme A, but it can be arranged at a position other than the N-terminal. In that case, the position needs to be arranged on the N-terminal side from the intersection unit.
- the triangular unit is selected from amino acids and amino acid analogs, and has a functional group that cyclizes with the crossing unit in the side chain.
- the black circle unit and the square unit are selected from amino acids or amino acid analogs. Also included are chemical structures that can be produced by post-translational modification after translation with an amino acid or amino acid analog (eg, the structure of the straight chain portion 2).
- the black circle unit is not particularly limited, but is preferably selected from a drug-like amino acid or an amino acid that has a reactive functional group and is chemically reacted by post-translational modification to be converted into a drug-like functional group. (For example, lysine is mentioned as the amino acid residue of the straight chain part 2).
- the black circle unit is not particularly limited, but preferably a drug-like amino acid analog or an amino acid analog having various reactive functional groups on the side chain is converted into a reactive functional group by post-translational modification. Selected from amino acid analogs that are chemically modified and converted to drug-like amino acid analogs.
- the number of straight chain portions is not particularly limited, and may be one as shown in Scheme A-1 or two or more as shown in Scheme A-2. Further, the compound may be a compound in which the number of square units in Scheme A-1 is 0, or may be a compound in which the number of straight chain portions 1 is 0 in the square units in Scheme A-2.
- By having a linear part it is possible to strengthen the function of the peptide compound having a cyclic part of the present invention. For example, when the peptide compound of the present invention is used to inhibit the binding between a certain receptor and a ligand, the peptide compound has a linear portion. The binding activity to a receptor or a ligand can be increased.
- the linear portion of the present invention can be provided at a desired position of the cyclic portion, for example, according to the method described later, and the linear portion is provided at the optimum position for obtaining a higher function.
- the obtained peptide compound can be obtained (hereinafter, linear part 2).
- linear portions are also preferable for obtaining a peptide compound having a desired activity more efficiently from a library of peptide compounds having a cyclic portion.
- Peptide compounds having a desired activity include those having binding activity to the target substance, those having an action of inhibiting the function of the target substance, those having an action of activating the function of the target substance, Examples include those having the function of changing the function, and the function can be selected from these according to the purpose.
- the peptide compound having a cyclic part of the present invention has binding activity to a target substance, these peptide compounds are excellent in membrane permeability and lipid stability. For example, by labeling the peptide compound In addition to in vivo, it is possible to monitor the distribution of target substances in cells in real time.
- the target substance when it is a causative factor of the disease, it may be used for diagnosis of the disease.
- the peptide compound having a cyclic part of the present invention has an effect of inhibiting, activating or changing the function of the target substance, for example, when the target substance is a causative factor of the disease, Available.
- the peptide compound having a cyclic part of the present invention has the straight chain part 2, it is possible to improve the acquisition ratio of the inhibitory compound as compared with the case where there is no linear part.
- the peptide contact site in protein A to which the peptide compound is bound is determined. This is shown on the right side of FIG. Since it is a cyclic compound, when the contact region of peptide and protein A is approximated by a circle, it can be determined that the diameter is about 3 to 5 residues.
- protein B binds to this contact area, it is possible to effectively inhibit the protein-protein interaction between protein A and protein B.
- protein B binds to protein A outside this contact area Therefore, there is a possibility that effective inhibition cannot be obtained (so-called allosteric inhibition examples are rarely reported as protein-protein interaction inhibition examples).
- FIG. 82-1 shows the same 13-residue cyclic peptide as above, but a peptide in which a cyclic structure was constructed with 10 residues and the remaining 3 were branched.
- the straight chain part 2 can be added to any position of the peptide compound according to the method described later, not only the four branch points in FIG. Points can be acquired, but if four are overlapped, the contact area is expanded to the area shown on the left in FIG. In spite of acting at the same place (hole), by overlapping with the binding region of protein A and protein B, the ratio of obtaining a peptide compound having a function such as an inhibitor is increased.
- amino acid and “amino acid analog” constituting a peptide compound may be referred to as “amino acid residue” and “amino acid analog residue”, respectively.
- Amino acids are ⁇ , ⁇ , and ⁇ amino acids, and natural amino acids (in this application, natural amino acids refer to 20 kinds of amino acids contained in proteins. Specifically, Gly, Ala, Ser, Thr, Val, Leu, It is not limited to Ile, Phe, Tyr, Trp, His, Glu, Asp, Gln, Asn, Cys, Met, Lys, Arg, and Pro), and may be an unnatural amino acid.
- an ⁇ -amino acid it may be an L-type amino acid, a D-type amino acid, or an ⁇ , ⁇ -dialkyl amino acid.
- the selection of the amino acid side chain is not particularly limited, but can be freely selected from, for example, an alkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, and a cycloalkyl group in addition to a hydrogen atom.
- Each may be provided with a substituent, and these substituents are also freely selected from arbitrary functional groups including, for example, N atom, O atom, S atom, B atom, Si atom, and P atom.
- amino acid and amino acid analogs constituting the peptide compound include all isotopes corresponding to each.
- the isotopes of “amino acids” and “amino acid analogs” are those in which at least one atom is replaced with an atom having the same atomic number (number of protons) and a different mass number (sum of protons and neutrons). is there.
- Examples of isotopes contained in the “amino acid” and “amino acid analog” constituting the peptide compound of the present invention include a hydrogen atom, a carbon atom, a nitrogen atom, an oxygen atom, a phosphorus atom, a sulfur atom, a fluorine atom, and a chlorine atom. And 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F, 36Cl, etc. are included.
- substituents examples include a halogen-derived substituent such as fluoro (—F), chloro (—Cl), bromo (—Br), iodo (—I) and the like.
- halogen-derived substituent such as fluoro (—F), chloro (—Cl), bromo (—Br), iodo (—I) and the like.
- alkyl group examples include an alkyl group, a cycloalkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group and the like, which may be substituted with one or more halogens.
- Examples of the substituent for forming an ether as the substituent derived from the O atom include an alkoxy group (—OR), and the alkoxy group is an alkylalkoxy group, a cycloalkylalkoxy group, an alkenylalkoxy group, an alkynylalkoxy group, an arylalkoxy group. , A heteroarylalkoxy group, an aralkylalkoxy group, and the like.
- Examples of the substituent (—CO 2 H) that forms a carboxylic acid include a carboxyl group, and the substituent that forms an ester group includes an oxycarbonyl group (—O—C ⁇ O—R) and a carbonylalkoxy group (—C ⁇
- the carbonylalkoxy group includes an alkyloxycarbonyl group, a cycloalkyloxycarbonyl group, an alkenyloxycarbonyl group, an alkynyloxycarbonyl group, an aryloxycarbonyl group, a heteroaryloxycarbonyl group, an aralkyloxycarbonyl group, and the like.
- the oxycarbonyl group is selected from alkylcarbonyloxy group, cycloalkylcarbonyloxy group, alkenylcarbonyloxy group, alkynylcarbonyloxy group, arylcarbonyloxy group, heteroarylcarbonyloxy group.
- Shi group selected from among an aralkyl carbonyl group.
- Examples of the substituent for forming a thioester include a mercaptocarbonyl group (—S—C ⁇ O—R) and a carbonylalkyl mercapto group (—C ⁇ O—SR), and the like. Selected from alkenylcarbonyl group, mercaptoalkynylcarbonyl group, mercaptoarylcarbonyl group, mercaptoheteroarylcarbonyl group, mercaptoaralkylcarbonyl group, etc., or carbonylalkyl mercapto group, carbonylcycloalkyl mercapto group, carbonylalkenyl mercapto group, carbonyl Alkynyl mercapto group, carbonyl aryl mercapto group, carbonyl heteroaryl mercapto group, carbonyl aralkyl mercapto group It is.
- Examples of the substituent that forms an amide group include an aminoalkylcarbonyl group (—NH—CO—R), an aminocycloalkylcarbonyl group, an aminoalkenylcarbonyl group, an aminoalkynylcarbonyl group, an aminocycloalkylcarbonyl group, an aminoarylcarbonyl group, Aminoheteroarylcarbonyl group, aminoaralkylcarbonyl group and the like, or carbonylalkylamino group (—CO—NHR), carbonylcycloalkylamino group, carbonylalkenylamino group, carbonylalkynylamino group, carbonylarylamino group, carbonylhetero An arylamino group, a carbonylaralkylamino group, etc.
- an aminoalkylcarbonyl group —NH—CO—R
- an aminocycloalkylcarbonyl group an aminoalkenylcarbonyl group, an aminoalkynylcarbonyl group,
- Examples of the substituent that forms the carbamate group include an aminoalkyl carbamate group (—NH—CO—OR), an aminocycloalkyl carbamate group, an aminoalkenyl carbamate group, an aminoalkynyl carbamate group, an aminocycloalkyl carbamate group, an aminoaryl carbamate group, An aminoheteroaryl carbamate group, an aminoaralkyl carbamate group, etc. are mentioned. Further, compounds in which an H atom bonded to an N atom is replaced with an alkyl group, a cycloalkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, or an aralkyl group are also included.
- Examples of the substituent that forms the sulfonamide group include an aminoalkylsulfonyl group (—NH—SO 2 —R), an aminocycloalkylsulfonyl group, an aminoalkenylsulfonyl group, an aminoalkynylsulfonyl group, an aminocycloalkylsulfonyl group, an aminoarylsulfonyl Group, aminoheteroarylsulfonyl group, aminoaralkylsulfonyl group, etc., or sulfonylalkylamino group (—SO 2 —NHR), sulfonylcycloalkylamino group, sulfonylalkenylamino group, sulfonylalkynylamino group, sulfonylarylamino group Sulfonylheteroarylamino group, sulfonylaralkylamino group, and the like.
- Examples of the substituent that forms the sulfamide group include aminoalkylsulfamoyl group (—NH—SO 2 —NHR), aminocycloalkylsulfamoyl group, aminoalkenylsulfamoyl group, aminoalkynylsulfamoyl group, aminocycloalkyl Examples thereof include a sulfamoyl group, an aminoarylsulfamoyl group, an aminoheteroarylsulfamoyl group, and an aminoaralkylsulfamoyl group.
- the H atom bonded to the N atom is an alkyl group, cycloalkyl group, alkenyl group, alkynyl group, aryl group, heteroaryl group, aralkyl group, any two of the same or different, or these form a ring
- a substituent selected from substituents that may be substituted is substituted.
- Examples of the substituent that forms thiocarboxylic acid include a thiocarboxylic acid group (—C ( ⁇ O) —SH), and the functional group that forms keto acid includes a keto acid group (—C ( ⁇ O) —CO 2 H). Is mentioned.
- examples of the substituent that forms a thiol group as a substituent derived from an S atom include a thiol group (—SH), such as alkylthiol, cycloalkylthiol, alkenylthiol, alkynylthiol, arylthiol, heteroarylthiol, aralkyl. Thiol is formed.
- the substituent forming thioether is selected from alkyl mercapto group, cycloalkyl mercapto group, alkenyl mercapto group, alkynyl mercapto group, aryl mercapto group, heteroaryl mercapto group, aralkyl mercapto group, etc.
- the substituent for forming a sulfoxide group includes an alkyl sulfoxide group, a cycloalkyl sulfoxide group, an alkenyl sulfoxide group, an alkynyl sulfoxide group, an aryl sulfoxide group, a heteroaryl sulfoxide group, an aralkyl sulfoxide group, and the like.
- examples of the substituent to form a sulfone group (-S (O) 2 -R) , alkyl sulfone group, a cycloalkyl sulfone group, alkenyl sulfone group, alkynyl sulfone group, Arirusu Hong group, heteroaryl sulfonic group, selected from among such aralkyl sulfone group, sulfonic acid group (-SO 3 H) are mentioned as the substituent to form a sulfonic acid.
- Examples of the substituent derived from the N atom include an azide group (—N 3 ), a nitrile group (—CN), and the substituent that forms a primary amine includes an amino group (—NH 2 ).
- Examples of the substituent for forming the secondary amine (—NH—R) include an alkylamino group, a cycloalkylamino group, an alkenylamino group, an alkynylamino group, an arylamino group, a heteroarylamino group, an aralkylamino group, and the like.
- Substituents for forming tertiary amines include alkyl (aralkyl) amino groups, alkyl groups, cycloalkyl groups, alkenyl groups, alkynyl groups, aryl groups, heteroaryl groups, aralkyl groups, etc.
- Examples of the substituent that forms a urea group include an aminocarbamoyl group (—NR—CO—NR′R ′′).
- R, R ′, and R ′′ each represent a hydrogen atom, an alkyl group, a cycloalkyl group, an alkenyl group, Examples include an alkynyl group, an aryl group, a heteroaryl group, and an aralkyl group, any three of the same or different ones, or a substituent selected from substituents that may form a ring.
- examples of the functional group derived from B atom include alkylborane (—BR (R ′)) and alkoxyborane (—B (OR) (OR ′)). These two substituents are an alkyl group, a cycloalkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, etc. And substituents selected from substituents that may form a ring.
- one or more various functional groups including O atom, N atom, S atom, B atom, P atom, Si atom, and halogen atom, which are usually used in low molecular compounds such as halogen groups, are imparted. May be. That is, one or more further substituents may be added to the alkyl group, cycloalkyl group, alkenyl group, alkynyl group, aryl group, heteroaryl group, or aralkyl group represented by one of these substituents. The condition of the functional group satisfying all of these is defined as freely selecting a substituent.
- any steric configuration is allowed as in the case of ⁇ -amino acids, and the selection of side chains thereof is not particularly limited, as in the case of ⁇ -amino acids.
- the amino acid main chain of the amino acid may be free (NH 2 group) or N-alkylated such as N-methylation (NHR group: R is an optionally substituted alkyl group, alkenyl group, alkynyl) A group, an aryl group, a heteroaryl group, an aralkyl group, or a cycloalkyl group, and a carbon atom from the N atom and a carbon atom from the ⁇ -position may form a ring as in the case of proline.
- “translated amino acid” or “translatable amino acid” refers to an “amino acid” having a side chain that can be translated.
- a halogen group a hydroxyl group (—OH), an alkoxy group (—OR), an ester group (—C ( ⁇ O) —OR), a thioester group (—C ( ⁇ O) —SR), Carboxyl group (—CO 2 H), amide group (—CO—NRR ′′ or —NR—CO—R ′), thiol group (—SH), alkylthio group (—SR), sulfoxide group (—S ( ⁇ O)) -R), sulfone group (-SO 2 -R), amino group (-NH 2 ), mono-substituted amino group (-NHR), di-substituted amino group (-NRR '), azido group (-N 3 ), nitrile An alkyl group, alkenyl group, alkyny
- L having a cycloalkyl group Type ⁇ -amino acids, N-methylated L-type ⁇ -amino acids, C1-C4 alkyl substitutions such as N-ethylation and N-propylation, and N-aralkyl substituted glycine derivatives such as N-benzylation, , L-type ⁇ -amino acids having various highly functional functional groups that can be utilized for substituents having a reaction auxiliary group such as a thiol group, triangular units such as amino groups, and crossing units, and D-tyrosine.
- Some D-type ⁇ -amino acids, ⁇ -amino acids such as ⁇ -alanine, and ⁇ , ⁇ -dialkyl amino acids such as ⁇ -methyl-alanine (Aib) are also included.
- the “drug-like amino acid” is the same skeleton as the “amino acid”, ie, ⁇ , ⁇ and ⁇ amino acids, and is one of two hydrogen atoms of the main chain amino group (NH 2 group). And one or two hydrogen atoms of the methylene group (—CH 2 — group) may be substituted with an alkyl group, a cycloalkyl group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, or the like. Good. A group in which a hydrogen atom of CH 2 group is substituted with the above group is defined as a side chain.
- substituents include those further substituted with a substituent that functions as a component of a drug-like peptide compound. These are preferably selected from the substituents defined above separately, for example, a hydroxyl group (—OH), an alkoxy group (—OR), an amide group (—NR—CO—R ′ or —CO—NRR ′). ), Sulfone group (—SO2-R), sulfoxide group (—SO—R), halogen group, hydroxylamino group (—NR—OR ′), aminohydroxy group (—O—NRR ′), etc. Optionally substituted by one or more substituents.
- any of those corresponding to L-type amino acid, D-type amino acid, ⁇ , ⁇ -dialkylamino acid may be used.
- Drug-like amino acids need not necessarily be translatable.
- Side-chain part of the peptide obtained from “translated amino acid” for example, when hit compound is obtained with D-tyrosine, hit compound is obtained with ⁇ -alanine, a D-type amino acid chemically modified therefrom.
- it includes ⁇ -amino acids chemically modified therefrom, or all amino acids that can be chemically synthesized by optimizing the structure of N-substituted moieties by chemical conversion of N-methyl amino acids.
- these amino acids function as components of a drug-like peptide compound, they are selected from a range in which the peptide compound obtained by chemical modification performed after translation becomes drug-like.
- lysine having an aminoalkyl group is not included in the drug-like amino acid when the amino group does not participate in post-translational modification.
- the amino group of lysine is used as a reactive functional group for post-translational modification (for example, a crossing unit)
- the lysine unit is included as a drug-like amino acid unit.
- drug-like amino acid is determined by the functional group after conversion by post-translational modification.
- substituents may include, for example, an ester group (—CO—OR), a thioester group (—CO—SR), a thiol group (—SH), or a protection group among the substituents defined above.
- Thiol group amino group (—NH 2), mono-substituted amino group (—NH—R) or di-substituted amino group (—NRR ′), protected amino group, substituted sulfonylamino group (—NH—SO 2 —) R), an alkylborane group (—BRR ′), an alkoxyborane group (—B (OR) (OR ′)), an azide group (—N3), a keto acid group (—CO—CO2H), a thiocarboxylic acid group (—CO -SH), phosphoryl ester group (-CO-PO (R) (R ')), acylhydroxylamino group (-NH-O-CO-R) and the like.
- the amino acid analog of the present invention preferably means ⁇ -hydroxycarboxylic acid.
- the side chain of ⁇ -hydroxycarboxylic acid may have various substituents in addition to hydrogen atoms as in the case of amino acids (may have free substituents).
- the steric structure of the ⁇ -hydroxycarboxylic acid may correspond to the L-type or D-type of amino acid, and there is no particular restriction on the selection of the side chain. , An alkynyl group, an aryl group, a heteroaryl group, an aralkyl group, a cycloalkyl group, and the like.
- the number of substituents is not limited to one and may be two or more. For example, it may have an S atom and further have a functional group such as an amino group or a halogen group.
- “translated amino acid analog” or “translatable amino acid analog” means an amino acid analog that can be translated among “amino acid analogs”. Specifically, for example, a compound in which the main chain amino group of the L-type amino acid is replaced with a hydroxyl group can be mentioned. Examples include L-lactic acid, ⁇ -hydroxyacetic acid, L or D-phenyllactic acid.
- the “drug-like amino acid analog” is not particularly limited as long as it functions as a component of a drug-like peptide compound among the “amino acid analogs”. This range is the same as the definition of the side chain or N-substituted moiety of the drug-like amino acid described above.
- L or D-lactic acid, or various drug-like substituents for example, halogen group, hydroxyl group, optionally substituted alkyl group, alkenyl group, alkynyl group) on the side chain methyl group , A cycloalkyl group, an aralkyl group, an aryl group, a heteroaryl group, etc.
- ⁇ -hydroxyacetic acid L or D-phenyllactic acid, or various drug-like substituents on the side chain benzyl group (for example, , Halogen groups, hydroxyl groups, optionally substituted alkyl groups, alkenyl groups, alkynyl groups, cycloalkyl groups, aralkyl groups, aryl groups, heteroaryl groups, etc.).
- a drug-like amino acid analog is not necessarily translatable.
- the amino acid analogue is also included in the “drug-like amino acid analogue”.
- amino acid analogs include ⁇ -hydroxycarboxylic acids having an SH group attached to the side chain and ⁇ -hydroxycarboxylic acids having an amino group or a protected amine moiety attached to the side chain.
- the SH group can be removed by desulfurization after post-translational modification, and the amino group can be converted to amide or the like by post-translational modification.
- Specific examples include R-2-hydroxy-3-sulfanylpropanoic acid.
- the N-terminal carboxylic acid analog of the present invention is a compound having an amino group and a carboxyl group at the same time and having 3 or more atoms between them, various carboxylic acid derivatives having no amino group, It may be a peptide formed from 4 residues, or an amino acid whose main chain amino group is chemically modified by an amide bond with a carboxylic acid. Moreover, you may have a boric acid and boric-ester part which can be used for the cyclization in a curve part. Moreover, the carboxylic acid which has a double bond site
- the portion other than the functional group that defines these compounds is also widely selected from alkyl groups, aralkyl groups, aryl groups, cycloalkyl groups, heteroaryl groups, alkenyl groups, alkynyl groups, etc. that may be substituted (free Substituents).
- translated N-terminal carboxylic acid analog or “translatable N-terminal carboxylic acid analog” refers to an N-terminal carboxylic acid analog that can be translated among “N-terminal carboxylic acid analogs”. Means the body.
- a compound in which a double bond and a carboxylic acid are connected by an alkyl group (but-3-enoic acid, penta-4-enoic acid, etc.), L having an N-terminal amidated by acetylation or the like Type amino acids (Ac-Phe, Ac-Ala, Ac-Leu, etc.), ⁇ -hydroxycarboxylic acid derivatives in which the OH group is alkylated, dipeptides, tripeptides and the like.
- the “drug-like N-terminal carboxylic acid analog” is not particularly limited as long as it functions as a component of a drug-like peptide compound among the “N-terminal carboxylic acid analogs”.
- substituents include the same substituents as defined in the side chain of the drug-like amino acid. Specifically, for example, in a compound in which a double bond and a carboxylic acid are connected by an alkyl group (but-3-enoic acid, penta-4-enoic acid, etc.), the substituent is within a drug-like range from the carbon atom.
- the acetyl group, the side chain or the ⁇ -position hydrogen atom is dragged Among the compounds substituted in the like range, ⁇ -hydroxycarboxylic acid derivatives in which the OH group is alkylated, the alkyl group of the OH group, the side chain of the hydroxycarboxylic acid, the ⁇ -position hydrogen atom, etc.
- Examples include substituted compounds, dipeptides substituted in a drug-like range, and tripeptides.
- a drug-like N-terminal carboxylic acid analog does not necessarily need to be translatable.
- the N-terminal carboxylic acid analog is also included in the “drug-like N-terminal carboxylic acid analog”. It is.
- Examples of such N-terminal carboxylic acid analogs include dipeptides with an amino group remaining at the N-terminus, ⁇ -aminocarboxylic acid, and ⁇ -aminocarboxylic acid.
- the total number of amino acids contained in the peptide compound is preferably 13 or less.
- the CLogP computer-based
- the CLogP computer-based
- the calculated partition coefficient (calculated using Daylight Version 4.9 of Daylight Chemical Information Systems, Inc.) exceeds 6.
- the amino acid side chain is more preferably selected from drug-like substituents.
- an optionally substituted alkyl group, cycloalkyl group, alkenyl group, alkynyl group, aralkyl group, aryl group, heteroaryl group for example, a halogen group, a hydroxyl group (—OH), an amide group ( -CO-NRR 'or -NR-CO-R'), a sulfone group (-SO 2 -R), an ether group (-OR), or the like is preferred (R and R 'are similarly substituted by these)
- aryl group part of the aryl group and aralkyl group examples include a phenyl group, a basic group such as a pyridine group, two or more heteroatom-containing groups such as a thiazole group, a hydrogen atom donor such as an imidazole group, and an indole group. Fused aromatic rings are also acceptable.
- N-alkylated compounds such as N-methylated or cyclized with ⁇ -position carbon atoms such as proline may be used as amino acids or amino acid analogs constituting peptide compounds.
- the number is 2 or more per peptide molecule.
- at least one amide bond that is not N-alkylated is present per peptide molecule.
- N-alkylation includes all chemical modifications other than NH and may therefore be substituted (selection of substituents is similar to amino acid side chain substituents to ensure membrane permeability above) It is selected from alkyl groups, cycloalkyl groups, alkenyl groups, alkynyl groups, aryl groups, heteroaryl groups, and aralkyl groups. N-alkylation includes those that form a ring structure between the N atom and the ⁇ -carbon, such as proline.
- the C-terminal part of the peptide compound is not a carboxylic acid but is chemically modified.
- an optionally substituted alkyl group, alkenyl group, alkynyl group, cycloalkyl group, aryl group, heteroaryl group, aralkyl group amide compound (—CO—NRR) such as piperidine amide obtained by reacting a carboxylic acid site with piperidine, etc.
- R and R' may be a hydrogen atom, the other may be chemically modified, both R and R 'may be chemically modified, and R and R' form a ring, for example, It may be chemically modified such as piperidine amide, R and R ′ may be both hydrogen atoms, or a carboxylic acid group may be substituted with a methyl group or a trifluoromethyl group. It is preferably converted into various nonionic functional groups such as a group, an alkenyl group, an alkynyl group, an aryl group, a heteroaryl group, and an aralkyl group. The selection of the substituent is the same as the substituent on the amino acid side chain for ensuring the membrane permeability above. Moreover, the amino acid which comprises a peptide compound can also optimize the compound synthesize
- optimization means chemical modification by converting the structure of each amino acid in a compound synthesized by translation from “translated amino acids” to become more drug-like peptide compounds.
- the peptide compound is chemically modified to have a peptide and / or chemically modified to be a peptide compound in which toxicity is further avoided.
- preferable toxicities to be avoided or avoided for example, hERG inhibition (heart toxicity), AMES test (carcinogenicity test), CYP inhibition (drug interaction test), CYP induction, GSH Binding ability measurement test (covalent bond formation test of peptide or peptide metabolite with glutathione) and the like.
- Such conversion is greatly different from conventional chemical modification by N-methylation or cyclization of natural peptides in that it can be carried out without greatly impairing the three-dimensional structure of the translationally synthesized compound.
- naturally-occurring peptides often have extremely ionized arginine and lysine residues contributing to the activity, so that it is difficult to easily convert them into drug-like functional groups.
- functional groups previously limited only by drug-like functional groups have acquired activity against medicinal targets and are cyclized by drug-like cyclization methods.
- clinical candidate compounds can be obtained with the same accuracy and success rate as small molecule chemistry.
- the CLogP value which is an index of fat solubility
- the binding activity to the target can be enhanced by alkylation or halogenation, which is usually performed even in the optimization of low molecular weight compounds. Usually, an improvement in activity of 10 to 50 times can be expected.
- These normal conversions can also increase the CLogP value at the same time. For example, it is possible to increase about 0.7 by chlorination and about 0.5 by methylation. If it has extremely ionized functional groups, increasing the CLogP value during the optimization process is not sufficient to obtain excellent membrane permeability, but if it does not have extremely ionized functional groups, A CLogP value sufficient for membrane permeation can be obtained during the optimization process.
- Confirmation of membrane permeability of the peptide compound of the present invention can be achieved by known methods such as rat intestinal tract method, cultured cell (Caco-2, MDCK, HT-29, LLC-PK1, etc.) monolayer method, Immobilized Artificial Membrane chromatography (Immobilized artificial membrane chromatography) method, method using a partition coefficient, ribosome membrane method, PAMPA (Parallel Artificial Membrane Permeation Assay) method and the like can be used.
- PAMPA Paraallel Artificial Membrane Permeation Assay
- the iPAMPA Pe values are 0.6 ⁇ 10 ⁇ 6 , 1.5 ⁇ 10 ⁇ 6 and 2.9 ⁇ 10 ⁇ 5 , respectively.
- the membrane permeability of the peptide compound of the present invention was able to obtain membrane permeability that can be used as a pharmaceutical when the value of iPAMPA Pe when measured by the PAMPA method is usually 1.0 x 10 -6 or more. It is possible to think, in is preferably 1.0x 10 -6 or more, more preferably 1.0 ⁇ 10 -5 or more, more preferably particularly not less 1.5 ⁇ 10 -5 or more, 2.0x10 -5 or higher Even more preferably.
- the total number of amino acids contained in the peptide compound is preferably 9 or more, more preferably 11 or more. If the total number of amino acids is a peptide compound of 9 or more, the above-described conditions for having membrane permeability do not affect the metabolic stability of the peptide compound.
- the metabolic stability of the peptide compound of the present invention can be confirmed using a known method, for example, hepatocytes, small intestinal cells, liver microsomes, small intestine microsomes, liver S9 and the like.
- a known method for example, hepatocytes, small intestinal cells, liver microsomes, small intestine microsomes, liver S9 and the like.
- the stability of peptide compounds in liver microsomes is described in LL von Moltke et al. (Midazolam hydroxylation by human liver microsomes in vitro: inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents. J Clin Pharmacol , 36 (9), 783-791). More specifically, it can be confirmed according to the method described in Example 18-2.
- Metabolic stability is determined when the value of liver clearance (CLh ⁇ ⁇ ⁇ int ( ⁇ L / min / mg protein)) is 150 or less when the stability in liver microsomes is measured according to the above method. It can be considered that metabolic stability that can be used as a pharmaceutical product has been obtained, and is preferably 100 or less.
- the peptide compound having a cyclic portion of the present invention can be produced using the method described below.
- the manufacturing method containing the following manufacturing methods can be mentioned.
- 1) An acyclic peptide compound composed of an amino acid residue and / or an amino acid analog residue, or an amino acid residue and / or an amino acid analog residue and an N-terminal carboxylic acid analog is encoded with the peptide compound A process of translating and synthesizing from the nucleic acid
- the acyclic peptide compound comprises an amino acid residue or amino acid analog residue having a reactive site on one of the side chains on the C-terminal side, and an amino acid residue or amino acid analog having another reactive site on the N-terminal side.
- a body residue or an N-terminal carboxylic acid analog 1) Combine the reaction point of the amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog on the N-terminal side with the reaction point of the amino acid residue or amino acid analog residue in the side chain on the C-terminal side And forming an amide bond or a carbon-carbon bond.
- a known method can be used for the translational synthesis of the present invention.
- N-terminal introduction of a translatable amino acid, a translatable amino acid analog or a translatable N-terminal carboxylic acid analog is generally translated with methionine as the N-terminal amino acid as the translation initiation amino acid, but other desired It is also possible to use a method in which the N-terminus is translated as a desired amino acid by translating the amino acid with an aminoacylated translation initiation tRNA. It is known that the introduction of an N-terminal amino acid has an amino acid tolerance higher than that at the time of extension, and uses amino acids and amino acid analogs that are significantly different in structure from natural amino acids (non-patent literature: J Am Chem Soc 2009 Apr 15; 131 (14): 5040-1.
- a translatable amino acid other than methionine, a translatable amino acid analog, or a translatable N-terminal carboxylic acid derivative into the N-terminus.
- a translation initiation tRNA a tRNA acylated with a triangle unit is added to a translation system from which methionine, formyl donor, or methionyl transferase has been removed, and the translation initiation codon (for example, ATG) is encoded and translated to translate the end unit.
- a peptide compound or peptide compound library before cyclization as a triangular unit is constructed.
- various combinations of a translation initiation tRNA whose anticodon is not CAU and a codon corresponding to the anticodon are used as a combination of the translation initiation tRNA and the initiation codon, diversity can be provided at the N-terminus.
- a desired amino acid, amino acid analog or N-terminal carboxylic acid analog is aminoacylated to multiple types of translation initiation tRNAs having different anticodons, and an mRNA or mRNA library having the corresponding codon as the start codon is translated.
- the peptide compound before cyclization or a library of peptide compounds can be prepared in which the N-terminal residue is not limited to one type.
- Anticodon sequence mutants of Escherichia coli initiator tRNA effects of overproduction of aminoacyl-tRNA synthetases, methionyl-tRNA formyltransferase, and initiation factor 2 on activity in initiation. Biochemistry. 2003, 42, 4787-99.
- Peptide compounds or peptide compound libraries can be generated.
- reaction point of the amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog on the N-terminal side and the reaction point of the amino acid residue or amino acid analog residue in the side chain on the C-terminal side are combined.
- the amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog having an amino group and a reaction auxiliary group in the N-terminal side chain, and an amino acid having an active ester group in the side chain A method of obtaining a cyclic compound by amide-bonding a residue or amino acid analog residue, having an amino group at the N-terminal amino acid residue, amino acid analog residue or N-terminal carboxylic acid analog, and an active ester group
- the present invention is not limited to the above example, and for example, an amino acid residue on the N-terminal side, an amino acid analog on the N-terminal side, or an active ester group on the N-terminal carboxylic acid side, and an amino group on the side chain on the C-terminal side
- a functional group arrangement opposite to the above may be used, such as amide bonding (which may have a reaction auxiliary group).
- reaction design taking the case of using amide cyclization as an example. It may be necessary to improve the reactivity of the amine to be reacted with respect to other functional groups in order to obtain reaction selectivity with the basic functional group that is not desired to react with the amino group to be reacted with the triangular unit. possible.
- a reaction auxiliary group can be introduced in the vicinity of the amine.
- the reaction auxiliary group is not particularly limited as long as it can activate the amine to such an extent that RNA does not react.
- a mercaptoethyl group or a mercaptopropyl group can be introduced from a terminal amine, and cysteine or the like can be used.
- RNA to which the SH group of the obtained cyclic peptide is added with a reagent such as TCEP (tris (2-carboxylethyl) phosphine) and VA-044 (2,2′-azobis-2- (2-imidazolin-2-yl) propane)
- TCEP tris (2-carboxylethyl) phosphine
- VA-044 2,2′-azobis-2- (2-imidazolin-2-yl) propane
- a thioester is preferred as the active ester of the crossing unit to be translated.
- an SH group is used as the reaction auxiliary group, for example, the following combinations are performed.
- an aspartic acid derivative is a crossing unit. In this case, all the black circle units and the square units can be arbitrarily selected from translated amino acids and translated amino acid analogs.
- the square unit on the C-terminal side of the aspartic acid thioester is an N-alkylated amino acid (such as proline or N-methyl alanine). Is preferably selected from.
- an amino acid having a reaction auxiliary group in the vicinity of an amine as a triangular unit an N-terminal carboxylic acid analog, for example, an N-terminal amino acid can be written as compound N-1 or N-2.
- Substituents of these compounds N-1 and N-2 are preferably defined as described above, among the substituents represented by R, except for protecting groups (R1, R23, trityl group, etc.) This is the same as the definition of the side chain of the drug-like amino acid.
- R1 is selected from among the protecting groups of SH group represented by a hydrogen atom or the like, or S-R23 group, or C (Phe) 3 group (trityl group).
- R23 is an alkyl group such as a methyl group, an ethyl group, an isopropyl group or a tert-butyl group, an aryl group such as a phenyl group, a p-trifluoromethylphenyl group or a p-fluorophenyl group, an aralkyl group such as a benzyl group or a phenethyl group. Selected from a group, a heteroaryl group, an alkenyl group, an alkynyl group, and the like.
- These groups are selected from substituents that enable the resulting compound N-1 or N-2 to be translationally synthesized.
- substituents that enable the resulting compound N-1 or N-2 to be translationally synthesized.
- an N, N-dimethylaminoethyl mercapto group in which the ethyl group of R23 is substituted with a dimethylamino group may be used.
- R1 has a protecting group (other than H atom)
- the protecting group selected is selected from protecting groups capable of translational synthesis, and deprotected in a translation synthesis solution, and cyclized. If it becomes a hydrogen atom before making it, it will not specifically limit and can be selected.
- protecting groups such as the S—R23 group are slowly deprotected in the translation synthesis solution, they can be deprotected without the need to positively define separate deprotection conditions. If necessary, a deprotecting agent can be added under various reaction conditions described in the present specification. (Description of protecting group of SH group.)
- R2 and R3 are defined similarly to the definition of the side chain of a drug-like amino acid.
- R2 and R3 preferably represent a hydrogen atom, an optionally substituted alkyl group, alkenyl group, alkynyl group, aryl group, heteroaryl group, aralkyl group, or cycloalkyl group, or R2 And R3 represents a substituent that forms a ring, or R2 or R3 represents a substituent that forms a ring with R4. More preferably, it is selected from a hydrogen atom, a C1-C4 alkyl group which may be substituted with a C1-C4 alkyl group, an alkoxy group, a halogen group or the like.
- the R3 group can accept both L-type amino acids and D-type amino acids.
- the steric structure of the R3 group corresponds to the L-type amino acid when the R3 group is assumed to be a hydrogen atom. *
- R4 is a unit that links an S (sulfur) atom and an amino acid site.
- the typical structure is shown below. Both units may be substituted methylene group (partial structure N-3, C1 unit), may be substituted ethylene group (partial structure N-4, C2 unit), may be substituted propylene group (partial structure) N1 and C6 units such as N-5 and C3 units).
- any of the derivatives when any of the derivatives is translationally synthesized, all other derivatives are included in this definition even if they are not translationally synthesized.
- R13, R14, R15, R16, R17, R18, R19, R20, R21, R22 and the like have the same definition as R4, but are substituted with, for example, a hydrogen atom, a C1-C4 alkyl group, an alkoxy group, a halogen atom, etc.
- C1-C4 alkyl groups which may be substituted.
- a cyclized structure may be formed between them. Particularly preferably, it is selected from a hydrogen atom and a methyl group. It can also be directly linked from the aryl carbon of the aromatic compound (partial structure N-6). Further, they can be linked with an aralkyl structure (partial structures N-7 and N-8).
- any of the divalent groups may be on the nitrogen atom side or on the sulfur atom side.
- the connecting position is limited to ortho, but not limited to ortho, meta, para, etc. are possible.
- an aryl group is represented by a phenyl group
- the phenyl group may be substituted with a substituent such as a halogen group, an alkoxy group, or a trifluoromethyl group, and an aryl group other than a phenyl group (that is, a heteroaryl group is included)
- a substituent such as a halogen group, an alkoxy group, or a trifluoromethyl group
- an aryl group other than a phenyl group that is, a heteroaryl group is included
- Various aromatic rings may be used.
- R11 and R12 are also selected from the same partial structure as R4. For example, it can be selected from the partial structures N-3, N-4, N-5, N-6, N-7, and N-8. R12 can also contain a C0 unit (when the linking site is directly attached).
- R12 site of compound N-1 is a C0 unit and the linking site is directly linked.
- the R11 site of compound N-2 is linked by a C1 unit (corresponding to partial structure N-3).
- the R11 site of compound N-2 is linked by a C2 unit (corresponding to partial structure N-4).
- the R11 site of compound N-2 is linked by a C3 unit (corresponding to partial bond N-5).
- R5 to R10 are the same as defined above for R13 to R18.
- compound N-13 the R4 site of compound N-9 is linked by a C1 unit (corresponding to partial structure N-3).
- compound N-14 the R4 site of compound N-10 is linked by a C2 unit (corresponding to partial structure N-4).
- compound N-15 the R4 site of compound N-11 is linked by a C2 unit (corresponding to partial structure N-4).
- compound N-16 the R4 site of compound N-12 is linked by a C2 unit (corresponding to partial structure N-4).
- compound N-17 the R4 site of compound N-9 is linked by a C2 unit (partial structure N-4).
- compound N-18 the R4 site of compound N-10 is linked by a C3 unit (partial structure N-5).
- compound N-19 the R4 site of compound N-11 is linked by a C3 unit (partial structure N-5).
- compound N-20 the R4 site of compound N-12 is linked by a C3 unit (partial structure N-5).
- the definition of the triangular unit containing the reaction auxiliary group SH is not limited thereto. That is, it has an amino group which is a unit to be reacted with the crossing unit and a reaction auxiliary group, and is selected from arbitrary structures among structures capable of translational synthesis.
- the amino group may be derived from a main chain or a side chain.
- the triangular unit is not necessarily present at the N-terminal, and a square unit (straight chain part) may be present on the N-terminal side of the triangular unit.
- a chemical structure in which the positional relationship between the SH group and the amino group is in the range of 2 to 6 linking atoms such as ⁇ (two linking atoms between two functional groups), ⁇ (three linking atoms) is preferable. More preferably, the positional relationship between the SH group and the amino group is ⁇ or ⁇ . Moreover, even if the unit which has an active ester functional group is arrange
- an active ester means a carboxylic acid derivative that can be reacted with an amino group site directly or via a reaction auxiliary group, and is not particularly limited as long as it is an active ester or an active thioester having such properties.
- R25 is selected from a hydrogen atom or an active ester group.
- the active ester is represented by, for example, N-hydroxysuccinimide (ONSu) group, OAt group, OBt group, methylthioester, arylthioester, aralkylthioester and the like as widely used.
- These active ester moieties are generally used as compound substituents (for example, halogen groups, nitro groups, trifluoromethyl groups, nitrile groups, etc., which are often used for the purpose of increasing reactivity).
- Electron-withdrawing groups alkoxy groups such as methoxy groups, which are often used for the purpose of lowering the reaction and increasing the reaction selectivity, electron-donating groups such as alkyl groups such as methyl groups, t-butyl groups and isopropyl
- alkyl groups such as methyl groups, t-butyl groups and isopropyl
- Examples include bulky substituents typified by groups, sulfonic acid groups, disubstituted amino groups such as dimethylamino groups, etc., taking into consideration the affinity with water after being carried out in water.
- Examples include high-fat-soluble groups such as long-chain alkyl groups in consideration of affinity with lipophilicity.) All derivatives having similar reactivity are also included.
- R2 and R3 are as defined above for the amine moiety.
- R26 is the same as the definition of R4, and its typical structure is shown below. Both units can be connected by C1 to C6 units such as methylene group (partial structure N-3), ethylene group (partial structure N-4), propylene group (partial structure N-5) and the like. In R13, R14, R15, R16, R17, R18, R19, R20, R21, R22, these substituents are preferably the same as the definition of the side chain of the drug-like amino acid defined above. Even if the derivative itself is not translationally synthesized, it includes those in which the analog is translated and synthesized.
- a hydrogen atom for example, it is selected from a hydrogen atom, a C1-C4 alkyl group, a C1-C4 alkyl group which may be substituted with a halogen atom and the like.
- a cyclized structure may be formed between them. More preferably, it is selected from a C4 unit, a C5 unit and a C-6 unit in addition to a methylene group (C1 unit, partial structure N-3). More preferably, a C1 unit (partial structure N-3) is selected. It can also be directly linked from the aryl carbon of the aromatic compound (partial structure N-6). Further, they can be linked with an aralkyl structure (partial structures N-7 and N-8).
- the connecting position is limited to ortho, but not limited to ortho, meta, para, etc. are possible.
- the phenyl group is shown as the aryl group, the phenyl group may be substituted with a substituent such as a halogen group or an alkoxy group, or an aryl group other than the phenyl group may be used.
- R27 represents a hydrogen atom, an alkyl group that may be substituted, an alkenyl group that may be substituted, an alkynyl group that may be substituted, an aryl group that may be substituted, a heteroaryl group that may be substituted, Or an aralkyl group to which an optionally substituted alkyl group may be provided.
- substituents are not particularly limited as long as compound C-2 obtained as a result of selection of the substituent can be translationally synthesized.
- an electron withdrawing group such as a halogen group, a nitro group, a trifluoromethyl group, or a nitrile group, which is often used for the purpose of increasing the reactivity, or a purpose of increasing the reaction selectivity by lowering the reaction
- Alkoxy groups such as methoxy groups
- electron donating groups such as alkyl groups such as methyl groups
- bulky substituents typified by t-butyl groups and isopropyl groups, etc.
- a sulfonic acid group considering the affinity with water a disubstituted amino group such as a dimethylamino group, and a highly lipophilic group such as a long-chain alkyl group considering lipophilicity are selected.
- a disubstituted amino group such as a dimethylamino group
- a highly lipophilic group such as a long-chain alkyl group considering lipophilicity are selected.
- it is selected from an optionally substituted alkyl group, an optionally substituted cycloalkyl group, and an optionally substituted aralkyl group. More preferably, it is selected from an alkyl group and an aralkyl group in which the aryl moiety may be substituted.
- R3 is defined similarly to the definition of the side chain of a drug-like amino acid, and is selected from, for example, a C1-C4 alkyl group, a C1-C4 alkyl group which may be substituted with a halogen, etc., and particularly preferably a hydrogen atom .
- the R3 group conforms to both L-type and D-type amino acids when the R3 group is assumed to be a hydrogen atom, those corresponding to L-type amino acids are preferred.
- R3 is selected from a hydrogen atom, a C1-C4 alkyl group, a C1-C4 alkyl group optionally substituted with halogen, etc., and a hydrogen atom is particularly preferred.
- the R3 group conforms to both L-type and D-type amino acids when the R3 group is assumed to be a hydrogen atom, those corresponding to L-type amino acids are preferred.
- the compound COH-1, Compound COH-2, and Compound COH-3 can also be selected.
- the reaction with Compound N-1 or Compound N-2 can be allowed to proceed gently and selectively.
- the reaction can proceed smoothly even in a translation solution (for example, 37 ° C. and pH around 7.3). Removal of the reaction auxiliary group can be easily performed under reaction conditions in which RNA is stable.
- an amine unit having a reaction auxiliary group may be arranged on the C-terminal side (crossing unit).
- an amino group and a thiol group are arranged in the side chain of the intersection unit. These may be protected at the translation stage, but are deprotected immediately before the reaction.
- the amino group and the thiol group are not particularly limited as long as they are present in the vicinity, but the relationship between the two is preferably the ⁇ -position or ⁇ -position.
- a drug-like ring is formed by an amide condensation reaction with an active ester such as a thioester on the side chain of the triangular unit and an amino group of the side chain (having a reaction auxiliary group such as thiol in the vicinity) on the other (crossing unit). All the techniques for making them into are also included in the present application, and either functional group may be arranged in a triangular unit or a crossing unit.
- Ra-7 can be selected from Na-7, Na-8, and Na-9 groups.
- the Na-7 group, Na-8 group and Na-9 group are selected by being limited to only one of Ra-7 or Ra20 to Ra25.
- Ra20 to Ra25 other than the substituent in which the Na-7 group, Na-8 group, or Na-9 group is selected include a hydrogen atom, an alkyl group that may be substituted with a drug-like functional group, an aryl group, A heteroaryl group, an aralkyl group, and the like are selected from. Preferably, it is selected from a hydrogen atom and an alkyl group.
- a unit having an active ester such as a thioester group is selected on the triangular unit (N-terminal side)
- these include, for example, Compound C-1, Compound C-2, Compound C-3, Compound Ca-1, Compound COH -1, compound COH-2, and compound COH-3.
- Compound C-1 and the like retain the main chain amino group after cyclization, whereas compound Ca-1, compound COH-1, compound COH-2, and compound COH-3 have no amino group. Therefore, it is more preferable because it becomes more like a drug.
- the crossing unit is selected from compound Na-10 (Na-7 group or Na-8 group), compound Na-11 (Na-7 group or Na-8 group), and the like.
- These compounds may be translated in a protected state.
- the protecting group that can be translated and the deprotection conditions under reaction conditions stable to RNA can use the methods described herein. More preferably, the use of the compound Na-7 group is more preferable than the use of the Na-8 group because of higher metabolic stability.
- Compound C-1, Compound C-2, Compound C-3, Compound COH-1, Compound COH-2, Compound COH-3, etc. having an active ester group at the intersection unit (C-terminal side) were selected.
- the triangular unit (on the N-terminal side) can be selected from Compound Na-10 and Compound Na-11 in addition to Compound N-1 and Compound N-2 already described.
- compound Na-10 (Na-8 group and Na-9 group) or Na-11 (Na-8 group, And Na-9 groups) are preferred. This is because when the Na-7 group is used, the main chain amine is retained and the drug likeness is lowered.
- compound Na-10 Na-7 group and Na-8 group
- compound Na-11 Na-7 group and Na-8 group
- the Na-7 group is derived from an ⁇ amino acid skeleton, but it may be a ⁇ amino acid skeleton.
- the method of activation from an amine is not limited to the one containing an SH group as the above-mentioned reaction auxiliary group.
- a method of activation from an amine it is possible to introduce a hetero atom directly into the amine to improve the reactivity of the amine.
- hydroxyamine compound F-1, compound F-4, compound F-5, compound F-7
- alkoxyamine compound F-2, compound F-14, compound F-15, compound F-16
- azide Compound F-3, Compound F-9, Compound F-10, Compound F-11
- R101, R102, R103, R104, R105, R106, and R107 are commonly used amino acid side chains, and are substituents not limited to natural amino acids. That is, a hydrogen atom, an optionally substituted alkyl group, an optionally substituted alkenyl group, an optionally substituted alkynyl group, an optionally substituted aryl group, an optionally substituted heteroaryl group, and optionally substituted Selected from good aralkyl groups. More preferably, one of R101 and R102 is a hydrogen atom, one of R103 and R104 is a hydrogen atom, and one of R106 and R107 is a hydrogen atom.
- the hydrogen atoms are preferably arranged so that they have the same steric configuration as the L-type amino acid.
- R105 is selected from an optionally substituted alkyl group, an optionally substituted alkenyl group, an optionally substituted alkynyl group, an optionally substituted aryl group, and an optionally substituted aralkyl group.
- an active ester which is a candidate for a crossing unit corresponding to the case where an activated amine as described above is selected
- a combination of an amine having a thioester and a thiol in the vicinity, an ⁇ ketoester and an azide, and the like can be considered.
- Initiation read through generally means that proteins and peptides are translated from methionine, which is the translation initiation amino acid encoded as an AUC codon. It means a phenomenon in which a translation product from an amino acid encoded by the second and subsequent codons is generated when an attempt is made to start translation from a low unnatural amino acid added to a translation initiation tRNA.
- a triangular unit is encoded at the second codon next to the start codon of mRNA encoding the peptide, and is translated by a translation system that does not contain methionine or translation initiation methionine tRNA.
- a method for preparing a peptide or peptide library having a terminal trigonal unit can be used.
- a method is known in which an N-terminal methionine of a peptide is scraped by the action of an enzyme such as peptide-deformylase and Methionine-aminopeptidase (Non-patent literature: Meinnel, T). ., Et al.
- a triangular unit can be encoded by a plurality of codons, so that the degree of freedom is expanded to two or more types and one variable part is added. Since it is possible to use two or more types of amino acids or amino acid analogs as a triangular unit, when preparing the peptide compound or peptide compound library of the present invention, the peptide compound or peptide compound live having increased diversity. A rally can be produced.
- the random region means a region where an amino acid or an amino acid analog can be freely selected in the peptide compound of the present invention. Except for this method, a region other than the crossing unit and the triangular unit of Scheme A (that is, a black circle). In this method, triangular units are also random areas. The structural diversity of the black circle unit and the square unit can be secured while maintaining the reactivity with the cross unit of the triangle unit.
- the number of residues having drug likeness is 13 or less, so that the number of units to be fixed decreases from 2 to 1, the number of units in the random region from 11 to 12 Means to increase.
- the value of increasing the number of residues that can be randomized by one is very high in the sense that it maximizes the degree of freedom under limited conditions to maintain drug likeness.
- carboxylic acid or carboxylic acid active ester can be translated and introduced into the crossing unit (amino acid intersecting linear part, cyclic part and triangular unit) side chain, so from fixed crossing unit and random amino acid
- An amide bond can be formed between selected triangular units (Scheme B).
- the crossing unit does not have to be one type, and can be selected from two or more types.
- a codon encoding each crossing unit, (amino) acylated tRNA is prepared, and a library can be constructed using mRNA in which the crossing unit codon is arranged at a desired position as a template.
- an example of amide cyclization can be given (this method can also be used when the N-terminus is fixed).
- An aspartic acid derivative is taken as an example of the crossing unit, but is not limited thereto, and is selected from, for example, a compound group represented by Compound C-1, Compound C-2, and Compound C-3. Any amino acid or amino acid analog having a carboxylic acid in the side chain, such as an N-alkylated product such as N-methylaspartic acid or a glutamic acid derivative, may be used.
- the key to realizing this method is to select a method in which only the carboxylic acid site is active esterified and RNA (such as a nucleic acid site) is not reacted.
- RNA such as a nucleic acid site
- An amino acid having a carboxylic acid active ester in the side chain can be introduced by translation, and the active ester can be reacted with an amine.
- an active ester such as compound E-2 is preliminarily translated and synthesized.
- a benzylthioester such as compound E-3, an arylthioester such as compound E-4, an alkylthioester, and the like can also be introduced by translation.
- a phenyl group is taken as an example, but there is no limitation as long as it is an aryl group or a heteroaryl group.
- aryl groups and heteroaryl groups can be substituted with, for example, electron-withdrawing groups such as halogen groups, nitro groups, trifluoromethyl groups, and nitrile groups, alkoxy groups such as methoxy groups, and alkyl groups such as methyl groups. Such as an electron donating group.
- substituents include bulky substituents typified by t-butyl group and isopropyl group, such as a sulfonic acid group and a dimethylamino group that have been used in water and have an affinity for water. Selected from di-substituted amino groups and conversely lipophilic groups such as long-chain alkyl groups in consideration of lipophilicity. Many kinds of substituents such as nitro group, trifluoromethyl group, and halogen may be introduced at the same time. Like compound E-4, a thioaryl active ester activated more than compound E-3 may be introduced in advance.
- the thioester can be selected from alkyl thioesters in addition to such aralkyl and aryl thioesters.
- the key to the realization of this method is to have two properties that are stable during translational synthesis and have sufficient reactivity with an amine having no reaction auxiliary group.
- Translation of active esters that can ensure sufficient stability during translation synthesis from thioesters, etc., and adding additives after translation to generate more active active esters in the system and react It is also possible to carry out a cyclization reaction with an amine having no auxiliary group.
- a more active thioester is generated in the system by adding thioester having a more electron deficiency to the thioester introduced by translation from the outside, and cyclized with an amine.
- thioester such as compound E-3
- it may be reacted directly with an amino group after translation, but a more reactive thiol such as trifluoromethylphenylthiol may be added to the translation system.
- the activated ester E-4 may be substituted with a more activated ester and then reacted with an amino group.
- various raw materials known to form active esters such as HOBt, HOAt, and HONSu can be added.
- One type of additive may be selected from these, or two or more types may be selected.
- An advantage of adding two or more additives is improved reactivity. It is energetically disadvantageous to convert an active ester that is sufficiently stable and translatable in a translation solution into an active ester that is sufficiently reactive with any amino group, and such a reaction may be difficult in one step. . In such a case, once the translatable stable active ester is returned to a more active active ester that can be exchanged, it can be further returned to an active ester sufficiently reactive with any amino group. Become. By using the activation of the active ester in such a multi-stage, an amidation reaction with an amino group having low reactivity may be possible. Substituents may be introduced in these additives such as active esters.
- substituents such as halogen groups, nitro groups, trifluoromethyl groups, nitrile groups and other electron-withdrawing groups, methoxy groups and the like It may have an electron donating group such as an alkyl group such as an alkoxy group or a methyl group.
- Bulky substituents typified by t-butyl and isopropyl groups, such as sulfonic acid groups, carboxyl groups, hydroxy groups, and dimethylamino groups in consideration of affinity with water It is selected from a di-substituted amino group, or a highly lipophilic group such as a long-chain alkyl group in consideration of lipophilicity.
- a stable active ester is introduced in the same manner as in (iii), a chemical reaction (for example, deprotection reaction) is carried out after translation, and after activation by an intramolecular reaction, an amine having no reaction auxiliary group It is also possible to cause a cyclization reaction.
- a chemical reaction for example, deprotection reaction
- a more stable thioester is translated and introduced, and after translation, the SS bond is deprotected, and is converted into a more reactive arylthiophenol by intramolecular reaction. May be reacted with an amine.
- this problem may be achieved by combining two or more of the concepts (i) to (iv). In this way, as one method for effectively utilizing the Initiation read through method, a more diverse Display library having a druglike cyclization site by reacting an amino group commonly present at the N-terminus with a crossover unit. Can be built.
- alkylthioesters or benzylthioesters such as alkylthioesters or benzylthioesters, which can ensure sufficient stability during translation synthesis, use active esters that can be translated, and add additives after translation to obtain more active active esters.
- active ester used for translational synthesis for example, aspartic acid side chain carboxylic acid is alkylthioester such as methylthioester (Asp (SMe)) or benzyl Aralkylthioesters such as thioester (Asp (SBn)) can be used.
- the electron-withdrawing group include a trifluoromethyl group, a nitro group, and a fluoro group, and an electron-withdrawing group of about the trifluoromethyl group is preferable.
- the addition amount of these thiols is not particularly limited, but is preferably more than 10 mM for the purpose of sufficiently increasing the reactivity, and less than 10 M for the purpose of dissolving the additive.
- the range of 50 mM to 5 M is more preferable, but 200 mM to 2 M is more preferable.
- the additive may be added as thiol (acidic), but the acidic part is preferably neutralized by adding an equivalent number of a base such as triethylamine and added as neutral conditions.
- Active esters with higher activity may react with various reagents present in the translation reaction system.
- the amine component (trishydroxymethylaminoethane) in the commonly used Tris buffer can be one of them, so translation synthesis and a buffer for chemical reaction are added in a buffer that does not contain a reactive amine component. It is preferable to do.
- a buffer examples include HEPES buffer and phosphate buffer.
- the side reaction is accompanied by an oxidation reaction in the air (SS formation reaction), avoiding a decrease in the amount of thiol necessary for the progress of the reaction, and the basicity is high and hydrolysis is dominant.
- SS formation reaction an oxidation reaction in the air
- a buffer to the reaction translation solution.
- a reducing agent such as tris (2-carboxyethyl) phosphine. It is also effective to keep the cyclization reaction away from oxygen in the air as much as possible.
- the pH of the solvent during the chemical reaction is preferably 2 to 10 for the purpose of stably presenting RNA.
- the reaction conditions may be carried out alone in a reaction translation solution such as PureSystem, and an organic solvent such as DMF or NMP may be added thereto.
- a reaction translation solution such as PureSystem
- an organic solvent such as DMF or NMP may be added thereto.
- the reaction temperature is not particularly limited as long as it is a range in which a chemical reaction can be usually carried out.
- the amino group can be either a primary amine or a secondary amine (for example, an N-alkyl group such as N-methyl). Is not limited. In particular, when the carbon atom next to the amino group is unsubstituted (CH2), either a primary amine or a secondary amine is acceptable. Among the secondary amines, a methyl group is more preferable. When a substituent is present at the carbon atom next to the amino group, a primary amine is more preferable. As the substituent portion, it is more preferable that the ⁇ -position is CH2 such as Ala or Phe, such as Val or Thr.
- CH2 such as Ala or Phe, such as Val or Thr.
- amino acids in which a nitrogen atom and an ⁇ -position carbon atom form a 5-membered ring such as proline, and cyclic secondary amines such as a 4-membered ring and a 6-membered ring are also preferable.
- a unit having an active ester may be arranged in the triangular unit, an amine side chain may be arranged on the crossing unit side, and the active ester and the amino group may be arranged between the triangular unit and the crossing unit. It may be arranged in either.
- the crossing unit is selected from compound C-1, compound C-2, compound C-3, or compound COH-1, compound COH-2, and compound COH-3. You can also choose. Whenever these six kinds of compounds are selected, it is preferable that the amino acid immediately after the C-terminal side is selected from N-alkylated units. This limitation is for the purpose of avoiding side reactions of aspartimide formation and is a common preferred choice when these six compounds are selected. From the viewpoint of metabolic stability, Compound C-1, or Compound C-2, or Compound C-3 is more preferable. In such a case, the triangular unit can be selected from Compound Na-1, Compound Na-2, and Compound Na-3.
- the N-terminal (triangular unit) When the N-terminal (triangular unit) is not fixed as a triangular unit, a plurality of these compounds can be selected simultaneously.
- the side chain amino group may be protected in any of Compound Na-1, Compound Na-2, and Compound Na-3.
- the cyclization reaction is carried out after deprotection at the same time or in advance of the cyclization reaction.
- the method described in this specification can be used for the protecting group or deprotection conditions.
- Ra13 of compound Na-1 can be selected from a hydrogen atom, a C1-C6 alkyl group or an aralkyl group. These may be substituted with a functional group defined by drug-like, such as a hydroxyl group, a fluoro group, and an ether group.
- a hydrogen atom or a methyl group, an ethyl group, an npropyl group, or a benzyl group is preferable.
- Compound Ra-2 Ra1 can be selected in the same manner as Ra13. Particularly preferably, it is selected from a hydrogen atom or a methyl group.
- Ra2 can be selected in the same manner as Ra13, but preferably a hydrogen atom or a methyl group. Particularly preferred is a hydrogen atom.
- a hydrogen atom When a hydrogen atom is selected, the steric configuration of the L-type and D-type is allowed as an amino acid. An L-shaped configuration is more preferable.
- Ra3 can be selected in the same manner as R13. Ra3 and Ra4 may form a ring. Particularly preferred is a hydrogen atom.
- Ra4 can be selected from a hydrogen atom, an alkyl group, a cycloalkyl group, an aralkyl group, a heteroaryl group, and an aryl group. These may be substituted with a functional group defined by drug-like. Ra4 may form a ring together with Ra1. For example, proline and the like correspond to this ring.
- Ra11 of compound Na-3 can be selected in the same manner as Ra13, and particularly preferably a hydrogen atom, or a methyl group, an ethyl group, an npropyl group, or a benzyl group. Particularly preferred is a hydrogen atom.
- Ra9 can be selected in the same way as Ra4. It is preferably selected from a hydrogen atom, a C1-C6 alkyl group or an aralkyl group. These may be substituted with a functional group defined by drug-like. Further, Ra9 and Ra11 may form a ring.
- the ring size is preferably a 3- to 8-membered ring. As the ring formation, a 5-membered ring and a 6-membered ring are particularly preferable.
- Ra10 and Ra12 can be selected in the same manner as Ra4. Preferably, it can select like Ra13. More preferably, either Ra10 or Ra12 is a hydrogen atom. Particularly preferably, both Ra10 and Ra12 are hydrogen atoms.
- the N-terminal is preferably selected from an amino acid derivative or an N-terminal carboxylic acid derivative. The presence of an amino group in the N-terminal main chain is disadvantageous in terms of reaction selectivity, and the amino group is retained even after completion of translation modification, resulting in a decrease in drug likeness.
- the crossing unit is selected from Compound C-1, Compound C-2, or Compound C-3, as a triangular unit, if the triangular unit is placed at a position other than the N-terminal and the triangular unit is fixed, the triangular unit is It can be selected from Compound Na-4 or Compound Na-5.
- Ra-4 Ra5 can be selected in the same manner as Ra1.
- Ra6 can be selected in the same manner as Ra2.
- Ra7 can be selected in the same way as Ra4. More preferred is a hydrogen atom or a methyl group. Particularly preferred is a hydrogen atom.
- Ra8 is the same as R4, alkylene group of C1-C6 units such as partial structures N-3, N-4, N-5, N-6, N-7, N-8 (not only ortho-substituted but also meta-substituted , Including para substitution).
- the substituents R13 to R22 can be selected from functional groups that do not react with an active ester or amino group among functional groups selected by drug-like.
- it is selected from a C4 to C6 alkylene unit which may have a substituent, or a partial structure N-6, N-7, N-8 or the like having an aryl group.
- ⁇ Ra5 of Compound Na-5 can be selected in the same manner as Ra1.
- Ra6 can be selected in the same manner as Ra2.
- Ra7 can be selected in the same way as Ra4. More preferred is a hydrogen atom or a methyl group. Particularly preferred is a hydrogen atom.
- Ra8 is the same as R4, alkylene group of C1-C6 units such as partial structures N-3, N-4, N-5, N-6, N-7, N-8 (not only ortho-substituted but also meta-substituted , Including para substitution).
- the substituents R13 to R22 can be selected from functional groups that do not react with an active ester or amino group among functional groups selected by drug-like.
- it is selected from a C4 to C6 alkylene unit which may have a substituent, or a partial structure N-6, N-7, N-8 or the like having an aryl group.
- the side chain amino group of either Compound Na-4 or Compound Na-5 may be protected.
- the cyclization reaction is carried out after deprotection at the same time or in advance of the cyclization reaction.
- the method described in this specification can be used for the protecting group or deprotection conditions.
- the triangular unit can be selected from a wide group of compounds having an amino group and a carboxyl group in addition to the compound Na-4 and the compound Na-5.
- various units can be translationally synthesized as N-terminal carboxylic acid analogs. There is no particular limitation as long as it has an amino group to be amide cyclized and a carboxylic acid to be translated into a peptide.
- the functional group of the divalent unit that joins the amino group and the carboxylic acid group is selected from among drug-like functional groups.
- An example of such a compound is Compound Na-6.
- Ra9 of Compound Na-6 may be selected from an alkyl group, a cycloalkyl group, an aryl group, a heteroaryl group, and an aralkyl group that may be substituted with a drug-like functional group.
- -NRCOR 'group R and R' are drug-like substituents
- -OR R is a drug-like substituent
- NR groups including R amino acids, dipeptides, tripeptides, etc.
- the triangular unit includes Compound C-1, Compound C-2, Compound C-3, Compound COH-1, Compound COH-2, Compound COH-3, etc. Can also be selected.
- a straight chain portion is present also on the N-terminal side from the triangular unit, and the N-terminal is selected from N-terminal carboxylic acid derivatives.
- Compound Na-4 or Compound Na-5 is selected, Compound COH-1, Compound COH-2, Compound COH-3, etc. can be selected as the triangular unit.
- the triangular unit may be present at the N-terminal, or that the straight-chain part is also present on the N-terminal side of the triangular unit and that the N-terminal is selected from N-terminal carboxylic acid derivatives It is preferable from the point of like.
- compound Ca-1 can also be selected as the triangular unit.
- an amino acid analog capable of forming an ester bond by translation synthesis without having an amino group in the main chain such as ⁇ -hydroxycarboxylic acid, and the like
- the protecting group is not particularly limited as long as it acts as a protecting group for the amino group and gives an amino acid to be translated and synthesized
- Post-modification techniques can be used (Scheme E). When applied to a display library, it is selected from the units to be translated.
- peptide compounds obtained by subsequent optimization include those obtained by post-translational modification after translation synthesis even if the peptide compound itself is not translationally synthesized.
- the side chain of the ⁇ -hydroxycarboxylic acid moiety is not particularly limited, but in the method described in Scheme E, Re1 is particularly a hydrogen atom (glycolic acid (HO Gly) itself) or a thiol in the side chain in terms of translational synthesis. Those having a group (SH group) or a protected thiol group are preferred.
- the configuration of the side chain is not particularly limited, but when a hydrogen atom is present at the ⁇ -position, it is more preferable to adopt the same configuration as that of the L-type amino acid.
- the arrangement of the thiol group or the protected thiol group is not particularly limited, but is arranged at the ⁇ -position or ⁇ -position of the OH group (that is, the mercaptomethyl group or mercaptoethyl group where Re1 may be protected). Is particularly preferred.
- These thiol groups or protected thiol groups are arranged from an optionally substituted alkyl group, alkenyl group, alkynyl group, cycloalkyl group, aralkyl group, aryl group, heteroaryl group of the ⁇ -hydroxycarboxylic acid side chain (Added).
- These ⁇ -hydroxycarboxylic acid side chain substituents are more preferably substituents defined on the side chain of a drug-like amino acid except for the added thiol group or protected thiol group.
- the type of amino acid having an amino side chain is also an alkylamino group, an alkenylamino group, an alkynylamino group, an aralkylamino group, an arylamino group, a heteroarylamino group, which may be substituted if it has an amino group,
- Any of the cycloalkylamino groups is possible and is not particularly limited, but may have a thiol group (SH group) or a protected thiol group in the side chain at the same time.
- These substituents are preferably substituents defined in the side chain of a drug-like amino acid, except for a reaction auxiliary group (for example, SH group). Furthermore, it is more preferable to select from a substituent capable of translational synthesis.
- One of the hydrogen atoms at the site represented by NH2 in the scheme is specified as a hydrogen atom, and the other hydrogen atom may be substituted with an alkyl group, an alkenyl group, an alkynyl group, an aralkyl group.
- An aryl group, a heteroaryl group, and a cycloalkyl group which may be substituted with a reaction auxiliary group, preferably an NH2 group.
- These substituents are preferably substituents defined in the side chain of a drug-like amino acid, except for a reaction auxiliary group (for example, SH group). Furthermore, it is more preferable to select from a substituent capable of translational synthesis.
- a codon encoding an amino acid analog capable of forming an ester bond without an amino group such as ⁇ -hydroxycarboxylic acid (A), an acylated tRNA, a codon encoding an amino acid having an amino group side chain (B),
- An aminoacylated tRNA is prepared, and mRNA in which a desired number (preferably 0 to 7, more preferably 0 to 2) of random codons is arranged between codon (A) and codon (B).
- the peptide obtained by translation into a template is first cyclized by the above method or another method. Codon A is arranged N-terminal from codon B.
- the obtained cyclized peptide (for example, the method of Scheme B or Scheme C can be used) is deprotected as needed when a side chain amino group has a protecting group, and is hydrolyzed or added from the outside.
- the ester site is activated by adding an agent, the amide bond is not hydrolyzed, but the ester bond can be hydrolyzed or activated (active esterification, active thioesterification).
- the peptide having the straight chain part 2 which is a desired branched peptide can be obtained by intramolecular cyclization reaction of the obtained main chain carboxylic acid or active (thio) ester and side chain amine.
- the additive added from the outside includes a compound group that forms various active esters such as thiol compounds, HONSu, HOBt, and HOAt, or a mixture of two or more of these.
- active esters such as thiol compounds, HONSu, HOBt, and HOAt, or a mixture of two or more of these.
- Scheme E an example was given in which a Re1-substituted hydroxycarboxylic acid was used as codon A and lysine was used as codon B.
- the triangular unit may be selected to have a reaction auxiliary group at the amine site. You may choose what you do not have.
- a unit having a reaction assisting group is selected as a triangular unit (for example, Compound N-1 or Compound N-2)
- the first cyclization reaction can easily proceed.
- a reaction additive may or may not be added at a pH of around 7 (translation conditions).
- a unit having no reaction auxiliary group is selected as a triangular unit (for example, when Ala or Phe contained in compound Na-2 is selected)
- trifluoromethylthiophenol it is more preferable to add an additive such as More preferably, the pH is raised to around 7.8 and a reaction time of about 6 to 10 hours is applied at a reaction temperature of about 37 ° C. to 50 ° C.
- the amine site may have no reaction auxiliary group, but it is more preferable to have a reaction auxiliary group at the amine site, in which case the NH group must be protected. Is preferred. In this case, it is preferable to add thiol as an ester activator.
- an alkyl thiol is preferable, and an alkyl thiol having a water-soluble substituent is particularly preferable from the viewpoint of solubility of the additive, and 2-dimethylaminoethane thiol and 2-mercaptoethane sulfonic acid are particularly preferable.
- the addition amount of these thiols is not particularly limited, but is preferably more than 10 mM for the purpose of sufficiently increasing the reactivity, and less than 10 M for the purpose of dissolving the additive.
- the range of 100 mM to 5 M is more preferable, but the range of 500 mM to 4 M is more preferable.
- the selection of the amine moiety is not particularly limited, but the reaction conditions in the case of having deprotection are the same as those described in Scheme F2.
- the obtained thioester easily reacts with an amine having a reaction assisting group, a peptide having a desired branched peptide (linear portion 2) can be obtained.
- the SH group of the obtained branched peptide containing an SH group can be easily desulfurized under mild reaction conditions such that RNA does not participate in chemical reactions.
- an additive may be added from the outside for the purpose of activating the (thio) ester.
- the cyclization reaction of the post-translation cyclization part may be performed either before or after the linear chain part 2 formation reaction.
- reaction auxiliary group such as an SH group or a protected SH group at both the hydroxycarboxylic acid and the amine side chain site, or may have a reaction auxiliary group only on one of them. Or both of them may not have a reaction auxiliary group. Moreover, it may have a protecting group in the amine side chain site
- an additive for accelerating the reaction may be added. Examples of the additive include an optionally substituted alkylthiol, an optionally substituted alkenylthiol group, an optionally substituted alkynylthiol group, an optionally substituted aralkylthiol, and an optionally substituted aryl.
- a thiol an optionally substituted heteroaryl thiol group, or an optionally substituted cycloalkyl thiol group.
- a reagent that is usually active esterified such as HOBt, HONSu, HOAt, paranitrophenol, or a derivative thereof may be used.
- HOBt HOBt
- HOAt HONSu
- HOAt paranitrophenol
- a free substituent can be selected as defined in the side chain of the “amino acid”.
- the method for generating the straight chain part 2 is the same in that a thioester is generated by using an ester functional group such as ⁇ -hydroxycarboxylic acid as a foothold, but two sequences immediately before the N-terminal side of ⁇ -hydroxycarboxylic acid are used.
- a technique in which Cys and Pro are arranged, for example, a thioester is generated from a Cys-Pro- HO Gly sequence can be used (see Scheme F2).
- an active thioester generating sequence for branching has Cys-Pro followed by ⁇ -hydroxycarboxylic acid having Rf5 in the side chain and lysine having an amino group in the side chain as an amino acid to be reacted therewith.
- a codon encoding an amino acid analog capable of forming an ester bond without an amino group such as ⁇ -hydroxycarboxylic acid (A), an acylated tRNA, a codon encoding an amino acid having an amino group side chain (B),
- An aminoacylated tRNA is prepared, and mRNA in which a desired number (preferably 0 to 7, more preferably 0 to 2) of random codons are arranged between codon (A) and codon (B).
- the peptide obtained by translation into a template is first cyclized by the above method or another method (for example, Scheme B or Scheme C). Codon A is arranged N-terminal from codon B.
- Branching sites such as Cys-Pro- HO Gly exist without causing a chemical reaction stably under the first cyclization reaction conditions, and then thiols such as 4-trifluoromethylphenylthiol as necessary.
- pH is adjusted to the basic side (for example, 9) to newly generate an active thioester, and then a side chain amino group of lysine etc. via a more active trifluoromethylphenylthioester
- a branched peptide in which an amide bond is formed when a protecting group is present on the side chain amino group, a deprotection reaction is preceded.
- the ⁇ -position (Rf5) of the ⁇ -hydroxycarboxylic acid moiety is selected from a hydrogen atom, an optionally substituted alkyl group, an aralkyl group, a heteroaryl group, a cycloalkyl group, an aryl group, and the like.
- the substituent is preferably a drug-like functional group.
- the thioester generation site is preferably stable under the conditions of the first cyclization and maintains a precursor (eg, Cys-Pro- HO Gly) structure.
- Aralkyl groups such as are preferred.
- any of those corresponding to the L-type and D-type amino acids as a steric structure when the main chain OH group is replaced with an amino group is allowed, but in particular, in terms of translation efficiency, L What corresponds to the solid of a type
- a unit that can be translated by ARS is preferable because translation efficiency can be improved.
- An example of this is Lac.
- the side chain of the amino acid having an amino group side chain that reacts with this thioester to form a branch is not particularly limited as long as it has an amino group and an optionally protected amino group, but is not protected.
- the amino group may be a secondary amine or a primary amine, but a primary amine is more preferable for the purpose of allowing the branching reaction to proceed efficiently.
- a reaction assisting group such as an SH group may be present in the vicinity of the amino group or the amino group which may be protected, and these may also be protected.
- any of an alkylamino group, an aralkylamino group, an arylamino group, a heteroarylamino group, and a cycloalkylamino group that may be substituted may be used.
- the side chain may have a thiol group (SH group) or a protected thiol group at the same time.
- These substituents are preferably substituents defined in the side chain of a drug-like amino acid, except for a reaction auxiliary group (for example, SH group). Furthermore, it is more preferable to select from a substituent capable of translational synthesis.
- One of the hydrogen atoms at the site represented by NH2 in the scheme is specified as a hydrogen atom, and the other hydrogen atom may be substituted with an alkyl group, an alkenyl group, an alkynyl group, an aralkyl group.
- An aryl group, a heteroaryl group, and a cycloalkyl group which may be substituted with a reaction auxiliary group, preferably an NH2 group.
- a reaction auxiliary group preferably an NH2 group.
- substituents are preferably substituents defined in the side chain of a drug-like amino acid, except for a reaction auxiliary group (for example, SH group). Furthermore, it is more preferable to select from a substituent capable of translational synthesis.
- the triangular unit does not have an amino acid, amino acid derivative, N-terminal carboxylic acid derivative or reaction auxiliary group having a reaction auxiliary group represented by compound N-1 or compound N-2. It is selected from Compound Na-1, Compound Na-2, Compound Na-3, Compound Na-4, Na-5, Na-6 and the like.
- the crossing unit is selected from active esters represented by Compound C-1.
- ⁇ -hydroxycarboxylic acid derivative represented by compound e1 or Cys-Pro- ⁇ -hydroxycarboxylic acid shown as compound F5 is selected from sites consisting of elements or active esters such as compound C-1 (Cys, Pro, ⁇ -hydroxycarboxylic acid is translated as a separate unit by tRNA, but after translation modification Cys and Pro Are separated from the black circle unit and the square unit, and ⁇ -hydroxycarboxylic acid becomes a square unit).
- the site having an amino group in this branching reaction is compound Na-4, compound Na-10, or compound Na-11 (and these H atoms represented by NH are protected during translation synthesis).
- Selected from the compounds represented by Examples thereof include compounds tk100, tk101, tk102, tk103, tk104, tk34, tk7, tk14, tk105, tk106, tk107, tk108, etc., in addition to unprotected amines such as lysine.
- the value of this method is not limited to the case where the first cyclization reaction is a drug-like cyclization, but the second cyclization reaction forms a drug-like cyclization. If so, the first cyclization reaction may be any cyclization reaction.
- the present method in which the first cyclization reaction is not limited to the drug-like cyclization reaction is useful in that various structures are formed. In such a case, all techniques for generating an active ester from a translation are within the scope of this technique.
- Examples thereof include compound F5, compound e1, compound C-1, compound C-2, compound C-3 and the like.
- a unit having a reaction auxiliary group is selected as a triangular unit (for example, Compound N-1 or Compound N-2), the first cyclization reaction can be easily advanced.
- a reaction additive may or may not be added at a pH of around 7 (translation conditions). In this case, the selection of the part used for the second branching becomes large. That is, compound e1 can be present stably, and in compound F5, Rf5 is preferably other than a hydrogen atom, but even a hydrogen atom can be present stably and is allowed.
- an amino group that does not have a reaction auxiliary group such as Lys, which is a unit in which the amino group is not protected, is also acceptable. Further, the amino group may be protected, or may be protected with an amine having a reaction auxiliary group. All these cases can be used. After removing the protecting group as necessary, the desired branched peptide can be obtained through a branching reaction, followed by a step of removing the reaction auxiliary group.
- the selection of the Rf1 group of the compound e1 is as described above, but it is more preferably selected from a hydrogen atom and an optionally protected mercaptoalkyl group (the SH group is protected). Particularly preferably, it is selected from a hydrogen atom, a mercaptomethyl group or a 2-mercaptoethyl group which may be protected.
- the Rf2 group of compound F5 is selected from mercaptoalkyl groups which may be protected.
- the steric configuration of the amino acid containing Rf2 is preferably L-type. More preferably, it is selected from a mercaptomethyl group or a 2-mercaptoethyl group which may be protected.
- the Rf3 group can be selected in the same manner as Ra13.
- a ring with a methyl group or Rf4.
- the ring size is preferably a 3- to 8-membered ring, more preferably a 4- to 6-membered ring.
- the carbon atoms forming these rings may be substituted with a substituent defined in the side chain of the drug-like amino acid.
- Proline and the like are preferable as a representative example of a 5-membered ring.
- Rf4 can be selected in the same manner as Ra4.
- a unit having no reaction auxiliary group is selected as a triangular unit (for example, when Ala or Phe contained in compound Na-2 is selected)
- trifluoromethylthiophenol it is more preferable to add an additive such as More preferably, the pH is raised to around 7.8 and a reaction time of about 6 to 10 hours is applied at a reaction temperature of about 37 ° C. to 50 ° C. Therefore, it is preferable that the branching site is present stably without undergoing a chemical reaction under such reaction conditions. Therefore, it is preferable not to select a unit having an unprotected amino group such as Lys at the amino group site involved in amide bond formation in the branching reaction.
- the NH group (including the reaction auxiliary group) of Compound Na-4, Compound Na-10, and Compound Na-11 is protected.
- it may have a reaction auxiliary group that may be protected (for example, compound tk100) or may not have (for example, tk104).
- These protecting groups need to be stably present during translation and the first cyclization reaction. Examples of such protecting groups include trifluoroacetyl groups, 4-azidobenzyloxycarbonyl groups, 3-nitro-2-pyridinesulfenyl groups, and thiazolidine ring protecting groups. The trifluoroacetyl group is selectively deprotected only when the pH is greater than 8.
- the 4-azidobenzyloxycarbonyl group is selectively deprotected only when the reducing agent tris (2-carboxyethyl) phosphine is added.
- the 3-nitro-2-pyridinesulfenyl group is selectively deprotected only at pH 4 when the additive 2-mercaptopyridine is added.
- the protecting group by the thiazolidine ring is selectively deprotected only by adding tris (2-carboxyethyl) phosphine after opening the thiazolidine ring by adding the additive dithiodipyridine at pH4.
- Rf5 is preferably other than a hydrogen atom.
- Rf5 is a methyl group or a benzyl group.
- the ⁇ -position (Rf5) of the hydroxyl site is a hydrogen atom
- thioester formation is unavoidable and the first cyclization reaction cannot be performed selectively, whereas a carbon atom is added at this position.
- the production of thioester can be suppressed under the first cyclization reaction conditions.
- the branching site is selected as described above, the deprotection reaction of the amine site or thioester site is carried out as necessary, and then the pH is increased to 8.2 or more to cause branching.
- the branched peptide is obtained by allowing it to stably exist during the first cyclization reaction, performing the branching reaction through the deprotection step, and finally removing the reaction auxiliary group as necessary. Can do.
- Protecting group of amino group used for chemical modification of peptide compound-nucleic acid complex obtained by translational synthesis and its deprotection method is a single primary amine or secondary amine
- structures that simultaneously inactivate multiple amino groups or other heteroatoms with one functional group are defined as protecting groups.
- a thiazolidine ring, a thiazinane ring, an oxazolidine ring, and an imidazolidine ring are also included as amino-protecting groups.
- the carbon atoms that form these protecting groups may be substituted.
- the above-mentioned protecting groups are 1) protecting groups that are removed under acidic conditions, 2) protecting groups that are removed under basic conditions, 3) protecting groups that are removed under oxidative conditions, 4) protecting groups that are removed under reducing conditions, and 5) light irradiation. 6) A protecting group that can be deprotected by combining any one of 1) to 6) or 1) to 6) among protecting groups removed by addition of a nucleophile.
- the protecting group that is removed under acidic conditions is a protecting group that is removed in the range of pH 1 to pH 7, preferably a protecting group that can be deprotected in the range of pH 2 to pH 6.
- Non-patent literature i) Green's Protective Groups in Organic Synthesis, Fourth Edition, ii) Chemical Reviews, 2009, 109 (6), 2455-2504) Carbons that may form these protective groups may be substituted. .
- the protecting group that is removed under basic conditions is a protecting group that is removed in the range of pH 7 to pH 14, and preferably a protecting group that can be deprotected in the range of pH 7 to pH 10, for example, an electron-withdrawing group that has been extremely attached.
- And-[phenyl (methyl) sulfonio] ethoxycarbonyl group for example, a trichloroacetyl group into which one or a plurality of halogen groups, nitro groups, trifluoro groups and the like are introduced may be mentioned. Specifically, it is a trifluoroacetyl group (Tfa) shown below.
- Non-patent literature i) Green's Protective Groups in Organic Synthesis, Fourth Edition, ii) Chemical Reviews, 2009, 109 (6), 2455-2504).
- Non-Patent Literature i Greene's Protective Groups in Organic Synthesis, Fourth Edition, ii) The Journal of Organic Chemistry, 1997, 62, 778-36, 1977-79, 1977-79, 1977-721
- the protecting group that is removed under reducing conditions is, for example, a protecting group that can be deprotected in the presence of tris (2-carboxyethyl) phosphine (TCEP) or dithiothreitol (DTT).
- TCEP 2,-carboxyethyl
- DTT dithiothreitol
- Examples of protecting groups removed by light irradiation include o-nitrobenzyloxycarbonyl group (oNz), 4,5-dimethoxy-2-nitrobenzoxy) carbonyl group (Nvoc), and 2- (2-nitrophenyl) propyloxycarbonyl. Group (Nppoc).
- Non-Patent Literature i) Chemical Reviews, 2009, 109 (6), 2455-2504, ii) The Journal of Organic Chemistry, 1997, 62, 778-779, iii) Bioorganic & m ) The carbon atoms forming these protecting groups may be substituted.
- the protecting group removed by addition of a nucleophile is, for example, a protecting group that is deprotected in the presence of a thiol, for example, o-nitrobenzenesulfonyl group (o-NBS), 2,4-dinitrobenzenesulfonyl group (dNBS) , A dithiosuccinoyl group (Dts).
- o-NBS o-nitrobenzenesulfonyl group
- dNBS 2,4-dinitrobenzenesulfonyl group
- Dts dithiosuccinoyl group
- a protective group that can be deprotected by combining a plurality of 1) to 6) is, for example, a deprotection method combining 1) and 6), and deprotecting by adding a thiol in the range of pH 2 to pH 6.
- Possible protecting groups such as 3-nitro-2-pyridinesulfenyl group (Npys) (non-patent literature i) International Journal of Peptide and Protein Research, 1990, 35, 545-549, ii) International Journal of Peptide tide. , 1980, 16, 392-401), 2-nitrophenylsulfenyl group (Nps) (non-patent document Chemical Reviews, 2009, 109 (6), 2455- 504) is.
- the carbon atoms that form these protecting groups may be substituted.
- the protecting group applied to the deprotecting method combining 1) and 3) is, for example, a protecting group that can be deprotected by adding a disulfide compound in the range of pH 2 to pH 6, such as a thiazolidine ring or a thiazinane ring.
- a disulfide compound in the range of pH 2 to pH 6, such as a thiazolidine ring or a thiazinane ring.
- the carbon atoms that form these protecting groups may be substituted.
- NCL Native Chemical Ligation
- Non-Patent Document v1 Angewandte Chemie, International Edition, 2006, 45 (24), 3985-3988
- non-patent document v2 Journal of the American Chemical Society, 2011, 133, 11418-11421
- an ester in the peptide compound described in this patent or in a structure containing a thioester, there is a concern that when a deprotection reaction or the next reaction is carried out in one pot, an alkoxyamide produced by the reaction of methoxyamine with an ester moiety is produced as a side reaction.
- the present inventors have developed a method of adding a disulfide compound as a method for deprotecting a thiazolidine ring and a thiazinane ring without using methoxyamine.
- This method is characterized by opening a thiazolidine ring and a thiazinane ring under acidic conditions to lead to an amino disulfide structure, which can be led to an aminothiol by the subsequent addition of a reducing agent.
- This procedure is performed in water, in a buffer solution, or in an organic solvent mixed with water.
- the organic solvent to be used is selected so that it is miscible with water and does not react or precipitate with the substrate.
- N, N-dimethylacetamide, N, N-dimethylformamide, acetonitrile is used.
- the ratio of water and organic solvent is determined by the solubility of the substrate and the disulfide compound.
- 5% or more of N, N-dimethyl is used from the viewpoint of the solubility of dithiodipyridine.
- Preference is given to using acetamide.
- the pH range for opening the thiazolidine ring is preferably pH 1 to 5 for the purpose of completing the reaction within 12 hours. Further, when handling a compound having an acidic and unstable thiazolidine ring, pH 4 to 5 is preferred.
- the reaction temperature for opening the thiazolidine ring is preferably 15 ° C. or higher for the purpose of completing the reaction within 12 hours. Further, when a compound having a thermally unstable thiazolidine ring is handled, the range of 15 to 50 ° C. is preferable.
- disulfide compound to be added for example, dialkyl disulfide or diaryl disulfide can be used, but diaryl disulfide is preferable, and dithiodipyridine is more preferable.
- the amount of dithiodipyridine used is determined by the amount of thiazolidine derivative used. For example, when 30 mM dithiodipyridine is used for 1.0 mM thiazolidine derivative and the reaction is carried out at pH 4.2 and 36 ° C., within 12 hours. Opening of the thiazolidine ring is complete.
- alkylphosphine, arylphosphine, and a thiol compound having a reducing power can be used, for example, TCEP or DTT is used.
- TCEP is preferred when the conversion to aminothiol proceeds rapidly.
- the amount of TCEP used depends on the amount of thiazolidine derivative and dithiodipyridine used. For example, when 1 mM thiazolidine derivative is converted to 2- (2-pyridyldithio) ethylamine with 30 mM dithiodipyridine, 40 mM When TCEP is used and the reaction is carried out at pH 4.5 and 24 ° C., induction into the aminoethanethiol form is completed within 1 hour.
- the technology for forming a branched structure from a linear peptide sequence translated from the primary sequence information of mRNA is the construction of an mRNA display library of highly diverse structural groups of peptides having a branched structure that cannot be achieved by conventional techniques. Is possible.
- the first cyclization reaction is not limited to the cyclization reaction described above, but enables a branching reaction (because it is stable during the first cyclization reaction and is branched to RNA under stable reaction conditions). It can be applied to all cyclization reactions that can be applied under reaction conditions.
- Compound F5 or Compound E1 is used as the second active ester generation site, and Compound Na-4, Compound Na-5, Compound Na-10 (Na-7 group or Na-8 group) or Compound Na is used as the second amino group site. -11 (Na-7 group or Na-8 group).
- a desired branched peptide (straight chain part 2) can be obtained by placing a hydroxycarboxylic acid and an amino acid having an amine moiety at a desired position and carrying out an appropriate chemical reaction.
- the selection of hydroxycarboxylic acid and amine, each amino acid or amino acid analog is free as long as it has a necessary functional group, but the hydroxycarboxylic acid moiety is preferably arranged on the N-terminal side.
- Hydroxycarboxylic acid is not limited to ⁇ -hydroxycarboxylic acid, and can take various hydroxycarboxylic acids represented by ⁇ and ⁇ -hydroxycarboxylic acid.
- a branched peptide forming reaction can also be carried out by translating and introducing thiocarboxylic acid instead of hydroxycarboxylic acid and using thioester instead of ester as a foothold. This method realizes chemical-space expansion, which is indispensable for creating hits in medium molecules with a limited number of amino acids.
- phosphine ligands As ligands used in ordinary Pd catalytic reactions, phosphine ligands, phosphine oxide ligands, ligands consisting of nitrogen atoms, ligands consisting of arsenic atoms, carbene type ligands, etc. can be used. It is. These may be monodentate ligands having one functional group capable of coordinating in the molecule, or bidentate ligands combining two of these functional groups capable of coordinating with Pd. Since the reaction needs to be performed in water, a ligand having a water-soluble functional group may be used.
- a ligand that forms a strong coordinate bond with Pd is preferable because Pd is inactivated by complex formation by an RNA component, GTP, ATP, or other nucleic acid component contained in the translation solution.
- RNA component GTP, ATP, or other nucleic acid component contained in the translation solution.
- bidentate phosphine ligands such as 5-bis (diphenylphosphino) -9,9-dimethylxanthene and 1,3-bis (diphenylphosphino) propane, and their solubility in aqueous solvents.
- the amount of Pd used is preferably 1 nmol or more, more preferably 60 nmol, relative to a translational compound containing 1 pmol of m-RNA.
- Use of micelles as described in the examples described later is preferable because acceleration of the reaction can be obtained.
- Usable micelles can be any of anionic, cationic, zwitterionic and nonionic, but in particular the nonionic micelle polyoxyethanyl sebacate- ⁇ -tocopheryl (PTS) is used. Is preferred.
- the polyoxyethanyl sebacate- ⁇ -tocopheryl can be used at any concentration, but it is preferably at a final concentration of 1% or more, more preferably at a final concentration of 7.5% or more. It is necessary to use a base for the progress of the reaction, and it is preferable to use a buffer composed of a component that does not coordinate to Pd, and a phosphate buffer, a carbonate buffer, or the like can be used.
- the pH of the reaction solvent is preferably adjusted to a range in which m-RNA can stably exist and the cyclization reaction proceeds, more preferably 7 or more and 10 or less, and even more preferably 7.5 or more and 8.5 or less. preferable.
- the reaction temperature is not particularly limited as long as m-RNA can stably exist and can usually carry out a chemical reaction, but it is preferably 15 ° C. to 80 ° C., more preferably 40 ° C. to 60 ° C.
- the CC bond cyclization reaction is not limited to the Heck reaction using Pd.
- Various reactions using Pd such as the Suzuki reaction and the Sonogashira reaction, can be similarly performed. In this case, selection of a ligand or a base may be important.
- the transition metal is not limited to Pd, and the CC bonding reaction can be carried out with various metals such as Ni, Ru, Co (Non-patent document Matthew S. Sigman et al. Advances in Transition Metal (Pd, Ni, Fe) -Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners.Chem.
- the amino acid, amino acid analog, and N-terminal carboxylic acid analog that are triangular units constituting Scheme C-2 are not particularly limited as long as they have a reactive functional group. However, if an amino acid is selected, an amine remains at the N-terminus, which is disadvantageous for membrane permeation compared to the case where no amino acid remains. For this reason, as a triangular unit, what has few number of heteroatoms is more preferable.
- Examples of such N-terminal carboxylic acid analogs include compounds described in compounds CC-1 to CC-4. In compound CC-1, a carbon-carbon double bond is the reaction site. In compound CC-2, a carbon-carbon triple bond is the reaction site. In compound CC-3, X is the reaction site.
- X is preferably halogen, and is selected from Cl, Br, I, and F. From the viewpoint of reactivity, Br and I are preferable, and I is most preferable.
- the borate ester moiety becomes the reaction site.
- R301, R302, and R303 are selected from a hydrogen atom, an optionally substituted alkyl group, alkenyl group, alkynyl group, cycloalkyl group, aryl group, heteroaryl group, and aralkyl group. These substituents are not particularly limited as long as the compounds CC-1 to CC-4 obtained as a result of the substitution can be translationally synthesized. Examples of such a substituent include a halogen group and an alkoxy group.
- R304 is a unit that links reaction points and translation points (carboxylic acid sites).
- the typical structure is shown below. Both units can be connected by C1 to C6 units such as a methylene group (partial structure N-3), an ethylene group (partial structure N-4), and a propylene group (partial structure N-5). It can also be directly linked from the aryl carbon of the aromatic compound (partial structure N-6). It can also be linked with an aralkyl structure (partial structures N-7, N-8).
- the connecting position is not limited to ortho, and may be meta or para, or may be a substituted aryl group or a substituted aralkyl group other than a phenyl group. Examples of the substituent include a halogen group and an alkoxy group.
- examples of the amino acid of the triangular unit constituting Scheme C-2 are not particularly limited as long as the amino acid has a double bond, triple bond, halogen, or borate ester in the side chain.
- L-type amino acids, D-type amino acids, and ⁇ - ⁇ -dialkyl amino acids may be used, but L-type amino acids are particularly preferred.
- the N-terminal amino group is preferably substituted. Alkylation such as N-methyl is also possible, but rather it is preferable to introduce a substituent that loses the basicity of the nitrogen atom, such as amidation such as acylation.
- the amino acid analog or N-terminal carboxylic acid analog of the triangular unit constituting Scheme C-2 may be a hydroxycarboxylic acid having a double bond, triple bond, halogen, or borate ester in the side chain.
- the selection of the side chain is not particularly limited. Besides ⁇ -hydroxycarboxylic acid, ⁇ or ⁇ -hydroxycarboxylic acid may be used. Further, it may be a dipeptide or tripeptide having a double bond, triple bond, halogen, or borate ester in the side chain.
- Examples of the crossing unit ( ⁇ unit) constituting Scheme C-2 include an amino acid or ⁇ -hydroxycarboxylic acid having a double bond, triple bond, halogen, or borate ester in the side chain.
- the side chain of the amino acid or ⁇ -hydroxycarboxylic acid is not particularly limited as long as these reactive functional groups are present. It is selected from an optionally substituted alkyl group, alkenyl group, alkynyl group, cycloalkyl group, aryl group, heteroaryl group, aralkyl group and the like substituted with each reactive functional group. Examples of these substituents include a halogen group and an alkoxy group. Any of L-type, D-type and ⁇ - ⁇ -dialkyl type is acceptable, but when a hydrogen atom is present at the ⁇ -position, an L-type amino acid or amino acid analog is preferred.
- the combination of the triangular unit and the crossing unit ( ⁇ ) unit is not particularly limited as long as it is a combination that can undergo a condensation reaction with a transition metal such as Pd.
- the peptide compounds of the present invention can also be produced by chemical synthesis.
- Fmoc synthesis there is a method called Fmoc synthesis and a method called Boc synthesis.
- Fmoc synthesis the main chain amino group is protected with an Fmoc group, the side chain functional group is protected with a basic non-cleavable protecting group such as piperidine as necessary, and an amino acid that does not protect the main chain carboxylic acid as a basic unit.
- a dipeptide may be used as a basic unit.
- the basic unit arranged at the N-terminus may be other than the Fmoc amino acid.
- it may be a Boc amino acid or a carboxylic acid analog having no amino group.
- the main chain carboxylic acid group is supported on the solid phase by a chemical reaction with the functional group of the solid phase carrier. Subsequently, the Fmoc group is deprotected with a base such as piperidine or DBU, and a peptide bond is generated by a condensation reaction between a newly generated amino group and a protected amino acid having a basic unit carboxylic acid added subsequently. .
- the desired peptide sequence can be generated by repeating the de-Fmoc group and the subsequent peptide bond formation reaction. After the desired sequence is obtained, excision from the solid phase and deprotection of the protecting group of the introduced side chain functional group are performed as necessary. It is also possible to perform structural transformation or cyclization of the peptide before excision from the solid phase. The excision from the solid phase and the deprotection may be performed under the same conditions, for example, 90:10 TFA / H 2 O, or may be deprotected under different conditions as necessary.
- Steps such as cyclization can also be performed during or at the end of these steps.
- the side chain carboxylic acid and the amino group of the N-terminal main chain can be condensed, or the side chain amino group and the C-terminal main chain carboxylic acid can be condensed.
- the protecting group is selected in consideration of the orthogonality of the protecting group.
- the reaction product thus obtained can be purified by a reverse phase column, a molecular sieve column or the like. These details are described, for example, in a solid-phase synthesis handbook issued by Merck on May 1, 2002.
- the present invention further provides a method for producing a drug-like peptide compound or peptide compound-nucleic acid complex having a desired activity.
- Examples of a method for producing a drug-like peptide compound-nucleic acid complex having a desired activity include a production method including the following steps: (i) A compound in which a non-cyclic peptide compound having 9 to 13 residues of amino acids and amino acid analogs is translated and synthesized, and the non-cyclic peptide compound and the nucleic acid sequence encoding it are linked via a linker Forming a non-cyclic peptide compound-nucleic acid complex comprising (ii) The acyclic peptide compound of the complex translated and synthesized in step (i) is cyclized by an amide bond or a carbon-carbon bond, and the total number of amino acids in the cyclic part and amino acid analogs is 5 to 12.
- Forming a cyclic compound comprising (iii) A step of contacting the peptide compound-nucleic acid complex library having a circular portion obtained in step (ii) with an in vivo molecule and selecting a complex having binding activity to the in vivo molecule.
- a drug-like peptide compound having a desired activity can be produced from the complex selected in the above step.
- a production method including the following steps can be mentioned: (iv) obtaining sequence information of the peptide compound from the nucleic acid sequence of the complex selected in the step (iii), and (v) a step of chemically synthesizing the peptide compound based on the sequence information obtained in the step (iv).
- the acyclic peptide compound may be ⁇ -hydroxycarboxylic acid and protected.
- An amino acid or amino acid having an amino group in the side chain and an amino acid or amino acid analog having an amino group in a good side chain and before or after the step of forming the cyclic compound in step (ii) A step of chemically reacting the analog site to form a branched site may be included. By this process, it is possible to produce peptide compounds having the above-mentioned straight chain part 2 at various positions of the cyclic part.
- the total number of amino acids and amino acid analogs described in step (i) above and the total number of amino acids and amino acid analogs in the cyclic part described in step (ii) are determined by post-translational modification. Amino acids or amino acid analogs that are excluded are not included.
- the Cys-Pro sequence that is eliminated from the peptide during post-translational modification during the production of the peptide compound having the linear portion 2 is the amino acid and amino acid analog of steps (i) and (ii).
- the Gly-Ser repeat structure is fixed or fixed
- the amino acid region, the peptide compound having the cyclic part of the present invention, particularly the drug-like peptide compound part and the target part for binding the nucleic acid are contained in the linker, and the amino acids and amino acid analogs of steps (i) and (ii) Not included in the number.
- the number of residues in the ring portion after cyclization in step (ii) is not 5 to 11 but longer than that, but after the reaction to form the straight chain portion 2, the residues in the ring portion The number should be 5 to 11.
- the target substance is preferably an in vivo molecule.
- the ⁇ in vivo molecule '' in the present invention is not particularly limited as long as it is an in vivo molecule, but is preferably a molecule that is a target for treatment of a disease, and in particular, a hole to which a conventional low molecular weight compound having a molecular weight of less than 500 can bind ( Molecules that do not have a cavity), intracellular proteins, nucleic acids, intracellular regions of membrane proteins or transmembrane domains of membrane proteins that are inaccessible to high molecular compounds such as antibodies are preferred.
- transcription factors such as STAT, AP1, CREB, and SREBP, muscarinic acetylcholine receptors, cannabinoid receptors, GLP-1 receptors, PTH receptors and other G protein-coupled receptors (GPCR), TNF , TNFR, IL-6, IL-6R and other cytokines and their receptors, P2X receptors, ion channel receptors such as nicotinic acetylcholine receptor, ion channels, transporters, miRNAs such as miR-21 and miR206 Can be mentioned.
- the cyclic portion can be formed, for example, using the cyclization reaction described above, and an amide bond or a carbon-carbon bond can be formed by the cyclization reaction.
- a known technique such as a cell-free translation system can be used. Specifically, it can be produced using the following method.
- Transfer RNA is an RNA consisting of 73 to 93 bases with a molecular weight of 25000 to 30000 and a CCA sequence at the 3 ′ end, and an aminoacylated tRNA ester-linked to the carboxy terminus of the amino acid via the 3 ′ end.
- Polypeptide elongation factor (EF-Tu) which forms a triple complex with GTP, is transported to ribosome, and in translating mRNA nucleotide sequence information into amino acid sequence by ribosome, the anticodon in the tRNA sequence and the codon of mRNA RNA involved in codon discrimination by base pairing.
- a tRNA biosynthesized in a cell contains a covalently modified base, which may affect tRNA conformation and anticodon base pairing, thereby assisting in tRNA codon discrimination.
- TRNA synthesized by general in vitro transcription consists of so-called nucleobases adenine, uracil, guanine, and cytosine, but those prepared from cells and chemically synthesized include other methylated products. Some of them contain modified bases such as sulfur derivatives, deaminated derivatives, adenosine derivatives containing isopentenyl groups and threonine, and sometimes contain deoxy bases when using the pdCpA method.
- RNA polymerase suitable for the promoter such as T7 RNA polymerase and T3,3SP6 RNA polymerase.
- I can do it. It is also possible to extract and purify tRNA from the cells, and extract the target generated tRNA using a probe having a sequence complementary to the sequence of tRNA. At this time, cells transformed with the expression vector of the target tRNA can also be used as a source. RNA of the target sequence can also be synthesized by chemical synthesis.
- an aminoacyl-tRNA can be obtained by binding a tRNA obtained by removing CA from the 3'-terminal CCA sequence thus obtained and an aminoacylated pdCpA separately prepared with RNA ligase (pdCpA method).
- Aminoacylation by flexizyme which is a ribozyme that prepares full-length tRNA and supports active esters of various unnatural amino acids on tRNA, is also possible.
- acylated tRNA can be obtained by using the method described later.
- Protein factors required for translation of E. coli (methionyl tRNA transformylase, EF-G, RF1, RF2, RF3, RRF, IF1, IF2, IF3, EF-Tu, EF-Ts, ARS (AlaRS, ArgRS, AsnRS, AspRS, CysRS, GlnRS, GluRS, GlyRS, HisRS, IleRS, LeuRS, LysRS, MetRS, PheRS, ProRS, SerRS, ThrRS, TrpRS, TyrRS, ValRS (choose what you need) , Inorganic pyrophosphatase, nucleoside diphosphate kinase, E.
- coli derived tRNA creatine phosphate, potassium glutamate, HEPES-KOH pH7.6, magnesium acetate, spermidine, dithiothreitol, GTP, ATP, CTP, UTP, etc. Can be translated into peptides by adding mRNA to the PUREsystem.
- T7-RNA-polymerase is added, transcription and translation from a template DNA containing a T7 promoter can be performed in a coupled manner.
- a peptide containing an unnatural amino acid group can be translated and synthesized by adding a desired acylated tRNA group or an unnatural amino acid group allowed by ARS (for example, F-Tyr) to the system (Kawakami T, et al . Ribosomal synthesis of polypeptoids and peptoid-peptide hybrids. J Am Chem Soc. 2008, 130, 16861-3., Kawakami T, et al. Diverse backbone-cyclized peptides via codon reprogramming. Nat 2009-Chem Biol.
- ARS for example, F-Tyr
- an mRNA display library is obtained by chemically synthesizing a DNA library in which a desired sequence is arranged downstream of a promoter such as a T7 promoter, and using this as a template to make a double-stranded DNA by a primer extension reaction. Using this as a template, it is transcribed into mRNA using RNA polymerase such as T7 RNA polymerase. A linker (spacer) to which the antibiotic puromycin, which is an analog of aminoacyl tRNA, is connected to the 3 ′ end of this RNA is bound.
- RNA polymerase such as T7 RNA polymerase
- a display library comprising a complex of mRNA and its product in which mRNA and its product are associated can be constructed.
- molecules that bind to the target can be concentrated (panning).
- PCR amplification and analysis of the base sequence can reveal the sequence of the bound peptide.
- a cyclized peptide compound-nucleic acid complex display library is constructed, and further, a cyclized peptide compound (a peptide compound having a cyclic portion) or a cyclized branched peptide that binds to a target from the constructed display library.
- a cyclized peptide compound (a peptide compound having a cyclic portion) or a cyclized branched peptide that binds to a target from the constructed display library.
- the peptide compound further having a linear part 2 in the cyclic part) can be obtained by, for example, the methods [I] to [XVII] shown below.
- the method for producing a peptide compound having a cyclic part according to the present invention may include the following one or more steps.
- step D) A step of providing a cell-free translation system that contains the tRNA of step C) and does not contain any of methionine, methionyl tRNA synthetase (MetRS), translation initiation tRNA for methionine, formyl donor, or methionyl tRNA transferase.
- MethodRS methionyl tRNA synthetase
- Step E) A step of providing a template DNA library encoding a peptide sequence containing a codon corresponding to the anticodon of the tRNA of step C) downstream of the promoter, followed by a cysteine codon UGU or UGC following the translation initiation ATG
- Step of providing an mRNA library from the template DNA library of step E) G) A step of attaching a spacer to the 3 ′ end of the mRNA library in step F)
- H) Step of providing a pre-cyclization peptide compound-mRNA complex display library by adding and translating the mRNA library to which the spacer of Step G) is bound to the cell-free translation system of Step D)
- I) Step of forming a cyclic structure In the formation of a cyclic structure, a desulfurization reaction can be performed as necessary.
- a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library may be included.
- the method of the present invention can include the following steps. J) Step of concentrating the mRNA library bound to the target substance by panning K) Step of synthesizing cDNA with reverse transcriptase L) Process of analyzing the base sequence
- the manufacturing method of the peptide compound which has a cyclic part of this invention can include the following 1 or several processes.
- Step of providing a cell-free translation system comprising the tRNA of step C)
- Step of forming a cyclic structure In the formation of a cyclic structure, a desulfurization reaction can be performed as necessary.
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step H) to step I).
- the method of the present invention can include the following steps. J) Step of concentrating the mRNA library bound to the target substance by panning K) Step of synthesizing cDNA with reverse transcriptase L) Process of analyzing the base sequence
- the manufacturing method of the peptide compound which has a cyclic part of this invention can include the following 1 or several processes.
- MetalRS methionyl tRNA synthetase
- Step E) A template DNA library having a cysteine codon next to the translation initiation codon ATG downstream of the promoter and further having a codon corresponding to the anticodon of the tRNA of step C), which encodes the peptide sequence
- Step F) Step of providing an mRNA library from the template DNA library of step E)
- G) A step of attaching a spacer to the 3 ′ end of the mRNA library in step F)
- step G) a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library may be included. it can.
- the method of the present invention can include the following steps. J) Step of concentrating the mRNA library bound
- the manufacturing method of the peptide compound which has a cyclic part of this invention can include the following 1 or several processes.
- Providing a template DNA library F) Step of providing an mRNA library from the template DNA library of step E)
- step G a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library may be included. it can. Furthermore, the method of the present invention can include the following steps. J) Step of concentrating the mRNA library bound to the target substance by panning K) Step of synthesizing cDNA with reverse transcriptase L) Process of analyzing the base sequence
- N-terminal introduction of amino acids other than methionine, amino acid analogs or N-terminal carboxylic acid analogs, or a method for producing a peptide compound having a cyclic moiety of the present invention includes The present invention relates to a method for constructing an amide cyclized peptide compound library that can be carried out by setting it to the N-terminus, which comprises the following one or more steps.
- A) Step of providing pdCpA obtained by aminoacylating tBSSEtGABA or tBSSEt ⁇ Ala B) Providing an initiation tRNA lacking the 3′-terminal CA C) linking the pdCpA of step A) and the start tRNA of step B) to provide a start tRNA aminoacylated with tBSSEtGABA or tBSSEt ⁇ Ala, D) Step of providing pdCpA obtained by aminoacylating compound C-1 E) Step of providing tRNA lacking 3′-terminal CA F) linking the pdCpA of step D) and the tRNA at the 3 ′ end of step E) to provide a tRNA in which compound C-1 is aminoacylated, G) A cell-free translation system that contains the start tRNA of step C) and the tRNA of step F) and does not contain any of methionine, methionyl tRNA synthetase (MetRS), translation start tRNA for methionine
- ATG is the first codon downstream of the promoter, and further, a codon corresponding to the anticodon of the tRNA in step F) is further downstream, and N- that serves as a substrate for proline or other ARS on its 3 ′ side.
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step J).
- the method of the present invention can include the following steps. M) Step of concentrating mRNA library bound to target substance by panning N) Step of synthesizing cDNA with reverse transcriptase O) Process of analyzing the base sequence
- N-terminal introduction of amino acids other than methionine, amino acid analogs or N-terminal carboxylic acid analogs, or the method for producing a peptide compound having a cyclic moiety of the present invention comprises tBSSEtGABA and tBSSEt ⁇ Ala, which are difficult to tolerate in a translation elongation reaction.
- the present invention relates to a method for constructing an amide cyclized peptide compound library that can be carried out by setting it to the N-terminus, which comprises the following one or more steps.
- Step of providing pdCpA obtained by aminoacylating tBSSEtGABA or tBSSEt ⁇ Ala B) Providing an initiation tRNA lacking the 3′-terminal CA C) linking the pdCpA of step A) and the start tRNA of step B) to provide a start tRNA aminoacylated with tBSSEtGABA or tBSSEt ⁇ Ala, D) Step of providing pdCpA obtained by aminoacylating compound C-1
- ATG is the first codon downstream of the promoter, and further downstream is a codon corresponding to the anticodon of the tRNA of step F), and a codon for the N-methylaminoacylated tRNA of step F) on the 3 ′ side thereof.
- Providing a template DNA library encoding a peptide sequence comprising I) Step of providing an mRNA library from the template DNA library of Step H) J) A step of attaching a spacer to the 3 ′ end of the mRNA library in step I) K) Step G) providing the pre-cyclization peptide compound-mRNA complex display library by adding the mRNA library to which the spacer of Step J) is bound to the cell-free translation system of Step G) and translating L) Circular site
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step J).
- the method of the present invention can include the following steps. M) Step of concentrating mRNA library bound to target substance by panning N) Step of synthesizing cDNA with reverse transcriptase O) Process of analyzing the base sequence
- a method for producing an N-terminal amino acid other than methionine, an amino acid analog or an N-terminal carboxylic acid analog, or a method for producing a peptide compound having a cyclic moiety of the present invention comprises tBSSEtGABA and tBSSEt ⁇ Ala, which are difficult to tolerate in a translation elongation reaction.
- the present invention relates to a method for constructing an amide cyclized peptide compound library that can be carried out by setting it to the N-terminus, which comprises the following one or more steps.
- A) Step of providing pdCpA obtained by aminoacylating tBSSEtGABA or tBSSEt ⁇ Ala B) Providing an initiation tRNA lacking the 3′-terminal CA C) linking the pdCpA of step A) and the start tRNA of step B) to provide a start tRNA aminoacylated with tBSSEtGABA or tBSSEt ⁇ Ala, D) Step of providing pdCpA obtained by aminoacylating Asp (SBn) E) Step of providing tRNA lacking 3′-terminal CA F) A step of linking the pdCpA of step D) and the tRNA at the 3 ′ end of step E) to provide a tRNA in which Asp (SBn) is aminoacylated, G) A cell-free translation system that contains the start tRNA of step C) and the tRNA of step F) and does not contain any of methionine, methionyl tRNA synthetase (MetRS
- ATG is the first codon downstream of the promoter, and further, a codon corresponding to the anticodon of the tRNA in step F) is further downstream, and N- that serves as a substrate for proline or other ARS on its 3 ′ side.
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step J).
- the method of the present invention can include the following steps. M) Step of concentrating mRNA library bound to target substance by panning N) Step of synthesizing cDNA with reverse transcriptase O) Process of analyzing the base sequence
- N-terminal introduction of amino acids other than methionine, amino acid analogs or N-terminal carboxylic acid analogs, or a method for producing a peptide compound having a cyclic moiety of the present invention comprises tBSSEtGABA and tBSSEt ⁇ Ala, which are difficult to tolerate in a translation elongation reaction.
- the present invention relates to a method for constructing an amide cyclized peptide compound library that can be carried out by setting it to the N-terminus, which comprises the following one or more steps.
- Step of providing pdCpA obtained by aminoacylating tBSSEtGABA or tBSSEt ⁇ Ala B) Providing an initiation tRNA lacking the 3′-terminal CA C) linking the pdCpA of step A) and the start tRNA of step B) to provide a start tRNA aminoacylated with tBSSEtGABA or tBSSEt ⁇ Ala, D) Step of providing pdCpA obtained by aminoacylating Asp (SBn) E) Step of providing tRNA lacking 3′-terminal CA F) After ligating the pdCpA of step D) and the tRNA at the 3 ′ end of step E) to provide a tRNA in which Asp (SBn) is aminoacylated and to provide pdCpA in which any N-methylamino acid is aminoacylated, Providing a tRNA lacking the 3′-terminal CA, linking these pdCpA and tRNA, and providing a
- ATG is the first codon downstream of the promoter, and further downstream is a codon corresponding to the anticodon of the tRNA of step F), and a codon for the N-methylaminoacylated tRNA of step F) on the 3 ′ side thereof.
- Providing a template DNA library encoding a peptide sequence comprising I) Step of providing an mRNA library from the template DNA library of Step H) J) A step of attaching a spacer to the 3 ′ end of the mRNA library in step I) K) Step G) providing the pre-cyclization peptide compound-mRNA complex display library by adding the mRNA library to which the spacer of Step J) is bound to the cell-free translation system of Step G) and translating L) Circular site
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step J).
- the method of the present invention can include the following steps. M) Step of concentrating mRNA library bound to target substance by panning N) Step of synthesizing cDNA with reverse transcriptase O) Process of analyzing the base sequence
- N-terminal introduction of amino acids other than methionine, amino acid analogs or N-terminal carboxylic acid analogs, or the method for producing a peptide compound having a cyclic moiety of the present invention comprises the step of The present invention relates to a method for constructing an amide cyclized peptide library that can be carried out as described above, which comprises the following one or more steps.
- MethodRS methion
- step J) a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library can be included.
- the method of the present invention can include the following steps. M) Step of concentrating mRNA library bound
- N-terminal introduction of amino acids other than methionine, amino acid analogs or N-terminal carboxylic acid analogs, or a method for producing a peptide compound having a cyclic moiety of the present invention comprises the step of The present invention relates to a method for constructing an amide cyclized peptide library that can be carried out as described above, which comprises the following one or more steps.
- A) Step of providing pdCpA obtained by aminoacylating compound N-2 B) Providing an initiation tRNA lacking the 3′-terminal CA C) linking pdCpA of step A) and the start tRNA of step B) to provide an aminotylated start tRNA in compound N-1 or compound N-2; D) Providing pdCpA obtained by aminoacylating compound C-1 E) Step of providing tRNA lacking 3′-terminal CA F) a step of linking the pdCpA of step D) and the tRNA of step E) to provide a tRNA in which compound C-1 is aminoacylated, G) A cell-free translation system that contains the start tRNA of step C) and the tRNA of step F) and does not contain any of methionine, methionyl tRNA synthetase (MetRS), translation start tRNA for methionine, formyl donor, or methionyl tRNA transferase.
- MethodRS methi
- a codon corresponding to the anticodon of the translation initiation tRNA of step C) is provided as the first codon downstream of the promoter, and a codon corresponding to the anticodon of the tRNA of step F) is further downstream of the promoter.
- step J) a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library can be included.
- the method of the present invention can include the following steps. M) Step of concentrating mRNA library bound to target substance by panning N) Step of synthesizing cDNA with reverse transcriptase O) Process of analyzing the base sequence
- N-terminal introduction of amino acids other than methionine, amino acid analogs or N-terminal carboxylic acid analogs, or a method for producing a peptide compound having a cyclic moiety of the present invention comprises the step of The present invention relates to a method for constructing an amide cyclized peptide library that can be carried out as described above, which comprises the following one or more steps.
- A) Step of providing pdCpA obtained by aminoacylating compound N-2 B) Providing an initiation tRNA lacking the 3′-terminal CA C) linking pdCpA of step A) and the start tRNA of step B) to provide an aminotylated start tRNA in compound N-1 or compound N-2; D) Providing pdCpA obtained by aminoacylating compound C-1 E) Step of providing tRNA lacking 3′-terminal CA F) linking pdCpA of step D) and tRNA of step E) to provide a tRNA obtained by aminoacylating compound C-1, and further providing a pdCpA obtained by aminoacylating any N-methylamino acid; Providing a tRNA lacking the 3′-terminal CA, further linking these pdCpA and tRNA to provide a tRNA aminoacylated with N-methylamino acid, G) A cell-free translation system that contains the start tRNA of step C) and the tRNA of
- a codon corresponding to the anticodon of the translation initiation tRNA of step C) is provided as the first codon downstream of the promoter, and a codon corresponding to the anticodon of the tRNA of step F) is further downstream of the promoter.
- Step F) Providing a template DNA library encoding a peptide sequence comprising a codon for the N-methylaminoacylated tRNA of step F)
- Step of providing an mRNA library from the template DNA library of Step H) J) A step of attaching a spacer to the 3 ′ end of the mRNA library in step I)
- step J) a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library can be included.
- the method of the present invention can include the following steps. M) Step of concentrating mRNA library bound to target substance by panning
- N) Step of synthesizing cDNA with reverse transcriptase O) Process of analyzing the base sequence
- Cyclization of peptides having amino acids other than methionine at the N-terminus or the method for producing a peptide compound having a cyclic moiety of the present invention can be carried out by simultaneously translating a plurality of types of peptides having different N-terminal amino acids.
- the present invention relates to a method for constructing an amide cyclized peptide library having different cyclization site structures, which comprises one or more of the following steps.
- A) Compound CX (In this specification, “Compound CX” refers to any compound selected from Compound C-1, Compound C-2 and Compound C-3.
- Step of forming a cyclic structure In the formation of a cyclic structure, a desulfurization reaction can be performed as necessary.
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step H) to step I).
- the method of the present invention can include the following steps. J) Step of concentrating the mRNA library bound to the target substance by panning K) Step of synthesizing cDNA with reverse transcriptase L) Process of analyzing the base sequence
- Cyclization of a peptide having glycine, alanine, phenylalanine at the N-terminus, or the method for producing a peptide compound having a cyclic moiety of the present invention can be carried out by simultaneously translating a plurality of types of peptides having different N-terminal amino acids.
- the present invention relates to a method for constructing an amide cyclized peptide library having different cyclization site structures, comprising one or more of the following steps.
- Step of forming a cyclic structure In the formation of a cyclic structure, a desulfurization reaction can be performed as necessary.
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step H) to step I).
- the method of the present invention can include the following steps. J) Step of concentrating the mRNA library bound to the target substance by panning K) Step of synthesizing cDNA with reverse transcriptase L) Process of analyzing the base sequence
- Cyclization of a peptide having an amino acid other than methionine at the N-terminus, or the method for producing a peptide compound having a cyclic moiety of the present invention can be carried out by simultaneously translating a plurality of types of peptides having different N-terminal amino acids.
- the present invention relates to a method for constructing an amide cyclized peptide library having a different cyclization site structure, which comprises the following one or more steps.
- Step of forming a cyclic structure In the formation of a cyclic structure, a desulfurization reaction can be performed as necessary.
- the present invention may include a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library after step H) to step I).
- the method of the present invention can include the following steps. J) Step of concentrating the mRNA library bound to the target substance by panning K) Step of synthesizing cDNA with reverse transcriptase L) Process of analyzing the base sequence
- step J mRNA live A step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the rally.
- the method of the present invention can include the following steps. O) Step of concentrating mRNA library bound to target substance by panning P) Step of synthesizing cDNA with reverse transcriptase Q) Process for analyzing nucleotide sequences
- J) A step of attaching a spacer to the 3 ′ end of the mRNA library in step I)
- Steps K and L can be performed simultaneously by adding peptide deformylase and methionine aminopeptidase to the system during translation.
- the manufacturing method of the peptide compound which has a cyclic part of this invention is a method of constructing
- the method for producing a peptide compound having a cyclic moiety of the present invention relates to a method for constructing a cyclic branched peptide library, which comprises the following one or more steps.
- Step L) Template DNA library of Step K) Step M) of providing an mRNA library from Step N) Step of attaching a spacer to the 3 ′ end of the mRNA library N) Step of M) Cell-free translation system of Step J) Spacer library
- step of reacting methionine aminopeptidase Steps K and L can be performed simultaneously by adding peptide deformylase and methionine aminopeptidase to the system during translation.
- Step of forming a cyclic structure Q) Compound Na-4 or Compound Na-10 (Na-7 group) or Compound Na-11 (Na-7 group) in which the amino group and optionally the thiol group are also protected Step of deprotection R) Step of generating straight chain moiety 2
- a desulfurization reaction can be performed as necessary.
- step J a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library can be included.
- the method of the present invention can include the following steps. S) Step of concentrating mRNA library bound to target substance by panning T) Step of synthesizing cDNA with reverse transcriptase U) Step of analyzing base sequence
- a method for constructing a cyclic branched peptide library by cyclization of a peptide having an amino acid other than methionine at the N-terminus which comprises the following one or more steps.
- the method for producing a peptide compound having a cyclic part according to the present invention is a method for constructing an amide cyclized peptide library having different cyclization site structures that can be carried out by simultaneously translating plural types of peptides having different N-terminal amino acids.
- the present invention relates to a method including one or more of the following steps.
- Process E Step of providing tRNA lacking 3′-terminal CA F) Compound Na-4 or Compound Na-10 (Na-7 group) or Compound Na-11 in which the pdCpA of Step D) and the tRNA of Step E) are linked and the amino group and optionally the thiol group are also protected.
- a desulfurization reaction can be performed as necessary.
- a step of synthesizing cDNA with a primer that anneals to the 3 ′ region of the mRNA library can be included.
- the method of the present invention can include the following steps. O) Step of concentrating mRNA library bound to target substance by panning P) Step of synthesizing cDNA with reverse transcriptase Q) Normal leucine can be added in place of methionine in step G) of analyzing the base sequence.
- a method for constructing a cyclic branched peptide library by cyclization of a peptide having an amino acid other than methionine at the N-terminus which comprises one or more of the following steps.
- the method for producing a peptide compound having a cyclic part according to the present invention is a method for constructing an amide cyclized peptide library having different cyclization site structures that can be carried out by simultaneously translating plural types of peptides having different N-terminal amino acids.
- the present invention relates to a method including one or more of the following steps.
- A) Step of providing pdCpA obtained by aminoacylating compound C-1 B) Step of providing tRNA lacking 3′-terminal CA C) a step of linking the pdCpA of step A) and the tRNA of step B) to provide a tRNA obtained by aminoacylating compound C-1; D) Provided is a pdCpA obtained by aminoacylating Compound Na-4 or Compound Na-10 (Na-7 group) or Compound Na-11 (Na-7 group) in which the compound amino group and optionally the thiol group are also protected.
- Process E Step of providing tRNA lacking 3′-terminal CA F) Compound Na-4 or Compound Na-10 (Na-7 group) or Compound Na— in which the pdCpA of Step D) and the tRNA of Step E) are ligated to protect the compound amino group and, if necessary, the thiol group.
- Step of forming a cyclic structure In the formation of the cyclic structure, a desulfurization reaction can be performed as necessary.
- N) Step of deprotecting the side chain protecting group of Compound Na-4 or Compound Na-10 (Na-7 group) or Compound Na-11 (Na-7 group), and active thioester from Cys, Pro and lactic acid units Process to generate O) The step of forming an active thioesterification and an amide bond with the side chain amino group to form a linear site 2
- a primer that anneals to the 3 ′ region of the mRNA library A step of synthesizing cDNA can be included.
- the method of the present invention can include the following steps.
- an “alkyl group” is a monovalent group derived by removing an arbitrary hydrogen atom from an aliphatic hydrocarbon, and contains a heteroatom or an unsaturated carbon-carbon bond in the skeleton. It has a subset of hydrocarbyl or hydrocarbon group structures containing hydrogen and carbon atoms. The length n of the carbon chain is in the range of 1-20.
- the “alkenyl group” is a monovalent group having at least one double bond (two adjacent SP2 carbon atoms). Depending on the arrangement of the double bonds and substituents (if present), the geometry of the double bond can take the Enthogen (E) or Tuzanmen (Z), cis or trans configurations.
- Examples of the alkenyl group include straight-chain or branched-chain groups, and include straight chains containing internal olefins. A C2-C10 alkenyl group is preferable, and a C2-C6 alkenyl group is more preferable.
- alkenyl includes, for example, vinyl group, allyl group, 1-propenyl group, 2-propenyl group, 1-butenyl group, 2-butenyl group (including cis and trans), and 3-butenyl group. , Pentenyl group, hexenyl group and the like.
- alkynyl is a monovalent group having at least one triple bond (two adjacent SP carbon atoms). Examples include linear or branched alkynyl groups, including internal alkylene. A C2-C10 alkynyl group is preferable, and a C2-C6 alkynyl group is more preferable. Specific examples of alkynyl include, for example, ethynyl group, 1-propynyl group, propargyl group, 3-butynyl group, pentynyl group, hexynyl group, 3-phenyl-2-propynyl group, and 3- (2′-fluorophenyl).
- Examples include -2-propynyl group, 2-hydroxy-2-propynyl group, 3- (3-fluorophenyl) -2-propynyl group, and 3-methyl- (5-phenyl) -4-pentynyl group.
- the C1-C6 alkyl group optionally having halogen as a substituent means a “C1-C6 alkyl group” substituted with one or more halogen atoms.
- aryl group means a monovalent aromatic hydrocarbon ring, preferably C5-C10 aryl.
- aryl include phenyl group and naphthyl (for example, 1-naphthyl group and 2-naphthyl group).
- the “aryl group” may include the following “heteroaryl”.
- C5-C10 aryl C1-C6 alkyl group refers to an alkyl group in which one of the hydrogen atoms of the C1-C6 alkyl group is substituted with the C5-C10 aryl group.
- An arylalkyl group is a group containing both an aryl group and an alkyl group. For example, it means a group in which at least one hydrogen atom of the alkyl group is substituted with an aryl group, preferably “C5 -C10 aryl C1-C6 alkyl group ". For example, a benzyl group etc. are mentioned.
- the “optionally substituted C5-C10 aryl C1-C6 alkyl group” means that at least one hydrogen atom of the aryl group and / or alkyl group of the “C5-C10 aryl C1-C6 alkyl group” is And represents a group substituted by a substituent. Examples of these substituents include various substituents defined as substituents on the side chain of “amino acid”.
- Alkoxy group means a group in which a hydrogen atom of a hydroxyl group is substituted with the alkyl group, and examples thereof include “C1-C6 alkoxy group”.
- nucleic acid in the present invention can also include deoxyribonucleic acid (DNA), ribonucleic acid (RNA), or a nucleotide derivative having an artificial base. Moreover, a peptide nucleic acid (PNA) can also be included.
- the nucleic acid of the present invention can be any of these nucleic acids or a hybrid as long as the desired genetic information is retained. That is, chimeric nucleic acids in which different nucleic acids such as DNA-RNA hybrid nucleotides and DNA and RNA are linked in a single strand are also included in the nucleic acids of the present invention.
- a nucleic acid library such as a display library containing these nucleic acids as templates can be suitably used.
- cDNA By synthesizing cDNA from mRNA, which is a tag containing genetic information attached to a molecule selected by panning, PCR amplification and analysis of the base sequence can reveal the sequence of the bound protein.
- the portion where the amino acid residue of the DNA library that is the template of the library is not fixed can be obtained by mixing with a base and synthesizing. Repeating 3 multiples of a mixture of 4 bases of A, T, G, and C (N), or N for the first and second letters of the codon and 2 bases such as W, M, K, and S for the third letter
- the third character is made into one type of base as long as the number of amino acids to be synthesized is further reduced to 16 or less.
- the frequency of appearance of amino acid residues can be freely adjusted by preparing a codon unit corresponding to three codon characters as in the Examples, mixing them at an arbitrary ratio, and using them for synthesis.
- these nucleic acid libraries can be translated using the cell-free translation system described below.
- a cell-free translation system it is preferable to include a sequence encoding a spacer downstream of the target nucleic acid.
- the spacer sequence include, but are not limited to, sequences containing glycine and serine.
- a linker formed of RNA, DNA, or a polymer of hexaethylene glycol (spc18) (for example, five polymers) is inserted between the compound incorporated into the peptide during translation by ribosome, such as puromycin and its derivatives, and the nucleic acid library. It is preferable to include.
- the cell-free translation system is obtained by crushing the cells of the material and adding tRNA, amino acid, ATP, etc. to the extract prepared by centrifugation, dialysis or the like.
- Escherichia coli MetalsmolEnzymol. 1983; 101: 674-90. Prokaryotic coupled transcription-translation. Chen HZ, Zubay G.
- yeast J. Biol. Chem. 1979 254: 3965-3969.
- E Gasior, F Herrera, I Sadnik, C S McLaughlin, and K Moldave wheat germ (Methods Enzymol.translation.
- An aminoacyl-tRNA can be obtained by binding an aminoacylated pdCpA obtained by removing the CA from the CCA sequence at the 3 ′ end of tRNA and a separately prepared aminoacylated pdCpA (Biochemistry. 1984; 23: 1468-73.T4 RNA ligase mediated preparation of novel “chemically misacylated” tRNAPheS.Heckler TG, Chang LH, Zama Y, Naka T, Chorghade MS, Hecht SM.). There is also aminoacylation by flexizyme, a ribozyme that carries active esters of various unnatural amino acids on tRNA (J Am Chem Soc.
- the library composed of peptide compounds having a cyclic part according to the present invention is composed of peptide compounds having a cyclic part consisting of 9 to 13 residues of amino acids and / or amino acid analogs. Is counted from the amino acid up to the 13th residue, with the N-terminal amino acid immediately before the random region of the display library starts as 1 (counted as the number of units after all post-translational modifications have been completed).
- the average number of N-methyl amino acids is 3 to 10, preferably 4 to 9, and more preferably 5 to 8 so that the random region contains It is necessary to consider the proportion of amino acids containing N-alkyl amino acids and amino acids having NH2.
- it is necessary to select the type of amino acid contained in the random region so that the average ClogP value of the 13-residue peptide is set to 4 or more, preferably 5 or more, more preferably 6 or more.
- the acid preferably has a pKa of 3.5 or more. More preferably, it is 4.5 or more, and more preferably 5 or more.
- pKa of the side chain carboxyl group of aspartic acid is 3.9
- pKa of the side difference phenolic hydroxyl group of tyrosine is 10. Tetrazole has a pKa of 5.6.
- pKa is 9 or less, More preferably, it is 7.5 or less, More preferably, it is 6.5 or less.
- one of the two fixed amino acids is N-alkyl
- the C-terminal side of the crossing unit is also selected from N-alkyl amino acids, so the average number of N-alkyl amino acids is 6.0. calculated. Similarly, the average ClogP values of both were 5.97 and 6.12.
- Non-natural amino acids that can be used for translational synthesis
- Non-natural amino acids (translated amino acids) that can be used in the present invention are exemplified below, but are not limited thereto. Many of these unnatural amino acids are purchased with the side chain protected or unprotected and the amine moiety protected or unprotected. Those not purchased are synthesized by known methods.
- N-Me amino acids can be used as non-natural amino acids.
- N-alkyl amino acids can also be used as non-natural amino acids.
- ⁇ , ⁇ -dialkyl amino acids can also be used as non-natural amino acids.
- amino acids can also be used as non-natural amino acids.
- aminoacyl-tRNA amino acids and amino acid analogs can be converted, for example, into active esters for reaction with pdCpA. Many of these active esters can be isolated as cyanomethyl esters.
- Amide bond formation reaction that can be utilized for cyclization after translational synthesis
- the amide bond formation reaction by the reaction of carboxylic acid with primary and secondary amines is widely used in organic synthetic chemistry in general.
- a method in which a carboxylic acid is isolated as a previously activated active ester and then mixed with an amine, or a method in which a carboxylic acid is directly mixed and then activated to form an amide bond is used. be able to.
- active forms of carboxylic acids acid halide groups such as acid chloride and acid aodide, and active ester groups such as HOBt, HOAt, HOSu, HOPfp, and thioester are widely known.
- Preferred examples of the thioester include those represented by —C ( ⁇ O) SR1.
- the acid halide can be isolated by a purification operation after mixing a carboxylic acid and a halide agent such as SOCl 2 . Since the obtained acid halide is highly reactive, when used in a translation system such as PureSystem, PPh 3 and I are used as mild reaction conditions that may cause the carboxylic acid to be acid halide after translation by PureSystem. A method of activating from 2 to acid iodide (Lakshman et al., Eur. J. Org. Chem. 2010, 2709) can be used.
- a technique for introducing a reaction auxiliary group in the vicinity of an amino group for example, a technique for introducing a thiol group in the vicinity of an amino group can be mentioned.
- the number of atoms (number of carbon chains) connecting the amino group and the thiol group is particularly preferably 2 or 3.
- N, O-benzylidene acetal can be rapidly formed selectively with a substrate having an alcohol moiety in the vicinity of the amino group. When this is transferred under acidic conditions, an amide bond is formed.
- This active ester can be selectively reacted with serine in the presence of phenylthioester.
- serine containing an OH group can be selectively reacted with the active ester in the presence of lysine.
- Azide can also be used as an amine equivalent (Vilarrasa et al. J. Org. Chem. 2009, 74, 2203).
- Carboxylic acid is activated in the reaction system in the presence of a tertiary phosphine with a dithiocatalyst or a diseleno catalyst to activate the thioester or selenoester.
- the azide moiety is activated by reacting with a tertiary phosphine and reacting with an active ester.
- a condensation reaction with a palladium complex catalyst of aldehyde and amine (Yoshida et al. S. Synthesis, 1983, 474) and a rhodium complex catalyst (Beller et al. Eur. J. Org. Chem. 2001, 423) can also be used.
- the present invention provides a pharmaceutical composition containing the peptide compound produced by the method of the present invention.
- the pharmaceutical composition of the present invention can be formulated by a known method by introducing a pharmaceutically acceptable carrier in addition to the peptide compound produced by the method of the present invention.
- Excipients, binders, lubricants, colorants, flavoring agents, and if necessary stabilizers, emulsifiers, absorption promoters, surfactants, pH adjusters, preservatives, Antioxidants and the like can be used.
- ingredients that are used as raw materials for pharmaceutical preparations are blended to prepare a preparation by a conventional method.
- the compound according to the present invention or a pharmacologically acceptable salt and excipient thereof in order to produce an oral preparation, the compound according to the present invention or a pharmacologically acceptable salt and excipient thereof, and optionally a binder, a disintegrant, a lubricant, a coloring agent, a flavoring agent.
- a binder a disintegrant, a lubricant, a coloring agent, a flavoring agent.
- these components include animal and vegetable oils such as soybean oil, beef tallow and synthetic glycerides; hydrocarbons such as liquid paraffin, squalane and solid paraffin; ester oils such as octyldodecyl myristate and isopropyl myristate; cetostearyl alcohol and behenyl alcohol Higher alcohols; silicone resins; silicone oils; surfactants such as polyoxyethylene fatty acid esters, sorbitan fatty acid esters, glycerin fatty acid esters, polyoxyethylene sorbitan fatty acid esters, polyoxyethylene hydrogenated castor oil, polyoxyethylene polyoxypropylene block copolymers Water-soluble such as hydroxyethylcellulose, polyacrylic acid, carboxyvinyl polymer, polyethylene glycol, polyvinylpyrrolidone, methylcellulose Polymers; lower alcohols such as ethanol and isopropanol; polyhydric alcohols such as glycerin, propylene glycol, dipropylene glycol
- excipient examples include lactose, corn starch, sucrose, glucose, mannitol, sorbitol, crystalline cellulose, silicon dioxide and the like.
- disintegrant examples include starch, agar, gelatin powder, crystalline cellulose, calcium carbonate, sodium bicarbonate, calcium citrate, dextrin, pectin, carboxymethylcellulose / calcium and the like.
- lubricant examples include magnesium stearate, talc, polyethylene glycol, silica, and hardened vegetable oil.
- these tablets and granules may be coated with sugar coating and other coatings as necessary.
- a liquid preparation such as a syrup or an injectable preparation
- a compound according to the present invention or a pharmacologically acceptable salt thereof, a pH adjuster, a solubilizer, an isotonic agent, etc. are necessary. Add a solubilizing agent, stabilizer, etc. accordingly, and formulate it by a conventional method.
- a pharmacologically acceptable carrier or medium specifically, sterilized water, physiological saline, vegetable oil, emulsifier, suspension, surfactant, stabilizer, flavoring agent, excipient, vehicle, preservative It is conceivable to formulate by combining with a binder or the like as appropriate and mixing in a unit dosage form generally required for pharmaceutical practice.
- Aqueous solutions for injection include, for example, isotonic solutions containing physiological saline, glucose and other adjuvants such as D-sorbitol, D-mannose, D-mannitol and sodium chloride.
- Suitable solubilizers such as Alcohols, specifically ethanol, polyalcohols such as propylene glycol, polyethylene glycol, nonionic surfactants such as polysorbate 80 (registered trademark), HCO-50 may be used in combination.
- oily liquid examples include sesame oil and soybean oil, which may be used in combination with benzyl benzoate or benzyl alcohol as a solubilizing agent.
- oily liquid examples include sesame oil and soybean oil, which may be used in combination with benzyl benzoate or benzyl alcohol as a solubilizing agent.
- buffer for example, phosphate buffer, sodium acetate buffer, a soothing agent, for example, procaine hydrochloride, stabilizer, for example, benzyl alcohol, phenol, antioxidant.
- the prepared injection solution is usually filled into a suitable ampoule.
- Administration is preferably oral administration, but the administration method is not limited to oral administration.
- Specific examples of parenteral administration include injection, nasal administration, pulmonary administration, and transdermal administration.
- the injection form it can be administered systemically or locally by, for example, intravenous injection, intramuscular injection, intraperitoneal injection, subcutaneous injection, or the like.
- the administration method can be appropriately selected depending on the age and symptoms of the patient.
- the dose of the pharmaceutical composition containing the peptide compound produced by the method of the present invention can be selected, for example, in the range of 0.0001 mg to 1000 mg per kg of body weight at a time. Alternatively, for example, the dose can be selected in the range of 0.001 to 100,000 mg / body per patient, but is not necessarily limited to these values.
- the dose and administration method vary depending on the weight, age, symptoms, etc. of the patient, but can be appropriately selected by those skilled in the art.
- a dehydrated solvent or an anhydrous solvent purchased from Kanto Chemical, Wako Pure Chemical, etc. was used.
- the LCMS analysis conditions are as follows.
- the reaction mixture was filtered, and the obtained filtrate was concentrated and diluted with ethyl acetate, and then washed with a saturated aqueous ammonium chloride solution, a saturated aqueous sodium bicarbonate solution, and a saturated aqueous sodium chloride solution. Thereafter, the organic extract was dried over magnesium sulfate and concentrated under reduced pressure.
- Buffer A was prepared as follows. Acetic acid is added to an aqueous solution of N, N, N-trimethylhexadecane-1-aminium chloride (6,40 g, 20 mmol) and imidazole (6.81 g, 100 mmol), pH 8, 20 mM N, N, N-trimethylhexadecane- Buffer solution A (1 L) of 1-aminium and 100 mM imidazole was obtained.
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran Using 2-yl) methyl (Compound 1h) (50 mg, 0.079 mol), 4- (benzylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (Compound 1g- 4-oxo-2- (pent-4-enamido) -4- (phenethylthio) butanoic acid (S) -cyanomethyl (compound 1g-IE
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran Using 2-yl) methyl (Compound 1h) (50 mg, 0.079 mmol), 4- (benzylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (Compound 1g- 4- (isopropylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (compound 1g-
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran Using 2-yl) methyl (Compound 1h) (50 mg, 0.079 mmol), 4- (benzylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (Compound 1g- 4- (tert-butylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (compound 1g- 4- (tert-butylthio) -4-
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran Using 2-yl) methyl (compound 1h) (150 mg, 0.236 mmol), 4- (benzylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (compound 1 g- 4- (methylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (compound 1g-IA)
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran Using 2-yl) methyl (compound 1h) (200 mg, 0.314 mmol), 4- (benzylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (compound 1g- 5- (methylthio) -5-oxo-2- (pent-4-enamido) pentanoic acid (S) -cyanomethyl (compound 1g-IIA
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran Using 2-yl) methyl (compound 1h) (150 mg, 0.236 mmol), 4- (benzylthio) -4-oxo-2- (pent-4-enamido) butanoic acid (S) -cyanomethyl (compound 1 g- (ID) instead of 5-oxo-2- (pent-4-enamido) -5- (phenylthio) pentanoic acid (S) -cyanomethyl (
- SEQ ID NO: R-1 (SEQ ID NO: 30) tRNAEnAsnGAG (-CA) RNA sequence: GGCUCUGUAGUUCAGUCGGUAGAACGGCGGACUgagAAUCCGUAUGUCACUGGUUCGAGUCCAGUCAGAGCCGC
- Example 4 Translational synthesis using amino acids in which side chain carboxylic acids are active esterified A tRNA aminoacylated with various amino acids is added to a cell-free translation system to initiate translation, thereby producing desired non-naturally occurring amino acids. Translational synthesis of a polypeptide containing amino acids was performed.
- the translation system used was a PURE system, which is a prokaryotic-derived reconstituted cell-free protein synthesis system including a transcription system from template DNA.
- transcription translation solution 5% (v / v) T7 RNA polymerase RiboMAX Enzyme Mix (Promega, P1300), 2 mM GTP, 2 mM ATP, 1 mM CTP, 1 mM UTP, 20 mM creatine phosphate, 50 mM HEPES-KOH pH 7.6, 100 mM potassium glutamate, 12 mM magnesium acetate, 2 mM spermidine, 1 mM dithiothreitol, 1.5 mg / ml E.
- coli MRE600 (RNase negative) -derived tRNA (Roche), 4 ⁇ g / ml creatine kinase, 3 ⁇ g / ml myokinase , 2 unit / ml inorganic pyrophosphatase, 1.1 ⁇ g / ml nucleoside diphosphate kinase, 0.6 ⁇ M methionyl tRNA transformylase, 0.26 ⁇ M EF-G, 0.25 ⁇ M RF1, 0.24 ⁇ M F2, 0.17 ⁇ M RF3, 0.5 ⁇ M RRF, 2.7 ⁇ M IF1, 0.4 ⁇ M IF2, 1.5 ⁇ M IF3, 40 ⁇ M EF-Tu, 84 ⁇ M EF-Ts, 1.2 ⁇ M ribosome, 0.73 ⁇ M AlaRS, 0.03 ⁇ M ArgRS, 0.38 ⁇ M AsnRS, 0.13 ⁇ M AspRS, 0.02 ⁇ M CysRS, 0.06
- the translation product was identified by measuring a MALDI-MS spectrum using ⁇ -cyano-4-hydroxycinnamic acid as a matrix.
- the peak of the mass spectrum derived from the translated peptide in the molecular weight range of 799 to 2000 could not be confirmed by MALDI-MS.
- the peaks detected in the vicinity of molecular weights 882, 918, and 1256 are not peaks derived from the translated peptide because they are also observed when analyzing a PURESystem without adding template DNA as a negative control.
- SEQ ID NO: D-2 (SEQ ID NO: 2) Mtyg_R DNA sequence GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgACTACAACGCGActttactaccgtcgtggcggcTAATAAATAGATAG
- SEQ ID NO: D-3 (SEQ ID NO: 3) Mtryg3 DNA sequence GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgACTACAACGCGActttactaccgtggcggcTAGTAGATAGATAG
- tRNA CA-deficient
- tRNAfMetCAU lacking 3′-end CA by in vitro transcription using RiboMAX Large Scale RNA production System T7 (Promega, P1300) -CA) (SEQ ID NO: R-5) was synthesized and purified by RNeasy Mini kit (Qiagen).
- SEQ ID NO: D-5 (SEQ ID NO: 5): GGCGTAATACGACTCACTATAGGCGGGGTGGAGCAGCCTGGTAGCTCGTCGGGCTCATAACCCGAAGATCGTCGGTTCAAATCCGGCCCCCGCAAC
- SEQ ID NO: R-5 (SEQ ID NO: 32): GGCGGGGUGGAGCAGCCUGGUAGCUCGUCGGGCUCAUAACCCGAAGAUCGUCGGUUCAAAUCCGGCCCCCGCAAC
- acylated tRNA (compound AT-2) by ligation of acylated pdCpA (compound 7c) and tRNA (CA deficient) (SEQ ID NO: R-5) not containing the N-terminal amino group 50 ⁇ M Transcription tRNAfMetCAU (-CA) ( SEQ ID NO: R-5)
- 10 ⁇ ligation buffer 500 mM HEPES-KOH pH 7.5, 200 mM MgCl 2 , 10 mM ATP
- Nuclease free water were added, heated at 95 ° C. for 2 minutes, and then at room temperature.
- the tRNA was refolded by leaving it for 5 minutes. 2 ⁇ L of 10 unit / ⁇ L T4 RNA ligase (New England Bio Lab.) And 2 ⁇ L of a 5 mM N-terminal amino group-containing acylated pdCpA (compound 7c) in DMSO were added, and a ligation reaction was performed at 15 ° C. for 45 minutes. It was. Acylated tRNA (compound AT-2) was recovered by phenol precipitation and ethanol precipitation. The acylated tRNA (Compound AT-2) was dissolved in 1 mM sodium acetate just prior to addition to the translation mixture. Compound AT-2 MeOFlac-tRNAfMetCAU (SEQ ID NO: 33)
- MS corresponding to the de-BnSH peptide was mainly produced from full-length peptide P-7 containing thioester. Observed as a product (translated peptide P-8). Although MeOFlac was used for translation initiation and a peptide without an amine at the N-terminal was translated, a decrease in molecular weight was observed, and thus a reaction with an N-terminal amine was also denied (FIG. 6).
- SEQ ID NO: D-6 (SEQ ID NO: 6) KA03 DNA sequence GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgACTAGAACTaaggcgTACTGGAGCcttCCGggcggctaa
- LCMS (ESI) m / z 448 (M + H) + Retention time: 1.09 minutes (analysis condition SQDAA05)
- LCMS (ESI) m / z 498 (M + H) + Retention time: 1.12 minutes (analysis condition SQDAA05)
- LCMS (ESI) m / z 469 (M + H) + Retention time: 0.99 minutes (analysis condition SQDAA05)
- the 0.4M tris (2-carboxyethyl) phosphine (TCEP) aqueous solution was prepared as follows. To a solution of tris (2-carboxyethyl) phosphine (TCEP) hydrochloride (1.0 g, 3.49 mmol) in water (6.8 ml) was added triethylamine (1.64 ml) to pH 7, and 0.4M tris (2-carboxyl An ethyl) phosphine (TCEP) aqueous solution was obtained.
- LCMS (ESI) m / z 485 (M + H) + Retention time: 2.80 minutes (analysis condition ZQAA05)
- LCMS (ESI) m / z 485 (M + H) + Retention time: 2.80 minutes (analysis condition ZQAA05)
- the reactivity with the thioester was greatly different between the Cys derivative which is a model compound of an amine having a reaction auxiliary group and the Gly derivative which is a model of an amine having no reaction auxiliary group. It was revealed that the Cys derivative having a reaction auxiliary group has sufficiently high cyclization reactivity under stable conditions of RNA. In addition, since amines with reaction auxiliary groups have sufficient selectivity for amines without reaction auxiliary groups, selective post-translational cyclization with amines with reaction auxiliary groups from multiple reaction points. It can be judged that a proper reaction is possible.
- thioester is higher in the case of thioaryl ester than in the case of thioaryl and thioalkyl or thioaralkyl. In this case, it is clear that the amino group having no reaction auxiliary group also has good reactivity. It became.
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran 2-yl) methyl (compound 1h) (112 mg, 0.176 mmol) was used and 2-((tert-butoxycarbonyl) amino) -3- (tert-butyldisulfanyl) propanoic acid (R) -cyanomethyl (compound 2-((tert-butoxycarbonyl) (methyl) amino) -3- (tert-butyldisulfanyl) propanoic acid (R -Cy
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran Using 2-yl) methyl (compound 1h) (200 mg, 0.314 mmol), 2-((tert-butoxycarbonyl) amino) -3- (tert-butyldisulfanyl) propanoic acid (R) -cyanomethyl (compound 2- (tert-butyldisulfanyl) -2- (pent-4-enamido) propanoic acid (R) -cyanomethyl (2l-C
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran- 2-yl) methyl (compound 1h) (150 mg, 0.236 mmol) was used and 2-((tert-butoxycarbonyl) amino) -3- (tert-butyldisulfanyl) propanoic acid (R) -cyanomethyl (compound 3- (methyldisulfanyl) -2- (pent-4-enamido) propanoic acid (R) -cyanomethyl (compound 21-D)
- dihydrogen phosphate ((2R, 3S, 5R) -5- (4-amino-2-oxopyrimidin-1 (2H) -yl) -3-(( (((2R, 3S, 4R, 5R) -5- (6-Amino-9H-purin-9-yl) -3,4-dihydroxytetrahydrofuran-2-yl) methoxy) (hydroxy) phosphoryl) oxy) tetrahydrofuran- 2-yl) methyl (compound 1h) (150 mg, 0.236 mmol) was used and 2-((tert-butoxycarbonyl) amino) -3- (tert-butyldisulfanyl) propanoic acid (R) -cyanomethyl (compound 2- (tert-butyldisulfanyl) -2-((((4,5-dimethoxy-2-nitrobenzyl) oxy) carbonyl instead of
- a saturated aqueous ammonium chloride solution (3 ml) was added to the reaction mixture, and the mixture was diluted with ethyl acetate, and the organic layer was washed with water. Thereafter, the organic extract was dried over magnesium sulfate.
- a saturated aqueous ammonium chloride solution (3 ml) was added to the reaction mixture, and the mixture was diluted with ethyl acetate, and the organic layer was washed with water. Thereafter, the organic extract was dried over magnesium sulfate.
- Example 7 Synthesis of aminoacylated tRNA having a Cys derivative 50 ⁇ M Transcription tRNAfMetCAU (-CA) (SEQ ID NO: R-5) 10 ⁇ l was mixed with 10 ⁇ ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP) 2 ⁇ l and 4 ⁇ l of Nuclease free water were added, heated at 95 ° C. for 2 minutes, and then allowed to stand at room temperature for 5 minutes to refold tRNA.
- 10 ligation buffer 500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP
- Peptide sequence P-10 [Cys (StBu)] ThrThrThrArgLeuTyrTyrArgGlyGly MALDI-MS: m / z: Not detected (Calc. 1345.7)
- DNA sequence (D-7) AKC17 DNA sequence (same as SEQ ID NO: D-31) (SEQ ID NO: 7) GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGACTAGAACTGCCTACTGGAGCcttTGCGGCAGCGGCAGCGGCAGC
- the following experiment was conducted to examine the deprotection of the side chain StBu of pdCpA-Cys and the stability of the deprotected compound 2e-A using the compound 2n-A which is a deprotection precursor of the compound 2n-A side chain StBu. It was.
- the side chain was deprotected at room temperature under the conditions of 250 ⁇ M compound 2n-A, pH 7 50 mM Tris ⁇ HCl buffer, DTT 10 mM, and the change with time was observed using LC-MS (SQD).
- the target compound 2e-A was not observed, and pdCpA from which the amino acid was hydrolyzed and eliminated was observed.
- the remaining% of compound 2n-A after 30 hours at pH 7 in HEPES® buffer was 30%.
- the compound 2e-A with the side chain deprotected showed a result of increased stability by protecting the side chain compared to the result of rapid hydrolysis when deprotected. .
- Example 10 Efficient translation introduction of N-terminal Cys using Initiation read through method The phenomenon (Intiation read through) in which translation initiation from the second codon observed in Example 9 was efficiently obtained was positive By utilizing this method, introduction of Cys at the N-terminus of the peptide was attempted as follows.
- SEQ ID NO: D-8 (SEQ ID NO: 8) Mctryg3 DNA sequence GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgtgcACTACAACGCGTctttactaccgtggcggcTAGTAGATAGATAG
- IniRt_XXX_am peptide sequence (translated peptide numbers P-15 to P-33) CysXaaThrThrThrLeuTyrTyrArgGlyGly (SEQ ID NO: 39 to 57)
- Example 11 Synthesis of non-natural amino acid with SH group other than Cys and its aminoacylated pdCpA
- the natural amino acid Cys was efficiently translated and introduced by the initiation read through method using ARS. Since there is a limit to introducing ARS into natural amino acids, among aminoacylated pdCpAs, compounds designed to slow down the hydrolysis rate of the active ester moiety were synthesized and evaluated. That is, a series of compound groups in which the SH group was further separated from the carboxylic acid active ester site was synthesized and evaluated.
- Example 12 Synthesis of aminoacylated tRNA using stable aminoacylated pdCpA having an SH group 50 ⁇ M Transcription tRNAfMetCAU (-CA) (SEQ ID NO: R-5)
- 10X ligation buffer 500 mM HEPES-KOH pH 7.5, 200 2 ⁇ l of mM MgCl2, 10 mM ATP
- 4 ⁇ l of Nuclease free water were added, heated at 95 ° C. for 2 minutes, and then allowed to stand at room temperature for 5 minutes to refold tRNA.
- Example 13 Translation synthesis using SH group-containing stabilized aminoacylated tRNA As shown in the following experiment, when the SH group or main chain amino group of Cys was protected, the translation solution of aminoacylated pdCpA compared to Cys itself It was shown that the stability in the model system (HEPES buffer) was improved and the translation synthesis efficiency was also improved. In addition, the transduction efficiency of various non-natural amino acids containing various new SH groups in which the position of the SH group is located far from the ⁇ -carboxylic acid site was also improved.
- SEQ ID NO: D-31 (SEQ ID NO: 7) AKC17 DNA sequence GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGACTAGAACTGCCTACTGGAGCcttTGCGGCAGCGGCAGCGGCAGC
- SEQ ID NO: D-33 (identical to SEQ ID NO: D-1) (SEQ ID NO: 1) (SEQ ID NO: 1) tRNAEnAsnGAG (-CA) DNA sequence: GGCGTAATACGACTCACTATAGGCTCTGTAGTTCAGTCGGTAGAACGGCGGACTgagAATCCGTATGTCACTGGTTCGAGTCCAGTCAGAGCCGCGC
- SEQ ID NO: R-33 (identical to SEQ ID NO: R-1) (SEQ ID NO: 30) tRNAEnAsnGAG (-CA) RNA sequence: GGCUCUGUAGUUCAGUCGGUAGAACGGCGGACUgagAAUCCGUAUGUCACUGGUUCGAGUCCAGUCAGAGCCGC
- Example 15 Translation and cyclization using Intioation read through method 1. Synthesis of amide-type cyclized peptide via NCL (Native Chemical Ligation) A translation reaction was performed using KA02.5U DNA sequence (SEQ ID NO: D-32) as template DNA. To the template DNA, the above-mentioned transcription translation solution, 17 mM amino acids of 0.3 mM each except Met, Ala and Leu, 3 mM lactic acid, and 50 ⁇ M Asp (SRex2) -tRNAEnAsnGAG (Rex2: (Benzyl, methyl, ethyl, isopropyl, tertiary butyl, phenethyl) (Compound Nos. AT-1-IA2, IB2, IC2, ID2, IE2, IG2) were added and translated at 37 ° C. for 3 hours. As a result, the cyclized full-length peptide was confirmed by MS spectrum (FIGS. 28 to 29).
- SEQ ID NO: D-32 (SEQ ID NO: 28) KA02.5U DNA sequence: GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG tgcACTAGAACTaaggcgTACTGGAGCcttCCGggctaa
- the non-natural amino acid (including protein amino acids) following the thioester has an N-alkyl group. You don't have to worry about amino acids. This is because an amide is formed via a reaction with the SH group immediately after the translation. When an amino acid with a protected SH group is translated, aspartimide is formed within the time from the completion of the translation process to the deprotection process.
- Peptide sequence P-53 (SEQ ID NO: 59) CysThrThrThrArg [Asp (SMe)] TyrTyrArgGlyGly Cys main chain amino group and Asp side chain carboxylic acid compound with intramolecular amide cyclization mass spectrum calc. 1273.6
- Fmoc-Cys (Mmt) -OH, Fmoc-Thr (tBu) -OH, and Fmoc-Arg (Pbf) -OH (all purchased from Watanabe Chemical Industry) were used as Fmoc amino acids.
- a 2-chlorotrityl resin 250 mg, 13 columns, 1.36 mmol to which Fmoc-Asp-OBn was bound was set in the synthesizer, and the peptide was synthesized on a solid phase.
- the resin in the tube was filtered through a synthesis column to remove the resin, and 2-iodoacetamide (252 mg, 1.36 mmol), DIPEA (15 mL), DMF (15 mL) were added to the reaction mixture, and S Was alkylated.
- Fmoc-Gly-OH, Fmoc-Tyr (tBu) -OH, Fmoc-Arg (Pbf) -OH (all purchased from Watanabe Chemical Industries) were used as Fmoc amino acids, and Fmoc-Gly-OH was bound to the synthesizer 2 -Chlorotrityl resin (250 mg) was set, and a peptide having the sequence H-Tyr (tBu) -Tyr (tBu) -Arg (Pbf) -Gly-Gly-OH (peptide P-59) was synthesized on a solid phase.
- Example 16 Translation synthesis, desulfurization reaction after cyclization While an amide cyclization reaction proceeded in translation synthesis, removal of the SH group as a reaction auxiliary group is necessary for our purpose. Since SH groups are known to form covalent bonds with various proteins, it is likely that SH group-dependent binding compounds will be obtained, while such compounds become drugs due to their high reactivity. Hateful. Removal of the SH group requires an intermolecular reaction. As already described, the degree of difficulty in realizing intermolecular reactions at a concentration of 1 uM and in the presence of various reactive functional groups is high. The following two methods are conceivable as methods for realizing such a reaction.
- Reagents used in excess generate only a reversible reaction (for example, coordination bond) other than the desired reaction point, and are removed by post-treatment after completion of the reaction.
- Reagents used in excess do not affect RNA at all. The results of various investigations of reaction substrates and reaction conditions that can satisfy these various conditions are shown below.
- Tris (2-carboxyethyl) phosphine (TCEP) aqueous solution Triethylamine (TCG) hydrochloride (1.0 g, 3.49 mmol) in water (6.8 ml) 1.64 ml) was added to adjust the pH to 7, and a 0.4 M tris (2-carboxyethyl) phosphine (TCEP) aqueous solution was obtained.
- the 0.4M tris (2-carboxyethyl) phosphine (TCEP) aqueous solution was prepared as follows. To a solution of tris (2-carboxyethyl) phosphine (TCEP) hydrochloride (1.0 g, 3.49 mmol) in water (6.8 ml) was added triethylamine (1.64 ml) to pH 7, and 0.4M tris (2-carboxyl An ethyl) phosphine (TCEP) aqueous solution was obtained.
- the 1M 2,2′-azobis-2- (2-imidazolin-2-yl) propane (VA-044) aqueous solution was prepared as follows. Water (0.309 ml) was added to 2,2′-azobis-2- (2-imidazolin-2-yl) propane (VA-044) hydrochloride (100 mg, 0.309 mmol) and 1M 2,2′-azobis-2 An aqueous solution of-(2-imidazolin-2-yl) propane (VA-044) was prepared.
- an aspartic acid derivative having a thioester in the side chain was translated and introduced as a carboxylic acid derivative, Cys or a derivative thereof was translated and introduced as an amino group into the N-terminal, and then a method was cyclized.
- Cys was introduced at the N-terminus by the initiator-read-through method, the target cyclic peptide was obtained by translating Asp (SBn) into the peptide.
- RNA stability evaluation in desulfurization reaction The RNA compound was subjected to desulfurization reaction conditions to confirm the stability.
- 2-1.5′-AGCUUAGUCA-Puromycin-3 ′ (Compound RP-1, (SEQ ID NO: 178)) Synthesis Using Puromycin CPG (22.7 mg, 0.999 ⁇ mol) manufactured by Glen Research, Inc. RNA binding elongation was performed by a machine.
- the amidite reagent used for the extension reaction of A, G, C, U uses Glen Research A-TOM-CE phosphoramidide, G-TOM-CE phosphoramidide, C-TOM-CE phosphoramidide, U-TOM-CE phosphoramidide, This was carried out using 5-benzylthio-1H-tetrazole as the condensation activator.
- Example 17 Evaluation of protein denaturation under cyclization desulfurization reaction Protein denaturation by a desulfurization reaction solution was evaluated (FIG. 34).
- As a model system the influence of desulfurization reaction conditions on IL-6R in an IL-6 and IL-6R interaction detection system by electrochemiluminescence immunoassay was evaluated.
- 750 ng of anti-IL-6R antibody clone number 17506 was stored overnight at 4 degrees in MSD Multi-array 384 well (MSD), and the antibody was immobilized. The plate was washed three times with 80 ⁇ l PBST to remove unreacted antibodies, and then blocked with 2% skim milk for 1 hour.
- the plate was washed 3 times with 80 ⁇ l of PBST blocking agent, 10 ⁇ l of soluble human IL-6R (IL-6R) was added at 25 nM, and the mixture was shaken at room temperature for 50 minutes.
- the plate was washed 3 times with 80 ⁇ l of PBST, desulfurization reaction solution (25 mM HEPES-K (pH 7.6), 200 mM TCEP, 250 mM VA-044, 10 mM Cys) was added, and the mixture was shaken at room temperature for 50 minutes.
- the plate was washed 3 times with 80 ⁇ l PBST, 10 ⁇ l human IL-6-BAP (500 nM) was added and shaken at room temperature for 50 minutes.
- the plate was washed 3 times with 80 ⁇ l PBST, 10 ⁇ L SULFO-TAG StAv (MSD, cat # R32AD-5, 1 ⁇ g / mL) was added, and the mixture was shaken at room temperature for 50 minutes.
- the plate was washed three times with 80 ⁇ l of PBST, 35 ⁇ l of 2X read buffer (MSD, R92SC-3) was added, and measurement was performed with a SECTOR Imager 2400 (MSD) (FIG. 34).
- MSD SECTOR Imager 2400
- Example 18 Comparison with thioetherification method The thioether cyclization method and the cyclization method of the present invention were compared. A highly lipid-soluble thioether cyclized peptide, which is thought to have a high lipid membrane permeation rate, was synthesized. 1.
- the resin in the tube is filtered through a synthesis column to remove the resin, and the reaction solution is added to a tube containing diisopropylethylamine (1 mL) solution to cyclize the chloroacetyl group and cysteine. went.
- the solvent was distilled off.
- the resulting crude product was purified by adding piperidine (1.9 eq.) O- (7-other 1Hbenzotriazol-1-yl) -N, N, N, N tetramethyluronium hexafluorophosphate (HATU) in DMF. Piperidine amidation of the C-terminal carboxylic acid was carried out at (1.7 eq.).
- the solvent was distilled off with Genevac.
- the obtained crude product was dissolved in dimethyl sulfoxide, and the obtained peptide solution was purified by high-speed reverse phase chromatography (HPLC).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicinal Chemistry (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
一方、ペプチドの環化方法としてドラックライクな環化法であるアミド環化が実現できた画期的な報告もなされている(非特許文献21、非特許文献25、非特許文献26、非特許文献27)が、それらはいずれも、主鎖アミド結合を切断させた結果得られる活性種とアミノ酸主鎖アミノ基を化学反応させた構造を生成させるため、本方法を、そのままDisplay libraryにおける環化方法としては適用することはできない。これらの手法は、シクロスポリンAなど多くの天然物の、主鎖のカルボン酸と主鎖のアミノ基で縮合環化させる手法として有用である。一方、Display libraryでは、主鎖カルボン酸末端はmRNAと結合させる必要があるため、このように主鎖アミド結合の分解により活性種を発生させる手法を用いることはできない。
また、環化部位の構造を変化させると、当該環化部位を有するペプチドが持つ活性(薬効の強度)が大きく低下してしまうことが報告されている(非特許文献24)。この例では、環化部位を有するペプチドを取得した後、膜透過性と代謝安定性に優れたペプチドとするために、その環化部位を改変することが困難であることが示されている。
医薬品の開発に利用できる膜透過性と代謝安定性に優れた環状部位を有するペプチドライブラリーが求められるが、そのようなペプチドライブラリーの確立には様々な点で改善の余地がある。
一方、中分子ペプチドdisplayの限られた規模(ドラックライクネスを考慮するとペプチド鎖長に制限があることを指す)を有効に利用するためには、ライブラリーに全く異なる構造のペプチド(非天然型アミノ酸を含んだペプチド)を多く含ませることが肝要である。側鎖の性質の異なるアミノ酸を多種類導入することも然ることながら、制限された分子量の中で固定された部位を少なくして可変領域を拡大すること、主鎖構造の異なるペプチドを含ませることにより様々な標的に対して結合ペプチドが得られる可能性が高められることになり価値がある。この観点から有効な方法として例えば、様々な環化部位を有するペプチド、分枝ペプチドをディスプレイさせることなどが挙げられる。
本発明はこのような状況に鑑みてなされたものであり、本発明はペプチドディスプレイライブラリー構築のための、ペプチド化合物の新規な環化方法、分枝ペプチド合成法を提供し、また、これらを用いてドラッグライクなディスプレイライブラリーおよびドラッグライクなペプチド化合物を提供することを課題とする。
〔1〕
環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、C末端側に側鎖の1つに反応点を有するアミノ酸残基又はアミノ酸類縁体残基、及び、N末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)N末端側のアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点と、C末端側の側鎖に有するアミノ酸残基又はアミノ酸類縁体残基の反応点とを結合させ、アミド結合又は炭素-炭素結合を形成させる工程
を含む、前記方法。
〔2〕
環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基、及び、アミン近傍に反応補助基を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)前記反応補助基を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体と、活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基をアミド結合させて環状化合物を得る工程
を含む、〔1〕に記載の方法。
〔3〕
活性エステルがチオエステルである、〔2〕に記載の方法。
〔4〕
前記反応補助基が、SH基である、〔2〕又は〔3〕に記載の方法。
〔5〕
前記環状化合物を得る工程の後、反応補助基を除去する工程を含む、〔3〕または〔4〕に記載の方法。
〔6〕
前記アミン近傍に反応補助基を有するアミノ酸、アミノ酸類縁体又はN末端カルボン酸類縁体が、下記一般式で表される化合物N-1または化合物N-2である、〔2〕から〔5〕のいずれかに記載の方法;

R2、R3は、それぞれ独立して、水素原子、または、置換基を有していてもよい、アルキル基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基、アラルキル基もしくはシクロアルキル基を示し;またはR2とR3が環を形成した置換基、またはR2もしくはR3がR4と環を形成した置換基を示し;
R4は、置換基を有していてもよいアルキレン基、置換基を有していてもよいアリーレン基または置換基を有していてもよい2価のアラルキル基を示し;
R11、R12は、それぞれ独立して、単結合、置換基を有していてもよいアルキレン基、置換基を有していてもよいアリーレン基または置換基を有していてもよい2価のアラルキル基を示す。)。
〔7〕
前記活性エステル基を側鎖に有するアミノ酸又はアミノ酸類縁体が、下記一般式で表される化合物C-1である、〔2〕~〔6〕のいずれかに記載の方法;

R26は、置換基を有していてもよいアルキレン基、置換基を有していてもよいアリーレン基または置換基を有していてもよい2価のアラルキル基を示し;
R2、R3は、それぞれ独立して、水素原子または置換基を有していてもよいアルキル基を示す。)。
〔8〕
前記環状部を有するペプチド化合物の環状部を構成する、アミノ酸残基及び/又はアミノ酸類縁体残基、あるいは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体の総数が、5~12である、〔1〕から〔7〕のいずれかに記載の方法。
〔9〕
前記環状部を有するペプチド化合物を構成する、アミノ酸残基及び/又はアミノ酸類縁体残基、あるいは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体の総数が、9~13である、〔1〕~〔8〕のいずれかに記載の方法。
〔10〕
環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、あるいは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基、及び、N末端の主鎖アミノ基を有するアミノ酸残基、あるいは、主鎖もしくは側鎖にアミノ基を有するアミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)N末端のアミノ酸残基、N末端のアミノ酸類縁体残基又はN末端カルボン酸類縁体と活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体をアミド結合させて環状化合物を得る工程
を含む、〔1〕に記載の方法。
〔11〕
活性エステル基がアルキルチオエステル基又はアラルキルチオエステル基であって、工程1)の翻訳合成後に活性化剤を添加させることにより、より活性な活性エステル基に変換させる工程を含む、〔10〕に記載の方法。
〔12〕
活性剤が、アリールチオール又はN-ヒドロキシスクシンイミドである、〔11〕に記載の方法。
〔13〕
翻訳されたチオエステルとの反応性が高い活性化剤と、環化させるアミンとの反応性が高い活性化剤を添加させ、より活性の高い活性エステル基に変換させる工程である、〔12〕に記載の方法。
〔14〕
アリールチオエステルで活性エステル基に変換させた後、オキシマおよびその誘導体で更に活性の高いエステル基に変換させる工程である、〔13〕に記載の方法。
〔15〕
工程1)のN末端部位の翻訳合成が、フォルミルメチオニン以外の翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体をアシル化した翻訳開始tRNAを用いて導入する方法にて行われる、〔1〕から〔14〕のいずれかに記載の方法。
〔16〕
工程1)のN末端部位の翻訳合成が、開始コドンを読み飛ばし、N末端にMet以外の翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体を導入する方法を用いて行われる、〔10〕から〔14〕のいずれかに記載の方法。
〔17〕
工程1)のN末端部位の翻訳合成が、アミノペプチダーゼにてN末端のアミノ酸、アミノ酸類縁体又はカルボン酸類縁体を切断する方法を用いて行われる、〔10〕から〔14〕のいずれかに記載の方法。
〔18〕
前記N末端部位の翻訳合成が、メチオニン アミノペプチダーゼにて処理することによりN末端のフォルミルMetを除去しN末端に他のの翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体を導入する方法を用いて行われる、〔17〕に記載の方法。
〔19〕
前記N末端部位の翻訳合成が、Metの代わりにノルロイシンを含む翻訳系にて翻訳され、メチオニン アミノペプチダーゼにて処理することによりN末端のフォルミルノルロイシンを除去しN末端に他のの翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体を導入する方法を用いて行われる、〔10〕~〔14〕のいずれかに記載の方法。
〔20〕
前記N末端のアミノ酸、アミノ酸類縁体又はカルボン酸類縁体の除去に、さらに、ペプチドデフォルミラーゼが用いられる、〔17〕から〔19〕のいずれかに記載の方法。
〔21〕
前記環状部を有するペプチド化合物が、さらに直鎖部を有する、〔1〕から〔2〕0のいずれかに記載の製造方法。
〔22〕
前記非環状ペプチド化合物がα-ヒドロキシカルボン酸、及び、側鎖に保護されていてもよいアミノ基を有するアミノ酸又はアミノ酸類縁体を含み、工程2)の環状化合物を形成する工程の後で、工程3)α―ヒドロキシカルボン酸部位と側鎖に保護されていてもよいアミノ基を有するアミノ酸又はアミノ酸類縁体部位を化学反応させて分枝部位を形成させる工程を含む、〔1〕から〔21〕のいずれかに記載の方法。
〔23〕
環状部と直鎖部とを有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基、N末端カルボン酸類縁体並びにα-ヒドロキシカルボン酸により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、該非環状のペプチド化合物が、
i)C末端側に側鎖の1つに反応点を有するアミノ酸残基(又はアミノ酸類縁体残基)、及び、N末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含み、
ii)前記i)に記載の2つの反応点の間にα位にRf5を有するα-ヒドロキシカルボン酸(Rf5は水素原子、置換されてもよいアルキル基、アラルキル基、ヘテロアリール基、シクロアルキル基、アリケニル基、アルキニル基から選択される)、及び、非環状ペプチド化合物中に保護されていてもよいアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基を含む、
非環状のペプチド化合物である、工程;
2)N末端側のアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点と、C末端側の側鎖に有するアミノ酸残基又はアミノ酸類縁体残基の反応点とを結合させる、環化反応工程;
3)工程1)のii)に記載のα-ヒドロキシカルボン酸のエステル結合を切断してチオエステル基を発生させる工程
4)工程3)において発生させたチオエステル基と工程1)のii)に記載のアミノ基を結合させる環化反応工程、
を含む、方法。
〔24〕
工程1)のii)に記載のα-ヒドロキシカルボン酸とアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基の間に含まれるアミノ酸残基及び/又はアミノ酸類縁体残基の数が7つ以下である、〔23〕に記載の方法。
〔25〕
工程1)のii)に記載のα-ヒドロキシカルボン酸が、Cys-Pro-α-ヒドロキシカルボン酸として非環状ペプチド化合物中に含まれる、〔23〕又は〔24〕に記載の方法。
〔26〕
工程1)のi)に記載のN末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体が反応補助基を有している、〔23〕から〔25〕のいずれかに記載の方法。
〔27〕
工程1)のi)に記載のN末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体が反応補助基を有さず、ii)に記載のアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基のアミノ基が保護基を有し、活性化剤を添加することによって、工程2)の環化反応が行われ、工程2)の環化反応後、工程3)の環化反応前に、前記ii)に記載のアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基のアミノ基の保護基を除去する工程を含む、〔23〕から〔26〕のいずれかに記載の方法。
〔28〕
環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、側鎖の1つに反応点を有するアミノ酸残基又はアミノ酸類縁体残基、及び、N末端にもう1つの反応点を有するアミノ酸、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)N末端のアミノ酸残基、N末端のアミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点と、側鎖に1つの反応点を有するアミノ酸残基又はアミノ酸類縁体の反応点とを炭素‐炭素結合させる工程
を含む、〔1〕に記載の方法。
〔29〕
N末端のアミノ酸残基、N末端のアミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点として炭素‐炭素2重結合を選択し、側鎖に1つの反応点を有するアミノ酸残基又はアミノ酸類縁体残基の反応点としてアリールハライドを選択して、遷移金属を触媒とした炭素-炭素結合反応により環化反応を実施する工程を含む、〔28〕に記載の方法。
〔30〕
遷移金属を触媒とした炭素-炭素結合反応が、Pdを触媒としたHeck型の化学反応である、〔29〕に記載の方法。
〔31〕
前記環状部を有するペプチド化合物の環状部を形成するアミノ酸又はアミノ酸類縁体の合計が5~12となる位置に環化反応を実施するための反応点を有する、〔1から〔30〕のいずれかに記載の方法。
〔32〕
前記環状部を有するペプチド化合物のアミノ酸及びアミノ酸類縁体残基の総数が9~13である、〔31〕に記載の方法。
〔33〕
ペプチド化合物のC末端と翻訳合成に用いた鋳型とがスペーサーを介して結合しているペプチド化合物-核酸複合体を製造することを特徴とする、〔1〕~〔32〕のいずれかに記載の方法。
〔34〕
ペプチド化合物-核酸複合体が、翻訳合成に用いられる非環状ペプチド化合物をコードする核酸の3'末端にリンカーを介してピューロマイシンが結合している核酸を用いて合成される、〔33〕に記載の方法。
〔35〕
スペーサーがペプチド、RNA、DNA、またはヘキサエチレングリコールのポリマー、又は、これらの組合せである、〔33〕又は〔34〕に記載の方法。
〔36〕
ペプチド化合物を、互いに異なる配列を有する複数の核酸から成る核酸ライブラリーを翻訳して製造することを特徴とする、〔1〕から〔35〕のいずれかに記載の方法。
〔37〕
〔1〕から〔36〕のいずれかに記載の製造方法により作製されるペプチド化合物又はペプチド化合物-核酸複合体。
〔38〕
異なる構造を有する複数の〔37〕記載のペプチド化合物又はペプチド化合物-核酸複合体からなるライブラリー。
〔39〕
以下の(i)~(iv)を有することを特徴とする、環状部を有するペプチド化合物;
(i)アミノ酸及びアミノ酸類縁体残基数の合計が5~12からなる環状部を含み、アミノ酸及びアミノ酸類縁体の総数が9~13である、
(ii)N置換アミノ酸を少なくとも2つ含み、N置換されていないアミノ酸を少なくとも1つ含む、
(iii)ClogP値が6以上である、
(iv)環状部を形成しているアミノ酸又はアミノ酸類縁体の結合が、アミノ酸又はアミノ酸類縁体の側鎖にある活性エステル基と、他のアミノ酸又はアミノ酸類縁体のアミン基とからなる結合を少なくとも1つ有する。
〔40〕
ペプチド化合物に含まれるアミノ酸及びアミノ酸類縁体が、翻訳合成可能なアミノ酸又はアミノ酸類縁体、若しくは、翻訳可能なアミノ酸又はアミノ酸類縁体の側鎖またはN置換部位を化学修飾することによって得られるアミノ酸又はアミノ酸誘導体から選択されるアミノ酸又はアミノ酸類縁体である、〔39〕に記載のペプチド化合物。
〔41〕
さらに、アミノ酸及びアミノ酸類縁体残基数の合計が1~8からなる直鎖部を少なくとも1つ含む、〔39〕又は〔40〕に記載のペプチド化合物。
〔42〕
環状部を形成しているアミノ酸又はアミノ酸類縁体の結合がアミド結合又は炭素-炭素結合である、〔39〕から〔41〕のいずれかに記載のペプチド化合物。
〔43〕
環状部が下記一般式(I)で示される交差ユニットを含む、〔39〕から〔42〕のいずれかに記載のペプチド化合物;

R51は、置換基を有していても良いC1-C6アルキル基、C5-C10アリール基、アラルキル基、エステル基、又は式1で示されるアミドであり、
R52は、置換基を有していても良いC1-C6アルキル基、アリール基、またはアラルキル基であり、
R53は、置換基を有していても良いC1-C6アルキル基、もしくは水素原子であるか、または、R53とR51とが互いに結合してC3-C5のアルキレン基を形成し窒素原子を含む5員~7員環を形成していてもよく、
R54は、0-8アミノ酸残基より構成されるペプチドであり、
R55は、置換基を有していても良いC1-C6アルキル基、C5-C10アリール基、アラルキル基、エステル基、置換基を有していても良いアミド基であり、
*は、環状部における結合部位を示す。)。
〔44〕
〔39〕から〔43〕のいずれかに記載のペプチド化合物を含む医薬組成物。
〔45〕
前記医薬組成物が経口剤である、〔44〕の医薬組成物。
〔46〕
〔39〕から〔45〕のいずれかに記載のペプチド化合物の製造方法であって、工程(i)~(v)を含む方法:
(i)アミノ酸及びアミノ酸類縁体の総数が9~13である非環状ペプチド化合物を翻訳合成して、当該非環状ペプチド化合物とそれをコードする核酸配列を有する複合体がリンカーを介して結合している非環状ペプチド化合物-核酸複合体を形成する工程;
(ii)工程(i)で翻訳合成された複合体の非環状ペプチド化合物をアミド結合又は炭素-炭素結合によって環化し、環状部のアミノ酸及びアミノ酸類縁体の残基数の合計が5~12となる環状化合物を形成する工程;および、
(iii)工程(ii)で得られた環状部を有するペプチド化合物-核酸複合体ライブラリーと生体内分子とを接触させ、当該生体内分子に対して結合活性を有する複合体を選択する工程。
〔47〕
さらに、以下の工程を含む、〔46〕に記載の製造方法;
(iv)前記工程(iii)で選択された複合体の核酸配列からペプチド化合物の配列情報を得る工程、および、
(v)前記工程(iv)で得られた配列情報に基づきペプチド化合物を化学合成する工程。
〔48〕
前記非環状ペプチド化合物がα-ヒドロキシカルボン酸、及び、保護されていてもよい側鎖にアミノ基を有するアミノ酸又はアミノ酸類縁体を含み、工程(ii)の環状化合物を形成する工程の後で、α―ヒドロキシカルボン酸部位と側鎖にアミノ基を有するアミノ酸又はアミノ酸類縁体部位を化学反応させて分枝部位を形成させる工程を含む、〔46〕又は〔47〕に記載の方法。
〔49〕
生体内分子が、分子量500未満の化合物が結合し得る領域を有していない分子である、〔46〕から〔48〕いずれかに記載の製造方法。
〔50〕
生体内分子に対して結合活性を有する複合体が、さらに該生体内分子が他の生体内分子との結合を阻害する活性を有する、〔46〕から〔49〕のいずれかに記載の製造方法。
〔51〕
環化反応するN末端側のアミノ酸又はアミノ酸類縁体が、前記化合物N-1または化合物N-2で表わされる化合物から選択されるアミノ酸又はアミノ酸類縁体であり、C末端側のアミノ酸又はアミノ酸類縁体が前記化合物C-1で表わされる化合物から選択されるアミノ酸又はアミノ酸類縁体であり、工程(ii)の環状化合物を得る工程の後、反応補助基を除去する工程を含む、〔46〕から〔50〕のいずれかに記載の製造方法。
〔52〕
核酸配列が3'末端にスペーサーを有し、翻訳合成されるペプチド化合物のC末端が、当該スペーサーを介して、当該核酸配列と複合体を形成する、〔46〕から〔51〕のいずれかに記載の製造方法。
〔53〕
ペプチド化合物-核酸複合体が、翻訳合成に用いられる非環状ペプチド化合物をコードする核酸の3'末端にリンカーを介してピューロマイシンが結合している核酸を用いて合成される、〔52〕に記載の方法。
〔54〕
スペーサーがペプチド、RNA、DNA、またはヘキサエチレングリコールのポリマーである、〔52〕又は〔53〕に記載の製造方法。
・環状部を有するペプチド化合物
本発明の環状部を有するペプチド化合物とは、アミノ酸あるいはアミノ酸類縁体がアミド結合あるいはエステル結合して形成される化合物であり、環状部がアミド結合あるいは炭素‐炭素結合形成反応などの共有結合を介して環化された化合物である。当該化合物をさらに化学修飾して得られる化合物も本発明のペプチド化合物に含まれる。本発明のペプチド化合物は直鎖部を有していてもよく、例えば、スキームA(スキームA-1、A-2)のように記載することができる。環状部を有するペプチド化合物は、さらに、直鎖部を有していてもよい。アミド結合あるいはエステル結合の数(アミノ酸又はアミノ酸類縁体の数・長さ)を特に限定しないが、直鎖部を有する場合、環状部と直鎖部を併せて30残基以内が好ましい。高い膜透過性を獲得するためには、環化部位と直鎖部位を併せた総アミノ酸数は13残基以下であることがより好ましい。高い代謝安定性を獲得するためには、総アミノ酸数が9以上であることがより好ましい。膜透過性と代謝安定性の両立(ドラックライクネス)を考慮すると上記に加えて環状部を構成するアミノ酸及びアミノ酸類縁体の数は5~12であることが好ましい。さらに、上の記載に加えて環状部を構成するアミノ酸及びアミノ酸類縁体の数はより好ましくは5~11、さらに7~11残基が好ましい。特に9~11残基が好ましい。直鎖部のアミノ酸及びアミノ酸類縁体の数(ユニットの数)は0~8であることが好ましい。さらに、0~3が好ましい。尚、本願では特に限定しない限り、アミノ酸にはアミノ酸類縁体も含まれる場合があるものとする。また、ここで、「ドラックライクネス」あるいは「ドラックライク」とは、ペプチド化合物が、少なくとも経口剤、あるいは、細胞内蛋白、核酸、膜蛋白の細胞内領域又は膜蛋白の膜貫通ドメインを標的とした場合に、医薬品として利用できる程度の膜透過性と代謝安定性を有することを意味する。
翻訳後修飾とは、翻訳後にリボゾームの作用以外で自動的あるいは別試薬を添加させて生じさせる化学反応を指し、例えば環化反応や脱保護反応を挙げることができる。
翻訳後環化とは、翻訳後修飾の中で環形成反応を伴うものである。(スキームA:本発明のペプチド化合物を説明したスキームである。白丸ユニット、黒丸ユニット、三角ユニット、四角ユニットはそれぞれアミノ酸あるいはアミノ酸類縁体を意味する。また、それぞれのユニットは同一あるいは別々のアミノ酸あるいはアミノ酸類縁体を意味する。三角ユニットはN末端カルボン酸類縁体の場合もあり得る。例えば、8個の黒丸ユニットはそれぞれ別種類のアミノ酸あるいはアミノ酸類縁体であってもよく、その中のいくつか、あるいは全てが同一のものであってもよい。また、翻訳後に実施可能な化学修飾により、アミノ酸あるいはアミノ酸類縁体は、別骨格を有する化合物へ化学変換や骨格変換されていてもよい。ここで、1ユニットとは、翻訳後修飾が終了した時点でのアミノ酸あるいはアミノ酸類縁体が、これに該当するが、1つのtRNAによって翻訳されたアミノ酸あるいはアミノ酸類縁体が、翻訳後修飾により、別骨格を有する化合物へ化学変換や骨格変換されたものも、これに含まれる。ユニットの数についても同様に計算される。尚、本願では特に限定しない限り、アミノ酸にはアミノ酸類縁体も含まれるものとする。また、本明細書中において、翻訳後環化は単に環化という場合がある。
アミノ酸とはα、βおよびγアミノ酸であり、天然型アミノ酸(本願では天然型アミノ酸とはタンパク質に含まれる20種類のアミノ酸を指す。具体的にはGly、Ala、Ser、Thr、Val、Leu、Ile、Phe、Tyr、Trp、His、Glu、Asp、Gln、Asn、Cys、Met、Lys、Arg、Proを指す。)に限定されず、非天然型アミノ酸であってもよい。α-アミノ酸の場合、L型アミノ酸でもD型アミノ酸でもよく、α,α-ジアルキルアミノ酸でもよい。アミノ酸側鎖の選択は特に制限を設けないが、水素原子の他にも例えばアルキル基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基、アラルキル基、シクロアルキル基から自由に選択される。それぞれには置換基が付与されていてもよく、それら置換基も例えば、N原子、O原子、S原子、B原子、Si原子、P原子を含む任意の官能基の中から自由に選択される(すなわち、置換されていてもよいアルキル基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基、アラルキル基、シクロアルキル基など)。
ペプチド化合物を構成する「アミノ酸」、「アミノ酸類縁体」にはそれぞれに対応する全ての同位体を含む。「アミノ酸」、「アミノ酸類縁体」の同位体は、少なくとも1つの原子が、原子番号(陽子数)が同じで,質量数(陽子と中性子の数の和)が異なる原子で置換されたものである。本発明ペプチド化合物を構成する「アミノ酸」、「アミノ酸類縁体」に含まれる同位体の例としては、水素原子、炭素原子、窒素原子、酸素原子、リン原子、硫黄原子、フッ素原子、塩素原子などがあり、それぞれ、2H、3H、13C、14C、15N、17O、18O、31P、32P、35S、18F、36Cl等が含まれる。
本発明のペプチド化合物が良好な膜透過性を有するためには、ペプチド化合物中に含まれる総アミノ酸数が13以下であることが好ましい。
それぞれのユニット(アミノ酸残基)の選択に特に制限はないが、翻訳後化学修飾が全て完了した形式の分子の形(主鎖構造)に考慮し直して、形成される分子のCLogP(コンピューターで計算した分配係数であって、Daylight Chemical Information Systems, Inc. 社のDaylight Version 4.9を用いて計算した)が6を超えるように選択することが好ましい。特に良好な膜透過性を確保するために、CLogPは8を超え、15を超えないように選択することが好ましい。
本発明のペプチド化合物が良好な代謝安定性を有するためには、ペプチド化合物中に含まれる総アミノ酸数が9以上であることが好ましく、11以上であることがより好ましい。総アミノ酸数が9以上のペプチド化合物であれば、上述の膜透過性を有するための条件は、当該ペプチド化合物の代謝安定性に影響しない。

本発明の環状部を有するペプチド化合物は、以下に記載の方法を利用して製造することが可能である。
例えば、以下の製造方法を含む製造方法を挙げることができる。
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、C末端側に側鎖の1つに反応点を有するアミノ酸残基又はアミノ酸類縁体残基、及び、N末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)N末端側のアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点と、C末端側の側鎖に有するアミノ酸残基又はアミノ酸類縁体残基の反応点とを結合させ、アミド結合又は炭素-炭素結合を形成させる工程
を含む、前記方法。
本発明の翻訳合成には、公知の方法を利用することができる。
本来は、一般的に翻訳開始アミノ酸としてメチオニンをN末端アミノ酸として翻訳されるが、他の所望のアミノ酸をアミノアシル化した翻訳開始tRNAを用いて翻訳させることによってN末端を所望のアミノ酸として翻訳させる方法を使用することもできる。非天然アミノ酸のN末端導入ではアミノ酸の許容度が伸長時よりも高く、天然型アミノ酸と大きく構造の異なるアミノ酸、アミノ酸類縁体を利用することが知られている(非特許文献:J Am Chem Soc. 2009 Apr 15;131(14):5040-1.Translation initiation with initiator tRNA charged with exotic peptides.Goto Y, Suga H.)。例えば、本発明において、メチオニン以外の翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸誘導体をN末端に導入する方法としては、以下の方法がある。翻訳開始tRNAとして、三角ユニットをアシル化したtRNAをメチオニン、フォルミルドナーもしくはメチオニルトランスフェラーゼを抜いた翻訳系に添加し、翻訳開始コドン(例えばATG)に三角ユニットをコードさせ翻訳させることによって末端を三角ユニットとする環化前のペプチド化合物又はペプチド化合物ライブラリーを構築する。アンチコドンがCAUでない翻訳開始tRNAと当該アンチコドンに対応するコドンの様々な組み合わせを翻訳開始tRNA及び開始コドンの組み合わせとして利用するとN末端に多様性をもたせることが可能である。つまり、アンチコドンの異なる複数種類の翻訳開始tRNAに所望のアミノ酸、アミノ酸類縁体又はN末端カルボン酸類縁体をそれぞれアミノアシル化し、これに対応するコドンを開始コドンとしたmRNA又はmRNAライブラリーを翻訳させることによってN末端残基が一種類に限定されない環化前のペプチド化合物又はペプチド化合物のライブラリーを作製することができる。具体的には例えば、Mayer C,et al. Anticodon sequence mutants of Escherichia coli initiator tRNA: effects of overproduction of aminoacyl-tRNA synthetases, methionyl-tRNA formyltransferase, and initiation factor 2 on activity in initiation.Biochemistry. 2003, 42, 4787-99.(CAU以外のanticodonを持つ開始tRNAの変異大腸菌によるf-Met以外のアミノ酸からの翻訳開始と同codonを途中に含む蛋白の発現。)に記載の方法を用いて環化前のペプチド化合物又はペプチド化合物ライブラリーを作製することができる。





R3はドラックライクなアミノ酸の側鎖の定義と同様に定義されるが、例えばC1-C4アルキル基、ハロゲンなどで置換されてもよいC1-C4アルキル基から選択されるが、特に水素原子が好ましい。なお、R3基の立体はR3基が水素原子と仮定した場合のL型、D型アミノ酸に対応するもの両方が許容されるが、L型アミノ酸に対応するものが好ましい。
化合物C-1、化合物C-2、化合物C-3と同様に、化合物COH-1、化合物COH-2、化合物COH-3から選択することもできる。


R101、R102のうち1つが水素原子、R103、R104のうち1つが水素原子、R106、R107のうち1つが水素原子であることがより好ましい。また水素原子の配置はこれらがL型アミノ酸と同じ立体配置になるように配置されることが好ましい。





スキームB


交差ユニットに活性エステル基を有するユニットが選択される場合、交差ユニットは化合物C-1、あるいは化合物C-2、化合物C-3やあるいは化合物COH-1、化合物COH-2、化合物COH-3から選択することもできる。これら6種類の化合物を選択する場合には常に、そのC末端側直後のアミノ酸はN-アルキル化されたユニットの中から選択することが好ましい。この制限は、アスパルチミド形成の副反応を避ける目的であり、これら6種類の化合物が選択された場合の共通の好ましい選択となる。代謝安定性の観点から、化合物C-1、あるいは化合物C-2、化合物C-3がより好ましい。このような場合、三角ユニットは化合物Na-1、化合物Na-2、化合物Na-3の中から選択することもできる。三角ユニットとして、N末端(三角ユニット)を固定させない場合には、これらの化合物から複数個を同時に選択することもできる。
化合物Na-1および化合物Na-2、化合物Na-3のどちらでも側鎖アミノ基は保護されていてもよい。保護されている場合には、環化反応と同時あるいは事前に脱保護させてから環化反応を行う。保護基あるいは脱保護の条件は本明細書に記載の方法を利用することができる。
例えば化合物Na-4や化合物Na-5が選択された場合、三角ユニットには化合物COH-1、化合物COH-2、化合物COH-3などを選択することもできる。この場合、三角ユニットがN末端に存在してもよく、あるいは三角ユニットよりN末端側にも直鎖部が存在して、かつN末端にはN末端カルボン酸誘導体から選択されることが、ドラックライクの点から好ましい。

直鎖部2を発生させる手法として、α―ヒドロキシカルボン酸などの主鎖にアミノ基を持たずにエステル結合を翻訳合成により形成できるアミノ酸類縁体と保護されていてもよいアミノ基側鎖(保護基はアミノ基の保護基として作用し、翻訳合成されるアミノ酸を与えるものであれば、特に限定されない)を有するアミノ酸の両方を翻訳導入して翻訳後修飾させる手法を用いることが可能である(スキームE)。ディスプレイライブラリーへの適用時には、翻訳されるユニットから選択される。しかし、その後の最適化により得られるペプチド化合物では、当該ペプチド化合物そのものが翻訳合成されなくても、翻訳合成後の翻訳後修飾により得られるものも含まれる。α―ヒドロキシカルボン酸部位の側鎖は特に限定はされないが、スキームEに記載の手法では、特に、翻訳合成の点ではRe1が水素原子(グリコール酸(HOGly)そのもの)あるいは、側鎖にチオール基(SH基)あるいは保護されたチオール基を有しているものが好ましい。側鎖の立体配置も特に限定されないが、α位に水素原子が存在する場合にはL型アミノ酸と同様な立体配置を取ることがより好ましい。チオール基あるいは保護されたチオール基の配置も特に限定されないが、OH基のβ位、もしくはγ位に配置されていること(すなわち、Re1が保護されていてもよいメルカプトメチル基あるいはメルカプトエチル基)が特に好ましい。これらのチオール基もしくは保護されたチオール基は、α―ヒドロキシカルボン酸側鎖の置換されてもよいアルキル基、アルケニル基、アルキニル基、シクロアルキル基、アラルキル基、アリール基、ヘテロアリール基、から配置(付加)される。これらα―ヒドロキシカルボン酸側鎖の置換基としては、付加されたチオール基あるいは保護されたチオール基を除けば、ドラックライクなアミノ酸の側鎖で定義された置換基であることがより好ましい。

上記のようにヒドロキシカルボン酸とアミン側鎖部位の両方にSH基あるいは保護されたSH基などの反応補助基を有してもよく、もしくはいずれか片方のみに反応補助基を有していてもよく、もしくは両方ともに反応補助基を有していなくともよい。また、アミン側鎖部位に保護基を有していてもよく、保護基を有していなくてもよい。
またいずれの場合でも、反応を加速させる添加剤を加えてもよい。添加剤としては、例えば置換していてもよいアルキルチオール、置換されていてもよいアルケニルチオール基、置換されてもよいアルキニルチオール基、置換されていてもよいアラルキルチオール、置換されていてもよいアリールチオール、置換されてもよいヘテロアリールチオール基、置換されてもよいシクロアルキルチオール基でもよい。また、HOBt、HONSu、HOAt、パラニトロフェノールなど通常活性エステル化させる試薬、あるいはその誘導体でもよい。添加剤を加える場合には、翻訳後環化部の環化反応を実施後に本直鎖部2形成反応を実施することが好ましい。添加剤の置換基は、前記"アミノ酸"の側鎖で定義したのと同様、自由な置換基を選択することができる。
化合物F5のRf2基は保護されても良いメルカプトアルキル基から選択される。Rf2を含むアミノ酸の立体配置はL型が好ましい。さらに好ましくは保護されてもよいメルカプトメチル基あるいは2-メルカプトエチル基から選択される。Rf3基はRa13と同様に選択させることができる。好ましくはRa1と同様に選択させることができる。特に好ましくはメチル基あるいは、Rf4とで環を形成することが好ましい。環のサイズは3~8員環が好ましく、より好ましくは4~6員環である。これら環を形成する炭素原子はドラックライクなアミノ酸の側鎖で定義される置換基にて置換されていてもよい。5員環の代表例としてプロリンなどが好ましい。Rf4はRa4と同様に選択させることができる。
ここで記載されるアミノ基の保護基とは、単一の1級アミンもしくは2級アミンの反応性を不活性化もしくは低下させる官能基だけでなく、1つの官能基で複数のアミノ基もしくは他のヘテロ原子を同時に不活性する構造も保護基として定義する。例えば、チアゾリジン環、チアジナン環、オキサゾリジン環、イミダゾリジン環もアミノ基の保護基として含まれる。これらの保護基を形成する炭素原子は置換されていてもよい。




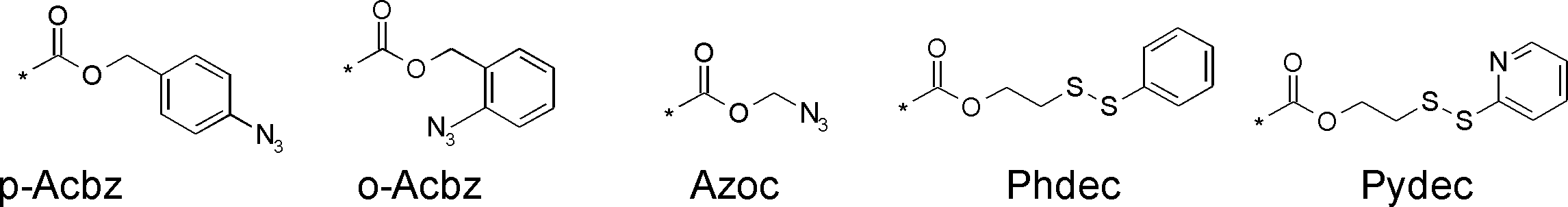
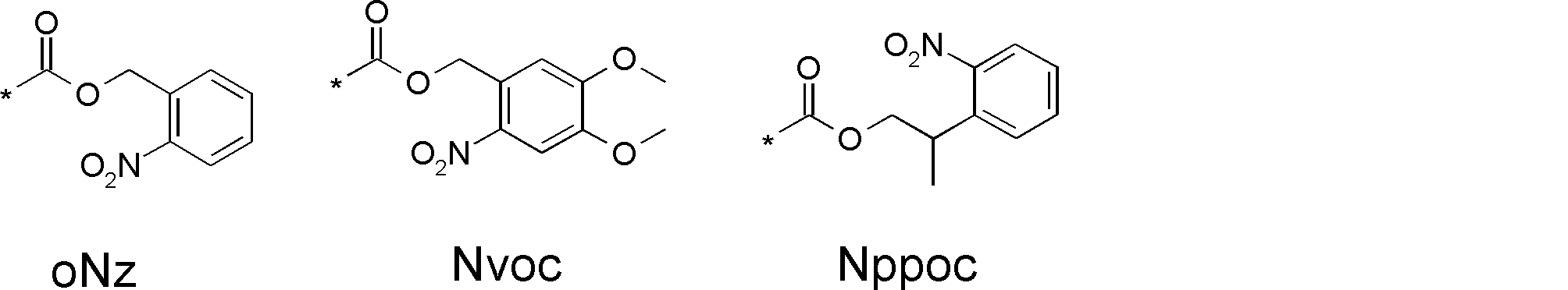
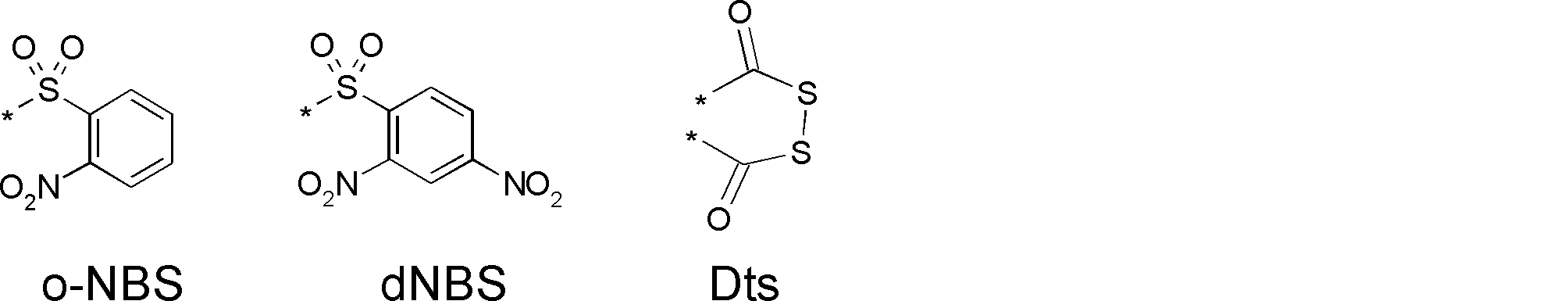


チオールの求核性を利用するNative Chemical Ligation(NCL)によるアミド化反応は、ペプチドおよびタンパク質の化学合成において有用な手法である。連続的なアミド化反応や任意のタイミングでNCLを行う場合、アミノ基およびチオール基にあらかじめ保護をしておくのは有用な手段であり、例えば非特許文献v1に記載されているペプチド鎖の多段階アミド化によるタンパク質合成や、非特許文献v2に記載されているタンパク質の化学修飾では、システインのアミノ基およびチオール基を同時に保護する構造としてチアゾリジン環が報告されている。
本手法は、酸性条件下でチアゾリジン環およびチアジナン環を開環し、アミノジスルフィド構造へと導くことを特徴としており、続く還元剤の添加によりアミノチオールへと導くことが可能である。
本手法は、水中、緩衝液中、もしくは水と混和した有機溶媒中で行う。使用する有機溶媒は、水と混和が可能で、基質と反応もしくは析出しないものを選択する。例えばN,N-ジメチルアセトアミド、N、N-ジメチルホルムアミド、アセトニトリルを使用する。水と有機溶媒の比率は、基質およびジスルフィド化合物の溶解性によって決まるが、例えば10mMのジチオジピリジンで反応を行う場合、ジチオジピリジンの溶解性の観点から、5%以上のN,N-ジメチルアセトアミドを使用するのが好ましい。
チアゾリジン環を開環するための反応温度は、反応を12時間以内に完結させることを目的とした場合、15℃以上が好ましい。更に熱に不安定なチアゾリジン環を有する化合物を取り扱う場合、15~50℃の範囲が好ましい。
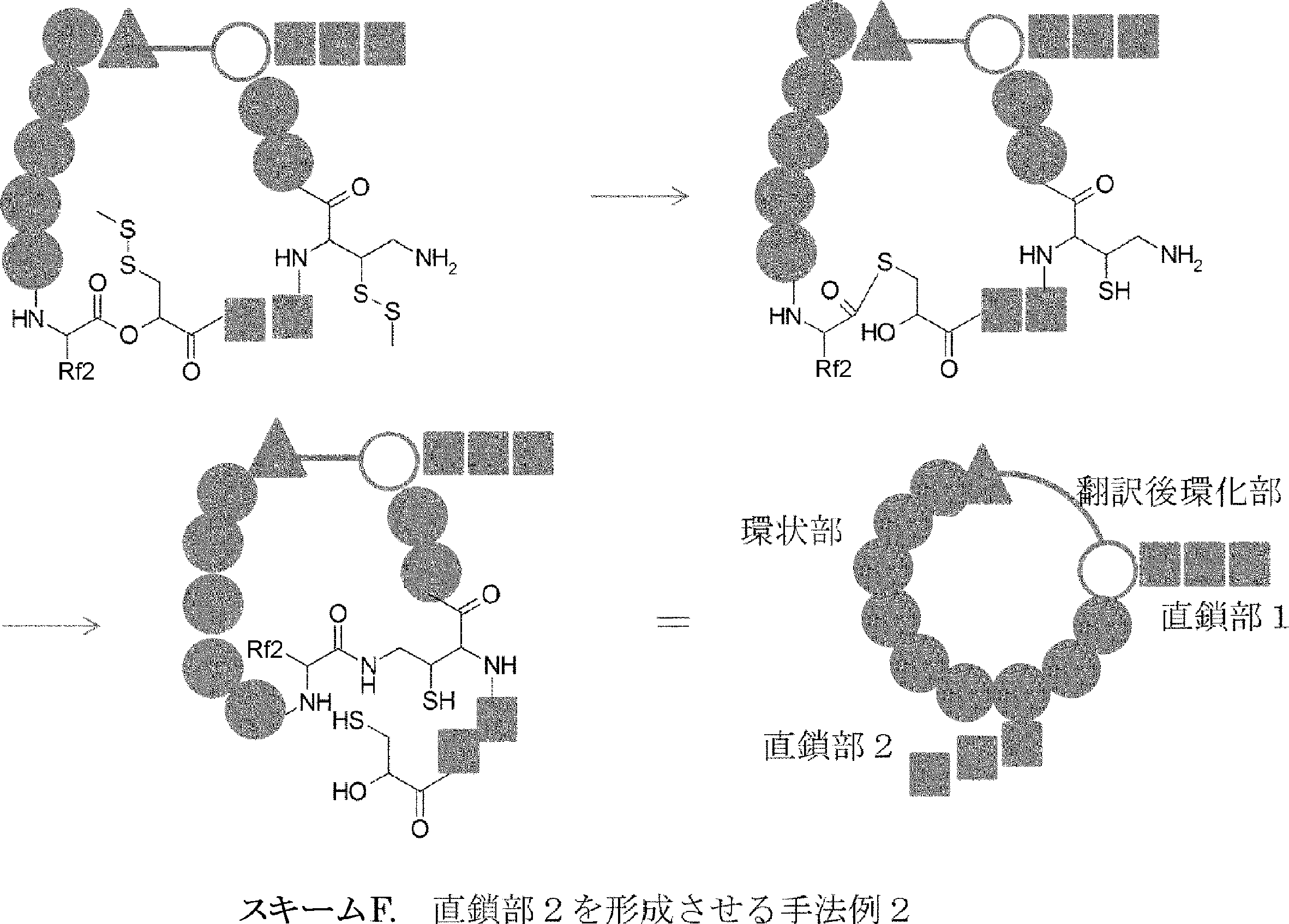

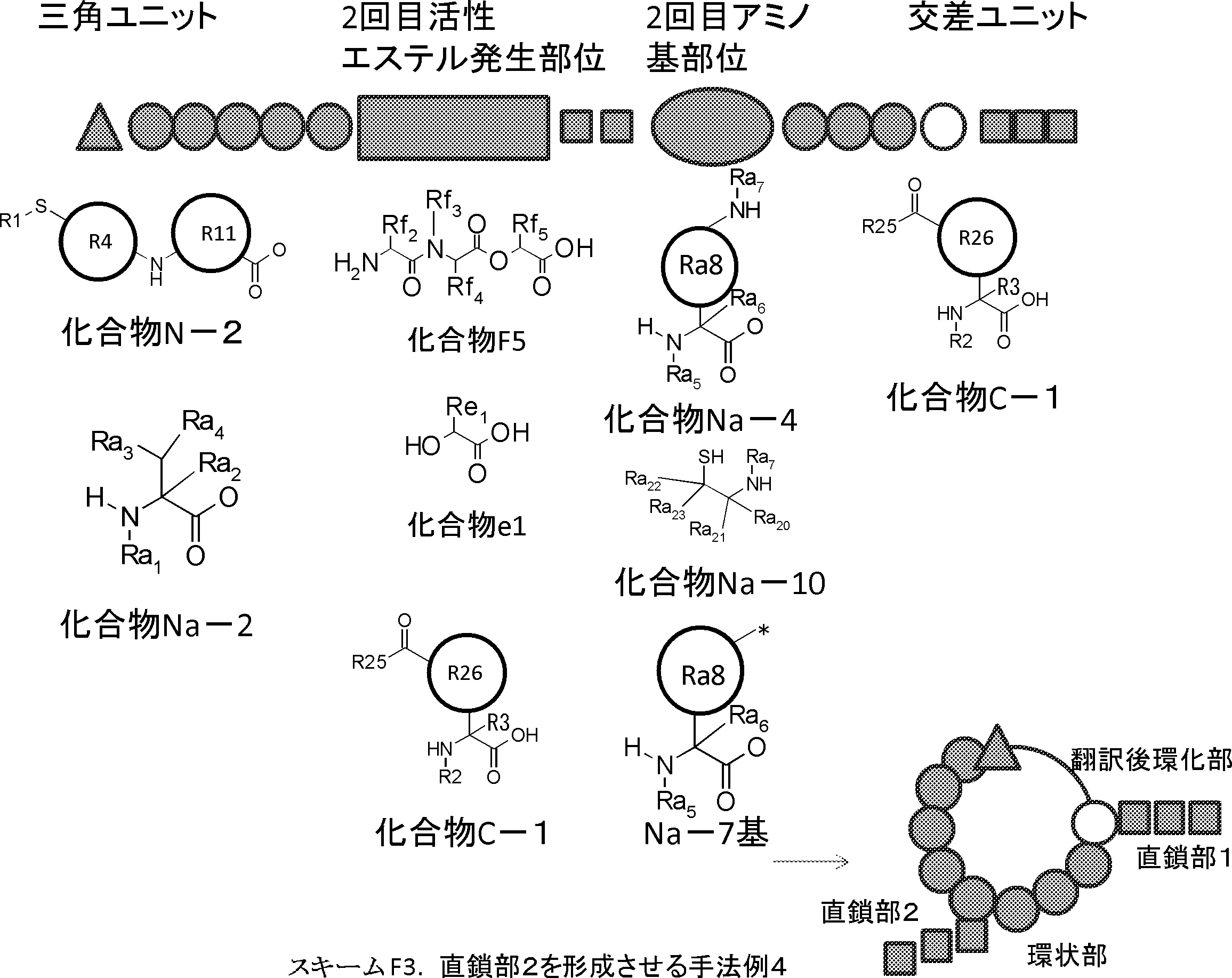

Products and Designed Biomolecules Synthesis. Chem. Rev. 2008, 108, 3054)。

L型、D型、α―α―ジアルキル型のいずれも許容されるが、α位に水素原子が存在する場合にはL型アミノ酸もしくはアミノ酸類縁体が好ましい。
本発明のペプチド化合物は、化学合成することによって製造することも可能である。
例えば、Fmoc合成と呼ばれる方法とBoc合成と呼ばれる方法が挙げられる。Fmoc合成では、主鎖アミノ基をFmoc基により保護され、側鎖官能基を必要に応じてピペリジンなどの塩基性で切断されない保護基で保護され、主鎖カルボン酸を保護しないアミノ酸を基本単位とする。その他、Fmoc保護されたアミノ基とカルボン酸基を有する組み合わせであれば、特に限定されない。例えばジペプチドを基本単位としてもよい。N末端に配置させる基本単位には、Fmocアミノ酸以外であってもよい。例えば、Bocアミノ酸であってもよく、アミノ基を有しないカルボン酸類縁体であってもよい。主鎖カルボン酸基を固相担体の官能基との化学反応により固相に担持させる。続いてピペリジンやDBUなどの塩基によりFmoc基を脱保護し、新たに生じたアミノ基と続いて添加される基本単位のカルボン酸を有する保護アミノ酸とを縮合反応させることで、ペプチド結合を生成させる。縮合反応では、DICとHOBtの組み合わせ、DICとHOAtの組み合わせ、HATUとDIPEAとの組み合わせなど様々な組み合わせが可能である。脱Fmoc基とそれに続くペプチド結合生成反応の繰り返しにより、望みのペプチド配列を生成させることができる。望みの配列が得られた後、固相からの切り出しと必要に応じて導入した側鎖官能基の保護基の脱保護が行われる。また、固相からの切り出しを行う前にペプチドの構造変換や環化を行うことも可能である。固相からの切り出しと、脱保護は同一の条件、例えば90:10のTFA/H2Oで行っても良いし、必要に応じて別々の条件で脱保護しても良い。固相からの切り出しは、1% TFAなどの弱酸での切り出しが可能なものもあれば、保護基としてPdなどを利用して、両者の化学反応の直行性を利用することが可能である。これらの工程の間もしくは最後に、環化などの工程を実施することもできる。例えば、側鎖カルボン酸とN末端主鎖のアミノ基とを縮合させることや、側鎖アミノ基とC末端主鎖のカルボン酸を縮合させることができる。この際、C末端側のカルボン酸と環化させる側鎖カルボン酸の間、もしくはN末端側の主鎖アミノ基またはヒドロキシル基と環化させる側鎖アミノ基の間で反応直行性が必要であり、前述のとおり保護基の直行性を考慮して保護基の選択が行われる。このようにして得られた反応物を逆相カラムや、分子ふるいカラムなどで精製することができる。これらの詳細は、例えばメルク株式会社が平成14年5月1日に発行した固相合成ハンドブックなどに記載されている。
さらに本発明は、所望の活性を有するドラックライクなペプチド化合物又はペプチド化合物-核酸複合体を製造する方法を提供する。
(i)アミノ酸及びアミノ酸類縁体の総数が9~13残基である非環状ペプチド化合物を翻訳合成して、当該非環状ペプチド化合物とそれをコードする核酸配列がリンカーを介して結合している複合体からなる非環状ペプチド化合物-核酸複合体を形成する工程
(ii)工程(i)で翻訳合成された複合体の非環状ペプチド化合物をアミド結合又は炭素-炭素結合によって環化し、環状部のアミノ酸及びアミノ酸類縁体の残基数の合計が5~12となる環状化合物を形成する工程
(iii)工程(ii)で得られた環状部を有するペプチド化合物-核酸複合体ライブラリーと生体内分子とを接触させ、当該生体内分子に対して結合活性を有する複合体を選択する工程。
例えば以下の工程を含む製造方法を挙げることができる:
(iv)前記工程(iii)で選択された複合体の核酸配列からペプチド化合物の配列情報を得る工程、および、
(v)前記工程(iv)で得られた配列情報に基づきペプチド化合物を化学合成する工程
さらに、上記の製造工程において、前記非環状ペプチド化合物がα-ヒドロキシカルボン酸、及び、保護されていてもよい側鎖にアミノ基を有するアミノ酸又はアミノ酸類縁体を含み、工程(ii)の環状化合物を形成する工程の前又は後で、α―ヒドロキシカルボン酸部位と側鎖にアミノ基を有するアミノ酸又はアミノ酸類縁体部位を化学反応させて分枝部位を形成させる工程を含んでいてもよい。当該工程により、環状部の様々な位置に上述の直鎖部2を有するペプチド化合物を製造することが可能である。
具体的には例えば、STAT, AP1, CREB, SREBPなどの転写因子、ムスカリン性アセチルコリン受容体, カンナビノイド受容体、GLP-1受容体、PTH受容体などのGタンパク質共役型受容体(GPCR)、TNF、TNFR、IL-6、IL-6R等のサイトカインやそのレセプター、P2X受容体, ニコチン性アセチルコリン受容体などのイオンチャネル型受容体、イオンチャネル、トランスポーター、miR-21, miR206などのmicroRNAなどが挙げられる。
また、環状部の形成には、例えば、上述の環化反応を利用して形成することが可能であり、当該環化反応によって、アミド結合や炭素-炭素結合を形成させることが可能である。
〔I〕 本発明の環状部を有するペプチド化合物の製造方法は、以下の1又は複数の工程を含むことができる。
A) 化合物C-1をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと前記の工程B)のtRNAを連結させ、化合物C-1をアミノアシル化した開始tRNAを提供する工程
D) 工程C)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
E) プロモーターの下流に、翻訳開始ATGに続いてシステインコドンUGUもしくはUGCを、その下流に工程C)のtRNAのアンチコドンに対応するコドンを含むペプチド配列をコードした鋳型DNAライブラリーを提供する工程
F) 工程E)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
G) 工程F)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
H) 工程D)の無細胞翻訳系に工程G)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
I) 環状構造を形成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。また本発明においては、工程G)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。
さらに本発明の方法は、以下の工程を含むことができる。
J) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
K) 逆転写酵素にてcDNAを合成する工程
L) 塩基配列を解析する工程
A) 化合物C-1をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと前記の工程B)のtRNAを連結させ、化合物C-1をアミノアシル化した開始tRNAを提供する工程
D) 工程C)のtRNAを含む無細胞翻訳系を提供する工程
E) プロモーターの下流に、翻訳開始ATGに続いてシステインコドンUGUもしくはUGCを、その下流に工程C)のtRNAのアンチコドンに対応するコドンを含むペプチド配列をコードした鋳型DNAライブラリーを提供する工程
F) 工程E)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
G) 工程F)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
H) 工程D)の無細胞翻訳系に工程G)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
I) 工程H)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
H,Iの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
J) 環状構造を形成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。また本発明においては、工程H)ないし工程I)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。
さらに本発明の方法は、以下の工程を含むことができる。
J) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
K) 逆転写酵素にてcDNAを合成する工程
L) 塩基配列を解析する工程
A) 化合物C-3(R2=R3=R28=R29=H、L-アスパラギン酸誘導体)をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと工程B)のtRNAを連結させ、化合物C-3をアミノアシル化した開始tRNAを提供する工程
D) 工程C)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
E) プロモーターの下流に、翻訳開始コドンATGの次にシステインコドンを有し、さらにその下流に、工程C)のtRNAのアンチコドンに対応するコドンを有する鋳型DNAライブラリーであって、ペプチド配列をコードする鋳型DNAライブラリーを提供する工程
F) 工程E)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
G) 工程F)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
H) 工程D)の無細胞翻訳系に工程G)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
I) 環状構造を形成させた後、脱硫反応をさせる工程
本発明においては、工程G)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。J) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
K) 逆転写酵素にてcDNAを合成する工程
L) 塩基配列を解析する工程
A) 化合物C-3(R2=R3=R28=R29=H、L-アスパラギン酸誘導体)をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと工程B)のtRNAを連結させ、化合物C-3をアミノアシル化した開始tRNAを提供する工程
D) 工程C)のtRNAを含む無細胞翻訳系を提供する工程
E) プロモーターの下流に、翻訳開始コドンATGの次にシステインコドンを有し、さらにその下流に、工程C)のtRNAのアンチコドンに対応するコドンを有する鋳型DNAライブラリーであって、ペプチド配列をコードする鋳型DNAライブラリーを提供する工程
F) 工程E)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
G) 工程F)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
H) 工程D)の無細胞翻訳系に工程G)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
I) 工程H)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
H,Iの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
I) 環状構造を形成させた後、脱硫反応をさせる工程
本発明においては、工程G)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。J) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
K) 逆転写酵素にてcDNAを合成する工程
L) 塩基配列を解析する工程
あるいは本発明の環状部を有するペプチド化合物の製造方法は、翻訳伸長反応で許容されにくいtBSSEtGABA、tBSSEtβAlaをN末端とすることで実施可能となるアミド環化ペプチド化合物ライブラリーを構築する方法であって、以下の1又は複数の工程を含む方法に関する。
A) tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)のpdCpAと工程B)の開始tRNAを連結させ、tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化した開始tRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)の3'末端のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、ATGを1番目のコドンとして有し、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドンを、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程G)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程L) 環状部位および直鎖部位2を形成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
M) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
N) 逆転写酵素にてcDNAを合成する工程
O) 塩基配列を解析する工程
あるいは本発明の環状部を有するペプチド化合物の製造方法は、翻訳伸長反応で許容されにくいtBSSEtGABA、tBSSEtβAlaをN末端とすることで実施可能となるアミド環化ペプチド化合物ライブラリーを構築する方法であって、以下の1又は複数の工程を含む方法に関する。
A) tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)のpdCpAと工程B)の開始tRNAを連結させ、tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化した開始tRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)の3'末端のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供し、任意のN-メチルアミノ酸をアミノアシル化したpdCpAを提供した後、3'末端のCAを欠損するtRNAを提供し、これらpdCpAとtRNAを連結させ、N-メチルアミノ酸をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、ATGを1番目のコドンとして有し、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、その3'側に工程F)のN-メチルアミノアシル化tRNAに対するコドンをを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程G)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程L) 環状部位および直鎖部位2を形成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
M) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
N) 逆転写酵素にてcDNAを合成する工程
O) 塩基配列を解析する工程
あるいは本発明の環状部を有するペプチド化合物の製造方法は、翻訳伸長反応で許容されにくいtBSSEtGABA、tBSSEtβAlaをN末端とすることで実施可能となるアミド環化ペプチド化合物ライブラリーを構築する方法であって、以下の1又は複数の工程を含む方法に関する。
A) tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)のpdCpAと工程B)の開始tRNAを連結させ、tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化した開始tRNAを提供する工程、
D) Asp(SBn)をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)の3'末端のtRNAを連結させ、Asp(SBn)をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、ATGを1番目のコドンとして有し、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドンを、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程G)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程L) 環状部位および直鎖部位2を形成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
M) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
N) 逆転写酵素にてcDNAを合成する工程
O) 塩基配列を解析する工程
あるいは本発明の環状部を有するペプチド化合物の製造方法は、翻訳伸長反応で許容されにくいtBSSEtGABA、tBSSEtβAlaをN末端とすることで実施可能となるアミド環化ペプチド化合物ライブラリーを構築する方法であって、以下の1又は複数の工程を含む方法に関する。
A) tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)のpdCpAと工程B)の開始tRNAを連結させ、tBSSEtGABAもしくはtBSSEtβAlaをアミノアシル化した開始tRNAを提供する工程、
D) Asp(SBn)をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)の3'末端のtRNAを連結させ、Asp(SBn)をアミノアシル化したtRNAを提供し、任意のN-メチルアミノ酸をアミノアシル化したpdCpAを提供した後、3'末端のCAを欠損するtRNAを提供し、これらpdCpAとtRNAを連結させ、N-メチルアミノ酸をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、ATGを1番目のコドンとして有し、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、その3'側に工程F)のN-メチルアミノアシル化tRNAに対するコドンをを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程G)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程L) 環状部位および直鎖部位2を形成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
M) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
N) 逆転写酵素にてcDNAを合成する工程
O) 塩基配列を解析する工程
あるいは本発明の環状部を有するペプチド化合物の製造方法は、翻訳伸長反応で許容されにくいアミノ酸をN末端とすることで実施可能となるアミド環化ペプチドライブラリーを構築する方法であって、以下の1又は複数の工程を含む方法に関する。
A) 化合物N-1をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)の pdCpAと工程B)の開始tRNAを連結させ、化合物N-1又は化合物N-2の中をアミノアシル化した開始tRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程 D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程C)の翻訳開始tRNAのアンチコドンに対応するコドンを1番目のコドンとして有し、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程G)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
L) 環状部位を形成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
M) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
N) 逆転写酵素にてcDNAを合成する工程
O) 塩基配列を解析する工程
あるいは本発明の環状部を有するペプチド化合物の製造方法は、翻訳伸長反応で許容されにくいアミノ酸をN末端とすることで実施可能となるアミド環化ペプチドライブラリーを構築する方法であって、以下の1又は複数の工程を含む方法に関する。
A) 化合物N-2をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)の pdCpAと工程B)の開始tRNAを連結させ、化合物N-1又は化合物N-2の中をアミノアシル化した開始tRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程 D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程C)の翻訳開始tRNAのアンチコドンに対応するコドンを1番目のコドンとして有し、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンをを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程G)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
L) 環状部位を形成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
M) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
N) 逆転写酵素にてcDNAを合成する工程
O) 塩基配列を解析する工程
あるいは本発明の環状部を有するペプチド化合物の製造方法は、翻訳伸長反応で許容されにくいアミノ酸をN末端とすることで実施可能となるアミド環化ペプチドライブラリーを構築する方法であって、以下の1又は複数の工程を含む方法に関する。
A) 化合物N-2をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)の pdCpAと工程B)の開始tRNAを連結させ、化合物N-1又は化合物N-2の中をアミノアシル化した開始tRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程 D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、さらに、任意のN-メチルアミノ酸をアミノアシル化したpdCpAを提供する工程、さらに3'末端のCAを欠損するtRNAを提供する工程、さらにこれらpdCpAとtRNAを連結させ、N-メチルアミノ酸をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程C)の翻訳開始tRNAのアンチコドンに対応するコドンを1番目のコドンとして有し、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、その3'側に工程F)のN-メチルアミノアシル化tRNAに対するコドンをを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程G)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
L) 環状部位を形成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
M) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
N) 逆転写酵素にてcDNAを合成する工程
O) 塩基配列を解析する工程
A) 化合物C-X(本明細書において「化合物C-X」とは化合物C-1、化合物C-2および化合物C―3から選択されるいずれかの化合物を指す。本明細において以下同じ)をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと前記の工程B)のtRNAを連結させ、化合物C-Xをアミノアシル化した開始tRNAを提供する工程
D) 工程C)のtRNAを含む無細胞翻訳系を提供する工程
E) プロモーターの下流に、工程C)のtRNAのアンチコドンに対応するコドンを、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを途中に含むペプチド配列をコードした鋳型DNAライブラリーを提供する工程
F) 工程E)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
G) 工程F)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
H) 工程D)の無細胞翻訳系に工程G)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
I) 工程H)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
H,Iの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
J) 環状構造を形成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。また本発明においては、工程H)ないし工程I)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。
さらに本発明の方法は、以下の工程を含むことができる。
J) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
K) 逆転写酵素にてcDNAを合成する工程
L) 塩基配列を解析する工程
A) 化合物C-Xをアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと前記の工程B)のtRNAを連結させ、化合物C-Xをアミノアシル化した開始tRNAを提供する工程
D) 工程C)のtRNAを含む無細胞翻訳系を提供する工程
E) プロモーターの下流に、翻訳開始ATGの直後にグリシン、アラニン、フェニルアラニンのいずれかのコドンを、工程C)のtRNAのアンチコドンに対応するコドンを、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを途中に含むペプチド配列をコードした鋳型DNAライブラリーを提供する工程
F) 工程E)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
G) 工程F)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
H) 工程D)の無細胞翻訳系に工程G)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
I) 工程H)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
H,Iの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
J) 環状構造を形成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。また本発明においては、工程H)ないし工程I)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。
さらに本発明の方法は、以下の工程を含むことができる。
J) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
K) 逆転写酵素にてcDNAを合成する工程
L) 塩基配列を解析する工程
A) Asp(SBn)をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと前記の工程B)のtRNAを連結させ、Asp(SBn)をアミノアシル化した開始tRNAを提供する工程
D) 工程C)のtRNAを含む無細胞翻訳系を提供する工程
E) プロモーターの下流に、工程C)のtRNAのアンチコドンに対応するコドン、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを途中に含むペプチド配列をコードした鋳型DNAライブラリーを提供する工程
F) 工程E)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
G) 工程F)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
H) 工程D)の無細胞翻訳系に工程G)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ディスプレイライブラリーを提供する工程
I) 工程H)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
H,Iの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
J) 環状構造を形成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。また本発明においては、工程H)ないし工程I)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程を含むことができる。
さらに本発明の方法は、以下の工程を含むことができる。
J) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
K) 逆転写酵素にてcDNAを合成する工程
L) 塩基配列を解析する工程
A) 化合物N-1又は化合物N-2をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損する開始tRNAを提供する工程
C) 工程A)のpdCpAと工程B)の開始tRNAを連結させ、化合物N-1又は化合物N-2をアミノアシル化した開始tRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) 工程C)の開始tRNAと工程F)のtRNAを含みメチオニン、メチオニルtRNA合成酵素(MetRS)、メチオニン用翻訳開始tRNA、フォルミルドナー、メチオニルtRNAトランスフェラーゼのいずれかを含まない無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程C)の翻訳開始tRNAのアンチコドンに対応するコドンを1番目のコドンとして有し、その下流にHOGlyのコドンとリジンのコドンとで挟んだ0個から2個の任意のコドンを(アラニンがよりN末端側)、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、化合物N-1もしくは化合物N-2のSH基が保護されている場合には、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程J)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ペプチドディスプレイライブラリーを提供する工程
L) 環を形成させる工程
M) HOGlyと直前のN末端側のアミノ酸とで形成されるエステルを活性化させてチオエステルを生成させる工程
N) リジン側鎖アミノ基とアミド結合形成させて直鎖部位2を生成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
O) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
P) 逆転写酵素にてcDNAを合成する工程
Q) 塩基配列を解析する工程
A) 化合物N-1又は化合物N-2をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと工程B)のtRNAを連結させ、化合物N-1又は化合物N-2をアミノアシル化したtRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) 工程C)のtRNAと工程F)のtRNAと乳酸を含む無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程C)のtRNAのアンチコドンに対応するコドンを2番目のコドンとして有し、その下流にシステイン、プロリン、乳酸と連なるコドンを、さらにその下流にリジンを、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、化合物N-1もしくは化合物N-2のSH基が保護されている場合には、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程J)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ペプチドディスプレイライブラリーを提供する工程
L) 環を形成させる工程
M) システイン、プロリン、乳酸部位から活性チオエステルを発生させる工程
N) 生成した活性チオエステルを、リジン側鎖アミノ基とアミド結合形成させて直鎖部位2を生成させる工程、さらに続いて脱硫反応を実施させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
O) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
P) 逆転写酵素にてcDNAを合成する工程
Q) 塩基配列を解析する工程
A) 化合物N-1又は化合物N-2をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと工程B)のtRNAを連結させ、化合物N-1又は化合物N-2をアミノアシル化したtRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) 工程C)のtRNAと工程F)のtRNAと乳酸を含む無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程C)のtRNAのアンチコドンに対応するコドンを2番目のコドンとして有し、その下流にシステイン、プロリン、乳酸と連なるコドンを、その下流にリジンのコドンを、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程J)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ペプチドディスプレイライブラリーを提供する工程
L) 工程K)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
K, Lの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
M) 環状構造を形成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。
N) システイン、プロリン、乳酸ユニットから活性チオエステルを発生させる工程
O) 生成したカルボン酸を活性エステル化し、リジン側鎖アミノ基とアミド結合形成させて直鎖部位2を生成させる工程、続いて必要に応じて脱硫反応させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
P) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
P) 逆転写酵素にてcDNAを合成する工程
Q) 塩基配列を解析する工程
A) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) 工程C)のtRNAと工程F)のtRNAと乳酸を含む無細胞翻訳系を提供する工程
H) プロモーターの下流に、システインコドンを2番目のコドンとして有し、その下流にシステイン、プロリン、乳酸と連なるコドンを、さらに保護されたアミン化合物Na-4のコドンを、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンをを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程J)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ペプチドディスプレイライブラリーを提供する工程
L) 環を形成させる工程
M) 化合物Na-4の側鎖アミノ基を脱保護させる工程、およびシステイン、プロリン、乳酸ユニットから、活性チオエステルを発生させる工程
N) 生成した活性チオエステルを化合物Na-4の側鎖アミノ基とアミド結合形成させて直鎖部位2を生成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
O) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
P) 逆転写酵素にてcDNAを合成する工程
Q) 塩基配列を解析する工程
A) 化合物N-1又は化合物N-2をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと工程B)のtRNAを連結させ、化合物N-1又は化合物N-2をアミノアシル化したtRNAを提供する工程、
D) 化合物C-1をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
G) アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)をアミノアシル化したpdCpAを提供する工程
H) 3'末端のCAを欠損するtRNAを提供する工程
I) 工程G)のpdCpAと工程H)のtRNAを連結させ、アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)をアミノアシル化したtRNAを提供する工程、
J) 工程C)のtRNAと工程F)のtRNAと工程I)のtRNAを含む無細胞翻訳系を提供する工程
K) プロモーターの下流に、工程C)のtRNAのアンチコドンに対応するコドンを2番目のコドンとして有し、その下流のCys、Pro、乳酸のtRNAのアンチコドンに対応するコドン、その下流に工程G)のtRNAのアンチコドンに対応するコドンを、さらにその下流に工程D)のtRNAのアンチコドンに対応するコドン、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
L) 工程K)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
M) 工程L)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
N) 工程M)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ペプチドディスプレイライブラリーを提供する工程
O) 工程K)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
K, Lの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
P) 環状構造を形成させる工程
Q) アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)を脱保護する工程
R) 直鎖部位2を生成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
S) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
T) 逆転写酵素にてcDNAを合成する工程
U) 塩基配列を解析する工程
A) 化合物C-1をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと工程B)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
D) 化合物アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)のtRNAを連結させ、アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)をアミノアシル化したtRNAを提供する工程、
G) 工程C)のtRNAと工程F)のtRNAと工程I)のtRNAを含む無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程F)のtRNAのアンチコドンに対応するコドンを、その下流にCys、Pro、乳酸のtRANのアンチコドンに対応するコドンを、さらにその下流に工程C)のtRNAのアンチコドンに対応するコドン、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程J)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ペプチドディスプレイライブラリーを提供する工程
L) 工程K)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
K, Lの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
L) 環状構造を形成させる工程
M) アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)を脱保護する工程
N) 直鎖部位2を生成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
O) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
P) 逆転写酵素にてcDNAを合成する工程
Q) 塩基配列を解析する工程
工程G)にてメチオニンの代わりにノルマルロイシンを加えることもできる。
A) 化合物C-1をアミノアシル化したpdCpAを提供する工程
B) 3'末端のCAを欠損するtRNAを提供する工程
C) 工程A)のpdCpAと工程B)のtRNAを連結させ、化合物C-1をアミノアシル化したtRNAを提供する工程、
D) 化合物アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)をアミノアシル化したpdCpAを提供する工程
E) 3'末端のCAを欠損するtRNAを提供する工程
F) 工程D)のpdCpAと工程E)のtRNAを連結させ、化合物アミノ基および必要に応じてチオール基も保護された化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)をアミノアシル化したtRNAを提供する工程、
G) 工程C)のtRNAと工程F)のtRNAと乳酸を含む無細胞翻訳系を提供する工程
H) プロモーターの下流に、工程C)のtRNAのアンチコドンに対応するコドンを2番目のコドンとして有し、その下流にシステイン、プロリン、乳酸と連なるコドンを、さらにその下流に工程F)のtRNAのアンチコドンに対応するコドン、その3'側にプロリンもしくは他のARSの基質となるN-メチルアミノ酸のコドンを含むペプチド配列をコードする鋳型DNAライブラリーを提供する工程
I) 工程H)の鋳型DNAライブラリーからmRNAライブラリーを提供する工程
J) 工程I)のmRNAライブラリーの3'末端にスペーサーを結合させる工程
K) 工程J)の無細胞翻訳系に工程J)のスペーサーを結合したmRNAライブラリーを加え、翻訳することで、環化前ペプチド化合物-mRNA複合体ペプチドディスプレイライブラリーを提供する工程
L) 工程K)のライブラリーにペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを作用させる工程
K, Lの工程は翻訳時にペプチドデフォルミラーゼ、メチオニン アミノペプチダーゼを系に加えることにより同時に実施することもできる。
M) 環状構造を形成させる工程
環状構造の形成においては、必要に応じて脱硫反応を行うことができる。
N) 化合物Na-4もしくは化合物Na-10(Na-7基)もしくは化合物Na-11(Na-7基)の側鎖保護基を脱保護させる工程、およびCys、Pro、乳酸ユニットから活性チオエステルを発生させる工程
O) 生成した活性チオエステル化と側鎖アミノ基とアミド結合形成させて直鎖部位2を生成させる工程
本発明においては、工程J)の後、mRNAライブラリーの3'領域にアニールするプライマーにてcDNAを合成する工程する工程を含むことができる。さらに本発明の方法は、以下の工程を含むことができる。
P) パニングにて、標的物質に結合したmRNAライブラリーを濃縮する工程
Q) 逆転写酵素にてcDNAを合成する工程
R) 塩基配列を解析する工程
工程G)にてメチオニンの代わりにノルマルロイシンを加えることもできる。
アミノアシルtRNAの調製は、pdCpAの利用に限らずアミノアシルtRNA合成酵素、フレキシザイム、カチオンミセル中超音波混合法、PNAアミノ酸活性エステル法などの利用も含む。
アスパラギン酸型のチオエステルを翻訳にて導入する場合、C末端側直後のアミド結合に水素原子と反応してアスパルチミドを形成してしまうため、アスパラギン酸型のチオエステルを翻訳にて導入する際に直後のアミノ酸残基をN-アルキル基を有するアミノ酸(例えばプロリン)にすることによって望みのチオエステルを含む全長ペプチドを翻訳合成する方法
本明細書において前記「アルキル基」は、下記の「アルケニル基」、「アルキニル基」が含まれる場合がある。
本明細書において前記「アリール基」は、下記の「ヘテロアリール」が含まれる場合がある。
ヘテロアリールとしては具体的には、たとえば、フリル基、チエニル基、ピロリル基、イミダゾリル基、ピラゾリル基、チアゾリル基、イソチアゾリル基、オキサゾリル基、イソオキサゾリル基、オキサジアゾリル基、チアジアゾリル基、トリアゾリル基、テトラゾリル基、ピリジル基、ピリミジル基、ピリダジニル基、ピラジニル基、トリアジニル基、ベンゾフラニル基、ベンゾチエニル基、ベンゾチアジアゾリル基、ベンゾチアゾリル基、ベンゾオキサゾリル基、ベンゾオキサジアゾリル基、ベンゾイミダゾリル基、インドリル基、イソインドリル基、インダゾリル基、キノリル基、イソキノリル基、シンノリニル基、キナゾリニル基、キノキサリニル基、ベンゾジオキソリル基、インドリジニル基、イミダゾピリジル基などが挙げられる。
「通常アミン」とは、反応補助基により活性化されていないアミンを意味する。
本発明における「核酸」には、デオキシリボ核酸(DNA)、リボ核酸(RNA)あるいは、人工塩基を有するヌクレオチド誘導体を含むこともできる。また、ペプチド核酸(PNA)を含むこともできる。本発明の核酸は、目的とする遺伝情報が保持される限り、これらの核酸のいずれか、あるいは混成とすることもできる。すなわち、DNA-RNAのハイブリッドヌクレオチドやDNAとRNAのような異なる核酸が1本鎖に連結されたキメラ核酸も本発明における核酸に含まれる。
本発明においては、これら核酸を鋳型として含む、displayライブラリーなどの核酸ライブラリーを好適に用いることができる。
本発明のペプチド化合物の製造方法では、無細胞翻訳系などのタンパク質製造システムを使用することが好ましい。無細胞翻訳系とは、細胞より抽出したribosomeの他、翻訳に関与する蛋白因子群、tRNA、アミノ酸、ATPなどのエネルギー源、その再生系を組み合わせたものでmRNAを蛋白に翻訳することが出来る。本発明の無細胞翻訳系には、これら以外にも、開始因子、伸長因子、解離因子、アミノアシルtRNA合成酵素、メチオニルtRNAトランスフォルミラーゼなどを含むことができる。これらの因子は、種々の細胞の抽出液から精製することによって得ることができる。因子を精製するための細胞は、例えば原核細胞、または真核細胞を挙げることができる。原核細胞としては、大腸菌細胞、高度好熱菌細胞、または枯草菌細胞を挙げることができる。真核細胞としては、酵母細胞、小麦胚芽、ウサギ網状赤血球、植物細胞、昆虫細胞、または動物細胞を材料にしたものが知られている。
ライブラリーを構成するペプチドに含まれるN-メチルアミノ酸の数は、DNAライブラリーを合成する際にN-メチルアミノ酸に当てたコドンの出現頻度にて調整することができる。例えば10のアミノ酸残基可変箇所を含むペプチドのライブラリーではアミノ酸の種類が20種類から選択されると仮定された場合、N-Meアミノ酸を10種類選択すると、N-Meアミノ酸のコドンユニットが可変箇所1箇所当り10/20(50%)を占めるように合成することによってライブラリーのペプチドに含まれるN-メチルアミノ酸の数は平均5個になる。
実際に我々の実施例24に記載の通り、このルールに則ると、可変領域のアミノ酸の種類は21種類であり、そのうち10種類がNメチルなどのNアルキルアミノ酸が選定された。Initiation read through法では固定アミノ酸2つのうち、両方ともがNH2であったため、平均のN-アルキル数は4.4と計算された。Inhitiation suppression法では2つの固定アミノ酸のうち1つがNアルキルであり、また交差ユニットのC末端側もN-アルキルアミノ酸の中から選定されたこともあり平均のN-アルキルアミノ酸数は6.0と計算された。
同様に両者の平均のCLogP値は、それぞれ5.97と6.12であった。
本発明に使用できる非天然型アミノ酸(翻訳アミノ酸)を以下に例示するが、それらに限定されない。これらの非天然型アミノ酸の多くは側鎖が保護あるいは無保護、アミン部位が保護あるいは無保護の状態で購入される。購入されないものは、既知の方法によって合成される。
N-メチルアラニン、N-メチルグリシン、N-メチルフェニルアラニン、N-メチルチロシン、N-メチルー3-クロロフェニルアラニン、N-メチルー4―クロロフェニルアラニン、N-メチルー4-メトキシフェニルアラニン、N-メチルー4-チアゾールアラニン、N-メチルヒスチジン、N-メチルセリン、N-メチルアスパラギン酸、
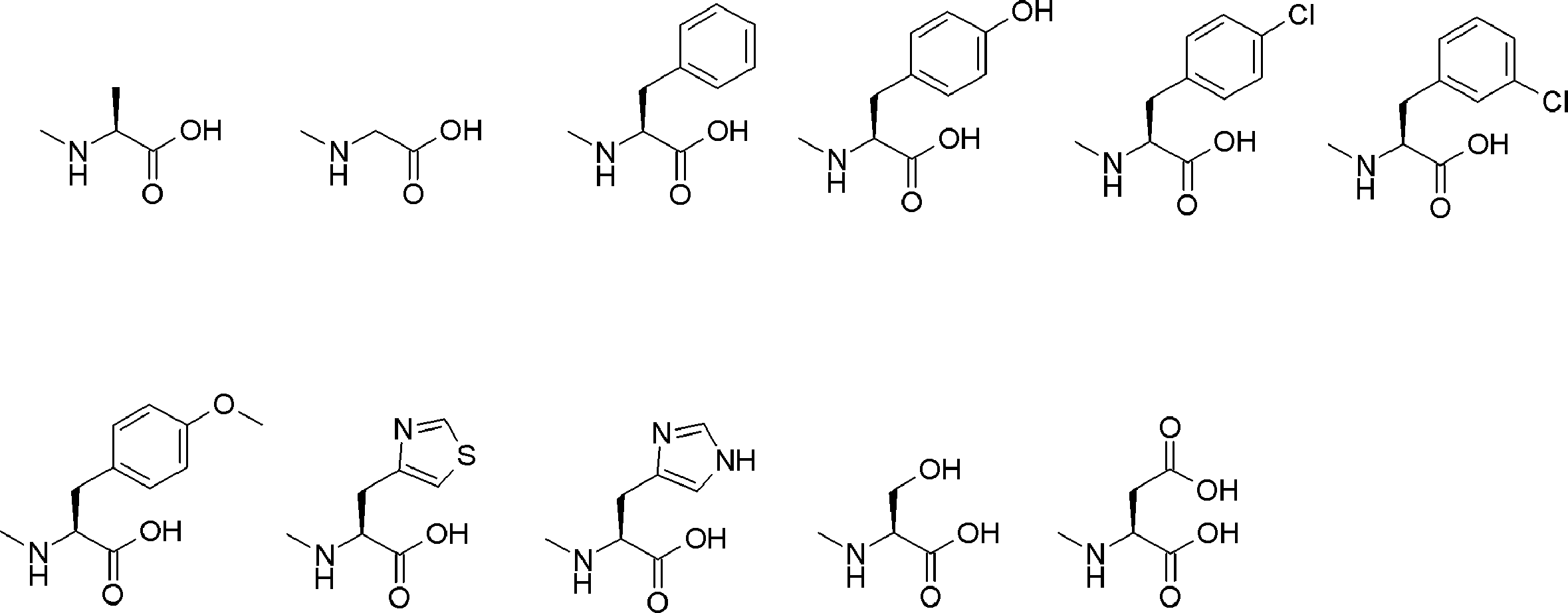
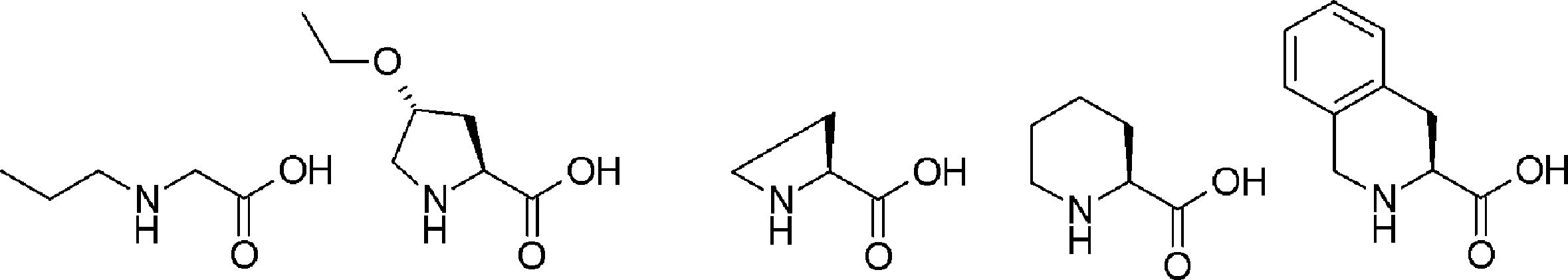


アミノ酸及びアミノ酸類縁体は、pdCpAと反応させるために、例えば活性エステルに変換することができる。これらの活性エステルは多くはシアノメチルエステルとして単離することができる。
カルボン酸と一級および二級アミンとの反応によるアミド結合形成反応は、広く有機合成化学一般に使用されている。例えば、カルボン酸を予め活性化された活性エステルなどとして単離してからアミンと混合させる方法、直接カルボン酸とアミンを混合させた後にカルボン酸を活性化させてアミド結合を形成させる方法などを用いることができる。カルボン酸の活性体として酸クロライドや酸アオイダイドなどの酸ハライド群や、HOBt、HOAt、HOSu、HOPfp、チオエステルなどの活性エステル群が広く知られている。
チオエステルとしては、好ましくは、-C(=O)SR1で表されるものが挙げられる。
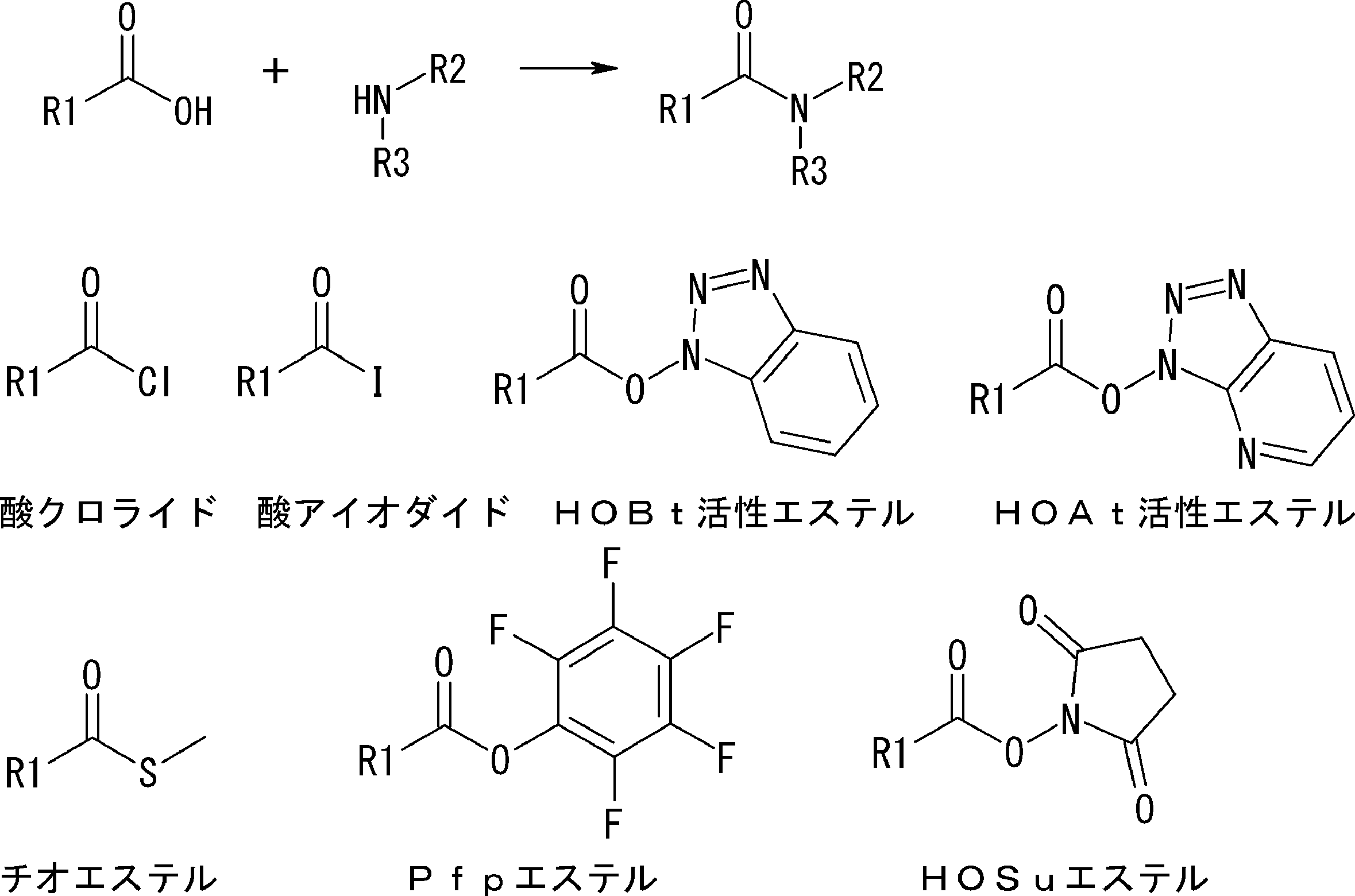
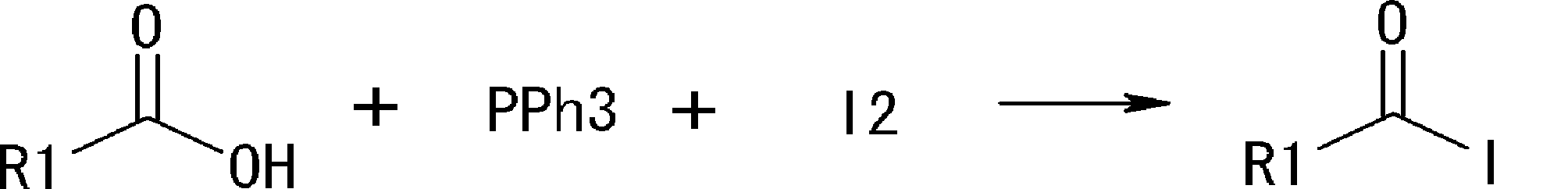
また、望みの位置で選択的にカルボン酸とアミノ基を反応させるため、カルボン酸とアミノ基側のいずれか、あるいは両方に反応補助基を導入させることも可能である。
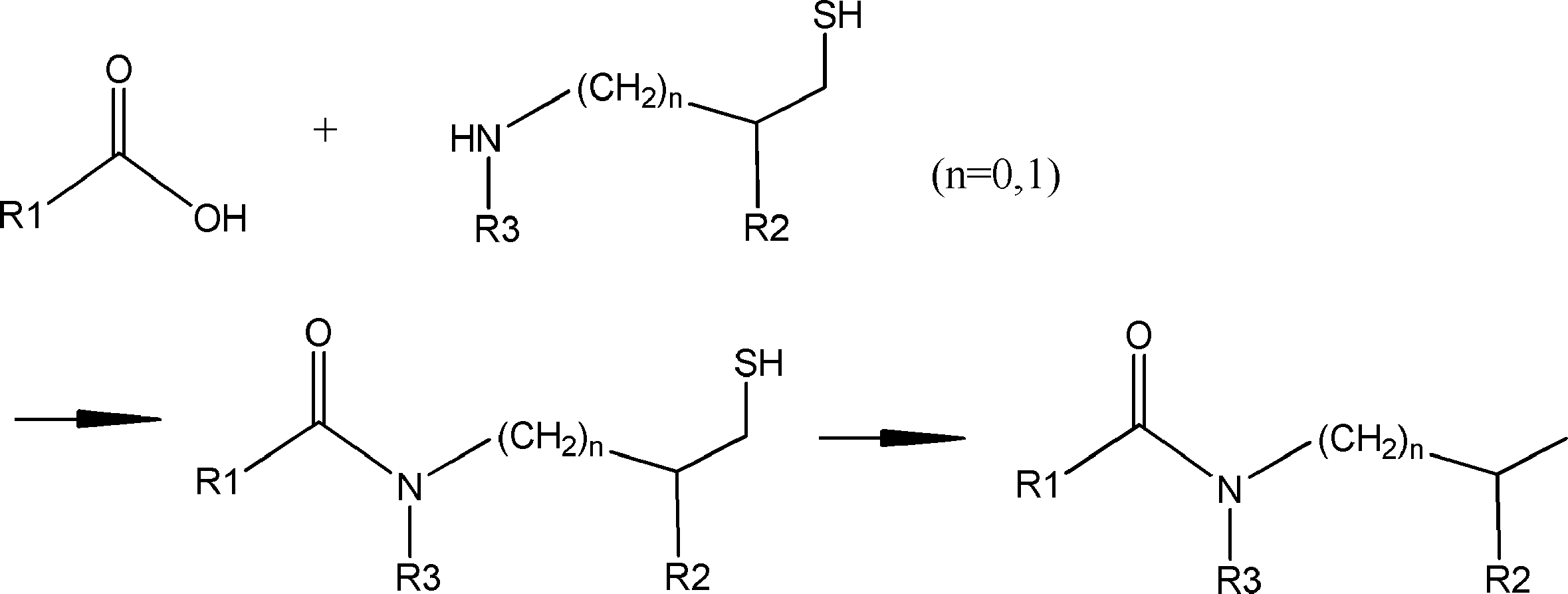
カルボン酸側とアミノ基側の両方に反応補助基を導入させる例として、以下スキームが挙げられる(Liら, Org. Lett. 2010, 12, 1724)。

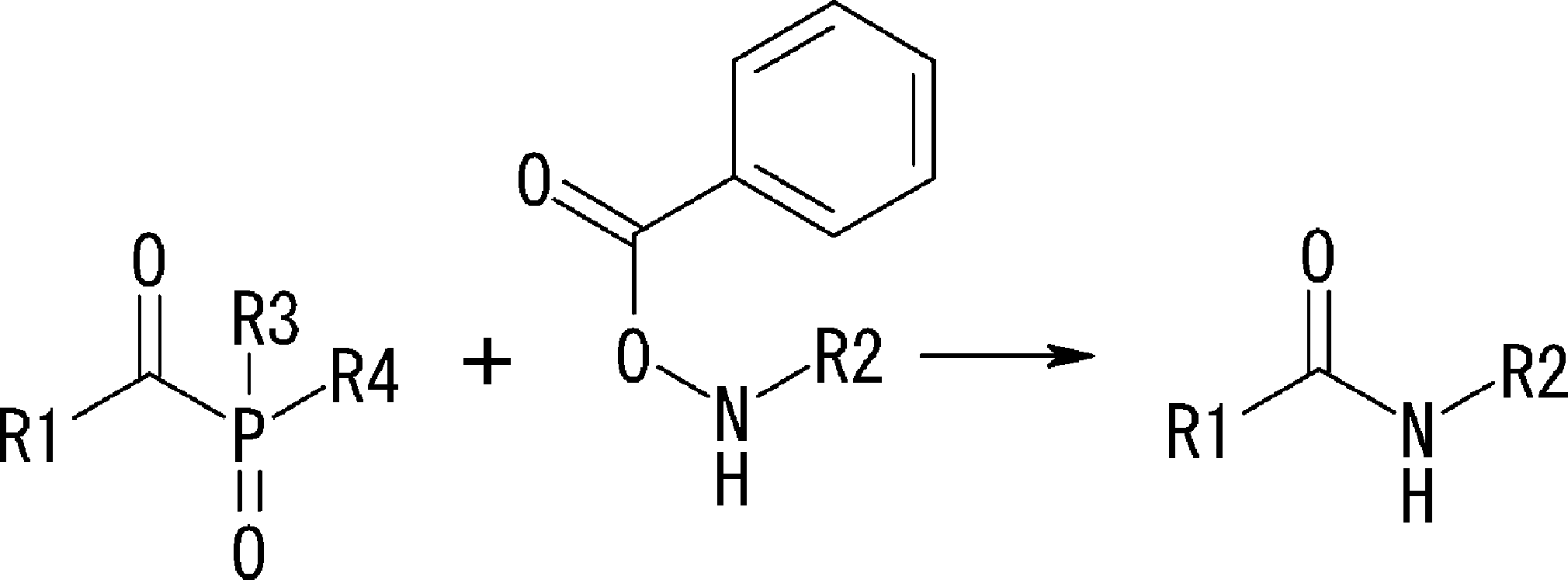
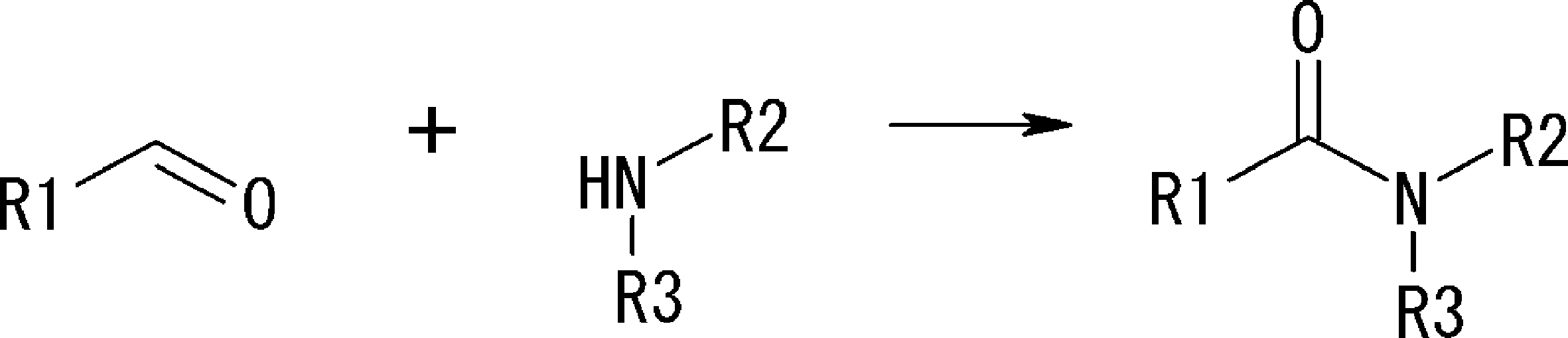
本発明は、本発明の方法で製造されたペプチド化合物を含有する医薬組成物を提供する。
本発明の医薬組成物は、本発明の方法で製造されたペプチド化合物に加えて医薬的に許容し得る担体を導入し、公知の方法で製剤化することが可能である。製剤化には通常用いられる賦形剤、結合剤、滑沢剤、着色剤、矯味矯臭剤や、および必要により安定化剤、乳化剤、吸収促進剤、界面活性剤、pH調製剤、防腐剤、抗酸化剤などを使用することができ、一般に医薬品製剤の原料として用いられる成分を配合して常法により製剤化される。
例えば、経口製剤を製造するには、本発明にかかる化合物またはその薬理学的に許容される塩と賦形剤、さらに必要に応じて結合剤、崩壊剤、滑沢剤、着色剤、矯味矯臭剤などを加えた後、常法により散剤、細粒剤、顆粒剤、錠剤、被覆錠剤、カプセル剤等とする。
注射のための無菌組成物は注射用蒸留水のようなベヒクルを用いて通常の製剤実施に従って処方することができる。
チオエステルを有する一連の活性エステル(化合物1g)の合成を実施し、Rex1=Me、Et、iPr、tBu、Bn、Ph、Phenetyl基の合成を図1の方法に従って行った。
なお、実施例中では以下の略号を使用した。
DCM ジクロロメタン
DIC N,N-ジイソプロピルカルボジイミド
DIPEA N,N-ジイソプロピルエチルアミン
DMAP 4-ジメチルアミノピリジン
DMF ジメチルホルムアミド
DMSO ジメチルスルホキシド
DTT ジチオスレイロール
FA ギ酸
TFA トリフルオロ酢酸
THF テトラヒドロフラン
HFIP 1,1,1,3,3,3-ヘキサフルオロ-2-プロパノール
HOBT 1H-ベンゾ[d][1,2,3]トリアゾール-1-オール
WSCI・HCl 1-エチル-3-(3-ジメチルアミノプロピル)カルボジイ
ミド塩酸塩
TCEP トリス(2-カルボキシエチル)ホスフィン
NMP N-メチル‐2‐ピロリドン
DBU 1,8-ジアザビシクロ[5.4.0]-7-ウンデセン
なお、ペプチド合成及び、固相合成に用いる反応溶媒はペプチド合成用(関東化学、和光純薬あるいは渡辺化学から購入)を用いた。例えばDCM、DMF、DMSO、2% DBU in DMF,20% piperidine in DMFなどである。また、水を溶媒として加えない反応では脱水溶媒や無水溶媒(関東化学、和光純薬などから購入)を用いた。


1-1.(S)-4-(tert-ブトキシ)-4-オキソ-3-(ペンタ-4-エンアミド)ブタン酸 (化合物1b-I)の合成

LCMS(ESI) m/z = 322 (M+H)+
保持時間:0.78分(分析条件SQDAA05)

LCMS(ESI) m/z = 361 (M+H)+
保持時間:0.91分(分析条件SQDAA05)
2-1.(S)-4-オキソ-2-(ペンタ-4-エンアミド)-4-(フェネチルチオ)ブタン酸 (化合物1f-IE)の合成
LCMS(ESI) m/z = 336 (M+H)+
保持時間:0.83分(分析条件SQDAA05)
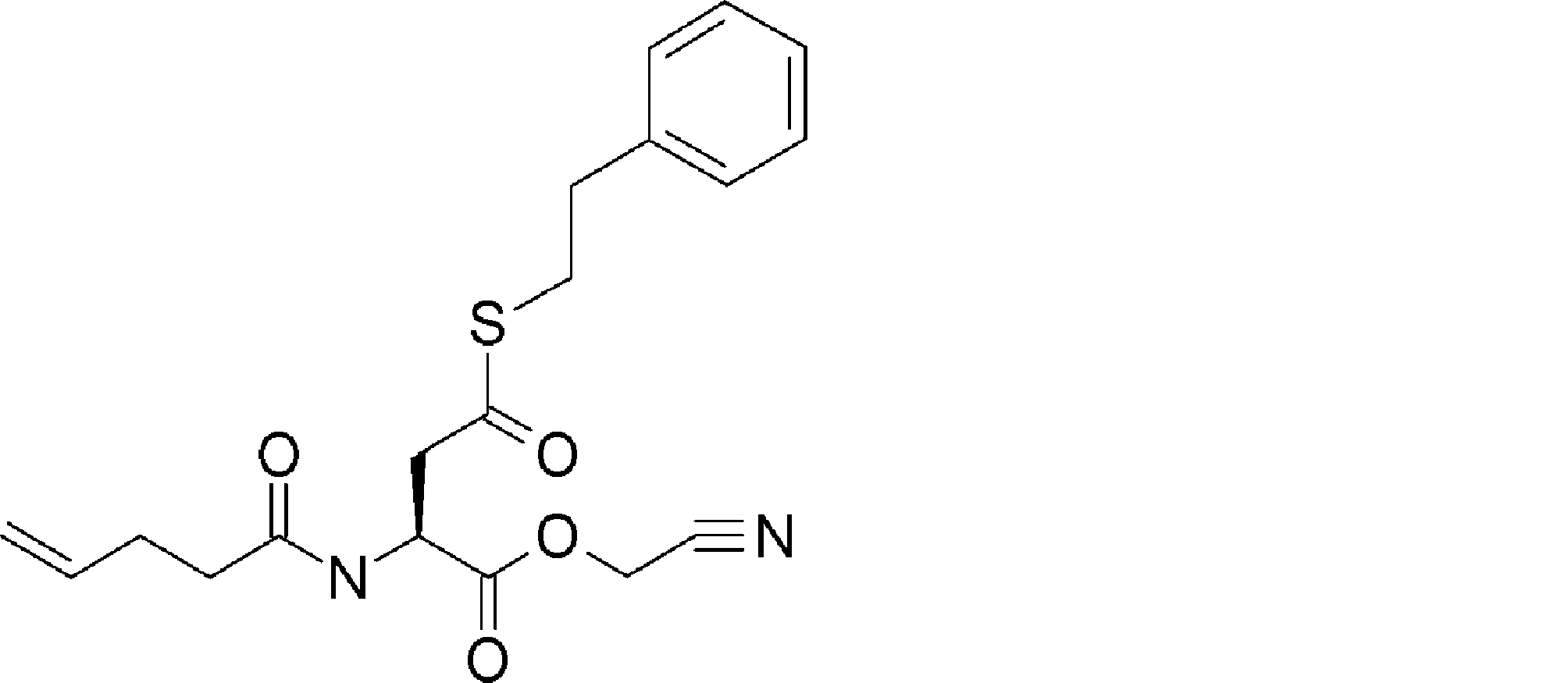
LCMS(ESI) m/z = 375 (M+H)+
保持時間:0.95分(分析条件SQDAA05)
3-1. (S)-4-(エチルチオ)-4-オキソ-2-(ペンタ-4-エンアミド)ブタン酸 (化合物1f-IG)の合成

LCMS(ESI) m/z = 260 (M+H)+
保持時間:0.58分(分析条件SQDAA05)
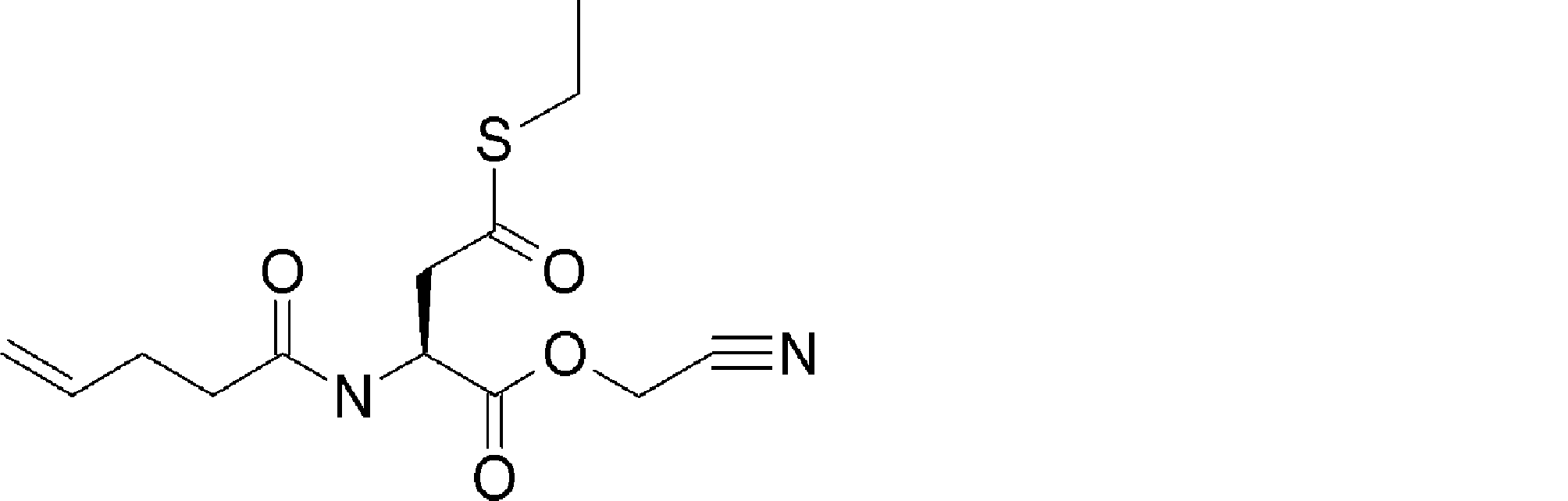
LCMS(ESI) m/z =299(M+H)+
保持時間:0.69分(分析条件SQDFA05)
4-1. (S)-4-(イソプロピルチオ)-4-オキソ-2-(ペンタ-4-エンアミド)ブタン酸(化合物1f-IB)の合成
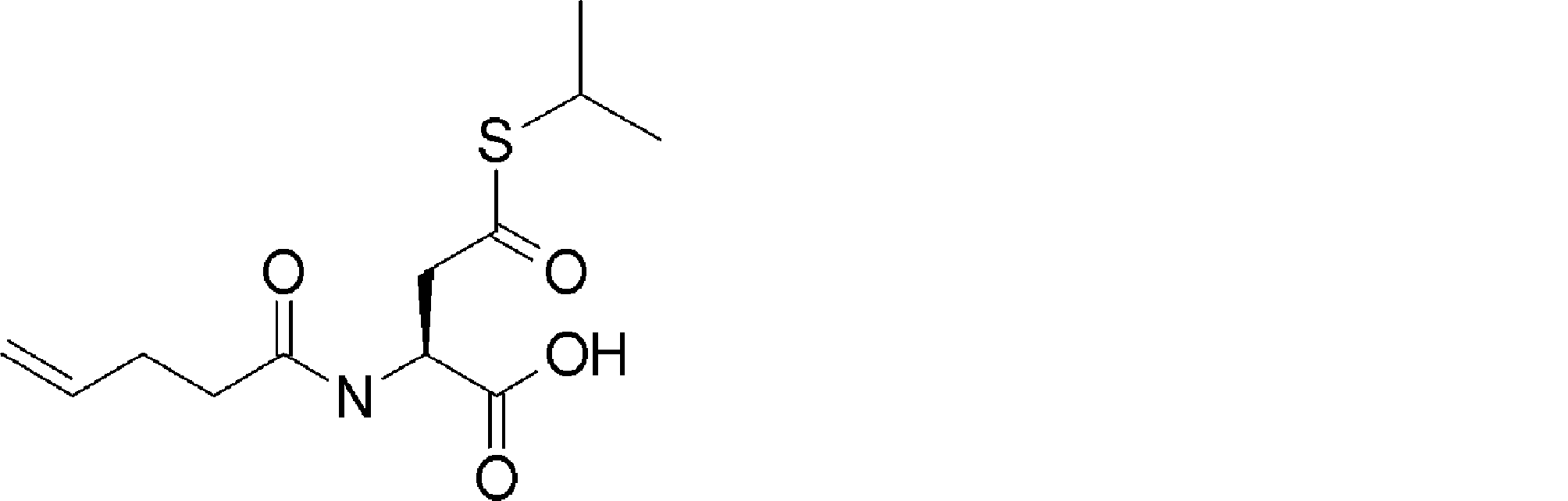
LCMS(ESI) m/z = 274 (M+H)+
保持時間:0.70分(分析条件SQDAA05)

LCMS(ESI) m/z = 313 (M+H)+
保持時間:0.75分(分析条件SQDFA05)
5-1.(S)-4-(tert-ブチルチオ)-4-オキソ-2-(ペンタ-4-エンアミド)ブタン酸(化合物1f-IC)の合成

LCMS(ESI) m/z = 288 (M+H)+
保持時間:0.79分(分析条件SQDAA05)
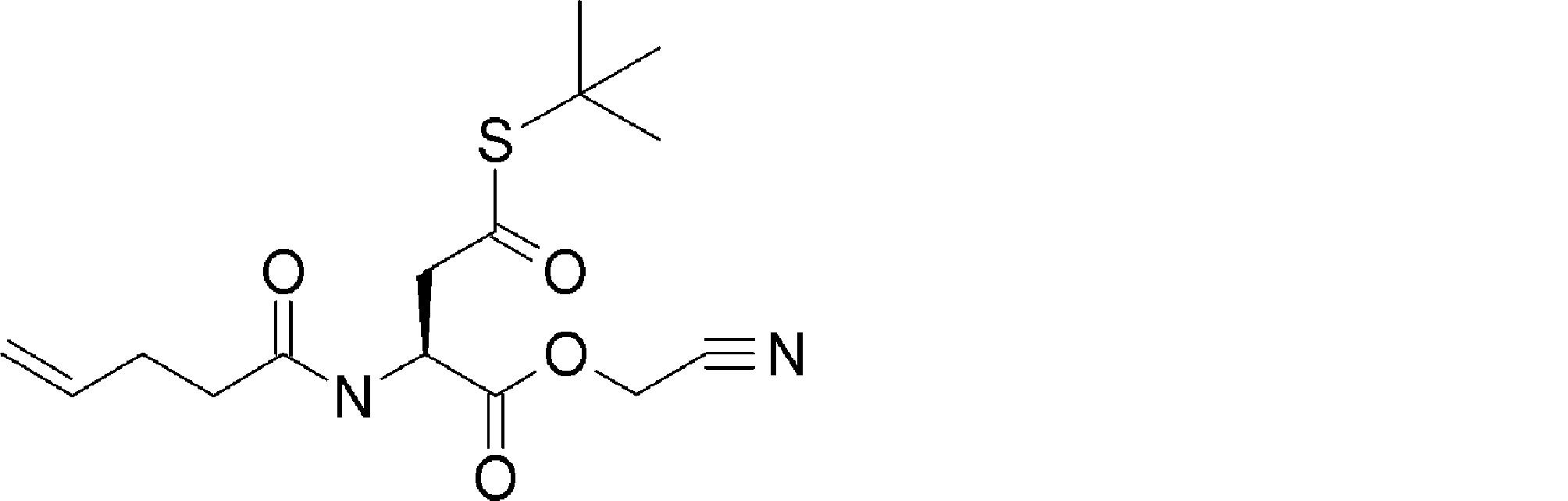
LCMS(ESI) m/z = 327 (M+H)+
保持時間:0.90分(分析条件SQDAA05)
6-1.4-オキソ-2-(ペンタ-4-エンアミド)-4-(フェニルチオ)ブタン酸 (S)-tert-ブチル(化合物1e-IF)の合成
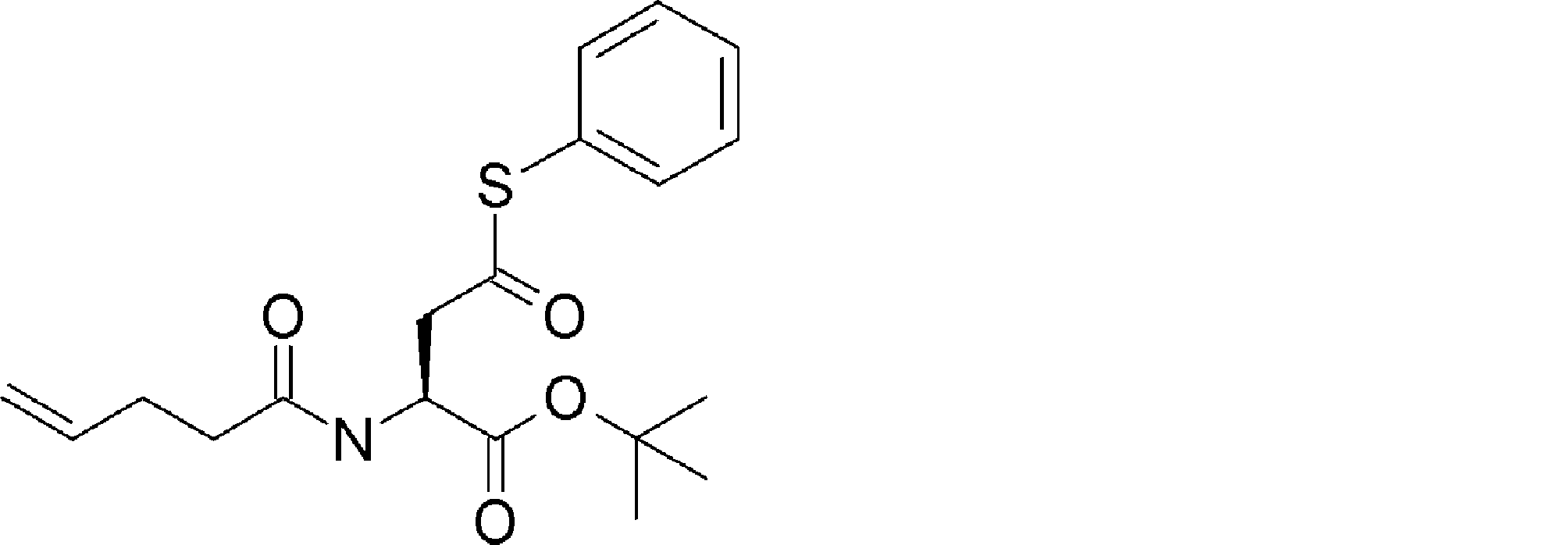
LCMS(ESI) m/z = 364 (M+H)+
保持時間:1.01分(分析条件SQDAA05)
7-1. 4-(メチルチオ)-4-オキソ-2-(ペンタ-4-エンアミド)ブタン酸 (S)-tert-ブチル(化合物1e-IA)の合成
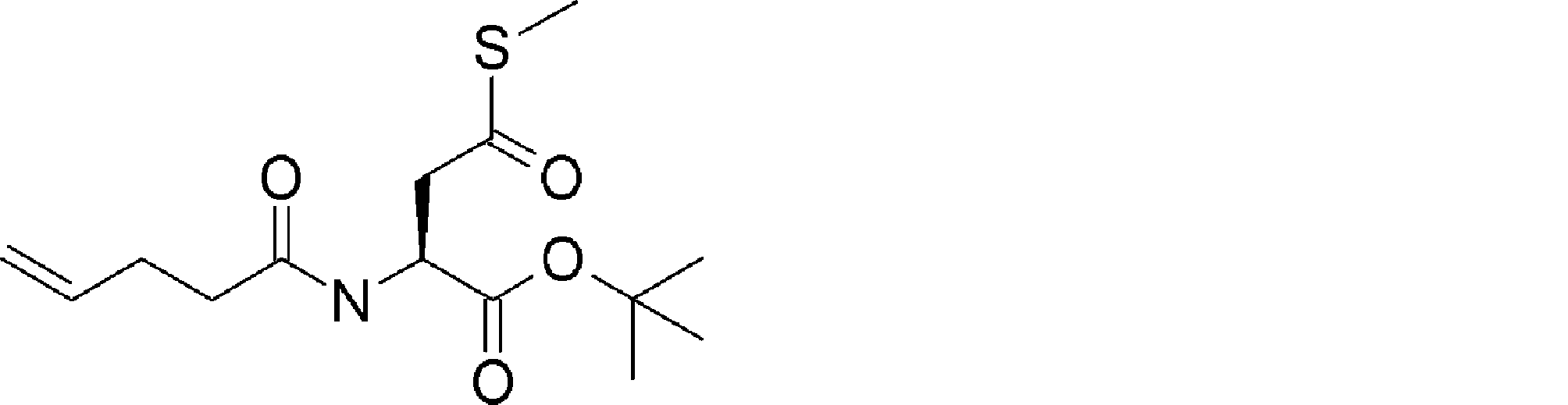
LCMS(ESI) m/z = 302 (M+H)+
保持時間:0.90分(分析条件SQDAA05)

LCMS(ESI) m/z = 246 (M+H)+
保持時間:0.51分(分析条件SQDAA05)
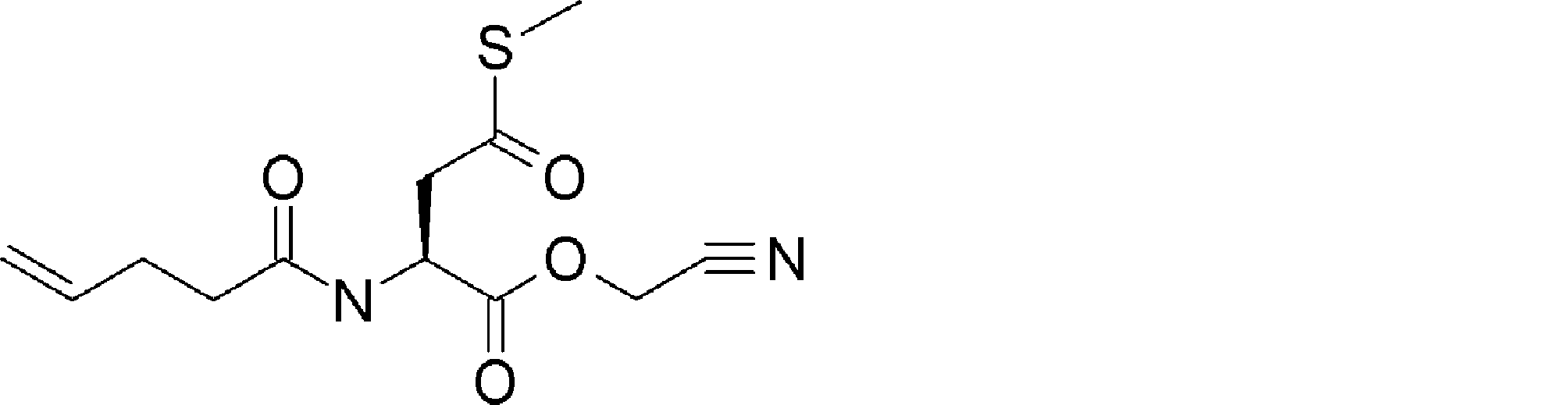
LCMS(ESI) m/z = 285 (M+H)+
保持時間:0.69分(分析条件SQDAA05)
8-1.(S)-5-(tert-ブトキシ)-5-オキソ-4-(ペンタ-4-エンアミド)ペンタン酸(化合物1b-II)の合成
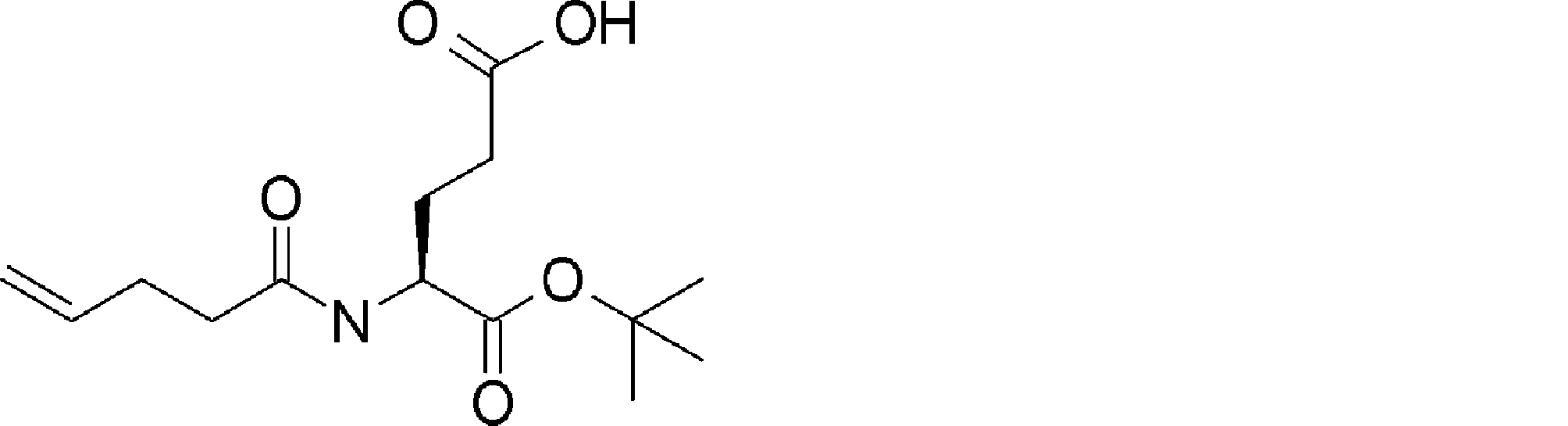
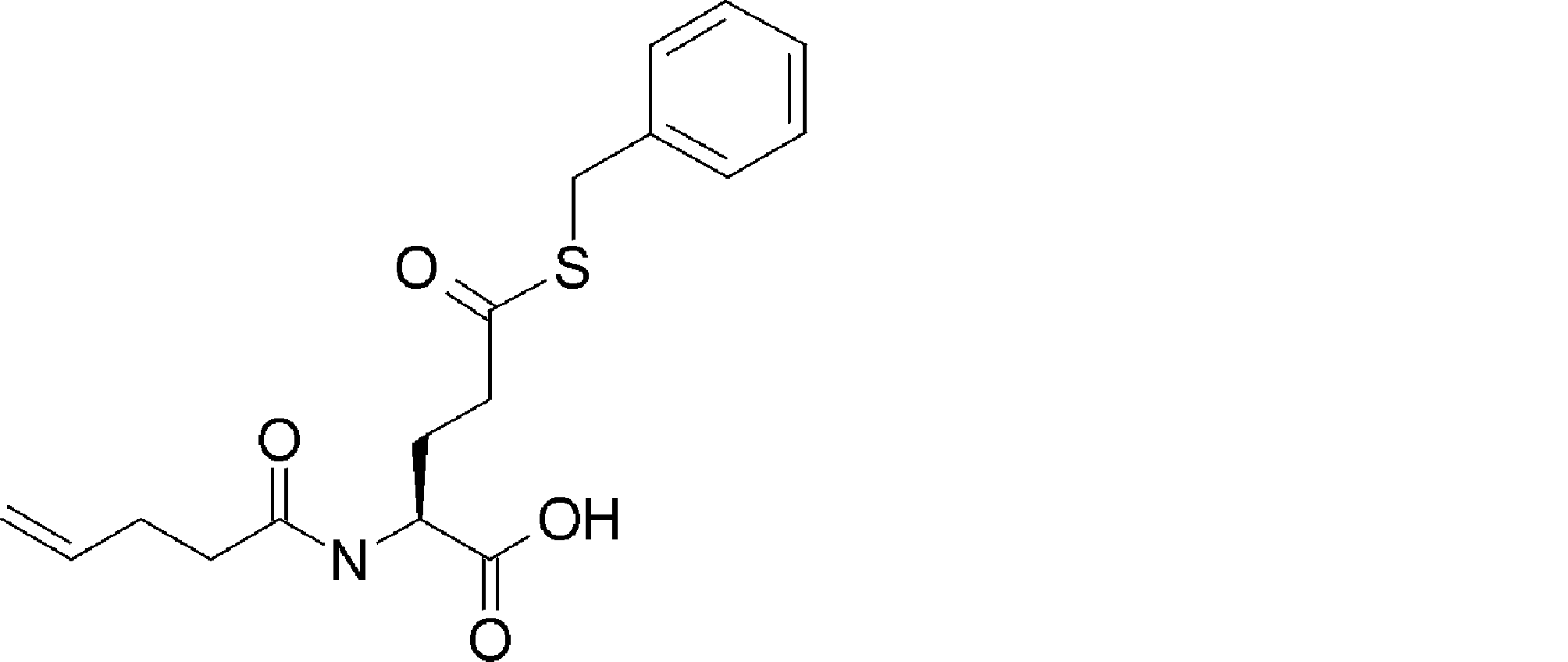
LCMS(ESI) m/z = 336 (M+H)+
保持時間:0.82分(分析条件SQDAA05)
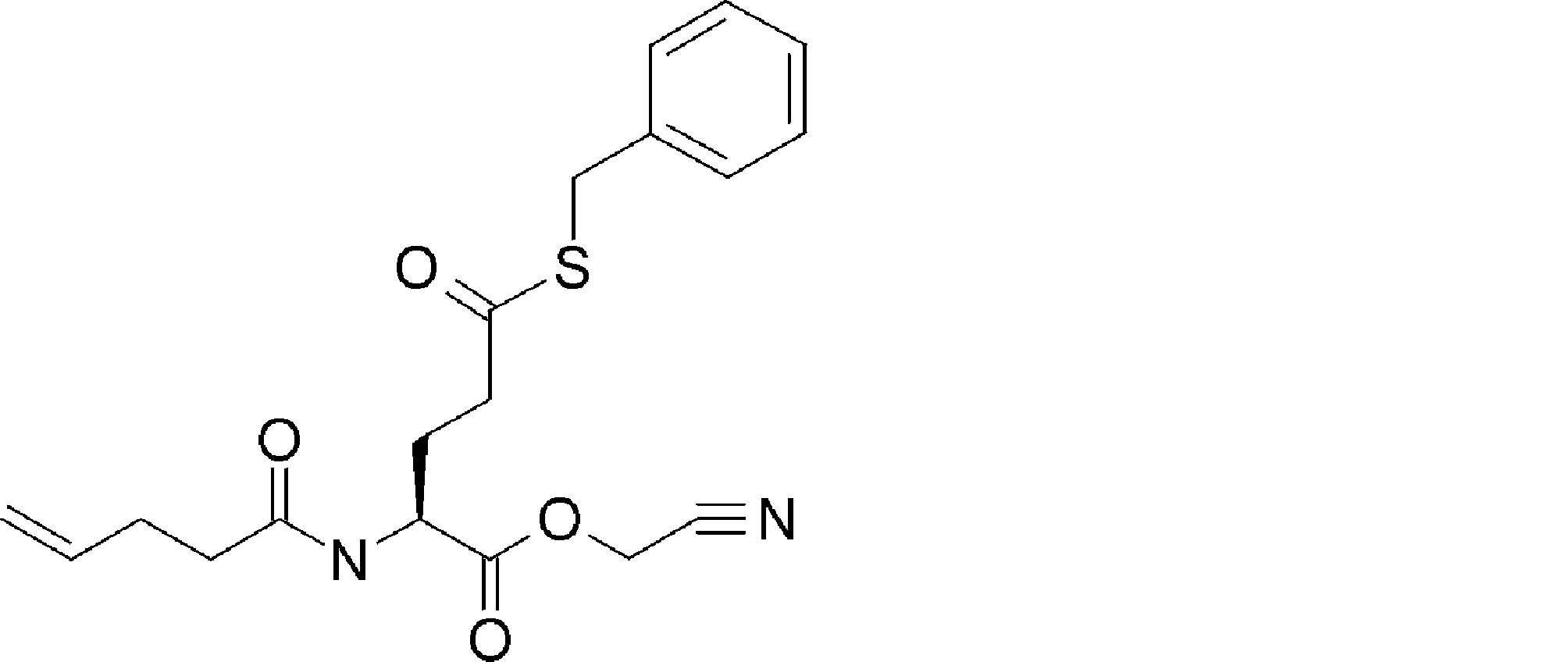
LCMS(ESI) m/z = 375 (M+H)+
保持時間:0.93分(分析条件SQDAA05)
9-1. 5-(メチルチオ)-5-オキソ-2-(ペンタ-4-エンアミド)ペンタン酸 (S)-tert-ブチル(化合物1e-IIA)の合成

LCMS(ESI) m/z = 316 (M+H)+
保持時間:0.92分(分析条件SQDAA05)

LCMS(ESI) m/z = 260(M+H)+
保持時間:0.54分(分析条件SQDAA05)
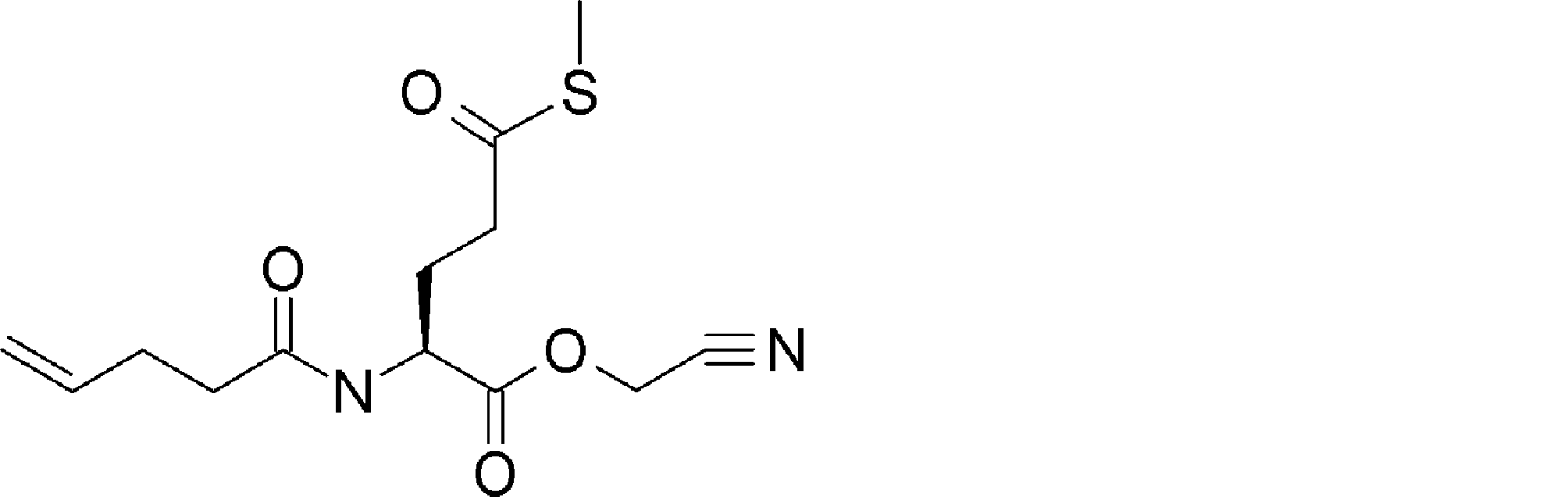
LCMS(ESI) m/z =299(M+H)+
保持時間:0.75分(分析条件SQDAA05)
10-1. 5-オキソ-2-(ペンタ-4-エンアミド)-5-(フェニルチオ)ペンタン酸 (S)-tert-ブチル(化合物1e-IIF)の合成

LCMS(ESI) m/z =378(M+H)+
保持時間:1.03分(分析条件SQDAA05)
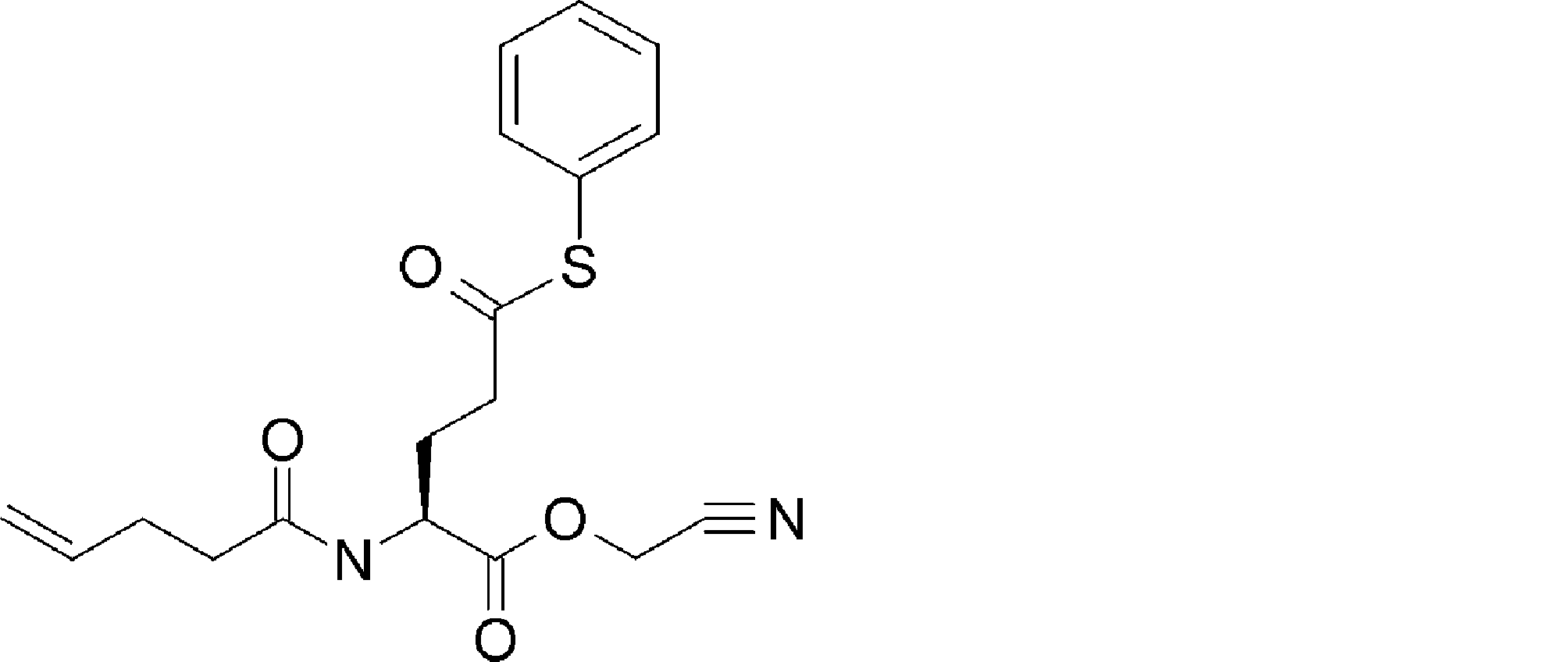
LCMS(ESI) m/z = 361 (M+H)+
保持時間:0.89分(分析条件SQDAA05)
実施例1で合成した側鎖カルボン酸が活性エステル化された化合物(化合物1g)を用いてアミノアシル化pdCpA(化合物1i)の合成を行った。
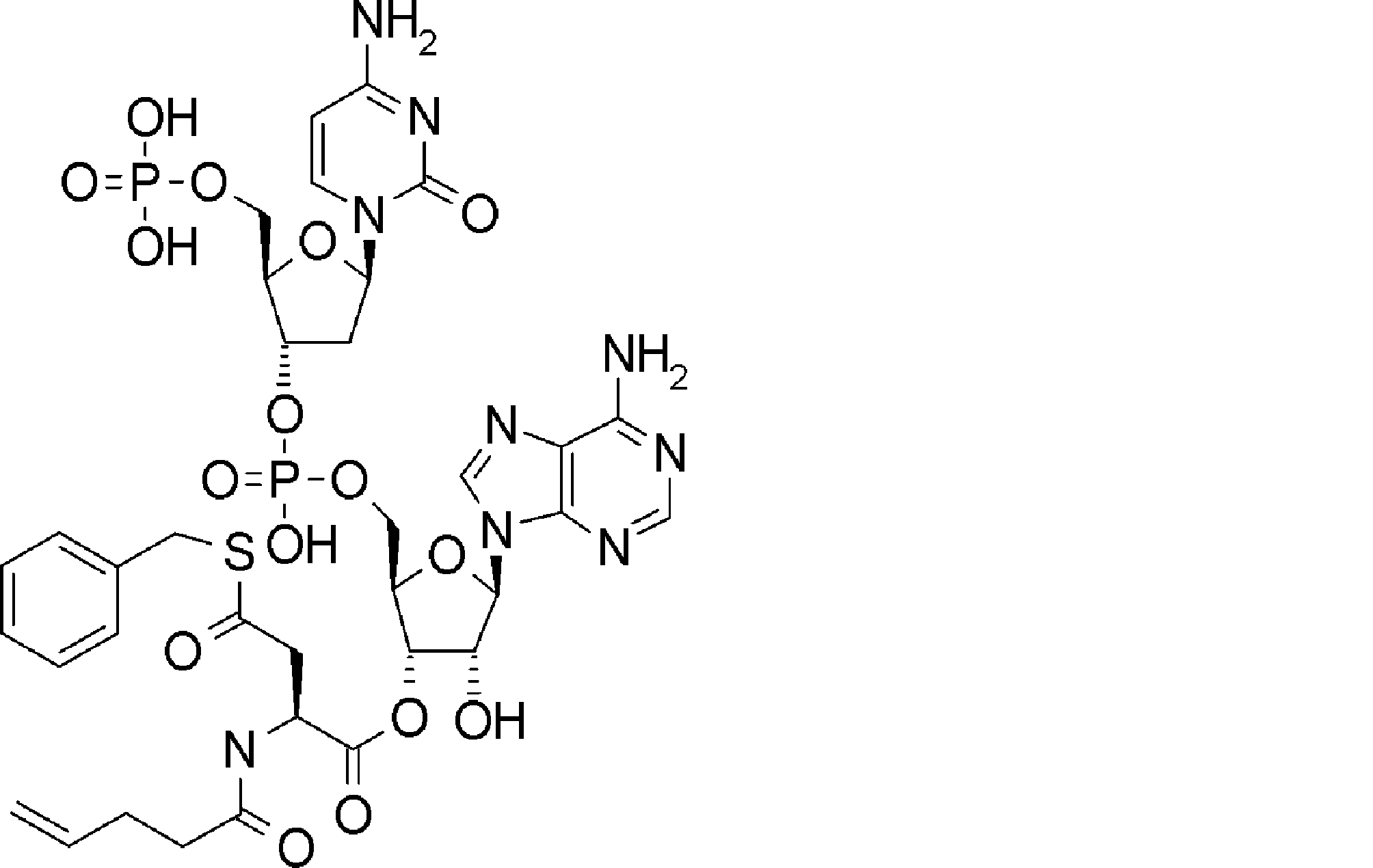
緩衝液A(113ml)にリン酸二水素 ((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-3-(((((2R,3S,4R,5R)-5-(6-アミノ-9H-プリン-9-イル)-3,4-ジヒドロキシテトラヒドロフラン-2-イル)メトキシ)(ヒドロキシ)ホスホリル)オキシ)テトラヒドロフラン-2-イル)メチル(化合物1h)(200mg、0.314mmol)の水溶液(6.25ml)と4-(ベンジルチオ)-4-オキソ-2-(ペンタ-4-エンアミド)ブタン酸 (S)-シアノメチル(化合物1g-ID)(454mg、1.26mmol)のテトラヒドロフラン(3.15ml)溶液を加え、室温で1時間攪拌した。その後、トリフルオロ酢酸(2.52ml、33.9mmol)を加えた後に、凍結乾燥させた。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.05%トリフルオロ酢酸水溶液/0.05%トリフルオロ酢酸アセトニトリル溶液=100/0→60/40)にて精製し、4-(ベンジルチオ)-4-オキソ-2-(ペンタ-4-エンアミド)ブタン酸 (2S)-(2R,3S,4R,5R)-2-((((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-((ホスホノオキシ)メチル)テトラヒドロフラン-3-イル)オキシ)(ヒドロキシ)ホスホリル)オキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イル(化合物1i-ID)(70.9mg、24%)を得た。
LCMS(ESI) m/z = 940 (M+H)+
保持時間:0.53分(分析条件SQDFA05)
N,N,N-トリメチルヘキサデカン-1-アミニウム 塩化物(6,40g、20mmol)とイミダゾール(6.81g、100mmol)の水溶液に酢酸を添加し、pH8、20mM N,N,N-トリメチルヘキサデカン-1-アミニウム、100mMイミダゾールの緩衝液A(1L)を得た。

LCMS(ESI) m/z =954(M+H)+
保持時間:0.80分(分析条件SMDmethod1)
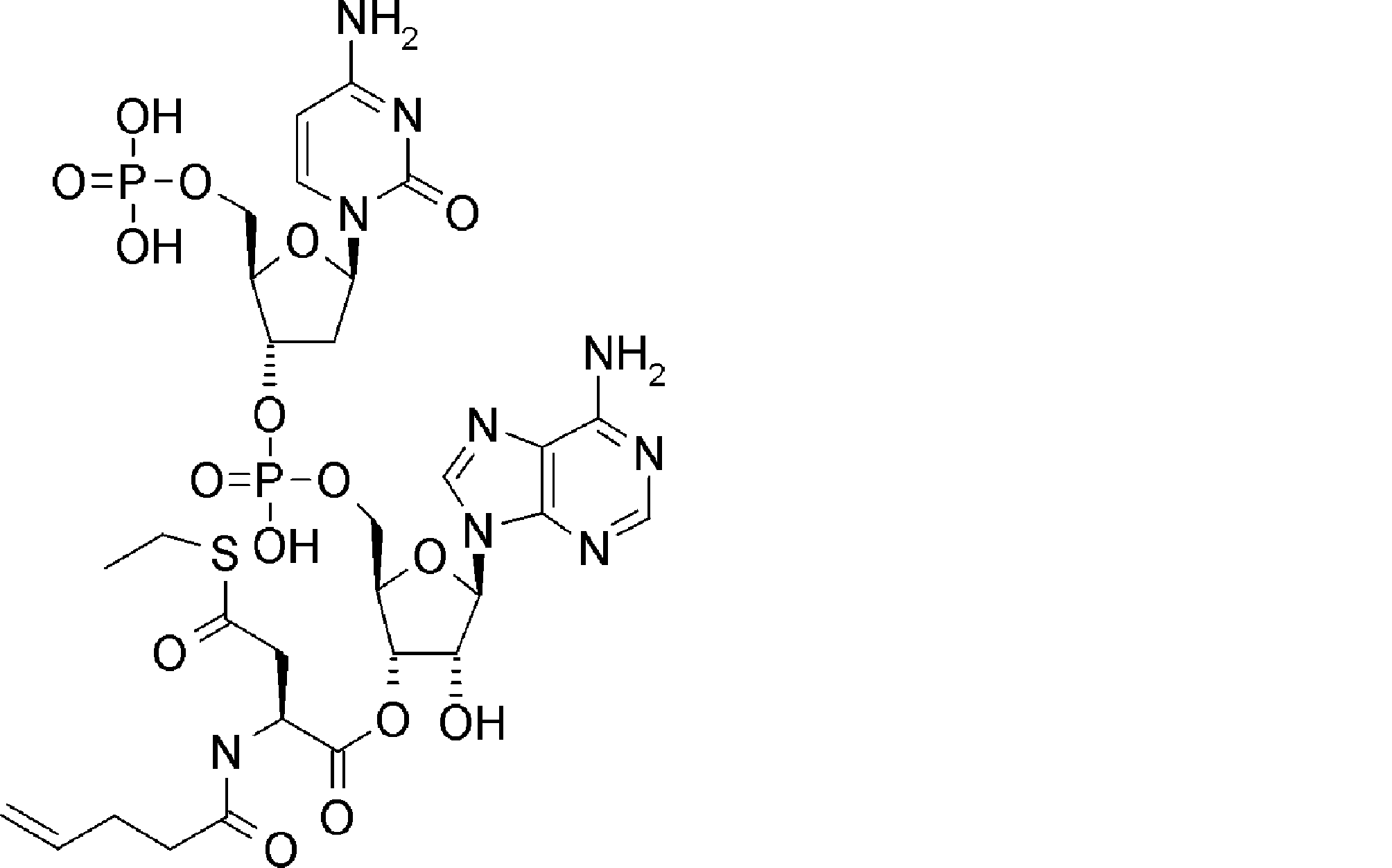
LCMS(ESI) m/z =878(M+H)+
保持時間:0.66分(分析条件SMDmethod1)
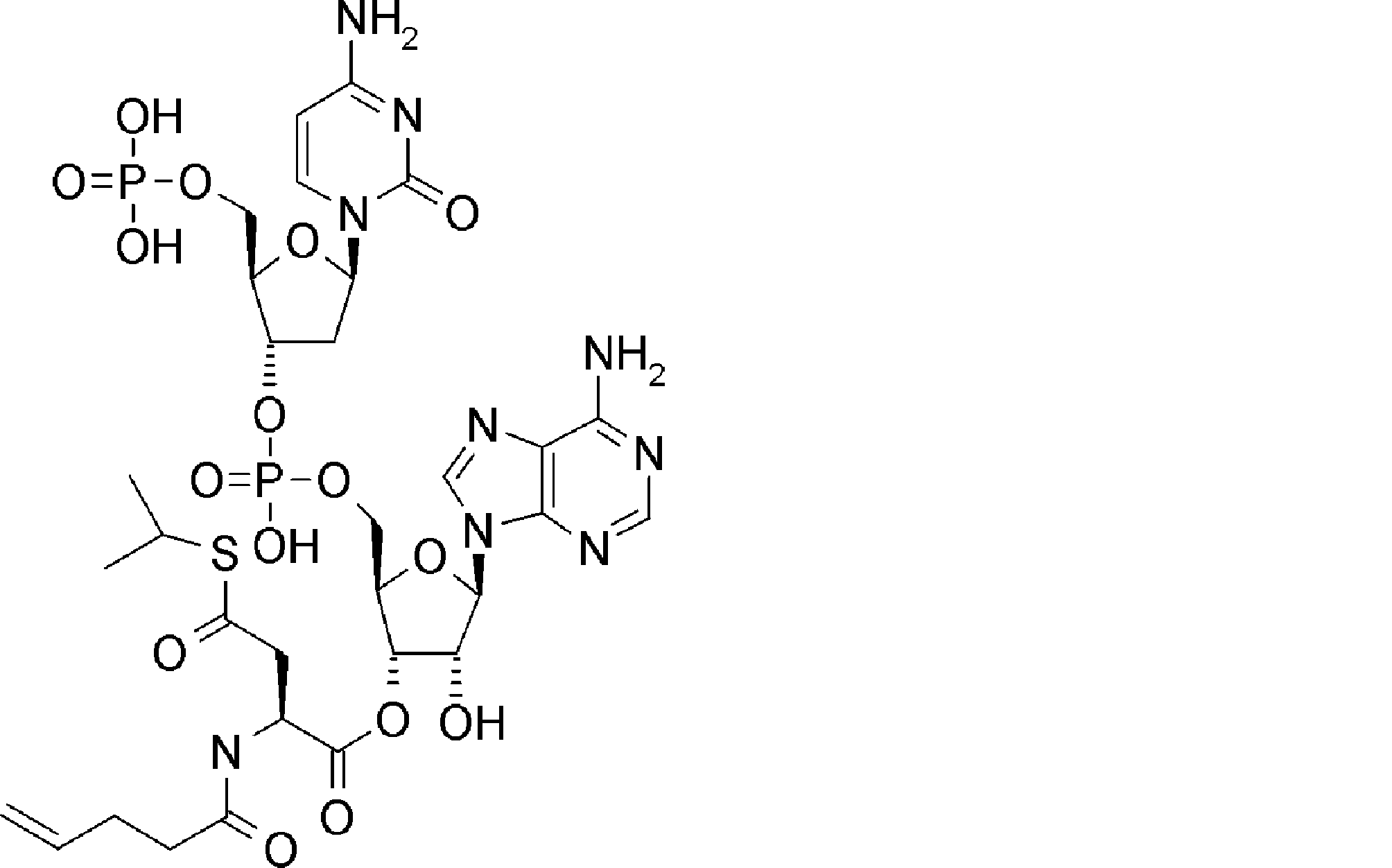
LCMS(ESI) m/z =892(M+H)+
保持時間:0.70分(分析条件SMDmethod1)
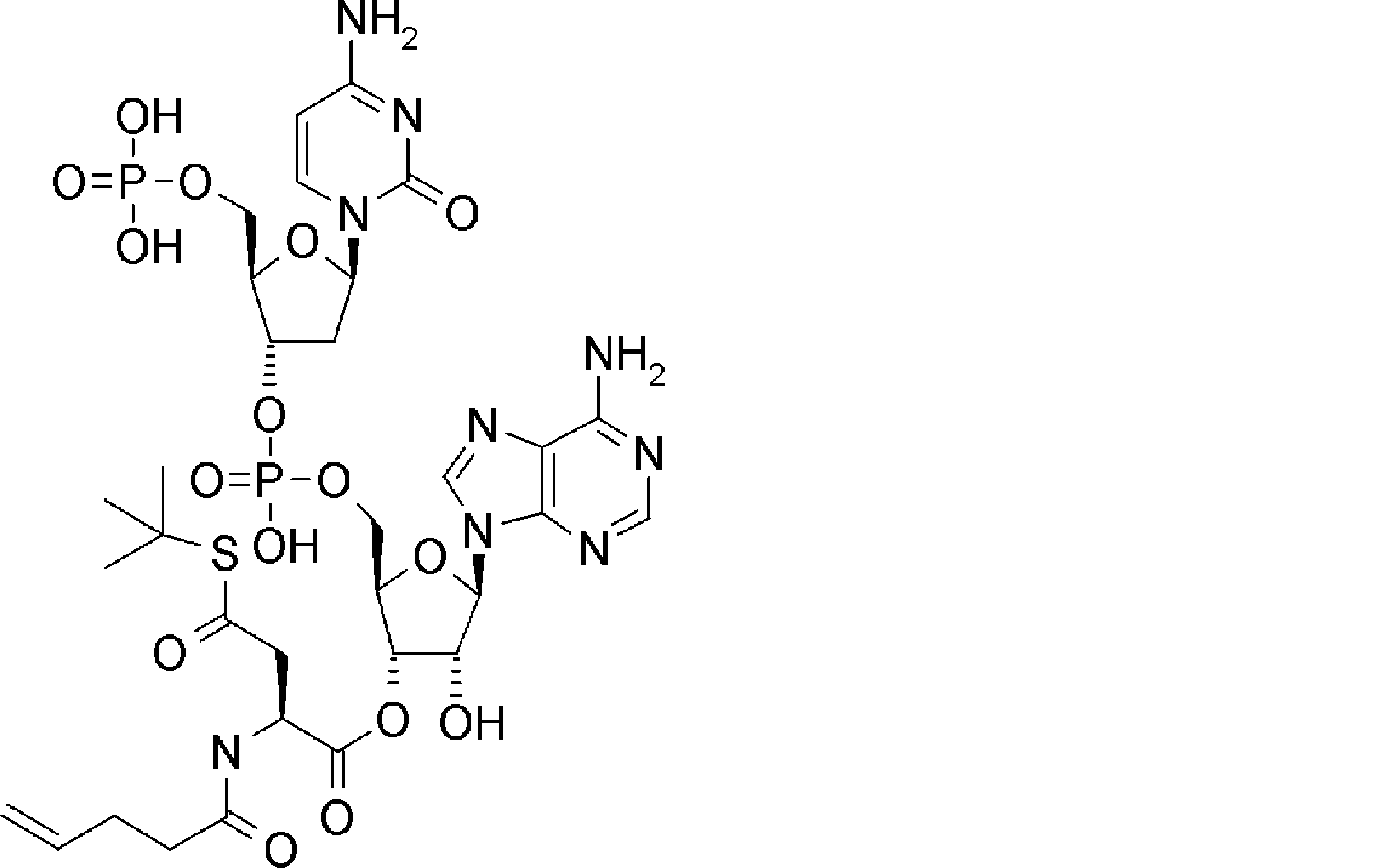
LCMS(ESI) m/z =906(M+H)+
保持時間:0.74分(分析条件SMDmethod1)

LCMS(ESI) m/z =864(M+H)+
保持時間:0.40分(分析条件SQDFA05)

LCMS(ESI) m/z =954(M+H)+
保持時間:0.55分(分析条件SQDFA05)
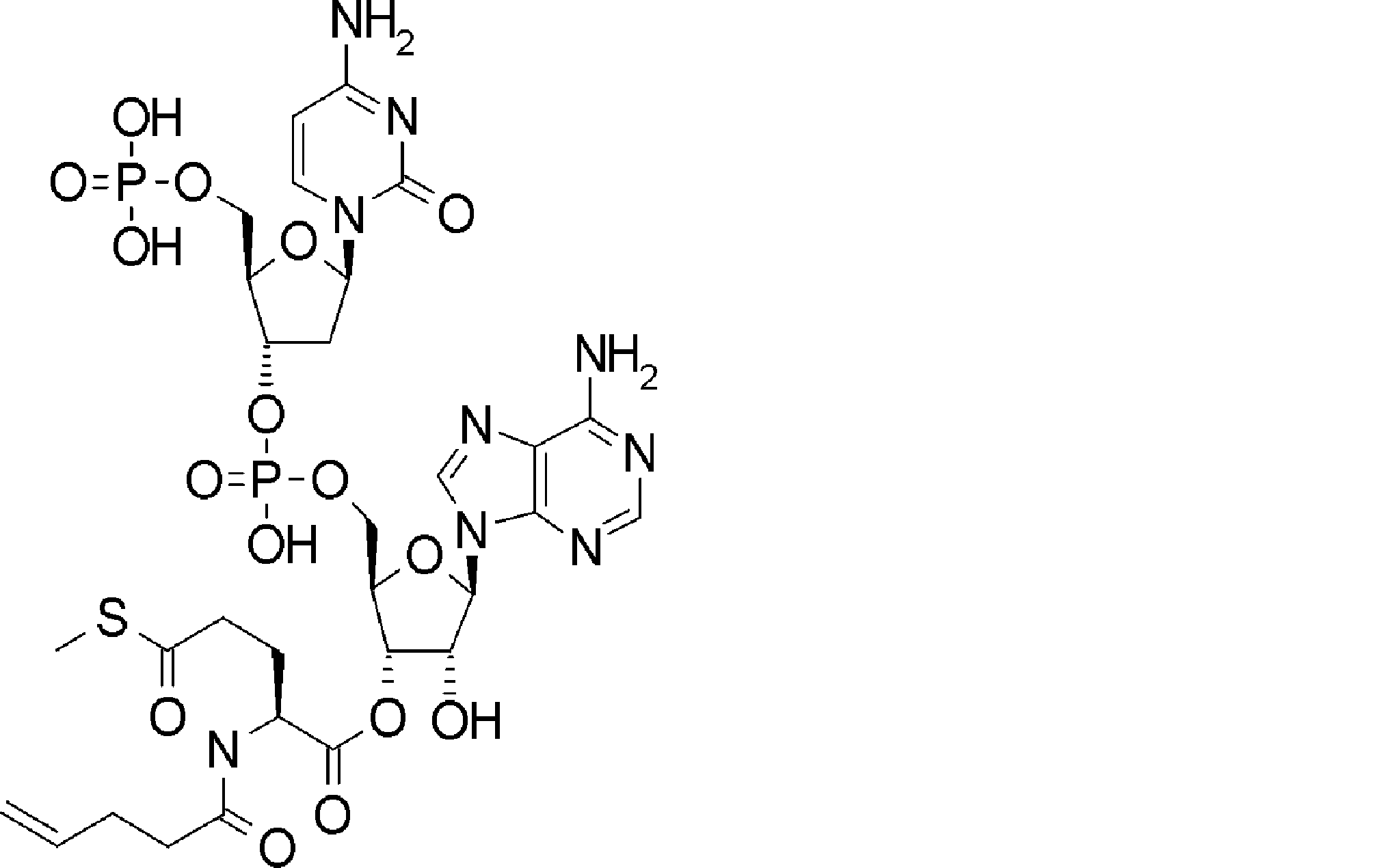
LCMS(ESI) m/z =878(M+H)+
保持時間:0.43分(分析条件SQDFA05)
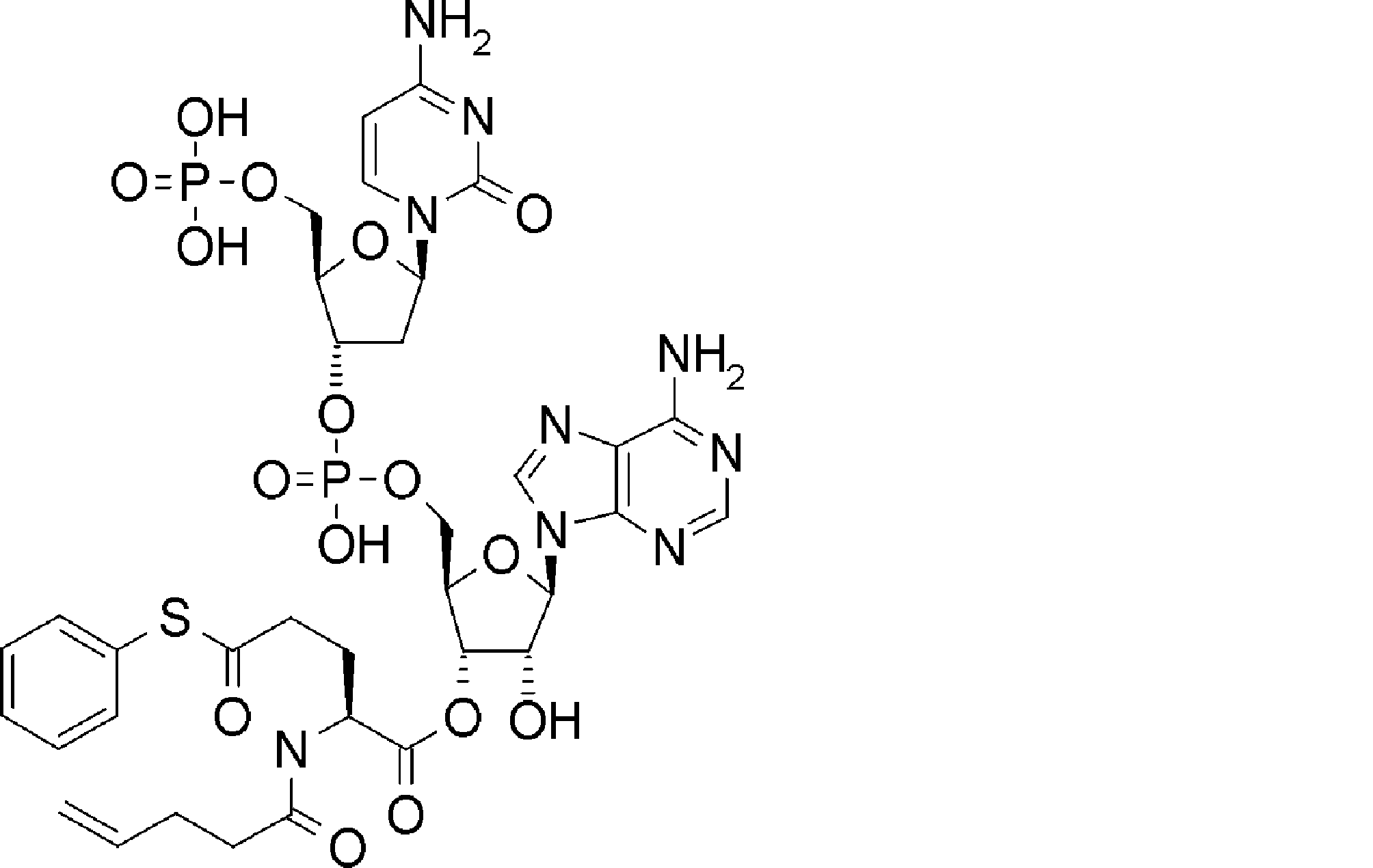
(化合物1i-IIF)
LCMS(ESI) m/z =940(M+H)+
保持時間:0.70分(分析条件SQDFA05)

LCMS(ESI) m/z =828(M-H)-
保持時間:0.65分(分析条件SQDFA05)
(1i-IIF-B2)
LCMS(ESI) m/z =828(M-H)-
保持時間:0.66分(分析条件SQDFA05)
これまで示してきたように、Rex1=Me、Et,iPr,Ph,Bz、Phenetyl基を有する側鎖カルボン酸が活性エステル化されたアミノアシル化pdCpA(化合物1i)を合成した。
以下の方法に従って、側鎖カルボン酸がチオエステル化されたアミノアシル化tRNAの合成を行なった。
鋳型DNA(配列番号D-1)から、RiboMAX Large Scale RNA production System T7(Promega社,P1300)を用いたin vitro の転写により3'端のCAを欠くtRNAEnAsnGAG (-CA)(配列番号R-1)を合成し、RNeasy Mini kit(Qiagen社)により精製した。
tRNAEnAsnGAG (-CA) DNA配列:
GGCGTAATACGACTCACTATAGGCTCTGTAGTTCAGTCGGTAGAACGGCGGACTgagAATCCGTATGTCACTGGTTCGAGTCCAGTCAGAGCCGC
tRNAEnAsnGAG (-CA) RNA配列:
GGCUCUGUAGUUCAGUCGGUAGAACGGCGGACUgagAAUCCGUAUGUCACUGGUUCGAGUCCAGUCAGAGCCGC
50μM 転写tRNAEnAsnGAG (-CA)(配列番号R-1) 10μLに、10X ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP) 2μL、Nuclease free water 4μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 20unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)2μLおよび、5mMの側鎖カルボン酸が活性エステル化されたアミノアシル化pdCpA(化合物1i)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。ライゲーション反応液 20μLに、3M酢酸ナトリウム 4μLと125mM ヨウ素(水:THF=1:1溶液)24μLを加え、室温で1時間、脱保護を行った。アミノアシル化tRNA(化合物AT-1)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-1)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Asp(SMe)-tRNAEnAsnGAG
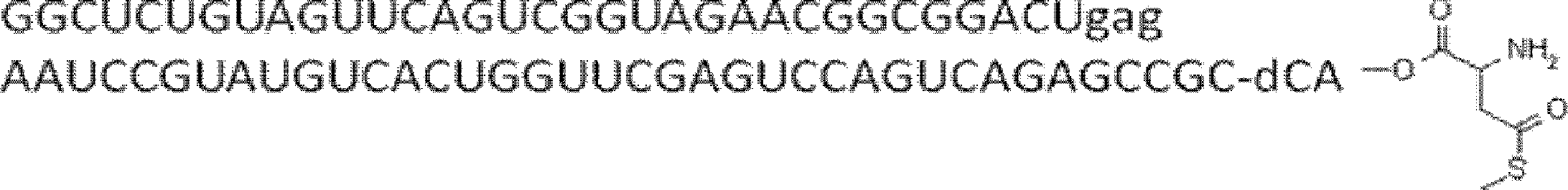
Asp(SiPr)-tRNAEnAsnGAG
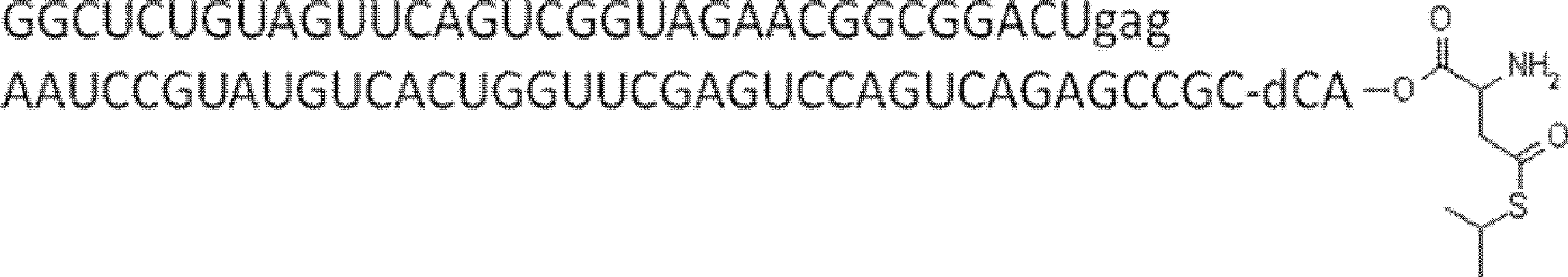
Asp(SEt)-tRNAEnAsnGAG
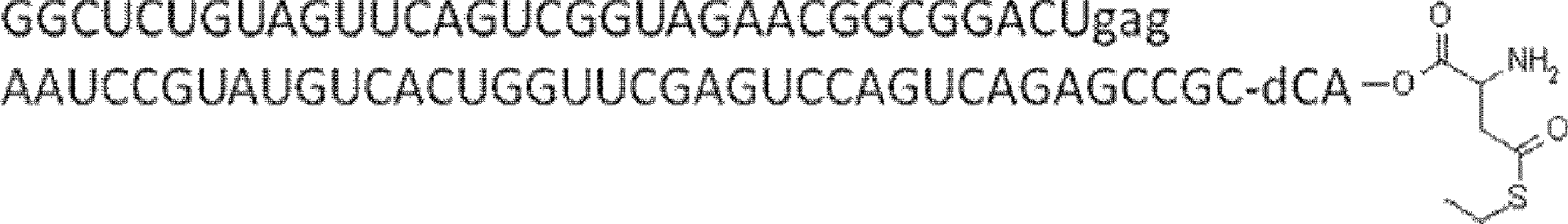
Asp(StBu)-tRNAEnAsnGAG
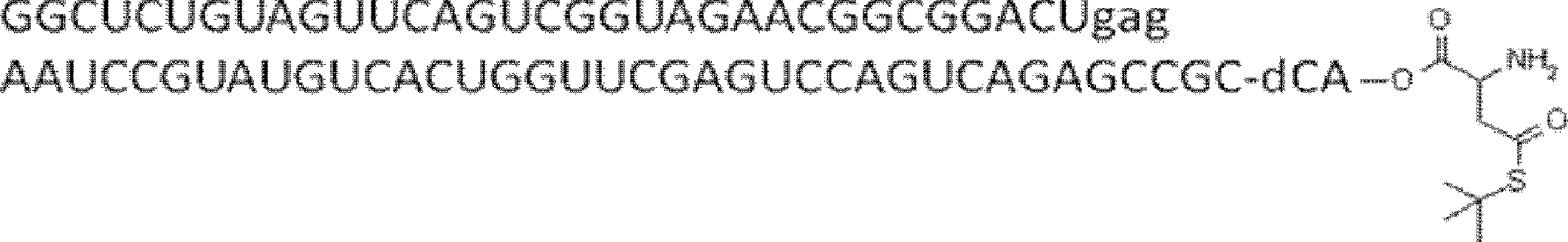
Asp(SBn)-tRNAEnAsnGAG
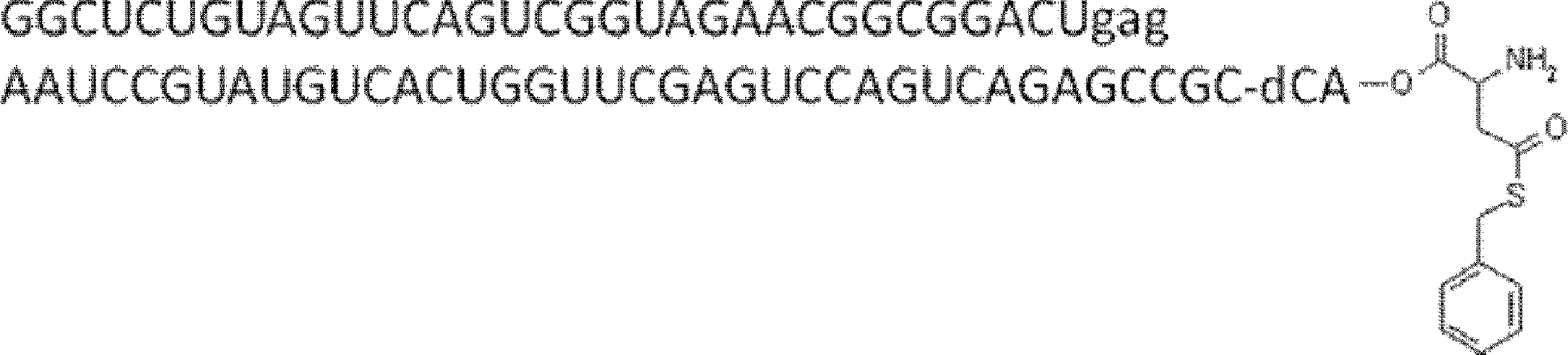
Asp(SPhenetyl)-tRNAEnAsnGAG
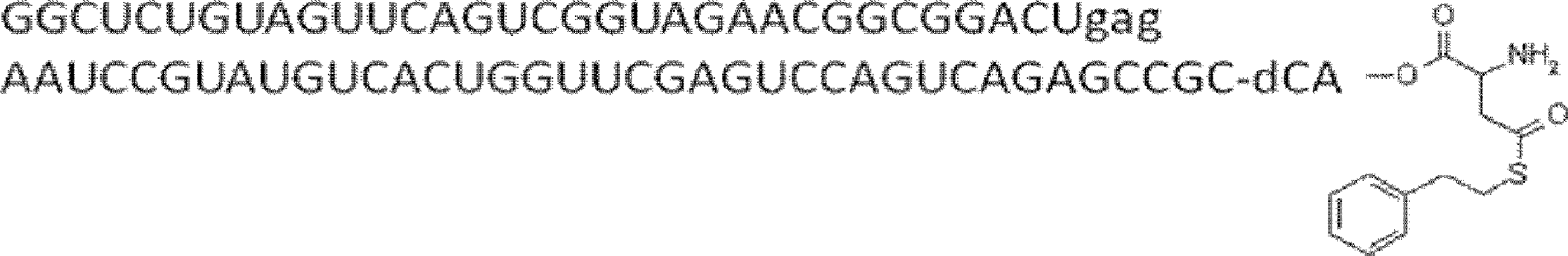
Glu(SMe)-tRNAEnAsnGAG
Glu(SBn)-tRNAEnAsnGAG
様々なアミノ酸によりアミノアシル化されたtRNAを、無細胞翻訳系に加えて翻訳を開始することにより、所望の非天然アミノ酸を含有するポリペプチドの翻訳合成を行った。翻訳系は、鋳型DNAからの転写系を含む原核生物由来の再構成無細胞タンパク質合成系であるPURE systemを用いた。具体的には、転写翻訳液(5%(v/v) T7 RNA polymerase RiboMAX Enzyme Mix (Promega社,P1300),2mM GTP,2mM ATP,1mM CTP,1mM UTP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM グルタミン酸カリウム,12mM 酢酸マグネシウム,2mMスペルミジン,1mM ジチオスレイトール,1.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.6μM メチオニルtRNAトランスフォルミラーゼ,0.26μM EF-G,0.25μM RF1,0.24μM RF2,0.17μM RF3,0.5μM RRF,2.7μM IF1,0.4μM IF2,1.5μM IF3,40μM EF-Tu,84μM EF-Ts,1.2μM リボソーム,0.73μM AlaRS,0.03μM ArgRS,0.38μM AsnRS,0.13μM AspRS,0.02μM CysRS,0.06μM GlnRS,0.23μM GluRS,0.09μM GlyRS,0.02μM HisRS,0.4μM IleRS,0.04μM LeuRS,0.11μM LysRS,0.03μM MetRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.02μM TyrRS,0.02μM ValRS(自家調製タンパクは基本的はHisタグ付加タンパクとして調製した))に、0.02μM 鋳型DNA、それぞれの鋳型DNAにコードされているタンパク質性アミノ酸群をそれぞれ300μMずつ、ならびに50μMの側鎖カルボン酸が活性エステル化されたアミノアシル化tRNA(化合物AT-1)を翻訳反応混合物に添加し、37℃で30分から1時間静置することで行った。
20nM 鋳型DNA Mtyg_R(配列番号D-2)に、0.3mM Gly,0.3mM Met,0.3mM Arg,0.3mM Thr,0.3mM Tyrおよび、50μM Glu(SBn)-tRNAEnAsnGAG(化合物AT-1-IID)を含む前述の転写翻訳液を37℃で60分間保温した。得られた翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析した。しかしながら、図2に示すように分子量799から2000の範囲に翻訳ペプチドに由来するマススペクトルのピークをMALDI-MSにて確認することが出来なかった。例えば、分子量882、918、1256付近に検出されたピークは、陰性対照として鋳型DNAを加えないPURESystemを分析したときにも観測されるピークであることから、翻訳ペプチド由来のピークではない。
Mtyg_R DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgACTACAACGCGActttactaccgtcgtggcggcTAATAAATAGATAG
MetThrThrThrArg[Glu(SBn)]TyrTyrArgArgGlyGly
Not detected (calc. 1595.7)
2-1.Asp(SMe)を含むモデルペプチドの翻訳合成
20nM 鋳型DNA Mtryg3(配列番号D-3)、Leuを除く各0.3mMの19種類のタンパク質性アミノ酸、50μM Asp(SMe)-tRNAEnAsnGAG(化合物AT-1-IA)を加えた転写翻訳液を、37℃で60分間保温した。SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析した。その結果、チオエステルを含む全長ペプチドから、脱MeSHされた分子量を示すマススペクトル(MS)が主生成物として観測された(図3)。
Mtryg3 DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgACTACAACGCGActttactaccgtggcggcTAGTAGATAGATAG
MetThrThrThrArg[Asp(SMe)]TyrTyrArgGlyGly
m/z: [H+M]+ = 1302.5 (チオエステル含む全長ペプチドCalc. 1349.6, 脱MeSHペプチドCalc. 1301.6) (観測された分子量1302の化合物をペプチドP-3とする)
20nM 鋳型DNA Ft02A_dR(配列番号D-4)に、前述の転写翻訳液に、Leuを除く各0.3mMの19種類のタンパク質性アミノ酸,および前記の方法で調製した50μM 化合物AT-1-ID(Asp(SBn)-tRNAEnAsnGAG)を加え、37℃で60分間翻訳した。翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析したところ、チオエステルアミノ酸を含む全長ペプチドP-5が生成した後に、脱BnSH化されたMSスペクトルが同様に主生成物として確認された(ペプチドP-6)。このことから脱BnSH化される反応点としてはフェノール性水酸基では無いことが確認された(図5)。
GTAATACGACTCACTATAGGGTTAACTTTAAgaaggagatatacatATGACTACAACGgcgggcggcCTTttttttggcggcAAATAATAA
MetThrThrThrAlaGlyGly[Asp(SBn)]PhePheGlyGlyLys
前述の翻訳反応物P-6を、333mM Bicine KOH pH9.0中で、95℃で15分間加水分解を行い、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析した。その結果、脱BnSHペプチドP-6が加水分解され18分子量の増加が確認された(ペプチド化合物P-4)。想定されるチオエステル部位の縮合反応はアミノ基とのアミド形成反応ではなく、比較的加水分解され易いOH基やSH基とのエステル化反応やチオエステル化反応などであると示唆された(図4)。
ペプチド化合物P-4
MALDI-MS:
m/z: [H+M]+ = 1289.6 (P-6の加水分解体Calc. 1288.7)
N末端にアミノ基を持たないペプチドの翻訳合成を行い、以下のように解析した。
2-4-1-1.(S)-2-メトキシ-3-フェニルプロパン酸の合成(化合物7a、MeOFlac)

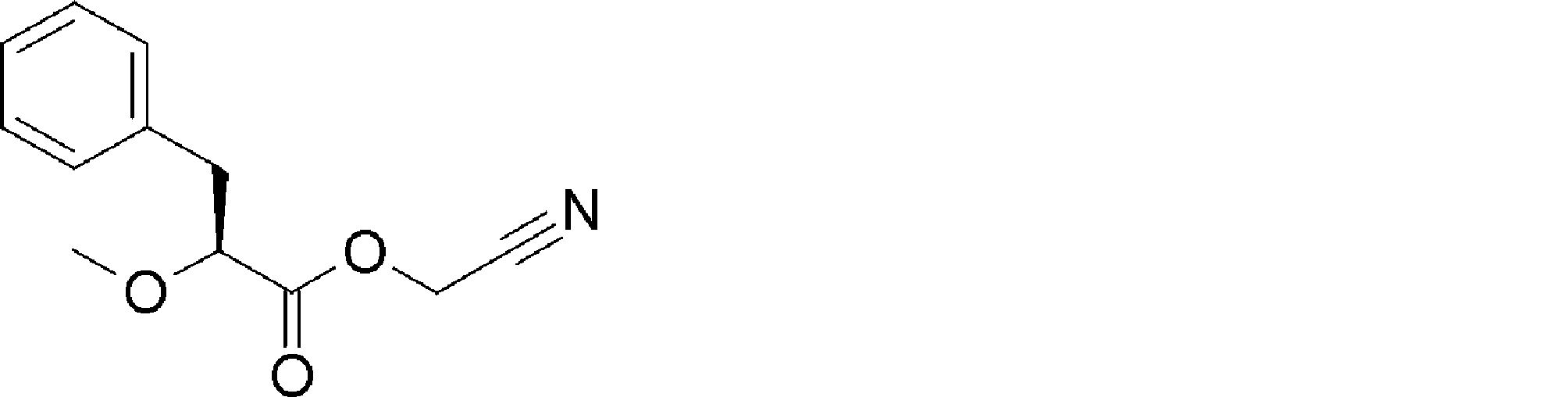
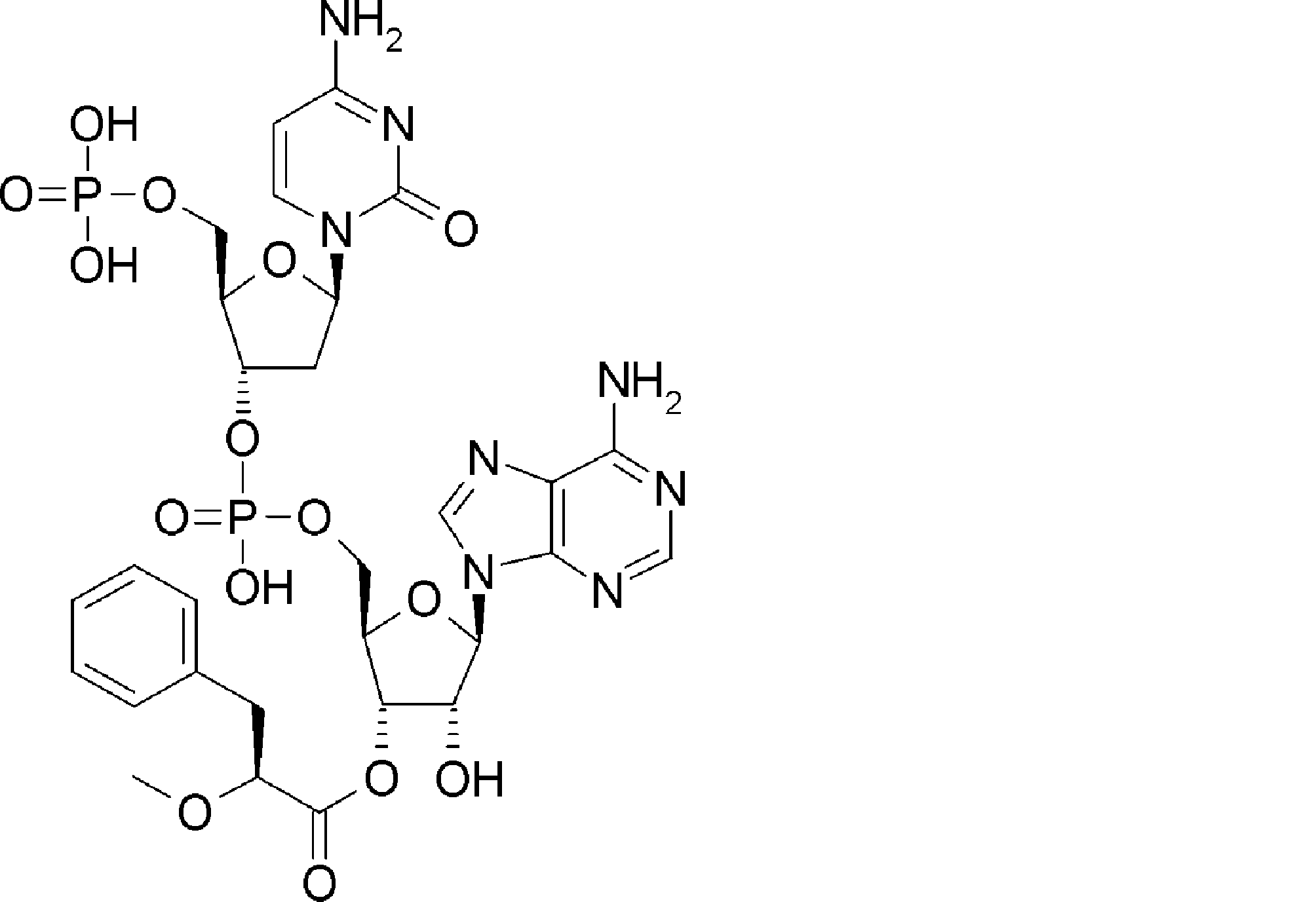
保持時間:0.609分 (分析条件 SMDmethod1)
鋳型DNA(配列番号D-5)から、RiboMAX Large Scale RNA production System T7(Promega社,P1300)を用いたin vitro の転写により3'端のCAを欠くtRNAfMetCAU(-CA)(配列番号R-5)を合成し、RNeasy Mini kit(Qiagen社)により精製した。
GGCGTAATACGACTCACTATAGGCGGGGTGGAGCAGCCTGGTAGCTCGTCGGGCTCATAACCCGAAGATCGTCGGTTCAAATCCGGCCCCCGCAAC
GGCGGGGUGGAGCAGCCUGGUAGCUCGUCGGGCUCAUAACCCGAAGAUCGUCGGUUCAAAUCCGGCCCCCGCAAC
50μM 転写tRNAfMetCAU(-CA)(配列番号R-5) 10μLに、10X ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP)2μL、Nuclease free water 4μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。10unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)2μLおよび、5mMのN末端アミノ基を含まないアシル化pdCpA(化合物7c)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。アシル化tRNA(化合物AT-2)は、フェノール抽出した後、エタノール沈殿により回収した。アシル化tRNA(化合物AT-2)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
化合物AT-2
MeOFlac-tRNAfMetCAU(配列番号:33)
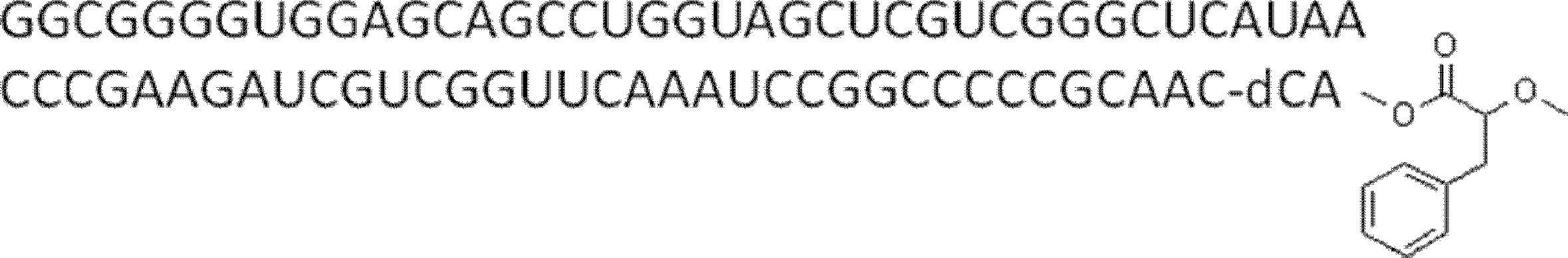
[MeOFlac]ThrThrThrArg[Asp(SBn)]TyrTyrArgArgGlyGly
MALDI-MS:
m/z: [H+M]+ = 1489.6 (ペプチド配列P-7から脱BnSH化されたペプチドP-8 Calc. 1488.6)
20nM 鋳型DNA KA03(配列番号 D-6)に、前述の転写翻訳液、0.1mM 10-HCO-H4folate(10-formyl-5, 6, 7, 8, -tetrahydrophilic acid、特開2003-102495参照)、Leuを除く各0.3mMの19種類のタンパク質性アミノ酸,および前記の方法で調製した50μM Asp(SMe)-tRNAEnAsnGAG(化合物AT-1-IA)を加え、37℃で60分間翻訳した。翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析したところ、チオエステルを含む全長ペプチドのMSが主生成物として観測され、脱MeSH化されたピークは認められなくなった。
以上の結果により、アスパラギン酸型のチオエステル残基は、直後のアミド結合に水素原子が存在する場合、これと反応してアスパルチミドを形成したことが明らかとなった(翻訳ペプチドP-3、P-6、P-8ではいずれもチオエステルのC末端側となりのアミド結合の水素原子と反応してアスパルチミドを形成していた)。一方で、直後のアミノ酸残基をN-アルキルのアミノ酸にすることによって望みのチオエステルを含む全長ペプチドの翻訳に成功した(図7)。
KA03 DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgACTAGAACTaaggcgTACTGGAGCcttCCGggcggctaa
MetThrArgThrLysAlaTyrTrpSer[Asp(SMe)]ProGlyGly
MALDI-MS: m/z: [H+M]+ = 1527.7 (翻訳ペプチドP-9 Calc. 1526.7,)
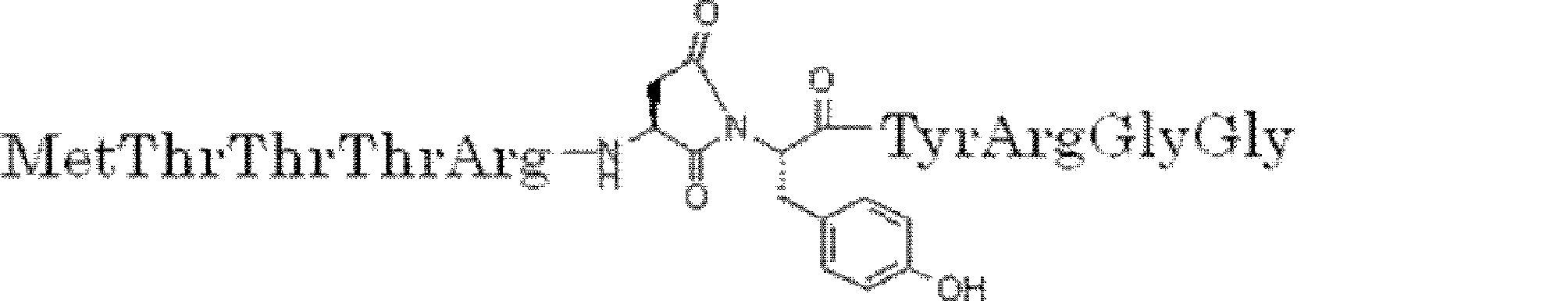
m/z: [H+M]+ = 1302.5 (チオエステル含む全長ペプチドCalc. 1348.6, 脱MeSHペプチドCalc. 1301.6) (観測された分子量1302の化合物をペプチドP-3とする)
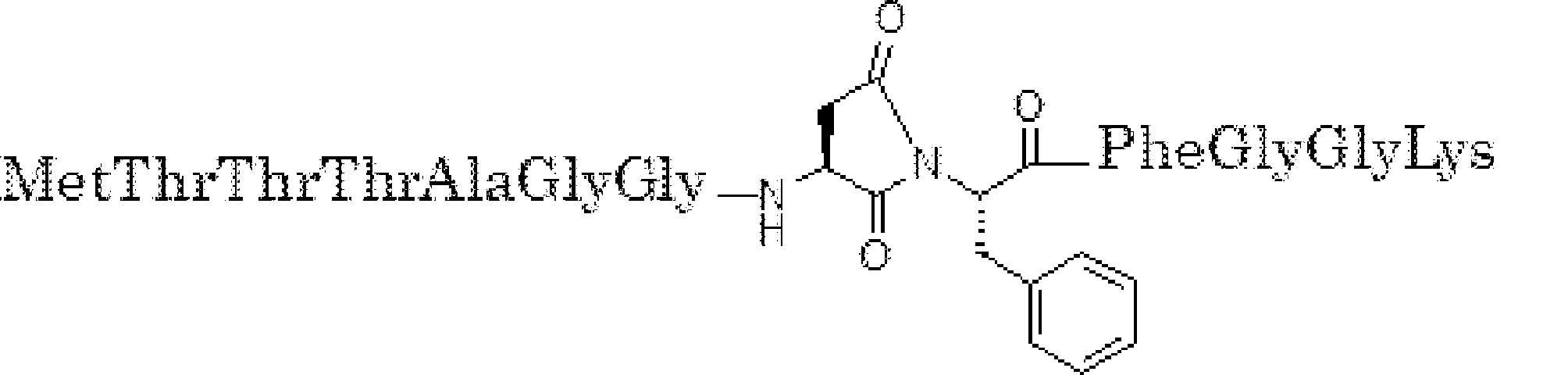
MALDI-MS: m/z: [H+M]+ = 1271.8 (ペプチドP-5が脱BnSH化されたペプチド Calc. 1270.6)

MALDI-MS:m/z: [H+M]+ = 1639.7 (Calc. 1638.7)
チオエステルの翻訳合成が可能となったため、これとアミド結合縮合させるアミノ基側のユニットとして良好な要件を特定するための実験を以下のような手順で行った。
図8に記載の方法に従って、アスパラギン酸もしくはグルタミン酸のチオエステルを有するペプチドモデルとして化合物5b-1および5b-2を合成した。
1-1. 4-(ベンジルチオ)-2-((tert-ブトキシカルボニル)アミノ)-4-オキソブタン酸 (S)-ベンジル(化合物5b-1)の合成
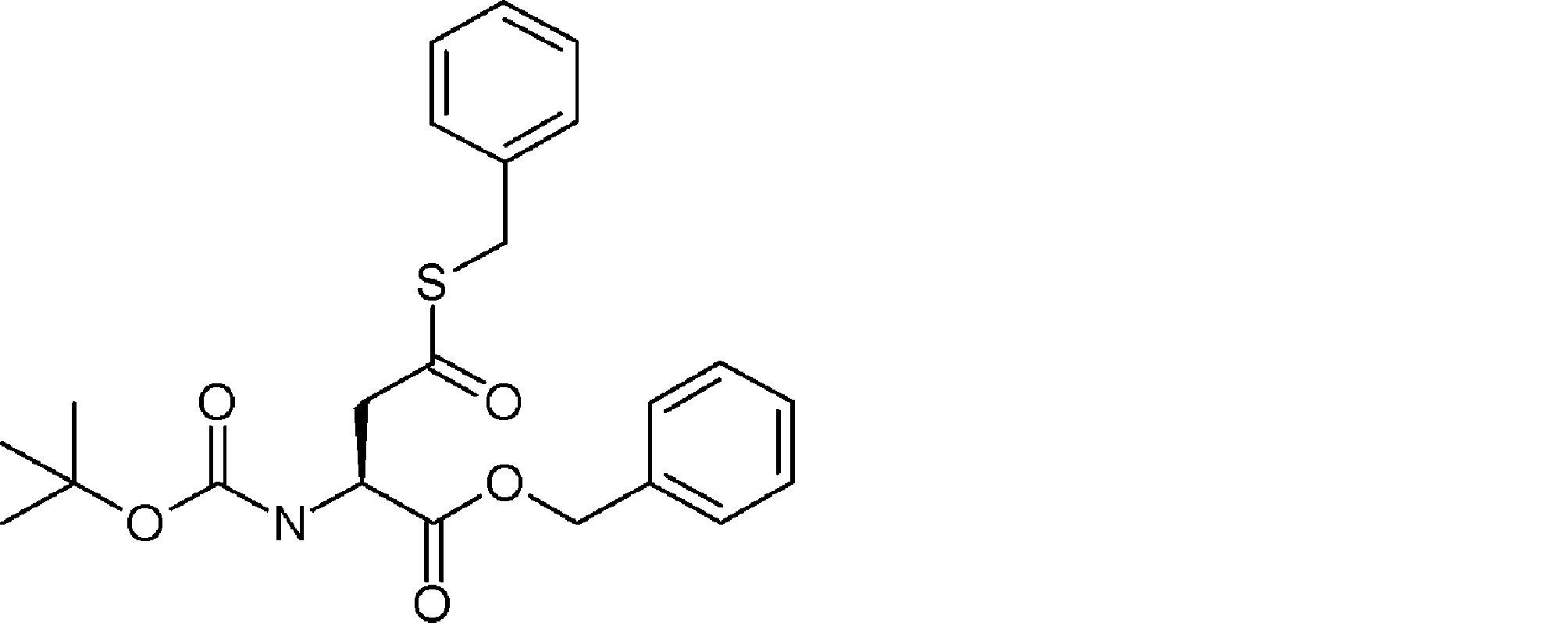
LCMS(ESI) m/z = 430 (M+H)+
保持時間:1.10分(分析条件SQDAA05)
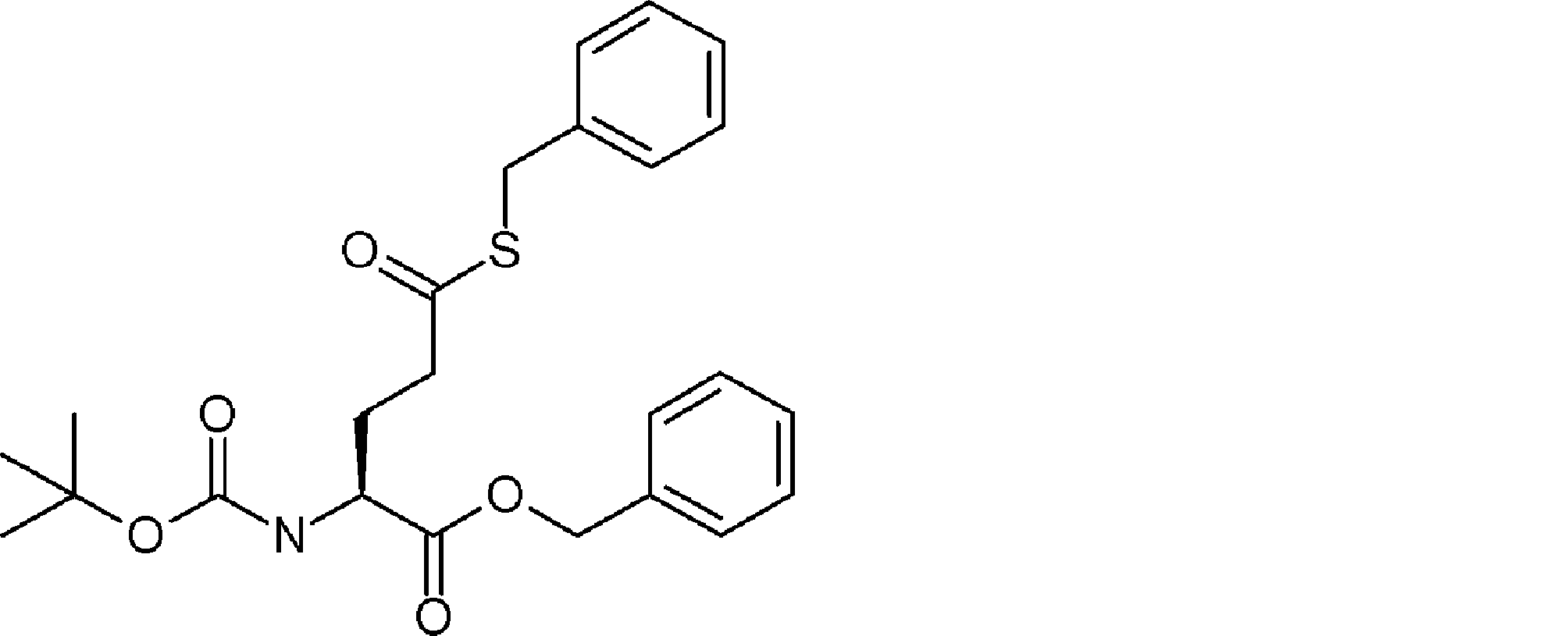
LCMS(ESI) m/z = 444 (M+H)+
保持時間:1.11分(分析条件SQDAA05)

LCMS(ESI) m/z = 448 (M+H)+
保持時間:1.09分(分析条件SQDAA05)
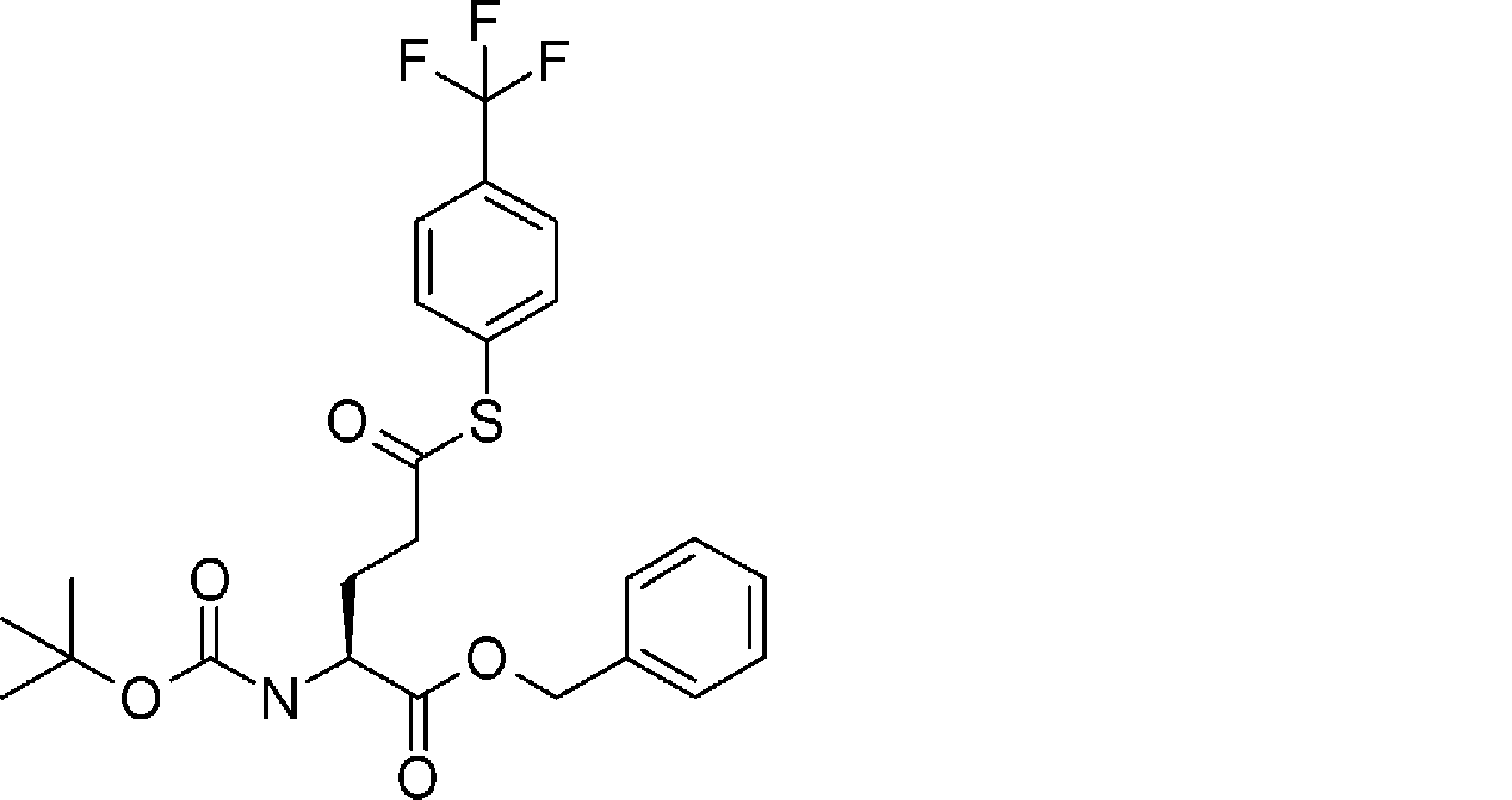
LCMS(ESI) m/z = 498 (M+H)+
保持時間:1.12分(分析条件SQDAA05)
図8に示すように、チオエステルモデル化合物5bまたは5e、5fと、システインあるいはグリシン誘導体モデル化合物とを反応させることで、チオエステルと容易にアミド結合を生成する基質を選択した。
2-1. 2-((tert-ブトキシカルボニル)アミノ)-4-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-4-オキソブタン酸 (S)-ベンジル(化合物5c-1)の合成

LCMS(ESI) m/z = 455 (M+H)+
保持時間:0.98分(分析条件SQDAA05)
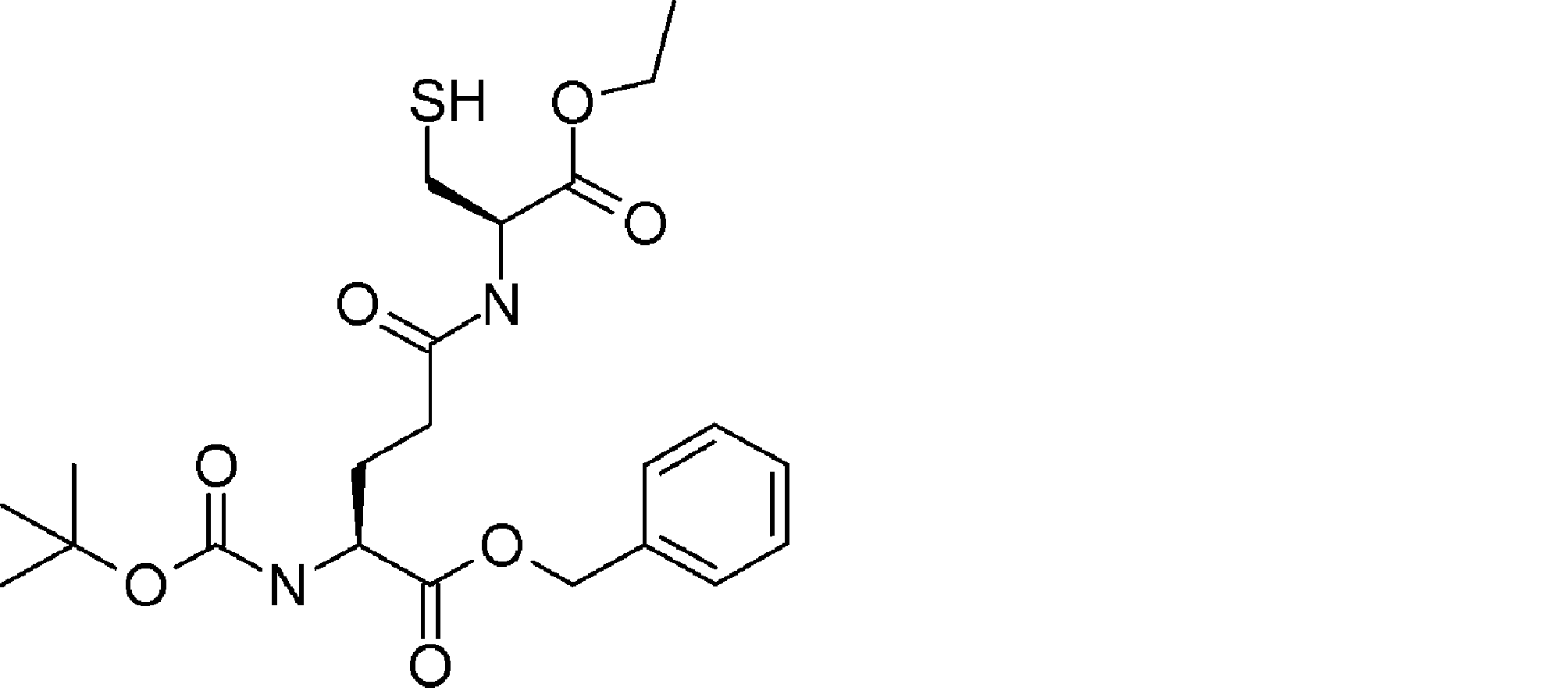
LCMS(ESI) m/z = 469 (M+H)+
保持時間:0.99分(分析条件SQDAA05)
トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(1.0g、3.49mmol)の水(6.8ml)溶液にトリエチルアミン(1.64ml)加え、pH7とし、0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液を得た。
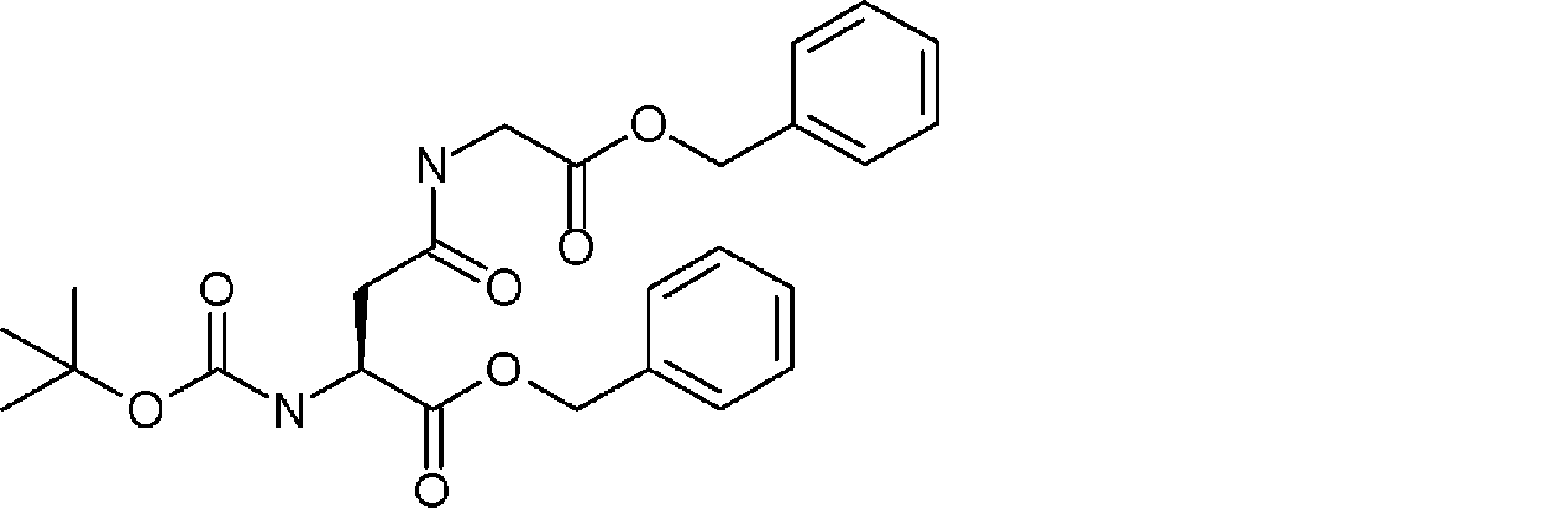
化合物5d-1
LCMS(ESI) m/z = 472 (M+H)+
保持時間:0.99分(分析条件SQDAA05)
化合物5d-1b
LCMS(ESI) m/z = 380(M+H)+
保持時間:0.90分(分析条件SQDAA05)

化合物5d-2
LCMS(ESI) m/z = 485 (M+H)+
保持時間:1.01分(分析条件SQDAA05)
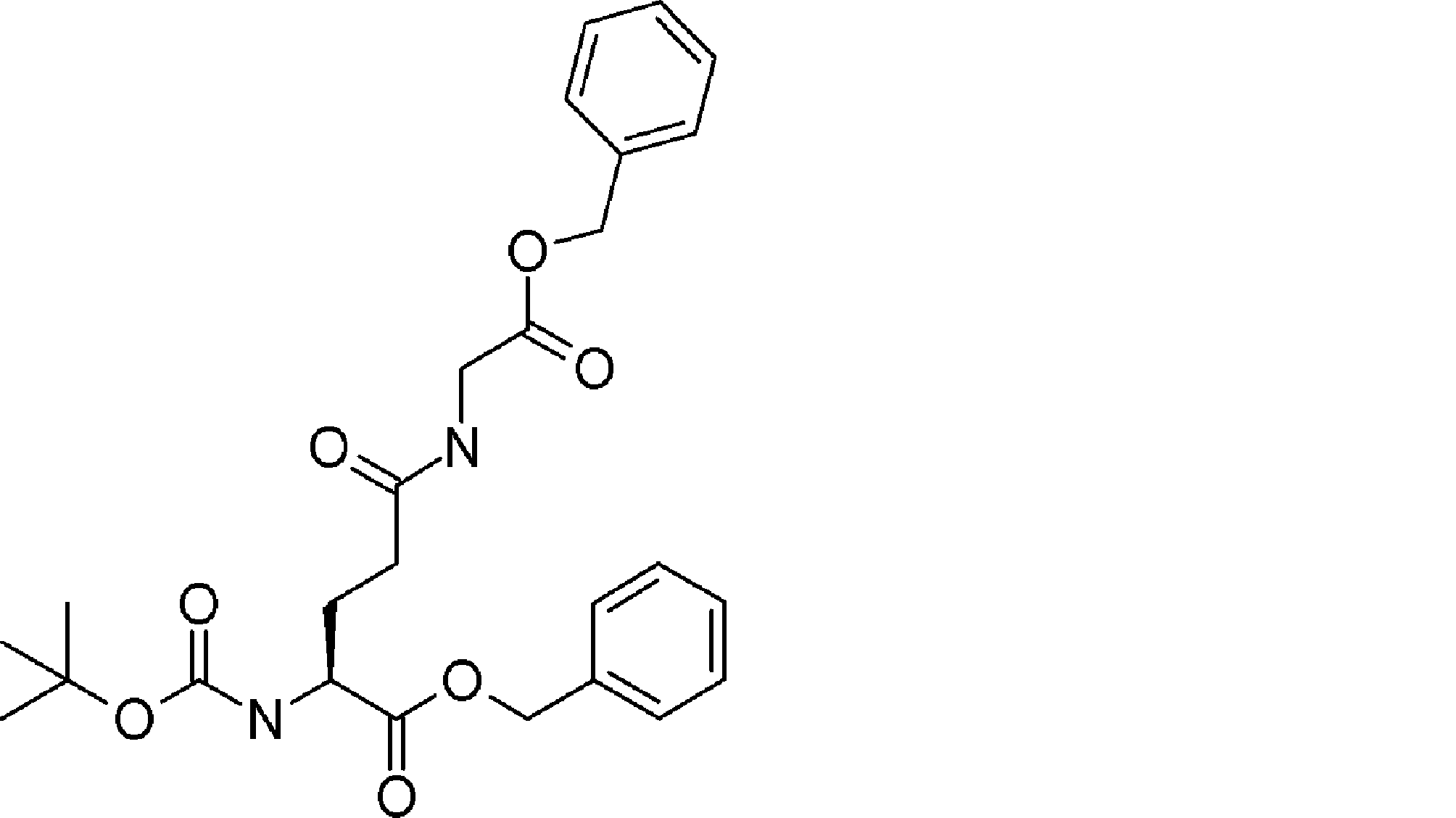
LCMS(ESI) m/z = 485 (M+H)+
保持時間:2.80分(分析条件ZQAA05)
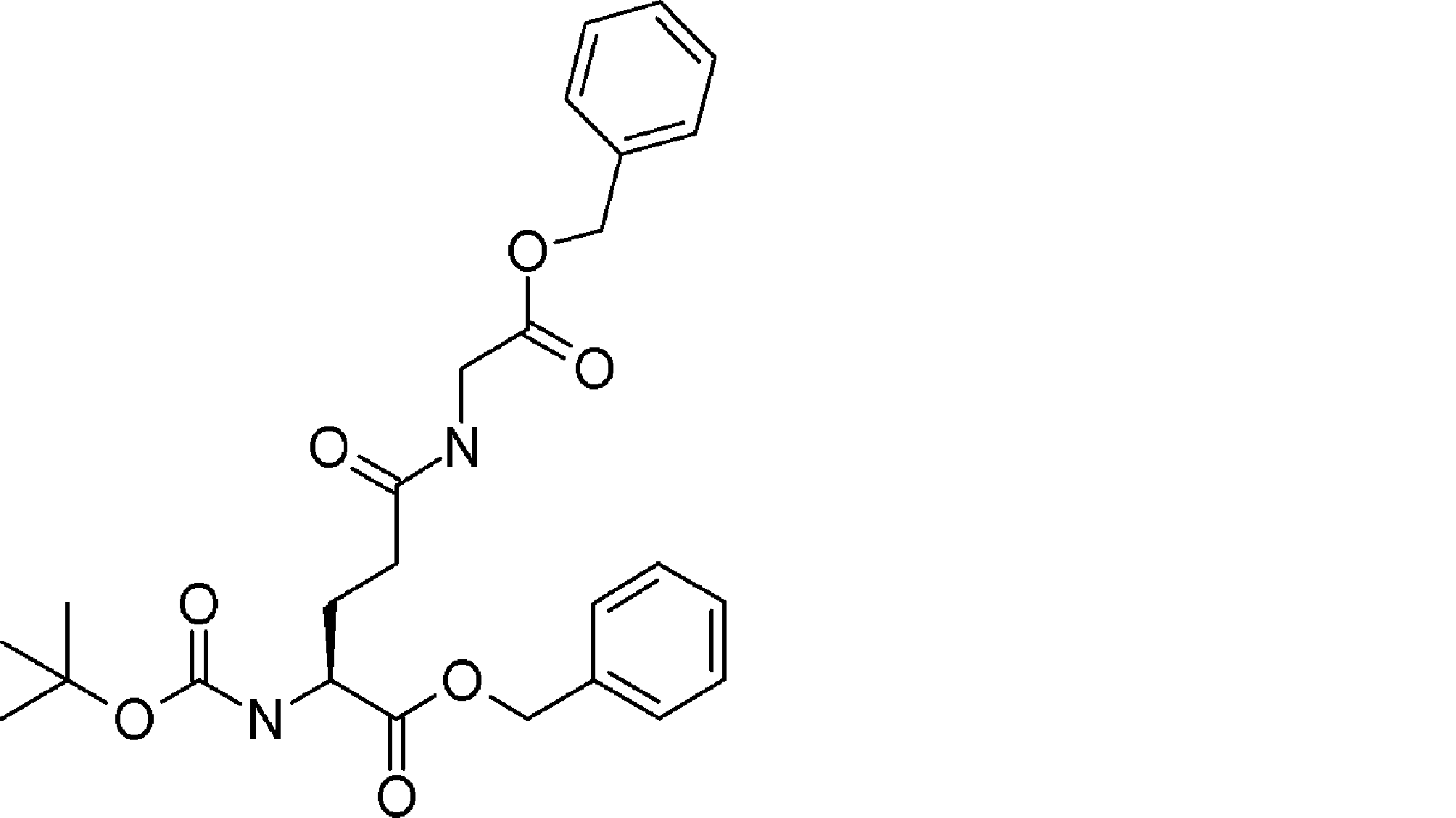
LCMS(ESI) m/z = 485 (M+H)+
保持時間:2.80分(分析条件ZQAA05)
Yangmeiらの文献(Journal of combinatorial chemistry2009, 11, 1066-1072)に従い、アセトニトリル-1.5Mイミダゾール水溶液(7:1)の条件下で反応を行った(図9)。
液性はpH6.4、pH7.4の2条件で、反応温度を37度、70度、100度と3条件行った。最も反応転化率が良好な反応条件(pH7.4、100度)では、望みのアミド化反応の進行が46LC area%程度確認された。
チオエステル部位と翻訳後環化させるN末端に使用するアミノ酸として、CysおよびMeCysなどのSH基にて活性化されたアミノ酸の合成及びそれらを用いたアミノアシル化pdCpAの合成を、図10に示す手法に従って以下のように行った。
1-1. 2-((tert-ブトキシカルボニル)アミノ)-3-(tert-ブチルジスルファニル)プロパン酸 (R)-シアノメチル(化合物2l-A)の合成

LCMS(ESI) m/z = 347 (M-H)-
保持時間:1.00分(分析条件SQDAA05)
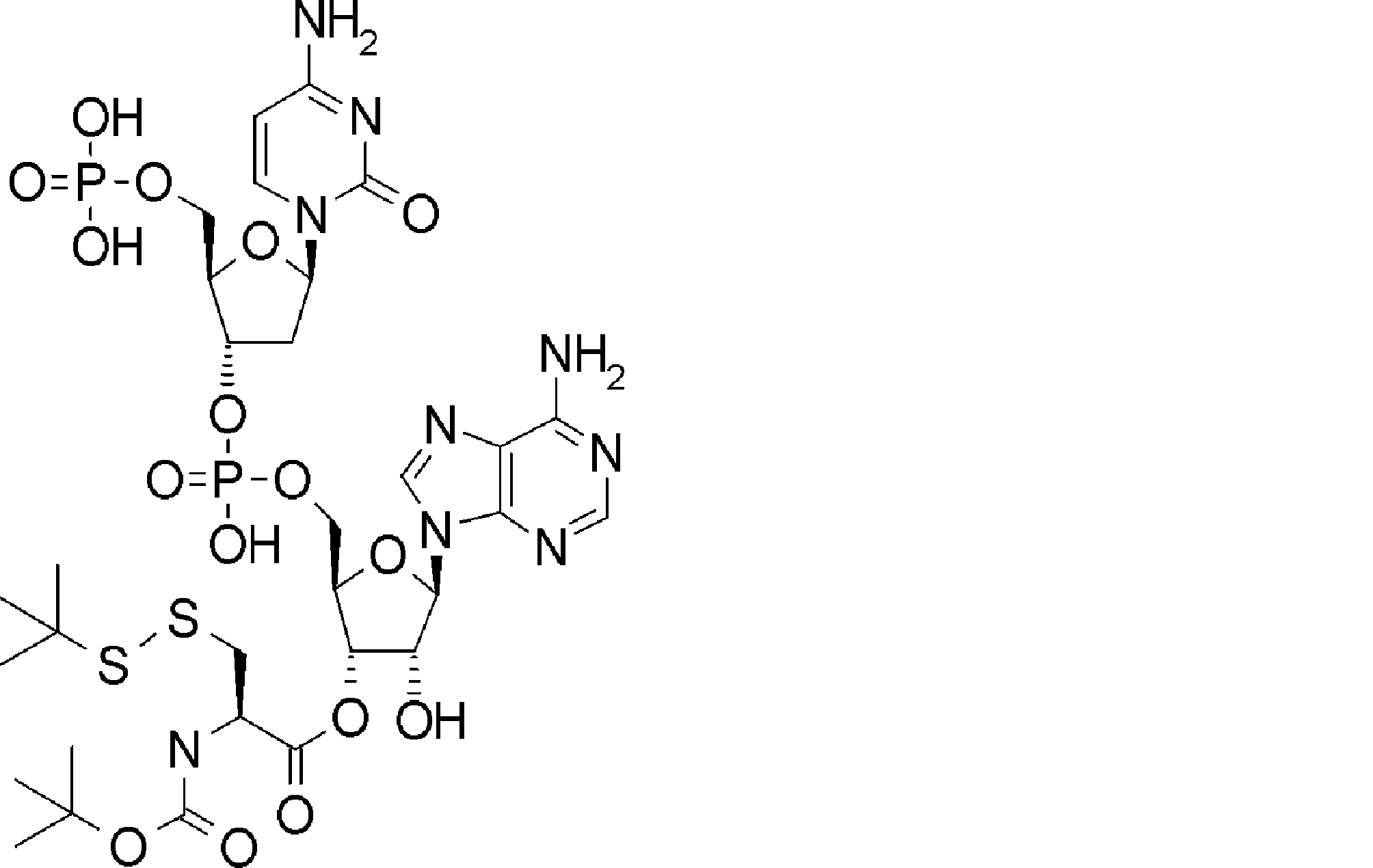
LCMS(ESI) m/z =928(M+H)+
保持時間:0.58分(分析条件SQDFA05)
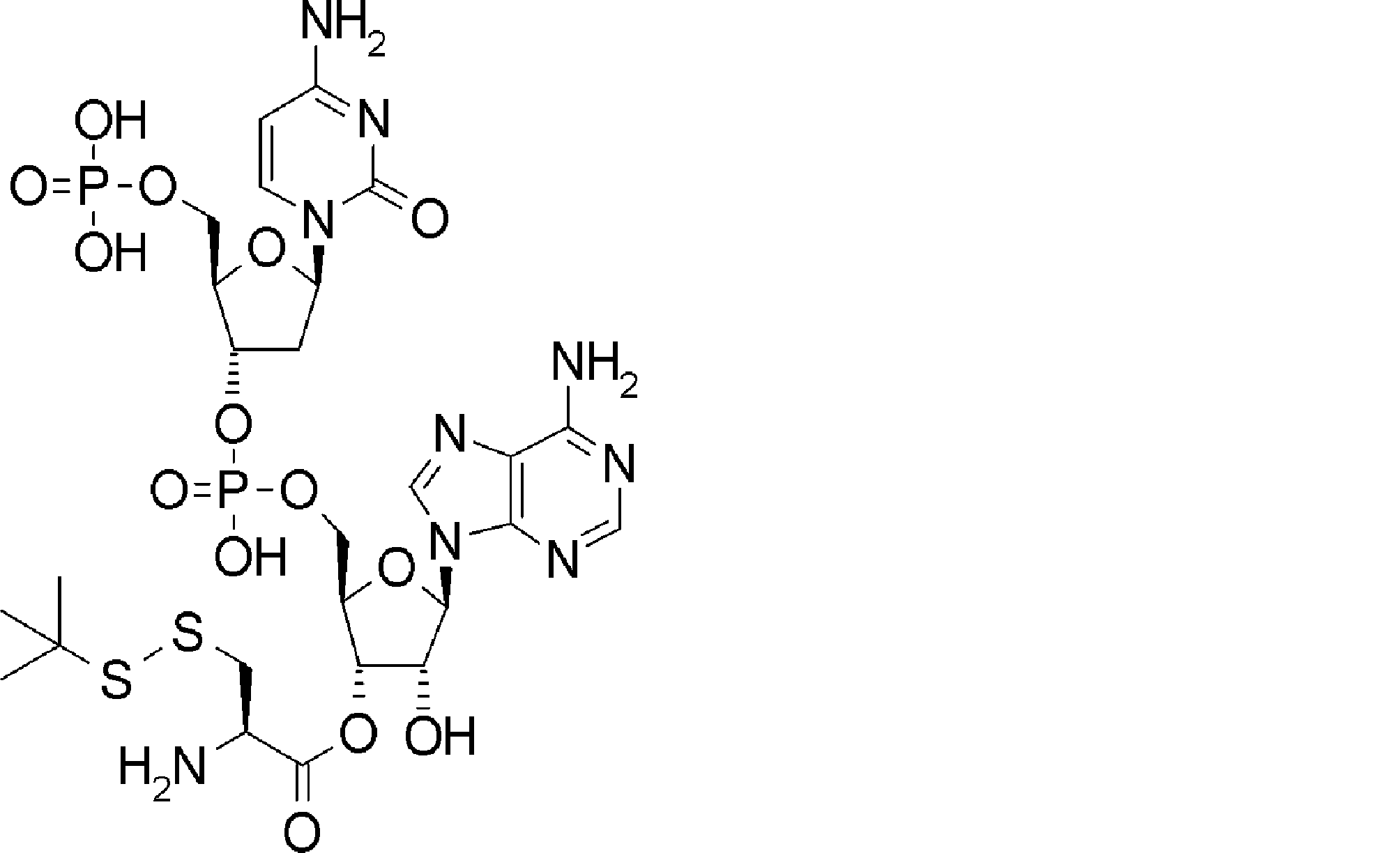
LCMS(ESI) m/z =828(M+H)+
保持時間:0.35分(分析条件SQDFA05)
2-1. 4-((tert-ブチルジスルファニル)メチル)-5-オキソオキサゾリジン-3-カルボン酸 (R)-(9H-フルオレン-9-イル)メチル(化合物2b-B)の合成
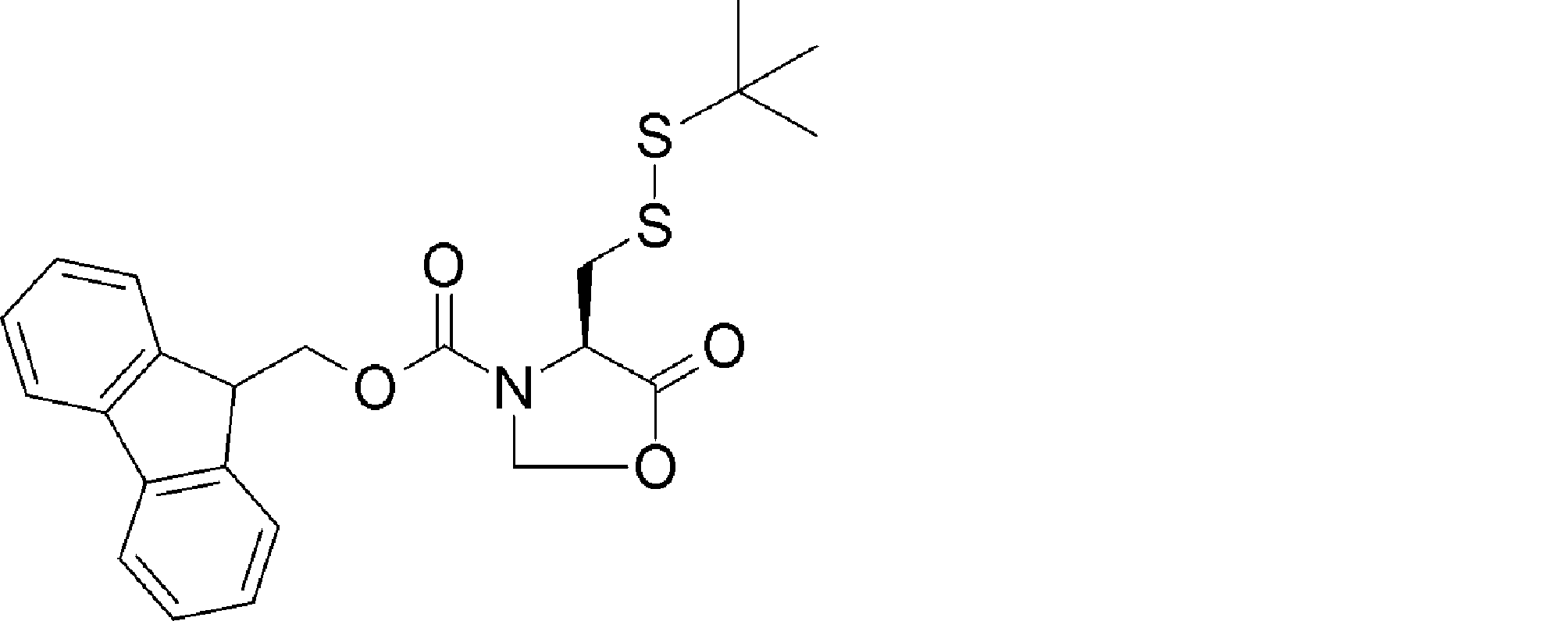
LCMS(ESI) m/z = 444 (M+H)+
保持時間:1.16分(分析条件SQDAA05)

LCMS(ESI) m/z = 446 (M+H)+
保持時間:1.02分(分析条件SQDAA05)
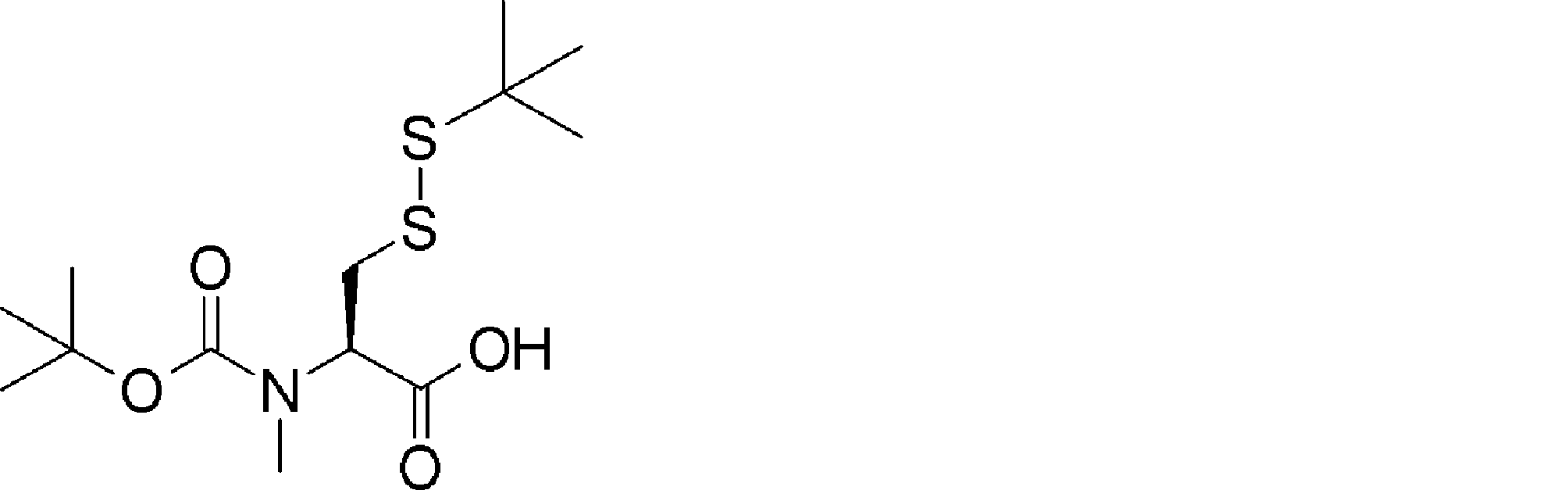
LCMS(ESI) m/z = 322 (M-H)-
保持時間:0.88分(分析条件SQDAA05)

LCMS(ESI) m/z =363(M+H)+
保持時間:1.05分(分析条件SQDAA05)

LCMS(ESI) m/z =942(M+H)+
保持時間:0.62分(分析条件SQDFA05)
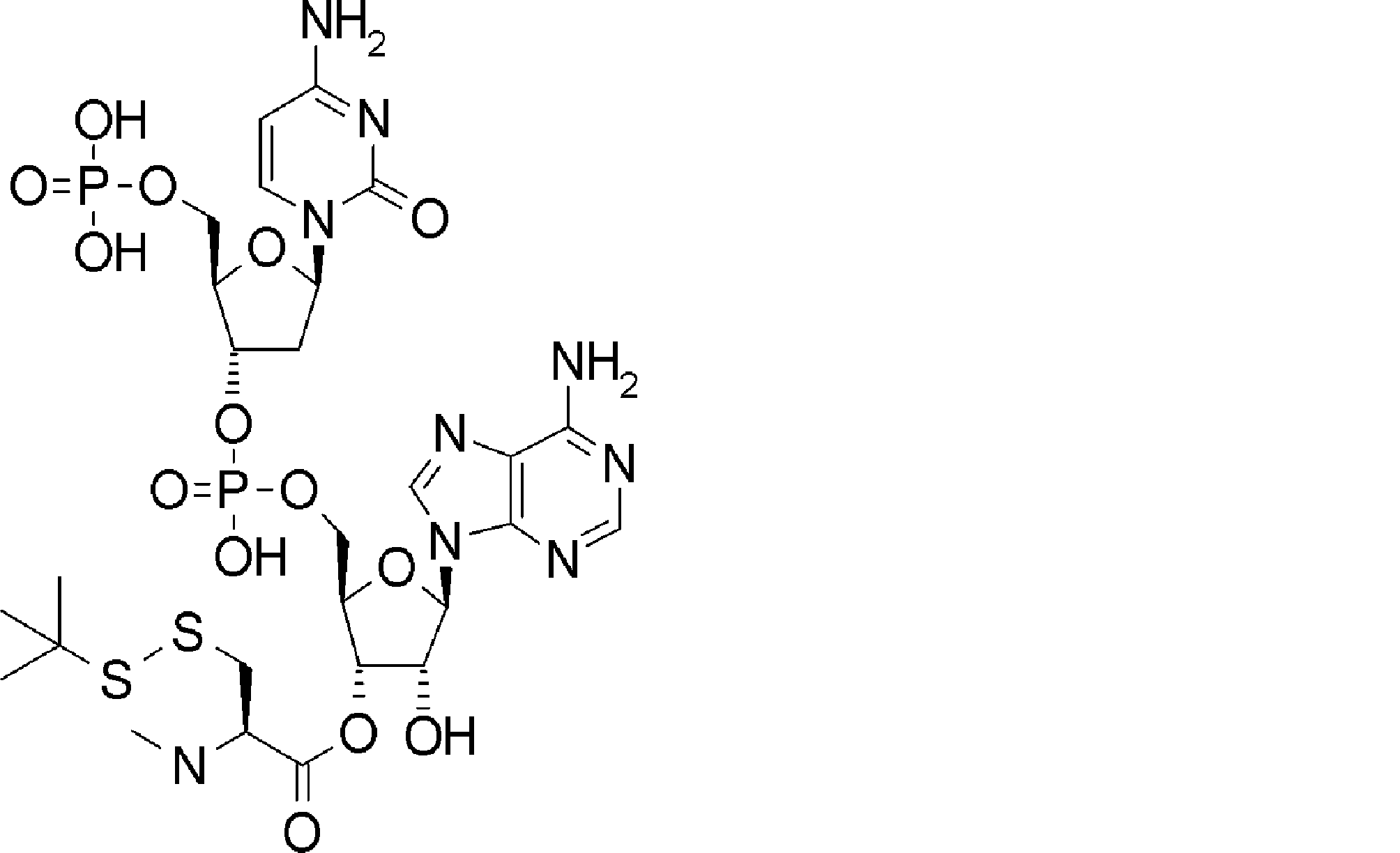
LCMS(ESI) m/z =842(M+H)+
保持時間:0.36分(分析条件SQDFA05)
3-1.(R)-3-(tert-ブチルジスルファニル)-2-(ペンタ-4-エンアミド)プロパン酸(化合物2k-C)の合成
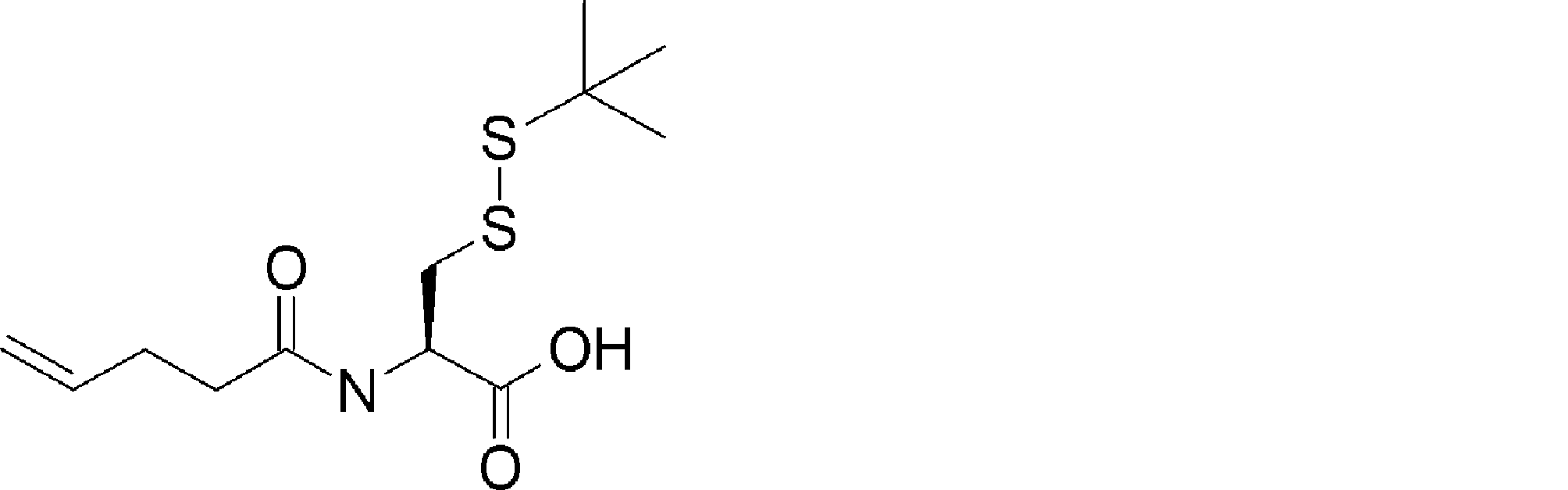
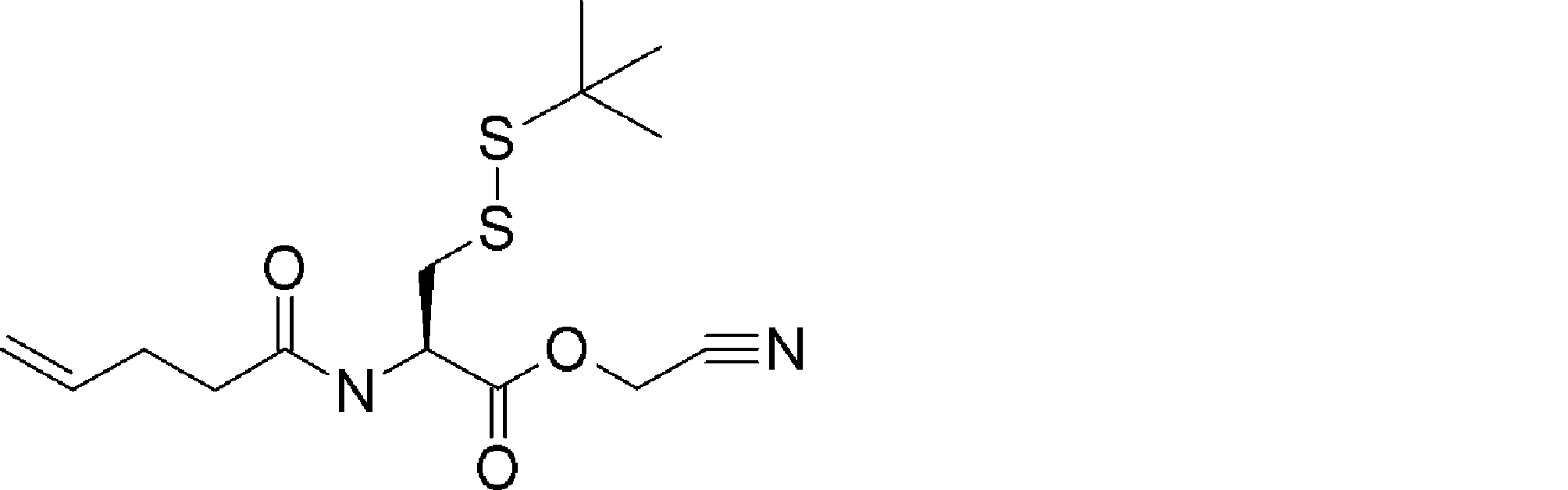
LCMS(ESI) m/z =331(M+H)+
保持時間:0.94分(分析条件SQDAA05)
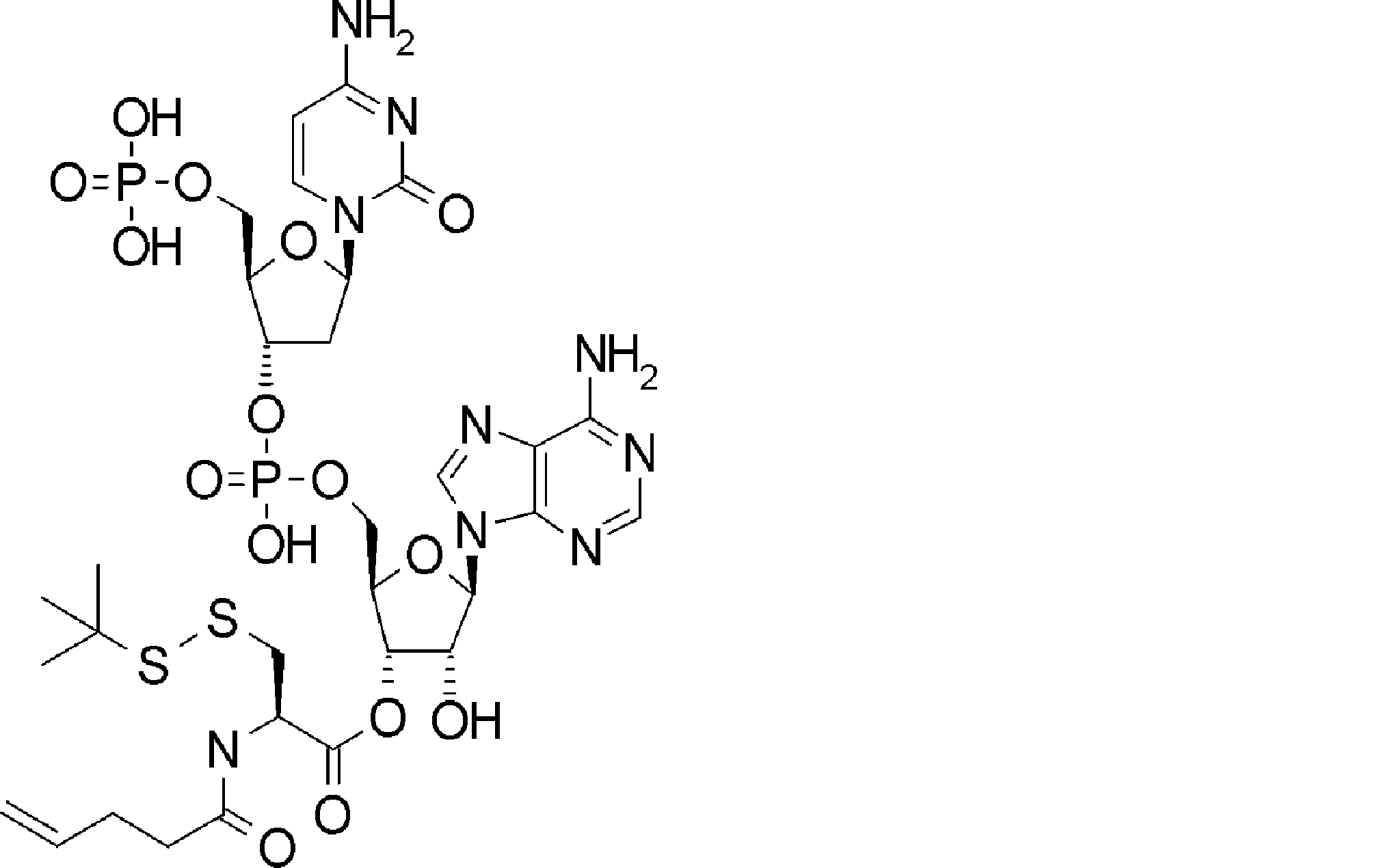
LCMS(ESI) m/z =910(M+H)+
保持時間:0.53分(分析条件SQDFA05)
4-1. (R)-2-(ペンタ-4-エンアミド)-3-(トリチルチオ)プロパン酸(化合物2h)の合成

LCMS(ESI) m/z = 444 (M-H)-
保持時間:0.95分(分析条件SQDAA05)
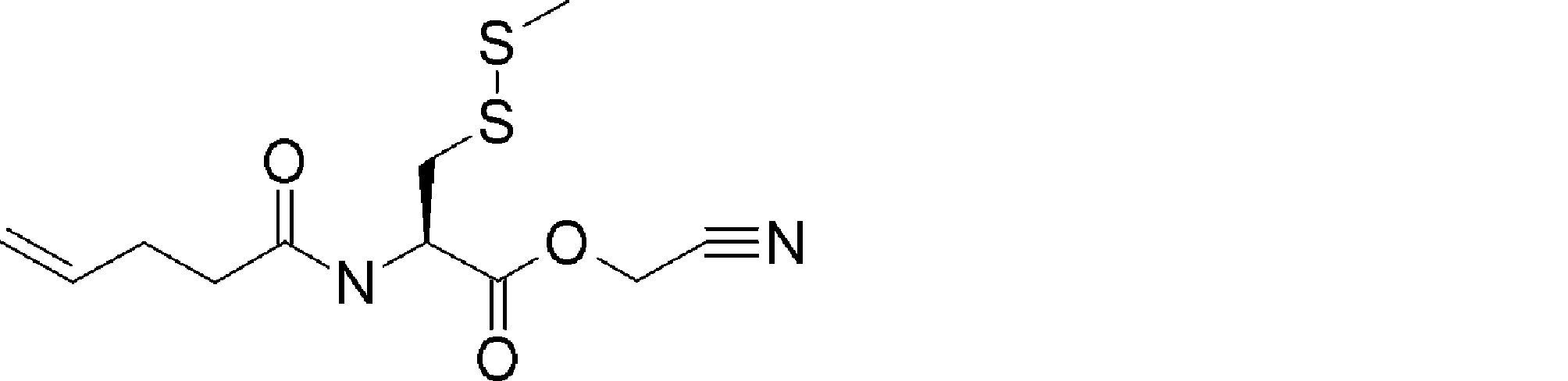
LCMS(ESI) m/z = 289 (M+H)+
保持時間:0.79分(分析条件SQDAA05)
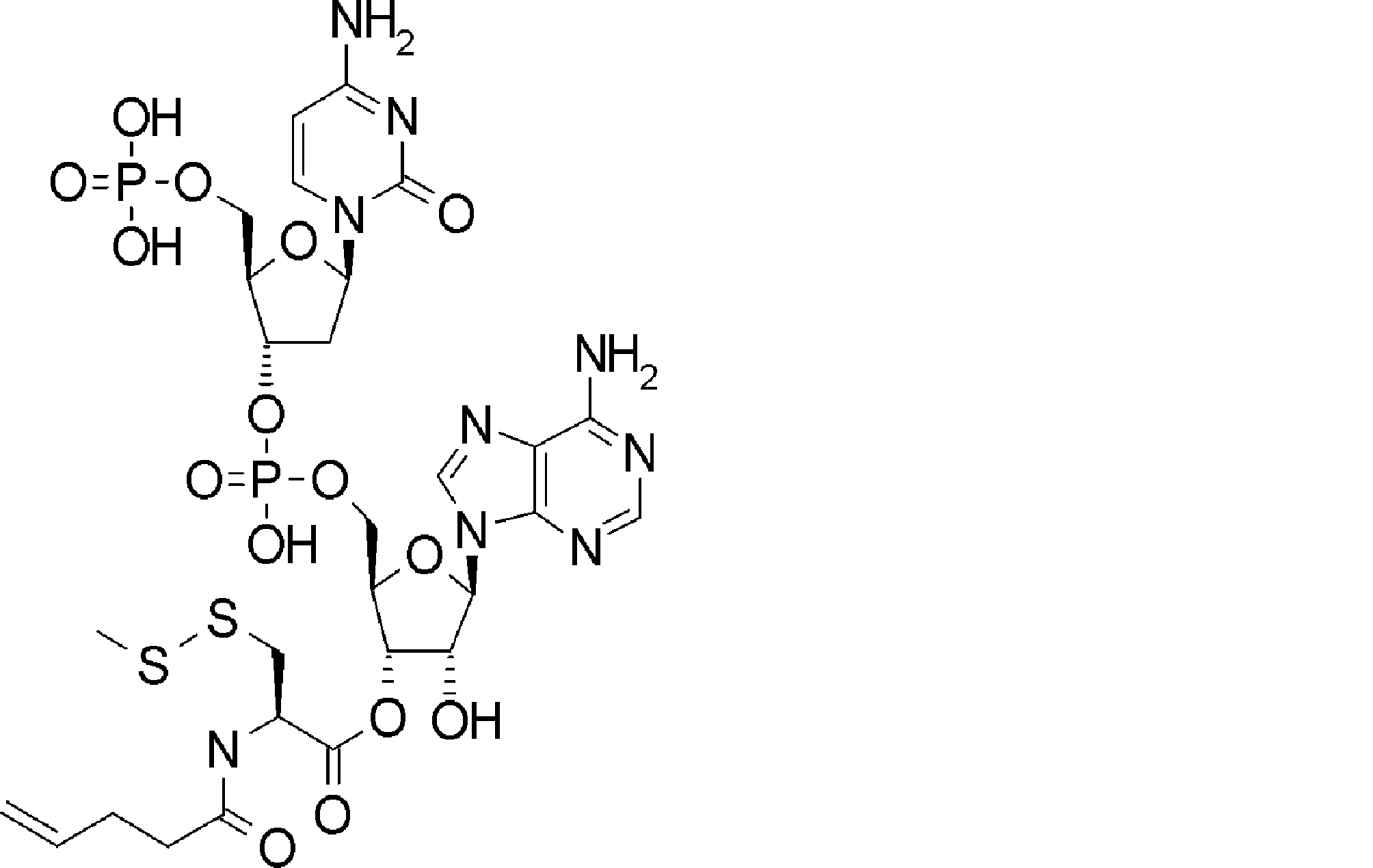
LCMS(ESI) m/z =868(M+H)+
保持時間:0.45分(分析条件SQDFA05)
5-1. 3-(tert-ブチルジスルファニル)-2-((((4,5-ジメトキシ-2-ニトロベンジル)オキシ)カルボニル)アミノ)プロパン酸 (R)-シアノメチル(化合物2l-E)の合成
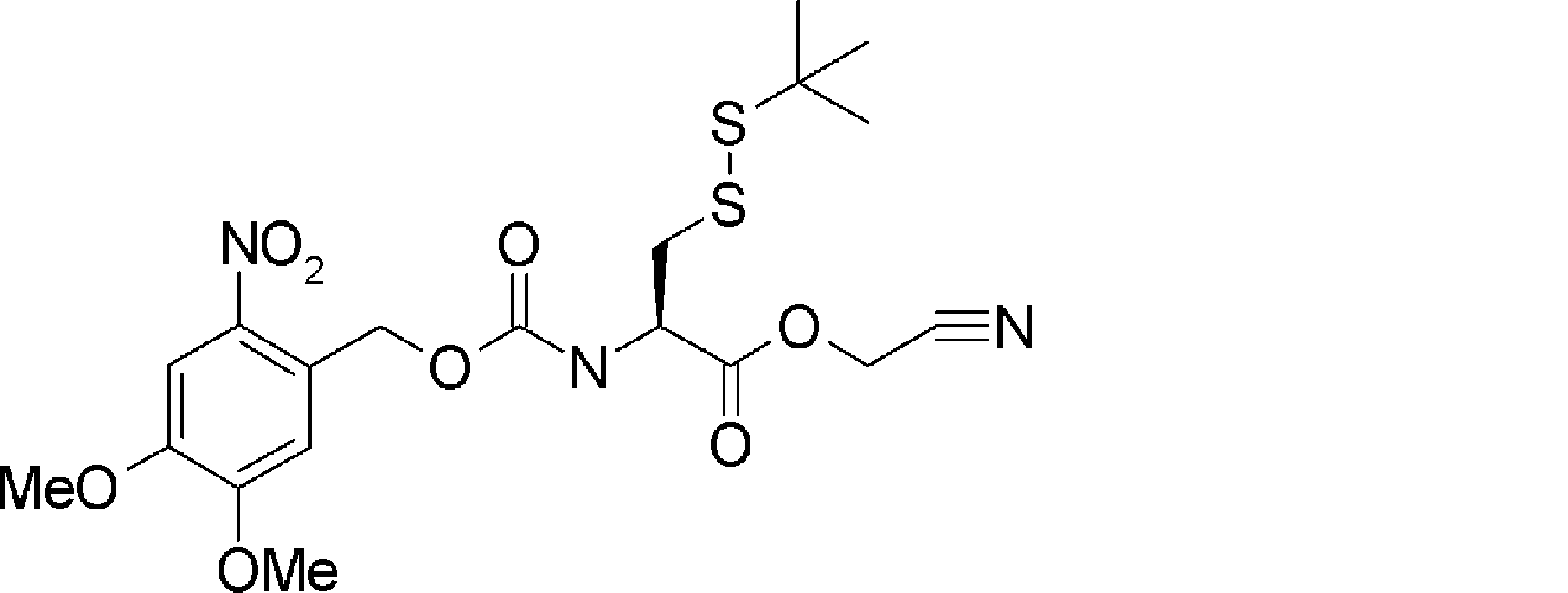
LCMS(ESI) m/z = 486 (M-H)-
保持時間:0.99分(分析条件SQDAA05)

LCMS(ESI) m/z =1067(M+H)+
保持時間:0.62分(分析条件SQDFA05)
6-1. 4-((エチルジスルファニル)メチル)-5-オキソオキサゾリジン-3-カルボン酸 (R)-tert-ブチル(2e-F)の合成

LCMS(ESI) m/z = 294(M+H)+
保持時間:0.98分(分析条件SQDAA05)

LCMS(ESI) m/z = 196 (M+H)+
保持時間:0.39分(分析条件SQDAA05)
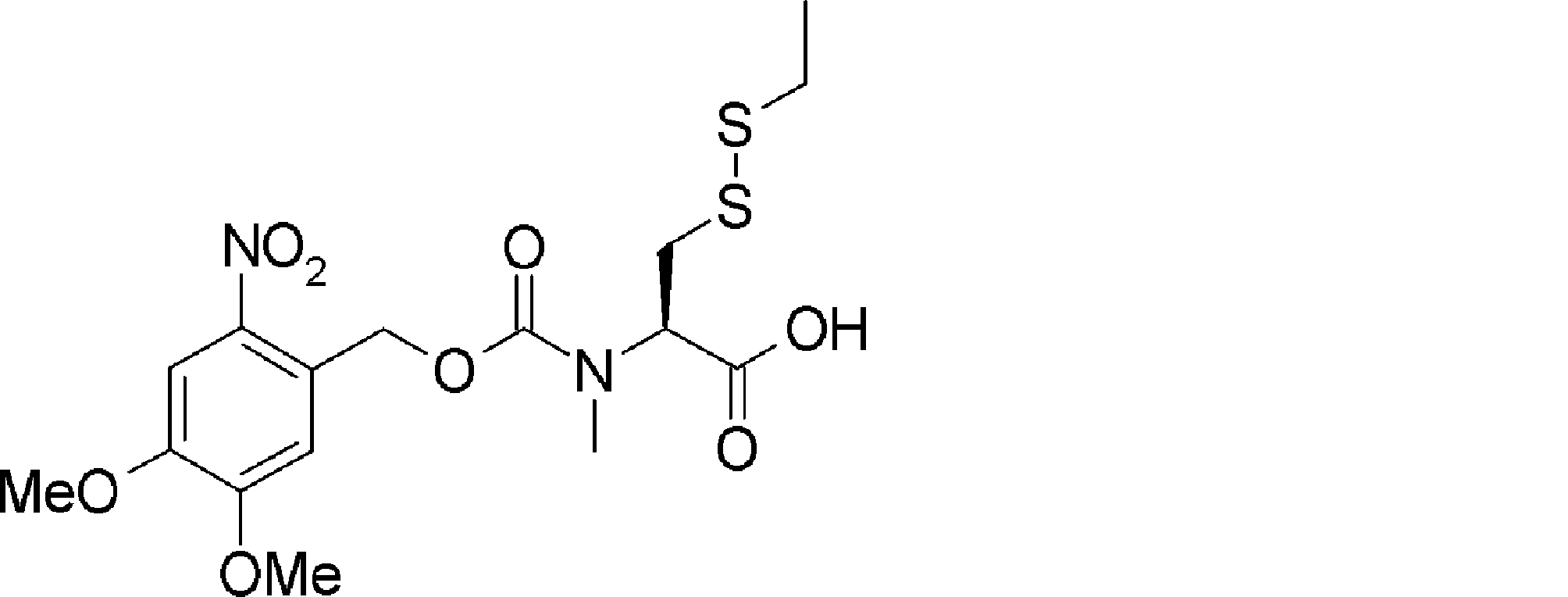
LCMS(ESI) m/z = 435 (M+H)+
保持時間:0.82分(分析条件SQDAA05)

LCMS(ESI) m/z = 474 (M+H)+
保持時間:0.97分(分析条件SQDAA05)

LCMS(ESI) m/z =1053(M+H)+
保持時間:0.59分(分析条件SQDFA05)
7-1. (R)-3-(tert-ブチルジスルファニル)-2-(メチルアミノ)プロパン酸(化合物2f-G)の合成

LCMS(ESI) m/z = 224 (M+H)+
保持時間:0.63分(分析条件SQDAA05)
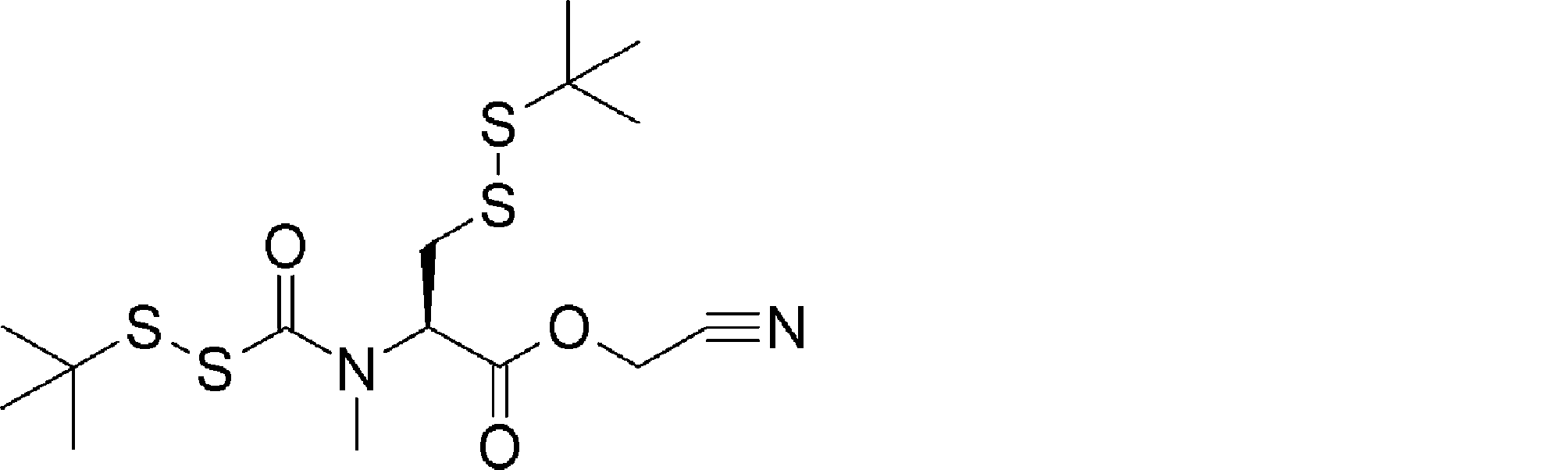
LCMS(ESI) m/z = 411 (M+H)+
保持時間:1.11分(分析条件SQDAA05)

LCMS(ESI) m/z =990(M+H)+
保持時間:0.65分(分析条件SQDFA05)
50μM 転写tRNAfMetCAU(-CA)(配列番号R-5) 10μlに、10X ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP) 2μl、Nuclease free water 4μlを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 10unit/μl T4 RNAリガーゼ(New england bio lab.社) 2μLおよび、5mMのCys(StBu)のアミノアシル化pdCpA (化合物2n-A)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。アミノアシル化tRNA(化合物AT-2-A)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-2-A)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Cys(StBu)-tRNAfMetCAU
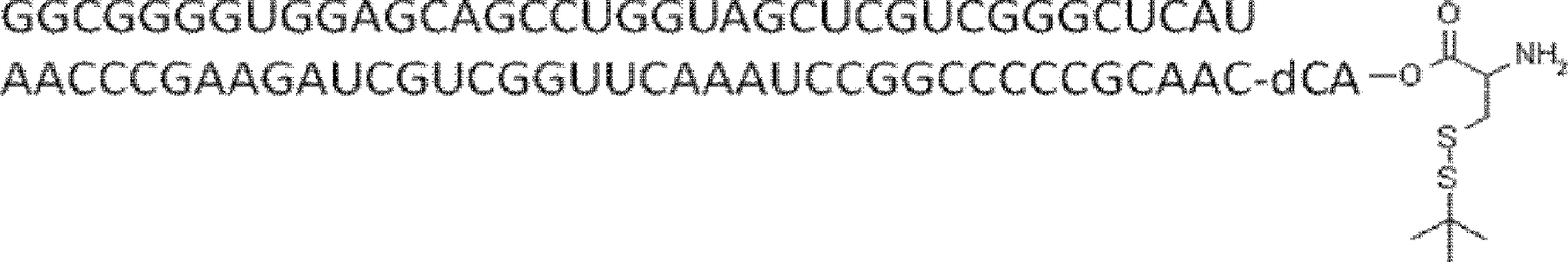
1.ペプチド配列P-10の翻訳合成
20nM 鋳型DNA Mtryg3(配列番号 D-3)に、前述の転写翻訳液、0.1mM 10-HCO-H4folate、0.3mM Gly,0.3mM Arg, 0.3mM Thr, 0.3mM Tyr, 0.3mM Leu,および前記の方法で調製した50μM Cys(StBu)-tRNAfMetCAU(化合物AT-2-A)を加え、37℃で60分間翻訳した。翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析したところ、標的分子であるペプチド配列P-10を観測することはできなかった(図12)。すなわち、開始AUGからの翻訳ペプチドは検出されなかった。Cys誘導体をN末端に配置させて翻訳導入させるには工夫が必要である。一方で、2番目コドンにコードされるアミノ酸であるThrから全長にわたって翻訳されたペプチド(ペプチドP-11)のMSが特異的に観測される興味深い現象が確認された。
[Cys(StBu)]ThrThrThrArgLeuTyrTyrArgGlyGly
MALDI-MS: m/z: Not detected (Calc. 1345.7)
ThrThrThrArgLeuTyrTyrArgGlyGly
MALDI-MS: m/z: [M+H]+ = 1300.7 (Calc. 1299.7)
20nM 鋳型DNA(配列番号 D-7)に、前述の転写翻訳液、0.1mM 10-HCO-H4folate、MetとLysを除く各0.3mMの18種類のタンパク質性アミノ酸,および前記の方法で調製した50μM Cys(StBu)-tRNAfMetCAU(化合物AT-2-A)を加え、37℃で60分間翻訳した。翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析したところ、標的分子であるペプチド配列P-12を観測することが出来た。一方で、2番目コドンにコードされるアミノ酸であるThrから全長にわたって翻訳されたペプチドP―13のMSが主生成物として観測された。Cys(StBu)をN末端に配置させたペプチド翻訳合成が可能であることが示されたが、その効率は低く改良検討を以下行った。
AKC17 DNA配列 (配列番号D-31と同一) (配列番号:7)
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGACTAGAACTGCCTACTGGAGCcttTGCGGCAGCGGCAGCGGCAGC
[Cys(StBu)]ThrArgThrAlaTyrTrpSerLeuCysGlySerGlySerGlySer
MALDI-MS:m/z: [M+H]+ = 1723.7 (Calc.1722.7)
ThrArgThrAlaTyrTrpSerLeuCysGlySerGlySerGlySer
MALDI-MS:m/z: [M+H]+ = 1532.7 (Calc.1531.7)
pdCpA法によるN末端Cys(StBu)導入効率が低い原因を特定するために以下の実験を行った。
前駆体である化合物2n-Aを用いてpdCpA-Cysの側鎖StBuの脱保護および脱保護された化合物2e-Aの安定性を調べる以下の実験を行った。250μM の化合物2n-A、pH7の50mM Tris・HCl buffer、DTT 10mMの条件下、室温で側鎖の脱保護を行い、その経時変化をLC-MS(SQD)を用いて観測した。目的の化合物2e-Aは観測されず、アミノ酸が加水分解し脱離したpdCpAが観測された。5分後、化合物2n-A 51%、化合物2e-A 0%,pdCpA49%であり、2時間後に脱保護は完了し化合物2n-Aが 0%になったが、化合物2e-Aは0%で、全てpdCpA100%に加水分解された(図13)。
以上のことから、pdCpA-Cys(化合物2e-A)は通常翻訳条件下で不安定であり、ほぼ生成と同時に加水分解されるため、翻訳に不利であることが判明した。
CysのSH基を保護した化合物2n-Aの安定性を評価した。以下の表2の通りに5条件の保存サンプルをそれぞれ調整し、LC-MSを用いて分析した。
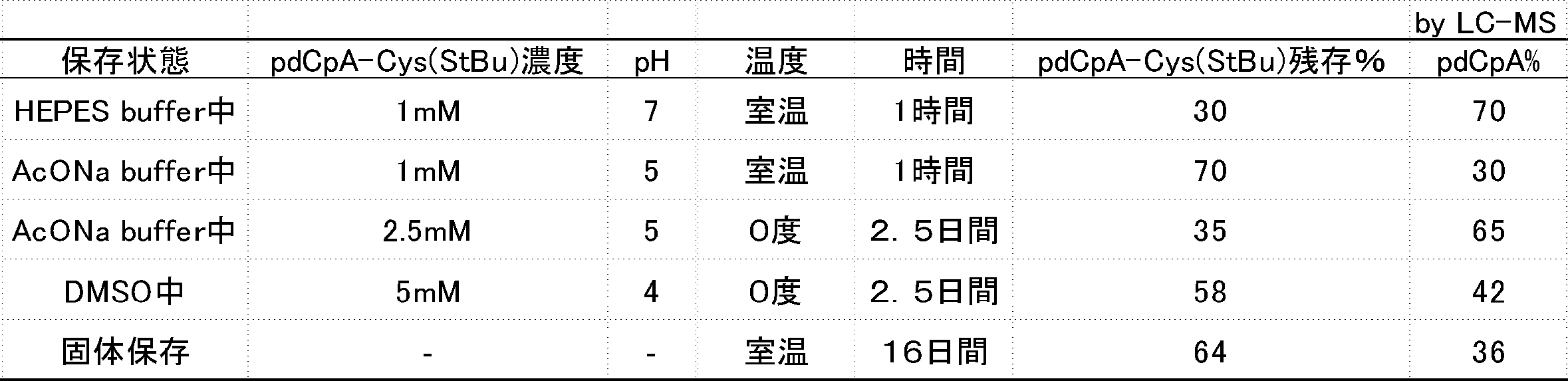
MeCysのSH基を保護した化合物2n-Bの安定性もあわせて評価した。以下の表3の通りに4条件の保存サンプルを調整し、LC-MSを用いて分析した。


以上のことからpdCpA法によるN末端Cys(StBu)導入効率が低い原因の一つとして、側鎖SH基の存在によるアミノアシル化tRNAの不安定化を特定した。
実施例9で観測された2番目のコドンからの翻訳開始が効率的に得られた現象(Intiation read through)を積極的に活用することで、ペプチドのN末端にCysの導入を以下のように試みた。
以下の方法により翻訳を行った。20nM 鋳型DNA Mctryg3(配列番号D-8)に、前述の転写翻訳液、0.3mM Gly,0.3mM Arg,0.3mM Thr, 0.3mM Tyr,0.3mM Leu、0.3mM Cysを加え、37℃で60分間翻訳した。翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析したところ、開始コドンであるAUGからではなく、AUGの次にコードされるコドン(TGC、Cysに相当)から翻訳された標的ペプチドP-14のMSが期待どおり主生成物として観測された(図14参照)。
Mctryg3 DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgtgcACTACAACGCGTctttactaccgtggcggcTAGTAGATAGATAG
CysThrThrThrArgLeuTyrTyrArgGlyGly
MALDI-MS: m/z: [M+H]+ = 1290.6 (Calc. 1289.6)
三文字目のコドンがinitiation read throughにどのように影響するか評価した。20nM 鋳型DNA IniRt_XXX_am(配列番号D-9からD-28まで)に、前述の大腸菌由来の再構成無細胞転写翻訳液、Metを除く各0.3mMの19種類のタンパク質性アミノ酸を加え、37℃で60分間翻訳した。対照実験としてMetを含む翻訳も別途行った。翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析したところ、開始のAUGの次にコードされるCysから翻訳されたペプチドのMSが観測された。
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGtgcXXXACTACAACGctttactaccgtggcggcTAGTAGATAGATAG
(XXX: TTT(配列番号D-9(配列番号:9)), TTG(配列番号D-10(配列番号:10)), TAC(配列番号D-11(配列番号:11)), TGC(配列番号D-12(配列番号:12)), TGG(配列番号D-13(配列番号:13)), CTT(配列番号D-14(配列番号:14)), CTA(配列番号D-15(配列番号:15)), CCG(配列番号D-16(配列番号:16)), CAT(配列番号D-17(配列番号:17)), CAG(配列番号D-18(配列番号:18) CGT(配列番号D-19(配列番号:19)), CGG(配列番号D-20(配列番号:20)), ATT(配列番号D-21(配列番号:21)), ACT(配列番号D-23(配列番号:22)), AAC(配列番号D-24(配列番号:23)), AGT(配列番号D-25(配列番号:24)), AGG(配列番号D-26(配列番号:25)), GTT(配列番号D-27(配列番号:26)), GCT(配列番号D-28(配列番号:27))
CysXaaThrThrThrLeuTyrTyrArgGlyGly(配列番号:39~57)

天然型アミノ酸であるCysは、ARSを活用したIntiation read through法により効率的に翻訳導入されたが、非天然型アミノ酸をARS導入するには限界があるため、アミノアシル化pdCpAのうち、活性エステル部分の加水分解速度を遅くする工夫をした化合物を合成・評価した。すなわち、SH基をカルボン酸活性エステル部位からより離した一連の化合物群を合成・評価した。
1.化合物6i-Aの合成
1-1.(2-(tert-ブチルジスルファニル)エチル)カルバミン酸 tert-ブチル(化合物6b-A)の合成

LCMS:m/z 266 (M+H)+
保持時間:1.05分(分析条件 SQDAA05)

LCMS:m/z 166 (M+H)+
保持時間:0.63分(分析条件 SQDAA05)
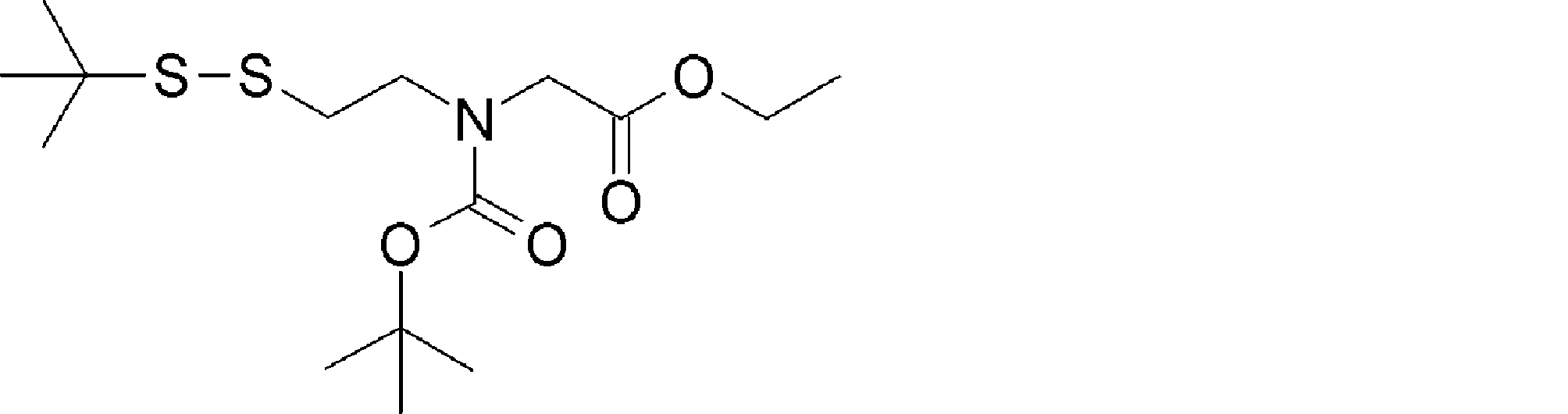
LCMS:m/z 352 (M+H)+
保持時間:1.11分(分析条件 SQDAA05)
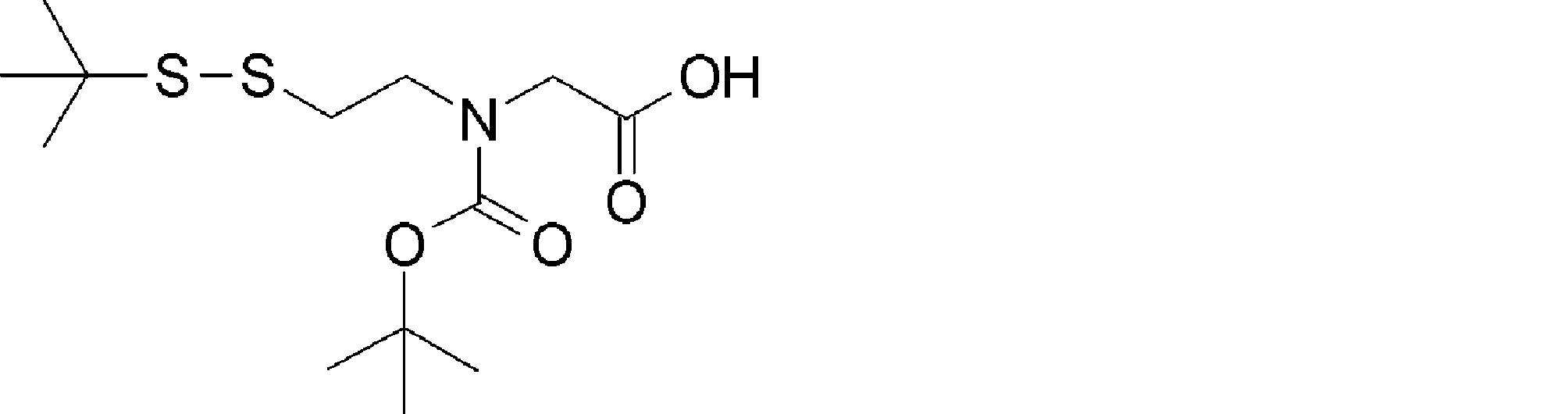
LCMS:m/z 322(M-H)-
保持時間:0.89分 (分析条件 SQDAA05)
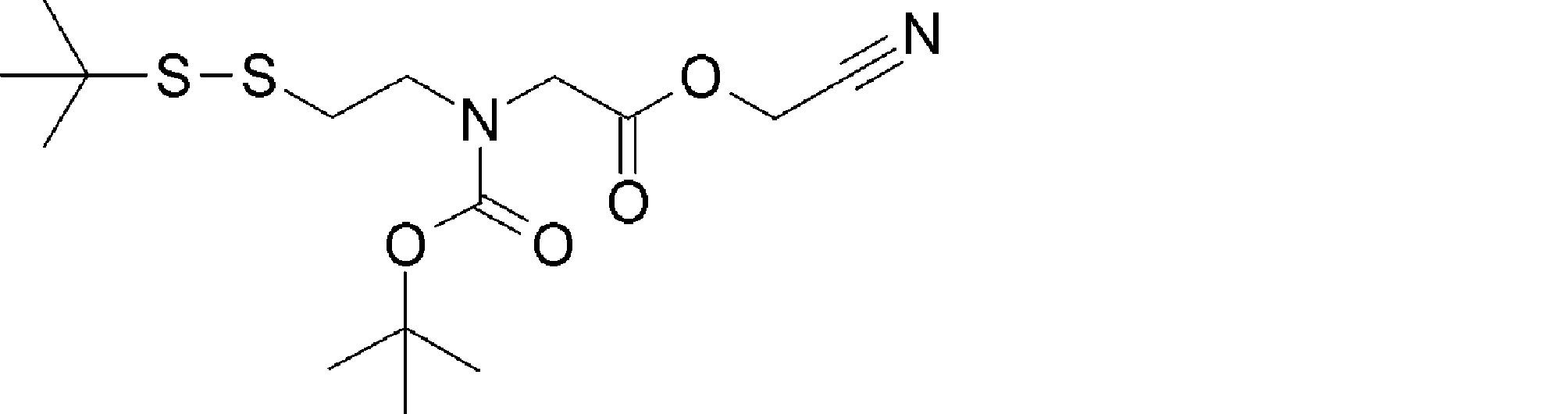
LCMS:m/z 363(M+H)+
保持時間:1.04分 (分析条件 SQDAA05)

LCMS:m/z 942(M+H)+
保持時間:0.89分 (分析条件 SMDmethod1)
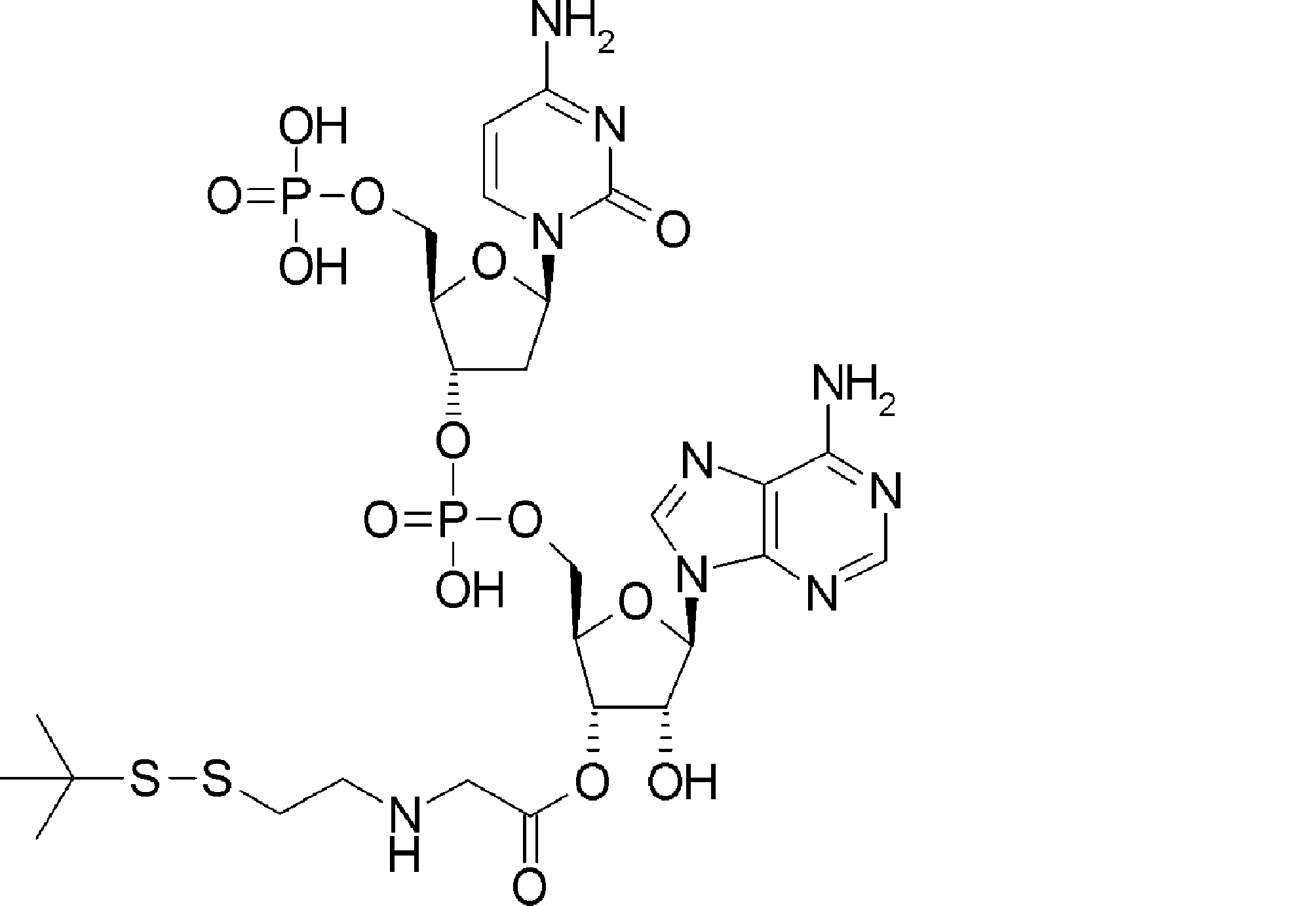
LCMS:m/z 842(M+H)+
保持時間:0.39分 (分析条件 SQDAA05)
2-1.3-((tert-ブトキシカルボニル)(2-(tert-ブチルジスルファニル)エチル)アミノ)プロパン酸メチル(化合物6e-B)の合成
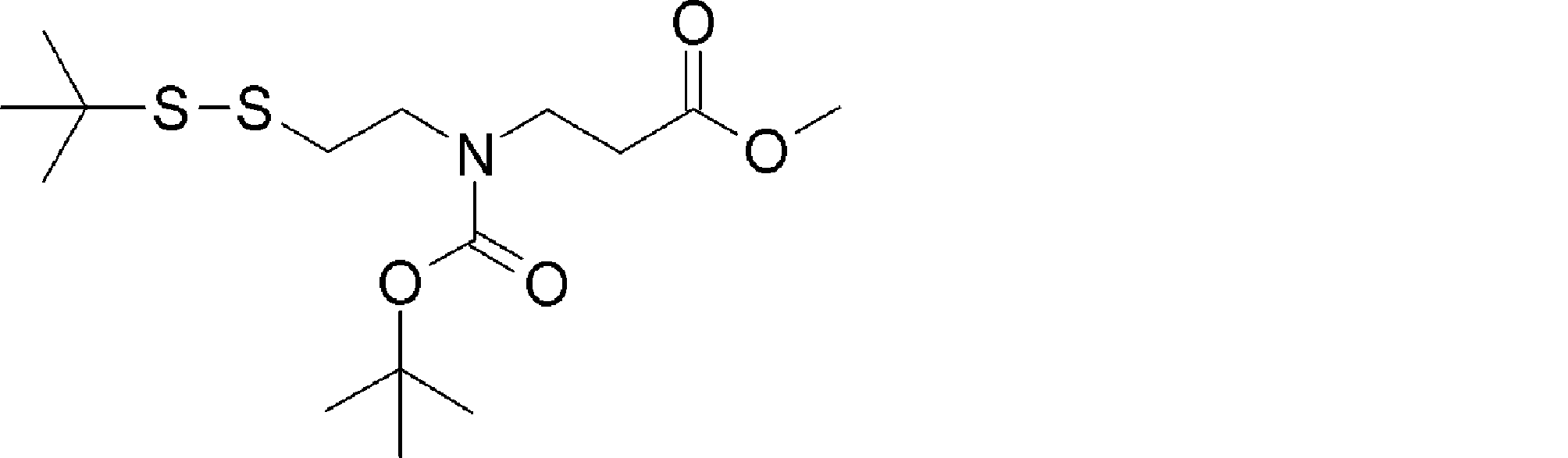
LCMS:m/z 352(M+H)+
保持時間:1.11分 (分析条件 SQDAA05)
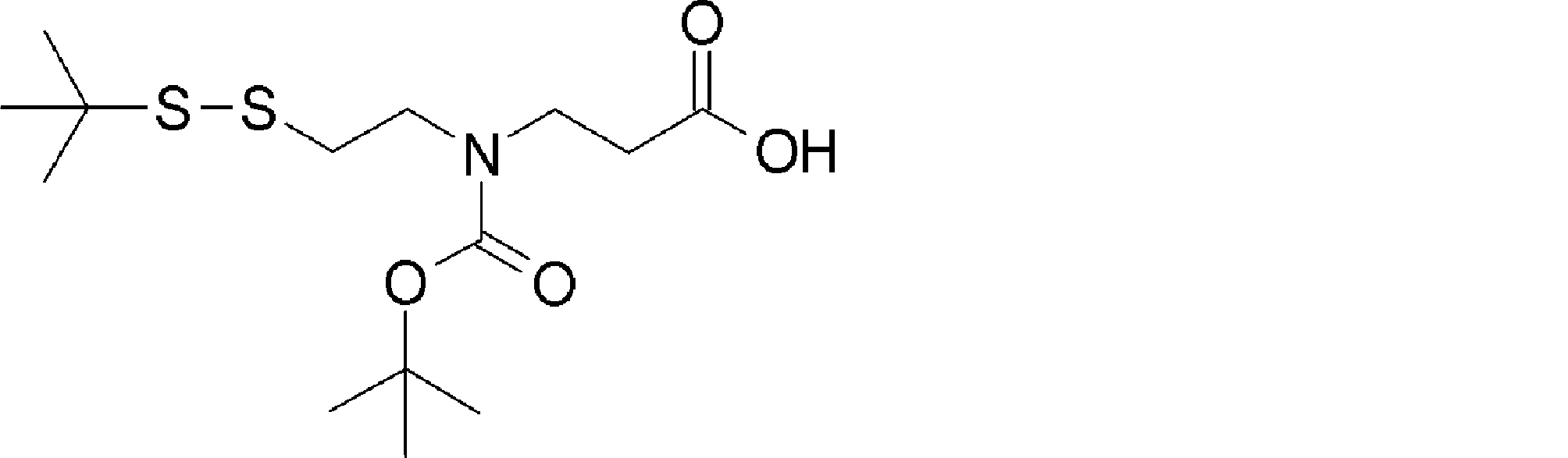
LCMS:m/z 336(M-H)-
保持時間:0.96分 (分析条件 SQDAA05)
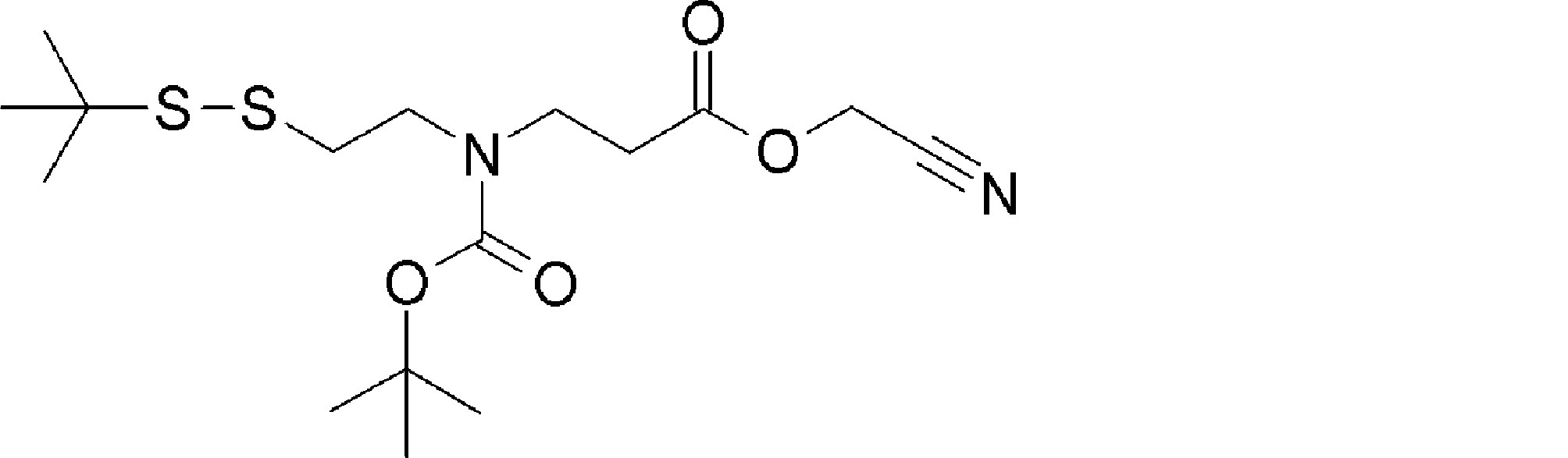
LCMS:m/z 377(M+H)+
保持時間:1.09分 (分析条件 SQDAA05)
LCMS:m/z 856(M+H)+
保持時間:0.629分 (分析条件 SMDmethod1)
3-1.4-((tert-ブトキシカルボニル)(2-(tert-ブチルジスルファニル)エチル)アミノ)ブタン酸エチル(化合物6e-C)の合成
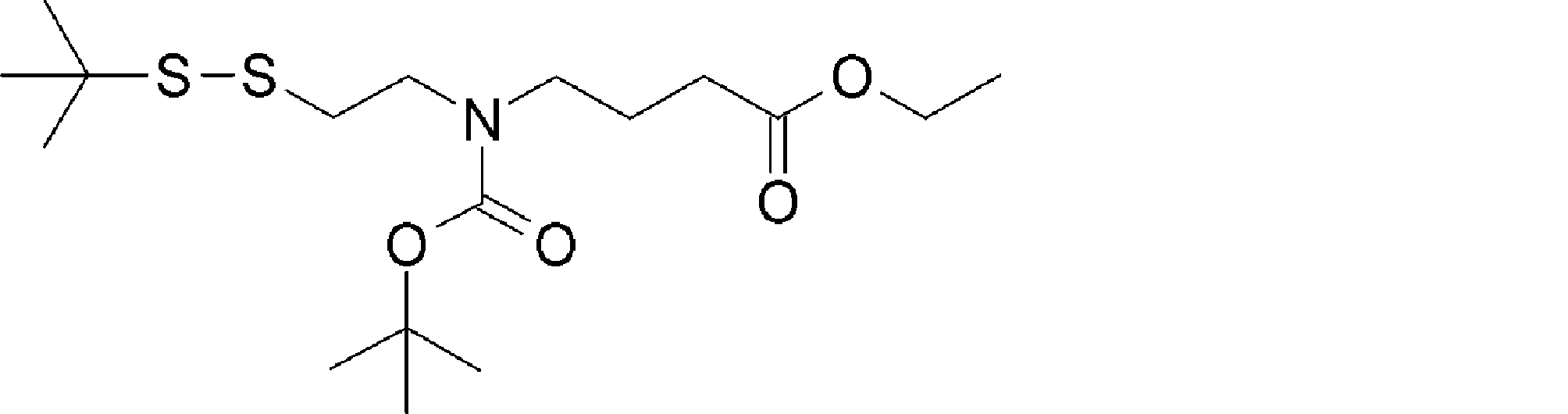
LCMS:m/z 380(M+H)+
保持時間:1.15分 (分析条件 SQDAA05)
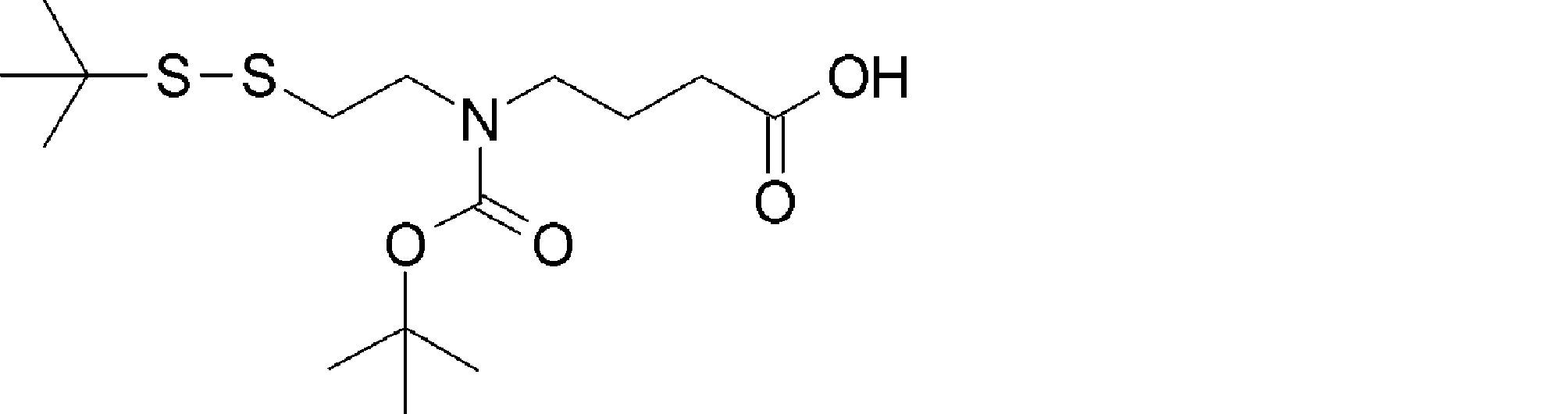
LCMS:m/z 352(M+H)+
保持時間:0.99分 (分析条件 SQDAA05)

LCMS:m/z 391(M+H)+
保持時間:1.05分 (分析条件 SQDAA05)

LCMS:m/z 870(M+H)+
保持時間:0.641分 (分析条件 SMDmethod1)
50μM 転写tRNAfMetCAU(-CA) (配列番号R-5) 10μlに、10X ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP)2μl、Nuclease free water 4μlを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。10unit/μl T4 RNAリガーゼ(New england bio lab.社)2μLおよび、5mMのアミノアシル化pdCpA(化合物6i-A、B、C、2n-A、2m-c、2m-E)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。アミノアシル化tRNA(化合物AT-2-A、C、E、AT-6-A、B、C)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-2-A、C、E、AT-6-A、B、C)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Cys(StBu)-tRNAfMetCAU

PenCys(StBu)-tRNAfMetCAU
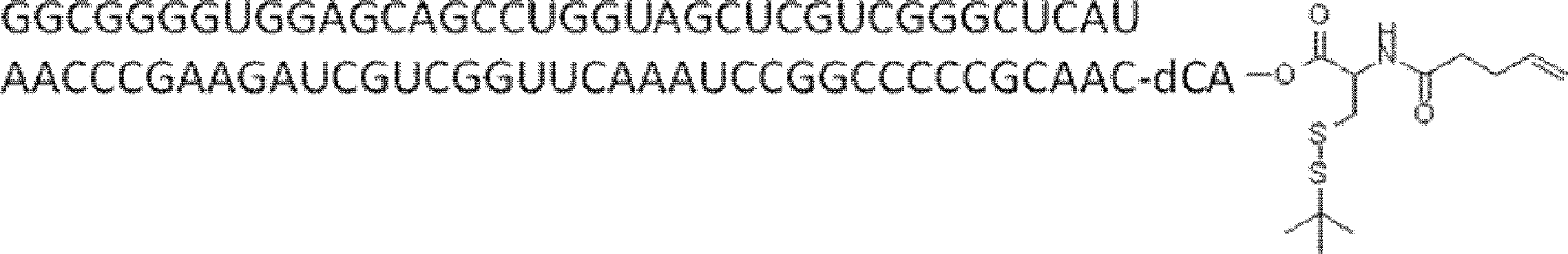
NVOCCys(StBu)-tRNAfMetCAU

tBuSSEtGly-tRNAfMetCAU
tBuSSEtβAla-tRNAfMetCAU
tBuSSEtGABA-tRNAfMetCAU
以下の実験で示すとおり、CysのSH基や主鎖アミノ基を保護すると、Cys自身と比較してアミノアシル化pdCpAの翻訳液モデル系中(HEPES buffer)の安定性が向上し、翻訳合成効率も向上することが示された。また、SH基の位置をα―カルボン酸部位から遠くに配置させた、各種新規SH基含有各種非天然型アミノ酸の翻訳導入効率も向上された。
結果、Cysと比較して、SH基やNを保護すると、pdCpA-アミノ酸体の安定性が向上し、かつ翻訳合成効率も向上することが示された。また、Cysをより安定化させたSH含有各種非天然型アミノ酸の翻訳導入効率も向上された(図25、26参照)。
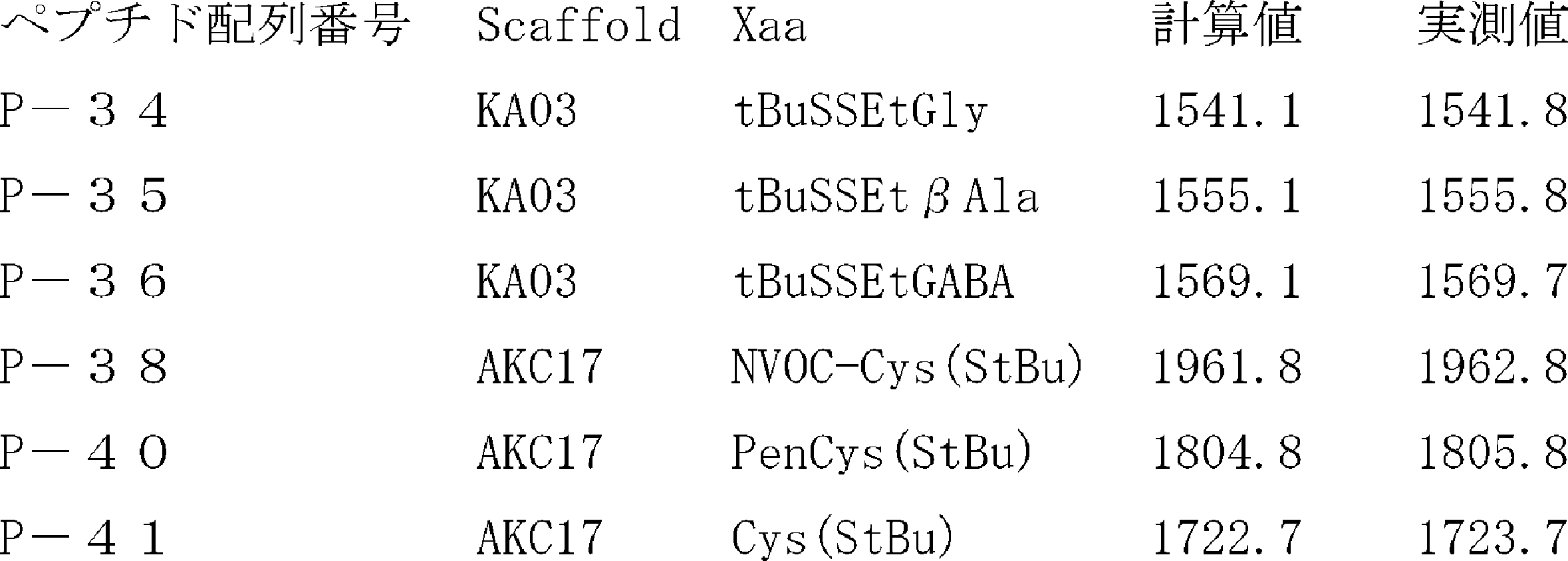
AKC17 DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGACTAGAACTGCCTACTGGAGCcttTGCGGCAGCGGCAGCGGCAGC
KA03 ペプチド配列
[Xaa]ThrArgThrLysAlaTyrTrpSerLeuProGlyGly
AKC17 ペプチド配列
[Xaa]ThrArgThrAlaTyrTrpSerLeuCysGlySerGlySerGlySer
1.側鎖カルボン酸が活性エステル化されたアミノアシルtRNAの合成
以下の方法に従って、側鎖カルボン酸がチオエステル化されたアミノアシル化tRNAの合成を行なった。
鋳型DNA(配列番号D-33)から、RiboMAX Large Scale RNA production System T7(Promega社,P1300)を用いたin vitro の転写により3'端のCAを欠くtRNAEnAsnGAG(-CA)(配列番号R-33)を合成し、RNeasy Mini kit(Qiagen社)により精製した。
tRNAEnAsnGAG(-CA) DNA配列:
GGCGTAATACGACTCACTATAGGCTCTGTAGTTCAGTCGGTAGAACGGCGGACTgagAATCCGTATGTCACTGGTTCGAGTCCAGTCAGAGCCGC
tRNAEnAsnGAG(-CA) RNA配列:
GGCUCUGUAGUUCAGUCGGUAGAACGGCGGACUgagAAUCCGUAUGUCACUGGUUCGAGUCCAGUCAGAGCCGC
50μM 転写tRNAEnAsnGAG(-CA)(配列番号R-33)10μLに、10X ligation buffer (500mM HEPES-KOH pH7.5,200mM MgCl2,10mM ATP)2μL、Nuclease free water 4μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。10unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)2μLおよび、5mMの側鎖カルボン酸が活性エステル化されたアミノアシル化pdCpA(化合物1i)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。ライゲーション反応液 20μLに、125mM ヨウ素(水:THF=1:1溶液)4μLを加え、室温で1時間、脱保護を行った。アミノアシル化tRNA(化合物AT-1)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-1)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Asp(SMe)-tRNAEnAsnGAG
Asp(SiPr)-tRNAEnAsnGAG
Asp(StBu)-tRNAEnAsnGAG
Asp(SBn)-tRNAEnAsnGAG
Asp(SPhenetyl)-tRNAEnAsnGAG
Asp(SEt)-tRNAEnAsnGAG
鋳型DNAとしてKA03 DNA配列(配列番号 D-6)を用いて翻訳反応を行った。
鋳型DNAに、前述の転写翻訳液、MetおよびLeuを除く各0.3mMの18種類のタンパク質性アミノ酸,および前記の方法で調製した25μM tBuSSEtGABA-tRNAfMetCAU(化合物番号 AT-6-C)と50μM Asp(SMe)-tRNAEnAsnGAG(化合物番号 AT-1-IA2)を加え37℃で3時間翻訳した。翻訳産物に、1/20(v/v)量の200mM TCEP(pH6.6)を添加し、37℃で15分反応させ、Asp(SMe)部位と分子内環化反応を行った。その結果、目的の環状ペプチド(ペプチド配列P-43)がMSスペクトルで確認された(図27)。チオエステルに続くアミノ酸をN-アルキルアミノ酸とすることで効率的に翻訳・環化が実現された。
[tBuSSEtGABA]ThrArgThrLysAlaTyrTrpSer[Asp(SMe)]ProGlyGly
MALDI-MS:m/z: [M+H]+ = 1601.7 (Calc.1601.0)
[HSEtGABA]ThrArgThrLysAlaTyrTrpSer[Asp(SMe)]ProGlyGlyのGABAの主鎖窒素原子とAspの側鎖カルボン酸でアミド環化した化合物

1.NCL(Native Chemical Ligation)を介したアミド型環化ペプチドの合成
鋳型DNAとしてKA02.5U DNA配列(配列番号 D-32)を用いて翻訳反応を行った。
鋳型DNAに、前述の転写翻訳液、MetおよびAla、Leuを除く各0.3mMの17種類のタンパク質性アミノ酸,3 mM乳酸、および前記の方法で調製した50μM Asp(SRex2)-tRNAEnAsnGAG (Rex2:ベンジル,メチル,エチル,イソプロピル、ターシャリーブチル、フェネチル)(化合物番号 AT-1-IA2、IB2、IC2、ID2、IE2,IG2)を加え37℃で3時間翻訳した。その結果、環化した全長ペプチドがMSスペクトルで確認された(図28~図29)。
KA02.5U DNA配列:
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG
tgcACTAGAACTaaggcgTACTGGAGCcttCCGggctaa
CysThrArgThrLys[Lac]TyrTrpSer[Asp(SRex2)]ProGly
(Rex2:ベンジル、メチル、エチル、イソプロピル、ターシャリーブチル、フェネチル、Lac:乳酸)
翻訳後、環化したペプチドの化学構造(ペプチド配列番号P-44からP-49)
CysThrArgThrLys[Lac]TyrTrpSer[Asp(SRex2)]ProGlyのCysの主鎖アミノ基とAspの側鎖カルボン酸との分子内アミド化された化合物


2-1.LC-MS分析用翻訳ペプチド調製
20nM 鋳型DNA Mctryg3(配列番号D-8)に、前述の転写翻訳液、0.1mM 10-HCO-H4folate(10-formyl-5, 6, 7, 8, -tetrahydrophilic acid)、MetおよびLeuを除く各0.3mMの18種類のタンパク質性アミノ酸,および前記の方法で調製した50μM Asp(SMe)-tRNAEnAsnGAG(化合物AT-1-IA)を加え、37℃で3時間翻訳した。翻訳反応物に1mM ジチオスレイトールを添加し、37℃で30分間還元を行った後、10mM ヨードアセトアミドを添加し、遮光・室温で40分間、チオールのカルボキシアミドメチル化を行った。
CysThrThrThrArg[Asp(SMe)]TyrTyrArgGlyGly
CysThrThrThrArg[Asp(SMe)]TyrTyrArgGlyGlyのCysの主鎖アミノ基とAspの側鎖カルボン酸が分子内アミド環化された化合物 massスペクトル calc. 1273.6
CysThrThrThrArg[Asp(SMe)]TyrTyrArgGlyGly(ペプチド配列P-52)のCysの主鎖アミノ基とAspの側鎖カルボン酸が分子内アミド環化されたペプチド配列P-53のCysのSH基がアセトアミド化された化合物

2-2-1.自動合成機によるペプチド固相合成の一般法
ペプチド合成機(Multipep RS; Intavis社製)を用いて、Fmoc法によりペプチド合成を行った。Fmoc-Cys(Mmt)-OHをノバビオケム社から購入し、Fmoc-Cys(Mmt)-OHを除く他のFmocアミノ酸を渡辺化学工業から購入した。なお、操作の詳細な手順については合成機に付属のマニュアルに従った。
合成機にC末端のFmocアミノが結合した2-クロロトリチルレジン(1カラムあたり250-300mg)と、各種Fmocアミノ酸(0.6mol/L)と1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)(0.375mol/L)のN,N-ジメチルホルムアミド溶液と、ジイソプロピルカルボジイミド(DIC)のN,N-ジメチルホルムアミド(DMF)溶液(10%v/v)をセットし、Fmoc脱保護溶液として、5%(wt/v)の尿素を含むピペリジンのN,N-ジメチルホルムアミド溶液(20%v/v)、またはジアザビシクロウンデセン(DBU)のN,N-ジメチルホルムアミド溶液(2%v/v)を用いて合成を行った。レジンはDMF溶液で洗浄した後、Fmoc脱保護に次いでFmocアミノ酸の縮合反応を1サイクルとし、このサイクルを繰り返すことでレジン表面上にペプチドを伸長させた。このような実験例としてRamon Subiros-funosasらの非特許文献(Org. Biomol. Chem. 2010, 8, 3665-3673)などを参考にして合成した。
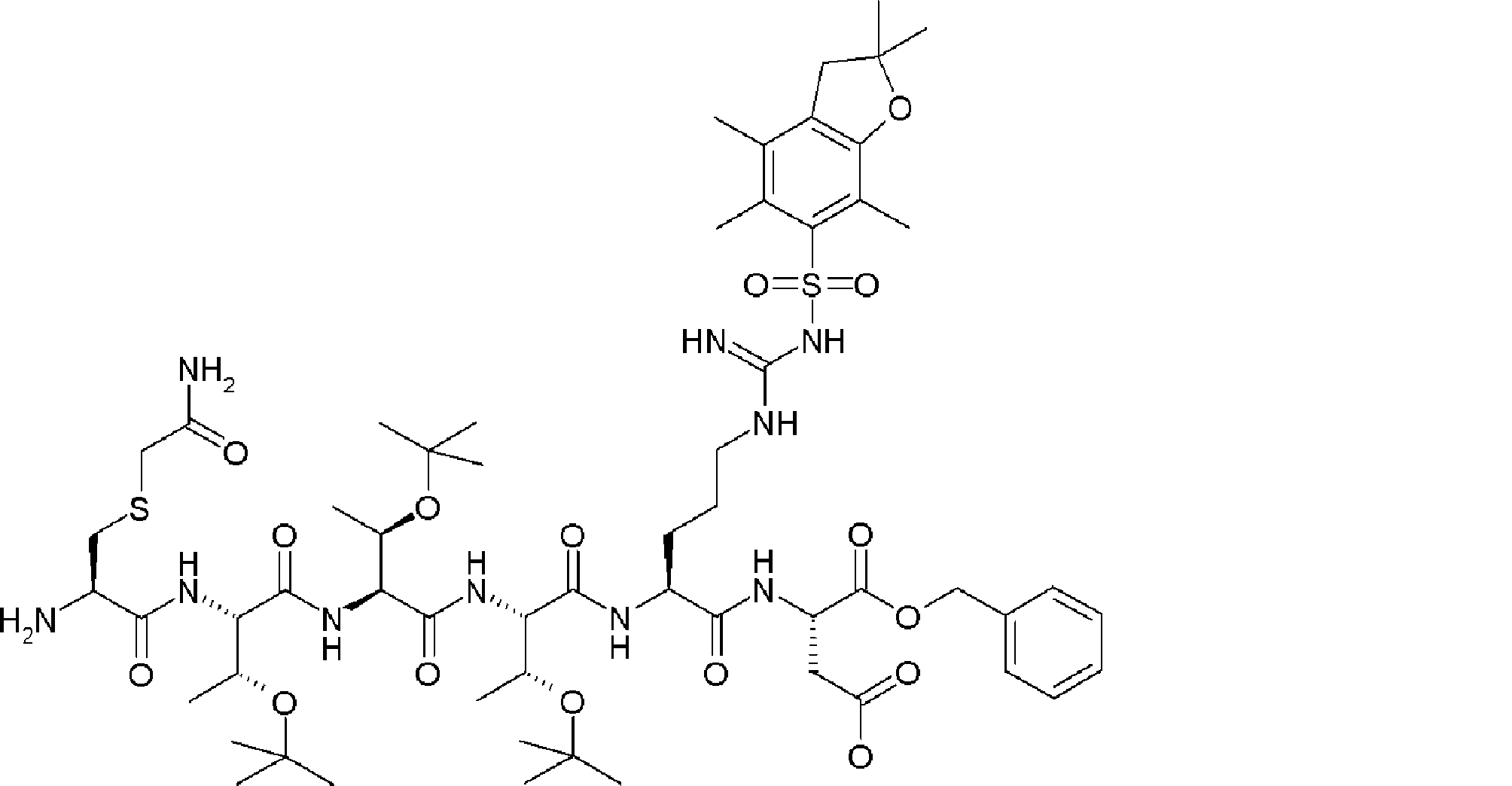
伸長終了後、レジンをジクロロメタンで洗浄し、トリフルオロ酢酸/トリイソプロピルシラン/ジクロロメタン(=2/5/93, 100mL)を加え、レジンからペプチドの切り出しとS-メトキシトリチル基の脱保護を行った。1.5時間後、チューブ内溶液を合成用カラムでろ過することによりレジンを除き、反応液に2-ヨードアセトアミド(252mg、1.36mmol),DIPEA(15mL),DMF(15mL)を加え、Sのアルキル化を行った。1.5時間後、減圧濃縮し得られた残渣を逆相シリカゲルクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液=95/5→0/100)にて精製し、直鎖ペプチドH-Cys(CH2CONH2)-Thr(tBu)-Thr(tBu)-Thr(tBu)-Asp-Arg(Pbf)-Asp-OBn(ペプチドP-55)を得た。(180mg、10%)
LCMS(ESI) m/z =1262(M-H)-
保持時間:0.71分(分析条件SQDFA05)

LCMS(ESI) m/z =1246(M+H)+
保持時間:1.02分(分析条件SQDFA05)

LCMS(ESI) m/z =1156(M+H)+
保持時間:0.93分(分析条件SQDFA05)
上記レジンに、c(Cys(CH2CONH2)-Thr(tBu)-Thr(tBu)-Thr(tBu)-Arg(Pbf)-Asp)-OH(ペプチドP-57、46mg、0.04mmol)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)(5.4mg、0.04mmol)ジイソプロピルカルボジイミド(DIC)(0.075mL0.048mmol)のN,N-ジメチルホルムアミド(DMF)溶液を加えた。17時間後、レジンをジクロロメタンで洗浄し、トリフルオロ酢酸/ジクロロメタン(=1/99, 4mL)を加え、レジンからペプチドの切り出しを行った。1.5時間後、チューブ内溶液を合成用カラムでろ過することによりレジンを除き、減圧濃縮し得られた残渣を逆相シリカゲルクロマトグラフィー(10mM酢酸アンモニウム水溶液/メタノール=50/50→0/100)にて精製し、C(Cys(CH2CONH2)-Thr(tBu)-Thr(tBu)-Thr(tBu)-Arg(Pbf)-Asp)-Tyr(tBu)-Tyr(tBu)-Arg(Pbf)-Gly-Gly-OH(ペプチドP-58)を得た。(6.2mg、7.3%)
LCMS:1059 m/z (M+2H)2+
保持時間:0.83分 (分析条件SQDAA50)
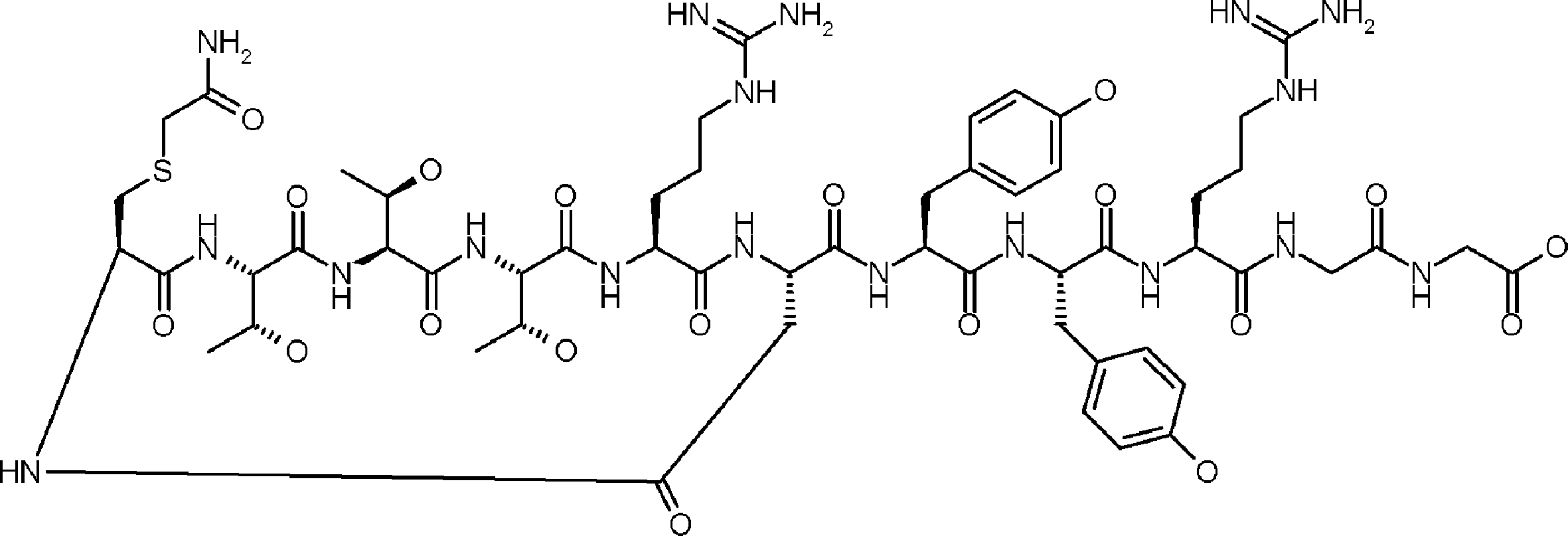
LCMS:664 m/z (M-2H)2-、1329.4(M-H)-
LCMS:1331.4 m/z (M+H)+
保持時間:0.49分 (分析条件SQDAA05)
翻訳合成品(ペプチドP-54)と同一の配列を有する有機合成品を作製したので、高分解能LC/MS装置(Orbitrap Velos、サーモフィッシャーサイエンティフィック社製、USA)を用いて比較分析した。翻訳品については、翻訳溶液約30uLに0.5%トリフルオロ酢酸60uLを加え攪拌後、遠心分離上清をOasis HLBカートリッジ(30mg、1cc、Waters Corporation、USA)にアプライした。0.5%トリフルオロ酢酸含有5%アセト二トリル溶液1mLで洗浄後、0.5%トリフルオロ酢酸含有60%アセト二トリル溶液1mLで溶出した。溶出液を窒素乾固し、0.5%トリフルオロ酢酸含有20%アセト二トリル溶液60uLに再溶解しLC/MS分析に付した。翻訳産物の精密質量をポジティブイオンモードで測定した結果、m/z 666.2937に2価のプロトン化分子[M+2H]2+を与え、理論値m/z 666.2935と良く一致していた。高速液体クロマトグラフにおける保持時間ならびにMS/MSスペクトルパターンを有機合成品と比較した結果、いずれも良く一致していた。以上の結果、翻訳合成産物は有機合成による環化ペプチドと同一のものであることが確認された(図30)。
翻訳合成にてアミド環化反応が進行した一方、我々の目的のためには反応補助基のSH基の除去が必要となる。SH基は様々なタンパクと共有結合を形成することが知られているため、SH基依存的な結合化合物が得られる可能性が高い一方で、そのような化合物は高い反応性のため薬剤になりにくい。
SH基の除去には分子間反応が必要である。既に述べたように分子間反応を1uM濃度にて、かつ様々な反応性官能基が共存する条件下での実現の難易度は高い。このような反応を実現させる手法として、以下の2つが考えられる。(i)過剰に用いる試薬は、望みの反応点以外では可逆的な反応(例えば配位結合)のみが生じ、反応完結後の後処理により除去される。(ii)過剰に用いる試薬はRNAには全く影響を与えない。
このような各種条件をクリアできる反応基質と反応条件を様々検討した結果を以下に示す。
モデル基質2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)を用いてWanら(Angew. Chem. Int. Ed. 2007, 46, 9248)と同様の方法でラジカル脱硫反応が進行することを確認した(図31および図32)。ラジカル脱硫反応としてMethodA及びMethodBを行った。
以下、図32に記載の実験を行った。
1-1-1.図32 Entry 1の実験
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b) の合成
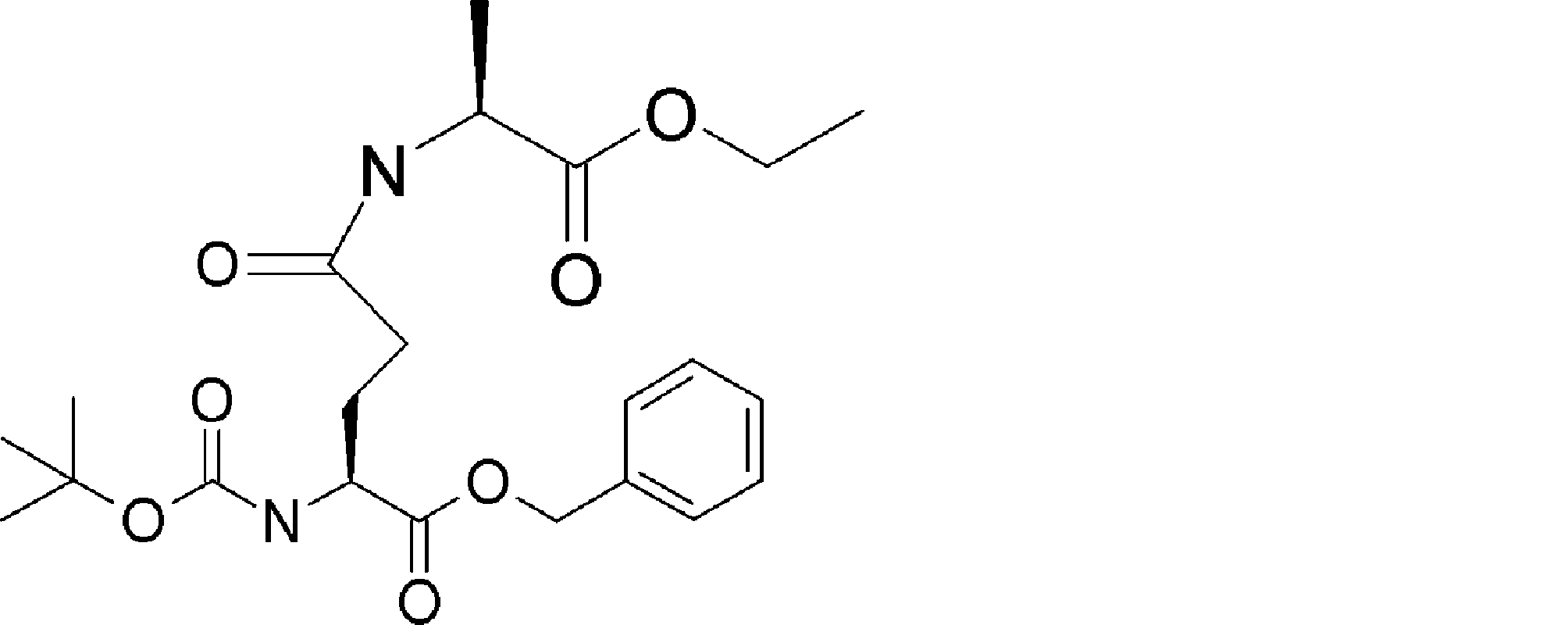
LCMS(ESI) m/z = 437 (M+H)+
保持時間:0.96分(分析条件SQDAA05)
0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液の調整
トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(1.0g、3.49mmol)の水(6.8ml)溶液にトリエチルアミン(1.64ml)加え、pH7とし、0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液を得た。
2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)塩酸塩(100mg、0.309mmol)に水(0.309ml)加え1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液を調整した。
2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)塩酸塩(100mg、0.309mmol)に水(3,09ml)加え0.1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液を調整した。
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(10.0mg、0.0213mmol)のDMF(0.5ml)あるいはメタノール(0.5ml)溶液に、グルタチオン(19.7mg、0.064mmol)と0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.214ml、0.0852mmol)を加え室温で10分間攪拌後、1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.0213ml、0.0213mmol) を室温で加え、50度で2時間攪拌した。反応の変化をLCMSを用いて観測したところ、entry2は2時間で原料化合物3aがすべて消失し、目的の化合物3bが観測された。
LCMS(ESI) m/z = 437 (M+H)+
保持時間:0.96分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(10.0mg、0.0213mmol)のメタノール(4.66ml)、水(2.06ml)溶液に0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.268ml、0.107mmol)を加え室温で10分間攪拌後、1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.0213ml、0.0213mmol) を室温で加え、50度で1時間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全には消失されなかったが、目的化合物3bが観測された。さらに、副生成物として、推定化合物3c、3dもまた観測された。その割合は、LCMS UVエリア強度比より、原料化合物3a:目的化合物3b:3c:3d=7:20:16:11であった。
化合物3b
LCMS(ESI) m/z =437(M+H)+
保持時間:0.96分(分析条件SQDAA05)
構造推定化合物3c
LCMS(ESI) m/z =453(M+H)+
保持時間:0.91分(分析条件SQDAA05)
構造推定化合物3d
LCMS(ESI) m/z =337(M+H)+
保持時間:0.87分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(10.0mg、0.0213mmol)のメタノール(4.66ml)、水(0.20ml)溶液にグルタチオン(196mg、0.639mmol)と0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.268ml、0.107mmol)を加え室温で10分間攪拌後、1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.0213ml、0.0213mmol) を室温で加え、50度で1時間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全には消失され、目的化合物3bが観測された。
LCMS(ESI) m/z =437(M+H)+
保持時間:0.96分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(10.0mg、0.0213mmol)のメタノール(4.66ml)、水(2.06ml)溶液にグルタチオン(19。6mg、0.0639mmol)と0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.268ml、0.107mmol)を加え室温で10分間攪拌後、1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.0213ml、0.0213mmol) を室温で加え、50度で1時間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全には消失され、目的化合物3bが観測された。さらに、副生成物として、推定化合物3c、3dもまた観測された。その割合は、LCMS UVエリア強度比より、原料化合物3a:目的化合物3b:3c:3d=0:79:14:6であった。
化合物3b
LCMS(ESI) m/z =437(M+H)+
保持時間:0.96分(分析条件SQDAA05)
構造推定化合物3c
LCMS(ESI) m/z =453(M+H)+
保持時間:0.91分(分析条件SQDAA05)
構造推定化合物3d
LCMS(ESI) m/z =337(M+H)+
保持時間:0.87分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(10.0mg、0.0213mmol)のメタノール(0.5ml)溶液に0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.268ml、0.107mmol)を加え室温で10分間攪拌後、1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.0213ml、0.0213mmol)を室温で加え、50度で1時間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全に消失され、目的化合物3bが観測された。さらに、副生成物として、推定化合物3c、3dもまた観測された。その割合は、LCMS UVエリア強度比より、原料化合物3a:目的化合物3b:3c:3d=0:24:32:19であった。
化合物3b
LCMS(ESI) m/z =437(M+H)+
保持時間:0.96分(分析条件SQDAA05)
構造推定化合物3c
LCMS(ESI) m/z =453(M+H)+
保持時間:0.91分(分析条件SQDAA05)
構造推定化合物3d
LCMS(ESI) m/z =337(M+H)+
保持時間:0.87分(分析条件SQDAA05)
Method Aとほぼ同様の製造方法であるが、2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)のMeOH-H2O溶液に0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液、グルタチオンを加え、目的の反応温度30度、40度あるいは50度に加熱後 1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液を加え、そのまま30度、40度あるいは50度の反応温度で攪拌した。反応の経時変化をLC-MSを用いて観測した。
トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(1.0g、3.49mmol)の水(6.8ml)溶液にトリエチルアミン(1.64ml)加え、pH7とし、0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液を得た。
2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)塩酸塩(100mg、0.309mmol)に水(0.309ml)加え1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液を調整した。
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(1.1mg、0.0023mmol)のメタノール(0.5ml),水(0.06ml)溶液に0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.192ml、0.092mmol)を加え50度で10分間攪拌後、0.1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.023ml、0.0023mmol) を50度で加え、その後、50度で30分間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全に消失され、目的化合物3bが観測された。
LCMS(ESI) m/z =437(M+H)+
保持時間:0.95分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(1.1mg、0.0023mmol)のメタノール(0.5ml),水(0.06ml)溶液にグルタチオン(21.2mg、0.069mmol)と0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.192ml、0.092mmol)を加え50度で10分間攪拌後、0.1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.023ml、0.0023mmol) を50度で加え、その後、50度で30分間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全に消失され、目的化合物3bが観測された。
LCMS(ESI) m/z =437(M+H)+
保持時間:0.95分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(1.1mg、0.0023mmol)のメタノール(0.5ml),水(0.06ml)溶液に0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.192ml、0.092mmol)を加え40度で15分間攪拌後、0.1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.023ml、0.0023mmol)を40度で加え、40度で30分間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全に消失され、目的化合物3bが観測された。
LCMS(ESI) m/z =437(M+H)+
保持時間:0.96分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(1.1mg、0.0023mmol)のメタノール(0.5ml),水(0.06ml)溶液にグルタチオン(21.2mg、0.069mmol)と0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.192ml、0.092mmol)を加え40度で15分間攪拌後、0.1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.023ml、0.0023mmol)を40度で加え、その後40度で30分間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全に消失され、目的化合物3bが観測された。
LCMS(ESI) m/z =437(M+H)+
保持時間:0.95分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(1.1mg、0.0023mmol)のメタノール(0.5ml),水(0.06ml)溶液に0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.192ml、0.092mmol)を加え30度で15分間攪拌後、0.1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.023ml、0.0023mmol)を30度で加え、その後30度で1時間30分間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全に消失され、目的化合物3bが観測された。
LCMS(ESI) m/z =437(M+H)+
保持時間:0.95分(分析条件SQDAA05)
2-((tert-ブトキシカルボニル)アミノ)-5-(((S)-1-エトキシ-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3b)
2-((tert-ブトキシカルボニル)アミノ)-5-(((R)-1-エトキシ-3-メルカプト-1-オキソプロパン-2-イル)アミノ)-5-オキソペンタン酸 (S)-ベンジル(化合物3a)(1.1mg、0.0023mmol)のメタノール(0.5ml),水(0.06ml)溶液にグルタチオン(21.2mg、0.069mmol)と0.4Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(0.192ml、0.092mmol)を加え30度で15分間攪拌後、0.1M 2,2'-アゾビス-2-(2-イミダゾリン-2-イル)プロパン(VA-044)水溶液(0.023ml、0.0023mmol)を30度で加え、その後30度で1時間30分間攪拌した。反応の変化をLCMSを用いて観測したところ、原料3aが完全に消失され、目的化合物3bが観測された。
LCMS(ESI) m/z =437(M+H)+
保持時間:0.95分(分析条件SQDAA05)
RNA化合物を脱硫反応の条件に付し、安定性の確認を行った。
グレンリサーチ社製ピューロマイシンCPG(22.7mg、0.999μmol)を用い、DNA合成機によりRNA結合伸長を行った。A、G、C、Uの伸長反応に用いるアミダイド試薬はグレンリサーチ社製A-TOM-CEホスホアミダイド、G-TOM-CEホスホアミダイド、C-TOM-CEホスホアミダイド、U-TOM-CEホスホアミダイドを使用し、縮合活性化剤として5-ベンジルチオ-1H-テトラゾールを使用し実施した。縮合後の固相担体を乾燥後、エタノール(0.25mL)、40%メチルアミン水溶液(0.25mL)を加え65℃にて1時間攪拌した。固相担体を濾別し、メタノール(0.5mL)で洗浄した。得られた溶液を濃縮しメタノール(1.0mL)に溶解し、そのうち0.8mLを濃縮しテトラメチルアンモニウムフルオライド水和物(50μL)に溶かし65℃にて15分間攪拌した。反応液に0.1M酢酸アンモニウム水溶液を加え逆相カラムで精製した。濃縮後、得られた化合物を再度逆相カラムで精製した。得られた溶液に水を加え逆相カラムに添着後、水(30mL)で洗浄したのちメタノール(50mL)で目的物を溶出した。得られた溶液を濃縮し水(1.0mL)に溶解し5´-AGCUUAGUCA-ピューロマイシン-3´(化合物RP-1)水溶液を得た。
LCMS(ESI) m/z = 1224.6 (M-3H)3-
保持時間:0.38分(分析条件SQDAA05)
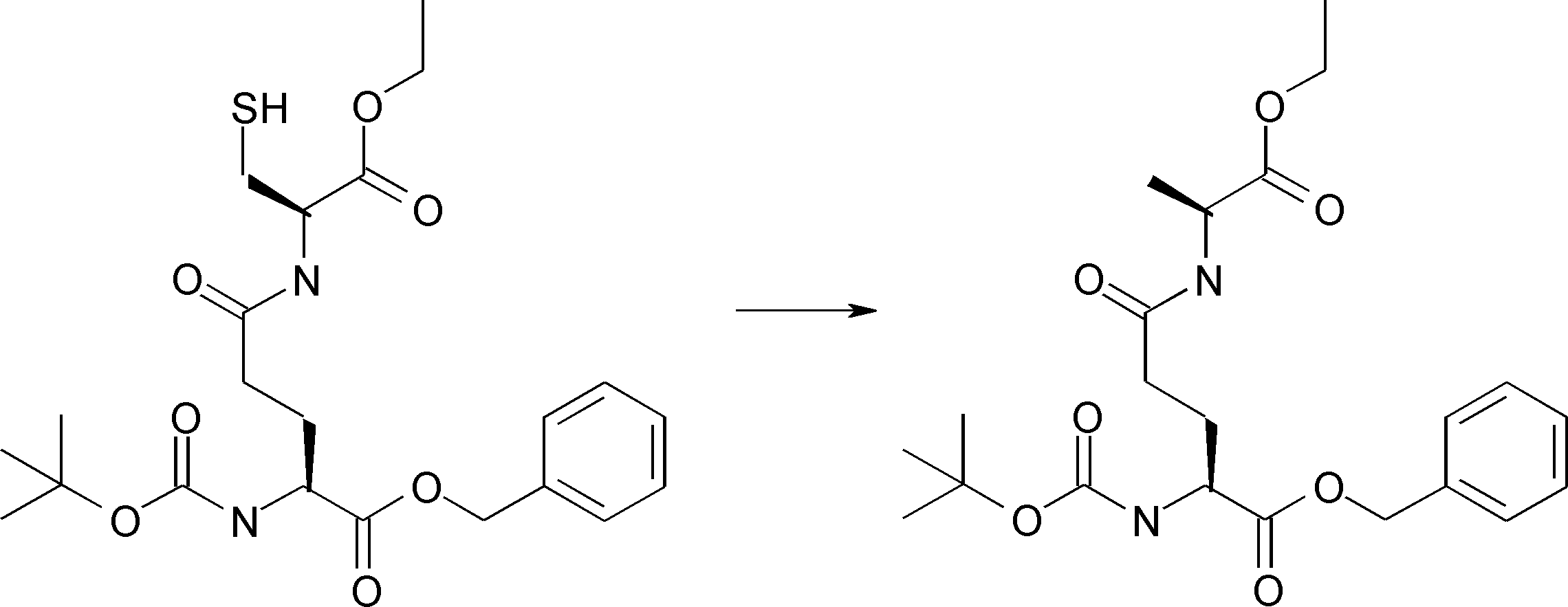
LCMS(ESI) m/z = 437.3 (M+H)+
保持時間:0.96分(分析条件SQDAA05)
以下の方法により翻訳を行った。
鋳型DNA配列番号D-34として以下の配列を用いて翻訳反応を行った。
Mctryg3_08U05U: GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGtgc
ACTACAACGCGTctttactaccgtggcggcTAGTAGATAGATA
CysThrThrThrArgLeuTyrTyrArgGlyGly
(Calc. 1289.6)
AlaThrThrThrArgLeuTyrTyrArgGlyGly
m/z: [M+H]+ = 1258.7 (Calc. 1257.7)
脱硫反応液によるタンパク質変性の評価を行った(図34)。モデル系として、電気化学発光免疫測定法によるIL-6およびIL-6Rの相互作用検出系において、脱硫反応条件が、IL-6Rに対して、どのような影響を及ぼすか評価した。まず、MSD Multi-array 384 well (MSD社)に750ngの抗IL-6R抗体クローン番号17506(R&D biosystems社)を4度で一晩保存し、抗体の固相化を行った。80μlのPBSTでプレートを三回洗浄し、未反応の抗体を除いた後、2%スキムミルクで1時間ブロッキングを行った。ブロッキング剤を80μlのPBSTでプレートを三回洗浄後、10μlのsoluble human IL-6R(IL-6R)を25nM加え、室温で50分間振とうした。80μlのPBSTでプレートを三回洗浄し、脱硫反応液(25mM HEPES-K(pH7.6),200mM TCEP,250mM VA-044,10mM Cys)を添加し、室温で50分間振とうした。80μlのPBSTでプレートを三回洗浄し、10μlのhuman IL-6-BAP(500nM)を添加し、室温で50分間振とうした。80μlのPBSTでプレートを三回洗浄し、10μL SULFO-TAG StAv(MSD社,cat#R32AD-5,1μg/mL)を添加後、室温で50分間振とうした。80μlのPBSTでプレートを三回洗浄し、35μlの2X read buffer (MSD社,R92SC-3)を添加後、SECTOR Imager 2400 (MSD社)で測定した(図34)。
その結果、脱硫反応液を添加することで、IL6-Rが影響を受け、IL-6との相互作用が弱まることが示された。また、脱硫反応液に、酸化型のDTT(DTTox)を添加することで、IL-6Rへの影響が弱まり、IL-6との相互作用が回復することが示された。
チオエーテル環化法と本発明の環化法の比較を行った。脂質膜透過速度が速いと考えられる脂溶性が高いチオエーテル環化ペプチドを合成した。
1.チオエーテル環化ペプチドの合成
Fmocアミノ酸として、Fmoc-MePhe-OH、Fmoc-MeAla-OH、Fmoc-MeLeu-OH、Fmoc-MeGly-OH、Fmoc-MeIle-OH、Fmoc-Ser(tBu)-OH、Fmoc-Thr(Trt)-OH、Fmoc-Ser(Trt)-OH、Fmoc-Phe-OH、Fmoc-Bip-OH、Fmoc-Leu-OH、Fmoc-Cha-OH、Fmoc-Ala-OH、Fmoc-Cys(Mmt)OHなどを用いてペプチドの伸長を行った(略語は渡辺化学カタログによる)。ペプチドの伸長後、N末端のFmoc基を脱保護し、HOAt,DICを縮合剤としてクロロ酢酸を縮合させた後、レジンをジクロロメタンにて洗浄した。レジンにトリフルオロ酢酸/ジクロロメタン/2,2,2-トリフルオロエタノール/トリイソプロピルシラン(=1/62/31/6,v/v/v/v、4mL)を加えて2時間反応させ、レジンからのペプチドの切り出しと同時にO-トリチル基、S-ジメトキシトリチル基の脱保護を行った。反応終了後、チューブ内溶液を合成用カラムでろ過することによりレジンを除き、反応液をジイソプロピルエチルアミン(1mL)溶液を入れたチューブに反応液を加え、クロロアセチル基とシステインとの環化反応を行った。反応終了後、溶媒を留去した。得られた租生成物を、DMF中、ピペリジン(1.9eq.)O-(7-アザー1Hベンゾトリアゾール-1-イル)-N,N,N,Nテトラメチルウロニウム ヘキサフルオロホスフェート(HATU)(1.7eq.)にてC末端カルボン酸のピペリジンアミド化を行った。反応終了後、Genevacにて溶媒留去した。得られた租生成物はジメチルスルホキシドに溶解し、得られたペプチド溶液は高速逆相クロマトグラフィー(HPLC)で精製した。
Ac*-MePhe-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Cys*-piperidine
(2つの*部位にて環化)

保持時間:0.77分 (分析条件 SQDAA50)
Ac*-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Cys*-piperidine
(2つの*部位にて環化)

保持時間:0.76分 (分析条件 SQDAA50)
Ac*-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine
(2つの*部位にて環化)

保持時間:2.72分 (分析条件 ZQAA50)
Ac*-MeLeu-Thr-MeGly-MeLeu-Ser-MeIle-Bip-Cys*-piperidine
(2つの*部位にて環化)
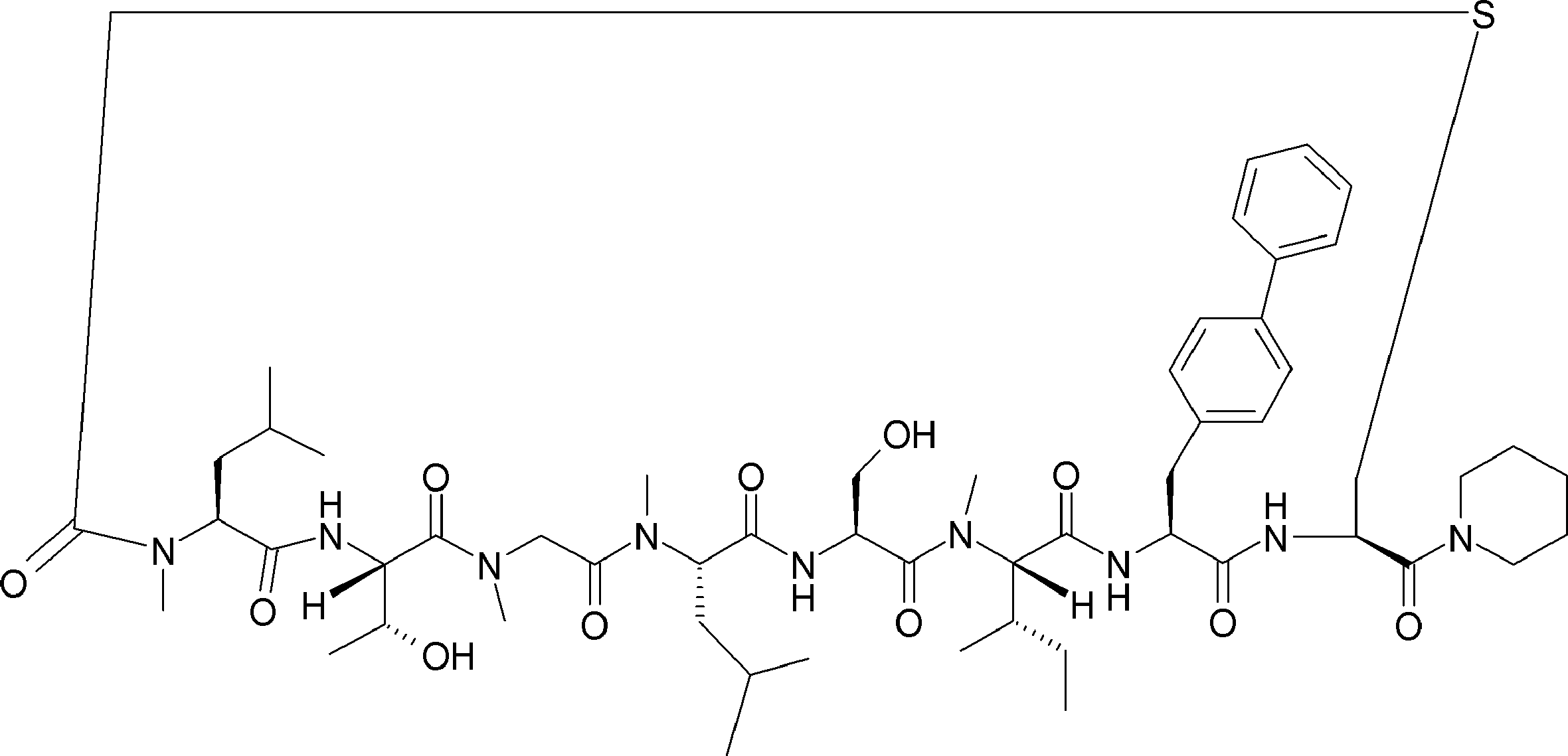
保持時間:2.52分 (分析条件 ZQAA50)
Ac*-MePhe-MeAla-Phe-MeLeu-Thr-MeGly-MeLeu-Cys*-piperidine
(2つの*部位にて環化)
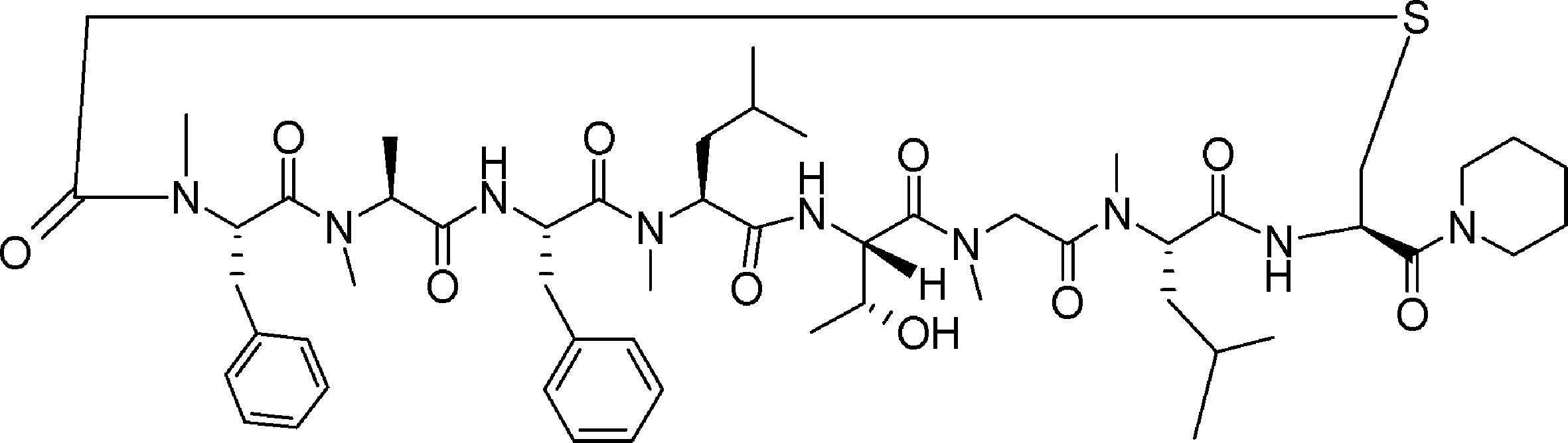
保持時間:0.77分 (分析条件 SQDAA50)
Ac*-MeAla-Phe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Cys*-piperidine
(2つの*部位にて環化)
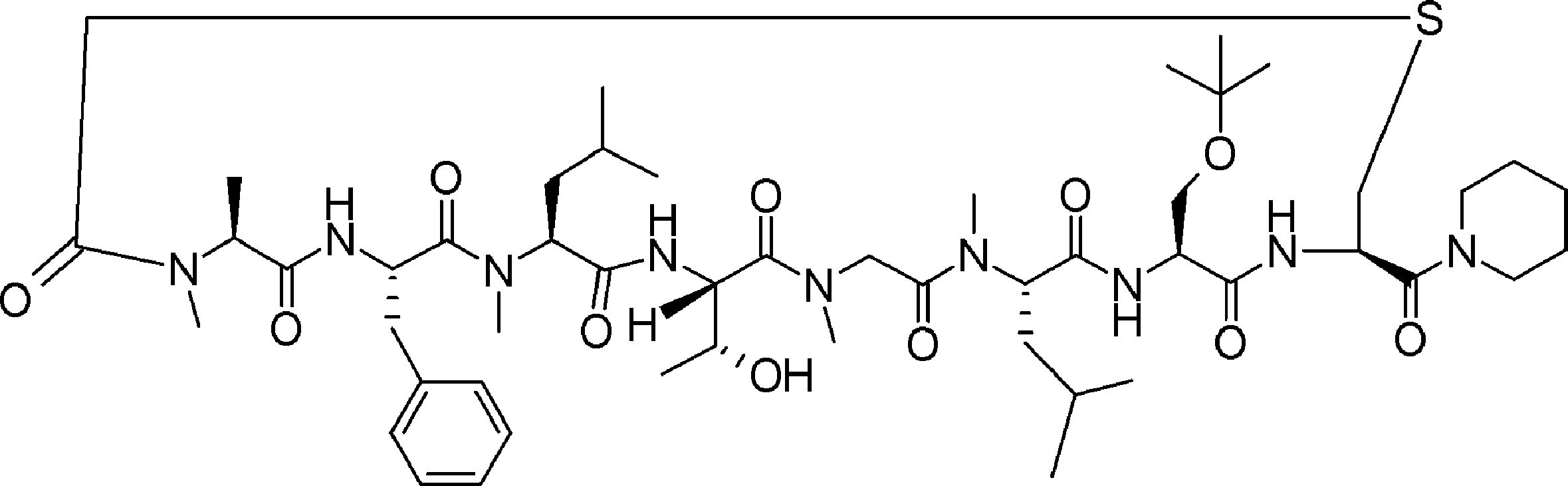
保持時間:0.74分 (分析条件 SQDAA50)
Ac*-Phe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine
(2つの*部位にて環化)
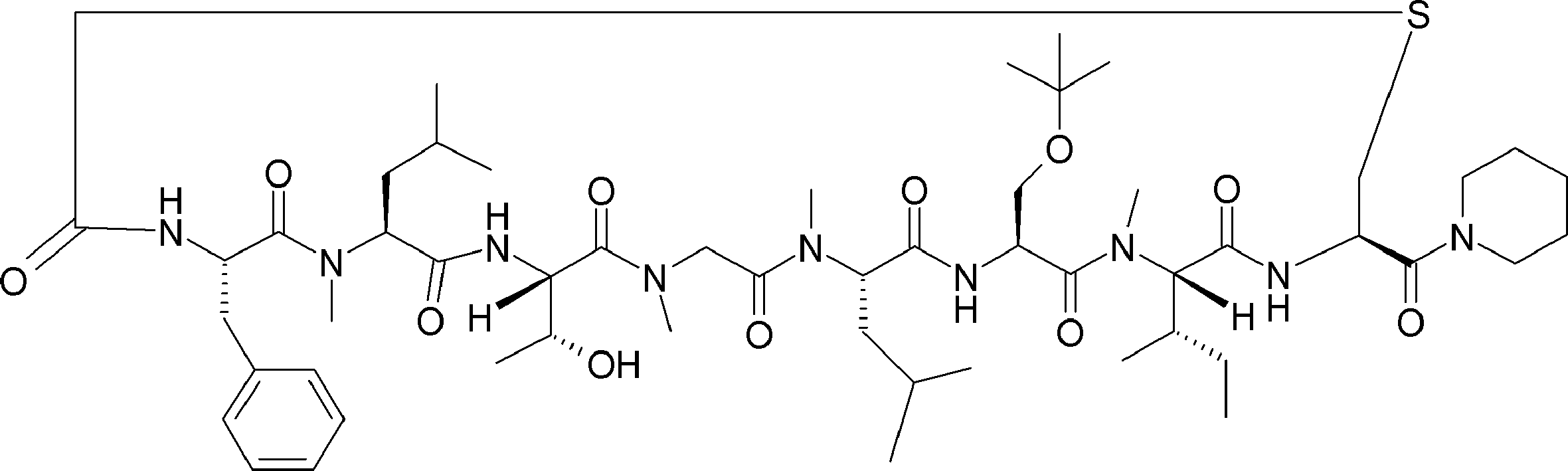
保持時間:0.82分 (分析条件 SQDAA50)
Ac*-MePhe-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Cys*-piperidine
(2つの*部位にて環化)
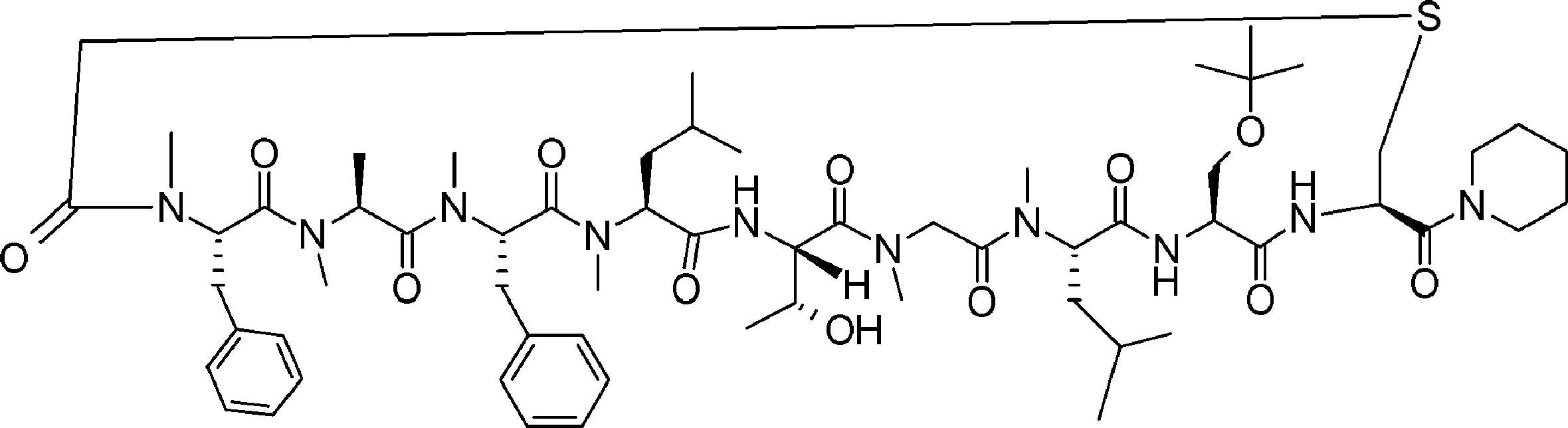
保持時間:2.60分 (分析条件 ZQAA50)
Ac*-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine
(2つの*部位にて環化)

保持時間:2.75分 (分析条件 ZQAA50)
Ac*-MePhe-MeAla-Phe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Cys*-piperidine
(2つの*部位にて環化)
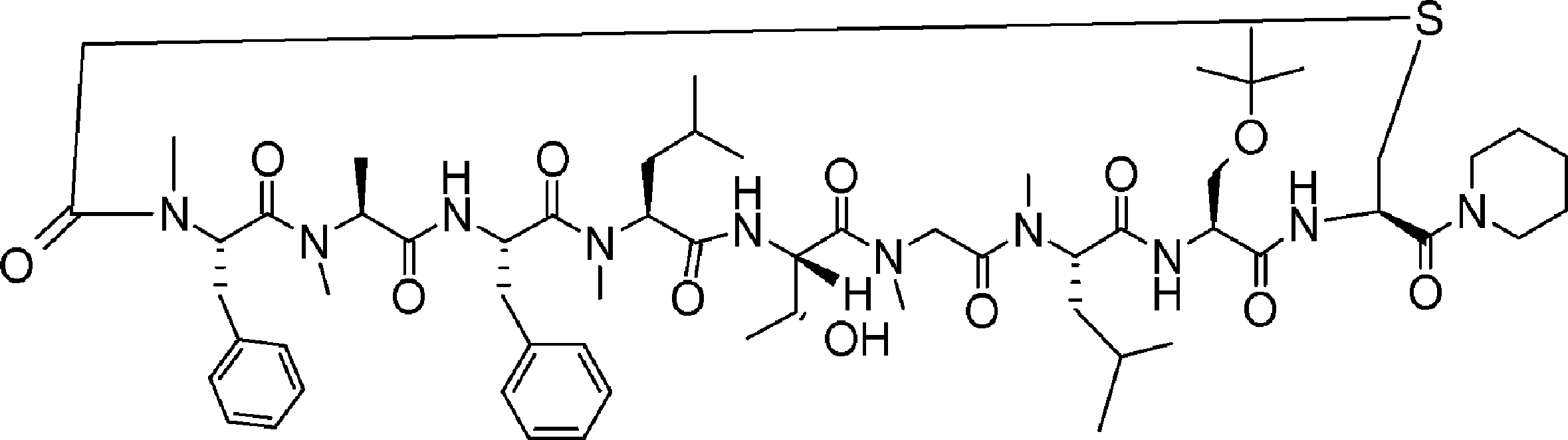
保持時間:0.81分 (分析条件 SQDAA50)
Ac*-MeAla-Phe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine
(2つの*部位にて環化)
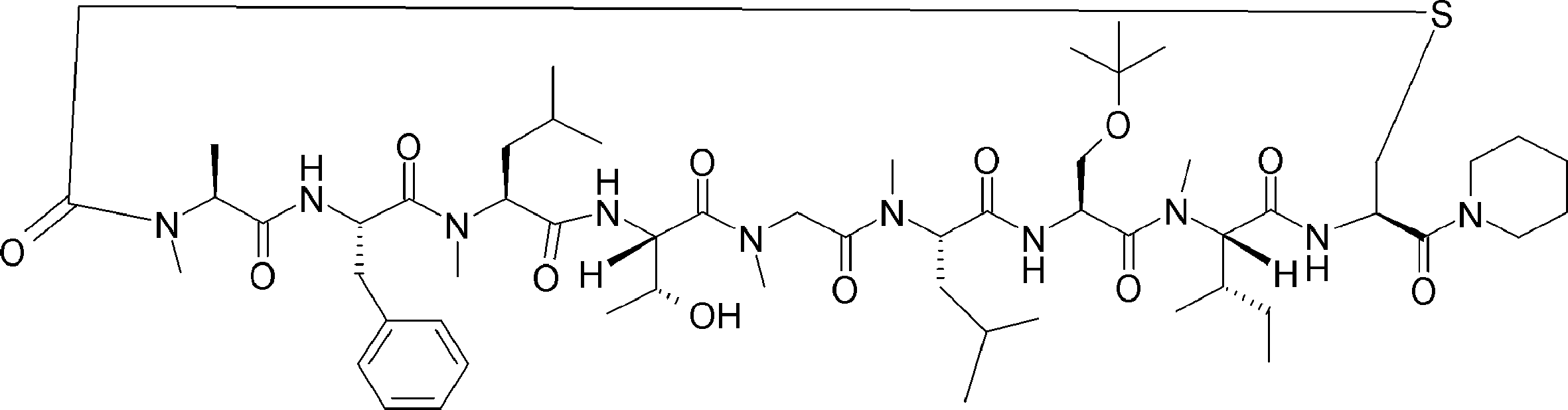
保持時間:2.75分 (分析条件 ZQAA50)
Ac*-MePhe-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine
(2つの*部位にて環化)

保持時間:2.87分 (分析条件 ZQAA05)
Ac*-Leu-MeAla-Cha-Thr-Thr-Ala-Cys*-Cha-piperidine
(2つの*部位にて環化)

保持時間:2.46分 (分析条件 ZQAA50)
2-1.チオエーテル環化ペプチドのSerum安定性、肝ミクロソーム中安定性試験
得られた化合物を2uMの濃度にてマウス血清(雄雌混合、Valley Biomedical社、USA)(33.5mM HEPES緩衝液によりpH7.4に調製)と2時間代謝反応させた。経時的にアセトニトリルによる除タンパク処理を施し、LC/MSを用いて残存する未変化体量を分析し、代謝半減期(t1/2)を算出した。NADPH存在下および非存在下、マウス肝ミクロソームならびに小腸ミクロソーム(雄、XENOTECH社、USA)を100mM リン酸緩衝液(pH7.4)を用いて0.5mg protein/mLになるよう調製したミクロソーム溶液と化合物を1uMの濃度にて30分間代謝反応させた。経時的にアセトニトリルによる除タンパク処理を施し、LC/MSを用いて残存する未変化体量を分析し,肝固有クリアランス(CLh,int)ならびに小腸固有クリアランス(CLg,int)を算出した。(表7)。これらのペプチドはマウス血清中並びにミクロソーム中においてペプチダーゼなどの加水分解による代謝を受ける速度は十分遅く、脂溶性の低い天然アミノ酸からなるペプチドと比較して対照的な結果となった。これらの結果と比較し、ミクロソーム中NADPH存在下における代謝速度は高く、代謝の主な経路は酸化代謝であることが明らかとなった。
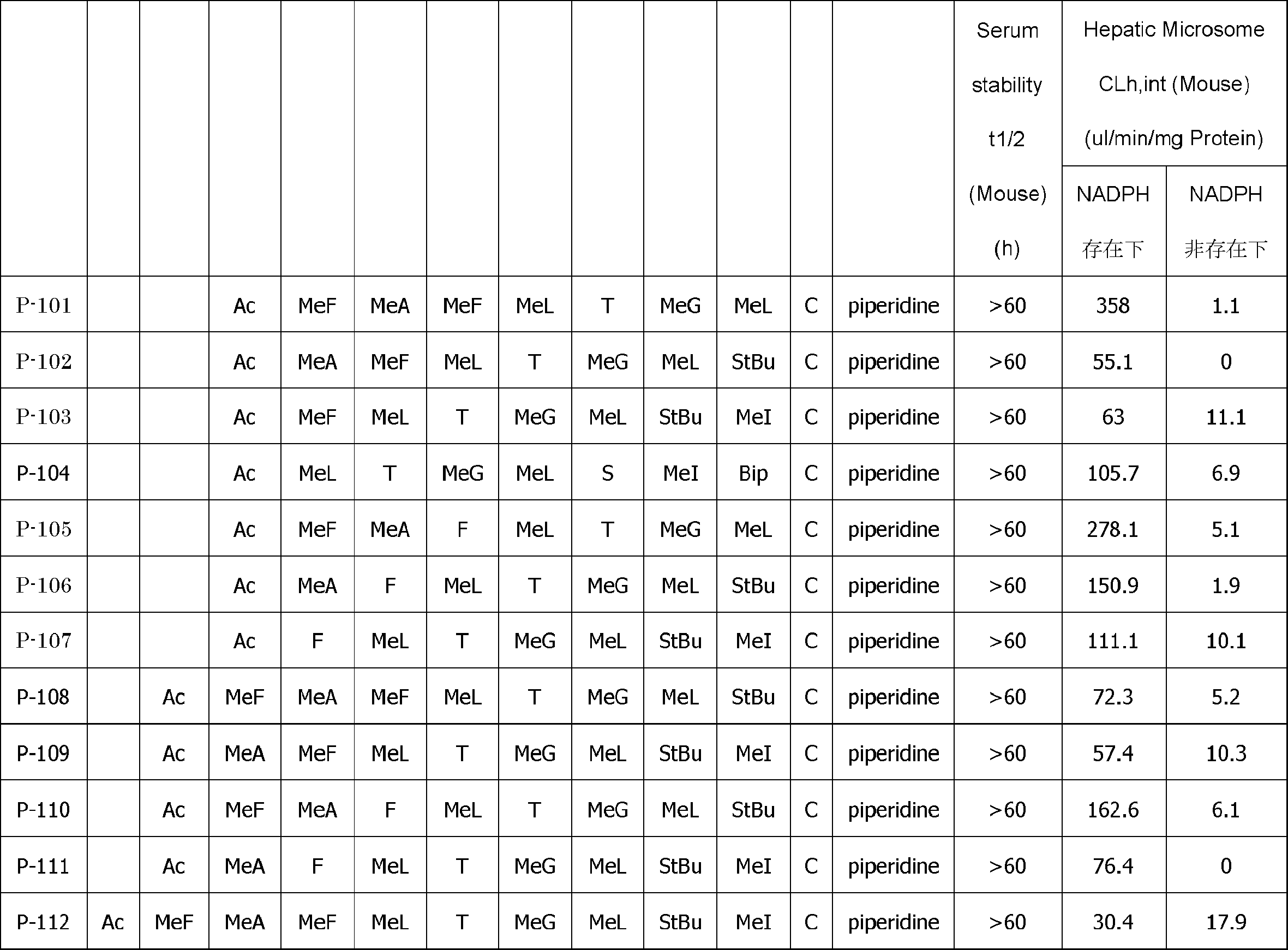
2-1.にて示したのと同様の実験にて、代謝安定性試験を行った。
マウスにて比較的代謝安定性の高かったチオエーテル化合物の小腸での代謝安定性試験と、ヒトでの肝ミクソーム中での代謝安定性試験を実施した(化合物P-112、103、102、109)。ヒトでの肝ミクロソームでの代謝安定性は、マウスの場合と比較しておよそ3倍程度悪化した。
続いて化合物P-112(配列 Ac*-MeA-MeF-MeL-Thr-MeG-MeL-SertBu-MeI-Cys*- piperidine)のS原子を酸化させたスルホキシド体(化合物P-114)の肝および小腸酸化代謝速度を測定した結果、化合物P-112と比較してそれぞれマウス肝0.6倍、ヒト肝およそ0.3倍、小腸0.2倍となり、ヒト肝および小腸代謝に改善が観測された(表8)。スルホン体P-115の小腸酸化代謝速度は0.5倍となった。
チオエーテルからスルホキシドへの変換での代謝安定化はマウスの肝ミクロソームでいくつか他の合成検体でも観測された(表9)。


化合物P-113環化ペプチドを10uMの濃度にてNADPH存在下、マウス肝臓(雄、XENOTECH社製、USA)を100mMリン酸緩衝液(pH7.4)を用いて0.5mg protein/mLになるよう調製したミクロゾーム溶液と1時間代謝反応させた(図35)。反応後アセト二トリルによる除タンパク処理を施し、得られた代謝産物を高分解能LC/MS装置(Q-TOF Ultima API、Waters Corporation社製、USA)にて分析した。代謝産物のマスクロマトグラムを以下に示した。比較的強度の強い代謝産物として、水酸化体に相当するピークが4本検出された。各ピークのMS/MSスペクトルを下記に示した。Peak1およびPeak2の代謝物については高い脂溶性側鎖のシクロヘキシル基を含む部分構造に水酸化を受けていることが推察された。Peak3の代謝物についてはピペリジン環に水酸化を受けた際の特徴的なイオンをm/z 102に与えたが、Peak1およびPeak2と類似のフラグメントイオンも与えており、Peak3中にはPeak1とPeak2の類縁体が存在するか、もしくは分離不十分によりPeak1およびPeak2が混入している可能性が示唆された。Peak4の代謝物はチオールを含む部位が酸化されていることが推察されたが、この部位での酸化容易度を考慮すると硫黄原子が代謝部位である可能性が高いと考えられた。
代謝部位をより特定させるために、得られたチオエーテル環化ペプチドを直接酸化反応した。チオエーテル体誘導体に対し、オキソンを用いて直接酸化させるとメチオニン誘導体(ペプチドP-130)が選択的に酸化され、一方トリプトファン誘導体(ペプチドP-132)は同一条件下で酸化反応が進行しないことを確認した。本条件を用いていくつかのチオエーテル体(ペプチドP-109、P-112、P-111、P-103)のチオエーテル部分を選択的に酸化させたペプチドP-114、P-115、P-116、P-117、P-118が得られた。
Ac*-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine化合物(2つの*にて環化、化合物P-109)のスルホキシド体
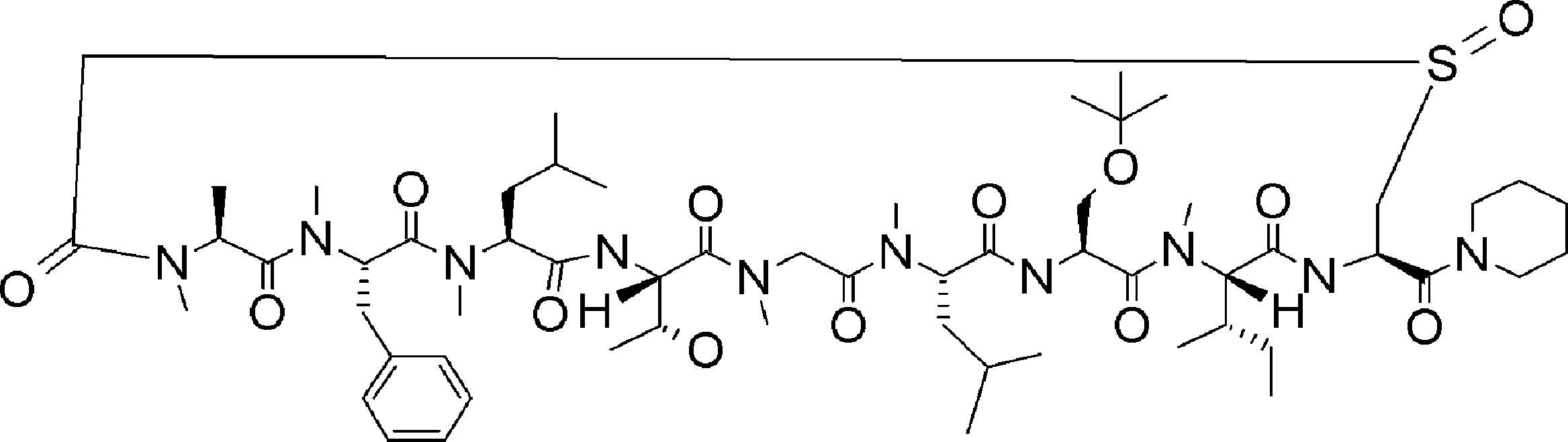
LCMS(ESI) m/z = 1187 (M+H)+
保持時間:2.48分、2.62分(分析条件ZQAA50)
Ac*-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)‐MeIle-Cys*-piperidine化合物(2つの*にて環化、化合物P-109)のスルホン体
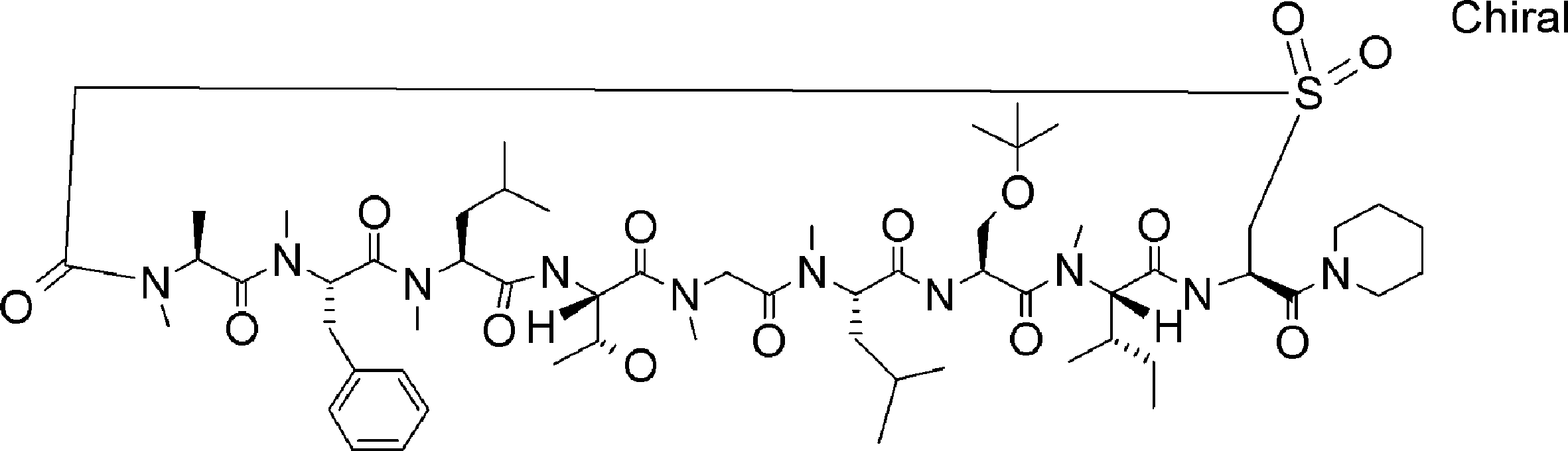
LCMS(ESI) m/z = 1203 (M+H)+
保持時間:0.82分(分析条件SQDAA50)
Ac*-MePhe-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine(化合物P-112)の直接酸化によるスルホキシド体

LCMS(ESI) m/z = 1348 (M+H)+
保持時間:0.84分(分析条件SQDAA50)
Ac*-MeAla-Phe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Cys*-piperidine(化合物P-111)の直接酸化によるスルホキシド体(化合物P-117)

LCMS(ESI) m/z = 1173 (M+H)+
保持時間:2.57分(分析条件ZQAA50)
化合物P-103の直接酸化反応による合成
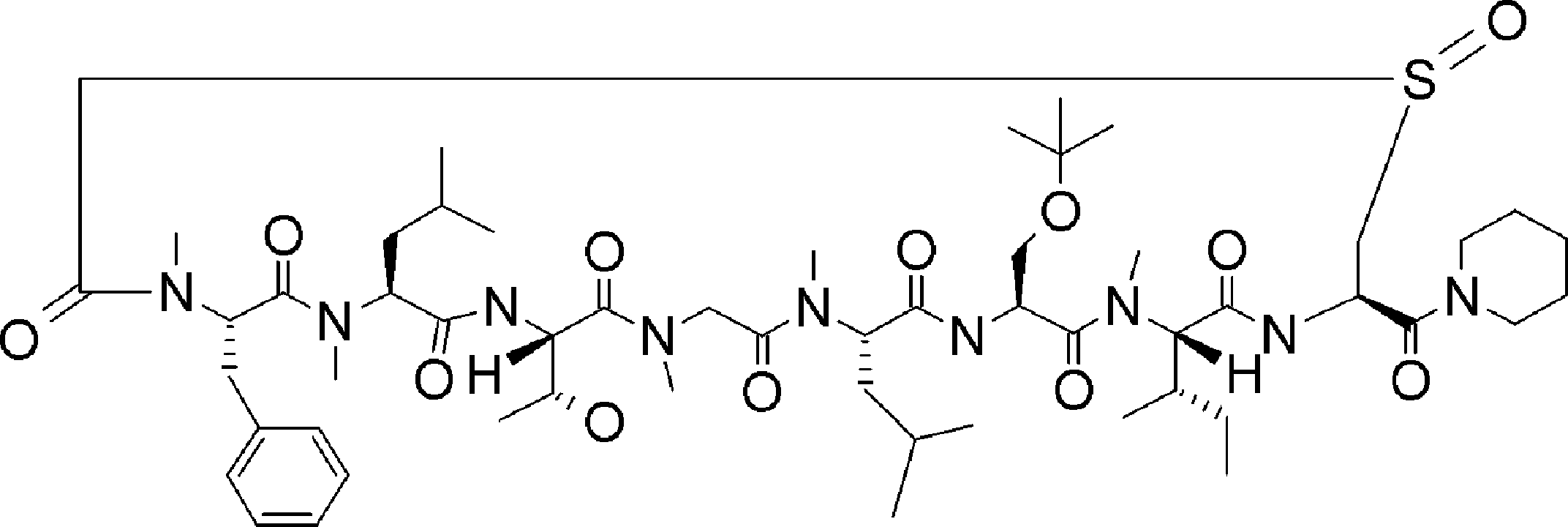
LCMS(ESI) m/z = 1102 (M+H)+
保持時間:0.80分(分析条件SQDAA50)
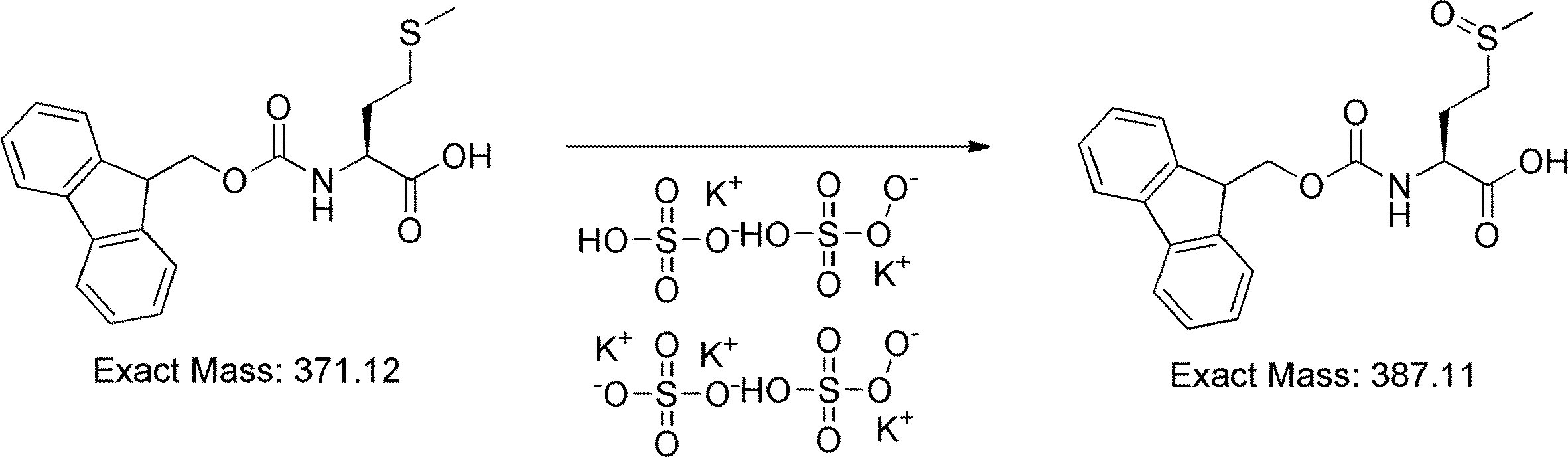
(S)-2-(9H-フルオレン-9-イルメトキシカルボニルアミノ)-4-メチルスルファニル-酪酸(ペプチドP-130)
LCMS(ESI) m/z = 372 (M+H)+
保持時間:0.94分(分析条件SQDAA05)
生成物P-131
LCMS(ESI) m/z = 388 (M+H)+
保持時間:0.86分(分析条件SQDAA05)
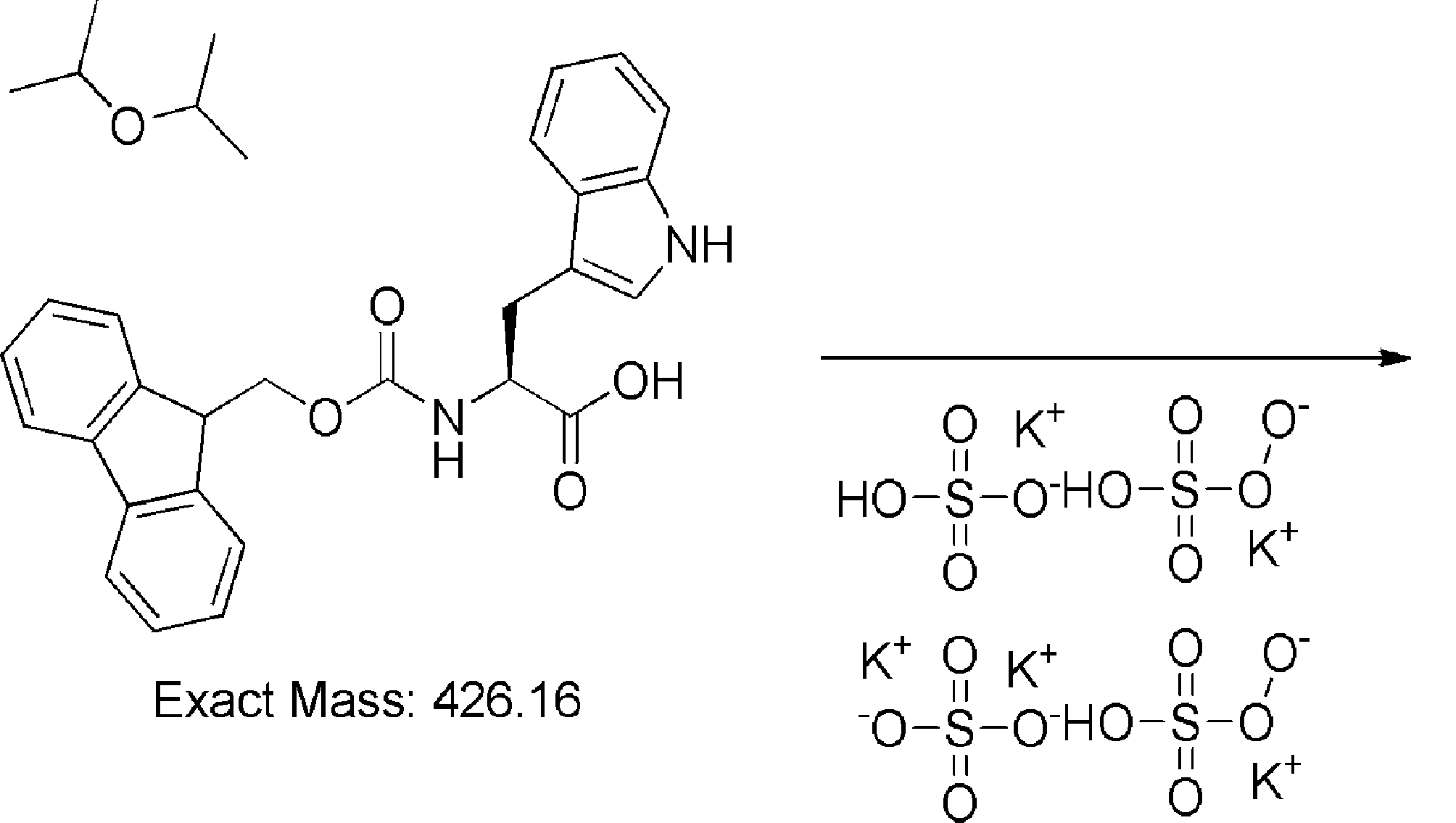
(S)-2-(9H-フルオレン-9-イルメトキシカルボニルアミノ)-3-(1H-インドール-3-イル)-プロピオン酸(ペプチドP-132)
LCMS(ESI) m/z = 427 (M+H)+
保持時間:0.95分(分析条件SQDAA05)
脂溶性が高いチオエーテル環化ペプチド群の中で、比較的代謝安定性の高かったチオエーテル3種類を選択し、環化部位以外は同じ配列(MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)、MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle、MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle)のアミド環化ペプチドを合成し、それらの代謝安定性を比較した。下表のようにマウス、ヒトともにミクロゾーム代謝安定性はマウスで2倍程度、ヒトで3倍程度向上した。一連の化合物で安定して代謝安定化が得られたことから、Displayされる全ての化合物に含まれていたチオエーテル部分構造が除去されたアミド環化法では、Displayされる化合物の多くがより代謝安定化された、よりDruglikeなDisplay手法であると考えられた。

Ala-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Asp-piperidine
N末端アミンとAsp側鎖がアミド環化
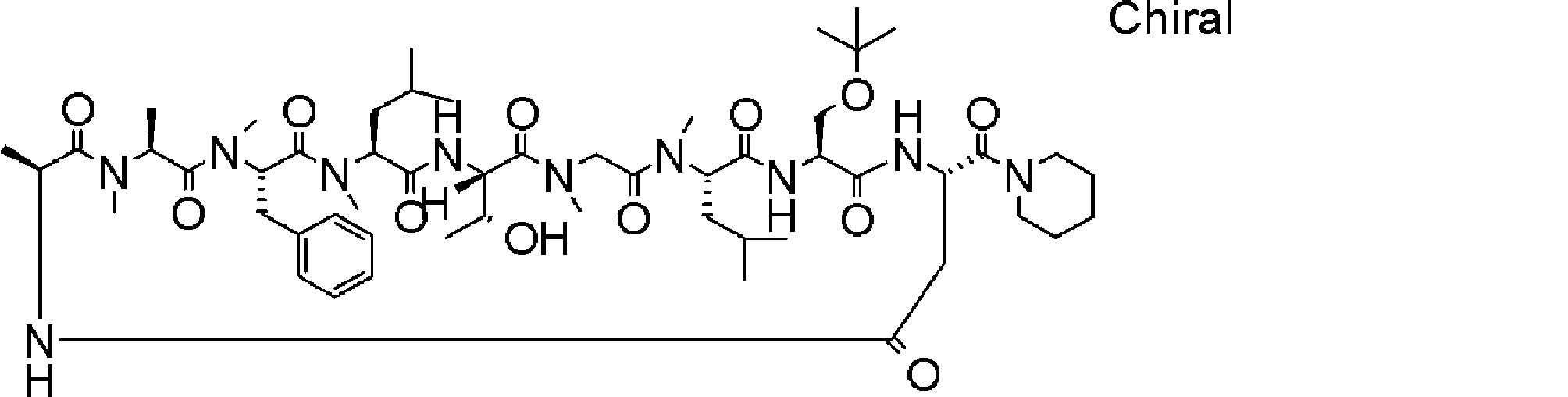
保持時間:0.72分 (分析条件 SQDAA50)
MeAla-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Asp-piperidine
N末端アミンとAsp側鎖がアミド環化
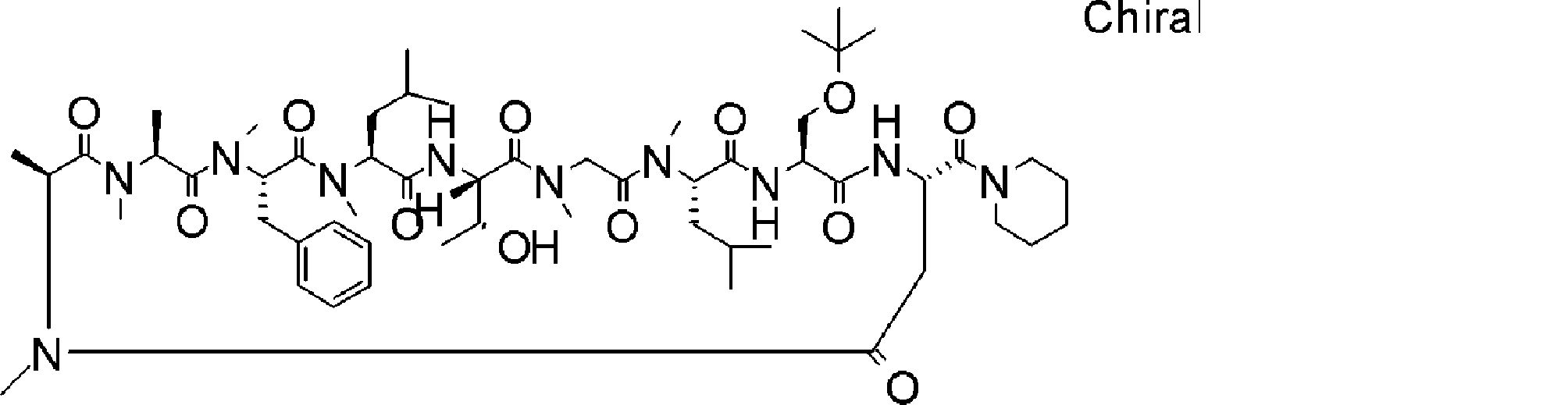
保持時間:0.73分 (分析条件 SQDAA50)
Ala-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Glu-piperidine
N末端アミンとGlu側鎖がアミド環化
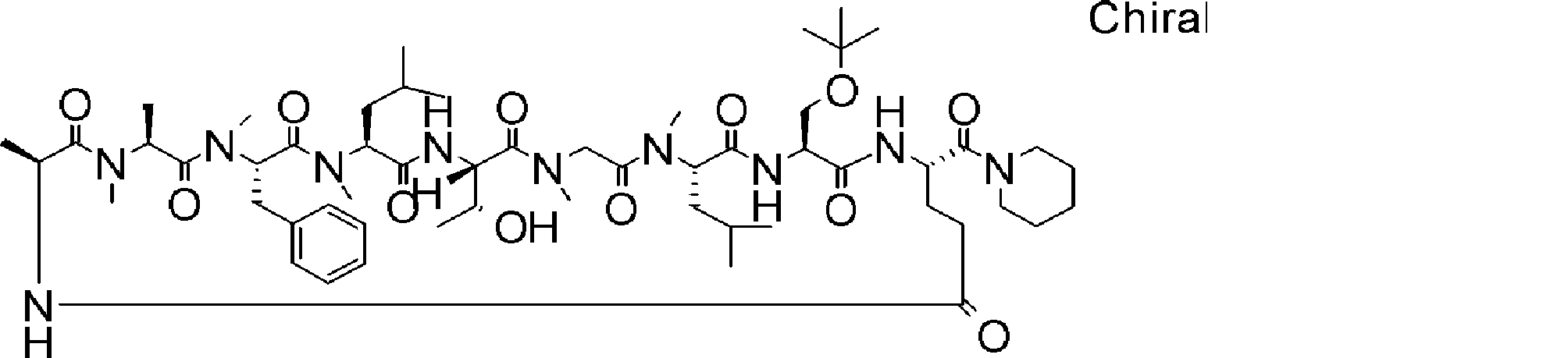
保持時間:0.73分 (分析条件 SQDAA50)
MeAla-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-Glu-piperidine
N末端アミンとGlu側鎖がアミド環化
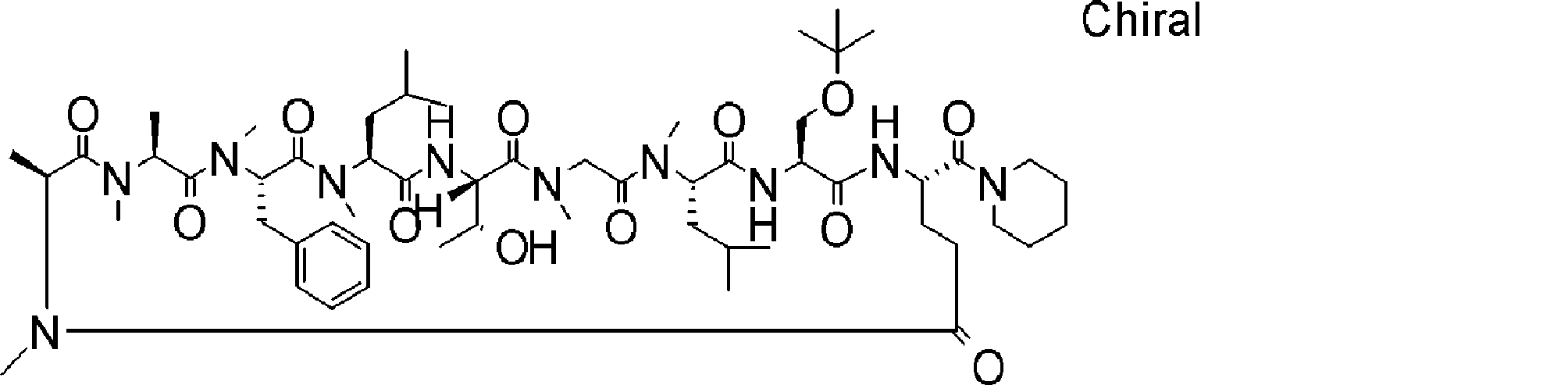
保持時間:0.73分 (分析条件 SQDAA50)
Ala-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Asp-piperidine
N末端アミンとAsp側鎖がアミド環化
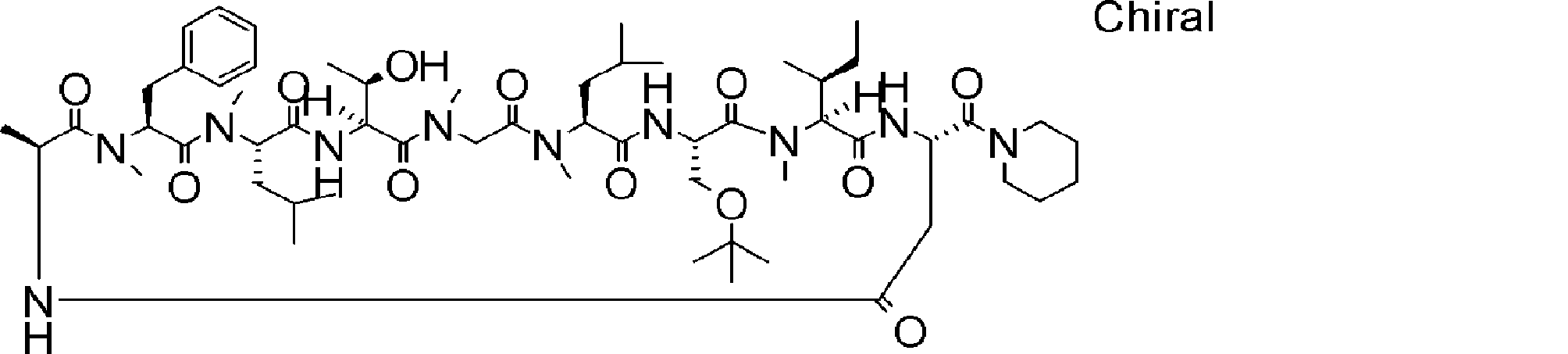
保持時間:0.82分 (分析条件 SQDAA50)
MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)- MeIle-Glu-piperidine
N末端アミンとGlu側鎖がアミド環化

保持時間:0.81分 (分析条件 SQDAA50)
Ala-MeAla-MePhe-MeLeu-Thr-MeGly-MeLeu-Ser(tBu)-MeIle-Asp-piperidine
N末端アミンとAsp側鎖がアミド環化

保持時間:0.81分 (分析条件 SQDAA50)
N末端でない位置のシステインと側鎖カルボン酸が活性化されたアミノ酸を含むペプチドを翻訳し、両官能基が反応したチオエステル環化ペプチドを調製した。続いて、同システインよりもN末端部に位置するアミノ酸を酵素的に除去することで、システイン残基のα-アミノ基を露出させ、それを用いたペプチドのアミド環化が進行するかを以下のように試みた。
1. 転写によるtRNA(CA欠損)の合成
鋳型DNA(配列番号DT-E1)から、RiboMAX Large Scale RNA production System T7(Promega社,P1300)を用いたin vitro の転写により3'端のCAを欠くtRNAGluAAG (-CA)(配列番号RT-E1)を合成し、RNeasy Mini kit(Qiagen社)により精製した。
tRNAGluAAG (-CA) DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTAAGACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluAAG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUAAGACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
50μM 転写tRNAGluAAG (-CA)(配列番号RT-E1) 10μLに、10X ligation buffer(500 mM HEPES-KOH pH 7.5, 200 mM MgCl2) 2μL、10 mM ATP 2μL、Nuclease free water 2.8μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 20unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)1.2μLおよび、5mMのアミノアシル化pdCpA(化合物1i-IA)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。ライゲーション反応液 20μLに、3M酢酸ナトリウム 4μLと125mM ヨウ素(水:THF=1:1溶液)24μLを加え、室温で1時間、脱保護を行った。アミノアシル化tRNA(化合物AT-E1)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-E1)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Asp(SMe)-tRNAGluAAG
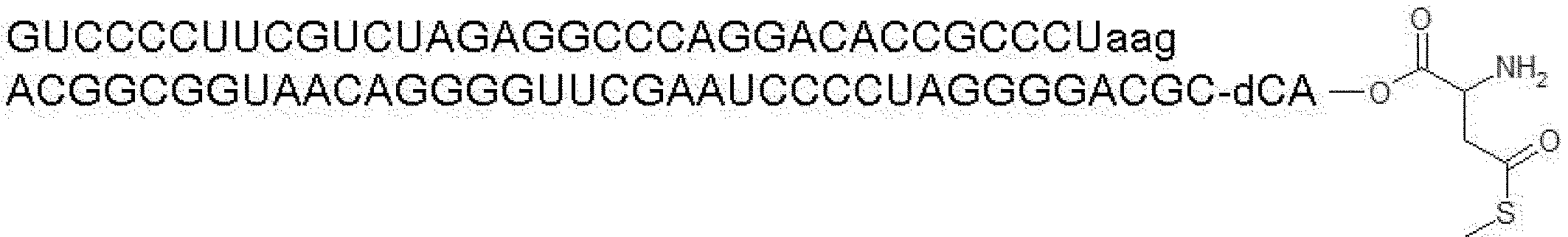
側鎖カルボン酸をチオエステルにより活性化したアスパラギン酸誘導体でアミノアシル化されたtRNAを、無細胞翻訳系に加えて翻訳を開始することにより、所望の非天然アミノ酸を含有するポリペプチドの翻訳合成を行った。翻訳系は、原核生物由来の再構成無細胞タンパク質合成系であるPURE systemを用いた。具体的には、翻訳液,1%(v/v) RNasein Ribonuclease inhibitor (Promega社,N2111), 1mM GTP,1mM ATP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,6mM 酢酸マグネシウム,2mMスペルミジン,2mM ジチオスレイトール,0.1mM 10-HCO-H4folate, 1.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.6μM メチオニルtRNAトランスフォルミラーゼ,0.26μM EF-G,0.24μM RF2,0.17μM RF3,0.5μM RRF,2.7μM IF1,0.4μM IF2,1.5μM IF3,40μM EF-Tu,93μM EF-Ts,1.2μM リボソーム,0.73μM AlaRS,0.03μM ArgRS,0.38μM AsnRS,0.13μM AspRS,0.02μM CysRS,0.06μM GlnRS,0.23μM GluRS,0.09μM GlyRS,0.4μM IleRS,0.04μM LeuRS,0.11μM LysRS,0.03μM MetRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.02μM TyrRS,0.02μM ValRS(自家調製タンパクは基本的にはHisタグ付加タンパクとして調製した))に、1μM 鋳型RNA、それぞれの鋳型DNAにコードされているタンパク質性アミノ酸群をそれぞれ250μMずつ、ならびに50μMの側鎖カルボン酸が活性エステル化されたアミノアシル化tRNA(化合物AT-E1)を翻訳反応混合物に添加し、37℃で1時間静置することで行った。
1μM 鋳型RNA OT43 RNA(配列番号RM-E1)に、0.25mM Met,0.25mM Cys,0.25mM Thr,0.25mM Arg,0.25mM Tyr,0.25mM Pro,0.25mM Glyおよび、50μM Asp(SMe)-tRNAGluAAG(化合物AT-E1)を含む前述の翻訳液を37℃で60分間保温した。得られた翻訳溶液1μLに9μLの0.2%トリフルオロ酢酸を加え、そのうち1μLをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート上乾固し、MALDI-MSで分析した。その結果、N末端フォルミルメチオニンを含み、側鎖チオール基とカルボン酸でチオエステル環化したペプチドP-E1に相当するピークが観測された(図62、ピークI)。続いて、上記翻訳反応物に終濃度がそれぞれ2μM、14μMとなるように酵素ペプチドデフォルミラーゼ、メチオニンアミノペプチダーゼを加え、37℃で5分間保温した。得られた反応物を上述したようにMALDI-MSで分析したところ、原料のチオエステル環化ペプチドのピークは小さくなり、その代わりに、N末端フォルミルメチオニンが除去され、その結果露出したN末端アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物P-E2に相当するピークが観測された(図62、ピークII)。このことから、チオエステル環化が進行した後でも、CysよりN末端部を除去することで、目的のアミド環化ペプチドが得られる事が示された。
OT43 RNA
GGGUUAACUUUAAGAAGGAGAUAUACAUaugUGCACUACAACGCGUCUUCCGUACCGUGGCGGCuaagcuucg
fMetCysThrThrThrArgAspProTyrArgGlyGlyの側鎖硫黄原子とAspの側鎖カルボン酸でチオエステル環化した化合物
MALDI-MS:m/z: [M+H]+ = 1367.5 (Calc.1367.6)
CysThrThrThrArgAspProTyrArgGlyGlyのN末端アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物
MALDI-MS:m/z: [M+H]+ = 1208.3 (Calc.1208.6)
1.アミド環化ペプチドの合成
以下に示す環化ペプチドを合成した(表11-1:合成したペプチド化合物例(計965化合物))。特に明示しない限り表中の1に位置するアミノ酸が前記スキームAなどで示した白丸○に対応する交差ユニットに対応し、アミノ酸配列中左端に表記したアミノ酸が同じく▲ユニットに対応する。これら2つの部位が結合を形成し環化ペプチドを構成する。また、表中H-1からH-6で示したアミノ酸は前記スキームAなどで示した直鎖部に相当する。ここで、表中右端に存在する部位がC末端を形成する。ここで表記したpipとはC末端カルボン酸がピペリジンとアミド結合を形成してピペリジンアミドを形成したことを意味している。また、H-1に何も記載されていない環化ペプチドはC末端カルボン酸が削除された化合物を意図する。この場合、1に位置するアミノ酸の持つ1つのカルボン酸とN末端のアミンとがアミド化反応して環化され、C末端カルボン酸部位が削除された誘導体、例えばメチル基やトリフルオロメチル基に置換された化合物がC末端に位置される。表11-1に示した略号は表11-2(アミノ酸略号が意図する構造との関連表)に表記したアミノ酸を意味する。
既に記載の方法と同様の手法を用いて表11-1に表記した環化ペプチドを合成し、それぞれの化合物の同定を行った(表11-3-1、表11-3-2:合成したペプチド化合物の同定(計965化合物))。
C末端部位の主鎖部位がアミドにて化学修飾(多くの実施例ではピペリジンアミド)され、かつアスパラギン酸の側鎖カルボン酸部位(交差ユニット)がN末端側のアミノ基(三角ユニット)とアミド結合させた様々な化合物を合成する目的で、以下のアミノ酸もしくはペプチドの担持レジンを合成した。以下に合成例のより詳細な内容を述べる。なお、以下の方法に限定されず、本明細書の他に記載の部分や、一般的なペプチドの合成法によっても合成できる。
LCMS(ESI) m/z =479.5(M+H)+
保持時間:1.10分(分析条件SQDAA05)

LCMS(ESI) m/z =423(M+H)+
保持時間:0.88分(分析条件SQDAA05)
なお、本明細書では、ポリマーやレジンと化合物が結合した場合、ポリマーやレジン部位を○にて表記する場合がある。また、レジン部位の反応点を明確にさせる目的で、○に接続させて反応部位の化学構造を表記させる場合がある。下の構造では、レジンの2-クロロトリチル基がAspの側鎖カルボン酸とエステル結合を介して結合している様子を示している。
(吸光度(301.2nm)x1000x50)/(13.42x7800)=0.267mmol/g
0.267mmol/gx100/(33.4/34.8)=27.8%
以下のスキームに従い、(3S)-3-(9H-フルオレン-9-イルメトキシカルボニルアミノ)-4-[メチル-[(2S)-1-オキソ-1-[[(2S)-1-オキソ-1-ピペリジン-1-イルプロパン-2-イル]アミノ]-3-フェニルプロパン-2-イル]アミノ]-4-オキソブタン酸-2-クロロトリチルレジン(化合物SP455,Fmoc-Asp(O-Trt(2-Cl)-Resin)-MePhe-Ala-pip)の合成を行った。
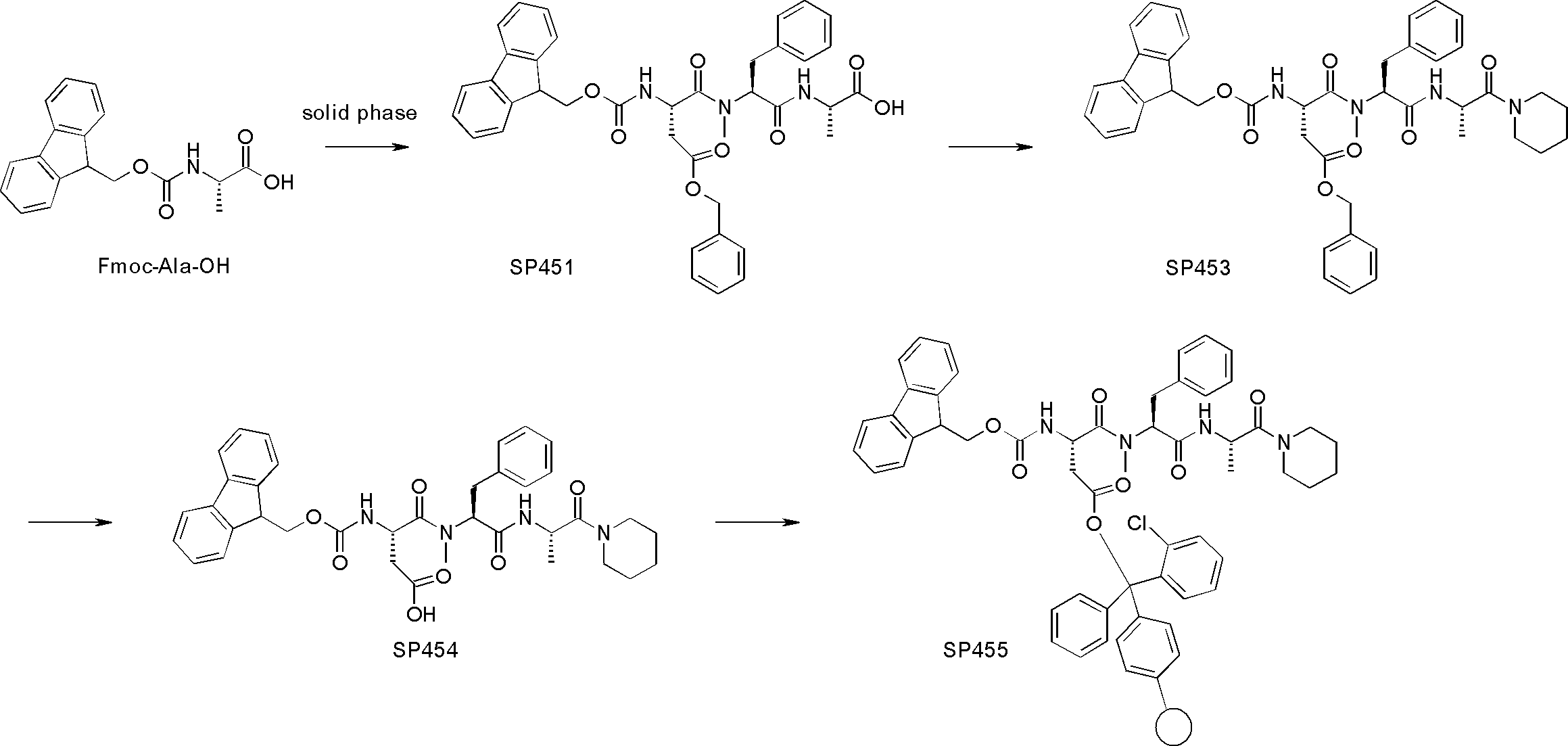

LCMS(ESI) m/z = 678.6 (M+H)+
保持時間:0.65分(分析条件SQDAA50)

LCMS(ESI) m/z = 745.7 (M+H)+
保持時間:0.82分(分析条件SQDAA50)
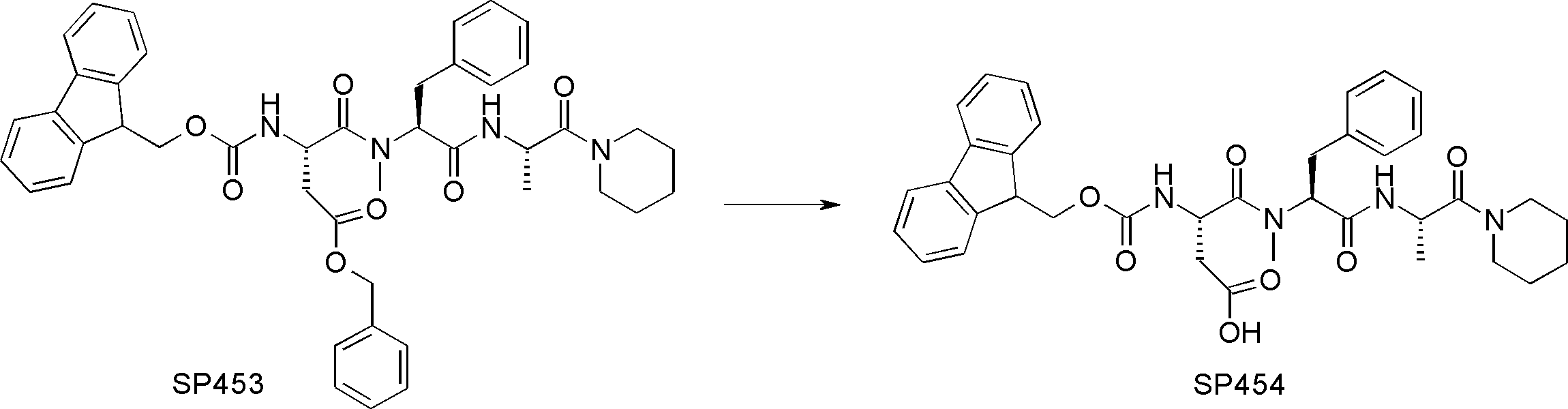
LCMS(ESI) m/z = 655.6 (M+H)+
保持時間:0.61分(分析条件SQDAA50)
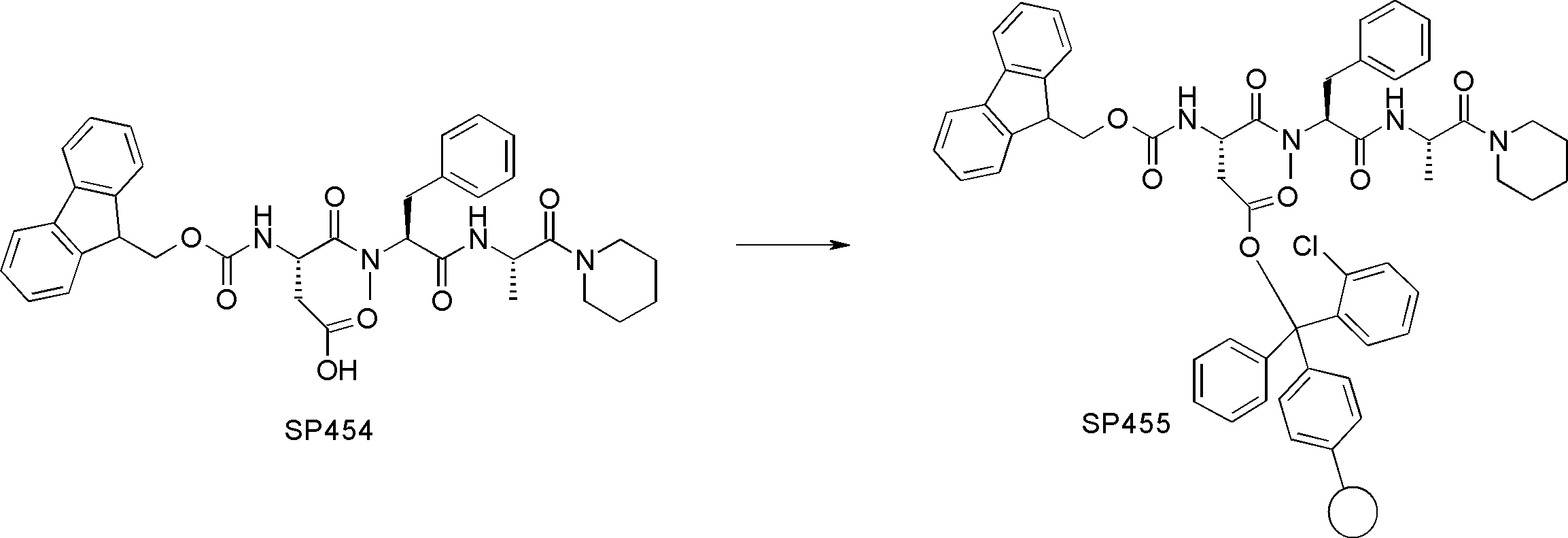
ローディング率:0.317mmol/g、25.7%

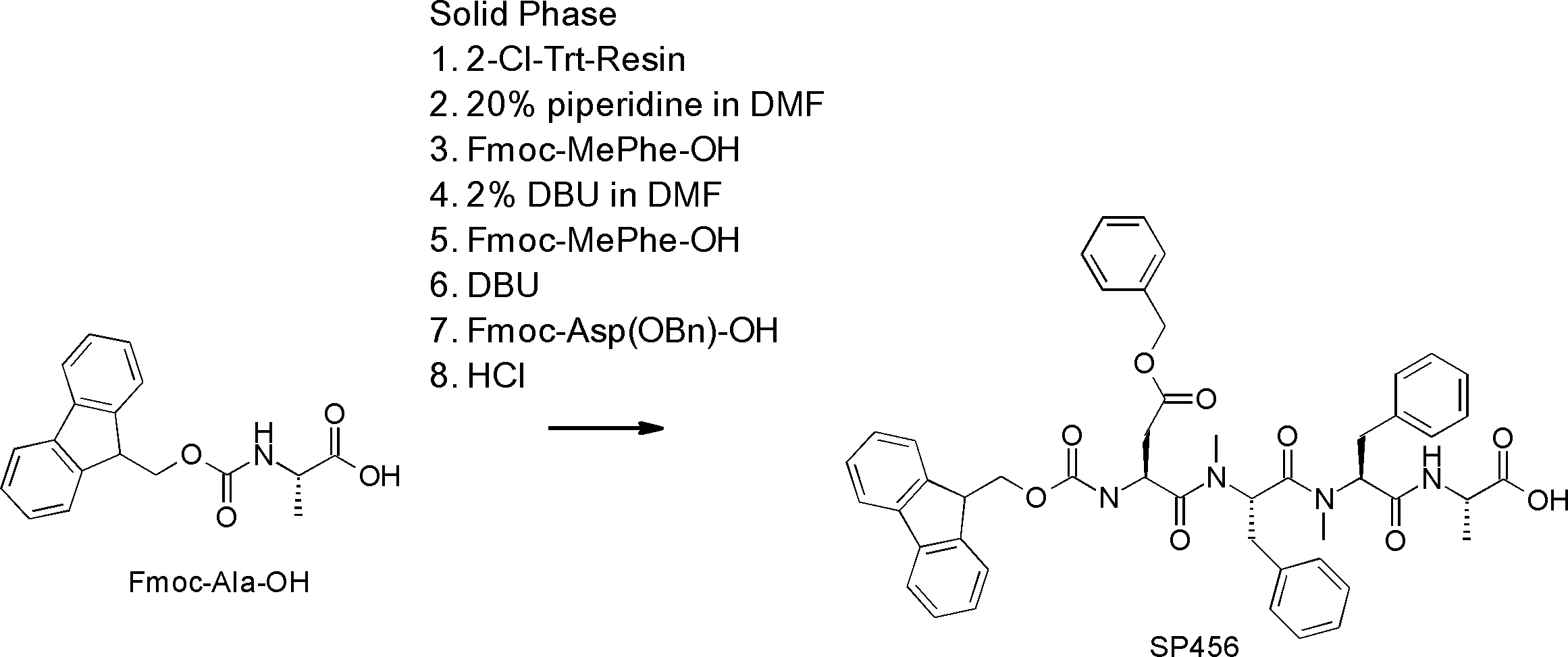
LCMS(ESI) m/z = 839.6(M+H)+
保持時間:0.71分(分析条件SQDAA50)

LCMS(ESI) m/z = 906.7 (M+H)+
保持時間:0.87分(分析条件SQDAA50)
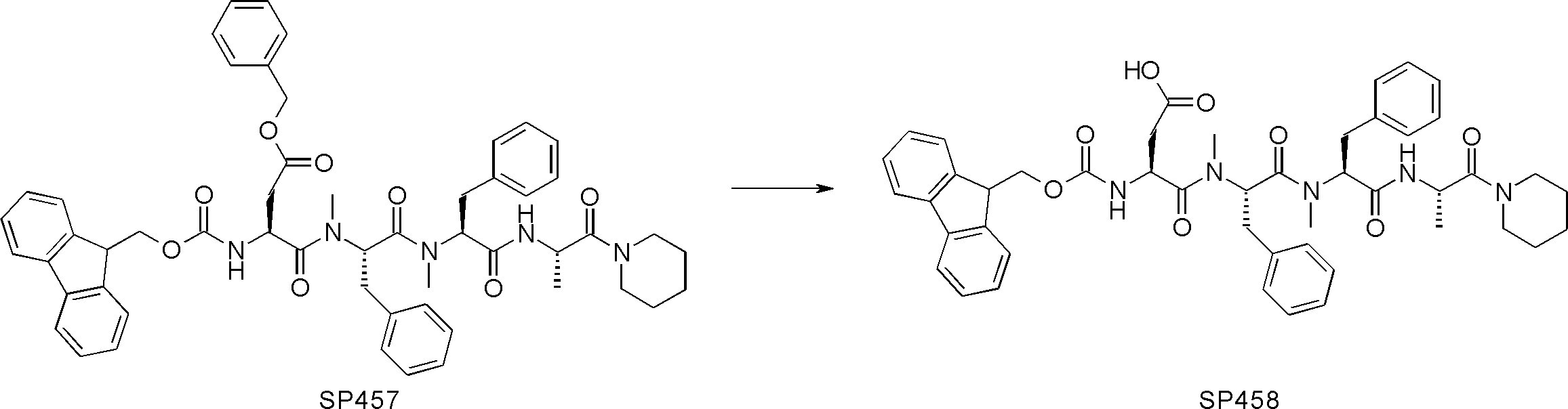
LCMS(ESI) m/z = 816.7 (M+H)+
保持時間:0.69分(分析条件SQDAA50)

ローディング率:0.285mmol/g、21.0%
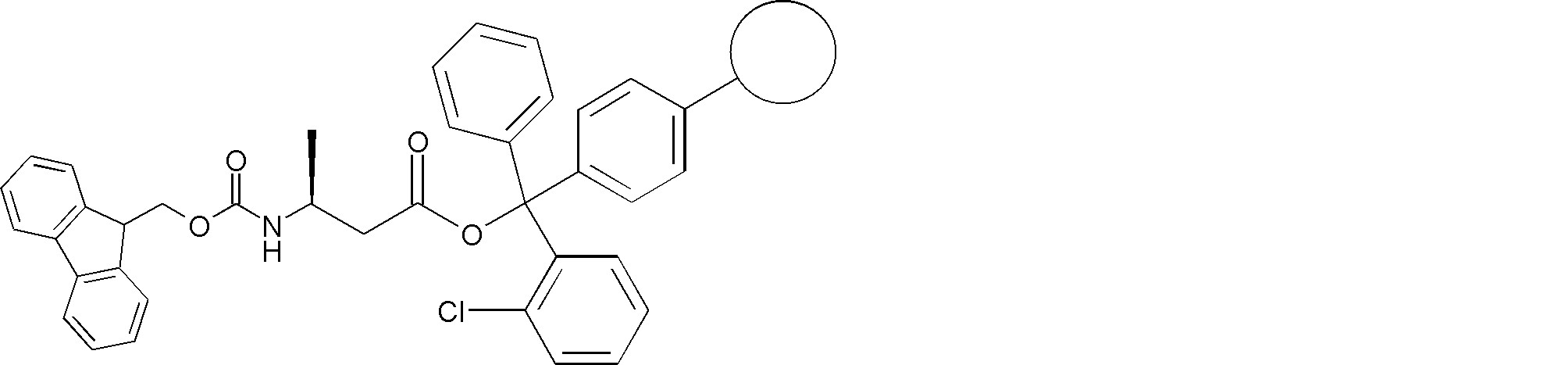
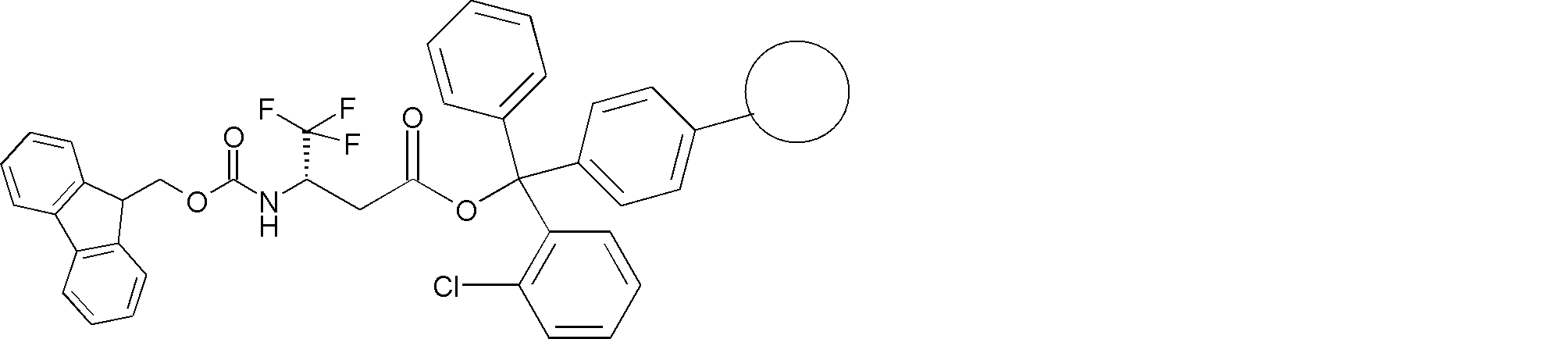
ローディング率:0.234mmol/g、15.6%
ドラックライクネスを評価するための多くのアミノ酸誘導体は購入可能もしくは文献既知であり、定法により合成することができる。以下には文献未知のアミノ酸誘導体の合成法を記載した。
以下のスキームに従い、合成を行った
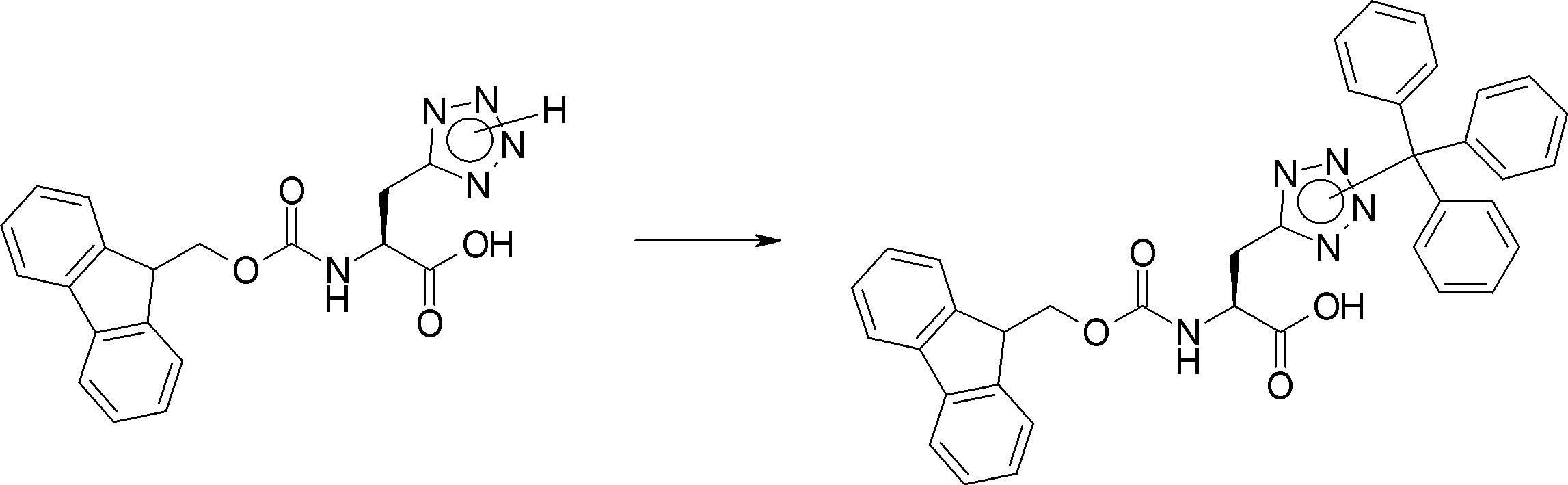
LCMS(ESI) m/z = 620.3 (M-H)-
保持時間:1.06分(分析条件SQDAA05)
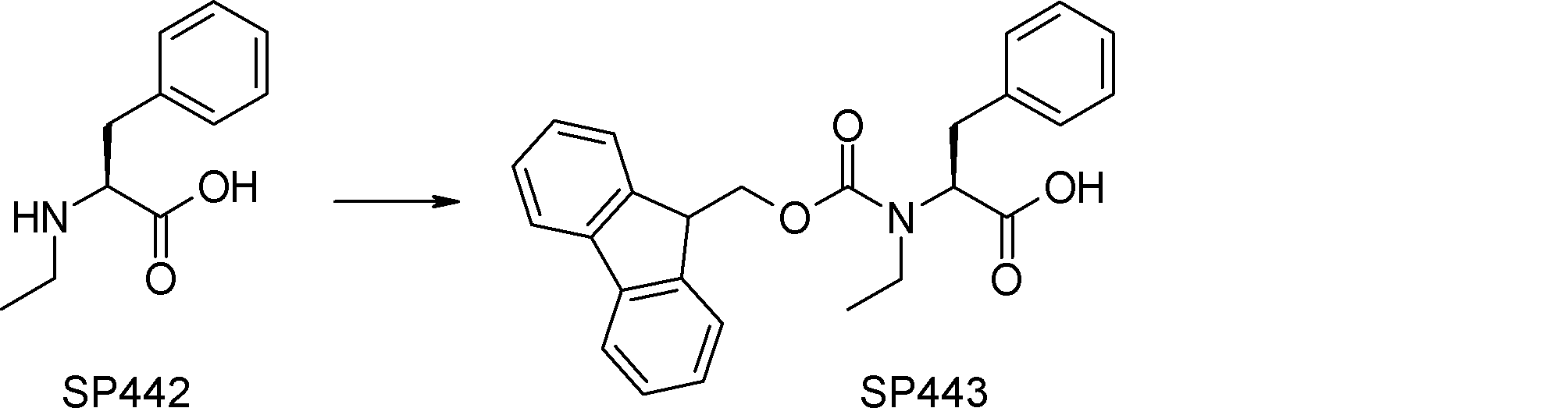
(S)-2-(エチルアミノ)-3-フェニルプロパン酸(化合物SP442)(4.00g、20.7mmol)を1,4-ジオキサン(100ml)と水(100ml)の混合液に溶解させ、炭酸カリウム(8.69g、62.9mmol)および炭酸 (2,5-ジオキソピロリジン-1-イル) (9H-フルオレン-9-イル)メチル(6.98g、20.7mol)を加え、室温で8時間攪拌した。水層を硫酸水素カリウム水溶液でpH4に調整し、減圧下で1,4-ジオキサンを留去した。得られた水溶液を酢酸エチルにて抽出し、得られた有機抽出物を飽和食塩水で洗浄し、硫酸ナトリウムで乾燥後に減圧濃縮した。得られた残渣をカラムクロマトグラフィ(酢酸エチル:石油エーテル=1:1)で精製し、(2S,4R)-1-(((9H-フルオレン-9-イル)メトキシ)カルボニル)-4-エトキシピロリジン-2-カルボン酸(化合物SP443)(3.2g、37%)を得た。
LCMS(ESI) m/z = 416 (M+H)+
保持時間:1.97分(分析条件SMDmethod11)
以下のスキームに従って合成した。
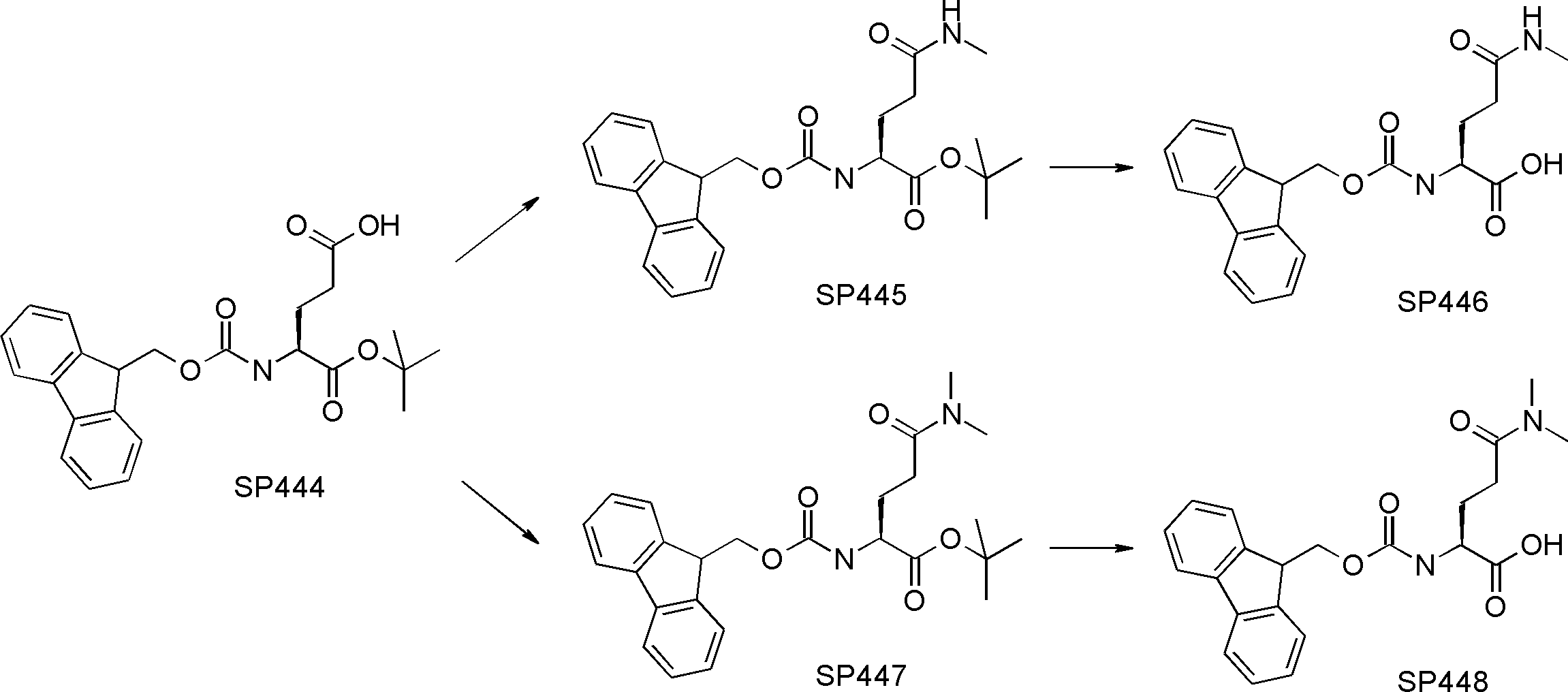
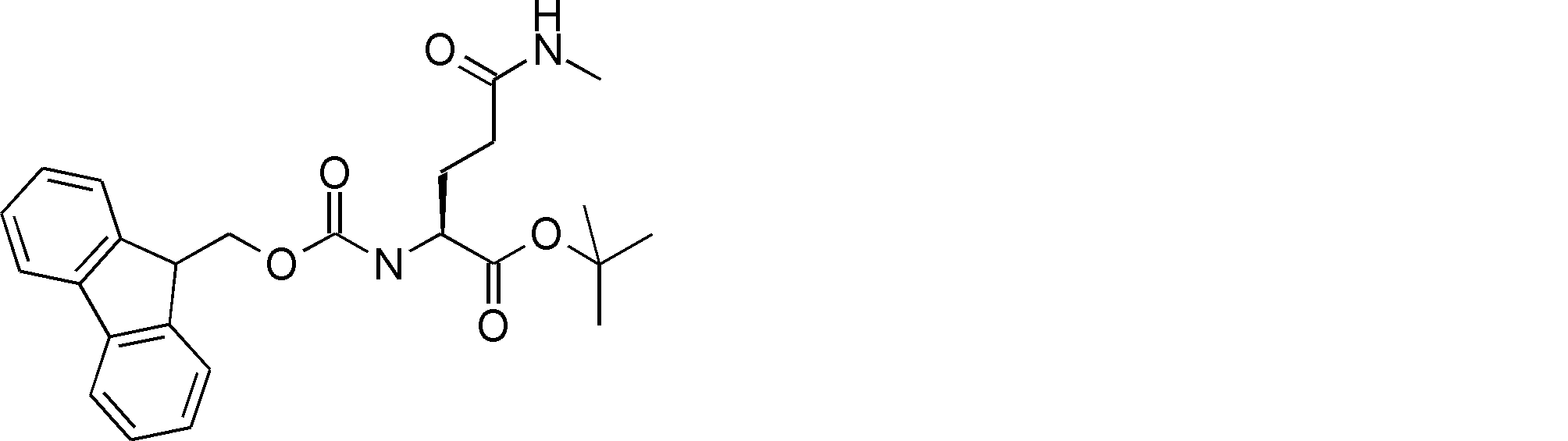
LCMS(ESI) m/z = 439 (M+H)+
保持時間:1.05分(分析条件SQDAA05)

LCMS(ESI) m/z = 383 (M+H)+
保持時間:2.07分(分析条件ZQAA05)
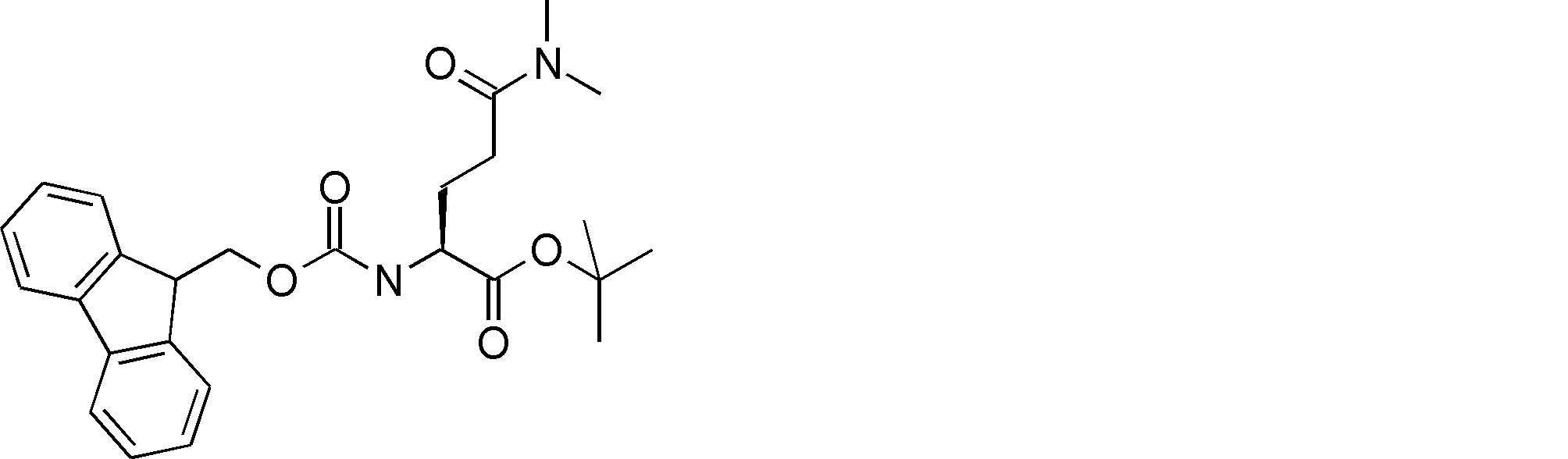
LCMS(ESI) m/z = 453 (M+H)+
保持時間:3.00分(分析条件SQDAA05)

LCMS(ESI) m/z = 397 (M+H)+
保持時間:2.18分(分析条件ZQAA05)
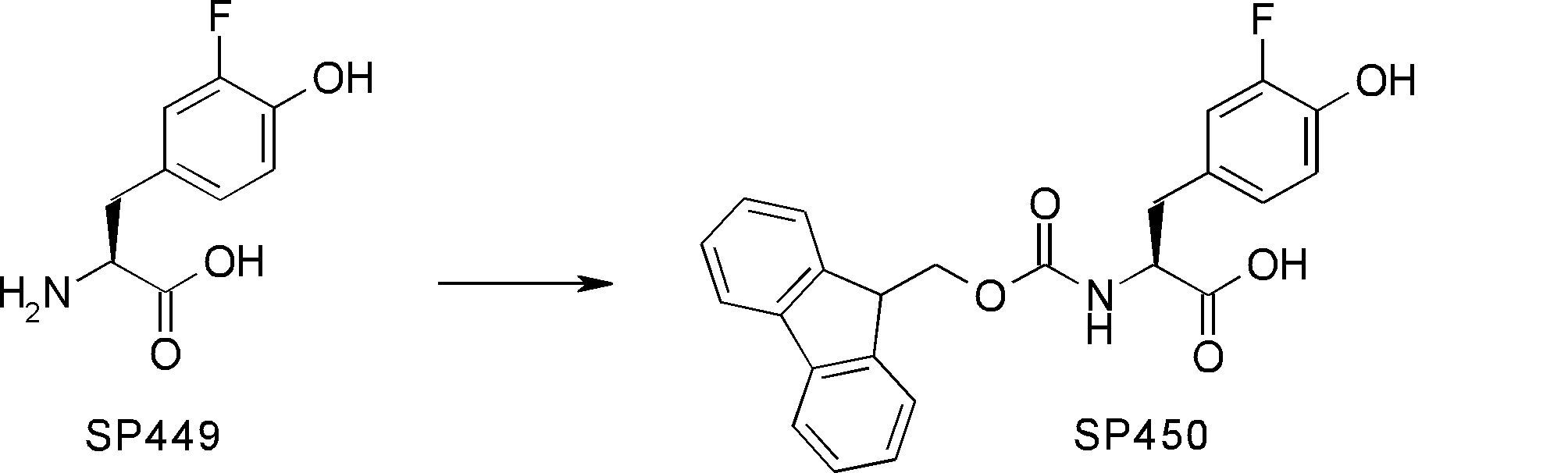
LCMS(ESI)m/z =420.3(M-H)-
保持時間:0.39分 (分析条件 SQDAA50)
1-3-1. C末端のAspが交差ユニットとして機能し、すなわち主鎖カルボン酸がピペリジンなどによりアミド化され、側鎖カルボン酸でN末端の主鎖アミン(三角ユニット)とアミド環化したアスパラギン酸を有するペプチド群の合成例


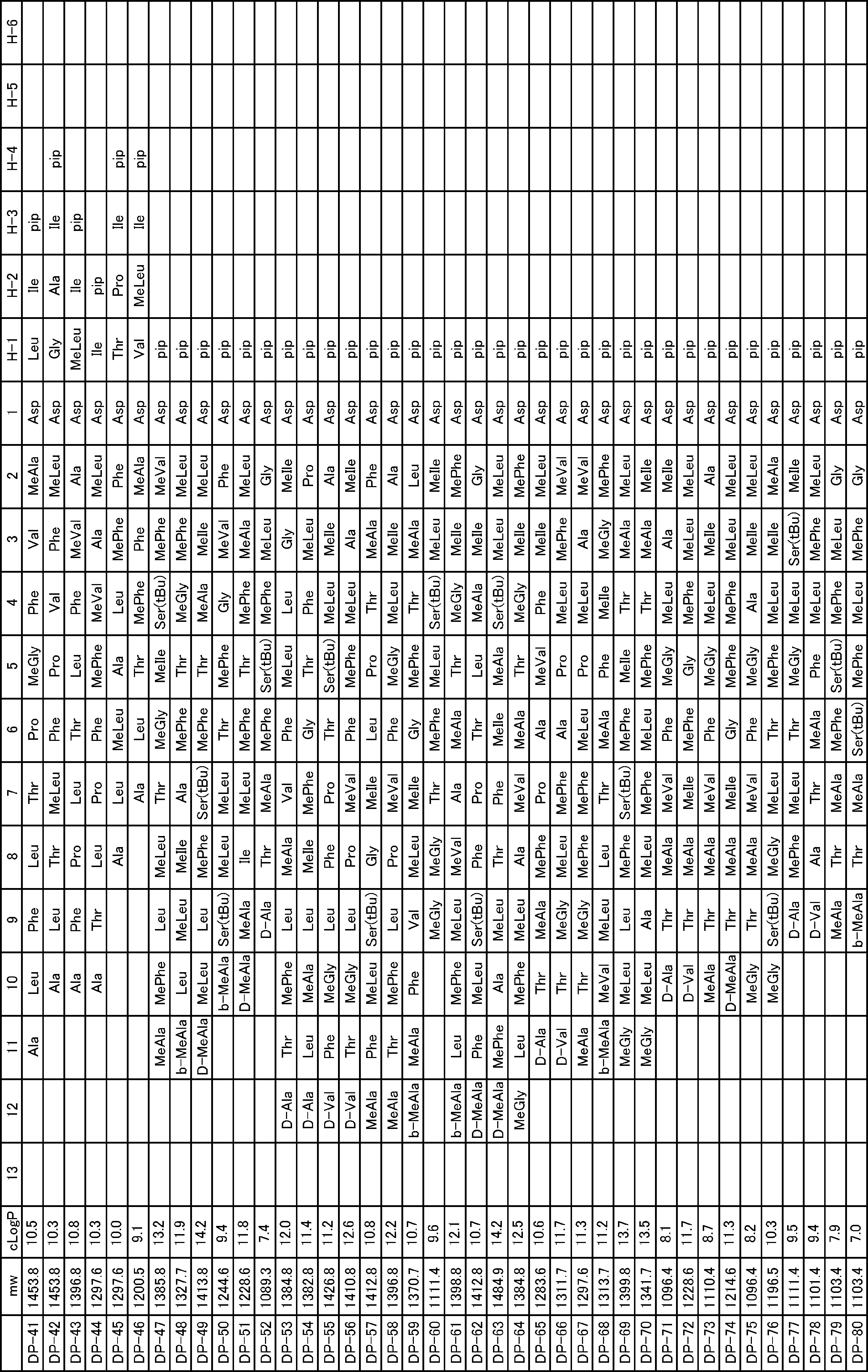
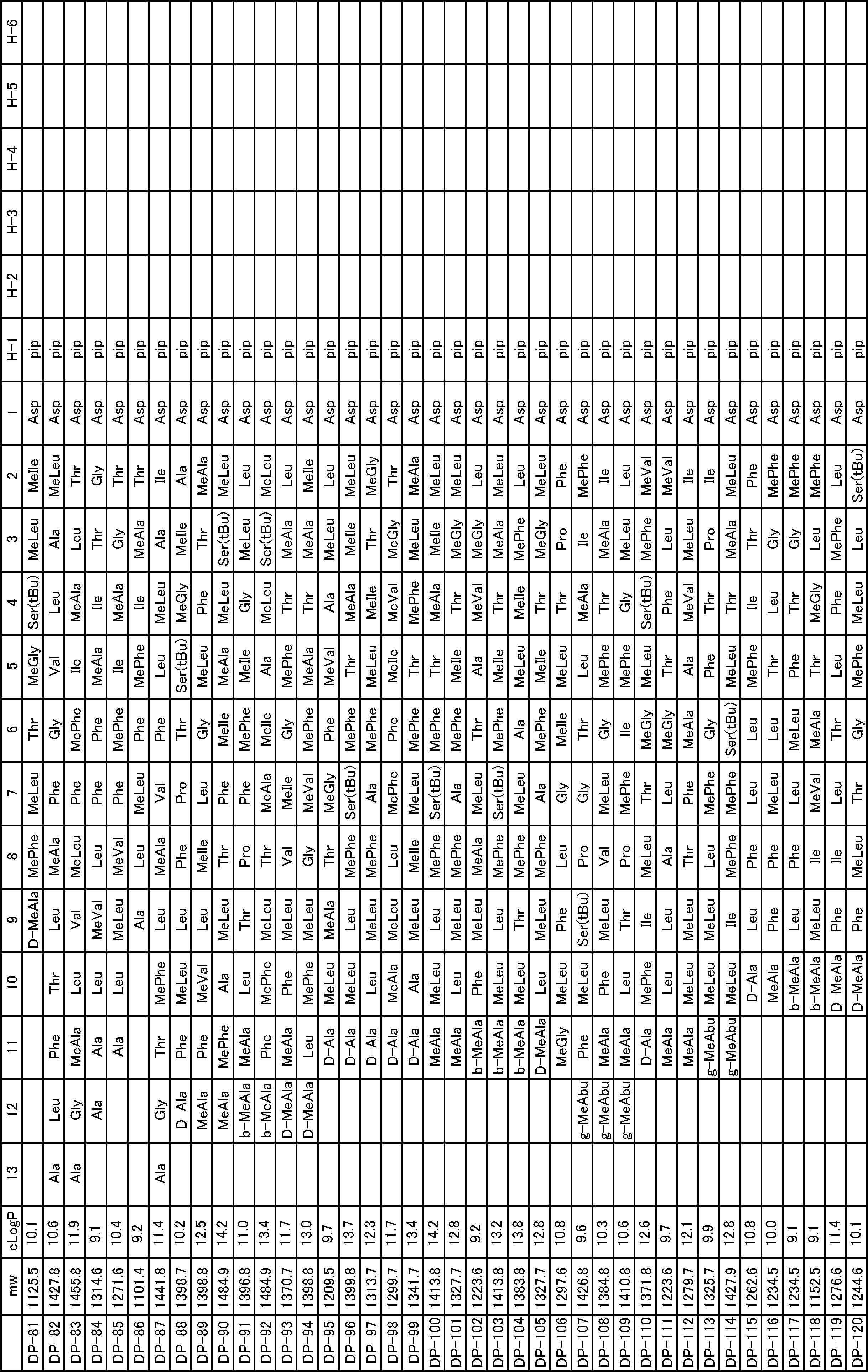



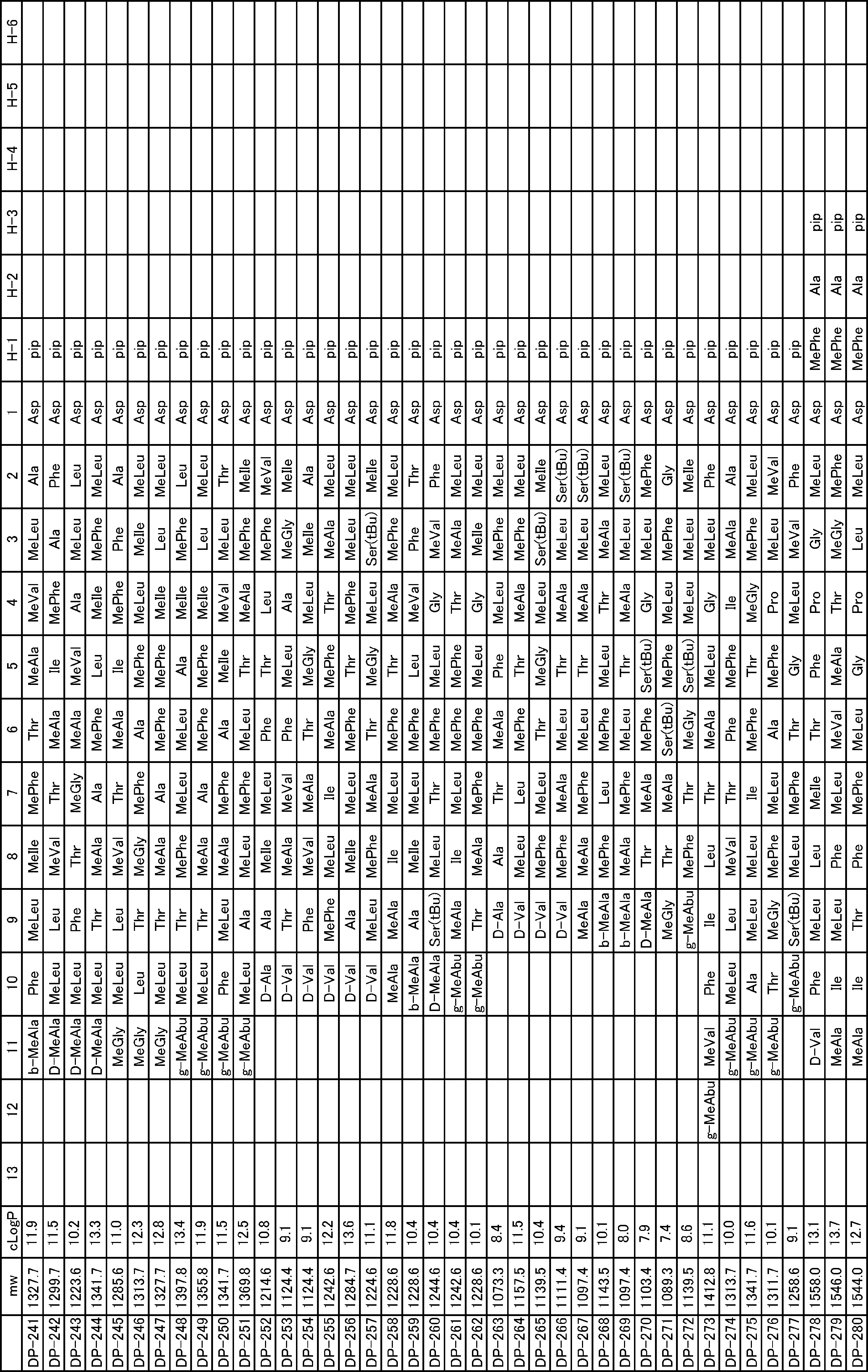

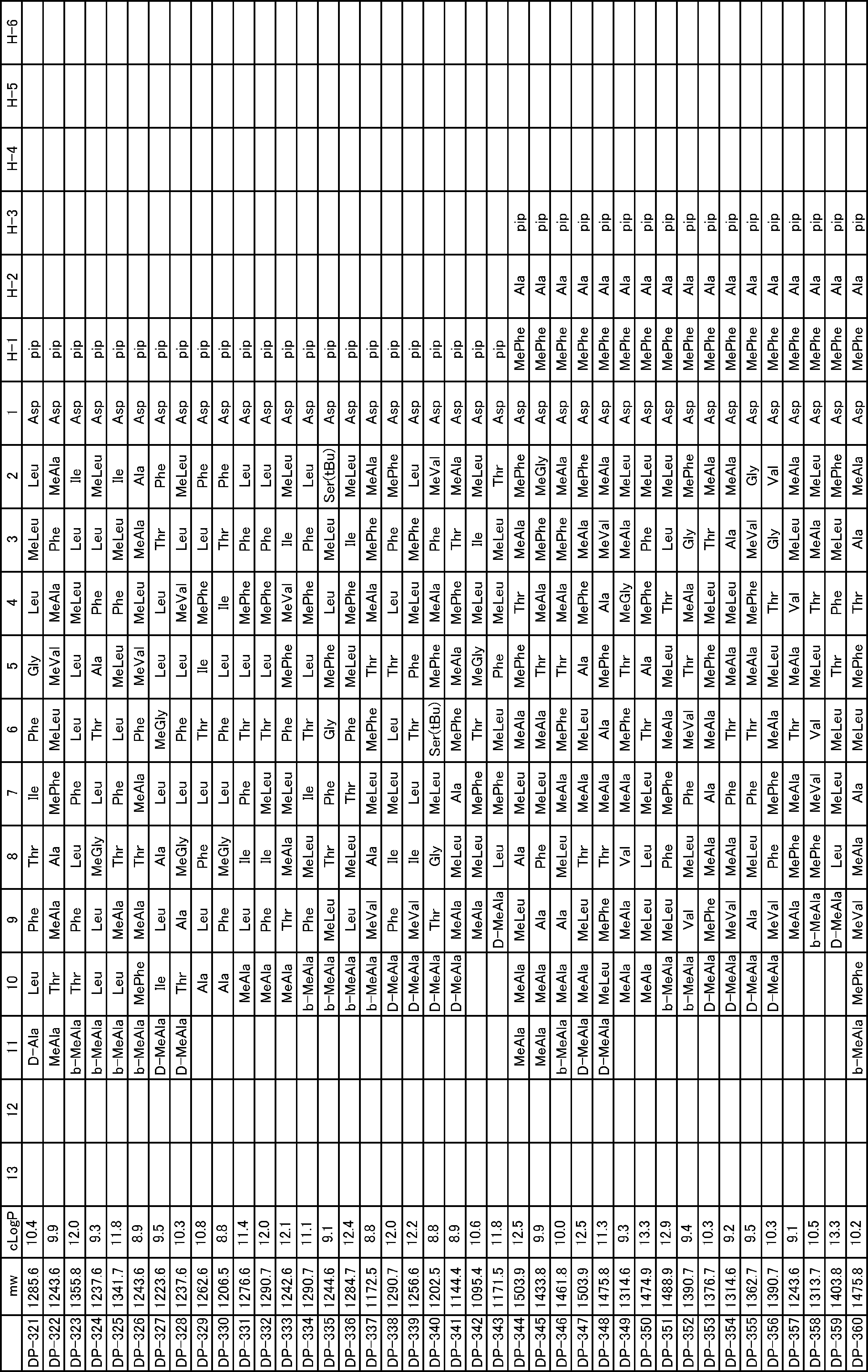


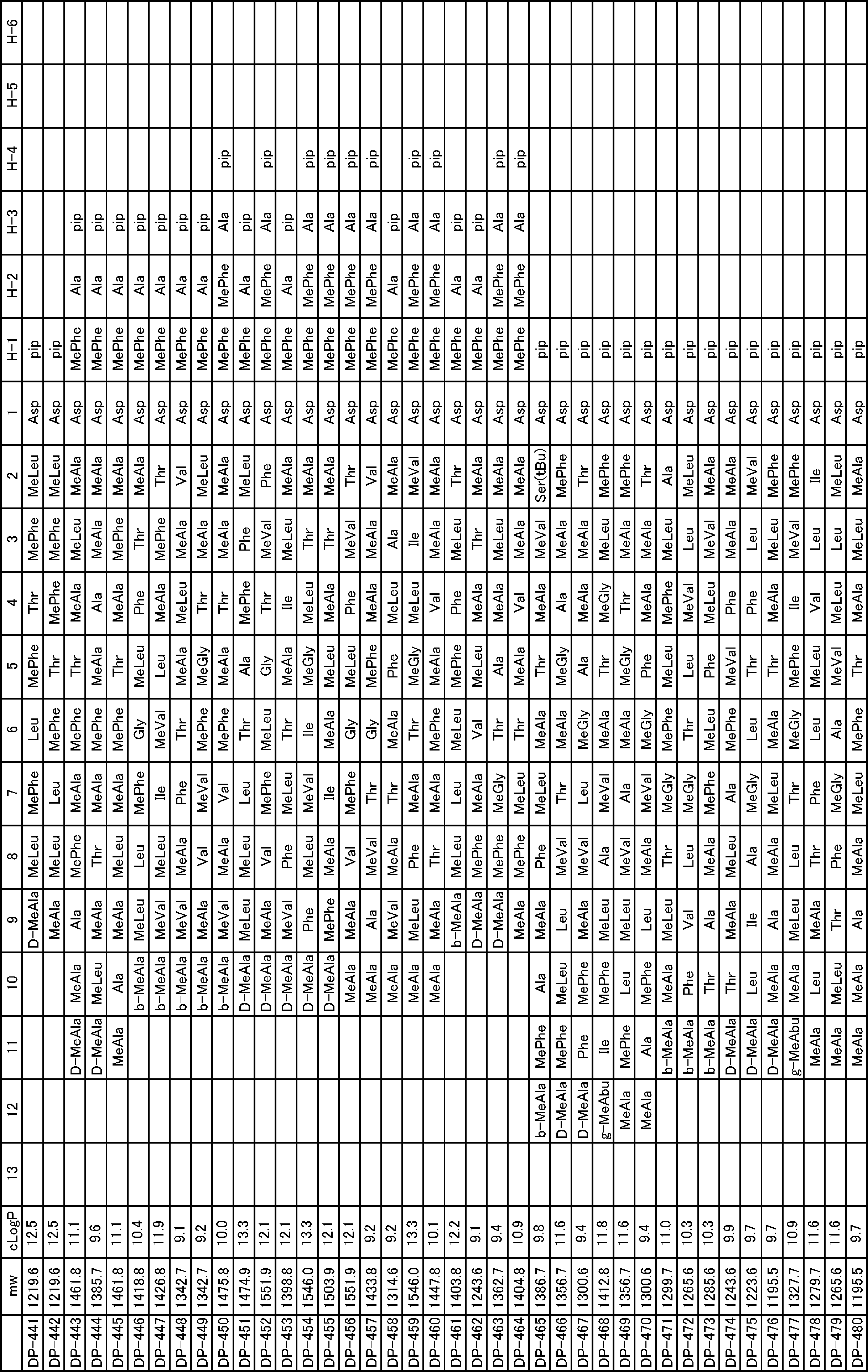

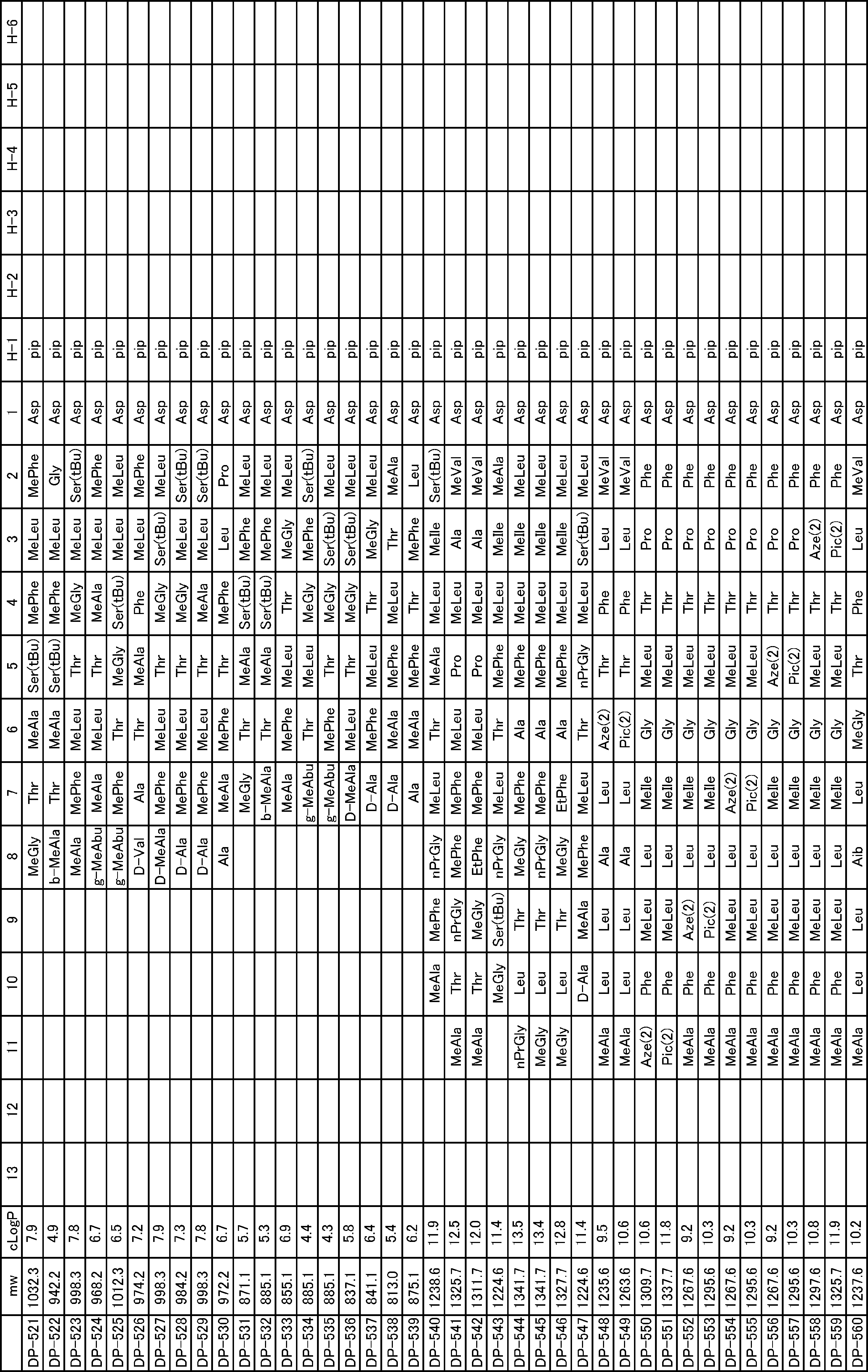


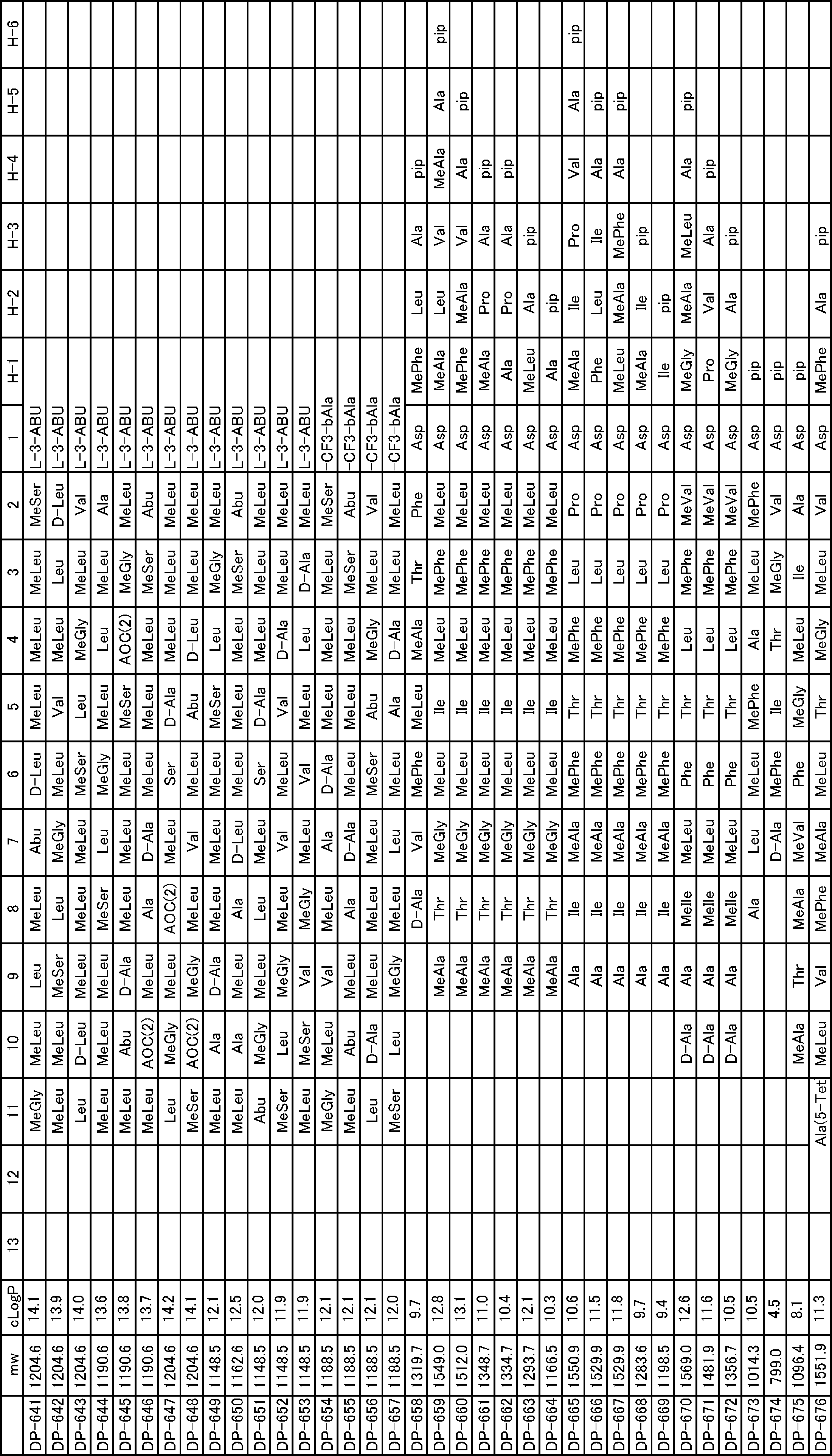






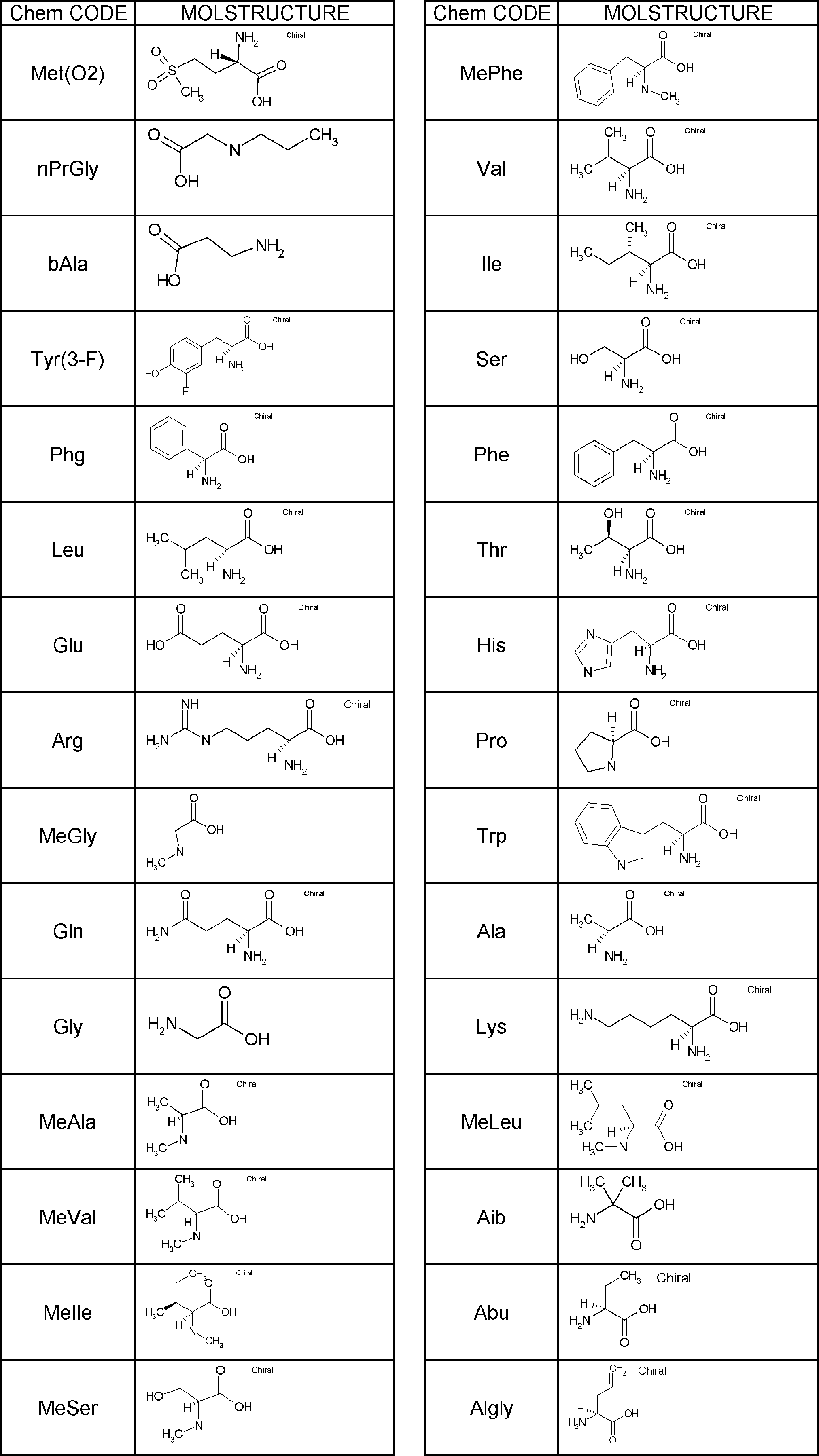
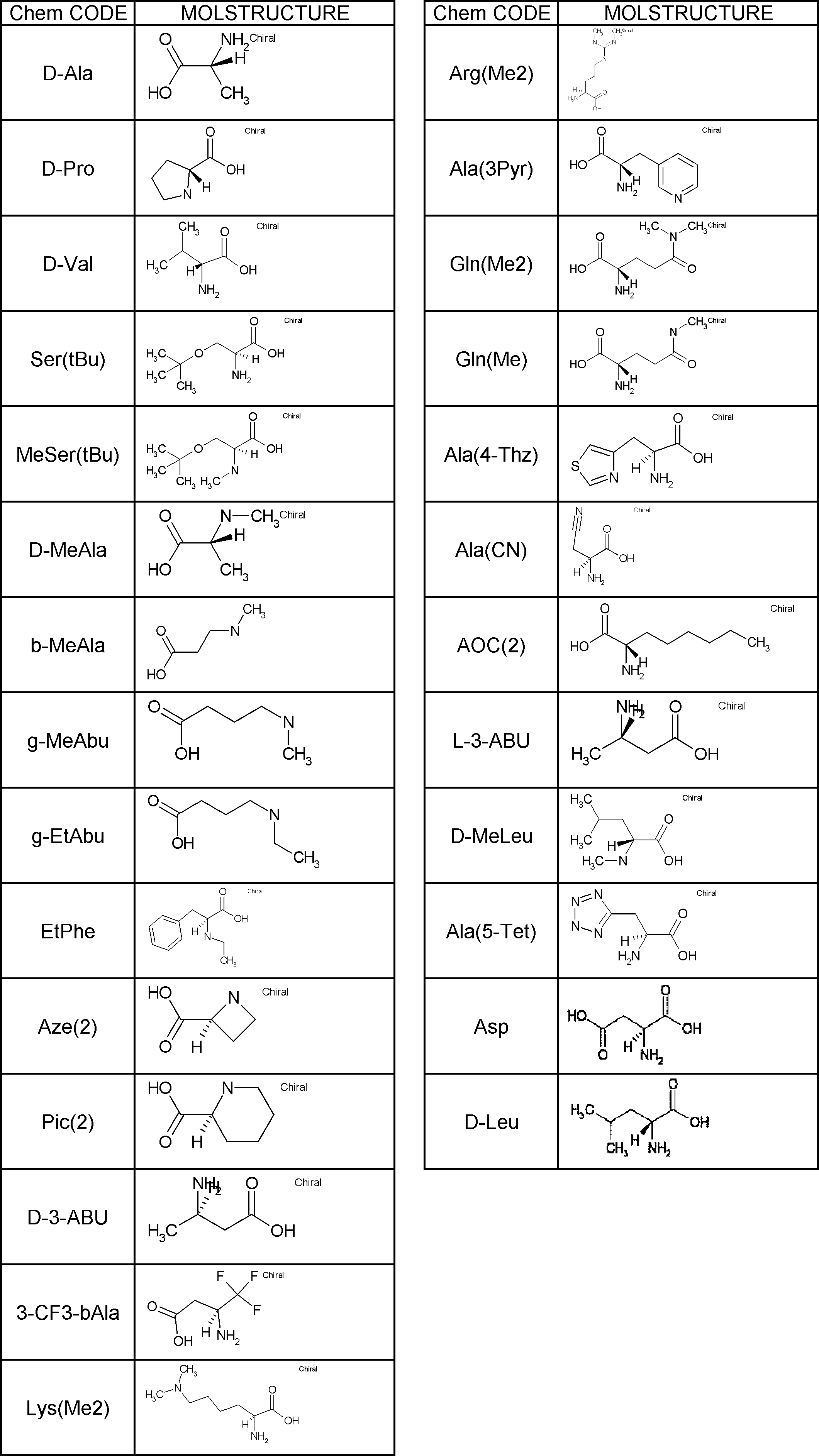
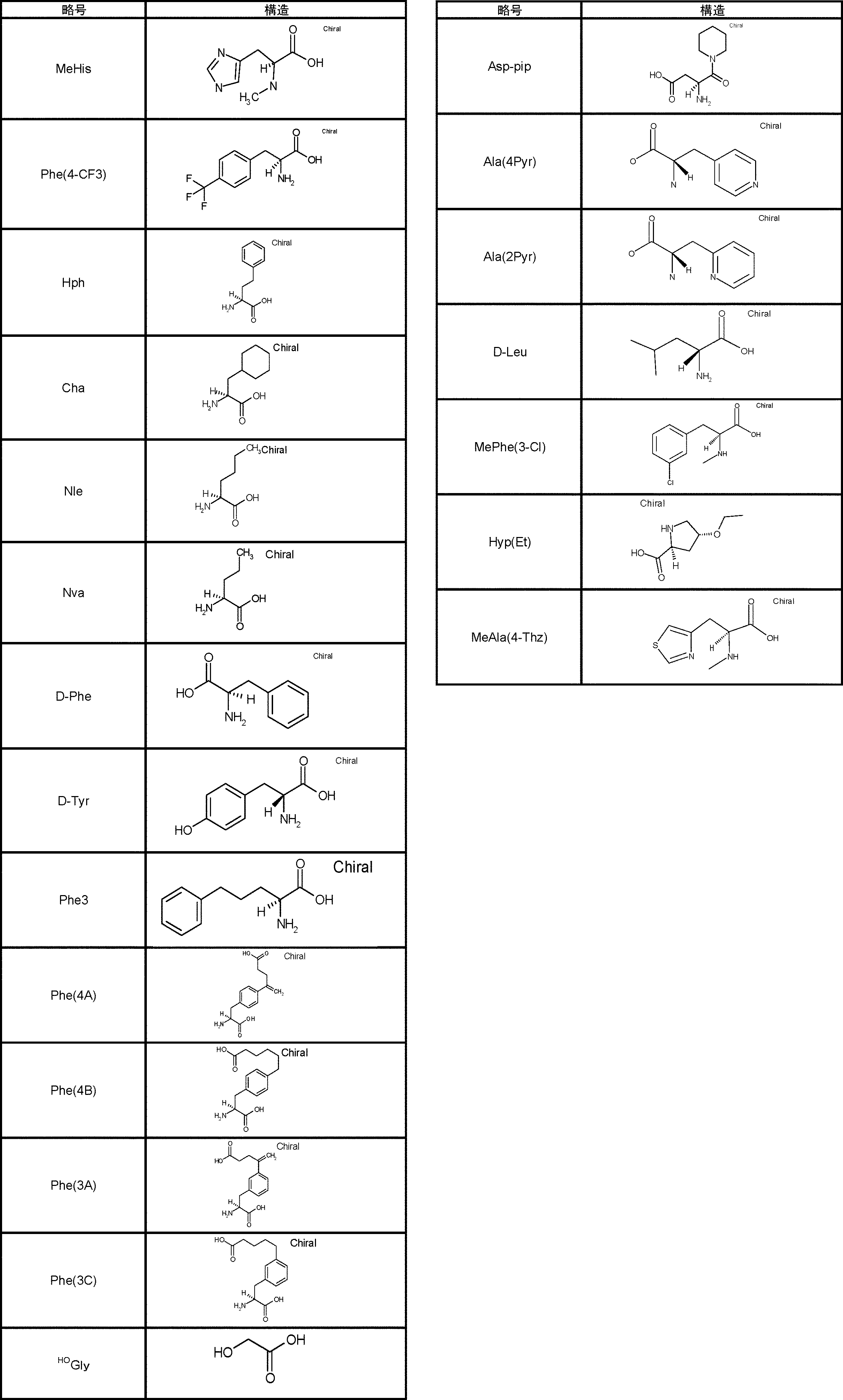

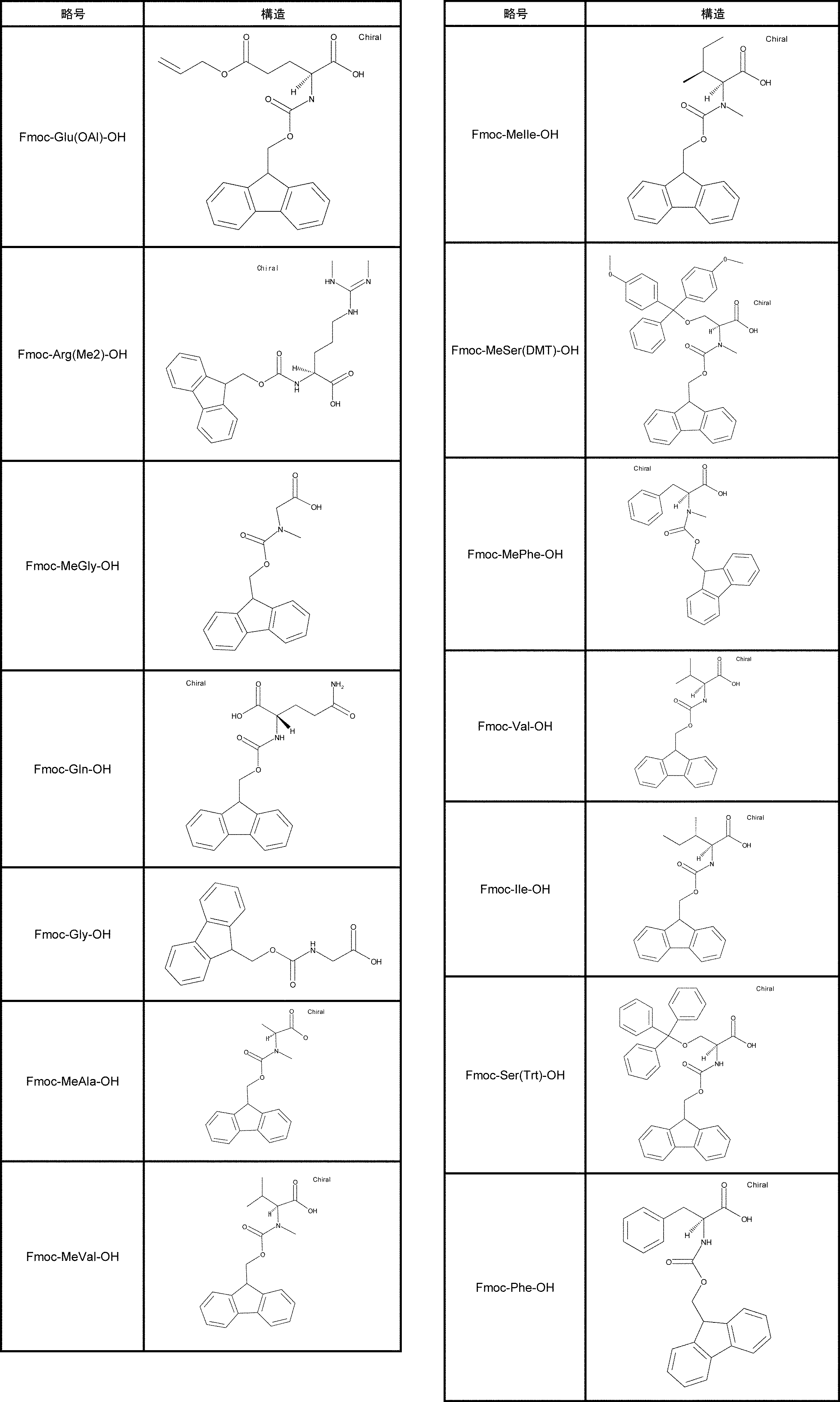

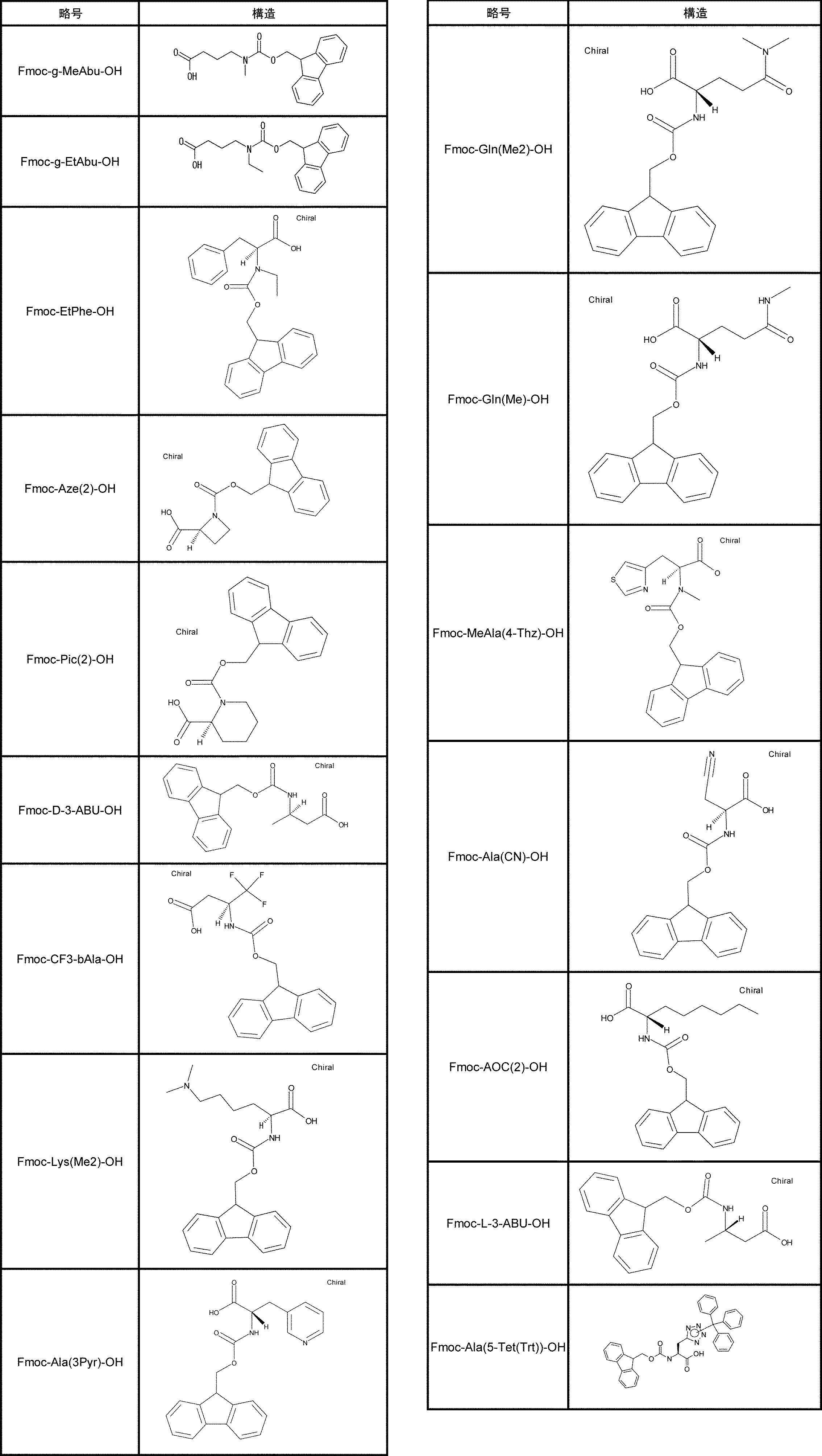
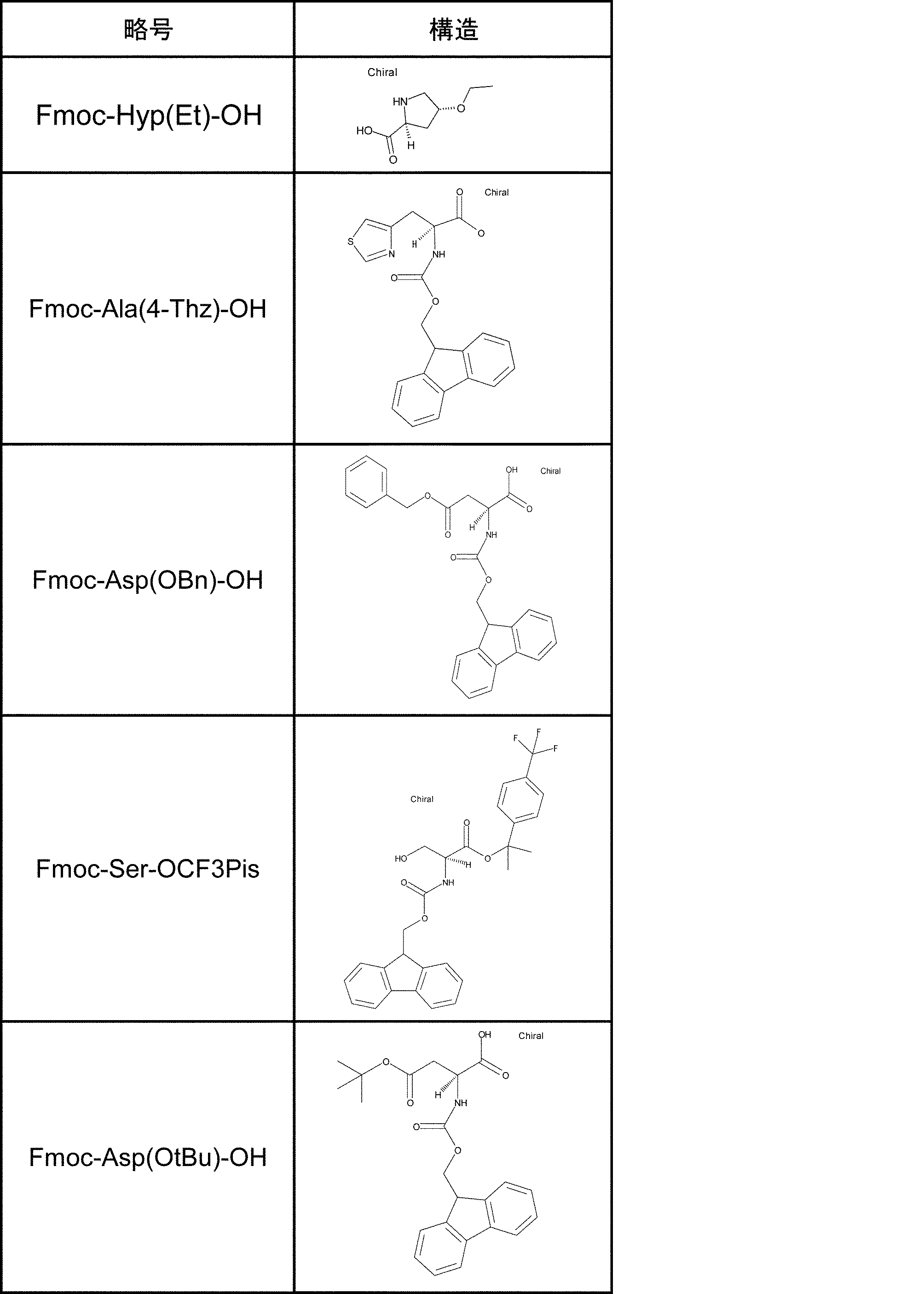


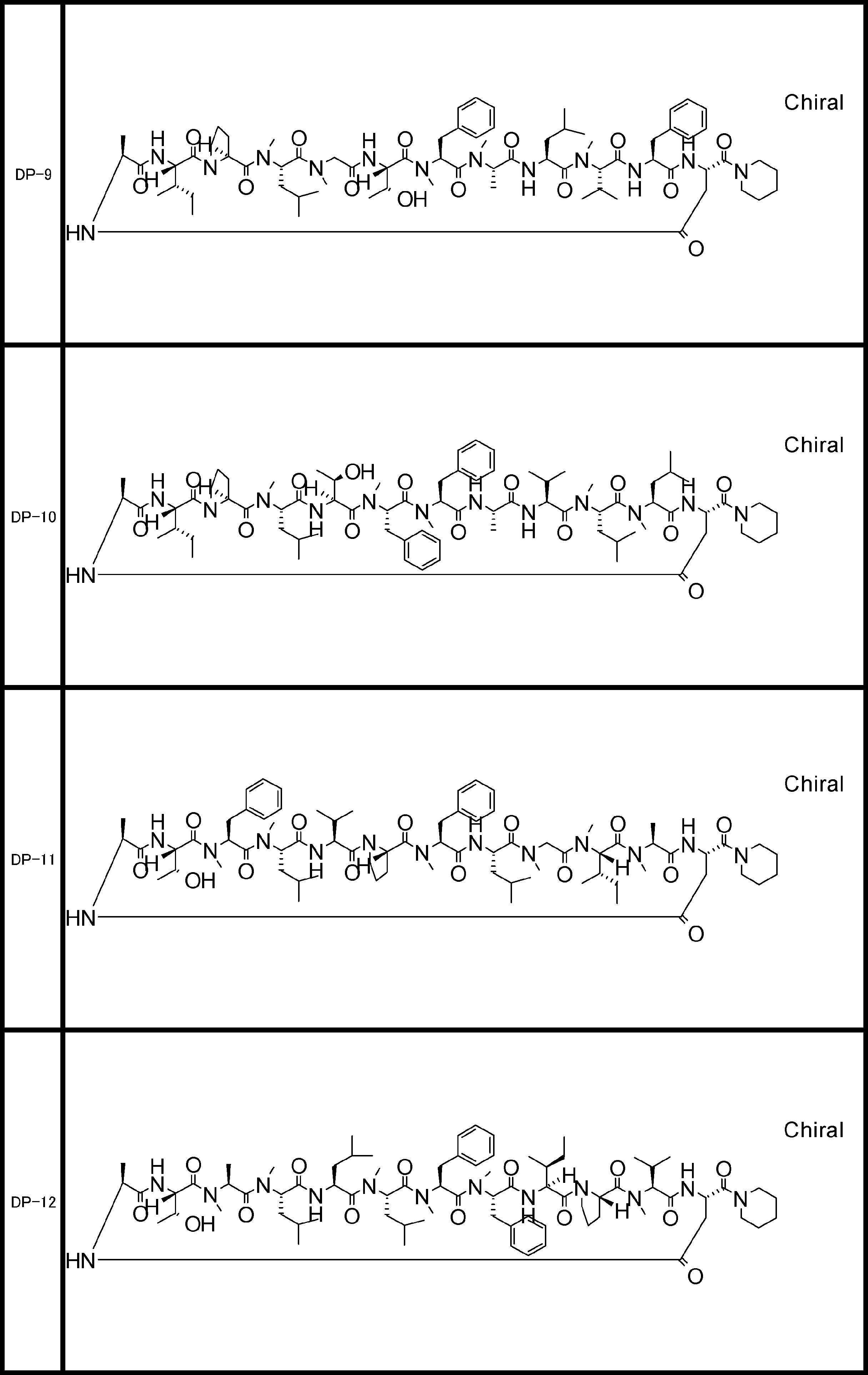

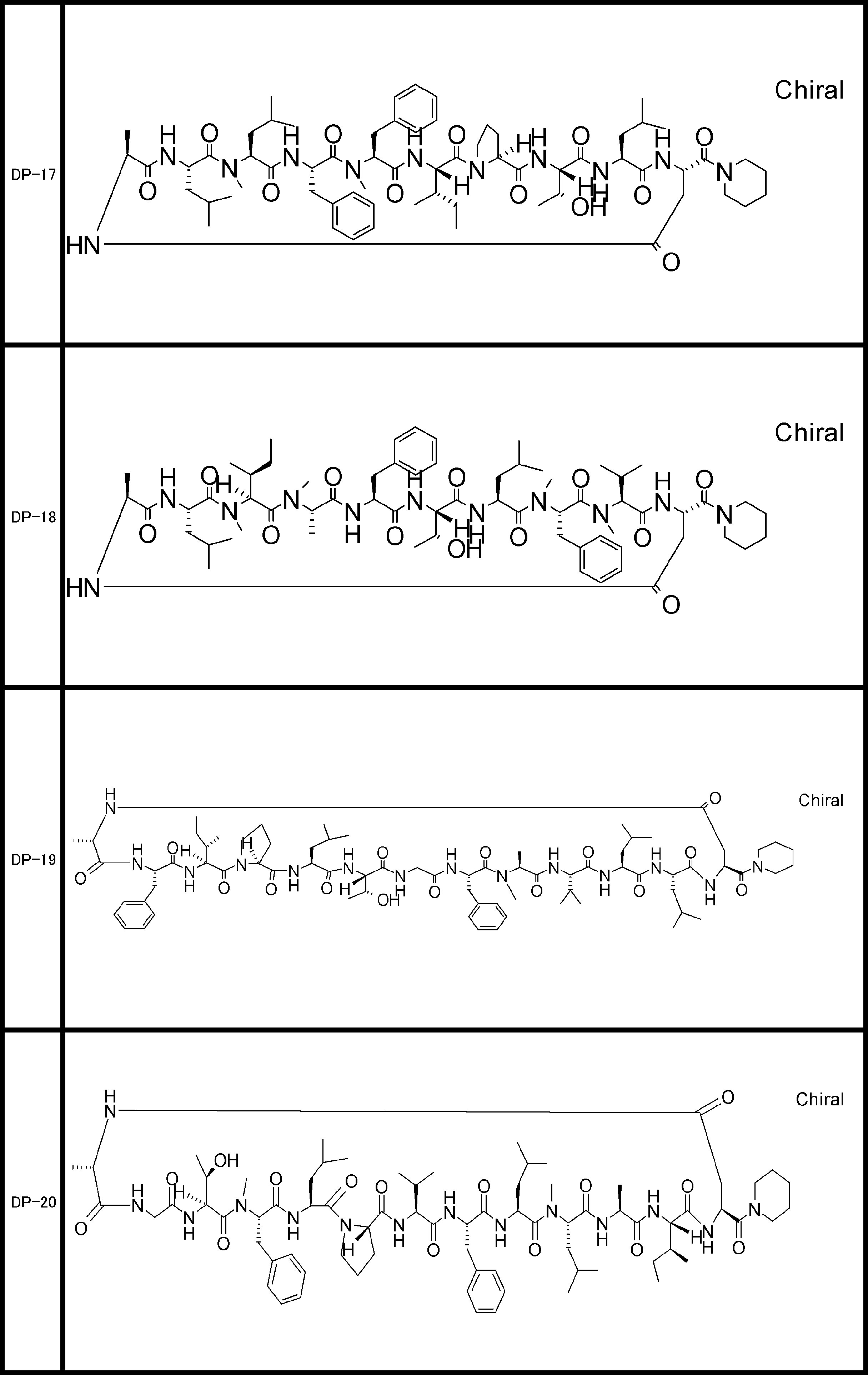
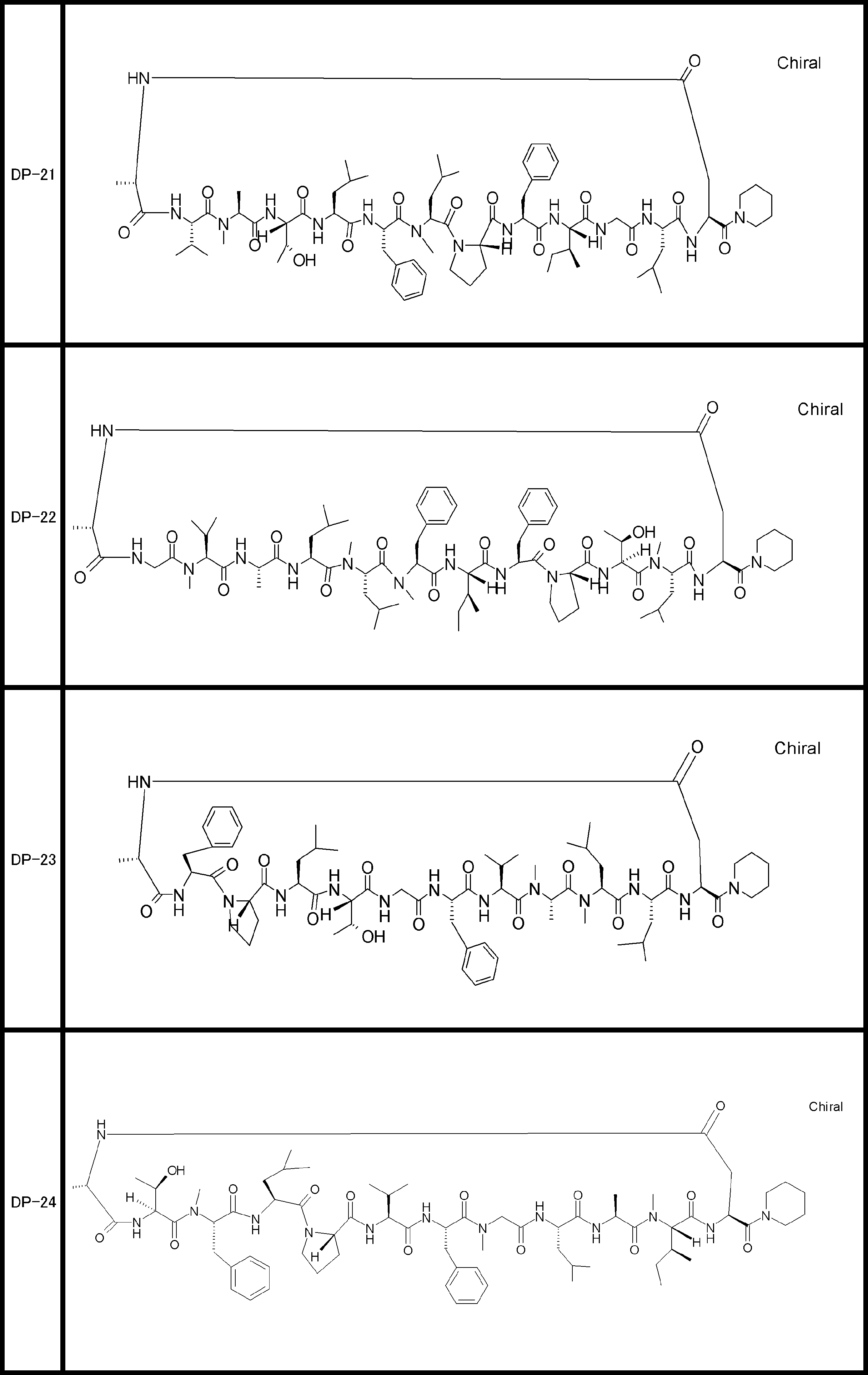

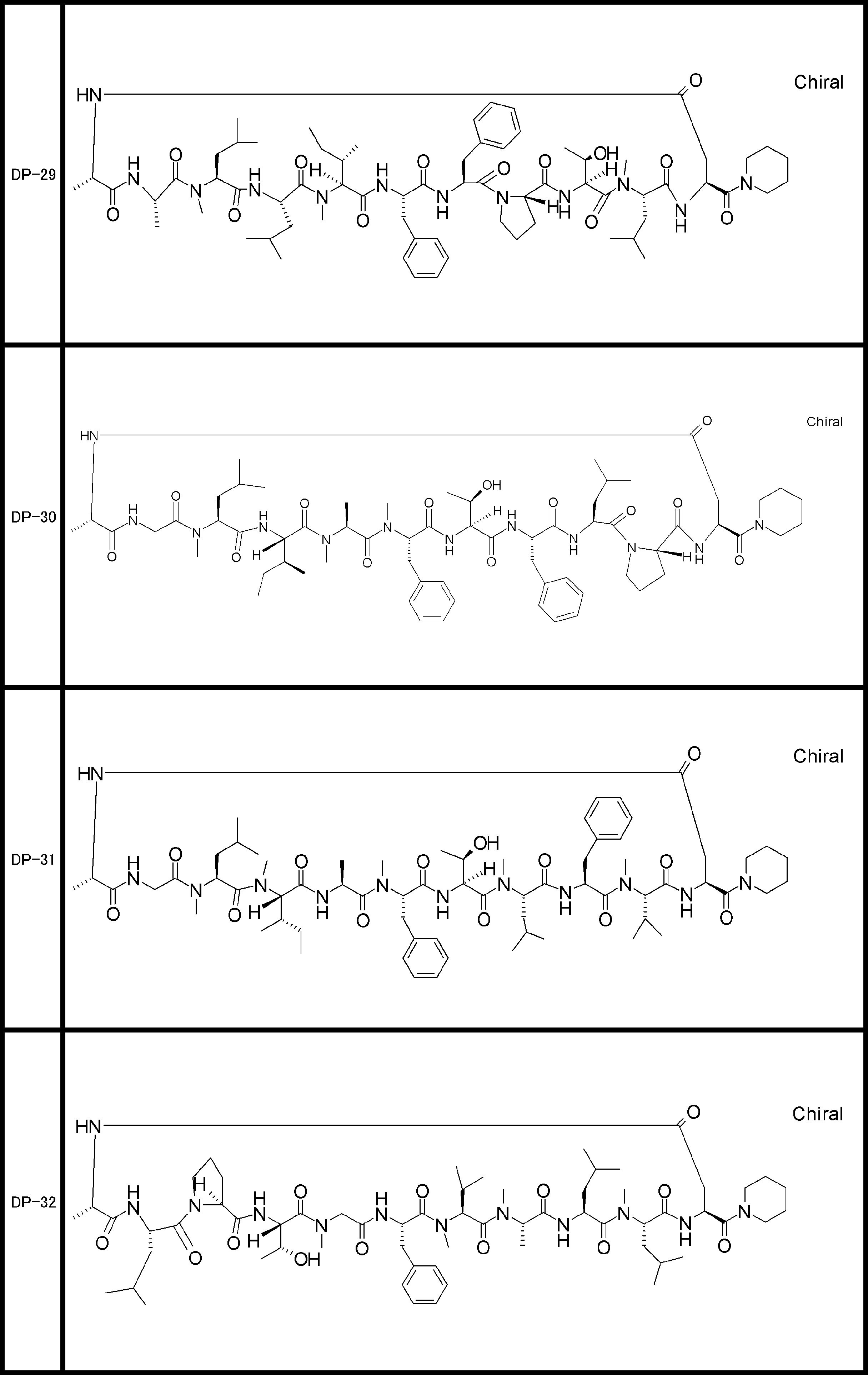


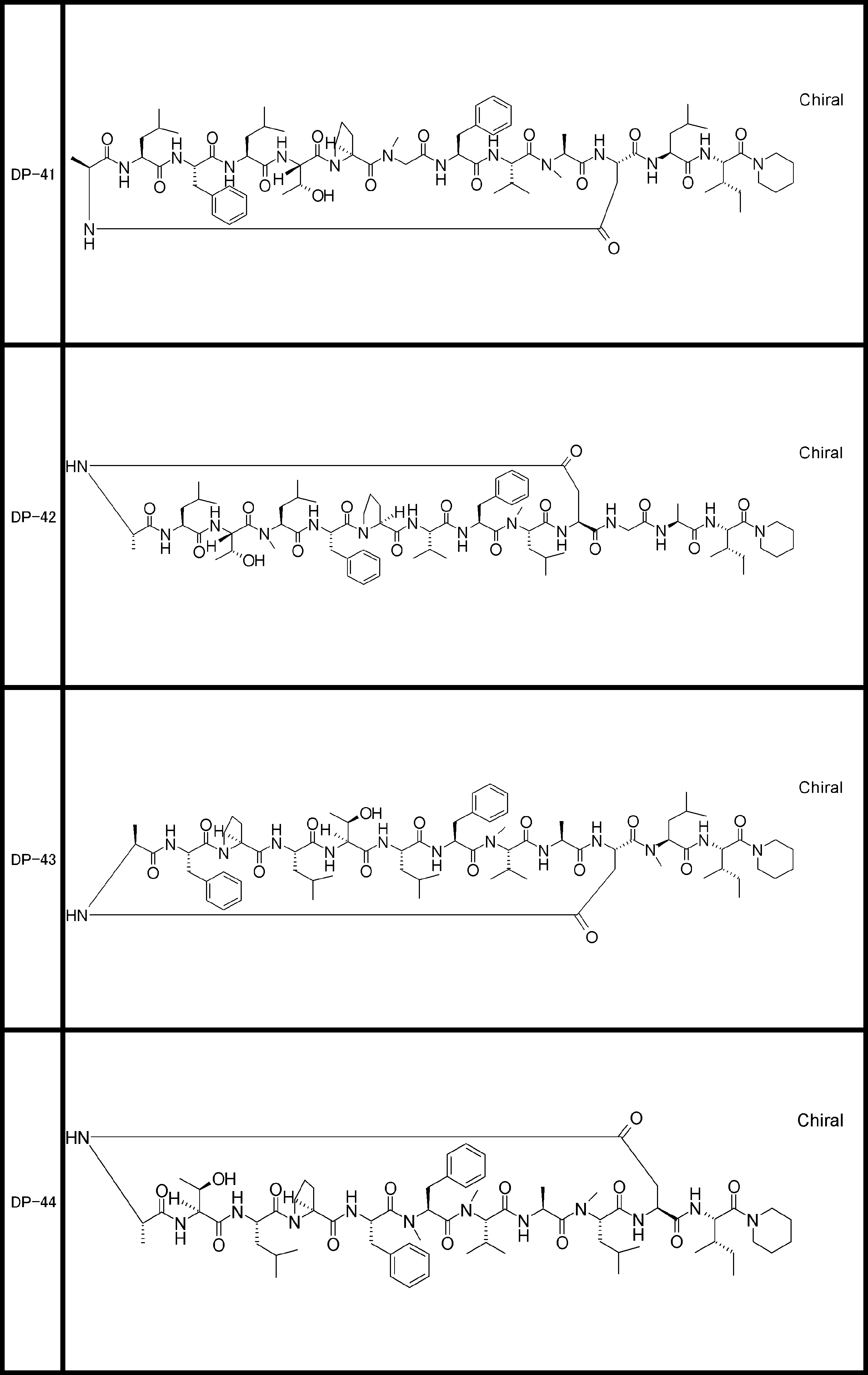
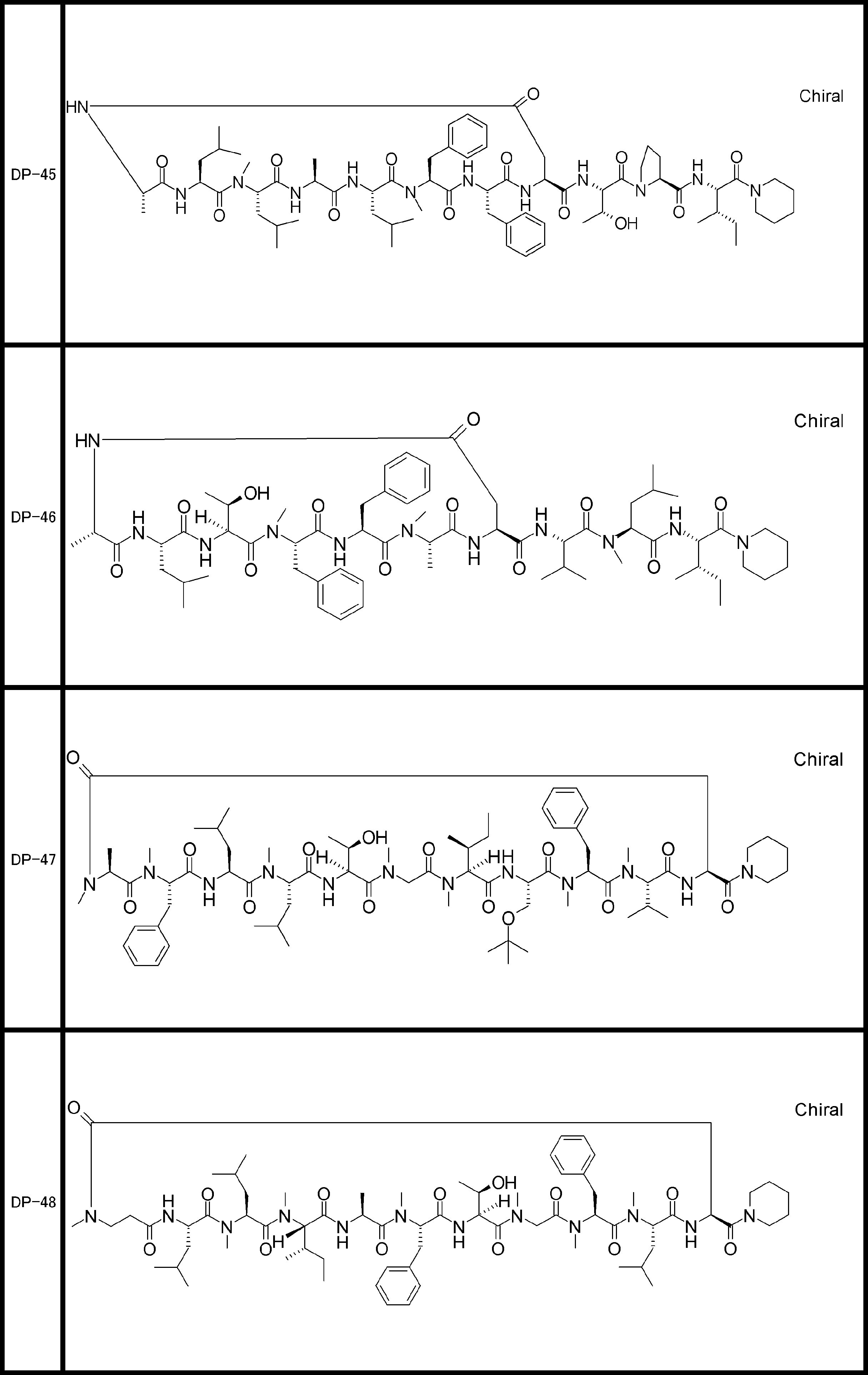
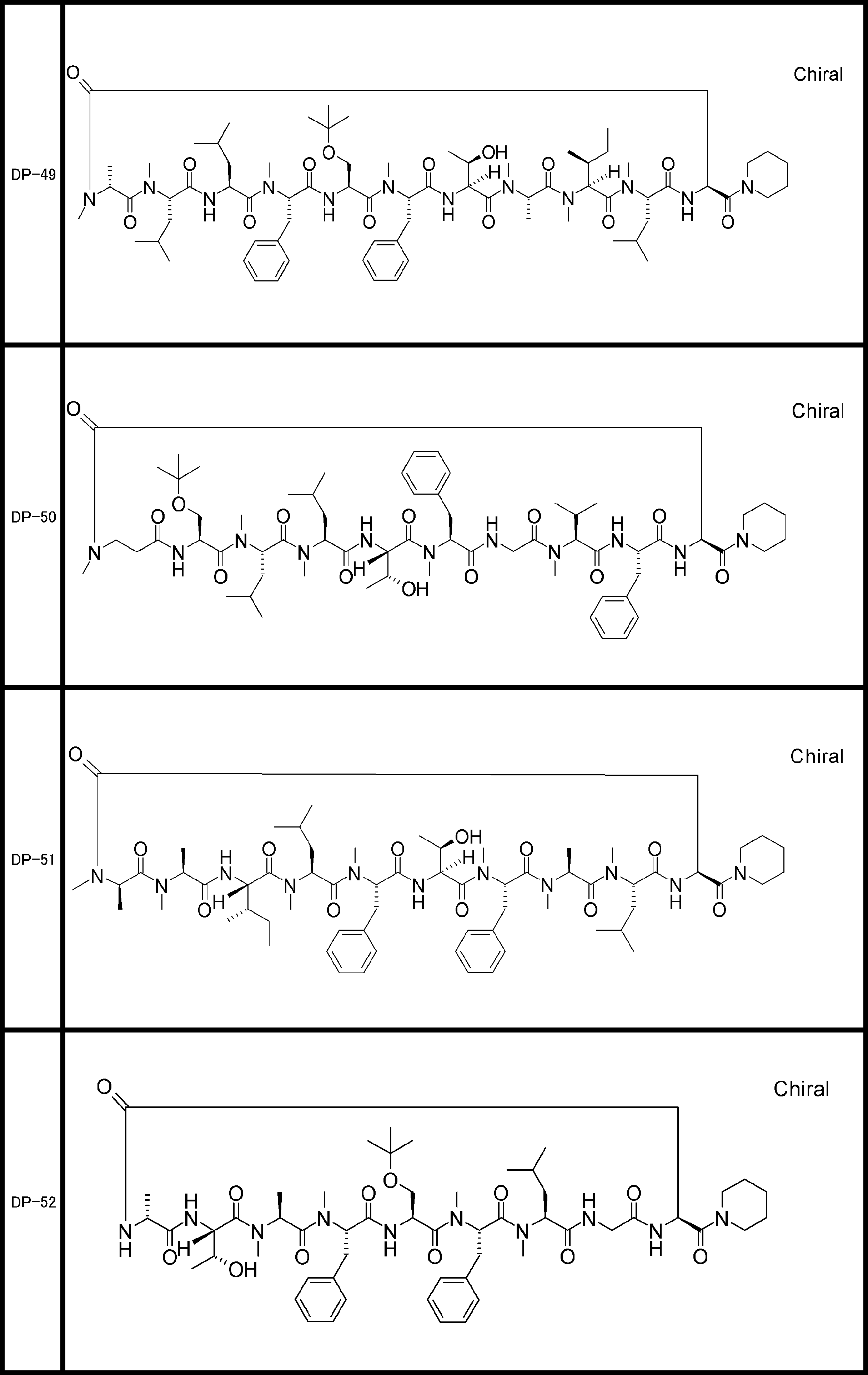

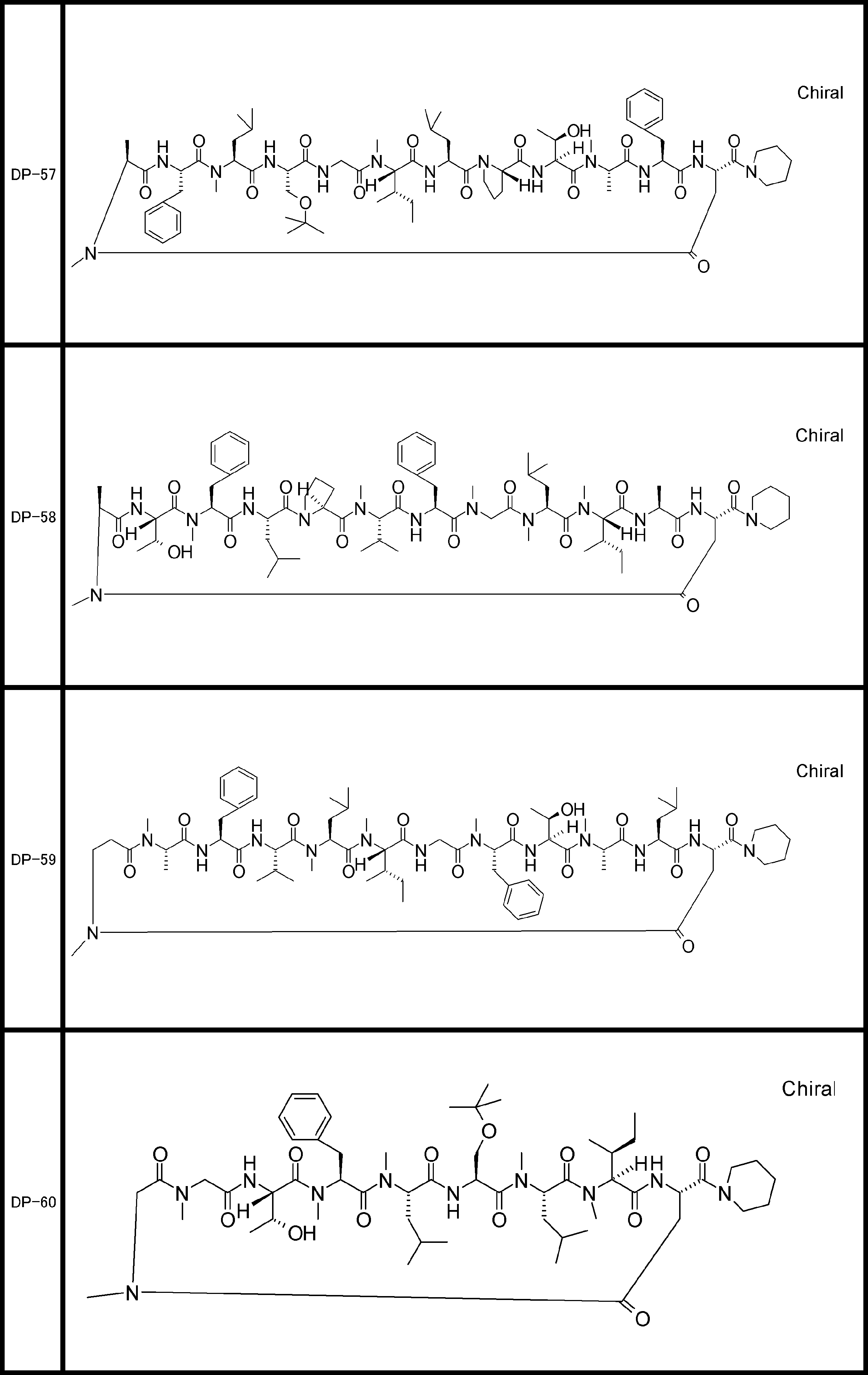
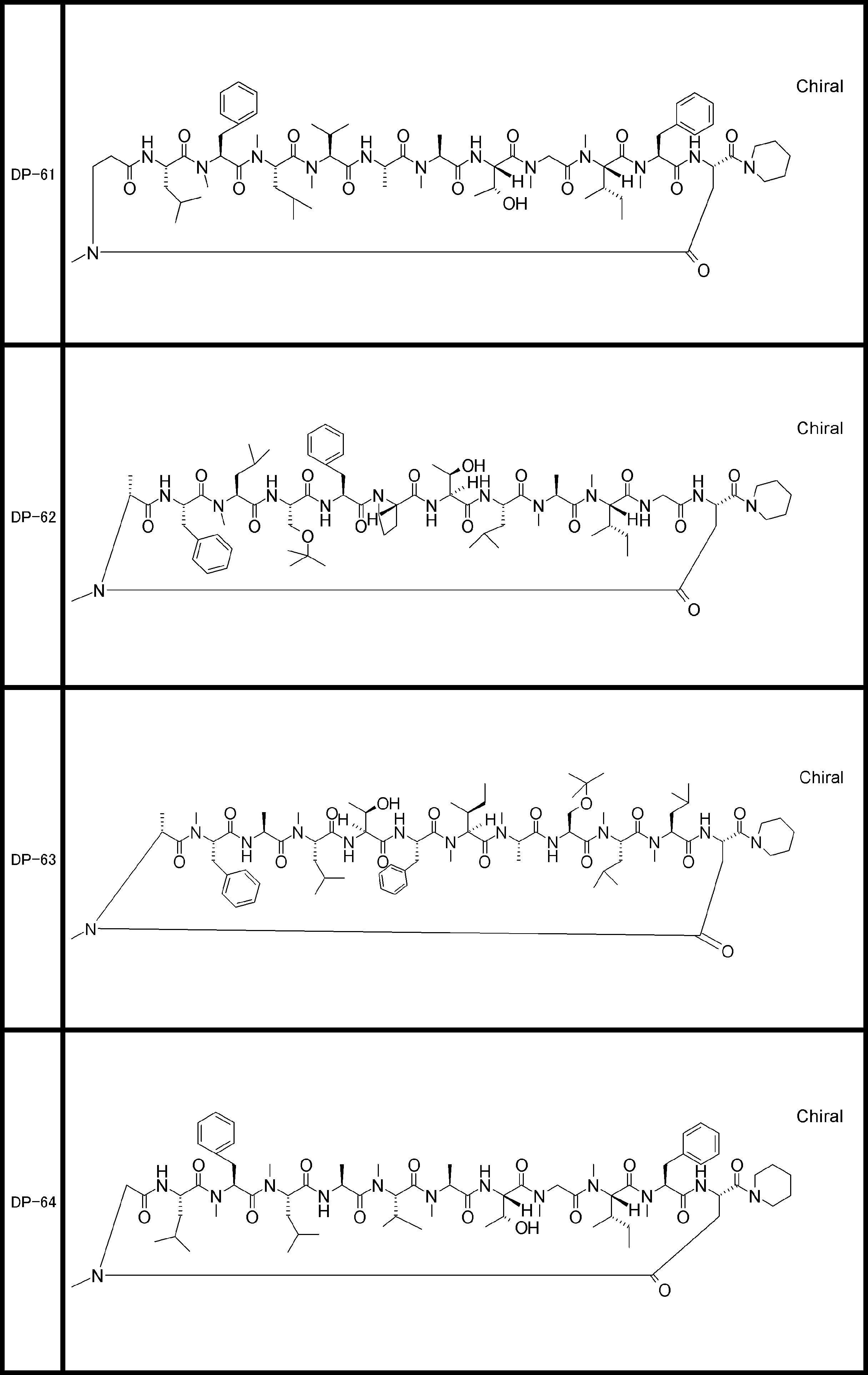


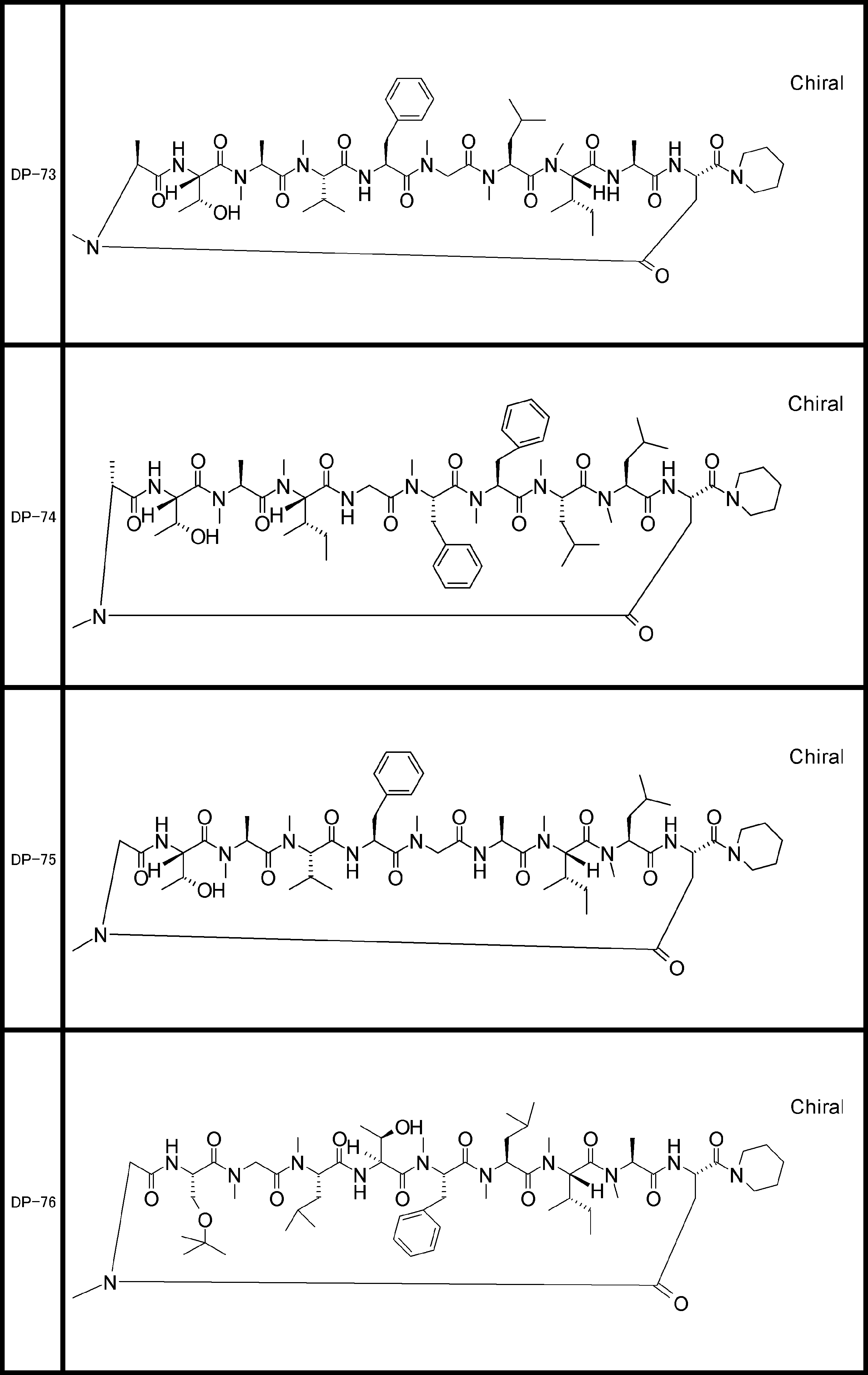
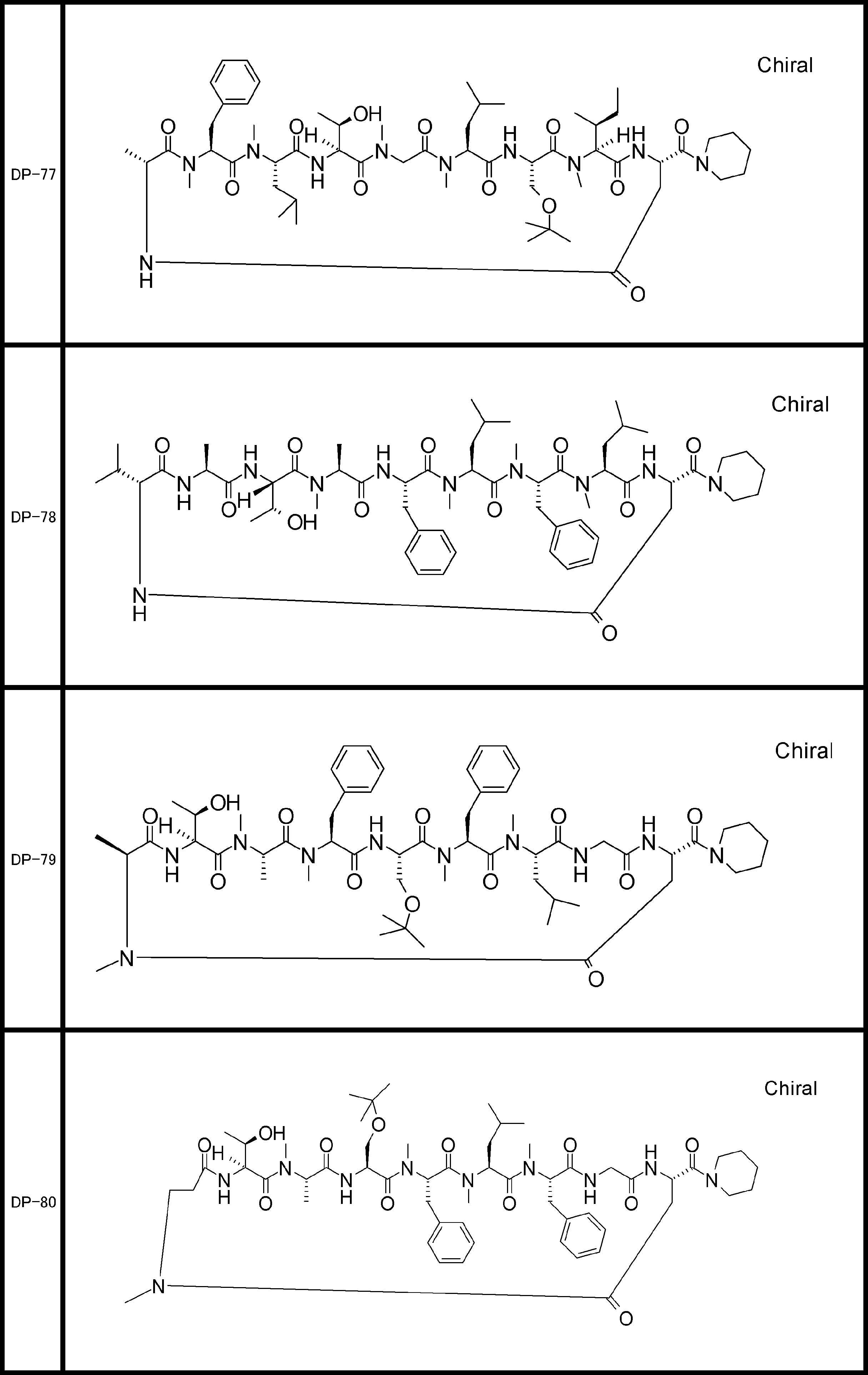


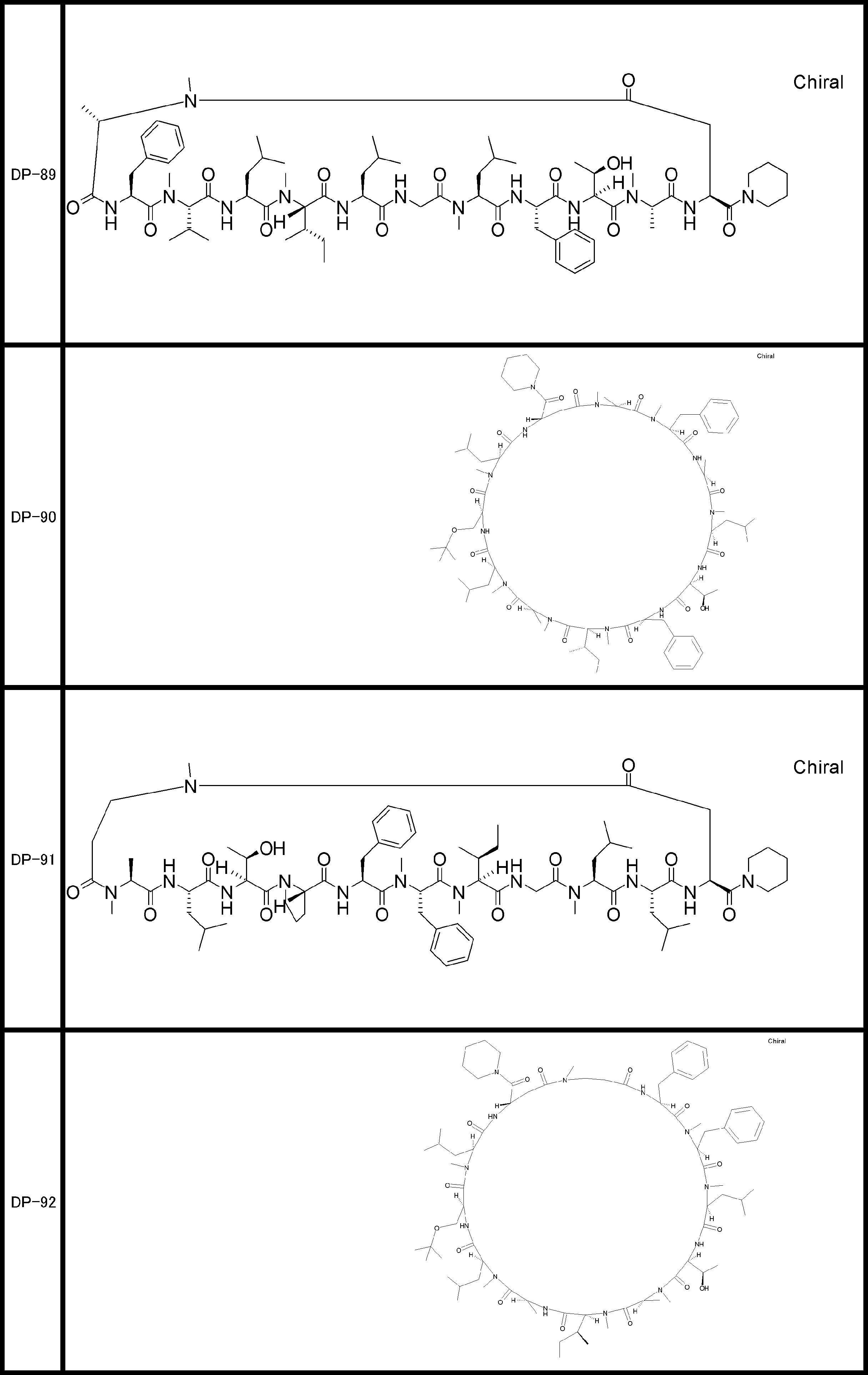
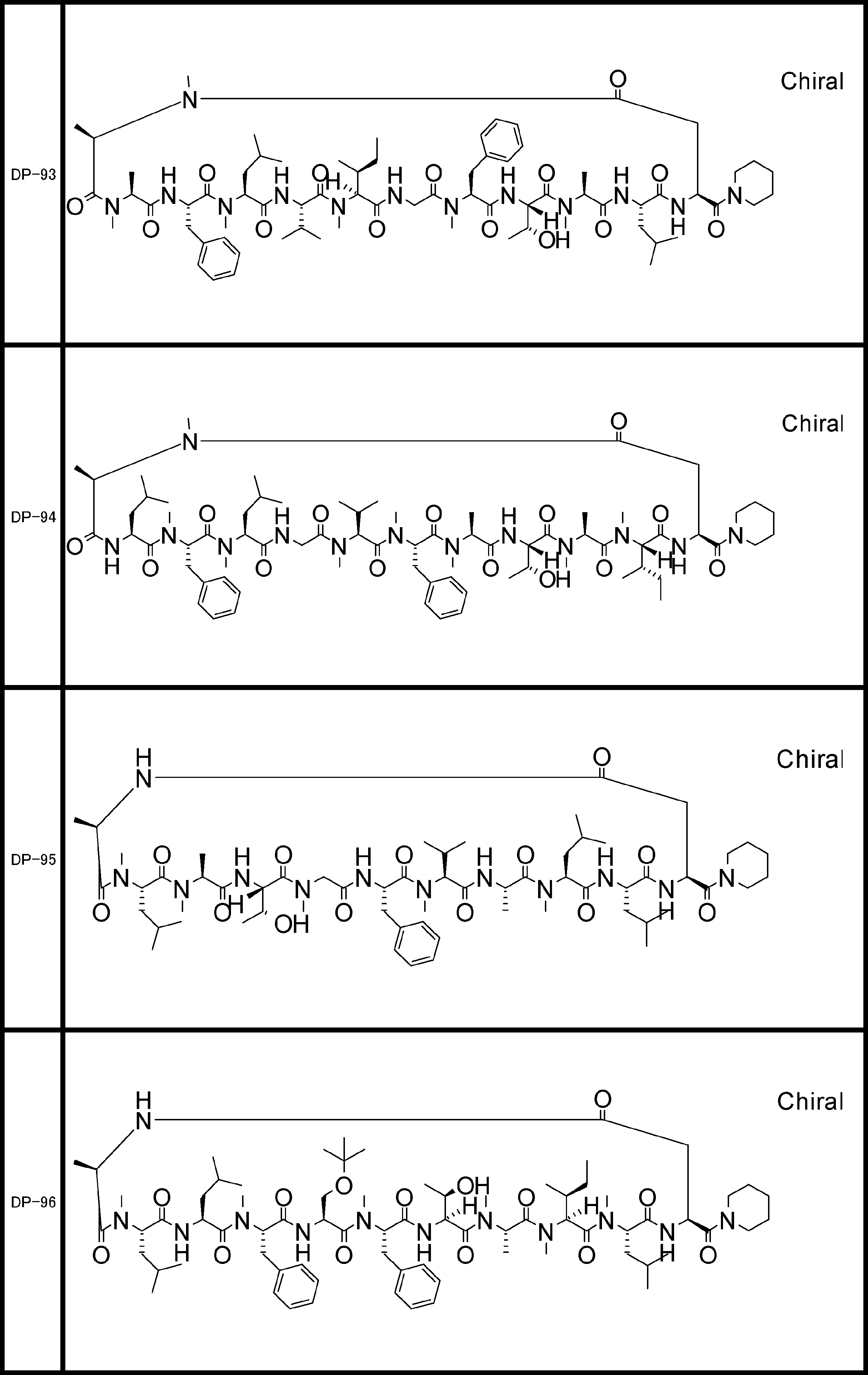
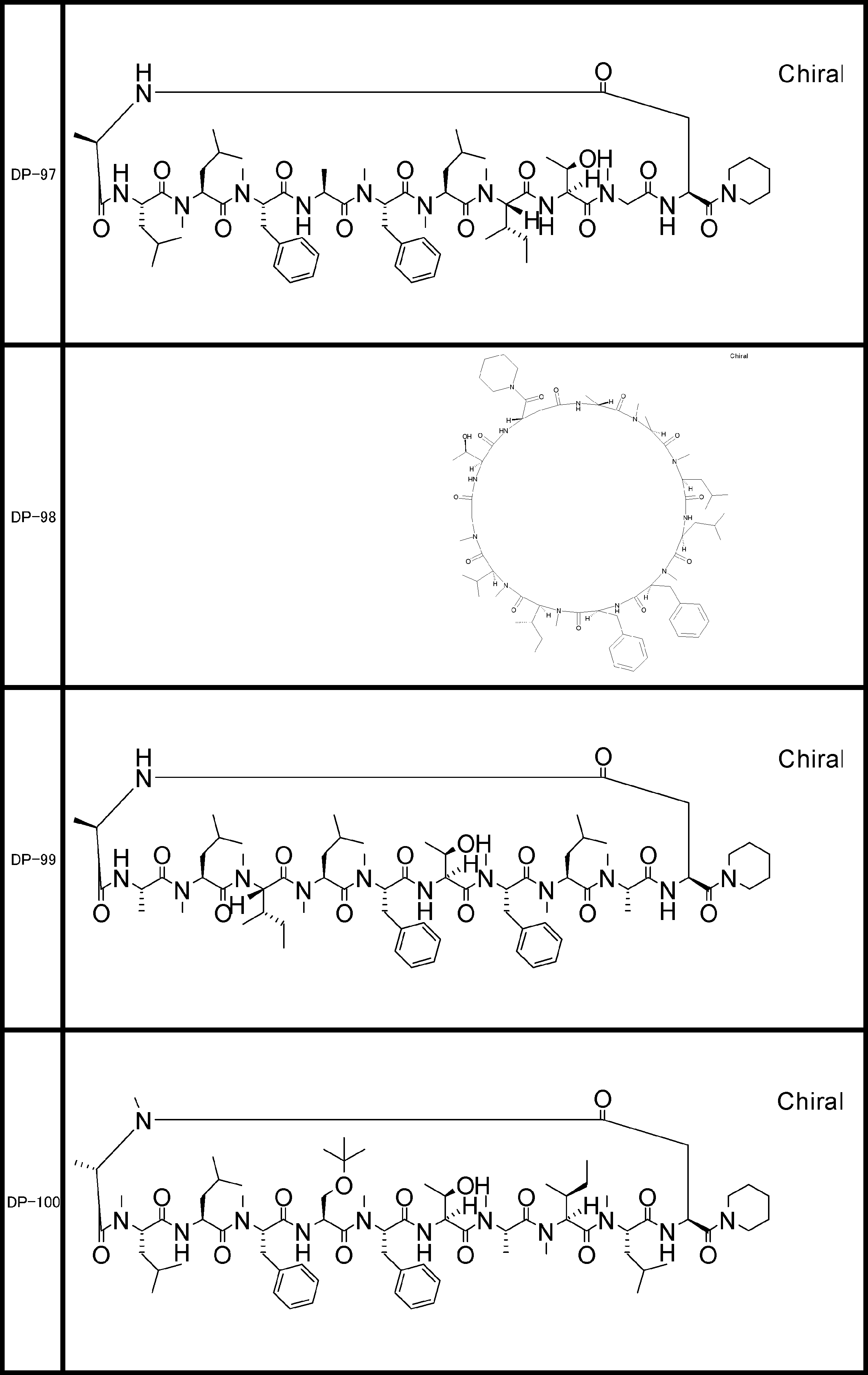
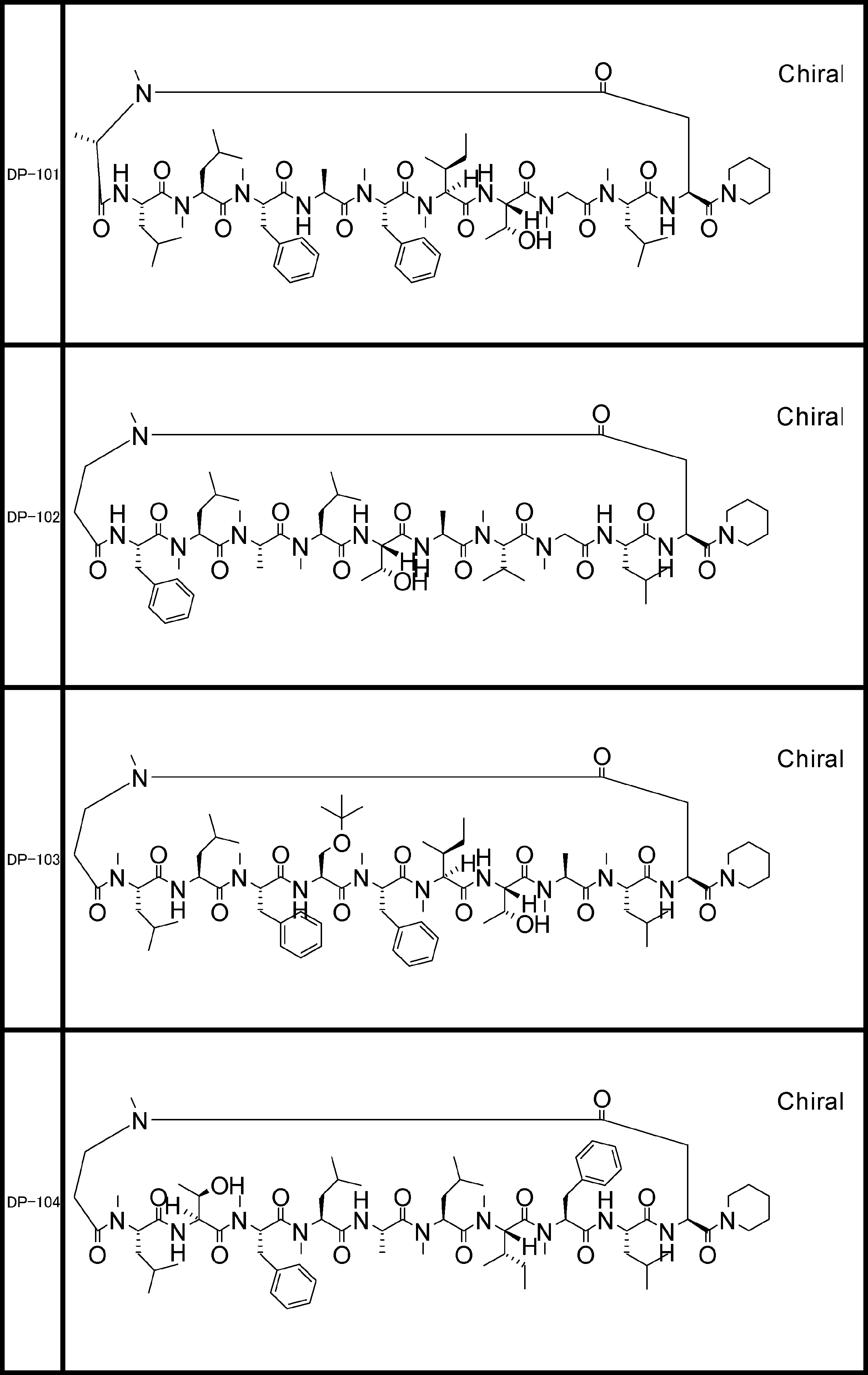
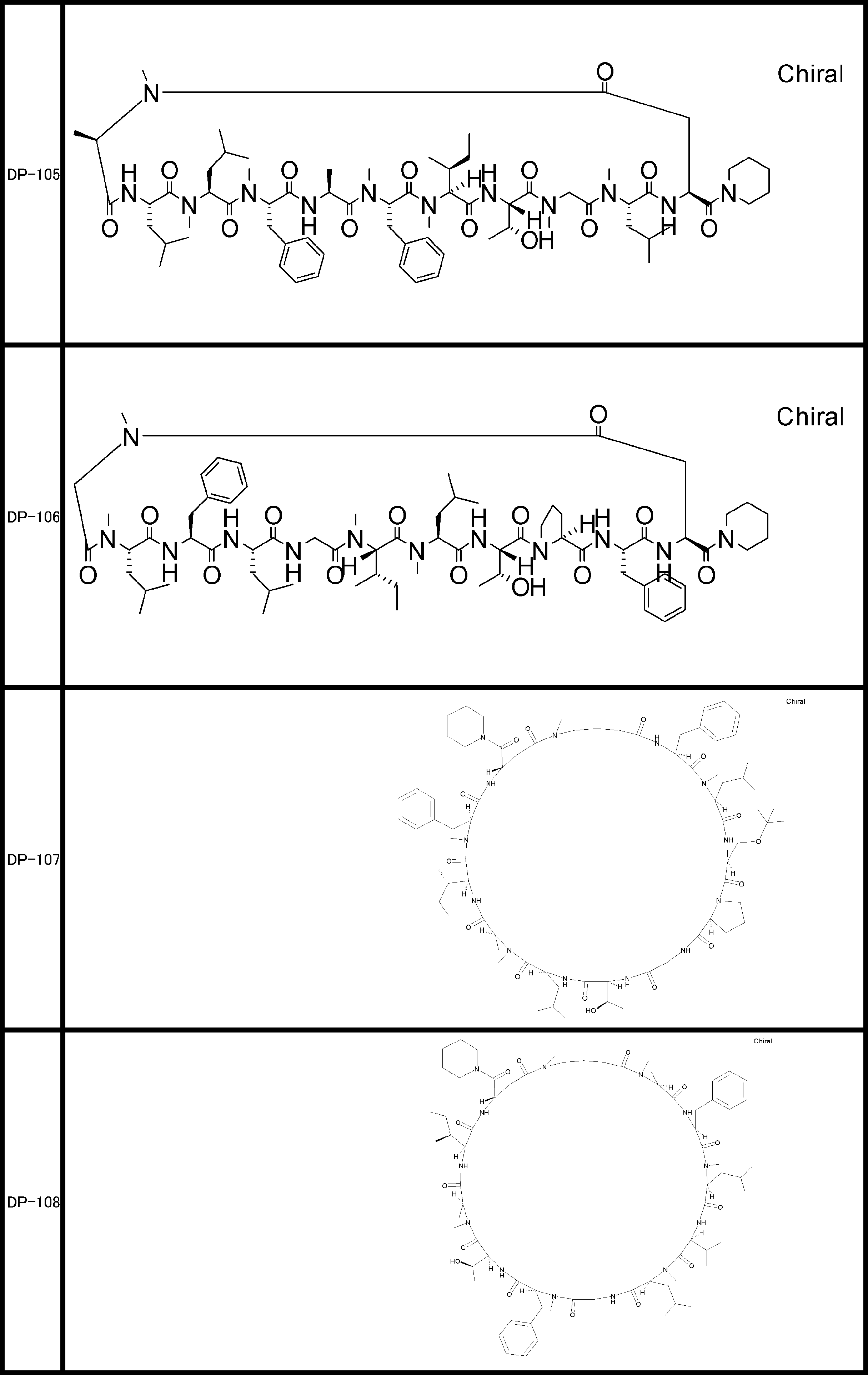

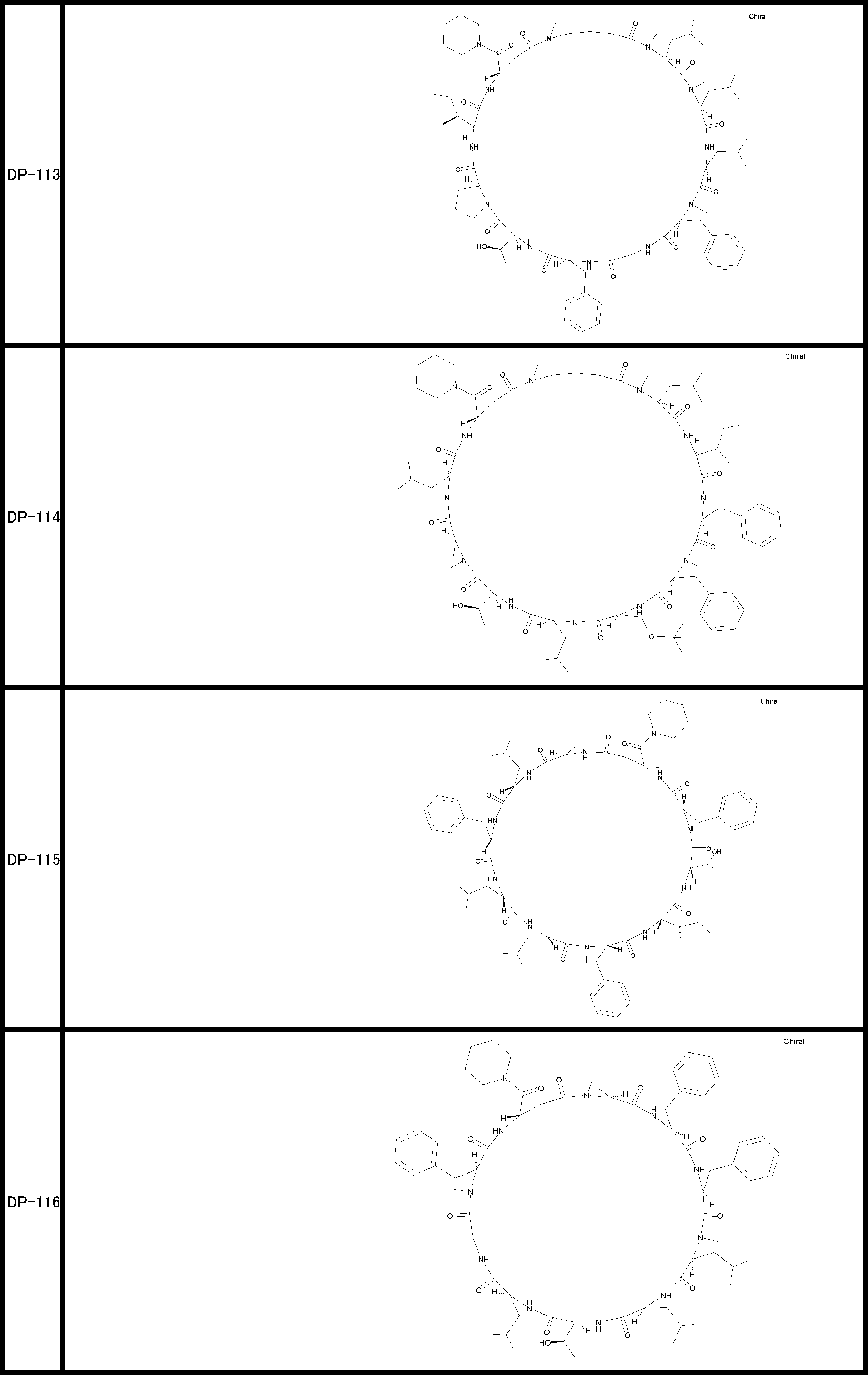
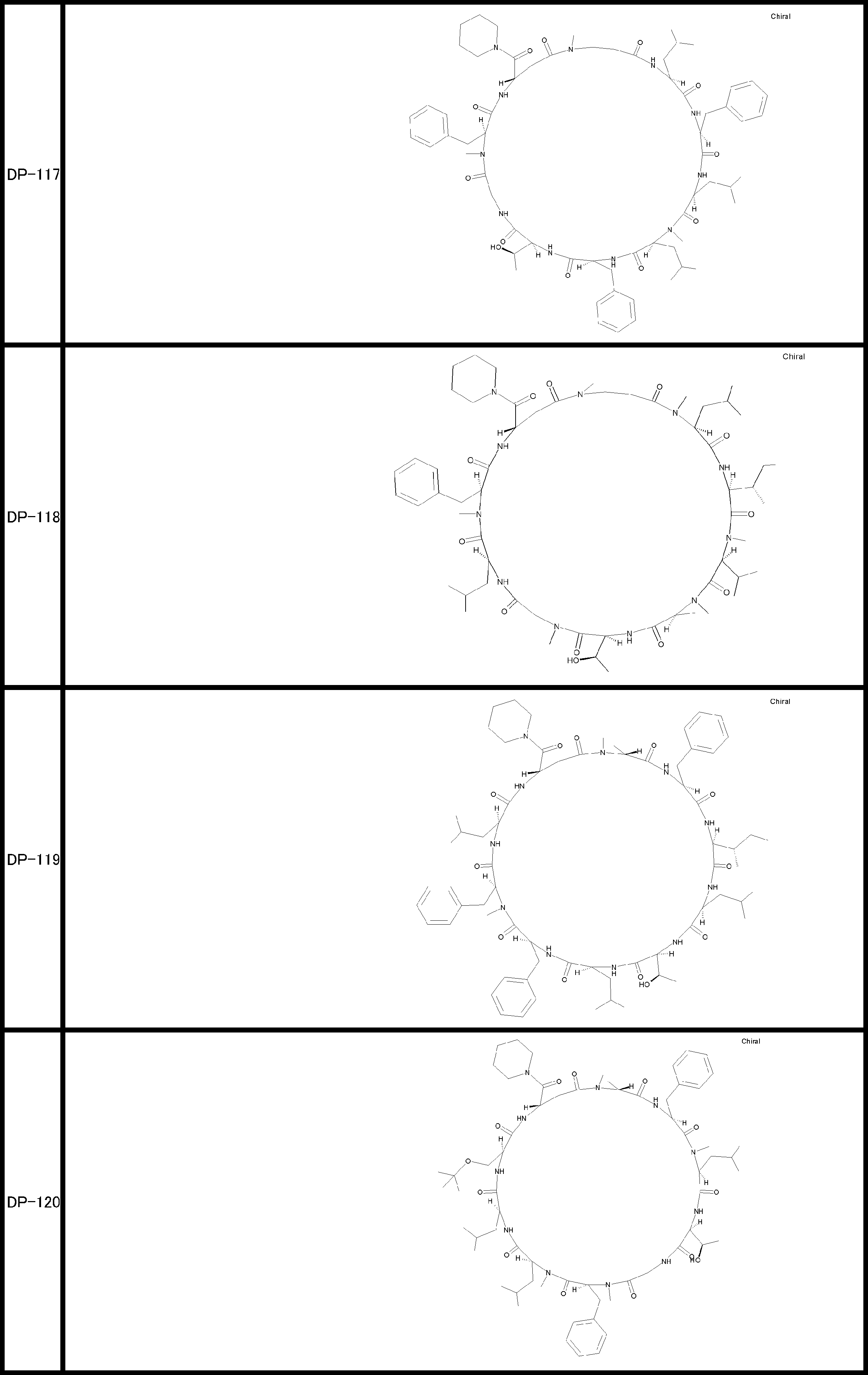




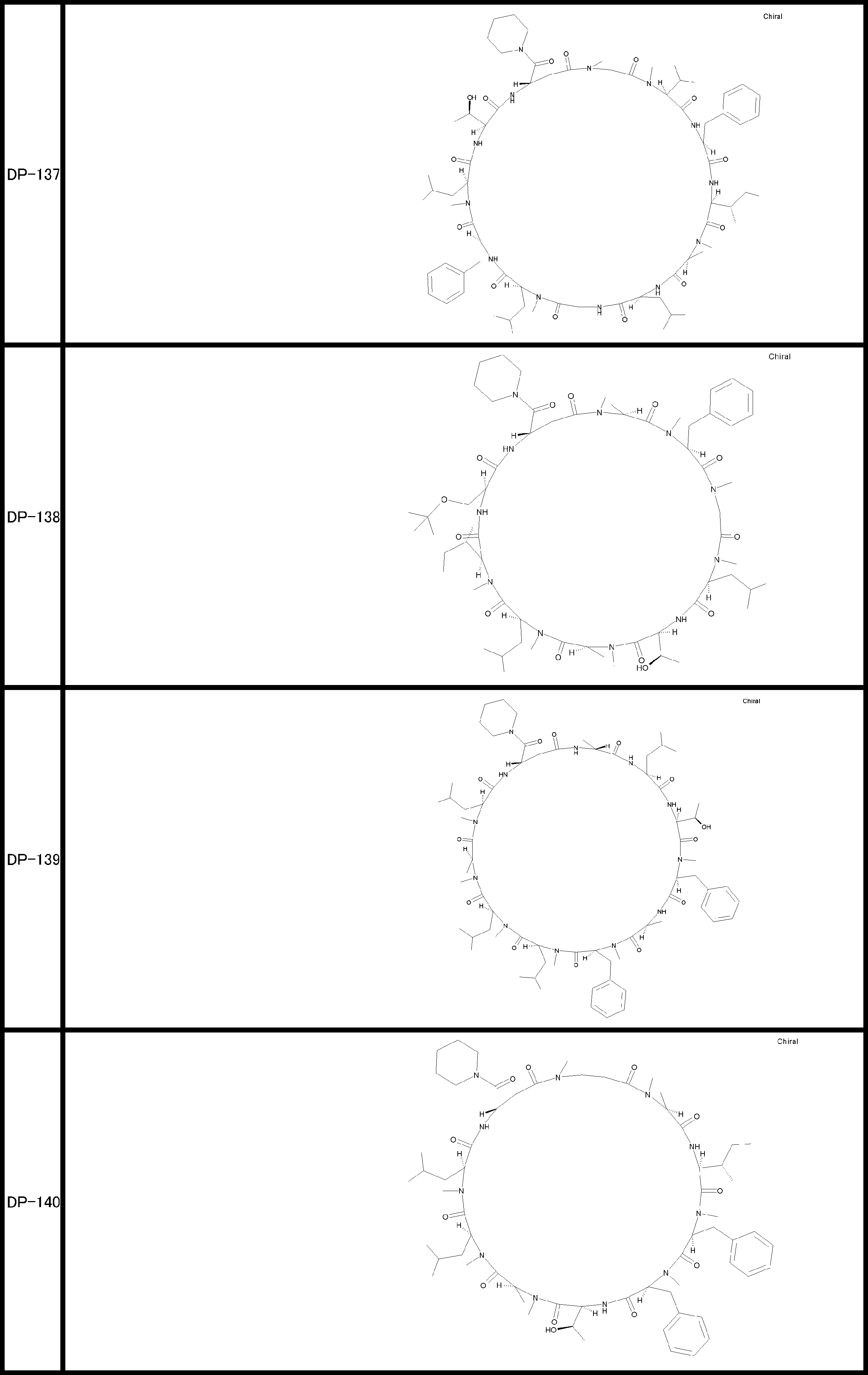








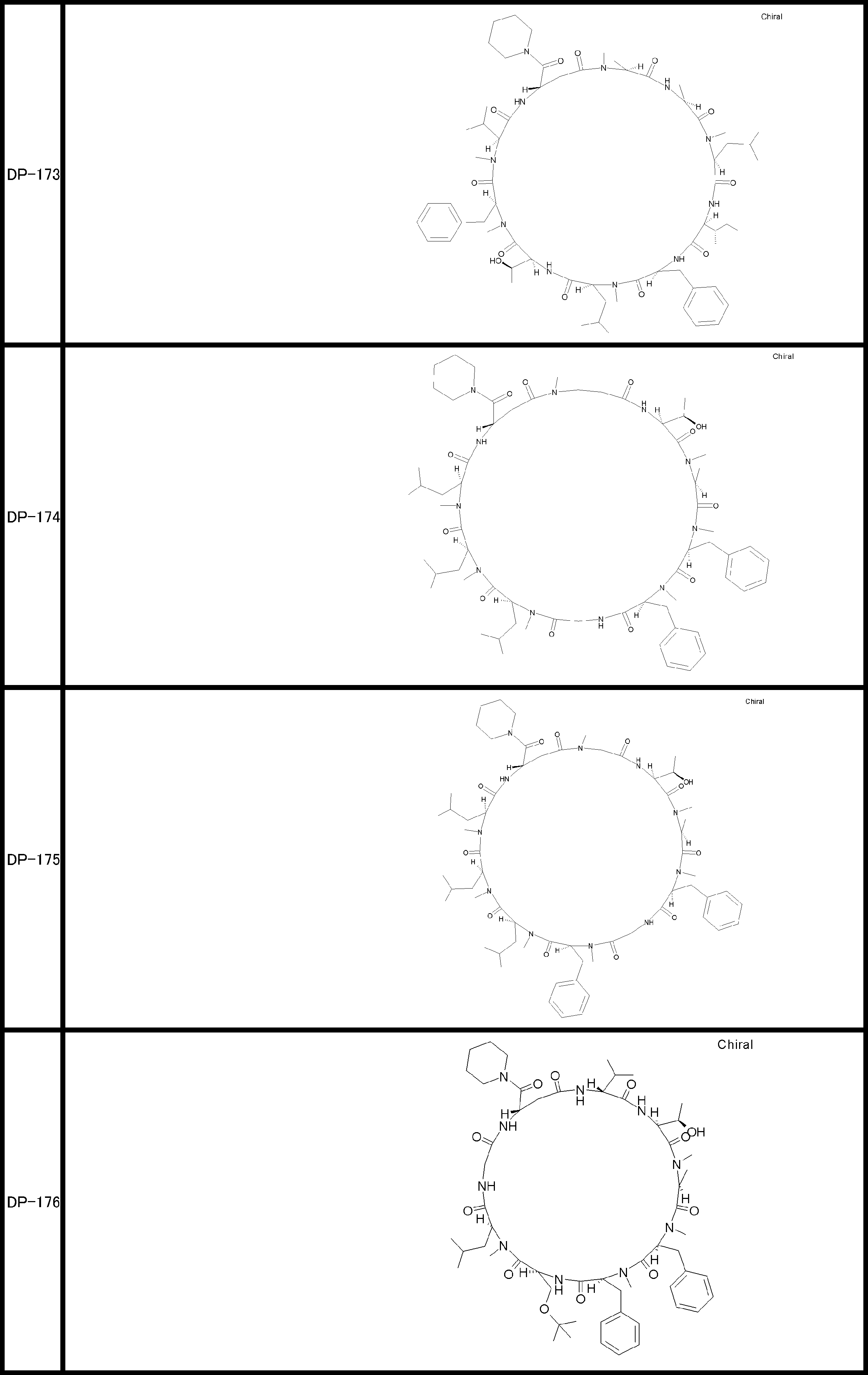


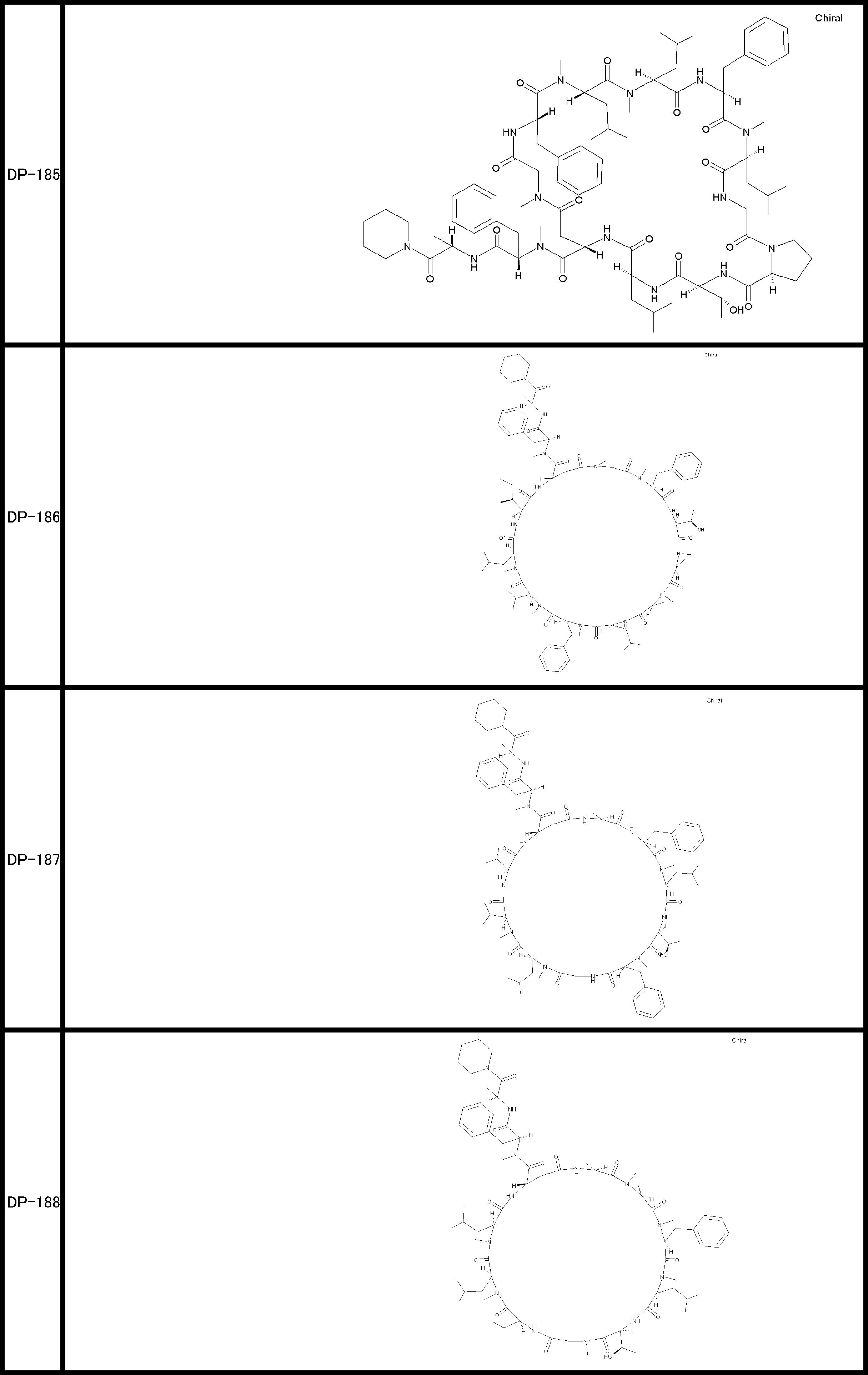




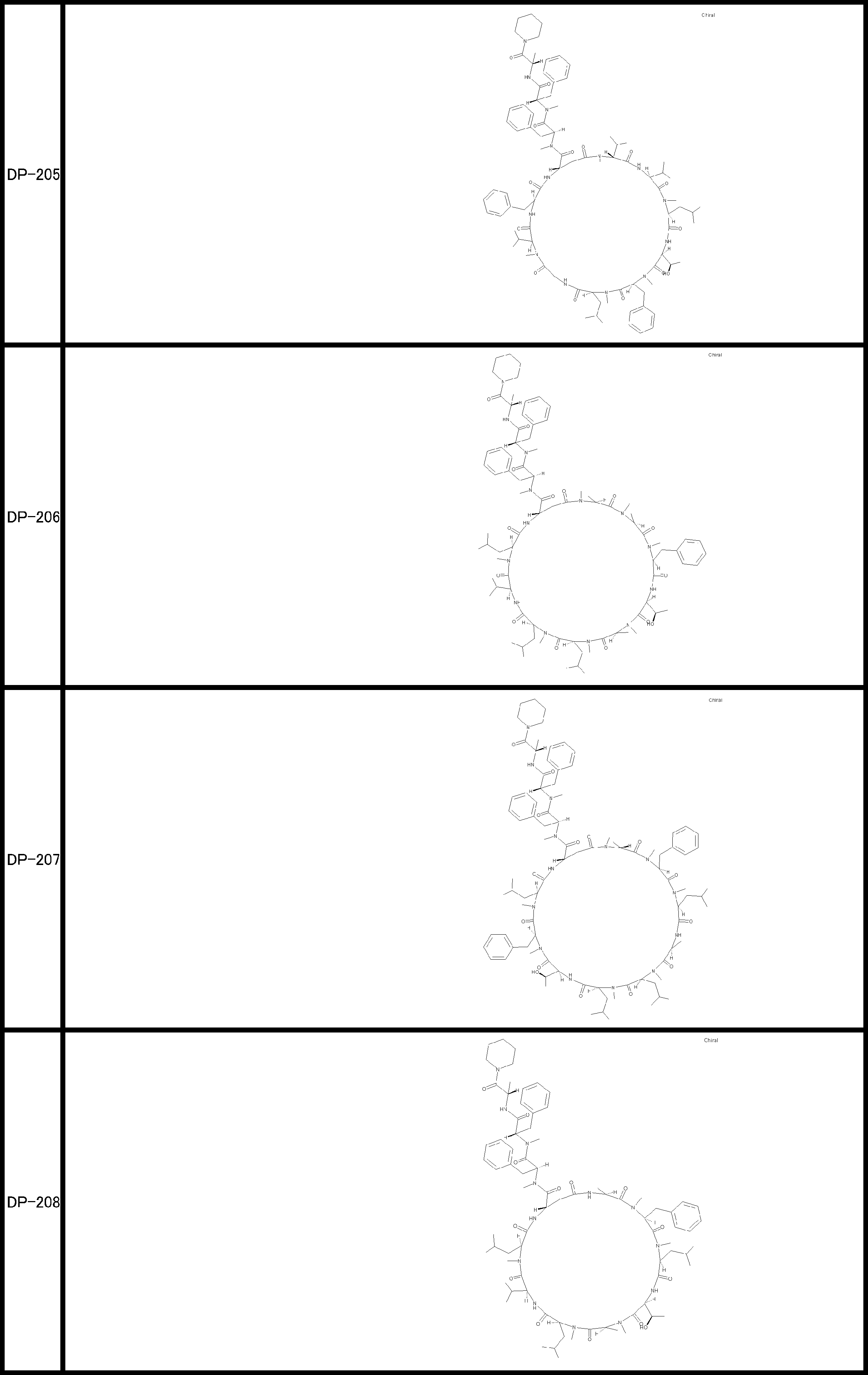

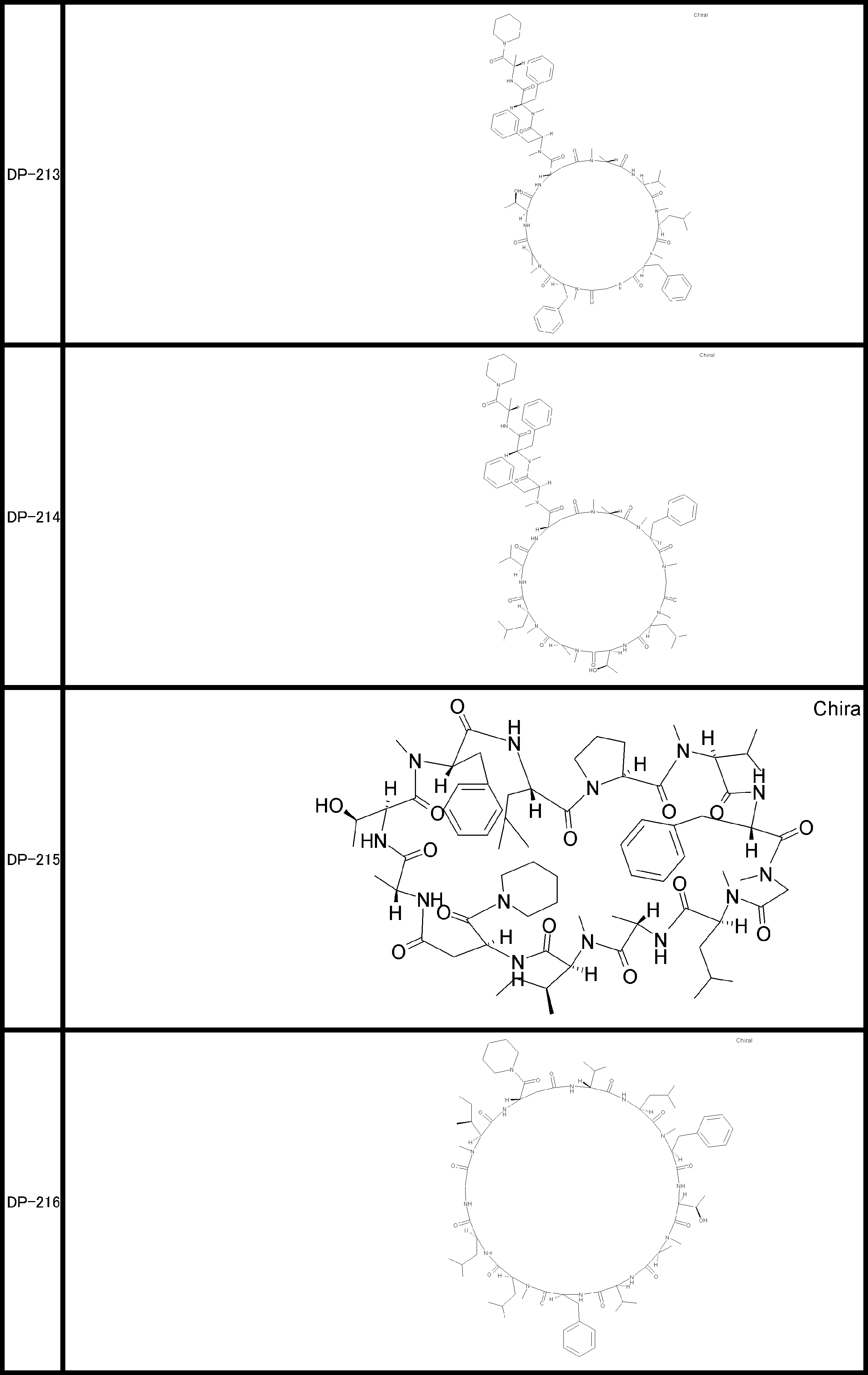
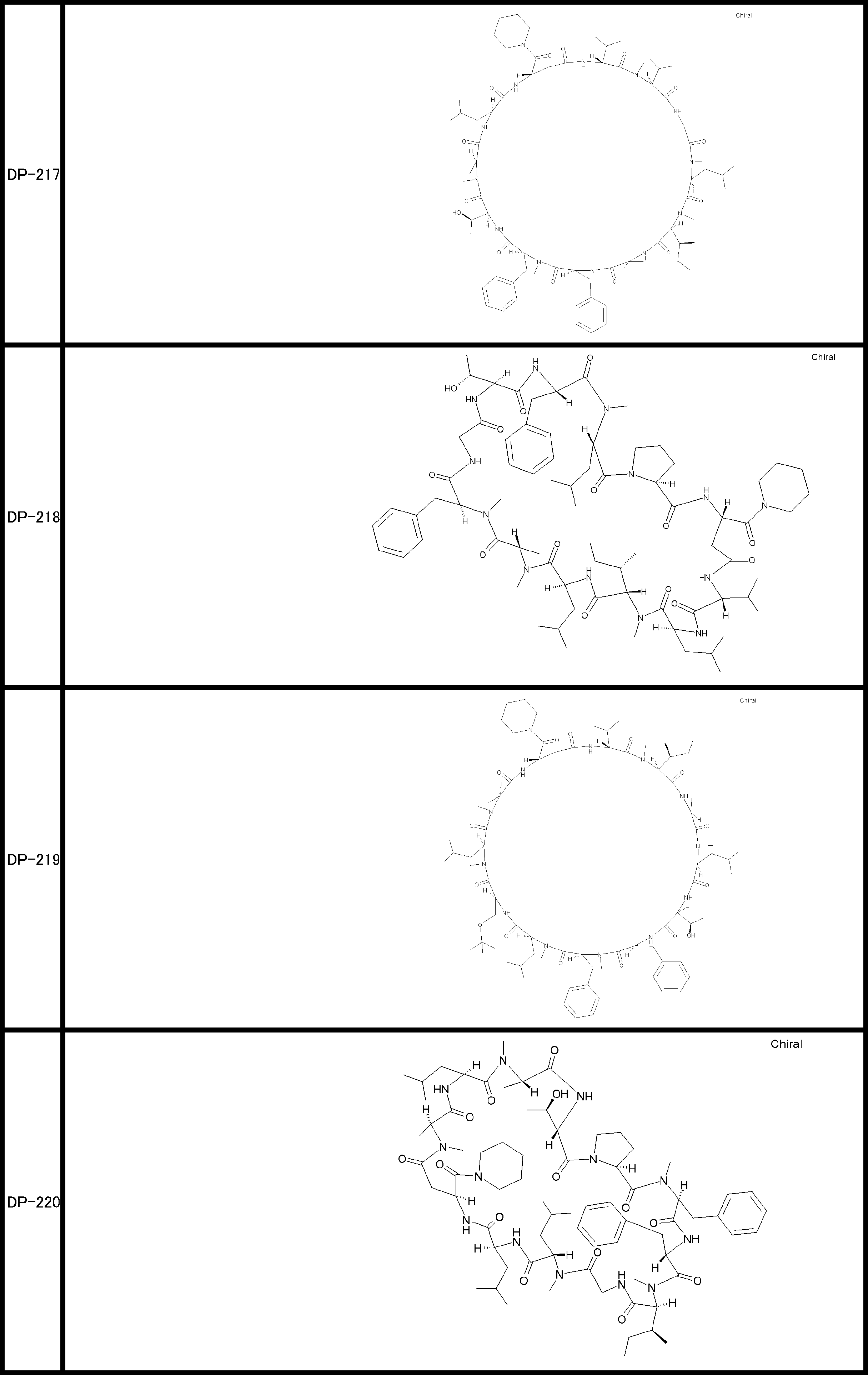


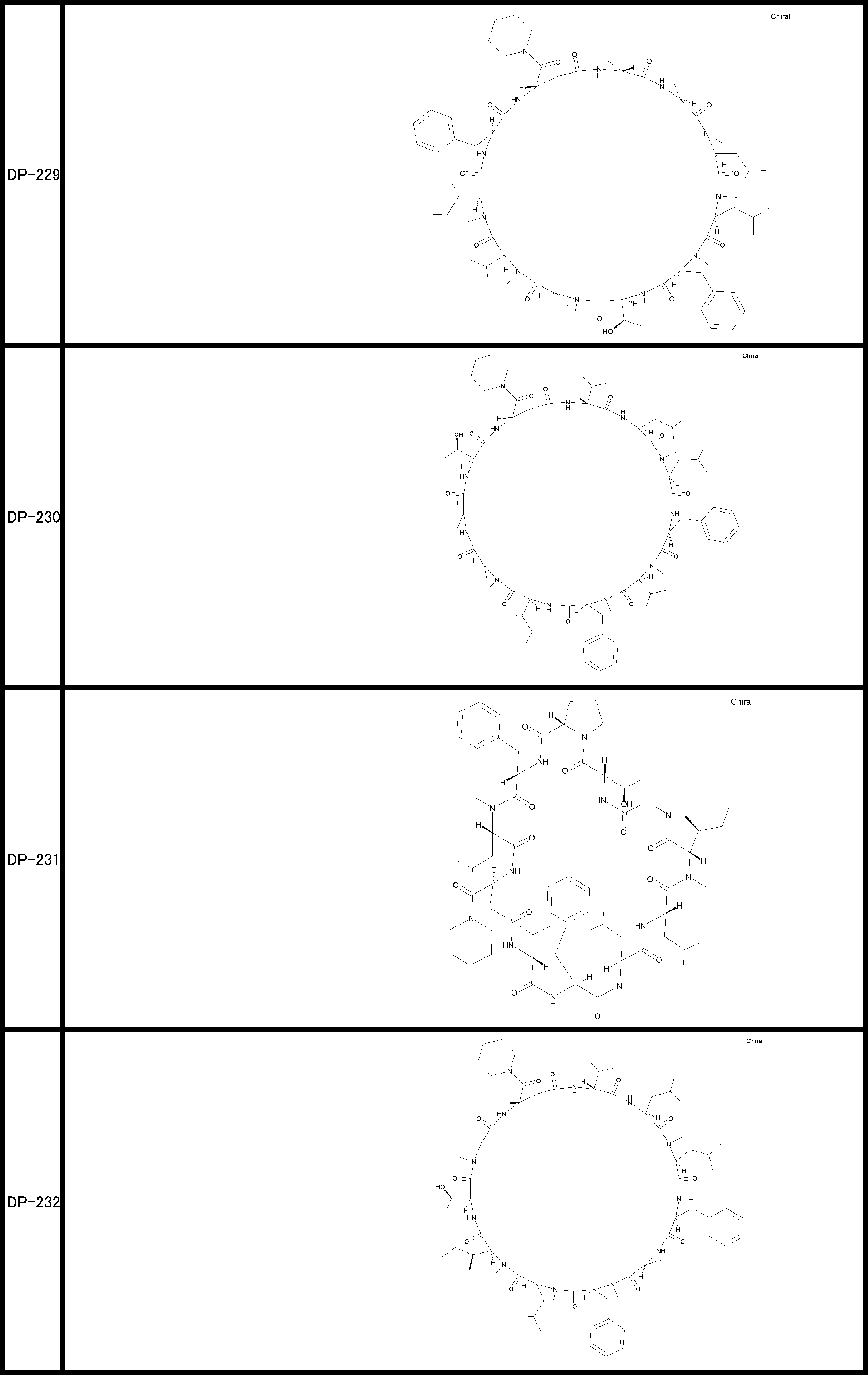

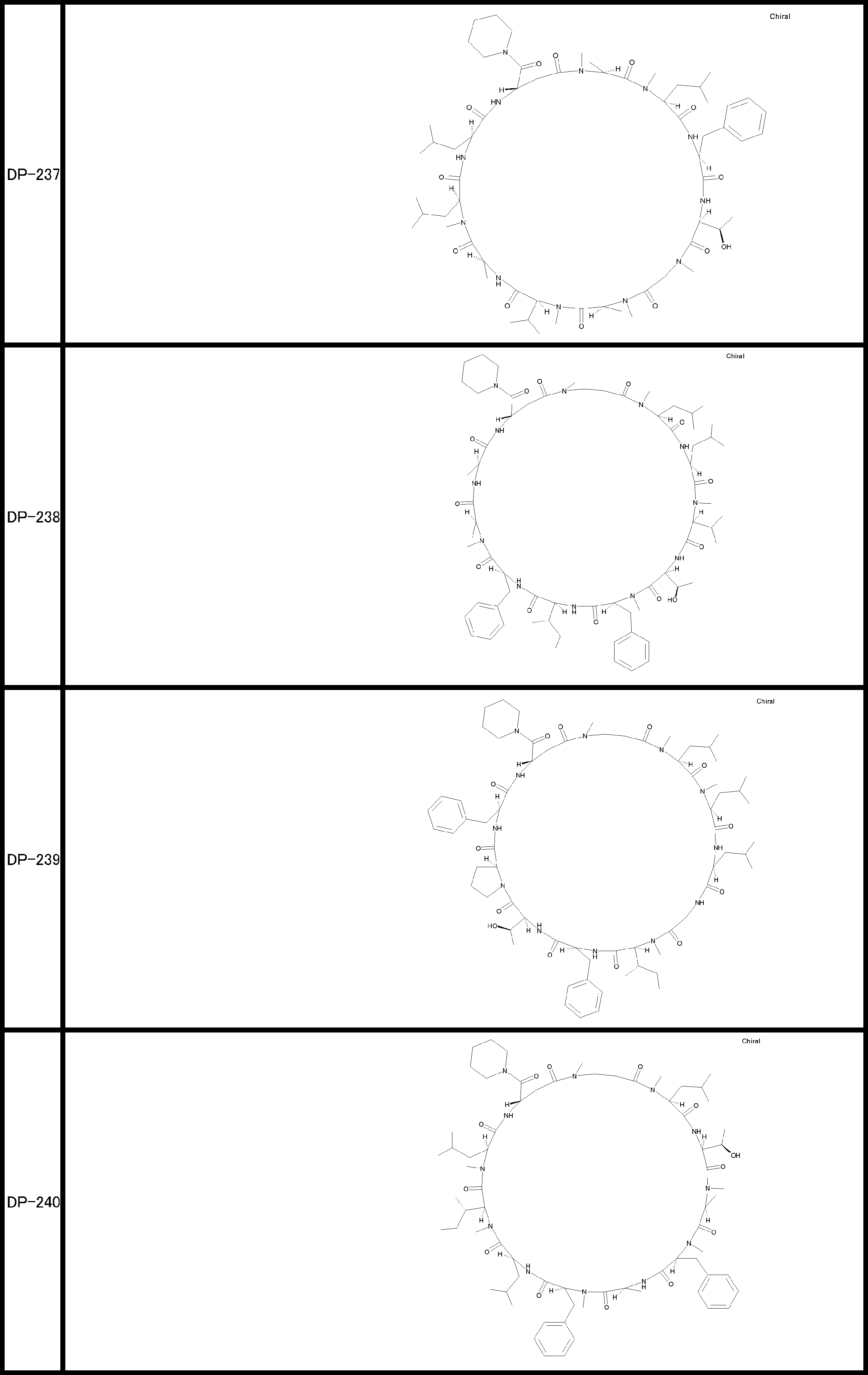
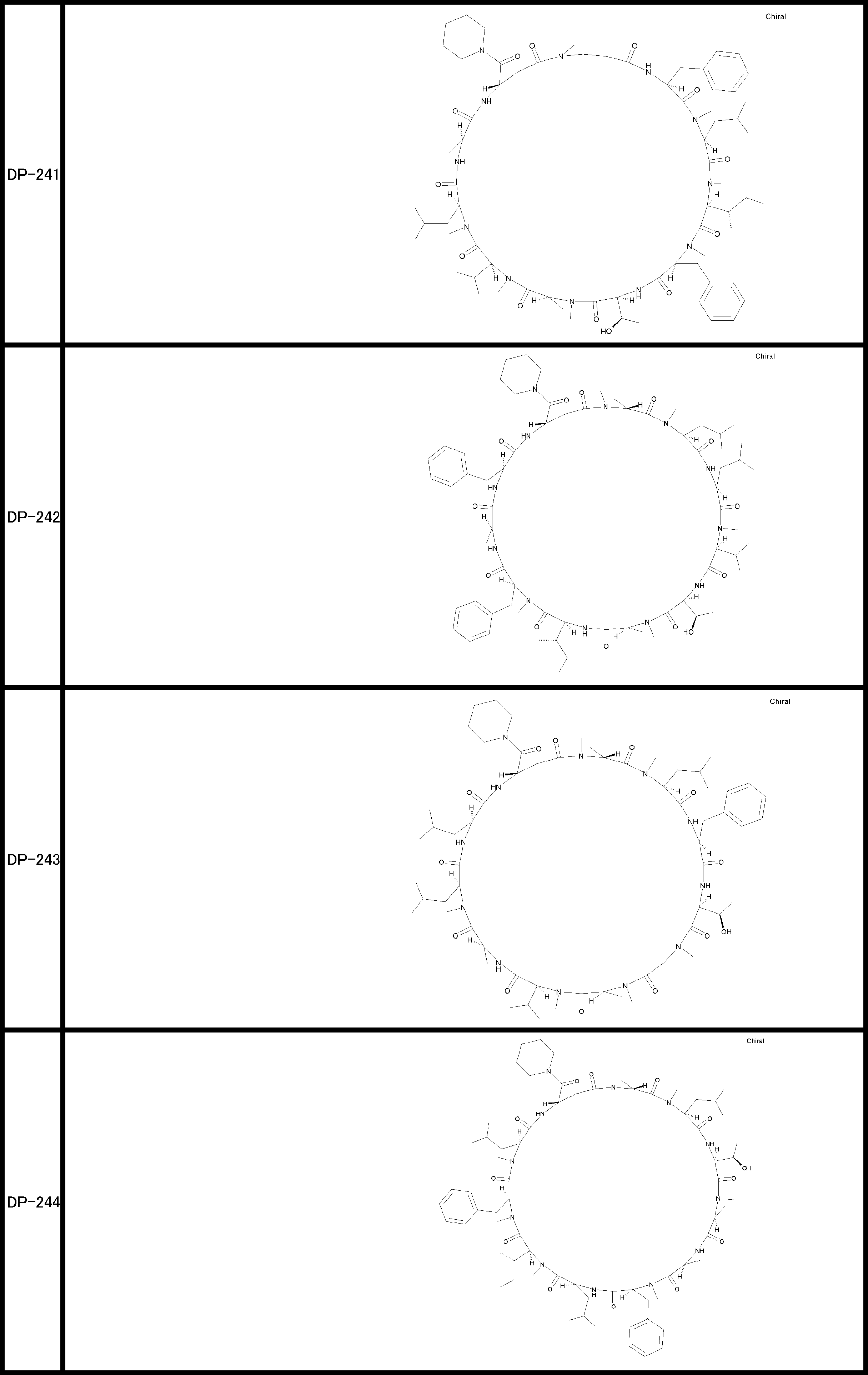


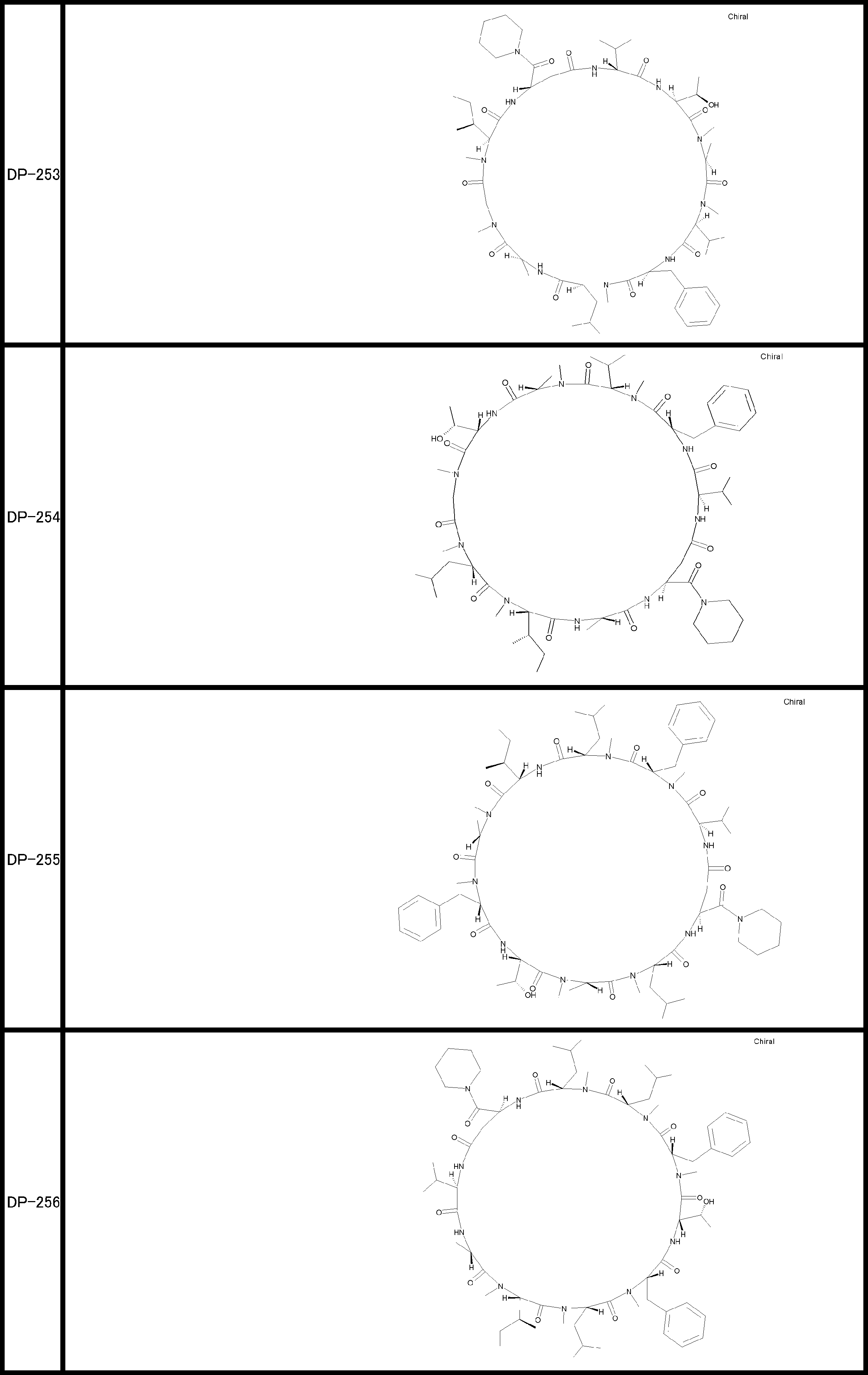





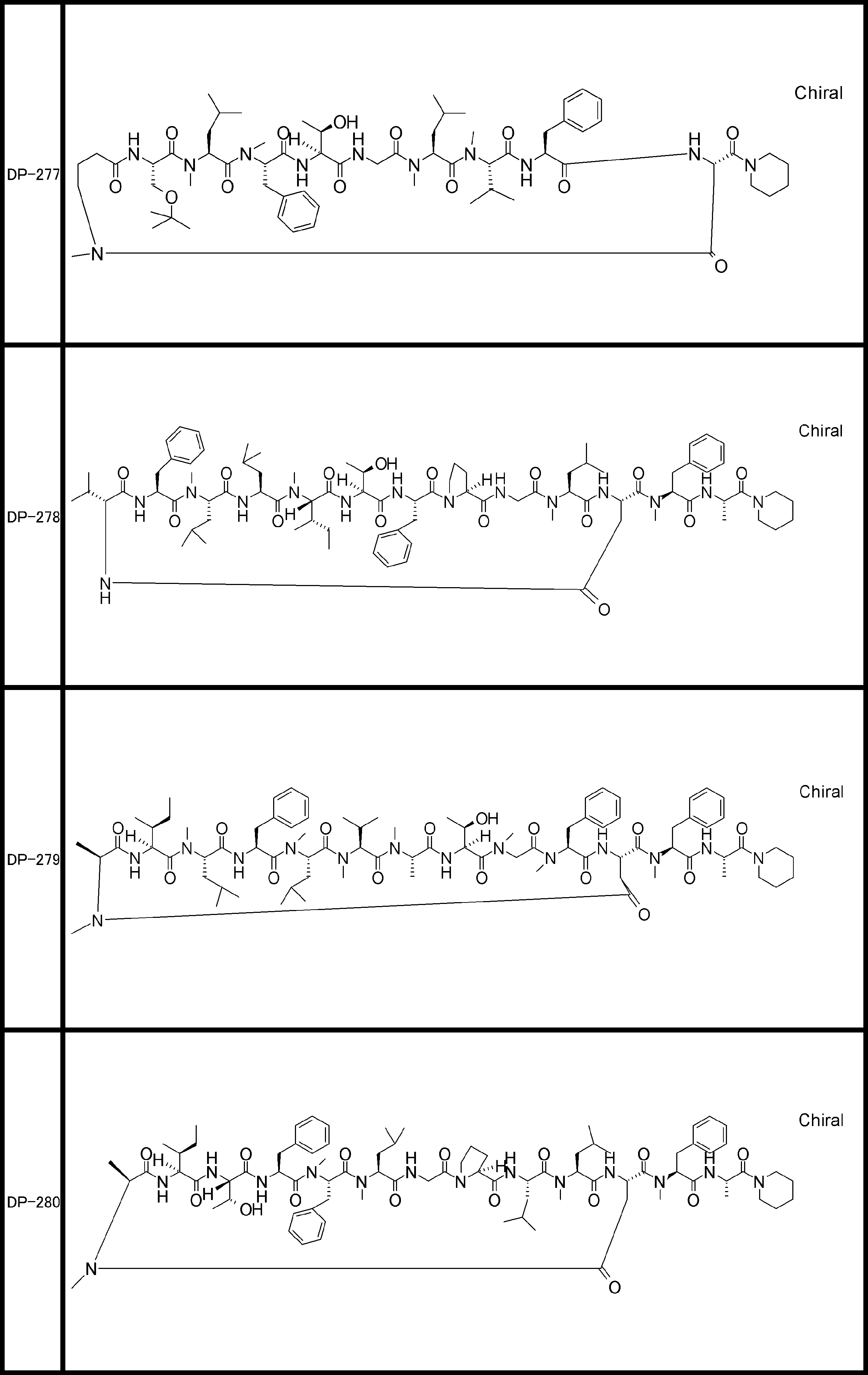

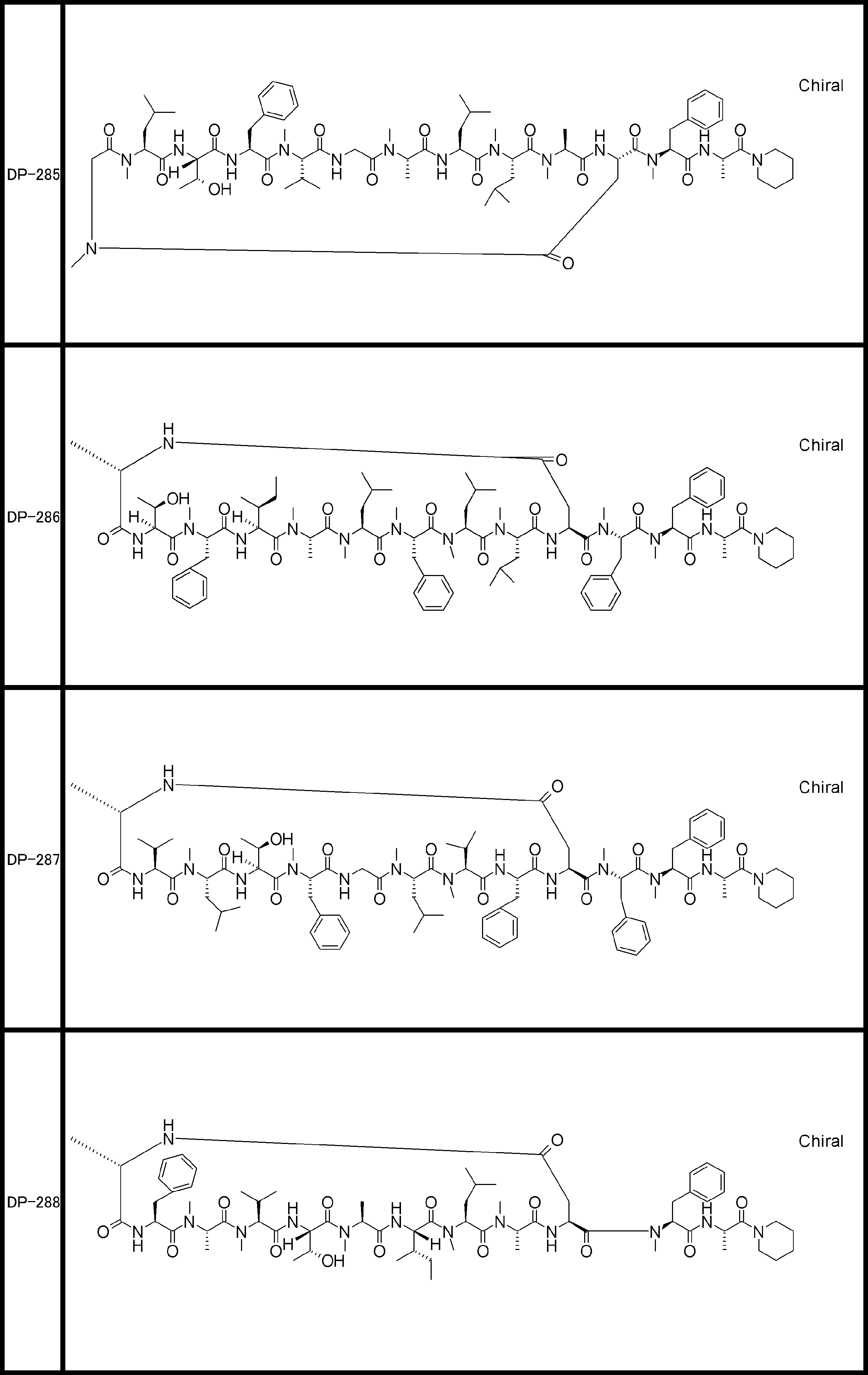
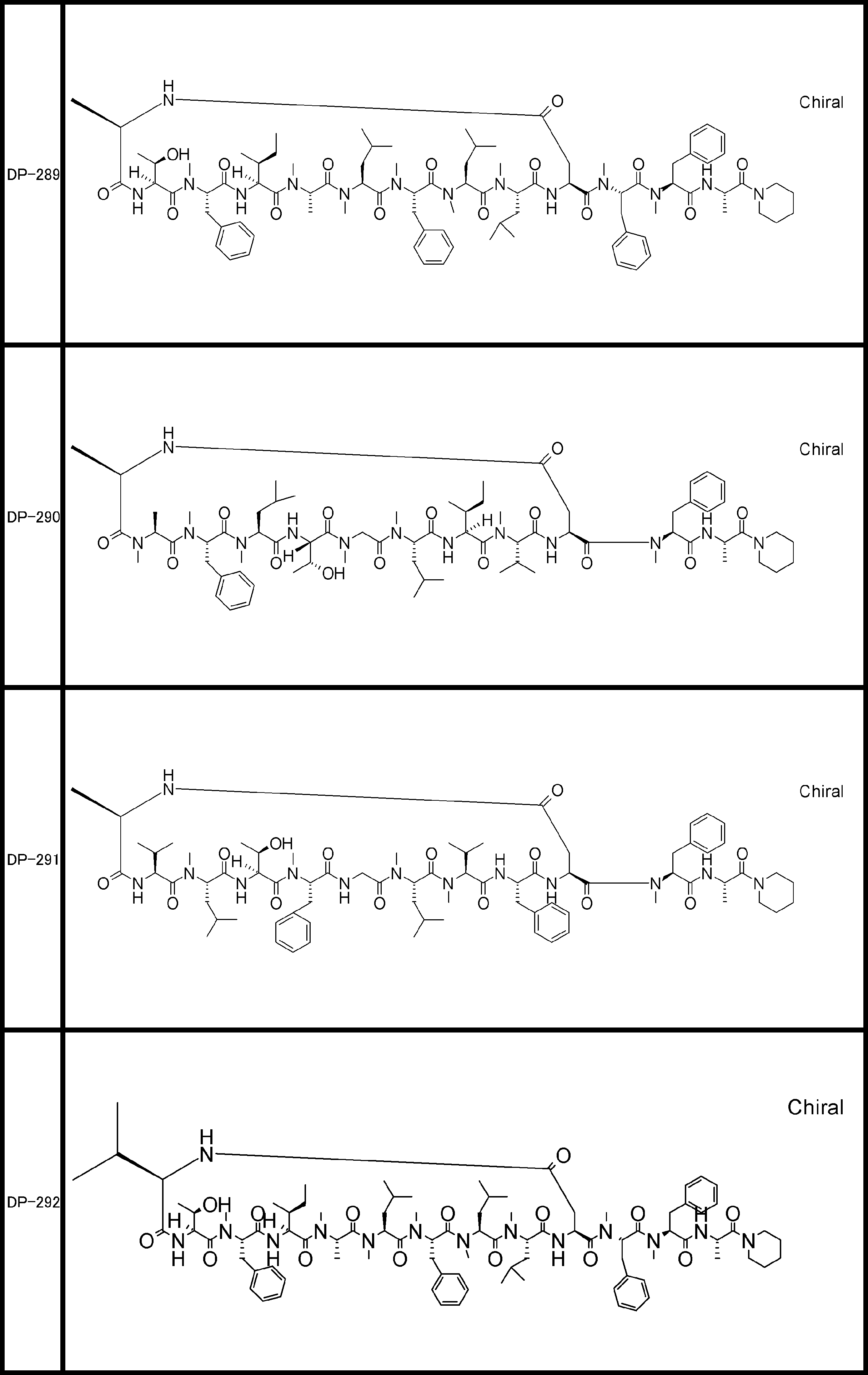

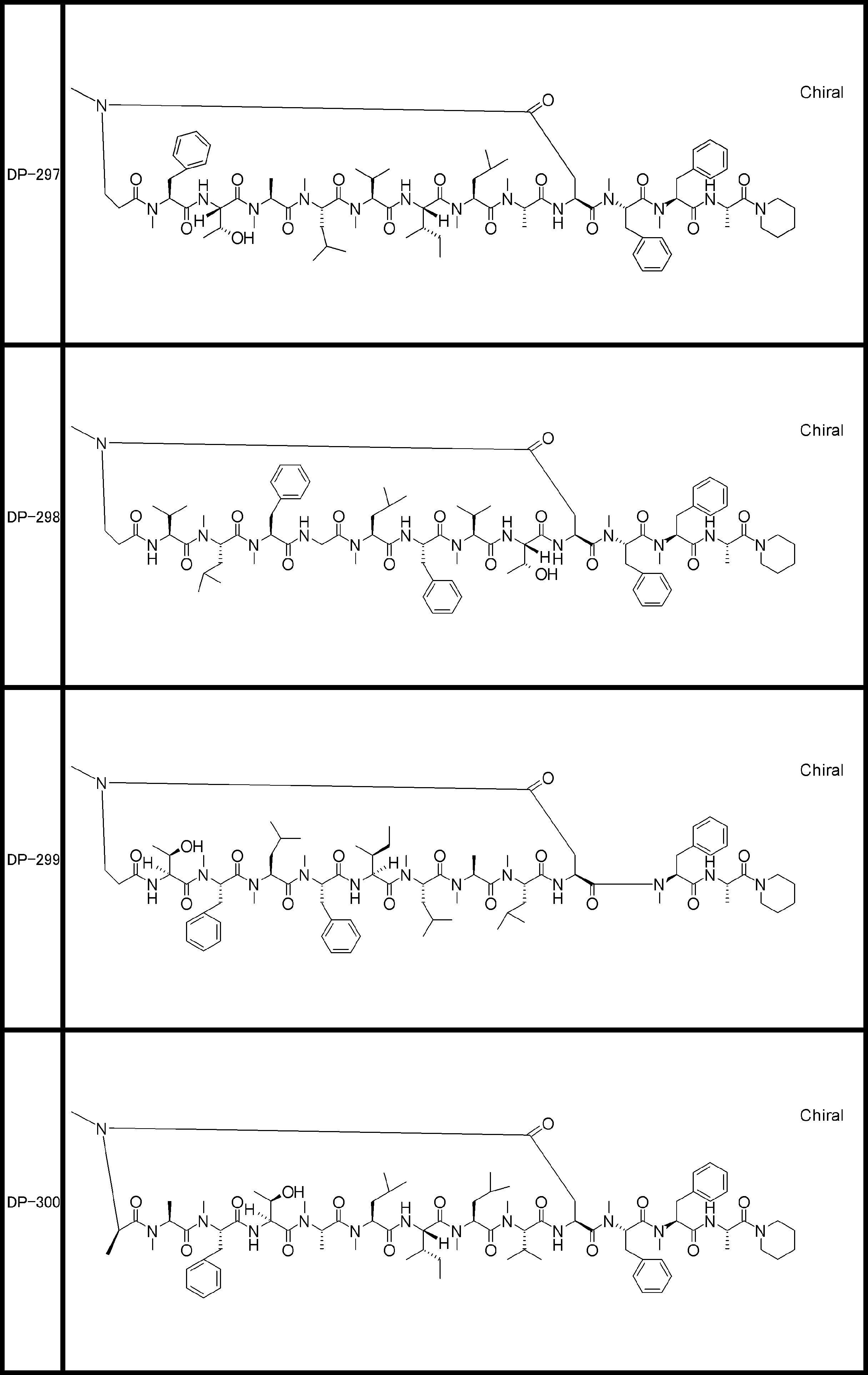

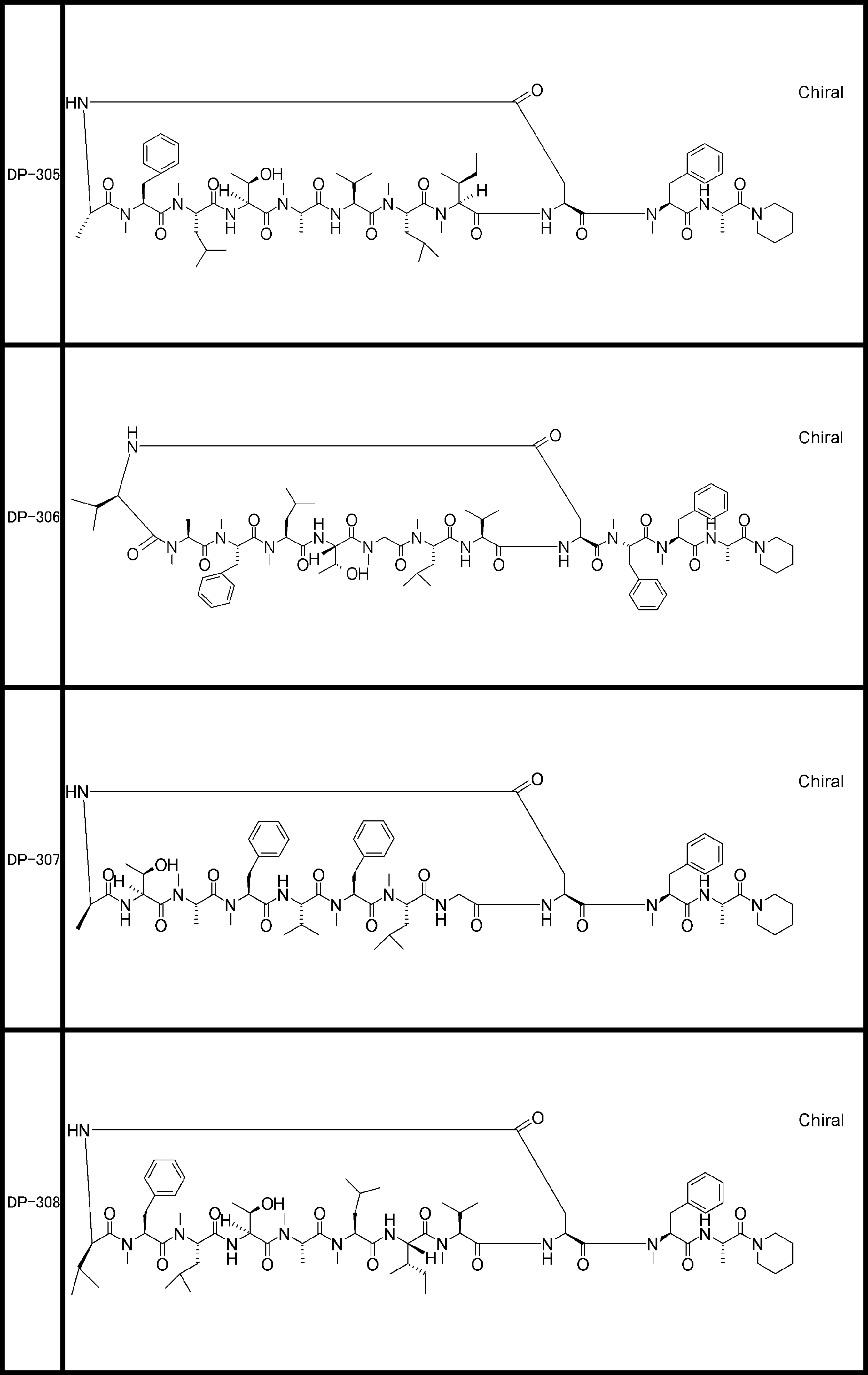



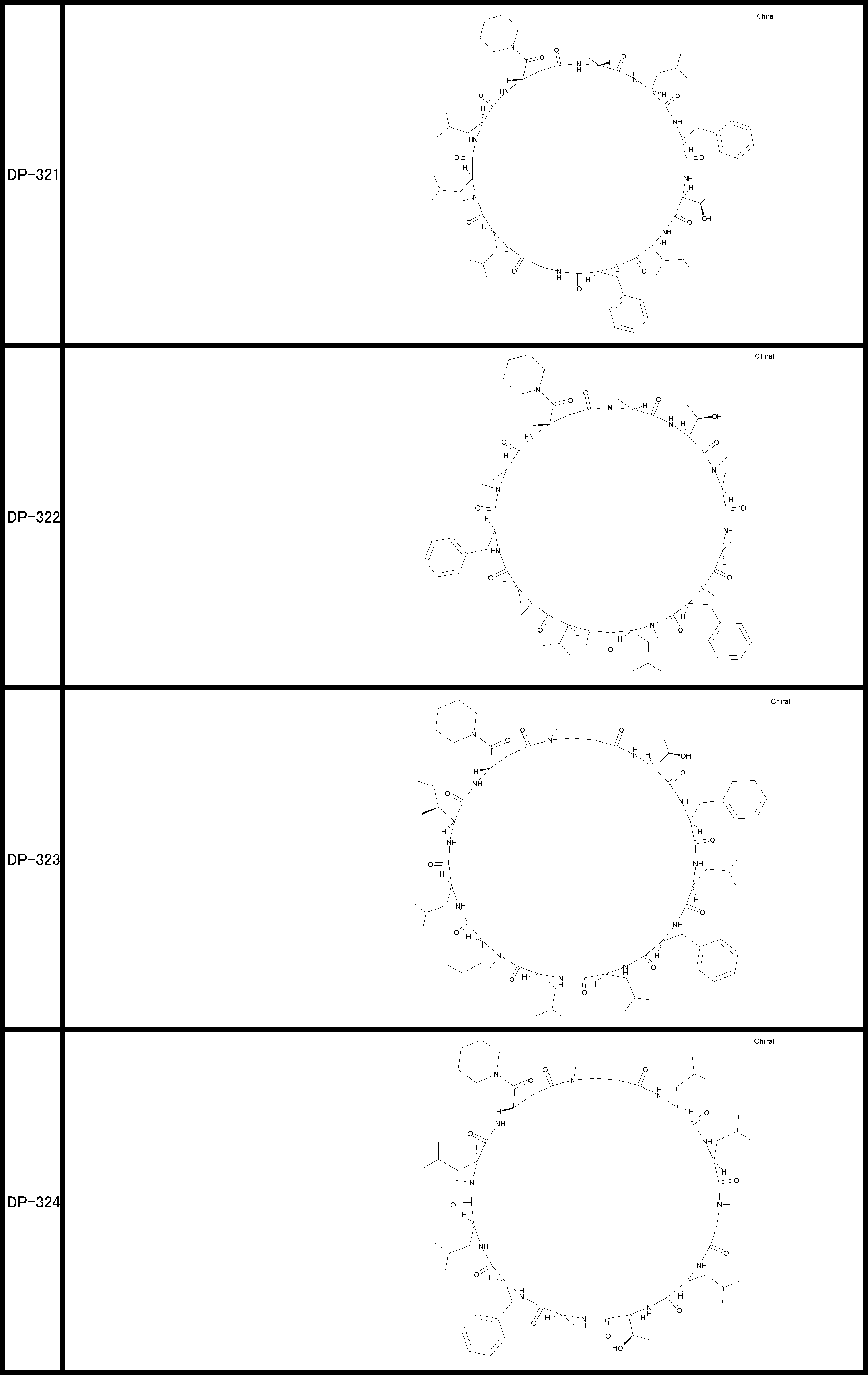


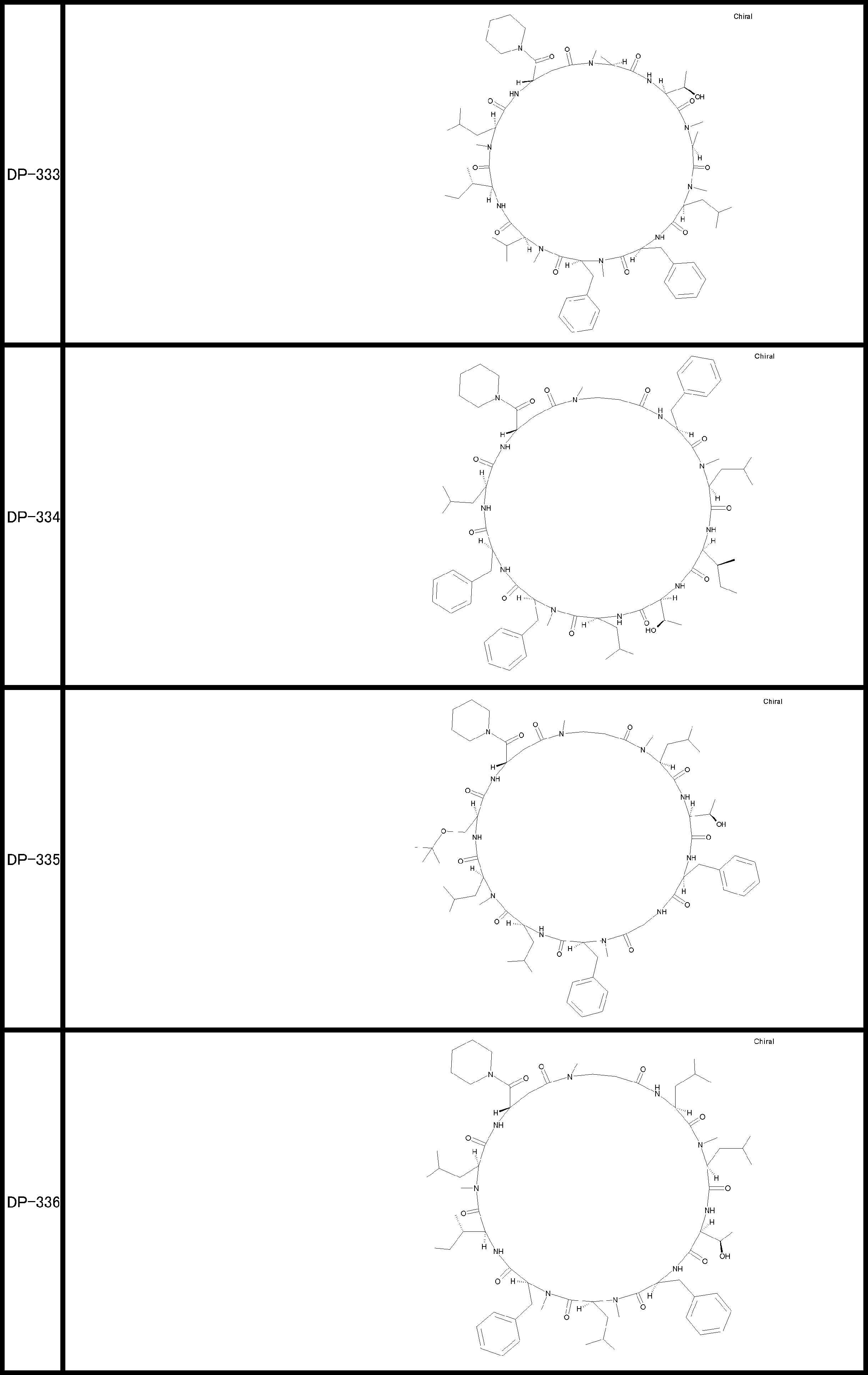



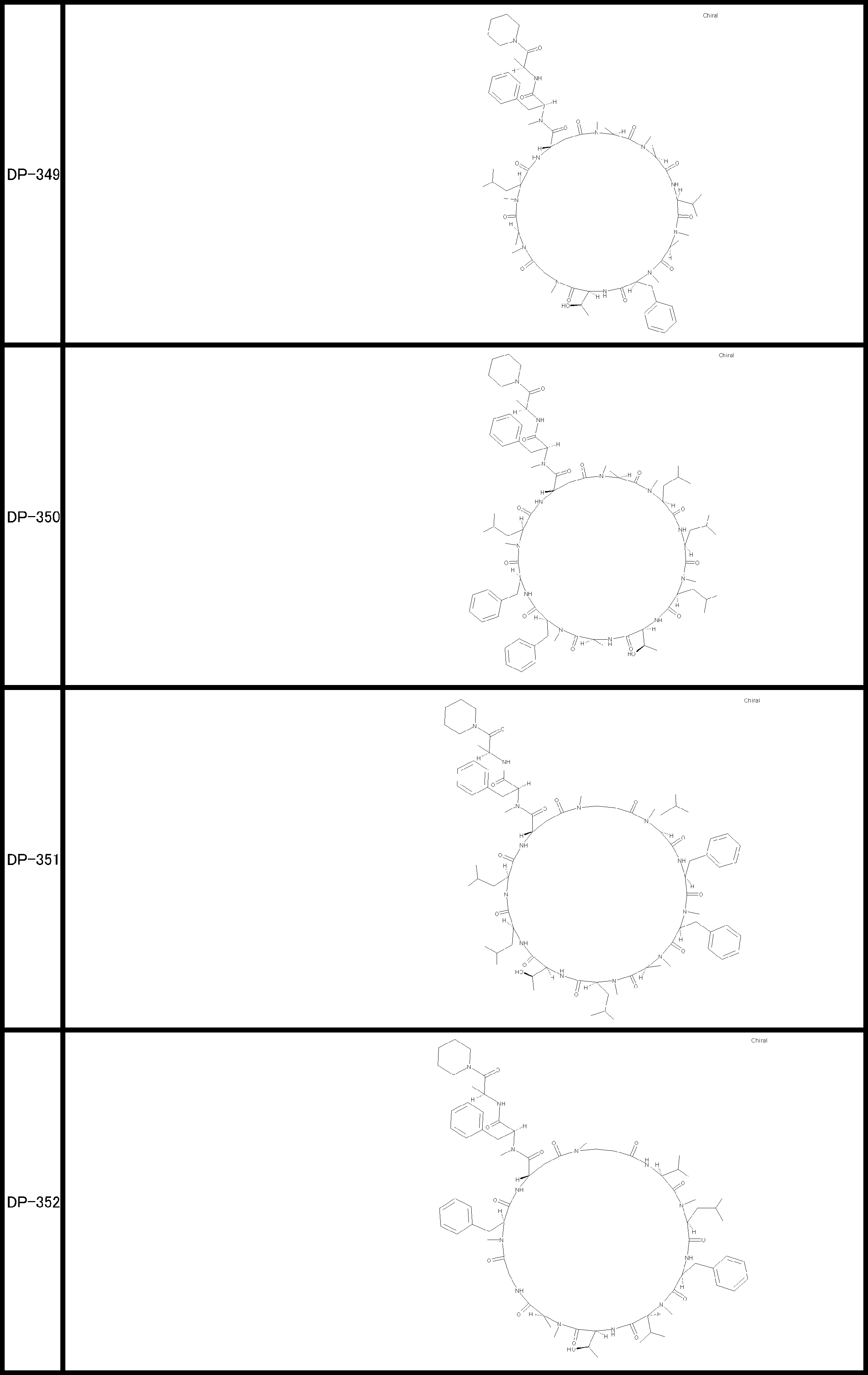






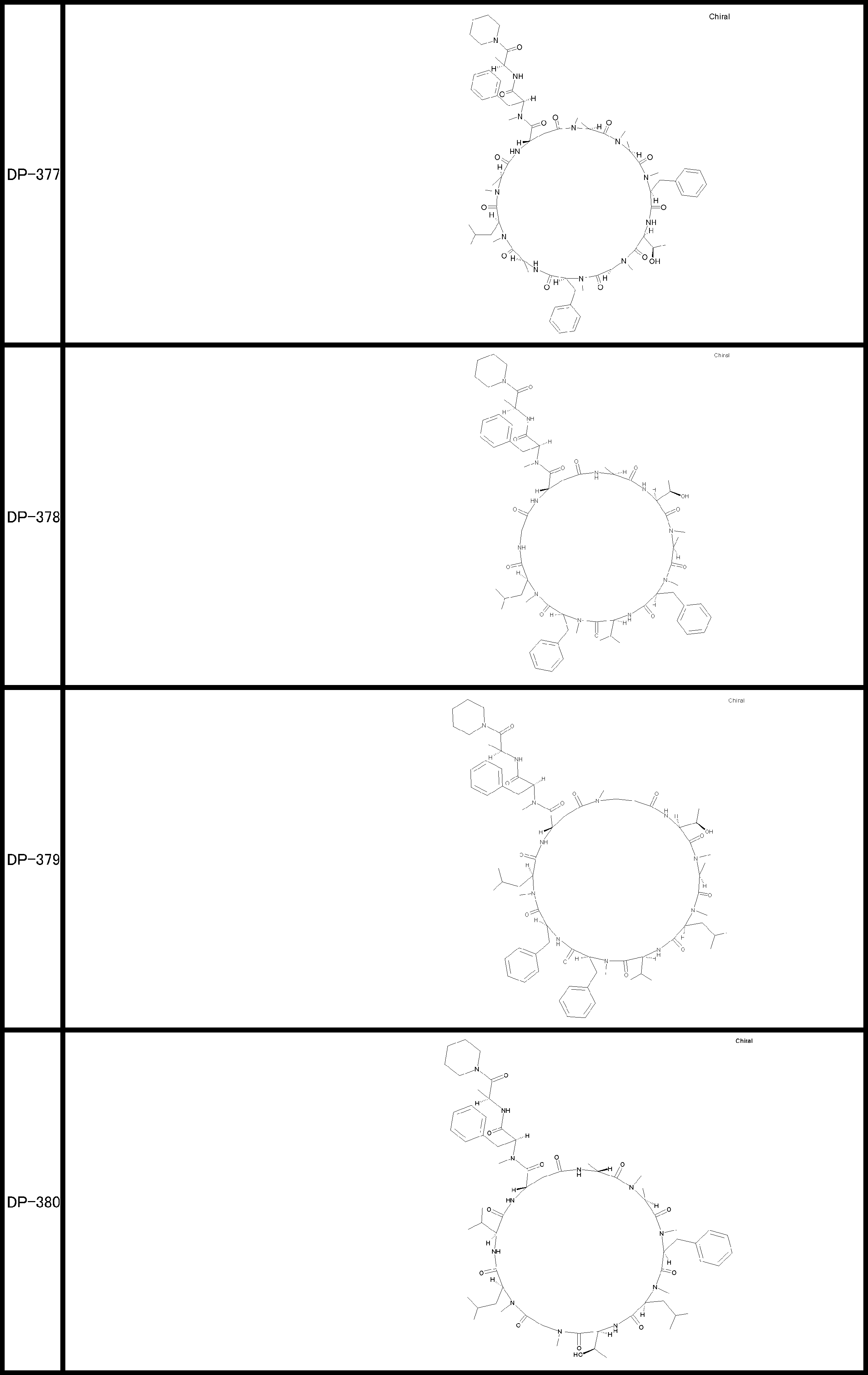





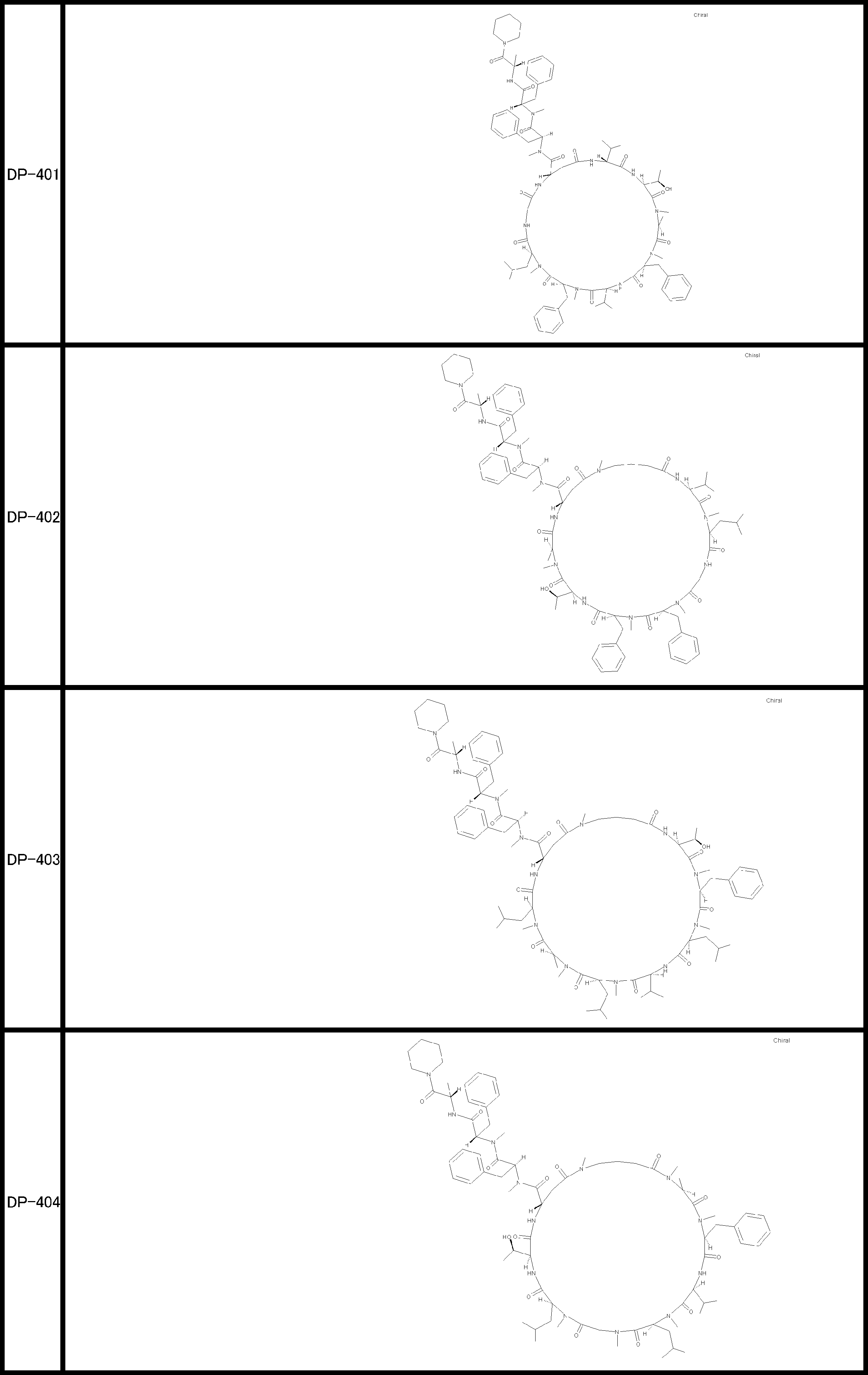

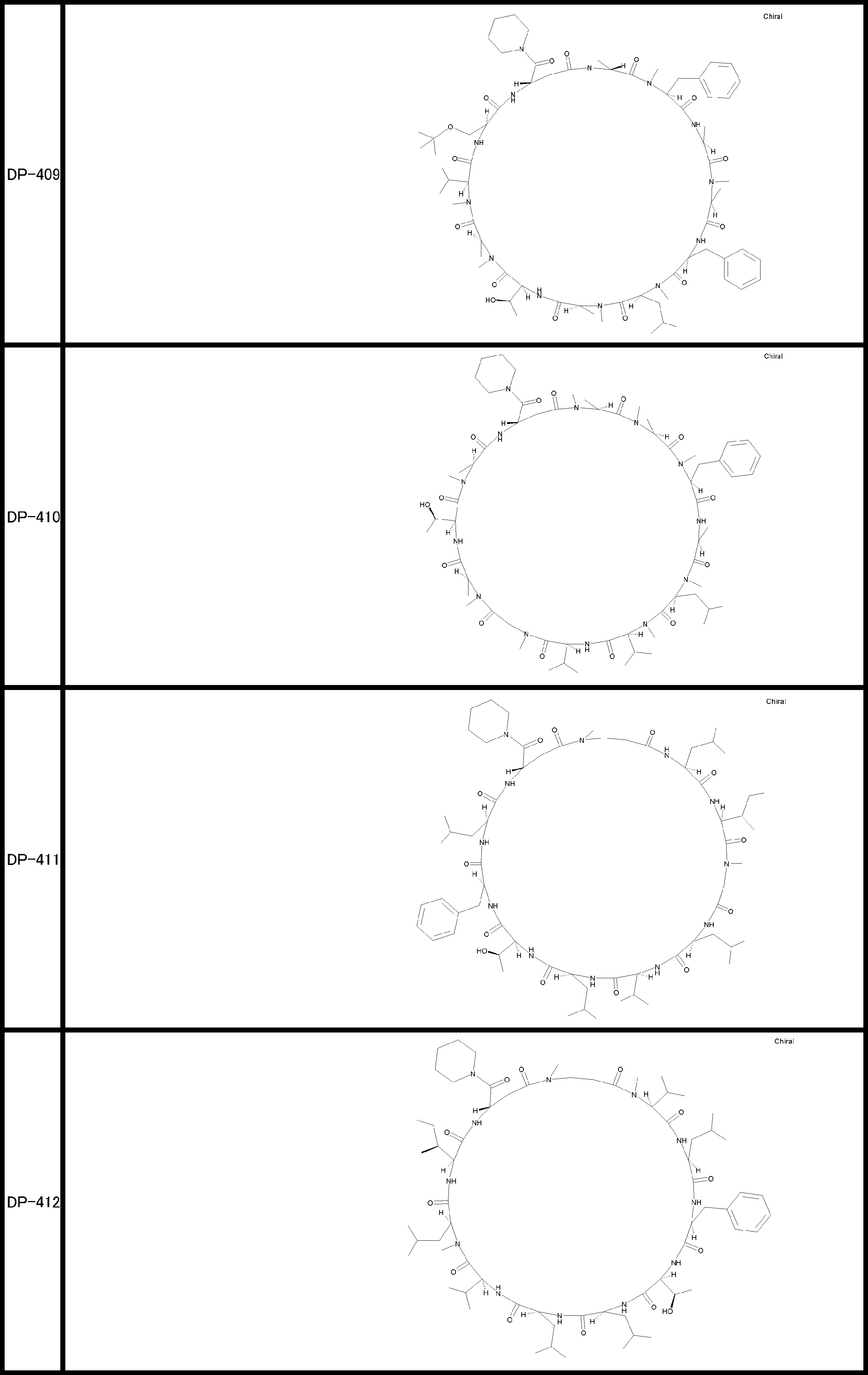



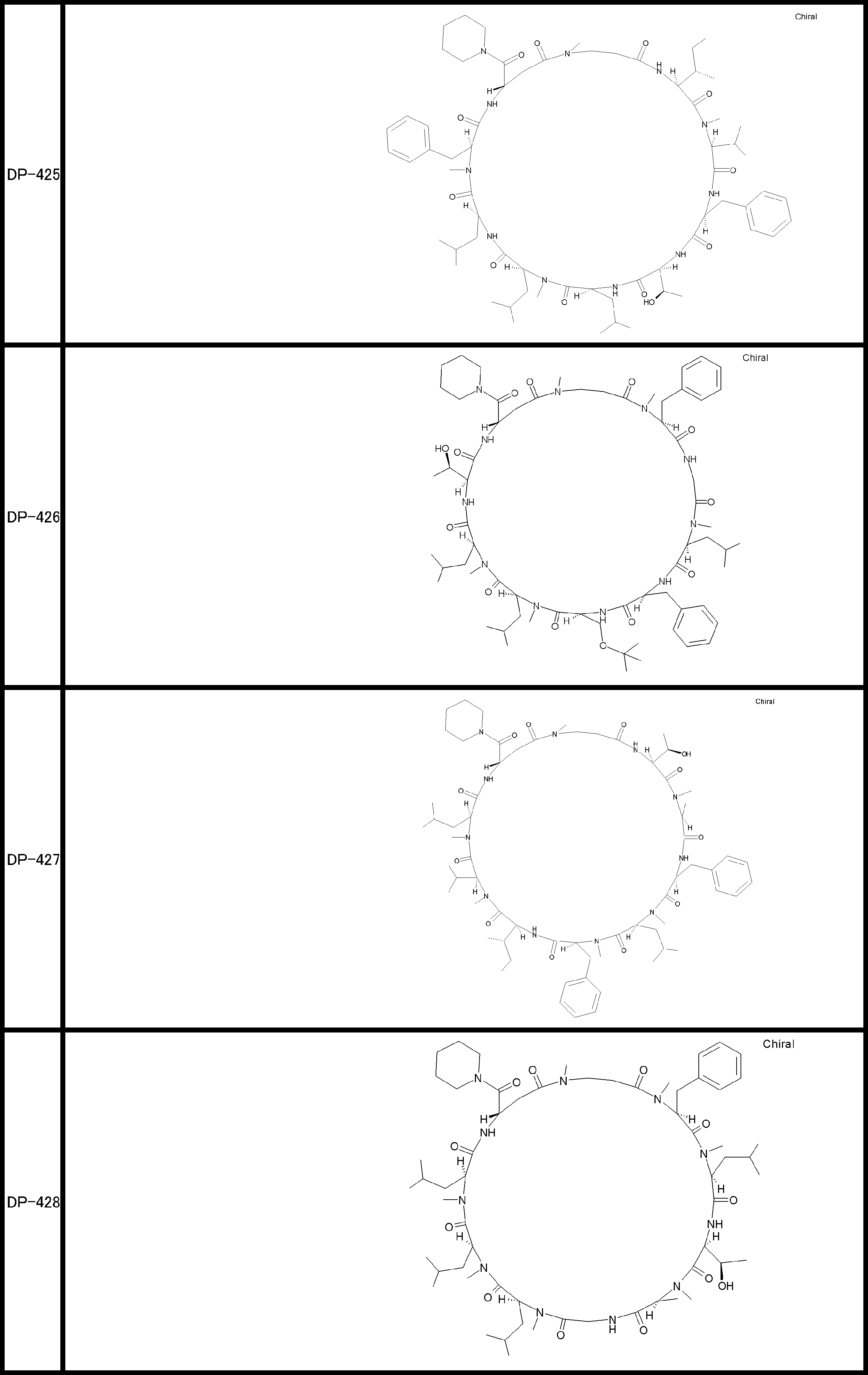

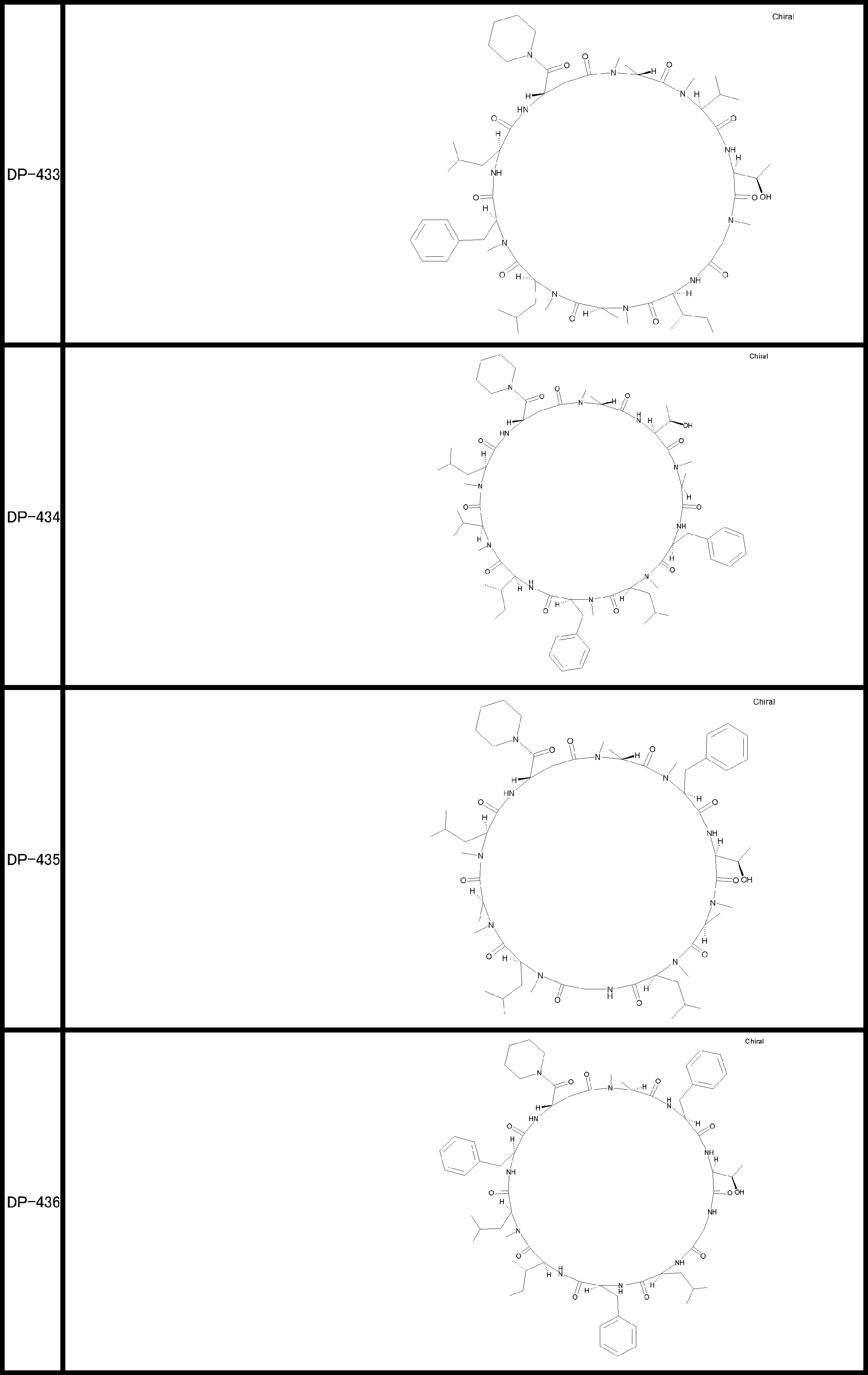





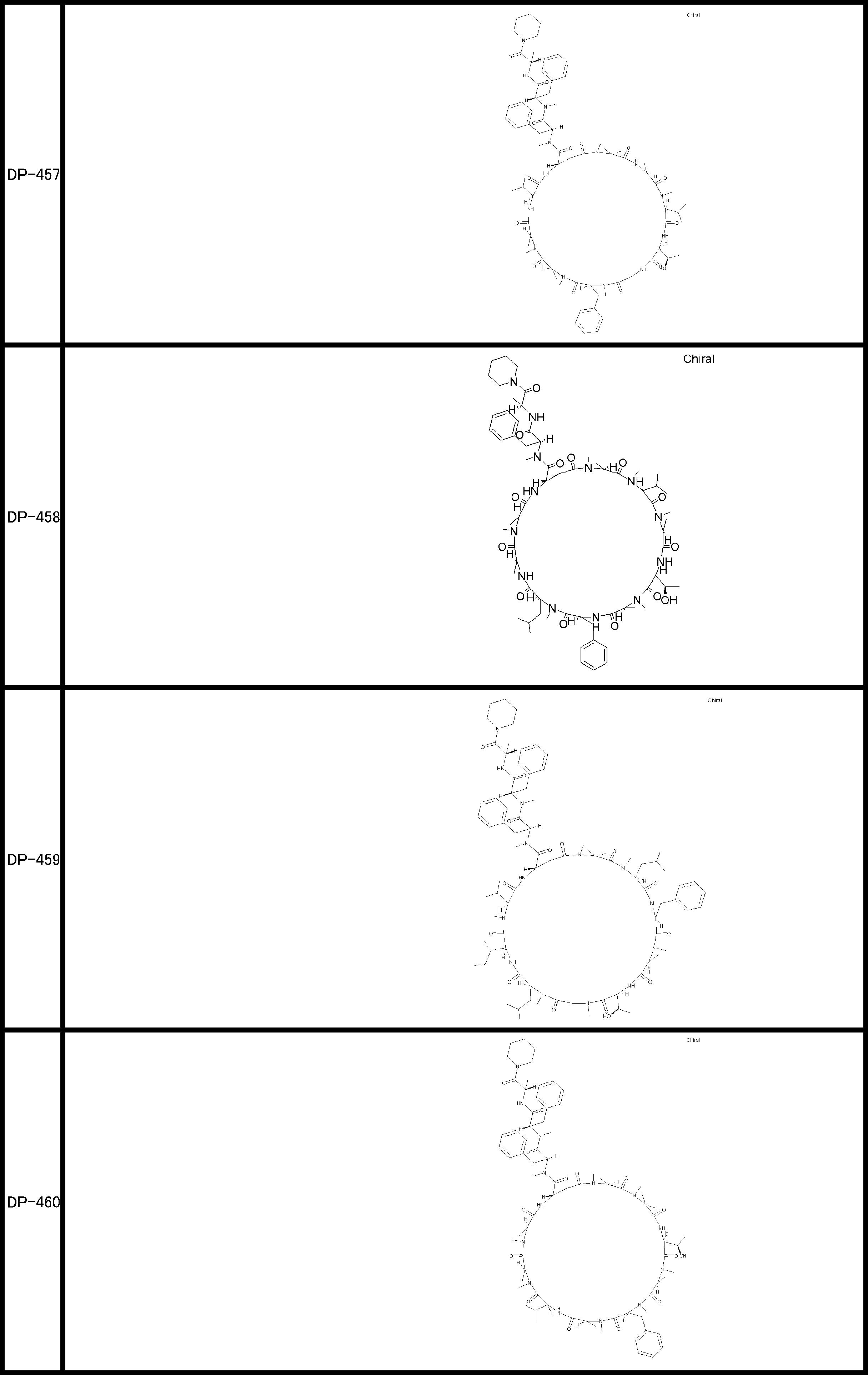


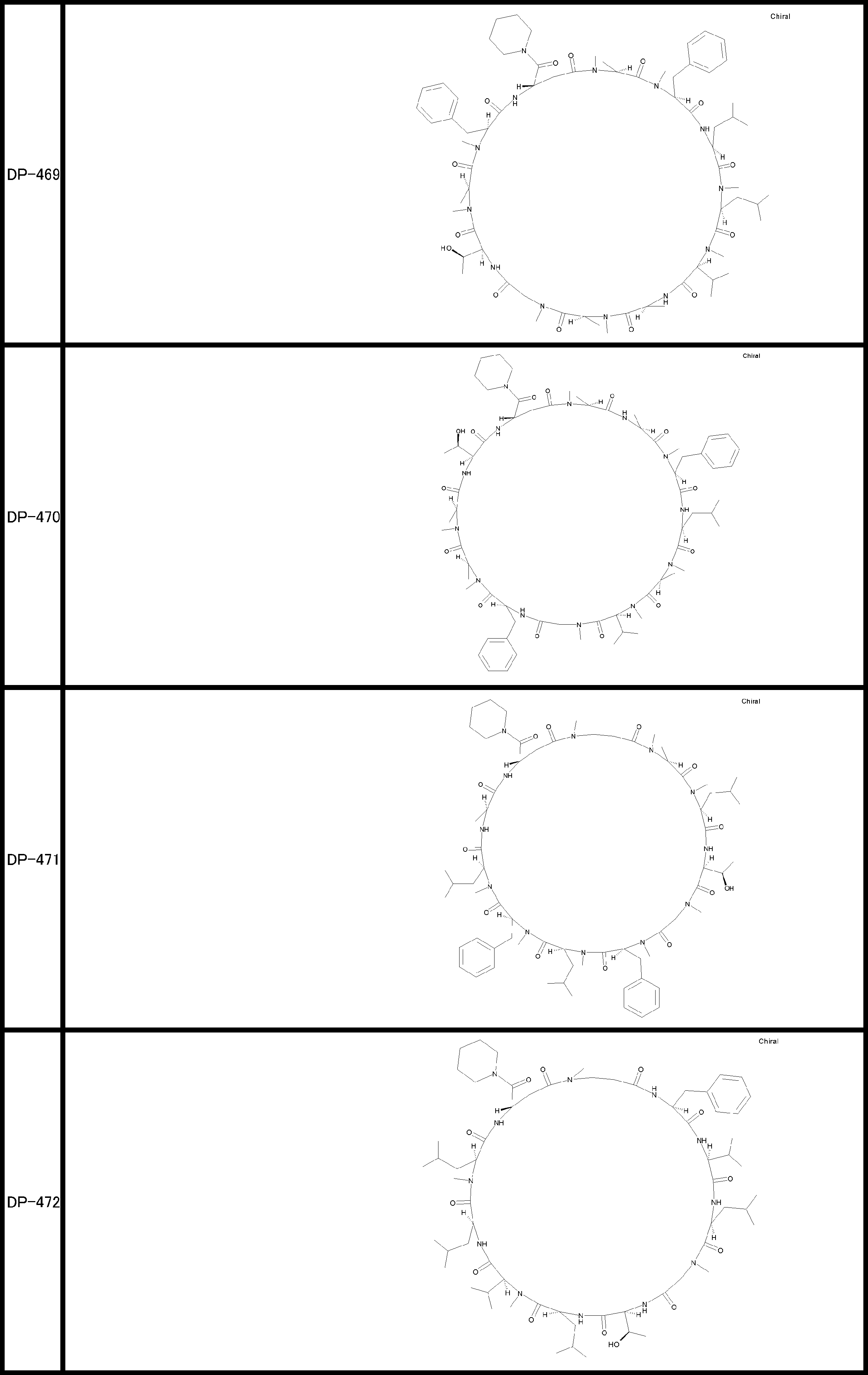



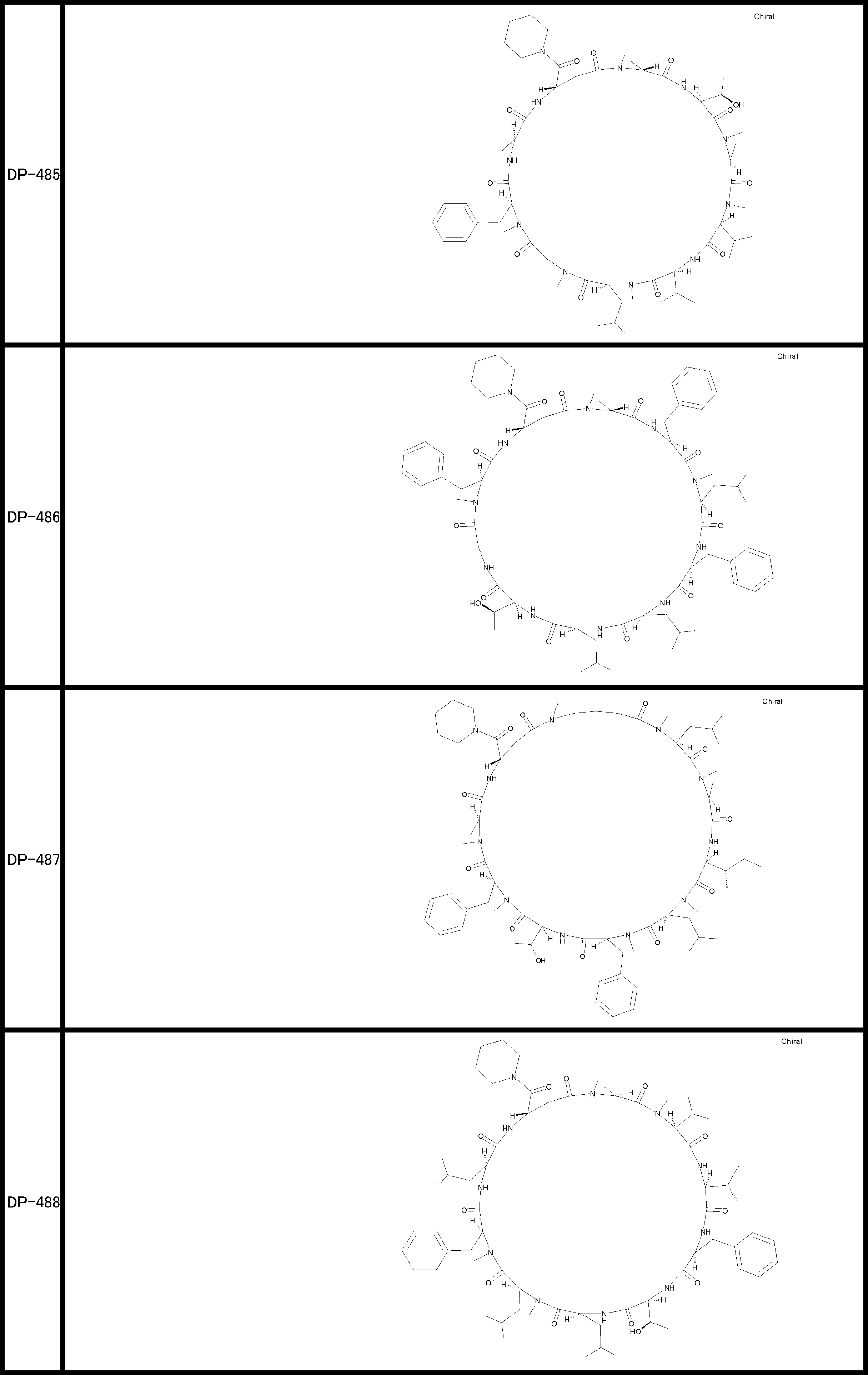


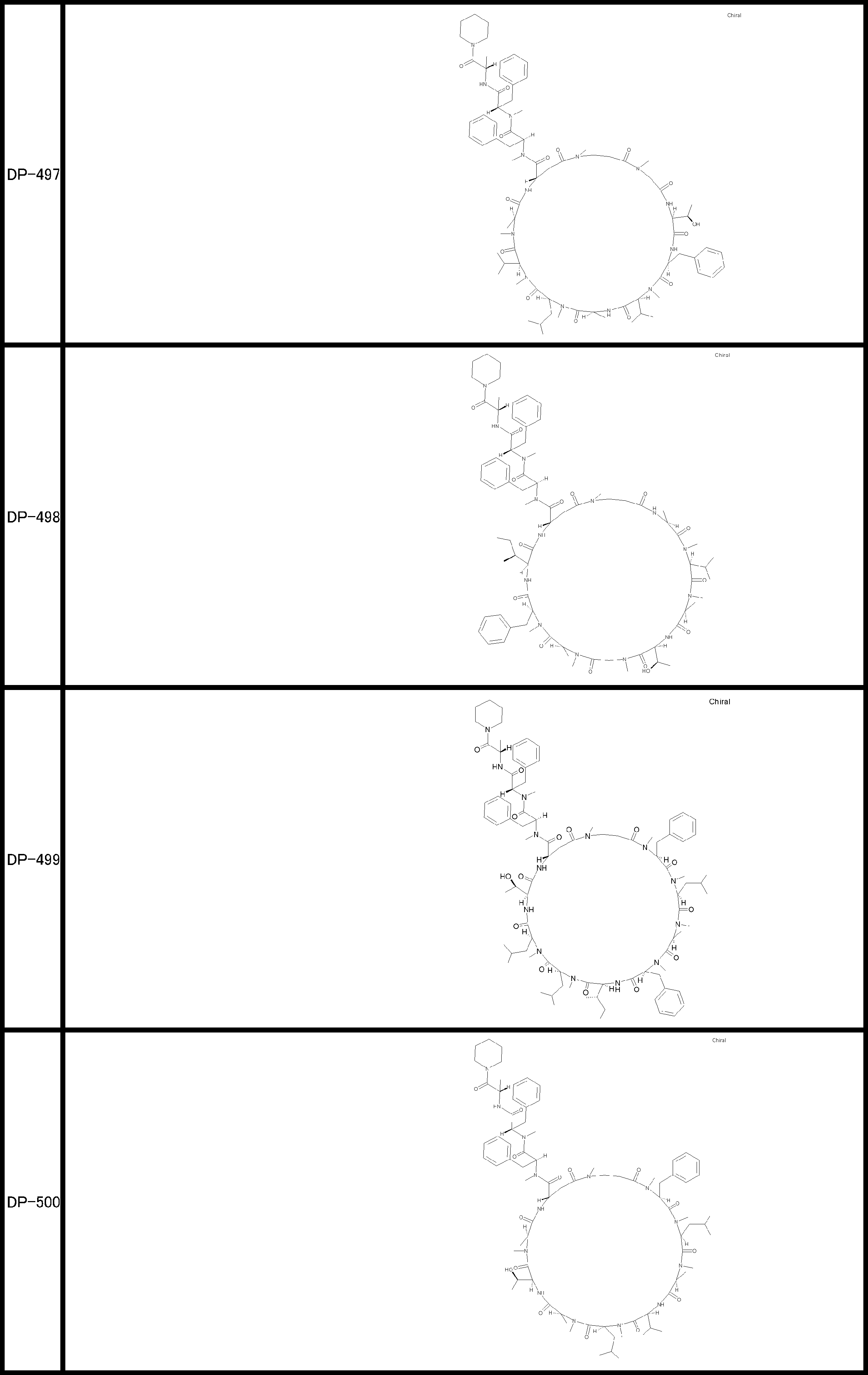
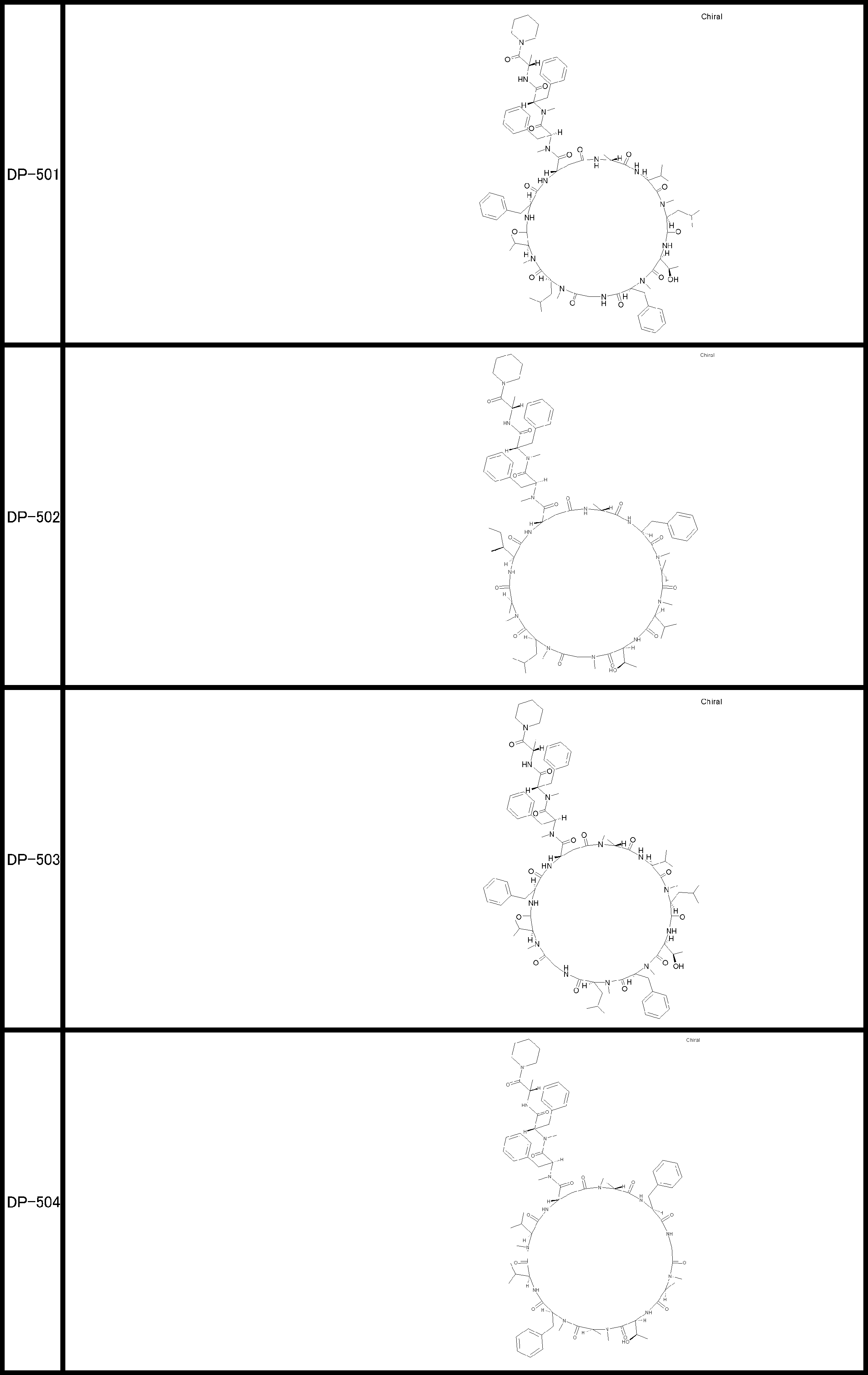


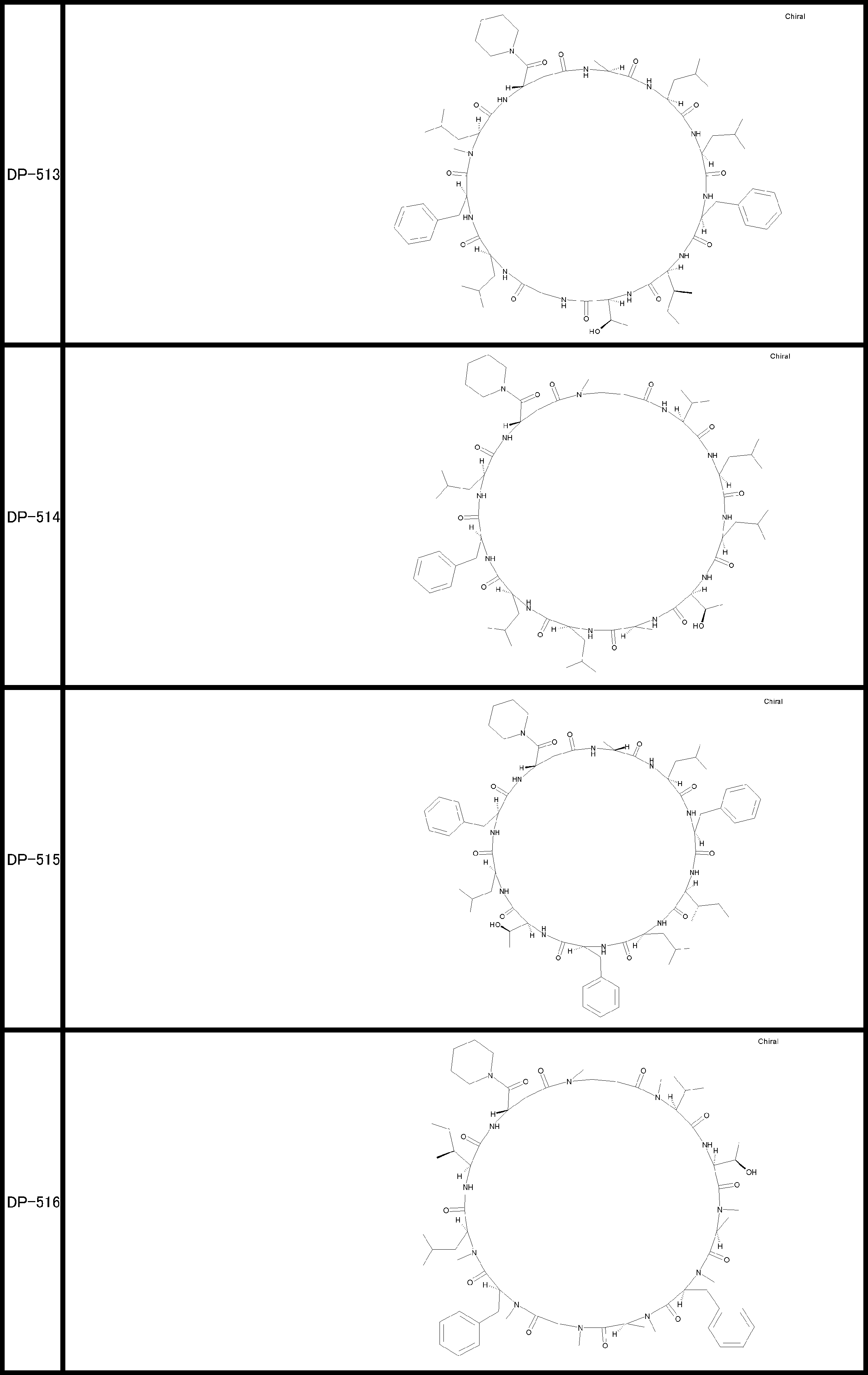
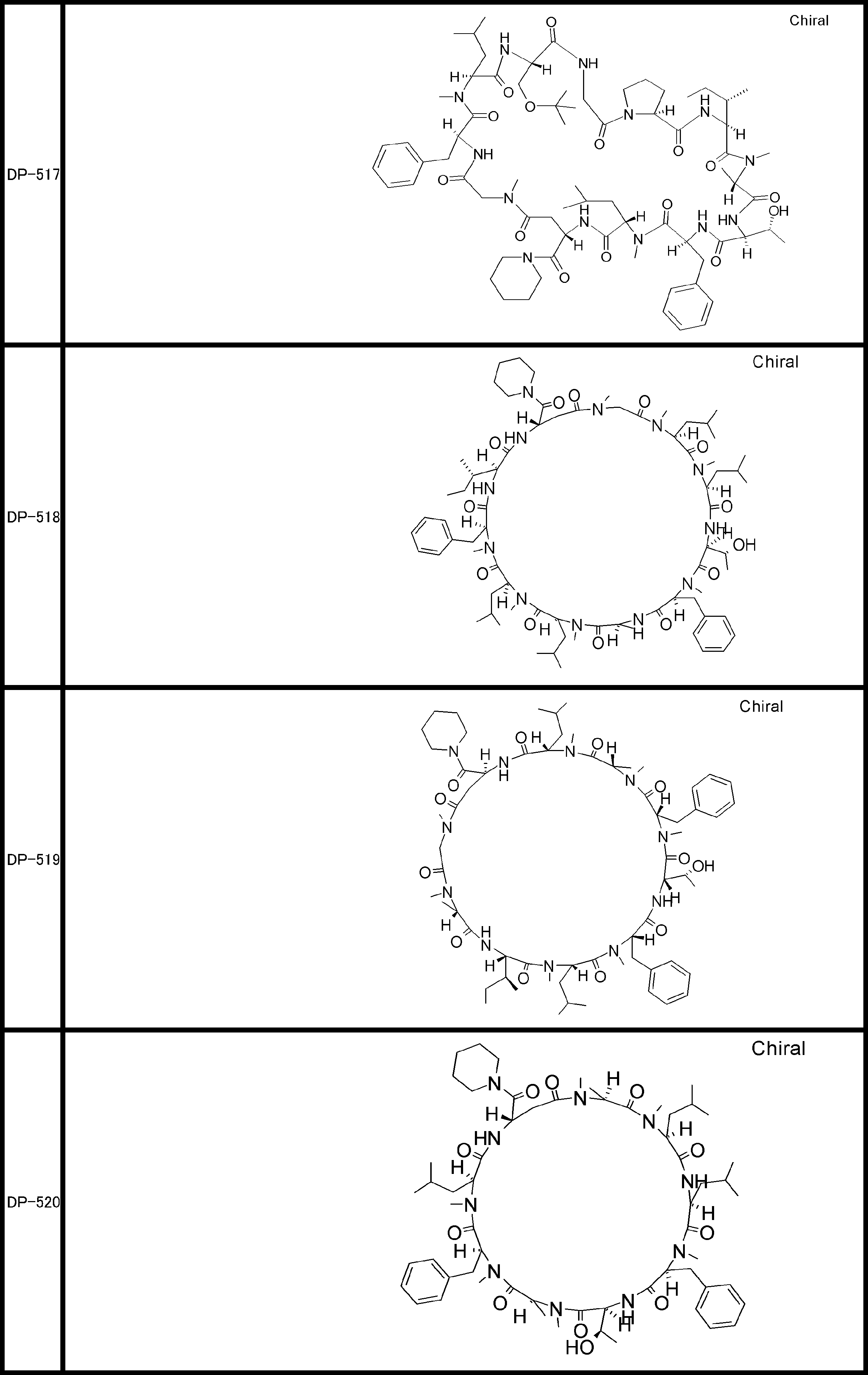
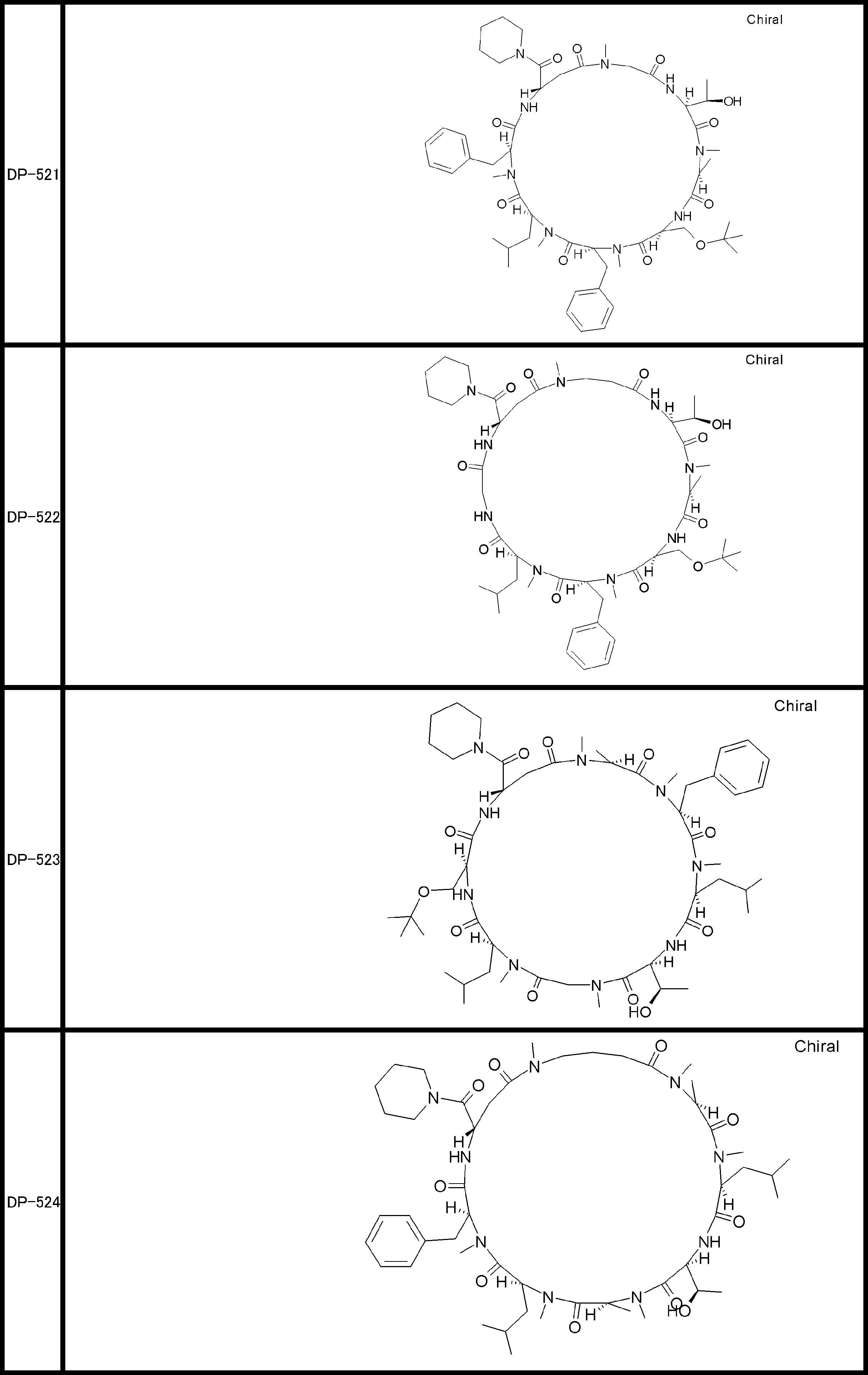

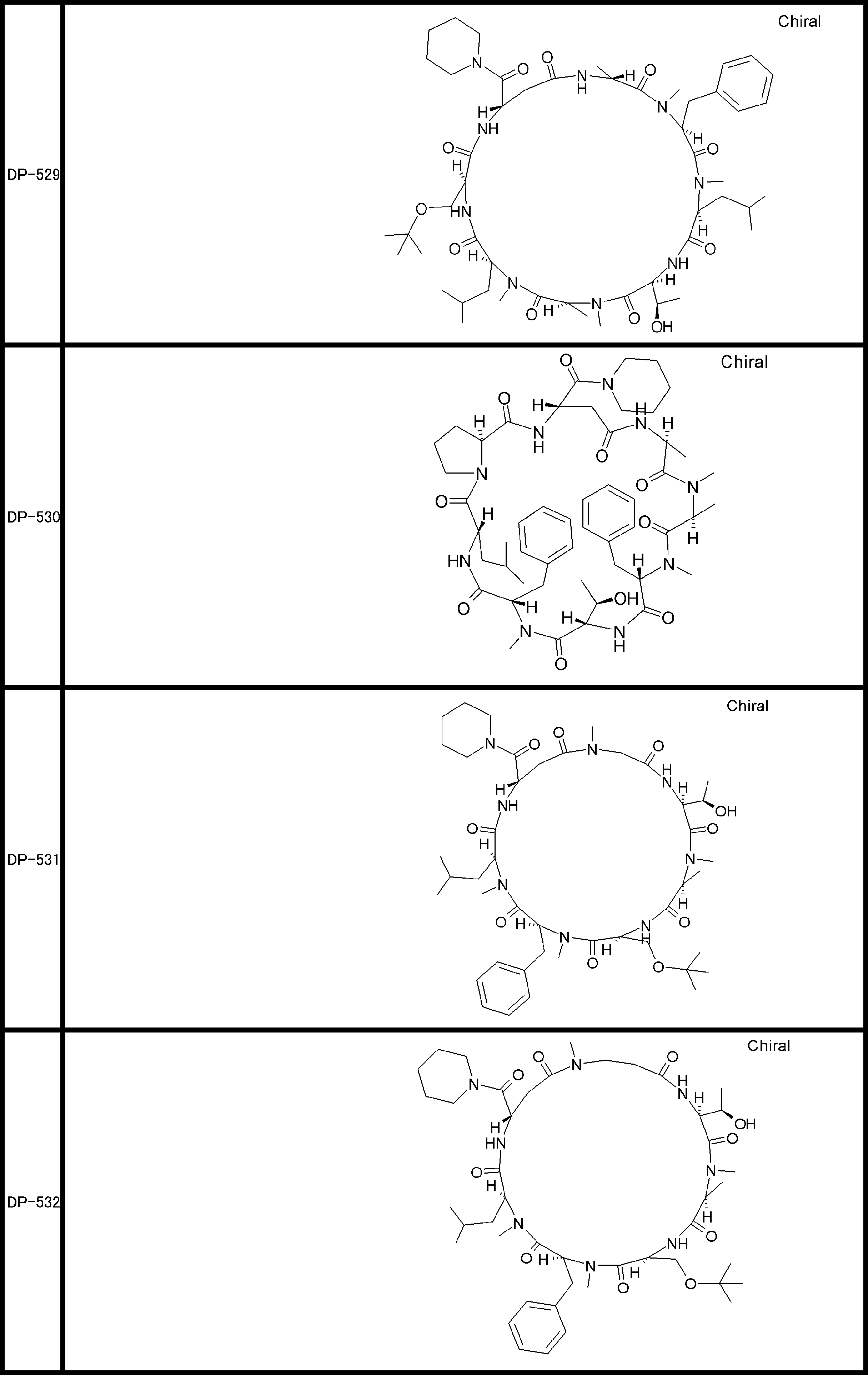

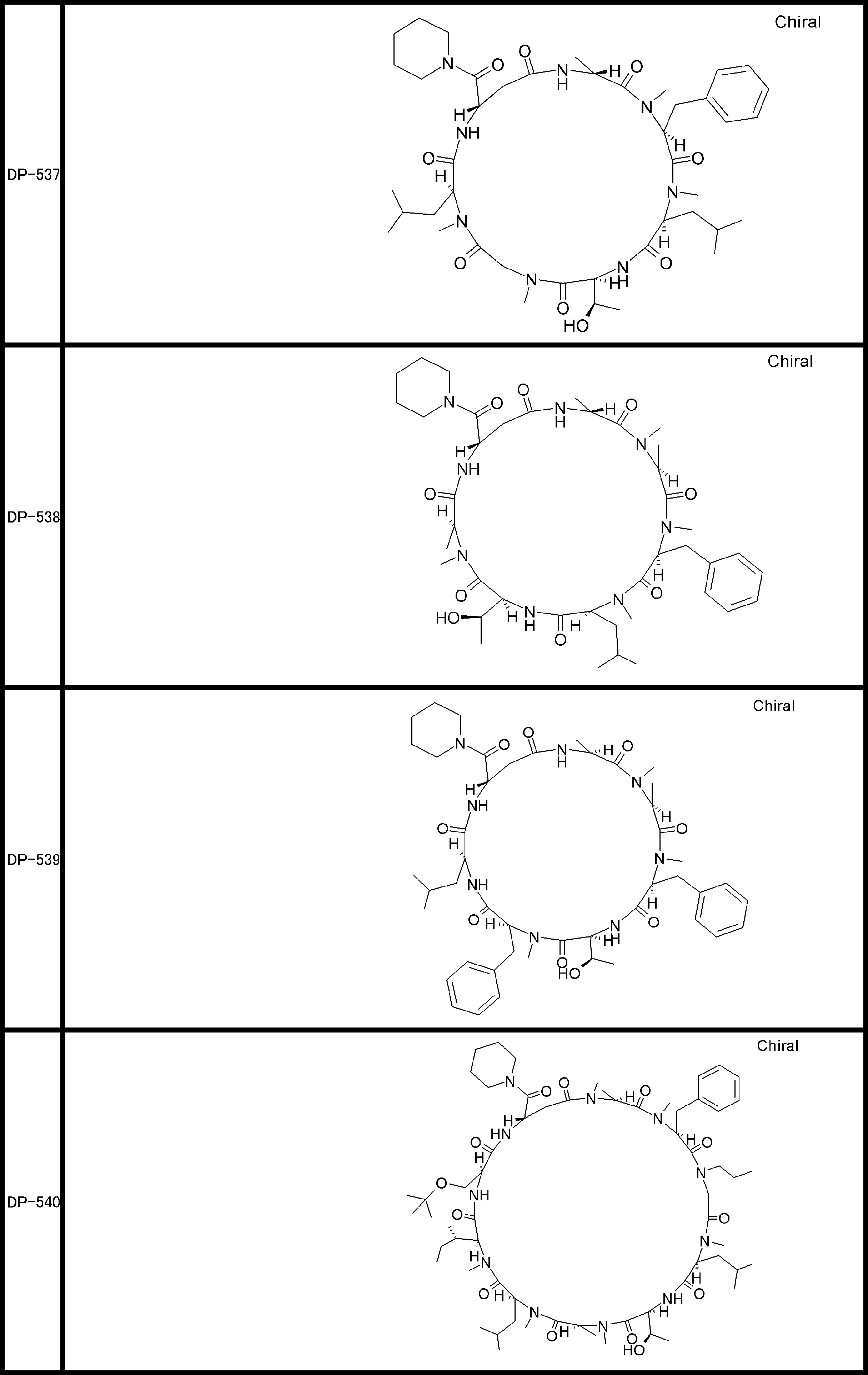


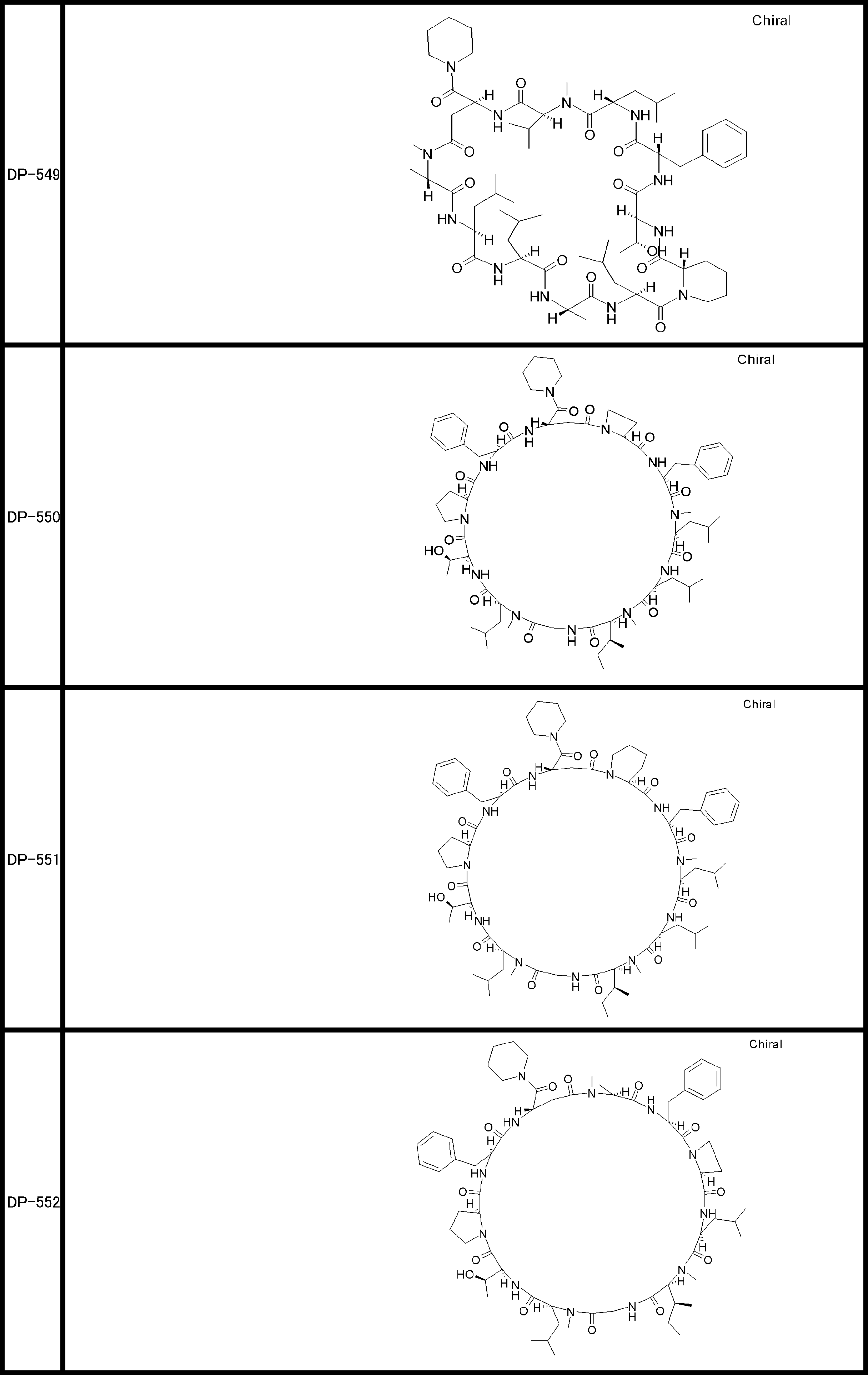


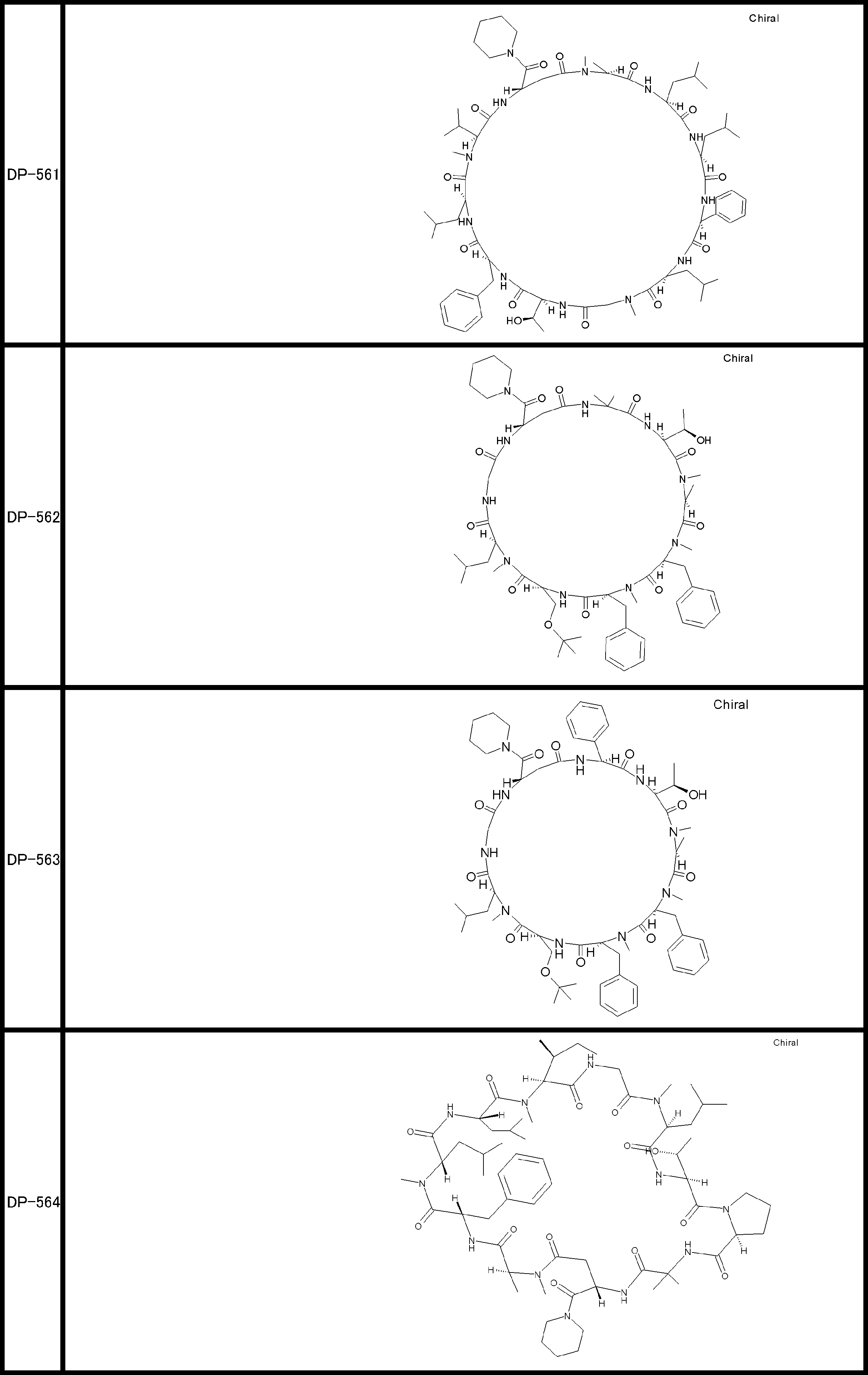
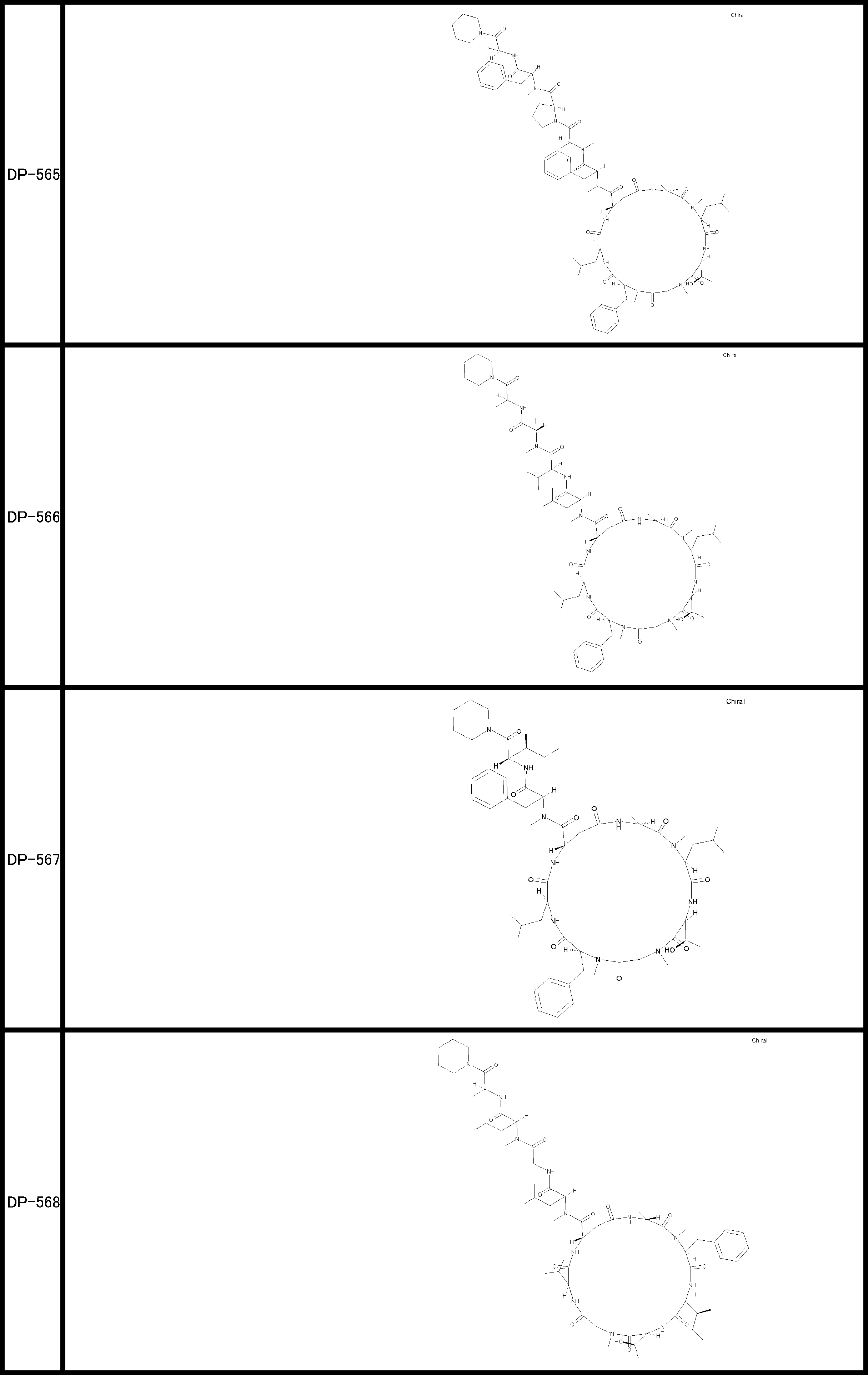

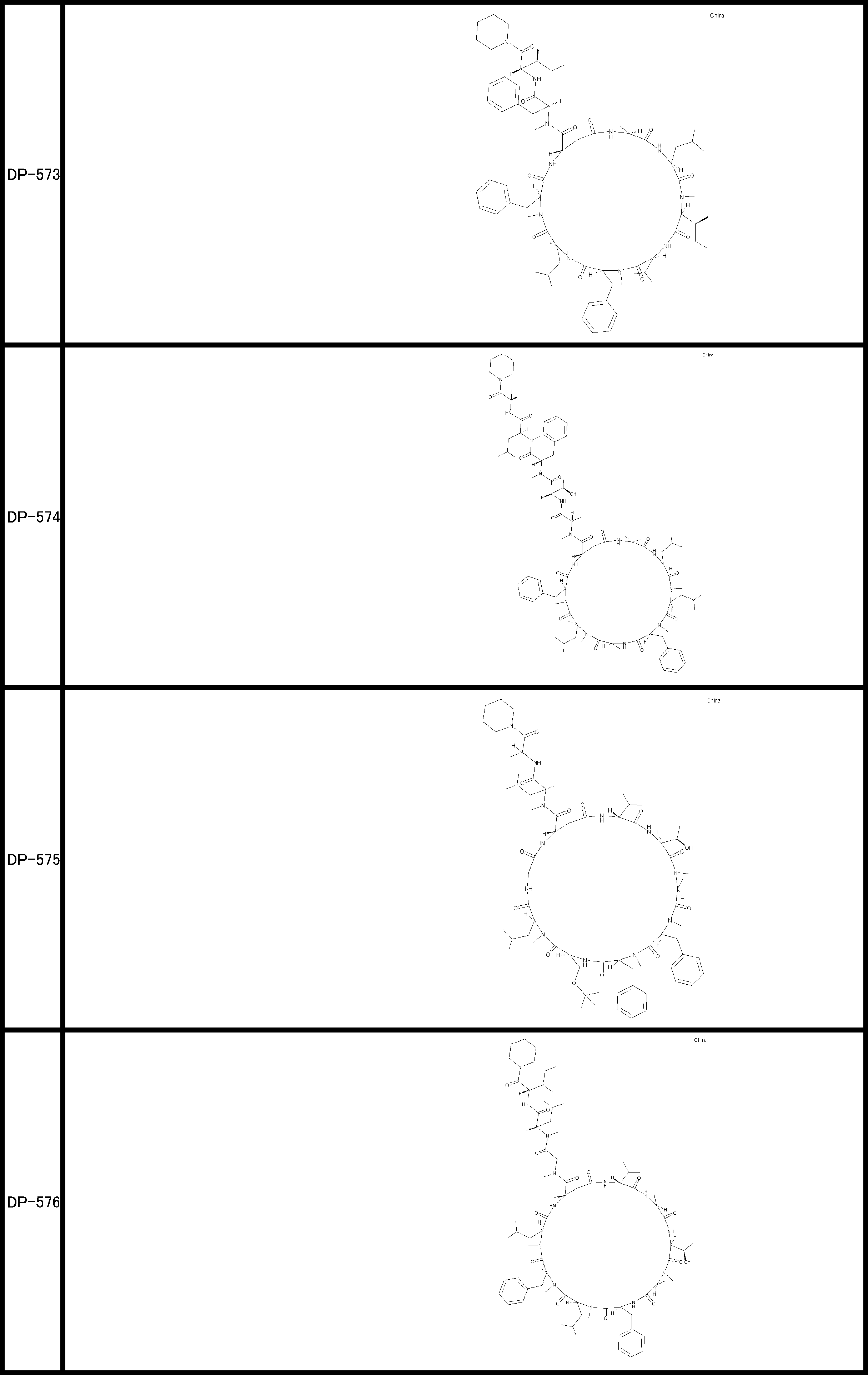






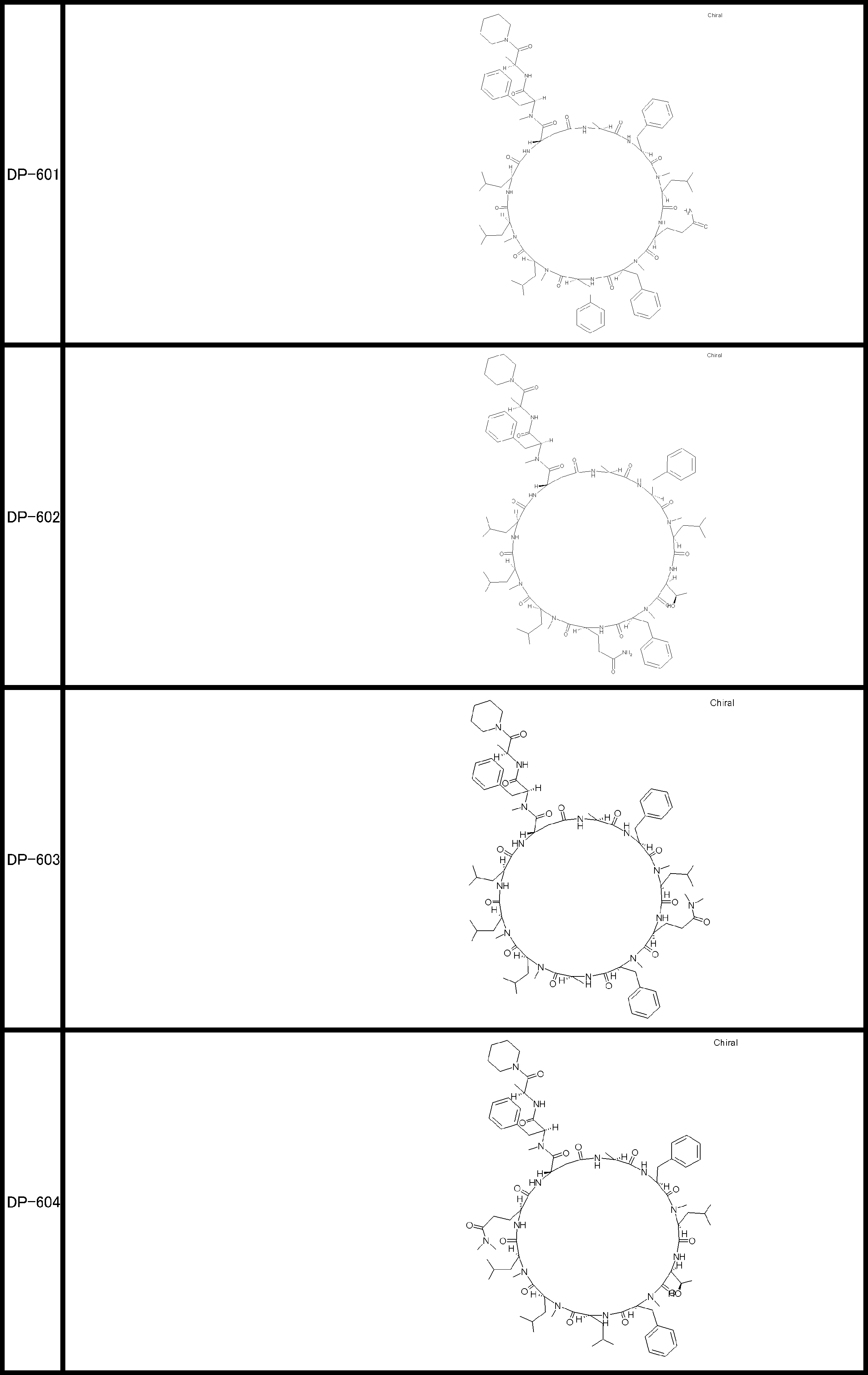
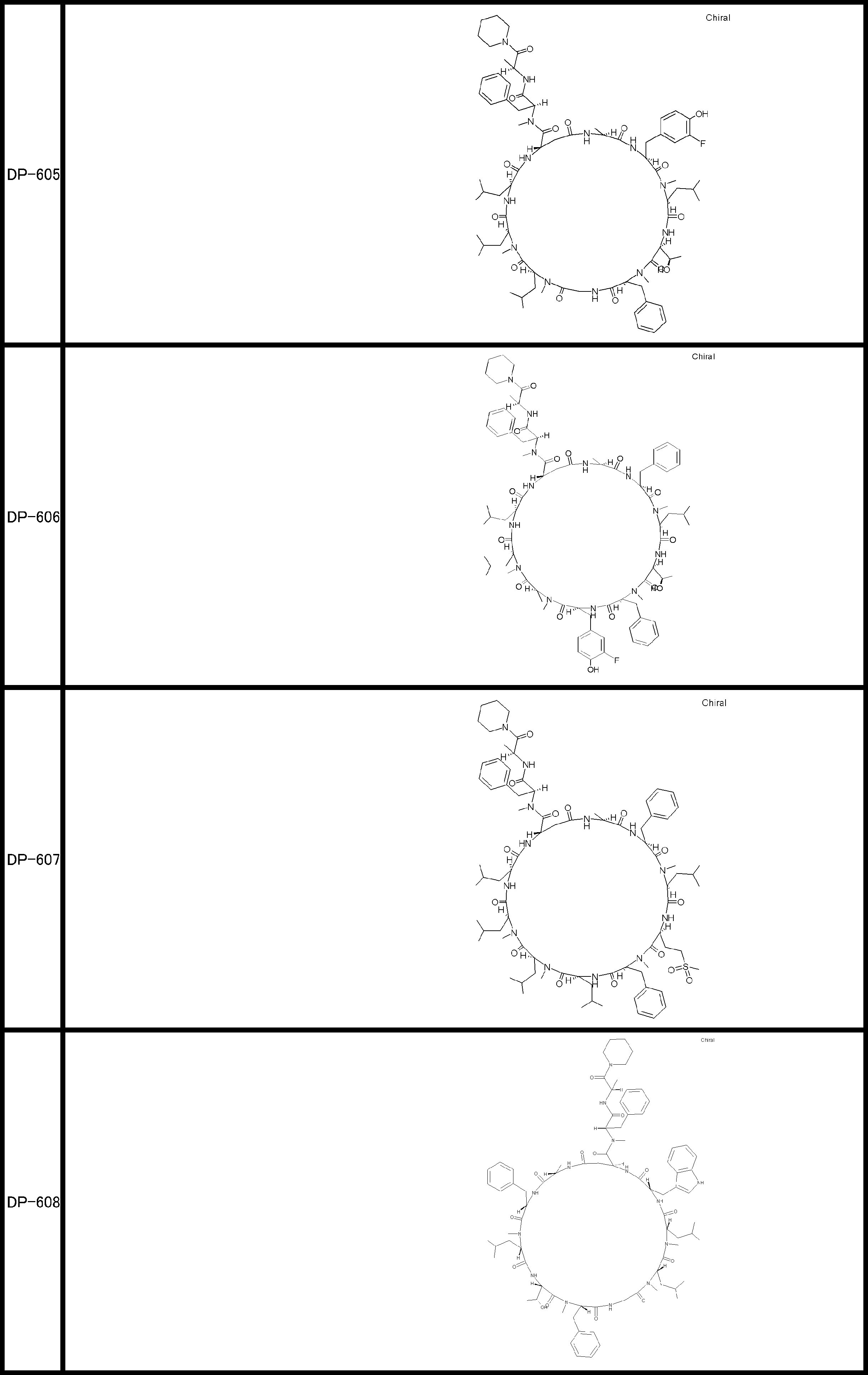

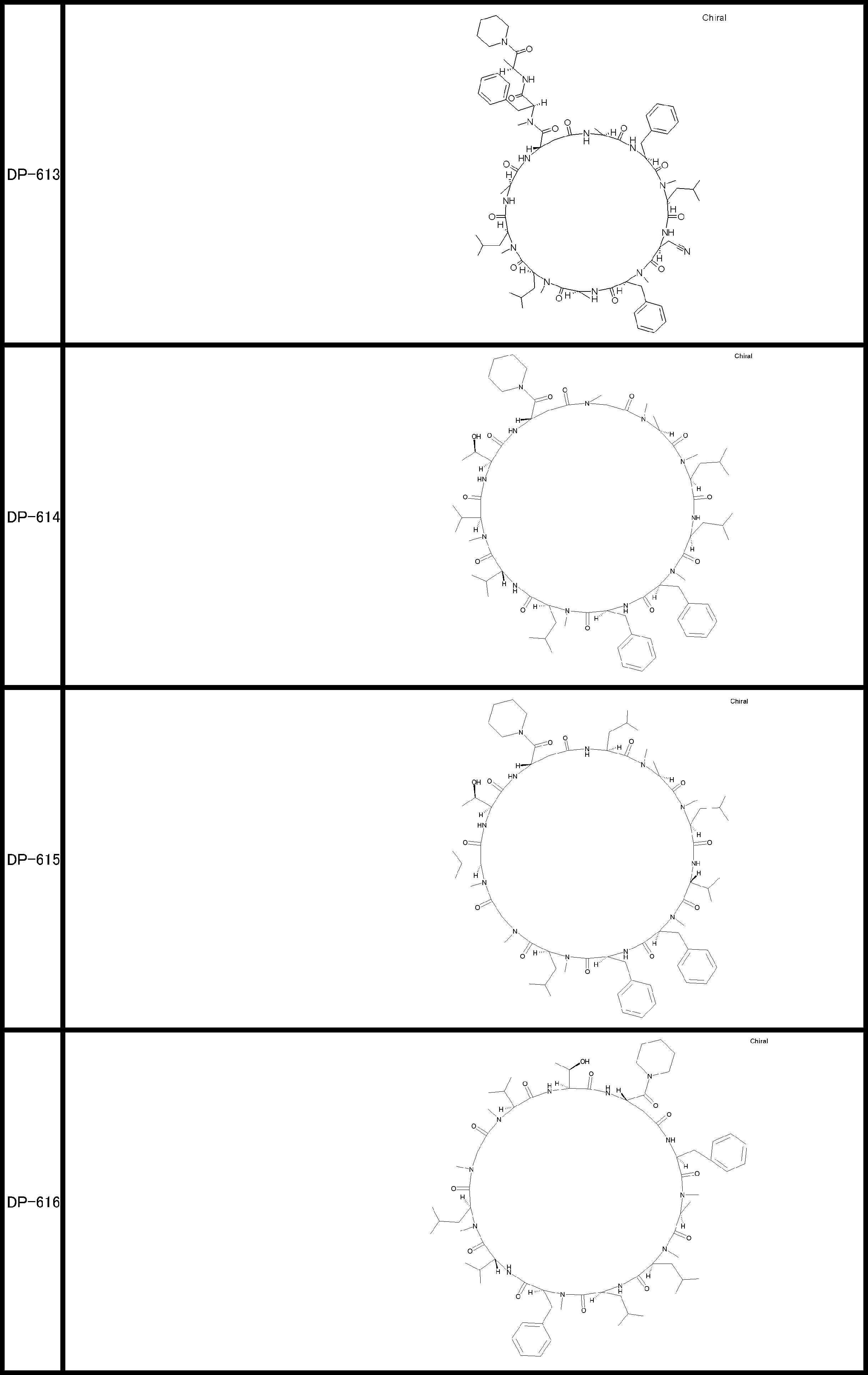
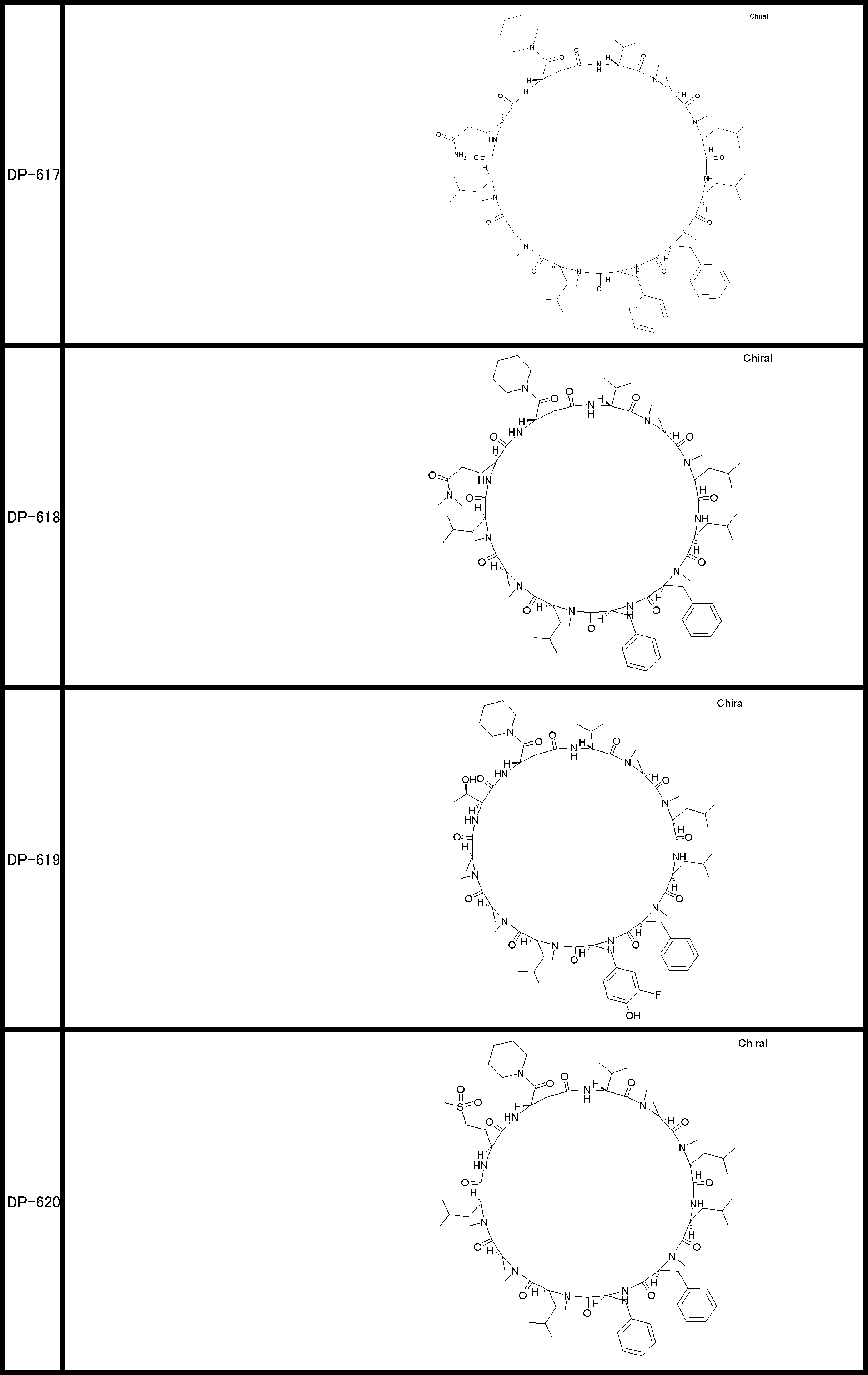
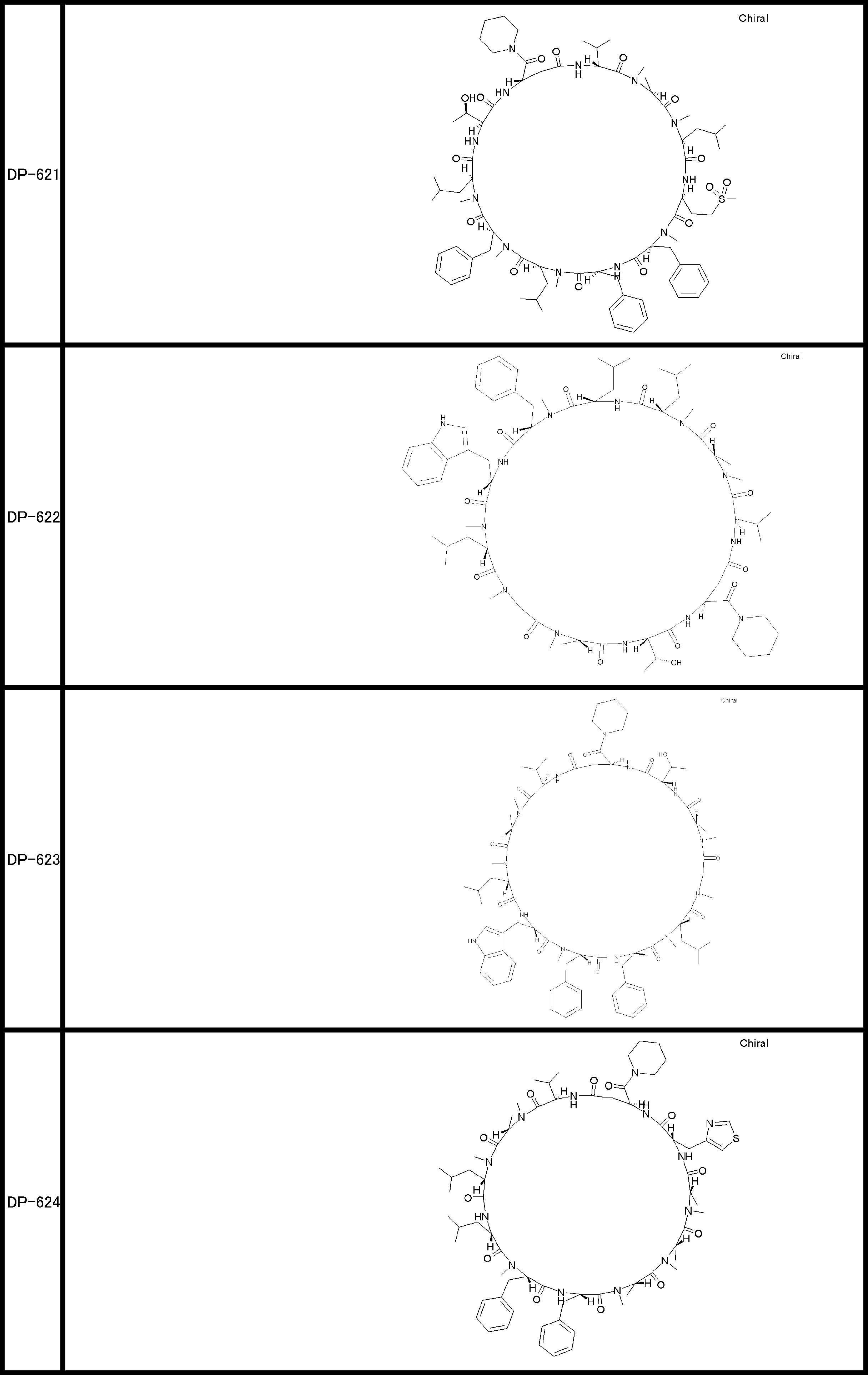
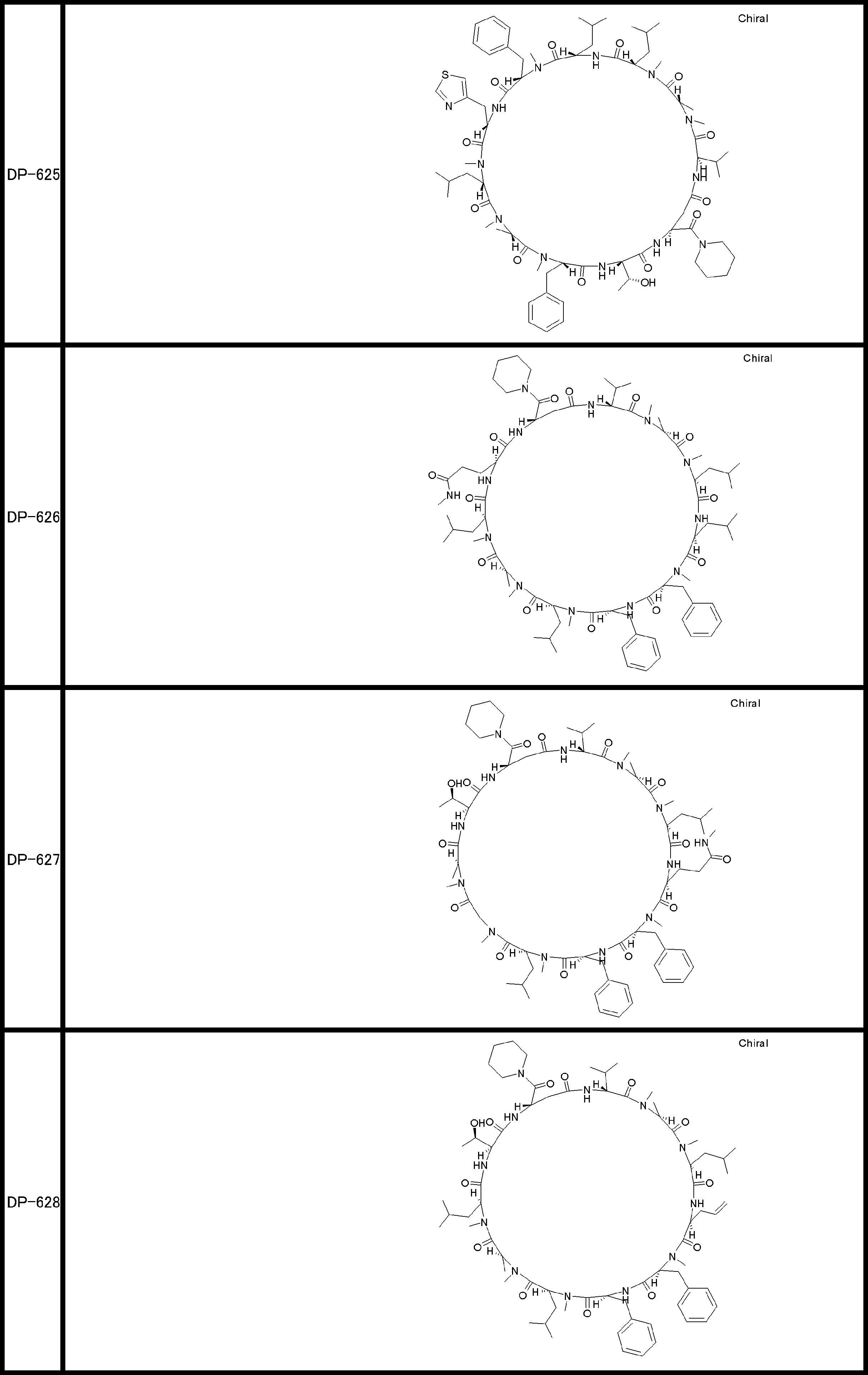
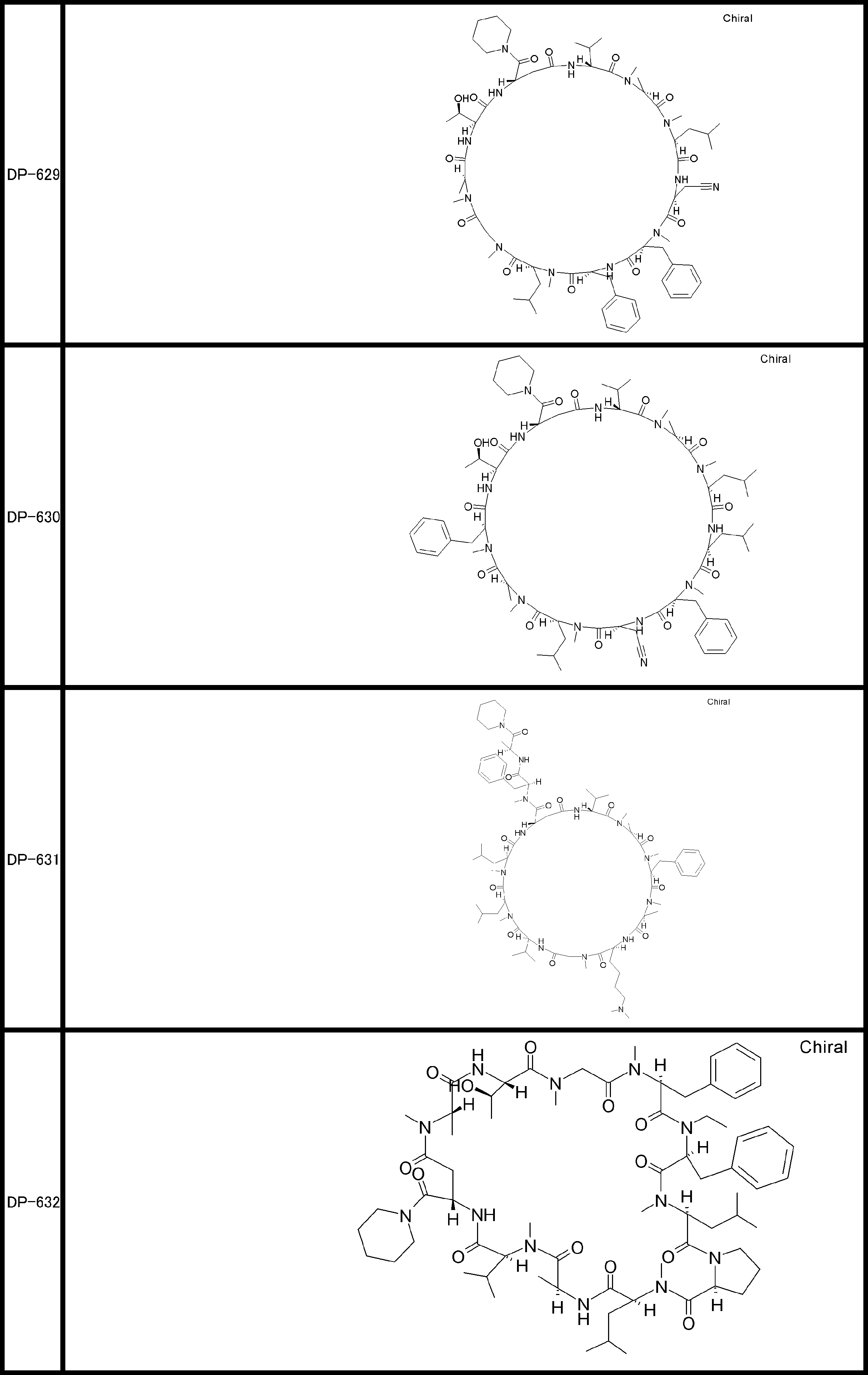
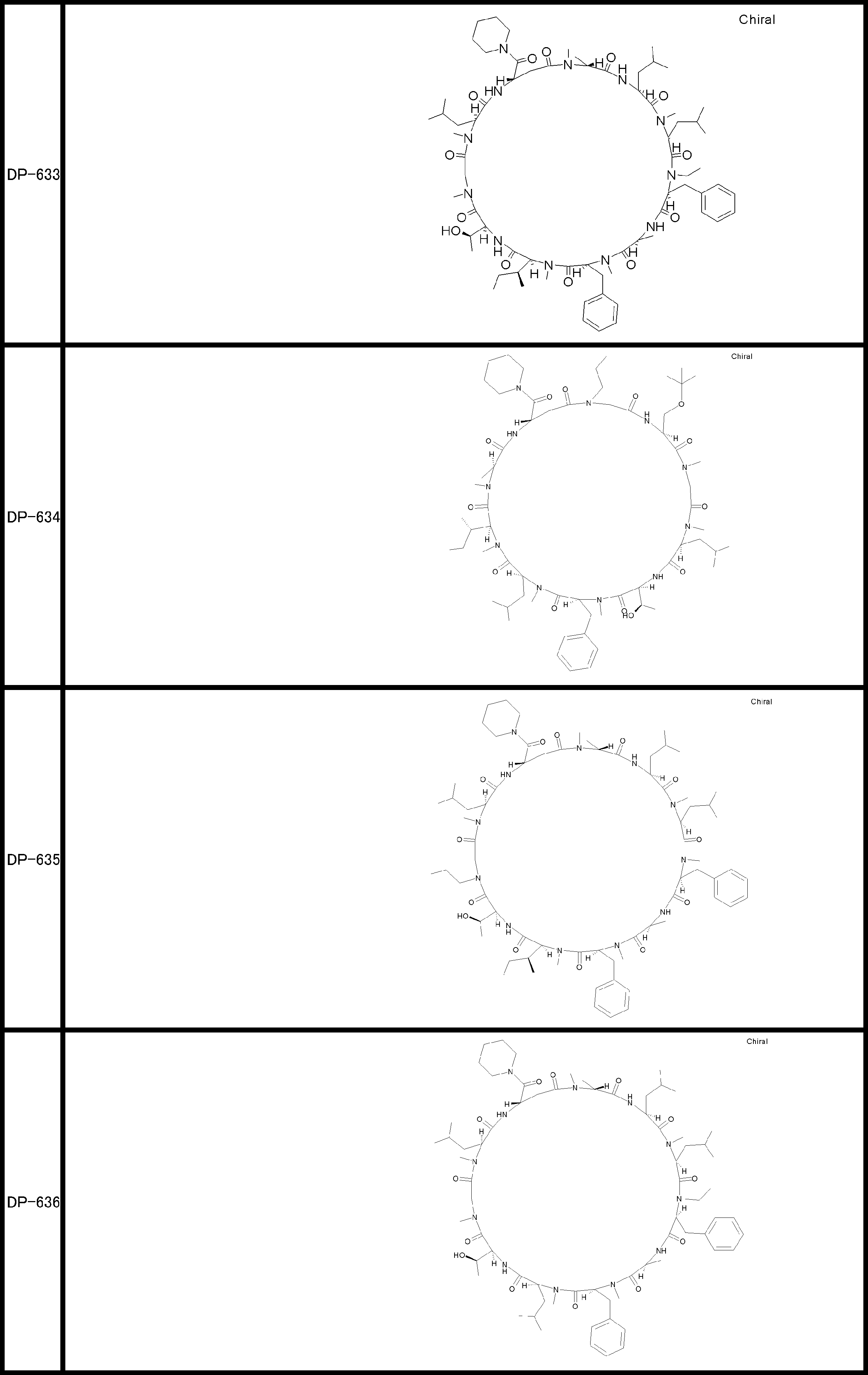
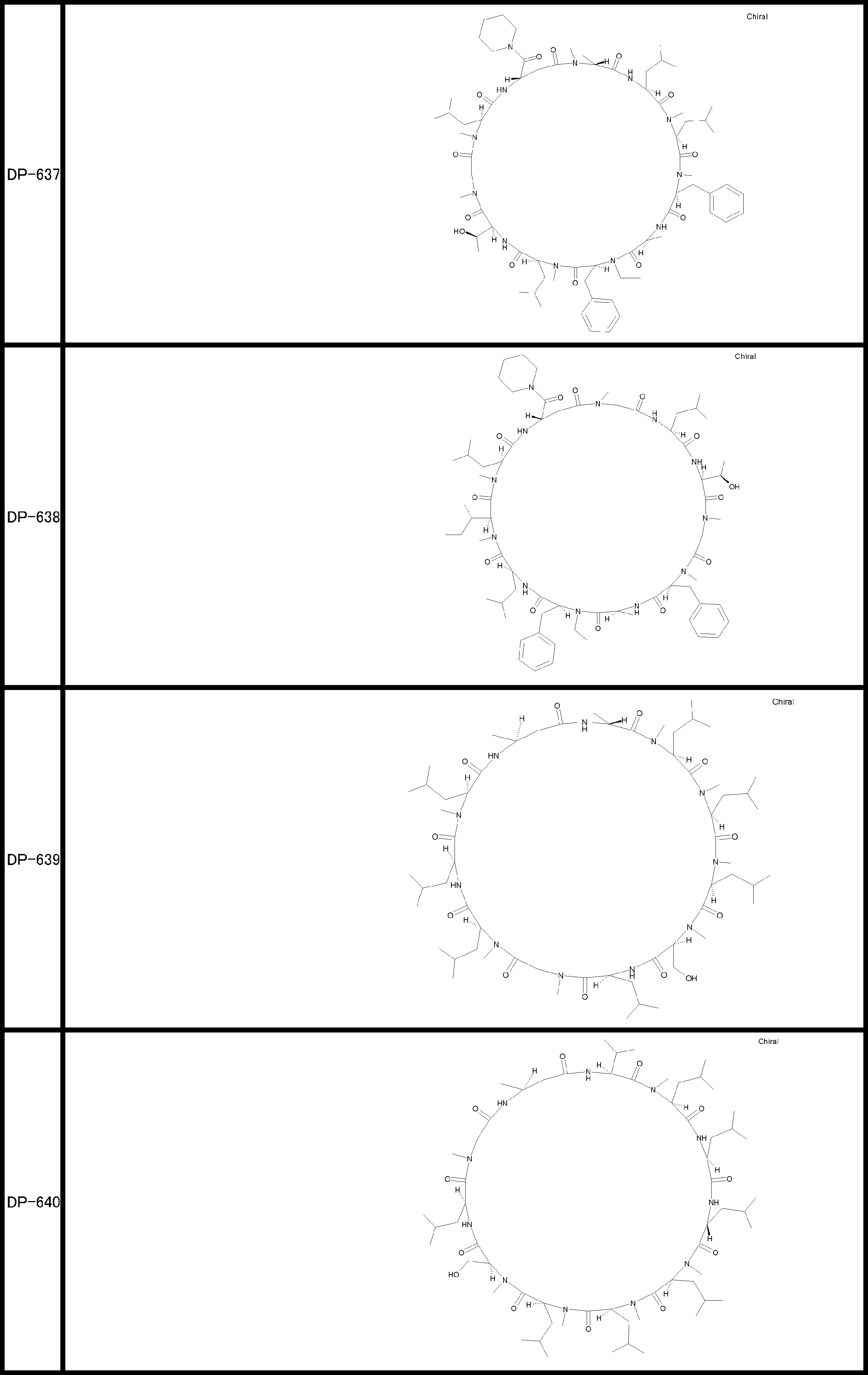






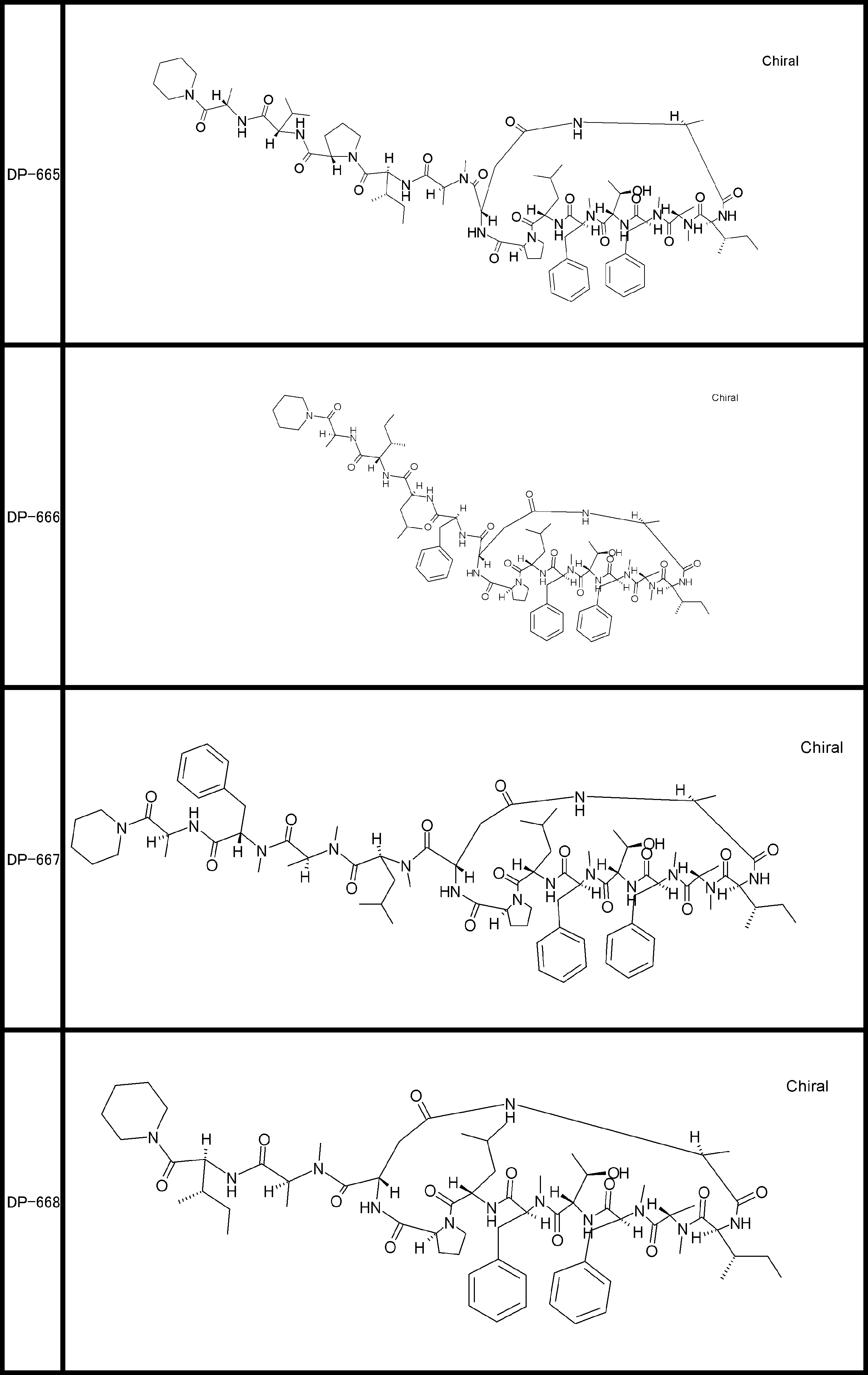

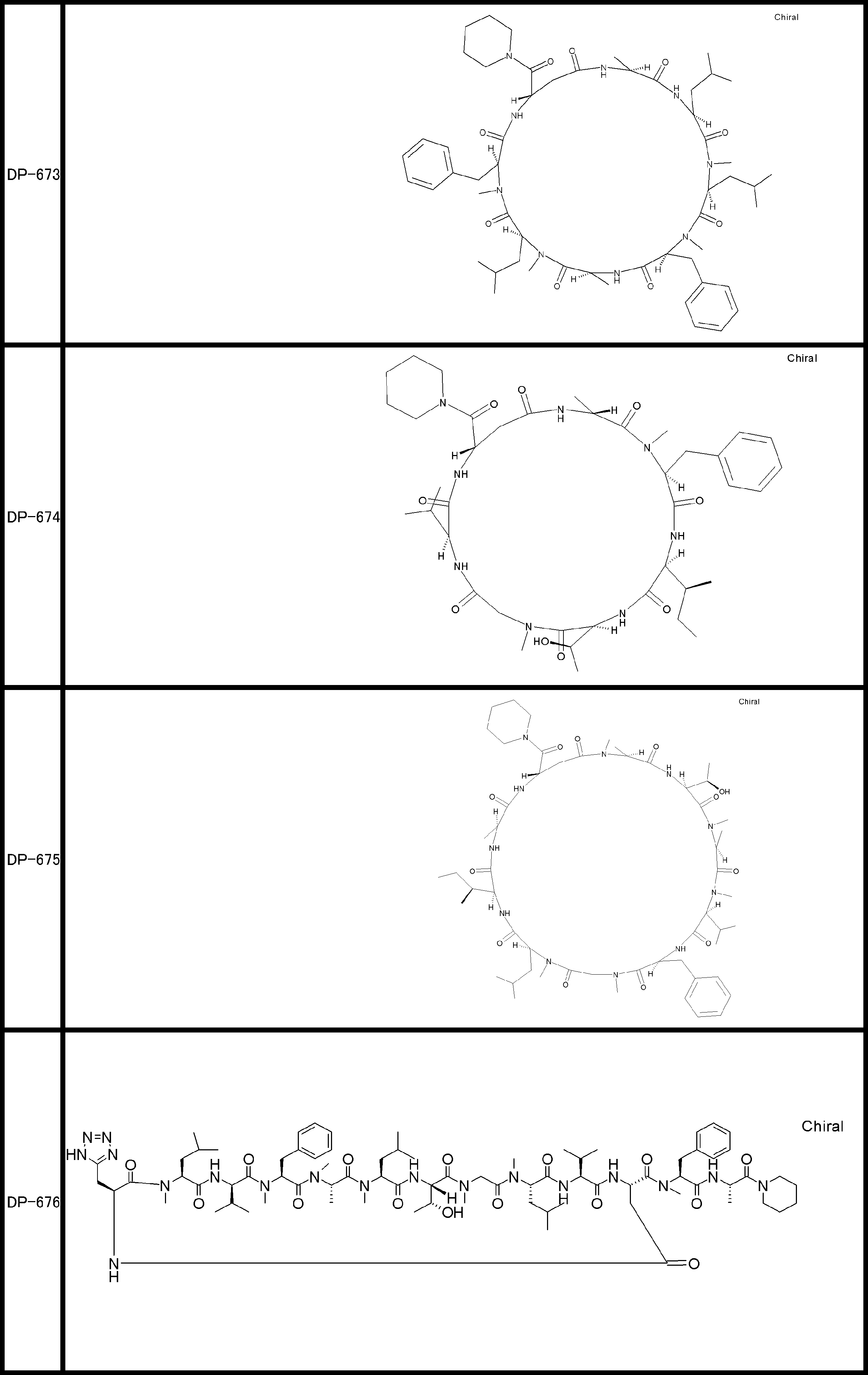

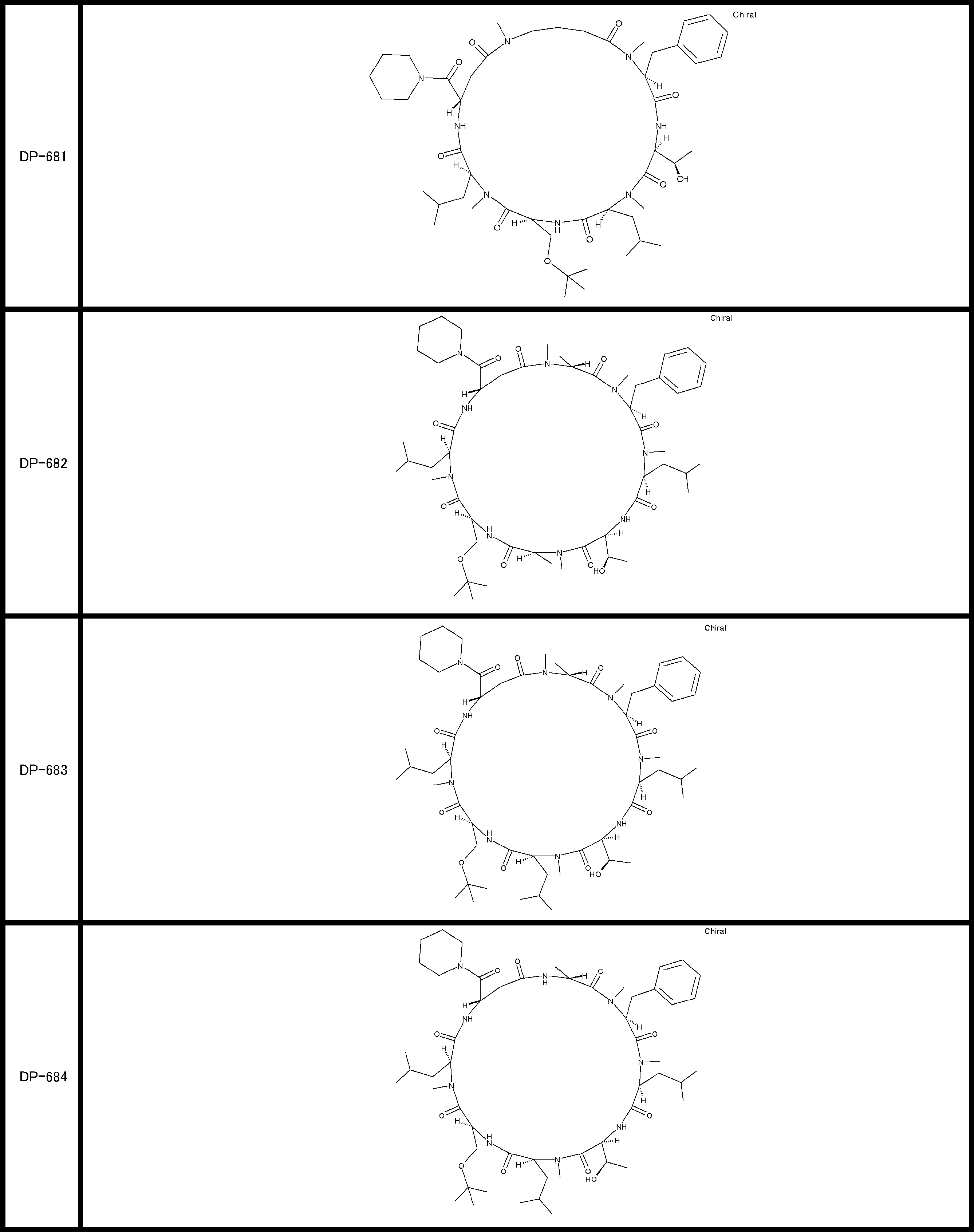
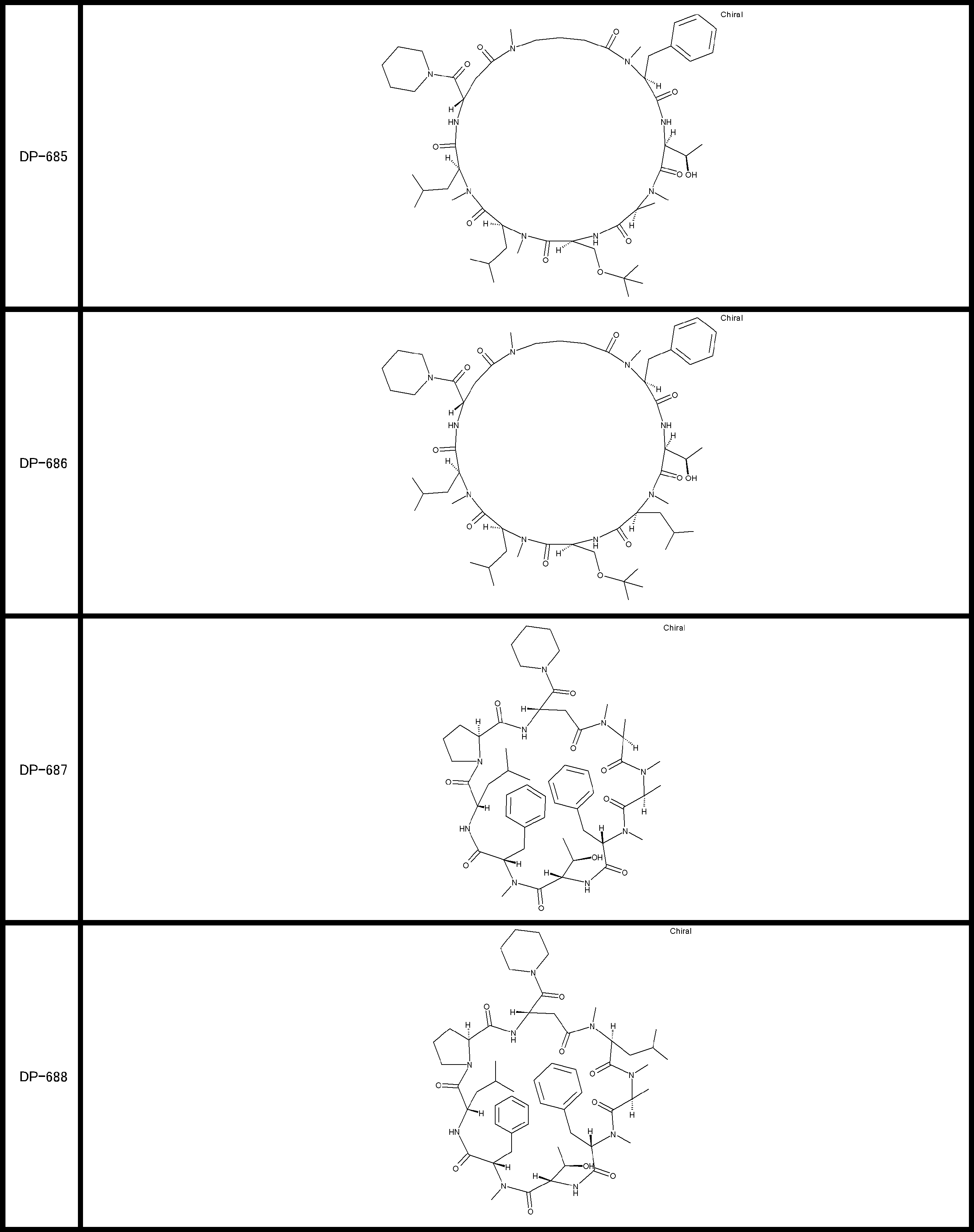
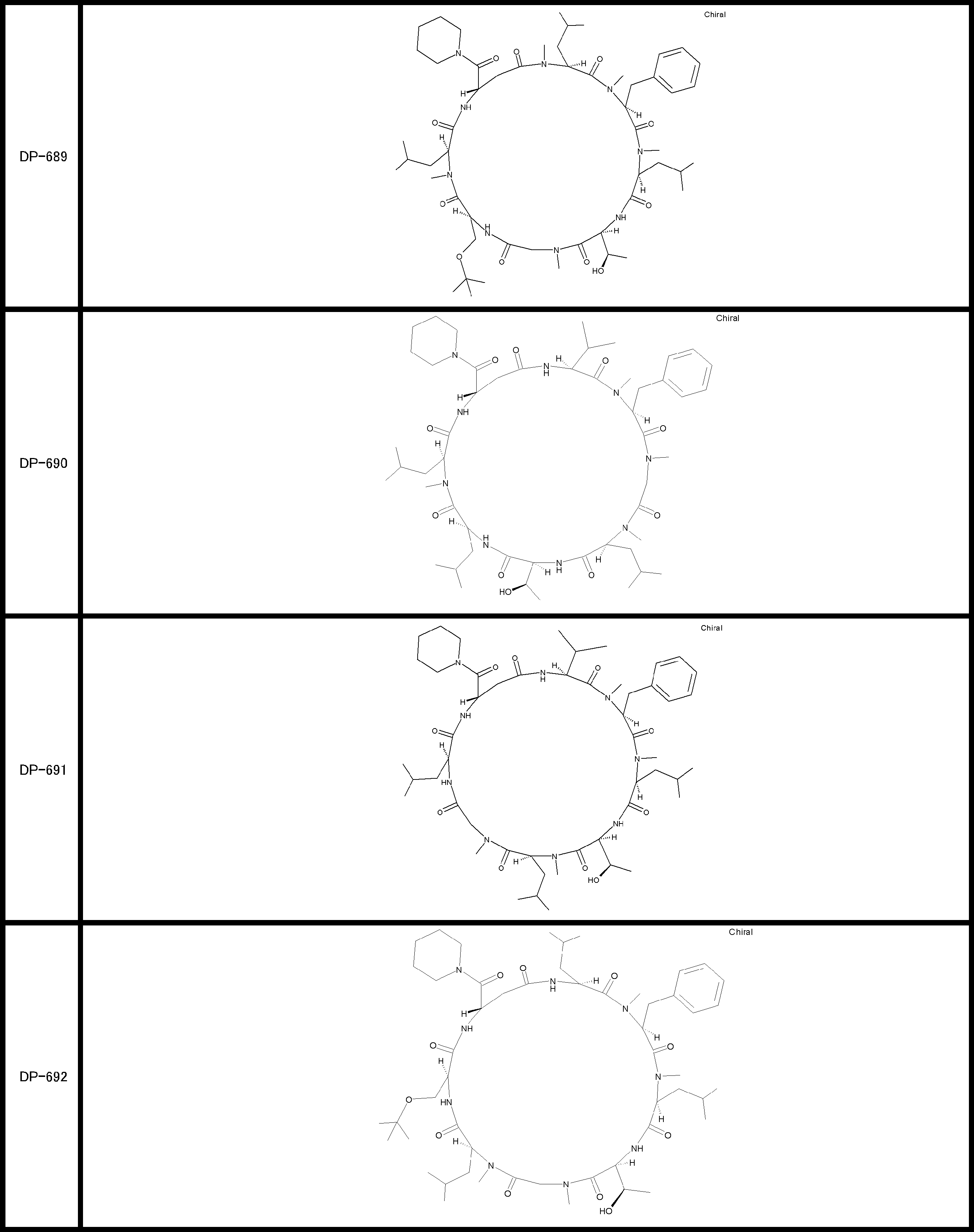
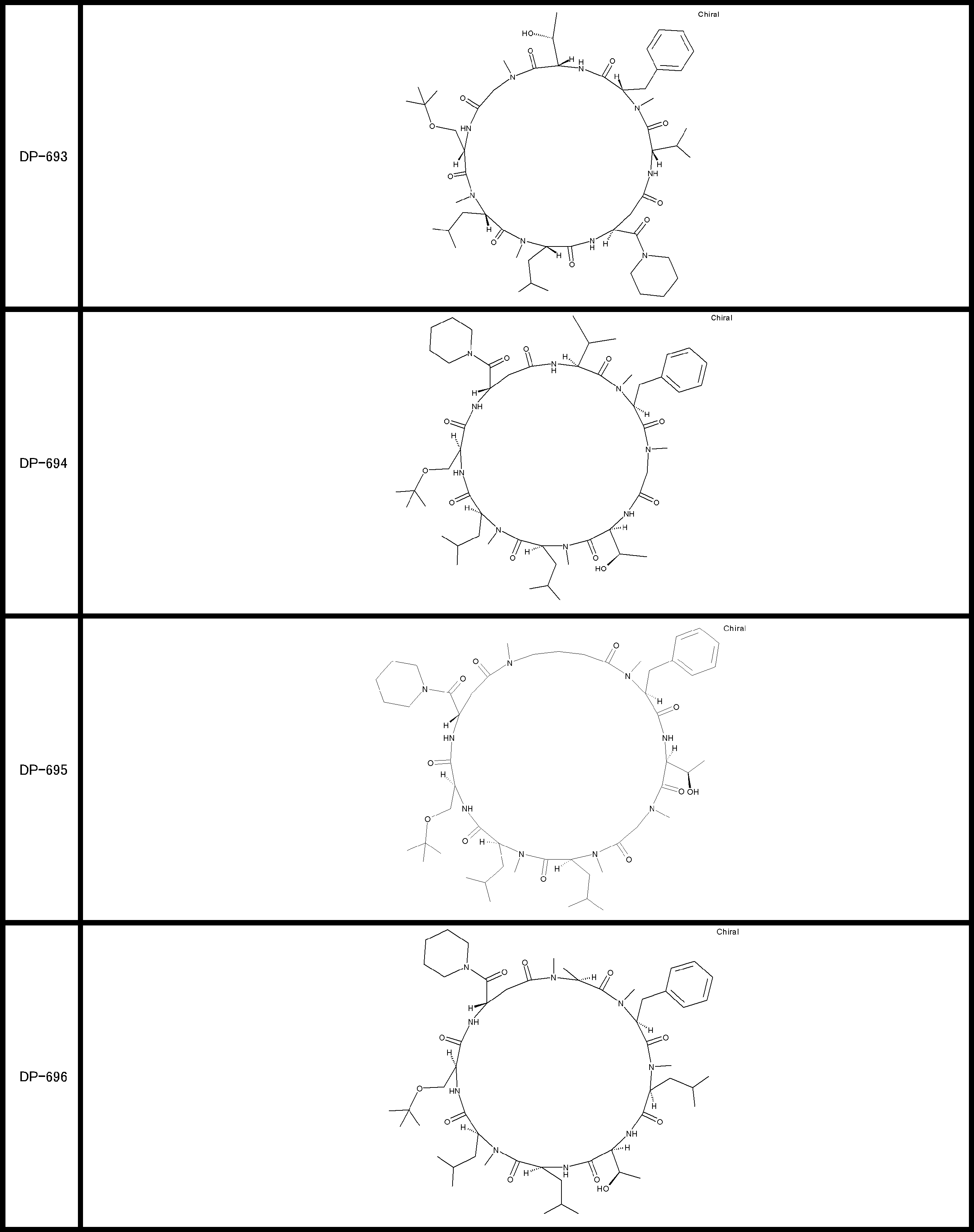


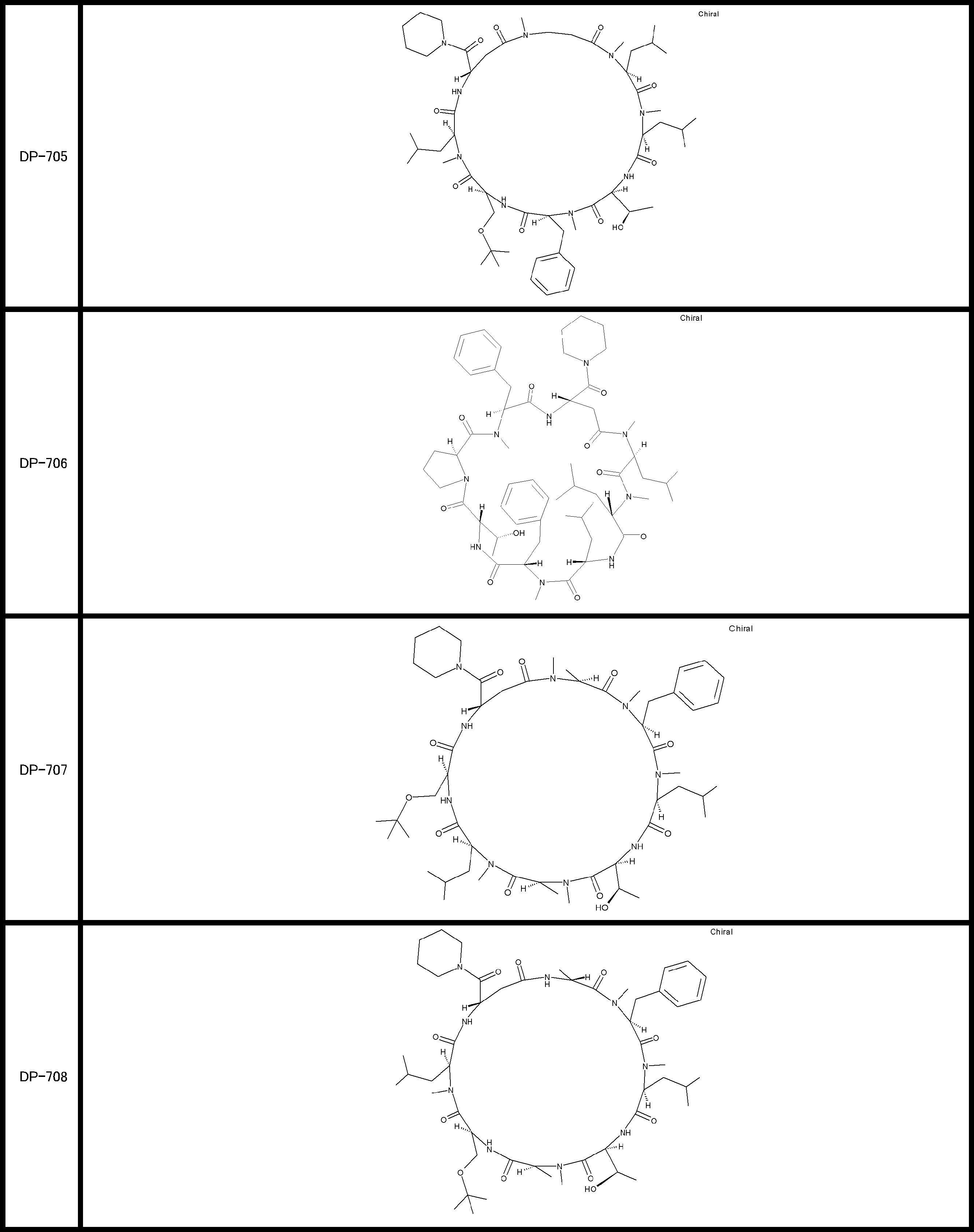

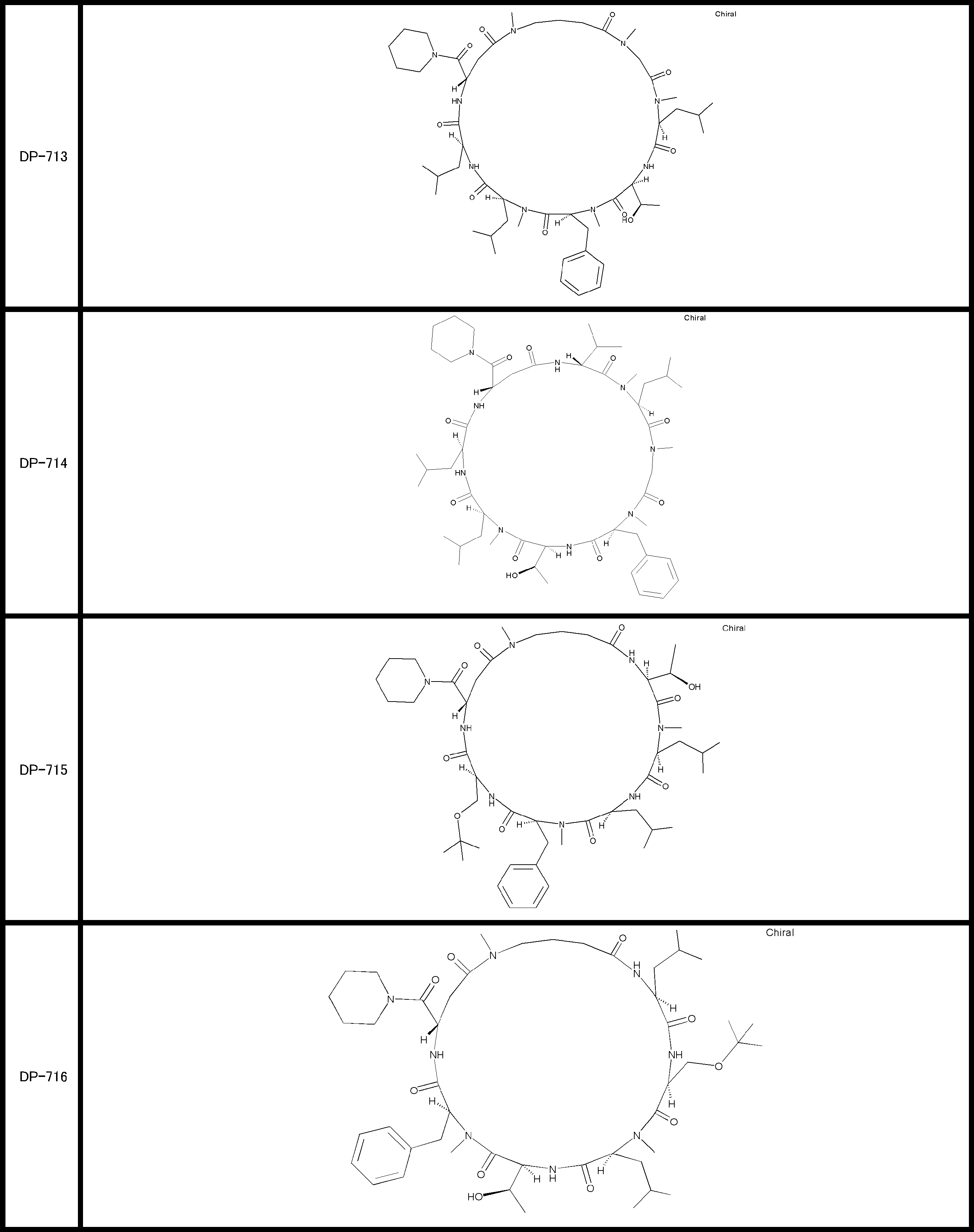
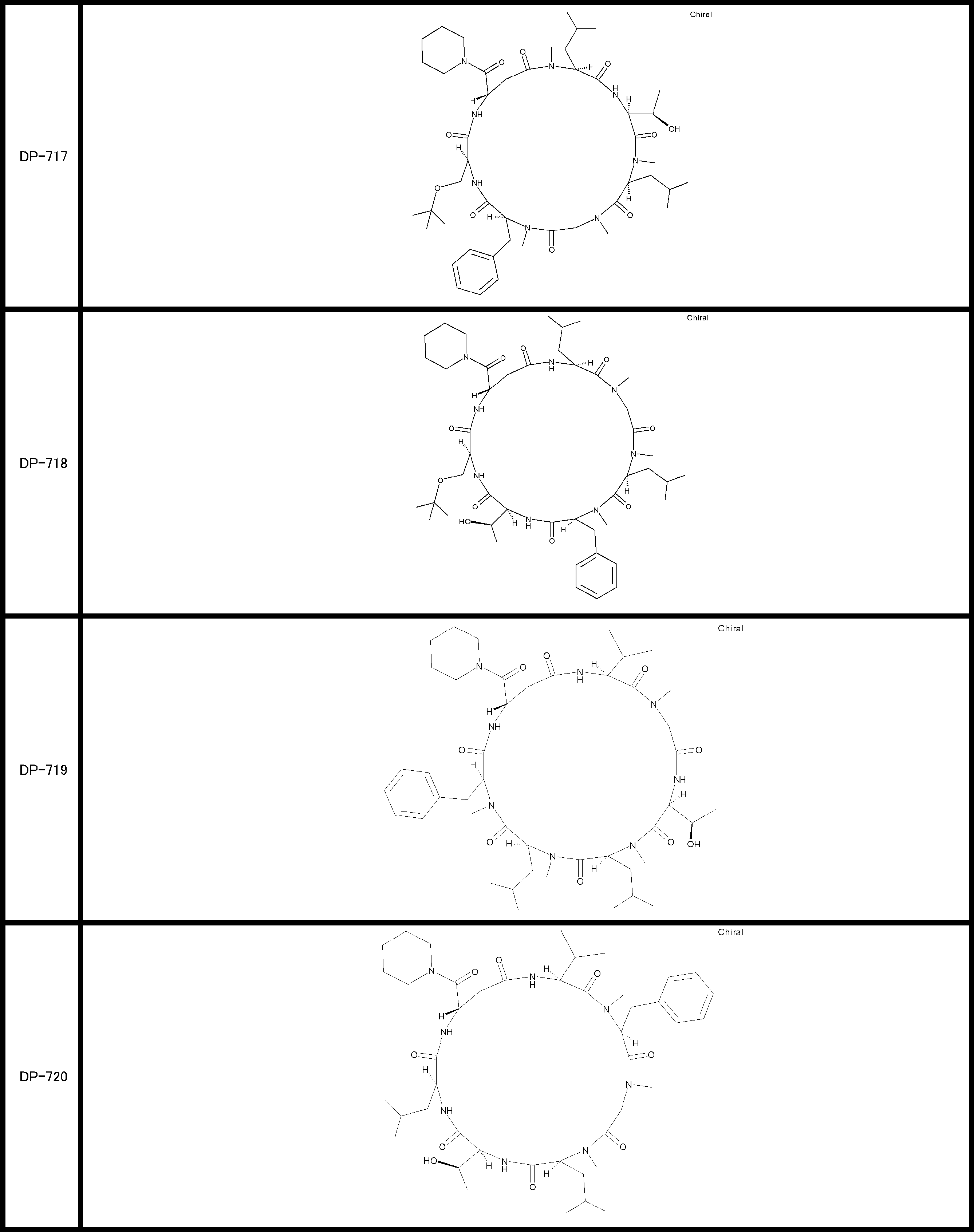
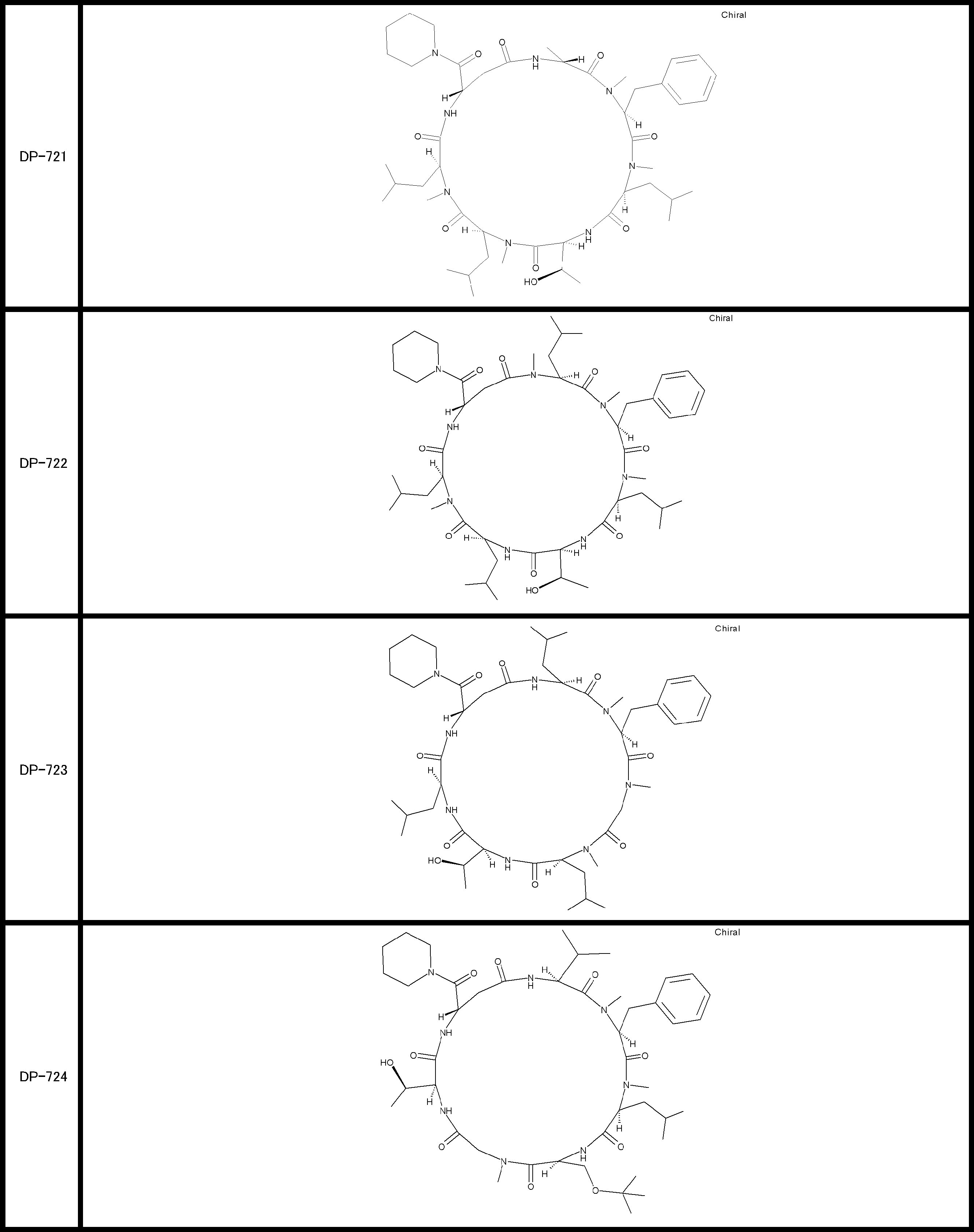



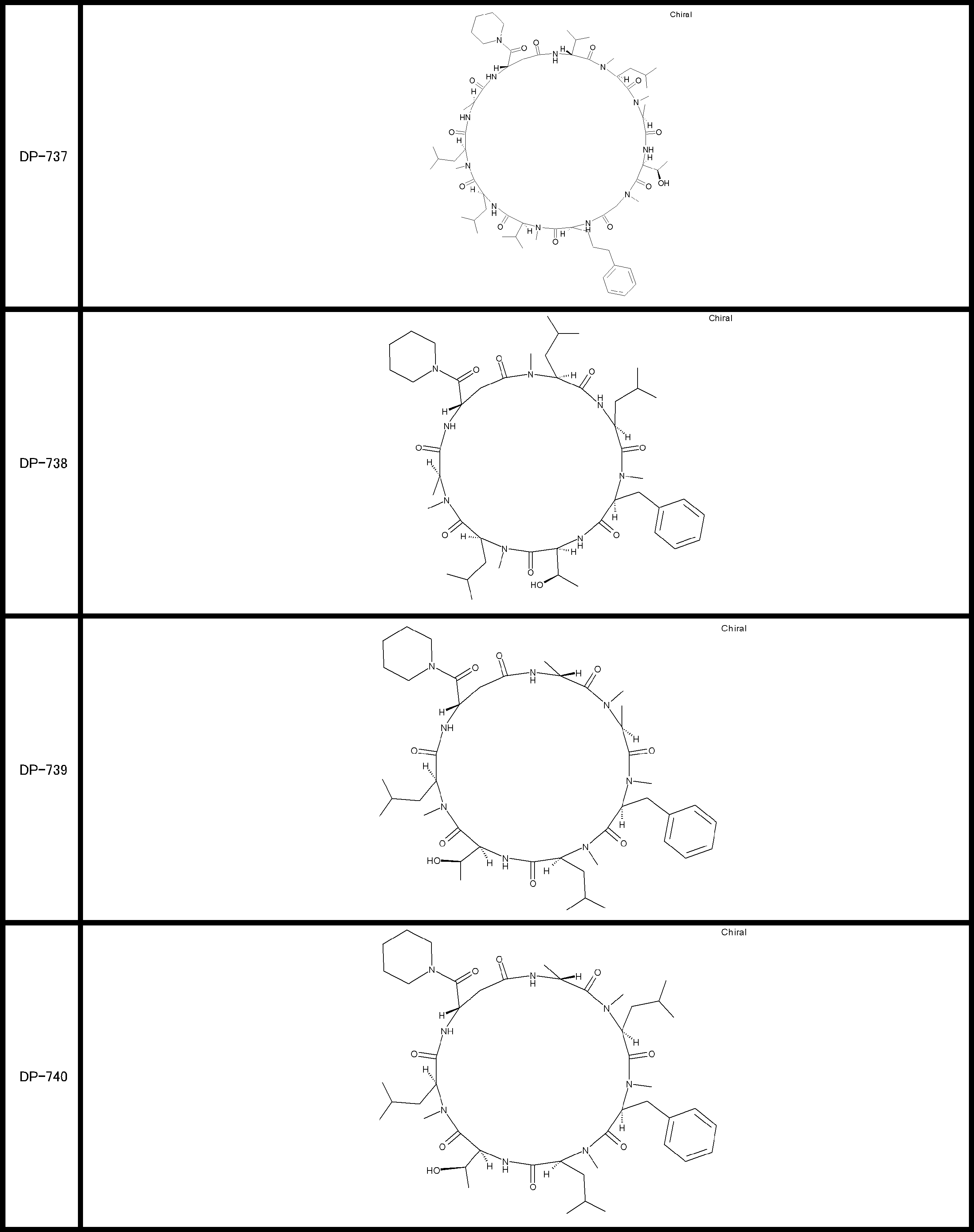
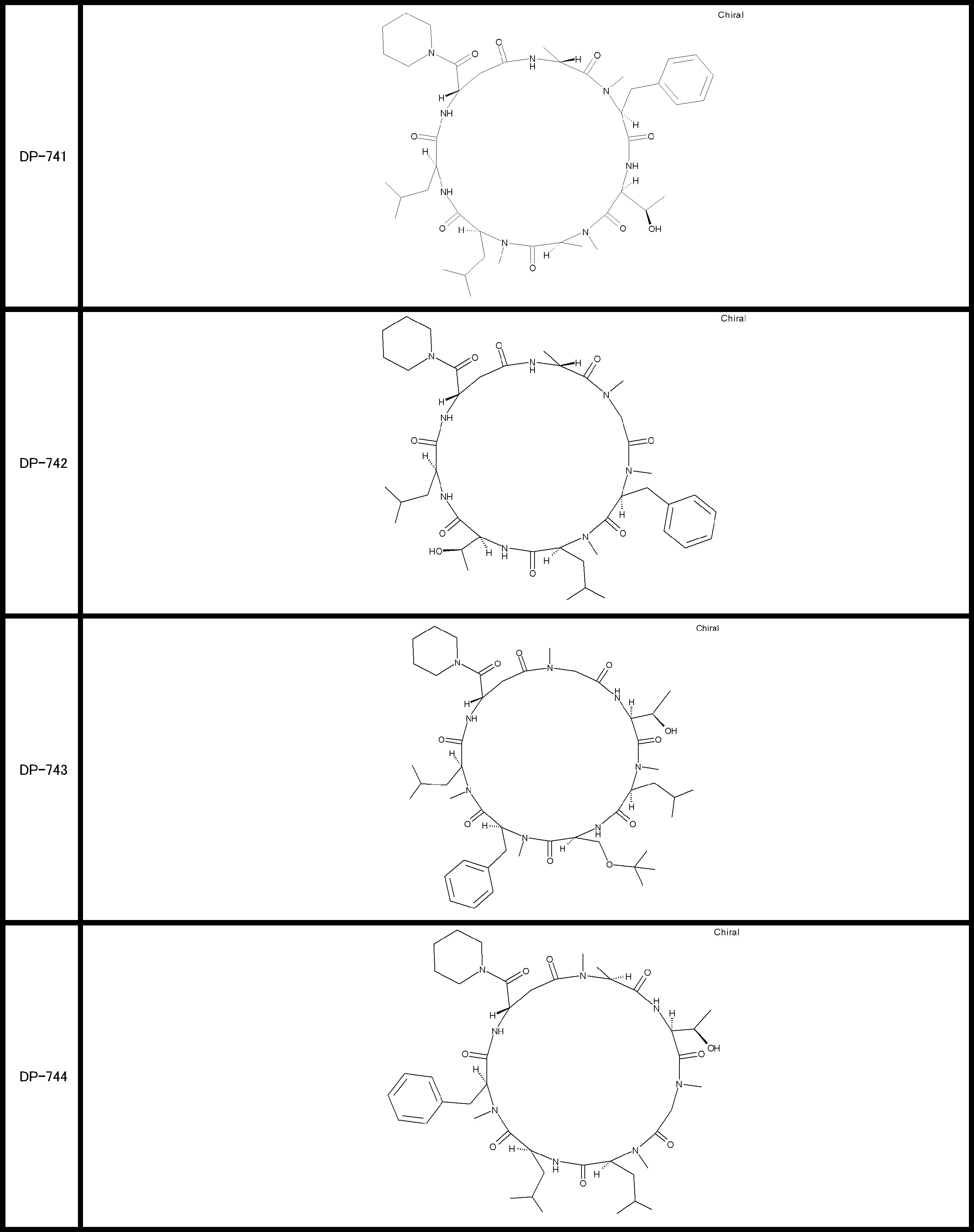
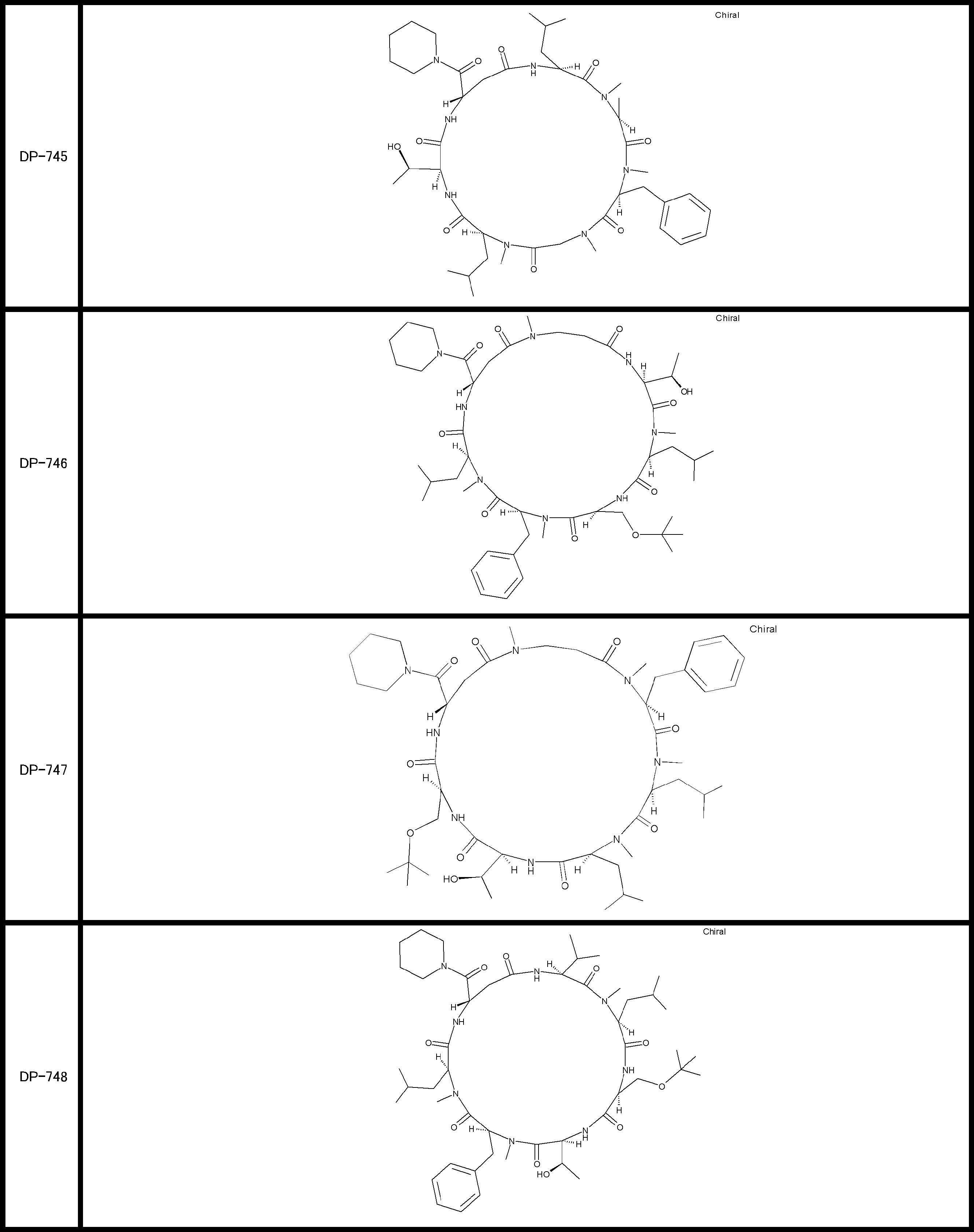











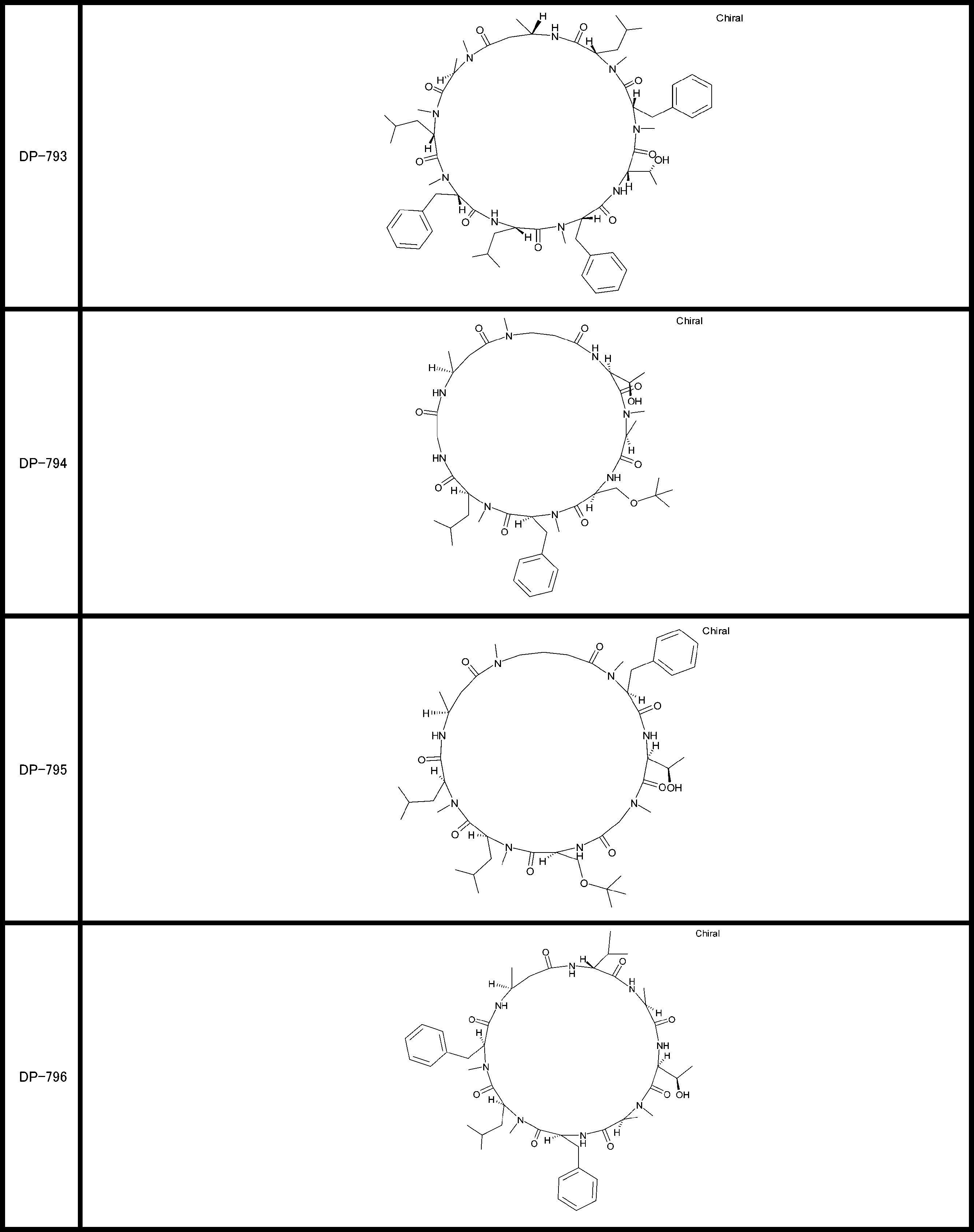

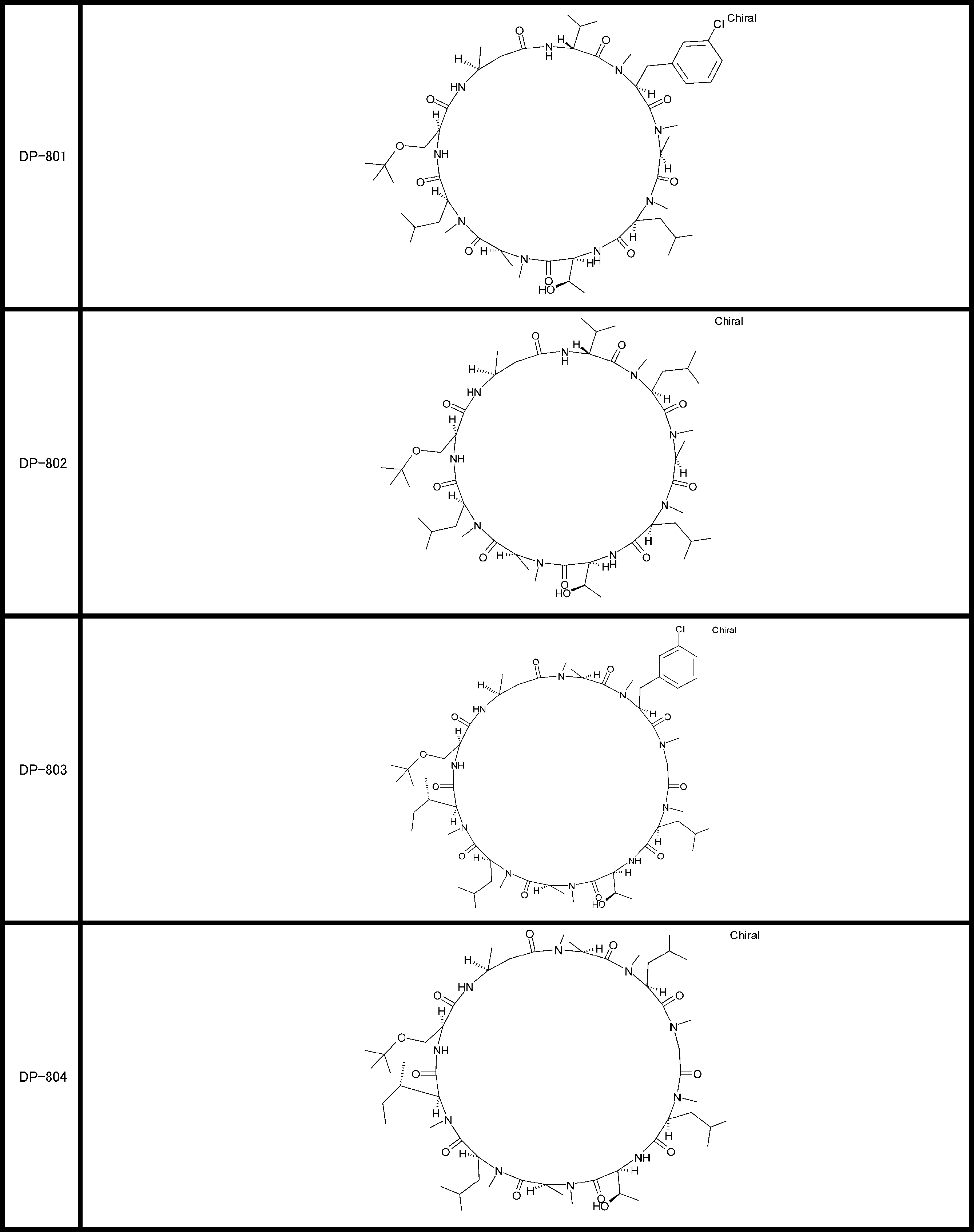

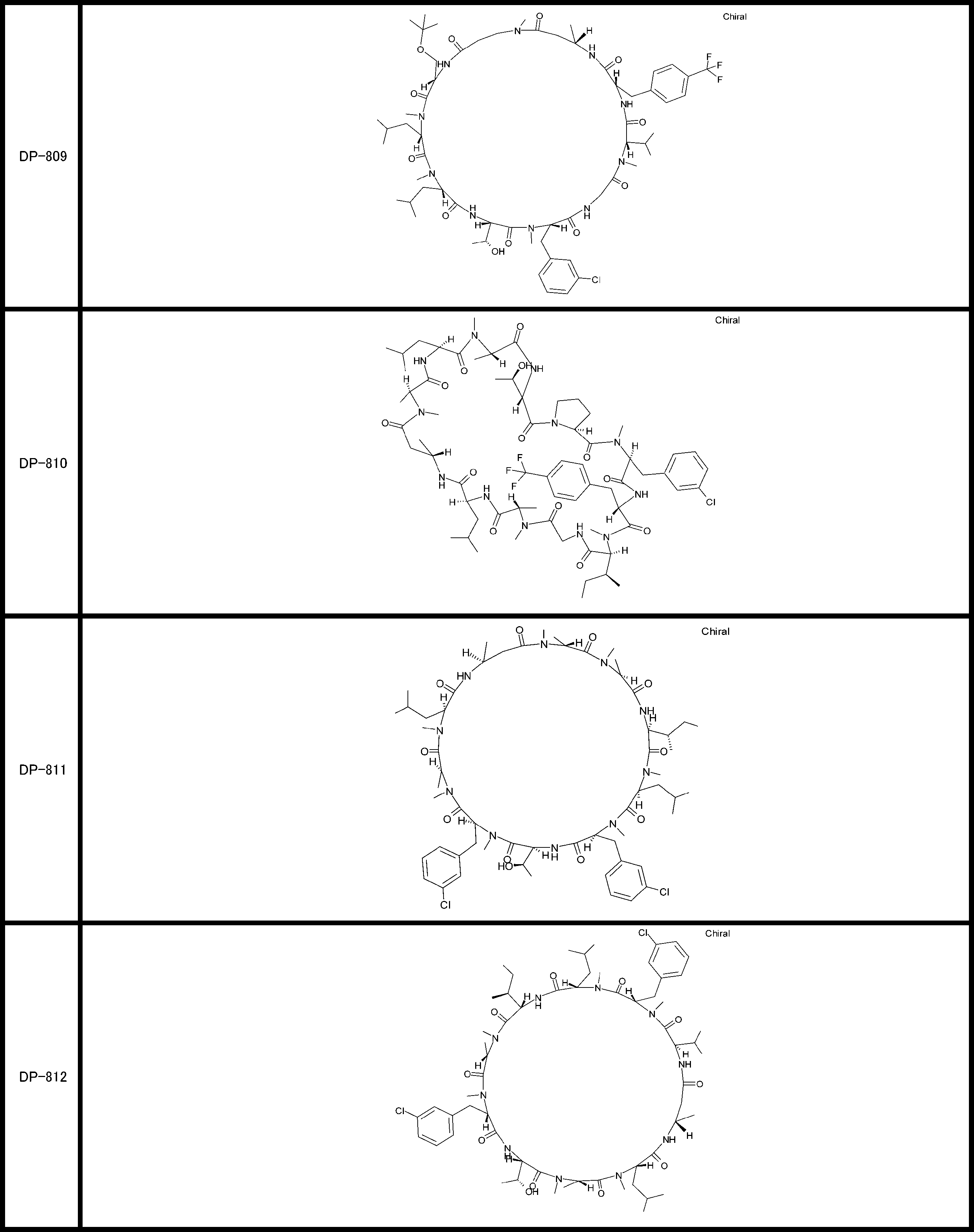



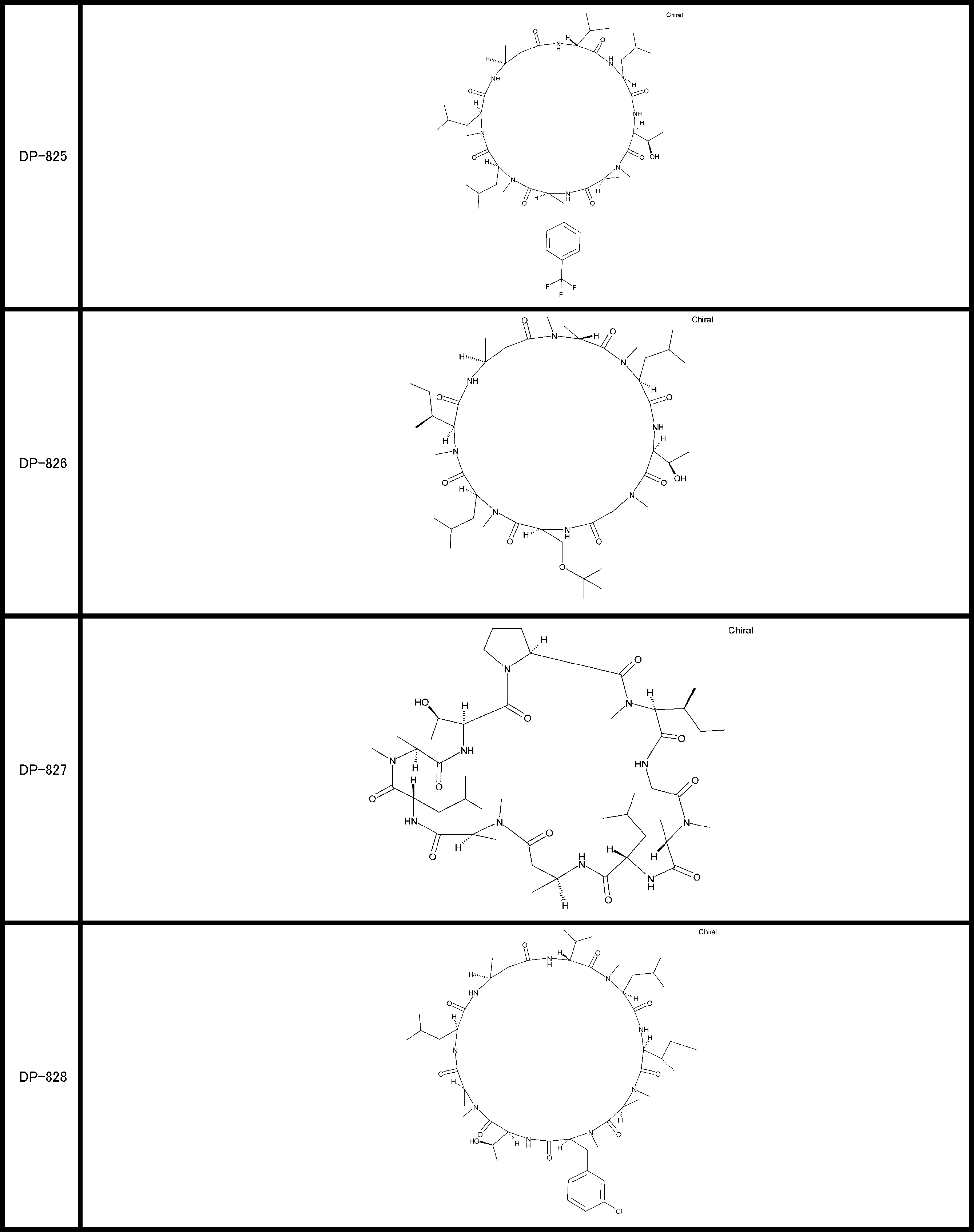
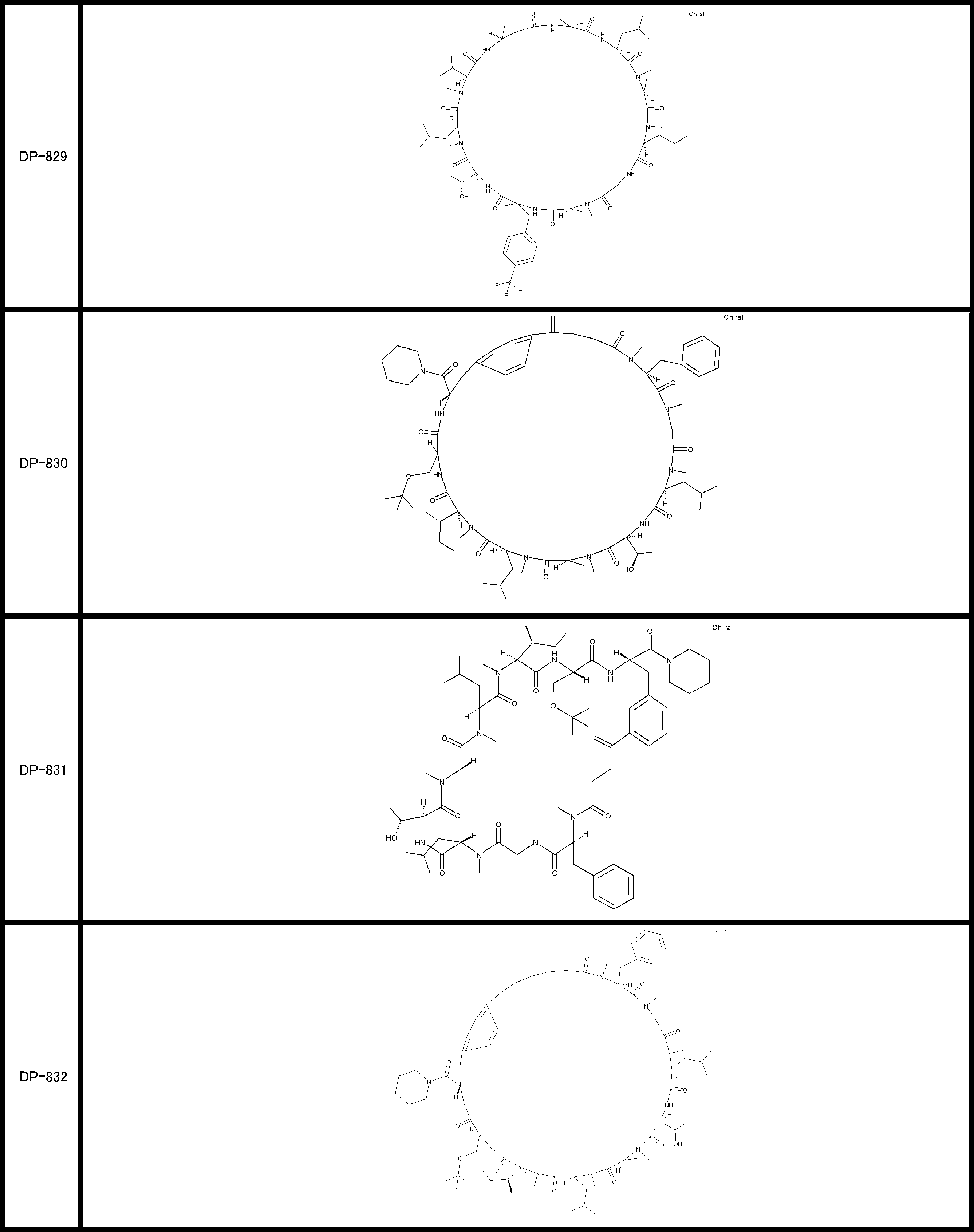

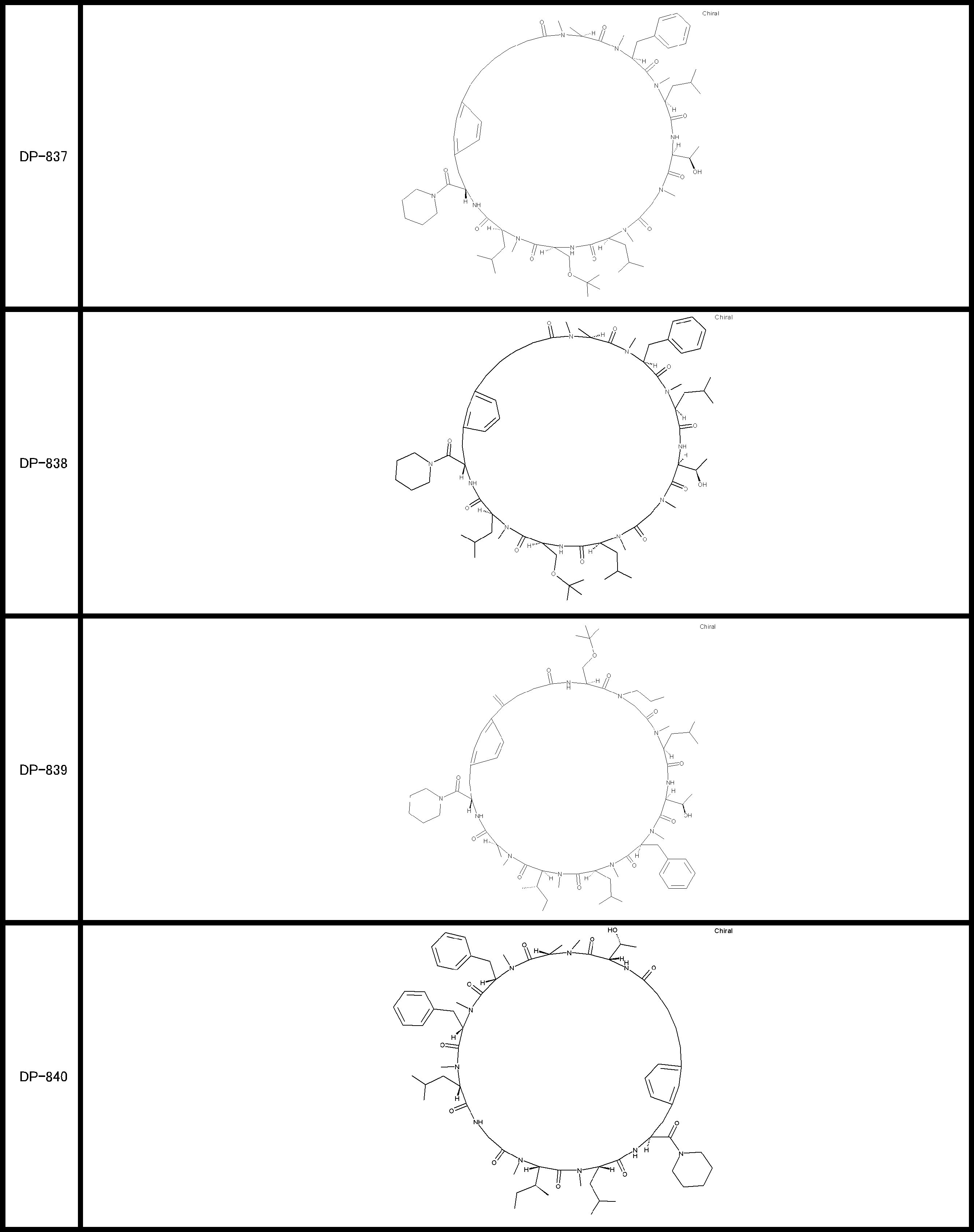
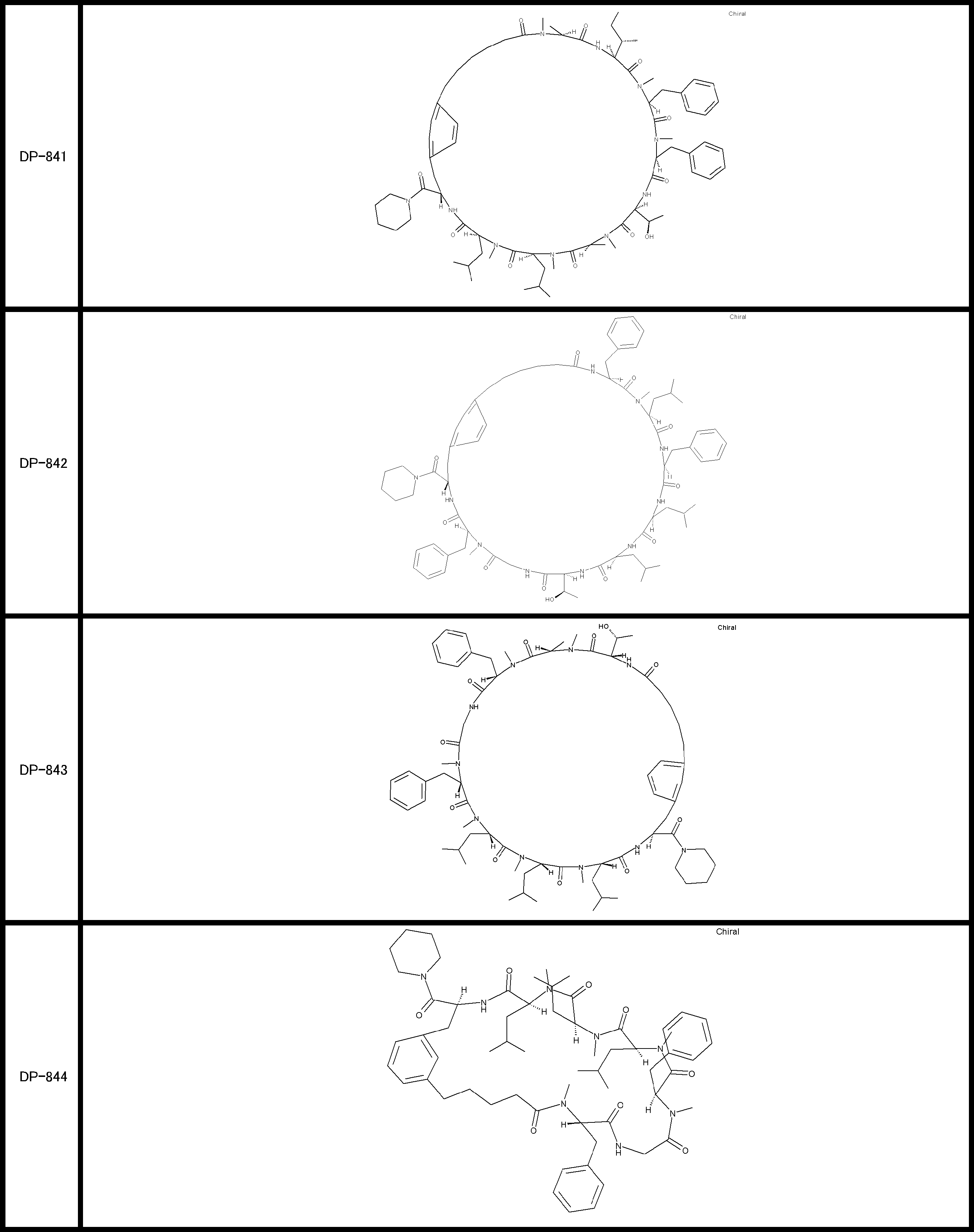

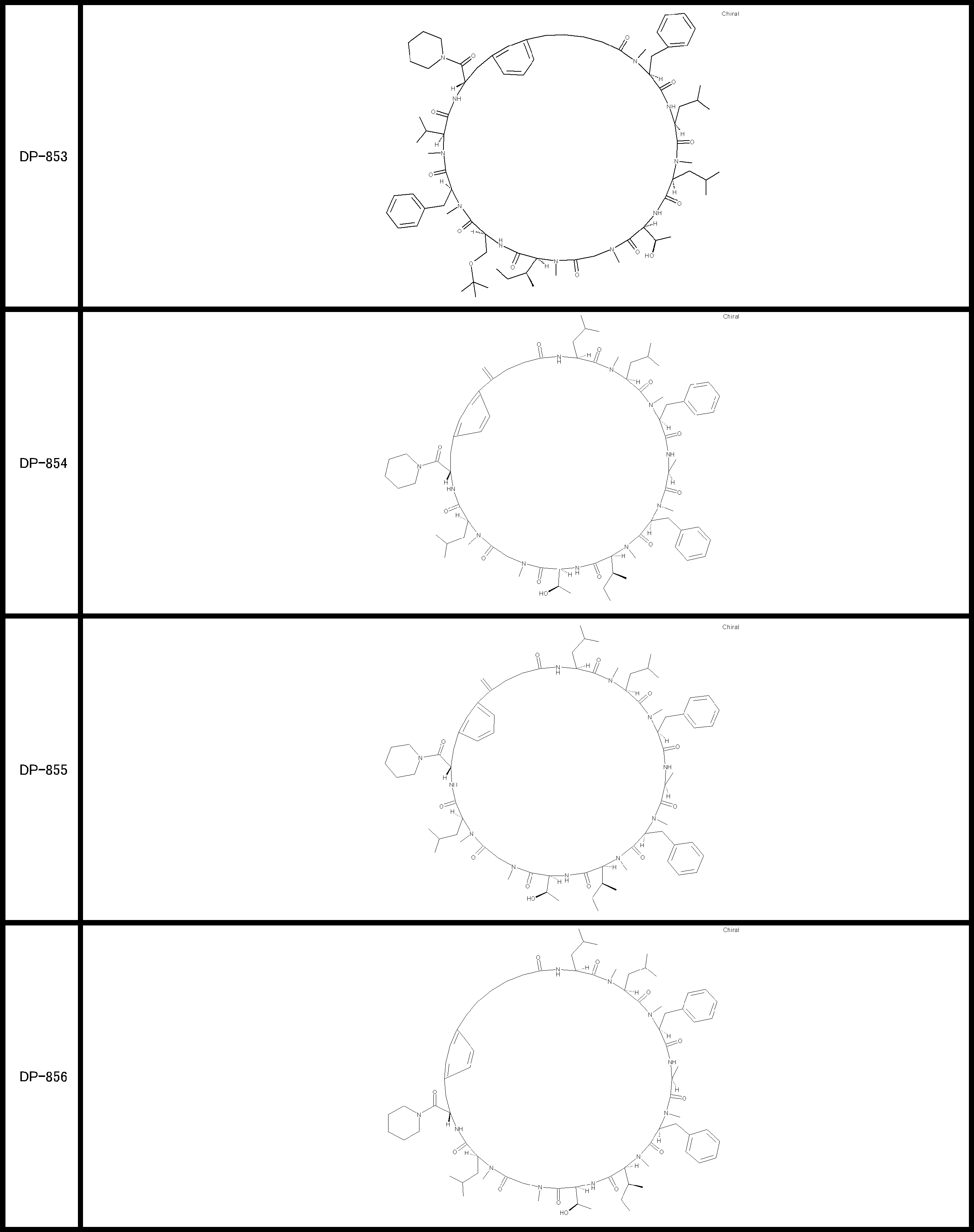
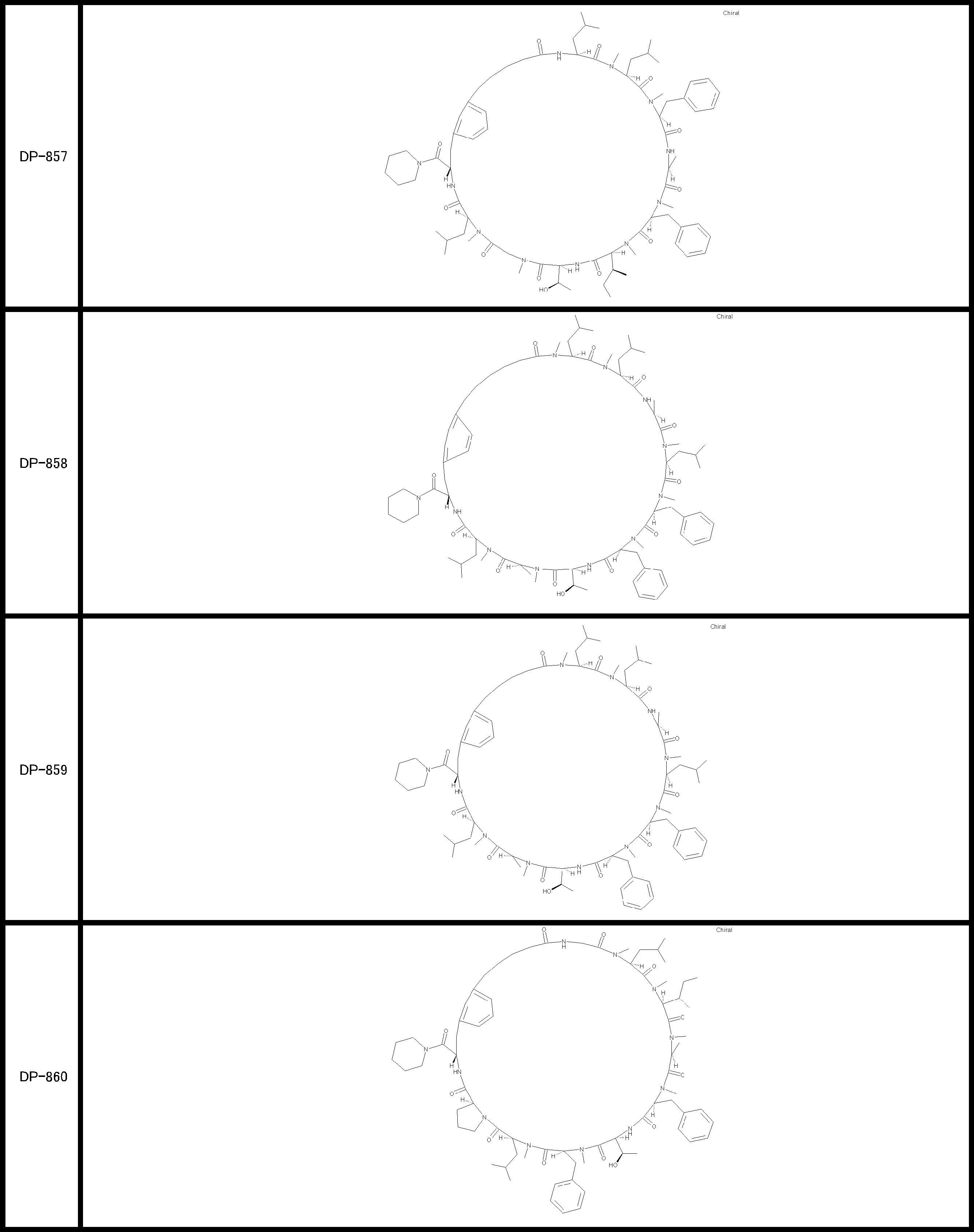


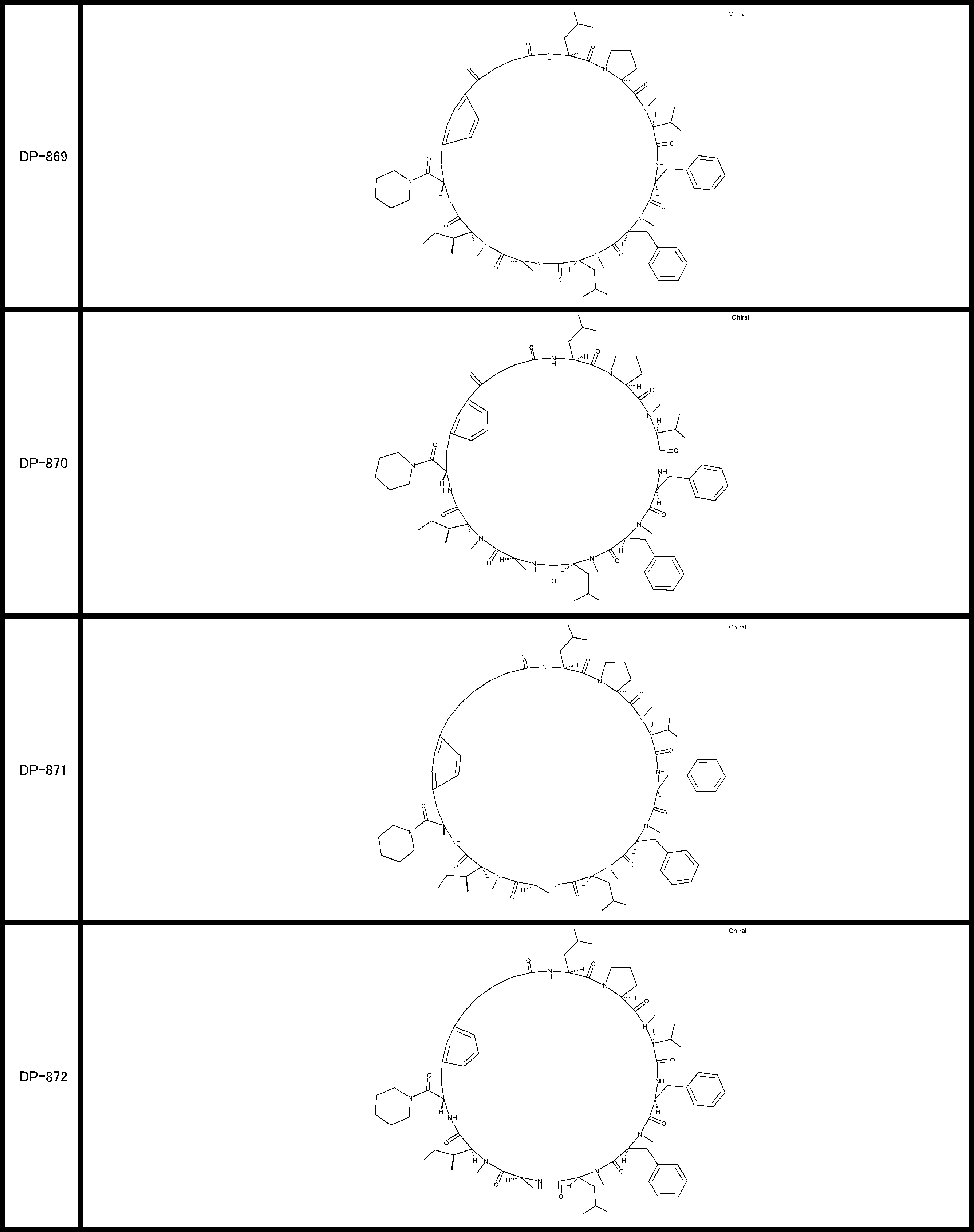
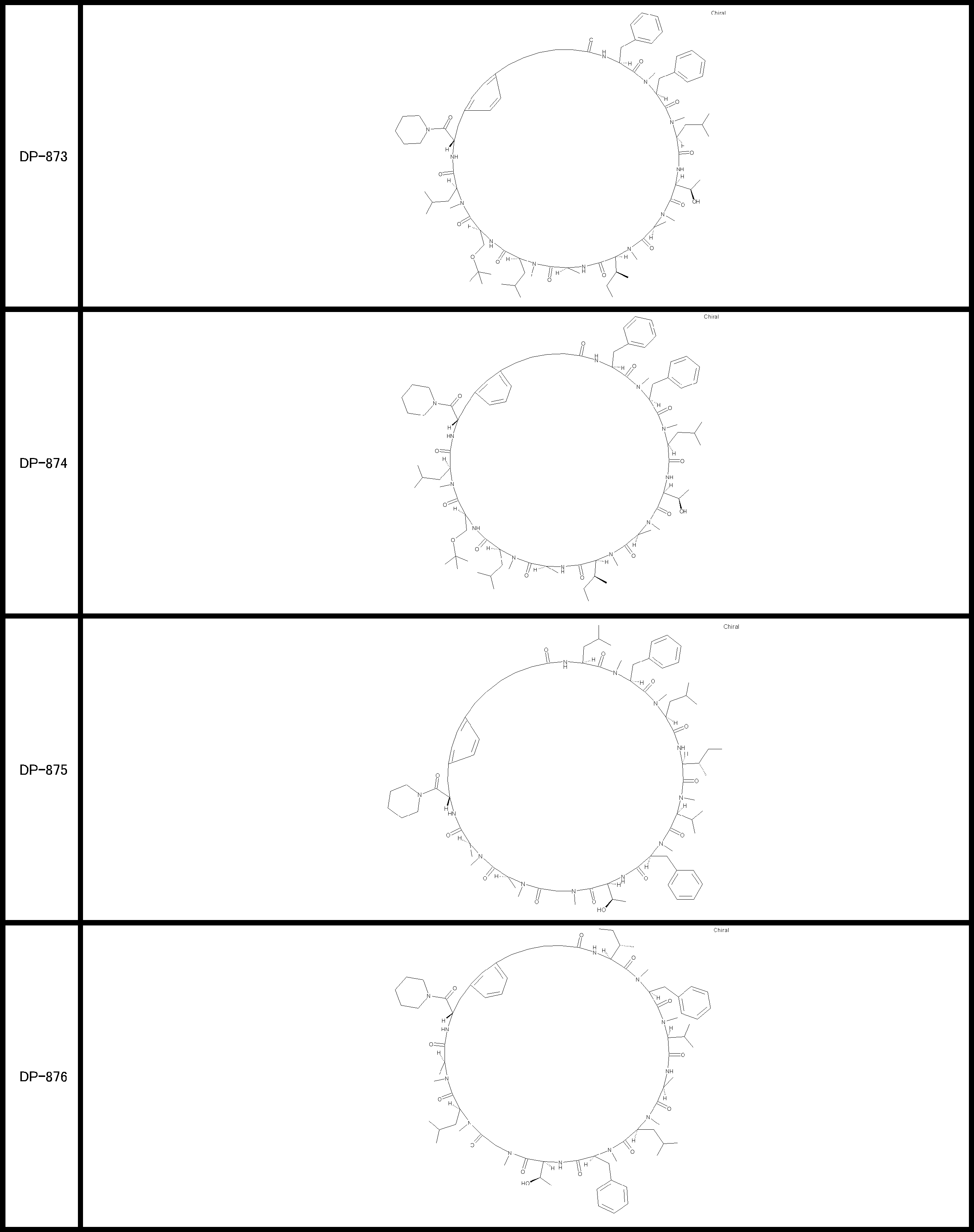

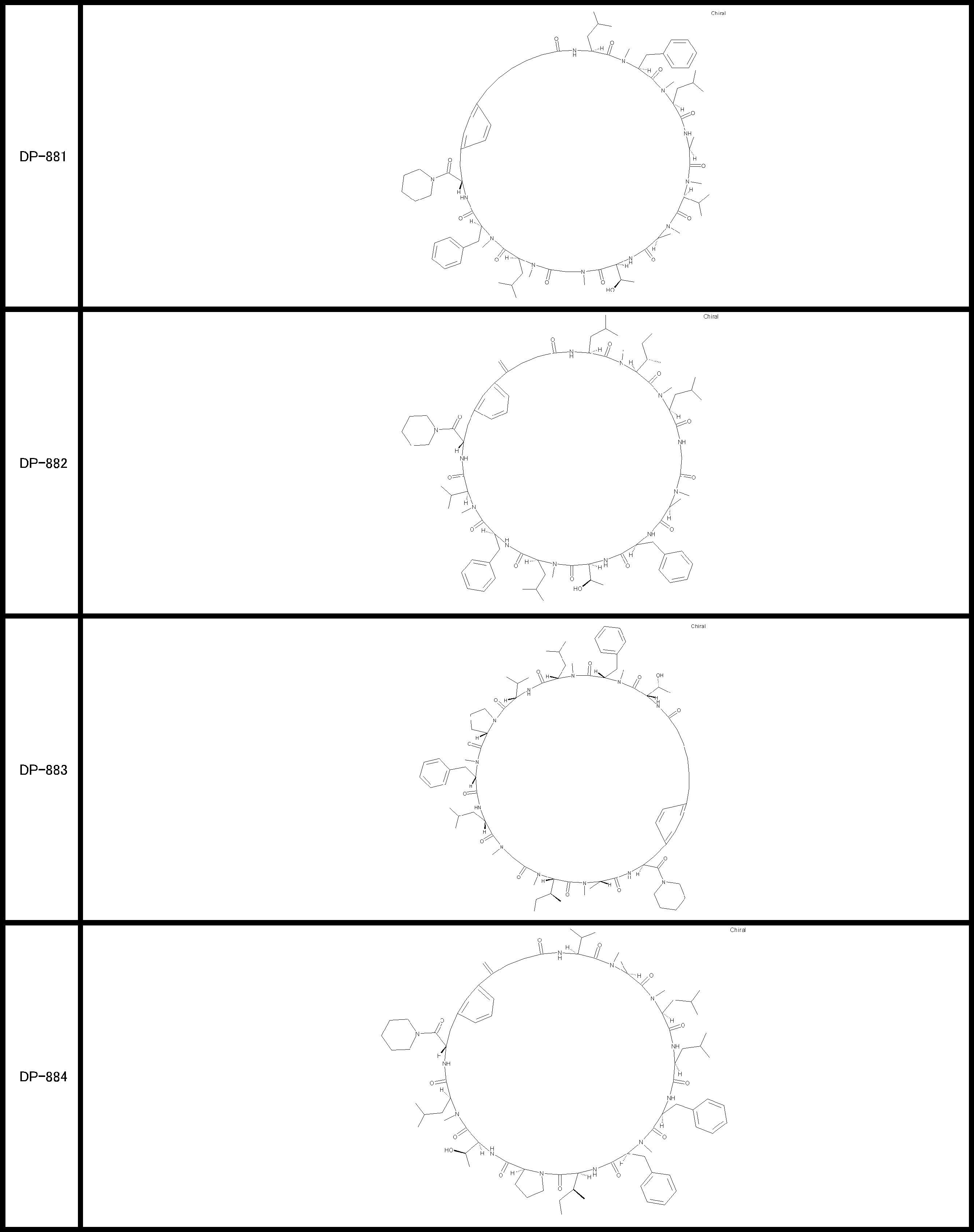
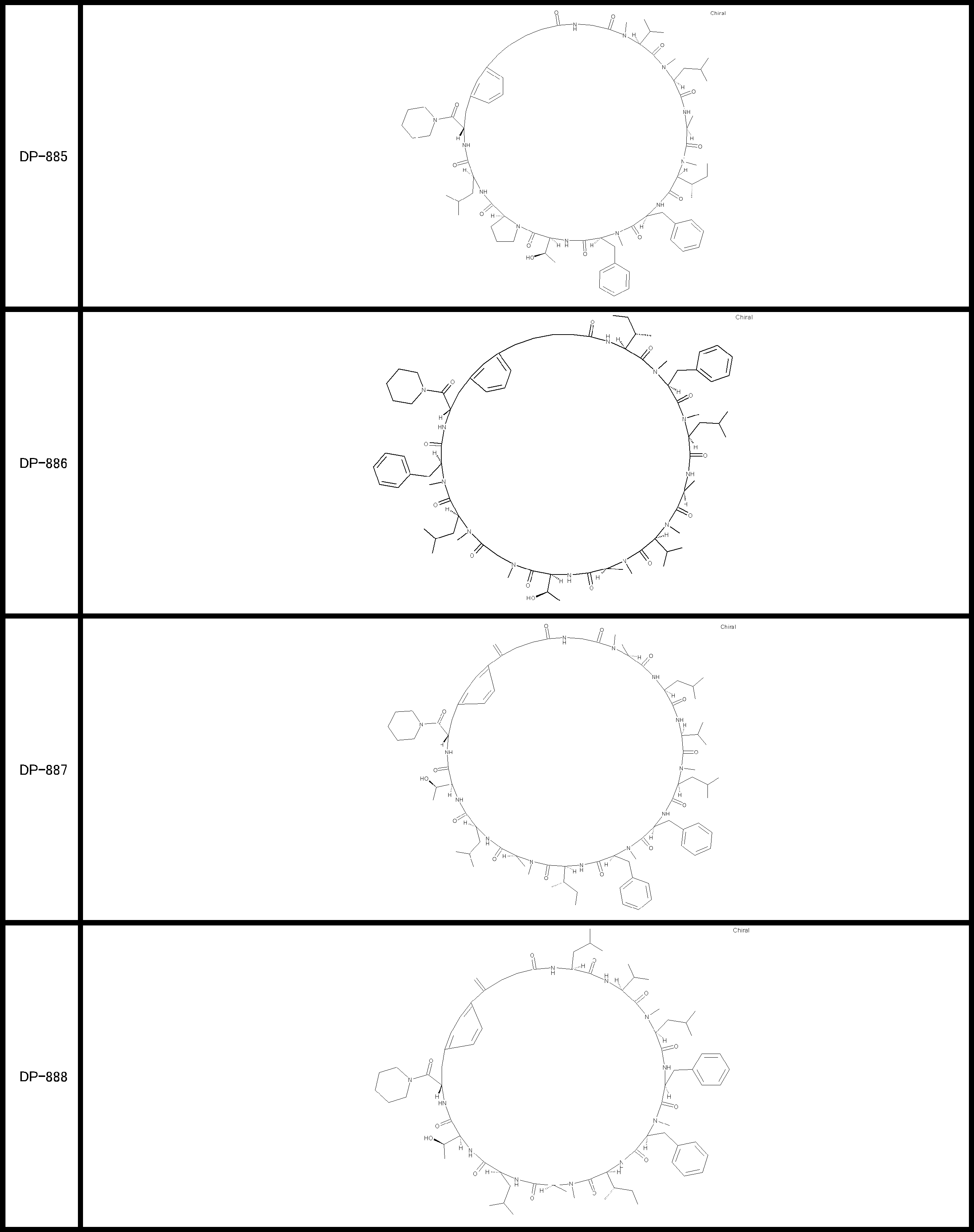
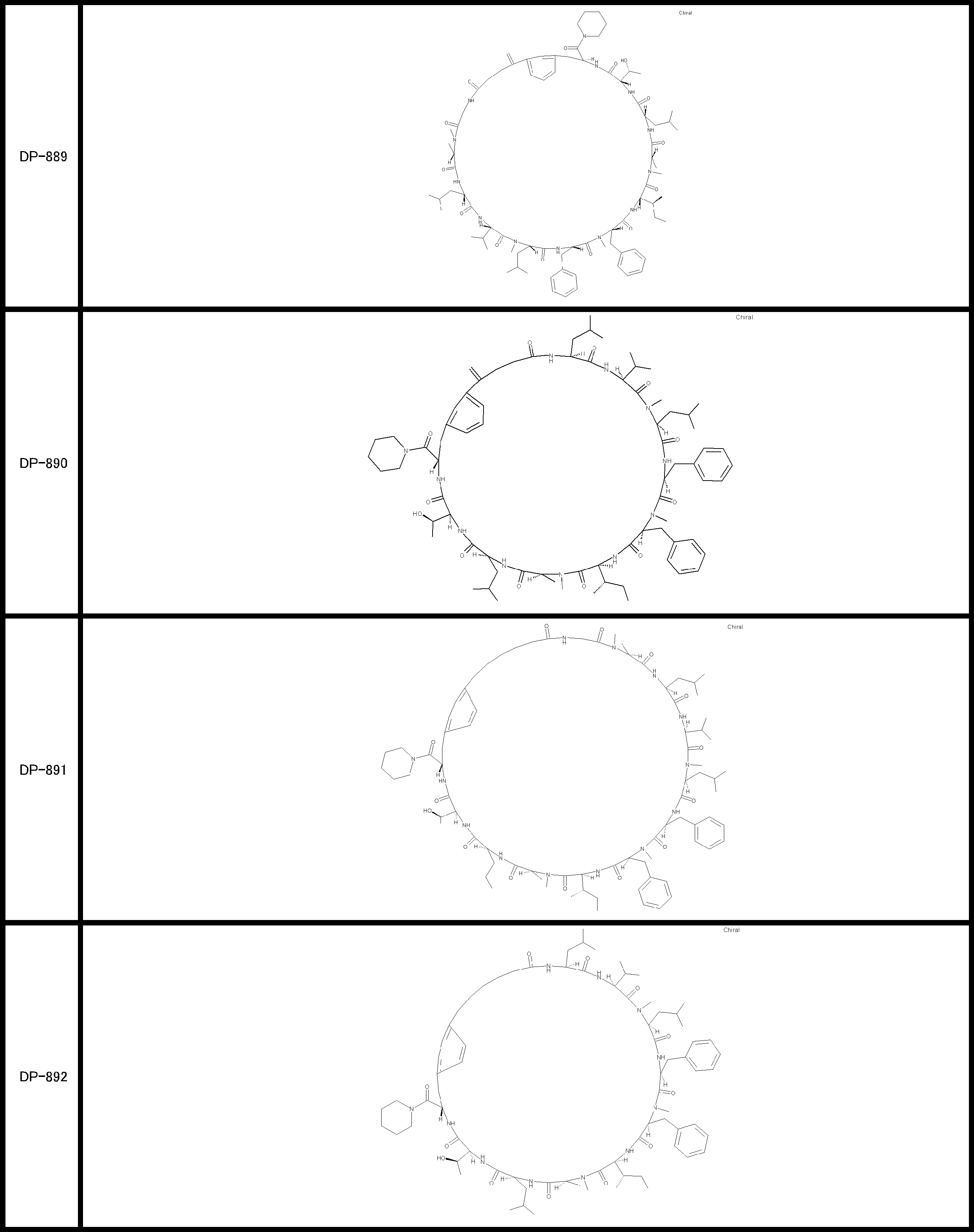

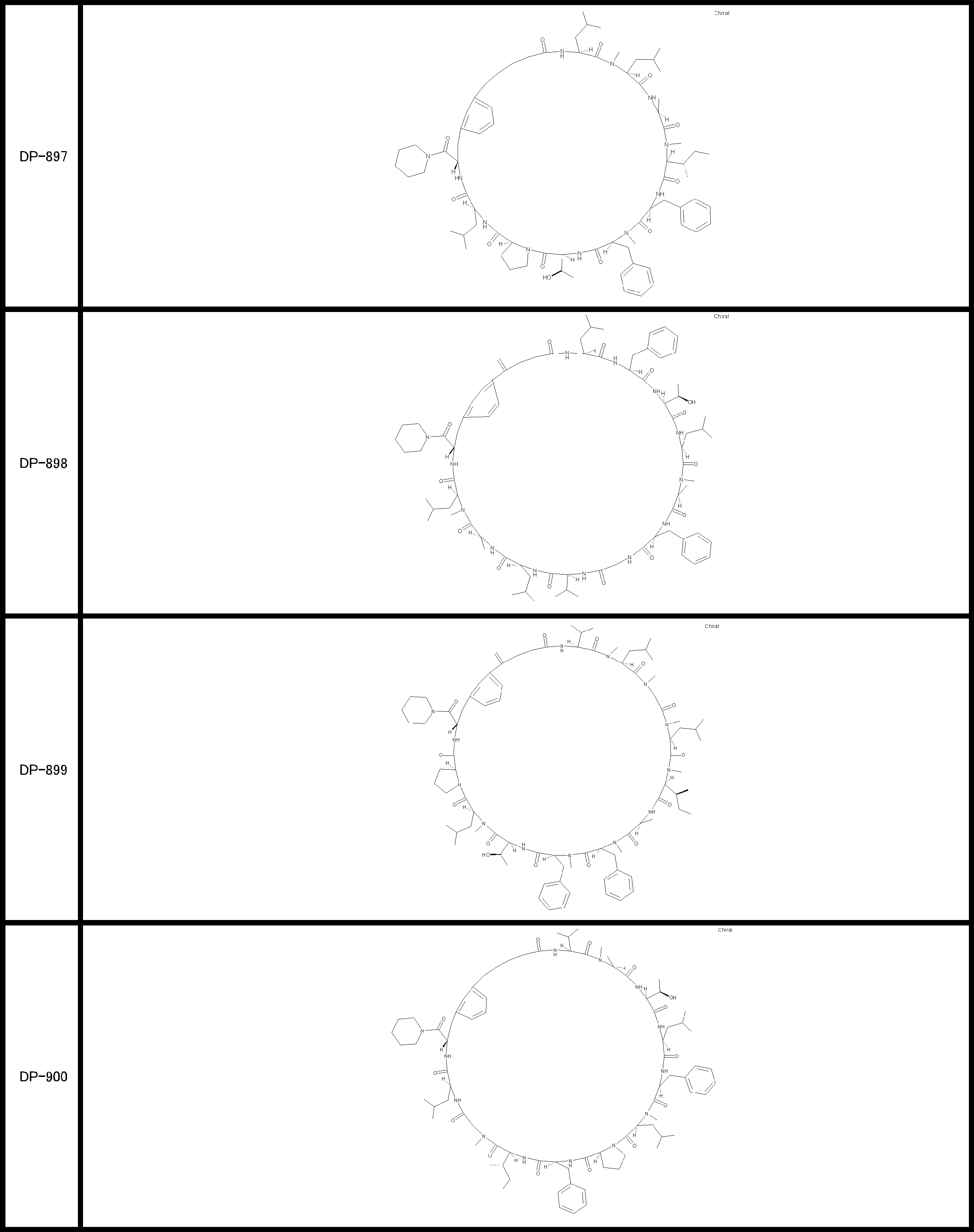

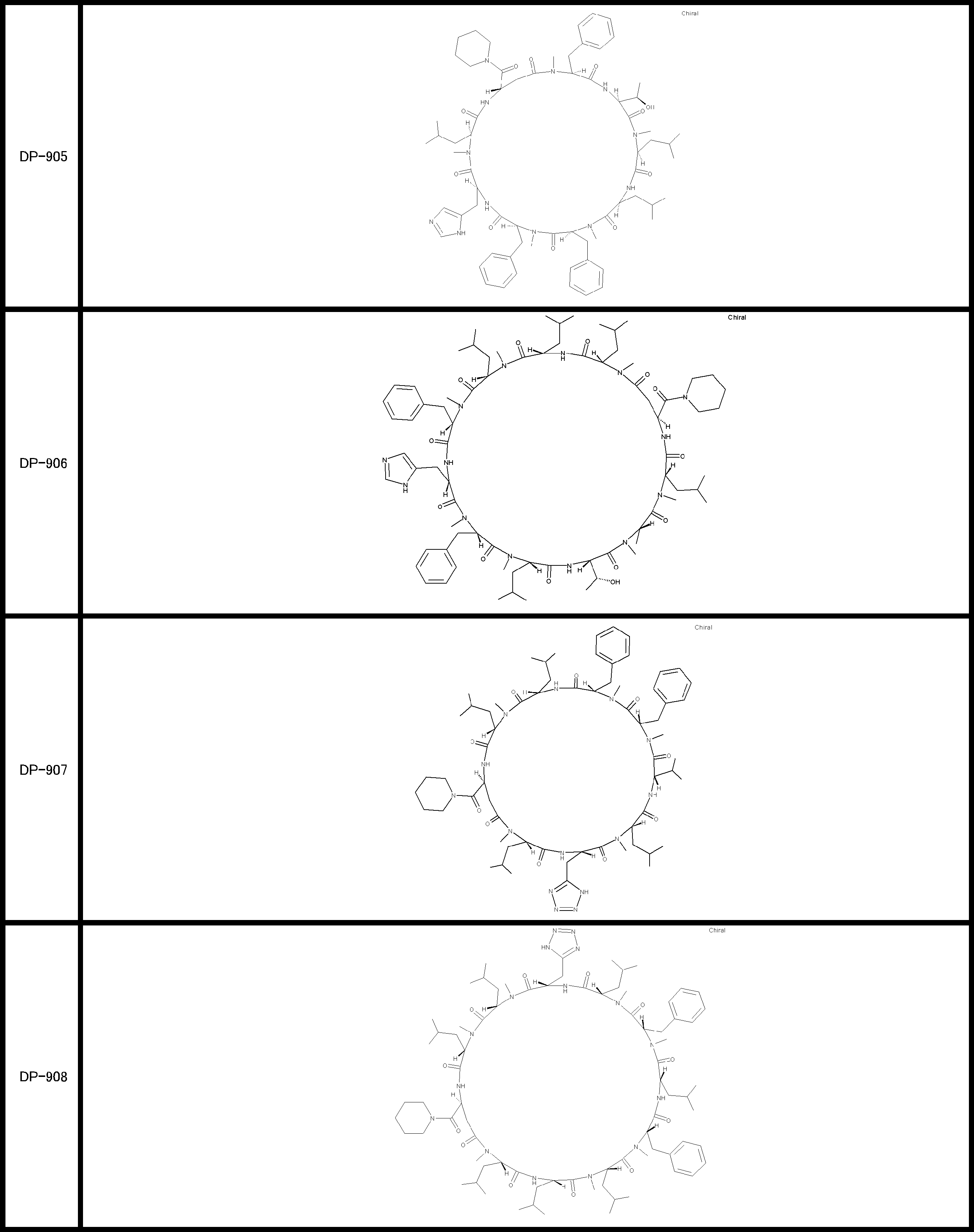






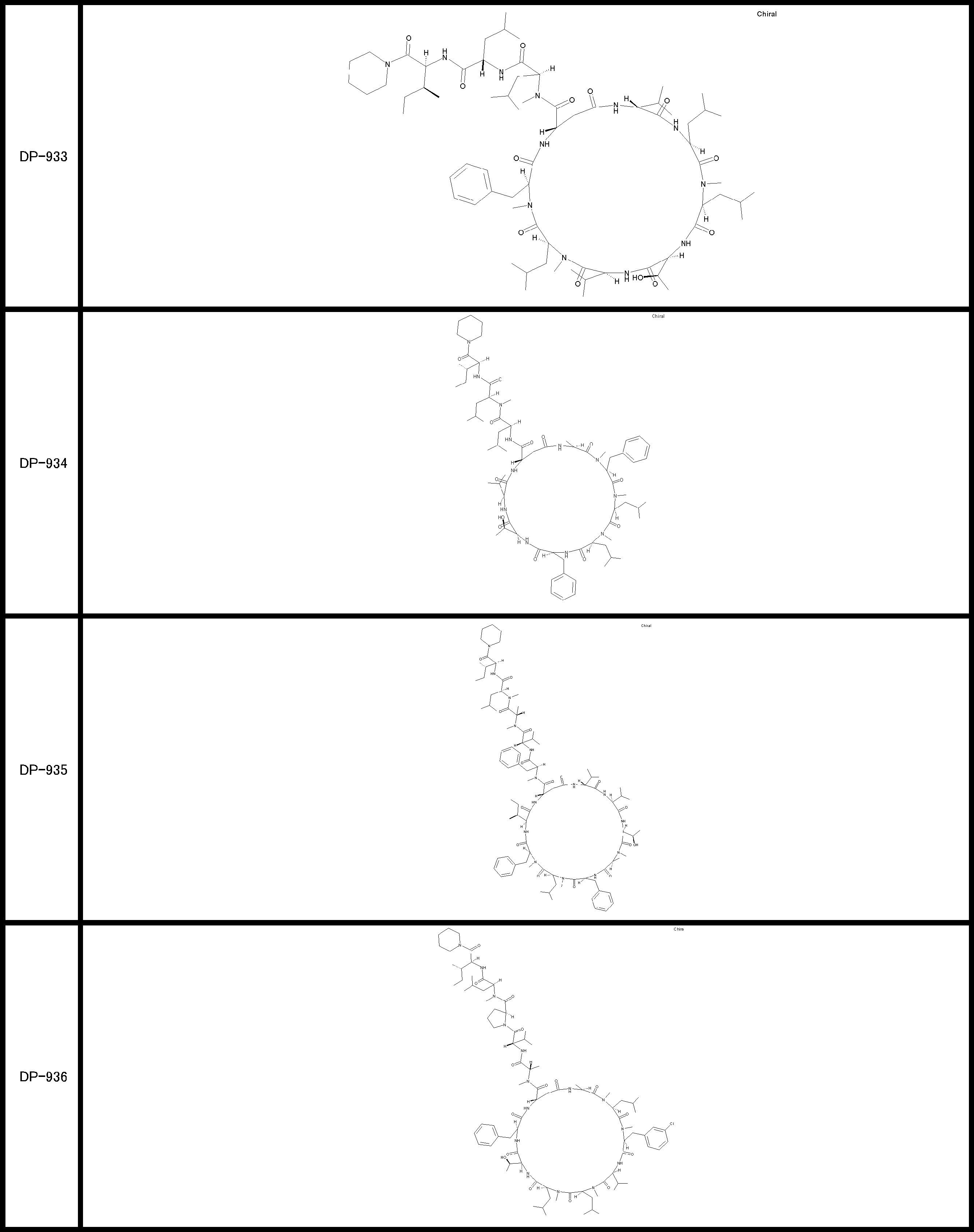





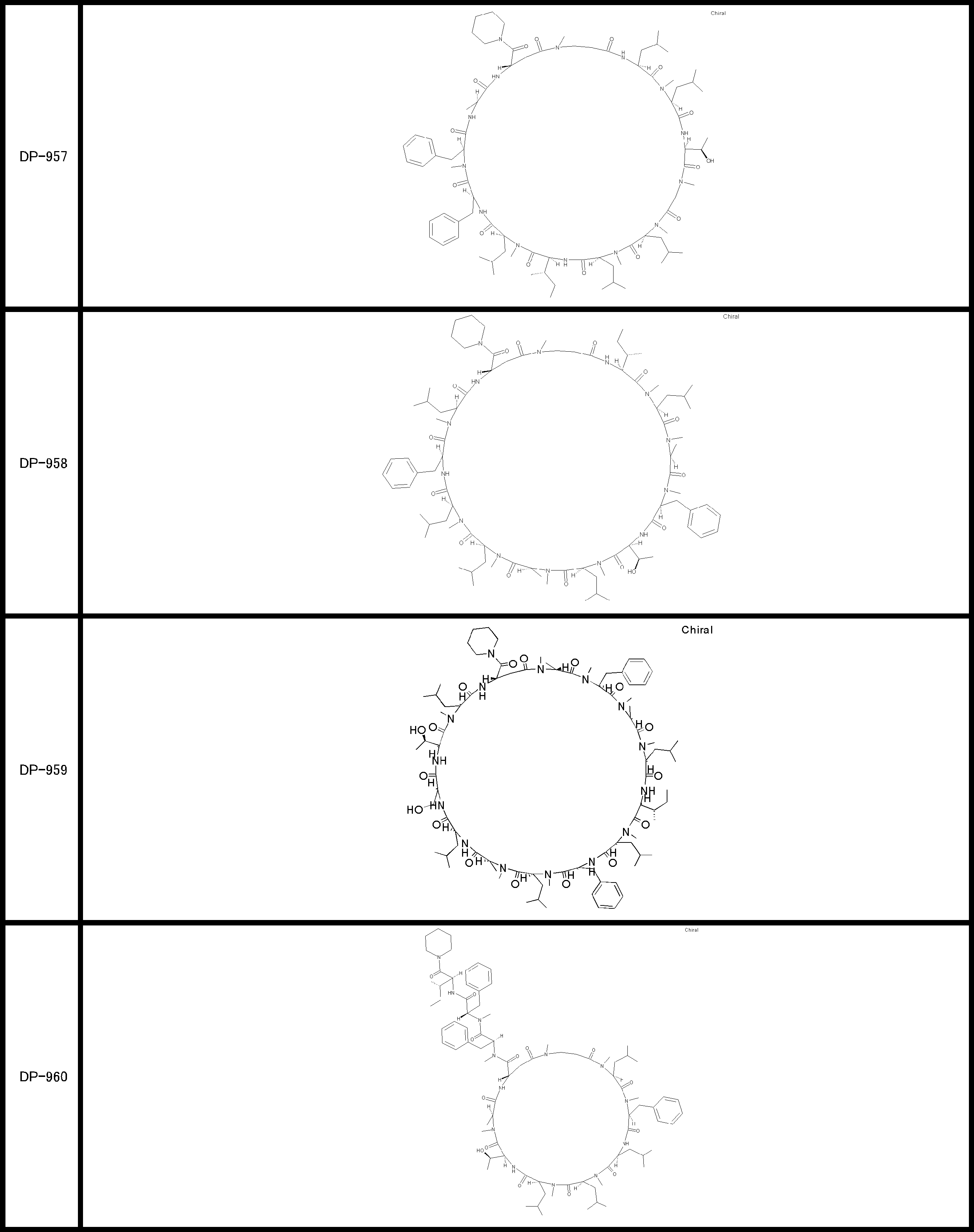
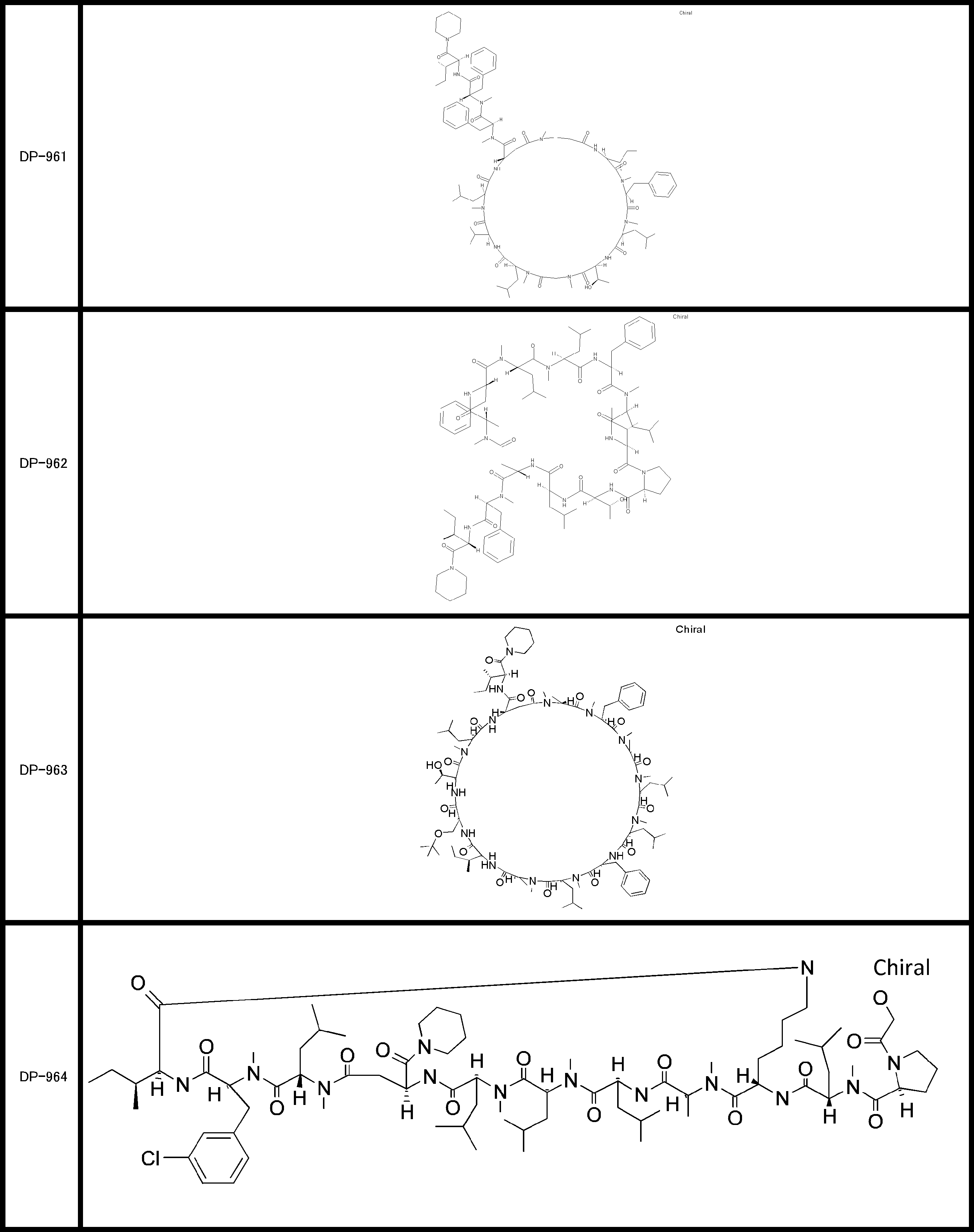
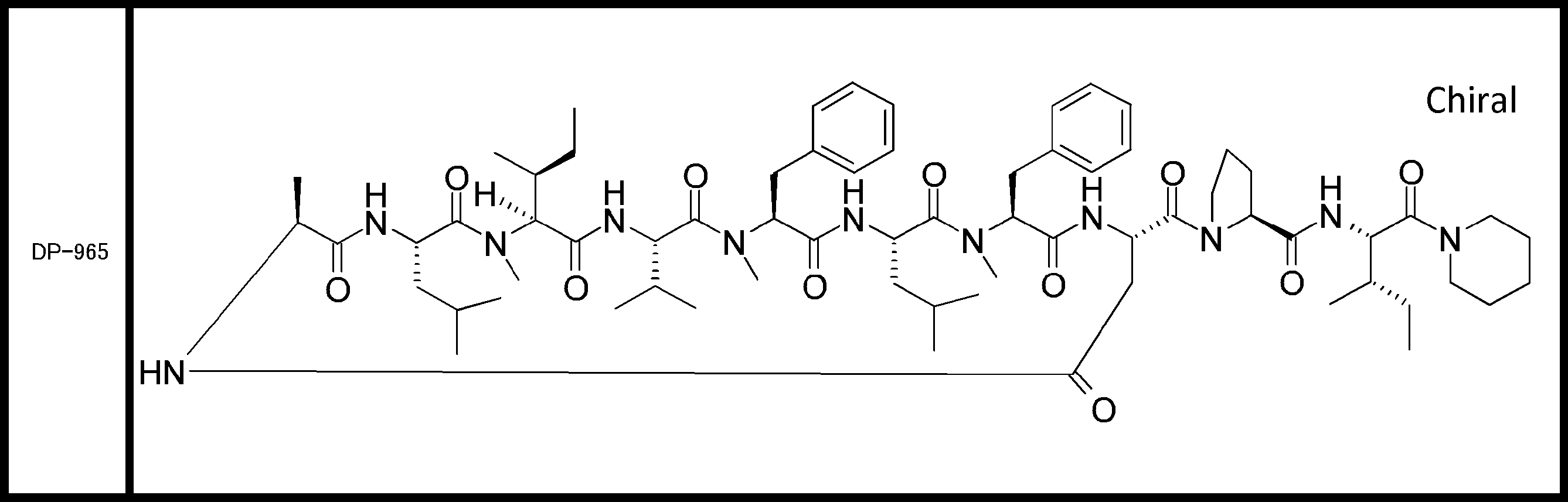

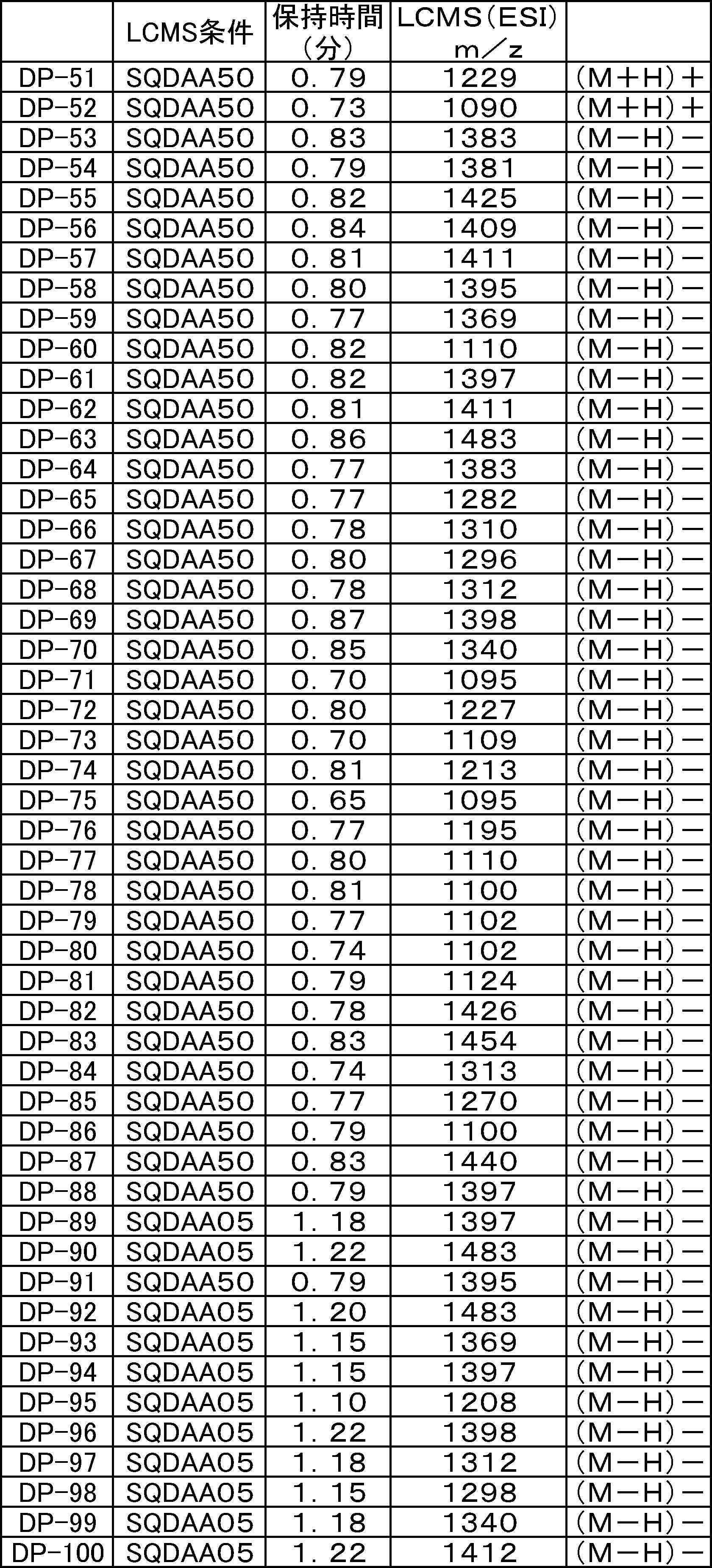
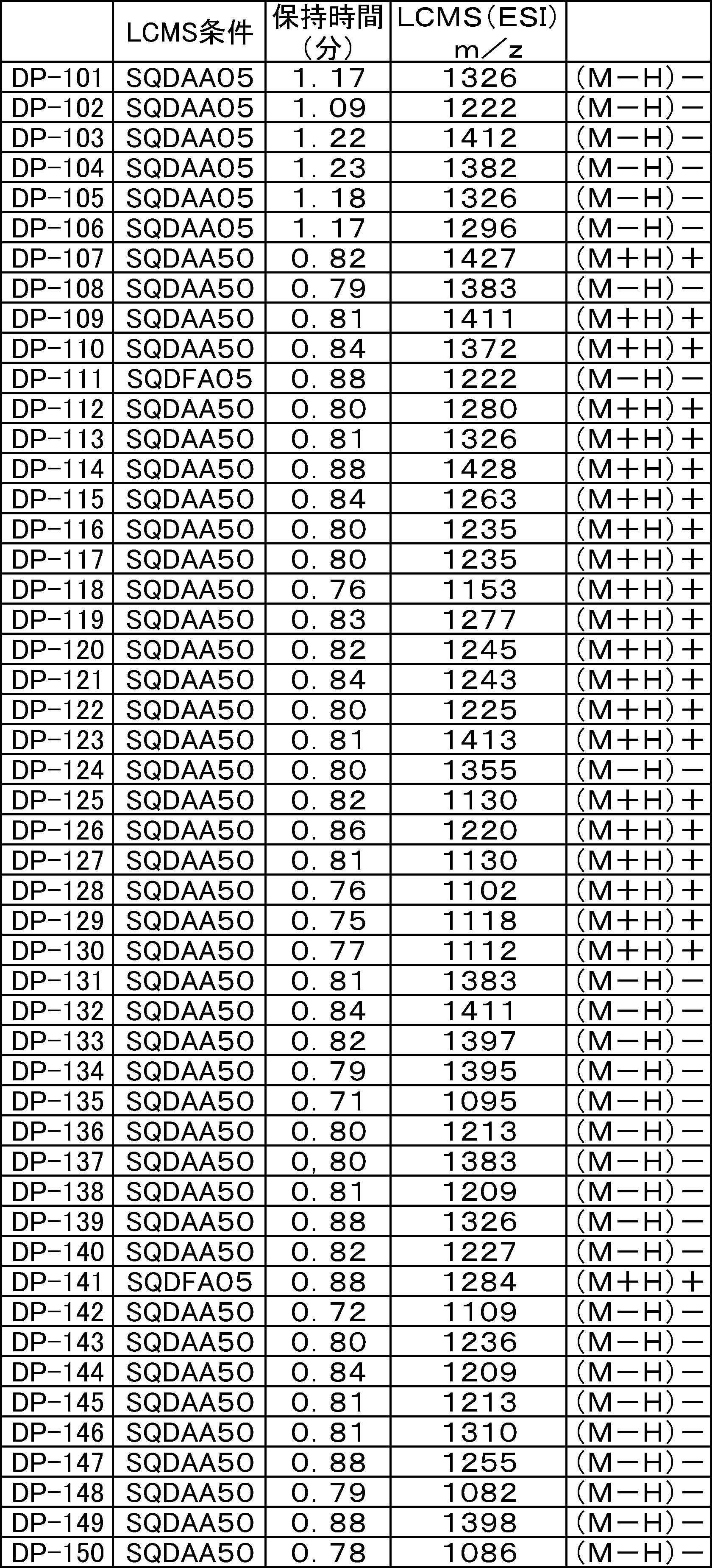

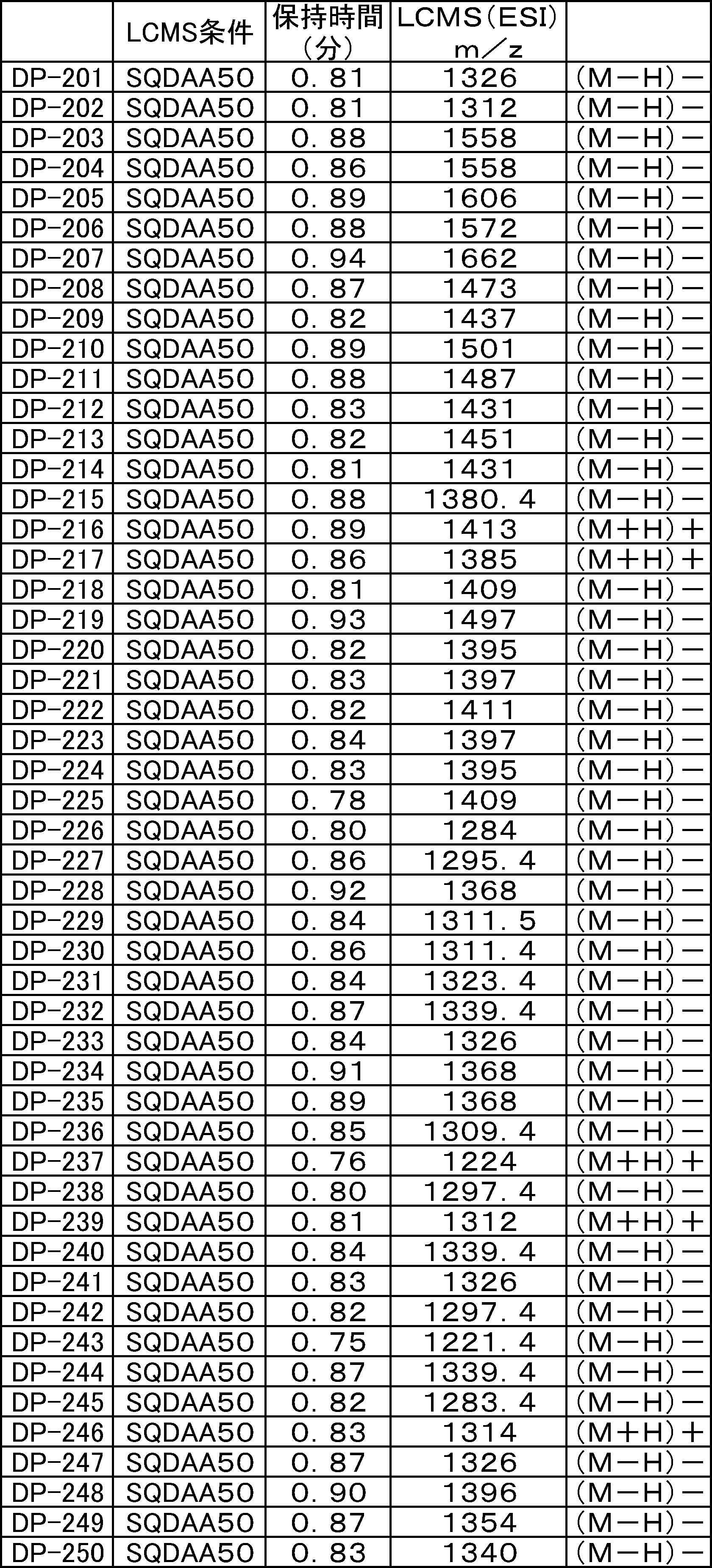

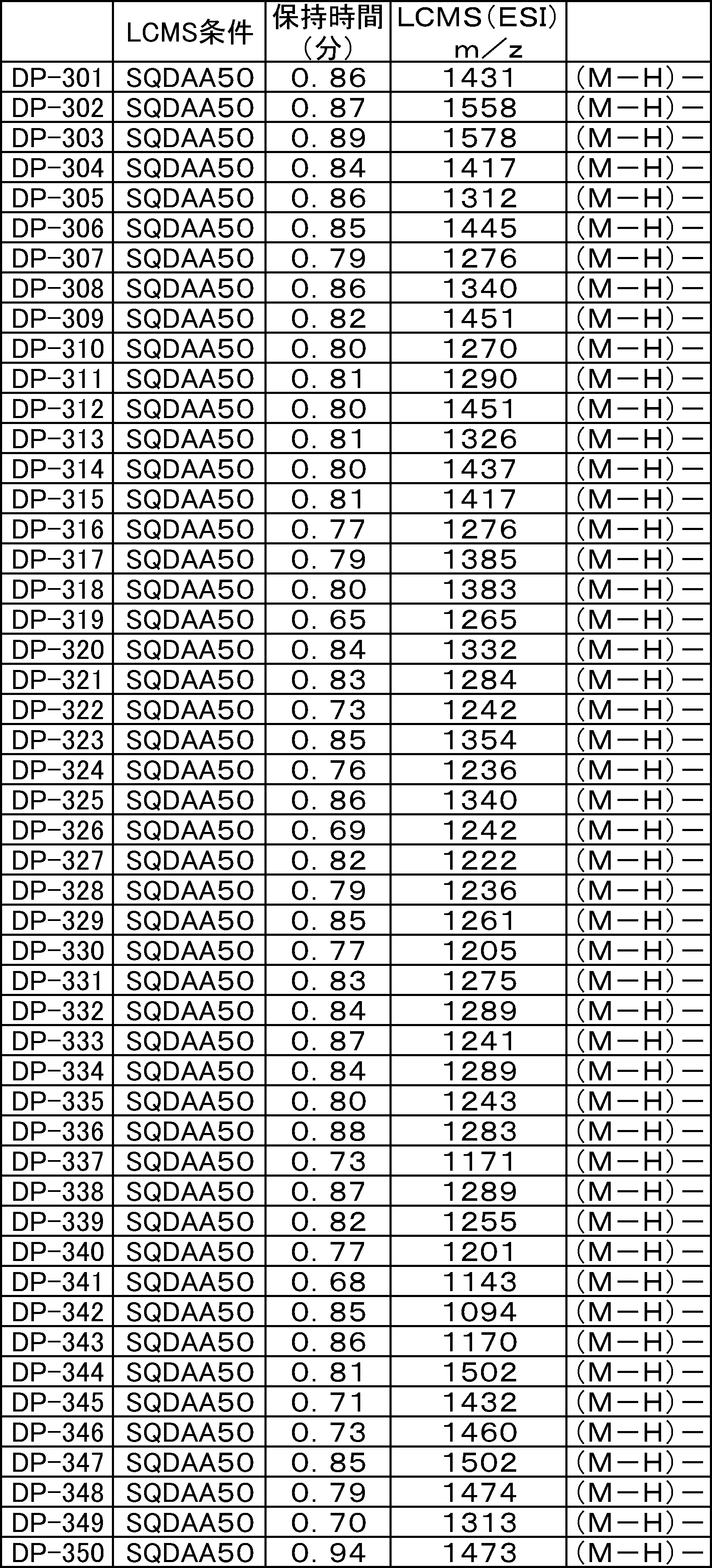
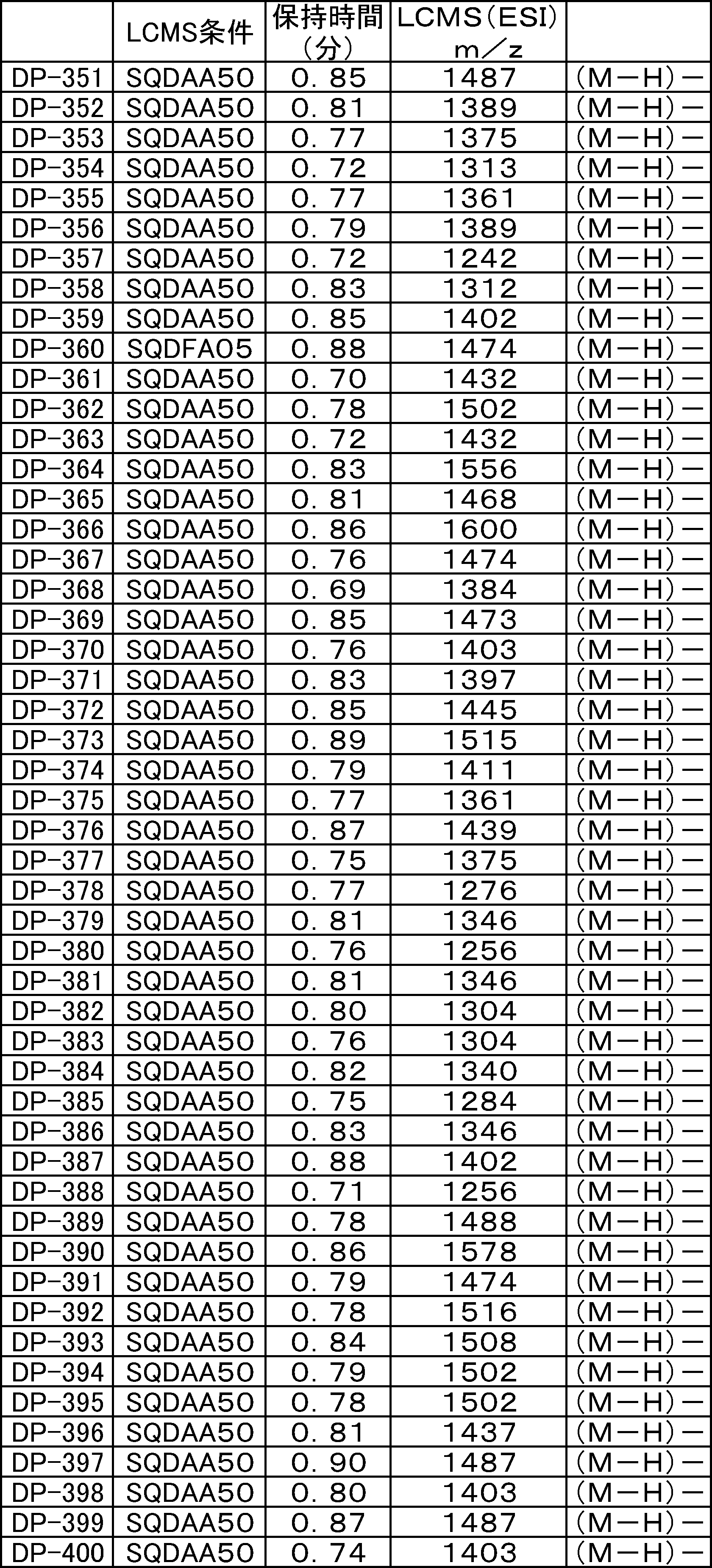



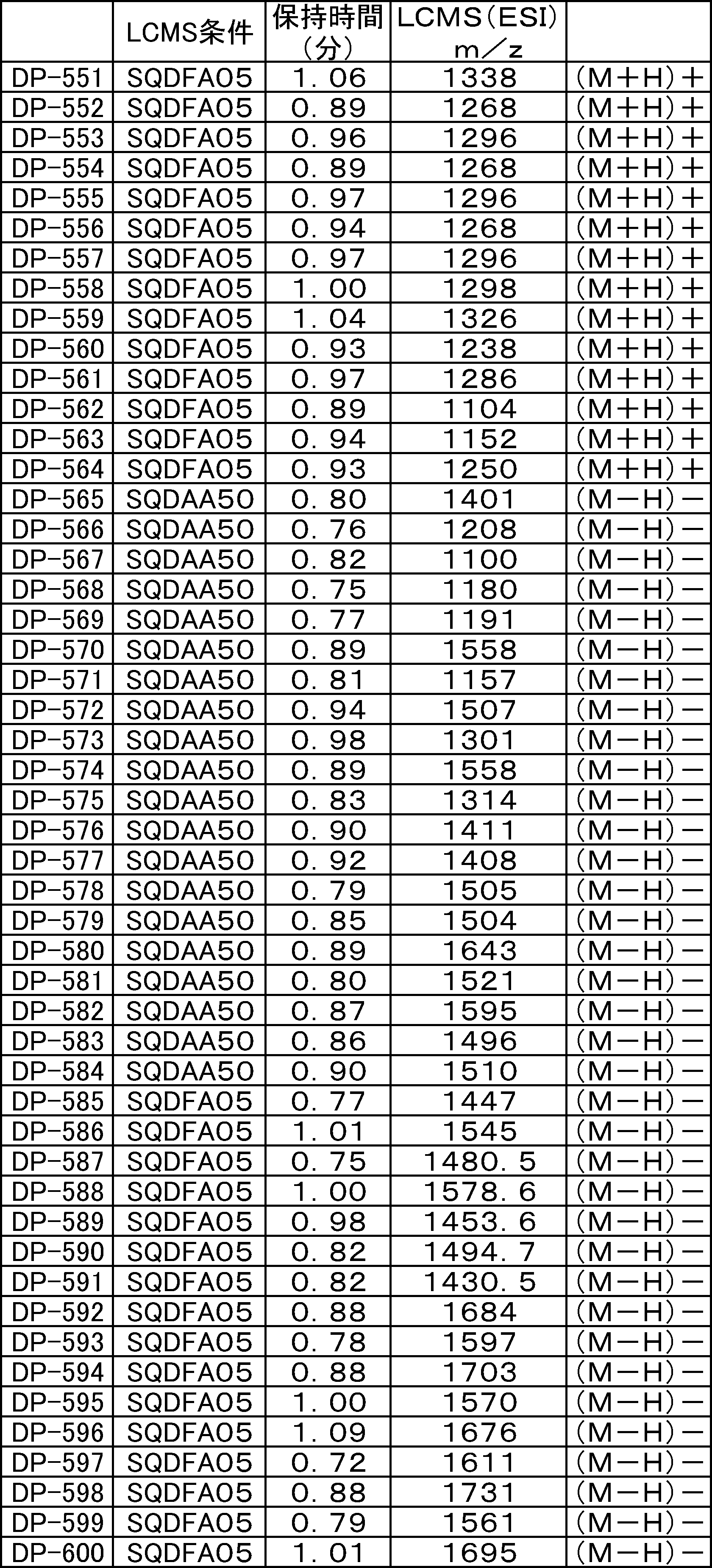
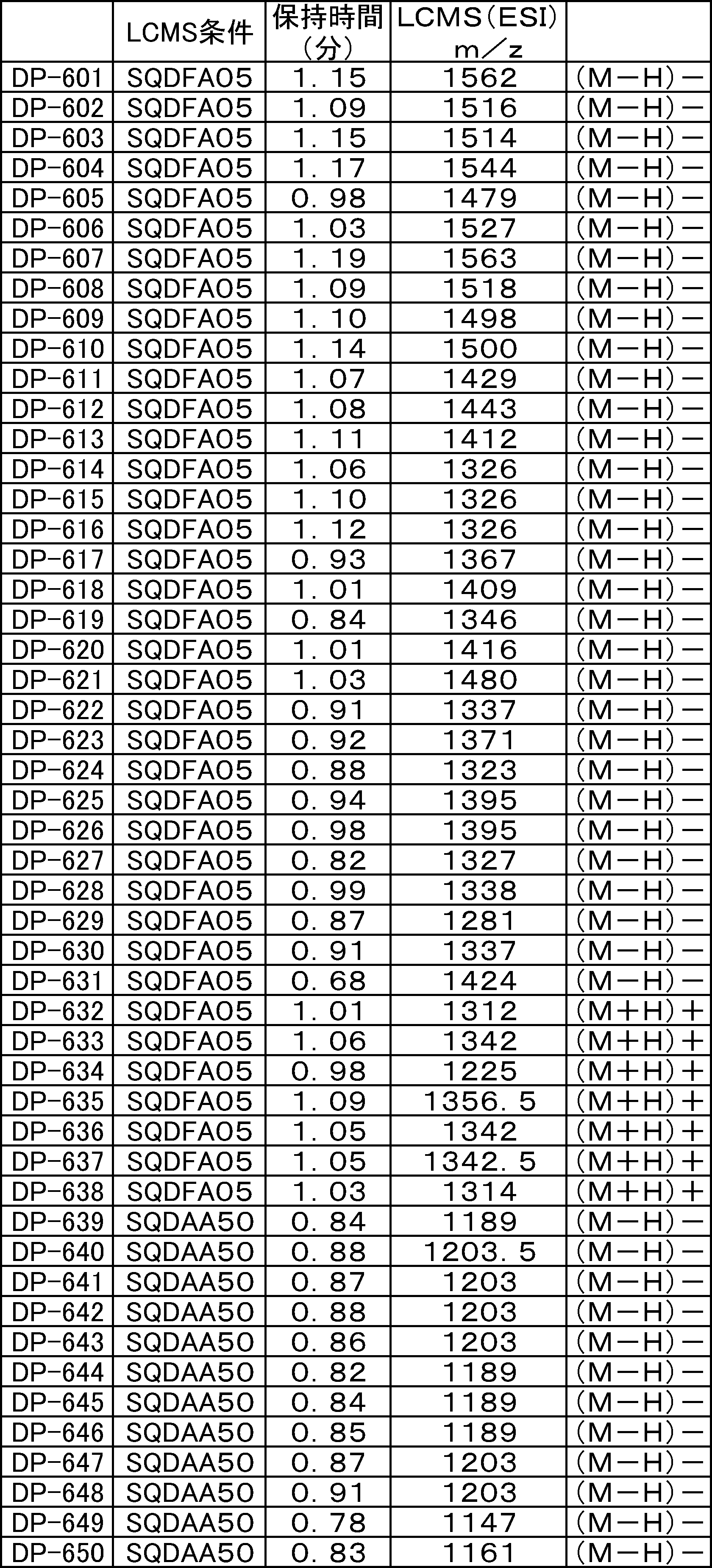





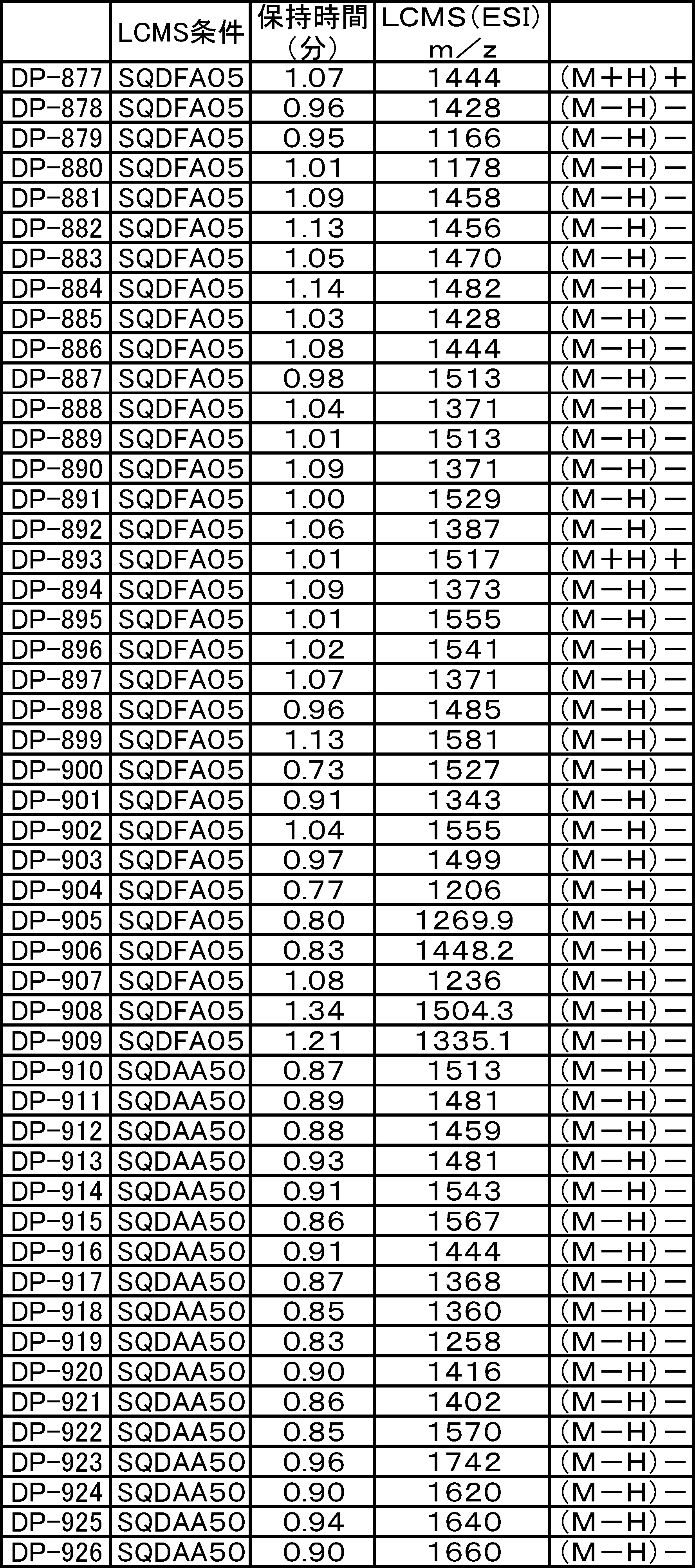


固定された直鎖部(MePhe-Ala-pip及び、MePhe-MePhe-Ala-pip)を有するペプチドの合成
C末端がアミド化(本例ではピペリジン化)され、C末端以外に配置されたアスパラギン酸側鎖カルボン酸とN末端アミノ基がアミド結合により環化された合成例を述べる。本例を1つの代表例として述べるが、ペプチド化学合成については、本明細書の異なる場所(例えば142、512(実施例15の2-2)、553(実施例18)など)の各所に記載されているどの手法を用いてもよい。以下のスキームXにその合成の一例を示した。
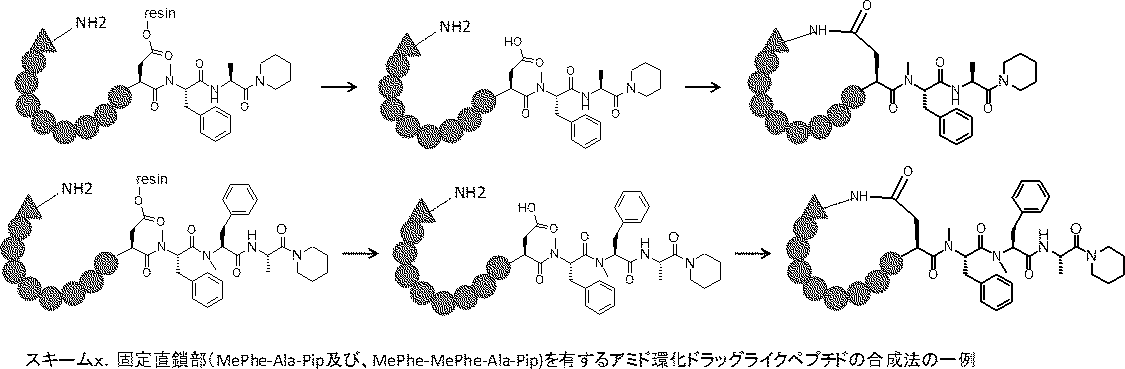
C末端がアミド化(本例ではピペリジン化)され、C末端以外に配置されたアスパラギン酸側鎖カルボン酸とN末端アミノ基がアミド結合により環化された合成例を述べる。本例を1つの代表例として述べるが、ペプチド化学合成については、本明細書の異なる場所の各所に記載されているどの手法を用いてもよい。以下のスキームG2にその合成の一例を示した。

C末端以外に配置されたアスパラギン酸側鎖カルボン酸がアミド化され(本例ではピペリジンであるが直鎖部1を構成するペプチドが結合していてもよい)、N末端以外に配置されたアミノ基側鎖を有するアミノ酸の側鎖アミノ基とC末端に配置されたアミノ酸のカルボン酸とで環化したペプチドの化学合成例を述べる。ここでは直鎖部1および2を有するペプチドの例として、交差ユニット(○ユニット)としてAsp-pip、アミノ基ソースとして保護基のついたアミノ基側鎖を有するアミノ酸、N末端にカルボン酸類縁体を用いたケースを代表例として示した。本例でN末端に用いたHOGlyはヒドロキシル基を保護した化合物を用いても良い。ペプチド化学合成については、本明細書の異なる場所の各所に記載されているどの手法を用いてもよい。以下のスキームG3にその合成の一例を示した。
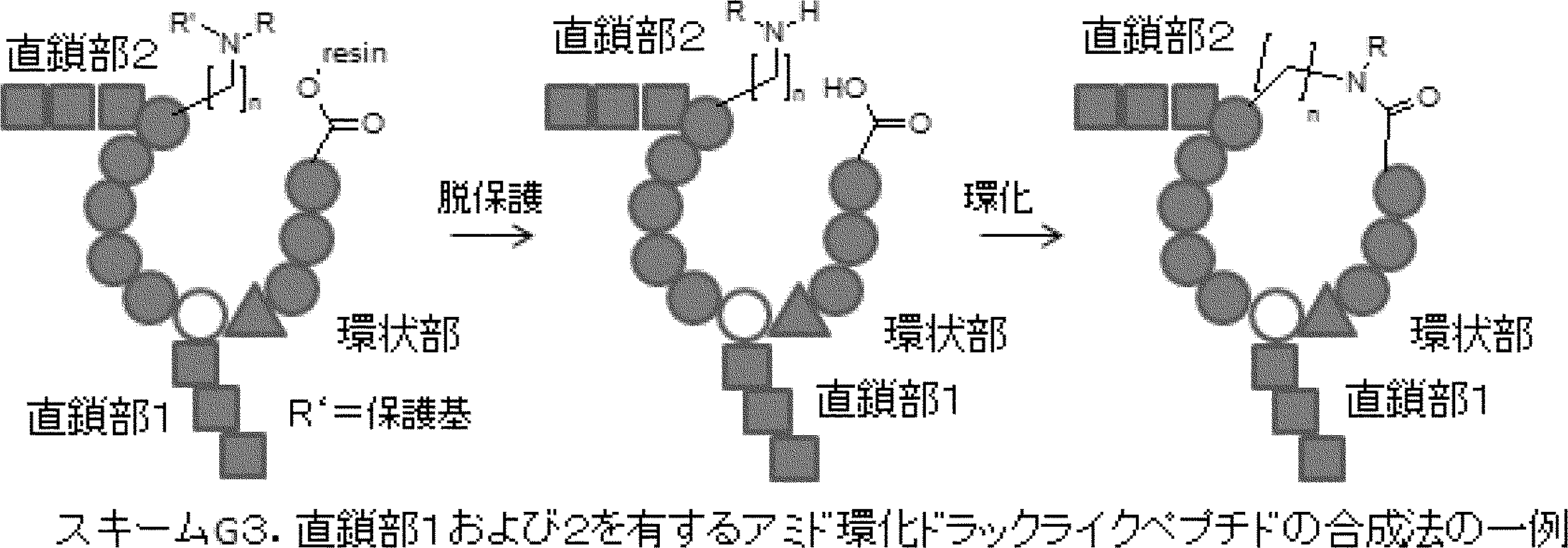
HOGly-Pro-MeLeu-*Lys-MeAla-Leu-MeLeu-MeLeu-Asp-piperidine-MeLeu-MePhe(3-Cl)-Ile*
(2つの*部位にて環化)
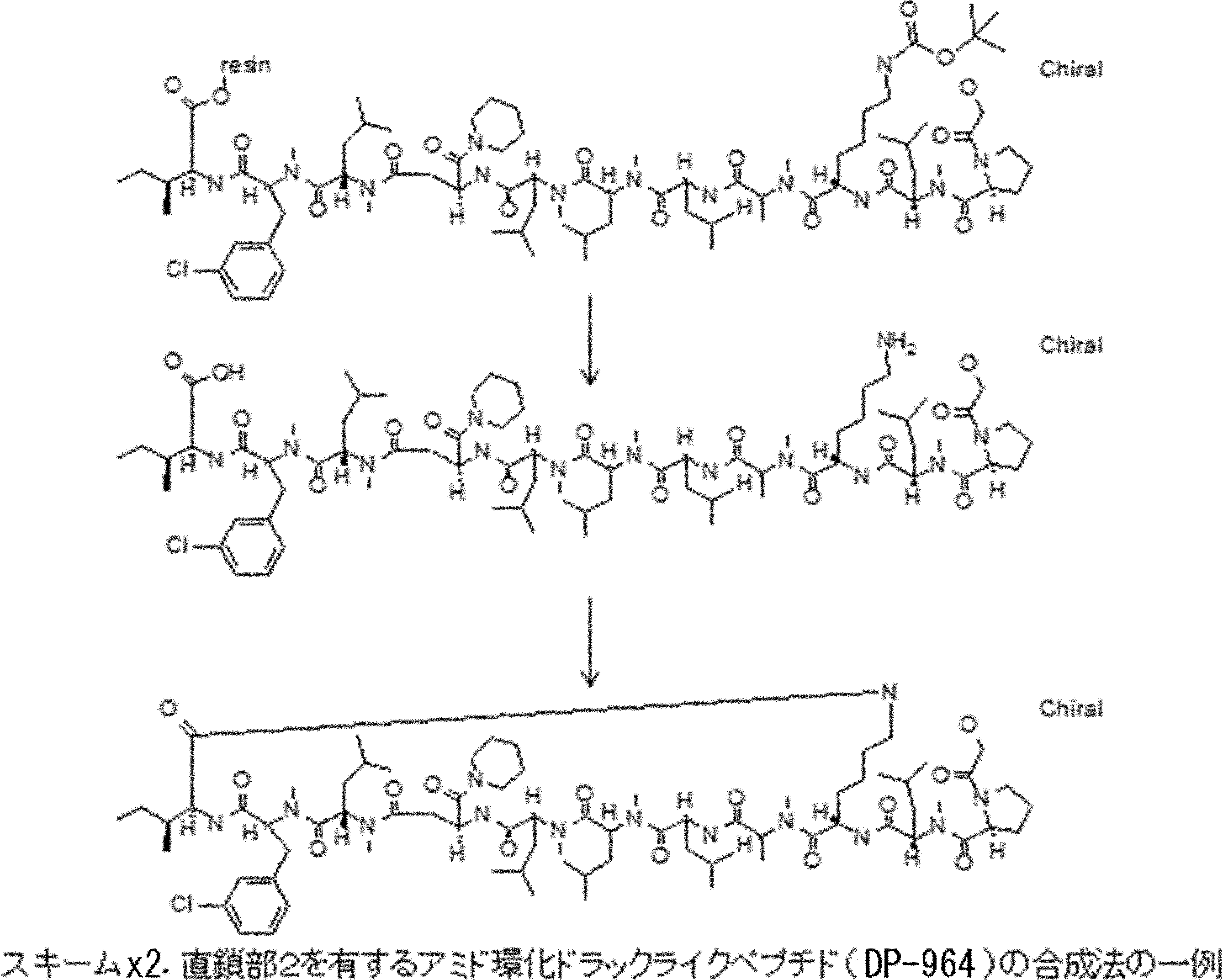
LCMS(ESI) m/z = 1479(M-H)-
保持時間:0.85分 (分析条件 SQDAA50)
化合物SP402と同様の手法にて合成したFmoc-Asp(O-Trt(2-Cl)-Resin)-pip(100mg)を用い、Fmocアミノ酸としてFmoc-MePhe-OH、Fmoc-MeAla-OH、Fmoc-MeLeu-OH、Fmoc-D-MeAla-OH、Fmoc-MeGly-OH、Fmoc-MeVal-OH、Fmoc-Pro-OH、Fmoc-Phe-OH、Fmoc-Val-OH、Fmoc-D-Val-OH、Fmoc-Ser(tBu)-OH、Fmoc-Thr(Trt)-OH、Fmoc-Leu-OH、Fmoc-D-Leu-OH、Fmoc-Ile-OH、Fmoc-Ala-OH、Fmoc-D-Ala-OH、Fmoc-Gly-OH、Fmoc-His(Mmt)-OH, Fmoc-Ala(5-Tet(Trt))-OH(化合物409)などを用いてペプチドの伸長を行った(アミノ酸の略語は表11-2に記載)。既に実施例に記載のFmoc法によるペプチド合成法に従い、ペプチドの伸長を行った。例えば、化合物DP-908の合成の場合、N末端のFmoc基を脱保護し、レジンにジクロロメタン/トリフルオロエタノール(=2/1、v/v、2ml)を加えて2時間振盪させ、レジンからのペプチドの切り出しを行った。反応終了後、チューブ内溶液を合成用カラムでろ過することによりレジンを除いた。さらにレジンにジクロロメタン/トリフルオロエタノール(=2/1、v/v、1ml)を加えて15分間振盪させ、上記の溶液とあわせて溶媒留去した。得られた残渣をDCM(8ml)に溶解し、DIPEA(17μL、0.096mmol)を加えた。これに1M-O-(7-アザー1Hベンゾトリアゾール-1-イル)-N,N,N,Nテトラメチルウロニウム ヘキサフルオロホスフェート(HATU)(30mg,0.08mmol)in DMSO(250μL)を加えてN末アミンとAsp側鎖カルボン酸との環化反応を行った。反応終了後、溶媒留去した。得られた残渣をトリフルオロエタノール(1.0ml)に溶解した。これにTFA/トリイソプロピルシラン/DCM(1/5/94、v/v/v、1.0ml)を加えて10分間攪拌し、保護基の脱保護を行った。反応終了後、DIPEA(50μL,0.286mmol)を加えてTFAを中和し、溶媒留去した。得られた粗生成物はジメチルスルホキシドに溶解し、得られたペプチド溶液は高速逆相クロマトグラフィー(HPLC)で精製し、溶媒留去した。得られた残渣をトリフルオロエタノール(1.5ml)に溶解し、水(0.5ml)とヘキサン(2ml)を加えて攪拌し、ヘキサン層を除去した。もう一度ヘキサン(2ml)を加えて攪拌し、ヘキサン層を除去した後に溶媒留去し,化合物DP-908(1.92mg,4.8%)を得た。上述と同様の方法、もしくはFmoc-Asp(O-Trt(2-Cl)-Resin)-pipの代わりに化合物SP455と同様に調製したFmoc-Asp(O-Trt(2-Cl)-Resin)-MePhe-Ala-pipを用い、表題のアミド環化ドラックライクペプチドの合成を行った。DP-585~586、DP-592、DP-676、DP-904~909が本合成法に準拠した方法で合成可能である。各化合物のマススペクトルの値と液体クロマトグラフィーの保持時間は表11-3-2に記載した。
化合物SP455と同様の手法にて合成したFmoc-Asp(O-Trt(2-Cl)-Resin)-MePhe-Ala-pip(100mg)を用い、Fmocアミノ酸としてFmoc-MePhe-OH、Fmoc-MeAla-OH、Fmoc-MeLeu-OH、Fmoc-MeGly-OH、Fmoc-MeVal-OH、Fmoc-Pro-OH、Fmoc-Phe-OH、Fmoc-Val-OH、Fmoc-D-Val-OH、Fmoc-Ser(tBu)-OH、Fmoc-Thr(Trt)-OH、Fmoc-Leu-OH、Fmoc-Ala-OH、Fmoc-D-Ala-OH、Fmoc-Gly-OH、Fmoc-Glu(OAl)-OHなどを用いてペプチドの伸長を行った(アミノ酸の略語は表11-2に記載)。既に実施例に記載のFmoc法によるペプチド合成法に従い、ペプチドの伸長を行った。例えば、化合物DP-595の合成の場合、N末端のFmoc基を脱保護し、レジンにジクロロメタン/トリフルオロエタノール(=2/1、v/v、2ml)を加えて2時間振盪させ、レジンからのペプチドの切り出しを行った。反応終了後、チューブ内溶液を合成用カラムでろ過することによりレジンを除いた。さらにレジンにジクロロメタン/トリフルオロエタノール(=2/1、v/v、1ml)を加えて15分間振盪させ、上記の溶液とあわせて溶媒留去した。得られた残渣をDCM(8ml)に溶解し、DIPEA(20μL、0.115mmol)を加えた。これに1M O-(7-アザー1Hベンゾトリアゾール-1-イル)-N,N,N,Nテトラメチルウロニウム ヘキサフルオロホスフェート(HATU)(37mg,0.096mmol)in DMSO(200μL)を加えてN末端アミンとAsp側鎖カルボン酸との環化反応を行った。反応終了後、溶媒留去した。得られた粗生成物をDMFに溶解し、テトラキス(トリフェニルホスフィン)パラジウム(0)(Pd(PPh3)4)(7mg、0.006mmol)のジクロロメタン(1ml)溶液を加えた。続いてフェニルシラン(7.4ul, 0.06mmol)を加え、室温で1時間攪拌した。反応液を濾過後、有機層を減圧濃縮し、得られた粗生成物をDMSOに溶解し、得られたペプチド溶液は高速逆相クロマトグラフィー(HPLC)で精製後、溶媒を留去し、化合物DP-595(14.7mg,22%)を得た。上述と同様の方法でアミド環化ドラックライクペプチドDP-589、DP-596の合成を行った。各化合物のマススペクトルの値と液体クロマトグラフィーの保持時間は表11-3-2に記載した。
C末端にベータアミノ酸誘導体を用い、C末端のベータアミノ酸誘導体とN末端アミノ基がアミド結合により環化された、直鎖部を有さない化合物の合成例を述べる。ペプチド化学合成については、本明細書の異なる場所の各所に記載されているどの手法を用いてもよい。
2-1.C-C環化ペプチドを合成するためのN末端カルボン酸誘導体の合成
2-1-1. ペンタ-4-イン酸メチル

1H-NMR(Varian 400-MR,400 MHz, CDCl3) δ ppm 3.72 (3H, s),2.50-2.60 (4H, m), 1.99 (1H, m)

1H-NMR(Varian 400-MR,400 MHz, CDCl3) δ ppm 6.02 (1H, dt,18, 5.6Hz), 5.48 (1H, d, 18Hz), 3.68 (3H, s), 2.40-2.53 (4H, m), 1.28 (12H, s)

1H-NMR(Varian 400-MR,400 MHz, D2O) δ ppm 6.59 (1H, m), 5.51 (1H, d, 18Hz), 2.44-2.51 (4H, m)
2-2-1. *Phe-MeLeu-MeVal-MeGly-Thr-MeAla-Ala-MeLeu-Leu-Phe*-piperidine(2つの*の間でC-C結合されている)(化合物DCC-1)の合成
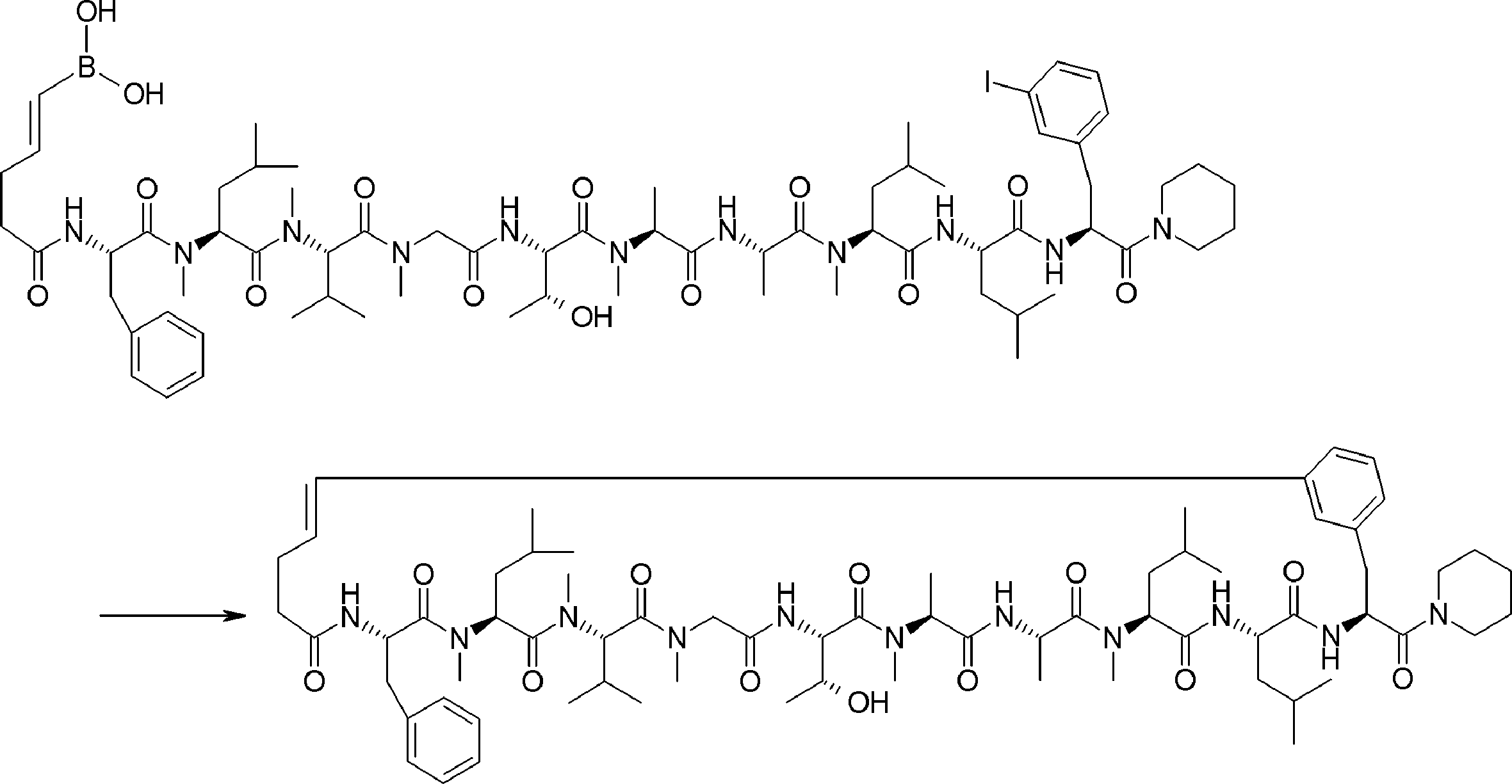
LCMS:m/z 1268.8(M+H)+
保持時間:0.75分 (分析条件 SQDAA50)
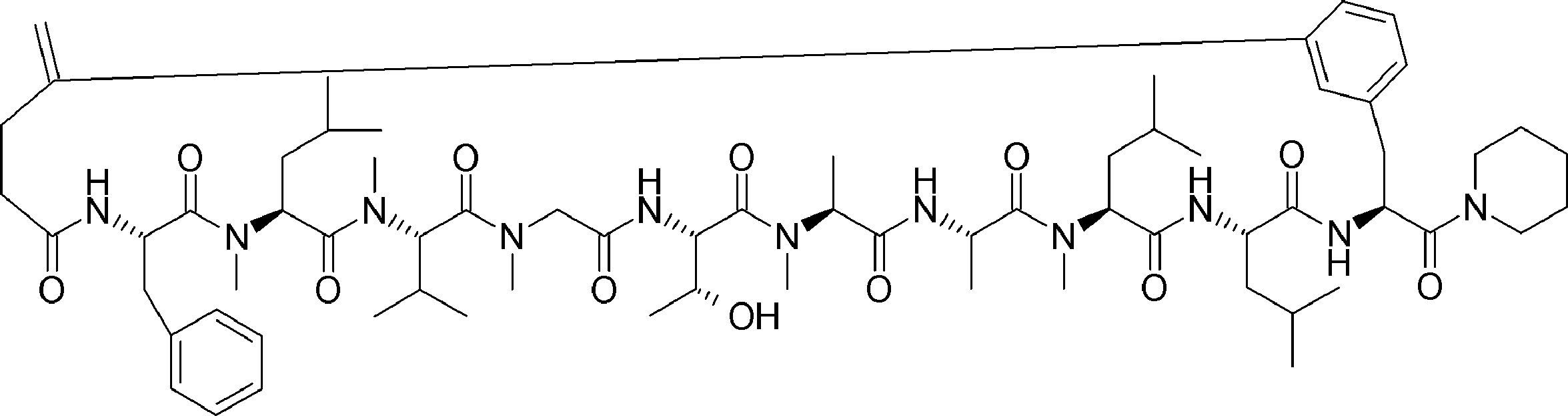
LCMS:m/z 1268.8(M+H)+
保持時間:0.78分 (分析条件 SQDAA50)
C-C結合環化ペプチド合成の別法も述べる。C-C結合環化を利用したDisplay libraryでは、例えばN末端側の三角ユニット部位に炭素-炭素二重結合、C末端側の交差ユニットのアミノ酸の側鎖にヨードフェニル基を有するペプチドを翻訳合成した後(スキームC-2)、Pdを使用したHeck反応によりC-C結合環化生成物を得ることができる。
上記手法で得られるC-C結合環化生成物のドラックライクネスを評価するために、C-C結合環化ペプチドの合成を行った。以下のスキームHにその合成の一例を示した。環化ペプチドの合成に際して、Pdによる環化反応ではなく、アミド化反応で環化ペプチドを合成した。すなわち、交差ユニットであるC末端フェニルアラニンの主鎖カルボン酸がアミドにて化学修飾(ピペリジンアミド化)され、かつ側鎖上のカルボン酸部位でレジンに結合させたフェニルアラニン誘導体を合成し、このレジンを用いて、Fmoc合成法に従いペプチドの伸張反応を行った。伸張後、ペプチドをレジンから切り出し、N末端側のアミノ基(三角ユニット)とC末端フェニルアラニン誘導体側鎖上のカルボン酸部位(交差ユニット)をアミド結合で縮合させて環化体とすることで、Display libraryの生成物に相当するC-C結合環化ペプチドの合成を行った。スキームHではC-C結合によりC-Cの2重結合が得られる場合のみを記載したが、C-Cの1重結合や3重結合も可能であり、その場合に応じて化合物群を合成した。

ドラックライクネス評価に使用した交差ユニット部位のフェニルアラニン誘導体として、以下に示すアミノ酸およびそのレジン結合化合物を合成した。

(S)-(3-(4-ヨードフェニル)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)カルバミン酸 tert-ブチル(化合物SP413、Boc-Phe(4-I)-pip)の合成

LCMS(ESI)m/z =459.5(M+H)+
保持時間:1.05分 (分析条件 SQDAA05)

LCMS(ESI)m/z =459.4(M+H)+
保持時間:0.99分 (分析条件 SQDFA05)
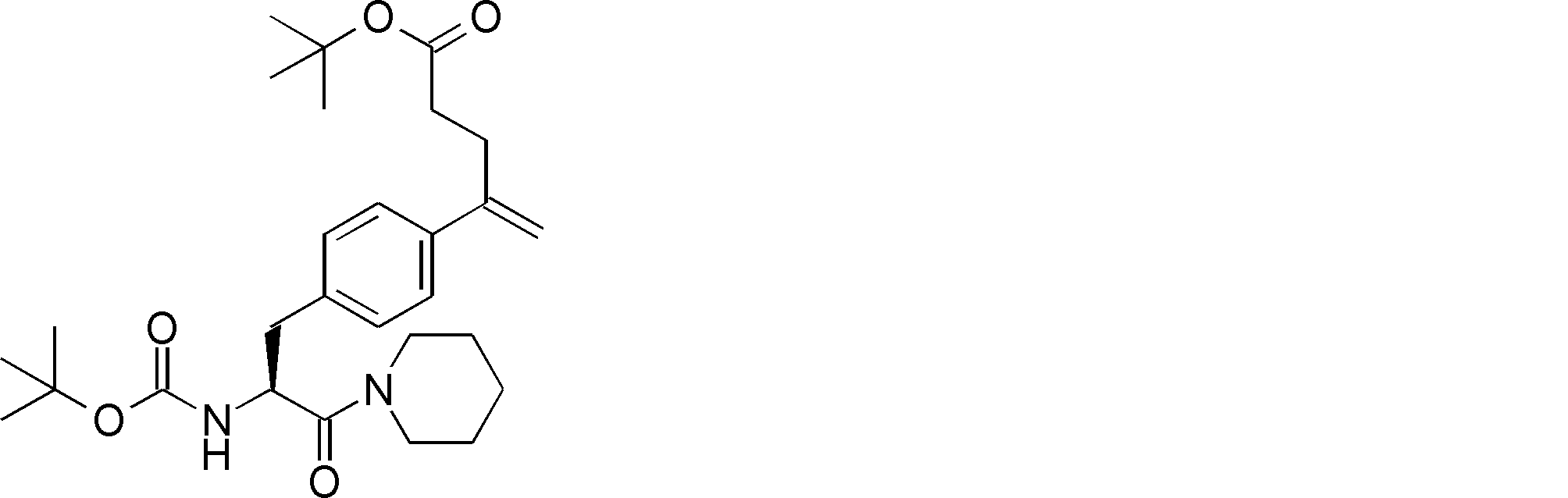
LCMS(ESI)m/z =487.6(M+H)+
保持時間:1.05分 (分析条件 SQDFA05)
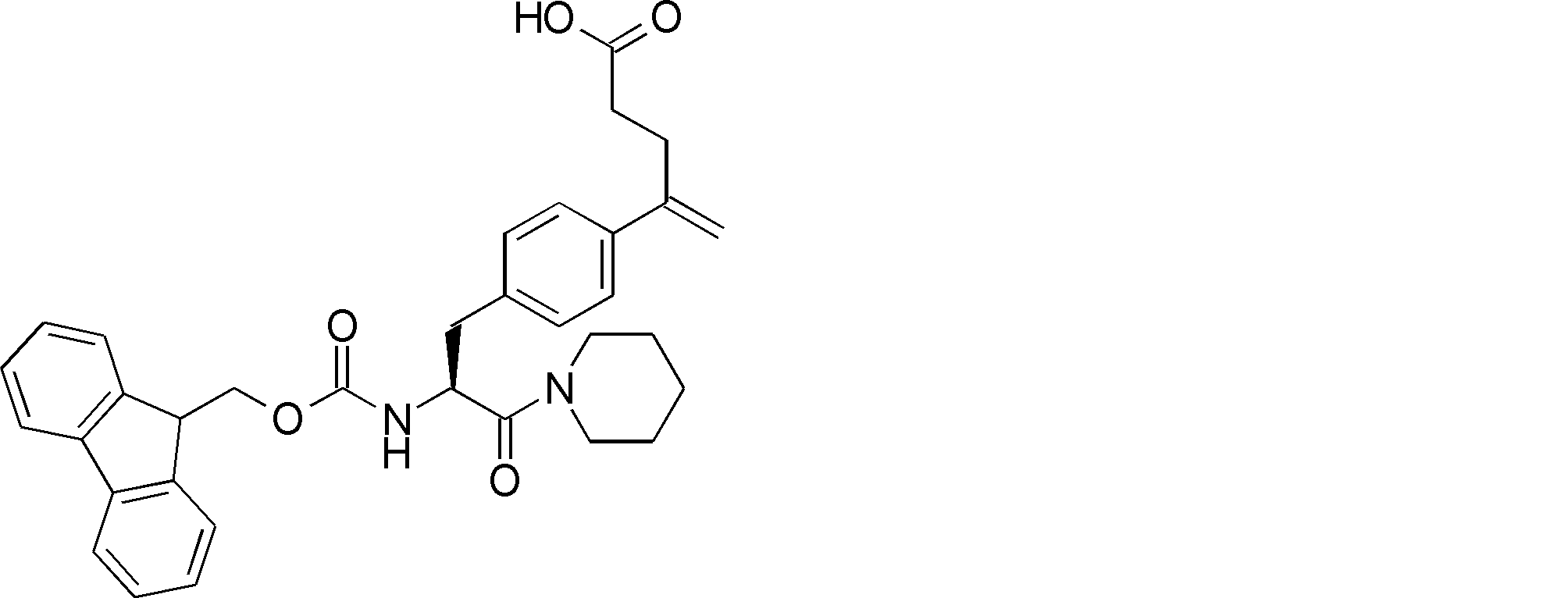
LCMS(ESI)m/z =553.4(M+H)+
保持時間:0.89分 (分析条件 SQDFA05)

(吸光度(301nm)x1000x50)/(用いたレジン量x7800)=0.265mmol/g
0.265mmol/gx100/(2.68/2.53)=25.0%

LCMS(ESI)m/z =459.2(M+H)+
保持時間:0.95分 (分析条件 SQDFA05)

LCMS(ESI)m/z =459.6(M+H)+
保持時間:0.99分 (分析条件 SQDFA05)

LCMS(ESI)m/z =487.6(M+H)+
保持時間:1.05分 (分析条件 SQDFA05)

LCMS(ESI)m/z =553.5(M+H)+
保持時間:0.90分 (分析条件 SQDFA05)
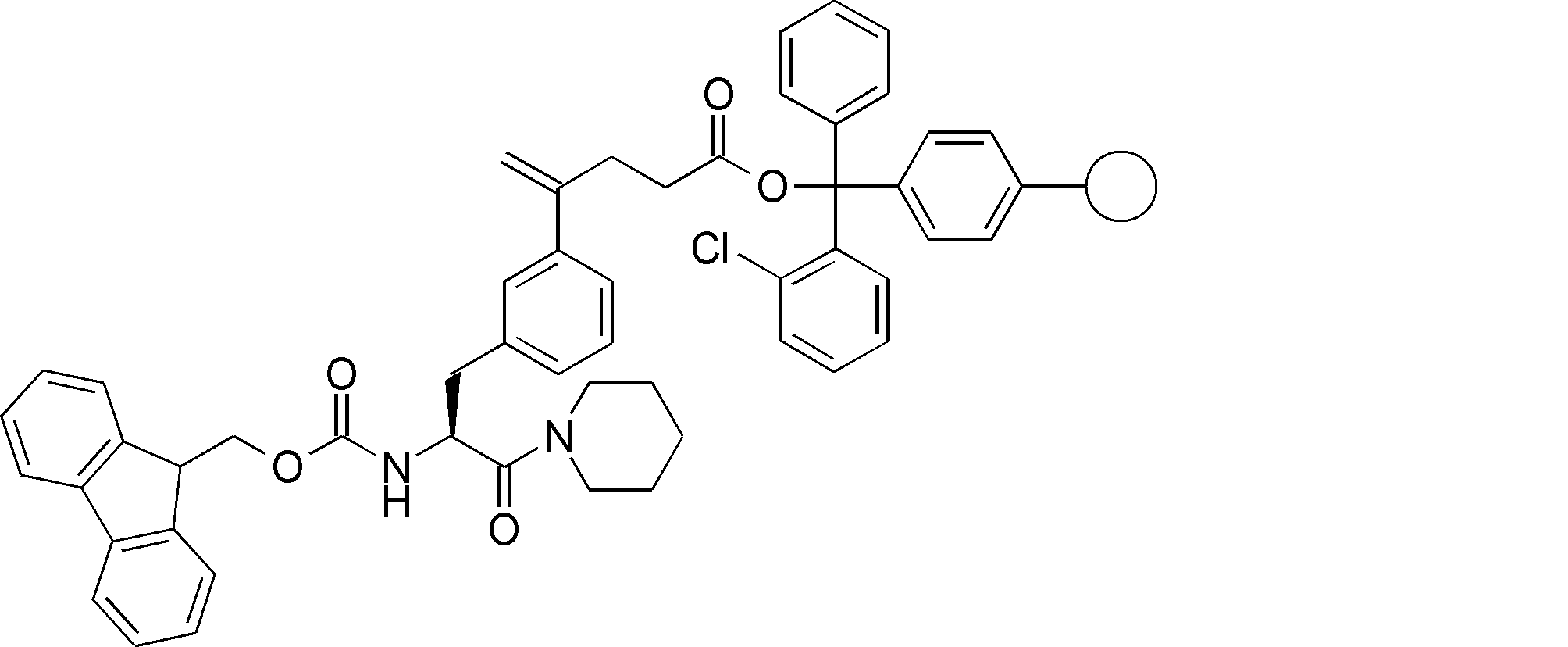
ローディング率:0.278mmol/g、26.1%

1H-NMR(Varian 400-MR, 400 MHz, CDCl3) δ ppm 2.39 (2H, t,7.2 Hz), 2.28 (2H, m), 2.08 (1H, s), 1.85 (2H, m), 1.49(9H, s)
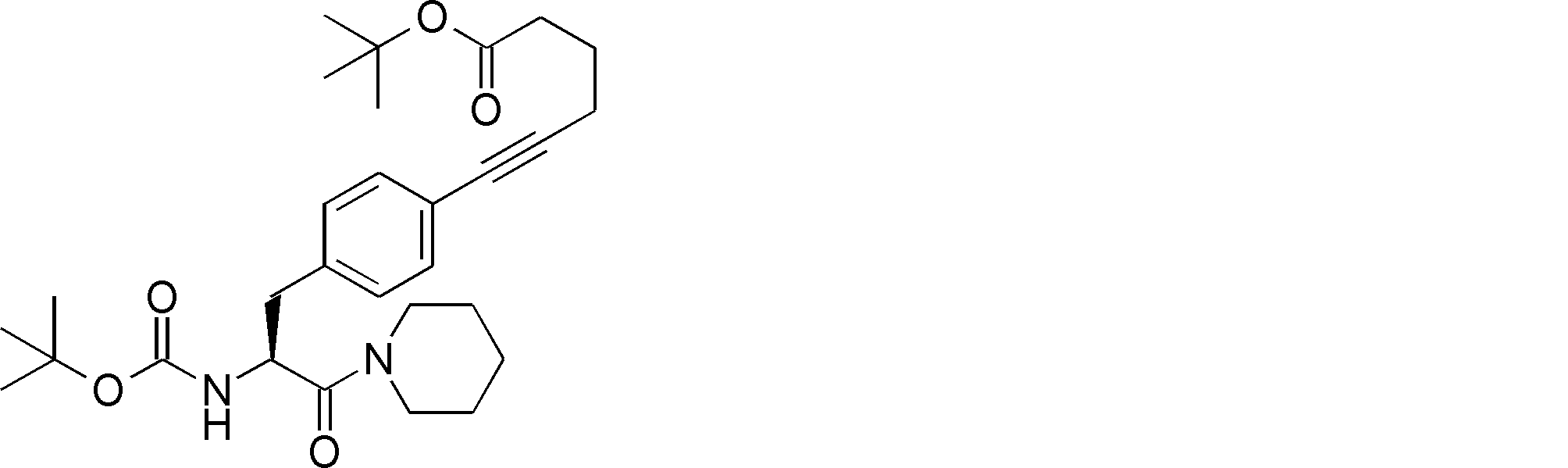
LCMS(ESI)m/z =499.4(M+H)+
保持時間:1.07分 (分析条件 SQDFA05)

LCMS(ESI)m/z =503.6(M+H)+
保持時間:1.14分 (分析条件 SQDAA05)
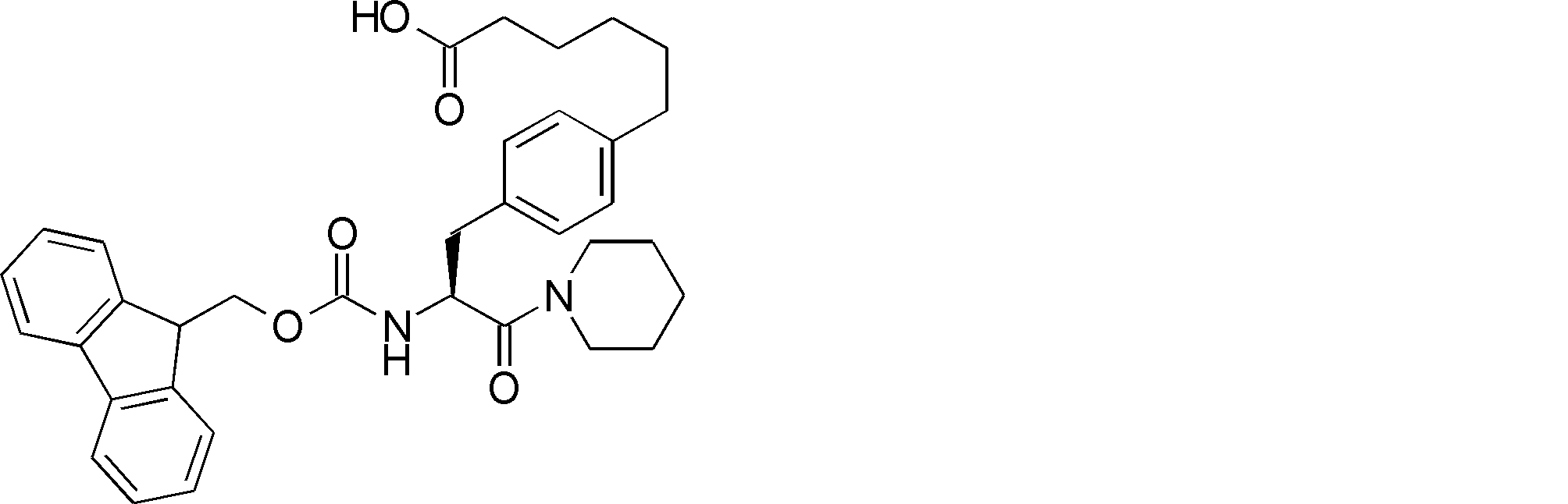
LCMS(ESI)m/z =569.6(M+H)+
保持時間:1.02分 (分析条件 SQDAA05)
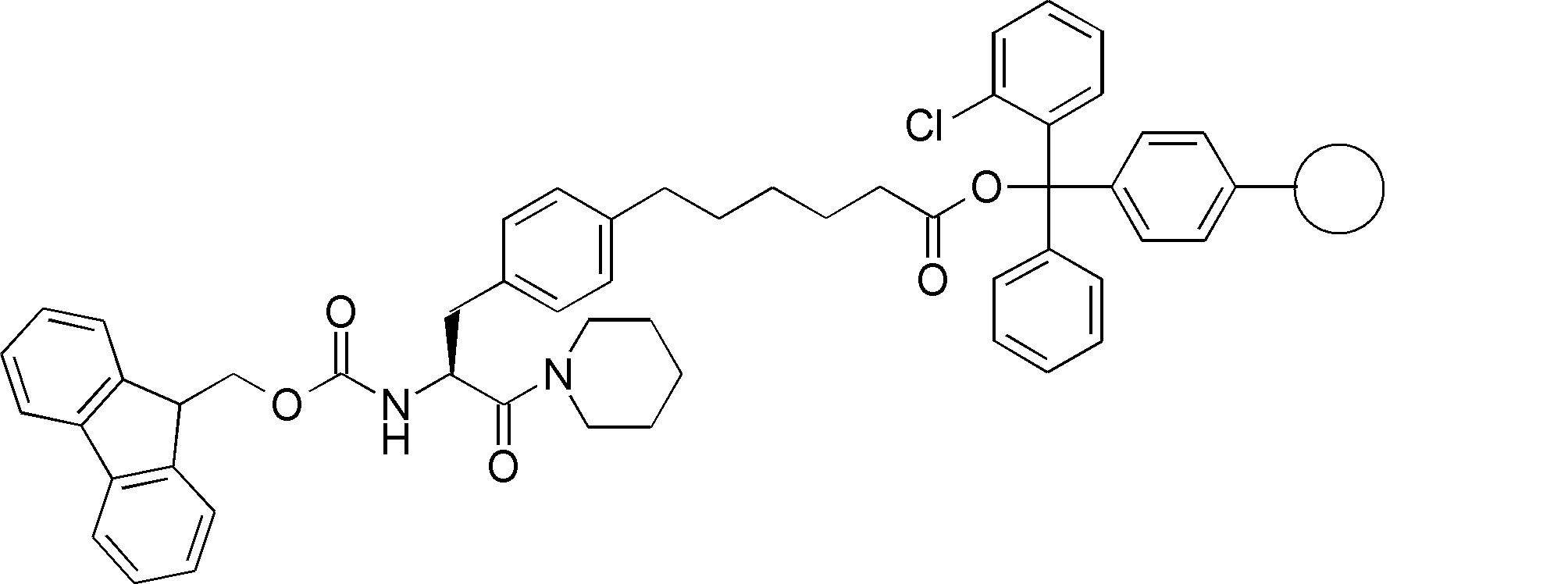
ローディング率:0.217mmol/g、21.3%

1H-NMR(Varian 400-MR, 400 MHz, CDCl3) δ ppm 2.48-2.50 (4H, m), 2.00 (1H, s), 1.49(9H, s)

LCMS(ESI)m/z =485.6(M+H)+
保持時間:1.03分 (分析条件 SQDFA05)

LCMS(ESI)m/z =489.6(M+H)+
保持時間:1.08分 (分析条件 SQDFA05)
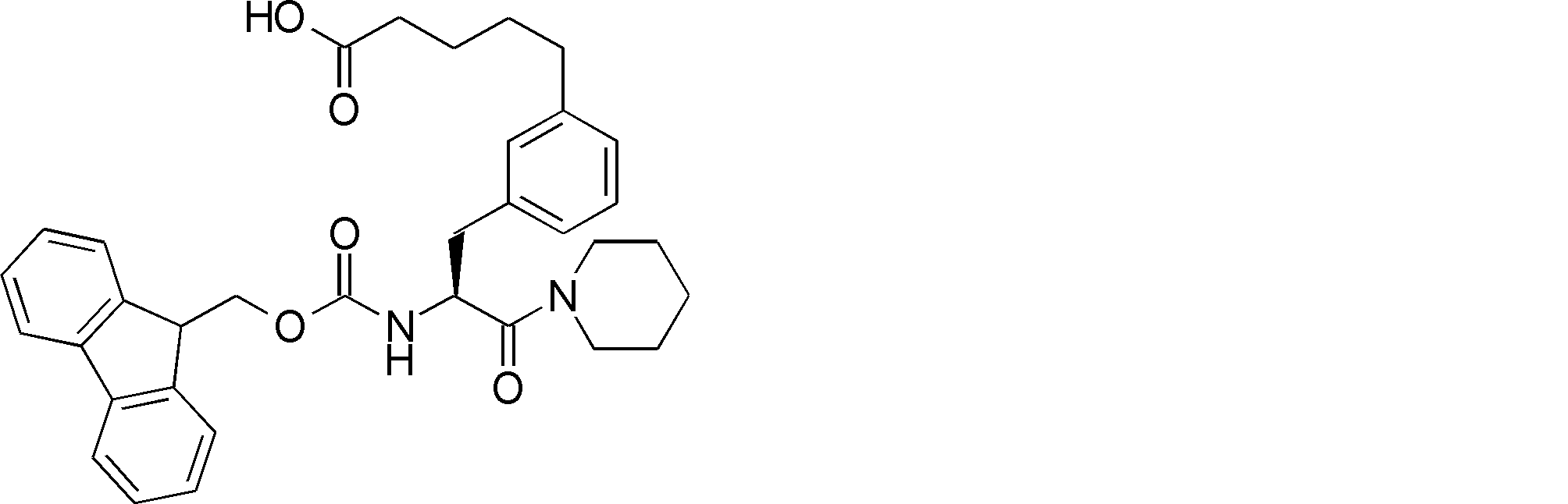
LCMS(ESI)m/z =555.6(M+H)+
保持時間:0.92分 (分析条件 SQDFA05)
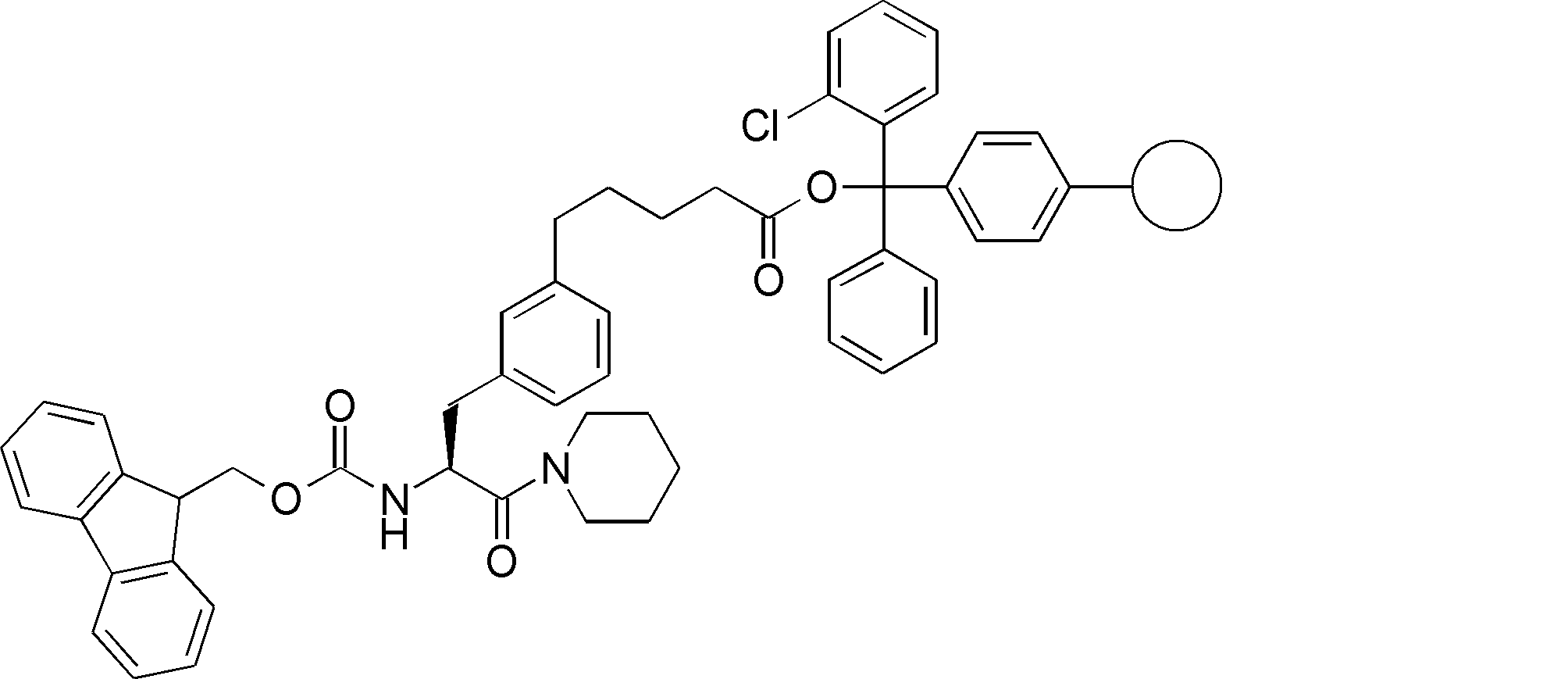
ローディング率:0.326mmol/g、35.3%
合成した環化ペプチドの膜透過性を比較検討する目的でPAMPA(Parallel Artificial Membrane Permeability Assay)による試験を実施した。
ミリポア96穴メンブレンフィルター(疎水性PVDF(ポリビニリデンジフルオリド)、pore size 0.45μm)(日本ミリポア社より購入)に、Egg lecithin10%(w/v)、コレステロール0.5%(w/v)、ドデカン(以上Fluka社より購入)からなるリン脂質有機溶媒溶液を4μL添加することで、人工リン脂質膜を作製した。

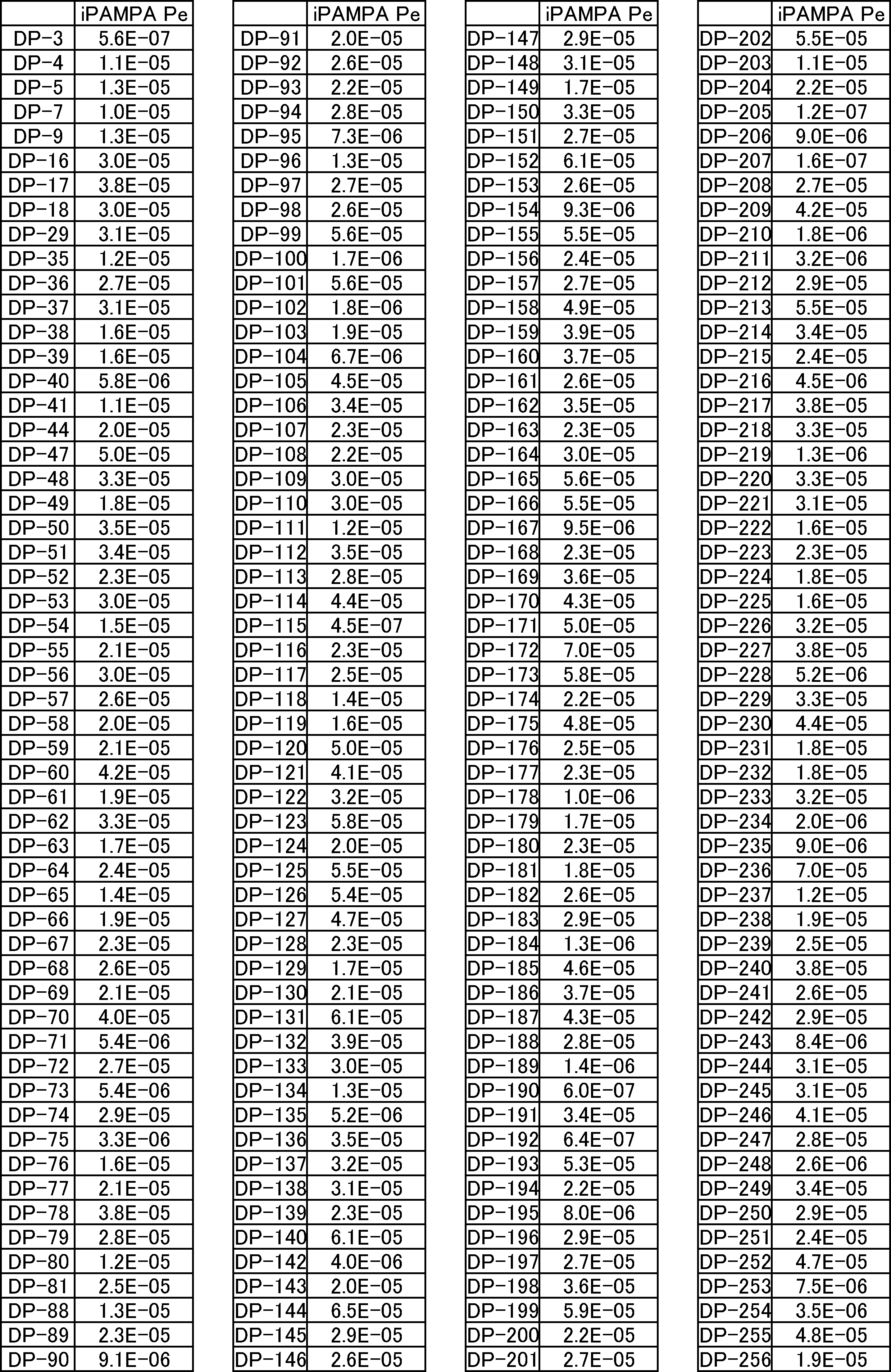




合成した環化ペプチドの代謝安定性を比較検討する目的で、ヒト肝ミクロソーム中代謝安定性試験を実施した。(方法は既に記載済み、結果は表11-6:ヒト肝ミクロソーム中代謝安定性試験結果)。


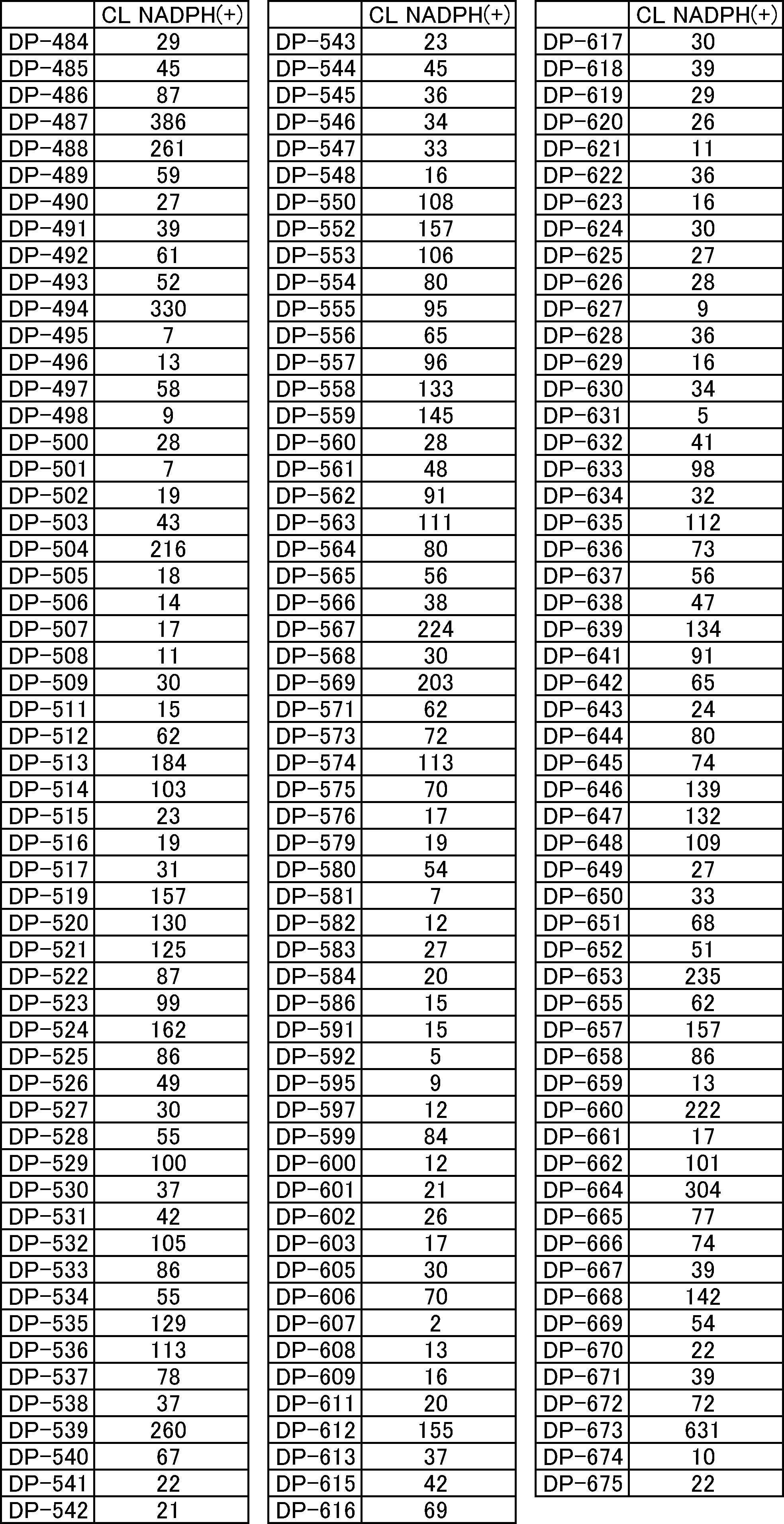
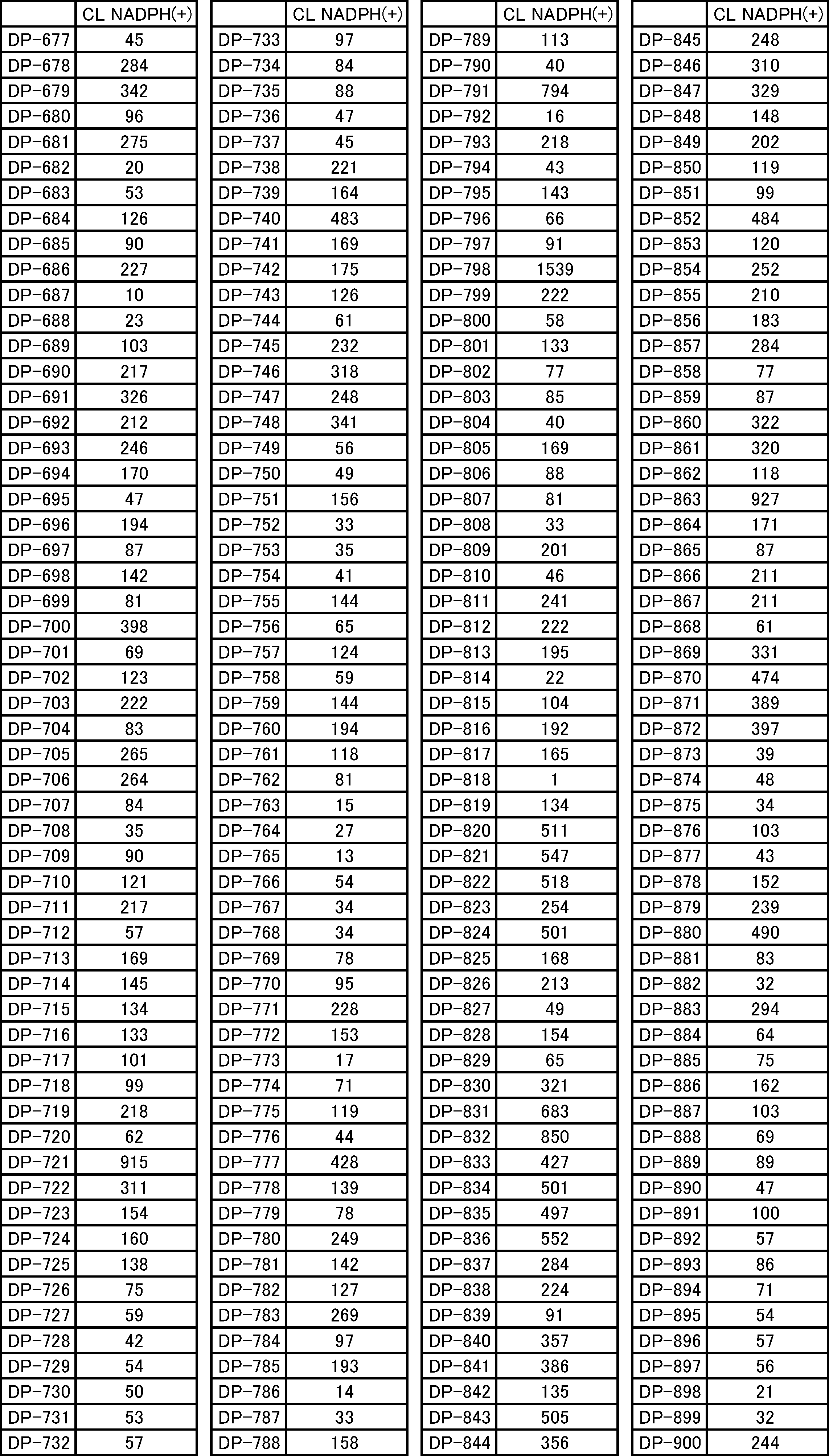

我々が明らかにしたドラッグライクを兼ね備えるためのペプチドの条件(鎖長、N-メチルアミノ酸数、ClogP)を満たす8種のペプチドのマウスにおける静脈内ならびに経口投与後の血漿中濃度推移を評価した。雄性マウス(C57BL/6J、6週齢、日本クレア社製:1群3匹)に化合物を20 mg/kgの用量で静脈内ならびに経口投与し、投与後24時間までの血液を抗凝固剤としてヘパリン処理済みのヘマトクリット管を用い、背中足静脈より経時的に採取した。血液は遠心分離により血漿を分離し、アセト二トリルによる除タンパク処理後、LC/MS/MS装置(API3200、AB SCIEX社製、USA)を用いて血漿中濃度を測定した。得られた血漿中濃度推移より、解析ソフトPhoenix WinNonlin 6.1(Pharsight Corporation社製、USA)を用いて、ノンコンパートメント解析により薬物動態パラメータを算出した。
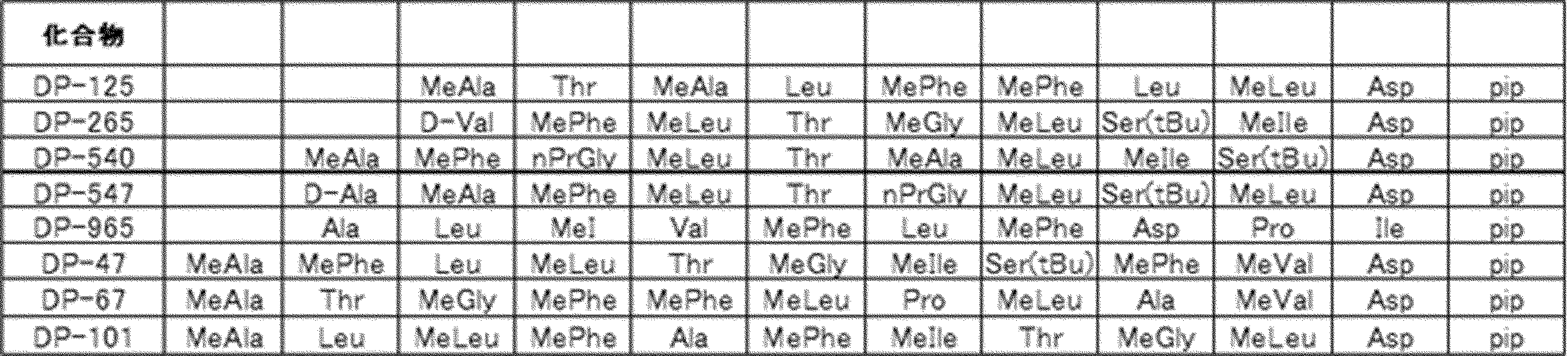
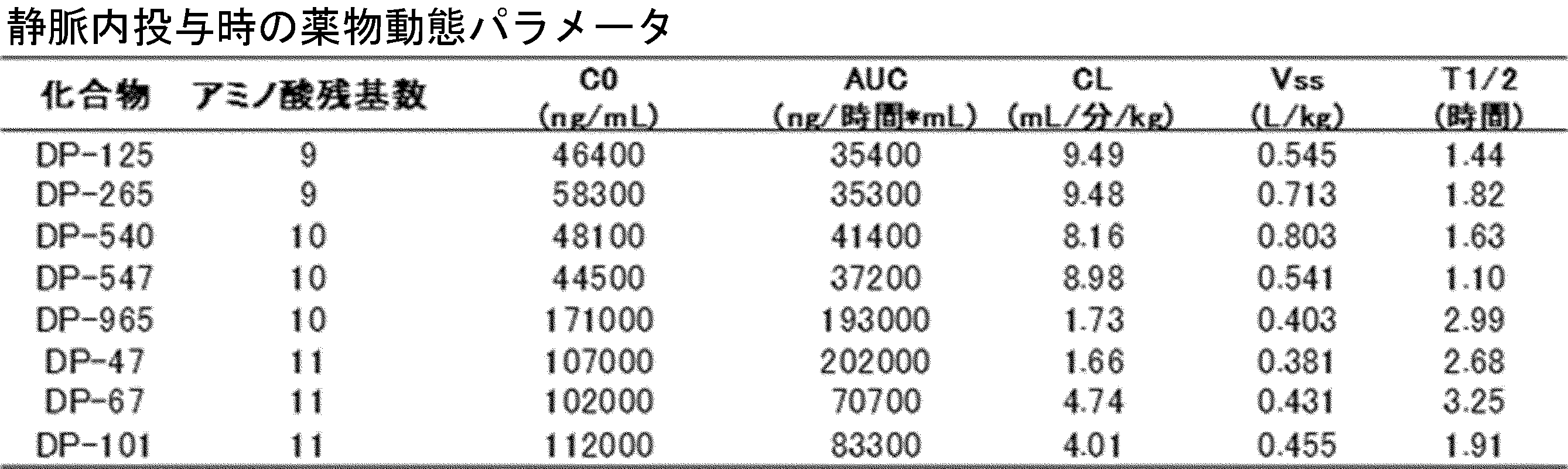
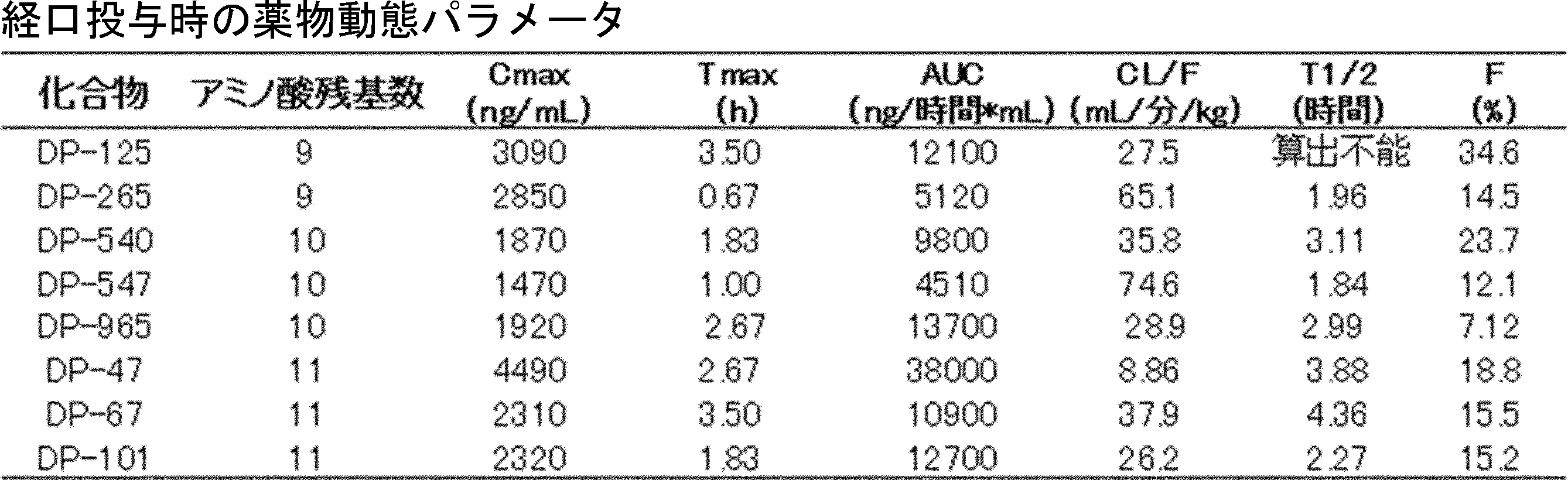
1.反応補助基を持たないN末端アミノ基と活性エステルとの翻訳液中でのアミド化反応
1-1.比較的安定な活性エステルを翻訳合成しておき、翻訳後に活性エステル部分を活性化させ、同時に系外から活性化剤を添加させて、反応補助基を有さないアミンと反応させる手法例
1-1-1.モデル反応原料化合物の合成
(9S,12S,18S,21S,24S,27S,30S)-30-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-(tert-ブトキシ)-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-12,27-ジベンジル-18-イソブチル-24-イソプロピル-2,2,9,11,17,21-ヘキサメチル-4,7,10,13,16,19,22,25,28-ノナオキソ-3-オキサ-5,8,11,14,17,20,23,26,29-ノナアザドトリアコンタン-32-酸(Boc-Gly-Ala- Me Phe-Gly- Me Leu-Ala-Val-Phe-Asp- Me Ala-Ser(tBu)-Gly-NH2)の合成
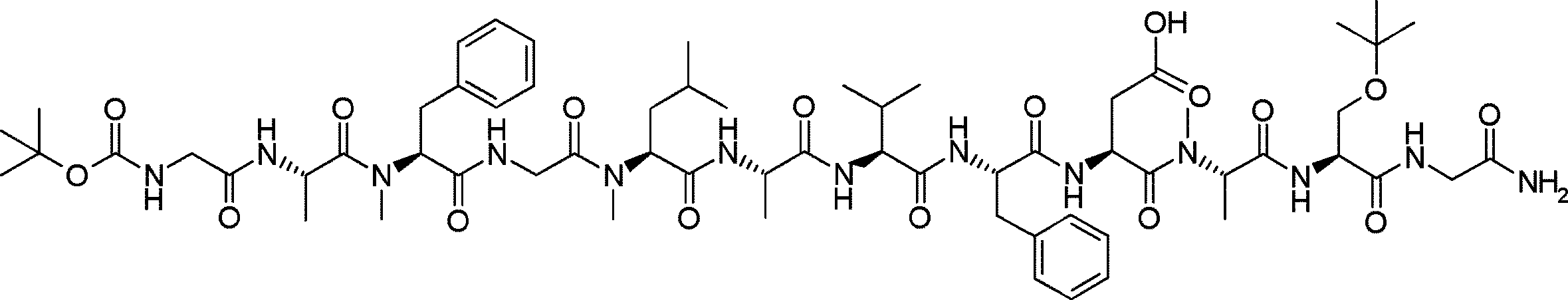
保持時間:1.09分 (分析条件 SQD AA05)
(9S,12S,18S,21S,24S,27S,30S)-30-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-(tert-ブトキシ)-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-12,27-ジベンジル-18-イソブチル-24-イソプロピル-2,2,9,11,17,21-ヘキサメチル-4,7,10,13,16,19,22,25,28-ノナオキソ-3-オキサ-5,8,11,14,17,20,23,26,29-ノナアザドトリアコンタン-32-チオ酸 S-ベンジル(Boc-Gly-Ala- Me Phe-Gly- Me Leu-Ala-Val-Phe-Asp(SBn)- Me Ala-Ser(tBu)-Gly-NH2)の合成
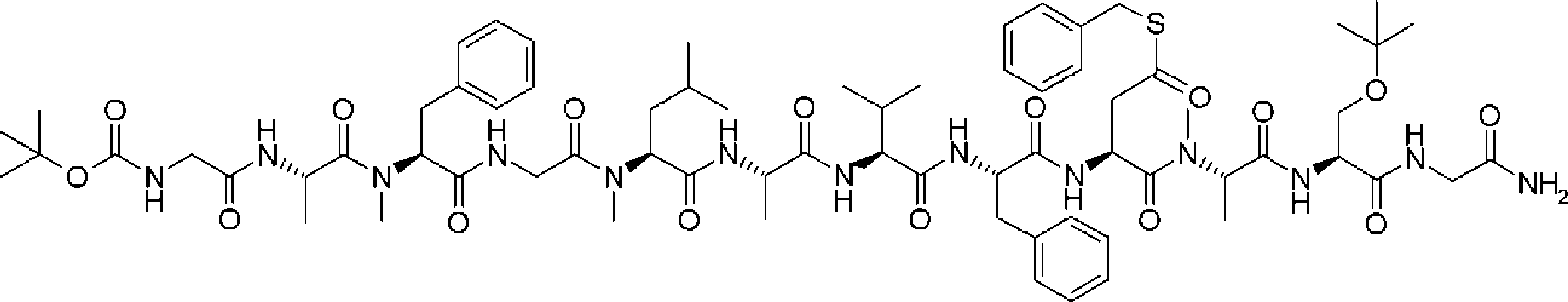
保持時間:1.08分 (分析条件 SQD AA05)
(4S,7S,13S,16S,19S,22S,25S)-1-アミノ-25-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-7,22-ジベンジル-13-イソブチル-19-イソプロピル-4,6,12,16-テトラメチル-2,5,8,11,14,17,20,23-オクタオキソ-3,6,9,12,15,18,21,24-オクタアザヘプタコサン-27-チオ酸 S-ベンジル(Gly-Ala- Me Phe-Gly- Me Leu-Ala-Val-Phe-Asp(SBn)- Me Ala-Ser-Gly-NH2)の合成
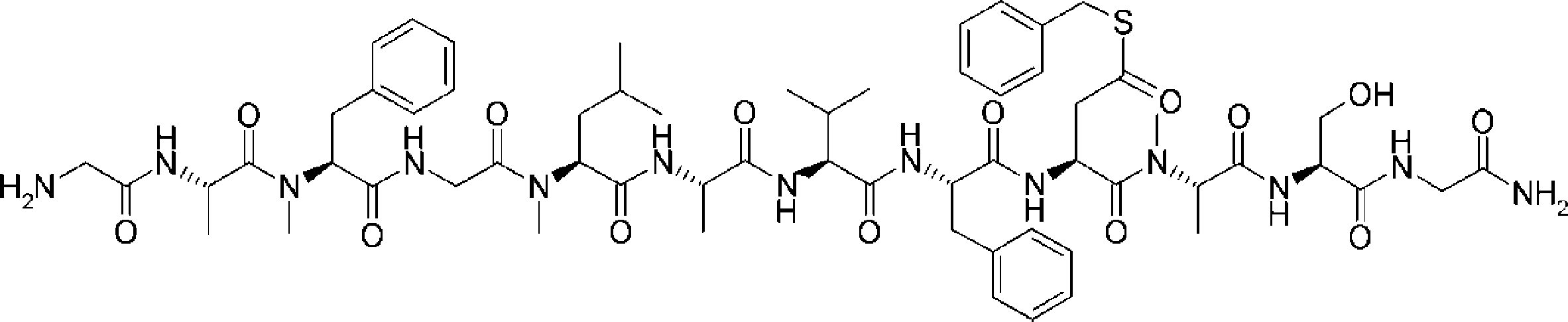
LCMS:m/z 1258.6(M+H)+
保持時間:0.58分 (分析条件 SQD FA05)
(6S,9S,12S,15S,18S,24S,27S,30S,33S)-18,30-ジベンジル-12,24-ジイソブチル-9,27-ジイソプロピル-2,2,6,11,17,23,26-ヘプタメチル-33-(メチル((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)カルバモイル)-4,7,10,13,16,19,22,25,28,31-デカオキソ-15-((R)-1-(トリチルオキシ)エチル)-3-オキサ-5,8,11,14,17,20,23,26,29,32-デカアザペンタトリアコンタン-35-酸(Boc-Ala-Val-MeLeu-Thr(Trt)-MePhe-Gly-MeLeu-MeVal-Phe-Asp-MePhe-Ala-piperidine)の合成
なお、本明細書ではAla-piperidineもしくはAla-pipとは、ピペリジンの窒素原子とAlaの主鎖カルボン酸部位とでアミド結合を形成した化合物とする。この部分構造をペプチド部位に有する場合にも、同様の表記を用いる。
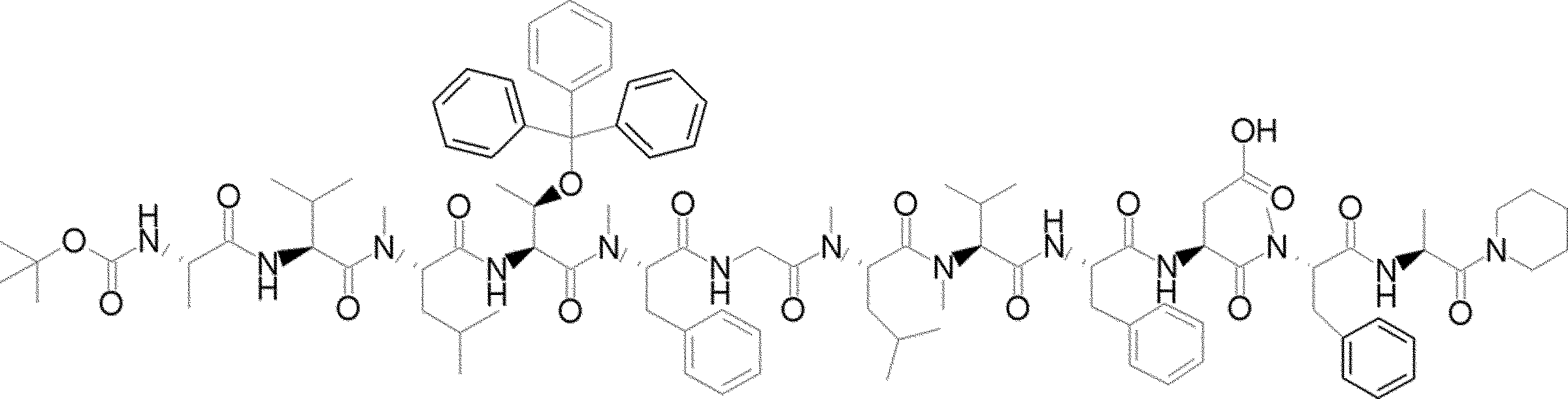
LCMS:m/z 1777.6(M-H)-
保持時間:0.86分 (分析条件 SQD AA50)
(6S,9S,12S,15S,18S,24S,27S,30S,33S)-18,30-ジベンジル-12,24-ジイソブチル-9,27-ジイソプロピル-2,2,6,11,17,23,26-ヘプタメチル-33-(メチル((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)カルバモイル)-4,7,10,13,16,19,22,25,28,31-デカオキソ-15-((R)-1-(トリチルオキシ)エチル)-3-オキサ-5,8,11,14,17,20,23,26,29,32-デカアザペンタトリアコンタン-35-チオ酸 S-ベンジル(Boc-Ala-Val-MeLeu-Thr(Trt)-MePhe-Gly-MeLeu-MeVal-Phe-Asp(SBn)-MePhe-Ala-piperidine)の合成
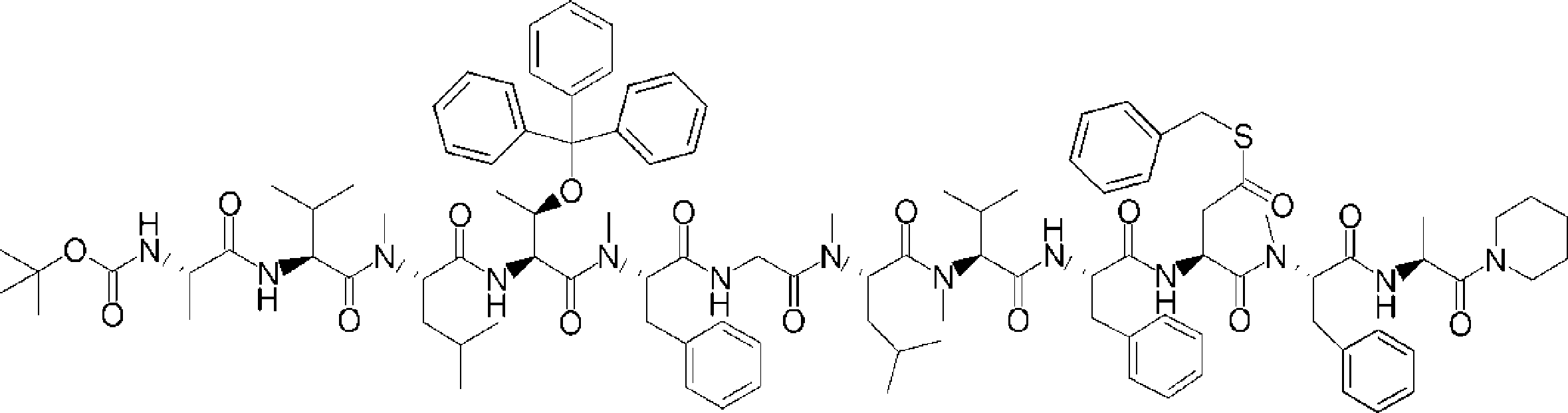
LCMS:m/z 1883.3(M-H)-
保持時間:0.93分 (分析条件 SQD AA50)
(3S,6S,9S,12S,18S,21S,24S,27S,30S)-30-アミノ-6,18-ジベンジル-21-((R)-1-ヒドロキシエチル)-12,24-ジイソブチル-9,27-ジイソプロピル-10,13,19,25-テトラメチル-3-(メチル((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)カルバモイル)-5,8,11,14,17,20,23,26,29-ノナオキソ-4,7,10,13,16,19,22,25,28-ノナアザヘントリアコンタン-1-チオ酸 S-ベンジル(Ala-Val-MeLeu-Thr-MePhe-Gly-MeLeu-MeVal-Phe-Asp(SBn)-MePhe-Ala-piperidine)の合成
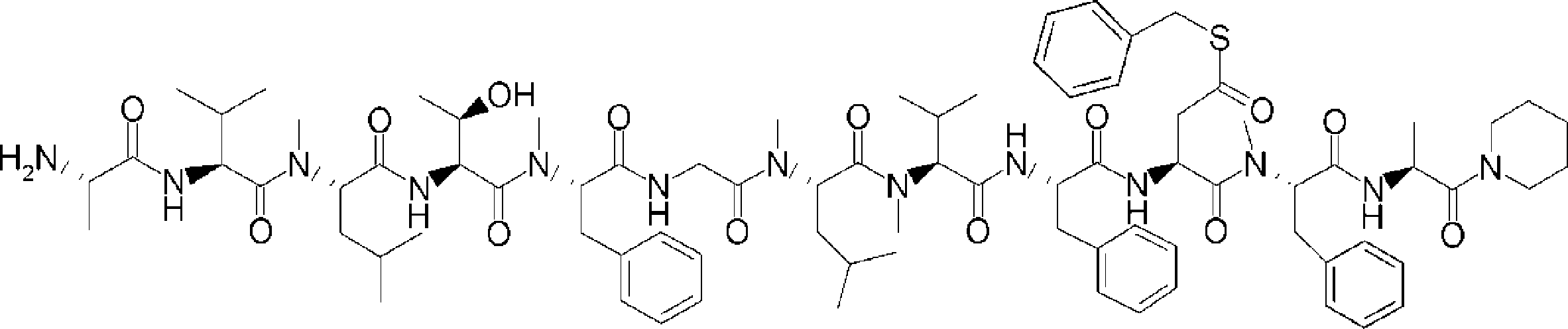
LCMS:m/z 1541.0(M-H)-
保持時間:0.77分 (分析条件 SQD FA05)
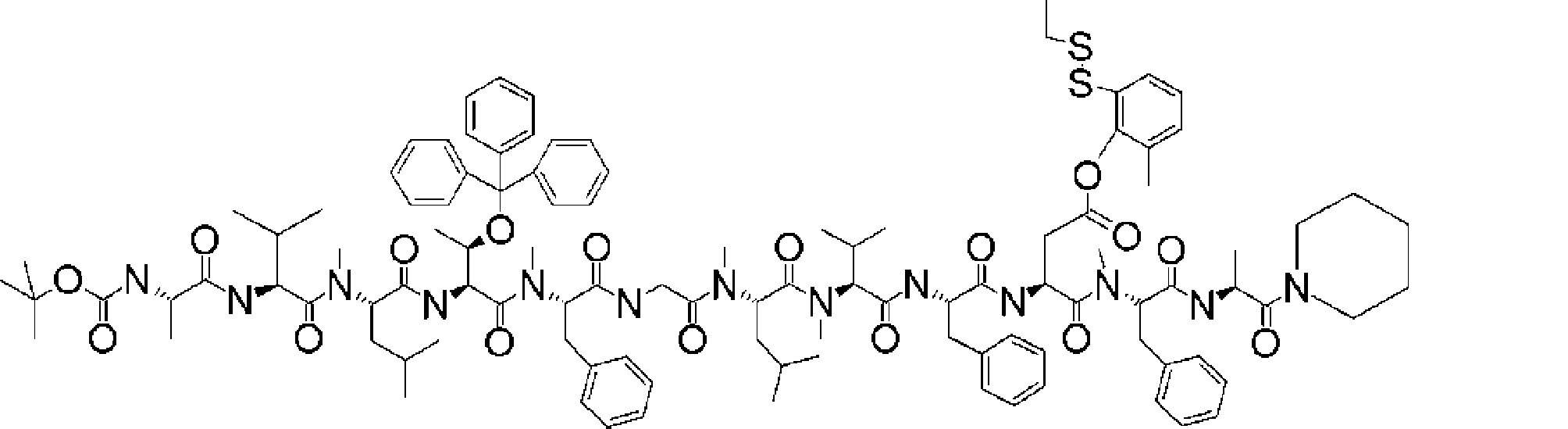
LCMS(ESI) m/z =1959.2(M―H)―
保持時間:1.05分(分析条件SQDAA50)

LCMS(ESI) m/z =1620(M+H)+
保持時間:0.88分(分析条件SQDAA50)
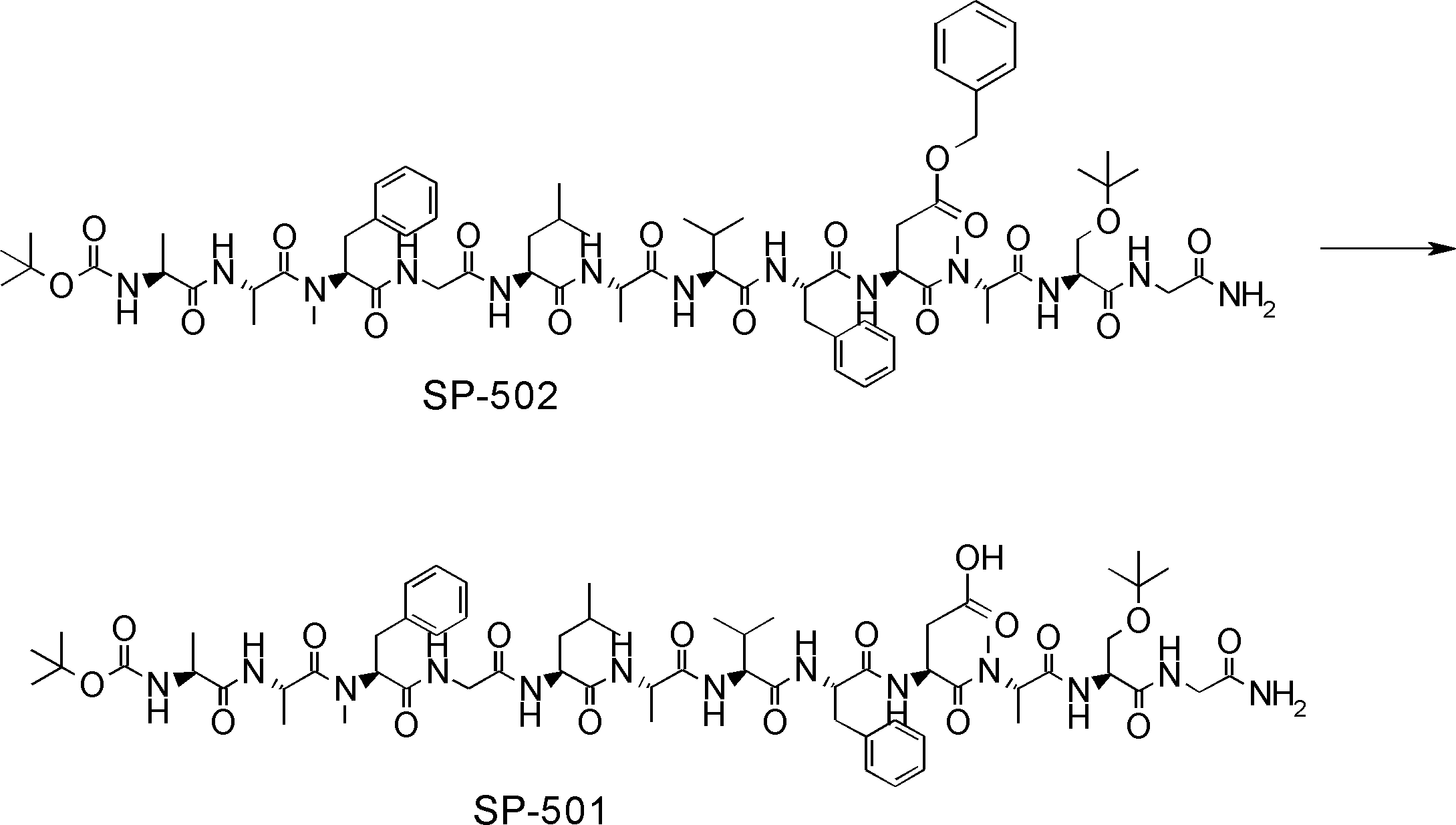
LCMS(ESI)m/z =1306.4(M-H)-
保持時間:0.74分 (分析条件 SQD FA05)
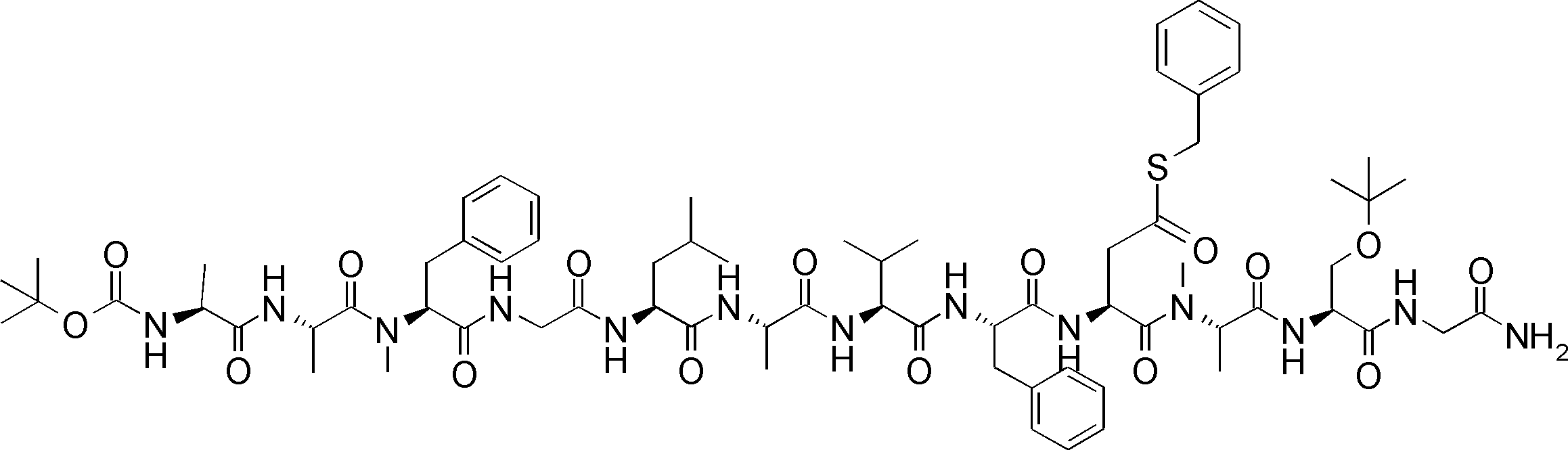
LCMS(ESI)m/z =1412.6(M-H)-
保持時間:0.87分 (分析条件 SQD FA05)
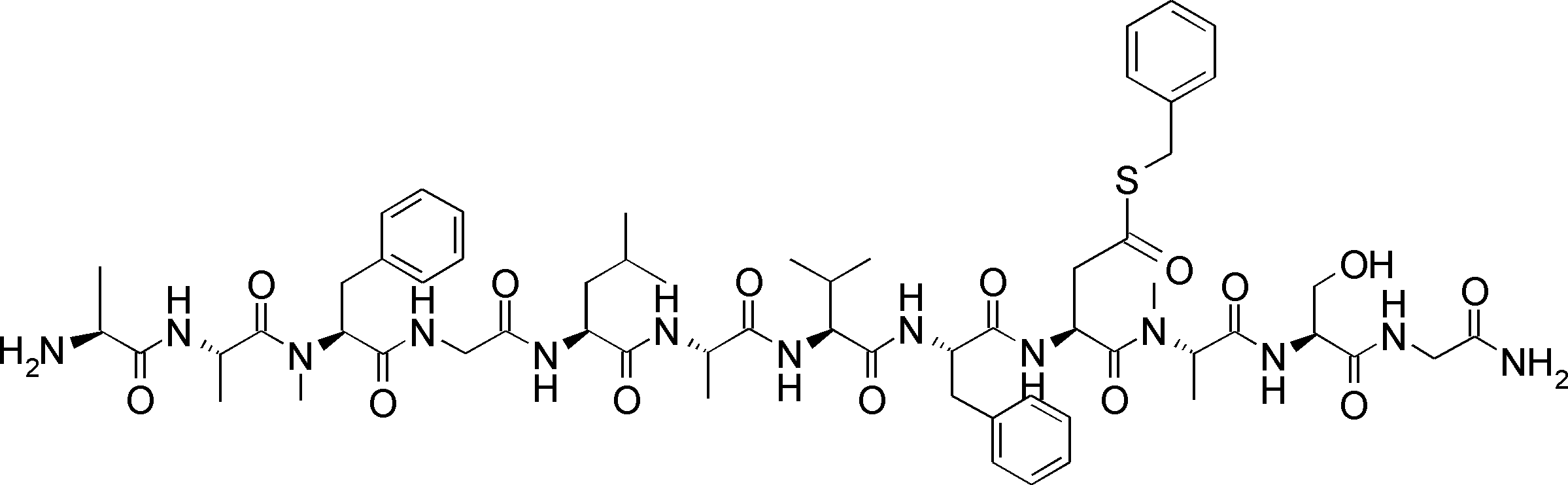
LCMS(ESI)m/z =1256.8(M-H)-
保持時間:0.58分 (分析条件 SQD FA05)
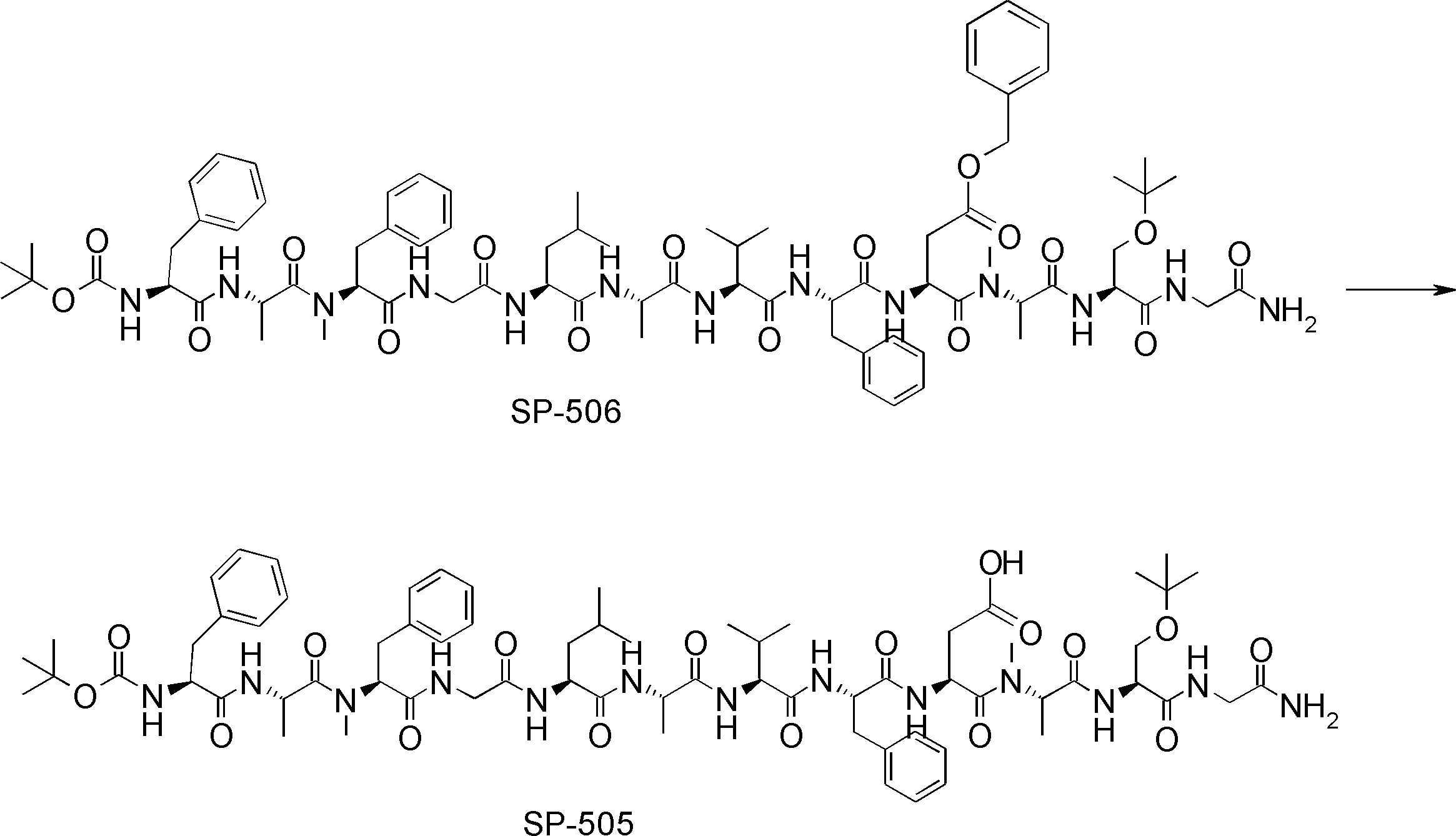
LCMS(ESI)m/z =1382.6(M-H)-
保持時間:0.80分 (分析条件 SQD FA05)
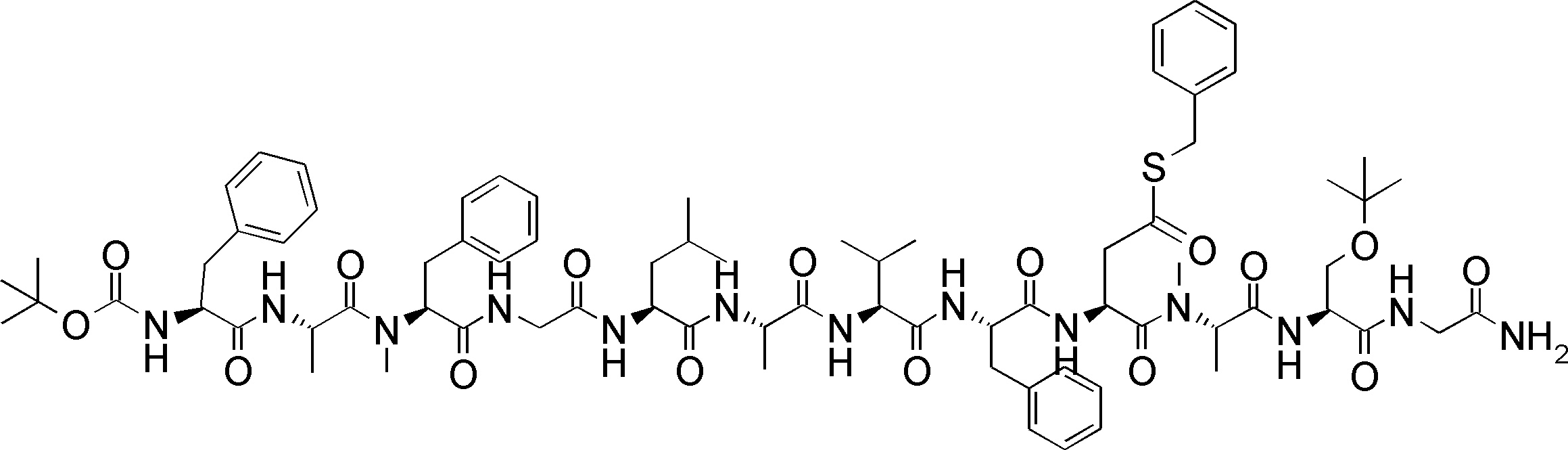
LCMS(ESI)m/z =1490.6(M+H)+
保持時間:0.93分 (分析条件 SQD FA05)
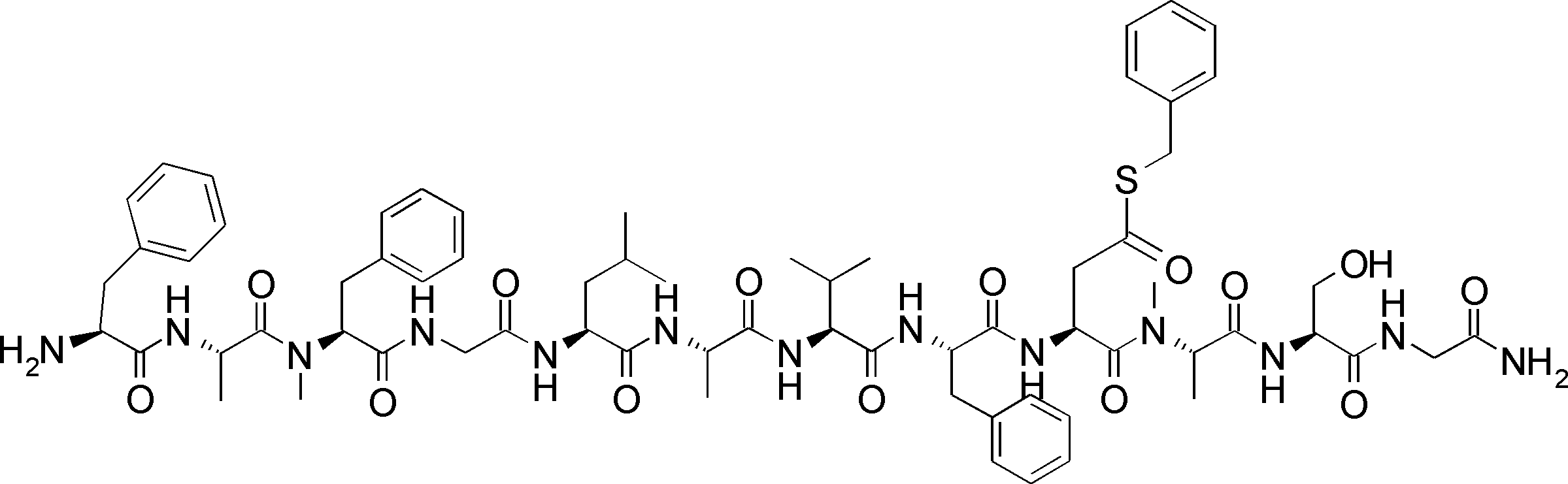
LCMS(ESI)m/z =1332.7(M-H)-
保持時間:0.60分 (分析条件 SQD FA05)
N末端がGlyである場合の反応例を示す。
(化合物P-136)
(5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-8,23-ジベンジル-14-イソブチル-20-イソプロピル-N,5,7,13,17-ペンタメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミドの合成
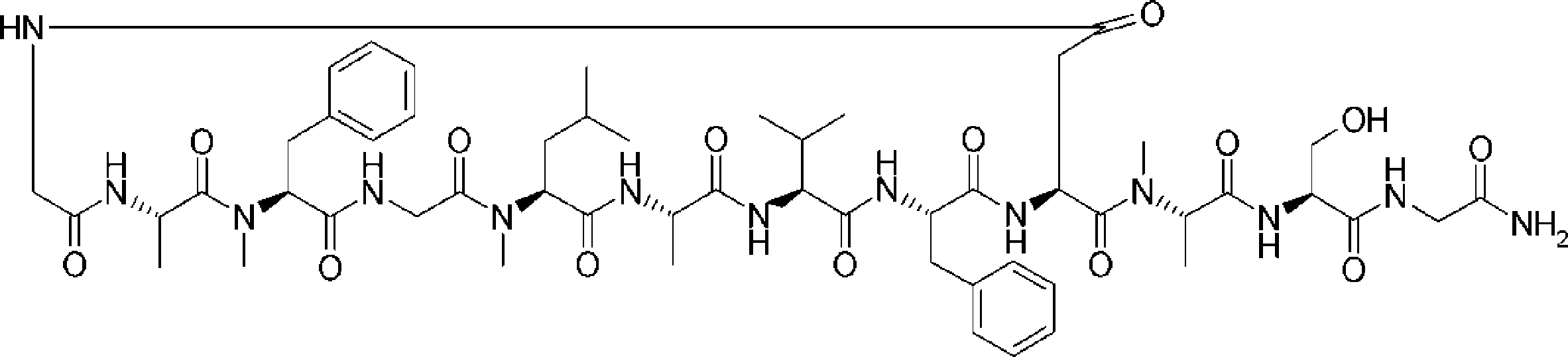
4-(トリフルオロメチル)ベンゼンチオール(0.018g、0.10mmol)およびトリエチルアミン(10.1mg、0.10mmol)を100mMのリン酸水素2ナトリウム水溶液(80μL)およびNMP(10μL)に溶解させた。この溶液に(4S,7S,13S,16S,19S,22S,25S)-1-アミノ-25-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-7,22-ジベンジル-13-イソブチル-19-イソプロピル-4,6,12,16-テトラメチル-2,5,8,11,14,17,20,23-オクタオキソ-3,6,9,12,15,18,21,24-オクタアザヘプタコサン-27-チオ酸 S-ベンジル(化合物P-147)のNMP溶液(10mM、10μL、0.10μmol)を加えて、室温で5時間攪拌した。反応液をLCMSで分析し、標題化合物の生成を確認した。なお、標題化合物と原料(保持時間0.58分、分析条件 SQDFA05)およびAspのチオエステルが加水分解されたカルボン酸体(保持時間:0.51分、分析条件 SQDFA05)の生成比はLCMSのUVエリア比でおおよそ74:11:15であった。結果を図36に示す。
LCMS:m/z 1134.6(M+H)+
保持時間:0.64分 (分析条件 SQDFA05)
ベンゼンチオール(0.110mg、0.001mmol)およびトリエチルアミン(0.202mg、0.002mmol)を100mMリン酸水素2ナトリウム水溶液(80μL)およびNMP(10μL)に溶解させた。この溶液に(4S,7S,13S,16S,19S,22S,25S)-1-アミノ-25-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-7,22-ジベンジル-13-イソブチル-19-イソプロピル-4,6,12,16-テトラメチル-2,5,8,11,14,17,20,23-オクタオキソ-3,6,9,12,15,18,21,24-オクタアザヘプタコサン-27-チオ酸 S-ベンジル(化合物P-147)のNMP溶液(10mM、10μL、0.10μmol)を加えて、室温で8時間攪拌した。反応液をLCMSで分析し、標題化合物の生成を確認した。なお、標題化合物と原料(保持時間0.58分、分析条件 SQDFA05)およびAspのチオエステルが加水分解されたカルボン酸体(保持時間:0.50分、分析条件 SQDFA05)の生成比はLCMSのUVエリア比でおおよそ89:8:3であった。結果を図37に示す。
LCMS:m/z 1134.6(M+H)+
保持時間:0.63分 (分析条件 SQDFA05)
(化合物P-137)
(2R,5S,8S,11S,14S,20S,23S,26S,29S)-14,26-ジベンジル-11-((R)-1-ヒドロキシエチル)-8,20-ジイソブチル-5,23-ジイソプロピル-N,2,7,13,19,22-ヘキサメチル-3,6,9,12,15,18,21,24,27,31-デカオキソ-N-((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)-1,4,7,10,13,16,19,22,25,28-デカアザシクロヘントリアコンタン-29-カルボキサミドの合成
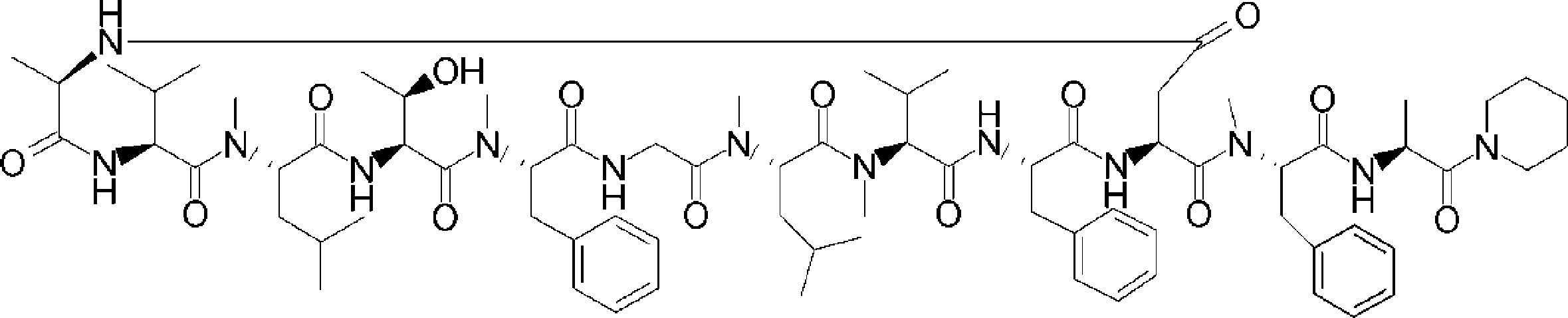
4-(トリフルオロメチル)ベンゼンチオール(8.91mg、0.05mmol)およびトリエチルアミン(5.06mg、0.05mmol)の水溶液(25μL)と(3S,6S,9S,12S,18S,21S,24S,27S,30S)-30-アミノ-6,18-ジベンジル-21-((R)-1-ヒドロキシエチル)-12,24-ジイソブチル-9,27-ジイソプロピル-10,13,19,25-テトラメチル-3-(メチル((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)カルバモイル)-5,8,11,14,17,20,23,26,29-ノナオキソ-4,7,10,13,16,19,22,25,28-ノナアザヘントリアコンタン-1-チオ酸 S-ベンジル(化合物P-135)のNMP溶液(5mM、20μL、0.1μmol)を混合し、さらに水(25μL)とNMP(30μL)を加え、50度で終夜攪拌した。LCMS測定により、原料の消失とともに標題化合物の生成を確認した。なお、標題化合物とAspのチオエステルが加水分解されたカルボン酸体(保持時間:0.68分、分析条件 SQDFA05)の生成比はLCMSのUVエリア比で、おおよそ72:28であった。結果を図38に示す。
LCMS:m/z 1417(M-H)-
保持時間:1.04分 (分析条件 SQDFA05)
(3S,6S,9S,12S,18S,21S,24S,27S,30S)-30-アミノ-6,18-ジベンジル-21-((R)-1-ヒドロキシエチル)-12,24-ジイソブチル-9,27-ジイソプロピル-10,13,19,25-テトラメチル-3-(メチル((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)カルバモイル)-5,8,11,14,17,20,23,26,29-ノナオキソ-4,7,10,13,16,19,22,25,28-ノナアザヘントリアコンタン-1-酸 2-(エチルジスルファニル)-6-メチルフェニル(0.162mg, 0.1umol)(化合物P-139)と1-ヒドロキシピロリジン-2,5-ジオン(0.575mg、5umol)のDMF-400mMリン酸緩衝溶液(pH8.5)1:1混合溶液(95ul)に1Mトリス(2-カルボキシエチル)ホスフィン(TCEP)水溶液(5ul)を加え、室温で30分攪拌した後、LCMSを用いて反応を観測したところ、環化前駆体は完全に消失し、標題化合物P-137と加水分解体P-140が3:1(LCMS:UVエリア比)で観測された。
LCMS(ESI) m/z =1417.2(M-H)―
保持時間:1.04分(分析条件SQDFA05)
(3S,6S,9S,12S,18S,21S,24S,27S,30S)-30-アミノ-6,18-ジベンジル-21-((R)-1-ヒドロキシエチル)-12,24-ジイソブチル-9,27-ジイソプロピル-10,13,19,25-テトラメチル-3-(メチル((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)カルバモイル)-5,8,11,14,17,20,23,26,29-ノナオキソ-4,7,10,13,16,19,22,25,28-ノナアザヘントリアコンタン-1-酸
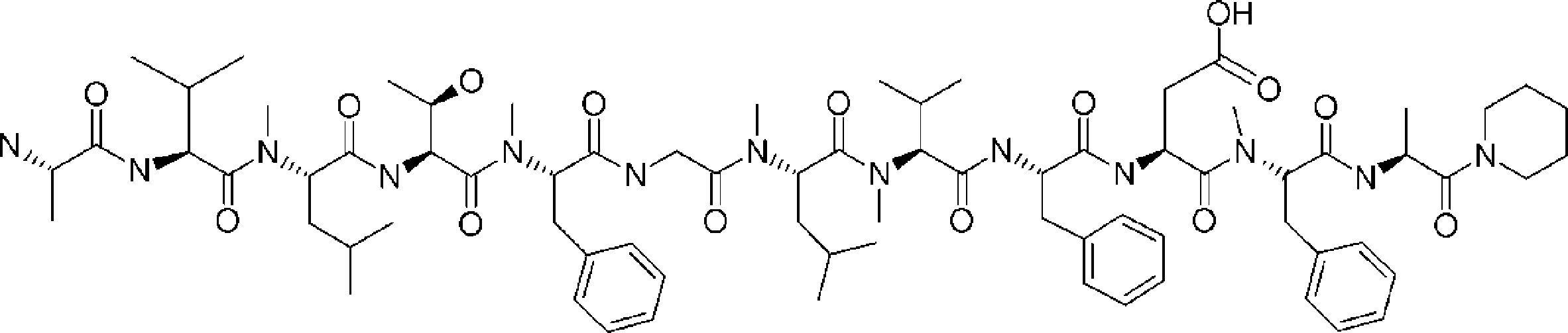
保持時間:0.67分(分析条件SQDFA05)
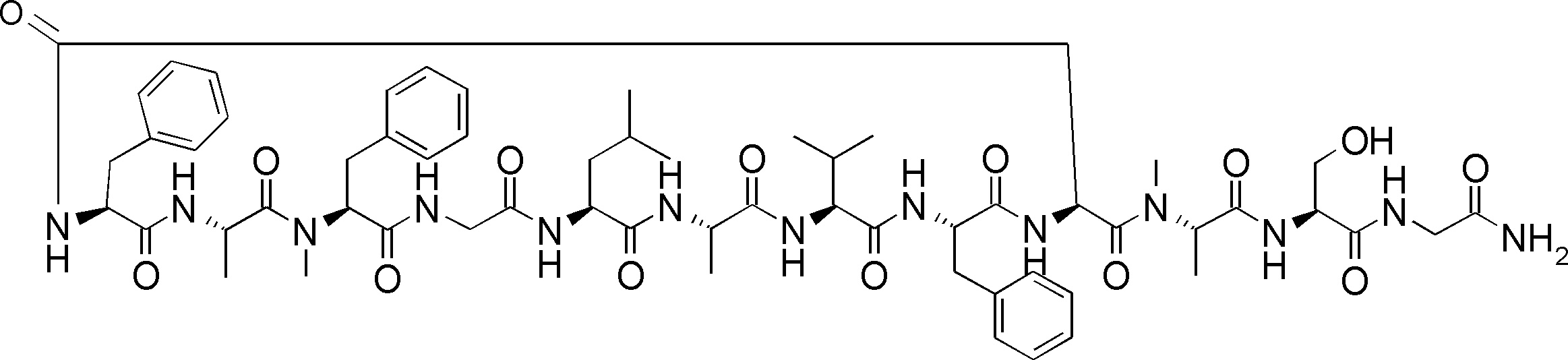
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成

・標題化合物
LCMS(ESI) m/z =1210.4(M+H)+
保持時間:1.04分(分析条件SMDmethod1)
(3S,6S,9S,12S,15S,21S,24S,27S)-27-アミノ-3-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-6,21-ジベンジル-15-イソブチル-9-イソプロピル-12,22,24-トリメチル-5,8,11,14,17,20,23,26-オクタオキソ-28-フェニル-4,7,10,13,16,19,22,25-オクタアザオクタコサン-1-酸
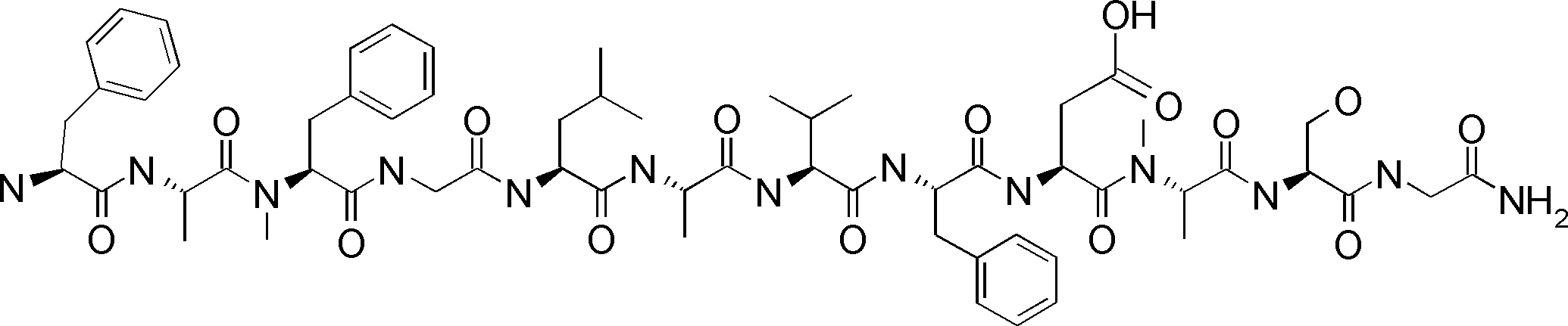
保持時間:0.89分(分析条件SMDmethod1)
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-8,23-ジベンジル-14-イソブチル-20-イソプロピル-N,2,5,7,17-ペンタメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-511)の合成

(3S,6S,9S,12S,15S,21S,24S,27S)-27-アミノ-3-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-6,21-ジベンジル-15-イソブチル-9-イソプロピル-12,22,24-トリメチル-5,8,11,14,17,20,23,26-オクタオキソ-4,7,10,13,16,19,22,25-オクタアザオクタコサン-1-チオ酸 S-ベンジル(化合物SP-504)のNMP溶液(10mM、5.0μl、0.050μmol)に内部標準として4-プロピル安息香酸のNMP溶液(11mM、2.25μl)を加えた。次に翻訳用緩衝液(6.25μl)、PURESYSTEM(r) classic II Sol. B(バイオコゥマー社、製品番号PURE2048C)(10μl)、20種の天然アミノ酸溶液(それぞれ5mM,2.5μl)を加えた。
なお翻訳用緩衝液の成分は8mM GTP,8mM ATP,160mMクレアチンリン酸,400mM HEPES-KOH pH7.6,800mM 酢酸カリウム,48mM 酢酸マグネシウム,16mMスペルミジン,8mM ジチオスレイトール,0.8mM 10-HCO-H4folate, 12mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社)である。これにトリス(2-カルボキシエチル)ホスフィン水溶液(pH=7.5、1.25M、2.0μl)および先に調整したチオール溶液を加えた。この時点での反応液のpHは7.8であった。
反応液を30℃で20時間攪拌した後、LC/MSで分析し、標題化合物の生成を確認した。20時間攪拌後の反応液のpHは9.4であった。また、標題化合物(SP-511)と加水分解体(化合物SP-512)との生成比はUVエリア比で1:1.6であった。
標題化合物
LCMS(ESI)m/z =1134.4(M+H)+
保持時間:0.64分(分析条件 SQDFA05)
加水分解体(化合物SP-512)
(3S,6S,9S,12S,15S,21S,24S,27S)-27-アミノ-3-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-6,21-ジベンジル-15-イソブチル-9-イソプロピル-12,22,24-トリメチル-5,8,11,14,17,20,23,26-オクタオキソ-4,7,10,13,16,19,22,25-オクタアザオクタコサン-1-酸

保持時間:0.48分(分析条件 SQDFA05)


LCMS(ESI) m/z = 167 (M+H)+
保持時間:0.81分(分析条件SQDFA05)

LCMS(ESI) m/z = 139 (M-H)-
保持時間:0.70分(分析条件SQDFA05)

LCMS(ESI) m/z = 277 (M-H)-
保持時間:0.95分(分析条件SQDFA05)

LCMS(ESI) m/z = 199 (M-H)-
保持時間:0.92分(分析条件SQDFA05)
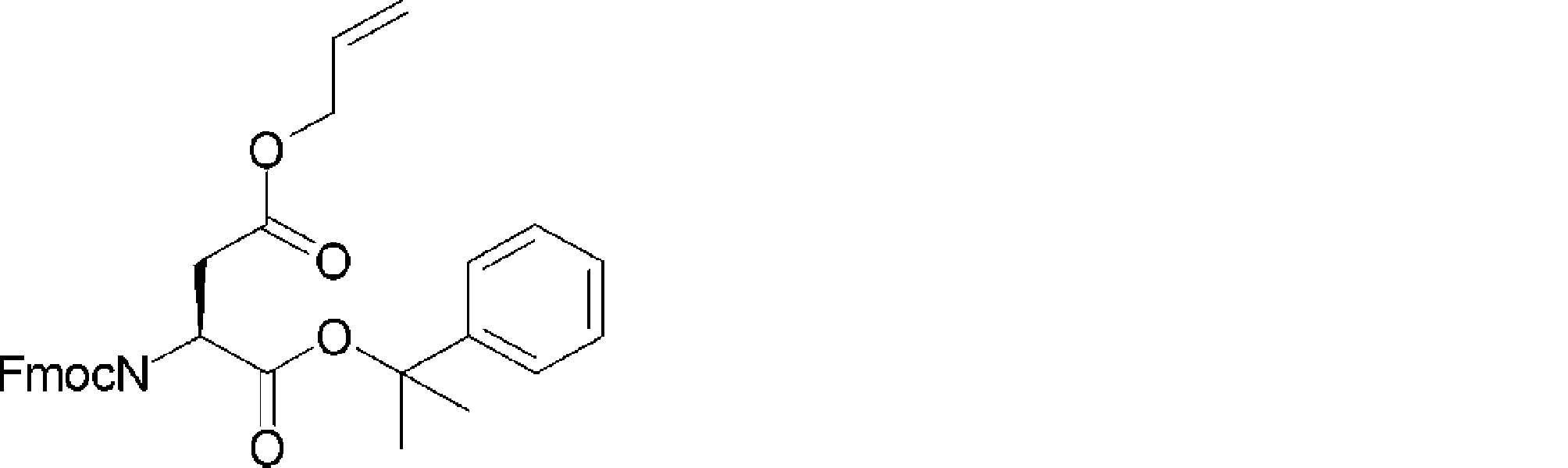
LC 保持時間:1.12分(分析条件SQDAA05)
1H-NMR(Varian 400-MR, 400 MHz, CDCl3) δ ppm 7.76 (2H, d, 7.6 Hz), 7.49 (2H, d, 7.2 Hz), 7.41-7.24. (9H, m),5.90(1H, m) 5.77 (1H,d, 8.4Hz), 5.29(2H, m), 4.62(3H, m), 4.37(2H, m), 4.21(1H, t, 6.8Hz),3.08(1H, dd, 17.2, 4.4Hz), 2.90(1H, dd, 17.2, 4.8Hz), 1.80 (3H, s), 1.78 (3H, s)
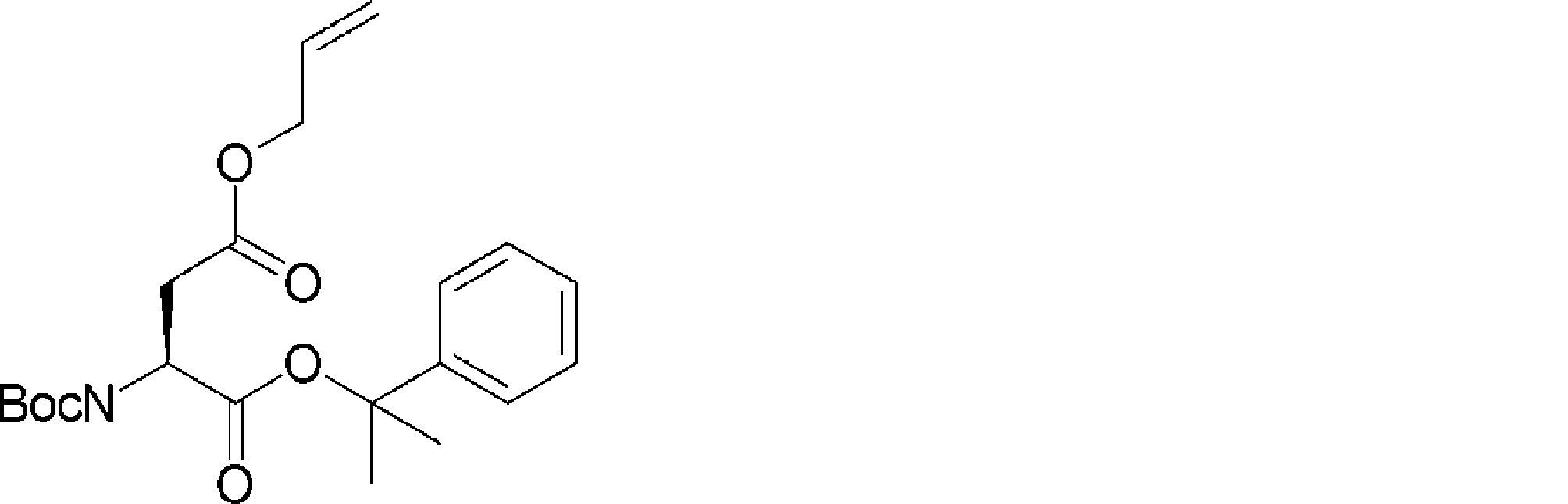
LCMS(ESI) m/z = 390(M-H)-
保持時間:0.63分(分析条件SQDAA50)
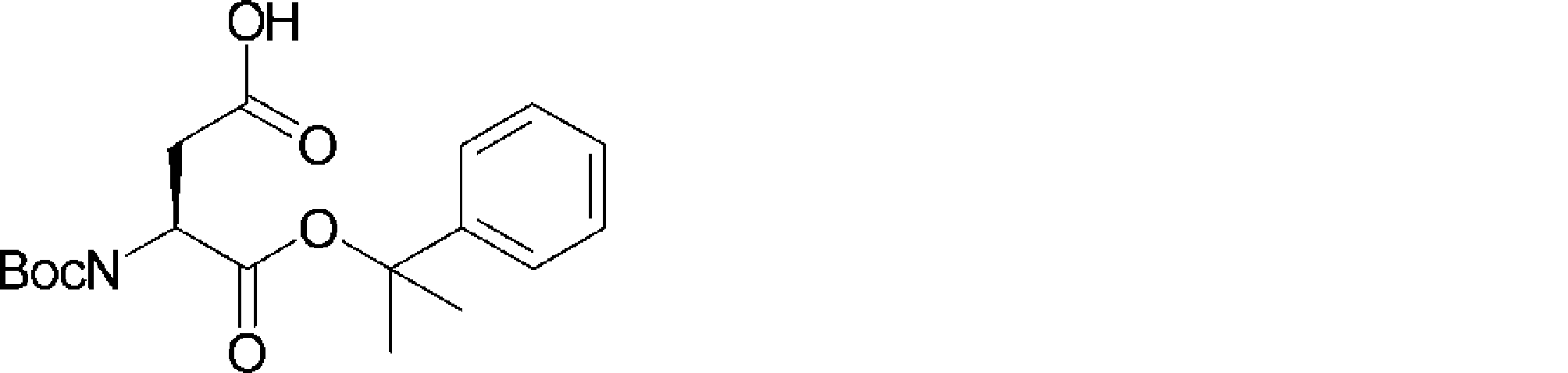
LCMS(ESI) m/z = 350(M-H)-
保持時間:0.80分(分析条件SQDAA05)
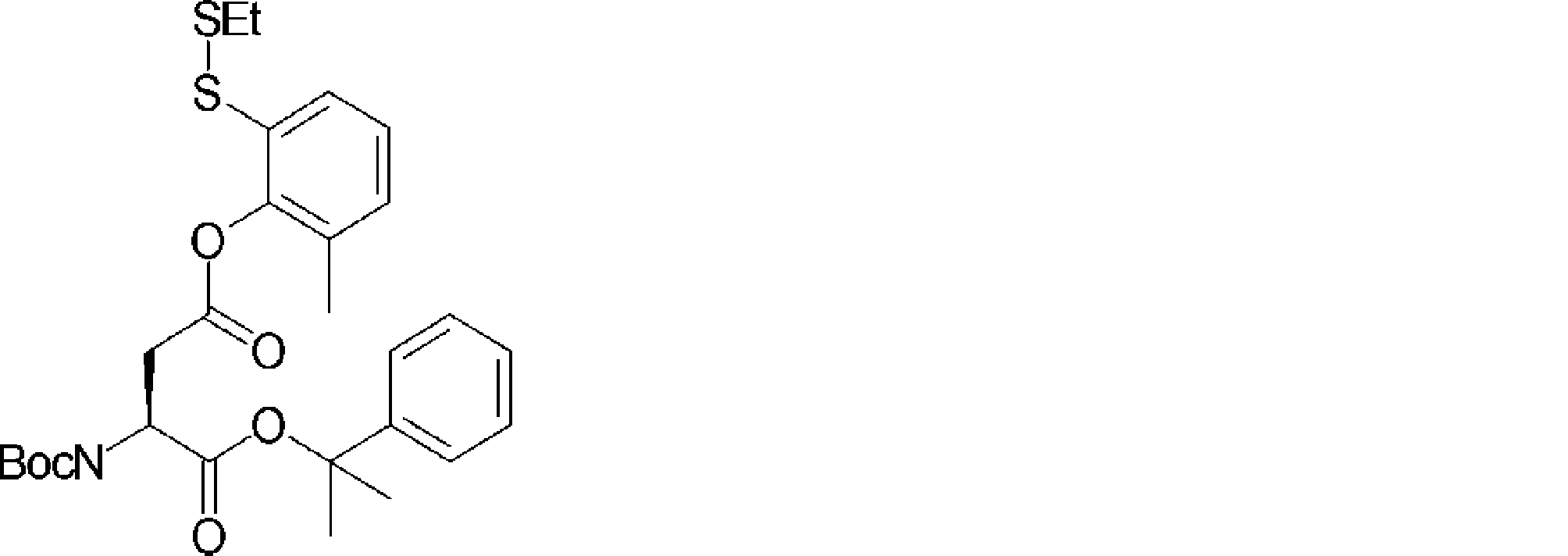
LCMS(ESI) m/z = 532(M-H)-
保持時間:1.15分(分析条件SQDAA05)

LCMS(ESI) m/z = 453(M-H)-
保持時間:1.04分(分析条件SQDAA05)
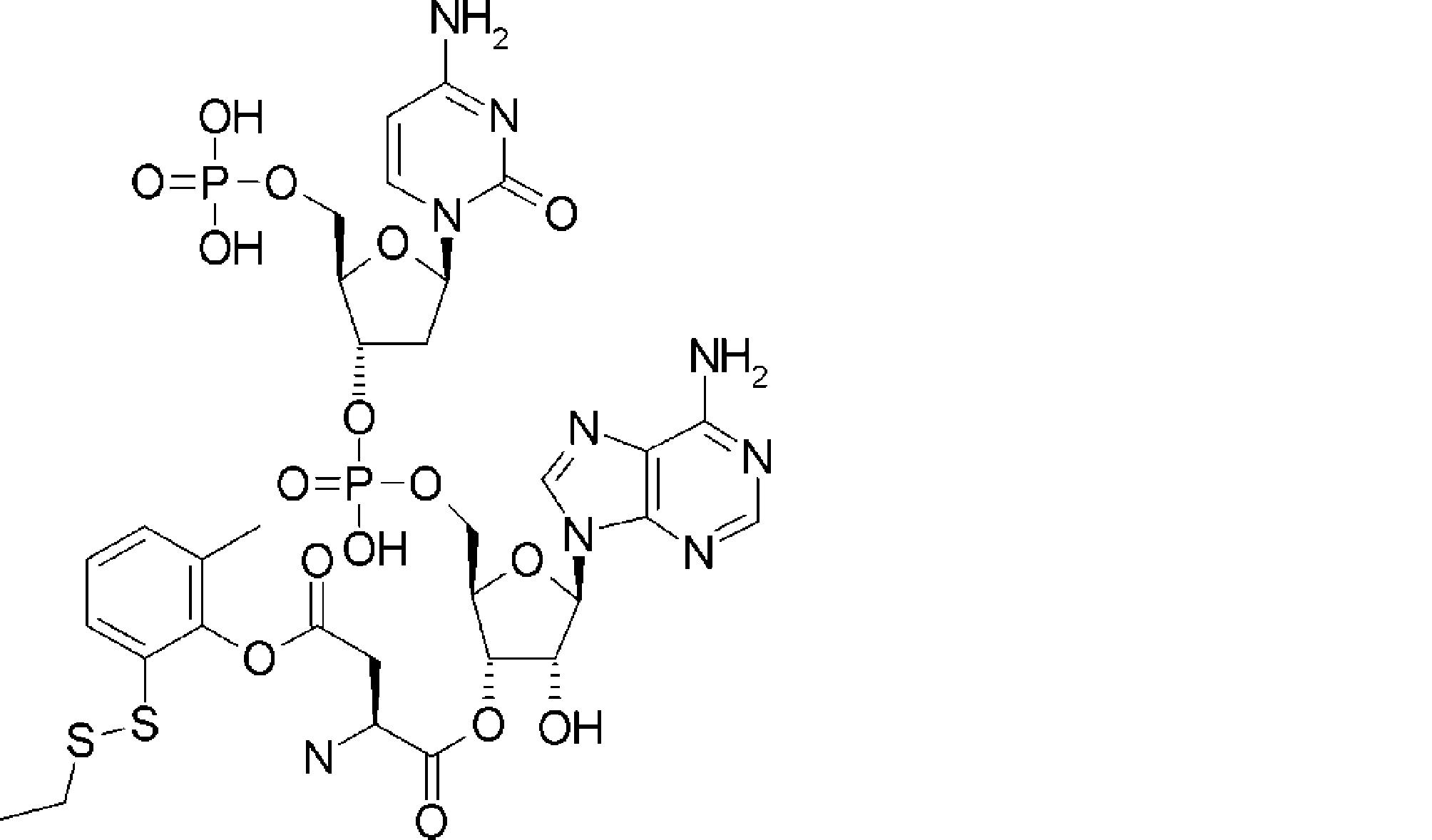
LCMS(ESI) m/z = 932 (M-H)-
保持時間:0.70分(分析条件SQDAA05)
翻訳合成後の反応補助基を利用しないアミド化反応をTOF-MSにて生成を確認した。
2-1. 転写によるtRNA(CA欠損)の合成
鋳型DNA(配列番号D-40)から、RiboMAX Large Scale RNA production System T7(Promega社,P1300)を用いたin vitro の転写により3'端のCAを欠くtRNAGluAAG (-CA)(配列番号R-40)を合成し、RNeasy Mini kit(Qiagen社)により精製した。
tRNAGluAAG (-CA) DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTAAGACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluAAG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUAAGACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
50μM 転写tRNAGluAAG (-CA)(配列番号R-40) 10μLに、10X ligation buffer(500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP)2μL、Nuclease free water 4μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 20unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)2μLおよび、5mMの側鎖カルボン酸が活性エステル化されたアミノアシル化pdCpA(化合物1i-ID)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。ライゲーション反応液 20μLに、3M酢酸ナトリウム 4μLと125mM ヨウ素(水:THF=1:1溶液)24μLを加え、室温で1時間、脱保護を行った。アミノアシル化tRNA(化合物AT-7-A)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-7-A)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Asp(SBn)-tRNAGluAAG
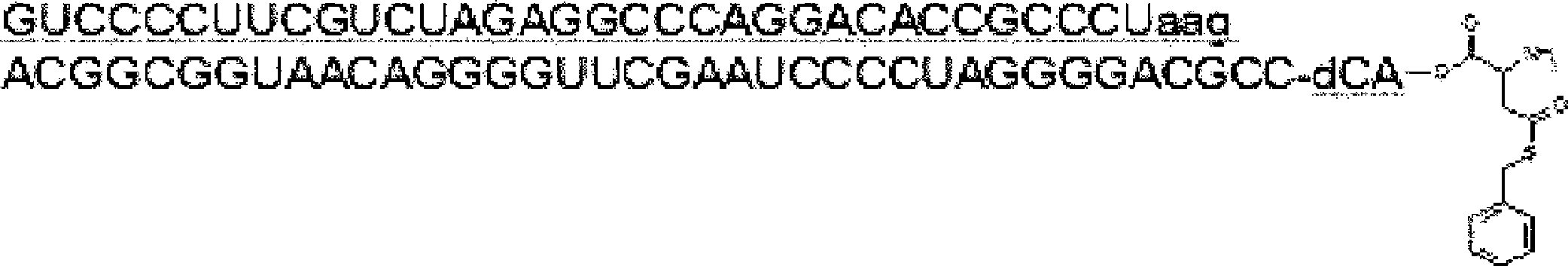
側鎖カルボン酸をチオエステルにより活性化したアスパラギン酸誘導体でアミノアシル化されたtRNAを、無細胞翻訳系に加えて翻訳を開始することにより、所望の非天然アミノ酸を含有するポリペプチドの翻訳合成を行った。翻訳系は、原核生物由来の再構成無細胞タンパク質合成系であるPURE systemを用いた。具体的には、翻訳液,1%(v/v) RNasein Ribonuclease inhibitor (Promega社,N2111), 1mM GTP,1mM ATP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,6mM 酢酸マグネシウム,2mMスペルミジン,1mM ジチオスレイトール,0.1mM 10-HCO-H4folate, 1.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.6μM メチオニルtRNAトランスフォルミラーゼ,0.26μM EF-G,0.24μM RF2,0.17μM RF3,0.5μM RRF,2.7μM IF1,0.4μM IF2,1.5μM IF3,40μM EF-Tu,93μM EF-Ts,1.2μM リボソーム,0.73μM AlaRS,0.03μM ArgRS,0.38μM AsnRS,0.13μM AspRS,0.02μM CysRS,0.06μM GlnRS,0.23μM GluRS,0.09μM GlyRS,0.02μM HisRS,0.4μM IleRS,0.04μM LeuRS,0.11μM LysRS,0.03μM MetRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.02μM TyrRS,0.02μM ValRS(自家調製タンパクは基本的にはHisタグ付加タンパクとして調製した))に、1μM 鋳型RNA、それぞれの鋳型DNAにコードされているタンパク質性アミノ酸群をそれぞれ250μMずつ、ならびに50μMの側鎖カルボン酸が活性エステル化されたアミノアシル化tRNA(化合物AT-7-A)を翻訳反応混合物に添加し、37℃で1時間静置することで行った。
1μM 鋳型RNAMgtp_R(配列番号R-41(配列番号:67))に、0.25mM Gly,0.25mM Pro,0.25mM Arg,0.25mM Thr,0.25mM Tyrおよび、50μM Asp(SBn)-tRNAGluAAG(化合物AT-7-A)を含む前述の翻訳液を37℃で60分間保温した。得られた翻訳反応物を、SPE C-TIP(日京テクノス社)で精製し、MALDI-MSで分析した。その結果、開始メチオニン直後のGlyから翻訳開始された、N末端α―アミノ基とチオエステルを含むペプチドP-141(図39、ピークI)、及びそのGly直後に位置するThrから翻訳開始されたペプチドP-142(図39、ピークII)が主生成物として観測された(図39)
Mgtp_R RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUaugGGUACUACAACGCGUCUUCCGUACCGUGGCGGCuaagcuucg
GlyThrThrThrArg[Asp(SBn)]ProTyrArgGlyGly
ThrThrThrArg[Asp(SBn)]ProTyrArgGlyGly
m/z: [H+M]+ = 1286.6(配列P-141に対応するペプチド。Calc.1286.6)
m/z: [H+M]+ = 1229.6(配列P-142に対応するペプチド。Calc.1229.6)
前述の翻訳反応物P-141を含む翻訳溶液3.5μL、1μLチオフェノール溶液(5M 4-トリフルオロメチルチオフェノール、5Mトリエチルアミンを等量混合したもの)、0.5μL 500mM トリカルボキシエチルホスフィン溶液 (pH7.5)を混合し、50℃で2時間保温した。その結果、出発物質であるP-141のピークは消失し、代わりにN末端α―アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物P-143に相当するピークを観測した(図40、ピークI)。
GlyThrThrThrArg[Asp(SBn)]ProTyrArgGlyGlyのN末端アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物
MALDI-MS:m/z: [M+H]+ = 1162.4 (Calc.1162.6)
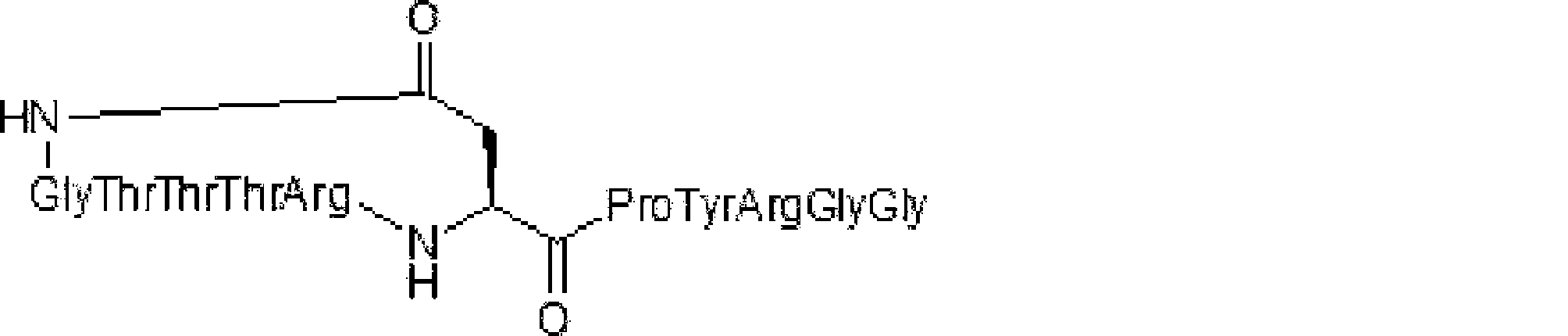
側鎖カルボン酸をチオエステルにより活性化したアスパラギン酸誘導体でアミノアシル化されたtRNAと、開始メチオニンを除いたタンパク質性アミノ酸混合物を無細胞翻訳系に加えて翻訳することで、所望の非天然アミノ酸を含有するポリペプチドの翻訳合成を行った。翻訳系は、原核生物由来の再構成無細胞タンパク質合成系であるPURE systemを用いた。具体的には、翻訳液,1%(v/v) RNasein Ribonuclease inhibitor (Promega社,N2111), 1mM GTP,1mM ATP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,6mM 酢酸マグネシウム,2mMスペルミジン,0.1mM 10-HCO-H4folate, 1.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.6μM メチオニルtRNAトランスフォルミラーゼ,0.26μM EF-G,0.24μM RF2,0.17μM RF3,0.5μM RRF,2.7μM IF1,0.4μM IF2,1.5μM IF3,40μM EF-Tu,93μM EF-Ts,1.2μM リボソーム,0.73μM AlaRS,0.03μM ArgRS,0.38μM AsnRS,0.13μM AspRS,0.02μM CysRS,0.06μM GlnRS,0.23μM GluRS,0.09μM GlyRS,0.4μM IleRS,0.04μM LeuRS,0.11μM LysRS,0.03μM MetRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.02μM TyrRS,0.02μM ValRS(自家調製タンパクは基本的にはHisタグ付加タンパクとして調製した))に、1μM 鋳型RNA、タンパク質性アミノ酸群をそれぞれ250μMずつ、ならびに50μMの側鎖カルボン酸が活性エステル化されたアミノアシル化tRNA(化合物AT-7-A)を翻訳反応混合物に添加し、37℃で1時間静置することで行った。
1μM 鋳型RNA OT89(配列番号RM-D1)に、0.25mM Phe,0.25mM Gly,0.25mM Pro,0.25mM Arg,0.25mM Thr,0.25mM Tyrおよび、50μM Asp(SBn)-tRNAGluAAG(化合物AT-7-A)を含む前述の翻訳液を37℃で60分間保温した。同様に別容器にて1μM 鋳型RNA OT90(配列番号RM-D2)に、0.25mM Ala,0.25mM Gly,0.25mM Pro,0.25mM Arg,0.25mM Thr,0.25mM Tyrおよび、50μM Asp(SBn)-tRNAGluAAG(化合物AT-7-A)を含む前述の翻訳液を37℃で60分間保温した。得られた2つの翻訳反応物1μLにそれぞれ9μLの0.2%トリフルオロ酢酸を加え、そのうち1μLずつをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート上乾固した。MALDI-MS分析の結果、RM-D1、RM-D2それぞれの鋳型から、開始メチオニン直後のPheもしくはAlaから翻訳開始された望みのペプチドP-D1(図44、ピークI)及びペプチドP-D2(図45、ピークI)が主生成物として観測された。
OT89 RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUaugUUUACUACAACGCGUCUUCCGUACCGUGGCGGCuaagcuucg
PheThrThrThrArg[Asp(SBn)]ProTyrArgGlyGly
OT90 RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUaugGCUACUACAACGCGUCUUCCGUACCGUGGCGGCuaagcuucg
AlaThrThrThrArg[Asp(SBn)]ProTyrArgGlyGly
m/z: [H+M]+ = 1376.4(配列P-D1に対応するペプチド。Calc.1376.6)
m/z: [H+M]+ = 1300.4(配列P-D2に対応するペプチド。Calc.1300.6)
前述の翻訳反応物P-D1を含む翻訳溶液3.5μLそれぞれに、1μLチオフェノール溶液(5M 4-トリフルオロメチルチオフェノール、5Mトリエチルアミンを等量混合したもの)、0.5μL 500mM トリカルボキシエチルホスフィン溶液 (pH7.5)を混合し、50℃で2時間保温した。得られた反応液2μLに12μLの2%トリフルオロ酢酸を加え、そのうち1μLをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート乾固した後、MALDI-MSにて解析を行った。その結果、出発物質であるP-D1のピークは消失し、代わりにN末端α―アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物P-D3に相当するピークを観測した(図44、ピークII)。前述の翻訳反応物P-D2を含む翻訳溶液についても、上記と同様な操作を行いMALDI-MSにて解析したところ、N末端α―アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物P-D4に相当するピークを観測した(図45、ピークII)
PheThrThrThrArg[Asp(SBn)]ProTyrArgGlyGlyのN末端アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物
MALDI-MS:m/z: [M+H]+ = 1252.3 (Calc.1252.6)
AlaThrThrThrArg[Asp(SBn)]ProTyrArgGlyGlyのN末端アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物
MALDI-MS:m/z: [M+H]+ = 1176.3 (Calc.1176.6)
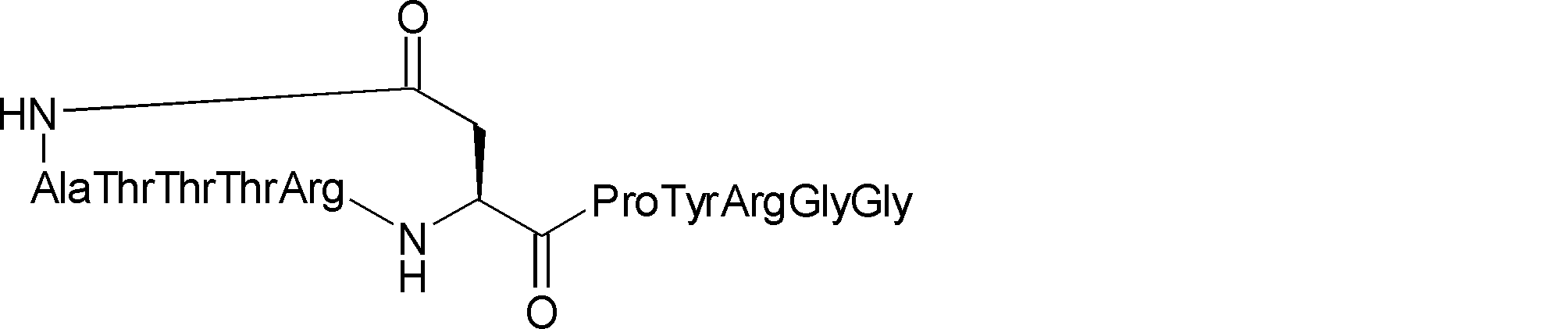
反応補助基を利用しないアミド化環化反応条件においてRNAが分解しないことを評価するため、反応条件に伏したRNAをゲル電気泳動により解析した。
1uM Mgtp_R RNA(配列番号R-41)を含む溶液(1mM GTP,1mM ATP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,6mM 酢酸マグネシウム,2mMスペルミジン,1mM ジチオスレイトール,0.1mM 10-HCO-H4folate,)3.5μL、1μLチオフェノール溶液(5M 4-トリフルオロメチルチオフェノール、5Mトリエチルアミンを等量混合したもの)、0.5μL 500mM トリカルボキシエチルホスフィン溶液 (pH7.6)を混合し、50℃で0.5~2時間保温した。その後反応液をRNeasy minelute(qiagen社)により精製した。環化条件に付していない対照実験として、保温時間をなくしたもの、チオフェノール溶液の代わりに水を加えたもの、それらに加えて精製操作を行わなかったものも同様の操作を施した。得られたRNA溶液は、6M尿素を含む10%ポリアクリルアミドゲルにて電気泳動を行った後、SYBR gold nucleic acid stain (Invitrogen社)により染色を行った。
その結果、対象実験として環化条件に付していないRNA (lane 1-3)と比べても、反応条件に付したRNA(lane 6)はバンドパターンやバンド濃さに変化がなく、環化反応条件においてもRNAが安定であることが明らかとなった(図46)。
翻訳ペプチドでの環化反応例と合成ペプチドでの翻訳合成液中での反応例では類似の結果を得たことから、合成ペプチドでの翻訳合成液中での反応収率向上が確認されれば、翻訳ペプチドの環化反応収率向上が得られたと解釈できる。以下、合成ペプチドでの翻訳液中での反応最適化検討を行った。
pHを9から10付近で反応を実施していたが、pH7.8, 8.1, 9.2の3点で環化反応を行った結果、pHが小さい方が環化:加水分解の比が目的の方向に向上することが明らかとなった。
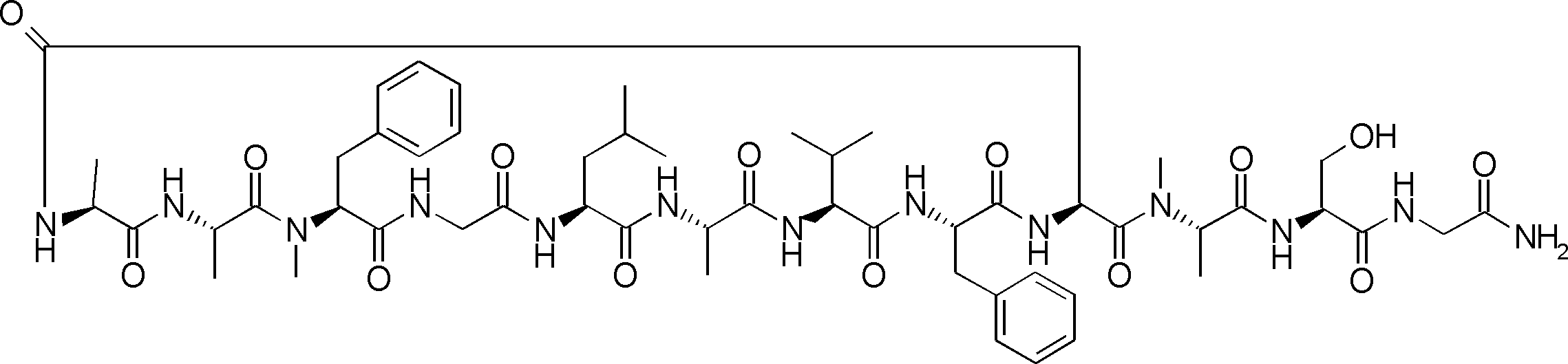
・標題化合物
LCMS(ESI)m/z =1132.3(M-H)-
保持時間:0.64分(分析条件SQDFA05)
・加水分解体(化合物SP-512)

保持時間:0.49分(分析条件SQDFA05)

従って、このような観点から、反応のpH上昇を抑えるため、反応液中の緩衝剤の濃度を62mMから500mMに上げ、還元剤としてトリス(2-カルボキシエチル)ホスフィン50mM を添加した条件で実験した。以下、環化体(標題化合物)/加水分解体の選択性に対するpHの影響を確認する為に、反応のpHは、pH7.8, 8.1, 9.2の3点で実施した。
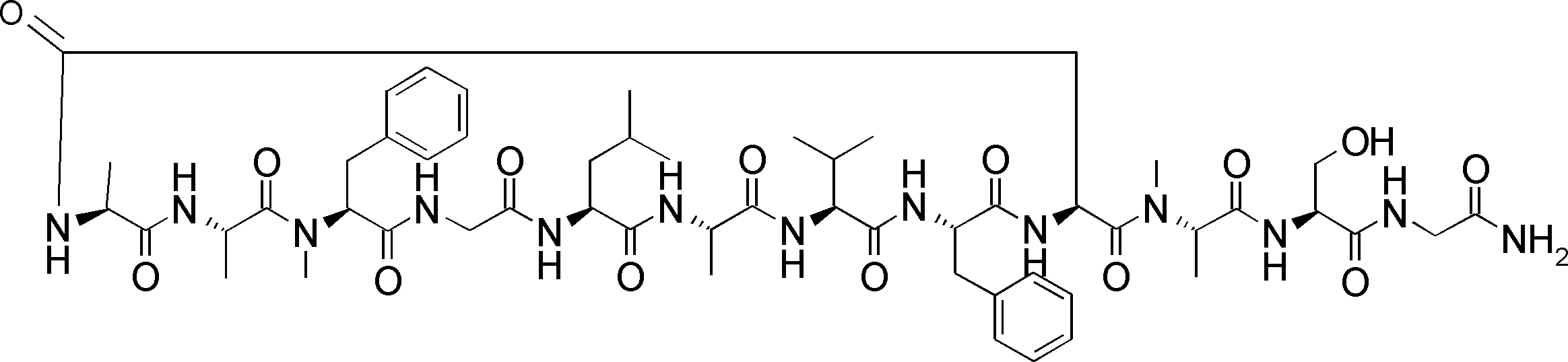
水(10.9ul)、4-(トリフルオロメチル)ベンゼンチオール(6.80ul、0.050mmol)とトリエチルアミン(6.97ul、0.050mmol)の混合溶液を調整した。その混合溶液に1.9M HEPES緩衝溶液(pH=7.5, 13.1ul)、0.5Mトリス(2-カルボキシエチル)ホスフィン水溶液(pH=7.6、5.0ul)を加えた。さらに、(3S,6S,9S,12S,15S,21S,24S,27S)-27-アミノ-3-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-6,21-ジベンジル-15-イソブチル-9-イソプロピル-12,22,24-トリメチル-5,8,11,14,17,20,23,26-オクタオキソ-28-フェニル-4,7,10,13,16,19,22,25-オクタアザオクタコサン-1-チオ酸 S-ベンジル(化合物SP-504)の10mM N-メチルピロリドン溶液5ulと11mMの内部標準(4-プロピル安息香酸)N-メチルピロリドン溶液(2.25ul)を加え30℃で6時間攪拌した。反応開始時のpHは7.8であった。LCMSを用いて反応の変化を観測したところ、標題化合物の生成を確認した。6時間で内部標準とのエリア面積比より転化率は96%であり、標題化合物(SP-511)と加水分解体(化合物SP-512)の生成比はLCMSのUVエリア比でおおよそ38:1であった。
・標題化合物
LCMS(ESI)m/z =1132.3(M-H)-
保持時間:0.63分(分析条件SQDFA05)
・加水分解体(化合物SP-512)
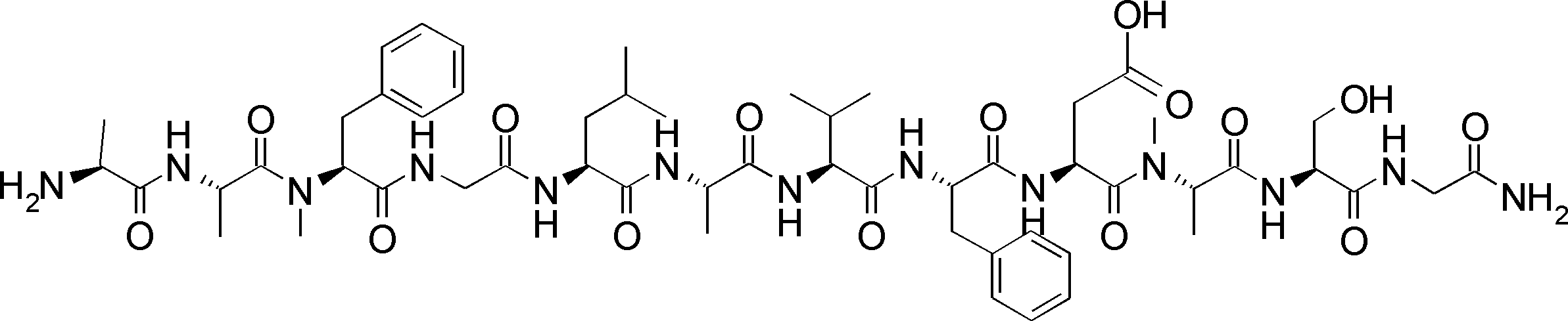
保持時間:0.48分(分析条件SQDFA05)
上記のpH7.8における化合物SP-511の合成と同様の方法で、1.9M HEPES緩衝溶液(pH=7.5, 13.1ul)の代わりに1.9M HEPES緩衝溶液(pH=8.1, 13.1ul)を用い反応を行った。30℃で6時間攪拌した。反応開始時のpHは8.1であった。LCMSを用いて反応の変化を観測したところ、標題化合物の生成を確認した。6時間で内部標準とのエリア面積比より転化率は93%であり、標題化合物(SP-511)と加水分解体(化合物SP-512)の生成比はLCMSのUVエリア比でおおよそ40:1であった。
・標題化合物(SP-511)
LCMS(ESI) m/z =1132.3(M-H)-
保持時間:0.63分(分析条件SQDFA05)
・加水分解体(化合物SP-512)
LCMS(ESI) m/z =1150.4(M-H)-
保持時間:0.48分(分析条件SQDFA05)
上記のpH7.8における化合物SP-511の合成と同様の方法で、1.9M HEPES緩衝溶液(pH=7.5, 13.1ul)の代わりに1.9M Bicine緩衝溶液(pH=9.5, 13.1ul)を用い反応を行った。30℃で6時間攪拌した。反応開始時のpHは9.2であった。LCMSを用いて反応の変化を観測したところ、標題化合物の生成を確認した。6時間で内部標準とのエリア面積比より転化率は87%であり、標題化合物(SP-511)と加水分解体(化合物SP-512)の生成比はLCMSのUVエリア比でおおよそ22:1であった。
・標題化合物(SP-511)
LCMS(ESI) m/z =1132.4(M-H)-
保持時間:0.63分(分析条件SQDFA05)
・加水分解体(化合物SP-512)
LCMS(ESI) m/z =1150.3(M-H)-
保持時間:0.48分(分析条件SQDFA05)
濃度1Mではなく100mMとした場合には、加水分解比率が増加することが明らかとなった。
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-8,23-ジベンジル-14-イソブチル-20-イソプロピル-N,2,5,7,17-ペンタメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-511)の合成

・標題化合物(SP-511)
LCMS(ESI) m/z =1132.3(M-H)-
保持時間:0.63分(分析条件SQDFA05)
・加水分解体(化合物SP-512)
LCMS(ESI) m/z =1150.2(M-H)-
保持時間:0.48分(分析条件SQDFA05)
有機溶媒比率は向上させる方が有利である結果を得た。
添加剤(4-(トリフルオロメチル)ベンゼンチオール)濃度1M、有機溶媒(NMP):水=50:50での反応例
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-8,23-ジベンジル-14-イソブチル-20-イソプロピル-N,2,5,7,17-ペンタメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-511)の合成
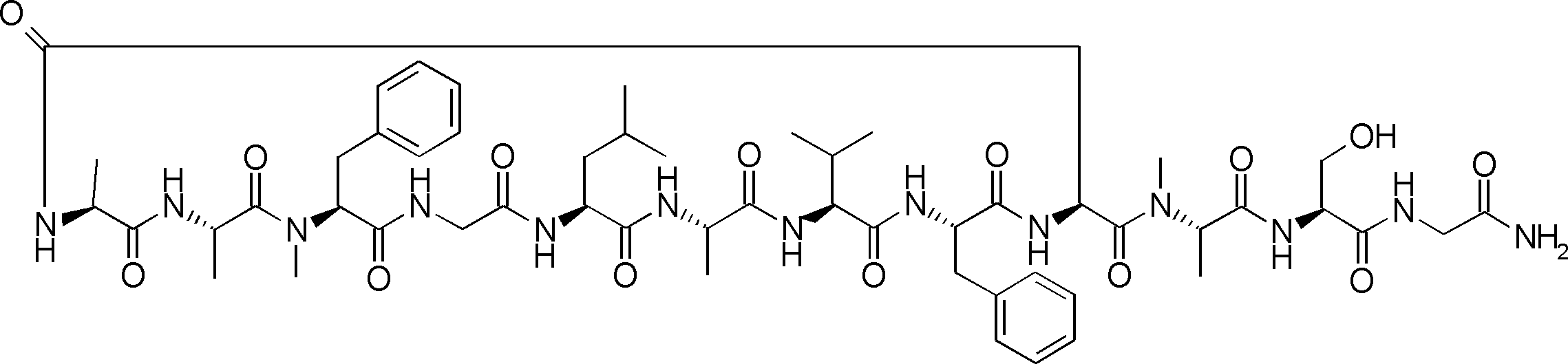
・標題化合物(SP-511)
LCMS(ESI)m/z =1134.5(M+H)+
保持時間:0.64分(分析条件SQDFA05)
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-8,23-ジベンジル-14-イソブチル-20-イソプロピル-N,2,5,7,17-ペンタメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-511)の合成
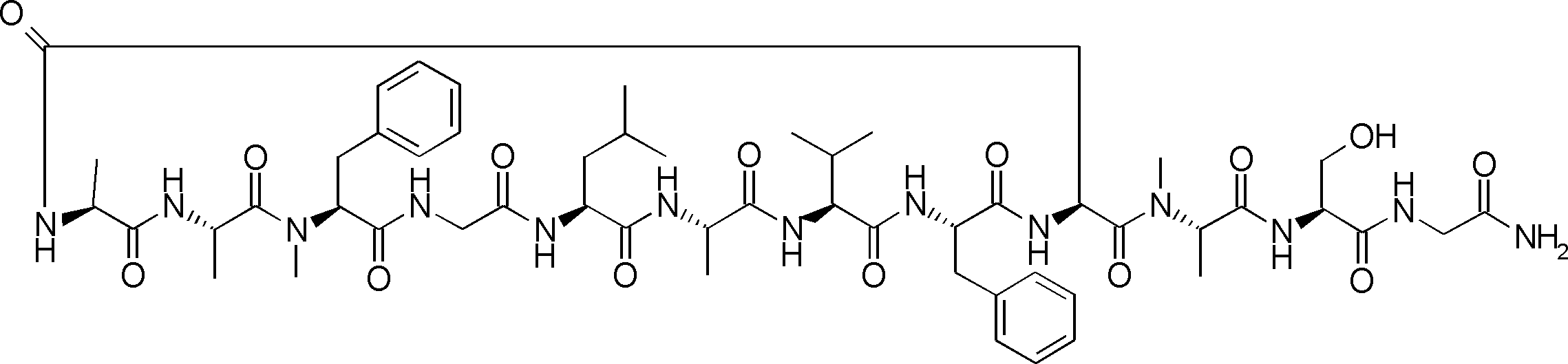
・標題化合物
LCMS(ESI)m/z =1134.5(M+H)+
保持時間:0.64分(分析条件SQDFA05)
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成
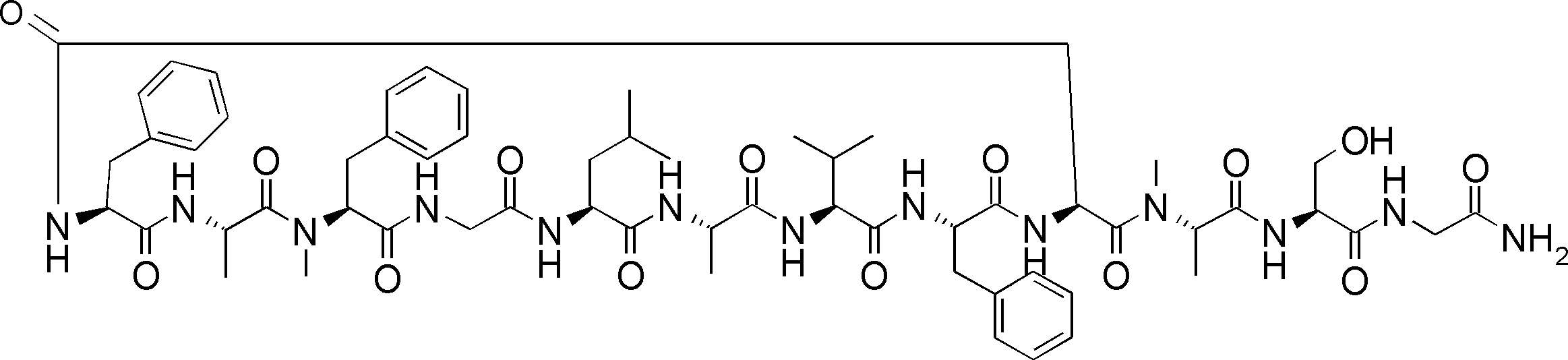
・標題化合物(SP-509)
LCMS(ESI)m/z =1210.4(M+H)+
・加水分解体(化合物SP-510)

(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成
・標題化合物(SP-509)
LCMS(ESI)m/z =1210.4(M+H)+
・加水分解体(化合物SP-510)
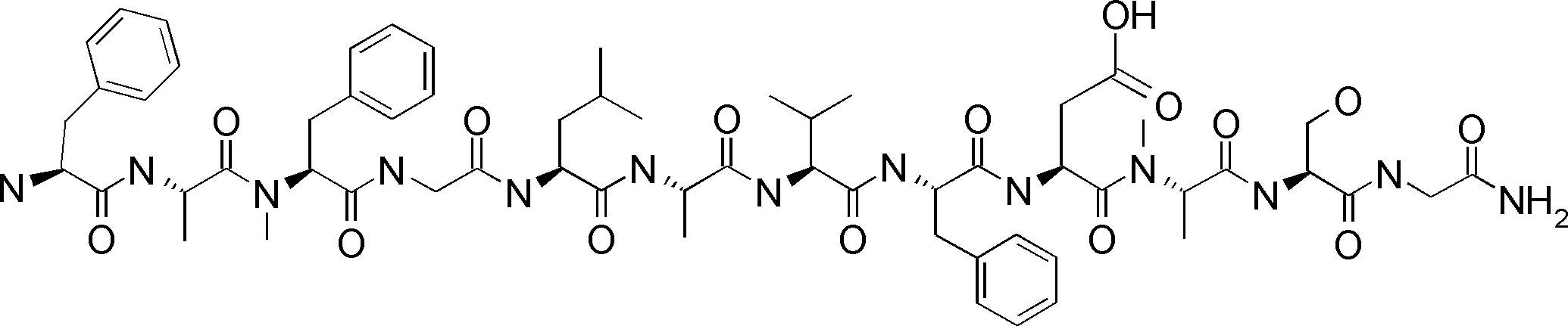
(3S,6S,9S,12S,15S,21S,24S,27S)-27-アミノ-3-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-6,21-ジベンジル-15-イソブチル-9-イソプロピル-12,22,24-トリメチル-5,8,11,14,17,20,23,26-オクタオキソ-28-フェニル-4,7,10,13,16,19,22,25-オクタアザオクタコサン-1-チオ酸 S-(4-ニトロフェニル)
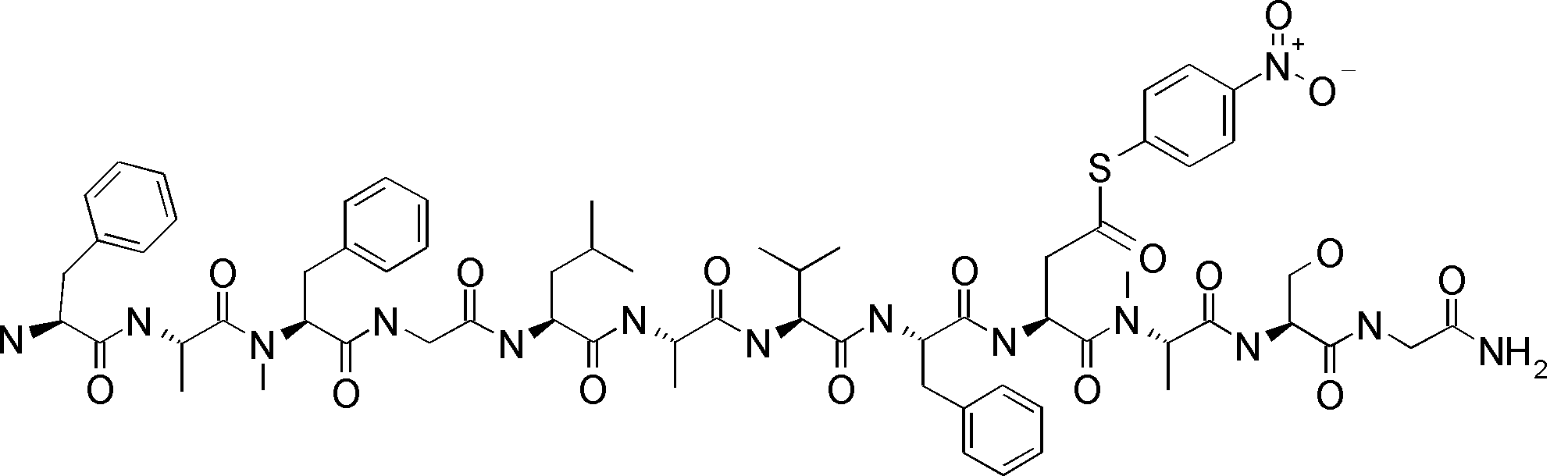
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成

・原料化合物(化合物SP-508)
LCMS(ESI)m/z =1334.4(M+H)+
・加水分解体(化合物SP-510)
LCMS(ESI)m/z =1228.4(M+H)+
・チオエステル交換体(化合物SP-514)

(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成
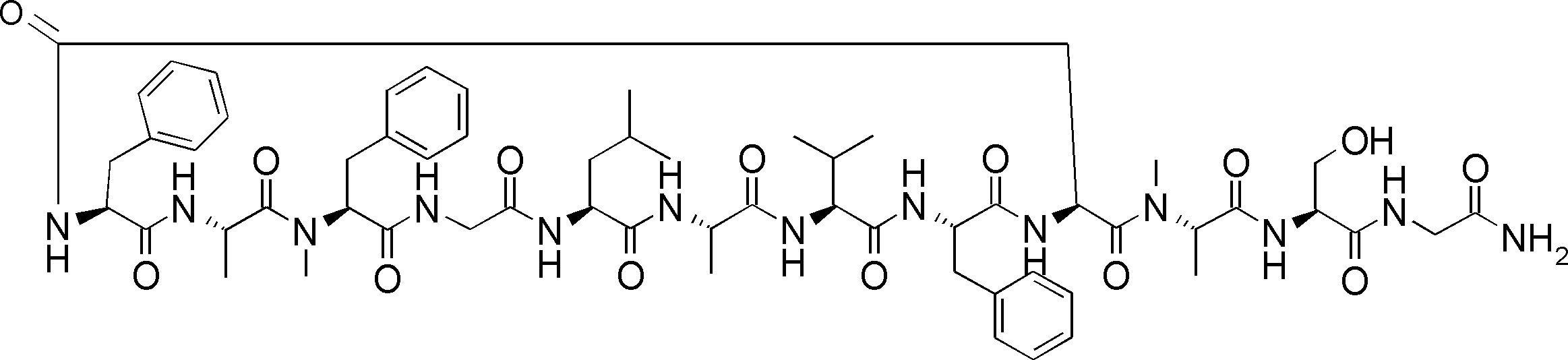
・標題化合物(SP-509)
LCMS(ESI)m/z =1208.9(M-H)-
・加水分解体(化合物SP-510)
LCMS(ESI)m/z =1226.3(M-H)-
・チオエステル交換体(化合物SP-515)
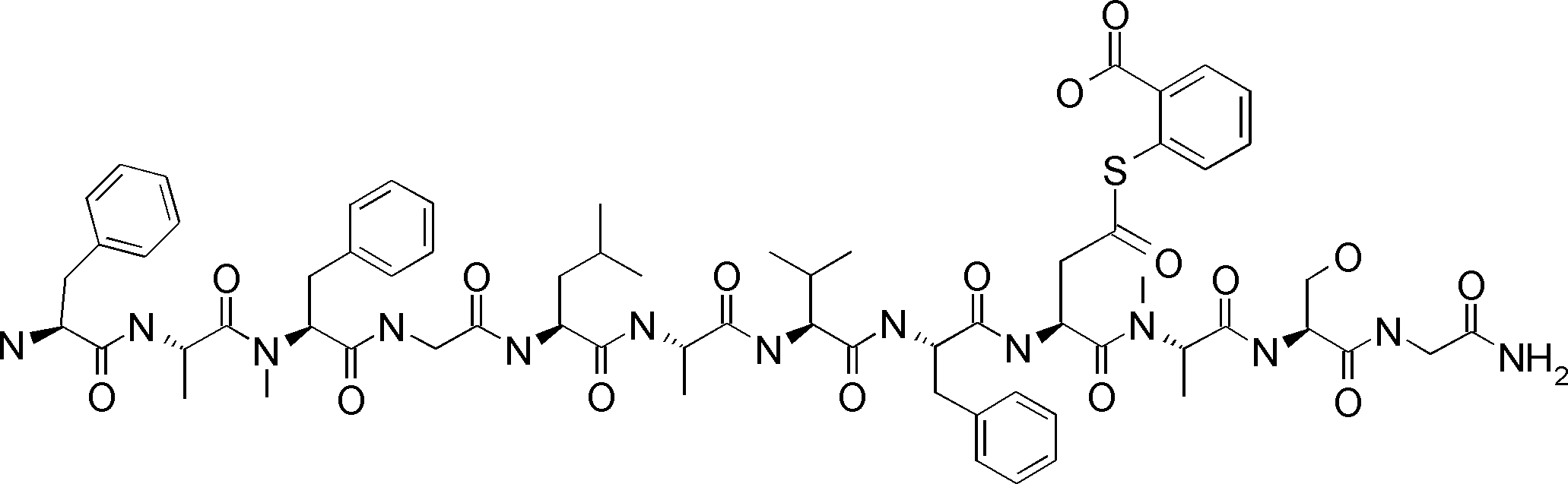
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成
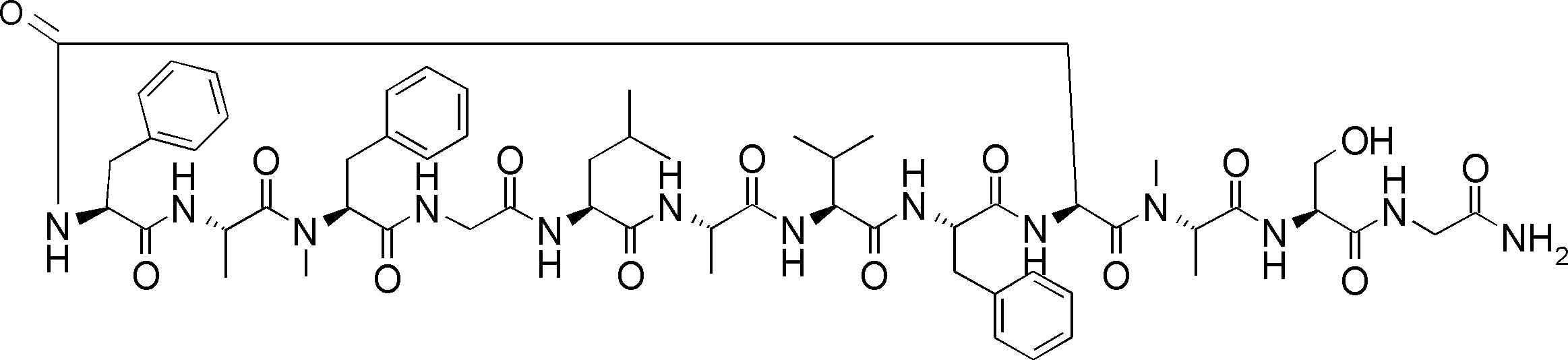
・標題化合物(SP-509)
LCMS(ESI)m/z =1210.4(M+H)+
・加水分解体(化合物SP-510)
LCMS(ESI)m/z =1228.4(M+H)+
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成
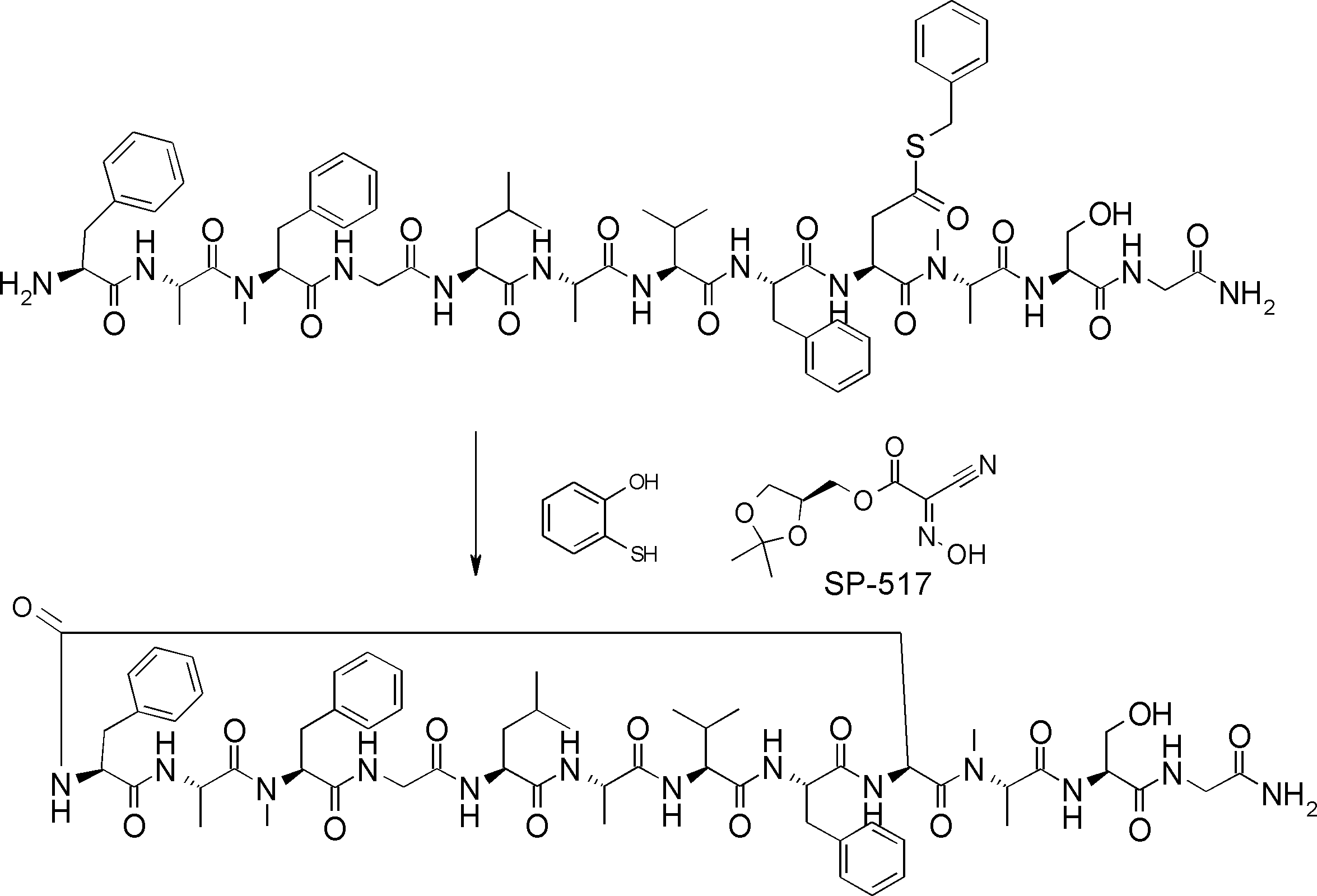
・標題化合物(SP-509)
LCMS(ESI)m/z =1208.1(M-H)-
・加水分解体(化合物SP-510)
LCMS(ESI)m/z =1226.2(M-H)-
・エステル交換体(化合物SP-516)
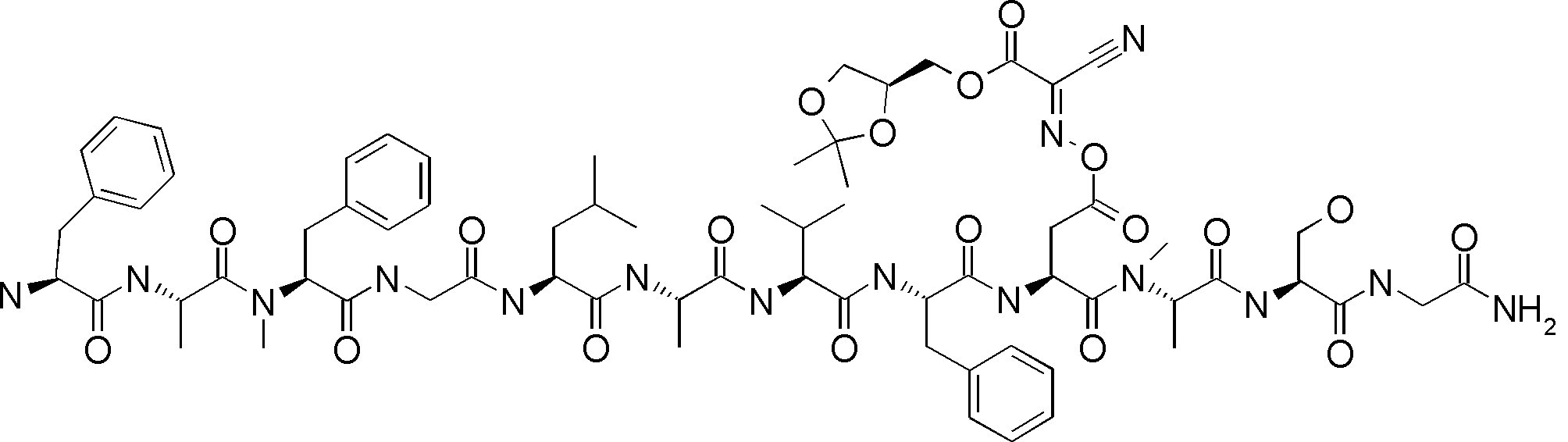
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-2,8,23-トリベンジル-14-イソブチル-20-イソプロピル-N,5,7,17-テトラメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-509)の合成

・加水分解体(化合物SP-510)
LCMS(ESI)m/z =1226.7(M-H)-
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-8,23-ジベンジル-14-イソブチル-20-イソプロピル-N,2,5,7,17-ペンタメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-511)の合成
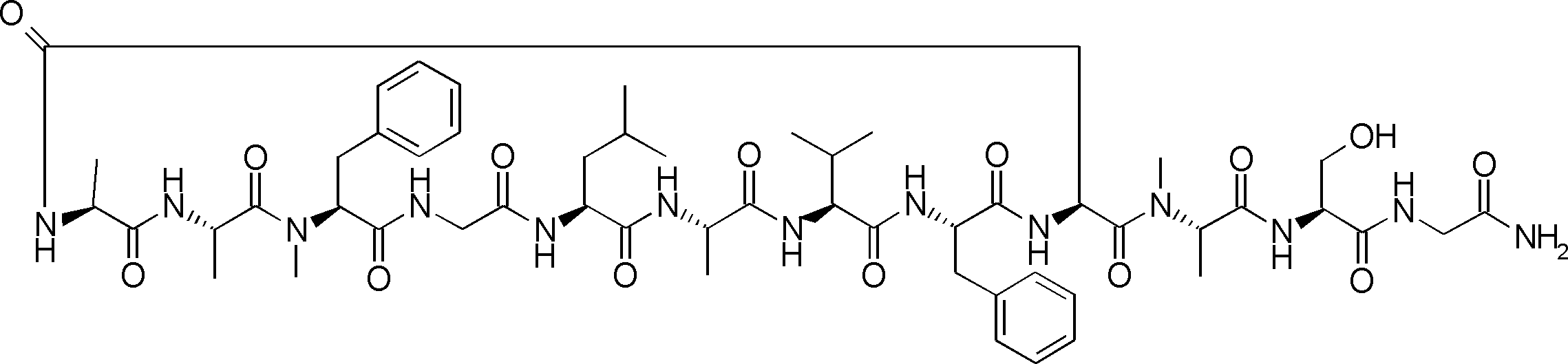
・標題化合物(SP-511)
LCMS(ESI) m/z =1132.3(M-H)-
保持時間:0.63分(分析条件SQDFA05)
・加水分解体(化合物SP-512)
LCMS(ESI) m/z =1150.2(M-H)-
保持時間:0.48分(分析条件SQDFA05)
(2S,5S,8S,14S,17S,20S,23S,26S)-N-((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)-8,23-ジベンジル-14-イソブチル-20-イソプロピル-N,2,5,7,17-ペンタメチル-3,6,9,12,15,18,21,24,28-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロオクタコサン-26-カルボキサミド(化合物SP-511)の合成

・標題化合物(SP-511)
LCMS(ESI) m/z =1132.3(M-H)-
保持時間:0.63分(分析条件SQDFA05)
・加水分解体(化合物SP-512)
LCMS(ESI) m/z =1150.4(M-H)-
保持時間:0.48分(分析条件SQDFA05)
また、活性エステルへの交換反応を単一の添加剤を加えて実施するのではなく、複数の添加剤を加えて実現させることも可能である。2-シアノ-2-(ヒドロキシイミノ)酢酸 (S,Z)-(2,2-ジメチル-1,3-ジオキソラン-4-イル)メチル(化合物SP-517)での活性化は、翻訳可能なチオエステルからの直接変換は達成できなかったが、まず、2-メルカプトフェノールにて翻訳可能なチオエステルを活性化した後、ここからのさらなる活性化が可能であることが明らかとなった。2-シアノ-2-(ヒドロキシイミノ)酢酸 (S,Z)-(2,2-ジメチル-1,3-ジオキソラン-4-イル)メチル(化合物SP-517)では、水中で高効率でN-メチル化されたアミノ酸やペプチドへの高効率なアミド化反応が報告されており(Organic Letter, 2012,14, 3372-3375)、2段階の活性化を経て本活性種への活性化が達成された事実は、注目に値する。
上に述べた反応条件最適化に伴い、反応翻訳液中で環化反応を実施した結果、翻訳ペプチドの環化実験実施条件と比較して加水分解比率を低下させ、目的化合物割合を向上させることができた。
(3S,6S,9S,12S,15S,21S,24S,27S)-27-アミノ-3-(((S)-1-(((S)-1-((2-アミノ-2-オキソエチル)アミノ)-3-ヒドロキシ-1-オキソプロパン-2-イル)アミノ)-1-オキソプロパン-2-イル)(メチル)カルバモイル)-6,21-ジベンジル-15-イソブチル-9-イソプロピル-12,22,24-トリメチル-5,8,11,14,17,20,23,26-オクタオキソ-4,7,10,13,16,19,22,25-オクタアザオクタコサン-1-チオ酸 S-ベンジル(化合物SP-504)のNMP溶液(10mM、5.0μl、0.050μmol)に内部標準として4-プロピル安息香酸のNMP溶液(11mM、2.25μl)を加えた。次に翻訳用緩衝液(6.25μl)、PURESYSTEM(r) classic II Sol. B(バイオコゥマー社、製品番号PURE2048C)(10μl)、20種の天然アミノ酸溶液(それぞれ5mM,2.5μl)を加えた。
反応液を30℃で22時間攪拌した後、LC/MSで分析し、標題化合物の生成を確認した。22時間攪拌後の反応液のpHは9.4であった。また、標題化合物(化合物SP-511)と加水分解体(化合物SP-512)との生成比はUVエリア比で2.6:1であった。
標題化合物(SP-511)
LCMS(ESI)m/z =1134.4(M+H)+
保持時間:0.64分(分析条件 SQDFA05)
加水分解体(化合物SP-512)
LCMS(ESI)m/z =1152.5(M+H)+
保持時間:0.48分(分析条件 SQDFA05)
N末端をAlaやPheに続き、N-アルキルアミノ酸であるMeAlaのモデル反応も実施し、環化体の生成を確認した。
(6S,9S,12S,15S,18S,24S,27S,30S,33S)-18,30-ジベンジル-12,24-ジイソブチル-9,27-ジイソプロピル-2,2,5,6,11,17,23,26-オクタメチル-33-(メチル((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)カルバモイル)-4,7,10,13,16,19,22,25,28,31-デカオキソ-15-((R)-1-(トリチルオキシ)エチル)-3-オキサ-5,8,11,14,17,20,23,26,29,32-デカアザペンタトリアコンタン-35-酸 (Boc-MeAla-Val-MeLeu-Thr(Trt)-MePhe-Gly-MeLeu-MeVal-Phe-Asp-MePhe-Ala-piperidine)(化合物SP-518)の合成
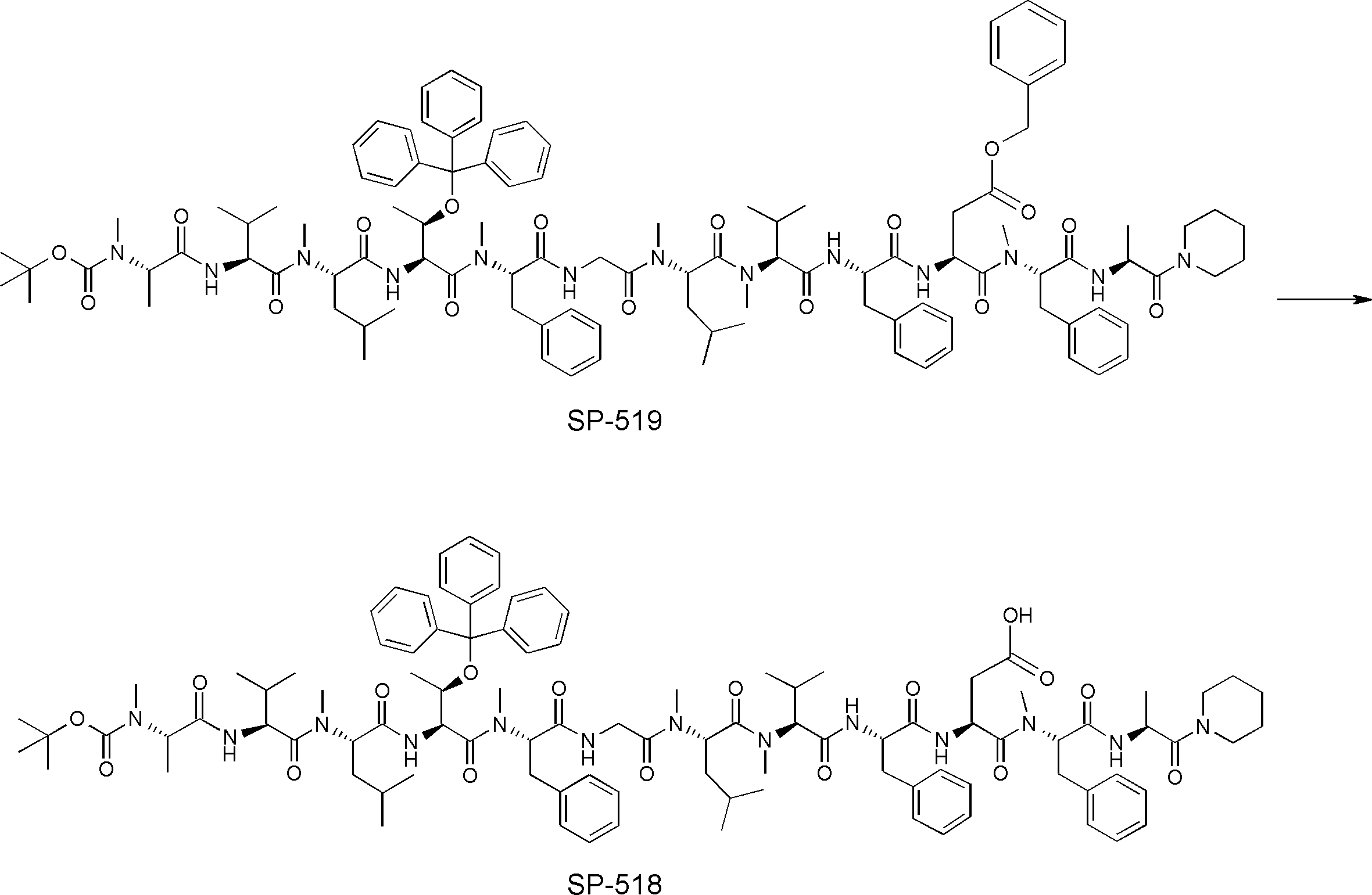
LCMS(ESI)m/z =1790.9(M-H)-
保持時間:0.87分(分析条件 SQDAA50)
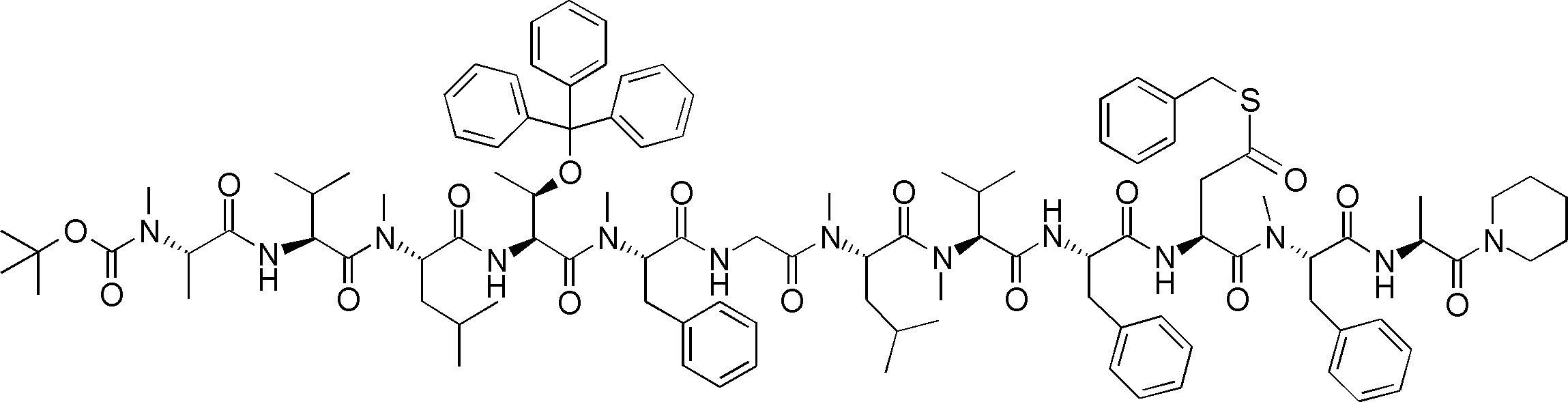
LCMS(ESI)m/z =1897.3(M-H)-
保持時間:0.99分(分析条件 SQDAA50)

LCMS(ESI)m/z =1555.1(M-H)-
保持時間:0.79分(分析条件 SQDFA05)
なお、本明細書では、Aspの側鎖カルボン酸が2-(エチルジスルファニル)-6-メチルフェニルエステル基にて置換された化合物をAsp(O(2-EtSS-6-Me-Ph))と記載する。この部位がペプチドに含まれる場合にも同様に記載することとする。
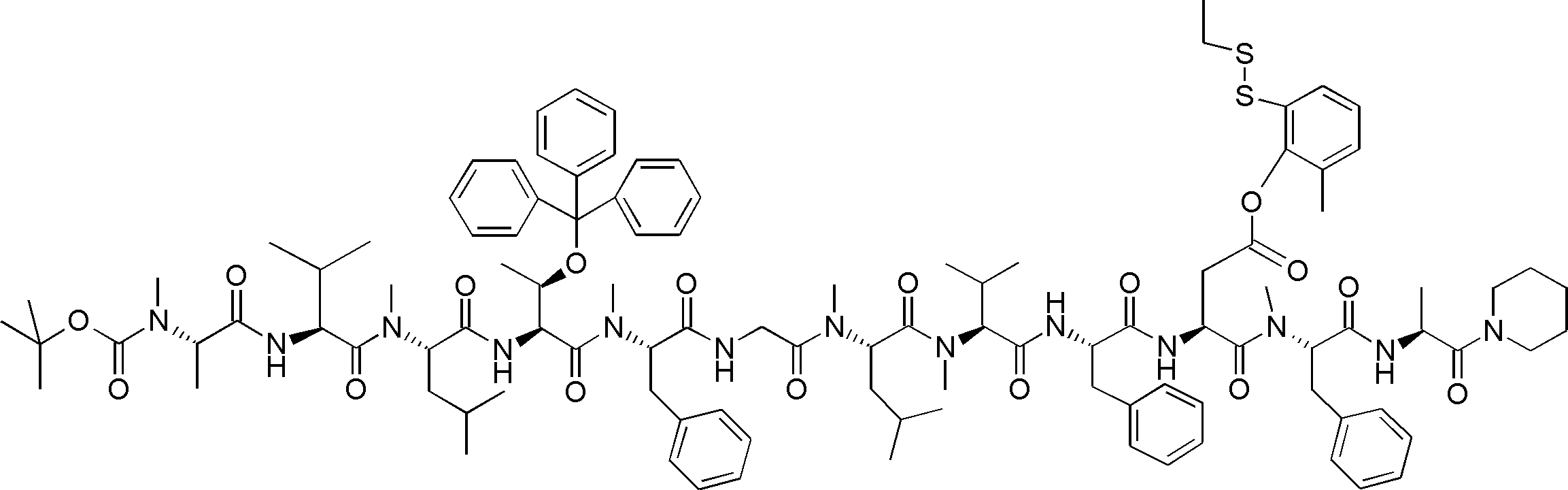
LCMS(ESI) m/z =1973.5(M―H)-
保持時間:1.01分(分析条件SQDAA50)

LCMS(ESI) m/z =1632.9(M+H)+
保持時間:0.83分(分析条件SQDFA05)
(2R,5S,8S,11S,14S,20S,23S,26S,29S)-14,26-ジベンジル-11-((R)-1-ヒドロキシエチル)-8,20-ジイソブチル-5,23-ジイソプロピル-N,1,2,7,13,19,22-ヘプタメチル-3,6,9,12,15,18,21,24,27,31-デカオキソ-N-((S)-1-オキソ-1-(((S)-1-オキソ-1-(ピペリジン-1-イル)プロパン-2-イル)アミノ)-3-フェニルプロパン-2-イル)-1,4,7,10,13,16,19,22,25,28-デカアザシクロヘントリアコンタン-29-カルボキサミドの(化合物SP-524)合成
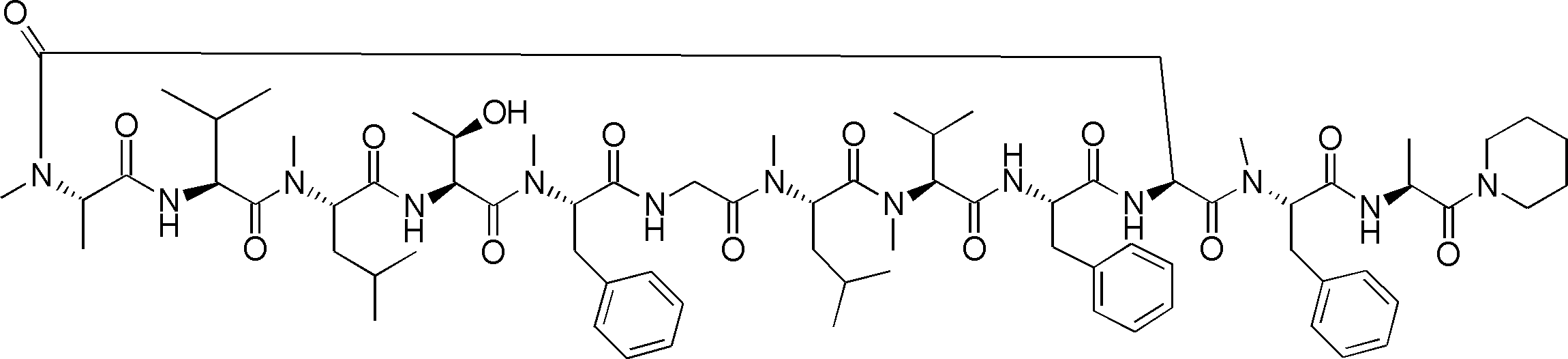
LCMS(ESI)m/z =1432.8(M+H)+
保持時間:1.06分(分析条件 SQDFA05)
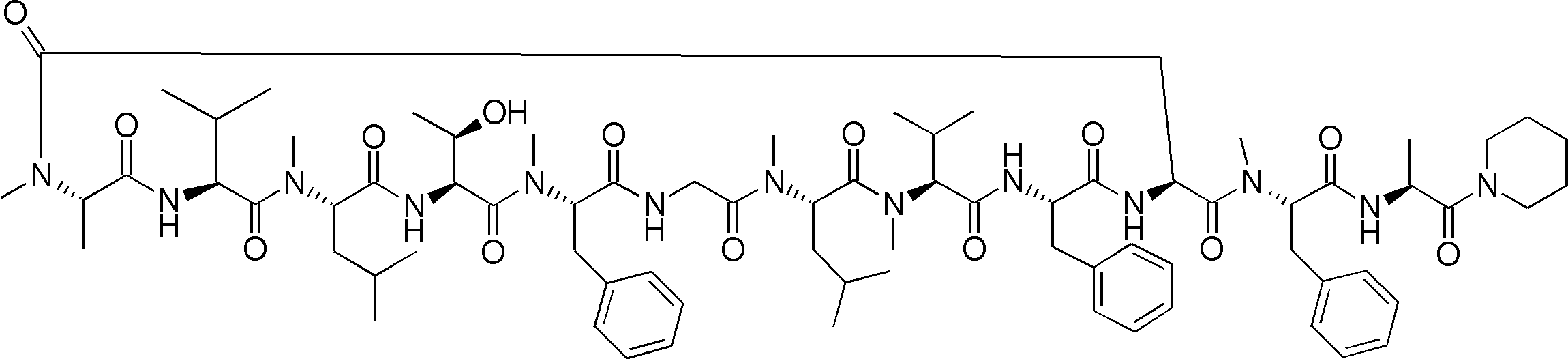
LCMS(ESI) m/z =1432.9(M+H)+
保持時間:1.09分(分析条件SQDFA05)
RNA-ペプチド複合体において反応補助基を利用しないペプチド環化反応を施し、生成物を電気泳動にて解析した。
PCRにより調製した3つのDNA(配列番号 DM-D1、DM-D2、DM-D3)を鋳型に、それぞれIn vitro転写によりmRNA(配列番号 RM-D3、R-D4、R-D5)を調整し、RNeasy mini kit (Qiagen社)を用いて精製した。各々に対し、10μMのmRNAに、50μMのピューロマイシンリンカー(Sigma社)(配列番号C-D1) 1X T4 RNAライゲースリアクションバッファー(NEB社)、1mM DTT、1mM ATP、0.02%BSA(Takara社)、510μM (PEG2000)(Wako社)、10% DMSO、1%(v/v) RNasein Ribonuclease inhibitor (Promega社,N2111)0.85 unit/μl T4 RNAライゲース(NEB社)を加え、15℃で一晩ライゲーション反応させた後、RNeasy MinElutekit(Qiagen社)により精製した。次に、上記で調製した3つのmRNA-ピューロマイシンリンカー連結体1 μMそれぞれを鋳型に、前述の無細胞翻訳液と0.25mM Ser、0.25mM Gly,0.25mM Pro,0.25mM Arg,0.25mM Thr,0.25mM Tyrおよび、50μM Asp(SBn)-tRNAGluAAG(化合物AT-7-A)を加え、37℃で60分間保温した後、続けて室温で12分間保温した。また、OT98RNA(配列番号 RM-D4)、OT99RNA(配列番号 RM-D5)由来のmRNA-ピューロマイシンを鋳型にする場合、それぞれ0.25mMのPhe, 0.25mM のAlaを加えて翻訳を行った。その後、反応液をRNeasy minelute(qiagen社)により精製し、RNA-ペプチド複合体を得た。
OT-97 DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGGGTACTACAACGCGTCTTCCGTACCGTAGCGGCTCTGGCTCTGGCTCTAAAAAAA
OT-98 DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTTTACTACAACGCGTCTTCCGTACCGTAGCGGCTCTGGCTCTGGCTCTAAAAAAA
OT-99 DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGGCTACTACAACGCGTCTTCCGTACCGTAGCGGCTCTGGCTCTGGCTCTAAAAAAA
OT-97 RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUAUGGGUACUACAACGCGUCUUCCGUACCGUAGCGGCUCUGGCUCUGGCUCUAAAAAAA
OT-98 RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUAUGUUUACUACAACGCGUCUUCCGUACCGUAGCGGCUCUGGCUCUGGCUCUAAAAAAA
OT-99 RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUAUGGCUACUACAACGCGUCUUCCGUACCGUAGCGGCUCUGGCUCUGGCUCUAAAAAAA
[P]dCdC [Fluorecein-dT][Spacer18][Spacer18][Spacer18][Spacer18]dCdC[puromycin] ([P]:5'リン酸化)
窒素雰囲気下、上記で調整したOT-97 RNA配列(配列番号RM-D3)に由来する70μLのRNA-ペプチド複合体に、20.1μLのチオフェノール溶液(5M 4-トリフルオロメチルチオフェノール水溶液、5Mトリエチルアミン水溶液を等量混合したもの)、10μL 500mM トリカルボキシエチルホスフィン溶液 (pH7.5)を混合し、50℃で2時間反応させた。また、HEPES緩衝液(pH7.6)を用いて5M 4-トリフルオロメチルチオフェノール溶液および5M トリエチルアミン溶液を調整し、これらを等量混合してチオフェノール溶液を調整した後、OT-98 RNA配列(配列番号RM-D4)、OT-99 RNA配列(配列番号RM-D5)に由来するRNA-ペプチド複合体70μLそれぞれに対し、20.1μLのNMP、上述のチオフェノール溶液20.1μL、500mM トリカルボキシエチルホスフィン溶液 (pH7.5) 10μL を窒素雰囲気下で混合し、30℃で22時間攪拌した。
得られた3つの反応液それぞれから、RNeasy minelute(qiagen社)を用いてペプチドーRNA複合体を精製し、純水70μLにてカラムから溶出した。続けて、8.4μLの10xRNase ONE ribonuclease反応バッファー(promega社)、2.8μLのRNase ONE ribonuclease (promega社)、2.8μLのRNase H (Life technologies社)を加え、37度にて一晩保温した。続いて、得られた反応溶液と何も反応させていないピューロマイシンリンカー(配列番号C-D1)をpeptide-PAGE mini(TEFCO社)にて電気泳動し、ピューロマイシンリンカーに由来するFluoreceinにてバンドを可視化した。その結果、いずれの反応溶液に関してもピューロマイシンリンカーにペプチドが結合したことに由来するバンド移動度の差が観測された((図47)。このことから、目的の環化ペプチド-RNA複合体の存在が示された。
翻訳後に、N末端のフォルミル化された翻訳開始アミノ酸をメチオニンアミノペプチダーゼ、及びペプチドデフォルミラーゼにて切断し、それにより露出したα-アミノ基を用いてペプチド環化反応を施し、MALDI-MSにて分析した。なお、翻訳開始アミノ酸としては、メチオニンの代わりに、酸化されやすい硫黄原子を含まないアミノ酸であるノルロイシン(CAS番号327-57-1)を使用した。
1μM 鋳型RNA Mgtp_R(配列番号R-41)に、0.25mM Pro,0.25mM Gly,0.25mM Thr,0.25mM Arg,0.25mM Tyr,2.5mM ノルロイシン(Nle)および、50μM Asp(SBn)-tRNAGluAAG(化合物AT-7-A)を含む前述の翻訳液を37℃で60分間保温した。得られた翻訳溶液1μLに9μLの0.2%トリフルオロ酢酸を加え、そのうち1μLをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート上乾固した。MALDI-MSの結果、フォルミルノルロイシンより翻訳開始された目的のペプチドP-D5に由来するピークが観測された(図48、ピークI)。続いて上記で得られた翻訳溶液4μLに、Hisタグ付加タンパクとして調製した150μM メチオニンアミノペプチダーゼ、20μM ペプチドデフォルミラーゼをそれぞれ0.5μLずつ加え、37℃で30分間保温した。得られた反応液1μLを前述したように前処理し、MALDI-MSにて分析したところ、開始フォルミルノルロイシンが外れ、N末端にGlyが露出したペプチドP-141に相当するピークが観測された(図48、ピークII)。最後に、このP-141を含む翻訳溶液3.5μL、1μLチオフェノール溶液(5M 4-トリフルオロメチルチオフェノール、5Mトリエチルアミンを等量混合したもの)、0.5μL 500mM トリカルボキシエチルホスフィン溶液 (pH7.6)を混合し、50℃で2時間保温した。得られた反応溶液2μLを使って、12μLの2%トリフルオロ酢酸を加え、そのうち1μLをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート乾固した後、MALDI-MSにて解析を行った。その結果N末端α―アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した目的の化合物P-143に相当するピークを観測した(図48、ピークIII)。
fNleGlyThrThrThrArg[Asp(SBn)]ProTyrArgGlyGly
m/z: [H+M]+ = 1427.4(配列P-D5に対応するペプチド。Calc.1427.7)
m/z: [H+M]+ = 1286.4(配列P-141に対応するペプチド。Calc.1286.6)
m/z: [H+M]+ = 1162.3(配列P-143に対応するペプチド。Calc1162.6)
1.分枝ペプチドの翻訳産物からの生成を可能とするユニットの選択および、反応条件検討
1-1.分子間モデル反応によるアミド化反応
以下に示す1-(2-メルカプトエチルアミノ)-1-オキソ-3-フェニルプロパン-2-イルカルバミン酸 (S)-tert-ブチル(化合物10)の合成検討の結果、エステル官能基を水中でチオールの添加によりチオエステルに活性化させることが出来、かつ活性化させたチオエステルからアミンとの縮合反応によりアミド結合を選択的に生成させることができることが明らかとなった。
以下の反応の結果を図41に示す。
2-メルカプトエタンスルホン酸ナトリウム(33mg、 0.200mmol)と2-アミノエタンチオール 塩酸塩(22.7mg、 0.200mmol)にpH7.7の0.2M HEPES緩衝液(1.60ml)、DMF(0.400ml)を加えpH7.4に調整し、室温で5分間攪拌した。その後、2-(tert-ブトキシカルボニルアミノ)-3-フェニルプロパン酸 (S)-2-アミノ-2-オキソエチル(化合物11)(6.4mg、 0.02mmol)を加え、50度で24時間攪拌した。
LCMSを用いて反応の変化を観測したところ、24時間で1-(2-メルカプトエチルアミノ)-1-オキソ-3-フェニルプロパン-2-イルカルバミン酸 (S)-tert-ブチル(化合物10)が生成していることを確認した。加水分解物と目的化合物10との生成比は、LCMSのUVエリア比で46:43であった。
2-メルカプトエタンスルホン酸ナトリウム(164mg、 1.000mmol)と2-アミノエタンチオール 塩酸塩(11.4mg、 0.100mmol)にpH7.0の0.2M HEPES緩衝液(0.250ml)、DMF(0.200ml)、水(0.500ml)を加え室温で5分間攪拌した。さらに1M 水酸化ナトリウム水溶液(0.050ml)を加えてpH7.6に調整した後に、2-(tert-ブトキシカルボニルアミノ)-3-フェニルプロパン酸 (S)-2-アミノ-2-オキソエチル(化合物11)(3.2mg、 0.01mmol)を加え、40度で24時間攪拌した。
LCMSを用いて反応の変化を観測したところ、24時間で1-(2-メルカプトエチルアミノ)-1-オキソ-3-フェニルプロパン-2-イルカルバミン酸 (S)-tert-ブチル(化合物10)が生成していることを確認した。加水分解物と目的化合物10との生成比は、LCMSのUVエリア比で24:60であった。
2-ジメチルアミノエタンチオール塩酸塩(142mg、 1.000mmol)と2-アミノエタンチオール 塩酸塩(11.4mg、 0.100mmol)にpH7.0の0.2M HEPES緩衝液(0.750ml)、DMF(0.200ml)、水(0.100ml)を加え室温で5分間攪拌した。さらに1M 水酸化ナトリウム水溶液(0.150ml)を加えてpH7.3に調整した後に、2-(tert-ブトキシカルボニルアミノ)-3-フェニルプロパン酸 (S)-2-アミノ-2-オキソエチル(化合物11)(3.2mg、 0.01mmol)を加え、40度で24時間攪拌した。
LCMSを用いて反応の変化を観測したところ、24時間で1-(2-メルカプトエチルアミノ)-1-オキソ-3-フェニルプロパン-2-イルカルバミン酸 (S)-tert-ブチル(化合物10)が生成していることを確認した。加水分解物と目的化合物10との生成比は、LCMSのUVエリア比で23:71であった。
2-ジメチルアミノエタンチオール塩酸塩(425mg、 3.000mmol)と2-アミノエタンチオール 塩酸塩(11.4mg、 0.100mmol)にpH7.0の0.5M HEPES緩衝液(0.300ml)、DMF(0.200ml)、水(0.200ml)を加え室温で5分間攪拌した。さらに1M 水酸化ナトリウム水溶液(0.300ml)を加えてpH6.9に調整した後に、2-(tert-ブトキシカルボニルアミノ)-3-フェニルプロパン酸 (S)-2-アミノ-2-オキソエチル(化合物11)(3.2mg、 0.01mmol)を加え、40度で24時間攪拌した。
LCMSを用いて反応の変化を観測したところ、24時間で1-(2-メルカプトエチルアミノ)-1-オキソ-3-フェニルプロパン-2-イルカルバミン酸 (S)-tert-ブチル(化合物10)が生成していることを確認した。加水分解物と目的化合物10との生成比は、LCMSのUVエリア比で15:84であった。
LCMS(ESI) m/z = 325 (M+H)+
保持時間:0.71分(分析条件SQDFA05)
既に環化されたペプチド(1回目のアミド環化)が生成後のペプチドを想定したモデルからの反応にて分枝化(直鎖部2の生成)を確認した。以下のスキームに従い、アミド環化(アミド環化部も主鎖環化を用いた)され、かつ主鎖にエステル官能基を有し、アミノ酸側鎖に反応補助基を有するアミノ基を有するモデル化合物の合成を行った。続いて分枝化実験を行い、環状ペプチドからRNAが安定に存在できる温和な反応条件にて水中にて分枝化が観測された。
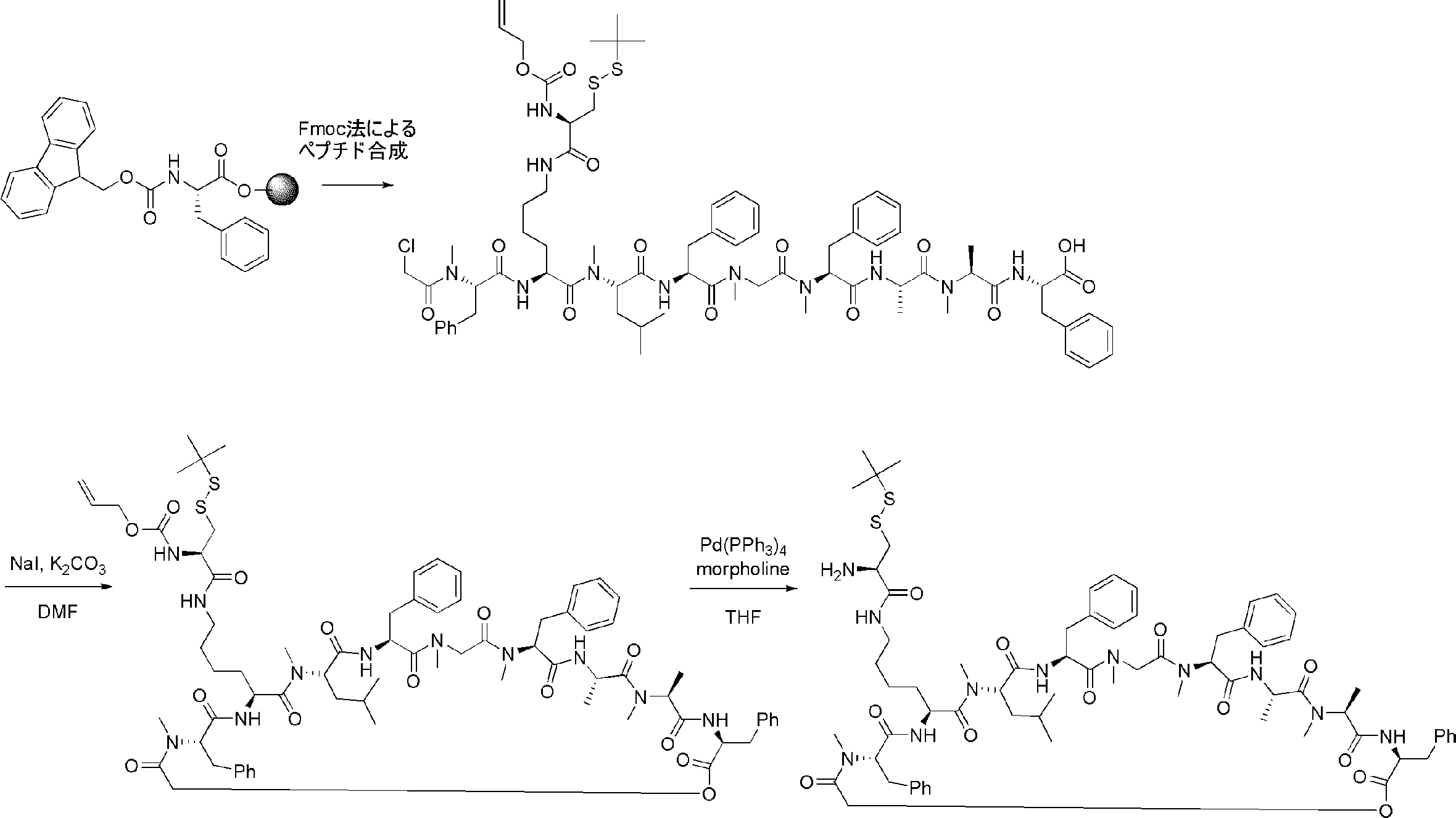
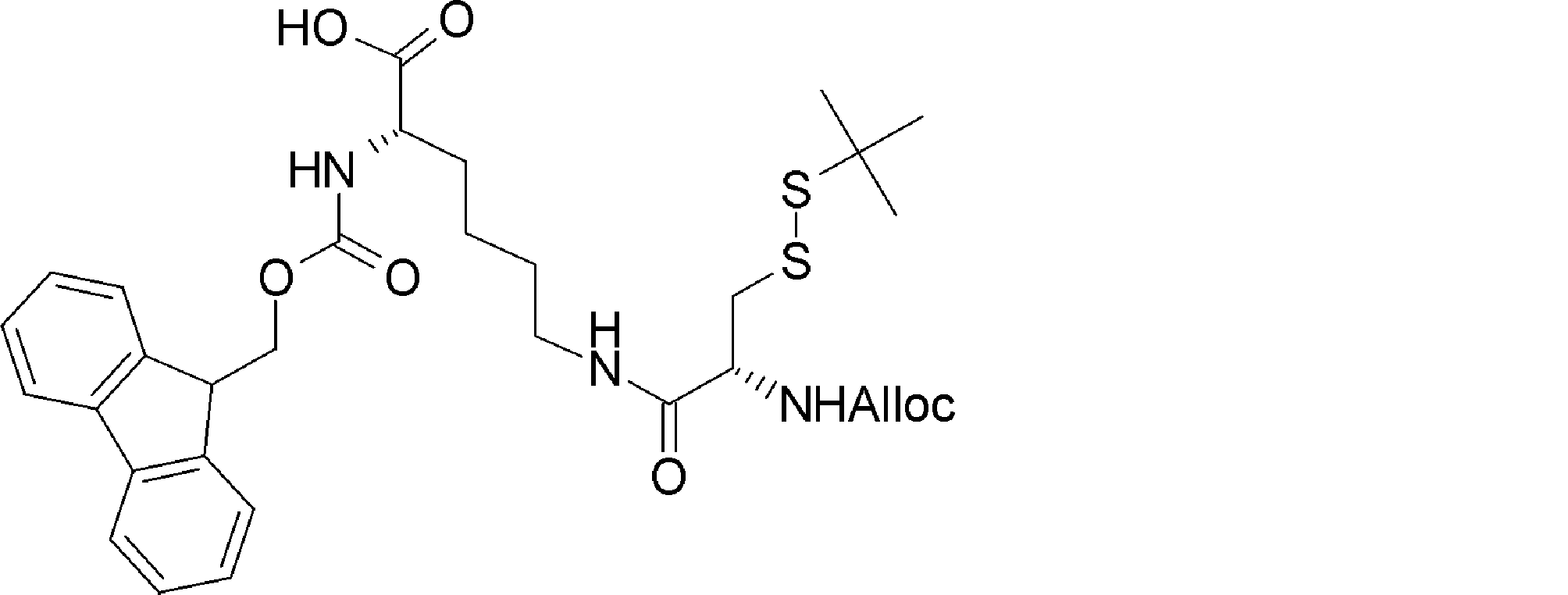
LCMS(ESI) m/z = 644.6 (M+H)+
保持時間:0.91分(分析条件SQD FA05)
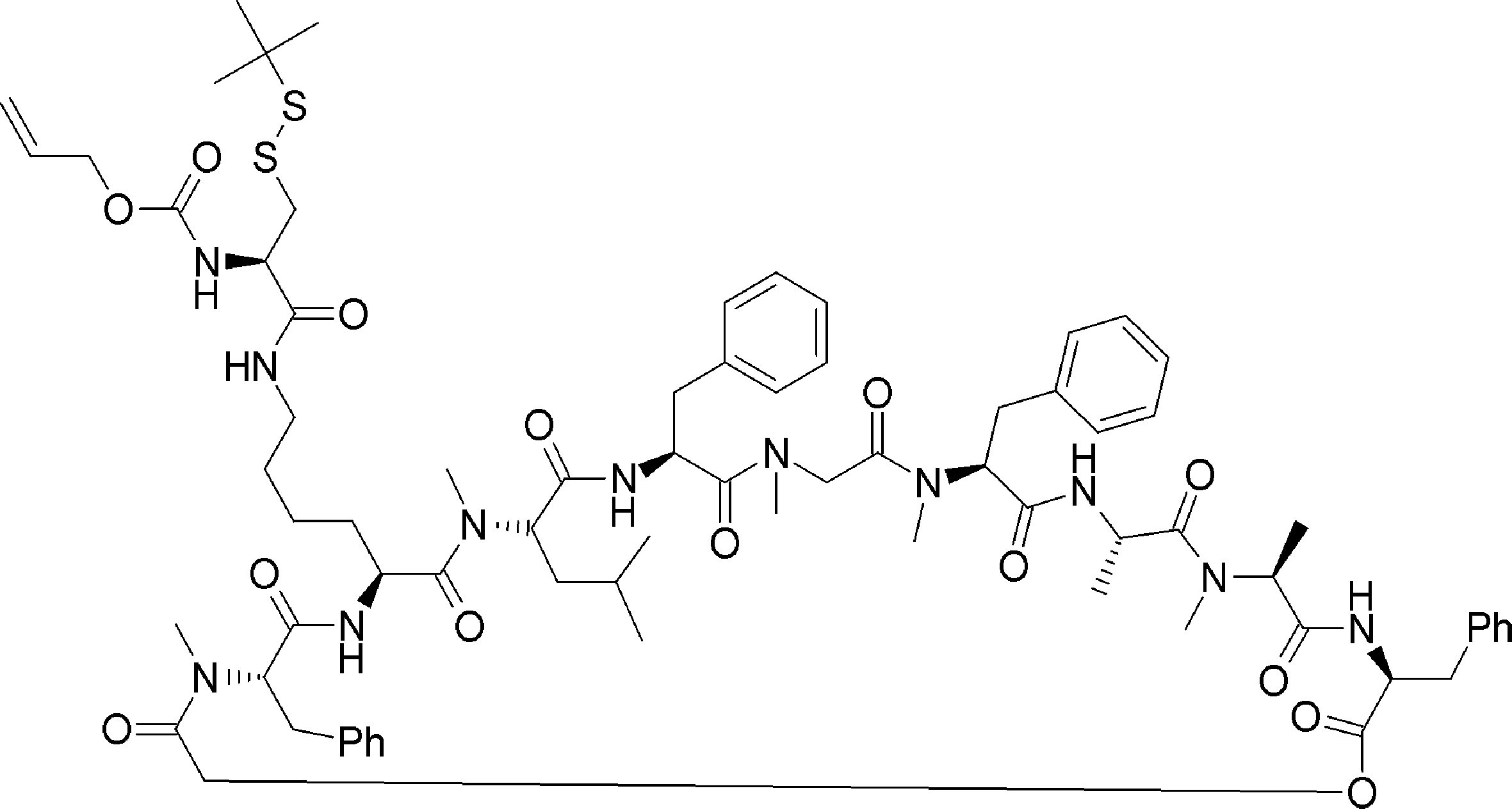
LCMS(ESI) m/z = 1433 (M+H)+
保持時間:0.66分(分析条件SQD FA50)
(R)-2-アミノ-3-(tert-ブチルジスルファニル)-N-(4-((5S,8S,11S,14S,20S,23S,26S,29S)-5,14,20,29-テトラベンジル-11-イソブチル-4,10,16,19,23,25,26-ヘプタメチル-3,6,9,12,15,18,21,24,27,30-デカオキソ-1-オキサ-4,7,10,13,16,19,22,25,28-ノナアザシクロトリアコンタン-8-イル)ブチル)プロパンアミドの合成

LCMS(ESI) m/z = 1349 (M+H)+
保持時間:0.74分(分析条件SQD FA05)
化合物P-151
(S)-2-(2-ヒドロキシ-N-メチルアセトアミド)-3-フェニル-N-((3R,6S,9S,12S,15S,21S,24S,27S)-6,15,21-トリベンジル-24-イソブチル-3-(メルカプトメチル)-9,10,12,16,19,25-ヘキサメチル-2,5,8,11,14,17,20,23,26-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロヘントリアコンタン-27-イル)プロパンアミドの合成
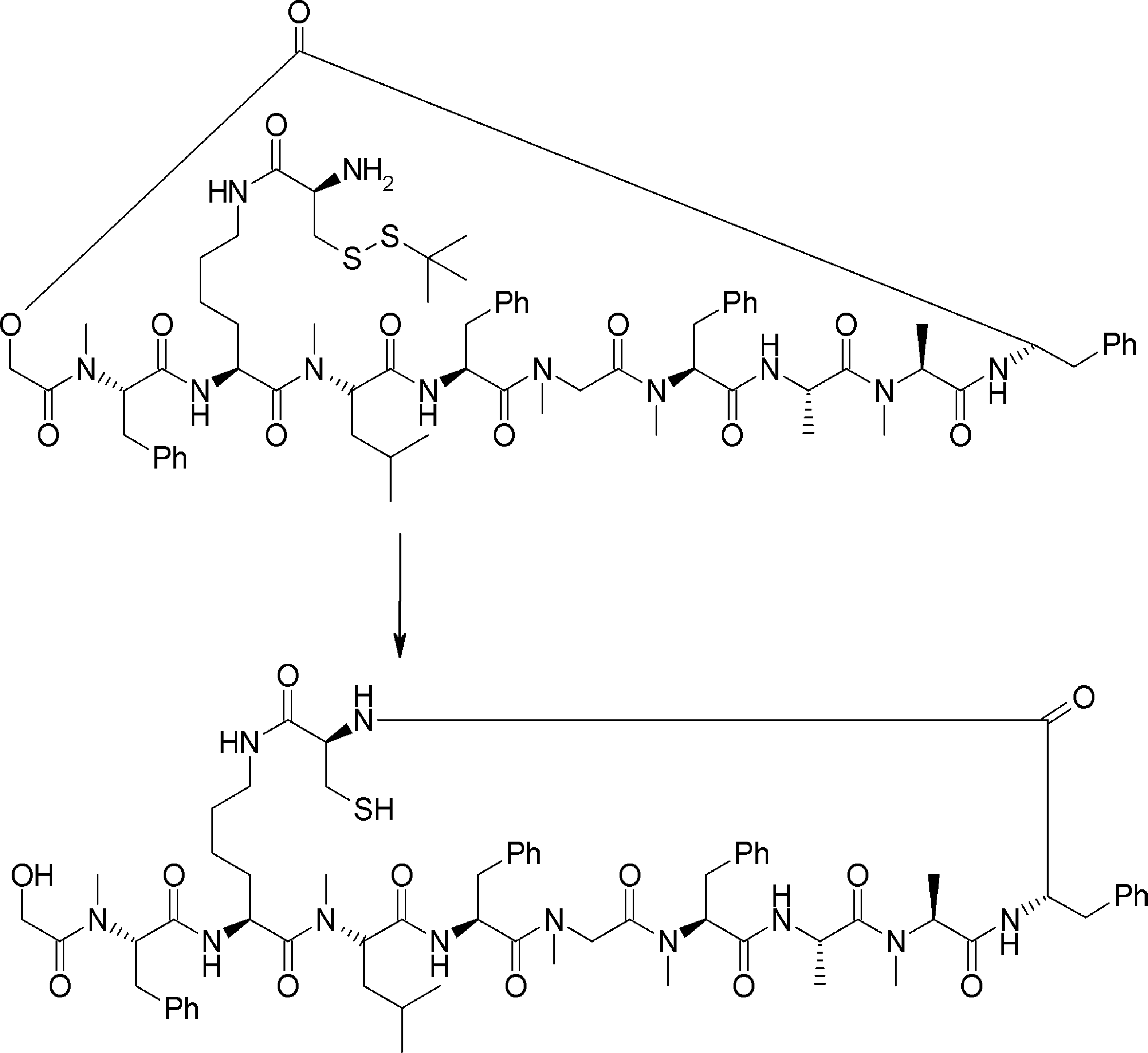
LCMS(ESI) m/z = 1261 (M+H)+
保持時間:0.87分(分析条件SQDFA05)
2-1.自動合成機による主鎖にエステル基を含むペプチドの固相合成一般法
合成機にSieber Amide resin(1カラムあたり160-200mg、Novabiochemより購入)と、各種Fmocアミノ酸(0.6mol/L)と1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)(0.375mol/L)のN-メチル-2-ピロリドン(NMP)溶液と、ジイソプロピルカルボジイミド(DIC)のN,N-ジメチルホルムアミド(DMF)溶液(10%v/v)をセットし、Fmoc脱保護溶液として、ピペリジンのN,N-ジメチルホルムアミド溶液(20%v/v)を用い、Fmoc脱保護反応時間を5分として合成を行った。DMF溶液で洗浄した後、Fmoc脱保護に次いでFmocアミノ酸の縮合反応を1サイクルとし、このサイクルを繰り返すことでレジン表面上にペプチドを伸長させた。このような実験例として相本らの非特許文献(Tetrahedron 2009, 65, 3871-3877)を参考にして合成することもできる。なお、本法は、他ペプチドの合成法としても、本明細書一般に渡って適切なケースに使用できる。
2-2-1.翻訳ペプチドモデル化合物SP605の合成
以下のスキームに従い、N末端にCysを有し、C末端側の1回目の環化ユニットとしてAsp(SBn)を有し、2回目の分枝化ユニットとして、活性チオエステル発生パートとしてCys-Pro-HOGlyを有し、側鎖アミン反応部位としてLysを有するモデルペプチド合成を行った。
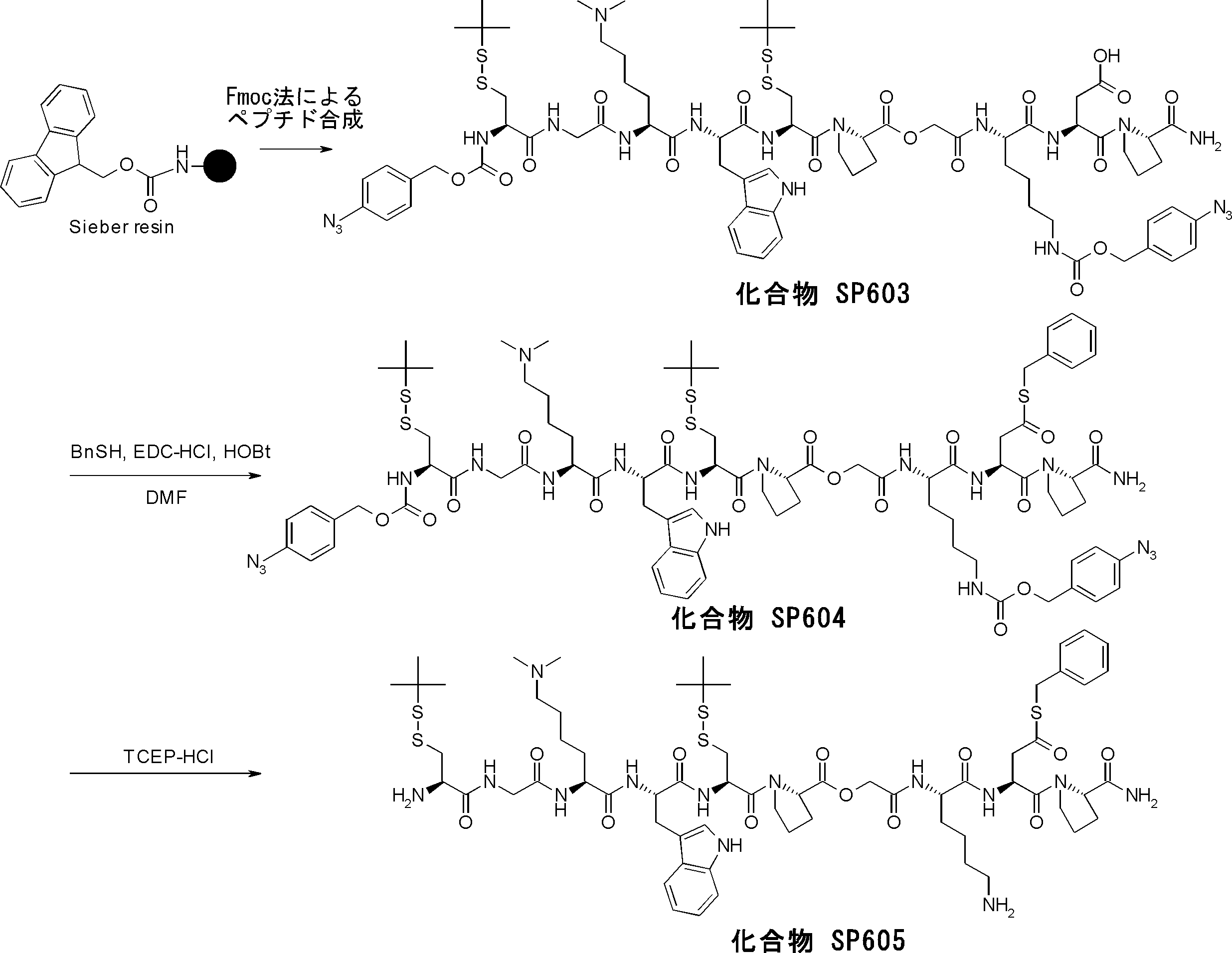

LCMS(ESI) m/z = 618 (M+H)+
保持時間:0.93分(分析条件SQD FA05)
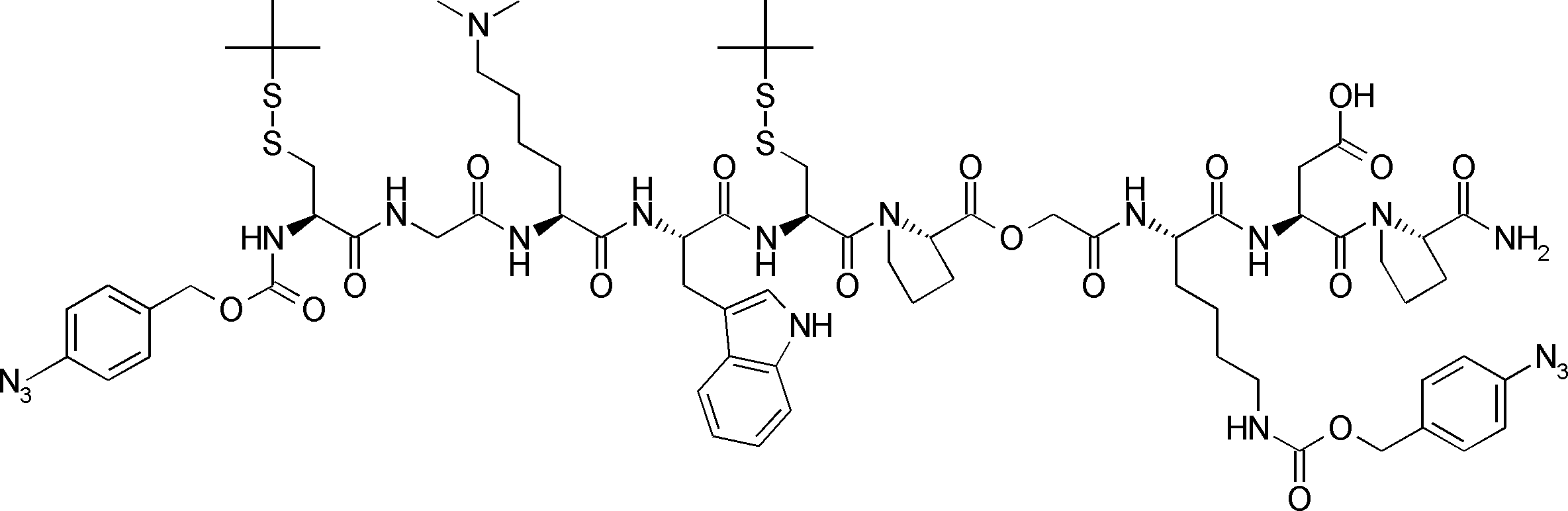
Acbz: 4-アジドベンジルオキシカルボニル基

Fmoc-Lys(Me2)-OH・HCl: N-α-(9-フルオレニルメトキシカルボニル)-N-ε,N-ε-ジメチル-L-リシン 塩酸塩
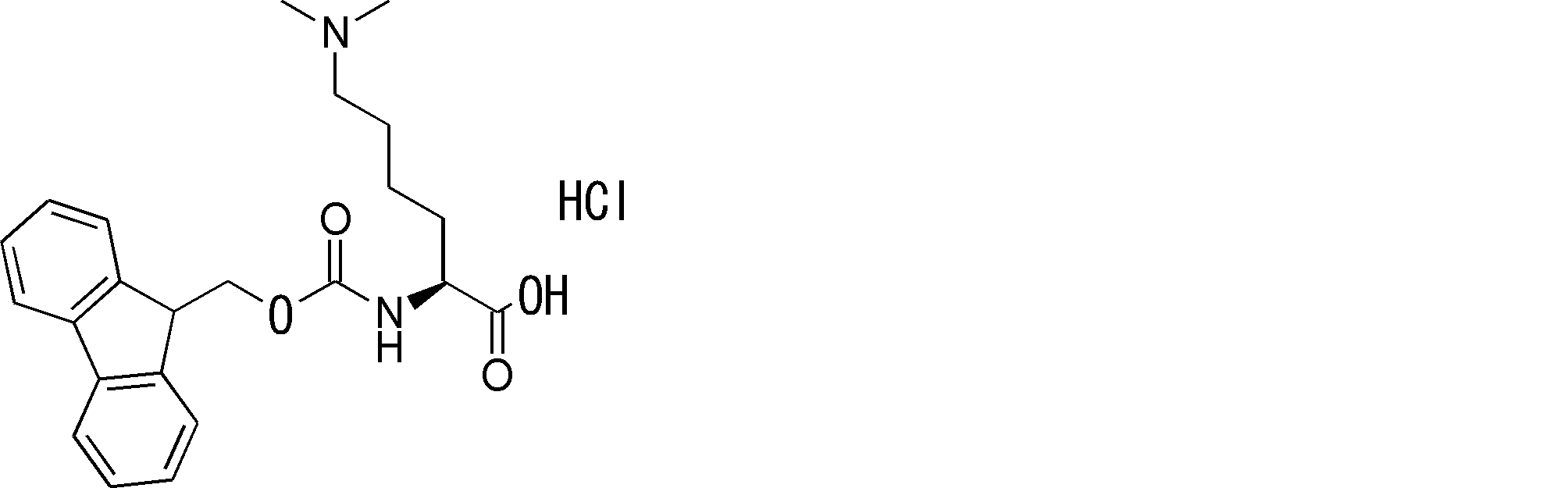
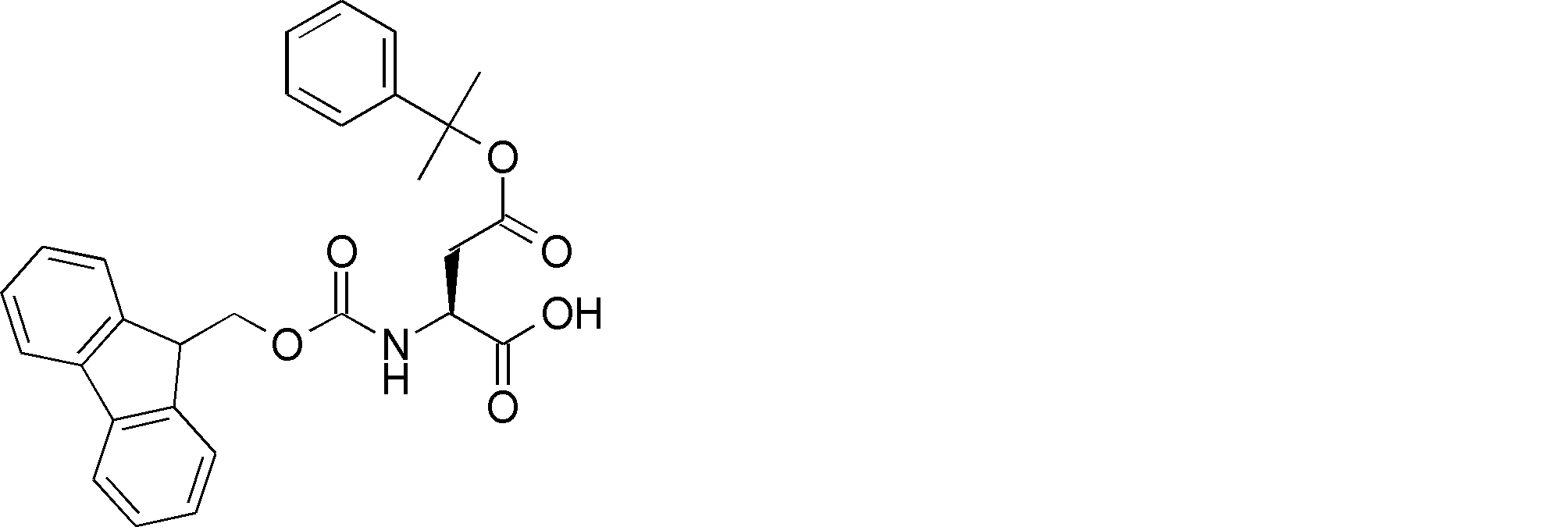
LCMS(ESI) m/z = 1645 (M+H)+
保持時間:0.75分(分析条件SQD FA05)

LCMS(ESI) m/z = 1751 (M+H)+
保持時間:0.78分(分析条件SQD FA05)
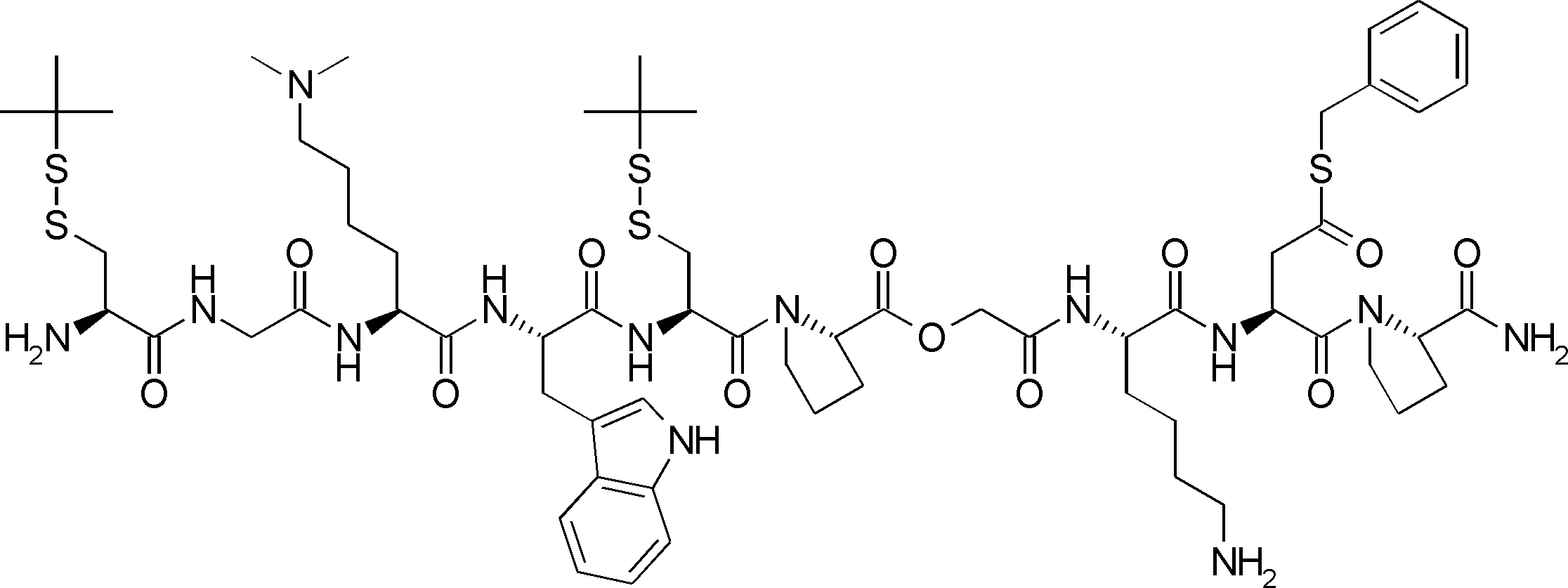
LCMS(ESI) m/z = 1401 (M+H)+
保持時間:0.46分(分析条件SQD FA05)
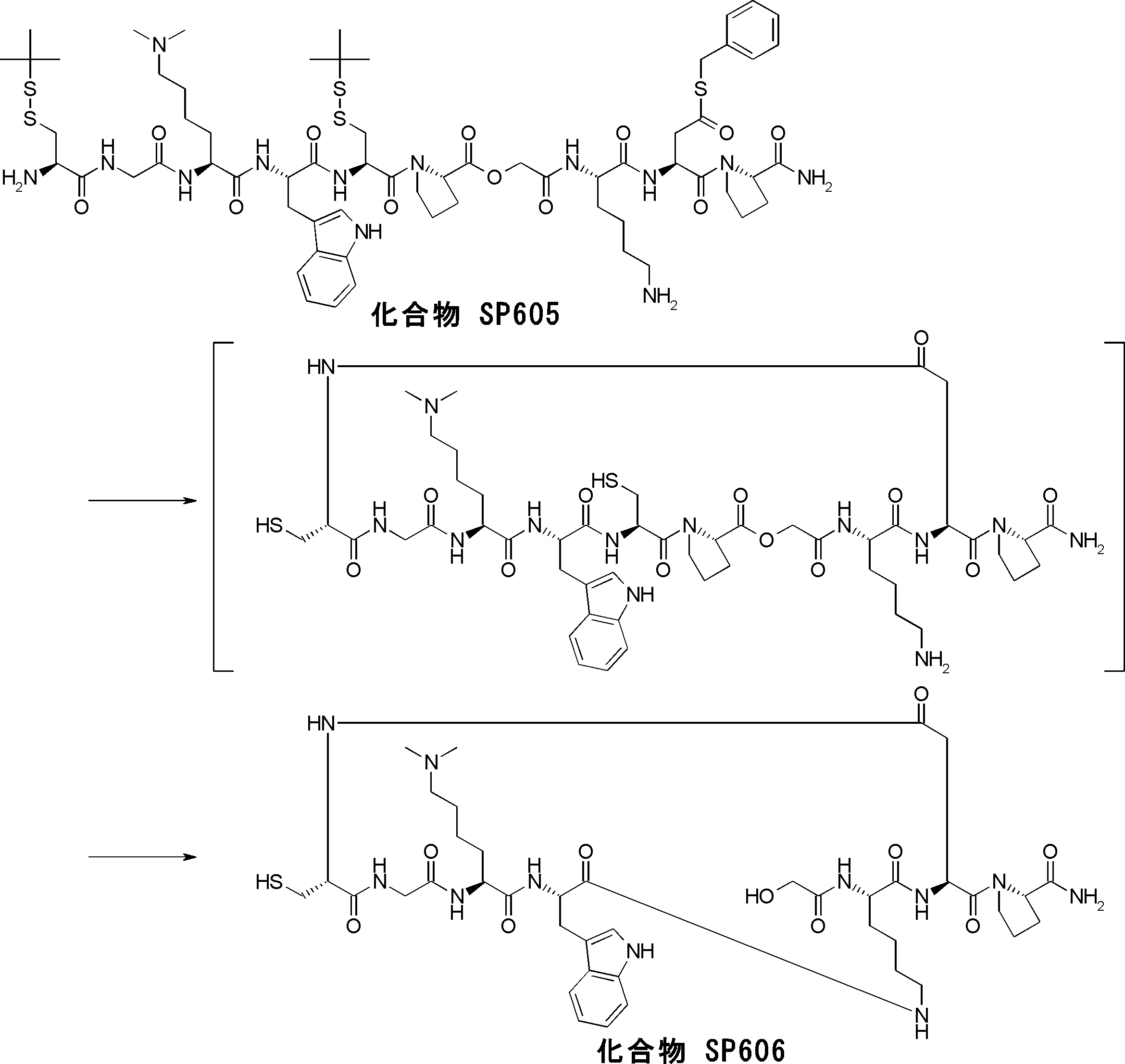
保持時間:0.36分(分析条件SQD FA05)
2-2にて合成されたペプチドが緩衝液中(水中)にてRNAが安定である温和な反応条件にて分枝させる反応条件が得られたため、翻訳合成後でも同様に分子内分枝ペプチドが生成することをMALDI-MSにて確認した。
2つの鋳型DNA(配列番号DT-H1、配列番号DT-H2)から、それぞれRiboMAX Large Scale RNA production System T7(Promega社,P1300)を用いたin vitro の転写により3'端のCAを欠くtRNAGluAAG (-CA)(配列番号RT-H1)、tRNAGluCUG (-CA)(配列番号RT-H2)を合成し、RNeasy Mini kit(Qiagen社)により精製した。
tRNAGluAAG (-CA) DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTAAGACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluCTG (-CA) DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTCTGACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluAAG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUAAGACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluCUG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUCUGACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
50μM 転写tRNAGluAAG (-CA)(配列番号RT-H1) 10μLに、10X ligation buffer(500 mM HEPES-KOH pH 7.5, 200 mM MgCl2) 2μL、10 mM ATP 2μL、Nuclease free water 2.8μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 20unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)1.2μLおよび、5mMのアミノアシル化pdCpA(化合物1i-IA)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。ライゲーション反応液 20μLに、3M酢酸ナトリウム 4μLと125mM ヨウ素(水:THF=1:1溶液)24μLを加え、室温で1時間、脱保護を行った。アミノアシル化tRNA(化合物AT-H1)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-H1)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Asp(SMe)-tRNAGluAAG(配列番号:66)
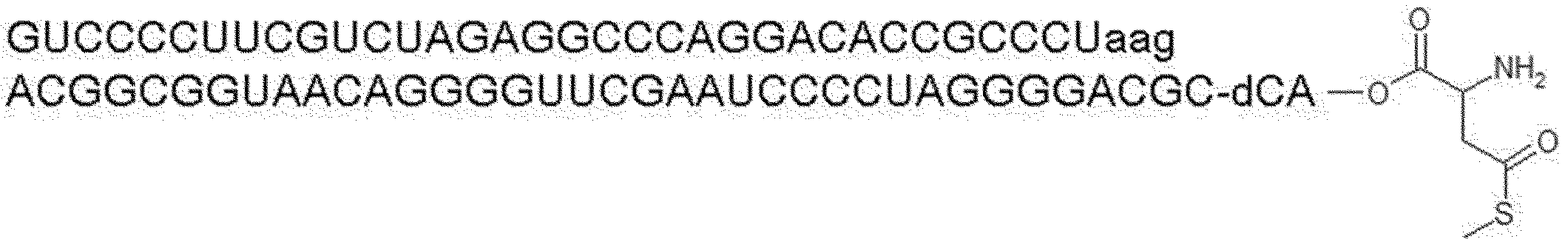
50μM 転写tRNAGluCTG (-CA)(配列番号RT-H2) 10μLに、10X ligation buffer(500 mM HEPES-KOH pH 7.5, 200 mM MgCl2) 2μL、10 mM ATP 2μL、Nuclease free water 2.8μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 20unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)1.2μLおよび、5mMのグリコール酸でアシル化されたpdCpA(化合物20)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。アミノアシル化tRNA(化合物AT-H2)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-H2)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
前述したアシル化されたtRNA(化合物AT-H1、化合物AT-H2)を、無細胞翻訳系に加えて翻訳を開始することにより、所望の非天然アミノ酸を含有するポリペプチドの翻訳合成を行った。翻訳系は、原核生物由来の再構成無細胞タンパク質合成系であるPURE systemを用いた。
OT86b RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUaugUGCACUACAUGGUUCCGUUGGUGCCCACAGUUCAAGUGGCUUCCUCGUAGUUAAGG
CysThrThrTrpPheArgTrpCysProHOGlyPheLysTrp[Asp(SMe)]ProArgSerのN末端アミノ基の窒素原子とAspの側鎖カルボン酸でアミド環化した化合物(HOGlyはグリコール酸を指す)
m/z: [H+M]+ = 2155.6(配列P-H1に対応するペプチド。Calc.2156.0)
前述の翻訳反応物P-H1を含む翻訳溶液5μLと、pH8.5に調整した5μLの環化反応試薬溶液(0.3M HEPES-KOH、0.1M TCEP、0.1M p-メルカプトフェニル酢酸)を混合し、30℃で17時間保温した。その後、得られた反応溶液を、SPE C-TIP(日京テクノス社)で精製し、マトリックスとしてα-シアノ-4-ヒドロキシケイ皮酸を用いてMALDI-MSで分析した。対象実験として、前述した鋳型RNAOT86b(配列番号RM-H1)を含まない翻訳溶液に対しても同様の操作を施し、両者を比較することで、鋳型RNA依存的に合成される翻訳産物由来のピークを判別し、解析を行った。その結果、鋳型RNAOT86b(配列番号RM-H1)を含む翻訳反応液には、目的の分子内分枝骨格をもつ化合物H1に相当するピークが観測され、翻訳ペプチドを用いた分子内分枝ペプチド(直鎖部2)の生成が確認された(図51、ピークI)。
化合物H1

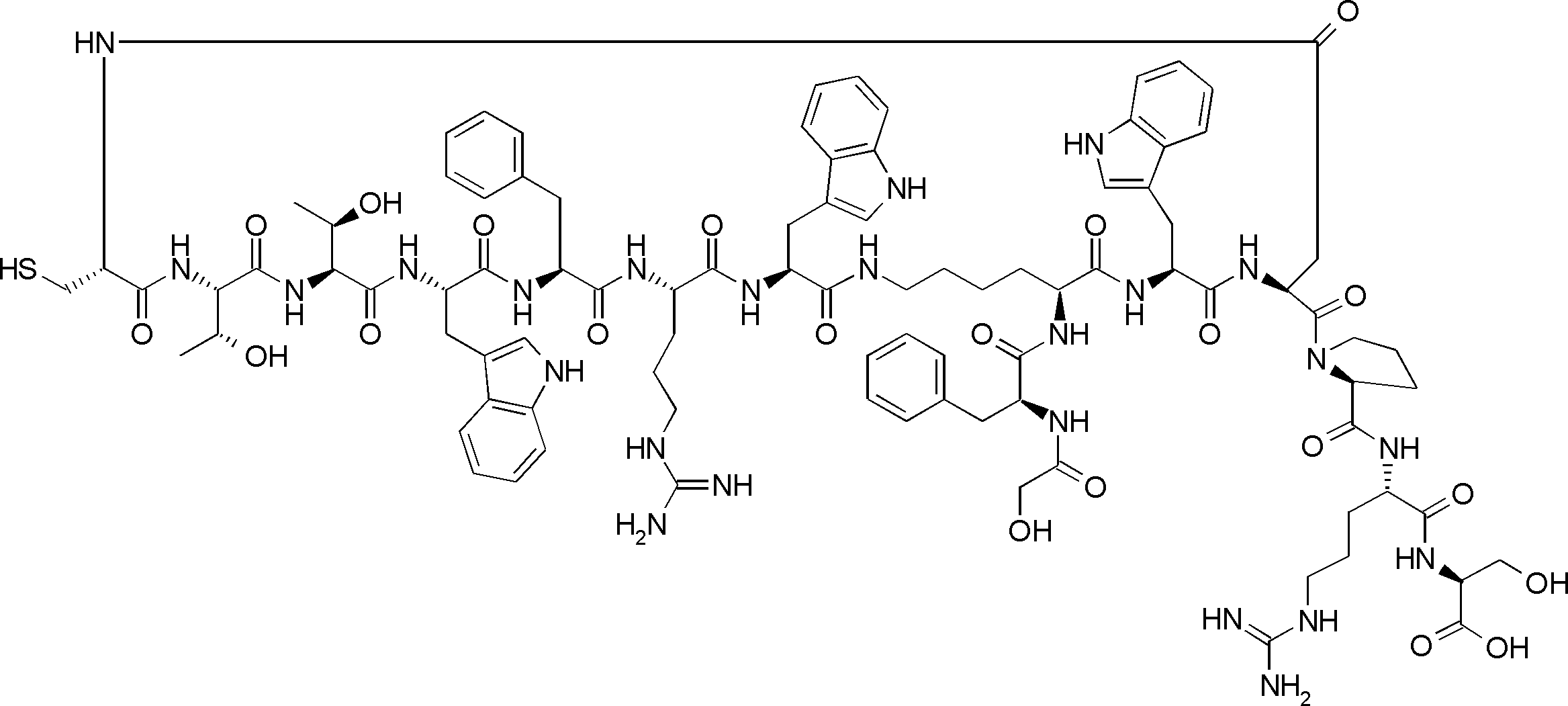
m/z: [H+M]+ = 1955.4(化合物H1に対応するペプチド。Calc.1955.9)
化合物H2

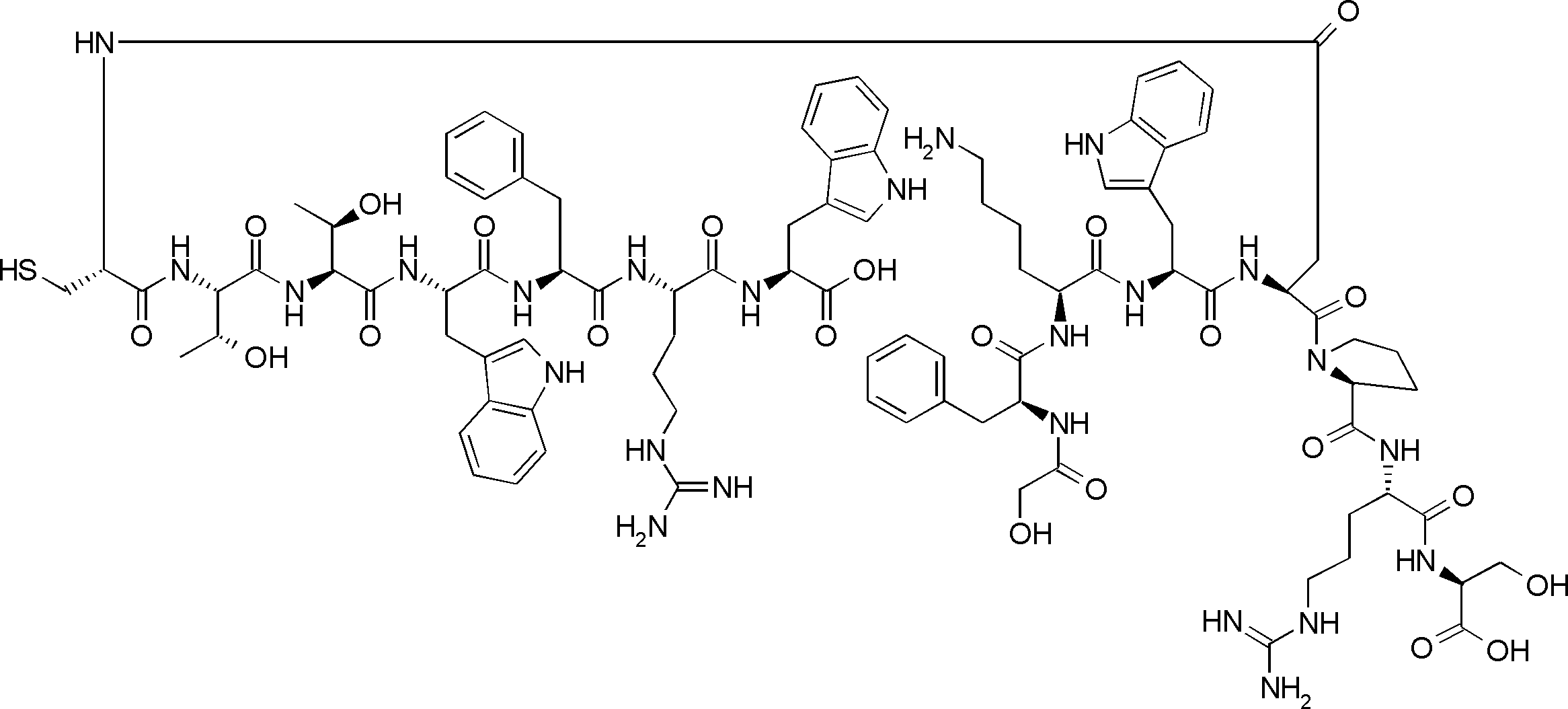
m/z: [H+M]+ = 1973.4(化合物H2に対応するペプチド。Calc.1973.9)
上に示したとおり、合成ペプチド化合物SP605から化合物SP606への変換にて設定した分枝化反応条件を翻訳ペプチドに適用した結果、翻訳ペプチドでも分枝化反応の進行が確認された。以下の実験では、化合物SP605から化合物SP606への合成ペプチドをモデルとした反応最適化である。水中での反応、および翻訳液中での反応を実施し、翻訳ペプチドによる分枝化反応の最適化を行った結果、反応選択性に優れた反応条件の設定に至った。
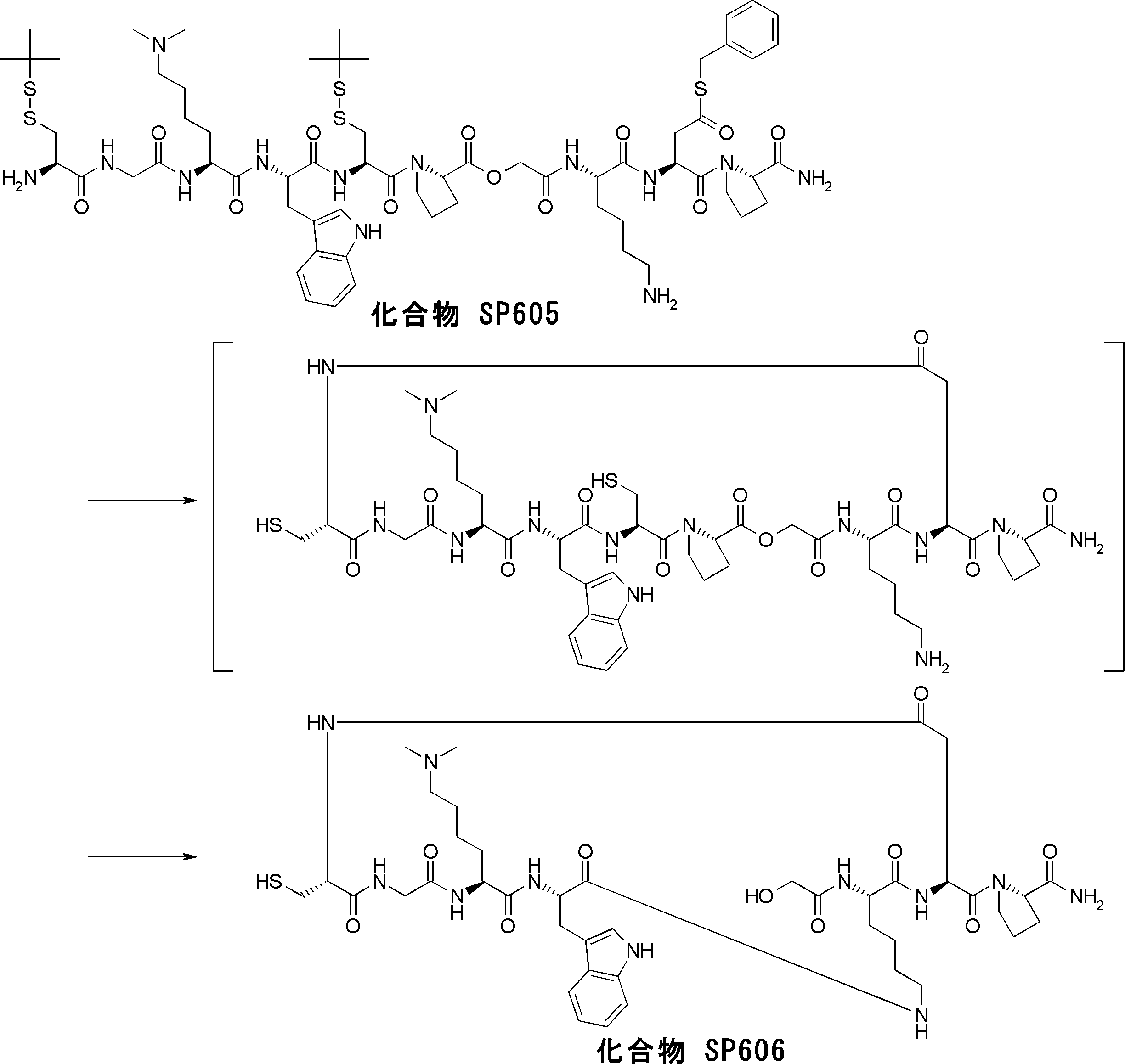
有機溶媒の効果の比較実験
変更点として、1回目の環化反応では有機溶媒(DMA)含有率を5%から50%へと増大させ、2回目の分枝化反応では2.5%から50%へと増大させた結果を示す。
以上の結果から、1回目の環化反応には有機溶媒含有率の向上が有利であることがわかり、加水分解が抑制されて目的化合物を得る選択性が高まった。一方、2回目の分枝化反応では、目的化合物を得るための化学反応速度が低下するため、さらに改良検討を継続した。
検討の結果、以下に示す反応条件が上の条件(2-2-2)よりも優れていることを見出した。変更点として、1回目の環化反応では有機溶媒含有率増大(50%)し、2回目の分枝化反応でも有機溶媒含有率増大(50%)と共に、添加剤のチオールを変更(4-(トリフルオロメチル)ベンゼンチオール)と共に添加剤濃度の増大(500mM)とした。さらに緩衝溶液としてHEPES 150mMから、Bicine(N,N-ビスー(2―ヒドロキシルエチル)グリシン)360mMに変更した。0.5M HEPES緩衝液(pH7.0, 10μl)、水(33μl)、1M水酸化ナトリウム水溶液(5μl)、N-メチル-2-ピロリドン(NMP)(45μl),0.5M TCEP塩酸塩水溶液(2μl)をそれぞれ加えて溶液を調製した。この溶液に、1-((6R,12S,15S,18R)-15-((1H-インドール-3-イル)メチル)-6-アミノ-18-((tert-ブチルジスルファニル)メチル)-12-(4-(ジメチルアミノ)ブチル)-2,2-ジメチル-7,10,13,16-テトラオキソ-3,4-ジチア-8,11,14,17-テトラアザノナデカン)ピロリジン-2-カルボン酸 (S)-2-((S)-6-アミノ-1-((S)-4-(ベンジルチオ)-1-((S)-2-カルバモイルピロリジン-1-イル)-1,4-ジオキソブタン-2-イルアミノ)-1-オキソヘキサン-2-イルアミノ)-2-オキソエチル(化合物SP605、 H-Cys(StBu)-Gly-Lys(Me2)-Trp-Cys(StBu)-Pro-HOGly-Lys-Asp(SBn)-Pro-NH2)(20mM, 0.10μmol)と内部標準として4-ブチル安息香酸(20mM, 0.10μmol)のNMP溶液(5μl)を室温下で添加し、反応液を37℃で30分間静置した。その後、4-(トリフルオロメチル)ベンゼンチオール(6.8μl, 50μmol)にNMP(40μl),2M Bicine(N,N-ビス-(2-ヒドロキシエチル)グリシン) 緩衝液(pH8.7, 18μl)、0.5M TCEP塩酸塩水溶液(10μl)をそれぞれ加え5M 水酸化ナトリウム水溶液(12μl)により溶液を調整した後に、反応液(20μl)を室温下で添加し(反応液を加えた後の混合溶液のpH 8.2)、反応液を30℃で24時間静置し、LCMSを用いて反応の変化を観測した。24時間で(S)-1-((3S,6S,12R,16S,19S)-3-((1H-インドール-3-イル)メチル)-6-(4-(ジメチルアミノ)ブチル)-19-(2-ヒドロキシアセトアミド)-12-(メルカプトメチル)-2,5,8,11,14,18-ヘキサオキソ-1,4,7,10,13,17-ヘキサアザシクロトリコサン-16-カルボニル)ピロリジン-2-カルボキサミド(化合物SP606)が生成していることを確認した。化合物SP606と加水分解物(副生成物)との生成比は、LCMSのUVエリア比で98:2であった。 (図52、加水分解化合物 保持時間:0.31分)
翻訳用緩衝液(6.25μl)、水(1.25μl)、PURESYSTEM(r) classic II Sol. B(バイオコゥマー社、製品番号PURE2048C)(10μl)、20種類の天然型アミノ酸溶液(それぞれ5mM,2.5μl)を混合しジメチルアセトアミド(DMA)(22.5μl), TCEP水溶液(100mM、5μl)、をそれぞれ加えて溶液を調製した。翻訳用緩衝液の成分は8mM GTP,8mM ATP,160mMクレアチンリン酸,400mM HEPES-KOH pH7.6,800mM 酢酸カリウム,48mM 酢酸マグネシウム,16mMスペルミジン,8mM ジチオスレイトール,0.8mM 10-HCO-H4folate, 12mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社)である。この溶液に、1-((6R,12S,15S,18R)-15-((1H-インドール-3-イル)メチル)-6-アミノ-18-((tert-ブチルジスルファニル)メチル)-12-(4-(ジメチルアミノ)ブチル)-2,2-ジメチル-7,10,13,16-テトラオキソ-3,4-ジチア-8,11,14,17-テトラアザノナデカン)ピロリジン-2-カルボン酸 (S)-2-((S)-6-アミノ-1-((S)-4-(ベンジルチオ)-1-((S)-2-カルバモイルピロリジン-1-イル)-1,4-ジオキソブタン-2-イルアミノ)-1-オキソヘキサン-2-イルアミノ)-2-オキソエチル(化合物SP605、H-Cys(StBu)-Gly-Lys(Me2)-Trp-Cys(StBu)-Pro-HOGly-Lys-Asp(SBn)-Pro-NH2)(20mM, 0.05μmol)と内部標準として4-ブチル安息香酸(20mM, 0.05μmol)のDMA溶液(2.5μl)を室温下で添加し、反応液を37℃で30分間静置した。4-(トリフルオロメチル)ベンゼンチオール(6.8μl, 50μmol)にDMA(40μl)、2M Bicine(N,N-ビス-(2-ヒドロキシエチル)グリシン) 緩衝液(pH8.7, 18μl)、0.5M TCEP塩酸塩水溶液(10μl)をそれぞれ加え5M 水酸化ナトリウム水溶液(12μl)により溶液を調整した後に、反応液(20μl)を室温下で添加し(反応液を加えた後の混合溶液のpH 8.2)、反応液を30℃で24時間静置し、LCMSを用いて反応の変化を観測した。24時間で(S)-1-((3S,6S,12R,16S,19S)-3-((1H-インドール-3-イル)メチル)-6-(4-(ジメチルアミノ)ブチル)-19-(2-ヒドロキシアセトアミド)-12-(メルカプトメチル)-2,5,8,11,14,18-ヘキサオキソ-1,4,7,10,13,17-ヘキサアザシクロトリコサン-16-カルボニル)ピロリジン-2-カルボキサミド(化合物SP606)が生成していることを確認した。化合物SP606と加水分解物(副生成物)との生成比は、LCMSのUVエリア比で91:9であった。(図53、加水分解化合物 保持時間:0.30分)
翻訳ペプチドでディスプレイライブラリーを実施する際には、翻訳液に直接試薬を追加した翻訳後修飾を実施することもできる一方、ペプチドーRNA複合体を一度精製した後に、一部あるいは全ての翻訳後修飾を実施することもできる。そのような精製過程の1つとして、RNeasy(r) MinEluteTM Cleanup Kit(キアゲン社)を用いた精製が挙げられる。そのような精製を実施した後の翻訳後修飾の例として以下の実験を実施した。
LCMS(ESI) m/z = 900 (M+H)+
保持時間:0.36分(分析条件SQDFA05)
三角ユニットに反応補助基を有するアミノ基が配置された場合、1回目の環化反応は高選択に進行して、2回目の分枝化のためのCys-Pro-HOGlyの活性化との選択性が獲得できた。特に水と混じり合う有機溶媒含有率が高いと反応選択性に優れる。2回目の分枝化では、添加剤としてチオール存在下(好ましくはアリールチオール)にて、水と混じるあう有機溶媒存在下が好ましく、反応溶液のpHが7.0から9.0の間が好ましく、反応温度は0℃から100℃(より好ましくは15℃から50℃)が好ましい。これまで示した結果から、本条件は緩衝液(水中)のみならず、翻訳液中(PureSystem)でも同様の結果を与える。
(S)-1-((3S,6S,12S,16S,19S)-3-((1H-インドール-3-イル)メチル)-6-(4-(ジメチルアミノ)ブチル)-19-(2-ヒドロキシアセトアミド)-12-メチル-2,5,8,11,14,18-ヘキサオキソ-1,4,7,10,13,17-ヘキサアザシクロトリコサン-16-カルボニル)ピロリジン-2-カルボキサミド(化合物SP607)の合成
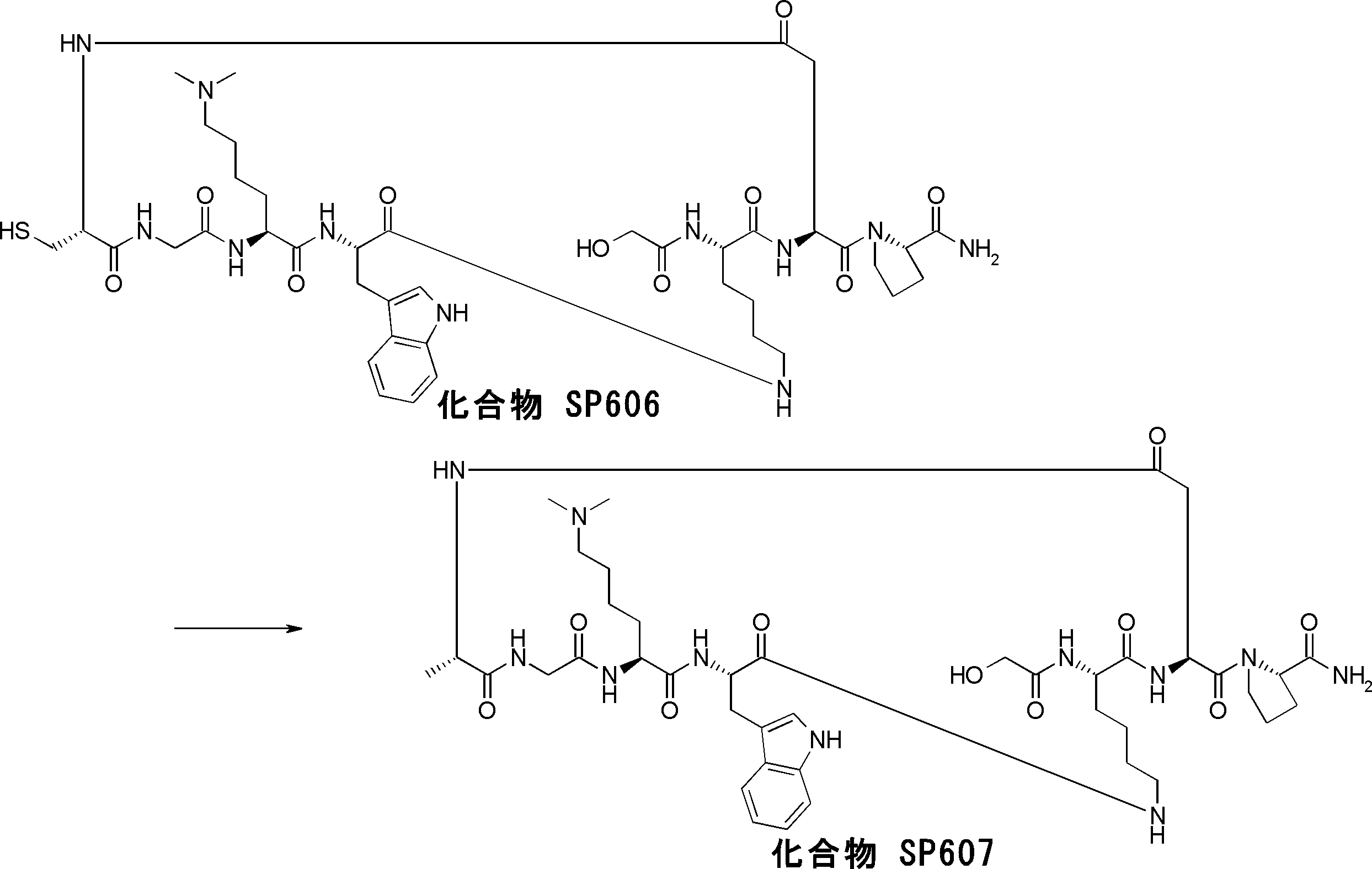
上述の3-2.に記載の翻訳ペプチドモデル化合物(化合物SP605)からの分子内分枝ペプチド生成反応に用いた反応溶液(10μl)に0.5M トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩水溶液(10μl)を加えた。混合溶液をヘキサン(200μl)にて洗浄した後に(ヘキサンによる洗浄を7回実施)、得られた水層に、500mM グルタチオン水溶液(4μl)、1M 2、2-アゾビス[2-(2-イミダゾリンー2-イル)プロパン]二塩酸塩(VA-044)水溶液(2μl)、5M 水酸化ナトリウム水溶液(2μl)を室温にて加え、40℃で3時間静置した。反応の変化をLCMSで追跡し、3時間後に原料である化合物SP606が完全に消費され、目的物である(S)-1-((3S,6S,12S,16S,19S)-3-((1H-インドール-3-イル)メチル)-6-(4-(ジメチルアミノ)ブチル)-19-(2-ヒドロキシアセトアミド)-12-メチル-2,5,8,11,14,18-ヘキサオキソ-1,4,7,10,13,17-ヘキサアザシクロトリコサン-16-カルボニル)ピロリジン-2-カルボキサミド(化合物SP607)に変換されていることを確認した。(図55、56)
LCMS(ESI) m/z = 868(M+H)+
保持時間:0.35分(分析条件SQD FA05)
LCMS(ESI) m/z = 868(M+H)+
保持時間:0.35分(分析条件SQD FA05)
3にて記載と同様の考え方で、有効性を確認した。
4-1.翻訳ペプチドモデル化合物(化合物SP616)の合成
以下のスキームに従い、1回目の環化としてN末端にCysを用い、C末端側にAsp(SBn)を用いてアミド化環化反応を行い、2回目の分枝化として、活性エステルをグリコール酸のエステルの活性化にて実施し、Lysの側鎖アミノ基にN原子とS原子を保護させたCysを配置させた化合物のS原子とN原子を脱保護してアミド化反応を行う例を実施する目的で、以下のモデル化合物SP616の合成を行った。
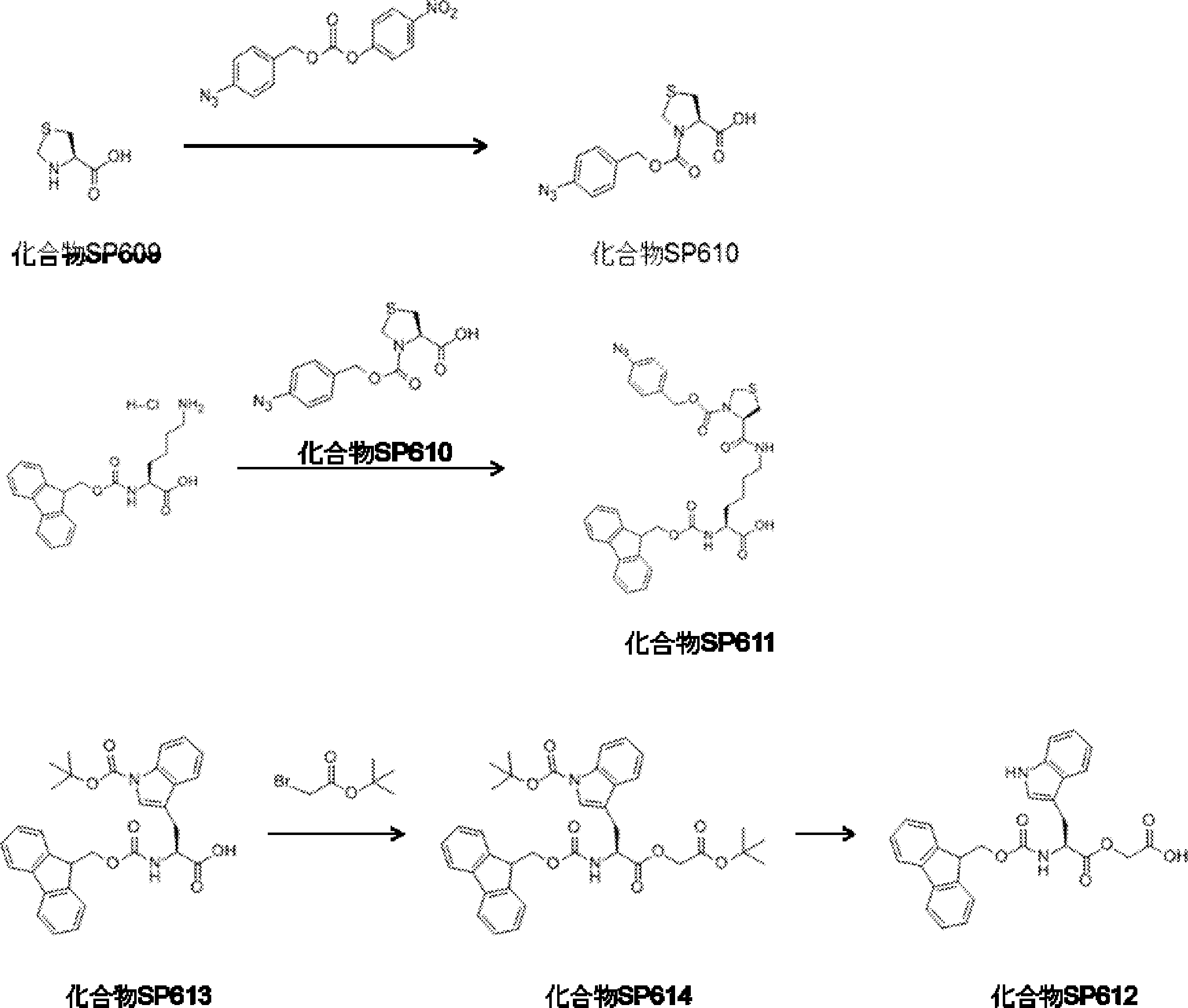
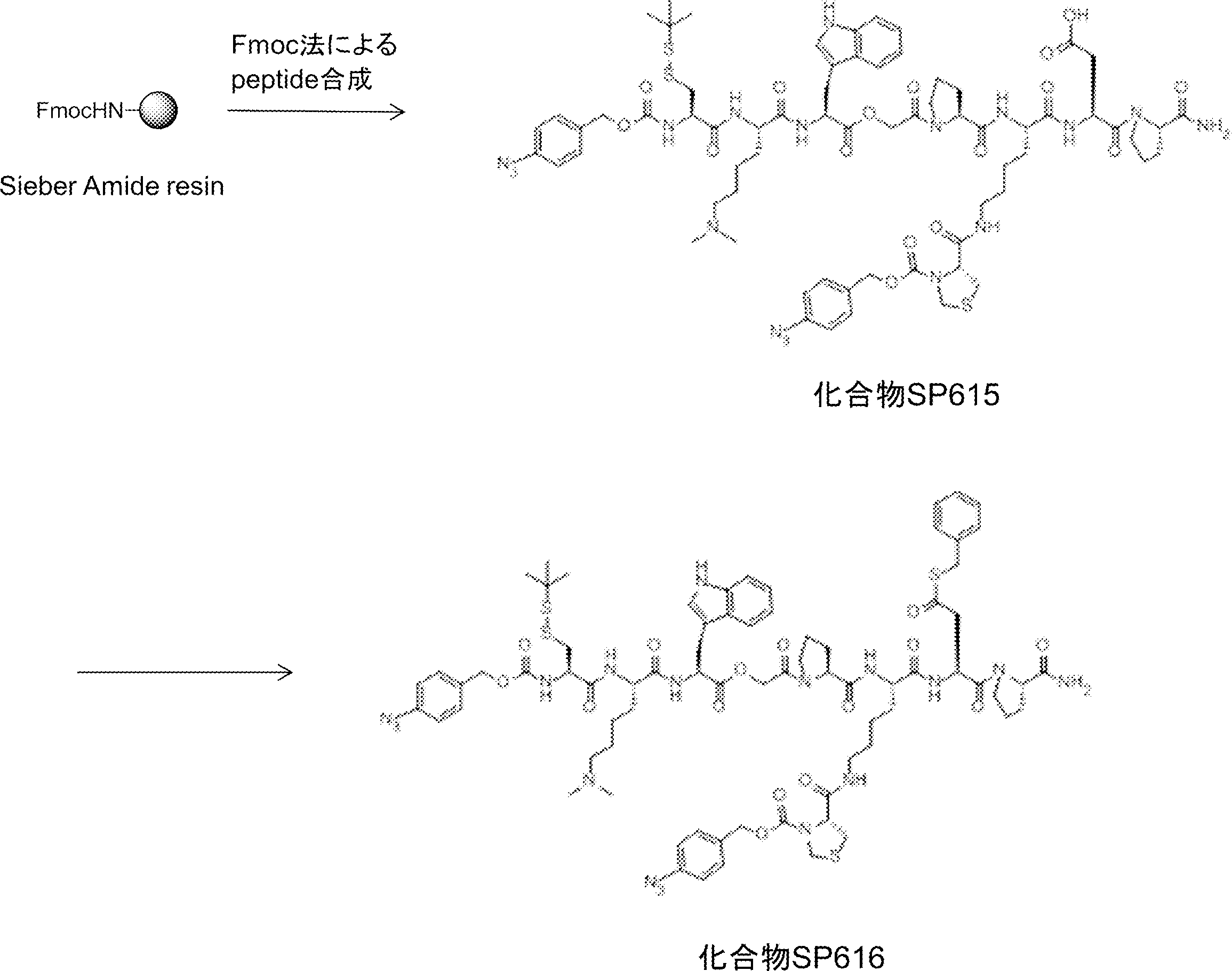

LCMS(ESI) m/z = 307 (M―H)―
保持時間:0.68分(分析条件SQDFA05)
なお、本明細書中では、他の例と同様Fmoc-Lys-OHの側鎖アミノ基とAcbz-Thz-OHのカルボキシル基がアミド結合した化合物をFmoc-Lys(Acbz-Thz)-OHと定義する。

LCMS(ESI) m/z = 659 (M+H)+
保持時間:0.87分(分析条件SQDFA05)

LCMS(ESI) m/z = 485 (M+H)+
保持時間:0.83分(分析条件SQDFA05)
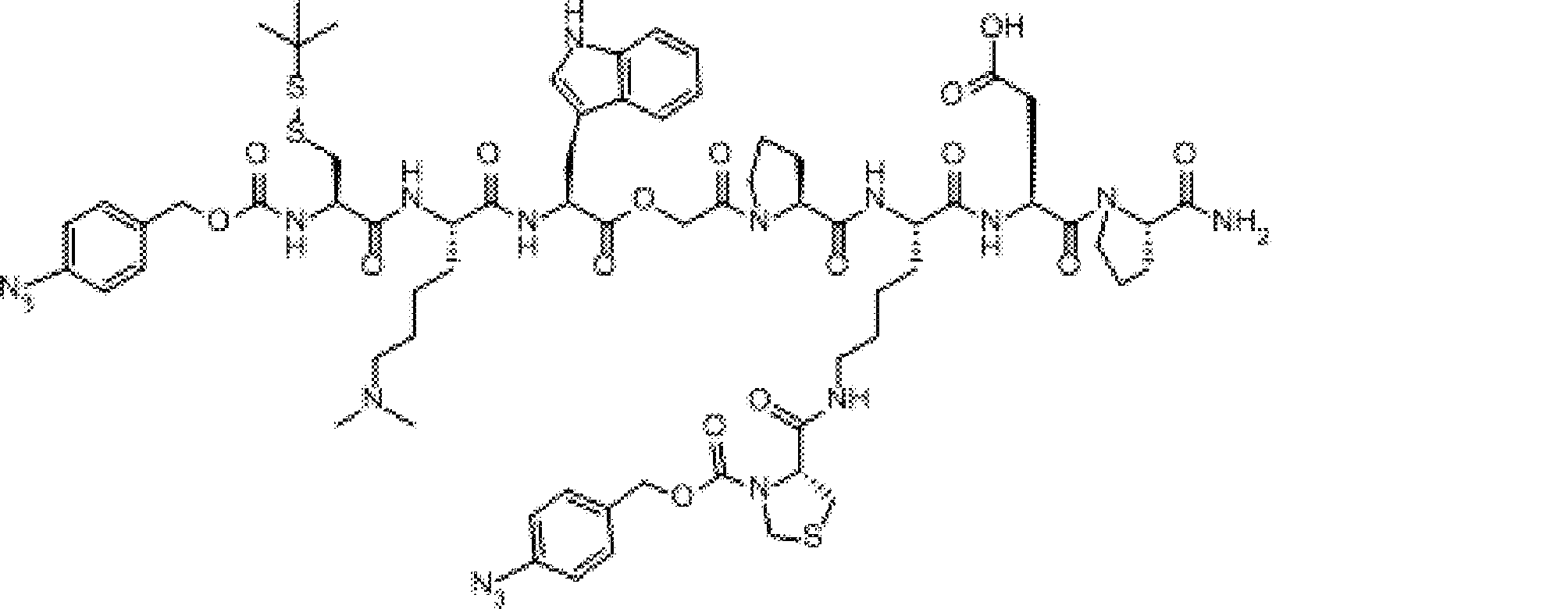
LCMS(ESI) m/z = 1511.4 (M+H)+
保持時間:0.69分(分析条件SQDFA05)
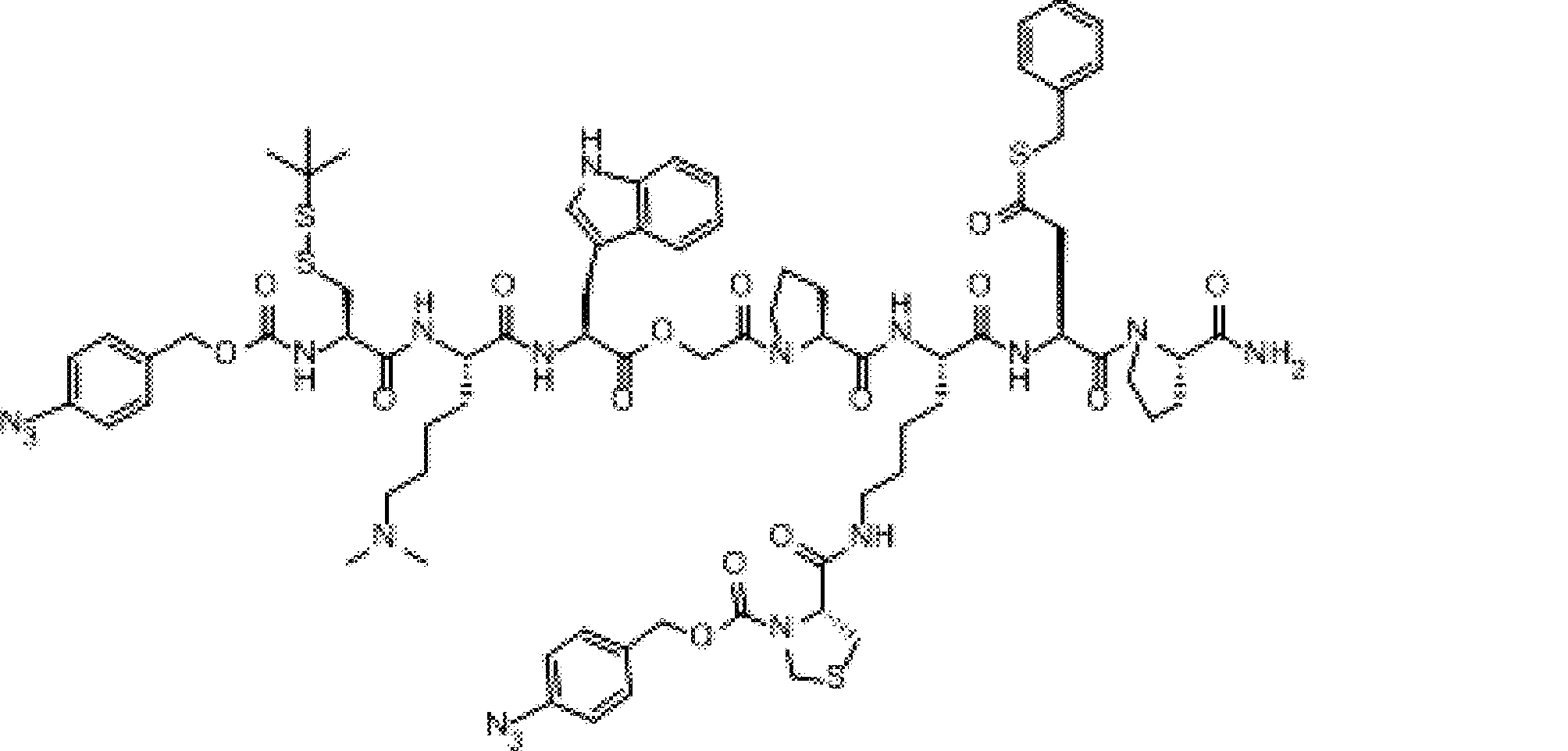
LCMS(ESI) m/z = 1617.4 (M+H)+
保持時間:0.74分(分析条件SQDFA05)
なお、本明細書中では他の例と同様、Lysの側鎖アミノ基とH-Cys-Lys(Me2)-Trp-Cys-OHのC末端のカルボキシル基がアミド結合した化合物を、H-Lys(H-Cys-Lys(Me2)-Trp-Cys)-OHと記載する。
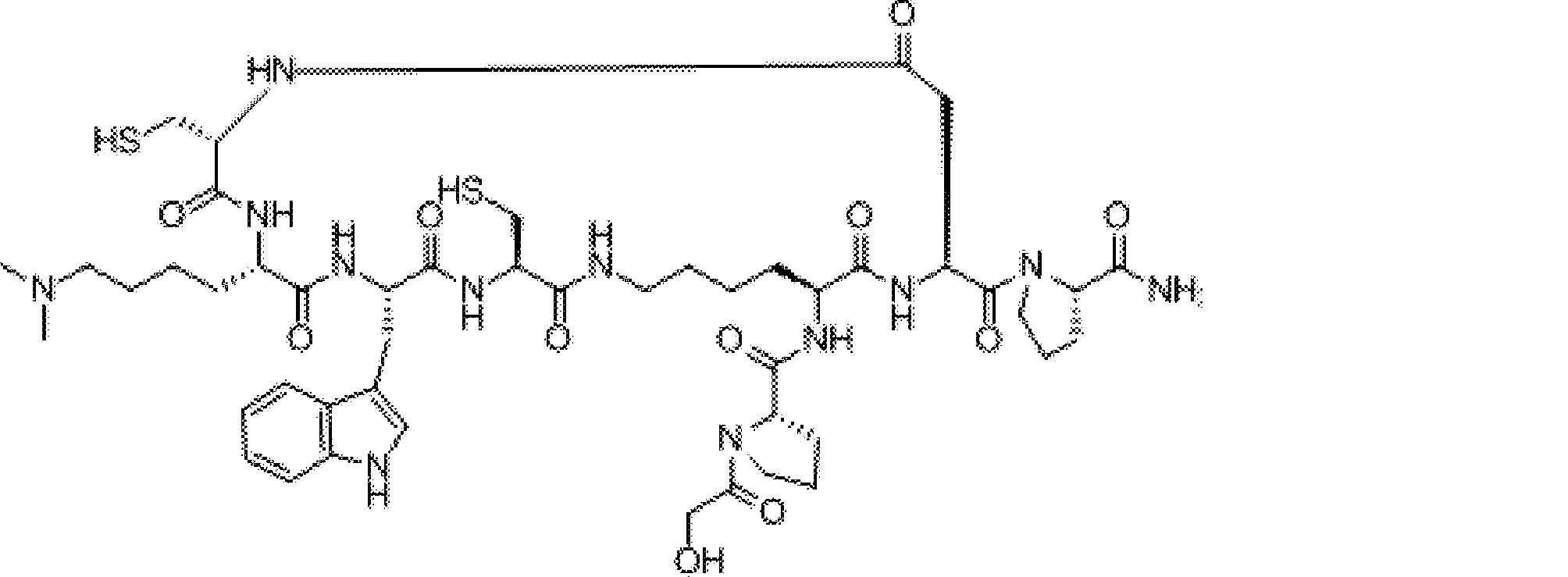
以下のスキームに従い反応を行った。すなわち、化合物SP616からステップ1では2つのアミノ基の保護基を脱保護した後(この時点で反応を構成するための各ユニットは翻訳ペプチドと同様の構造となる。C末端がカルボン酸ではなく、カルボン酸アミドとなっている他は翻訳ペプチドと同様)、そのまま1回目の環化反応を実施したところ、選択的に目的化合物SP618を得ることができた。続いて、化合物618から分枝化反応で用いるためのアミン側のユニットの脱保護をステップ2およびステップ3にて実施し、反応補助基を有するアミノ基の脱保護が達成された化合物SP620が得られた。RNAに安定な化学反応条件にて、アミンの保護基を脱保護することができた。エステル官能基から直接チオエステルを発生させる手法にて分枝化ペプチド化合物SP617が得られた。
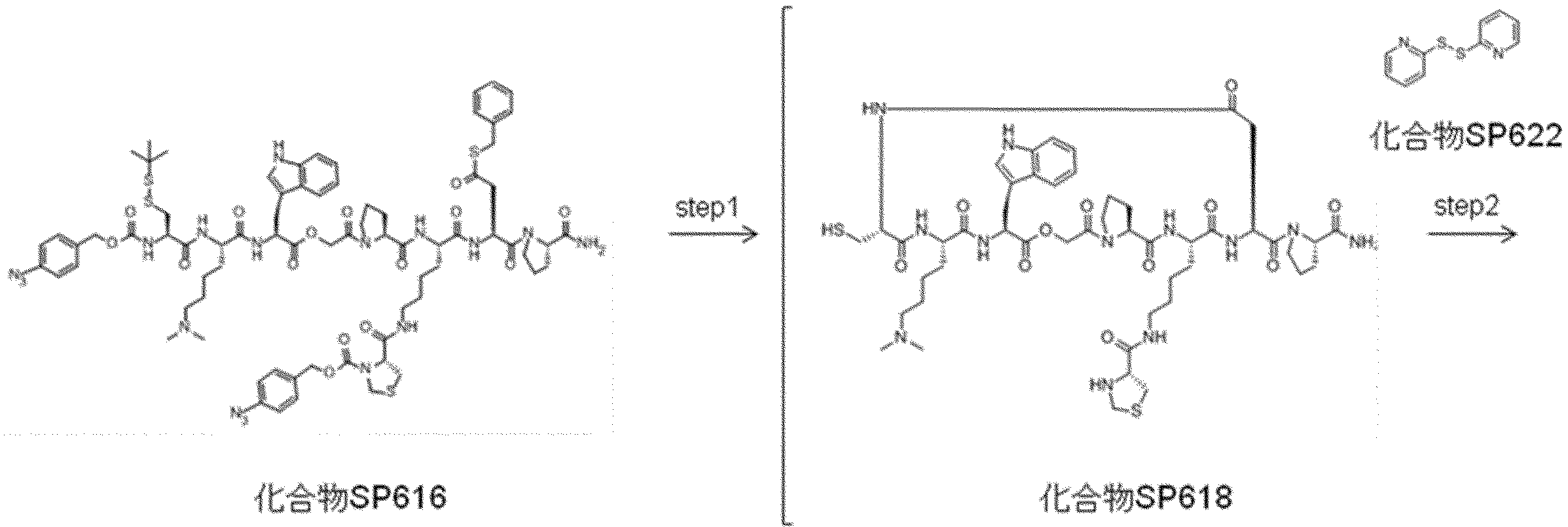
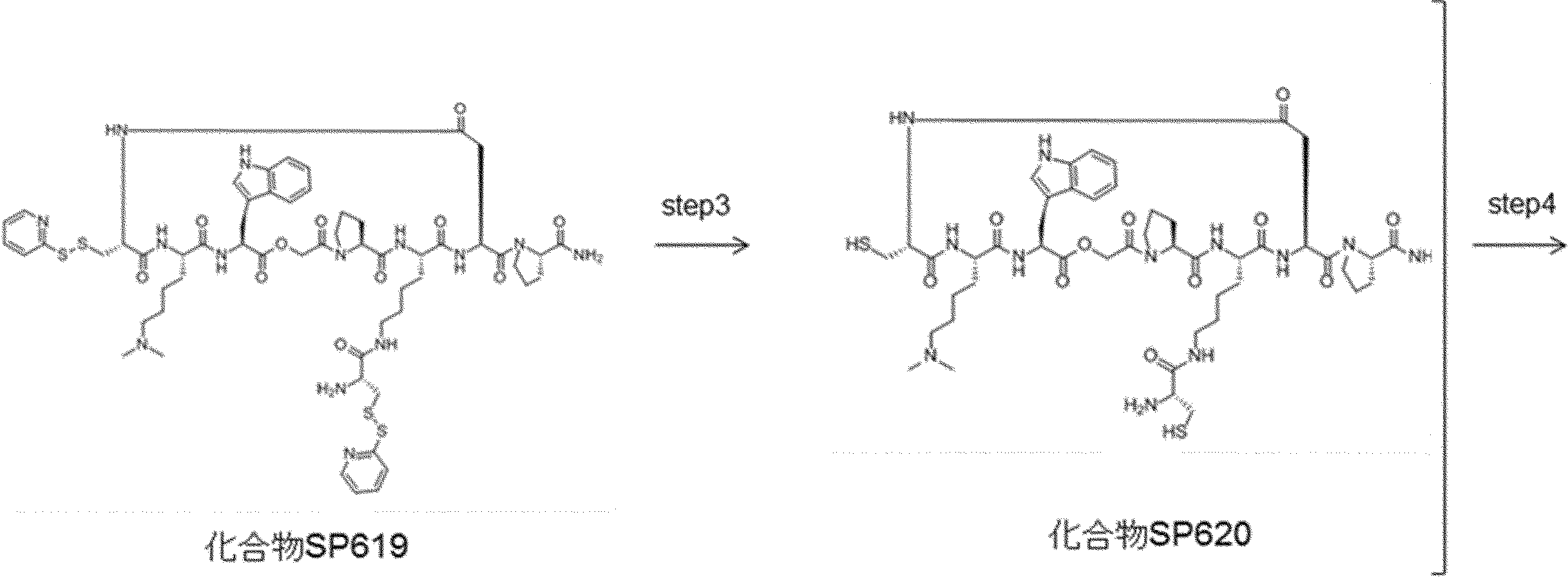

窒素雰囲気下、50mM HEPES緩衝液(70μl、pH=7.5)に4mM 4-(((S)-5-((S)-1-((6R,9S,12S)-12-((1H-インドール-3-イル)メチル)-6-((((4-アジドベンジル)オキシ)カルボニル)アミノ)-9-(4-(ジメチルアミノ)ブチル)-2,2-ジメチル-7,10,13-トリオキソ-14-オキサ-3,4-ジチア-8,11-ジアザヘキサデカン-16-オイル)ピロリジン-2-カルボキサミド)-6-(((S)-4-(ベンジルチオ)-1-((S)-2-カルバモイルピロリジン-1-イル)-1,4-ジオキソブタン-2-イル)アミノ)-6-オキソヘキシル)カルバモイル)チアゾリジン-3-カルボン酸(R)-4-アジドベンジル(化合物SP616、Acbz-Cys(StBu)-Lys(Me2)-Trp-HOGly-Pro-Lys(Acbz-Thz)-Asp(SBn)-Pro-NH2)のジメチルアセトアミド(DMA)溶液(25μl、0.1μmol)、内部標準として用いた20mM 2、4-ジメチル安息香酸のジメチルアセトアミド(DMA)溶液(5.0μl、0.1μmol)、別途調整した200mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液(10μl、2.0μmol、pH=7.4)を室温にて加え、pH=7.4にて、同温度で1時間30分静置した。反応の変化をLCMSで追跡し、1時間30分後目的物SP618が主要生成物であり、副反応は観測されなかった(目的化合物SP618(LCMS保持時間0.38分)と内部標準(LCMS保持時間0.64分)はLCMSのUVエリア比で54:46であった)。(図59)
LCMS(ESI) m/z = 1053 (M―H)―
保持時間:0.38分(分析条件SQDFA05)
窒素雰囲気下、step1で調整した反応液(90μl)に80mM 2,2'-ジチオジピリジン(化合物SP622、2,2'-PySSPy)溶液(90μl)を室温にて加え、pH=3.0にてサーマルサイクラー中、37度で12時間静置した。反応の変化をLCMSで追跡し、12時間後目的物が生成していることを確認した。目的化合物(化合物SP619、LCMS保持時間0.43分)と加水分解体(副生成物、LCMS保持時間0.41分)はLCMSのUVエリア比で97:3であった。(図60)
LCMS(ESI) m/z = 1261.3 (M+H)+
保持時間:0.43分(分析条件SQDFA05)
窒素雰囲気下、step2で調整した反応液(90μl)に別途調整した600mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液(10μl、6.0μmol、pH=7.2)を室温にて加え、pH=4.6にて同温度で1時間間静置した。反応の変化をLCMSで追跡し、1時間後目的物(化合物SP620)が生成していることを確認した。
600mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液の調整法は以下の通りである。トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(13.8mg、48μmol)に2N 水酸化ナトリウム水溶液(80μl)を加え、pH=7.2に調整した。
LCMS(ESI) m/z = 1041 (M―H)―(図61)
保持時間:0.36分(分析条件SQDFA05)
窒素雰囲気下、step3で調整した反応液(30μl)に別途調整した5.0M 2-メルカプトエタンスルホン酸ナトリウム溶液(30μl、pH=8.5)を室温にて加え、pH=8.5にてサーマルサイクラー中、30度で15時間間静置した。反応の変化をLCMSで追跡し、15時間後目的物である(S)-N-((3R,6S,9S,12R,16S,19S)-6-((1H-インドール-3-イル)メチル)-16-((S)-2-カルバモイルピロリジン-1-カルボニル)-9-(4-(ジメチルアミノ)ブチル)-3,12-ビス(メルカプトメチル)-2,5,8,11,14,18-ヘキサオキソ-1,4,7,10,13,17-ヘキサアザシクロトリコサン-19-イル)-1-(2-ヒドロキシアセチル)ピロリジン-2-カルボキサミド(HOGly-Pro-Lys(*Cys-Lys(Me2)-Trp-Cys)-Asp*-Pro-NH2、2つの*部位にて環化)(化合物SP617)が生成していることを確認した。
LCMS(ESI) m/z = 1043 (M+H)+
保持時間:0.40分(分析条件SQDFA05)
実際に行われる翻訳条件を模擬して、以下の条件で反応を行った。脱保護(化合物SP620)までPURE SYSTEMの翻訳系において反応を実施した。その後の分枝ペプチド生成反応において、ディスプレイライブラリー実験ではRNeasy(r) MinEluteTM Cleanup Kit(キアゲン社)での精製を行うこともできる。この精製過程で、RNAとペプチドの複合体の精製が可能で、特にたんぱく成分や低分子成分が除去される。本実験では、化合物SP620が得られた時点で、精製を実施することを仮定して化合物SP620を単離し、分枝化反応を実施した。そのために、使用する反応溶媒としてディスプレイライブラリーでの精製を考慮してPURE SYSTEMの翻訳系溶媒をRNeasy(r) MinEluteTM Cleanup Kit(キアゲン社)を用い精製した溶出液を用いた。なお、化合物SP620の精製中にペプチドに存在する2つのSH基が分子内にてS-S結合を形成した化合物SP623に変換された。よって化合物SP623から、再びディスプレイライブラリー模擬中で化合物SP620を再発生させてから分枝化反応を行い、化合物SP617を得た。
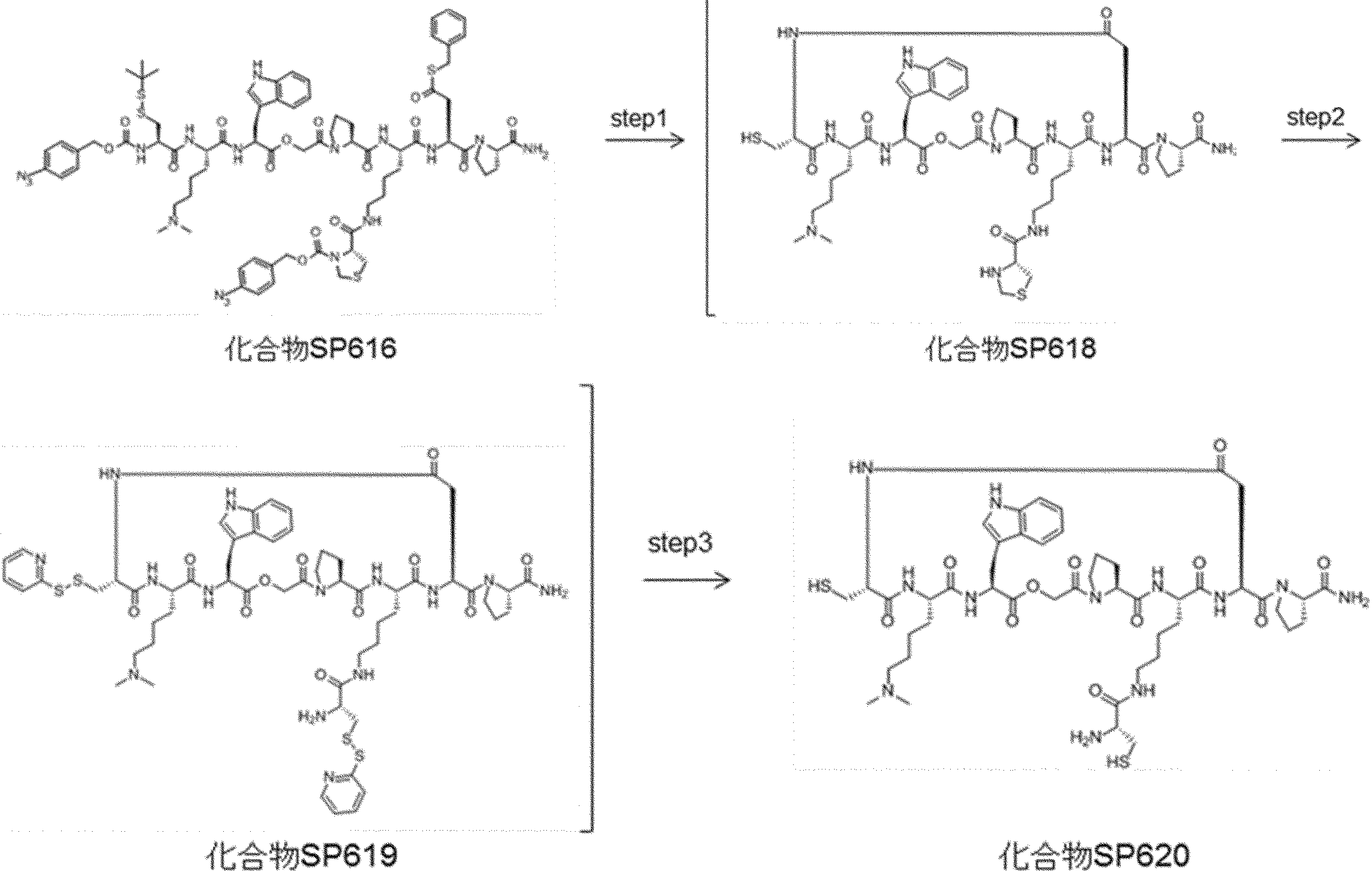
窒素雰囲気下、翻訳用緩衝液(12.5μl)、PURESYSTEM(r) classic II Sol. B(バイオコゥマー社、製品番号PURE2048C)(20μl)、20種の天然アミノ酸溶液(それぞれ5mM,5.0μl)、水(32.5μl)を混合し、ジメチルアセトアミド(DMA)(26.25μl)を加えて溶液を調製した。4mM 4-(((S)-5-((S)-1-((6R,9S,12S)-12-((1H-インドール-3-イル)メチル)-6-((((4-アジドベンジル)オキシ)カルボニル)アミノ)-9-(4-(ジメチルアミノ)ブチル)-2,2-ジメチル-7,10,13-トリオキソ-14-オキサ-3,4-ジチア-8,11-ジアザヘキサデカン-16-オイル)ピロリジン-2-カルボキサミド)-6-(((S)-4-(ベンジルチオ)-1-((S)-2-カルバモイルピロリジン-1-イル)-1,4-ジオキソブタン-2-イル)アミノ)-6-オキソヘキシル)カルバモイル)チアゾリジン-3-カルボン酸(R)-4-アジドベンジル(Acbz-Cys(StBu)-Lys(Me2)-Trp-HOGly-Pro-Lys(Acbz-Thz)-Asp(SBn)-Pro-NH2)(化合物SP616)のジメチルアセトアミド(DMA)溶液(2.5μl、0.01μmol)、内部標準として用いた8mM 2、4-ジメチル安息香酸のジメチルアセトアミド(DMA)溶液(1.25μl、0.01μmol)、別途調整した100mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液(10μl、1.0μmol、pH=7.6)を室温にて加え、pH=7.5にて、同温度で1時間静置した。反応の変化をLCMSで追跡し、1時間後目的物(化合物SP618)が生成していることを確認した。目的化合物(LCMS保持時間0.37分)と内部標準(LCMS保持時間0.64分)はLCMSのUVエリア比で45:55であったことから、翻訳液中でも緩衝液と同等の反応の選択性と速度が得られていることがわかる。(図63)
LCMS(ESI) m/z = 1053 (M―H)―
保持時間:0.37分(分析条件SQDFA05)
窒素雰囲気下、step1で調整した反応液(80μl)に80mM 2,2'-ジチオジピリジン(化合物SP622、2,2'-PySSPy)溶液(80μl)を室温にて加え、500mM 水酸化ナトリウム水溶液(2.0μl)を室温にて加え、pH=4.0にてサーマルサイクラー中、37度で13時間静置した。反応の変化をLCMSで追跡し、13時間後目的物(化合物SP619)が生成していることを確認した。目的化合物(LCMS保持時間0.44分)と加水分解体(LCMS保持時間0.40分)はLCMSのUVエリア比で90:10であった。(図64)
80mM 2,2'-ジチオジピリジン(化合物SP622、2,2'-PySSPy)溶液の調整法は以下の通りである。400mM 2,2'-ジチオジピリジン(化合物SP622、2,2'-PySSPy)のジメチルアセトアミド(DMA)溶液(20μl、8.0μmol)にジメチルアセトアミド(DMA)(10μl)230mMの塩酸水溶液(70μl)を加えた。
LCMS(ESI) m/z = 1261.3 (M+H)+
保持時間:0.44分(分析条件SQDFA05)
窒素雰囲気下、step2で調整した反応液(81μl)に別途調整した600mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液(9.0μl、pH=7.2)を室温にて加え、pH=5.1にて同温度で1時間間静置した。反応の変化をLCMSで追跡し、1時間後目的物(化合物SP620)が生成していることを確認した。HEPES 緩衝液中での反応と比較し、PURE SYSTEMの使用による新たな副生成物は観測されなかった。(図65)
600mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液の調整法は以下の通りである。トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(17.6mg、61μmol)に2N 水酸化ナトリウム水溶液(102μl)を加え、pH=7.2に調整した。
LCMS(ESI) m/z = 1041 (M―H)―保持時間:0.36分(分析条件SQDFA05)
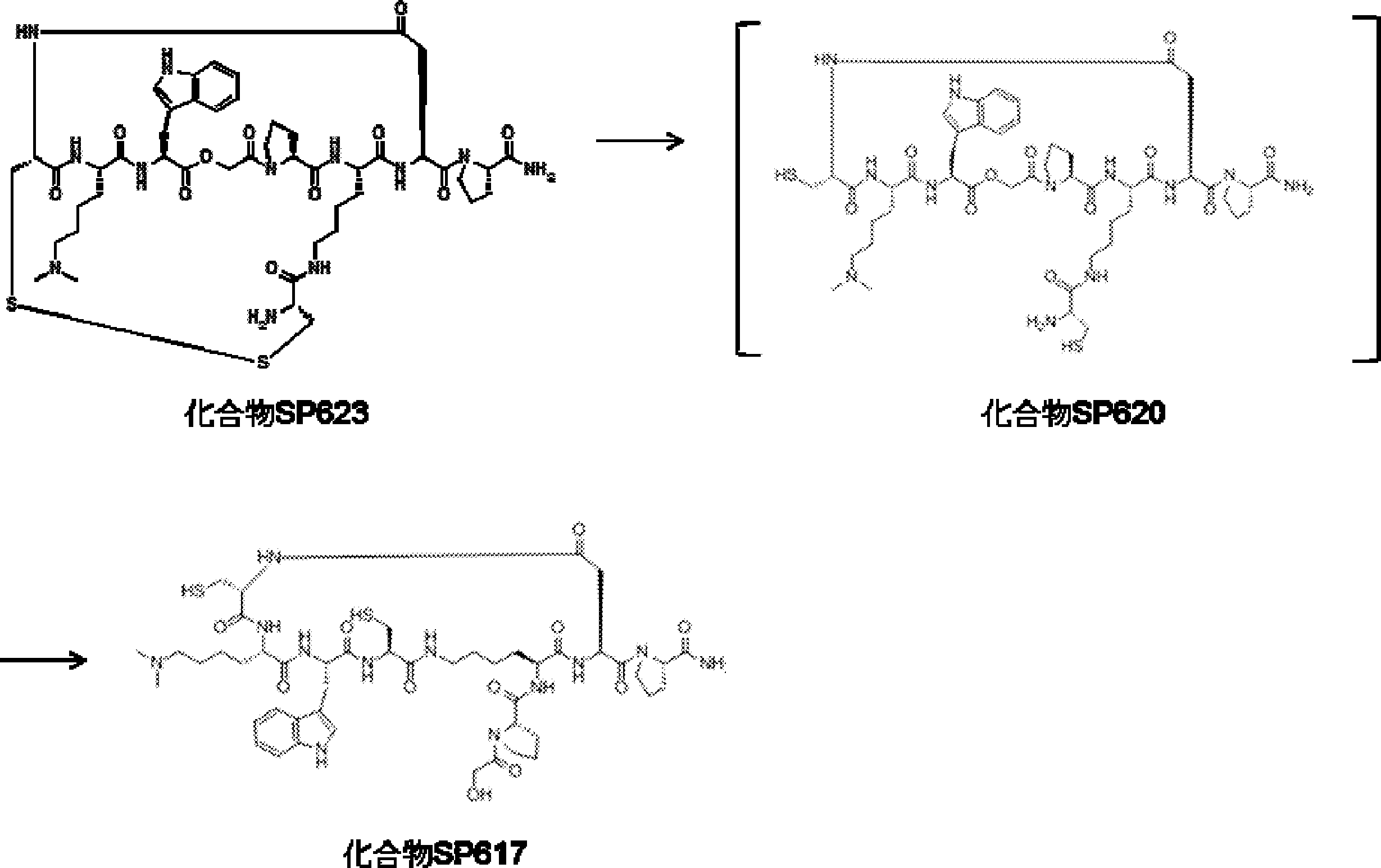
LCMS(ESI) m/z = 1043 (M+H)+
保持時間:0.40分(分析条件SQDFA05)
4-2-2で用いた、化合物617を含むPure SYSTEMの翻訳系溶媒をRNeasy(登録商標)MinElute(登録商標) Cleanup Kit(キアゲン社)を用い精製した溶出液中での分枝ペプチド精製反応で用いた反応溶液に対し脱硫反応を行った。
(S)-N-((3S,6S,9S,12S,16S,19S)-6-((1H-インドール-3-イル)メチル)-16-((S)-2-カルバモイルピロリジン-1-カルボニル)-9-(4-(ジメチルアミノ)ブチル)-3,12-ジメチル-2,5,8,11,14,18-ヘキサオキソ-1,4,7,10,13,17-ヘキサアザシクロトリコサン-19-イル)-1-(2-ヒドロキシアセチル)ピロリジン-2-カルボキサミド(化合物SP624、 HO Gly-Pro-Lys(*Ala-Lys(Me 2 )-Trp-Ala)-Asp*-Pro-NH 2 、2つの*部位にて環化)の合成
なお、本明細書中では他の例と同様、Lysの側鎖アミノ基とH-Ala-Lys(Me2)-Trp-Ala-OHのC末端のカルボキシル基がアミド結合した化合物を、H-Lys(H-Ala-Lys(Me2)-Trp-Ala)-OHと記載する。
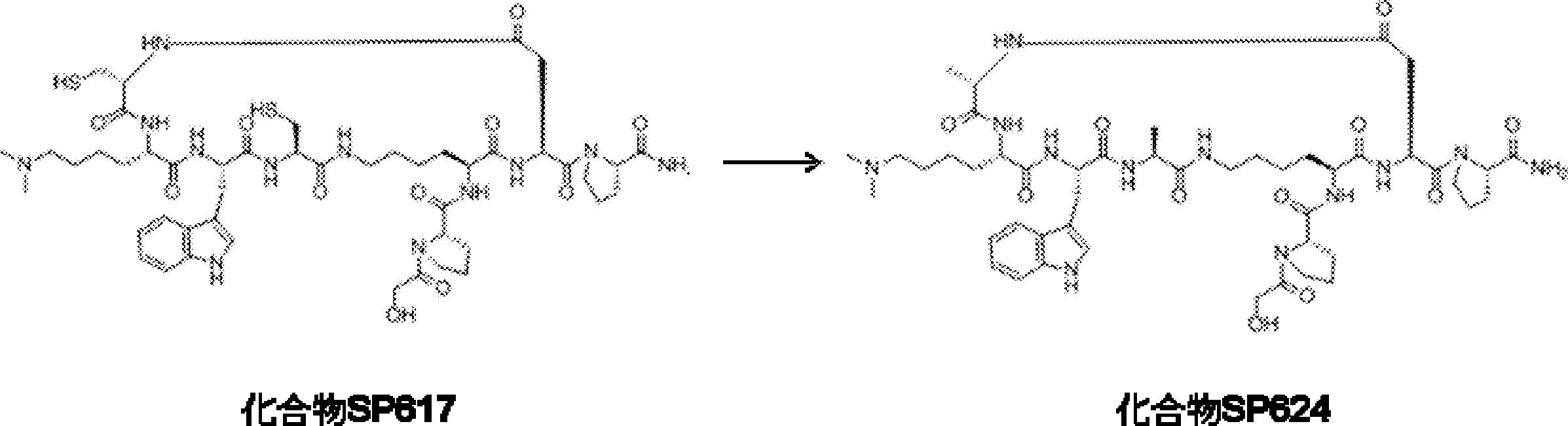
1.5M トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液の調整法は以下の通りである。トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(24.5mg、85.5μmol)5N 水酸化ナトリウム水溶液(57μl)を加え、pH=7.4に調整した。
LCMS(ESI) m/z = 979(M+H)+
保持時間:0.36分(分析条件SQDFA05)
分枝ペプチド生成反応に使用した(S)-1-((3S,10R,15R,18S,21S,29aS,33S)-21-((1H-インドール-3-イル)メチル)-10-アミノ-18-(4-(ジメチルアミノ)ブチル)-1,9,16,19,22,25,31,35-オクタオキソヘキサコサヒドロ-15,3-(エピミノプロパノイミノメタノ)ピロロ[2,1-x][1,11,12,4,7,16,22,25]オキサジチアペンタアザシクロヘプタコシン-33-カルボニル)ピロリジン-2-カルボキサミド(*Cys#-Lys(Me 2 )-Trp- HO Gly-Pro-Lys(H-Cys#)-Asp*-Pro-NH 2 、2つの*部位、#部位にて環化、#部位で2つのSH基がジスルフィド結合を形成している)(化合物SP623)の合成
精製した結果、SP620のままでは精製できず、化合物SP623となった。化合物SP623は容易にSP620へと還元条件にて再び変換可能なため、化合物SP623を精製し、単離した。なお、精製には化合物重量が多く必要なため、上記実験とは別に再合成を行った。

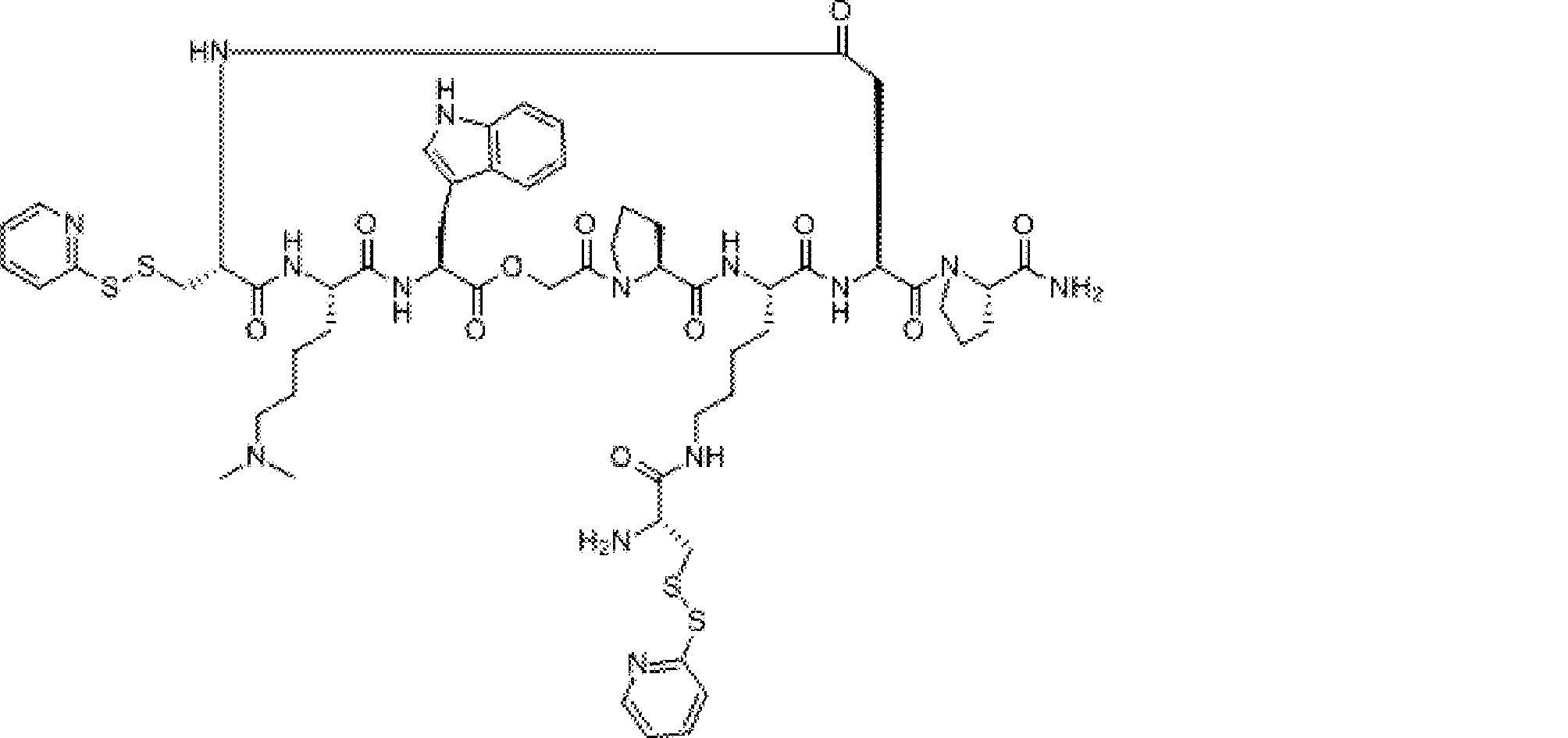
1M トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液の調整法は以下の通りである。トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(39mg、0.136mmol)を5N 水酸化ナトリウム水溶液(91μl)、水(45μl)で溶解させ、pH=7.4に調整した。
330mM 2,2'-ジチオジピリジン(化合物SP622、2,2'-PySSPy)溶液の調整法は以下の通りである。400mM 2,2-ジチジオピリジン(化合物SP622、2,2'-PySSPy)のジメチルアセトアミド(DMA)溶液(1.5ml、0.6mmol)に2N 塩酸水溶液(238μl)、水(80μl)を加えた。
LCMS(ESI) m/z = 1261.4 (M+H)+
保持時間:0.43分(分析条件SQDFA05)
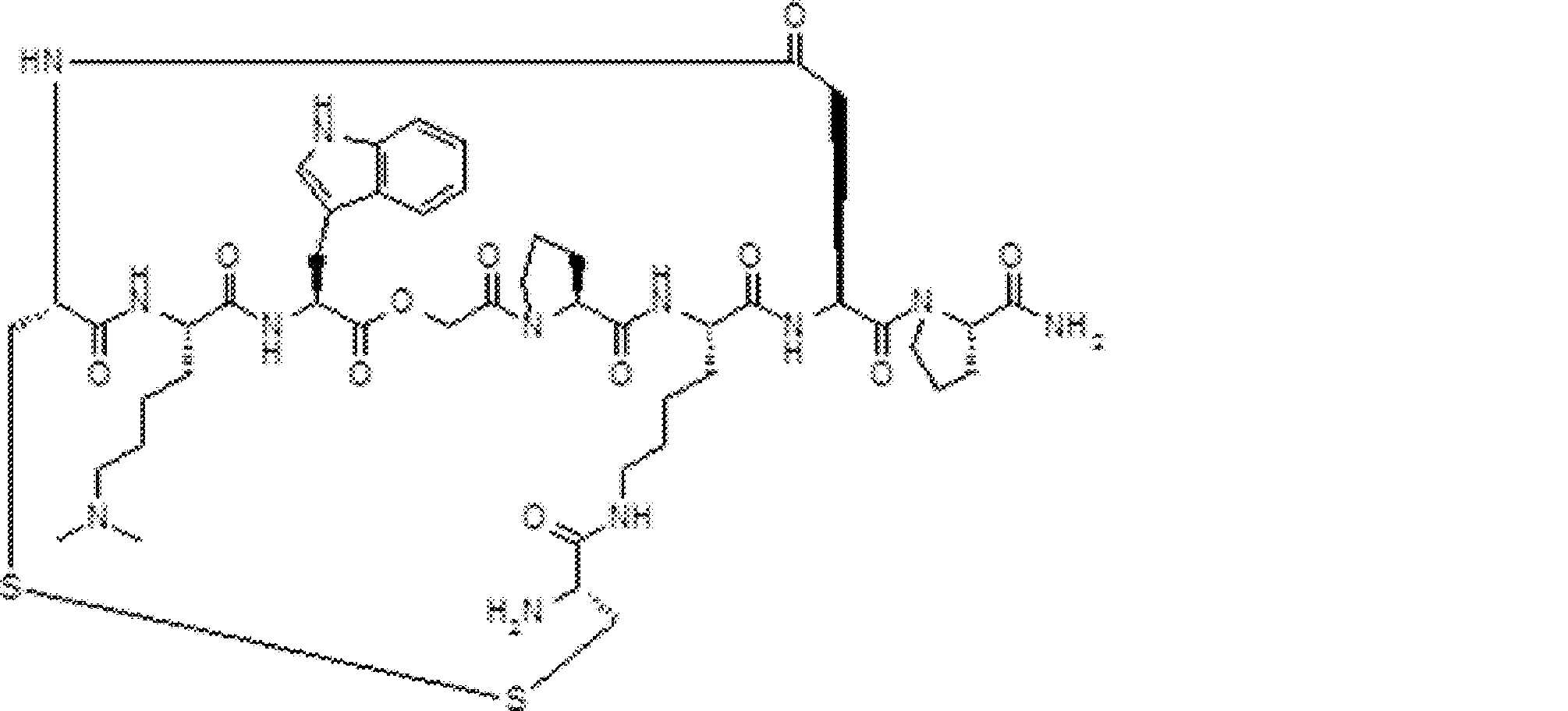
600mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液の調整法は以下の通りである。トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(22.5mg、78.5mmol)を1N 水酸化ナトリウム水溶液(131μl)で溶解させ、pH=3.7に調整した。
LCMS(ESI) m/z = 1039 (M―H)―
保持時間:0.34分(分析条件SQDFA05)
Cys-Pro-HOGlyからチオエステルが発生させる反応条件は温和でpH=7.3でもゆっくりと進行することが明らかとなった(後述)。1回目の環化反応との選択性を最大化する目的、さらには1回目の環化反応でより温和でない反応条件を使用できるようにすることを目的として、Cys-Pro-HOGlyよりも安定で、反応進行のスイッチオンーオフが可能なユニットを探索した結果、より好ましいユニットの条件が明らかとなった。望ましいCys-Pro-Lacユニットを用いて、翻訳液中での環化反応および分枝化反応が進行することを確認した。
Cys-Pro-HOGlyおよびその代替ユニットを含む5残基ペプチドの合成を、そのチオエステル発生の条件を得るためのモデルペプチドとした。そのモデル化合物を合成した。
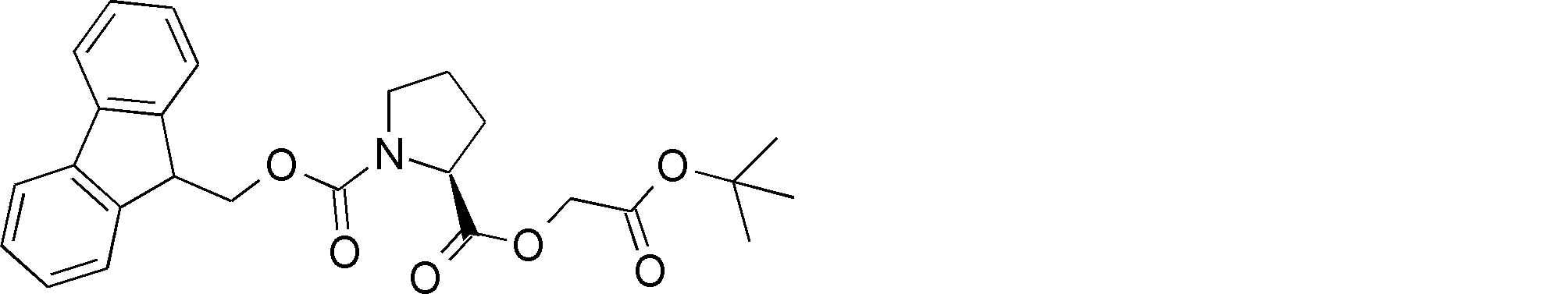
LCMS(ESI) m/z = 396 (M-tBu+H)+
保持時間:1.02分(分析条件SQDFA05)
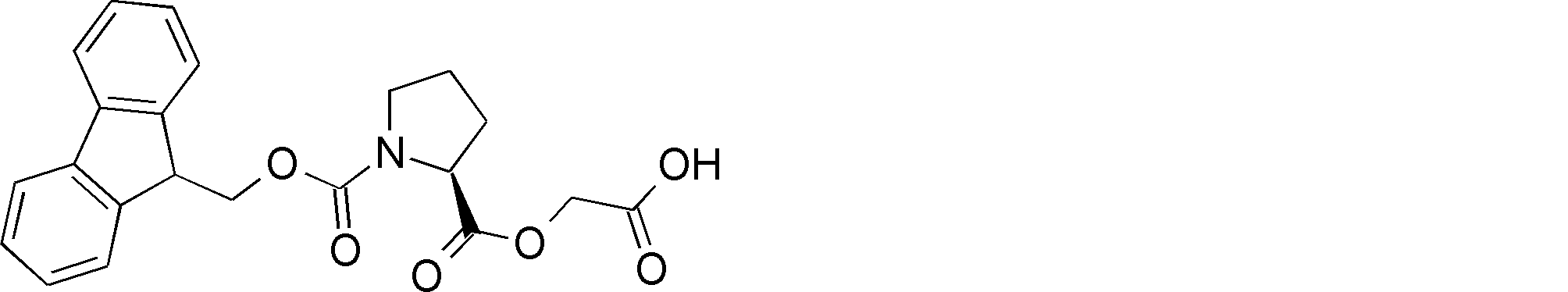
LCMS(ESI) m/z = 396 (M+H)+
保持時間:0.77分(分析条件SQDFA05)
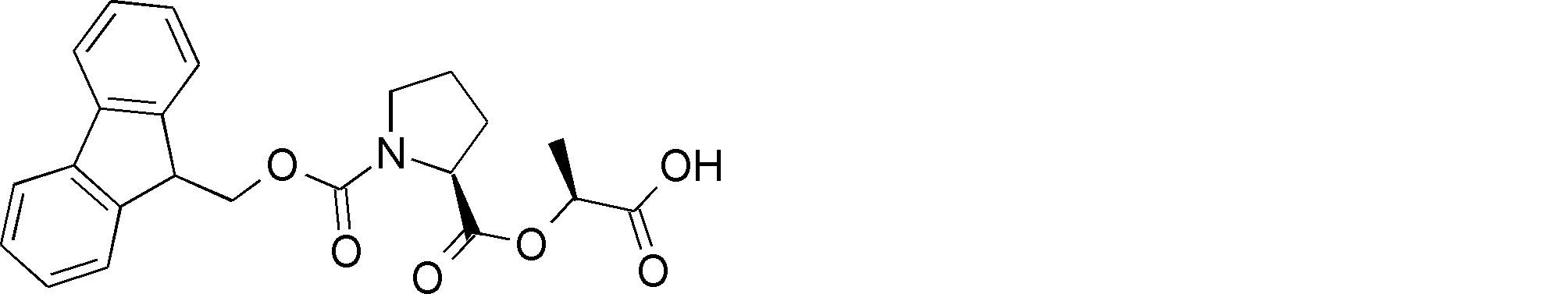
LCMS(ESI) m/z = 410 (M+H)+
保持時間:0.81分(分析条件SQDFA05)
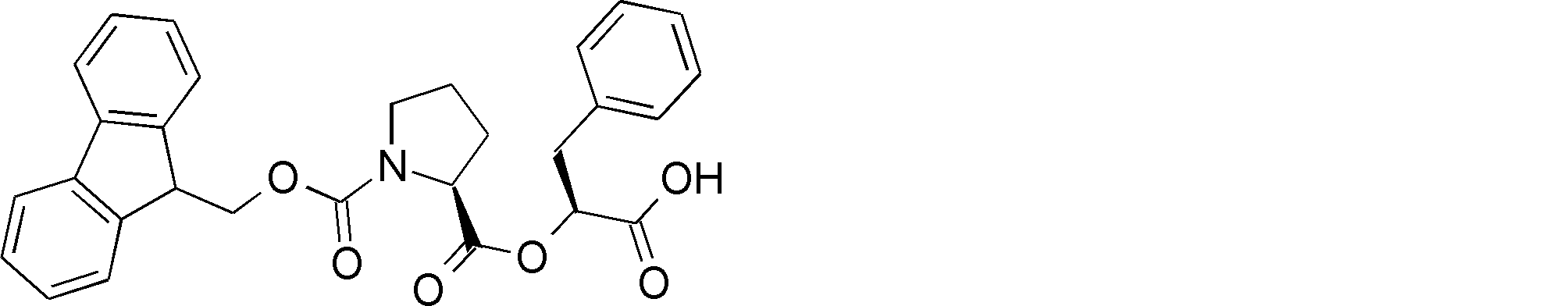
LCMS(ESI) m/z = 486 (M+H)+
保持時間:0.92分(分析条件SQDFA05)
なお、PhLacとは、本明細書では(S)-ヒドロキシ‐3-フェニルプロパン酸のヒドロキシル基とカルボン酸官能基を形成するヒドロキシル基を取り除いた部分構造とする。
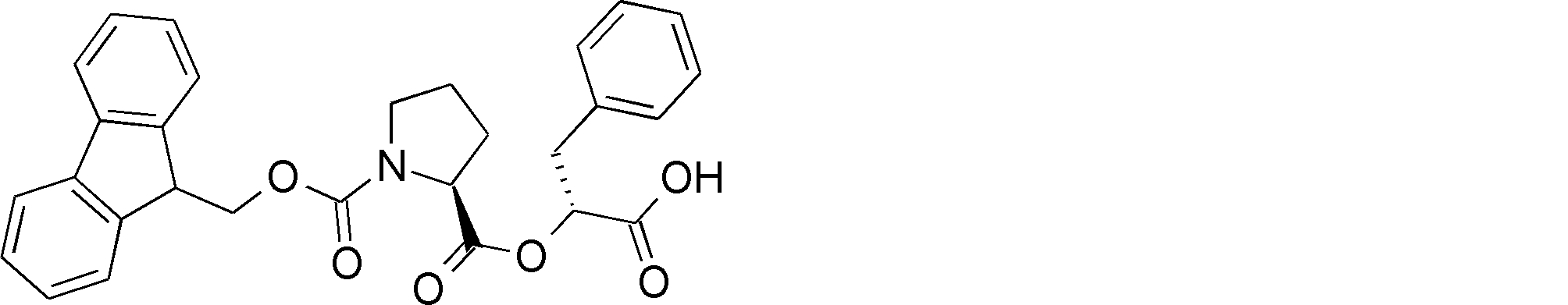
LCMS(ESI) m/z = 486 (M+H)+
保持時間:0.91分(分析条件SQDFA05)
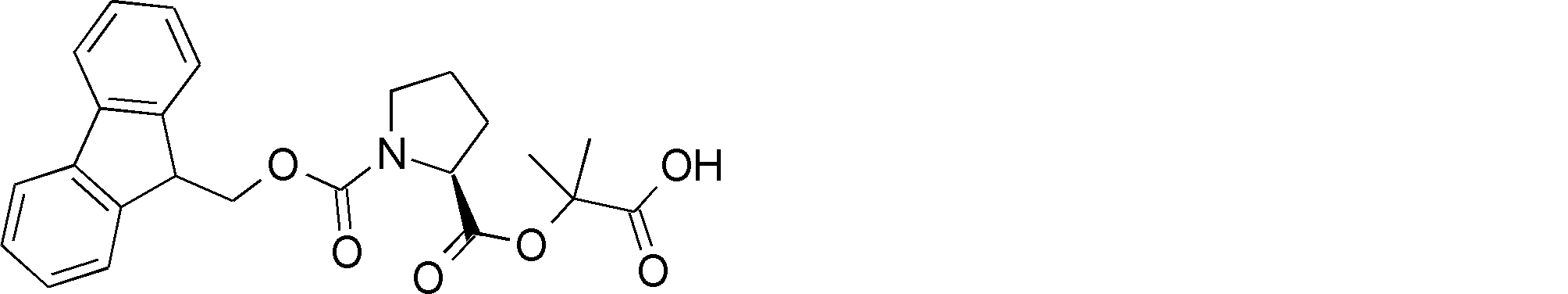
LCMS(ESI) m/z = 424 (M+H)+
保持時間:0.85分(分析条件SQDFA05)
なお、HOGly(Me)2とは、本明細書では2-ヒドロキシ‐2-メチルプロパン酸のヒドロキシル基とカルボン酸官能基を形成するヒドロキシル基を取り除いた部分構造とする。
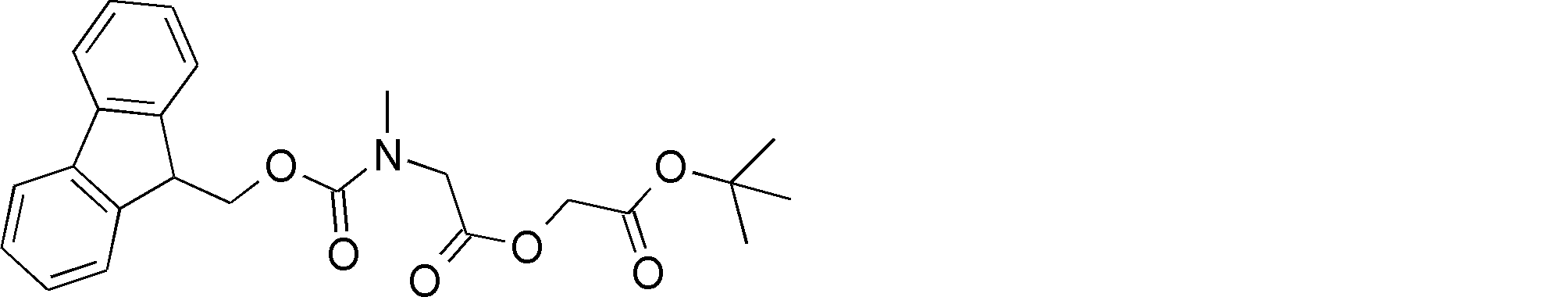
LCMS(ESI) m/z = 370 (M-tBu+H)+
保持時間:0.99分(分析条件SQDFA05)
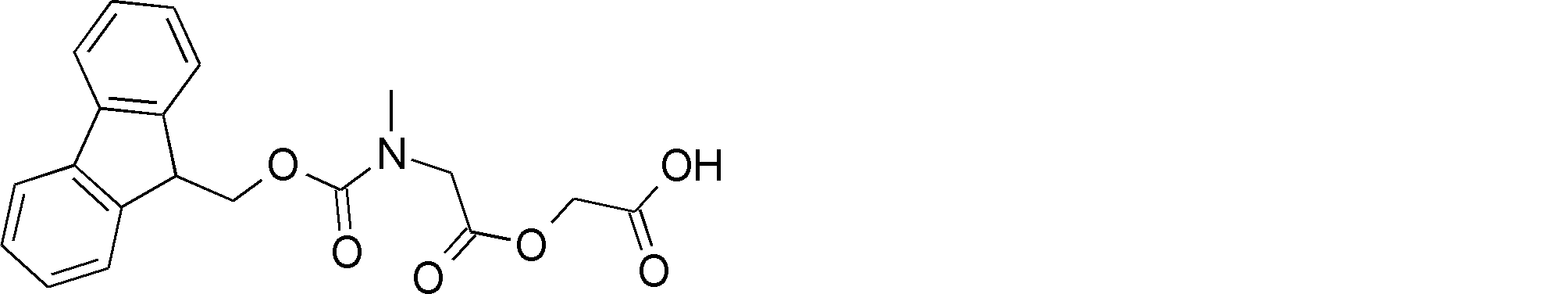
LCMS(ESI) m/z = 370 (M+H)+
保持時間:0.75分(分析条件SQDFA05)

LCMS(ESI) m/z = 396 (M+H)+
保持時間:0.49分(分析条件SQDFA05)

LCMS(ESI) m/z = 438 (M+H)+
保持時間:0.66分(分析条件SQDFA05)

LCMS(ESI) m/z = 210 (M+H)+
保持時間:0.56分(分析条件SQDAA05)

LCMS(ESI) m/z = 618 (M+H)+
保持時間:0.94分(分析条件SQDFA05)

LCMS(ESI) m/z = 396 (M+H)+
保持時間:0.83分(分析条件SQDAA05)

LCMS(ESI) m/z = 438 (M+H)+
保持時間:0.67分(分析条件SQDFA05)

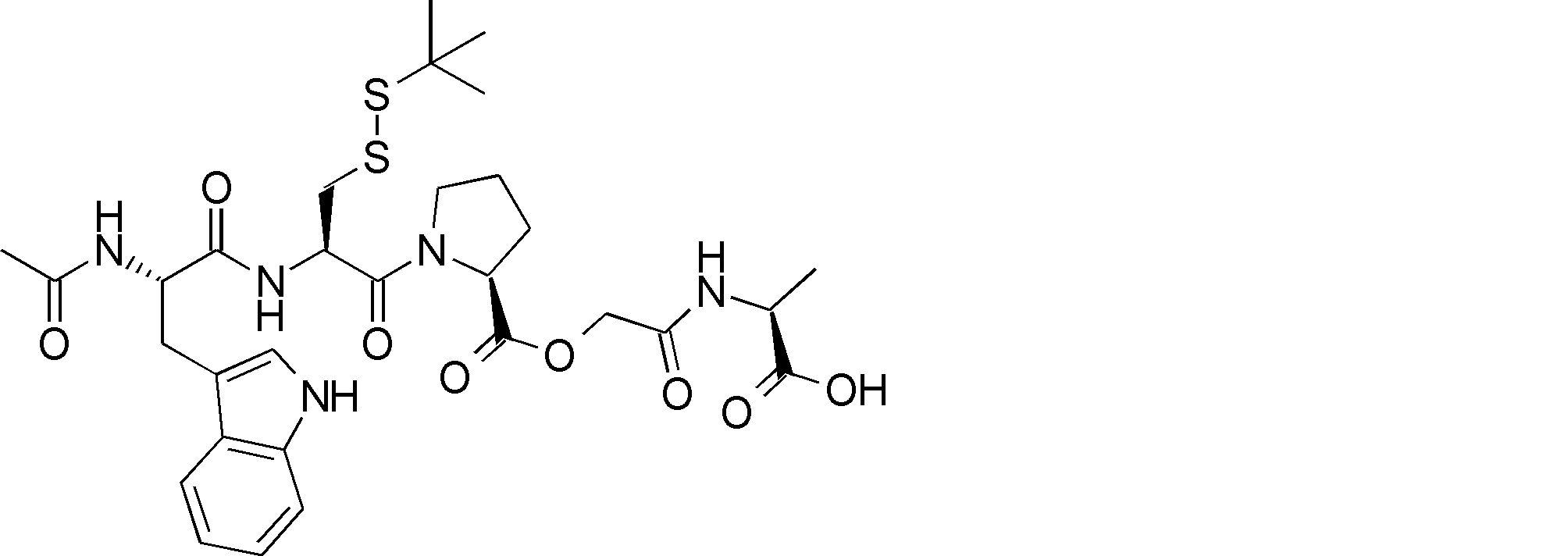
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度洗浄した後、5%ピペリジン/N,N-ジメチルホルムアミド溶液(0.8mL)を混合し、室温にて1時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.8mL)にて4度洗浄した後、(R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-Cys(StBu)-OH) (化合物SP640)(170mg、0.388mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt(39.6mg、0.243mmol)のNMP溶液(0.64mL)とジイソプロピルカルボジイミド(DIC)(65μL、0.417mmol)のN,N-ジメチルホルムアミド溶液(0.52mL)を混合し、室温にて15時間振とうした。
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度、続いて塩化メチレン(0.8mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(1.0mL)を混合し、室温にて2時間振とうした。
固相と液相を分離し、固相を塩化メチレン(1.0mL)で洗浄し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(S)-2-(2-(((S)-1-((R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパノイル)ピロリジン-2-カルボニル)オキシ)アセトアミド)プロパン酸(Ac-Trp-Cys(StBu)-Pro-HOGly-Ala-OH) (化合物SP646)(37.8mg、57μmol、70%)を得た。
LCMS(ESI) m/z = 664 (M+H)+
保持時間:0.80分(分析条件SQDAA05)
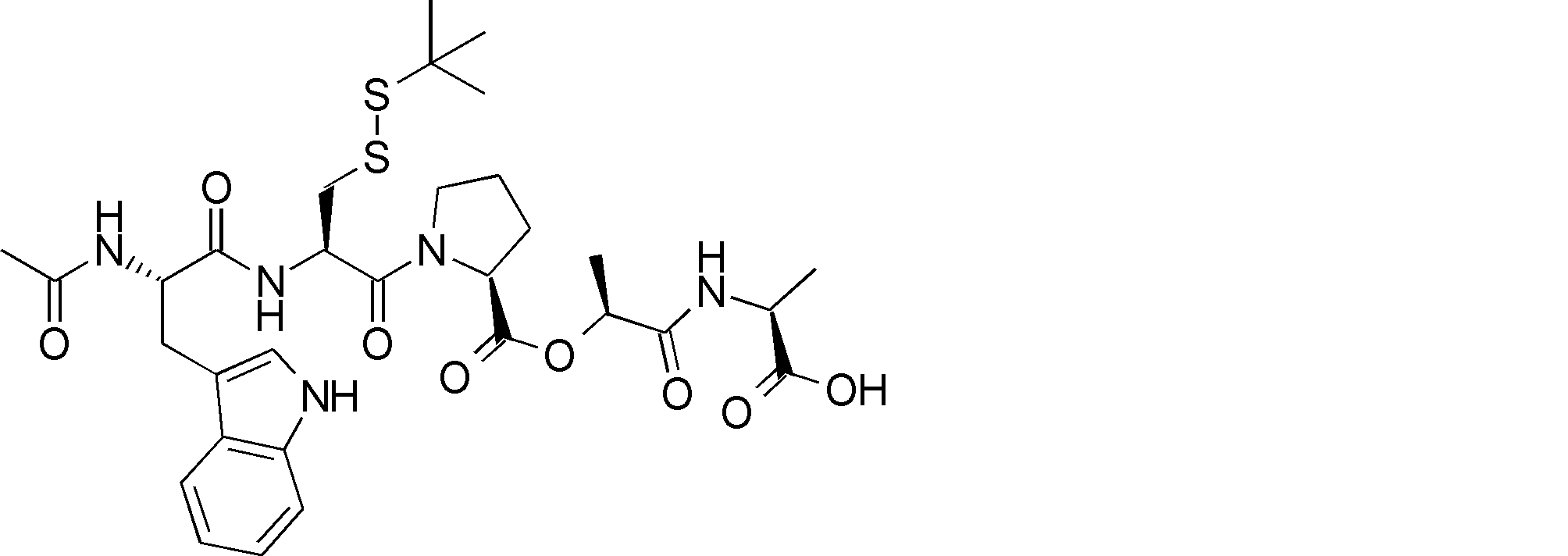
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.8mL)にて4度洗浄した後、(R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-Cys(StBu)-OH) (化合物SP640)(170mg、0.388mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt)(39.6mg、0.243mmol)のNMP溶液(0.64mL)とジイソプロピルカルボジイミド(DIC)(65μL、0.417mmol)のN,N-ジメチルホルムアミド溶液(0.52mL)を混合し、室温にて15時間振とうした。
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度、続いて塩化メチレン(0.8mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(1.0mL)を混合し、室温にて2時間振とうした。
固相と液相を分離し、固相を塩化メチレン(1.0mL)で洗浄し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(S)-2-((S)-2-(((S)-1-((R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパノイル)ピロリジン-2-カルボニル)オキシ)プロパンアミド)プロパン酸(Ac-Trp-Cys(StBu)-Pro-Lac-Ala-OH) (化合物SP647)(51.4mg、76μmol、94%)を得た。
LCMS(ESI) m/z = 678 (M+H)+
保持時間:0.82分(分析条件SQDAA05)
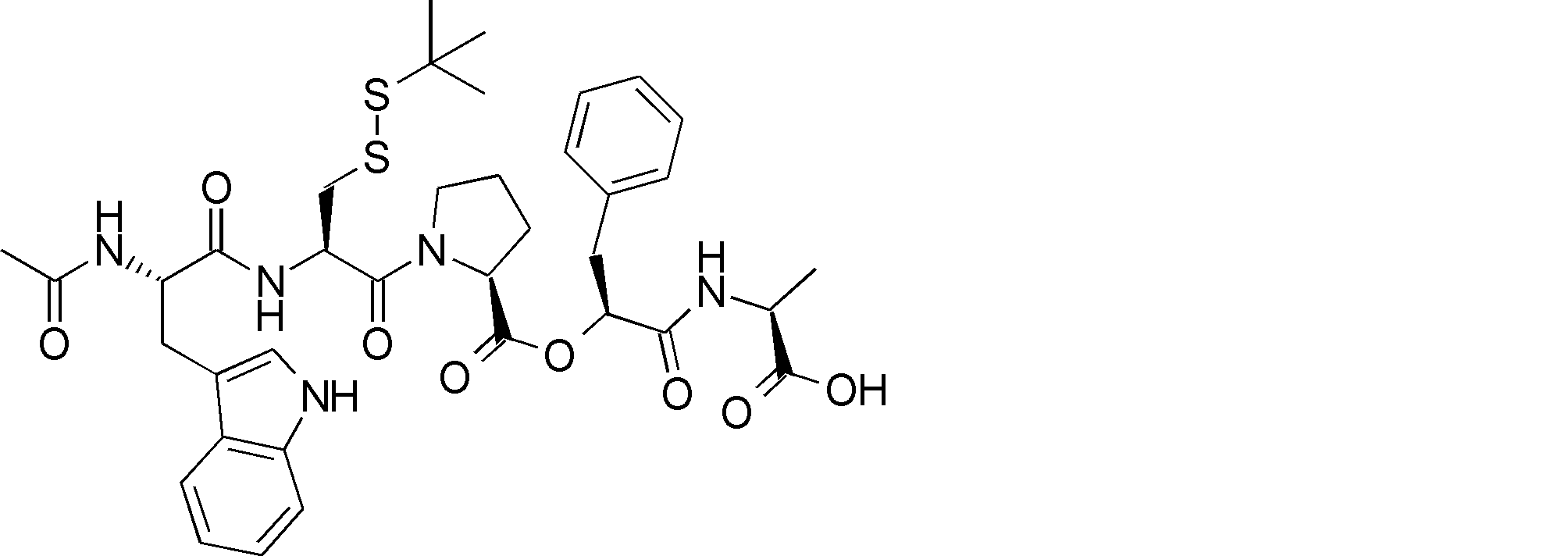
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度洗浄した後、5%ピペリジン/N,N-ジメチルホルムアミド溶液(0.8mL)を混合し、室温にて1時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.8mL)にて4度洗浄した後、(R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-Cys(StBu)-OH) (化合物SP640)(170mg、0.388mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt(39.6mg、0.243mmol)のNMP溶液(0.64mL)とジイソプロピルカルボジイミド(DIC)(65μL、0.417mmol)のN,N-ジメチルホルムアミド溶液(0.52mL)を混合し、室温にて15時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度、続いて塩化メチレン(0.8mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(1.0mL)を混合し、室温にて2時間振とうした。
固相と液相を分離し、固相を塩化メチレン(1.0mL)で洗浄し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(S)-2-((S)-2-(((S)-1-((R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパノイル)ピロリジン-2-カルボニル)オキシ)-3-フェニルプロパンアミド)プロパン酸(Ac-Trp-Cys(StBu)-Pro-PhLac-Ala-OH) (化合物SP648)(51.2mg、68μmol、84%)を得た。
LCMS(ESI) m/z = 754 (M+H)+
保持時間:0.92分(分析条件SQDAA05)
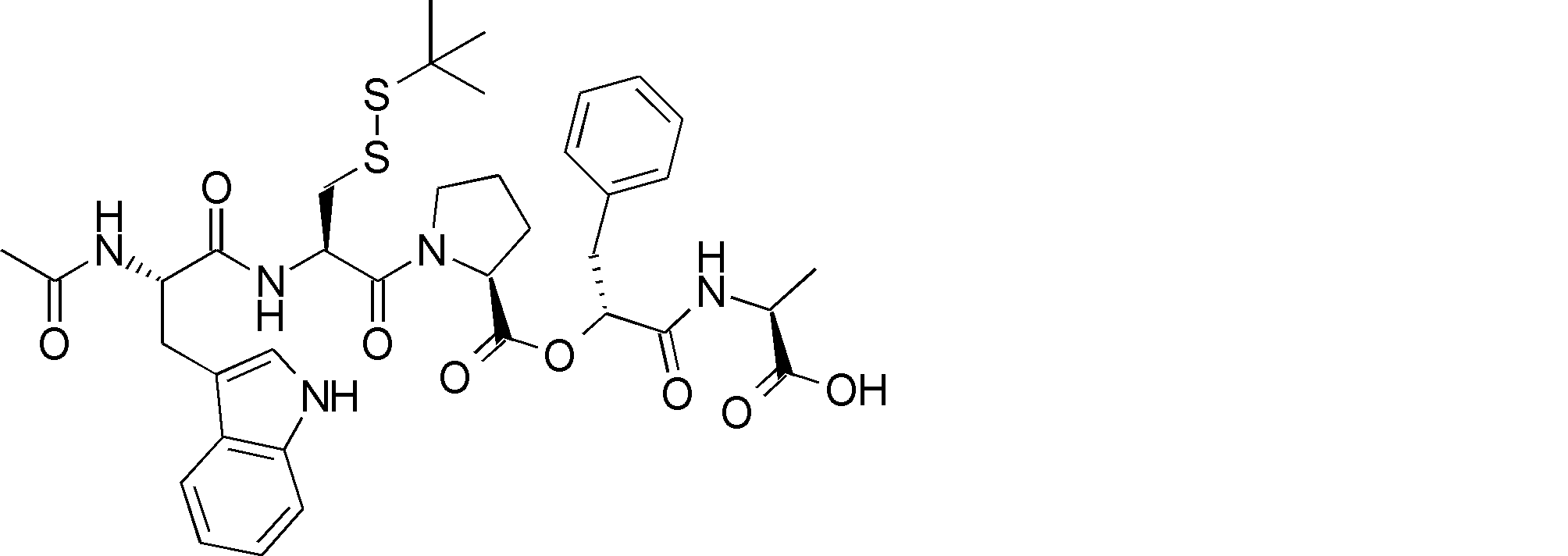
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度洗浄した後、5%ピペリジン/N,N-ジメチルホルムアミド溶液(0.8mL)を混合し、室温にて1時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.8mL)にて4度洗浄した後、(R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-Cys(StBu)-OH) (化合物SP640)(170mg、0.388mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt)(39.6mg、0.243mmol)のNMP溶液(0.64mL)とジイソプロピルカルボジイミド(DIC)(65μL、0.417mmol)のN,N-ジメチルホルムアミド溶液(0.52mL)を混合し、室温にて15時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度、続いて塩化メチレン(0.8mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(1.0mL)を混合し、室温にて2時間振とうした。
固相と液相を分離し、固相を塩化メチレン(1.0mL)で洗浄し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(S)-2-((R)-2-(((S)-1-((R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパノイル)ピロリジン-2-カルボニル)オキシ)-3-フェニルプロパンアミド)プロパン酸(Ac-Trp-Cys(StBu)-Pro-D-PhLac-Ala-OH) (化合物SP649)(50.0mg、66.0μmol、82%)を得た。
LCMS(ESI) m/z = 754 (M+H)+
保持時間:0.93分(分析条件SQDAA05)
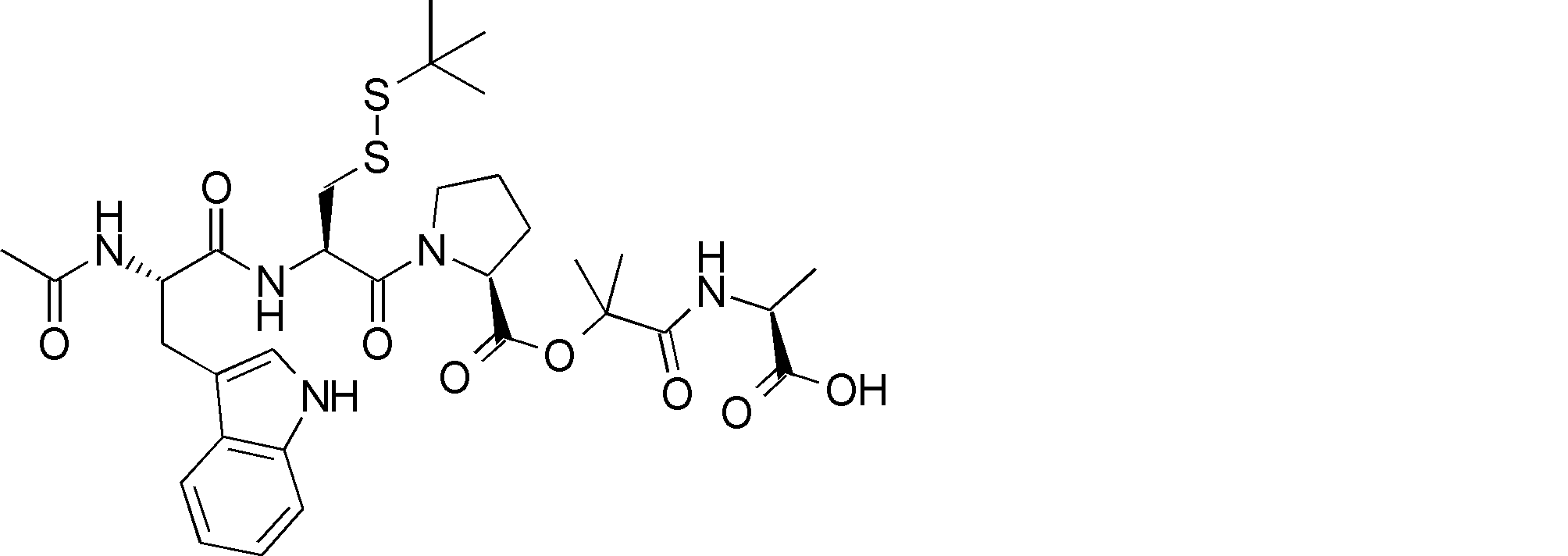
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.8mL)にて4度洗浄した後、(R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-Cys(StBu)-OH) (化合物SP640)(170mg、0.388mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt)(39.6mg、0.243mmol)のNMP溶液(0.64mL)とジイソプロピルカルボジイミド(DIC)(65μL、0.417mmol)のN,N-ジメチルホルムアミド溶液(0.52mL)を混合し、室温にて15時間振とうした。
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度、続いて塩化メチレン(0.8mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(1.0mL)を混合し、室温にて2時間振とうした。固相と液相を分離し、固相を塩化メチレン(1.0mL)で洗浄し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(S)-2-(2-(((S)-1-((R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパノイル)ピロリジン-2-カルボニル)オキシ)-2-メチルプロパンアミド)プロパン酸(Ac-Trp-Cys(StBu)-Pro-HOGly(Me)2-Ala-OH) (化合物SP650)(46.4mg、67μmol、83%)を得た。
LCMS(ESI) m/z = 692 (M+H)+
保持時間:0.84分(分析条件SQDAA05)

固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度洗浄した後、5%ピペリジン/N,N-ジメチルホルムアミド溶液(0.8mL)を混合し、室温にて1時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.8mL)にて4度洗浄した後、(R)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-Cys(StBu)-OH)(化合物SP640)(170mg、0.388mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt)(39.6mg、0.243mmol)のNMP溶液(0.64mL)とジイソプロピルカルボジイミド(DIC)(65μL、0.417mmol)のN,N-ジメチルホルムアミド溶液(0.52mL)を混合し、室温にて15時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度、続いて塩化メチレン(0.8mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(1.0mL)を混合し、室温にて2時間振とうした。固相と液相を分離し、固相を塩化メチレン(1.0mL)で洗浄し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(4S,7R,16S)-4-((1H-インドール-3-イル)メチル)-7-((tert-ブチルジスルファニル)メチル)-9,16-ジメチル-2,5,8,11,14-ペンタオキソ-12-オキサ-3,6,9,15-テトラアザヘプタデカン-17-酸(Ac-Trp-Cys(StBu)-MeGly-HOGly-Ala-OH)(化合物SP651)(26.7mg、42μmol、52%)を得た。
LCMS(ESI) m/z = 638 (M+H)+
保持時間:0.79分(分析条件SQDAA05)

固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度洗浄した後、5%ピペリジン/N,N-ジメチルホルムアミド溶液(0.8mL)を混合し、室温にて1時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.8mL)にて4度洗浄した後、(S)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-D-Cys(StBu)-OH)(化合物SP644)(170mg、0.388mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt)(39.6mg、0.243mmol)のNMP溶液(0.64mL)とジイソプロピルカルボジイミド(DIC)(65μL、0.417mmol)のN,N-ジメチルホルムアミド溶液(0.52mL)を混合し、室温にて15時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.8mL)にて4度、続いて塩化メチレン(0.8mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(1.0mL)を混合し、室温にて2時間振とうした。固相と液相を分離し、固相を塩化メチレン(1.0mL)で洗浄し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(4S,7S,16S)-4-((1H-インドール-3-イル)メチル)-7-((tert-ブチルジスルファニル)メチル)-9,16-ジメチル-2,5,8,11,14-ペンタオキソ-12-オキサ-3,6,9,15-テトラアザヘプタデカン-17-酸(Ac-Trp-D-Cys(StBu)- MeGly-HOGly-Ala-OH) (化合物SP652)(36.5mg、57μmol、71%)を得た。
LCMS(ESI) m/z = 638 (M+H)+
保持時間:0.81分(分析条件SQDAA05)
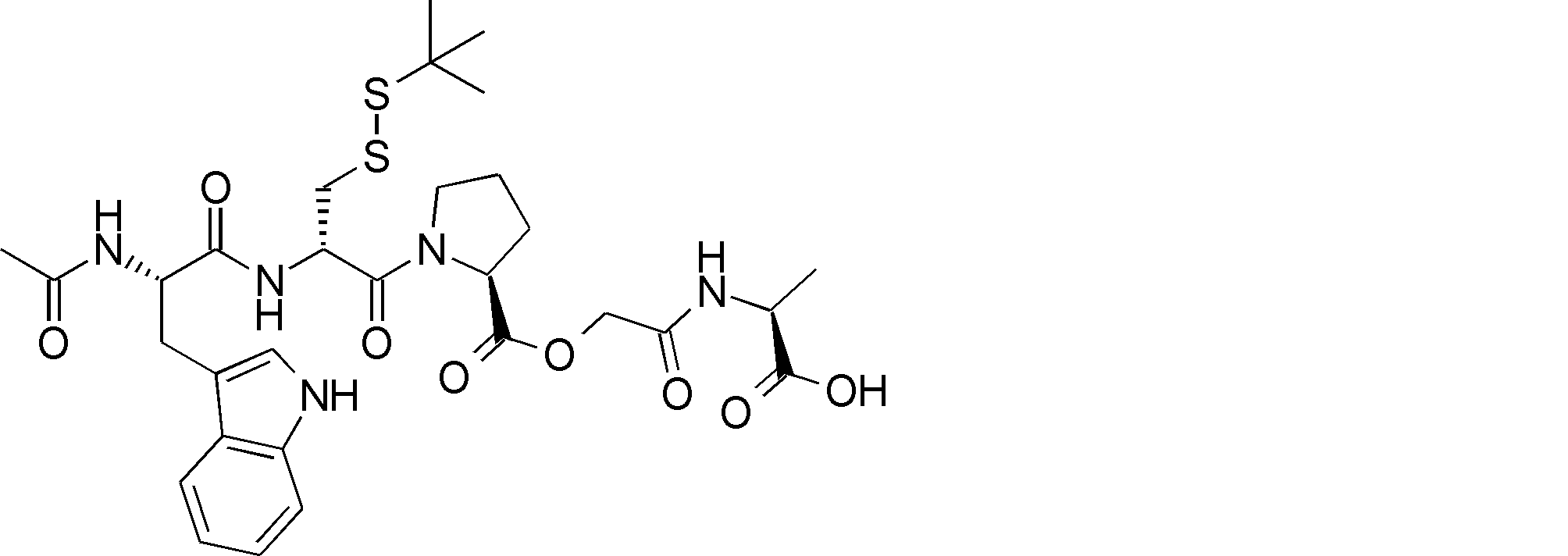
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.2mL)にて4度洗浄した後、(S)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-D-Cys(StBu)-OH)(化合物SP644)(42.5mg、0.097mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt)(9.9mg、60.8mol)のNMP溶液(0.160mL)とジイソプロピルカルボジイミド(DIC)(16μL、0.104mmol)のN,N-ジメチルホルムアミド溶液(0.13mL)を混合し、室温にて15時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.2mL)にて4度、続いて塩化メチレン(0.2mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(0.25mL)を混合し、室温にて2時間振とうした。固相と液相を分離し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(10mM酢酸アンモニウム水溶液/メタノール)にて精製し、(S)-2-(2-(((S)-1-((S)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパノイル)ピロリジン-2-カルボニル)オキシ)アセトアミド)プロパン酸(Ac-Trp-D-Cys (StBu)-Pro-HOGly-Ala-OH)(化合物SP653)(3.0mg、4.5μmol、22%)を得た。
LCMS(ESI) m/z = 664 (M+H)+
保持時間:0.85分(分析条件SQDAA05)
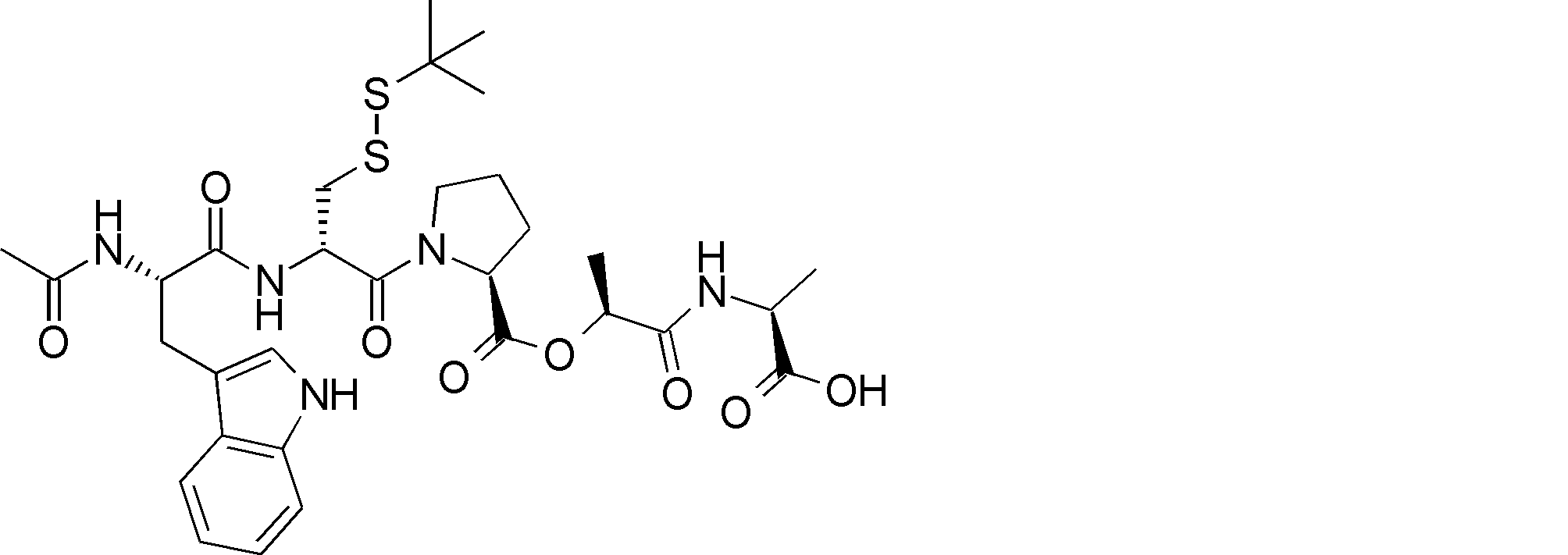
固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.2mL)にて4度洗浄した後、5%ピペリジン/N,N-ジメチルホルムアミド溶液(0.2mL)を混合し、室温にて1時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド溶液(0.2mL)にて4度洗浄した後、(S)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパン酸(Ac-Trp-D-Cys(StBu)-OH) (化合物SP644)(42.5mg、0.097mmol)と3,4-ジヒドロ-3-ヒドロキシー4-オキソー1,2,3-ベンゾトリアジン(HOObt)(9.9mg、60.8mol)のNMP溶液(0.160mL)とジイソプロピルカルボジイミド(DIC)(16μL、0.104mmol)のN,N-ジメチルホルムアミド溶液(0.13mL)を混合し、室温にて15時間振とうした。固相と液相を分離し、得られた樹脂をN,N-ジメチルホルムアミド(0.2mL)にて4度、続いて塩化メチレン(0.2mL)にて4度洗浄した後、50%2,2,2-トリフルオロエタノール/塩化メチレン溶液(0.5mL)を混合し、室温にて2時間振とうした。固相と液相を分離し、得られた液相を減圧濃縮した。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(10mM酢酸アンモニウム/メタノール)にて精製し、(S)-2-((S)-2-(((S)-1-((S)-2-((S)-2-アセトアミド-3-(1H-インドール-3-イル)プロパンアミド)-3-(tert-ブチルジスルファニル)プロパノイル)ピロリジン-2-カルボニル)オキシ)プロパンアミド)プロパン酸(Ac-Trp-D-Cys (StBu)-Pro-Lac-Ala-OH) (化合物SP654)(3.0mg、4.4μmol、22%)を得た。
LCMS(ESI) m/z = 678 (M+H)+
保持時間:0.86分(分析条件SQDAA05)
各pH中での安定性(1回目の環化反応中では反応せず安定に存在することが望まれる)および、pHを向上させた場合の反応選択性(メルカプトエチルアミンとの反応にて確認した)を評価した。
各pH中での安定性(1回目の環化反応中では反応せず安定に存在することが望まれる。)および、pHを向上させた場合の反応選択性(2-アミノエタンチオールとの反応にて確認した)を評価した。
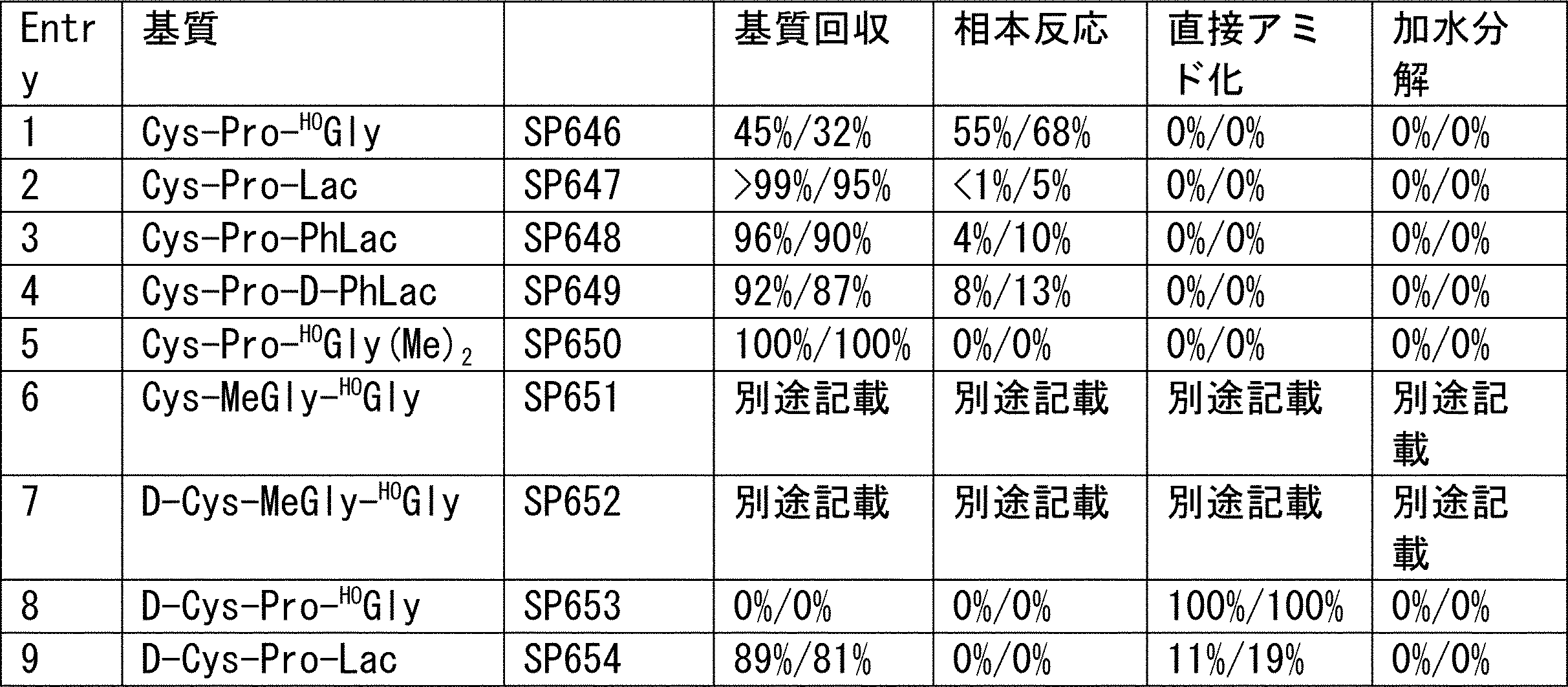
従って、指定されたpHにて1回目の環化反応が実施されることが想定される場合、基質回収割合が高いほど望ましい。また、指定されたpHにて2回目の環化反応が想定される場合には相本反応の割合が高いほど望ましい。
Entry 6
(8時間)基質回収:27% 相本反応:39% 直接アミド化:33% 加水分解:0%
(24時間)基質回収:13% 相本反応:31% 直接アミド化:56% 加水分解:0%
Entry 7
(8時間)基質回収:12% 相本反応:50% 直接アミド化:38% 加水分解:0%
(24時間)基質回収:0% 相本反応:50% 直接アミド化:50% 加水分解:0%
10mM Ac-Trp-Cys(StBu)-Pro-RRR-Ala-OH/25%DMA水溶液(5.0μL)、10mM4-ペンチル安息香酸/25%DMA水溶液(5.0μL、内部標準として使用)、100mMTCEP/50mMHEPES緩衝液溶液(pH7.6、5.0μL)を混合させ、システイン残基側鎖保護基を素早く脱保護し、窒素雰囲気下、室温にて1時間静置した。続いて、翻訳用緩衝液(9.5μL)、PURESYSTEM(登録商標) classic II Sol. B(バイオコゥマー社、製品番号PURE2048C)(10μL)、50mM1,2-ジチアン-4,5-ジオール水溶液(10.5μL)、500mM2-アミノエタンチオール/50mMHEPES緩衝液溶液(pH7.6、5.0μL)を混合し、さらに1N水酸化ナトリウム水溶液(1.0μL)を混合して反応液をpH7.8に調整して、窒素雰囲気下、37度にて静置した。なお翻訳用緩衝液の成分の反応溶液中での濃度はそれぞれ次のとおりである。2mM GTP,2mM ATP,1mM CTP,1mM UTP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,14mM 酢酸マグネシウム,2mMスペルミジン,1mM ジチオスレイトール,0.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),アミノ酸(Ala, Cys, Phe, Gly, His, Ile, Leu, Met, Pro, Ser, Thr, Val, Trpがそれぞれ0.3mM)。反応の進行状況はLCMS測定によって確認した。

10mM Ac-Trp-Cys(StBu)-Pro-RRR-Ala-OH/25%DMA水溶液(5.0μL)、10mM4-ペンチル安息香酸/25%DMA水溶液(5.0μL、内部標準として使用)、100mMTCEP/50mMHEPES緩衝液溶液(pH7.8、5.0μL)を混合させ、システイン残基側鎖保護基を素早く脱保護し、窒素雰囲気下、室温にて1時間静置した。続いて、100mMBicine緩衝液(pH8.9、19.5μL)、50mM1,2-ジチアン-4,5-ジオール/100mMBicine緩衝液溶液(pH8.9、10.5μL)、500mM2-アミノエタンチオール/100mMBicine緩衝液溶液(pH8.5、5.0μL)を混合し、さらに0.5N水酸化ナトリウム水溶液(1.0μL)を混合して反応液をpH8.5に調整して、窒素雰囲気下、37度もしくは57度にて静置した。反応の進行状況はLCMS測定によって確認した。
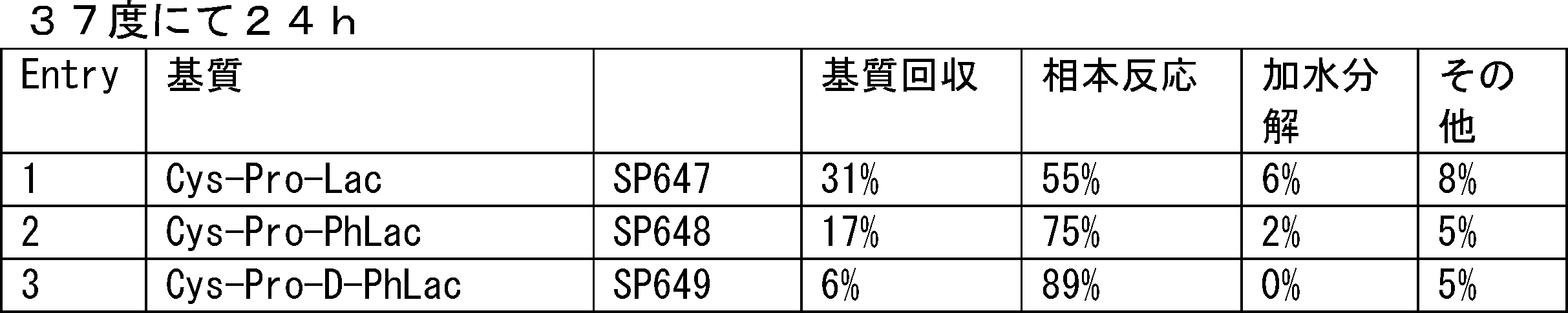
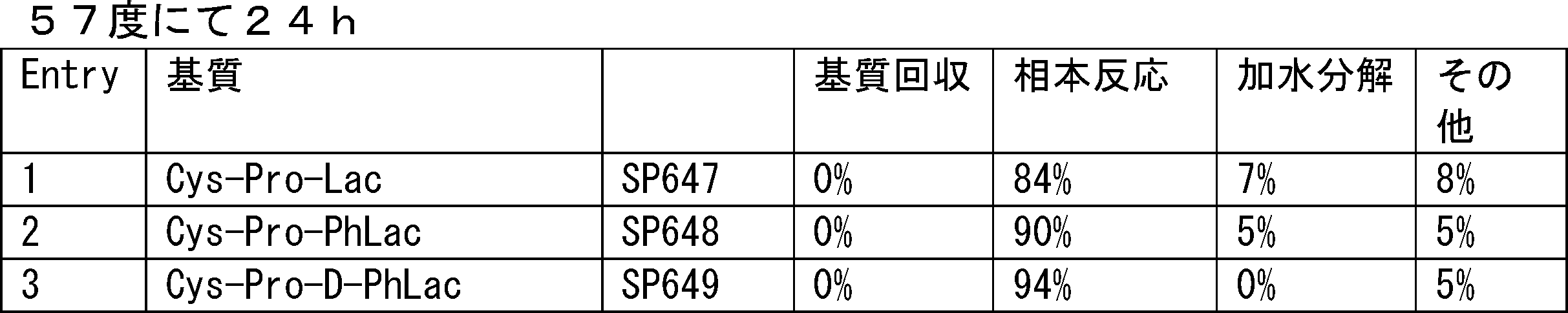
10mM Ac-Trp-Cys(StBu)-Pro-RRR-Ala-OH/25%DMA水溶液(5.0μL)、10mM4-ペンチル安息香酸/25%DMA水溶液(5.0μL、内部標準として使用)、100mMTCEP/50mMHEPES緩衝液溶液(pH7.8、5.0μL)を混合させ、システイン残基側鎖保護基を素早く脱保護し、窒素雰囲気下、室温にて1時間静置した。続いて、100mMBicine緩衝液(pH8.9、19.5μL)、50mM1,2-ジチアン-4,5-ジオール/100mMBicine緩衝液溶液(pH8.9、10.5μL)、500mM2-アミノエタンチオール/100mMBicine緩衝液溶液(pH8.5、5.0μL)を混合し、さらに0.5N水酸化ナトリウム水溶液(2.0μL)を混合して反応液をpH9.0に調整して、窒素雰囲気下、37度もしくは57度にて静置した。反応の進行状況はLCMS測定によって確認した。


5-3-1.翻訳ペプチドモデル化合物SP655の合成
以下のスキームに従い、1回目の環化としてN末端にCysを用い、C末端側にAsp(SBn)を用いてアミド化環化反応を行い、2回目の分枝化として、活性チオエステル発生パートとしてCys-Pro-Lacを有し、Lysの側鎖アミノ基にN原子とS原子を保護させたCysを配置させた化合物のS原子とN原子を脱保護してアミド化反応を行う例を実施する目的で、以下のモデル化合物SP655の合成を行った。

本明細書では、Lysの側鎖のアミノ基とAcbz-Thzの有するカルボキシル基とでアミド結合した化合物をLys(Acbz-Thz)と表記する。
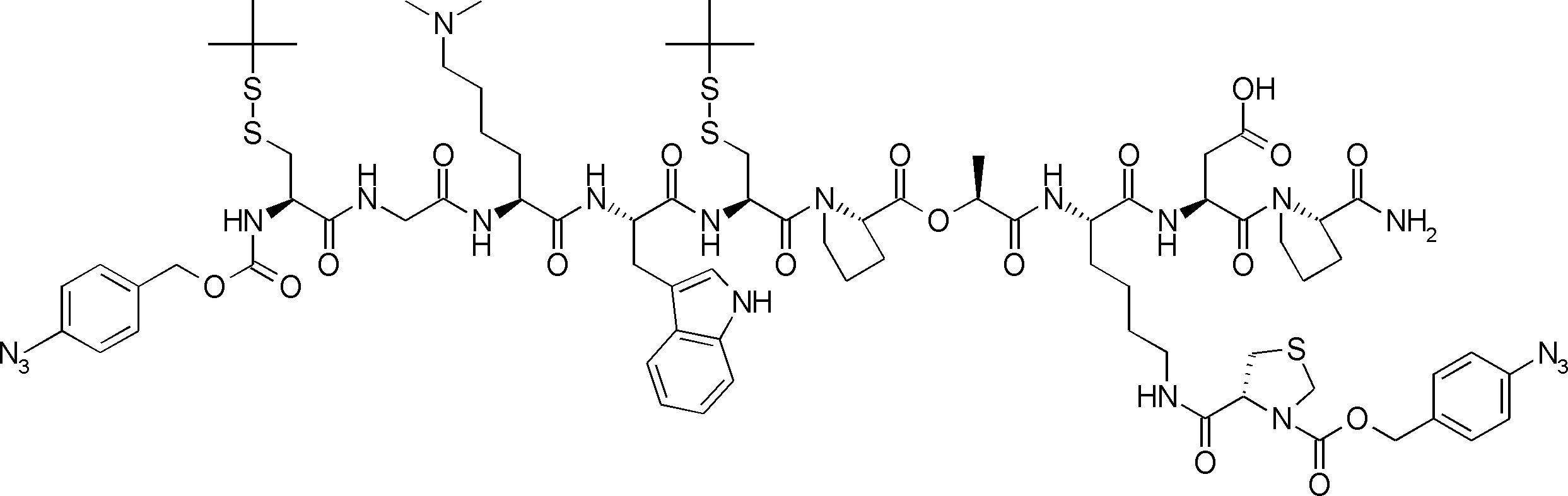
ペプチドの伸長後、レジンをジメチルホルムアミド(DMF)、ジクロロメタン(DCM)で洗浄した。レジンに2%トリフルオロ酢酸(TFA)のジクロロメタン(DCM)/2,2,2-トリフルオロエタノール(TFE)(=1/1、v/v、4.0ml)溶液を加えて室温にて3時間反応させ、レジンからのペプチドの切り出しを行った。反応終了後、チューブ内溶液を合成用カラムでろ過することによりレジンを除き、レジンをジクロロメタン(DCM)/2,2,2-トリフルオロエタノール(TFE)(=1/1,v/v、4.0mL)にて4回洗浄した。得られた溶液を減圧濃縮し、残渣を逆相シリカゲルクロマトグラフィー(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)にて精製し、(S)-3-((S)-2-((S)-2-((S)-1-((6R,12S,15S,18R)-15-((1H-インドール-3-イル)メチル)-6-((4-アジドベンジルオキシ)カルボニルアミノ)-18-((tert-ブチルジスルファニル)メチル)-12-(4-(ジメチルアミノ)ブチル)-2,2-ジメチル-7,10,13,16-テトラオキソ-3,4-ジチア-8,11,14,17-テトラアザノナデカン)ピロリジン-2-カルボニルオキシ)プロパンアミド)-6-((R)-3-((4-アジドベンジルオキシ)カルボニル)チアゾリジン-4-カルボキサミド)ヘキサンアミド)-4-((S)-2-カルバモイルピロリジン-1-イル)-4-オキソブタン酸(Acbz-Cys(StBu)-Gly-Lys(Me2)-Trp-Cys(StBu)-Pro-Lac-Lys(Acbz-Thz)-Asp-Pro-NH2)(化合物SP681)(184.6mg、24%)を得た。
LCMS(ESI) m/z = 1773.9 (M+H)+
保持時間:0.76分(分析条件SQDFA05)

LCMS(ESI) m/z = 1880.0 (M+H)+
保持時間:0.80分(分析条件SQDFA05)
Cys-Pro-Lacユニットでは、1回目の環化反応中、反応補助基の有無にかかわらず安定に存在することが可能であり、また2回目の分枝化反応では高い選択性で望みの分枝化反応が実現できることが明らかとなったため、モデルペプチドにて分枝化反応を翻訳液中にて確認した。
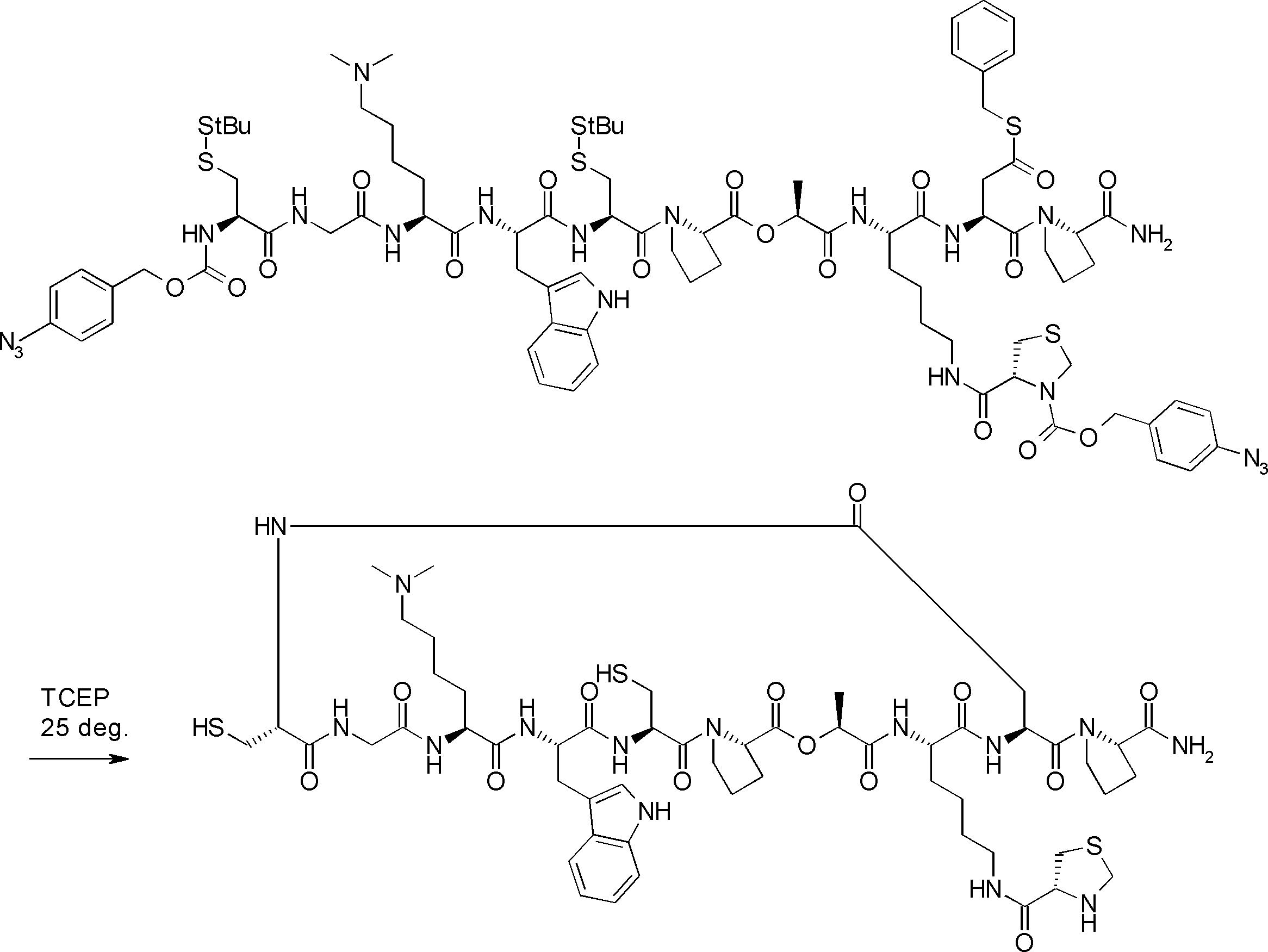
LCMS(ESI) m/z = 1227 (M-H)-
保持時間:0.76分(分析条件SQDAA05)
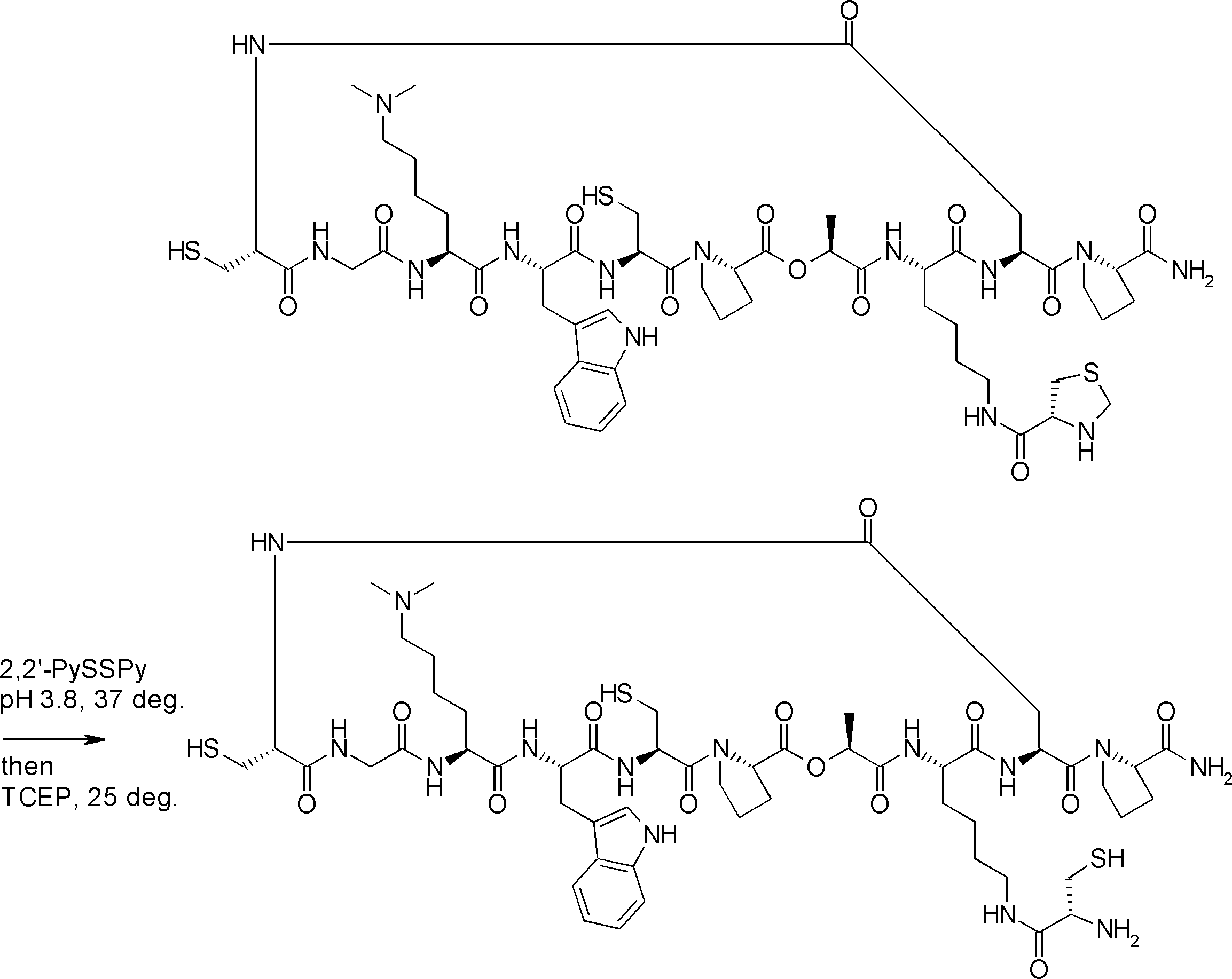
LCMS(ESI) m/z = 1215 (M-H)-(図69)
保持時間:0.73分(分析条件SQDAA05)

LCMS(ESI) m/z = 1017 (M+H)+
保持時間:0.65分(分析条件SQDAA05)(図70)
2回目の分枝化のコンセプト証明のための別実験である。1回目の環化が終了済であることを想定している。従って、Aspの側鎖のカルボン酸とN末端のアミノ基によるアミド環化を実施せず、主鎖環化したモデル化合物とした。2回目の分枝化にてCys-Pro-HOGlyから活性エステルを発生させ、その前後にて保護されたアミンを脱保護させて分枝化させるモデル反応性を以下のように実施した。
以下のスキームに従い、合成を行った。
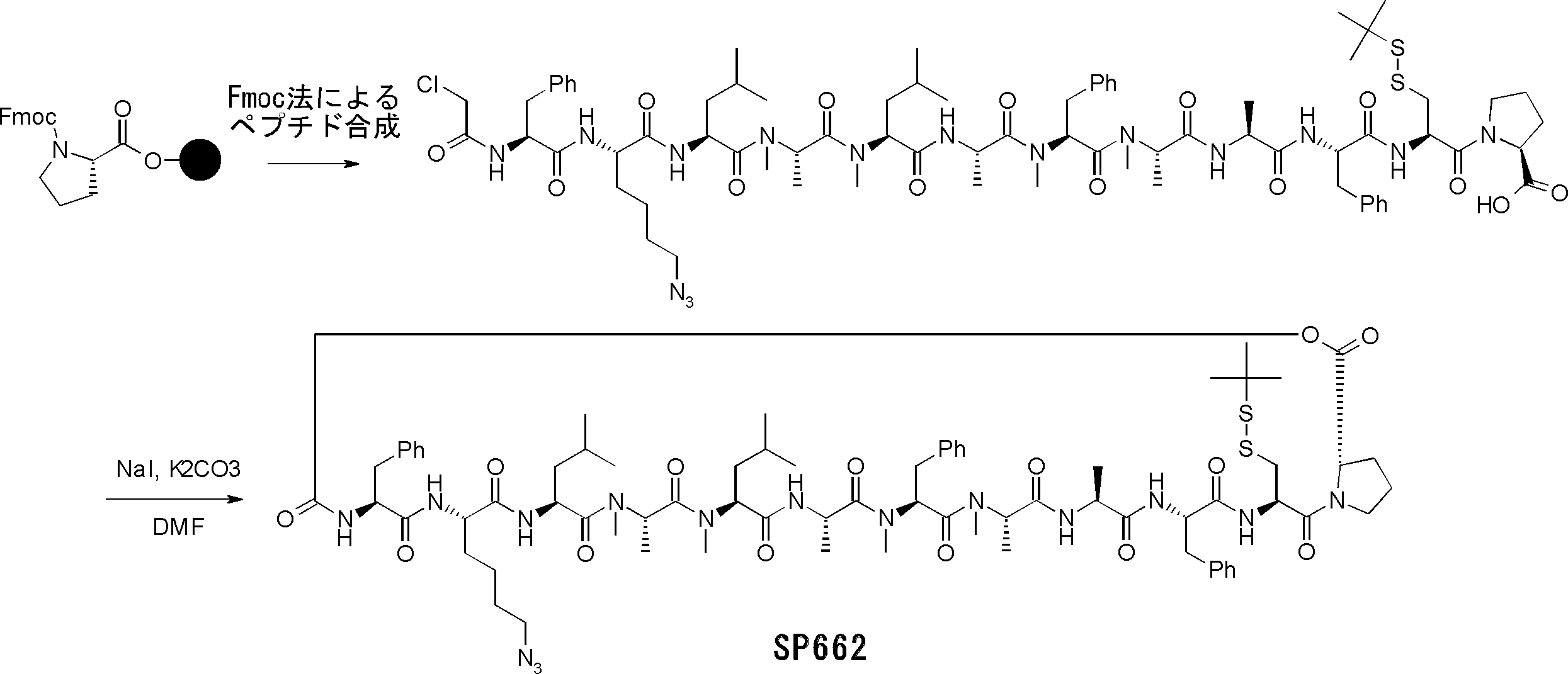
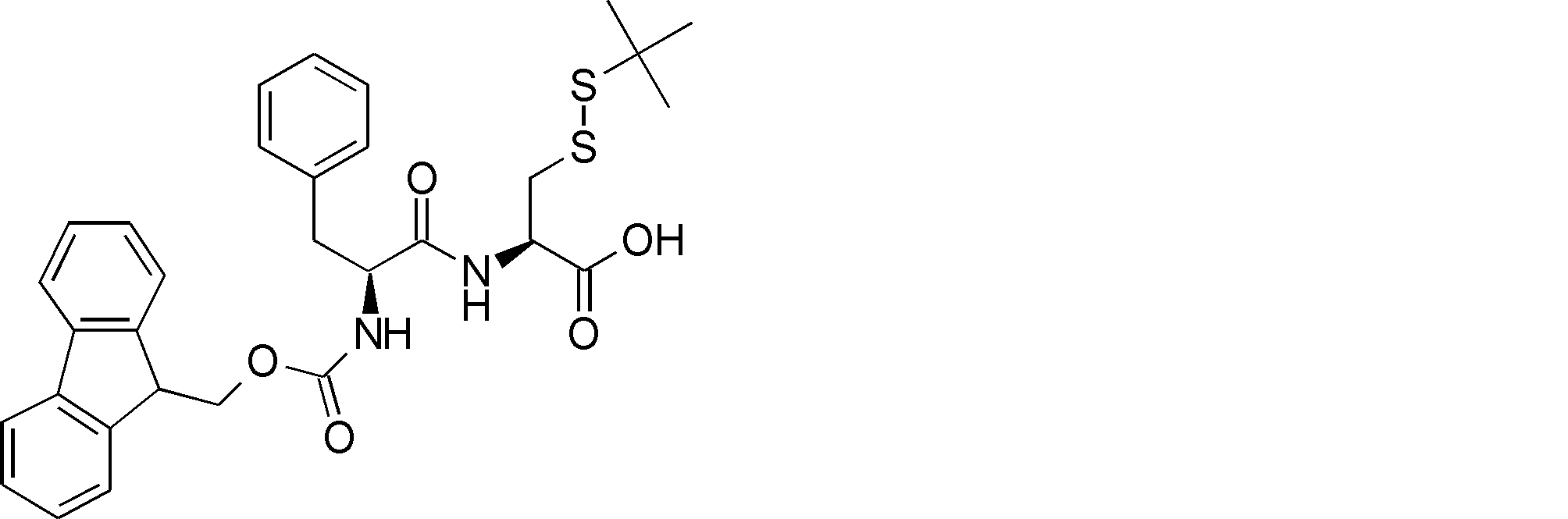
LCMS(ESI) m/z = 579 (M+H)+
保持時間:0.97分(分析条件SQD FA05)
(HOGlyはグリコール酸を示す)
用語の定義
Fmoc-Lys(N3)-OH: (S)-2-(((9H-フルオレン-9-イル)メトキシ)カルボニルアミノ)-6-アジドヘキサン酸
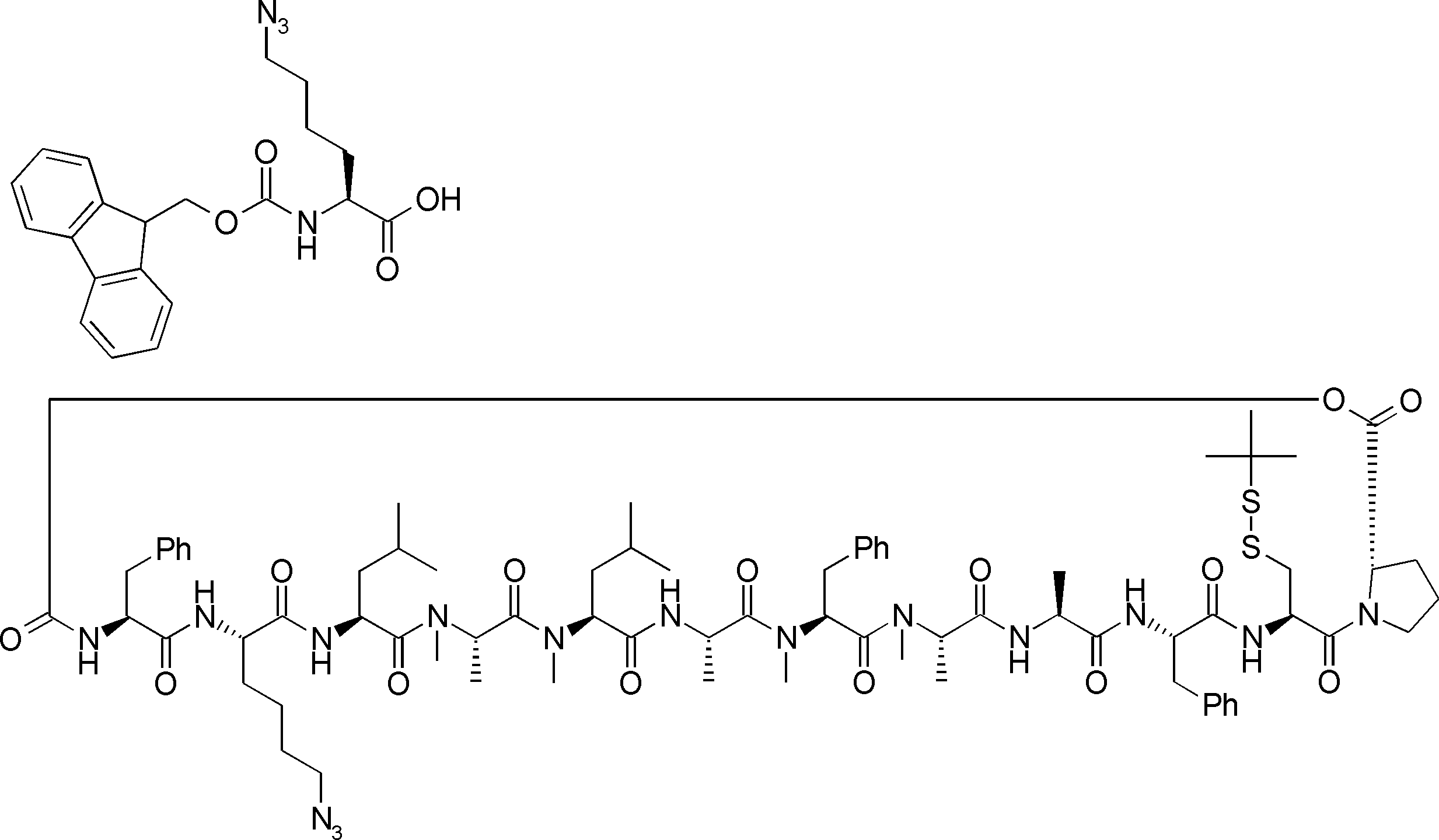
LCMS(ESI) m/z = 1509 (M+H)+
保持時間:0.63分(分析条件SQD FA50)
(S)-N-((3S,6S,9S,12S,15S,18S,21S,24S,27S)-3,12-ジベンジル-18,24-ジイソブチル-6,9,10,13,15,19,21,22-オクタメチル-2,5,8,11,14,17,20,23,26-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロヘントリアコンタン-27-イル)-2-(2-ヒドロキシアセトアミド)-3-フェニルプロパンアミド(化合物SP664)の合成
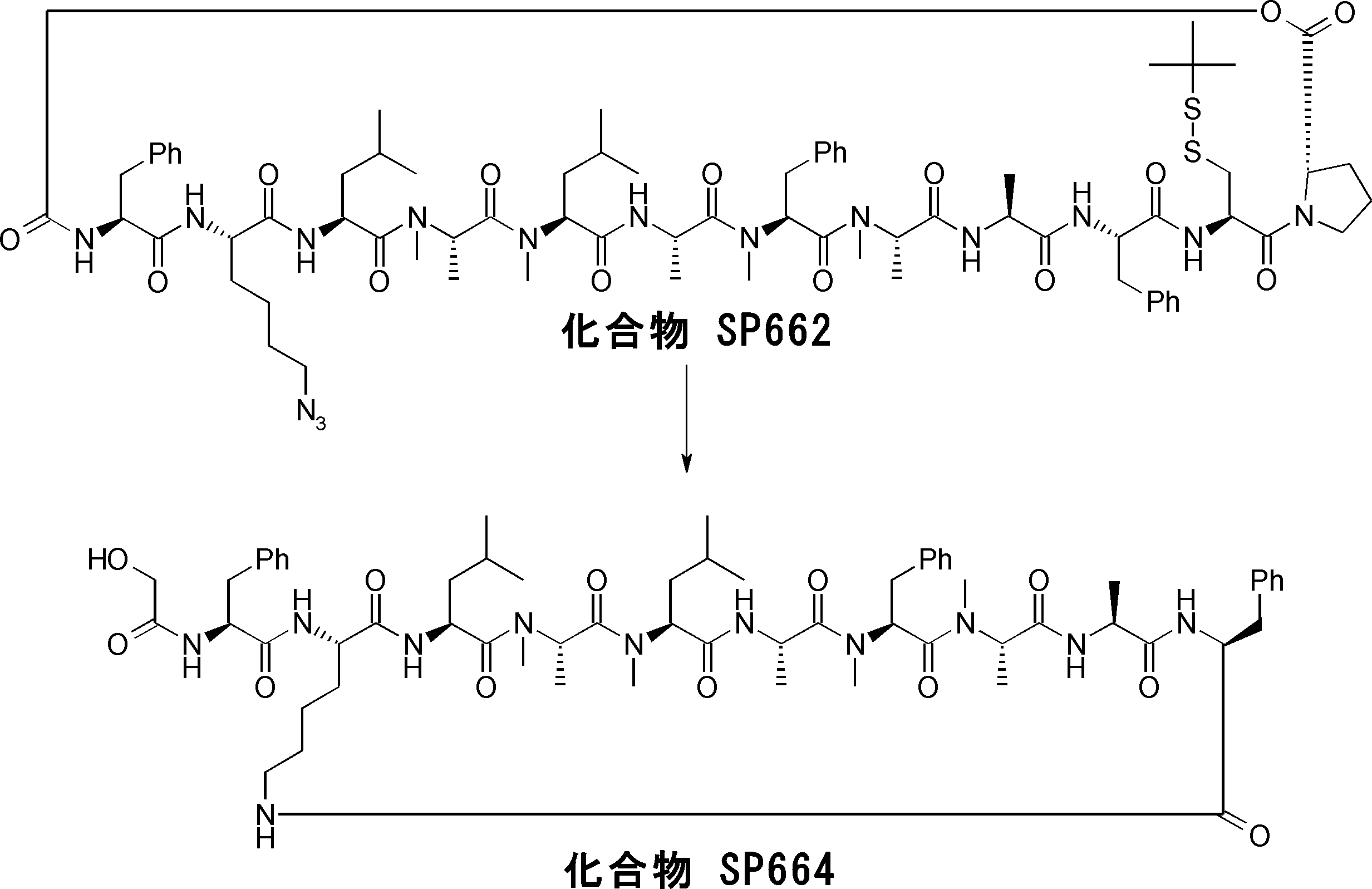
0.5M HEPES緩衝液(pH7.0、 60μl)と1,3-ジメチル-2-イミダゾリジノン(DMI)(40μl)の混合溶液に、TCEP塩酸塩(トリス(2-カルボキシエチル)ホスフィン塩酸塩)(2.9mg、 0.010mmol)、2-(4-メルカプトフェニル)酢酸(1.7mg、 0.010mmol)を加えた。さらにこの溶液に2N水酸化ナトリウム水溶液(35μl)と水(45μl)を加えてpH8.5に調整し、室温で5分攪拌した。
その後、(6S,9S,12S,15S,18S,21S,24S,27S,30S,33S,36R,41aS)-9-(4-アジドブチル)-6,24,33-トリベンジル-36-((tert-ブチルジスルファニル)メチル)-12,18-ジイソブチル-14,15,17,21,23,26,27,30-オクタメチルヘキサコサヒドロピロロ[2,1-c][1,4,7,10,13,16,19,22,25,28,31,34,37]オキサドデカアザシクロノナトリアコンチン-1,4,7,10,13,16,19,22,25,28,31,34,37(3H)-トリデカオン(c(HOGly-Phe-Lys(N3)-Leu-MeAla-MeLeu-Ala-MePhe-MeAla-Ala-Phe-Cys(StBu)-Pro)) (化合物SP662)の0.01M 1,3-ジメチル-2-イミダゾリジノン(DMI)溶液(20μl、 0.2μmol)を加え、反応液を30℃で24時間攪拌した。LCMSを用いて反応の変化を観測したところ、24時間で(S)-N-((3S,6S,9S,12S,15S,18S,21S,24S,27S)-3,12-ジベンジル-18,24-ジイソブチル-6,9,10,13,15,19,21,22-オクタメチル-2,5,8,11,14,17,20,23,26-ノナオキソ-1,4,7,10,13,16,19,22,25-ノナアザシクロヘントリアコンタン-27-イル)-2-(2-ヒドロキシアセトアミド)-3-フェニルプロパンアミド(化合物SP664)が生成していることを確認した。加水分解物と化合物SP664との生成比は、LCMSのUVエリア比で12:88であった。(図71、加水分解化合物 保持時間:0.62分)
LCMS(ESI) m/z = 1195 (M+H)+
保持時間:0.77分(分析条件SQD FA05)
コンセプト証明のための別実験である。1回目の環化反応終了後を想定した。従ってAspの側鎖カルボン酸とN末端アミンとの環化ではなく、C末端Alaのカルボン酸とN末端アミンとをアミド環化させた。2回目の分枝化のためのアミノ基の保護基もディスプレーライブラリーでの使用を想定したものではない。反応補助基を有するエステルからの分枝化反応が進行することを確認することを目的としたコンセプト証明のためのモデル実験である。
以下のスキームに従い、合成を行った。

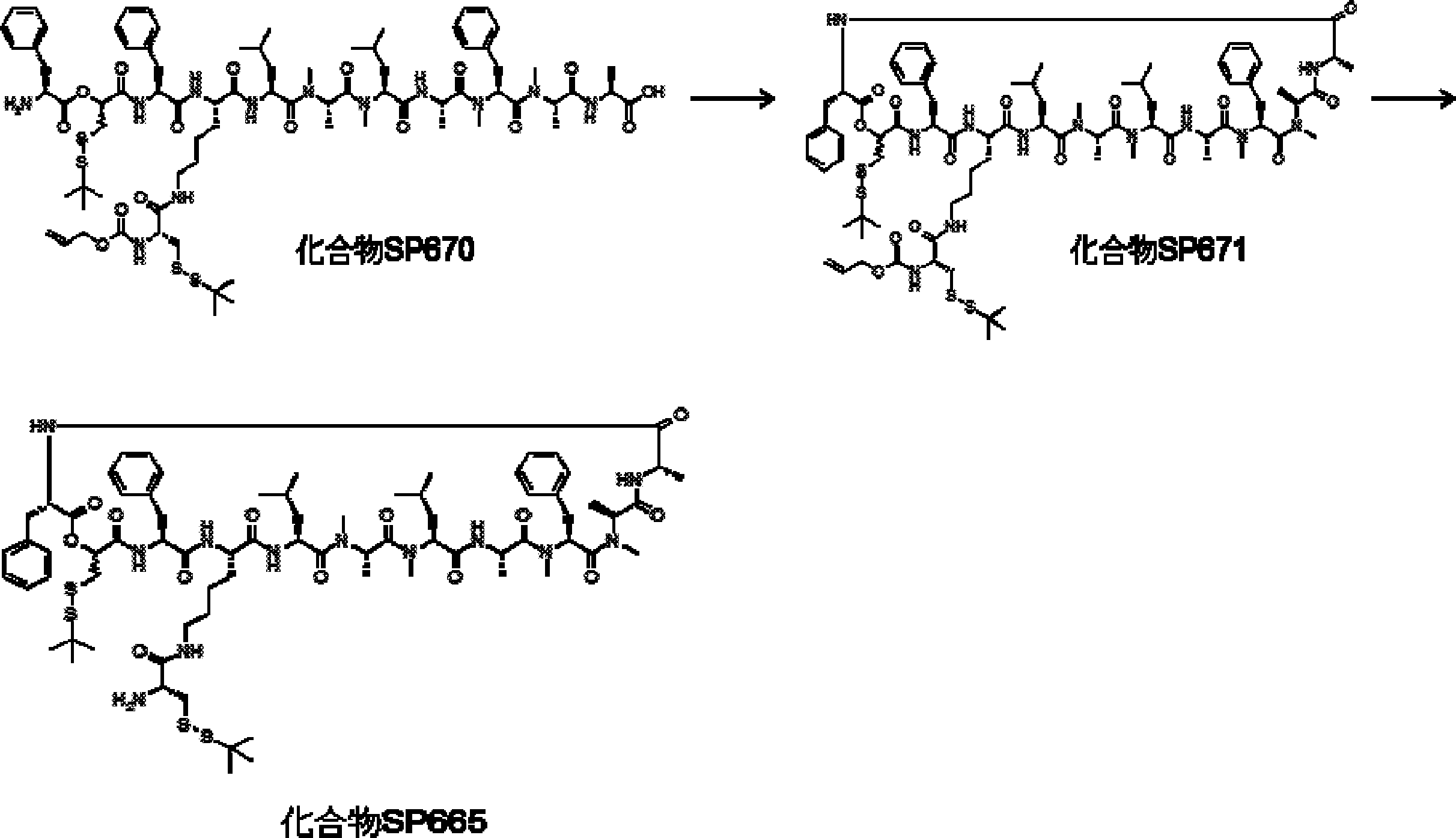

なお、本明細書中では、化合物SP666のヒドロキシル基上の水素原子とカルボン酸上のヒドロキシル基を除いた2価の構造をtBuSSlacとする。
LCMS(ESI) m/z = 209 (M―H)―
保持時間:0.60分(分析条件SQDFA05)

LCMS(ESI) m/z = 580.5 (M+H)+
保持時間:1.05分(分析条件SQDFA05)
なお、本明細書中では、他の例と同様H-Lys-OHの側鎖アミノ基とAlloc-Cys(StBu)-OHのカルボキシル基とがアミド結合した化合物をH-Lys(Alloc-Cys(StBu))-OHと記載する。
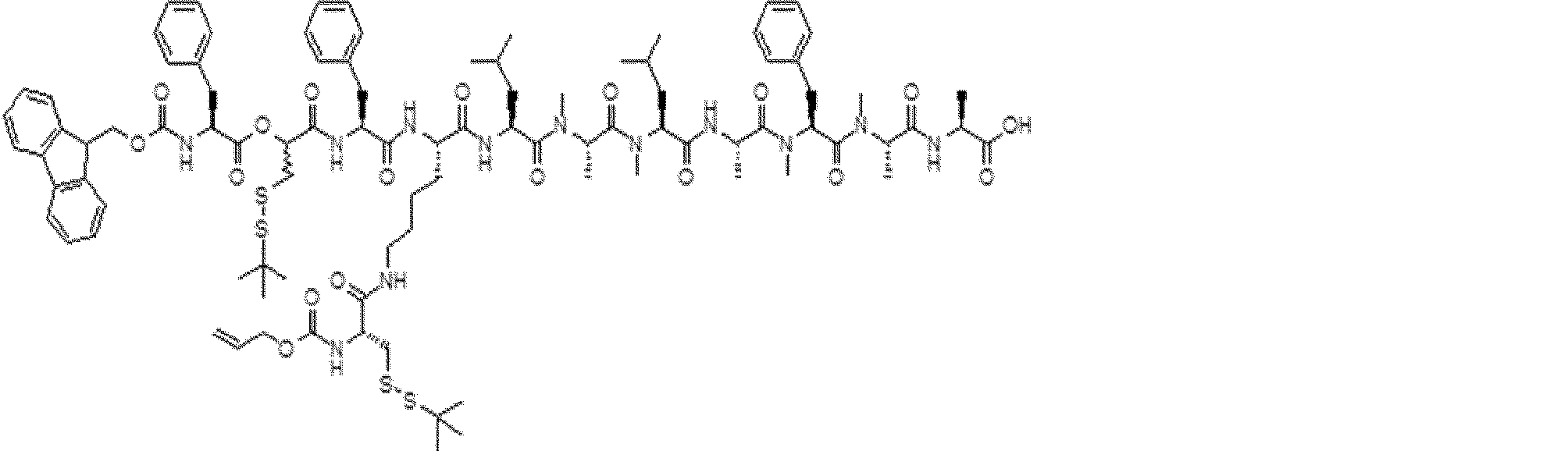
LCMS(ESI) m/z = 1844.6 (M+H)+
保持時間:0.84分(分析条件SQDAA50)
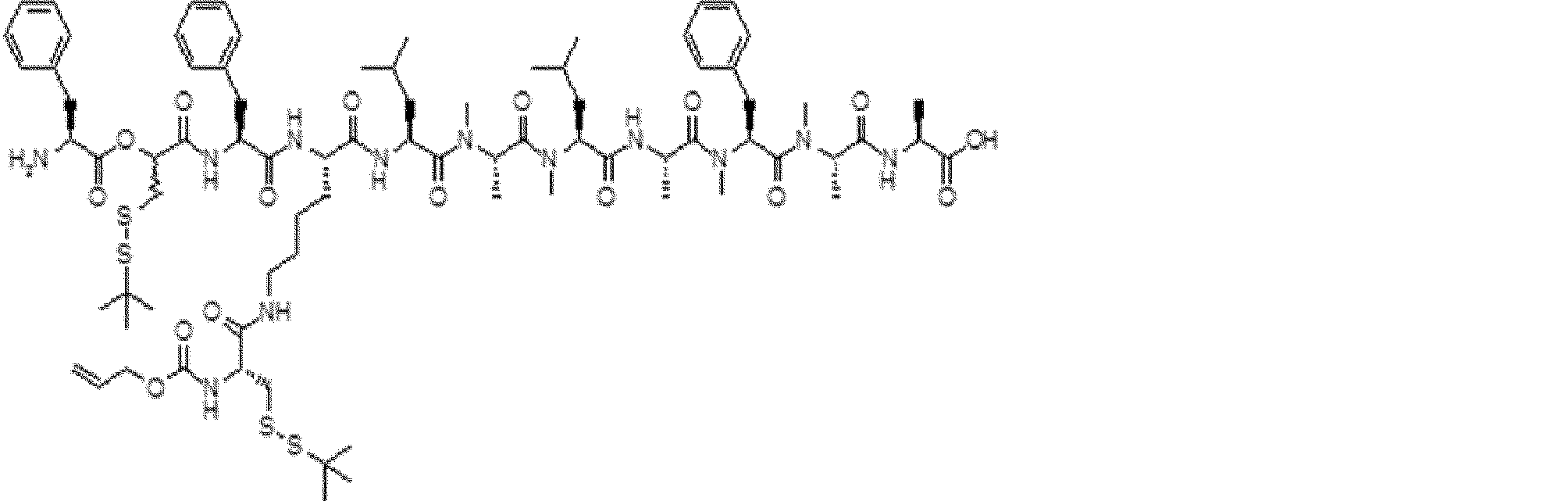
LCMS(ESI) m/z = 1622 (M+H)+
保持時間:0.30分、0.34分(分析条件SQDFA50)
tBuSSlac部位が光学異性体混合物であるため、化合物としてジアステレオマーが存在し、2本のピークを観測した。
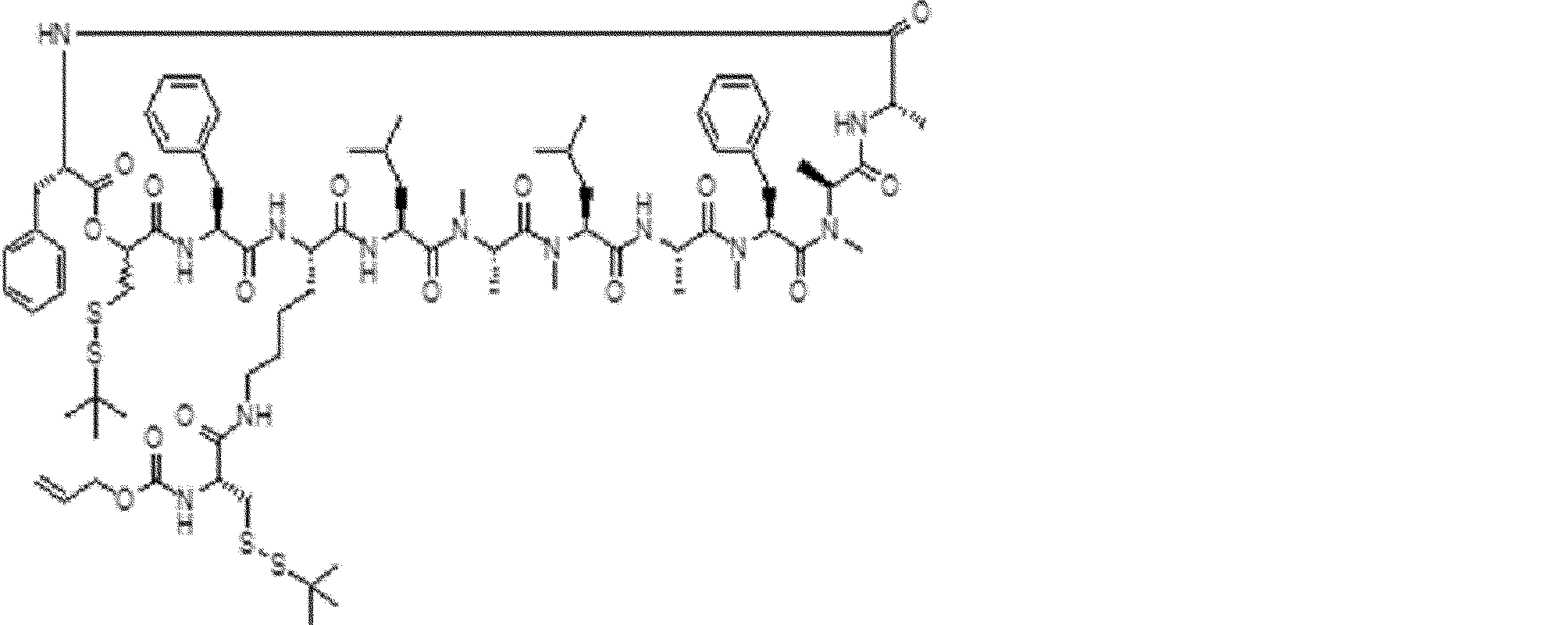
LCMS(ESI) m/z = 1604.5 (M+H)+
保持時間:0.79分(分析条件SQDFA50)
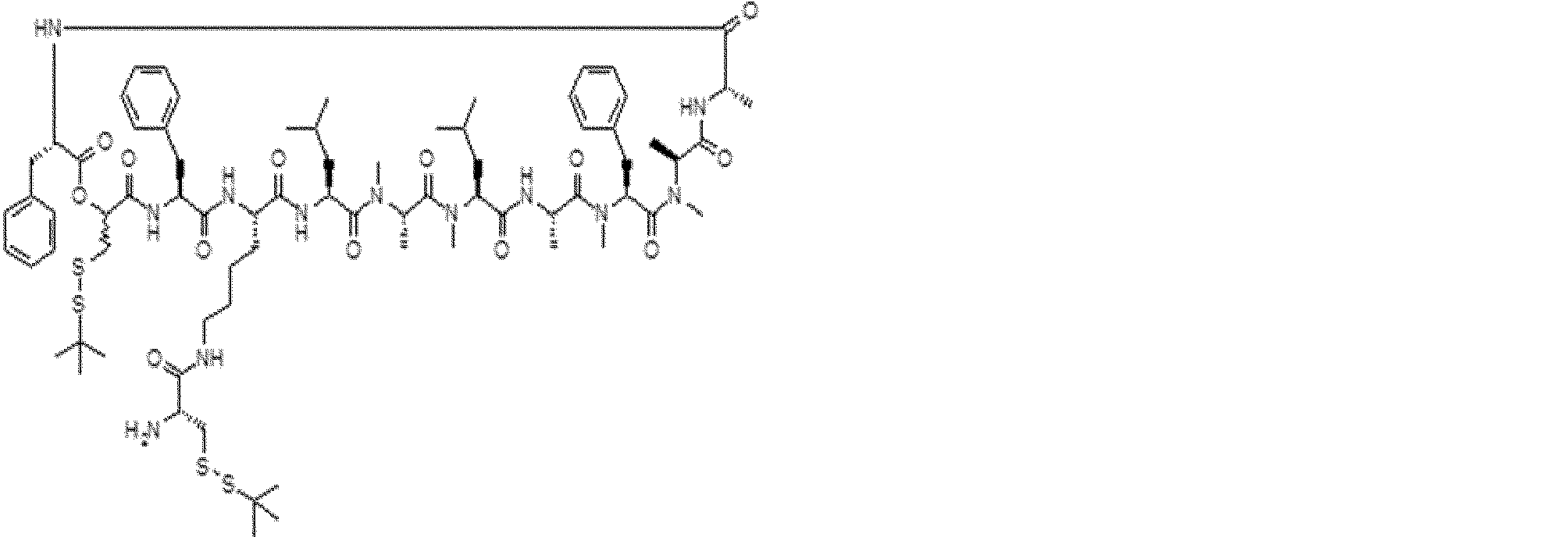
LCMS(ESI) m/z = 1520 (M+H)+
保持時間:0.83分(分析条件SQDFA05)
(2S)-N-((3R,6S,9S,12S,15S,18S,21S,24S,27S,30S)-6,15-ジベンジル-21,27-ジイソブチル-3-(メルカプトメチル)-9,12,13,16,18,22,24,25-オクタメチル-2,5,8,11,14,17,20,23,26,29-デカオキソ-1,4,7,10,13,16,19,22,25,28-デカアザシクロテトラトリアコンタン-30-イル)-2-(2-ヒドロキシ-3-メルカプトプロパンアミド)-3-フェニルプロパンアミド(H-HSlac-Phe-Lys(*Cys)-Leu-MeAla-MeLeu-Ala-MePhe-MeAla-Ala-Phe*、2つの*部位にて環化)(化合物SP672)の合成
なお、本明細書では、HSlacとは化合物SP666の側鎖tBuS基が脱保護されたもので、ヒドロキシル基上の水素原子とカルボン酸上のヒドロキシル基を除いた2価の構造を意味する。
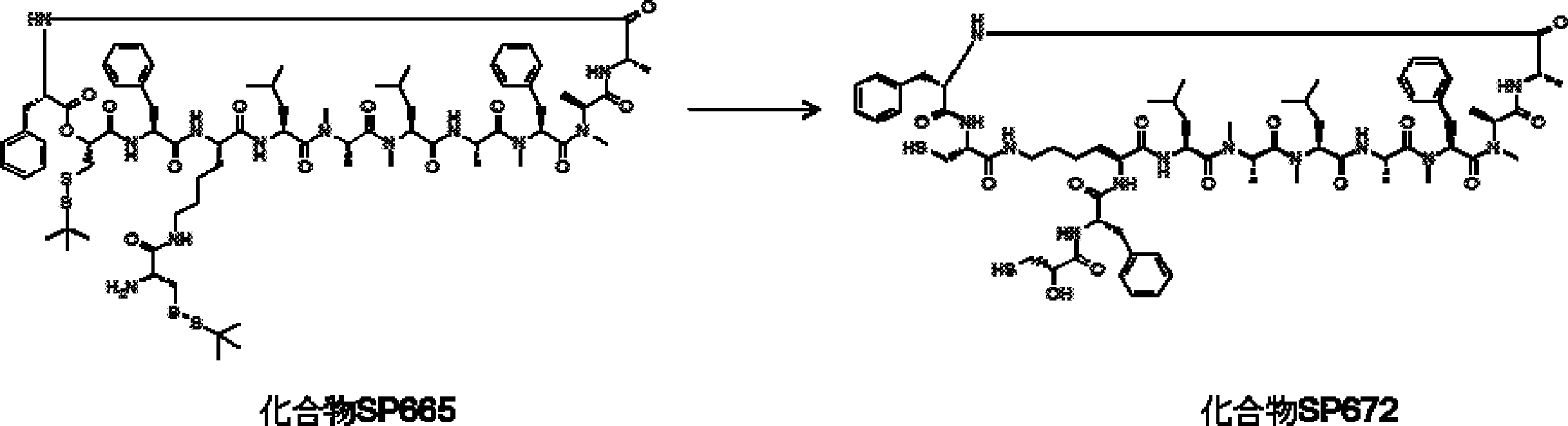
LCMS(ESI) m/z = 1342 (M―H)―
保持時間:0.81分(分析条件SQDFA05)
(2S)-N-((3S,6S,9S,12S,15S,18S,21S,24S,27S,30S)-6,15-ジベンジル-21,27-ジイソブチル-3,9,12,13,16,18,22,24,25-ノナメチル-2,5,8,11,14,17,20,23,26,29-デカオキソ-1,4,7,10,13,16,19,22,25,28-デカアザシクロテトラトリアコンタン-30-イル)-2-(2-ヒドロキシプロパンアミド)-3-フェニルプロパンアミド(H-lac-Phe-Lys(*Ala)-Leu-MeAla-MeLeu-Ala-MePhe-MeAla-Ala-Phe*、2つの*部位にて環化)(化合物SP673)の合成

670mM トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩溶液の調整法は以下の通りである。トリス(2-カルボキシエチル)ホスフィン(TCEP)塩酸塩(29mg、101μmol)に水(100μl)、トリエチルアミン(Et3N)(50μl)を加え、pH=7.1に調整した。
LCMS(ESI) m/z = 1280(M+H)+
保持時間:0.77分(分析条件SQDFA05)
8.ペプチドーRNA複合体を用いた分子内分枝ペプチド(直鎖部2)生成反応
RNA-ペプチド複合体において分子内分枝ペプチド反応を施し、生成物を電気泳動により分析した。
PCRにより調製したDNA(配列番号 DM-H1)を鋳型に、In vitro転写によりmRNA(配列番号RM-H2)を調整し、RNeasy mini kit (Qiagen社)を用いて精製した。10μMのmRNAに、15μMのピューロマイシンリンカー(Sigma社)(配列番号C-H1) 1X T4 RNAライゲースリアクションバッファー(NEB社)、1mM ATP、10% DMSO、0.63 unit/μl T4 RNAライゲース(NEB社)を加え、室温で30分間ライゲーション反応させた後、RNeasy MiniElutekit(Qiagen社)により精製した。次に、前述した翻訳系の鋳型RNAOT86b(配列番号RM-H1)の代わりに上記で調製した1 μMのmRNA-ピューロマイシンリンカー連結体を鋳型として用い、さらに0.25mM Glyを追加した翻訳反応液を調製し、37℃で60分間保温した後、続けて室温で12分間保温した。その後、反応液をRNeasy minelute(qiagen社)により精製し、ペプチド-RNA複合体分子を得た。
OT-104 DNA配列
GGCGTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATatgTGCACTACATGGTTCCGTTGGTGCCCACAGTTCAAGTGGCTTCCTCGTAGTGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
OT-104 RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUaugUGCACUACAUGGUUCCGUUGGUGCCCACAGUUCAAGUGGCUUCCUCGUAGUGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
[P]CCCGTCCCCGCCGCCC [Fluorecein-dT][Spacer18][Spacer18][Spacer18][Spacer18][Spacer18]CC[Puromycin] ([P]:5'リン酸化)
上記で調製したペプチドーRNA複合体溶液100μLに対して100μLのN, N‐ジメチルアセトアミド(DMA)、5.5μLの2M HEPES-KOH(pH7.6)、2.1μLの1M トリス(2-カルボキシエチル)ホスフィン(TCEP)(pH7.6)を加え、37℃で2時間保温した。続けて848μLの試薬溶液(59mMトリス(2-カルボキシエチル)ホスフィン塩酸塩、425mM N,N-ビス-(2-ヒドロキシエチル)グリシン(Bicine)(pH8.7)、590mM 水酸化ナトリウム、47%(v/v)N,N-ジメチルアセトアミド(DMA)、593mM 4-(トリフルオロメチル)ベンゼンチオール)を反応溶液に加え、37℃でさらに20時間保温した。得られた反応液から、RNeasy minelute (qiagen社)を用いてペプチド-RNA複合体を精製し、純水100μLにてカラムから溶出した。続けて、12μLの10xRNase ONE ribonuclease反応バッファー(promega 社)、6μLのRNase ONEribonuclease (promega社)、6μLのRNase H (Life Technologies社)を加え、室温にて3日間保温した。得られた反応溶液と何も反応させていないピューロマイシンリンカー(配列番号C-H1)をpeptide-PAGE mini(TEFCO社)にて電気泳動し、ピューロマイシンリンカーに由来するFluoreceinにてバンドを可視化した(図74)。その結果、分枝ペプチド(直鎖部2) 生成反応を行ったサンプルに関して、ピューロマイシンリンカーにペプチドが 結合したことに由来するバンド移動度の差が観測された(図74、レーン2、バンドI)。このことから、目的の分子内分枝ペプチド-RNA複合体の存在が示された。
なお、本明細書では、アミノ酸あるいはアミノ酸誘導体、アミノ酸類縁体の主鎖カルボン酸とpdCpAのヒドロキシル基(2位あるいは3位)とエステル結合を形成した化合物をアミノ酸-pdCpAと記載する。アミノ酸、アミノ酸誘導体、アミノ酸類縁体部位は略称で示しており、各略称は以下のように定義する。
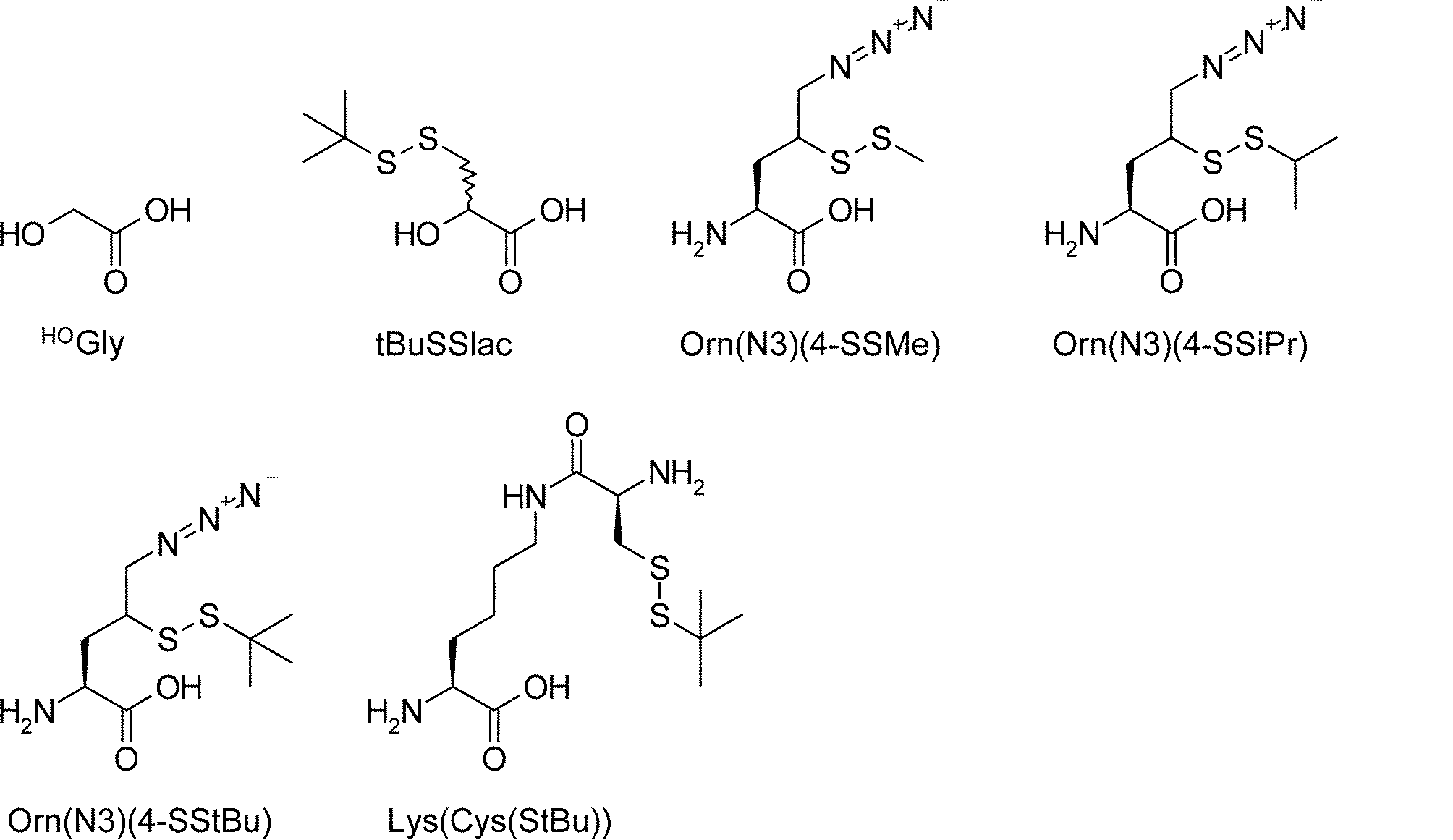
化合物21
2-(tert-ブチルジメチルシリルオキシ)酢酸シアノメチルの合成
粗生成物(1.53g)にアセトニトリル(6.7ml)とDMF(6.7ml)を加えて、混合物を0度に冷却した後に、ジイソプロピルエチルアミン(3.51mL、20.1mmol)と2-ブロモアセトニトリル(0.93ml、13.4mmol)を加え、反応液を室温で6時間攪拌した。反応混合物をジエチルエーテル/水で抽出操作を行い、有機層を25wt%食塩水で洗浄した。有機層を無水硫酸ナトリウムで乾燥させ、ろ過後、減圧濃縮した。得られた残渣を順相シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)にて精製し、2-(tert-ブチルジメチルシリルオキシ)酢酸シアノメチル(化合物21)(556mg、52%)を得た。
LCMS(ESI) m/z = 230 (M+H)+
保持時間:0.54分(分析条件SQDAA50)
2-(tert-ブチルジメチルシリルオキシ)酢酸 (2R,3S,4R,5R)-2-((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-(ホスホノオキシメチル)テトラヒドロフラン-3-イルオキシ)(ヒドロキシ)ホスホリルオキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イルの合成

LCMS(ESI) m/z = 809.5 (M+H)+
保持時間:0.52分(分析条件SQDFA05)
2-ヒドロキシ酢酸 (2R,3S,4R,5R)-2-((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-(ホスホノオキシメチル)テトラヒドロフラン-3-イルオキシ)(ヒドロキシ)ホスホリルオキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イル( HO Gly-pdCpA)の合成
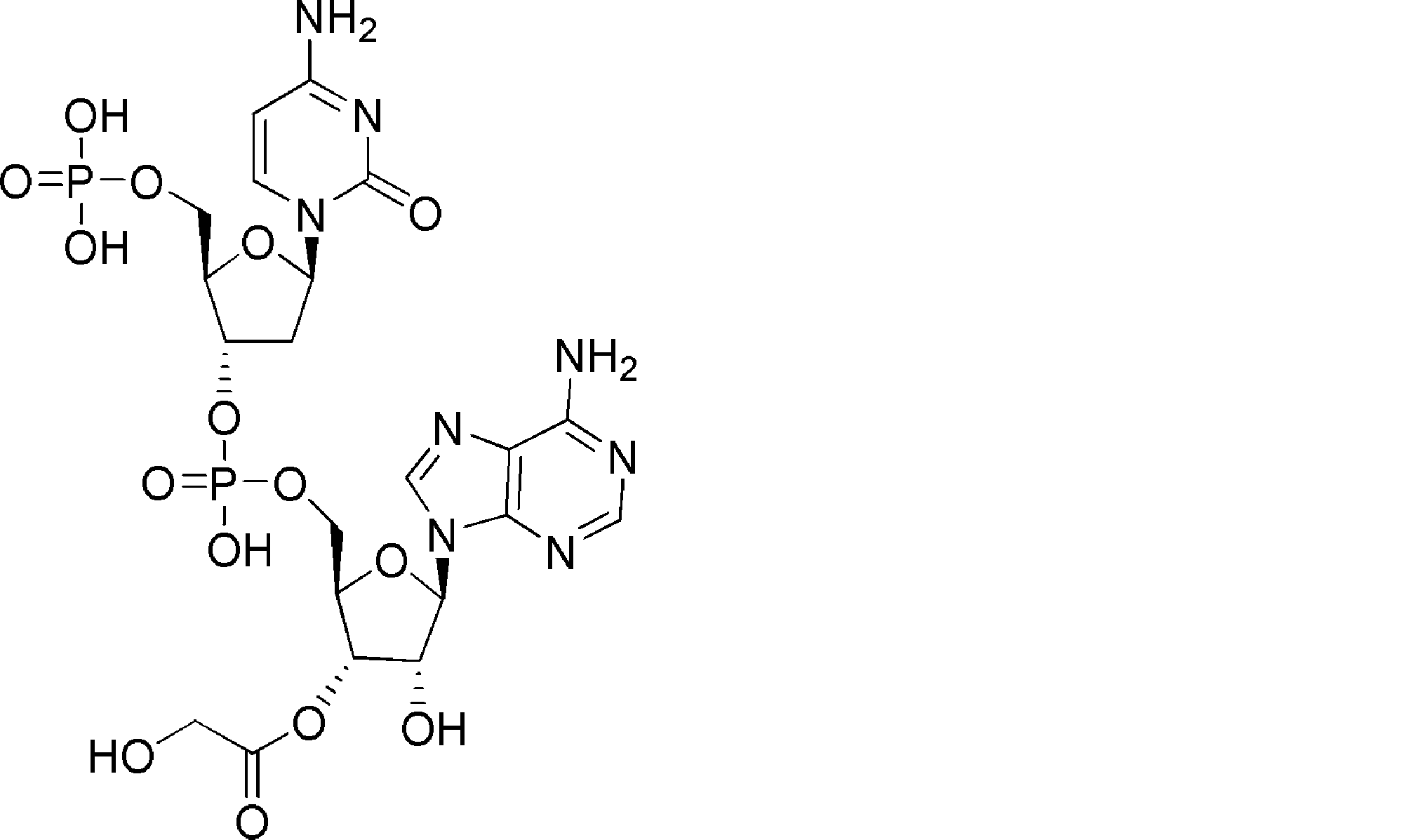
LCMS(ESI) m/z = 695 (M+H)+
保持時間:0.28分(分析条件SQDAA05)
以下のスキームに従い、合成を行った。


LCMS(ESI) m/z = 364 (M+H)+
保持時間:0.84分(分析条件SQDAA50)

LCMS(ESI) m/z = 829 (M+H)+
保持時間:0.41分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。


LCMS(ESI) m/z = 294 (M+Na)+
保持時間:1.05分(分析条件SQDAA05)

LCMS(ESI) m/z = 310 (M+Na)+
保持時間:1.03分(分析条件SQDAA05)

LCMS(ESI) m/z = 326 (M+Na)+
保持時間:0.59分(分析条件SQDAA50)

LCMS(ESI) m/z = 430 (M+H)+
保持時間:0.74分(分析条件SQDAA50)

LCMS(ESI) m/z = 415 (M+Na)+
保持時間:0.80分(分析条件SQDAA50)

LCMS(ESI) m/z = 237 (M+H)+
保持時間:0.36分(分析条件SQDFA05)

LCMS(ESI) m/z = 335 (M-H)-
保持時間:0.78分(分析条件SQDFA05)

LCMS(ESI) m/z = 374 (M-H)-
保持時間:0.87分(分析条件SQDFA05)
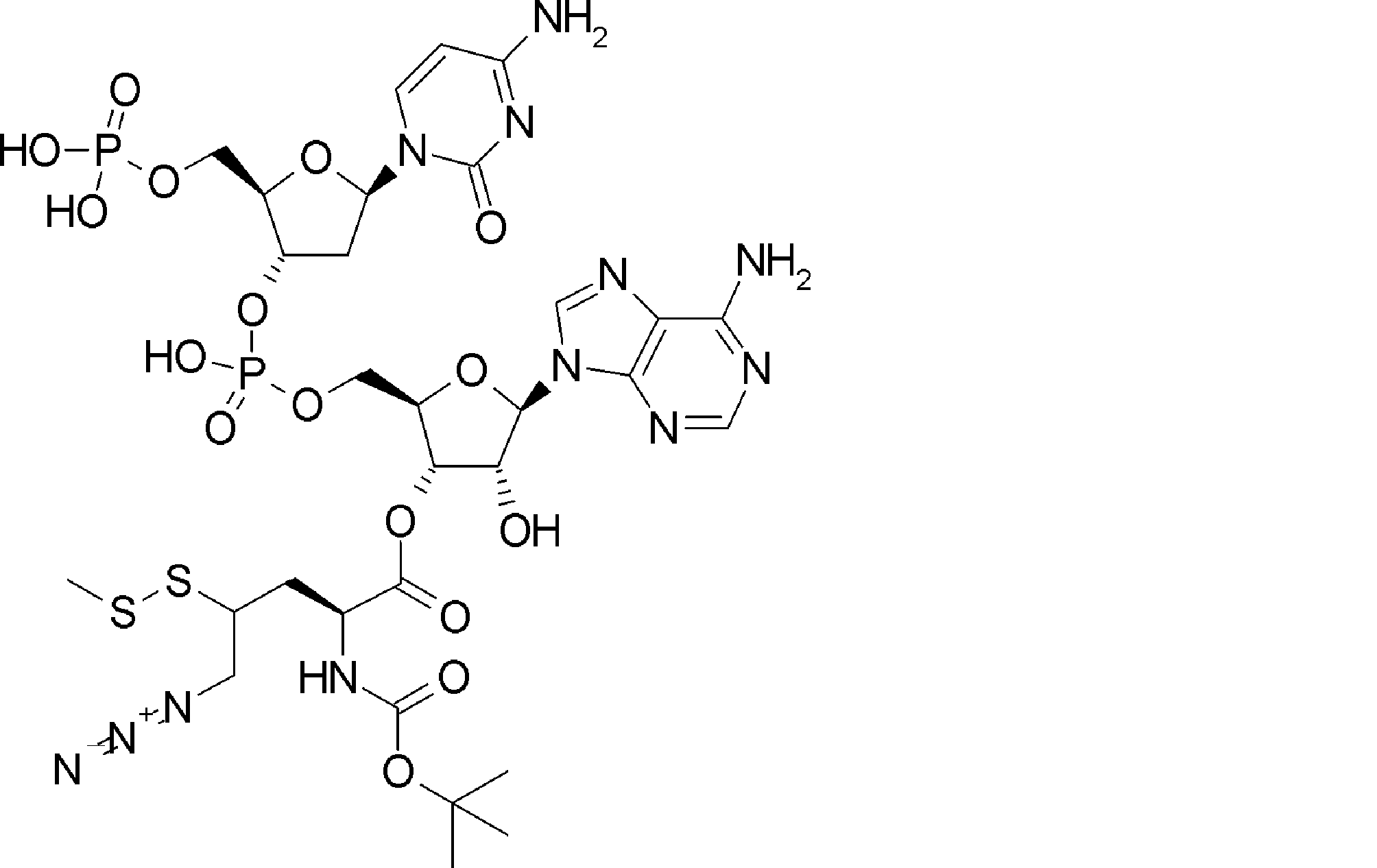
LCMS(ESI) m/z = 955 (M+H)+
保持時間:0.55分(分析条件SQDFA05)
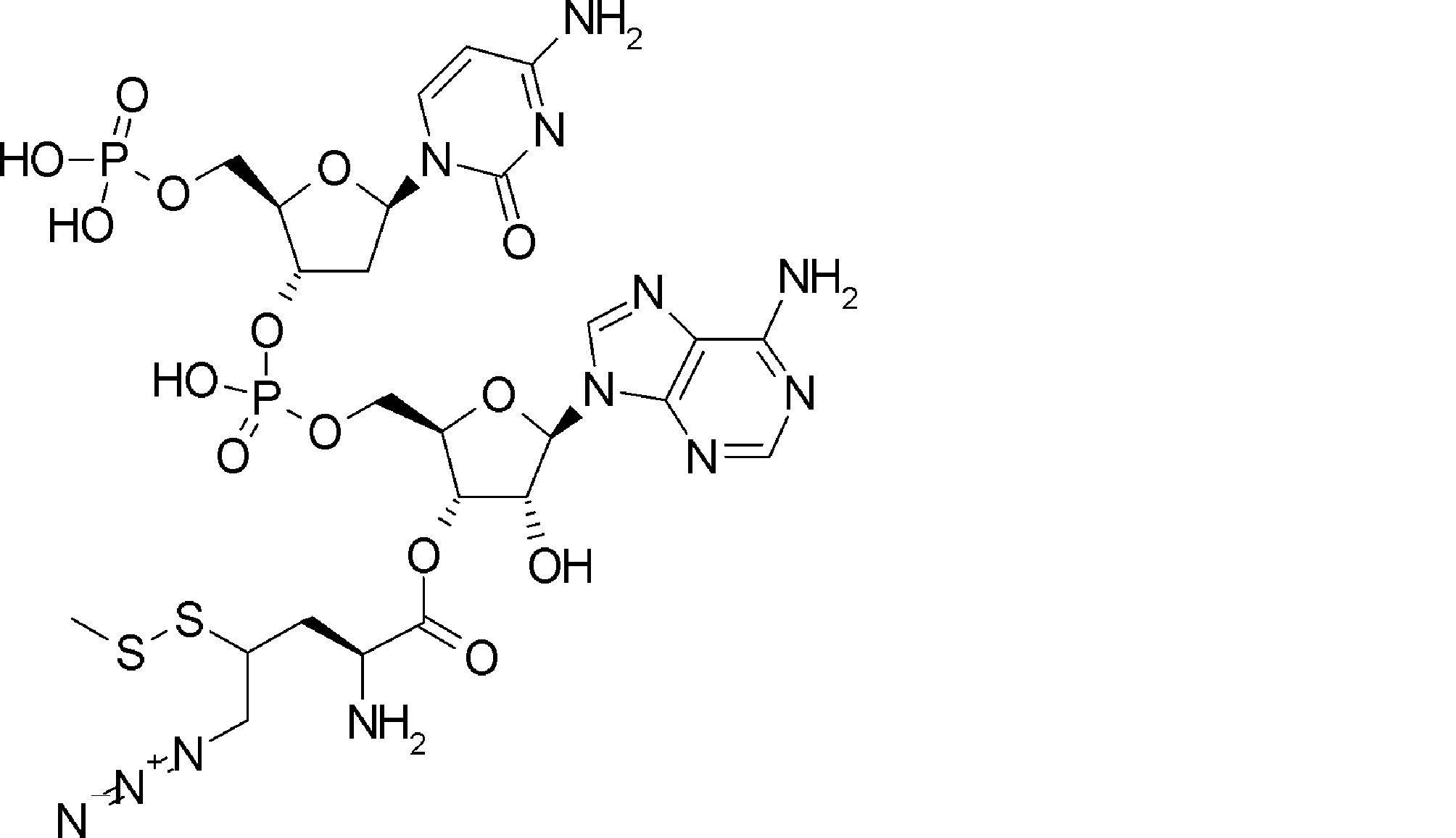
LCMS(ESI) m/z = 855 (M+H)+
保持時間:0.32分(分析条件SQDFA05)
(2S,2'S)-ジ-tert-ブチル 4,4'-ジスルファンジイルビス(5-アジド-2-((tert-ブトキシカルボニル)アミノ)ペンタノエート) (化合物38)の合成
LCMS(ESI) m/z = 691.7 (M+H)+
保持時間:0.80分(分析条件SQDAA50)
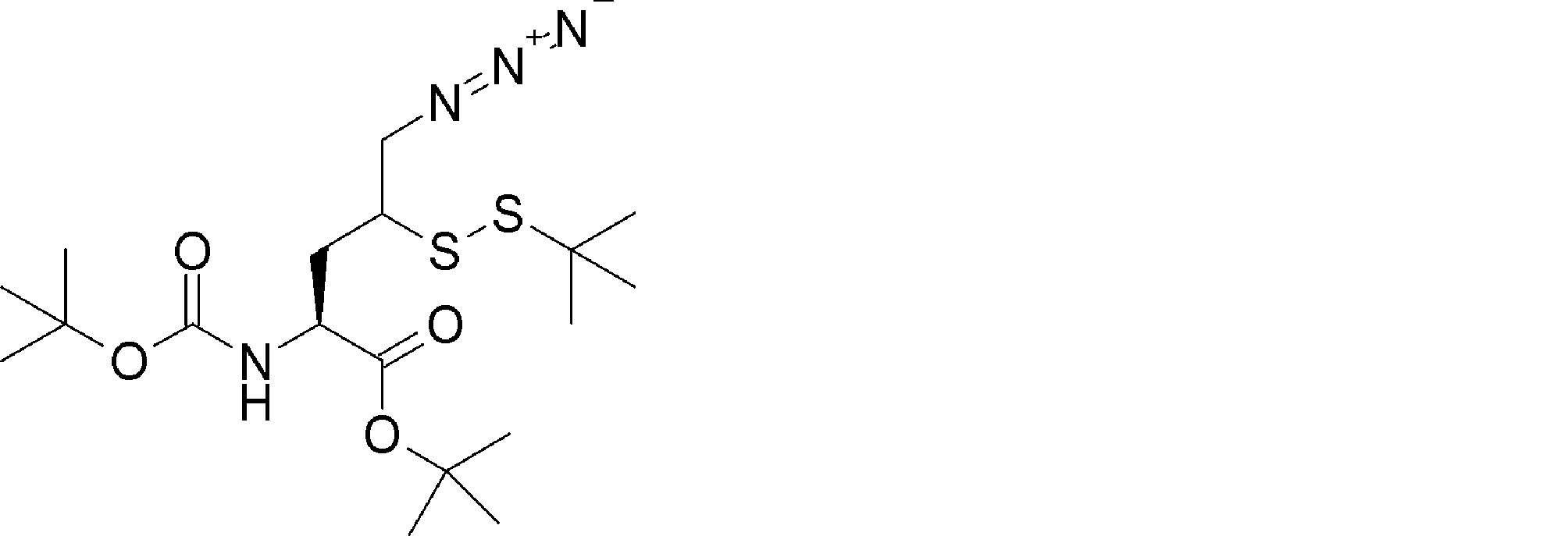
LCMS(ESI) m/z = 435.5 (M+H)+
保持時間:0.77分(分析条件SQDAA50)

LCMS(ESI) m/z = 279 (M+H)+
保持時間:0.78分(分析条件SQDAA05)

LCMS(ESI) m/z = 377 (M-H)-
保持時間:0.89分(分析条件SQDFA05)
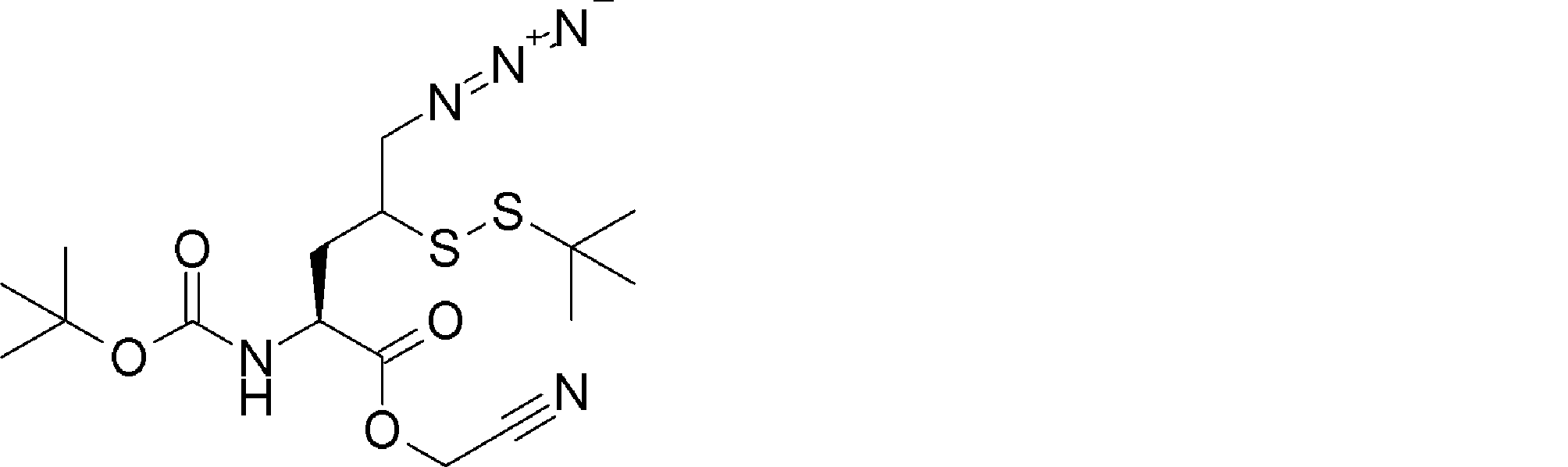
LCMS(ESI) m/z = 416 (M-H)-
保持時間:0.97分(分析条件SQDFA05)
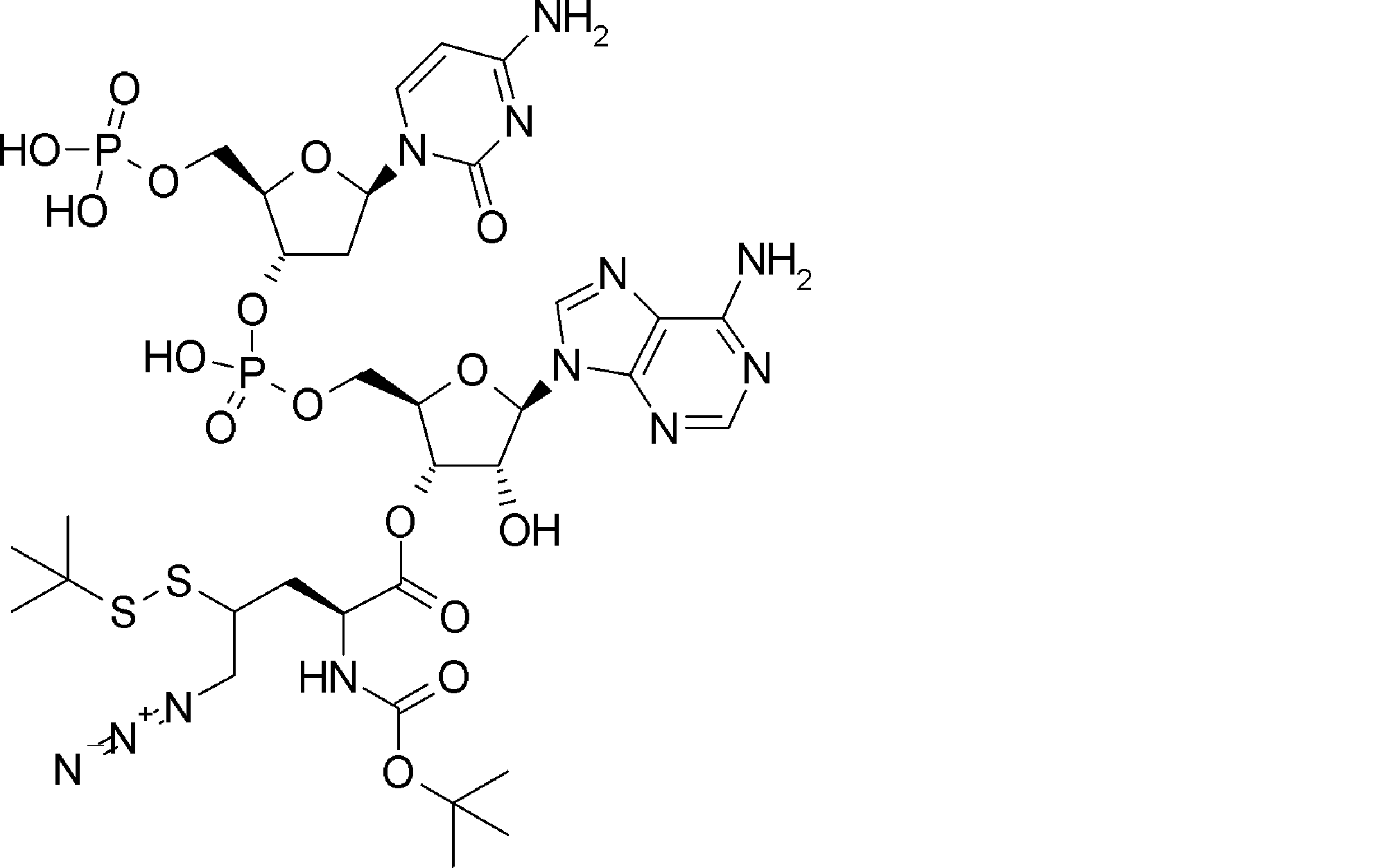
LCMS(ESI) m/z = 997 (M+H)+
保持時間:0.64分(分析条件SQDFA05)
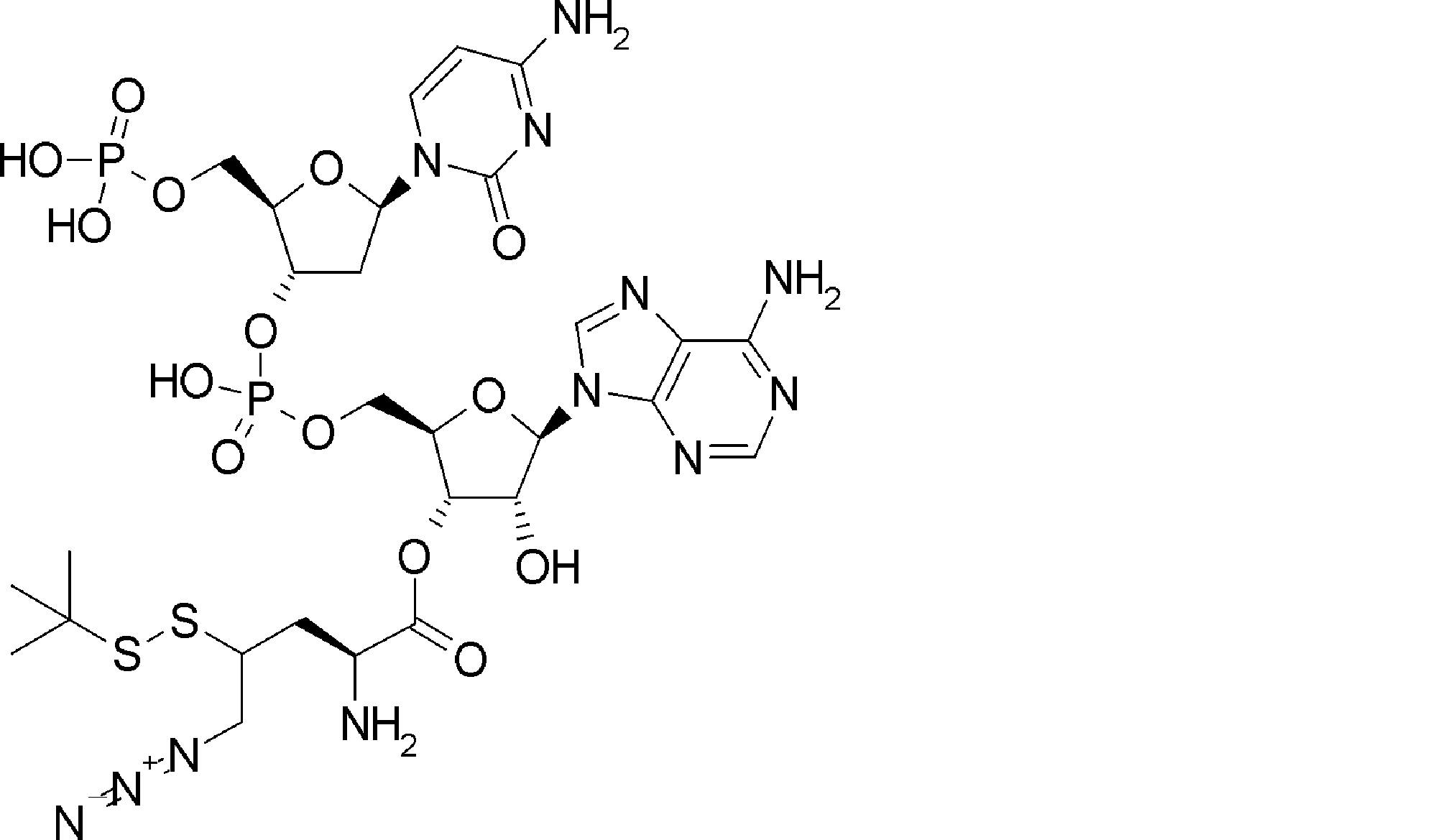
LCMS(ESI) m/z = 897 (M+H)+
保持時間:0.41分(分析条件SQDFA05)
以下に述べる手法により、化合物36のSHの保護基を1工程にて変更でき、容易にpdCpA誘導体を合成する手法を確立した。

LCMS(ESI) m/z = 402 (M-H)-
保持時間:0.61分(分析条件SQDAA50)
5-アジド-2-((tert-ブトキシカルボニル)アミノ)-4-(イソプロピルジスルファニル)ペンタン酸 (2S)-(2R,3S,4R,5R)-2-((((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-((ホスホノオキシ)メチル)テトラヒドロフラン-3-イル)オキシ)(ヒドロキシ)ホスホリル)オキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イルの合成
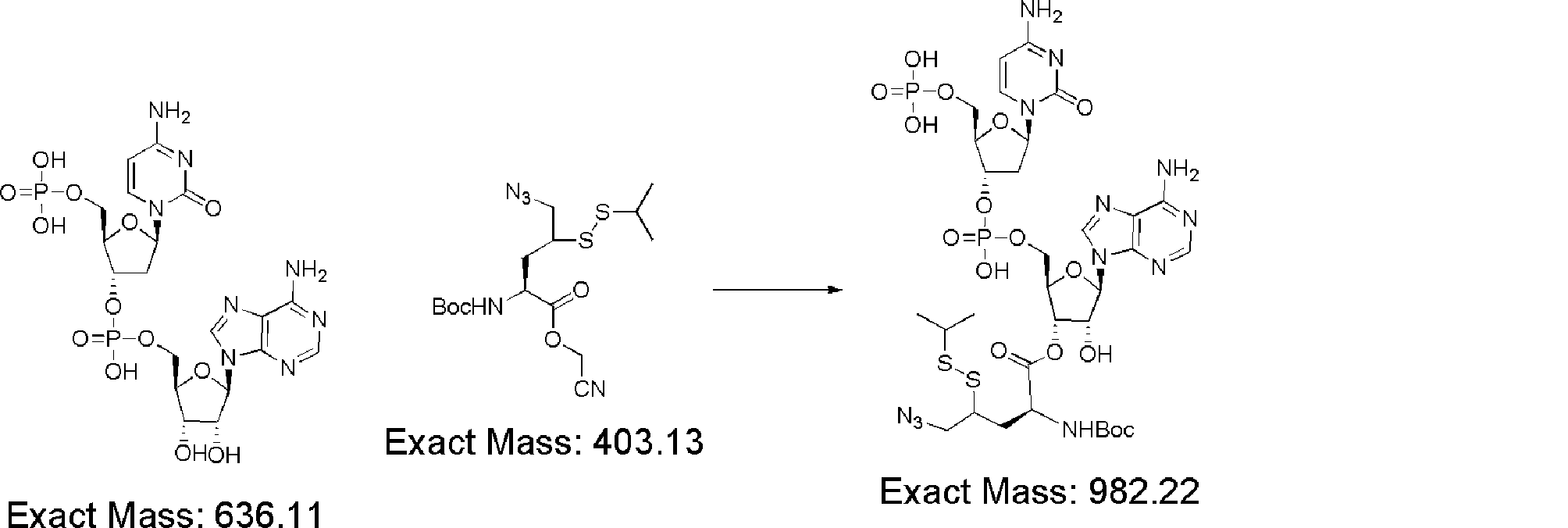
LCMS(ESI) m/z = 981.6 (M-H)-
保持時間:0.58分(分析条件SQDFA05)
2-アミノ-5-アジド-4-(イソプロピルジスルファニル)ペンタン酸 (2S)-(2R,3S,4R,5R)-2-((((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-((ホスホノオキシ)メチル)テトラヒドロフラン-3-イル)オキシ)(ヒドロキシ)ホスホリル)オキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イル(Orn(N3)(4-SSiPr)-pdCpA)の合成
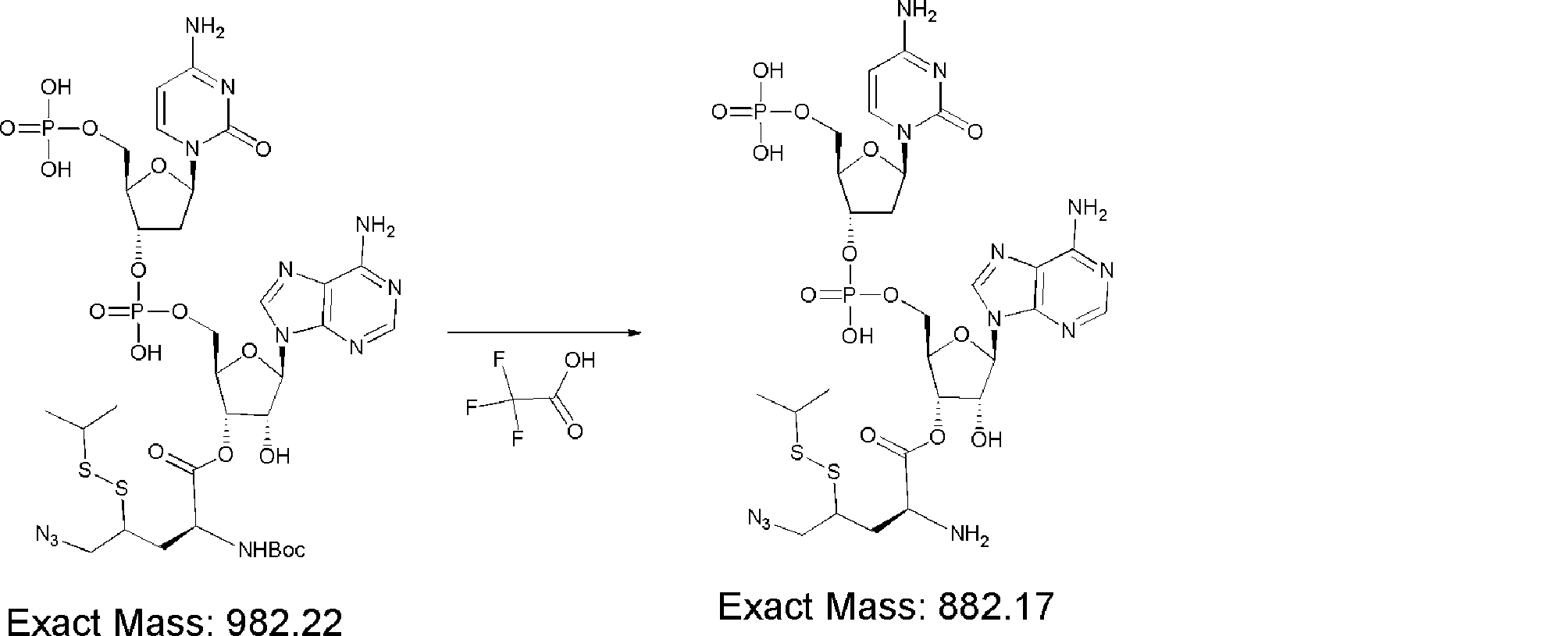
LCMS(ESI) m/z = 881.4 (M-H)-
保持時間:0.39分(分析条件SQDFA05)
以下のスキームに従い合成を行った。

LCMS(ESI) m/z = 265 (M-H)-
保持時間:0.43分(分析条件SQDFA05)
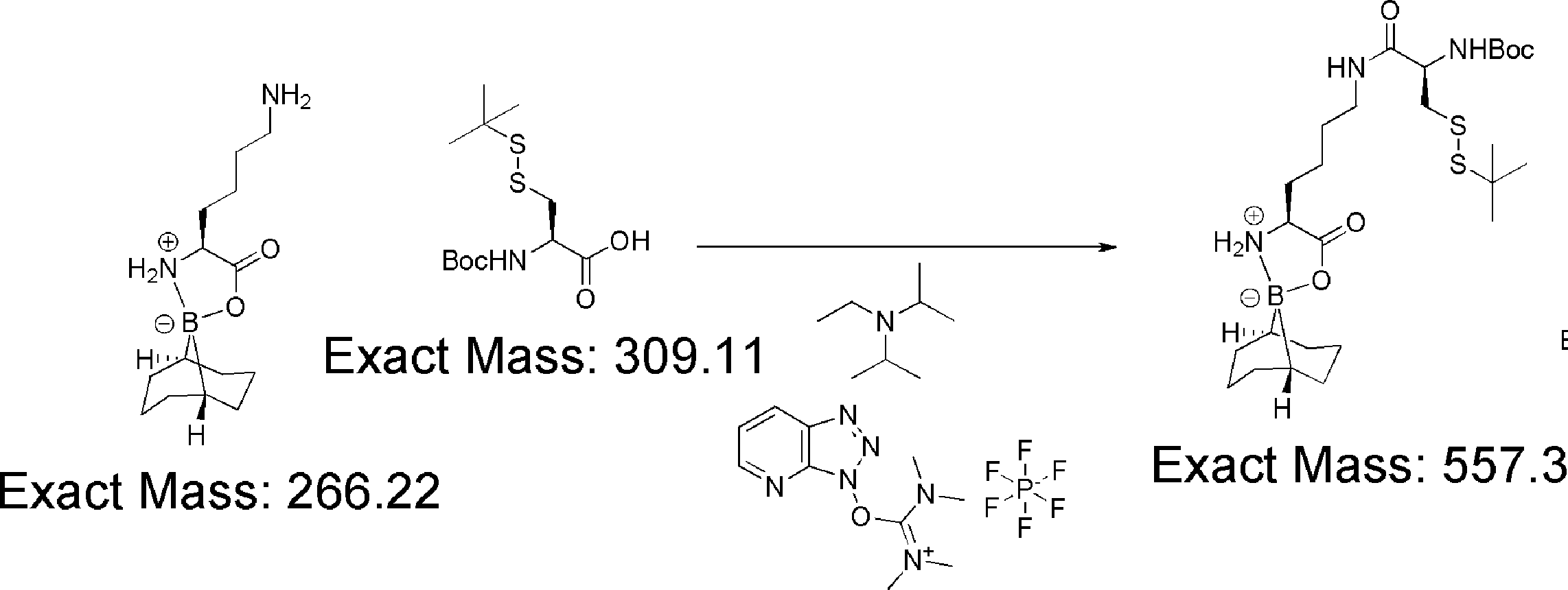
LCMS(ESI) m/z = 558.5 (M+H)+
保持時間:0.98分(分析条件SQDFA05)
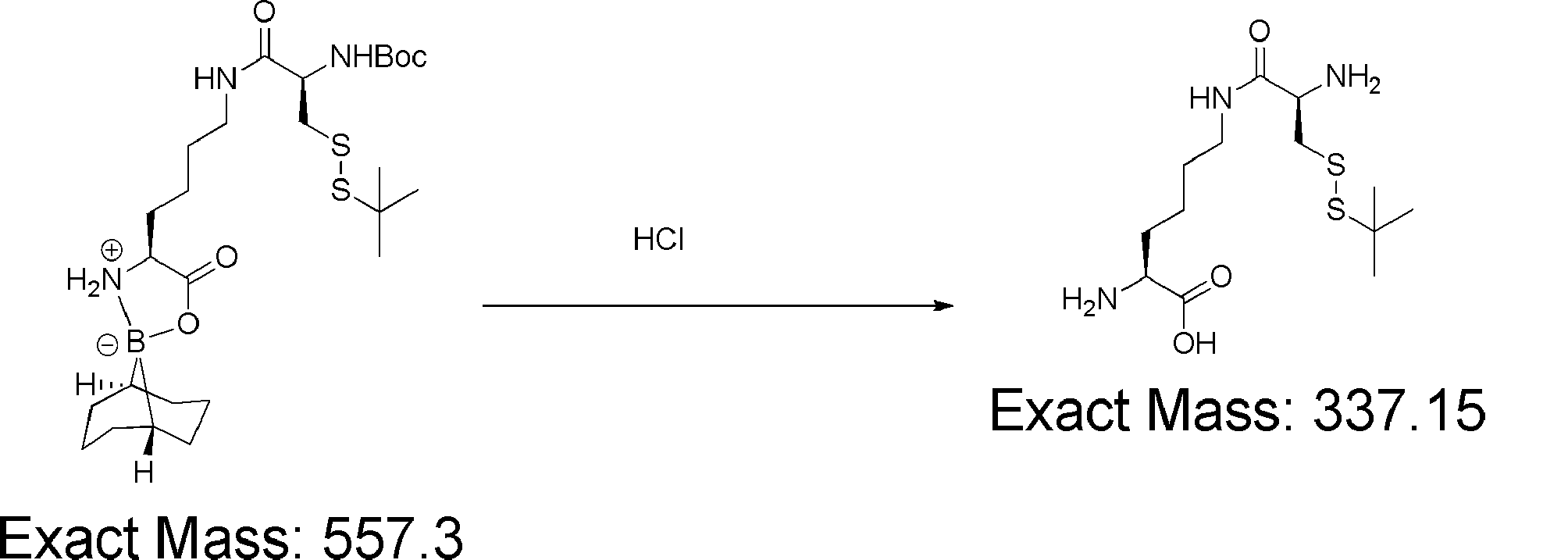
LCMS(ESI) m/z = 336 (M-H)-
保持時間:0.67分(分析条件SQDAA05)
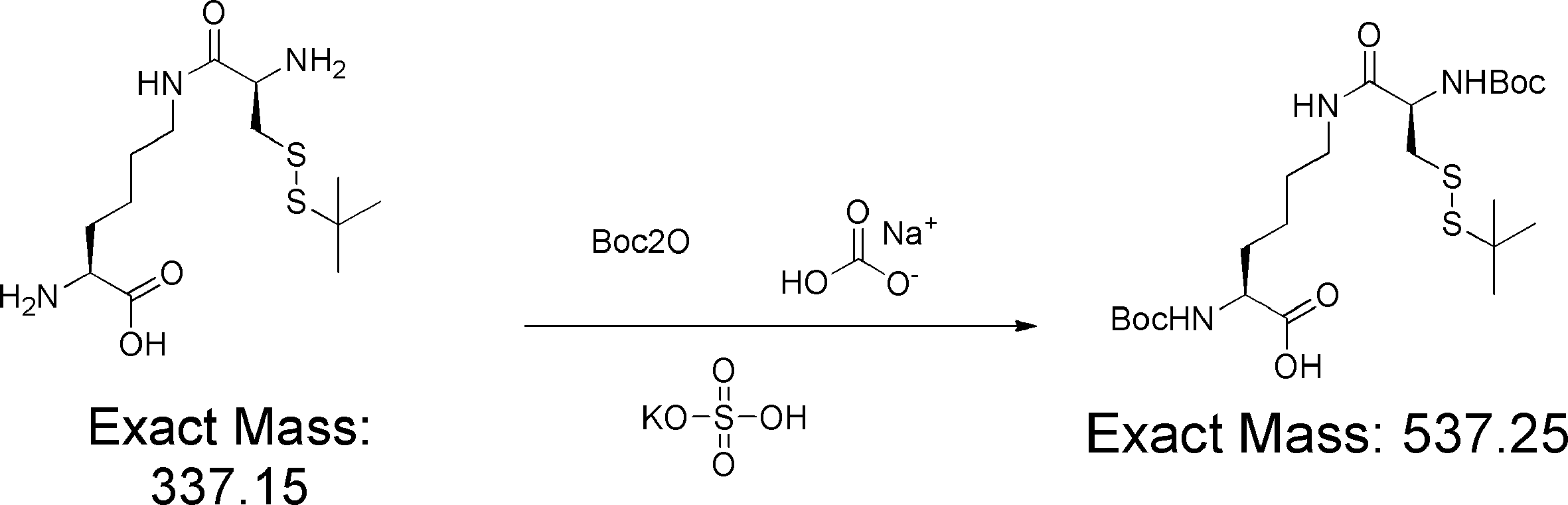
LCMS(ESI) m/z = 536 (M-H)-
保持時間:0.86分(分析条件SQDFA05)
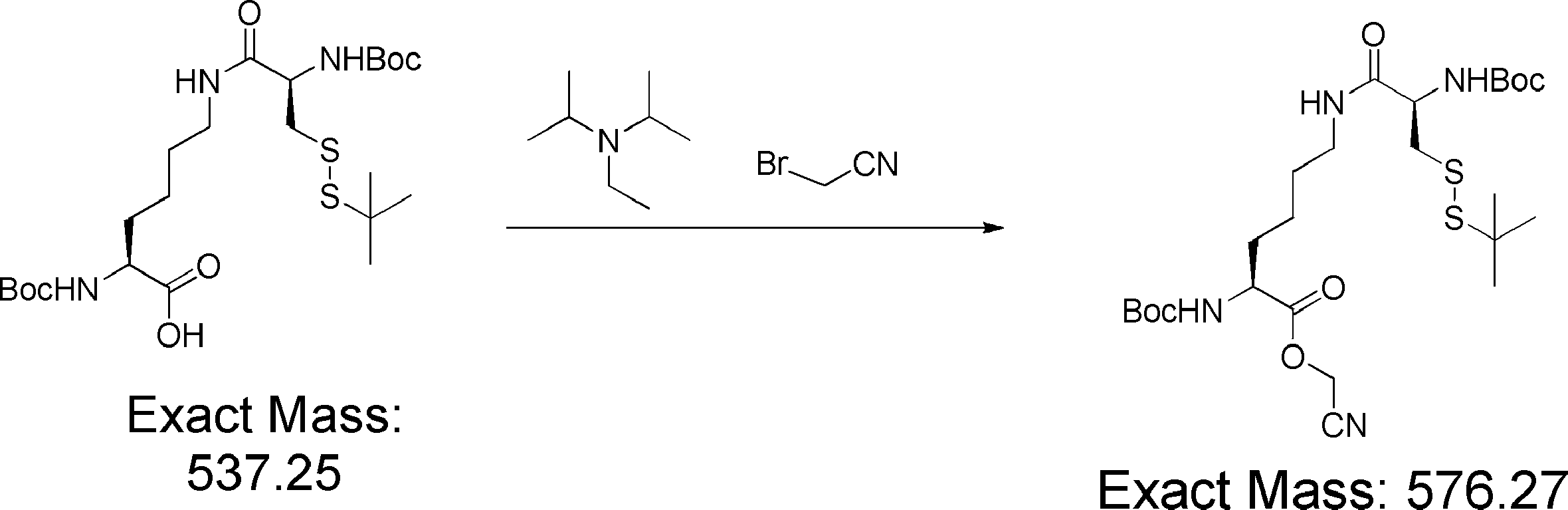
LCMS(ESI) m/z = 577.6 (M+H)+
保持時間:0.92分(分析条件SQDFA05)

LCMS(ESI) m/z = 1156.7 (M+H)+
保持時間:0.63分(分析条件SQDFA05)

LCMS(ESI) m/z = 956 (M+H)+
保持時間:0.29分(分析条件SQDFA05)
翻訳合成可能であり、かつRNAに安定な反応条件にて脱保護できる保護基の探索、およびアミノ酸ユニットとの組み合わせ例を以下に示す。また、以下に示すように、本明細書中に使用する各アミノ酸の略称も化合物番号と共に示した。これらの略称はペプチド内に含有されても、あるいはpdCpAなどのRNAやDNAに含有されても同じユニットが含まれるものとする。また、例えばH-Gly-OHなどの場合、N末端もしくはC末端が化学修飾されてない場合には、HやOH基の記載を省略して単にGlyなどとすることもある。


10-1-1.保護基を有するアミン部位の探索:アミノアシル化pdCpA化合物tk5の合成
以下のスキームに従い、合成を行った。


LCMS(ESI) m/z = 389 (M-H)-
保持時間:0.83分(分析条件SQDAA05)
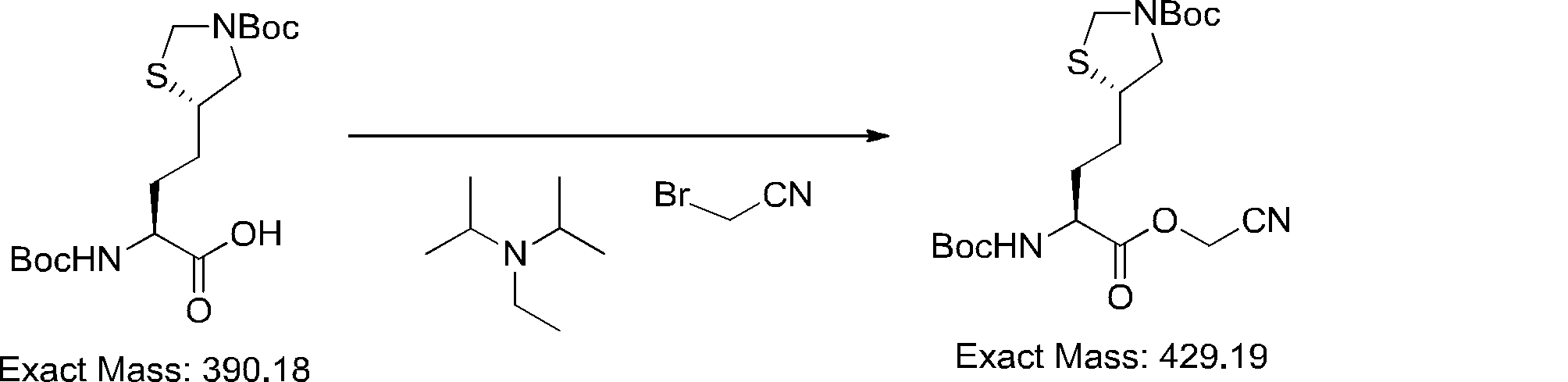
LCMS(ESI) m/z = 428.1 (M-H)-
保持時間:0.85分(分析条件SQDFA05)

LCMS(ESI) m/z = 1007.7 (M-H)-
保持時間:0.54分(分析条件SQDFA05)
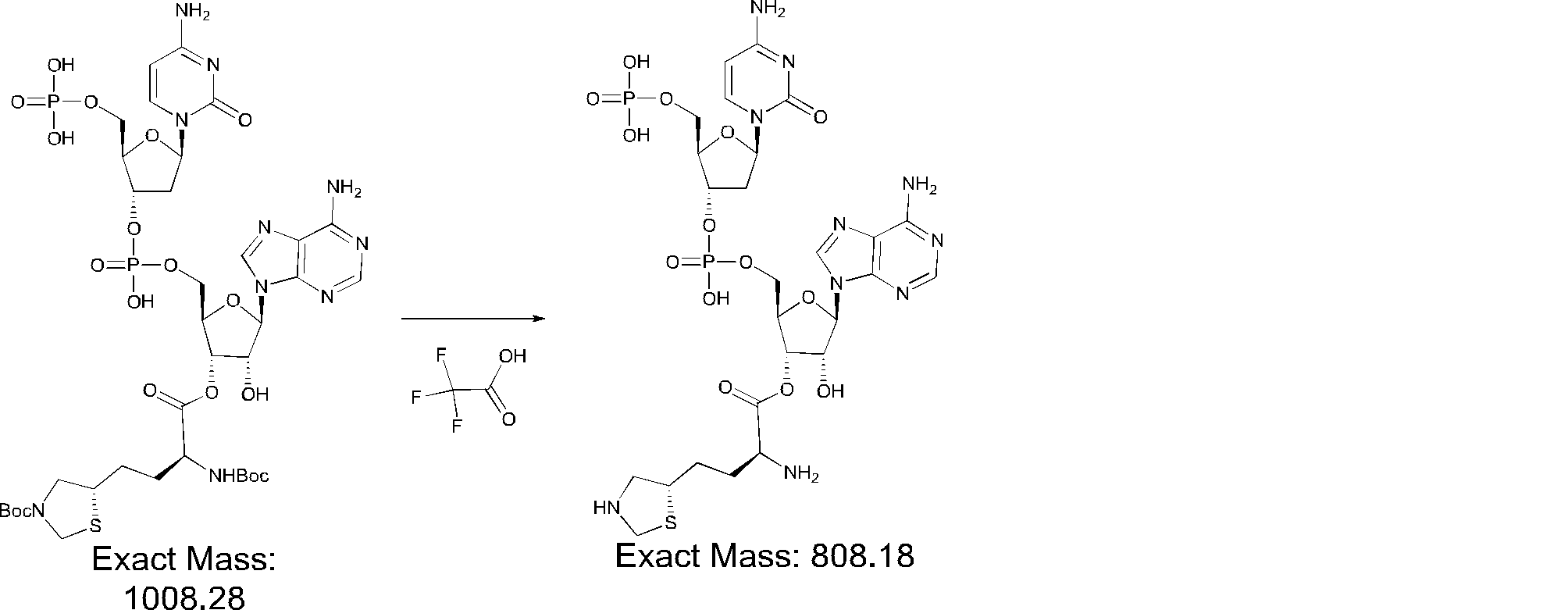
LCMS(ESI) m/z = 807.5 (M-H)-
保持時間:0.14分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
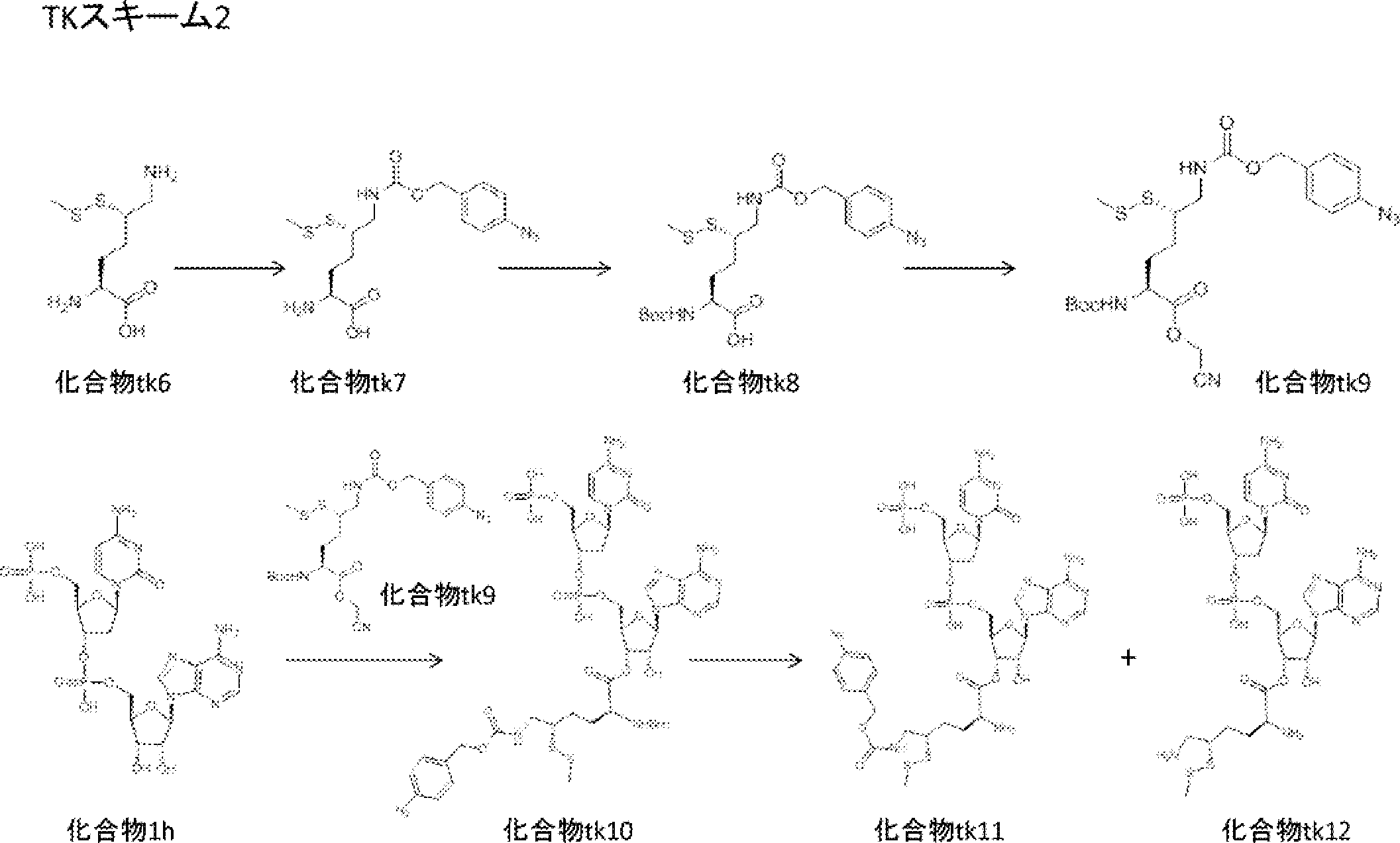
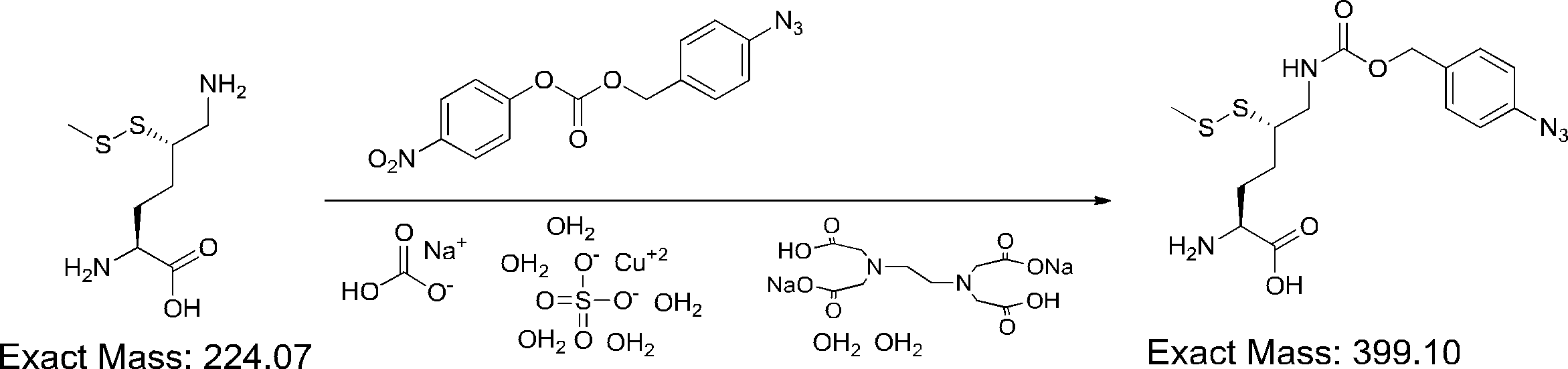
LCMS(ESI) m/z = 398 (M-H)-
保持時間:0.85分(分析条件SQDAA05)
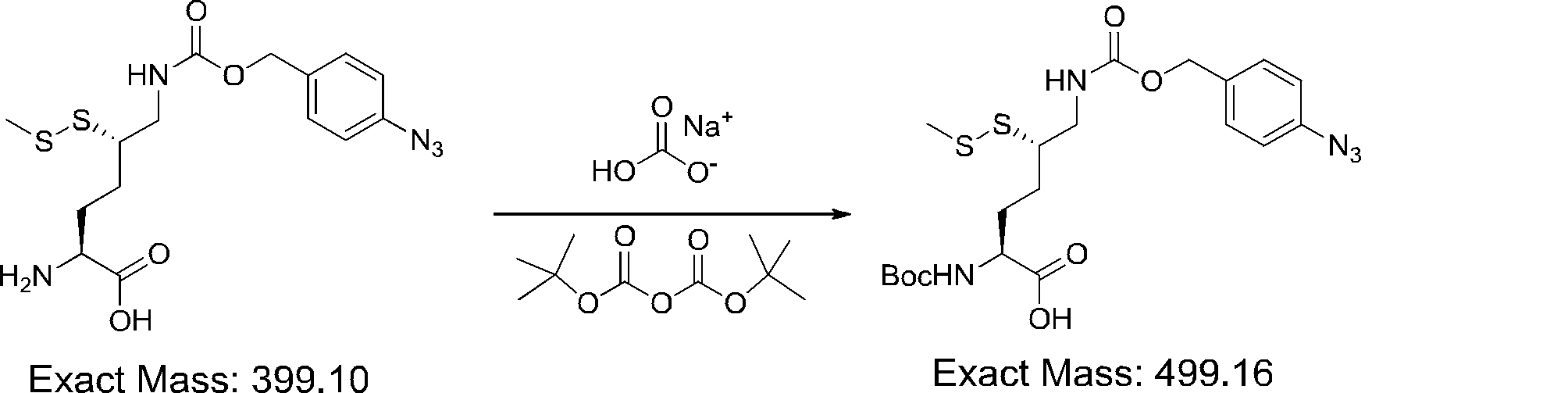
LCMS(ESI) m/z = 498.4 (M-H)-
保持時間:0.91分(分析条件SQDAA05)
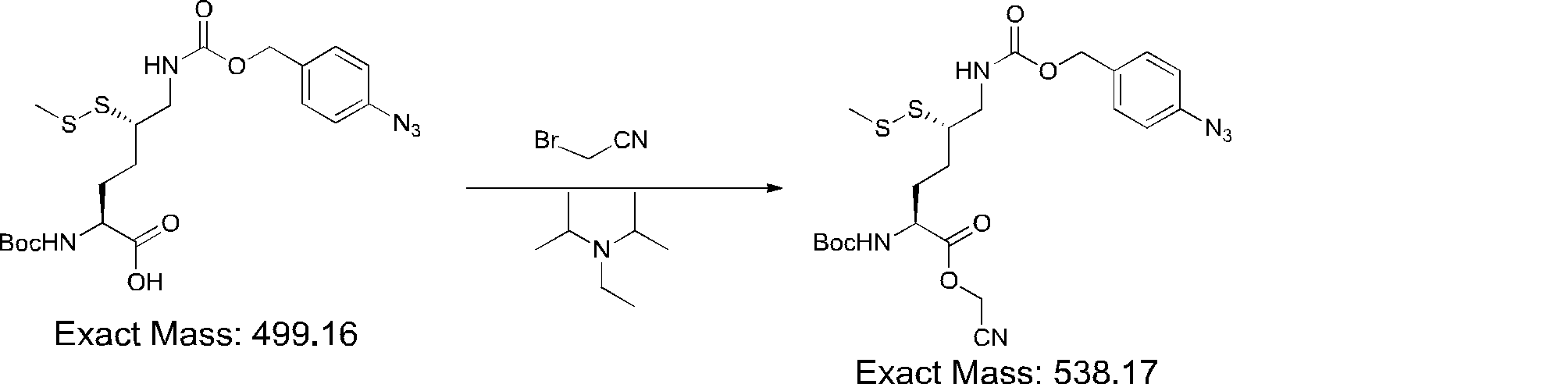
LCMS(ESI) m/z = 537.6 (M-H)-
保持時間:0.91分(分析条件SQDFA05)
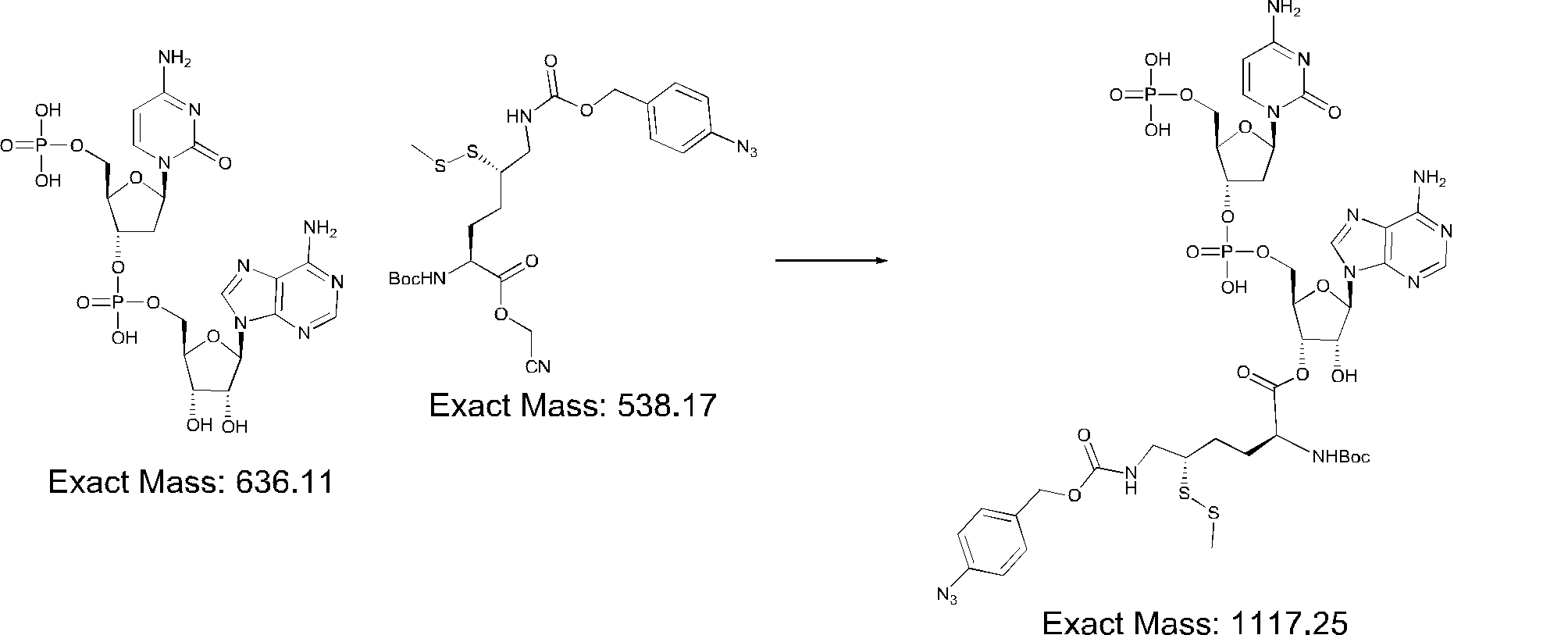
LCMS(ESI) m/z = 1116.7 (M-H)-
保持時間:0.62分(分析条件SQDFA05)

LCMS(ESI) m/z = 1016.5 (M-H)-
保持時間:0.45分(分析条件SQDFA05)
LCMS(ESI) m/z = 841.4 (M-H)-
保持時間:0.20分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
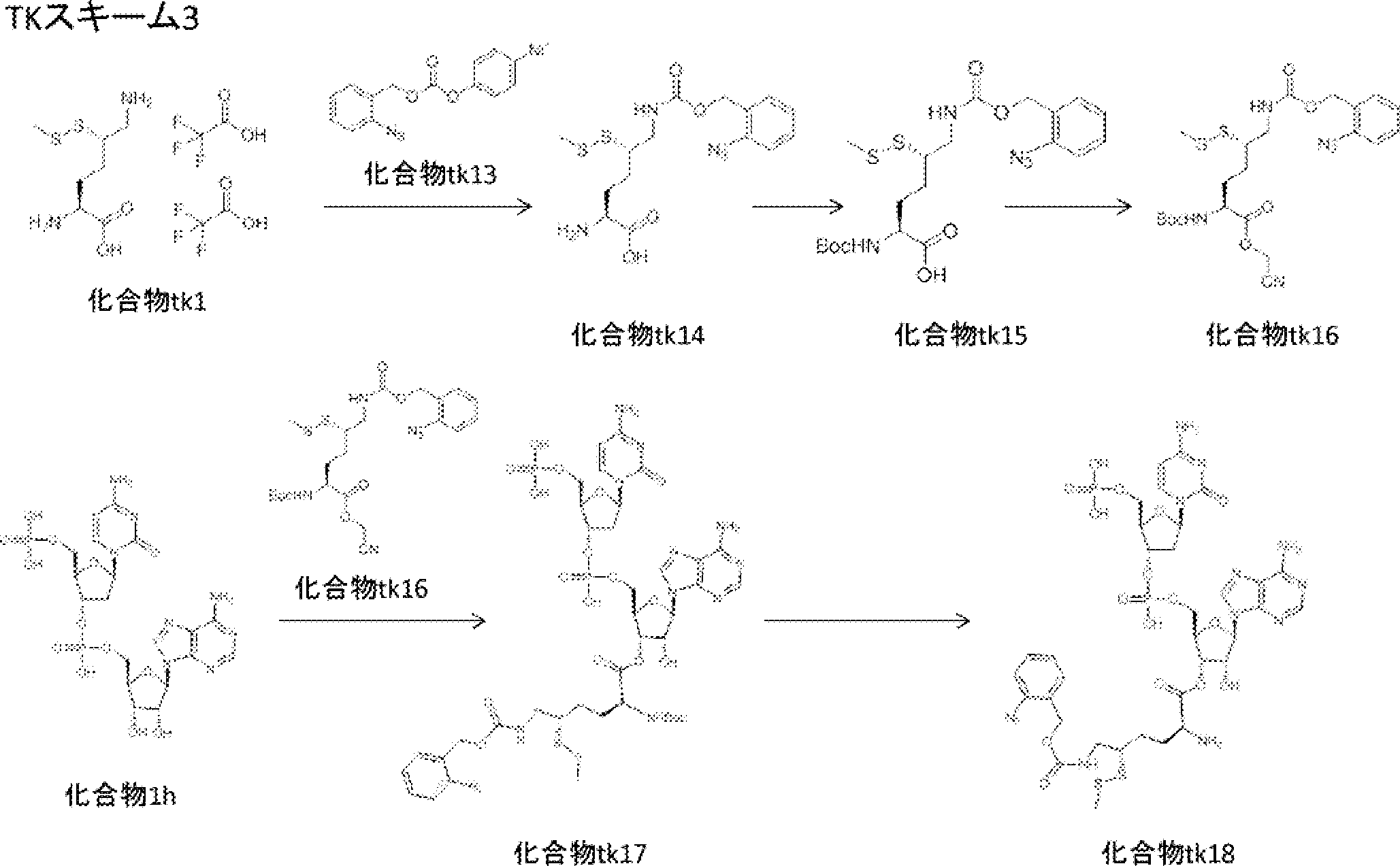

LCMS(ESI) m/z = 138 (HOC6H4NO2-H)-
保持時間:1.03分(分析条件SQDAA05)
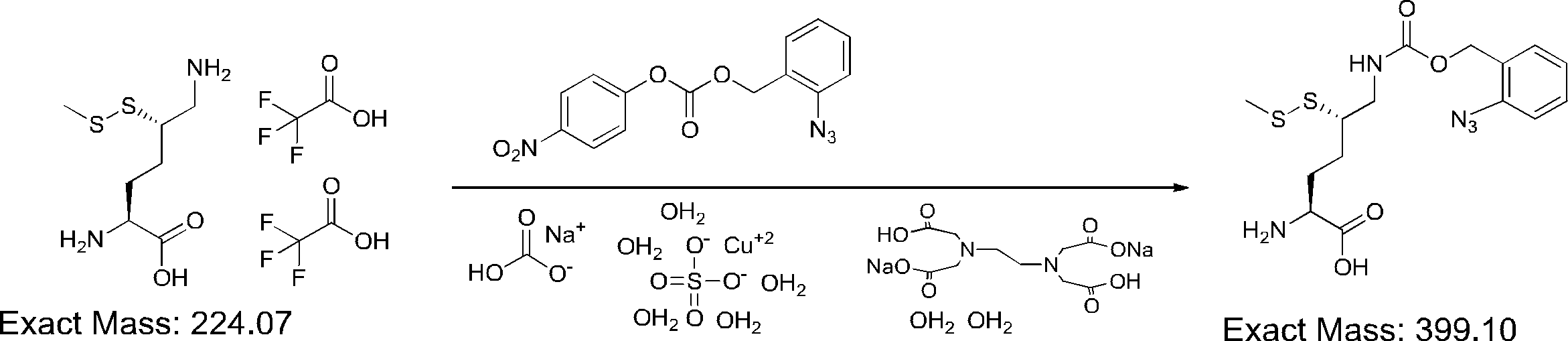
LCMS(ESI) m/z = 398 (M-H)-
保持時間:0.85分(分析条件SQDAA05)
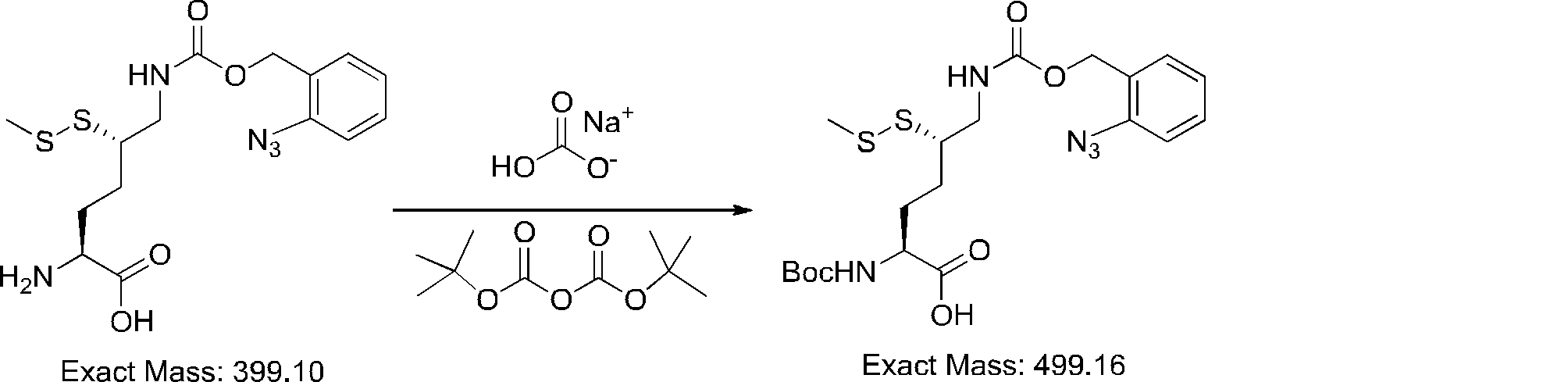
LCMS(ESI) m/z = 498 (M-H)-
保持時間:0.90分(分析条件SQDAA05)
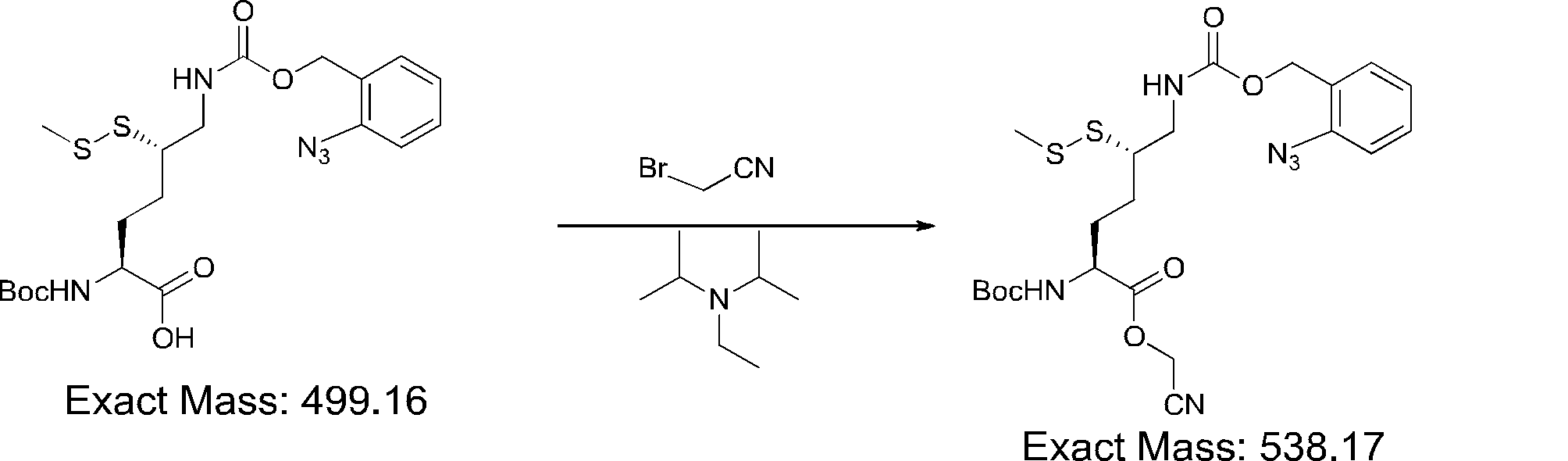
LCMS(ESI) m/z = 539.5 (M+H)+
保持時間:0.92分(分析条件SQDFA05)
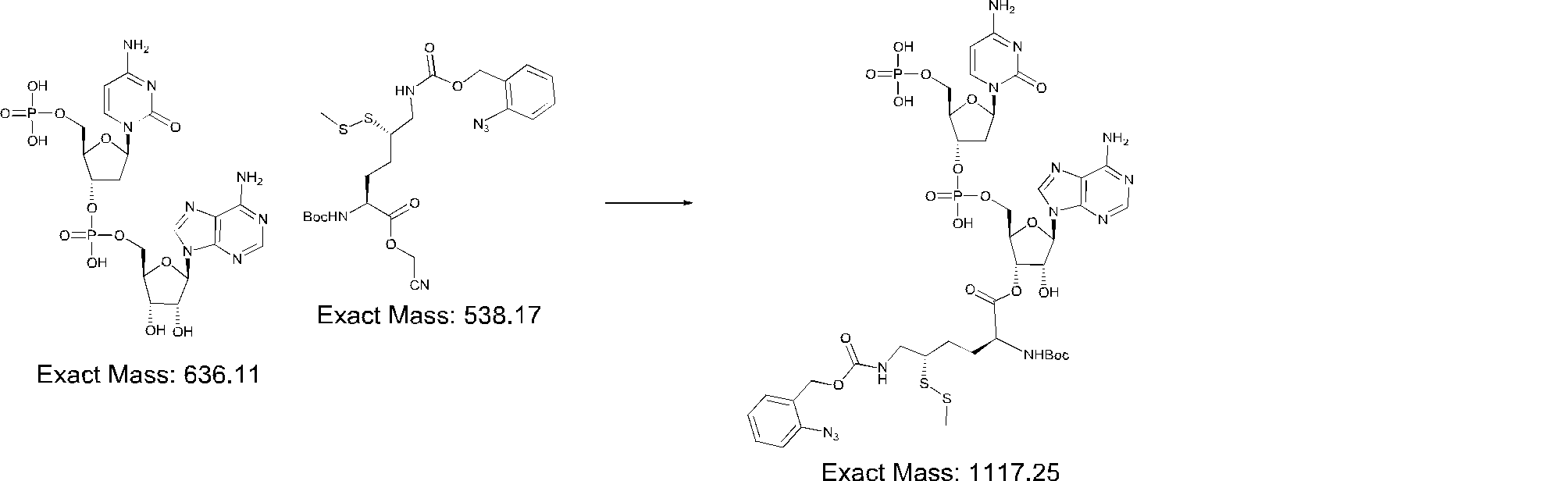
LCMS(ESI) m/z = 1116.2 (M-H)-
保持時間:0.65分(分析条件SQDFA05)

LCMS(ESI) m/z = 1016.0 (M-H)-
保持時間:0.44分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。


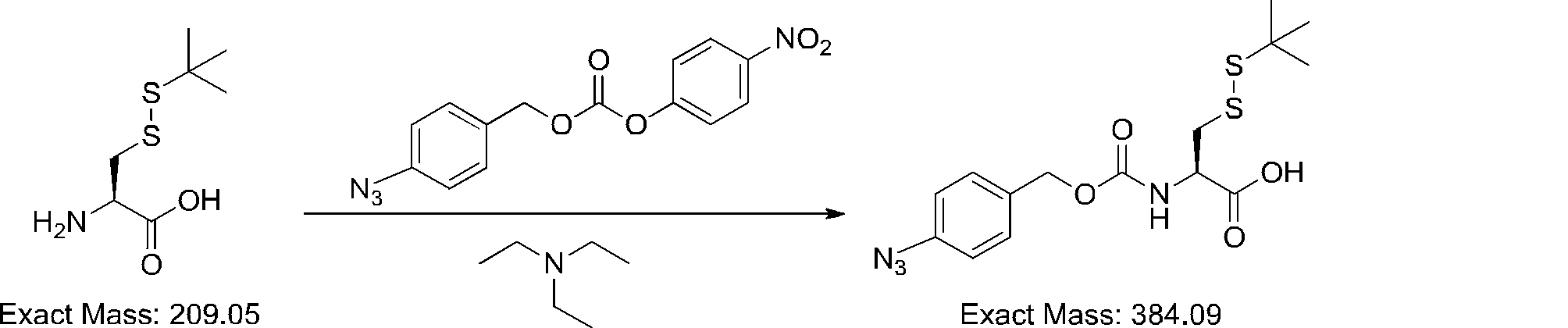
LCMS(ESI) m/z = 383 (M-H)-
保持時間:0.84分(分析条件SQDFA05)
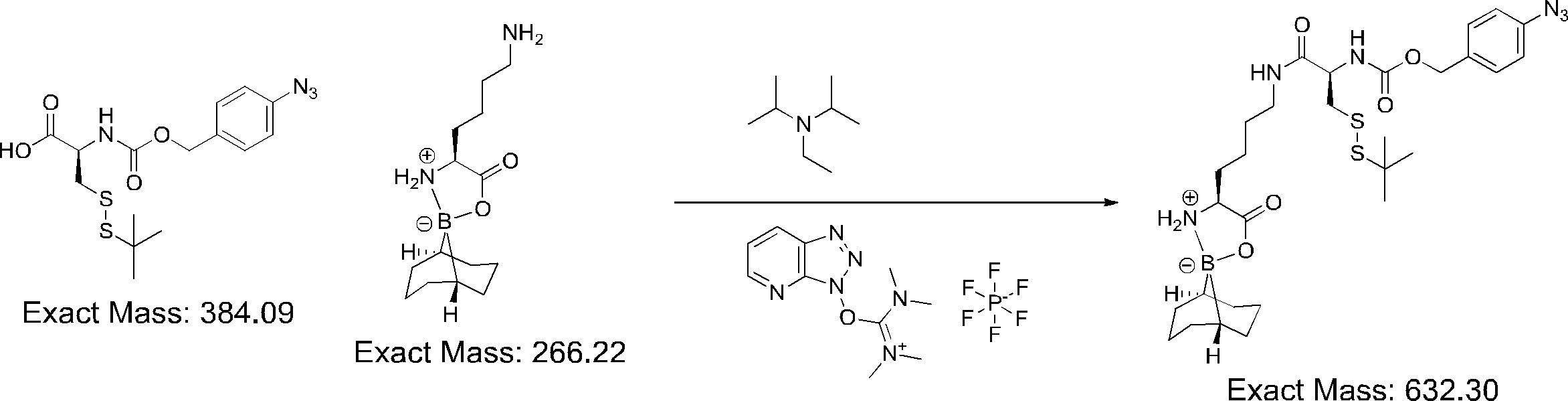
LCMS(ESI) m/z = 631.5 (M-H)-
保持時間:1.01分(分析条件SQDFA05)
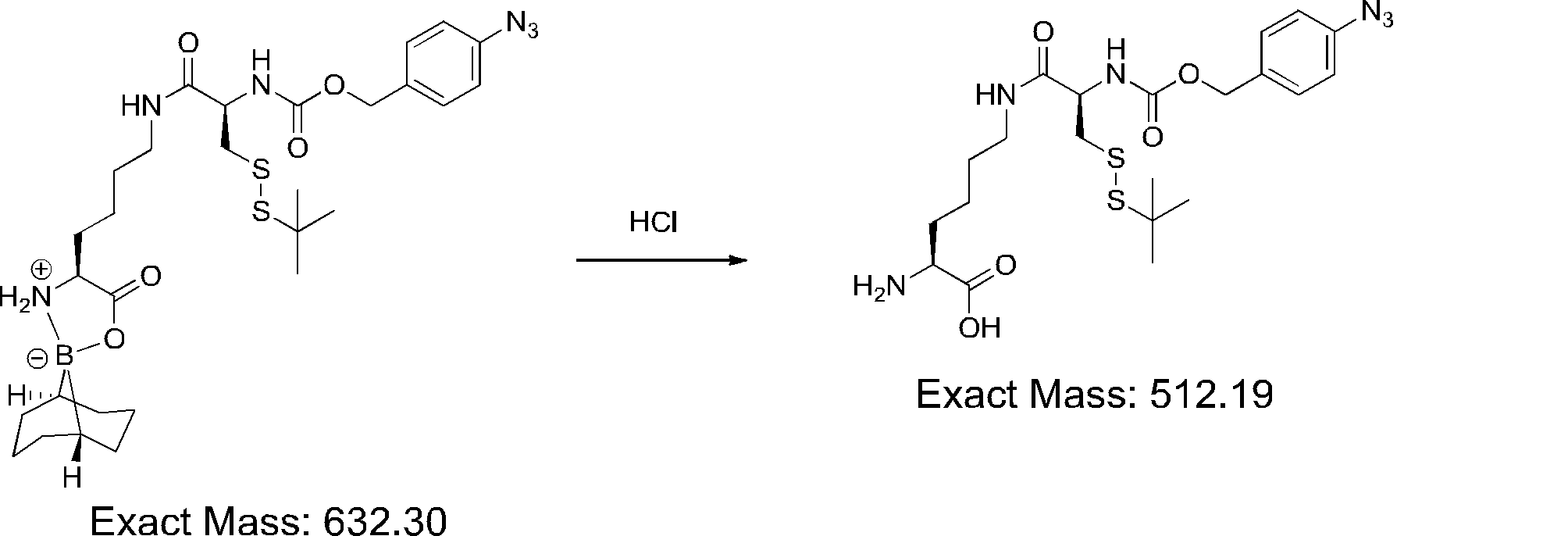
LCMS(ESI) m/z = 513 (M+H)+
保持時間:0.61分(分析条件SQDFA05)
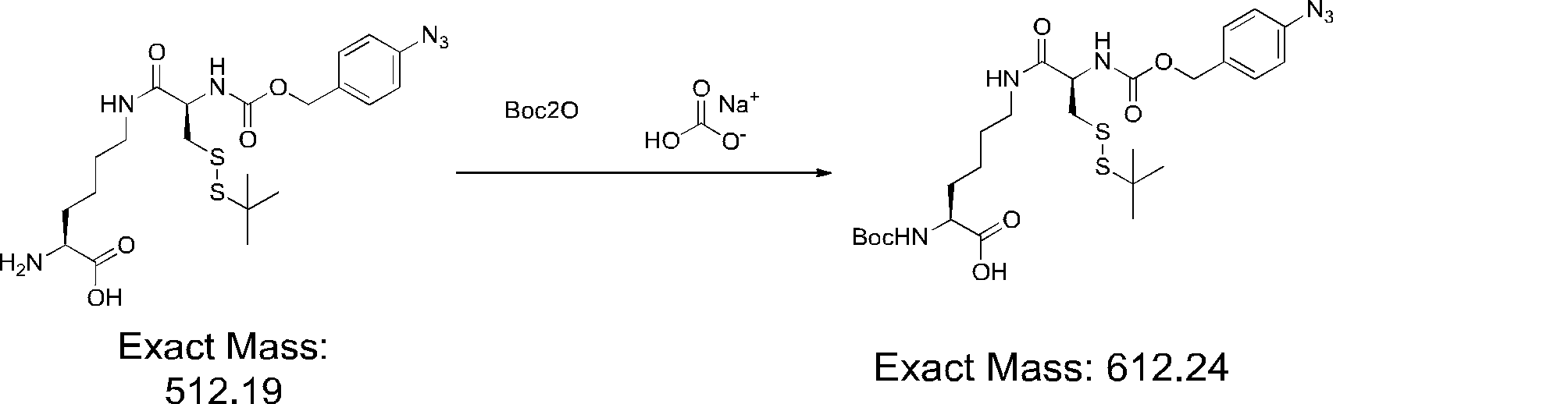
LCMS(ESI) m/z = 611 (M-H)-
保持時間:0.88分(分析条件SQDFA05)
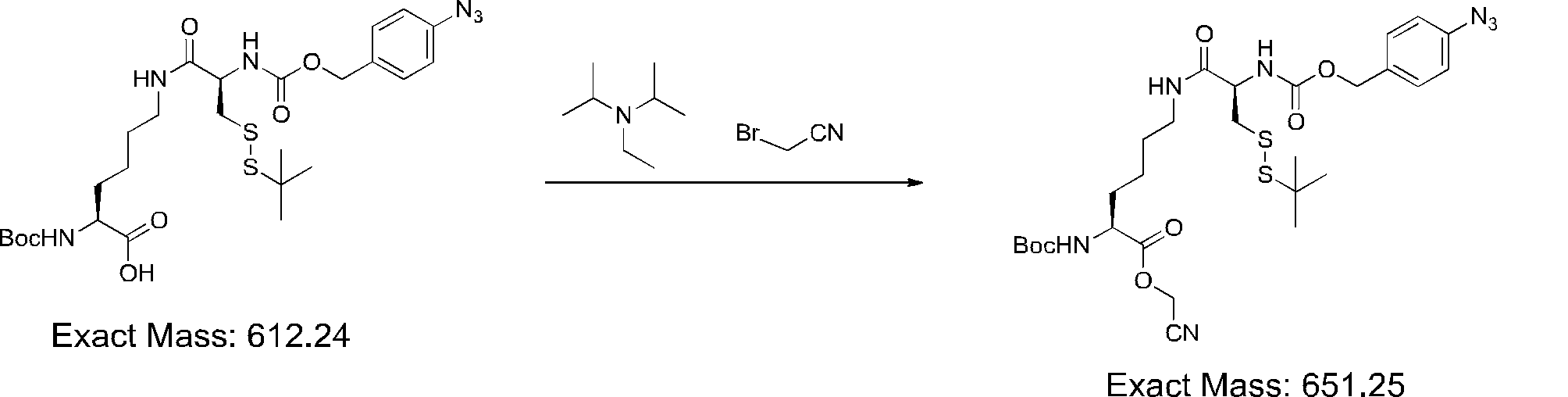
LCMS(ESI) m/z = 650.6 (M-H)-
保持時間:0.95分(分析条件SQDFA05)
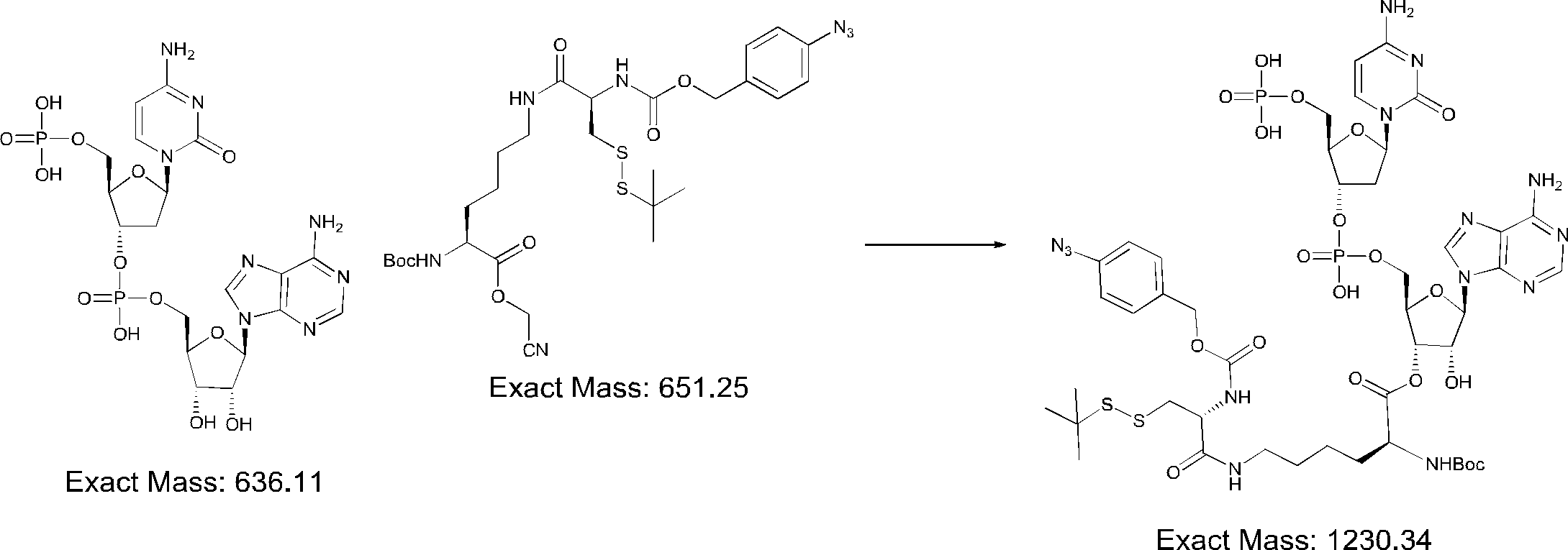
LCMS(ESI) m/z = 1231.7 (M+H)+
保持時間:0.90分(分析条件SQDAA05)
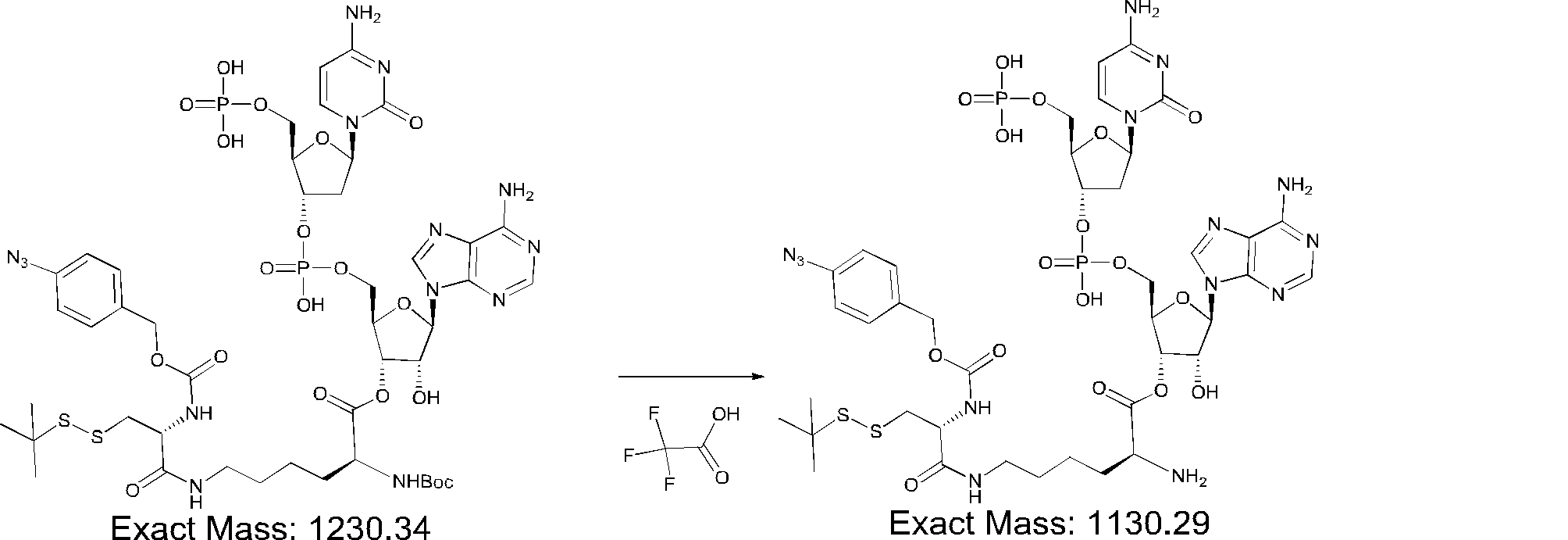
LCMS(ESI) m/z = 1130.1 (M-H)-
保持時間:0.54分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
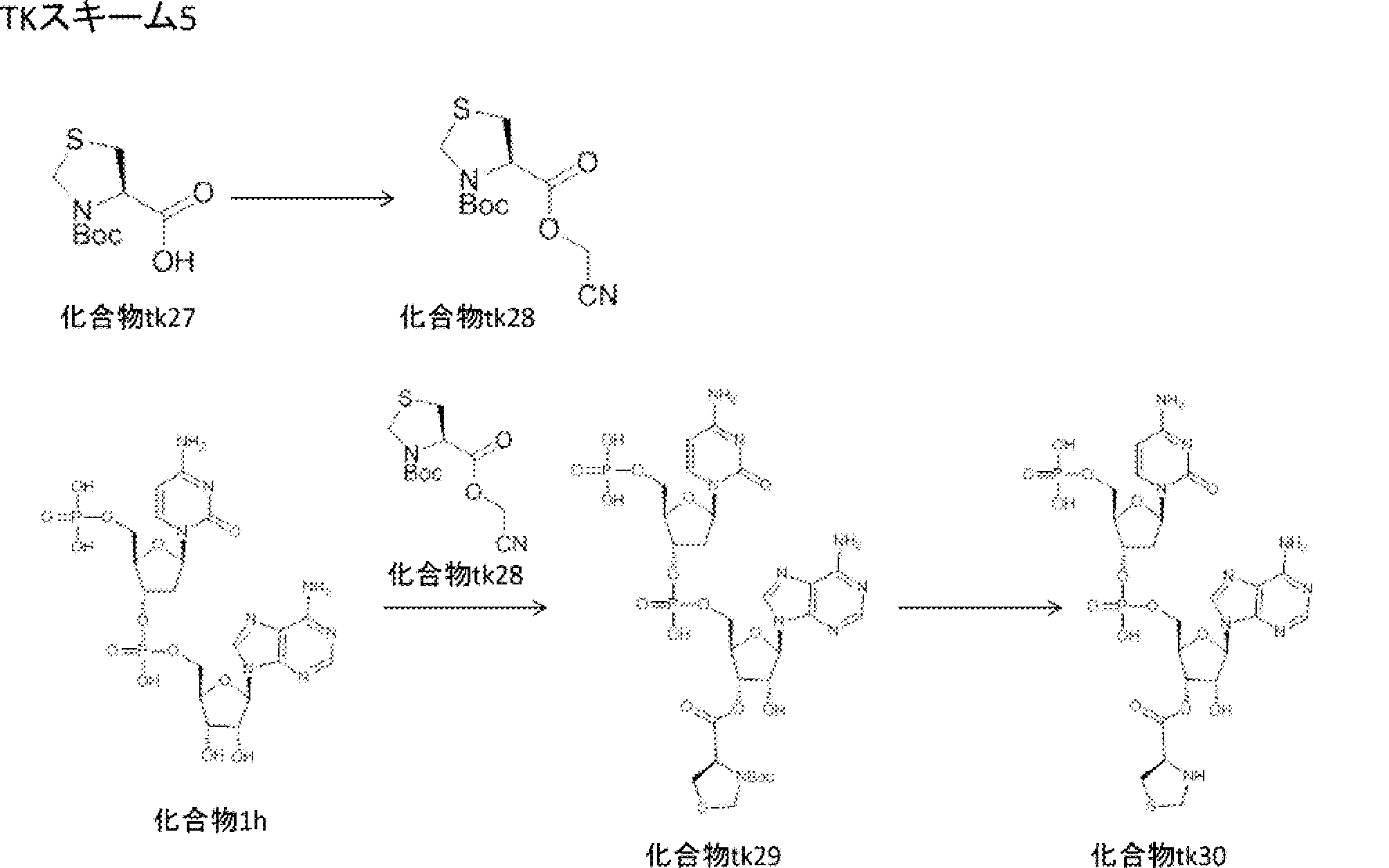
LCMS(ESI) m/z = 273.3 (M+H)+
保持時間:0.71分(分析条件SQDFA05)
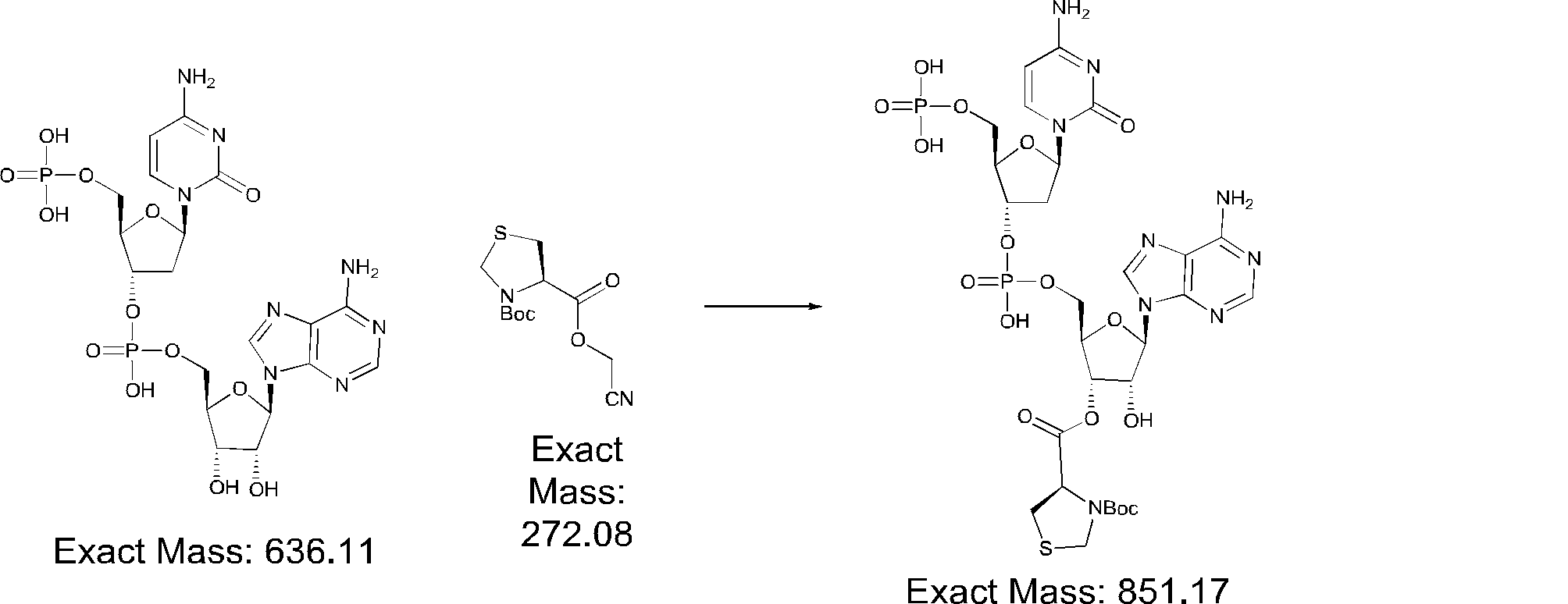
LCMS(ESI) m/z = 850.3 (M-H)-
保持時間:0.41分(分析条件SQDFA05)

LCMS(ESI) m/z = 750.4 (M-H)-
保持時間:0.34分(分析条件SQDAA05)
以下のスキームに従い、合成を行った。

LCMS(ESI) m/z = 542 (M-H)-
保持時間:0.89分(分析条件SQDFA05)
LCMS(ESI) m/z = 320 (M-H)-
保持時間:0.47分(分析条件SQDFA05)

LCMS(ESI) m/z = 420 (M-H)-
保持時間:0.76分(分析条件SQDFA05)
LCMS(ESI) m/z = 459 (M-H)-
保持時間:0.84分(分析条件SQDFA05)
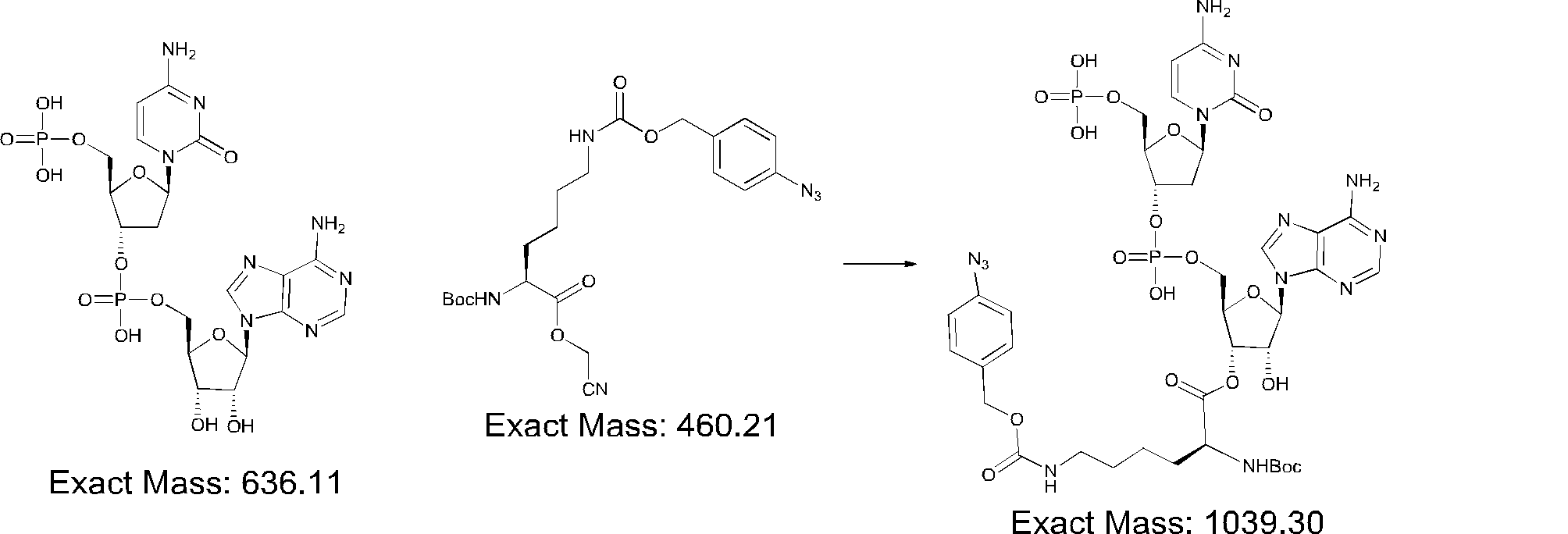
LCMS(ESI) m/z = 1040.8 (M+H)+
保持時間:0.56分(分析条件SQDFA05)

LCMS(ESI) m/z = 938.5 (M-H)-
保持時間:0.39分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
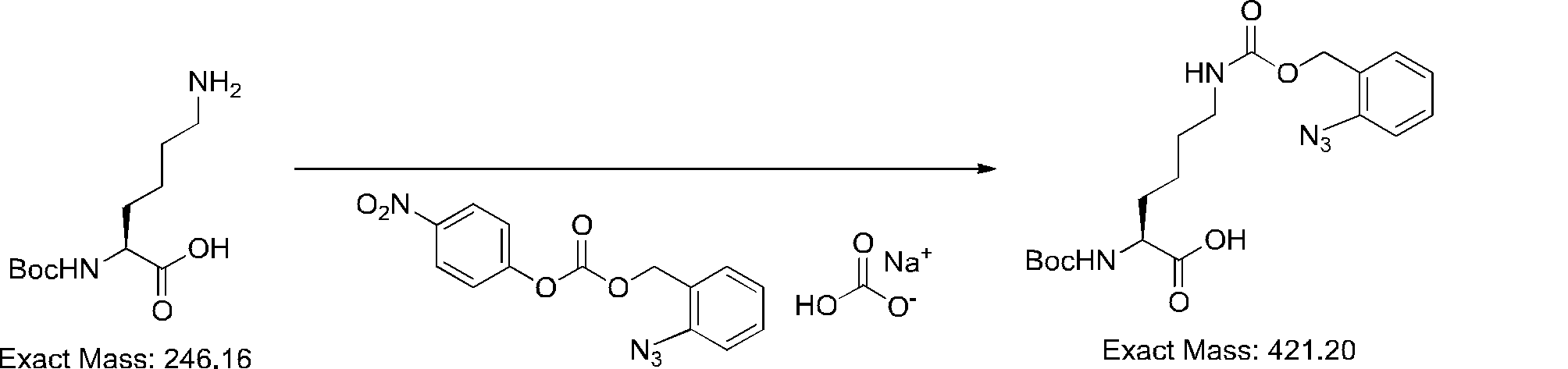
LCMS(ESI) m/z = 420 (M-H)-
保持時間:0.76分(分析条件SQDFA05)
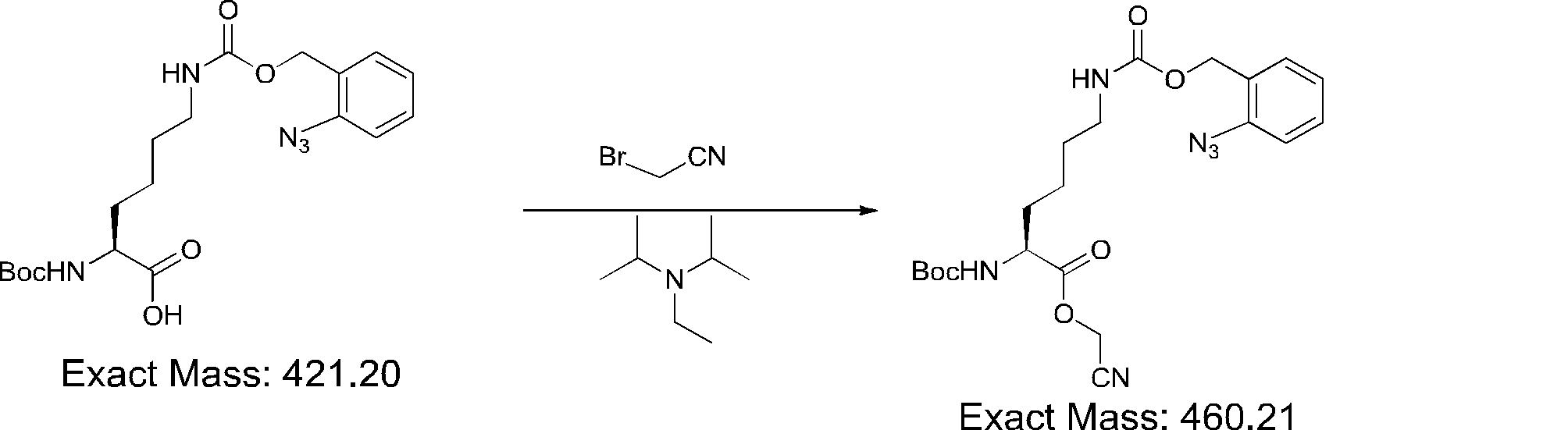
LCMS(ESI) m/z = 459.5 (M-H)-
保持時間:0.84分(分析条件SQDFA05)
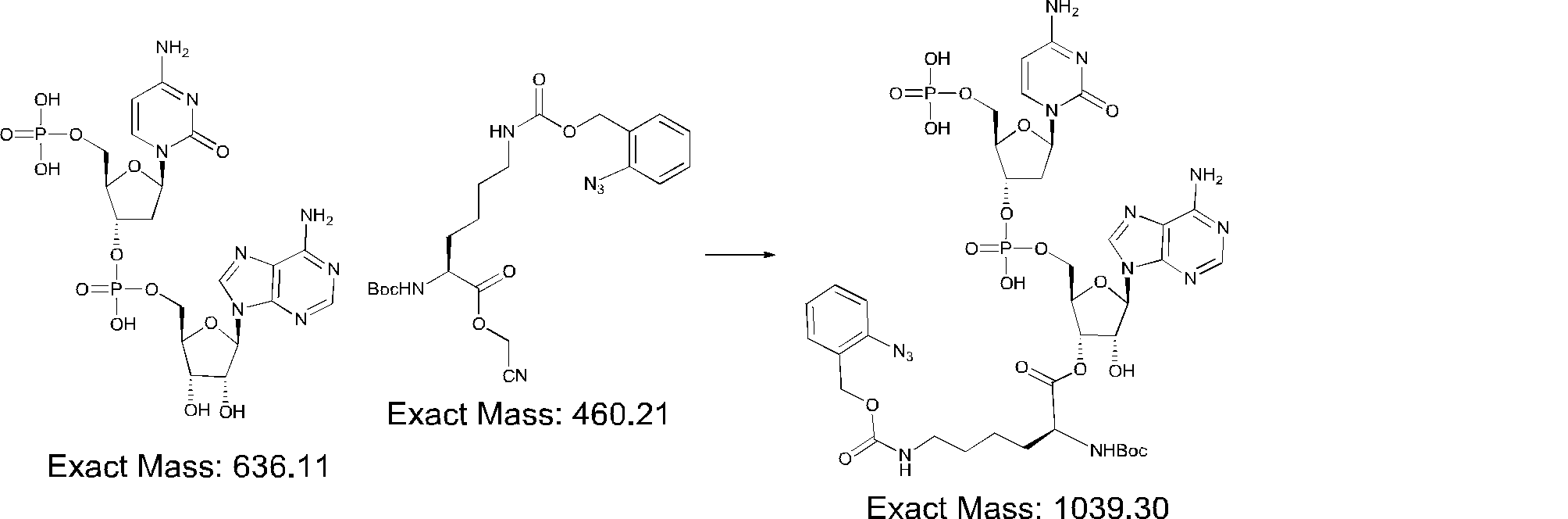
LCMS(ESI) m/z = 1038.6 (M-H)-
保持時間:0.58分(分析条件SQDFA05)

LCMS(ESI) m/z = 938.5 (M-H)-
保持時間:0.39分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
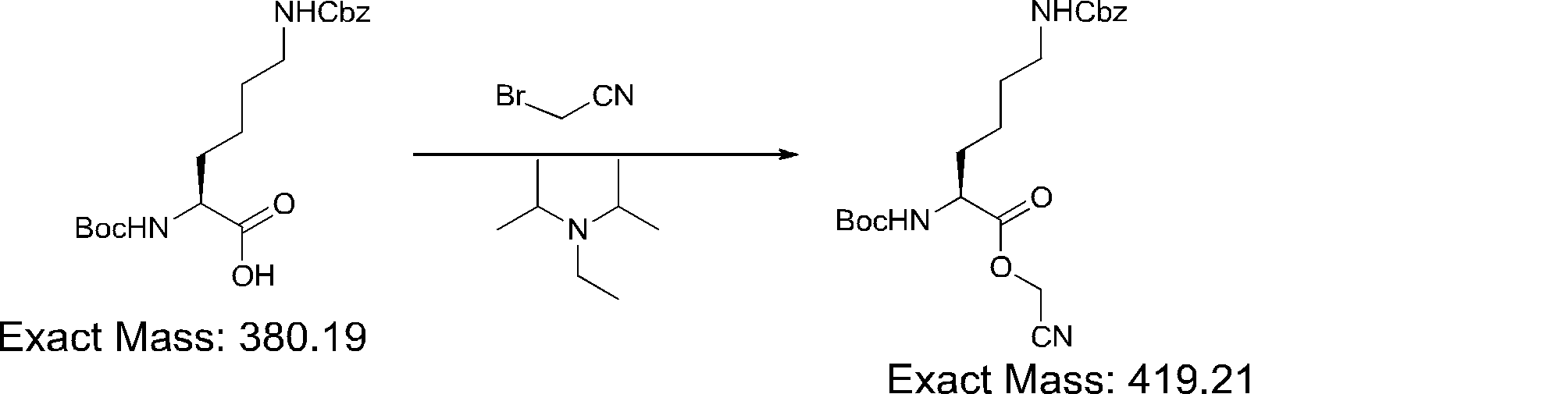
LCMS(ESI) m/z = 418 (M-H)-
保持時間:0.79分(分析条件SQDFA05)
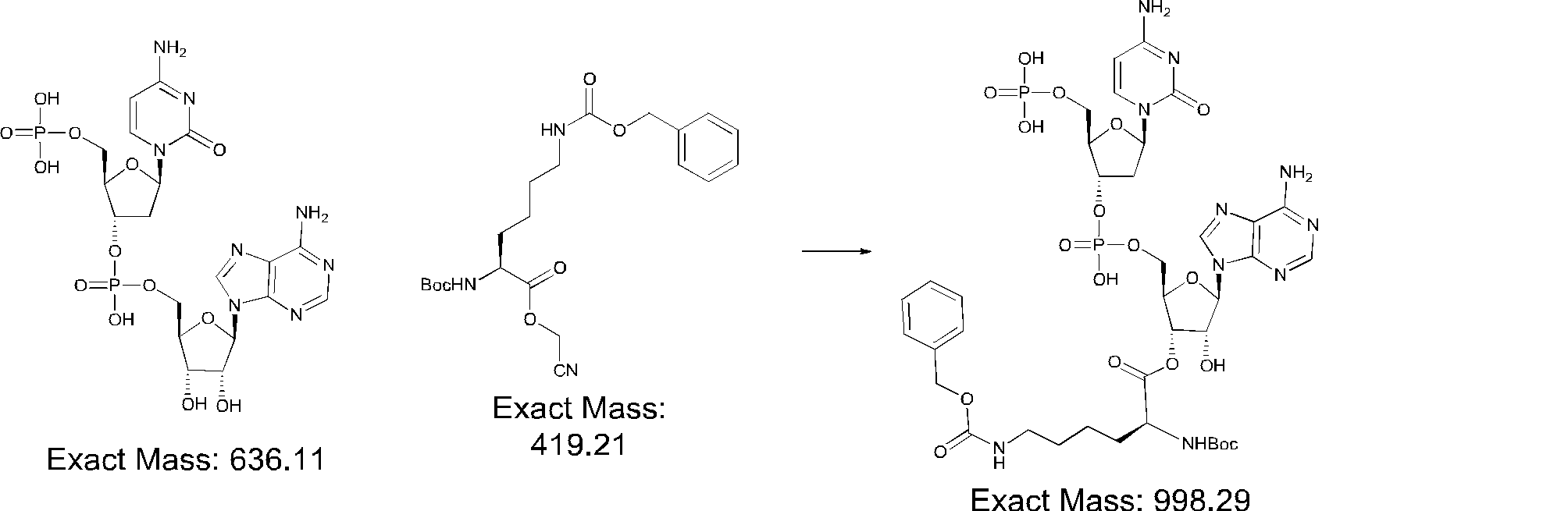
LCMS(ESI) m/z = 999.7 (M+H)+
保持時間:0.53分(分析条件SQDFA05)
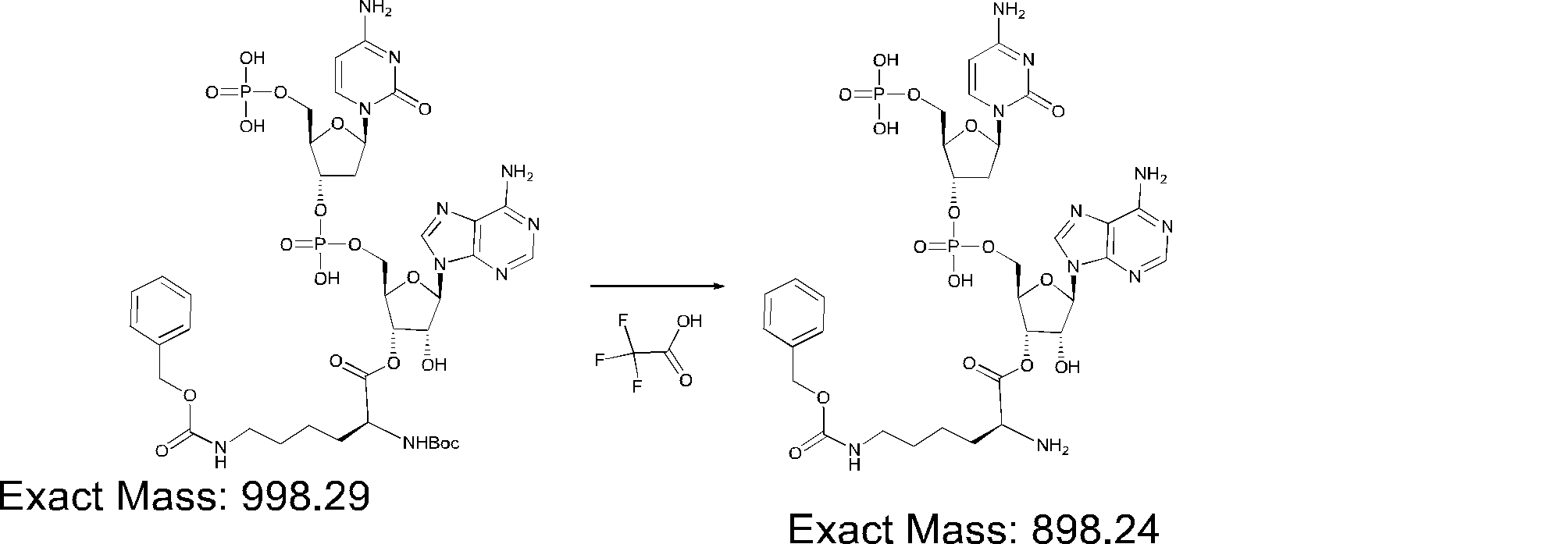
LCMS(ESI) m/z = 897.4 (M-H)-
保持時間:0.35分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。

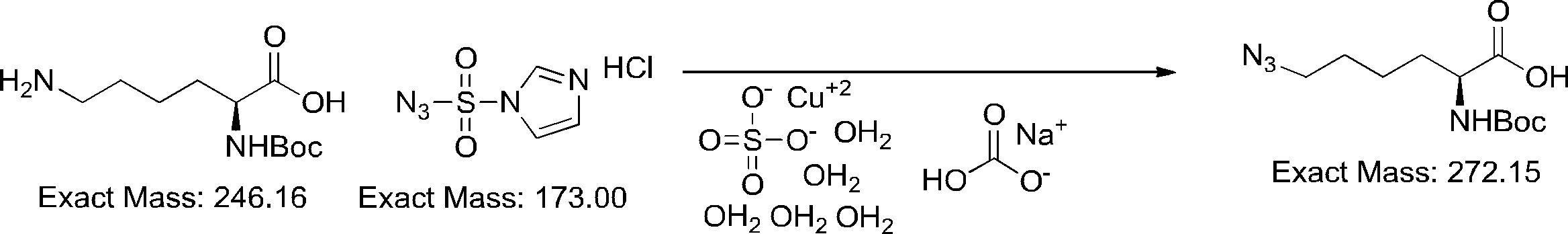
LCMS(ESI) m/z = 271 (M-H)-
保持時間:0.66分(分析条件SQDFA05)
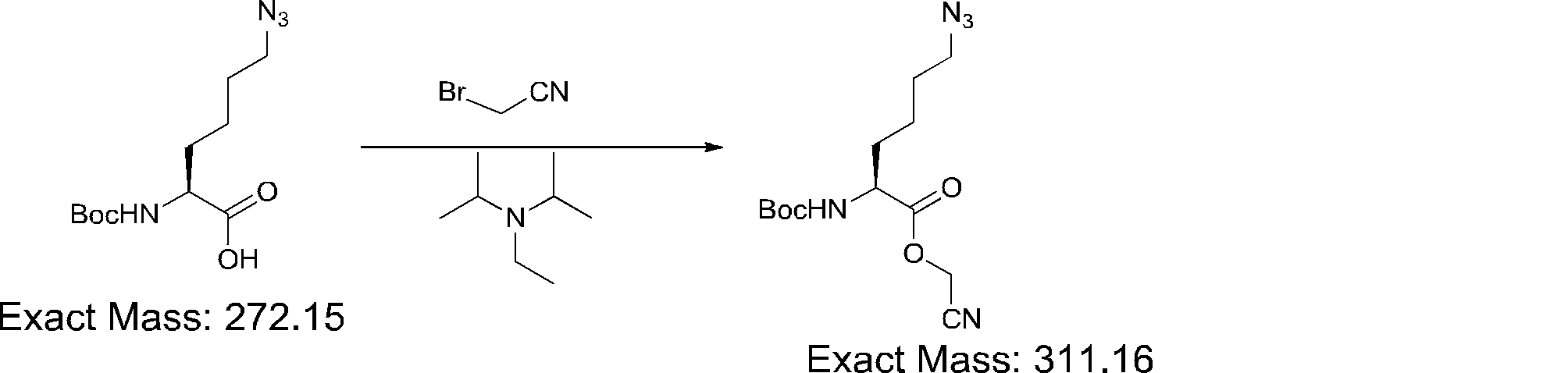
LCMS(ESI) m/z = 310 (M-H)-
保持時間:0.77分(分析条件SQDFA05)

LCMS(ESI) m/z = 889.4 (M-H)-
保持時間:0.47分(分析条件SQDFA05)
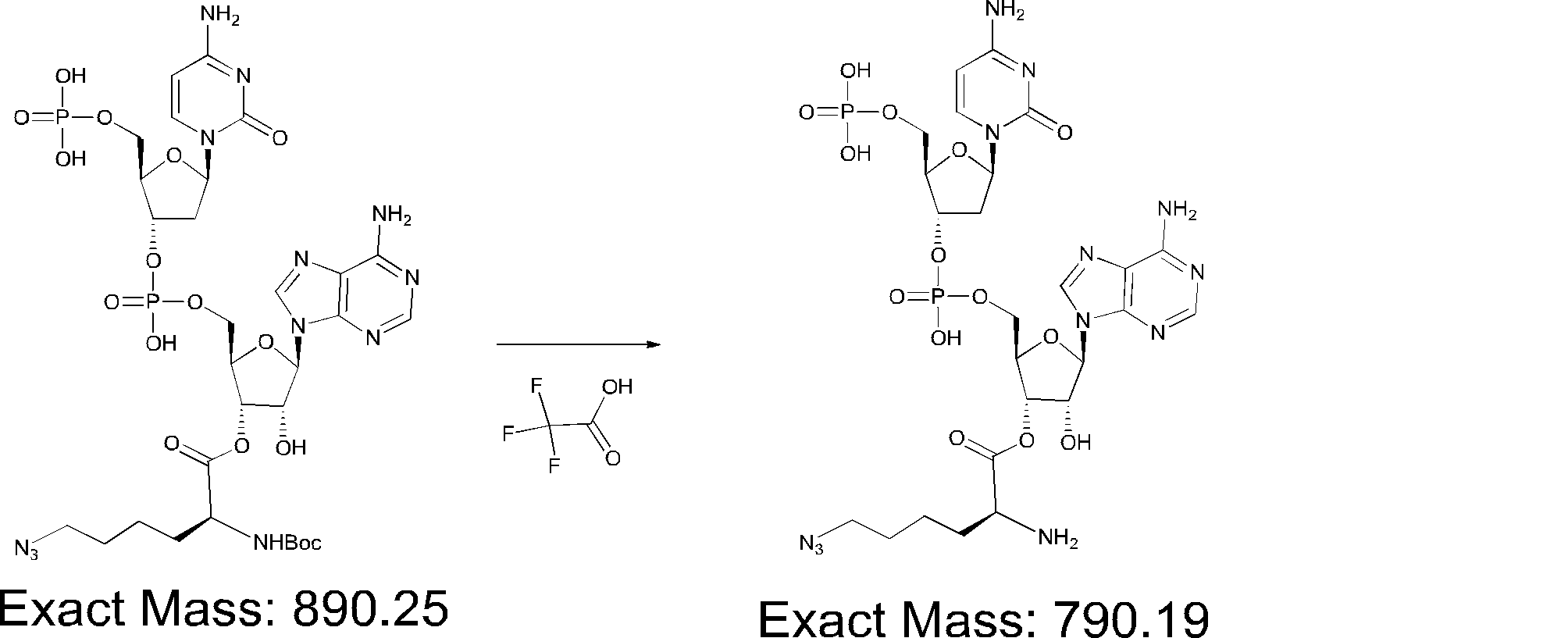
LCMS(ESI) m/z = 789.4 (M-H)-
保持時間:0.27分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
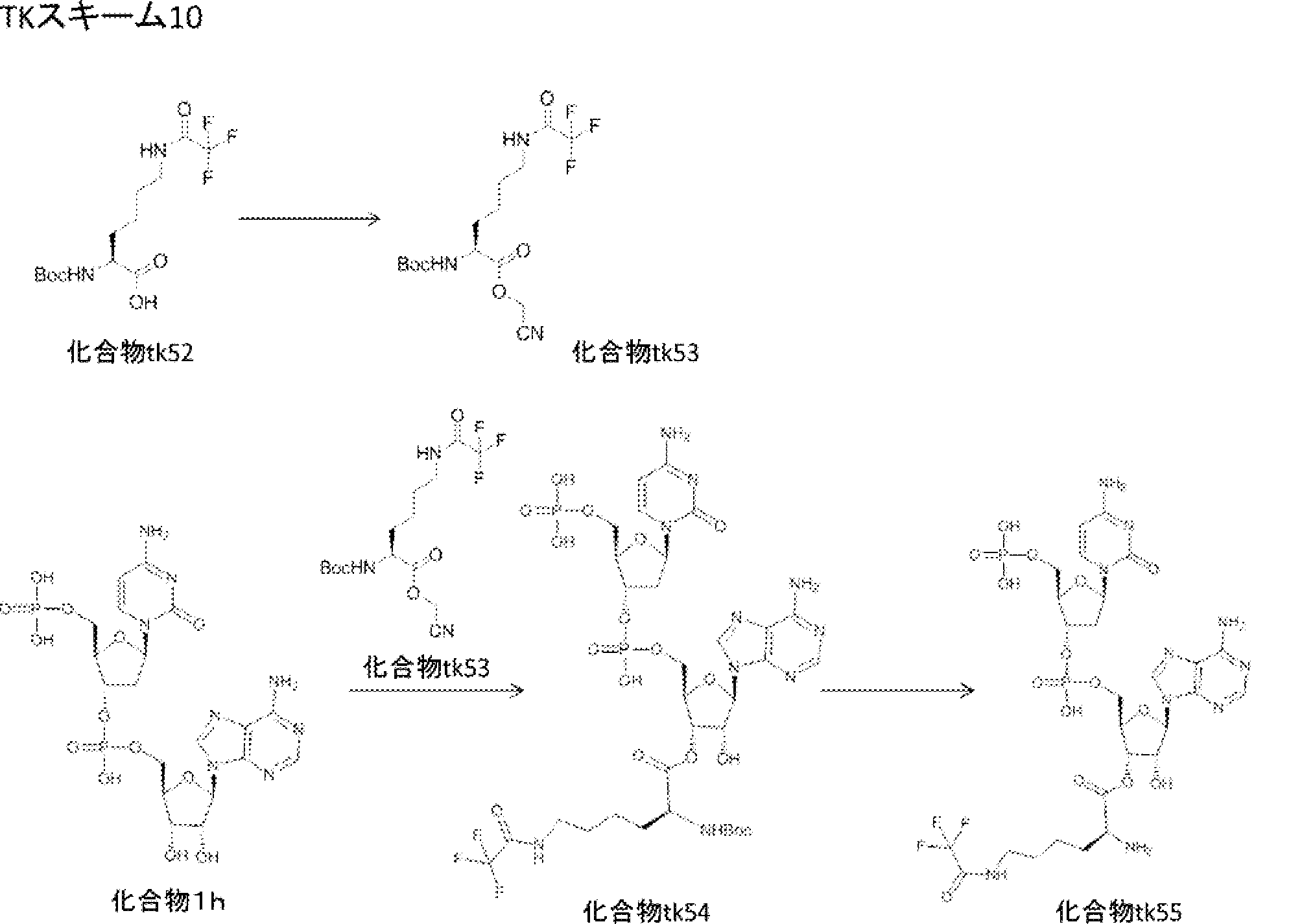

LCMS(ESI) m/z = 379.9 (M-H)-
保持時間:0.70分(分析条件SQDFA05)
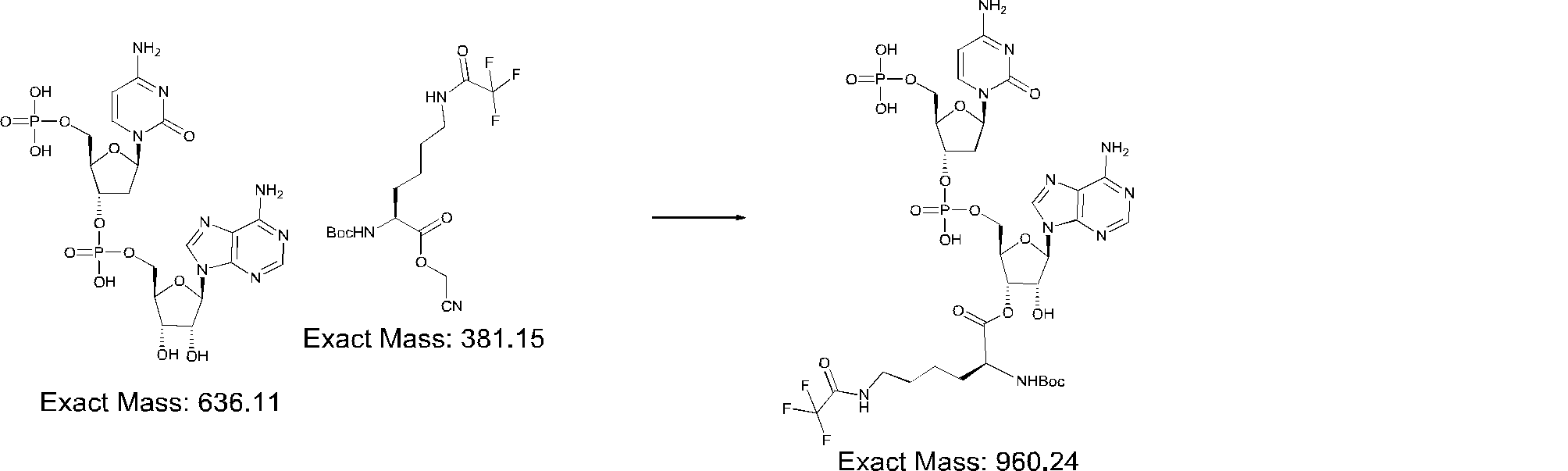
LCMS(ESI) m/z = 959.5 (M-H)-
保持時間:0.44分(分析条件SQDFA05)

LCMS(ESI) m/z = 859.4 (M-H)-
保持時間:0.26分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
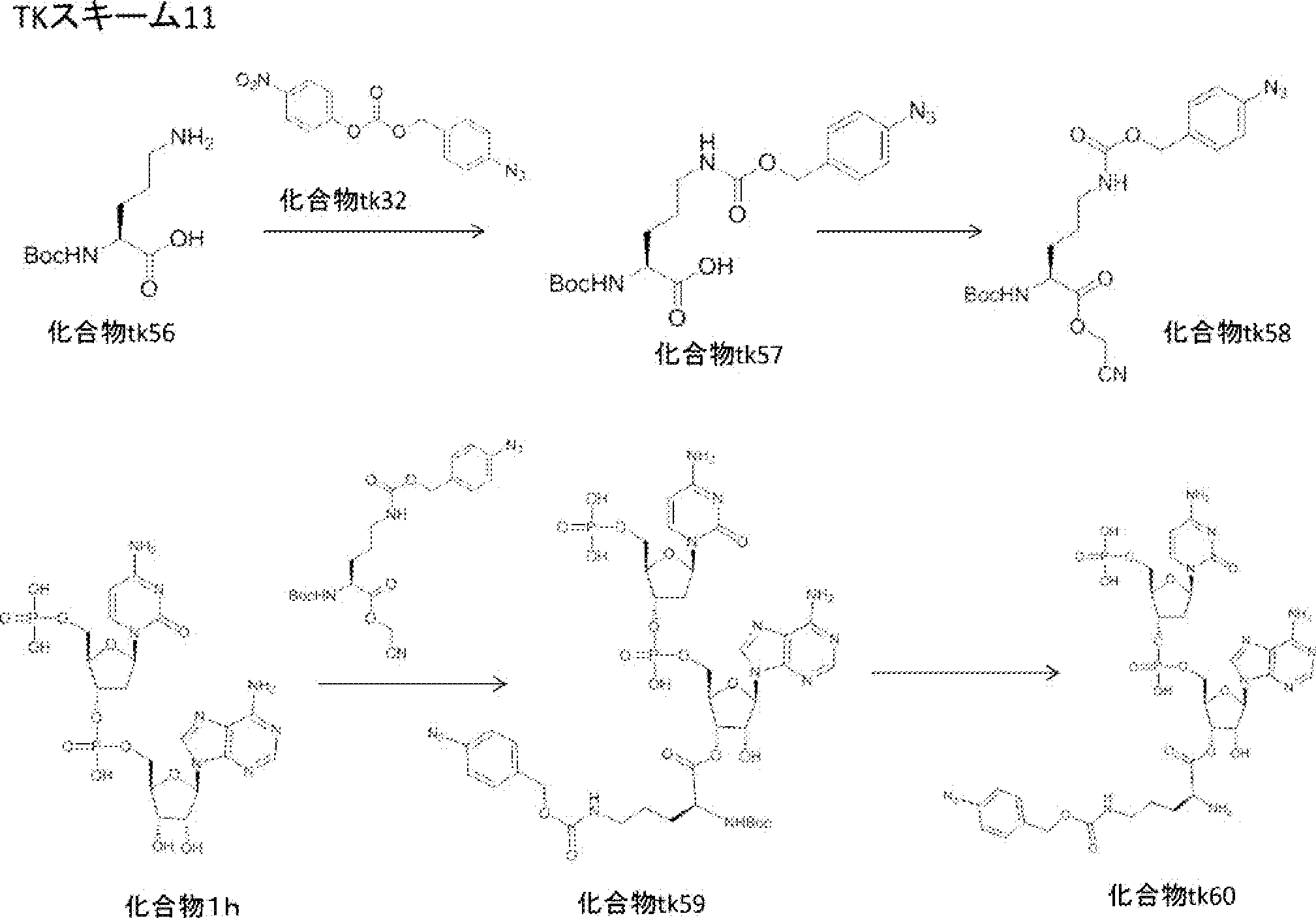

LCMS(ESI) m/z = 406 (M-H)-
保持時間:0.73分(分析条件SQDFA05)

LCMS(ESI) m/z = 445 (M-H)-
保持時間:0.82分(分析条件SQDFA05)
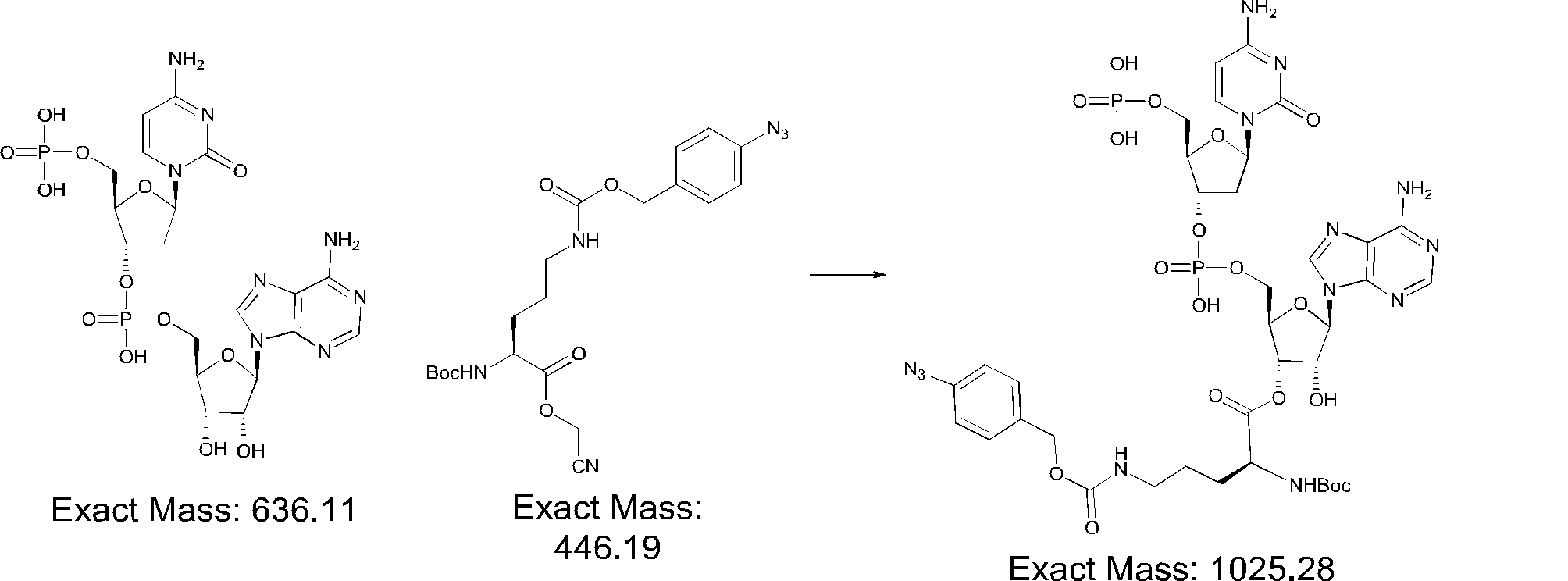
LCMS(ESI) m/z = 1024.5 (M-H)-
保持時間:0.54分(分析条件SQDFA05)

LCMS(ESI) m/z = 924.3 (M-H)-
保持時間:0.37分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
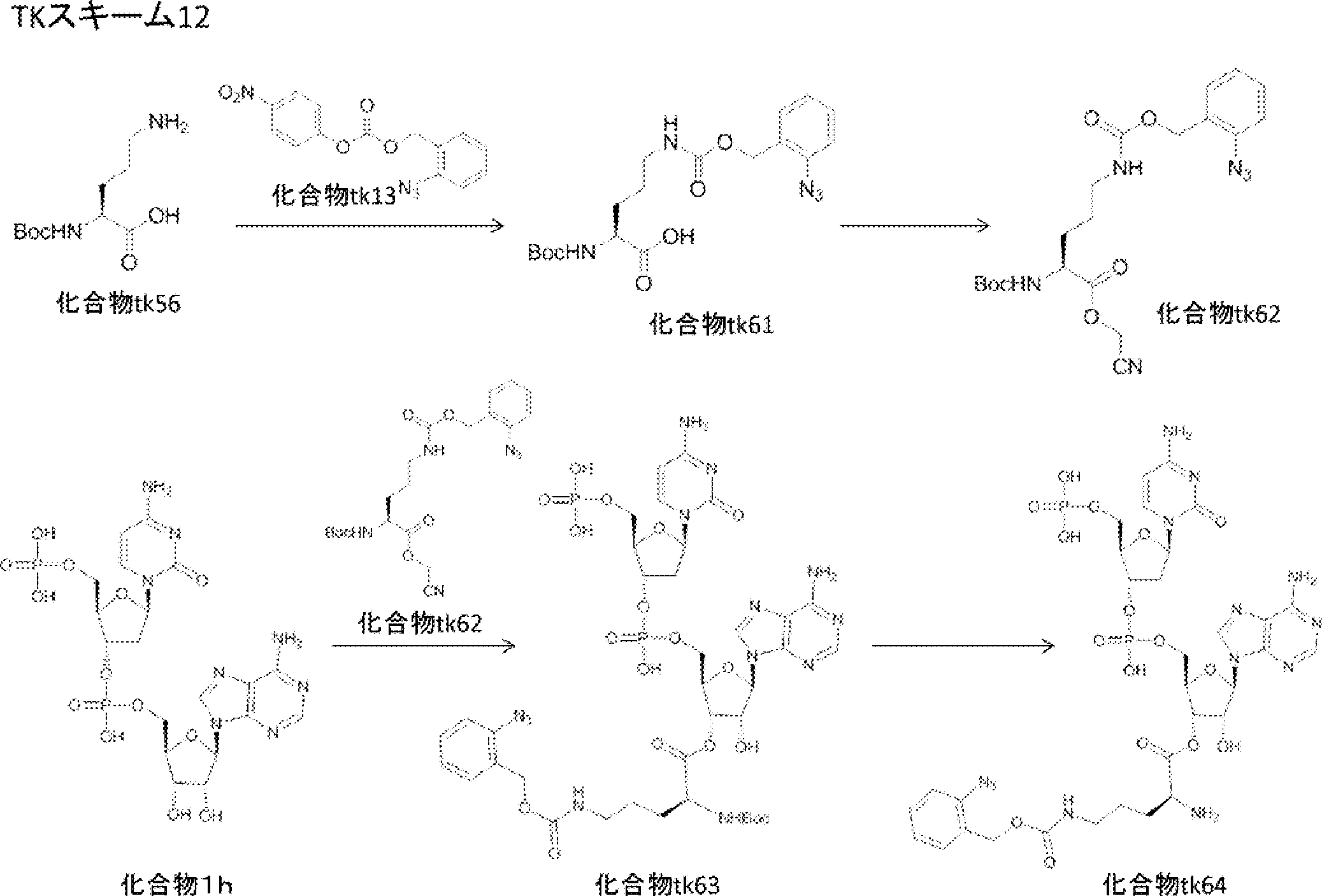

LCMS(ESI) m/z = 406 (M-H)-
保持時間:0.73分(分析条件SQDFA05)
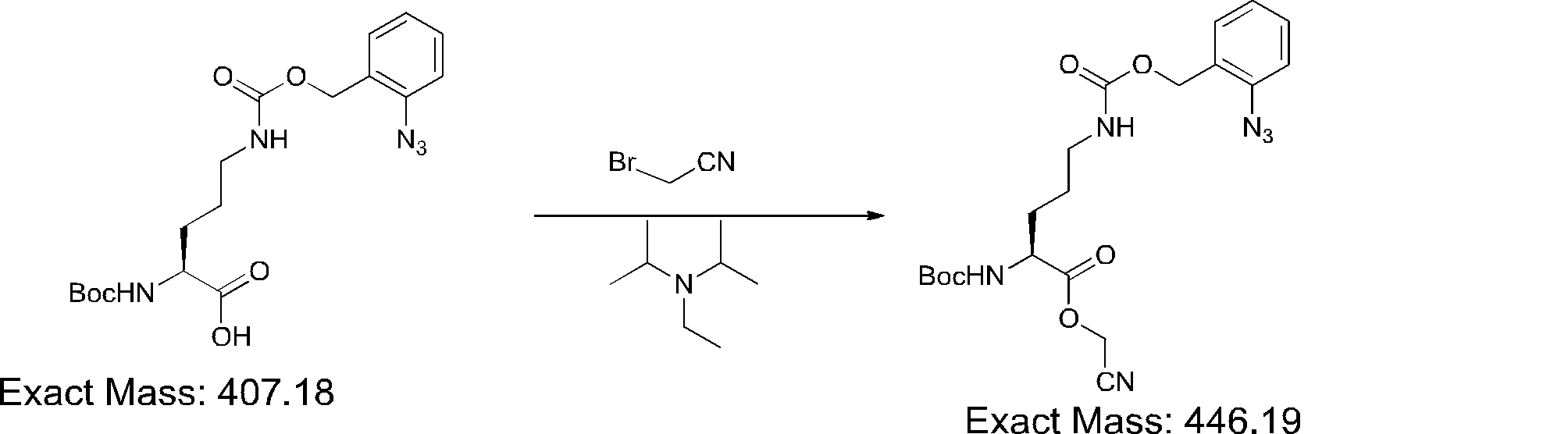
LCMS(ESI) m/z = 445.4 (M-H)-
保持時間:0.81分(分析条件SQDFA05)
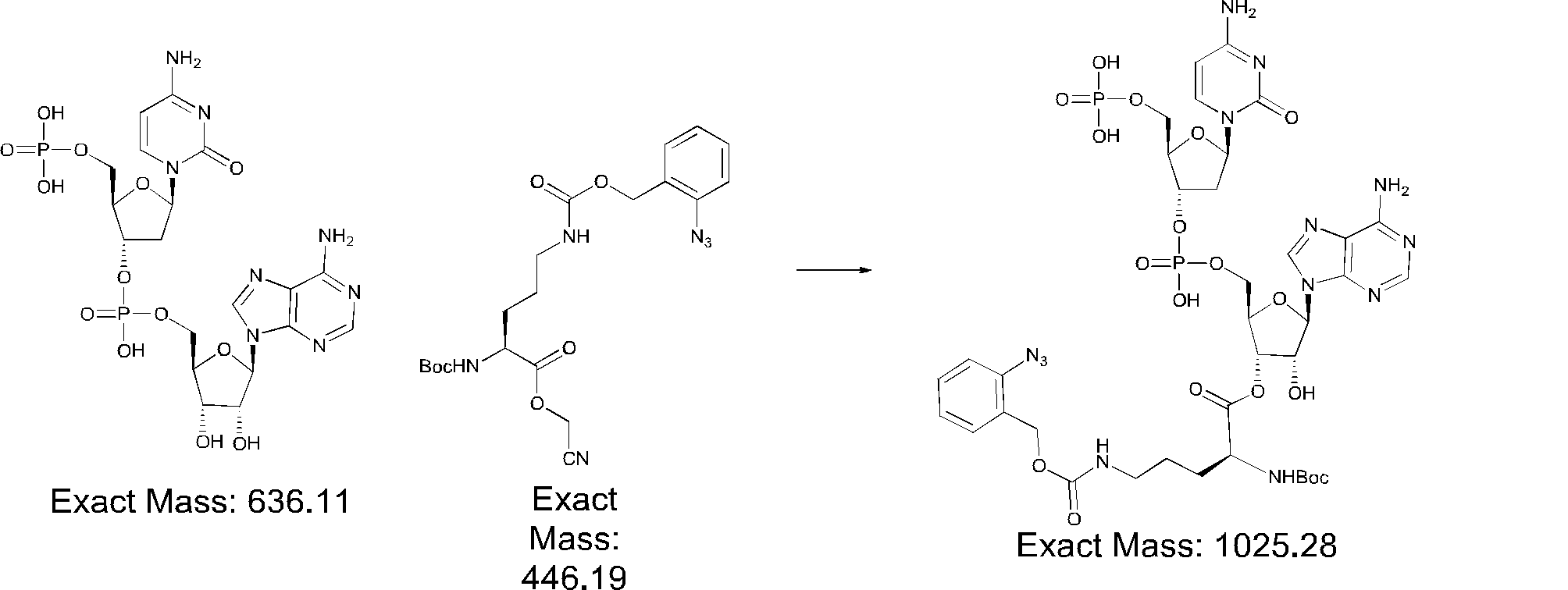
LCMS(ESI) m/z = 1024.5 (M-H)-
保持時間:0.54分(分析条件SQDFA05)

LCMS(ESI) m/z = 924.4 (M-H)-
保持時間:0.37分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
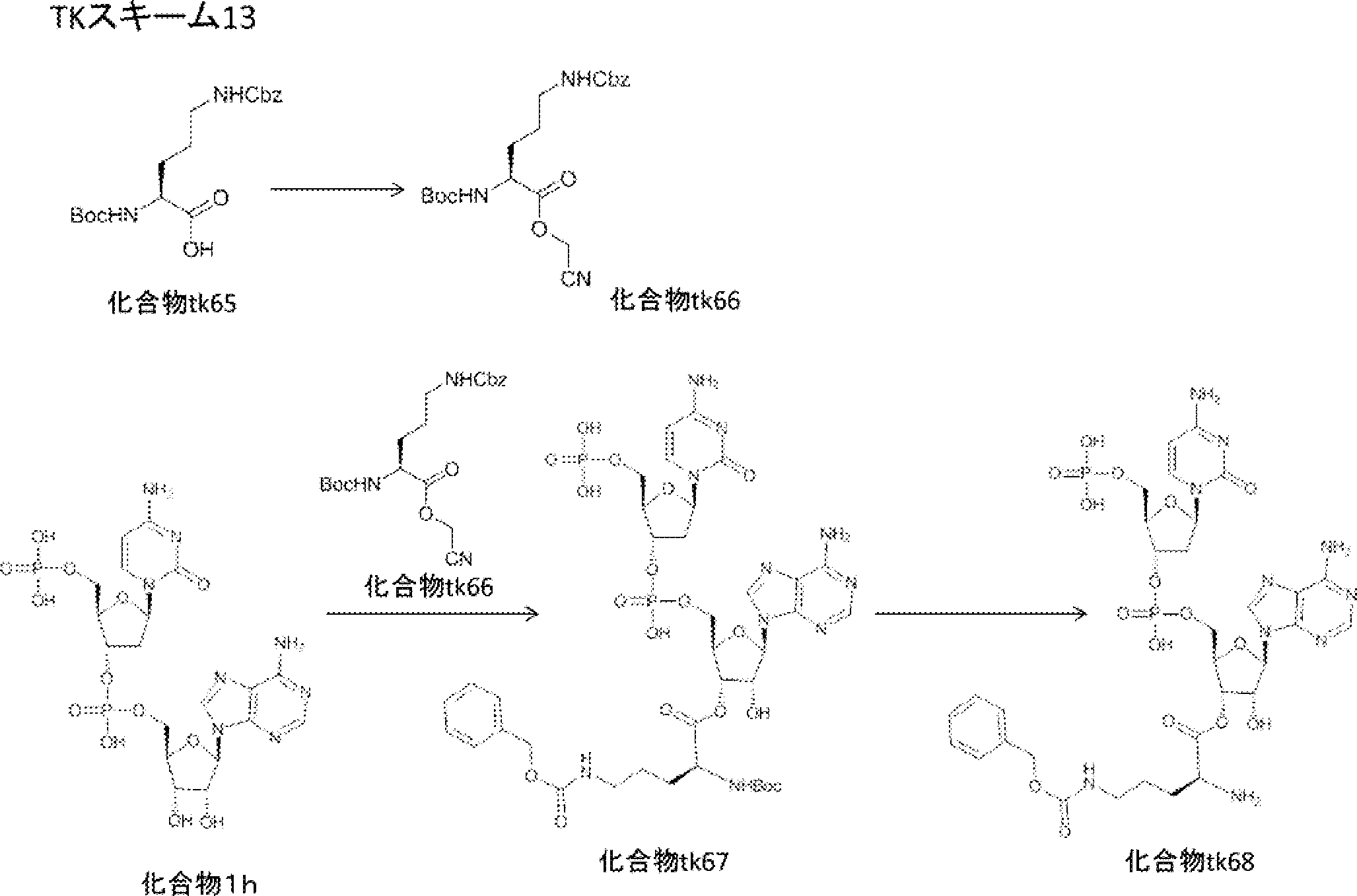

LCMS(ESI) m/z = 404 (M-H)-
保持時間:0.77分(分析条件SQDFA05)
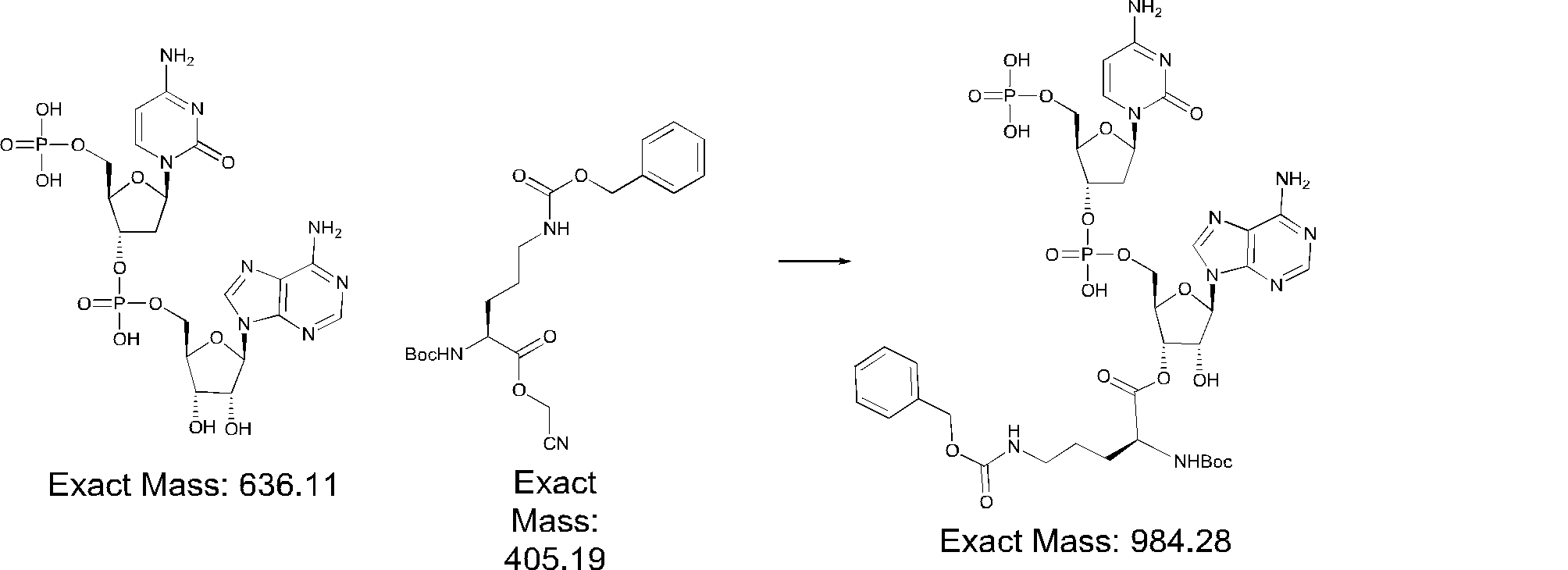
LCMS(ESI) m/z = 983.4 (M-H)-
保持時間:0.54分(分析条件SQDFA05)
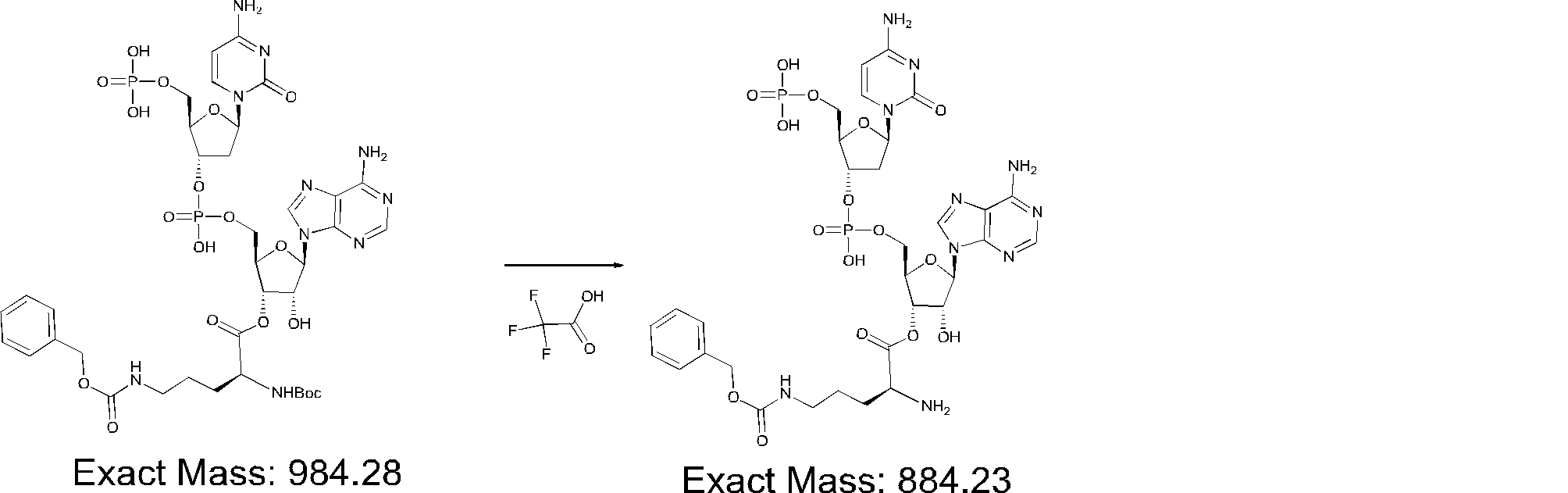
LCMS(ESI) m/z = 883.3 (M-H)-
保持時間:0.33分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
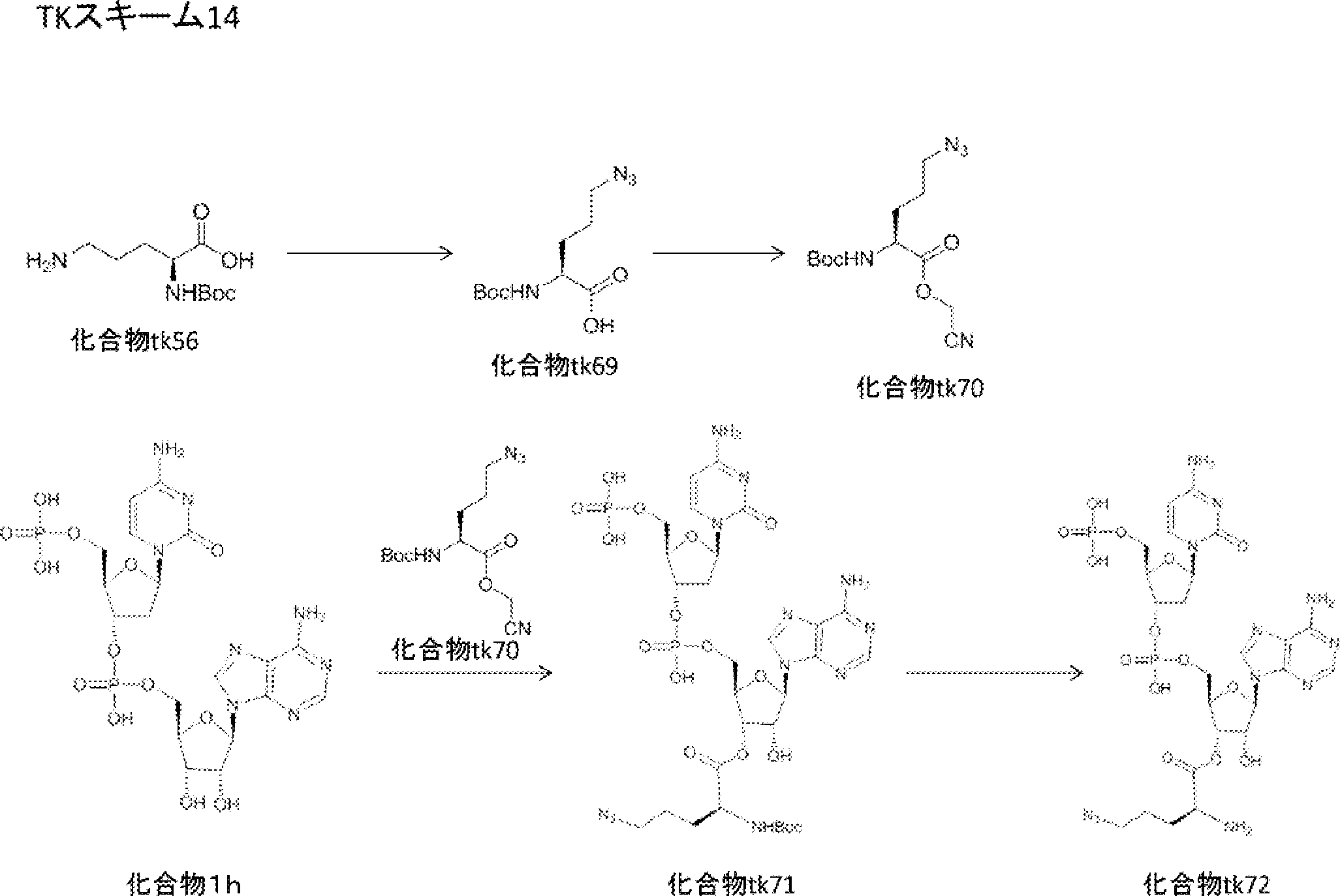
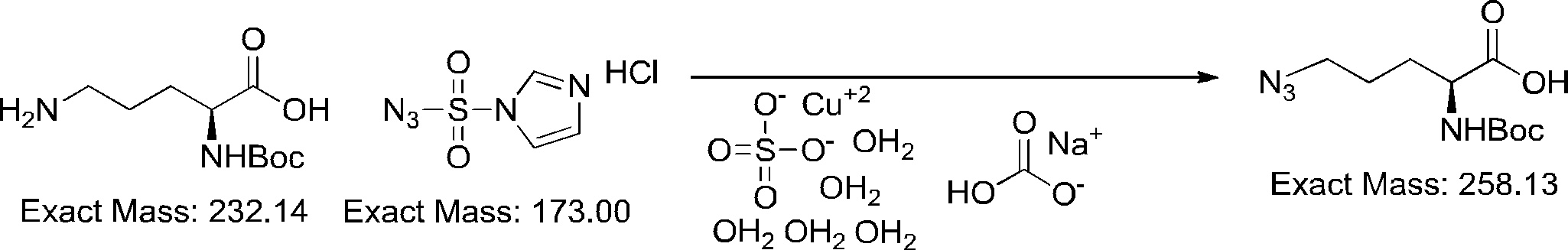
LCMS(ESI) m/z = 257 (M-H)-
保持時間:0.61分(分析条件SQDFA05)
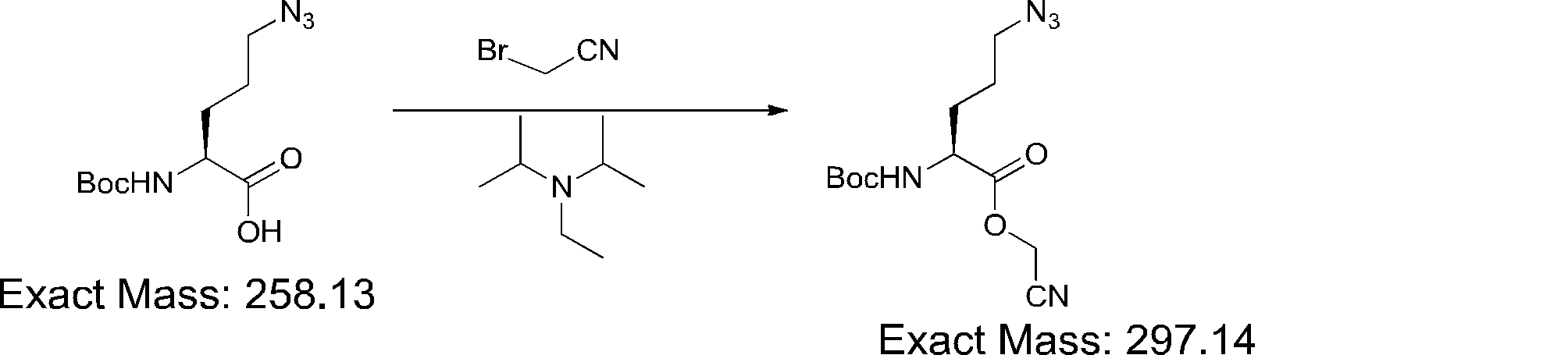
LCMS(ESI) m/z = 296 (M-H)-
保持時間:0.73分(分析条件SQDFA05)
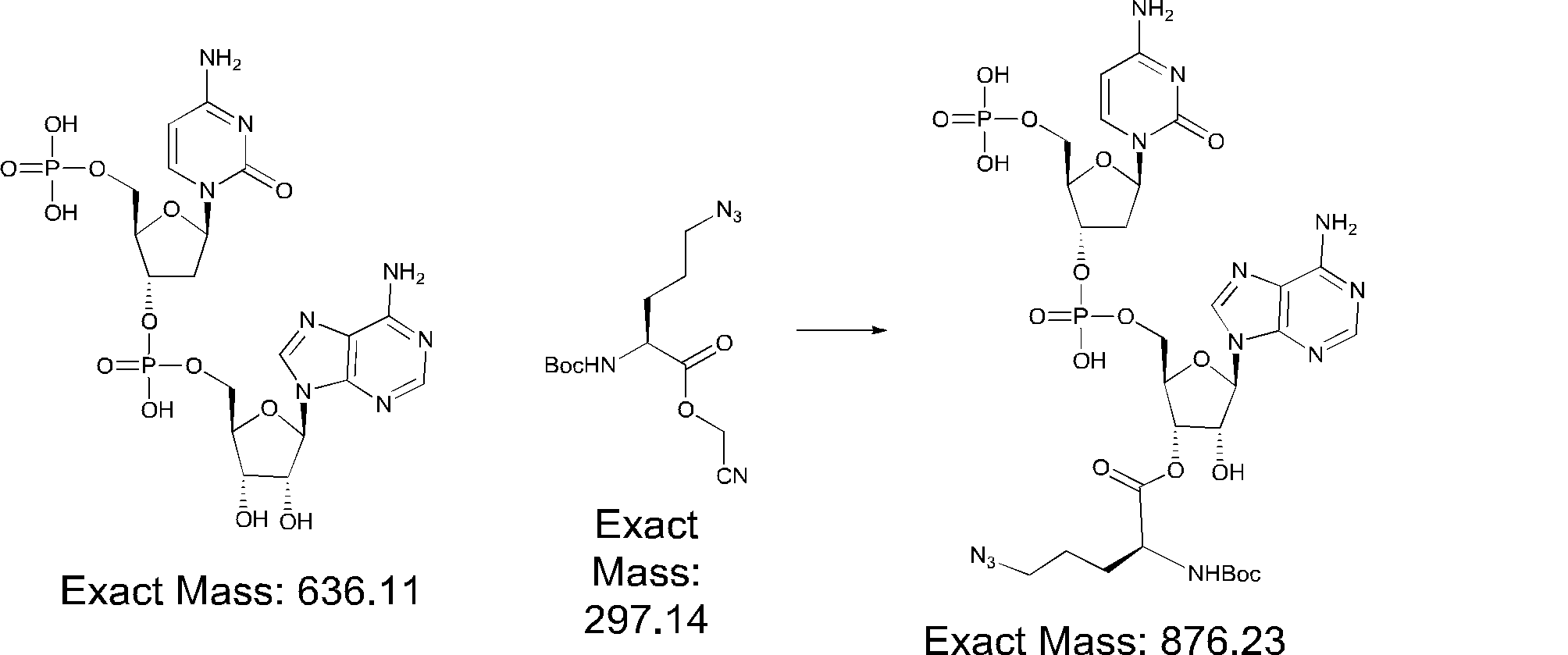
LCMS(ESI) m/z = 875.4 (M-H)-
保持時間:0.44分(分析条件SQDFA05)
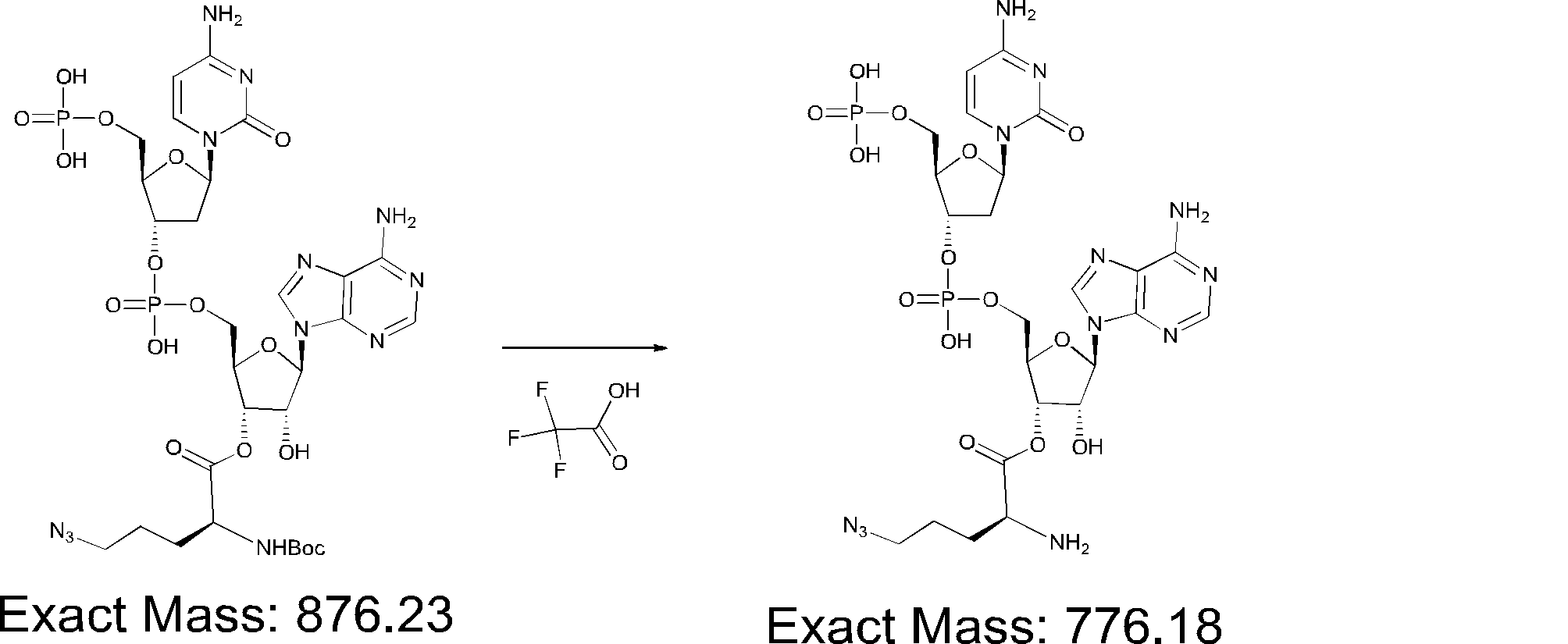
LCMS(ESI) m/z = 775.4 (M-H)-
保持時間:0.25分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。
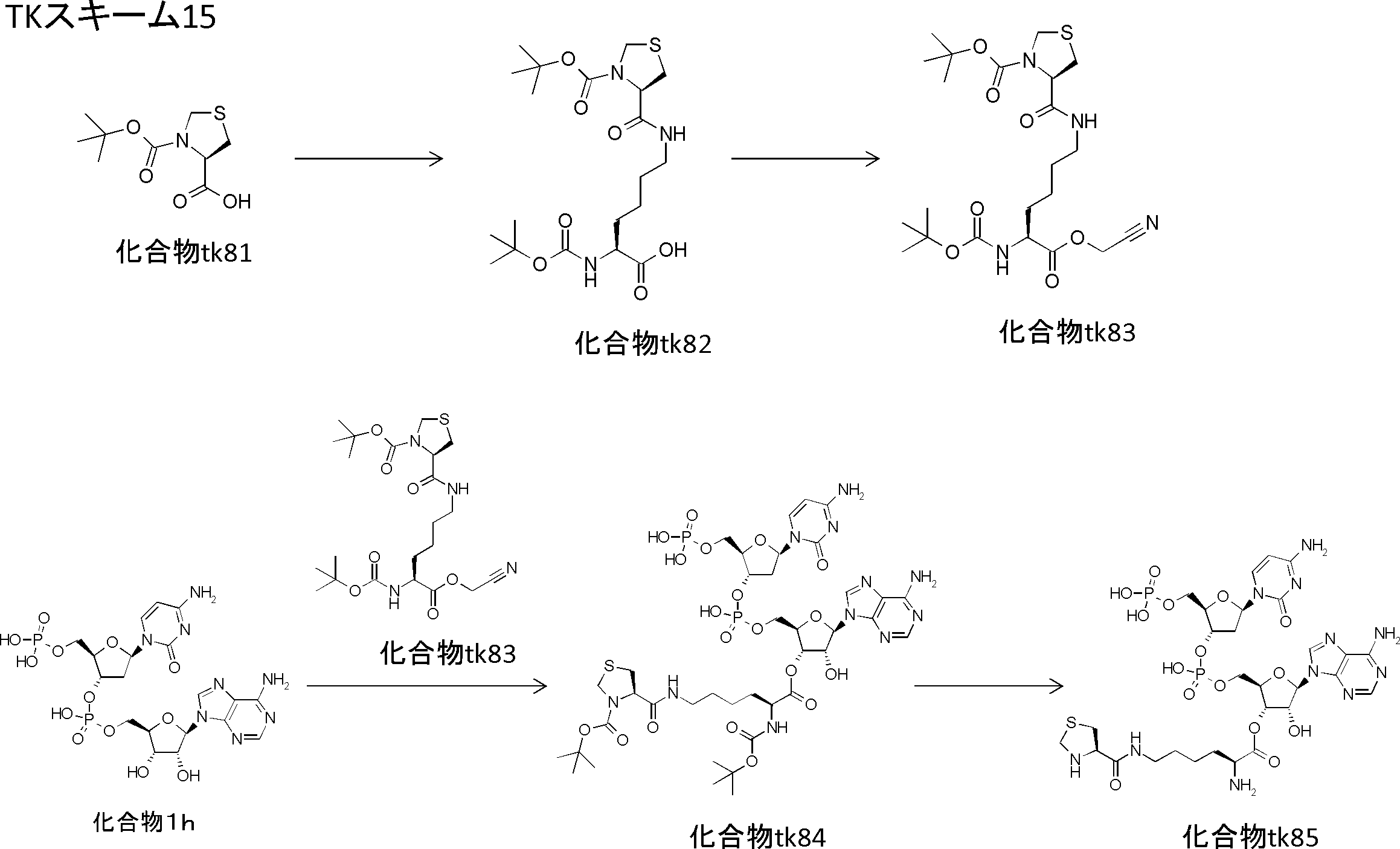

LCMS(ESI) m/z = 462 (M+H)+
保持時間:0.66分(分析条件SQDFA05)

LCMS(ESI) m/z = 501.5(M+H)+
保持時間:0.76分(分析条件SQDFA05)
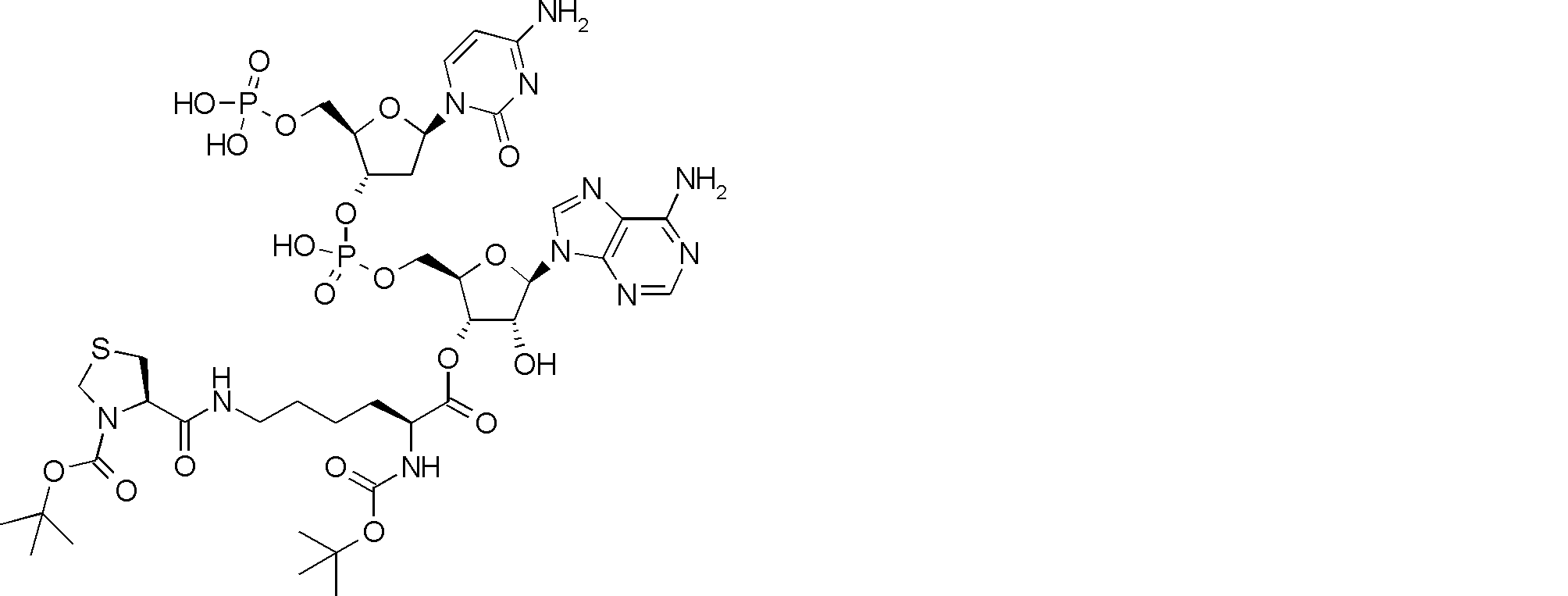
LCMS(ESI) m/z = 1078(M-H)-
保持時間:0.55分(分析条件SQDFA05)

LCMS(ESI) m/z = 878(M-H)-
保持時間:0.17分(分析条件SQDFA05)
以下のスキームに従い、合成を行った。


LCMS(ESI) m/z = 401(M+H)+
保持時間:0.74分(分析条件SQDFA05)

LCMS(ESI) m/z = 440(M+H)+
保持時間:0.83分(分析条件SQDFA05)
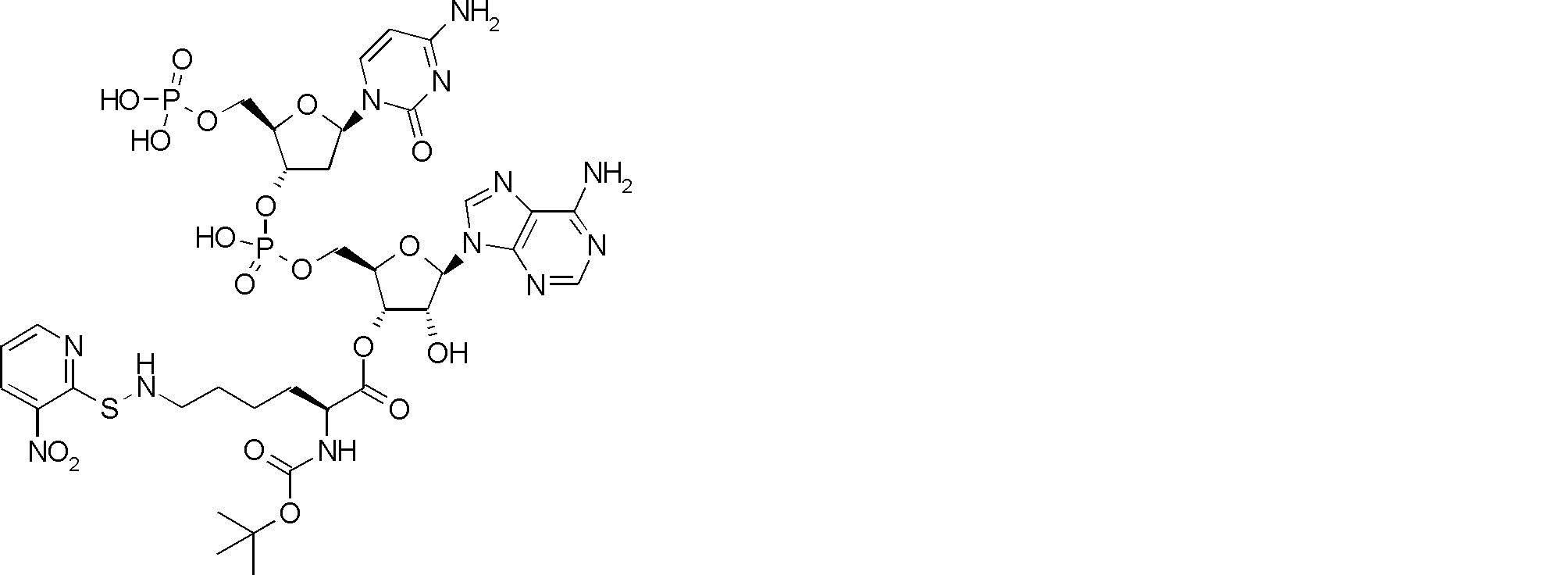
LCMS(ESI) m/z = 1017(M-H)-
保持時間:0.54分(分析条件SQDFA05)
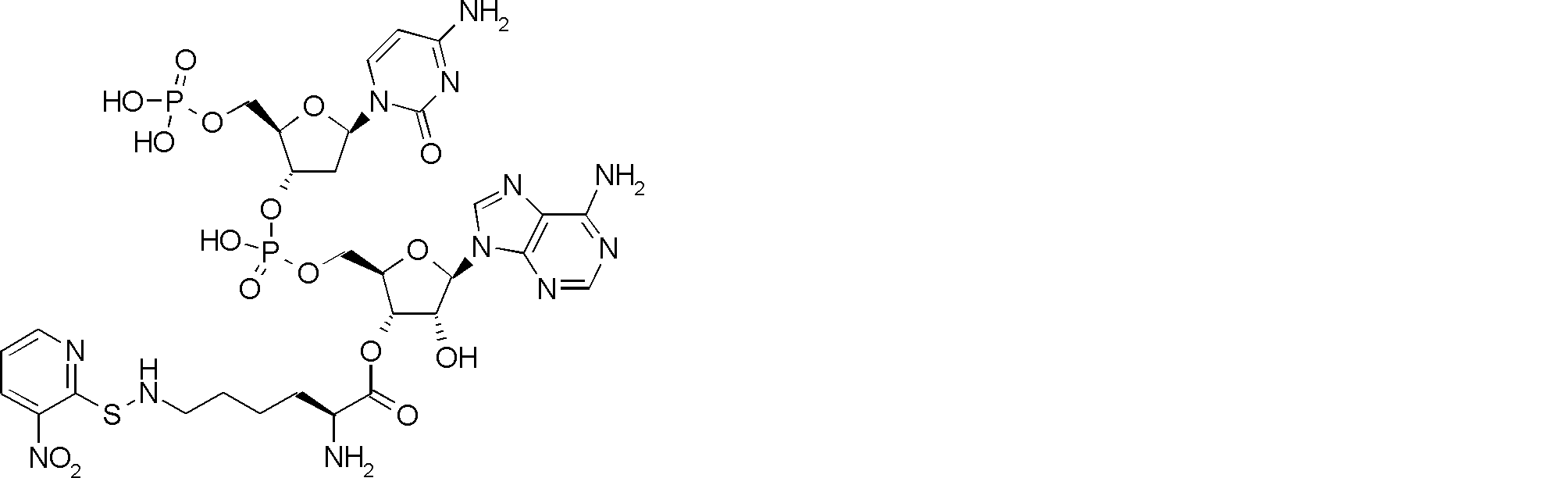
LCMS(ESI) m/z = 917(M-H)-
保持時間:0.34分(分析条件SQDFA05)
RNAが安定な反応条件にてアミノ基保護基が脱保護されることを確認、もしくは新条件を探索する目的で以下の実験を行い、RNAが安定に存在する反応条件でのアミノ基脱保護反応を得た。
以下のスキームに従い、合成を行った。
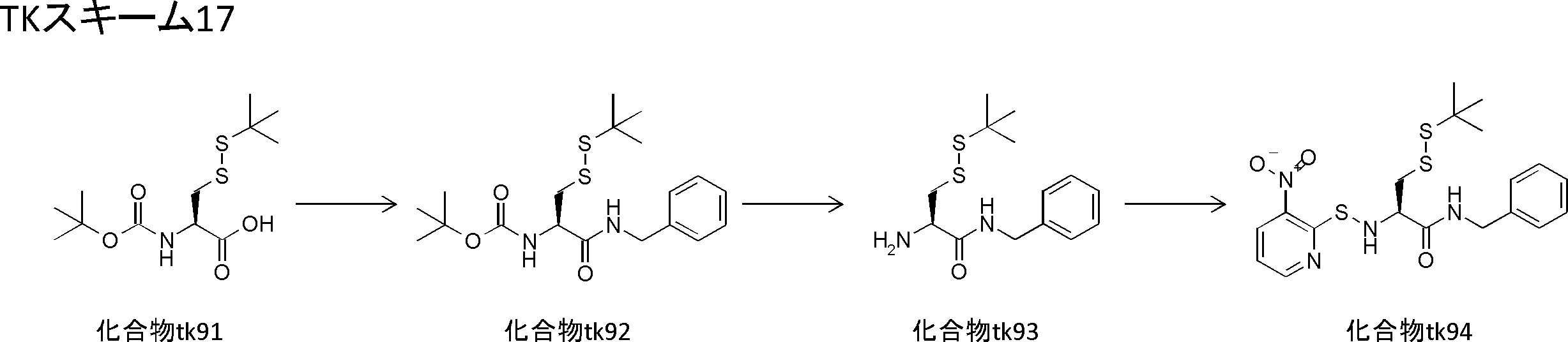

LCMS(ESI) m/z = 399 (M+H)+
保持時間:0.93分(分析条件SQDFA05)

LCMS(ESI) m/z = 297 (M-H)-
保持時間:0.93分(分析条件SQDAA05)

LCMS(ESI) m/z = 453 (M+H)+
保持時間:1.03分(分析条件SQDAA05)

LCMS(ESI) m/z = 299 (M+H)+
保持時間:0.90分(分析条件SQDAA05)
既知のNpys基の脱保護手法として、有機溶媒中、求核剤2-メルカプトピリジンを用い、更に反応液中に酢酸を添加することで脱保護反応を加速させる条件が報告されている(非特許文献 International journal of peptide and protein research, 1990, 35, 545-549)。本発明者らは、従来法の改良を行った結果、RNAが安定して存在する条件で、かつNpys基の脱保護反応が容易に進行する手法を見出した。実際にRNAを本反応条件に伏した結果、RNAは安定性して存在することが確認され(表19 反応条件E、および図80 レーン5)、本反応条件の有用性が証明された。
以下のスキームに従い、合成を行った。
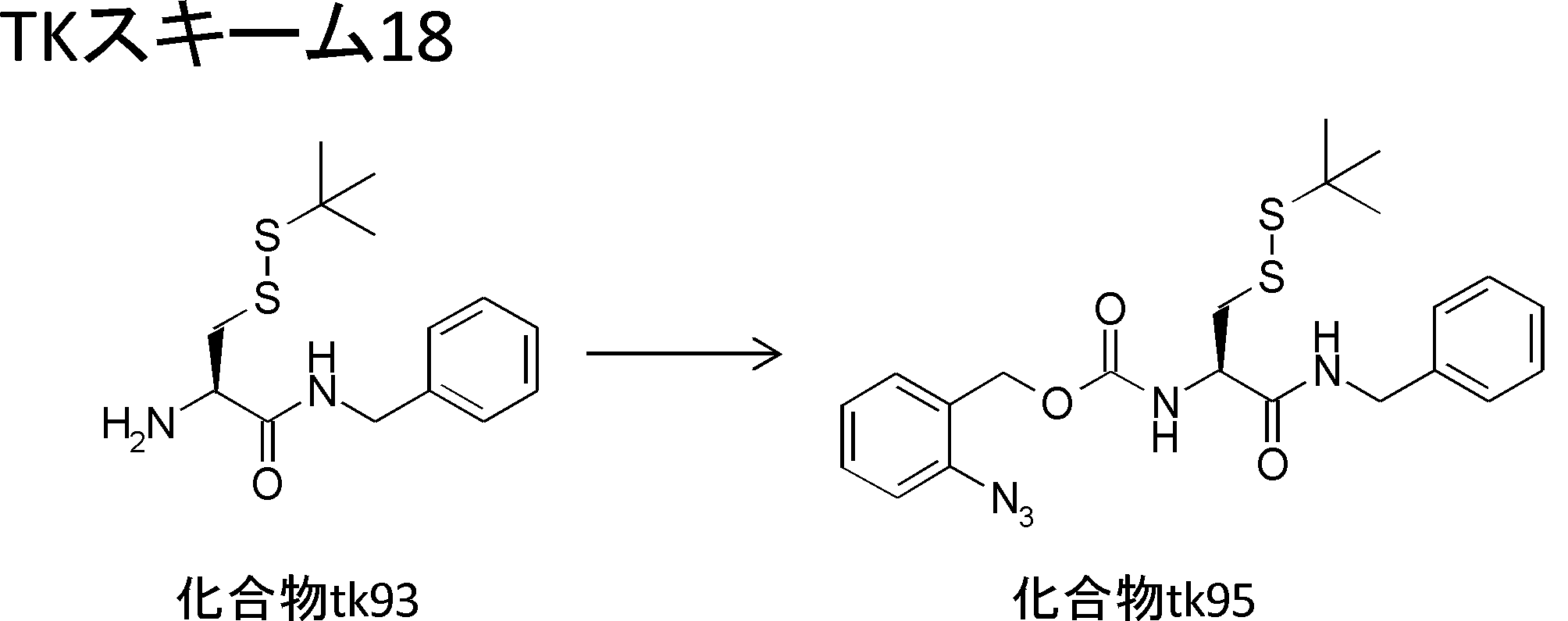

LCMS(ESI) m/z = 474.4 (M+H)+
保持時間:0.97分(分析条件SQDFA05)
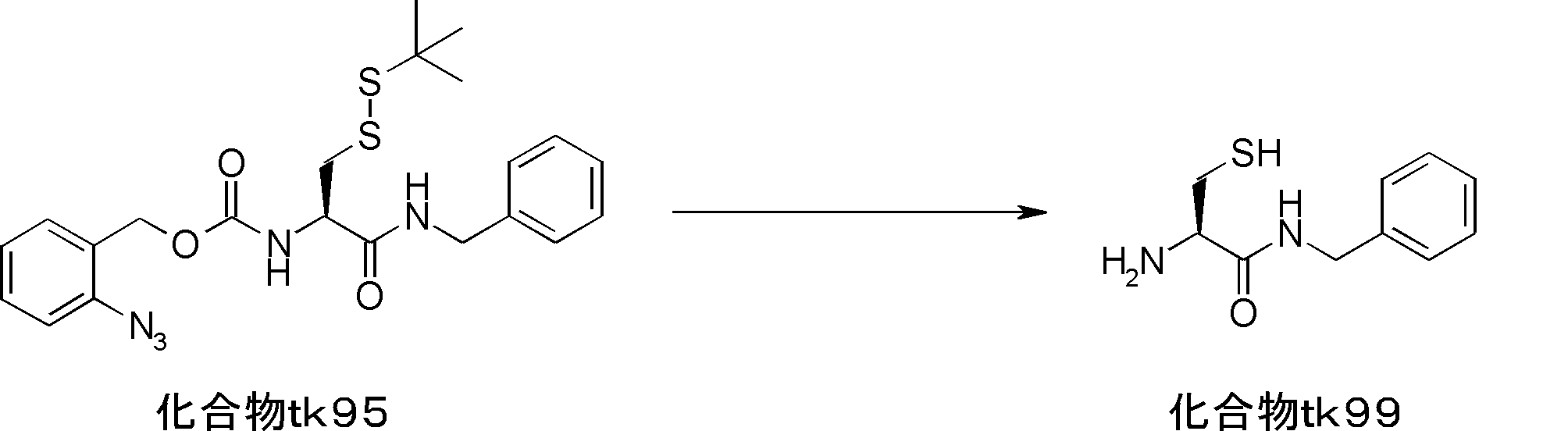
化合物tk99のLCMSの保持時間と質量電荷比は以下のとおりである。
LCMS(ESI) m/z = 211(M+H)+
保持時間:0.61分(分析条件SQDAA05)
以下のスキームに従い、合成を行った。
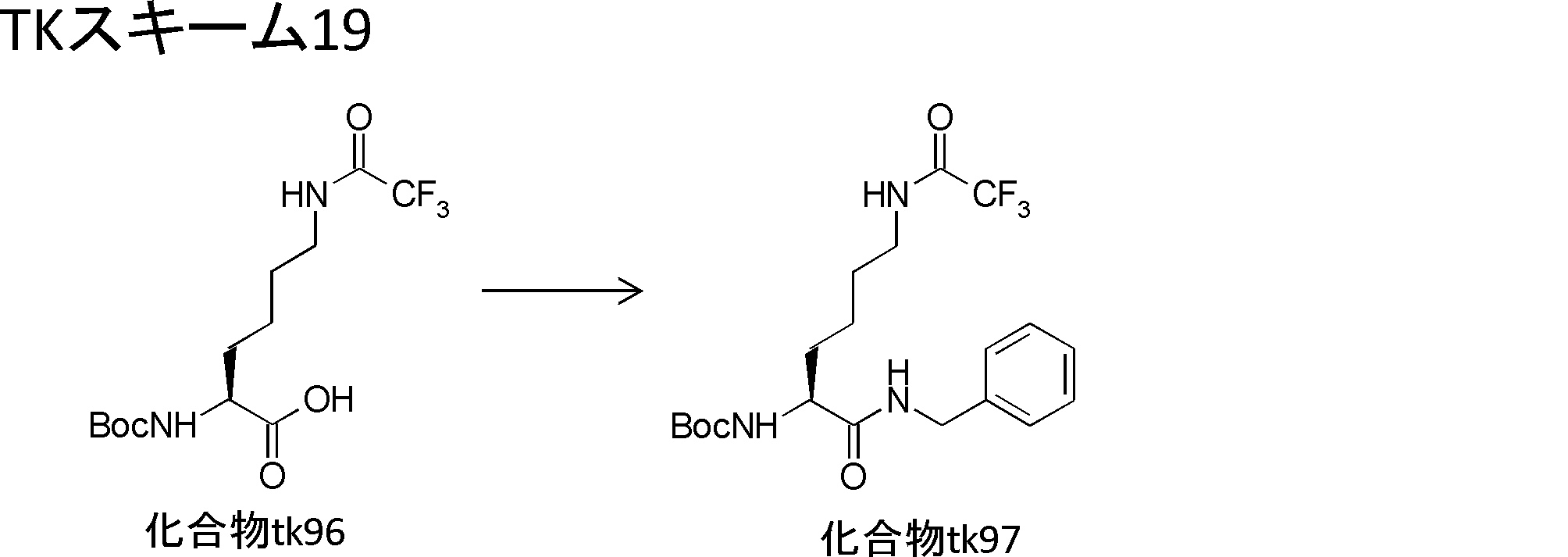
窒素雰囲気下、Boc-Lys(Tfa)-OH(化合物tk96)(104mg、0.304mmol)及び1H-ベンゾ[d][1,2,3]トリアゾール-1-オール(HOBT)(61.6mg、0.456mmol)のDMF(0.5mL)溶液を氷浴で冷却後、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(WSCI・HCl)(87mg、0.456mmol)を添加した。反応混合物を同温度で5分間撹拌後、ベンジルアミン(40μL、0.365mmol)を添加した。反応混合物を室温にて17.5時間撹拌後、酢酸エチル、ヘキサン及び食塩水を添加した。得られた混合物を酢酸エチルにて抽出し、有機相を食塩水(1mLで2回)にて洗浄後、硫酸ナトリウムで乾燥した。減圧濃縮して得られた残渣を順相シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)にて精製し、(1-(ベンジルアミノ)-1-オキソ-6-(2,2,2-トリフルオロアセトアミド)ヘキサン-2-イル)カルバミン酸 (S)-tert-ブチル(化合物tk97)(134.4mg、quant.)を得た。
LCMS(ESI) m/z = 430.4 (M-H)-
保持時間:0.73分(分析条件SQDFA05)
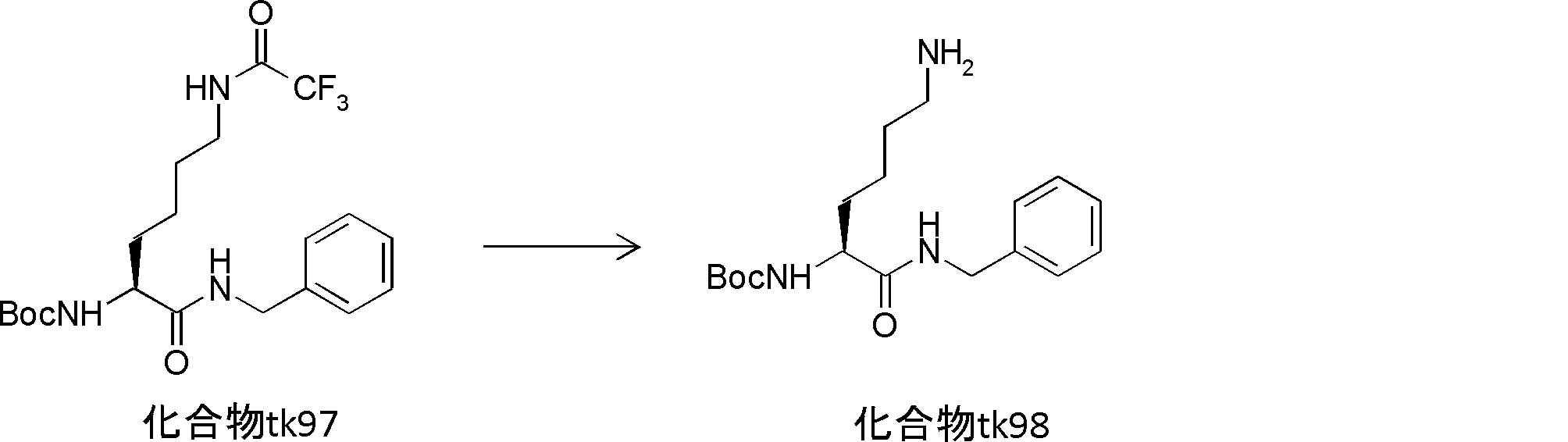
17時間後:化合物tk98:化合物tk97=19:81
93.5時間後:化合物tk98:化合物tk97=70:30
LCMS(ESI) m/z = 336.4 (M+H)+
保持時間:0.74分(分析条件SQDAA05)
以下に示す化合物SP616から化合物618への変換反応の実験にて、別途証明済である
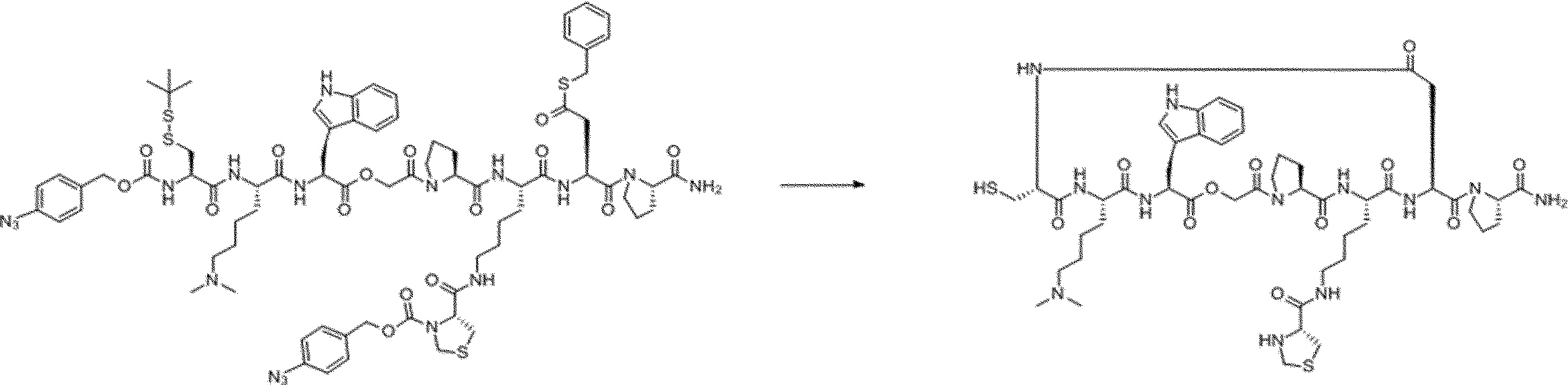
以下に示す化合物SP618から化合物620への変換反応の実験にて、別途証明済である
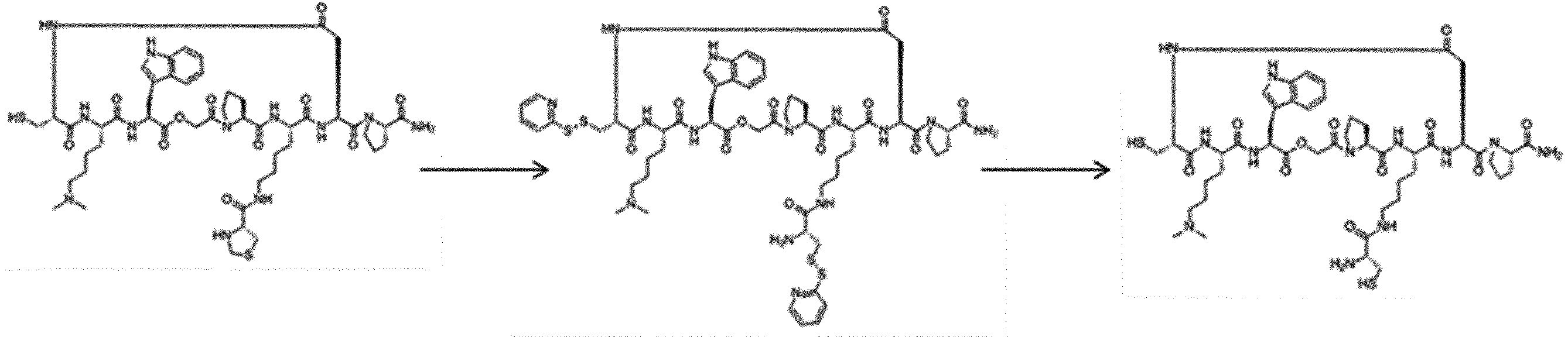
solA: 次の成分を含んだ混合物 8mM GTP,8mM ATP,160mMクレアチンリン酸,400mM HEPES-KOH pH7.6,800mM 酢酸カリウム,48mM 酢酸マグネシウム,16mMスペルミジン,8mM ジチオスレイトール,0.8mM 10-HCO-H4folate, 12mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社)
solB: PURESYSTEM(r) classic II Sol. B(バイオコゥマー社、製品番号PURE2048C)
20種天然アミノ酸溶液: 20種の天然アミノ酸溶液(それぞれ5mM)
HO Gly:グリコール酸
Lac:L-(+)-乳酸
PhLac:(S)-2-ヒドロキシ-3-フェニルプロパン酸
D PhLac: (R)-2-ヒドロキシ-3-フェニルプロパン酸
HO Gly(Me) 2 :2-ヒドロキシ-2-メチルプロパン酸
環状化反応と続く分枝骨格生成反応の二つの反応を厳密に制御することを可能にする、温和な条件下で脱保護可能な側鎖アミノ基を含むアミノ酸の翻訳導入、及びMALDI-MSによる解析を行った。
3-1. 転写によるtRNA(CA欠損)の合成
鋳型DNA(配列番号DT-H3)から、RiboMAX Large Scale RNA production System T7(Promega社,P1300)を用いたin vitro の転写により3'端のCAを欠くtRNAAsn-E2GUU(-CA)(配列番号RT-H3)を合成し、RNeasy Mini kit(Qiagen社)により精製した。
tRNAAsn-E2GUU (-CA) DNA配列:
GGCGTAATACGACTCACTATAGGCTCTGTAGTTCAGTCGGTAGAACGGCGGACTgttAATCCGTATGTCACTGGTTCGAGTCCAGTCAGAGCCGC
tRNAAsn-E2GUU (-CA) RNA配列:
GGCUCUGUAGUUCAGUCGGUAGAACGGCGGACUguuAAUCCGUAUGUCACUGGUUCGAGUCCAGUCAGAGCCGC
50μM 転写tRNAAsn-E2GUU (-CA)(配列番号RT-H3) 20μLに、10X ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2) 4μL、, 10 mM ATP 4μL、Nuclease free water 5.6μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 T4 RNAリガーゼ(New England Bio Lab.社)2.4μLおよび、5mMの側鎖アミノ基が保護されたアミノアシル化pdCpA(化合物tk5、tk60、tk64、tk90、tk38、tk11のいずれか一種類)のDMSO溶液 4μLを加え、16℃で45分間ライゲーション反応を行った。アミノアシル化tRNA(化合物AT-H3)は、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-H3)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Ala(Tzm)-tRNAAsn-E2GUU(配列番号:96)
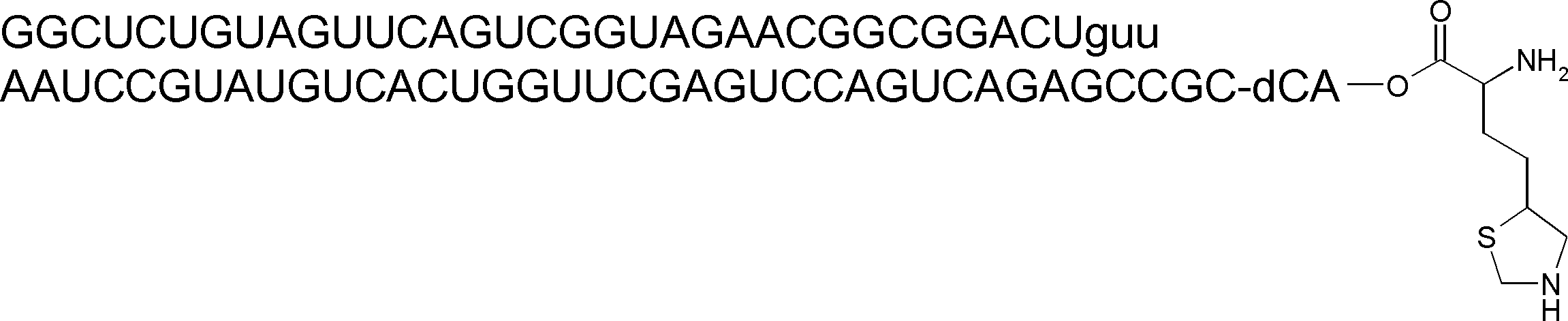
Orn(Acbz)-tRNAAsn-E2GUU(配列番号:96)
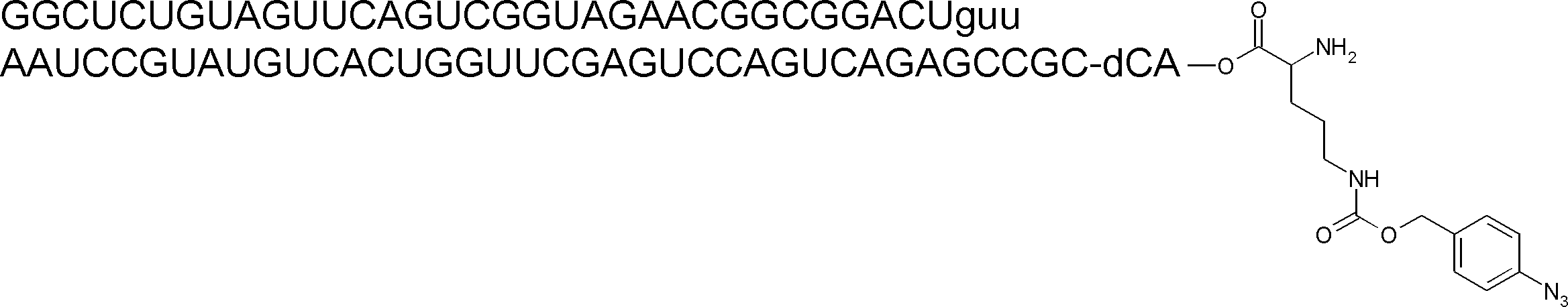
Orn(oAcbz)-tRNAAsn-E2GUU(配列番号:96)

Lys (Npys)-tRNAAsn-E2GUU(配列番号:96)

Lys(Acbz)-tRNAAsn-E2GUU(配列番号:96)

Lys(5S-SSMe)(Acbz)-tRNAAsn-E2GUU(配列番号:96)

様々なアミノ酸によりアミノアシル化されたtRNA(化合物AT-H3)を、無細胞翻訳系に加えて翻訳を開始することにより、所望の非天然アミノ酸を含有するポリペプチドの翻訳合成を行った。翻訳系は、原核生物由来の再構成無細胞タンパク質合成系であるPURE systemを用いた。具体的には、翻訳液,1%(v/v) RNasein Ribonuclease inhibitor (Promega社,N2111), 1mM GTP,1mM ATP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,6mM 酢酸マグネシウム,2mMスペルミジン,0.1mM 10-HCO-H4folate, 1.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.6μM メチオニルtRNAトランスフォルミラーゼ,0.26μM EF-G,0.24μM RF2,0.17μM RF3,0.5μM RRF,2.7μM IF1,0.4μM IF2,1.5μM IF3,40μM EF-Tu,93μM EF-Ts,1.2μM リボソーム,0.73μM AlaRS,0.03μM ArgRS,0.38μM AsnRS,0.13μM AspRS,0.02μM CysRS,0.06μM GlnRS,0.23μM GluRS,0.09μM GlyRS,0.4μM IleRS,0.04μM LeuRS,0.11μM LysRS,0.03μM MetRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.02μM TyrRS,0.02μM ValRS(自家調製タンパクは基本的にHisタグ付加タンパクとして調製した))に、1μM 鋳型RNA、タンパク質性アミノ酸群をそれぞれ250μMずつ、ならびに50μMずつアシル化tRNAを翻訳反応混合物に添加し、37℃で1時間静置することで行った。
1μM 鋳型RNA CT32(配列番号RM-H3)、0.25mM Met、0.25mM Arg,0.25mM Asp、0.25mM Tyr、0.25mM Lys、1mM ジチオスレイトール、50μM Ala(Tzm)-tRNAAsn-E2GUU(化合物AT-H3A)を含む前述の翻訳液を37℃で60分間保温した。得られた翻訳反応物1μLに9μLの0.2%トリフルオロ酢酸を加え、そのうち1μLをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート上乾固した。MALDI-MS分析の結果、Ala(Tzm)を含む全長ペプチド(ペプチド配列P-H2)が主生成物として観測された(図75、ピークI)。
CT32 RNA配列
GGGUUAACUUUAACAAGGAGAAAAACAUGCGUaacCGUGACUACAAGGACGACGACGACAAGUAAGCUUCG
fMetArg[Ala(Tzm)] ArgAspTyrLysAspAspAspAspLys
m/z: [H+M]+ = 1656.4 (Calc. 1656.7)
1μM 鋳型RNA CT32(配列番号RM-H3)、0.25mM Met、0.25mM Arg,0.25mM Asp、0.25mM Tyr、0.25mM Lys、及び前記の方法で調製した50μMのOrn(Acbz)-tRNAAsn-E2GUU(化合物AT-H3B)、Orn(oAcbz)-tRNAAsn-E2GUU(化合物AT-H3C)、Lys (Npys)-tRNAAsn-E2GUU(化合物AT-H3D)をそれぞれ1種類含む前述の翻訳液、計3反応溶液を37℃で60分間保温した。Orn(Acbz)-tRNAAsn-E2GUU(化合物AT-H3B)を含む翻訳サンプルに関して、得られた翻訳溶液1μLに9μLの0.2%トリフルオロ酢酸を加え、そのうち1μLをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート乾固した後、MALDI-MSにて解析を行った。その結果、目的の全長ペプチドに由来するピークでなく、側鎖Acbz基が外れたペプチドP-H3Aに対応するピークが主生成物として観測された(図75ピークII)。これは上述の測定前処理操作あるいは測定中のレーザー照射により側鎖脱保護反応が進行したことに由来すると考え、測定をLC-ESI-MSに変更して続けて同翻訳産物の解析をおこなった。上述のOrn(Acbz)-tRNAAsn-E2GUU(化合物AT-H3B)を含む翻訳溶液5μLと水5μLを混合したものを用いLC-MS分析した結果、Acbz基を有する全長の目的ペプチド(Orn(Acbz):ペプチド配列P-H3)に由来するピークを確認することができた(図76 ピークI)。その一方で、側鎖脱保護されたP-H3Aに由来するピークは観測されなかったことから、上述したように側鎖Acbz基の脱保護反応はMALDI測定あるいは分析前処理過程で起こっていることが明らかとなった(図77)。同様に、Orn(oAcbz)-tRNAAsn-E2GUU(化合物AT-H3C)、Lys (Npys)-tRNAAsn-E2GUU(化合物AT-H3D)を含む翻訳溶液に関しても、LC-MSによる分析を行った結果、目的どおりOrn(oAcbz):ペプチド配列P-H4、Lys (Npys):ペプチド配列P-H5)に相当するピークが観測された(図78ピークI及び図79ピークI)。
fMetArg[Orn(Acbz)] ArgAspTyrLysAspAspAspAspLys
保持時間:4.99分(分析条件 OrbitrapHFIP-Et3N-3)
fMetArgOrn ArgAspTyrLysAspAspAspAspLys
m/z: [H+M]+ = 1598.5 (Calc. 1598.7)
fMetArg[Orn(oAcbz)] ArgAspTyrLysAspAspAspAspLys
保持時間:5.23分(分析条件 OrbitrapHFIP-Et3N-3)
fMetArg[Lys(Npys)] ArgAspTyrLysAspAspAspAspLys
保持時間:4.34分(分析条件 OrbitrapHFIP-Et3N-3)
1μM 鋳型RNA CT32(配列番号RM-H3)、0.25mM Met、0.25mM Arg,0.25mM Asp、0.25mM Tyr、0.25mM Lys、及び前記の方法で調整した50μMのLys(Acbz)-tRNAAsn-E2GUU(化合物AT-H3E)、Lys(5S-SSMe)( Acbz)-tRNAAsn-E2GUU(化合物AT-H3F)、をそれぞれ1種類含む前述の翻訳液、計3反応溶液を37℃で60分間保温した。得られた翻訳反応物1μLに9μLの0.2%トリフルオロ酢酸を加え、そのうち1μLをMALDIターゲットプレート上に載せた後、1μLのCHCA溶液(10mg/ml α-シアノ-4-ヒドロキシケイ皮酸、50%アセトニトリル、0.1%トリフルオロ酢酸溶液)と混和し、プレート上乾固した。MALDI-MS分析の結果、いずれにおいても側鎖Acbz基がMALDI測定中及び前処理中に外れたことに由来するペプチド配列P-H6,P-H7が観測された(それぞれ図75ピークIII、IV)。
fMetArgLysArgAspTyrLysAspAspAspAspLys(配列番号:98)
m/z: [H+M]+ = 1612.4(Calc. 1612.7)
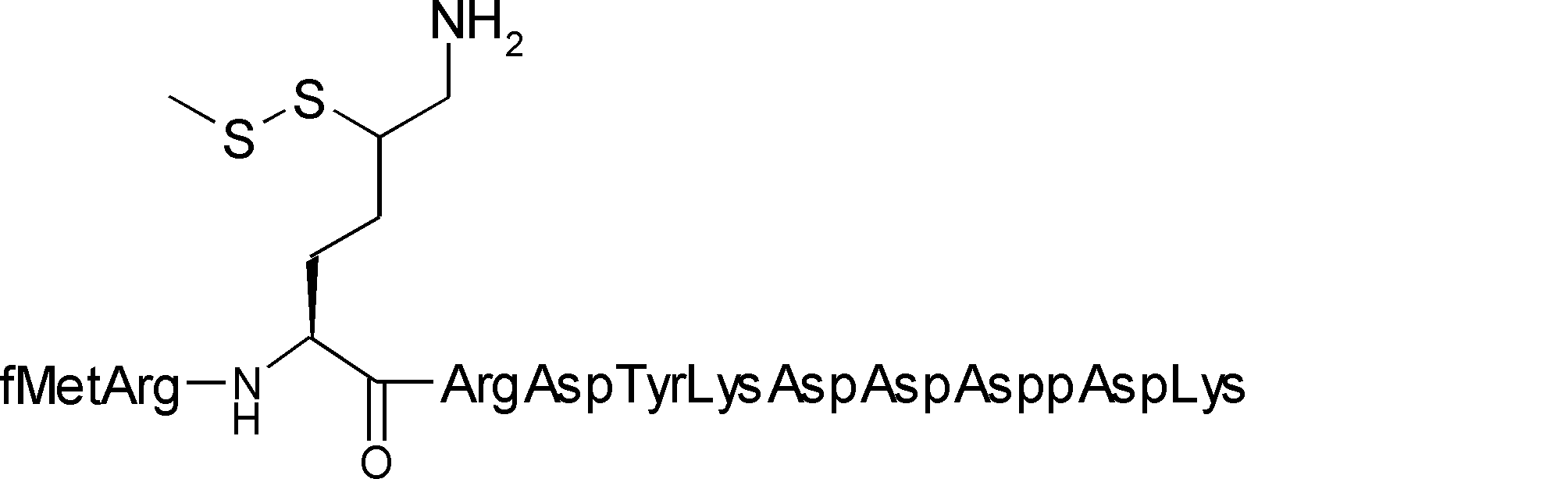
m/z: [H+M]+ = 1690.1 (Calc. 1690.7)
前述した側鎖アミノ基の保護基各々の脱保護条件下においてRNAが安定に存在するかを確認するため、RNAをそれぞれの脱保護条件に伏した後、ゲル電気泳動による解析を行った。
表19に示す反応条件にあるように各反応溶液を調製し、各々の反応温度、反応時間に従ってRNAを保温した。反応条件B-Eに伏した反応溶液それぞれに対しては、その後RNeasy minelute(qiagen社)を用いてRNAを精製し、10μLの水にて溶出した。これらと、反応溶液Aの各々10μLの溶液に対して10μLのTBE-Ureaサンプルバッファー(2x)(Invitrogen社)を加え、そのうち1μLを使って10%TBE-ウレアゲルにて電気泳動を行い、SYBR gold nucleic acid stain (Invitrogen社)により染色を行った(図80)。
その結果、対照実験として電気泳動した反応条件AのRNAと比べ反応条件B-Eに伏したRNAはバンドパターンやバンド濃さに大きな変化は観測されず、(図80、レーン1vsレーン2-5)、RNAがこれらの反応条件にて安定に存在することが示された。なおB-Eの条件とは、それぞれ、B:4-アジドベンジルオキシカルボニル基(Acbz)、C:2-アジドベンジルオキシカルボニル基(oAcbz)、D:チアゾリジン環、E:3-ニトロ-2-ピリジンスルフェニル基(Npys)の脱保護条件を模している。

OT95 RNA配列
GGGUUAACUUUAAGAAGGAGAUAUACAUAUGUGCACUACAACGCGUCUUCCGUACCGUAGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
実際にディスプレイライブラリー構築で合成される化合物はmRNAの長さが約100のmRNA-ペプチドフュージョンであり、そのような高分子化合物の反応の詳細な解析は困難である。そこで分子内に分子量分析可能な長さのRNA構造を有し、かつ分子内に金属触媒による環化反応が可能なペプチド構造を有するモデル化合物を用いmRNA-ペプチドフュージョンでの環化反応条件特定を実施した。
Pdを用いた翻訳後修飾でディスプレイライブラリーに適用させるためにはRNAに影響を与えることなく、望みの化学修飾反応を進行させる必要がある。その目的を達するため、以下のような実験を行った。(1)tRNAなどが混在した系(PureSystem)中でのHeck反応を実施し、tRNAに影響を与えることなく、Heck反応が進行する反応条件を見出した。(2)ペプチドとRNAのコンジュゲートを作成の上、Heck反応を実施し、ペプチドとRNAが結合していてもRNAに影響を与えることなくHeck反応が進行する反応条件を見出した。
モデル化合物として使用したのは以下の3種類の化合物である。
4-mer RNA-ペプチドコンジュゲート
10-mer RNA-ペプチドコンジュゲート
20-mer RNA-ペプチドコンジュゲート
以下の一般製法‐1に従って、モデル反応実施のための原料合成を行った。
(一般製法-1)RNA-ペプチドコンジュゲートの合成
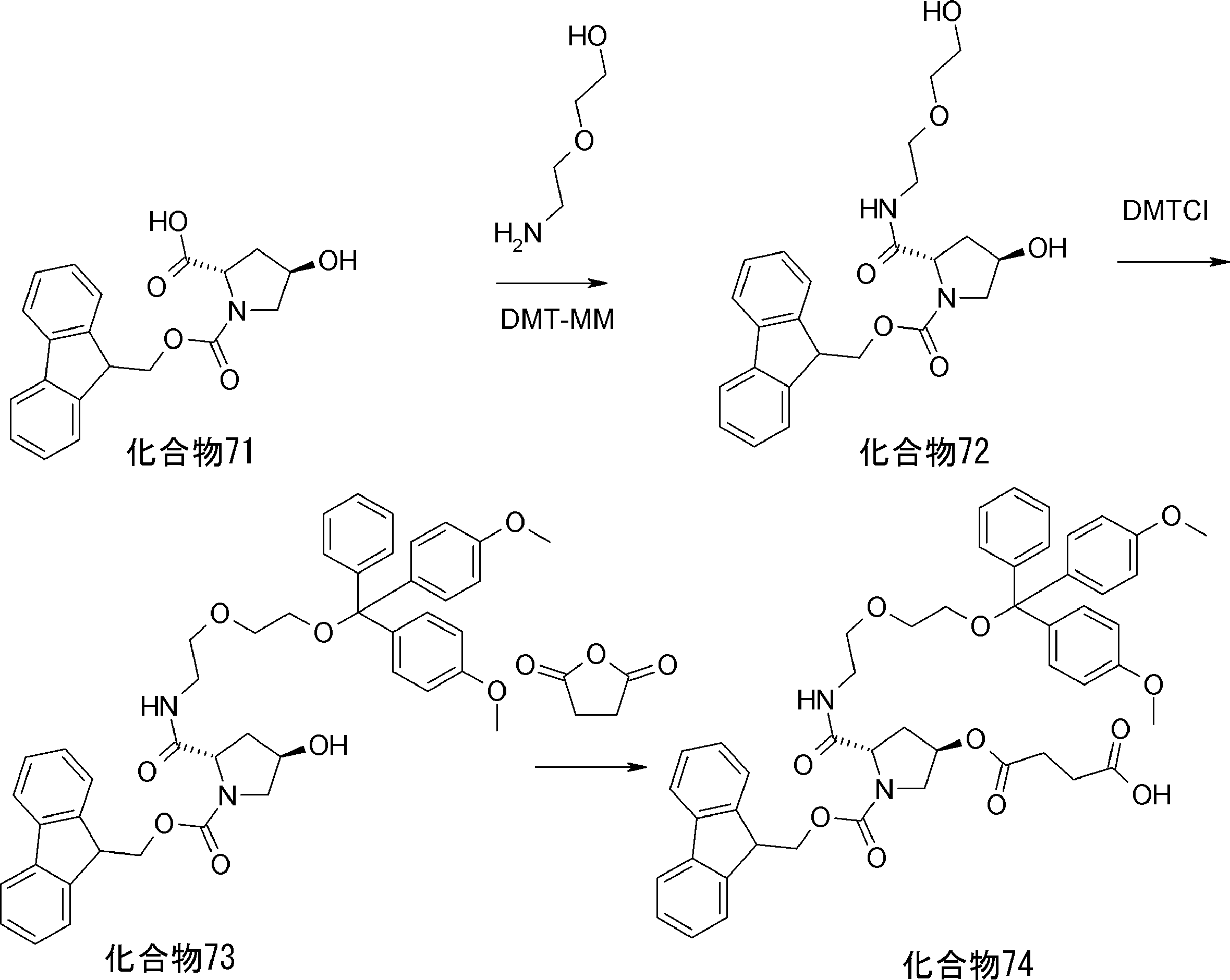
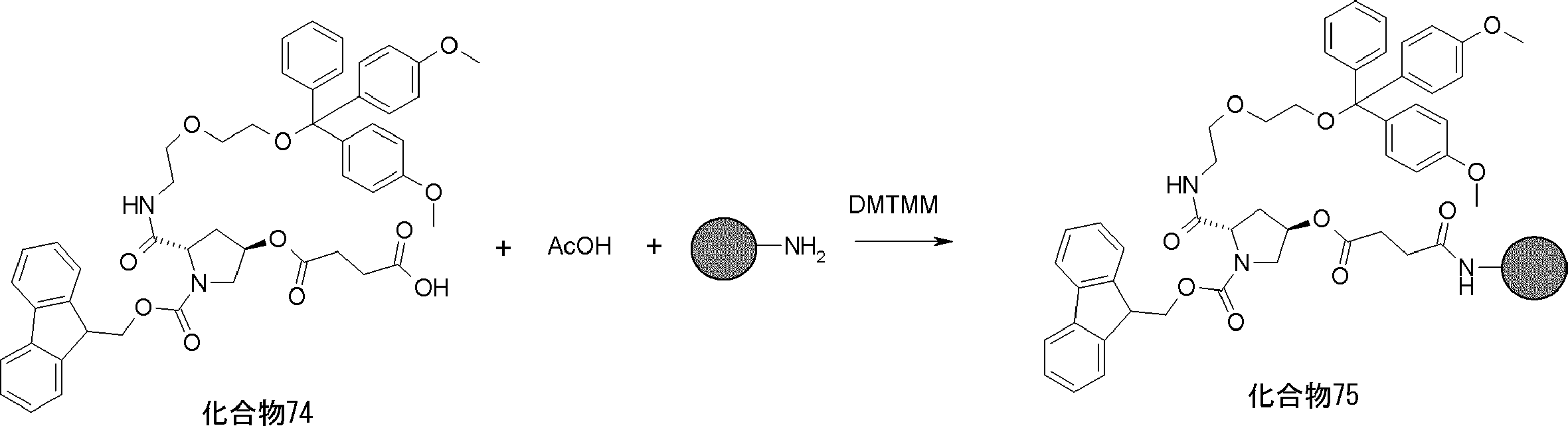

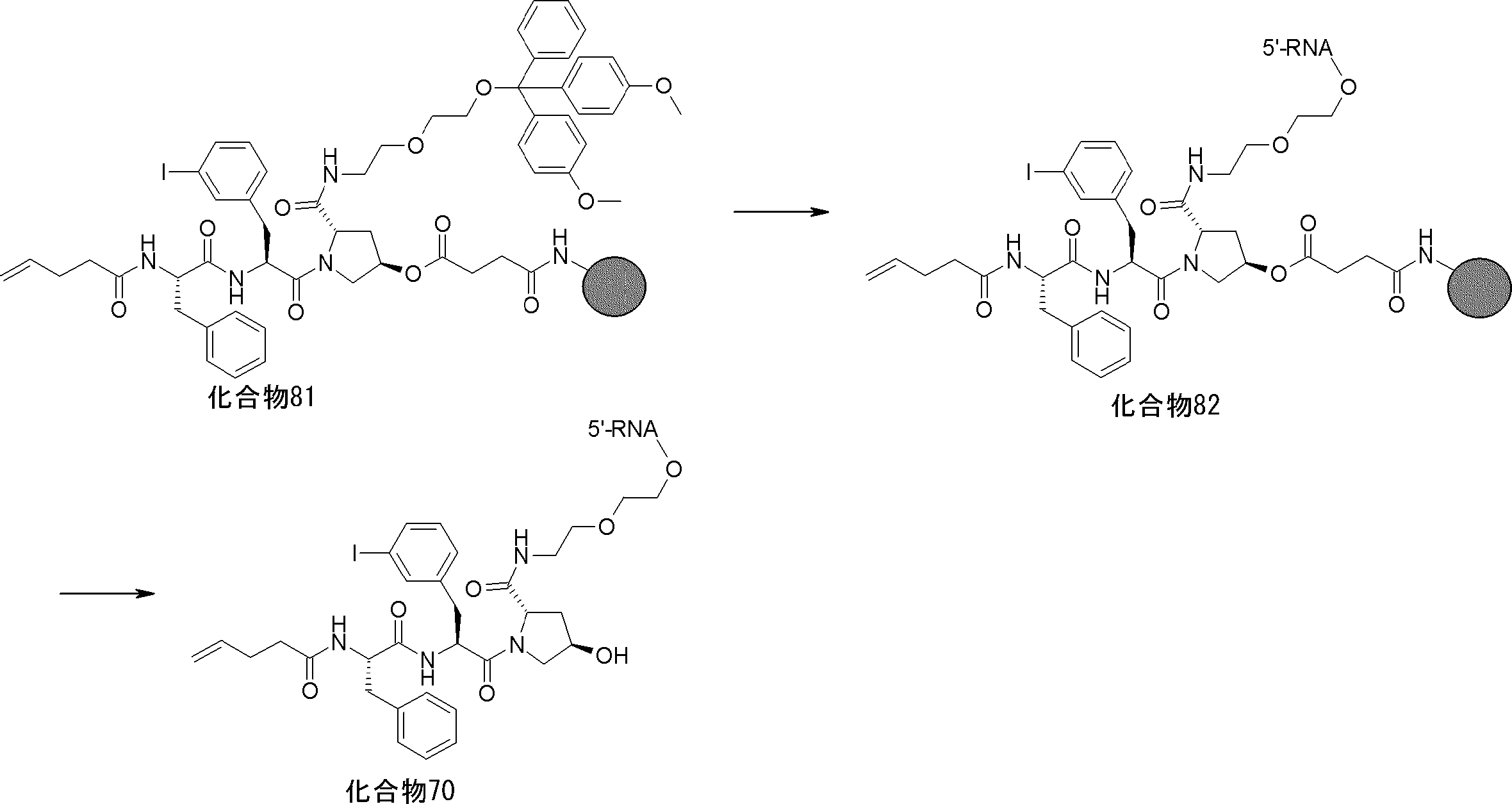
(2S、4R)-4-ヒドロキシ-2-[2-(2-ヒドロキシ―エトキシ)-エチルカルバモイル]-ピロリジン-1-カルボン酸 9H-フルオレン―9-イルメチルエステル(化合物72)の合成
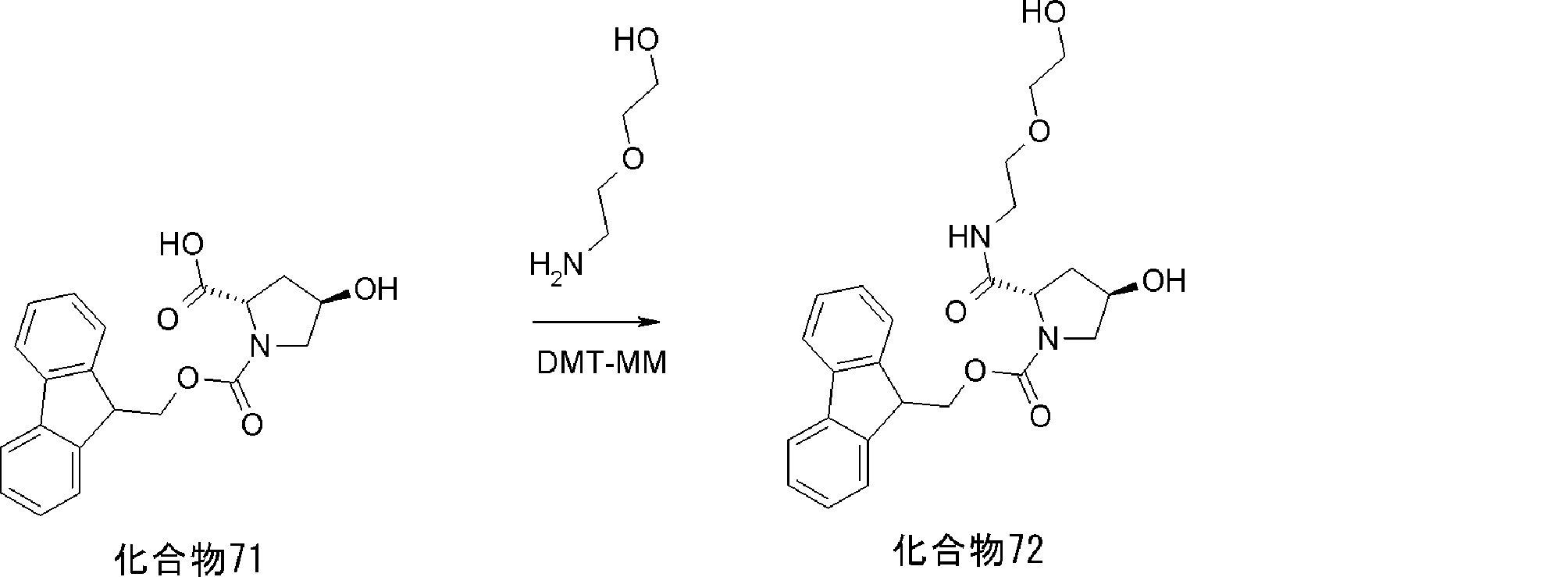
LCMS(ESI) 441.2(M+H)+
保持時間:1.57分(分析条件ZQAA05)

LCMS(ESI) 760.8(M+NH4)+、765.7(M+Na)+
保持時間:1.15分(分析条件SQDAA05)

LCMS(ESI) 860.5(M+NH4)+、865.5(M+Na)+
保持時間:1.11分(分析条件SQDAA05)
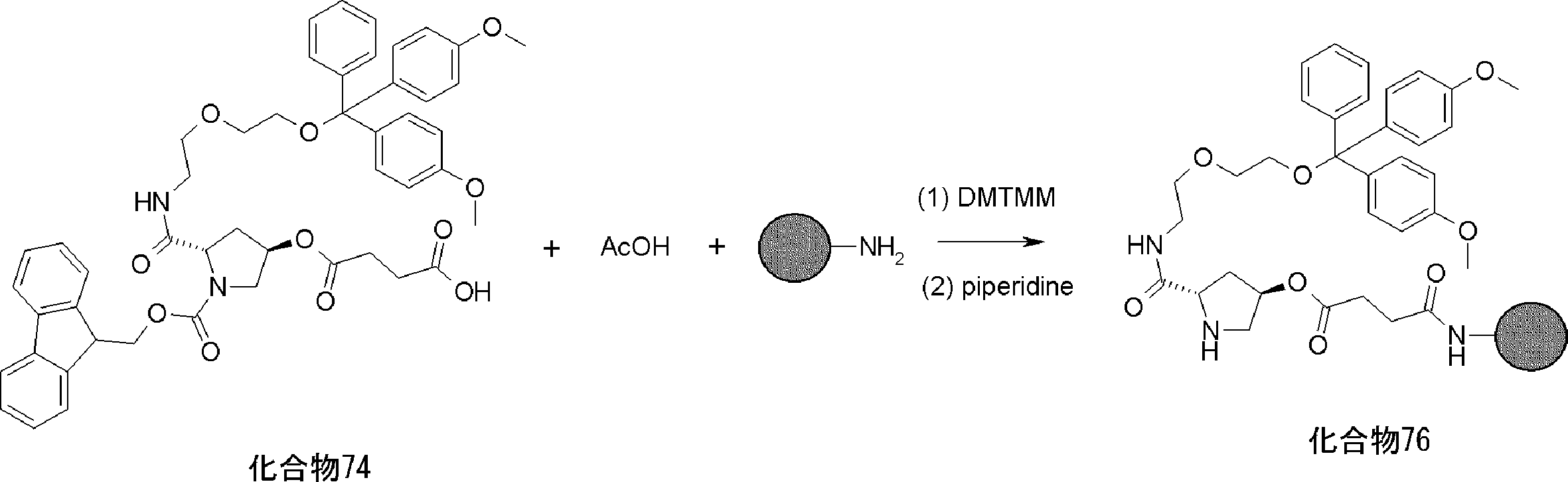
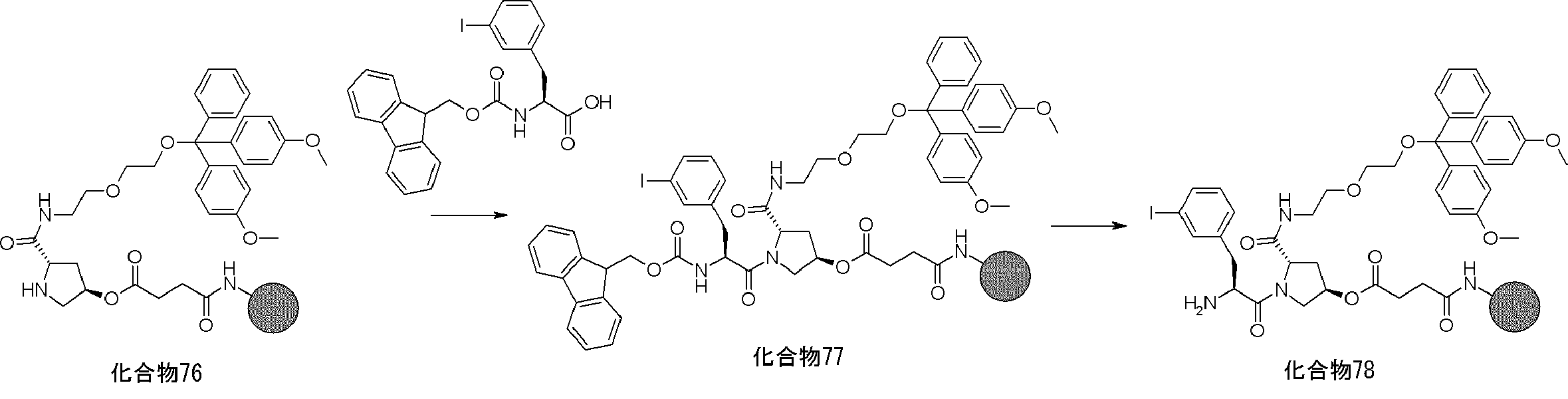
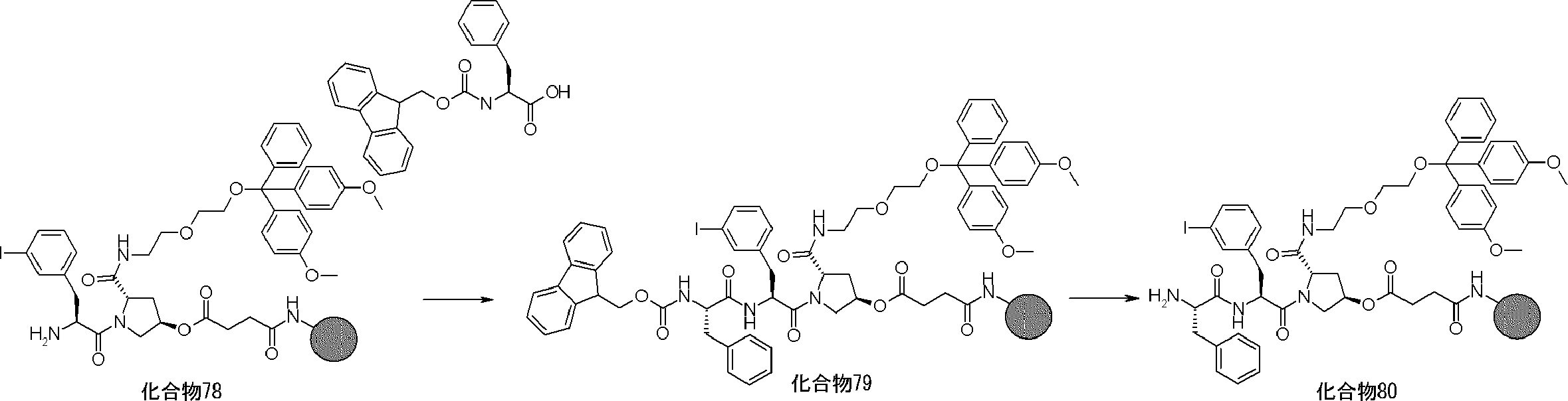
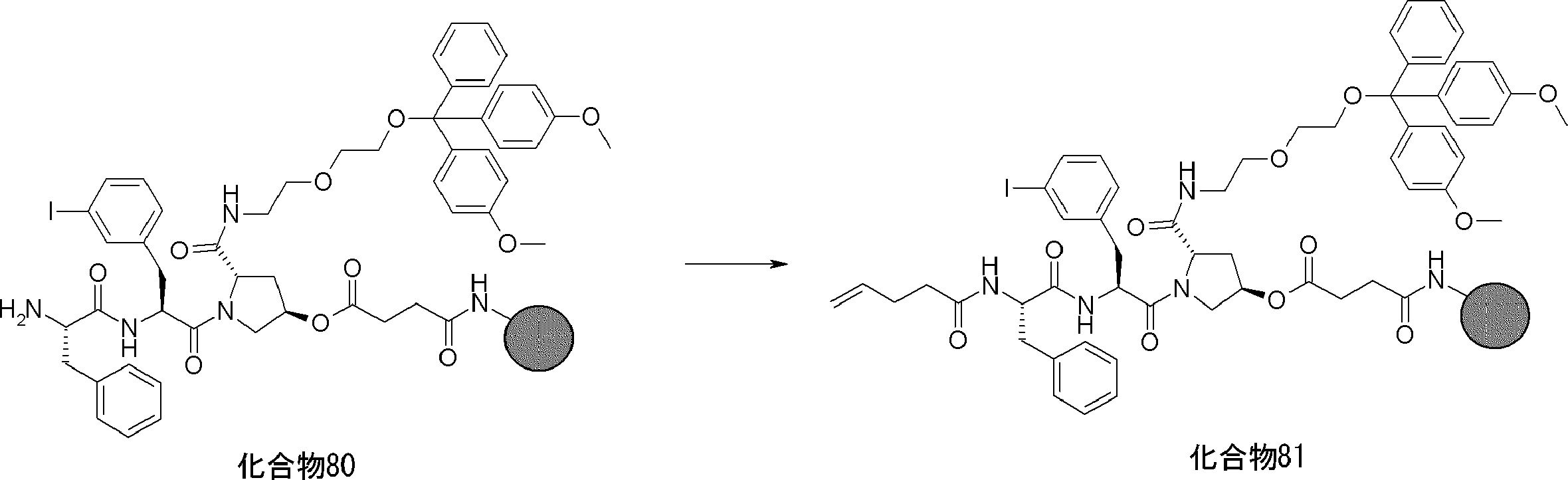

LCMS(ESI) 668.0(M-3H)3-、1002.4(M-2H)2-
保持時間:5.15分(分析条件ZQHFIP-Et3N)
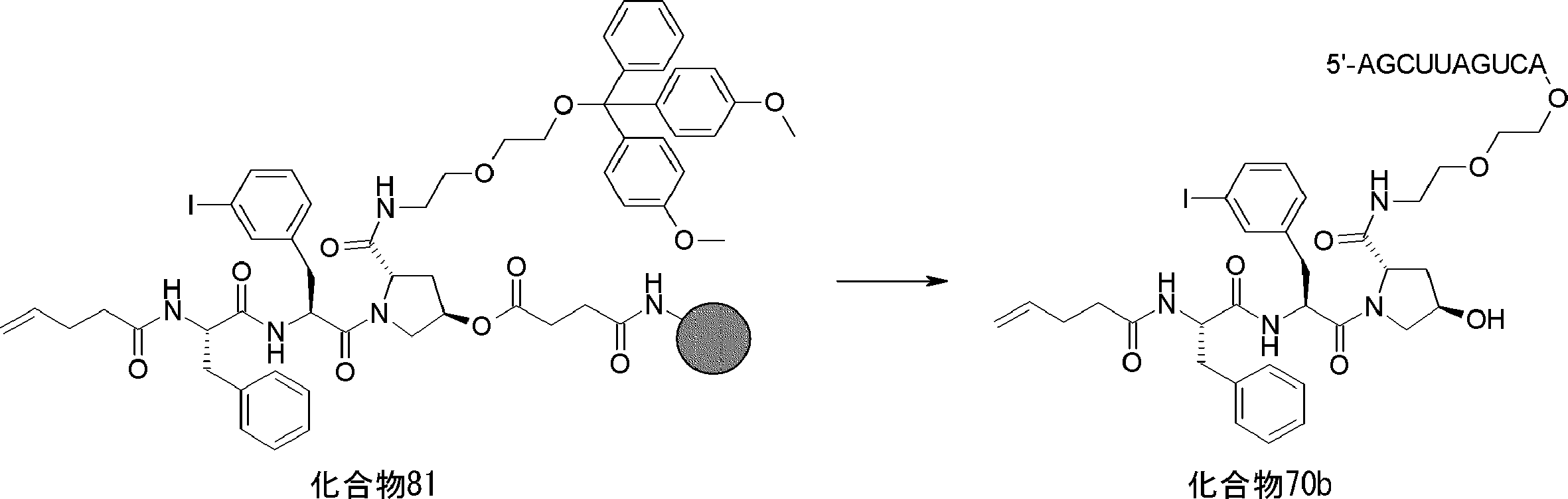
LCMS(ESI) 784.8(M-5H)5-、981.2(M-4H)4-、1308.5(M-3H)3-
保持時間:4.87分(分析条件ZQHFIP-Et3N)
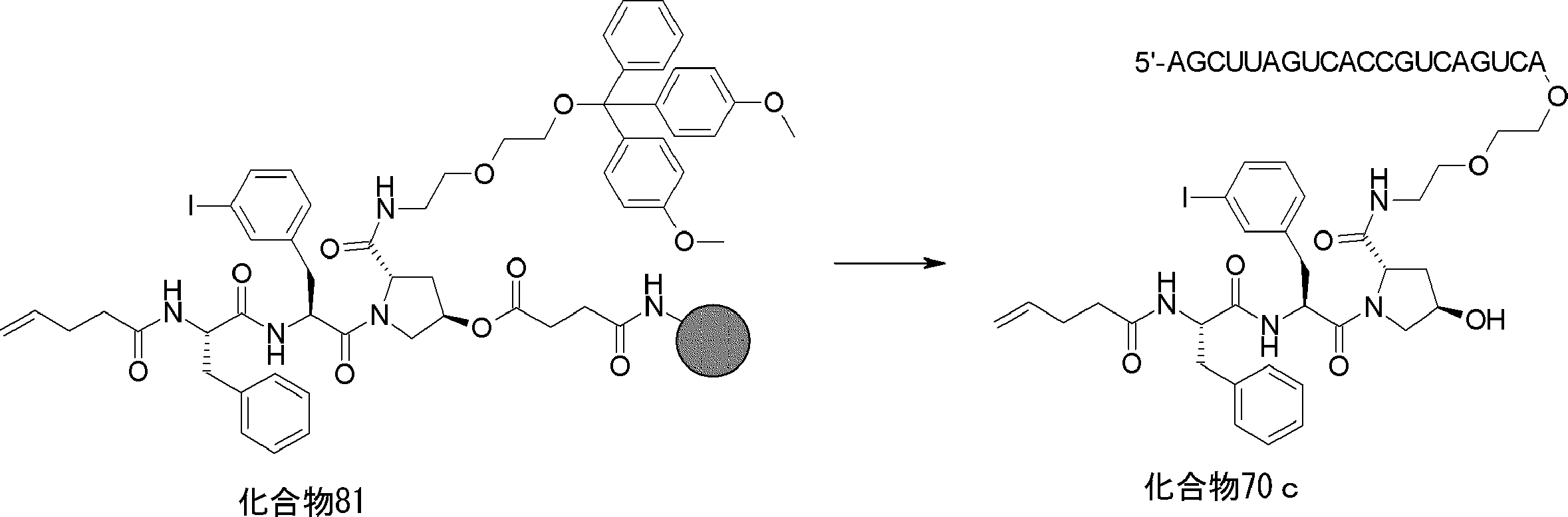
LCMS(ESI) 887.9(M-8H)8-、1015.0(M-7H)7-、1184.3(M-6H)6-、1421.0(M-5H)5-、1776.6(M-4H)4-
保持時間:4.60分(分析条件ZQHFIP-Et3N)
2-1-1. 10-mer RNA-ペプチドコンジュゲート(化合物70b)のHeck環化反応(化合物83b)の合成
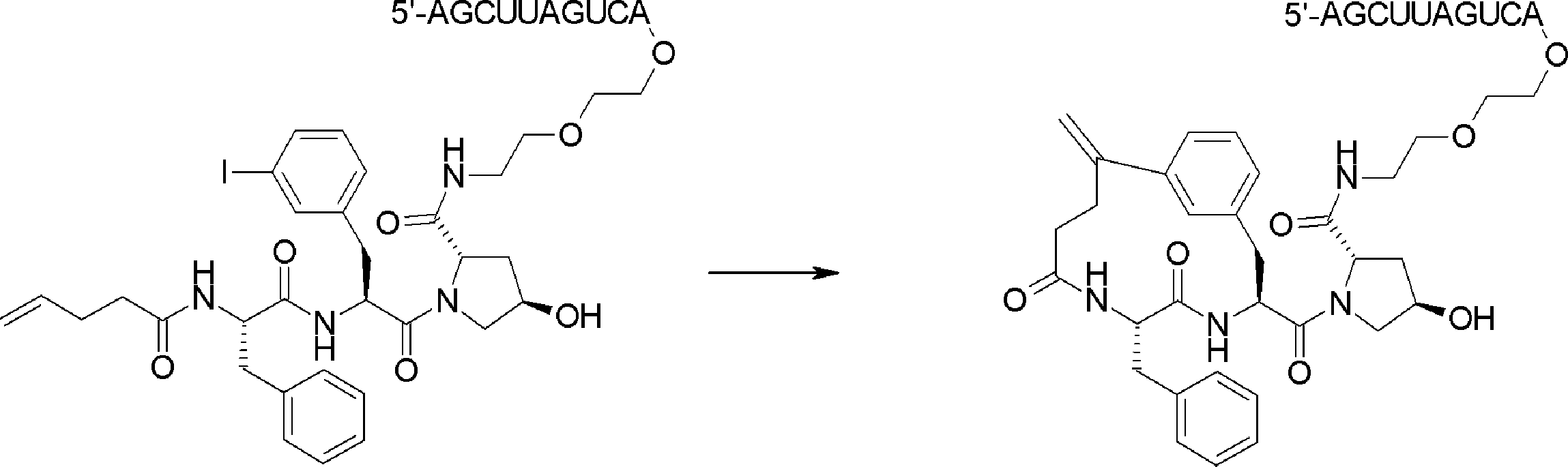
LCMS(ESI) 759.4(M-5H)5-、949.5(M-4H)4-、1265.9(M-3H)3-
保持時間:3.40分(分析条件ZQHFIP-Me2NEt)

LCMS(ESI) 997.0(M-7H)7-、1163.0(M-6H)6-、1395.5(M-5H)5-、1745.0(M-4H)4-
保持時間:4.08分(分析条件ZQHFIP-Et3N)
1.翻訳合成に活用させるためのC-C結合ユニットの合成
1-1.pdCpAアミノアシル化化合物85の合成
1-1-1.ブタ-3-エン酸シアノメチル(化合物86)の合成
1H-NMR(Bruker, avanceIII, 400 MHz, CDCl3) δ ppm 5.92 (1H, m), 5.22-5.26 (2H, m), 4.75 (2H, s), 3.21 (2H, m)
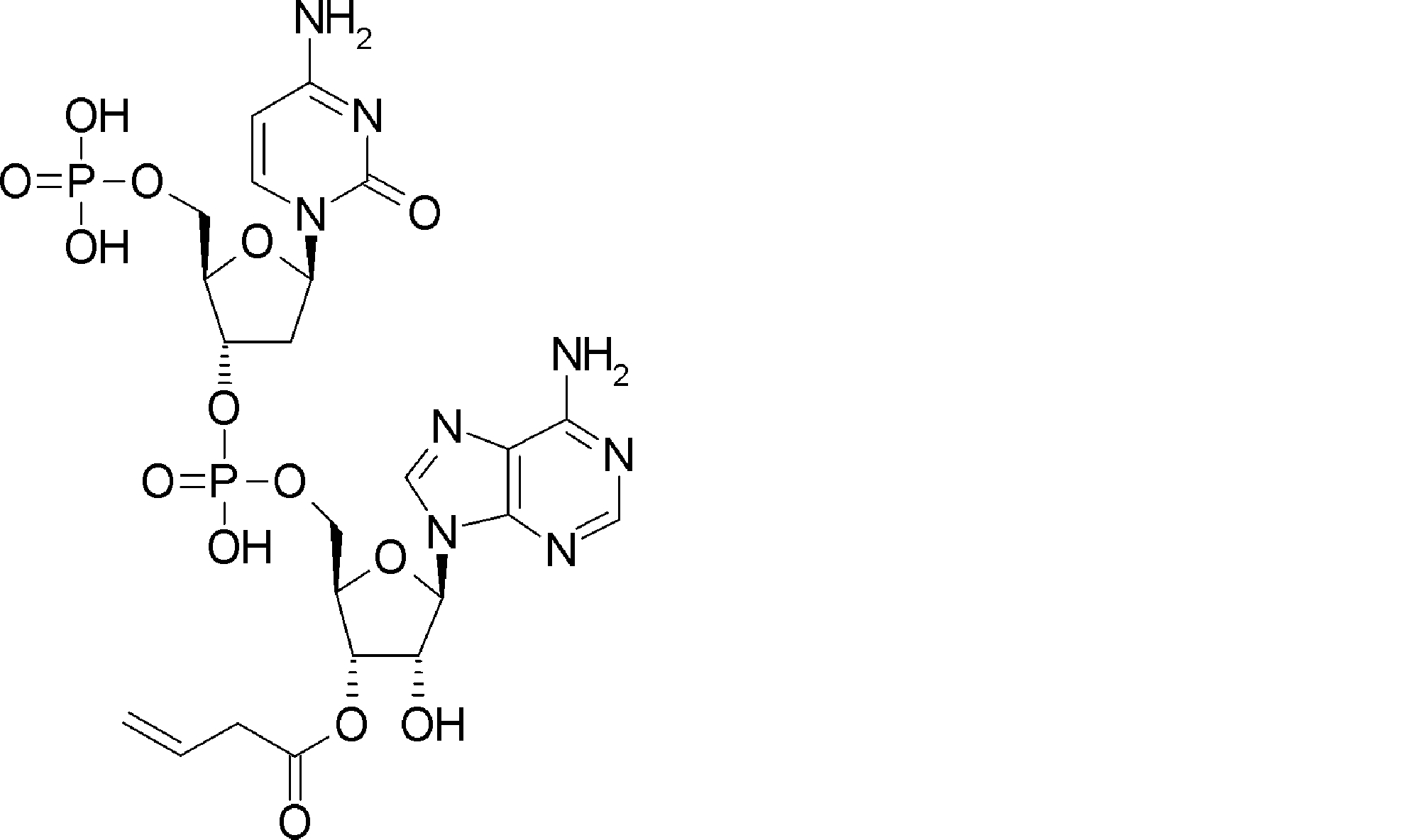
LCMS:m/z 705(M+H)+
保持時間:0.461、0.488分 (分析条件 SMDmethod1)
1-2-1.ペンタ-4-エン酸シアノメチル(化合物88)の合成
1H-NMR(Bruker, avanceII, 300 MHz, CDCl3) δ ppm 5.82 (1H, m), 5.04-5.32 (2H, m), 4.74 (2H, s), 2.39-2.57 (4H, m)
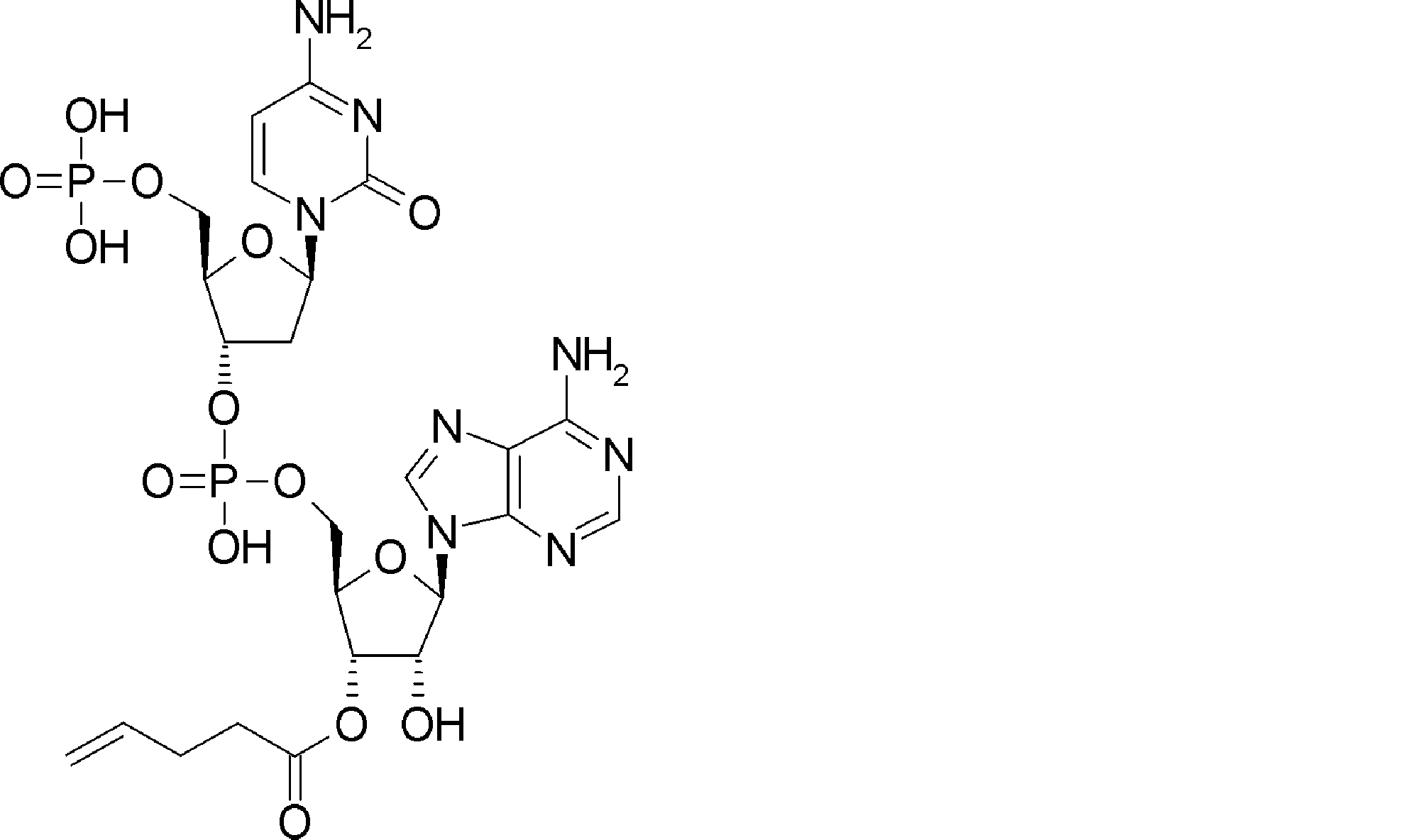
LCMS:m/z 719(M+H)+
保持時間:0.506、0.523分 (分析条件 SMDmethod1)
1-3-1. 2-(アリルオキシ)酢酸 (2R,3S,4R,5R)-2-((((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-((ホスホノオキシ)メチル)テトラヒドロフラン-3-イル)オキシ)(ヒドロキシ)ホスホリル)オキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イル(化合物89)の合成
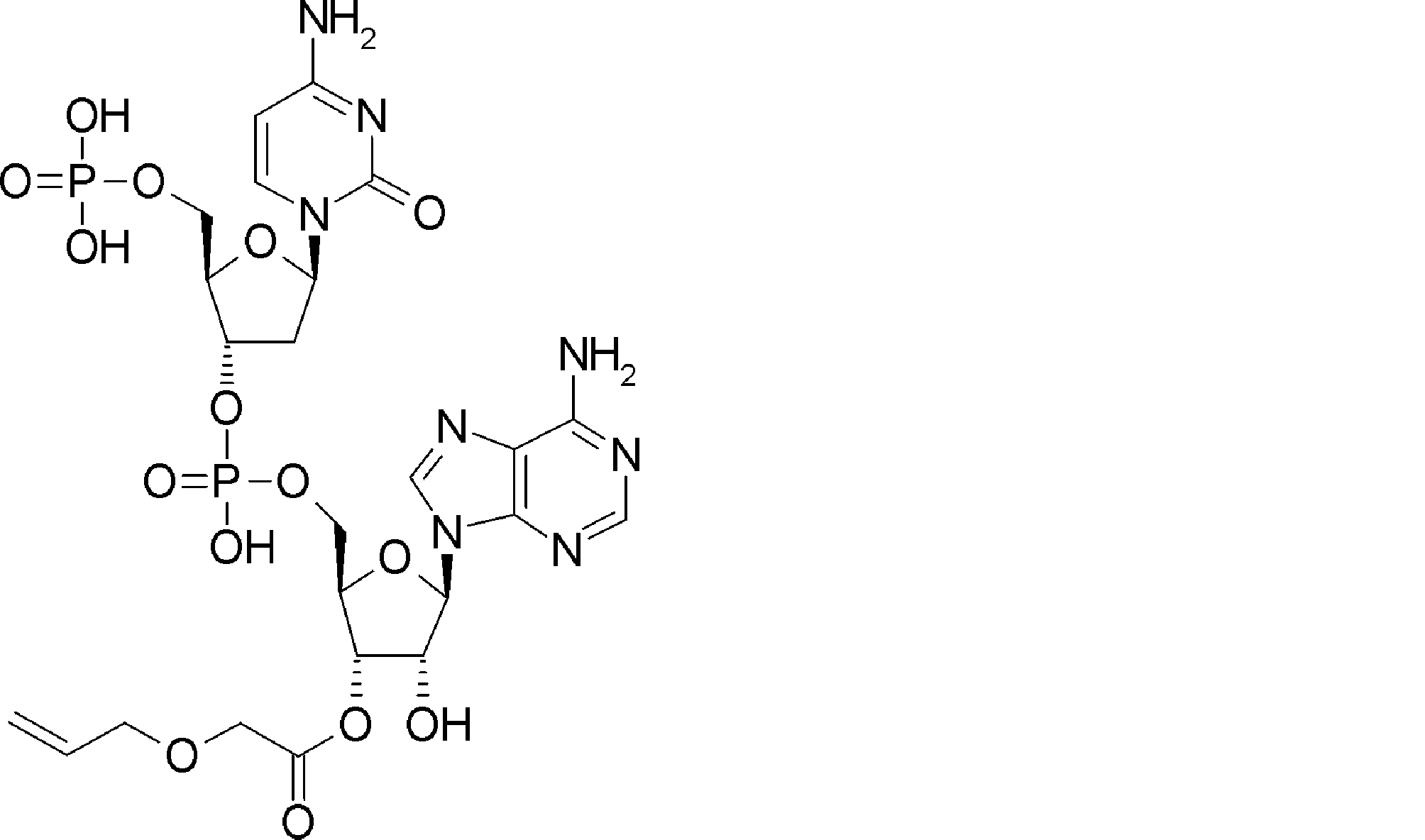
イミダゾール(1.0g、15.7mmol)およびN,N,N-トリメチルヘキサデカン-1-アミニウム 塩化物(1.0g、3.14mmol)を溶かし、酢酸でpH8に調節した緩衝液(100ml)にリン酸二水素 ((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-3-(((((2R,3S,4R,5R)-5-(6-アミノ-9H-プリン-9-イル)-3,4-ジヒドロキシテトラヒドロフラン-2-イル)メトキシ)(ヒドロキシ)ホスホリル)オキシ)テトラヒドロフラン-2-イル)メチル(化合物1h)(100mg、0.157mmol)を加えた後、上記方法で得られた2-(アリルオキシ)酢酸シアノメチル(93mg、0.628mmol)のTHF(3.0ml)溶液を加えて室温で30分攪拌した。反応液に酢酸を加えて凍結乾燥し、得られた残渣をpreparative-HPLC(0.05%TFA水溶液:アセトニトリル)で精製し、標題化合物89(24.7mg、22%)を得た。
LCMS:m/z 735(M+H)+
保持時間:0.473、0.491分 (分析条件 SMDmethod1)
1-4-1.2-((tert-ブトキシカルボニル)アミノ)-3-(4-ヨードフェニル)プロパン酸 (S)-シアノメチル(化合物91)の合成
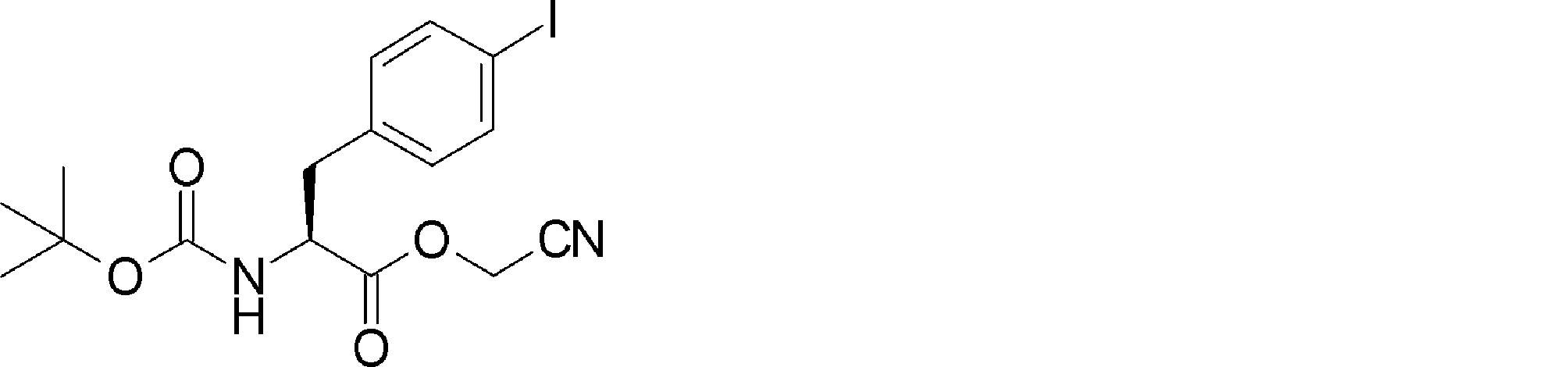
保持時間:1.59分 (分析条件 SMDmethod4)
1H-NMR(Bruker, avanceII, 300 MHz, CDCl3) δ ppm 7.67 (2H, d,8.1 Hz), 6.93 (2H, d, 8.1 Hz), 4.62-4.94 (4H, m), 2.98-3.14 (2H, m), 1.44 (9H, s)
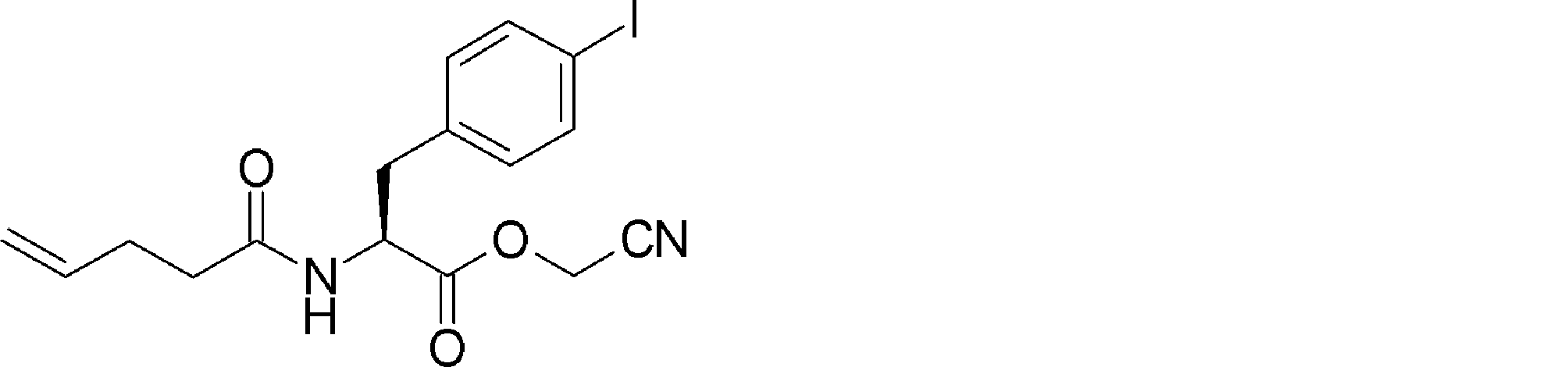
窒素雰囲気下、2-アミノ-3-(4-ヨードフェニル)プロパン酸 (S)-シアノメチル(0.65g、1.78mmol)をジクロロメタン(25.0ml)に溶解し、氷冷下でトリエチルアミン(0.45g、4.45mmol)を滴下した。その後、氷冷下で塩化ペンタ-4-エノイル(0.25g, 2.14mmol)のジクロロメタン溶液(25.0ml)を滴下し、室温で2時間半攪拌した。反応液を減圧濃縮して得られた残渣をカラムクロマトグラフィ(石油エーテル:酢酸エチル=50:50~33:67)で精製し、3-(4-ヨードフェニル)-2-(ペンタ-4-エンアミド)プロパン酸 (S)-シアノメチル(化合物92)(0.57g、78%)を得た。
LCMS:m/z 413.2(M+H)+
保持時間:1.51分 (分析条件 SMDmethod5)

LCMS:m/z 990(M-H)-
保持時間:1.45分 (分析条件 ZQFA05)
1-5-1. 3-(3-ヨードフェニル)-2-(ペンタ-4-エンアミド)プロパン酸 (2S)-(2R,3S,4R,5R)-2-((((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-((ホスホノオキシ)メチル)テトラヒドロフラン-3-イル)オキシ)(ヒドロキシ)ホスホリル)オキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イル(化合物93、pdCpA-Phe(3-I))の合成
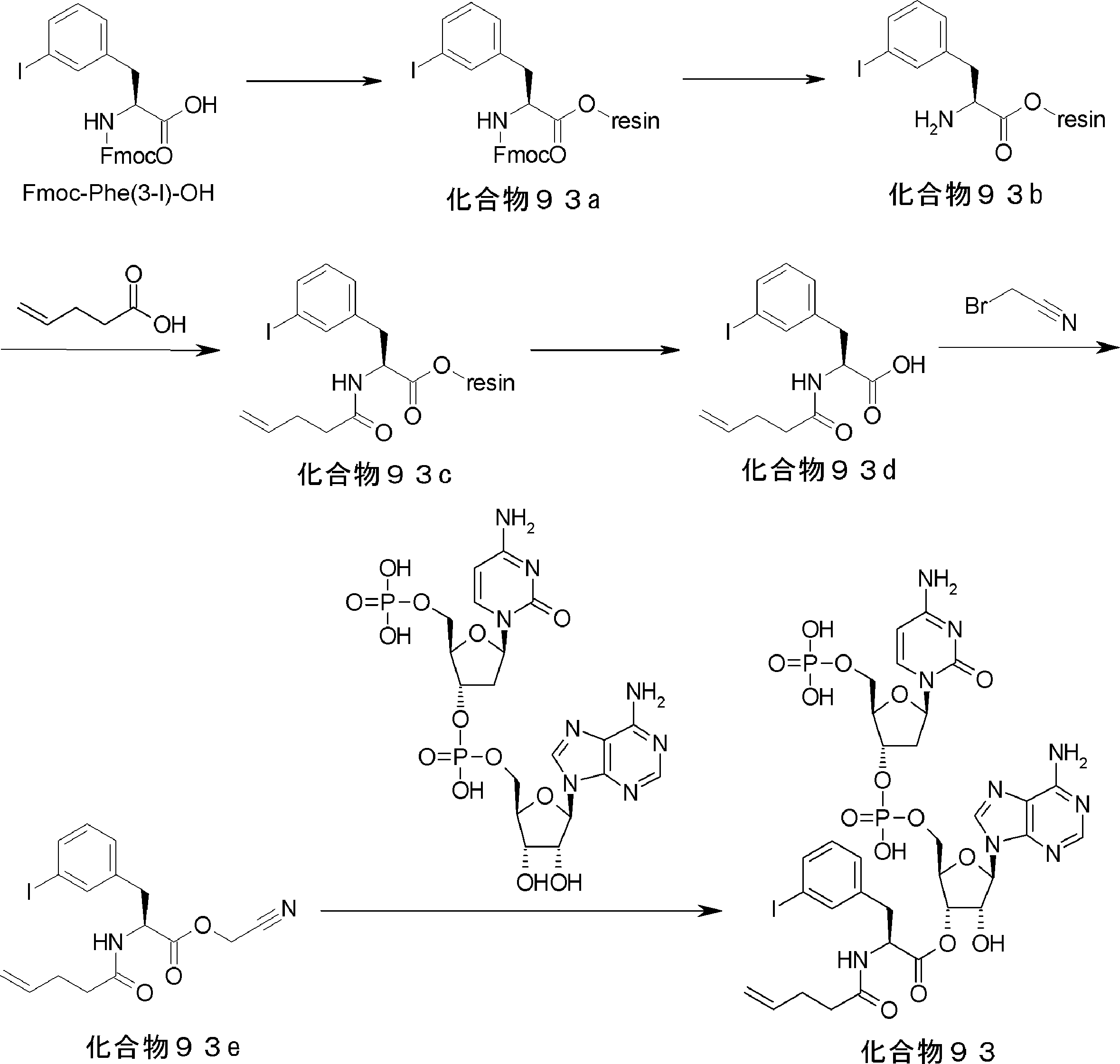
Fmoc基の脱保護のために、化合物93aに20%ピペリジンのDMF溶液(20ml)を加えて室温で2時間攪拌した後、反応液を除去した。このレジンをDMF(30ml×4回)で洗浄し、化合物93bを得た。
化合物93bにペンタ-4-エン酸(0.39g、3.90mmol)、DIC(0.550g、4.29mmol)、HOAt(0.870g、4.29mmol)のDMF溶液(20ml)を加えて室温で5時間攪拌した。反応液を除去し、レジンをDMF(30ml×4回)およびジクロロメタン(50ml×4回)で洗浄して化合物93cを得た。
化合物93cに2%TFAのジクロロメタン溶液(20ml)を加えて、室温で1時間攪拌し、レジンをろ過して除いた。同様の操作をさらに3回繰り返し、得られた反応液をまとめて減圧濃縮した。残渣をカラムクロマトグラフィ(ジクロロメタン:メタノール=30:1)で精製し、(S)-3-(3-ヨードフェニル)-2-(ペンタ-4-エンアミド)プロパン酸(化合物93d)(0.494g、68%)を得た。
化合物93d(0.494g、1.324mmol)をDMF(10ml)に溶解し、2-ブロモアセトニトリル(0.63g、5.30mmol)およびDIPEA(0.341g、2.65mmol)を室温でゆっくり加えた。室温で30分攪拌した後、反応液を減圧濃縮して得られた残渣をカラムクロマトグラフィ(石油エーテル:酢酸エチル=5:1)で精製し、(S)-3-(3-ヨード-フェニル)-2-ペンタ-4-エノイルアミノ-プロピオン酸 シアノメチル エステル(化合物93e)(0.392g、72%)を得た。
イミダゾール(476.6mg、7.0mmol)およびN,N,N-トリメチルヘキサデカン-1-アミニウム 塩化物(448.0mg、1.4mmol)を溶かし、酢酸でpH8に調節した緩衝液(70ml)にリン酸二水素 ((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-3-(((((2R,3S,4R,5R)-5-(6-アミノ-9H-プリン-9-イル)-3,4-ジヒドロキシテトラヒドロフラン-2-イル)メトキシ)(ヒドロキシ)ホスホリル)オキシ)テトラヒドロフラン-2-イル)メチル(化合物1h、pdCpA)(70mg、0.11mmol)を加えた後、(S)-3-(3-ヨード-フェニル)-2-ペンタ-4-エノイルアミノ-プロピオン酸 シアノメチル エステル(化合物93e)(181.3mg、0.44mmol)のTHF溶液(3.0ml)を加えて室温で2時間攪拌した。反応液にTFA(1.0ml)を加えて凍結乾燥した後、得られた残渣をpreparative-HPLC(0.05%TFA水溶液:アセトニトリル=80:20~60:40)で精製し、標題化合物93(17.9mg、16%)を得た。
LCMS:m/z 992(M+H)+
保持時間:0.75分 (分析条件 SQDAA05)
1-6-1.3-(2-ヨードフェニル)-2-(ペンタ-4-エンアミド)プロパン酸 (2S)-(2R,3S,4R,5R)-2-((((((2R,3S,5R)-5-(4-アミノ-2-オキソピリミジン-1(2H)-イル)-2-((ホスホノオキシ)メチル)テトラヒドロフラン-3-イル)オキシ)(ヒドロキシ)ホスホリル)オキシ)メチル)-5-(6-アミノ-9H-プリン-9-イル)-4-ヒドロキシテトラヒドロフラン-3-イルの合成

LCMS:m/z 992(M+H)+
保持時間:0.71、0.73分 (分析条件 SQDAA05)
pdCpA-PenteA(化合物87)とtRNA(CA欠損)(配列番号:R-5)のligationによるアシル化tRNA(化合物AT-2-IIIA)の合成
50μM 転写tRNAfMetCAU(-CA)(配列番号R-5) 10μLに、10X ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP)2μL、Nuclease free water 4μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。10unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)2μLおよび、5mMのpdCpA-PenteA(化合物87)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。アシル化tRNA(化合物AT-2-IIIA)は、フェノール抽出した後、エタノール沈殿により回収した。アシル化tRNA(化合物AT-2-IIIA)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
PenteA-tRNAfMetCAU

50μM 転写tRNAEnAsnGAG (-CA)(配列番号R-1) 10μLに、10X ligation buffer (500 mM HEPES-KOH pH 7.5, 200 mM MgCl2, 10 mM ATP) 2μL、Nuclease free water 4μLを加え、95℃で2分間加熱した後、室温で5分間放置し、tRNAのリフォールディングを行った。 20unit/μLのT4 RNAリガーゼ(New England Bio Lab.社)2μLおよび、5mMのpdCpA-Phe(3-I)(化合物93)のDMSO溶液 2μLを加え、15℃で45分間ライゲーション反応を行った。ライゲーション反応液 20μLに、3M酢酸ナトリウム 4μLと125mM ヨウ素(水:THF=1:1溶液)24μLを加え、室温で1時間、脱保護を行った。アミノアシル化tRNAは、フェノール抽出した後、エタノール沈殿により回収した。アミノアシル化tRNA(化合物AT-1-IIIA)は、翻訳混合物に添加する直前に1mM酢酸ナトリウムに溶解した。
Phe(3-I)-tRNAEnAsnGAG(配列番号:33)
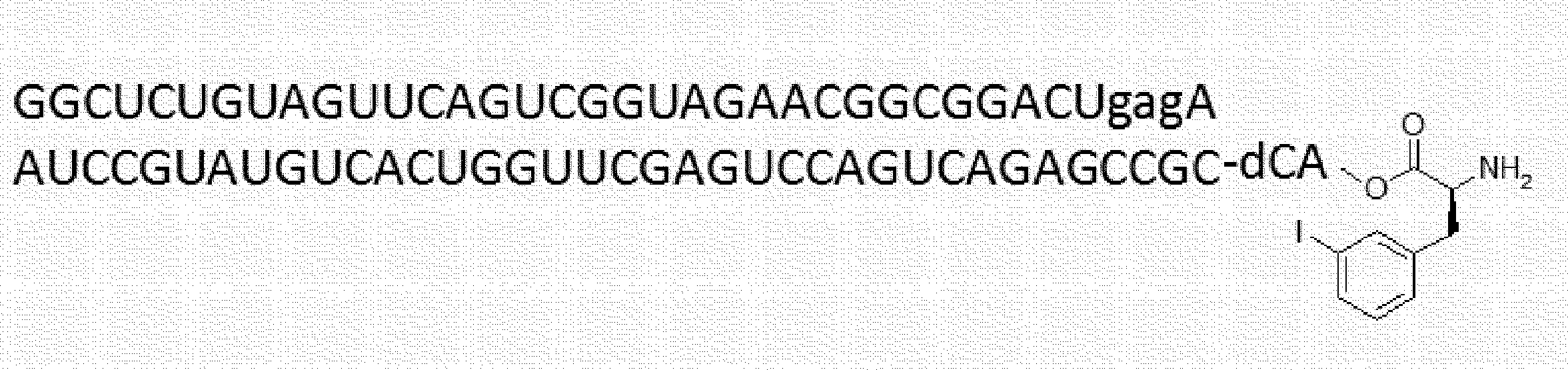
GGGUUAACUUUAAgaaggagauauacauAUGUUUCUUCCGagcggcucuggcucuggcucuUAGGGCGGCGGGGACAAA
GGGUUAACUUUAAgaaggagauauacauAUGACUACAACGagcggcucuggcucuUUUCUUCCGggcucuUAGGGCGGCGGGGACAAA
GGGUUAACUUUAAgaaggagauauacauAUGAACAACAACAACAACACUACAACGggcCUUCCGggcucuUAGGGCGGCGGGGACAAA
[PenteA]Phe[Phe(3-I)]ProSerGlySerGlySerGlySer
[PenteA] ThrThrThrSerGlySerGlySerPhe[Phe(3-I)]ProGlySer
[PenteA]AsnAsnAsnAsnAsnThrThrThrGly[Phe(3-I)]ProGlySer
Met、Leuを除く各0.3mMの18種類のタンパク質性アミノ酸、20μM Phe(3-I)-tRNAEnAsnGAG(化合物AT―1-IIIA)、20μM PenteA-tRNAfMetCAU(化合物AT-2-IIIA)を加えた翻訳液に、1μM 鋳型mRNA Hc1、Hc2、もしくはHc3(配列番号R-41'、R-42、R-43)を添加し、37℃で60分間保温した。続いて、以下に示す方法でHeck型の環化反応を行った。
リン酸バッファー(K2HPO4 1.0mmolとK3PO4 0.2mmolの水10mLの水溶液 80μL)、5%PTS(Polyoxyethanyl―α―tocopheryl sebacate)水溶液(200μL)を混合し配列番号R-41'により得られた翻訳生成物(Hc1, 20μL)を加え窒素置換した。得られた混合物にPdCl2(MeCN)2(1.0mg、3.9μmol)、2,2'-ジフェニルフォスフィノ-1,1'-ビフェニル(6.2mg、12μmol)をN-メチルピロリジノン(0.2mL)に窒素雰囲気下溶解したPd溶液(60.0μL)を加え50℃にて窒素雰囲気下12時間攪拌した。反応液に0.2Mチオシアヌル酸 1.5カリウム塩水溶液(73.5μL)を加えた。
得られた反応溶液に水を加え2mLとし、遠心を行った(10000rpm、6分間、室温)。上清について凍結乾燥を行い、得られた残渣を水:アセトニトリル=9:1溶液 100μLに再溶解させLC/MS分析を行い、環化体の生成をを確認した。
LC―HRMS:m/z 1007.4149(M-H)-,(Calcd for C46H59N10O16:1007.4116)
保持時間:6.43分 (分析条件 OrbitrapHFIP-Et3N)
Hc1の場合と同様の方法により環化反応を実施した。得られた生成物をHc1の場合と同様の方法によりLC/MS分析を行い、環化体を示す二種類の化合物の生成を確認した。
LC―HRMS:m/z 1310.5579(M-H)-,(Calcd for C58H80N13O22:1310.5546)
保持時間:4.91分
LC―HRMS:m/z 1310.5570(M-H)-,(Calcd for C58H80N13O22:1310.5546)
保持時間:5.17分
(分析条件 OrbitrapHFIP-Et3N)
Hc1の場合と同様の方法により環化反応を実施した。得られた生成物をHc1の場合と同様の方法によりLC/MS分析を行い、環化体の生成を確認した。
LC―HRMS:m/z 1415.5860(M-H)-, Calcd for C58H83N18O24:1415.5833)
保持時間:3.35分 (分析条件 OrbitrapHFIP-Et3N)
mRNA-ペプチドフュージョンをHeck環化反応条件に付し回収したmRNA-ペプチドフュージョンを逆転写し逆転写が問題なく進行することを確認しmRNAがHeck環化反応条件において安定であることを確認した。
PCRにより調製したDNA(配列番号 D-50)を鋳型に、In vitro転写によりmRNAを調整し、RNeasy mini kit (Qiagen社)を用いて精製した。1μMのmRNAに、1.5 μMのピューロマイシンリンカー(Sigma社)(配列番号C-1) 1X T4 RNAライゲースリアクションバッファー(NEB社)、20% DMSO、2 unit/μl T4 RNAライゲース(NEB社)を加え、37℃で30分間ライゲーション反応させた後、RNeasy MiniElutekit(Qiagen社)により精製した。次に、1 μMのmRNA-ピューロマイシンリンカー連結体(以下、mRNA-Pu)を鋳型に、前述の無細胞翻訳液からRF1を除いたものを加え、37℃で30分間翻訳し、mRNA-ペプチドフュージョン分子(化合物Fusion-1)を得た。
KA03S2 DNA配列:
gtaatacgactcactataGGGTTAACTTTAAGAAGGAGATATACATatgACTAGAACTaaggcgTACTGGAGCcttCCGggcggcAGCGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
GGGUUAACUUUAAGAAGGAGAUAUACAUaUgACUAGAACUaaggcgUACUGGAGCcUUCCGggcggcAGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
S2PuFLin配列:
[P]CCCGTCCCCGCCGCCC[Fluorecein-dT][Spacer18][Spacer18][Spacer18][Spacer18][Spacer18]CC[Puromycin] ([P]:5'リン酸化)
得られたmRNA-ペプチドフュージョン分子(化合物Fusion-1, 11.0μL)に10-mer RNA-ペプチドコンジュゲート(化合物70b)(0.33mM水溶液 5.0μL、1.65nmol)、リン酸バッファー(K2HPO4 1.0mmolとK3PO4 0.20mmolの水10mLの水溶液 40μL)、5%PTS(Polyoxyethanyl―α―tocopheryl sebacate)水溶液(100μL)を混合し、PdCl2(MeCN)2(1.0mg、3.9μmol)、2,2'-ジフェニルフォスフィノ-1,1'-ビフェニル(6.2mg、12μmol)をN-メチルピロリジノン(0.2mL)に窒素雰囲気下溶解したPd溶液(30.0μL)を加え50℃にて窒素雰囲気下12時間攪拌した。得られた反応液に0.2Mチオシアヌル酸 1.5カリウム塩水溶液(37.0μL)を加え室温にて30分間放置し、環化反応液を得た。反応液(5.0μL)を水(10.0μL)で希釈しLC分析サンプルとし分析し10-mer RNA-ペプチドコンジュゲート(化合物70b)の消失および10-mer RNA-ペプチドコンジュゲートの環化体(化合物83b)の生成を確認した。
LCMS(ESI) 1265.9(M-3H)3-
保持時間:3.48分(分析条件ZQHFIP-Me2NEt)
環化反応後のmRNA-ペプチドフュージョン分子をRNeasy MiniElute Kit(Qiagen社)で精製した。0.5 μM mRNA-ペプチドフュージョン分子を鋳型に、逆転写反応液(1X M-MLV逆転写反応バッファー(Promega社), 各0.5 mMのdNTP mix, 4 unit/μl M-MLV逆転写酵素RNaseHマイナス点変異, 3 μM 逆転写プライマー(配列番号C-2))を添加し、42°Cで20分間逆転写反応を行った。反応産物を、TGX gel anykD(Biorad社)を用いて電気泳動を行い、ピューロマイシンリンカーに付加されたフルオレセインの蛍光を検出した。その結果、環化反応の有無にかかわらず、効率良く逆転写反応が進行していることが確認された。結果を図43に示す。
逆転写プライマー
TTTGTCCCCGCCGCCCTAAGAGCCAGAGCCAGAGCCGCT
RNA-複合体のHeck反応
mRNA-ペプチド複合体溶液調製
6μMのmRNA(Hc1 mRNA配列(配列番号R-41))に、9 μMのピューロマイシンリンカー(Sigma社)(配列番号C-1) 1X T4 RNAライゲースリアクションションバッファー(New England Biolab社、カタログ番号M0204L)、10% DMSO、0.63 unit/μl T4 RNAライゲース(New England Biolab社、カタログ番号M0204L)を加え、37℃で30分間ライゲーション反応させた後、RNeasy MiniElute kit(Qiagen社)によりmRNA-ピューロマイシンリンカー連結体(以下、mRNA-Pu)を精製した。精製した1 μMのmRNA-Puを鋳型に、無細胞翻訳液(1mM GTP,1mM ATP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,9mM 酢酸マグネシウム,2mMスペルミジン,1mM ジチオスレイトール,0.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.6μM メチオニルtRNAトランスフォルミラーゼ,0.26μM EF-G,0.24μM RF2,0.17μM RF3,0.5μM RRF,2.7μM IF1,0.4μM IF2,1.5μM IF3,40μM EF-Tu,84μM EF-Ts,1.2μM リボソーム,0.73μM AlaRS,0.03μM ArgRS,0.38μM AsnRS,0.13μM AspRS,0.02μM CysRS,0.06μM GlnRS,0.23μM GluRS,0.09μM GlyRS,0.02μM HisRS,0.4μM IleRS,0.04μM LeuRS,0.11μM LysRS,0.03μM MetRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.02μM TyrRS,0.02μM ValRS(自家調製タンパクは基本的はHisタグ付加タンパクとして調製した),各300μMのメチオニンとロイシンを除く18種類のタンパク質性アミノ酸),20μMの化合物AT-1-IIIA、20μM 4-PenteA-tRNAfMetCAU(化合物AT-2-IIIA)を加え、37℃で30分間翻訳し、18mM EDTA pH8.0を加え、mRNA-ペプチド複合体を得た。
得られたmRNA-ペプチド複合体(20.0μL)に、リン酸バッファー(K2HPO4 1.0mmolとK3PO4 0.40mmolの水10mLの水溶液 120μL)、15%PTS(Polyoxyethanyl―α―tocopheryl sebacate)水溶液(200μL)を混合し、PdCl2(MeCN)2(4.0mg、15.4μmol)、2,2'-ジフェニルフォスフィノ-1,1'-ビフェニル(24.8mg、47.5μmol)をN-メチルピロリジノン(0.8mL)に窒素雰囲気下溶解したPd溶液(60.0μL)を加え50℃にて窒素雰囲気下24時間攪拌した。得られた反応液に0.2Mチオシアヌル酸 1.5カリウム塩水溶液(73.5μL)を加え室温にて30分間放置し、環化反応液を得た。
上記の環化反応液を卓上遠心機で遠心分離し、上澄みをRNeasy miniElute Cleanup kit(Qiagen社、Catalog No.74204)で精製し、RNaseフリー水で溶出した。溶出液を、1×Reaction buffer (Promega社、Catalog No.M217A、10mM TrisHCl pH7.5, 5mM EDTA, 0.2M AcONa)、0.5unit/μl RNaseONE ribonuclease(Promega社、Catalog No.M425A)、0.1Unit/μl RNaseH(LifeTechnologies社Catalog No.18021-014)を用いて37℃で3時間反応させ、以下の化合物(化合物Fusion-2)を得た。

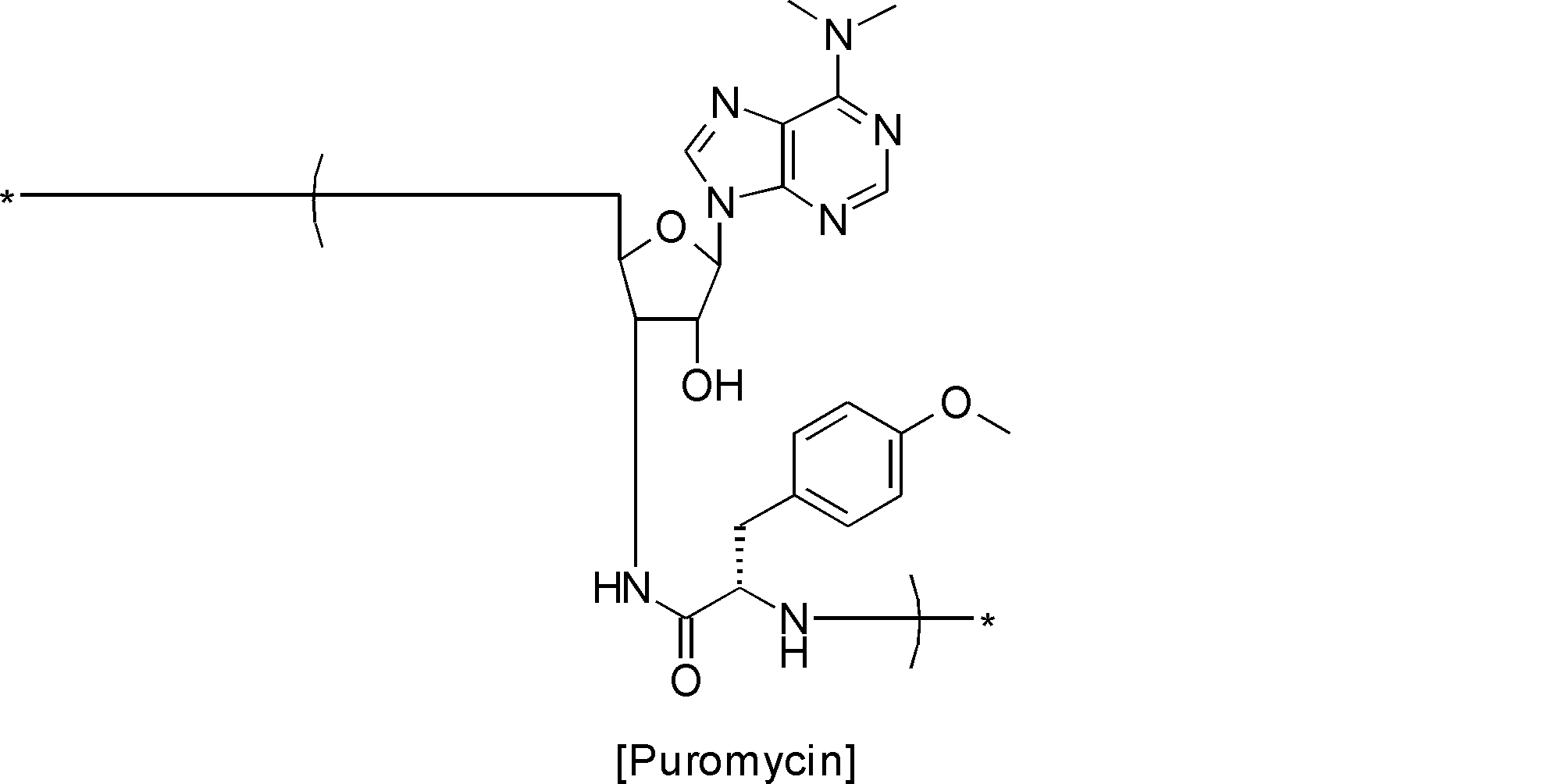

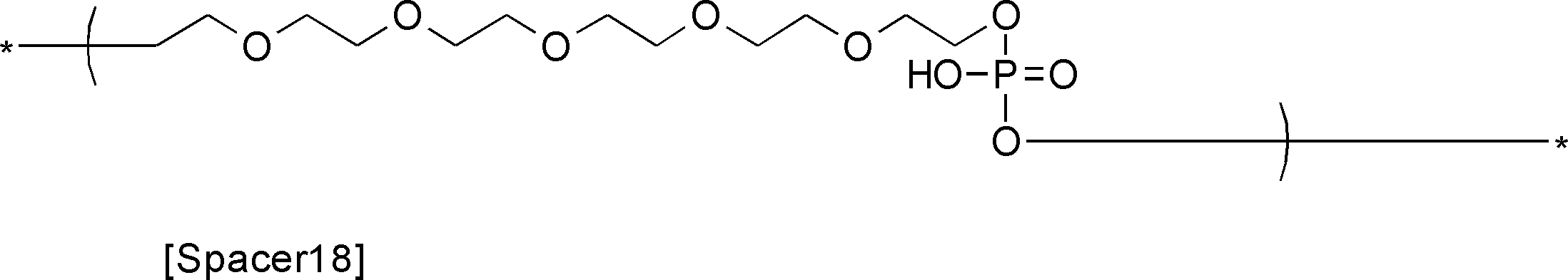
RNase処理済みサンプル(10μL)に400mM HFIP、15mM トリエチルアミン水溶液を90μL加えた後、Oasis HLB μElution(Waters社)を用いて固相抽出を行った。50%アセトニトリル溶出画分について凍結乾燥を行い、水に再溶解したサンプルについてLC/MS分析を行い、ペプチドフュージョン環化生成物の生成を確認した。(図81)
LC―HRMS:m/z 666.2124(M-14H)14-,621.7293(M-15H)15-,582.8100(M-16H)16-(Calcd for C333H466N80O188P24:Molecular Weight 9341)
保持時間:29.74分 (分析条件 OrbitrapHFIP-Et3N-2)
pdCpA-アミノ酸群の合成、続いてtRNA-アミノ酸複合体の合成により、導入される非天然型アミノ酸のうち、ARSによらずに導入されるものの準備を行った。続いて、多様性が高いランダムDNAの準備を行った。転写、翻訳、化学修飾によるRNAと環化ペプチドとの複合体作製に続き、パニングを実施してTNFα、TNFR1、IL-6Rに対する結合ペプチドを取得するための実験を行った
pdCpAとの複合体として、以下の非天然型アミノ酸(MeGly、Phg、Phe(4-CF3)、Met(O2)、βAla、MeAla(4-Thz)、MePhe(3-Cl)、nPrGly、MeSer、Hyp(Et))を翻訳導入するための合成を行った。すなわち、以下のスキームに従い、化合物SP705(Pen-MeGly-pdCpA)、化合物SP710(Pen-Phg-pdCpA)、化合物SP715(Pen-Phe(4-CF3)-pdCpA)、化合物SP720(Pen-Met(O2)-pdCpA)、化合物SP725(Pen-βAla-pdCpA)、化合物SP731(Pen-MeAla(4-Thz)-pdCpA)、化合物SP737(Pen-MePhe(3-Cl)-pdCpA)、化合物SP742(Pen-nPrGly-pdCpA)、化合物SP754(Pen-Hyp(Et)-pdCpA)を合成した。化合物6i-C(tBuSSEtGABA)、化合物1i-IAの合成は既に記載済である。

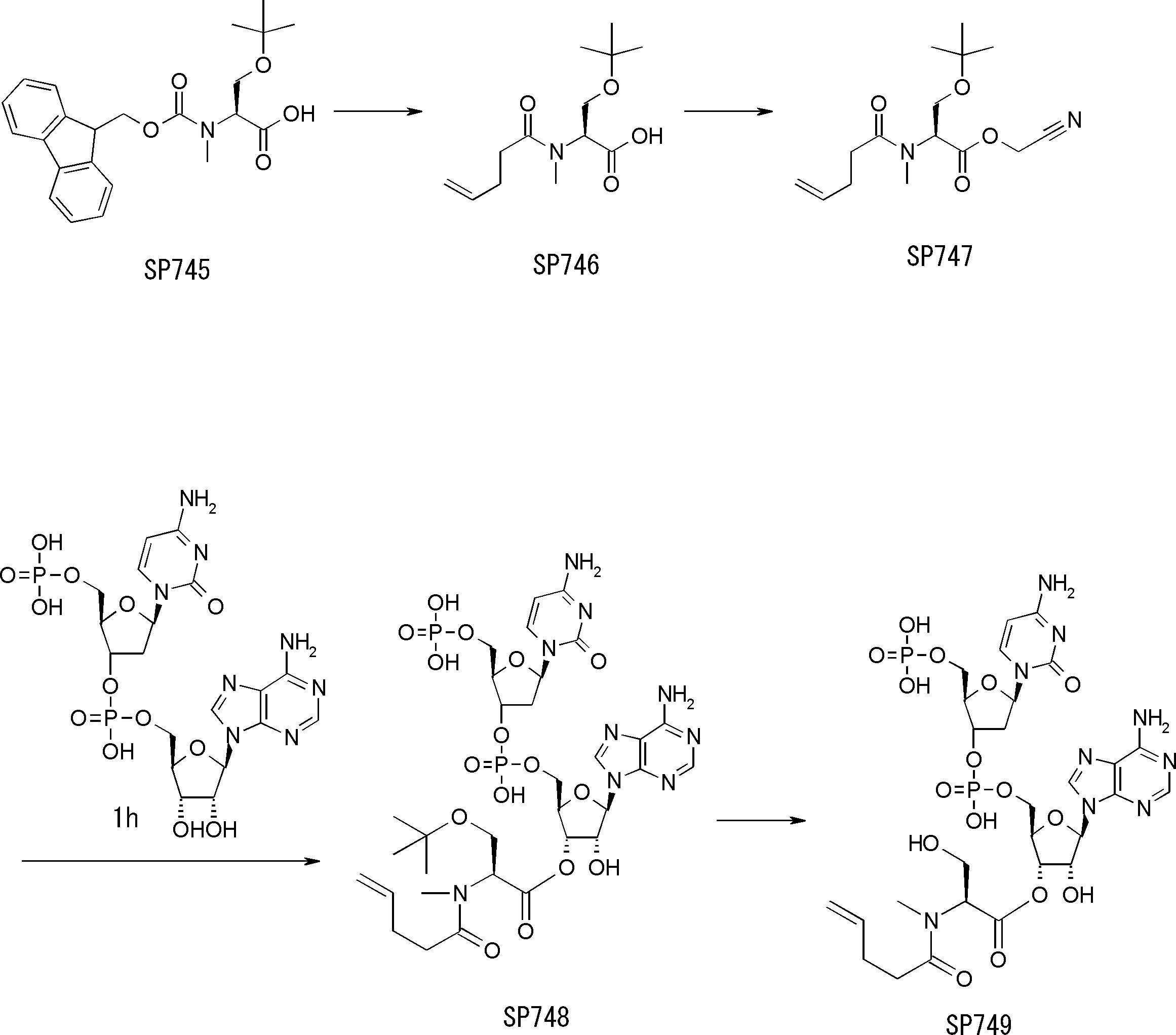

LCMS(ESI) m/z = 251 (M+Na)+
保持時間:1.82分(分析条件SMDmethod6)

なお、本明細書では、4-ペンテノイル基をPen基と略称し、シアノメチル基をCH2CNと記載する。
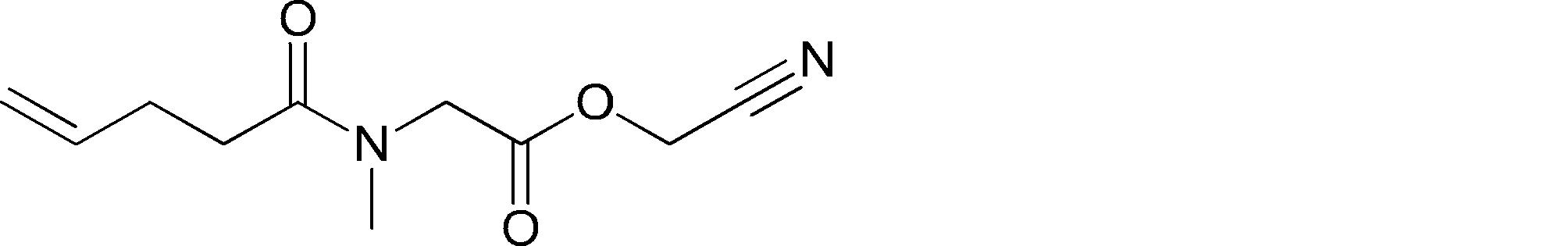
LCMS(ESI) m/z = 211 (M+H)+
保持時間:1.50分(分析条件SMDmethod6)
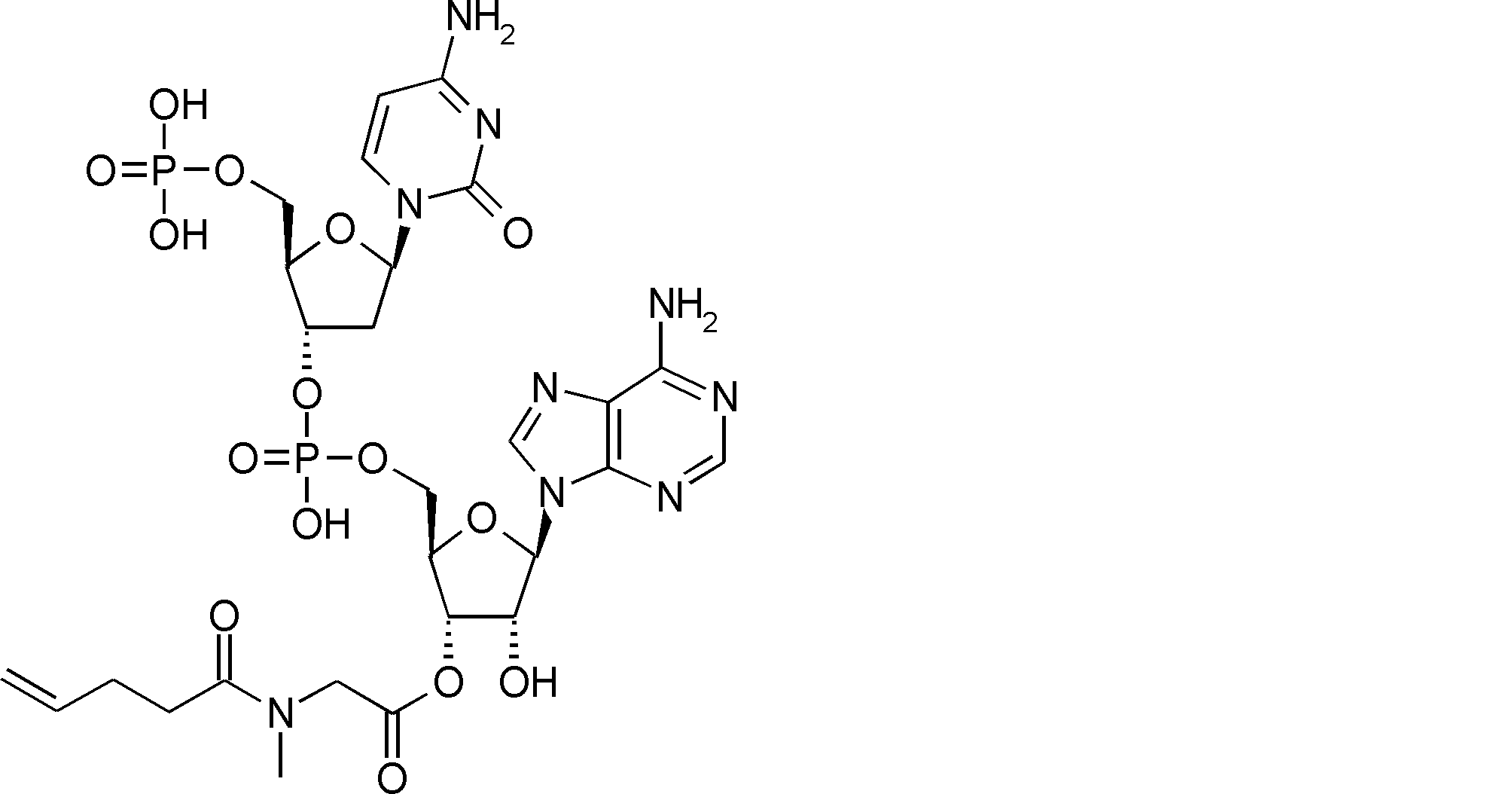
LCMS(ESI) m/z = 790.5 (M+H)+
保持時間:0.46分(分析条件SQDAA05)

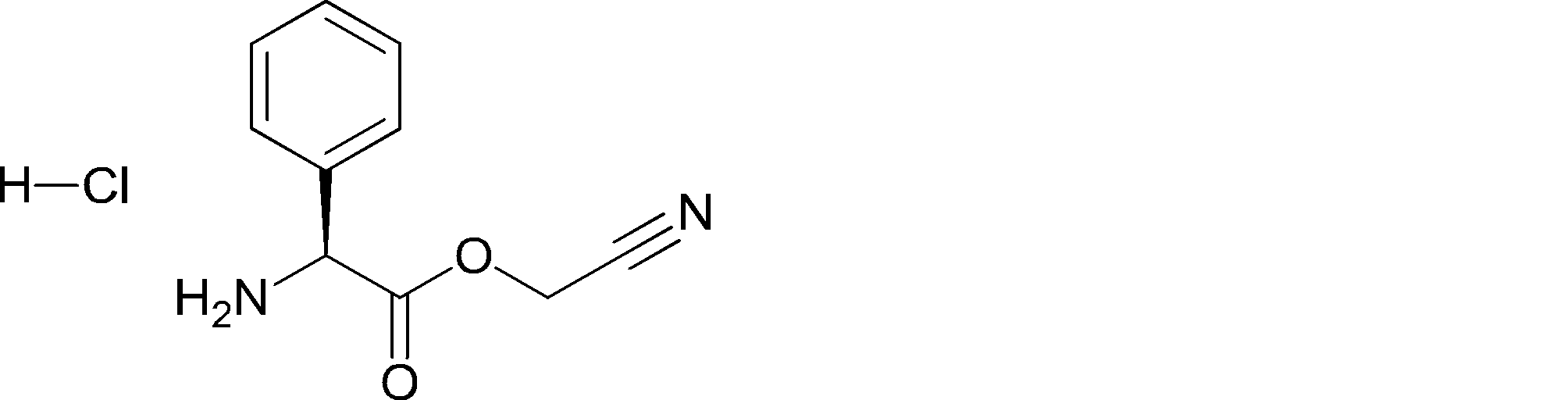
LCMS(ESI) m/z = 273 (M+H)+
保持時間:1.88分(分析条件SMDmethod7)

LCMS(ESI) m/z = 852.5 (M+H)+
保持時間:0.42分(分析条件SQDFA05)

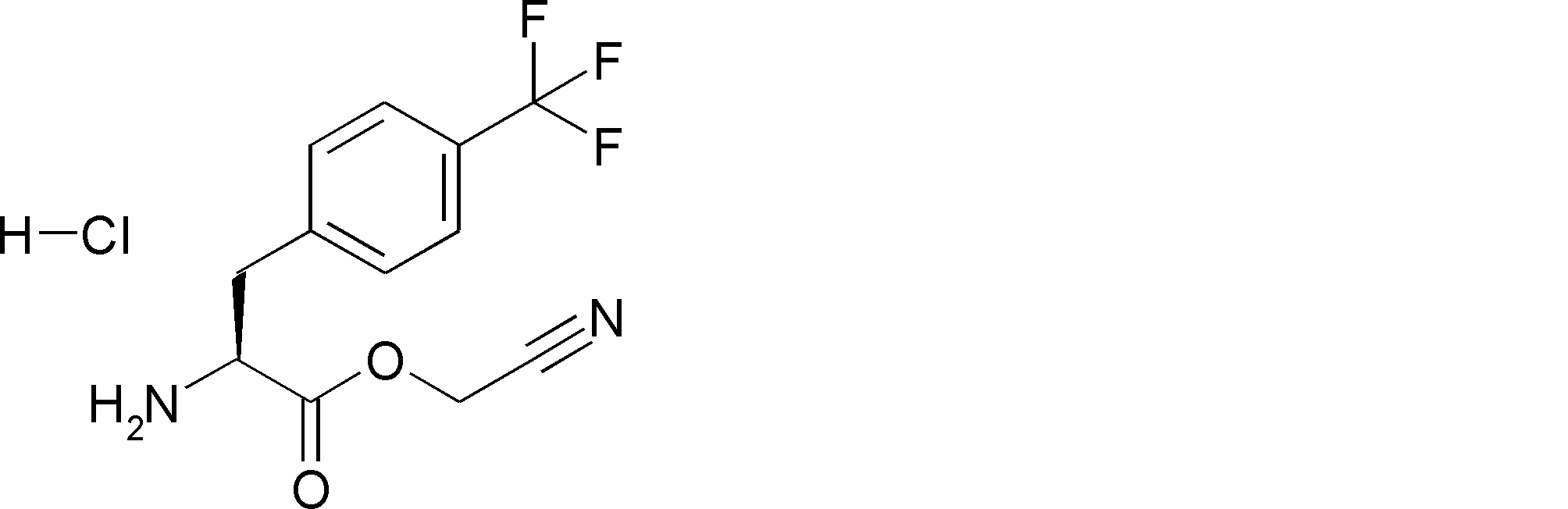

LCMS(ESI) m/z = 355 (M+H)+
保持時間:1.47分(分析条件SMDmethod7)
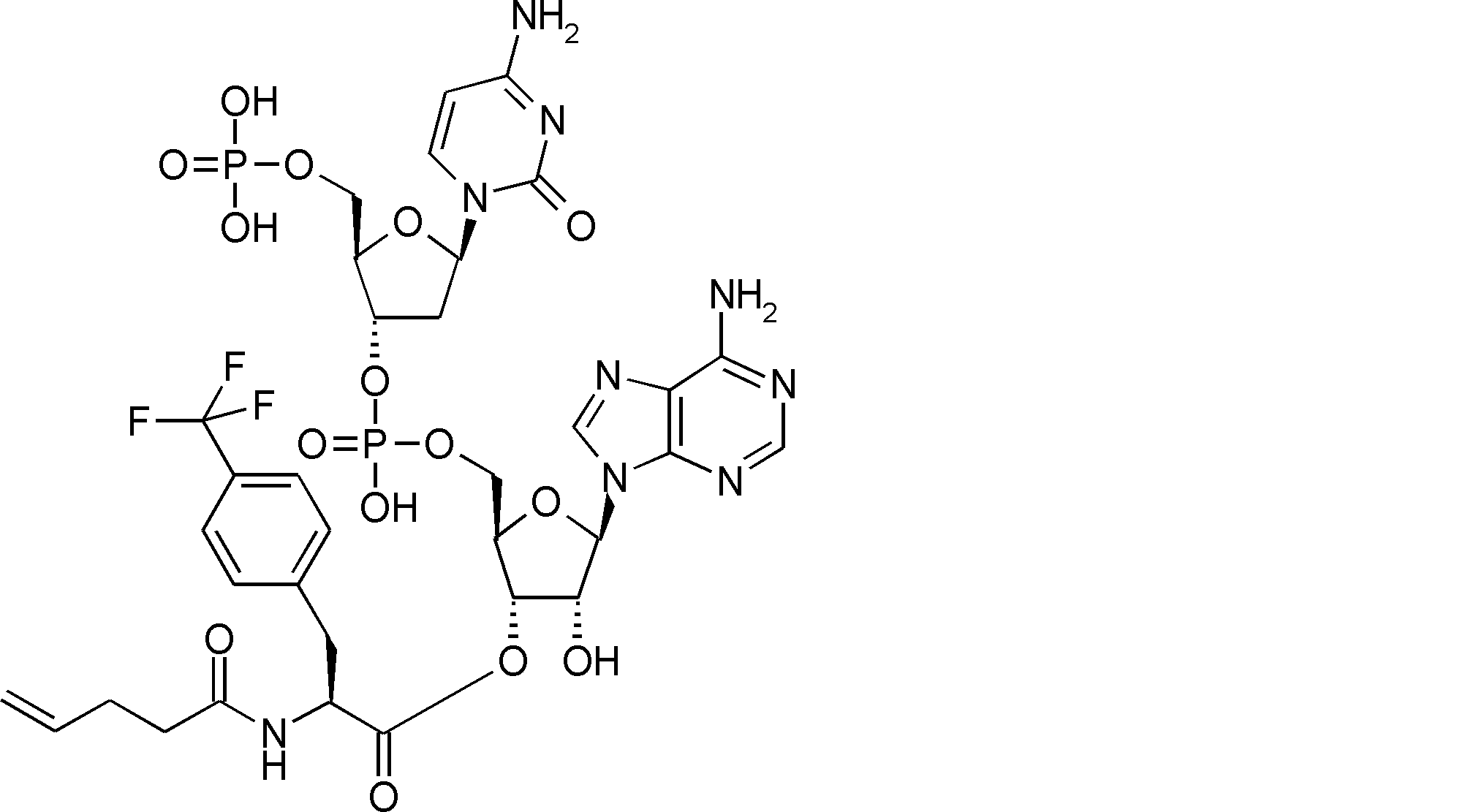
LCMS(ESI) m/z = 934.6 (M+H)+
保持時間:0.73分(分析条件SQDAA05)



LCMS(ESI) m/z = 303 (M+H)+
保持時間:1.28分(分析条件SMDmethod7)
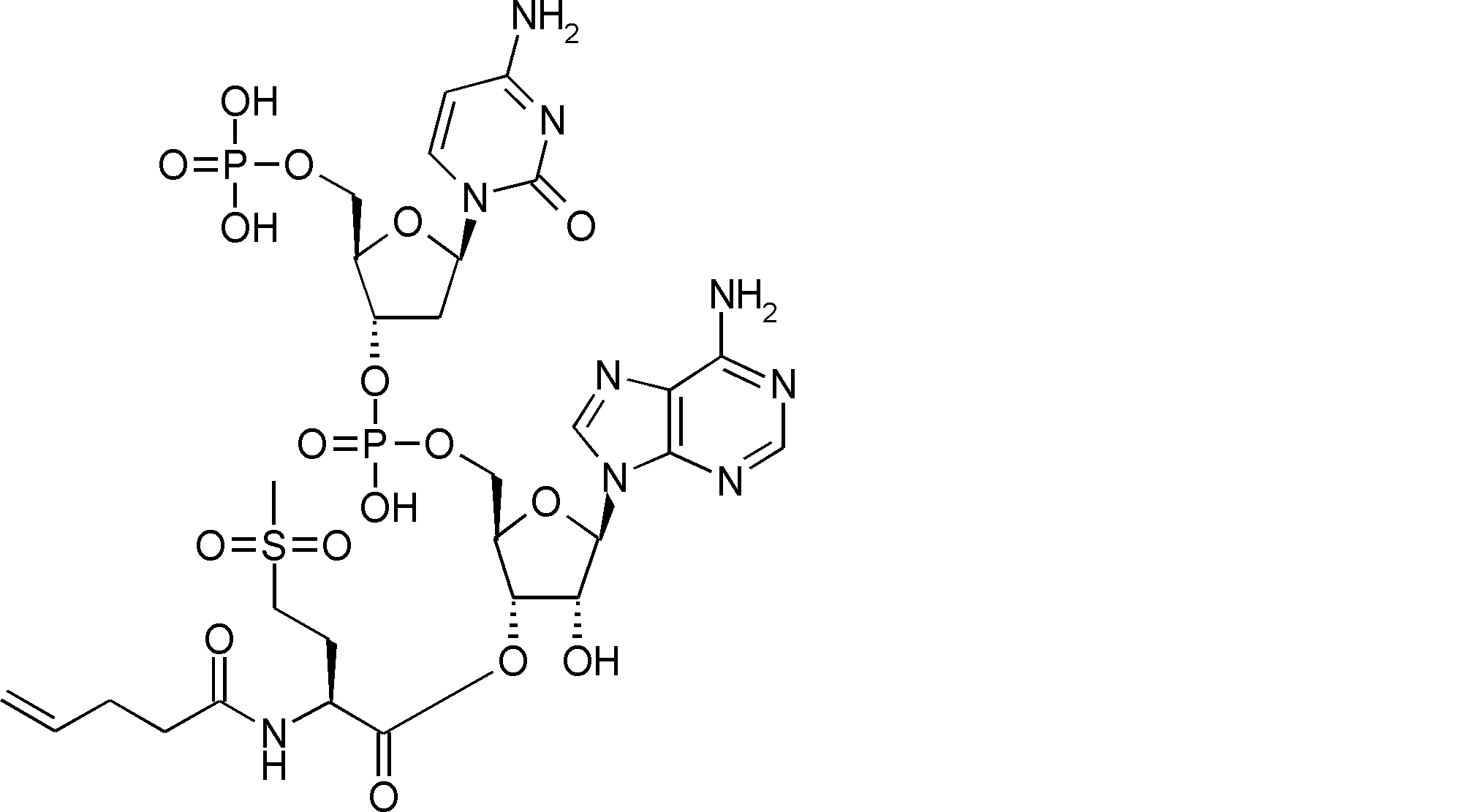
LCMS(ESI) m/z = 882.3 (M+H)+
保持時間:0.40分(分析条件SQDAA05)

LCMS(ESI) m/z = 211 (M+H)+
保持時間:1.37分(分析条件SMDmethod8)
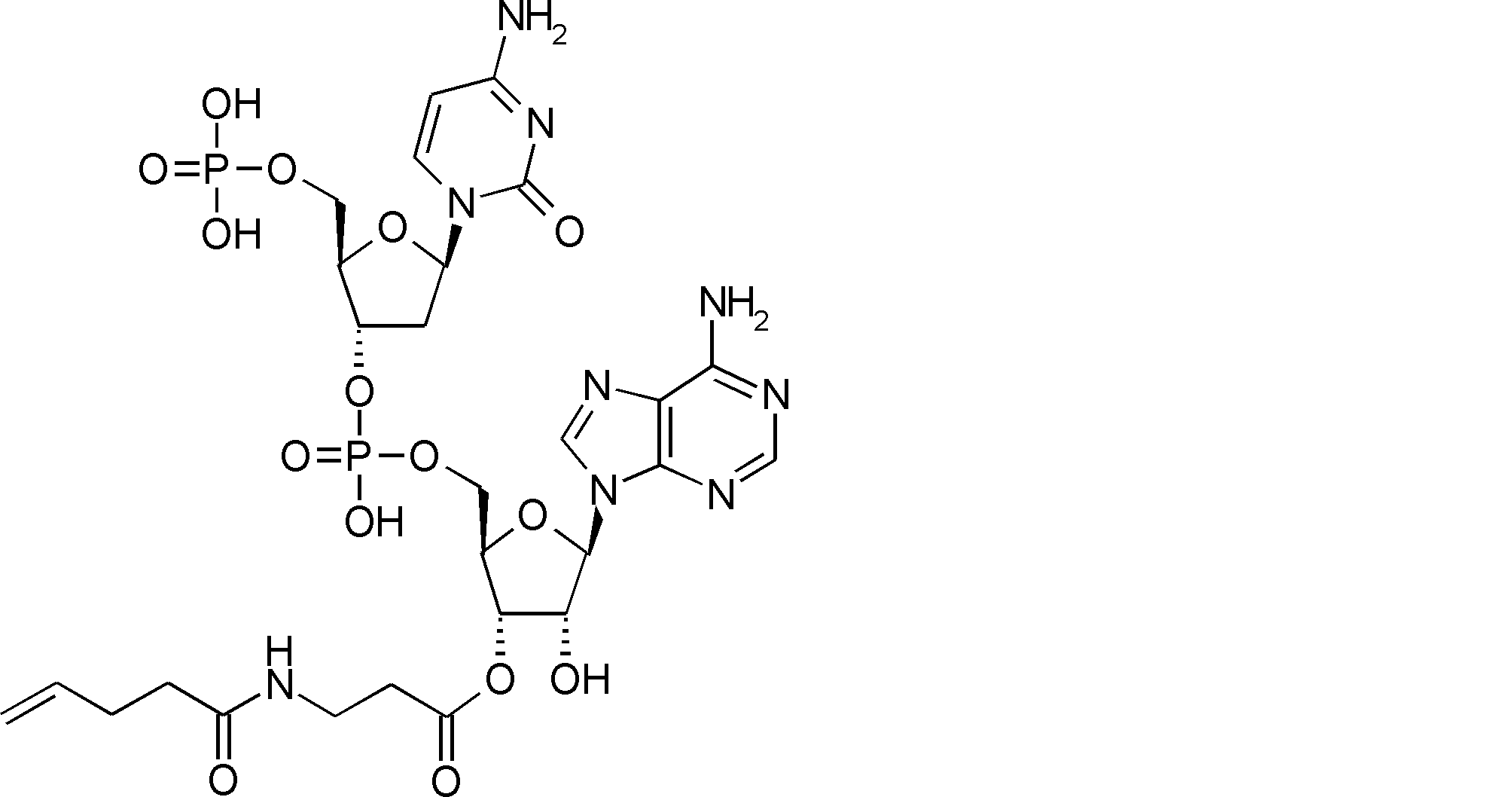
LCMS(ESI) m/z = 790.6 (M+H)+
保持時間:0.41分(分析条件SQDAA05)

LCMS(ESI) m/z = 287 (M+H)+
保持時間:1.24分(分析条件SMDmethod7)
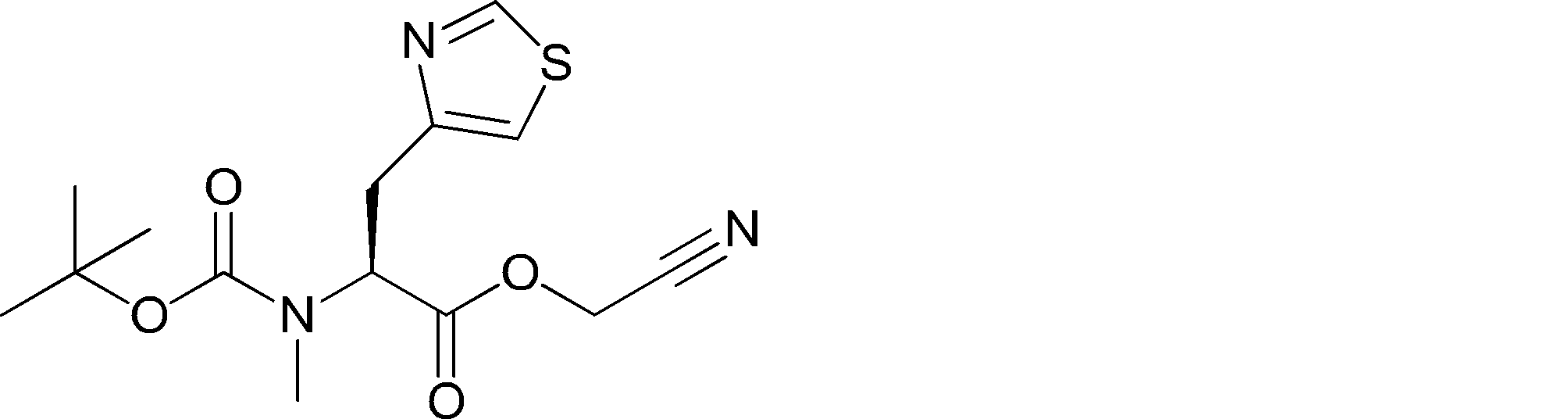

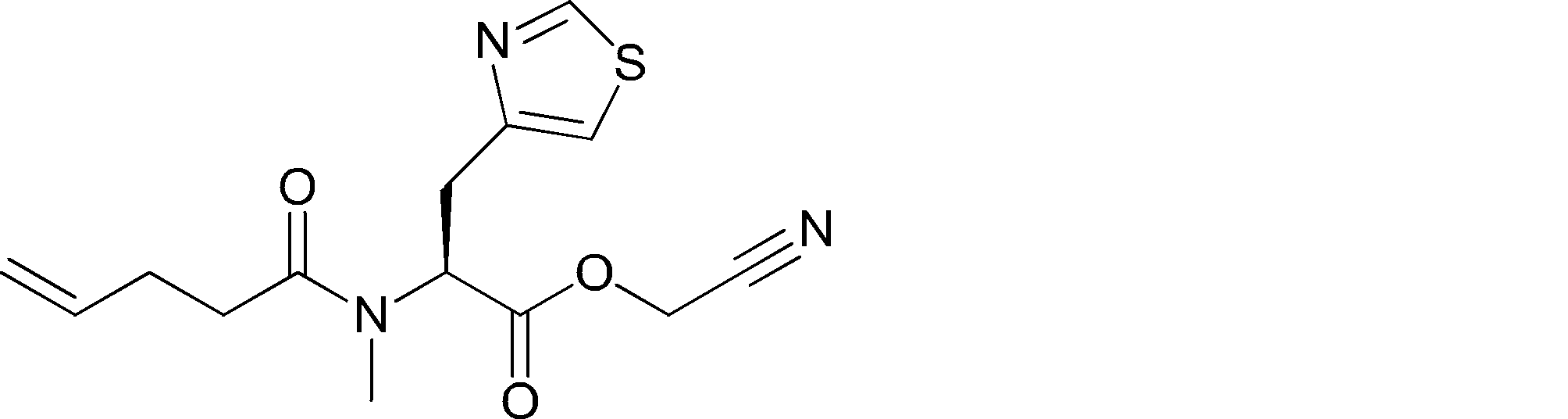
LCMS(ESI) m/z = 308 (M+H)+
保持時間:1.28分(分析条件SMDmethod7)
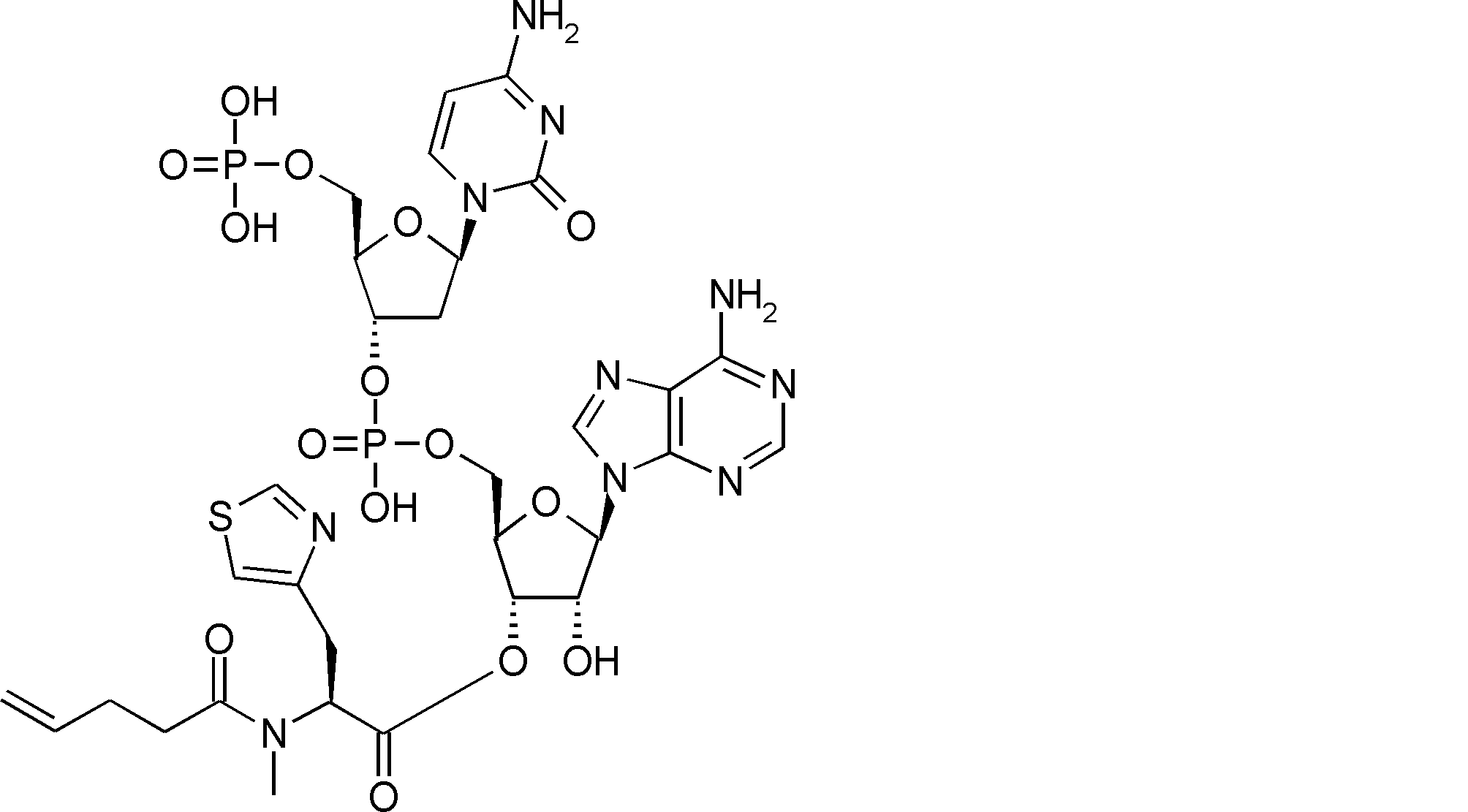
LCMS(ESI) m/z = 887.4 (M+H)+
保持時間:0.40分(分析条件SQDFA05)

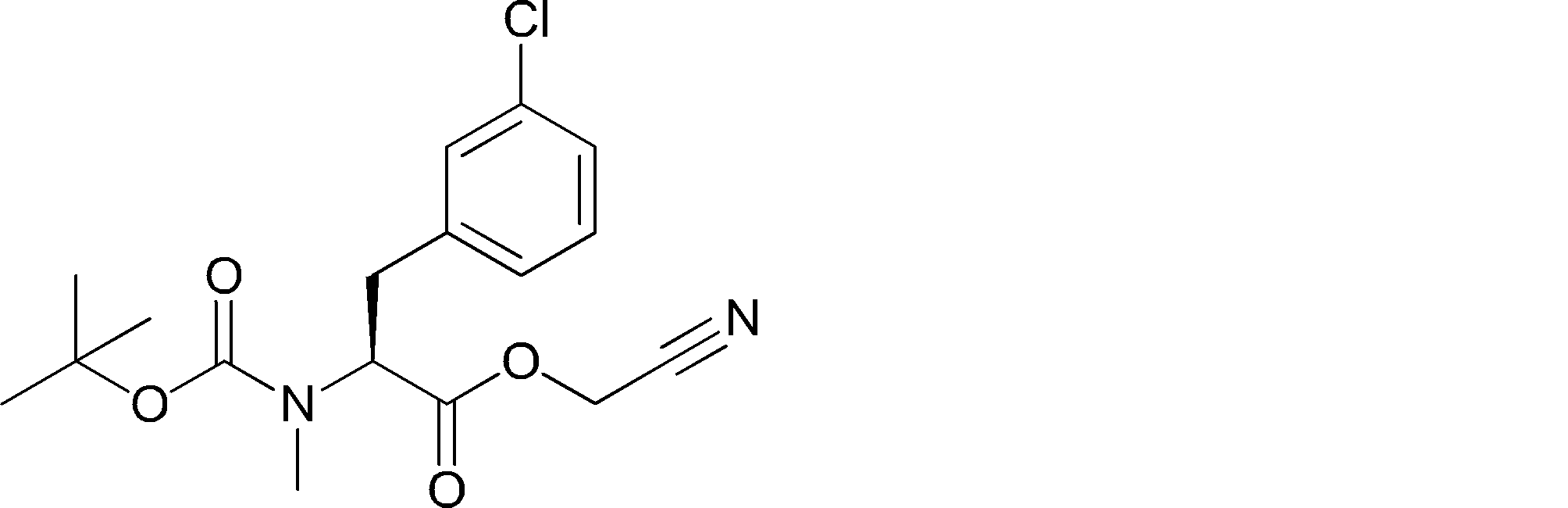
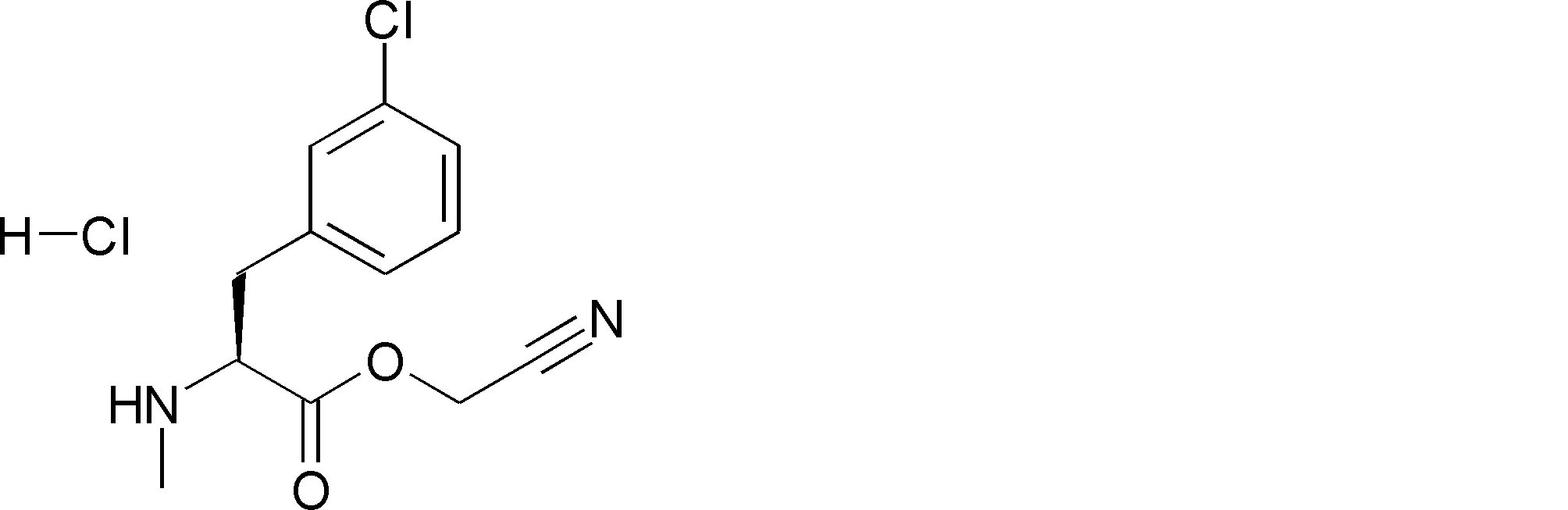

LCMS(ESI) m/z = 335 (M+H)+
保持時間:1.67分(分析条件SMDmethod4)

LCMS(ESI) m/z = 914.6 (M+H)+
保持時間:0.75分(分析条件SQDAA05)
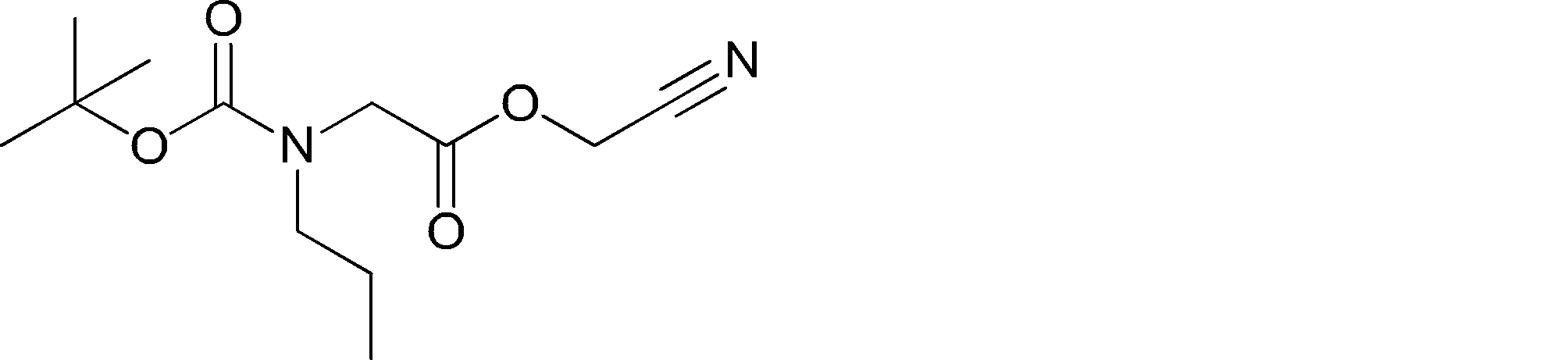


1H-NMR(Bruker AVANCE II、300MHz、CDCl3) δ ppm 6.19-6.32 (1H、m)、5.38-5.49 (2H、m)、5.17-5.22 (2H、m)、4.49-4.55 (2H、m)、3.70-3.78 (2H、m)、2.78-2.90 (4H、m)、1.84-2.07 (2H、m)、1.34 (3H、t、J = 8.1 Hz)
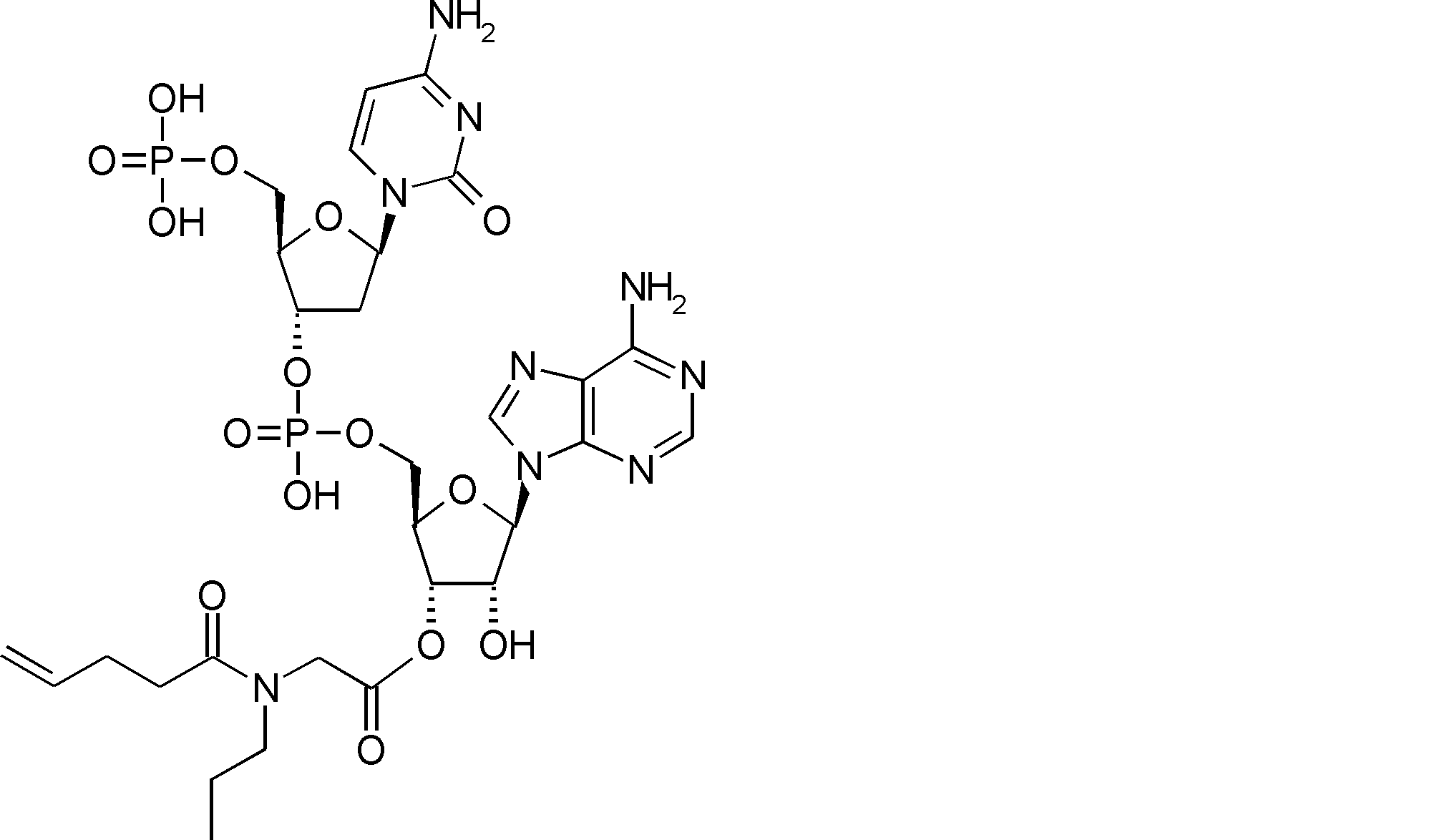
LCMS(ESI) m/z = 818.4 (M+H)+
保持時間:0.57分(分析条件SQDAA05)
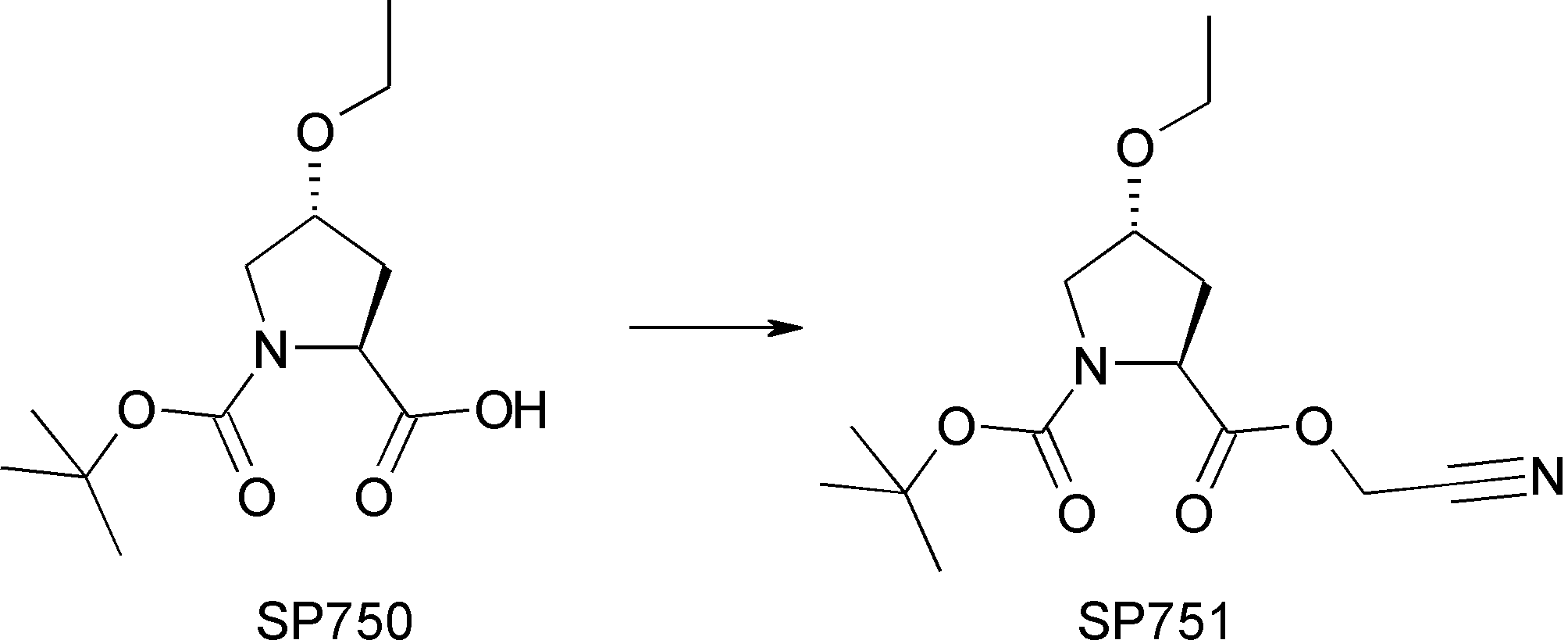

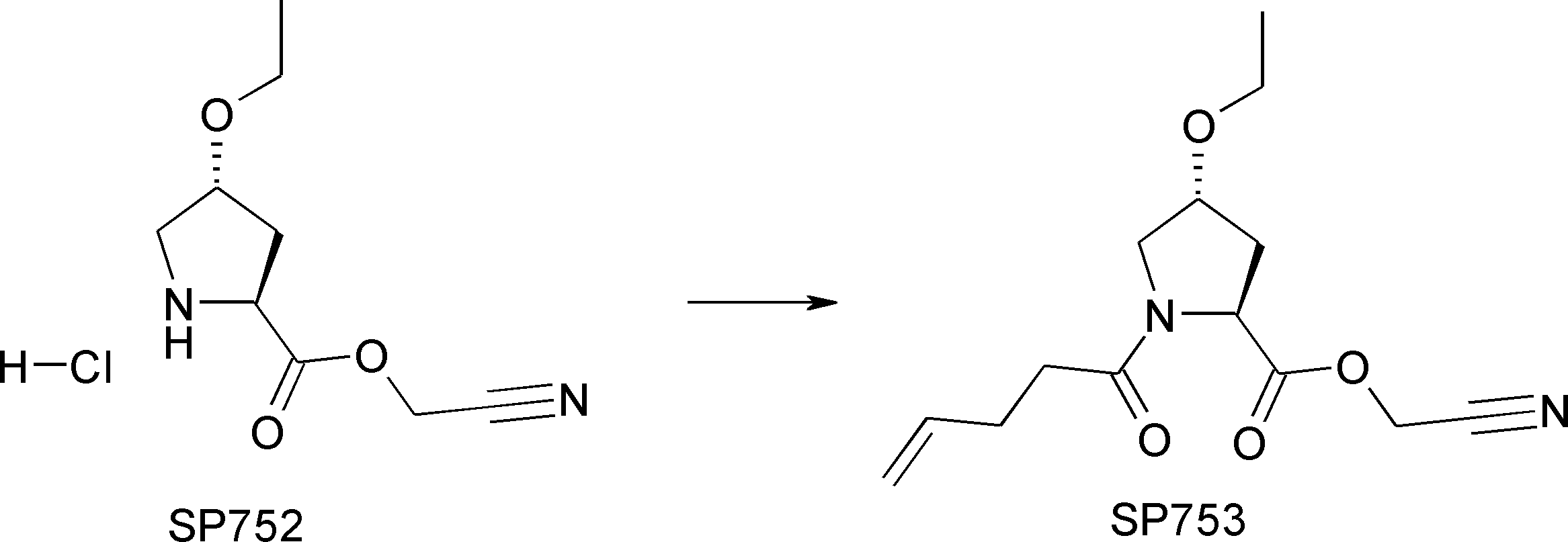
LCMS(ESI) m/z = 281 (M+H)+
保持時間:1.45分(分析条件SMDmethod4)
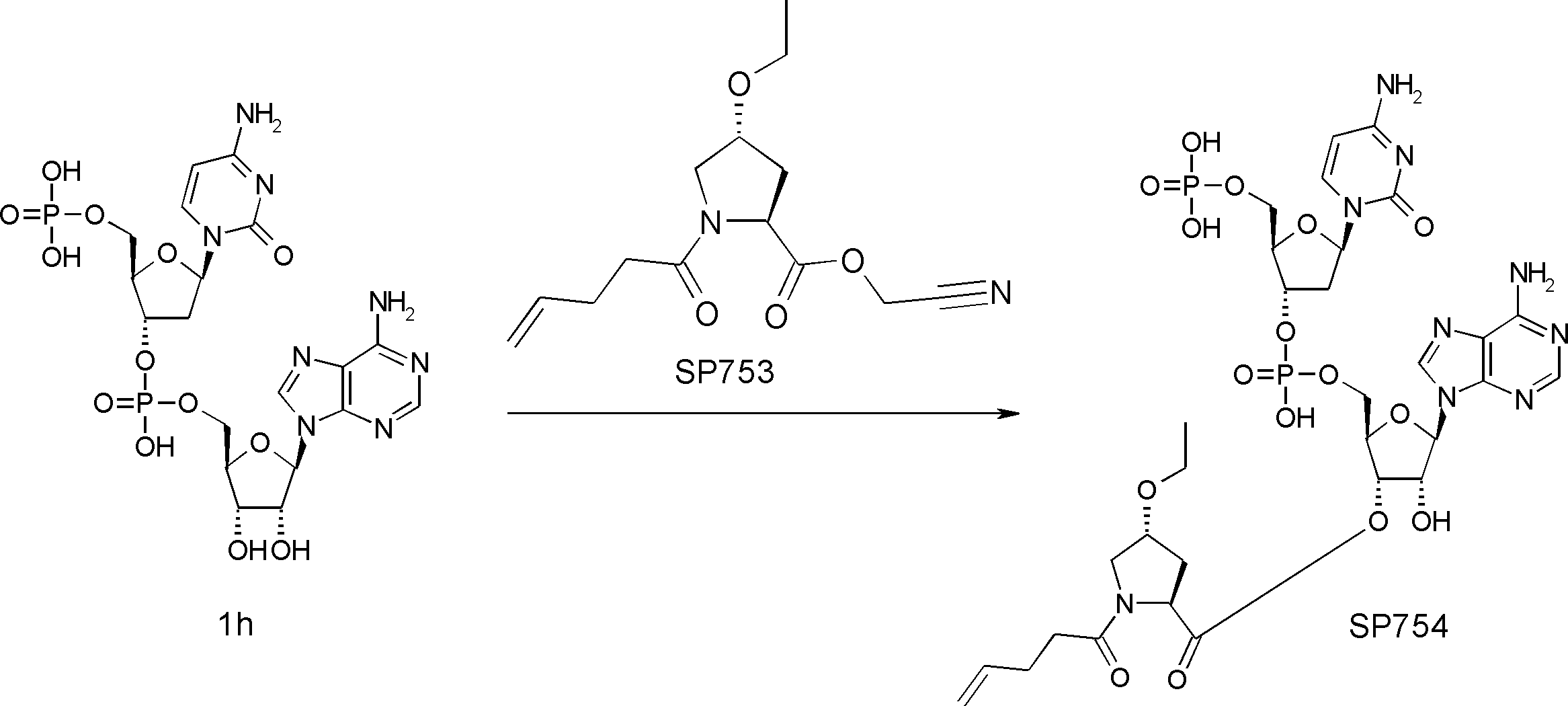
LCMS(ESI) m/z = 860.4 (M+H)+
保持時間:0.55分(分析条件SQDAA05)
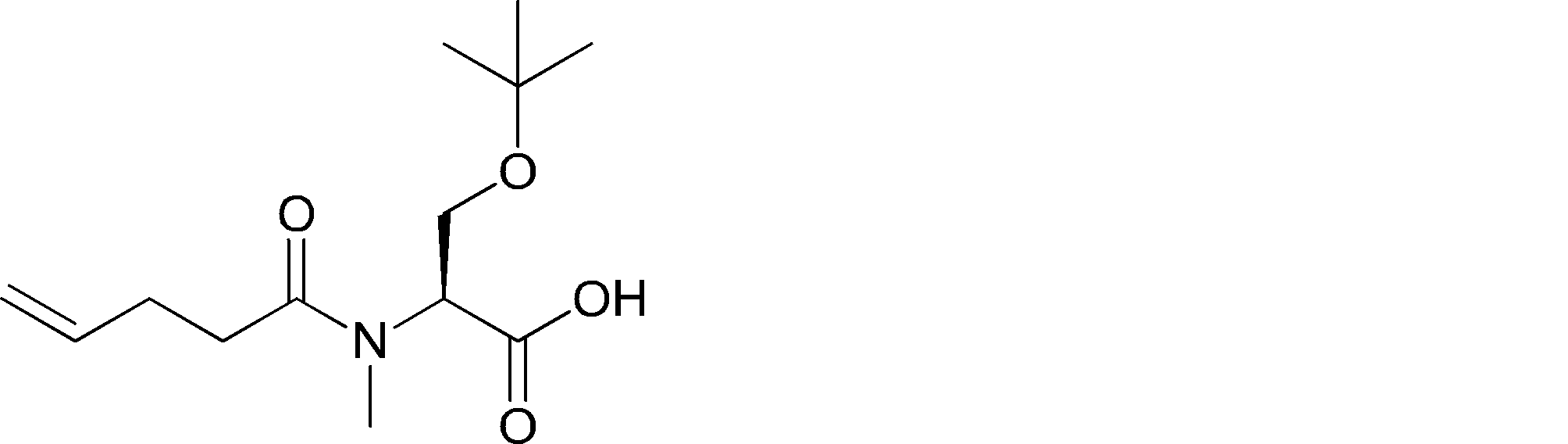
LCMS(ESI) m/z = 258 (M+H)+
保持時間:0.71分(分析条件SQDAA05)
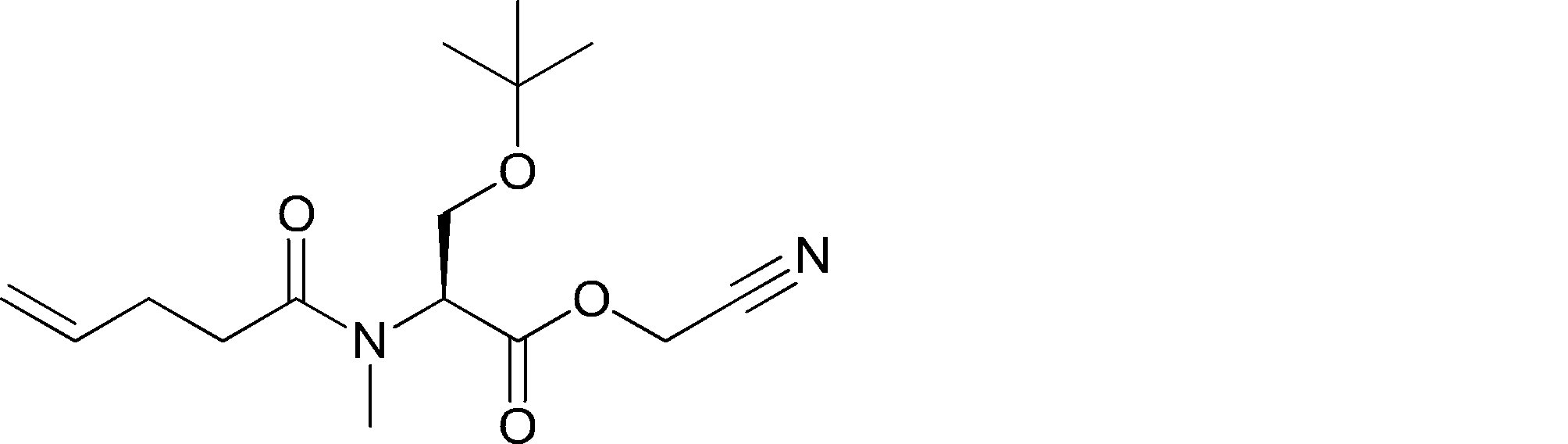
LCMS(ESI) m/z = 297.6 (M+H)+
保持時間:0.77分(分析条件SQDFA05)
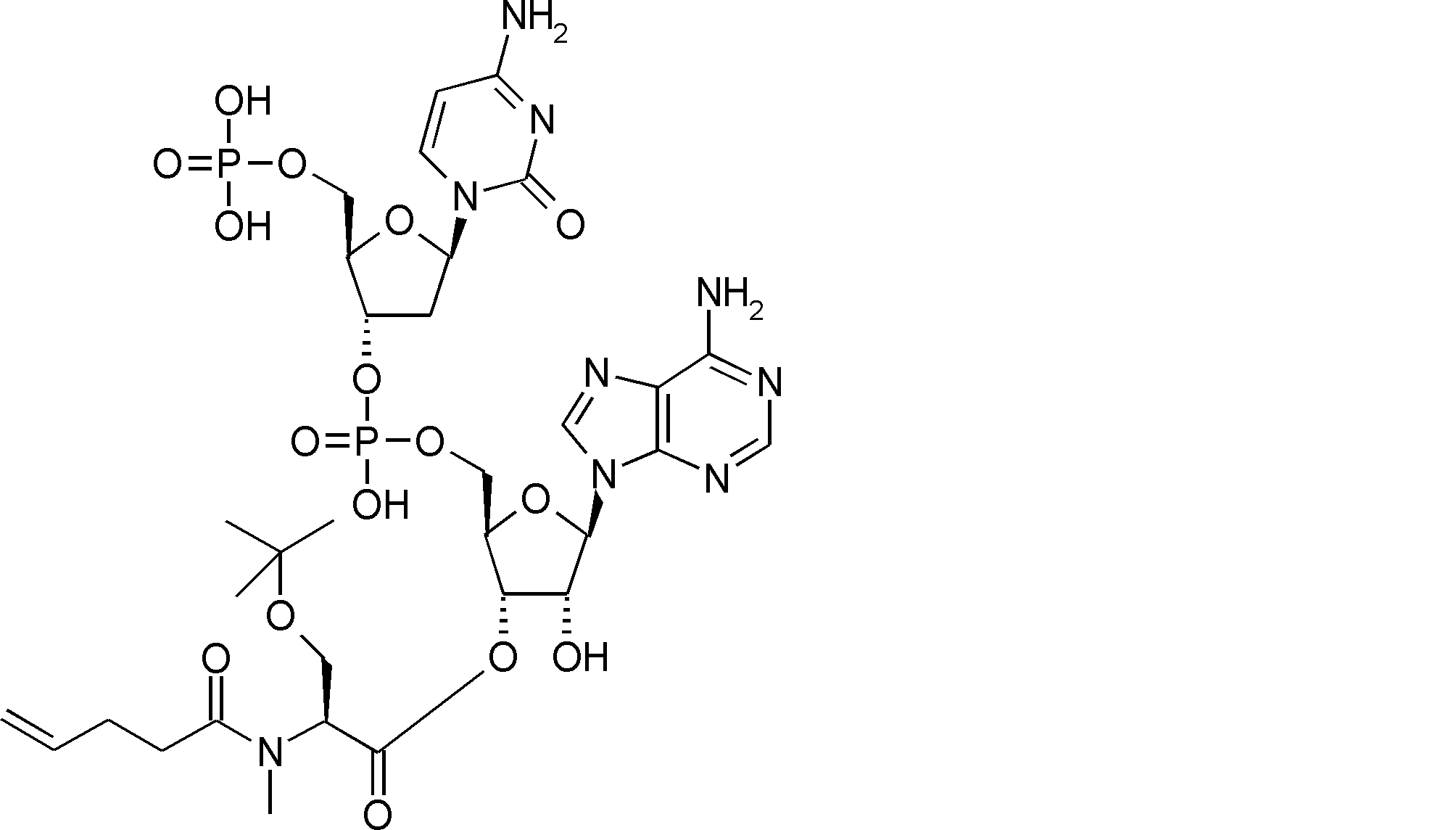
LCMS(ESI) m/z = 876.7 (M+H)+
保持時間:0.47分(分析条件SQDFA05)
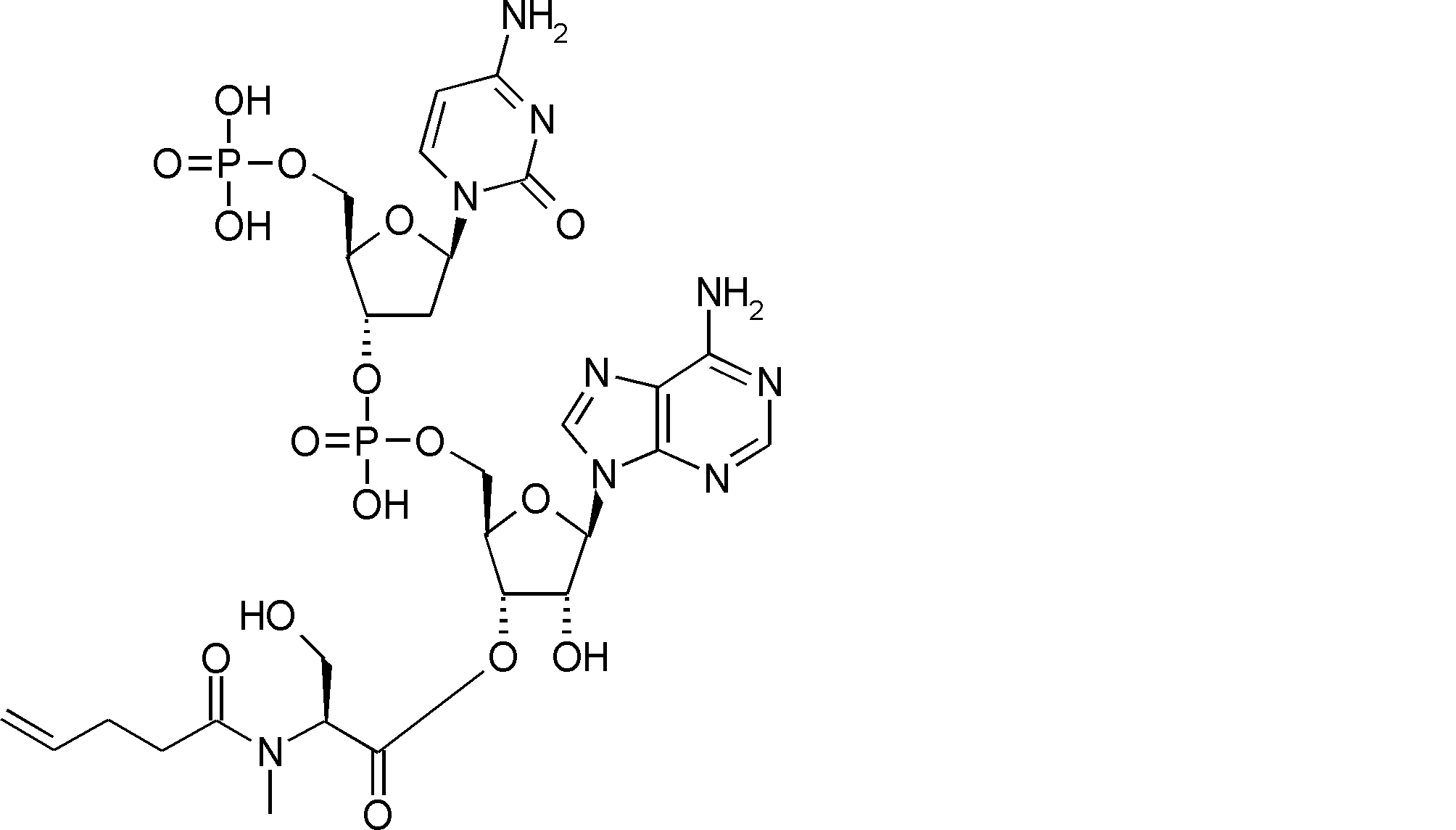
LCMS(ESI) m/z = 820.5 (M+H)+
保持時間:0.31分(分析条件SQDFA05)
パニングに使用する転写tRNAおよび転写tRNA(CA欠損)を以下の通り調製した。
tRNAGluCUG (-CA) CAG DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTctgACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluACG (-CA) CGU DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTacgACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluUUC (-CA) GAA DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTttcACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluCCU (-CA) AGG DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTcctACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluCAA (-CA) UUG DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTcaaACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluUAG (-CA) CUA DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTtagACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluCUA (-CA) UAG DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTctaACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluCCG (-CA) CGG DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTccgACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluAAG (-CA) CUU DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTaagACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluGCA (-CA) UGC DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTgcaACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAGluCAU (-CA) AUG DNA配列:
GGCGTAATACGACTCACTATAGTCCCCTTCGTCTAGAGGCCCAGGACACCGCCCTcatACGGCGGTAACAGGGGTTCGAATCCCCTAGGGGACGC
tRNAAsn-E2GUU (-CA) AAC DNA配列:
GGCGTAATACGACTCACTATAGGCTCTGTAGTTCAGTCGGTAGAACGGCGGACTgttAATCCGTATGTCACTGGTTCGAGTCCAGTCAGAGCCGC
tRNAAla1B DNA配列:
GGCGTAATACGACTCACTATAGGGGCTATAGCTCAGCTGGGAGAGCGCCTGCTTAGCACGCAGGAGGTCTGCGGTTCGATCCCGCATAGCTCCACCA
tRNATyr1 DNA配列:
GGCGTAATACGACTCACTATAGGTGGGGTTCCCGAGCGGCCAAAGGGAGCAGACTGTAAATCTGCCGTCATCGACTTCGAAGGTTCGAATCCTTCCCCCACCACCA
tRNAGluCUG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUcugACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluACG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUacgACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluUUC (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUuucACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluCCU (-CA) RNA配列:(配列番号:116)
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUccuACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluCAA (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUcaaACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluUAG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUuagACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluCUA (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUcuaACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluCCG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUccgACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluAAG (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUaagACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluGCA (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUgcaACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAGluCAU (-CA) RNA配列:
GUCCCCUUCGUCUAGAGGCCCAGGACACCGCCCUcauACGGCGGUAACAGGGGUUCGAAUCCCCUAGGGGACGC
tRNAAsn-E2GUU (-CA) RNA配列:
GGCUCUGUAGUUCAGUCGGUAGAACGGCGGACUguuAAUCCGUAUGUCACUGGUUCGAGUCCAGUCAGAGCCGC
tRNAAla1B RNA配列:
GGGGCUAUAGCUCAGCUGGGAGAGCGCCUGCUUAGCACGCAGGAGGUCUGCGGUUCGAUCCCGCAUAGCUCCACCA
tRNATyr1 RNA配列:
GGUGGGGUUCCCGAGCGGCCAAAGGGAGCAGACUGUAAAUCUGCCGUCAUCGACUUCGAAGGUUCGAAUCCUUCCCCCACCACCA
まず、個別にアミノアシル化tRNA(化合物ATL-1~13)を調製した。ライゲーションは、ligation buffer (50mM HEPES-KOH pH7.5, 20mM MgCl2, 1mM ATP)、25μM転写tRNA (-CA)(配列番号MTL-1~12、R-5)、0.6U/μl T4 RNAリガーゼ(New england bio lab.社)、0.5mM アミノアシル化pdCpAの条件で行った。25μM転写tRNA (-CA) と0.5 mM アミノアシル化pdCpAの組み合わせは、以下の表20~22の通りに行った。
ライゲーションを行う前に、ligation buffer、転写tRNA (-CA)(配列番号MTL-1~12、R-5)およびNuclease free waterを混合したものを、95℃で2分間加熱した後、室温で5分間放置し、予めtRNAのリフォールディングを行った。その後、T4 RNAリガーゼ、アミノアシル化pdCpAのDMSO溶液を上述の通り加え、15℃で45分間ライゲーション反応を行った。
ライゲーション反応液に、0.3M 酢酸ナトリウムと、62.5mMヨウ素(水:THF=1:1溶液)を加え、室温で1時間(ただし、化合物SP749を用いる際は室温で5分間)、ペンテノイル基の脱保護を行い、化合物ATL-1~12を得た。ペンテノイル基を持たない化合物6i-Cを用いる際は、脱保護は行わず、ライゲーション反応まで行い、化合物ATL-13を得た。
次に、翻訳液S用アミノアシル化tRNA 混合物(Initiation suppression翻訳に使用)、翻訳液S用Initiatorアミノアシル化tRNA(Initiation suppression翻訳に使用)、および翻訳液T用アミノアシル化tRNA混合物(Read through翻訳に使用)を調製した。得られた化合物 ATL-1~13を、表20~22に示す混合比で混合し、翻訳液S用アミノアシル化tRNA混合物、翻訳液S用Initiatorアミノアシル化tRNA、翻訳液T用アミノアシル化tRNA混合物として、フェノール抽出した後、エタノール沈殿により回収した。調製したアミノアシル化tRNAは、翻訳液に添加する直前に1mM酢酸ナトリウムに溶解した。以降、翻訳で使用する際には、表20~22に示す最終濃度となるよう翻訳液を調製した。
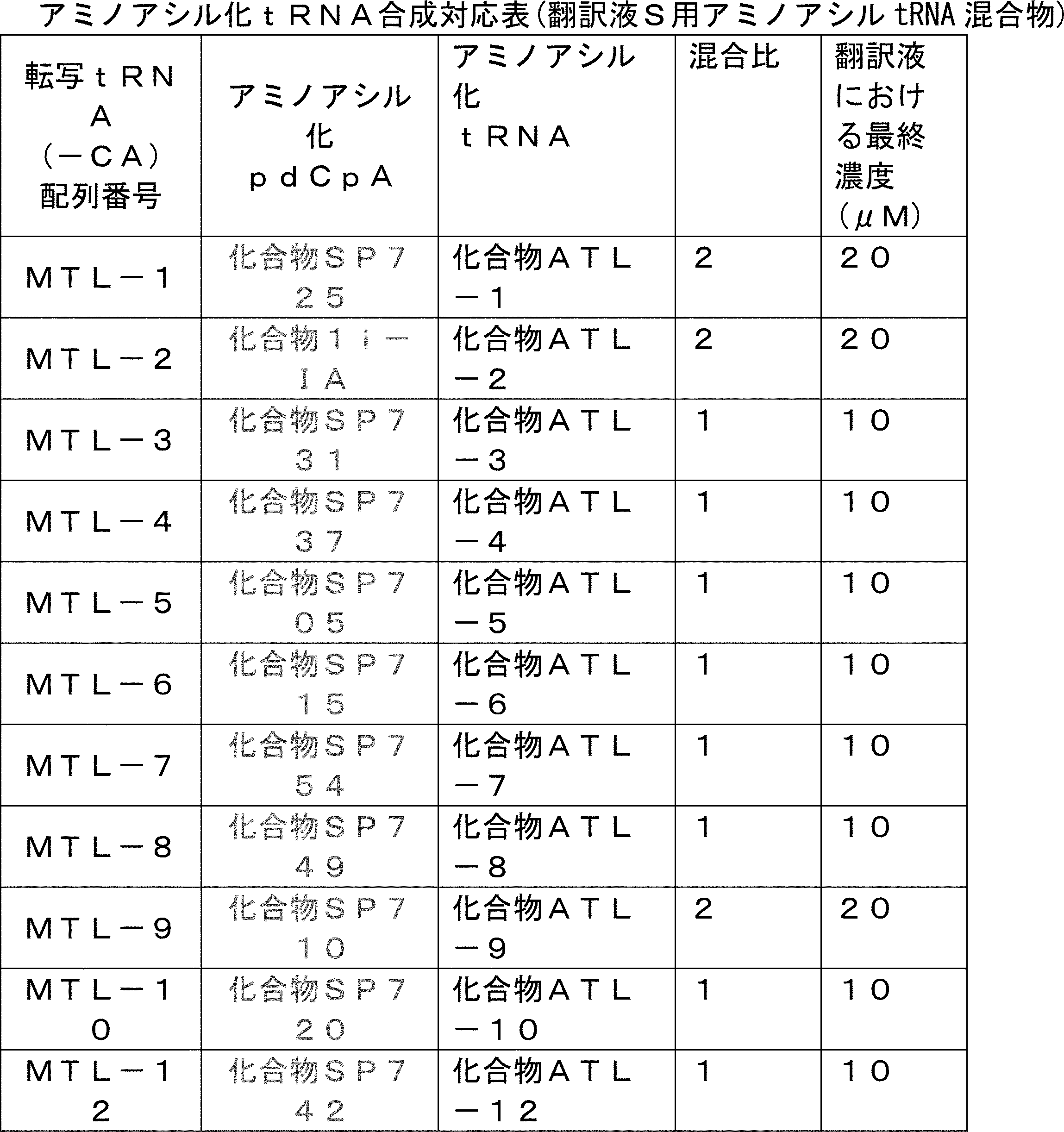
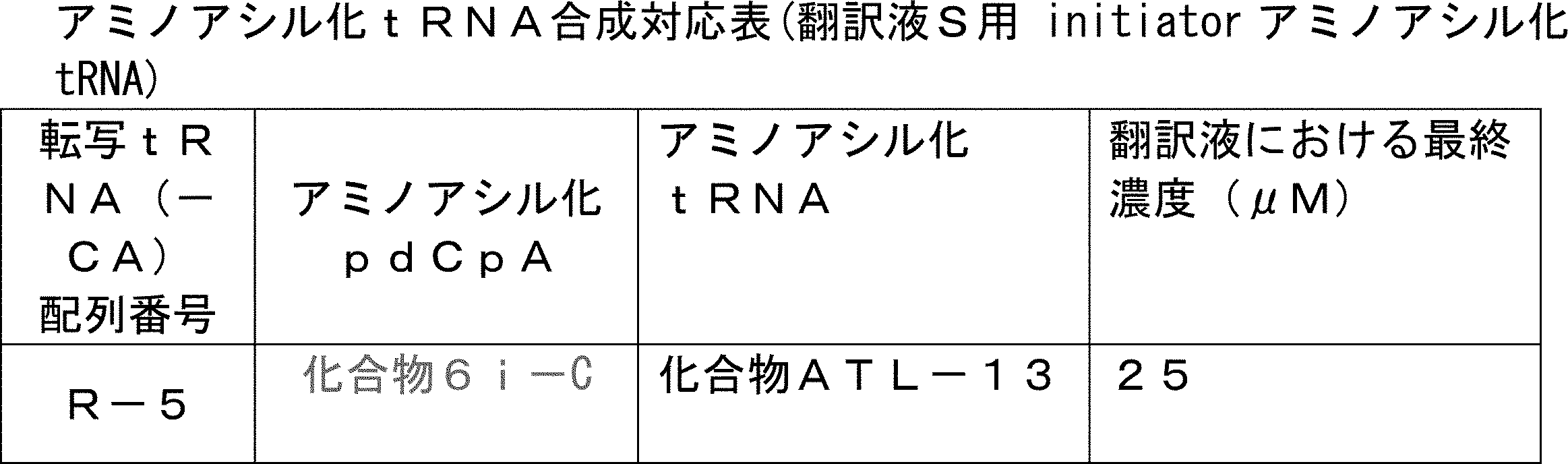
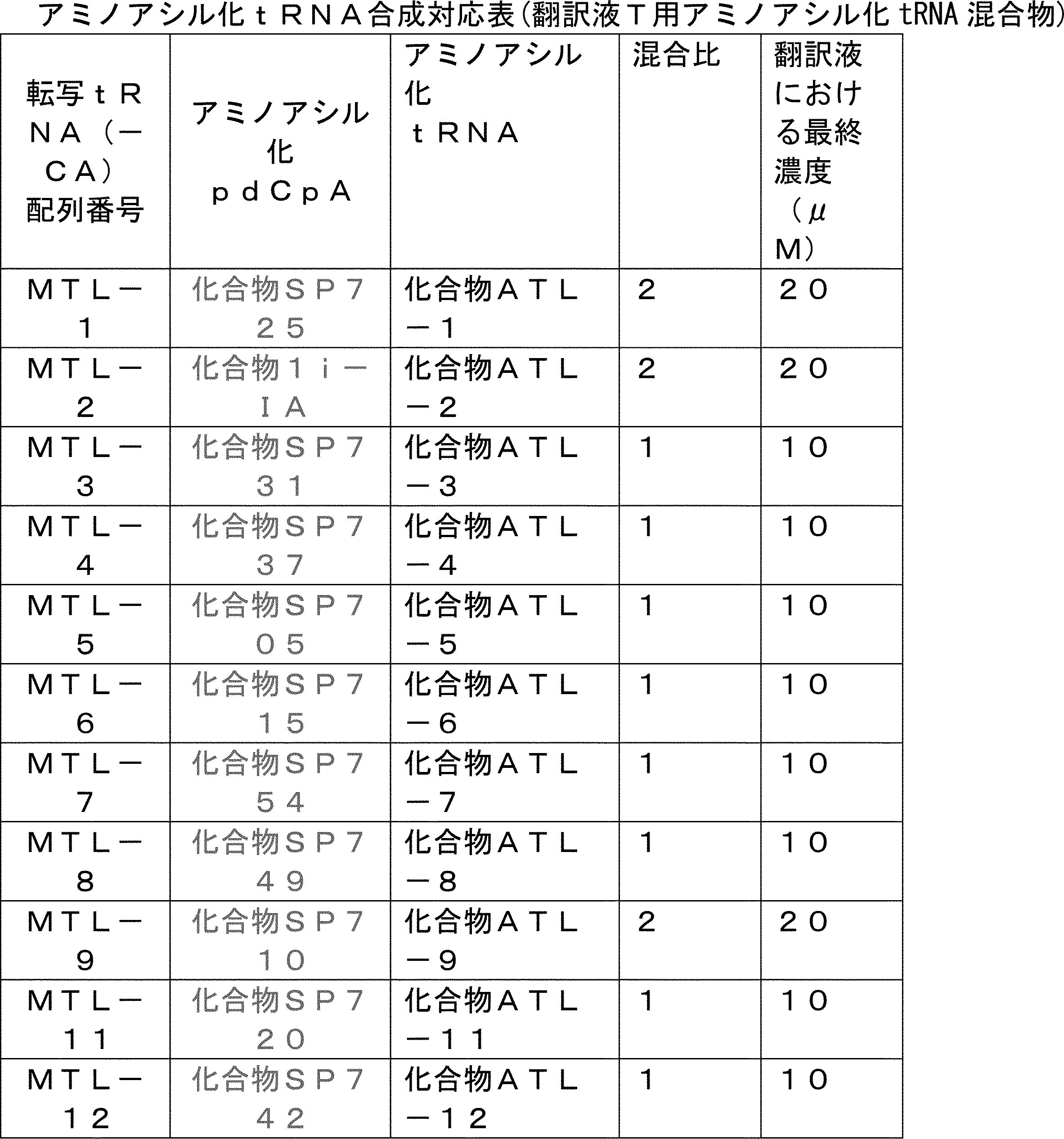
βAla-tRNAGluCUG (-CA)
Asp(SMe)-tRNAGluACG (-CA)
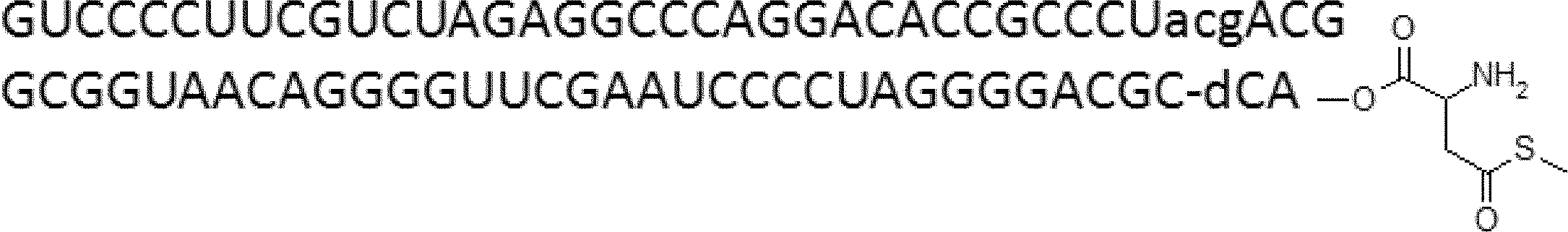
MeAla(4-Thz)-tRNAGluUUC (-CA)
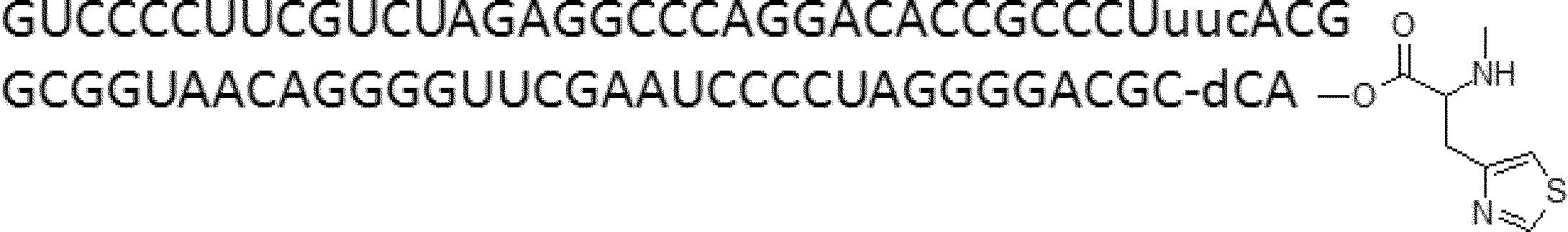
MePhe(3-Cl)-tRNAGluCCU (-CA)
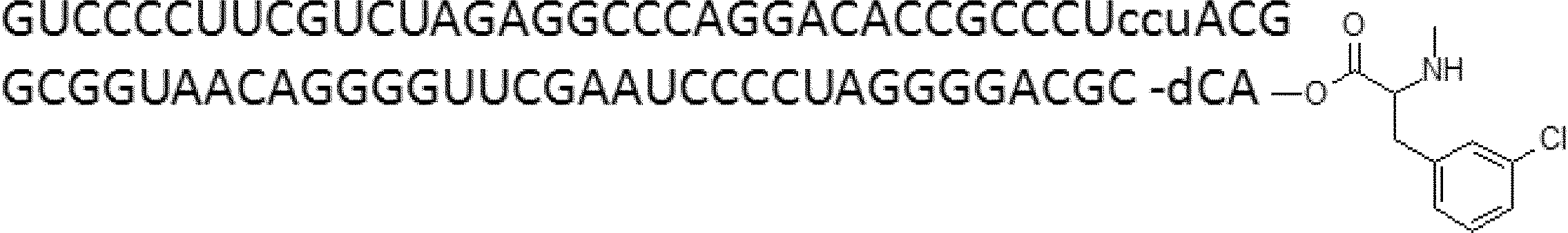
MeGly-tRNAGluCAA (-CA)
Phe(4-CF3)-tRNAGluUAG (-CA)
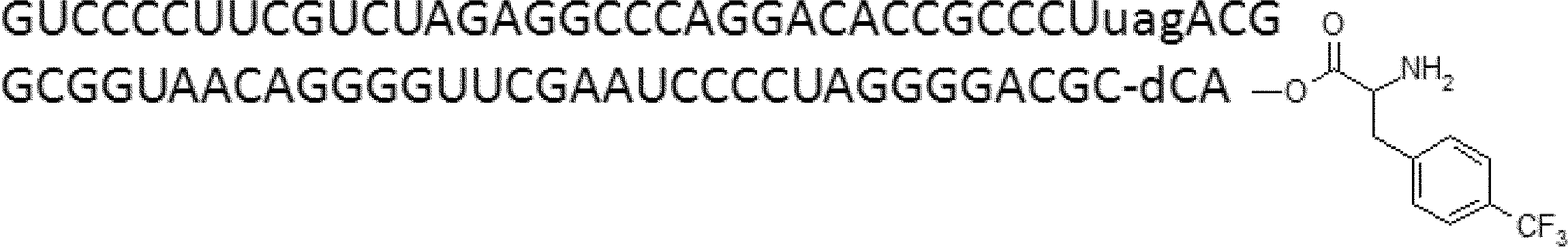
Hyp(Et)-tRNAGluCUA (-CA)

MeSer-tRNAGluCCG (-CA)

Phg-tRNAGluAAG (-CA)

Met(O2)-tRNAGluGCA (-CA)

Met(O2)-tRNAGluCAU (-CA)
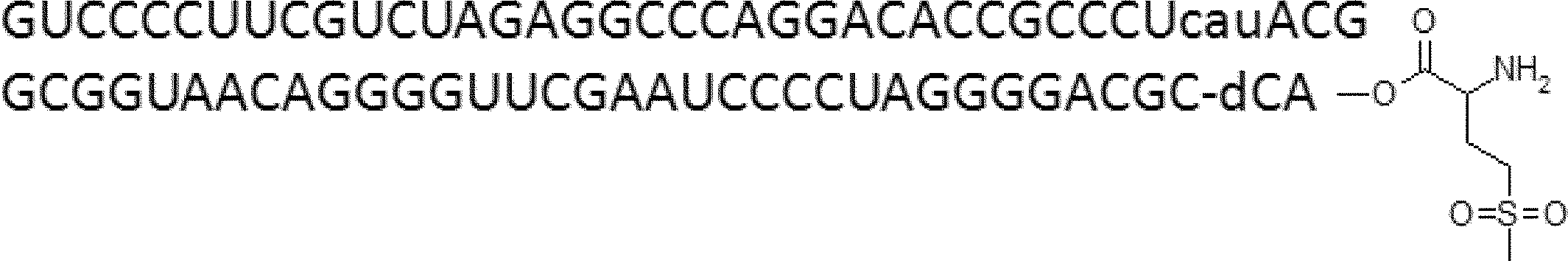
nPrGly-tRNAAsn-E2GUU (-CA)
tBuSSEtGABA-tRNAfMetCAU (-CA)
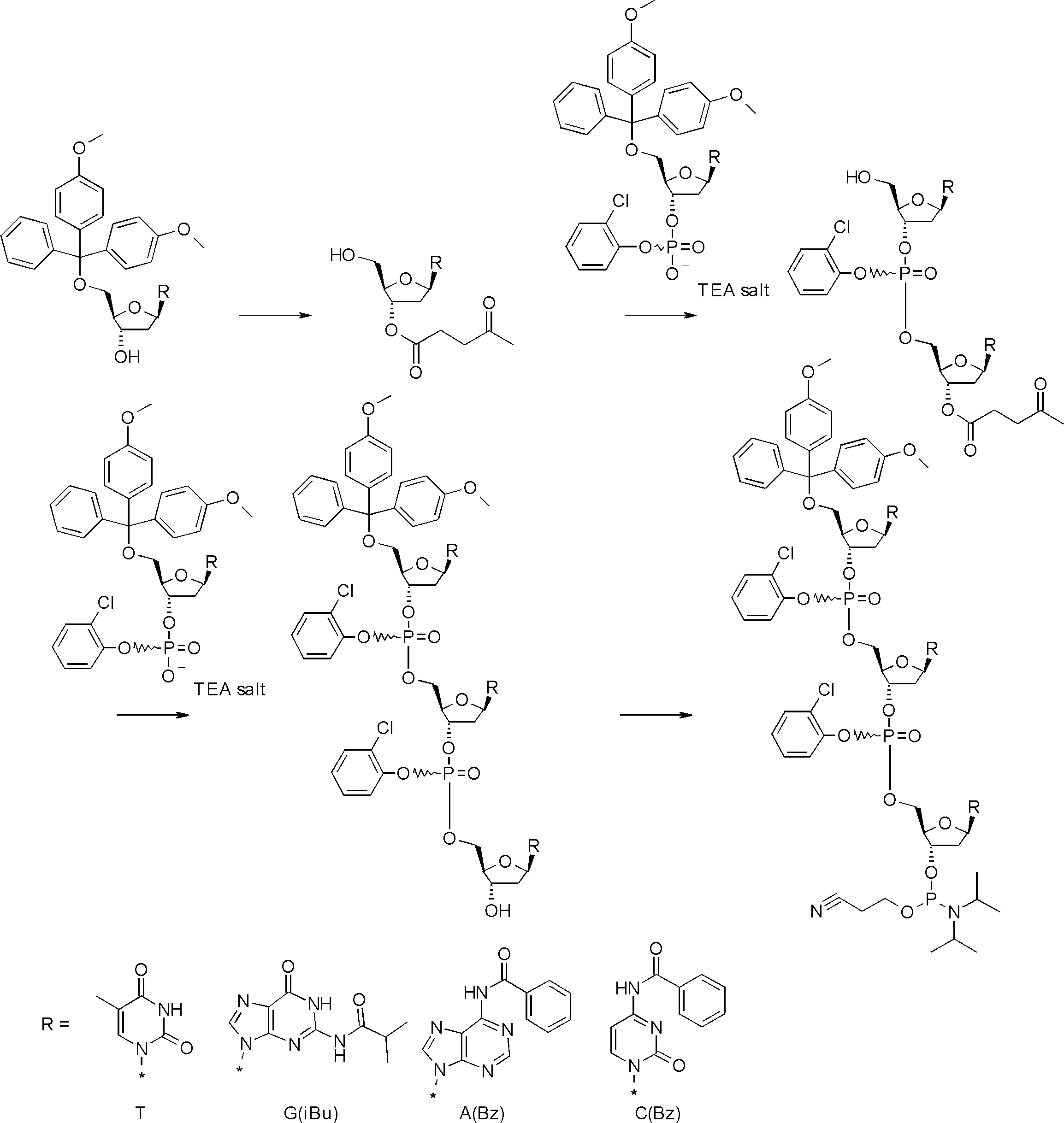

LCMS(ESI) m/z = 341.3 (M+H)+
保持時間:0.52分(分析条件 SQDAA05)

LCMS(ESI) m/z = 850.4 (M+H)+
保持時間:0.62分(分析条件 SQDFA05)
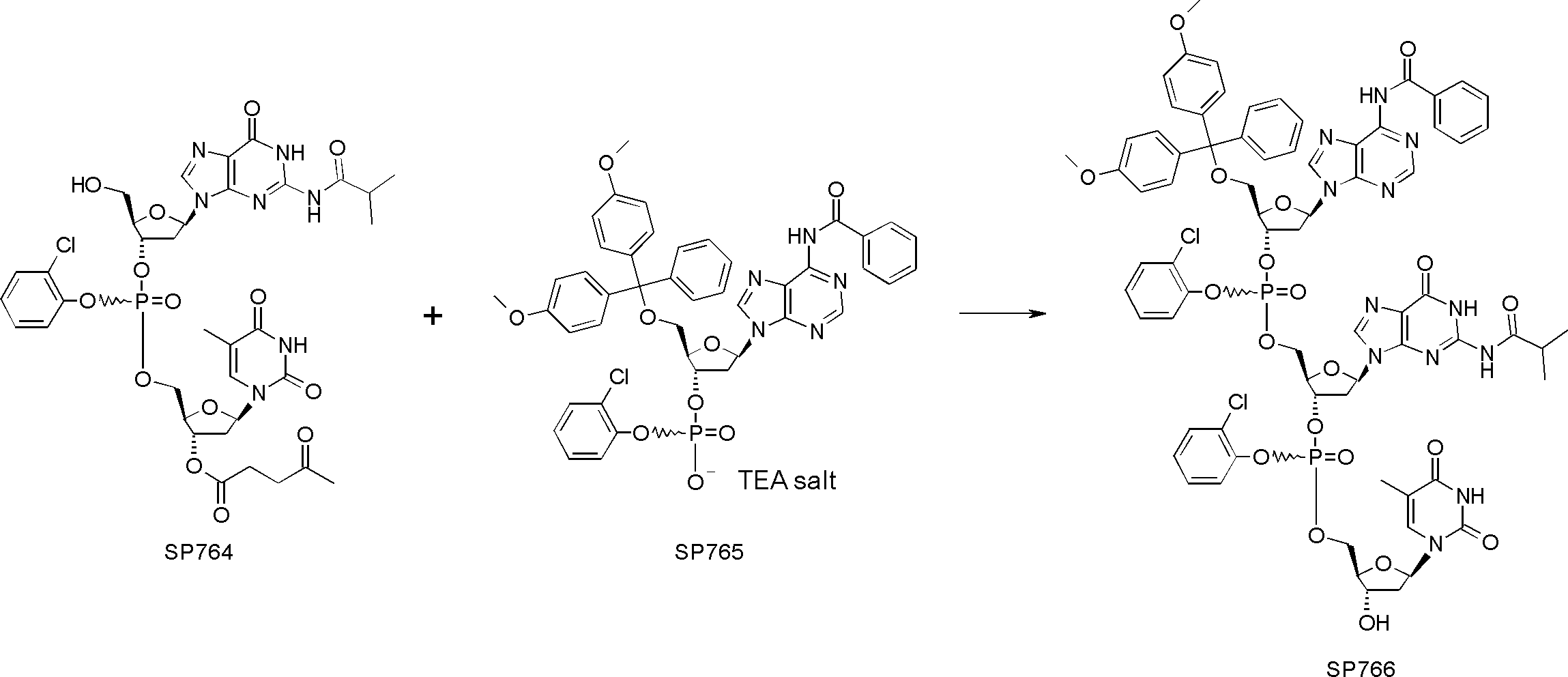
LCMS(ESI) m/z = 1581.8 (M+H)+
保持時間:1.11分(分析条件 SQDAA05)
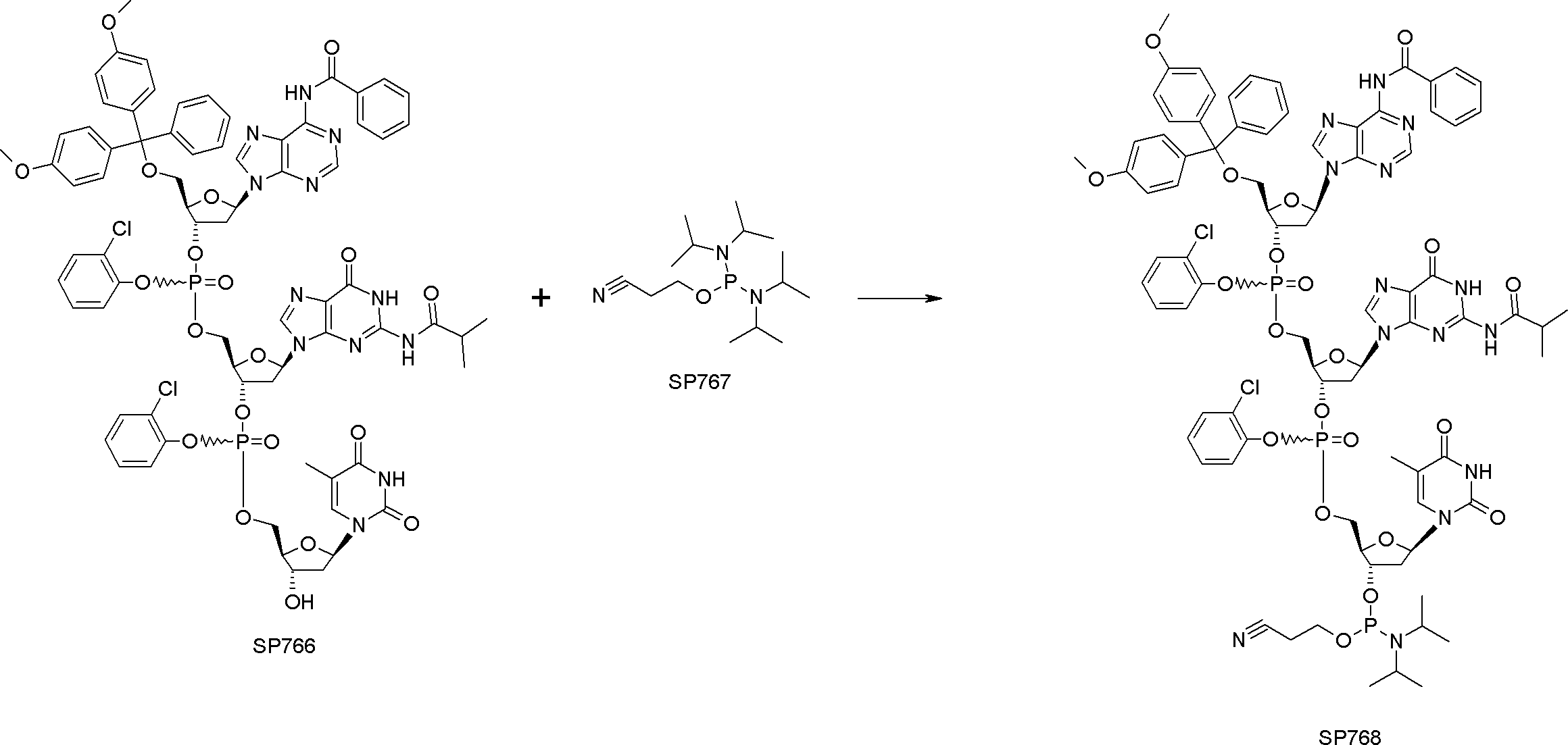
保持時間:1.17分(分析条件 SQDAA05)
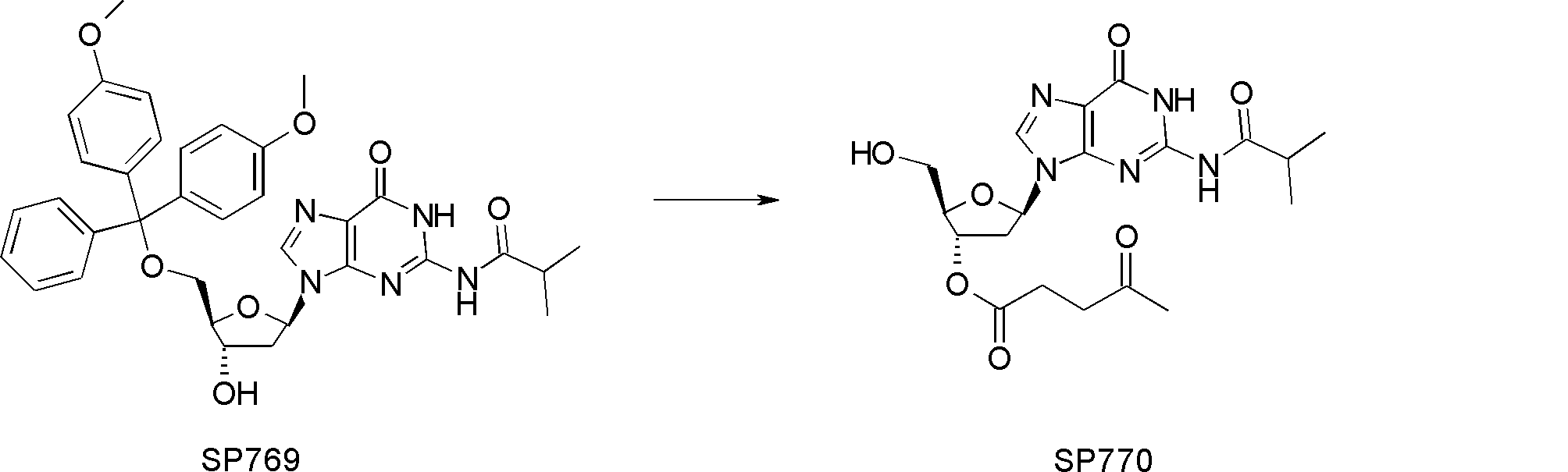
LCMS(ESI) m/z = 435.9 (M+H)+
保持時間:1.205分(分析条件 SMDMethod9)
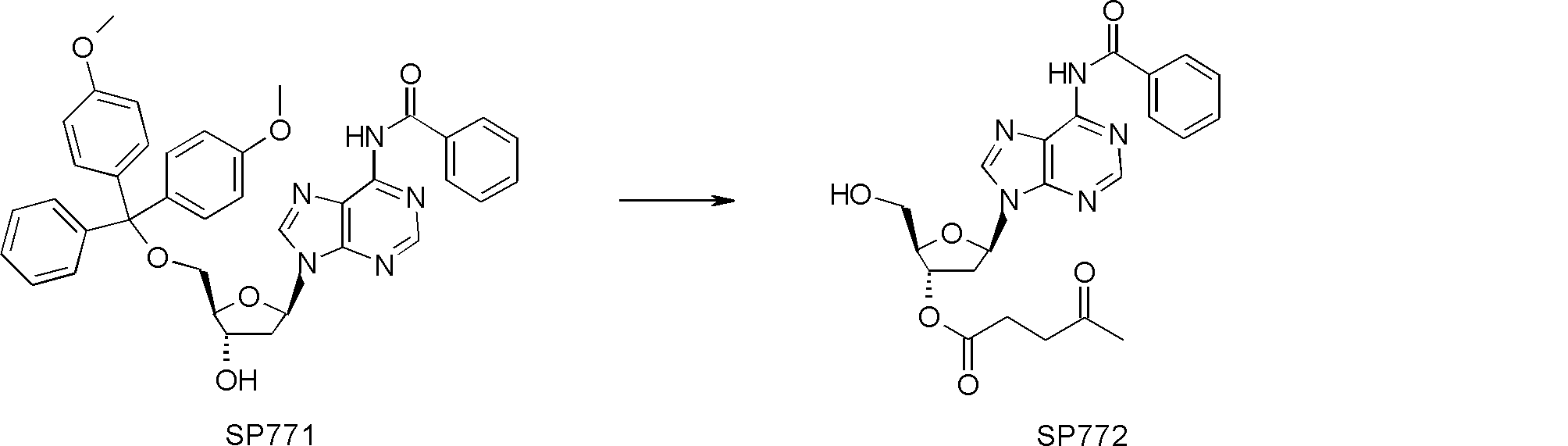
LCMS(ESI) m/z = 454.2 (M+H)+
保持時間:1.177分(分析条件 SMDMethod4)

LCMS(ESI) m/z = 430.2 (M+H)+
保持時間:3.193分(分析条件 SMDMethod10)

LCMS(ESI) m/z = 1668.8 (M+H)+
保持時間:1.15分(分析条件 SQDAA05)
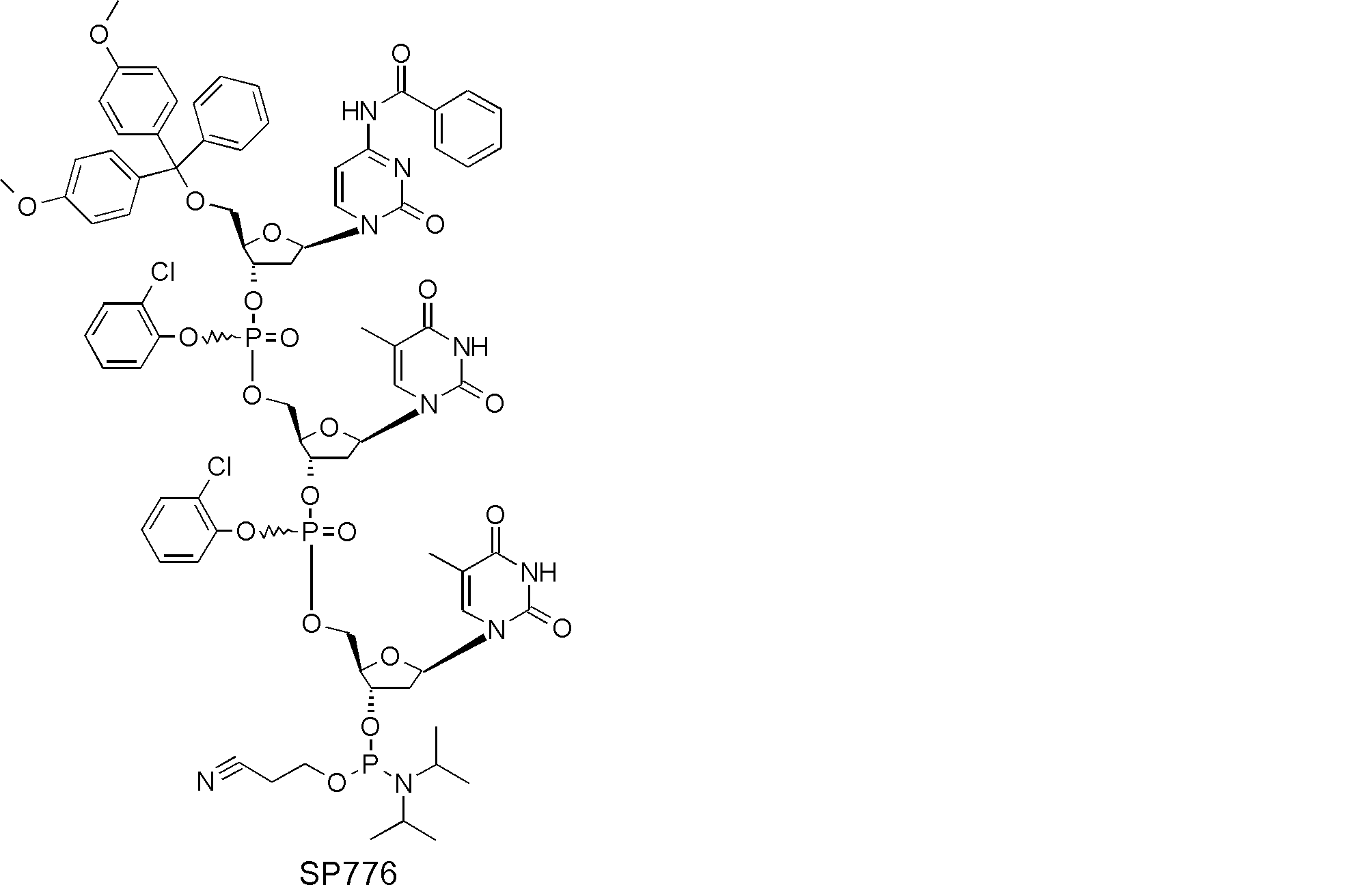
LCMS(ESI) m/z = 1662.4 (M+H)+
保持時間:1.18分(分析条件 SQDAA05)
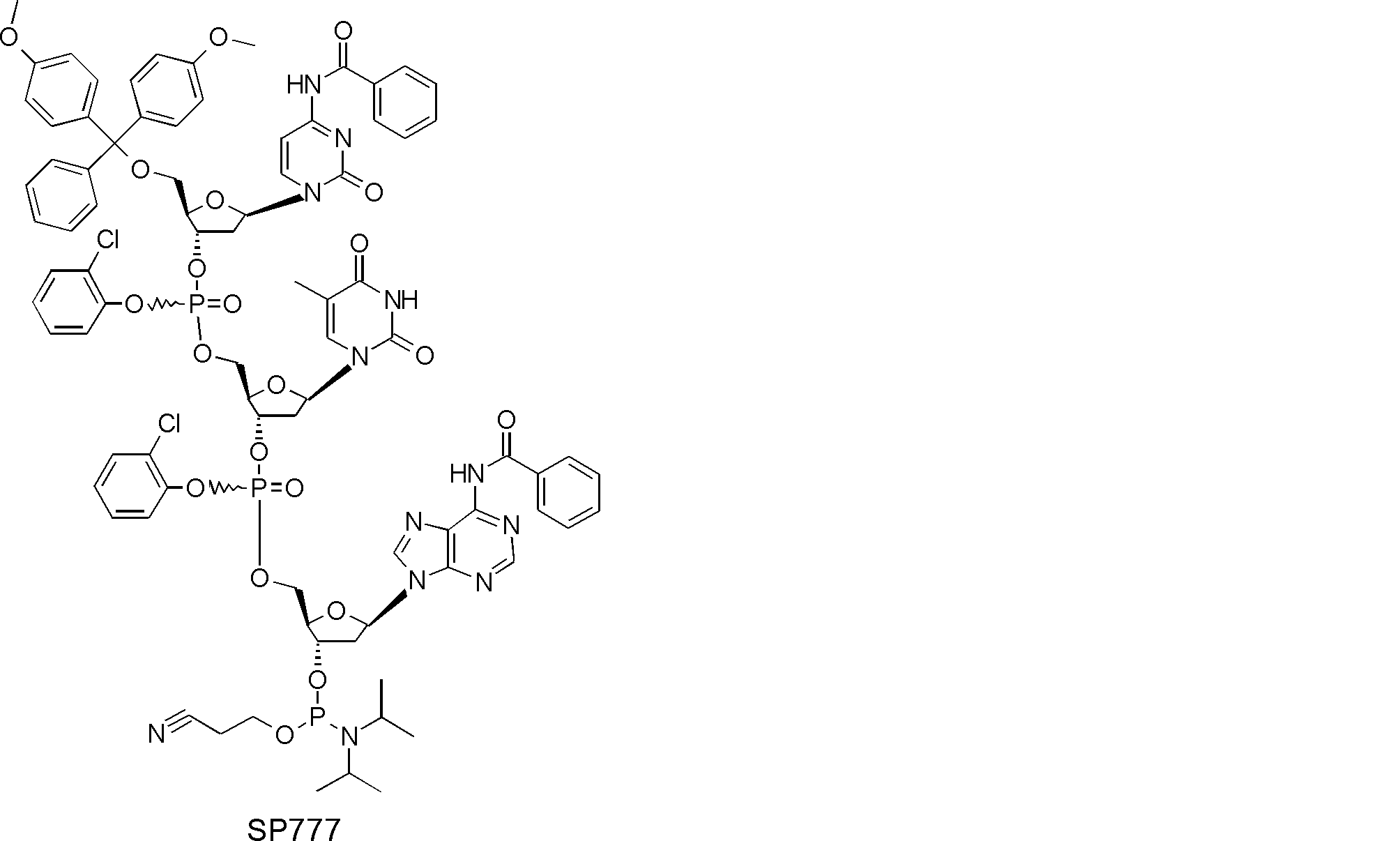
LCMS(ESI) m/z = 1775.9 (M+H)+
保持時間:1.20分(分析条件 SQDAA05)

LCMS(ESI) m/z = 1781.9 (M+H)+
保持時間:1.17分(分析条件SQDAA05)
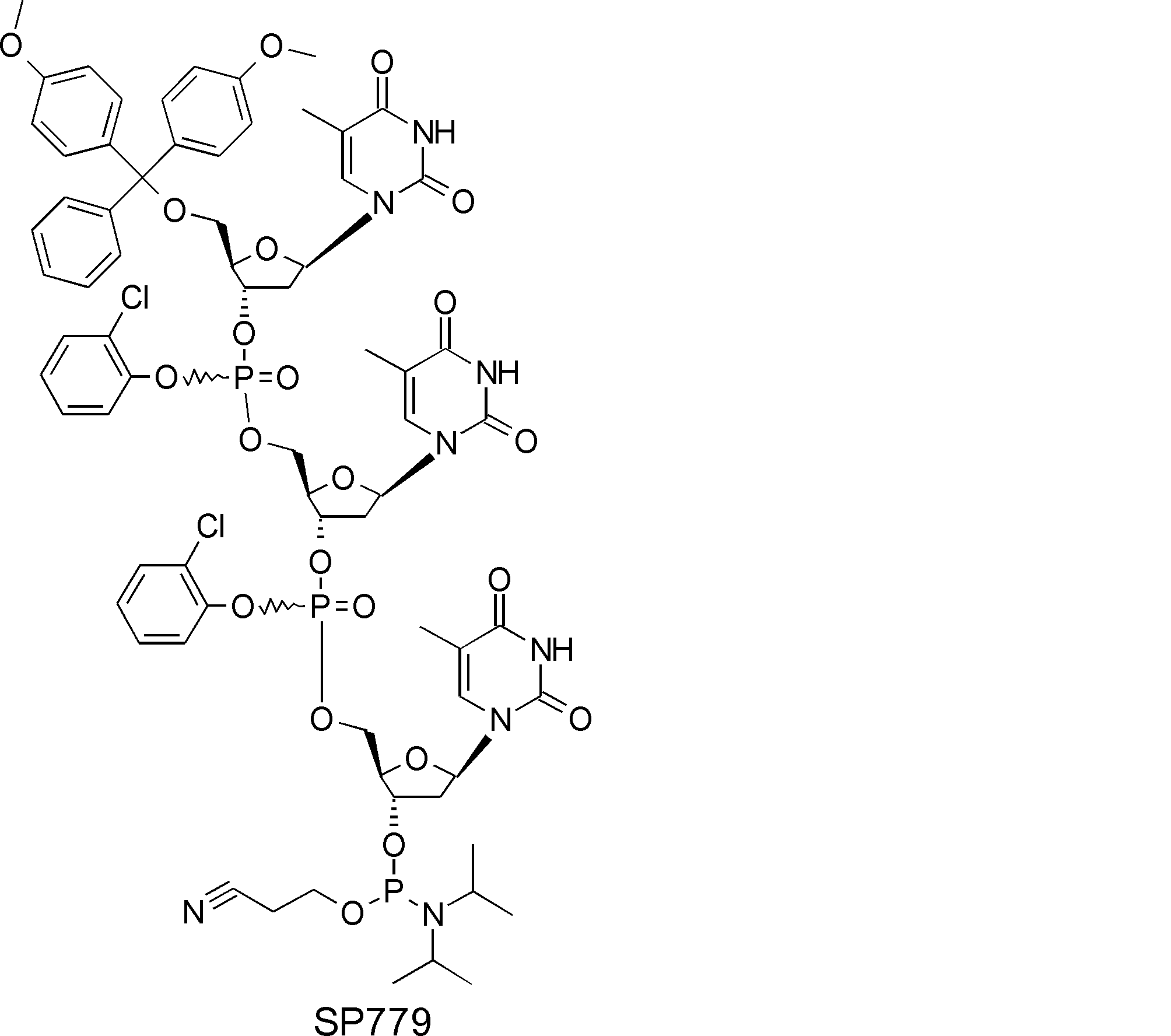
LCMS(ESI) m/z = 1573.8 (M+H)+
保持時間:1.15分(分析条件 SQDAA05)
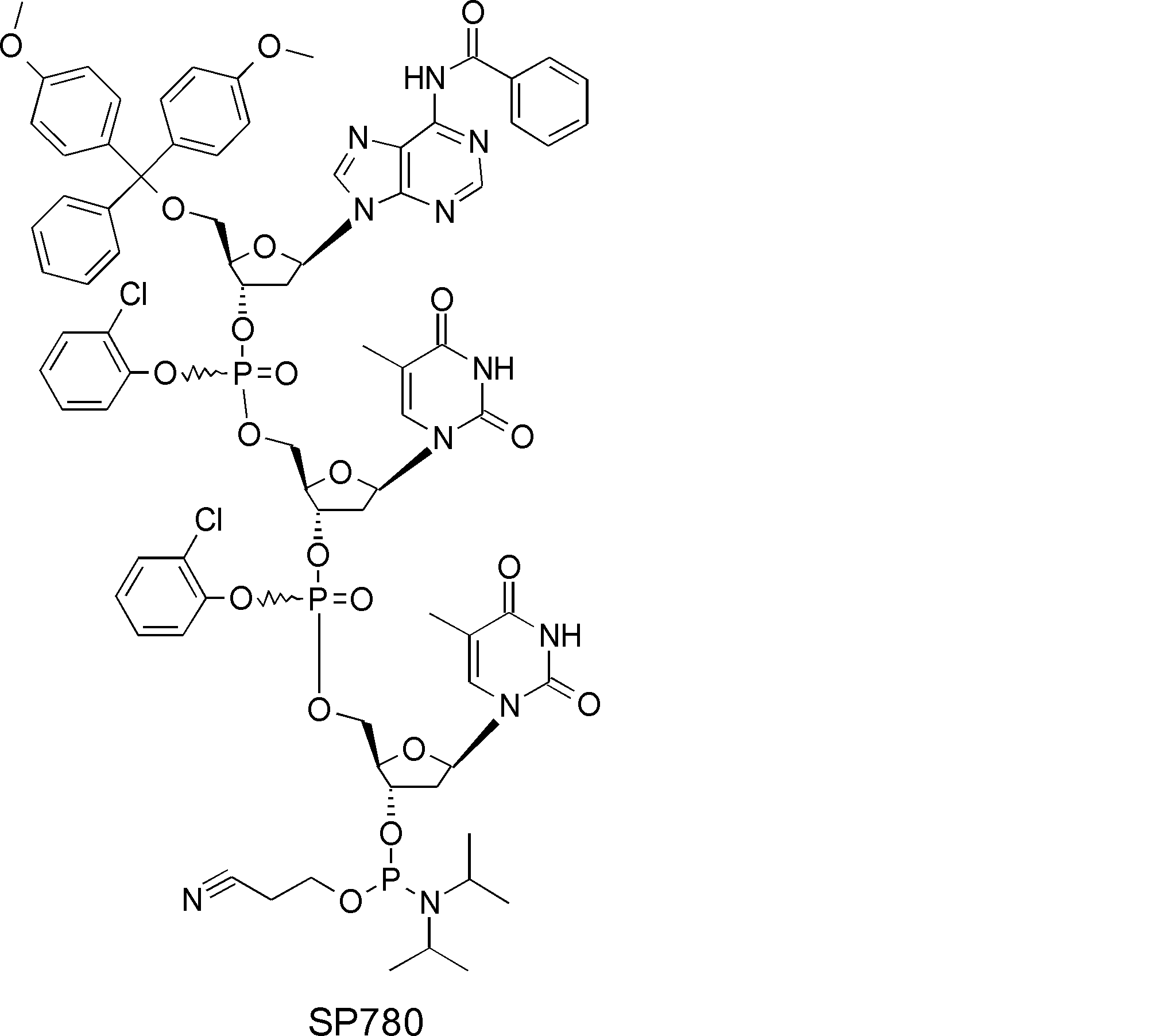
LCMS(ESI) m/z = 1686.4 (M+H)+
保持時間:1.16分(分析条件 SQDAA05)
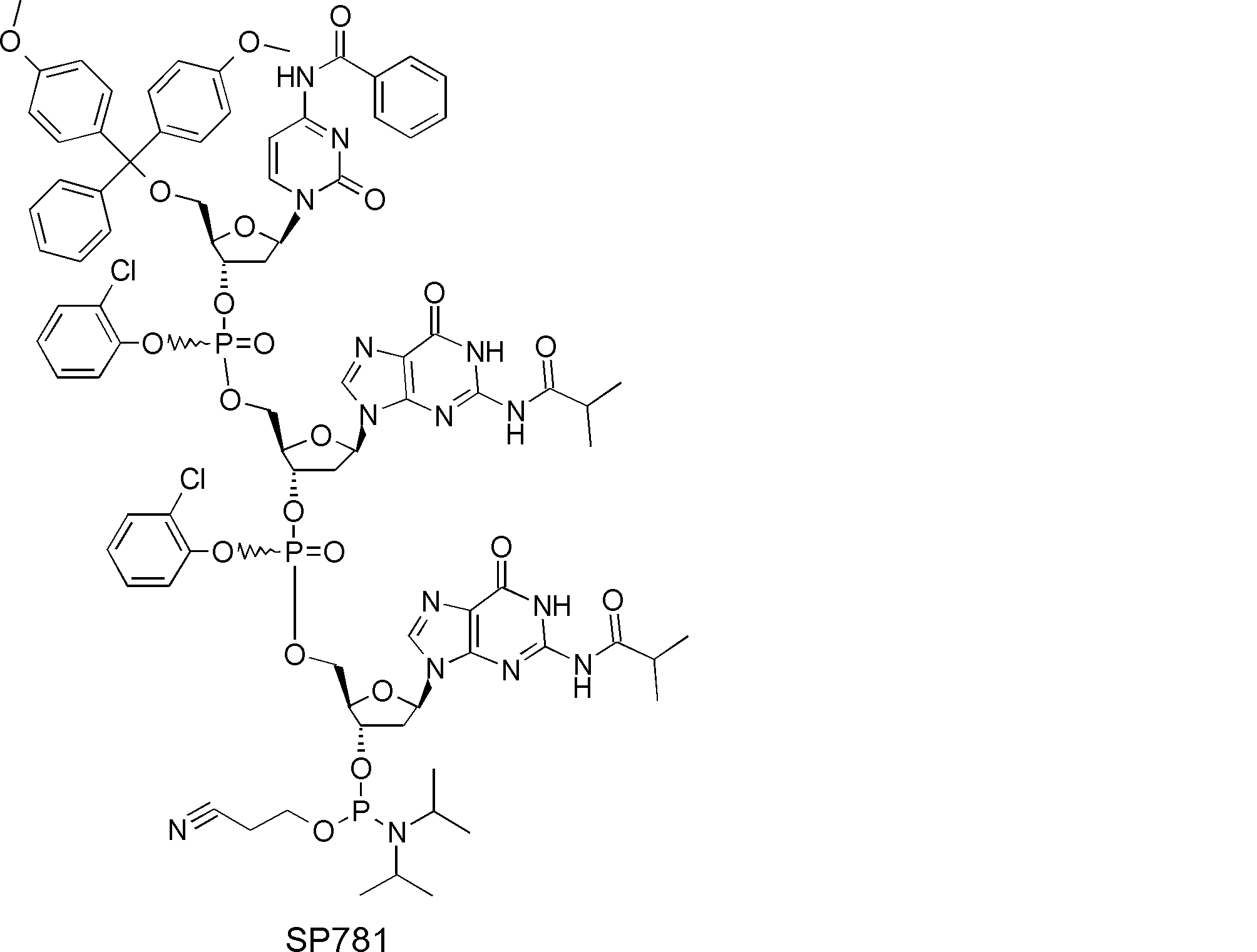
LCMS(ESI) m/z = 1852.5 (M+H)+
保持時間:0.87分(分析条件 SQDAA50)
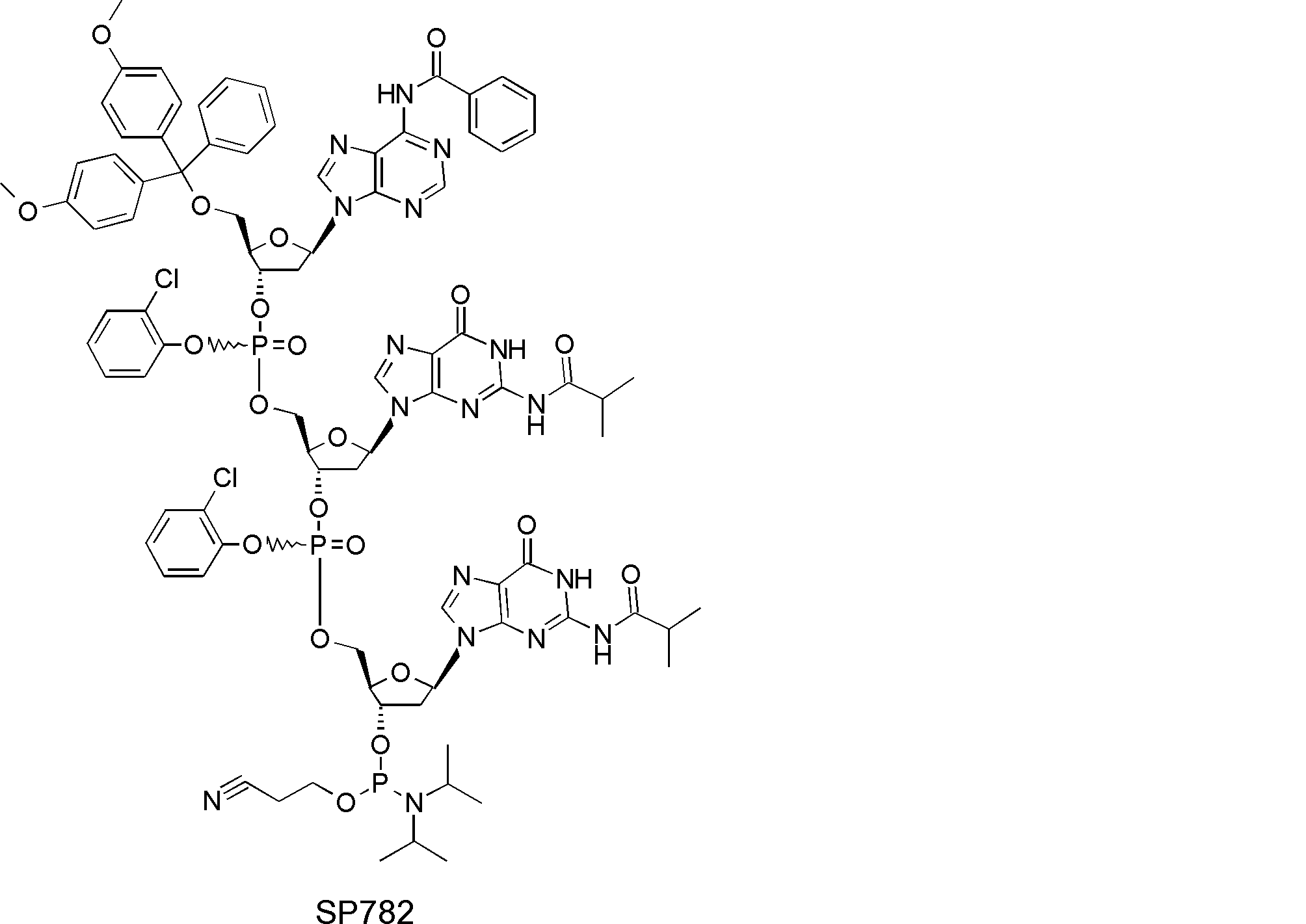
LCMS(ESI) m/z = 1876.5 (M+H)+
保持時間:0.84分(分析条件 SQDAA50)
上記載の方法もしくは購入して得たコドンユニットを用いて、以下のような手法でランダム化された多様性の高いDNAを合成した。
化合物SP791
5'-GAA GGA GAT ATA CAT ATG (PPP)7 CGT (XXX) (PPP) AGC GGC TCT GGC TCT GGC TCT-3'(配列番号:140)
DNA/RNA合成機(NTS H-6、日本テクノサービス株式会社製)を用いて、ホスホロアミダイト法によりランダムDNA合成を行った。脱水アセトニトリルおよびデブロッキング溶液-1(3w/v% トリクロロ酢酸 ジクロロメタン溶液)は和光純薬から購入し、ホスホロアミダイト試薬(dA, dC, dG, dT-CE phosphoramidite)、Cap Mix A(THF/Pyridine/Acetic anhydride)、Cap Mix B(16% 1-Me-Imidazole/THF)、Oxidizing Solution(0.02M I2 in THF/Pyridine/H2O)、Activator(5-Benzylthio-1H-tetrazole in MeCN)、トリマーホスホロアミダイト試薬(GTT、CCG、ACT、ATG、GCT、CAT、TGG、GGT、TAC、CAG、AAC、TGC、GAA Trimer Phosphoramidite)についてはGlen Research社から購入した。また、トリマーホスホロアミダイト試薬(TTT(化合物SP779)、ATT(化合物SP780)、AGT(化合物SP768)、CGG(化合物SP781)、AGG(化合物SP782)、TTG(化合物SP775)、CTT(化合物SP776)、CTA(化合物SP777)、TAG(化合物SP778) Trimer Phosphoramidite)については、上記載のとおり合成したものを用いた。
なお、操作の詳細な手順については合成機に付属のマニュアルに従った。
伸長反応終了後、固相担体をスクリューキャップ付きガラスチューブに移し、30%アンモニア水溶液(0.4ml)を加え、60℃にて6時間攪拌することによりCPGからDNAの切り出しと脱保護を行い、反応混合液を分取HPLCにて精製した(分析条件LC05)。HPLCフラクションを凍結乾燥して得られた残渣を水(0.8ml)に溶かし、これに酢酸(3ml)を加え、室温にて10分間攪拌することにより、5'末端水酸基のDMT基を脱保護した。反応液を水(10ml)で希釈し、酢酸エチル(10ml)で5回抽出操作を行った後、得られた水層を凍結乾燥することにより、目的とするランダム化DNA(化合物SP791)(6.7%)を得た。なお収率については260nmにおける吸光度から算出した。
保持時間:8.55分(分析条件LC04)
5'-GAA GGA GAT ATA CAT ATG (PPP)8 CGT (XXX) (PPP) AGC GGC TCT GGC TCT GGC TCT-3'(配列番号:141)
保持時間:8.71分(分析条件LC04)
5'-GAA GGA GAT ATA CAT ATG (PPP)9 CGT (XXX) (PPP) AGC GGC TCT GGC TCT GGC TCT-3'(配列番号:142)
保持時間:8.67分(分析条件LC04)
5'-GAA GGA GAT ATA CAT ATG TGC (QQQ)7 CGT (QQQ)2 AGC GGC TCT GGC TCT GGC TCT-3'(配列番号:143)
保持時間:8.56分(分析条件LC04)
5'-GAA GGA GAT ATA CAT ATG TGC (QQQ)8 CGT (QQQ)2 AGC GGC TCT GGC TCT GGC TCT-3'(配列番号:144)
保持時間:8.54分(分析条件LC04)
5'-GAA GGA GAT ATA CAT ATG TGC (QQQ)9 CGT (QQQ)2 AGC GGC TCT GGC TCT GGC TCT-3'(配列番号:145)
保持時間:8.80分(分析条件LC04)

合成オリゴDNA(化合物SP791、SP792、SP793、SP794、SP795、SP796)と合成オリゴDNA(配列番号 DOL-1)を用いて、ExTaq (Takara社)により伸長反応を行った。95℃で二分間変性した後、50℃で一分、72℃で一分のサイクルを5ないし10回繰り返した。続いて、この伸長反応産物を鋳型に合成オリゴDNA(配列番号DOL-1)と合成オリゴDNA(配列番号DOL-2)を用いて、ExTaq (Takara社)によりPCRを行った。95℃で二分間変性した後、95℃で一分、50℃で一分、72℃で一分のサイクルを5から10回繰り返し、DNAライブラリー(配列番号DML-1~DML-6)を構築した。
配列中に(PPP)と示した箇所は、TTT、ATT、GTT、AGT、CCG、ACT、GCT、CAT、TGG、GGT、TAC、CAG、AAC、CGG、AGG、TTG、CTT、CTA、TAG、TGC、GAA がランダムに出現することを、(XXX)と示した箇所には、TTT、CCG、GCT、CAT、AAC、CGG、AGG、TTG、TAG、GAAがランダムに出現することを、(QQQ)と示した箇所には、TTT、ATT、ATG、GTT、AGT、CCG、ACT、GCT、CAT、TGG、GGT、TAC、CAG、AAC、CGG、AGG、TTG、CTT、CTA、TAG、GAAがランダムに出現することを意味する。
TTTGTCCCCGCCGCCCTAAGAGCCAGAGCCAGAGCCGCT
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG(PPP)7 CGT(XXX)(PPP)AGCGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG(PPP)8CGT(XXX)(PPP)AGCGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG(PPP)9CGT(XXX)(PPP)AGCGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGC(QQQ)7CGT(QQQ)2AGCGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGC(QQQ)8CGT(QQQ)2AGCGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGC(QQQ)9CGT(QQQ)2AGCGGCTCTGGCTCTGGCTCTTAGGGCGGCGGGGACAAA
PCRにより調製したDNAライブラリー(配列番号 DML-1~DML-3(Initiation suppression翻訳用)、DML-4~DML-6(Read throuth翻訳用)) を鋳型に、in vitro転写により以下のmRNA(配列番号MML-1~MML-3(Initiation suppression翻訳用)、配列番号MML-4~MML-6(Read throuth翻訳用))を調製し、RNeasy mini kit (Qiagen社)を用いて精製した。10μMのmRNAに、15μMのピューロマイシンリンカー (Sigma社)(配列番号C-1)、 1X T4 RNAライゲースリアクションバッファー(New england bio lab.社)、1mM ATP、10% DMSO、0.625unit/μl T4 RNAライゲース(New england bio lab.社)を加え、37℃で30分間ライゲーション反応させた後、RNeasy Mini kit(Qiagen社)により精製した。配列番号MML-1とMML-4は30μlスケール、配列番号MML-2とMML-5は60μlスケール、配列番号MML-3とMML-6は240μlスケールで、ライゲーションを行い、精製した。ピューロマイシンリンカー(配列番号C-1)と連結したMML-1,MML-2,MML-3をそれぞれモル比1:21:441となるように、ピューロマイシンリンカー(配列番号C-1)と連結したMML-4,MML-5,MML-6をそれぞれモル比1:21:441となるように混合し、前者をinitiation suppression翻訳用mRNA-ピューロマイシンリンカーライゲーション産物混合物と、後者をRead throuth 翻訳用mRNA-ピューロマイシンリンカーライゲーション産物混合物とした。
GGGUUAACUUUAAGAAGGAGAUAUACAUAUG(RRR)7CGU(YYY)(RRR)AGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
GGGUUAACUUUAAGAAGGAGAUAUACAUAUG(RRR)8CGU(YYY)(RRR)AGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
GGGUUAACUUUAAGAAGGAGAUAUACAUAUG(RRR)9CGU(YYY)(RRR)AGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
GGGUUAACUUUAAGAAGGAGAUAUACAUAUGUGC(SSS)7CGU(SSS)2 AGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
GGGUUAACUUUAAGAAGGAGAUAUACAUAUGUGC(SSS)8CGU(SSS)2 AGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
GGGUUAACUUUAAGAAGGAGAUAUACAUAUGUGC(SSS)9CGU(SSS)2 AGCGGCUCUGGCUCUGGCUCUUAGGGCGGCGGGGACAAA
パニングに使用する標的タンパク質として、インターロイキン6受容体(IL-6R)、Tumor Necrosis Factor-α(TNFα)、Tumor Necrosis Factor Receptor1(TNFR1)を用いた。IL-6Rのビオチン化は非特許文献BMC biotechnology, 2008,8,41、および非特許文献Protein Science,1999,8,921-929の方法を用いた。IL-6Rの調製は非特許文献J Biochem. 1990;108(4):673-6.に従って行った。TNFαはProspec社のRcombinant Human Tumor Necrosis Factor-alpha(Catalog Number #CYT-223)を、TNFR1はSinoBiological社のRecombinant Human TNFR1/Fc Chimera(Catalog Number 10872-H03H)を購入した。それぞれThermo scientific社のEZ-Link NHS-PEG4-Biotin(Catalog Number 21329)を用いてビオチン化を行った。
パニングに使用した翻訳液Sは以下の組成で構成される。1mM GTP,1mM ATP,20mMクレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,10mM 酢酸マグネシウム,2mMスペルミジン,1mM ジチオスレイトール,0.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.26μM EF-G,2.7μM IF1,0.4μM IF2,1.5μM IF3,40μM EF-Tu,84μM EF-Ts,1.2μM リボソーム,2.73μM AlaRS,0.09μM GlyRS,0.4μM IleRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.22μM TyrRS,0.02μM ValRS、3μM 転写tRNA Ala1B(配列番号MTL-13)、3μM 転写tRNA Tyr1(配列番号MTL-14)、250μMグリシン、250μMイソロイシン、250μMプロリン、250μMスレオニン、250μMトリプトファン、250μMバリン、100μMセリン、5mM N-メチルアラニン、5mM N-メチルフェニルアラニン、2mM D-チロシン。さらに翻訳液S用アミノアシル化tRNA混合物および翻訳液S用Initiatorアミノアシル化tRNAを加えて調製した。また翻訳液Tは上記の翻訳液S用アミノアシル化tRNA混合物および翻訳液S用Initiatorアミノアシル化tRNAを含まない翻訳液Sに0.02μM CysRS、250μMシステイン、および翻訳液T用アミノアシル化tRNA混合物を加えて調製した。
1μM initiation suppression翻訳用もしくはRead throuth 翻訳用mRNA-ピューロマイシンリンカーライゲーション産物混合物を含む前述のS用、T用それぞれ2ml翻訳液を調製し、37℃で120分間保温後、室温で12分間放置した。この翻訳混合物に対して、200μlの200mM EDTA溶液(pH8.0、Nacalai社、14362-24)および100μlの200mM TCEP溶液(pH7.0)を加え、37℃で120分間保温し、環化反応を行った。その後、翻訳混合物に対して、260μlの250mM Cys溶液および4000μlの400mM TCEP溶液(pH7.0)を加え、40℃で5分間保温した。続けて、1600μlの1M VA-044(Wako社、CAS No.27776-21-2)を加えて、40℃で1時間脱硫反応後、RNeasy mini kit(Qiagen社)により精製し、1mlのペプチド-mRNA複合体溶液を調製した。この溶液に、3μM プライマー(配列番号DOL-3)、M-MLV reverse transcriptase reaction buffer(Promega社、M368B)、0.5mM dGTP,0.5mM dATP,0.5mM dCTP,0.5mM dTTP、8U/μl M-MLV Reverse transcriptase (H-) (Promega社、M368B)を加えNuclease free waterで2mlにメスアップし、42℃、1時間保温し、逆転写反応を行った。逆転写溶液に対してブロッキング剤SuperBlock T20(Pierce社、37516)を等量加え、4mlのペプチド-mRNA複合体溶液とした。
TTTGTCCCCGCCGCCCTAAGAGCCAGAGCCAGAGCCGCT
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGA
TTTGTCCCCGCCGCCcta
ラウンド1で増幅したcDNAから、RiboMAX Express Large Scale RNA Production System (Promega社,P1320)を用いてmRNAを合成し、RNeasy MinElute kit(Qiagen社)により精製した。次にT4 RNA ligase(NEB, M0204L)を利用してmRNAとピューロマイシンリンカーを連結し、RNeasy MinElute kit(Qiagen社)により精製した。1μM mRNA-ピューロマイシンリンカーライゲーション産物を含む前述の100μl翻訳液を調製し、37℃で60分間保温後、室温で12分間放置した。この翻訳混合物に対して環化反応のために、10μlの200mM EDTA溶液(pH8.0、Nacalai社、14362-24)および5μlの200mM TCEP溶液(pH7.0)を加え、37℃で120分間保温した。翻訳液Sに0.5M KOHを加えて、pH10に調整後、42℃で30分間保温した。その後、0.5M HClを加えて、pH7に調整した。その後、それぞれの翻訳混合物に対して、13μlの250mM Cys溶液および200μlの400mM TCEP溶液(pH7.0)を加え、40℃で1分間保温した。続けて、80μlの1M VA-044(Wako社、CAS No.27776-21-2)を加えて、40℃で1時間脱硫反応後、RNeasy MinElute kit(Qiagen社)により精製し、100μlのペプチド-mRNA複合体溶液を調製した。この溶液に、3μM プライマー(配列番号DOL-3)、M-MLV reverse transcriptase reaction buffer(Promega社、M368B)、0.5mM dGTP,0.5mM dATP,0.5mM dCTP,0.5mM dTTP、8U/μl M-MLV Reverse transcriptase (H-) (Promega社、M368B)を加えNuclease free waterでメスアップし、200μl溶液で42℃、15分間保温し、逆転写反応を行った。逆転写溶液に対してブロッキング剤SuperBlock T20(Pierce社、37516)を等量加え、400μlのペプチド-mRNA複合体溶液とした。
ラウンド2で増幅したcDNAから、RiboMAX Express Large Scale RNA Production System (Promega社,P1320)を用いてmRNAを合成し、RNeasy MinElute kit(Qiagen社)により精製した。次にT4 RNA ligase(NEB, M0204L)を利用してmRNAとピューロマイシンリンカーを連結し、RNeasy MinElute kit(Qiagen社)により精製した。1μM mRNA-ピューロマイシンリンカーライゲーション産物を含む前述の10μl翻訳液を調製し、37℃で60分間保温後、室温で12分間放置した。この翻訳混合物に対して環化反応のために、1μlの200mM EDTA溶液(pH8.0、Nacalai社、14362-24)および0.5μlの200mM TCEP溶液(pH7.0)を加え、37℃で120分間保温した。翻訳液Sに0.5M KOHを加えて、pH10に調整後、42℃で30分間保温した。その後、0.5M HClを加えて、pH7に調整した。その後、それぞれの翻訳混合物に対して、1.3μlの250mM Cys溶液および20μlの400mM TCEP溶液(pH7.0)を加え、40℃で1分間保温した。続けて、8μlの1M VA-044(Wako社、CAS No.27776-21-2)を加えて、40℃で1時間脱硫反応後、RNeasy MinElute kit(Qiagen社)により精製し、10μlのペプチド-mRNA複合体溶液を調製した。この溶液に、3μM プライマー(配列番号DOL-3)、M-MLV reverse transcriptase reaction buffer(Promega社、M368B)、0.5mM dGTP,0.5mM dATP,0.5mM dCTP,0.5mM dTTP、8U/μl M-MLV Reverse transcriptase (H-)(Promega社、M368B)を加えNuclease free waterで200μlにメスアップし、42℃、15分間保温し、逆転写反応を行った。逆転写溶液に対してブロッキング剤SuperBlock T20(Pierce社、37516)を等量加え、40μlのペプチド-mRNA複合体溶液とした。
ラウンド4-6においても、ラウンド3と同じ操作を繰り返すことで標的タンパク質特異的な結合を示すcDNAの濃縮を行った。濃縮されたDNAプールの塩基配列解析を行い、濃縮されたペプチド配列を同定した。
計算機によるシミュレーションを活用した仮想ライブラリによるCLOGP値、NMeアミノ酸数、分子量の平均値および分布の推測(図66、67)
今回設計されたディスプレイライブラリーのドラックライクネスの程度を評価した。すなわち、選択されたアミノ酸が均等にランダムにディスプレイされた場合のCLogP値、NMeアミノ酸が1つのペプチドに含まれる数の分布を計算によりシミュレーションした。
パニングにて濃縮されたクローンの一部について、各クローンの翻訳産物ペプチドを用いたイムノアッセイを実施した。その際、低濃度のペプチド産物を高感度に検出するため、電気化学発光法(ECL)測定装置(SECTOR Imager 2400)を利用した。その結果、IL-6R、TNFα、TNFR1に対する結合ペプチドが同定された。
dC-Puromycin CPG (化合物SP802)の合成
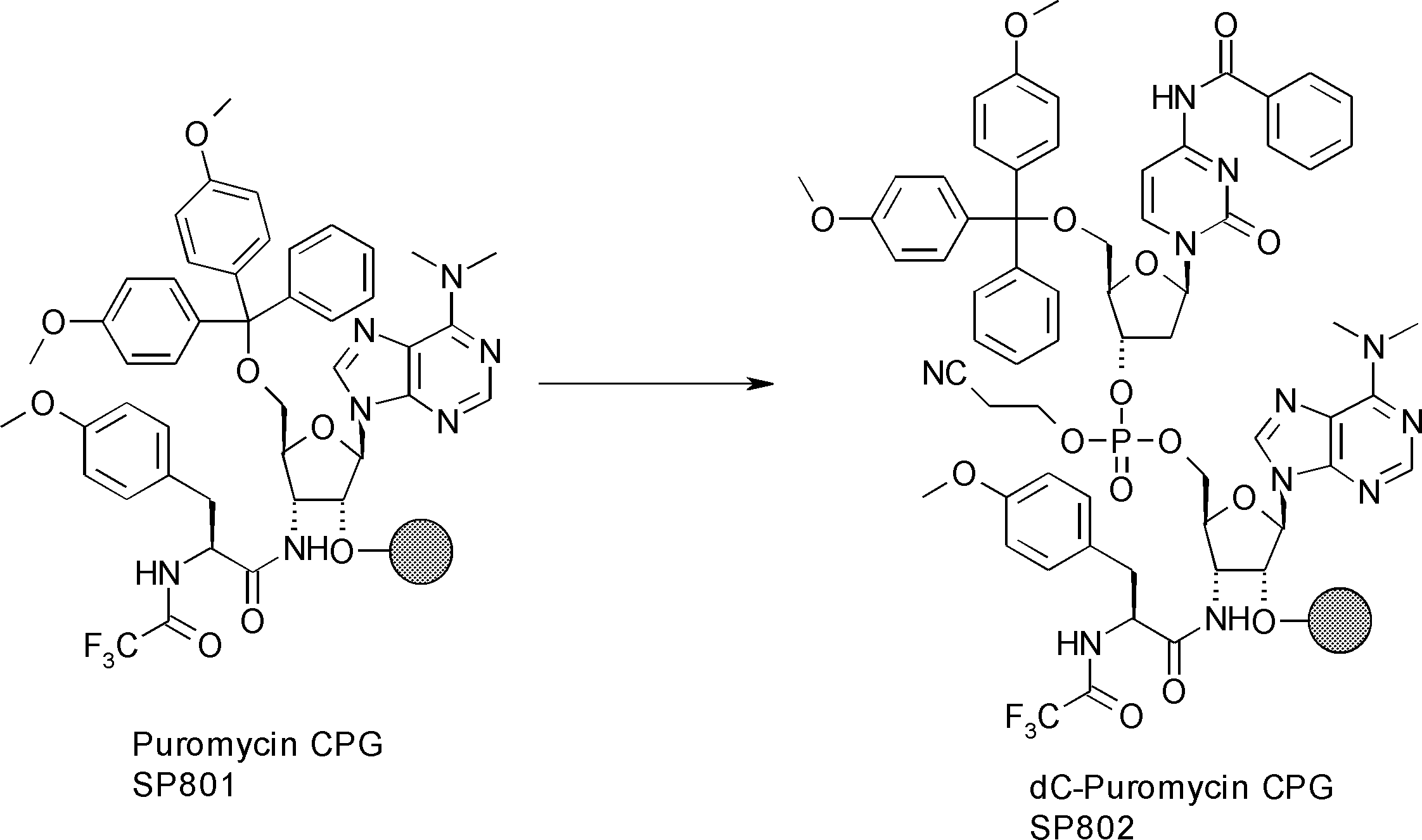
得られたCPG担体にdC-CE Phosphoramidite(Glen Research社製、250mg、0.300mmol)のアセトニトリル溶液(3.0ml)とActivator(Glen Research社製、5-Benzylthio-1H-tetrazole in Acetonitrile、3ml)を加えて室温で10分間振とうさせた後、反応液を除去し、CPG担体をアセトニトリルで洗浄(3ml×4)し、乾燥させた。
次いでOxidizing Solution(Glen Research社製、0.02M Iodine in THF/Pyridine/Water、3ml)を加えて室温で振とうさせた後、反応液を除去した。担体をアセトニトリルで洗浄(3ml×4)後、乾燥させて標題のdC-Puromycin CPG(化合物SP802)(450mg)を得た。
少量のdC-Puromycin CPGに25%アンモニア水溶液を加えて60℃で1時間半攪拌して担体から切り出し、反応液をLC/MSで分析することで、反応の進行、およびdC-Puromycin(化合物SP803)の生成を確認した。

保持時間:0.53分 (分析条件 SQDAA50)
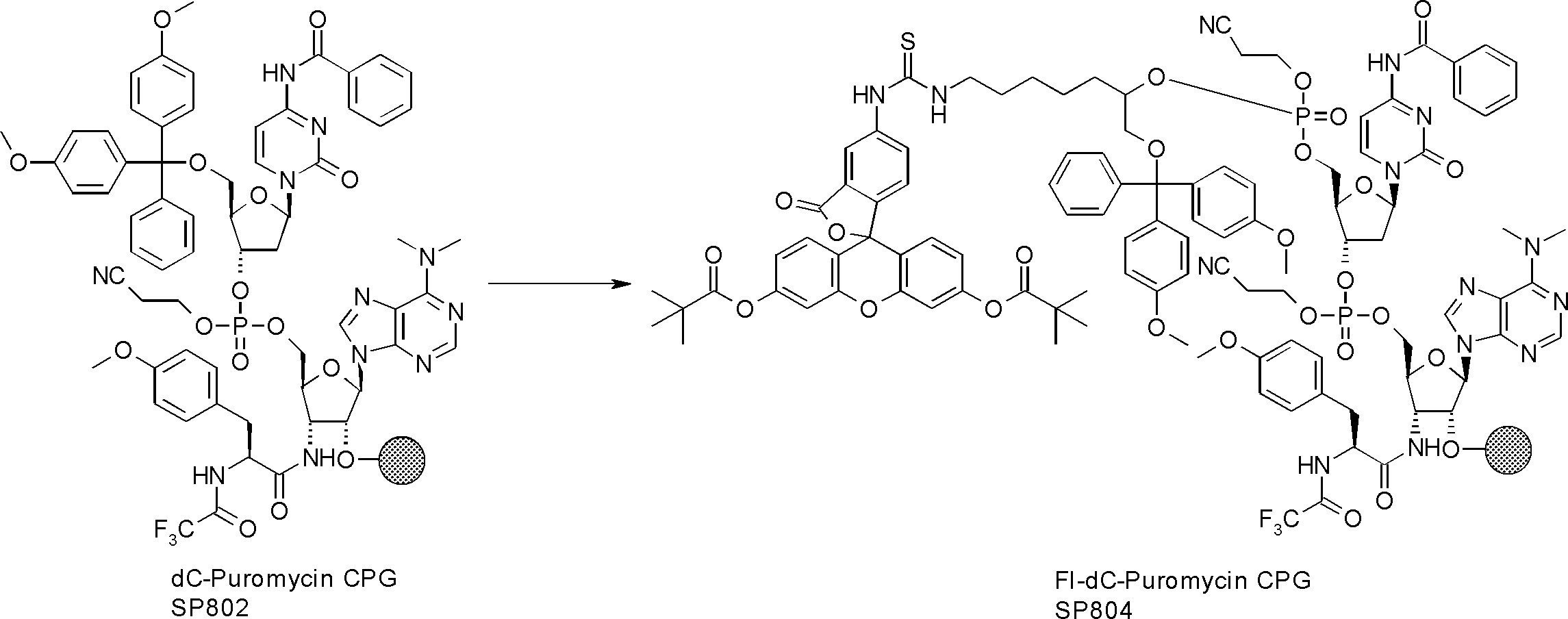
得られたCPG担体にFluorescein Phosphoramidite(Glen Research社製、125mg、0.104mmol)のアセトニトリル溶液(1.0ml)とActivator(Glen Research社製、5-Benzylthio-1H-tetrazole in Acetonitrile、1.0ml)を加えて室温で30分間振とうさせた後、反応液を除去し、CPG担体をアセトニトリルで洗浄し、乾燥させた。
次いでOxidizing Solution(Glen Research社製、0.02M Iodine in THF/Pyridine/Water、3ml)を加えて室温で振とうさせた後、反応液を除去した。CPG担体をアセトニトリルで洗浄後、乾燥させて標題のFl-dC-Puromycin CPG(450mg)を得た。
少量のFl-dC-Puromycin CPGに25%アンモニア水溶液を加えて60℃で1時間半攪拌して担体から切り出し、反応液をLC/MSで分析することで、反応の進行、およびFl-dC-Puromycin(化合物SP805)の生成を確認した。
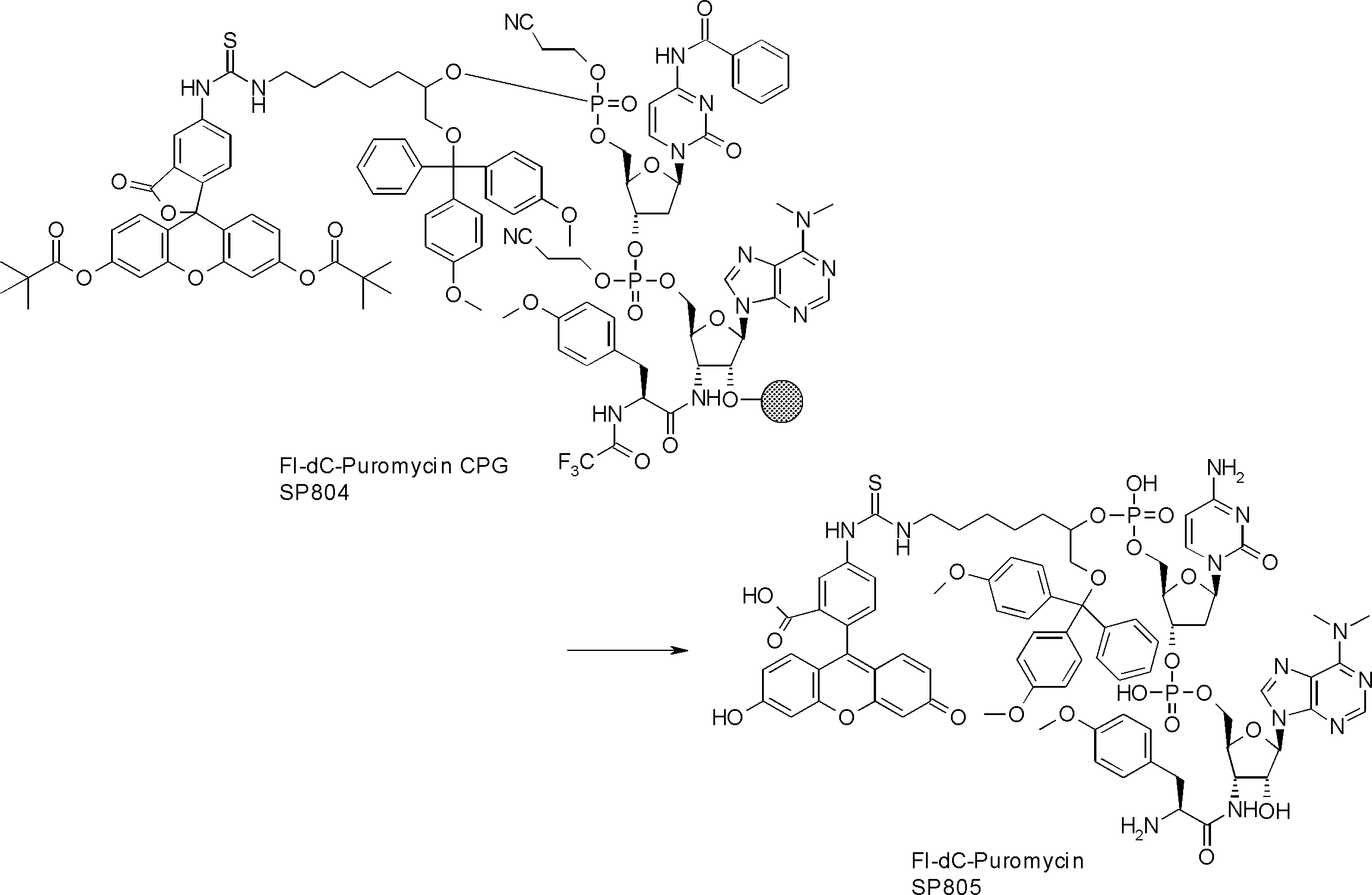
保持時間:0.94分 (分析条件 SQDAA05)

その後、CPG担体をDeblocking Mix(Glen Research社製、3%トリクロロ酢酸/DCM)で処理(3ml×4)した後、ジクロロメタンで洗浄し、乾燥させた。
次いでCPG担体に25%アンモニア水溶液(1.0ml)を加えて60℃で1時間半攪拌した。放冷後、反応液をカラムクロマトグラフィ(0.1%ギ酸水溶液/0.1%ギ酸アセトニトリル溶液)で精製し、標題化合物(化合物SP806)(2.1mg、33.8%)を黄色固体として得た。
LCMS(ESI)m/z =1341.8(M-H)-
保持時間:0.44分 (分析条件 SQDFA05)
配列解析の結果をもとに、配列番号DME-2からDME-11,DME-13からDME-15に示す鋳型DNAを合成した。これらをECL用転写翻訳液に加えて翻訳によるペプチド合成を行った。ECL用転写翻訳液の組成は下記の通りである。5%(v/v) T7 RNA polymerase RiboMAX Enzyme Mix (Promega社,P1300),2mM GTP,2mM ATP,1mM CTP,1mM UTP,20mM クレアチンリン酸,50mM HEPES-KOH pH7.6,100mM 酢酸カリウム,15mM 酢酸マグネシウム,2mM スペルミジン,1mM ジチオスレイトール,0.5mg/ml E.coli MRE600(RNaseネガティブ)由来tRNA(Roche社),3μM 転写tRNA Ala1B(配列番号MTL-13)、3μM 転写tRNA Tyr1(配列番号MTL-14)、4μg/ml クレアチンキナーゼ,3μg/ml ミオキナーゼ,2unit/ml 無機ピロフォスファターゼ,1.1μg/ml ヌクレオシド二リン酸キナーゼ,0.26μM EF-G,0.24μM RF2,0.17μM RF3,0.5μM RRF,2.7μM IF1,0.4μM IF2,1.5μM IF3,53μM EF-Tu,112μM EF-Ts,1.2μM リボソーム,2.73μM AlaRS,0.09μM GlyRS,0.4μM IleRS,0.68μM PheRS,0.16μM ProRS,0.04μM SerRS,0.09μM ThrRS,0.03μM TrpRS,0.22μM TyrRS,0.02μM ValRS,250μM グリシン、250μM イソロイシン、250μM プロリン、250μM スレオニン、250μM トリプトファン、250μM バリン、100μM セリン、5mM N-メチルアラニン、5mM N-メチルフェニルアラニン、2mM D-チロシン,さらにS用ECL転写翻訳液には、翻訳液S用アミノアシル化tRNA混合物および翻訳液S用Initiatorアミノアシル化tRNAを、T用ECL転写翻訳液には0.02μM CysRS、250μMシステイン、および翻訳液T用アミノアシル化tRNA混合物を加えて調製した。
各サンプルの測定値を表24に示した。
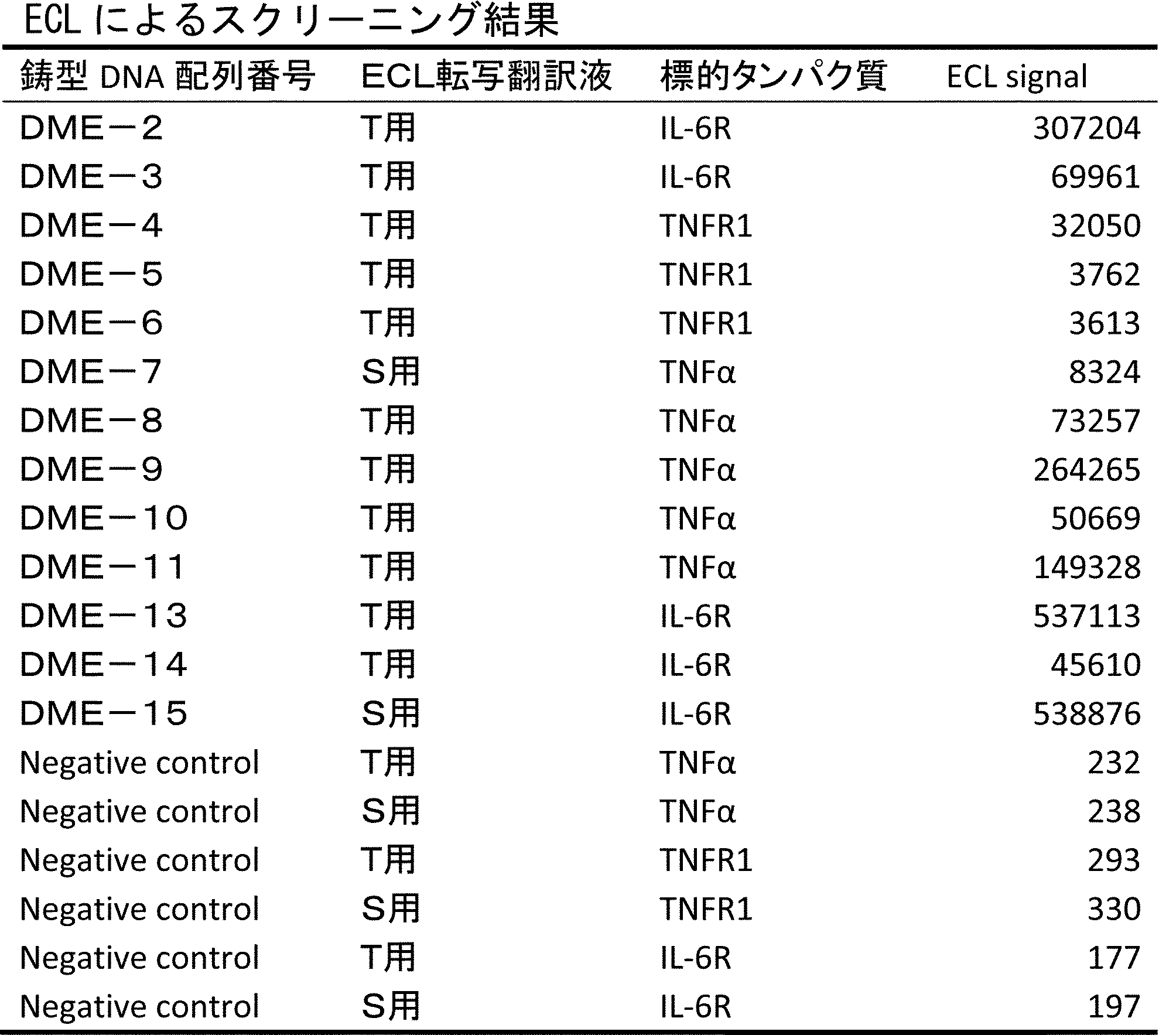
配列番号DME-2(配列番号:164)
(IL-6R binder) DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCCCGATTATTTTTATGCCGAGGTACGTTCGTAGTACTAGCGGCTCTGGCTCTGGCTCT
(IL-6R binder) DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCCCGATTATTTGGAGGATGCCGAGGTACCGTGTTTACAGCGGCTCTGGCTCTGGCTCT
(TNFR1 binder) DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCTACATTTGGATGAGTATGAGGGTTTTTCGTACTAGGAGCGGCTCTGGCTCTGGCTCT
(TNFR1 binder) DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCCCGTTGATTATTGTTAGTCGGCTTCTTCGTGAAACTAGCGGCTCTGGCTCTGGCTCT
(TNFR1 binder) DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCCCGTTGGTTATTACTGTTCGGCTTAGTCGTGCTAGGAGCGGCTCTGGCTCTGGCTCT
TNFα binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGACTCTTATTGAATGGTGGCTATACAGGCGTCCGTTGAGCGGCTCTGGCTCTGGCTCT
TNFα binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCCTATGGTGGGTTTTGGGTCCGTAGAGTCGTCGGATGAGCGGCTCTGGCTCTGGCTCT
TNFα binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCGAAATTCCGGTTTGGTGGCTAATGGTTCGTTGGGAAAGCGGCTCTGGCTCTGGCTCT
TNFα binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCACTATTCCGTACTGGTGGCTAATGGTTCGTTGGGAAAGCGGCTCTGGCTCTGGCTCT
TNFα binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCACTTGGATTTTTCTTTGGCAGCTACTTCGTGTTACTAGCGGCTCTGGCTCTGGCTCT
IL-6R binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCCCGGTTATTTTTATGCCGAGGGTTATGCGTCCGTTGAGCGGCTCTGGCTCTGGCTCT
IL-6R binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGTGCATTATTGAATGGCCGAGGATGTACCAGCGTCCGAGGAGCGGCTCTGGCTCTGGCTCT
IL-6R binder DNA配列
GTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGATTGTTTGGAGGTGCCCGAGGTACTGCCGTGAAGCTAGCGGCTCTGGCTCTGGCTCT
MePhe(3-Cl):(S)-3-(3-クロロフェニル)-2-(メチルアミノ)プロパン酸
Hyp(Et):(2S,4R)-4-エトキシピロリジン-2-カルボン酸
γEtAbu:4-(エチルアミノ)ブタン酸
nPrGly:2-(プロピルアミノ)酢酸
MePhe:(S)-2-(メチルアミノ)-3-フェニルプロパン酸
MeAla:(S)-2-(メチルアミノ)プロパン酸
MeGly:2-(メチルアミノ)酢酸
Pro:(S)-ピロリジン-2-カルボン酸
Thr:(2S,3R)-2-アミノ-3-ヒドロキシブタン酸
Phg:(S)-2-アミノ-2-フェニル酢酸
Ile:(2S,3S)-2-アミノ-3-メチルペンタン酸
Val:(S)-2-アミノ-3-メチルブタン酸
Asp:(S)-2-アミノコハク酸
Trp:(S)-2-アミノ-3-(1H-インドール-3-イル)プロパン酸
DTyr:(R)-2-アミノ-3-(4-ヒドロキシフェニル)プロパン酸
Phe(4-CF3):(S)-2-アミノ-3-(4-(トリフルオロメチル)フェニル)プロパン酸
Ser:(S)-2-アミノ-3-ヒドロキシプロパン酸
Met(O2):(S)-2-アミノ-4-(メチルスルホニル)ブタン酸
βAla:3-アミノプロパン酸
Ala:(S)-2-アミノプロパン酸
Gly:2-アミノ酢酸
MeSer:(S)-3-ヒドロキシ-2-(メチルアミノ)プロパン酸
FlPu(urea):化合物SP806
(IL-6R binder) ペプチド配列
Ala-Pro-Ile-Ile-MePhe-Met(O2)-Pro-MePhe(3-Cl)-DTyr-Val-Asp-Ser-Thr-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(IL-6R binder) ペプチド配列
Ala-Pro-Ile-Ile-Trp-MePhe(3-Cl)-Met(O2)-Pro-MePhe(3-Cl)-DTyr-Asp-Val-DTyr-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFR1 binder)ペプチド配列
Ala-DTyr-Ile-Trp-Met(O2)-Ser-Met(O2)-MePhe(3-Cl)-Val-MePhe-Asp-Thr-MePhe(3-Cl)-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFR1 binder)ペプチド配列
Ala-Pro-MeGly-Ile-Ile-Val-Ser-MeSer-Phg-Phg-Asp-MeAla(4-Thz)-Thr-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFR1 binder) ペプチド配列
Ala-Pro-MeGly-Val-Ile-Thr-Val-MeSer-Phg-Ser-Asp-MeAla-MePhe(3-Cl)-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFα binder) ペプチド配列
γEtAbu-Thr-Phg-Ile-MeAla(4-Thz)-Trp-Trp-Phe(4-CF3)-DTyr-MePhe(3-Cl)-Asp-Pro-MeGly-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFα binder) ペプチド配列
Ala-Phe(4-CF3)-Trp-Trp-Val-MeGly-Gly-Pro-Hyp(Et)-Ser-Asp-MeSer-Met(O2)-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFα binder) ペプチド配列
Ala-MeAla(4-Thz)-Ile-Pro-Val-Trp-Trp-Phe(4-CF3)-Met(O2)-Val-Asp-Trp-MeAla(4-Thz)-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFα binder) ペプチド配列
Ala-Thr-Ile-Pro-DTyr-Trp-Trp-Phe(4-CF3)-Met(O2)-Val-Asp-Trp-MeAla(4-Thz)-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(TNFα binder) ペプチド配列
Ala-Thr-Trp-Ile-MePhe-Phg-Trp-βAla-Phe(4-CF3)-Phg-Asp-Val-Thr-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(IL-6R binder) ペプチド配列
Ala-Pro-Val-Ile-MePhe-Met(O2)-Pro-MePhe(3-Cl)-Val-Met(O2)-Asp-Pro-MeGly-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(IL-6R binder) ペプチド配列
Ala-Ile-Ile-MeAla(4-Thz)-Trp-Pro-MePhe(3-Cl)-Met(O2)-DTyr-βAla-Asp-Pro-MePhe(3-Cl)-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
(IL-6R binder)ペプチド配列
γEtAbu-Ile-Val-Trp-MePhe(3-Cl)-Met(O2)-Pro-MePhe(3-Cl)-DTyr-Met(O2)-Asp-MeAla(4-Thz)-MeAla-Ser-Gly-Ser-Gly-Ser-Gly-Ser-FlPu(urea)
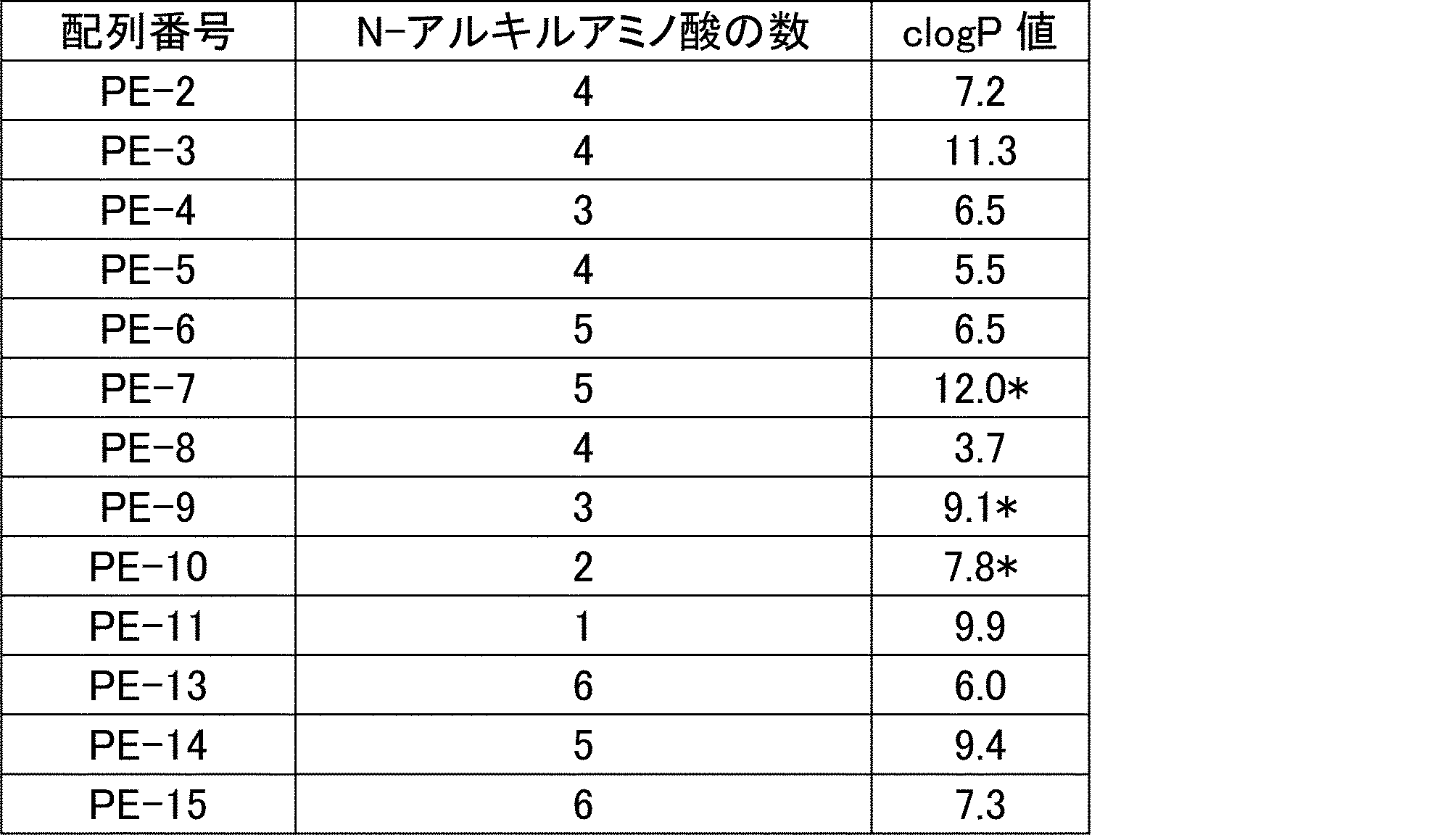
ペプチド合成はスキームJ-1に示すFmoc固相ペプチド合成法に従って行った。実施例中のアミノ酸の略号は、通常文献や商品カタログ(渡辺化学)などに記載の略号を用いているが、詳細を以下に示す。
MeAla(4-Thz):(S)-2-(メチルアミノ)-3-(チアゾール-4-イル)プロパン酸
MePhe(3-Cl):(S)-3-(3-クロロフェニル)-2-(メチルアミノ)プロパン酸
Hyp(Et):(2S,4R)-4-エトキシピロリジン-2-カルボン酸
γEtAbu:4-(エチルアミノ)ブタン酸
nPrGly:2-(プロピルアミノ)酢酸
MePhe:(S)-2-(メチルアミノ)-3-フェニルプロパン酸
MeAla:(S)-2-(メチルアミノ)プロパン酸
MeGly:2-(メチルアミノ)酢酸
Pro:(S)-ピロリジン-2-カルボン酸
Thr:(2S,3R)-2-アミノ-3-ヒドロキシブタン酸
Phg:(S)-2-アミノ-2-フェニル酢酸
Ile:(2S,3S)-2-アミノ-3-メチルペンタン酸
Val:(S)-2-アミノ-3-メチルブタン酸
Asp:(S)-2-アミノコハク酸
Trp:(S)-2-アミノ-3-(1H-インドール-3-イル)プロパン酸
DTyr:(R)-2-アミノ-3-(4-ヒドロキシフェニル)プロパン酸
Phe(4-CF3):(S)-2-アミノ-3-(4-(トリフルオロメチル)フェニル)プロパン酸
Ser:(S)-2-アミノ-3-ヒドロキシプロパン酸
Met(O2):(S)-2-アミノ-4-(メチルスルホニル)ブタン酸
βAla:3-アミノプロパン酸
Ala:(S)-2-アミノプロパン酸
Gly:2-アミノ酢酸
MeSer:(S)-3-ヒドロキシ-2-(メチルアミノ)プロパン酸
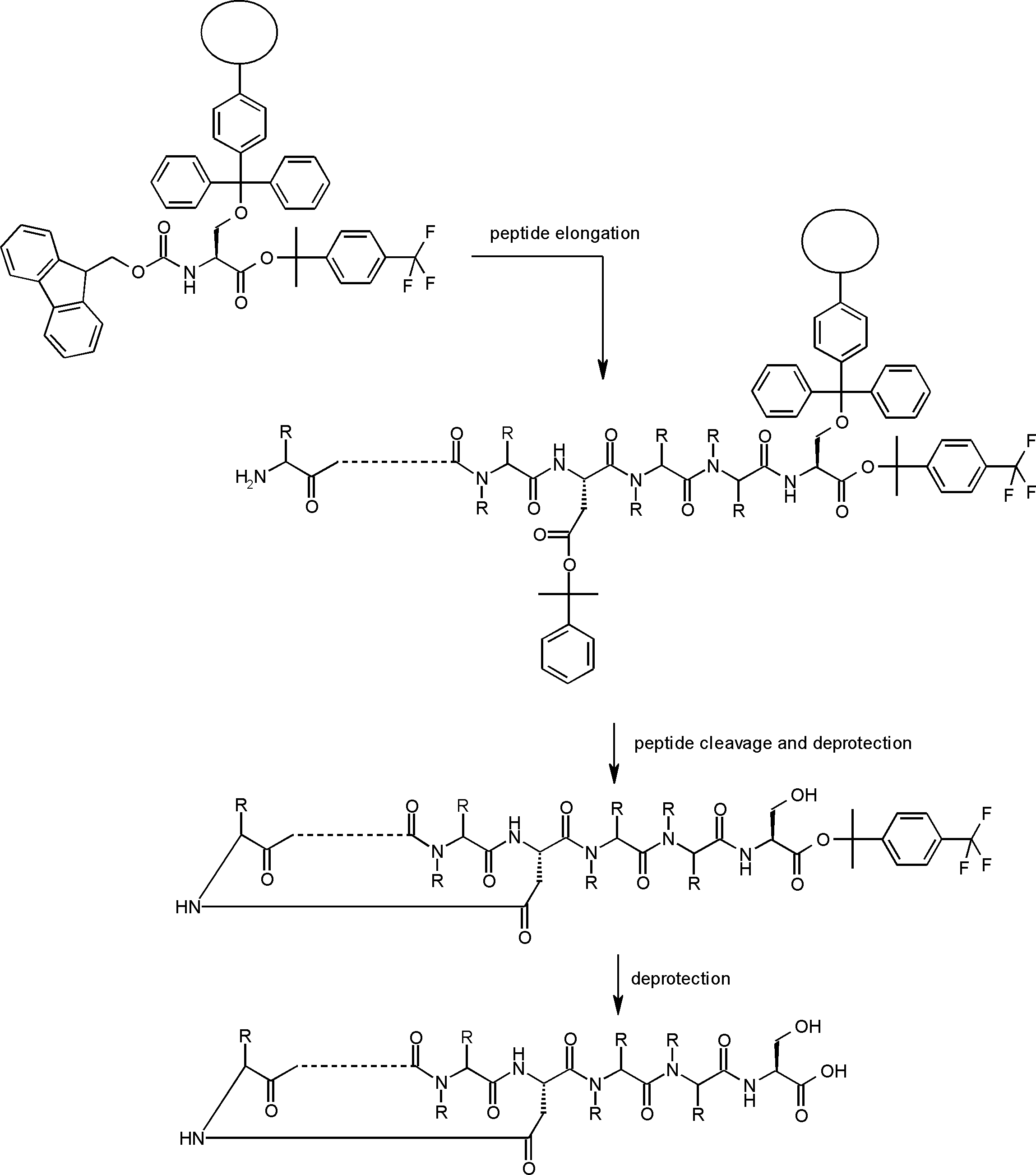
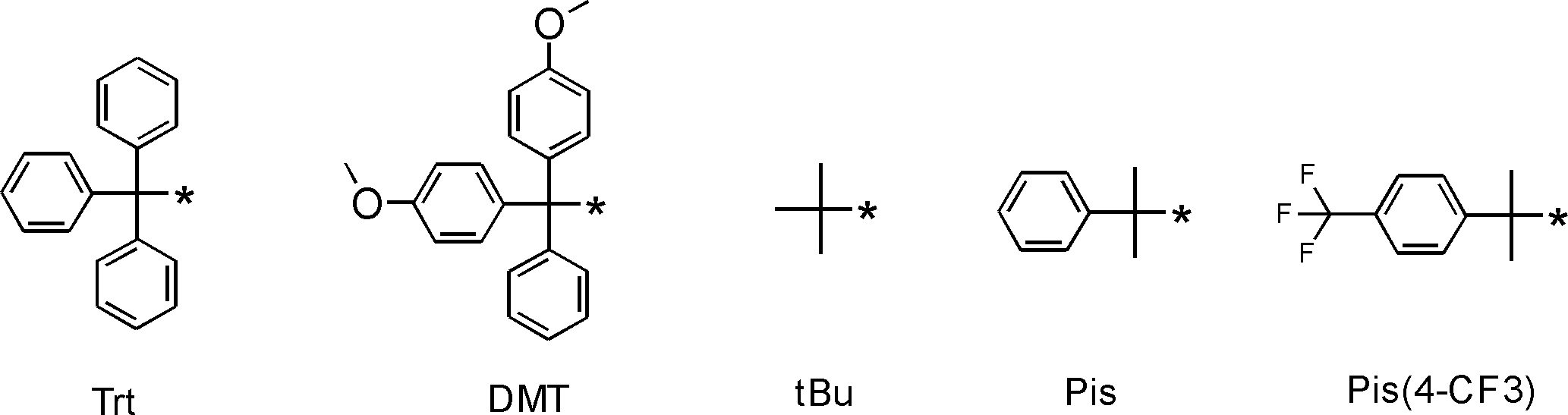





LCMS(ESI) m/z = 409 (M+H)+
保持時間:0.44分(分析条件SQDAA50)


LCMS(ESI) m/z = 436 (M+H)+
保持時間:0.66分(分析条件SQDAA50)

LCMS(ESI) m/z = 382 (M+H)+
保持時間:0.92分(分析条件SQDAA05)

LCMS(ESI) m/z = 354 (M+H)+
保持時間:0.93分(分析条件SQDAA05)
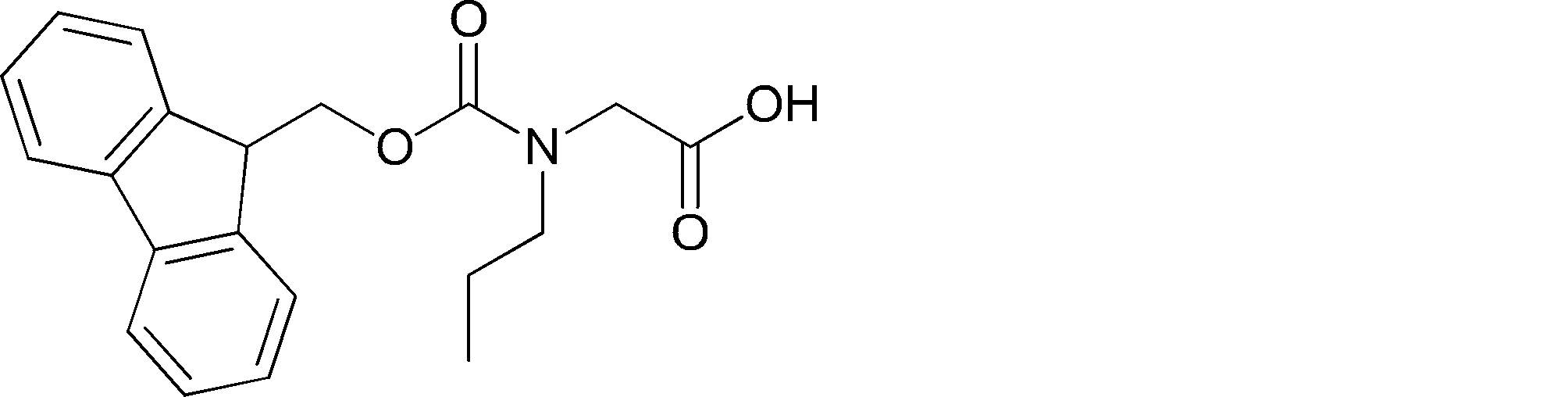
LCMS(ESI) m/z = 340 (M+H)+
保持時間:0.94分(分析条件SQDAA05)
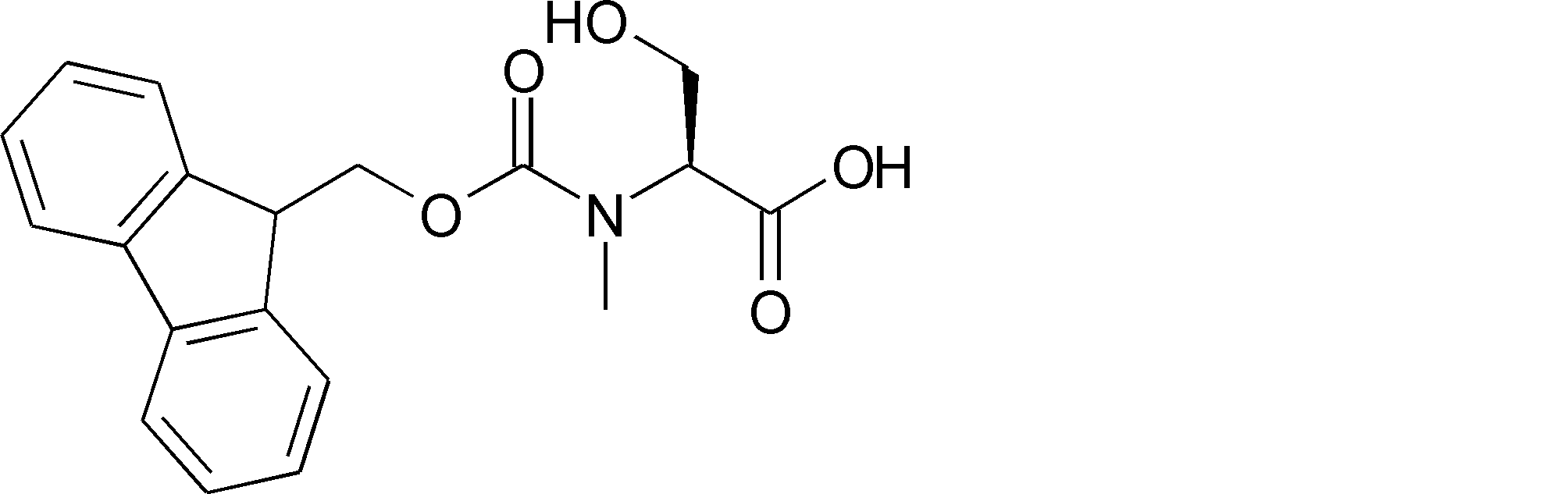
LCMS(ESI) m/z = 342 (M+H)+
保持時間:0.67分(分析条件SQDFA05)
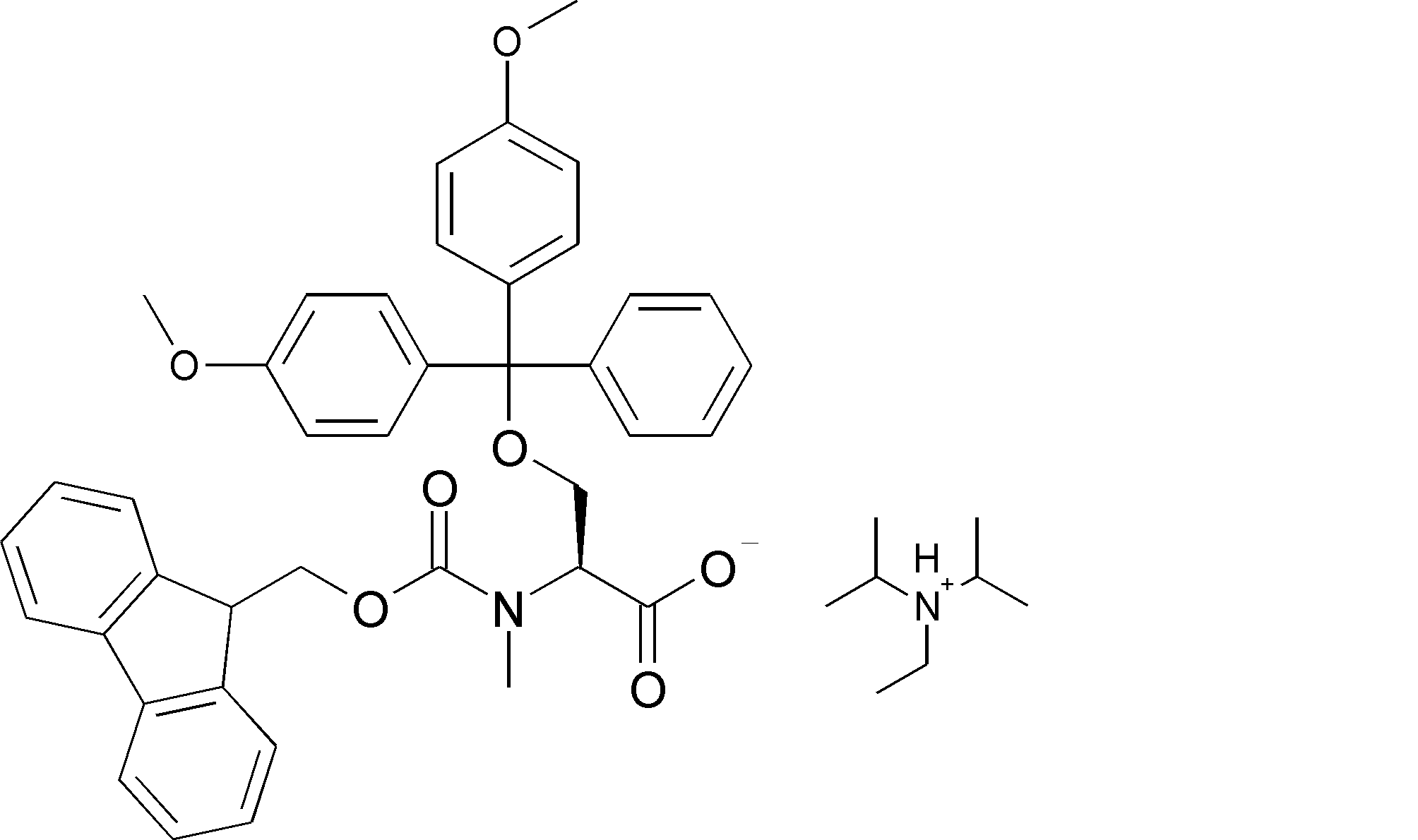
LCMS(ESI) m/z = 642 (M-H)-
保持時間:0.72分(分析条件SQDAA50)
スキームL


保持時間:0.73分(分析条件SQDFA05)
1H-NMR (Varian 400-MR,400 MHz,DMSO-D6) δ ppm 7.69 (2H, d, 8.7 Hz), 7.65(2H,8.7Hz),5.24 (1H, s), 1.45 (6H, s)

保持時間:0.75分(分析条件SQDAA50)
1H-NMR (Varian 400-MR,400 MHz,DMSO-D6) δ ppm 9.17 (1H, s), 7.73 (2H, d, 8.5 Hz), 7.63 (2H, d, 8.5 Hz), 1.84 (6H, s)

LCMS(ESI) m/z = 536 (M+Na)+
保持時間:0.72分(分析条件SQDAA50)
なお、本明細書ではポリマー(例えばレジン)と化合物を結合させた場合、構造式ではポリマー部を○で示す場合がある。○が有する反応性官能基(下の場合にはトリチル基)を示してポリマーと化合物の反応点が理解しやすいように記載する場合がある。以下の例ではレジンのトリチル基とセリンのヒドロキシル基がエーテル結合にて結合しているため、レジンのトリチル基とセリンのエーテル結合部位を記載した。


ポリスチレンレジン(化合物SP828)を用い、Fmocアミノ酸として、Fmoc-MeAla(4-Thz)-OH(化合物SP811)、Fmoc-MePhe(3-Cl)-OH(化合物SP812)、Fmoc-MePhe-OH、Fmoc-MeSer(DMT)-OH(化合物SP816)、Fmoc-MeAla-OH、Fmoc-nPrGly-OH(化合物SP815)、Fmoc-MeGly-OH、Fmoc-Hyp(Et)-OH(化合物SP813)、Fmoc-Pro-OH、Fmoc-Thr(Trt)-OH、Fmoc-Phg-OH、Fmoc-Ile-OH、Fmoc-Val-OH、Fmoc-Asp(Pis)-OH、Fmoc-Trp-OH、Fmoc-DTyr(tBu)-OH、Fmoc-Phe(4-CF3)-OH、Fmoc-Ser(Trt)-OH、Fmoc-Met(O2)-OH、Fmoc-γEtAbu-OH(化合物SP814)、Fmoc-βAla-OH、Fmoc-Ala-OH、Fmoc-Gly-OHを用いて合成を行った。
得られた残渣をジクロロエタンに溶解し、t-ブチルメチルエーテルを加え、ペプチドを沈殿させた。遠心分離にて上清を除き、残渣にトリフルオロ酢酸/トリイソプロピルシラン/ジクロロエタン(=5/5/90)を加え、Pis(4-CF3)を含む側鎖保護基の脱保護を行った。減圧下で溶媒留去し、得られた粗生成物を高速逆相クロマトグラフィー(HPLC)で精製を行った。
配列番号PE-7に由来するペプチド
γEtAbu*-Thr-Phg-Ile-MeAla(4-Thz)-Trp-Trp-Phe(4-CF3)-DTyr-MePhe(3-Cl)-Asp*-Pro-MeGly-Ser-OH
(2つの*部位にて環化)

保持時間:0.81分(分析条件SQDFA05)
配列番号PE-8に由来するペプチド
Ala*-Phe(4-CF3)-Trp-Trp-Val-MeGly-Gly-Pro-Hyp(Et)-Ser-Asp*-MeSer-Met(O2)-Ser-OH
(2つの*部位にて環化)

保持時間:0.64分(分析条件SQDFA05)
配列番号PE-9に由来するペプチド
Ala*-MeAla(4-Thz)-Ile-Pro-Val-Trp-Trp-Phe(4-CF3)-Met(O2)-Val-Asp*-Trp-MeAla(4-Thz)-Ser-OH
(2つの*部位にて環化)

保持時間:0.82分(分析条件SQDFA05)
配列番号PE-10に由来するペプチド
Ala*-Thr-Ile-Pro-DTyr-Trp-Trp-Phe(4-CF3)-Met(O2)-Val-Asp*-Trp-MeAla(4-Thz)-Ser-OH
(2つの*部位にて環化)
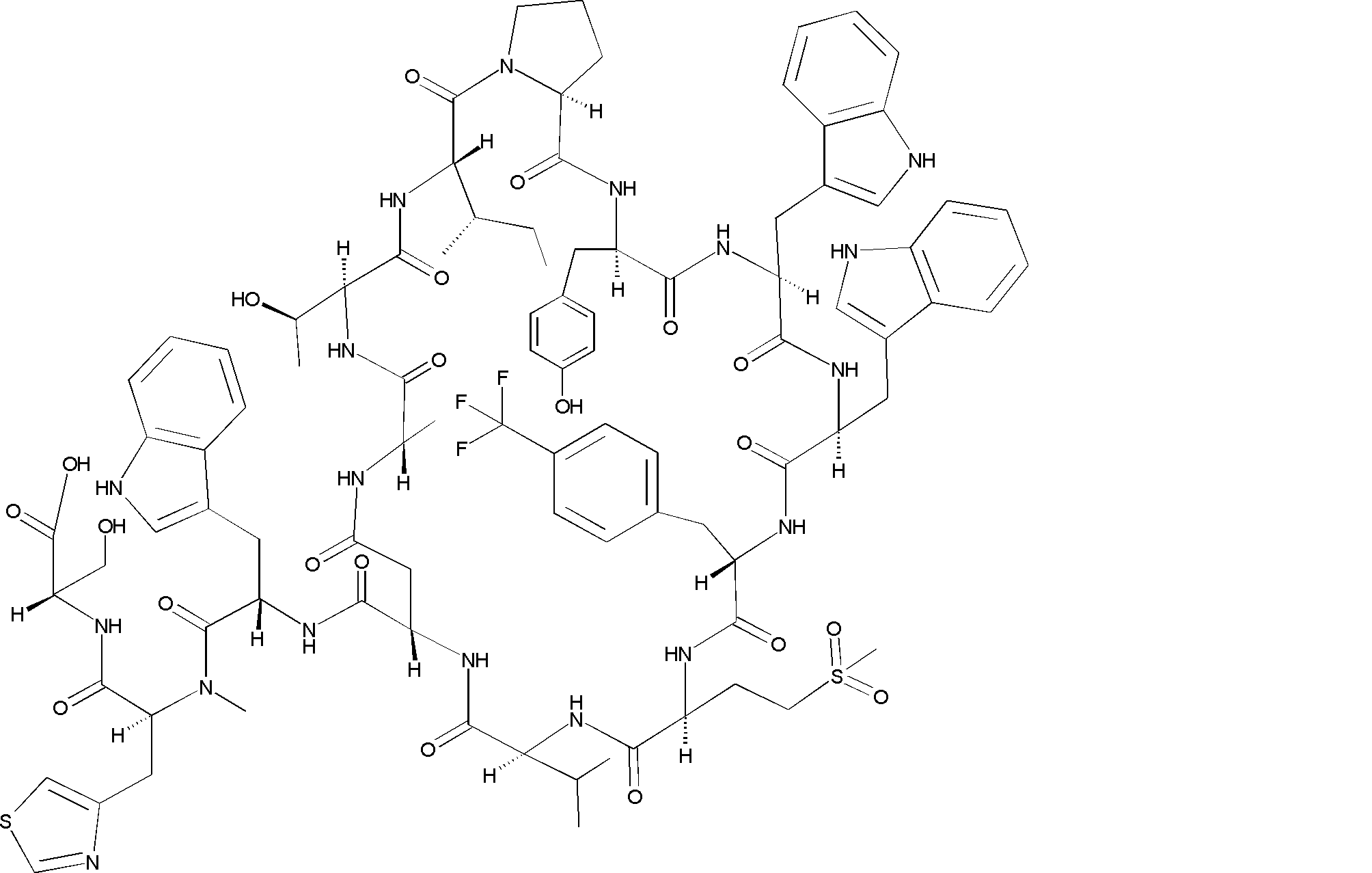
保持時間:0.75分(分析条件SQDFA05)
配列番号PE-11に由来するペプチド
Ala*-Thr-Trp-Ile-MePhe-Phg-Trp-βAla-Phe(4-CF3)-Phg-Asp*-Val-Thr-Ser-OH
(2つの*部位にて環化)
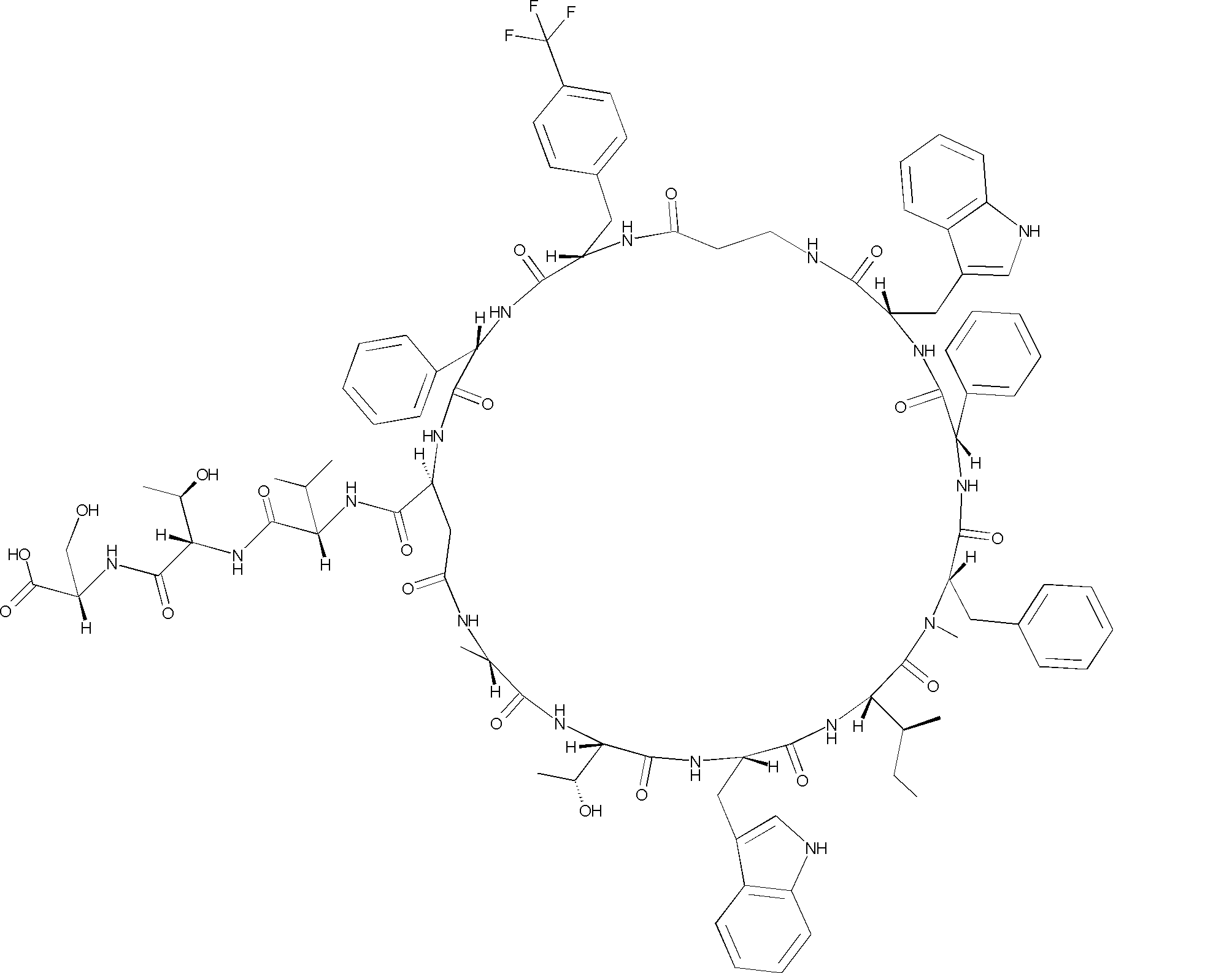
保持時間:0.81分(分析条件SQDFA05)
配列番号PE-13に由来するペプチド
Ala*-Pro-Val-Ile-MePhe-Met(O2)-Pro-MePhe(3-Cl)-Val-Met(O2)-Asp*-Pro-MeGly-Ser-OH
(2つの*部位にて環化)

保持時間:0.70分(分析条件SQDFA05)
配列番号PE-14に由来するペプチド
Ala*-Ile-Ile-MeAla(4-Thz)-Trp-Pro-MePhe(3-Cl)-Met(O2)-DTyr-βAla-Asp*-Pro-MePhe(3-Cl)-Ser-OH
(2つの*部位にて環化)

保持時間:0.80分(分析条件SQDFA05)
配列番号PE-15に由来するペプチド
γEtAbu*-Ile-Val-Trp-MePhe(3-Cl)-Met(O2)-Pro-MePhe(3-Cl)-DTyr-Met(O2)-Asp*-MeAla(4-Thz)-MeAla-Ser-OH
(2つの*部位にて環化)
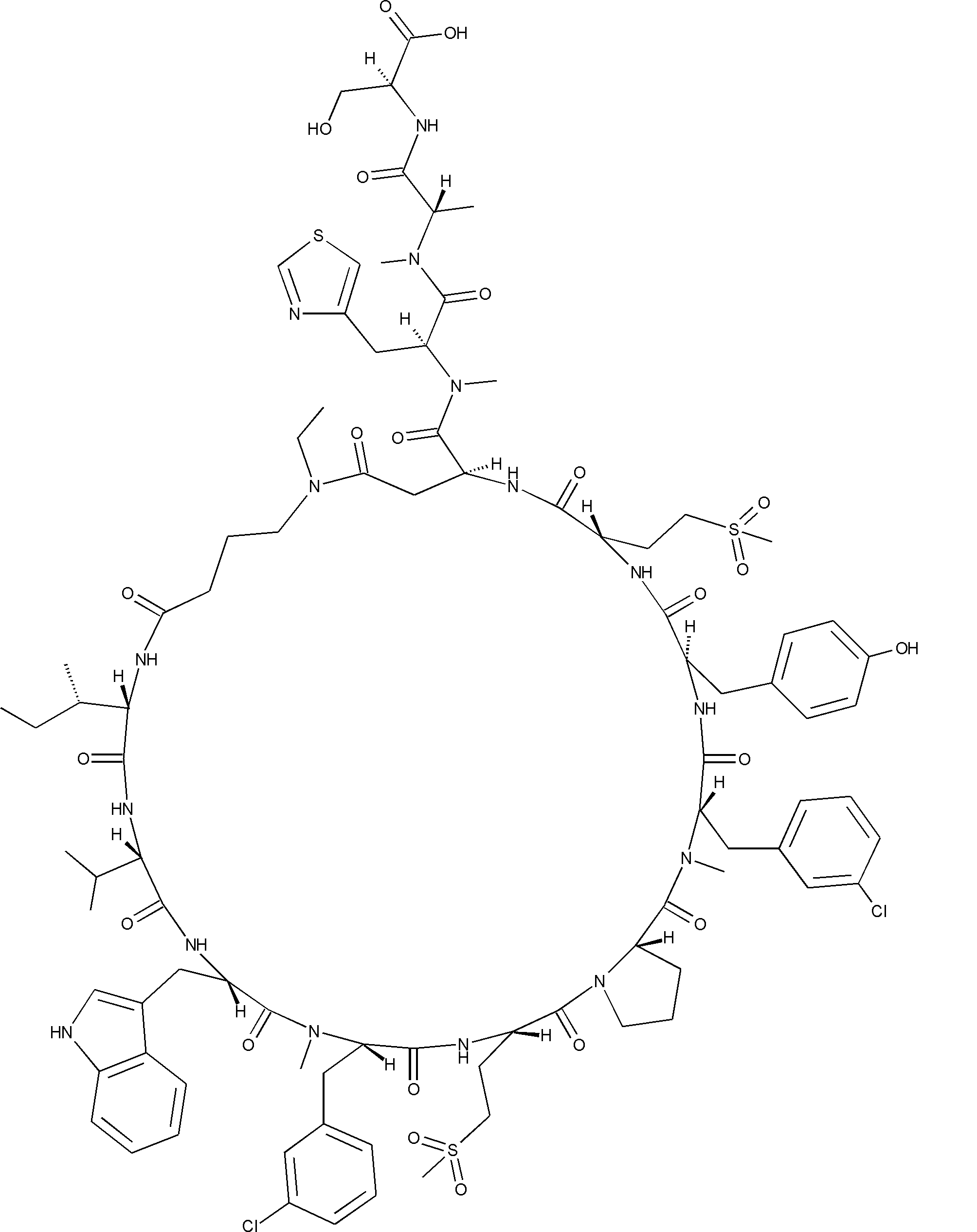
保持時間:0.76分(分析条件SQDFA05)
Ala*-Pro-Val-Ile-MePhe-Met(O2)-Pro-MePhe(3-Cl)-Val-Met(O2)-Asp*-Pro-MeGly-Ser-NH(CH2)2NMe2
(2つの*部位にて環化)
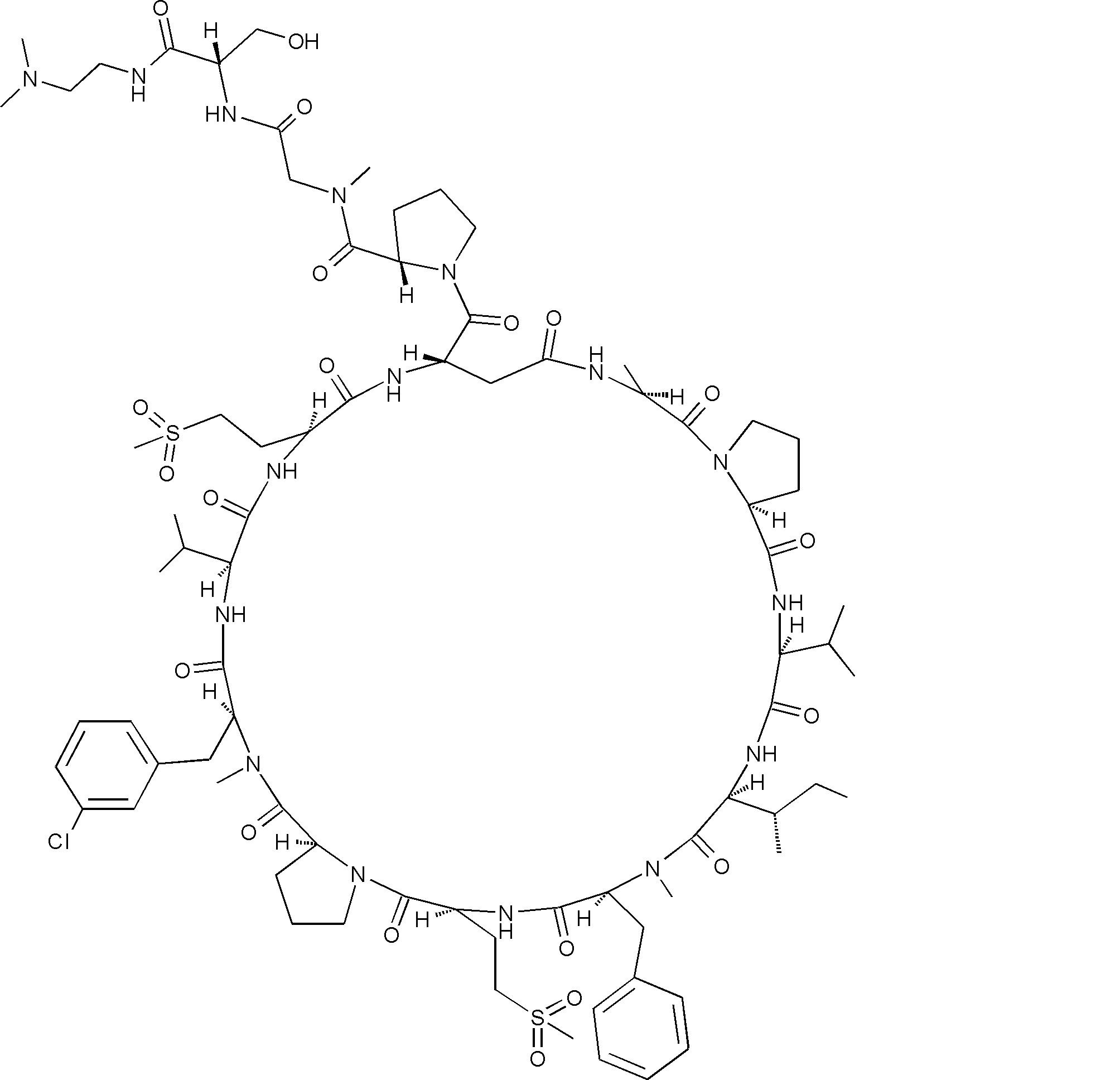
LCMS(ESI) m/z = 1698 (M-H)-
保持時間:0.59分(分析条件SQDFA05)
4-1.TNFαへの結合評価
試薬として、以下のものを使用した。Dimethylsulfoxyde (DMSO), phosphate buffered saline (以上Sigma-Aldrich Co. LLC.),surfactant P-20 (GE healthcare)
合成ペプチドとTNFαとの相互作用解析のSPR実験はBiacoreT200 (GE healthcare) を用いて、20 oCにて実施した。固定化したタンパク質に対して、合成ペプチドを添加し、相互作用を評価した。
ランニングバッファーとして上述のphosphate buffered salineにSurfactant P-20およびDMSOをそれぞれ終濃度0.01vol%, 5vol%となるように添加したものを用いた。ビアコア用センサーチップ Series S sensor chip CAP (GE healthcare) 上に取扱指示書にしたがってCAP reagentを固定化し, ビオチン化したTNFαを固定化させた。解離定数(KD)を測定するため、各合成ペプチドを複数の濃度で添加し、固定化TNFαに対する結合のセンサーグラムを得た。
以上のように解析して得られたKDを以下の表26に示す。

試薬として、以下のものを使用した。Dimethylsulfoxyde (DMSO) (Sigma-Aldrich Co. LLC.), HBS-EP, Surfactant P-20 (以上GE healthcare)
合成ペプチドとIL-6Rとの相互作用解析のSPR実験は、BiacoreT200 (GE healthcare) を用いて、20 oCにて実施した。固定化したIL-6Rに対して、合成ペプチドを添加し、相互作用を評価した。
ランニングバッファーとして、上述のHBS-EPにSurfactant P-20およびDMSOをそれぞれ終濃度0.01vol%, 1vol%となるように添加したものを用いた。
ビアコア用センサーチップ Series S sensor chip CAP, (GE healthcare) 上に取扱指示書にしたがってCAP reagentを固定化し, さらにビオチン化IL-6Rを固定化した。
解離定数(KD)を測定するため、各合成ペプチドを複数の濃度で添加し、固定化IL-6Rに対する結合のセンサーグラムを得た。
得られたセンサーグラムの解析は、T200 evaluation software (GE healthcare)またはScrubber2 (Biologic software) を用いて行った。まず、DMSOに対する溶媒補正およびダブルリファレンス (IL-6Rが固定化されていないフローセルへのセンサーグラム、およびバッファーを添加した場合のセンサーグラムを差し引くことによる補正) を行い、次に、補正後のセンサーグラムに対するカーブフィッティングから、結合速度定数(kon)、解離速度定数(koff)を算出し、KDをKD=koff/konの関係式を用いて決定した。
以上のように解析して得られたKDを以下の表27に示す。

[実施例27]Human gp130発現Ba/F3細胞を用いた合成ペプチドの中和活性評価
実施例26にて確認されたhuman IL-6 receptor(hIL-6R)結合活性を有する合成ペプチドの阻害活性は、human gp130 cDNAを導入し強制発現させることによってhIL-6及び可溶型hIL-6R(shIL-6R)用量依存的に増殖するBa/F3細胞株(gp130 Ba/F3細胞)を用いて評価した。また、カウンターアッセイとしてgp130 Ba/F3細胞のmouse IL-3 (mIL-3)用量依存的な増殖に対する合成ペプチドの影響を評価した。
hIL-6、shIL-6R、mIL-3非存在下かつ合成ペプチド非存在下の細胞増殖算出値の平均値(A)を100%阻害、hIL-6及びshIL-6RまたはmIL-3存在下かつ合成ペプチド非存在下の細胞増殖算出値の平均値(B)を0%阻害として、hIL-6及びshIL-6RまたはmIL-3存在下かつ合成ペプチド存在下の細胞増殖算出値(C)から合成ペプチドの各濃度の阻害率を算出した。
阻害率(%)=(B-C)/(B-A) x 100
その結果、実施例26にて選択、合成した合成ペプチドはgp130 Ba/F3細胞のhIL-6及びshIL-6Rによる細胞増殖を濃度依存的に抑制することが明らかになり、ヒトIL-6シグナリングに対する特異的な阻害活性を有することが示された(図73)。
Claims (54)
- 環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、C末端側に側鎖の1つに反応点を有するアミノ酸残基又はアミノ酸類縁体残基、及び、N末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)N末端側のアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点と、C末端側の側鎖に有するアミノ酸残基又はアミノ酸類縁体残基の反応点とを結合させ、アミド結合又は炭素-炭素結合を形成させる工程
を含む、前記方法。 - 環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基、及び、アミン近傍に反応補助基を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)前記反応補助基を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体と、活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基をアミド結合させて環状化合物を得る工程
を含む、請求項1に記載の方法。 - 活性エステルがチオエステルである、請求項2に記載の方法。
- 前記反応補助基が、SH基である、請求項2又は3に記載の方法。
- 前記環状化合物を得る工程の後、反応補助基を除去する工程を含む、請求項3または4に記載の方法。
- 前記アミン近傍に反応補助基を有するアミノ酸、アミノ酸類縁体又はN末端カルボン酸類縁体が、下記一般式で表される化合物N-1または化合物N-2である、請求項2から5のいずれかに記載の方法;
(式中、R1は、水素原子、S-R23(R23は、置換基を有していてもよいアルキル基、アリール基もしくはアラルキル基を示す。)、またはHS基の保護基を示し;
R2、R3は、それぞれ独立して、水素原子、または、置換基を有していてもよい、アルキル基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基、アラルキル基もしくはシクロアルキル基を示し;またはR2とR3が環を形成した置換基、またはR2もしくはR3がR4と環を形成した置換基を示し;
R4は、置換基を有していてもよいアルキレン基、置換基を有していてもよいアリーレン基または置換基を有していてもよい2価のアラルキル基を示し;
R11、R12は、それぞれ独立して、単結合、置換基を有していてもよいアルキレン基、置換基を有していてもよいアリーレン基または置換基を有していてもよい2価のアラルキル基を示す。)。 - 前記活性エステル基を側鎖に有するアミノ酸又はアミノ酸類縁体が、下記一般式で表される化合物C-1である、請求項2~6のいずれかに記載の方法;
(式中、R25は、OH、ハロゲン原子、OR、またはSR1を示し(RはBt、At、NSuまたはPfpを示し、R1は、水素原子、置換基を有していてもよいアルキル基、置換基を有していてもよいアリール基または置換基を有していてもよいアラルキル基、置換基を有していてもよいシクロアルキル基、置換基を有していてもよいヘテロアリール基、置換基を有していてもよいアルケニル基、置換基を有していてもよいアルキレン基を示す。);
R26は、置換基を有していてもよいアルキレン基、置換基を有していてもよいアリーレン基または置換基を有していてもよい2価のアラルキル基を示し;
R2、R3は、それぞれ独立して、水素原子または置換基を有していてもよいアルキル基を示す。)。 - 前記環状部を有するペプチド化合物の環状部を構成する、アミノ酸残基及び/又はアミノ酸類縁体残基、あるいは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体の総数が、5~12である、請求項1から7のいずれかに記載の方法。
- 前記環状部を有するペプチド化合物を構成する、アミノ酸残基及び/又はアミノ酸類縁体残基、あるいは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体の総数が、9~13である、請求項1~8のいずれかに記載の方法。
- 環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、あるいは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基、及び、N末端の主鎖アミノ基を有するアミノ酸残基、あるいは、主鎖もしくは側鎖にアミノ基を有するアミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)N末端のアミノ酸残基、N末端のアミノ酸類縁体残基又はN末端カルボン酸類縁体と活性エステル基を側鎖に有するアミノ酸残基又はアミノ酸類縁体をアミド結合させて環状化合物を得る工程
を含む、請求項1に記載の方法。 - 活性エステル基がアルキルチオエステル基又はアラルキルチオエステル基であって、工程1)の翻訳合成後に活性化剤を添加させることにより、より活性な活性エステル基に変換させる工程を含む、請求項10に記載の方法。
- 活性剤が、アリールチオール又はN-ヒドロキシスクシンイミドである、請求項11に記載の方法。
- 翻訳されたチオエステルとの反応性が高い活性化剤と、環化させるアミンとの反応性が高い活性化剤を添加させ、より活性の高い活性エステル基に変換させる工程である、請求項12に記載の方法。
- アリールチオエステルで活性エステル基に変換させた後、オキシマおよびその誘導体で更に活性の高いエステル基に変換させる工程である、請求項13に記載の方法。
- 工程1)のN末端部位の翻訳合成が、フォルミルメチオニン以外の翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体をアシル化した翻訳開始tRNAを用いて導入する方法にて行われる、請求項1から14のいずれかに記載の方法。
- 工程1)のN末端部位の翻訳合成が、開始コドンを読み飛ばし、N末端にMet以外の翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体を導入する方法を用いて行われる、請求項10から14のいずれかに記載の方法。
- 工程1)のN末端部位の翻訳合成が、アミノペプチダーゼにてN末端のアミノ酸、アミノ酸類縁体又はカルボン酸類縁体を切断する方法を用いて行われる、請求項10から14のいずれかに記載の方法。
- 前記N末端部位の翻訳合成が、メチオニン アミノペプチダーゼにて処理することによりN末端のフォルミルMetを除去しN末端に他のの翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体を導入する方法を用いて行われる、請求項17に記載の方法。
- 前記N末端部位の翻訳合成が、Metの代わりにノルロイシンを含む翻訳系にて翻訳され、メチオニン アミノペプチダーゼにて処理することによりN末端のフォルミルノルロイシンを除去しN末端に他のの翻訳可能なアミノ酸、翻訳可能なアミノ酸類縁体又は翻訳可能なN末端カルボン酸類縁体を導入する方法を用いて行われる、請求項10~14のいずれかに記載の方法。
- 前記N末端のアミノ酸、アミノ酸類縁体又はカルボン酸類縁体の除去に、さらに、ペプチドデフォルミラーゼが用いられる、請求項17から19のいずれかに記載の方法。
- 前記環状部を有するペプチド化合物が、さらに直鎖部を有する、請求項1から20のいずれかに記載の製造方法。
- 前記非環状ペプチド化合物がα-ヒドロキシカルボン酸、及び、側鎖に保護されていてもよいアミノ基を有するアミノ酸又はアミノ酸類縁体を含み、工程2)の環状化合物を形成する工程の後で、工程3)α―ヒドロキシカルボン酸部位と側鎖に保護されていてもよいアミノ基を有するアミノ酸又はアミノ酸類縁体部位を化学反応させて分枝部位を形成させる工程を含む、請求項1から21のいずれかに記載の方法。
- 環状部と直鎖部とを有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基、N末端カルボン酸類縁体並びにα-ヒドロキシカルボン酸により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、該非環状のペプチド化合物が、
i)C末端側に側鎖の1つに反応点を有するアミノ酸残基(又はアミノ酸類縁体残基)、及び、N末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含み、
ii)前記i)に記載の2つの反応点の間にα位にRf5を有するα-ヒドロキシカルボン酸(Rf5は水素原子、置換されてもよいアルキル基、アラルキル基、ヘテロアリール基、シクロアルキル基、アリケニル基、アルキニル基から選択される)、及び、非環状ペプチド化合物中に保護されていてもよいアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基を含む、
非環状のペプチド化合物である、工程;
2)N末端側のアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点と、C末端側の側鎖に有するアミノ酸残基又はアミノ酸類縁体残基の反応点とを結合させる、環化反応工程;
3)工程1)のii)に記載のα-ヒドロキシカルボン酸のエステル結合を切断してチオエステル基を発生させる工程
4)工程3)において発生させたチオエステル基と工程1)のii)に記載のアミノ基を結合させる環化反応工程、
を含む、方法。 - 工程1)のii)に記載のα-ヒドロキシカルボン酸とアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基の間に含まれるアミノ酸残基及び/又はアミノ酸類縁体残基の数が7つ以下である、請求項23に記載の方法。
- 工程1)のii)に記載のα-ヒドロキシカルボン酸が、Cys-Pro-α-ヒドロキシカルボン酸として非環状ペプチド化合物中に含まれる、請求項23又は24に記載の方法。
- 工程1)のi)に記載のN末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体が反応補助基を有している、請求項23から25のいずれかに記載の方法。
- 工程1)のi)に記載のN末端側にもう1つの反応点を有するアミノ酸残基、アミノ酸類縁体残基又はN末端カルボン酸類縁体が反応補助基を有さず、ii)に記載のアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基のアミノ基が保護基を有し、活性化剤を添加することによって、工程2)の環化反応が行われ、工程2)の環化反応後、工程3)の環化反応前に、前記ii)に記載のアミノ基を側鎖に有するアミノ酸残基又はアミノ酸類縁体残基のアミノ基の保護基を除去する工程を含む、請求項23から26のいずれかに記載の方法。
- 環状部を有するペプチド化合物の製造方法であって、
1)アミノ酸残基及び/又はアミノ酸類縁体残基、もしくは、アミノ酸残基及び/又はアミノ酸類縁体残基並びにN末端カルボン酸類縁体により構成される非環状のペプチド化合物を、該ペプチド化合物をコードする核酸から翻訳して合成する工程であって、
該非環状のペプチド化合物は、側鎖の1つに反応点を有するアミノ酸残基又はアミノ酸類縁体残基、及び、N末端にもう1つの反応点を有するアミノ酸、アミノ酸類縁体残基又はN末端カルボン酸類縁体を含む、工程;
2)N末端のアミノ酸残基、N末端のアミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点と、側鎖に1つの反応点を有するアミノ酸残基又はアミノ酸類縁体の反応点とを炭素‐炭素結合させる工程
を含む、請求項1に記載の方法。 - N末端のアミノ酸残基、N末端のアミノ酸類縁体残基又はN末端カルボン酸類縁体の反応点として炭素‐炭素2重結合を選択し、側鎖に1つの反応点を有するアミノ酸残基又はアミノ酸類縁体残基の反応点としてアリールハライドを選択して、遷移金属を触媒とした炭素-炭素結合反応により環化反応を実施する工程を含む、請求項28に記載の方法。
- 遷移金属を触媒とした炭素-炭素結合反応が、Pdを触媒としたHeck型の化学反応である、請求項29に記載の方法。
- 前記環状部を有するペプチド化合物の環状部を形成するアミノ酸又はアミノ酸類縁体の合計が5~12となる位置に環化反応を実施するための反応点を有する、請求項1から30のいずれかに記載の方法。
- 前記環状部を有するペプチド化合物のアミノ酸及びアミノ酸類縁体残基の総数が9~13である、請求項31に記載の方法。
- ペプチド化合物のC末端と翻訳合成に用いた鋳型とがスペーサーを介して結合しているペプチド化合物-核酸複合体を製造することを特徴とする、請求項1~32のいずれかに記載の方法。
- ペプチド化合物-核酸複合体が、翻訳合成に用いられる非環状ペプチド化合物をコードする核酸の3'末端にリンカーを介してピューロマイシンが結合している核酸を用いて合成される、請求項33に記載の方法。
- スペーサーがペプチド、RNA、DNA、またはヘキサエチレングリコールのポリマー、又は、これらの組合せである、請求項33又は34に記載の方法。
- ペプチド化合物を、互いに異なる配列を有する複数の核酸から成る核酸ライブラリーを翻訳して製造することを特徴とする、請求項1から35のいずれかに記載の方法。
- 請求項1から36のいずれかに記載の製造方法により作製されるペプチド化合物又はペプチド化合物-核酸複合体。
- 異なる構造を有する複数の請求項37記載のペプチド化合物又はペプチド化合物-核酸複合体からなるライブラリー。
- 以下の(i)~(iv)を有することを特徴とする、環状部を有するペプチド化合物;
(i)アミノ酸及びアミノ酸類縁体残基数の合計が5~12からなる環状部を含み、アミノ酸及びアミノ酸類縁体の総数が9~13である、
(ii)N置換アミノ酸を少なくとも2つ含み、N置換されていないアミノ酸を少なくとも1つ含む、
(iii)ClogP値が6以上である、
(iv)環状部を形成しているアミノ酸又はアミノ酸類縁体の結合が、アミノ酸又はアミノ酸類縁体の側鎖にある活性エステル基と、他のアミノ酸又はアミノ酸類縁体のアミン基とからなる結合を少なくとも1つ有する。 - ペプチド化合物に含まれるアミノ酸及びアミノ酸類縁体が、翻訳合成可能なアミノ酸又はアミノ酸類縁体、若しくは、翻訳可能なアミノ酸又はアミノ酸類縁体の側鎖またはN置換部位を化学修飾することによって得られるアミノ酸又はアミノ酸誘導体から選択されるアミノ酸又はアミノ酸類縁体である、請求項39に記載のペプチド化合物。
- さらに、アミノ酸及びアミノ酸類縁体残基数の合計が1~8からなる直鎖部を少なくとも1つ含む、請求項39又は40に記載のペプチド化合物。
- 環状部を形成しているアミノ酸又はアミノ酸類縁体の結合がアミド結合又は炭素-炭素結合である、請求項39から41のいずれかに記載のペプチド化合物。
- 環状部が下記一般式(I)で示される交差ユニットを含む、請求項39から42のいずれかに記載のペプチド化合物;
(式中の記号は以下の意味を示す;
R51は、置換基を有していても良いC1-C6アルキル基、C5-C10アリール基、アラルキル基、エステル基、又は式1で示されるアミドであり、
R52は、置換基を有していても良いC1-C6アルキル基、アリール基、またはアラルキル基であり、
R53は、置換基を有していても良いC1-C6アルキル基、もしくは水素原子であるか、または、R53とR51とが互いに結合してC3-C5のアルキレン基を形成し窒素原子を含む5員~7員環を形成していてもよく、
R54は、0-8アミノ酸残基より構成されるペプチドであり、
R55は、置換基を有していても良いC1-C6アルキル基、C5-C10アリール基、アラルキル基、エステル基、置換基を有していても良いアミド基であり、
*は、環状部における結合部位を示す。)。 - 請求項39から43のいずれかに記載のペプチド化合物を含む医薬組成物。
- 前記医薬組成物が経口剤である、請求項44の医薬組成物。
- 請求項39から45のいずれかに記載のペプチド化合物の製造方法であって、工程(i)~(v)を含む方法:
(i)アミノ酸及びアミノ酸類縁体の総数が9~13である非環状ペプチド化合物を翻訳合成して、当該非環状ペプチド化合物とそれをコードする核酸配列を有する複合体がリンカーを介して結合している非環状ペプチド化合物-核酸複合体を形成する工程;
(ii)工程(i)で翻訳合成された複合体の非環状ペプチド化合物をアミド結合又は炭素-炭素結合によって環化し、環状部のアミノ酸及びアミノ酸類縁体の残基数の合計が5~12となる環状化合物を形成する工程;および、
(iii)工程(ii)で得られた環状部を有するペプチド化合物-核酸複合体ライブラリーと生体内分子とを接触させ、当該生体内分子に対して結合活性を有する複合体を選択する工程。 - さらに、以下の工程を含む、請求項46に記載の製造方法;
(iv)前記工程(iii)で選択された複合体の核酸配列からペプチド化合物の配列情報を得る工程、および、
(v)前記工程(iv)で得られた配列情報に基づきペプチド化合物を化学合成する工程。 - 前記非環状ペプチド化合物がα-ヒドロキシカルボン酸、及び、保護されていてもよい側鎖にアミノ基を有するアミノ酸又はアミノ酸類縁体を含み、工程(ii)の環状化合物を形成する工程の後で、α―ヒドロキシカルボン酸部位と側鎖にアミノ基を有するアミノ酸又はアミノ酸類縁体部位を化学反応させて分枝部位を形成させる工程を含む、請求項46又は47に記載の方法。
- 生体内分子が、分子量500未満の化合物が結合し得る領域を有していない分子である、請求項46から48いずれかに記載の製造方法。
- 生体内分子に対して結合活性を有する複合体が、さらに該生体内分子が他の生体内分子との結合を阻害する活性を有する、請求項46から49のいずれかに記載の製造方法。
- 環化反応するN末端側のアミノ酸又はアミノ酸類縁体が、前記化合物N-1または化合物N-2で表わされる化合物から選択されるアミノ酸又はアミノ酸類縁体であり、C末端側のアミノ酸又はアミノ酸類縁体が前記化合物C-1で表わされる化合物から選択されるアミノ酸又はアミノ酸類縁体であり、工程(ii)の環状化合物を得る工程の後、反応補助基を除去する工程を含む、請求項46から50のいずれかに記載の製造方法。
- 核酸配列が3'末端にスペーサーを有し、翻訳合成されるペプチド化合物のC末端が、当該スペーサーを介して、当該核酸配列と複合体を形成する、請求項46から51のいずれかに記載の製造方法。
- ペプチド化合物-核酸複合体が、翻訳合成に用いられる非環状ペプチド化合物をコードする核酸の3'末端にリンカーを介してピューロマイシンが結合している核酸を用いて合成される、請求項52に記載の方法。
- スペーサーがペプチド、RNA、DNA、またはヘキサエチレングリコールのポリマーである、請求項52又は53に記載の製造方法。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DK12861199.3T DK2813512T3 (da) | 2011-12-28 | 2012-12-28 | Fremgangsmåde til cyklisering af en peptidforbindelse |
| EP12861199.3A EP2813512B1 (en) | 2011-12-28 | 2012-12-28 | Peptide-compound cyclization method |
| EP21165014.8A EP3974563A1 (en) | 2011-12-28 | 2012-12-28 | Cyclic peptides |
| US14/368,564 US9409952B2 (en) | 2011-12-28 | 2012-12-28 | Peptide-compound cyclization method |
| JP2013551857A JP6294080B2 (ja) | 2011-12-28 | 2012-12-28 | ペプチド化合物の環化方法 |
| US15/166,550 US20160311858A1 (en) | 2011-12-28 | 2016-05-27 | Peptide-compound cyclization method |
| US17/011,815 US11891457B2 (en) | 2011-12-28 | 2020-09-03 | Peptide-compound cyclization method |
| US18/460,300 US12415835B2 (en) | 2011-12-28 | 2023-09-01 | Peptide-compound cyclization method |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011288865 | 2011-12-28 | ||
| JP2011-288865 | 2011-12-28 | ||
| JP2012156943 | 2012-07-12 | ||
| JP2012-156943 | 2012-07-12 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/368,564 A-371-Of-International US9409952B2 (en) | 2011-12-28 | 2012-12-28 | Peptide-compound cyclization method |
| US15/166,550 Continuation US20160311858A1 (en) | 2011-12-28 | 2016-05-27 | Peptide-compound cyclization method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013100132A1 true WO2013100132A1 (ja) | 2013-07-04 |
Family
ID=48697609
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/084103 Ceased WO2013100132A1 (ja) | 2011-12-28 | 2012-12-28 | ペプチド化合物の環化方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (4) | US9409952B2 (ja) |
| EP (2) | EP2813512B1 (ja) |
| JP (6) | JP6294080B2 (ja) |
| DK (1) | DK2813512T3 (ja) |
| HU (1) | HUE054006T2 (ja) |
| TW (6) | TWI787670B (ja) |
| WO (1) | WO2013100132A1 (ja) |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017150732A1 (ja) * | 2016-03-03 | 2017-09-08 | 中外製薬株式会社 | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド-核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド-核酸複合体ライブラリーの製造方法 |
| JP2018515448A (ja) * | 2015-04-23 | 2018-06-14 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | ペプチド環化およびプロテアーゼ処理のための方法および組成物 |
| WO2018124162A1 (ja) | 2016-12-28 | 2018-07-05 | 中外製薬株式会社 | 化合物の膜透過性を改善するための自己乳化型製剤 |
| WO2018143145A1 (ja) | 2017-01-31 | 2018-08-09 | 中外製薬株式会社 | 無細胞翻訳系におけるペプチドの合成方法 |
| WO2018207904A1 (ja) | 2017-05-12 | 2018-11-15 | 中外製薬株式会社 | 環状有機化合物の製造方法 |
| WO2018225851A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | N-置換アミノ酸を含むペプチドの合成方法 |
| WO2018225864A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| WO2019117274A1 (ja) | 2017-12-15 | 2019-06-20 | 中外製薬株式会社 | ペプチドの製造方法、及び塩基の処理方法 |
| WO2019235581A1 (ja) | 2018-06-06 | 2019-12-12 | 国立大学法人大阪大学 | Regnase-1が関与する疾患の治療および/または予防方法 |
| WO2019235420A1 (ja) | 2018-06-04 | 2019-12-12 | 中外製薬株式会社 | 複合体を検出する方法 |
| WO2020111238A1 (ja) | 2018-11-30 | 2020-06-04 | 中外製薬株式会社 | ペプチド化合物、またはアミド化合物の脱保護法および固相反応における脱樹脂方法、並びにペプチド化合物の製造方法 |
| WO2020122182A1 (ja) | 2018-12-12 | 2020-06-18 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| WO2020138336A1 (ja) | 2018-12-26 | 2020-07-02 | 中外製薬株式会社 | コドン拡張のための変異tRNA |
| JP2020527576A (ja) * | 2017-07-20 | 2020-09-10 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | N−((1R,2S,5R)−5−(tert−ブチルアミノ)−2−((S)−3−(7−tert−ブチルピラゾロ[1,5−A][1,3,5]トリアジン−4−イルアミノ)−2−オキソピロリジン−1−イル)シクロヘキシル)アセトアミドの製造方法 |
| US10815489B2 (en) | 2015-03-13 | 2020-10-27 | Chugai Seiyaku Kabushiki Kaisha | Modified aminoacyl-tRNA synthetase and use thereof |
| WO2020241816A1 (ja) | 2019-05-31 | 2020-12-03 | 国立大学法人大阪大学 | 新規な消化器がん治療剤およびそのスクリーニング方法 |
| WO2021090856A1 (ja) | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | 立体障害の大きなアミノ酸を含むペプチド化合物の製造方法 |
| WO2021090855A1 (ja) * | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
| WO2021117848A1 (ja) | 2019-12-12 | 2021-06-17 | 中外製薬株式会社 | 非天然アミノ酸を含むペプチドの製造方法 |
| WO2021132546A1 (ja) | 2019-12-26 | 2021-07-01 | 中外製薬株式会社 | 翻訳用組成物及びペプチドの製造方法 |
| JP2021534199A (ja) * | 2018-08-20 | 2021-12-09 | ウントレサーチ・エービー | Wntヘキサペプチドの液相合成 |
| WO2021261577A1 (ja) | 2020-06-25 | 2021-12-30 | 中外製薬株式会社 | 改変された遺伝暗号表を有する翻訳系 |
| WO2022097540A1 (ja) | 2020-11-05 | 2022-05-12 | 中外製薬株式会社 | ジケトピペラジン形成による欠損を抑制するペプチド合成方法 |
| WO2022138892A1 (ja) | 2020-12-25 | 2022-06-30 | 中外製薬株式会社 | 複数の標的分子と共に複合体を形成し得る候補分子のスクリーニング方法 |
| WO2022138891A1 (ja) | 2020-12-25 | 2022-06-30 | 中外製薬株式会社 | N-置換-アミノ酸残基を含むペプチド化合物の製造方法 |
| WO2022145444A1 (ja) | 2020-12-28 | 2022-07-07 | 中外製薬株式会社 | アミノ酸の固相合成用樹脂への担持方法 |
| JP7165289B1 (ja) * | 2021-05-07 | 2022-11-02 | 中外製薬株式会社 | N-置換アミノ酸残基を含む環状化合物の製造方法 |
| WO2022234853A1 (ja) | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物 |
| WO2022234852A1 (ja) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | 環状ペプチド化合物の医薬用途 |
| WO2022234850A1 (ja) | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | 環状ペプチド化合物を含む製剤及びその製造方法 |
| WO2022234851A1 (ja) | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | 変異型ras(g12d)に対する選択的結合性を示す結合分子 |
| WO2022234864A1 (ja) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | N-置換アミノ酸残基を含む環状化合物の製造方法 |
| KR20230019120A (ko) | 2020-06-03 | 2023-02-07 | 추가이 세이야쿠 가부시키가이샤 | 고난도 서열의 효율적 펩티드 축합법 |
| WO2023054636A1 (ja) | 2021-09-30 | 2023-04-06 | 中外製薬株式会社 | 光開裂性部位を有する化合物、連結体及びこれを用いたスクリーニング方法 |
| EP4166133A1 (en) | 2021-10-13 | 2023-04-19 | Chugai Seiyaku Kabushiki Kaisha | Composition containing peptide compound and surfactant |
| WO2023190283A1 (ja) | 2022-03-28 | 2023-10-05 | 中外製薬株式会社 | ポリオキシエチレン構造を含む油性成分を含有する化合物製剤 |
| WO2023195516A1 (ja) | 2022-04-08 | 2023-10-12 | 中外製薬株式会社 | ペプチド化合物の製造方法 |
| WO2023214576A1 (ja) | 2022-05-06 | 2023-11-09 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物 |
| WO2023214577A1 (ja) | 2022-05-02 | 2023-11-09 | 中外製薬株式会社 | ジケトピペラジン形成による欠損を抑制するペプチド合成方法 |
| WO2023214509A1 (ja) | 2022-05-02 | 2023-11-09 | 中外製薬株式会社 | 界面活性剤と併用するためのペプチド化合物を含む組成物 |
| WO2023234425A1 (ja) | 2022-06-03 | 2023-12-07 | ペプチドリーム株式会社 | アミノ酸活性エステル及びその塩 |
| WO2024024965A1 (ja) | 2022-07-29 | 2024-02-01 | 中外製薬株式会社 | O-置換セリン誘導体の製造方法 |
| US11891457B2 (en) | 2011-12-28 | 2024-02-06 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
| WO2024043251A1 (ja) | 2022-08-23 | 2024-02-29 | 富士フイルム株式会社 | 環状ペプチドの細胞膜透過性の予測方法 |
| WO2024043250A1 (ja) | 2022-08-23 | 2024-02-29 | 富士フイルム株式会社 | 環状ペプチドまたはその塩、およびmdmx阻害剤 |
| WO2024043249A1 (ja) | 2022-08-23 | 2024-02-29 | 富士フイルム株式会社 | 環状ペプチドまたはその塩、およびmdmx阻害剤 |
| WO2024085235A1 (ja) | 2022-10-20 | 2024-04-25 | 中外製薬株式会社 | 環状ペプチドの結晶の製造方法 |
| WO2024096023A1 (ja) | 2022-11-01 | 2024-05-10 | 中外製薬株式会社 | ジベンゾフルベンまたはジベンゾフルベン誘導体の除去方法 |
| WO2024101386A1 (ja) | 2022-11-09 | 2024-05-16 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物 |
| WO2024101402A1 (ja) | 2022-11-09 | 2024-05-16 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物を含む医薬組成物 |
| KR20240082196A (ko) | 2022-11-09 | 2024-06-10 | 추가이 세이야쿠 가부시키가이샤 | Hras 및 nras에 대해서 선택적인 kras 저해 작용을 갖는 환상 화합물을 포함하는 의약 조성물 |
| WO2024128195A1 (ja) | 2022-12-12 | 2024-06-20 | 中外製薬株式会社 | 特定位置に酸素原子を有するアミノ酸を翻訳する工程を含む、ペプチドの製造方法 |
| WO2024126687A1 (en) * | 2022-12-16 | 2024-06-20 | Silence Therapeutics Gmbh | Nucleic acids conjugated to an albumin binding moiety |
| WO2024143514A1 (ja) | 2022-12-28 | 2024-07-04 | 中外製薬株式会社 | 環状ペプチドの精製法及び製造法 |
| US12071396B2 (en) | 2019-03-15 | 2024-08-27 | Chugai Seiyaku Kabushiki Kaisha | Method for preparing aromatic amino acid derivative |
| WO2024195801A1 (ja) | 2023-03-20 | 2024-09-26 | 中外製薬株式会社 | 環状ペプチドの共結晶の製造方法 |
| WO2024209857A1 (ja) * | 2023-04-03 | 2024-10-10 | 富士フイルム株式会社 | 核酸ディスプレイライブラリの作製方法、スクリーニング方法、及びポリペプチドの作製方法 |
| WO2024214825A1 (ja) | 2023-04-14 | 2024-10-17 | 中外製薬株式会社 | 複数種類の標的分子との複合体を形成し得る目的分子のスクリーニング方法、及び当該スクリーニング方法を含む目的分子の製造方法 |
| WO2024219446A1 (ja) | 2023-04-19 | 2024-10-24 | 中外製薬株式会社 | 環状ペプチド化合物及び界面活性剤を含む組成物 |
| JP2025503500A (ja) * | 2021-12-24 | 2025-02-04 | セントセル リミテッド | セストリン-mapk複合体阻害剤 |
| CN119708146A (zh) * | 2024-12-30 | 2025-03-28 | 五邑大学 | 一种环肽Lagunamide D的制备方法 |
| WO2025070753A1 (ja) | 2023-09-28 | 2025-04-03 | 中外製薬株式会社 | 混合酸無水物法を用いたペプチド化合物の製造方法 |
| WO2025079650A1 (ja) | 2023-10-11 | 2025-04-17 | 中外製薬株式会社 | 複数のライブラリを用いたペプチドのスクリーニング方法 |
| WO2025100405A1 (ja) | 2023-11-06 | 2025-05-15 | 中外製薬株式会社 | 細胞膜透過性と代謝安定性を有する大環状ペプチド化合物、及びこれを含むライブラリ |
| US12312297B2 (en) | 2018-11-07 | 2025-05-27 | Chugai Seiyaku Kabushiki Kaisha | O-substituted serine derivative production method |
| WO2025143077A1 (ja) | 2023-12-27 | 2025-07-03 | 中外製薬株式会社 | スタンドアローン型縮合剤を用いたペプチド化合物の製造方法 |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10011631B2 (en) * | 2015-12-01 | 2018-07-03 | The University Of Hong Kong | Stapled helical peptides and methods of synthesis |
| US10738338B2 (en) | 2016-10-18 | 2020-08-11 | The Research Foundation for the State University | Method and composition for biocatalytic protein-oligonucleotide conjugation and protein-oligonucleotide conjugate |
| CN109983026B (zh) * | 2016-12-09 | 2023-09-19 | 富士胶片株式会社 | 混合模式亲和层析用载体 |
| WO2018201050A1 (en) * | 2017-04-28 | 2018-11-01 | Board Of Trustees Of Michigan State University | Tryptathionine biosynthesis by flavin mono-oxygenase 1 (fmo1) |
| US11814621B2 (en) | 2018-06-01 | 2023-11-14 | Northwestern University | Expanding the chemical substrates for genetic code reprogramming |
| US20210207188A1 (en) * | 2018-09-20 | 2021-07-08 | The University Of Hong Kong | Beta-lactam compounds and methods of use thereof |
| WO2020169658A2 (en) | 2019-02-20 | 2020-08-27 | Novo Nordisk A/S | Aminoacyl-trna synthetases and uses hereof |
| CN109824622B (zh) * | 2019-02-27 | 2022-12-30 | 中国科学技术大学 | 一类细胞内形成纳米结构杀死癌细胞的前体药物及其制法 |
| US12565517B2 (en) | 2019-10-08 | 2026-03-03 | Genefrontier Corporation | Cyclic peptide having CTLA-4 inhibitory activity and use thereof |
| IL301342A (en) * | 2020-10-21 | 2023-05-01 | Provincial Health Services Authority | Novel cxcr4-targeting compounds |
| US20240124910A1 (en) * | 2021-02-02 | 2024-04-18 | Northwestern University | Ribosome-mediated polymerization of novel chemistries |
| CN115286671B (zh) * | 2022-07-13 | 2023-06-23 | 康龙化成(宁波)科技发展有限公司 | 一种寡聚核酸-噻唑烷类化合物、合成方法、应用 |
| AU2023353878A1 (en) * | 2022-09-28 | 2025-04-10 | Massachusetts Institute Of Technology | Evolution of fluorogenic sensors |
| WO2024151615A1 (en) * | 2023-01-10 | 2024-07-18 | The University Of North Carolina At Chapel Hill | Mrna display libraries and methods of use |
| EP4731636A2 (en) * | 2023-06-22 | 2026-04-29 | Dana-Farber Cancer Institute, Inc. | Methods for the synthesis and linearization of peptidomimetic macrocycles |
| WO2025226861A1 (en) * | 2024-04-24 | 2025-10-30 | Circle Pharma, Inc. | Cyclin inhibitors |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003102495A (ja) | 2000-12-28 | 2003-04-08 | Post Genome Institute Co Ltd | invitro転写/翻訳系によるペプチド等の製造方法 |
| WO2008117833A1 (ja) | 2007-03-26 | 2008-10-02 | The University Of Tokyo | 環状ペプチド化合物の合成方法 |
Family Cites Families (112)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57159747A (en) | 1981-03-27 | 1982-10-01 | Kenji Okawa | Synthesis of amino acid derivative |
| JPS60169451A (ja) | 1984-02-13 | 1985-09-02 | Mitsui Toatsu Chem Inc | L−フエニルアラニンの分離精製法 |
| FR2583432B1 (fr) | 1985-06-13 | 1988-11-04 | Inst Francais Du Petrole | Procede de production enzymatique de l-a-aminoacides a partir d'a-cetoacides |
| ES8607214A1 (es) | 1985-10-16 | 1986-06-01 | Gema Sa | Procedimiento enzimatico de resolucion de n-fenilacetil-ami-nocompuestos y purificacion de sus isomeros opticamente ac- tivos |
| US4859736A (en) | 1987-03-30 | 1989-08-22 | Ciba-Geigy Corporation | Synthetic polystyrene resin and its use in solid phase peptide synthesis |
| DE3733198A1 (de) | 1987-10-01 | 1989-04-13 | Kernforschungsanlage Juelich | Enzymatisches verfahren zur herstellung von dipeptiden |
| JPH0681759B2 (ja) | 1988-03-31 | 1994-10-19 | 信越化学工業株式会社 | ポリペプチドの製造方法 |
| GB9022788D0 (en) | 1990-10-19 | 1990-12-05 | Cortecs Ltd | Pharmaceutical formulations |
| US7045363B2 (en) * | 1996-05-01 | 2006-05-16 | Fujirebio Inc. | Nucleic acid-bound polypeptide method of producing nucleic acid-bound polypeptide and immunoassay using the polypeptide |
| DK1712623T3 (da) * | 1997-01-21 | 2012-02-06 | Gen Hospital Corp | Udvælgelse af proteiner ved anvendelse af RNA-proteinfusioner |
| US20020068301A1 (en) | 1997-05-28 | 2002-06-06 | Hung-Sen Lai | Cyclic peptide libraries and methods of use thereof to identify binding motifs |
| EP1014979A4 (en) | 1997-06-09 | 2003-08-20 | Bridge Pharma Inc | CONNECTIONS WITH COMBINED ANTIHISTAMINIC AND FAST-CELL STABILIZING ACTIVITY FOR OPHTHALMOLOGICAL USE |
| US6281175B1 (en) | 1997-09-23 | 2001-08-28 | Scimed Life Systems, Inc. | Medical emulsion for lubrication and delivery of drugs |
| US6977083B1 (en) | 1998-10-02 | 2005-12-20 | Jenapharm Gmbh & Co. Kg | Bioadhesive tablet containing testosterone/testosterone ester mixtures and method for producing a predetermined testosterone time-release profile with same |
| PT1123084E (pt) | 1998-10-02 | 2005-05-31 | Schering Ag | Utilizacao de esteres de testosterona e/ou testosterona para a preparacao de sistemas bioadesivos aplicaveis por via bucal com agentes activos medicamentosos de accao retardada |
| US6057289A (en) | 1999-04-30 | 2000-05-02 | Pharmasolutions, Inc. | Pharmaceutical composition comprising cyclosporin in association with a carrier in a self-emulsifying drug delivery system |
| JP4502293B2 (ja) | 1999-06-04 | 2010-07-14 | 長瀬産業株式会社 | 軸不斉を有する光学活性な4級アンモニウム塩、その製法およびα−アミノ酸誘導体の不斉合成への応用 |
| DE19942624A1 (de) * | 1999-08-28 | 2001-03-08 | Chemotopix Gmbh | Verfahren zur Herstellung von zyklischen Peptidomimetika |
| US7732404B2 (en) | 1999-12-30 | 2010-06-08 | Dexcel Ltd | Pro-nanodispersion for the delivery of cyclosporin |
| GB0001449D0 (en) | 2000-01-21 | 2000-03-08 | Cortendo Ab | Compositions |
| AU2001253621A1 (en) * | 2000-04-19 | 2001-11-07 | Schering Corporation | Macrocyclic NS3-serine protease inhibitors of hepatitis C virus comprising alkyl and aryl alanine P2 moieties |
| HK1049331A1 (en) | 2000-04-21 | 2003-05-09 | Nagase & Co., Ltd. | Optically active quaternary ammonium salt with axial asymmetry, its preparation method, and application of asymmetric synthesis of α amino acid derivatives |
| KR100401296B1 (ko) | 2000-12-27 | 2003-10-11 | 드림바이오젠 주식회사 | 수식물질에 의해 수식된 단백질 변이체 및 이것의 제조방법 |
| US7083970B2 (en) | 2001-04-19 | 2006-08-01 | The Scripps Research Institute | Methods and compositions for the production of orthogonal tRNA-aminoacyl tRNA synthetase pairs |
| EP1424395B1 (en) | 2001-08-01 | 2011-07-27 | Japan Science and Technology Agency | Tyrosyl-trna synthase variants |
| US7288372B2 (en) | 2002-01-17 | 2007-10-30 | Ambergen, Inc. | Methods for the preparation of chemically misaminoacylated tRNA via protective groups |
| AU2003225085A1 (en) * | 2002-04-19 | 2003-11-03 | California Institute Of Technology | Unnatural amino acid containing display libraries |
| CA2461654A1 (en) | 2002-07-18 | 2004-01-29 | Protein Express Co., Ltd. | Non-natural labeled amino acid and method for producing a conjugate of said amino acid and trna |
| JP4490663B2 (ja) | 2003-09-22 | 2010-06-30 | 株式会社東京大学Tlo | イソロイシンtRNA(tRNAIle)のライシジン合成酵素(TilS)としてのmesJ遺伝子産物及びその相同性遺伝子(COG0037) |
| ES2295961T3 (es) | 2003-12-31 | 2008-04-16 | F. Hoffmann-La Roche Ag | Proceso para la sintesis peptidica utilizacndo una cantidad reducida de agente de desproteccion. |
| WO2005076744A2 (en) | 2004-02-18 | 2005-08-25 | Frutarom Ltd. | Method for the preparation of peptide-oligonucleotide conjugates |
| MX2007006183A (es) | 2004-11-24 | 2007-09-11 | Merck & Co Inc | Formulaciones farmaceuticas liquidas y semisolidas para administracion oral de una amida sustituida. |
| US8188260B2 (en) | 2005-12-06 | 2012-05-29 | The University Of Tokyo | Versatile acylation catalytic RNAs and uses thereof |
| AU2007224019A1 (en) | 2006-03-03 | 2007-09-13 | California Institute Of Technology | Site-specific incorporation of amino acids into molecules |
| EP2010556B1 (en) | 2006-04-11 | 2015-09-23 | Sloan-Kettering Institute for Cancer Research | Homogeneous erythropoietin and method for their preparation |
| JP2007319064A (ja) | 2006-05-31 | 2007-12-13 | Japan Science & Technology Agency | 修飾化アミノ酸を部位特異的に導入したタンパク質を発現させる方法 |
| JP5200241B2 (ja) | 2006-11-17 | 2013-06-05 | 国立大学法人 東京大学 | N末端に非天然骨格をもつポリペプチドの翻訳合成とその応用 |
| JP2009096791A (ja) | 2007-09-26 | 2009-05-07 | Kaneka Corp | アミノ酸の製造方法 |
| WO2009084021A2 (en) | 2007-10-16 | 2009-07-09 | Sun Pharma Advanced Research Company Limited | Novel ophthalmic compositions |
| EP2177533A1 (en) | 2008-10-09 | 2010-04-21 | TU München | Multiple N-methylated cyclic hexapeptides for treatment of neurogenic inflammation |
| TWI468375B (zh) | 2008-10-27 | 2015-01-11 | Janssen Pharmaceutica Nv | 製備經保護之l-丙胺酸衍生物之方法 |
| WO2010053050A1 (ja) | 2008-11-04 | 2010-05-14 | 株式会社カネカ | O-アルキルセリンおよびn-ベンジル-o-アルキルセリンの製造法 |
| CN102239176A (zh) | 2008-12-03 | 2011-11-09 | 弗·哈夫曼-拉罗切有限公司 | 用于制备治疗性肽的方法 |
| WO2010125079A2 (en) | 2009-05-01 | 2010-11-04 | F. Hoffmann-La Roche Ag | Insulinotropic peptide synthesis using solid and solution phase combination techniques |
| US20120208720A1 (en) | 2009-10-22 | 2012-08-16 | Kenji Kashiwagi | Rapid display method in translational synthesis of peptide |
| GB0919194D0 (en) | 2009-11-02 | 2009-12-16 | Lytix Biopharma As | Compounds |
| US20110118314A1 (en) | 2009-11-16 | 2011-05-19 | Weiya Yun | Piperidine analogs as glycogen synthase activators |
| JP2011139667A (ja) | 2010-01-07 | 2011-07-21 | Tottori Univ | プロリンおよびβ−アラニンをN末端に有するジペプチド、及びその環化ジペプチドの酵素合成法 |
| CA2786953A1 (en) | 2010-01-12 | 2011-07-21 | Florian Anders Foeger | Pharmaceutical compositions for oral administration of insulin peptides |
| US20130065272A1 (en) | 2010-03-24 | 2013-03-14 | Medical Research Council | Synthesis of Site Specifically-Linked Ubiquitin |
| EP2380596A1 (en) | 2010-04-20 | 2011-10-26 | Technische Universität München | Cyclopentapeptide derivatives and uses thereof |
| JP5725467B2 (ja) | 2010-08-27 | 2015-05-27 | 国立大学法人 東京大学 | 新規人工翻訳合成系 |
| JP5818237B2 (ja) * | 2010-09-09 | 2015-11-18 | 国立大学法人 東京大学 | N−メチルアミノ酸およびその他の特殊アミノ酸を含む特殊ペプチド化合物ライブラリーの翻訳構築と活性種探索法 |
| US10195578B2 (en) * | 2010-12-03 | 2019-02-05 | The University Of Tokyo | Peptide library production method, peptide library, and screening method |
| JP6239979B2 (ja) | 2011-03-04 | 2017-11-29 | ニューヨーク・ユニバーシティ | Rasの調節因子としての水素結合代替大環状分子 |
| US9133245B2 (en) | 2011-06-13 | 2015-09-15 | Trustees Of Boston College | Cyclic lactadherin peptide mimetics and their uses |
| TWI787670B (zh) | 2011-12-28 | 2022-12-21 | 日商中外製藥股份有限公司 | 具有環狀部之胜肽化合物及其醫藥組成物 |
| GB201215538D0 (en) | 2012-08-31 | 2012-10-17 | Stetsenko Dmitry | Method and compositions for removing acid-labile protecting groups |
| WO2014044803A1 (en) | 2012-09-24 | 2014-03-27 | Dsm Sinochem Pharmaceuticals Netherlands B.V. | Method for producing a cyclic peptide |
| JP6440055B2 (ja) | 2013-05-10 | 2018-12-19 | 国立大学法人 東京大学 | ペプチドライブラリの製造方法、ペプチドライブラリ、及びスクリーニング方法 |
| CN105980350B (zh) | 2013-08-05 | 2018-11-27 | 米迪缪尼有限公司 | 氨基酸衍生物 |
| US10745692B2 (en) | 2013-08-05 | 2020-08-18 | The University Of Tokyo | Production method for charged non-protein amino acid-containing peptide |
| JP6754997B2 (ja) | 2013-08-26 | 2020-09-16 | 国立大学法人 東京大学 | 大環状ペプチド、その製造方法、及び大環状ペプチドライブラリを用いるスクリーニング方法 |
| CN106029684A (zh) | 2014-04-08 | 2016-10-12 | 诺华股份有限公司 | 用于生产大环缩酚酸肽和新中间体的新型缩醛类方法 |
| TW201622746A (zh) | 2014-04-24 | 2016-07-01 | 諾華公司 | 改善或加速髖部骨折術後身體復原之方法 |
| US20170112896A1 (en) | 2014-05-20 | 2017-04-27 | Ohio State Innovation Foundation | Small molecule rac or rho inhibitors |
| EP3152226B1 (en) | 2014-06-06 | 2019-05-29 | Technische Universität München | Modified cyclopentapeptides and uses thereof |
| SG11201609352TA (en) | 2014-06-19 | 2017-01-27 | Solural Pharma ApS | Solid oral dosage form of lipophilic compounds |
| WO2016115168A1 (en) | 2015-01-12 | 2016-07-21 | Synthorx, Inc. | Incorporation of unnatural nucleotides and methods thereof |
| DK3269809T3 (da) | 2015-03-13 | 2022-09-12 | Chugai Pharmaceutical Co Ltd | MODIFICERET AMINOACYL-tRNA-SYNTETASE OG ANVENDELSE DERAF |
| EP3274459A4 (en) | 2015-03-27 | 2018-08-22 | The University of Queensland | Platform for non-natural amino acid incorporation into proteins |
| JP2017043615A (ja) | 2015-08-27 | 2017-03-02 | 塩野義製薬株式会社 | 環状ペプチドの膜透過性および/または代謝安定性を改善するシクロプロパンアミノ酸ユニット |
| MX2018009405A (es) | 2016-02-04 | 2018-11-09 | Univ Johns Hopkins | Sintesis y composicion de bibliotecas de rapafucina. |
| WO2017150732A1 (ja) | 2016-03-03 | 2017-09-08 | 中外製薬株式会社 | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド-核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド-核酸複合体ライブラリーの製造方法 |
| US10954287B2 (en) | 2016-04-15 | 2021-03-23 | Ra Pharmaceuticals, Inc. | Ras binding peptides and methods of use |
| IT201600122363A1 (it) | 2016-12-02 | 2018-06-02 | Dolores Fregona | Composti di coordinazione, sintesi, nanoformulazione ed uso degli stessi in oncologia |
| EP4039250A1 (en) | 2016-12-28 | 2022-08-10 | Chugai Seiyaku Kabushiki Kaisha | Self-emulsifying drug formulation for improving membrane permeability of compound |
| US12391971B2 (en) | 2017-01-31 | 2025-08-19 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptides in cell-free translation system |
| GB201702938D0 (en) | 2017-02-23 | 2017-04-12 | Univ Southampton | Methods of generating and screening compartmentalised peptides libraries |
| WO2018225851A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | N-置換アミノ酸を含むペプチドの合成方法 |
| JP7232758B2 (ja) | 2017-06-09 | 2023-03-06 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| EP3725796A4 (en) | 2017-12-15 | 2021-09-15 | Chugai Seiyaku Kabushiki Kaisha | METHOD FOR MANUFACTURING PEPTIDE AND METHOD FOR PROCESSING BASES |
| EP3878836B1 (en) | 2018-11-07 | 2025-07-23 | Chugai Seiyaku Kabushiki Kaisha | O-substituted serine derivative production method |
| JP7568510B2 (ja) | 2018-11-30 | 2024-10-16 | 中外製薬株式会社 | ペプチド化合物、またはアミド化合物の脱保護法および固相反応における脱樹脂方法、並びにペプチド化合物の製造方法 |
| WO2020122182A1 (ja) | 2018-12-12 | 2020-06-18 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| WO2020138336A1 (ja) | 2018-12-26 | 2020-07-02 | 中外製薬株式会社 | コドン拡張のための変異tRNA |
| KR20210139366A (ko) | 2019-03-15 | 2021-11-22 | 추가이 세이야쿠 가부시키가이샤 | 방향족 아미노산 유도체의 제조 방법 |
| US12404299B2 (en) | 2019-11-07 | 2025-09-02 | Chugai Seiyaku Kabushiki Kaisha | Method for producing peptide compound comprising highly sterically hindered amino acid |
| PE20221323A1 (es) | 2019-11-07 | 2022-09-09 | Chugai Pharmaceutical Co Ltd | Compuesto de peptidos ciclicos que tiene accion inhibidora de kras |
| US20230069218A1 (en) | 2019-12-12 | 2023-03-02 | Chugai Seiyaku Kabushiki Kaisha | Method for producing peptide containing non-natural amino acid |
| JP7744244B2 (ja) | 2019-12-26 | 2025-09-25 | 中外製薬株式会社 | 翻訳用組成物及びペプチドの製造方法 |
| JP7709920B2 (ja) | 2019-12-27 | 2025-07-17 | 中外製薬株式会社 | ペプチド化合物の合成法 |
| EP4144747A4 (en) | 2020-06-03 | 2024-05-29 | Chugai Seiyaku Kabushiki Kaisha | EFFICIENT PEPTIDE CONDENSATION PROCESS FOR DIFFICULT SEQUENCES |
| TW202208623A (zh) | 2020-06-25 | 2022-03-01 | 日商中外製藥股份有限公司 | 具有經改變之遺傳密碼表的轉譯系統 |
| US20230391818A1 (en) | 2020-11-05 | 2023-12-07 | Chugai Seiyaku Kabushiki Kaisha | Peptide synthesis method for suppressing defect caused by diketopiperazine formation |
| EP4248951A4 (en) | 2020-12-22 | 2024-10-23 | Chugai Seiyaku Kabushiki Kaisha | METHOD FOR THE MANUFACTURE OF MEDICAMENTS WITH A MIXED SOLVENT FREEZE-DRYING STEP |
| JPWO2022138891A1 (ja) | 2020-12-25 | 2022-06-30 | ||
| US20240067674A1 (en) | 2020-12-28 | 2024-02-29 | Chugai Seiyaku Kabushiki Kaisha | Method for loading amino acid on resin for solid-phase synthesis |
| AU2022270498A1 (en) | 2021-05-07 | 2023-10-05 | Chugai Seiyaku Kabushiki Kaisha | Preparation containing cyclic peptide compound and method for producing same |
| KR102666765B1 (ko) | 2021-05-07 | 2024-05-16 | 추가이 세이야쿠 가부시키가이샤 | 환상 펩타이드 화합물의 의약 용도 |
| TW202309065A (zh) | 2021-05-07 | 2023-03-01 | 日商中外製藥股份有限公司 | 包含n-取代胺基酸殘基之環狀化合物的製造方法 |
| AR125796A1 (es) | 2021-05-07 | 2023-08-16 | Chugai Pharmaceutical Co Ltd | Compuesto cíclico que tiene una acción inhibitoria selectiva en kras sobre hras y nras |
| WO2022234639A1 (ja) | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | 変異型ras(g12d)に対する選択的結合性を示す結合分子 |
| JPWO2023054636A1 (ja) | 2021-09-30 | 2023-04-06 | ||
| TW202330014A (zh) | 2021-10-13 | 2023-08-01 | 日商中外製藥股份有限公司 | 含有胜肽化合物及界面活性劑之組成物 |
| MX2024008002A (es) | 2021-12-28 | 2024-07-12 | Chugai Pharmaceutical Co Ltd | Metodo para producir n-alquilaminoacido y peptido que incluye n-alquilaminoacido. |
| WO2023190283A1 (ja) | 2022-03-28 | 2023-10-05 | 中外製薬株式会社 | ポリオキシエチレン構造を含む油性成分を含有する化合物製剤 |
| EP4488283A1 (en) | 2022-04-08 | 2025-01-08 | Chugai Seiyaku Kabushiki Kaisha | Production method for peptide compound |
| US20250353875A1 (en) | 2022-05-02 | 2025-11-20 | Chugai Seiyaku Kabushiki Kaisha | Peptide synthesis method for suppressing defect caused by diketopiperazine formation |
| KR20250002095A (ko) | 2022-05-02 | 2025-01-07 | 추가이 세이야쿠 가부시키가이샤 | 계면활성제와 병용하기 위한 펩타이드 화합물을 포함하는 조성물 |
| CA3256645A1 (en) | 2022-05-06 | 2025-04-23 | Chugai Seiyaku Kabushiki Kaisha | CYCLIC COMPOUND HAVING A SELECTIVE KRAS INHIBITORY EFFECT ON HRAS AND NRAS |
| TW202430539A (zh) | 2022-10-13 | 2024-08-01 | 日商中外製藥股份有限公司 | 包含n-取代胺基酸殘基的環狀胜肽化合物的製造方法 |
-
2012
- 2012-12-28 TW TW109139943A patent/TWI787670B/zh active
- 2012-12-28 TW TW113126390A patent/TW202446781A/zh unknown
- 2012-12-28 US US14/368,564 patent/US9409952B2/en active Active
- 2012-12-28 TW TW107130626A patent/TWI713889B/zh active
- 2012-12-28 TW TW101150926A patent/TWI579296B/zh active
- 2012-12-28 TW TW106103188A patent/TWI640541B/zh active
- 2012-12-28 EP EP12861199.3A patent/EP2813512B1/en active Active
- 2012-12-28 TW TW111144798A patent/TWI852183B/zh active
- 2012-12-28 JP JP2013551857A patent/JP6294080B2/ja active Active
- 2012-12-28 HU HUE12861199A patent/HUE054006T2/hu unknown
- 2012-12-28 DK DK12861199.3T patent/DK2813512T3/da active
- 2012-12-28 WO PCT/JP2012/084103 patent/WO2013100132A1/ja not_active Ceased
- 2012-12-28 EP EP21165014.8A patent/EP3974563A1/en active Pending
-
2016
- 2016-05-27 US US15/166,550 patent/US20160311858A1/en not_active Abandoned
-
2018
- 2018-02-15 JP JP2018024856A patent/JP2018118968A/ja not_active Ceased
-
2020
- 2020-03-31 JP JP2020062647A patent/JP6961742B2/ja active Active
- 2020-09-03 US US17/011,815 patent/US11891457B2/en active Active
-
2021
- 2021-10-13 JP JP2021167863A patent/JP7297027B2/ja active Active
-
2023
- 2023-06-13 JP JP2023096892A patent/JP7749619B2/ja active Active
- 2023-09-01 US US18/460,300 patent/US12415835B2/en active Active
-
2025
- 2025-09-24 JP JP2025158195A patent/JP2025186468A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003102495A (ja) | 2000-12-28 | 2003-04-08 | Post Genome Institute Co Ltd | invitro転写/翻訳系によるペプチド等の製造方法 |
| WO2008117833A1 (ja) | 2007-03-26 | 2008-10-02 | The University Of Tokyo | 環状ペプチド化合物の合成方法 |
Non-Patent Citations (111)
| Title |
|---|
| "Greene's Protective Groups in Organic Synthesis" |
| ANDERSON JC; SCHULTZ PG.: "Adaptation of an orthogonal archaeal leucyl-tRNA and synthetase pair for four-base, amber, and opal suppression", BIOCHEMISTRY, vol. 42, 2003, pages 9598 - 608, XP002479161, DOI: doi:10.1021/bi034550w |
| ANGEWANDTE CHEMIE, INTERNATIONAL EDITION, vol. 45, no. 24, 2006, pages 3985 - 3988 |
| BARTON DJ; MORASCO BJ; FLANEGAN JB.: "Assays for poliovirus polymerase, 3D(Pol), and authentic RNA replication in HeLa S10 extracts", METHODS ENZYMOL., vol. 275, 1996, pages 35 - 57 |
| BELLER ET AL., EUR. J. ORG. CHEM., 2001, pages 423 |
| BIOCHEMISTRY, vol. 45, 2006, pages 15541 - 51 |
| BIOORGANIC & MEDICINAL CHEMISTRY, vol. 20, 2012, pages 2679 - 2689 |
| BODE ET AL., ANGEW. CHEM. INT. ED., vol. 45, 2006, pages 1248 |
| CELL-FREE TRANSLATION RECONSTITUTED WITH PURIFIED COMPONENTS; SHIMIZU Y; INOUE A; TOMARI Y; SUZUKI T; YOKOGAWA T; NISHIKAWA K; UED, NAT BIOTECHNOL., vol. 19, 2001, pages 751 - 5 |
| CHATTERJEE J ET AL.: "N-methylation of peptides: a new perspective in medicinal chemistry", ACC CHEM RES, vol. 41, no. 10, 2008, pages 1331 - 1342, XP055164292 * |
| CHATTERJEE, J.; GILON, C.; HOFFMAN, A.; KESSLER, H.: "N-Methylation of peptides: A new perspective in medicinal chemistry", vol. 41, 2008, pages: 1331 |
| CHE ET AL., J. AM. CHEM. SOC., vol. 128, 2006, pages 14796 |
| CHEM. REV., vol. 111, 2011, pages 1417 - 1492 |
| CHEMBIOCHEM, vol. 10, 2009, pages 1186 - 1192 |
| CHEMICAL REVIEWS, vol. 109, no. 6, 2009, pages 2455 - 2504 |
| CHEN HZ; ZUBAY G: "Prokaryotic coupled transcription-translation", METHODS ENZYMOL., vol. 101, 1983, pages 674 - 90 |
| CHEN S ET AL.: "Structurally diverse cyclisation linkers impose different backbone conformations in bicyclic peptides", CHEMBIOCHEM., vol. 13, 2012, pages 1032 - 8, XP055205952, DOI: doi:10.1002/cbic.201200049 |
| DAS ET AL., J. ORG. CHEM., vol. 68, 2003, pages 1165 |
| DAWSON P E ET AL.: "Synthesis of proteins by native chemical ligation", SCIENCE, vol. 266, 1994, pages 776 - 779, XP000889577 * |
| DOI Y ET AL.: "Elongation factor Tu mutants expand amino acid tolerance of protein biosynthesis system", J AM CHEM SOC., vol. 129, 2007, pages 14458 - 62 |
| E GASIOR; F HERRERA; I SADNIK; C S MCLAUGHLIN; K MOLDAVE: "The preparation and characterization of a cell-free system from Saccharomyces cerevisiae that translates natural messenger ribonucleic acid", J. BIOL. CHEM., vol. 254, 1979, pages 3965 - 3969 |
| ERICKSON AH; BLOBEL G.: "Cell-free translation of messenger RNA in a wheat germ system", METHODS ENZYMOL., vol. 96, 1983, pages 38 - 50, XP008071019, DOI: doi:10.1016/S0076-6879(83)96007-X |
| FORSTER, A. C.: "Specificity of translation for N-alkyl amino acids", J. AM. CHEM. SOC., vol. 129, 2007, pages 11316, XP008184753, DOI: doi:10.1021/ja0734871 |
| GANESAN, A: "The impact of natural products upon modern drug discovery", CURR. OPIN. CHEM. BIO., vol. 12, 2008, pages 306, XP022717315, DOI: doi:10.1016/j.cbpa.2008.03.016 |
| GOSUKE HAYASHI ET AL.: "Ribosomal synthesis of nonstandard cyclic peptides and its application to drug discovery", JOURNAL OF JAPANESE BIOCHEMICAL SOCIETY, vol. 82, no. 6, 25 June 2010 (2010-06-25), pages 505 - 514, XP008167839 * |
| GOTO Y ET AL.: "Translation initiation with initiator tRNA charged with exotic peptides", J AM CHEM SOC, vol. 131, no. 14, 2009, pages 5040 - 5041, XP002564833 * |
| GOTO Y: "Flexizymes for genetic code reprogramming", NAT PROTOC., vol. 6, 2011, pages 779 - 90, XP009158277, DOI: doi:10.1038/nprot.2011.331 |
| GRACIA, S. R.; GAUS, K.; SEWALD, N.: "Synthesis of chemically modified bioactive peptides: recent advances, challenges and developments for medicinal chemistry", FUTURE MED. CHEM., vol. 1, 2009, pages 1289 |
| GWILHERM EVANO ET AL.: "Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis", CHEM. REV., vol. 108, 2008, pages 3054, XP008151521, DOI: doi:10.1021/cr8002505 |
| H. FISCHER ET AL.: "Permeation of permanently positive charged molecules through artificial membranes-influence of physic-chemical properties", EUR J. PHARM. SCI., vol. 31, 2007, pages 32 - 42, XP022036116 |
| HACKENBERGER ET AL., ANGEW. CHEM. INT. ED., vol. 47, 2008, pages 5984 |
| HARTMAN MC; JOSEPHSON K; SZOSTAK JW: "Enzymatic aminoacylation of tRNA with unnatural amino acids", PROC NATL ACAD SCI U S A., vol. 103, 2006, pages 4356 - 61, XP055311246, DOI: doi:10.1073/pnas.0509219103 |
| HASHIMOTO N; NINOMIYA K; ENDO T; SISIDO M.: "Simple and quick chemical aminoacylation of tRNA in cationic micellar solution under ultrasonic agitation", CHEM COMMUN (CAMB, 2005, pages 4321 - 3 |
| HECKLER TG; CHANG LH; ZAMA Y; NAKA T; CHORGHADE MS; HECHT SM.: "T4 RNA ligase mediated preparation of novel ''chemically misacylated'' tRNAPheS", BIOCHEMISTRY, vol. 23, 1984, pages 1468 - 73 |
| HEINIS, C.; RUTHERFORD, T.; FREUND, S.; WINTER, G.: "Phage-encoded combinatorial chemical libraries based on bicyclic peptides", NATURE CHEM. BIO., vol. 5, 2009, pages 502 |
| INTERNATIONAL JOURNAL OF PEPTIDE AND PROTEIN RESEARCH, vol. 16, 1980, pages 392 - 401 |
| INTERNATIONAL JOURNAL OF PEPTIDE AND PROTEIN RESEARCH, vol. 35, 1990, pages 545 - 549 |
| J AM CHEM SOC., vol. 131, no. 14, 15 April 2009 (2009-04-15), pages 5040 - 1 |
| JACKSON RJ; HUNT T.: "Preparation and use of nuclease-treated rabbit reticulocyte lysates for the translation of eukaryotic messenger RNA", METHODS ENZYMOL., vol. 96, 1983, pages 50 - 74, XP008071020, DOI: doi:10.1016/S0076-6879(83)96008-1 |
| JOSEPHSON K; HARTMAN MC; SZOSTAK JW: "Ribosomal synthesis of unnatural peptides", J AM CHEM SOC., vol. 127, 2005, pages 11727 - 35, XP055236675, DOI: doi:10.1021/ja0515809 |
| JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 133, 2011, pages 11418 - 11421 |
| JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS, vol. 1, 1996, pages 1205 - 1211 |
| KATOH T ET AL.: "Ribosomal synthesis of backbone macrocyclic peptides", CHEM COMMUN, vol. 47, 15 July 2011 (2011-07-15), pages 9946 - 9958, XP055076618 * |
| KATOH, TAKAYUKI; GOTO, YUKI; REZA, MD. SHAMIM; SUGA, HIROAKI, CHEMICAL COMMUNICATIONS (CAMBRIDGE, UNITED KINGDOM, vol. 47, no. 36, 2011, pages 9946 - 9958 |
| KAWAKAMI T ET AL.: "Diverse backbone-cyclized peptides via codon reprogramming", NAT CHEM BIOL, vol. 5, no. 12, 2009, pages 888 - 890, XP055076621 * |
| KAWAKAMI T ET AL.: "Diverse backbone-cyclized peptides via codon reprogramming", NAT CHEM BIOL., vol. 5, 2009, pages 888 - 90, XP055143411, DOI: doi:10.1038/nchembio.259 |
| KAWAKAMI T ET AL.: "Ribosomal synthesis of polypeptoids and peptoid-peptide hybrids", J AM CHEM SOC., vol. 130, 2008, pages 16861 - 3 |
| KAWAKAMI T ET AL.: "Sequential peptide ligation by using a controlled cysteinyl prolyl ester (CPE) autoactivating unit", TETRAHEDRON LETT, vol. 48, no. 11, 12 March 2007 (2007-03-12), pages 1903 - 1905, XP005890679 * |
| KAWAKAMI T: "Diverse backbone-cyclized peptides via codon reprogramming", NAT CHEM BIOL., vol. 5, 2009, pages 888 - 90, XP055143411, DOI: doi:10.1038/nchembio.259 |
| KESSLER, H.: "Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation", ANTI-CANCER AGENTS IN MEDICINAL CHEMISTRY, vol. 10, 2010, pages 753, XP055034886, DOI: doi:10.2174/187152010794728639 |
| KESSLER, H.: "Improvement of drug-like properties of peptides: the somatostatin paradigm", EXPERT OPIN. DRUG DISCOV., vol. 5, 2010, pages 655 |
| KESSLER, H.: "The impact of amino acid side chain mutations in conformational design of peptides and proteins", CHEM. EUR. J., vol. 16, 2010, pages 5385 |
| KIGA D; SAKAMOTO K; KODAMA K; KIGAWA T; MATSUDA T; YABUKI T; SHIROUZU M; HARADA Y; NAKAYAMA H; TAKIO K: "An expanded eukaryotic genetic code", SCIENCE, vol. 301, 2003, pages 964 - 7 |
| KLEINEWEISCHEDE R ET AL.: "Chemoselective peptide cyclization by traceless Staudinger ligation", ANGEW CHEM INT ED, vol. 47, 2008, pages 5984 - 5988, XP055076643 * |
| KWON I ET AL.: "Breaking the degeneracy of the genetic code", J AM CHEM SOC., vol. 125, 2003, pages 7512 - 3 |
| LAKSHMAN ET AL., EUR. J. ORG. CHEM., 2010, pages 2709 |
| LI ET AL., ORG. LETT., vol. 12, 2010, pages 1724 |
| LI X ET AL.: "Salicylaldehyde ester-induced chemoselective peptide ligations: enabling generation of natural peptidic linkages at the serine/threonine sites", ORG LETT, vol. 12, no. 8, 2010, pages 1724 - 1727, XP055076642 * |
| LOKEY, R. S.: "On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds", NATURE CHEM. BIO., vol. 7, 2011, pages 810, XP055332542, DOI: doi:10.1038/nchembio.664 |
| LOKEY, R. S.: "Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers", J. AM. CHEM. SOC., vol. 128, 2006, pages 2510 |
| LUTZ ACKERMANN ET AL.: "Ruthenium-Catalyzed Direct Arylations Through C-H Bond Cleavages", TOP CURR CHEM., vol. 292, 2010, pages 21 |
| M. KATO ET AL.: "The intestinal first-pass metabolism of substances of CYP3A4 and P-glycoprotein-quantitative analysis based on information from the literature", DRUG METAB. PHARMACOKINET, vol. 18, no. 6, 2003, pages 365 - 372 |
| MATTHEAKIS LC; BHATT RR; DOWER WJ: "An in vitro polysome display system for identifying ligands from very large peptide libraries", PROC NATL ACAD SCI U S A., vol. 91, 1994, pages 9022 - 6, XP002043737, DOI: doi:10.1073/pnas.91.19.9022 |
| MAYER C ET AL.: "Anticodon sequence mutants of Escherichia coli initiator tRNA: effects of overproduction of aminoacyl-tRNA synthetases, methionyl-tRNA formyltransferase, and initiation factor 2 on activity in initiation", BIOCHEMISTRY, vol. 42, 2003, pages 4787 - 99, XP003022641, DOI: doi:10.1021/bi034011r |
| MEINNEL, T. ET AL., BIOCHIMIE, vol. 75, 1993, pages 1061 - 1075 |
| MERRYMAN, C.; GREEN, R.: "Transformation of aminoacyl tRNAs for the in vitro selection of ''Drug-like'' molecules", CHEM. BIO., vol. 11, 2004, pages 575, XP025916606, DOI: doi:10.1016/j.chembiol.2004.03.009 |
| METHOD, vol. 36, 2005, pages 245 - 251 |
| MILSTEIN ET AL., SCIENCE, vol. 317, 2007, pages 790 |
| MOLTKE ET AL.: "Midazolam hydroxylation by human liver microsomes in vitro: inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents", J CLIN PHARMACOL, vol. 36, no. 9, 1996, pages 783 - 791 |
| MURAHASHI ET AL., J. AM. CHEM. SOC., vol. 108, 1986, pages 7846 |
| MURAKAMI H; BONZAGNI NJ; SUGA H.: "Aminoacyl-tRNA synthesis by a resin-immobilized ribozyme", J AM CHEM SOC., vol. 124, 2002, pages 6834 - 5 |
| MURAKAMI H; KOUROUKLIS D; SUGA H.: "Using a solid-phase ribozyme aminoacylation system to reprogram the genetic code", CHEM BIOL., vol. 10, 2003, pages 1077 - 84, XP003013895, DOI: doi:10.1016/j.chembiol.2003.10.010 |
| NAOHIRO TERASAKA ET AL.: "Taisha o Tsukuru 4. Idenshi Ango no Reprogramming ni yoru Tokushu Peptide Library no Kochiku to Seiri Kassei Peptide Tansaku", EXPERIMENTAL MEDICINE, vol. 29, no. 7, 1 May 2011 (2011-05-01), pages 1063 - 1070, XP008174822 * |
| NEMOTO N; MIYAMOTO-SATO E; HUSIMI Y; YANAGAWA H.: "In vitro virus: bonding of mRNA bearing puromycin at the 3'-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro", FEBS LETT, vol. 414, 1997, pages 405 - 8, XP004366358, DOI: doi:10.1016/S0014-5793(97)01026-0 |
| NINOMIYA K; MINOHATA T; NISHIMURA M; SISIDO M.: "In situ chemical aminoacylation with amino acid thioesters linked to a peptide nucleic acid", J AM CHEM SOC., vol. 126, 2004, pages 15984 - 9, XP002337183, DOI: doi:10.1021/ja048200o |
| ODEGRIP R; COOMBER D; ELDRIDGE B; HEDERER R; KUHLMAN PA; ULLMAN C; FITZGERALD K; MCGREGOR D: "CIS display: In vitro selection of peptides from libraries of protein-DNA complexes", PROC NATL ACAD SCI U S A., vol. 101, 2004, pages 2806 - 10, XP002364430, DOI: doi:10.1073/pnas.0400219101 |
| OHTA A: "Synthesis of polyester by means of genetic code reprogramming", CHEM BIOL., vol. 14, 2007, pages 1315 - 22, XP022392655 |
| ORGANIC LETTERS, vol. 9, no. 11, 2007, pages 2223 - 2225 |
| PARK HS ET AL.: "Expanding the genetic code of Escherichia coli with phosphoserine", SCIENCE, vol. 333, 2011, pages 1151 - 4, XP055014139, DOI: doi:10.1126/science.1207203 |
| PARTHASARATHY, R; SUBRAMANIAN, S.; BODER, E. T., BIOCONJUGATE CHEM., vol. 18, 2007, pages 469 - 476 |
| PAUL KNOCHEL ET AL.: "Pd-, Ni-, Fe-, and Co-Catalyzed Cross-Couplings Using Functionalized Zn-, Mg-, Fe", ORGANOMETALLICS. ISR. J. CHEM., vol. 50, 2010, pages 547 |
| PROC NATL ACAD SCI U S A., vol. 99, 2002, pages 9715 - 20 |
| RAINES ET AL., ORG. LETT., vol. 3, 2001, pages 9 |
| REDDY P R ET AL.: "Synthesis of small cyclic peptides via intramolecular Heck reactions", TETRAHEDRON LETT, vol. 44, 2003, pages 353 - 356, XP004397193 * |
| REIERSEN H; LOBERSLI I; LOSET GA; HVATTUM E; SIMONSEN B; STACY JE; MCGREGOR D; FITZGERALD K; WELSCHOF M; BREKKE OH: "Covalent antibody display--an in vitro antibody-DNA library selection system", NUCLEIC ACIDS RES., vol. 33, 2005, pages E10 |
| ROBERTS RW; SZOSTAK JW.: "RNA-peptide fusions for the in vitro selection of peptides and proteins", PROC NATL ACAD SCI USA., vol. 94, 1997, pages 12297 - 302, XP002913434, DOI: doi:10.1073/pnas.94.23.12297 |
| ROBERTS, R. W. ET AL.: "Encodamers: Unnatural peptide oligomers encoded in RNA", CHEM. BIO., vol. 10, 2003, pages 1043, XP002323510, DOI: doi:10.1016/j.chembiol.2003.11.004 |
| ROBERTS, R.: "Design of cyclic peptides that bind protein surfaces with antibody-like affinity", ACS CHEM. BIO., vol. 9, 2007, pages 625 |
| ROBERTS, R.: "J. Am. Chem. Soc.", vol. 124, 2002, article "In vitro selection of mRNA display libraries containing an unnatural amino acid", pages: 9972 |
| SATYANARAYANAJOIS, S. D.; HILL, R. A.: "Medicinal chemistry for 2020", FUTURE MED. CHEM., vol. 3, 2011, pages 1765 |
| SHIMIZU Y; UEDA T.: "PURE technology", METHODS MOL BIOL., vol. 607, 2010, pages 11 - 21 |
| SHUKLA GS; KRAG DN: "Phage-displayed combinatorial peptide libraries in fusion to beta-lactamase as reporter for an accelerated clone screening: Potential uses of selected enzyme-linked affinity reagents in downstream applications", COMB CHEM HIGH THROUGHPUT SCREEN, vol. 13, 2010, pages 75 - 87, XP055448553, DOI: doi:10.2174/138620710790218258 |
| SUBTELNY AO; HARTMAN MC; SZOSTAK JW.: "Ribosomal synthesis of N-methyl peptides", J AM CHEM SOC., vol. 130, 2008, pages 6131 - 6, XP055376645, DOI: doi:10.1021/ja710016v |
| SUGA, H.: "Messenger RNA-programmed incorporation of multiple N-methyl-amino acids into linear and cyclic peptides", CHEM. BIO., vol. 15, 2008, pages 32, XP022428215, DOI: doi:10.1016/j.chembiol.2007.12.008 |
| SWERDEL MR; FALLON AM: "Cell-free translation in lysates from Spodoptera frugiperda (Lepidoptera: Noctuidae) cells", COMP BIOCHEM PHYSIOL B., vol. 93, 1989, pages 803 - 6, XP025830067, DOI: doi:10.1016/0305-0491(89)90049-7 |
| SZOSTAK, J. W. ET AL.: "Ribosomal synthesis of N-methyl peptides", J. AM. CHEM. SOC., vol. 130, 2008, pages 6131, XP055376645, DOI: doi:10.1021/ja710016v |
| TAKESHI HIGUCHI ET AL.: "Programmed Synthesis of Natural Product-like Non-standard Peptides Using the Translation System and Its Application", JOURNAL OF SYNTHETIC ORGANIC CHEMISTRY, vol. 68, no. 3, 1 March 2010 (2010-03-01), JAPAN, pages 217 - 227, XP008168060 * |
| TAWFIK DS; GRIFFITHS AD: "Man-made cell-like compartments for molecular evolution", NAT BIOTECHNOL., vol. 16, 1998, pages 652 - 6, XP002669764, DOI: doi:10.1038/nbt0798-652 |
| THE JOURNAL OF ORGANIC CHEMISTRY, vol. 62, 1997, pages 778 - 779 |
| TSUKIJI S.; NAGAMUNE T., CHEMBIOCHEM, vol. 10, 2009, pages 787 - 798 |
| VILARRASA ET AL., J. ORG. CHEM., vol. 74, 2009, pages 2203 |
| VRIES ET AL., TET. LETT., vol. 41, 2000, pages 2467 |
| WELLS, J. A.; MCCLENDON, C. L.: "Nature", vol. 450, 2007, article "Reaching for high-hanging fruit in drug discovery at protein-protein interfaces", pages: 1001 |
| WILLIAMS ET AL., J. AM. CHEM. SOC., vol. 125, 2003, pages 7754 |
| WILLIAMS ET AL., TET. LETT., vol. 50, 2009, pages 4262 |
| XUE-FENG ZHU; A. IAN SCOTT, NUCLEOSIDES, NUCLEOTIDES & NUCLEIC ACIDS, vol. 20, no. 3, 2001, pages 197 - 211 |
| YAKUBUTSU TAISHAGAKU: "Iryo yakugaku/dokuseigaku no kiso to shite" |
| YAMAGISHI Y. ET AL.: "Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library", CHEM BIOL., vol. 18, 2011, pages 1562 - 70, XP028348893, DOI: doi:10.1016/j.chembiol.2011.09.013 |
| YAMAGUCHI J; NAIMUDDIN M; BIYANI M; SASAKI T; MACHIDA M; KUBO T; FUNATSU T; HUSIMI Y; NEMOTO N.: "cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions", NUCLEIC ACIDS RES., vol. 37, no. 16, 2009, pages E108, XP008154704, DOI: doi:10.1093/nar/gkp514 |
| YOSHIDA ET AL., SYNTHESIS, 1983, pages 474 |
| YUKI GOTO ET AL.: "Ribosomal synthesis of combinatorial polypeptides containing unusual amino acid blocks", KAGAKU KOGYO, vol. 58, no. 4, 2007, pages 255 - 262, XP008169057 * |
Cited By (152)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11891457B2 (en) | 2011-12-28 | 2024-02-06 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
| US12415835B2 (en) | 2011-12-28 | 2025-09-16 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
| US12163134B2 (en) | 2015-03-13 | 2024-12-10 | Chugai Seiyaku Kabushiki Kaisha | Modified aminoacyl-tRNA synthetase and use thereof |
| US10815489B2 (en) | 2015-03-13 | 2020-10-27 | Chugai Seiyaku Kabushiki Kaisha | Modified aminoacyl-tRNA synthetase and use thereof |
| EP4166664A1 (en) | 2015-03-13 | 2023-04-19 | Chugai Seiyaku Kabushiki Kaisha | Modified aminoacyl-trna synthetase and use thereof |
| JP2018515448A (ja) * | 2015-04-23 | 2018-06-14 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | ペプチド環化およびプロテアーゼ処理のための方法および組成物 |
| WO2017150732A1 (ja) * | 2016-03-03 | 2017-09-08 | 中外製薬株式会社 | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド-核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド-核酸複合体ライブラリーの製造方法 |
| JP7351968B2 (ja) | 2016-03-03 | 2023-09-27 | 中外製薬株式会社 | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド-核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド-核酸複合体ライブラリーの製造方法 |
| JP2022106965A (ja) * | 2016-03-03 | 2022-07-20 | 中外製薬株式会社 | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド-核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド-核酸複合体ライブラリーの製造方法 |
| JP7073250B2 (ja) | 2016-03-03 | 2022-05-23 | 中外製薬株式会社 | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド-核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド-核酸複合体ライブラリーの製造方法 |
| JPWO2017150732A1 (ja) * | 2016-03-03 | 2018-12-27 | 中外製薬株式会社 | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド−核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド−核酸複合体ライブラリーの製造方法 |
| EP4039250A1 (en) | 2016-12-28 | 2022-08-10 | Chugai Seiyaku Kabushiki Kaisha | Self-emulsifying drug formulation for improving membrane permeability of compound |
| JP2023078216A (ja) * | 2016-12-28 | 2023-06-06 | 中外製薬株式会社 | 化合物の膜透過性を改善するための自己乳化型製剤 |
| JPWO2018124162A1 (ja) * | 2016-12-28 | 2019-10-31 | 中外製薬株式会社 | 化合物の膜透過性を改善するための自己乳化型製剤 |
| JP7173870B2 (ja) | 2016-12-28 | 2022-11-16 | 中外製薬株式会社 | 化合物の膜透過性を改善するための自己乳化型製剤 |
| WO2018124162A1 (ja) | 2016-12-28 | 2018-07-05 | 中外製薬株式会社 | 化合物の膜透過性を改善するための自己乳化型製剤 |
| US12391971B2 (en) | 2017-01-31 | 2025-08-19 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptides in cell-free translation system |
| JPWO2018143145A1 (ja) * | 2017-01-31 | 2019-11-21 | 中外製薬株式会社 | 無細胞翻訳系におけるペプチドの合成方法 |
| WO2018143145A1 (ja) | 2017-01-31 | 2018-08-09 | 中外製薬株式会社 | 無細胞翻訳系におけるペプチドの合成方法 |
| JP7526243B2 (ja) | 2017-01-31 | 2024-07-31 | 中外製薬株式会社 | 無細胞翻訳系におけるペプチドの合成方法 |
| JP7187323B2 (ja) | 2017-01-31 | 2022-12-12 | 中外製薬株式会社 | 無細胞翻訳系におけるペプチドの合成方法 |
| JP2023021170A (ja) * | 2017-01-31 | 2023-02-09 | 中外製薬株式会社 | 無細胞翻訳系におけるペプチドの合成方法 |
| US11807664B2 (en) | 2017-05-12 | 2023-11-07 | Chugai Seiyaku Kabushiki Kaisha | Method for producing cyclic organic compound |
| EP4570371A2 (en) | 2017-05-12 | 2025-06-18 | Chugai Seiyaku Kabushiki Kaisha | Method for producing cyclic organic compound |
| JP7634933B2 (ja) | 2017-05-12 | 2025-02-25 | 中外製薬株式会社 | 環状有機化合物の製造方法 |
| WO2018207904A1 (ja) | 2017-05-12 | 2018-11-15 | 中外製薬株式会社 | 環状有機化合物の製造方法 |
| KR20200007863A (ko) | 2017-05-12 | 2020-01-22 | 추가이 세이야쿠 가부시키가이샤 | 환상 유기 화합물의 제조 방법 |
| JP7232758B2 (ja) | 2017-06-09 | 2023-03-06 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| KR20250007021A (ko) | 2017-06-09 | 2025-01-13 | 추가이 세이야쿠 가부시키가이샤 | N-치환 아미노산을 포함하는 펩타이드의 합성 방법 |
| JP2023062125A (ja) * | 2017-06-09 | 2023-05-02 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| US11787836B2 (en) | 2017-06-09 | 2023-10-17 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptide containing N-substituted amino acid |
| KR20230169447A (ko) | 2017-06-09 | 2023-12-15 | 추가이 세이야쿠 가부시키가이샤 | N-치환 아미노산을 포함하는 펩타이드의 합성 방법 |
| US11542299B2 (en) | 2017-06-09 | 2023-01-03 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptide containing N-substituted amino acid |
| US12281141B2 (en) | 2017-06-09 | 2025-04-22 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptide containing N-substituted amino acid |
| WO2018225851A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | N-置換アミノ酸を含むペプチドの合成方法 |
| JP2025038213A (ja) * | 2017-06-09 | 2025-03-18 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| JPWO2018225864A1 (ja) * | 2017-06-09 | 2020-04-09 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| WO2018225864A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| JP7611279B2 (ja) | 2017-06-09 | 2025-01-09 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| CN110869544A (zh) * | 2017-06-09 | 2020-03-06 | 中外制药株式会社 | 膜透过性高的环状肽化合物及包含其的文库 |
| KR20200016941A (ko) | 2017-06-09 | 2020-02-17 | 추가이 세이야쿠 가부시키가이샤 | N-치환 아미노산을 포함하는 펩타이드의 합성 방법 |
| CN110869544B (zh) * | 2017-06-09 | 2024-03-08 | 中外制药株式会社 | 膜透过性高的环状肽化合物及包含其的文库 |
| JP2020527576A (ja) * | 2017-07-20 | 2020-09-10 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | N−((1R,2S,5R)−5−(tert−ブチルアミノ)−2−((S)−3−(7−tert−ブチルピラゾロ[1,5−A][1,3,5]トリアジン−4−イルアミノ)−2−オキソピロリジン−1−イル)シクロヘキシル)アセトアミドの製造方法 |
| US11492369B2 (en) | 2017-12-15 | 2022-11-08 | Chugai Seiyaku Kabushiki Kaisha | Method for producing peptide, and method for processing bases |
| WO2019117274A1 (ja) | 2017-12-15 | 2019-06-20 | 中外製薬株式会社 | ペプチドの製造方法、及び塩基の処理方法 |
| WO2019235420A1 (ja) | 2018-06-04 | 2019-12-12 | 中外製薬株式会社 | 複合体を検出する方法 |
| WO2019235581A1 (ja) | 2018-06-06 | 2019-12-12 | 国立大学法人大阪大学 | Regnase-1が関与する疾患の治療および/または予防方法 |
| JP7531913B2 (ja) | 2018-08-20 | 2024-08-13 | ウントレサーチ・エービー | Wntヘキサペプチドの液相合成 |
| JP2024119861A (ja) * | 2018-08-20 | 2024-09-03 | ウントレサーチ・エービー | Wntヘキサペプチドの液相合成 |
| JP2021534199A (ja) * | 2018-08-20 | 2021-12-09 | ウントレサーチ・エービー | Wntヘキサペプチドの液相合成 |
| US12312297B2 (en) | 2018-11-07 | 2025-05-27 | Chugai Seiyaku Kabushiki Kaisha | O-substituted serine derivative production method |
| US11732002B2 (en) | 2018-11-30 | 2023-08-22 | Chugai Seiyaku Kabushiki Kaisha | Deprotection method and resin removal method in solid-phase reaction for peptide compound or amide compound, and method for producing peptide compound |
| WO2020111238A1 (ja) | 2018-11-30 | 2020-06-04 | 中外製薬株式会社 | ペプチド化合物、またはアミド化合物の脱保護法および固相反応における脱樹脂方法、並びにペプチド化合物の製造方法 |
| US12331072B2 (en) | 2018-11-30 | 2025-06-17 | Chugai Seiyaku Kabushiki Kaisha | Deprotection method and resin removal method in solid-phase reaction for peptide compound or amide compound, and method for producing peptide compound |
| WO2020122182A1 (ja) | 2018-12-12 | 2020-06-18 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| JPWO2020122182A1 (ja) * | 2018-12-12 | 2021-10-28 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| JP7531401B2 (ja) | 2018-12-12 | 2024-08-09 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| CN113454058B (zh) * | 2018-12-12 | 2024-03-22 | 中外制药株式会社 | 具有能形成分子内氢键的官能团的氨基酸、包含该氨基酸的肽化合物、及其制备方法 |
| KR20210102326A (ko) | 2018-12-12 | 2021-08-19 | 추가이 세이야쿠 가부시키가이샤 | 분자내 수소 결합 가능한 작용기를 갖는 아미노산과 그들 아미노산을 포함하는 펩타이드 화합물, 및 그들의 제조 방법 |
| CN113454058A (zh) * | 2018-12-12 | 2021-09-28 | 中外制药株式会社 | 具有能形成分子内氢键的官能团的氨基酸、包含该氨基酸的肽化合物、及其制备方法 |
| EP3896056A4 (en) * | 2018-12-12 | 2023-03-15 | Chugai Seiyaku Kabushiki Kaisha | AMINO ACID WITH FUNCTIONAL GROUP FOR INTERMOLECULAR HYDROGEN BONDING, PEPTIDE COMBINATION THEREOF AND PROCESS FOR THEIR PREPARATION |
| WO2020138336A1 (ja) | 2018-12-26 | 2020-07-02 | 中外製薬株式会社 | コドン拡張のための変異tRNA |
| US12071396B2 (en) | 2019-03-15 | 2024-08-27 | Chugai Seiyaku Kabushiki Kaisha | Method for preparing aromatic amino acid derivative |
| WO2020241816A1 (ja) | 2019-05-31 | 2020-12-03 | 国立大学法人大阪大学 | 新規な消化器がん治療剤およびそのスクリーニング方法 |
| KR20220081327A (ko) | 2019-11-07 | 2022-06-15 | 추가이 세이야쿠 가부시키가이샤 | Kras 저해 작용을 갖는 환상 펩티드 화합물 |
| WO2021090855A1 (ja) * | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
| US12404299B2 (en) | 2019-11-07 | 2025-09-02 | Chugai Seiyaku Kabushiki Kaisha | Method for producing peptide compound comprising highly sterically hindered amino acid |
| JP2021119176A (ja) * | 2019-11-07 | 2021-08-12 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
| US20230151060A1 (en) * | 2019-11-07 | 2023-05-18 | Chugai Seiyaku Kabushiki Kaisha | Cyclic peptide compound having kras inhibitory action |
| KR20210064234A (ko) | 2019-11-07 | 2021-06-02 | 추가이 세이야쿠 가부시키가이샤 | Kras 저해 작용을 갖는 환상 펩티드 화합물 |
| JP6880352B1 (ja) * | 2019-11-07 | 2021-06-02 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
| KR102368948B1 (ko) | 2019-11-07 | 2022-03-02 | 추가이 세이야쿠 가부시키가이샤 | Kras 저해 작용을 갖는 환상 펩티드 화합물 |
| IL292140B1 (en) * | 2019-11-07 | 2025-10-01 | Chugai Pharmaceutical Co Ltd | A cyclic peptide compound with KRAS inhibitory activity |
| KR20250160521A (ko) | 2019-11-07 | 2025-11-13 | 추가이 세이야쿠 가부시키가이샤 | Kras 저해 작용을 갖는 환상 펩티드 화합물 |
| WO2021090856A1 (ja) | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | 立体障害の大きなアミノ酸を含むペプチド化合物の製造方法 |
| US12371454B2 (en) | 2019-11-07 | 2025-07-29 | Chugai Seiyaku Kabushiki Kaisha | Cyclic peptide compound having Kras inhibitory action |
| IL292140B2 (en) * | 2019-11-07 | 2026-02-01 | Chugai Pharmaceutical Co Ltd | A cyclic peptide compound with KRAS inhibitory activity |
| CN114729006A (zh) * | 2019-11-07 | 2022-07-08 | 中外制药株式会社 | 具有Kras抑制作用的环状肽化合物 |
| WO2021117848A1 (ja) | 2019-12-12 | 2021-06-17 | 中外製薬株式会社 | 非天然アミノ酸を含むペプチドの製造方法 |
| WO2021132546A1 (ja) | 2019-12-26 | 2021-07-01 | 中外製薬株式会社 | 翻訳用組成物及びペプチドの製造方法 |
| KR20230019120A (ko) | 2020-06-03 | 2023-02-07 | 추가이 세이야쿠 가부시키가이샤 | 고난도 서열의 효율적 펩티드 축합법 |
| WO2021261577A1 (ja) | 2020-06-25 | 2021-12-30 | 中外製薬株式会社 | 改変された遺伝暗号表を有する翻訳系 |
| WO2022097540A1 (ja) | 2020-11-05 | 2022-05-12 | 中外製薬株式会社 | ジケトピペラジン形成による欠損を抑制するペプチド合成方法 |
| KR20230124935A (ko) | 2020-12-25 | 2023-08-28 | 추가이 세이야쿠 가부시키가이샤 | N-치환-아미노산 잔기를 포함하는 펩타이드 화합물의제조 방법 |
| WO2022138892A1 (ja) | 2020-12-25 | 2022-06-30 | 中外製薬株式会社 | 複数の標的分子と共に複合体を形成し得る候補分子のスクリーニング方法 |
| WO2022138891A1 (ja) | 2020-12-25 | 2022-06-30 | 中外製薬株式会社 | N-置換-アミノ酸残基を含むペプチド化合物の製造方法 |
| KR20230125807A (ko) | 2020-12-28 | 2023-08-29 | 추가이 세이야쿠 가부시키가이샤 | 아미노산의 고상 합성용 수지에의 담지 방법 |
| WO2022145444A1 (ja) | 2020-12-28 | 2022-07-07 | 中外製薬株式会社 | アミノ酸の固相合成用樹脂への担持方法 |
| WO2022234852A1 (ja) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | 環状ペプチド化合物の医薬用途 |
| WO2022234850A1 (ja) | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | 環状ペプチド化合物を含む製剤及びその製造方法 |
| KR102582225B1 (ko) | 2021-05-07 | 2023-09-22 | 추가이 세이야쿠 가부시키가이샤 | 환상 펩타이드 화합물을 포함하는 제제 및 그의 제조 방법 |
| KR20230017916A (ko) | 2021-05-07 | 2023-02-06 | 추가이 세이야쿠 가부시키가이샤 | 환상 펩타이드 화합물의 의약 용도 |
| KR20230003614A (ko) | 2021-05-07 | 2023-01-06 | 추가이 세이야쿠 가부시키가이샤 | N-치환 아미노산 잔기를 포함하는 환상 화합물의 제조 방법 |
| JP7181442B1 (ja) * | 2021-05-07 | 2022-11-30 | 中外製薬株式会社 | 環状ペプチド化合物を含む製剤及びその製造方法 |
| JP7179241B1 (ja) * | 2021-05-07 | 2022-11-28 | 中外製薬株式会社 | 環状ペプチド化合物の医薬用途 |
| WO2022234864A1 (ja) * | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | N-置換アミノ酸残基を含む環状化合物の製造方法 |
| KR102666765B1 (ko) | 2021-05-07 | 2024-05-16 | 추가이 세이야쿠 가부시키가이샤 | 환상 펩타이드 화합물의 의약 용도 |
| WO2022234851A1 (ja) | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | 変異型ras(g12d)に対する選択的結合性を示す結合分子 |
| US12312379B2 (en) | 2021-05-07 | 2025-05-27 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing cyclic compounds comprising N-substituted amino acid residues |
| KR20230169072A (ko) | 2021-05-07 | 2023-12-15 | 추가이 세이야쿠 가부시키가이샤 | N-치환 아미노산 잔기를 포함하는 환상 화합물의 제조방법 |
| KR20240074875A (ko) | 2021-05-07 | 2024-05-28 | 추가이 세이야쿠 가부시키가이샤 | 환상 펩타이드 화합물의 의약 용도 |
| KR20230020548A (ko) | 2021-05-07 | 2023-02-10 | 추가이 세이야쿠 가부시키가이샤 | 환상 펩타이드 화합물을 포함하는 제제 및 그의 제조 방법 |
| KR20230169093A (ko) | 2021-05-07 | 2023-12-15 | 추가이 세이야쿠 가부시키가이샤 | 환상 펩타이드 화합물을 포함하는 제제 및 그의 제조 방법 |
| KR20240005777A (ko) | 2021-05-07 | 2024-01-12 | 추가이 세이야쿠 가부시키가이샤 | Hras 및 nras에 대해서 선택적인 kras 저해 작용을 갖는 환상 화합물 |
| WO2022234853A1 (ja) | 2021-05-07 | 2022-11-10 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物 |
| JP7165289B1 (ja) * | 2021-05-07 | 2022-11-02 | 中外製薬株式会社 | N-置換アミノ酸残基を含む環状化合物の製造方法 |
| KR20240082357A (ko) | 2021-09-30 | 2024-06-10 | 추가이 세이야쿠 가부시키가이샤 | 광개열성 부위를 갖는 화합물, 연결체 및 이를 이용한 스크리닝 방법 |
| WO2023054636A1 (ja) | 2021-09-30 | 2023-04-06 | 中外製薬株式会社 | 光開裂性部位を有する化合物、連結体及びこれを用いたスクリーニング方法 |
| EP4609875A2 (en) | 2021-10-13 | 2025-09-03 | Chugai Seiyaku Kabushiki Kaisha | Composition containing peptide compound and surfactant |
| EP4609875A3 (en) * | 2021-10-13 | 2025-12-03 | Chugai Seiyaku Kabushiki Kaisha | Composition containing peptide compound and surfactant |
| EP4166133A1 (en) | 2021-10-13 | 2023-04-19 | Chugai Seiyaku Kabushiki Kaisha | Composition containing peptide compound and surfactant |
| KR20240070705A (ko) | 2021-10-13 | 2024-05-21 | 추가이 세이야쿠 가부시키가이샤 | 펩타이드 화합물 및 계면활성제를 포함하는 조성물 |
| JP2025503500A (ja) * | 2021-12-24 | 2025-02-04 | セントセル リミテッド | セストリン-mapk複合体阻害剤 |
| WO2023190283A1 (ja) | 2022-03-28 | 2023-10-05 | 中外製薬株式会社 | ポリオキシエチレン構造を含む油性成分を含有する化合物製剤 |
| KR20240169679A (ko) | 2022-04-08 | 2024-12-03 | 추가이 세이야쿠 가부시키가이샤 | 펩타이드 화합물의 제조 방법 |
| WO2023195516A1 (ja) | 2022-04-08 | 2023-10-12 | 中外製薬株式会社 | ペプチド化合物の製造方法 |
| EP4406546A4 (en) * | 2022-05-02 | 2025-10-08 | Chugai Pharmaceutical Co Ltd | COMPOSITION CONTAINING A PEPTIDE COMPOUND FOR USE WITH A SURFACTANT |
| KR20250002095A (ko) | 2022-05-02 | 2025-01-07 | 추가이 세이야쿠 가부시키가이샤 | 계면활성제와 병용하기 위한 펩타이드 화합물을 포함하는 조성물 |
| WO2023214509A1 (ja) | 2022-05-02 | 2023-11-09 | 中外製薬株式会社 | 界面活性剤と併用するためのペプチド化合物を含む組成物 |
| WO2023214577A1 (ja) | 2022-05-02 | 2023-11-09 | 中外製薬株式会社 | ジケトピペラジン形成による欠損を抑制するペプチド合成方法 |
| KR20250008758A (ko) | 2022-05-06 | 2025-01-15 | 추가이 세이야쿠 가부시키가이샤 | Hras 및 nras에 대한 선택적인 kras 억제 효과를 갖는 고리 화합물 |
| WO2023214576A1 (ja) | 2022-05-06 | 2023-11-09 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物 |
| US12410212B2 (en) | 2022-05-06 | 2025-09-09 | Chugai Seiyaku Kabushiki Kaisha | Cyclic compound having selective KRAS inhibitory effect on HRAS and NRAS |
| WO2023234425A1 (ja) | 2022-06-03 | 2023-12-07 | ペプチドリーム株式会社 | アミノ酸活性エステル及びその塩 |
| WO2024024965A1 (ja) | 2022-07-29 | 2024-02-01 | 中外製薬株式会社 | O-置換セリン誘導体の製造方法 |
| WO2024043249A1 (ja) | 2022-08-23 | 2024-02-29 | 富士フイルム株式会社 | 環状ペプチドまたはその塩、およびmdmx阻害剤 |
| WO2024043251A1 (ja) | 2022-08-23 | 2024-02-29 | 富士フイルム株式会社 | 環状ペプチドの細胞膜透過性の予測方法 |
| WO2024043250A1 (ja) | 2022-08-23 | 2024-02-29 | 富士フイルム株式会社 | 環状ペプチドまたはその塩、およびmdmx阻害剤 |
| KR20250088544A (ko) | 2022-10-20 | 2025-06-17 | 추가이 세이야쿠 가부시키가이샤 | 고리형 펩티드 결정을 제조하는 방법 |
| WO2024085235A1 (ja) | 2022-10-20 | 2024-04-25 | 中外製薬株式会社 | 環状ペプチドの結晶の製造方法 |
| WO2024096023A1 (ja) | 2022-11-01 | 2024-05-10 | 中外製薬株式会社 | ジベンゾフルベンまたはジベンゾフルベン誘導体の除去方法 |
| WO2024101386A1 (ja) | 2022-11-09 | 2024-05-16 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物 |
| WO2024101402A1 (ja) | 2022-11-09 | 2024-05-16 | 中外製薬株式会社 | Hrasおよびnrasに対して選択的なkras阻害作用を有する環状化合物を含む医薬組成物 |
| KR20240082196A (ko) | 2022-11-09 | 2024-06-10 | 추가이 세이야쿠 가부시키가이샤 | Hras 및 nras에 대해서 선택적인 kras 저해 작용을 갖는 환상 화합물을 포함하는 의약 조성물 |
| WO2024128195A1 (ja) | 2022-12-12 | 2024-06-20 | 中外製薬株式会社 | 特定位置に酸素原子を有するアミノ酸を翻訳する工程を含む、ペプチドの製造方法 |
| EP4628592A1 (en) | 2022-12-12 | 2025-10-08 | Chugai Seiyaku Kabushiki Kaisha | Peptide production method comprising step for translating amino acid having oxygen atom at specific position |
| WO2024126687A1 (en) * | 2022-12-16 | 2024-06-20 | Silence Therapeutics Gmbh | Nucleic acids conjugated to an albumin binding moiety |
| EP4631955A1 (en) | 2022-12-28 | 2025-10-15 | Chugai Seiyaku Kabushiki Kaisha | Purification method and production method of cyclic peptide |
| WO2024143514A1 (ja) | 2022-12-28 | 2024-07-04 | 中外製薬株式会社 | 環状ペプチドの精製法及び製造法 |
| WO2024195801A1 (ja) | 2023-03-20 | 2024-09-26 | 中外製薬株式会社 | 環状ペプチドの共結晶の製造方法 |
| KR20250160895A (ko) | 2023-03-20 | 2025-11-14 | 추가이 세이야쿠 가부시키가이샤 | 고리형 펩타이드의 공결정을 제조하는 방법 |
| EP4663653A1 (en) | 2023-03-20 | 2025-12-17 | Chugai Seiyaku Kabushiki Kaisha | Method for producing eutectic of cyclic peptide |
| WO2024209857A1 (ja) * | 2023-04-03 | 2024-10-10 | 富士フイルム株式会社 | 核酸ディスプレイライブラリの作製方法、スクリーニング方法、及びポリペプチドの作製方法 |
| WO2024214825A1 (ja) | 2023-04-14 | 2024-10-17 | 中外製薬株式会社 | 複数種類の標的分子との複合体を形成し得る目的分子のスクリーニング方法、及び当該スクリーニング方法を含む目的分子の製造方法 |
| EP4674859A1 (en) | 2023-04-14 | 2026-01-07 | Chugai Seiyaku Kabushiki Kaisha | Method for screening molecules of interest capable of forming complexes with plurality of types of target molecules, and method for producing molecules of interest including screening method |
| WO2024219446A1 (ja) | 2023-04-19 | 2024-10-24 | 中外製薬株式会社 | 環状ペプチド化合物及び界面活性剤を含む組成物 |
| EP4663198A1 (en) | 2023-04-19 | 2025-12-17 | Chugai Seiyaku Kabushiki Kaisha | Composition containing cyclic peptide compound and surfactant |
| WO2025070753A1 (ja) | 2023-09-28 | 2025-04-03 | 中外製薬株式会社 | 混合酸無水物法を用いたペプチド化合物の製造方法 |
| WO2025079650A1 (ja) | 2023-10-11 | 2025-04-17 | 中外製薬株式会社 | 複数のライブラリを用いたペプチドのスクリーニング方法 |
| WO2025100405A1 (ja) | 2023-11-06 | 2025-05-15 | 中外製薬株式会社 | 細胞膜透過性と代謝安定性を有する大環状ペプチド化合物、及びこれを含むライブラリ |
| WO2025143077A1 (ja) | 2023-12-27 | 2025-07-03 | 中外製薬株式会社 | スタンドアローン型縮合剤を用いたペプチド化合物の製造方法 |
| CN119708146A (zh) * | 2024-12-30 | 2025-03-28 | 五邑大学 | 一种环肽Lagunamide D的制备方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7297027B2 (ja) | ペプチド化合物の環化方法 | |
| JP7351968B2 (ja) | チオール基をアミノ基近傍に有するアミノ酸をn末端に持つ非環状ペプチド-核酸複合体、そのライブラリー、およびそれから誘導される環状ペプチド-核酸複合体ライブラリーの製造方法 | |
| JP7526243B2 (ja) | 無細胞翻訳系におけるペプチドの合成方法 | |
| CN110869544B (zh) | 膜透过性高的环状肽化合物及包含其的文库 | |
| JP6754997B2 (ja) | 大環状ペプチド、その製造方法、及び大環状ペプチドライブラリを用いるスクリーニング方法 | |
| Chen et al. | Synthetic strategies for polypeptides and proteins by chemical ligation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12861199 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013551857 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14368564 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012861199 Country of ref document: EP |