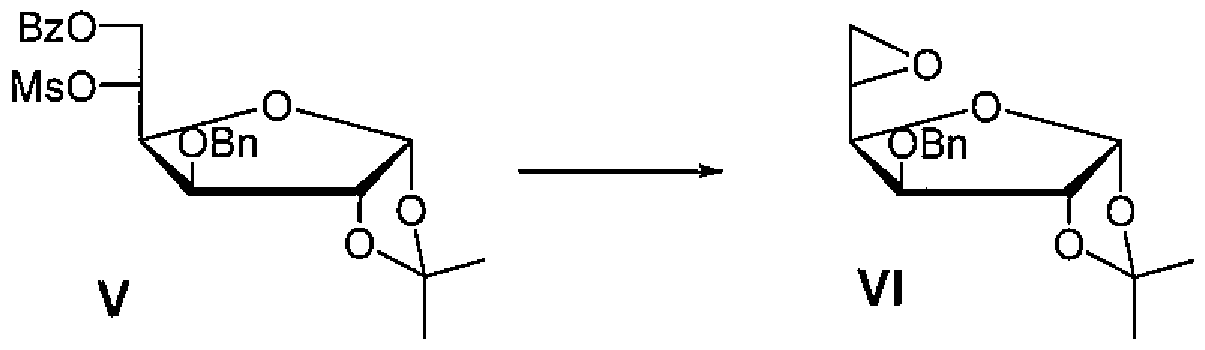—种制备 3-0-苄基 -1, 2-0-异亚丙基 -α-L-呋喃艾杜糖的方法 技术领域 本发明涉及制药领域,更具体地说,本发明涉及制备 3-0-苄基 -1,2-0- 异亚丙基 -a-L-呋喃艾杜糖的新方法。 背景技术 哺乳动物体内的粘多糖包括肝素、 硫酸肝素、 和^酸皮肤素等, 在多种生理过程中起重要的作用, L-艾杜糖醛酸单元是它们的关键组 成部分。 L-艾杜糖片段的合成一直是糖化学中的难点, 制约着肝素类 分子人工合成研究工作的开展,也成为药物磺达肝癸钠合成中的瓶颈 之一 0
现有的 L-艾杜糖^■生物的制备方法中, 包括通过对葡萄糖 5,6 位环外默键的硼氢化氧化翻转 5位羟基(Hinou, Hiroshi; Kurosawa, Hidehiro; Matsuoka, Koji; Terunuma, Daiyo; Kuzuhara, Hiroyoshi; Tetrahedron Lett. 1999, 40, 1501; Hung, S. C; Thopate, S. R.; Chi, F. C; Chang, S. W.; Lee, J. C; Wang, C. C; Wen, Y. S. J. Am. Chem. Soc. 2001: 123, 3153 ); 5 位溴代葡萄糖醛酸的自由基反应异构化来翻转构型 (Chiba, T.; Sinay, P. Carbohydr. Res. 1986, 151, 379); 利用氰基, (PhS)3CLi 等负离子对醛的不对称加成构建 5 位手性 (Lubineau, A.;
Gavard, 0.; Alais, J,; Bonnaffe, D. Tetrahedron Lett. 2000, 41, 307;Jarosz, S. Carbohydr. Res. 1987, 166, 211;
Kapeller, D. C; Hammerschmidt, F. Tetrahedron 2010, 66, 591);葡 萄糖或葡萄糖醛酸酯 5 位羟基三氟曱烷磺酸酯或曱烷磺酸酯的亲核 取代反应翻转 5 位羟基 (Orgueira, H. A.; Bartolozzi, A.; Schell, P.; Litjens, R. E. J. N.; Palmacci, E. R.; Seeberger, P. H. Chem. Eur. J. 2003, 9, 140; Ke, W.; Whitfield, D. M.; Gill, M.; Larocque, S.; Yu, S. Tetrahedron Lett. 2003, 44, 7767; Tadano, K., Idogaki, Y., Yamada, H., Suami, T. J. Org. Chem. 1987, 52, 1201; Ban-oca, N.; Jacquinet, J. C. Carbohydr. Res. 2000, 329, 667.)> 这些方法产率低, 路线长, 不易操 作, 成本高, 后续区分性的保护基操作也比较困难。 也有利用环氧的 酸性开环翻转 5位羟基( Van Boeckel, C. A. A.; Beetz, T.; Vos, J. N.; De Jong, A. J. M.; Van Aelst, S. R; Van den Bosch, R. H.; Mertens, J. M. R.; Van der Vlugt, F. A. J. Carbohydr. Chem. 1985, 4, 293 ), 但在翻转后的 环氧开环上未能找到有效, 简便, 适合大规模工业化生产的方法。 发明内容 本发明提供了一种 3-0-苄基 -1,2-0-异亚丙基 -α-L-呋喃艾杜糖 VII 的制备方法, 谅方法包括: 将化合物 VI在碱性条件下选择性开环得 到化合物 VII:
在一些实施方案中, 化合物 VI在碱性条件下开环得到化合物 VII 的反应中, 其中反应温度可以为 - 30°C ~+100°C, 优选 0°C~+10(TC, 更优选 +6( +80Γ。 碱可以优选无机碱, 无机碱优选氢氧化钾、 氢氧 化钠、 氢氧化锂, 氢氧化镁、 氢氧化钙、 氢氧化钡, 反应溶剂优选为 水、 曱醇、 乙醇、 丙醇、 叔丁醇、 四氢呋喃、 二氧六环、 乙腈、 Ν,Ν- 二甲基甲酰胺、 二甲亚砜, 或者它们的任意混合物。 粗产品可经重结 晶精制
在更进一步实施方案中,化合物 VI可以通过以下方法进行制备: 化合物 V在碱性条件下进行环化反应, 得到 5位构型翻转的 5, 6位 环氧化合物 VI:
在一些实施方案中, 化合物 V在碱性条件下形成 5位构型翻转 的 5,6位环氧化合物 VI的反应中, 其中反应温度可以为 -30°C ~+60 °C, 优选 0°C~+40°C, 更优选 +20°C ~+30°C。 碱的用量可以为化合 物 V的 1~ 10倍(物质的量比), 碱可以优选无机碱、 有机碱, 其中 无机碱优选氢氧化钾、 氢氧化钠、 氢氧化锂, 氢氧化镁、 氢氧化钙、 氢氧化钡, 其中有机碱优选吡啶、 三乙胺、 C^C4低级醇钠或 -
低级醇钾, 其中 低级醇钠或 crc4低级醇钟优选曱醇钠、 乙醇 钠、 叔丁醇钠、 叔丁醇钟, 反应溶剂优选为水、 甲醇、 乙醇、 丙醇、 叔丁醇、 四氢呋喃、二氧六环、 乙腈、 Ν,Ν-二曱基曱酰胺、二甲亚砜、 二氯甲烷, 或者它们的任意混合物。反应产物无需后处理可以直接进 行下一步反应, 或者后处理的粗产品可以直接用于下一步反应。在一 些实施方案中, 所述反应可以与上述开环反应在一个反应体系中进 行。
在更进一步实施方案中, 化合物 V可以通过以下方法进行制备: 利用空间位阻, 先对化合物 III的 6位羟基在碱性条件下进行苯曱酰 基保护得到化合物 IV, 然后对化合物 IV的 5位羟基在碱性条件下进 行甲烷磺酰基保护得到化合物 V:
在一些实施方案中, 选择性的在化合物 III 的 6位羟基在碱性条 件下进行苯甲酰基保护制备化合物 IV的反应中, 其中反应温度可以 为 -30°C ~ +60°C ,苯曱酰氯的用量可以为化合物 ΠΙ的 0.6 - 2.0倍(物 质的量比), 分批次加入; 反应所用的碱优选有机碱, 更优选吡啶、 取代吡啶、 哌啶, 或 d-C4脂肪胺, 反应的溶剂可以优选 -C6单卤
或多 [¾炕烃、 四氢呋喃、 乙腈、吡啶、二氧六环、 N,N-二甲基甲酰胺、 二甲亚砜, 或者它们的任意混合物。
在一些实施方案中, 化合物 IV的 5位羟基在碱性条件下进行甲 烷磺酰基保护制备化合物 V的反应中, 其中反应温度可以为 -3CTC - 60°C , 曱烷横酰氯的用量可以为化合物 III的 0.6 ~ 3倍(物质的量 比), 反应所用的碱优选有机碱, 更优选吡啶、 取代吡啶、 哌啶, 或 CrC4脂肪胺, 反应的溶剂可以优选 CrC6单! ¾或多! ¾烷烃、 四氢呋 喃、 乙腈、 吡啶、 二氧六环、 Ν,Ν-二甲基甲酰胺、 二甲亚砜, 或者它 们的任意混合物。 粗产品可经重结晶精制。
其中所述的化合物 ΠΙ的合成可参考文献 J. Org. Chem. 2003, 68, 7559的方法制备。
在本发明的上下文中, Bn表示苄基, Ms表示甲坑磺酰基, Bz 表示苯曱酰基。
本发明的一个目的在于提供一种 3-0-苄基 -1,2-0-异亚丙基 -a-L- 呋喃艾杜糖的制备方法, 包括以下步骤:
( 1 )利用空间位阻, 先对化合物 ΙΠ的 6位羟基在碱性条件下进
行苯曱酰基保护得到化合物 IV, 然后对化合物 IV的 5位羟基在碱性 条件下进行曱烷磺酰基保护得到化合物 V;
( 2 )化合物 V在碱性条件下进行环化反应, 得到 5位构型翻转 的 5, 6位环氧化合物 VI;
( 3 )化合物 VI在碱性条件下选择性开环得到化合物 VII。
在一些实施方案中, 在步骤(1 )中, 选择性的在化合物 III 的 6 位羟基在碱性条件下进行苯曱酰基保护制备化合物 IV的反应中 , 其 中反应温度为 -30°C ~ +60°C,苯甲酰氯的用量为化合物 ΠΙ的 0.6 - 2.0 倍(物质的量比), 分批次加入; 反应所用的碱优选有机碱, 更优选 吡啶、取代吡啶、哌啶, 或 CrC4脂肪胺, 反应的溶剂可以优选 d-C6 单卤或多卤烷烃、 四氢呋喃、 乙腈、 吡啶、 二氧六环、 Ν,Ν-二甲基曱 酰胺、 二曱亚砜, 或者它们的任意混合物。 将所得的化合物 IV的 5 位羟基在碱性奈件下进行曱烷磺酰基保护制备化合物 V的反应中, 其中反应温度为 -30 °C ~ 60 °C , 曱烷磺酰氯的用量为化合物 ΠΙ 的 0.6 ~ 3倍(物质的量比), 反应所用的碱优选有机碱, 更优选吡啶、 取代吡啶、 哌啶, 或 -C4脂肪胺, 反应的溶剂可以优选 Cr C6单卤 或多卤烷烃、 四氢呋喃、 乙腈、吡啶、二氧六环、 N,N-二甲基甲酰胺、 二甲亚砜, 或者它们的任意混合物。 粗产品可经重结晶精制。
在一些实施方案中, 在步骤(2 ) 中, 化合物 V在碱性条件下形 成 5位构型翻转的 5,6位环氧化合物 VI的所应中,其中反应温度为 -30 ~ ÷60°C , 碱的用量为化合物 V的 1 ~ 10倍(物质的量比), 碱可 以优选无机碱、 有机碱, 其中无机碱优选氢氧化钾、 氢氧化钠、 氢氧
化锂, 氢氧化镁、 氢氧化钙、 氢氧化钡, 其中有机碱优选吡啶、 三乙 胺、 d_C4低级醇钠或 Cr C4低级醇钾, 其中 C C4低级醇钠或 crc4 低级醇钾优选甲醇钠、 乙醇钠、 叔丁醇钠、 叔丁醇钾, 反应溶剂优选 为水、 曱醇、 乙醇、 丙醇、 叔丁醇、 四氢呋喃、 二氧六环、 乙腈、
Ν,Ν-二甲基曱酰胺、 二曱亚砜、 二氯曱烷, 或者它们的任意混合物。 反应溶液无需后处理可以直接进行下一步反应,或者后处理的粗产品 直接用于下一步反应。
在一些实施方案中, 在步骤(3 )中, 化合物 VI在碱性条件下开 环得到化合物 VII的反应中, 其中反应温度为 -30°C ~ +100°C, 碱的 用量为化合物 V的 1 ~ 10倍(物质的量比), 碱可以优选无机碱, 无 机碱优选氢氧化钾、 氢氧化钠、 氢氧化锂, 氢氧化鎂、 氢氧化钙、 氢 氧化钡, 反应溶剂优选为水、 曱醇、 乙醇、 丙醇、叔丁醇、 四氢呋喃、 二氧六环、 乙腈、 N,N-二曱基曱酰胺、 二甲亚砜, 或者它们的任意混 合物。 粗产品可经重结晶精制。
其中步骤( 1 )所述的化合物 ΠΙ的合成可参考文献 J. Org. Chem. 2003, 68, 7559的方法制备。 相对于现有技术, 本发明的优点在于:
在现有的合成艾杜糖衍生物的技术中, 通过对葡萄糖 5,6位环外 双键的硼氢化氧化翻转 5位羟基的方法路线长, 操作繁瑣, 产率低, 纯化困难; 5位溴代葡萄糖醛酸的自由基反应异构化来翻转构型的方 法效率低, 使用了昂贵且毒性大的试剂; 利用氰基, (PhS)3CLi 等负
离子对醛的不对称加成构建 5位手性的方法路线长,需要苛刻的反应 条件, 而且反应的立体选择性不理想, 产物純化困难; 葡萄糖或葡萄 糖醛酸酯 5 位羟基三氟甲烷磺酸酯或曱烷磺酸酯的亲核取代反应翻 转 5位羟基的方法反应产率低, 副反应严重, 纯化困难; 利用环氧的 酸性开环翻转 5位羟基的方法在翻转后的环氧开环上未能找到有效, 简便, 适合大规模工业化生产的方法, 产物纯化困难。 本发明的有益 效果是: 工艺路线短, 产率高, 反应条件温和, 操作过程简单, 避免 使用昂贵且毒性大的试剂, 成本低廉, 中间体及产品可通过重结晶的 方法纯化, 适合于工业化大生产。 具体实施方式 实施例一:
制备 3-0-苄基 -1,2-0-异亚丙基- cx -D-呋喃葡萄糖 III
a步骤:制备 3-0-苄基 -1, 2: 5, 6-二 -0-异亚丙基 - CC -D-呋喃葡萄糖 氩气保护下, 在 5 L的四口反应瓶内依次加入 1升四氢呋喃, 64 克 60%的氢化钠,冰水浴冷却至 0。C至 5°C。滴加 1, 2: 5, 6-二 -0-异亚 丙基- cc -D-呋喃葡萄糖 315克和 1升四氢呋喃的混合溶液, 控温 0°C 至 10°C , 在 0°C左右保温反应五小时。 滴加 220毫升溴化苄, 控温 0
°c至 lcrc, 加入 20克四丁基溴化铵, 撤去水水浴, 自然升至室温, 继续搅拌直至反应完全。滴加少量曱醇淬灭反应,减压浓缩除去溶剂, 加入乙酸乙酯溶解, 并用水洗涤 3次, 无水疏酸钠干燥, 浓缩得粗产 品 540克, 直接投下一步反应。
b步骤: 制备 3- O-苄基- 1,2-0-异亚丙基- a -D-呋喃葡萄糖 III 称取 a步骤所得的 3-0-苄基 -1, 2: 5, 6-二- 0-异亚丙基 - α -D-呋喃 葡萄糖粗产品 540克于 3升单口瓶中, 注入 60% 的乙酸水溶液 1.8 升, 室温搅拌过夜, TLC监测反应完全。 加入 1.8升水, 石油醚洗涤 三次, 除去低极性杂质, 水相用碳酸氢钠固体中和, 再用乙酸乙酯萃 取三次,无水硫酸钠干燥,浓缩得粗产品 342克( HPLC纯度为 91% ), 两步反应产率合计 91%。 无须进一步纯化, 直接进行下一步反应。 少 量样品柱层析纯化后作 MS、 ^ NMR和 13C NMR分析。
ESI- MS ( m/z ) : 333.1 [M+Na]+; lR NMR (400 MHz, CDC13 ): δ 7.32-7.24 (m, 5 H), 5.88 (d, J = 3.2 Hz, 1 H), 4.61 (m, 2 H), 4.56 (s, 1 H), 4.12- 4.08 (m, 2 H), 4.00 (m, 1 H), 3.80- 3.60 (m, 2 H), 3.27 (br, 2 H), 1.45 (s, 3 H), 1.27 (s, 3 H); 13C NMR (100 MHz, CDC13): δ 137.46, 128.54, 127.98, 127.72, 111.71, 106.09, 82.19, 81.88, 79.96, 77.52, 76.88, 72.23, 68.96, 64.26, 26.88, 26.17. 实施例二:
制备 6-0-苯甲酰基- 3-0-苄基 -1,2-0-异亚丙基 - a 呋喃葡萄糖 IV和 6-0-苯曱酰基 -3-0-苄基 -1,2-0-异亚丙基 -5-0-甲烷磺酰基 - a -D- 呋喃葡萄糖 V
将 96.5克化合物 III溶于 500毫升吡啶中, 冰盐浴冷却至 -10°C , 分批滴加 36.89 亳升苯曱酰氯的 40毫升二氯曱烷溶液。 反应温度控 制在 -10 ~ 0°C。 反应完全后, 反应混合物无须分离纯化直接投下一步
反应。
冰水浴冷却至 o°C ,向上述反应混和液中緩慢滴加甲烷磺酰氯 26 亳升, 搅拌过夜。 反应完全后, 将反应液倒入约 2升 55 °C ~ 6(TC温 水中, 冷却后析出白色晶体, 过滤干燥, 再用乙醇重结晶。 干燥得 114.8克化合物 V, 两步收率为 75%。
化合物 V的 MS、 'Η NM 和 13C NMR测定数椐如下:
ESI-MS ( m/z ) : 493.1 [Μ+Η]+; 'Η NMR (400 MHz, CDC13): δ 8.07 (d, J = 7.2 Hz, 2 H), 7.56 (t, J = 7.6 Hz, 1 H), 7.28-7.43 (m, 7 H), 5.91 (d, J = 3.6 Hz, 1 H), 5.41 (m, 1 H), 4.92 (d, J = 12.4 Hz, 1 H), 4.70 (d, J = 10.8 Hz, 1 H), 4.62 (s, 1 H), 4.60 (d, J = 7.6 Hz, 1 H), 4.50- 4.40 (m, 2 H), 4.14 (d, J =2.8 Hz, 1 H), 2.98 (s, 3 H), 1.50 (s, 3 H), 1.31 (s,3 H); 13C NMR (100 MHz, CDC13): δ 165.57, 137.16, 133.31, 129.75, 129.69, 128.53, 128.41 , 128.36, 128.12, 128.08, 112.30, 106.41 , 81.58, 81.15, 78.29, 77.46, 76.14, 76.82, 75.37, 72.37, 64.10, 39.12, 26.88, 26.32. 实施例三:
制备 5,6-环氧- 3-0-苄基 -1,2-0-异亚丙基- a -L-呋喃艾杜糖 VI 将 50克化合物 V溶于 300 毫升二氧六环中,室温下往反应液中 緩慢加入 29克氢氧化钾和 100毫升水的溶液, 滴加完成后在室温搅 拌直至反应完全。将反应混合物用冰水浴冷却,緩慢滴加盐酸中和至 中性。 减压除去低沸点溶剂, 所得混合物用乙酸乙酯萃取。 合并有机 相, 依次用饱和碳酸氢钠溶液、 饱和氯化钠溶液洗涤, 用无水硫酸钠
干燥, 浓缩后得化合物 VI粗品 30.5克。 该粗品无需进一步纯化直接 进行下一步反应。 实施例四
制备 5,6-环氧 -3-0-苄基 -1,2-0-异亚丙基- cc -L-呋喃艾杜糖 VI 将 20克化合物 V溶于 200 毫升二氯甲烷和 100亳升叔丁醇的混 合溶剂中, 冰水浴冷却, 往反应液中緩慢加入 10.5克叔丁醇钾, 完 成后在室温搅拌直至反应完全。将反应混合物用冰水浴冷却,緩慢滴 加盐酸中和至中性。 所得混合物用乙酸乙酯萃取。 合并有机相, 依次 用饱和碳酸氢钠溶液、 饱和氯化钠溶液洗涤, 用无水硫酸钠干燥, 浓 缩后柱层析纯化得 11.4克化合物 VI。
化合物 VI的 MS、 iH NMR和 13C NMR测定数据如下:
ESI-MS { ml 7. ) : 331.0 [M+K]+; !H NMR (400 MHz, CDC13): δ 7.37-7.30 (m, 5 H), 6.00 (d, J = 4.0 Hz, 1 H), 4.74 (d, J = 12.4 Hz, 1 H), 4.64 (d, J = 4.0 Hz, 1 H), 4.51 (d, J = 12.4 Hz, 1 H), 3.97 (d, J = 3.2 Hz, 1 H), 3.80 (dd, Ji = 6.0 Hz, J2 = 3.2 Hz, 1 H), 3.27 (m, 1 H), 2.75 (t, J = 4.4 Hz, 1 H), 2.53(m5 1 H), 1.45 (s, 3 H), 1.32 (s,3 H); ,3C NMR (100 MHz, CDC13): δ 137.16, 128.39, 127.92, 127.56, 111.77, 105.33, 82.58, 82.29, 81.99, 71.80, 50.05, 43.02, 26.73, 26.20. 实施例五
制备 3-0-苄基 -1,2-0-异亚丙基- a -L-呋喃艾杜糖 VII
将实施例三所得化合物 VI粗品 30.5克溶解于 200毫升二曱亚砜 中, 緩慢加入 30.3克氢氧化钾和 100毫升水的溶液。 滴加完成后加 热至 60 - 70 °C反应, 直至反应完全。 用水稀释, 二氯曱烷萃取, 饱 和氯化钠溶液洗涤,无水硫酸钠干燥,减压除去溶剂。粗产品重结晶, 干燥后得 25.2克化合物 VII。
化合物 VII的 MS、 'H MR和 13C NMR测定数据如下:
ESI-MS ( m/z ) : 333.1 [M+Na]+; Ή NMR (400 MHz, CDC13 ): δ 7.34- 7.27 (m, 5 H), 5.95 (d, J = 3.2 Hz, 1 H), 4.63-4.44 (m, 3 H), 4.20 (m,
1 H), 4.04 (m, 1 H), 3.97 (d, J = 3.2 Hz, 1 H,), 3.65-3.50 (m, 2 H), 3.51 (s,
2 H), 1.45 (s, 3 H), 1.29 (s, 3 H); 13C NMR (100 MHz, CDC13): δ 138.88, 128.66, 128.10, 127.83, 111.76, 104.72, 82.40, 82.17, 80.17, 77.69, 76.98, 71.72, 70.60, 63.22, 26.70, 26.26. 实施例六
制备 5,6-环氧 -3-0-苄基 -1,2-0-异亚丙基 - cc -L-呋喃艾杜糖 VI和 3-0-苄基 -1,2-0-异亚丙基- -L-呋喃艾杜糖 VII
将 114.8克化合物 V溶于 500 毫升的 Ν,Ν-二曱基甲酰胺中, 室 温下往反应液中緩慢加入 65.37克氢氧化钾和 262毫升水的溶液, 反 应液由澄清变棕黑色, 直至反应完全。反应产物无须分离純化直接进 行下一步反应。
将上述反应液直接升温至 60 ~ 70°C , 继续进行反应, 直至反应 完全。 用水稀释, 二氯甲烷萃取, 饱和氯化钠溶液洗涤, 无水硫酸钠
干燥, 减压除去溶剂。 粗产品重结晶, 干燥后得 55.61克化合物 VII。 化合物 VII的 MS、 Ή NMR和 13C NMR测定数据如下:
ESI-MS ( m/z ) : 333.1 [M+Na]+; Ή NMR (400 MHz, CDC13 }: δ
7.34- 7.27 (m, 5 H), 5.95 (d, J = 3.2 Hz, 1 H), 4.63-4.44 (m, 3 H), 4.20 (m,
1 H), 4.04 (m, 1 H), 3.97 (d, J = 3.2 Hz, 1 H,), 3.65-3.50 (m, 2 H), 3.51 (s,
2 H), 1.45 (s, 3 H), 1.29 (s, 3 H); 13C NMR (100 MHz, CDC13): δ 138.88, 128.66, 128.10, 127.83, 111.76, 104.72, 82,40, 82.17, 80.17, 77.69, 76.98, 71.72, 70.60, 63.22, 26.70, 26.26. 实施例七
制备 5,6-环氧 -3-0-苄基 -1,2-0-异亚丙基 - α -L-呋喃艾杜糖 VI和 3-0-苄基 -1,2-0-异亚丙基- a 呋喃艾杜糖 VII
将 80克化合物 V溶于 350 亳升的二曱亚砜中,室温下往反应液 中緩慢加入 46克氢氧化钾和 180毫升水的溶液。 室温搅拌直至反应 完全。 反应产物无须分离纯化直接进行下一步反应。
将上述反应液直接升温至 60 ~ 70°C , 继续进行反应, 直至反应 完全。 用水稀释, 二氯曱烷萃取, 饱和氯化钠溶液洗涤, 无水硫酸钠 干燥, 减压除去溶剂。 粗产品重结晶, 干燥后得 41克化合物 VII。
化合物 VII的 MS、 'H NMR和 13C NMR测定数据如下:
ESI-MS ( m/z ) : 333.1 [M+Na]+; ]H NMR (400 MHz, CDC13 ): δ 7.34- 7.27 (m, 5 H), 5.95 (d, J = 3.2 Hz, 1 H), 4.63-4.44 (m, 3 H), 4.20 (m, 1 H), 4.04 (m, 1 H), 3.97 (d, J = 3.2 Hz, 1 H,), 3.65-3.50 (m, 2 H), 3.51 (s,
2 H), 1.45 (s, 3 H), 1.29 (s, 3 H); 13C NMR (100 MHz, CDC13): δ 138.88, 128.66, 128.10, 127.83, 111.76, 104.72, 82.40, 82.17, 80.17, 77.69, 76.98, 71.72, 70.60, 63.22, 26.70, 26.26. 实施例八
制备 5,6-环氧 -3-0-苄基 -1,2- Ο-异亚丙基- α -L-呋喃艾杜糖 VI和 3-0-苄基- 1 ,2- 0-异亚丙基- oc -L-呋喃艾杜糖 VII
将 82克化合物 V溶于 350 毫升的二氧六环中,室温下往反应液 中緩慢加入 46克氢氧化钾和 180毫升水的溶液。 室温搅拌直至反应 完全。 反应产物无须分离纯化直接进行下一步反应。
将上述反应液直接升温至 60 ~ 70°C , 继续进行反应, 直至反应 完全。 用水稀释, 二氯甲炕萃取, 饱和氯化钠溶液洗涤, 无水硫酸钠 干燥, 减压除去溶剂。 粗产品重结晶, 干燥后得 38克化合物 VII。
化合物 VII的 MS、 iH NMR和 13C NMR测定数据如下:
ESI-MS C m/z ) : 333.1 [M+Na]+; Ή NMR (400 MHz, COC δ 7.34- 7.27 (m, 5 H), 5.95 (d, J = 3.2 Hz, 1 H), 4.63-4.44 (m, 3 H), 4.20 (m,
1 H), 4.04 (m, 1 H), 3.97 (d, J = 3.2 Hz, 1 H,), 3.65-3.50 (m, 2 H), 3.51 (s,
2 H), 1.45 (s, 3 H), 1.29 (s, 3 H); 13C NMR (100 MHz, CDC13): δ 138.88, 128.66, 128.10, 127.83, 111.76, 104,72, 82.40, 82.17, 80: 17, 77.69, 76.98, 71.72, 70.60, 63.22, 26.70, 26.26.