WO2013152608A1 - 一种二盐酸沙丙喋呤的合成方法 - Google Patents
一种二盐酸沙丙喋呤的合成方法 Download PDFInfo
- Publication number
- WO2013152608A1 WO2013152608A1 PCT/CN2012/087732 CN2012087732W WO2013152608A1 WO 2013152608 A1 WO2013152608 A1 WO 2013152608A1 CN 2012087732 W CN2012087732 W CN 2012087732W WO 2013152608 A1 WO2013152608 A1 WO 2013152608A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- molar ratio
- ratio
- acid
- hours
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
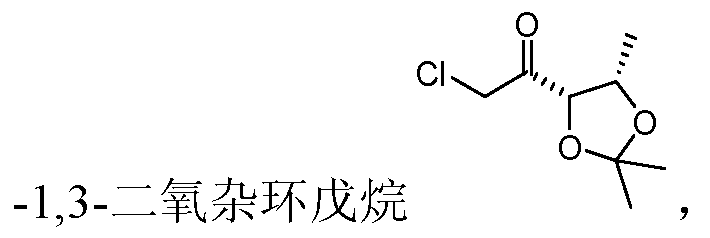
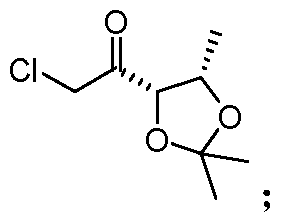
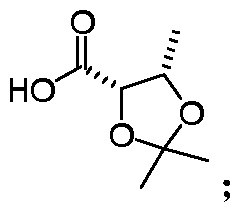
- 0 C[C@@]1OC(C)(C)OC1C(C*)=O Chemical compound C[C@@]1OC(C)(C)OC1C(C*)=O 0.000 description 5
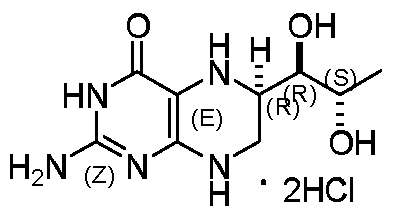
- PMGGEBQILGCQNW-NVNXEXLPSA-N C[C@@H]([C@@H](C(CN1)=NC2=C1N=C(NC(C)=O)NC2=O)O)O Chemical compound C[C@@H]([C@@H](C(CN1)=NC2=C1N=C(NC(C)=O)NC2=O)O)O PMGGEBQILGCQNW-NVNXEXLPSA-N 0.000 description 2
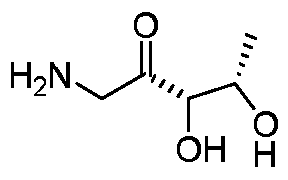
- PPFTYFJBDQDNLS-WWLRPLJCSA-N C[C@@H](C(C(CN)=O)O)O Chemical compound C[C@@H](C(C(CN)=O)O)O PPFTYFJBDQDNLS-WWLRPLJCSA-N 0.000 description 1
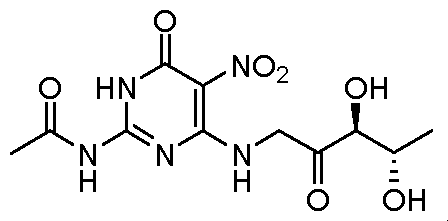
- YQEWJHNYUVUHAK-NVNXEXLPSA-N C[C@@H]([C@@H](C(CNC(N=C(NC(C)=O)NC1=O)=C1[N+]([O-])=O)=O)O)O Chemical compound C[C@@H]([C@@H](C(CNC(N=C(NC(C)=O)NC1=O)=C1[N+]([O-])=O)=O)O)O YQEWJHNYUVUHAK-NVNXEXLPSA-N 0.000 description 1
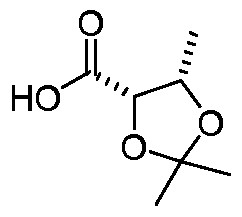
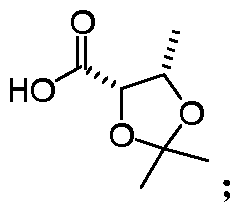
- HNLGBHQJNORDKW-WHFBIAKZSA-N C[C@@H]1OC(C)(C)O[C@@H]1C(O)=O Chemical compound C[C@@H]1OC(C)(C)O[C@@H]1C(O)=O HNLGBHQJNORDKW-WHFBIAKZSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
- C07D475/02—Heterocyclic compounds containing pteridine ring systems with an oxygen atom directly attached in position 4
- C07D475/04—Heterocyclic compounds containing pteridine ring systems with an oxygen atom directly attached in position 4 with a nitrogen atom directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/62—Preparation of compounds containing amino groups bound to a carbon skeleton by cleaving carbon-to-nitrogen, sulfur-to-nitrogen, or phosphorus-to-nitrogen bonds, e.g. hydrolysis of amides, N-dealkylation of amines or quaternary ammonium compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C221/00—Preparation of compounds containing amino groups and doubly-bound oxygen atoms bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C225/00—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones
- C07C225/02—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton
- C07C225/04—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being saturated
- C07C225/06—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being saturated and acyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/50—Three nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/48—Compounds containing oxirane rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/14—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D317/26—Radicals substituted by doubly bound oxygen or sulfur atoms or by two such atoms singly bound to the same carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/14—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D317/28—Radicals substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/32—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
Definitions
- the present invention relates to a method for synthesizing a medicament for treating phenylketonuria, and more particularly to a method for synthesizing sapropidine dihydrochloride.
- BACKGROUND OF THE INVENTION Sodium bromide hydrochloride, chemical name (6R)-2-amino-6-[(lR,2S)-l,2-dihydroxypropyl]-5,6,7,8-tetrahydro- 4 ( 1H) - hydrazine dihydrochloride, the molecular formula C 9 H 15 N 5 0 3 .2HC1, CAS registration number 69056-38-28.
- Sapropene dihydrochloride is a synthetic product of tetrahydrobiopterin (BH 4 ) dihydrochloride.
- BH 4 is a cofactor of phenylalanine hydroxylase (PAH). Phenylalanine (Phe) undergoes hydroxylation reaction under the action of PAH to obtain tyrosine. PAH activity in PKU patients is weak or even inactive, BH 4 can activate PAH, promote the normal oxidative metabolism of Phe in vivo, and reduce the level of Phe in some patients.
- PAH activity in PKU patients is weak or even inactive, BH 4 can activate PAH, promote the normal oxidative metabolism of Phe in vivo, and reduce the level of Phe in some patients.
- the US FDA approved BioMarin Pharmaceuticals' oxaprofen dihydrochloride tablets for the treatment of phenylketonuria. Due to the effective activity of sapropene dihydrochloride, it is necessary to select a route suitable for industrial
- the present invention provides a method for synthesizing a sapropium dihydrochloride compound, which shortens the synthetic route of sapropene dihydrochloride, and uses a chiral resolving reagent to resolve a racemic intermediate or a low enantiomer
- the intermediates of the structure are obtained as intermediates with high enantiomeric isomeric values, and the raw materials are cheap and easy to obtain, which greatly reduces the cost, and provides an effective idea for large-scale industrial production of sapropium dihydrochloride.
- Technical Solution of the Invention A method for synthesizing sapropene dihydrochloride, characterized in that the specific steps are as follows:
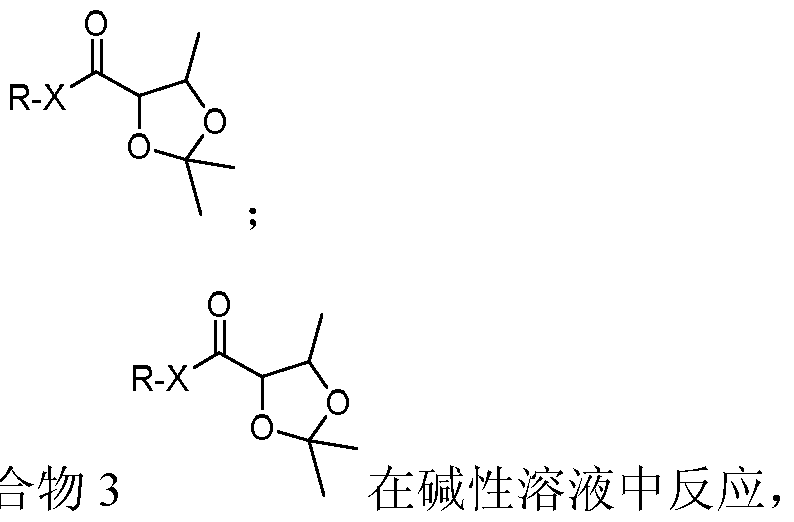
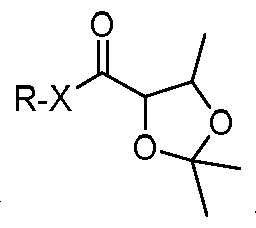
- compound 2 the structural formula is RX ⁇ ; when X is NH, compound 1 is crotonic acid alkyl or benzyl amide, compound 2 is 2,3 epoxy-butyric acid alkylamide; when X is oxygen, compound 1 is alkyl crotonate or benzyl crotonate, and compound 2 is 2,3 epoxy-butyric acid alkyl ester or 2,3 epoxy-butyric acid benzyl ester;
- R-x ⁇ ⁇ and the polar solvent are used in an amount of lg/5 ⁇ 20ml, R_x ⁇ and oxidizing agent
- the molar ratio is 1 : 1 to 3, and the molar ratio of R-x ⁇ to the strong base is 1 : 1 to 3, and the step (1) can also be carried out by referring to the method in Patent 200910070240.8;
- compound 3 is 2,3-propionketone-alkylbutanamide or 2,3-propionketone-benzylbutyramide.
- X oxygen
- compound 3 is 2,3-propane. a ketal-alkyl butyrate or a 2,3-propionketone-butyl butyrate; wherein, the molar ratio of the compound 2 to acetone is 1:3 to 15, and the molar ratio of the compound 2 to the Lewis acid is 1: 0.1. ⁇ 1, the molar ratio of the compound 2 to the inorganic base is 1: 0.5-3;
- Keto-butyric acid alkyl aniline salt, , n 0, l ; wherein, the ratio of the compound 3 to the polar solvent is lg / 3 ⁇ 10ml, the molar ratio of the compound 3 to the pure water is 1: 0.5-3, the basic substance of the compound 3 and the alkaline solution a molar ratio of 1: 0.5 ⁇ 2, the ratio of the amount of the compound 3 to the polar solvent of the dissolution cake is lg / 2 ⁇ 10ml, the molar ratio of the compound 3 to the resolving agent is 1: 1 ⁇ 5;
- the structural formula is wherein, the ratio of the compound 4 to the ether solvent is lg / 3 ⁇ 10ml, and the molar ratio of the compound 4 to hydrazine, hydrazine-diisopropylethylamine is 1: 0.8-3;
- the structural formula is Wherein, the ratio of the compound 5 to the ether solvent is lg/5 1515 ml; the molar ratio of the compound 5 to hydrazine, hydrazine-diisopropylethylamine is 1:1 to 5; the molar ratio of the compound 5 to the chloroformate a ratio of 1: 1 to 3; a molar ratio of the compound 5 to hydrogen chloride in a solution of hydrogen chloride is 1: 1 to 5;
- the compound 8 is (3S, 4S)-1-amino-3,4-dihydroxy-2-pentanone, the structural formula is 0H 3; wherein the ratio of the compound 7 to the ether solvent is lg/5 ⁇ 15ml; The molar ratio of compound 7 to triphenylphosphine is 1: 0.1 to 3; the ratio of compound 7 to water is 1: 0.1-3; mass ratio of compound 7 to 5% palladium carbon or 10% palladium carbon or Raney nickel 1: 0.05 ⁇ 0.6, hydrogen is introduced to the system pressure of 0.4 ⁇ 0.9MPa;
- Nitro-3H-pyrimidin-4-one the structural formula is , Compound 8, an alkaline reagent, the system is incubated at 30 to 80 ° C for 4 to 8 hours, a buffer solution is added to adjust the pH of the system to 6 to 8, and the system is filtered to obtain a compound 9,
- the structural formula is wherein, the ratio of the compound 8 to the alcohol solvent is lg/5 ⁇ 15ml; the ratio of the compound 8 to the pure water is lg/l ⁇ 5ml; the molar ratio of the compound 8 to the compound A is 1:1 ⁇ 1.5; The molar ratio of the catalyst to the catalyst is 1: 0.05-0.5; the molar ratio of the compound 8 to the alkaline reagent is 1: 3 ⁇ 8;
- Base-7,8-dihydropterin Wherein the ratio of the amount of the compound 9 to the polar solvent is lg / 20 ⁇ 50ml; the mass ratio of the compound 9 to the catalyst is 1: 0.05 - 0.6; (10) In the presence of the solution of the compound 10 obtained in the step (9), adding a catalyst, introducing hydrogen gas to a system pressure of 0.4 to 0.9 MPa, controlling the temperature of the system by 10 to 30 ° C, and controlling the pressure to 0.4 to 0.9 MPa. After the reaction is completed for 72 to 84 hours, the reaction is completed, and quenched to 10% to 20% of dilute hydrochloric acid.
- the system is suction filtered and dried to give the compound 11, which is the crude product of the target product, sapropion hydrochloride, and further alcohol.
- the solvent or the ketone solvent is purified by crystallization at 0 to 40 ° C to obtain a pure product of sapropene dihydrochloride; wherein the mass ratio of the compound 10 to the catalyst is 1: 0.05 to 0.6; and the molar ratio of the compound 10 to hydrochloric acid is 1: 3 to 10;
- the ratio of the amount of the compound 10 to the alcohol solvent or the ketone solvent is lg/5 to 25 ml.
- the polar solvent is water, methanol, ethanol or isopropanol, preferably water, methanol or ethanol, most preferably water;
- the oxidizing agent is N-bromosuccinimide, Chloroperoxybenzoic acid, 35% hydrogen peroxide or 50% t-butyl hydroperoxide in toluene, preferably N-bromosuccinimide, m-chloroperoxybenzoic acid or 50% t-butyl hydroperoxide
- the toluene solution is most preferably N-bromosuccinimide;
- the strong base is sodium hydroxide or potassium hydroxide, preferably sodium hydroxide;
- the ratio of R-x ⁇ to polar solvent is lg/5 ⁇ 15ml, preferably lg/6 ⁇ 12ml; R"X
- the molar ratio to the oxidizing agent is from 1:1 to 2.5, preferably from 1:1 to 2; and the molar ratio of Rx ⁇ to the strong base is from 1:1 to 2.5, preferably from 1:1 to 2.
- the Lewis acid is aluminum trichloride, ferric chloride, zinc chloride, 47% boron trifluoride diethyl ether solution, zinc bromide or lithium chloride, preferably trichlorinated.
- the polar solvent is tetrahydrofuran, methanol or ethanol, preferably tetrahydrofuran or methanol, most preferably methanol;
- the resolving agent is L-a-phenylethylamine or L-a-amphetamine, preferably left-handed A-phenethylamine;
- the alkaline solution is 29% sodium methoxide in methanol, 20% potassium hydroxide aqueous solution or 20% sodium hydroxide aqueous solution, preferably 29% sodium methoxide in methanol or 20% hydrogen
- An aqueous solution of potassium oxide preferably a methanol solution of 29% sodium methoxide;
- the ratio of the amount of the compound 3 to the polar solvent is lg / 3 to 8 ml, preferably lg / 4 to 8 ml;
- the molar ratio of the compound 3 to pure water is 1: 0.5 - 1.8, preferably 1: 0.5 - 1.5;
- the ether solvent is tetrahydrofuran, 2-methyltetrahydrofuran, methyl tert-butyl ether, 1,4-dioxycyclohexane or diethyl ether, preferably tetrahydrofuran, 2-methyltetrahydrofuran, Methyl tert-butyl ether or 1,4-dioxane, most preferably 2-methyltetrahydrofuran or 1,4-dioxane;
- the inorganic acid is sulfuric acid, hydrochloric acid or phosphoric acid, preferably sulfuric acid or hydrochloric acid, most Preferably, it is sulfuric acid;
- the ratio of the compound 4 to the ether solvent is lg/3 ⁇ 8ml, preferably lg/3 ⁇ 6ml; the molar ratio of the compound 4 to hydrazine, hydrazine-diisopropylethylamine is 1: 0.8-2.5 Preferably, it is 1:
- the ether solvent is tetrahydrofuran, 2-methyltetrahydrofuran, methyl tert-butyl ether or 1,4-dioxane or diethyl ether, preferably tetrahydrofuran, 2-methyltetrahydrofuran or Methyl tert-butyl ether, preferably tetrahydrofuran or 2-methyltetrahydrofuran;
- the chloroformate is methyl chloroformate, ethyl chloroformate or propyl chloroformate, preferably methyl chloroformate or ethyl chloroformate.
- ethyl chloroformate is triethylamine, sodium carbonate, potassium carbonate, sodium hydrogencarbonate, potassium hydrogencarbonate, sodium hydroxide or potassium hydroxide, preferably triethylamine, sodium carbonate, potassium carbonate, Sodium hydrogencarbonate or potassium hydrogencarbonate, most preferably triethylamine;
- the ratio of compound 5 to ether solvent is lg / 6 ⁇ 12ml, preferably lg / 8 ⁇ 12ml; compound 5 and hydrazine, hydrazine - diisopropyl
- the molar ratio of ethylamine is 1: 1.5 to 4; preferably 1: 2 to 4; the molar ratio of compound 5 to chloroformate is 1: 1-2.5, preferably 1: 1 to 2; The molar ratio is 1:1.5-4.5, preferably 1:2 ⁇ 4.
- the polar solvent is acetonitrile, methanol, ethanol, acetone or tetrahydrofuran, preferably methanol, ethanol or acetone, most preferably acetone;
- the catalyst is sodium iodide or potassium iodide, preferably sodium iodide.
- the azide is sodium azide or trimethylsilyl azide, preferably sodium azide;
- the ratio of compound 6 to polar solvent is lg / 6 ⁇ 12ml, preferably lg / 8 ⁇ 12ml; compound 6 and stack
- the molar ratio of the nitrogen is 1:1 to 3, preferably 1: 1-2.5; and the molar ratio of the compound 6 to the catalyst is 1: 0.05 to 0.6, preferably 0.1 to 0.5.
- the ether solvent is tetrahydrofuran, 2-methyltetrahydrofuran, methyl tert-butyl ether, 1,4-dioxane or diethyl ether; preferably tetrahydrofuran, 2-methyltetrahydrofuran, Methyl tert-butyl ether or 1,4-dioxane; optimally tetrahydrofuran; acid reagents are citric acid, p-toluenesulfonic acid, benzenesulfonic acid, formic acid, acetic acid, salt Acid, sulfuric acid or phosphoric acid, preferably citric acid, p-toluenesulfonic acid, benzenesulfonic acid, hydrochloric acid or sulfuric acid, most preferably citric acid or hydrochloric acid; the ratio of compound 7 to ether solvent is lg/5 ⁇ 12ml, preferably Lg / 6 ⁇ 12ml
- the alcohol solvent is methanol, ethanol, propanol or isopropanol, preferably methanol, ethanol or isopropanol, most preferably isopropanol or ethanol;
- the catalyst is sodium iodide or Potassium iodide, preferably sodium iodide;
- alkaline reagent is triethylamine, diisopropylethylamine, diisopropylamine, pyridine, sodium hydrogencarbonate, potassium hydrogencarbonate, sodium carbonate, potassium carbonate or cesium carbonate, preferably triethyl Amine, pyridine, sodium hydrogencarbonate, potassium hydrogencarbonate, sodium carbonate or potassium carbonate, most preferably triethylamine;
- buffer solution is sodium dihydrogen phosphate-dihydrogen phosphate aqueous solution, potassium dihydrogen phosphate-dipotassium hydrogen phosphate solution or
- the ammonium formate-ammonia aqueous solution is preferably an a
- the dosage ratio is lg/6 ⁇ 12ml, preferably lg/6 ⁇ 10ml; the ratio of compound 8 to pure water is lg/l ⁇ 4ml, preferably lg/l ⁇ 3ml; the molar ratio of compound 8 to compound A is 1 : 1 to 1.4, preferably 1: 1 to 1.2; Compound 8 and reminder
- the molar ratio of the chemical agent is 1:0.1 to 0.4, preferably 1:0.1 to 0.3; and the molar ratio of the compound 8 to the alkaline agent is 1:4 to 7, preferably 1:4 to 6.
- the catalyst is Raney nickel, 5% palladium carbon, 10% palladium carbon, platinum dioxide or 20% palladium carbon, preferably Raney nickel, 5% palladium carbon or 10% palladium carbon.
- the most preferred is Raney nickel;
- the polar solvent is pure water, methanol or ethanol, preferably pure water or methanol, most preferably pure water;
- the alkaline reagent is sodium hydroxide, potassium hydroxide, sodium carbonate or potassium carbonate, Sodium hydroxide or sodium carbonate; most preferably sodium hydroxide;
- the ratio of compound 9 to polar solvent is lg / 25 ⁇ 45ml, preferably lg / 30 ⁇ 40ml;
- the mass ratio of compound 9 to catalyst is 1: 0.05- 0.5, preferably 1: 0.1 to 0.4.
- the catalyst is Raney nickel, 5% palladium carbon, 10% palladium carbon, platinum dioxide or 20% palladium carbon, preferably Raney nickel, platinum dioxide or 20% palladium carbon.
- the solvent is preferably 20% palladium carbon;
- the alcohol solvent is methanol, ethanol, isopropanol or n-butanol, preferably methanol, ethanol or isopropanol, most preferably methanol;
- the ketone solvent is acetone or methyl ethyl ketone, preferably Acetone;
- the mass ratio of the compound 10 to the catalyst is 1: 0.05 to 0.5, preferably 1: 0.1 to 0.4;
- the molar ratio of the compound 10 to hydrochloric acid is 1: 4 to 9, preferably 1: 5 to 8;
- the solvent or ketone solvent is used in an amount of from lg/5 to 20 ml, preferably from lg/10 to 20 ml.
- the raw materials used in the synthesis method are easily available, and the cost is greatly reduced compared with the current process; 2.
- the route of the invention is simple, and the synthetic route of sapropene dihydrochloride is greatly shortened; 3.
- Process conditions Stable the whole operation process is simple, the output of three wastes is small, and the pollution is small, which provides an effective idea for the large-scale industrial production of sapropene dihydrochloride.
- the present invention utilizes chiral separation reagent to separate the middle of the race. Intermediates with lower enantiomerically isomeric values, intermediates with higher enantiomeric isomeric values, complemented by chiral routes; 5.
- Fig. 1 is a flow chart showing the chiral preparation process of a sapropene dihydrochloride compound according to the present invention.
- DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS It should be noted that the embodiments of the present invention and the features of the embodiments may be combined with each other without conflict. The invention will be described in detail below with reference to the drawings in conjunction with the embodiments. The range of intervals that appear in the embodiment is due to the fact that temperature and pH will fluctuate with the progress of the reaction in one test.
- step (10) 1.7 kg of acetamido-7,8-dihydropterin obtained in step (9)
- 0.255 kg (0.15 g/g) of 20% palladium carbon was added, hydrogen gas was introduced to the reactor pressure of 0.6 ⁇ 0.05 MPa, and the system was controlled at a temperature of 20 ⁇ 5 ° C and a pressure of 0.6 ⁇ 0.05 MPa.
- the crude product was purified by recrystallization from L (14.7 ml / g) methanol at 20 ⁇ 5 ° C to give 0.95 kg of pure product, yield 50%, purity 98.5%, enantiomeric excess value 99.2%.
- Example 2 The main raw material is
- Trimethyl-1,3-dioxolan-4-carboxylic acid amphetamine salt Then, 10% dilute sulfuric acid aqueous solution was added to the system to adjust the pH to 3 ⁇ 0.5, the temperature was controlled at 10 °C ⁇ 5 °C, and the reaction was carried out for 1 hour. The organic phase was separated, and 6.1 kg (3 eq) of hydrazine was added to the organic phase. , ⁇ -amine, system concentration to obtain 1.3kg of (4S, 5S)-2,2,5-three
- Trimethyl 1,3-dioxolane-4- 8kg (5eq) of hydrazine, hydrazine-diisopropylethylamine, cooled to 0 ⁇ 5 °C, add 2.9kg C3eq) of propyl chloroformate, keep the reaction for 1-2 hours, and dilute methane gas for 2 hours.
- Add 12.7kg (5eq) of 20% hydrogen chloride ethanol solution react for 2 hours, add sodium carbonate to adjust the pH value to 9 ⁇ 0.5, extract, separate and concentrate to obtain 1.3kg (4S, 5S)-2,2 ,5-trimethyl-5-chloroacetyl-1,3-
- step (10) 2.5kg obtained in step (9)
- 1.5 kg (0.6 g / g) of Raney nickel was added, hydrogen gas was introduced until the system pressure was 0.9 ⁇ 0.05 MPa, and the system was controlled at a temperature of 30 ⁇ 5 ° C, a pressure of 0.9 ⁇ 0.05 MPa, and the reaction was 84. After the reaction is completed, it is quenched into dilute hydrochloric acid, and the system is suction filtered and dried to obtain the target product sapropamine hydrochloride.
- the crude product was purified by recrystallization from 62.5 L (25 ml / g) of acetone at 40 ⁇ 5 ° C to give 1.31 kg of pure product, yield 47%, purity 98.1%, and enantiomeric excess of 98.9%.
- the reaction was kept for 5 hours, and 610 kg of a 5% carbon q solution was added to the system, and the system was subjected to liquid separation.
- Ketone-alkylformamide Warm up to 25 ⁇ 5 ° C, add 4.23 kg (0.5 eq) of pure water and 47 kg (0.5 eq) of 20% sodium hydroxide solution, keep the reaction for 3 hours, centrifuge, filter cake with 122.6L (2ml / g) The ethanol was dissolved, and 56.9 kg (leq) of L-a-phenylethylamine was added, and the mixture was dried at 15 ⁇ 5 ° C to obtain 32.3 kg.
- (4S,5S)-2,2,5-trimethyl1,3-dioxolane-4-carboxylic acid 3.7kg ( leq) of hydrazine, hydrazine-diisopropylethylamine, cooled to -30 ⁇ 5 °C, add 3.1kg (l eq) of methyl chloroformate, keep the reaction for 1 hour, pass diazomethane Gas for 1 hour, add 2kg (leq) of 20% hydrogen chloride ethanol solution, react for 1 hour, add potassium bicarbonate to adjust the pH value of 7 ⁇ 0. Liquid, concentrate to get 5kg of (4S, 5S)-2,2,5 -Trimethyl-5-chloroethyl Yield 81%;
- Butyric acid The temperature was raised to 35 ⁇ 5 °C, 9.3 kg (1.8 eq) of pure water and 144.5 kg (1.8 eq) of 20% potassium hydroxide aqueous solution were added, the reaction was incubated for 6.5 hours, centrifuged, and the filter cake was used for 398 L (8 ml/ g) the methanol is dissolved, Add 154.7kg (4eq) °C for 4.5 hours, centrifuge and dry to obtain 21.1kg.
- Trimethyl-1,3-dioxolane-4-carboxylic acid 9.7kg (4eq) of hydrazine, hydrazine-diisopropylethylamine, cooled to -25 ⁇ 5 °C, add 4.4kg (2.5eq) of methyl chloroformate, keep the reaction for 1.5 hours, pass diazomethane After 1.5 hours, a solution of 15.3 kg (4.5 eq) of 20% hydrogen chloride in ethanol was added for 1.5 hours. Triethylamine was added to adjust the pH to 8.5 ⁇ 0.5, and concentrated to obtain 3.0 kg of (4S, 5S)-2,2. ,5-trimethyl-5-chloro
- the pH of the system was adjusted to 7.5 ⁇ 0.5 by adding sodium dihydrogen phosphate-sodium hydrogen phosphate aqueous solution; the system was subjected to amido-5-nitro-6-((3S,4S)-3 , 3-dihydroxy-2-oxo-pentylamino) -
- step (10) 1.5kg obtained in step (9)
- 0.7 kg (0.5 g/g) of 5% palladium carbon was added, hydrogen gas was introduced to the reactor pressure of 0.8 ⁇ 0.05 MPa, and the system was controlled at a temperature of 25 ⁇ 5 ° C and a pressure of 0.8 ⁇ 0.05 MPa.
- the system was suction filtered and dried to give the target product
- the crude product was purified by recrystallization from 29 L (20 ml/g) methanol at 35 ⁇ 5 ° C to give 0.8 kg.
- Example 5 The main raw material is,
- the pH of the system was adjusted to 7.5 ⁇ 0.5 by adding potassium dihydrogen phosphate-dipotassium hydrogen phosphate solution; the system was subjected to -acetylamino-5-nitro-6-((3S,4S)- 3,3-dihydroxy-2-oxo-pentylamino) -
- step (10) 1.1kg obtained in step (9)
- 0.44 kg (0.4 g / g) of 10% palladium carbon was added, hydrogen gas was introduced to the reactor pressure of 0.8 ⁇ 0.05 MPa, and the system was controlled at a temperature of 25 ⁇ 5 ° C and a pressure of 0.8 ⁇ 0.05 MPa. After reacting for 80 hours, after completion of the reaction, it was quenched into hydrochloric acid, and the system was suction filtered and dried to obtain the desired product, sapropamine hydrochloride.
- Ketal-butyric acid isopropylamide warmed to 27 ⁇ 5 ° C, added 2.9 kg (0.5 eq) of pure water and
- Heterocyclic pentane-4-carboxylic acid 8.5kg (2eq) of hydrazine, hydrazine-diisopropylethylamine, cooled to -10 ⁇ 5 °C, add 4kg (21.0eq) of propyl chloroformate, keep the reaction for 2 hours, pass diazomethane gas 2 hours, add 12kg (2eq) of 20% hydrogen chloride ethanol solution, react for 2 hours, add sodium hydroxide to adjust the pH value to 7.5 ⁇ 0.5, and concentrate to obtain 5.1kg of (4S, 5S)-2,2,5- Trimethyl-5-chloroacetyl-1,3-dioxolane , yield 81%;
- step (10) 1.7kg obtained in step (9)
- 0.2kg (0.1g/g) of platinum dioxide is added, hydrogen is introduced to the reactor pressure of 0.6 ⁇ 0.05MPa, the system is controlled at a temperature of 15 ⁇ 5 °C, and the pressure is 0.6 ⁇ 0.05MPa.
- the crude product was purified by recrystallization from 17 L (10 ml/g) butanone at 15 ⁇ 5 ° C to give 0.6 kg of pure product, yield 43%, purity 98.4%, and an enantiomeric excess of 98.9%.
- the synthesis of a bismuth dihydrochloride salt disclosed in the method of the present invention can obtain a target product of high purity, high enantiomeric excess value, and the product yield is high.
- the raw materials used in the synthesis method are easy to obtain, greatly shorten the synthetic route of sapropene dihydrochloride, the process conditions are stable, and the whole process is less polluted, which provides an effective method for large-scale industrial production of sapropene dihydrochloride.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description

























































































Claims
























Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RS20170360A RS55853B1 (sr) | 2012-04-10 | 2012-12-27 | Postupak za sintezu sapropterin dihidrohlorida |
| US14/394,079 US9371326B2 (en) | 2012-04-10 | 2012-12-27 | Method for synthesizing sapropterin dihydrochloride |
| LTEP12874086.7T LT2837629T (lt) | 2012-04-10 | 2012-12-27 | Sapropterino dihidrochlorido sintetinimo būdas |
| ES12874086.7T ES2624500T3 (es) | 2012-04-10 | 2012-12-27 | Método para sintetizar diclorhidrato de sapropterina |
| DK12874086.7T DK2837629T3 (da) | 2012-04-10 | 2012-12-27 | Fremgangsmåde til syntetisering af sapropterindihydrochlorid |
| EP12874086.7A EP2837629B1 (en) | 2012-04-10 | 2012-12-27 | Method for synthesizing sapropterin dihydrochloride |
| SI201230925A SI2837629T1 (sl) | 2012-04-10 | 2012-12-27 | Postopek za sintetiziranje sapropterinijevega dihidroklorida |
| PL12874086T PL2837629T3 (pl) | 2012-04-10 | 2012-12-27 | Sposób syntetyzowania dichlorowodorku sapropteryny |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210102879.1 | 2012-04-10 | ||
| CN201210102879.1A CN102633799B (zh) | 2012-04-10 | 2012-04-10 | 一种从消旋体中间体拆分路线合成二盐酸沙丙蝶呤的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013152608A1 true WO2013152608A1 (zh) | 2013-10-17 |
Family
ID=46618392
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2012/087732 Ceased WO2013152608A1 (zh) | 2012-04-10 | 2012-12-27 | 一种二盐酸沙丙喋呤的合成方法 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9371326B2 (zh) |
| EP (1) | EP2837629B1 (zh) |
| CN (1) | CN102633799B (zh) |
| DK (1) | DK2837629T3 (zh) |
| ES (1) | ES2624500T3 (zh) |
| HU (1) | HUE033497T2 (zh) |
| LT (1) | LT2837629T (zh) |
| PL (1) | PL2837629T3 (zh) |
| PT (1) | PT2837629T (zh) |
| RS (1) | RS55853B1 (zh) |
| SI (1) | SI2837629T1 (zh) |
| WO (1) | WO2013152608A1 (zh) |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9096537B1 (en) | 2014-12-31 | 2015-08-04 | Mahesh Kandula | Compositions and methods for the treatment of mucositis |
| US9102649B1 (en) | 2014-09-29 | 2015-08-11 | Mahesh Kandula | Compositions and methods for the treatment of multiple sclerosis |
| US9108942B1 (en) | 2014-11-05 | 2015-08-18 | Mahesh Kandula | Compositions and methods for the treatment of moderate to severe pain |
| US9150557B1 (en) | 2014-11-05 | 2015-10-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of hyperglycemia |
| US9174931B2 (en) | 2013-06-04 | 2015-11-03 | Cellix Bio Private Limited | Compositions for the treatment of diabetes and pre-diabetes |
| US9173877B1 (en) | 2014-11-05 | 2015-11-03 | Cellix Bio Private Limited | Compositions and methods for the treatment of local pain |
| US9175008B1 (en) | 2014-11-05 | 2015-11-03 | Cellix Bio Private Limited | Prodrugs of anti-platelet agents |
| US9187427B2 (en) | 2012-08-03 | 2015-11-17 | Cellix Bio Private Limited | N-substituted nicotinamide compounds and compositions for the treatment migraine and neurologic diseases |
| US9206111B1 (en) | 2014-12-17 | 2015-12-08 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological diseases |
| US9227974B2 (en) | 2012-05-23 | 2016-01-05 | Cellex Bio Private Limited | Compositions and methods for the treatment of respiratory disorders |
| US9233161B2 (en) | 2012-05-10 | 2016-01-12 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological conditions |
| US9242939B2 (en) | 2012-05-10 | 2016-01-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of respiratory disorders |
| US9266823B2 (en) | 2012-05-08 | 2016-02-23 | Cellix Bio Private Limited | Compositions and methods for the treatment of parkinson's disease |
| US9273061B2 (en) | 2012-05-10 | 2016-03-01 | Cellix Bio Private Limited | Compositions and methods for the treatment of chronic pain |
| US9284287B1 (en) | 2014-11-05 | 2016-03-15 | Cellix Bio Private Limited | Compositions and methods for the suppression of carbonic anhydrase activity |
| US9290486B1 (en) | 2014-11-05 | 2016-03-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of epilepsy |
| US9303038B2 (en) | 2011-09-06 | 2016-04-05 | Cellix Bio Private Limited | Compositions and methods for the treatment of epilepsy and neurological diseases |
| US9309233B2 (en) | 2012-05-08 | 2016-04-12 | Cellix Bio Private Limited | Compositions and methods for the treatment of blood clotting disorders |
| US9315478B2 (en) | 2012-05-10 | 2016-04-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic syndrome |
| US9315461B2 (en) | 2012-05-10 | 2016-04-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurologic diseases |
| US9321775B2 (en) | 2012-05-10 | 2016-04-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of moderate to severe pain |
| US9321716B1 (en) | 2014-11-05 | 2016-04-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic syndrome |
| US9333187B1 (en) | 2013-05-15 | 2016-05-10 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammatory bowel disease |
| US9339484B2 (en) | 2012-05-10 | 2016-05-17 | Cellix Bio Private Limited | Compositions and methods for the treatment of restless leg syndrome and fibromyalgia |
| US9346742B2 (en) | 2012-05-10 | 2016-05-24 | Cellix Bio Private Limited | Compositions and methods for the treatment of fibromyalgia pain |
| US9394288B2 (en) | 2012-05-10 | 2016-07-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of asthma and allergy |
| US9399634B2 (en) | 2012-05-07 | 2016-07-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of depression |
| US9403857B2 (en) | 2012-05-10 | 2016-08-02 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic syndrome |
| US9403826B2 (en) | 2012-05-08 | 2016-08-02 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammatory disorders |
| US9434729B2 (en) | 2012-05-23 | 2016-09-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of periodontitis and rheumatoid arthritis |
| US9434704B2 (en) | 2012-05-08 | 2016-09-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological degenerative disorders |
| US9492409B2 (en) | 2012-05-23 | 2016-11-15 | Cellix Bio Private Limited | Compositions and methods for the treatment of local pain |
| US9498461B2 (en) | 2012-05-23 | 2016-11-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammatory bowel disease |
| US9499527B2 (en) | 2012-05-10 | 2016-11-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of familial amyloid polyneuropathy |
| US9499526B2 (en) | 2012-05-10 | 2016-11-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurologic diseases |
| US9522884B2 (en) | 2012-05-08 | 2016-12-20 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic disorders |
| US9573927B2 (en) | 2012-05-10 | 2017-02-21 | Cellix Bio Private Limited | Compositions and methods for the treatment of severe pain |
| US9580383B2 (en) | 2012-05-23 | 2017-02-28 | Cellix Bio Private Limited | Compositions and methods for the treatment of multiple sclerosis |
| US9624168B2 (en) | 2012-09-06 | 2017-04-18 | Cellix Bio Private Limited | Compositions and methods for the treatment inflammation and lipid disorders |
| US9642915B2 (en) | 2012-05-07 | 2017-05-09 | Cellix Bio Private Limited | Compositions and methods for the treatment of neuromuscular disorders and neurodegenerative diseases |
| US9670153B2 (en) | 2012-09-08 | 2017-06-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammation and lipid disorders |
| US9725404B2 (en) | 2014-10-27 | 2017-08-08 | Cellix Bio Private Limited | Compositions and methods for the treatment of multiple sclerosis |
| US9738631B2 (en) | 2012-05-07 | 2017-08-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological disorders |
| US9765020B2 (en) | 2012-05-23 | 2017-09-19 | Cellix Bio Private Limited | Dichlorophenyl-imino compounds and compositions, and methods for the treatment of mucositis |
| US9771355B2 (en) | 2014-09-26 | 2017-09-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of epilepsy and neurological disorders |
| US9932294B2 (en) | 2014-12-01 | 2018-04-03 | Cellix Bio Private Limited | Compositions and methods for the treatment of multiple sclerosis |
| US10208014B2 (en) | 2014-11-05 | 2019-02-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological disorders |
| US10227301B2 (en) | 2015-01-06 | 2019-03-12 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammation and pain |
| CN109776540A (zh) * | 2017-11-15 | 2019-05-21 | 北京启慧医疗器械有限公司 | 一种盐酸沙丙蝶呤的制备方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102627644B (zh) | 2012-04-10 | 2014-04-16 | 凯莱英医药集团(天津)股份有限公司 | 一种通过直接手性合成方法制备二盐酸沙丙蝶呤的方法 |
| CN102633799B (zh) | 2012-04-10 | 2014-06-25 | 凯莱英医药集团(天津)股份有限公司 | 一种从消旋体中间体拆分路线合成二盐酸沙丙蝶呤的方法 |
| WO2014041446A2 (en) * | 2012-09-17 | 2014-03-20 | Mahesh Kandula | Compositions and methods for the treatment of metabolic diseases |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS574990A (en) * | 1980-05-09 | 1982-01-11 | Bisukonchiini Matsukusu | Polyacylated tetrahydropterin derivative and manufacture |
| EP0153696A2 (en) * | 1984-02-23 | 1985-09-04 | Kanegafuchi Kagaku Kogyo Kabushiki Kaisha | Process for preparing 5,6,7,8-tetrahydro-6-(l-erythro-1',2'-dihydroxypropyl) pterin |
| JPS60204786A (ja) * | 1984-03-29 | 1985-10-16 | Univ Nagoya | (6r)―テトラヒドロビオプテリンの合成法 |
| EP0191335B1 (en) * | 1985-01-28 | 1991-08-14 | Shiratori Pharmaceutical Co. Ltd. | Preparation process of (6r)-tetrahydro-l-biopterin |
| WO2005063752A1 (en) * | 2003-12-30 | 2005-07-14 | Vasopharm Biotech Gmbh | 4-amino-7,8-dihydropteridines, pharmaceutical compositions containing them and their use for the treatment of diseases which are caused by an increased nitric oxide level |
| US20060142573A1 (en) * | 2004-12-27 | 2006-06-29 | Shiratori Pharmaceutical Co., Ltd. | Method for producing L-biopterin |
| WO2009088979A1 (en) * | 2008-01-07 | 2009-07-16 | Biomarin Pharmaceutical Inc. | Method of synthesizing tetrahydrobiopterin |
| CN102633799A (zh) * | 2012-04-10 | 2012-08-15 | 凯莱英医药集团(天津)股份有限公司 | 一种从消旋体中间体拆分路线合成二盐酸沙丙蝶呤的方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2575781B2 (ja) * | 1988-02-29 | 1997-01-29 | 日清製粉株式会社 | 2,3−ジアシルオキシ−4−ヒドロキシ−トペンタナールおよびその製造方法 |
| JP2882116B2 (ja) | 1991-09-13 | 1999-04-12 | 日本電気株式会社 | ヒートシンク付パッケージ |
| JP5184783B2 (ja) * | 2003-11-17 | 2013-04-17 | バイオマリン ファーマシューティカル インコーポレイテッド | テトラヒドロビオプテリン、及びテトラヒドロビオプテリン類似体の製造方法 |
| JP2011011976A (ja) * | 2007-10-10 | 2011-01-20 | Shiratori Pharmaceutical Co Ltd | プテリジン化合物及びl−ビオプテリンの製造方法 |
| CN102627644B (zh) | 2012-04-10 | 2014-04-16 | 凯莱英医药集团(天津)股份有限公司 | 一种通过直接手性合成方法制备二盐酸沙丙蝶呤的方法 |
-
2012
- 2012-04-10 CN CN201210102879.1A patent/CN102633799B/zh active Active
- 2012-12-27 SI SI201230925A patent/SI2837629T1/sl unknown
- 2012-12-27 DK DK12874086.7T patent/DK2837629T3/da active
- 2012-12-27 PT PT128740867T patent/PT2837629T/pt unknown
- 2012-12-27 RS RS20170360A patent/RS55853B1/sr unknown
- 2012-12-27 PL PL12874086T patent/PL2837629T3/pl unknown
- 2012-12-27 ES ES12874086.7T patent/ES2624500T3/es active Active
- 2012-12-27 US US14/394,079 patent/US9371326B2/en not_active Expired - Fee Related
- 2012-12-27 HU HUE12874086A patent/HUE033497T2/en unknown
- 2012-12-27 EP EP12874086.7A patent/EP2837629B1/en not_active Not-in-force
- 2012-12-27 WO PCT/CN2012/087732 patent/WO2013152608A1/zh not_active Ceased
- 2012-12-27 LT LTEP12874086.7T patent/LT2837629T/lt unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS574990A (en) * | 1980-05-09 | 1982-01-11 | Bisukonchiini Matsukusu | Polyacylated tetrahydropterin derivative and manufacture |
| EP0153696A2 (en) * | 1984-02-23 | 1985-09-04 | Kanegafuchi Kagaku Kogyo Kabushiki Kaisha | Process for preparing 5,6,7,8-tetrahydro-6-(l-erythro-1',2'-dihydroxypropyl) pterin |
| JPS60204786A (ja) * | 1984-03-29 | 1985-10-16 | Univ Nagoya | (6r)―テトラヒドロビオプテリンの合成法 |
| EP0191335B1 (en) * | 1985-01-28 | 1991-08-14 | Shiratori Pharmaceutical Co. Ltd. | Preparation process of (6r)-tetrahydro-l-biopterin |
| WO2005063752A1 (en) * | 2003-12-30 | 2005-07-14 | Vasopharm Biotech Gmbh | 4-amino-7,8-dihydropteridines, pharmaceutical compositions containing them and their use for the treatment of diseases which are caused by an increased nitric oxide level |
| US20060142573A1 (en) * | 2004-12-27 | 2006-06-29 | Shiratori Pharmaceutical Co., Ltd. | Method for producing L-biopterin |
| WO2009088979A1 (en) * | 2008-01-07 | 2009-07-16 | Biomarin Pharmaceutical Inc. | Method of synthesizing tetrahydrobiopterin |
| CN102633799A (zh) * | 2012-04-10 | 2012-08-15 | 凯莱英医药集团(天津)股份有限公司 | 一种从消旋体中间体拆分路线合成二盐酸沙丙蝶呤的方法 |
Non-Patent Citations (4)
| Title |
|---|
| E.L.PATTERSON ET AL., J.AM.CHEM.SOC., vol. 78, 1956, pages 5868 |
| MATSUURA ET AL., BUII.CHEM.SOC.JPN., vol. 48, 1975, pages 3767 |
| MATSUURA ET AL., CHEMISTRY OF ORGANIC SYNTHESIS,ML/G, vol. 46, no. 6, 1988, pages 570 |
| See also references of EP2837629A4 * |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9303038B2 (en) | 2011-09-06 | 2016-04-05 | Cellix Bio Private Limited | Compositions and methods for the treatment of epilepsy and neurological diseases |
| US9738631B2 (en) | 2012-05-07 | 2017-08-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological disorders |
| US9642915B2 (en) | 2012-05-07 | 2017-05-09 | Cellix Bio Private Limited | Compositions and methods for the treatment of neuromuscular disorders and neurodegenerative diseases |
| US9399634B2 (en) | 2012-05-07 | 2016-07-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of depression |
| US9266823B2 (en) | 2012-05-08 | 2016-02-23 | Cellix Bio Private Limited | Compositions and methods for the treatment of parkinson's disease |
| US9522884B2 (en) | 2012-05-08 | 2016-12-20 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic disorders |
| US9434704B2 (en) | 2012-05-08 | 2016-09-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological degenerative disorders |
| US9403826B2 (en) | 2012-05-08 | 2016-08-02 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammatory disorders |
| US9309233B2 (en) | 2012-05-08 | 2016-04-12 | Cellix Bio Private Limited | Compositions and methods for the treatment of blood clotting disorders |
| US9394288B2 (en) | 2012-05-10 | 2016-07-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of asthma and allergy |
| US9339484B2 (en) | 2012-05-10 | 2016-05-17 | Cellix Bio Private Limited | Compositions and methods for the treatment of restless leg syndrome and fibromyalgia |
| US9242939B2 (en) | 2012-05-10 | 2016-01-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of respiratory disorders |
| US9499526B2 (en) | 2012-05-10 | 2016-11-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurologic diseases |
| US9273061B2 (en) | 2012-05-10 | 2016-03-01 | Cellix Bio Private Limited | Compositions and methods for the treatment of chronic pain |
| US9499527B2 (en) | 2012-05-10 | 2016-11-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of familial amyloid polyneuropathy |
| US9403857B2 (en) | 2012-05-10 | 2016-08-02 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic syndrome |
| US9573927B2 (en) | 2012-05-10 | 2017-02-21 | Cellix Bio Private Limited | Compositions and methods for the treatment of severe pain |
| US9346742B2 (en) | 2012-05-10 | 2016-05-24 | Cellix Bio Private Limited | Compositions and methods for the treatment of fibromyalgia pain |
| US9315478B2 (en) | 2012-05-10 | 2016-04-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic syndrome |
| US9315461B2 (en) | 2012-05-10 | 2016-04-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurologic diseases |
| US9321775B2 (en) | 2012-05-10 | 2016-04-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of moderate to severe pain |
| US9233161B2 (en) | 2012-05-10 | 2016-01-12 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological conditions |
| US9765020B2 (en) | 2012-05-23 | 2017-09-19 | Cellix Bio Private Limited | Dichlorophenyl-imino compounds and compositions, and methods for the treatment of mucositis |
| US9492409B2 (en) | 2012-05-23 | 2016-11-15 | Cellix Bio Private Limited | Compositions and methods for the treatment of local pain |
| US9227974B2 (en) | 2012-05-23 | 2016-01-05 | Cellex Bio Private Limited | Compositions and methods for the treatment of respiratory disorders |
| US9498461B2 (en) | 2012-05-23 | 2016-11-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammatory bowel disease |
| US9434729B2 (en) | 2012-05-23 | 2016-09-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of periodontitis and rheumatoid arthritis |
| US9580383B2 (en) | 2012-05-23 | 2017-02-28 | Cellix Bio Private Limited | Compositions and methods for the treatment of multiple sclerosis |
| US9403793B2 (en) | 2012-07-03 | 2016-08-02 | Cellix Bio Private Limited | Compositions and methods for the treatment of moderate to severe pain |
| US9187427B2 (en) | 2012-08-03 | 2015-11-17 | Cellix Bio Private Limited | N-substituted nicotinamide compounds and compositions for the treatment migraine and neurologic diseases |
| US9624168B2 (en) | 2012-09-06 | 2017-04-18 | Cellix Bio Private Limited | Compositions and methods for the treatment inflammation and lipid disorders |
| US9670153B2 (en) | 2012-09-08 | 2017-06-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammation and lipid disorders |
| US9333187B1 (en) | 2013-05-15 | 2016-05-10 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammatory bowel disease |
| US9174931B2 (en) | 2013-06-04 | 2015-11-03 | Cellix Bio Private Limited | Compositions for the treatment of diabetes and pre-diabetes |
| US9840472B2 (en) | 2013-12-07 | 2017-12-12 | Cellix Bio Private Limited | Compositions and methods for the treatment of mucositis |
| US9771355B2 (en) | 2014-09-26 | 2017-09-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of epilepsy and neurological disorders |
| US9102649B1 (en) | 2014-09-29 | 2015-08-11 | Mahesh Kandula | Compositions and methods for the treatment of multiple sclerosis |
| US9988340B2 (en) | 2014-09-29 | 2018-06-05 | Cellix Bio Private Limited | Compositions and methods for the treatment of multiple sclerosis |
| US9725404B2 (en) | 2014-10-27 | 2017-08-08 | Cellix Bio Private Limited | Compositions and methods for the treatment of multiple sclerosis |
| US10208014B2 (en) | 2014-11-05 | 2019-02-19 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological disorders |
| US9284287B1 (en) | 2014-11-05 | 2016-03-15 | Cellix Bio Private Limited | Compositions and methods for the suppression of carbonic anhydrase activity |
| US9150557B1 (en) | 2014-11-05 | 2015-10-06 | Cellix Bio Private Limited | Compositions and methods for the treatment of hyperglycemia |
| US9175008B1 (en) | 2014-11-05 | 2015-11-03 | Cellix Bio Private Limited | Prodrugs of anti-platelet agents |
| US9321716B1 (en) | 2014-11-05 | 2016-04-26 | Cellix Bio Private Limited | Compositions and methods for the treatment of metabolic syndrome |
| US9173877B1 (en) | 2014-11-05 | 2015-11-03 | Cellix Bio Private Limited | Compositions and methods for the treatment of local pain |
| US9290486B1 (en) | 2014-11-05 | 2016-03-22 | Cellix Bio Private Limited | Compositions and methods for the treatment of epilepsy |
| US9108942B1 (en) | 2014-11-05 | 2015-08-18 | Mahesh Kandula | Compositions and methods for the treatment of moderate to severe pain |
| US9932294B2 (en) | 2014-12-01 | 2018-04-03 | Cellix Bio Private Limited | Compositions and methods for the treatment of multiple sclerosis |
| US9206111B1 (en) | 2014-12-17 | 2015-12-08 | Cellix Bio Private Limited | Compositions and methods for the treatment of neurological diseases |
| US9096537B1 (en) | 2014-12-31 | 2015-08-04 | Mahesh Kandula | Compositions and methods for the treatment of mucositis |
| US10227301B2 (en) | 2015-01-06 | 2019-03-12 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammation and pain |
| US10343994B2 (en) | 2015-01-06 | 2019-07-09 | Mahesh Kandula | Compositions and methods for the treatment of inflammation and pain |
| CN109776540A (zh) * | 2017-11-15 | 2019-05-21 | 北京启慧医疗器械有限公司 | 一种盐酸沙丙蝶呤的制备方法 |
| CN109776540B (zh) * | 2017-11-15 | 2021-07-06 | 北京启慧生物医药有限公司 | 一种盐酸沙丙蝶呤的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2837629A4 (en) | 2015-11-25 |
| CN102633799A (zh) | 2012-08-15 |
| RS55853B1 (sr) | 2017-08-31 |
| PT2837629T (pt) | 2017-06-09 |
| DK2837629T3 (da) | 2017-06-19 |
| CN102633799B (zh) | 2014-06-25 |
| US9371326B2 (en) | 2016-06-21 |
| EP2837629A1 (en) | 2015-02-18 |
| LT2837629T (lt) | 2017-06-12 |
| EP2837629B1 (en) | 2017-03-22 |
| PL2837629T3 (pl) | 2017-12-29 |
| US20150119573A1 (en) | 2015-04-30 |
| SI2837629T1 (sl) | 2017-05-31 |
| HUE033497T2 (en) | 2017-12-28 |
| ES2624500T3 (es) | 2017-07-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013152608A1 (zh) | 一种二盐酸沙丙喋呤的合成方法 | |
| CN108699068A (zh) | 一种三氟甲基取代的吡喃衍生物制备方法 | |
| KR101540435B1 (ko) | 발리올아민의 입체선택적 합성 | |
| CN103709221B (zh) | 一种虫草素的制备方法 | |
| CN102229538B (zh) | 一种达泊西汀的合成方法 | |
| CN113444083B (zh) | 一种小分子酪氨酸激酶抑制剂的制备方法 | |
| CN110078736B (zh) | 吡唑并嘧啶衍生物、其制备方法及其应用 | |
| WO2013152609A1 (zh) | 一种二盐酸沙丙蝶呤的合成方法 | |
| CN105949153B (zh) | 一种他司美琼的合成方法 | |
| CN115448848A (zh) | 一种抗真菌化合物制备方法 | |
| CN114195712B (zh) | 一种能够用来制备盐酸丙卡特罗的中间体及其制备方法 | |
| CN107540575B (zh) | 一种西他列汀中间体的制备方法 | |
| CN114105872B (zh) | 一种用于制备盐酸丙卡特罗的中间体及其制备方法 | |
| CN106749235B (zh) | 多取代喹啉并吡咯衍生物的制备方法 | |
| CN110804022B (zh) | 一种右丙亚胺的制备方法 | |
| CN105348285B (zh) | 一种低成本高收率制备腺嘌呤的方法 | |
| CN107382898B (zh) | 一种基于anpz含能母体结构的含能材料及其合成方法 | |
| CN119161348B (zh) | 一种依鲁替尼及其合成方法 | |
| CN117886751B (zh) | 一种伐尼克兰中间体的制备方法 | |
| CN114213323B (zh) | 一种盐酸丙卡特罗的合成新工艺 | |
| CN121850950A (zh) | 一种2-氰基嘧啶及2-甲胺基嘧啶盐酸盐的制备方法 | |
| CN102491954B (zh) | 利奈唑胺的制备方法 | |
| CN111793071B (zh) | 奥格列汀的合成工艺 | |
| US20260015324A1 (en) | Processes for Preparing 5-Bromo-3,4-dimethylpyridin-2-amine and 6-Bromo-7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridine | |
| CN102020615B (zh) | 一种吗啉酮衍生物及其制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12874086 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012874086 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012874086 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14394079 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2017/0360 Country of ref document: RS |