WO2013157612A1 - 炭素繊維束および炭素繊維の製造方法 - Google Patents
炭素繊維束および炭素繊維の製造方法 Download PDFInfo
- Publication number
- WO2013157612A1 WO2013157612A1 PCT/JP2013/061535 JP2013061535W WO2013157612A1 WO 2013157612 A1 WO2013157612 A1 WO 2013157612A1 JP 2013061535 W JP2013061535 W JP 2013061535W WO 2013157612 A1 WO2013157612 A1 WO 2013157612A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fiber bundle
- carbon fiber
- dtex
- less
- gpa
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/02—Fibres or whiskers
- C08K7/04—Fibres or whiskers inorganic
- C08K7/06—Elements
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F6/00—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof
- D01F6/28—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from copolymers obtained by reactions only involving carbon-to-carbon unsaturated bonds
- D01F6/38—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from copolymers obtained by reactions only involving carbon-to-carbon unsaturated bonds comprising unsaturated nitriles as the major constituent
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F9/00—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments
- D01F9/08—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material
- D01F9/12—Carbon filaments; Apparatus specially adapted for the manufacture thereof
- D01F9/14—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments
- D01F9/20—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products
- D01F9/21—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds
- D01F9/22—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds from polyacrylonitriles
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F9/00—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments
- D01F9/08—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material
- D01F9/12—Carbon filaments; Apparatus specially adapted for the manufacture thereof
- D01F9/14—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments
- D01F9/20—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products
- D01F9/21—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds
- D01F9/22—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds from polyacrylonitriles
- D01F9/225—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds from polyacrylonitriles from stabilised polyacrylonitriles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
- C08K3/046—Carbon nanorods, nanowires, nanoplatelets or nanofibres
Definitions
- the present invention relates to a carbon fiber bundle, a manufacturing method thereof, and a composite material using the carbon fiber bundle.
- a method for producing a carbon fiber bundle from a polyacrylonitrile-based precursor fiber bundle using a polymer containing acrylonitrile as a main component as a raw material is widely known.
- Patent Document 1 discloses that 1 to 10% of a specific polymerizable unsaturated carboxylic acid alkyl ester and a specific polymerizable unsaturated carboxylic acid for the purpose of efficiently producing and providing a high-quality and high-performance carbon fiber bundle.
- a secondary product comprising a copolymer obtained by copolymerizing an acid in an amount of 0.25 to 5% and flameproofed at 260 ° C. for 5 minutes and further at 280 ° C. for 5 minutes in air at normal pressure.
- SIMS ion mass spectrometry
- a carbon bundle fiber of 389 kgf / mm 2 or more has been proposed.
- This example uses a 2.0 denier polyacrylonitrile-based precursor fiber bundle made of a copolymer of 92.5% acrylonitrile, 1.5% itaconic acid and 6% normal butyl methacrylate.
- the fiber was flame-resistant in air at 240 to 260 ° C. for 30 minutes and heat-treated in a nitrogen stream up to 1,300 ° C. to obtain a carbon fiber bundle, having a strand strength of 501 kgf / mm 2 and a strand modulus of 26 tonf / mm 2 .
- Carbon fiber bundles have been proposed. This significantly lowers the acrylonitrile ratio, thereby reducing the flame resistance and producing a carbon fiber bundle with relatively high tensile strength in a short flame resistance time, even if the fineness of the single fiber is relatively large. Has succeeded.
- Patent Documents 2 and 3 a flame resistant polymer having a polyacrylonitrile-based polymer as a precursor is spun to obtain a flame resistant fiber bundle having a single fiber fineness of 2 dtex or more, and then the flame resistant fiber bundle is carbonized.
- a carbon fiber bundle having a high fineness can be efficiently produced, and after obtaining a flame resistant fiber bundle by heat-treating a polyacrylonitrile-based precursor fiber bundle in a liquid phase, the flame resistant fiber bundle is carbonized.
- a carbon fiber bundle having a strand tensile strength of 4 GPa or more and a strand tensile modulus of 200 GPa or more has been proposed.
- a polyacrylonitrile precursor fiber having a single fiber fineness of 1.7 dtex or more and 4.2 dtex or less comprising a polyacrylonitrile copolymer containing 96 to 99 mol% of acrylonitrile units and 1 to 4 mol% of hydroxyalkyl methacrylate units. 30 ° C. or higher and 450 ° C. measured at a heating rate of 10 ° C./min in an air stream of 100 ml / min at 30 ° C.
- the amount of heat Jb obtained by integrating the heat generation rate of 260 ° C. or more and 290 ° C. or less is 600 kJ / kg or more and 1000 kJ / kg or less.
- the fiber bundle obtained from the heat treatment step is subjected to a carbonization treatment in which the maximum temperature is 1,000 ° C. to 1,700 ° C. in an inert gas atmosphere, and the strand strength required by the ASTM D4018 method is 4
- a polyacrylonitrile precursor fiber having a single fiber fineness of 1.7 dtex or more and 4.6 dtex or less comprising a polyacrylonitrile copolymer containing 96 to 99 mol% of acrylonitrile units and 1 to 4 mol% of hydroxyalkyl methacrylate units. 30 ° C. or higher and 450 ° C. measured at a heating rate of 10 ° C./min in an air stream of 100 ml / min at 30 ° C.
- the fiber bundle obtained from the heat treatment step is subjected to a carbonization treatment in which heat treatment is performed at a maximum temperature of 1,500 ° C. or higher in an inert gas atmosphere, and a carbonization having a strand modulus of 240 GPa or more required by the ASTM D4018 method is performed.
- the manufacturing method of the carbon fiber bundle which obtains the carbon fiber bundle as described in (3) or (4) which has the process made into a fiber bundle.
- the present invention it is possible to increase the single fiber fineness of the polyacrylonitrile-based precursor fiber bundle, thereby reducing the cost and providing a carbon fiber bundle excellent in mechanical properties. Moreover, according to this invention, the composite material using such a carbon fiber bundle is provided.
- the strand strength and the strand elastic modulus are obtained by the ASTM D4018 method.
- a carbon fiber bundle having a single fiber fineness of 0.8 dtex or more and 2.1 dtex or less, a strand strength of 4.9 GPa or more and a strand elastic modulus of 200 GPa or more.
- the fluctuation rate of the single fiber elastic modulus in the single fiber tensile test is preferably 20% or less.
- a heat treatment step is performed by heat-treating the polyacrylonitrile-based precursor fiber bundle in an oxidizing atmosphere in which the temperature is raised within a temperature range of 220 to 300 ° C. for 80 minutes or more and 240 minutes or less.
- a process for producing the carbon fiber bundle (a carbon fiber bundle having a strand strength of 4.9 GPa or more and a strand elastic modulus of 200 GPa or more).
- polyacrylonitrile-based precursor fiber bundle one or a plurality of polyacrylonitrile-based precursors selected from the group consisting of the following polyacrylonitrile-based precursor fiber bundles of ia and polyacrylonitrile-based precursor fiber bundles of ia Fiber bundles can be used.
- a certain type of precursor fiber bundle may correspond to both ia and ia, or may correspond to only one of ia and ia.
- a polyacrylonitrile precursor fiber bundle composed of a polyacrylonitrile copolymer containing 96 to 99 mol% of acrylonitrile units and 1 to 4 mol% of hydroxyalkyl methacrylate units.
- the single fiber fineness is 1.7 dtex or more and 4.2 dtex or less.
- the amount of heat Jb obtained by integrating the heat generation rate of 260 ° C. or more and 290 ° C. or less is 600 kJ / kg or more and 1000 kJ / kg or less.
- a carbon fiber bundle having a single fiber fineness of 0.8 dtex or more and 2.5 dtex or less, a strand strength of 3.0 GPa or more and a strand elastic modulus of 240 GPa or more.
- the fluctuation rate of the single fiber elastic modulus in the single fiber tensile test is preferably 20% or less.
- a heat treatment step (flameproofing) is performed by heat-treating the polyacrylonitrile-based precursor fiber bundle in an oxidizing atmosphere in which the temperature is raised within a temperature range of 220 to 300 ° C. for 80 minutes or more and 240 minutes or less.
- a carbonization treatment in which heat treatment is performed at a maximum temperature of 1,500 ° C. or higher in an inert gas atmosphere to obtain a carbonized fiber bundle having a strand elastic modulus of 240 GPa or more.
- a process for producing the carbon fiber bundle (a carbon fiber bundle having a strand strength of 3.0 GPa or more and a strand elastic modulus of 240 GPa or more).
- polyacrylonitrile-based precursor fiber bundle one or a plurality of polyacrylonitrile-based precursors selected from the group consisting of the polyacrylonitrile-based precursor fiber bundle of ib and the polyacrylonitrile-based precursor fiber bundle of ib shown below Fiber bundles can be used.
- a certain type of precursor fiber bundle may correspond to both ib and iib, or may correspond to only one of ib and iib.
- a polyacrylonitrile-based precursor fiber bundle comprising a polyacrylonitrile-based copolymer containing 96 to 99 mol% of acrylonitrile units and 1 to 4 mol% of hydroxyalkyl methacrylate units.
- the single fiber fineness is 1.7 dtex or more and 4.6 dtex or less.
- the amount of heat Jb obtained by integrating the heat generation rate of 260 ° C. or more and 290 ° C. or less is 600 kJ / kg or more and 1000 kJ / kg or less.
- the “polyacrylonitrile-based precursor fiber bundle” is a fiber bundle for carbon fibers made of a polymer containing acrylonitrile as a main component.
- the ratio of the acrylonitrile unit in the monomer unit constituting the polymer is, for example, 94 mol% or more, and further 96 mol% or more.
- the polyacrylonitrile polymer that can be used as a raw material for the polyacrylonitrile-based precursor fiber bundle which is a raw material for the carbon fiber bundle of the present invention, includes a polyacrylonitrile homopolymer or copolymer, or a mixture thereof.
- the copolymer is preferably a copolymer containing a hydroxyalkyl methacrylate unit, and the content of the hydroxyalkyl methacrylate unit is preferably 1 to 4 mol%.
- the carboxylic acid ester group of the hydroxyalkyl methacrylate unit is thermally decomposed into a carboxylic acid group at a high temperature of 250 ° C. or higher. If the content of the hydroxyalkyl methacrylate unit in the copolymer is 1 mol% or more, when the carboxylic acid ester group of the hydroxyalkyl methacrylate unit becomes a carboxylic acid group in the flameproofing step, the flameproofing reaction is performed. Sufficient effects to promote can be easily obtained. On the other hand, if it is 4 mol% or less, runaway of the flameproofing reaction can be easily suppressed. Furthermore, it is easy to suppress a decrease in carbonization yield due to elimination of the hydroxyalkyl group in the flameproofing step.
- the lower limit of the content of the hydroxyalkyl methacrylate unit is preferably 1.2 mol% or more from the viewpoint of securing the denseness of the polyacrylonitrile-based precursor fiber bundle (hereinafter referred to as “precursor fiber bundle” as appropriate), and has higher performance.
- the amount of 1.5 mol% or more is more preferable in that a carbon fiber can be obtained.
- the upper limit of the content of the hydroxyalkyl methacrylate unit is preferably 4.0 mol% or less from the viewpoint of suppressing a runaway reaction in the flameproofing step, and 3.0 from the viewpoint of suppressing a decrease in carbonization yield. The mol% or less is more preferable.
- hydroxyalkyl methacrylate used as a raw material for the hydroxyalkyl methacrylate unit examples include 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, 4-hydroxybutyl methacrylate, monoglyceryl methacrylate, and tetrahydrofurfuryl methacrylate. It is done. Furthermore, these hydroxyalkyl methacrylates may be used in combination.
- 2-hydroxyethyl methacrylate has a hydroxyethyl group elimination temperature of 240 ° C. or higher in the flameproofing process, has a sufficient bulk to improve oxygen permeability, and the hydroxyethyl group is eliminated. It is suitable as a constituent component of the copolymer used in the present invention from the viewpoints of little decrease in mass when it is produced and industrial availability.
- the polyacrylonitrile-based copolymer contains acrylonitrile units and hydroxyalkyl methacrylate units, but may contain other monomer units as necessary. Alternatively, the polyacrylonitrile-based copolymer may be composed of acrylonitrile units and hydroxyalkyl methacrylate units. Instead of the hydroxyalkyl methacrylate unit, for example, a hydroxyalkyl acrylate unit can be used.
- a vinyl monomer copolymerizable with acrylonitrile is preferable.
- (meth) acrylate esters such as methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, butyl (meth) acrylate, hexyl (meth) acrylate, chloride
- Vinyl halides such as vinyl, vinyl bromide, vinylidene chloride, acids such as (meth) acrylic acid, itaconic acid, crotonic acid and their salts, maleic imide, phenylmaleimide, (meth) acrylamide, styrene, ⁇ - Examples include methylstyrene and vinyl acetate. These may be used alone or in combination of two or more.
- the content of other monomer units in the copolymer is preferably 3.0 mol% or less in consideration of the content of acrylonitrile units and hydroxyalkyl methacrylate units.
- an acrylonitrile-based polymer that can be used as a raw material for the polyacrylonitrile-based precursor fiber bundle, which is a raw material for the carbon fiber bundle of the present invention
- a polymerization method for obtaining an acrylonitrile-based polymer that can be used as a raw material for the polyacrylonitrile-based precursor fiber bundle which is a raw material for the carbon fiber bundle of the present invention
- redox polymerization in an aqueous solution, suspension in a heterogeneous system examples thereof include polymerization and emulsion polymerization using a dispersant, but are not limited thereto.
- a polyacrylonitrile-based precursor fiber bundle for carbon fiber bundles is obtained by dissolving a spinning stock solution having a polymer concentration of 15 to 30% by mass obtained by dissolving a polyacrylonitrile-based polymer in a solvent, and an aqueous solution having a solvent concentration of 30 to 70% by mass.
- a coagulated yarn is obtained by discharging it into a coagulation bath having a temperature of 20 to 50 ° C., and the coagulated yarn is drawn. The spinning method will be described below.
- ⁇ Preparation of spinning dope> The above polymer is dissolved in a solvent by a known method to obtain a spinning dope.
- a solvent organic solvents such as dimethylacetamide, dimethylsulfoxide, dimethylformamide, and aqueous solutions of inorganic compounds such as zinc chloride and sodium thiocyanate can be used.
- An organic solvent is preferable in that the precursor fiber does not contain a metal and the process is simplified.
- dimethylacetamide is preferably used in that the density of the precursor fiber bundle is high.
- the spinning dope preferably has a polymer concentration of a certain level or more so as to obtain a dense coagulated yarn and to have an appropriate viscosity and fluidity.
- the concentration of the polymer in the spinning dope is preferably in the range of 15 to 30% by mass, more preferably in the range of 18 to 25% by mass.
- the coagulation bath liquid is generally an aqueous solution of the same solvent as the spinning dope. At this time, water functions as a poor solvent for the polymer.
- a spinning method of the polyacrylonitrile-based precursor fiber bundle of the carbon fiber bundle obtained in the present invention a known method can be adopted, and specific examples include a wet spinning method, a dry wet spinning method, a dry spinning method, and the like. Among these, the wet spinning method is preferably used from the viewpoint of productivity.
- a coagulated yarn can be obtained by discharging the above spinning solution into a coagulation bath through a spinneret and spinning it.
- the coagulation bath condition is important for obtaining a dense structure necessary for the precursor fiber for carbon fiber and ensuring coagulability that enables high productivity.
- As the coagulation bath conditions a solvent concentration of 30% by mass to 70% by mass and a temperature of 20 ° C. or more and 50 ° C. or less are preferable. If the coagulation bath conditions are within this range, a precursor fiber bundle can be obtained while maintaining an appropriate coagulation rate. Moreover, the roundness of the single fiber of the precursor fiber bundle described later can be controlled in the coagulation process in the spinning process.
- the solvent concentration in the coagulation bath is 70% by mass or less, the exchange rate of the solvent and water on the surface of the spinning dope discharged into the coagulation bath exceeds the diffusion rate of water into the spinning dope, Precursor fibers can be easily obtained, and furthermore, adhesion between single fibers of the precursor fiber bundle can be easily suppressed.
- the solvent concentration is preferably 67% by mass or less and more preferably 50% by mass or less from the viewpoint of further suppressing adhesion between single fibers. preferable.
- the concentration of the solvent in the coagulation bath is 30% by mass or more, the exchange rate of the solvent and water on the surface of the spinning dope discharged into the coagulation bath is significantly higher than the diffusion rate of water into the spinning dope. This can be easily suppressed, and a dense precursor fiber bundle can be obtained within a range in which rapid shrinkage of the coagulated yarn does not occur, preferably 35% by mass or more, and more preferably 40% by mass or more. .
- the cross-sectional shape of the single fiber of the precursor fiber bundle varies depending on the coagulation bath conditions. When the concentration is in the range of 30% by weight to 70% by weight, the roundness representing the cross-sectional shape is maintained within a good range in terms of performance expression of the carbon fiber bundle and resin impregnation.
- the exchange rate of the solvent and water on the surface of the spinning dope discharged into the coagulation bath is significantly higher than the diffusion rate of water into the spinning dope.
- a dense precursor fiber bundle can be easily obtained within a range that can be easily suppressed and abrupt shrinkage of the coagulated yarn does not occur.
- it is 20 degreeC or more, the replacement
- the coagulation bath temperature is preferably 25 ° C. or higher, more preferably 35 ° C. or higher.
- the coagulation bath temperature is preferably 45 ° C. or lower.
- the single fiber fineness of the polyacrylonitrile-based precursor fiber bundle in the present invention is preferably 1.7 dtex or more and 4.6 dtex or less. If it is 1.7 dtex or more, it is easy to produce a carbon fiber bundle having the desired strand strength and strand elastic modulus with good productivity. On the other hand, if the single fiber fineness of the precursor fiber bundle is 4.6 dtex or less, the cross-sectional double structure is not remarkable in the flameproofing process, and it becomes possible to stably produce a carbon fiber bundle of uniform quality, One of the objects of the present invention is to easily produce a carbon fiber bundle having a strand strength of 3.0 GPa or more and a strand elastic modulus of 240 GPa or more.
- the single fiber fineness of the precursor fiber bundle is 4.2 dtex or less
- another object of the present invention is to easily form a carbon fiber bundle having a strand strength of 4.9 GPa or more and a strand elastic modulus of 200 GPa or more.
- the single fiber fineness is preferably 1.8 dtex or more, and more preferably 1.9 dtex or more.
- the single fiber fineness is preferably 3.5 dtex or less, and more preferably 3.0 dtex or less.
- the carbon fiber bundle of the present invention preferably has 6,000 or more and 50,000 or less single fibers. If the number of single fibers in the carbon fiber bundle is 50,000 or less, the structural non-uniformity in the cross-sectional direction of the carbon fiber bundle between the single fibers generated in the carbon fiber firing step can be easily reduced, and sufficient mechanical performance can be obtained. It is because it becomes easy to express. Moreover, if it is 6,000 or more, it will become easy to ensure productivity.
- the number of carbon fiber bundles is preferably 9,000 or more and 36,000 or less, and more preferably 12,000 or more and 30,000 or less. Further, when the single fiber fineness of the polyacrylonitrile-based precursor fiber bundle exceeds 3.1 dtex, the total fineness does not exceed 110,000 dtex. It is preferable in terms of reducing the number.
- the method for producing the carbon fiber of the present invention will be described.
- the polyacrylonitrile-based precursor fiber bundle is subjected to flameproofing treatment at a temperature of 220 ° C. or higher and 300 ° C. or lower in an oxidizing atmosphere to form a flameproof fiber bundle.
- “under an oxidizing atmosphere” means in the air containing an oxidizing substance such as nitrogen dioxide, sulfur dioxide and oxygen.
- the flameproofing treatment time is preferably 80 to 240 minutes. If the flameproofing treatment time is 80 minutes or more, it is easy to sufficiently diffuse oxygen into the single fibers constituting the precursor fiber bundle, and the strand strength depends on the heat treatment conditions in the subsequent carbonization step. A carbon fiber bundle of 4.9 GPa or more or a carbonized fiber bundle having a strand elastic modulus of 240 GPa or more can be easily obtained. By taking the flameproofing treatment time, it is easy to flameproof up to the inside of the fiber bundle. Moreover, if the flameproofing treatment time is 240 minutes or less, the carbon fiber bundle can be efficiently produced without causing the flameproofing treatment process to impair the productivity in the production process of the carbon fiber bundle.
- the flameproofing time is more preferably 85 minutes to 220 minutes, and still more preferably 95 minutes to 200 minutes.
- the flame strength treatment time is one of the embodiments of the present invention
- the strand strength is 4.9 GPa or more and the strand elastic modulus is 200 GPa or more. It becomes easy to obtain the carbon fiber bundle which becomes.
- the elongation of the polyacrylonitrile-based precursor fiber bundle is ⁇ 5% or less and 5% or more in the process from the start of the flameproofing process to the end of the flameproofing process, the quality of the carbon fiber bundle, particularly the strand The strength does not decrease and sufficient strength is easily developed. If the stretch rate at this time is ⁇ 5% or more and 5% or less, it is easy to stably produce a flame resistant fiber bundle without causing yarn breakage or the like. From the viewpoint of strength development when the fineness is large, the elongation percentage is preferably 0% or more, and more preferably 3% or more.
- the polyacrylonitrile-based precursor fiber bundle is measured at a heating rate of 10 ° C./min in an air stream of 100 ml / min (standard: 30 ° C., 0.10 MPa) using a heat flux type differential scanning calorimeter. It is preferable that the constant temperature heating exothermic curve from 30 ° C. to 450 ° C. satisfies the following conditions.
- the initial processing temperature is a temperature equal to or higher than the temperature at which the flameproofing reaction is started, and the precursor fiber. It is set within a temperature range below the temperature at which the bundle does not melt.
- a higher processing temperature can be set in order to efficiently perform the flameproofing process.
- the present inventors set this temperature range around 260 ° C. in the first half of the flame resistance process and the second half of the flame resistance process.
- the calorific value of 230 ° C or higher and 260 ° C or lower is defined as the calorific value Ja
- the calorific value of 260 ° C or higher and 290 ° C or lower is defined as the calorific value Jb. The quality and performance of fiber bundles were compared.
- the flameproofing reaction and oxygen diffusion are performed in a well-balanced manner, and the cross-sectional double structure of the flameproofing fiber is suppressed in the high-speed flameproofing treatment, and high quality and performance It was found that a carbon fiber bundle with good expression was efficiently obtained, and a precursor fiber bundle having a large single fiber fineness could be uniformly flame-resistant.
- the flameproofing treatment temperature when manufacturing an actual carbon fiber bundle is affected by the equipment used and the treatment time, so the temperature setting during the flameproofing treatment should be raised within the range of 220 to 300 ° C. What is necessary is just to set it as the optimal temperature setting in order to flame-proof the precursor fiber bundle.
- the flameproofing reaction proceeds appropriately in the first half of the flameproofing process, and the precursor fiber bundle can be easily passed through without being melted by heat.
- Ja is 200 kJ / kg or less
- the flameproofing reaction does not proceed at a stretch, and it becomes easy to uniformly flameproof the precursor fiber bundle having a large single fiber fineness.
- the amount of heat Ja is more preferably 150 kJ / kg or more from the viewpoint of productivity.
- 190 kJ / kg or less is more preferable, and 180 kJ / kg or less is more preferable from the viewpoint of more uniformly flameproofing a precursor fiber bundle having a large single fiber fineness. Is particularly preferred.
- the amount of heat Jb is 600 kJ / kg or more, the precursor fiber bundle can be easily flameproofed to the target flameproof fiber density without impairing productivity in the flameproofing step. Further, if it is 1000 kJ / kg or less, the flameproofing reaction proceeds slowly in the flameproofing process, and thus it becomes easy to uniformly flameproof the precursor fiber bundle having a large single fiber fineness, thereby forming a double cross section structure. It becomes easy to suppress.
- the amount of heat Jb is preferably 620 kJ / kg or more from the viewpoint of improving productivity, and more preferably 640 kJ / kg or more from the viewpoint of further improving productivity. Moreover, 900 kJ / kg or less is preferable from a viewpoint of performing a flameproofing treatment on a precursor fiber bundle having a large single fiber fineness more uniformly.
- the amount of heat Ja can be used as an index of flameproofing reactivity in the first half of the flameproofing process
- the amount of heat Jb can be used as an index of flameproofing reactivity in the second half of the flameproofing process.
- the amount of heat Ja and the amount of heat Jb can only be used as an index of the flame resistance reactivity of the precursor fiber bundle
- the processing temperature range applied to the actual flame resistance process is the amount of heat Ja or the amount of heat Jb. It may or may not include a temperature range (ie 230 to 260 ° C. or 260 to 290 ° C.), and 220 to 300 depending on the precursor fiber bundle used, the equipment used, and the processing time. It can adjust suitably in the range of ° C.
- the heat quantity Ja obtained by integrating the heat generation rate of 230 ° C. or higher and 260 ° C. or lower of the constant-temperature heating exothermic curve is integrated, and the heat generation rate of 140 kJ / kg or higher and 200 kJ / kg or lower and 260 ° C. or higher and 290 ° C. or lower is integrated.
- the flame resistant fiber having a calorific value Jb of 600 kJ / kg or more and 1000 kJ / kg or less has a small difference in internal and external structure of the single fiber, and when the flame resistant yarn is fired, a uniform carbon fiber is easily obtained as a single fiber. Is obtained.
- the density of the flameproof fiber bundle obtained by the flameproofing treatment is preferably 1.34 to 1.43 g / cm 3 . If it is 1.34 g / cm 3 or more, it is easy to produce a carbon fiber bundle without reducing the yield of the carbon fiber bundle. In general, it is known that the higher the density of the flame-resistant fiber, the higher the yield of the carbon fiber bundle obtained, but the performance of the carbon fiber bundle decreases.
- the density of the flame-resistant fiber bundle is 1.43 g. If it is / cm 3 or less, it is easy to improve the yield of the obtained carbon fiber bundle while suppressing the performance degradation of the carbon fiber bundle. From the viewpoint of maintaining the performance and improving the yield of the obtained carbon fiber bundle, the density of the flameproof fiber bundle is more preferably 1.34 to 1.38 g / cm 3 .
- Carbon fiber bundles can be obtained by carbonizing fiber bundles obtained from the flameproofing process. After the flameproofing treatment and before the carbonization treatment, a precarbonization treatment can be performed in which the obtained flameproofed fiber bundle is treated in an inert gas at a maximum temperature of 550 ° C or higher and 800 ° C or lower.
- a carbon fiber bundle can be produced by subjecting the obtained flame resistant fiber bundle to a carbonization treatment in an inert gas at a temperature of 800 ° C. or higher and 2,800 ° C. or lower.
- the temperature is set according to the desired mechanical properties of the carbon fiber bundle.
- the maximum temperature of the carbonization treatment is preferably low, 1,000 ° C. or more. 1700 degrees C or less is preferable. From the viewpoint of mechanical properties, 1,100 ° C. or higher and 1600 ° C. or lower is preferable, and 1,200 ° C. or higher and 1400 ° C.
- the elastic modulus can be increased by increasing the processing time, the maximum temperature can be lowered as a result. Furthermore, by increasing the processing time, the temperature gradient can be set gently, which is effective in suppressing defect point formation.
- the temperature gradient is not particularly limited, but it is preferable to set a linear gradient.
- the maximum carbonization temperature is 1,500 ° C. or more. It is preferable to perform the treatment.
- the maximum temperature is preferably 1,800 ° C. or more, more preferably 2,200 ° C. or more.
- a surface treatment may be performed before the sizing treatment step.
- the main components of the sizing agent in the sizing treatment liquid are epoxy resin, epoxy-modified polyurethane resin, polyester resin, phenol resin, polyamide resin, polyurethane resin, polycarbonate resin, polyetherimide resin, polyamideimide resin, polyimide resin, bismaleimide Resins, urethane-modified epoxy resins, polyvinyl alcohol resins, polyvinyl pyrrolidone resins, polyether sulfone resins and the like can be mentioned, and are not particularly limited.
- the content of the sizing agent in the sizing treatment liquid is not particularly limited, but is preferably 0.2 to 20% by mass, more preferably 3 to 10% by mass.
- the content of the sizing agent in the sizing treatment liquid is not particularly limited, but is preferably 0.2 to 20% by mass, more preferably 3 to 10% by mass.
- the solvent or dispersion medium used for the sizing treatment liquid is not particularly limited, it is preferable to use water from the viewpoint of handleability and safety.
- the adhesion amount of the sizing agent with respect to 100% by mass of the carbon fiber bundle is preferably 0.3 to 5% by mass, and more preferably 0.4 to 3% by mass.
- the adhesion amount of the sizing agent is 0.3% by mass or more, it becomes easy to sufficiently impart the desired function to the carbon fiber bundle.
- the adhesion amount of the sizing agent 3% by mass or less, the impregnation property of the matrix resin into the carbon fiber bundle at the time of manufacturing the composite material which is a subsequent process is easily improved.
- the solvent or dispersion medium of the sizing process liquid is removed by drying.
- the conditions at this time are preferably in the range of 10 to 10 minutes at a temperature of 120 to 300 ° C., more preferably in the range of 30 to 4 minutes at a temperature of 150 to 250 ° C.
- the drying temperature By setting the drying temperature to 120 ° C. or higher, the solvent can be easily removed sufficiently.
- the quality of the sizing-treated carbon fiber bundle can be easily maintained by setting the drying temperature to 300 ° C. or less.
- the drying method is not particularly limited, and examples thereof include a method in which the carbon fiber bundle is brought into contact with a hot roll using steam as a heat source and a method in which the carbon fiber bundle is dried in an apparatus in which hot air is circulated. be able to.
- a carbon fiber bundle having a strand strength of 3.0 GPa or more and a strand elastic modulus of 200 GPa or more can be applied to many existing composite materials, but the strand strength is 4.9 GPa or more.
- the strand strength is preferably 5.0 GPa or more, and more preferably 5.1 GPa or more.
- the carbon fiber bundle has a strand elastic modulus of 240 GPa or more, it becomes possible to reduce the mixing ratio of fibers in the composite material in a structure material of a wind turbine blade that requires rigidity of the material. This is preferable in terms of weight reduction. Furthermore, it is more preferable that the strand elastic modulus is 265 GPa or more.
- the single fiber fineness of the carbon fiber bundle is 0.00 due to the restriction of the single fiber fineness of the polyacrylonitrile-based precursor fiber bundle described above.
- the range is 8 to 2.1 dTex.
- the average single fiber fineness is in the range of 0.8 to 2.1 dtex, a high-strength carbon fiber bundle can be easily obtained even if the single fiber fineness is relatively large.
- the single fiber fineness of the carbon fiber bundle having a strand strength of 4.9 GPa or more and a strand elastic modulus of 200 GPa or more is preferably 0.90 dTex or more and 1.8 dTex or less, and 1.0 dTex or more and 1.4 dTex or less. Is more preferable.
- the single fiber fineness of the carbon fiber bundle is 0.00 due to the restriction of the single fiber fineness of the polyacrylonitrile-based precursor fiber bundle.
- the range is 8 to 2.5 dTex.
- the average single fiber fineness is in the range of 0.8 to 2.5 dtex, a high elastic modulus carbon fiber bundle can be easily obtained even if the single fiber fineness is relatively large.
- the single fiber fineness of the carbon fiber bundle having a strand strength of 3.0 GPa or more and a strand elastic modulus of 240 GPa or more is preferably 0.90 dTex or more and 2.3 dTex or less, and more preferably 1.0 dTex or more and 1.8 dTex or less.
- the polyacrylonitrile-based precursor fiber bundle is oxidized in a temperature range of 220 to 300 ° C.
- the polyacrylonitrile precursor fiber bundle of ib described above is used as the polyacrylonitrile precursor fiber bundle, and Production using one or more polyacrylonitrile-based precursor fiber bundles selected from the group consisting of iib polyacrylonitrile-based precursor fiber bundles Law by variation of the single fiber elastic modulus at a single fiber tensile test can be obtained carbon fiber bundle is 20% or less.
- the cross-sectional shape of the single fiber of the carbon fiber bundle of the present invention is not particularly limited, and may be any of a circular shape, an elliptical shape, and an empty bean shape.
- roundness is adopted as an index representing the cross-sectional shape of a single fiber.
- the roundness of a perfect circle is 1.00, and this value decreases as the shape moves away from the perfect circle.
- the roundness of the elliptical shape and the empty bean shape is smaller than 1.00.
- the numerical value becomes smaller as the ratio of the major axis to the minor axis increases.
- the cross-sectional shape of the single fiber of the precursor fiber bundle approximately matches the cross-sectional shape of the single fiber of the carbon fiber bundle, it can be considered as a form derived from the precursor fiber.
- the solvent concentration of the coagulation bath liquid is low, the coagulation rate is relatively fast, so that a precursor fiber bundle having a small roundness and a sparse structure is obtained.
- the structure of the precursor fiber bundle is dense, a carbon fiber bundle having a high strength is easily obtained.
- the carbon fiber bundle having a small roundness has a larger gap between the single fibers, and as a result, the impregnation property of the resin can be further improved. Therefore, it is easy to form a composite with a resin, and a composite material with higher performance can be obtained.
- the carbon fiber having a large roundness has a high rotational symmetry of the shape. Therefore, in the comparison of carbon fibers having the same fineness, the minimum stiffness value of the second moment of section is the largest, and the single fiber The straightness is higher. As a result, the strength is excellent.
- the cross-sectional shape of the precursor fiber can be selected depending on the properties and performance of the target composite material.
- the cross-sectional shape of a single fiber of a preferable carbon fiber bundle has a roundness of 0.70 to 0.99. If the roundness is 0.70 or more, the denseness of the precursor fiber bundle is likely to be obtained, and a tendency to easily obtain a high-strength carbon fiber bundle is strong. If it is 0.99 or less, the oxygen diffusibility inside the fiber bundle and further inside the single fiber in the flameproofing process can be easily set to a sufficient level. A more preferable range is 0.79 or more and 0.98 or less.
- ⁇ Variation rate of single fiber elastic modulus in single fiber tensile test One single fiber is taken out from the carbon fiber bundle, and the elasticity of the single fiber is tested using a universal testing machine (Instron 5500 (trade name) manufactured by Instron) under the test conditions of a test length of 5 mm and a pulling speed of 0.5 mm / min. The modulus is measured, and this is repeated until 100 test results are obtained from the same carbon fiber bundle sample.
- a universal testing machine Instron 5500 (trade name) manufactured by Instron
- Fluctuation rate (%) (standard deviation / average value) ⁇ 100
- the variation rate was determined according to the above, and this was taken as the variation rate of the elastic modulus between fibers (the variation rate of the single fiber elastic modulus in the single fiber tensile test).
- the cross-sectional area of the carbon fiber used for calculating the elastic modulus is obtained from the density of the carbon fiber bundle and the single fiber fineness of the carbon fiber: The cross-sectional area of the carbon fiber was calculated according to the single fiber fineness of the carbon fiber (mass per unit length) / density of the carbon fiber bundle.
- ⁇ Constant temperature heating exothermic curve of precursor fiber bundle was measured by a heat flux type differential scanning calorimeter as follows. First, the precursor fiber bundle was cut into a length of 4.0 mm, 4.0 mg was precisely weighed, and packed in 50 ⁇ l (product name: P / N SSC000E030) made of a sealed sample container Ag manufactured by SII, The lid was covered with a mesh cover made by SII (trade name: P / N 50-037) (450 ° C./15 minutes, heat-treated in air).
- This acrylonitrile copolymer was dissolved in dimethylacetamide to prepare a 21% by mass spinning dope.
- the spinning dope is discharged through a spinneret (spinning nozzle) having a hole number of 24,000 and a hole diameter of 60 ⁇ m into a coagulation bath composed of an aqueous dimethylacetamide solution having a concentration of 45 mass% and a temperature of 35 ° C., and the discharge linear velocity from the spinneret surface
- the second oil bath continues to have the same composition and concentration as the first oil bath
- the oil agent treatment liquid was again applied to the fiber bundle.
- the fiber bundle to which the oil agent treatment liquid was applied again was dried using a heating roll, and dry heat drawing was performed 1.34 times between the heating rolls whose rotation speed was adjusted to a predetermined condition.
- the total draw ratio from the swollen yarn at this time was 7.4 times.
- the moisture content was adjusted by applying water to the fiber bundle with a touch roll to obtain a precursor fiber bundle having a single fiber fineness of 2.5 dtex.
- the precursor fiber bundle was subjected to a flameproofing treatment at a stretch rate of 5% under a temperature distribution of 220 to 260 ° C. for 107 minutes to obtain a flameproof fiber bundle having a density of 1.35 g / cm 3 .
- the obtained flame-resistant fiber bundle was further pre-carbonized in a nitrogen atmosphere at 700 ° C. and 3.0% elongation for 1.4 minutes, and then in a nitrogen atmosphere at 1,300 ° C. and ⁇ 4.0%.
- the carbon fiber bundle was obtained by carbonizing at an elongation rate of 1.0 min. Thereafter, the carbon fiber bundle was subjected to a surface treatment by an electrolytic oxidation method, and then a sizing treatment.
- the sizing agent used was 80 parts by mass of “Epicoat 828 (trade name)” manufactured by Japan Epoxy Resin Co., Ltd. as the main agent, and 20 parts by mass of “Pluronic F88 (trade name)” manufactured by Asahi Denka Co., Ltd. as the emulsifier. And an aqueous dispersion prepared by phase inversion emulsification. 1% by mass of this sizing agent was attached to 100% by mass of the carbon fiber bundle, and after a drying treatment, a carbon fiber bundle was obtained. When the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.3 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex
- the variation rate of the single fiber elastic modulus between the single fibers was 16.5%
- the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg
- the heat quantity Jb was 725 kJ / kg.
- a carbon fiber bundle was obtained in the same manner as in Example 1 except that it was changed to (mol%).
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.2 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.26 dtex
- the variation rate of the single fiber elastic modulus between the single fibers was 13.3%
- the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 168 kJ / kg
- the heat quantity Jb was 722 kJ / kg.
- Example 3 A carbon fiber bundle was obtained in the same manner as in Example 1 except that the carbonization temperature was 1,550 ° C.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.1 GPa and the strand elastic modulus was 256 GPa.
- the single fiber fineness of the carbon fiber was 1.21 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 17.2%, and the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg, and the heat quantity Jb was 725 kJ / kg.
- Example 4 A carbon fiber bundle was obtained in the same manner as in Example 1 except that the coagulation bath concentration was 45% by mass, the coagulation bath temperature was 25 ° C., and the flameproofing time was 100 minutes.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.3 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex, the fluctuation rate of the single fiber elastic modulus between the single fibers was 15.2%, and the roundness was 0.79.
- the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 175 kJ / kg, and the heat quantity Jb was 740 kJ / kg.
- Example 5 A carbon fiber bundle was obtained in the same manner as in Example 2 except that the coagulation bath concentration was 50% by mass and the coagulation bath temperature was 35 ° C.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.3 GPa and the strand elastic modulus was 232 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 17.0%, and the roundness was 0.86.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg, and the heat quantity Jb was 725 kJ / kg.
- Example 6 A carbon fiber bundle was obtained in the same manner as in Example 2 except that the coagulation bath concentration was 50% by mass and the coagulation bath temperature was 40 ° C.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.2 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.26 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 17.3%, and the roundness was 0.88.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 172 kJ / kg, and the heat quantity Jb was 727 kJ / kg.
- Example 7 A carbon fiber bundle was obtained in the same manner as in Example 1 except that the coagulation bath concentration was 60% by mass and the coagulation bath temperature was 45 ° C.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.2 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex, the fluctuation rate of the single fiber elastic modulus between single fibers was 17.5%, and the roundness was 0.93.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 168 kJ / kg, and the heat quantity Jb was 722 kJ / kg.
- Example 8 A carbon fiber bundle was obtained in the same manner as in Example 1 except that the coagulation bath concentration was 67% by mass and the coagulation bath temperature was 35 ° C.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.2 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.26 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 17.7%, and the roundness was 0.95.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 163 kJ / kg, and the heat quantity Jb was 710 kJ / kg.
- the carbon fiber bundle was obtained in the same manner as in Example 1 except that the coagulation bath concentration was 67% by mass and the coagulation bath temperature was 45 ° C.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.2 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.26 dtex
- the variation rate of the single fiber elastic modulus between the single fibers was 17.8%
- the roundness was 0.98.
- the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 159 kJ / kg
- the heat quantity Jb was 698 kJ / kg.
- Example 10 The spinning stock solution is discharged through a spinneret (spinning nozzle) having a pore number of 36,000 and a hole diameter of 60 ⁇ m into a coagulation bath composed of a dimethylacetamide aqueous solution having a concentration of 45% by mass and a temperature of 35 ° C., and the discharge linear velocity from the spinneret surface.
- a precursor fiber bundle was obtained in the same manner as in Example 1 except that a fiber bundle (swelling yarn) was obtained by taking it up at a rate 0.45 times as high as. Using this precursor fiber bundle, carbon fibers were obtained in the same manner as in Example 1 except that the flameproofing treatment time was 85 minutes and the flameproofing elongation was ⁇ 4%.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.8 GPa and the strand elastic modulus was 235 GPa. Further, the single fiber fineness of the carbon fiber was 1.04 dtex, the variation rate of the single fiber elastic modulus between single fibers was 11.6%, and the roundness was 0.82. Further, the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 190 kJ / kg, and the heat quantity Jb was 745 kJ / kg.
- Example 11 The spinning dope is discharged through a spinneret (spinning nozzle) having a hole number of 24,000 and a hole diameter of 60 ⁇ m into a coagulation bath composed of an aqueous dimethylacetamide solution having a concentration of 45 mass% and a temperature of 35 ° C., and the discharge linear velocity from the spinneret surface
- a precursor fiber bundle was obtained in the same manner as in Example 1 except that a fiber bundle (swelling yarn) was obtained by taking it up at a rate 0.44 times that of.
- carbon fibers were obtained in the same manner as in Example 1 except that the flameproofing treatment time was 85 minutes and the flameproofing elongation was set to -2%.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.7 GPa and the strand elastic modulus was 235 GPa. Further, the single fiber fineness of the carbon fiber was 0.95 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 12.2%, and the roundness was 0.82. Further, the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 185 kJ / kg, and the heat quantity Jb was 740 kJ / kg.
- the oil treatment liquid is guided to the first oil bath made of the oil treatment liquid dispersed in water at a concentration of 1%, applied to the fiber bundle, and once squeezed with a guide, then the second oil composition having the same composition and concentration as the first oil bath It led to the oil bath and the oil agent treatment liquid was again applied to the fiber bundle.
- This fiber bundle was dried using a heating roll, and dry-heat-stretched 1.7 times between heating rolls whose rotation speed was adjusted to a predetermined condition. The total draw ratio from the swollen yarn at this time is 9.0 times. Except for these, a precursor fiber bundle with a single fiber fineness of 2.3 dtex was obtained in the same manner as in Example 1.
- Carbon fibers were obtained in the same manner as in Example 1 except that the above precursor fiber bundle was used, the flameproofing treatment time was 85 minutes, and the flameproofing elongation was 1.0%.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.4 GPa and the strand elastic modulus was 235 GPa.
- the single fiber fineness of the carbon fiber was 1.12 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 12.8%, and the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 175 kJ / kg, and the heat quantity Jb was 730 kJ / kg.
- Example 13 The spinning stock solution is discharged through a spinneret (spinning nozzle) having a hole number of 15,000 and a hole diameter of 60 ⁇ m into a coagulation bath composed of an aqueous dimethylacetamide solution having a concentration of 45% by mass and a temperature of 35 ° C., and the discharge linear velocity from the spinneret surface
- a precursor fiber bundle having a single fiber fineness of 3.5 dtex was obtained in the same manner as in Example 1 except that the fiber was taken up at a rate 0.23 times as high as that in Example 1.
- the above precursor fiber bundle was subjected to a flame resistance treatment at a stretch rate of 5.0% under a temperature distribution of 220 to 260 ° C. for 240 minutes to obtain a flame resistant fiber bundle.
- the obtained flame-resistant fiber bundle was further pre-carbonized in a nitrogen atmosphere at 700 ° C. and an elongation of 3.0% for 2.7 minutes, and then in a nitrogen atmosphere at 1,300 ° C. and ⁇ 3.5%.
- a carbon fiber bundle was obtained in the same manner as in Example 1 except that the carbonization treatment was performed at an elongation rate of 2.5 minutes for 2.5 minutes.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.1 GPa and the strand elastic modulus was 235 GPa.
- the single fiber fineness of the carbon fiber was 1.69 dtex
- the fluctuation rate of the single fiber elastic modulus between the single fibers was 18.5%
- the roundness was 0.84.
- the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 150 kJ / kg
- the heat quantity Jb was 690 kJ / kg.
- Example 14 A carbon fiber was obtained in the same manner as in Example 1 except that the flameproofing treatment time was 160 minutes and the flameproofing elongation was 5.0%.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.2 GPa and the strand elastic modulus was 235 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex, the fluctuation rate of the single fiber elastic modulus between the single fibers was 11.3%, and the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg, and the heat quantity Jb was 725 kJ / kg.
- Example 15 A carbon fiber was obtained in the same manner as in Example 1 except that the flameproofing treatment time was 240 minutes and the flameproofing elongation was 5.0%.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.3 GPa and the strand elastic modulus was 238 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex, the fluctuation rate of the single fiber elastic modulus between the single fibers was 9.5%, and the roundness was 0.85.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg, and the heat quantity Jb was 725 kJ / kg.
- a carbon fiber bundle was obtained in the same manner as in Example 1 except that it was changed to (mol%).
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 5.2 GPa and the strand elastic modulus was 233 GPa.
- the single fiber fineness of the carbon fiber was 1.26 dtex
- the variation rate of the single fiber elastic modulus between the single fibers was 18.2%
- the roundness was 0.85.
- the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 198 kJ / kg
- the heat quantity Jb was 850 kJ / kg.
- Example 17 A carbon fiber bundle was obtained in the same manner as in Example 1 except that the flameproof elongation was -6.0%.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 4.9 GPa and the strand elastic modulus was 230 GPa.
- the single fiber fineness of the carbon fiber was 1.41 dtex, the variation rate of the single fiber elastic modulus between single fibers was 16.8%, and the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg, and the heat quantity Jb was 725 kJ / kg.
- Example 18 The spinning stock solution is discharged through a spinneret (spinning nozzle) having a pore number of 40,000 and a hole diameter of 60 ⁇ m into a coagulation bath composed of an aqueous dimethylacetamide solution having a concentration of 45 mass% and a temperature of 35 ° C., and the discharge linear velocity from the spinneret surface
- a carbon fiber was produced in the same manner as in Example 1 except that a fiber bundle (swelling yarn) was obtained by taking it up at a rate 0.32 times that of.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 4.9 GPa and the strand elastic modulus was 225 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex
- the single fiber elastic modulus variation rate was 26.3%
- the roundness was 0.83.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg
- the heat quantity Jb was 725 kJ / kg.
- Example 1 A carbon fiber bundle was obtained in the same manner as in Example 1 except that the flameproofing treatment time was 70 minutes and the flameproofing elongation was 5.0%.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 4.2 GPa and the strand elastic modulus was 232 GPa.
- the single fiber fineness of the carbon fiber was 1.27 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 23.2%, and the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 170 kJ / kg, and the heat quantity Jb was 725 kJ / kg.
- the oil treatment liquid is introduced into the first oil bath made of the oil treatment liquid dispersed in the water and applied to the fiber bundle, and after squeezing with a guide, the oil composition treatment liquid continues to the second oil bath having the same composition and concentration as the first oil bath. Then, the oil treatment liquid was again applied to the fiber bundle.
- the fiber bundle to which the oil agent treatment liquid was applied again was dried using a heating roll, and dry heat drawing was performed 1.3 times between the heating rolls whose rotation speed was adjusted to a predetermined condition. The total draw ratio from the swollen yarn at this time was 7.3 times. Thereafter, the moisture content was adjusted by applying water to the fiber bundle with a touch roll to obtain a precursor fiber bundle having a single fiber fineness of 2.5 dtex.
- the above precursor fiber bundle was subjected to a flameproofing treatment at a stretch rate of 5.0% under a temperature distribution of 220 to 260 ° C. for 300 minutes to obtain a flameproofed fiber bundle.
- the obtained flame-resistant fiber bundle was further pre-carbonized in an atmosphere of nitrogen at 700 ° C. and an elongation of 3.0% for 3.7 minutes, followed by 1,300 ° C. and ⁇ 4.0% in a nitrogen atmosphere. Was carbonized for 3.2 minutes.
- a carbon fiber bundle was obtained in the same manner as Example 1 except for these. When the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 3.8 GPa and the strand elastic modulus was 231 GPa.
- the single fiber fineness of the carbon fiber was 1.37 dtex
- the variation rate of the single fiber elastic modulus between single fibers was 27.0%
- the roundness was 0.85.
- the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 190 kJ / kg
- the heat quantity Jb was 1151 kJ / kg.
- Example 3 A carbon fiber bundle was obtained in the same manner as in Example 13 except that the flameproofing treatment was performed at a stretch rate of 5.0% under a temperature distribution of 220 to 260 ° C. for 70 minutes.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 3.8 GPa and the strand elastic modulus was 235 GPa.
- the single fiber fineness of the carbon fiber was 1.75 dtex, the fluctuation rate of the single fiber elastic modulus between the single fibers was 25.1%, and the roundness was 0.82.
- the heat quantity Ja obtained from the heat flux type differential scanning calorimetry was 150 kJ / kg, and the heat quantity Jb was 690 kJ / kg.
- the spinning stock solution is discharged through a spinneret (spinning nozzle) having a pore number of 12,000 and a hole diameter of 60 ⁇ m into a coagulation bath composed of an aqueous dimethylacetamide solution having a concentration of 45 mass% and a temperature of 35 ° C., and the discharge linear velocity from the spinneret surface
- a precursor fiber bundle having a single fiber fineness of 4.5 dtex was obtained in the same manner as in Example 1 except that the fiber was taken up at a rate of 0.18 times the above.
- the above precursor fiber bundle was subjected to a flame resistance treatment at a stretch rate of 5.0% under a temperature distribution of 220 to 260 ° C. for 240 minutes to obtain a flame resistant fiber bundle.
- the obtained flame-resistant fiber bundle was further pre-carbonized in an atmosphere of nitrogen at 700 ° C. and an elongation of 3.0% for 3.7 minutes, followed by 1,300 ° C. and ⁇ 4.0% in a nitrogen atmosphere.
- a carbon fiber bundle was obtained in the same manner as in Example 1 except that the carbonization treatment was performed at an elongation rate of 3.2 minutes for 3.2 minutes.
- the strand strength was 3.5 GPa and the strand elastic modulus was 230 GPa.
- the single fiber fineness of the carbon fiber was 2.43 dtex
- the variation rate of the single fiber elastic modulus between the single fibers was 18.3%
- the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 135 kJ / kg
- the heat quantity Jb was 660 kJ / kg.
- a carbon fiber bundle was obtained in the same manner as in Example 1 except that it was changed to (mol%).
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 4.0 GPa and the strand elastic modulus was 229 GPa.
- the single fiber fineness of the carbon fiber was 1.21 dtex
- the variation rate of the single fiber elastic modulus between the single fibers was 9.2%
- the roundness was 0.82.
- the heat quantity Ja determined by heat flux type differential scanning calorimetry was 139 kJ / kg
- the heat quantity Jb was 650 kJ / kg.
- a precursor fiber bundle having a single fiber fineness of 2.5 dtex produced by the same method as in Example 1 is a carbonization furnace (first carbonization) in a nitrogen atmosphere at a maximum temperature of 1,600 ° C. and an elongation of -3.8%. Furnace) for 2.0 minutes, followed by a carbonization furnace (second carbonization furnace) in a nitrogen atmosphere with a maximum temperature of 2,400 ° C and an elongation of -3.8%. Then, carbon fibers were obtained in the same manner as in Example 1 except that the second carbonization treatment was performed for 2.0 minutes.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 3.7 GPa and the strand elastic modulus was 343 GPa.
- the single fiber fineness of the carbon fiber was 1.17 dtex
- the variation rate of the single fiber elastic modulus between single fibers was 17.0%
- the roundness was 0.82.
- Example 20 A precursor fiber bundle having a single fiber fineness of 3.5 dtex produced by the same method as in Example 13 was obtained in the same manner as in Example 19 to obtain carbon fibers.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 3.4 GPa and the strand elastic modulus was 314 GPa.
- the single fiber fineness of the carbon fiber was 1.60 dtex, the variation rate of the single fiber elastic modulus between the single fibers was 18.8%, and the roundness was 0.84.
- Example 21 Carbon fibers were obtained in the same manner as in Example 19 using a precursor fiber bundle with a single fiber fineness of 4.5 dtex produced by the same method as in Comparative Example 4.
- the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 3.0 GPa and the strand elastic modulus was 294 GPa.
- the single fiber fineness of the carbon fiber was 2.26 dtex, the variation rate of the single fiber elastic modulus between single fibers was 19.1%, and the roundness was 0.85.
- a precursor fiber bundle with a single fiber fineness of 2.5 dtex produced by the same method as in Example 1 is a carbonization furnace (first carbonization furnace) in a nitrogen atmosphere at a maximum temperature of 1,500 ° C. and an elongation of -3.8%. ) In a carbonization furnace (second carbonization furnace) in a nitrogen atmosphere at a maximum temperature of 1,800 ° C. and an elongation of ⁇ 3.8%. Carbon fibers were obtained in the same manner as in Example 1 except that the second carbonization treatment was performed for 0 minute. When the strand physical property of the obtained carbon fiber bundle was measured, the strand strength was 4.0 GPa and the strand elastic modulus was 290 GPa. Moreover, the single fiber fineness of the carbon fiber was 1.21 dtex, the variation rate of the single fiber elastic modulus between single fibers was 17.5%, and the roundness was 0.82.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Textile Engineering (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Inorganic Fibers (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description
(A)等速昇温発熱曲線の230℃以上260℃以下の発熱速度を積分して求めた熱量Jaが140kJ/kg以上200kJ/kg以下、
(B)260℃以上290℃以下の発熱速度を積分して求めた熱量Jbが600kJ/kg以上1000kJ/kg以下。
(C)140×ポリアクリロニトリル系前駆体繊維束の単繊維繊度(dtex)-100≧熱処理時間T1(分)≧140×ポリアクリロニトリル系前駆体繊維束の単繊維繊度(dtex)-270。
(A)等速昇温発熱曲線の230℃以上260℃以下の発熱速度を積分して求めた熱量Jaが140kJ/kg以上200kJ/kg以下、
(B)260℃以上290℃以下の発熱速度を積分して求めた熱量Jbが600kJ/kg以上1000kJ/kg以下、
かつ、前記熱処理工程から得られる繊維束に、不活性ガス雰囲気下で最高温度が1,500℃以上で熱処理する炭素化処理を施し、ASTM D4018法で求められるストランド弾性率が240GPa以上の炭素化繊維束とする工程を有する、(3)または(4)に記載の炭素繊維束を得る炭素繊維束の製造方法。
(A)等速昇温発熱曲線の230℃以上260℃以下の発熱速度を積分して求めた熱量Jaが140kJ/kg以上200kJ/kg以下、
(B)260℃以上290℃以下の発熱速度を積分して求めた熱量Jbが600kJ/kg以上1000kJ/kg以下。
(A)等速昇温発熱曲線の230℃以上260℃以下の発熱速度を積分して求めた熱量Jaが140kJ/kg以上200kJ/kg以下、
(B)260℃以上290℃以下の発熱速度を積分して求めた熱量Jbが600kJ/kg以上1000kJ/kg以下。
上述の重合体を溶剤に公知の方法で溶解して、紡糸原液とする。溶剤としては、ジメチルアセトアミド、ジメチルスルホキシド、ジメチルホルムアミドなどの有機溶剤や、塩化亜鉛、チオシアン酸ナトリウムなどの無機化合物の水溶液を用いることができる。前駆体繊維中に金属を含有せず、また、工程が簡略化される点で有機溶剤が好ましく、その中でも前駆体繊維束の緻密性が高いという点で、ジメチルアセトアミドを用いることが好ましい。
紡糸原液は、緻密な凝固糸を得るため、また、適正な粘度、流動性を有するように、ある程度以上の重合体濃度を有することが好ましい。紡糸原液における重合体の濃度は、15~30質量%の範囲にあることが好ましく、より好ましくは18~25質量%の範囲である。凝固浴液は、一般に紡糸原液と同じ溶剤の水溶液が用いられる。この際、水が重合体の貧溶媒として機能する。
本発明で得られる炭素繊維束のポリアクリロニトリル系前駆体繊維束の紡糸方法としては、公知の方法を採用でき、具体的には湿式紡糸法、乾湿式紡糸法、乾式紡糸法などが挙げられる。これらの中でも生産性の観点から湿式紡糸法が好ましく用いられる。
また、後述する前駆体繊維束の単繊維の真円度は、紡糸工程における凝固工程において制御することが可能である。
(C)140×ポリアクリロニトリル系前駆体繊維束の単繊維繊度(dtex)-100≧熱処理時間T1(分)≧140×ポリアクリロニトリル系前駆体繊維束の単繊維繊度(dtex)-270。
本発明において、ポリアクリロニトリル系前駆体繊維束は、熱流束型示差走査熱量計を用いて100ml/分(基準:30℃、0.10MPa)の空気気流中、昇温速度10℃/分で測定したときの30℃から450℃までの等速昇温発熱曲線が以下の条件を満たすものが好ましい。
(A)等速昇温発熱曲線の230℃以上260℃以下の発熱速度を積分して求めた熱量Jaが140kJ/kg以上200kJ/kg以下、かつ、
(B)等速昇温発熱曲線の260℃以上290℃以下の発熱速度を積分して求めた熱量Jbが600kJ/kg以上1000kJ/kg以下。
上述の等速昇温発熱曲線は、前駆体繊維束中で耐炎化反応が進行する時に発生する熱量を示している。
伸長率(%)=(A-B)/A×100・・・・式(1)
により計算される。
真円度=4πS/L2・・・(2)
真円の真円度は1.00であり、真円から形状が離れるに従いこの数値は小さくなる。したがって、楕円形状、空豆形状の真円度は1.00よりも小さく、例えば楕円形状においては、その長径と短径の比率が高くなれば数値はより小さくなる。また、前駆体繊維束の単繊維の断面形状は、おおよそ炭素繊維束の単繊維の断面形状と一致することから、前駆体繊維由来の形態と考えることができる。単繊維の断面形状決定に大きな影響を及ぼす製造工程として凝固過程がある。凝固浴液の溶剤濃度が高い場合は、凝固速度は比較的遅いため、真円度が大きく、構造が緻密な前駆体繊維束を得ることができる。一方、凝固浴液の溶剤濃度が低い場合は、凝固速度は比較的速いため、真円度が小さく、構造が疎な前駆体繊維束を得る。一般に前駆体繊維束の構造が緻密な場合、炭素繊維束は高強度のものが得られやすい。
ストランド強度及びストランド弾性率は、ASTM D4018の方法に準拠してエポキシ樹脂含浸ストランドの引張物性を測定した。
炭素繊維束より単繊維を1本取り出し、万能試験機(インストロン社製 インストロン5500(商品名))を用いて試長5mm、引張り速度0.5mm/minの試験条件にて単繊維の弾性率を測定し、これを同一の炭素繊維束試料より100本の試験結果が得られるまで繰り返し行い、100本の試験結果により得られた弾性率の平均値と標準偏差より次式:
変動率(%)=(標準偏差/平均値)×100
に従って変動率を求め、これを繊維間の弾性率変動率(単繊維引張り試験における単繊維弾性率の変動率)とした。また、弾性率の計算に用いる炭素繊維の断面積に関しては、炭素繊維束の密度と炭素繊維の単繊維繊度より次式:
炭素繊維の断面積=炭素繊維の単繊維繊度(単位長さ当たりの質量)/炭素繊維束の密度
に従って算出した。
(1)サンプルの作製
長さ5cmに切断した炭素繊維束をエポキシ樹脂(エポマウント主剤:エポマウント硬化剤=100:9(質量比))に包埋し、2cmに切断して横断面を露出させ、鏡面処理した。
更に、繊維の外形を明瞭にするために、サンプルの横断面を次の方法でエッチング処理した。
・使用装置:日本電子(株)JP-170(商品名) プラズマエッチング装置、
・処理条件:(雰囲気ガス:Ar/O2=75/25(体積比)、プラズマ出力:50W、真空度:約120Pa、処理時間:5min。)。
前記(1)及び(2)により得られたサンプルの横断面を、SEM(PHILIPS FEI-XL20(商品名))を用いて観察し、画面上に5個以上の繊維断面が写っている写真を任意に5枚撮影した。
画像解析ソフトウェア(日本ローパー(株)製、製品名:Image-Pro PLUS)を用いて繊維断面の外形をトレースし、周長Lおよび面積Sを計測した。各サンプルについて5枚の写真から任意に20個、ただし、1枚の写真から3個以上の繊維断面を選んで計測し、LおよびSの平均値を求め、次式:
真円度=4πS/L2
により真円度を算出した。
前駆体繊維束の等速昇温発熱曲線は、熱流束型示差走査熱量計により、以下のようにして測定した。先ず、前駆体繊維束を4.0mmの長さに切断し、4.0mgを精秤して、エスアイアイ社製の密封試料容器Ag製50μl(商品名:P/N SSC000E030)中に詰め、エスアイアイ社製メッシュカバーCu製(商品名:P/N 50-037)(450℃/15分間、空気中で熱処理済)で蓋をした。次いで、熱流束型示差走査熱量計:エスアイアイ社製DSC/220(商品名)を用いて、10℃/分の昇温速度、エアー供給量100ml/min(エアー供給量の基準:30℃、0.10MPa)の条件で、室温(30℃)から450℃まで測定した。得られた等速昇温発熱曲線の230℃以上260℃以下の発熱量を熱量Jaとし、260℃以上290℃以下の発熱量を熱量Jbとした。
アクリロニトリル、メタクリル酸2-ヒドロキシエチル、過硫酸アンモニウム-亜硫酸水素アンモニウムおよび硫酸鉄の存在下、水系懸濁重合により共重合し、アクリロニトリル単位/メタクリル酸2-ヒドロキシエチル単位=98.5/1.5(モル%)からなるアクリロニトリル系共重合体を得た。このアクリロニトリル系共重合体をジメチルアセトアミドに溶解し、21質量%の紡糸原液を調製した。孔数24,000、孔直径60μmの紡糸口金(紡糸ノズル)を通して、濃度45質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に紡糸原液を吐出させ、紡糸口金面からの吐出線速度の0.32倍の速度で引き取ることで繊維束(膨潤糸条)を得た。ついで、この繊維束を水洗と同時に5.4倍に延伸し、さらにアミノ変性シリコン/ポリオキシエチレン(6)ラウリルエーテル=91/9(質量比)の油剤組成物が1.5質量%の濃度で水中に分散した油剤処理液からなる第一油浴槽に導き、油剤処理液を繊維束に付与し、ガイドで一旦絞った後、引き続き第一油浴槽と同じ組成・濃度からなる第二油浴槽に導き、再度油剤処理液を繊維素束に付与した。再度油剤処理液を付与した繊維束を加熱ロールを用いて乾燥し、回転速度を所定の条件に調節した加熱ロール間で1.34倍に乾熱延伸をした。この時の膨潤糸条からの全延伸倍率は7.4倍であった。その後、タッチロールにて繊維束に水を付与することで水分率を調整し、単繊維繊度2.5dtexの前駆体繊維束を得た。
アクリロニトリル、メタクリル酸2-ヒドロキシエチル、過硫酸アンモニウム-亜硫酸水素アンモニウムおよび硫酸鉄の存在下、水系懸濁重合により共重合し、アクリロニトリル単位/メタクリル酸2-ヒドロキシエチル単位=98.0/2.0(モル%)にした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.2GPa、ストランド弾性率233GPaであった。また、炭素繊維の単繊維繊度1.26dtex、単繊維間の単繊維弾性率の変動率13.3%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは168kJ/kgであり、熱量Jbは722kJ/kgであった。
炭素化温度を1,550℃とした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.1GPa、ストランド弾性率256GPaであった。また、炭素繊維の単繊維繊度1.21dtex、単繊維間の単繊維弾性率の変動率17.2%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは170kJ/kgであり、熱量Jbは725kJ/kgであった。
凝固浴濃度45質量%、凝固浴温度25℃、耐炎化時間を100分とした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.3GPa、ストランド弾性率233GPaであった。また、炭素繊維の単繊維繊度1.27dtex、単繊維間の単繊維弾性率の変動率15.2%、真円度0.79であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは175kJ/kgであり、熱量Jbは740kJ/kgであった。
凝固浴濃度50質量%、凝固浴温度35℃とした以外は実施例2と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.3GPa、ストランド弾性率232GPaであった。また、炭素繊維の単繊維繊度1.27dtex、単繊維間の単繊維弾性率の変動率17.0%、真円度0.86であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは170kJ/kgであり、熱量Jbは725kJ/kgであった。
凝固浴濃度50質量%、凝固浴温度40℃とした以外は実施例2と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.2GPa、ストランド弾性率233GPaであった。また、炭素繊維の単繊維繊度1.26dtex、単繊維間の単繊維弾性率の変動率17.3%、真円度0.88であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは172kJ/kgであり、熱量Jbは727kJ/kgであった。
凝固浴濃度60質量%、凝固浴温度45℃とした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.2GPa、ストランド弾性率233GPaであった。また、炭素繊維の単繊維繊度1.27dtex、単繊維間の単繊維弾性率の変動率17.5%、真円度0.93であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは168kJ/kgであり、熱量Jbは722kJ/kgであった。
凝固浴濃度67質量%、凝固浴温度35℃とした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.2GPa、ストランド弾性率233GPaであった。また、炭素繊維の単繊維繊度1.26dtex、単繊維間の単繊維弾性率の変動率17.7%、真円度0.95であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは163kJ/kgであり、熱量Jbは710kJ/kgであった。
アクリロニトリル、メタクリル酸2-ヒドロキシエチル、過硫酸アンモニウム-亜硫酸水素アンモニウムおよび硫酸鉄の存在下、水系懸濁重合により共重合し、アクリロニトリル単位/メタクリル酸2-ヒドロキシエチル単位=97.5/2.5(モル%)とし、また、凝固浴濃度67質量%、凝固浴温度45℃とした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.2GPa、ストランド弾性率233GPaであった。また、炭素繊維の単繊維繊度1.26dtex、単繊維間の単繊維弾性率の変動率17.8%、真円度0.98であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは159kJ/kgであり、熱量Jbは698kJ/kgであった。
孔数36,000、孔直径60μmの紡糸口金(紡糸ノズル)を通して、濃度45質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に紡糸原液を吐出させ、紡糸口金面からの吐出線速度の0.45倍の速度で引き取ることで繊維束(膨潤糸条)を得た以外は実施例1と同じ方法で前駆体繊維束を得た。この前駆体繊維束を用い、耐炎化処理時間85分、耐炎化伸張率を-4%とした以外は実施例1と同様にして炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.8GPa、ストランド弾性率235GPaであった。また、炭素繊維の単繊維繊度1.04dtex、単繊維間の単繊維弾性率の変動率11.6%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは190kJ/kgであり、熱量Jbは745kJ/kgであった。
孔数24,000、孔直径60μmの紡糸口金(紡糸ノズル)を通して、濃度45質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に紡糸原液を吐出させ、紡糸口金面からの吐出線速度の0.40倍の速度で引き取ることで繊維束(膨潤糸条)を得た以外は実施例1と同じ方法で前駆体繊維束を得た。この前駆体繊維束を用い、耐炎化処理時間85分、耐炎化伸張率を-2%とした以外は実施例1と同様にして炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.7GPa、ストランド弾性率235GPaであった。また、炭素繊維の単繊維繊度0.95dtex、単繊維間の単繊維弾性率の変動率12.2%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは185kJ/kgであり、熱量Jbは740kJ/kgであった。
濃度45質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に紡糸原液を吐出させ、紡糸原液の吐出線速度の0.35倍の速度で引き取ることで繊維束(膨潤糸条)を得た。ついで、この繊維束に対して水洗と同時に5.3倍の延伸を行い、さらにアミノ変性シリコン/ポリオキシエチレン(6)ラウリルエーテル=91/9(質量比)の油剤組成物が1.5質量%の濃度で水中に分散した油剤処理液からなる第一油浴槽に導き油剤処理液を繊維束に付与し、ガイドで一旦絞った後、引き続き第一油浴槽と同じ組成・濃度からなる第二油浴槽に導き、再度油剤処理液を繊維素束に付与した。この繊維束を加熱ロールを用いて乾燥し、回転速度を所定の条件に調節した加熱ロール間で1.7倍に乾熱延伸をした。この時の膨潤糸条からの全延伸倍率は9.0倍である。これら以外は実施例1と同様の方法で、単繊維繊度2.3dtexの前駆体繊維束を得た。
孔数15,000、孔直径60μmの紡糸口金(紡糸ノズル)を通して、濃度45質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に紡糸原液を吐出させ、紡糸口金面からの吐出線速度の0.23倍の速度で引き取ること以外は実施例1と同様にして単繊維繊度3.5dtexの前駆体繊維束を得た。
耐炎化処理時間160分、耐炎化伸張率を5.0%とした以外は実施例1と同様にして炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.2GPa、ストランド弾性率235GPaであった。また、炭素繊維の単繊維繊度1.27dtex、単繊維間の単繊維弾性率の変動率11.3%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは170kJ/kgであり、熱量Jbは725kJ/kgであった。
耐炎化処理時間240分、耐炎化伸張率を5.0%とした以外は実施例1と同様にして炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.3GPa、ストランド弾性率238GPaであった。また、炭素繊維の単繊維繊度1.27dtex、単繊維間の単繊維弾性率の変動率9.5%、真円度0.85であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは170kJ/kgであり、熱量Jbは725kJ/kgであった。
アクリロニトリル、アクリル酸2-ヒドロキシエチル、過硫酸アンモニウム-亜硫酸水素アンモニウムおよび硫酸鉄の存在下、水系懸濁重合により共重合し、アクリロニトリル単位/アクリル酸2-ヒドロキシエチル単位=98.5/1.5(モル%)にした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度5.2GPa、ストランド弾性率233GPaであった。また、炭素繊維の単繊維繊度1.26dtex、単繊維間の単繊維弾性率の変動率18.2%、真円度0.85であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは198kJ/kgであり、熱量Jbは850kJ/kgであった。
耐炎化伸張率を-6.0%とした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度4.9GPa、ストランド弾性率230GPaであった。また、炭素繊維の単繊維繊度1.41dtex、単繊維間の単繊維弾性率の変動率16.8%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは170kJ/kgであり、熱量Jbは725kJ/kgであった。
孔数40,000、孔直径60μmの紡糸口金(紡糸ノズル)を通して、濃度45質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に紡糸原液を吐出させ、紡糸口金面からの吐出線速度の0.32倍の速度で引き取ることで繊維束(膨潤糸条)を得た以外は実施例1と同様の方法で炭素繊維を製造した。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度4.9GPa、ストランド弾性率225GPaであった。また、炭素繊維の単繊維繊度1.27dtex単繊維間の単繊維弾性率の変動率26.3%、真円度0.83であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは170kJ/kgであり、熱量Jbは725kJ/kgであった。
耐炎化処理時間を70分間、耐炎化伸張率を5.0%とした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度4.2GPa、ストランド弾性率232GPaであった。また、炭素繊維の単繊維繊度1.27dtex、単繊維間の単繊維弾性率の変動率23.2%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは170kJ/kgであり、熱量Jbは725kJ/kgであった。
アクリロニトリル、アクリルアミド、及びメタクリル酸を、過硫酸アンモニウム-亜硫酸水素アンモニウムおよび硫酸鉄の存在下、水系懸濁重合により共重合し、アクリロニトリル単位/アクリルアミド単位/メタクリル酸単位=96/3/1(モル%)からなるアクリロニトリル系共重合体を得た。このアクリロニトリル系共重合体をジメチルアセトアミドに溶解し、21質量%の紡糸原液を調製した。孔数24,000、孔直径60μmの紡糸口金(紡糸ノズル)を通して、濃度60質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に吐出させ、紡糸口金面からの吐出線速度の0.32倍の速度で引き取ることで繊維束(膨潤糸条)を得た。ついで、この繊維束を水洗と同時に5.4倍に延伸し、さらにアミノ変性シリコン/ポリオキシエチレン(6)ラウリルエーテル=91/9(質量比)の油剤組成物が1.5質量%の濃度で水中に分散した油剤処理液からなる第一油浴槽に導き油剤処理液を繊維束に付与し、ガイドで一旦絞った後、引き続き第一油浴槽と同じ組成・濃度からなる第二油浴槽に導き、再度油剤処理液を繊維素束に付与した。再度油剤処理液を付与した繊維束を加熱ロールを用いて乾燥し、回転速度を所定の条件に調節した加熱ロール間で1.3倍に乾熱延伸をした。この時の膨潤糸条からの全延伸倍率は7.3倍であった。その後、タッチロールにて繊維束に水を付与することで水分率を調整し、単繊維繊度2.5dtexの前駆体繊維束を得た。
220~260℃の温度分布の下、伸張率5.0%で70分間耐炎化処理した以外は実施例13と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度3.8GPa、ストランド弾性率235GPaであった。また、炭素繊維の単繊維繊度1.75dtex、単繊維間の単繊維弾性率の変動率25.1%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは150kJ/kgであり、熱量Jbは690kJ/kgであった。
孔数12,000、孔直径60μmの紡糸口金(紡糸ノズル)を通して、濃度45質量%、温度35℃のジメチルアセトアミド水溶液からなる凝固浴中に紡糸原液を吐出させ、紡糸口金面からの吐出線速度の0.18倍の速度で引き取ること以外は実施例1と同様にして単繊維繊度4.5dtexの前駆体繊維束を得た。
アクリロニトリル、メタクリル酸2-ヒドロキシエチル、過硫酸アンモニウム-亜硫酸水素アンモニウムおよび硫酸鉄の存在下、水系懸濁重合により共重合し、アクリロニトリル単位/メタクリル酸2-ヒドロキシエチル単位=95.0/5.0(モル%)にした以外は実施例1と同様にして炭素繊維束を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度4.0GPa、ストランド弾性率229GPaであった。また、炭素繊維の単繊維繊度1.21dtex、単繊維間の単繊維弾性率の変動率9.2%、真円度0.82であった。更に、熱流束型示差走査熱量測定より求められる熱量Jaは139kJ/kgであり、熱量Jbは650kJ/kgであった。
実施例1と同様の方法で製造した単繊維繊度2.5dtexの前駆体繊維束を、最高温度1,600℃、伸長率-3.8%で、窒素雰囲気の炭素化炉(第1炭素化炉)中にて2.0分間の炭素化処理を付し、続いて最高温度2,400℃、伸長率-3.8%で、窒素雰囲気の炭素化炉(第2炭素化炉)中にて2.0分間、第二の炭素化処理に付す以外は実施例1と同様にして炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度3.7GPa、ストランド弾性率343GPaであった。また、炭素繊維の単繊維繊度1.17dtex、単繊維間の単繊維弾性率の変動率17.0%、真円度0.82であった。
実施例13と同様の方法で製造した単繊維繊度3.5dtexの前駆体繊維束を実施例19と同様にして、炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度3.4GPa、ストランド弾性率314GPaであった。また、炭素繊維の単繊維繊度1.60dtex、単繊維間の単繊維弾性率の変動率18.8%、真円度0.84であった。
比較例4と同様の方法で製造した単繊維繊度4.5dtexの前駆体繊維束を用い、実施例19と同様にして、炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度3.0GPa、ストランド弾性率294GPaであった。また、炭素繊維の単繊維繊度2.26dtex、単繊維間の単繊維弾性率の変動率19.1%、真円度0.85であった。
実施例1と同様の方法で製造した単繊維繊度2.5dtexの前駆体繊維束を、最高温度1,500℃、伸長率-3.8%で、窒素雰囲気の炭素化炉(第1炭化炉)中にて2.0分間の炭素化処理を付し、続いて最高温度1,800℃、伸長率-3.8%で、窒素雰囲気の炭素化炉(第2炭化炉)中にて2.0分間、第二の炭素化処理に付す以外は実施例1と同様にして炭素繊維を得た。得られた炭素繊維束のストランド物性を測定したところ、ストランド強度4.0GPa、ストランド弾性率290GPaであった。また、炭素繊維の単繊維繊度1.21dtex、単繊維間の単繊維弾性率の変動率17.5%、真円度0.82であった。

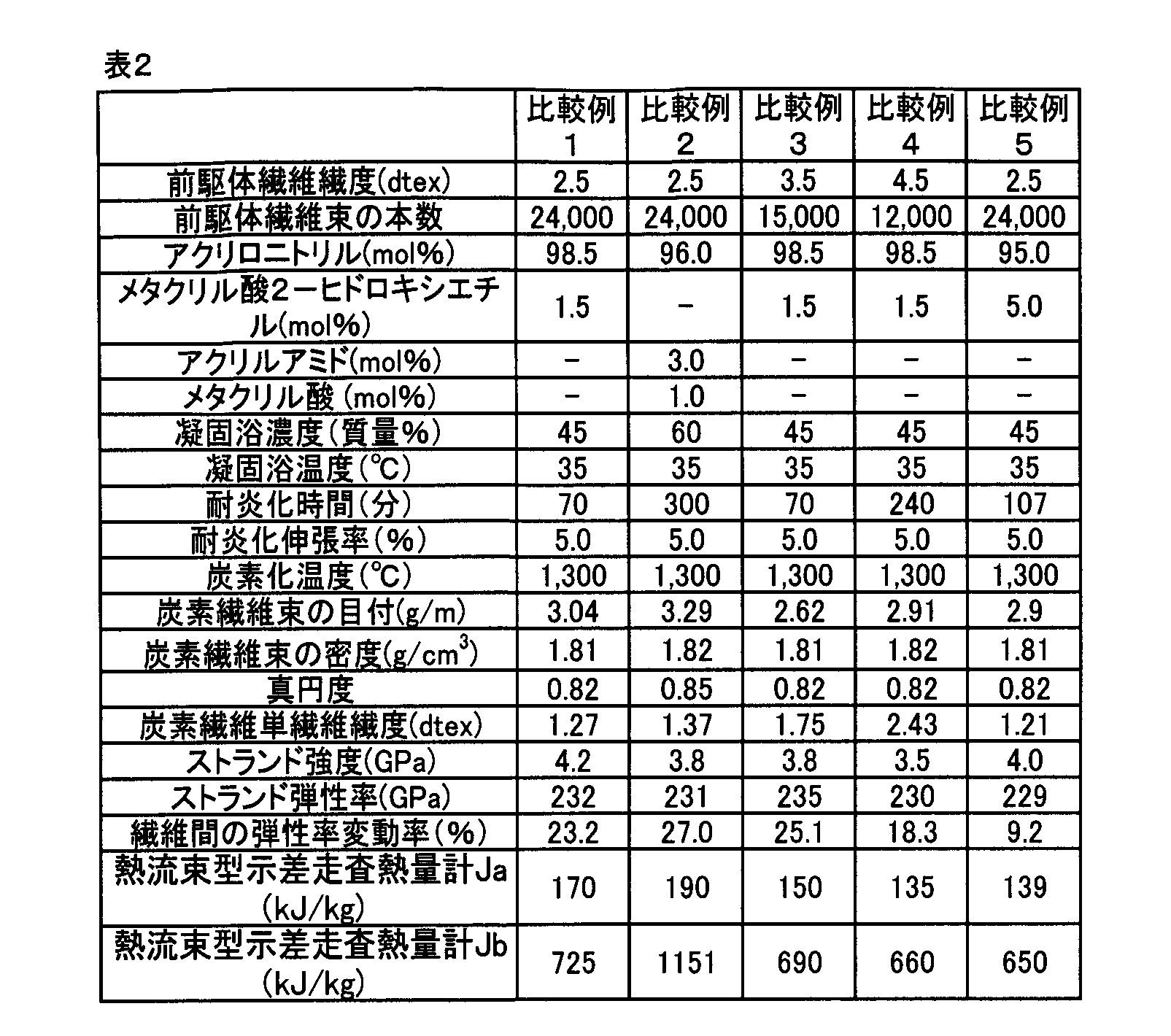

Claims (18)
- 炭素繊維束の単繊維繊度が0.8dtex以上2.1dtex以下、ASTM D4018法で求められるストランド強度が4.9GPa以上かつ前記方法で求められるストランド弾性率が200GPa以上である炭素繊維束。
- 単繊維引張り試験における単繊維弾性率の変動率が20%以下である請求項1に記載の炭素繊維束。
- 炭素繊維束の単繊維繊度が0.8dtex以上2.5dtex以下、ASTM D4018法で求められるストランド強度が3.0GPa以上かつ前記方法で求められるストランド弾性率が240GPa以上である炭素繊維束。
- 単繊維引張り試験における単繊維弾性率の変動率が20%以下である請求項3に記載の炭素繊維束。
- 前記ASTM D4018法で求められるストランド弾性率が265GPa以上である請求項4に記載の炭素繊維束。
- 前記単繊維繊度が1.1dtex以上である請求項1~5のいずれか1項に記載の炭素繊維束。
- 単繊維本数が6,000本以上50,000本以下である請求項1~5のいずれか1項に記載の炭素繊維束。
- 単繊維本数が36,000本以下である請求項7に記載の炭素繊維束。
- 総繊度が4800dtex以上56000dtex以下である請求項1~5のいずれか1項に記載の炭素繊維束。
- アクリロニトリル単位96~99モル%とメタクリル酸ヒドロキシアルキル単位1~4モル%を含むポリアクリロニトリル系共重合体からなる単繊維繊度が1.7dtex以上4.2dtex以下のポリアクリロニトリル系前駆体繊維束、および/または、熱流束型示差走査熱量計を用いて、30℃、0.10MPa基準の流量で100ml/分の空気気流中、昇温速度10℃/分で測定した30℃以上450℃以下の等速昇温発熱曲線が以下の(A)および(B)の条件を満たす単繊維繊度が1.7dtex以上4.2dtex以下のポリアクリロニトリル系前駆体繊維束を、220~300℃の温度範囲内で昇温する酸化性雰囲気下で、80分以上240分以下熱処理する熱処理工程を有する、請求項1または2に記載の炭素繊維束を得る炭素繊維束の製造方法:
(A)等速昇温発熱曲線の230℃以上260℃以下の発熱速度を積分して求めた熱量Jaが140kJ/kg以上200kJ/kg以下、
(B)260℃以上290℃以下の発熱速度を積分して求めた熱量Jbが600kJ/kg以上1000kJ/kg以下。 - 前記熱処理工程における熱処理時間T1(分)が、以下の(C)の条件を満たす請求項10に記載の炭素繊維束の製造方法:
(C)140×ポリアクリロニトリル系前駆体繊維束の単繊維繊度(dtex)-100≧熱処理時間T1(分)≧140×ポリアクリロニトリル系前駆体繊維束の単繊維繊度(dtex)-270。 - 前記熱処理工程から得られる繊維束に、不活性ガス雰囲気下で最高温度が1,000℃~1,700℃で熱処理する炭素化処理を施し、ASTM D4018法で求められるストランド強度が4.9GPa以上の炭素化繊維束とする請求項10または11に記載の炭素繊維束の製造方法。
- アクリロニトリル単位96~99モル%とメタクリル酸ヒドロキシアルキル単位1~4モル%を含むポリアクリロニトリル系共重合体からなる単繊維繊度が1.7dtex以上4.6dtex以下のポリアクリロニトリル系前駆体繊維束、および/または、熱流束型示差走査熱量計を用いて、30℃、0.10MPa基準の流量で100ml/分の空気気流中、昇温速度10℃/分で測定した30℃以上450℃以下の等速昇温発熱曲線が以下の(A)および(B)の条件を満たす単繊維繊度が1.7dtex以上4.6dtex以下のポリアクリロニトリル系前駆体繊維束を、220~300℃の温度範囲内で昇温する酸化性雰囲気下で、80分以上240分以下熱処理する熱処理工程を有し、
(A)等速昇温発熱曲線の230℃以上260℃以下の発熱速度を積分して求めた熱量Jaが140kJ/kg以上200kJ/kg以下、
(B)260℃以上290℃以下の発熱速度を積分して求めた熱量Jbが600kJ/kg以上1000kJ/kg以下、
かつ、前記熱処理工程から得られる繊維束に、不活性ガス雰囲気下で最高温度が1,500℃以上で熱処理する炭素化処理を施し、ASTM D4018法で求められるストランド弾性率が240GPa以上の炭素化繊維束とする工程を有する、
請求項3または4に記載の炭素繊維束を得る炭素繊維束の製造方法。 - 前記最高温度が1,800℃以上である請求項13に記載の炭素繊維束の製造方法。
- 前記ポリアクリロニトリル系前駆体繊維束の単繊維本数が、6,000本以上50,000本以下である請求項10~14のいずれか一項に記載の炭素繊維束の製造方法。
- 前記熱処理工程におけるポリアクリロニトリル系前駆体繊維束の伸長率が-5%以上5%以下である請求項10~14のいずれか一項に記載の炭素繊維束の製造方法。
- 単繊維引張り試験における単繊維弾性率の変動率が20%以下の炭素繊維束とする請求項10~16のいずれか一項に記載の炭素繊維束の製造方法。
- 請求項1~9のいずれか一項に記載の炭素繊維束を含有する樹脂系複合材料。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020147029176A KR101658298B1 (ko) | 2012-04-18 | 2013-04-18 | 탄소 섬유속 및 탄소 섬유의 제조 방법 |
| JP2013519881A JP5765420B2 (ja) | 2012-04-18 | 2013-04-18 | 炭素繊維束および炭素繊維の製造方法 |
| EP19178945.2A EP3572564A1 (en) | 2012-04-18 | 2013-04-18 | Carbon fiber bundle and method of producing carbon fibers |
| KR1020167024975A KR101813433B1 (ko) | 2012-04-18 | 2013-04-18 | 탄소 섬유속 및 탄소 섬유의 제조 방법 |
| CN201380020045.9A CN104246032B (zh) | 2012-04-18 | 2013-04-18 | 碳纤维束以及碳纤维的制造方法 |
| US14/395,185 US9873777B2 (en) | 2012-04-18 | 2013-04-18 | Carbon fiber bundle and method of producing carbon fibers |
| EP13778358.5A EP2840172B1 (en) | 2012-04-18 | 2013-04-18 | Carbon fiber bundle and method of producing carbon fibers |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012094551 | 2012-04-18 | ||
| JP2012-094551 | 2012-04-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013157612A1 true WO2013157612A1 (ja) | 2013-10-24 |
Family
ID=49383564
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/061535 Ceased WO2013157612A1 (ja) | 2012-04-18 | 2013-04-18 | 炭素繊維束および炭素繊維の製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9873777B2 (ja) |
| EP (2) | EP2840172B1 (ja) |
| JP (1) | JP5765420B2 (ja) |
| KR (2) | KR101813433B1 (ja) |
| CN (1) | CN104246032B (ja) |
| HU (1) | HUE046228T2 (ja) |
| TW (1) | TWI527947B (ja) |
| WO (1) | WO2013157612A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160168761A1 (en) * | 2013-07-30 | 2016-06-16 | Toray Industries, Inc. | Carbon fiber bundle and stabilized fiber bundle |
| JP2017020142A (ja) * | 2015-07-14 | 2017-01-26 | 三菱レイヨン株式会社 | 炭素繊維とその製造方法 |
| JP2018145292A (ja) * | 2017-03-06 | 2018-09-20 | 三井化学株式会社 | ポリアミド樹脂組成物及びその成形品 |
| JP2018145541A (ja) * | 2017-03-02 | 2018-09-20 | 三菱ケミカル株式会社 | 炭素繊維束及びその製造方法 |
| US10189985B2 (en) | 2013-12-23 | 2019-01-29 | Cytec Industries Inc. | Polyacrylonitrile (PAN) polymers with low polydispersity index (PDI) and carbon fibers made therefrom |
| WO2019244830A1 (ja) | 2018-06-18 | 2019-12-26 | 東レ株式会社 | 炭素繊維およびその製造方法 |
| KR20210079410A (ko) | 2016-07-22 | 2021-06-29 | 미쯔비시 케미컬 주식회사 | 다공질 기재, 다공질 전극, 탄소 섬유지, 탄소 섬유지의 제조 방법, 다공질 기재의 제조 방법 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101813433B1 (ko) | 2012-04-18 | 2017-12-28 | 미쯔비시 케미컬 주식회사 | 탄소 섬유속 및 탄소 섬유의 제조 방법 |
| JP2019500511A (ja) | 2015-12-31 | 2019-01-10 | ユーティー−バテル, エルエルシー | 多目的商用繊維からカーボン繊維を製造する方法 |
| US11286583B2 (en) * | 2016-06-30 | 2022-03-29 | Toray Industries, Inc. | Carbon fiber bundle and method of manufacturing same |
| CN111868315A (zh) * | 2018-03-27 | 2020-10-30 | 东丽株式会社 | 丙烯腈系纤维束的制造方法及碳纤维束的制造方法 |
| JP7341648B2 (ja) * | 2018-10-05 | 2023-09-11 | 帝人株式会社 | 前駆体繊維束の製造方法及び炭素繊維束の製造方法並びに炭素繊維束 |
| KR20210034432A (ko) | 2019-09-20 | 2021-03-30 | 윤준혁 | 초음파 센서를 이용한 스크린도어 |
| CN111621878B (zh) * | 2020-03-13 | 2023-04-28 | 北京化工大学 | 一种具有表面沟槽结构的大直径高强中模、高强高模碳纤维及其制备方法 |
| KR102534975B1 (ko) * | 2021-08-03 | 2023-05-26 | ㈜제이에스페브릭스 | 탄소 섬유를 포함하는 습식 부직포 및 이의 제조방법 |
| CN114481367A (zh) * | 2021-12-30 | 2022-05-13 | 吉林宝旌炭材料有限公司 | 一种35k大丝束碳纤维及其制备方法 |
| CN116876115B (zh) * | 2023-07-18 | 2026-01-09 | 中国科学院宁波材料技术与工程研究所 | 一种高强度超高模量的聚丙烯腈基碳纤维及其制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0931758A (ja) | 1988-04-26 | 1997-02-04 | Toray Ind Inc | 炭素繊維 |
| JP2004300600A (ja) | 2003-03-31 | 2004-10-28 | Toray Ind Inc | 耐炎化繊維、炭素繊維およびそれらの製造方法 |
| JP2008202207A (ja) | 2007-01-26 | 2008-09-04 | Toray Ind Inc | 炭素繊維束およびその製造方法 |
| JP2011046942A (ja) * | 2009-07-31 | 2011-03-10 | Mitsubishi Rayon Co Ltd | ポリアクリロニトリル系共重合体、炭素繊維用ポリアクリロニトリル系前駆体繊維、および炭素繊維の製造方法 |
| WO2012050171A1 (ja) * | 2010-10-13 | 2012-04-19 | 三菱レイヨン株式会社 | 炭素繊維前駆体繊維束、炭素繊維束、及びそれらの利用 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5742925A (en) | 1980-08-22 | 1982-03-10 | Toho Rayon Co Ltd | Production of high-performance carbon fiber strand |
| TW459075B (en) | 1996-05-24 | 2001-10-11 | Toray Ind Co Ltd | Carbon fiber, acrylic fiber and preparation thereof |
| JPH11124743A (ja) | 1997-10-20 | 1999-05-11 | Toray Ind Inc | 炭素繊維および炭素繊維強化複合材料 |
| JP2003328271A (ja) | 2002-05-08 | 2003-11-19 | Toho Tenax Co Ltd | 炭素繊維ストランド、及び炭素繊維強化樹脂 |
| JP2004244258A (ja) | 2003-02-13 | 2004-09-02 | Toray Ind Inc | 炭素繊維強化炭素複合材料用炭素繊維およびその製造方法 |
| JP4360233B2 (ja) * | 2004-03-11 | 2009-11-11 | 東レ株式会社 | ゴルフシャフト |
| US7749479B2 (en) * | 2006-11-22 | 2010-07-06 | Hexcel Corporation | Carbon fibers having improved strength and modulus and an associated method and apparatus for preparing same |
| KR101813433B1 (ko) | 2012-04-18 | 2017-12-28 | 미쯔비시 케미컬 주식회사 | 탄소 섬유속 및 탄소 섬유의 제조 방법 |
-
2013
- 2013-04-18 KR KR1020167024975A patent/KR101813433B1/ko active Active
- 2013-04-18 CN CN201380020045.9A patent/CN104246032B/zh active Active
- 2013-04-18 US US14/395,185 patent/US9873777B2/en active Active
- 2013-04-18 KR KR1020147029176A patent/KR101658298B1/ko active Active
- 2013-04-18 EP EP13778358.5A patent/EP2840172B1/en active Active
- 2013-04-18 EP EP19178945.2A patent/EP3572564A1/en active Pending
- 2013-04-18 HU HUE13778358A patent/HUE046228T2/hu unknown
- 2013-04-18 TW TW102113803A patent/TWI527947B/zh active
- 2013-04-18 JP JP2013519881A patent/JP5765420B2/ja active Active
- 2013-04-18 WO PCT/JP2013/061535 patent/WO2013157612A1/ja not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0931758A (ja) | 1988-04-26 | 1997-02-04 | Toray Ind Inc | 炭素繊維 |
| JP2004300600A (ja) | 2003-03-31 | 2004-10-28 | Toray Ind Inc | 耐炎化繊維、炭素繊維およびそれらの製造方法 |
| JP2008202207A (ja) | 2007-01-26 | 2008-09-04 | Toray Ind Inc | 炭素繊維束およびその製造方法 |
| JP2011046942A (ja) * | 2009-07-31 | 2011-03-10 | Mitsubishi Rayon Co Ltd | ポリアクリロニトリル系共重合体、炭素繊維用ポリアクリロニトリル系前駆体繊維、および炭素繊維の製造方法 |
| WO2012050171A1 (ja) * | 2010-10-13 | 2012-04-19 | 三菱レイヨン株式会社 | 炭素繊維前駆体繊維束、炭素繊維束、及びそれらの利用 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160168761A1 (en) * | 2013-07-30 | 2016-06-16 | Toray Industries, Inc. | Carbon fiber bundle and stabilized fiber bundle |
| US11105022B2 (en) * | 2013-07-30 | 2021-08-31 | Toray Industries, Inc. | Carbon fiber bundle and stabilized fiber bundle |
| US10189985B2 (en) | 2013-12-23 | 2019-01-29 | Cytec Industries Inc. | Polyacrylonitrile (PAN) polymers with low polydispersity index (PDI) and carbon fibers made therefrom |
| TWI675853B (zh) * | 2013-12-23 | 2019-11-01 | 美商塞特工業公司 | 具有低聚合度分佈性指數(pdi)的聚丙烯腈(pan)聚合物及從其製成的碳纖維 |
| US10647844B2 (en) * | 2013-12-23 | 2020-05-12 | Cytec Industries Inc. | Polyacrylonitrile (PAN) polymers with low polydispersity index (PDI) and carbon fibers made therefrom |
| JP2017020142A (ja) * | 2015-07-14 | 2017-01-26 | 三菱レイヨン株式会社 | 炭素繊維とその製造方法 |
| KR20210079410A (ko) | 2016-07-22 | 2021-06-29 | 미쯔비시 케미컬 주식회사 | 다공질 기재, 다공질 전극, 탄소 섬유지, 탄소 섬유지의 제조 방법, 다공질 기재의 제조 방법 |
| JP2018145541A (ja) * | 2017-03-02 | 2018-09-20 | 三菱ケミカル株式会社 | 炭素繊維束及びその製造方法 |
| JP2018145292A (ja) * | 2017-03-06 | 2018-09-20 | 三井化学株式会社 | ポリアミド樹脂組成物及びその成形品 |
| WO2019244830A1 (ja) | 2018-06-18 | 2019-12-26 | 東レ株式会社 | 炭素繊維およびその製造方法 |
| KR20210019029A (ko) | 2018-06-18 | 2021-02-19 | 도레이 카부시키가이샤 | 탄소 섬유 및 그의 제조 방법 |
Also Published As
| Publication number | Publication date |
|---|---|
| HUE046228T2 (hu) | 2020-02-28 |
| EP2840172A4 (en) | 2015-04-22 |
| TWI527947B (zh) | 2016-04-01 |
| US9873777B2 (en) | 2018-01-23 |
| CN104246032A (zh) | 2014-12-24 |
| EP2840172A1 (en) | 2015-02-25 |
| KR20140135257A (ko) | 2014-11-25 |
| KR101813433B1 (ko) | 2017-12-28 |
| TW201402893A (zh) | 2014-01-16 |
| US20150094401A1 (en) | 2015-04-02 |
| CN104246032B (zh) | 2017-12-05 |
| EP2840172B1 (en) | 2019-06-12 |
| JP5765420B2 (ja) | 2015-08-19 |
| KR20160110563A (ko) | 2016-09-21 |
| KR101658298B1 (ko) | 2016-09-20 |
| JPWO2013157612A1 (ja) | 2015-12-21 |
| EP3572564A1 (en) | 2019-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5765420B2 (ja) | 炭素繊維束および炭素繊維の製造方法 | |
| US11970791B2 (en) | Carbon fiber bundle and method of producing carbon fiber bundle | |
| JP2012188781A (ja) | 炭素繊維およびその製造方法 | |
| KR102197333B1 (ko) | 폴리아크릴로니트릴계 내염화 섬유, 탄소섬유 및 그의 제조방법 | |
| JP4875238B2 (ja) | 炭素繊維およびその前駆体の製造方法並びに油剤付着方法 | |
| JP2013181264A (ja) | 炭素繊維束 | |
| JP2004060126A (ja) | 炭素繊維及びその製造方法 | |
| JP2004183194A (ja) | 炭素繊維束、炭素繊維用アクリロニトリル系前駆体繊維及びその製造方法 | |
| JP5842343B2 (ja) | 炭素繊維前駆体アクリル繊維束の製造方法 | |
| JP2020128614A (ja) | 炭素繊維束およびその製造方法 | |
| JP2007023476A (ja) | 炭素繊維用アクリロニトリル系前駆体繊維の製造方法 | |
| JP2012211814A (ja) | 炭素繊維前駆体アクリル繊維の品質管理方法 | |
| JP2006233360A (ja) | 炭素繊維および炭素繊維の製造方法 | |
| JPH06228810A (ja) | 炭素繊維用前駆体繊維の製造方法 | |
| JP2005248358A (ja) | 炭素繊維前駆体繊維 | |
| JP2007284807A (ja) | アクリロニトリル系炭素繊維前駆体繊維束及びその製造方法、並びに炭素繊維束 | |
| JPH03241014A (ja) | 低比重炭素繊維 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2013519881 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13778358 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20147029176 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14395185 Country of ref document: US Ref document number: 2013778358 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |