WO2014103704A1 - P1,p4-ジ(ウリジン5'-)テトラホスフェートの製造法 - Google Patents
P1,p4-ジ(ウリジン5'-)テトラホスフェートの製造法 Download PDFInfo
- Publication number
- WO2014103704A1 WO2014103704A1 PCT/JP2013/083100 JP2013083100W WO2014103704A1 WO 2014103704 A1 WO2014103704 A1 WO 2014103704A1 JP 2013083100 W JP2013083100 W JP 2013083100W WO 2014103704 A1 WO2014103704 A1 WO 2014103704A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- ump
- utp
- phosphoric acid
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
- A61K31/7072—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid having two oxo groups directly attached to the pyrimidine ring, e.g. uridine, uridylic acid, thymidine, zidovudine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/10—Expectorants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
Definitions
- the present invention relates to an efficient new process for the production of P 1 , P 4 -di (uridine 5 ′-) tetraphosphate.
- P 1 , P 4 -di (uridine 5 ′-) tetraphosphate represented by the following formula [I] (hereinafter referred to as “UP 4 U”) or a salt thereof is a treatment for keratoconjunctival epithelial disorder associated with dry eye. It is used as a medicine. In addition, it is a compound that has an action of inducing excretion and is expected to be developed as an expectorant or a pneumonia therapeutic agent.
- Non-patent document 1 a method of reacting uridine 5′-cyclic triphosphate prepared by dehydration condensation reaction of uridine 5′-triphosphate (UTP) with uridine 5′-monophosphate (UMP) ( Non-patent document 1) and its improved method (patent document 1) have been reported.
- UP 4 U can be synthesized at a high yield of 80% or more at the laboratory level, but there is a problem in mass-producing the method on an industrial scale.
- UTP as a raw material is converted into a form of a tertiary amine salt such as tri-n-butylamine. It is necessary to use in. For this reason, a process of forming a salt with a tertiary amine after making UTP trisodium salt into free UTP (UTP free) by ion exchange column chromatography in advance is necessary.
- UTP-free is a very unstable substance, and it is very difficult to use UTP-free as a raw material in mass production on an industrial scale.
- the method of Patent Document 1 using UTP as a starting material is not an optimal method for industrial mass production of UP 4 U. Therefore, the present invention can avoid a decrease in synthesis efficiency due to decomposition of raw materials by not using UTP free and avoiding the dehydration operation of UTP salt for synthesizing uridine 5′-cyclic triphosphate.
- the purpose is to develop a new manufacturing method.
- the one-pot synthesis method and the old pyrophosphoric acid diimidazolide method were both carried out in an anhydrous environment using an organic solvent such as dimethylformamide (DMF) as a solvent for the UP 4 U synthesis reaction.
- an organic solvent such as dimethylformamide (DMF)
- UMP, UTP, pyrophosphoric acid, and the like used as reaction raw materials are all highly hydrophilic compounds, and in order to use these raw materials for the reaction, a dehydration step has to be performed in each step.
- pyrophosphate has poor solubility in organic solvents, and there is a problem that it needs to be handled with care.
- the inventor further examined the above two improved synthesis methods.
- the one-pot synthesis method was examined in detail, as shown in the examples described later, the synthesis yield of the target product UP 4 U was greatly reduced to about 40%.
- the method of patent document 1 was implemented as a one-pot synthesis method, since various by-products were produced, it turned out that the refinement
- the present inventor has (a) uridine 5′-diphosphate (UDP), UMP or pyrophosphate. And a phosphate active compound synthesized by condensing a compound selected from the group consisting of imidazole, benzimidazole or 1,2,4-triazole, which may have a substituent, and (b) UMP , UDP, UTP, and a phosphate compound selected from the group consisting of pyrophosphate or a salt thereof (excluding UTP-free), and as a catalyst, iron (II) ion, iron (III) ion, trivalent aluminum ion Simple reaction in water or a hydrophilic organic solvent in the presence of a metal ion selected from the group consisting of trivalent lanthanum ion and trivalent cerium ion By the way, you can avoid dehydration of UTP-free use and UTP salts, addition-
- the present invention relates to a phosphoric acid active compound represented by the following formula [II] or [III] and a phosphoric acid compound selected from the group consisting of UMP, UDP, UTP and pyrophosphoric acid or a salt thereof (however, UTP-free).
- a metal ion selected from the group consisting of iron (II) ion, iron (III) ion, trivalent aluminum ion, trivalent lanthanum ion and trivalent cerium ion in water or a hydrophilic organic solvent
- the present invention provides a process for producing P 1 , P 4 -di (uridine 5 ′-) tetraphosphate, which comprises a step of reacting with
- R 1 represents a uridyl group bonded at the 5′-position;
- X represents one heterocyclic group selected from the group consisting of an imidazolyl group, a benzimidazolyl group and a 1,2,4-triazolyl group
- N represents an integer of 1 or 2.
- X represents one heterocyclic group selected from the group consisting of an imidazolyl group, a benzimidazolyl group and a 1,2,4-triazolyl group
- UP 4 U can be synthesized in a high yield while avoiding UTP-free use and UTP salt dehydration which are not suitable for industrial mass production. Further, in the production method of the present invention, almost no by-products are produced, so that the synthesized UP 4 U can be easily purified. Furthermore, a complicated dehydration step can be omitted by carrying out the reaction under hydrophilic conditions. That is, the production method of the present invention is a very suitable method for industrial mass synthesis of UP 4 U.
- the HPLC chart of the analysis of products in a reaction mixture shown.
- the peak No. 10 indicates UP 4 U which is the target product
- the peak No. 6 indicates UDP which is the raw material.
- the peak No. 19 indicates UP 4 U which is the target product
- the peak No. 5 indicates UMP which is the raw material.
- the peak No. 16 shows the target product UP 4 U, and the peak No. 6 shows the raw material UMP.
- Pyrophosphate imidazolide method former law (without catalyst) at the time of synthesizing the UP 4 U, shows the HPLC chart of the analysis of the products in the reaction mixture.
- the peak No. 17 indicates the target product UP 4 U
- the No. 5 peak indicates the raw material UMP.
- pyrophosphate diimidazolide method former law catalogalyst ZnCl 2
- it shows the HPLC chart of the analysis of the products in the reaction mixture.
- the peak No. 16 shows UP 4 U which is a target organism, and the peak No.
- UMP which is a raw material.
- Pyrophosphate diimidazolide method former law catalyst tetrazole
- the peak No. 20 indicates the target product UP 4 U
- the No. 5 peak indicates the raw material UMP.
- the phosphoric acid active compound used in the present invention is a compound represented by the formula [II] or [III], which may have UMP, UDP or pyrophosphoric acid and a substituent, imidazole, benzimidazole or 1 , 2,4-triazole is synthesized by condensation.
- R 1 represents a uridyl group bonded at the 5′-position;
- X represents one heterocyclic group selected from the group consisting of an imidazolyl group, a benzimidazolyl group and a 1,2,4-triazolyl group
- N represents an integer of 1 or 2.
- X represents one heterocyclic substituent selected from the group consisting of an imidazolyl group, a benzimidazolyl group, and a 1,2,4-triazoyl group
- heterocyclic substituent represented by X examples include an imidazolyl group, a benzimidazolyl group, and a 1,2,4-triazoyl group.
- these heterocyclic groups may have a substituent in the heterocyclic group, and examples of the substituent include a C 1-6 alkyl group, a nitro group, and a cyano group.
- Such a phosphoric acid active compound examples include UDP-phosphate imidazolide, UDP-phosphate triazolide, UMP-phosphate imidazolide, and pyrophosphate diimidazolide. These compounds are known methods (Nucleic Acids Research). 4, 2843 (1977), Journal of American Chemical Society, vol. 126, 9548 (2004)).
- a synthetic solution of a phosphoric acid active compound such as UDP-phosphate imidazolide, UDP-phosphate triazolide, UMP-phosphate imidazolide, or pyrophosphate diimidazolide, or a concentrated solution thereof is used as it is or as necessary. It can be purified and used.
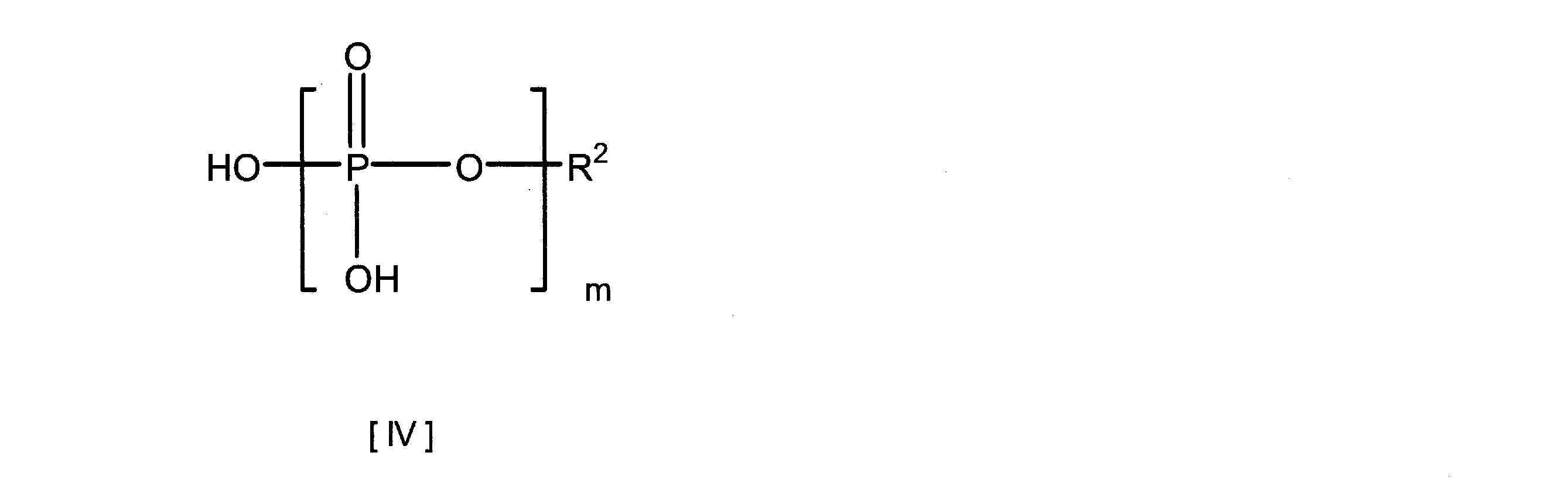
- the phosphoric acid compound used in the present invention is a compound represented by the following formula (IV) or a salt thereof, specifically, UMP, UDP, UTP, pyrophosphoric acid or a salt thereof excluding UTP free. Can be mentioned.
- R 2 represents a hydrogen atom or a uridyl group bonded at the 5 ′ position; m represents an integer of 1 to 3)
- the phosphoric acid compound can be used in the form of an alkali metal salt such as sodium or potassium, a tertiary ammonium salt such as triethylammonium or tributylammonium, or a quaternary ammonium salt such as tetraethylammonium salt or tetrabutylammonium.
- an alkali metal salt such as sodium or potassium
- a tertiary ammonium salt such as triethylammonium or tributylammonium
- quaternary ammonium salt such as tetraethylammonium salt or tetrabutylammonium.
- Such a combination of the raw material compounds of the phosphoric acid active compound and the phosphoric acid compound may be determined appropriately, and preferably used in the following combinations.
- this molar ratio is, for example, (1) When the above combination 1 is used, the molar ratio is 3: 8, 4: 7, 5: 6, 6: 5, 7: 4, 8: 3, 9: 2, 10: 1, (2) In the case of Combination 2, the molar ratio is 1:10, 2: 9, 3: 8, 4: 7, 5: 6, 6: 5, 7: 4, or 8: 3, (3) In combination 3, any of 4: 7, 5: 6, 6: 5, 7: 4, 8: 3, 9: 2, 10: 1, (4) For combination 4, any of 4: 7, 5: 6, 6: 5, 7: 4, 8: 3, 9: 2, 10: 1, It may be within the range of any two numerical values exemplified here.
- the metal ion used in the present invention can be supplied to the reaction system by being added to the solution in the form of a metal salt containing the target metal ion to form a metal ion in water or a hydrophilic organic solvent.
- a metal salt containing the target metal ion to form a metal ion in water or a hydrophilic organic solvent.
- metal salts metal halides, metal inorganic acid salts, metal organic acid salts and the like can be presented. Further specific examples include (i) ferrous chloride, ferric chloride, ferric bromide, aluminum trichloride, cerium trichloride, lanthanum trichloride, etc.
- metal halides examples include metal inorganic and acid salts include inorganic acid salts such as sulfuric acid, nitric acid, perchloric acid, etc. selected from the group consisting of iron (divalent), iron (trivalent), aluminum, cerium, and lanthanum, (iii) metal organic Examples of acid salts include trifluoromethanesulfonate, acetate, trifluoroacetate, citrate and the like of metals selected from the group consisting of iron (divalent), iron (trivalent), aluminum, cerium, and lanthanum Is preferable because the yield of UP 4 U is improved.
- inorganic acid salts such as sulfuric acid, nitric acid, perchloric acid, etc. selected from the group consisting of iron (divalent), iron (trivalent), aluminum, cerium, and lanthanum
- metal organic Examples of acid salts include trifluoromethanesulfonate, acetate, trifluoroacetate, citrate and the like of metals
- ferric salts are preferable and ferric chloride is particularly preferable in terms of synthesis yield and ease of handling.
- the metal salt used may be an anhydride or a hydrate.
- iron (III) ion is particularly preferable to use in the case of the combination 4, it is particularly preferable to use iron (III) ion, trivalent aluminum ion, or the like.
- the metal salt as the metal ion source is preferably 0.001 to 10 times the molar amount relative to the total number of moles of the phosphoric acid compound used in the reaction.
- a 0.001 to 1-fold molar amount can be used.
- the amount of this metal salt is, for example, 0.001, 0.005, 0.01, 0.05, 0.1, 0.5, 1-fold mole relative to the total number of moles of the phosphoric acid compound used in the reaction. It may be any of the quantities and may be within the range of any two numerical values exemplified here.
- the condensation reaction between the phosphoric acid active compound and the phosphoric acid compound is carried out using water or a hydrophilic organic solvent as a solvent.
- hydrophilic organic solvents include alcohols having 6 or less carbon atoms such as methanol and ethanol, ketones such as acetone, ethers such as dioxane, nitriles such as acetonitrile, and dimethyl. Amides such as formamide can be used.
- the reaction pH is preferably 7 or less, particularly preferably around pH 1 to 4.
- the reaction temperature is preferably 0 ° C. to 60 ° C. in order to improve the yield of UP 4 U.
- the reaction temperature is particularly preferably 20 to 30 ° C.
- the reaction temperature is particularly preferably 0 to 20 ° C.
- the condensation reaction time is preferably about 1 to 36 hours, particularly preferably 2 to 20 hours, in order to perform a necessary and sufficient condensation reaction.
- the UP produced by appropriately combining methods used for isolation and purification of general nucleotides for example, crystallization method, ion exchange column chromatography, adsorption column chromatography, activated carbon column chromatography, etc. 4 U can be separated and purified and, if necessary, can be converted into a salt form.
- general nucleotides for example, crystallization method, ion exchange column chromatography, adsorption column chromatography, activated carbon column chromatography, etc.
- ion exchange resins used in ion exchange column chromatography include strongly basic anion exchange resins (for example, Amberlite IRA402 (Rohm & Haas), Diaion PA-312, Diaion SA-11A [Mitsubishi Chemical Corporation). )], Weakly basic anion exchange resin (for example, Amberlite IRA67 (Rohm & Haas), Diaion WA-30 (Mitsubishi Chemical)), strongly acidic cation exchange resin (for example, Diaion PK) -216 (Mitsubishi Chemical Corporation)) or weakly acidic cation exchange resin (Diaion WK-30 (Mitsubishi Chemical Corporation)) or the like can be used.
- strongly basic anion exchange resins for example, Amberlite IRA402 (Rohm & Haas), Diaion PA-312, Diaion SA-11A [Mitsubishi Chemical Corporation).
- Weakly basic anion exchange resin for example, Amberlite IRA67 (Rohm & Haas), Diai
- activated carbon As the activated carbon, activated carbon for chromatography formed in a crushed or granular form may be used.
- commercially available products such as those manufactured by Wako Pure Chemical Industries, Ltd., Nimura Chemical Co., Ltd., etc. can be used.
- a known method may be used for crystallization, and for example, it is obtained by adding a hydrophilic organic solvent to the obtained UP 4 U or a salt thereof to precipitate crystals.
- a hydrophilic organic solvent to be used include alcohols having 6 or less carbon atoms such as methanol and ethanol, ketones such as acetone, ethers such as dioxane, nitriles such as acetonitrile, and amides such as dimethylformamide. Alcohols are preferable, and ethanol is particularly preferable.
- uridine 5′-diphosphate UDP
- UMP uridine 5′-diphosphate
- imidazole imidazole
- benzimidazole 1,2,4-triazole which may have a substituent
- a phosphate compound selected from the group consisting of UMP, UDP, UTP and pyrophosphate, or a salt thereof (provided that UTP)
- a metal ion selected from the group consisting of iron (II) ion, iron (III) ion, trivalent aluminum ion, trivalent lanthanum ion, and trivalent cerium ion.
- the conventionally known method of Patent Document 1 can synthesize UP 4 U at a high yield of 80% or more at the laboratory level, but the method is mass-produced on an industrial scale.
- UTP as a raw material is used in the form of a tertiary amine salt such as tri-n-butylamine. Need to use. For this reason, a process of forming a salt with a tertiary amine after making UTP trisodium salt into free UTP (UTP free) by ion exchange column chromatography in advance is necessary.
- UTP-free is a very unstable substance, and it is very difficult to use UTP-free as a raw material in mass production on an industrial scale. Further, in order to efficiently synthesize the 5′-cyclic triphosphorylation reaction of UTP, it is necessary to dehydrate the UTP tri-n-butylamine salt immediately before the reaction. However, as shown in the comparative test examples to be described later, when UTP tri-n-butylamine salt is azeotropically dehydrated in mass production on an industrial scale, a UTP thermal decomposition reaction occurs, so UTP As a result, the synthesis efficiency was lowered.
- Patent Document 2 it was possible to synthesize UP 4 U without using the UTP-free process, but as shown in the comparative test described later, The yield to UMP was significantly reduced. Moreover, it produces a lot of by-products, and it cannot be said that the production method is suitable for industrial large-scale production.
- the production method of the present invention is a very suitable method for industrial mass synthesis of UP 4 U.
- the phosphoric acid active compound is preferably UDP-phosphoric acid imidazolide, and the phosphoric acid compound is preferably UDP or a UDP salt.
- the phosphoric acid compound is preferably UDP or a UDP salt.
- the phosphate active compound is UMP-phosphate imidazolide and the phosphate compound is UTP or UTP salt. Also in this case, it is demonstrated in the examples described later that UP 4 U can be synthesized with good yield while avoiding UTP-free use and UTP salt dehydration which are not suitable for industrial mass production. Because it is.
- the phosphoric acid active compound is preferably UMP-phosphoric acid imidazolide, and the phosphoric acid compound is preferably pyrophosphoric acid or pyrophosphate. Also in this case, it is demonstrated in the examples described later that UP 4 U can be synthesized with good yield while avoiding UTP-free use and UTP salt dehydration which are not suitable for industrial mass production. Because it is.
- a phosphoric acid active compound is pyrophosphate diimidazolide, and a phosphoric acid compound is UMP or a UMP salt. Also in this case, it is demonstrated in the examples described later that UP 4 U can be synthesized with good yield while avoiding UTP-free use and UTP salt dehydration which are not suitable for industrial mass production. Because it is.
- the metal ions are preferably selected from the group consisting of iron (II) ions, iron (III) ions, or aluminum ions.
- the phosphate active compound is pyroimidazole diimidazolide and the phosphate compound is UMP-free or UMP salt, it is preferably selected from the group consisting of iron (III) ions or aluminum ions. This is because, in these cases, it has been proved by a comparative test described later that UP 4 U can be synthesized more efficiently than in the case of using other metal ions.
- the metal ions are preferably supplied in the form of a salt selected from the group consisting of metal chlorides, bromides, nitrates, sulfates and acetates. This is also because UP 4 U can be synthesized efficiently in this case.
- the synthetic solution was concentrated to 10 ml and dissolved by adding sodium monohydrogen phosphate (4.2 mmol) to precipitate iron phosphate.
- the suspension was allowed to stand at 4 ° C. overnight, and then the precipitate was removed by centrifugation.
- the supernatant was adjusted to pH 2.5 and then used as a column adsorbent as a 30 ml solution.
- the column adsorbed solution was desalted by activated carbon column chromatography (resin amount: 20 ml), and the obtained recovered solution was adjusted to pH 7 and concentrated, followed by membrane filtration. Further concentration was performed, ethanol was added dropwise to the residue, and crystallization was performed overnight at room temperature. The crystals were collected by filtration and vacuum dried at 50 ° C. for 3 hours to obtain 2.12 g (yield 75% as a tetrahydrate) of the desired product UP 4 U.
- UDP-2 sodium (0.53 g, 1.0 mmol) was added to the UDP-phosphate imidazolide solution, and the mixture was adjusted to pH 3.9 with a 6 M aqueous hydrochloric acid solution. Further, 1M aqueous ferric chloride solution (15 ⁇ l, 0.02 mmol) was added, and the mixture was stirred at 10 ° C. for 27 hours. The reaction solution was adjusted to pH 10 by adding 7.5M aqueous sodium hydroxide solution, and stirred for 1 hour under ice cooling. Under ice cooling, 10 ml of ethanol was added to the reaction solution, and the mixture was allowed to stand at 4 ° C. overnight.
- the precipitate was collected, prepared in a 20 ml aqueous solution having a pH of 7.5, and quantified by HPLC analysis to calculate a synthetic yield of UP 4 U of 94%. That is, it was found that the reaction proceeds well even when the amount of iron chloride catalyst used is greatly reduced.
- UMP-tributylamine salt solution was dissolved in dimethylacetamide (1.5 ml), carbonyldiimidazole (486 mg, 3.0 mmol) was added, and the mixture was stirred at room temperature for 1 hour. Water (0.2 ml) was added to the reaction solution under ice-cooling, and 0.5 M aqueous hydrochloric acid solution (4.0 ml) was further added dropwise (pH 7.2). The reaction solution was concentrated under reduced pressure to prepare a UMP-phosphate imidazolide solution.
- the obtained UMP-tributylamine salt solution was dissolved in dimethylacetamide (1.5 ml), carbonyldiimidazole (486 mg, 3.0 mmol) was added, and the mixture was stirred at room temperature for 1 hour. Water (0.2 ml) was added to the reaction solution under ice-cooling, and 0.5 M aqueous hydrochloric acid solution (4.0 ml) was further added dropwise (pH 7.2). The reaction solution was concentrated under reduced pressure to prepare a UMP-phosphate imidazolide solution (this operation was performed twice).
- Test Example 1 Measurement of Stability of UTP and Related Compounds
- UTP-free an aqueous solution of UTP sodium salt (UTP-Na) and dimethyl of UTP-tributylamine salt (UTP-TBA)
- UTP-TBA dimethyl of UTP-tributylamine salt
- the stability of the acetamide solution was investigated.
- As a comparative control the stability of aqueous solutions of UDP-free and UDP-Na was also measured.
- the solution of each substance was allowed to stand at ⁇ 20 ° C., 25 ° C. or 50 ° C. for a fixed time, the concentration of each substance was measured by HPLC, and the decomposition rate was calculated.
- the decomposition rate was defined as [Formula 1] below.
- UTP-free and UTP-TBA have clearer stability during storage than other compounds such as UTP sodium salt, UDP-free and UDP sodium salt. It became clear that it was low.
- the method of the present invention produces less impurities than the one-pot synthesis method, and the yield is remarkably improved, and is extremely suitable as an industrial mass production method of UP 4 U. It was shown that.
- anhydrous UMP-tributylamine solution was prepared. This was added to a pyrophosphoric anhydride-diimidazolide solution, concentrated under reduced pressure, and stirred at 25 ° C. for 48 hours. Water was added to the reaction solution to prepare a 50 ml aqueous solution, which was quantified by HPLC analysis to calculate a synthetic yield of UP 4 U of 10%.
- anhydrous UMP-tributylamine solution was prepared. This was added to an anhydrous pyrophosphoric acid-diimidazolide solution, and further zinc chloride (1.34 g, 9.8 mmol) was added, followed by concentration under reduced pressure and stirring at 25 ° C. for 5 hours. Water was added to the reaction solution to prepare a 50 ml aqueous solution, which was quantified by HPLC analysis to calculate a UP 4 U synthesis yield of 17%.
- Tributylamine (0.95 ml, 4.0 mmol) and formamide (2.0 ml) were added to an aqueous solution (1.86 ml, 2.0 mmol) of 1.09 M pyrophosphate-triethylamine salt. Azeotropic dehydration was performed 4 times with dimethylformamide. The obtained residue was dissolved in dimethylformamide (8.0 ml), carbonyldiimidazole (0.97 g, 6.0 mmol) was added, and the mixture was stirred at room temperature for 1 hour.
- anhydrous UMP-tributylamine solution was prepared. This was added to a pyrophosphoric anhydride-diimidazolide solution, 1H-tetrazole (0.5 g, 7.2 mmol) was further added, and the mixture was concentrated under reduced pressure and stirred at 25 ° C. for 19 hours. Water was added to the reaction solution to prepare a 50 ml aqueous solution, which was quantified by HPLC analysis to calculate a synthetic yield of UP 4 U of 9%.
- the method of the present invention produces less impurities than the old method of pyrophosphoric acid imidazolide method, and the yield is remarkably improved, and is an industrial mass production method of UP 4 U. It was shown to be very suitable.
- an aqueous ferric chloride solution was used as an aqueous solution containing metal ions, but there is no particular limitation.
- metal halides include ferrous chloride, ferric chloride, odor Ferric chloride, aluminum trichloride, cerium trichloride, lanthanum trichloride, etc.
- metal inorganic acid salts from the group consisting of iron (divalent), iron (trivalent), aluminum, cerium, lanthanum Inorganic acid salts of selected metals such as sulfuric acid, nitric acid, perchloric acid, etc.
- examples of metal organic acid salts the group consisting of iron (divalent), iron (trivalent), aluminum, cerium, lanthanum
- An aqueous solution in which a trifluoromethanesulfonate, acetate, trifluoroacetate, citrate, or the like of a selected metal is dissolved can be suitably used. It can be easily understood by those skilled in the art that the yield of UP 4 U is improved in the same manner as in the above examples even when an aqueous solution containing these metal ions is used.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pulmonology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description

具体的には、特許文献1の方法では、5’-UTPの5’-環状トリリン酸化反応を行うために、原料となるUTPを、トリ-n-ブチルアミンのような三級アミンの塩の形態で使用する必要がある。このため、UTPの3ナトリウム塩を事前にイオン交換カラムクロマトグラフィーにより遊離型のUTP(UTPフリー)とした後、三級アミンとの塩形成を行うという工程が必要であった。しかしながら後述する試験例でも検証しているように、UTPフリーは非常に不安定な物質であり、工業的な規模の大量製造においてUTPフリーを原料として使用することは非常に困難である。
(A)出発原料としてUTPではなくUMPを用い、反応系内でUMPからUTPを調製した後、これを精製することなく、特許文献1の方法に適用してUP4Uを合成する方法(以下「ワンポット合成法」)。
(B)ピロリン酸をイミダゾールによって活性化してピロリン酸ジイミダゾリドを合成し、これをUMPと縮合させることによってUP4Uを合成する方法(以下「ピロリン酸ジイミダゾリド法旧法」、特許文献2、非特許文献2)。


本発明で使用するリン酸活性化合物は、式[II]または[III]で表される化合物であり、UMP、UDPまたはピロリン酸と、置換基を有しても良い、イミダゾール、ベンズイミダゾールまたは1,2,4-トリアゾールとを縮合させることで合成される。

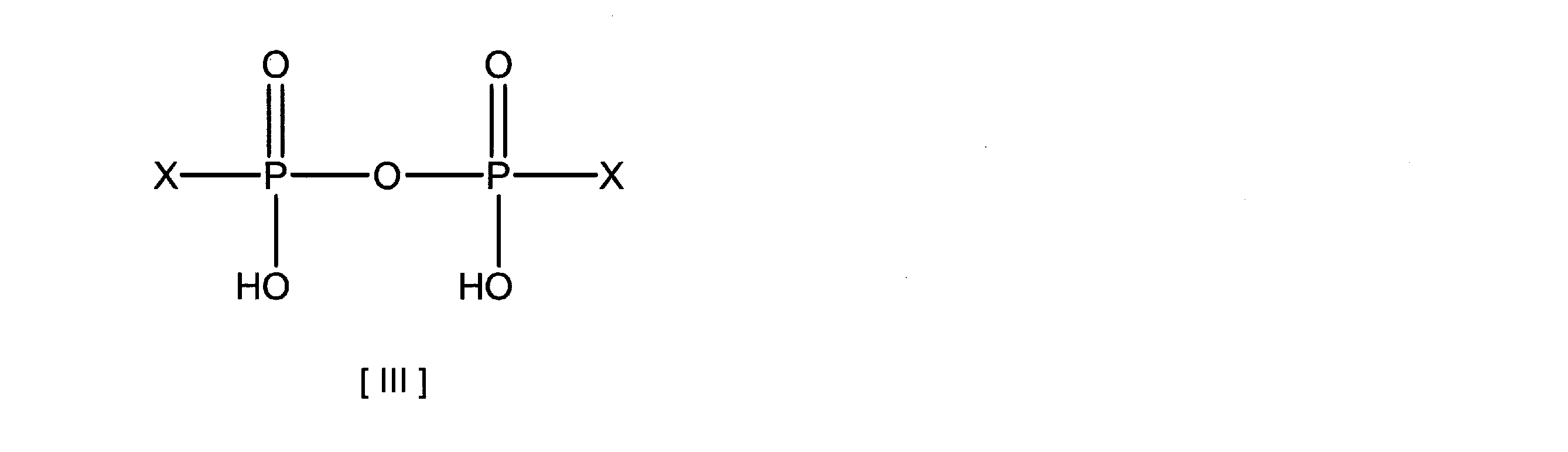
本発明で使用するリン酸化合物は、下記式(IV)で表される化合物またはその塩であり、具体的には、UTPフリーを除く、UMP、UDP、UTP、ピロリン酸、またはそれらの塩を挙げることができる。
このようなリン酸活性化合物とリン酸化合物との原料化合物の組み合わせとしては、合目的的に決定すればよく、好ましくは以下の組み合わせで用いるのが好適である。
組み合わせ1:UDP活性化合物とUDPまたはUDP塩の組み合わせ
組み合わせ2:UMP活性化合物とUTP塩の組み合わせ
組み合わせ3:UMP活性化合物とピロリン酸またはピロリン酸塩の組み合わせ
組み合わせ4:ピロリン酸活性化合物と、UMPまたはUMP塩の組み合わせ。
いずれの組み合わせにおいても、縮合反応における、リン酸活性化合物とリン酸化合物とのモル比は、1:10~10:1の範囲に設定すれば、UP4Uの収率が向上するため好ましい。もっとも、このモル比は、例えば、
(1)上記組み合わせ1のとき、モル比3:8、4:7、5:6、6:5、7:4、8:3、9:2、10:1のいずれか、
(2)組み合わせ2のときにはモル比1:10、2:9、3:8、4:7、5:6、6:5、7:4、8:3のいずれか、
(3)組み合わせ3のときには4:7、5:6、6:5、7:4、8:3、9:2、10:1のいずれか、さらに、
(4)組み合わせ4のときには4:7、5:6、6:5、7:4、8:3、9:2、10:1のいずれか、
であり、ここで例示した何れか2つの数値の範囲内であってもよい。
本発明に使用する金属イオンは、目的の金属イオンを含む金属塩の形態で溶液に添加することによって、水または親水性有機溶媒中で金属イオンとなり、反応系に供給することができる。金属塩の種類としては、金属ハロゲン化物、金属無機酸塩、金属有機酸塩等を提示することができる。さらなる具体例としては、(i)金属ハロゲン化物の例として塩化第一鉄、塩化第二鉄、臭化第二鉄、三塩化アルミニウム、三塩化セリウム、三塩化ランタニウム等を、(ii)金属無機酸塩の例として鉄(2価)、鉄(3価)、アルミニウム、セリウム、ランタンから成る群より選択される金属の硫酸、硝酸、過塩素酸等の無機酸塩を、(iii)金属有機酸塩の例として、鉄(2価)、鉄(3価)、アルミニウム、セリウム、ランタンから成る群より選択される金属のトリフルオロメタンスルホン酸塩、酢酸塩、トリフルオロ酢酸塩、クエン酸塩等を用いることが、UP4Uの収率が向上するため好ましい。中でも、合成収率および取り扱い易さの点で、第二鉄塩が好ましく、塩化第二鉄が特に好ましい。なお、用いる金属塩は、無水物であっても水和物であっても良い。ただし、上記リン酸活性化合物とリン酸化合物の組み合わせのうち、特に組み合わせ4の場合は、鉄(III)イオン、3価アルミニウムイオン等を用いることが特に好ましい。
本発明におけるリン酸活性化合物とリン酸化合物との縮合反応は、水または親水性有機溶剤を溶媒として実施する。合成収率および取り扱い易さの点から、親水性有機溶剤としては、メタノール、エタノール等の炭素数6以下のアルコール類、アセトン等のケトン類、ジオキサン等のエーテル類、アセトニトリル等のニトリル類、ジメチルホルムアミド等のアミド類等を使用することができる。
これまで説明したとおり、本発明では、(a)ウリジン5’-ジリン酸(UDP)、UMPまたはピロリン酸と、置換基を有しても良い、イミダゾール、ベンズイミダゾールまたは1,2,4-トリアゾールから成る群より選択される化合物とを縮合させることで合成されるリン酸活性化合物と、(b)UMP、UDP、UTPおよびピロリン酸から成る群より選択されるリン酸化合物またはその塩(ただしUTPフリーを除く)とを組み合わせ、触媒として、鉄(II)イオン、鉄(III)イオン、3価アルミニウムイオン、3価ランタンイオン、3価セリウムイオンからなる群より選択される金属イオンの存在下、水または親水性有機溶媒中で反応させるという方法を用いることによって、後述の実施例に示すように、簡便な工程でUP4Uの工業的な大量製造を行うことができる。
0.32M UDP-トリブチルアミン塩の水溶液(1.26ml,4.0mmol)にジメチルアセトアミド(6.0ml)を加え、共沸脱水を4回行った。得られた残渣をジメチルアセトアミド(6.0ml)で溶解し、カルボニルジイミダゾール(1.95g,12.0mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水(0.2ml)を加え、さらに0.5M塩酸水溶液(12.0ml)を滴下した(pH7.0)。反応液を減圧下で濃縮し、UDP-リン酸イミダゾリド溶液を調製した。
0.94M UDPナトリウム水溶液(1.0ml)に5.64M イミダゾール-ジメチルアセトアミド溶液(0.5ml)を加え、N,N-ジイソプロピルカルボジイミド(0.73ml,4.7mmol)を加え室温で5時間、50℃で2時間攪拌し、UDPとUDP-リン酸イミダゾリド溶液の混合液を調製した。
0.17M UDP-トリブチルアミン塩の水溶液 (11.8ml,2.0mmol)にプロピオニトリル(3.0ml)を加え共沸脱水を4回行った。得られた残渣をプロピオニトリル(3.0ml)で溶解した。これを1,1’-カルボニルジイミダゾール(0.97g,6.0mmol)のプロピオニトリル(2.0ml)懸濁液に滴下した。室温で30分間攪拌した後、反応液を減圧下で濃縮し、UDP-リン酸イミダゾリド溶液を調製した。
UDP-2ナトリウム塩(0.5g,0.94mmol)を水2.7mlに溶解し、ベンズイミダゾール(0.33g,2.82mmol)のジメチルアセトアミド溶液(1.8ml)、およびN,N-ジイソプロピルカルボジイミド(0.44ml,2.82mmol)を加え、室温で一晩攪拌した。減圧下で溶媒を留去し、UDP-2ナトリウムとUDP-リン酸ベンズイミダゾリドの混合物を調製した。
0.37M UDP-トリブチルアミン塩の水溶液 (1.6ml,0.6mmol)にジメチルアセトアミド(2.0ml)を加え共沸脱水を4回行った。得られた残渣をジメチルアセトアミド(2.0ml)で溶解し、1,1’-カルボニルジ-(1,2,4-トリアゾール)(0.25g,1.5mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水 (0.2ml)を加え、さらに0.5M塩酸水溶液を加えてpH7.4とした。反応液を減圧下で濃縮し、UDP-リン酸トリアゾリド溶液を調製した。
2.05M UMPフリー水溶液(0.49ml,1.0mmol)にトリブチルアミン(0.25ml,1.1mmol)を加え、室温で20分間攪拌した後、ジメチルアセトアミド(1.5ml)をさらに加え共沸脱水を3回行った。得られたUMP-トリブチルアミン塩溶液をジメチルアセトアミド(1.5ml)で溶解し、カルボニルジイミダゾール(486mg,3.0mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水(0.2ml)を加え、さらに0.5M塩酸水溶液(4.0ml)を滴下した(pH7.2)。反応液を減圧下で濃縮し、UMP-リン酸イミダゾリド溶液を調製した。
2.05M UMPフリー水溶液(0.49ml,1.0mmol)にトリブチルアミン(0.25ml,1.1mmol)を加え室温で20分間攪拌した後、ジメチルアセトアミド(1.5ml)を加え共沸脱水を3回行った。得られたUMP-トリブチルアミン塩溶液をジメチルアセトアミド(1.5ml)で溶解し、カルボニルジイミダゾール(486mg,3.0mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水 (0.2ml)を加え、さらに0.5M塩酸水溶液(4.0ml)を滴下した(pH7.2)。反応液を減圧下で濃縮し、UMP-リン酸イミダゾリド溶液を調製した(本操作を2回行った)。
1.09M ピロリン酸-トリエチルアミン塩の水溶液(1.86ml,2.0mmol)にトリブチルアミン(0.95ml,4.0mmol)、ホルムアミド(2.0ml)を加え、ジメチルホルムアミドで共沸脱水を4回行った。得られた残渣をジメチルホルムアミド(8.0ml)で溶解し、カルボニルジイミダゾール(0.97g,6.0mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水(0.2ml)を加え、さらに2.0M塩酸水溶液(1.5ml)を滴下した(pH7)。反応液を減圧下で濃縮し、ピロリン酸-ジイミダゾリド溶液を調製した。
氷冷下でピロリン酸-ジイミダゾリド溶液にUMP-フリー水溶液(2.03M,2.47ml,5.0mmol)を加えた。さらに1M塩化第二鉄水溶液(2ml,2.0mmol)を加え、2M塩酸水溶液を加えてpHを2.4に調整し、0℃で5時間攪拌した。反応液に6.0M水酸化ナトリウム水溶液を加え、pH6.3とした。これに水を加えて50ml水溶液に調製し、HPLC分析で定量してUP4Uの合成収率51%を算出した。
UTPフリーの安定性を評価するため、UTPフリー、UTPナトリウム塩(UTP-Na)の水溶液およびUTP-トリブチルアミン塩(UTP-TBA)のジメチルアセトアミド溶液の安定性を調べた。また、比較対照としてUDP-フリーおよびUDP-Naの水溶液の安定性も測定した。各物質の溶液を-20℃、25℃または50℃で一定時間静置し、それぞれの物質の濃度をHPLCで測定し、分解率を算出した。なお、分解率は以下[数1]のように定義した。


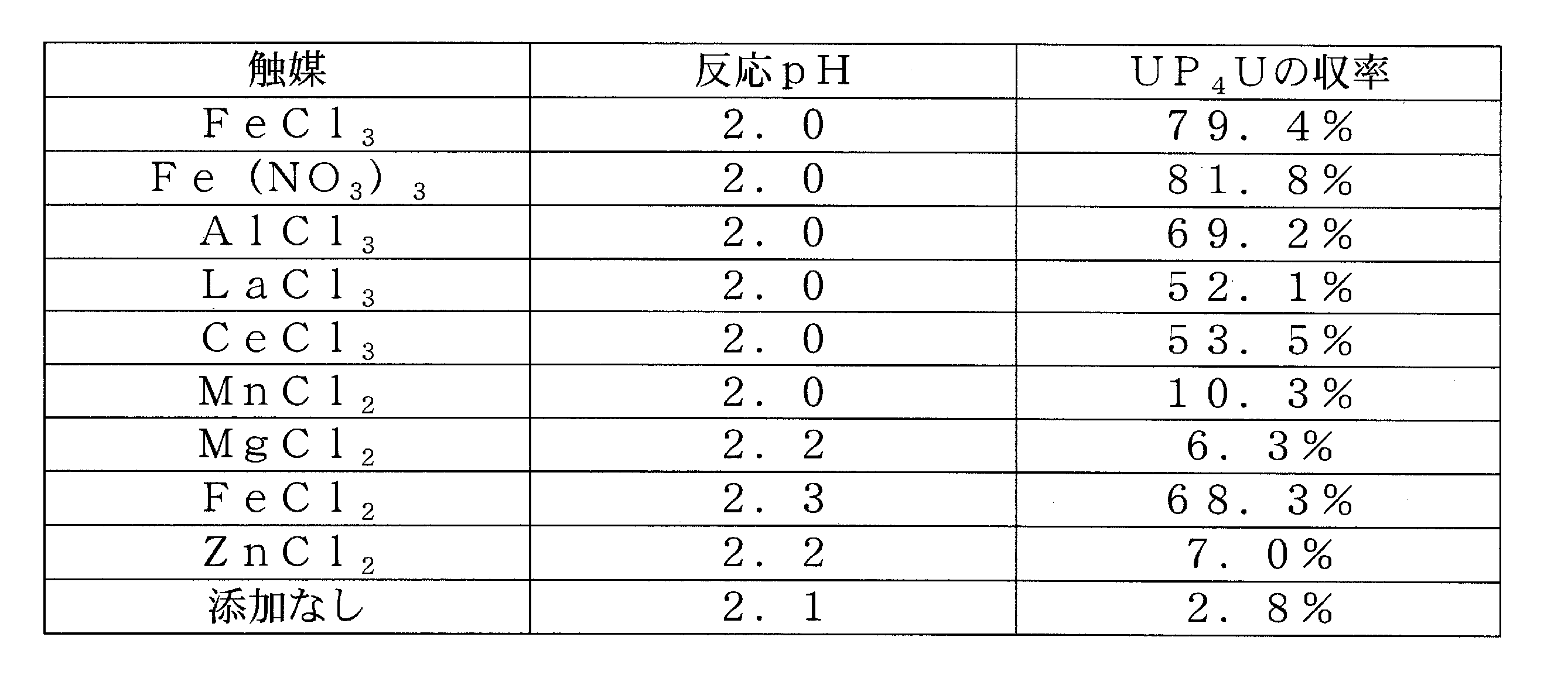
実施例1の方法で調製されたUDP-リン酸イミダゾリドとUDPナトリウム塩とのモル比1:1の混合水溶液(各0.15M)に各種金属触媒(0.06M)を添加し、2M塩酸水溶液でpH2±0.3として、25℃で6時間後のUP4Uの生成量をHPLCで定量した。結果を表3に示す。
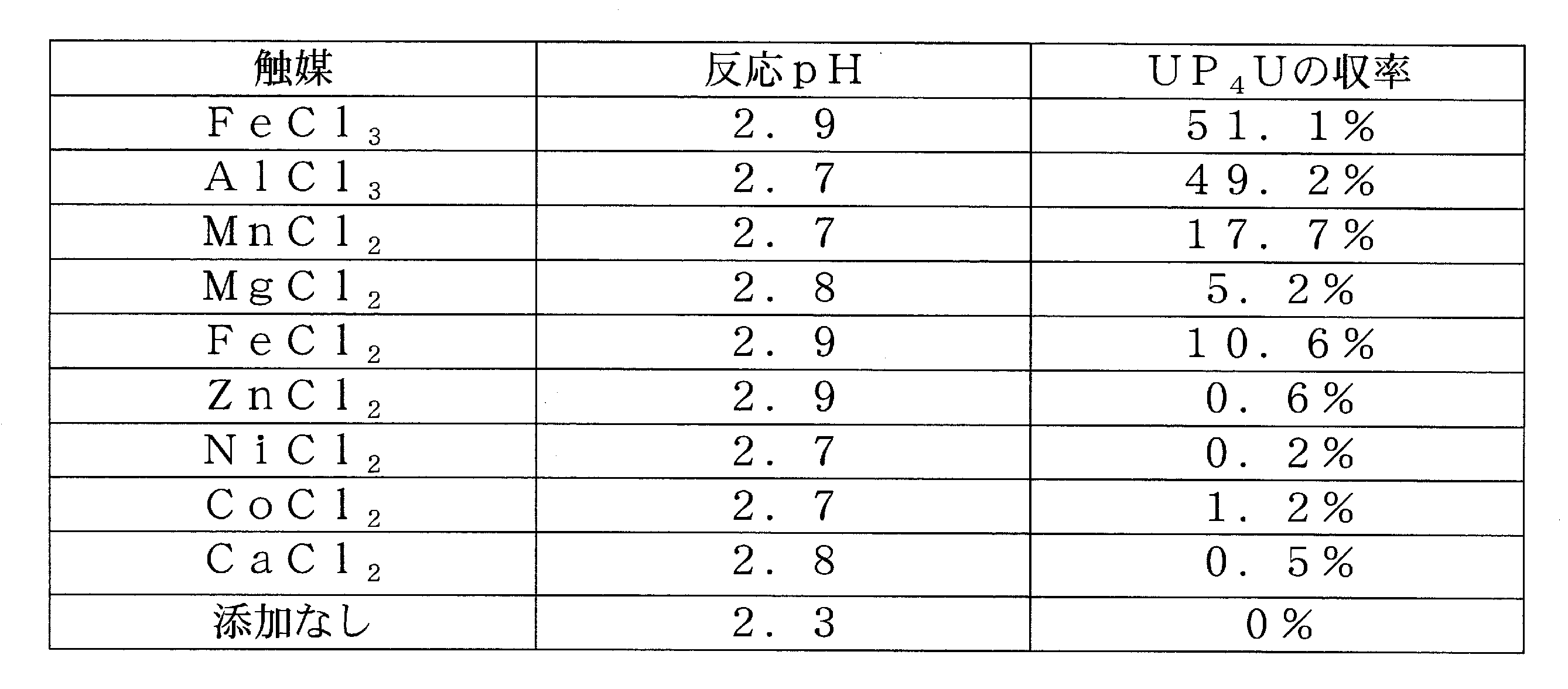
実施例8の方法で調製されたピロリン酸-ジイミダゾリドとUMPフリーとのモル比2:5の混合水溶液(ピロリン酸-ジイミダゾリドとして0.4mmol)に各種金属触媒(0.4mmol)を添加し、2M塩酸水溶液でpH2.5±0.5として、0℃で5時間後のUP4Uの生成量をHPLCで定量した。結果を[表4]に示す。
従来法を改善し、UTPフリーを使う工程を回避した合成法であるワンポット合成法と、本実施例の方法を用いてそれぞれUP4Uを合成した際の、最終産物の収率をそれぞれ比較した。本実施例の方法としては、上記実施例1の方法に従った。従来の方法を変更したワンポット合成法での合成は、以下の工程によって行った。
2.05MのUMPフリー水溶液(2.1ml,4.3mmol)にトリ-n-ブチルアミン(2.2ml,9.2mmol)を加え20分間攪拌した後、N,N-ジメチルアセトアミド(DMAC)で4回共沸脱水した。残渣をDMAC(5.5ml)に溶解し、10℃でクロロリン酸ジフェニル(DPC)(1.1ml,5.2mmol)を加え、室温で1時間攪拌した。トリ-n-ブチルアミン(4.1ml,17.2mmol)を加え室温で10分間攪拌し、これを合成液1とした。
0.95Mピロリン酸-トリエチルアミン塩水溶液(9.0ml,8.5mmol)にトリ-n-ブチルアミン(2.2ml,9.2mmol)、DMAC(3.0ml)を加えた後、ピリジンで3回共沸脱水した。残渣にトリ-n-ブチルアミン(0.4ml,2.8mmol)、ピリジン(4.0ml)を加えて、これを合成液2とした。
2.05MのUMPフリー水溶液(3.1ml,6.4mmol)にトリ-n-ブチルアミン(1.7ml,6.9mmol)を加え20分間攪拌した後、N,N-ジメチルアセトアミド(DMAC)で4回共沸脱水した。残渣をDMAC(3.5ml)に溶解し、UMP-TBA溶液を調製した。
室温下で合成液2に合成液1を滴下し、室温で1時間攪拌した(HPLC分析でUTPが約60%生成していることを確認した)。N,N-ジイソプロピルカルボジイミド(2.3ml,17.0mmol)を加え室温で3時間攪拌した後、0℃でUMP-TBA溶液および塩化マグネシウム-6水和物(1.7g,8.4mmol)を加え、0℃で1時間、室温で19時間攪拌した。0℃で水(20ml)を加えた後、室温で1時間攪拌した。反応液を減圧濃縮した後、水(50ml)を加え室温で2時間静置した。沈殿物をろ過して除きUP4U合成液を調製した。HPLCによりUP4Uを定量し、UMPからの合成収率を42%と算出した。
その結果、本実施例の方法でUP4Uを合成した際には、上述の実施例1で示したように、UDPからの合成収率87%であるのに対し、ワンポット合成法ではUMPからの合成収率は42%に過ぎず、本発明の方法はワンポット合成法に比較して、著しく高い収率を達成するものであることが判明した。
さらに、本実施例の方法とワンポット合成法での不純物の生成度合の差を比較するため、それぞれの方法でUP4Uを合成後の反応液中の生成物をHPLCで分析した。HPLCチャートを[図1][図2]に示す。図1及び2から明らかなように、ワンポット合成法では、産物であるUP4Uおよび原料のUMP以外の副生成物がHPLCチャートの面積の割合で約48%生じていた上に、副生成物の種類数も多いのに対して、本発明の方法では、UP4Uおよび原料であるUDP以外の副生成物の割合は4.8%と明らかに低下しており、副生成物の種類も極めて少なくなっていることから、本発明の方法では副生成物の発生を著しく低下させることができ、もって収率よく合成できるとともに、単離精製も容易である。
イミダゾリド法旧法と本実施例の方法を用いてそれぞれUP4Uを合成した際の、最終産物の収率をおよび副生成物の生成をそれぞれ比較した。本実施例の方法としては、上記実施例8の方法に従った。イミダゾリド法旧法での合成は以下の工程によって行った。また反応時に用いる触媒に関しては、公知の方法(特許文献2および非特許文献2参照)を参考に、(1)反応時に触媒を添加しなかった場合、触媒として(2)塩化亜鉛または(3)テトラゾールを使用した場合について検討を行った。
1.09M ピロリン酸-トリエチルアミン塩の水溶液(1.86ml,2.0mmol)にトリブチルアミン(0.95ml,4.0mmol)、ホルムアミド(2.0ml)を加え、ジメチルホルムアミドで共沸脱水を4回行った。得られた残渣をジメチルホルムアミド(8.0ml)で溶解し、カルボニルジイミダゾール(0.97g,6.0mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水(0.2ml)を加え、さらに2.0M塩酸水溶液(1.5ml)を滴下した(pH7)。反応液を減圧下で濃縮し、さらにジメチルホルムアミドで2回共沸脱水して、無水ピロリン酸-ジイミダゾリド溶液を調製した。
UMP-フリー水溶液(2.03M,2.47ml,5.0mmol)にトリブチルアミン(3.56ml,15.0mmol)を加え、ジメチルホルムアミドで4回共沸脱水し、ジメチルホルムアミド(2.0ml)を加えて、無水UMP-トリブチルアミン溶液を調製した。これを無水ピロリン酸-ジイミダゾリド溶液に加え、減圧下で濃縮し、25℃で48時間攪拌した。反応液に水を加えて50ml水溶液に調製し、HPLC分析で定量してUP4Uの合成収率10%を算出した。
1.09M ピロリン酸-トリエチルアミン塩の水溶液(1.86ml,2.0mmol)にトリブチルアミン(0.95ml,4.0mmol)、ホルムアミド(2.0ml)を加え、ジメチルホルムアミドで共沸脱水を4回行った。得られた残渣をジメチルホルムアミド(8.0ml)で溶解し、カルボニルジイミダゾール(0.97g,6.0mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水(0.2ml)を加え、さらに2.0M塩酸水溶液(1.5ml)を滴下した(pH7)。反応液を減圧下で濃縮し、さらにジメチルホルムアミドで2回共沸脱水して、無水ピロリン酸-ジイミダゾリド溶液を調製した。
UMP-フリー水溶液(2.03M,2.47ml,5.0mmol)にトリブチルアミン(3.56ml,15.0mmol)を加え、ジメチルホルムアミドで4回共沸脱水し、ジメチルホルムアミド(2.0ml)を加えて、無水UMP-トリブチルアミン溶液を調製した。これを無水ピロリン酸-ジイミダゾリド溶液に加え、さらに塩化亜鉛(1.34g,9.8mmol)を加えた後、減圧下で濃縮し、25℃で5時間攪拌した。反応液に水を加えて50ml水溶液に調製し、HPLC分析で定量してUP4Uの合成収率17%を算出した。
1.09M ピロリン酸-トリエチルアミン塩の水溶液(1.86ml,2.0mmol)にトリブチルアミン(0.95ml,4.0mmol)、ホルムアミド(2.0ml)を加え、ジメチルホルムアミドで共沸脱水を4回行った。得られた残渣をジメチルホルムアミド(8.0ml)で溶解し、カルボニルジイミダゾール(0.97g,6.0mmol)を加え室温で1時間攪拌した。氷冷下で反応液に水(0.2ml)を加え、さらに2.0M塩酸水溶液(1.5ml)を滴下した(pH7)。反応液を減圧下で濃縮し、さらにジメチルホルムアミドで2回共沸脱水して、無水ピロリン酸-ジイミダゾリド溶液を調製した。
UMP-フリー水溶液(2.03M,2.47ml,5.0mmol)にトリブチルアミン(3.56ml,15.0mmol)を加え、ジメチルホルムアミドで4回共沸脱水し、ジメチルホルムアミド(2.0ml)を加えて、無水UMP-トリブチルアミン溶液を調製した。これを無水ピロリン酸-ジイミダゾリド溶液に加え、さらに1H-テトラゾール(0.5g,7.2mmol)を加えた後、減圧下で濃縮し、25℃で19時間攪拌した。反応液に水を加えて50ml水溶液に調製し、HPLC分析で定量してUP4Uの合成収率9%を算出した。
ピロリン酸ジイミダゾリド法旧法と本発明の方法におけるUP4Uの合成収率をまとめると、下記[表5]のようになった。すなわち、公知の方法であるリン酸イミダゾリド法旧法と比較して、本発明の方法は著しく高い収率を達成するものであることが判明した。

さらに、本発明の方法とピロリン酸ジイミダゾリド法旧法での不純物の生成度合の差を比較するため、それぞれの方法におけるUP4Uを合成後の反応液中の生成物をHPLCで分析した。得られたHPLCチャートを[図3]~[図6]に示す。また、ピロリン酸ジイミダゾリド法旧法と本実施例の方法において、HPLCチャートの面積における産物であるUP4Uおよび原料のUMP以外の副生成物の割合をまとめると、下記[表6]のようになった。

Claims (8)
- 下記式[II]または[III]で示されるリン酸活性化合物と、UMP、UDP、UTPおよびピロリン酸から成る群より選択されるリン酸化合物またはその塩(ただしUTPフリーを除く)とを、鉄(II)イオン、鉄(III)イオン、3価アルミニウムイオン、3価ランタンイオンおよび3価セリウムイオンからなる群より選択される金属イオンの存在下、水または親水性有機溶媒中で反応させる工程を含むことを特徴とする、P1,P4-ジ(ウリジン5’-)テトラホスフェートの製造法。


- リン酸活性化合物がUDP-リン酸イミダゾリドであり、リン酸化合物がUDPまたはUDP塩である、請求項1記載の製造法。
- リン酸活性化合物がUMP-リン酸イミダゾリドであり、リン酸化合物がUTP塩である、請求項1記載の製造法。
- リン酸活性化合物がUMP-リン酸イミダゾリドであり、リン酸化合物がピロリン酸またはピロリン酸塩である、請求項1記載の製造法。
- リン酸活性化合物がピロリン酸ジイミダゾリドであり、リン酸化合物がUMPまたはUMP塩である、請求項1記載の製造法。
- 金属イオンが鉄(II)イオン、鉄(III)イオンおよびアルミニウムイオンから成る群より選ばれる、請求項1~4のいずれか1項に記載の製造法。
- 金属イオンが鉄(III)イオンまたはアルミニウムイオンである、請求項5記載の製造法。
- 金属イオンが、金属の塩化物、臭化物、硝酸化物、硫酸化物または酢酸化物の形態で供給される請求項1記載の製造法。
Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/758,102 US10179799B2 (en) | 2012-12-28 | 2013-12-10 | Method for producing P1,P4-di(uridine 5′-) tetraphosphate |
| JP2014554298A JP6049760B2 (ja) | 2012-12-28 | 2013-12-10 | P1,p4−ジ(ウリジン5’−)テトラホスフェートの製造法 |
| SI201330744T SI2940030T1 (sl) | 2012-12-28 | 2013-12-10 | Postopek za izdelavo P1, P4-di(uridin 5'-)tetrafosfata |
| SM20170405T SMT201700405T1 (it) | 2012-12-28 | 2013-12-10 | Metodo per produrre p1,p4-di(uridina 5'-)tetrafosfato |
| HRP20171333TT HRP20171333T1 (hr) | 2012-12-28 | 2013-12-10 | Postupak za proizvodnju p1, p4-di(uridin 5'-) tetrafosfata |
| RS20170824A RS56227B1 (sr) | 2012-12-28 | 2013-12-10 | Postupak proizvodnje p1,p4-di(uridin 5'-)tetrafosfata |
| CA2896455A CA2896455C (en) | 2012-12-28 | 2013-12-10 | Method for producing p1,p4-di(uridine 5'-)tetraphosphate |
| ES13869663.8T ES2629743T3 (es) | 2012-12-28 | 2013-12-10 | Procedimiento para producir P1,P4-di(uridina 5'-)tetrafosfato |
| LTEP13869663.8T LT2940030T (lt) | 2012-12-28 | 2013-12-10 | P1,p4-di (uridino 5`-) tetrafosfato gamybos būdas |
| DK13869663.8T DK2940030T3 (en) | 2012-12-28 | 2013-12-10 | METHOD OF PREPARING P1, P4-DI (URIDIN 5 '-) TETRAPHOSPHATE |
| KR1020157017372A KR102024700B1 (ko) | 2012-12-28 | 2013-12-10 | P1,p4-디(우리딘 5'-)테트라포스페이트의 제조법 |
| EP13869663.8A EP2940030B1 (en) | 2012-12-28 | 2013-12-10 | Method for producing p1,p4-di(uridine 5'-)tetraphosphate |
| CN201380074088.5A CN105026414B (zh) | 2012-12-28 | 2013-12-10 | P1,p4‑二(尿苷5’‑)四磷酸酯的制造方法 |
| CY20171100946T CY1119372T1 (el) | 2012-12-28 | 2017-09-07 | Μεθοδος παραγωγης p1, p4-δι(ουριδινο 5'-) τετραφωσφορικης ενωσης |
| US16/206,380 US11208428B2 (en) | 2012-12-28 | 2018-11-30 | Method for producing P1,P4-di(uridine 5′-) tetraphosphate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-287578 | 2012-12-28 | ||
| JP2012287578 | 2012-12-28 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/758,102 A-371-Of-International US10179799B2 (en) | 2012-12-28 | 2013-12-10 | Method for producing P1,P4-di(uridine 5′-) tetraphosphate |
| US16/206,380 Continuation US11208428B2 (en) | 2012-12-28 | 2018-11-30 | Method for producing P1,P4-di(uridine 5′-) tetraphosphate |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014103704A1 true WO2014103704A1 (ja) | 2014-07-03 |
Family
ID=51020794
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/083100 Ceased WO2014103704A1 (ja) | 2012-12-28 | 2013-12-10 | P1,p4-ジ(ウリジン5'-)テトラホスフェートの製造法 |
Country Status (18)
| Country | Link |
|---|---|
| US (2) | US10179799B2 (ja) |
| EP (1) | EP2940030B1 (ja) |
| JP (1) | JP6049760B2 (ja) |
| KR (1) | KR102024700B1 (ja) |
| CN (1) | CN105026414B (ja) |
| CA (1) | CA2896455C (ja) |
| CY (1) | CY1119372T1 (ja) |
| DK (1) | DK2940030T3 (ja) |
| ES (1) | ES2629743T3 (ja) |
| HR (1) | HRP20171333T1 (ja) |
| HU (1) | HUE035675T2 (ja) |
| LT (1) | LT2940030T (ja) |
| PL (1) | PL2940030T3 (ja) |
| PT (1) | PT2940030T (ja) |
| RS (1) | RS56227B1 (ja) |
| SI (1) | SI2940030T1 (ja) |
| SM (1) | SMT201700405T1 (ja) |
| WO (1) | WO2014103704A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018079503A1 (ja) | 2016-10-25 | 2018-05-03 | ヤマサ醤油株式会社 | P1,p4-ジ(ウリジン5'-)テトラホスフェートの精製方法 |
| CN108164577A (zh) * | 2017-04-28 | 2018-06-15 | 广东众生药业股份有限公司 | 一种p1,p4-二(尿苷-5’-四磷酸)钠盐的工业化制备方法 |
| KR20180091672A (ko) | 2017-06-21 | 2018-08-16 | 주식회사 종근당 | 디뉴클레오사이드 폴리포스페이트 화합물의 제조방법 |
| CN111662350A (zh) * | 2020-07-07 | 2020-09-15 | 南京宸翔医药研究有限责任公司 | 一种绿色化智能化高纯度地夸磷索四钠的制备方法 |
| KR20200106738A (ko) | 2019-03-05 | 2020-09-15 | 주식회사 파마코스텍 | P1,p4-디(우리딘 5'-)테트라포스페이트, 이의 염 또는 이의 수화물의 신규한 제조방법 |
| WO2022212442A1 (en) | 2021-03-31 | 2022-10-06 | Modernatx, Inc. | Synthesis of trinucleotide and tetranucleotide caps for mrna production |
| JP2023511656A (ja) * | 2019-12-18 | 2023-03-22 | チョングンダン ファーマスーティカル株式会社 | ウリジン5’-二リン酸(udp)、その塩またはその水和物の製造方法 |
| WO2024185734A1 (ja) | 2023-03-03 | 2024-09-12 | 株式会社ナティアス | ポリリン酸化ヌクレオシド、ポリリン酸化ヌクレオシドの合成に用いられるリン酸部活性化ヌクレオチド及びその合成方法、ならびにリン酸部活性化ヌクレオチドを用いたポリリン酸化ヌクレオシドの合成方法 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107056859A (zh) * | 2017-04-28 | 2017-08-18 | 广东众生药业股份有限公司 | 一种高纯度p1,p4‑二(尿苷‑5’‑四磷酸)盐的制备方法 |
| CN110218233B (zh) * | 2018-03-01 | 2023-11-14 | 江苏恒瑞医药股份有限公司 | 一种p1,p4-二(尿苷5`-)四磷酸盐的制备方法 |
| CN109021049B (zh) * | 2018-06-14 | 2020-12-08 | 扬子江药业集团北京海燕药业有限公司 | 一种尿苷5′-二磷酸-苯并咪唑二钠的合成方法 |
| CN110655545B (zh) * | 2018-06-28 | 2022-09-09 | 上海致根医药科技有限公司 | P1,p4-二(尿苷5’-)四磷酸酯的制备方法 |
| CN110590887B (zh) * | 2019-09-04 | 2020-12-15 | 江苏金殳医药科技有限公司 | 一种磷酸酯的制备方法 |
| CN115011113B (zh) * | 2019-12-18 | 2023-07-21 | 江苏集萃先进高分子材料研究所有限公司 | 一种阻燃材料及其制备方法 |
| CN113527394A (zh) * | 2020-04-18 | 2021-10-22 | 上海键合医药科技有限公司 | 一种地夸磷索的纯化方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008012949A1 (en) | 2006-07-26 | 2008-01-31 | Yamasa Corporation | Process for producing di(pyrimidine nucleoside 5'-)polyphosphate |
| WO2008024169A1 (en) | 2006-07-21 | 2008-02-28 | Glsynthesis, Inc. | Reactive pyrophosphoric and bisphosphonic acid derivatives and methods of their use |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5051422B2 (ja) | 2006-07-21 | 2012-10-17 | アイシン精機株式会社 | タイヤ空気圧監視システム |
-
2013
- 2013-12-10 US US14/758,102 patent/US10179799B2/en active Active
- 2013-12-10 RS RS20170824A patent/RS56227B1/sr unknown
- 2013-12-10 PL PL13869663T patent/PL2940030T3/pl unknown
- 2013-12-10 EP EP13869663.8A patent/EP2940030B1/en active Active
- 2013-12-10 DK DK13869663.8T patent/DK2940030T3/en active
- 2013-12-10 LT LTEP13869663.8T patent/LT2940030T/lt unknown
- 2013-12-10 SI SI201330744T patent/SI2940030T1/sl unknown
- 2013-12-10 HU HUE13869663A patent/HUE035675T2/hu unknown
- 2013-12-10 KR KR1020157017372A patent/KR102024700B1/ko active Active
- 2013-12-10 JP JP2014554298A patent/JP6049760B2/ja active Active
- 2013-12-10 CN CN201380074088.5A patent/CN105026414B/zh active Active
- 2013-12-10 ES ES13869663.8T patent/ES2629743T3/es active Active
- 2013-12-10 CA CA2896455A patent/CA2896455C/en active Active
- 2013-12-10 HR HRP20171333TT patent/HRP20171333T1/hr unknown
- 2013-12-10 PT PT138696638T patent/PT2940030T/pt unknown
- 2013-12-10 SM SM20170405T patent/SMT201700405T1/it unknown
- 2013-12-10 WO PCT/JP2013/083100 patent/WO2014103704A1/ja not_active Ceased
-
2017
- 2017-09-07 CY CY20171100946T patent/CY1119372T1/el unknown
-
2018
- 2018-11-30 US US16/206,380 patent/US11208428B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008024169A1 (en) | 2006-07-21 | 2008-02-28 | Glsynthesis, Inc. | Reactive pyrophosphoric and bisphosphonic acid derivatives and methods of their use |
| WO2008012949A1 (en) | 2006-07-26 | 2008-01-31 | Yamasa Corporation | Process for producing di(pyrimidine nucleoside 5'-)polyphosphate |
Non-Patent Citations (7)
| Title |
|---|
| BIOORG. & MEDICINAL CHEMISTRY LETTERS, vol. 11, 2001, pages 157 - 160 |
| HUANG, KAISHENG ET AL.: "Engineering Human Fhit, a Diadenosine Triphosphate Hydrolase, into an Efficient Dinucleoside Polyphosphate Synthase", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 126, no. 31, 2004, pages 9548 - 9549, XP055261676 * |
| JOURNAL OF AMERICAN CHEMICAL SOCIETY, vol. 126, 2004, pages 9548 |
| NUCLEIC ACIDS RESEARCH, vol. 4, 1977, pages 2843 |
| ORG. BIOMOL. CHEM., vol. 2011, no. 9, 2011, pages 730 - 738 |
| See also references of EP2940030A4 |
| SHIMAZU, MASAMITSU ET AL.: "Facile synthesis of nucleotides containing polyphosphates by manganese(II) and cadmium(II) ion-catalyzed pyrophosphate bond formation in aqueous solution", TETRAHEDRON LETTERS, vol. 31, no. 2, 1990, pages 235 - 238, XP002636622 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10815263B2 (en) | 2016-10-25 | 2020-10-27 | Yamasa Corporation | Method for purifying P1,P4-di(uridine 5′-)tetraphosphate |
| WO2018079503A1 (ja) | 2016-10-25 | 2018-05-03 | ヤマサ醤油株式会社 | P1,p4-ジ(ウリジン5'-)テトラホスフェートの精製方法 |
| KR102259309B1 (ko) | 2016-10-25 | 2021-06-01 | 야마사 쇼유 가부시키가이샤 | P1, p4-디(우리딘 5'-)테트라포스페이트의 정제 방법 |
| KR20190067162A (ko) * | 2016-10-25 | 2019-06-14 | 야마사 쇼유 가부시키가이샤 | P1, p4-디(우리딘 5'-)테트라포스페이트의 정제 방법 |
| JPWO2018079503A1 (ja) * | 2016-10-25 | 2019-09-19 | ヤマサ醤油株式会社 | P1,p4−ジ(ウリジン5’−)テトラホスフェートの精製方法 |
| CN108164577A (zh) * | 2017-04-28 | 2018-06-15 | 广东众生药业股份有限公司 | 一种p1,p4-二(尿苷-5’-四磷酸)钠盐的工业化制备方法 |
| JP7028727B2 (ja) | 2017-06-21 | 2022-03-02 | チョン クン ダン ファーマシューティカル コーポレーション | ジヌクレオシドポリリン酸化合物の製造方法{Method for Preparing a Dinucleoside Polyphosphate Compound} |
| JP7373598B2 (ja) | 2017-06-21 | 2023-11-02 | チョン クン ダン ファーマシューティカル コーポレーション | ジヌクレオシドポリリン酸化合物の製造方法 |
| JP2022065110A (ja) * | 2017-06-21 | 2022-04-26 | チョン クン ダン ファーマシューティカル コーポレーション | ジヌクレオシドポリリン酸化合物の製造方法 |
| JP2019006771A (ja) * | 2017-06-21 | 2019-01-17 | チョン クン ダン ファーマシューティカル コーポレーション | ジヌクレオシドポリリン酸化合物の製造方法{Method for Preparing a Dinucleoside Polyphosphate Compound} |
| KR20180091672A (ko) | 2017-06-21 | 2018-08-16 | 주식회사 종근당 | 디뉴클레오사이드 폴리포스페이트 화합물의 제조방법 |
| JP7028727B6 (ja) | 2017-06-21 | 2022-03-14 | チョン クン ダン ファーマシューティカル コーポレーション | ジヌクレオシドポリリン酸化合物の製造方法{Method for Preparing a Dinucleoside Polyphosphate Compound} |
| KR20200106738A (ko) | 2019-03-05 | 2020-09-15 | 주식회사 파마코스텍 | P1,p4-디(우리딘 5'-)테트라포스페이트, 이의 염 또는 이의 수화물의 신규한 제조방법 |
| JP2023511656A (ja) * | 2019-12-18 | 2023-03-22 | チョングンダン ファーマスーティカル株式会社 | ウリジン5’-二リン酸(udp)、その塩またはその水和物の製造方法 |
| JP7405991B2 (ja) | 2019-12-18 | 2023-12-26 | チョングンダン ファーマスーティカル株式会社 | ウリジン5’-二リン酸(udp)、その塩またはその水和物の製造方法 |
| CN111662350A (zh) * | 2020-07-07 | 2020-09-15 | 南京宸翔医药研究有限责任公司 | 一种绿色化智能化高纯度地夸磷索四钠的制备方法 |
| CN111662350B (zh) * | 2020-07-07 | 2022-06-07 | 南京宸翔医药研究有限责任公司 | 一种绿色化智能化高纯度地夸磷索四钠的制备方法 |
| WO2022212442A1 (en) | 2021-03-31 | 2022-10-06 | Modernatx, Inc. | Synthesis of trinucleotide and tetranucleotide caps for mrna production |
| WO2024185734A1 (ja) | 2023-03-03 | 2024-09-12 | 株式会社ナティアス | ポリリン酸化ヌクレオシド、ポリリン酸化ヌクレオシドの合成に用いられるリン酸部活性化ヌクレオチド及びその合成方法、ならびにリン酸部活性化ヌクレオチドを用いたポリリン酸化ヌクレオシドの合成方法 |
| KR20250157528A (ko) | 2023-03-03 | 2025-11-04 | 나티아스 인크. | 폴리인산화 뉴클레오시드, 폴리인산화 뉴클레오시드의 합성에 사용되는 인산부 활성화 뉴클레오티드 및 그 합성 방법, 및 인산부 활성화 뉴클레오티드를 사용한 폴리인산화 뉴클레오시드의 합성 방법 |
| EP4667479A1 (en) | 2023-03-03 | 2025-12-24 | Natias Inc. | Polyphosphorylated nucleoside, phosphate-activated nucleotide used for synthesis of polyphosphorylated nucleoside and method for synthesis of same, and method for synthesis of polyphosphorylated nucleoside using phosphate-activated nucleotide |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105026414B (zh) | 2017-06-23 |
| US10179799B2 (en) | 2019-01-15 |
| PT2940030T (pt) | 2017-09-11 |
| CA2896455A1 (en) | 2014-07-03 |
| CY1119372T1 (el) | 2018-02-14 |
| RS56227B1 (sr) | 2017-11-30 |
| HUE035675T2 (hu) | 2018-05-28 |
| SI2940030T1 (sl) | 2017-09-29 |
| KR102024700B1 (ko) | 2019-09-24 |
| SMT201700405T1 (it) | 2017-09-07 |
| JP6049760B2 (ja) | 2016-12-21 |
| DK2940030T3 (en) | 2017-07-24 |
| US20160194347A1 (en) | 2016-07-07 |
| CA2896455C (en) | 2021-04-20 |
| US20190092803A1 (en) | 2019-03-28 |
| LT2940030T (lt) | 2017-07-10 |
| EP2940030A1 (en) | 2015-11-04 |
| ES2629743T3 (es) | 2017-08-14 |
| KR20150100708A (ko) | 2015-09-02 |
| EP2940030B1 (en) | 2017-06-14 |
| JPWO2014103704A1 (ja) | 2017-01-12 |
| HRP20171333T1 (hr) | 2017-11-03 |
| EP2940030A4 (en) | 2016-09-14 |
| PL2940030T3 (pl) | 2017-11-30 |
| CN105026414A (zh) | 2015-11-04 |
| US11208428B2 (en) | 2021-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6049760B2 (ja) | P1,p4−ジ(ウリジン5’−)テトラホスフェートの製造法 | |
| JP5193040B2 (ja) | ジ(ピリミジンヌクレオシド5’−)ポリホスフェートの製造法 | |
| JP7373598B2 (ja) | ジヌクレオシドポリリン酸化合物の製造方法 | |
| JP3247685B2 (ja) | ジウリジンテトラホスフェート又はその塩の結晶及びその製造法、並びに該化合物の製造法 | |
| US10815263B2 (en) | Method for purifying P1,P4-di(uridine 5′-)tetraphosphate | |
| CN101896496B (zh) | 4-脱氧-4-氟-d-葡萄糖衍生物的制造方法 | |
| AU2010288524A1 (en) | Method for producing pyrazole glycoside derivatives | |
| JP2023511656A (ja) | ウリジン5’-二リン酸(udp)、その塩またはその水和物の製造方法 | |
| KR20210110055A (ko) | P1, p4-디(우리딘 5'-)테트라포스페이트 나트륨염 4 수화물 결정형 a의 제조방법 | |
| JPS591719B2 (ja) | チミン誘導体の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201380074088.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13869663 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014554298 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2896455 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14758102 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20157017372 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013869663 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013869663 Country of ref document: EP |