WO2014189139A1 - オキシメチレン共重合体の製造方法 - Google Patents
オキシメチレン共重合体の製造方法 Download PDFInfo
- Publication number
- WO2014189139A1 WO2014189139A1 PCT/JP2014/063733 JP2014063733W WO2014189139A1 WO 2014189139 A1 WO2014189139 A1 WO 2014189139A1 JP 2014063733 W JP2014063733 W JP 2014063733W WO 2014189139 A1 WO2014189139 A1 WO 2014189139A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymerization
- trioxane
- oxymethylene copolymer
- production method
- copolymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/17—Amines; Quaternary ammonium compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2/00—Addition polymers of aldehydes or cyclic oligomers thereof or of ketones; Addition copolymers thereof with less than 50 molar percent of other substances
- C08G2/04—Polymerisation by using compounds which act upon the molecular weight, e.g. chain-transferring agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2/00—Addition polymers of aldehydes or cyclic oligomers thereof or of ketones; Addition copolymers thereof with less than 50 molar percent of other substances
- C08G2/06—Catalysts
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2/00—Addition polymers of aldehydes or cyclic oligomers thereof or of ketones; Addition copolymers thereof with less than 50 molar percent of other substances
- C08G2/10—Polymerisation of cyclic oligomers of formaldehyde
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2/00—Addition polymers of aldehydes or cyclic oligomers thereof or of ketones; Addition copolymers thereof with less than 50 molar percent of other substances
- C08G2/18—Copolymerisation of aldehydes or ketones
- C08G2/24—Copolymerisation of aldehydes or ketones with acetals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G4/00—Condensation polymers of aldehydes or ketones with polyalcohols; Addition polymers of heterocyclic oxygen compounds containing in the ring at least once the grouping —O—C—O—
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/13—Phenols; Phenolates
- C08K5/134—Phenols containing ester groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L59/00—Compositions of polyacetals; Compositions of derivatives of polyacetals
- C08L59/04—Copolyoxymethylenes
Definitions
- the present invention relates to a method for producing an oxymethylene copolymer with few unstable parts suitable for extrusion molding.
- Polyoxymethylene resin is excellent in mechanical and thermal performance. Especially, oxymethylene copolymer has better thermal stability and moldability than oxymethylene homopolymer, so it is used as an engineering plastic. It has been. Regarding the method for producing an oxymethylene copolymer, trioxane and 1,3-dioxolane are mixed with boron trifluoride or its distribution using a continuous polymerization apparatus in which a continuous polymerization apparatus and a terminator mixer connected thereto are connected in series.
- the polymerization is preferably carried out by bringing the oxymethylene copolymer produced without a washing step into contact with the polymerization terminator in the terminator mixer.
- a method of continuously polymerizing an oxymethylene copolymer to be stopped is known (for example, see Patent Document 1).
- Patent Document 1 discloses a technique for producing an oxymethylene copolymer that suppresses the generation of unstable moieties using trioxane, 1,3-dioxolane, and boron trifluoride, which are industrially inexpensive and easy to handle, as raw materials. Has been. This technique is excellent in that the monomer recovery cost can be reduced because the polymerization yield is high and washing is not required when the polymerization is stopped.
- Patent Document 1 discloses that when 1,3-dioxolane is used as a comonomer in the production of polyoxymethylene resin, the base instability is reduced to about half compared to the case of using ethylene oxide. It is described that the conversion rate is also reduced by about half. Thus, 1,3-dioxolane and ethylene oxide behave significantly differently as comonomer of polyoxymethylene resin.
- a sterically hindered phenol having a molecular weight of 350 or more is contained in the monomer in an amount of 0.001 to 2.2.
- a technique for carrying out copolymerization by adding 0% by weight is known (for example, see Patent Document 2).
- Patent Document 2 discloses that when trioxane and 1,3-dioxolane are copolymerized using a boron trifluoride ether coordination compound as a catalyst, the copolymerization is carried out in the presence of a sterically hindered phenol to perform alkali decomposition. The technique which improved the rate and the weight reduction rate by heating is disclosed.
- trioxane and 1,3-dioxolane are copolymerized using a boron trifluoride ether coordination compound as a catalyst
- copolymerization is performed using 1,3-dioxolane to which a sterically hindered phenol having a molecular weight of 350 or more is added.
- the technique of performing is known (see, for example, Patent Documents 3 and 4).
- the polymerization yield is 85% or less, and washing is performed simultaneously with the termination of the polymerization, which requires a large amount of energy to recover the unreacted monomer, which is economically disadvantageous. .
- polyoxymethylene resin is also an engineering plastic with an excellent balance of mechanical properties, and is generally used as a cutting material for so-called round bars and plates.
- the polyoxymethylene resin has high crystallinity, the shrinkage at the time of solidification is large, so when molding after melting once, it is called void (hereinafter referred to as nest, microvoid, white core, whitening, etc.) inside the molded product (It is a generic term for defective portions) and is disadvantageous in terms of physical properties and appearance.
- nest microvoid
- white core white core
- a round bar having a large diameter is generally formed by a so-called solidification extrusion method in which it is solidified in a cooled die, but in such a case, a void is likely to occur in the central portion.
- the polyoxymethylene for cutting made of a polyoxymethylene resin having an isothermal crystallization speed at 150 ° C. of 200 seconds or more is assumed.
- Resin materials have been proposed (see, for example, Patent Document 9).
- it is necessary to increase the molecular weight of the polyoxymethylene resin that is, to use a resin having a melt index of 2.0 g / 10 min or less as a means for achieving a delay in the isothermal crystallization rate.
- the isothermal crystallization rate is delayed by increasing the molecular weight of the resin, it has a serious industrial disadvantage.
- the polyoxymethylene resin in the polyoxymethylene resin, impurities tend to act as a chain terminator during polymerization, and it is extremely difficult to achieve a high molecular weight of a melt index of 2.0 g / 10 min or less without the introduction of a crosslinking or branched structure. Even if it can be achieved, it is only an industrially unfavorable method in which the production rate is significantly reduced. Further, the high molecular weight polyoxymethylene resin has the disadvantage that the load on the extruder during molding is increased due to an increase in viscosity, resulting in a decrease in moldability, and thermal decomposition is likely to occur due to shear heat generation during plasticization.
- JP-A-8-325341 Japanese Examined Patent Publication No. 3-63965 Japanese Patent Laid-Open No. 7-242652 Japanese Patent Laid-Open No. 11-269165 JP-A-6-212054 JP-A-7-207117 JP 7-292216 A JP-A-9-066753 JP 9-052926 A JP 2003-064141 A
- the present invention suppresses the generation of a structure unstable to heat and hydrolysis by having a formate structure, improves the polymer quality such as reducing the amount of formaldehyde generated, and extrusion molding.
- An object of the present invention is to continuously produce an oxymethylene copolymer excellent in properties, particularly void reduction effect, at a high polymerization yield.
- the inventors of the present invention copolymerized a monomer raw material containing trioxane and 1,3-dioxolane using boron trifluoride as a catalyst to obtain an oxymethylene copolymer.
- trioxane and a specific amount of 1,3-dioxolane are copolymerized in the presence of a specific amount of a sterically hindered phenol, and when the polymerization yield is 92% or more, Polymerization is terminated by bringing the polymer into contact with a polymerization terminator to obtain a crude oxymethylene copolymer, and the crude oxymethylene copolymer is heated and melted in the presence of a specific amount of an antioxidant and a nitrogen-containing compound.
- the inventors have found that the above object can be achieved by heat stabilization, and have completed the present invention.
- the present invention provides the following production method: A monomer raw material containing trioxane and 5 to 7% by mass of 1,3-dioxolane with respect to trioxane was mixed with 0.025 to 0.07 mmol of boron trifluoride per mole of trioxane and 0.03 with respect to trioxane. A step of carrying out copolymerization in the presence of 006 to 2.0% by mass of a sterically hindered phenol; when the polymerization yield of the copolymer becomes 92% or more, a polymerization terminator is added to the reaction system for polymerization.
- a process for producing an oxymethylene copolymer comprising:
- trioxane and 1,3-dioxolane are copolymerized using boron trifluoride as a catalyst, they are insensitive to heat and hydrolysis having a formate structure. Since the production
- the obtained copolymer has excellent extrudability and has a feature that the formation of voids in the extrudate is extremely small.
- productivity can be greatly improved, and a molded product extruded from the copolymer or a resin composition containing the copolymer can be cut as a polyoxymethylene resin material to obtain high quality. It provides electrical, electronic parts, automobile parts and various industrial parts and is very useful.
- the method for producing an oxymethylene copolymer of the present invention comprises a monomer raw material containing trioxane and a specific amount of 1,3-dioxolane in the presence of a specific amount of boron trifluoride and a specific amount of sterically hindered phenol. And when the polymerization yield is 92% or more, the produced copolymer and a polymerization terminator are brought into contact with each other to terminate the polymerization to obtain a crude oxymethylene copolymer, and a specific amount. And a step of thermally stabilizing the crude oxymethylene copolymer in the presence of an antioxidant and a nitrogen-containing compound.
- the trioxane (1,3,5-trioxane) used as a monomer in the present invention is a cyclic trimer of formaldehyde, which is commercially available or can be prepared by a production method known to those skilled in the art. Is not particularly limited.
- amines are usually contained in an amount of 0.00001 to 0.003 mmol, preferably 0.00001 to 0.0005 mmol, more preferably 0.00001 to 0.0003 mmol, per mol of trioxane, relative to trioxane. If the content of amines is higher than this, it will cause adverse effects such as deactivation of the catalyst, and if it is less, it will cause adverse effects such as generation of paraformaldehyde during storage of trioxane.
- amines to be contained in trioxane primary amines, secondary amines, tertiary amines, amine compounds having an alcoholic hydroxyl group in the molecule, alkylated melamines, and hindered amine compounds are used alone or as a mixture.
- Primary amines are n-propylamine, isopropylamine, n-butylamine, etc.
- Secondary amines are diethylamine, di-n-propylamine, diisopropylamine, di-n-butylamine, piperidine, piperazine, 2-methylpiperazine , Morpholine, N-methylformoline, N-ethylformoline, etc.
- tertiary amines include triethylamine, tri-n-propylamine, triisopropylamine, tri-n-butylamine, etc.
- amine-based compounds include monoethanolamine, diethanolamine, triethanolamine, N-methylethanolamine, N, N-dimethylethanolamine, N-ethylethanolamine, N, N-diethylethanolamine, N- ( ⁇ -Ami Ethyl) isopropanolamine, hydroxyethyl piperazine and the mono as the alkylated melamine methoxymethyl substituents melamine, di-, tri-, tetra, penta or hexa methoxymethyl melamine, or mixtures thereof and the like are preferably used.
- hindered amine compounds include bis (2,2,6,6-tetramethyl-4-piperidinyl) sebacate, bis (1,2,2,6,6-pentamethyl-4-piperidinyl) sebacate, 1,2,3, 4-Butanetetracarboxylic acid tetrakis (2,2,6,6-tetramethyl-4-piperidinyl) ester, poly [[6- (1,1,3,3-tetramethylbutyl) amino-1,3,5 -Triazine-2,4-diyl] [(2,2,6,6-tetramethyl-4-piperidinyl) imino] hexamethylene [(2,2,6,6-tetramethyl-4-piperidinyl) imino]] 1,2,2,6,6-pentamethylpiperidine, dimethyl succinate 1- (2-hydroxyethyl) -4-hydroxy-2,2,6,6-tetramethylpiperidine polycondensate Or N, N′-bis (3-aminopropyl) ethylene
- the 1,3-dioxolane used as a comonomer in the present invention is commercially available or can be prepared by a production method known to those skilled in the art.
- the manufacturing method is not particularly limited.
- 1,3-dioxolane is used in an amount of 5 to 7% by mass based on trioxane.
- the mechanical strength of the extruded product for example, impact strength, bending strength, tensile strength, and creep properties are weakened, and when it is small, the rate of semicrystallization is increased. Therefore, it is inferior to extrusion moldability.
- the chain transfer agent In order to adjust the molecular weight of the obtained copolymer to adjust the MVR (Melute Volume Rate) and the intrinsic viscosity, 0.01 to 0.3 mol% of the chain transfer agent can be used with respect to trioxane.
- the chain transfer agent include carboxylic acid, carboxylic acid anhydride, ester, amide, imide, phenol, acetal compound and the like.
- phenol, 2,6-dimethylphenol, methylal, and polyoxymethylene dimethoxide are preferably used. Most preferred is methylal.
- the chain transfer agent is used as is or in the form of a solution.
- examples of the solvent include aliphatic hydrocarbons such as hexane, heptane, and cyclohexane; aromatic hydrocarbons such as benzene, toluene, and xylene; and halogenated hydrocarbons such as methylene dichloride and ethylene dichloride.
- Impurities such as water, formic acid, methanol, and formaldehyde contained in trioxane are inevitably generated when trioxane is produced industrially, but the total amount is preferably 100 ppm or less in trioxane. Preferably it is 70 ppm or less, Most preferably, it is 50 ppm or less. In particular, water is preferably 50 ppm or less, more preferably 20 ppm or less, and most preferably 10 ppm or less.
- impurities such as water, formic acid and formaldehyde present in 1,3-dioxolane are preferably 1000 ppm or less, more preferably 200 ppm or less in total.
- the boron trifluoride used in the present invention is preferably a coordination compound thereof, which is commercially available or can be prepared by a production method known to those skilled in the art.
- a coordination compound of boron trifluoride a coordination compound with an organic compound having an oxygen atom or a sulfur atom can be given.
- the organic compound include alcohol, phenol, acid, ether, acid anhydride, ester, ketone, aldehyde, dialkyl, and sulfide.
- the coordination compound of boron trifluoride is preferably an etherate, and preferred specific examples include diethyl etherate and dibutyl etherate of boron trifluoride.
- the amount added is generally 0.025 to 0.07 mmol, preferably 0.030 to 0.06 mmol, and most preferably 0.035 to 0.005 mol per mol of trioxane of the main monomer. It is in the range of 055 mmol.
- the amount of boron trifluoride added is larger than this, an adverse effect such as an increase in the formate structure is caused, and when it is less, an adverse effect such as a decrease in the polymerization conversion rate is caused.
- Boron trifluoride is used as is or in the form of a solution.
- examples of the solvent include aliphatic hydrocarbons such as hexane, heptane, and cyclohexane; aromatic hydrocarbons such as benzene, toluene, and xylene; and halogenated hydrocarbons such as methylene dichloride and ethylene dichloride.
- the polymerization time is usually 0.25 to 120 minutes, preferably 1 to 60 minutes, more preferably 1 to 30 minutes, and most preferably 2 to 15 minutes. If the polymerization time is longer than this, unstable parts increase, and if it is shorter, the polymerization yield may decrease.
- the polymerization reaction may be solution polymerization carried out in the presence of an inert solvent, but bulk polymerization under substantially no solvent is preferable because the cost of solvent recovery is unnecessary and the effect of sterically hindered phenol is large.
- a solvent aliphatic hydrocarbons such as hexane, heptane, and cyclohexane; aromatic hydrocarbons such as benzene, toluene, and xylene; and halogenated hydrocarbons such as methylene dichloride and ethylene dichloride.
- the copolymerization reaction is preferably performed using a continuous polymerization apparatus.
- a method in which two or more continuous polymerization machines are connected in series is suitable.
- a continuous polymerization machine a kneading machine having at least two horizontal rotation shafts and screw-type or paddle-type rotation blades on the rotation shafts can be mentioned as a preferable example.
- copolymerization is carried out in the presence of a sterically hindered phenol, and the addition amount of the sterically hindered phenol is usually 0.006 to 2.0% by mass, preferably 0.01% with respect to trioxane. To 0.5% by mass, more preferably 0.02 to 0.1% by mass. If the amount of sterically hindered phenol used is greater than this, it will cause adverse effects such as a decrease in the molecular weight of the resulting crude oxymethylene copolymer and a decrease in the polymerization yield.
- the unstable portion such as the formic acid ester structure increases, which causes adverse effects such as a decrease in heat or hydrolysis stability.
- the sterically hindered phenol is added as it is or in the form of a solution.
- the solvent include aliphatic hydrocarbons such as hexane, heptane and cyclohexane; aromatic hydrocarbons such as benzene, toluene and xylene; and halogenated hydrocarbons such as methylene dichloride and ethylene dichloride. It is done.
- the monomer trioxane and the comonomer 1,3-dioxolane may be used as the solvent.
- a part or all of the sterically hindered phenol as it is or in the form of a solution at the continuous polymerization apparatus inlet.
- a predetermined amount can also be dissolved in advance in trioxane charged into the polymerization machine.
- the sterically hindered phenol used in the polymerization in the present invention is, for example, dibutylhydroxytoluene, triethylene glycol-bis-3- (3-tert-butyl-4-hydroxy-5-methylphenyl) propionate, pentaerythrityl-tetrakis -3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate, hexamethylenebis [3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate], 2,2 ' -Methylenebis (6-t-butyl-4-methylphenol), 3,9-bis ⁇ 2- [3- (3-t-butyl-4-hydroxy-5-methylphenyl) propionyloxy] -1,1- Dimethylethyl ⁇ -2,4,8,10-tetraoxaspiro [5.5] undecane, N, N′-hexane-1,6- Irbis [3- (3,5-di-
- a polymerization terminator is added to deactivate the catalyst (boron trifluoride). Stop the polymerization.
- Trioxane and 1,3-dioxolane are copolymerized in the presence of boron trifluoride and sterically hindered phenol, the resulting copolymer is contacted with a polymerization terminator to terminate the polymerization, and crude oxymethylene copolymer
- the step of obtaining a coalescence when the polymerization is stopped at a polymerization yield of less than 92%, the amount of the unstable portion of the crude oxymethylene copolymer having a formate structure that is unstable with respect to heat and hydrolysis is small and steric hindrance is caused.
- the polymerization is terminated at a polymerization yield of 92% or more, the recovery cost of the unreacted monomer is reduced, but in the prior art, the crude oxymethylene copolymer is unstable to heat and hydrolysis having a formate structure. Part is generated suddenly.
- the formate ester in the crude oxymethylene copolymer is present by the presence of a specific amount of boron trifluoride and a specific amount of sterically hindered phenol during the copolymerization and termination of the polymerization at a polymerization yield of 92% or more. It has been clarified that the amount of unstable parts having a structure can be greatly reduced.
- the formate group content in the polymer after termination of the polymerization is desirably 10 ⁇ mol or less per 1 g of the polymer, and 9 ⁇ mol It is more desirable that it is less than or equal to 8 ⁇ mol or less.
- the polymerization reaction is stopped by bringing it into contact with a copolymer that has produced a polymerization terminator.
- the polymerization terminator is used as it is or in the form of a solution or suspension, but the contact method is to continuously add a small amount of polymerization terminator, or a solution or suspension of the polymerization terminator to the reaction system, It is desirable to grind and contact.
- a washing step such as introducing a copolymer into a solution or suspension of a large amount of the polymerization terminator, a solvent recovery step or a solvent removal step in the subsequent stage is required. Is complicated and leads to an increase in utilities, which is industrially disadvantageous.
- a method in which a small amount of a polymerization terminator is added to a reaction system containing a copolymer when the polymerization is terminated is industrially more preferable.
- a polymerization terminator in the reaction system it is preferable to mix with a mixer after the addition.
- a continuous mixer such as a single or twin screw or a paddle type mixer can be used.
- a mixer connected in series with the continuous polymerization machine used in a copolymerization reaction is preferable to use a mixer connected in series with the continuous polymerization machine used in a copolymerization reaction.
- primary amine include n-propylamine, isopropylamine, n-butylamine, etc.
- Secondary amines include diethylamine, di-n-propylamine, diisopropylamine, di-n-butylamine, piperidine, morpholine, etc. Examples of amines include triethylamine, tri-n-propylamine, triisopropylamine, and tri-n-butylamine.
- alkylated melamine examples include mono, di, tri, tetra, penta, or hexamethoxy, which are methoxymethyl substitutes of melamine. Methylmelamine or a mixture thereof is preferably used.
- hindered amine compounds include bis (2,2,6,6-tetramethyl-4-piperidinyl) sebacate, bis (1,2,2,6,6-pentamethyl-4-piperidinyl) sebacate, 1,2,3, 4-Butanetetracarboxylic acid tetrakis (2,2,6,6-tetramethyl-4-piperidinyl) ester, poly [[6- (1,1,3,3-tetramethylbutyl) amino-1,3,5 -Triazine-2,4-diyl] [(2,2,6,6-tetramethyl-4-piperidinyl) imino] hexamethylene [(2,2,6,6-tetramethyl-4-piperidinyl) imino]] 1,2,2,6,
- hindered amine compounds trivalent organic phosphorus compounds, and alkylated melamines are preferred in terms of hue.
- hindered amine compounds include bis (2,2,6,6-tetramethyl-4-piperidinyl) sebacate, bis (1,2,2,6,6-pentamethyl-4-piperidinyl) sebacate, dimethyl succinate 1- ( 2-hydroxyethyl) -4-hydroxy-2,2,6,6-tetramethylpiperidine polycondensate, N, N′-bis (3-aminopropyl) ethylenediamine ⁇ 2,4-bis [N-butyl-N -(1,2,2,6,6-pentamethyl-4-piperidyl) amino] -1,3,5-triazine condensate is a trivalent organophosphorus compound, triphenylphosphine as alkylated melamine Hexamethoxymethylmelamine is most preferably used.
- the solvent used is not particularly limited, but other than water and alcohols, acetone, methyl ethyl ketone, hexane, cyclohexane, heptane, benzene, toluene, xylene, Various aliphatic and aromatic organic solvents such as methylene dichloride and ethylene dichloride can be used. Among these, preferred are water, alcohols, and aliphatic and aromatic organic solvents such as acetone, methyl ethyl ketone, hexane, cyclohexane, heptane, benzene, toluene, and xylene.
- the crude oxymethylene copolymer after the termination of polymerization is obtained in a high yield, it can be sent to the stabilization step as it is.
- the stabilization methods described in (A) and (B) below can be employed.
- the thermal stabilization method (A) has a simpler process than the method (B) and is preferable as an industrial method. That is, when the method (A) is adopted, the crude oxymethylene copolymer is melt-kneaded under a pressure of 760 to 0.1 mmHg in the temperature range from the melting temperature to a temperature 100 ° C. higher than the melting temperature. Is preferred. When the heat stabilization treatment temperature is lower than the melting temperature of the crude oxymethylene copolymer, the decomposition reaction of the unstable portion becomes insufficient, and the stabilization effect cannot be obtained.
- the temperature is higher than the melting temperature of 100 ° C.
- yellowing occurs, the main chain of the polymer is decomposed due to heat, and at the same time, an unstable portion is generated and the thermal stability is impaired.
- a more preferable range is 170 to 250 ° C., and a most preferable range is 180 to 235 ° C.
- the pressure during the stabilization process is higher than 760 mmHg, the effect of removing the decomposition gas generated by the decomposition of the unstable portion is low, and a sufficient stabilization effect cannot be obtained.
- a more preferable range is 740 to 10 mmHg, most preferably 400 to 50 mmHg. Further, the treatment time is appropriately selected within the range of 5 minutes to 1 hour.
- a single-screw or twin-screw or more extruder with a vent can be used as the apparatus used for the thermal stabilization treatment.
- a method of combining a highly degassing effect such as a WSK ZSK extruder or a ZDS extruder is a more advantageous method.
- a method of combining a surface renewal type mixer with the above-described extruder as shown in Examples described later is the most effective method.
- a stabilizer such as an antioxidant or a heat stabilizer can be added during the melt kneading of the crude oxymethylene copolymer.
- the stabilization treatment can be carried out in the presence of 0.05 to 5% by mass of an antioxidant and 0.005 to 5% by mass of a nitrogen-containing compound based on the crude oxymethylene copolymer.
- Antioxidants that can be used in the thermal stabilization treatment include triethylene glycol-bis-3- (3-tert-butyl-4-hydroxy-5-methylphenyl) propionate, pentaerythrityl-tetrakis-3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate, 2,2′-methylenebis (6-tert-butyl-4-methylphenol), 3,9-bis ⁇ 2- [3- (3 -T-butyl-4-hydroxy-5-methylphenyl) propionyloxy] -1,1-dimethylethyl ⁇ -2,4,8,10-tetraoxaspiro [5.5] undecane, N, N′-hexane -1,6-diylbis [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionamide], 3,5-bis (1,1-dimethylethyl)- - one or more sterically hindered phenols, such as hydroxy
- Heat stabilizers include amine-substituted triazines such as melamine, methylolmelamine, benzoguanamine, cyanoguanidine, N, N-diarylmelamine, polyamides, urea derivatives, nitrogen-containing compounds such as urethanes, and sodium, potassium, calcium, magnesium , Barium inorganic acid salt, hydroxide, organic acid salt and the like.
- amine-substituted triazines such as melamine, methylolmelamine, benzoguanamine, cyanoguanidine, N, N-diarylmelamine, polyamides, urea derivatives, nitrogen-containing compounds such as urethanes, and sodium, potassium, calcium, magnesium , Barium inorganic acid salt, hydroxide, organic acid salt and the like.
- the MVR of the stabilization process after the oxymethylene copolymer is regulated to 0.5 ⁇ 20cm 3/10 minutes, preferably 0.5 ⁇ 6cm 3/10 min, more preferably 1 ⁇ 3 cm 3 Adjusted to / 10 minutes.
- the isothermal half crystallization time of the oxymethylene copolymer at 150 ° C. is adjusted to 150 to 2000 seconds depending on the amount of catalyst and the amount of impurities during the polymerization, preferably 200 to 1000 seconds, more preferably 300. Adjusted to ⁇ 500 seconds.
- the isothermal half crystallization time is less than 150 seconds (high half crystallization speed), the voids of the molded product obtained by extrusion molding become remarkable.
- the isothermal half crystallization time means that it is measured under the conditions described in the Examples below.
- the oxymethylene copolymer obtained by the method of the present invention described in detail has the same excellent properties as the oxymethylene copolymer obtained by the conventional method, and can be used for the same application.
- the oxymethylene copolymer produced by the method of the present invention includes a coloring agent, a nucleating agent, a plasticizer, a release agent, an optical brightener, an antistatic agent such as polyethylene glycol and glycerin, and a benzophenone compound.
- Additives such as light stabilizers such as hindered amine compounds can be added as desired.
- the oxymethylene copolymer produced by the method of the present invention can be used for extrusion molding.
- the oxymethylene copolymer produced by the method of the present invention is supplied thereto, and melt kneaded at 180 to 240 ° C.
- the resin material for cutting can be obtained by extruding while cooling the molded product continuously discharged from the mold and taking it out with a roller or the like.
- the resin material for cutting obtained by such extrusion molding typically includes a round bar having a diameter of 10 mm or more, a plate material having a thickness of 5 mm or more, and a deformed cross-section having a thickness of 10 mm or more.
- These resin materials for cutting are machined by usual means such as cutting with a lathe or a milling machine, and are processed into machine parts, automobile parts, and the like.
- Specific processing parts include screws, bearings, nozzle parts, sensor parts, gears, rollers, axles, pallets, and the like.
- Crude oxymethylene copolymer An oxymethylene copolymer after the termination of polymerization and before the stabilization step is referred to as a crude oxymethylene copolymer.
- Formate ester group content about 12 mg of crude oxymethylene copolymer powder before being subjected to stabilization step, weighed about 12 mg, and dissolved in 1 g of hexafluoroisopropanol-d 2 solvent, a nuclear magnetic resonance apparatus (JNM LA500 ; Japan was measured by an electronic Ltd.), 1 H-NMR chart formic ester 1 H peak and 4.9ppm methylene main crude oxymethylene copolymer appeared in the vicinity of the group appeared in the vicinity of 8.0ppm of The formate group content was determined from the area ratio of the chain to the 1 H peak. The content of formate group in 1 g of the polymer was expressed in micromol.
- Examples 1 to 8 and Comparative Examples 1 to 10 An oxymethylene copolymer was produced by batch polymerization using a desktop type biaxial kneader having a volume of 1 L having a jacket and two Z-shaped blades as a polymerization apparatus. 85 ° C. warm water was circulated through the jacket, and the interior was further heated and dried with high-temperature air, then a lid was attached, and the system was purged with nitrogen.
- 1,3,5-trioxane 320 g (containing 0.00025 mmol of triethanolamine per mole of trioxane as a stabilizer), a predetermined amount of comonomer (1,3-dioxolane) and a predetermined amount of steric hindrance from the raw material inlet
- a phenol triethylene glycol-bis-3- (3-t-butyl-4-hydroxy-5-methylphenyl) propionate (trade name; Irganox 245 manufactured by BASF)
- a predetermined amount of boron trifluoride diethyl etherate as a catalyst was added as a benzene solution (solution concentration: 0.6 mmol / g) to initiate polymerization.
- triphenylphosphine corresponding to twice the amount of the catalyst used was added as a benzene solution (solution concentration: 5 mmol / ml) into the polymerization apparatus using a syringe and mixed for another 15 minutes.
- the polymerization was stopped to obtain a crude oxymethylene copolymer.
- the crude oxymethylene copolymer was measured for polymerization yield and formate group content.
- the isothermal half crystallization time of the pellet obtained by putting a predetermined amount of antioxidant and a nitrogen-containing compound into the crude oxymethylene copolymer and melt-kneading was measured.
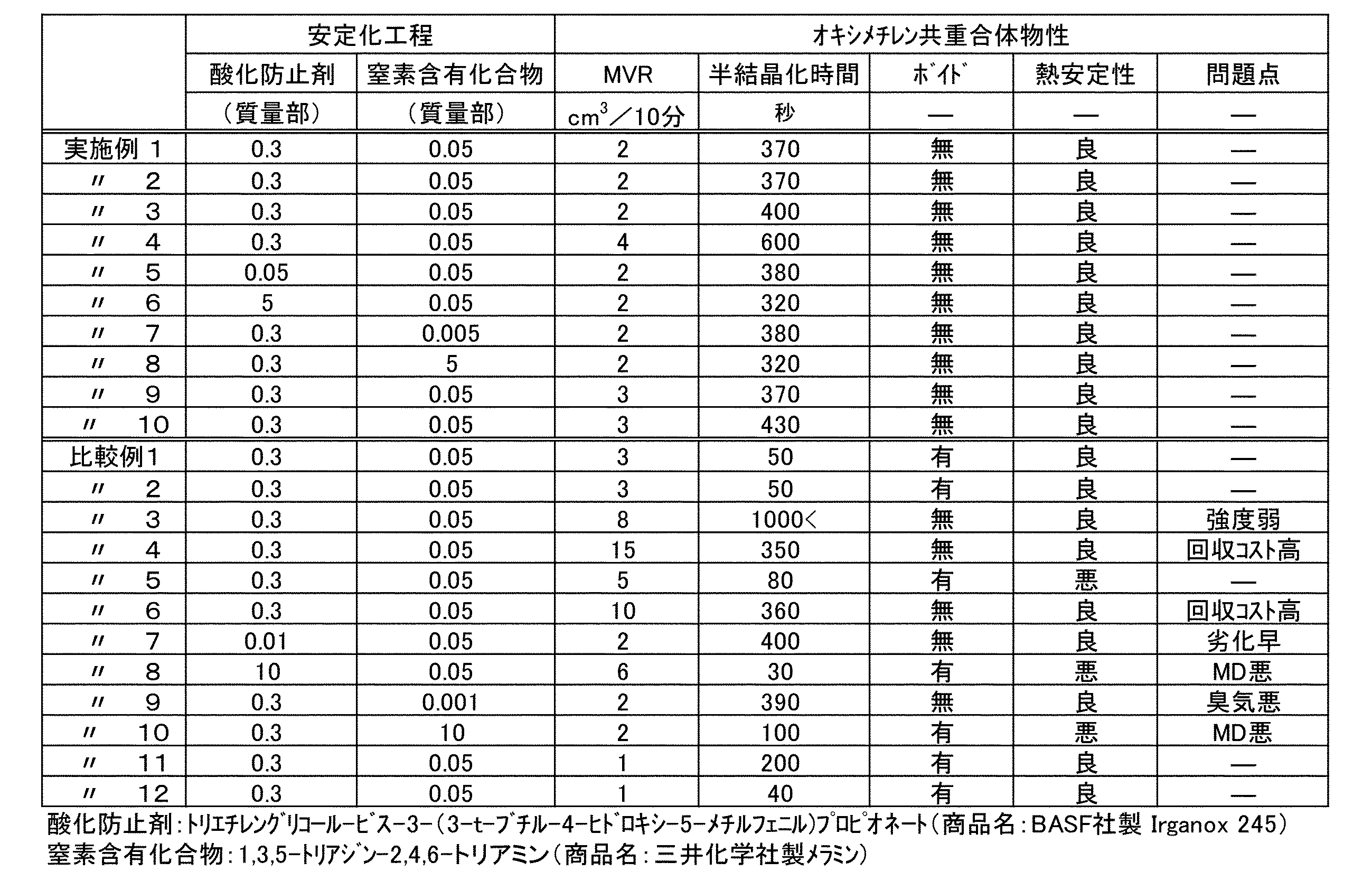
- Tables 1 and 2 show the types and predetermined amounts of the comonomer, catalyst, sterically hindered phenol, antioxidant and nitrogen-containing compound used, as well as the measurement results.
- Examples 9 to 10 and Comparative Examples 11 to 12 As a continuous polymerization apparatus, a pair of shafts are provided in a long case having an inner cross section in which two circles partially overlap each other, a long diameter of the inner cross section being 100 mm, and having a jacket around it. A large number of convex lens type paddle blades engaged with each other were fitted, and two continuous polymerization machines capable of cleaning the inner surface of the case and the surface of the opposite convex lens type paddle blade at the tip of the convex lens type paddle blade were connected in series.
- a continuous mixing system having a structure similar to that of the second-stage continuous polymerization machine as a polymerization terminator mixer, injecting a solution containing the polymerization terminator from the supply port and continuously mixing with the polymer.
- the machine was connected in series with the second-stage continuous polymerization machine to produce an oxymethylene copolymer.
- trioxane containing 0.00025 mmol of triethanolamine per mole of trioxane as a stabilizer
- Tables 1 and 2 the kind and amount of steric compounds shown in Tables 1 and 2 at the inlet of the first-stage continuous polymerization machine
- the sterically hindered phenol was fed as an 11% by weight 1,3-dioxolane solution so that the hindered phenol was fed. Further, 1,3-dioxolane was continuously supplied from another line, and the total supply amount of 1,3-dioxolane was adjusted to 8 to 12 kg / hr.
- the first stage shaft rotation speed of the continuous polymerization machine is about 35 rpm
- the second stage shaft rotation speed is about 60 rpm
- the first stage jacket temperature is 85 ° C.
- the second stage jacket temperature is 85 ° C.
- the polymerization operation was carried out with the jacket temperature of the polymerization terminator mixer set at 15 ° C.
- the polymerization time was about 10 minutes. Measure the polymerization yield, formate group content, and half-crystallization time of the resulting crude oxymethylene copolymer, and then melt knead the crude oxymethylene copolymer with the specified antioxidant and nitrogen-containing compound. The isothermal half crystallization time of the pellets thus obtained was measured.
- Tables 1 and 2 show the types and predetermined amounts of the comonomer, catalyst, sterically hindered phenol, antioxidant and nitrogen-containing compound used, as well as the respective measurement results.
- Examples 1 to 8 in which 5 to 7% by mass of 1,3-dioxolane was polymerized in the presence of a sterically hindered phenol with respect to trioxane, and the polymerization was terminated at a polymerization yield of 92% or more, a formate group was contained. It can be seen that the amount is small and there is no void or white core. In contrast to Comparative Examples 1 and 12, the formate group content is high when there is no sterically hindered phenol. In Comparative Examples 2 and 13 in which 1,3-dioxolane was 4 parts by mass, the crystallization time was short and white core was generated.
- Comparative Example 3 where 1,3-dioxolane was 13 parts by mass, the mechanical strength of the molded piece was deteriorated.
- Comparative Example 4 in which boron trifluoride diethyl etherate was 0.01 mmol per mole of trioxane and in Comparative Example 6 in which a polymerization terminator was added at a stage where the polymerization did not proceed sufficiently, the polymerization yield did not increase, and trioxane was recovered. The cost has increased.
- Comparative Example 5 in which boron trifluoride diethyl etherate was 0.07 mmol or more per mol of trioxane, the formate group content increased even when the polymerization yield was 92% or more.
- Comparative Example 7 with little antioxidant at the time of stabilization is significantly deteriorated by oxidation by air, and Comparative Example 9 with little nitrogen-containing compound at the time of stabilization has a worse formaldehyde odor.
- Comparative Examples 8 and 10 when there are many antioxidants or nitrogen-containing compounds at the time of stabilization (Comparative Examples 8 and 10), there are many mold stains (MD) at the time of molding.
- Examples 9 to 10 show, for comparison with Comparative Examples 11 to 12, the formate ester group reduction effect and the extrudability improvement effect due to the addition of sterically hindered phenol when carried out under conditions assuming actual production equipment. Yes.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
Abstract
Description
トリオキサンと、トリオキサンに対して5~7質量%の1,3-ジオキソランとを含むモノマー原料を、トリオキサン1モル当たり0.025~0.07ミリモルの三フッ化ホウ素と、トリオキサンに対して0.006~2.0質量%の立体障害性フェノールとの存在下に共重合を行う工程;該共重合の重合収率が92%以上となった時点で反応系中に重合停止剤を加えて重合を停止させ、粗オキシメチレン共重合体を得る工程;ならびに粗オキシメチレン共重合体に対して0.05~5質量%の酸化防止剤および0.005~5質量%の窒素含有化合物の存在下で、粗オキシメチレン共重合体を加熱溶融する熱安定化工程、
を含む、オキシメチレン共重合体の製造方法。
(A)上記で得られた粗オキシメチレン共重合体を加熱溶融して、不安定部分を除去する熱安定化方法。
(B)上記で得られた粗オキシメチレン共重合体を水性媒体中で加水分解して、不安定部分を除去する方法。
これらの方法により安定化した後、ペレット化し、安定化された成形可能なオキシメチレン共重合体を得ることができる。
重合収率=M1/M0×100
M0 ;アセトン洗浄前の質量
M1 ;アセトン洗浄、乾燥後の質量
(i)サンプルを入れたパンを試料台にのせ、温度を30℃に設定し、
(ii)30℃から210℃まで、320℃/分の一定速度にて昇温し、
(iii)210℃で5分間保持した後、
(iv)-80℃/分の一定速度にて150℃まで冷却し、
(v)サンプルの温度が150℃になった時点を0秒とし、半結晶化による発熱ピークのピークトップまでの時間を半結晶化時間とした。
重合装置としてジャケットと2枚のZ型翼を有する内容積1Lの卓上型二軸混練機を用い、バッチ式の重合によりオキシメチレン共重合体の製造を実施した。ジャケットに85℃温水を循環させ、さらに内部を高温空気で加熱乾燥した後、蓋を取り付け、系内を窒素置換した。原料投入口より1,3,5-トリオキサン320g(安定剤としてトリオキサン1モル当たり0.00025ミリモルのトリエタノールアミンを含有する)、所定量のコモノマー(1,3-ジオキソラン)および所定量の立体障害性フェノール(トリエチレングリコール-ビス-3-(3-t-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピオネート(商品名; BASF社製 Irganox 245))を仕込み、Z型翼によって撹拌しながら、触媒として所定量の三フッ化ホウ素ジエチルエーテラートをベンゼン溶液(溶液濃度:0.6ミリモル/g)として添加し重合を開始した。15分間重合させたのち、使用した触媒量の2倍モルに相当するトリフェニルホスフィンをベンゼン溶液(溶液濃度:5ミリモル/ml)としてシリンジを用いて重合装置内に添加し、さらに15分間混合して重合を停止し、粗オキシメチレン共重合体を収得した。この粗オキシメチレン共重合体について重合収率、ギ酸エステル基含有量を測定した。また、粗オキシメチレン共重合体に所定量の酸化防止剤および窒素含有化合物を入れて溶融混練して得たペレットの等温半結晶化時間を測定した。使用したコモノマー、触媒、立体障害性フェノール、酸化防止剤および窒素含有化合物の種類および所定量等、ならびに各測定結果を表1および2に示した。
連続重合装置として、二つの円が一部重なった内断面を有し、内断面の長径が100mmであり、周囲にジャケットを有する、長いケース内に1対のシャフトを備え、それぞれのシャフトには互いにかみ合う凸レンズ型パドル翼が多数はめ込まれ、凸レンズ型パドル翼の先端でケース内面および相手の凸レンズ型パドル翼の表面をクリーニングできる連続重合機を2台直列に連結した。それに続いて、重合停止剤混合機として前記2段目の連続重合機と類似の構造を有し、供給口部分から重合停止剤を含む溶液を注入し、連続的に重合体と混合せしめる連続混合機を前記2段目の連続重合機に直列に接続し、オキシメチレン共重合体の製造を実施した。1段目の連続重合機の入口に、200kg/hrのトリオキサン(安定剤としてトリオキサン1モル当たり0.00025ミリモルのトリエタノールアミンを含有する)および、表1および2に示した種類、量の立体障害性フェノールが供給されるように立体障害性フェノールを11質量%の1,3-ジオキソラン溶液として供給した。さらに、1,3-ジオキソランを別のラインから連続的に供給し、1,3-ジオキソランの供給量合計が8ないし12kg/hrとなるように調整した。同時に触媒としてトリオキサン1モル当たり0.04ミリモルの三フッ化ホウ素ジエチルエーテラートを連続的に供給した。三フッ化ホウ素ジエチルエーテラートおよびメチラールはそれぞれベンゼン溶液として添加した。ベンゼンの合計使用量はトリオキサンに対して1質量%以下であった。また、重合停止剤混合機の入口より、使用した触媒量の2倍モルの表2に示した重合停止剤をベンゼン溶液で連続的に供給して重合反応を停止し、出口より粗オキシメチレン共重合体を収得した。なお、連続重合装置は、連続重合機の1段目シャフト回転数を約35rpm、2段目シャフト回転数を約60rpmとし、また1段目ジャケット温度を85℃、2段目ジャケット温度を85℃、重合停止剤混合機のジャケット温度を15℃に設定して重合運転を行った。重合時間は約10分であった。得られた粗オキシメチレン共重合体の重合収率、ギ酸エステル基含有量、半結晶化時間を測定し、さらに粗オキシメチレン共重合体に所定の酸化防止剤並びに窒素含有化合物を入れて溶融混練して得たペレットの等温半結晶化時間を測定した。使用したコモノマー、触媒、立体障害性フェノール、酸化防止剤および窒素含有化合物の種類および所定量等、ならびに各測定結果を表1および2に示した。

Claims (11)
- トリオキサンと、トリオキサンに対して5~7質量%の1,3-ジオキソランとを含むモノマー原料を、トリオキサン1モル当たり0.025~0.07ミリモルの三フッ化ホウ素と、トリオキサンに対して0.006~2.0質量%の立体障害性フェノールとの存在下に共重合反応を行う工程;
該共重合反応の重合収率が92%以上となった時点で、反応系中に重合停止剤を加えて重合を停止させ、粗オキシメチレン共重合体を得る工程;ならびに
粗オキシメチレン共重合体に対して0.05~5質量%の酸化防止剤および0.005~5質量%の窒素含有化合物の存在下で、粗オキシメチレン共重合体を加熱溶融する熱安定化工程
を含む、オキシメチレン共重合体の製造方法。 - トリオキサンが、トリオキサン1モル当たり0.00001~0.003ミリモルのアミン類を含有する、請求項1に記載の製造方法。
- 酸化防止剤が、立体障害性フェノールである、請求項1または2に記載の製造方法。
- 窒素含有化合物が、メラミン、メチロールメラミン、ベンゾグアナミン、シアノグアニジン、アミン置換トリアジン類、ポリアミド類、尿素誘導体およびウレタン類からなる群から選ばれる一種以上である、請求項1~3のいずれか一項に記載の製造方法。
- 重合停止剤が、トリフェニルホスフィン、ヒンダードアミン化合物およびアルキル化メラミンからなる群から選択される1種または2種以上である、請求項1~4のいずれか一項に記載の製造方法。
- 共重合の重合収率が97%以上となった時点で、反応系中に重合停止剤を加えて重合を停止させる、請求項1~5のいずれか一項に記載の製造方法。
- 連続重合機と重合停止剤混合機を直列に接続した連続重合装置を用いて行う、請求項1~6のいずれか一項に記載の製造方法。
- 立体障害性フェノールの一部または全部を、連続重合機入口で添加する、請求項7に記載の製造方法。
- 熱安定化工程が、粗オキシメチレン共重合体を、その溶融温度から溶融温度より100℃高い温度までの範囲の温度で、760~0.1mmHgの圧力下で溶融混練することにより行われる、請求項1~8のいずれか一項に記載の製造方法。
- 熱安定化工程が、単軸または2軸以上のベント付押出機と、表面更新型の混合機を組み合わせた装置で行われる、請求項1~9のいずれか一項に記載の製造方法。
- 粗オキシメチレン共重合体が、そのまま熱安定化工程に供される、請求項1~10のいずれか一項に記載の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015518303A JPWO2014189139A1 (ja) | 2013-05-24 | 2014-05-23 | オキシメチレン共重合体の製造方法 |
| EP14800583.8A EP3006476A4 (en) | 2013-05-24 | 2014-05-23 | Method for producing oxymethylene copolymer |
| KR1020157031222A KR20160012123A (ko) | 2013-05-24 | 2014-05-23 | 옥시메틸렌 공중합체의 제조 방법 |
| US14/892,319 US20160102190A1 (en) | 2013-05-24 | 2014-05-23 | Method for producing an oxymethylene copolymer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-109937 | 2013-05-24 | ||
| JP2013109937 | 2013-05-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014189139A1 true WO2014189139A1 (ja) | 2014-11-27 |
Family
ID=51933689
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/063733 Ceased WO2014189139A1 (ja) | 2013-05-24 | 2014-05-23 | オキシメチレン共重合体の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20160102190A1 (ja) |
| EP (1) | EP3006476A4 (ja) |
| JP (1) | JPWO2014189139A1 (ja) |
| KR (1) | KR20160012123A (ja) |
| TW (1) | TW201446822A (ja) |
| WO (1) | WO2014189139A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020026781A1 (ja) * | 2018-08-01 | 2020-02-06 | 三菱瓦斯化学株式会社 | オキシメチレン共重合体の製造方法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101827789B1 (ko) * | 2016-10-17 | 2018-02-09 | 한국엔지니어링플라스틱 주식회사 | 폴리옥시메틸렌 수지 조성물 |
| CN108484850A (zh) * | 2018-04-08 | 2018-09-04 | 开封龙宇化工有限公司 | 一种聚甲醛树脂及其制备方法 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01269165A (ja) | 1988-04-20 | 1989-10-26 | Mitsubishi Electric Corp | 自動取引カード |
| JPH0363965B2 (ja) | 1983-06-08 | 1991-10-03 | Polyplastics Kk | |
| JPH06212054A (ja) | 1993-01-20 | 1994-08-02 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| JPH07207117A (ja) | 1994-01-25 | 1995-08-08 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| JPH07242652A (ja) | 1994-03-07 | 1995-09-19 | Polyplastics Co | 環状ホルマールの変質防止方法 |
| JPH07292216A (ja) | 1994-04-28 | 1995-11-07 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| JPH08325341A (ja) | 1995-05-31 | 1996-12-10 | Mitsubishi Gas Chem Co Inc | オキシメチレン共重合体の製造方法 |
| JPH0952926A (ja) | 1995-08-10 | 1997-02-25 | Toyo Plast Seiko Kk | 切削加工用ポリオキシメチレン樹脂素材およびその加工品 |
| JPH0967503A (ja) | 1995-06-20 | 1997-03-11 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| WO2002077049A1 (en) * | 2001-03-27 | 2002-10-03 | Mitsubishi Gas Chemical Company, Inc. | Polyoxymethylene copolymer and molded article thereof |
| JP2003064141A (ja) | 2001-08-29 | 2003-03-05 | Polyplastics Co | 切削加工用ポリオキシメチレン樹脂素材 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3137896B2 (ja) * | 1996-03-12 | 2001-02-26 | 旭化成株式会社 | ポリオキシメチレンコポリマーの製造方法 |
| US20030105199A1 (en) * | 2001-11-16 | 2003-06-05 | Mitsubishi Gas Chemical Company, Inc. | Polyoxymethylene resin composition and molded article thereof |
-
2014
- 2014-05-23 JP JP2015518303A patent/JPWO2014189139A1/ja active Pending
- 2014-05-23 EP EP14800583.8A patent/EP3006476A4/en not_active Withdrawn
- 2014-05-23 US US14/892,319 patent/US20160102190A1/en not_active Abandoned
- 2014-05-23 WO PCT/JP2014/063733 patent/WO2014189139A1/ja not_active Ceased
- 2014-05-23 KR KR1020157031222A patent/KR20160012123A/ko not_active Withdrawn
- 2014-05-23 TW TW103118089A patent/TW201446822A/zh unknown
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0363965B2 (ja) | 1983-06-08 | 1991-10-03 | Polyplastics Kk | |
| JPH01269165A (ja) | 1988-04-20 | 1989-10-26 | Mitsubishi Electric Corp | 自動取引カード |
| JPH06212054A (ja) | 1993-01-20 | 1994-08-02 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| JPH07207117A (ja) | 1994-01-25 | 1995-08-08 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| JPH07242652A (ja) | 1994-03-07 | 1995-09-19 | Polyplastics Co | 環状ホルマールの変質防止方法 |
| JPH07292216A (ja) | 1994-04-28 | 1995-11-07 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| JPH08325341A (ja) | 1995-05-31 | 1996-12-10 | Mitsubishi Gas Chem Co Inc | オキシメチレン共重合体の製造方法 |
| JPH0967503A (ja) | 1995-06-20 | 1997-03-11 | Asahi Chem Ind Co Ltd | ポリオキシメチレン樹脂組成物 |
| JPH0952926A (ja) | 1995-08-10 | 1997-02-25 | Toyo Plast Seiko Kk | 切削加工用ポリオキシメチレン樹脂素材およびその加工品 |
| WO2002077049A1 (en) * | 2001-03-27 | 2002-10-03 | Mitsubishi Gas Chemical Company, Inc. | Polyoxymethylene copolymer and molded article thereof |
| JP2003064141A (ja) | 2001-08-29 | 2003-03-05 | Polyplastics Co | 切削加工用ポリオキシメチレン樹脂素材 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3006476A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020026781A1 (ja) * | 2018-08-01 | 2020-02-06 | 三菱瓦斯化学株式会社 | オキシメチレン共重合体の製造方法 |
| JP6673539B1 (ja) * | 2018-08-01 | 2020-03-25 | 三菱瓦斯化学株式会社 | オキシメチレン共重合体の製造方法 |
| US10954332B2 (en) | 2018-08-01 | 2021-03-23 | Mitsubishi Gas Chemical Company, Inc. | Method for producing oxymethylene copolymer |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014189139A1 (ja) | 2017-02-23 |
| US20160102190A1 (en) | 2016-04-14 |
| EP3006476A1 (en) | 2016-04-13 |
| KR20160012123A (ko) | 2016-02-02 |
| EP3006476A4 (en) | 2017-01-18 |
| TW201446822A (zh) | 2014-12-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6024749B2 (ja) | オキシメチレン共重合体の製造方法 | |
| WO2009081517A1 (ja) | ポリアセタール樹脂組成物 | |
| JP2009286874A (ja) | ポリアセタール樹脂組成物及びその成形品 | |
| WO2014097808A1 (ja) | ポリアセタール樹脂組成物 | |
| WO2014189139A1 (ja) | オキシメチレン共重合体の製造方法 | |
| JP3309641B2 (ja) | オキシメチレン共重合体の製造方法 | |
| JP5389468B2 (ja) | ポリアセタール樹脂組成物の製造方法 | |
| WO2015008537A1 (ja) | オキシメチレン共重合体の製造方法 | |
| WO2014196385A1 (ja) | オキシメチレン共重合体の製造方法 | |
| EP3460002B1 (en) | Method for producing polyacetal resin composition | |
| WO2020261693A1 (ja) | ポリアセタ-ル樹脂組成物及びその製造方法 | |
| JP5445019B2 (ja) | ポリアセタール共重合体 | |
| EP4032944B1 (en) | Method for producing oxymethylene copolymer resin composition, and oxymethylene copolymer resin composition | |
| JP3745267B2 (ja) | 押出し成形用ポリアセタール樹脂組成物およびそれを用いた成形品 | |
| JP2005225973A (ja) | ポリオキシメチレン共重合体の製造方法 | |
| JP7687818B2 (ja) | 樹脂組成物、および、押出成形体 | |
| JP5066832B2 (ja) | 高靭性ポリアセタール樹脂組成物の製造方法。 | |
| JP2006291001A (ja) | 低ホルムアルデヒド化ポリアセタール樹脂組成物の製造方法。 | |
| JP2011116906A (ja) | ポリアセタール共重合体 | |
| JP5403234B2 (ja) | ポリアセタール樹脂組成物 | |
| JP2013129749A (ja) | 樹脂組成物および成形体 | |
| JP2010180312A (ja) | ポリアセタール樹脂組成物 | |
| JP2011116905A (ja) | ポリアセタール共重合体 | |
| JP2011116907A (ja) | ポリアセタール樹脂組成物 | |
| JP2004352913A (ja) | ポリオキシメチレン樹脂組成物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14800583 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015518303 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20157031222 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14892319 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014800583 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014800583 Country of ref document: EP |