WO2014203855A2 - 2-アミノ-2-ヒドロキシイミノ-n-アルコキシアセトイミドイルシアニドの製造方法およびその製造中間体 - Google Patents
2-アミノ-2-ヒドロキシイミノ-n-アルコキシアセトイミドイルシアニドの製造方法およびその製造中間体 Download PDFInfo
- Publication number
- WO2014203855A2 WO2014203855A2 PCT/JP2014/065891 JP2014065891W WO2014203855A2 WO 2014203855 A2 WO2014203855 A2 WO 2014203855A2 JP 2014065891 W JP2014065891 W JP 2014065891W WO 2014203855 A2 WO2014203855 A2 WO 2014203855A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- reaction
- general formula
- amino
- represented
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *ON=C(C(N)=NO)C#N Chemical compound *ON=C(C(N)=NO)C#N 0.000 description 2
- USSJTOBUSKIOLC-QHHAFSJGSA-N N/C(/C(/C#N)=N/O)=N\O Chemical compound N/C(/C(/C#N)=N/O)=N\O USSJTOBUSKIOLC-QHHAFSJGSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/63—Carboxylic acid nitriles containing cyano groups and nitrogen atoms further bound to other hetero atoms, other than oxygen atoms of nitro or nitroso groups, bound to the same carbon skeleton
- C07C255/64—Carboxylic acid nitriles containing cyano groups and nitrogen atoms further bound to other hetero atoms, other than oxygen atoms of nitro or nitroso groups, bound to the same carbon skeleton with the nitrogen atoms further bound to oxygen atoms
Definitions
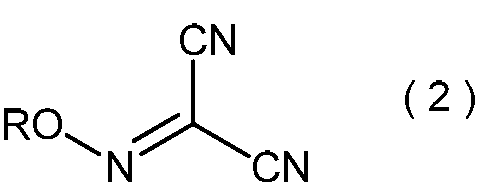
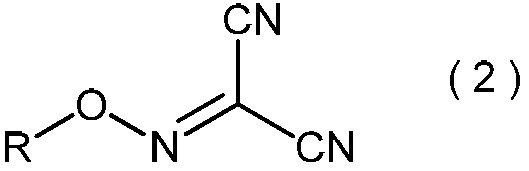
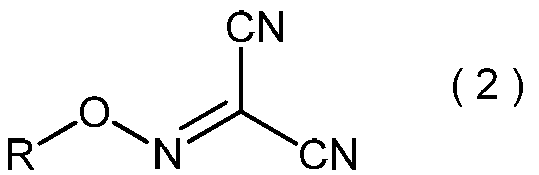
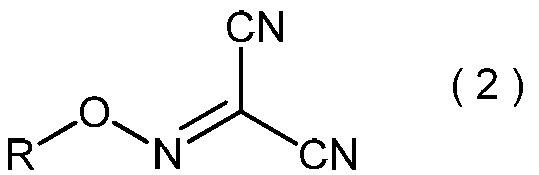
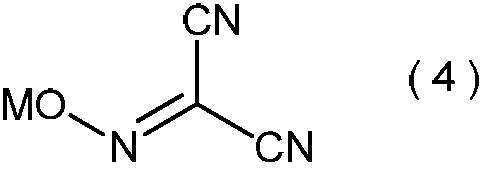
- the present invention is directed to general formula (2): (Wherein R represents an alkyl group having 3 to 6 carbon atoms), and a process for producing the 2-alkoxyiminopropanedinitrile.
- the 2-alkoxyiminopropanedinitrile represented by the general formula (2) produces the 2-amino-2-hydroxyimino-N-alkoxyacetimidoylcyanide represented by the general formula (3) in the method of the present invention. Is a useful intermediate for.
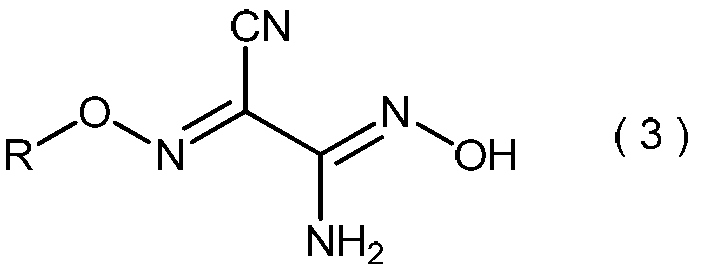
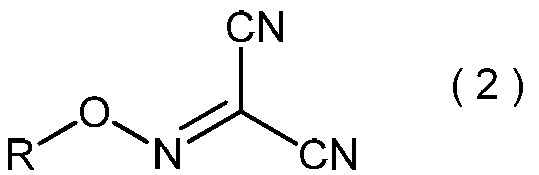
- the invention also relates to general formula (3): (Wherein R is as defined above), and relates to a process for producing 2-amino-2-hydroxyimino-N-alkoxyacetimidoylcyanide.
- the 2-amino-2-hydroxyimino-N-alkoxyacetimidoylcyanide represented by the general formula (3) is useful as a pesticide by itself and as a production intermediate for other pesticides.
- 2-Amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3) is a useful compound having excellent pest control activity (see Patent Document 1).
- 2-amino-2-hydroxyimino-N-isopropoxyacetimidoylcyanide exhibits excellent pest control activity.
- 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by general formula (3) is converted to 2-amino-2-alkoxyimino represented by general formula (5).
- —N-alkoxyacetimidoyl cyanide can be prepared.
- 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3) can be produced.
- Patent Document 1 discloses that 2-amino-2-alkoxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (5) also has excellent pest control activity.
- 2-amino-2-isopropoxyimino-N-isopropoxyacetimidoylcyanide, 2-amino-2-isopropoxyiminoimido-N-propoxyacetimidoylcyanide and 2-amino-2-propoxy Imino-N-propoxyacetimidoylcyanide exhibits excellent pest control activity.
- 2-amino-2-alkoxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (5) can be used as an intermediate for producing the alkoxyimino derivative disclosed in Patent Document 2.
- 2-amino-2-alkoxyimino-N-alkoxyacetimidoyl cyanide represented by the formula (5) is disclosed in Patent Document 2 by a method known to those skilled in the art. Some of the alkoxyimino derivatives can be produced.
- the target compound in Example 1 (2) of Patent Document 2 is the above 2-amino-2-isopropoxy compound in the same manner as in Example 1 of Patent Document 3, comprising diazotization followed by chlorination. It can be prepared from imino-N-isopropoxyacetimidoyl cyanide.
- the target compound in Example 23 (4) of Patent Document 2 can be produced from 2-amino-2-propoxyimino-N-propoxyacetimidoyl cyanide described above in the same manner.
- the target compound in Example 1 (2) of Patent Document 2 is an intermediate for producing various alkoxyimino derivatives, as specifically described in other Examples of Patent Document 2. Further, the target compound in Example 23 (4) of Patent Document 2 is an intermediate for producing some alkoxyimino derivatives, as specifically described in other Examples of Patent Document 2.
- the alkoxyimino derivative disclosed in Patent Document 2 is also known to be a useful compound having excellent pest control activity.
- a pest control agent containing an alkoxyimino derivative as an active ingredient exhibits an excellent control effect against a wide range of pests in the fields of agriculture and horticulture, and particularly, brown planthopper, cotton aphid, etc. Highly effective against stink bugs.
- a pest control agent containing an alkoxyimino derivative as an active ingredient can also control resistant pests.
- alkoxyimino derivative produced from the target compound of Example 1 (2) or Example 23 (4) of Patent Document 2 described above and exhibiting excellent pest control activity include: 1- (2-cyano-1,2-diisopropoxyiminoethyl) -1H-1,2,4-triazole, 1- (2-carbamoyl-1,2-diisopropoxyiminoethyl) -1H-1,2,4-triazole, 1- (2-cyano-1,2-dipropoxyiminoethyl-1H-1,2,4-triazole, 1- (2-carbamoyl-1,2-dipropoxyiminoethyl] -1H-1,2,4-triazole, etc.
- 1- (2-carbamoyl-1,2-diisopropoxyiminoethyl) -1H-1,2,4-triazole 1- (2-carbamoyl-1,2-dipropoxyiminoethyl] -1H-1,2,4-triazole, Particularly preferred is 1- (2-carbamoyl-1,2-diisopropoxyiminoethyl) -1H-1,2,4-triazole.
- 2-amino-2-hydroxyimino-N-alkoxyacetimidoylcyanide represented by the general formula (3) is not only useful as an active ingredient of a pest control agent per se, but also Patent Documents 1 and 2 It is also useful as an intermediate for the production of a series of active ingredients of the pest control agent disclosed in.
- Non-Patent Document 1 shows thermal behavior in which differential scanning calorimetry of 2-amino-2-hydroxyimino-N-hydroxyacetimidoyl cyanide represented by formula (6) is extremely dangerous, that is, exothermic decomposition reaction. I suggest that.
- Non-Patent Document 1 discloses that a monohydrate of 2-amino-2-hydroxyimino-N-hydroxyacetimidoyl cyanide represented by the formula (6) has an exothermic onset temperature of 124 ° C. and 10.3 kJ / It is specifically disclosed to show the calorific value of g. These values are extremely risky and 2-amino-2-hydroxyimino-N-hydroxyacetimidoyl cyanide represented by formula (6) is a dangerous compound. A large exotherm of 10.3 kJ / g suggests that the decomposition reaction is extremely exothermic, resulting in an uncontrollable state in the reactor. In addition, a low exothermic starting temperature of 124 ° C.
- the present inventors unexpectedly made the 2-amino-2-hydroxyimino represented by the general formula (3) from the 2-alkoxyiminopropanedinitrile represented by the general formula (2). It has been found that —N-alkoxyacetimidoyl cyanide can be produced safely. (In the formula, any R is as defined above.)
- the alkylating agent capable of achieving a high yield is limited to alkylating agents having high reactivity such as dimethyl sulfate, ⁇ -halocarboxylic acid ester and benzyl bromide ( (See Patent Documents 4, 5, 6 and 7, and Non-Patent Document 2).
- alkylating agents having high reactivity such as dimethyl sulfate, ⁇ -halocarboxylic acid ester and benzyl bromide
- a less reactive alkylating agent such as 2-bromofluoro-1,1-ethane
- Patent Document 9 discloses that the reaction of a silver salt of 2-hydroxyiminopropanedinitrile with cyclopropylmethyl bromide has achieved a yield of 64%. However, since silver salts are expensive, this method is not industrially preferable.
- 2-alkoxyiminopropanedinitrile represented by the general formula (2) may be useful as an intermediate in the method of the present invention.
- an industrially satisfactory production method of 2-alkoxyiminopropanedinitrile represented by the general formula (2) has not been known.
- An object of the present invention is to provide an economically preferable production method suitable for industrialization of 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3).
- the object of the present invention is to use a thermally dangerous compound such as 2-amino-2-hydroxyimino-N-hydroxyacetimidoyl cyanide represented by the formula (6) as a production intermediate.
- An object of the present invention is to provide a safer method for producing 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3), which is not used.
- the present invention provides 2-alkoxyiminopropanedinitrile represented by the general formula (2) as such a safer production intermediate, but its industrial It is necessary to provide a production method that satisfies the above requirements. That is, another object of the present invention is to provide an industrially preferable method for producing 2-alkoxyiminopropanedinitrile represented by the general formula (2). For example, it is necessary to provide a method capable of producing 2-alkoxyiminopropanedinitriles in a high yield even when the above-described highly reactive alkylating agent is not used. Furthermore, there has been a demand for a production method thereof that does not use expensive raw materials such as silver salts.
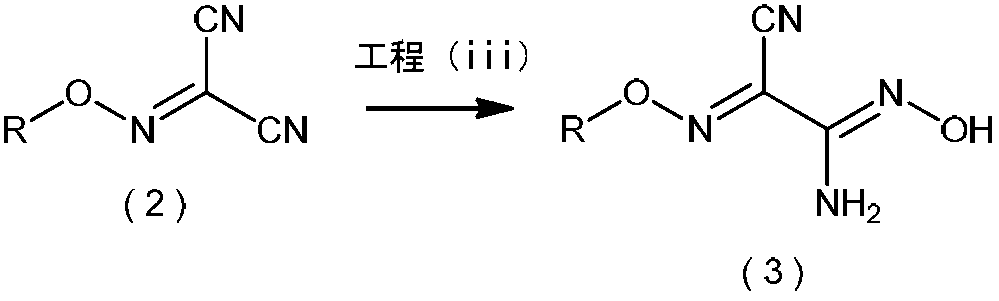
- the inventors surprisingly have the formula (1): Is reacted with an alkali metal salt of nitrous acid in the presence of a protonic acid, Subsequently, after reacting the resulting product with a C3-C6 alkyl halide in the presence of a phase transfer catalyst, General formula (2) obtained: (Wherein R is as defined above.) By reacting a compound represented by the formula: 2-alkoxyiminopropanedinitrile with a hydroxylamine compound, General formula (3): (Wherein R represents a C3-C6 alkyl group) It was found that the compound represented by the above formula, that is, 2-amino-2-hydroxyimino-N-alkoxyacetimidoylcyanide, can be produced safely and in a high yield. Based on this finding, the present inventors have completed the present invention.
- the present invention provides a novel industrially applicable production method for 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3).
- the present invention provides a novel 2-alkoxyiminopropanedinitrile represented by the general formula (2) and an industrially satisfactory production method thereof.
- the use of a thermally dangerous compound as a production intermediate is avoided, and the desired 2-amino-2-hydroxyimino-N-alkoxyacetimidoylsia represented by the general formula (3) is used. Nido can be produced.
- the dangerous production intermediate of the prior art is 2-amino-2-hydroxyimino-N-hydroxyacetimidoyl cyanide represented by the formula (6). That is, according to the present invention, in the differential scanning calorimetry, even the monohydrate is expressed by the equation (6) indicating an exothermic decomposition of an exothermic onset temperature of 124 ° C. and an exothermic amount of 10.3 kJ / g. Production of the desired 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3) without using amino-2-hydroxyimino-N-hydroxyacetimidoylcyanide Can do.
- the production intermediate used in the method of the present invention is 2-alkoxyiminopropanedinitrile represented by the general formula (2).
- Specific examples of the 2-alkoxyiminopropanedinitrile represented by the general formula (2) include 2-isopropoxyiminopropanedinitrile. Differential scanning calorimetry of 2-isopropoxyiminopropanedinitrile gave an exotherm starting temperature of 225 ° C. and an exotherm of 0.6 kJ / g.
- the exothermic onset temperature of 2-isopropoxyiminopropanedinitrile is significantly higher than that of 2-amino-2-hydroxyimino-N-hydroxyacetimidoylcyanide represented by formula (6), which is a production intermediate of the prior art. high.
- This exothermic onset temperature implies a much better thermal stability than that of the prior art described above.
- the value of 2-isopropoxyiminopropanedinitrile is 2-amino-2-hydroxyimino-N-hydroxyacetimidoylsia represented by the formula (6), which is a production intermediate in the prior art. Much smaller than that of Nido. This value means a greatly increased safety of the method according to the invention.
- the 2-alkoxyiminopropanedinitrile represented by the general formula (2) which is a novel production intermediate of the present invention is converted into a 2-amino-2- represented by the formula (6) which is a production intermediate of the prior art. It was found to be dramatically safer than hydroxyimino-N-hydroxyacetimidoylcyanide.
- malononitrile represented by the formula (1) is reacted with an alkali metal salt of nitrous acid in the presence of a protonic acid, and then 2-alkoxyimino represented by the general formula (2).
- 2-hydroxyiminopropanedinitrile represented by the following formula (8) and / or a salt thereof is produced as a precursor of propanedinitrile.
- Specific examples of the salt of 2-hydroxyiminopropanedinitrile represented by the formula (8) include sodium salt of 2-hydroxyiminopropanedinitrile.
- 2-hydroxyiminopropanedinitrile represented by the formula (8) an exothermic starting temperature of 147 ° C. and a calorific value of 1.6 kJ / g were found.
- the exothermic onset temperature of this 2-hydroxyiminopropanedinitrile is higher than that of 2-amino-2-hydroxyimino-N-hydroxyacetimidoylcyanide represented by formula (6), which is a production intermediate of the prior art. High, ie safer.
- the value of 2-hydroxyiminopropanedinitrile is much smaller than that in the above prior art and is sufficient to maintain safety.
- the sodium salt of 2-hydroxyiminopropanedinitrile shows an endothermic onset temperature of 222 ° C. and an endothermic amount of ⁇ 0.9 kJ / g by differential scanning calorimetry.
- This starting temperature also implies better thermal stability than that of the prior art described above.
- the thermal event is endothermic rather than exothermic, ie much safer than that of the prior art described above.
- the physical properties of the production intermediate in the present invention show the significantly improved safety of the method of the present invention, that is, the superiority of the method of the present invention, as compared with the prior art. In other words, they inherently reduce risks such as dangerous decomposition during industrial production. Thus, the method of the present invention can be safely applied to large scale manufacturing such as pilot plants or industrial production.
- the alkylating agent when a compound obtained after reacting malononitrile and an alkali metal salt of nitrous acid is reacted with an alkylating agent, the alkylating agent is not limited to a highly reactive (ie, Regardless of the type, 2-alkoxyiminopropanedinitrile represented by the general formula (2) can be produced in high yield without using an expensive silver salt. Furthermore, according to the present invention, a 2-alkoxy represented by the general formula (2) using a recyclable and inexpensive solvent (for example, an organic solvent such as an ether solvent or an aromatic hydrocarbon derivative solvent). Iminopropanedinitrile can be produced. Therefore, the present invention also provides a novel industrially satisfactory production method for 2-alkoxyiminopropanedinitrile represented by the general formula (2).
- the desired 2-amino-2-hydroxyimino-N-alkoxyacetimidoylcyanide represented by the general formula (3) and its production intermediate having preferable thermal properties are obtained.
- a 2-alkoxyiminopropanedinitrile represented by a general formula (2) can be produced by using a reagent and a solvent which can be obtained easily and dramatically at a low cost.
- the method of the present invention has a high yield and can be carried out efficiently on an industrial scale by simple operation under mild conditions without using a special reaction apparatus.
- the amount of waste is small and the nature of the waste is considered favorable.
- the present invention is economical, friendly to the environment, and has a high industrial utility value.
- step (ii) is carried out in a solvent system comprising an aromatic hydrocarbon derivative and water or a solvent system comprising an ether and water.
- step (ii) The method according to [6], wherein the reaction in step (ii) is carried out in a solvent system comprising one or more aromatic hydrocarbon derivatives selected from the group consisting of toluene, xylene and chlorobenzene and water. .
- step (ii) The method according to [6], wherein the reaction in step (ii) is carried out in a solvent system comprising toluene and water.
- step (ii) The method according to [6], wherein the reaction in step (ii) is carried out in a solvent system comprising tetrahydrofuran and water.
- step (ii) is carried out in a solvent system comprising an ether and water.
- step (ii) is carried out in a solvent system comprising one or more aromatic hydrocarbon derivatives selected from the group consisting of toluene, xylene and chlorobenzene and water.
- step (ii) The method according to [26], wherein the reaction in step (ii) is carried out in a solvent system comprising an aromatic hydrocarbon derivative and water or a solvent system comprising an ether and water.
- step (ii) is carried out in a solvent system comprising one or more aromatic hydrocarbon derivatives selected from the group consisting of toluene, xylene and chlorobenzene and water.
- step (ii) is carried out in a solvent system comprising toluene and water.
- step (ii) is carried out in a solvent system comprising tetrahydrofuran and water.
- step (ii) is carried out in a solvent system comprising an aromatic hydrocarbon derivative and water or a solvent system comprising an ether and water.
- step (ii) is carried out in a solvent system comprising one or more aromatic hydrocarbon derivatives selected from the group consisting of toluene, xylene and chlorobenzene and water.
- Ca to Cb means that the number of carbon atoms is a to b.
- C3 to C6 in “C3 to C6 alkyl” means that the alkyl has 3 to 6 carbon atoms.
- alkyl examples include C3-C6 alkyl, preferably C3 alkyl.
- C3-C6 alkyl means a straight-chain or branched alkyl having 3-6 carbon atoms.
- Specific examples of the C3-C6 alkyl include propyl, isopropyl, butyl, sec-butyl, isobutyl, tert-butyl, pentyl, hexyl and the like, preferably propyl, isopropyl, butyl, sec-butyl, isobutyl, tert- Examples include but are not limited to butyl, more preferably propyl and isopropyl.
- C3 alkyl means a straight or branched alkyl having 3 carbon atoms. Specific examples of C3 alkyl include propyl and isopropyl.
- alkoxy examples include C3 to C6 alkoxy, preferably C3 alkoxy.
- C3-C6 alkoxy means (C3-C6 alkyl) -O- (wherein C3-C6 alkyl has the same meaning as described above).
- Specific examples of the C3-C6 alkoxy include propoxy, isopropoxy, butoxy, sec-butoxy, isobutoxy, tert-butoxy, pentyloxy, hexyloxy and the like, preferably propoxy, isopropoxy, butoxy, sec-butoxy, Examples include, but are not limited to, isobutoxy, tert-butoxy, more preferably propoxy, isopropoxy.
- C3 alkoxy means (C3 alkyl) -O- (wherein C3 alkyl has the same meaning as described above).
- Specific examples of C3 alkoxy include propoxy and isopropoxy.
- aromatic hydrocarbon derivatives for example, benzene, toluene, xylene, ethylbenzene, cumene, trimethylbenzene, methylisopropylbenzene, methylnaphthalene, dimethylnaphthalene, ethylnaphthalenechlorobenzene, dichlorobenzene, trichlorobenzene, tetrachlorobenzenenitrobenzene, etc. are preferable. Is toluene, xylene, chlorobenzene, dichlorobenzene, nitrobenzene, more preferably toluene, xylene, chlorobenzene, and still more preferably toluene.
- Examples of the aliphatic hydrocarbons include hexane, heptane, octane, isooctane, nonane, decane, undecane, dodecane, isododecane, tridecane, tetradecane, pentadecane, hexadecane, isohexadecane, heptadecane, octadecane, nonadecane, eicosane, isoeicosane, tria.
- halogenated aliphatic hydrocarbons include dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, 1,3-dichloropropane, and the like.
- ethers examples include tetrahydrofuran (THF), 2-methyltetrahydrofuran, diethyl ether, di-n-propyl ether, diisopropyl ether, dibutyl ether, di-tert-butyl ether, diphenyl ether, cyclopentyl methyl ether (CPME), methyl- tert-butyl ether, 1,2-dimethoxyethane (DME), diglyme, triglyme, 1,4-dioxane, etc., preferably tetrahydrofuran (THF), diisopropyl ether, dibutyl ether, cyclopentyl methyl ether (CPME) , Methyl-tert-butyl ether, more preferably tetrahydrofuran.
- THF tetrahydrofuran
- 2-methyltetrahydrofuran diethyl ether
- di-n-propyl ether di
- alcohols examples include methanol, ethanol, propanol, 2-propanol, butanol, tert-butyl alcohol, pentanol, hexanol, heptanol, octanol, cyclohexanol, ethylene glycol, propylene glycol and the like.
- nitriles examples include acetonitrile, propionitrile, benzonitrile and the like.
- amides include N, N-dimethylformamide (DMF), N, N-diethylformamide, N, N-dimethylacetamide (DMAC), N, N-diethylacetamide, N-methylpyrrolidone (NMP) and the like. Can be mentioned.
- alkylureas examples include tetramethylurea, N, N′-dimethylimidazolidinone (DMI), and the like.
- sulfoxides examples include dimethyl sulfoxide (DMSO).
- sulfones examples include sulfolane and dimethyl sulfone.
- carbonate esters examples include ethylene carbonate and propylene carbonate.
- ketones include acetone, ethyl methyl ketone, isopropyl methyl ketone, isobutyl methyl ketone (MIBK), and cyclohexanone.
- carboxylic acid esters examples include methyl acetate, ethyl acetate, propyl acetate, butyl acetate and the like.
- carboxylic acids examples include formic acid, acetic acid, propionic acid, and the like.
- nitrated aromatic hydrocarbons examples include nitrobenzene.
- aromatic heterocycles examples include pyridine.
- alkali metal hydroxide examples include lithium hydroxide, sodium hydroxide, potassium hydroxide and the like.
- alkaline earth metal hydroxide examples include magnesium hydroxide, calcium hydroxide, barium hydroxide and the like.
- alkali metal carbonate examples include lithium carbonate, sodium carbonate, and potassium carbonate.
- alkaline earth metal carbonates examples include magnesium carbonate, calcium carbonate, and barium carbonate.
- alkali metal hydrogen carbonate examples include lithium hydrogen carbonate, sodium hydrogen carbonate, and potassium hydrogen carbonate.
- alkaline earth metal hydrogen carbonate examples include magnesium hydrogen carbonate, calcium hydrogen carbonate, barium hydrogen carbonate and the like.
- Examples of the phosphate include sodium phosphate, potassium phosphate, and calcium phosphate.
- hydrogen phosphate examples include sodium hydrogen phosphate, potassium hydrogen phosphate, calcium hydrogen phosphate, and the like.
- pyridines examples include pyridine, 4-dimethylaminopyridine, 4-pyrrolidinopyridine, 2,6-lutidine and the like.
- quinolines examples include quinoline and the like.
- isoquinolines examples include isoquinoline and the like.
- tertiary amines examples include trimethylamine, triethylamine, tributylamine, diisopropylethylamine, triisopropylamine and the like.
- Secondary amines include, for example, diethylamine, dipropylamine, diisopropylamine and the like.
- Examples of primary amines include propylamine and butylamine.
- aromatic amines examples include aniline, N, N-dimethylaniline, N, N-diethylaniline and the like.
- cyclic amines examples include pyrrolidine, piperidine, morpholine, N-methylmorpholine, piperazine, 1,8-diazabicyclo [5.4.0] -7-undec-7-ene (DBU), 1,5-diazabicyclo [4.3.0] non-5-ene (DBN), 1,4-diazabicyclo [2.2.2] Octane (DABCO) and the like.
- alkali metal carboxylate examples include sodium formate, potassium formate, lithium acetate, sodium acetate, potassium acetate, sodium propionate, and potassium propionate.
- alkaline earth metal carboxylate examples include magnesium acetate, magnesium propionate, calcium acetate, calcium propionate and the like.
- step (i) will be described.
- Step (i) comprises formula (1): Is a step of reacting an alkali metal salt of nitrous acid in the presence of a protonic acid.
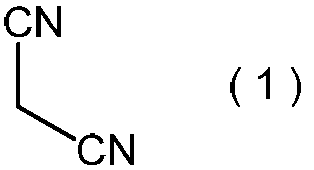
- the malononitrile represented by the above formula (1) is used as a raw material for the method of the present invention.
- Malononitrile is a known compound, is industrially available at a relatively low cost, and is also preferable as an industrial raw material from the viewpoint of handling.
- alkali metal salt of nitrous acid examples include, but are not limited to, sodium nitrite, potassium nitrite, lithium nitrite and the like. From the viewpoint of price and availability, sodium nitrite or potassium nitrite is preferable, and sodium nitrite is more preferable.
- the form of the alkali metal salt of nitrous acid may be any form as long as the reaction proceeds.
- Examples of the form of the alkali metal salt of nitrous acid include a solid containing only the alkali metal salt of nitrous acid, an aqueous solution having an arbitrary concentration, or a solution of a solvent other than water.
- the alkali metal salt of nitrous acid may be used alone or in combination of two or more in any ratio.
- the amount of alkali metal nitrite used may be any amount as long as the reaction proceeds. From the viewpoints of yield, by-product suppression, economic efficiency, etc., usually 0.8 to 2.0 mol, preferably 0.9 to 1.5 mol, per 1 mol of malononitrile represented by the formula (1) More preferably, the range is 0.9 to 1.2 mol.
- Examples of the protonic acid include, but are not limited to, hydrohalic acid, sulfuric acid, nitric acid, phosphoric acid, polyphosphoric acid, carboxylic acid, sulfonic acid, and the like.
- hydrohalic acid examples include hydrochloric acid, hydrobromic acid, hydrofluoric acid, and the like.
- carboxylic acid examples include formic acid, acetic acid, propionic acid, trifluoroacetic acid, benzoic acid and the like.
- sulfonic acid examples include methanesulfonic acid, ethanesulfonic acid, trifluoromethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, and the like.
- hydrohalic acid or carboxylic acid is preferable, and hydrochloric acid or acetic acid is more preferable.
- the form of the protonic acid may be any form as long as the reaction proceeds.
- Examples of the form of the protonic acid include a solid or liquid containing only a protonic acid, an aqueous solution having an arbitrary concentration, or a solution of a solvent other than water.
- Protonic acids may be used alone or in combination of two or more at any ratio.
- Protonic acid may be used in any amount as long as the reaction proceeds. From the viewpoints of yield, by-product suppression, economic efficiency, etc., it is usually 0.005 to 3.0 mol, preferably 0.01 to 2.0 mol, per 1 mol of malononitrile represented by the formula (1). More preferred is a range of 0.05 to 1.5 mol, still more preferably 0.05 to 1.3 mol.
- the particularly preferable range of the amount of proton acid used varies depending on the type of acid.
- the protic acid is hydrochloric acid
- a range of 0.9 to 1.2 mol can be particularly preferably exemplified.
- the protic acid is acetic acid
- a range of 0.05 to 1.2 mol can be particularly preferably exemplified.
- the reaction in step (i) may be performed without a solvent, but a solvent may be used from the viewpoint of smooth progress of the reaction.
- the solvent in step (i) may be any solvent as long as the reaction in step (i) proceeds and does not adversely affect the reactions in steps (ii) and (iii).
- Examples of the solvent that can be used in the step (i) include water, ethers, alcohols, nitriles, amides, alkylureas, sulfoxides, sulfones, carbonate esters, ketones, and carboxylic acid esters. , Carboxylic acids, nitrated aliphatic hydrocarbons, aromatic heterocycles, and the like, and mixed solvents of any mixing ratio thereof, but are not limited thereto.
- the solvent used in the step (i) include ethers, alcohols, nitriles, amides, alkylureas, sulfoxides, sulfones, water, and the like. More preferably, ethers, alcohols, nitriles, amides, water, and mixed solvents thereof can be mentioned.
- the solvent used in the step (i) include tetrahydrofuran (THF), dipropyl ether, diisopropyl ether, dibutyl ether, cyclopentyl methyl ether (CPME), methyl-tert-butyl ether, methanol, ethanol, propanol, 2-propanol, butanol, acetonitrile, N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAC), N-methylpyrrolidone (NMP), N, N′-dimethylimidazolidinone (DMI) dimethyl sulfoxide (DMSO), sulfolane, water, and mixed solvents thereof, more preferably tetrahydrofuran (THF), diisopropyl ether, dibutyl ether, cyclopentyl methyl ether (CPME), methyl Ru-tert-butyl ether, methanol, ethanol, propan
- the amount of the solvent used in step (i) may be any amount as long as the reaction system can be sufficiently stirred. From the viewpoint of reactivity, suppression of by-products and economic efficiency, etc., it is usually 0 (zero) to 10.0 L (liter), preferably 0.01 to 5 with respect to 1 mol of malononitrile represented by the formula (1). A range of 0.0 L, more preferably 0.05 to 3.0 L, and still more preferably 0.1 to 2.0 L can be exemplified.
- the reaction temperature in step (i) is not particularly limited. From the viewpoint of yield, by-product suppression, economic efficiency, etc., it is usually ⁇ 30 ° C. (minus 30 ° C.) to 90 ° C., preferably ⁇ 20 ° C. (minus 20 ° C.) to 70 ° C., more preferably ⁇ 10 ° C. ( A range of minus 10 ° C.) to 30 ° C., more preferably ⁇ 5 ° C. (minus 5 ° C.) to 20 ° C. can be exemplified.
- the reaction time in step (i) is not particularly limited. From the viewpoint of yield, by-product suppression, economic efficiency, etc., a range of usually 0.5 hours to 48 hours, preferably 0.5 hours to 24 hours, more preferably 0.5 hours to 12 hours can be exemplified. .
- the form of “product of step (i)” in the present invention is a crude product in which the reaction mixture of step (i) and, if possible, the reaction product of step (i) are isolated and not purified. Products and substances in which the reaction product of step (i) is isolated and purified, but are not limited thereto. Therefore, the form of the product of step (i) used for step (ii) is not limited, but from the viewpoint of economic efficiency and safety, the “product of step (i)” is preferably step (i). ) Is used for step (ii).
- the reaction mechanism in the present invention is not clear. However, when the present invention is considered after the present invention is completed, the product of step (i) is 2-hydroxyiminopropanedinitrile represented by the following formula (8) or a salt thereof, or a mixture thereof. It is estimated to be.
- the product of step (i) has the general formula (4): (In the formula, M represents hydrogen or an alkali metal, or a mixture thereof.) It is estimated that it is a compound represented by.
- alkali metal examples include sodium, potassium, lithium and the like.
- the description of the form of the compound represented by the general formula (4) is in accordance with the description of the “product of step (i)”.
- step (ii) will be described.
- Step (ii) comprises reacting the product of step (i) with a C3-C6 alkyl halide in the presence of a phase transfer catalyst, (Wherein R represents a C3-C6 alkyl group) Is a step of producing a compound represented by the formula:
- phase transfer catalyst examples include, but are not limited to, quaternary ammonium salts, quaternary phosphonium salts, crown ethers, and the like.
- Examples of the quaternary ammonium salt include tetramethylammonium chloride, tetramethylammonium bromide, tetraethylammonium chloride, tetraethylammonium bromide, tetrabutylammonium fluoride, tetrabutylammonium chloride, tetrabutylammonium bromide, tetrabutylammonium iodide, sulfuric acid.
- crown ethers examples include 12-crown-4, 15-crown-5, and 18-crown-6.
- Examples of the quaternary phosphonium salt include tetrabutylphosphonium bromide, tetraoctylphosphonium bromide, and tetraphenylphosphonium bromide.
- the phase transfer catalyst is preferably a quaternary ammonium salt, more preferably tetrabutylammonium bromide, tetrabutylammonium hydrogen sulfate, benzyltrimethylammonium chloride, benzyltrimethylammonium bromide, Octylmethylammonium chloride, trioctylmethylammonium bromide, benzyllauryldimethylammonium chloride, myristyltrimethylammonium bromide, benzyldimethylstearylammonium chloride, benzyldimethylstearylammonium bromide, etc., more preferably tetrabutylammonium bromide, benzyltrimethylammonium chloride, particularly preferred Is tetrabutylammonium bromide.
- the form of the phase transfer catalyst may be any form as long as the reaction proceeds.
- Examples of the form of the phase transfer catalyst include a solid or liquid containing only the phase transfer catalyst, an aqueous solution having an arbitrary concentration, or a solution of a solvent other than water.
- the phase transfer catalyst may be used alone or in combination of two or more at any ratio.
- the amount of the phase transfer catalyst used may be any amount as long as the reaction proceeds. From the viewpoints of yield, by-product suppression and economic efficiency, etc., usually 0.001 to 1.0 mol, preferably 0.005 to 0.3 mol, based on 1 mol of malononitrile of the formula (1), A range of 0.01 to 0.2 mol can be exemplified.
- C3-C6 alkyl halide means a linear or branched alkyl halide having 3 to 6 carbon atoms.
- Examples of the C3-C6 alkyl halide include propyl chloride, isopropyl chloride, butyl chloride, sec-butyl chloride, isobutyl chloride, tert-butyl chloride, pentyl chloride, 1-methylbutyl chloride, 2-methylbutyl chloride, 3-methyl Butyl chloride, 1-ethylpropyl chloride, 1,1-dimethylpropyl chloride, 1,2-dimethylpropyl chloride, 2,2-dimethylpropyl chloride, hexyl chloride, 1-methylpentyl chloride, 2-methylpentyl chloride, 3- Methylpentyl chloride, 4-methylpentyl chloride, 1,2-dimethylbutyl chloride, 1,3-dimethylbutyl chloride, 1-ethylbutyl chloride,
- the C3-C6 alkyl halide is preferably propyl chloride, isopropyl chloride, butyl chloride, sec-butyl chloride, isobutyl chloride, tert-butyl chloride, propyl.
- the amount of C3-C6 alkyl halide used may be any amount as long as the reaction proceeds. From the viewpoints of yield, by-product suppression, economic efficiency, etc., usually 0.8 to 3.0 mol, preferably 0.9 to 2.0 mol, more preferably 1 to 1 mol of malononitrile of the formula (1) A range of 0.9 to 1.5 mol is preferable.
- step (ii) the pH of the solvent system may be adjusted to allow the reaction to proceed smoothly.
- the pH may or may not be adjusted.
- the pH may be adjusted at any time in step (ii).
- the pH may be adjusted before adding the C3 to C6 alkyl halide, or may be performed during the dropwise addition of the C3 to C6 alkyl halide.
- the pH of the solvent system in step (ii) may be in any range as long as the reaction proceeds. From the viewpoint of yield, by-product suppression, economic efficiency, etc., the pH is usually in the range of 4-9, preferably pH 4-8, more preferably pH 5-8, further preferably pH 5-7, particularly preferably pH 6-7. It can be illustrated.
- a base can be used.
- the reaction mechanism in the present invention is not clear. However, as a result of considering the present invention after the completion of the present invention, it was presumed as follows. It was speculated that it was necessary to use a base when the protonic acid used in step (i) was a strong acid of acetic acid or higher. In particular, it is necessary to use a base when the protic acid used in step (i) is a strong acid of acetic acid or higher and the amount of the protic acid used is more than 0.1 equivalent per 1 mol of malononitrile. It was speculated.
- Examples of the base that can be used in the step (ii) include alkali metal hydroxides, alkaline earth metal hydroxides, alkali metal carbonates, alkaline earth metal carbonates, alkali metal hydrogen carbonates, alkaline earths.
- Inorganic bases such as metal hydrogen carbonates, pyridines, quinolines, isoquinolines, tertiary amines, secondary amines, primary amines, aromatic amines, cyclic amines, carboxylic acid alkali metal salts, carvone Examples include, but are not limited to, organic bases such as acid alkaline earth metal salts.
- preferable examples of the base used in step (ii) include alkali metal hydroxides, alkali metal carbonates, alkali metal hydrogen carbonates, tertiary amines.
- Carboxylic acid alkali metal salts more preferably alkali metal hydroxides, alkali metal carbonates, alkali metal hydrogen carbonates, more preferably alkali metal hydroxides.
- Specific preferred examples of the base used in the step (ii) include sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, triethylamine, sodium acetate, potassium acetate, more preferably water.
- the form of the base used in step (ii) may be any form as long as the reaction proceeds.
- Examples of the form of the base used in the step (ii) include a solid or liquid containing only a base, an aqueous solution having an arbitrary concentration, or a solution of a solvent other than water.
- the base used for a process (ii) may be used individually or in mixture of 2 or more types in arbitrary ratios.
- the base in step (ii) may or may not be used as long as the reaction proceeds.
- the amount of the base used is usually that the pH of the solvent system is pH 4-9, preferably pH 4-8, The amount adjusted to pH 5 to 8, more preferably pH 5 to 7, particularly preferably pH 6 to 7 can be mentioned.
- Solvent systems that can be used in step (ii) include, for example, aromatic hydrocarbon derivatives, aliphatic hydrocarbons, halogenated aliphatic hydrocarbons, ethers, alcohols, nitriles, amides, alkyls Examples include ureas, sulfoxides, sulfones, ketones, carboxylic esters, carboxylic acids, aromatic heterocycles, water, and solvent systems consisting of them in any mixing ratio.
- preferred examples of the solvent system used in step (ii) include aromatic hydrocarbon derivatives, halogenated aliphatic hydrocarbons, ethers, alcohols. , Nitriles, amides, alkylureas, sulfoxides, sulfones, water, and solvent systems comprising them, more preferably aromatic hydrocarbon derivatives, halogenated aliphatic hydrocarbons, ethers, alcohols Nitriles, amides, water, and solvent systems composed of these, more preferably solvent systems composed of aromatic hydrocarbon derivatives and water, and solvent systems composed of ethers and water.
- solvent system used in step (ii) include toluene, xylene, chlorobenzene, dichlorobenzene, nitrobenzene, dichloromethane, tetrahydrofuran (THF), diisopropyl ether, dibutyl ether, cyclopentyl methyl ether (CPME), methyl- tert-butyl ether, methanol, ethanol, propanol, 2-propanol, butanol, acetonitrile, N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAC), N-methylpyrrolidone (NMP), N, N ′ Dimethyl imidazolidinone (DMI), dimethyl sulfoxide (DMSO), sulfolane, water, and solvent systems comprising them, more preferably toluene, xylene, chlorobenzene, dichloro Lobenzene, nitrobenzene, dichloromethane,
- the amount of the solvent used to form the solvent system in step (ii) may be any amount as long as the reaction system can be sufficiently stirred.
- the amount of water is usually 0 (zero) to 10.0 L (liter), preferably 0.01, relative to 1 mol of malononitrile of the formula (1).
- a range of ⁇ 10.0 L, more preferably 0.1-5.0 L, and even more preferably 0.2-3.0 L can be exemplified.
- the amount of the above-mentioned solvent other than water is usually from 0 (zero) to 10.0 L (liter), preferably from 0.01 to 10.3, per 1 mol of malononitrile of the formula (1).
- a range of 0 L, more preferably 0.1 to 5.0 L, and still more preferably 0.2 to 3.0 L can be exemplified.
- the mixing ratio of water and a solvent other than water may be any ratio as long as the reaction proceeds.
- the mixing ratio of the two or more kinds of solvents other than water may be any ratio as long as the reaction proceeds.
- the reaction temperature in step (ii) is not particularly limited. From the viewpoints of yield, by-product suppression, economic efficiency, etc., it is usually in the range of 10 ° C. to 100 ° C., preferably 40 ° C. to 95 ° C., more preferably 50 ° C. to 95 ° C., further preferably 55 ° C. to 90 ° C. Can be illustrated.
- the reaction time in step (ii) is not particularly limited. From the viewpoint of yield, by-product suppression, economic efficiency, etc., a range of usually 0.5 hours to 48 hours, preferably 0.5 hours to 24 hours, more preferably 1 hour to 12 hours can be exemplified.
- the 2-alkoxyiminopropanedinitrile represented by the general formula (2) obtained in the step (ii) is a novel compound.
- Specific examples of the 2-alkoxyiminopropanedinitrile represented by the general formula (2) include, for example, 2-propoxyiminopropanedinitrile, 2-isopropoxyiminopropanedinitrile, 2-butoxyiminopropanedinitrile, 2-sec-butoxyiminopropanedinitrile, 2-isobutoxyiminopropanedinitrile, 2-tert-butoxyiminopropanedinitrile, 2-pentyloxyiminopropanedinitrile, 2- (1-methylbutyloxyimino) propanedinitrile, 2- (2-methylbutyloxyimino) propanedinitrile, 2- (3-methylbutyloxyimino) propanedinitrile, 2- (1-ethylpropyloxyimino) propanedinitrile, 2- (1,1-dimethylpropyloxyimino)
- 2-propoxyiminopropanedinitrile, 2-isopropoxyiminopropanedinitrile, 2-butoxyiminopropanedinitrile, 2-sec-butoxyiminopropanedinitrile, 2-isobutoxyiminopropane Dinitrile and 2-tert-butoxyiminopropanedinitrile are preferred, and 2-propoxyiminopropanedinitrile and 2-isopropoxyiminopropanedinitrile are more preferred.
- the compound represented by the above general formula (2) used in the step (iii), that is, the form of 2-alkoxyiminopropanedinitrile is a substance obtained by isolating and purifying the reaction product of the step (ii), the step Examples include, but are not limited to, crude products in which the reaction product of (ii) is isolated and not purified, the reaction mixture of step (ii), and the like. Therefore, the compound represented by the above general formula (2), that is, 2-alkoxyiminopropanedinitrile, may be used in step (iii) either isolated or not. In addition, the compound represented by the above general formula (2), that is, 2-alkoxyiminopropanedinitrile, may be used in step (iii) with or without purification.
- the yield through step (i) and step (ii) is usually 60% or more, preferably 65 to 100%, more preferably 70 to 100%.
- the yields through the steps (i) and (ii) are as follows.
- step (iii) will be described.
- Step (iii) is represented by the general formula (2): (Wherein R represents a C3-C6 alkyl group)
- the compound represented by general formula (3) is reacted with a hydroxylamine compound: (Wherein R represents a C3-C6 alkyl group) Is a step of producing a compound represented by the formula:
- Examples of the hydroxylamine compound include hydroxylamine and hydroxylamine salt.
- Examples of the hydroxylamine salt include, but are not limited to, hydroxylamine sulfate or hydroxylamine hydrochloride.
- an aqueous solution of hydroxylamine is usually used in consideration of danger.
- the concentration of the aqueous solution is usually preferably 60% or less.
- the form of the hydroxylamine salt may be any form as long as the reaction proceeds.
- Examples of the form of the hydroxylamine salt include a solid containing only the hydroxylamine salt, an aqueous solution having an arbitrary concentration, or a solution of a solvent other than water. From the viewpoint of price, availability and safety, the hydroxylamine salt form is preferably an aqueous solution.
- the hydroxylamine compounds may be used alone or in combination of two or more in any ratio.
- the amount of the hydroxylamine compound used may be any amount as long as the reaction proceeds. From the viewpoints of yield, suppression of by-products and economic efficiency, etc., usually 0.8 to 2.0 mol, preferably 0.9 to 1.5 mol, per 1 mol of the compound represented by the formula (2) More preferred is a range of 0.9 to 1.3 mol, and still more preferred is a range of 0.9 to 1.2 mol.
- a hydroxylamine salt is used as the hydroxylamine compound, it is preferable to use a base.
- Examples of the base that can be used in the step (iii) include alkali metal hydroxides, alkaline earth metal hydroxides, alkali metal carbonates, alkaline earth metal carbonates, alkali metal bicarbonates, and alkaline earths.
- Inorganic bases such as metal hydrogen carbonates, pyridines, quinolines, isoquinolines, tertiary amines, secondary amines, primary amines, aromatic amines, cyclic amines, carboxylic acid alkali metal salts, carvone Examples include, but are not limited to, organic bases such as acid alkaline earth metal salts.
- preferred examples of the base used in step (iii) include alkali metal hydroxides, alkali metal carbonates, alkali metal bicarbonates, alkali carboxylates. Metal salts, more preferably alkali metal hydroxides, alkali metal carbonates, alkali metal hydrogen carbonates, and more preferably alkali metal carbonates and alkali metal hydrogen carbonates.
- Specific preferred examples of the base used in the step (iii) include sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, sodium acetate, potassium acetate, more preferably sodium hydroxide. Potassium hydroxide, sodium carbonate, potassium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, more preferably sodium carbonate, potassium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate.
- the form of the base used in step (iii) may be any form as long as the reaction proceeds.
- Examples of the form of the base used in the step (iii) include a solid or liquid containing only a base, an aqueous solution having an arbitrary concentration, or a solution of a solvent other than water.
- the base used for a process (iii) may be used individually or in mixture of 2 or more types in arbitrary ratios.
- the amount of base used in step (iii) may be any amount as long as the reaction proceeds. From the viewpoints of yield, by-product suppression, economic efficiency, etc., the amount is usually 0.8 to 2.0 equivalents, preferably 0.9 to 1.5 equivalents, relative to 1 mol of the compound of the formula (2). The range is preferably 0.9 to 1.3 equivalents, more preferably 0.9 to 1.2 equivalents.
- the reaction in step (iii) may be carried out without solvent, but it is preferable to use a solvent from the viewpoint of smooth progress of the reaction.
- the solvent in step (iii) may be any solvent as long as the reaction in step (iii) proceeds.
- Examples of the solvent that can be used in the step (iii) include water, ethers, alcohols, nitriles, amides, alkylureas, sulfoxides, sulfones, carbonates, ketones, and carboxylic acid esters. , Carboxylic acids, nitrated aliphatic hydrocarbons, aromatic heterocycles, and the like, and mixed solvents of any mixing ratio thereof, but are not limited thereto.
- preferred examples of the solvent used in the step (iii) include ethers, alcohols, nitriles, amides, alkylureas, sulfoxides, sulfones, water, and the like. More preferably, ethers, alcohols, nitriles, amides, water, and mixed solvents thereof, more preferably alcohols, water, and mixed solvents thereof.
- the solvent used in the step (iii) include tetrahydrofuran (THF), diisopropyl ether, dibutyl ether, cyclopentyl methyl ether (CPME), methyl-tert-butyl ether, methanol, ethanol, propanol, 2-propanol, Butanol, acetonitrile, N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAC), N-methylpyrrolidone (NMP), N, N′-dimethylimidazolidinone (DMI), dimethyl sulfoxide (DMSO) , Sulfolane, water, and mixed solvents thereof, more preferably tetrahydrofuran (THF), diisopropyl ether, dibutyl ether, cyclopentyl methyl ether (CPME), methyl-tert- Butyl ether, methanol, ethanol, propanol, 2-propanol
- the amount of the solvent used in step (iii) may be any amount as long as the reaction system can be sufficiently stirred. From the viewpoints of reactivity, suppression of by-products and economic efficiency, etc., it is usually 0 (zero) to 10.0 L (liter), preferably 0.01 to 10.0 L with respect to 1 mol of malononitrile of the formula (1). A range of 0.1 to 5.0 L is more preferable, and a range of 0.2 to 3.0 L is more preferable.
- the reaction temperature in step (iii) is not particularly limited. From the viewpoint of yield, by-product suppression, economic efficiency, etc., it is usually ⁇ 30 ° C. (minus 30 ° C.) to 100 ° C., preferably ⁇ 20 ° C. (minus 20 ° C.) to 70 ° C., more preferably ⁇ 10 ° C. ( Examples include a range of minus 10 ° C.) to 50 ° C., more preferably ⁇ 10 ° C. (minus 10 ° C.) to 35 ° C.
- reaction time in step (iii) is not particularly limited. From the viewpoint of yield, by-product suppression, economic efficiency, etc., a range of usually 0.5 hours to 48 hours, preferably 0.5 hours to 24 hours, more preferably 1 hour to 12 hours can be exemplified.
- 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3) obtained in step (iii) include, for example, 2-amino-2-hydroxyimino-N-propoxyacetimidoyl cyanide, 2-amino-2-hydroxyimino-N-isopropoxyacetimidoyl cyanide, 2-amino-2-hydroxyimino-N-butoxyacetimidoyl cyanide, 2-amino-2-hydroxyimino-N-sec-butoxyacetimidoyl cyanide, 2-amino-2-hydroxyimino-N-isobutoxyacetimidoyl cyanide, 2-amino-2-hydroxyimino-N-tert-butoxyacetimidoyl cyanide, 2-amino-2-hydroxyimino-N-pentyloxyacetimidoyl cyanide, 2-amino-2-hydroxyimino-N-pent
- 2-amino-2-hydroxyimino-N-propoxyacetimidoylcyanide 2-amino-2-hydroxyimino-N-isopropoxyacetimidoylcyanide
- 2-amino-2- Hydroxyimino-N-butoxyacetimidoylcyanide 2-amino-2-hydroxyimino-N-sec-butoxyacetimidoylcyanide
- 2-amino-2-hydroxyimino-N-isobutoxyacetimidoylcyanide and 2- Amino-2-hydroxyimino-N-tert-butoxyacetimidoylcyanide is preferred, 2-amino-2-hydroxyimino-N-propoxyacetimidoylcyanide and 2-amino-2-hydroxyimino-N-isopropoxyacetate More imidoyl cyanide Masui.
- the yield of step (iii) is usually 70% or more, preferably 80 to 100%, more preferably 85 to 100%.
- room temperature indicates 10 ° C to 35 ° C.
- 1 H nuclear magnetic resonance spectral analysis ( 1 H-NMR) was performed using model: Oxford NMR300 (manufactured by Varian Medical Systems) and an internal standard substance: tetramethylsilane.
- PH was measured with a glass electrode type hydrogen ion concentration indicator.
- a glass electrode type hydrogen ion concentration indicator for example, model HM-20P manufactured by Toa DKK Corporation can be used.
- Differential scanning calorimetry was performed using a model: DSC-60 (manufactured by Shimadzu Corporation) at a heating rate of 10 ° C./min in a temperature range of 40 to 400 ° C.
- DSC-60 manufactured by Shimadzu Corporation
- the following documents can be referred to as necessary.
- the reaction vessel was charged with 100 ml of water, 66 g (1.00 mol) of malononitrile and 6 g (0.10 mol) of acetic acid. While stirring the mixture under ice cooling, 272 ml (1.05 mol) of 28% aqueous sodium nitrite solution was added dropwise thereto. After completion of the dropwise addition, the mixture was stirred for 2 hours under ice cooling. Subsequently, 534 ml of toluene, 184.5 g (1.50 mol) of isopropyl bromide and 32.2 g (0.10 mol) of tetrabutylammonium bromide were charged in another reaction vessel, and the reaction obtained above was stirred at 60 ° C.
- the mixture was added dropwise. Thereafter, the mixture was stirred at the same temperature for 5 hours.
- the resulting reaction mixture was partitioned between toluene and water, and the toluene layer was separated.
- the obtained toluene solution was analyzed by HPLC absolute calibration curve method. As a result, the yield of 2-isopropoxyiminopropanedinitrile was 80% of the theoretical amount calculated from the amount of malononitrile used.
- the toluene solution was worked up by a conventional method to obtain 2-isopropoxyiminopropanedinitrile.
- the resulting 2-isopropoxyiminopropanedinitrile was identified by NMR analysis.
- the reaction vessel was charged with 100 ml of water, 66 g (1.00 mol) of malononitrile and 6 g (0.10 mol) of acetic acid. While stirring the mixture under ice cooling, 272 ml (1.05 mol) of 28% aqueous sodium nitrite solution was added dropwise thereto. After completion of the dropwise addition, the mixture was stirred for 2 hours under ice cooling. Subsequently, 534 ml of toluene, 184.5 g (1.50 mol) of isopropyl bromide and 32.2 g (0.10 mol) of tetrabutylammonium bromide were charged into another reaction vessel, and the reaction obtained above was stirred at 70 ° C.
- the mixture was added dropwise. During the dropwise addition, the pH of the solution was adjusted to about 6-7 using a 25% aqueous sodium hydroxide solution. Thereafter, the mixture was stirred at the same temperature for 3 hours. The resulting reaction mixture was partitioned between toluene and water, and the toluene layer was separated. The obtained toluene solution was analyzed by HPLC absolute calibration curve method. As a result, the yield of 2-isopropoxyiminopropanedinitrile was 75% of the theoretical amount calculated from the amount of malononitrile used.
- a reaction vessel was charged with 6.20 g (45 mmol) of 2-isopropoxyiminopropanedinitrile and 40 ml of methanol. While stirring the mixture under ice cooling, a solution of 3.30 g (20 mmol) of hydroxylamine sulfate and 8 ml of water and 2.10 g (20 mmol) of sodium carbonate were added. The mixture was stirred for 1 hour under ice-cooling, and then stirred at room temperature for 2 hours. The solvent was distilled off under reduced pressure. The resulting residue was dissolved by adding water and diethyl ether. The two layers were separated, the organic layer was dried over sodium sulfate, and sodium sulfate was removed by filtration. The filtrate was concentrated under reduced pressure, and the resulting residue was dried under reduced pressure to obtain 6.12 g of 2-amino-2-hydroxyimino-N-isopropoxyacetimidoylcyanide in a yield of 90%.
- 2-Hydroxyiminopropanedinitrile is a known compound, and was produced by a conventional method known to those skilled in the art and subjected to differential scanning calorimetry.
- the sodium salt of 2-hydroxyiminopropanedinitrile is a known compound, which was produced by a conventional method known to those skilled in the art, and was subjected to differential scanning calorimetry.
- an industrially suitable method for producing 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3) is useful as a pesticide itself and is also useful as an intermediate for producing other pesticides. . *
- the use of a thermally dangerous compound in the prior art as a production intermediate is avoided, and the target 2-amino-2-hydroxyimino-N-alkoxy represented by the general formula (3) is avoided.
- Acetimidyl cyanide can be produced. That is, according to the present invention, there is no need to use 2-amino-2-hydroxyimino-N-hydroxyacetimidoyl cyanide represented by the formula (6), which is considered to exhibit extremely dangerous exothermic decomposition.
- a 2-amino-2-hydroxyimino-N-alkoxyacetimidoyl cyanide represented by the general formula (3) can be produced.
- the novel 2-alkoxyiminopropanedinitrile represented by the general formula (2) provided by the present invention has a target 2-amino-2- represented by the general formula (3) as compared with that of the prior art. It is a much safer intermediate for the production of hydroxyimino-N-alkoxyacetimidoylcyanides. Other production intermediates in the present invention are also significantly safer than those of the prior art. That is, the method of the present invention has dramatically increased safety compared to the prior art. Thus, the method of the present invention can be safely applied to large scale manufacturing such as pilot plants or industrial production.
- the present invention provides a novel industrially satisfactory production method of 2-alkoxyiminopropanedinitrile represented by the general formula (2) which is a useful intermediate of the present invention.
- 2-alkoxyiminopropanedinitrile represented by the general formula (2) can be produced in high yield without using an expensive silver salt.
- a 2-alkoxy represented by the general formula (2) using a recyclable and inexpensive solvent for example, an organic solvent such as an ether solvent or an aromatic hydrocarbon derivative solvent. Iminopropanedinitrile can be produced.
- the method of the present invention has a high yield and can be carried out efficiently on an industrial scale by simple operation under mild conditions without using a special reaction apparatus.
- the amount of waste is small and the nature of the waste is considered favorable.
- the present invention is environmentally friendly and has high industrial utility value.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
【課題】本発明の目的は、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの工業化に適したかつ経済的に好ましい製造方法を提供することにある。 【解決手段】本発明は、 式(1):で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させ、続けて、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させた後、得られる新規な一般式(2):(式中、RはC3~C6アルキル基を示す。) で表される化合物すなわち2-アルコキシイミノプロパンジニトリルを、ヒドロキシルアミン化合物と反応させることにより、 一般式(3):(式中、Rは前記と同意義である。) で表わされる化合物すなわち2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを、安全にかつ高収率で製造できることを見出し、完成するに至った。
Description
本発明は、一般式(2):

(式中、Rは3から6個の炭素原子を有するアルキル基を示す。)で表わされる2-アルコキシイミノプロパンジニトリル、およびその製造方法に関する。一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルは、本発明の方法における、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造するための有用な中間体である。
(式中、Rは3から6個の炭素原子を有するアルキル基を示す。)で表わされる2-アルコキシイミノプロパンジニトリル、およびその製造方法に関する。一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルは、本発明の方法における、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造するための有用な中間体である。
本発明は、また一般式(3):
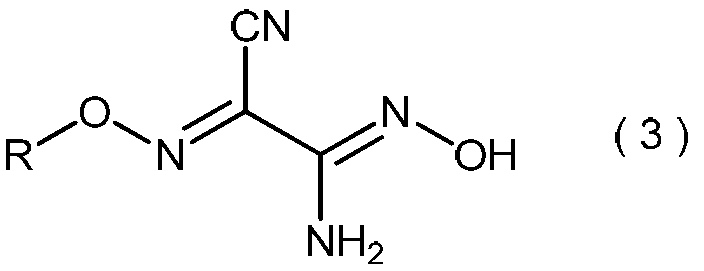
(式中、Rは前記と同意義である。)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの製造方法に関する。一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドは、それ自身が農薬として有用であり、そして他の農薬の製造中間体としても有用である。
(式中、Rは前記と同意義である。)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの製造方法に関する。一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドは、それ自身が農薬として有用であり、そして他の農薬の製造中間体としても有用である。
一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドは、優れた有害生物防除活性を有する有用な化合物である(特許文献1参照)。例えば、具体的に、2-アミノ-2-ヒドロキシイミノ-N-イソプロポキシアセトイミドイルシアニドは優れた有害生物防除活性を示す。
加えて、下図に示すように、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドから一般式(5)で表わされる2-アミノ-2-アルコキシイミノ-N-アルコキシアセトイミドイルシアニドを製造することができる。

例えば、特許文献1の実施例1(2)および実施例2に具体的に記載されるように、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドのアルキル化により、一般式(5)で表わされる2-アミノ-2-アルコキシイミノ-N-アルコキシアセトイミドイルシアニドは製造することができる。
さらに、一般式(5)で表わされる2-アミノ-2-アルコキシイミノ-N-アルコキシアセトイミドイルシアニドもまた優れた有害生物防除活性を有することが、特許文献1には開示されている。
例えば、具体的に、2-アミノ-2-イソプロポキシイミノ-N-イソプロポキシアセトイミドイルシアニド、2-アミノ-2-イソプロポキシイミノ-N-プロポキシアセトイミドイルシアニドおよび2-アミノ-2-プロポキシイミノ-N-プロポキシアセトイミドイルシアニドは優れた有害生物防除活性を示す。
例えば、具体的に、2-アミノ-2-イソプロポキシイミノ-N-イソプロポキシアセトイミドイルシアニド、2-アミノ-2-イソプロポキシイミノ-N-プロポキシアセトイミドイルシアニドおよび2-アミノ-2-プロポキシイミノ-N-プロポキシアセトイミドイルシアニドは優れた有害生物防除活性を示す。
一方で、一般式(5)で表わされる2-アミノ-2-アルコキシイミノ-N-アルコキシアセトイミドイルシアニドは、特許文献2に開示されるアルコキシイミノ誘導体を製造する中間体として使用することができる。言い換えれば、下図に示すように、当業者に知られた方法により、式(5)で表わされる2-アミノ-2-アルコキシイミノ-N-アルコキシアセトイミドイルシアニドから、特許文献2に開示されるアルコキシイミノ誘導体のいくつかを製造することができる。

例えば、特許文献2の実施例1(2)における目的化合物は、ジアゾ化とそれに続く塩素化からなる、特許文献3の実施例1と同様の方法で、上記の2-アミノ-2-イソプロポキシイミノ-N-イソプロポキシアセトイミドイルシアニドから製造することができる。別の例としては、特許文献2の実施例23(4)における目的化合物は、同じ方法で、上記の2-アミノ-2-プロポキシイミノ-N-プロポキシアセトイミドイルシアニドから製造することができる。
そして、特許文献2の実施例1(2)における目的化合物は、特許文献2の他の実施例で具体的に記載されるように、種々のアルコキシイミノ誘導体を製造する中間体である。また、特許文献2の実施例23(4)における目的化合物は、特許文献2の他の実施例で具体的に記載されるように、いくつかのアルコキシイミノ誘導体を製造する中間体である。
ここで、特許文献2に開示されるアルコキシイミノ誘導体もまた、優れた有害生物防除活性を有する有用な化合物であると知られている。特許文献2に開示されるように、アルコキシイミノ誘導体を有効成分として含有する有害生物防除剤は、農業および園芸分野における広範囲の有害生物に対して優れた防除効果を示し、特にトビイロウンカ、ワタアブラムシ等のカメムシ目害虫に高い効果を示す。加えて、アルコキシイミノ誘導体を有効成分として含有する有害生物防除剤は抵抗性の有害生物をも防除することができる。上述の特許文献2の実施例1(2)または実施例23(4)の目的化合物から製造されており、かつ優れた有害生物防除活性を示すアルコキシイミノ誘導体としては、具体的には例えば、
1-(2-シアノ-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール、
1-(2-カルバモイル-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール、
1-(2-シアノ-1,2-ジプロポキシイミノエチル-1H-1,2,4-トリアゾール、
1-(2-カルバモイル-1,2-ジプロポキシイミノエチル]-1H-1,2,4-トリアゾール等、
好ましくは
1-(2-カルバモイル-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール、
1-(2-カルバモイル-1,2-ジプロポキシイミノエチル]-1H-1,2,4-トリアゾール、
特に好ましくは
1-(2-カルバモイル-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール
が挙げられる。
1-(2-シアノ-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール、
1-(2-カルバモイル-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール、
1-(2-シアノ-1,2-ジプロポキシイミノエチル-1H-1,2,4-トリアゾール、
1-(2-カルバモイル-1,2-ジプロポキシイミノエチル]-1H-1,2,4-トリアゾール等、
好ましくは
1-(2-カルバモイル-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール、
1-(2-カルバモイル-1,2-ジプロポキシイミノエチル]-1H-1,2,4-トリアゾール、
特に好ましくは
1-(2-カルバモイル-1,2-ジイソプロポキシイミノエチル)-1H-1,2,4-トリアゾール
が挙げられる。
要するに、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドは、それ自体が有害生物防除剤の活性成分として有用なだけではなく、特許文献1および2に開示される、有害生物防除剤の一連の活性成分を製造するための中間体としても有用である。
従来、下図に示すように、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造する方法として、式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドを中間体として使用する方法が知られている(特許文献1参照)。

しかしながら、非特許文献1は、式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドの示差走査熱量分析が極めて危険な熱的挙動、すなわち発熱分解反応を示すことを示唆する。実際に、非特許文献1は、式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドの一水和物が、124℃の発熱開始温度と10.3kJ/gの発熱量を示すことを具体的に開示する。これらの値は非常にリスクが高く、式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドは危険な化合物である。10.3kJ/gもの大きな発熱量は、分解反応が極めて高い発熱性であるため、反応器中に制御不能な状態を生じることを示唆する。加えて、124℃の低い発熱開始温度は、当該発熱分解反応が比較的低い温度で開始することを意味する。したがって、反応の暴走および爆発が起こる可能性があるため、この方法は生産プラントの安全性を保障できない。つまり、安全性の欠如のため、式(6)の2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドを使用するこの方法は、工業生産に好ましくない。
上記の状況で、本発明者らが、式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドのような危険な化合物を使用しない、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの製造方法を研究した。その結果、本発明者らは、新規な下記一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルの製造に成功した。(式中、Rは3から6個の炭素原子を有するアルキル基を示す。)
さらに、下図に示すように、本発明者らは、意外にも、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルから一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを安全に製造できることを見出した。(式中、いずれのRも前記と同意義である。)
その一方で、当該2-アルコキシイミノプロパンジニトリルの類縁体のいくつかを製造する方法は知られていた。これまでに、当該2-アルコキシイミノプロパンジニトリルの類縁体を製造する方法としては、マロノニトリルと亜硝酸のアルカリ金属塩を反応させた後、得られる化合物をアルキル化剤と反応させる方法が知られている。
しかしながら、従来から知られているこの方法では、高収率を達成できるアルキル化剤は、ジメチル硫酸、α-ハロカルボン酸エステルおよびベンジルブロミドなどの高い反応性を有するアルキル化剤に限られている(特許文献4、5、6および7、および非特許文献2参照)。実際に、2-ブロモフルオロ-1,1-エタンのような反応性が低いアルキル化剤が用いられた場合には、反応がN,N-ジメチルホルムアミド溶媒中で行われているにもかかわらず、収率が24%と悪いことが報告されている(特許文献8参照)。
また、特許文献9は、2-ヒドロキシイミノプロパンジニトリルの銀塩とシクロプロピルメチルブロミドとの反応が64%の収率を達成したことを開示する。しかしながら、銀塩は高価であるため、この方法は工業的に好ましくない。
要するに、本発明者らは、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルが本発明の方法における中間体として有用であろう可能性を見出した。しかしながら、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルの工業的に満足できる製造方法は知られていなかった。
Journal of Organic Chemistry,2000年,65巻,1139-1143頁
Bulletin of the Chemical Society of Japan,76巻,1063-1070頁 (2003年)
本発明の目的は、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの工業化に適したかつ経済的に好ましい製造方法を提供することにある。
具体的には、例えば、本発明の目的は、式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドのような熱的に危険な化合物を製造中間体として使用しない、より安全な一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの製造方法を提供することにある。
上記のより安全な方法という目的を達成するためには、より安全な製造中間体を提供する必要がある。
加えて、工業的に好ましい、そのようなより安全な製造中間体を製造する方法を提供する必要もある。言い換えれば、本明細書中で説明するように、本発明はそのようなより安全な製造中間体として一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルを提供するが、しかしながらその工業的に満足できる製造方法を提供する必要がある。つまり、本発明の他の目的は、工業的に好ましい、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルの製造方法を提供することにある。例えば、上述の高い反応性を有するアルキル化剤が用いられないときでも、高収率で2-アルコキシイミノプロパンジニトリル類を製造できる方法を提供する必要がある。さらに、銀塩のような高価な原料などを使用しない、それらの製造方法が望まれていた。
上記のような状況に鑑み、本発明者らが一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの製造方法について鋭意研究した。その結果、本発明者らは、意外にも、新規な一般式(2):

(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物すなわち2-アルコキシイミノプロパンジニトリルを使用することにより、そして新規な工業的に満足できるその製造方法を同時に提供することにより、上記課題の解決が可能であることを見出した。言い換えれば、本発明者らは、意外にも、式(1):
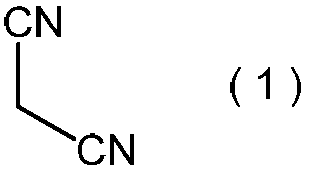
で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させ、
続けて、得られる生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させた後、
得られる一般式(2):
(式中、Rは前記と同意義である。)
で表わされる化合物すなわち2-アルコキシイミノプロパンジニトリルを、ヒドロキシルアミン化合物と反応させることにより、
一般式(3):

(式中、RはC3~C6アルキル基を示す)
で表わされる化合物すなわち2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを、安全にかつ高収率で製造できることを見出した。本発明者らはこの知見に基づき本発明を完成するに至った。
で表わされる化合物すなわち2-アルコキシイミノプロパンジニトリルを使用することにより、そして新規な工業的に満足できるその製造方法を同時に提供することにより、上記課題の解決が可能であることを見出した。言い換えれば、本発明者らは、意外にも、式(1):
続けて、得られる生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させた後、
得られる一般式(2):
で表わされる化合物すなわち2-アルコキシイミノプロパンジニトリルを、ヒドロキシルアミン化合物と反応させることにより、
一般式(3):
で表わされる化合物すなわち2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを、安全にかつ高収率で製造できることを見出した。本発明者らはこの知見に基づき本発明を完成するに至った。
本発明により、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの新規な工業的に適用できる製造方法が提供される。
同時に、本発明により、新規な一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルおよびその工業的に満足できる製造方法が提供される。
本発明によれば、熱的に危険な化合物を製造中間体として使用することを回避し、目的とする一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造することができる。ここで、上記のように、先行技術の危険な製造中間体とは、式(6)でわ表される2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドである。すなわち、本発明によれば、示差走査熱量分析において、その一水和物でさえ124℃の発熱開始温度と10.3kJ/gの発熱量の発熱分解を示す式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドを使用せず、目的とする一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造することができる。
その一方で、本発明方法において使用される製造中間体は、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルである。一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルとしては、具体的には例えば、2-イソプロポキシイミノプロパンジニトリルが挙げられる。2-イソプロポキシイミノプロパンジニトリルの示差走査熱量測定は、225℃の発熱開始温度と0.6kJ/gの発熱量を与えた。2-イソプロポキシイミノプロパンジニトリルの発熱開始温度は、先行技術の製造中間体である式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドのそれよりも著しく高い。この発熱開始温度は上記の先行技術のそれよりはるかに良い熱的安定性を意味する。さらに、当該発熱量については、2-イソプロポキシイミノプロパンジニトリルの値は、先行技術における製造中間体である式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドのそれよりも、はるかに小さい。この値は大幅に増加された本発明方法の安全性を意味する。
したがって、本発明の新規な製造中間体である一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルが、先行技術の製造中間体である式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドより、劇的に安全であることが見出されたのである。
加えて、本発明方法においては、式(1)で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させた後、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルの前駆体として、下記式(8)で表わされる2-ヒドロキシイミノプロパンジニトリルおよび/またはその塩が生成していると推定される。
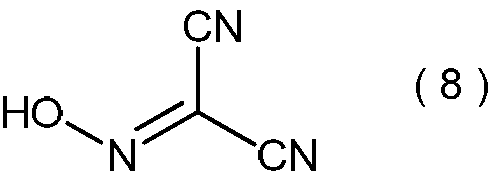
式(8)で表わされる2-ヒドロキシイミノプロパンジニトリルの塩としては、具体的には例えば、2-ヒドロキシイミノプロパンジニトリルのナトリウム塩が挙げられる。これらの化合物もまた本発明方法における製造中間体と言える。これらの化合物の示差走査熱量測定でも、下記のように改善された熱的性質が観察された。式(8)で表わされる2-ヒドロキシイミノプロパンジニトリルについては、147℃の発熱開始温度と1.6kJ/gの発熱量が見出された。この2-ヒドロキシイミノプロパンジニトリルの発熱開始温度は、先行技術の製造中間体である式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドのそれよりも比較的高く、すなわちより安全である。さらに、当該発熱量については、2-ヒドロキシイミノプロパンジニトリルの値は、上記の先行技術におけるそれよりもはるかに小さく、安全を維持するために十分である。さらに、2-ヒドロキシイミノプロパンジニトリルのナトリウム塩は、示差走査熱量測定で、222℃の吸熱開始温度と-0.9kJ/gの吸熱量を示す。この開始温度も上記の先行技術のそれより良い熱的安定性を意味する。加えて、その熱的事象は発熱ではなく吸熱であり、すなわち上記の先行技術のそれより極めて安全である。
上記のように、本発明における製造中間体の物性は、先行技術に比較して、本発明方法の有意に改善された安全性、すなわち本発明方法の優位性を示している。
言い換えれば、それらは工業的な生産中の危険な分解などのリスクを本質的に減らす。したがって、本発明方法は、パイロットプラントまたは工業的な生産のような大きなスケールでの製造に安全に適用できる。
言い換えれば、それらは工業的な生産中の危険な分解などのリスクを本質的に減らす。したがって、本発明方法は、パイロットプラントまたは工業的な生産のような大きなスケールでの製造に安全に適用できる。
本発明によれば、マロノニトリルと亜硝酸のアルカリ金属塩を反応させた後に得られる化合物をアルキル化剤と反応させるときに、高い反応性を有するアルキル化剤に限られずに(すなわちアルキル化剤の種類に関わらず)、高価な銀塩を使用せず、高収率で、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルを製造することができる。さらに、本発明によれば、リサイクルが可能なかつ安価な溶媒(例えば、エーテル類溶媒または特に芳香族炭化水素誘導体類溶媒などの有機溶媒)を用いて、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルを製造することができる。したがって、本発明により、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルの新規な工業的に満足できる製造方法もまた提供される。
工業的な利用価値などの観点からは、このような一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルの工業的に満足できる生産方法が見出されたときに、上述の危険な製造中間体を使用しない、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの製造ルートが、初めて現実のものとなったのである。
このように、本発明によれば、目的とする一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニド、及び好ましい熱的特性を有するその製造中間体である一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルを、劇的に安全に、低価格で容易に入手可能な試薬と溶媒を使用して、製造することができる。
さらに、本発明方法は、高収率であり、工業的なスケールで、簡便な操作により、穏やかな条件下で、特殊な反応装置を用いることなく、効率的に実施できる。加えて、本発明では、廃棄物の量は少なく、廃棄物の性質は好ましいと考えられる。
したがって、本発明は経済的であり、環境にも優しく、高い工業的な利用価値を有する。
以下、本発明について詳細に説明する。
本発明は、下記〔1〕から〔47〕項に記載の発明を提供することにより前記課題を解決したものである。
本発明は、下記〔1〕から〔47〕項に記載の発明を提供することにより前記課題を解決したものである。
〔1〕一般式(2):
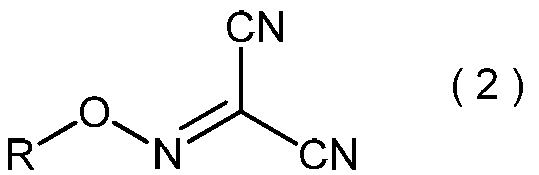
(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物。
で表わされる化合物。
〔2〕Rがイソプロピルまたはプロピルである、〔1〕に記載の化合物。
〔3〕Rがイソプロピルである、〔1〕に記載の化合物。
〔4〕Rがプロピルである、〔1〕に記載の化合物。
〔5〕一般式(2):

(式中、Rは前記と同意義である。)
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、 一般式(3):

(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物の製造方法。
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、 一般式(3):
で表わされる化合物の製造方法。
〔6〕(i) 式(1):
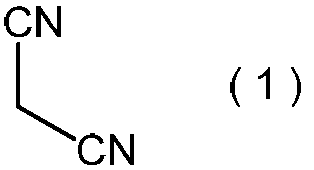
で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させる工程;
(ii) 工程(i)の生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させ、一般式(2):
(式中、Rは前記と同意義である。)
で表わされる化合物を製造する工程および
(iii) 得られる一般式(2)
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、一般式(3):
(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物の製造方法。
(ii) 工程(i)の生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させ、一般式(2):
で表わされる化合物を製造する工程および
(iii) 得られる一般式(2)
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、一般式(3):
で表わされる化合物の製造方法。
〔7〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系またはエーテル類と水からなる溶媒系で行われる、〔6〕に記載の方法。
〔8〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔6〕に記載の方法。
〔9〕工程(ii)の反応が、エーテル類と水からなる溶媒系で行われる、〔6〕に記載の方法。
〔10〕工程(ii)の反応が、トルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔6〕に記載の方法。
〔11〕工程(ii)の反応が、トルエンと水からなる溶媒系で行われる、〔6〕に記載の方法。
〔12〕工程(ii)の反応が、テトラヒドロフランと水からなる溶媒系で行われる、〔6〕に記載の方法。
〔13〕Rがイソプロピルまたはプロピルである、〔6〕から〔12〕のいずれか1項に記載の方法。
〔14〕Rがイソプロピルである、〔6〕から〔12〕のいずれか1項に記載の方法。
〔15〕Rがプロピルである、〔6〕から〔12〕のいずれか1項に記載の方法。
〔16〕(i) 式(1):
で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させる工程および
(ii) 工程(i)の生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させる工程を含むことを特徴とする、一般式(2):
(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物の製造方法。
(ii) 工程(i)の生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させる工程を含むことを特徴とする、一般式(2):
で表わされる化合物の製造方法。
〔17〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系またはエーテル類と水からなる溶媒系で行われる、〔16〕に記載の方法。
〔18〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔16〕に記載の方法。
〔19〕工程(ii)の反応が、エーテル類と水からなる溶媒系で行われる、〔16〕に記載の方法。
〔20〕工程(ii)の反応が、トルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔16〕に記載の方法。
〔21〕工程(ii)の反応が、トルエンと水からなる溶媒系で行われる、〔16〕に記載の方法。
〔22〕工程(ii)の反応が、テトラヒドロフランと水からなる溶媒系で行われる、〔16〕に記載の方法。
〔23〕Rがイソプロピルまたはプロピルである、〔16〕から〔22〕のいずれか1項に記載の方法。
〔24〕Rがイソプロピルである、〔16〕から〔22〕のいずれか1項に記載の方法。
〔25〕Rがプロピルである、〔16〕から〔22〕のいずれか1項に記載の方法。
〔26〕(i) 式(1):
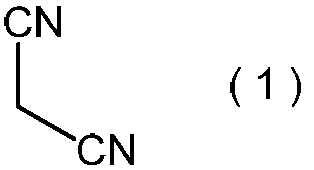
で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させて、一般式(4):
(式中、Mは水素またはアルカリ金属、あるいはそれらの混合物を示す。)
で表わされる化合物を製造する工程;
(ii) 一般式(4)で表わされる化合物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させ、一般式(2)
(式中、Rは前記と同意義である。)
で表わされる化合物を製造する工程および
(iii) 工程(ii)で得られる一般式(2)
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、
一般式(3):
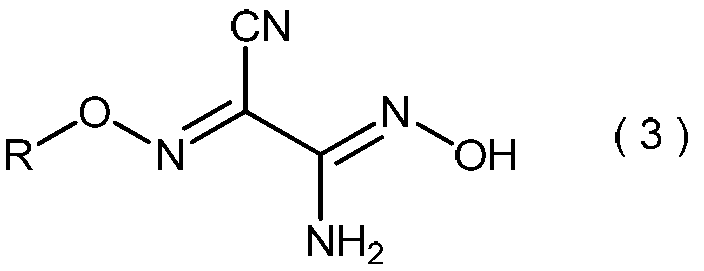
(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物の製造方法。
で表わされる化合物を製造する工程;
(ii) 一般式(4)で表わされる化合物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させ、一般式(2)
で表わされる化合物を製造する工程および
(iii) 工程(ii)で得られる一般式(2)
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、
一般式(3):
で表わされる化合物の製造方法。
〔27〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系またはエーテル類と水からなる溶媒系で行われる、〔26〕に記載の方法。
〔28〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔26〕に記載の方法。
〔29〕工程(ii)の反応が、エーテル類と水からなる溶媒系で行われる、〔26〕に記載の方法。
〔30〕工程(ii)の反応が、トルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔26〕に記載の方法。
〔31〕工程(ii)の反応が、トルエンと水からなる溶媒系で行われる、〔26〕に記載の方法。
〔32〕工程(ii)の反応が、テトラヒドロフランと水からなる溶媒系で行われる、〔26〕に記載の方法。
〔33〕Rがイソプロピルまたはプロピルである、〔26〕から〔32〕のいずれか1項に記載の方法。
〔34〕Rがイソプロピルである、〔26〕から〔32〕のいずれか1項に記載の方法。
〔35〕Rがプロピルである、〔26〕から〔32〕のいずれか1項に記載の方法。
〔36〕(i) 式(1):

で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させて、一般式(4):

(式中、Mは水素またはアルカリ金属、あるいはそれらの混合物を示す。)
で表わされる化合物を製造する工程および
(ii) 一般式(4)で表わされる化合物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させる工程
を含むことを特徴とする、一般式(2):

(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物の製造方法。
で表わされる化合物を製造する工程および
(ii) 一般式(4)で表わされる化合物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させる工程
を含むことを特徴とする、一般式(2):
で表わされる化合物の製造方法。
〔37〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系またはエーテル類と水からなる溶媒系で行われる、〔36〕に記載の方法。
〔38〕工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔36〕に記載の方法。
〔39〕工程(ii)の反応が、エーテル類と水からなる溶媒系で行われる、〔36〕に記載の方法。
〔40〕工程(ii)の反応が、トルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系で行われる、〔36〕に記載の方法。
〔41〕工程(ii)の反応が、トルエンと水からなる溶媒系で行われる、〔36〕に記載の方法。
〔42〕工程(ii)の反応が、テトラヒドロフランと水からなる溶媒系で行われる、〔36〕に記載の方法。
〔43〕Rがイソプロピルまたはプロピルである、〔36〕から〔42〕のいずれか1項に記載の方法。
〔44〕Rがイソプロピルである、〔36〕から〔42〕のいずれか1項に記載の方法。
〔45〕Rがプロピルである、〔36〕から〔42〕のいずれか1項に記載の方法。
〔46〕工程(ii)の溶媒系のpHが6から7の範囲である、〔6〕から〔45〕のいずれか1項に記載の方法。
〔47〕塩基を加えることにより、工程(ii)の溶媒系のpHを6から7の範囲に調整する、〔6〕から〔45〕のいずれか1項に記載の方法。
本明細書において用いられる用語および記号について以下に説明する。
「Ca~Cb」とは炭素原子数がa~b個であることを意味する。例えば、「C3~C6アルキル」の「C3~C6」とは、アルキルの炭素原子数が3~6であることを意味する
アルキルとしては、例えば、C3~C6アルキル、好ましくはC3アルキルが挙げられる。C3~C6アルキルとは、3~6個の炭素原子を有する直鎖または分岐鎖のアルキルを意味する。C3~C6アルキルとしては、具体的には例えば、プロピル、イソプロピル、ブチル、sec-ブチル、イソブチル、tert-ブチル、ペンチル、ヘキシル等、好ましくはプロピル、イソプロピル、ブチル、sec-ブチル、イソブチル、tert-ブチル、より好ましくはプロピル、イソプロピルが挙げられるが、これらに限定されるものではない。C3アルキルとは、3個の炭素原子を有する直鎖または分岐鎖のアルキルを意味する。C3アルキルとしては、具体的には例えば、プロピル、イソプロピルが挙げられる。
アルコキシとしては、例えば、C3~C6アルコキシ、好ましくはC3アルコキシが挙げられる。C3~C6アルコキシとは、(C3~C6アルキル)-O-を意味する(ここで、C3~C6アルキルは前述と同じ意味を有する。)。C3~C6アルコキシとしては、具体的には例えば、プロポキシ、イソプロポキシ、ブトキシ、sec-ブトキシ、イソブトキシ、tert-ブトキシ、ペンチルオキシ、ヘキシルオキシ等、好ましくはプロポキシ、イソプロポキシ、ブトキシ、sec-ブトキシ、イソブトキシ、tert-ブトキシ、より好ましくはプロポキシ、イソプロポキシが挙げられるが、これらに限定されるものではない。C3アルコキシとは、(C3アルキル)-O-を意味する(ここで、C3アルキルは前述と同じ意味を有する。)。C3アルコキシとしては、具体的には例えば、プロポキシ、イソプロポキシが挙げられる。
芳香族炭化水素誘導体類としては、例えば、ベンゼン、トルエン、キシレン、エチルベンゼン、クメン、トリメチルベンゼン、メチルイソプロピルベンゼン、メチルナフタレン、ジメチルナフタレン、エチルナフタレンクロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、テトラクロロベンゼンニトロベンゼン等、好ましくはトルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ニトロベンゼン、より好ましくはトルエン、キシレン、クロロベンゼン、さらに好ましくはトルエンが挙げられる。
脂肪族炭化水素類としては、例えば、ヘキサン、ヘプタン、オクタン、イソオクタン、ノナン、デカン、ウンデカン、ドデカン、イソドデカン、トリデカン、テトラデカン、ペンタデカン、ヘキサデカン、イソヘキサデカン、ヘプタデカン、オクタデカン、ノナデカン、エイコサン、イソエイコサン、トリアコンタン、シクロヘキサン、エチルシクロヘキサン、メチルイソプロピルシクロヘキサン、メチルデカリン、ジメチルデカリン、エチルデカリン等が挙げられる。
ハロゲン化脂肪族炭化水素類としては、例えば、ジクロロメタン、クロロホルム、四塩化炭素、1,2‐ジクロロエタン、1,3‐ジクロロプロパン等が挙げられる。
エーテル類としては、例えば、テトラヒドロフラン(THF)、2-メチルテトラヒドロフラン、ジエチルエーテル、ジ-n-プロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジ-tert-ブチルエーテル、ジフェニルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、1,2-ジメトキシエタン(DME)、ジグリム(diglyme)、トリグリム(triglyme)、1,4-ジオキサン等、好ましくはテトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、より好ましくはテトラヒドロフランが挙げられる。
アルコール類としては、例えば、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、tert-ブチルアルコール、ペンタノール、ヘキサノール、ヘプタノール、オクタノール、シクロヘキサノール、エチレングリコール、プロピレングリコール等が挙げられる。
ニトリル類としては、例えば、アセトニトリル、プロピオニトリル、ベンゾニトリル等が挙げられる。
アミド類としては、例えば、N,N-ジメチルホルムアミド(DMF)、N,N-ジエチルホルムアミド、N,N-ジメチルアセトアミド(DMAC)、N,N-ジエチルアセトアミド、N-メチルピロリドン(NMP)等が挙げられる。
アルキル尿素類としては、例えば、テトラメチル尿素、N,N’-ジメチルイミダゾリジノン(DMI)等が挙げられる。
スルホキシド類としては、例えば、ジメチルスルホキシド(DMSO)等が挙げられる。
スルホン類としては、例えば、スルホラン、ジメチルスルホン等が挙げられる。
炭酸エステル類としては、例えば、エチレンカーボネート、プロピレンカーボネート等が挙げられる。
ケトン類としては、例えば、アセトン、エチルメチルケトン、イソプロピルメチルケトン、イソブチルメチルケトン(MIBK)、シクロヘキサノン等が挙げられる。
カルボン酸エステル類としては、例えば、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸ブチル等が挙げられる。
カルボン酸類としては、例えば、ギ酸、酢酸、プロピオン酸等が挙げられる。
ニトロ化芳香族炭化水素類としては、例えば、ニトロベンゼン等が挙げられる。
芳香族複素環類としては、例えば、ピリジン等が挙げられる。
アルカリ金属水酸化物としては、例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム等が挙げられる。
アルカリ土類金属水酸化物としては、例えば、水酸化マグネシウム、水酸化カルシウム、水酸化バリウム等が挙げられる。
アルカリ金属炭酸塩としては、例えば、炭酸リチウム、炭酸ナトリウム、炭酸カリウム等が挙げられる。
アルカリ土類金属炭酸塩としては、炭酸マグネシウム、炭酸カルシウム、炭酸バリウム等が挙げられる。
アルカリ金属炭酸水素塩としては、例えば、炭酸水素リチウム、炭酸水素ナトリウム、炭酸水素カリウム等が挙げられる。
アルカリ土類金属炭酸水素塩としては、例えば、炭酸水素マグネシウム、炭酸水素カルシウム、炭酸水素バリウム等が挙げられる。
リン酸塩としては、例えば、リン酸ナトリウム、リン酸カリウム、リン酸カルシウム等が挙げられる。
リン酸水素塩としては、例えば、リン酸水素ナトリウム、リン酸水素カリウム、リン酸水素カルシウム等が挙げられる。
ピリジン類としては、例えば、ピリジン、4-ジメチルアミノピリジン、4-ピロリジノピリジン、2,6-ルチジン等が挙げられる。
キノリン類としては、例えば、キノリン等が挙げられる。
イソキノリン類としては、例えば、イソキノリン等が挙げられる。
3級アミン類としては、例えば、トリメチルアミン、トリエチルアミン、トリブチルアミン、ジイソプロピルエチルアミン、トリイソプロピルアミン等が挙げられる。
2級アミン類としては、例えば、ジエチルアミン、ジプロピルアミン、ジイソプロピルアミン等が挙げられる。
1級アミン類としては、例えば、プロピルアミン、ブチルアミン等が挙げられる。
芳香族アミン類としては、例えば、アニリン、N,N-ジメチルアニリン、N,N-ジエチルアニリン等が挙げられる。
環状アミン類としては、例えば、ピロリジン、ピペリジン、モルホリン、N-メチルモルホリン、ピペラジン、
1,8-ジアザビシクロ[5.4.0]-7-ウンデカ-7-エン(DBU)、1,5-ジアザビシクロ[4.3.0]ノナ-5-エン(DBN)、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)等が挙げられる。
1,8-ジアザビシクロ[5.4.0]-7-ウンデカ-7-エン(DBU)、1,5-ジアザビシクロ[4.3.0]ノナ-5-エン(DBN)、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)等が挙げられる。
カルボン酸アルカリ金属塩としては、例えば、ギ酸ナトリウム、ギ酸カリウム、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、プロピオン酸ナトリウム、プロピオン酸カリウム等が挙げられる。
カルボン酸アルカリ土類金属塩としては、例えば、酢酸マグネシウム、プロピオン酸マグネシウム、酢酸カルシウム、プロピオン酸カルシウム等が挙げられる。
(工程(i))
まず、工程(i)について説明する。
工程(i)は、式(1):
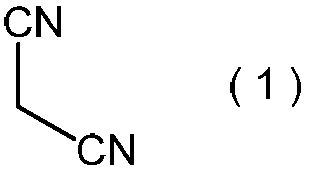
で表わされるマロノニトリルを、プロトン酸の存在下で、亜硝酸のアルカリ金属塩と反応させる工程である。
(原料;マロノニトリル)
本発明方法の原料として、上記式(1)で表わされるマロノニトリルを用いる。マロノニトリルは、公知化合物であり、工業的に比較的安価に入手可能であり、さらに取り扱いの面からも工業的原材料として好ましい。
(亜硝酸のアルカリ金属塩)
亜硝酸のアルカリ金属塩としては、例えば、亜硝酸ナトリウム、亜硝酸カリウム、亜硝酸リチウム等が挙げられるが、これらに限定されるものではない。価格および入手性等の観点から、亜硝酸ナトリウムまたは亜硝酸カリウムが好ましく、亜硝酸ナトリウムがより好ましい。
亜硝酸のアルカリ金属塩の形態は、反応が進行する限りは如何なる形態でもよい。亜硝酸のアルカリ金属塩の形態としては、例えば、亜硝酸のアルカリ金属塩のみの固体、または任意の濃度の水溶液若しくは水以外の溶媒の溶液等を挙げられる。また、亜硝酸のアルカリ金属塩は単独でまたは2種以上を任意の割合で混用しても良い。
(亜硝酸のアルカリ金属塩の使用量)
亜硝酸のアルカリ金属塩の使用量は、反応が進行する限りは何れの量でもよい。収率、副生成物抑制および経済効率等の観点から、式(1)で表わされるマロノニトリル1モルに対して、通常は0.8~2.0モル、好ましくは0.9~1.5モル、より好ましくは0.9~1.2モルの範囲を例示できる。
(プロトン酸)
プロトン酸としては、例えば、ハロゲン化水素酸、硫酸、硝酸、リン酸、ポリリン酸、カルボン酸、スルホン酸等が挙げられるが、これらに限定されるものではない。
ハロゲン化水素酸としては、例えば、塩酸、臭化水素酸、フッ化水素酸等が挙げられる。
カルボン酸としては、例えば、ギ酸、酢酸、プロピオン酸、トリフルオロ酢酸、安息香酸等が挙げられる。
スルホン酸としては、例えば、メタンスルホン酸、エタンスルホン酸、トリフルオロメタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸等が挙げられる。
価格、入手性、および反応性等の観点から、ハロゲン化水素酸またはカルボン酸が好ましく、塩酸または酢酸がより好ましい。
プロトン酸の形態は、反応が進行する限りは如何なる形態でもよい。プロトン酸の形態としては、例えば、プロトン酸のみの固体若しくは液体、または任意の濃度の水溶液若しくは水以外の溶媒の溶液等を挙げられる。また、プロトン酸は単独でまたは2種以上を任意の割合で混用しても良い。
(プロトン酸の使用量)
プロトン酸の使用量は、反応が進行する限りは何れの量でもよい。収率、副生成物抑制および経済効率等の観点から、式(1)で表わされるマロノニトリル1モルに対して、通常は0.005~3.0モル、好ましくは0.01~2.0モル、より好ましくは0.05~1.5モル、さらに好ましくは0.05~1.3モルの範囲を例示できる。また、プロトン酸の使用量の特に好ましい範囲は、酸の種類によって異なる。プロトン酸が塩酸であるときは、特に好ましくは0.9~1.2モルの範囲を例示できる。プロトン酸が酢酸であるときは、特に好ましくは0.05~1.2モルの範囲を例示できる。
(工程(i)の溶媒)
工程(i)の反応は、無溶媒で実施してもよいが、円滑な反応の進行などの観点から、溶媒を用いてもよい。工程(i)の溶媒は、工程(i)の反応が進行してかつ工程(ii)および(iii)の反応へ悪影響を及さない限りは、如何なる溶媒でもよい。工程(i)に用いることができる溶媒としては、例えば、水、エーテル類、アルコール類、ニトリル類、アミド類、アルキル尿素類、スルホキシド類、スルホン類、炭酸エステル類、ケトン類、カルボン酸エステル類、カルボン酸類、ニトロ化脂肪族炭化水素類、芳香族複素環類等、およびそれらの任意の混合割合の混合溶媒が挙げられるが、これらに限定されるものではない。
価格および取り扱いの容易さ等の観点から、工程(i)に用いる溶媒の好ましい例としては、エーテル類、アルコール類、ニトリル類、アミド類、アルキル尿素類、スルホキシド類、スルホン類、水、およびそれらの混合溶媒が挙げられ、より好ましくはエーテル類、アルコール類、ニトリル類、アミド類、水、およびそれらの混合溶媒が挙げられる。
工程(i)に用いる溶媒の具体的な好ましい例としては、テトラヒドロフラン(THF)、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、N,N’-ジメチルイミダゾリジノン(DMI)ジメチルスルホキシド(DMSO)、スルホラン、水、およびそれらの混合溶媒、より好ましくはテトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、水、およびそれらの混合溶媒、さらに好ましくは水が挙げられる。
工程(i)に用いる溶媒の具体的な好ましい例としては、テトラヒドロフラン(THF)、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、N,N’-ジメチルイミダゾリジノン(DMI)ジメチルスルホキシド(DMSO)、スルホラン、水、およびそれらの混合溶媒、より好ましくはテトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、水、およびそれらの混合溶媒、さらに好ましくは水が挙げられる。
(工程(i)の溶媒の使用量)
工程(i)の溶媒の使用量は、反応系の撹拌が充分にできる限りは何れの量でもよい。反応性、副生成物抑制および経済効率等の観点から、式(1)で表わされるマロノニトリル1モルに対して、通常は0(ゼロ)~10.0L(リットル)、好ましくは0.01~5.0L、より好ましくは0.05~3.0L、さらに好ましくは0.1~2.0Lの範囲を例示できる。
(工程(i)の反応温度)
工程(i)における反応温度は、特に制限されない。
収率、副生成物抑制および経済効率等の観点から、通常は-30℃(マイナス30℃)~90℃、好ましくは-20℃(マイナス20℃)~70℃、より好ましくは-10℃(マイナス10℃)~30℃、さらに好ましくは-5℃(マイナス5℃)~20℃の範囲を例示できる。
収率、副生成物抑制および経済効率等の観点から、通常は-30℃(マイナス30℃)~90℃、好ましくは-20℃(マイナス20℃)~70℃、より好ましくは-10℃(マイナス10℃)~30℃、さらに好ましくは-5℃(マイナス5℃)~20℃の範囲を例示できる。
(工程(i)の反応時間)
工程(i)における反応時間は、特に制限されない。収率、副生成物抑制および経済効率等の観点から、通常は0.5時間~48時間、好ましくは0.5時間~24時間、より好ましくは0.5時間~12時間の範囲を例示できる。
(工程(i)の生成物)
本発明における「工程(i)の生成物」の形態は、工程(i)の反応混合物、並びに可能である場合は工程(i)の反応生成物が単離されてかつ精製されていない粗生成物および工程(i)の反応生成物が単離されてかつ精製された物質等を包含するが、これらに限定されるものではない。したがって、工程(ii)へ用いられる工程(i)の生成物の形態も限定されないが、経済効率および安全性等の観点から、「工程(i)の生成物」としては、好ましくは工程(i)の反応混合物が工程(ii)へ用いられる。
本発明における反応機構等は明らかではない。しかしながら、本発明が完成した後で本発明を考察したときに、工程(i)の生成物は下記式(8)で表わされる2‐ヒドロキシイミノプロパンジニトリルまたはその塩、あるいはそれらの混合物であると推定される。
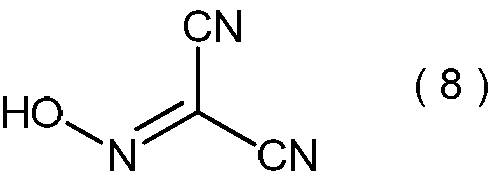
本発明における反応機構等は明らかではない。しかしながら、本発明が完成した後で本発明を考察したときに、工程(i)の生成物は下記式(8)で表わされる2‐ヒドロキシイミノプロパンジニトリルまたはその塩、あるいはそれらの混合物であると推定される。
言い換えれば、工程(i)の生成物は、一般式(4):
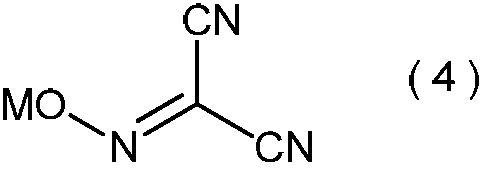
(式中、Mは水素またはアルカリ金属、あるいはそれらの混合物を示す。)
で表わされる化合物であると推定される。
で表わされる化合物であると推定される。
アルカリ金属としては、例えば、ナトリウム、カリウム、リチウム等が挙げられる。
上記の一般式(4)で表わされる化合物の形態についての説明は、「工程(i)の生成物」についての説明に準じる。
上記の一般式(4)で表わされる化合物の形態についての説明は、「工程(i)の生成物」についての説明に準じる。
(工程(ii))
次に、工程(ii)について説明する。
工程(ii)は、工程(i)の生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させ、

(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物を製造する工程である。
で表わされる化合物を製造する工程である。
(相間移動触媒)
相間移動触媒としては、例えば、四級アンモニウム塩、四級ホスホニウム塩、クラウンエーテル類等が挙げられるが、これらに限定されるものではない。
四級アンモニウム塩としては、例えば、テトラメチルアンモニウムクロリド、テトラメチルアンモニウムブロミド、テトラエチルアンモニウムクロリド、テトラエチルアンモニウムブロミド、テトラブチルアンモニウムフルオリド、テトラブチルアンモニウムクロリド、テトラブチルアンモニウムブロミド、テトラブチルアンモニウムヨージド、硫酸水素テトラブチルアンモニウム、水酸化テトラブチルアンモニウム、ベンジルトリメチルアンモニウムクロリド、ベンジルトリメチルアンモニウムブロミド、水酸化ベンジルトリメチルアンモニウム、ベンジルトリエチルアンモニウムクロリド、ベンジルトリエチルアンモニウムブロミド、オクチルトリメチルアンモニウムクロリド、オクチルトリメチルアンモニウムブロミド、トリオクチルメチルアンモニウムクロリド、トリオクチルメチルアンモニウムブロミド、トリオクチルエチルアンモニウムクロリド、トリオクチルエチルアンモニウムブロミド、ラウリルトリメチルアンモニウムクロリド(ドデシルトリメチルアンモニウムクロリド)、ラウリルトリメチルアンモニウムブロミド(ドデシルトリメチルアンモニウムブロミド)、ベンジルラウリルジメチルアンモニウムクロリド(ベンジルドデシルジメチルアンモニウムクロリド)、ベンジルラウリルジメチルアンモニウムブロミド(ベンジルドデシルジメチルアンモニウムブロミド)、ミリスチルトリメチルアンモニウムクロリド(テトラデシルトリメチルアンモニウムクロリド)、ミリスチルトリメチルアンモニウムブロミド(テトラデシルトリメチルアンモニウムブロミド)、ベンジルジメチルステアリルアンモニウムクロリド(ベンジルオクタデシルジメチルアンモニウムクロリド)、ベンジルジメチルステアリルアンモニウムブロミド(ベンジルオクタデシルジメチルアンモニウムブロミド)、等が挙げられる。
クラウンエーテル類としては、例えば、12-クラウン-4、15-クラウン-5、18-クラウン-6等が挙げられる。
四級ホスホニウム塩としては、例えば、テトラブチルホスホニウムブロミド、テトラオクチルホスホニウムブロミド、テトラフェニルホスホニウムブロミド等が挙げられる。
価格、入手性、および反応性等の観点から、相間移動触媒は、好ましくは四級アンモニウム塩、より好ましくはテトラブチルアンモニウムブロミド、硫酸水素テトラブチルアンモニウム、ベンジルトリメチルアンモニウムクロリド、ベンジルトリメチルアンモニウムブロミド、トリオクチルメチルアンモニウムクロリド、トリオクチルメチルアンモニウムブロミド、ベンジルラウリルジメチルアンモニウムクロリド、ミリスチルトリメチルアンモニウムブロミド、ベンジルジメチルステアリルアンモニウムクロリド、ベンジルジメチルステアリルアンモニウムブロミド等、さらに好ましくはテトラブチルアンモニウムブロミド、ベンジルトリメチルアンモニウムクロリド、特に好ましくはテトラブチルアンモニウムブロミドである。
相間移動触媒の形態は、反応が進行する限りは如何なる形態でもよい。相間移動触媒の形態としては、例えば、相間移動触媒のみの固体若しくは液体、または任意の濃度の水溶液若しくは水以外の溶媒の溶液等を挙げられる。また、相間移動触媒は単独でまたは2種以上を任意の割合で混用しても良い。
(相間移動触媒の使用量)
相間移動触媒の使用量は、反応が進行する限りは何れの量でもよい。収率、副生成物抑制および経済効率等の観点から、式(1)のマロノニトリル1モルに対して、通常は0.001~1.0モル、好ましくは0.005~0.3モル、より好ましくは0.01~0.2モルの範囲を例示できる。
(C3~C6アルキルハライド)
C3~C6アルキルハライドとは、3~6個の炭素原子を有する直鎖または分岐鎖のアルキルハライドを意味する。C3~C6アルキルハライドとしては、例えば、プロピルクロリド、イソプロピルクロリド、ブチルクロリド、sec-ブチルクロリド、イソブチルクロリド、tert-ブチルクロリド、ペンチルクロリド、1-メチルブチルクロリド、2-メチルブチルクロリド、3-メチルブチルクロリド、1-エチルプロピルクロリド、1,1-ジメチルプロピルクロリド、1,2-ジメチルプロピルクロリド、2,2-ジメチルプロピルクロリド、ヘキシルクロリド、1-メチルペンチルクロリド、2-メチルペンチルクロリド、3-メチルペンチルクロリド、4-メチルペンチルクロリド、1,2-ジメチルブチルクロリド、1,3-ジメチルブチルクロリド、1-エチルブチルクロリド、2-エチルブチルクロリド、1-メチル-2,2-ジメチルプロピルクロリド、1,1-ジメチル-2-メチルプロピルクロリド、1-エチル-1-メチルプロピルクロリド、プロピルブロミド、イソプロピルブロミド、ブチルブロミド、sec-ブチルブロミド、イソブチルブロミド、tert-ブチルブロミド、ペンチルブロミド、1-メチルブチルブロミド、2-メチルブチルブロミド、3-メチルブチルブロミド、4-メチルペンチルブロミド、1-エチルプロピルブロミド、1,1-ジメチルプロピルブロミド、1,2-ジメチルプロピルブロミド、2,2-ジメチルプロピルブロミド、ヘキシルブロミド、1-メチルペンチルブロミド、2-メチルペンチルブロミド、3-メチルペンチルブロミド、1,2-ジメチルブチルブロミド、1,3-ジメチルブチルブロミド、1-エチルブチルブロミド、2-エチルブチルブロミド、1-メチル-2,2-ジメチルプロピルブロミド、1,1-ジメチル-2-メチルプロピルブロミド、1-エチル-1-メチルプロピルブロミド、プロピルヨージド、イソプロピルヨージド、ブチルヨージド、sec-ブチルヨージド、イソブチルヨージド、tert-ブチルヨージド、ペンチルヨージド、1-メチルブチルヨージド、2-メチルブチルヨージド、3-メチルブチルヨージド、1-エチルプロピルヨージド、1,1-ジメチルプロピルヨージド、1,2-ジメチルプロピルヨージド、2,2-ジメチルプロピルヨージド、ヘキシルヨージド、1-メチルペンチルヨージド、2-メチルペンチルヨージド、3-メチルペンチルヨージド、4-メチルペンチルヨージド、1,2-ジメチルブチルヨージド、1,3-ジメチルブチルヨージド、1-エチルブチルヨージド、2-エチルブチルヨージド、1-メチル-2,2-ジメチルプロピルヨージド、1,1-ジメチル-2-メチルプロピルヨージド、1-エチル-1-メチルプロピルヨージド等が挙げられる。
価格、入手性、反応性、生成物の有用性等の観点から、C3~C6アルキルハライドは、好ましくはプロピルクロリド、イソプロピルクロリド、ブチルクロリド、sec-ブチルクロリド、イソブチルクロリド、tert-ブチルクロリド、プロピルブロミド、イソプロピルブロミド、ブチルブロミド、sec-ブチルブロミド、イソブチルブロミド、tert-ブチルブロミド、より好ましくはプロピルクロリド、イソプロピルクロリド、プロピルブロミド、イソプロピルブロミド、さらに好ましくはプロピルブロミド、イソプロピルブロミドである。
(C3~C6アルキルハライドの使用量)
C3~C6アルキルハライドの使用量は、反応が進行する限りは何れの量でもよい。収率、副生成物抑制および経済効率等の観点から、式(1)のマロノニトリル1モルに対して、通常は0.8~3.0モル、好ましくは0.9~2.0モル、より好ましくは0.9~1.5モルの範囲を例示できる。
(工程(ii)のpH)
工程(ii)においては、反応を円滑に進行させるために、溶媒系のpHを調整してもよい。反応が円滑に進行する限りは、当該pHの調整は行ってもよく、また行わなくてもよい。さらに、反応が円滑に進行する限りは、当該pHの調整は、工程(ii)のいつ行ってもよい。例えば、当該pHの調整は、C3~C6アルキルハライドを加える前に行ってもよく、C3~C6アルキルハライドの滴下中に行ってもよい。
工程(ii)の溶媒系のpHは、反応が進行する限りは何れの範囲でもよい。収率、副生成物抑制および経済効率等の観点から、通常はpH4~9、好ましくはpH4~8、より好ましくはpH5~8、さらに好ましくはpH5~7、特に好ましくはpH6~7の範囲を例示できる。
(工程(ii)の塩基)
上記のように工程(ii)において溶媒系のpHを調整するためには、塩基を用いることができる。本発明における反応機構等は明らかではない。しかしながら、本発明が完成した後で本発明を考察した結果、次のように推測された。工程(i)で使用されるプロトン酸が酢酸以上の強酸である場合に、塩基を使用する必要があると推測された。特に、工程(i)で使用されるプロトン酸が酢酸以上の強酸であり、かつ当該プロトン酸の使用量がマロノニトリル1モルに対して0.1当量より多い場合に、塩基を使用する必要があると推測された。
工程(ii)に用いることができる塩基としては、例えば、アルカリ金属水酸化物、アルカリ土類金属水酸化物、アルカリ金属炭酸塩、アルカリ土類金属炭酸塩、アルカリ金属炭酸水素塩、アルカリ土類金属炭酸水素塩等の無機塩基類、ピリジン類、キノリン類、イソキノリン類、3級アミン類、2級アミン類、1級アミン類、芳香族アミン類、環状アミン類、カルボン酸アルカリ金属塩、カルボン酸アルカリ土類金属塩等の有機塩基類が挙げられるが、これらに限定されるものではない。
反応性、収率、価格および取り扱いの容易さ等の観点から、工程(ii)に用いる塩基の好ましい例としては、アルカリ金属水酸化物、アルカリ金属炭酸塩、アルカリ金属炭酸水素塩、3級アミン類、カルボン酸アルカリ金属塩、より好ましくはアルカリ金属水酸化物、アルカリ金属炭酸塩、アルカリ金属炭酸水素塩さらに好ましくはアルカリ金属水酸化物が挙げられる。工程(ii)に用いる塩基の具体的な好ましい例としては、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、トリエチルアミン、酢酸ナトリウム、酢酸カリウム、より好ましくは水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、さらに好ましくは水酸化ナトリウム、水酸化カリウムが挙げられる。
工程(ii)に用いる塩基の形態は、反応が進行する限りは如何なる形態でもよい。工程(ii)に用いる塩基の形態としては、例えば、塩基のみの固体若しくは液体、または任意の濃度の水溶液若しくは水以外の溶媒の溶液等を挙げられる。また、工程(ii)に用いる塩基は単独でまたは2種以上を任意の割合で混用しても良い。
(工程(ii)の塩基の使用量)
工程(ii)の塩基は、反応が進行する限りは、使用してもよく、また使用しなくてもよい。工程(ii)で塩基を使用する場合、収率、副生成物抑制および経済効率等の観点から、塩基の使用量としては、溶媒系のpHが通常はpH4~9、好ましくはpH4~8、より好ましくはpH5~8、さらに好ましくはpH5~7、特に好ましくはpH6~7に調整される量が言及できる。
(工程(ii)の溶媒系)
工程(ii)の溶媒について説明する。工程(ii)に用いることができる溶媒系としては、例えば、芳香族炭化水素誘導体類、脂肪族炭化水素類、ハロゲン化脂肪族炭化水素類、エーテル類、アルコール類、ニトリル類、アミド類、アルキル尿素類、スルホキシド類、スルホン類、ケトン類、カルボン酸エステル類、カルボン酸類、芳香族複素環類、水、および任意の混合割合のそれらからなる溶媒系が挙げられる。
価格、取り扱いの容易さ、反応性および収率等の観点から、工程(ii)に用いる溶媒系の好ましい例としては、芳香族炭化水素誘導体類、ハロゲン化脂肪族炭化水素類、エーテル類、アルコール類、ニトリル類、アミド類、アルキル尿素類、スルホキシド類、スルホン類、水、およびそれらからなる溶媒系、より好ましくは芳香族炭化水素誘導体類、ハロゲン化脂肪族炭化水素類、エーテル類、アルコール類、ニトリル類、アミド類、水、およびそれらからなる溶媒系、さらに好ましくは芳香族炭化水素誘導体類と水からなる溶媒系、エーテル類と水からなる溶媒系が挙げられる。
工程(ii)に用いる溶媒系の具体的な好ましい例としては、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ニトロベンゼン、ジクロロメタン、テトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、N,N’-ジメチルイミダゾリジノン(DMI)、ジメチルスルホキシド(DMSO)、スルホラン、水、およびそれらからなる溶媒系、より好ましくはトルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ニトロベンゼン、ジクロロメタン、テトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、水、およびそれらからなる溶媒系、さらに好ましくはトルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ニトロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系、テトラヒドロフラン、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテルからなる群より選択される1種以上のエーテル類と水からなる溶媒系、さらに好ましくはトルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系、テトラヒドロフランと水からなる溶媒系、さらに好ましくはトルエンと水からなる溶媒系が挙げられる。
工程(ii)に用いる溶媒系の具体的な好ましい例としては、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ニトロベンゼン、ジクロロメタン、テトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、N,N’-ジメチルイミダゾリジノン(DMI)、ジメチルスルホキシド(DMSO)、スルホラン、水、およびそれらからなる溶媒系、より好ましくはトルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ニトロベンゼン、ジクロロメタン、テトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、水、およびそれらからなる溶媒系、さらに好ましくはトルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ニトロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系、テトラヒドロフラン、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテルからなる群より選択される1種以上のエーテル類と水からなる溶媒系、さらに好ましくはトルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系、テトラヒドロフランと水からなる溶媒系、さらに好ましくはトルエンと水からなる溶媒系が挙げられる。
(工程(ii)の溶媒の使用量)
工程(ii)の溶媒系を形成する溶媒の使用量としては、反応系の撹拌が充分にできる限りは何れの量でもよい。反応性、副生成物抑制および経済効率等の観点から、式(1)のマロノニトリル1モルに対して、水の量が通常は0(ゼロ)~10.0L(リットル)、好ましくは0.01~10.0L、より好ましくは0.1~5.0L、さらに好ましくは0.2~3.0Lの範囲を例示できる。さらに、同様の観点から、式(1)のマロノニトリル1モルに対して、水以外の上記した溶媒の量が通常は0(ゼロ)~10.0L(リットル)、好ましくは0.01~10.0L、より好ましくは0.1~5.0L、さらに好ましくは0.2~3.0Lの範囲を例示できる。なお、水および水以外の溶媒の混合割合は、反応が進行する限りはいずれの割合でもよい。2種以上の水以外の溶媒を用いるときは、2種以上の水以外の溶媒の混合割合は、反応が進行する限りはいずれの割合でもよい。
(工程(ii)の反応温度)
工程(ii)における反応温度は、特に制限されない。収率、副生成物抑制および経済効率等の観点から、通常は10℃~100℃、好ましくは40℃~95℃、より好ましくは50℃~95℃、さらに好ましくは55℃~90℃の範囲を例示できる。
(工程(ii)の反応時間)
工程(ii)における反応時間は、特に制限されない。収率、副生成物抑制および経済効率等の観点から、通常は0.5時間~48時間、好ましくは0.5時間~24時間、より好ましくは1時間~12時間の範囲を例示できる。
(工程(ii)の生成物;一般式(2)で表わされる2-アルコキシイミノプロパンジニトリル)
工程(ii)で得られる一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルは新規化合物である。
一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルとしては、具体的には例えば、
2-プロポキシイミノプロパンジニトリル、
2-イソプロポキシイミノプロパンジニトリル、
2-ブトキシイミノプロパンジニトリル、
2-sec-ブトキシイミノプロパンジニトリル、
2-イソブトキシイミノプロパンジニトリル、
2-tert-ブトキシイミノプロパンジニトリル、
2-ペンチルオキシイミノプロパンジニトリル、
2-(1-メチルブチルオキシイミノ)プロパンジニトリル、
2-(2-メチルブチルオキシイミノ)プロパンジニトリル、
2-(3-メチルブチルオキシイミノ)プロパンジニトリル、
2-(1-エチルプロピルオキシイミノ)プロパンジニトリル、
2-(1,1-ジメチルプロピルオキシイミノ)プロパンジニトリル、
2-(1,2-ジメチルプロピルオキシイミノ)プロパンジニトリル、
2-(2,2-ジメチルプロピルオキシイミノプロパンジニトリル、
2-ヘキシルオキシイミノプロパンジニトリル、
2-(1-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(2-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(3-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(4-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(1,2-ジメチルブチルオキシイミノ)プロパンジニトリル、
2-(1,3-ジメチルブチルオキシイミノ)プロパンジニトリル、
2-(1-エチルブチルオキシイミノ)プロパンジニトリル、
2-(2-エチルブチルオキシイミノ)プロパンジニトリル、
2-(1-メチル-2,2-ジメチルプロピルオキシイミノ)プロパンジニトリル、
2-(1,1-ジメチル-2-メチルプロピルオキシイミノ)プロパンジニトリル、
2-(1-エチル-1-メチルプロピルオキシイミノ)プロパンジニトリル
等が挙げられる。
化合物の有用性等の観点から、2-プロポキシイミノプロパンジニトリル、2-イソプロポキシイミノプロパンジニトリル、2-ブトキシイミノプロパンジニトリル、2-sec-ブトキシイミノプロパンジニトリル、2-イソブトキシイミノプロパンジニトリル、および2-tert-ブトキシイミノプロパンジニトリルが好ましく、2-プロポキシイミノプロパンジニトリルおよび2-イソプロポキシイミノプロパンジニトリルがより好ましい。
一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルとしては、具体的には例えば、
2-プロポキシイミノプロパンジニトリル、
2-イソプロポキシイミノプロパンジニトリル、
2-ブトキシイミノプロパンジニトリル、
2-sec-ブトキシイミノプロパンジニトリル、
2-イソブトキシイミノプロパンジニトリル、
2-tert-ブトキシイミノプロパンジニトリル、
2-ペンチルオキシイミノプロパンジニトリル、
2-(1-メチルブチルオキシイミノ)プロパンジニトリル、
2-(2-メチルブチルオキシイミノ)プロパンジニトリル、
2-(3-メチルブチルオキシイミノ)プロパンジニトリル、
2-(1-エチルプロピルオキシイミノ)プロパンジニトリル、
2-(1,1-ジメチルプロピルオキシイミノ)プロパンジニトリル、
2-(1,2-ジメチルプロピルオキシイミノ)プロパンジニトリル、
2-(2,2-ジメチルプロピルオキシイミノプロパンジニトリル、
2-ヘキシルオキシイミノプロパンジニトリル、
2-(1-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(2-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(3-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(4-メチルペンチルオキシイミノ)プロパンジニトリル、
2-(1,2-ジメチルブチルオキシイミノ)プロパンジニトリル、
2-(1,3-ジメチルブチルオキシイミノ)プロパンジニトリル、
2-(1-エチルブチルオキシイミノ)プロパンジニトリル、
2-(2-エチルブチルオキシイミノ)プロパンジニトリル、
2-(1-メチル-2,2-ジメチルプロピルオキシイミノ)プロパンジニトリル、
2-(1,1-ジメチル-2-メチルプロピルオキシイミノ)プロパンジニトリル、
2-(1-エチル-1-メチルプロピルオキシイミノ)プロパンジニトリル
等が挙げられる。
化合物の有用性等の観点から、2-プロポキシイミノプロパンジニトリル、2-イソプロポキシイミノプロパンジニトリル、2-ブトキシイミノプロパンジニトリル、2-sec-ブトキシイミノプロパンジニトリル、2-イソブトキシイミノプロパンジニトリル、および2-tert-ブトキシイミノプロパンジニトリルが好ましく、2-プロポキシイミノプロパンジニトリルおよび2-イソプロポキシイミノプロパンジニトリルがより好ましい。
工程(iii)へ用いられる上記の一般式(2)で表わされる化合物すなわち2-アルコキシイミノプロパンジニトリルの形態は、工程(ii)の反応生成物が単離されてかつ精製された物質、工程(ii)の反応生成物が単離されてかつ精製されていない粗生成物、工程(ii)の反応混合物等を包含するが、これらに限定されるものではない。したがって、上記の一般式(2)で表わされる化合物すなわち2-アルコキシイミノプロパンジニトリルは、単離されてまたは単離されないで、工程(iii)へ用いられてもよい。加えて、上記の一般式(2)で表わされる化合物すなわち2-アルコキシイミノプロパンジニトリルは、精製されてまたは精製されないで、工程(iii)へ用いられてもよい。
(工程(i)と工程(ii)の収率)
工程(i)と工程(ii)を通した収率としては、通常は60%以上、好ましくは65~100%、より好ましくは70~100%の範囲を例示できる。
工程(i)と工程(ii)を通した収率は、原料としての式(1)で表わされるマロノニトリルのモル数に対する、得られる一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルのモル数から計算することができる。
すなわち、この収率は、次の式で表される;
収率(%)=100×{(得られた一般式(2)で表わされる化合物のモル数)/(原料としての式(1)で表わされるマロノニトリルのモル教)}
すなわち、この収率は、次の式で表される;
収率(%)=100×{(得られた一般式(2)で表わされる化合物のモル数)/(原料としての式(1)で表わされるマロノニトリルのモル教)}
(工程(iii))
次に、工程(iii)について説明する。
工程(iii)は、一般式(2):

(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物を、ヒドロキシルアミン化合物と反応させ、一般式(3):
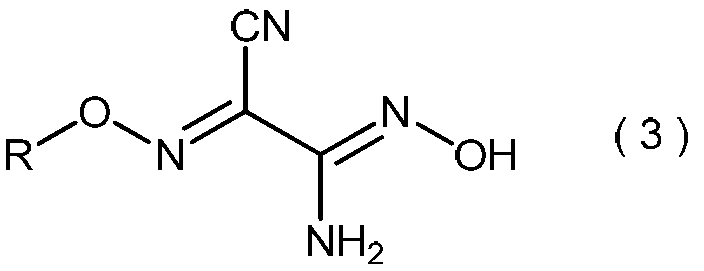
(式中、RはC3~C6アルキル基を示す。)
で表わされる化合物を製造する工程である。
で表わされる化合物を、ヒドロキシルアミン化合物と反応させ、一般式(3):
で表わされる化合物を製造する工程である。
(ヒドロキシルアミン化合物)
ヒドロキシルアミン化合物としては、例えば、ヒドロキシルアミンおよびヒドロキシルアミン塩が挙げられる。ヒドロキシルアミン塩としては、例えば、ヒドロキシルアミン硫酸塩またはヒドロキシルアミン塩酸塩が挙げられるが、これらに限定されるものではない。ヒドロキシルアミンについては、危険性を考慮して、通常はヒドロキシルアミンの水溶液が使用される。当該水溶液の濃度は、通常60%以下が好ましい。ヒドロキシルアミン塩の形態は、反応が進行する限りは如何なる形態でもよい。ヒドロキシルアミン塩の形態としては、例えば、ヒドロキシルアミン塩のみの固体、または任意の濃度の水溶液若しくは水以外の溶媒の溶液等を挙げられる。価格、入手性および安全性の観点から、ヒドロキシルアミン塩の形態は水溶液が好ましい。また、ヒドロキシルアミン化合物は単独でまたは2種以上を任意の割合で混用しても良い。
(ヒドロキシルアミン化合物の使用量)
ヒドロキシルアミン化合物の使用量は、反応が進行する限りは何れの量でもよい。収率、副生成物抑制および経済効率等の観点から、式(2)で表わされる化合物1モルに対して、通常は0.8~2.0モル、好ましくは0.9~1.5モル、より好ましくは0.9~1.3モル、さらに好ましくは0.9~1.2モルの範囲を例示できる。
(工程(iii)の塩基)
ヒドロキシルアミン化合物としてヒドロキシルアミン塩を使用するときには、塩基を使用することが好ましい。
工程(iii)に用いることができる塩基としては、例えば、アルカリ金属水酸化物、アルカリ土類金属水酸化物、アルカリ金属炭酸塩、アルカリ土類金属炭酸塩、アルカリ金属炭酸水素塩、アルカリ土類金属炭酸水素塩等の無機塩基類、ピリジン類、キノリン類、イソキノリン類、3級アミン類、2級アミン類、1級アミン類、芳香族アミン類、環状アミン類、カルボン酸アルカリ金属塩、カルボン酸アルカリ土類金属塩等の有機塩基類が挙げられるが、これらに限定されるものではない。
反応性、収率、価格および取り扱いの容易さ等の観点から、工程(iii)に用いる塩基の好ましい例としては、アルカリ金属水酸化物、アルカリ金属炭酸塩、アルカリ金属炭酸水素塩、カルボン酸アルカリ金属塩、より好ましくはアルカリ金属水酸化物、アルカリ金属炭酸塩、アルカリ金属炭酸水素塩、さらに好ましくはアルカリ金属炭酸塩、アルカリ金属炭酸水素塩が挙げられる。工程(iii)に用いる塩基の具体的な好ましい例としては、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、酢酸ナトリウム、酢酸カリウム、より好ましくは水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、さらに好ましくは炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウムが挙げられる。
工程(iii)に用いる塩基の形態は、反応が進行する限りは如何なる形態でもよい。工程(iii)に用いる塩基の形態としては、例えば、塩基のみの固体若しくは液体、または任意の濃度の水溶液若しくは水以外の溶媒の溶液等を挙げられる。また、工程(iii)に用いる塩基は単独でまたは2種以上を任意の割合で混用しても良い。
(工程(iii)の塩基の使用量)
工程(iii)の塩基の使用量は、反応が進行する限りは何れの量でもよい。収率、副生成物抑制および経済効率等の観点から、式(2)の化合物1モルに対して、通常は0.8~2.0当量、好ましくは0.9~1.5当量、より好ましくは0.9~1.3当量、さらに好ましくは0.9~1.2当量の範囲を例示できる。
(工程(iii)の溶媒)
工程(iii)の反応は、無溶媒で実施してもよいが、円滑な反応の進行などの観点から、溶媒を使用することが好ましい。工程(iii)の溶媒は、工程(iii)の反応が進行する限りは、如何なる溶媒でもよい。工程(iii)に用いることができる溶媒としては、例えば、水、エーテル類、アルコール類、ニトリル類、アミド類、アルキル尿素類、スルホキシド類、スルホン類、炭酸エステル類、ケトン類、カルボン酸エステル類、カルボン酸類、ニトロ化脂肪族炭化水素類、芳香族複素環類等、およびそれらの任意の混合割合の混合溶媒が挙げられるが、これらに限定されるものではない。
価格および取り扱いの容易さ等の観点から、工程(iii)に用いる溶媒の好ましい例としては、エーテル類、アルコール類、ニトリル類、アミド類、アルキル尿素類、スルホキシド類、スルホン類、水、およびそれらの混合溶媒、より好ましくはエーテル類、アルコール類、ニトリル類、アミド類、水、およびそれらの混合溶媒、さらに好ましくはアルコール類、水、およびそれらの混合溶媒が挙げられる。
工程(iii)に用いる溶媒の具体的な好ましい例としては、テトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、N,N’-ジメチルイミダゾリジノン(DMI)、ジメチルスルホキシド(DMSO)、スルホラン、水、およびそれらの混合溶媒、より好ましくはテトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、水、およびそれらの混合溶媒、さらに好ましくはメタノール、エタノール、プロパノール、2-プロパノール、水、およびそれらの混合溶媒、特に好ましくはメタノールおよび水の混合溶媒が挙げられる。
工程(iii)に用いる溶媒の具体的な好ましい例としては、テトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、N,N’-ジメチルイミダゾリジノン(DMI)、ジメチルスルホキシド(DMSO)、スルホラン、水、およびそれらの混合溶媒、より好ましくはテトラヒドロフラン(THF)、ジイソプロピルエーテル、ジブチルエーテル、シクロペンチルメチルエーテル(CPME)、メチル-tert-ブチルエーテル、メタノール、エタノール、プロパノール、2-プロパノール、ブタノール、アセトニトリル、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド(DMAC)、N-メチルピロリドン(NMP)、水、およびそれらの混合溶媒、さらに好ましくはメタノール、エタノール、プロパノール、2-プロパノール、水、およびそれらの混合溶媒、特に好ましくはメタノールおよび水の混合溶媒が挙げられる。
(工程(iii)の溶媒の使用量)
工程(iii)の溶媒の使用量は、反応系の撹拌が充分にできる限りは何れの量でもよい。反応性、副生成物抑制および経済効率等の観点から、式(1)のマロノニトリル1モルに対して、通常は0(ゼロ)~10.0L(リットル)、好ましくは0.01~10.0L、より好ましくは0.1~5.0L、さらに好ましくは0.2~3.0Lの範囲を例示できる。
(工程(iii)の反応温度)
工程(iii)における反応温度は、特に制限されない。収率、副生成物抑制および経済効率等の観点から、通常は-30℃(マイナス30℃)~100℃、好ましくは-20℃(マイナス20℃)~70℃、より好ましくは-10℃(マイナス10℃)~50℃、さらに好ましくは-10℃(マイナス10℃)~35℃の範囲を例示できる。
(工程(iii)の反応時間)
工程(iii)における反応時間は、特に制限されない。収率、副生成物抑制および経済効率等の観点から、通常は0.5時間~48時間、好ましくは0.5時間~24時間、より好ましくは1時間~12時間の範囲を例示できる。
(工程(iii)の生成物;一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニド)
工程(iii)で得られる一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドとしては、具体的には例えば、
2-アミノ-2-ヒドロキシイミノ-N-プロポキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-イソプロポキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-ブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-sec-ブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-イソブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-tert-ブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-ペンチルオキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-メチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2-メチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(3-メチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-エチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,1-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,2-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2,2-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-ヘキシルオキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(3-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(4-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,2-ジメチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,3-ジメチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-エチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2-エチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-メチル-2,2-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,1-ジメチル-2-メチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-エチル-1-メチルプロピルオキシ)アセトイミドイルシアニド
等が挙げられる。
2-アミノ-2-ヒドロキシイミノ-N-プロポキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-イソプロポキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-ブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-sec-ブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-イソブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-tert-ブトキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-ペンチルオキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-メチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2-メチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(3-メチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-エチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,1-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,2-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2,2-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-ヘキシルオキシアセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(3-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(4-メチルペンチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,2-ジメチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,3-ジメチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-エチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(2-エチルブチルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-メチル-2,2-ジメチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1,1-ジメチル-2-メチルプロピルオキシ)アセトイミドイルシアニド、
2-アミノ-2-ヒドロキシイミノ-N-(1-エチル-1-メチルプロピルオキシ)アセトイミドイルシアニド
等が挙げられる。
化合物の有用性等の観点から、2-アミノ-2-ヒドロキシイミノ-N-プロポキシアセトイミドイルシアニド、2-アミノ-2-ヒドロキシイミノ-N-イソプロポキシアセトイミドイルシアニド、2-アミノ-2-ヒドロキシイミノ-N-ブトキシアセトイミドイルシアニド、2-アミノ-2-ヒドロキシイミノ-N-sec-ブトキシアセトイミドイルシアニド、2-アミノ-2-ヒドロキシイミノ-N-イソブトキシアセトイミドイルシアニドおよび2-アミノ-2-ヒドロキシイミノ-N-tert-ブトキシアセトイミドイルシアニドが好ましく、2-アミノ-2-ヒドロキシイミノ-N-プロポキシアセトイミドイルシアニドおよび2-アミノ-2-ヒドロキシイミノ-N-イソプロポキシアセトイミドイルシアニドがより好ましい。
(工程(iii)の収率)
工程(iii)の収率としては、通常は70%以上、好ましくは80~100%、より好ましくは85~100%、の範囲を例示できる。
工程(iii)の収率は、本工程の原料としての一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルのモル数に対する、得られる一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドのモル数から計算することができる。
すなわち、この収率は、次の式で表される;
収率(%)=100×{(得られた一般式(3)で表わされる化合物のモル数)/(原料としての一般式(2)で表わされる化合物のモル教)}
すなわち、この収率は、次の式で表される;
収率(%)=100×{(得られた一般式(3)で表わされる化合物のモル数)/(原料としての一般式(2)で表わされる化合物のモル教)}
次に、実施例を挙げて本発明の製造方法を具体的に説明するが、本発明は、これら実施例によって何ら限定されるものではない。
本明細書中、室温は10℃から35℃を示す。
(1H核磁気共鳴スペクトル(1H-NMR))
1H核磁気共鳴スペクトル分析(1H-NMR)は、機種:Oxford NMR300(バリアンメディカルシステムズ社製)、および内部基準物質:テトラメチルシランを用いて実施された。
(pHの測定方法)
pHはガラス電極式水素イオン濃度指示計により測定した。ガラス電極式水素イオン濃度指示計としては、例えば、東亜ディーケーケー株式会社製、形式:HM-20Pが使用できる。
(示差走査熱量測定法)
示差走査熱量分析は、機種:DSC-60(株式会社島津製作所社製)を用いて、40~400℃の温度範囲において10℃/minの加熱速度で行われた。示差走査熱量測定方法に関しては、必要に応じて、以下の文献を参照することができる。
(a):(社)日本化学会編、「第4版実験化学講座4 熱、圧力」、第57~93頁(1992年)、発行者 海老原熊雄、丸善株式会社
(b):(社)日本化学会編、「第5版実験化学講座6 温度・熱、圧力」、第203~205頁(2005年)、発行者 村田誠四郎、丸善株式会社
(a):(社)日本化学会編、「第4版実験化学講座4 熱、圧力」、第57~93頁(1992年)、発行者 海老原熊雄、丸善株式会社
(b):(社)日本化学会編、「第5版実験化学講座6 温度・熱、圧力」、第203~205頁(2005年)、発行者 村田誠四郎、丸善株式会社
(HPLC分析方法)
HPLC分析方法に関しては、必要に応じて、以下の文献を参照することができる。
(a):(社)日本化学会編、「新実験化学講座9 分析化学 II」、第86~112頁(1977年)、発行者 飯泉新吾、丸善株式会社(例えば、カラムに使用可能な充填剤-移動相の組合せに関しては、第93~96頁を参照することができる。)
(b):(社)日本化学会編、「実験化学講座20-1 分析化学」第5版、第130~151頁(2007年)、発行者 村田誠四郎、丸善株式会社(例えば、逆相クロマトグラフィー分析の具体的な使用方法・条件に関しては、第135~137頁を参照することができる。)
(a):(社)日本化学会編、「新実験化学講座9 分析化学 II」、第86~112頁(1977年)、発行者 飯泉新吾、丸善株式会社(例えば、カラムに使用可能な充填剤-移動相の組合せに関しては、第93~96頁を参照することができる。)
(b):(社)日本化学会編、「実験化学講座20-1 分析化学」第5版、第130~151頁(2007年)、発行者 村田誠四郎、丸善株式会社(例えば、逆相クロマトグラフィー分析の具体的な使用方法・条件に関しては、第135~137頁を参照することができる。)
実施例1
2-イソプロポキシイミノプロパンジニトリルの製造
反応容器に水100ml、マロノニトリル66g(1.00mol)および酢酸6g(0.10mol)を仕込んだ。混合物を氷冷下で撹拌しながら、そこへ28%亜硝酸ナトリウム水溶液272ml(1.05mol)を滴下した。滴下終了後、混合物を氷冷下で2時間撹拌した。続いて、他の反応容器にトルエン534ml、イソプロピルブロミド184.5g(1.50mol)およびテトラブチルアンモニウムブロミド32.2g(0.10mol)を仕込み、60℃で撹拌しながら、上記で得られた反応混合物を滴下した。その後、混合物を同温度で5時間撹拌した。得られた反応混合物をトルエンと水に分配し、トルエン層を分離した。得られたトルエン溶液をHPLC絶対検量線法により分析した。その結果、2-イソプロポキシイミノプロパンジニトリルの収率は、使用したマロノニトリルの量から計算される理論量の80%であった。トルエン溶液は常法により後処理され、2-イソプロポキシイミノプロパンジニトリルを得た。得られた2-イソプロポキシイミノプロパンジニトリルはNMR分析により同定された。
1H-NMR(300MHz,CDCl3)δ(ppm):4.85(sep,J=6.3Hz,1H),1.43(d,J=6.3Hz,6H).
屈折率(nD 20):1.4405
屈折率(nD 20):1.4405
示差走査熱量測定; 発熱開始温度:225℃、発熱量:0.6kJ/g
実施例2
2-イソプロポキシイミノプロパンジニトリルの製造
反応容器に水100ml、マロノニトリル66g(1.00mol)および酢酸6g(0.10mol)を仕込んだ。混合物を氷冷下で撹拌しながら、そこへ28%亜硝酸ナトリウム水溶液272ml(1.05mol)を滴下した。滴下終了後、混合物を氷冷下で2時間撹拌した。続いて、他の反応容器にトルエン534ml、イソプロピルブロミド184.5g(1.50mol)およびテトラブチルアンモニウムブロミド32.2g(0.10mol)を仕込み、70℃で撹拌しながら、上記で得られた反応混合物を滴下した。滴下中、25%水酸化ナトリウム水溶液を用いて溶液のpHを約6~7に調整した。その後、混合物を同温度で3時間撹拌した。得られた反応混合物をトルエンと水に分配し、トルエン層を分離した。得られたトルエン溶液をHPLC絶対検量線法により分析した。その結果、2-イソプロポキシイミノプロパンジニトリルの収率は、使用したマロノニトリルの量から計算される理論量の75%であった。
実施例3
2-イソプロポキシイミノプロパンジニトリルの製造
反応容器に水30ml、マロノニトリル6.60g(100mmol)および亜硝酸ナトリウム7.45g(105mmol)を仕込んだ。混合物を氷冷下で撹拌しながら、そこへ6規定塩酸17ml(102mmol)をゆっくり滴下した。滴下終了後、混合物を氷冷下で1時間撹拌した。反応混合物をテトラヒドロフラン(THF)50mlで抽出した。得られたテトラヒドロフラン溶液へ水20mlを加え、25%水酸化ナトリウム水溶液を用いて溶液のpHを約6~7に調整した。続けて、そこへイソプロピルブロミド18.70g(110mmol)、テトラブチルアンモニウムブロミド3.22g(10mmol)を加えた後、その混合物を70℃で2時間撹拌した。得られた反応混合物をテトラヒドロフランと水に分配し、テトラヒドロフラン層を分離した。得られたテトラヒドロフラン溶液をHPLC絶対検量線法により分析した。その結果、2-イソプロポキシイミノプロパンジニトリルの収率は、使用したマロノニトリルの量から計算される理論量の78%であった。
実施例4
2-プロポキシイミノプロパンジニトリルの製造
反応容器に水6ml、マロノニトリル1.32g(20mmol)および亜硝酸ナトリウム1.49g(21mmol)を仕込んだ。混合物を氷冷下で撹拌しながら、そこへ6規定塩酸3.8ml(23mmol)をゆっくり滴下した。滴下終了後、混合物を氷冷下で1時間撹拌した。反応混合物をテトラヒドロフラン(THF)5mlで2回抽出した。得られたテトラヒドロフラン溶液へ水4mlを加え、25%水酸化ナトリウム水溶液を用いて溶液のpHを約6~7に調整した。続けて、そこへプロピルブロミド2.71g(22mmol)、テトラブチルアンモニウムブロミド645mg(2mmol)を加えた後、混合物を85℃で3時間撹拌した。得られた反応混合物をテトラヒドロフランと水に分配し、テトラヒドロフラン層を分離した。得られたテトラヒドロフラン溶液をHPLC絶対検量線法により分析した。その結果、2-プロポキシイミノプロパンジニトリルの収率は、使用したマロノニトリルの量から計算される理論量の70%であった。テトラヒドロフラン溶液は常法により後処理され、2-プロポキシイミノプロパンジニトリルを得た。得られた2-プロポキシイミノプロパンジニトリルはNMR分析により同定された。
1H-NMR(300MHz,CDCl3)δ(ppm):4.53(t,J=6.8Hz,2H),1.85(m,2H),1.00(t,J=6.8Hz,3H).
屈折率(nD 20):1.4470
屈折率(nD 20):1.4470
実施例5
2-アミノ-2-ヒドロキシイミノ-N-イソプロポキシアセトイミドイルシアニドの製造
反応容器に2-イソプロポキシイミノプロパンジニトリル6.20g(45mmol)およびメタノール40mlを仕込んだ。混合物を氷冷下で撹拌しながら、ヒドロキシルアミン硫酸塩3.30g(20mmol)と水8mlの溶液、および炭酸ナトリウム2.10g(20mmol)を加えた。混合物を氷冷下で1時間撹拌した後、室温で2時間撹拌した。減圧下で溶媒を留去した。得られた残渣を水とジエチルエーテルを加えることにより溶解した。2層を分離し、有機層を硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ過により除去した。ろ液を減圧下で濃縮し、得られた残渣を減圧乾燥することにより、6.12gの2-アミノ-2-ヒドロキシイミノ-N-イソプロポキシアセトイミドイルシアニドを収率90%で得た。
1H-NMR(300MHz,CDCl3)δ(ppm):8.60(brs,1H),5.03(brs,2H),4.63(sep,J=4.7Hz,1H),1.38(d,J=4.7Hz,6H).
融点:155-157℃
融点:155-157℃
実施例6
2-アミノ-2-ヒドロキシイミノ-N-プロポキシアセトイミドイルシアニドの製造
反応容器に2-プロポキシイミノプロパンジニトリル(HPLC純度=93%)738.1mg(5.0mmol)およびメタノール5mlを仕込んだ。混合物を氷冷下で撹拌しながら、ヒドロキシルアミン硫酸塩410.4mg(2.5mmol)と水1mlの溶液、および炭酸ナトリウム265.0mg(2.5mmol)を加えた。混合物を氷冷下で2時間撹拌した。減圧下で溶媒を留去した。得られた残渣を水とジエチルエーテルを加えることにより溶解した。2層を分離し、有機層を硫酸ナトリウムで乾燥し、硫酸ナトリウムをろ過により除去した。ろ液を減圧下で濃縮し、得られた残渣を減圧乾燥することにより、815.4mgの2-アミノ-2-ヒドロキシイミノ-N-プロポキシアセトイミドイルシアニドを収率95%で得た。
1H-NMR(300MHz,CDCl3)δ(ppm):8.37(brs,1H),5.00(brs,2H),4.34(t,J=5.1Hz,2H),1.79(m,2H),1.00(t,J=5.5Hz,3H).
融点:110-113℃
融点:110-113℃
実施例7
2-ヒドロキシイミノプロパンジニトリルの示差走査熱量測定
2-ヒドロキシイミノプロパンジニトリルは、公知化合物であり、当業者に知られた常法により製造され、示差走査熱量測定にかけられた。
示差走査熱量測定; 発熱開始温度:147℃、発熱量:1.6kJ/g
実施例8
2-ヒドロキシイミノプロパンジニトリルのナトリウム塩の示差走査熱量測定
2-ヒドロキシイミノプロパンジニトリルのナトリウム塩は、公知化合物であり、当業者に知られた常法により製造され、示差走査熱量測定にかけられた。
示差走査熱量測定; 発熱開始温度:222℃、発熱量:-0.9kJ/g(マイナス0.9kJ/g)
参考例1
2-プロポキシイミノプロパンジニトリルの製造
特公平06-004647号の製造例10と同様の方法
マロノニトリル(13.21g、200mmol)および酢酸(2.40g、40mmol)のアセトニトリル(100mL)溶液に、亜硝酸ナトリウム(15.18g、220mmol)を60~65℃で加えた。同温度で1時間撹拌した後、プロピルブロミド27.06g(220mmol)を65℃で加えた。5時間還流した後、混合物を室温まで冷却して一晩放置した。得られた混合物を水(180ml)とジエチルエーテル(180ml)の混合物中に注いだ。有機層を分離し、水および飽和食塩水で洗浄し、硫酸ナトリウムで乾燥した。減圧下で溶媒を留去した。得られた残渣を減圧蒸留により精製し、2-プロポキシイミノプロパンジシアニド11.52gを収率42%で得た。
上記のように、特公平06-004647号の製造例10と同様の方法で2-プロポキシイミノプロパンジニトリルを製造した。しかしながら、実施例4と比較して、収率が大幅に低かった。
本発明によれば、工業的に適した一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドの製造方法が提供される。なお、一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドは、それ自身が農薬として有用であり、そして他の農薬の製造中間体としても有用である。
本発明によれば、先行技術における熱的に危険な化合物を製造中間体として使用することを回避し、目的とする一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造することができる。すなわち、本発明によれば、非常に危険な発熱分解を示すと考えられる式(6)で表わされる2-アミノ-2-ヒドロキシイミノ-N-ヒドロキシアセトイミドイルシアニドを使用せず、目的とする一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造することができる。
本発明により提供される新規な一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルは、先行技術のそれと比較して、目的とする一般式(3)で表わされる2-アミノ-2-ヒドロキシイミノ-N-アルコキシアセトイミドイルシアニドを製造するためのはるかに安全な中間体である。本発明におけるその他の製造中間体も、先行技術のそれよりも有意に安全である。つまり、本発明方法は、先行技術と比較して、劇的に安全性を増したのである。したがって、本発明方法は、パイロットプラントまたは工業的な生産のような大きなスケールでの製造に安全に適用できる。
同時に、本発明は、本発明の有用な中間体である一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルの新規な工業的に満足できる製造方法を、提供する。
本発明によれば、マロノニトリルと亜硝酸のアルカリ金属塩を反応させた後に得られる化合物をアルキル化剤と反応させるときに、高い反応性を有するアルキル化剤に限られずに、すなわちアルキル化剤の種類に関わらず、高価な銀塩を使用せず、高収率で、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルを製造することができる。さらに、本発明によれば、リサイクルが可能なかつ安価な溶媒(例えば、エーテル類溶媒または特に芳香族炭化水素誘導体類溶媒などの有機溶媒)を用いて、一般式(2)で表わされる2-アルコキシイミノプロパンジニトリルを製造することができる。
さらに、本発明方法は、高収率であり、工業的なスケールで、簡便な操作により、穏やかな条件下で、特殊な反応装置を用いることなく、効率的に実施できる。加えて、本発明では、廃棄物の量は少なく、廃棄物の性質は好ましいと考えられる。
したがって、本発明は環境にも優しく、そして高い工業的な利用価値を有する。
Claims (25)
- 一般式(2):
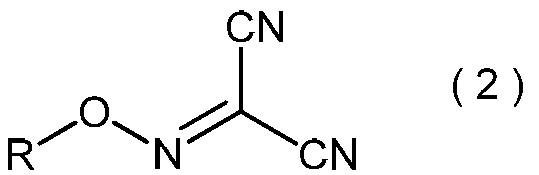
で表わされる化合物。 - Rがイソプロピルまたはプロピルである、請求項1に記載の化合物。
- Rがイソプロピルである、請求項1に記載の化合物。
- Rがプロピルである、請求項1に記載の化合物。
- 一般式(2):
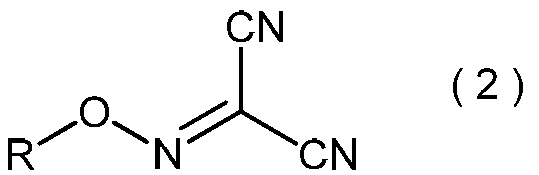
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、一般式(3):
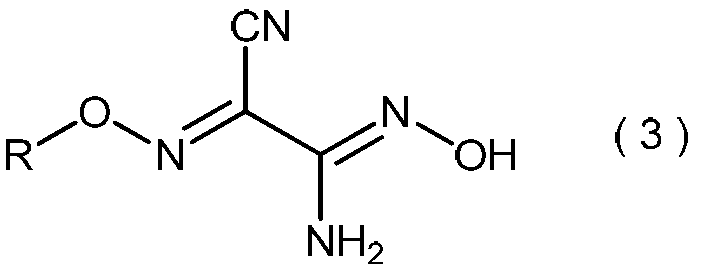
で表わされる化合物の製造方法。 - (i) 式(1):
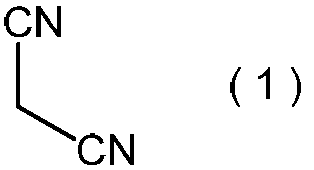
(ii) 工程(i)の生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させ、一般式(2):

で表わされる化合物を製造する工程および
(iii) 得られる一般式(2)
で表わされる化合物を、ヒドロキシルアミン化合物と反応させる工程
を含むことを特徴とする、
一般式(3):
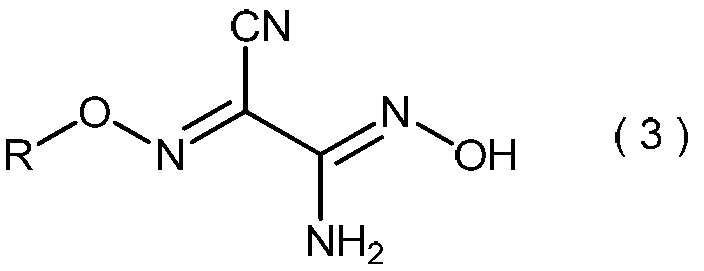
で表わされる化合物の製造方法。 - 工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系またはエーテル類と水からなる溶媒系で行われる、請求項6に記載の方法。
- 工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系で行われる、請求項6に記載の方法。
- 工程(ii)の反応が、エーテル類と水からなる溶媒系で行われる、請求項6に記載の方法。
- 工程(ii)の反応が、トルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系で行われる、請求項6に記載の方法。
- 工程(ii)の反応が、トルエンと水からなる溶媒系で行われる、請求項6に記載の方法。
- 工程(ii)の反応が、テトラヒドロフランと水からなる溶媒系で行われる、請求項6に記載の方法。
- Rがイソプロピルまたはプロピルである、請求項6から請求項12のいずれか1項に記載の方法。
- Rがイソプロピルである、請求項6から請求項12のいずれか1項に記載の方法。
- Rがプロピルである、請求項6から請求項12のいずれか1項に記載の方法。
- (i) 式(1):
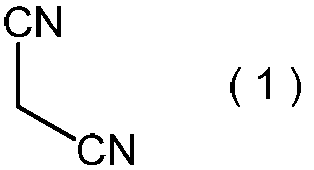
(ii) 工程(i)の生成物を、相間移動触媒の存在下で、C3~C6アルキルハライドと反応させる工程を含むことを特徴とする、 一般式(2):
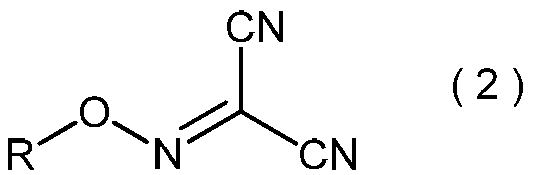
で表わされる化合物の製造方法。 - 工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系またはエーテル類と水からなる溶媒系で行われる、請求項16に記載の方法。
- 工程(ii)の反応が、芳香族炭化水素誘導体類と水からなる溶媒系で行われる、請求項16に記載の方法。
- 工程(ii)の反応が、エーテル類と水からなる溶媒系で行われる、請求項16に記載の方法。
- 工程(ii)の反応が、トルエン、キシレンおよびクロロベンゼンからなる群より選択される1種以上の芳香族炭化水素誘導体類と水からなる溶媒系で行われる、請求項16に記載の方法。
- 工程(ii)の反応が、トルエンと水からなる溶媒系で行われる、請求項16に記載の方法。
- 工程(ii)の反応が、テトラヒドロフランと水からなる溶媒系で行われる、請求項16に記載の方法。
- Rがイソプロピルまたはプロピルである、請求項16から請求項22のいずれか1項に記載の方法。
- Rがイソプロピルである、請求項16から請求項22のいずれか1項に記載の方法。
- Rがプロピルである、請求項16から請求項22のいずれか1項に記載の方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201480034948.7A CN105377812B (zh) | 2013-06-21 | 2014-06-16 | 2-氨基-2-肟基-n-烷氧基亚氨代乙酰基氰化物的制备方法及其制备中间体 |
| EP14813379.6A EP3012247B1 (en) | 2013-06-21 | 2014-06-16 | Manufacturing method for 2-amino-2-hydroxyimino-n-alkoxy acetoimidoyl cyanide, and manufacturing intermediate thereof |
| JP2015522910A JP6327754B2 (ja) | 2013-06-21 | 2014-06-16 | 2−アミノ−2−ヒドロキシイミノ−n−アルコキシアセトイミドイルシアニドの製造方法およびその製造中間体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013130079 | 2013-06-21 | ||
| JP2013-130079 | 2013-06-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2014203855A2 true WO2014203855A2 (ja) | 2014-12-24 |
| WO2014203855A3 WO2014203855A3 (ja) | 2015-01-29 |
Family
ID=52105435
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/065891 Ceased WO2014203855A2 (ja) | 2013-06-21 | 2014-06-16 | 2-アミノ-2-ヒドロキシイミノ-n-アルコキシアセトイミドイルシアニドの製造方法およびその製造中間体 |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP3012247B1 (ja) |
| JP (1) | JP6327754B2 (ja) |
| CN (1) | CN105377812B (ja) |
| WO (1) | WO2014203855A2 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110713443B (zh) | 2018-07-13 | 2022-01-28 | 沈阳中化农药化工研发有限公司 | 制备丙二腈肟醚类化合物的方法及中间体化合物 |
| WO2021088756A1 (zh) * | 2019-11-04 | 2021-05-14 | 沈阳中化农药化工研发有限公司 | 一种肟醚类化合物及其用途 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0150609A2 (en) | 1983-12-30 | 1985-08-07 | Glaxo Group Limited | Cephalosporin antibiotics |
| EP0069872B1 (en) | 1981-06-22 | 1987-10-28 | Fujisawa Pharmaceutical Co., Ltd. | New cephem compounds and processes for preparation thereof |
| JPH04187667A (ja) | 1990-11-20 | 1992-07-06 | Takeda Chem Ind Ltd | 2―置換ヒドロキシイミノプロパンジニトリルの製造法 |
| EP0517041A1 (en) | 1991-06-07 | 1992-12-09 | Fujisawa Pharmaceutical Co., Ltd. | New Cephem Compounds |
| JPH064647A (ja) | 1991-12-30 | 1994-01-14 | Schlumberger Technol Inc | 選択したic導体が隠蔽されていない視野の探索 |
| JPH064647B2 (ja) | 1986-09-22 | 1994-01-19 | 藤沢薬品工業株式会社 | 新規セフェム化合物およびその製造法 |
| EP0397511B1 (en) | 1989-05-11 | 1996-09-04 | Lucky, Ltd. | Novel cephalosporin compounds and processes for preparation thereof |
| US20060258719A1 (en) | 2005-05-10 | 2006-11-16 | Combs Andrew P | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| WO2011161945A1 (ja) | 2010-06-24 | 2011-12-29 | クミアイ化学工業株式会社 | アルコキシイミノ誘導体及び有害生物防除剤 |
| WO2013080479A1 (ja) | 2011-11-30 | 2013-06-06 | クミアイ化学工業株式会社 | グリオキシム誘導体及び有害生物防除剤 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3234266A (en) * | 1963-08-13 | 1966-02-08 | Du Pont | N-hydrocarbyloxyiminomalononitriles and process for preparing them |
| GB2094794B (en) * | 1981-03-06 | 1985-02-20 | Fujisawa Pharmaceutical Co | Processes for preparing 2-substituted hydroxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)acetic acid or its salt and intermediates thereof |
| JPH0477477A (ja) * | 1990-07-18 | 1992-03-11 | Takeda Chem Ind Ltd | チアジアゾール酢酸誘導体の製造法 |
| CN1850823A (zh) * | 2006-05-19 | 2006-10-25 | 中国科学院上海药物研究所 | 一类含有肟基的喹诺酮类化合物及其制备方法和用途 |
-
2014
- 2014-06-16 CN CN201480034948.7A patent/CN105377812B/zh not_active Expired - Fee Related
- 2014-06-16 WO PCT/JP2014/065891 patent/WO2014203855A2/ja not_active Ceased
- 2014-06-16 JP JP2015522910A patent/JP6327754B2/ja not_active Expired - Fee Related
- 2014-06-16 EP EP14813379.6A patent/EP3012247B1/en not_active Not-in-force
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0069872B1 (en) | 1981-06-22 | 1987-10-28 | Fujisawa Pharmaceutical Co., Ltd. | New cephem compounds and processes for preparation thereof |
| EP0150609A2 (en) | 1983-12-30 | 1985-08-07 | Glaxo Group Limited | Cephalosporin antibiotics |
| JPH064647B2 (ja) | 1986-09-22 | 1994-01-19 | 藤沢薬品工業株式会社 | 新規セフェム化合物およびその製造法 |
| EP0397511B1 (en) | 1989-05-11 | 1996-09-04 | Lucky, Ltd. | Novel cephalosporin compounds and processes for preparation thereof |
| JPH04187667A (ja) | 1990-11-20 | 1992-07-06 | Takeda Chem Ind Ltd | 2―置換ヒドロキシイミノプロパンジニトリルの製造法 |
| EP0517041A1 (en) | 1991-06-07 | 1992-12-09 | Fujisawa Pharmaceutical Co., Ltd. | New Cephem Compounds |
| JPH064647A (ja) | 1991-12-30 | 1994-01-14 | Schlumberger Technol Inc | 選択したic導体が隠蔽されていない視野の探索 |
| US20060258719A1 (en) | 2005-05-10 | 2006-11-16 | Combs Andrew P | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| WO2011161945A1 (ja) | 2010-06-24 | 2011-12-29 | クミアイ化学工業株式会社 | アルコキシイミノ誘導体及び有害生物防除剤 |
| WO2013080479A1 (ja) | 2011-11-30 | 2013-06-06 | クミアイ化学工業株式会社 | グリオキシム誘導体及び有害生物防除剤 |
Non-Patent Citations (7)
| Title |
|---|
| "Experimental Chemistry Course", 1992, MARUZEN COMPANY, LIMITED, article "The Chemical Society of Japan", pages: 57 - 93 |
| "Experimental Chemistry Course", 2005, MARUZEN COMPANY, LIMITED, pages: 203 - 205 |
| "Jikkenkagaku Koza", 2007, MARUZEN COMPANY, LIMITED, pages: 130 - 151 |
| "Shin Jikkenkagaku Koza", 1977, MARUZEN COMPANY, LIMITED, pages: 86 - 112 |
| BULL. CHEM. SOC. JPN., vol. 76, 2003, pages 1063 - 1070 |
| J. ORG. CHEM., vol. 65, 2000, pages 1139 - 1143 |
| See also references of EP3012247A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3012247A4 (en) | 2017-01-25 |
| EP3012247B1 (en) | 2018-01-10 |
| JP6327754B2 (ja) | 2018-05-23 |
| CN105377812B (zh) | 2018-09-18 |
| JPWO2014203855A1 (ja) | 2017-02-23 |
| EP3012247A2 (en) | 2016-04-27 |
| WO2014203855A3 (ja) | 2015-01-29 |
| CN105377812A (zh) | 2016-03-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9000210B2 (en) | Preparation of substituted 2-fluoroacrylic acid derivatives | |
| WO2008053043A1 (de) | Verfahren zur herstellung von difluormethylpyrazolylcarboxylaten | |
| KR102623135B1 (ko) | Dipea 염기 존재 하의 아릴피롤 화합물의 제조 | |
| BR102013032003A2 (pt) | Processo para a preparação de 4-amino-5-fluoro-3-cloro-6-(substituído)picolinato | |
| CN101878192A (zh) | 4-全氟异丙基苯胺类的制造方法 | |
| JP6327754B2 (ja) | 2−アミノ−2−ヒドロキシイミノ−n−アルコキシアセトイミドイルシアニドの製造方法およびその製造中間体 | |
| KR102720359B1 (ko) | 공기 산화에 의한 3-메틸-2-니트로벤조산의 제조 공정 | |
| CN115806497A (zh) | 制备邻氨基苯甲酸酯化合物的改进方法 | |
| CN109071411B (zh) | 制造硝基苯化合物的方法 | |
| EP3645509B1 (en) | Method for preparing substituted 4-aminoindane derivatives | |
| KR20250044809A (ko) | 치환된 피라졸의 제조 및 안트라닐아미드 전구체로서의 그의 용도 | |
| AU2006227484B2 (en) | Conversion of 2-pyrazolines to pyrazoles using bromine | |
| CN109071428B (zh) | 用于制备顺式-烷氧基取代的螺环1-h-吡咯烷-2,4-二酮衍生物的方法 | |
| KR0172014B1 (ko) | 히드록실아민에테르와 그 염의 제조 방법 및 이를 위한 중간체 | |
| AU2023288874A1 (en) | Method for producing sulfone derivative using haloacetic acid | |
| US7141693B2 (en) | Process for producing β-oxonitrile compound or alkali metal salt thereof | |
| JP2012193124A (ja) | 2,3−ジクロロピリジンの製造方法 | |
| EP2119693B1 (en) | Process for producing 2-isopropenyl-5-methyl-4-hexene-1-yl- 3-methyl-2-butenoate | |
| EP1873145B1 (en) | Method for producing nicotinic acid derivative or salt thereof | |
| EP1121344A2 (en) | Chemical processes | |
| CN120774857A (zh) | 一种1-取代基-5-羟基吡唑的制备方法 | |
| CA3053443C (en) | Production of arylpyrrol compounds in the presence of dipea base | |
| WO2016086722A1 (zh) | 一种异噁唑类化合物及其中间体的制备方法 | |
| WO2007000412A1 (de) | Verfahren zur herstellung von substituierten phenylmalonestern, zwischenverbindungen sowie deren verwendung zur herstellung von 5 , 7-dihydr0xy-6- (2,4, 6-trifluorphenyl) -(1,2 , 4) triazolo (1 , 5-a) pyrimidinen | |
| WO2007076835A1 (de) | VERFAHREN ZUR HERSTELLUNG VON α,β-UNGESÄTTIGTEN CARBONSÄUREDERIVATEN |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14813379 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2015522910 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014813379 Country of ref document: EP |