WO2015007073A1 - 奎宁类化合物、其光学异构体及其制备方法和医药用途 - Google Patents
奎宁类化合物、其光学异构体及其制备方法和医药用途 Download PDFInfo
- Publication number
- WO2015007073A1 WO2015007073A1 PCT/CN2014/000669 CN2014000669W WO2015007073A1 WO 2015007073 A1 WO2015007073 A1 WO 2015007073A1 CN 2014000669 W CN2014000669 W CN 2014000669W WO 2015007073 A1 WO2015007073 A1 WO 2015007073A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- ethoxy
- hydroxy
- phenyl
- azabicyclo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 O[C@@](CO[C@@]1C(CC2)CCN2C1)(*1CCCCC1)c1ccccc1 Chemical compound O[C@@](CO[C@@]1C(CC2)CCN2C1)(*1CCCCC1)c1ccccc1 0.000 description 1
- IVLICPVPXWEGCA-SSDOTTSWSA-N O[C@H]1C(CC2)CCN2C1 Chemical compound O[C@H]1C(CC2)CCN2C1 IVLICPVPXWEGCA-SSDOTTSWSA-N 0.000 description 1
- RIHJDNCSHZBSKC-RTWAWAEBSA-N O[C@](CO[C@H]1C(CC2)CCN2C1)(C1CCCCC1)c1ccccc1 Chemical compound O[C@](CO[C@H]1C(CC2)CCN2C1)(C1CCCCC1)c1ccccc1 RIHJDNCSHZBSKC-RTWAWAEBSA-N 0.000 description 1
- APJIIEDYGZSPNQ-UHFFFAOYSA-N [O-][NH+](C1CCCC1)c1ccc[o]1 Chemical compound [O-][NH+](C1CCCC1)c1ccc[o]1 APJIIEDYGZSPNQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0043—Nose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
- A61K9/008—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy comprising drug dissolved or suspended in liquid propellant for inhalation via a pressurized metered dose inhaler [MDI]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4808—Preparations in capsules, e.g. of gelatin, of chocolate characterised by the form of the capsule or the structure of the filling; Capsules containing small tablets; Capsules with outer layer for immediate drug release
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/14—Antitussive agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a quinine-based compound, an optical isomer thereof, a process for the preparation thereof, and a composition for medicinal purposes containing the same, and in particular to a novel M receptor blocker which selectively acts on the M receptor subtype
- M receptor blockers have a strong effect on M 3 and receptor subtypes and have no significant effect on M 2 receptor subtypes.
- French patent FR2012964 discloses the following structure: R is a H atom, a hydroxyl group or an alkyl group of 1-4 carbon; is a phenyl or thiophene; R 2 is cyclohexanyl, cyclopentamidine or thiophene.
- the compounds of the present invention overcome the shortcomings of the above compounds, especially in the treatment of chronic bronchitis, high airway, asthma, and chronic obstructive pulmonary disease, which have the characteristics of longer drug efficacy, quick onset, and low toxic side effects compared with the prior art.
- the compound of the present invention is suitable for preparing an inhalant for treating chronic obstructive pulmonary disease once a day, and is particularly suitable for preparing a solution-type quantitative inhalation aerosol for once-a-day administration.
- the present invention relates to the synthesis of the compound, Preparation of pharmaceutical compositions of the compounds and their pharmaceutical use.
- the compound of the present invention can also be used in combination with a 02 agonist, a hormone, an antiallergic, an anti-inflammatory, an anti-infective, a phospholipase iv antagonist, etc. for the treatment of the above respiratory diseases such as rhinitis, post-cold rhinitis, chronic trachea Inflammation, high airway, asthma, chronic obstructive pulmonary disease, etc.
- novel M receptor subtype selective receptor antagonist compounds of the present invention can be represented by the structure of formula (I):
- n is selected from 1 to 7, preferably from 1 to 3, and most preferably 1.
- 1 is a hydrocarbon group of 3 - (: 7 , the above group may be unsubstituted, and may be, but not limited to, optionally substituted by halogen, an anthraceneoxy group, an alkoxy hydrocarbon group, a heterocyclic ring, an aryl group;
- the substituted cycloalkyl group is most preferably a cyclopentyl group and a cyclohexyl group.
- R 2 is an aryl group, that is, a phenyl group, a heteroaryl group containing one or more hetero atoms (the hetero atom may be 1 ⁇ , 0, S), a naphthyl group, or a biphenyl group, and the above group may be unsubstituted. , may also be optionally substituted, the substituent may be one or more, such as halogen, hydroxy, phenyl, - 0R 6 , - SR e , - NR 6 R 7 , - NHC0R 6 , - C0NR 6 R 7 , - CN, -N0 2 , - C00R 6 , - CF 3 or d - C 4
- R 6 and R 7 may be a hydrogen atom, a linear or branched hydrocarbon group of CC, or a combination of a cyclic hydrocarbon group.
- the hydroxy, halogen, alkoxy or acyloxy group, the above alkoxy or acyloxy group may be unsubstituted, but may also be, but not limited to, optionally halogen, hydroxy, decyloxy, hydrocarbyl, decyloxy , a cycloalkyl group, a heterocyclic ring, an aryl group; preferably a hydroxyl group and a methoxy group, most preferably a hydroxyl group.
- R 5 may or may not be present, and when present, may, but are not limited to, a halogen, a hydroxyl group, a hydrocarbyloxy group, a hydrocarbyl group, a hydrocarbyloxy hydrocarbyl group, a heterocyclic ring, an aryl group, and the like, respectively.
- Y is a linear or branched alkyl group of dG or -(CH 2 -0-CH 2 ) m -, ra is 1-3, and these groups may be optionally substituted, preferably by halogen, hydroxy, decyloxy And an alkyloxy group, an unsaturated hydrocarbon group, a cyclic hydrocarbon group, a heterocyclic ring; preferably a methyl group, an ethyl group, a propyl group and a -(CH 2 -0-CH 2 )-; most preferably an ethyl group and a propyl group.
- X is an acid or hydroxide, a pharmaceutically acceptable acid radical
- examples include salts derived from inorganic acids, inorganic acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, a phosphate, a hydrogen phosphate, a dihydrogen phosphate, a sulfate, a hydrogen sulfate, a sulfite, a bisulfite, a phosphite, or the like; a salt derived from a relatively non-toxic organic acid, for example, the organic acid But not limited to acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid , methanesulfonic acid, glu
- the compound represented by the formula (I) may contain one or more chiral centers, and a single optical isomer or a mixture of various optical isomers is required by the scope of the invention.
- the present invention provides a process route for synthesizing a compound of formula (I) as follows:
- Step 1 According to the literature 2) (1. Wen Guangzhen, Wu Peijin. Improvement of the synthesis method of 3-(2-phenyl-2-cyclopentylethoxy)quinuclidin hydrochloride. Journal of Academy of Military Medical Sciences , 1988: 470 to 402; 2. Wu Peijin, ⁇ . Synthesis of anticholinergic 2-(1-naphthyl)-2 cyclopentyl-2-hydroxyethoxycycloalkylamine compound.
- Step 2 Preparation of 3-[(2- 2-R 2 -2-hydroxy)ethoxy]- 1-azabicyclo[2, 2, 2]octane free base intermediate 1 with quinolol (or R 4 Substituted Quinol) Intermediate 2 can be obtained under NaH conditions.
- Step 3 3-[(2-1 ⁇ -2- R 2 - 2-hydroxy)ethoxy]- 1-azabicyclo[2, 2, 2] octopine free base column chromatography purification and related purification treatment
- the intermediate 2 will be a mixture containing different optical structures, such as quinolol in the S configuration, the intermediate 2 will contain (2R, 3S), (2S, 3S) two configurations, in order of elution Can be purified into (2R, 3S) and (2S, 3S) two free bases; if quinolol is in the R configuration, intermediate 2 will contain (2R, 3R), (2S, 3R) two configurations, The elution can be purified into two free bases (2S, 3R) and (2R, 3R); if quinolol is a racemate, intermediate 2 will contain (2R, 3S), (2S, 3S), (2R , 3R), (2S, 3R), (2S, 3R), (2S, 3R), (2S, 3R), (2S, 3R), (2S, 3R
- Phenol was added to a three-necked flask, and sodium hydroxide was added thereto, and a solution of ruthenium-iridium-ruthenium (here, ruthenium atom) and absolute ethanol was added thereto, and the mixture was heated under reflux in an oil bath to precipitate a white solid.
- the compound of the above formula (I) is obtained by reacting with Ag 2 0 to remove a halogenated hydroxide, and reacting with other acids to convert it into another acid salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid
- compositions containing one or more compounds of the formula (I) is within the scope of the claimed invention and may be administered, for example, orally, topically, intravenously, intramuscularly, intraarterially, intraperitoneally, Rectal, vaginal, intranasal, inhalation.
- the formulations of the invention can be designed to be fast-acting, immediate-release or long-acting. Additionally, the compounds may be administered by topical rather than systemic means.
- the compositions of the invention may be formulated as a medicament for administration to a mammal, preferably a human.
- a composition comprising one or more compounds of the invention and a suitable excipient may be administered repeatedly, or may be replaced by a continuous replacement page (Rule 26) To the composition.
- Suitable sites for administration include, but are not limited to, the nasal cavity, the lungs, the blood vessels, the muscles, the bronchi, the gastrointestinal tract.
- the preparation may be in the form of a liquid dosage form, a lyophilized powder form, a solid or a semi-solid, such as a solution, a suspension, an emulsion, a tablet, a pill, a capsule, a powder, a suppository, a retention enema, an aerosol, a powder, etc.
- it is suitable for simply administering a precise dosage unit dosage form.
- excipients include, but are not limited to, water, saline, lactose, glucose, sucrose, sorbitol, mannitol, starch, gum arabic, calcium phosphate, alginate, tragacanth, gelatin, calcium silicate, microcrystalline Cellulose, polyvinylpyrrolidone, cellulose, syrup, methylcellulose, ethylcellulose, hydroxypropylmethylcellulose, and polyacrylic acid.
- composition may also contain lubricants such as talc, magnesium stearate and mineral oil; wetting agents; emulsifiers; suspending agents; preservatives such as methyl-, ethyl- and propyl-hydroxy-benzoates; Regulators such as inorganic and organic acids and bases; sweeteners; and flavoring agents.
- lubricants such as talc, magnesium stearate and mineral oil
- wetting agents such as talc, magnesium stearate and mineral oil
- emulsifiers such as methyl-, ethyl- and propyl-hydroxy-benzoates
- preservatives such as methyl-, ethyl- and propyl-hydroxy-benzoates
- Regulators such as inorganic and organic acids and bases; sweeteners; and flavoring agents.
- compositions may be in the form of a sterile injectable solution and a sterile packaged powder.
- the injection is prepared in 114. 5-7.
- the pharmaceutical composition of the present invention may also be in any orally acceptable dosage form, including tablets, capsules, cachets, emulsions, suspensions, solutions, syrups, elixirs, sprays, pills, troches, powders. , granules and sustained release preparations.
- Suitable excipients for oral administration include pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharin, talc, cellulose, glucose, gelatin, sucrose, magnesium carbonate and the like.
- conventional carriers include lactose and microcrystalline cellulose, usually with a lubricant such as magnesium stearate; for capsules, useful diluents include lactose and dried corn starch;
- the active ingredient is mixed with the emulsifying and suspending agent, and some sweeteners, flavoring agents or coloring agents may also be added as appropriate.
- a pharmaceutical preparation in the form of a liquid suspension or solution can be prepared with a sterile liquid such as oil, water, ethanol and a combination thereof; for pulmonary inhalation, nasal spray, oral or parenteral administration, a suitable pharmaceutical agent can be added.
- the suspension may contain an oil such as peanut oil, sesame oil, cottonseed oil, corn oil and olive oil; the suspension preparation may also contain an ester of a fatty acid such as ethyl oleate or myristic acid.
- the composition may be in the form of a pill, tablet or capsule, and thus, the composition may contain one or more diluents such as lactose, sucrose, dicalcium phosphate, etc.; a disintegrant such as starch or a derivative thereof; Agents such as magnesium stearate and the like; and/or binders such as starch, gum arabic, polyvinylpyrrolidone, gelatin, cellulose and derivatives thereof. Pills, tablets or capsules can be prepared by any method known to those skilled in the art.
- diluents such as lactose, sucrose, dicalcium phosphate, etc.
- a disintegrant such as starch or a derivative thereof
- Agents such as magnesium stearate and the like
- binders such as starch, gum arabic, polyvinylpyrrolidone, gelatin, cellulose and derivatives thereof. Pills, tablets or capsules can be prepared by any method known to those skilled in the art.
- compositions of the invention may be in the form of a suppository for rectal administration. It can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and thereby releases the drug in the rectum.
- compositions suitable for rectal administration may also contain rectal enema units containing one or more compounds of the invention and a pharmaceutically acceptable vehicle (for example, a 50% aqueous solution of ethanol or saline), such vehicles and rectal and / or the colon is physiologically compatible.
- a pharmaceutically acceptable vehicle for example, a 50% aqueous solution of ethanol or saline
- the rectal enema unit comprises an applicator tip protected by an inert cover, preferably composed of polyethylene, lubricated with a lubricant such as white petrolatum, preferably with a one-way valve to prevent backflow of the ejected drug.
- the rectal enema unit is also of sufficient length, preferably 2 inches, to be inserted through the anus into the colon.
- compositions of the present invention may also be in the form of topical administration, especially when the therapeutic target includes areas or organs readily accessible by topical application, and diseases of these organs include diseases of the lungs, nasal mucosa, and trachea. Suitable topical formulations for use in these areas or organs are readily prepared.
- compositions containing one or more compounds of the invention may be in the form of nasal sprays, solution inhalers, metered powder mist inhalers, metered solution inhalers, metered suspension inhalants, and the like.
- a dry powder or liquid form of the composition can be delivered by a nebulizer.
- Such compositions are prepared according to techniques known in the art of pharmaceutical formulation, and may be employed in saline, with benzyl alcohol or other suitable preservatives, bioavailability absorption enhancers, fluorocarbons and/or other conventional solubilizing agents or A dispersant prepares a composition in the form of a solution.
- a solution-type metered dose inhalation aerosol containing one or more compounds of formula (I), the latent solvent comprising one or more of anhydrous ethanol, glycerol, glycols, wherein the glycol comprises However, it is not limited to ethylene glycol, propylene glycol, polyethylene glycol 200, polyethylene glycol 300, polyethylene glycol 400, polyethylene glycol 600, polyethylene glycol 800, and the like.
- the propellant includes one of tetrafluoroethane (HFA-134a), heptafluoropropene (HFA-227ea), or a mixture thereof.
- Surfactants include oleic acid; oligomeric lactic acid (0LA); sorbitan, such as span20, span65, span80, span85 ; polyoxyethylene sorbitan, such as Tween 20, Tween 80; polyoxyethylene fat Alcohols such as Bri j30, Bri j35, Creraophor; polyoxyethylene polyoxypropylene copolymers such as Pluronic F-68; polyethylene glycol stearates such as Solutol HS15; phospholipids such as soybean phospholipids, lecithin One or several of them. Oleic acid, lecithin or a mixture of the two is preferred. 5% ⁇ The 5% by weight of the 5% by weight.
- the content of the latent solvent is from 5 to 40% by weight, preferably 17. 5-29. 975%. 005 ⁇ 2% ⁇ The content of the surfactant is 0. 005 ⁇ 2%.
- the propellant content in the inhalation aerosol is from 54 to 90% by weight, preferably from 70 to 80% by weight.
- a metered powder mist inhalant comprising one or more compounds of formula (I), said inert carrier comprising a diluent and a lubricant, said diluent being dextran, arabinose, lactose, mannitol, mannitol, A mixture of one or more of xylitol, sucrose, fructose, sorbitol, maltose, amino acid, and glucose, the lubricant being magnesium stearate or sodium benzoate.
- a metered nasal or nasal spray comprising one or more compounds of formula (I) selected from the group consisting of benzalkonium chloride, benzalkonium bromide, benzyl alcohol, benzoic acid, chlorobutanol, A mixture of one or more of parabens, sorbic acid, phenol, thymol, and volatile oils.
- the invention also provides the use of the pharmaceutical composition, which can be used for the preparation of various acute and chronic airway obstructive diseases, such as chronic obstructive pulmonary disease, bronchial asthma, and acute and chronic rhinitis, for the prevention and treatment of mammals and humans.
- various acute and chronic airway obstructive diseases such as chronic obstructive pulmonary disease, bronchial asthma, and acute and chronic rhinitis, for the prevention and treatment of mammals and humans.
- a drug for rhinitis after a cold is a cold.
- Compounds of formula (I) with other active ingredients can be used in a variety of acute and chronic Treatment of airway obstructive diseases such as chronic obstructive pulmonary disease, bronchial asthma and various rhinitis.
- active ingredients such as beclomethasone dipropionate, chlorpheniramine, nemetazoline or fenoterol fenoterol
- the compound of the present invention in a daily dose of 10-1000 w g, preferably 40-500 y g.
- Step 1 Preparation of 1-phenyl-1-cyclopentyl epoxy acetamidine
- Step 3 3-[(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2 2 2]octane free radical column chromatography purification and related purification treatment
- Literature 3. Bingdahl B, Resul B and Dahlbom R. Facile preparation of the enantiomers of 3-acetoxyquinuclidine and 3-quinuclidinol. Acta Pharm Suec, 1979; 16:281-283), Literature ( 4 — " (4. Gao Jianhua, Wen Guangxuan, Zhang Qiqi. Synthesis and separation of optically pure hydroxyether compounds. Acta Pharmacologica Sinica, 1987;22(9):708-710; 5. Han Xiangyu. Stereoselective Synthesis of M Receptor Chiral Antagonists ⁇ . Report of the Postdoctoral Mobile Station of the Academy of Military Medical Sciences, p39-40 2005, Beijing.
- the compound of Example 1 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reacting with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galact
- Steps 1, 2, 3, and 4 are the same as in [Example 1] Steps 1, 2, 3, and 4
- Step 5 (2S,3S) -3-[(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)-bromide-1-nitrogen Heterobicyclo[2, 2,
- Example 2 The compound of Example 2 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with an acid to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid, and examples include salts derived from inorganic acids, inorganic acids.
- Salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzenesulfonate Acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic acid. Also included are salts of amino acids such as arginine and the like.
- Step 1 Same as [Example 1] Step 1
- Step 2 Preparation of R-3-quinolol 3- [(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1 -azabicyclo[2 2 2]octyl free base
- (2S.3R) (2R, 3R) Take commercially available R-3-quinucin 18.721g ( 147 ol), add 190 ml of DMSO to dissolve, and force sodium hydride 4. 558g ( 190 ol 20- 60 ° C reaction 0. 5-12h, cooled to room temperature, add p-phenyl-1-cyclopentyl oxirane (home-made) 35. 75g (190mmol) and 45ml DMSO solution, drip, oil bath heated 20- 70 ° C reaction 0. 5_ 12h, under ice bath conditions, add 120ml of ice water, keep the internal temperature below 30 ° C.
- Step 3 Purification and related purification of 3-[(2-cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octane free base column chromatography deal with
- Step 4 Same as [Example 1] Step 4
- Step 5 (2S,3R)-3-[(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)-brominated- 1-nitrogen Heterobicyclo[2, 2, 2]
- the compound of Example 3 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reacting with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galact
- Step 1 Same as [Example 1] Step 1
- Steps 2, 3 Same as the example [3] Steps 2, 3
- Step 4 Same as [Example 1] Step 4
- Step 5 ( 2R, 3R) -3- [(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy] - 1-(3-phenoxypropyl)-bromide-1-nitrogen Heterobicyclo[2, 2, 2]
- the compound of Example 4 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, gluconic acid or galactonic
- Step 1 with the same embodiment [1] Step 1
- Step 2 Preparation of racemic quinolol 3- [(2-cyclopentyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2, 2,
- Step 3 3-[(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2] octopine free base column chromatography purification and related purification deal with
- the sample of the above step 2 was separated on a silica gel column, and the purity of the sample was monitored by using a TLC plate using ammoniated methylene chloride or chloroform with methanol as a mobile phase.
- the sample (2R, 3S), (2R) , 3R) , (2S, 3R) , (2S, 3S) Four configurations 3- [(2-cyclopentyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2 , 2, 2] Xinzhi free base can be purified into (2R, 3S), (2S, 3R) and (2R, 3R), (2S, 3S) two racemic free bases after elution.
- the cleavage moiety is (2R,3S), (2S, 3R) racemate configuration 3-[(2-cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2 , 2, 2] octane free base 22.05 g, yield 81.12%: post-column elution fraction is (2R, 3R), (2S, 3S) configuration 3-[(2-cyclopentyl-2-hydroxyl) - 2-phenyl) ethoxy]-1-azabicyclo[2,2,2]octyl free base 21.29g, yield 78.33%
- Step 4 Same as [Example 1] Step 4
- the compound of Example 5 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reacting with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galact
- Step 1 Same as [Example 1] Step 1
- Steps 2, 3 Same as the example [5] Steps 2, 3
- Step 4 Same as Embodiment 1 Step 4
- Step 4 (2R, 3R), (2S, 3S) configuration 3- [(2-cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl) -Bromo-1-azabicyclo[2, 2, 2] octyl
- Quaternization will produce 3-[(2-cyclopentyl-2-hydroxy-2-) with different ratios (2R, 3S), (2R, 3R), (2S, 3R) (2S, 3S) configuration Phenyl)ethoxy]_1 -(3-phenoxypropyl)-bromo-1-azabicyclo[2 2 , 2]octane mixture.
- the compound of Example 6 removes a bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid, and examples include a salt derived from a mineral acid, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or gal
- the desiccant was filtered off, the pump was depressurized (40-42 ° C -0.095 MPa) to remove the solvent, and then the oil pump was used to collect 291.13 g of the 117-127'C/3 mmHg fraction under reduced pressure.
- Step 3 3-[(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2, 2, 2]octane free base column chromatography purification and related purification treatment
- Step 4 Same as [Example 1] Step 4
- Step 5 (2R,3S)-3-[(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy (3-phenoxypropyl)-bromide-1-azabicyclo[2, 2,
- the compound of Example 7 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reacting with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galact
- Steps 1, 2, 3, and 4 are the same [Example 7] Steps 1, 2, 3, 4
- Step 4 is the same as [Example 1] Step 4
- Step 5 (2S,3S) -3-[(2-Cyclohexyl-2-hydroxy-2-indolyl)ethoxy]-1-(3-phenoxypropyl)-bromo-1-aza Double loop [2, 2,
- the compound of Example 8 removes a bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reacting with other acids, a salt of a pharmaceutically acceptable acid, and examples include a salt derived from a mineral acid, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or
- Step 1 Same as [Example 7] Step 1
- Step 2 Preparation of 3-[(2-cyclohexyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octanyl free base by R- 3-quinol
- R-3-quinucin 18.72g (147 leg ol), dissolved in 190 ml of DMSO, and added 7.591 g (190 mmol) of sodium amide.
- Step 3 3-[(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octane free base column chromatography purification and related purification treatment
- Step 4 Same as [Example 1] Step 4
- Step 5 (2S,3R) Configuration 3-[(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy]- 1_(3-phenoxypropyl)-bromo-1-aza Double ring [2, 2, 2]
- the compound of Example 9 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic
- Step 1 Same as the example [7] Step 1
- Steps 2 and 3 are the same as the examples [9] Steps 2, 3
- Step 4 Same as [Example 1] Step 4
- Step 5 ( 2R, 3R) -3- [(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy]-1-(3-phenoxypropyl)-bromo-1-aza Double ring [2, 2, 2]
- reaction solution was removed at 25-40 ° C under a reduced pressure of water, and the solvent was evaporated to give a yellow oil, diethyl ether was added to precipitate a solid, and the solid was filtered to give an off-white solid (2R. 3R) -3 - [(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy]-1-(3-phenoxypropyl)-bromo-1-azabicyclo[2, 2, 2]octane 4. 205g,. Yield 84. 95%.
- the compound of Example 10 removes a bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid, examples include salts derived from inorganic acids, inorganic Acid salt such as hydrochloride
- Step 1 Same as [Example 7] Step 1
- Step 2 Preparation of racemic quinolol 3- [(2-cyclohexyl-2-hydroxy-2-phenyl)ethoxy azabicyclo [2, 2, 2] octopine free base
- Step 3 3-[(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2,2,2]octane free base column chromatography purification and related purification deal with
- step 2 sample is separated on a silica gel column, and ammoniated methylene chloride or trichloromethane is added to methanol as a mobile phase.
- Step 4 Same as [Example 1] Step 4
- Step 5 (2R, 3S), (2S.3R) Configuration 3- [(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)- Bromide-1-azabicyclo
- Example 11 The compound of Example 11 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucur
- Step 1 Same as [Example 7] Step 1
- Steps 2, 3 Same as [Example 11] Steps 2, 3
- Step 4 Same as [Example 1] Step 4
- Step 5 (2R, 3R), (2S, 3S) Configuration 3-[(2-Cyclohexyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)- Bromide-1-azabicyclo[2, 2, 2] octane
- Example 12 The compound of Example 12 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, sulfuric acid
- Hydrogen salt, phosphite, etc. a salt derived from a relatively non-toxic organic acid such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, Fumar Acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic acid. Also included are salts of amino acids such as arginine and the like.
- the filter is filtered off, the pump is depressurized (40-42 ° C, -0. 095 MPa) to remove the solvent, and then the oil pump is used to collect 113-126 ° C / 3 mmHg fraction 282. 7 g, yield 82. 37% .
- Step 3 Purification and related purification of 3-[(2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2,2,2]octane free base column chromatography deal with
- step 2 The above sample of step 2 was separated on a silica gel column, and the purity of the sample was monitored by TLC plate using ammoniated dichloromethane or chloroform with methanol as the mobile phase.
- the sample (2R, 3S), (2S) , 3S) Two configurations of 3-[(2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2, 2, 2]octyl free base by elution It can be purified into (2R, 3S), and (2S, 3S) two free bases to obtain (2R, 3S)-3-([2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy - 1-Azabicyclo[2,2,2]octyl free base 17.5548g, yield 80.42%; (2S, 3S) -3- [(2-cyclobutyl-2-hydroxy-2-) Phenyl) ethoxy]-1-azabicyclo[2,2,2]octane
- Step 4 Same as [Example 1] Step 4
- Example 13 The compound of Example 13 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucur
- Steps 1, 2, and 3 are the same [Example 13] Steps 1, 2, 3
- Step 4 is the same as [Example 1] Step 4
- Step 5 ( 2S. 3S ) -3- [(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-( 3-phenoxypropyl)-bromide-1-nitrogen Heterobicyclo[2, 2,
- reaction solution was removed under reduced pressure at 25-40 ° C under a reduced pressure of water to give a yellow oil, and diethyl ether was added to precipitate a solid, and the solid was filtered to give an off-white solid (2S. 3S ) -3 - [(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]-1-(3-phenoxypropyl)-bromo-1-azabicyclo[2, 2, 2] octyl 726 3. 726g, yield 79. 34%.
- the compound of Example 14 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reacting with other acids, a salt of a pharmaceutically acceptable acid, examples include salts derived from inorganic acids, inorganic Acid salt such as hydrochloride
- Step 1 is the same as [Example 13] Step 1
- Step 2 Preparation of 3-[(2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octane free from R-3-quinol
- Step 3 3-[(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2, 2, 2]octane free base column chromatography purification and related purification deal with
- the sample of the above step 2 was separated on a silica gel column, and the purity of the sample was monitored by TLC plate using ammoniated methylene chloride or trichloromethane and methanol as a mobile phase.
- the sample (2R, 3R), 2S, 3R) Two configurations of 3-[(2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octyl free base by washing It can be purified into (2R, 3R), and (2S, 3R) two free bases to obtain (2R, 3R)-3-([2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy
- Step 4 Same as [Example 1] Step 4
- Step 5 ( 2S. 3R) - 3- [(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)-brominated-1_aza Bicyclo[2, 2, 2]octane
- reaction solution was removed at 25-40 ° C under a reduced pressure of water, and the solvent was evaporated to give a yellow oil, and diethyl ether was added to precipitate a solid, and the solid was filtered to give an off-white solid (2S. 3R) -3 - [(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]-1-(3-phenoxypropyl)-bromo-1-azabicyclo[2, 2, 2] octyl 790 ⁇ Yield: 80. 72%.
- Example 15 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or gal
- Step 1 Same as the example [13] Step 1
- Step 4 Same as [Example 1] Step 4
- Step 5 ( 2R, 3R) - 3- [(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy] -1_(3-phenoxypropyl)-bromo-1-aza Double ring [2, 2, 2] Xin ⁇ replacement page (Article 26)
- the compound of Example 16 removes a bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galact
- Step 1 Same as the example [13] Step 1
- Step 2 Preparation of racemic quinolol 3- [(2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2 2 2]octyl free base
- Step 3 3-[(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2] octopine free base column chromatography purification and related purification deal with
- the sample of the above step 2 was separated on a silica gel column, and the purity of the sample was monitored by using a TLC plate using ammoniated methylene chloride or chloroform with methanol as a mobile phase.
- the sample (2R, 3S), (2R) , 3R) , (2S, 3R) , (2S, 3S) Four configurations 3-[(2-cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2 , 2, 2] Xinzhi free base can be purified into (2R, 3S), (2S, 3R) and (2R, 3R), (2S, 3S) two racemic free bases, which are washed first.
- Step 4 Same as [Example 1] Step 4
- Step 5 (2R, 3S), (2S.3R) - 3-[(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)- Bromo-1-azabicyclo[2, 2, 2]octane
- reaction solution is removed at 25-40 ° C under a reduced pressure of water, and the solvent is evaporated to give a yellow oil, and diethyl ether is added to precipitate a solid, and the solid is filtered to give an off-white solid (2R, 3S) ( 2S, 3R) Configuration 3-[(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)-bromo-1-azabicyclo[ 2, 2, 2] ⁇ 3. 916g, yield 83. 38%.
- the compound of Example 17 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic
- Steps 1, 2, and 3 are the same as the examples [17] Steps 1, 2, 3
- Step 4 Same as Embodiment 1 Step 4
- Step 5 (2R, 3R), (2S, 3S) - 3-[(2-Cyclobutyl-2-hydroxy-2-phenyl)ethoxy]- 1_(3-phenoxypropyl)-bromo -1-azabicyclo[2, 2, 2] xin
- Example 18 The compound of Example 18 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucur
- the filter is filtered off, the pump is depressurized (40-42 ° C, -0. 095 MPa) to remove the solvent, and then the oil pump is used to collect 111-124 ° C / 3 mmHg fraction 275. 8 g, yield 83. 75% .
- the sample of the above step 2 was separated on a silica gel column, and the purity of the sample was monitored by using a TLC plate using ammoniated methylene chloride or trichloromethane and methanol as a mobile phase.
- the sample (2R, 3S), 2S, 3S ) Two configurations of 3-[(2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2, 2, 2]octyl free base by washing Purification can be purified into (2R, 3S), and (2S, 3S) two free bases to obtain (2R, 3S) -3-[(2-cyclopropyl-2-hydroxy-2-phenyl) ethoxylate yl] - 1-azabicyclo [2, 2, 2] oct-free base embankment 16.
- Step 4 Same as [Example 1] Step 4
- Step 5 ( 2R. 3S ) -3- [(2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy]-1-(3-phenoxypropyl)-brominated 1-nitrogen Heterobicyclo[2, 2,
- Example 19 The compound of Example 19 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucur
- Steps 1, 2, and 3 are the same as in [Example 19] Steps 1, 2, and 3
- Step 4 is the same as [Example 1] Step 4
- Step 5 ( 2S. 3S ) - 3- [(2-Cyclopropyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)-bromide-1-nitrogen Heterobicyclo[2, 2,
- the compound of Example 20 removes a bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galact
- Step 1 is the same as [Example 19] Step 1
- Step 2 Preparation of 3-[(2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octane free from R-3-quinol Alkali
- Step 3 Purification and related purification of 3-[(2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2,2,2]octanyl free base column chromatography deal with
- the sample of the above step 2 was separated on a silica gel column, and the purity of the sample was monitored by using a TLC plate using ammoniated chloroform or chloroform and methanol as a mobile phase.
- the sample (2R, 3R), 2S, 3R) Two configurations 3-[(2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2] octopine free base by washing It can be purified into (2R, 3R), and (2S, 3R) two free bases to obtain (2R,3R)-3-([2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy • base]- 1-azabicyclo[2,2,2]octyl free base 16.45 g, yield 80.25%; (2S, 3R) configuration 3-[(2-cyclopropyl-2-hydroxyl) -2-Phenyl)ethoxy]- 1-azabicyclo[2,2,2]octyl free base
- Step 4 Same as [Example 1] Step 4
- Step 5 (2S.3R) - 3-[(2-Cyclopropyl-2-hydroxy-2-phenyl)ethoxy]-1-(3-phenoxypropyl)-brominated- 1-nitrogen Heterobicyclo[2, 2, 2]
- Example 21 The compound of Example 21 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucur
- Step 1 with the same embodiment [19] Step 1
- Steps 2 and 3 are the same as the examples [21] Steps 2, 3
- Step 4 Same as [Example 1] Step 4
- Step 4 ( 2R, 3R) -3- [(2-Cyclopropyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl)-bromo- 1-nitrogen Heterobicyclo[2, 2, 2]
- reaction mixture was evaporated to dryness under reduced pressure of 25-40 <[>&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
- the compound of Example 22 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a hydroxide by reacting with other acids.
- salts of pharmaceutically acceptable acids examples include salts derived from inorganic acids, inorganic acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphoric acid a salt, a hydrogen phosphate, a dihydrogen phosphate, a sulfate, a hydrogen sulfate, a phosphite, or the like; a salt derived from a relatively non-toxic organic acid such as acetic acid, propionic acid, isobutyric acid, or C.
- inorganic acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphoric acid a salt, a hydrogen phosphate, a dihydrogen phosphate, a sulfate, a hydrogen sulfate, a phosphite, or the like
- a salt derived from a relatively non-toxic organic acid such as ace
- Step 1 with the same embodiment [19] Step 1
- Step 2 Preparation of 3-[(2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy]-1-azabicyclo[2,2,2]octanyl free base by racemic quinolol
- Step 3 3-[(2-Cyclopropyl-2-hydroxy-2-phenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octyl free radical column chromatography purification and related purification deal with
- Step 4 Same as [Example 1] Step 4
- Step 5 (2R, 3S), (2S. 3R) Configuration 3- [(2-Cyclopropyl-2-hydroxy-2-phenyl)ethoxy]- 1-(3-phenoxypropyl) -Bromo-1-azabicyclo[2,2,2]octane
- Example 23 The compound of Example 23 is reacted with Ag 2 to remove a bromine atom to form a hydroxide, and reacted with other acids to be converted into a corresponding salt, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucur
- Step 1 with the same embodiment [19] Step 1
- Step 2 3 with the same example [23] Step 2 3
- Step 4 Same as Embodiment 1 Step 4
- Step 5 (2R, 3R), (2S, 3S) configuration 3- [(2-cyclopropyl-2-hydroxy-2-phenyl)ethoxy]-1 - (phenylphenoxypropyl) -Bromo-1-azabicyclo[2 2 2]octane
- the compound of Example 24 removes the bromine atom into a hydroxide by reacting with Ag 20 , and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid, and examples include a salt derived from a mineral acid, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, sulfuric acid
- inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, sulfuric acid
- Hydrogen salt, phosphite, etc. a salt derived from a relatively non-toxic organic acid such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, Fumar Acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic acid. Also included are salts of amino acids such as arginine and the like.
- Step 1 Same as [Example 1] Step 1
- Step 5 ( 2S, 3R) -3- [(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]- 1-(2-phenoxyethyl)-bromide-1-nitrogen Heterobicyclo[2, 2, 2] xin
- the compound of Example 25 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic
- Step 1 Same as [Example 1] Step 1
- Steps 2 and 3 are the same as [Example 3] steps 2, 3
- Step 4 is the same as [Example 25] Step 4
- Step 5 (2R,3R)-3-[(2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]-1-(2-phenoxyethyl)-brominated- 1-nitrogen Heterobicyclo[2, 2, 2]
- the compound of Example 26 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid, and examples include salts derived from inorganic acids, inorganic Acid salt such as hydrochloride
- Step 1 Same as the example [1] Step 1
- Step 4 (2R,3R)-3-([2-Cyclopentyl-2-hydroxy-2-phenyl)ethoxy]-1-phenoxymethyl-bromide-1-azabicyclo[2 , 2, 2] octane
- the compound of Example 27 removes the bromine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic
- Step 1 with the same embodiment [1] Step 1
- the compound of Example 28 removes a chlorine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen hydride, dihydrogen phosphate, sulphate, hydrogen sulphate, phosphite, etc.; a relatively non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, Benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic acid
- Step 1 230 g of 1-naphthyl-1-cyclopentylethylene oxide was obtained according to the method of the literature (2) .
- Step 2 Preparation of 3-[(2-cyclopentyl-2-hydroxy-2-naphthalenyl)ethoxy]- 1-azabicyclo[2, 2, 2]octane free from R-3-quinol
- Step 3 3-[(2-Cyclopentyl-2-hydroxy-2-naphthalenyl)ethoxy]- 1-azabicyclo[2,2,2]octane free base column chromatography purification and related purification deal with
- the sample of the above step 2 was separated on a silica gel column, and the purity of the sample was monitored by a TLC plate using ammoniated methylene chloride or trichloromethane and methanol as a mobile phase.
- the sample (2R, 3R), (2S) , 3R) Two configurations of 3-[(2-cyclopentyl-2-hydroxy-2-naphthyl)ethoxy]- 1-azabicyclo[2, 2, 2]octane free base It can be purified into (2R, 3R), and (2S, 3R) two free bases to obtain (2R, 3R) configuration 3_[(2-cyclopentyl-2-hydroxy-2-naphthyl)ethoxy - 1-Azabicyclo[2,2,2]octyl free base 22.36 g, yield 86.5%; (2S, 3R) configuration 3-[(2-cyclopentyl-2) 55% ⁇ The hydroxy- 2-naphthyl)
- Step 4 Same as [Example 1] Step 4
- Step 5 ( 2R. 3R) - 3- [(2-Cyclopentyl-2-hydroxy-2-naphthyl)ethoxy]_1-(3-phenoxypropyl)-bromo-1-aza Double loop [2, 2,
- 033g (51. 3mmol) was added to the (2R, 3R) configuration base 3.
- 322g (9. lmmol) was added to a 100ml bottle, added with chloroform 18ml, dissolved to give a yellow transparent solution, added 3-bromopropoxybenzene 11.
- the compound of Example 29 removes a chlorine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, lemon
- Citric acid tartaric acid, methanesulfonic acid, glucuronic acid or galactonic acid. Also included are salts of amino acids such as arginine and the like.
- Step 1 1-o-chlorophenyl-1-cyclopentyl ethylene oxide
- the remaining solid ice bath was added to 0-5 ° C, and ice water was added to 2630 ml (the internal temperature was controlled at 0- 5°C), dilute in about 1 hour, extract with isopropyl ether (650ml X 3 times), combine the ether layer, wash with water to neutral (pH of the water washing solution is 7.0), ether layer with anhydrous sodium sulfate Dry overnight.
- the filter is filtered off, the pump is depressurized (40-42 ° C, -0. 095 MPa) to remove the solvent, and then the oil pump is used to collect 119-132 ° C / 3 mmHg fraction 315. 8 g, yield 83. 36% .
- Step 2 Preparation of R-3-quinuclol 3- [(2-cyclopentyl-2-hydroxy-2-o-chlorophenyl)ethoxy]- 1-azabicyclo[2, 2, 2] octane Free base
- Extract with isopropyl ether 100 ml ⁇ 3 times; wash the combined ether layer with saturated aqueous NaCl solution, 100 ml*3 times.
- the organic layer is dried over anhydrous sodium sulfate and filtered.
- the solvent was removed under reduced pressure to give 3-[(2-cyclopentyl-2-hydroxy-2-o-chlorophenyl)ethoxy]-1-azabicyclo[2 2, 2] hydrazine free.
- the base is 49.48 g, the yield is 96.31%.
- Step 3 Purification and purification of 3-[(2-cyclopentyl-2-hydroxy-2-o-chlorophenyl)ethoxy]- 1-azabicyclo[2 2 2]octyl free base column chromatography deal with
- Step 5 (2R 3R) - 3- [(2-Cyclopentyl-2-hydroxy-2-o-chlorophenyl)ethoxy]- 1-(3-phenoxypropyl)-bromide-1- Azabicyclo[2 2, 2]octane
- the compound of Example 30 removes a chlorine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a reaction with other acids.
- salts of pharmaceutically acceptable acids examples include salts derived from inorganic acids, inorganic acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphoric acid a salt, a hydrogen phosphate, a dihydrogen phosphate, a sulfate, a hydrogen sulfate, a phosphite, or the like; a salt derived from a relatively non-toxic organic acid such as acetic acid, propionic acid, isobutyric acid, or C.
- inorganic acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphoric acid a salt, a hydrogen phosphate, a dihydrogen phosphate, a sulfate, a hydrogen sulfate, a phosphite, or the like
- a salt derived from a relatively non-toxic organic acid such as ace
- the bromocyclopentanthene 78. 28 g is added to the granules of the granules of the granules of the granules. It was added dropwise to the reaction solution, and about 5 minutes after the reaction was completely started, the reaction liquid gradually changed from light reddish brown to colorless, and the internal temperature rose to 63-65 °C. Another 547. 93 g of bromocyclopentanone was added dropwise, and about 35 tnin was dropped, and the reflux temperature was gradually increased to 75_77 °C. The oil bath was refluxed for 2 hours.
- Step 2 Preparation of l-(3-pyridyl)-1-cyclopentyl ethylene oxide
- the remaining solid ice bath was incubated at 0-5 ° C, and ice water was added to 2630 ml (the internal temperature was controlled at 0-5). °C), after about 1 hour, was extracted with isopropyl ether (650 ml X 3 times), the ether layer was combined, washed with water to neutral (pH 7.0 of water), and the ether layer was dried over anhydrous sodium sulfate overnight.
- the desiccant was filtered off, the pump was depressurized (40-42 Torr, -0.095 MPa) to remove the solvent, and then an oil pump was used to collect 285.5 g of a 121- 13 rC/3 mmHg fraction.
- Step 3 Preparation of 3-[(2-cyclopentyl-2-hydroxy-2-(3-pyridyl))ethoxy]-1-azabicyclo[2, 2, 2
- Step 4 3-[(2-Cyclopentyl-2-hydroxy-2-(3-pyridyl))ethoxy]- 1-azabicyclo[2, 2, 2] octane free base column chromatography Purification and related purification treatment
- the sample of the above step 3 was separated on a silica gel column, and the purity of the sample was monitored by using a TLC plate using ammoniated chloroform or chloroform and methanol as a mobile phase.
- the sample (2R, 3R), 2S, 3R) Two configurations of 3-[(2-cyclopentyl-2-hydroxy-2-(3-pyridyl))ethoxy]-1-azabicyclo[2, 2, 2]xin
- the free base can be purified to (2R, 3R), and (2S, 3R) two free bases to obtain (2R,3R)-3-([2-cyclopentyl-2-hydroxy-2- ( 3-pyridyl
- Step 5 Same as [Example 1] Step 4
- Step 6 (2R, 3 ) - 3_[(2-Cyclopentyl-2-hydroxy-2-(3 pyridyl))ethoxy]-1-(3-phenoxypropyl)-bromide-1 - azabicyclo[2,
- the compound of Example 31 removes a chlorine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen dihydrogenate, sulfate, hydrogen sulfate, phosphite, etc.; a relatively non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, Benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or gal
- the mixture was heated under reflux for 3 hours, and the organic layer was separated, cooled and extracted with isopropyl ether ( 200 ral X 3 times), the organic layers were combined, washed with 1% N C0 3 (500 ⁇ 3 times) and water (500 ⁇ 3 times), and the organic layer was dried over anhydrous magnesium sulfate overnight, filtered and evaporated. 6 ⁇
- the solvent 42 ° C, -0. 095MPa), and then distilled under reduced pressure with an oil pump, collecting 108-119 ° C / 6-7 mmHg oil 272.6 g.
- Step 2 Preparation of 1-(2-furyl)-1-cyclopentyl ethylene oxide
- Step 3 Preparation of R-3- 3-quinolol 3- [(2-Cyclopentyl-2-hydroxy-2-(2-furyl))ethoxy]- 1-azabicyclo[2, 2, 2 Octane free base
- R- 3-quinucin 18.72 g (147 mmol) was dissolved in 190 ml of DMSO, and sodium hydride 7.59 g (190 mmol), 20-60.
- C reaction 0.5-12h, cooled to room temperature, adding 1-(2-furyl) 1-cyclopentyl ethylene oxide (home-made) 33.88g (190nmiol) and 45ml DMSO solution, drip, oil bath heating
- the reaction was carried out at 20-70 ° C for 0.5-12 h, and 120 ml of ice water was added under ice bath, and the internal temperature was kept below 30 Torr.
- Step 4 3-[(2-Cyclopentyl-2-hydroxy-2-(2-furyl))ethoxy]- 1 -azabicyclo[2, 2, 2]octane free base column chromatography Purification and related purification treatment
- Step 5 Same as [Example 1] Step 3
- Step 6 (2R.3R) - 3-[(2-Cyclopentyl-2-hydroxy-2-(2-furyl))ethoxy]-1-(3-phenoxypropyl)-bromination -1- azabicyclo[2,
- the compound of Example 32 removes a chlorine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid
- examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid, phthalic acid, benzene Sulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid, methanesulfonic acid, glucuronic acid or galactonic
- Step 5 Same as [Example 1] Step 4
- Step 6 Same as [Example 31] Step 6
- Step 7 ( 2R. 3R) -3- [( 2 -Cyclopentyl-2-methoxy-2-(3 pyridyl))ethoxy] - 1 -( 3-phenoxypropyl)-bromo -1-azabicyclo[2, 2, 2] xin
- Example 33 removes a chlorine atom into a hydroxide by reacting with Ag 2 0, and can be converted into a corresponding salt by reaction with other acids, a salt of a pharmaceutically acceptable acid, examples include salts derived from inorganic acids, inorganic Acid salts such as hydrochloride, bromide, iodide, nitrate, carbonate, bicarbonate, phosphate, hydrogen phosphate, dihydrogen phosphate, sulfate, hydrogen sulfate, phosphite, etc.; a non-toxic organic acid-derived salt, such as acetic acid, propionic acid, isobutyric acid, malonic acid, benzoic acid, succinic acid, suberic acid, fumaric acid, mandelic acid
- Example compound antagonizes the intensity of contractile response of guinea pig tracheal smooth muscle induced by carbachol (CCh)
- guinea pig isolated tracheal smooth muscle specimens Guinea pigs were anesthetized with urethane and quickly removed the trachea between the larynx and the carina, and placed in a Krebs-Henseleit (K-H) solution of 5% ⁇ 2 and 95 % 0 2 mixture.
- K-H Krebs-Henseleit
- the tracheal slices with a width of about 3 mm and a length of 20 mm were separated by surgical scissors, and the ends were ligated with 4 to 0 silk wires, and placed in a 37 ° C constant temperature bath containing 5 ml of KH solution ( pH 7. 4), continuously containing a mixture of 5% CO 2 and 95% 02 .
- the upper end of the muscle tension transducer was given a resting tension of 1.0 g. The muscle tension was recorded, and the nutrient solution was changed every 20 min. The experiment was started after 60 min of equilibration.
- Method of administration After the trachea is stabilized, add 3 X l (T 6 mol/LCCh to the bath, and after the peak tension of the trachea reaches a peak, respectively, l (T 9 ⁇ l (T 5 m 0 l cumulative dose plus The method of adding the example compound, ipratropium bromide and tiotropium bromide in the wheat bath to observe the tracheal strip relaxation, if there is no reaction (threshold concentration), continue to add the next dose in sequence, such as the reaction After the diastolic platform is reached, add the next dose. Repeat the above procedure until the contraction curve reaches a minimum. Finally add 1(T)1/L isoproterenol to maximize relaxation and record the curve.
- CCh 3 X 10- 6 mol/L CCh can produce a long-lasting stable contractile response in guinea pig tracheal smooth muscle.
- Cumulative dosing method was added to the wheat bath to the example compound and the reference drugs ipratropium bromide and tiotropium bromide.
- Each of the example compounds and the control drug can relax the contraction of the tracheal smooth muscle caused by CCh. 2.
- Table 2 show that the compounds of each example have a significant blocking effect on M, among which (2R, 3R) type compounds have the strongest effect, and the strength of various compounds and positive control agents ipratropium bromide and tiotropium quite.
- Both the compound of the present invention and ipratropium bromide have a faster onset time than tiotropium bromide, whereas tiotropium bromide has a slower onset of action.
- the compound of the present invention reacts with tiotropium bromide for a long period of time.
- Method Transfected Chinese hamster egg cells (CHO) rai , m 2 , and ra 3 cells in DMEM medium (containing 153 ⁇ 4 fetal bovine serum, L-glutamine, 1% non-essential amino acids, 1% antibiotic) / Antifungal), cultured at 37 ° C, 5% C0 2 incubator.
- the cells in the culture flask grow and proliferate to form a monolayer of cells, and when the bottom of the bottle is about 90%, the medium is discarded, and PBS (pH 7.4) is used.
- the buffer was washed twice, and then the cells were scraped off with ice-cold phosphate buffer (pH 7.7, containing 5 mmol/LMgCl 2 ).
- the collected cells were homogenized with a Teflon glass homogenizer, and the homogenate was centrifuged at 20000 r X for 20 min, and the reaction buffer (pH 7.7, containing 5ramol/LMgCl 2 ) was homogenized to form a membrane protein suspension.
- the amount of protein added to each reaction tube was: (about 0.05 rag), m 2 (0. 05 mg), m 3 (0. lrag); [3 ⁇ 4]- QNB concentration was 0. 1-2.
- each tube was: ra 3 ( 1. 042nmol/L), ra 2 ( 1. 81nraol/L); Concentration of non-labeled competitor Pirenzepine (PZ, nu selective competitor) or Gallamine (GI, m 2 selective competitor) or 4-DAMP (m 3 selective competitor) or a compound of the invention, with a final concentration of 10 - KTmol / a total of 11 doses; the same amount of membrane protein samples were added; the total volume of the reaction was 300 ⁇ , and the competitive binding reaction was carried out.
- a non-specific binding tube was also provided, and non-specific binding was measured with a large amount of atropine (final concentration of 1 Mmol/L).
- the reaction was allowed to proceed at 25 ° C for 30 min, and the reaction was stopped with ice-cold reaction buffer, and then collected on a glass fiber filter using a multi-head cell harvester. After drying at 80 ° C, the filter was placed in a liquid flash bottle, 5 ml of liquid scintillator was added, and the light was measured overnight, and the cpm was measured with a liquid scintillation meter.
- the Ki value of each of the three M receptor subtypes of the competitor was calculated by Graphpad prism software, and converted to pKi value to compare the selectivity of PZ, GI, 4-DAMP and the test compound to the M receptor subtype.
- CHO cells transfected with three M receptor subtype cDNAs of m 2 and m3 were used to identify the M receptor subtype selectivity of the compounds of the present invention.
- the results showed that these three cells can be used for the selective identification of drug receptors.
- the experimental results show that the tested compounds have the highest selectivity to the m 3 receptor, followed by the nu receptor, and the m 2 receptor has the lowest selectivity; among them: 3 ⁇ 4 and nu
- Tidal volume, airway velocity, and transpulmonary pressure Ulatan 1. 5g/kg intraperitoneal injection of anesthetized guinea pigs. The guinea pig is fixed on the back, the tracheal intubation is performed, and the external jugular vein is inserted into the indwelling needle; the guinea pig is placed in the body plethysmograph box, and the blunt needle of the thoracic cannula is inserted into the intercostal space of the guinea pig front chest 4th to 5th, and the chest can be measured. Internal pressure (the sign is that the water gauge has a negative water column and fluctuates with the guinea pig's breathing).
- the MedLab biosignal acquisition and treatment system was used to record the tidal volume, airway velocity and transpulmonary pressure value of Mch pre-guinea pigs as the base value. Intravenous injection of 10 pg/kg body weight Mch. The changes in airway velocity, tidal volume and transpulmonary pressure in guinea pigs were observed within 5 minutes. Calculation of ⁇ and : Calculate the change in R a , the percentage increase in value and the percentage decrease in C dyn after aerosol inhalation of Mch.
- Dose-effect relationship Each of the 27 compounds of the guinea pigs was randomly divided into 3 groups: vehicle control group 15 guinea pigs, example compounds 4, 10, 14, 26 1 ⁇ g/kg, example compounds 4, 10, 14, 26 3 ⁇ g / kg and the compound of the example compound 4, 10, 14, 26 10 g / kg; airway drip into the above concentration of drugs for 30min, intravenous injection of 10 ⁇ g / kg body weight Mch excitation, determination of airway within 5min Resistance and lung dynamic compliance.
- Dose-effect relationship The compound doses of the compounds of Examples 4, 10, 14, and 26 were lug/kg, 3 ⁇ g/kg, 10 ⁇ g/kg, respectively. After intravenous instillation for 30 minutes, 10 ⁇ g/kg body weight was administered intravenously. Mch excitation, determination of airway resistance (? consult) within 5 min and lung dynamic compliance 1 ⁇ 2,found), the results are shown in the table below -
- Example compounds 4, 10, 14, 26 antagonize Mch-induced bronchial contractile response in guinea pigs.
- the guinea pig airway was instilled into the compounds of Examples 4, 10, 14, and 26 at 1, 3, and 10 ⁇ g/kg to inhibit the increase in airway resistance and the decrease in lung dynamic compliance in a dose-dependent manner.
- Example Compound 4 The inhibition rate of airway resistance increase in the three dose groups was 64. 2% (p ⁇ 0.01), 86. ⁇ % ⁇ p ⁇ 0. 001) and 90. %% ⁇ p ⁇ 0 001); The inhibition rate of lung dynamic compliance decreased by 11.2% (p>0.05), 46.6% ( ⁇ 0. and 50.0% ( ⁇ 0. 001).
- the inhibitory rate of airway resistance was increased by 63.7% ( ⁇ 0.01), 84.5% ( ⁇ 0.0011) and 91.3% ( ⁇ 0. 001); 9% ( ⁇ >0.05), 45.9% ( ⁇ 0. 001), and 49.8% ( ⁇ 0. 001), respectively.
- the inhibition rate of airway resistance increase in the three dose groups of Example Compound 14 was 63.5% ⁇ 0. 01), 85.4% ( ⁇ 0. 001) and 90.5% ⁇ 0 001);
- the inhibition rate of lung dynamic compliance decreased by 11. 0% ( ⁇ >0.05), 46.1% ⁇ 0. 001), and 49.5% ⁇ 0. 001).
- the inhibitory rate of the increase in airway resistance was 6.4% ( ⁇ 0.01), 87.
- Time-effect relationship Intravenous injection of Mch 10 ⁇ g/kg, guinea pig airway resistance increased by 328%. Guinea pig airway instillation [Example compounds 4, 10, 14, 26] 10 ug / kg 0. 25h, the inhibition rate of airway resistance increase is 80% or more; lh can reach the maximum inhibition rate of 90% or more . 001. 001. 001. 001. 001. 001. 001.
- the doses of the compounds of Examples 4, 10, 14, 26 were reduced to 5 yg/kg, and the results showed that the guinea pig airway was instilled into the example compounds 4, 10, 14, 26 5 ug/ After kg, the airway resistance inhibition rate was 85% or more after 12h P ⁇ 0. 001, and the airway resistance was suppressed after 24h.
- the active ingredient example compound 10 and the bacteriostatic agent are dissolved in 0, filled in a small brown bottle, and have a spindle-type cap, which can be used for dropping nose.
- the single nasal drops can dissolve 2. 0-20mg per 10ml of water, and can be formulated into different concentrations of nasal drops; the amount of the bacteriostatic agent benzalkonium chloride can be varied within the range of 1- 5mg.
- the active ingredients (Example Compound 10 and fenoterol) and the bacteriostatic agent (benzalkonium chloride) are dissolved in 0, filled in small brown bottles, and have a spindle-type cap. , the cap can be used to drip the nose.
- the amount of the bacteriostatic agent benzalkonium chloride can vary from 1 to 5 mg.
- the compound nose drops are 2 - 3 drops (0.1 ml - 0.15 ml) / nostril, about 25 u g / nostril dose.
- the above components are dissolved in pure 0, and placed in a bottle, and a fixed-purpose pump suitable for nasal spray is installed, which is 70-90 ⁇ l per spray. For every 1-2 sprays / nostrils, about 21-60 u g / nostril dose.
- the amount of Compound 4 can be increased.
- Example Compound 4 After dissolving Example Compound 4 with ethanol and oleic acid, one-step filling was carried out to charge HFA-134a. Under the production conditions of 100,000 grades, it is then mixed with the surfactant and the propellant 1-fluoro-3chloromethane, 2-fluoro-2chloroformamidine and 4-fluoro-2chloroacetamidine in proportion, and then passed through the press machine. Potting in a quantitative pressure vessel is sufficient. Bottle 10g, per puff 100mg, Example 4 containing compound to embodiment 20 ⁇ ⁇ .
- Capsule powder consists of the following elements
- the pharmaceutical composition for treating respiratory diseases lactose is a diluent, and the diluent may also be selected from arabinose, dextran, mannitol, mannitol, xylitol, sucrose, fructose, sorbitol, maltose, amino acid. Or glucose, etc.; sodium benzoate is a lubricant, and magnesium stearate can also be used as a lubricant.
- the compound 6 was mixed with lactose and microcrystalline cellulose, and further mixed uniformly after adding magnesium stearate, and the tablet was dry-dried, and the tablet weight was 100 mg.
- Compound 10 was dissolved in 800 ml of physiological saline, transferred to a 1000 ml volumetric flask, supplemented with physiological saline to the mark, and dispensed in ampoules, 1 ml/branch, and heated at 115 ° C for 30 min.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Otolaryngology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description















































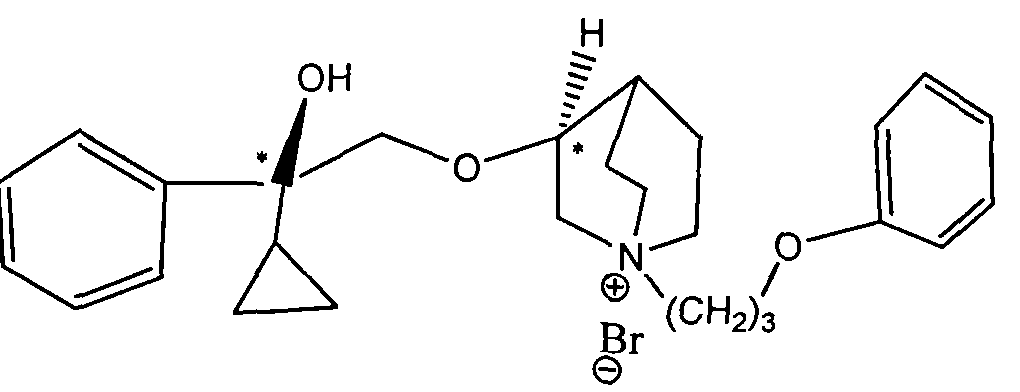
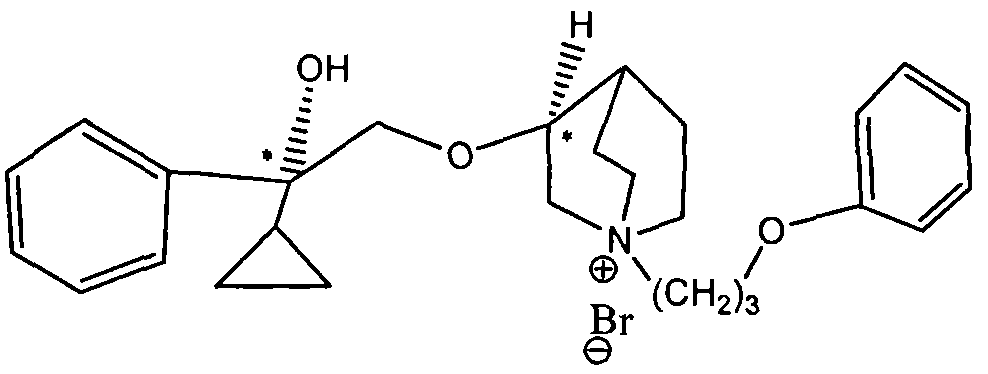



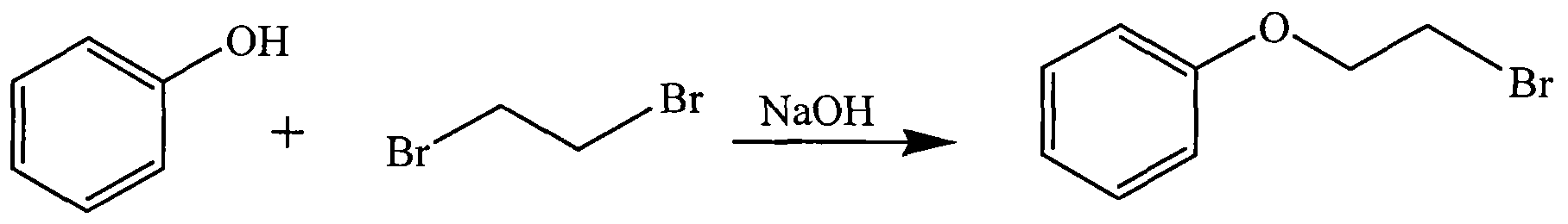


























Claims




Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201480020342.8A CN105431432B (zh) | 2013-07-13 | 2014-07-11 | 奎宁类化合物、其光学异构体及其制备方法和医药用途 |
| EP14825941.9A EP3023424B1 (en) | 2013-07-13 | 2014-07-11 | Quinine compounds, and optical isomers, preparation method and medical use thereof |
| BR112016000629-1A BR112016000629B1 (pt) | 2013-07-13 | 2014-07-11 | Compostos de quinina e isômeros ópticos, método de preparação e utilização médica dos mesmos |
| AU2014292722A AU2014292722B2 (en) | 2013-07-13 | 2014-07-11 | Quinine compounds, and optical isomers, preparation method and medical use thereof |
| RU2016104437A RU2641285C2 (ru) | 2013-07-13 | 2014-07-11 | Соединения хинина, способ их получения и их медицинское применение |
| JP2016524657A JP6218940B2 (ja) | 2013-07-13 | 2014-07-11 | キニン系化合物、その光学異性体及びその製造方法と医薬用途 |
| ES14825941T ES2726664T3 (es) | 2013-07-13 | 2014-07-11 | Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos |
| CA2921621A CA2921621C (en) | 2013-07-13 | 2014-07-11 | Quinine compounds, and optical isomers, preparation method and medical use thereof |
| KR1020167003638A KR101823451B1 (ko) | 2013-07-13 | 2014-07-11 | 퀴논계 화합물, 이의 광학 이성질체 및 이의 제조 방법과 의약 용도 |
| US14/904,766 US9751875B2 (en) | 2013-07-13 | 2014-07-11 | Quinine compounds, and optical isomers, preparation method and medical use thereof |
| ZA2016/00997A ZA201600997B (en) | 2013-07-13 | 2016-02-12 | Quinine compounds, and optical isomers, preparation method and medical use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310297901 | 2013-07-13 | ||
| CN201310297901.7 | 2013-07-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015007073A1 true WO2015007073A1 (zh) | 2015-01-22 |
Family
ID=52345746
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/000669 Ceased WO2015007073A1 (zh) | 2013-07-13 | 2014-07-11 | 奎宁类化合物、其光学异构体及其制备方法和医药用途 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US9751875B2 (zh) |
| EP (1) | EP3023424B1 (zh) |
| JP (1) | JP6218940B2 (zh) |
| KR (1) | KR101823451B1 (zh) |
| CN (1) | CN105431432B (zh) |
| AU (1) | AU2014292722B2 (zh) |
| BR (1) | BR112016000629B1 (zh) |
| CA (1) | CA2921621C (zh) |
| ES (1) | ES2726664T3 (zh) |
| RU (1) | RU2641285C2 (zh) |
| TR (1) | TR201906532T4 (zh) |
| WO (1) | WO2015007073A1 (zh) |
| ZA (1) | ZA201600997B (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016140136A1 (ja) * | 2015-03-02 | 2016-09-09 | 株式会社Lttバイオファーマ | キヌクリジン誘導体 |
| WO2018108089A1 (zh) | 2016-12-14 | 2018-06-21 | 北京硕佰医药科技有限责任公司 | 一类具有季铵盐结构的双功能化合物 |
| WO2021218833A1 (zh) | 2020-04-26 | 2021-11-04 | 北京硕佰医药科技有限责任公司 | 一种m受体拮抗剂的晶体、制备方法及其应用 |
| WO2023138478A1 (zh) * | 2022-01-19 | 2023-07-27 | 北京硕佰医药科技有限责任公司 | 一种治疗鼻炎的溶液型鼻喷雾剂及制备方法 |
| CN116514800A (zh) * | 2023-05-06 | 2023-08-01 | 湖南一格制药有限公司 | 盐酸戊乙奎醚的制备方法 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109824661B (zh) * | 2019-03-30 | 2020-06-05 | 山东博洛德生物科技有限公司 | 一种盐酸戊乙奎醚中杂质的制备方法 |
| CN109824662A (zh) * | 2019-03-30 | 2019-05-31 | 山东博洛德生物科技有限公司 | 一种奎宁类化合物及其制备方法 |
| CN109776521A (zh) * | 2019-03-30 | 2019-05-21 | 山东博洛德生物科技有限公司 | 一种含有季铵基团的奎宁类化合物及其制备方法 |
| CN114306331B (zh) * | 2020-10-10 | 2023-07-18 | 远大生命科学(武汉)有限公司 | 戊乙奎醚在治疗或预防视力损伤性眼部疾病中的用途 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2012964A1 (zh) | 1968-07-15 | 1970-03-27 | Sogeras | |
| US5654314A (en) | 1991-03-15 | 1997-08-05 | Boehringer Ingelheim Kg | Esters of bi- and tricyclic amino alcohols and their use in pharmaceutical compositions |
| CN1675206A (zh) * | 2002-06-21 | 2005-09-28 | 阿尔米雷尔普罗迪斯制药有限公司 | 奎宁环衍生物及含有相同化合物的药用组合物 |
| CN1954938A (zh) * | 2005-10-17 | 2007-05-02 | 日信工业株式会社 | 车辆用支承结构体的制造方法 |
| CN102040602A (zh) * | 2004-04-27 | 2011-05-04 | 葛兰素集团有限公司 | 毒蕈碱性乙酰胆碱受体拮抗剂 |
| CN102070631A (zh) * | 2009-11-20 | 2011-05-25 | 中国人民解放军军事医学科学院毒物药物研究所 | 戊乙奎醚光学异构体的季铵盐、药物组合物及其医药用途 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NO2005012I1 (no) * | 1994-12-28 | 2005-06-06 | Debio Rech Pharma Sa | Triptorelin og farmasoytisk akseptable salter derav |
| ES2165768B1 (es) * | 1999-07-14 | 2003-04-01 | Almirall Prodesfarma Sa | Nuevos derivados de quinuclidina y composiciones farmaceuticas que los contienen. |
| CZ294251B6 (cs) * | 2000-06-27 | 2004-11-10 | Laboratorios S. A. L. V. A. T., S. A. | Karbamáty a jejich použití pro výrobu farmaceutického prostředku |
| IL156558A0 (en) * | 2000-12-28 | 2004-01-04 | Almirall Prodesfarma Ag | Novel quinuclidine derivatives and medicinal compositions containing the same |
| ES2204295B1 (es) * | 2002-07-02 | 2005-08-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de quinuclidina-amida. |
| CN1951938A (zh) * | 2005-10-21 | 2007-04-25 | 刘丽娅 | 戊乙奎醚季铵盐及其衍生物 |
| AU2007336054A1 (en) * | 2006-12-19 | 2008-06-26 | Astrazeneca Ab | Quinuclidinol derivatives as muscarinic receptor antagonists |
| KR20090107567A (ko) * | 2007-02-09 | 2009-10-13 | 아스텔라스세이야쿠 가부시키가이샤 | 아자 가교환 화합물 |
| CN102850344A (zh) * | 2011-06-28 | 2013-01-02 | 中国人民解放军军事医学科学院毒物药物研究所 | 戊乙奎醚光学异构体衍生物用于抗肿瘤的医药用途 |
-
2014
- 2014-07-11 EP EP14825941.9A patent/EP3023424B1/en active Active
- 2014-07-11 RU RU2016104437A patent/RU2641285C2/ru active
- 2014-07-11 US US14/904,766 patent/US9751875B2/en active Active
- 2014-07-11 CN CN201480020342.8A patent/CN105431432B/zh active Active
- 2014-07-11 AU AU2014292722A patent/AU2014292722B2/en active Active
- 2014-07-11 TR TR2019/06532T patent/TR201906532T4/tr unknown
- 2014-07-11 ES ES14825941T patent/ES2726664T3/es active Active
- 2014-07-11 CA CA2921621A patent/CA2921621C/en active Active
- 2014-07-11 JP JP2016524657A patent/JP6218940B2/ja active Active
- 2014-07-11 KR KR1020167003638A patent/KR101823451B1/ko active Active
- 2014-07-11 BR BR112016000629-1A patent/BR112016000629B1/pt active IP Right Grant
- 2014-07-11 WO PCT/CN2014/000669 patent/WO2015007073A1/zh not_active Ceased
-
2016
- 2016-02-12 ZA ZA2016/00997A patent/ZA201600997B/en unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2012964A1 (zh) | 1968-07-15 | 1970-03-27 | Sogeras | |
| US5654314A (en) | 1991-03-15 | 1997-08-05 | Boehringer Ingelheim Kg | Esters of bi- and tricyclic amino alcohols and their use in pharmaceutical compositions |
| CN1675206A (zh) * | 2002-06-21 | 2005-09-28 | 阿尔米雷尔普罗迪斯制药有限公司 | 奎宁环衍生物及含有相同化合物的药用组合物 |
| CN102040602A (zh) * | 2004-04-27 | 2011-05-04 | 葛兰素集团有限公司 | 毒蕈碱性乙酰胆碱受体拮抗剂 |
| CN1954938A (zh) * | 2005-10-17 | 2007-05-02 | 日信工业株式会社 | 车辆用支承结构体的制造方法 |
| CN102070631A (zh) * | 2009-11-20 | 2011-05-25 | 中国人民解放军军事医学科学院毒物药物研究所 | 戊乙奎醚光学异构体的季铵盐、药物组合物及其医药用途 |
Non-Patent Citations (7)
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102570702B1 (ko) * | 2015-03-02 | 2023-08-24 | 가부시키가이샤 엘티티 바이오파마 | 퀴누클리딘 유도체 |
| KR20170122739A (ko) * | 2015-03-02 | 2017-11-06 | 가부시키가이샤 엘티티 바이오파마 | 퀴누클리딘 유도체 |
| JPWO2016140136A1 (ja) * | 2015-03-02 | 2018-01-18 | 株式会社Lttバイオファーマ | キヌクリジン誘導体 |
| US10155755B2 (en) | 2015-03-02 | 2018-12-18 | Ltt Bio-Pharma Co., Ltd. | Quinuclidine derivative |
| AU2016227080B2 (en) * | 2015-03-02 | 2019-10-03 | Ltt Bio-Pharma Co., Ltd. | Quinuclidine derivative |
| WO2016140136A1 (ja) * | 2015-03-02 | 2016-09-09 | 株式会社Lttバイオファーマ | キヌクリジン誘導体 |
| WO2018108089A1 (zh) | 2016-12-14 | 2018-06-21 | 北京硕佰医药科技有限责任公司 | 一类具有季铵盐结构的双功能化合物 |
| WO2021218833A1 (zh) | 2020-04-26 | 2021-11-04 | 北京硕佰医药科技有限责任公司 | 一种m受体拮抗剂的晶体、制备方法及其应用 |
| CN115397823A (zh) * | 2020-04-26 | 2022-11-25 | 北京硕佰医药科技有限责任公司 | 一种m受体拮抗剂的晶体、制备方法及其应用 |
| AU2021264829B2 (en) * | 2020-04-26 | 2024-02-01 | Beijing Showby Pharmaceutical Co., Ltd. | Crystal of M receptor antagonist as well as preparation method therefor and application thereof |
| US12358908B2 (en) | 2020-04-26 | 2025-07-15 | Beijing Showby Pharmaceutical Co., Ltd. | Crystals, preparation method and application of a muscarinic receptor antagonist |
| CN115397823B (zh) * | 2020-04-26 | 2025-11-11 | 北京硕佰医药科技有限责任公司 | 一种m受体拮抗剂的晶体、制备方法及其应用 |
| WO2023138478A1 (zh) * | 2022-01-19 | 2023-07-27 | 北京硕佰医药科技有限责任公司 | 一种治疗鼻炎的溶液型鼻喷雾剂及制备方法 |
| CN118555954A (zh) * | 2022-01-19 | 2024-08-27 | 北京硕佰医药科技有限责任公司 | 一种治疗鼻炎的溶液型鼻喷雾剂及制备方法 |
| CN116514800A (zh) * | 2023-05-06 | 2023-08-01 | 湖南一格制药有限公司 | 盐酸戊乙奎醚的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112016000629B1 (pt) | 2023-10-31 |
| CA2921621A1 (en) | 2015-01-22 |
| CA2921621C (en) | 2018-08-28 |
| EP3023424A4 (en) | 2017-01-18 |
| CN105431432B (zh) | 2018-01-09 |
| EP3023424A1 (en) | 2016-05-25 |
| ES2726664T3 (es) | 2019-10-08 |
| US9751875B2 (en) | 2017-09-05 |
| KR20160033146A (ko) | 2016-03-25 |
| CN105431432A (zh) | 2016-03-23 |
| KR101823451B1 (ko) | 2018-01-30 |
| EP3023424B1 (en) | 2019-02-27 |
| BR112016000629A2 (zh) | 2017-07-25 |
| RU2641285C2 (ru) | 2018-01-17 |
| TR201906532T4 (tr) | 2019-05-21 |
| ZA201600997B (en) | 2018-05-30 |
| RU2016104437A (ru) | 2017-08-16 |
| JP6218940B2 (ja) | 2017-10-25 |
| US20160244439A1 (en) | 2016-08-25 |
| AU2014292722A1 (en) | 2016-03-03 |
| AU2014292722B2 (en) | 2017-03-09 |
| JP2016528199A (ja) | 2016-09-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015007073A1 (zh) | 奎宁类化合物、其光学异构体及其制备方法和医药用途 | |
| CN113272298B (zh) | 用于治疗呼吸系统疾病的化合物和组合物 | |
| CN102070631B (zh) | 戊乙奎醚光学异构体的季铵盐、药物组合物及其医药用途 | |
| WO2021202986A1 (en) | Compositions and methods for the prevention and/or treatment of mitochondrial disease, including friedreich's ataxia | |
| JP2023134493A (ja) | 呼吸器疾患の治療のための化合物 | |
| TW202345833A (zh) | 一種次黃鹼衍化合物在製備治療肺纖維化藥物中的應用 | |
| EP4520752A1 (en) | Carebastine salt and use thereof | |
| CN111655675B (zh) | 氨基烷基化合物 | |
| CN102850345A (zh) | 季铵盐及其药物组合物及其医药用途 | |
| WO2010040315A1 (zh) | 1-丁基-2-羟基芳烷哌嗪衍生物及作为抗抑郁剂的应用 | |
| EP2730575B1 (en) | Crystal form i of salt of dipeptidyl peptidase-iv inhibitor and preparation method and use thereof | |
| WO2004031125A1 (ja) | スピロ化合物、それを含有する医薬組成物及び該化合物の中間体 | |
| US9662318B2 (en) | Bitopic muscarinic agonists and antagonists and methods of synthesis and use thereof | |
| JP3571114B2 (ja) | 麻薬拮抗剤 | |
| WO2019015640A1 (zh) | 一种氮杂环酰胺衍生物的盐、其晶型及其制备方法和用途 | |
| WO2024131672A1 (zh) | 二苯基吡嗪类衍生物、其制备方法及应用 | |
| WO2025007744A1 (zh) | 一种曲前列环素衍生物、其制备方法及应用 | |
| HK40114273A (zh) | 一种曲前列环素衍生物、其制备方法及应用 | |
| WO2025168119A1 (zh) | 四唑类化合物、其药物组合物及其应用 | |
| WO2020094634A1 (en) | Thiophene prodrugs of naltroxene for long-acting injectable compositions and related methods | |
| WO2008086678A2 (en) | The medical use of levo-phencynonate as neuroprotective agent | |
| HK1192231B (zh) | 二肽基肽酶-iv抑制剂的盐的晶型i及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480020342.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14825941 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016524657 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016000629 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014825941 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20167003638 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2016104437 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2921621 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2014292722 Country of ref document: AU Date of ref document: 20140711 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14904766 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 112016000629 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160112 |