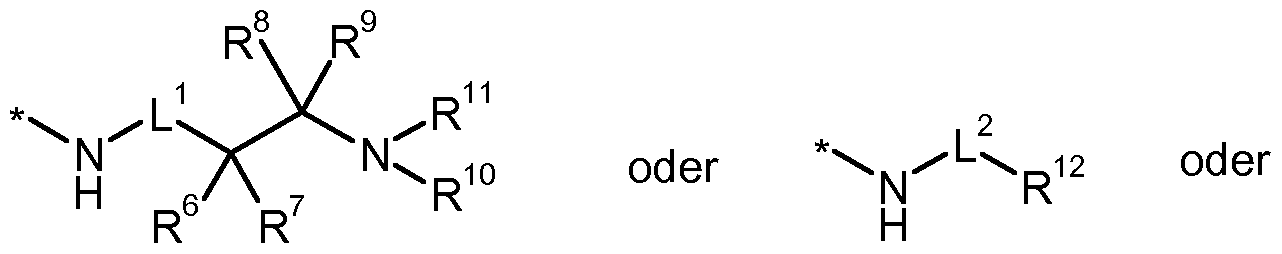,
Substituierte Imidazo[l,2-alpyrazincarboxamide und ihre Verwendung
Die vorliegende Anmeldung betrifft neue substituierte Imidazo[l,2-a]pyrazincarboxamide, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Be- handlung und/oder Prophylaxe von Krankheiten, insbesondere zur Behandlung und/oder Prophylaxe von kardiovaskulären Erkrankungen.
Eines der wichtigsten zellulären Übertragungssysteme in Säugerzellen ist das cyclische Guanosin- monophosphat (cGMP). Zusammen mit Stickstoffmonoxid (NO), das aus dem Endothel freigesetzt wird und hormonelle und mechanische Signale überträgt, bildet es das NO/cGMP-System. Die Guanylatcyclasen katalysieren die Biosynthese von cGMP aus Guanosintriphosphat (GTP). Die bisher bekannten Vertreter dieser Familie lassen sich sowohl nach strukturellen Merkmalen als auch nach der Art der Liganden in zwei Gruppen aufteilen: Die partikulären, durch natriuretische Peptide stimulierbaren Guanylatcyclasen und die löslichen, durch NO stimulierbaren Guanylatcyclasen. Die löslichen Guanylatcyclasen bestehen aus zwei Untereinheiten und enthalten höchst- wahrscheinlich ein Häm pro Heterodimer, das ein Teil des regulatorischen Zentrums ist. Dieses hat eine zentrale Bedeutung für den Aktivierungsmechanismus. NO kann an das Eisenatom des Häms binden und so die Aktivität des Enzyms deutlich erhöhen. Hämfreie Präparationen lassen sich hingegen nicht durch NO stimulieren. Auch Kohlenmonoxid (CO) ist in der Lage, an das Eisen- Zentralatom des Häms zu binden, wobei die Stimulierung durch CO deutlich geringer ist als die durch NO.
Durch die Bildung von cGMP und der daraus resultierenden Regulation von Phosphodiesterasen, Ionenkanälen und Proteinkinasen spielt die Guanylatcyclase eine entscheidende Rolle bei unterschiedlichen physiologischen Prozessen, insbesondere bei der Relaxation und Proliferation glatter Muskelzellen, der Plättchenaggregation und -adhäsion, der neuronalen Signalübertragung sowie bei Erkrankungen, welche auf einer Störung der vorstehend genannten Vorgänge beruhen. Unter pathophysiologischen Bedingungen kann das NO/cGMP-System supprimiert sein, was zum Beispiel zu Bluthochdruck, einer Plättchenaktivierung, einer vermehrten Zellproliferation, endothelialer Dysfunktion, Atherosklerose, Angina pectoris, Herzinsuffizienz, Myokardinfarkt, Thrombosen, Schlaganfall und sexueller Dysfunktion führen kann. Eine auf die Beeinflussung des cGMP-Signalweges in Organismen abzielende NO-unabhängige Behandlungsmöglichkeit für derartige Erkrankungen ist aufgrund der zu erwartenden hohen Effizienz und geringen Nebenwirkungen ein vielversprechender Ansatz.
Zur therapeutischen Stimulation der löslichen Guanylatcyclase wurden bisher ausschließlich Verbindungen wie organische Nitrate verwendet, deren Wirkung auf NO beruht. Dieses wird durch
Biokonversion gebildet und aktiviert die lösliche Guanylatcyclase durch Angriff am Eisen-Zentralatom des Häms. Neben den Nebenwirkungen gehört die Toleranzentwicklung zu den entscheidenden Nachteilen dieser Behandlungsweise.
In den letzten Jahren wurden einige Substanzen beschrieben, die die lösliche Guanylatcyclase direkt, d.h. ohne vorherige Freisetzung von NO stimulieren, wie beispielsweise 3-(5'-Hydroxy- methyl-2'-furyl)-l -benzylindazol [YC-1 ; Wu et al., Blood 84 (1994), 4226; Mülsch et al., Brit. J. Pharmacol. 120 (1997), 681 ], Fettsäuren [Goldberg et al., J. Biol. Chem. 252 (1977), 1279], Diphenyliodonium-hexafluorphosphat [Pettibone et al., Eur. J. Pharmacol. 1 16 (1985), 307], Iso- liquiritigenin [Yu et al., Brit. J. Pharmacol. 1 14 (1995), 1587] sowie verschiedene substituierte Pyrazol-Derivate (WO 98/16223).
Unter anderem in WO 89/03833-A1 und WO 96/34866-A1 sind verschiedene Imidazo[l ,2- a]pyrazin-Derivate beschrieben, die zur Behandlung von Erkrankungen verwendet werden können.
Aufgabe der vorliegenden Erfindung war die Bereitstellung neuer Substanzen, die als Stimulatoren der löslichen Guanylatcyclase wirken, und als solche zur Behandlung und/oder Prophylaxe von Krankheiten geeignet sind.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)
A für CH2, CD2 oder CH(CH3) steht,
R für (C4-C6)-Alkyl, (C3-C7)-Cycloalkyl, Pyridyl oder Phenyl steht, wobei (C4-C6)-Alkyl bis zu sechsfach mit Fluor substituiert sein kann, wobei (C3-Cv)-Cycloalkyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl und (Ci-C i)-Alkyl substituiert sein kann,
wobei Pyridyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano und (Ci-C i)-Alkyl substituiert ist, und wobei Phenyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Monofluormethyl, Difluormethyl, Trifluormethyl, (Ci-C i)-Alkyl, (C2-C3)-Alkinyl, (Ci-C i)-Alkoxy, (C3-Cs)-Cyclopropyl, Difluormethoxy und Trifluormethoxy substituiert sein kann oder an zwei benachbarten Kohlenstoffatomen des Phenyls mit einer Difluormethylendioxy-Brücke substituiert sein kann, für Wasserstoff, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxymethyl, Cyclopropyl, Monofluormethyl, Difluormethyl oder Trifluormethyl steht, eine Gruppe der Formel
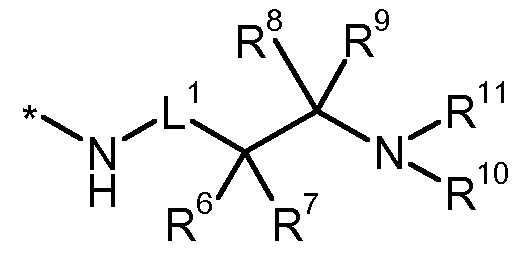
* für die Anknüpfstelle an die Carbonylgruppe steht, L1 für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht, worin Methandiyl und 1 ,2-Ethandiyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl, (C3- Cs)-Cycloalkyl, Hydroxy und (Ci-C4)-Alkoxy substituiert sein können,
L2 für eine Bindung oder (Ci-C4)-Alkandiyl steht, worin (Ci-C4)-Alkandiyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl, (C3-C5)- Cycloalkyl, Hydroxy und (Ci-C4)-Alkoxy substituiert sein können,
für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht, worin Methandiyl oder 1 ,2-Ethandiyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl, (C3- Cv)-Cycloalkyl, Hydroxy und (Ci-C4)-Alkoxy substituiert sein können, R6 für Wasserstoff, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-C6)-Alkinyl, (C3-C7)-
Cycloalkyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Difluormethoxy, Trifluormethoxy, Hydroxy, (Ci-C4)-Alkoxy, (Ci-C4)-Alkoxycarbonyl, (Ci-C4)-Alkylthio, (Ci-C4)-Alkyl- sulfonyl, Phenyl, Phenoxy und Benzyloxy substituiert sein kann, worin Phenyl, Phenoxy und Benzyloxy mit 1 bis 3 Substituenten Halogen substituiert sein können, worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl und (C1-C4)- Alkoxy substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Nitro, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy, Difluormethoxy, Trifluormethoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können,
R7 für Wasserstoff oder (Ci-C6)-Alkyl steht, oder
Pv6 und R7 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C4)-Alkyl substituiert sein können,
R8 für Wasserstoff, (Ci-C6)-Alkyl, (C3-C7)-Cycloalkyl, (C2-C6)-Alkenyl, (C2-C6)- Alkinyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Difluormethyl, Trifluormethyl, Difluormethoxy, Trifluormethoxy, Hydroxy, (Ci-C4)-Alkoxy, Benzyloxy, Phenoxy und Phenyl substituiert sein kann, worin Benzyloxy, Phenoxy und Phenyl mit 1 bis 3 Substituenten Halogen substituiert sein können, worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können,
R9 für Wasserstoff oder (Ci-C6)-Alkyl steht, oder
R8 und R9 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C4)-Alkyl substituiert sein können, oder
R6 und R8 zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R6 und R7, R8 und R9 bzw. R6 und R8 einen Carbo- oder Heterocyclus bildet,
r
- 6 - mit der Maßgabe, dass die Reste R6 und R8 nicht beide gleichzeitig für Phenyl oder 5- oder 6-gliedriges Heteroaryl stehen,
R10 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy und (Ci-C i)-Alkoxy substituiert sein kann,
R11 für Wasserstoff, (Ci-C6)-Alkyl, (C3-C7)-Cycloalkyl, Phenyl oder Benzyl steht, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, (Ci-C i)-Alkoxy und Phenoxy substituiert sein kann, und worin Phenyl und Benzyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen und Trifluormethyl substituiert sein können, oder R10und R11 zusammen mit dem Sticktoffatom, an das sie gebunden sind, einen 4- bis 7- gliedrigen Aza-Heterocyclus bilden,
R12 für 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes Aza- Heterocyclyl steht, worin 5- bis 9-gliedriges Aza-Heterocyclyl mit 1 bis 5 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl, (C3-
Cv)-Cycloalkyl und Benzyl substituiert sein kann, und worin 5- bis 9-gliedriges Aza-Heterocyclyl mit einem Phenyl-Ring anneliert sein kein, der seinerseits mit 1 oder 2 Substituenten ausgewählt aus Halogen, (C1-C4)- Alkyl und Trifluormethyl substituiert sein kann,
R13 für Wasserstoff, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-C6)-Alkinyl, (C3-C7)- Cycloalkyl, (Ci-C4)-Alkoxycarbonyl, -(C=0)NR23R24, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht,
worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Difluormethoxy, Trifluormethoxy, Hydroxy, (Ci-C4)-Alkoxy, (Ci-C4)-Alkoxycarbonyl, (Ci-C4)-Alkylthio, (Ci-C4)-Alkyl- sulfonyl, Phenyl, Phenoxy und Benzyloxy substituiert sein kann, worin Phenyl, Phenoxy und Benzyloxy ihrerseits mit 1 bis 3 Substituenten
Halogen substituiert sein können, worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl und (Ci-C4)- Alkoxy substituiert sein kann, worin R23 für Wasserstoff, (Ci-C4)-Alkyl, (C3-C7)-Cycloalkyl, Aryl oder Naphthyl steht, worin R24 für Wasserstoff oder Methyl steht, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano,
Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können,
R14 für Wasserstoff oder (Ci-C6)-Alkyl steht, worin (Ci-C4)-Alkyl mit Hydroxy substituiert sein kann, oder
R13 und R14 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C4)-Alkyl substituiert sein können,
R für Wasserstoff, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-C6)-Alkinyl, (C3-C7)- Cycloalkyl, (Ci-C4)-Alkoxycarbonyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht,
worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Difluormethoxy, Trifluormethoxy, Hydroxy, (Ci-C4)-Alkoxy, (Ci-C4)-Alkoxycarbonyl, (Ci-C4)-Alkylthio, (Ci-C4)-Alkyl- sulfonyl, Phenyl, Phenoxy und Benzyloxy substituiert sein kann, worin Phenyl, Phenoxy und Benzyloxy ihrerseits mit 1 bis 3 Substituenten
Halogen substituiert sein können, worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl und (Ci-C4)- Alkoxy substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können, R16 für Wasserstoff oder (Ci-C6)-Alkyl steht, worin (Ci-C4)-Alkyl mit Hydroxy substituiert sein kann, oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C4)-Alkyl substituiert sein können, mit der Maßgabe, dass die Reste R13 und R15 nicht beide gleichzeitig für Phenyl oder 5- oder 6-gliedriges Heteroaryl stehen, oder
R und R zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden,
mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R13 und R14, R15 und R16 bzw. R13 und R15 einen Carbo- oder Heterocyclus bildet,
R17 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, m für 0, 1 oder 2 steht, n für 0 oder 1 steht,
R18 für Wasserstoff, Cyano oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann,
R19 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann,
R20 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann,
R21 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, oder
R18 und R19 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C4)-Alkyl substituiert sein können, oder
R20 und R21 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden,
worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können, oder
R18 und R20 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R18 und R19, R20 und R21 bzw. R18 und R20 einen Carbo- oder Heterocyclus bildet, R22 für (Ci-C6)-Alkyl, 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes
Heterocyclyl, 5- bis 9-gliedriges Carbocyclyl, Phenyl, Indanyl oder 5- bis 10- gliedriges Heteroaryl steht, wobei (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl und Cyano substituiert sein kann, wobei Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, Difluormethyl, (Ci-C6)-Alkyl, (Ci- C4)-Alkylcarbonyl, (Ci-C4)-Alkoxycarbonyl, Hydroxycarbonyl, -(C=0)NR25R26, (Ci-C4)-Alkylsulfonyl, (C3-C6)-Cycloalkylsulfonyl, (Ci-C4)-Alkylthio, (Ci-C4)- Alkoxy, Trifluormethoxy, Difluormethoxy, Phenoxy, Hydroxy, 5- bis 10-gliedriges Heteroaryl und (C3-Cv)-Cycloalkyl substituiert sein kann, worin (Ci-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethoxy, (Ci-C4)-Alkylcarbonyl, -(C=0)NR25R26, (Ci-C4)-Alkoxy, (C3-C6)-Cycloalkyl, Morpholinyl, Piperidinyl, Pyrrolidinyl, Piperazinyl, Phenyl, Hydroxy und Amino substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten Halogen substituiert sein kann,
worin Amino mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus (Ci-C6)-Alkyl, (Ci-C i)-Alkylcarbonyl, (C3-Ce)- Cycloalkylsulfonyl, (Ci-C -Alkylsulfonyl und Methoxy-(Ci-C4)- alkyl substituiert sein kann, worin (C3-C6)-Cycloalkyl mit Amino oder Hydroxy substituiert sein kann, und worin
R25 und R26 jeweils unabhängig voneinander für Wasserstoff,
(Ci-C4)-Alkyl oder (C3-C7)-Cycloalkyl stehen, wobei Indanyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl und Hydroxy substituiert sein kann, wobei 5- bis 10-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Cyano, (Ci-C6)-Alkyl, Trifluormethyl, (Ci-C i)-Alkoxy, Amino, (Ci-C i)-Alkoxycarbonyl, Hydroxy- carbonyl, -(C=0)NR25R26, Phenyl, Pyridyl, Pyrimidyl, l ,3-Thiazol-5-yl und (C3-
Cv)-Cycloalkyl substituiert sein kann, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Difluormethyl, (Ci-C4)-Alkylsulfonyl, (Ci-C4)-Alkyl- carbonyl, (Ci-C4)-Alkoxycarbonyl, Hydroxycarbonyl, (Ci-C4)-Alkylthio,
(Ci-C4)-Alkoxy, Trifluormethoxy, Difluormethoxy, Phenoxy, Phenyl, Pyridyl, Pyrimidyl, 5-gliedriges Heteroaryl, Tetrahydrothiophenyl-1 , 1 - dioxid, (C3-Cv)-Cycloalkyl, Morpholinyl, Piperidinyl, Pyrrolidinyl, 2- Oxopyrro lidin- 1 -yl, Piperazinyl, Tetrahydrothiophenyl- 1 , 1 -dioxid, Thiomorpholinyl- 1 , 1 -dioxid und Azetidin substituiert sein kann, worin 5-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, (Ci-C4)-Alkyl und (Ci- C4)-Alkoxy substituiert sein kann, worin Piperidinyl mit 1 bis 4 Substituenten Fluor substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, (Ci-C4)-Alkyl und (Ci-C4)-Alkoxy substituiert sein kann,
worin Azetidin mit Hydroxy substituiert sein kann, worin Piperazinyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe (Ci-C i)-Alkyl, (C3-Cv)-Cycloalkyl und Trifluormethyl substituiert sein kann, und worin
R25 und R26 jeweils unabhängig voneinander für Wasserstoff, (C1-C4)- Alkyl oder (C3-Cv)-Cycloalkyl stehen, wobei 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes, Heterocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Oxo, Fluor, Hydroxy und (Ci-C4)-Alkyl substiuiert sein kann, und wobei 5- bis 9-gliedriges Carbocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Trifluormethyl, Fluor, Hydroxy, Hydroxycarbonyl, (Ci-C4)-Alkoxycarbonyl und (Ci-C4)-Alkyl substiuiert sein kann,
R4 für Wasserstoff steht,
R5 für Wasserstoff, Halogen, Cyano, Difluormethyl, Trifluormethyl, (Ci-C4)-Alkyl, (C3-C7)- Cycloalkyl, (C2-C4)-Alkinyl, (Ci-C4)-Alkylamino, Difluormethoxy, Trifluormethoxy, (Ci- C4)-Alkoxy, Amino, 4- bis 7-gliedriges Heterocyclyl oder 5- oder 6-gliedriges Heteroaryl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)
(I),
in welcher für CH2, CD2 oder CH(CH3) steht,
R1 für (C4-C6)-Alkyl, (C3-C7)-Cycloalkyl, Pyridyl oder Phenyl steht, wobei (C4-Ce)-Alkyl bis zu sechsfach mit Fluor substituiert sein kann, wobei (C3-Cv)-Cycloalkyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl und (Ci-C i)-Alkyl substituiert sein kann, wobei Pyridyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano und (Ci-C i)-Alkyl substituiert ist,
wobei Phenyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Monofluormethyl, Difluormethyl, Trifluormethyl, (Ci-C i)-Alkyl, (C2-C3)-Alkinyl, (Ci-C i)-Alkoxy, (C3-Cs)-Cyclopropyl, Difluormethoxy und Trifluormethoxy substituiert sein kann oder an zwei benachbarten Kohlenstoffatomen des Phenyls mit einer Difluormethylendioxy- Brücke substituiert sein kann, R für Wasserstoff, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxymethyl, Cyclopropyl, Monofluormethyl, Difluormethyl oder Trifluormethyl steht, eine Gruppe der Formel
* für die Anknüpfstelle an die Carbonylgruppe steht,
L für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht,
worin Methandiyl und 1 ,2-Ethandiyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl, (C3- C5)-Cycloalkyl, Hydroxy und (Ci-C4)-Alkoxy substituiert sein können, für eine Bindung oder (Ci-C4)-Alkandiyl steht, worin (Ci-C4)-Alkandiyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl, (C3-C5)- Cycloalkyl, Hydroxy und (Ci-C4)-Alkoxy substituiert sein können, für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht, worin Methandiyl oder 1 ,2-Ethandiyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl, (C3- Cv)-Cycloalkyl, Hydroxy und (Ci-C4)-Alkoxy substituiert sein können, für Wasserstoff, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-C6)-Alkinyl, (C3-C7)- Cycloalkyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Difluormethoxy, Trifluormethoxy, Hydroxy, (C1-C4)- Alkoxy, (Ci-C4)-Alkoxycarbonyl, (Ci-C4)-Alkylthio, (Ci-C4)-Alkyl-sulfonyl, Phenyl, Phenoxy und Benzyloxy sowie bis zu sechsfach mit Fluor substituiert sein kann, worin Phenyl, Phenoxy und Benzyloxy mit 1 bis 3 Substituenten Halogen substituiert sein können, worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C4)-Alkyl und (C1-C4)- Alkoxy substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Nitro, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy, Difluormethoxy, Trifluormethoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können, für Wasserstoff oder (Ci-C6)-Alkyl steht, oder
R6 und R7 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können,
R8 für Wasserstoff, (Ci-C6)-Alkyl, (C3-C7)-Cycloalkyl, (C2-C6)-Alkenyl, (C2-C6)- Alkinyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Trifluormethyl, Difluormethoxy, Trifluormethoxy, Hydroxy, (Ci-C4)-Alkoxy, Benzyloxy, Phenoxy und Phenyl sowie bis zu sechsfach mit Fluor substituiert sein kann, worin Benzyloxy, Phenoxy und Phenyl mit 1 bis 3 Substituenten Halogen substituiert sein können, worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano,
Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können,
R9 für Wasserstoff oder (Ci-C6)-Alkyl steht, oder
R8 und R9 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander
1 r
- 16 - ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können, oder
R6 und R8 zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R6 und R7, R8 und R9 bzw. R6 und R8 einen Carbo- oder Heterocyclus bildet, mit der Maßgabe, dass die Reste R6 und R8 nicht beide gleichzeitig für Phenyl oder 5- oder 6-gliedriges Heteroaryl stehen,
R10 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy und (Ci-C i)-Alkoxy substituiert sein kann, R11 für Wasserstoff, (Ci-C6)-Alkyl, (C3-C7)-Cycloalkyl, Phenyl oder Benzyl steht, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy, (Ci-C i)-Alkoxy und Phenoxy substituiert sein kann, und worin Phenyl und Benzyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen und Trifluormethyl substituiert sein können, oder
R10und R11 zusammen mit dem Sticktoffatom, an das sie gebunden sind, einen 4- bis 7- gliedrigen Aza-Heterocyclus bilden, R für 5- bis 10-gliedriges, über ein Ring- Kohlenstoffatom gebundenes Aza-
Heterocyclyl steht, worin 5- bis 10-gliedriges, über ein Ring- Kohlenstoffatom gebundenes Aza- Heterocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus
der Gruppe Trifluormethyl, (C3-Cv)-Cycloalkyl, Oxo und Benzyl sowie bis zu vierfach mit (Ci-C i)-Alkyl und bis zu zweifach mit Fluor substituiert sein kann, und worin 5- bis 10-gliedriges, über ein Ring-Kohlenstoffatom gebundenes Aza- Heterocyclyl mit einem Phenyl-Ring anneliert sein kein, der seinerseits mit 1 oder 2 Substituenten ausgewählt aus Halogen, (Ci-C i)-Alkyl und Trifluormethyl substituiert sein kann, oder für Amino stehen kann, wenn L2 für eine Bindung steht, worin Amino mit (Ci-Cio)-Alkyl, (Ci-C i)-Alkylcarbonyl, (C3-C6)-Carbocyclyl, 4- bis 7-gliedriges Heterocyclyl, Phenyl oder 5- oder 6-gliedriges Heteroaryl substituiert sein kann, worin (Ci-C i)-Alkylcarbonyl mit Mono-alkylamino oder Di-alkylamino substituiert sein kann, worin (C3-C6)-Carbocyclyl und 4- bis 7-gliedriges Heterocyclyl mit Hydroxy substituiert sein können, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, (Ci-C4)- Alkyl und Trifluormethyl substituiert sein können, für Wasserstoff, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-C6)-Alkinyl, (C3-C7)- Cycloalkyl, (Ci-C4)-Alkoxycarbonyl, -(C=0)NR23R24, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Difluormethoxy, Trifluormethoxy, Hydroxy, (C1-C4)- Alkoxy, (Ci-C4)-Alkoxycarbonyl, (Ci-C4)-Alkylthio, (Ci-C4)-Alkyl-sulfonyl, Phenyl, Phenoxy und Benzyloxy sowie bis zu sechsfach mit Fluor substituiert sein kann, worin Phenyl, Phenoxy und Benzyloxy ihrerseits mit 1 bis 3 Substituenten Halogen substituiert sein können,
worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C i)-Alkyl und (Ci-C4)- Alkoxy substituiert sein kann, worin R23 für Wasserstoff, (Ci-C4)-Alkyl, (C3-C7)-Cycloalkyl, Aryl oder Naphthyl steht, worin R24 für Wasserstoff oder Methyl steht, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können,
R14 für Wasserstoff oder (Ci-C6)-Alkyl steht, worin (Ci-C4)-Alkyl mit Hydroxy substituiert sein kann, oder R13 und R14 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C4)-Alkyl substituiert sein können,
R15 für Wasserstoff, (Ci-C6)-Alkyl, (C2-C6)-Alkenyl, (C2-C6)-Alkinyl, (C3-C7)- Cycloalkyl, (Ci-C4)-Alkoxycarbonyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Difluormethoxy, Trifluormethoxy, Hydroxy, (C1-C4)- Alkoxy, (Ci-C4)-Alkoxycarbonyl, (Ci-C4)-Alkylthio, (Ci-C4)-Alkyl-sulfonyl, Phenyl, Phenoxy und Benzyloxy sowie bis zu sechsfach mit Fluor substituiert sein kann,
worin Phenyl, Phenoxy und Benzyloxy ihrerseits mit 1 bis 3 Substituenten Halogen substituiert sein können, worin (C3-Cv)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, (Ci-C i)-Alkyl und (C1-C4)- Alkoxy substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy und (Ci-C4)-Alkylsulfonyl substituiert sein können,
R16 für Wasserstoff oder (Ci-C6)-Alkyl steht, worin (Ci-C4)-Alkyl mit Hydroxy substituiert sein kann, oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können, mit der Maßgabe, dass die Reste R13 und R15 nicht beide gleichzeitig für Phenyl oder 5- oder 6-gliedriges Heteroaryl stehen, oder
R13 und R15 zusammen mit den Kohlenstoffatomen, an die sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R13 und R14, R15 und R16 bzw. R13 und R15 einen Carbo- oder Heterocyclus bildet, für Wasserstoff oder (Ci-C i)-Alkyl steht,
worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, m für 0, 1 oder 2 steht, n für 0 oder 1 steht,
R18 für Wasserstoff, Cyano oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, R19 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, R20 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, R21 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, oder
R18 und R19 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können, oder
R20 und R21 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können,
oder
R18 und R20 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 7-gliedrigen Carbocyclus oder einen 4- bis 7-gliedriger Heterocyclus bilden, worin der 3- bis 7-gliedrige Carbocyclus und der 4- bis 7-gliedrige
Heterocyclus ihrerseits mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und (Ci-C i)-Alkyl substituiert sein können, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R18 und R19, R20 und R21 bzw. R18 und R20 einen Carbo- oder Heterocyclus bildet,
R22 für (Ci-C6)-Alkyl, Cyano, (Ci-C6)-Alkoxy, 5- bis 9-gliedriges, über ein Ring- Kohlenstoffatom gebundenes Heterocyclyl, 5- bis 9-gliedriges Carbocyclyl, Phenyl, Indanyl oder 5- bis 10-gliedriges Heteroaryl steht, worin (Ci-C6)-Alkyl mit Cyano, sowie bis zu sechsfach mit Fluor substituiert sein kann, worin (Ci-C6)-Alkoxy mit Hydroxy, Amino, Mono-alkylamino, Di-alkylamino, Cyclopropyl, Phenyl oder (C2-C i)-Alkenyl substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, Difluormethyl, (Ci-C6)-Alkyl, (Ci- C4)-Alkylcarbonyl, (Ci-C4)-Alkoxycarbonyl, Hydroxycarbonyl, -(C=0)NR25R26,
(Ci-C4)-Alkylsulfonyl, (C3-C6)-Cycloalkylsulfonyl, (Ci-C4)-Alkylthio, (Ci-C4)- Alkoxy, Trifluormethoxy, Difluormethoxy, Phenoxy, Hydroxy, 5- bis 10-gliedriges Heteroaryl und (C3-Cv)-Cycloalkyl substituiert sein kann, worin (Ci-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethoxy, (Ci-C4)-Alkylcarbonyl,
-(C=0)NR25R26, (Ci-C4)-Alkoxy, (C3-C6)-Cycloalkyl, Morpholinyl, Piperidinyl, Pyrrolidinyl, Piperazinyl, Phenyl, Hydroxy und Amino substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten Halogen substituiert sein kann,
„„
- 22 - worin Amino mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus (Ci-C6)-Alkyl, (Ci-C i)-Alkylcarbonyl, (C3-Ce)- Cycloalkylsulfonyl, (Ci-C4)-Alkylsulfonyl und Methoxy-(Ci-C4)- alkyl substituiert sein kann, worin (C3-C6)-Cycloalkyl mit Amino oder Hydroxy substituiert sein kann, und worin
R25 und R26 jeweils unabhängig voneinander für Wasserstoff,
(Ci-C4)-Alkyl oder (C3-C7)-Cycloalkyl stehen, worin Indanyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl und Hydroxy substituiert sein kann, worin 5- bis 10-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Cyano, (Ci-C6)-Alkyl, Trifluormethyl, (Ci-C4)-Alkoxy, Amino, (Ci-C4)-Alkoxycarbonyl, Hydroxy- carbonyl, -(C=0)NR25R26, Phenyl, Pyridyl, Pyrimidyl, l ,3-Thiazol-5-yl und (C3-
Cv)-Cycloalkyl substituiert sein kann, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Difluormethyl, (Ci-C4)-Alkylsulfonyl, (Ci-C4)-Alkyl- carbonyl, (Ci-C4)-Alkoxycarbonyl, Hydroxycarbonyl, (Ci-C4)-Alkylthio,
(Ci-C4)-Alkoxy, Trifluormethoxy, Difluormethoxy, Phenoxy, Phenyl, Pyridyl, Pyrimidyl, 5-gliedriges Heteroaryl, Tetrahydrothiophenyl-1 , 1 - dioxid, (C3-Cv)-Cycloalkyl, Morpholinyl, Piperidinyl, Pyrrolidinyl, 2- Oxopyrro lidin- 1 -yl, Piperazinyl, Tetrahydrothiophenyl- 1 , 1 -dioxid, Thiomorpholinyl- 1 , 1 -dioxid und Azetidin substituiert sein kann, worin 5-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, (Ci-C4)-Alkyl und (Ci- C4)-Alkoxy substituiert sein kann, worin Piperidinyl mit 1 bis 4 Substituenten Fluor substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, (Ci-C4)-Alkyl und (Ci-C4)-Alkoxy substituiert sein kann,
worin Azetidin mit Hydroxy substituiert sein kann, worin Piperazinyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe (Ci-C4)-Alkyl, (C3-C7)-Cycloalkyl und Trifluormethyl substituiert sein kann, und worin
R25 und R26 jeweils unabhängig voneinander für Wasserstoff, (C1-C4)- Alkyl oder (C3-Cv)-Cycloalkyl stehen, worin 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes, Heterocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Oxo, Fluor, Hydroxy und (Ci-C4)-Alkyl substiuiert sein kann, und worin 5- bis 9-gliedriges Carbocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Trifluormethyl, Fluor, Cyano, Hydroxy, Hydroxycarbonyl, (Ci-C4)-Alkoxycarbonyl und (Ci-C4)-Alkyl substiuiert sein kann,
R4 für Wasserstoff steht,
R5 für Wasserstoff, Halogen, Cyano, Difluormethyl, Trifluormethyl, (Ci-C4)-Alkyl, (C3-C7)- Cycloalkyl, (C2-C4)-Alkinyl, (Ci-C4)-Alkylamino, Difluormethoxy, Trifluormethoxy, (Ci- C4)-Alkoxy, Amino, 4- bis 7-gliedriges Heterocyclyl oder 5- oder 6-gliedriges Heteroaryl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische
„ Λ
- 24 -
Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasser- stoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethansulfon- säure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Ameisensäure, Essigsäure, Trifluoressigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in unterschiedlichen stereoisomeren Formen existieren, d.h. in Gestalt von Konfigurationsisomeren oder gegebenenfalls auch als Konformations isomere (Enantiomere und/oder Diastereomere, einschließlich solcher bei Atrop isomeren). Die vorliegende Erfindung umfasst deshalb die Enantiomere und Dia- stereomere und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/ oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren; vorzugsweise werden hierfür chromatographische Verfahren verwendet, insbesondere die HPLC-Chromatographie an achiraler bzw. chiraler Phase.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Die vorliegende Erfindung umfasst auch alle geeigneten isotopischen Varianten der erfindungsgemäßen Verbindungen. Unter einer isotopischen Variante einer erfindungsgemäßen Verbindung wird hierbei eine Verbindung verstanden, in welcher mindestens ein Atom innerhalb der erfindungsgemäßen Verbindung gegen ein anderes Atom der gleichen Ordnungszahl, jedoch mit
^ einer anderen Atommasse als der gewöhnlich oder überwiegend in der Natur vorkommenden Atommasse ausgetauscht ist. Beispiele für Isotope, die in eine erfindungsgemäße Verbindung inkorporiert werden können, sind solche von Wasserstoff, Kohlenstoff, Stickstoff, Sauerstoff, Phosphor, Schwefel, Fluor, Chlor, Brom und Iod, wie 2H (Deuterium), 3H (Tritium), 13C, 14C, 15N, 170, 180, 32P, 33P, 33S, 34S, 35S, 36S, 18F, 36C1, 82Br, 123I, 124I, 129I und 131I. Bestimmte isotopische Varianten einer erfindungsgemäßen Verbindung, wie insbesondere solche, bei denen ein oder mehrere radioaktive Isotope inkorporiert sind, können von Nutzen sein beispielsweise für die Untersuchung des Wirkmechanismus oder der Wirkstoff- Verteilung im Körper; aufgrund der vergleichsweise leichten Herstell- und Detektierbarkeit sind hierfür insbesondere mit 3H- oder 14C- Isotopen markierte Verbindungen geeignet. Darüber hinaus kann der Einbau von Isotopen, wie beispielsweise von Deuterium, zu bestimmten therapeutischen Vorteilen als Folge einer größeren metabolischen Stabilität der Verbindung führen, wie beispielsweise eine Verlängerung der Halbwertszeit im Körper oder eine Reduktion der erforderlichen Wirkdosis; solche Modifikationen der erfindungsgemäßen Verbindungen können daher gegebenenfalls auch eine bevorzugte Ausfüh- rungsform der vorliegenden Erfindung darstellen. Isotopische Varianten der erfindungsgemäßen Verbindungen können nach den dem Fachmann bekannten Verfahren hergestellt werden, so beispielsweise nach den weiter unten beschriebenen Methoden und den bei den Ausführungsbeispielen wiedergegebenen Vorschriften, indem entsprechende isotopische Modifikationen der jeweiligen Reagentien und/oder Ausgangsverbindungen eingesetzt werden. Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" bezeichnet hierbei Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Alkyl steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkylrest mit der jeweils angegebenen Anzahl an Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, iso-Butyl, 1 -Methylpropyl, tert.-Butyl, n-Pentyl, iso-Pentyl, 1- Ethylpropyl, 1 -Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, n-Hexyl, 1 -Methylpentyl, 2- Methylpentyl, 3 -Methylpentyl, 4-Methylpentyl, 3,3-Dimethylbutyl, 1 -Ethylbutyl, 2-Ethylbutyl.
Cycloalkyl bzw. Carbocyclus bzw. Carbocyclyl steht in Rahmen der Erfindung für einen monocyclischen, gesättigten Alkylrest mit der jeweils angegebenen Anzahl an Ring- Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
_
- 26 -
Alkenyl steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkenylrest mit 2 bis 6 Kohlenstoffatomen und einer oder zwei Doppelbindungen. Bevorzugt ist ein linearer oder verzweigter Alkenylrest mit 2 bis 4 Kohlenstoffatomen und einer Doppelbindung. Beispielhaft und vorzugsweise seien genannt: Vinyl, Allyl, Isopropenyl und n-But-2-en-l-yl. Alkinyl steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkinylrest mit 2 bis 6 Kohlenstoffatomen und einer Dreifachbindung. Beispielhaft und vorzugsweise seien genannt: Ethinyl, n-Prop-l-in-l-yl, n-Prop-2-in-l-yl, n-But-2-in- 1 -yl und n-But-3-in-l-yl.
Alkandiyl steht im Rahmen der Erfindung für einen linearen oder verzweigten divalenten Alkylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methylen, 1,2- Ethylen, Ethan-l,l-diyl, 1,3-Propylen, Propan-l,l-diyl, Propan-l,2-diyl, Propan-2,2-diyl, 1,4- Butylen, Butan- 1,2-diyl, Butan-l,3-diyl und Butan-2,3-diyl.
Alkoxy steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy, 1 -Methylpropoxy, n-Butoxy, iso-Butoxy und tert.-Butoxy. Alkoxycarbonyl stehen im Rahmen der Erfindung für einen linearen oder verzweigten Alkoxyrest mit 1 bis 4 Kohlenstoffatomen und einer am Sauerstoffatom angebundenen Carbonylgruppe. Beispielhaft und vorzugsweise seien genannt: Methoxycarbonyl, Ethoxycarbonyl, n- Propoxycarbonyl, Isopropoxycarbonyl und tert.-Butoxycarbonyl.
Alkylsulfonyl steht in Rahmen der Erfindung für einen linearen oder verzweigten Alkylrest mit 1 bis 4 Kohlenstoffatomen, der über eine Sulfonylgruppe gebunden ist. Beispielhaft und vorzugsweise seinen genannt: Methylsulfonyl, Ethylsulfonyl, n-Propylsulfonyl, iso-Propylsulfonyl, n-Butylsulfonyl und tert.-Butylsulfonyl.
Ein 4- bis 7-gliedriger Heterocyclus steht im Rahmen der Erfindung für einen monocyclischen, gesättigten Heterocyclus mit insgesamt 4 bis 7 Ringatomen, der ein oder zwei Ring-Heteroatome aus der Reihe N, O, S, SO und/oder SO2 enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls ein Ring-Stickstoffatom verknüpft ist. Beispielhaft seien genannt: Azetidinyl, Oxetanyl, Pyrrolidinyl, Pyrazolidinyl, Tetrahydrofuranyl, Thiolanyl, Piperidinyl, Piperazinyl, Tetrahydro- pyranyl, Tetrahydrothiopyranyl, Morpholinyl, Thiomorpholinyl, Hexahydroazepinyl und Hexahydro-l,4-diazepinyl. Bevorzugt sind Azetidinyl, Oxetanyl, Pyrrolidinyl, Tetrahydrofuranyl, Piperidinyl, Piperazinyl, Tetrahydropyranyl und Morpholinyl.
Ein 4- bis 7-gliedriger Aza-Heterocyclus steht im Rahmen der Erfindung für einen monocyclischen, gesättigten Heterocyclus mit insgesamt 4 bis 7 Ringatomen, der ein Stickstoffatom enthält und darüberhinaus ein weiteres Ring-Heteroatom aus der Reihe N, O, S, SO
oder SO2 enthalten kann und über ein Ring-Stickstoffatom verknüpft ist. Beispielhaft seien genannt: Azetidinyl, Pyrrolidinyl, Pyrazolidinyl, Piperidinyl, Piperazinyl, Morpholinyl, Thio- morpholinyl, 1 , 1 -Dioxothiomorpholinyl, Hexahydroazepinyl und Hexahydro-l,4-diazepinyl.
5- bis 9-gliedriges Aza-Heterocyclyl steht im Rahmen der Erfindung für einen monocyclischen oder bicyclischen, gesättigten oder teilweise ungesättigten Heterocyclus mit insgesamt 5 bis 9 Ringatomen, der ein Stickstoffatom enthält und darüberhinaus ein oder zwei weitere Ring- Heteroatome aus der Reihe N, O, S, SO und/oder SO2 enthalten kann und über ein Ring- Kohlenstoffatom verknüpft ist. Beispielhaft seien genannt: Pyrrolidinyl, Pyrazolidinyl, Piperidinyl, Piperazinyl, Morpholinyl, Thiomorpholinyl, 1 , 1 -Dioxothiomorpholinyl, Hexahydroazepinyl, Hexahydro-l,4-diazepinyl, 1,2,3,4-Tetrahydroisochinolinyl, 1,2,3,4-Tetrahydrochinolinyl, Indolinyl, 8-Azabicyclo[3.2.1]octanyl, 9-Azabicyclo[3.3.1]nonanyl, 3-Azabicyclo[4.1.0]heptanyl und Quinuclidinyl.
Heteroaryl steht im Rahmen der Erfindung für einen monocyclischen aromatischen Heterocyclus (Heteroaromaten) mit insgesamt 5 oder 6 Ringatomen, der bis zu drei gleiche oder verschiedene Ring-Heteroatome aus der Reihe N, O und/oder S enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls über ein Ring-Stickstoffatom verknüpft ist. Beispielhaft und vorzugsweise seien genannt: Furyl, Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thiazolyl, Oxazolyl, Isoxazolyl, Iso- thiazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl und Triazinyl. Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und Iod ein. Bevorzugt sind Chlor oder Fluor.
In der Formel der Gruppe, für die R3 bzw. R1 stehen kann, steht der Endpunkt der Linie, an dem das Zeichen * und # steht, nicht für ein Kohlenstoffatom beziehungsweise eine CH2-Gruppe, sondern ist Bestandteil der Bindung zu dem jeweils bezeichneten Atom, an das R3 bzw. R1 gebunden ist.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevorzugt. Im Sinne der vorliegenden Erfindung umfasst der Begriff "Behandlung" oder "behandeln" ein Hemmen, Verzögern, Aufhalten, Lindern, Abschwächen, Einschränken, Verringern, Unterdrücken, Zurückdrängen oder Heilen einer Krankheit, eines Leidens, einer Erkrankung, einer Verletzung oder einer gesundheitlichen Störung, der Entfaltung, des Verlaufs oder des Fortschreitens solcher
Zustände und/oder der Symptome solcher Zustände. Der Begriff "Therapie" wird hierbei als synonym mit dem Begriff "Behandlung" verstanden.
Die Begriffe "Prävention", "Prophylaxe" oder "Vorbeugung" werden im Rahmen der vorliegenden Erfindung synonym verwendet und bezeichnen das Vermeiden oder Vermindern des Risikos, eine Krankheit, ein Leiden, eine Erkrankung, eine Verletzung oder eine gesundheitliche Störung, eine Entfaltung oder ein Fortschreiten solcher Zustände und/oder die Symptome solcher Zustände zu bekommen, zu erfahren, zu erleiden oder zu haben.
Die Behandlung oder die Prävention einer Krankheit, eines Leidens, einer Erkrankung, einer Verletzung oder einer gesundheitlichen Störung können teilweise oder vollständig erfolgen. Bevorzugt sind im Rahmen der vorliegenden Erfindung Verbindungen der Formel (I), in welcher
A für CH2 oder CD2 steht,
R1 für (C3-C6)-Cycloalkyl, Pyridyl oder Phenyl steht, wobei (C3-C6)-Cycloalkyl mit 1 bis 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Methyl und Ethyl substituiert sein kann, wobei Pyridyl mit 1 oder 2 Substituenten F substituiert ist, und wobei Phenyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Difluormethyl, Trifluormethyl, (Ci-C i)-Alkyl, (Ci-C i)-Alkoxy und (C3-Cs)-Cyclopropyl substituiert sein kann,
R 2 für Wasserstoff, (Ci-C4)-Alkyl, Cyclopropyl, Difluormethyl oder Trifluormethyl steht,
R eine Gruppe der Formel
* für die Anknüpfstelle an die Carbonylgruppe steht,
L für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht,
L 2 für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht,
L für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht, für Wasserstoff, (Ci-C6)-Alkyl, (C3-C7)-Cycloalkyl, Phenyl oder 5- oder gliedriges Heteroaryl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom, Cyano, Trifluormethyl, Methyl, Ethyl, Methoxy oder Ethoxy substituiert sein können, für Wasserstoff oder (Ci-C i)-Alkyl steht, oder
R6 und R7 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind,
bis 5-gliedrigen Carbocyclus bilden, für Wasserstoff, (Ci-C6)-Alkyl, (C3-Cs)-Cycloalkyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, worin (Ci-C6)-Alkyl mit (Ci-C i)-Alkoxy, Benzyloxy oder Phenoxy substituiert sein kann, worin Benzyloxy und Phenoxy mit 1 bis 3 Substituenten unabhängig voneineander ausgewählt aus der Gruppe Fluor, Chlor und Brom substituiert sein können, worin (C3-C5)-Cycloalkyl mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, und
3Q worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom, Cyano, Trifluormethyl, Methyl, Ethyl, Methoxy oder Ethoxy substituiert sein können,
R9 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R8 und R9 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, oder
R6 und R8 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein können, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R6 und R7, R8 und
R9 bzw. R6 und R8 einen Carbo- oder Heterocyclus bildet, und mit der Maßgabe, dass die Reste R6 und R8 nicht beide gleichzeitig für Phenyl oder 5- oder 6-gliedriges Heteroaryl stehen, R10 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann,
R11 für Wasserstoff, (Ci-C6)-Alkyl oder (C3-C7)-Cycloalkyl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, oder R10und R11 zusammen mit dem Sticktoffatom, an das sie gebunden sind, einen 4- bis 7- gliedrigen Aza-Heterocyclus bilden,
R12 für 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes Aza- Heterocyclyl steht, worin 5- bis 9-gliedriges Aza-Heterocyclyl mit 1 bis 5 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Ethyl substituiert sein kann,
R13 für Wasserstoff, (Ci-C6)-Alkyl, (C3-C5)-Cycloalkyl, -(C=0)NR23R24, 5- oder 6- gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Difluormethoxy, Trifluormethoxy, Hydroxy und (Ci-C i)-Alkoxy substituiert sein kann, worin R23 für Wasserstoff, (Ci-C i)-Alkyl, Aryl oder Naphthyl steht, worin R24 für Wasserstoff steht, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom,
Trifluormethyl, Methyl und Ethyl substituiert sein können,
R14 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R13 und R14 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden,
R15 für Wasserstoff, (Ci-C6)-Alkyl oder (C3-C5)-Cycloalkyl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, und worin (C3-Cs)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy und (Ci-C i)-Alkyl substituiert sein kann,
R für Wasserstoff oder (Ci-C4)-Alkyl steht,
„„
- 32 - oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, oder
R13 und R15 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus und der 4- bis 7-gliedrige
Heterocyclus mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, mit der Maßgabe, dass die Reste R13 und R15 nicht beide gleichzeitig für Phenyl stehen, und mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R13 und R14, R15 und R16 bzw. R13 und R15 einen Carbo- oder Heterocyclus bildet,
R17 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, m für 0 oder 1 steht, n für 0 oder 1 steht,
R18 für Wasserstoff, Cyano oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, R19 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, R20 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann,
R21 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, oder
R18 und R19 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, worin der 3- bis 5-gliedrige Carbocyclus mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Ethyl substituiert sein kann, oder R20 und R21 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, worin der 3- bis 5-gliedrige Carbocyclus mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Ethyl substituiert sein kann, oder
R18 und R20 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus, worin der 3- bis 5-gliedrige Carbocyclus mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Ethyl substituiert sein kann, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R18 und R19, R20 und R21 bzw. R18 und R20 einen Carbo- oder Heterocyclus bildet,
R22 für (Ci-C6)-Alkyl, 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes Heterocyclyl, 5- bis 9-gliedriges Carbocyclyl, Phenyl, Indanyl oder 5- bis 10- gliedriges Heteroaryl steht, wobei (Ci-C6)-Alkyl mit Cyano oder bis zu dreifach mit Fluor substituiert sein kann,
wobei Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, Difluormethyl, (Ci-C i)-Alkyl, (Ci- C i)-Alkoxy und 5- bis 10-gliedriges Heteroaryl substituiert sein kann, worin (Ci-C4)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethoxy, (Ci-C i)-Alkoxy, (C3- C6)-Cycloalkyl, Hydroxy und Amino substituiert sein kann, wobei Indanyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl und Hydroxy substituiert sein kann, wobei 5- bis 10-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Cyano, Trifluormethyl, (Ci-C i)-Alkyl, (Ci-C i)-Alkoxy, Amino und Hydroxy substituiert sein kann, worin (Ci-C4)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt ausgewählt aus der Gruppe Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Difluormethyl, (Ci-C4)-Alkoxy, Trifluormethoxy, Difluormethoxy und Phenyl substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten Halogen substituiert sein kann, wobei 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes, Heterocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt ausgewählt aus der Gruppe Oxo, Fluor, Hydroxy und (Ci-C4)-Alkyl substiuiert sein kann, und wobei 5- bis 9-gliedriges Carbocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt ausgewählt aus der Gruppe Trifluormethyl, Fluor, Hydroxy, Hydroxycarbonyl, (Ci-C4)-Alkoxycarbonyl und (Ci-C4)-Alkyl substiuiert sein kann, für Wasserstoff steht, für Wasserstoff, Fluor, Chlor, Brom, Cyano, Difluormethyl, Trifluormethyl, (Ci-C4)-Alkyl, (C2-C4)-Alkinyl oder (C3-C5)-Cycloalkyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
„
- 35 -
Bevorzugt sind im Rahmen der vorliegenden Erfindung Verbindungen der Formel (I), in welcher
A für CH2 oder CD2 steht,
R1 für (C3-C6)-Cycloalkyl, Pyridyl oder Phenyl steht, wobei (C3-C6)-Cycloalkyl mit 1 bis 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Methyl und Ethyl substituiert sein kann, wobei Pyridyl mit 1 oder 2 Substituenten Fluor substituiert ist, und wobei Phenyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Difluormethyl, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C i)-Alkoxy und (C3-Cs)-Cyclopropyl substituiert sein kann,
R2 für Wasserstoff, (Ci-C i)-Alkyl, Cyclopropyl, Difluormethyl oder Trifluormethyl steht,
R3 für eine Gruppe der Formel
steht, wobei * für die Anknüpfstelle an die Carbonylgruppe steht,
L1 für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht,
L2 für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht,
L3 für eine Bindung, Methandiyl oder 1 ,2-Ethandiyl steht,
R6 für Wasserstoff, (Ci-C6)-Alkyl, (C3-C7)-Cycloalkyl, Phenyl oder 5- oder 6- gliedriges Heteroaryl steht,
„„
- 36 - worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom, Cyano, Trifluormethyl, Methyl, Ethyl, Methoxy oder Ethoxy substituiert sein können,
R7 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R6 und R7 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, R8 für Wasserstoff, (Ci-C6)-Alkyl, (C3-C5)-Cycloalkyl, 5- oder 6-gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit (Ci-C i)-Alkoxy, Benzyloxy oder Phenoxy sowie bis zu fünffach mit Fluor substituiert sein kann, worin Benzyloxy und Phenoxy mit 1 bis 3 Substituenten unabhängig voneineander ausgewählt aus der Gruppe Fluor, Chlor und Brom substituiert sein können, worin (C3-C5)-Cycloalkyl mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom, Cyano, Trifluormethyl, Methyl, Ethyl, Methoxy oder Ethoxy substituiert sein können,
R9 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen bis 5-gliedrigen Carbocyclus bilden, oder
„
- 37 -
R6 und R8 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein können, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R6 und R7, R8 und R9 bzw. R6 und R8 einen Carbo- oder Heterocyclus bildet, und mit der Maßgabe, dass die Reste R6 und R8 nicht beide gleichzeitig für Phenyl oder 5- oder
6-gliedriges Heteroaryl stehen,
R10 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl bis zu fünffach mit Fluor substituiert sein kann,
R11 für Wasserstoff, (Ci-C6)-Alkyl oder (C3-C7)-Cycloalkyl steht, worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, oder
R10und R11 zusammen mit dem Sticktoffatom, an das sie gebunden sind, einen 4- bis 7- gliedrigen Aza-Heterocyclus bilden,
R12 für 5- bis 10-gliedriges, über ein Ring- Kohlenstoffatom gebundenes Aza- Heterocyclyl steht, worin 5- bis 10-gliedriges Aza-Heterocyclyl mit 1 bis 5 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Ethyl substituiert sein kann, oder für Amino stehen kann, wenn L2 für eine Bindung steht, worin Amino mit (Ci-C4)-Alkyl, (Ci-C i)-Alkylcarbonyl, (C3-C6)-Carbocyclyl, 4- bis 7-gliedriges Heterocyclyl, Phenyl oder 5- oder 6-gliedriges Heteroaryl substituiert sein kann,
worin (Ci-C i)-Alkylcarbonyl mit Mono-alkylamino oder Di-alkylamino substituiert sein kann, worin (C3-C6)-Carbocyclyl und 4- bis 7-gliedriges Heterocyclyl mit Hydroxy substituiert sein können, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Methyl und Trifluormethyl substituiert sein können, für Wasserstoff, (Ci-C6)-Alkyl, (C3-C5)-Cycloalkyl, -(C=0)NR23R24, 5- oder 6- gliedriges Heteroaryl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Difluormethoxy, Trifluormethoxy, Hydroxy und (Ci- C i)-Alkoxy sowie bis zu sechsfach mit Fluor substituiert sein kann, worin R23 für Wasserstoff, (Ci-C i)-Alkyl, Aryl oder Naphthyl steht, worin R24 für Wasserstoff steht, und worin Phenyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom, Trifluormethyl, Methyl und Ethyl substituiert sein können,
R14 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R13 und R14 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden,
R15 für Wasserstoff, (Ci-C6)-Alkyl oder (C3-C5)-Cycloalkyl steht, worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, und
^ worin (C3-Cs)-Cycloalkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl, Hydroxy und (Ci-C i)-Alkyl substituiert sein kann,
R16 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, oder
R13 und R15 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus oder einen 4- bis 7-gliedrigen Heterocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus und der 4- bis 7-gliedrige Heterocyclus mit 1 oder 2 Substituenten Fluor oder (Ci-C i)-Alkyl substituiert sein kann, mit der Maßgabe, dass die Reste R13 und R15 nicht beide gleichzeitig für Phenyl stehen, und mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R13 und R14, R15 und R16 bzw. R13 und R15 einen Carbo- oder Heterocyclus bildet,
R17 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, m für 0 oder 1 steht, n für 0 oder 1 steht, R18 für Wasserstoff, Cyano oder (Ci-C4)-Alkyl steht, worin (Ci-C4)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann,
R für Wasserstoff oder (Ci-C4)-Alkyl steht,
_
- 40 - worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, R20 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, R21 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C i)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, oder
R18 und R19 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, worin der 3- bis 5-gliedrige Carbocyclus mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und
Ethyl substituiert sein kann, oder
R20 und R21 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, worin der 3- bis 5-gliedrige Carbocyclus mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Ethyl substituiert sein kann, oder
R18 und R20 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus, worin der 3- bis 5-gliedrige Carbocyclus mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Methyl und Ethyl substituiert sein kann, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R18 und R19, R20 und R21 bzw. R18 und R20 einen Carbo- oder Heterocyclus bildet, für (Ci-C6)-Alkyl, Cyano, (Ci-C6)-Alkoxy, 5- bis 9-gliedriges, über ein Ring- Kohlenstoffatom gebundenes Heterocyclyl, 5- bis 9-gliedriges Carbocyclyl, Phenyl, Indanyl oder 5- bis 10-gliedriges Heteroaryl steht,
worin (Ci-C6)-Alkyl mit Cyano oder bis zu fünffach mit Fluor substituiert sein kann, worin (Ci-Ce)-Alkoxy mit Hydroxy oder (C2-C i)-Alkenyl substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Trifluormethyl, Difluormethyl, (Ci-C i)-Alkyl, (Ci- C i)-Alkoxy und 5- bis 10-gliedriges Heteroaryl substituiert sein kann, worin (Ci-C4)-Alkyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethoxy, (Ci-C4)-Alkoxy, (C3- C6)-Cycloalkyl, Hydroxy und Amino substituiert sein kann, worin Indanyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Trifluormethyl und Hydroxy substituiert sein kann, worin 5- bis 10-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Cyano, Trifluormethyl, (Ci-C4)-Alkyl, (Ci-C4)-Alkoxy, Amino und Hydroxy substituiert sein kann, worin (Ci-C4)-Alkyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt ausgewählt aus der Gruppe Halogen, Cyano, Hydroxy, Amino, Trifluormethyl, Difluormethyl, (Ci-C4)-Alkoxy, Trifluormethoxy, Difluormethoxy und Phenyl substituiert sein kann, worin Phenyl mit 1 bis 3 Substituenten Halogen substituiert sein kann, worin 5- bis 9-gliedriges, über ein Ring-Kohlenstoffatom gebundenes, Heterocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt ausgewählt aus der Gruppe Oxo, Fluor, Hydroxy und (Ci-C4)-Alkyl substiuiert sein kann, und worin 5- bis 9-gliedriges Carbocyclyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt ausgewählt aus der Gruppe Trifluormethyl, Fluor, Hydroxy, Hydroxycarbonyl, (Ci-C4)-Alkoxycarbonyl und (Ci-C4)-Alkyl substiuiert sein kann, serstoff steht,
R5 für Wasserstoff, Fluor, Chlor, Brom, Cyano, Difluormethyl, Trifluormethyl, (Ci-C4)-Alkyl, (C2-C4)-Alkinyl oder (C3-C5)-Cycloalkyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Bevorzugt sind im Rahmen der vorliegenden Erfindung Verbindungen der Formel (I), in welcher
A für CH2 steht,
R1 für Cyclohexyl, Pyridyl oder Phenyl steht, wobei Cyclohexyl mit 1 bis 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und Methyl substituiert sein kann, wobei Pyridyl mit 1 oder 2 Substituenten Fluor substituiert ist, und wobei Phenyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Methyl, Methoxy und Cyclopropyl substituiert sein kann,
R 2 für Methyl, Cyclopropyl oder Trifluormethyl steht,
R für eine Gruppe der Formel
* für die Anknüpfstelle an die Carbonylgruppe steht, L1 für eine Bindung steht, L2 für eine Bindung steht,
,„
- 43 -
L3 für eine Bindung steht,
R6 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, und worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Trifluormethyl, Methyl oder Methoxy substituiert sein kann,
R7 für Wasserstoff, Methyl oder Ethyl steht,
R8 für Wasserstoff, (Ci-C6)-Alkyl, Cyclopropyl oder Cyclobutyl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann,
R9 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R8 und R9 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus bilden, R10 für Wasserstoff, Methyl oder Ethyl steht, worin Ethyl mit 1 bis 3 Substituenten Fluor substituiert sein kann,
R11 für Wasserstoff, (Ci-C4)-Alkyl oder (C3-C5)-Cycloalkyl steht, oder
R10und Ru zusammen mit dem Sticktoffatom, an das sie gebunden sind, einen
Morpholinyl-Ring oder Piperidinyl-Ring bilden,
R12 für 9-Azabicyclo[3.3.1 ]nonan-3-yl oder Piperidin-4-yl steht, worin 9-Azabicyclo[3.3.1 ]nonan-3-yl mit Methyl substituiert ist, worin Piperidin-4-yl mit 1 bis 5 Substituenten Methyl substituiert ist,
R13 für Wasserstoff, (Ci-C6)-Alkyl, -(C=0)NR23R24 oder Phenyl steht,
worin (Ci-C6)-Alkyl mit einem Rest Hydroxy oder Methoxy oder bis zu fünffach mit Fluor substituiert sein kann, worin R23 für Aryl oder Naphthyl steht, worin R24 für Wasserstoff steht und worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Trifluormethyl und Methyl substituiert sein kann,
R14 für Wasserstoff oder (Ci-C4)-Alkyl steht,
R15 für Wasserstoff, (Ci-C6)-Alkyl, Cyclopropyl oder Cyclobutyl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, worin Cyclopropyl und Cyclobutyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor oder Methyl substituiert sein können,
R16 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus mit 1 oder 2 Substituenten Fluor oder Methyl substituiert sein kann,
R17 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C3)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, m für 0 oder 1 steht, n für 0 oder 1 steht,
R18 für Wasserstoff, Cyano oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann,
R für Wasserstoff oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, R20 für Wasserstoff oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, R21 für Wasserstoff oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, oder
R18 und R19 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, oder
R18 und R20 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen
Cyclopropyl-Ring bilden, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R18 und R19 bzw. R18 und R20 einen Carbocyclus bildet,
R22 für (Ci-C6)-Alkyl, 2-Oxopyrrolidin-3-yl, 2-Oxotetrahydrofuran-3-yl, Cyclopentyl, Cyclohexyl, Phenyl, Indanyl, l ,2,4-Oxadiazol-5-yl, lH-Imidazol-2-yl, lH-Pyrazol- 4-yl, Pyridin-3-yl, Pyrimidin-5-yl, Chinolin-4-yl oder Pyrazolo[l ,5-a]pyridin-3-yl, steht, worin (Ci-C6)-Alkyl mit einem Rest Cyano oder bis zu dreifach mit Fluor substituiert sein kann, wobei Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Cyano, Trifluormethyl, Methyl, Ethyl, Methoxy und Pyridyl substituiert sein kann, wobei Indanyl mit Hydroxy substituiert ist, wobei l ,2,4-Oxadiazol-5-yl, lH-Imidazol-2-yl, lH-Pyrazol-4-yl, Pyridin-3-yl, Pyrimidin-5-yl, Chinolin-4-yl oder Pyrazolo[l ,5-a]pyridin-3-yl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Trifluormethyl, (Ci-C3)-Alkyl, Amino und Hydroxy substituiert sein können,
worin (Ci-C3)-Alkyl mit Fluor, Hydroxy, Amino oder Trifluormethyl, substituiert sein kann, wobei Cyclopentyl und Cyclohexyl mit Methoxycarbonyl oder Ethoxycarbonyl substiuiert sind, R für Wasserstoff steht,
R5 für Wasserstoff, Methyl oder Ethyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung Verbindungen der Formel (I), in welcher A für CH2 steht, R1 für Cyclohexyl, Pyridyl oder Phenyl steht, wobei Cyclohexyl mit 1 bis 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor und Methyl substituiert sein kann, wobei Pyridyl mit 1 oder 2 Substituenten Fluor substituiert ist, und wobei Phenyl mit 1 bis 4 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Methyl, Methoxy und Cyclopropyl substituiert sein kann,
R 2 für Methyl, Cyclopropyl oder Trifluormethyl steht,
R eine Gruppe der Formel
- 47 -
* für die Anknüpfstelle an die Carbonylgruppe steht, L1 für eine Bindung steht, L2 für eine Bindung steht, L3 für eine Bindung steht, R6 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, und worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Trifluormethyl, Methyl oder Methoxy substituiert sein kann,
R7 für Wasserstoff, Methyl oder Ethyl steht,
R8 für Wasserstoff, (Ci-C6)-Alkyl, Cyclopropyl oder Cyclobutyl steht, worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, R9 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R8 und R9 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus bilden,
R10 für Wasserstoff, Methyl oder Ethyl steht, worin Ethyl bis zu dreifach mit Fluor substituiert sein kann, R11 für Wasserstoff, (Ci-C4)-Alkyl oder (C3-C5)-Cycloalkyl steht, oder
R10und Ru zusammen mit dem Sticktoffatom, an das sie gebunden sind, einen
Morpholinyl-Ring oder Piperidinyl-Ring bilden,
R12 für 9-Azabicyclo[3.3.1 ]nonan-3-yl oder Piperidin-4-yl steht, worin 9- Azabicyclo [3.3.1 ]nonan-3 -yl mit Methyl substituiert ist,
_
- 48 - worin Piperidin-4-yl mit 1 bis 5 Substituenten Methyl substituiert ist,
R13 für Wasserstoff, (Ci-C6)-Alkyl, -(C=0)NR23R24 oder Phenyl steht, worin (Ci-C6)-Alkyl mit einem Rest Hydroxy oder Methoxy oder bis zu fünffach mit Fluor substituiert sein kann, worin R23 für Aryl oder Naphthyl steht, worin R24 für Wasserstoff steht und worin Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Trifluormethyl und Methyl substituiert sein kann, R14 für Wasserstoff oder (Ci-C4)-Alkyl steht,
R15 für Wasserstoff, (Ci-C6)-Alkyl, Cyclopropyl oder Cyclobutyl steht, worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, worin Cyclopropyl und Cyclobutyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor oder Methyl substituiert sein können,
R16 für Wasserstoff oder (Ci-C4)-Alkyl steht, oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus bilden, worin der 3- bis 6-gliedrige Carbocyclus mit 1 oder 2 Substituenten Fluor oder Methyl substituiert sein kann,
R17 für Wasserstoff oder (Ci-C4)-Alkyl steht, worin (Ci-C3)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, m für 0 oder 1 steht, n für 0 oder 1 steht,
R für Wasserstoff, Cyano oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, R19 für Wasserstoff oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, R20 für Wasserstoff oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, R21 für Wasserstoff oder Methyl steht, worin Methyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, oder
R18 und R19 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 5-gliedrigen Carbocyclus bilden, oder
R18 und R20 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen
Cyclopropyl-Ring bilden, mit der Maßgabe, dass gleichzeitig nicht mehr als eines der Reste-Paare R18 und R19 bzw. R18 und R20 einen Carbocyclus bildet,
R22 für (Ci-C6)-Alkyl, Cyano, 2-Oxopyrrolidin-3-yl, 2-Oxotetrahydrofuran-3-yl, Cyclopentyl, Cyclohexyl, Phenyl, Indanyl, l ,2,4-Oxadiazol-5-yl, lH-Imidazol-2- yl, lH-Pyrazol-4-yl, Pyridin-3-yl, Pyrimidin-5-yl, Chinolin-4-yl oder Pyrazolo[l ,5- a]pyridin-3-yl, steht, worin (Ci-C6)-Alkyl mit einem Rest Cyano oder bis zu dreifach mit Fluor substituiert sein kann, wobei Phenyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Cyano, Trifluormethyl, Methyl, Ethyl, Methoxy und Pyridyl substituiert sein kann, wobei Indanyl mit Hydroxy substituiert ist,
5Q wobei l ,2,4-Oxadiazol-5-yl, lH-Imidazol-2-yl, lH-Pyrazol-4-yl, Pyridin-3-yl, Pyrimidin-5-yl, Chinolin-4-yl oder Pyrazolo[l ,5-a]pyridin-3-yl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Trifluormethyl, (Ci-C3)-Alkyl, Amino und Hydroxy substituiert sein können, worin (Ci-C3)-Alkyl mit Fluor, Hydroxy, Amino oder Trifluormethyl, substituiert sein kann, wobei Cyclopentyl und Cyclohexyl mit Cyano, Methoxycarbonyl oder Ethoxy- carbonyl substiuiert sind,
R4 für Wasserstoff steht, R5 für Wasserstoff, Methyl oder Ethyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Besonders bevorzugt sind im Rahmen der vorliegenden Erfindung Verbindungen der Formel (I), in welcher
A für CH2 steht, eine Phenyl-Gruppe der Formel
# für die Anknüpfstelle an A steht, und R27 für Wasserstoff oder Fluor steht,
R28 für Fluor steht, R29 für Fluor steht, R2 für Methyl steht,
für eine Gruppe der Formel
wobei für die Anknüpfstelle an die Carbonylgruppe steht,
L1 für eine Bindung steht, L3 für eine Bindung steht, R6 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl mit 1 bis 5 Substituenten Fluor substituiert sein kann, und worin Phenyl mit 1 bis 2 Substituenten Chlor oder Fluor substituiert sein kann,
R7 für Wasserstoff, Methyl oder Ethyl steht,
R für Wasserstoff, (Ci-C6)-Alkyl, Trifluormethyl oder Cyclopropyl steht, worin (Ci-C6)-Alkyl mit 1 bis 3 Substituenten Fluor substituiert sein kann,
R9 für Wasserstoff, Methyl oder Ethyl steht, R10 für Wasserstoff steht, R11 für Wasserstoff steht, R13 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl mit einem Rest Hydroxy oder bis zu fünffach mit Fluor substituiert sein kann, und worin Phenyl mit 1 oder 2 Substituenten Fluor substituiert sein kann,
R14 für Wasserstoff, Methyl oder Ethyl steht,
R15 für Wasserstoff oder (Ci-C6)-Alkyl steht, R16 für Wasserstoff, Methyl oder Ethyl steht, oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen 3- bis 6-gliedrigen Carbocyclus bilden,
R17 für Wasserstoff steht,
R4 für Wasserstoff steht,
R5 für Wasserstoff oder Methyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Besonders bevorzugt sind im Rahmen der vorliegenden Erfindung Verbindungen der Formel (I), in welcher
A für CH2 steht,
R1 für eine Phenyl-Gruppe der Formel
# für die Anknüpfstelle an A steht, und
R27 für Wasserstoff oder Fluor steht,
R28 für Fluor steht,
R
29 für Fluor steht, für Methyl steht, für eine Gruppe der Formel
wobei für die Anknüpfstelle an die Carbonylgruppe steht,
L1 für eine Bindung steht, L3 für eine Bindung steht, R6 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, und worin Phenyl mit 1 bis 2 Substituenten Chlor oder Fluor substituiert sein kann,
R7 für Wasserstoff, Methyl oder Ethyl steht, R8 für Wasserstoff, (Ci-C6)-Alkyl, Trifluormethyl oder Cyclopropyl steht, worin (Ci-C6)-Alkyl bis zu dreifach mit Fluor substituiert sein kann,
R9 für Wasserstoff, Methyl oder Ethyl steht, R10 für Wasserstoff steht, R11 für Wasserstoff steht, R13 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl mit einem Rest Hydroxy oder bis zu fünffach mit Fluor substituiert sein kann, und worin Phenyl mit 1 oder 2 Substituenten Fluor substituiert sein kann,
R für Wasserstoff, Methyl oder Ethyl steht,
R für Wasserstoff oder (Ci-C6)-Alkyl steht,
R16 für Wasserstoff, Methyl oder Ethyl steht, oder
R15 und R16 zusammen mit dem Kohlenstoffatom, an das sie gebunden sind, einen bis 6-gliedrigen Carbocyclus bilden, R17 für Wasserstoff steht,
R4 für Wasserstoff steht,
R5 für Wasserstoff oder Methyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), welcher
R1 für eine Phenyl-Gruppe der Formel
# für die Anknüpfstelle an A steht, und
R27 für Wasserstoff oder Fluor steht,
R28 für Fluor steht,
R29 für Fluor steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), welcher
R2 für Methyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R3 für eine Gruppe der Formel
* für die Anknüpfstelle an die Carbonylgruppe steht,
L1 für eine Bindung steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), welcher
R3 für eine Gruppe der Formel
* für die Anknüpfstelle an die Carbonylgruppe steht, R6 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl bis zu fünffach mit Fluor substituiert sein kann, und worin Phenyl mit 1 bis 2 Substituenten Chlor oder Fluor substituiert sein kann, ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
r
- 56 -
Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R
3 für eine Gruppe der Formel
steht, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht,
R7 für Wasserstoff steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R
3 für eine Gruppe der Formel
steht, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht,
und
R
8 für Wasserstoff, (Ci-C6)-Alkyl, Trifluormethyl oder Cyclopropyl steht, worin (Ci-C6)-Alkyl bis zu dreifach mit Fluor substituiert sein kann, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R eine Gruppe der Formel
steht, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht,
und
R9 für Wasserstoff oder Methyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R
3 für eine Gruppe der Formel
steht, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht,
und
R10 für Wasserstoff steht,
R11 für Wasserstoff steht,
sowie ihre .V-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R
3 für eine Gruppe der Formel
stellt, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht,
L3 für eine Bindung steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in
welcher
R3 für eine Gruppe der Formel
* für die Anknüpfstelle an die Carbonylgruppe steht,
R13 für Wasserstoff, (Ci-C6)-Alkyl oder Phenyl steht, worin (Ci-C6)-Alkyl mit einem Rest Hydroxy oder bis zu fünffach mit Fluor substituiert sein kann, und worin Phenyl mit 1 oder 2 Substituenten Fluor substituiert sein kann, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R
3 für eine Gruppe der Formel
steht, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht,
R14 für Wasserstoff steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), welcher
R
1 für eine Gruppe der Formel
steht, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht,
und
R15 für Wasserstoff oder (Ci-C6)-Alkyl steht,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), welcher
R
3 für eine Gruppe der Formel
rn - 60 - steht, wobei
* für die Anknüpfstelle an die Carbonylgruppe steht, und
R16 für Wasserstoff oder Methyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R3 für eine Gruppe der Formel
* für die Anknüpfstelle an die Carbonylgruppe steht, und
R17 für Wasserstoff steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze. Bevorzugt sind im Rahmen der vorliegenden Erfindung auch Verbindungen der Formel (I), in welcher
R5 für Wasserstoff oder Methyl steht, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im Einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
„
- 61 -
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Verbindungen der Formel (I) dadurch gekennzeichnet, dass man
[A] eine Verbindung der Formel (II)
in welcher A, R
1, R
2, R
4 und R
5 jeweils die oben angegebenen Bedeutungen haben und T
1 für (Ci-C
4)-Alkyl oder Benzyl steht, in einem inerten Lösungsmittel in Gegenwart einer geeigneten Base oder Säure
Carbonsäure der Formel (III)
in welcher A, R
1, R
2, R
4 und R
5 jeweils die oben angegebenen Bedeutungen haben, umsetzt und diese in der Folge in einem inerten Lösungsmittel unter Amidkupplungsbedmgungen mit einem Amin der Formel (IV- A), (IV-B), (IV-C) oder (IV-D)
_
- 62 -
(IV-C) (IV-D), in welchem L1, L2, L3, R6, R7, R8, R9, R12, R13, R14, R15, R16, R17, R18, R19, R20, R21 und R22 jeweils die oben angegebenen Bedeutungen haben und R10A und RUA die oben für R10 und R11 angegebenen Bedeutungen haben oder für eine Amino- Schutzgruppe, wie beispielsweise tert.-Butoxycarbonyl, Benzyloxycarbonyl oder Benzyl stehen, umsetzt, anschliessend gegebenenfalls vorhandene Schutzgruppen abspaltet, und die resultierenden Verbindungen der Formel (I) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Säuren oder Basen in ihre Solvate, Salze und/oder Solvate der Salze überführt.
Das beschriebene Herstellverfahren kann durch das folgende Syntheseschema (Schema 1) beispielhaft verdeutlicht werden:
Schema 1 :
[a) 1 N wässriges Natriumhydroxid, 1,4-Dioxan, RT; b) HATU, NN-Diisopropylethylamin, DMF, RT].
Die Verbindungen der Formeln (IV-A), (IV-B), (IV-C) und (IV-D) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaturbekannten Verfahren hergestellt werden.
Inerte Lösungsmittel für die Verfahrensschritte (III) + (IV)— »■ (I) sind beispielsweise Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, 1 ,2-Dichlorethan, Tri- chlorethylen oder Chlorbenzol, oder andere Lösungsmittel wie Aceton, Essigsäureethylester, Acetonitril, Pyridin, Dimethylsulfoxid, NN-Dimethylformamid, NN-Dimethylacetamid, NN- Dimethylpropylenharnstoff (DMPU) oder N-Methylpyrrolidon (ΝΜΡ). Ebenso ist es möglich, Ge- mische der genannten Lösungsmittel zu verwenden. Bevorzugt sind Dichlormethan, Tetrahydrofuran, Dimethylformamid oder Gemische dieser Lösungsmittel.
Als Kondensationsmittel für die Amidbildung in den Verfahrensschritte (III) + (IV)—> (I) und eignen sich beispielsweise Carbodiimide wie NN'-Diethyl-, NN'-Dipropyl-, NN'-Diisopropyl-, NN'-Dicyclohexylcarbodiimid (DCC) oder N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimid- Hydrochlorid (EDC), Phosgen-Derivate wie NN'-Carbonyldiimidazol (CDI), 1 ,2-Oxazolium- verbindungen wie 2-Ethyl-5-phenyl-l,2-oxazolium-3-sulfat oder 2-fert.-Butyl-5-methyl-isoxazo- lium-perchlorat, Acylaminoverbindungen wie 2-Ethoxy-l-ethoxycarbonyl-l,2-dihydrochinolin, oder Isobutylchlorformiat, Propanphosphonsäureanhydrid (T3P), l-Chlor-N,N,2-trimethylpropl- en-l-amin, Cyanophosphonsäurediethylester, Bis-(2-oxo-3-oxazolidinyl)-phosphorylchlorid, Benzotriazol- 1 -yloxy-tris(dimethylamino)phosphonium-hexafluorphosphat, Benzotriazol- 1 -yloxy- tris(pyrrolidino)phosphonium-hexafluorphosphat (PyBOP), 0-(Benzotriazol- 1 -yl)-N,N,N',N- tetramethyluronium-tetrafluorborat (TBTU), 0-(Benzotriazol- 1 -yl)-N,N,N',N'-tetramethyluronium-
hexafluorphosphat (HBTU), 2-(2-Oxo-l-(2H)-pyridyl)-l,l,3,3-tetramethyluronium-tetrafluorborat (TPTU), 0-(7-Azabenzotriazol- 1 -yl)-N,N,N',N'-tetramethyluronium-hexafluorphosphat (HATU) oder 0-(lH-6-Chlorbenzotriazol-l-yl)-l,l,3,3-tetramethyluronium-tetrafluorborat (TCTU), gegebenenfalls in Kombination mit weiteren Hilfsstoffen wie 1 -Hydroxybenzotriazol (HOBt) oder N-Hydroxysuccinimid (HOSu), sowie als Basen Alkalicarbonate, z.B. Natrium- oder Kalium- carbonat oder -hydrogencarbonat, oder organische Basen wie Trialkylamine, z.B. Triethylamin, N-Methylmorpholin, N-Methylpiperidin oder NN-Diisopropylethylamin. Bevorzugt wird TBTU in Verbindung mit N-Methylmorpholin, HATU in Verbindung mit NN-Diisopropylethylamin oder 1- Chlor-NN,2-trimethylprop- 1 -en- 1 amin verwendet. Die Kondensationen (III) + (IV)—> (I) und wird im Allgemeinen in einem Temperaturbereich von - 20°C bis +100°C, bevorzugt bei 0°C bis +60°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder bei erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Alternativ kann die Carbonsäure der Formel (III) auch zunächst in das entsprechende Carbonsäurechlorid überführt werden und dieses dann direkt oder in einer separaten Umsetzung mit einem Amin der Formel (IV) zu den erfindungsgemäßen Verbindungen umgesetzt werden. Die Bildung von Carbonsäurechloriden aus Carbonsäuren erfolgt nach den dem Fachmann bekannten Methoden, beispielsweise durch Behandlung mit Thionylchlorid, Sulfurylchlorid oder Oxalylchlorid in Gegenwart einer geeigneten Base, beispielsweise in Gegenwart von Pyridin, sowie optional unter Zusatz von Dimethylformamid, optional in einem geeigneten inerten Lösemittel.
Die Hydrolyse der Ester-Gruppe T1 der Verbindungen der Formel (II) erfolgt nach üblichen Methoden, indem man die Ester in inerten Lösungsmitteln mit Säuren oder Basen behandelt, wobei bei letzterem die zunächst entstehenden Salze durch Behandeln mit Säure in die freien Carbonsäuren überführt werden. Im Falle der tert.-Butylester erfolgt die Esterspaltung bevorzugt mit Säuren. Im Falle der Benzylester erfolgt die Esterspaltung bevorzugt hydrogenolytisch mit Palladium auf Aktivkohle oder Raney-Nickel. Als inerte Lösungsmittel eignen sich für diese Reaktion Wasser oder die für eine Esterspaltung üblichen organischen Lösungsmittel. Hierzu gehören bevorzugt Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder tert.- Butanol, oder Ether wie Diethylether, Tetrahydrofuran, 2-Methyltetrahydrofuran, Dioxan oder Glykoldimethylether, oder andere Lösungsmittel wie Aceton, Dichlormethan, Dimethylformamid oder Dimethylsulfoxid. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Im Falle einer basischen Ester-Hydrolyse werden bevorzugt Gemische von Wasser mit Dioxan, Tetrahydrofuran, Methanol und/oder Ethanol eingesetzt.
Als Basen für die Ester-Hydrolyse sind die üblichen anorganischen Basen geeignet. Hierzu gehören bevorzugt Alkali- oder Erdalkalihydroxide wie beispielsweise Natrium-, Lithium-, Kalium- oder Bariumhydroxid, oder Alkali- oder Erdalkalicarbonate wie Natrium-, Kalium- oder Calciumcarbonat. Besonders bevorzugt sind Natrium- oder Lithiumhydroxid. Als Säuren eignen sich für die Esterspaltung im Allgemeinen Schwefelsäure, Chlorwasserstoff/ Salzsäure, Bromwasserstoff/Bromwasserstoffsäure, Phosphorsäure, Essigsäure, Trifluoressigsäure, Toluolsulfonsäure, Methansulfonsäure oder Trifluormethansulfonsäure oder deren Gemische gegebenenfalls unter Zusatz von Wasser. Bevorzugt sind Chlorwasserstoff oder Trifluoressigsäure im Falle der tert.-Butylester und Salzsäure im Falle der Methylester. Die Esterspaltung erfolgt im Allgemeinen in einem Temperaturbereich von 0°C bis +100°C, bevorzugt bei +0°C bis +50°C.
Die genannten Umsetzungen können bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man jeweils bei Normaldruck.
Als Amino- Schutzgruppe wird bevorzugt tert. -Butoxycarbonyl (Boc) oder Benzyloxycarbonyl (Z) verwendet. Als Schutzgruppe für eine Hydroxy- oder Carboxyl-Funktion wird vorzugsweise tert.- Butyl oder Benzyl eingesetzt. Die Abspaltung dieser Schutzgruppen wird nach üblichen Methoden, vorzugsweise durch Reaktion mit einer starken Säure wie Chlorwasserstoff, Bromwasserstoff oder Trifluoressigsäure in einem inerten Lösungsmittel wie Dioxan, Diethylether, Dichlormethan oder Essigsäure durchgeführt; gegebenenfalls kann die Abspaltung auch ohne ein zusätzliches inertes Lösungsmittel erfolgen. Im Falle von Benzyl und Benzyloxycarbonyl als Schutzgruppe können diese auch durch Hydrogenolyse in Gegenwart eines Palladium-Katalysators entfernt werden. Die Abspaltung der genannten Schutzgruppen kann gegebenenfalls simultan in einer Eintopf-Reaktion oder in separaten Reaktionschritten vorgenommen werden.
Die Verbindungen der Formel (II) sind literaturbekannt oder können hergestellt werden, indem eine Verbindung der Formel (V)
in welcher R
4 und R
5 oben angegebene Bedeutung haben,
r r
- 66 - in einem inerten Lösungsmittel in Gegenwart einer geeigneten Base mit einer Verbindung Formel (VI)
X1 (VI), in welcher A und R1 die oben angegebene Bedeutung haben und
X1 für Hydroxy steht, zu einer Verbindung der Formel (VII)
in welcher A, R , R und R jeweils die oben angegebenen Bedeutungen haben, umgesetzt wird, und diese anschliessend in einem inerten Lösungsmittel mit einer Verbindung der Formel (VIII)
in welcher R
2und T
1 jeweils die oben angegebenen Bedeutungen haben, umgesetzt wird.
Das beschriebene Verfahren wird durch das nachfolgende Schema (Schema 2) beispielhaft verdeutlicht:
Schema 2:
[(a) Kalium-tert-butylat, 1 ,2-Dimethoxyethan, 80°C; (b) Ethanol, Molekularsieb, Rückfluss].
Die gezeigte Synthesesequenz kann dahingehend modifiziert werden, dass die jeweiligen Reaktionsschritte in einer veränderten Reihenfolge durchlaufen werden. Ein Beispiel für eine solche modifizierte Synthesesequenz ist in Schema 3 gezeigt.
Schema 3 :
[a): EtOH, Molekularsieb, Rückfluss; b): Kalium-tert-butylat, 1 ,2-Dimethoxyethan, 80°C]. Inerte Lösungsmittel für den Verfahrensschritt (V) + (VI) ->· (VII) bzw. (X) + (VI) ->· (II) sind beispielsweise Ether wie Diethylether, Dioxan, Tetrahydrofuran, Dimethoxymethan, Glykol- dimethylether oder Diethylenglykoldimethylether oder andere Lösungsmittel wie Aceton, Methylethylketon, Essigsäureethylester, Acetonitril, NN-Dimethylformamid, NN-Di- methylacetamid, Dimethylsulfoxid, NN'-Dimethylpropylenharnstoff (DMPU), N-Methylpyrroli- don (ΝΜΡ). Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt wird Dimethoxyethan verwendet.
Als Basen für den Verfahrensschritt (V) + (VI) ->■ (VII) bzw. (X) + (VI) ->■ (II) eignen sich die üblichen anorganischen oder organischen Basen. Hierzu gehören bevorzugt Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkali- oder Erdalkalicarbonate wie Lithium-, Natrium-, Kalium-, Calcium- oder Cäsiumcarbonat gegebenenfalls unter Zusatz eines Alkaliiodids wie beispielsweise Natriumiodid oder Kaliumiodid, Alkali-Alkoholate wie Natriumoder Kaliummethanolat, Natrium- oder Kaliumethanolat oder Natrium- oder Kalium-tert.-butylat, Alkalihydride wie Natrium- oder Kaliumhydrid, Amide wie Natriumamid, Lithium- oder Kalium- bis(trimethylsilyl)amid oder Lithiumdiisopropylamid, oder organische Amine wie Triethylamin, N-Methylmorpholin, N-Methylpiperidin, NN-Diisopropylethylamin, Pyridin, 4-(NN- Dimethylamino)-pyridin (DMAP), l,5-Diazabicyclo[4.3.0]non-5-en (DBN), 1,8- Diazabicyclo[5.4.0]undec-7-en (DBU) oder l,4-Diazabicyclo[2.2.2]octan (DABCO®). Bevorzugt wird Natrium- oder Kalium-tert.-butylat verwendet.
Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von 0°C bis +120°C, bevorzugt bei +20°C bis +80°C, gegebenenfalls in einer Mikrowelle. Die Umsetzung kann bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar).
Inerte Lösungsmittel für den Ringschluss zum Imidazo[l,2-a]pyrazin-Grundgerüst (VII) + (VIII) — > (II) bzw. (VIII) + (IX)— > (X) sind die üblichen organischen Lösungsmittel. Hierzu gehören bevorzugt Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol, n-Pentanol oder tert.-Butanol, oder Ether wie Diethylether, Tetrahydrofuran, 2-Methyltetrahydrofuran, Dioxan oder Glykoldimethylether, oder andere Lösungsmittel wie Aceton, Dichlormethan, 1 ,2-Dichlorethan, Acetonitril, Dimethylformamid oder Dimethylsulfoxid. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt wird Ethanol verwendet.
Der Ringschluss erfolgt im Allgemeinen in einem Temperaturbereich von +50°C bis +150°C, bevorzugt bei +50°C bis +100°C, gegebenenfalls in einer Mikrowelle. Der Ringschluss (VII) + (VIII) -> (II) bzw. (VIII) + (IX) -> (X) erfolgt optional in Gegenwart wasserziehender Reaktionszusätze, beispielsweise in Gegenwart von Molekularsieb (3Ä oder 4Ä Porengröße) oder mittels Wasserabscheider. Die Umsetzung (VII) + (VIII)— > (II) bzw. (VIII) + (IX) — > (X) erfolgt unter Verwendung eines Überschusses des Reagenzes der Formel (VIII), beispielsweise mit 1 bis 20 Äquivalenten des Reagenzes (VIII), gegebenenfalls unter Zusatz von Basen (wie z.B. Natriumhydrogencarbonat) wobei die Zugabe dieses Reagenzes einmalig oder in mehreren Portionen erfolgen kann.
Weitere erfindungsgemäße Verbindungen können gegebenenfalls auch hergestellt werden durch Umwandlungen von funktionellen Gruppen einzelner Substituenten, insbesondere den unter R3 aufgeführten, ausgehend von den nach obigen Verfahren erhaltenen Verbindungen der Formel (I).
^
- 69 -
Diese Umwandlungen werden nach üblichen, dem Fachmann bekannten Methoden durchgeführt und umfassen beispielsweise Reaktionen wie nukleophile und elektrophile Substitutionen, Oxidationen, Reduktionen, Hydrierungen, Übergangsmetall-katalysierte Kupplungsreaktionen, Eliminierungen, Alkylierung, Aminierung, Veresterung, Esterspaltung, Veretherung, Etherspaltung, Bildung von Carbonamiden, sowie Einführung und Entfernung temporärer Schutzgruppen.
Die erfindungsgemäßen Verbindungen besitzen wertvolle pharmakologische Eigenschaften und können zur Vorbeugung und Behandlung von Erkrankungen bei Menschen und Tieren verwendet werden. Die erfindungsgemäßen Verbindungen eröffnen eine weitere Behandlungsalternative und stellen somit eine Bereicherung der Pharmazie dar.
Die erfindungsgemäßen Verbindungen bewirken eine Gefäßrelaxation und eine Hemmung der Thrombozytenaggregation und führen zu einer Blutdrucksenkung sowie zu einer Steigerung des koronaren Blutflusses. Diese Wirkungen sind über eine direkte Stimulation der löslichen Guanylat- cyclase und einen intrazellulären cGMP-Anstieg vermittelt. Außerdem verstärken die erfindungs- gemäßen Verbindungen die Wirkung von Substanzen, die den cGMP-Spiegel steigern, wie beispielsweise EDRF (endothelium-derived relaxing factor), NO-Donatoren, Protoporphyrin IX, Arachidonsäure oder Phenylhydrazin-Derivate.
Die erfindungsgemäßen Verbindungen eignen sich zur Behandlung und/oder Prophylaxe von kardiovaskulären, pulmonalen, thromboembolischen und fibrotischen Erkrankungen. Die erfindungsgemäßen Verbindungen können daher in Arzneimitteln zur Behandlung und/oder Prophylaxe von kardiovaskulären Erkrankungen wie beispielsweise Bluthochdruck (Hypertonie), resistente Hypertonie, akute und chronische Herzinsuffizienz, koronare Herzerkrankung, stabile und instabile Angina pectoris, periphere und kardiale Gefäßerkrankungen, Arrhythmien, Rhythmusstörungen der Vorhöfe und der Kammern sowie Überleitungsstörungen wie beispielsweise atrio-ventrikuläre Blockaden Grad I-III (AB-Block I-III), supraventrikuläre Tachyarrhythmie, Vorhofflimmern, Vorhoffflattern, Kammerflimmern, Kammerflattern, ventrikuläre Tachyarrhytmie, Torsade de pointes-Tachykardie, Extrasystolen des Vorhoffs und des Ventrikels, AV-junktionale Extrasystolen, Sick-Sinus Syndrom, Synkopen, AV-Knoten- Reentrytachykardie, Wolff-Parkinson-White-Syndrom, von akutem Koronarsyndrom (ACS), autoimmune Herzerkrankungen (Perikarditis, Endokarditis, Valvo litis, Aortitis, Kardiomyopathien), Schock wie kardiogenem Schock, septischem Schock und anaphylaktischem Schock, Aneurysmen, Boxerkardiomyopathie (premature ventricular contraction (PVC)), zur Behandlung und/oder Prophylaxe von thromboembolischen Erkrankungen und Ischämien wie myokardiale Ischämie, Myokardinfarkt, Hirnschlag, Herzhypertrophie, transistorischen und ischämischen Attacken, Präeklampsie, entzündliche kardiovaskuläre Erkrankungen, Spasmen der Koronararterien
„
- 70 - und peripherer Arterien, Ödembildung wie beispielsweise pulmonales Ödem, Hirnödem, renales Ödem oder Herzinsuffizienz-bedingtes Ödem, peripheren Durchblutungsstörungen, Reperfusionsschäden, arterielle und venöse Thrombosen, Mikroalbuminurie, Herzmuskelschwäche, endotheliale Dysfunktion, zur Verhinderung von Restenosen wie nach Thrombolysetherapien, per- cutan-transluminalen Angioplastien (PTA), transluminalen Koronarangioplastien (PTCA), Herztransplantationen und Bypass-Operationen, sowie mikro- und makrovaskuläre Schädigungen (Vasculitis), erhöhte Spiegel von Fibrinogen und von LDL geringer Dichte sowie erhöhte Konzentrationen von Plasminogenaktivator-Inhibitor 1 (PAI-1), sowie zur Behandlung und/oder Prophylaxe von erektiler Dysfunktion und weiblicher sexueller Dysfunktion eingesetzt werden. Im Sinne der vorliegenden Erfindung umfasst der Begriff Herzinsuffizienz sowohl akute als auch chronische Erscheinungsformen der Herzinsuffizienz, wie auch spezifischere oder verwandte Krankheitsformen wie akut dekompensierte Herzinsuffizienz, Rechtsherzinsuffizienz, Linksherzinsuffizienz, Globalinsuffizienz, ischämische Kardiomyopathie, dilatative Kardiomyopathie, hypertrophe Kardiomyopathie, idiopathische Kardiomyopathie, angeborene Herzfehler, Herz- Insuffizienz bei Herzklappenfehlern, Mitralklappenstenose, Mitralklappeninsuffizienz, Aortenklappenstenose, Aortenklappeninsuffizienz, Trikuspidalstenose, Trikuspidalinsuffizienz, Pulmonal- klappenstenose, Pulmonalklappeninsuffizienz, kombinierte Herzklappenfehler, Herzmuskelentzündung (Myokarditis), chronische Myokarditis, akute Myokarditis, virale Myokarditis, diabetische Herzinsuffizienz, alkoholtoxische Kardiomyopathie, kardiale Speichererkrankungen, diastolische Herzinsuffizienz sowie systolische Herzinsuffizienz und akute Phasen der Verschlechterung einer bestehenden chronischen Herzinsuffizienz (worsening heart failure).
Darüber hinaus können die erfindungsgemäßen Verbindungen auch zur Behandlung und/oder Prophylaxe von Arteriosklerose, Lipidstoffwechselstörungen, Hypolipoproteinämien, Dyslipi- dämien, Hypertriglyceridämien, Hyperlipidämien, Hypercholesterolämien, Abetelipoproteinämie, Sitosterolämie, Xanthomatose, Tangier Krankheit, Fettsucht (Adipositas), Fettleibigkeit (Obesitas) und von kombinierten Hyperlipidämien sowie des Metabolischen Syndroms eingesetzt werden.
Außerdem können die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von primärem und sekundärem Raynaud-Phänomen, von Mikrozirkulationsstörungen, Claudicatio, peripheren und autonomen Neuropathien, diabetischen Mikroangiopathien, diabetischer Retinopathie, diabetischen Geschwüren an den Extremitäten, Gangren, CREST-Syndrom, Erythematose, Onychomykose, rheumatischen Erkrankungen sowie zur Förderung der Wundheilung verwendet werden.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen zur Behandlung urologischer Erkrankungen wie beispielsweise benignes Prostata- Syndrom (BPS), benigne Prostata-Hyperplasie (BPH), benigne Prostata Vergrösserung (BPE), Blasenentleerungsstörung (BOO), untere
- 71 -
Harnwegssyndrome (LUTS, einschließlich Feiines Urologisches Syndrom (FUS)), Erkrankungen des Urogenital- Systems einschliesslich neurogene überaktive Blase (OAB) und (IC), Inkontinenz (UI) wie beispielsweise Misch-, Drang-, Stress-, oder Überlauf-Inkontinenz (MUI, UUI, SUI, OUI), Beckenschmerzen, benigne und maligne Erkrankungen der Organe des männlichen und weiblichen Urogenital-Systems.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Nierenerkrankungen, insbesondere von aktuer und chronischer Niereninsuffizienz, sowie von akutem und chronischem Nierenversagen. Im Sinne der vorliegenden Erfindung umfasst der Begriff Niereninsuffizienz sowohl akute als auch chronische Erscheinungsformen der Nieren- Insuffizienz, wie auch zugrundeliegende oder verwandte Nierenerkrankungen wie renale Hypoper- fusion, intradialytische Hypotonie, obstruktive Uropathie, Glomerulopathien, Glomerulonephritis, akute Glomerulonephritis, Glomerulosklerose, tubulointerstitielle Erkrankungen, nephropathische Erkrankungen wie primäre und angeborene Nierenerkrankung, Nierenentzündung, immunologische Nierenerkrankungen wie Nierentransplantatabstoßung, Immunkomplex-induzierte Nierener- krankungen, durch toxische Substanzen induzierte Nephropathie, Kontrastmittel-induzierte Nephropathie, diabetische und nicht-diabetische Nephropathie, Pyelonephritis, Nierenzysten, Nephrosklerose, hypertensive Nephrosklerose und nephrotisches Syndrom, welche diagnostisch beispielsweise durch abnorm verminderte Kreatinin- und/oder Wasser- Ausscheidung, abnorm erhöhte Blutkonzentrationen von Harnstoff, Stickstoff, Kalium und/oder Kreatinin, veränderte Aktivität von Nierenenzymen wie z.B. Glutamylsynthetase, veränderte Urinosmolarität oder Urinmenge, erhöhte Mikroalbuminurie, Makroalbuminurie, Läsionen an Glomerula und Arteriolen, tubuläre Dilatation, Hyperphosphatämie und/oder die Notwendigkeit zur Dialyse charakterisiert werden können. Die vorliegende Erfindung umfasst auch die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Folgeerscheinungen einer Niereninsuffizienz, wie beispielsweise Lungenödem, Herzinsuffizienz, Urämie, Anämie, Elektrolytstörungen (z.B. Hyperkalämie, Hyponaträmie) und Störungen im Knochen- und Kohlenhydrat-Metabolismus.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen auch zur Behandlung und/oder Prophylaxe von asthmatischen Erkrankungen, pulmonaler arterieller Hypertonie (PAH) und anderen Formen der pulmonalen Hypertonie (PH), umfassend mit Linksherzerkrankung, HIV, Sichelzellanämie, Thromboembolien (CTEPH), Sarkoidose, COPD oder Lungenfibrose assoziierte pulmonale Hypertonie, der chronisch-obstruktive Lungenerkrankung (COPD), des akuten Atemwegs Syndrom (ARDS), der akuten Lungenschädigung (ALI), der alpha- 1 -Antitrypsin- Defizienz (AATD), der Lungenfibrose, des Lungenemphysem (z.B. durch Zigarettenrauch induziertes Lungenemphysem) und der zystischen Fibrose (CF).
„
- 72 -
Die in der vorliegenden Erfindung beschriebenen Verbindungen stellen auch Wirkstoffe zur Bekämpfung von Krankheiten im Zentralnervensystem dar, die durch Störungen des NO/cGMP- Systems gekennzeichnet sind. Insbesondere sind sie geeignet zur Verbesserung der Wahrnehmung, Konzentrationsleistung, Lernleistung oder Gedächtnisleistung nach kognitiven Störungen, wie sie insbesondere bei Situationen/Krankheiten/Syndromen auftreten wie "Mild cognitive impairment", altersassoziierten Lern- und Gedächtnisstörungen, altersassoziierten Gedächtnisverlusten, vaskulärer Demenz, Schädel-Hirn-Trauma, Schlaganfall, Demenz, die nach Schlaganfällen auftritt ("post stroke dementia"), post-traumatischem Schädel-Hirn-Trauma, allgemeinen Konzentrationsstörungen, Konzentrationsstörungen bei Kindern mit Lern- und Gedächtnisproblemen, Alzhei- mer'scher Krankheit, Demenz mit Lewy-Körperchen, Demenz mit Degeneration der Frontallappen einschliesslich des Pick's-Syndroms, Parkinson'scher Krankheit, progressiver nuclear palsy, Demenz mit corticobasaler Degeneration, Amyolateralsklerose (ALS), Huntington'scher Krankheit, Demyelinisation, Multipler Sklerose, Thalamischer Degeneration, Creutzfeld- Jacob-Demenz, HIV- Demenz, Schizophrenie mit Demenz oder Korsakoff-Psychose. Sie eignen sich auch zur Behandlung und/oder Prophylaxe von Erkrankungen des Zentralnervensystems wie Angst-, Spannungs- und Depressionszuständen, zentral-nervös bedingten Sexualdysfunktionen und Schlafstörungen sowie zur Regulierung krankhafter Störungen der Nahrungs-, Genuss- und Suchtmittelaufnahme.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen auch zur Regulation der cerebralen Durchblutung und stellen wirkungsvolle Mittel zur Bekämpfung von Migräne dar. Auch eignen sie sich zur Prophylaxe und Bekämpfung der Folgen cerebraler Infarktgeschehen (Apoplexia cerebri) wie Schlaganfall, cerebraler Ischämien und des Schädel-Hirn-Traumas. Ebenso können die erfindungsgemäßen Verbindungen zur Bekämpfung von Schmerzzuständen und Tinnitus eingesetzt werden. Zudem besitzen die erfindungsgemäßen Verbindungen antiinflammatorische Wirkung und können daher als entzündungshemmende Mittel zur Behandlung und/oder Prophylaxe von Sepsis (SIRS), multiplem Organversagen (MODS, MOF), entzündlichen Erkrankungen der Niere, chronischen Darmentzündungen (IBD, Crohn's Disease, UC), Pankreatitis, Peritonitis, rheumatoiden Erkrankungen, entzündlichen Hauterkrankungen sowie entzündlichen Augenerkrankungen eingesetzt werden.
Desweiteren können die erfindungsgemäßen Verbindungen ebenfalls zur Behandlung und/ oder Prophylaxe von Autoimmunerkrankungen eingesetzt werden.
Weiterhin sind die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe fibrotischer Erkrankungen der inneren Organe, wie beispielsweise der Lunge, des Herzens, der Niere, des Knochenmarks und insbesondere der Leber, sowie dermatologischer Fibrosen und
„
- 73 - fibrotischer Erkrankungen des Auges, geeignet. Im Sinne der vorliegenden Erfindungen umfasst der Begriff fibrotischer Erkrankungen insbesondere die folgenden Begriffe Leberfibrose, Leberzirrhose, Lungenfibrose, Endomyocardfibrose, Nephropathie, Glomerulonephritis, interstitielle Nierenfibrose, fibrotische Schäden in Folge von Diabetes, Knochenmarksfibrose und ähnliche fibrotische Erkrankungen, Sklerodermie, Morphaea, Keloide, hypertrophe Narbenbildung (auch nach chirurgischen Eingriffen), Naevi, diabetische Retinopathie, proliferative Vitroretinopathie und Erkrankungen des Bindegewebes (z.B. Sarkoidose).
Weiterhin eignen sich die erfindungsgemäßen Verbindungen zur Bekämpfung postoperativer Narbenbildung, z.B. in Folge von Glaukom-Operationen. Die erfindungsgemäßen Verbindungen können ebenfalls kosmetisch bei alternder und verhornender Haut eingesetzt werden.
Außerdem sind die erfindungsgemäßen Verbindungen zur Behandlung und/ oder Prophylaxe von Hepatitis, Neoplasma, Osteoporose, Glaukom und Gastroparese geeignet.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Ver- bindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, Niereninsuffizienz, thrombo- embolischen Erkrankungen, fibrotischen Erkrankungen und Arteriosklerose.
Weiterer Gegenstand der vorliegenden Erfindung sind die erfindungsgemäßen Verbindungen zur Verwendung in einem Verfahren zur Behandlung und/ oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, Niereninsuffizienz, thromboembolischen Erkrankungen, fibrotischen Erkrankungen und Arteriosklerose.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Ver- bindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen,
Λ
- 74 -
Niereninsuffizienz, thromboembolischen Erkrankungen, fibrotischen Erkrankungen und Arteriosklerose.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwen- dung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, Niereninsuffizienz, thromboembolischen Erkrankungen, fibrotischen Erkrankungen und Arteriosklerose, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prophylaxe der zuvor genannten Er- krankungen. Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
• organische Nitrate und NO-Donatoren, wie beispielsweise Natriumnitroprussid, Nitroglycerin, Isosorbidmononitrat, Isosorbiddinitrat, Molsidomin oder SIN-1, sowie inhalatives NO;
• Verbindungen, die den Abbau von cyclischem Guanosinmonophosphat (cGMP) inhibieren, wie beispielsweise Inhibitoren der Phosphodiesterasen (PDE) 1, 2 und/oder 5, insbesondere PDE 5- Inhibitoren wie Sildenafil, Vardenafil und Tadalafil;
• antithrombotisch wirkende Mittel, beispielhaft und vorzugsweise aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien oder der profibrinolytischen Substanzen;
• den Blutdruck senkende Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, Endothelin-Antago- nisten, Renin-Inhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker, Mineralocorti- coid-Rezeptor- Antagonisten sowie der Diuretika; und/oder
• den Fettstoffwechsel verändernde Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie beispielhaft und vorzugsweise HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, CETP- Inhibitoren, MTP-Inhibitoren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta-Agonisten,
Cholesterin-Absorptionshemmer, Lipase-Inhibitoren, polymeren Gallensäureadsorber, Gallensäure-Reabsorptionshemmer und Lipoprotein(a)-Antagonisten.
Unter antithrombotisch wirkenden Mittel werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien oder der profibrinolytischen Substanzen verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombin- Inhibitor, wie beispielhaft und vorzugsweise Ximela- gatran, Dabigatran, Melagatran, Bivalirudin oder Clexane, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem GPIIb/IIIa- Antagonisten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Riva- roxaban (BAY 59-7939), DU- 176b, Apixaban, Otamixaban, Fidexaban, Razaxaban, Fondaparinux, Idraparinux, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten, Renin-Inhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker, Mineralocorticoid-Rezep- tor- Antagonisten sowie der Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Calcium- Antagonisten, wie beispielhaft und vorzugsweise Nife- dipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
„
- 76 -
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem alpha- 1 -Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem beta-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Meti- pranolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol, Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucin- dolol, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin AII-Antagonisten, wie beispielhaft und vorzugsweise Losartan, Candesartan, Valsartan, Telmisartan oder Embursatan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Lisinopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Endothelin- Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Renin-Inhibitor, wie beispielhaft und vorzugsweise Aliskiren, SPP-600 oder SPP-800, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Mineralocorticoid-Rezeptor- Antagonisten, wie beispielhaft und vorzugsweise Spironolacton oder Eplerenon, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Schleifendiuretikum, wie beispielsweise Furosemid, Torasemid, Bumetanid und Piretanid, mit kaliumsparenden Diuretika wie beispielsweise Amilorid und Triamteren, mit Aldosteronantagonisten, wie beispielsweise Spironolacton, Kaliumcanrenoat und Eplerenon sowie Thiaziddiuretika, wie beispielsweise Hydrochlorothiazid, Chlorthalidon, Xipamid, und Indapamid, verabreicht.
Unter den Fettstoffwechsel verändernden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der CETP-Inhibitoren, Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, MTP-Inhibi- toren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta-Agonisten, Cholesterin-Absorptions- hemmer, polymeren Gallensäureadsorber, Gallensäure-Reabsorptionshemmer, Lipase-Inhibitoren sowie der Lipoprotein(a)-Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP-Inhibitor, wie beispielhaft und vorzugsweise Dalcetrapib, BAY 60-5521, Anacetrapib oder CETP -Vaccine (CETi-1), verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidrezeptor-Agonisten, wie beispielhaft und vorzugsweise D-Thyroxin, 3,5,3'-Triiodothyronin (T3), CGS 23425 oder Axitirome (CGS 26214), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem HMG-CoA-Reduktase-Inhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK-475, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS-201038, R-103757 oder JTT-130, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PP AR- gamma- Agonisten, wie beispielhaft und vorzugsweise Pioglitazone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem PPAR-delta-Agonisten, wie beispielhaft und vorzugsweise GW 501516 oder BAY 68-5042, verabreicht.
„
- 78 -
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterin- Absorptionshemmer, wie beispielhaft und vorzugsweise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimid, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugsweise ASBT (= IBAT)-Inhibitoren wie z.B. AZD-7806, S-8921, AK-105, BARI-1741, SC-435 oder SC-635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Lipoprotein(a)- Antagonisten, wie beispielhaft und vorzugsweise Gemcabene calcium (CI-1027) oder Nicotinsäure, verabreicht.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otisch oder als Implantat bzw. Stent. Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende
„
- 79 -
Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulver- Inhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augen- präparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents. Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige Poly- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien. Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.001 bis 2 mg/kg, vorzugsweise etwa 0.001 bis 1 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden
muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt. Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
_
A. Beispiele
Abkürzungen und Akronyme: abs. absolutiert (= getrocknet) aq. wässrige Lösung br. Verbreitertes Signal (NMR Kupplungsmuster)
CAS-Nr. Chemical Abstracts Service Nummer δ Verschiebung im NMR Spektrum (Angabe in ppm) d Dublett (NMR-Kupplungsmuster)
DC Dünnschichtchromatographie
DCI direkte chemische Ionisation (bei MS)
DMAP 4-NN-Dimethylaminopyridin
DMF Dimethylformamid
DMSO Dimethylsulfoxid d. TL der Theorie (bei Ausbeute) eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl h Stunde(n)
HATU N-[(Dimethylamino)(3H-[l,2,3]triazolo[4,5-b]-pyridin-3- yloxy)methylen]-N-methylmethanaminiumhexafluorophosphat
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
HRMS hochaufgelöste Massenspektrometrie konz. konzentriert
LC-MS Flüssigchromatographie-gekoppelte Massenspektrometrie
LiHMDS Lithiumhexamethyldisilazid
m Multiple« (NMR Kupplungsmuster)
Me Methyl
min Minute(n)
MS Massenspektrometrie
NMR Kernresonanzspektrometrie
Ph Phenyl q Quartett (NMR Kupplungsmuster)
quint. Quintett (NMR Kupplungsmuster)
RF Retentionsfaktor (bei Dünnschichtchromatographie)
RT Raumtemp er atur
Rt Retentionszeit (bei HPLC)
s Singulett (NMR Kupplungsmuster)
t Triplett (NMR Kupplungsmuster)
THF Tetrahydrofuran
TB TU (Benzotriazol- 1 -yloxy)bisdimethylaminomethyliumfluorborat
UV Ultraviolett-Spektrometrie
v/v Volumen zu Volumen- Verhältnis (einer Lösung)
LC-MS- und HPLC-Methoden:
Methode 1 (LC-MS):
Instrument: Micromass Quattro Premier mit Waters UPLC Acquity; Säule: Thermo Hypersil GOLD 1.9 μ 50 x 1 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 90% A -> 0.1 min 90% A -> 1.5 min 10% A -> 2.2 min 10% A Ofen: 50°C; Fluss: 0.33 ml/min; UV-Detektion: 210 nm
Methode 2 (LC-MS):
Instrument: Waters ACQUITY SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1.8 μ 50 x 1 mm; Eluent A: 1 1 Wasser + 0.25 ml 99%>ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%ige Ameisensäure; Gradient: 0.0 min 90% A -> 1.2 min 5% A -> 2.0 min 5% A Ofen: 50°C; Fluss: 0.40 ml/min; UV-Detektion: 210 - 400 nm.
Methode 3 (LC-MS):
Instrument: Micromass Quattro Premier mit Waters UPLC Acquity; Säule: Thermo Hypersil GOLD 1.9 μ 50 x 1 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%>ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 97% A -> 0.5 min 97% A -> 3.2 min 5% A -> 4.0 min 5% A Ofen: 50°C; Fluss: 0.3 ml/min; UV-Detektion: 210 nm.
Methode 4 (präparative HPLC):
Säule: Chromatorex C18 10 μ 250 x 20mm Gradient: A= Wasser + 0.5% Ameisensäure, B= Acetonitril, 0 min = 5% B, 3 min = 5% B vorspülen ohne Substanz, dann Injektion, 5 min = 5% B, 25 mi = 30% B, 38 mi = 30% B, 38.1 min = 95% B, 43 min = 95% B, 43.01 min= 5% B, 48.0 min= 5% B Flussrate 20 ml min. Wellenlänge 210 nm.
Methode 5 (präparative HPLC):
Säule: Chromatorex C18 10 μ 250x20 mm Gradient: A= Wasser + 0,5% Ameisensäure, B= ACetonitril, 0 min = 5% B, 3 min = 5% B vorspülen ohne Substanz, dann Injektion, 5 min = 5% B, 25 min = 50% B, 38 min = 50% B, 38.1 min = 95% B, 43 min= 95% B, 43.01 min= 5% B, 48.0 min= 5% B F hissrate 20 ml min. Wellenlänge 210 nm.
Methode 6 (präparative HPLC):
Säule: X Bridge Prep. C18 5μ 50 x 19 mm Gradient: A = Wasser + 0.5% Ammoniumhydroxid, B= Acetonitril, 0 min = 5% B, 3 min = 5% B vorspülen ohne Substanz, dann Injektion, 5 min = 5% B,
„ Λ
- 84 -
25 min = 50% B, 38 min = 50% B, 38.1 min = 95% B, 43 min= 95% B, 43.01min= 5% B, 48.0 min- 5% B Flussrate 15 ml/min, Wellenlänge 210 um.
Methode 7 (MS):
Instrument: Thermo Fisher-Scientific DSQ; chemische Ionisierung; Reaktantgas Ammoniak; Quellentemperatur: 200°C; Ionisierungsenergie 70eV.
Methode 8 (LC-MS):
Instrument: Waters ACQUITY SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1.8 μ 30 x 2 mm; Eluent A: 1 1 Wasser + 0.25 ml 99%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%ige Ameisensäure; Gradient: 0.0 min 90% A -> 1.2 min 5% A -> 2.0 min 5% A Ofen: 50°C; Fluss: 0.60 ml/min; UV-Detektion: 208 - 400 nm.
Methode 9 (präparative HPLC):
Instrument MS: Waters; Instrument HPLC: Waters; Säule Waters X-Bridge C18, 18 mm x 50 mm, 5 μιη, Eluent A: Wasser + 0.05% Triethylamin, Eluent B: Acetonitril (ULC) + 0.05% Triethylamin, mit Gradient; Fluss: 40 ml/min; UV-Detektion: DAD; 210 - 400 nm. bzw. :
Instrument MS: Waters; Instrument HPLC: Waters; Säule Phenomenex Luna 5μ C18 100A, AXIA Tech. 50 x 21.2 mm, Eluent A: Wasser + 0.05%> Ameisensäure, Eluent B: Acetonitril (ULC) + 0.05%) Ameisensäure, mit Gradient; Fluss: 40 ml/min; UV-Detektion: DAD; 210 - 400 nm. bzw. : Instrument MS: Waters, Instrument HPLC: Waters (Säule Waters X-Bridge C18, 19 mm x 50 mm, 5 μιη, Eluent A: Wasser + 0.05%> Ammoniak, Eluent B: Acetonitril (ULC) mit Gradient; Fluss: 40 ml/min; UV-Detektion: DAD; 210 - 400 nm).
Methode 10 (LC-MS):
Instrument MS: Waters SQD; Instrument HPLC: Waters UPLC; Säule: Zorbax SB-Aq (Agilent), 50 mm x 2.1 mm, 1.8 μιη; Eluent A: Wasser + 0.025%) Ameisensäure, Eluent B: Acetonitril (ULC) + 0.025% Ameisensäure; Gradient: 0.0 min 98% A - 0.9 min 25% A - 1.0 min 5% A - 1.4 min 5% A - 1.41 min 98% A - 1.5 min 98% A; Ofen: 40°C; Fluss: 0.600 ml/min; UV-Detektion: DAD; 210 nm.
Methode 11 (MS):
Instrument: Waters ZQ 2000; Elektrospray-lonisierung; Eluent A: 1 1 Wasser + 0.25 ml 99%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%ige Ameisensäure; 25% A, 75% B; Fluss: 0.25 ml/min.
Methode 12 (GC-MS): Instrument: Thermo Scientific DSQII, Thermo Scientific Trace GC Ultra; Säule: Restek RTX- 35MS, 15 m x 200 μιη x 0.33 μιη; konstanter Fluss mit Helium: 1.20 ml/min; Ofen: 60°C; Inlet: 220°C; Gradient: 60°C, 30°C/min -> 300°C (3.33 min halten).
Methode 13 (LC-MS):
Gerätetyp MS: Waters Synapt G2S; Gerätetyp UPLC: Waters Acquity I-CLASS; Säule: Waters, HSST3, 2.1 x 50 mm, C18 1.8 μηι; Eluent A: 1 1 Wasser + 0.01% Ameisensäure; Eluent B: 1 1 Acetonitril + 0.01% Ameisensäure; Gradient: 0.0 min 10% B -> 0.3 min 10% B -> 1.7 min 95% B -> 2.5 min 95% B; Ofen: 50°C; Fluss: 1.20 ml/min; UV-Detektion: 210 nm
Methode 14 (LC-MS):
Instrument: Waters ACQUITY SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1.8 μ 50 x 1 mm; Eluent A: 1 1 Wasser + 0.25 ml 99%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%ige Ameisensäure; Gradient: 0.0 min 95% A— > 6.0 min 5% A— > 7.5 min 5% A Ofen:
50°C; Fluss: 0.35 ml/min; UV-Detektion: 210 - 400 nm.
Methode 15 (LC-MS):
Instrument MS: Waters (Micromass) QM; Instrument HPLC: Agilent 1100 Serie; Säule : Agient ZORBAX Extend-C18 3.0x50mm 3.5-Micron; Eluent A: 1 1 Wasser + 0.01 mol Ammoniumcarbonat, Eluent B: 1 1 Acetonitril; Gradient: 0.0 min 98% A— > 0.2min 98% A— > 3.0 min 5% A^ 4.5 min 5% A ; Ofen: 40°C; Fluss: 1.75 ml/min; UV-Detektion: 210 nm.
Methode 16 (LC-MS):
Gerätetyp MS: Waters (Micromass) Quattro Micro; Gerätetyp HPLC: Agilent 1100 Serie; Säule : Thermo Hypersil GOLD 3 μ 20 x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%ige Ameisensäure; Gradient: 0.0 min 100%) A -> 3.0 min 10% A -> 4.0 min 10% A; Ofen: 50°C; Fluss: 2 ml/min; UV-Detektion: 210 nm.
Bei Aufreinigungen von erfindungsgemäßen Verbindungen per präparativer HPLC nach den oben beschriebenen Methoden, in denen die Elutionsmittel Zusatzstoffe wie beispielsweise Trifluoressigsäure, Ameisensäure oder Ammoniak enthalten, können die erfindungsgemäßen Verbindungen in Salz-Form, beispielsweise als Trifluoracetat, Formiat oder Ammonium-Salz anfallen, sofern die erfindungsgemäßen Verbindungen eine ausreichend basische bzw. saure Funktionalität enthalten. Ein solches Salz kann durch verschiedene dem Fachmann bekannte Methoden in die entsprechende freie Base bzw. Säure überführt werden.
Salze können unter- oder überstöchiometrisch vorliegen, insbesondere bei Vorliegen eines Amins oder einer Carbonsäure. Zusätzlich können bei den vorliegenden Imidazopyrazine unter sauren Bedingungen stets Salze, auch unterstöchiometrisch, vorliegen, ohne dass diese im 'H-NMR erkenntlich sind und ohne besondere Angabe und Kennzeichnung dieser in den jeweiligen IUPAC- Namen und Strukturformeln.
Die in den folgenden Paragraphen angegebenen Multiplizitäten von Protonensignalen in 'H-NMR- Spektren geben die jeweils beobachtete Signalform wieder und berücksichtigen keine Signalphänomene höherer Ordnung.
Ausgangsverbindungen und Intermediate: Beispiel 1A
3 - [(2,6-Difluorbenzyl)oxy] -5-methylpyrazin-2-
Zu einer Lösung von 2.71 g (2,6-Difluorphenyl)methanol [CAS-Nr.: 19064-18-7] (18.8 mmol, 1.3 eq.) in 120 ml 1 ,2-Dimethoxyethan wurden 4.86 g Kalium-tert-butylat (43.3 mmol, 3.0 eq.) gegeben und das Gemisch wurde 60 min bei RT gerührt. Anschließend wurden 2.60 g 2-Amino-3- chlor-5-methylpyrazin Hydrochlorid [CAS-Nr.: 89182-14-9] (14.4 mmol, 1.0 eq.) zugegeben und die Mischung wurde über Nacht bei 80°C gerührt. Nach Abkühlen auf Raumtem eratur wurde gesättigte wässrige Natriumhydrogencarbonatlösung zugegeben und die wässrige Phase dreimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter wässriger Natriumchloridlösung gewaschen, mit Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Biotage Isolera (340 g Kieselgelkartusche, Cyclohexan /Essigsäureethylester Gradient, 10% -> 72% Essigsäureethylester) gereingt. Es wurden 1.77 g der Titelverbindung (39% d. TL, Reinheit 85%) erhalten.
LC-MS (Methode 2): Rt = 0.94 min MS (ESpos): m/z = 252 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 2.20 (s, 3H), 5.35 (s, 2H), 5.88 (s, 2H), 7.09 - 7.23 (m, 2H), 7.37 (s, 1H), 7.46 - 7.57 (m, 1H). Beispiel 2A
Ethyl-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carboxylat
Eine Lösung von 1.77 g 3-[(2,6-Difluorbenzyl)oxy]-5-methylpyrazin-2-amin (7.05 mmol, 1.0 eq.) aus Beispiel 1A in 50 ml Ethanol wurde mit Molekularsieb 4A und 11.1 g Ethyl-2- chloroacetoacetat [CAS-Nr.: 609-15-4] (70.5 mmol, 10 eq.) versetzt und über Nacht auf Rückfluss erhitzt. Anschließend wurden 11.1 g Ethyl-2-chloroacetoacetat (70.5 mmol, 10.0 eq.) zugegeben und es wurde über Nacht auf Rückfluss erhitzt. Dann wurde abfiltriert, das Filtrat eingeengt, der erhaltene Rückstand mit Diethylether ausgerührt, abfiltriert und das Filtrat eingeengt. Der Rückstand wurde zweimal mittels Biotage Isolera (120 g Kieselgelkartusche, Cyclohexan/Essigsäureethylester Gradient) gereinigt. Es wurden 0.81 g der Titelverbindung (16% d. TL, Reinheit 52%) isoliert.
LC-MS (Methode 2): Rt = 1.28 min
MS (ESpos): m/z = 362 (M+H)+
Beispiel 3A
8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure
Eine Lösung von 800 mg Ethyl-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3- carboxylat (Reinheit 52%, 1.15 mmol, 1.0 eq.) aus Beispiel 2A in 10 ml Dioxan wurde mit 5.8 ml 1 N wässriger Natronlauge (5.8 mmol, 5 eq.) versetzt und 2 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt, der Rückstand in Wasser aufgenommen und der unlösliche Feststoff wurde abfiltriert. Das Filtrat wurde mit 1 N wässriger Salzsäure angesäuert, der gebildete Feststoff wurde abfiltriert und getrocknet. Es wurden 354 mg der Titelverbindung (83% d. Th., Reinheit 90%>) isoliert.
LC-MS (Methode 2): Rt = 0.99 min
MS (ESpos): m/z = 334 (M+H)+ ' H-NM R (400 M Hz, DMSO-de): δ [ppm] = 2.41 (s, 3H), 2.54 (s, 3H verborgen unter Lösemittelsigna! ). 5.55 (s, 2H), 7.12 - 7.28 (m, 2H), 7.49 - 7.64 (m, 1H), 8.64 (s, 1H), 13.20 - 13.66 (br s, 1H).
Beispiel 4A rac-B enzyl- (2-cyanbutan-2 -yl)carbamat
5.00 g (50.94 mmol) 2-Amino-2-methylbutanonitril [Synthese beschrieben in: Lonza AG, US 5698704 (1997); Deng, S. L. et al. Synthesis 2001 , 2445; Hjorringgaard, C. U. et al. J. Org. Chem. 2009, 74, 1329; Ogrel, A. et al. Eur. J. Org. Chem. 2000, 857] wurden in 50 ml THF und 6.5 ml Wasser vorgelegt, mit 21.83 g (157.92 mmol) Kaliumcarbonat versetzt und bei 0°C langsam mit 7.9 ml (56.04 mmol) Benzylchlorocarbonat (Chlorameisensäurebenzylester) versetzt. Nach Zugabe von 8 ml THF und 3 ml Wasser wurde das Reaktionsgemisch langsam auf RT kommend über Nacht gerührt. Dann wurde mit Wasser versetzt und dreimal mit Essigsäureethylester extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet und eingeengt. Der Rückstand wurde in Diethylether gelöst und mit Petrolether ausgefällt. Das Produkt wurde abfiltriert, der Feststoff mit etwas Petrolether gewaschen und im Hochvakuum getrocknet. Es wurden 11.35 g der Zielverbindung (93%> d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.97 min
MS (ESpos): m/z = 233 (M+H)
'H-NMR (400 MHz, DMSO-de): δ = 0.95 (t, 3H), 1.51 (s, 3H), 1.75 - 1.95 (m, 2H), 5.07 (s, 2H), 7.30 - 7.43 (m, 4H), 7.88 - 8.03 (m, 1H).
Beispiel 5A eni-Benzyl-(2-cyanbutan-2-yl)carbamat (Enantiomer A)
8 g raoBenzyl-(2-cyanbutan-2-yl)carbamat aus Beispiel 4A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralcel OJ-H, 5 μηι, 250 x 20 mm, Eluent: 50% Iso-Hexan, 50% Isopropanol, Fluß: 20 ml/min; 40°C, Detektion: 220 nm].
Enantiomer A: Ausbeute: 3.23 g (>99% ee) Rt = 6.69 min [Daicel Chiralcel OJ-H, 5 μηι, 250 x 4.6 mm; Eluent: 50% Iso-Hexan, 50% Isopropanol; Fluss 1.0 ml/min; 30°C; Detektion: 220 nm].
Beispiel 6A e«i-Benzyl-(2-cyanbutan-2-yl)carbamat (Enantiomer B)
8 g raoBenzyl-(2-cyanbutan-2-yl)carbamat aus Beispiel 4A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralcel OJ-H, 5 μηι, 250 x 20 mm, Eluent: 50% Iso-Hexan, 50% Isopropanol, Fluß: 20 ml/min; 40°C, Detektion: 220 nm].
Enantiomer B: Ausbeute: 3.18 g (>99%> ee)
Rt = 8.29 min [Daicel Chiralcel OJ-H, 5 μηι, 250 x 4.6 mm; Eluent: 50% Iso-Hexan, 50% Isopropanol; Fluss 1.0 ml/min; 30°C; Detektion: 220 nm].
Beispiel 7A eni-Benzyl-(l-amino-2-methylbutan-2-yl)carbamat (Enantiomer A)
4.00 g (17.22 mmol) eni-Benzyl-(2-cyanbutan-2-yl)carbamat aus Beispiel 5A wurden in 50 ml einer 7 N Lösung von Ammoniak in Methanol gelöst, mit 5.33 g Raney-Nickel versetzt und 24 h bei ca. 25 bar bei RT hydriert. Es wurde über Celite abfiltriert, mit Methanol gewaschen und eingeengt. Das Rohprodukt wurde mittels Kieselgelchromatographie gereinigt (Laufmittel: Dichlormethan/2N Ammoniak in Methanol = 10/0.5). Es wurden 2.20 g der Zielverbindung (54% d. Th.) erhalten. LC-MS (Methode 2): Rt = 0.56 min
MS (ESpos): m/z = 237 (M+H)+
Beispiel 8A eni-Benzyl-(l-amino-2-methylbutan-2-yl)carbamat (Enantiomer B)
4.00 g (17.22 mmol) e«i-Benzyl-(2-cyanbutan-2-yl)carbamat aus Beispiel 6A wurden in 50 ml 7 N ammoniakalische Methanol-Lösung gelöst, mit 5.33 g Raney-Nickel versetzt und 24 h bei ca. 25 bar bei RT hydriert. Das Reaktionsgemisch wurde über Celtite abfiltriert, gut mit Methanol gespült und eingeengt. Das Rohprodukt wurde mittels Kieselgelchromatographie gereinigt (Laufmittel: Dichlormethan/2N Ammoniak in Methanol = 10/0.5). Es wurden 3.56 g der Zielverbindung (87% d. Th.) erhalten.
LC-MS (Methode 3): Rt = 1.40 min MS (ESpos): m/z = 237 (M+H)+
Beispiel 9A eni-Benzyl- { 1 -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- yl}carbonyl)amino]-2-methylbutan-2-yl}carbamat (Enantiomer A)
Eine Mischung von 100 mg 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3- carbonsäure (0.30 mmol, 1.0 eq.), 106 mg eni-Benzyl-(l -amino-2-methylbutan-2-yl)carbamat (Enantiomer A, Beispiel 7A) (0.450 mmol, 1.5 eq.) und 0.26 ml N,N-Diisopropylethylamin (1.50 mmol, 5.0 eq.) in 1.0 ml DMF wurde mit 148 mg HATU (0.39 mmol, 1.3 eq) versetzt und für 1 h bei RT gerührt. Dann wurden 70 ml Wasser zugegeben und das Rohprodukt abfiltriert. Anschließend wurde das Rohprodukt mittels Biotage Isolera (10 g Kieselgelkartusche, Cyclohexan/Essigsäureethylester Gradient) gereinigt, wobei 74 mg der Titelverbindung (41% d. Th., Reinheit 92%) isoliert wurden.
DC (Kieselgel, Cyclohexan/Essigsäureethylester 1 : 1): RF = 0.53 LC-MS (Methode 2): Rt = 1.31 min, MS (ESpos): m/z = 552 (M+H)+ Beispiel 10A rac-Methyl-N-[(benzyloxy)carbonyl]norleucinat
12 g (66.05 mmol) rac-Methylnorleucinathydrochlorid wurden in 974 ml Wasser/THF (8: 1) vorgelegt und mit 28.3 g (204.77 mmol) Kaliumcarbonat versetzt. Das Reaktionsgemisch wurde auf 0°C abgekühlt. 12.3 ml (72.66 mmol) Chlorameisensäurebenzylester wurden langsam zugetropft und die Reaktionsmischung über Nacht bei RT gerührt. Das Gemisch wurde mit 480 ml Wasser verdünnt und dreimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, abfiltriert und eingeengt. Der Rückstand wurde mittels Kieselgelchromatographie (Cyclohexan/Essigsäureethylester = 4:1) gereinigt. Man erhielt 18 g (97% d. Th.) der Zielverbindung. LC-MS (Methode 2): Rt = 1.10 min.
MS (ESIpos): m/z = 280 (M+H)+.
'H-NMR (400 MHz, DMSO-de): δ = 0.79 - 0.90 (m, 3H), 1.21 - 1.35 (m, 4H), 1.52 - 1.73 (m, 2H), 3.63 (s, 3H), 3.95 - 4.05 (m, 1H), 4.97 - 5.11 (m, 2H), 7.24 - 7.42 (m, 5H), 7.74 (d, 1H).
Beispiel IIA
-Benzyl-(2-hydroxy-2-methylheptan-3-yl)carbamat
16.9 g (60.39 mmol) rac-Methyl-N-[(benzyloxy)carbonyl]norleucinat aus Beispiel 10A wurden unter Argon in 584 ml THF vorgelegt. Das Reaktionsgemisch wurde auf 0°C abgekühlt, 70.5 ml (211.38 mmol) 3 M Methylmagnesiumbromid in Diethylether wurden zugetropft und es wurde 15
min bei 0°C nachgerührt. Dann ließ man langsam auf RT erwärmen und rührte über Nacht bei Raumtem eratur. Das Reaktionsgemisch wurde vorsichtig mit 1 N wässriger Salzsäure angesäuert, die Reaktionslösung mit Celite versetzt und der Feststoff abfiltriert. Es wurde gut mit THF gewaschen und das Filtrat eingeengt. Der Rückstand wurde zwischen Dichlormethan und Wasser verteilt, die organische Phase zweimal mit Wasser gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Kieselgel-Chromatographie (Cyclohexan/Essigsäureethylester = 9: 1 nach 7:3) gereinigt und die Produktfraktionen wurden eingeengt. Man erhielt 15.8 g (94% d. Th.) der Zielverbindung.
LC-MS (Methode 2): Rt = 0.98 min. MS (ESIpos): m/z = 280 (M+H)+.
'H-NMR (400 MHz, DMSO-de): δ = 0.80 - 0.89 (m, 3H), 0.98 (s, 3H), 1.05 (s, 3H), 1.09 - 1.37 (m, 5H), 1.58 - 1.74 (m, 1H), 3.25 - 3.32 (m, 1H), 4.24 (s, 1H), 4.99 - 5.08 (m, 2H), 6.85 (d, 1H), 7.26 - 7.40 (m, 5H).
Beispiel 12A eni-Benzyl-(2-hydroxy-2-methylheptan-3-yl)carbamat (Enantiomer A)
15.8 g der Verbindung aus Beispiel I IA wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AD-H, 5 μιη, 250 x 20 mm, Eluent: 80% isoHexan, 20% Ethanol, Fluß: 20 ml/min; 35°C, Detektion: 210 um].
Enantiomer A:
Ausbeute: 5.4 g (97% ee)
Rt = 5.93 min [Daicel Chiralpak AD-H, 5μηι, 250 x 4.6 mm; Eluent: 80% iso-Hexan, 20% Ethanol; Fluss 1.0 ml/min; 40°C; Detektion: 220 um].
Beispiel 13A eni-3-Amino-2-methylheptan-2-ol Hydrochlorid (Enantiomer A)
x HCl
H3C CH3
1 g (3.58 mmol) eni-Benzyl-(2-hydroxy-2-methylheptan-3-yl)carbamat (Enantiomer A) aus Beispiel 12A wurden unter Argon in Ethanol (25 ml) vorgelegt, mit 381 mg (0.36 mmol) 10%igem Palladium auf Aktivkohle und 10.9 ml (107.38 mmol) Cyclohexen versetzt und die Reaktionsmischung 3 h bei Rückfluss gerührt. Das Gemisch wurde über einen Millipore®-Filter filtriert und es wurde mit Ethanol nachgewaschen. Das Filtrat wurde mit 3.6 ml (7.16 mmol) 2 N wässriger Salzsäure in Diethylether versetzt, dann eingeengt und am Hochvakuum getrocknet. Man erhielt 801 mg (123% d. Th.) der Zielverbindung. Das Produkt wurde ohne weitere Reinigung in die nächste Reaktion eingesetzt. DCI-MS (Methode 7): m/z = 146 (M-HC1+H)+
'H-NMR (400 MHz, DMSO-de): δ = 0.88 (t, 3H), 1.07 (s, 3H), 1.18 (s, 3H), 1.21 - 1.58 (m, 6H), 2.73 -2.83 (m, 1H), 7.69 - 7.84 (m, 2H).
Beispiel 14A rac-Methyl-6,6,6-trifluornorleucinat Hydrochlorid
2.7 g (14.58 mmol) rac-6,6,6-Trifluornorleucin wurden in 27.6 ml gesättigter Salzsäure in Methanol vorgelegt und 4 h unter Rückfluss gerührt. Dann wurden nochmals 10 ml gesättigte Salzsäure in Methanol zu der Reaktionslösung gegeben und weitere 4 h bei Rückfluss gerührt. Die Reaktionslösung wurde eingeengt und der Rückstand am Hochvakuum getrocknet. Es wurden 3.8 g der Zielverbindung (99% d. Th.; Reinheit 90%>) erhalten.
DCI-MS (Methode 7): (ESpos): m/z = 200 (M-HC1+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.48-1.73 (m, 2H), 1.80-1.96 (m, 2H), 2.24-2.38 (m, 2H), 3.76 (s, 3H), 4.06-4.14 (m, 1H), 8.49-8.68 (br. s, 3H).
Beispiel 15A rac-Methyl-N-[(benzyloxy)carbonyl]-6,6,6-trifluornorleucinat
3.8 g (14.39 mmol, Reinheit 90%) rac-Methyl-6,6,6-trifluornorleucinat Hydrochlorid aus Beispiel 14A wurden in 212 ml Wasser/THF (8:1) vorgelegt und mit 6.2 g (44.64 mmol) Kaliumcarbonat versetzt. Das Reaktionsgemisch wurde auf 0°C abgekühlt und es wurden 2.7 ml (15.84 mmol) Chlorameisensäurebenzylester langsam zugetropft und dann über Nacht bei RT gerührt. Das Gemisch wurde mit 100 ml Wasser verdünnt und dreimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, abfiltriert und eingeengt. Der Rückstand wurde mittels Kieselgelchromatographie (Cyclohexan/ Essigsäureethylester 4: 1) gereinigt. Man erhielt 3.6 g (76% d. Th.) der Zielverbindung. LC-MS (Methode 3): Rt = 2.32 min.
MS (ESIpos): m/z = 334 (M+H)+.
'H-NMR (400 MHz, DMSO-de): δ = 1.47 - 1.59 (m, 2H), 1.61 - 1.72 (m, 1H), 1.73 - 1.85 (m, 1H), 2.14 - 2.34 (m, 2H), 3.64 (s, 3H), 4.04 - 4.12 (m, 1H), 5.04 (s, 2H), 7.25 - 7.40 (m, 5H), 7.81 (d, 1H). Beispiel 16A rac-Benzyl-(7,7,7-trifluor-2-hydroxy-2-methylheptan-3-yl)carbamat
3.2 g (9.70 mmol) rac-Methyl-N-[(benzyloxy)carbonyl]-6,6,6-trifluornorleucinat aus Beispiel 15A wurde unter Argon in 94 ml THF vorgelegt. Das Reaktionsgemisch wurde auf 0°C abgekühlt, 11.3 ml (33.96 mmol) 3M Methylmagnesiumbromid in Diethylether wurden zugetropft und 15 min bei 0°C nachgerührt. Man ließ langsam auf RT erwärmen und rührte über Nacht bei Raumtemperatur. Das Reaktionsgemisch wurde vorsichtig mit gesättigter, wässriger Ammoniumchlorid-Lösung und anschließend mit Celite versetzt. Der Feststoff wurde abfiltriert, gut mit THF gewaschen und das
Filtrat wurde eingeengt. Der wässrige Rückstand wurde zwischen Dichlormethan und Wasser verteilt. Die organische Phase wurde noch zweimal mit Wasser gewaschen, über Natriumsulfat getrocknet, filtriert und das Filtrat eingeengt. Der Rückstand wurde mittels Kieselgel- Chromatographie (Cyclohexan/Essigsäureethylester 7:3) gereinigt und die Produktfraktionen wurden eingeengt. Man erhielt 3 g (90% d. Th.) der Zielverbindung.
LC-MS (Methode 8): Rt = 1.02 min.
MS (ESIpos): m/z = 334 (M+H)+.
'H-NMR (400 MHz, DMSO-de): δ = 0.99 (s, 3H), 1.06 (s, 3H), 1.25 - 1.45 (m, 2H), 1.47 - 1.60 (m, 1H), 1.67 - 1.80 (m, 1H), 2.06 - 2.35 (m, 2H), 3.29 - 3.32 (m, 1H, teilweise verdeckt durch Wasser- Peak), 4.32 (s, 1H), 5.05 (q, 2H), 6.95 (d, 1H), 7.26 - 7.38 (m, 5H).
Beispiel 17A eni-Benzyl-(7,7,7-trifluor-2-hydroxy-2-methylheptan-3-yl)carbamat (Enantiomer A)
1.9 g der Verbindung aus Beispiel 16A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AY-H, 5 μηι, 250 x 20 mm, Eluent: 90%> isoHexan, 10% Ethanol, Fluß: 15 ml/min; 35°C, Detektion: 220 um].
Enantiomer A:
Ausbeute: 766 mg (99% ee)
Rt = 5.12 min [Daicel Chiralpak AY-H, 5μηι, 250 x 4.6 mm; Eluent: 90% iso-Hexan, 10% Ethanol; Fluss 1.0 ml/min; 30°C; Detektion: 220 um].
Beispiel 18A rac-3-Amino-7,7,7-trifluor-2-methylheptan-2-ol Hydrochlorid
xHCI
1 g (3.00 mmol) rac-Benzyl-(7,7,7-trifluor-2-hydroxy-2-methylheptan-3-yl)carbamat aus Beispiel 16A wurden unter Argon in Ethanol (21 ml) vorgelegt, mit 319 mg (0.30 mmol, 10%igem Palladium auf Aktivkohle und 9.1 ml (89.99 mmol) Cyclohexen versetzt und die Reaktionsmischung über Nacht bei Rückfluss gerührt. Das Gemisch wurde über einen Millipore®- Filter filtriert und es wurde mit Ethanol nachgewaschen. Das Filtrat wurde mit 3 ml (6.00 mmol) 2 N wässriger Salzsäure in Diethylether versetzt, eingeengt und am Hochvakuum getrocknet. Man erhielt 785 mg (111% d. Th.) der Zielverbindung. Das Produkt wurde ohne weitere Reinigung in die nächste Reaktion eingesetzt. MS (Methode 7) : m/z = 200 (M-HC1+H)+.
'H-NMR (400 MHz, DMSO-de): δ = 1.08 (s, 3H), 1.19 (s, 3H), 1.40 - 1.59 (m, 2H), 1.60 - 1.82 (m, 2H), 2.15 - 2.41 (m, 2H), 2.80 - 2.91 (m, 1H), 5.17 - 5.35 (br. s, 1H), 7.65 - 7.93 (br. s, 2H).
Beispiel 19A eni-3-Amino-7,7,7-trifluor-2-methylheptan-2-olhydrochlorid (Enantiomer A) xHCI
765 mg (2.29 mmol) eni-Benzyl-(7,7,7-trifluor-2-hydroxy-2-methylheptan-3-yl)carbamat (Enantiomer A) aus Beispiel 17A wurden unter Argon in Ethanol (16.1 ml) vorgelegt, mit 244 mg (0.23 mmol) 10%> igem Palladium auf Aktivkohle und 7.0 ml (68.85 mmol) Cyclohexen versetzt und die Reaktionsmischung 3 h bei Rückfluss gerührt. Das Gemisch wurde über einen Millipore®- Filter filtriert und mit Ethanol gewaschen. Das Filtrat wurde mit 2.3 ml (4.59 mmol) 2 N wässriger Salzsäure in Diethylether versetzt, eingeengt und am Hochvakuum getrocknet. Man erhielt 559 mg (99% d. Th.) der Zielverbindung.
DCI-MS (Methode 7): m/z = 200 (M-HC1+H)
^
'H-NMR (400 MHz, DMSO-de): δ = 1.08 (s, 3H), 1.19 (s, 3H), 1.40 - 1.59 (m, 2H), 1.60 - 1.69 (m, 1H), 1.70 - 1.82 (m, 1H), 2.15 - 2.27 (m, 1H), 2.28 - 2.42 (m, 1H), 2.80 - 2.91 (m, 1H), 5.17 - 5.35 (br. s, 1H), 7.73 - 7.97 (br. s, 2H).
Beispiel 20A 3 ,3 ,4,4,4-Pentafluorbutyltrifluormethansulfonat
Unter Argon wurden 198.49 g (703.51 mmol) Trifluormethansulfonsäureanhydrid vorgelegt. Der Reaktionskolben wurde in ein 70°C heißes Ölbad getaucht und auf 56°C Innentemperatur erhitzt. 88.2 ml (738.68 mmol) 3,3,4,4,4-Pentafluorobutanol wurden innerhalb von 35 min zum Reaktionsgemisch getropft und das Gemisch wurde zwei Stunden bei 70-73°C Badtemperatur und 69°C Innentemperatur gerührt. Das Reaktionsgemisch wurde am Rotationsverdampfer aufkonzentriert und der Rückstand in 1500 ml Dichlormethan aufgenommen. Es wurde einmal mit 300 ml kaltem Wasser, einmal mit 300 ml kalter, wässriger gesättigter Natriumhydrogencarbonat- Lösung und einmal mit 300 ml kaltem Wasser gewaschen. Die organische Phase wurde mit Magnesiumsulfat getrocknet, ab filtriert und eingeengt. Man erhielt 192.86 g (92.6% d. Th.) der Zielverbindung.
'H-NMR (400 MHz, DMSO-de): δ = 2.71 - 2.89 (m, 2H), 4.58 (t, 2H). Beispiel 21A rac-Methyl-5,5,6,6,6-pentafluornorleucinathydrochlorid (Racemat)
Unter Argon wurden 132 g (521.0 mmol) Methyl-N-(diphenylmethylen)glycinat [beschrieben in: WO2010/123792 A I , 2010; S 11-13] in 1000 ml THF (wasserfrei) vorgelegt und auf -40°C abgekühlt. 625.2 ml (625.20 mmol) Bis-(trimethylsilyl)-lithiumamid (1 M in THF) wurden innerhalb von 30 min zugetropft. Nach 10 min bei -40°C wurde innerhalb von 35 min die
Innentemperatur auf 0°C ansteigen gelassen. 192.86 g (651.25 mmol) 3,3,4,4,4- Pentafluorbutyltrifluormethansulfonat aus Beispiel 20A, gelöst in 400 ml THF wurden bei 0°C in die Reaktionslösung getropft. Nach 10 min wurde das Kältebad entfernt und das Gemisch wurde 3 Tage bei RT gerührt. Anschließend wurde die Reaktionsmischung auf 0°C abgekühlt und mit 410 ml (1.33 mol) 3 N wässriger Salzsäure tropfenweise versetzt. Das Kältebad wurde entfernt und die Reaktionslösung wurde zwei Stunden bei RT gerührt. Das Gemisch wurde anschließend eingeengt. Man erhielt 141.5 g der Zielverbindung als Rohgemisch (Reinheit unbekannt), welche ohne weitere Reinigung in die Folgestufe eingesetzt wurde.
Beispiel 22A rac-Methyl-N-[(benzyloxy)carbonyl]-5,5,6,6,6-pentafluornorleucinat (Racemat)
141.5 g (520.99 mmol) rac-Methyl-5,5,6,6,6-pentafluornorleucinathydrochlorid aus Beispiel 21 A wurden unter Argon in 850 ml THF und 850 ml Wasser aufgenommen und vorsichtig mit 223.2 g (1.62 mol) Kaliumcarbonat bei RT versetzt. Anschließend wurden 82 ml (573.09 mmol) Chlorameisensaeurebenzylester zugetropft und die Suspension über Nacht bei RT gerührt. Das Reaktionsgemisch wurde zweimal mit 500 ml Essigsäurethylester extrahiert, die organische Phase wurde mit Magnesiumsulfat getrocknet, abfiltriert und eingeengt. Der Rückstand wurde in 50 ml Dichlormethan verdünnt und mittels Kieselgel-Chromatographie (Laufmittel: Cyclohexan/Essigsäurethylester 9/1 nach 4/1) gereinigt. Die isolierten Produktfraktionen wurden nochmals mittels präparativer HPLC gereinigt [Säule: Daiso C18 Ι Ομιη Bio 300 x 100mm, neutral; Eluent: Acetonitril/Wasser-Gradient; Fluss: 250 ml/min; Temperatur: RT; Wellenlänge: 210 nm). Man erhielt 27.4 g (14% d. TL, Reinheit 97%) der Zielverbindung.
LC-MS (Methode 2): Rt = 1.09 min. MS (ESIpos): m/z = 370 (M+H)+.
'H-NMR (400 MHz, DMSO-de): δ = 1.78 - 1.91 (m, 1H), 1.93 - 2.05 (m, 1H), 2.10 - 2.30 (m, 1H), 2.30 - 2.46 (m, 1H), 3.66 (s, 3H), 4.18 - 4.26 (m, 1H), 5.05 (s, 2H), 7.27 - 7.40 (m, 5H), 7.89 (d, 1H).
Beispiel 23A rac-Benzyl-(6,6,7,7,7-pentafluor-2-hydroxy-2-methylheptan-3 -yl)carbamat (Racemat)
1.7 g (3.68 mmol, 80% rein) rac-Methyl-N-[(benzyloxy)carbonyl]-5,5,6,6,6-pentafluornorleucinat (Racemat) aus Beispiel 22A wurden unter Argon in THF vorgelegt und das Reaktionsgemisch wurde auf 0°C abgekühlt. 4.3 ml (12.89 mmol) 3M Methylmagnesiumbromid in Diethylether wurden zugetropft und es wurde 15 min bei 0°C nachgerührt. Dann ließ man das Gemisch langsam auf RT erwärmen und rührte über Nacht bei Raumtemperatur. Das Reaktionsgemisch wurde vorsichtig mit gesättigter wässriger Ammoniumchlorid-Lösung versetzt und dann die Reaktionslösung zur Hälfte eingeengt. Der Rückstand wurde zwischen Dichlormethan und Wasser verteilt, die organische Phase zweimal mit Wasser gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Kieselgel-Chromatographie (Cyclohexan/Essigsäurethylester 10:1 nach 7:3) gereinigt. Man erhielt 1.31 g (96% d. Th.) der Zielverbindung.
LC-MS (Methode 2): Rt = 1.03 min.
MS (ESIpos): m/z = 370 (M+H)+. 'H-NMR (400 MHz, DMSO-de): δ = 1.01 (s, 3H), 1.08 (s, 3H), 1.43 - 1.56 (m, 1H), 1.92 - 2.01 (m, 1H), 2.01 - 2.19 (m, 2H), 3.36 - 3.44 (m, 1H), 4.48 (s, 1H), 4.99 - 5.12 (m, 2H), 7.11 (d, 1H), 7.27 - 7.38 (m, 5H).
Beispiel 24A eni-Benzyl-(6,6,7,7,7-pentafluor-2-hydroxy-2-methylheptan-3 -yl)carbamat (Enantiomer A)
1.31 g von Beispiel 23A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AY-H, 5 μιη, 250 x 20 mm, Eluent: 90% iso-Hexan, 10%> Ethanol, Fluß: 15 ml/min; 35°C; Detektion: 220 nm]. Enantiomer A:
Ausbeute: 459 mg (99% ee)
Rt = 4.31 min [Daicel Chiralpak AY-H, 5μηι, 250 x 4.6 mm; Eluent: 90% iso-Hexan, 10% Ethanol; Fluss: 1.0 ml/min; 30°C; Detektion: 220 nm].
Beispiel 25A eni-3-Amino-6,6,7,7,7-pentafluor-2-methylheptan-2-olhydrochlorid (Enantiomer A)
455 mg (1.23 mmol) eni-Benzyl-(6,6,7,7,7-pentafluor-2-hydroxy-2-methylheptan-3-yl)carbamat (Enantiomer A) aus Beispiel 24A wurden in 8.6 ml Ethanol vorgelegt, mit 131 mg Palladium auf Kohle (10%oig) und 3.74 ml (36.96 mmol) Cyclohexen versetzt und 3 h unter Rückfluss gerührt. Das Reaktionsgemisch wurde über einen Millipore-Filter filtriert und mit Ethanol gewaschen. Das Filtrat wurde mit 1.23 ml Chlorwasserstoff (2 N in Diethylether) versetzt, eingeengt und im Hochvakuum getrocknet. Es wurden 335 mg (98% d. Th.) der Zielverbindung erhalten.
MS (Methode 11): m/z = 236 (M-HC1+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.11 (s, 3H), 1.22 (s, 3H), 1.58 - 1.72 (m, 1H), 1.80 - 1.92 (m, 1H), 2.27 - 2.46 ( m, 2H, teilweise verdeckt durch DMSO-Peak), 2.94 - 3.04 (m, 1H), 5.35 (s, 1H), 7.80 - 8.01 (m, 3H).
Beispiel 26A rac-2-Amino-3-fluor-2-methylpropanonitril
Die Titelverbindung ist literaturbekannt:
1) McConathy, J. et al., Journal of Medicinal Chemistry 2002, 45, 2240-2249.
2) Bergmann, E.D. et al., Journal of the Chemical Society 1963, 3462-3463.
Weitere Methode:
1.0 g (0.94 ml; 13.15 mmol) Fluoraceton wurden in 11 ml 2 N Ammoniak in Methanol vorgelegt. Bei RT wurden nacheinander 721 mg (14.72 mmol) Natriumcyanid und 788 mg (14.72 mmol) Ammoniumchlorid zugeben und das Gemisch wurde 2 Stunden bei Rückfluss gerührt. Die Reaktionslösung wurde abgekühlt, filtriert und mit Methylenchlorid gewaschen. Aus der Mutterlauge fiel ein Feststoff aus, der abfiltiert wurde. Aus der Mutterlauge wurden Methylenchlorid und Methanol bei Normaldruck abdestilliert. Es wurden 1.32 g der Zielverbindung (89% d. Th., Reinheit ca. 90%) erhalten. Das Produkt wurde ohne weitere Reinigung in die nächste Reaktion eingesetzt.
GC-MS (Methode 12): Rt = 1.64 min MS (EIpos): m/z = 87 (M-CH3)+
Beispiel 27A rac-Benzyl-(2-cyan- 1 -fluorpropan-2-yl)carbamat
1.34 g (11.83 mmol, ca. 90%ig) rac-2-Amino-3-fluor-2-methylpropanonitril aus Beispiel 26A wurden in 29 ml THF/Wasser (9/1) mit 5.07 g (36.67 mmol) Kaliumcarbonat versetzt. Bei 0°C wurden langsam 1.69 ml (11.83 mmol) Chlorameisensäurebenzylester zugetropft und die Reaktionsmischung wurde über Nacht bei RT gerührt. Das Lösungsmittel wurde abdekantiert und die wässrige Phase zweimal mit THF ausgeschüttelt und das THF anschließend abdekantiert. Die vereinigten organischen Phasen wurden mit Natriumsulfat getrocknet, abfiltriert und eingeengt. Der Rückstand wurde mittels Kieselgelchromatographie getrennt (Laufmittel: Cyclohexan/Essigsäureethylester-Gradient) und die Produktfraktionen wurden am Rotationsverdampfer eingedampft. Es wurden 1.89 g der Zielverbindung (66% d. Th.; Reinheit 97%) erhalten.
LC-MS (Methode 2): Rt = 0.89 min MS (ESpos): m/z = 237 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.58 (d, 3H), 4.47 - 4.78 (m, 2H), 5.10 (s, 2H), 7.30 - 7.43 (m, 5H), 8.34 (br. s, 1H).
Beispiel 28A eni-Benzyl-(2-cyan-l-fluorpropan-2-yl)carbamat (Enantiomer A)
3.0 g (12.69 mmol) rac-Benzyl-(2-cyan-l -fluorpropan-2-yl)carbamat aus Beispiel 27A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AY-H, 5 μηι, 250 x 20 mm, Eluent: 80% Iso-Hexan, 20% Isopropanol, Fluß: 15 ml/min; 40°C, Detektion: 220 um]. Enantiomer A: Ausbeute: 1.18 g (>99% ee)
Rt = 5.37 min [Daicel Chiralcel AY-H, 5 μηι, 250 x 4.6 mm; Eluent: 70% iso-Hexan, 30% 2- Propanol; Fluss 1.0 ml/min; 40°C; Detektion: 220 um].
Beispiel 29A eni-Benzyl-(2-cyan-l -fluorpropan-2-yl)carbamat (Enantiomer B)
3.0 g (12.69 mmol) rac-Benzyl-(2-cyan-l -fluorpropan-2-yl)carbamat aus Beispiel 27A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AY-H, 5 μηι, 250 x 20 mm, Eluent: 80% Iso-Hexan, 20% Isopropanol, Fluß: 15 ml/min; 40°C, Detektion: 220 um]. Enantiomer B: Ausbeute: 1.18 g (>99%> ee)
Rt = 6.25 min [Daicel Chiralcel AY-H, 5 μηι, 250 x 4.6 mm; Eluent: 70% iso-Hexan, 30% 2- Propanol; Fluss 1.0 ml/min; 40°C; Detektion: 220 um].
Beispiel 30A rac-Benzyl-(l -amino-3-fluor-2-methylpropan-2-yl)carbamat
Unter Argon wurden 1.2 g (5.08 mmol) rac-Benzyl-(2-cyan-l-fluorpropan-2-yl)carbamat aus Beispiel 27A in 14.9 ml 7 N Ammoniak in Methanol mit 1.55 g Raney-Nickel (wässrige Aufschlämmung) versetzt und 24 Stunden bei ca. 25 bar Wasserstoff-Druck und RT hydriert. Das Reaktionsgemisch wurde über Kieselgur filtriert, mit Methanol gewaschen und eingeengt. Es wurden 1.2 g der Zielverbindung (98% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.49 min
MS (ESpos): m/z = 241 (M+H)+
Beispiel 31 A eni-Benzyl-(l -amino-3-fluor-2-methylpropan-2-yl)carbamat (Enantiomer A)
Unter Argon wurden 1.2 g (5.08 mmol) eni-Benzyl-(2-cyan-l-fluorpropan-2-yl)carbamat (Enantiomer A) aus Beispiel 28A in 14.9 ml 7 N Ammoniak in Methanol mit 1.55 g Raney-Nickel (wässrige Aufschlämmung) versetzt und 24 Stunden bei ca. 25 bar Wasserstoff-Druck und RT hydriert. Das Reaktionsgemisch wurde über Kieselgur filtriert, mit Methanol gewaschen und eingeengt. Es wurden 700 mg der Zielverbindung (57% d. Th.; Reinheit ca. 85%) erhalten.
LC-MS (Methode 2): Rt = 0.52 min
MS (ESpos): m/z = 241 (M+H)
Beispiel 32A eni-Benzyl-(l -amino-3-fluor-2-methylpropan-2-yl)carbamat (Enantiomer B)
Unter Argon wurden 1.2 g (5.08 mmol) eni-Benzyl-(2-cyan-l-fluorpropan-2-yl)carbamat (Enantiomer B) aus Beispiel 29A in 14.9 ml 7 N Ammoniak in Methanol mit 1.55 g Raney-Nickel (wässrige Aufschlämmung) versetzt und 24 Stunden bei ca. 25 bar Wasserstoff-Druck und RT hydriert. Das Reaktionsgemisch wurde über Kieselgur filtriert, mit Methanol gewaschen und eingeengt. Es wurden 1.2 g der Zielverbindung (98% d. Th.; Reinheit ca. 85%) erhalten.
LC-MS (Methode 2): Rt = 0.50 min MS (ESpos): m/z = 241 (M+H)+
Beispiel 33A rac-2- Amino-5 , 5 , 5 -trifluor-2-methylpentanonitril
8.0 g (57.1 mmol) 5,5,5-Trifluorpentan-2-on [CAS Registry Nummer: 1341078-97-4; käuflich erhältlich oder das Methylketon kann nach literaturbekannten Methoden, welche dem Fachmann bekannt sind z. B. über a) zwei Stufen aus 4,4,4-Trifluorobutanal nach Y. Bai et al. Angewandte Chemie 2012, 51, 4112-4116; K. Hiroi et al. Synlett 2001, 263-265; K. Mikami et al. 1982 Chemistry Letters, 1349-1352; b) oder aus 4,4,4-Trifluorbutansäure nach A. A. Wube et al. Bioorganic and Medicinal Chemistry 201 1, 19, 567-579; G. M. Rubottom et al. Journal of Organic Chemistry 1983, 48, 1550-1552; T. Chen et al. Journal of Organic Chemistry 1996, 61, 4716-4719, hergestellt werden. Die Isolierung des Produktes kann durch Destillation oder Chromatographie erfolgen] wurden in 47.8 ml 2 N Ammoniak in Methanol vorgelegt und bei Raumtemperatur mit
3.69 g (75.4 mmol) Natriumcyanid sowie 4.03 g (75.4 mmol) Ammoniumchlorid versetzt und 4 Stunden unter Rückfluss gerührt. Das Reaktionsgemisch wurde abgekühlt, mit Diethylether versetzt und der enthaltene Feststoff wurde abfiltriert. Das Lösungsmittel aus dem Filtrat wurde unter Normaldruck abdestilliert. Es wurden 8.7 g der Titelverbindung (92 % d. Th.) als Rückstand erhalten, der ohne weitere Reinigung in die Folgestufe eingesetzt wurde.
GC-MS (Methode 12): Rt = 1.90 min
MS (ESpos): m/z = 151 (M-CH3)+
Beispiel 34A rac-Benzyl-(2-cyan-5,5,5-trifluorpentan-2-yl)carbamat
8.7 g (52.36 mmol) rflc-2-Amino-5,5,5-trifluor-2-methylpentanonitril aus Beispiel 33A wurden in 128 ml Tetrahydrofuran/Wasser = 9/1 vorgelegt und mit 22.43 g (162.3 mmol) Kaliumcarbonat versetzt. Bei 0°C wurden langsam 8.93 g (52.36 mmol) Chlorameisensäurebenzylester zugetropft. Dann ließ man unter Rühren langsam auf Raumtemperatur erwärmen und rührte über Nacht bei Raumtemperatur. Das überstehende Lösungsmittel wurde abdekantiert, der Rückstand zweimal mit je 100 ml Tetrahydrofuran verrührt wonach das überstehende Lösungsmittel jeweils abdekantiert wurde. Die vereinigten organischen Phasen wurde eingeengt und das Rohprodukt wurde mittels Kieselgelchromatographie gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester-Gradient 9/1 bis 4/1). Es wurden 11.14 g der Titelverbindung (68 % d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.01 min
MS (ESpos): m/z = 301 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.58 (s, 3H), 2.08 - 2.21 (m, 2H), 2.24 - 2.52 (m, 2H), 5.09 (s, 2H), 7.29 - 7.41 (m, 5H), 8.17 (br. s, 1H).
Beispiel 35A eni-Benzyl-(2-cyan-5,5,5-trifluorpentan-2-yl)carbamat (Enantiomer A)
11.14 g rac-Benzyl-(2-cyan-5,5,5-trifluorpentan-2-yl)carbamat aus Beispiel 34A wurden durch präparative Trennung an der chiralen Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AZ-H, 5 μηι, SFC, 250 x 50 mm, Eluent: 94% Kohlendioxid, 6% Methanol, Fluss: 200 ml/min, Temperatur: 38°C, Druck: 135 bar; Detektion: 210 um].
Enantiomer A: 4.12 g (ca. 79% ee)
Rt = 1.60 min [SFC, Daicel Chiralpak AZ-H, 250 x 4.6 mm, 5 μιη, Eluent: 90% Kohlendioxid, 10% Methanol, Fluss: 3 ml/min, Temperatur: 30°C, Detektion: 220 um].
LC-MS (Methode 2): Rt = 1.01 min
MS (ESpos): m/z = 301 (M+H)+
Beispiel 36A eni-Benzyl-(2-cyan-5,5,5-trifluorpentan-2-yl)carbamat (Enantiomer B)
11.14 g rflc-Benzyl-(2-cyan-5,5,5-trifluorpentan-2-yl)carbamat aus Beispiel 34A wurden durch präparative Trennung an der chiralen Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AZ-H, 5 μηι, SFC, 250 x 50 mm, Eluent: 94% Kohlendioxid, 6% Methanol, Fluss: 200 ml/min, Temperatur: 38°C, Druck: 135 bar; Detektion: 210 um]. Enantiomer B: 4.54 g (ca. 70% ee; Reinheit ca. 89%)
Rt = 1.91 min [SFC, Daicel Chiralpak AZ-H, 250 x 4.6 mm, 5 μιη, Eluent: 90% Kohlendioxid, 10% Methanol, Fluss: 3 ml/min, Temperatur: 30°C, Detektion: 220 um].
LC-MS (Methode 2): Rt = 1.01 min
MS (ESpos): m/z = 301 (M+H)+ Beispiel 37A eni-Benzyl-(l -amino-5,5,5-trifluor-2-methylpentan-2-yl)carbamat (Enantiomer A)
4.12 g (13.17 mmol) eni-Benzyl-(2-cyan-5,5,5-trifluorpentan-2-yl)carbamat (Enantiomer A) aus Beispiel 35A wurden in 39 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 4 g Raney-Nickel (50%>ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde über Nacht im Autoklaven bei 20-30 bar hydriert. Es wurden nochmals 1 g Raney-Nickel (50%>ige wässrige Aufschlämmung) hinzugegeben und das Reaktionsgemisch wurde 5 h im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Kieselgur abfiltriert, mit Methanol gespült und eingeengt. Es wurden 3.35 g (56% d. Th.; Reinheit ca. 67%) der Zielverbindung erhalten, welche ohne weitere Reinigung in die Folgestufe eingesetzt wurde.
LC-MS (Methode 8): Rt = 1.68 min MS (ESpos): m/z = 305 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.13 (s, 3H), 1.40 (br. s, 2H), 1.70 - 1.80 (m, 1H), 1.83 - 1.95 (m, 1H), 2.08 - 2.2 (m, 2H), 4.98 (s, 2H), 6.85 (br. s, 1H), 7.28 - 7.41 (m, 5H).
Beispiel 38A eni-Benzyl-(l -amino-5,5,5-trifluor-2-methylpentan-2-yl)carbamat (Enantiomer B)
4.54 g (13.45 mmol; Reinheit ca. 89%) eni-Benzyl-(2-cyan-5,5,5-trifluorpentan-2-yl)carbamat (Enantiomer B) aus Beispiel 36A wurden in 39 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 5 g Raney-Nickel (50%ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde 3 h im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Kieselgur ab filtriert, mit Methanol gespült und eingeengt. Es wurden 4.20 g (97% d. Th.; Reinheit ca. 95%) der Zielverbindung erhalten, welche ohne weitere Reinigung in die Folgestufe eingesetzt wurde.
LC-MS (Methode 15): Rt = 2.19 min
MS (ESpos): m/z = 305 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.13 (s, 3H), 1.40 (br. s, 2H), 1.69 - 1.80 (m, 1H), 1.83 - 1.96 (m, 1H), 2.07 - 2.22 (m, 2H), 4.98 (s, 2H), 6.85 (br. s, 1H), 7.27 - 7.40 (m, 5H). Beispiel 39A raoBenzyl-(2-cyanpentan-2-yl)carbamat
20 g (178.3 mmol) rac-2-Amino-2-methylpentanonitril (beschrieben in: Deng, S L. et al., Synthesis 2001, 2445-2449; Freifelder, M. et al., J. Am. Chem. Soc. 1960, 696-698) wurden in 2.63 1 THF/Wasser (8/1) vorgelegt und mit 76.4 g (552.7 mmol) Kaliumcarbonat versetzt. Dann wurden bei 0°C 27.6 ml (196.1 mmol) Chlorameisensäurebenzylester langsam zugetropft und über Nacht bei RT gerührt. Die Reaktionsmischung wurde eingeengt, der Rückstand mit Wasser versetzt und
zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Kieselgel- Chromatographie (Laufmittel: Cyclohexan/Essigsäureethylester = 4/1) gereinigt. Es wurden 43.84 g der Zielverbindung (76% d. Th., Reinheit 76%) erhalten. LC-MS (Methode 2): Rt = 1.02 min
MS (ESpos): m/z = 247 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 0.90 (t, 3H), 1.31 - 1.48 (m, 2H), 1.52 (s, 3H), 1.70 - 1.88 (m, 2H), 5.07 (s, 2H), 7.30 - 7.42 (m, 5H), 8.00 (br. s, 1H).
Beispiel 40A e«i-Benzyl-(2-cyanpentan-2-yl)carbamat (Enantiomer A)
43.8 g (135.3 mmol) rac-Benzyl-(2-cyanpentan-2-yl)carbamat aus Beispiel 39A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: SFC Chiralpak AZ-H, 5 μιη, 250 x 50 mm, Eluent: 85% C02, 15% Methanol, Fluss: 250 ml/min; Temperatur: 28°C, backpressure: 100 bar, Detektion: 220 um].
Enantiomer A: Ausbeute: 13.13 g (>99% ee)
Rt = 2.76 min [SFC Chiralpak AZ-H, 5 μιη, 250 x 4.6 mm; Eluent: 90% C02, 10% Methanol; Fluss: 3 ml/min; Detektion: 220 nm].
Beispiel 41A e«i-Benzyl-(2-cyanpentan-2-yl)carbamat (Enantiomer B)
43.8 g (135.3 mmol) rac-Benzyl-(2-cyanpentan-2-yl)carbamat aus Beispiel 39A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: SFC Chiralpak AZ-H, 5 μηι, 250 x 50 mm, Eluent: 85% C0
2, 15% Methanol, Fluss: 250 ml/min; Temperatur: 28°C, backpressure: 100 bar, Detektion: 220 um]. Enantiomer B: Ausbeute: 13.48 g (ca. 90.4% ee)
Rt = 3.93 min [SFC Chiralpak AZ-H, 5 μηι, 250 x 4.6 mm; Eluent: 90% C02, 10% Methanol; Fluss: 3 ml/min; Detektion: 220 um].
Beispiel 42A eni-Benzyl-(l -amino-2-methylpentan-2-yl)carbamat (Enantiomer A)
13.1 g (53.31 mmol) e«i-Benzyl-(2-cyanpentan-2-yl)carbamat (Enantiomer A) aus Beispiel wurden in 155 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 16.5 g Raney-Nickel (50%ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde über Nacht im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Celite abfiltriert, mit Methanol, Dichlormethan/2 N Ammoniak in Methanol (20/1) gespült und eingeengt. Der Rückstand wurde mittels Kieselgel-Chromatographie (Laufmittel: Dichlormethan/Methanol 40/lnach 20/1) gereinigt. Es wurden 9.85 g der Zielverbindung (63% d. Th., Reinheit 86%) erhalten.
LC-MS (Methode 2): Rt = 0.58 min
MS (ESpos): m/z = 251 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 0.83 (t, 3H), 1.1 1 (s, 3H), 1.15 - 1.24 (m, 2H), 1.37 (br. 2H), 1.42 - 1.51 (m, 1H), 1.53 - 1.63 (m, 1H), 2.46 (d, 1H), 2.66 (d, 1H), 4.97 (s, 2H), 6.69 (br. 1H), 7.26 - 7.40 (m, 5H).
Beispiel 43A eni-Benzyl-(l-amino-2-methylpentan-2-yl)carbamat (Enantiomer B)
13.5 g (54.73 mmol) eni-Benzyl-(2-cyanpentan-2-yl)carbamat (Enantiomer B) aus Beispiel 41A wurden in 159 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 16.95 g Raney- Nickel (50%ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde über Nacht im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Celite abfiltriert, mit Methanol, Dichlormethan/2 N Ammoniak in Methanol (10/1) gespült und eingeengt. Der Rückstand wurde mittels Kieselgel-Chromatographie (Laufmittel: Dichlormethan/Methanol 40/lnach 20/1) gereinigt. Es wurden 9.46 g der Zielverbindung (61% d. Th., Reinheit 88%) erhalten.
LC-MS (Methode 2): Rt = 0.58 min MS (ESpos): m/z = 251 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 0.83 (t, 3H), 1.11 (s, 3H), 1.15 - 1.24 (m, 2H), 1.37 (br. s, 2H), 1.42 - 1.51 (m, 1H), 1.53 - 1.63 (m, 1H), 2.46 (d, 1H), 2.66 (d, 1H), 4.97 (s, 2H), 6.69 (br. s., 1H), 7.26 - 7.40 (m, 5H).
Beispiel 44A rac-2-[(Diphenylmethylen)amino]-4-fluorbutanonitril
16.5 g (74.91 mmol) [(Diphenylmethylen)amino]acetonitril wurden in 495 ml abs. THF vorgelegt und bei -78°C unter Argon mit 35.96 ml (89.89 mmol) n-Butyllithium (2.5 N in Hexan) versetzt und 15 min bei -78°C gerührt. Anschließend wurde die Reaktionslösung auf 0°C erwärmt. Es wurden 13.03 g (74.91 mmol) l-Iodo-2-fluoroethan zu der Reaktionslösung getropft und 15 min bei 0°C nachgerührt. Die Reaktionslösung wurde bei 0°C mit Wasser gequenscht, mit Ethylacetat versetzt und mit gesättigter, wässriger Natriumchloridlösung gewaschen. Die wässrige Phase wurde zweimal mit Ethylacetat reextrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel- Chromatographie (Laufmittel: Dichlormethan/Cyclohexan = 1/1 bis 2/1) gereinigt. Es wurden 18.7 g der Zielverbindung (80% d. Th., Reinheit 85%) erhalten.
LC-MS (Methode 3): Rt = 2.42 min
MS (ESpos): m/z = 267 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 2.13 - 2.41 (m, 2H), 4.40 (t, 1H), 4.43 - 4.71 (m, 2H), 7.25 - 7.30 (m, 2H), 7.33 - 7.63 (m, 8H).
Beispiel 45A rac-2-[(Diphenylmethylen)amino]-4,4-difluorbutanonitril
18 g (81.72 mmol) [(Diphenylmethylen)amino]acetonitril wurden in 500 ml abs. THF vorgelegt und bei -78°C unter Argon mit 39.22 ml (98.06 mmol) n-Butyllithium (2.5 N in Hexan) versetzt
und 15 min bei -78°C nachgerührt. Anschließend wurde die Reaktionslösung auf 0°C erwärmt. Es wurden 17.25 g (89.89 mmol) 1 , 1 -Difluor-2-iodethan zu der Reaktionslösung getropft und 15 min bei 0°C nachgerührt. Die Reaktionslösung wurde bei 0°C mit Wasser gequenscht, mit Ethylacetat versetzt und dreimal mit halbgesättigter, wässriger Natriumchloridlösung gewaschen. Die vereinigten wässrigen Phasen wurden zweimal mit Ethylacetat reextrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel-Chromatographie (Laufmittel: Dichlormethan/Cyclohexan = 1/1) gereinigt. Es wurden 13.57 g der Zielverbindung (49% d. Th., Reinheit 84%) erhalten.
LC-MS (Methode 3): Rt = 2.48 min MS (ESpos): m/z = 285 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 2.53 - 2.61 (m, 2H; z.T. überlagert mit Lösungsmittelpeak), 4.50 (t, 1H), 6.08 - 6.41 (m, 1H), 7.23 - 7.33 (m, 2H), 7.38 - 7.47 (m, 2H), 7.49 - 7.67 (m, 6H).
Beispiel 46A rac-2- [(Diphenylmethylen)amino] -5 -fluorpentanonitril
18 g (81.72 mmol) [(Diphenylmethylen)amino]acetonitril wurden in 500 ml abs. THF vorgelegt und bei -78°C unter Argon mit 39.22 ml (98.06 mmol) n-Butyllithium (2.5 N in Hexan) versetzt und 15 min bei -78°C nachgerührt. Anschließend wurde die Reaktionslösung auf 0°C erwärmt, 16.9 g (89.89 mmol) l-Fluor-3-iodpropan zu der Reaktionslösung getropft und 15 min bei 0°C nachgerührt. Die Reaktionslösung wurde bei 0°C mit Wasser gequenscht, mit Ethylacetat versetzt und mit gesättigter, wässriger Natriumchloridlösung gewaschen. Die wässrige Phase wurde zweimal mit Ethylacetat reextrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel- Chromatographie (Laufmittel: Toluol 100%), Nachreinigung mit Dichlormethan/Cyclohexan = 1/1 bis 2/1) gereinigt. Es wurden in Summe 16.73 g der Zielverbindung (73% d. Th.) erhalten.
LC-MS (Methode 3): Rt
MS (ESpos): m/z = 281 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.66 - 1.85 (m, 2H), 1.87 - 2.00 (m, 2H), 4.26 - 4.41 (m, 2H), 4.43 - 4.55 (m, 1H), 7.20 - 7.33 (m, 2H), 7.38 - 7.48 (m, 2H), 7.48 - 7.63 (m, 6H). Beispiel 47A rac-2-[(Diphenylmethylen)amino]-4-fluor-2-methylbutanonitril
19.94 g (63.64 mmol, Reinheit 85%) rac-2-[(Diphenylmethylen)amino]-4-fluorbutanonitril aus Beispiel 44A wurden in 421 ml abs. THF vorgelegt und bei -78°C unter Argon mit 25.71 ml (64.28 mmol) n-Butyllithium (2.5 N in Hexan) versetzt und 10 min bei -78°C nachgerührt. Anschließend wurde die Reaktionslösung bei -78°C mit 36.1 g (254.57 mmol) Iodmethan versetzt. Das Reaktionsgemisch wurde langsam über 4.5 h auf 0°C gebracht. Nach vollständiger Abreaktion des Startmaterials wurde die Reaktionslösung bei 0°C mit Wasser gequenscht, mit Ethylacetat versetzt und zweimal mit gesättigter, wässriger Natriumchloridlösung gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel-Chromatographie (Laufmittel: Cyclohexan/Ethylacetat = 15/1) gereinigt. Es wurden 17.2 g der Zielverbindung (78% d. Th., Reinheit 81%) erhalten.
LC-MS (Methode 3): Rt = 2.46 min MS (ESpos): m/z = 281 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.65 - 1.67 (s, 3H), 2.30 - 2.47 (m, 2H), 4.55 - 4.84 (m, 2H), 7.27 - 7.32 (m, 2H), 7.37 - 7.42 (m, 2H), 7.43 - 7.52 (m, 6H).
Beispiel 48A rflc-2-[(Diphenylmethylen)amino]-4,4-difluor-2-methylbutanonitril
13.07 g (38.62 mmol) rac-2-[(Diphenylmethylen)amino]-4,4-difluorbutanonitril aus Beispiel 45A wurden in 255 ml abs. THF vorgelegt und bei -78°C unter Argon mit 15.6 ml (39.0 mmol) n- Butyllithium (2.5 N in Hexan) versetzt und 10 min bei -78°C nachgerührt. Anschließend wurde die Reaktionslösung bei -78°C mit 22.6 g (154.46 mmol) lodmethan versetzt. Das Reaktionsgemisch wurde langsam über 3.5 h auf 0°C gebracht. Nach vollständiger Abreaktion des Startmaterials wurde die Reaktionslösung bei 0°C mit Wasser gequenscht, mit Ethylacetat versetzt und zweimal mit gesättigter, wässriger Natriumchloridlösung gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel- Chromatographie (Laufmittel: Cyclohexan/Ethylacetat = 15/1) gereinigt. Es wurden 11.4 g der Zielverbindung (91% d. Th., Reinheit 92%) erhalten.
LC-MS (Methode 3): Rt = 2.52 min MS (ESpos) : m/z = 299 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.67 (s, 3H), 2.55 - 2.77 (m, 2H), 6.14 - 6.48 (m, 1H), 7.28 - 7.34 (m, 2H), 7.36 - 7.44 (m, 2H), 7.44 - 7.54 (m, 6H).
Beispiel 49A rac-2- [(Diphenylmethylen)amino] -5 -fluor-2-methylpentanonitril
16.73 g (59.68 mmol) rflc-2-[(Diphenylmethylen)amino]-5-fluorpentanonitril aus Beispiel 46A wurden in 394 ml abs. THF vorgelegt und bei -78°C unter Argon mit 24.11 ml (60.27 mmol) n- Butyllithium (2.5 N in Hexan) versetzt und 10 min bei -78°C nachgerührt. Anschließend wurde die Reaktionslösung bei -78°C mit 34.93 g (238.70 mmol) Iodmethan versetzt. Das Reaktionsgemisch wurde langsam über 4.5 h auf 0°C gebracht. Die Reaktionslösung wurde bei 0°C mit Wasser gequenscht, mit Ethylacetat versetzt und zweimal mit gesättigter, wässriger Natriumchloridlösung gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel-Chromatographie (Laufmittel: Cyclohexan/Ethylacetat = 15/1) gereinigt. Es wurden 18.94 g der Zielverbindung (95% d. Th., Reinheit 88%) erhalten.
LC-MS (Methode 3): Rt = 2.55 min
MS (ESpos): m/z = 295 (M+H)+
1H-NMR (400 MHz, DMSO-de): δ = 1.62 (s, 3H), 1.73 - 1.90 (m, 2H), 1.94 - 2.03 (m, 1H), 2.04 - 2.18 (m, 1H), 4.47 (t, 1H), 4.58 (t, 1H), 7.23 - 7.33 (m, 2H), 7.35 - 7.43 (m, 2H), 7.44 - 7.56 (m, 6H).
Beispiel 50A rac-2-Amino-4-fluor-2-methylbutanonitrilhydrochlorid
17.45 g (50.45 mmol; Reinheit 81%>) rac-2-[(Diphenylmethylen)amino]-4-fluor-2- methylbutanonitril aus Beispiel 47A wurden in 235.6 ml Tetrahydrofuran und 9.1 ml Wasser gelöst, mit 111 ml (55.46 mmol) Chlorwasserstoff-Lösung (0.5 N in Diethylether) versetzt und über Nacht bei Raumtemperatur gerührt. Die leicht trübe Reaktionslösung wurde mit 25.21 ml
_
- 120 -
(50.42 mmol) Chlorwasserstoff-Lösung (2 N in Diethylether) versetzt und einrotiert. Das isolierte Rohprodukt wurde direkt ohne weitere Reinigung weiter umgesetzt.
LC-MS (Methode 3): Rt = 0.22 min
MS (ESpos): m/z = 117 (M-HC1+H)+ Beispiel 51A rac-Benzyl-(2-cyan-4-fluorbutan-2-yl)carbamat
Das Rohprodukt rac-2-Amino-4-fluor-2-methylbutanonitrilhydrochlorid aus Beispiel 50A wurde in 165 ml Tetrahydrofuran/Wasser (1 : 1) vorgelegt, mit 28.57 g (206.71 mmol) Kaliumcarbonat und 9.46 g (55.46 mmol) Chlorameisensäurebenzylester versetzt. Das Reaktionsgemisch (Zwei-Phasen- Gemisch) wurde über Nacht bei Raumtemperatur gerührt. Es wurden erneut 1.72 g (10.1 mmol) Chlorameisensäurebenzylester zu der Reaktion gegeben und weitere 2 h bei Raumtemperatur gerührt. Anschließend wurde das Zweiphasensystem voneinander getrennt und die wässrige Phase zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter, wässriger Natriumchloridlösung gewaschen und anschließend über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel-Chromatographie gereinigt (Laufmittel: Cyclohexan/Ethylacetat-Gradient 20/1 bis 5/1). Es wurden 5.04 g der Zielverbindung (38 % d. Th. über zwei Stufen; Reinheit 96%) erhalten.
LC-MS (Methode 3): Rt = 1.95 min MS (ESpos): m/z = 251 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.59 (s, 3H), 2.20 - 2.43 (m, 2H), 4.55 (t, 1H), 4.67 (t, 1H), 5.08 (s, 2H), 7.28 - 7.45 (m, 5H), 8.12 (br. s, 1H).
Beispiel 52A rac-2-Amino-4,4-difluor-2-methylbutanonitrilhydrochlorid
10.84 g (33.43 mmol; Reinheit 92%) rac-2-[(Diphenylmethylen)amino]-4,4-difluor-2- methylbutanonitril aus Beispiel 48A wurden in 156 ml Tetrahydrofuran und 6 ml Wasser gelöst, mit 73.5 ml (36.77 mmol) Chlorwasserstoff-Lösung (0.5 N in Diethylether) versetzt und über Nacht bei Raumtemperatur gerührt. Die Reaktionslösung wurde mit 16.71 ml (33.43 mmol) Chlorwasserstoff-Lösung (2 N in Diethylether) versetzt und einrotiert. Das isolierte Rohprodukt wurde direkt ohne weitere Reinigung weiter umgesetzt.
LC-MS (Methode 3): Rt = 0.32 min
MS (ESpos): m/z = 135 (M-HC1+H)+ Beispiel 53A rac-Benzyl-(2-cyan-4,4-difluorbutan-2-yl)carbamat
Das Rohprodukt rac-2-Amino-4,4-difluor-2-methylbutanonitrilhydrochlorid aus Beispiel 52A wurde in 109 ml Tetrahydrofuran/Wasser (1 :1) vorgelegt, mit 18.94 g (137.06 mmol) Kaliumcarbonat und 6.27 g (36.77 mmol) Chlorameisensäurebenzylester versetzt. Das Reaktionsgemisch (Zwei-Phasen-Gemisch) wurde über Nacht bei Raumtemperatur gerührt. Es wurden erneut 1.14 g (6.69 mmol) Chlorameisensäurebenzylester zu der Reaktion gegeben und weitere 2 h bei Raumtemperatur gerührt. Anschließend wurde das Zweiphasensystem voneinander getrennt und die wässrige Phase zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter, wässriger Natriumchloridlösung gewaschen und anschließend über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel-Chromatographie gereinigt (Laufmittel: Cyclohexan/Ethylacetat-Gradient 20/1 bis 5/1). Es wurden 7.68 g der Zielverbindung (61 % d. Th. über zwei Stufen; Reinheit 71%) erhalten.
LC-MS (Methode 3): Rt = 2.04 min MS (ESpos): m/z = 269 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.65 (s, 3H), 2.51 - 2.65 (m, 2H), 5.10 (s, 2H), 6.08 6.41 (m, 1H), 7.27 - 7.44 (m, 5H), 8.24 (br. s, 1H). Beispiel 54A rac-2-Amino-5-fluor-2-methylpentanonitrilhydrochlorid
18.94 g (56.62 mmol; Reinheit 88%) rac-2-[(Diphenylmethylen)amino]-5-fluor-2- methylpentanonitril aus Beispiel 49A wurden in 264.6 ml Tetrahydrofuran und 10.2 ml Wasser gelöst, mit 124.6 ml (62.28 mmol) Chlorwasserstoff-Lösung (0.5 N in Diethylether) versetzt und über Nacht bei Raumtemperatur gerührt. Die Reaktionslösung wurde mit 28.3 ml (56.62 mmol) Chlorwasserstoff-Lösung (2 N in Diethylether) versetzt und einrotiert. Das isolierte Rohprodukt wurde direkt ohne weitere Reinigung weiter umgesetzt.
LC-MS (Methode 3): Rt = 0.25 min MS (ESpos): m/z = 131 (M-HC1+H)+
Beispiel 55A rac-Benzyl-(2-cyan-5-fluorpentan-2-yl)carbamat
Das Rohprodukt rflc-2-Amino-5-fluor-2-methylpentanonitrilhydrochlorid aus Beispiel 54A wurde in 185 ml Tetrahydrofuran/Wasser (1/1) vorgelegt, mit 32.09 g (232.18 mmol) Kaliumcarbonat und 10.63 g (62.29 mmol) Chlorameisensäurebenzylester versetzt. Das Reaktionsgemisch (Zwei- Phasen-Gemisch) wurde über Nacht bei Raumtemperatur gerührt. Es wurden erneut 1.93 g (1 1.33 mmol) Chlorameisensäurebenzylester zu der Reaktion gegeben und weitere 2 h bei Raumtemperatur gerührt. Anschließend wurde das Zweiphasensystem voneinander getrennt und die wässrige Phase zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter, wässriger Natriumchloridlösung gewaschen und anschließend über Natriumsulfat getrocknet, filtriert und einrotiert. Der Rückstand wurde mittels Kieselgel- Chromatographie gereinigt (Laufmittel: Cyclohexan/Ethylacetat-Gradient 20/1 bis 5/1). Es wurden 1 1.77 g der Zielverbindung (72 % d. Th. über zwei Stufen, Reinheit 92%) erhalten.
LC-MS (Methode 3): Rt = 2.03 min
MS (ESpos): m/z = 265 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.55 (s, 3H), 1.66 - 1.85 (m, 2H), 1.86 - 2.04 (m, 2H), 4.40 (t, 1H), 4.52 (t, 1H), 5.08 (s, 2H), 7.28 - 7.44 (m, 5H), 8.05 (br. s, 1H).
Beispiel 56A eni-Benzyl-(2-cyan-4,4-difluorbutan-2-yl)carbamat (Enantiomer A)
7.68 g (20.33 mmol, Reinheit 71%) rac-Benzyl-(2-cyan-4,4-difluorbutan-2-yl)carbamat aus Beispiel 53A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AY-H, 5 μηι, 250 x 20 mm, Eluent: 80% iso-Hexan, 20% Isopropanol, Fluss: 25 ml/min; Temperatur: 22°C, Detektion: 210 um].
Enantiomer A: Ausbeute: 2.64 g (>99% ee)
Rt = 6.67 min [Chiralpak AY-H, 5 μηι, 250 x 4.6 mm; Eluent: 80% iso-Hexan, 20% Isopropanol; Fluss: 3 ml/min; Detektion: 220 um].
Beispiel 57A eni-Benzyl-(2-cyan-4,4-difluorbutan-2-yl)carbamat (Enantiomer B)
7.68 g (20.33 mmol, Reinheit 71%) rac-Benzyl-(2-cyan-4,4-difluorbutan-2-yl)carbamat aus Beispiel 53A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: Daicel Chiralpak AY-H, 5 μηι, 250 x 20 mm, Eluent: 80% iso-Hexan, 20% Isopropanol, Fluss: 25 ml/min; Temperatur: 22°C, Detektion: 210 um].
Enantiomer B: Ausbeute: 2.76 g (93% ee)
Rt = 7.66 min [Chiralpak AY-H, 5 μηι, 250 x 4.6 mm; Eluent: 80% iso-Hexan, 20% Isopropanol; Fluss: 3 ml/min; Detektion: 220 um].
Beispiel 58A eni-Benzyl-(2-cyan-5-fluorpentan-2-yl)carbamat (Enantiomer A)
1 1.77 g (40.97 mmol, Reinheit 92%) rac-Benzyl-(2-cyan-5-fluorpentan-2-yl)carbamat aus Beispiel 55A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: SFC Daicel Chiralpak AZ-H, 5 μηι, 250 x 30 mm, Eluent: 90% C02, 10% Methanol, Fluss: 100 ml/min; Temperatur: 40°C, Detektion: 210 um].
Enantiomer A: Ausbeute: 5.7 g (>99% ee)
Rt = 1.76 min [SFC Chiralpak AZ-3, 3 μηι, 50 x 4.6 mm; Eluent: C02/Methanol-Gradient (5% bis 60% Methanol); Fluss: 3 ml/min; Detektion: 220 um].
Beispiel 59A eni-Benzyl-(2-cyan-5-fluorpentan-2-yl)carbamat (Enantiomer B)
1 1.77 g (40.97 mmol, Reinheit 92%) rac-Benzyl-(2-cyan-5-fluorpentan-2-yl)carbamat aus Beispiel 55A wurden durch präparative Trennung an chiraler Phase in die Enantiomere getrennt [Säule: SFC Daicel Chiralpak AZ-H, 5 μηι, 250 x 30 mm, Eluent: 90% C02, 10% Methanol, Fluss: 100 ml/min; Temperatur: 40°C, Detektion: 210 um]. Enantiomer B: Ausbeute: 5.0 g (>99% ee)
Rt = 1.97 min [SFC Chiralpak AZ-3, 3 μηι, 50 x 4.6 mm; Eluent: C02/Methanol-Gradient (5% bis 60%) Methanol); Fluss: 3 ml/min; Detektion: 220 um].
Beispiel 60A eni-Benzyl-(l -amino-4,4-difluor-2-methylbutan-2-yl)carbamat (Enantiomer A)
2.3 g (8.57 mmol) eni-Benzyl-(2-cyan-4,4-difluorbutan-2-yl)carbamat (Enantiomer A) aus Beispiel 56A wurden in 75 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 2.66 g Raney-Nickel (50%>ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde für 1.5 h im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Celite abfiltriert, mit
Methanol und 2 N Ammoniak in Methanol gespült und eingeengt. Es wurden 2.23 g der Zielverbindung (94% d. TL, Reinheit 98%) erhalten.
LC-MS (Methode 3): Rt = 1.48 min
MS (ESpos): m/z = 273 (M+H)+ 'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.19 (s, 3H), 1.48 (br. s, 2H), 2.08 - 2.40 (m, 2H), 2.53 - 2.72 (m, 2H; z.T. überlagert mit Lösungsmittelpeak), 5.00 (s, 2H), 5.90 - 6.23 (m, 1H), 6.95 (br. s, 1H), 7.25 - 7.41 (m, 5H).
Beispiel 61A eni-Benzyl-(l -amino-4,4-difluor-2-methylbutan-2-yl)carbamat (Enantiomer B)
2.76 g (10.29 mmol) eni-Benzyl-(2-cyan-4,4-difluorbutan-2-yl)carbamat (Enantiomer B) aus Beispiel 57A wurden in 90 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 3.19 g Raney-Nickel (50%>ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde für 1.5 h im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Celite ab filtriert, mit Methanol und 2 N Ammoniak in Methanol gespült und eingeengt. Es wurden 2.64 g der Zielverbindung (88%> d. Th., Reinheit 93%>) erhalten.
LC-MS (Methode 3): Rt = 1.49 min
MS (ESpos): m/z = 273 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.19 (s, 3H), 1.48 (br. s, 2H), 2.08 - 2.40 (m, 2H), 2.53 - 2.73 (m, 2H; z.T. überlagert mit Lösungsmittelpeak), 5.00 (s, 2H), 5.90 - 6.24 (m, 1H), 6.95 (br. s, 1H), 7.25 - 7.41 (m, 5H).
Beispiel 62A eni-Benzyl-(l -amino-5-fluor-2-methylpentan-2-yl)carbamat (Enantiomer A)
5.7 g (21.57 mmol) eni-Benzyl-(2-cyan-5-fluorpentan-2-yl)carbamat (Enantiomer A) aus Beispiel 58A wurden in 125 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 6.68 g Raney-Nickel (50%ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde für 4.5 h im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Celite abfiltriert, mit Methanol und 2 N Ammoniak in Methanol gespült und eingeengt. Es wurden 5.22 g der Zielverbindung (77% d. Th., Reinheit 85%) erhalten.
LC-MS (Methode 3): Rt = 1.51 min
MS (ESpos): m/z = 269 (M+H)+ Beispiel 63A eni-Benzyl-(l -amino-5-fluor-2-methylpentan-2-yl)carbamat (Enantiomer B)
5.0 g (18.92 mmol) eni-Benzyl-(2-cyan-5-fluorpentan-2-yl)carbamat (Enantiomer B) aus Beispiel 59A wurden in 110 ml 7 N Ammoniak-Lösung in Methanol gelöst und unter Argon mit 5.86 g Raney-Nickel (50%>ige wässrige Aufschlämmung) versetzt. Das Reaktionsgemisch wurde für 4.5 h im Autoklaven bei 20-30 bar hydriert. Die Reaktionsmischung wurde über Celite abfiltriert, mit Methanol und 2 N Ammoniak in Methanol gespült und eingeengt. Es wurden 4.6 g der Zielverbindung (84% d. Th., Reinheit 93%) erhalten.
LC-MS (Methode 3): Rt = 1.47 min MS (ESpos): m/z = 269 (M+H)
Beispiel 64A rac-4-Fluor-2-methylbutan- 1 ,2-diamin Dihydrochlorid
1.00 g (4.00 mmol) rac-Benzyl-(2-cyan-4-fluorbutan-2-yl)carbamat aus Beispiel 51A wurden in 1 14 ml Ethanol Eisessig (1/1) gelöst und mit 0.85 g Palladium auf A-Kohle (10%ig) versetzt. Das Reaktionsgemisch wurde für 3 h im Autoklaven bei 30-50 bar hydnert. Die Reaktionsmischung wurde einen Faltenfilter abfiltriert, mit Ethanol gespült und anschließend nochmal über einen Millipore-Filter filtriert. Das Filtrat wurde mit 10 ml Chlorwasserstoff-Lösung (2 N in Diethylether) versetzt und anschließend eingedampft. Es wurden 1 .04 g der Zielverbindung erhalten, welche ohne weitere Reinigung in die Folgestufe eingesetzt wurde.
LC-MS ( Methode 3): R, = 0.19 min MS (ESpos): m z = 121 ( -2HC1+H ) " Beispiel 65A eni-Benzyl- { 1 -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- yl}carbonyl)amino]-2-methylpentan-2-yl}carbamat Trifluoracetat (Enantiomer B)
50 mg (0.15 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 74 mg (0.20 mmol) HATU und 0.13 ml (0.75 mmol) N,N-
Diisopropylethylamin in 0.5 ml DMF vorgelegt und 20 min bei Raumtemperatur gerührt. Anschließend wurden 49 mg (0.17 mmol; Reinheit 88%) eni-Benzyl-(l-amino-2-methylpentan-2- yl)carbamat aus Beispiel 43A (Enantiomer B) zur Reaktionslösung gegeben und über Nacht bei RT gerührt. Dann wurde mit Acetonitril und Wasser verdünnt, mit TFA versetzt und mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1%) TFA). Die Produktfraktionen wurden vereinigt, eingeengt und lyophilisiert. Es wurden 66 mg der Zielverbindung (65% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.31 min
MS (ESpos): m/z = 566 (M-TFA+H)+ Beispiel 66A eni-Benzyl- { 1 -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- yl}carbonyl)amino]-3-fluor-2-methylpropan-2-yl}carbamat (Enantiomer B)
60 mg (0.18 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 75 mg (0.20 mmol) HATU und 0.094 ml (0.54 mmol) N,N- Diisopropylethylamin in 0.53 ml DMF vorgelegt, 10 min vorgerührt, dann mit 55 mg (0.19mmol, 85 %>ig) eni-Benzyl-(l-amino-3-fluor-2-methylpropan-2-yl)carbamat (Enantiomer B) aus Beispiel 32A versetzt und 2.5 h bei RT gerührt. Die Reaktionslösung wurde mit Acetonitril, Wasser und TFA versetzt und mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1%> TFA). Die Produktfraktionen wurden vereinigt und eingeengt. Anschließend wurde der Rückstand in Dichlormethan und wenig Methanol aufgenommen und zweimal mit gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan extrahiert. Die vereinigten organischen
Phasen wurden über Natriumsulfat getrocknet, filtriert, eingeengt und lyophilisiert. Es wurden 62 mg der Zielverbindung (62% d. Th.) erhalten.
LC-MS (Methode 13): Rt = 1.55 min
MS (ESpos): m/z = 556 (M+H)+ Beispiel 67A eni-Benzyl- { 1 -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- yl}carbonyl)amino]-5,5,5-trifluor-2-methylpentan-2-yl}carbamat (Enantiomer B)
60 mg (0.18 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 75 mg (0.20 mmol) HATU und 0.094 ml (0.54 mmol) N,N- Diisopropylethylamin in 0.53 ml DMF vorgelegt und 15 min gerührt. Anschließend wurden 69 mg (0.22 mmol; Reinheit ca. 95%) eni-Benzyl-(l-amino-5,5,5-trifluor-2-methylpentan-2-yl)carbamat (Enantiomer B) aus Beispiel 38A hinzugegeben und das Gemisch wurde 2.5 h bei RT gerührt. Die Reaktionslösung wurde mit Acetonitril, Wasser und TFA versetzt und mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1 % TFA). Die Produktfraktionen wurden vereinigt und eingeengt. Anschließend wurde der Rückstand in Dichlormethan und wenig Methanol aufgenommen und zweimal mit gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert, eingeengt und lyophilisiert. Es wurden 86 mg der Zielverbindung (77% d. Th.) erhalten.
LC-MS (Methode 2): Rt
MS (ESpos): m/z = 620 (M+H)+ Beispiel 68A eni-Benzyl- { 1 -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- yl}carbonyl)amino]-5-fluor-2-methylpentan-2-yl}carbamat Trifluoracetat (Enantiomer B)
80 mg (0.24 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 119 mg (0.31 mmol) HATU und 0.21 ml (1.20 mmol) N,N- Diisopropylethylamin in 0.80 ml DMF vorgelegt und 20 min gerührt. Anschließend wurden 91 mg (0.31 mmol; Reinheit 93%) eni-Benzyl-(l-amino-5-fluor-2-methylpentan-2-yl)carbamat (Enantiomer B) aus Beispiel 63A hinzugegeben und das Gemisch wurde 0.5 h bei RT gerührt. Die Reaktionslösung wurde mit Acetonitril, Wasser und TFA versetzt und mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1%> TFA). Es wurden 109 mg der Zielverbindung (56% d. Th.; Reinheit ca. 86%) erhalten. LC-MS (Methode 2): Rt = 1.28 min
MS (ESpos): m/z = 584 (M-TFA+H)+
Beispiel 69A eni-Benzyl- { 1 -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- yl}carbonyl)amino]-4,4-difluor-2-methylbutan-2-yl}carbamat (Enantiomer A)
80 mg (0.24 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 119 mg (0.31 mmol) HATU und 0.21 ml (1.20 mmol) N,N- Diisopropylethylamin in 0.80 ml DMF vorgelegt und 20 min gerührt. Anschließend wurden 85 mg (0.30 mmol; Reinheit 98%) eni-Benzyl-(l-amino-4,4-difluor-2-methylbutan-2-yl)carbamat (Enantiomer A) aus Beispiel 60A hinzugegeben und das Gemisch wurde 0.5 h bei RT gerührt. Die Reaktionslösung wurde mit Wasser versetzt und 30 min bei RT verrührt. Der erhaltene Feststoff wurde ab filtriert, gut mit Wasser gewaschen und getrocknet. Es wurden 127 mg der Zielverbindung (90% d. Th.) erhalten. LC-MS (Methode 14): Rt = 4.07 min
MS (ESpos): m/z = 588 (M+H)+
Beispiel 70A eni-Benzyl- { 1 -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- yl}carbonyl)amino]-4,4-difluor-2-methylbutan-2-yl}carbamat Trifluoracetat (Enantiomer B)
80 mg (0.24 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 119 mg (0.31 mmol) HATU und 0.21 ml (1.20 mmol) N,N- Diisopropylethylamin in 0.80 ml DMF vorgelegt und 20 min gerührt. Anschließend wurden 85 mg (0.29 mmol; Reinheit 93%) eni-Benzyl-(l-amino-4,4-difluor-2-methylbutan-2-yl)carbamat (Enantiomer B) aus Beispiel 61 A hinzugegeben und das Gemisch wurde 0.5 h bei RT gerührt. Die Reaktionslösung wurde mit Wasser versetzt und 30 min bei RT verrührt. Der erhaltene Feststoff wurde ab filtriert, gut mit Wasser gewaschen und mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1%> TFA). Es wurden 93 mg der Zielverbindung (55% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.22 min
MS (ESpos): m/z = 588 (M-TFA+H)+
Beispiel 71A
Ethyl-8-chlor-2-methylimidazo[l,2-a]pyrazin-3-carboxylat
Unter Argon wurde 1 g (7.72 mmol) 3-Chlorpyrazin-2-amin in 35 ml Ethanol gelöst und mit 6.35 g (38.6 mmol) Ethyl-2-chlor-3-oxobutanoat versetzt. Das Reaktionsgemisch wurde unter Rückfluss für ca. 40 h gerührt. Es wurde abgekühlt und das Gemisch wurde eingedampft (Trockeneiskühlung; Ölpumpe bei ca. 0.4 mbar; 60°C Wasserbadtemperatur). Der Rückstand wurde mit Acetonitril versetzt und ausgerührt. Das Filtrat wurde eingedampft und mittels Kieselgelchromatographie gereinigt (Laufmittel: Cyclohexan/Ethylacetat-Gradient). Es wurden 140 mg der Zielverbindung (6% d. Th.) erhalten.
LC-MS (Methode 16): Rt = 1.70 min
MS (ESpos): m/z = 240 (M+H)+ Beispiel 72A
8-Chlor-2-methylimidazo[l,2-a]pyrazin-3-carbonsäure
140 mg (0.47 mmol) Ethyl-8-chlor-2-methylimidazo[l,2-a]pyrazin-3-carboxylat aus Beispiel 71A wurden in 1.9 ml 1,4-Dioxan gelöst, mit 1.9 ml 2 N Natronlauge versetzt und über Nacht bei RT gerührt. Das Reaktionsgemisch wurde mit 6 N Salzsäure angesäuert und das Gemisch wurde eingedampft. Der Rückstand wurde mit wenig Wasser versetzt, kurz gerührt und anschließend wurde der ausgefallene Niederschlag abfiltriert. Es wurden 68 mg der Zielverbindung (68% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.50 min MS (ESpos): m/z = 212 (M+H)
Beispiel 73A
8-Chlor-N-(3,4-difluorbenzyl)-2-methylimidazo[l,2-a]pyrazin-3-carboxamid
Eine Lösung von 65 mg (0.31 mmol) 8-Chlor-2-methylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 72A in 3.5 ml Dichlormethan und 0.1 ml DMF wurden mit 99 mg (0.31 mmol) (Benzotriazol-l-yloxy)bisdimethylaminomethyliumfluoroborat (TBTU), 44 mg (0.31 mmol) 1- (3,4-Difluorphenyl)methanamin und 0.17 ml (1.54 mmol) 4-Methylmorpholin versetzt und über Nacht bei RT gerührt. Das Gemisch wurde mit 2 ml Wasser versetzt, kurz gerührt und anschließend über eine Extrelut-Kartusche filtriert. Es wurde gut mit Dichlormethan/Essigester nachgewaschen und das Filtrat wurde eingedampft und der Rückstand mittels präparativer HPLC gereinigt (Macherey-Nagel, VP50/21 Nucleodur C18 Gravity, 5 μιη, 21x50 mm, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1 % konz. wässriger Ammoniak-Lösung). Es wurden 50 mg der Zielverbindung (49% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.91 min
MS (ESpos): m/z = 337 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 2.67 (s, 3H), 4.54 (d, 2H), 7.21 - 7.28 (m, 1H), 7.36 - 7.49 (m, 2H), 7.84 (d, 1H), 8.74 (t, 1H), 8.92 (d, 1H).
Beispiel 74A
3-(Cyclobutylmethoxy)pyrazin-2-amin
- 136 -
Eine Mischung von 1.05 g (26.25 mmol; Reinheit 60%) Natriumhydrid in 15 ml DMF wurde mit 1.33 g (15.44 mmol) Cyclobutylmethanol versetzt und 1 h bei RT gerührt. Anschließend wurde eine Mischung von 1.0 g (7.72 mmol) 3-Chlorpyrazin-2-amin in 10 ml DMF hinzugegeben und das Reaktionsgemisch wurde auf 100°C erwärmt. Nach 20 h wurde das Gemisch mit Wasser versetzt und mehrfach mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit gesättigter Natriumchlorid-Lsg. gewaschen, über Natriumsulfat getrocknet, filtriert und eingedampft. Der erhaltene Rückstand wurde mittels Kieselgelchromatograpie aufgereinigt (Laufmittel: Cyclohexan/Ethylacetat-Gradient). Es wurden 1.25 g der Titelverbindung (89% d. Th.; Reinheit 98%) erhalten.
LC-MS (Methode 2): Rt = 0.80 min MS (ESpos): m/z = 180 (M+H)+ Beispiel 75A
Ethyl-8-(cyclobutylmethoxy)-2-methylimidazo[l,2-a]pyrazin-3-carboxylat
Unter Argon wurden 150 mg (0.84 mmol) 3-(Cyclobutylmethoxy)pyrazin-2-amin aus Beispiel 74A in 6 ml Ethanol gelöst und mit 689 mg (38.6 mmol) Ethyl-2-chlor-3-oxobutanoat versetzt. Das Reaktionsgemisch wurde unter Rückfluss für ca. 48 h gerührt. Es wurde abgekühlt und das Gemisch wurde eingedampft (Trockeneiskühlung; Ölpumpe bei ca. 0.4 mbar; 60°C Wasserbadtemperatur). Der Rückstand wurde in wenig Ethylacetat aufgenommen und mittels Kieselgelchromatograpie aufgereinigt (Laufmittel: Cyclohexan/Ethylacetat-Gradient). Es wurden 75 mg der Zielverbindung (30% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.19 min
MS (ESpos): m/z = 290 (M+H)
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.37 (t, 3H), 1.80 - 1.98 (m, 4H), 2.05 - 2.16 (m, 2H), 2.62 (s, 3H), 2.75 - 2.88 (m, 1H), 4.39 (q, 2H), 4.45 (d, 2H), 7.63 (d, 1H), 8.68 (d, 1H).
Beispiel 76A
8-(Cyclobutylmethoxy)-2-methylimidazo[l,2-a]pyrazin-3-carbonsäure
72 mg (0.25 mmol) Ethyl-8-(cyclobutylmethoxy)-2-methylimidazo[l,2-a]pyrazin-3-carboxylat aus Beispiel wurden in 1.5 ml 1,4-Dioxan gelöst, mit 1 ml 2 N Natronlauge versetzt und 3 h bei RT gerührt. Das Reaktionsgemisch wurde mit 6 N Salzsäure angesäuert und mit Dichlormethan versetzt und über eine Extrelut-Kartusche filtriert. Es wird gut mit Dichlormethan/Ethylacetat nachgewaschen. Das Filtrat wurde eingedampft und im Hochvakuum getrocknet. Es wurden 55 mg der Zielverbindung (85% d. Th.) erhalten.
LC-MS (Methode 1): Rt = 0.96 min
MS (ESpos): m/z = 262 (M+H)
Ausführungsbeispiele Beispiel 1 en N-[(2S)-2-Amino-2-methylbutyl]-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-carboxamid
Eine Mischung von 74.0 mg eni-Benzyl- {l-[( {8-[(2,6-difluorbenzyl)oxy]-2,6- dimethylimidazo[l,2-a]pyrazin-3-yl}carbonyl)amino]-2-methylbutan-2-yl}carbamat (0.134 mmol, 1.0 eq.) aus Beispiel 9A und 9.4 mg Palladiumhydroxid 20% auf Kohle (0.01 mmol, 0.1 eq.) in Ethanol wurde für 2 h bei Normaldruck hydriert. Anschließend wurde das Gemisch über Kieselgur filtriert, nachgewaschen, und das Filtrat eingeengt. Das Rohprodukt wurde mittels präperativer HPLC (Methode 6) gereinigt, wobei 39 mg der Titelverbindung (66% d. Th.) erhalten wurden.
LC-MS (Methode 2): Rt = 0.81 min
MS (ESpos): m/z = 418 (M+H)+
1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.86 (t, 3H), 0.97 (s, 3H), 1.24 - 1.53 (m, 4H), 2.37 (s, 3H), 2.54 (s, 3H verborgen unter Lösemittelsigna! ). 3.14 - 3.27 (m, 2H), 5.54 (s, 2H), 7.16 - 7.28 (m, 2H), 7.50 - 7.63 (m, 1H), 7.69 - 8.01 (br s, 1H), 8.38 (d, 1H).
Beispiel 2
8-[(2,6-Difluorbenzyl)oxy]-N-[(2S)-hexan-2-yl]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carboxamid
Eine Mischung von 60.0 mg 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3- carbonsäure (0.180 mmol, 1.0 eq) aus Beispiel 3A, 27.0 mg (2S)-Hexan-2-amin [CAS-Nr.: 70492- 67-0] (0.270 mmol, 1.5 eq.) und 0.16 ml N,N-Diisopropyethylamin (0.90 mmol, 5 eq.) in 0.60 ml DMF wurde mit 89.0 mg HATU (0.234 mmol, 1.3 eq.) versetzt und über Nacht bei RT gerührt. Anschließend wurde mit Wasser ausgerührt, der Feststoff abfiltriert, mit Wasser gewaschen und im Hochvakuum getrocknet. Es wurden 46 mg der Titelverbindung (58% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.31 min
MS (ESpos): m/z = 417 (M+H)+ 'H-NMR (400 MHz, DMSO-de): δ [ppm] = 0.83 - 0.92 (m, 3H), 1.18 (d, 3H), 1.22 - 1.63 (m, 6H), 2.37 (s, 3H), 2.49 (s, 3H überlagert mit Lösemittelsigna! ). 3.76 - 4.25 (m, 1H), 5.54 (s, 2H), 7. 17 - 7.26 (m, 2H), 7.48 - 7.67 (m, 1H), 7.90 - 7.96 (m, 1H), 8.26 (s, 1H).
Beispiel 3
8-[(2,6-Difluorbenzyl)oxy]-N-[(2R)-l-hydroxyhexan-2-yl]-2,6-dimethylimidazo[l,2-a]pyrazin-3- carboxamid
Eine Mischung von 60.0 mg 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3- carbonsäure (0.180 mmol, 1.0 eq) aus Beispiel 3A, 31.6 mg (2R)-2-Aminohexan-l-ol [CAS-Nr.: 80696-28-2] (0.27 mmol, 1.5 eq.) und 0.16 ml N,N-Diisopropyethylamin (0.90 mmol, 5 eq.) in 0.60 ml DMF wurde mit 89.0 mg HATU (0.234 mmol, 1.3 eq.) versetzt und über Nacht bei RT gerührt Anschließend wurde mit Wasser ausgerührt, der Feststoff abfiltriert, mit Wasser gewaschen und im Hochvakuum getrocknet. Es wurden 59.0 mg der Titelverbindung (72% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.12 min
MS (ESpos): m/z = 433 (M+H)+ 'H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.80 - 0.93 (m, 3H), 1.20 - 1.70 (m, 6H), 2.37 (s, 3H), 2.54 (s, 3H verborgen unter Lösemittelsigna! ). 3.39 - 3.55 (m, 2H), 3.88 - 4.03 ( m, 1H), 4.65 - 4.83 (m, 1H), 5.54 (s, 2H), 7.17 - 7.26 (m, 2H), 7.46 - 7.63 (m, 1H), 7.71 - 7.83 (m, 1H), 8.27 (s, 1H).
Beispiel 4
8-[(2,6-Difluorbenzyl)oxy]-N-[l -(3,4-difluorphenyl)cyclopropyl]-2,6-dimethylimidazo[l,2- a]pyrazin-3-carboxamid
Eine Mischung von 60.0 mg 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3- carbonsäure (0.180 mmol, 1.0 eq) aus Beispiel 3A, 45.7 mg l -(3,4-Difluorphenyl)cyclopropanamin [CAS-Nr.: 474709-85-8] (0.27 mmol, 1.5 eq.) und 0.16 ml N,N-Diisopropyethylamin (0.90 mmol, 5 eq.) in 0.60 ml DMF wurde mit 89.0 mg HATU (0.234 mmol, 1.3 eq.) versetzt und über Nacht bei RT gerührt. Anschließend wurde mit Wasser ausgerührt, der Feststoff abfiltriert, mit Wasser gewaschen und im Hochvakuum getrocknet. Es wurden 61.0 mg der Titelverbindung (69% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.26 min MS (ESpos): m/z = 485 (M+H)+
' H-NM R (400 M Hz, DMSO-d6): δ [ppm] = 1.33 (s, 4H), 2.36 (s, 3H), 2.54 (s, 3H verborgen unter Lösemittelsigna! ). 5.54 (s, 2H), 7.07 - 7.42 (m, 5H), 7.51 - 7.66 (m, 1H), 8.30 (s, 1H), 8.81 (s, 1H).
Beispiel 5 rac-N-(2-Amino-2,4-dimethylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-carboxamid
70 mg (0.21 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 96 mg (0.25 mmol) HATU und 0.18 ml (1.05 mmol) N,N- Diisopropylethylamin in 2.1 ml DMF vorgelegt, 10 min gerührt, anschließend bei RT mit 33 mg (0.25 mmol) rac-2,4-Dimethylpentan-l,2-diamin versetzt und über Nacht bei RT gerührt. Das Reaktionsgemisch wurde mit Dichlormethan extrahiert, die organische Phase wurde über Natriumsulfat getrocknet, abfiltriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA) gereinigt. Die Produktfraktion wurde mit gesättigter, wässriger Natriumhydrogencarbonatlösung und Dichlormethan gewaschen, über Natriumsulfat getrocknet, ab filtriert, eingeengt und lyophilisiert. Es wurden 69 mg der Zielverbindung (72% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.87 min
MS (ESpos): m/z = 446 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 0.91 (d, 3H), 0.94 (d, 3H), 1.02 (s, 3H), 1.20 - 1.33 (m, 2H), 1.51 (br. s, 2H), 1.75 - 1.88 (m, 1H), 2.36 (s, 3H), 2.55 (s, 3H; überlagert mit Lösungmittelpeak), 3.21 (q, 2H), 5.54 (s, 2H), 7.17 - 7.27 (m, 2H), 7.51 - 7.62 (m, 1H), 7.76 - 7.96 (m, 1H), 8.38 (s, 1H).
Beispiel 6 eni-8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethyl-N-(7,7,7-trifluor-2-hydroxy-2-methylheptan-3- yl)imidazo[l,2-a]pyrazin-3-carboxamid
70 mg (0.21 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 96 mg (0.25 mmol) HATU und 0.18 ml (1.05 mmol) N,N- Diisopropylethylamin in 2.1 ml DMF vorgelegt, 10 min gerührt, anschließend bei RT mit 50 mg (0.25 mmol) eni-3-Amino-7,7,7-trifluor-2-methylheptan-2-ol aus Beispiel 19A versetzt und über Nacht bei RT gerührt. Das Reaktionsgemisch wurde mit Dichlormethan extrahiert, die organische Phase wurde über Natriumsulfat getrocknet, abfiltriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA) gereinigt. Die Produktfraktion wurde mit gesättigter, wässriger Natriumhydrogencarbonatlösung und Dichlormethan gewaschen, über Natriumsulfat getrocknet, abfiltriert, eingeengt und lyophilisiert. Es wurden 98 mg der Zielverbindung (86% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.18 min
MS (ESpos): m/z = 515 (M+H)
'H-NMR (400 MHz, DMSO-de): δ = 1.12 (s, 3H), 1.17 (s, 3H), 1.42 - 1.63 (m, 3H), 1.77 - 1.87 (m, 1H), 2.17 - 2.30 (m, 1H), 2.30 - 2.45 (m, 4H), 2.52 (s, 3H, verdeckt durch Lösungsmittel-Peak), 3.91 - 4.01 (m, 1H), 4.58 (s, 1H), 5.55 (s, 2H), 7.18 - 7.26 (m, 2H), 7.52 - 7.61 (m, 1H), 7.68 (d, 1H), 8.23 (s, 1H).
Beispiel 7 eni-8-[(2,6-Difluorbenzyl)oxy]-N-(2-hydroxy-2-methylheptan-3-yl)-2,6-dimethylimidazo[l,2- a]pyrazin-3-carboxamid
70 mg (0.21 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 96 mg (0.25 mmol) HATU und 0.18 ml (1.05 mmol) N,N- Diisopropylethylamin in 2.1 ml DMF vorgelegt, 10 min gerührt, anschließend bei RT mit 36.6 mg (0.25 mmol) eni-3-Amino-2-methylheptan-2-ol aus Beispiel 13A versetzt und über Nacht bei RT gerührt. Es wurden nochmals 24 mg (0.06 mmol) HATU hinzugegeben und das Gemisch wurde 1.5 Stunden bei RT gerührt. Die Reaktionslösung wurde mit Wasser und Dichlormethan versetzt. Die Phasen wurden getrennt, die organische Phase wurde über Natriumsulfat getrocknet, ab filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA) gereinigt. Die Produktfraktion wurde mit gesättigter, wässriger Natriumhydrogencarbonatlösung und Dichlormethan gewaschen, die organische Phase wurde über Natriumsulfat getrocknet, abfiltriert, eingeengt und lyophilisiert. Es wurden 59 mg der Zielverbindung (59% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.20 min
MS (ESpos): m/z = 461 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 0.86 (t, 3H), 1.10 (s, 3H), 1.16 (s, 3H), 1.21 - 1.47 (m, 5H), 1.68 - 1.81 (m, 1H), 2.37 (s, 3H), 2.52 (s, 3H, verdeckt durch Lösungsmittel-Peak), 3.87 - 3.96 (m, 1H), 4.49 (s, 1H), 5.55 (s, 2H), 7.17 - 7.26 (m, 2H), 7.52 - 7.63 (m, 2H), 8.23 (s, 1H).
Beispiel 8 Methyl-irani'-4- {[({8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazm-3- yl} carbonyl)amino]methyl} cyclohexancarboxylat
50 mg (0.15 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 68.4 mg (0.18 mmol) HATU und 0.13 ml (0.75 mmol) N,N- Diisopropylethylamin in 2 ml DMF vorgelegt und 10 min bei RT gerührt. Anschließend wurden 46.7 mg (0.23 mmol) Methyl-iraft -4-aminomethylcyclohexanecarboxylat Hydrochlorid zur Reaktionslösung gegeben und über Nacht bei RT gerührt. Das Reaktionsgemisch wurde mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.05% Ameisensäure) gereinigt. Es wurden 50 mg der Zielverbindung (73% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.18 min MS (ESpos): m/z = 487 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 0.95 - 1.09 (m, 2H), 1.24 - 1.38 (m, 2H), 1.47 - 1.62 (m, 1H), 1.76 - 1.85 (m, 2H), 1.88 - 1.97 (m, 2H), 2.21 - 2.31 (m, 1H), 2.36 (s, 3H), 2.52 (s, 3H, überlagert durch Lösungsmittel-Peak), 3.17 (t, 2H), 3.58 (s, 3H), 5.54 (s, 2H), 7.17 - 7.26 (m, 2H), 7.52 - 7.61 (m, 1H), 8.08 (t, 1H), 8.32 (s, 1H). Beispiel 9
8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethyl-N-[(3S)-2-oxopyrrolidin-3-yl]imidazo[l,2-a]pyrazin-3- carboxamid
50 mg (0.15 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 68.4 mg (0.18 mmol) HATU und 0.13 ml (0.75 mmol) N,N- Diisopropylethylamin in 2 ml DMF vorgelegt und 10 min bei RT gerührt. Anschließend wurden 22.5 mg (0.22 mmol) (S)-3-Aminopyrrolidin-2-on zur Reaktionslösung gegeben und über Nacht bei RT gerührt. Das Reaktionsgemisch wurde mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.05% Ameisensäure) gereinigt. Es wurden 50 mg der Zielverbindung (80% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.89 min MS (ESpos) : m/z = 416 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.98 - 2.1 1 (m, 1H), 2.37 (s, 3H), 2.39 - 2.44 (m, 1H), 2.52 (s, 3H), 3.25 (dd, 2H), 4.57 (q, 1H), 5.54 (s, 2H), 7.17 - 7.26 (m, 2H), 7.52 - 7.62 (m, 1H), 7.92 (s, 1H), 8.30 (d, 1H), 8.36 (s, 1H).
Beispiel 10 8-[(2,6-Difluorbenzyl)oxy]-N-(6-fluorchinolin-4-yl)-2,6-dimethylimidazo[l ,2-a]pyrazin-3- carboxamid
50 mg (0.15 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 74 mg (0.20 mmol) HATU und 0.03 ml (0.30 mmol) 4- Methylmorpholin in 2 ml DMF vorgelegt und 30 min bei RT gerührt. Anschließend wurden 36.5 mg (0.23 mmol) 6-Fluorchinolin-4-amin zur Reaktionslösung gegeben und 1 Stunde bei RT gerührt. Das Reaktionsgemisch wurde mittels präparativer HPLC (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.05% Ameisensäure) gereinigt. Es wurden 22 mg der Zielverbindung (31% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.09 min MS (ESpos) : m/z = 478 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 2.40 (s, 3H), 2.69 (s, 3H), 5.59 (s, 2H), 7.20 - 7.28 (m, 2H), 7.54 - 7.63 (m, 1H), 7.70 - 7.77 (m, 1H), 8.03 - 8.09 (m, 1H), 8.10 - 8.16 (m, 2H), 8.41 (s, 1H), 8.89 (d, 1H), 10.43 (s, 1H).
Beispiel 11 irani'-4- {[({8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazm-3- yl} carbonyl)amino]methyl} cyclohexancarbonsäurehydrochlorid
43 mg (0.088 mmol) Methyl-irara-4- {[({8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-yl}carbonyl)amino]methyl}cyclohexancarboxylat aus Beispiel 8 wurden in 3 ml THF/Methanol (5/1) gelöst, mit 0.44 ml (0.44 mmol) einer 1 N wässrigen Lithiumhydroxidlösung versetzt und 6 h bei RT gerührt. Es wurden 0.44 ml (0.44 mmol) einer 1 N wässrigen Lithiumhydroxidlösung zugegeben und 4 h bei RT gerührt. Dann wurden 0.44 ml (0.44 mmol) einer 1 N wässrigen Lithiumhydroxidlösung zugegeben und erneut 4 h bei RT gerührt. Dann wurde abermals mit 0.44 ml (0.44 mmol) einer 1 N wässrigen Lithiumhydroxidlösung versetzt und 4 h bei RT gerührt. Die Reaktionslösung wurde mit 1 N wässriger Salzsäure angesäuert und das organische Lösungsmittel wurde abdestilliert. Der entstandene Niederschlag wurde abfiltriert, mit Wasser gewaschen und im Hochvakuum getrocknet. Anschließend wurde der Rückstand lyophilisiert. Es wurden 31 mg der Zielverbindung (65% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.02 min
MS (ESpos): m/z = 473 (M-HC1+H)+ 'H-NMR (500 MHz, DMSO-d6): δ = 0.95 - 1.06 (m, 2H), 1.24 - 1.36 (m, 2H), 1.48 - 1.61 (m, 1H), 1.77 - 1.84 (m, 2H), 1.88 - 1.96 (m, 2H), 2.10 - 2.19 (m, 1H), 2.37 (s, 3H), 2.52 (s, 3H, überlagert durch Lösungsmittel-Peak), 3.18 (t, 2H), 5.54 (s, 2H), 7.18 - 7.25 (m, 2H), 7.53 - 7.60 (m, 1H), 8.08 (t, 1H), 8.33 (s, 1H).
Beispiel 12 8-[(2,6-Difluorbenzyl)oxy]-N-[2-(l -hydroxycyclopentyl)ethyl]-2,6-dimethylimidazo[l ,2- a]pyrazin-3-carboxamid
12.9 mg (0.1 mmol) 1 -(2-Aminoethyl)cyclopentanol wurden in einer 96er deep well Multititerplatte vorgelegt. Eine Lösung von 33.3 mg (0.1 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6- dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A in 0.3 ml DMF und eine Lösung von 45.6 mg (0.12 mol) HATU in 0.3 ml DMF wurden nacheinander hinzugefügt. Nach Zugabe von 20.2 mg (0.2 mmol) 4-Methylmorpholin wurde das Gemisch bei RT über Nacht geschüttelt. Dann wurde filtriert und aus dem Filtrat die Zielverbindung per präparativer LC-MS (Methode 9) isoliert. Die produkthaltigen Fraktionen wurden mittels Zentrifugaltrockner im Vakuum eingeengt. Der Rückstand der Produktfraktionen wurde in je 0.6 ml DMSO gelöst. Diese wurden vereint und abschließend im Zentrifugaltrockner vom Lösemittel befreit. Es wurden 10.5 mg (21.5% d. Th.; Reinheit 91%) erhalten.
LC-MS (Methode 10): Rt = 1.10 min
MS (ESpos): m/z = 445 (M+H)+
In Analogie zu Beispiel 12 wurden die in Tabelle 1 gezeigten Beispielverbindungen hergestellt, indem 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A mit den entsprechenden, kommerziell erhältlichen oder zuvor beschriebenen Aminen unter den beschriebenen Bedingungen umgesetzt wurden:
Tabelle 1:
BeiIUPAC-Name/Struktur
Analytische Daten spiel (Ausbeute)
Beispiel 37 e^N-(2-Amino-3-fluor-2-methylpropyl)-8-[(2,6^
a]pyrazin-3-carboxamid (Enantiomer B)
Unter Argon wurden 62 mg (0.11 mmol) eni-Benzyl- {l-[( {8-[(2,6-difluorbenzyl)oxy]-2,6- dimethylimidazo [ 1 ,2-a]pyrazin-3 -yl} carbonyl)amino] -3 -fluor-2-methylpropan-2-yl} carbamat (Enantiomer B) aus Beispiel 66A in 2.9 ml Ethanol gelöst, mit 6 mg Palladium auf Aktivkohle (10%ig) versetzt und die Reaktionsmischung 45 min bei Normaldruck bei RT hydriert. Das Reaktionsgemisch wurde über Celite abfiltriert, gut mit Ethanol gewaschen und das Filtrat anschließend eingeengt. Der Rückstand wurde mittels Dickschichtchromatographie gereinigt (Laufmittel: Dichlormethan/2M Ammoniaklösung in Methanol = 20/1). Es wurden 34 mg der Zielverbindung (47% d. TL, Reinheit 98%) erhalten.
LC-MS (Methode 2): Rt = 0.70 min
MS (ESpos): m/z = 422 (M+H)+ 'H-NMR (500 MHz, DMSO-d6): δ = 1.03 (s, 3H), 1.68 (br. s, 2H), 2.37 (s, 3H), 2.51 (s, 3H; überlagert mit Lösungsmittel-Peak), 3.25 - 3.38 (m, 2H; überlagert mit Wasser-Peak), 4.08 - 4.15 (m, 1H), 4.18 - 4.25 (m, 1H), 5.53 (s, 2H), 7.18 - 7.25 (m, 2H), 7.54 - 7.62 (m, 1H), 7.87 - 7.94 (m, 1H), 8.38 (s, 1H).
Beispiel 38 eni-N-(2-Amino-5,5,5-trifluor-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2,6- dimethylimidazo[l,2-a]pyrazin-3-carboxamid (Enantiomer B)
Eine Mischung von 86 mg (0.14 mmol) eni-Benzyl- { l -[( {8-[(2,6-difluorbenzyl)oxy]-2,6- dimethylimidazo[l ,2-a]pyrazin-3-yl}carbonyl)amino]-5,5,5-trifluor-2-methylpentan-2-yl}carbamat (Enantiomer B) aus Beispiel 67A und 7 mg Palladium auf Aktivkohle (10%ig) in 3.2 ml Ethanol wurden für 1.5 h bei Raumtemperatur und Normaldruck hydriert. Anschließend wurde über einen Millipore-Filter abfiltriert, mit Ethanol gewaschen und das Filtrat eingeengt. Das Rohprodukt wurde mit Acetonitril/Wasser und TFA versetzt und mittels präparativer HPLC gereinigt (RP-C18, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA). Die Produktfraktionen wurden in Dichlormethan aufgenommen und zweimal mit gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die vereinten wässrigen Phasen wurden zweimal mit Dichlormethan extrahiert. Die vereinten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingedampft. Es wurden 10 mg der Titelverbindung (15% d. Th.; Reinheit 98%) erhalten.
LC-MS (Methode 2): Rt = 0.79 min MS (ESpos): m/z = 486 (M+H)+
'H-NMR (500 MHz, DMSO-de): δ [ppm] = 1.02 (s, 3H), 1.59 - 1.70 (m, 2H), 2.30 - 2.47 (m, 5H), 2.50 (s, 3H; überlagert vom Lösungsmittelpeak), 3.20 - 3.40 (m, 2H; überlagert mit Wasserpeak), 5.54 (s, 2H), 7.18 - 7.25 (m, 2H), 7.54 - 7.62 (m, 1H), 8.04 - 8.15 (m, 1H), 8.32 (s, 1H).
Beispiel 39 eni-N-(2-Amino-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2-a]pyrazin-3- carboxamid (Enantiomer B)
66 mg (0.10 mmol) eni-Benzyl- {l -[({8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-yl}carbonyl)amino]-2-methylpentan-2-yl}carbamat Trifluoracetat (Enantiomer B) aus Beispiel 65A wurden in 2.5 ml Ethanol gelöst, mit 3,1 mg Palladium auf Aktivkohle (10%ig) versetzt und insgesamt 100 min bei Normaldruck hydriert. Die Reaktionslösung wurde mittels Millipore-Filter filtriert und das Filtrat einrotiert. Der Rückstand wurde mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA). Die Produktfraktionen wurden vereinigt und eingeengt. Alle Produktfraktionen wurden vereinigt und eingeengt. Anschließend wurde der Rückstand in Dichlormethan und wenig Methanol aufgenommen und mit wenig gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan reextrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert, eingeengt und lyophilisiert. Es wurden 31 mg der Zielverbindung (73% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.77 min MS (ESpos) : m/z = 432 (M+H)
'H-NMR (400 MHz, DMSO-de): δ = 0.86 (t, 3H), 0.99 (s, 3H), 1.20 - 1.42 (m, 4H), 1.70 (br. s, 2H), 2.35 (s, 3H), 2.55 (s, 3H; überlagert mit Lösungsmittelpeak), 3.15 - 3.30 (m, 2H), 5.54 (s, 2H), 7.16 - 7.25 (m, 2H), 7.52 - 7.61 (m, 1H), 7.82 (br. s, 1H), 8.37 (s, 1H).
Beispiel 40 eni-N-(2-Amino-5-fluor-2-methylpentyl)-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-carboxamid (Enantiomer B)
109 mg (0.13 mmol, Reinheit 86%) eni-Benzyl- {l-[( {8-[(2,6-difluorbenzyl)oxy]-2,6- dimethylimidazo[l,2-a]pyrazin-3-yl}carbonyl)amino]-5-fluor-2-methylpentan-2-yl}carbamat Trifluoracetat (Enantiomer B) aus Beispiel 68A wurden in 3.4 ml Ethanol gelöst, mit 4.3 mg Palladium auf Aktivkohle (10%ig) versetzt und insgesamt 2.5 Stunden bei Normaldruck hydriert. Die Reaktionslösung wurde mittels Milliporfilter filtriert und das Filtrat einrotiert. Der Rückstand wurde mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA). Alle Produktfraktionen wurden vereinigt und eingeengt. Anschließend wurde der Rückstand in Dichlormethan und wenig Methanol aufgenommen und zweimal mit gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan re- extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert, eingeengt und lyophilisiert. Es wurden 42 mg der Zielverbindung (68% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.69 min MS (ESpos) : m/z = 450 (M+H)
'H-NMR (400 MHz, DMSO-de): δ = 1.00 (s, 3H), 1.32 - 1.88 (m, 8H), 2.35 (s, 3H), 2.54 (s, 3H; überlagert mit Lösungsmittelpeak), 3.18 - 3.30 (m, 2H), 4.35 (t, 1H), 4.48 (t, 1H), 5.53 (s, 2H), 7.18 - 7.28 (m, 2H), 7.52 - 7.63 (m, 1H), 7.88 (br. s, 1H), 8.35 (s, 1H).
Beispiel 41 en N-(2-Amino-4,4-difluor-2-methylbutyl)-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-carboxamid (Enantiomer A)
127 mg (0.22 mmol) eni-Benzyl- { l -[( {8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2- a]pyrazin-3-yl}carbonyl)amino]-4,4-difluor-2-methylbutan-2-yl}carbamat (Enantiomer A) aus Beispiel 69A wurden in 5.5 ml Ethanol gelöst, mit 33 μΐ TFA und 7 mg Palladium auf Aktivkohle (10%ig) versetzt und 2.5 Stunden bei Normaldruck hydriert. Die Reaktionslösung wurde mittels Millipore-Filter filtriert und das Filtrat einrotiert. Der Rückstand wurde mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA). Die Produktfraktionen wurden vereinigt und eingeengt. Alle Produktfraktionen wurden vereinigt und eingeengt. Anschließend wurde der Rückstand in Dichlormethan und wenig Methanol aufgenommen und mit wenig gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan re- extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert, eingeengt und lyophilisiert. Es wurden 75 mg der Zielverbindung (75% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.74 min MS (ESpos) : m/z = 454 (M+H)
'H-NMR (500 MHz, DMSO-de): δ = 1.08 (s, 3H), 1.72 (br. s, 2H), 1.87 - 1.99 (m, 2H), 2.36 (s, 3H), 2.56 (s, 3H), 3.22 - 3.32 (m, 2H; überlagert mit Lösungsmittelpeak), 5.55 (s, 2H), 6.1 1 - 6.39 (m, 1H), 7.18 - 7.25 (m, 2H), 7.53 - 7.62 (m, 1H), 7.92 - 8.01 (m, 1H), 8.34 (s, 1H).
Beispiel 42 en N-(2-Amino-4,4-difluor-2-methylbutyl)-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l ,2- a]pyrazin-3-carboxamid (Enantiomer B)
93 mg (0.13 mmol) eni-Benzyl- {l -[({8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-yl}carbonyl)amino]-4,4-difluor-2-methylbutan-2-yl}carbamat Trifluoracetat (Enantiomer B) aus Beispiel 70A wurden in 3.4 ml Ethanol gelöst, mit 4.2 mg Palladium auf Aktivkohle (10%ig) versetzt und 70 min bei Normaldruck hydriert. Die Reaktionslösung wurde mittels Millipore-Filter filtriert und das Filtrat eingedampft. Der Rückstand wurde mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA). Die Produktfraktionen wurden vereinigt und eingeengt. Anschließend wurde der Rückstand in Dichlormethan und wenig Methanol aufgenommen und zweimal mit wenig gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan reextrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert, eingedampft und lyophilisiert. Es wurden 47 mg der Zielverbindung (78% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.74 min MS (ESpos) : m/z = 454 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.08 (s, 3H), 1.71 (br. s, 2H), 1.86 - 2.00 (m, 2H), 2.36 (s, 3H), 2.56 (s, 3H), 3.22 - 3.32 (m, 2H; überlagert mit Lösungsmittelpeak), 5.56 (s, 2H), 6.08 - 6.42 (m, 1H), 7.18 - 7.26 (m, 2H), 7.53 - 7.62 (m, 1H), 7.91 - 8.01 (m, 1H), 8.34 (s, 1H).
Beispiel 43 rac-N-(2-Amino-4-fluor-2-methylbutyl)-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2- a]pyrazin-3-carboxamid
100 mg (0.30 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 137 mg (0.36 mmol) HATU und 0.21 ml (1.20 mmol) N,N- Diisopropylethylamin in 1.4 ml DMF vorgelegt und 20 min gerührt. Eine Lösung von 164 mg (0.63 mmol bei Annahme einer Reinheit von ca. 75%) rac-4-Fluor-2-methylbutan-l,2-diamin Dihydrochlorid aus Beispiel 64A in 0.7 ml DMF und und 0.31 ml (1.80 mmol) N,N- Diisopropylethylamin wurde zum ersten Reaktionsgemisch hinzugegeben und das Gemisch wurde 0.5 h bei RT gerührt. Die Reaktionslösung wurde mit Acetonitril, Wasser und TFA versetzt und mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1 % TFA). Die Produktfraktionen wurden vereinigt und eingeengt. Anschließend wurde der Rückstand in Dichlormethan und wenig Methanol aufgenommen und zweimal mit gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert, eingeengt und lyophilisiert. Es wurden 65 mg der Zielverbindung (49% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 0.73 min MS (ESpos): m/z = 436 (M+H)+
'H-NMR (500 MHz, DMSO-de): δ = 1.05 (s, 3H), 1.66 - 2.15 (m, 4H), 2.35 (s, 3H), 2.56 (s, 3H), 3.19 - 3.32 (m, 2H; z. T. überlagert mit Lösungsmittelpeak), 4.55 - 4.63 (m, 1H), 4.66 - 4.73 (m, 1H), 5.54 (s, 2H), 7.18 - 7.24 (m, 2H), 7.53 - 7.61 (m, 1H), 7.91 (br. s, 1H), 8.36 (s, 1H).
Beispiel 44
8-[(2,6-Difluorbenzyl)oxy]-N-(2-hydroxyethoxy)-2,6-dimethylimidazo[l,2-a]pyrazin-3- carboxamid
33 mg (0.10 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden in einer 96er deep well Multititerplatte vorgelegt. Eine Lösung von 8 mg (0.10 mmol) 2-(Aminooxy)ethanol in 0.4 ml DMF sowie eine Lösung von 45.6 mg (0.12 mol) HATU in 0.4 ml DMF wurden nacheinander hinzugegeben. Nach Zugabe von 20.2 mg (0.20 mmol) 4-Methylmorpholin wurde das Gemisch bei RT über Nacht geschüttelt. Dann wurde filtriert und aus dem Filtrat die Zielverbindung per präparativer LC-MS (Methode 9) isoliert. Die produkthaltigen Fraktionen wurden mittels Zentrifugaltrockner im Vakuum eingeengt. Der Rückstand der Produktfraktionen wurde in je 0.6 ml DMSO gelöst. Diese wurden vereint und abschließend im Zentrifugaltrockner vom Lösemittel befreit. Es wurden 0.4 mg (1% d. Th.) erhalten. LC-MS (Methode 10): Rt = 0.94 min MS (ESpos): m/z = 393 (M+H)+
In Analogie zu Beispiel 44 wurden die in Tabelle 2 gezeigten Beispielverbindungen hergestellt, indem die entsprechenden Carbonsäuren mit den entsprechenden, kommerziell erhältlichen oder zuvor beschriebenen Aminen [Hydrazine] unter den beschriebenen Bedingungen umgesetzt wurden:
8-(Cyclohexylmethoxy)-N-(3,4-difluorbenzyl)-2-methylimidazo[l,2-a]pyrazin-3-carboxamid
Eine Mischung von 3.3 mg (0.08 mmol; Reinheit 60%) Natriumhydrid in 0.25 ml DMF wurde mit 8.5 mg (0.74 mmol) Cyclohexylmethanol versetzt und 1 h bei RT gerührt. Anschließend wurde eine Mischung von 25 mg (0.074 mmol) 8-Chlor-N-(3,4-difluorbenzyl)-2-methylimidazo[l,2- a]pyrazin-3-carboxamid aus Beispiel 73A in 0.25 ml DMF hinzugegeben und das Reaktionsgemisch wurde auf 100°C erwärmt. Nach 1.5 h wurde das Gemisch mit Wasser versetzt und eingedampft und mittels präparativer HPLC gereinigt (RP-C18, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% Ameisensäure). Es wurden 7 mg der Titelverbindung (22% d. Th.; Reinheit 95%) erhalten.
LC-MS (Methode 2): Rt = 1.26 min
MS (ESpos): m/z = 415 (M+H)+ 'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.00 - 1.33 (m, 5H), 1.61 - 1.90 (m, 6H), 2.61 (s, 3H), 4.27 (d, 2H), 4.52 (d, 2H), 7.20 - 7.27 (m, 1H), 7.36 - 7.47 (m, 2H), 7.48 (d, 1H), 8.49 (d, 1H), 8.60 (t, 1H).
Beispiel 48
8-(Cyclobutylmethoxy)-N-(3,4-difluorbenzyl)-2-methylimidazo[l,2-a]pyrazin-3-carboxamid
Eine Lösung von 27 mg (0.10 mmol) 8-(Cyclobutylmethoxy)-2-methylimidazo[l,2-a]pyrazin-3- carbonsäure aus Beispiel 76A in 1.4 ml Dichlormethan und 0.1 ml DMF wurden mit 37 mg (0.12 mmol) (Benzotriazol-l-yloxy)bisdimethylaminomethyliumfluoroborat (TBTU), 17 mg (0.12 mmol) l-(3,4-Difluorphenyl)methanamin und 0.057 ml (0.52 mmol) 4-Methylmorpholin versetzt und über Nacht bei RT gerührt. Das Gemisch wurde mit 2 ml 10 %iger Zitronensäure versetzt, kurz gerührt und anschließend über eine Extrelut-Kartusche filtriert. Es wurde gut mit Dichlormethan/Essigester nachgewaschen und das Filtrat wurde eingedampft und der Rückstand mittels präparativer HPLC gereinigt (Macherey-Nagel, VP50/21, Nucleosil 100-5 C18 Nautilus, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1 % Ameisensäure). Es wurden 26 mg der Zielverbindung (66% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.16 min
MS (ESpos): m/z = 387
'H-NMR (400 MHz, DMSO-de): δ [ppm] = 1.79 - 1.98 (m, 4H), 2.04 - 2.16 (m, 2H), 2.60 (s, 3H), 2.75 - 2.87 (m, 1H), 4.44 (d, 2H), 4.51 (d, 2H), 7.20 - 7.27 (m, 1H), 7.37 - 7.47 (m, 2H), 7.49 (d, 1H), 8.49 (d, 1H), 8.60 (t, 1H).
„
- 173 -
Beispiel 49
N-(5-Cyanpentyl)-8-[(2,6-difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carboxamid
30 mg (0.09 mmol) 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A wurden mit 44 mg (0.12 mmol) HATU und 0.05 ml (0.27 mmol) N,N- Diisopropylethylamin in 0.3 ml DMF vorgelegt und 20 min bei Raumtemperatur gerührt. Anschließend wurden 13 mg (0.12 mmol) 6-Aminohexannitril zur Reaktionslösung gegeben und das Gemisch wurde 30 min bei RT gerührt. Das Reaktionsgemisch wurde mit Wasser versetzt und für 30 min bei Raumtemperatur verrührt. Der enthaltene Feststoff wurde abfiltriert und gut mit Wasser gewaschen und anschließend getrocknet. Es wurden 34 mg der Zielverbindung (87% d. Th.) erhalten.
LC-MS (Methode 2): Rt = 1.03 min MS (ESpos): m/z = 428 (M+H)+
'H-NMR (400 MHz, DMSO-de): δ = 1.39 - 1.49 (m, 2H), 1.54 - 1.68 (m, 4H), 2.36 (s, 3H), 3.27 - 3.36 (m, 2H; überlagert mit Lösungsmittelpeak), 5.54 (s, 2H), 7.17 - 7.25 (m, 2H), 7.53 - 7.63 (m, 1H), 8.09 (t, 1H), 8,36 (s, 1H), [weitere Signale unter Lösungsmittelpeak].
In Analogie zu Beispiel 49 wurden die in Tabelle 3 gezeigten Beispielverbindungen hergestellt, indem 8-[(2,6-Difluorbenzyl)oxy]-2,6-dimethylimidazo[l,2-a]pyrazin-3-carbonsäure aus Beispiel 3A mit den entsprechenden, kommerziell erhältlichen oder zuvor beschriebenen Aminen [Hydrazine, Hydrazide, Hydroxylamine] (1.1 - 5 Äquivalente), HATU (1.1 - 4.5 Äquivalente) und NN-Diisopropylethylamin (3 - 12 Äquivalente) unter den beschriebenen Reaktionsbedingungen (Reaktionszeit: 0.5 - 48 h; Temperatur: 0°C - RT, -20°C, RT oder 60°C) umgesetzt wurden.
Beispielhafte Aufarbeitung der Reaktionsmischung:
Die Reaktionslösung wurde mit Wasser versetzt und der ausgefallene Feststoff ca. 30 min bei Raumtem eratur verrührt. Anschließend wurde der Feststoff abfiltriert, gut mit Wasser gewaschen und im Hochvakuum getrocknet.
Alternativ dazu wurde das Reaktionsgemisch mit Wasser/TFA verdünnt und mittels präparativer HPLC gereinigt (RP18 Säule, Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% TFA oder 0.05%) Ameisensäure). Das Rohprodukt wurde ggf. zusätzlich oder alternativ mittels Kieselgelchromatographie (Laufmittel: Dichlormethan/Methanol oder Cyclohexan/Ethylacetat) und/oder Dickschichtchromatographie (Laufmittel: Dichlormethan/Methanol) gereinigt.
Die produkthaltigen Fraktionen der präparativen HPLC wurden ggf. eingedampft, der Rückstand in Dichlormethan aufgenommen und mit gesättigter, wässriger Natriumhydrogencarbonatlösung gewaschen. Die wässrige Phase wurde zweimal mit Dichlormethan extrahiert, die vereinigten organischen Phasen über Natriumsulfat getrocknet, filtriert, eingedampft und lyophilisiert.
Tabelle 3 :
B. Bewertung der pharmakologischen Wirksamkeit
Es werden die folgenden Abkürzungen verwendet:
ATP Adenosintriphosphat
Brij35 Polyoxyethylen(23)laurylether
BSA Rinderserumalbumin
DTT Dithiothreitol
TEA Triethanolamin
Die pharmakologische Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
B-l. Vermessung von sGC Enzymaktivität mittels PPi Nachweis
Lösliche Guanylylcyclase (sGC) setzt unter Stimulation GTP zu cGMP und Pyrophosphat (PPi) um. PPi wird mit Hilfe des in WO 2008/061626 beschriebenen Verfahrens nachgewiesen. Das im Test entstehende Signal nimmt mit fortschreitender Umsetzung zu und dient als Maß für die sGC- Enzymaktivität. Mit Hilfe einer PPi Referenzkurve kann das Enzym in bekannter Weise charakterisiert werden, z.B. hinsichtlich Umsatzrate, Stimulierbarkeit oder Michaelis Konstante.
Durchführung des Tests
Zur Durchführung des Tests wurden 29 μΕ Enzymlösung (0-10 nM lösliche Guanylylcyclase (hergestellt nach Hönicka et al., Journal of Molecular Medicine 77(1999)14-23), in 50 mM TEA, 2 mM Magnesiumchlorid, 0.1% BSA (FraktionV), 0.005% Brij 35, pH 7.5) in die Mikroplatte vorgelegt und 1 μΕ der Stimulatorlösung (0-10 μΜ 3-Morpholinosydnonimine, SIN-1, Merck in DMSO) hinzugegeben. Es wurde 10 min bei RT inkubiert. Anschließend wurden 20 μΐ Detektionsmix (1,2 nM Firefly Luciferase {Photinus pyralis Luziferase, Promega), 29 μΜ Dehydro-Luziferin (hergestellt nach Bitler & McElroy, Arch. Biochem. Biophys. 72 (1957) 358), 122 μΜ Luziferin (Promega), 153 μΜ ATP (Sigma) und 0,4 mM DTT ( Sigma) in 50 mM TEA, 2 mM Magnesiumchlorid, 0.1% BSA (Fraktion V), 0.005% Brij 35, pH 7,5) zugegeben. Die Enzymreaktion wurde durch Zugabe von 20 μΐ Substratlösung (1.25 mM Guanosin-5'-triphosphat (Sigma) in 50 mM TEA, 2 mM Magnesiumchlorid, 0.1% BSA (Fraktion V), 0.005% Brij 35, pH 7.5) gestartet und kontinuierlich luminometrisch vermessen.
B-2. Wirkung an rekombinanter Guanylatcyclase-Reporterzelllinie
Die zelluläre Wirkung der erfindungsgemäßen Verbindungen wird an einer rekombinanten Guanylatcyclase-Reporterzelllinie, wie in F. Wunder et al., Anal. Biochem. 339, 104-112 (2005) beschrieben, bestimmt.
Repräsentative MEC-Werte (MEC = minimal effektive Konzentration) für die erfindungsgemäßen Verbindungen sind in der nachstehenden Tabelle wiedergegeben (zum Teil als Mittelwerte aus Einzelbestimmungen) :
Tabelle A:
Beispiel MEC [μΜ] Beispiel MEC [μΜ]
1 0.77 26 3
2 1 27 1
3 3 28 1
4 1 29 1
5 1 30 3
6 3 31 3
7 0.3 32 1
8 1 33 3
9 3 34 3
10 0.3 35 3
11 3 36 1
12 1 37 0.3
13 3 38 0.3
14 3 39 0.2
15 1 40 0.3
16 3 41 0.65
17 10 42 0.3
18 3 43 1
19 3 44 10
20 3 45 10
21 3 46 10
22 3 47 1.0
23 1 48 3.0
24 3 49 1.0
25 3 50 1.0
Beispiel MEC [μΜ] Beispiel MEC [μΜ]
51 3.0 53 3.0
52 0.3 54 1.0
B-3. Gefäßrelaxierende Wirkung in vitro
Kaninchen werden durch Nackenschlag betäubt und entblutet. Die Aorta wird entnommen, von anhaftendem Gewebe befreit, in 1.5 mm breite Ringe geteilt und einzeln unter einer Vorspannung in 5 ml-Organbäder mit 37°C warmer, Carbogen-begaster Krebs-Henseleit-Lösung folgender Zusammensetzung gebracht (jeweils mM): Natriumchlorid: 119; Kaliumchlorid: 4.8; Calciumchlorid- Dihydrat: 1 ; Magnesiumsulfat-Heptahydrat: 1.4; Kaliumdihydrogenphosphat: 1.2; Natriumhydrogencarbonat: 25; Glucose: 10. Die Kontraktionskraft wird mit Statham UC2-Zellen erfasst, verstärkt und über A/D-Wandler (DAS- 1802 HC, Keithley Instruments München) digitalisiert sowie parallel auf Linienschreiber registriert. Zur Erzeugung einer Kontraktion wird Phenylephrin dem Bad kumulativ in ansteigender Konzentration zugesetzt. Nach mehreren Kontrollzyklen wird die zu untersuchende Substanz in jedem weiteren Durchgang in jeweils steigender Dosierung zugesetzt und die Höhe der Kontraktion mit der Höhe der im letzten Vordurchgang erreichten Kontraktion verglichen. Daraus wird die Konzentration errechnet, die erforderlich ist, um die Höhe des Kontrollwertes um 50% zu reduzieren (ICso-Wert). Das Standardapplikationsvolumen beträgt 5 μΐ, der DMSO-Anteil in der Badlösung entspricht 0.1%.
B-4. Blutdruckmessung an narkotisierten Ratten
Männliche Wistar-Ratten mit einem Körpergewicht von 300 - 350 g werden mit Thiopental (100 mg/kg i.p.) anästhesiert. Nach der Tracheotomie wird in die Femoralarterie ein Katheter zur Blutdruckmessung eingeführt. Die zu prüfenden Substanzen werden als Lösungen entweder oral mittels Schlundsonde oder über die Femoralvene intravenös verabreicht (Stasch et al. Br. J. Pharmacol. 2002; 135: 344-355).
B-5. Radiotelemetrische Blutdruckmessung an wachen, spontan hypertensiven Ratten
Für die im Folgenden beschriebene Blutdruckmessung an wachen Ratten wird ein im Handel erhältliches Telemetriesystem der Firma DATA SCIENCES INTERNATIONAL DSI, USA eingesetzt.
Das System besteht aus 3 Hauptkomponenten: Implantierbare Sender (Physiotel® Telemetrietransmitter)
Empfänger (Physiotel® Receiver), die über einen Multiplexer (DSI Data Exchange Matrix ) mit einem
Datenakquisitionscomputer verbunden sind.
Die Telemetrieanlage ermöglicht eine kontinuierliche Erfassung von Blutdruck Herzfrequenz und Körperbewegung an wachen Tieren in ihrem gewohnten Lebensraum.
Tiermaterial
Die Untersuchungen werden an ausgewachsenen weiblichen spontan hypertensiven Ratten (SHR Okamoto) mit einem Körpergewicht von >200 g durchgeführt. SHR/NCrl von Okamoto Kyoto School of Medicine, 1963 wurden aus männlichen Wistar Kyoto Ratten mit stark erhöhtem Blutdruck und weiblichen mit leicht erhöhtem Blutdruck gekreuzt und in der Fl 3 an die U.S. National Institutes of Health abgegeben.
Die Versuchstiere werden nach Senderimplantation einzeln in Makroion - Käfigen Typ 3 gehalten. Sie haben freien Zugang zu Standardfutter und Wasser.
Der Tag - Nacht - Rhythmus im Versuchslabor wird per Raumbeleuchtung um 6:00 Uhr morgens und um 19:00 Uhr abends gewechselt.
Senderimplantation
Die eingesetzten Telemetriesender TAH PA - C40 werden den Versuchstieren mindestens 14 Tage vor dem ersten Versuchseinsatz unter aseptischen Bedingungen chirurgisch implantiert. Die so instrumentierten Tiere sind nach Abheilen der Wunde und Einwachsen des Implantats wiederholt einsetzbar.
Zur Implantation werden die nüchternen Tiere mit Pentobabital (Nembutal, Sanofi: 50mg/kg i.p. ) narkotisiert und an der Bauchseite weiträumig rasiert und desinfiziert. Nach Eröffnung des Bauchraumes entlang der Linea alba wird der flüssigkeitsgefüllte Meßkatheter des Systems oberhalb der Bifurcation nach cranial in die Aorta descendens eingesetzt und mit Gewebekleber (VetBonD TM, 3M) befestigt. Das Sendergehäuse wird intraperitoneal an der Bauchwandmuskulatur fixiert und die Wunde wird schichtweise verschlossen.
Postoperativ wird zur Infektionsprophylaxe ein Antibiotikum verabreicht (Tardomyocel COMP Bayer 1ml/kg s.c.)
Substanzen und Lösungen
Wenn nicht anders beschrieben werden die zu untersuchenden Substanzen jeweils einer Gruppe von Tieren (n = 6 ) per Schlundsonde oral verabreicht. Entsprechend einem Applikationsvolumen von 5 ml/kg Körpergewicht werden die Testsubstanzen in geeigneten Lösungsmittelgemischen gelöst oder in 0.5% iger Tylose suspendiert. Eine Lösungsmittel- behandelte Gruppe von Tieren wird als Kontrolle eingesetzt.
Versuchsablauf
Die vorhandene Telemetrie - Meßeinrichtung ist für 24 Tiere konfiguriert. Jeder Versuch wird unter einer Versuchsnummer registiert (VJahr Monat Tag).
Den in der Anlage lebenden instrumentierten Ratten ist jeweils eine eigene Empfangsantenne zugeordnet (1010 Receiver, DSI ).
Die implantierten Sender sind über einen eingebauten Magnetschalter von außen aktivierbar. Sie werden bei Versuchsvorlauf auf Sendung geschaltet. Die ausgestrahlten Signale können durch ein Datenakquisitionssystem (Dataquest TM A.R.T. for WINDOWS, DSI ) online erfasst und entsprechend aufgearbeitet werden. Die Ablage der Daten erfolgt jeweils in einem hierfür eröffneten Ordner der die Versuchsnummer trägt.
Im Standardablauf werden über je 10 Sekunden Dauer gemessen
Systolischer Blutdruck (SBP)
Diastolischer Blutdruck (DBP)
Arterieller Mitteldruck (MAP) Herzfrequenz (HR)
Aktivität (ACT)
Die Messwerterfassung wird rechnergesteuert in 5 Minuten Abständen wiederholt. Die als Absolutwert erhobenen Quelldaten werden im Diagramm mit dem aktuell gemessenen Barometerdruck (Ambient Pressure Reference Monitor; APR-1) korrigiert und in Einzeldaten abgelegt. Weitere technische Details sind der umfangreichen Dokumentation der Herstellerfirma (DSI) zu entnehmen.
Wenn nicht anders beschrieben erfolgt die Verabreichung der Prüfsubstanzen am Versuchstag um 9.00 Uhr. Im Anschluss an die Applikation werden die oben beschriebenen Parameter 24 Stunden gemessen.
Auswertung
Nach Versuchsende werden die erhobenen Einzeldaten mit der Analysis-Software (DATAQUEST TM A. R.T. TM ANALYSIS) sortiert. Als Leerwert werden hier 2 Stunden vor Applikation angenommen, so dass der selektierte Datensatz den Zeitraum von 7:00 Uhr am Versuchstag bis 9:00 Uhr am Folgetag umfasst.
Die Daten werden über eine voreinstellbare Zeit durch Mittelwertbestimmung geglättet (15 Minuten Average) und als Textdatei auf einen Datenträger übertragen. Die so vorsortierten und komprimierten Messwerte werden in Excel- Vorlagen übertragen und tabellarisch dargestellt. Die Ablage der erhobenen Daten erfolgt pro Versuchstag in einem eigenen Ordner, der die Versuchsnummer trägt. Ergebnisse und Versuchsprotokolle werden in Papierform nach Nummern sortiert in Ordnern abgelegt.
Literatur:
Klaus Witte, Kai Hu, Johanna Swiatek, Claudia Müssig, Georg Ertl and Björn Lemmer: Experimental heart failure in rats: effects on cardiovascular circadian rhythms and on myocardial ß-adrenergic signaling. Cardiovasc Res 47 (2): 203-405, 2000; Kozo Okamoto: Spontaneous hypertension in rats. Int Rev Exp Pathol 7: 227- 270, 1969; Maarten van den Buuse: Circadian Rhythms of Blood Pressure, Heart Rate, and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio-Telemetry. Physiology & Behavior 55(4): 783-787, 1994.
B-6. Bestimmung pharmakokinetischer Kenngrößen nach intravenöser und oraler Gabe Die pharmakokinetischen Parameter der erfindungsgemäßen Verbindungen werden in männlichen CD- 1 -Mäusen, männlichen Wistar-Ratten und weiblichen Beagle-Hunden bestimmt. Die intravenöse Gabe erfolgt bei Mäusen und Ratten mittels einer speziesspezifischen Plasma/DMSO- Formulierung und bei Hunden mittels einer Wasser/PEG400/Ethanol-Formulierung. Die orale Gabe der gelösten Substanz mittels Schlundsonde wird in allen Spezies basierend auf einer Wasser/PEG400/Ethanol-Formulierung durchgeführt. Den Ratten wird zur vereinfachten Blutabnahme vor der Substanzgabe ein Silikonkatheter in die rechte Vena jugularis externa gelegt. Die Operation erfolgt mindestens einen Tag vor dem Versuch unter Isofluran-Narkose und unter Gabe eines Analgetikums (Atropin/Rimadyl (3/1) 0.1 mL s.c). Die Blutabnahme (in der Regel mehr als 10 Zeitpunkte) erfolgt in einem Zeitfenster, welches terminale Zeitpunkte von mindestens 24 bis maximal 72 Stunden nach Substanzgabe beinhaltet. Das Blut wird bei der Entnahme in heparinisierte Röhrchen geleitet. So dann wird mittels Zentrifugation das Blutplasma gewonnen und gegebenenfalls bis zur weiteren Bearbeitung bei -20°C gelagert.
Den Proben der erfindungsgemäßen Verbindungen, Kalibrierproben und Qualifier wird ein interner Standard zugesetzt (dies kann auch eine chemisch nicht verwandte Substanz sein) und es folgt eine Proteinfällung mittels Acetonitril im Überschuss. Nach Zugabe einer Puffer-Lösung, die an die LC- Bedingungen angepasst ist, und folgendem Vortexen wird bei 1000 g zentrifugiert. Der Überstand wird mittels LC-MS/MS unter Verwendung von C18-reversed-phase-Säulen und variablen Eluenten-Gemischen vermessen. Die Quantifizierung der Substanzen erfolgt anhand der Peakhöhen oder -flächen aus extrahierten Ionenchromatogrammen spezifischer selected ion monitoring- Experimente.
Aus den ermittelten Plasmakonzentration-Zeit- Verläufen werden die pharmakokinetischen Kenngrößen wie AUC, Cmax, ti 2 (terminale Halbwertszeit), F (Bioverfügbarkeit), MRT (Mean Residence Time) und CL (Clearance) mittels eines validierten pharmakokinetischen Rechenprogramms berechnet.
Da die Substanzquantifizierung in Plasma durchgeführt wird, muss die Blut/Plasma- Verteilung der Substanz bestimmt werden, um die pharmakokinetischen Parameter entsprechend anpassen zu können. Dazu wird eine definierte Menge Substanz in heparinisiertem Vollblut der entsprechenden Spezies für 20 min im Taumelrollenmischer inkubiert. Nach Zentrifugation bei 1000g wird die Konzentration im Plasma gemessen (mittels LC-MS/MS; s.o.) und durch Quotientenbildung der CBiut/Cpksma-Wert ermittelt.
B-7. Metabolismus-Untersuchung Zur Bestimmung des Metabolismus-Profils der erfindungsgemäßen Verbindungen werden diese mit rekombinanten humanen Cytochrom P450 (CYP) Enzymen, Lebermikrosomen oder mit primären frischen Hepatozyten verschiedener Tierspezies (z.B. Ratte, Hund) als auch humanen Ursprungs inkubiert, um Informationen über einen möglichst kompletten hepatischen Phase I- und Phase II-Metabolismus sowie über die am Metabolismus beteiligten Enzyme zu erhalten und zu vergleichen.
Die erfindungsgemäßen Verbindungen wurden mit einer Konzentration von etwa 0.1-10 μΜ inkubiert. Dazu wurden Stammlösungen der erfindungsgemäßen Verbindungen mit einer Konzentration von 0.01-1 mM in Acetonitril hergestellt, und dann mit einer 1 :100 Verdünnung in den Inkubationsansatz pipettiert. Die Lebermikrosomen und rekombinanten Enzyme wurden in 50 mM Kaliumphosphatpuffer pH 7.4 mit und ohne NADPH-generierendem System, bestehend aus 1 mM NADP+, 10 mM Glucose-6-phosphat und 1 Unit Glucose-6-phosphat Dehydrogenase, bei 37°C inkubiert. Primäre Hepatozyten wurden in Suspension in Williams E Medium ebenfalls bei 37°C inkubiert. Nach einer Inkubationszeit von 0 - 4h wurden die Inkubationsansätze mit Acetonitril abgestoppt (Endkonzentration ca. 30%) und das Protein bei ca. 15000 x g
„ Λ
- 184 - abzentrifugiert. Die so abgestoppten Proben wurden entweder direkt analysiert oder bis zur Analyse bei -20°C gelagert.
Die Analyse erfolgt mittels Hochleistungsflüssigkeits-Chromatographie mit Ultraviolett- und massenspektrometrischer Detektion (HPLC-UV-MS/MS). Dazu werden die Überstände der Inku- bationsproben mit geeigneten C18-reversed-phase-Säulen und variablen Eluenten-Gemischen aus Acetonitril und 10 mM wässriger Ammoniumformiat-Lösung oder 0.05 % Ameisensäure chromatographiert. Die UV-Chromatogramme in Verbindung mit massenspektrometrischen Daten dienen zur Identifizierung, Strukturaufklärung und quantitativen Abschätzung der Metabolite, und der quantitativen metabolischen Abnahme der erfindungsgemäßen Verbindung in den Inkubationsansätzen.
B-8. Caco-2 Permeabilitäts-Test
Die Permeabilität einer Testsubstanz wurde mit Hilfe der Caco-2 Zelllinie, einem etablierten in vitro Modell für Permeabilitätsvorhersagen an der gastrointestinalen Barriere, bestimmt (Artursson, P. and Karlsson, J. (1991). Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys.175 (3), 880-885). Die Caco-2 Zellen (ACC No. 169, DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Deutschland) wurden in 24-Well Platen mit Einsatz ausgesät und 14 bis 16 Tage kultiviert. Für die Permeabilitätsstudien wurde die Testsubstanz in DMSO gelöst und mit Transportpuffer (Hanks Buffered Salt Solution, Gibco/Invitrogen, mit 19.9 mM Glukose und 9.8 mM HEPES) auf die finale Testkonzentration verdünnt. Um die Permeabilität von apikal nach basolateral (PappA-B) der Testsubstanz zu bestimmen, wurde die Lösung mit der Testsubstanz auf die apikale Seite des Caco-2 Zellmonolayers gegeben und Transportpuffer auf die basolaterale Seite. Um die Permeabilität von basolateral nach apikal (PappB-A) der Testsubstanz zu bestimmen, wurde die Lösung mit der Testsubstanz auf die basolaterale Seite des Caco-2 Zellmonolayers gegeben und Transportpuffer auf die apikale Seite. Zu Beginn des Experiments wurden Proben aus dem jeweiligen Donor-Kompartiment genommen, um die Massenbilanz sicher zu stellen. Nach einer Inkubation von zwei Stunden bei 37° C wurden Proben aus beiden Kompartimenten genommen. Die Proben wurden mittels LC-MS/MS analysiert und die apparenten Permeabilitätskoeffizienten (PapP) berechnet. Die Permeabilität von Lucifer Yellow wurde für jeden Zellmonolayer bestimmt, um die Integrität der Zellschicht sicher zu stellen. Die Permeabilität von Atenolol (Marker für niedrige Permeabilität) und Sulfasalazin (Marker für aktive Exkretion) wurde in jedem Testlauf als Qualitätskontrolle mitbestimmt.
B-9. hERG Kaliumstrom Assay.
Der sogenannte hERG (human ether-a-go-go related gene) Kaliumstrom trägt wesentlich zur Repolarisierung des humanen kardialen Aktionspotentials bei (Scheel et al., 2011). Eine
Inhibition dieses Stroms durch Pharmaka kann in seltenen Fällen potentiell letale Herzrhythmusstörungen zur Folge haben, und wird deshalb frühzeitig während der Arzneimittelentwicklung untersucht.
Der hier verwendete funktionelle hERG Assay basiert auf einer recombinanten HEK293 Zell- Linie, die das KCNH2(HERG)-Gen stabil exprimiert (Zhou et al., 1998). Diese Zellen werden mittels der "whole-cell voltage-clamp" Technik (Hamill et al., 1981) in einem automatisierten System (Patchliner™; Nanion, München, D) untersucht, welches die Membranspannung kontrolliert und den hERG Kalium-Strom bei Zimmertemperatur misst. Die PatchControlHT™ Software (Nanion) steuert Patchliner System, Datenerfassung und Datenanalyse. Die Spannungskontrolle erfolgt durch 2 EPC-10 quadro Verstärker unter Kontrolle der PatchMasterPro™ Software (beide: HEKA Elektronik, Lambrecht, D). NPC-16 Chips mit mittlerem Widerstand (~2 ΜΩ; Nanion) dienen als planares Substrat für die Voltage-Clamp Experimente.
NPC-16 Chips werden mit intra- und extrazellulärer Lösung (vgl. Himmel, 2007) sowie mit Zellsuspension befüllt. Nach Bildung eines Giga-Ohm-Seals und Herstellen des Ganzzell-Modus (einschliesslich mehrerer automatisierter Qualitätskontrollschritte) wird die Zellmembran auf das Haltepotential -80 mV geklemmt. Das nachfolgende Spannungsklemm-Protokoll ändert die Kommandospannung auf +20 mV (Dauer 1000 ms), -120 mV (Dauer 500 ms), und zurück zum Haltepotential -80 mV; dies wird alle 12 s wiederholt. Nach einer initialen Stabilisierungsphase (ca 5-6 Minuten) wird Testsubstanzlösung in aufsteigenden Konzentrationen (z.B. 0.1, 1, und 10 μιηοΙ/L) zupipettiert (Exposition ca 5-6 Minuten pro Konzentration), gefolgt von mehreren Auswaschschritten.
Die Amplitude des einwärtsgerichteten "Tail"-Stroms, der durch eine Potentialänderung von +20 mV auf -120 mV erzeugt wird, dient zur Quantifizierung des hERG Kaliumstroms, und wird als Funktion der Zeit dargestellt (IgorPro™ Software). Die Stromamplitude am Ende verschiedener Zeitabschnitte (z.B. Stabilisierungsphase vor Testsubstanz, erste/zweite/dritte Konzentration Testsubstanz) dient zur Erstellung einer Konzentrations-Wirkungs-Kurve, aus der die halbmaximale Hemmkonzentration IC50 der Testsubstanz errechnet wird. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pfluegers Arch 1981 ; 391 :85-100.
Himmel HM. Suitability of commonly used excipients for electrophysiological in-vitro safety pharmacology assessment of effects on hERG potassium current and on rabbit Purkinje fiber action potential. J Pharmacol Toxicol Methods 2007;56:145-158.
Scheel O, Himmel H, Rascher-Eggstein G, Knott T. Introduction of a modular automated voltage- clamp platform and its correlation with manual human ether-a-go-go related gene voltage-clamp data. Assay Drag Dev Technol 2011 ;9:600-607.
Zhou ZF, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of hERG Channels stably expressed in HEK293 cells studied at physiological temperature.
Biophys J 1998;74:230-241.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Tablette:
Zusammensetzung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung: 1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
„
- 187 -
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung: Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt. i.v. -Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die erhaltene Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.