WO2015019658A1 - Test d'amplification d'acide nucléique - Google Patents
Test d'amplification d'acide nucléique Download PDFInfo
- Publication number
- WO2015019658A1 WO2015019658A1 PCT/JP2014/058309 JP2014058309W WO2015019658A1 WO 2015019658 A1 WO2015019658 A1 WO 2015019658A1 JP 2014058309 W JP2014058309 W JP 2014058309W WO 2015019658 A1 WO2015019658 A1 WO 2015019658A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- nucleic acid
- dna polymerase
- amino acid
- pcna
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images













Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/12—Transferases (2.) transferring phosphorus containing groups, e.g. kinases (2.7)
- C12N9/1241—Nucleotidyltransferases (2.7.7)
- C12N9/1252—DNA-directed DNA polymerase (2.7.7.7), i.e. DNA replicase
Definitions
- the present invention relates to a nucleic acid amplification method by PCR.
- the present invention can be used not only for research but also for clinical diagnosis and environmental examination.
- PCR polymerase chain reaction
- DNA denaturation by heat treatment dissociation from double-stranded DNA to single-stranded DNA
- primer annealing of primer to template single-stranded DNA
- DNA polymerase This is a method of amplifying a target nucleic acid in a sample by repeating the cycle with three steps of extension of the primer using.
- Target nucleic acids can be amplified hundreds of thousands of times even from a small amount of sample such as several copies. It has come to be used.
- PCR is a very sensitive detection method, false positives caused by carry-over of previously performed PCR amplification products become a problem. Accordingly, PCR is performed using a substrate containing dUTP instead of dTTP, uracil base is incorporated into the amplification product, and uracil-N-glycosylase (UNG) treatment is performed in the next PCR, thereby allowing contamination (carrying).
- dUTP / UDG contamination removal method has been taken (Non-Patent Document 1).
- Non-patent Document 2 PCR is easily affected by contaminants such as pigments, proteins, and sugars, and the contaminants inhibit the reaction.
- a gene in a biological sample is amplified, purification of DNA is required.
- purification of DNA is complicated and requires time, and contamination occurs during the operation. There is a risk.
- the target nucleic acid content in the sample is small, it may not be recovered. Therefore, there has been a demand for a method for amplifying a target nucleic acid by bringing a biological sample into a PCR reaction solution containing dUTP without going through a purification step.
- Patent 4395377 Special table 2006-507012
- an object of the present invention is to provide an efficient method for amplifying a target nucleic acid in PCR containing dUTP as a substrate. More specifically, the present invention provides a nucleic acid amplification method capable of performing PCR amplification even in the presence of a large amount of contaminants derived from biological samples in PCR containing dUTP as a substrate. Furthermore, the other object of this invention is to provide the reagent kit suitable for said objective. In summary, an object of the present invention is to provide a PCR improvement method and a PCR reaction reagent suitable for amplification of genes present in animal and plant tissues and body fluids in the presence of dUTP.
- the nucleic acid amplification method of the present invention for achieving the above object uses the DNA polymerase belonging to Family B to amplify DNA in a biological sample in the presence of dUTP without going through a purification step. This is a nucleic acid amplification method.
- the present inventor uses a DNA polymerase belonging to family B in a nucleic acid amplification method in which a biological sample itself and a gene amplification reaction solution are mixed and reacted, so that even if a large amount of biological contaminants exist, Found that PCR is possible even when a biological sample such as blood or oral mucosa is directly added to the PCR reaction solution, and the present invention has been achieved.
- the representative invention of the present application is as follows.
- [1] A method of amplifying a target nucleic acid in a biological sample by adding a biological sample that has not undergone a purification step to a nucleic acid amplification reaction solution, wherein the enzyme used for the amplification is a DNA polymerase belonging to Family B, and deoxyuridine (dUTP ) In the reaction solution.
- [2] The nucleic acid amplification method according to [1], wherein the biological sample is any of animal and plant tissues, body fluids, excreta, cells, bacteria, and viruses.
- nucleic acid amplification method according to [1] or [2], wherein the enzyme used for nucleic acid amplification is an archaeal DNA polymerase mutant having reduced base analog detection activity.
- nucleic acid amplification method according to [3] or [4], wherein the archaeal DNA polymerase mutant is any one of the following (a) to (c): (A) at least one amino acid among the amino acids corresponding to positions 7, 36, 37, 90 to 97 and 112 to 119 of the amino acid sequence represented by SEQ ID NO: 1 or 2 (corresponding to the wild type sequence of Pfu) It is an amino acid sequence having the following modification. (B) A reduced base analog in which at least one amino acid is further modified in the amino acid sequence shown in (a), the amino acid sequence is 80% or more identical to the amino acid sequence shown in (a), and reduced An amino acid sequence encoding a DNA polymerase having detection activity.
- At least one amino acid is further modified in the amino acid sequence represented by (a), and one or several amino acids are deleted, substituted or added in the amino acid sequence represented by (a). And an amino acid sequence encoding a DNA polymerase having reduced base analog detection activity.
- the mutant of archaebacterial DNA polymerase has at least one amino acid modification among amino acids corresponding to positions 36 and 93 in SEQ ID NO: 1 or SEQ ID NO: 2, according to any of [3] to [5] Nucleic acid amplification method.
- the archaeal DNA polymerase variant is any one of SEQ ID NO: 1 or SEQ ID NO: 2, wherein Y7A / V93K, Y7A / V93R, Y7A / V93Q, Y7A / P36H, Y7A / P36R, P36H / V93K, P36K, P36R or P36H
- the nucleic acid amplification method according to any one of [3] to [6], wherein [8] The nucleic acid amplification according to any one of [3] to [7], wherein the archaebacterial DNA polymerase mutant has any one of Y7A / V93K, Y7A / P36H, and P36H in SEQ ID NO: 1 or SEQ ID NO: 2.
- the modification to the 3′-5 ′ exonuclease active region of DNA polymerase belonging to family B is any one of D141A / E143A, I142R, H147E, H147D, N210D or Y311F in SEQ ID NO: 1 or SEQ ID NO: 2 [1 ]
- PCNA nucleic acid amplification method according to any one of [1] to [13], wherein PCNA is a mutant that is loaded alone into DNA.
- PCNA is (a) N-terminal region consisting of amino acids corresponding to positions 82, 84 and 109 of the amino acid sequence shown in SEQ ID NO: 21 or SEQ ID NO: 22, and (b) amino acids corresponding to positions 139, 143 and 147
- [16] PCNA modified amino acid corresponding to position 143 in SEQ ID NO: 21 or SEQ ID NO: 22 to basic amino acid, or modified from both amino acids corresponding to positions 82 and 143 to neutral amino acid, equivalent to position 147
- Nucleic acid amplification method [17] [1] A reagent for carrying out the nucleic acid amplification method according to any one of [16]. [18] [1] A kit comprising a reagent for performing the nucleic acid amplification method according to any one of [16].
- the present invention eliminates the risk of loss and carryover during DNA purification, and further reduces time and cost. Also, in order to prevent contamination by the dUTP / UDG contamination removal method, not only in the research field, but also in the clinical field or forensic field such as genetic diagnosis in which the same sample is amplified many times, or in the microbiological examination in foods and the environment, etc. Can also be widely used
- PCNA PCR enhancement factor
- mutant type in the case of “mutant PCNA” means that it has an amino acid sequence different from the conventionally known PCNA, and it is due to artificial mutation or due to mutation in nature. There is no distinction.
- the nucleic acid amplification method of the present invention is a method of amplifying a target nucleic acid in a biological sample by adding a biological sample that has not undergone a purification step to a nucleic acid amplification reaction solution, and the enzyme used for amplification is a polymerase belonging to family B. And deoxyuridine (dUTP) is contained in the reaction solution.
- dUTP deoxyuridine
- nucleic acid in a biological sample that has not undergone a purification step is amplified without purification.
- Purification is a method for separating contaminants such as tissues and cell walls of biological samples from DNA in biological samples. Methods for separating DNA using phenol or phenol / chloroform, ion exchange resins, glass filters, etc. Alternatively, there is a method of separating DNA with a reagent having a protein aggregation action.
- nucleic acids to be amplified such as organs and cells are present in the tissue of the sample
- an act of destroying the tissue to extract the nucleic acid does not correspond to the purification referred to in the present invention.
- the act of diluting the sample obtained by the above method or the biological sample with a buffer solution does not correspond to the purification referred to in the present invention.
- the nucleic acid amplification method in the present invention is a method in which a biological sample is added to a nucleic acid amplification reaction solution and amplified without taking these purification methods.
- the “biological sample that has not undergone the purification step” means that the biological sample itself, or a liquid biological sample diluted with a solvent such as water, or a solid biological sample is added to a solvent such as water and heated. However, it is not limited to these as long as it has not undergone a purification process.
- blood resistance means that even when blood is present in a nucleic acid amplification reaction solution, it resists inhibition.
- the biological sample applied to the nucleic acid amplification method of the present invention is not particularly limited as long as it is a sample collected from a living body.
- it refers to animal and plant tissues, body fluids, excreta, cells, bacteria, viruses and the like.
- Body fluid includes blood and saliva, and cells include, but are not limited to, leukocytes separated from blood.
- the reaction solution contains dUTP.
- the route for obtaining dUTP is not particularly limited, but commercially available products can be used.
- the concentration of dUTP in the reaction solution is not particularly limited, but the preferable lower limit is 0.5 ⁇ M or more, more preferably 50 ⁇ M or more, and more preferably 0.1 mM or more from the viewpoint of the removal efficiency of contamination. From the viewpoint of PCR efficiency, dUTP may be contained at a high concentration.
- a preferable upper limit is 1 mM or less, more preferably 0.6 mM or less.
- dTTP and dUTP may be mixed from the viewpoint of PCR efficiency.
- the ratio of dTTP to dUTP is preferably 100: 1 to 1: 100. More preferably, it is 10: 1 to 1:10, and further preferably 1: 1, but is not limited thereto.
- the nucleic acid applied to the nucleic acid amplification method of the present invention is not limited by the difference in length, sequence, GC content, etc., but since dUTP is incorporated during amplification, the nucleic acid must contain T. It is preferable that 10 or more T is contained.
- the DNA polymerase used in the nucleic acid amplification method of the present invention is a DNA polymerase belonging to family B.
- the DNA polymerase belonging to the family B is preferably an archaea-derived DNA polymerase.
- DNA polymerases derived from archaea belonging to family B include DNA polymerases isolated from bacteria of the genera Pyrococcus and Thermococcus.
- DNA polymerase derived from the genus Pyrococcus include Pyrococcus furiosus and Pyrococcus sp. Including, but not limited to, DNA polymerases isolated from GB-D, Pyrococcus Wosei, Pyrococcus abyssi, Pyrococcus horikoshii.
- DNA polymerases derived from the genus Thermococcus include Thermococcus kodakaraensis, Thermococcus gorgonarius, Thermococcus litoralis, Thermococcus sp. JDF-3, Thermococcus sp. 9 degrees North-7 (Thermococcus sp. 9 ° N-7), Thermococcus sp. Including, but not limited to, DNA polymerase isolated from KS-1, Thermococcus celler, or Thermococcus sicili.
- PCR enzymes using these DNA polymerases are commercially available, Pfu (Staragene), KOD (Toyobo), Pfx (Life Technologies), Vent (New England Biolabs), Deep Vent (New England) , Tgo (Roche), Pwo (Roche).
- KOD DNA polymerase excellent in extensibility and thermal stability is preferable.
- the DNA polymerase belonging to family B used in the nucleic acid amplification method of the present invention may be a mutant having reduced base analog detection activity.
- Base analogs refer to bases other than adenine, cytosine, guanine, and thymine, and include uracil and inosine.
- a DNA polymerase belonging to Family B binds strongly when a base analog such as uracil or inosine is detected, and inhibits the polymerase function.
- the base analog detection activity refers to an activity that strongly binds to a base analog and inhibits the polymerase function.
- a DNA polymerase mutant belonging to family B having reduced base analog detection activity is a DNA polymerase mutant belonging to family B characterized by low binding ability to uracil and inosine.
- An example of such a mutant having a decreased base analog detection activity of a DNA polymerase belonging to Family B is an archaeal DNA polymerase mutant.
- the amino acid sequence (uracil-binding pocket) relating to the binding of uracil formed by amino acids 1 to 40 and amino acids 78 to 130 is modified, and compared to wild-type DNA polymerase, uracil and inosine An archaeal DNA polymerase variant characterized by low binding ability.
- the DNA polymerase mutant having reduced base analog detection activity used in the nucleic acid amplification method of the present invention preferably has a higher DNA synthesis rate than the wild type (WT).
- the wild-type polymerase belonging to family B has high accuracy. This is to amplify while confirming whether the incorporated base is correct. Examples of bases that cause errors include uracil that is generated by thermal decomposition of cytosine and paired with alanine. Polymerases belonging to Family B have a pocket that binds strongly to uracil, and amplify the nucleic acid to be amplified while checking for the presence of this uracil one by one.
- this pocket interacts strongly with uracil and slows (or stops) the reaction. This interaction is thought to extend to bases such as cytosine that are somewhat similar in structure, and thus the DNA synthesis rate may be suppressed.
- the DNA polymerase mutant having reduced base analog detection activity used in the nucleic acid amplification method of the present invention is modified to the uracil-binding pocket, it will not interact with not only uracil but also cytosine, etc. Conceivable.
- the extent to which the synthesis rate is increased by modifying the pocket that strongly binds to uracil is higher when the mutant that modified the exo region is further modified than when the wild type is modified.
- the DNA polymerase belonging to the family B or the DNA polymerase derived from archaea desirably has a DNA synthesis rate of 30 bases / second or more by the following measurement method.
- PCR is inhibited by strong binding between dUTP and polymerase.
- a polymerase having a higher DNA synthesis rate is less likely to cause dUTP binding and is less susceptible to dUTP in the presence of dUTP.
- the synthesis rate of the DNA polymerase used in the nucleic acid amplification method is measured as follows. If the enzyme activity is strong, samples should be stored in storage buffer (50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM dithiothreitol, 0.1% Tween 20, 0.1% Nonidet P40, 50% glycerin). Dilute and measure. (1) Mix 2.5 ⁇ l of the following A liquid, 2.5 ⁇ l of B liquid, 1.5 ⁇ l of C liquid, 4.8 ⁇ l of D liquid, and 4 ⁇ l of E liquid, 10 minutes at 95 ° C., 10 minutes at 37 ° C. Place and create substrate.
- storage buffer 50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM dithiothreitol, 0.1% Tween 20, 0.1% Nonidet P40, 50% glycerin.
- the DNA synthesis rate can be further increased by adding a PCR enhancing factor (for example, PCNA) described later.
- a PCR enhancing factor for example, PCNA
- a DNA polymerase having a DNA synthesis rate of 50 bases / second or more is preferable, but not limited thereto.
- Thermococcus gorgonarius (SEQ ID NO: 3), it is formed by amino acids 1 to 40 and amino acids 78 to 130.
- Thermococcus litoralis (SEQ ID NO: 4), it is formed by amino acids 1 to 40 and amino acids 78 to 130.
- Pyrococcus sp. GB-D (SEQ ID NO: 5), it is formed by amino acids 1 to 40 and amino acids 78 to 130.
- Thermococcus sp. JDF-3 (SEQ ID NO: 6), it is formed by amino acids 1 to 40 and amino acids 78 to 130.
- Thermococcus sp 9 ° N-7 (SEQ ID NO: 7), it is formed by amino acids 1 to 40 and amino acids 78 to 130.
- Thermococcus sp. KS-1 (SEQ ID NO: 8), it is formed by amino acids 1 to 40 and amino acids 78 to 130.
- Thermococcus cellar (SEQ ID NO: 9), it is formed by amino acids 1-40 and amino acids 78-130.
- Thermococcus cyclis (SEQ ID NO: 10), it is formed by amino acids 1 to 40 and amino acids 78 to 130.
- DNA polymerase mutant for use in the nucleic acid amplification method of the present invention is the seventh, thirty-six, thirty-seven, thirty-nine, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-seventh, ninety-nineth, thirty-seventh, ninety-nineth, thirty-seventh, thirty-seventh, ninety-nineth, thirty-seventh, ninety-nineth, thirty-seventh, ninety-nineth, thirty-seventh, ninety-seventh, thirty-seventh, ninety-seventh, thirty-seventh, ninety-nine
- the archaeal DNA polymerase mutant may be one represented by the following amino acid sequence (b).
- BLAST Basic local alignment search tool
- NCBI National Institute of Biotechnology Information
- ncbi. nlm. nih The amino acid sequence identity is calculated by using default (initial setting) parameters in gov / BLAST /.
- the archaeal DNA polymerase mutant may be one represented by the following amino acid sequence (c).
- “several” is not limited as long as “decreased base analog detection activity” is maintained, but is, for example, a number corresponding to less than about 20% of all amino acids, preferably less than about 15%. It is a corresponding number, more preferably a number corresponding to less than about 10%, even more preferably a number corresponding to less than about 5%, and most preferably a number corresponding to less than about 1%. More specifically, the number of amino acid residues to be mutated is, for example, 2 to 160, preferably 2 to 120, more preferably 2 to 80, still more preferably 2 to 40, and even more. The number is preferably 2-5.
- amino acids corresponding to positions 7, 36, 37, 90 to 97, and 112 to 119 in the amino acid sequence shown in SEQ ID NO: 1 are amino acid sequences that are not completely identical to the amino acid sequence shown in SEQ ID NO: 1.
- a position (order) on SEQ ID NO: 1 and a corresponding position are when the primary structure of the sequence is compared (alignment) , A position corresponding to the position of SEQ ID NO: 1.
- Various methods are known as methods for comparing the primary structures of sequences. For example, it can be calculated using an analysis tool that is commercially available or available through a telecommunication line (Internet).
- the archaeal DNA polymerase mutant having reduced base analog detection activity used in the nucleic acid amplification method of the present invention is more preferably selected from amino acids corresponding to amino acids Y7, P36, or V93 in SEQ ID NO: 1 or SEQ ID NO: 2. Having at least one amino acid modification.
- Y7 means a tyrosine (Y) residue that is the seventh amino acid, and one letter of the alphabet represents an abbreviation of a commonly used amino acid.
- the Y7 amino acid has tyrosine (Y) substituted with a nonpolar amino acid, specifically selected from the group consisting of Y7A, Y7G, Y7V, Y7L, Y7I, Y7P, Y7F, Y7M, Y7W, and Y7C. Amino acid substitution.
- the P36 amino acid is a proline (P) substituted with a positively charged polar amino acid, specifically a P36H, P36K, or P36R amino acid substitution.
- the V93 amino acid is a valine (V) having a positive charge and substituted with a polar amine acid, specifically an amino acid substitution of V93H, V93K, or V93R. More preferably, it is V93K.
- the modification is at least one amino acid modification selected from the group consisting of Y7A, P36H, P36K, P36R, V93Q, V93K, and V93R. More preferably, it is P36K, P36R or P36H. More preferably, it is P36H.
- the archaeal DNA polymerase mutant having reduced base analog detection activity in the present invention modifies two or more amino acids selected from amino acids corresponding to amino acids Y7, P36, or V93 in SEQ ID NO: 1 or SEQ ID NO: 2. What you did is fine. Specific examples include Y7A / V93K, Y7A / P36H, Y7A / P36R, Y7A / V93R, Y7A / V93Q or P36H / V93K, and preferred examples include Y7A / P36H or Y7A / V93K. It is not limited to.
- Patent Document 1 or 2 any of the amino acids 7, 36, 37, 90 to 97, and 112 to 119 assumed to be directly related to the interaction with uracil is modified.
- Several archaeal DNA polymerase mutants are exemplified, but not all variants have good properties to meet the challenges of the present application, some of which have lost activity .
- the base analog detection activity in the present invention can be evaluated by PCR.
- the base analog is typically uracil.
- a dUTP solution is added at a final concentration of 0.5 ⁇ M to 200 ⁇ M to a normal PCR reaction solution containing DNA as a template, buffer material, magnesium, dNTPs, primers, and a DNA polymerase to be evaluated, and thermal cycling is performed. .
- the presence or absence of a PCR product can be confirmed by ethidium bromide-stained agarose electrophoresis, and the detection activity of uracil can be evaluated by the allowable dUTP concentration.
- a DNA polymerase having a high uracil detection activity inhibits the extension reaction by adding a little dUTP, and the PCR product cannot be confirmed.
- DNA polymerase with low uracil detection activity can confirm gene amplification by PCR without problems even when a high concentration of dUTP is added.
- An archaeal DNA polymerase mutant having reduced base analog detection activity is a result of optimal thermal cycling using any primer and DNA as a template in an enzyme optimal reaction buffer. Compared with the wild-type without DNA, it means a DNA polymerase in which the extension reaction is not inhibited even when a high concentration of dUTP is added, and the PCR product can be confirmed. However, if it is difficult to compare with the wild type, the archaeal DNA polymerase mutant that can amplify PCR even when dUTP is added at a concentration of 0.5 ⁇ M is reduced compared to the wild type. It is presumed to have the activity of detecting a base analog.
- the evaluation of the base analog detection activity in the present invention follows the following method. KOD -Plus- Ver. 2 (Toyobo) attached 10 ⁇ PCR Buffer or Pfu DNA Polymerase (Agilent) attached 10 ⁇ PCR Buffer, 1 ⁇ PCR Buffer, 1.5 mM MgSO 4 , 0.2 mM dNTPs (dATP, dTTP, dCTP, dGTP), 15 pmol of the primer according to SEQ ID NOS: 13 and 14, which amplifies about 1.3 kb, 10 ng of human genomic DNA (Roche), and 1 U of each enzyme in a 50 ⁇ l reaction solution containing dUTP ( Roche) to a final concentration of 0.5, 5, 50, 100, 200 ⁇ M.
- PCR is performed with a schedule of repeating 30 cycles of 98 ° C., 10 seconds ⁇ 65 ° C., 30 seconds ⁇ 68 ° C., 1 minute 30 seconds.
- 5 ⁇ l of the reaction solution is subjected to agarose electrophoresis, ethidium bromide staining, and an amplified DNA fragment of about 1.3 kb is confirmed under ultraviolet irradiation to evaluate whether the base analog detection activity is reduced. .
- dUTP concentration at which PCR can be amplified the lower the base analog detection activity. In this specification, this is also expressed as “dUTP resistance is higher” as the dUTP concentration at which PCR can be amplified is higher.
- the DNA polymerase used in the nucleic acid amplification method of the present invention measures the activity as follows. If the enzyme activity is strong, samples should be stored in storage buffer (50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM dithiothreitol, 0.1% Tween 20, 0.1% Nonidet P40, 50% glycerin). Dilute and measure. (1) 25 ⁇ l of the following solution A, 5 ⁇ l of solution B, 5 ⁇ l of solution C, 10 ⁇ l of sterilized water, and 5 ⁇ l of enzyme solution are added to a microtube and reacted at 75 ° C. for 10 minutes.
- storage buffer 50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM dithiothreitol, 0.1% Tween 20, 0.1% Nonidet P40, 50% glycerin.
- A 40 mM Tris-HCl buffer (pH 7.5) 16 mM magnesium chloride 15 mM dithiothreitol 100 ⁇ g / mL BSA (bovine serum albumin) B: 1.5 ⁇ g / ⁇ l activated calf thymus DNA C: 1.5 mM dNTP (250 cpm / pmol [3H] dTTP) D: 20% trichloroacetic acid (2 mM sodium pyrophosphate) E: 1 mg / mL calf thymus DNA
- the modified DNA polymerase used in the nucleic acid amplification method of the present invention may further contain at least one amino acid modification in any of the amino acids constituting the 3′-5 ′ exonuclease active region.
- the 3′-5 ′ exoase activity refers to the ability to remove the incorporated nucleotide from the 3 ′ end of the DNA polymer, and the above 3′-5 ′ exonuclease region is a DNA polymerase belonging to family B and highly DNA polymerase (SEQ ID NO: 1) derived from Thermococcus kodakaraensis, DNA polymerase (SEQ ID NO: 2) derived from Pyrococcus furiosus, DNA derived from Thermococcus gorgonarius Polymerase (SEQ ID NO: 3), DNA polymerase derived from Thermococcus litoralis (SEQ ID NO: 4), DNA polymerase derived from Pyrococcus sp.
- SEQ ID NO: 1 derived from Thermococcus kodakaraensis
- SEQ ID NO: 2 DNA polymerase derived from Pyrococcus furiosus
- SEQ ID NO: 3 DNA polymerase derived from Therm
- GB-D (SEQ ID NO: 5), derived from Thermococcus sp. JDF-3 DNA polymerase (SEQ ID NO: 6), Thermoco DNA polymerase derived from Cass SP 9 ° N-7 (SEQ ID NO: 7), DNA polymerase derived from Thermococcus sp KS-1 (SEQ ID NO: 8), DNA polymerase derived from Thermococcus cellar (SEQ ID NO: 9)
- SEQ ID NO: 10 the amino acids 137 to 147, 206 to 222, and 308 to 318 are used.
- the present invention is also applicable to DNA polymerases other than the DNA polymerase specifically presenting the sequence.
- the 3′- consisting of amino acids 137 to 147, 206 to 222, and 308 to 318 of SEQ ID NO: 1 The region corresponding to the 5 ′ exonuclease region is shown.
- amino acids corresponding to positions 137 to 147, 206 to 222, and 308 to 318 shown in SEQ ID NO: 1 are DNA polymerases having an amino acid sequence that is not completely identical to the amino acid sequence shown in SEQ ID NO: 1.
- An expression comprising amino acid sequences corresponding to positions 137 to 147, 206 to 222, and 308 to 318 of SEQ ID NO: 1.
- the modification to the 3'-5 'exonuclease region can consist of substitutions, deletions or additions. Modifications to amino acids corresponding to positions 137 to 147, 206 to 222, and 308 to 318 in SEQ ID NO: 1 are shown.
- DNA polymerase in which the 3′-5 ′ exonuclease active region is modified one in which at least one of amino acids corresponding to positions 141, 142, 143, 210, 311 in SEQ ID NO: 1 or SEQ ID NO: 2 is modified is preferable.
- These modified DNA polymerases are deficient in 3′-5 ′ exonuclease activity. More preferably, it is a DNA polymerase deficient in 3′-5 ′ exonuclease activity, wherein the amino acid modification is any one selected from D141A, E143A, D141A / E143A, I142R, N210D, or Y311F.
- 3'-5 'exonuclease activity-deficient (exo (-)) DNA polymerase includes a complete lack of activity, for example, 0.03%, 0.05% compared to the parent enzyme , 0.1%, 1%, 5%, 10%, 20%, or a maximum of 50% or less exonuclease activity.
- DNA polymerase in which the 3'-5 'exonuclease active region has been modified is one in which the amino acid corresponding to position 147 in SEQ ID NO: 1 or SEQ ID NO: 2 has been modified. More preferably, it is any one selected from H147E and H147D. These modified DNA polymerases have improved PCR efficiency while maintaining exonuclease activity.
- mutants can be considered as the modified DNA polymerase used in the nucleic acid amplification method of the present invention.
- examples of such mutants include, but are not limited to, archaeal DNA polymerase mutants having any of the following modifications (1) to (4).
- Amino acid modification introduction method A method for producing a DNA polymerase in which the 3'-5 'exonuclease active region is modified and a method for analyzing the 3'-5' exonuclease activity are known. This is disclosed in Japanese Patent No. 6946273.
- a DNA polymerase with improved PCR efficiency refers to a modified DNA polymerase in which the amount of PCR product is increased compared to the parent enzyme.
- a method for analyzing whether the amount of the PCR product is increased as compared with the parent enzyme is described in Japanese Patent No. 3891330.
- a method for modifying the DNA polymerase used in the nucleic acid amplification method of the present invention has already been established in the art. Therefore, it can modify
- a site-specific mutagenesis method based on the Inverse PCR method can be used.
- KOD-Plus-Mutageness Kit manufactured by Toyobo
- the modified DNA polymerase gene is transferred to an expression vector as necessary, and, for example, E. coli as a host is transformed with the expression vector, and then applied to an agar medium containing a drug such as ampicillin to form colonies.
- the colony is inoculated into a nutrient medium such as LB medium or 2 ⁇ YT medium and cultured at 37 ° C. for 12 to 20 hours, and then the cells are crushed and the crude enzyme solution is extracted.
- a vector derived from pBluescript is preferable. Any known method may be used as a method for crushing bacterial cells. For example, ultrasonic treatment, a physical crushing method such as French press or glass bead crushing, or a lytic enzyme such as lysozyme can be used.
- the crude enzyme solution is heat-treated at 80 ° C. for 30 minutes to inactivate the host-derived polymerase, and the DNA polymerase activity is measured.
- any method may be used as a method for obtaining purified DNA polymerase from the strain selected by the above method, for example, the following method.
- the crude enzyme solution is obtained by crushing and extraction by enzymatic or physical crushing methods.
- the obtained crude enzyme extract is heat-treated, for example, at 80 ° C. for 30 minutes, and then the DNA polymerase fraction is recovered by ammonium sulfate precipitation.
- This crude enzyme solution can be desalted by a method such as gel filtration using Sephadex G-25 (manufactured by Amersham Pharmacia Biotech). After this operation, it can be separated and purified by heparin sepharose column chromatography to obtain a purified enzyme preparation.
- the purified enzyme preparation is purified by SDS-PAGE to such an extent that it shows almost a single band.
- the nucleic acid amplification method of the present invention is a method of adding a biological sample that has not undergone a purification step to a nucleic acid amplification reaction solution to amplify a target nucleic acid in the biological sample. It is not particularly limited except that it is a DNA polymerase belonging to family B and contains deoxyuridine (dUTP) in the reaction solution.
- dUTP deoxyuridine
- a typical example of a method that can be amplified by a DNA polymerase is PCR.
- PCR DNA polymerase
- DNA is used as a template, and one primer, dNTP (deoxyribonucleotide triphosphate) is reacted.
- dNTP deoxyribonucleotide triphosphate
- a primer extension method, a sequencing method, a conventional method that does not perform temperature cycling, and a cycle sequence method are included.
- Amplification target DNA obtained from an unpurified biological sample, (A) an archaeal DNA polymerase variant having reduced base analog detection activity (b) DNA synthesis comprising a pair of primers (c) dUTP in which one primer is complementary to the DNA extension product of the other primer A substrate (deoxynucleotide triphosphate (dNTP)) and (D) mixing a buffer solution containing magnesium ions, ammonium ions and / or potassium ions, By raising or lowering the temperature of the reaction solution using a thermal cycler or the like, a specific DNA fragment is amplified by repeating thermal cycles of (1) DNA denaturation, (2) primer annealing, and (3) primer extension.
- a PCR enhancing factor (described later), BSA, a nonionic surfactant, and the like may be further used as necessary.
- an antibody having the activity of suppressing the polymerase activity and / or 3'-5 'exonuclease activity of the thermostable DNA polymerase may be used.
- the antibody include a monoclonal antibody and a polyclonal antibody. This reaction composition is particularly effective for increasing the sensitivity of PCR and reducing nonspecific amplification.
- the nucleic acid amplification reaction in the present invention may contain a PCR enhancing factor.
- the enhancement factor refers to a complex or protein having polynucleotide polymerase enhancing activity, such as PEF, dUTPase, ssb, PCNA, RFC, helicase, etc. (Hogrefe et al., 1997, Strategies 10: 93-96; and US Pat. 6,183,997 etc.).
- PCR enhancing factors also include non-protein factors such as DMSO and betaine.
- the PCR enhancing factor used in the nucleic acid amplification method of the present invention is not limited to these, but PCNA is particularly preferred.
- the PCNA used in the nucleic acid amplification method of the present invention is desirably heat-resistant to withstand the thermal cycle of PCR, and preferably remains active after PCR. More preferably, it is soluble even after heat treatment at 80 ° C. for 30 minutes, and the activity remains at 50% or more, more preferably 70% or more, more preferably 90% or more.
- the PCNA used in the nucleic acid amplification method of the present invention more preferably includes PCNA isolated from bacteria of the genus Pyrococcus and Thermococcus.
- PCNAs derived from the genus Pyrococcus include Pyrococcus furiosus, Pyrococcus sp. Including, but not limited to, PCNA isolated from GB-D, Pyrococcus Wosei, Pyrococcus abyssi, Pyrococcus horikoshii.
- PCNAs derived from the genus Thermococcus include Thermococcus kodakaraensis (SEQ ID NO: 21), Thermococcus gorgonaris, Thermococcus literalis, Thermococcus sp. JDF-3, Thermococcus sp. 9 degrees North-7 (Thermococcus sp. 9 ° N-7), Thermococcus sp. Including, but not limited to, PCNA isolated from KS-1, Thermococcus celer, or Thermococcus siculi.
- PCNA used in the nucleic acid amplification method of the present invention may be a modified methionine corresponding to position 73 of SEQ ID NO: 21 or SEQ ID NO: 22 in order to increase the expression level. More preferably, it is modified to M73L, but is not limited thereto.
- the PCNA used in the nucleic acid amplification method of the present invention may be a mutant that is loaded alone into DNA.
- PCNA usually forms a multimer and has a ring-like structure. Loading to DNA indicates that the DNA is allowed to pass inside the ring structure of the PCNA multimer, and PCNA can be loaded into DNA only in combination with a factor usually called RFC.
- Mutants that load DNA alone are those that modify the sites involved in PCNA multimer formation and destabilize multimer formation, making it easier to pass DNA into PCNA multimers without RFC. Show.
- PCNA The site where PCNA is related to multimer formation is PCNA (SEQ ID NO: 21) derived from Thermococcus kodakaraensis, and PCNA of Pyrococcus furiosus (SEQ ID NO: 22) is an N consisting of amino acids 82, 84 and 109. Examples thereof include a terminal region and a C-terminal region consisting of amino acids 139, 143 and 147. The N-terminal region is positively charged, the C-terminal region is negatively charged, and multimers are formed by interaction.
- the PCNA variant that is loaded into DNA alone is more preferably the amino acid sequence represented by SEQ ID NO: 21 or SEQ ID NO: 22, which is involved in PCNA multimer formation.
- SEQ ID NO: 21 or SEQ ID NO: 22 which is involved in PCNA multimer formation.
- C-terminal region consisting of amino acids corresponding to positions 139, 143 and 147, having at least one modification and no RFC Both include mutants that load into DNA and promote the elongation reaction of DNA polymerase.
- the amino acid corresponding to the 143rd position of SEQ ID NO: 21 is changed to a basic amino acid
- the 82nd and 143rd positions are both changed to neutral amino acids
- the 147th position is changed to a neutral amino acid, or 109 And the like, in which both the 145th and 143rd are modified to neutral amino acids.
- neutral amino acids of the present invention include glycine, alanine, valine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, proline, serine, threonine, cysteine, methionine, asparagine, and glutamine.
- alanine has the least influence on the three-dimensional structure of the peripheral site of the substitution site.
- Examples of basic amino acids include arginine, histidine and lysine as long as they are natural. Arginine is preferable.
- a sequence in which the 147th amino acid residue is replaced with alanine (D147A), the 82nd and 143rd amino acid residues are alanine.
- 109th and 143rd amino acid residues changed to alanine SEQ ID NO: 22
- R109A / D143A, or R109A / E143A in the case of SEQ ID NO: 21 and the like, but is not limited thereto.
- Whether a PCNA variant can be loaded into DNA alone can be evaluated by PCR.
- PCR For example, to a normal PCR reaction solution containing DNA as a template, buffer material, magnesium, dNTPs, primers, and DNA polymerase belonging to Family B, PCNA to be evaluated is added, PCNA is not added, and wild-type PCNA is added
- the amount of amplification with that of DNA it can be confirmed whether it can be loaded into DNA alone (whether it has amplification enhancing activity). Even if wild-type PCNA or other PCNA that cannot be loaded into DNA alone is added, the PCR amplification amount does not change, but rather the amplification amount tends to decrease.
- a mutant that can be loaded into DNA alone can obtain an amplification amount superior to that without PCNA addition or with wild-type PCNA addition.
- the evaluation of “whether the PCNA mutant can be loaded into DNA alone (whether it has amplification enhancing activity)” is based on an archaeal DNA polymerase having reduced base analog detection activity in a reaction system containing dUTP. Evaluate using mutants. Specifically, the following “method for evaluating amplification enhancing activity” is followed. "Evaluation method of amplification enhancing activity” In this specification, (I) PCR KOD -Plus- Ver.
- PCR is performed on a schedule of 98 ° C., 10 seconds ⁇ 60 ° C., 30 seconds ⁇ 68 ° C., 1 minute 30 seconds repeated 35 cycles.
- (Ii) After completion of the PCR product analysis reaction, 5 ⁇ l of the reaction solution was subjected to agarose electrophoresis, stained with ethidium bromide, and under ultraviolet irradiation, the amplified DNA fragment was compared with the one not added with PCNA alone. Whether it can be loaded into DNA (whether it has amplification enhancing activity) can be evaluated. PCNA that can be loaded into DNA alone (high amplification enhancing activity) increases the amount of amplification upon addition.
- kit Reagent for carrying out nucleic acid amplification method of the present invention kit comprises DNA polymerase belonging to family B and deoxyuridine (dUTP) in the reaction solution
- dUTP deoxyuridine
- Other configurations are not particularly limited.
- the applied nucleic acid amplification method is not particularly limited.
- examples of the reagent and kit of the present invention include the following configurations (a) to (d), but are not limited thereto.
- the reagent and kit may further use other reagents, for example, a PCR enhancing factor, BSA, a nonionic surfactant and the like, if necessary.
- a PCR enhancing factor for example, BSA, a nonionic surfactant and the like, if necessary.
- Example 1 Production of KOD mutant A plasmid containing a modified thermostable DNA polymerase gene derived from Thermococcus kodakaraensis KOD1 strain was produced.
- a DNA template used for mutagenesis a modified thermostable DNA polymerase gene (SEQ ID NO: 11) (pKOD) derived from Thermococcus kodakaraensis KOD1 strain cloned in pBluescript was used. Mutation was introduced using KOD-Plus-Mutageness Kit (manufactured by Toyobo) according to the instruction manual. The mutant was confirmed by decoding the base sequence. Escherichia coli JM109 was transformed with the obtained plasmid and used for enzyme preparation.
- Example 2 Preparation of Pfu mutant A plasmid containing a modified thermostable DNA polymerase gene derived from Pyrococcus furiosus was prepared.
- a modified thermostable DNA polymerase gene SEQ ID NO: 12
- pPfu modified thermostable DNA polymerase gene derived from Pyrococcus furiosus cloned in pBluescript was used. Mutation was introduced using KOD-Plus-Mutageness Kit (manufactured by Toyobo) according to the instruction manual. The mutant was confirmed by decoding the base sequence. Escherichia coli JM109 was transformed with the obtained plasmid and used for enzyme preparation.
- pKOD SEQ ID NO: 11 pKOD Y7A 19th to 21st TAC of SEQ ID NO: 11 is replaced with GCC pKOD P36H 106th to 108th CCC of SEQ ID NO: 11 is replaced with CAC pKOD P36K 106th to 108th CCC of SEQ ID NO: 11 is replaced with AAA pKOD P36R PCOD V93Q Replaces C277 at 106-108 in SEQ ID NO: 11 with CGT pKOD V93Q Replaces GTC at 277-279 in SEQ ID NO: 11 with CAG pKOD V93K Replaces GTC at 277-279 in SEQ ID NO: 11 with AAA pKOD V93R SEQ ID NO: 11 277 to 279th GTC was replaced with CGT pKOD P115 ⁇ 343 to 345th CCC of SEQ ID NO: 11 was deleted pKOD
- PKOD P36H / D141A / E143A 106th-108th CCC of SEQ ID NO: 11 is replaced with CAC And 42 Replace 423rd to 423rd GAC with GCC and replace 427th to 429th GAA with GCA pKOD P36R / D141A / E143A
- Replace 106th to 108th CCC of SEQ ID NO: 11 with CGT, 421st to 423rd GAC with GCC, 427 ⁇ 429th GAA is replaced with GCA pKOD Y7A / P36H / D141A / E143A 19th to 21st TAC of SEQ ID NO: 11 to GCC
- 421st to 423rd GAC to GCC 427 to 429th GAA is replaced with GCA pKOD Y7A / P36R / D141A / E143A 19th to 21st TAC of SEQ ID
- Example 3 Production of modified thermostable DNA polymerase The cells obtained in Example 1 and Example 2 were cultured as follows. First, 80 mL of TB medium (Molecular cloning 2nd edition, p.A.2) containing sterilized 100 ⁇ g / mL ampicillin was dispensed into a 500 mL Sakaguchi flask. Escherichia cultivated for 16 hours at 37 ° C. in 3 mL of LB medium (1% bactotryptone, 0.5% yeast extract, 0.5% sodium chloride; Gibco) containing 100 ⁇ g / mL ampicillin in advance in this medium. Coli JM109 (plasmid transformant) (using a test tube) was inoculated and cultured at 37 ° C.
- TB medium Molecular cloning 2nd edition, p.A.2
- LB medium 1% bactotryptone, 0.5% yeast extract, 0.5% sodium chloride; Gibco
- the bacterial cells are collected from the culture solution by centrifugation, suspended in 50 mL of disruption buffer (30 mM Tris-HCl buffer (pH 8.0), 30 mM NaCl, 0.1 mM EDTA), and then subjected to sonication. By crushing, a cell lysate was obtained. Next, the cell lysate was treated at 80 ° C. for 15 minutes, and then the insoluble fraction was removed by centrifugation. Further, nucleic acid treatment using polyethyleneimine, ammonium sulfate precipitation, and heparin sepharose chromatography were performed.
- thermostable DNA polymerase 50 mM Tris-HCl buffer (pH 8.0), 50 mM potassium chloride, 1 mM dithiothreitol, 0 1% Tween 20, 0.1% nonidet P40, 50% glycerin
- Example 3-2 Evaluation of the reduced base analog detection activity of the modified thermostable DNA polymerase The uracil detection activity was carried out according to the “Method for evaluating base analog detection activity” described in (10) above.
- FIG. 1 shows a reaction system using a total of 8 types of wild type (KOD) and 7 types of KOD variants of Y7A, P36K, P36R, Y7A / P36K, Y7A / P36R, P36H, V93Q, and Y7A / P36H.
- KOD wild type
- FIG. 1 shows a reaction system using a total of 8 types of wild type (KOD) and 7 types of KOD variants of Y7A, P36K, P36R, Y7A / P36K, Y7A / P36R, P36H, V93Q, and Y7A / P36H.
- amplification products were confirmed in the seven mutants when dUTP having a final concentration of 0.5 ⁇ M was added. Among them, amplification products were confirmed in 6 ranges of P36K, P36R, Y7A / P36K, Y7A / P36R, P36H, V93Q, and Y7A / P36H in the entire dUTP concentration range of 0.5 to 200 ⁇ M.
- FIG. 2 shows that wild type (KOD), 3′-5 ′ exonuclease deleted by mutation of N210D, KOD (KOD N210D) and I142R deleted by 3′-5 ′ exonuclease were deleted.
- KOD I142R Three types of KOD (KOD I142R) are prepared, and each of KOD and KOD N210D is added with any of the mutations of V93K, V93R, V93Q, Y7A (8 types in total), and KOD I142R of V93K, Y7A Those with any mutation were prepared (two types).
- mutations in Y7, P36 or V93 to the uracil-binding pocket tend to increase dUTP resistance levels, and further to mutants carrying 3′-5 ′ exonucleases such as H147E (exo (+))
- the dUTP resistance concentration is higher when mutation is further added to a mutant lacking 3′-5 ′ exonuclease such as N210D, I142R or D141A / E143A (exo ( ⁇ )).
- N210D, I142R or D141A / E143A exo ( ⁇ )
- Table 2 shows both amino acid mutations in the exo region and amino acid mutations related to uracil bonds. Regarding amino acid mutations in the exo region, exo (+) is a mutation that retains 3'-5 'exonuclease, including wild type, and exo (-) is missing 3'-5' exonuclease. It shows that it is a lost mutation.
- PCR For PCR, KOD-Plus-Ver. 2 (manufactured by Toyobo), 1 ⁇ PCR Buffer, and 1.5 mM MgSO 4 , dNTPs (dATP, dUTP, dCTP, dGTP) in which 2 mM dTTP is replaced with dUTP, 15 pmol primer (1.3 kbp)
- dNTPs dATP, dUTP, dCTP, dGTP
- 15 pmol primer 15 pmol primer
- PCR was performed with PCR system GeneAmp9700 (Applied Biosystem) on a schedule of 35 cycles of 3 minutes for amplification of 3.6 kbp and 4 minutes for amplification.
- Taq DNA polymerase was manufactured by Toyobo and mixed with Anti-Taq High (manufactured by Toyobo). Reaction is 1 ⁇ BlendTaq with attached Buffer, 2 mM dTTP with dUTP (dATP, dUTP, dCTP, dGTP), 10 pmol primer (same as above), 10 ng human genomic DNA (Roche), mixed with antibody 50 ⁇ l of the reaction solution containing 2.5 U of the enzyme was added at 94 ° C.
- FIG. 3 shows that the wild type (KOD), V93K, Y7A / V93K, P36R, Y7A / P36R, P36H, Y7A / P36H, P36R / V93K, Y7A / P36R / V93K, P36H / V93K, Y7A / P36H / V93K
- PCR reaction was performed on human ⁇ -globin DNAs of different lengths in a reaction system containing 0.2 mM final concentration of dUTP (Roche) instead of dTTP. This is a result of electrophoresis of the obtained product.
- dUTP dUTP
- FIG. 4 shows that any of the mutations H147E, N210D, I142R, D141A / E143A was first added to the wild type (KOD) exo region, and V93K, Y7A / V93K, P36R, Y7A / P36R, P36H, Y7A were added to each.
- / P36H to which any of the mutations (total 24 types) were prepared, and these mutants were used to lengthen the reaction system containing 0.2 mM final concentration of dUTP (Roche) instead of dTTP.
- the results are obtained by performing a PCR reaction on different human ⁇ -globin DNAs and electrophoresing the obtained products.
- 1.3 kbp is Lane 1
- 2.8 kbp is Lane 2
- 3.6 kbp is Lane 3.
- wild-type DNA polymerases having mutations such as V93K and P36H and exo ( ⁇ ) mutants N210D, I142R and D141A / E143A are mutants such as V93K and P36H.
- the amount of amplification was larger when the exo ( ⁇ ) DNA polymerase was mutated.
- mutations such as V93K and P36H are performed on wild-type DNA polymerase, and mutations such as V93K and P36H are performed on H147E which is an exo (+) mutant that improves PCR efficiency.
- the exo (+) H147E mutant resulted in a larger amount of amplification.
- the evaluation results of the KOD mutant are summarized in Table 3.
- Table 3 the 11-step evaluation for dUTP resistance indicates that the closer to 0, the stronger the base analog detection activity, and the closer to 10, the lower the base analog detection activity.
- ⁇ indicates that the signal is sufficiently amplified
- ⁇ indicates that the signal is amplified to some extent
- ⁇ indicates that the signal is not amplified.
- Example 3-4 Comparison of synthesis rate
- the synthesis rate was determined according to ⁇ DNA polymerase synthesis rate measurement method> described in (8) above.
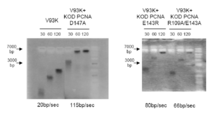
- FIG. 5 shows a total of 8 types of wild type (WT) and 7 types of KOD variants including Y7A / V93K, P36H, Y7A / P36H, Y7A / V93K / N210D, P36H / N210D, Y7A / P36H / N210D, and N210D.
- WT wild type
- “30” indicates that the PCR reaction was performed for 30 seconds.
- FIG. 5 shows that the longer the amplification product is detected at the top of the electrophoresis photograph, the longer the amplification product is taken and the faster the synthesis rate.
- 7000 bp and 3000 bp amplification products are detected at the respective arrows.
- the numbers shown at the bottom of the photograph are the synthesis rates of each DNA polymerase calculated based on the results.
- the Y7A / V93K, P36H, and Y7A / P36H mutants showed faster synthesis rates than the wild type (WT).
- the N210D mutant in which the exo region is modified has a faster synthesis rate than the wild type
- the mutant in which the N210D mutant is modified with Y7A / V93K, P36H, Y7A / P36H, etc. has a higher synthesis rate. It was confirmed to be faster.
- Example 3-5 Amplification from blood in PCR in the presence of dUTP Blood resistance was evaluated by changing the amount of blood added to the reaction solution.
- Taq DNA polymerase Taq DNA polymerase (Toyobo) mixed with KOD (wild type), KOD Y7A / V93K mutant, KOD Y7A / P36H / N210D mutant, and antibody mixed with 0.8 ⁇ g of KOD antibody per 1U.
- Anti-Taq High Toyobo
- 482 bp PCR of HBg was performed to compare the differences in amplification.
- KOD PCR was performed using KOD-Plus-Ver. 2 (Toyobo Co., Ltd.) attached Buffer, using MgSO 4, 1 ⁇ PCR Buffer, and 1.5 mM MgSO 4, 15 pmol of primer (SEQ ID NO: 19 and 20 in the amplification of 482 bp), each enzyme 1U mixed with antibody Of normal dNTPs (dATP, dTTP, dCTP, dGTP) added in 0.2 mM, and dNTPs (dATP, dUTP, dCTP, dGTP) in which dTTP is replaced with dUTP in 0.2 mM Each of these was added to be used, and blood itself and blood diluted with water were used as templates.
- dNTPs dATP, dTTP, dCTP, dGTP
- PCR system GeneAmp 9700 (Applied) with a schedule that repeats 35 cycles of 94 ° C., 2 minutes pre-reaction, 98 ° C., 10 seconds ⁇ 65 ° C., 10 seconds ⁇ 68 ° C., 1 minute per 1 bp (1 minute for 482 bp amplification) PCR was performed using Biosystem).
- Taq DNA polymerase PCR was performed by adding ordinary dNTPs (in a 50 ⁇ l reaction solution containing Buffer (Toyobo product) attached to 1 ⁇ BlendTaq, 10 pmol primer (same as above), 2.5 U enzyme mixed with antibody.
- dATP, dTTP, dCTP, dGTP added at 0.2 mM
- dNTPs with dTTP replaced by dUTP dATP, dUTP, dCTP, dGTP
- Blood itself and blood diluted with water were used.
- PCR system GeneAmp9700 (Applied Biosystem) with a schedule of 94 cycles at 94 ° C for 2 minutes followed by 35 cycles of 94 ° C, 30 seconds ⁇ 65 ° C, 30 seconds ⁇ 68 ° C, 1 minute per 1 bp (same as above) PCR was performed using After completion of the reaction, 5 ⁇ l of the reaction solution was subjected to agarose electrophoresis, stained with ethidium bromide, and the amplification amount of the amplified DNA fragment was confirmed under ultraviolet irradiation.
- FIG. 6 shows various DNA polymerases prepared by using blood as a sample and adjusting the reaction solution so that the proportion of blood in the reaction solution is 10, 5, 2, 0.2, 0.02, 0.002%.
- the result of having performed PCR reaction by and electrophoresis of the obtained product is shown.
- the DNA polymerases used are 4 types in total: Taq, KOD (wild type), and 2 types of KOD variants (Y7A / V93K, Y7A / P36H / N210D).
- the left side of each photograph uses dTTP, and the right side uses dUTP.
- Lane 1 is the case where the proportion of blood in the reaction solution is 10%, and the following 2-6 lanes are for 5%, 2%, 0.2%, 0.02% and 0.002%, respectively.
- KOD KOD
- Example 4 Preparation of KOD-PCNA mutant A plasmid containing a modified thermostable PCNA gene derived from Thermococcus kodakaraensis KOD1 strain was prepared.
- the DNA template used for mutagenesis was PCNA (SEQ ID NO: 23) (pKODPCNA) derived from Thermococcus kodakaraensis KOD1 strain cloned into pBluescript. Mutation was introduced using KOD-Plus-Mutageness Kit (manufactured by Toyobo) according to the instruction manual. The mutant was confirmed by decoding the base sequence. Escherichia coli DH5 ⁇ was transformed with the obtained plasmid and used for enzyme preparation.
- Example 4-2 Preparation of Pfu-PCNA mutant A plasmid containing a modified thermostable PCNA gene derived from Pyrococcus furiosus was prepared.
- PCNA SEQ ID NO: 24
- pPfuPCNA derived from Pyrococcus furiosus cloned in pBluescript was used. Mutation was introduced using KOD-Plus-Mutageness Kit (manufactured by Toyobo) according to the instruction manual. The mutant was confirmed by decoding the base sequence. Escherichia coli DH5 ⁇ was transformed with the obtained plasmid and used for enzyme preparation.
- pKOD PCNA SEQ ID NO: 23 pKOD PCNA M73L 217 to 219th ATG in SEQ ID NO: 23 is replaced with CTG pKOD PCNA M73L / E143A 217 to 219th ATG in SEQ ID NO: 23 is replaced with CTG 427 to 429th GAG is replaced with GCC pKOD PCNA M73L / E143F Substitution of 217-219th ATG of SEQ ID NO: 23 with CTG and 427-429th GAG with TTC pKOD PCNA M73L / E143R 217-219th ATG of SEQ ID NO: 23 with CTG, 427-429th GAG PKOD PCNA M73L / D147A pGOD PCNA M73L / R82A / E143A pKOD PCNA M73L / R82A / E143A of SEQ ID NO: 23:
- Example 4-3 Production of modified heat-resistant PCNA
- the cells obtained in Example 4-1 were cultured as follows. First, 80 mL of TB medium (Molecular cloning 2nd edition, p.A.2) containing sterilized 100 ⁇ g / mL ampicillin was dispensed into a 500 mL Sakaguchi flask. Escherichia cultivated for 16 hours at 37 ° C. in 3 mL of LB medium (1% bactotryptone, 0.5% yeast extract, 0.5% sodium chloride; Gibco) containing 100 ⁇ g / mL ampicillin in advance in this medium. E. coli DH5 ⁇ (plasmid transformant) (using a test tube) was inoculated and cultured at 37 ° C.
- TB medium Molecular cloning 2nd edition, p.A.2
- LB medium 1% bactotryptone, 0.5% yeast extract, 0.5% sodium chloride; Gibco
- the bacterial cells are collected from the culture solution by centrifugation, suspended in 50 mL of disruption buffer (30 mM Tris-HCl buffer (pH 8.0), 30 mM NaCl, 0.1 mM EDTA), and then subjected to sonication. By crushing, a cell lysate was obtained. Next, the cell lysate was treated at 80 ° C. for 15 minutes, and then the insoluble fraction was removed by centrifugation. Furthermore, the nucleic acid treatment using polyethyleneimine, ammonium sulfate precipitation, and Q sepharose chromatography were performed.
- a storage buffer 50 mM Tris-HCl buffer (pH 8.0), 50 mM potassium chloride, 1 mM dithiothreitol, 0 .1% Tween 20, 0.1% nonidet P40, 50% glycerin
- a storage buffer 50 mM Tris-HCl buffer (pH 8.0), 50 mM potassium chloride, 1 mM dithiothreitol, 0 .1% Tween 20, 0.1% nonidet P40, 50% glycerin
- PCNA PCR enhancement factor
- FIG. 7 shows the results of electrophoresis of the products obtained by adding 250 ng of various PCNA mutants and performing PCR reaction.
- the PCNA mutants used were M73L, M73L / E143R, M73L / E143A, M73L / R109A / E143A, M73L / D147A, M73L / R82A / E143A, and M73L / E143F.
- no PCNA was added
- the KOD-PCNA M73L mutant was used as the PCNA (lane 1)
- only a small band was found, but other KOD PCNA mutants were not detected.
- the amount of amplification increased and a clearer band was confirmed.
- PCNA forms a multimer and promotes a nucleic acid synthesis reaction. Usually, however, the reaction cannot proceed without loading into DNA without the action of RFC. M73L / E143R, M73L / E143A, M73L / R109A / E143A, M73L / D147A, M73L / R82A / E143A, and M73L / E143F are changes to sites involved in multimer formation, and multimer formation is moderately weakened. Therefore, it is conceivable that PCNA could be loaded onto DNA and the amount of PCR amplification was improved (FIG. 7). PCNA has also been reported to increase the synthesis rate and shorten the elongation time. However, the improvement in the synthesis rate shortens the time for the interaction between uracil and the binding pocket, and weakens the detection activity of uracil. It is possible.
- Example 4-5 Comparison of synthesis rate by adding PCNA
- KOD V93K was diluted with a storage buffer (50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM dithiothreitol, 0.1% Tween 20, 0.1% Nonidet P40, 50% glycerin).
- a storage buffer 50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM dithiothreitol, 0.1% Tween 20, 0.1% Nonidet P40, 50% glycerin.
- 250 ng of PCNA mutant was added to each reaction system, and the synthesis rate was measured. Reagents and methods were performed according to ⁇ DNA polymerase synthesis rate measurement method> described in (8) above.
- FIG. 8 shows the results of electrophoresis of products obtained by adding various PCNA mutants to the V93K mutant, performing PCR reaction for 30 seconds, 60 seconds, and 120 seconds.
- the PCNA mutant a PCNA mutant derived from KOD (D147A, E143R, R109A / E143A) was used.
- “30” indicates that the PCR reaction was performed for 30 seconds. The same applies to 60 and 120.
- FIG. 8 shows that the longer the amplified product is detected at the top of the electrophoretic photograph, the longer the amplified product is taken and the faster the synthesis rate.
- 7000 bp and 3000 bp amplification products are detected at the respective arrows.
- the numbers shown at the bottom of the photograph are the synthesis rates of each DNA polymerase calculated based on the results.
- Example 4-6 Effects of amplification from blood and addition of PCNA in PCR in the presence of dUTP PCNA was added to the evaluation of blood resistance to confirm the influence.
- Taq DNA polymerase Taq DNA polymerase (Toyobo) mixed with KOD (wild type), KOD Y7A / V93K mutant, KOD Y7A / P36H / N210D mutant, and antibody mixed with 0.8 ⁇ g of KOD antibody per 1U.
- PCA D147A mutant was added in an amount of 250 ng, and HBg 482 bp PCR was performed to compare the differences in the amplification using a mixture of equal amounts of Anti-Taq High (manufactured by Toyobo)).
- KOD PCR was performed using KOD-Plus-Ver. 2 (manufactured by Toyobo), using the attached Buffer, MgSO 4 , 50 ⁇ l containing 1 ⁇ PCR Buffer, and 1.5 mM MgSO 4 , 15 pmol primer (SEQ ID NOs: 19 and 20), 1 U of each enzyme mixed with the antibody
- 0.2 mM of normal dNTPs (dATP, dTTP, dCTP, dGTP) and dNTPs (dATP, dUTP, dCTP, dGTP) in which dTTP is replaced with dUTP are added to 0.2 mM.
- KOD-PCNA M73L / D147A mutant 250 ng of KOD-PCNA M73L / D147A mutant was added to each reaction solution.
- As the mold blood itself and blood diluted with water were used.
- PCR was performed using PCR system GeneAmp 9700 (Applied Biosystem) on a schedule of repeating 94 cycles of 94 ° C., 2 minutes and 98 cycles of 10 ° C., 10 seconds ⁇ 65 ° C., 10 seconds ⁇ 68 ° C., 1 minute.
- Taq DNA polymerase PCR was performed by adding ordinary dNTPs (in a 50 ⁇ l reaction solution containing Buffer (Toyobo product) attached to 1 ⁇ BlendTaq, 10 pmol primer (same as above), 2.5 U enzyme mixed with antibody.
- dATP, dTTP, dCTP, dGTP added at 0.2 mM
- dNTPs with dTTP replaced by dUTP dATP, dUTP, dCTP, dGTP
- Blood itself and blood diluted with water were used.
- PCR was performed using PCR system GeneAmp 9700 (Applied Biosystem) on a schedule of repeating 94 cycles of 94 ° C., 30 seconds ⁇ 65 ° C., 30 seconds ⁇ 68 ° C., 1 minute after 35 minutes at 94 ° C. After completion of the reaction, 5 ⁇ l of the reaction solution was subjected to agarose electrophoresis, stained with ethidium bromide, and the amplification amount of the amplified DNA fragment was confirmed under ultraviolet irradiation.
- FIG. 9 shows various DNA polymerases prepared by using blood as a sample and adjusting the reaction solution so that the proportion of blood in the reaction solution is 10, 5, 2, 0.2, 0.02, 0.002%.
- 2 shows the result of electrophoresis of the product obtained by performing the PCR reaction in the presence of KOD-derived PCNA mutant (D147A).
- the DNA polymerases used are 4 types in total: Taq, KOD (wild type), and 2 types of KOD variants (Y7A / V93K, Y7A / P36H / N210D).
- the left side of each photograph uses dTTP, and the right side uses dUTP.
- Lane 1 is the case where the proportion of blood in the reaction solution is 10%, and the following 2-6 lanes are for 5%, 2%, 0.2%, 0.02% and 0.002%, respectively.
- PCNA binds to a site called PIP motif of polymerase belonging to family B. Since polymerases belonging to family A such as Taq have no PIP motif, no effect was seen even when PCNA was added (FIG. 9).
- the amount of amplification of KOD Y7A / P36H / N210D to which PCNA was added and KOD Y7A / V93K to which PCNA was added increased in order of increasing synthesis rate, and the synthesis rate is also considered important for PCR in the presence of dUTP.
- KOD KOD
- PCNA is also a variant of KOD-PCNA M73L / E143R, M73L / R82A / E143A, M73L / R109A / E143A, Pfu-PCNA M73L / D143R, M73L / D147A, M73L / R82A / D143A, M73L / R109A / D143A.
- the same reaction was carried out, amplification was confirmed from the addition of 10% blood, and a solid band could be confirmed even in the presence of dUTP.
- Example 4-7 PCR from body tissues (nail, hair, oral mucosa) in the presence of dUTP
- body tissues nail, hair, oral mucosa
- PCR can be performed in the presence of dUTP using nails, hair, and oral mucosa as templates.
- a piece of nail cut with a nail clipper and one piece of hair were added to 180 ⁇ l of 50 mM NaOH, crushed by heat treatment at 95 ° C. for 10 minutes, and then neutralized by adding 20 ⁇ l of 1M Tris-HCl (pH 8.0). The supernatant was used as a template.
- the oral mucosa was obtained by suspending mucosa collected with a cotton swab in 200 ⁇ l of water as a template.
- the enzyme includes Taq DNA polymerase (Taq DNA polymerase (Toyobo), which is a mixture of KOD (wild type), KOD Y7A / V93K mutant, KOD Y7A / P36H / N210D mutant, and antibody mixed with 0.8 ⁇ g of KOD antibody per 1 U.
- Taq DNA polymerase Taq DNA polymerase (Toyobo)
- Toyobo Taq DNA polymerase
- HBg 482 bp PCR was carried out using a mixture of equal amounts of (manufactured) and Anti-Taq High (manufactured by Toyobo)) to compare the differences in amplification.
- KOD PCR was performed using KOD-Plus-Ver.
- Each reaction solution was also added with 250 ng of KOD-PCNA E143R mutant, which is a PCR enhancing factor.
- PCR was performed using PCR system GeneAmp 9700 (Applied Biosystem) on a schedule of repeating 94 cycles of 94 ° C., 2 minutes and 98 cycles of 10 ° C., 10 seconds ⁇ 65 ° C., 10 seconds ⁇ 68 ° C., 1 minute.
- Taq DNA polymerase PCR was performed by adding ordinary dNTPs (in a 50 ⁇ l reaction solution containing Buffer (Toyobo product) attached to 1 ⁇ BlendTaq, 10 pmol primer (same as above), 2.5 U enzyme mixed with antibody.
- PCR was performed using PCR system GeneAmp 9700 (Applied Biosystem) on a schedule of repeating 94 cycles of 94 ° C., 30 seconds ⁇ 65 ° C., 30 seconds ⁇ 68 ° C., 1 minute after 35 minutes at 94 ° C. After completion of the reaction, 5 ⁇ l of the reaction solution was subjected to agarose electrophoresis, stained with ethidium bromide, and the amplification amount of the amplified DNA fragment was confirmed under ultraviolet irradiation.
- FIG. 10 shows the results of electrophoresis of products obtained by performing PCR reaction with various DNA polymerases in the presence of KOD-derived PCNA mutant (E143R) using nail, hair and oral mucus as samples.
- the DNA polymerases used are 4 types in total: Taq, KOD (wild type), and 2 types of KOD variants (Y7A / V93K, Y7A / P36H / N210D).
- the left side of each photograph uses dTTP, and the right side uses dUTP.
- 1 lane is a nail sample
- 2 lane is hair
- 3 lane is oral mucosa.
- the respective “+” lanes indicate the presence of KOD-derived PCNA mutant (E143R) and “ ⁇ ” indicates no PCNA.
- KOD KOD
- Example 4-8 Amplification from plant lysate Amplification from plant lysate was performed in a PCR reaction system containing dUTP. For comparison, KOD Y7A / P36H / N210D mutant and Taq polymerase were used, and the difference in amplification amount of rbcL 1.3 kb was compared. The KOD Y7A / P36H / N210D mutant was also added with KOD-PCNA M73L / D147A added as a PCR enhancing factor.
- a rice leaf 3 mm square was added to 100 ⁇ l of Buffer A (100 mM Tris-HCl (pH 9.5), 1 M KCl, 10 mM EDTA) and heat-treated at 95 ° C. for 10 minutes, and used as a lysate.
- Buffer A 100 mM Tris-HCl (pH 9.5), 1 M KCl, 10 mM EDTA
- the PCR of KOD Y7A / P36H / N210D was performed using KOD-Plus-Ver.
- Taq DNA polymerase was manufactured by Toyobo and mixed with Anti-Taq High (manufactured by Toyobo). The reaction consists of Buffer attached to 1 ⁇ BlendTaq, 10 pmol primer (same as above), dNTPs (dATP, dUTP, dCTP, dGTP) in which 2 mM dTTP is replaced by dUTP, and 50 ⁇ l of 2.5 U enzyme mixed with antibody.
- Plant lysate was added to the reaction solution so as to be 2% of the reaction solution, and after 94 ° C. for 2 minutes, 94 ° C., 30 seconds ⁇ 65 ° C., 30 seconds ⁇ 68 ° C.
- the PCR was performed using a PCR system GeneAmp9700 (Applied Biosystem) on a schedule of repeating 35 cycles of minutes. After completion of the reaction, 5 ⁇ l of the reaction solution was subjected to agarose electrophoresis, stained with ethidium bromide, and the amplification amount of the amplified DNA fragment was confirmed under ultraviolet irradiation.
- FIG. 11 shows that plant lysate was used as a sample, and the reaction solution was prepared so that the ratio of lysate in the reaction solution was 2%, and KOD Y7A / P36H / N210D mutant, KOD Y7A / P36H / N210D mutant were subjected to KOD.
- KOD Y7A / P36H / N210D mutant KOD Y7A / P36H / N210D mutant
- KOD Y7A / V93K, P36H, P36K, P36R, V93K, V93R, Y7A / P36H, Y7A / P36R, Y7A / V93R, P36H / H147E, P36K / H147E, P36R / H147E, V93K / H147E, V93K / R147E / H147E, Y7A / P36H / H147E, Y7A / P36R / H147E, Y7A / V93K / H147E, Y7A / V93R / H147E, P36H / N210D, P36K / N210D, P36R / N210D, V93K / N210D, V93R7 / N210D, Y7A / V93K / N210D, Y7A / V93R / N210D, P36H / N
- PCNA is also a variant of KOD-PCNA M73L / E143R, M73L / R82A / E143A, M73L / R109A / E143A, Pfu-PCNA M73L / D143R, M73L / D147A, M73L / R82A / D143A, M73L / R109A / D143A.
- the same reaction was carried out, and it was confirmed that the amplification amount was improved as compared with the case where PCNA was not added.
- Example 4-9 It was evaluated whether amplified dUTP from stool and gene amplification can be performed in the presence of stool.
- As the enzyme KOD Y7A / P36H / N210D mutant and Taq polymerase were used, and the difference in amplification of about 700 bp of Salmonella invA gene was compared by real-time PCR using SYBR GREEN I and melting curves.
- the KOD Y7A / P36H / N210D mutant was also added with KOD-PCNA M73L / D147A added as a PCR enhancing factor.
- a 10% fecal suspension was heat-treated at 95 ° C. for 10 minutes.
- a buffer attached to KOD Dash manufactured by Toyobo
- stool was added to the reaction solution at 0, 0.
- the reaction is as follows: Buffer attached to 1 ⁇ Taq (Mg attachment type), 50 copies of Salmonella genome, 4 pmol primer (same as above), dNTPs with 2 mM dTTP replaced with dUTP (dATP, dUTP, dCTP, dGTP), 4 mM In 20 ⁇ l of a reaction solution containing 1 U of enzyme mixed with MgSO 4 , 1/30000 SYBR GREEN I, antibody, feces were added to the reaction solution at 0, 0.1, 0.25, 0.5, 1.0. , 1.5, 2.0, 2.5%, 95 ° C., 30 seconds pre-reaction, 98 ° C., 10 seconds ⁇ 60 ° C. 10 seconds ⁇ 68 ° C., 30 seconds repeated 50 cycles
- the schedule was performed using LightCycler 2.0 (Roche). After completion of the reaction, a target peak appearing in the latter half of 80 ° C. was confirmed by melting curve analysis.
- Table 5 shows the Cq value of Example 4-9.
- Table 5 shows Cq values (default settings of LightCycler 2.0) of real-time PCR performed in the presence of dUTP and feces.
- N. D. Indicates that no amplification was observed and no Cq value was obtained. As a result, amplification was not observed when 0.5% stool was added to Taq polymerase, but amplification was confirmed even when 2.5% stool was added to KOD Y7A / P36H / N210D. Further, comparing the presence or absence of PCNA, it was found that the one with PCNA added had a smaller Cq value and showed excellent PCR efficiency.
- FIG. 12 shows the results of melting curve analysis of the obtained amplification product using PCR in the presence of dUTP and feces.
- the total number of polymerases used was KOD Y7A / P36H / N210D mutant, KOD Y7A / P36H / N210D mutant with KOD-PCNA M73L / D147A added, and Taq polymerase.
- 1 shows the result of KOD Y7A / P36H / N210D
- 2 shows the result of adding KOD-PCNA M73L / D147A to KOD Y7A / P36H / N210D
- 3 shows the result of Taq polymerase.
- KOD Y7A / V93K, P36H, P36K, P36R, V93K, V93R, Y7A / P36H, Y7A / P36R, Y7A / V93R, P36H / H147E, P36K / H147E, P36R / H147E, V93K / H147E, V93K / R147E / H147E, Y7A / P36H / H147E, Y7A / P36R / H147E, Y7A / V93K / H147E, Y7A / V93R / H147E, P36H / N210D, P36K / N210D, P36R / N210D, V93K / N210D, V93R7 / N210D, Y7A / V93K / N210D, Y7A / V93R / N210D, P36H / N
- PCNA is also a variant of KOD-PCNA M73L / E143R, M73L / R82A / E143A, M73L / R109A / E143A, Pfu-PCNA M73L / D143R, M73L / D147A, M73L / R82A / D143A, M73L / R109A / D143A.
- the same reaction was performed, and it was confirmed that the PCR efficiency was improved as compared with the case where PCNA was not added.
- Example 4-10 Amplification from blood by addition of PCNA using reaction solution containing dUTP
- reaction solution containing dUTP As the enzyme, KOD Y7A / P36H / N210D and Taq polymerase were used, and the difference in the amount of amplification of 1.3 kb of HBg was compared.
- the KOD mutant was also added with Pfu-PCNA M73L / D147A.
- PCR for KOD mutants was performed using KOD-Plus-Ver. 2 (manufactured by Toyobo), using Buffer, MgSO 4 attached, 1 ⁇ PCR Buffer, and 1.5 mM MgSO 4 , 15 pmol primer (SEQ ID NO: 13 and 14), dNTPs (dATP, dUTP in which 2 mM dTTP was replaced with dUTP) , DCTP, dGTP), and 50 ⁇ l of the reaction solution containing 1 U of each enzyme mixed with KOD antibody, blood was added to 2% of the reaction solution, and 250 ng of Pfu-PCNA M73L / D147A without PCNA was added. The additions were compared.
- Taq DNA polymerase PCR was mixed with 1 ⁇ BlendTaq attached Buffer (Toyobo product), 10 pmol primer (same as above), dNTPs (dATP, dUTP, dCTP, dGTP) with 2 mM dTTP replaced with dUTP, and antibody.
- a 50 ⁇ l reaction solution containing 2.5 U of enzyme was used by adding 2% of blood to the reaction solution.
- the reaction was carried out using PCR system GeneAmp 9700 (Applied Biosystem) on a schedule of repeating 94 cycles at 94 ° C. for 2 minutes followed by 35 cycles of 98 ° C., 10 seconds ⁇ 65 ° C., 30 seconds ⁇ 68 ° C., 1.5 minutes. It was.
- 5 ⁇ l of the reaction solution was subjected to agarose electrophoresis, stained with ethidium bromide, and the amplification amount of the amplified DNA fragment was confirmed under ultraviolet irradiation.
- FIG. 13 shows a reaction solution containing 2% blood prepared by adding Pfu-PCNA M73L / D147A to KOD Y7A / P36H / N210D mutant and KOD Y7A / P36H / N210D mutant with Taq polymerase.
- the result of carrying out PCR reaction and electrophoresis of the obtained product is shown.
- -indicates no PCNA, and + indicates that PCNA was added.
- it was shown that even a relatively long amplification of 1.3 kb can be amplified from crude conditions including blood components.
- a solid band was confirmed in the case where the PCNA mutant was added compared to the case where the PCNA mutant was not added (FIG. 13).
- KOD P36H / N210D, P36K / N210D, P36R / N210D, V93K / N210D, V93R / N210D, Y7A / P36R / N210D, Y7A / V93K / N210D, Y7A / V93R / N210D, P36H / I142R, P36R / I142R, V93K / I142R, V93R / I142R, Y7A / P36H / I142R, Y7A / P36R / I142R, P36H / D141A / E143A, P36R / D141A / E143A, V93K / D141A / E143A, V93R / D141A / E / D141A / E143A, Y7A / P36R / D141A / E143A) under the same reaction conditions, the
- PCNA is also KOD-PCNA M73L / D147A, M73L / E143R, M73L / R82A / E143A, M73L / R109A / E143A, Pfu-PCNA M73L / D143R, M73L / R82A / D143A, M73L / R109A / D143A variants
- the same reaction was carried out, and it was confirmed that the amplification amount was improved as compared with the case where PCNA was not added.
- the present invention eliminates the risk of loss and carryover during DNA purification, and further reduces time and cost. Also, in order to prevent contamination by the dUTP / UDG contamination removal method, not only in the research field, but also in the clinical field or forensic field such as genetic diagnosis in which the same sample is amplified many times, or in the microbiological examination in foods and the environment, etc. Can also be widely used
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Microbiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
L'invention fournit un test d'amplification d'acide nucléique permettant une amplification en chaîne par polymérase (PCR), dans le cadre d'une amplification en chaîne par polymérase incluant une déoxyuridine triphosphate (dUTP) en tant que matériau de base, y compris en présence d'une grande quantité d'impuretés provenant d'un échantillon biologique. Plus précisément, l'invention concerne un procédé selon lequel un échantillon biologique est ajouté à un liquide de réaction d'amplification d'acide nucléique sans processus de purification préalable, et un acide nucléique cible est amplifié dans l'échantillon biologique. Ce test d'amplification d'acide nucléique est caractéristique en ce qu'un enzyme mis en œuvre dans l'amplification consiste en une ADN-polymérase appartenant à la famille B, et une déoxyuridine (dUTP) est contenue le liquide de réaction.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013162988 | 2013-08-06 | ||
| JP2013-162988 | 2013-08-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015019658A1 true WO2015019658A1 (fr) | 2015-02-12 |
Family
ID=52461007
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/058309 Ceased WO2015019658A1 (fr) | 2013-08-06 | 2014-03-25 | Test d'amplification d'acide nucléique |
Country Status (2)
| Country | Link |
|---|---|
| JP (3) | JP6428997B2 (fr) |
| WO (1) | WO2015019658A1 (fr) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210222162A1 (en) * | 2020-01-20 | 2021-07-22 | Tecan Genomics, Inc. | Depletion of abundant uninformative sequences |
| CN114381442A (zh) * | 2021-12-16 | 2022-04-22 | 大连博格林生物科技有限公司 | 一种可快速延伸的高保真dna聚合酶及其制备方法和应用 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6428997B2 (ja) * | 2013-08-06 | 2018-11-28 | 東洋紡株式会社 | 核酸増幅法 |
| JP6980998B2 (ja) * | 2016-11-28 | 2021-12-15 | 東洋紡株式会社 | 核酸増幅用組成物 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06277062A (ja) * | 1992-09-11 | 1994-10-04 | F Hoffmann La Roche Ag | 血液試料中の核酸の増幅および検出 |
| JP2002253265A (ja) * | 2000-05-11 | 2002-09-10 | Toyobo Co Ltd | 改変された耐熱性dnaポリメラーゼ |
| JP2003284576A (ja) * | 2003-03-14 | 2003-10-07 | Toyobo Co Ltd | 核酸増幅用dnaポリメラーゼ組成物 |
| JP2004283165A (ja) * | 2003-03-04 | 2004-10-14 | Shimadzu Corp | Hlaタイピング法 |
| JP2005526510A (ja) * | 2002-04-17 | 2005-09-08 | ニューキャッスル アポン タイン大学 | 古細菌からのdnaポリメラーゼの変異 |
| JP2006507012A (ja) * | 2002-10-25 | 2006-03-02 | ストラタジーン カリフォルニア | 減少した塩基類似体検出活性を有するdnaポリメラーゼ |
| WO2007004654A1 (fr) * | 2005-07-04 | 2007-01-11 | Celestar Lexico-Sciences, Inc. | Antigene pcna mutant |
| JP2008228589A (ja) * | 2007-03-16 | 2008-10-02 | Toppan Printing Co Ltd | 核酸の分析方法 |
| WO2014051031A1 (fr) * | 2012-09-28 | 2014-04-03 | 東洋紡株式会社 | Adn polymérase modifiée |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2742593C (fr) * | 2008-11-03 | 2020-05-05 | Kapa Biosystems, Inc. | Adn polymerases chimeriques |
| JP2014061741A (ja) * | 2012-09-20 | 2014-04-10 | Kawasaki Heavy Ind Ltd | 鞍乗型車両のフレーム構造 |
| JP6428997B2 (ja) * | 2013-08-06 | 2018-11-28 | 東洋紡株式会社 | 核酸増幅法 |
-
2014
- 2014-03-25 JP JP2014061741A patent/JP6428997B2/ja active Active
- 2014-03-25 WO PCT/JP2014/058309 patent/WO2015019658A1/fr not_active Ceased
-
2017
- 2017-12-12 JP JP2017237408A patent/JP6648749B2/ja active Active
-
2019
- 2019-11-19 JP JP2019208737A patent/JP2020036614A/ja active Pending
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06277062A (ja) * | 1992-09-11 | 1994-10-04 | F Hoffmann La Roche Ag | 血液試料中の核酸の増幅および検出 |
| JP2002253265A (ja) * | 2000-05-11 | 2002-09-10 | Toyobo Co Ltd | 改変された耐熱性dnaポリメラーゼ |
| JP2005526510A (ja) * | 2002-04-17 | 2005-09-08 | ニューキャッスル アポン タイン大学 | 古細菌からのdnaポリメラーゼの変異 |
| JP2006507012A (ja) * | 2002-10-25 | 2006-03-02 | ストラタジーン カリフォルニア | 減少した塩基類似体検出活性を有するdnaポリメラーゼ |
| JP2004283165A (ja) * | 2003-03-04 | 2004-10-14 | Shimadzu Corp | Hlaタイピング法 |
| JP2003284576A (ja) * | 2003-03-14 | 2003-10-07 | Toyobo Co Ltd | 核酸増幅用dnaポリメラーゼ組成物 |
| WO2007004654A1 (fr) * | 2005-07-04 | 2007-01-11 | Celestar Lexico-Sciences, Inc. | Antigene pcna mutant |
| JP2008228589A (ja) * | 2007-03-16 | 2008-10-02 | Toppan Printing Co Ltd | 核酸の分析方法 |
| WO2014051031A1 (fr) * | 2012-09-28 | 2014-04-03 | 東洋紡株式会社 | Adn polymérase modifiée |
| JP2014079236A (ja) * | 2012-09-28 | 2014-05-08 | Toyobo Co Ltd | 改変された耐熱性dnaポリメラーゼ |
| JP2014079237A (ja) * | 2012-09-28 | 2014-05-08 | Toyobo Co Ltd | 改変された耐熱性dnaポリメラーゼ |
| JP2014079235A (ja) * | 2012-09-28 | 2014-05-08 | Toyobo Co Ltd | 改変された耐熱性dnaポリメラーゼ |
Non-Patent Citations (4)
| Title |
|---|
| CHOI, J. J. ET AL.: "Unique Substrates Spectrum and PCT Application of Nanoarchaeum equitans Family B DNA Polymerase", APPLIED AND ENVIRONMENTAL MICROBIOLOGY, vol. 74, no. 21, 2008, pages 6563 - 6569 * |
| FOGG, M. J. ET AL.: "Structural basis for uracil recognition by archaeal family B DNA polymerases", NATURE STRUCTURAL BIOLOGY, vol. 9, no. 12, 2002, pages 922 - 927 * |
| SATO, T. ET AL.: "Targeted Gene Disruption by Homologous Recombination in the Hyperthermophilic Archaeon Thermococcus kodakaraensis KOD1", JOURNAL OF BACTERIOLOGY, vol. 185, no. 1, 2003, pages 210 - 220 * |
| TUBELEVICIUTE, A. ET AL.: "Compartmentalized self-replication (CSR) selection of Thermococcus litoralis Sh1B DNA polymerase for diminished uracil binding", PROTEIN ENG. DES. SEL., vol. 23, no. 8, 2010, pages 589 - 597 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210222162A1 (en) * | 2020-01-20 | 2021-07-22 | Tecan Genomics, Inc. | Depletion of abundant uninformative sequences |
| CN113278681A (zh) * | 2020-01-20 | 2021-08-20 | 帝肯基因组学公司 | 大量非信息性序列的耗竭 |
| US12600963B2 (en) * | 2020-01-20 | 2026-04-14 | Tecan Genomics, Inc. | Depletion of abundant uninformative sequences |
| CN114381442A (zh) * | 2021-12-16 | 2022-04-22 | 大连博格林生物科技有限公司 | 一种可快速延伸的高保真dna聚合酶及其制备方法和应用 |
| CN114381442B (zh) * | 2021-12-16 | 2023-12-01 | 大连博格林生物科技有限公司 | 一种可快速延伸的高保真dna聚合酶及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6648749B2 (ja) | 2020-02-14 |
| JP6428997B2 (ja) | 2018-11-28 |
| JP2015050994A (ja) | 2015-03-19 |
| JP2018042567A (ja) | 2018-03-22 |
| JP2020036614A (ja) | 2020-03-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12241095B2 (en) | Modified heat-resistant DNA polymerase | |
| JP6579204B2 (ja) | 改変された耐熱性dnaポリメラーゼ | |
| JP2020036614A (ja) | 核酸増幅法 | |
| JP6658796B2 (ja) | 核酸増幅方法 | |
| JP7014256B2 (ja) | 核酸増幅試薬 | |
| JP6741061B2 (ja) | 核酸増幅法 | |
| KR101230362B1 (ko) | 나노아케움 이퀴탄스 dna 중합효소의 단백질 트랜스 스플라이싱을 기반으로 한 핫―스타트 pcr 수행방법 | |
| JP7107345B2 (ja) | Pcr方法 | |
| JP6798594B2 (ja) | 核酸増幅の正確性を向上させる方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14834694 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14834694 Country of ref document: EP Kind code of ref document: A1 |