WO2015056679A1 - 多孔質セルロースビーズの製造方法及びそれを用いた吸着体 - Google Patents
多孔質セルロースビーズの製造方法及びそれを用いた吸着体 Download PDFInfo
- Publication number
- WO2015056679A1 WO2015056679A1 PCT/JP2014/077360 JP2014077360W WO2015056679A1 WO 2015056679 A1 WO2015056679 A1 WO 2015056679A1 JP 2014077360 W JP2014077360 W JP 2014077360W WO 2015056679 A1 WO2015056679 A1 WO 2015056679A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- beads
- porous cellulose
- cellulose beads
- adsorbent
- crosslinked
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images






Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3231—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the coating or impregnating layer
- B01J20/3242—Layers with a functional group, e.g. an affinity material, a ligand, a reactant or a complexing group
- B01J20/3268—Macromolecular compounds
- B01J20/3272—Polymers obtained by reactions otherwise than involving only carbon to carbon unsaturated bonds
- B01J20/3274—Proteins, nucleic acids, polysaccharides, antibodies or antigens
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/24—Naturally occurring macromolecular compounds, e.g. humic acids or their derivatives
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
- B01J20/267—Cross-linked polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28014—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their form
- B01J20/28016—Particle form
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/281—Sorbents specially adapted for preparative, analytical or investigative chromatography
- B01J20/282—Porous sorbents
- B01J20/285—Porous sorbents based on polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/281—Sorbents specially adapted for preparative, analytical or investigative chromatography
- B01J20/286—Phases chemically bonded to a substrate, e.g. to silica or to polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/3085—Chemical treatments not covered by groups B01J20/3007 - B01J20/3078
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3202—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the carrier, support or substrate used for impregnation or coating
- B01J20/3206—Organic carriers, supports or substrates
- B01J20/3208—Polymeric carriers, supports or substrates
- B01J20/3212—Polymeric carriers, supports or substrates consisting of a polymer obtained by reactions otherwise than involving only carbon to carbon unsaturated bonds
-
- C—CHEMISTRY; METALLURGY
- C02—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F1/00—Treatment of water, waste water, or sewage
- C02F1/28—Treatment of water, waste water, or sewage by sorption
- C02F1/286—Treatment of water, waste water, or sewage by sorption using natural organic sorbents or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/22—Affinity chromatography or related techniques based upon selective absorption processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/14—Preparation of compounds containing saccharide radicals produced by the action of a carbohydrase (EC 3.2.x), e.g. by alpha-amylase, e.g. by cellulase, hemicellulase
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2220/00—Aspects relating to sorbent materials
- B01J2220/50—Aspects relating to the use of sorbent or filter aid materials
- B01J2220/54—Sorbents specially adapted for analytical or investigative chromatography
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2220/00—Aspects relating to sorbent materials
- B01J2220/80—Aspects related to sorbents specially adapted for preparative, analytical or investigative chromatography
- B01J2220/82—Shaped bodies, e.g. monoliths, plugs, tubes, continuous beds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2201/00—Foams characterised by the foaming process
- C08J2201/02—Foams characterised by the foaming process characterised by mechanical pre- or post-treatments
- C08J2201/026—Crosslinking before of after foaming
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2201/00—Foams characterised by the foaming process
- C08J2201/04—Foams characterised by the foaming process characterised by the elimination of a liquid or solid component, e.g. precipitation, leaching out, evaporation
- C08J2201/046—Elimination of a polymeric phase
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2301/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2301/02—Cellulose; Modified cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/26—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof by elimination of a solid phase from a macromolecular composition or article, e.g. leaching out
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/50—Conditioning of the sorbent material or stationary liquid
- G01N30/52—Physical parameters
- G01N2030/524—Physical parameters structural properties
- G01N2030/525—Physical parameters structural properties surface properties, e.g. porosity
Definitions
- the present invention relates to a method for producing a porous cellulose bead and an adsorbent using the same, and more specifically, a method for producing a highly functional porous cellulose bead that can be used for producing an adsorbent excellent in adsorption amount and strength, and The present invention relates to an adsorbent using the same.
- the adsorbent using porous cellulose beads is advantageous in that it is safer than other synthetic polymers, has less nonspecific adsorption, and can be used in a wide pH range. Moreover, there is also an advantage that the mechanical strength is large while being a polysaccharide.
- Examples of applications of the adsorbent using porous cellulose beads include various medical adsorbents, chromatographic adsorbents, and affinity adsorbents.
- affinity adsorbents have been used as medical adsorbents and antibody drug purifying adsorbents because they can efficiently purify target substances and reduce the concentration of unwanted substances.
- an adsorbent for treating or treating rheumatism, hemophilia, dilated cardiomyopathy an adsorbent in which protein A is immobilized on a porous carrier as an affinity ligand has attracted attention (for example, Non-Patent Document 1).
- Non-Patent Document 2 Non-Patent Document 2
- an adsorbent in which protein A is immobilized on a porous carrier using an affinity ligand is attracting attention as an adsorbent for purifying antibody drugs that can specifically adsorb immunoglobulin (IgG).
- IgG immunoglobulin
- the present invention has been made in view of the above-described problems of conventional techniques, and an object thereof is to provide a simple method for producing highly functional porous cellulose beads having high mechanical strength. It is another object of the present invention to provide an adsorbent using the highly functional porous cellulose beads.
- the present inventor has intensively studied in view of the above problems. As a result, by acting a cellulolytic enzyme on the crosslinked porous cellulose beads, the strength is surprisingly maintained or rather improved, and in addition, when the affinity ligand is immobilized and the adsorbent is used, the amount of adsorption is reduced.
- the present invention was completed by finding a significant increase.
- a method for producing highly functional porous cellulose beads comprising a step of allowing a cellulolytic enzyme to act on crosslinked porous cellulose beads.
- An adsorbent is obtained by immobilizing a ligand that interacts with the target adsorbing substance on the high-performance porous cellulose beads produced by the method according to any one of [1] to [6] above.
- the manufacturing method of the adsorbent characterized by including a process.
- An adsorbent comprising a highly functional porous cellulose bead produced by the method according to any one of [1] to [6] above, and a ligand that interacts with a target adsorbing substance. .
- a purification method comprising a step of bringing the adsorbent according to [8] or [9] above into contact with a solution containing the target adsorbing substance.
- porous cellulose beads can be produced from porous cellulose beads by a simple method. Further, an adsorbent exhibiting high strength and high adsorption amount can be obtained by a simple method using the high-functional porous cellulose beads.
- the method for producing a highly functional porous cellulose bead according to the present invention includes a step of allowing a cellulolytic enzyme to act on the crosslinked porous cellulose bead.
- the cross-linked porous cellulose beads and the porous cellulose that is a precursor thereof may be used if they are commercially available, or may be produced from cellulose.
- the method of the present invention will be described step by step, including the step of producing crosslinked porous cellulose beads from cellulose.
- porous cellulose beads are obtained by adding a coagulation solvent to the cellulose emulsion.
- the “cellulose emulsion” refers to those in which droplets containing cellulose are dispersed in an oil-soluble solvent.
- the droplets containing cellulose may be a cellulose dispersion or a cellulose solution.
- Examples of the solvent for the cellulose dispersion include basic aqueous solutions such as an aqueous sodium hydroxide solution. Although cellulose is difficult to dissolve, it is hydrophilic and has many hydroxyl groups, so that it cannot be dissolved but can be dispersed in a basic aqueous solution. Examples of the solvent for the cellulose solution include ionic liquids such as 1-ethyl-3-methylimidazolium acetate. As the raw material cellulose, it is preferable to use cellulose powder because it is easily dispersed or dissolved.
- oil-soluble solvent constituting the cellulose emulsion examples include animal and vegetable oils and fats, hydrogenated animal and vegetable oils and fats, fatty acid triglycerides, aliphatic hydrocarbon solvents, and aromatic hydrocarbon solvents.
- a surfactant such as a nonionic surfactant may be used.
- the cellulose emulsion may be prepared by a conventional method. For example, it can be prepared by vigorously stirring a liquid dispersion or solution of cellulose, a mixed solution containing a fat-soluble solvent and a surfactant.
- the solvent is removed from the cellulose droplets to obtain porous cellulose beads. Since the cellulose emulsion may be unstable, it is preferable to add the coagulation solvent in a state where the droplets are vigorously stirred so that the droplets do not bind to each other.
- the coagulation solvent is not particularly limited as long as it has an affinity for the cellulose dispersion or solution solvent, and examples thereof include alcohols and aqueous alcohol solutions.
- examples of the alcohol include C 1-4 alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, s-butanol, and t-butanol.
- the coagulated porous cellulose beads may be separated by filtration or centrifugation, and washed with water or alcohol.
- the obtained porous cellulose beads may be classified using a sieve or the like in order to make the particle diameter uniform.
- porous cellulose beads may be used, and this step is optional.
- crosslinking step of porous cellulose beads In this step, the porous cellulose beads are crosslinked with a crosslinking agent to obtain crosslinked porous cellulose beads.
- the crosslinking agent refers to one having two or more reactive groups capable of forming a covalent bond with a hydroxyl group on cellulose and capable of crosslinking between cellulose molecules.
- crosslinking conditions or crosslinking agent of the crosslinked porous cellulose beads that can be used in the present invention.
- the method described in WO2008 / 146906 can be used.
- the porous cellulose beads are crosslinked with a glycidyl ether compound, a more remarkable effect may be exhibited.
- the surface pores of the porous cellulose beads may be clogged when the molecular weight is particularly high and / or when highly crosslinked using a polyfunctional crosslinking agent.
- the use of a glycidyl ether compound as a cross-linking agent may make it possible to generate surface pores in the beads.
- the strength of the beads does not decrease even though the cellulolytic enzyme is clearly acting.
- the glycidyl ether compound that can be used as a crosslinking agent is more preferably resorcinol diglycidyl ether, neopentyl glycol diglycidyl ether, 1,6-hexanediol diglycidyl ether, hydrogen Natobisphenol A diglycidyl ether, glycerol diglycidyl ether, trimethylolpropane diglycidyl ether, diglycidyl terephthalate, diglycidyl orthophthalate, ethylene glycol diglycidyl ether, diethylene glycol diglycidyl ether, propylene glycol diglycidyl ether, glycerol polyglycidyl ether, Trimethylolpropane polyglycidyl ether, pentaerythritol polyglycy Ethers, diglycerol polyglycidyl ether, polyglycerol polyglycidyl ether, polyglycerol
- the crosslinking agent may be used alone or in combination of two or more.
- other crosslinking agents such as epichlorohydrin and a glycidyl ether crosslinking agent can be mixed and used.
- the solvent for the reaction for cross-linking the porous cellulose beads with a cross-linking agent may be selected as appropriate.
- water miscibility such as alcohol solvents such as methanol, ethanol and isopropanol, and nitrile solvents such as acetonitrile, etc. Mention may be made of organic solvents. Further, two or more crosslinking reaction solvents may be mixed and used.
- the crosslinking reaction may be performed a plurality of times, and the reaction solvent and the crosslinking agent may be changed each time.
- the first crosslinking reaction may be performed in a water-miscible organic solvent
- the final crosslinking reaction may be performed in water.
- the intermediate solvent composition may be the same as or different from either the first time or the last time, or may be an intermediate composition thereof.
- all rounds may be carried out in an aqueous solvent.
- the crosslinking agent it is as above-mentioned that a special effect will be acquired if water is used as a reaction solvent, and when implementing a crosslinking reaction in multiple times, it is preferable to use all water as a reaction solvent.
- the crosslinking reaction is repeated a plurality of times, it is preferable to remove the crosslinking agent by washing the crosslinked porous cellulose with water or the like between the crosslinking reactions.
- a base may be added to the reaction solution.
- bases include alkali metal hydroxides such as sodium hydroxide and potassium hydroxide; alkali metal hydrogen carbonates such as sodium hydrogen carbonate and potassium hydrogen carbonate; alkali metal carbonates such as sodium carbonate and potassium carbonate; triethylamine and pyridine.
- organic bases such as
- the crosslinked porous cellulose beads are insoluble, and may be washed with a solvent such as water.
- cross-linked porous cellulose beads may be used, and this step is optional. However, this step is preferably carried out because highly functional porous cellulose beads having good characteristics can be obtained by using a glycidyl ether-based crosslinking agent as the crosslinking agent or using water as the solvent for the crosslinking reaction.
- the highly functional porous cellulose bead according to the present invention is produced by allowing a cellulose-degrading enzyme to act on a crosslinked porous cellulose bead, and has an excellent strength and an adsorbent exhibiting a high adsorption amount can be obtained.
- a cellulose-degrading enzyme to act on a crosslinked porous cellulose bead, and has an excellent strength and an adsorbent exhibiting a high adsorption amount can be obtained.
- the highly functional porous cellulose beads obtained according to the present invention can maintain the strength before the action even though the cellulolytic enzyme is allowed to act.
- the present inventors have found that the adsorbent using the highly functional porous cellulose beads according to the present invention has a larger amount of adsorption than the case where no cellulolytic enzyme is used. Perhaps cellulolytic enzymes liberate uncrosslinked glucose units from the cellulose molecules that make up the highly functional porous cellulose beads, increasing the pore volume within the porous cellulose beads, resulting in a highly functional porous When an adsorbent is produced from cellulose beads, it is considered that the adsorption site of the target product increases and the amount of adsorption increases.
- glucose unit when cross-linked, it is not recognized as a substrate by the cellulolytic enzyme, or even if attacked by the cellulolytic enzyme, it remains bound to the cellulose molecule by cross-linking, so that the strength of the beads is maintained. It is thought that.
- the cellulose-degrading enzyme that can be used in the present invention is not particularly limited, and examples thereof include endo-type cellulose-degrading enzymes such as endo- ⁇ -1,4-glucanase and carboxymethyl cellulase; exo- ⁇ -1,4-glucanase and the like. Exo-type cellulolytic enzymes can be mentioned.
- the cellulolytic enzyme selectively attacks uncrosslinked glucose units.
- Endo-type cellulose-degrading enzymes act randomly on cellulose to produce cellodextrin, cellobiose, and glucose. Therefore, if an endo-type cellulose-degrading enzyme is used, even if an uncrosslinked glucose unit is present at the non-terminal portion in the cellulose molecule, the glucose unit can be detached and the pores can be maintained while maintaining the strength. It can be said that highly functional porous cellulose beads having an increased capacity are easily obtained.
- An exo-type cellulose-degrading enzyme generally has an action of cleaving cellulose molecules from cell ends in cellobiose units.
- the exo-type cellulose-degrading enzyme is generally considered to have higher resolution than the end-type, and the glucose unit at the end of the cellulose molecule may not be crosslinked, It is also preferable to use an exo-type cellulolytic enzyme.
- cellulose degrading enzyme Only one kind of cellulose degrading enzyme may be used alone, or two or more kinds may be used in combination. For example, a mixture of endo-type cellulolytic enzyme and exo-type cellulolytic enzyme may be used. Furthermore, ⁇ -glucosidase that decomposes cellobiose into glycose may be used. Some cellulolytic enzymes exhibit both endo-type and exo-type actions.
- the origin of the cellulolytic enzyme is not particularly limited, and natural cellulase derived from higher plants, bacteria, filamentous fungi, wood-rotting fungi, termite symbiotic protozoa, etc. may be used, or artificially engineered by genetic engineering methods. You may use what was produced automatically.
- the origin of the cellulase includes Trichoderma, Aspergillus, Humicola, Staphylotrichum, Rhizopus, Mucor ( Preferred examples include Mucor bacteria, Acremonium bacteria, Chaetomium bacteria, Acidothermus bacteria, Cellulomonas bacteria, and the like. From the viewpoint of availability, Aspergillus and Trichoderma are more preferable, and Aspergillus is most preferable.
- the amount of cellulolytic enzyme used in the present invention is not particularly limited.
- the specific activity of the enzyme preparation is 20000 to 30000 u / g, particularly 22900 u / g, it is preferably 0.01 g or more per 1 g of wet beads. .
- the wet beads are beads in a state where the slurry of beads is filtered to remove excess solvent around the beads.
- water is usually used as a solvent for the enzyme reaction.
- the pH of the reaction solution on which the cellulolytic enzyme acts is not particularly limited, but is preferably 2 or more and 7 or less from the viewpoint of allowing the enzyme reaction to proceed efficiently.
- the pH is more preferably 3 or more and 6 or less, further preferably 4 or more and 5.5 or less, and most preferably 4.5 or more and 5.1 or less.
- the pH control method is not particularly limited, but it is preferable to use a substance having a pH buffering action.
- the substance having a pH buffering action is not particularly limited, and examples thereof include acetic acid, acetate, citric acid, citrate, phosphoric acid, phosphate, carbonic acid, carbonate, etc., and at least one kind is used. preferable.
- the amount of the substance having a pH buffering action is not particularly limited, but it is preferably 0.01% by weight or more of the reaction solution because it is easy to suppress fluctuations in pH, and 50% by weight or less from the viewpoint of cost. It is preferable that More preferably, they are 0.05 weight% or more and 10 weight% or less, More preferably, they are 0.1 weight% or more and 5 weight% or less.
- reaction liquid temperature when the cellulose-degrading enzyme is allowed to act, but it is preferably 0 ° C. or higher and 100 ° C. or lower in order to efficiently advance the reaction. More preferably, they are 10 degreeC or more and 80 degrees C or less, More preferably, they are 25 degreeC or more and 70 degrees C or less, Especially preferably, they are 35 degreeC or more and 60 degrees C or less, Most preferably, they are 40 degreeC or more and 55 degrees C or less.
- the reaction time is preferably 10 minutes or longer, more preferably 30 minutes or longer, particularly preferably 1 hour or longer, more preferably within 24 hours, further preferably within 12 hours, particularly preferably within 6 hours, Most preferably within 3 hours.
- highly functional porous cellulose capable of producing an adsorbent having high adsorption performance while maintaining strength can be obtained more reliably.
- the said reaction time is less than 10 hours, it will become possible to maintain the intensity
- the high-functionality porous cellulose beads obtained by this process maintain or increase the strength of the crosslinked porous cellulose beads, which are raw materials, despite the treatment with the cellulolytic enzyme.
- the ligand When the ligand is immobilized and used as an adsorbent, it exhibits excellent adsorption ability for the target adsorbed substance.
- 20% compressive stress is about 0.03 MPa or more and 0.4 MPa or less, preferably 0.08 MPa or more and 0.3 MPa or less.
- the 20% compressive stress refers to the stress exhibited by the beads when compressed so that the sedimentation volume of the beads is reduced by 20%.
- the high-performance porous cellulose beads according to the present invention have high strength, and when adsorbed by immobilizing a ligand that interacts with the target adsorbing substance, the adsorbing ability to the target adsorbing substance is improved. Are better.
- an adsorbent is obtained by immobilizing a ligand on the highly functional porous cellulose beads according to the present invention.
- the “ligand” means an affinity ligand that has a specific affinity for the target adsorbing substance and interacts with the target adsorbing substance.
- the target adsorbing substance is an antibody
- antigens, proteins, peptide fragments, etc. that specifically interact with the antibody can be exemplified.
- the ligand that can be used for the adsorbent according to the present invention is not particularly limited as long as it has a specific affinity for the target adsorbent to be purified using the adsorbent according to the present invention.
- the method for immobilizing the ligand on the highly functional porous cellulose beads according to the present invention is not particularly limited, and a conventional method can be used.
- a conventional method can be used.
- cyanogen bromide method, trichlorotriazine method, epoxy Method immobilization of amino group-containing ligands using methods such as tresyl chloride method, periodate oxidation method, divinyl sulfonic acid method, benzoquinone method, carbonyldiimidazole method, acyl azide method; epoxy method, diazo coupling method, etc.
- a method of immobilizing a hydroxyl group-containing ligand using a method a method of immobilizing a thiol group-containing ligand using an epoxy method, a tresyl chloride method, a divinyl sulfonic acid method, etc .; a carboxylic acid-containing ligand or a formyl group on an amination carrier
- immobilization methods such as a method of immobilizing the contained ligand can be mentioned. The entire contents of this document are incorporated herein by reference.
- the adsorbent according to the present invention can be used as an adsorbent for purification, but it can also be used as an adsorbent for purifying antibody drugs that has been attracting attention in recent years.
- the ligand when used in adsorbents for antibody drug purification, for example, antigens and proteins highly specific for antibodies, protein A, protein G, protein L and their variants, antibodies Examples thereof include amino group-containing ligands such as peptides having binding activity.
- an adsorbent capable of specifically adsorbing immunoglobulin (IgG) an adsorbent obtained by immobilizing protein A, protein G, or a variant thereof as a ligand on a porous carrier has attracted attention.
- the protein A or the like that can be used in the present invention is not particularly limited, and natural products and genetically modified products can be used without limitation.
- antibody binding domains, mutants thereof, those containing oligomers thereof, fusion proteins, and the like may be used.
- the number of polymerizations of the oligomer can be 2 or more and 10 or less.
- the adsorbent of the present invention in which protein A is immobilized can also be used as a therapeutic adsorbent that can be used for the treatment of dilated cardiomyopathy and the like.
- the adsorbent of the present invention in which dextran sulfate or the like is immobilized can be used as an adsorbent for treating hypercholesterolemia.
- the method for introducing the ligand into the high-performance porous cellulose beads can be selected from the various immobilization methods described above, but more preferably, the formyl group contained in the porous particle and the amino group of the ligand are combined. This is a method of immobilization using a reaction. For example, there is a method described in JP-A-1-278534. The entire contents of this publication are incorporated herein by reference.
- the amount of the ligand immobilized on the adsorbent of the present invention is not particularly limited, and can be, for example, 1 mg or more and 1000 mg or less per 1 mL of high-performance porous cellulose beads. If the said ratio is 1 mg or more, since the adsorption amount with respect to a target adsorption substance becomes large, it is preferable, since manufacturing cost can be suppressed if it is 1000 mg or less, it is preferable.
- the amount of the ligand introduced is preferably 2 mg or more, more preferably 4 mg or more, particularly preferably 5 mg or more, more preferably 500 mg or less, further preferably 250 mg or less, more preferably 200 mg or less per mL of highly functional porous cellulose beads. Is particularly preferable, and 100 mg or less is most preferable.
- the adsorbent according to the present invention is obtained using the high-performance porous cellulose beads produced by the method of the present invention.
- strength of a highly functional porous cellulose bead and an adsorbent body it can evaluate by compressive stress measurement as shown in Test example 2 of this application.
- the present invention is a very simple method in which a cellulose-degrading enzyme is allowed to act on crosslinked cellulose beads, while maintaining the strength or improving the strength while improving the surface porosity of the porous cellulose beads, When used as, there is an effect of increasing the adsorption amount.
- the adsorbent that can be used is not particularly limited, but can be suitably used as a medical adsorbent, particularly a therapeutic adsorbent that adsorbs and removes large-sized pathogenic substances (such as LDL cholesterol) because the surface porosity can be improved. be able to.
- it can be used as various chromatographic carriers, especially industrial chromatographic carriers packed in large-diameter columns.
- the adsorbent may be brought into contact with a solution containing the target adsorbent.
- the contact method is not particularly limited, and the adsorbent according to the present invention may be added to a solution containing the target adsorbent, or the column is filled with the adsorbent of the present invention and contains the target adsorbent.
- the target adsorbent may be selectively adsorbed to the adsorbent of the present invention by passing the solution. Since the adsorbent according to the present invention is high in strength, particularly when packed in a column, liquid can be passed at a high speed, and the target adsorbent can be purified efficiently.
- the adsorbent of the present invention in which the target adsorbing material is selectively adsorbed is separated from the solution by filtration or centrifugation.
- the target adsorbing substance and other substances can be separated.
- the target adsorbed material is separated from the adsorbent of the present invention using the eluate.
- the eluate for example, an acidic buffer having a pH of about 2.5 or more and 4.5 or less can be used.
- the freeze-dried beads were subjected to vapor deposition using gold / palladium as a vapor deposition source, and SEM observation images were taken with a scanning electron microscope (Hitachi, Ltd. “S-800”).
- Test Example 2 Measurement of 20% Compressive Stress (1) Sample Preparation Pure water was added to sample beads to prepare a slurry having a concentration of about 50% by volume. A homogenization / deaeration operation consisting of homogenization by stirring the slurry and subsequent deaeration by decompression for 30 minutes or more was repeated 3 times to obtain a deaeration slurry. Separately from this operation, the object to be treated was changed to pure water, and the homogenization / demethod operation was performed for 90 minutes or more to obtain deaerated water.
- PBS buffer solution having a pH of 7.4 was prepared using “Phosphate buffered saline” manufactured by Sigma and RO water (reverse osmosis membrane purified water).
- Solution B A 35 mM aqueous sodium acetate solution having a pH of 3.5 was prepared using acetic acid, sodium acetate and RO water.
- Solution D An IgG aqueous solution having a concentration of 1 mg / mL was prepared using a polyclonal antibody (“Gamma Guard” manufactured by Baxter) and the solution A.
- E solution 6M urea aqueous solution was prepared with urea and RO water.
- Neutralization solution A 2M tris (hydroxymethyl) aminomethane aqueous solution was prepared with tris (hydroxymethyl) aminomethane and RO water.
- the dynamic adsorption amount of IgG at RT 3 min was determined from the amount of IgG adsorbed on the adsorbent until the IgG broke through 5% and the adsorbent volume.
- the dynamic adsorption amount is abbreviated as “5% DBC”.
- Production Example 1 Production of Crosslinked Porous Cellulose Beads (1) Production of Alkaline Aqueous Solution Using sodium hydroxide and pure water manufactured by Wako Pure Chemical Industries, Ltd., a 33% by weight aqueous sodium hydroxide solution was produced, and the temperature was adjusted to 4 ° C. It was adjusted.
- Example 1 Production of High-Functional Porous Cellulose Beads
- the crosslinked cellulose beads produced in Production Example 1 were subjected to suction filtration for 15 minutes, and 6 g of the filtered beads were put into a reaction vessel.
- a 0.1 wt% citric acid monohydrate (Wako Pure Chemical Industries, Ltd.) aqueous solution and a 0.1 wt% trisodium citrate (Wako Pure Chemical Industries, Ltd.) aqueous solution were mixed to prepare a pH 5.0 buffer.
- Cellulase (derived from Aspergillus niger, Lot: YQ211-QM, specific activity: 22900 u / g) was added to this buffer so as to be 1 wt% and dissolved.
- Production Example 2 Production of Crosslinked Porous Cellulose Beads
- Crosslinked cellulose beads were obtained in the same manner as in Production Example 1 except that acetonitrile in the crosslinking agent solution in Production Example 1 was replaced with isopropyl alcohol.
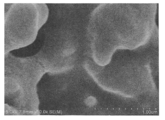
- An SEM observation image of the bead surface is shown in FIG.
- Example 2 Production of highly functional porous cellulose beads Highly functional porous cellulose beads treated with cellulase in the same manner as in Example 1 except that the crosslinked cellulose beads obtained in Production Example 2 were used. Obtained. An SEM observation image of the bead surface is shown in FIG.
- Production Example 3 Production of Crosslinked Porous Cellulose Beads 20 mL of uncrosslinked porous cellulose beads produced in the same manner as in Production Example 1 were weighed and temperature-controlled at 40 ° C. Then, it filtered on the glass filter and moved the bead after filtration to the reaction container. In this reaction vessel, 12.2 mL of 2M sodium hydroxide aqueous solution adjusted to 40 ° C. was added and stirred for 30 minutes.
- the obtained first cross-linked beads were subjected to the cross-linking operation from the above temperature control to 40 ° C. to washing with a large amount of pure water once more to obtain double cross-linked beads.
- the obtained second cross-linked beads and water slurry were transferred to a reaction vessel, the beads were allowed to settle naturally, and then the excess amount of water was extracted to adjust the liquid volume to 29 mL. This was heated to 40 ° C., 3.7 mL of 4M sodium hydroxide aqueous solution (made with pure water and sodium hydroxide manufactured by Wako Pure Chemical Industries, Ltd.), 7.6 g of mirabilite, and 2.6 g of epichlorohydrin were added. The mixture was stirred at 40 ° C. for 2 hours.
- FIG. 6 shows an SEM observation image of the fractured surface of this bead.
- the obtained beads had a 20% compressive stress of 0.084 MPa.
- Example 3 Production of highly functional porous cellulose beads Highly functional porous cellulose beads treated with cellulase were prepared in the same manner as in Example 1 except that the crosslinked cellulose beads obtained in Production Example 3 were used. Obtained.
- FIG. 7 shows an SEM observation image of the fractured surface of this bead.
- the beads before cellulase treatment can be said to have a relatively thick skeleton and a small pore volume, whereas in the highly functional porous cellulose beads according to the present invention treated with cellulase, It can be said that the skeleton is relatively thin and the pore volume is increased.
- the high-performance porous cellulose beads obtained have a 20% compressive stress of 0.140 MPa, and the strength is rather improved. It was.
- the filtrate was washed with RO water on a glass filter (“11GP100” manufactured by Shibata Co., Ltd.) until the electric conductivity of the filtrate was 1 ⁇ S / cm or less to obtain formyl group-containing crosslinked porous cellulose beads.
- the electrical conductivity of the washing filtrate was measured with a conductivity meter (“ECTester 10 Pure +” manufactured by EUTECH INSTRUMENTS).
- the obtained formyl group-containing crosslinked porous cellulose beads (5 mL) were transferred to a glass filter ("11GP100" manufactured by Shibata Co., Ltd.), and trisodium citrate dihydrate was dissolved in RO water to obtain 0.25M tricitrate citrate. 15 mL or more of an aqueous sodium solution was passed, and the liquid in the beads was replaced with the aqueous trisodium citrate solution. Using the 0.25M trisodium citrate aqueous solution, the substituted formyl group-containing crosslinked porous cellulose beads were placed in a centrifuge tube. After the formyl group-containing porous cellulose beads were settled, the supernatant was removed and adjusted so that the total volume became 7.5 mL.
- the beads after the reaction were washed with 3 times volume RO water of beads on a glass filter (“11GP100” manufactured by Shibata). Next, 3 times volume of 0.1M citric acid monohydrate obtained by dissolving citric acid monohydrate in RO water was added, and 0.1M citric acid monohydrate was added to the beads to make a total amount. was placed in a centrifuge tube and washed with acid while stirring at 25 ° C. for 30 minutes.
- the beads were washed with 3 times the volume of RO water of beads on a glass filter ("11GP100" manufactured by Shibata), and then 0.05M obtained by dissolving sodium hydroxide in RO water Sodium hydroxide and a 1M sodium sulfate aqueous solution obtained by dissolving sodium sulfate in RO water were each added in three times volume. Next, 0.05 M sodium hydroxide and 1 M sodium sulfate aqueous solution were added to the beads to make the total volume 30 mL or more, and the beads were placed in a centrifuge tube and washed with alkali while stirring at room temperature for 30 minutes.
- the beads were washed with 20 times volume of RO water on the glass filter (“11GP100” manufactured by Shibata).
- 0.1M sodium citrate aqueous solution obtained by dissolving trisodium citrate dihydrate in RO water was added 3 times the amount of beads, and after confirming that the filtrate was neutral,
- an adsorbent on which protein A was immobilized was obtained.
- the conductivity of the washing filtrate was measured with a conductivity meter (“ECTester 10 Pure +” manufactured by EUTECH INSTRUMENTS).
- the 5% DBC of this adsorbent at RT 3 min was 22.0 g / L.
- Example 4 Production of Adsorbent An adsorbent was obtained in the same manner as in Production Example 4 except that the highly functional porous cellulose beads obtained in Example 3 were used. The 5% DBC of this adsorbent at RT 3 min was 24.4 g / L. As can be seen from the results, the adsorption performance of the adsorbent containing the high-functionality porous cellulose beads subjected to the cellulase treatment was improved as compared with the normal adsorbent.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Zoology (AREA)
- Materials Engineering (AREA)
- Polymers & Plastics (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Microbiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Environmental & Geological Engineering (AREA)
- Hydrology & Water Resources (AREA)
- Water Supply & Treatment (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
本工程では、セルロースエマルションに凝固溶媒を加えることにより多孔質セルロースビーズを得る。
本工程では、多孔質セルロースビーズを架橋剤により架橋して、架橋多孔質セルロースビーズを得る。
本工程では、架橋多孔質セルロースビーズにセルロース分解酵素を作用させることにより、高機能多孔質セルロースビーズを得る。
本発明に係る高機能多孔質セルロースビーズは、高強度である上に、目的吸着物質と相互作用するリガンドを固定化して吸着体とした場合に、目的吸着物質に対する吸着能に優れている。本工程では、本発明に係る高機能多孔質セルロースビーズにリガンドを固定化することにより吸着体を得る。
各製造例と実施例で得られたビーズを5倍体積量の30%エタノールで洗浄し、ビーズに含まれる液体部分を30%エタノールで置換した。次いで、50%エタノール、70%エタノール、90%エタノール、特級エタノール、特級エタノール、特級エタノールを順に用いてビーズを同様に処理し、液体部分をエタノールで置換した。さらにt-ブチルアルコール/エタノール=3/7の混合液を用いてビーズを同様に処理した。次いで、t-ブチルアルコール/エタノール=5/5、7/7、9/1、10/0、10/0、10/0の混合液を順に用いてビーズを処理し、液体部分をt-ブチルアルコールで置換した後、凍結乾燥した。表面観察の場合は、凍結乾燥を行なったビーズに金/パラジウムを蒸着源とした蒸着処理を行い、走査型電子顕微鏡(日立製作所「S-800」)にてSEM観察像を撮影した。割断面観察の場合は、凍結乾燥を行ったビーズを割ってビーズ内部を露出させ、四塩化オスミウム結晶を蒸着源とした蒸着処理を行った後、走査型電子顕微鏡(CarlZeiss社製「ULTRAplus」)にてSEM観察像を撮影した。
(1) 試料調製
試料ビーズに純水を加えて濃度約50体積%のスラリーを調製した。このスラリーの攪拌による均質化と、それに続く30分以上の減圧による脱気とからなる均質・脱気操作を3回繰り返して実施し、脱気スラリーを得た。この操作とは別に、処理対象を純水に変えて、前記均質・脱法操作を90分以上実施し、脱気水を得た。
5mLのルアロック付ディスポーサブルシリンジ(HANKE SASS WOLF社製「商標名:NORM-JECT」)の先端にディスポーサブルフィルター(ザルトリウス社製,孔径:5.00μm,親水性)を取り付けた。シリンジのピストンを外し、シリンジ後端側から脱気水を約3mL投入し、この脱気水が0mLの標線を下回らないうちに、脱気スラリーを投入した。ディスポーザブルフィルターの二次側にアスピレーターを接続し、前記脱気スラリーを吸引した。ただし、ビーズがサクションドライ状にならないように注意し、ビーズの沈降体積+0.5mL程度まで液面が下がったところで吸引を停止した。以降の作業は、液面がビーズの沈降体積+0.5mLを下回らないよう、減圧脱気した純水を追加しながら実施した。振動を与えながら前記脱気スラリーを追加またはビーズを除去し、ビーズ充填高さを3mLの標線に合わせ、振動を与えてもビーズ充填高さが低下しないことを確認した。ビーズが舞い上がらないようゆっくりと脱気水を溢れるまで追加し、気泡が入らないように注意しながらピストンを挿入した。以下、このシリンジを「ビーズ充填シリンジ」という。
物性測定器(レオテック社製「FUDOH RHEO METER」)に10Kのロードセルを取り付け、変位速度のダイヤルを2cm/MINに合わせ、前記ビーズ充填シリンジをセットし、ピストンの変位を開始した。変位と応力との関係を記録し、下記式に基づき、20%圧縮応力を求めた。
試験例3 RT(Residence time)3分でのIgGの動的吸着量測定
(1) 溶液作成
下記A~E液および中和液を調製し、使用前に脱気した。
架橋ビーズにリガンドを固定化したものを吸着体試料とした。また、移動相には、RO水にNaClを加えて調製した0.2M NaCl水溶液を用いた。カラムクロマトグラフィー用装置として、AKTAexplorer100(GEヘルスケア社製)を用い、直径0.5cm、高さ15cmのカラムに、吸着体試料を3mL入れ、線速230cm/hで0.2MのNaCl水溶液を15分通液して充填した。フラクションコレクターに15mLの採取用チューブをセットし、溶出液の採取用チューブについては、あらかじめ中和液を入れておいた。
前記カラムにA液を15mL通液し、次いでD液を150mL通液した。次いで、A液を21mL通液後、B液を12mL通液してIgGを溶出させた。次にC液を6mL、E液を6mL、A液を15mL通液した。なお各液の流速は1mL/分とし、吸着体との接触時間が3分となるようにした。
IgGが5%破過するまでに吸着体に吸着したIgG量と吸着体体積からRT3minでのIgGの動的吸着量を求めた。以下、当該動的吸着量を「5%DBC」と略記する。
(1) アルカリ水溶液の作製
和光純薬社製の水酸化ナトリウムと純水を用いて、33重量%の水酸化ナトリウム水溶液を作製し、温度を4℃に調整した。
粉末状のセルロース(旭化成ケミカルズ社製「局方セルロース PH-F20JP」)76gと純水800gを混合し、攪拌しながら4℃に調整した。次いで設定温度と攪拌を維持しながら4℃に調整した前記アルカリ水溶液を400g投入し、30分間攪拌した。
4℃に調整されたセルロース分散液1276gと、4℃に調整したオルトジクロロベンゼン7801gと、4℃に調整したソルビタンモノオレエート(span80相当品)78gを混合し、ディスクタービン(Rushton turbine)翼2枚を取り付けた温調ジャケット付ステンレス容器内にて460rpm(Pv値:5.0kW/m3)で4℃、15分間攪拌し、エマルションを作製した。設定温度と攪拌を維持しながら4℃に調整されたメタノール592gを凝固溶媒として加えた。また凝固溶媒の添加所要時間は5秒であった。その後、攪拌数と設定温度を維持しながら30分間攪拌した。加圧濾過を行なった後、洗浄液としてメタノール3000gを用いて洗浄を行い、次いで3000gの水で洗浄を行い、多孔質セルロースビーズを得た。得られた多孔質セルロースビーズは、図1に示す通り、表面に良好な孔が空いていることが確認できた。
得られた多孔質セルロースビーズを、38μmと90μmの篩を用いて湿式分級した後、ビーズ体積として20mLに相当する多孔質セルロースビーズと水とのスラリーを反応容器に入れた。ビーズを自然沈降させた後、過剰の水を抜き取ることにより、液量を22mLに調整した。グリセロールポリグリシジルエーテル(ナガセケムテックス社製「デナコールEX-314」)とアセトニトリルを体積比1:1で混合した架橋剤溶液22mLを投入し、一晩攪拌した。
製造例1で作製した架橋されたセルロースビーズを15分間吸引濾過し、濾過後のビーズ6gを反応容器に投入した。0.1wt%のクエン酸一水和物(和光純薬社製)水溶液と0.1wt%のクエン酸三ナトリウム(和光純薬社製)水溶液を混合してpH5.0の緩衝液を作製した。この緩衝液にセルラーゼ(東京化成工業社製,Aspergillus niger由来,Lot:YQ211-QM,比活性:22900u/g)が1wt%になるように添加し、溶解した。このセルラーゼが溶解したクエン酸系緩衝液400mLを前記反応容器へ投入し、45℃で1時間攪拌した。反応容器の内容物を濾過後、大量の水で洗浄することにより、高機能多孔質セルロースビーズを取得した。得られたビーズ表面のSEM観察像を図3に示す。
製造例1における架橋剤溶液のアセトニトリルをイソプロピルアルコールに替えた以外は、製造例1と同様の方法で架橋セルロースビーズを得た。このビーズ表面のSEM観察像を図4に示した。
架橋セルロースビーズとして製造例2で得られたものを用いること以外は、実施例1と同様の方法で、セルラーゼで処理された高機能多孔質セルロースビーズを得た。このビーズ表面のSEM観察像を図5に示した。
製造例1と同様に作製された未架橋の多孔質セルロースビーズ20mLを計量し、40℃に温調した。その後、グラスフィルター上で濾過し、濾過後のビーズを反応容器に移した。この反応容器に40℃に温調した2M水酸化ナトリウム水溶液を12.2mL投入し、30分間攪拌した。次いで、水素化ホウ素ナトリウム(和光純薬社製)を24.4mg、グリセロールポリグリシジルエーテル(ナガセケムテックス社製「デナコールEX-314」)を0.5mL投入し10分間40℃で攪拌した。その後、芒硝を7.6g、グリセロールポリグリシジルエーテル(ナガセケムテックス社製「デナコールEX-314」)を6.1mL投入し、40℃で4時間50分攪拌した。濾過後、大量の純水で洗浄し、1回目架橋ビーズを得た。さらに、得られた1回目架橋ビーズに対して、上記の40℃への温調から大量の純水での洗浄までの架橋操作をもう1回繰り返し、架橋2回ビーズを得た。得られた2回目架橋ビーズと水のスラリーを反応容器に移し、ビーズを自然沈降させた後、過剰の水を抜き取ることにより、液量を29mLに調整した。これを40℃に加熱し、4Mの水酸化ナトリウム水溶液(純水と和光純薬社製の水酸化ナトリウムで作製)3.7mLと芒硝7.6gとエピクロロヒドリン2.6gを添加し、40℃で2時間攪拌した。濾過後、大量の純水で洗浄し、3回目架橋ビーズを得た。得られた3回目架橋ビーズを容器に移し、純水を加えて、全量をセルロースビーズの10倍体積量とし、オートクレーブを用いて120℃で60分間加温した。室温まで放冷した後、ビーズの5倍体積量以上の蒸留水で洗浄し、オートクレーブ済みの架橋3回ビーズを得て、これを架橋セルロースビーズとした。このビーズの割断面のSEM観察像を図6に示した。得られたビーズの20%圧縮応力は0.084MPaであった。
架橋セルロースビーズを製造例3で得られたものを用いること以外は、実施例1と同様の方法で、セルラーゼで処理された高機能多孔質セルロースビーズを得た。このビーズの割断面のSEM観察像を図7に示した。
(1) プロテインA調製工程
国際公開WO2011/118699号の実施例を参照して、改変型プロテインAとして、国際公開WO2011/118699号記載のアルカリ耐性を有する改変Cドメイン5連結体を調製した。
製造例3で得られた架橋多孔質セルロースビーズ5mLを遠沈管に入れ、RO水を加えて、全量を7.5mLとした。これを25℃にてミックスローター(アズワン社製「ミックスローターMR-3」)上に取り付けた後、撹拌した。次に、過ヨウ素酸ナトリウムをRO水に溶解して得られた12.84mg/mL過ヨウ素酸ナトリウム水溶液を2.5mL加え、25℃で1時間撹拌した。反応後、グラスフィルター(シバタ社製「11GP100」)上で、濾液の電気伝導度が1μS/cm以下となるまでRO水で洗浄し、ホルミル基含有架橋多孔質セルロースビーズを得た。洗浄濾液の電気伝導度は、導電率計(EUTECH INSTRUMENTS社製「ECTester10 Pure+」)で測定した。
高機能多孔質セルロースビーズを実施例3で得られたものを用いること以外は製造例4と同様に吸着体を得た。この吸着体のRT3minでの5%DBCは24.4g/Lであった。かかる結果のとおり、通常の吸着体に比べ、セルラーゼ処理を経た高機能多孔質セルロースビーズを含む吸着体の吸着性能は改善された。
Claims (10)
- 架橋多孔質セルロースビーズにセルロース分解酵素を作用させる工程を含むことを特徴とする、高機能多孔質セルロースビーズの製造方法。
- 前記セルロース分解酵素としてエンド型セルロース分解酵素を用いる請求項1に記載の高機能多孔質セルロースビーズの製造方法。
- 前記セルロース分解酵素として、さらにエキソ型セルロース分解酵素を用いる請求項2に記載の高機能多孔質セルロースビーズの製造方法。
- 前記セルロース分解酵素を、1時間以上、10時間以内、前記架橋多孔質セルロースビーズに作用させる請求項1~3のいずれか一項に記載の高機能多孔質セルロースビーズの製造方法。
- 前記架橋多孔質セルロースビーズが架橋剤としてグリシジルエーテル系化合物を用いて架橋されたものであることを特徴とする、請求項1~4のいずれか一項に記載の高機能多孔質セルロースビーズの製造方法。
- 前記架橋多孔質セルロースビーズが架橋反応の反応溶媒として水を用いて架橋されたものであることを特徴とする、請求項1~5のいずれか一項に記載の高機能多孔質セルロースビーズの製造方法。
- 請求項1~6のいずれか一項に記載の方法で製造された高機能多孔質セルロースビーズに、目的吸着物質と相互作用するリガンドを固定化することにより吸着体を得る工程を含むことを特徴とする吸着体の製造方法。
- 請求項1~6のいずれか一項に記載の方法で製造された高機能多孔質セルロースビーズ、および、目的吸着物質と相互作用するリガンドを含むことを特徴とする吸着体。
- 請求項1~6のいずれか一項に記載の方法で製造された高機能多孔質セルロースビーズに、目的吸着物質と相互作用するリガンドを固定化することにより製造されたものであることを特徴とする吸着体。
- 請求項8または9に記載の吸着体と、前記目的吸着物質を含む溶液とを接触させる工程を含むことを特徴とする精製方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14853421.7A EP3059320A4 (en) | 2013-10-15 | 2014-10-14 | Production method for porous cellulose beads, and absorbent employing same |
| US15/029,293 US10040053B2 (en) | 2013-10-15 | 2014-10-14 | Production method for porous cellulose beads, and adsorbent employing same |
| JP2015542618A JP6517145B2 (ja) | 2013-10-15 | 2014-10-14 | 多孔質セルロースビーズの製造方法及びそれを用いた吸着体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013215119 | 2013-10-15 | ||
| JP2013-215119 | 2013-10-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015056679A1 true WO2015056679A1 (ja) | 2015-04-23 |
Family
ID=52828122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/077360 Ceased WO2015056679A1 (ja) | 2013-10-15 | 2014-10-14 | 多孔質セルロースビーズの製造方法及びそれを用いた吸着体 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10040053B2 (ja) |
| EP (1) | EP3059320A4 (ja) |
| JP (1) | JP6517145B2 (ja) |
| WO (1) | WO2015056679A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110304715A (zh) * | 2019-06-21 | 2019-10-08 | 安吉国千环境科技有限公司 | 一种适用于生活污水的mbbr填料挂膜方法 |
| CN111247999A (zh) * | 2020-02-20 | 2020-06-09 | 宁夏新起点现代农业装备科技有限公司 | 蓄热块、其制备方法及蓄热温室大棚 |
| CN116120391A (zh) * | 2023-01-09 | 2023-05-16 | 苏州星谱生物科技有限公司 | 一种三层结构的蛋白质分离用多糖微球的制备方法及应用 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01278534A (ja) | 1988-04-28 | 1989-11-08 | Kanegafuchi Chem Ind Co Ltd | 分離材用バイモーダル粒子 |
| US5151350A (en) | 1982-10-27 | 1992-09-29 | Repligen Corporation | Cloned genes encoding recombinant protein a |
| JP2001139938A (ja) * | 1999-11-14 | 2001-05-22 | Keijiro Nakamura | 微生物、その代謝物あるいは両者を含有する油吸収材 |
| WO2003080655A1 (en) | 2002-03-25 | 2003-10-02 | Amersham Biosciences Ab | A mutated immunoglobulin-binding protein |
| WO2006004067A1 (ja) | 2004-07-06 | 2006-01-12 | Kaneka Corporation | ブレビバチルス属細菌を用いたプロテインa様蛋白質の生産方法 |
| JP2006304633A (ja) | 2005-04-26 | 2006-11-09 | Apro Life Science Institute Inc | イムノグロブリン結合タンパク質 |
| WO2008146906A1 (ja) | 2007-05-30 | 2008-12-04 | Kaneka Corporation | ホルミル基含有多孔質担体、それを用いた吸着体、およびそれらの製造方法 |
| JP2009247981A (ja) * | 2008-04-04 | 2009-10-29 | Saga Univ | 吸着剤及び貴金属の回収方法 |
| WO2010110288A1 (ja) | 2009-03-24 | 2010-09-30 | 株式会社カネカ | 免疫グロブリンに親和性を有するタンパク質および免疫グロブリン結合性アフィニティーリガンド |
| WO2011118699A1 (ja) | 2010-03-24 | 2011-09-29 | 株式会社カネカ | 免疫グロブリンに特異的に結合するタンパク質および免疫グロブリン結合性アフィニティーリガンド |
| JP2011252929A (ja) | 2011-09-05 | 2011-12-15 | Kaneka Corp | 多孔質担体、およびそれを用いた精製用吸着体、およびそれらの製造方法、およびそれらを用いた精製方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140011337A (ko) | 2011-03-08 | 2014-01-28 | 가부시키가이샤 가네카 | 다공질 셀룰로오스 비즈의 제조 방법 |
-
2014
- 2014-10-14 US US15/029,293 patent/US10040053B2/en active Active
- 2014-10-14 JP JP2015542618A patent/JP6517145B2/ja active Active
- 2014-10-14 EP EP14853421.7A patent/EP3059320A4/en not_active Withdrawn
- 2014-10-14 WO PCT/JP2014/077360 patent/WO2015056679A1/ja not_active Ceased
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5151350A (en) | 1982-10-27 | 1992-09-29 | Repligen Corporation | Cloned genes encoding recombinant protein a |
| JPH01278534A (ja) | 1988-04-28 | 1989-11-08 | Kanegafuchi Chem Ind Co Ltd | 分離材用バイモーダル粒子 |
| JP2001139938A (ja) * | 1999-11-14 | 2001-05-22 | Keijiro Nakamura | 微生物、その代謝物あるいは両者を含有する油吸収材 |
| WO2003080655A1 (en) | 2002-03-25 | 2003-10-02 | Amersham Biosciences Ab | A mutated immunoglobulin-binding protein |
| WO2006004067A1 (ja) | 2004-07-06 | 2006-01-12 | Kaneka Corporation | ブレビバチルス属細菌を用いたプロテインa様蛋白質の生産方法 |
| JP2006304633A (ja) | 2005-04-26 | 2006-11-09 | Apro Life Science Institute Inc | イムノグロブリン結合タンパク質 |
| WO2008146906A1 (ja) | 2007-05-30 | 2008-12-04 | Kaneka Corporation | ホルミル基含有多孔質担体、それを用いた吸着体、およびそれらの製造方法 |
| JP2009247981A (ja) * | 2008-04-04 | 2009-10-29 | Saga Univ | 吸着剤及び貴金属の回収方法 |
| WO2010110288A1 (ja) | 2009-03-24 | 2010-09-30 | 株式会社カネカ | 免疫グロブリンに親和性を有するタンパク質および免疫グロブリン結合性アフィニティーリガンド |
| WO2011118699A1 (ja) | 2010-03-24 | 2011-09-29 | 株式会社カネカ | 免疫グロブリンに特異的に結合するタンパク質および免疫グロブリン結合性アフィニティーリガンド |
| JP2011252929A (ja) | 2011-09-05 | 2011-12-15 | Kaneka Corp | 多孔質担体、およびそれを用いた精製用吸着体、およびそれらの製造方法、およびそれらを用いた精製方法 |
Non-Patent Citations (4)
| Title |
|---|
| "Annals of the New York Academy of Sciences", vol. 1051, 2005, pages: 635 - 646 |
| AMERICAN HEART JOURNAL, vol. 152, no. 4, 2006 |
| KENICHI KASAI ET AL.: "Affinity chromatography", 1991, TOKYO KAGAKUDOUJIN |
| See also references of EP3059320A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3059320A1 (en) | 2016-08-24 |
| US10040053B2 (en) | 2018-08-07 |
| JP6517145B2 (ja) | 2019-05-22 |
| EP3059320A4 (en) | 2017-06-21 |
| JPWO2015056679A1 (ja) | 2017-03-09 |
| US20160251394A1 (en) | 2016-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10457705B2 (en) | Carrier for ligand immobilization | |
| JP6506554B2 (ja) | 吸着体、及びそれを用いた精製方法 | |
| JP6440320B2 (ja) | 多孔質セルロースビーズの製造方法およびそれを用いた吸着体 | |
| JPWO2016167268A1 (ja) | 多孔質セルロースビーズの製造方法およびそれを用いた吸着体 | |
| Salimi et al. | β-Cyclodextrin modified PES hollow fiber membrane, a new strategy for bilirubin separation | |
| JP6517145B2 (ja) | 多孔質セルロースビーズの製造方法及びそれを用いた吸着体 | |
| JP5623357B2 (ja) | 多孔質担体、およびそれを用いた精製用吸着体、およびそれらを用いた精製方法 | |
| JP2008279366A (ja) | 多孔質担体、およびそれを用いた精製用吸着体、およびそれらの製造方法、およびそれらを用いた精製方法 | |
| Thirumavalavan et al. | A short review on chitosan membrane for biomolecules immobilization | |
| Xue et al. | Surface-modified anodic aluminum oxide membrane with hydroxyethyl celluloses as a matrix for bilirubin removal | |
| JP6442409B2 (ja) | 多孔質セルロースビーズの製造方法 | |
| WO2019220866A1 (ja) | 多孔質セルロースビーズおよび吸着体の製造方法 | |
| CN111818981A (zh) | 用于生物分离的复合材料 | |
| JP5883068B2 (ja) | 多孔質担体、およびそれを用いた精製用吸着体、およびそれらの製造方法、およびそれらを用いた精製方法 | |
| JP4606524B2 (ja) | ポリリジン、及びポリリジンの製造方法、及びポリリジン組成物、及びエンドトキシンを除去する医薬品の生産方法 | |
| Zhao et al. | Imidazolium-modified sulfonated polyetheretherketone for selective isolation of hemoglobin | |
| JPWO2018186222A1 (ja) | 多孔質セルロースビーズおよび吸着体 | |
| Wang | Functionalization of Cellulose-Based Monolithic | |
| CN111818996A (zh) | 用于生物分离的复合材料 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| REEP | Request for entry into the european phase |
Ref document number: 2014853421 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014853421 Country of ref document: EP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14853421 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015542618 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15029293 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |