WO2015093507A1 - Nouvelle protéine modifiée du domaine extracellulaire de la protéine g - Google Patents
Nouvelle protéine modifiée du domaine extracellulaire de la protéine g Download PDFInfo
- Publication number
- WO2015093507A1 WO2015093507A1 PCT/JP2014/083346 JP2014083346W WO2015093507A1 WO 2015093507 A1 WO2015093507 A1 WO 2015093507A1 JP 2014083346 W JP2014083346 W JP 2014083346W WO 2015093507 A1 WO2015093507 A1 WO 2015093507A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- protein
- region
- immunoglobulin
- amino acid
- domain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/22—Affinity chromatography or related techniques based upon selective absorption processes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/315—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Streptococcus (G), e.g. Enterococci
Definitions
- the present invention is a protein comprising a conventional wild type protein G / B domain (extracellular domain), or a variant of the protein, wherein the binding activity in the weakly acidic region to the Fc region of immunoglobulin G is reduced.
- the binding activity in the weakly acidic region to the protein having the Fc / Fab region of immunoglobulin G (hereinafter referred to as “pH-responsive variant”) is also reduced. For example).
- affinity chromatography is a method for purifying a target protein using specific affinity with the target protein.
- proteins can be easily and selectively recovered.
- affinity is very strong, in order to dissociate the protein adsorbed on the chromatographic packing material, the pH is generally about 2.5. Elution with acidic buffer is often required. Under such strongly acidic conditions, activity reduction such as protein denaturation is likely to occur, and purification under milder conditions is required.
- Protein G is a membrane protein present in the cell membrane of Streptococcus spp. And is known to have specific binding activity to the Fc region of immunoglobulin G, which is a kind of antibody (Non-patent Document 1, Patent Document 1). ). Protein G is a multidomain membrane protein composed of a plurality of domains, and shows a binding activity to a protein having an Fc region of immunoglobulin G (hereinafter referred to as “antibody binding activity”). It is an extracellular domain (Non-patent Document 2). For example, in the case of protein G derived from the G148 strain shown in FIG. 1 of Patent Document 8, three domains B1, B2, and B3 exhibit antibody binding activity (also referred to as C1, C2, and C3 domains in the literature).
- the antibody is selectively adsorbed by contacting with a phase support. Thereafter, washing with a neutral to weak acid solution (pH 5 to 8) is performed to remove components other than antibodies. Finally, it is common to add a strongly acidic solution of pH 2.4 to 3.5 to desorb the antibody from the immobilized protein G and to elute it together with the strongly acidic solution (Patent Document 3). Thereby, the antibody can be isolated, recovered and purified with high purity.
- Non-patent Document 4 Treatment in a weakly acidic region at a pH higher than 2.4-3.5 is attempted, but since the binding force between the extracellular domain of protein G and the antibody is strong, in the weakly acidic region the antibody is separated from protein G. It does not elute and a sufficient recovery amount cannot be obtained.
- Protein G extracellular domain is known to bind to Fab (Non-patent Document 2), and one antibody molecule can bind to protein G extracellular domain in two regions, Fc region and Fab region. is there. In such a binding state, the antibody and the extracellular domain of protein G cannot be easily dissociated, making it difficult to recover the antibody.
- protein stability having thermal stability, chemical stability against denaturing agents, resistance to proteolytic enzymes, etc.
- Improved protein comprising an extracellular domain mutant of the above (Patent Document 5 and Patent Document 6), and further, has a binding property to an immunoglobulin Fc region and / or a binding property to the Fab region in a weakly acidic region.
- Patent Document 7 A reduced improved protein was also developed (Patent Document 7). However, all of these improved proteins contain only one domain exhibiting antibody binding activity.
- the present inventors have developed a protein comprising a tandem multimer of the improved protein (Patent Document 8). Compared with the tandem multimer of the wild-type protein G / B1 domain, such a protein has a much lower binding property in the weakly acidic region with the Fc region of human immunoglobulin G of different subclasses IgG1 and IgG3.
- the captured human immunoglobulin G can be more easily obtained in a weakly acidic region (about pH 4 to 5) without denaturation. It became possible to elute.
- the problem to be solved by the present invention is that the binding to the Fc region of immunoglobulin and / or the Fab in the weakly acidic region further compared with the conventional wild-type protein G / B domain or its modified protein. It is to provide a novel protein having a reduced binding property to a region and a more acidic pH shifted to a slightly acidic side. Furthermore, the problem to be solved by the present invention is to provide a chromatography column for protein separation and purification, particularly an affinity chromatography column for antibody purification, which is packed with the capture agent.
- the present inventor has a positive charge that exists within a certain distance from the Fc region in the domain variant / Fc complex model structure of wild-type protein G. We found that by substituting amino acid residues near the residues with positively charged residues, the binding activity of these proteins to the Fc region of immunoglobulin G in the weakly acidic region can be significantly reduced. Completed the invention.
- each aspect of the present invention is as follows.
- [Aspect 1] Compared to the protein consisting of wild-type protein G and B domains, it has binding activity to the Fc region of immunoglobulin G. By substituting residues near the positively charged residues of protein G with positively charged residues A protein with reduced binding activity in the weakly acidic region to the Fc region of immunoglobulin G.
- [Aspect 2] The protein according to embodiment 1, wherein the amino acid residue after mutation is histidine.
- Aspect 19 A method for purifying an immunoglobulin G or a protein having an Fc region or Fab region of immunoglobulin G using the affinity chromatography for purification according to aspect 18.
- the present invention there is a residue near the positively charged residue of protein G in an acidic solution as compared with a protein having a binding activity to the Fc region of immunoglobulin G and consisting of wild-type protein G and B domains.
- a positively charged residue By substituting with a positively charged residue, it is possible to provide a protein having reduced binding activity in the weakly acidic region to the Fc region of immunoglobulin G.
- the captured antibody such as immunoglobulin G is more easily eluted in a weakly acidic region without denaturation. It becomes possible.
- Protein G a streptococcal protein, is known to have a specific binding activity to the Fc region of immunoglobulin G, which is a kind of antibody (Reference Document 1). It is a protein useful for purification, removal, diagnosis, treatment, testing, etc. using antibodies. Protein G is a multidomain membrane protein composed of a plurality of domains, and shows a binding activity to a protein having an Fc region of immunoglobulin G (hereinafter referred to as “antibody binding activity”). It is an extracellular domain (Reference Document 2). For example, in the case of protein G derived from the G148 strain, three domains B1, B2, and B3 exhibit antibody binding activity (also referred to as C1, C2, and C3 domains in the literature).
- the present invention has a binding activity to the Fc region of immunoglobulin G, and a residue near the positively charged residue of protein G as a positively charged residue compared to a protein consisting of wild type protein G and B domains. It relates to a protein whose binding activity in the weakly acidic region to the Fc region of immunoglobulin G is reduced by substitution.
- immunoglobulin G examples include human and non-human animals, particularly various antibodies of mammals such as rats, mice, hamsters, goats and rabbits, and various fragments of antibodies such as Fab fragments of human IgG. including.
- its structure or component is not particularly limited, and includes any of various types of antibody molecules and their fragment molecules known to those skilled in the art. That is, in addition to normal (complete) IgG type antibody molecules, for example, single chain antibodies (scFv), single chain antibody dimers, bispecific antibodies, diabody type bispecific antibodies, And multimerized low molecular weight antibodies, and various antibody fragments such as Fab fragments, F (ab ′) 2 and Fab ′.
- the binding activity in the weakly acidic region with respect to the Fc region of immunoglobulin G is reduced means that, for example, as shown in the Examples of the present specification, pH 4 is measured in the SPR method. This means that the antibody dissociation rate (%) in the solution is higher than that of the wild type protein G ⁇ B or its domain variant, preferably about 2 to 30 times higher than that of the domain variant.
- the column in which the protein of the present invention is immobilized has an elution peak of immunoglobulin G such as human IgG in pH gradient affinity chromatography as compared to a column in which wild type protein G • B or a domain variant thereof is immobilized. This means that it shifts to the neutral side (pH value is about 0.6 to 2.6).
- Substituting a residue in the vicinity of a positively charged residue of protein G in the protein of the present invention with a positively charged residue specifically includes one to several positively charged residues of protein G (in acidic solution)
- a positively charged residue specifically includes one to several positively charged residues of protein G (in acidic solution)
- the closest residue of the positively charged residue of protein G is changed to a positively charged residue. To replace.
- the distance from the positively charged amino acid residue substituted with the positively charged residue in the wild type protein G • B or its domain variant is the origin of the charged atom of the positively charged residue, and any substituted amino acid With the heavy atom in the side chain of the residue as the end point, the smallest value among these distances is defined as the distance between the residues.
- the type of amino acid of the mutated residue is not particularly limited, but the mutated amino acid residue is preferably histidine.
- the positively charged residue of wild type protein G or its domain variant is the same as the Fc region in the wild type protein G / Fc complex crystal structure or the wild type protein G domain variant / Fc complex model structure derived therefrom. From the viewpoint of the pH responsiveness of the protein of the present invention, it is preferably in the vicinity of the binding interface, for example, within 6 angstroms from any amino acid residue in the Fc region.
- the distance between the positively charged residue and the mutated positively charged amino acid residue is preferably within about 6 angstroms in the above crystal structure or model structure.
- the design concept (design procedure) of the structure of the protein of the present invention having such characteristics is as follows. [Example of design procedure using PG19 described in Patent Document 8 (see FIG. 7)] (1) Based on the wild-type PG / Fc region complex crystal structure (pdb, 1FCC) and the PG19 crystal structure (pdb, 2ZW1), the PG19 / Fc region complex structure is modeled. Complex modeling is performed by software superpose (non-patent literature @@ E. Krissinel and K. Henrick (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst. D60, 2256-2268) is used for molecular structure calculation by software MOE (Ryoka System).
- Information on the atoms selected for calculating the inter-atomic distance between the PG19 / Fc region molecules and the PG19 molecule using the software CONTACT is as follows. For positively charged residues ( ⁇ ) contained in PG19, select a charged nitrogen atom. For the residue in PG19 closest to the positively charged residue ( ⁇ ), the closest side chain atom (main chain atom in the case of glycine) is selected from the above positively charged nitrogen atom. For the residue in the Fc region closest to the positively charged residue ( ⁇ ), the closest side chain atom (main chain atom in the case of glycine) is selected from the above positively charged nitrogen atom.
- wild-type protein G ⁇ B domain examples include any of B1, B2 and B3 of protein G of Streptococcus genus Streptococcus.
- the protein of the present invention is a mutant of the wild type protein G ⁇ B1 domain, and is compared to a protein comprising the wild type protein G ⁇ B1 domain (amino acid sequence of SEQ ID NO: 1 described in Patent Document 8).
- a protein comprising the wild type protein G ⁇ B1 domain amino acid sequence of SEQ ID NO: 1 described in Patent Document 8.
- One or several amino acids in the amino acid sequence of a conventional improved protein having reduced binding in the weakly acidic region of binding to the Fab region of immunoglobulin G and / or binding to the Fc region Those containing mutants obtained by mutation are preferred.
- an improved protein of the wild type protein G • B1 domain described in Patent Document 8 (SEQ ID NOS: 13 to 20). Amino acid sequence).
- the amino acid sequences of these wild-type protein G ⁇ B1 domains and mutants thereof are shown as SEQ ID NOS: 30 to 38 in the sequence listing of the present specification.
- Such a mutation in the amino acid sequence of the improved protein can be performed by any method known to those skilled in the art, for example, as described in the examples of the present specification.
- amino acid sequence of the mutant obtained by further mutating the amino acid sequence of such an improved protein the amino acids at positions 25, 36 and 41 in the sequence of the protein G / B domain or its domain mutant Mention may be made of proteins in which at least one of the residues is mutated to histidine.
- the protein of the present invention can be a tandem multimer of these proteins.
- the multimer can be appropriately converted into, for example, a dimer, trimer, tetramer, or pentamer according to the wild type.
- each extracellular domain variant which comprises the multimer contained in the protein of this invention is mutually different, or is mutually the same.
- each domain variant may be linked by a linker sequence.
- a linker sequence can be appropriately designed and adjusted by those skilled in the art in consideration of the amino acid sequence of each variant.
- the protein of the present invention may be a fusion protein comprising a fused amino acid sequence in which the amino acid sequence of any other protein is linked to the N-terminal side or C-terminal side.
- Examples of other amino acid sequences used for such fusion proteins include the amino acid sequence of oxaloacetatetdecarboxylase alpha-subunit c-terminal domain (OXADac).
- OXADac-protein G mutant fusion protein can carry a plurality of functions of an avidin binding activity derived from the OXADac region and an antibody binding activity derived from the protein G variant region as a single molecule.
- the protein of the present invention when synthesized in the form of a His-tagged or fusion protein with another protein, the protein is sequenced between the tag and the mutant protein after synthesis or between the other protein and the protein of the present invention. Even if it is degraded with a specific proteolytic enzyme, one to several amino acid residues may remain on the N-terminal side or C-terminal side of the protein of the present invention. In production, methionine derived from an initiation codon may be added to the N-terminal side, but the addition of these amino acid residues does not change the activity of the protein of the present invention as shown below. In addition, the addition of these amino acid residues does not lose the effect of the designed mutation. Therefore, the protein of the present invention naturally includes these mutations.
- a protein produced using Escherichia coli or the like, and further using an enzyme such as methionylaminopeptidase It can be obtained by selectively cleaving amino acid residues (Reference Document 7) and separating and purifying the reaction mixture by chromatography or the like.
- the present invention further relates to a nucleic acid encoding the protein, a recombinant vector containing the nucleic acid, and a transformant into which the recombinant vector has been introduced.
- the above-mentioned protein is immobilized on a water-insoluble solid phase support, and is characterized by immunoglobulin G or a protein having an Fc region or Fab region of immunoglobulin G (also referred to as “immunoglobulin G etc.”) ), An antibody containing the capture agent, affinity chromatography for purifying immunoglobulin G and the like, a method for purifying immunoglobulin G and the like using the affinity chromatography for purification, and the like.
- “having affinity” means that, for example, immunoglobulin G can be adsorbed in chromatography.
- a filler formed by immobilizing a protein on a water-insoluble carrier (water-insoluble solid support) represented by agarose beads is glass. It is preferable to use an affinity column packed in a column such as a tube.
- a buffer having a neutral pH is used, and any salt species can be used as long as the pH can be adjusted. Typically, phosphate buffer, Tris buffer, sodium chloride, etc. The one in which the electrolyte is dissolved is used.
- the pH of the adsorption buffer is 9.0 to 6.5, preferably pH 8.0 to 7.0.
- the elution buffer may be in the pH range where the target immunoglobulin G or the like is eluted, and one having a pH of 6.5 to 2.0 is used.
- the type of elution buffer may be any known to those skilled in the art, and representative examples include phosphate buffer, citrate buffer, acetate buffer, glycine buffer, and the like.
- the operation itself in the purification method of the present invention can be carried out by ordinary operations known to those skilled in the art. That is, as in normal affinity purification, first, a sample solution containing immunoglobulin G or the like to be purified is injected into a column stabilized with an adsorption buffer, and immunoglobulin G or the like is adsorbed to the filler. Thereafter, after washing away non-adsorbed components remaining in the column with the adsorption buffer, the immunoglobulin G adsorbed with the elution buffer is eluted, and the immunoglobulin G is recovered in the eluate. It should be noted that other affinity purification conditions such as the flow rate (flow rate) of the adsorption buffer and elution buffer and the column temperature can be appropriately determined by those skilled in the art.
- any purification means can be used as long as it utilizes the affinity between a protein containing a domain mutant (artificially mutated domain) and immunoglobulin G or the like.
- any means known to those skilled in the art such as immunoprecipitation or immobilizing the protein on magnetic beads can be used.
- mutant protein described in Patent Document 8 As a preferable example of the mutant of the wild type protein G ⁇ B1 domain contained in the improved protein used as the basis for obtaining a suitable protein in the present invention, the mutant protein described in Patent Document 8 is mentioned. be able to. Such mutant proteins can be easily prepared by those skilled in the art according to the method described in Patent Document 7 or Patent Document 8, for example, by the following method.
- Production of protein (1) Production of protein by genetic engineering a. Gene encoding protein (variant)
- a genetic engineering method can be used to produce the above-designed protein.
- the gene used in such a method has a binding activity to a protein having an Fc region of immunoglobulin G, and at least to an Fc region of immunoglobulin G compared to a protein comprising a wild type protein G / B domain.
- the binding activity in the weakly acidic region is reduced, while the binding activity in the neutral region to immunoglobulin G is not reduced, such as any of SEQ ID NOS: 14 to 16 in Patent Document 8.
- the gene used in the present invention is a nucleic acid that hybridizes under stringent conditions with a nucleic acid comprising a sequence complementary to the base sequence of the above nucleic acid, and an antibody, immunoglobulin G, or immunoglobulin G.
- Examples also include a nucleic acid encoding the mutant protein having a reduced binding activity.
- stringent conditions refer to conditions in which a specific hybrid is formed and a non-specific hybrid is not formed.
- nucleic acids having high identity refers to conditions under which nucleic acids having high identity (identity is 60% or higher, preferably 80% or higher, more preferably 90% or higher, most preferably 95% or higher) hybridize. More specifically, it refers to conditions under which the sodium concentration is 150 to 900 mM, preferably 600 to 900 mM, and the temperature is 60 to 68 ° C., preferably 65 ° C.
- hybridization conditions are 65 ° C. and washing conditions are 0.1 ⁇ SSC containing 0.1% SDS at 65 ° C. for 10 minutes
- hybridization is performed by a conventional method such as Southern blotting or dot blot hybridization. When it is confirmed that the hybridization occurs, it can be said that the cells hybridize under stringent conditions.
- the gene encoding the protein of the present invention includes the above nucleic acids and nucleic acids encoding any of the above linker sequences, depending on the desired structure of the protein of the present invention.
- Nucleic acid encoding each mutant protein constituting a tandem multimer and a nucleic acid encoding a linker sequence may be linked in plural, or the nucleic acid and a nucleic acid encoding an amino acid sequence of an arbitrary protein may be linked. And may be designed to encode a fused amino acid sequence.
- the gene of the present invention described above can be synthesized by chemical synthesis, PCR, cassette mutagenesis, site-directed mutagenesis and the like. For example, a plurality of oligonucleotides up to about 100 bases having a complementary region of about 20 base pairs at the end are chemically synthesized, and the target gene is totally synthesized by combining these and performing the overlap extension method (Reference Document 8). be able to.
- the recombinant vector of the present invention can be obtained by linking (inserting) a gene containing the above-described base sequence to an appropriate vector.
- the vector used in the present invention is not particularly limited as long as it can be replicated in the host or can integrate the target gene into the host genome. For example, bacteriophage, plasmid, cosmid, phagemid and the like can be mentioned.
- plasmid DNA As plasmid DNA, plasmids derived from actinomycetes (eg pK4, pRK401, pRF31 etc.), plasmids derived from E. coli (eg pBR322, pBR325, pUC118, pUC119, pUC18 etc.), plasmids derived from Bacillus subtilis (eg pUB110, pTP5 etc.) Yeast-derived plasmids (eg, YEp13, YEp24, YCp50, etc.) and the like, and phage DNAs include ⁇ phage ( ⁇ gt10, ⁇ gt11, ⁇ ZAP, etc.).
- animal viruses such as retrovirus or vaccinia virus
- insect virus vectors such as baculovirus
- a method in which purified DNA is cleaved with a suitable restriction enzyme, inserted into a restriction enzyme site or a multicloning site of a suitable vector DNA, and linked to the vector is employed. .
- the gene needs to be integrated into the vector so that the mutant protein of the invention is expressed.
- the vector of the present invention includes a promoter, a base sequence of a gene, a cis element such as an enhancer, a splicing signal, a poly A addition signal, a selection marker, a ribosome binding sequence (SD sequence), an initiation codon, a termination codon, if desired. Etc. can be connected.
- a tag sequence for facilitating purification of the protein to be produced can also be linked.
- a base sequence encoding a known tag such as His tag, GST tag, MBP tag, BioEase tag can be used.
- Whether or not a gene has been inserted into a vector can be confirmed using a known genetic engineering technique. For example, in the case of a plasmid vector or the like, it can be confirmed by subcloning the vector using a competent cell, extracting the DNA, and then specifying the base sequence using a DNA sequencer. For other vectors that can be subcloned using bacteria or other hosts, the same technique can be used. In addition, vector selection using a selection marker such as a drug resistance gene is also effective.
- a transformant can be obtained by introducing the recombinant vector of the present invention into a host cell so that the mutant protein of the present invention can be expressed.
- the host used for transformation is not particularly limited as long as it can express a protein or polypeptide. Examples include bacteria (E. coli, Bacillus subtilis, etc.), yeast, plant cells, animal cells (COS cells, CHO cells, etc.) and insect cells.
- the recombinant vector When a bacterium is used as a host, the recombinant vector is autonomously replicable in the bacterium, and at the same time, it is composed of a promoter, a ribosome binding sequence, a start codon, a nucleic acid encoding the mutant protein of the present invention, and a transcription termination sequence. It is preferable.
- E. coli include Escherichia coli BL21
- Bacillus subtilis include Bacillus subtilis.
- the method for introducing a recombinant vector into bacteria is not particularly limited as long as it is a method for introducing DNA into bacteria. Examples thereof include a heat shock method, a method using calcium ions, and an electroporation method.
- yeast When yeast is used as a host, for example, Saccharomyces cerevisiae, Schizosaccharomyces pombe and the like are used.
- the method for introducing a recombinant vector into yeast is not particularly limited as long as it is a method for introducing DNA into yeast, and examples thereof include an electroporation method, a spheroplast method, and a lithium acetate method.
- monkey cells COS-7, Vero, Chinese hamster ovary cells (CHO cells), mouse L cells, rat GH3, human FL cells and the like are used.
- methods for introducing a recombinant vector into animal cells include an electroporation method, a calcium phosphate method, and a lipofection method.
- Sf9 cells and the like are used.
- the method for introducing a recombinant vector into insect cells include the calcium phosphate method, lipofection method, electroporation method and the like.
- PCR Southern hybridization
- Northern hybridization or the like.
- DNA is prepared from the transformant, PCR is performed by designing a DNA-specific primer. Subsequently, the PCR amplification product is subjected to agarose gel electrophoresis, polyacrylamide gel electrophoresis, capillary electrophoresis, or the like, stained with ethidium bromide, SyberGreen solution, etc., and the amplification product is detected as a single band. You can confirm that it has been converted.
- PCR can be performed using a primer previously labeled with a fluorescent dye or the like to detect an amplification product.
- the protein of the present invention can be obtained by culturing the above-described transformant and collecting it from the culture.
- the culture means any of culture supernatant, cultured cells, cultured cells, or disrupted cells or cells.
- the method for culturing the transformant of the present invention is carried out according to a usual method used for culturing a host.
- a medium for culturing transformants obtained using microorganisms such as Escherichia coli and yeast as a host contains a carbon source, nitrogen source, inorganic salts, etc. that can be assimilated by the microorganisms, and efficiently cultures the transformants.
- a carbon source include carbohydrates such as glucose, fructose, sucrose, and starch, organic acids such as acetic acid and propionic acid, and alcohols such as ethanol and propanol.
- Examples of the nitrogen source include ammonia, ammonium chloride, ammonium sulfate, ammonium acetate, ammonium salts of organic acids such as ammonium phosphate or other nitrogen-containing compounds, peptone, meat extract, corn steep liquor, and the like.
- Examples of the inorganic substance include monopotassium phosphate, dipotassium phosphate, magnesium phosphate, magnesium sulfate, sodium chloride, ferrous sulfate, manganese sulfate, copper sulfate, and calcium carbonate.
- the culture is usually performed at 20 to 37 ° C. for 12 hours to 3 days under aerobic conditions such as shaking culture or aeration and agitation culture.
- the protein of the present invention When the protein of the present invention is produced in cells or cells after culturing, the protein or cells are collected by crushing the cells or cells by sonication, repeated freeze-thawing, homogenizer treatment, etc. . When the protein is produced outside the cells or cells, the culture solution is used as it is, or the cells or cells are removed by centrifugation or the like. Thereafter, a general biochemical method used for protein isolation and purification, for example, ammonium sulfate precipitation, gel chromatography, ion exchange chromatography, affinity chromatography, etc. can be used alone or in appropriate combination in the culture. From the above, the protein of the present invention can be isolated and purified.
- the mutant protein of the present invention when using a so-called cell-free synthesis system in which only factors (enzymes, nucleic acids, ATP, amino acids, etc.) involved in protein biosynthesis reactions are mixed, the mutant protein of the present invention can be obtained from a vector without using living cells. Can be synthesized in vitro (Reference 9). Thereafter, using the same purification method as described above, the mutant protein of the present invention can be isolated and purified from the mixed solution after the reaction. In order to confirm whether the isolated and purified protein of the present invention is a protein having a desired amino acid sequence, a sample containing the protein is analyzed. As an analysis method, SDS-PAGE, Western blotting, mass spectrometry, amino acid analysis, amino acid sequencer, etc. can be used (Reference Document 10).
- the protein of the present invention can also be produced by organic chemical methods such as solid phase peptide synthesis. Protein production methods using such techniques are well known in the art and are briefly described below.
- the protection having the amino acid sequence of the protein of the present invention is preferably achieved by repeating the polycondensation reaction of the activated amino acid derivative using an automatic synthesizer.
- the polypeptide is synthesized on the resin.
- the protective polypeptide is cleaved from the resin and the side chain protecting group is cleaved simultaneously. This cleaving reaction is known to have an appropriate cocktail depending on the type of resin, protecting group, and amino acid composition (Reference 11).
- the crude protein is transferred from the organic solvent layer to the aqueous layer, and the target protein is purified.
- reverse phase chromatography or the like can be used (Ref. 11).
- the protein of the present invention can be used as a capture agent for antibodies and the like by utilizing its antibody binding property.
- the antibody capture agent can be used for purification and removal of antibodies, diagnosis, treatment, examination, etc. using antibodies.
- the antibody capture agent of the present invention may be in any form as long as it contains the protein of the present invention.
- the form of the mutant protein of the present invention immobilized on a water-insoluble solid support. Is appropriate.
- water-insoluble carrier used examples include inorganic carriers such as glass beads and silica gel, synthetic polymers such as crosslinked polyvinyl alcohol, crosslinked polyacrylate, crosslinked polyacrylamide, and crosslinked polystyrene, crystalline cellulose, crosslinked cellulose, crosslinked agarose, and crosslinked dextran.
- inorganic carriers such as glass beads and silica gel
- synthetic polymers such as crosslinked polyvinyl alcohol, crosslinked polyacrylate, crosslinked polyacrylamide, and crosslinked polystyrene, crystalline cellulose, crosslinked cellulose, crosslinked agarose, and crosslinked dextran.
- organic carriers composed of polysaccharides and organic-organic and organic-inorganic composite carriers obtained by a combination thereof.
- hydrophilic carriers have relatively little nonspecific adsorption, and antibodies or immunoglobulin G Alternatively, it is preferable because the protein having the Fc region of immunoglobulin G has good selectivity.
- hydrophilic carrier refers to a carrier having a contact angle with water of 60 ° or less when the compound constituting the carrier is formed into a flat plate shape.
- Such carriers include cellulose, chitosan, dextran and other polysaccharides, polyvinyl alcohol, saponified ethylene-vinyl acetate copolymer, polyacrylamide, polyacrylic acid, polymethacrylic acid, polymethyl methacrylate, polyacrylic acid grafting
- Representative examples include carriers made of polyethylene, polyacrylamide grafted polyethylene, glass and the like.
- porous cellulose gels GCL2000 and GC700 include porous cellulose gels GCL2000 and GC700, Sephacryl® S-1000 with allyl dextran and methylenebisacrylamide cross-linked covalently, acrylate-based carrier Toyopearl, agarose-based cross-linked carrier SepharoseCL4B, epoxy group
- Eupergit C250L which is polymethacrylamide activated by the above method
- the present invention is not limited to these carriers and activated carriers.
- Each of the above carriers may be used alone, or any two or more of them may be mixed.
- the water-insoluble carrier used in the present invention preferably has a large surface area in view of the purpose and method of use of the present antibody capture agent, and has a large number of pores of an appropriate size, that is, is porous. preferable.
- the form of the carrier can be any of beads, fibers, membranes (including hollow fibers), and any form can be selected.
- a bead shape is particularly preferably used because of easy preparation of a carrier having a specific exclusion limit molecular weight.
- the average particle size of beads is 10 to 2500 ⁇ m, and is particularly preferably in the range of 25 ⁇ m to 800 ⁇ m from the viewpoint of easy ligand immobilization reaction. Furthermore, if a functional group that can be used for the ligand immobilization reaction is present on the surface of the carrier, it is convenient for immobilization of the ligand.
- these functional groups include hydroxyl group, amino group, aldehyde group, carboxyl group, thiol group, silanol group, amide group, epoxy group, succinimide group, acid anhydride group, iodoacetyl group and the like.
- a hydrophilic spacer for example, a polyalkylene oxide derivative in which both ends are substituted with a carboxyl group, an amino group, an aldehyde group, an epoxy group or the like is preferably used.
- the method and conditions for immobilizing the mutant protein to be introduced into the carrier and the organic compound used as the spacer are not particularly limited, but the method generally employed when immobilizing a protein or peptide on the carrier is used. Illustrate.
- the carrier is reacted with cyanogen bromide, epichlorohydrin, diglycidyl ether, tosyl chloride, tresyl chloride, hydrazine, etc.
- the carrier compounds that are immobilized as ligands from the functional groups that the carrier originally has reacted
- a functional group that can be easily immobilized reacting with a compound to be immobilized as a ligand, a method of immobilizing, or a system in which a compound to be immobilized as a carrier and a ligand exists, such as a condensation reagent such as carbodiimide, or glutaraldehyde
- Performance confirmation test of protein and antibody capture agent The protein produced as described above (hereinafter also simply referred to as “protein”) and the antibody capture agent may be selected by performing the following performance confirmation test and selecting a good one. However, both the protein and the antibody capturing material of the present invention had good performance.
- the antibody binding of the protein of the present invention is confirmed and evaluated using Western blotting, immunoprecipitation, pull-down assay, ELISA (Enzyme-Linked ImmunoSorbent Assay), surface plasmon resonance (SPR) method, etc. can do.
- the SPR method allows the interaction between living organisms to be observed over time in real time without a label, so that the binding reaction of the mutant protein can be quantitatively evaluated from a kinetic viewpoint.
- the antibody binding property of the mutant protein immobilized on the water-insoluble solid phase support can be confirmed and evaluated by the above SPR method or liquid chromatography method. Among them, the liquid chromatography method can accurately evaluate the pH dependence on the antibody binding property.
- the thermal stability of the mutant protein of the present invention includes circular dichroism (CD) spectrum, fluorescence spectrum, infrared spectroscopy, differential scanning calorimetry, residual activity after heating. Etc. can be used for evaluation.
- CD spectrum is a spectroscopic analysis method that sharply reflects changes in the secondary structure of the protein, so the change in the three-dimensional structure with respect to the temperature of the mutant protein is observed, and the structural stability is quantified thermodynamically. Can be evaluated.
- Patent Document 8 The following contents are specifically disclosed in Patent Document 8.
- Example 1 the wild type amino acid sequence of the protein G • B1 domain represented by [SEQ ID NO: 1], the wild type amino acid sequence of the protein G • B2 domain represented by [SEQ ID NO: 2], and [SEQ ID NO: 3]
- a mutant protein contained in the protein used in the present invention in which a mutation is introduced into the B1, B2 or B3 domain of protein G (hereinafter referred to as "improved protein G")
- the amino acid residue to be substituted is specified and the amino acid residue to be substituted is specified.
- Example 2 the amino acid sequences of a plurality of improved protein Gs represented by [SEQ ID NO: 4] to [SEQ ID NO: 19] are obtained using the selected mutation target site and the above-identified amino acid residue information to be substituted.
- [SEQ ID NO: 13] to [SEQ ID NO: 20] were finally selected as specific amino acid sequences, and an improved protein G showing this sequence was actually synthesized and its molecular properties were evaluated. Are listed.
- Example 3 the base sequence of the nucleic acid encoding the amino acid sequence of the improved protein G ([SEQ ID NO: 13] to [SEQ ID NO: 20]) and the base sequence of Oxaloacetate decarboxylase alpha-subunit c-terminal domain (OXADac) No. 31]
- the nucleotide sequence of the mutant protein using cocoons is described.
- Example 4 a plasmid vector containing a gene encoding improved protein G was synthesized using the PG gene consisting of the nucleotide sequence of [SEQ ID NO: 21] to [SEQ ID NO: 29], and then Oxaloacetate decarboxylase using E. coli. It describes the production of a fusion protein of alpha-subunit c-terminal domain (OXADac) [SEQ ID NO: 31] and a mutant protein.
- OXADac alpha-subunit c-terminal domain
- Example 5 a plasmid vector containing a gene encoding improved protein G was synthesized using various primers ([SEQ ID NO: 32] to [SEQ ID NO: 35]), and then Met-added improved protein using Escherichia coli. The manufacture of G is described.
- Example 6 the purity of the improved protein G was confirmed by polyacrylamide gel electrophoresis.
- Example 7 the molecular weight of the improved protein G was measured by a MALDI-TOF mass spectrometer.
- Example 8 the protein was identified.
- pH gradient affinity chromatography was performed using a column on which the OXADac-PG fusion protein was immobilized, and the pH at which the monoclonal antibody was eluted was examined.
- Example 9 in Example 9, stepwise pH affinity chromatography was performed using a column immobilized with OXADac-PG fusion protein, and elution of monoclonal antibodies was examined at several pHs.
- Example 10 the protein dissociation property in the weakly acidic region of improved protein G was evaluated.
- Example 11 In G mutant was evaluated by surface plasmon resonance (SPR) method.
- the mutant was obtained in the neutral region and in the weakly acidic region where 95% or more of the histidine residues were protonated.
- the antibody binding property of the protein was evaluated by the surface plasmon resonance (SPR) method, the thermal stability of the mutant protein was evaluated in Example 12, the single crystal of the mutant protein was prepared in Example 13, It is described that the three-dimensional structure was determined by X-ray diffraction analysis.
- Example 14 a tandem type of a trimeric wild type PG (CGB01H-3D, [SEQ ID NO: 36]) ⁇ ⁇ added with a cysteine residue and a His tag on the carboxyl terminal side or a mutant PG which is a protein of the present invention.
- Example 15 describes a comparison of protein G extracellular domain mutant tandem multimers and the same monomers with respect to antibody binding dissociation with IgG1 type humanized monoclonal antibodies.
- Example 16 a mutant PG monomer mutant PG (CGB19H-1D, FIG. 4, [SEQ ID NO: 38]) having a cysteine residue and a His tag added to the carboxyl terminus, a tandem tetramer of the mutant PG Incorporating a gene encoding the body PG (CGB19H-4D, FIG. 4, [SEQ ID NO: 39]) and the tandem pentamer PG of the mutant PG (CGB19H-5D, FIG. 4, [SEQ ID NO: 40]) 3 It describes the production of a protein G extracellular domain mutant monomer, tandem tetramer, and pentamer using a species of artificially synthesized expression plasmid.
- each of the mutant proteins for the IgG1-type humanized monoclonal antibody was prepared by immobilizing the monomer and tandem-type multimer of the extracellular domain mutant of protein G to the solid phase through a cysteine residue at the carboxyl terminal. It is described that the antibody binding property of each was comparatively evaluated by the SPR method.
- N-terminus amino terminus
- C-terminus carboxyl terminus
- Non-patent Document 5 such as histidine residues of a fusion protein (SEQ ID NO: 1) containing an extracellular domain mutant of protein G (hereinafter referred to as “modified protein G”). It consists of an amino acid sequence in which a part is substituted with a wild-type amino acid residue.
- the site of mutagenesis introduction of histidine was determined as follows.
- the lysine residues that are positively charged residues of the improved protein G are identified from the three-dimensional structure information (PDB: 2ZW1) of protein G downloaded from the public database Protein Data Bank, and the residues present within 7 ⁇ ⁇ of the lysine residue Was identified as a candidate for mutagenesis.
- CONTACT of the structural analysis software CCP4 was used.
- Non-Patent Document 7 residues that have been found to be important for binding to Fc from the previously reported mutant analysis (Non-Patent Document 7), and the structure of protein G that has been determined from structural information Residues important for formation were excluded from the mutagenesis candidates, and residues that contact these important residues were also excluded from the candidates. As a result, nine residue positions of Asp1, Asn8, Glu15, Thr25, Glu36, Tyr33, Gly41, Thr55, and Glu56 were determined as mutation introduction sites.
- SEQ ID NO: 2 a fusion protein gene
- PG19 improved protein G
- Mutant gene encoding the amino acid sequence (SEQ ID NO: 21-29) of the fusion protein containing the body (PG19 T25H, PG19 Y33H, PG19 E36H, PG19 G41H, PG19 T55H, PG19 D1H, PG19 N8H, PG19 E15H, Each was produced.
- An expression plasmid containing the gene of each histidine additional mutant was isolated and purified, and transformed into E. coli BL21 (DE3) strain (Novagen) for expression.
- the histidine additional mutant of the present invention was immobilized on a solid phase via a cysteine residue at the carboxyl terminus, and the pH responsiveness of each histidine additional mutant was evaluated by the SPR method.
- improved protein G was also evaluated in the same manner.
- the improved protein G (PG19) and histidine additional mutant (PG19 T25H, PG19 Y33H, PG19 E36H, PG19 G41H, PG19 T55H, PG19 D1H, PG19 N8H, PG19 E15H and PG19 E56H) were immobilized by maleimide coupling method using EMCH (N- [ ⁇ -Maleimidocaproic acid] hydrazide, trifluoroacetic acid) (Thermo scientific).
- EMCH N- [ ⁇ -Maleimidocaproic acid] hydrazide, trifluoroacetic acid
- IgG1-type humanized monoclonal antibody was dissolved in a running buffer (10 mM HEPES pH7.4, 150 mM NaCl, 0.005% v / v Surfactant P20) to prepare a sample antibody solution of 1 mg / ml.
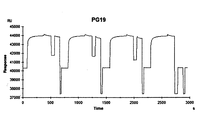
- SPR measurement was performed using a Biacore T100 (GE Healthcare) at a reaction temperature of 25 ° C., and the SPR response at each pH solution was measured. ( Figures 2-4).
- an extracellular domain mutant of protein G was produced, and a column using the protein was produced.
- To express the target protein 0.5 mM IPTG was added, and further cultured with shaking at 37 ° C. for 3 hours.
- the collected cells were suspended in PBS, subjected to ultrasonic disruption and then centrifuged, and the resulting supernatant was used as a total protein solution.
- Recombinant PG was adsorbed onto a 1 ml column of HisTrap FF (GE Healthcare Bioscience), washed with 20 mM imidazole, and eluted with 500 mM imidazole to obtain purified protein.
- 1 mL of the activated carrier was collected on a glass filter and washed with a coupling buffer (0.1 M sodium phosphate, 1.0 M sodium sulfate, 1 mM EDTA, pH 8.0). Transfer the activated carrier to the flask, add 1.4 ml of recombinant PG-containing solution containing 5 mg / ml recombinant PG (PG19 T25H) and 2 mL of coupling buffer, and shake for 16 hours at 37 ° C and 150 rpm. Of cysteine residues. The solution was filtered with a glass filter and washed with a coupling buffer.
- a coupling buffer 0.1 M sodium phosphate, 1.0 M sodium sulfate, 1 mM EDTA, pH 8.0.
- the carrier was transferred to a flask, 3 ml of a solution of 1 M thioglycerol, 0.1 M sodium phosphate, 1 mM EDTA, pH 8.0 was added, and the mixture was shaken at 37 ° C. and 150 rpm for 4 hours to mask unreacted active groups.
- the solution was filtered off with a glass filter, and washing solution 1 (0.1 M Tris-HCl, 0.5 M sodium chloride, pH 8.0) and washing solution 2 (0.1 M acetic acid, 0.5 M sodium chloride, pH 4.0) were alternately washed with 15 ml for 3 cycles.
- 1 ml of the immobilization carrier was washed with ultrapure water and packed in a Tricon 5/50 Column.
- PG19 E36H 921 ⁇ L of 7.6mg / mL recombinant PG-containing solution
- ⁇ PG19 G41H 897 ⁇ L of 7.8mg / mL recombinant PG solution -PG01: 853 ⁇ L of 8.2mg / mL recombinant PG containing solution ⁇ PG19: 1.4mL of 12.0mg / mL recombinant PG solution
- the antibody adsorption capacity of the recombinant PG-immobilized column prepared in Example 3 was measured.
- human IgG orientational yeast
- the injection was continued until the absorbance at 280 nm of the eluate reached 15% of the absorbance of the injected sample, and after washing with the adsorption buffer, the adsorption buffer was replaced with 20 mM citric acid (pH 2.4).
- the dynamic binding capacity was calculated from the amount of sample injected until the absorbance excluding non-adsorbed components at 280 nm of the eluate reached 10% of the absorbance of the injected sample.
- Table 3 shows the DBC of each immobilized column. PG19 G41H had a significantly reduced DBC compared to other columns.
- pH gradient affinity chromatography was performed using the recombinant PG-immobilized column prepared in Example 3.
- human IgG (oriental yeast) prepared to 1 mg / mL was injected.
- the solution was replaced with 20 mM citric acid buffer (pH 6.0) and continuously with 20 mM citric acid (pH 2.4) at a flow rate of 1.0 mL / min over 80 min.
- Human IgG elution peak is around pH3.9 for PG01 immobilized column, around pH4.4 for PG19 immobilized column, around pH5.0 for PG19 T25H immobilized column, around pH4.5 for PG19 E36H immobilized column, PG19 G41H It was around pH 6.5 in the immobilized column (FIG. 6). Therefore, the PG19 G41H immobilized column can elute human IgG on the neutral side from pH 6.0, and the PG19 T25H and PG19 E36H immobilized columns elute under mild acidic conditions compared to the PG19 immobilized column. It became clear that it was possible.
- the wild-type protein G extracellular domain is commercially available as an affinity chromatography carrier for antibody purification and a test reagent for antibody detection, and is widely used in each field of life science.
- the demand for these products has increased dramatically. Therefore, many protein G extracellular domain-containing products utilize the advantage that the binding activity in the weakly acidic region to the Fc region of immunoglobulin G is reduced by substituting the protein of the present invention with the wild type. This greatly contributes to technological development in a wide range of technical fields dealing with various antibodies.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Analytical Chemistry (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
La présente invention vise à fournir une nouvelle protéine, ou analogues, qui a une aptitude réduite à une liaison à la région Fc et/ou à la région Fab d'une immunoglobuline sur une gamme faiblement acide et qui a un pH d'élution qui a été décalé vers une acidité faible. La présente invention concerne une protéine qui a une activité de liaison avec la région Fc de l'immunoglobuline G et, en conséquence du remplacement du résidu positivement chargé par un résidu au voisinage d'un résidu positivement chargé de la protéine G, a une activité réduite de liaison à la région Fc de l'immunoglobuline G dans une gamme faiblement acide par comparaison avec une protéine comprenant un domaine B de la protéine G de type sauvage ; un fixateur d'immunoglobuline G ou d'une protéine ayant la région Fc ou la région Fab de l'immunoglobuline G, la protéine étant immobilisée sur un support en phase solide insoluble dans l'eau ; une chromatographie d'affinité pour la purification de l'immunoglobuline G ou d'une protéine ayant la région Fc ou la région Fab de l'immunoglobuline G, ladite chromatographie d'affinité comprenant le fixateur ; et un procédé qui utilise la chromatographie d'affinité pour la purification, et dans lequel l'immunoglobuline G ou une protéine ayant la région Fc ou la région Fab de l'immunoglobuline est purifiée.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013262097A JP2017029001A (ja) | 2013-12-19 | 2013-12-19 | プロテインgの細胞膜外ドメインの新規な改変型タンパク質 |
| JP2013-262097 | 2013-12-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015093507A1 true WO2015093507A1 (fr) | 2015-06-25 |
Family
ID=53402852
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/083346 Ceased WO2015093507A1 (fr) | 2013-12-19 | 2014-12-17 | Nouvelle protéine modifiée du domaine extracellulaire de la protéine g |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2017029001A (fr) |
| WO (1) | WO2015093507A1 (fr) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06153945A (ja) * | 1992-06-25 | 1994-06-03 | Yakult Honsha Co Ltd | 新規なプロテアーゼ及び新規なプロテアーゼ活性を有する微生物 |
| JP2009297018A (ja) * | 2008-05-16 | 2009-12-24 | National Institute Of Advanced Industrial & Technology | 弱酸性域での解離特性を改良した抗体結合性タンパク質及び抗体捕捉剤 |
| JP2012531439A (ja) * | 2009-06-26 | 2012-12-10 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | 天然の免疫グロブリン形式を有する容易に単離される二重特異性抗体 |
| WO2013018880A1 (fr) * | 2011-08-04 | 2013-02-07 | 独立行政法人産業技術総合研究所 | Nouvelle protéine modifiée comportant un multimère de type tandem d'un domaine extracellulaire mutant de protéine g |
| JP2013075916A (ja) * | 1999-01-15 | 2013-04-25 | Biogen Idec Ma Inc | Tweakのアンタゴニストおよびtweakレセプターのアンタゴニスト、ならびに免疫学的障害を処置するためのこれらの使用 |
-
2013
- 2013-12-19 JP JP2013262097A patent/JP2017029001A/ja active Pending
-
2014
- 2014-12-17 WO PCT/JP2014/083346 patent/WO2015093507A1/fr not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06153945A (ja) * | 1992-06-25 | 1994-06-03 | Yakult Honsha Co Ltd | 新規なプロテアーゼ及び新規なプロテアーゼ活性を有する微生物 |
| JP2013075916A (ja) * | 1999-01-15 | 2013-04-25 | Biogen Idec Ma Inc | Tweakのアンタゴニストおよびtweakレセプターのアンタゴニスト、ならびに免疫学的障害を処置するためのこれらの使用 |
| JP2009297018A (ja) * | 2008-05-16 | 2009-12-24 | National Institute Of Advanced Industrial & Technology | 弱酸性域での解離特性を改良した抗体結合性タンパク質及び抗体捕捉剤 |
| JP2012531439A (ja) * | 2009-06-26 | 2012-12-10 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | 天然の免疫グロブリン形式を有する容易に単離される二重特異性抗体 |
| WO2013018880A1 (fr) * | 2011-08-04 | 2013-02-07 | 独立行政法人産業技術総合研究所 | Nouvelle protéine modifiée comportant un multimère de type tandem d'un domaine extracellulaire mutant de protéine g |
Non-Patent Citations (1)
| Title |
|---|
| HIDEKI WATANABE ET AL.: "Optimizing pH response of affinity between protein G and IgG Fc: how electrostatic modulations affect protein- protein interactions.", J. BIOL. CHEM., vol. 284, no. 18, 2009, pages 12373 - 12383 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017029001A (ja) | 2017-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5229888B2 (ja) | 弱酸性域での易解離性を向上したプロテインa変異型タンパク質及び抗体捕捉剤 | |
| JP5812303B2 (ja) | 酸性域での親和性が低下したプロテインa変異型タンパク質及び抗体捕捉剤 | |
| EP3497118B1 (fr) | Protéines de liaison fc stables dans des conditions alcalines pour chromatographie d'affinité | |
| JP6181145B2 (ja) | 改善された特異性を有する新規免疫グロブリン結合タンパク質 | |
| AU2016293063B2 (en) | Novel immunoglobulin-binding proteins and their use in affinity purification | |
| WO2014021240A1 (fr) | Protéine contenant un agent désactivateur formée à partir d'un multimère de type tandem d'un domaine extracellulaire muté de la protéine g | |
| JP5858394B2 (ja) | 弱酸性域での解離特性を改良した抗体結合性タンパク質及び抗体捕捉剤 | |
| JPWO2015050153A1 (ja) | 免疫グロブリンG(IgG)のFc部分を有するタンパク質に対するアフィニティを有する複数のドメイン間をリンカーで結合させて成るタンパク質 | |
| US20140221613A1 (en) | Novel modified protein comprising tandem-type multimer of mutant extracellular domain of protein g | |
| JP5236311B2 (ja) | IgG−Fab断片抗体結合性ペプチド | |
| JP2015019615A (ja) | プロテインgの細胞膜外ドメイン変異体の新規な改変型タンパク質 | |
| WO2024055988A1 (fr) | Protéine de liaison à l'immunoglobuline et son utilisation | |
| JP2015003872A (ja) | マウスIgGの精製方法 | |
| WO2015093507A1 (fr) | Nouvelle protéine modifiée du domaine extracellulaire de la protéine g | |
| JP4481260B2 (ja) | 抗体結合性ペプチド |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14871779 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14871779 Country of ref document: EP Kind code of ref document: A1 |