WO2016125829A1 - 共重合ポリエステル樹脂、及びその製造方法 - Google Patents
共重合ポリエステル樹脂、及びその製造方法 Download PDFInfo
- Publication number
- WO2016125829A1 WO2016125829A1 PCT/JP2016/053225 JP2016053225W WO2016125829A1 WO 2016125829 A1 WO2016125829 A1 WO 2016125829A1 JP 2016053225 W JP2016053225 W JP 2016053225W WO 2016125829 A1 WO2016125829 A1 WO 2016125829A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- copolyester resin
- mol
- less
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/12—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/16—Dicarboxylic acids and dihydroxy compounds
- C08G63/18—Dicarboxylic acids and dihydroxy compounds the acids or hydroxy compounds containing carbocyclic rings
- C08G63/181—Acids containing aromatic rings
- C08G63/183—Terephthalic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/12—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/16—Dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
- C08G63/84—Boron, aluminium, gallium, indium, thallium, rare-earth metals, or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J167/00—Adhesives based on polyesters obtained by reactions forming a carboxylic ester link in the main chain; Adhesives based on derivatives of such polymers
- C09J167/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
Definitions
- the present invention relates to a copolyester resin having excellent heat resistance and color tone and reduced environmental load, and a method for producing the same.
- Polyesters represented by polyethylene terephthalate (PET), polybutylene terephthalate (PBT), polyethylene naphthalate (PEN) and the like are excellent in mechanical properties and chemical properties.
- highly functional copolyesters obtained by selecting a wide variety of acid components / alcohol components, for example, fibers for clothing and industrial materials, packaging, Used in a wide range of fields such as films and sheets for magnetic tape and optics, bottles that are hollow molded products, casings for electrical and electronic parts, paints, adhesives, ink binders, and other engineering plastic molded products. .
- polyesters are produced by producing an oligomer mixture by esterification or transesterification of dicarboxylic acid and / or its ester-forming derivative and diol and / or its ester-forming derivative, and using a catalyst at high temperature under vacuum. And liquid phase polycondensation.
- antimony compounds, germanium compounds, and titanium compounds have been widely used as polyester polycondensation catalysts used in such polycondensation of polyesters.
- Antimony trioxide is an inexpensive catalyst with excellent catalytic activity. However, if it is used in an amount of such a main component, that is, a practical polymerization rate, metal antimony is used during polycondensation. As a result of precipitation, darkening and foreign matter are generated in the polyester.
- Germanium compounds have already been put to practical use as catalysts that can obtain polyesters that have excellent catalytic activity other than antimony compounds and that do not have the above problems, but this catalyst is very expensive.
- this catalyst is very expensive.
- Polyester produced using a titanium compound typified by tetraalkoxy titanate has a problem that it is susceptible to thermal degradation during melt molding, and the polyester is markedly colored.
- polyesters with low copolymerization content of other components other than terephthalic acid and ethylene glycol represented by polyethylene terephthalate obtained by the above polycondensation catalyst system color tone, transparency and thermal stability are good, and the above requirements Is a response to
- copolyester the decrease in the polycondensation reaction rate is remarkable, and when the polycondensation reaction proceeds to the target molecular weight, the color tone of the polyester obtained and the molecular weight decrease due to the thermal decomposition reaction occur.
- the above requirements Is a response to
- Patent Document 2 As one method for increasing the polycondensation rate, in Patent Document 2, terephthalic acid is added to an oligomer obtained by esterification, and esterification is carried out again at a high temperature, whereby specific viscosity conditions, acid value conditions, hydroxyl value conditions, A production method is described in which an oligomer that satisfies molecular weight conditions and esterification conditions is obtained and the product is polycondensed.
- the solubility of terephthalic acid is poor, and thus esterification is performed at high temperature for a long time.
- the copolymer polyester has a problem that it is difficult to control the composition of the glycol component and the hydroxyl end groups of the oligomer by esterification at a high temperature for a long time. Further, there is a problem that the polyester is easily colored due to the influence of a small amount of water and oxygen contained in the dicarboxylic acid added later.
- Patent Document 4 polycondensation reaction is carried out by adjusting the ratio of hydroxyl group end groups to 55 to 75 mol% with respect to the total end groups of the reaction product obtained by the reaction of the dicarboxylic acid component and the diol component. Increasing speed is disclosed. However, the ratio of the terephthalic acid component to the total dicarboxylic acid component is 75 mol% or less, and in the case of a complex system of copolyester composed of two or more diol components, it is described in Patent Document 4. Even if the polycondensation reaction is carried out according to the above method, a copolymer polyester having a high molecular weight and a stable composition cannot be produced efficiently.
- the present inventors made various co-polymers consisting of terephthalic acid and one or more other dicarboxylic acids as the dicarboxylic acid component, and two or more diol components as the diol component.
- the color deterioration and thermal decomposition behavior of the copolymerized polyester resin itself depended on the time of the adhesive solution. It has been found that there are practical problems such as discoloration and deterioration of adhesiveness.
- the present invention is a copolyester resin produced using a polycondensation catalyst having a metal component other than antimony, germanium and titanium as the main metal component of the catalyst, and when used for an adhesive adapted for various applications Furthermore, the present invention provides a copolyester resin from which an adhesive resin having excellent color tone and durability can be obtained. Furthermore, the present invention provides a method for producing the copolyester resin, which has a high polycondensation rate and achieves both quality and productivity.
- G / A needs to be 1.50 or more in order to perform sufficient esterification to the extent that it does not affect the subsequent polycondensation reaction.
- a copolymerized polyester resin having a dicarboxylic acid component and a diol component of two or more types in which the proportion of the terephthalic acid component is 75 mol% or less, and the copolymerized polyester resin under a nitrogen atmosphere By using a copolyester resin in which the reduction in reduced viscosity after heat treatment at 275 ° C. for 2 hours is 0.20 dl / g or less and the color b value is 5 or less, sufficient adhesion and color tone are obtained. It was found that an adhesive having excellent stability can be obtained.
- the ratio (molar ratio, G / A) of the diol component (total molar amount, G) to the proportion of the dicarboxylic acid component (total molar amount, A) is 0.00. It was found that by setting the ratio in the range of 8 to 1.4, it is possible to suppress a decrease in the reaction rate of the subsequent polycondensation reaction and obtain a copolyester resin having a sufficient viscosity.
- this invention consists of the following structures.
- a copolymer polyester resin comprising a dicarboxylic acid component containing terephthalic acid in a proportion of 75 mol% or less and two or more diol components as constituents satisfies the following (1) and (2): Copolyester resin.
- (1) Reduction in reduced viscosity after heat treatment of copolymerized polyester resin at 275 ° C. for 2 hours in a nitrogen atmosphere is 0.20 dl / g or less
- Color b value is 5 or less
- the dicarboxylic acid component contains terephthalic acid in a proportion of 75 mol% or less, and a total of 80 mol of terephthalic acid and at least one of orthophthalic acid, isophthalic acid, adipic acid, sebacic acid and azelaic acid.
- the diol component is ethylene glycol, neopentyl glycol, 1,3-propylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,4-cyclohexane.
- the copolyester resin according to the above item 1 or 2 which contains 80 mol% or more in total of at least two kinds of dimethanol.
- a dicarboxylic acid component containing terephthalic acid in a proportion of 75 mol% or less of the total dicarboxylic acid component, and two types using a polymerization catalyst containing at least one selected from aluminum compounds and at least one selected from phosphorus compounds A method for producing a copolyester resin comprising the above diol component as a constituent component, which satisfies the following (3) and (4).
- the ratio (G / A) of the molar amount (A) of all dicarboxylic acid components and the molar amount (G) of all diol components at the start of the esterification reaction is 0.8 to 1.4 (4 )
- the reaction intermediate product has a carboxylic acid end group concentration of 500 to 1500 eq / ton and a hydroxyl end group concentration of 1500 to 3000 eq / ton.
- the dicarboxylic acid component contains terephthalic acid in a proportion of 75 mol% or less, and a total of 80 mol of terephthalic acid and at least one of orthophthalic acid, isophthalic acid, adipic acid, sebacic acid and azelaic acid.
- the diol component is ethylene glycol, neopentyl glycol, 1,3-propylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,4-cyclohexane. 6.
- a copolyester resin useful as a high-quality adhesive having excellent heat resistance and weather resistance can be obtained. Furthermore, since the esterification reaction can proceed at a low temperature for a short time, it is easy to control the end group composition of the product after the esterification reaction. In addition, a copolyester resin having a high polycondensation rate and excellent heat resistance and color tone can be obtained, and the productivity thereof is dramatically increased. In addition, since the distillate of the esterification reaction and polycondensation reaction can be minimized, the production loss can be reduced and the copolyester resin can be produced at a low cost. In addition, the amount of excess glycol discharged can be reduced and the environmental load can be reduced.
- the “copolyester resin” includes a polymerization catalyst compound described later. Although it can be said to be a kind of “composition” in that it contains a substance other than a chemical substance called “copolymerized polyester”, since the amount of the polymerization catalyst compound is very small, in the present invention, “copolyester resin” It expresses. In some cases, it may be simply referred to as “copolyester”.
- the copolyester referred to in the present invention is a polyester mainly composed of a dicarboxylic acid component and a diol component, and a dicarboxylic acid component containing terephthalic acid in a proportion of 75 mol% or less of the total dicarboxylic acid component, and two or more kinds of polyesters. It is formed from a diol component.
- terephthalic acid 75 mol% or less is for the reason described later. There is no particular lower limit to the proportion of terephthalic acid, and it may be 0 mol%.
- Dicarboxylic acids other than terephthalic acid include oxalic acid, malonic acid, succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, decanedicarboxylic acid, dodecanedicarboxylic acid, tetradecanedicarboxylic acid, hexadecanedicarboxylic acid 1,3-cyclobutanedicarboxylic acid, 1,3-cyclopentanedicarboxylic acid, 1,2-cyclohexanedicarboxylic acid, 1,3-cyclohexanedicarboxylic acid, 1,4-cyclohexanedicarboxylic acid, 2,5-norbornanedicarboxylic acid, Saturated aliphatic dicarboxylic acid exemplified by dimer acid, etc., fumaric acid, maleic acid, itaconic acid, unsaturated aliphatic dicarbox
- the dicarboxylic acid other than terephthalic acid preferably contains at least one of orthophthalic acid and isophthalic acid from the viewpoint of physical properties of the resulting polyester.
- terephthalic acid is contained in a proportion of 75 mol% or less, and “terephthalic acid” and “at least one of orthophthalic acid, isophthalic acid, adipic acid, sebacic acid and azelaic acid” are combined.
- the dicarboxylic acid component contains terephthalic acid in a proportion of 75 mol% or less, and a total of 80 mol% of terephthalic acid, at least one of orthophthalic acid and isophthalic acid is combined. It is preferable to occupy the above, and more preferably 90 mol% or more.
- a polyvalent carboxylic acid may be used in combination if the amount is small.
- the polyvalent carboxylic acid include ethanetricarboxylic acid, propanetricarboxylic acid, butanetetracarboxylic acid, pyromellitic acid, trimellitic acid, trimesic acid, and 3,4,3 ', 4'-biphenyltetracarboxylic acid.
- These polyvalent carboxylic acids are preferably 5 mol% or less with respect to 100 mol% of all dicarboxylic acid components.
- Diols include ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol, diethylene glycol, triethylene glycol, 1,2-butanediol, 1,3-butanediol, 2,3-butanediol, 1,4 -Butanediol, 1,5-pentanediol, neopentyl glycol, 1,6-hexanediol, 1,2-cyclohexanediol, 1,3-cyclohexanediol, 1,4-cyclohexanediol, 1,2-cyclohexanedimethanol 1,3-cyclohexanedimethanol, 1,4-cyclohexanedimethanol, 1,4-cyclohexanediethanol, 1,10-decamethylene glycol, 1,12-dodecanediol, polyethylene glycol, polytrimethyleneglycol Aliphatic glycols exemplified by polyte
- diols ethylene glycol, neopentyl glycol, 1,3-propylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,4-cyclohexanedimethanol are preferred.
- diol component include ethylene glycol, neopentyl glycol, 1,3-propylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,4-cyclohexanedimethanol.
- the total of at least two types preferably accounts for 80 mol% or more, more preferably 90 mol% or more.
- polyhydric alcohols may be used in combination if the amount is small.
- examples of the polyhydric alcohol include trimethylolmethane, trimethylolethane, trimethylolpropane, pentaerythritol, glycerol, hexanetriol, and the like. These polyhydric alcohols are preferably 5 mol% or less with respect to 100 mol% of all diol components.
- hydroxycarboxylic acid may be used in combination.
- examples of the hydroxycarboxylic acid include lactic acid, citric acid, malic acid, tartaric acid, hydroxyacetic acid, 3-hydroxybutyric acid, p-hydroxybenzoic acid, p- (2-hydroxyethoxy) benzoic acid, and 4-hydroxycyclohexanecarboxylic acid.
- These hydroxycarboxylic acids are preferably 5 mol% or less with respect to 100 mol% of all dicarboxylic acid components.
- cyclic esters examples include ⁇ -caprolactone, ⁇ -propiolactone, ⁇ -methyl- ⁇ -propiolactone, ⁇ -valerolactone, glycolide, lactide and the like. These cyclic esters are preferably 5 mol% or less with respect to 100 mol% of all dicarboxylic acid components.
- Dicarboxylic acid, polyvalent carboxylic acid and hydroxycarboxylic acid can be partially used as raw material monomers, and ester-forming derivatives can also be used.
- ester-forming derivatives include alkyl esters and hydroxylalkyl esters of these compounds. Is mentioned.
- an ester-forming derivative can be used as a raw material monomer, and examples of the diol ester-forming derivative include esters of a diol with a lower aliphatic carboxylic acid such as acetic acid.
- the proportion of terephthalic acid in the dicarboxylic acid component is 75 mol% or less and a dicarboxylic acid component having a low melting point of 400 ° C. or less is used as the other dicarboxylic acid.
- the proportion of the terephthalic acid component exceeds 75 mol%, the reactivity of the esterification reaction is significantly lowered, the polycondensation time becomes long, and a target high-viscosity polyester cannot be obtained, and the obtained polyester is colored.
- the low melting point dicarboxylic acid having a melting point of 400 ° C. or lower include orthophthalic acid, isophthalic acid, adipic acid, sebacic acid, azelaic acid and the like. It is also possible to use a small amount of a monovalent carboxylic acid.
- any of the diol components exemplified above can be used.
- Preferred examples of the diol component include ethylene glycol, neopentyl glycol, 1,3-propylene glycol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, and 1,4-cyclohexanedimethanol. Etc. It is also possible to use a small amount of a polyhydric alcohol component such as trimethylolpropane or pentaerythritol as long as the effects of the invention are not impaired.
- the most diol component of the two or more diol components is preferably 95 mol% or less, more preferably 90 mol% or less of the total diol components.
- a preferred method for producing the copolymerized polyester resin of the present invention is to obtain a reaction intermediate product by a direct esterification reaction of a dicarboxylic acid component and a diol component, and further to obtain a copolymerized polyester resin from the reaction intermediate product by a polycondensation reaction.
- Esterification in which the ratio (G / A) of the molar amount (A) of all dicarboxylic acid components to the total amount of diol components (G / A) at the start of the esterification reaction is 0.8 to 1.4 It goes through a process of reaction.
- the lower limit of the ratio (G / A, molar ratio) of the proportion of diol component (G, total molar amount) to the proportion of dicarboxylic acid component (A, total molar amount) is 0.8. However, it is preferably 0.85 or more, more preferably 0.9 or more. If G / A is less than 0.8, polycondensation becomes impossible due to poor esterification. Moreover, although the upper limit of G / A is 1.4, Preferably it is 1.3 or less, More preferably, it is 1.2 or less.
- the dicarboxylic acid component has a higher melting point and boiling point than the diol component. Therefore, when placed in a reduced pressure environment during polyester polymerization, the dicarboxylic acid component is the constituent molar ratio of the copolymerized polyester resin in which the charged molar ratio (the blending ratio of each monomer) is obtained as it is, while the diol component is reduced in pressure. It is necessary to consider the volatile matter in the environment. If G / A is 1.1 or less, there is almost no volatilization in a reduced pressure environment, so it can be considered that the charged molar ratio of the dicarboxylic acid component and the diol component is the constituent molar ratio.
- the mol% of the dicarboxylic acid component and the diol component described above represents the mol% in the resulting copolymerized polyester resin.
- the reduced viscosity of the copolyester resin of the present invention is preferably 0.45 dl / g or more. More preferably, it is 0.50 dl / g or more, More preferably, it is 0.60 dl / g or more.
- the reduced viscosity of the copolyester resin is preferably 1.2 dl / g or less, and more preferably 1.0 dl / g or less.
- the polymerization catalyst used in the present invention is a polymerization catalyst characterized by having an ability to promote esterification.
- a polymerization catalyst containing at least one selected from aluminum compounds and at least one selected from phosphorus compounds is preferable.
- the reduction in reduced viscosity after heat-treating the copolyester resin of the present invention at 275 ° C. for 2 hours in a nitrogen atmosphere is 0.20 dl / g or less, and the color b value is 5 or less. Therefore, the selection of this polymerization catalyst is very important.
- known aluminum compounds can be used without limitation.
- the aluminum compound examples include aluminum acetate, basic aluminum acetate, aluminum lactate, aluminum chloride, aluminum hydroxide, aluminum hydroxide chloride, aluminum acetylacetonate, organoaluminum compounds such as aluminum oxalate, and parts thereof.
- examples include hydrolysates.
- carboxylate, inorganic acid salt and chelate compound are preferable, and among these, aluminum acetate, basic aluminum acetate, aluminum lactate, aluminum chloride, aluminum hydroxide, aluminum hydroxide chloride and aluminum acetylacetonate are more preferable, Aluminum acetate, basic aluminum acetate, aluminum chloride, aluminum hydroxide and aluminum hydroxide chloride are more preferred, and aluminum acetate and basic aluminum acetate are most preferred.
- the copolymer polyester resin of the present invention preferably contains 1 to 80 ppm of aluminum atoms derived from an aluminum compound as a polymerization catalyst with respect to the total mass of the copolymer polyester resin.
- the amount of the aluminum compound used in the polymerization catalyst according to the present invention is preferably 1 to 80 ppm remaining (contained) as an aluminum atom with respect to the total mass of the resulting copolymerized polyester resin, more preferably It is 2 to 60 ppm, more preferably 3 to 50 ppm, particularly preferably 5 to 40 ppm, and most preferably 10 to 30 ppm. If it is less than the above, the catalyst activity may be poor, and if it exceeds the above, aluminum-based foreign matter may be generated. Even if the aluminum compound is placed in a reduced pressure environment at the time of polyester polymerization, since almost 100% of the used amount remains, it may be considered that the used amount becomes the remaining amount.
- the phosphorus compound used for the polymerization catalyst is not particularly limited, but the use of a phosphonic acid compound or a phosphinic acid compound is highly preferable for improving the catalytic activity. Among these, the use of a phosphonic acid compound is effective for improving the catalytic activity. Is particularly large and preferred.
- phosphorus compounds having a phenol moiety in the same molecule are preferred. It is not particularly limited as long as it is a phosphorus compound having a phenol structure, but it is a catalyst if one or more compounds selected from the group consisting of phosphonic acid compounds and phosphinic acid compounds having a phenol moiety in the same molecule are used.
- the effect of improving the activity is large and preferable.
- the use of a phosphonic acid compound having a phenol moiety in one or two or more of the same molecules is particularly preferable because the effect of improving the catalytic activity is particularly large.
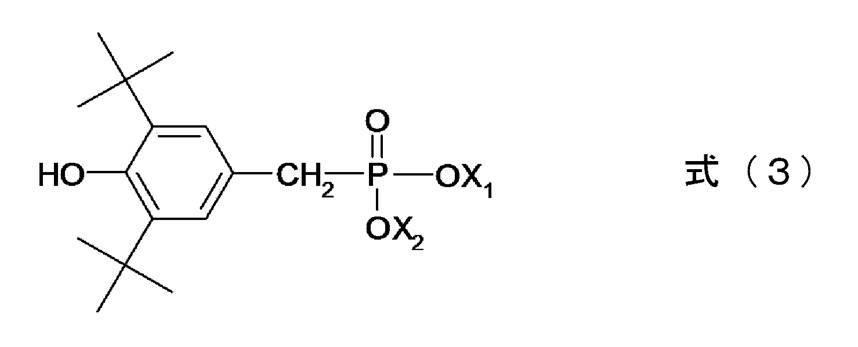
- examples of the phosphorus compound having a phenol moiety in the same molecule include compounds represented by the following general formulas (1) and (2).
- R 1 is a hydrocarbon group having 1 to 50 carbon atoms including a phenol part, a hydroxyl group, a halogen group, an alkoxyl group, an amino group or the like, and a carbon number 1 including a phenol part.
- R 4 represents a hydrocarbon group having 1 to 50 carbon atoms, including a substituent such as hydrogen, a hydrocarbon group having 1 to 50 carbon atoms, a hydroxyl group, a halogen group, an alkoxyl group, or an amino group.
- R 2 and R 3 each independently represents hydrogen, a hydrocarbon group having 1 to 50 carbon atoms, a hydrocarbon group having 1 to 50 carbon atoms including a substituent such as a hydroxyl group or an alkoxyl group.
- the group may contain a branched structure, an alicyclic structure such as cyclohexyl, or an aromatic ring structure such as phenyl or naphthyl, and the ends of R 2 and R 4 may be bonded to each other.
- Examples of the phosphorus compound having a phenol moiety in the same molecule include p-hydroxyphenylphosphonic acid, dimethyl p-hydroxyphenylphosphonate, diethyl p-hydroxyphenylphosphonate, diphenyl p-hydroxyphenylphosphonate, bis ( p-hydroxyphenyl) phosphinic acid, methyl bis (p-hydroxyphenyl) phosphinate, phenyl bis (p-hydroxyphenyl) phosphinate, p-hydroxyphenylphenylphosphinic acid, methyl p-hydroxyphenylphenylphosphinate, p-hydroxy Examples include phenyl phenylphenylphosphinate, p-hydroxyphenylphosphinic acid, methyl p-hydroxyphenylphosphinate, and phenyl p-hydroxyphenylphosphinate.
- Other examples include phosphorus compounds represented by the following general formula (3).
- X 1 and X 2 each represent hydrogen, an alkyl group having 1 to 4 carbon atoms, or a monovalent or higher metal. Moreover, X 1 is metal be two or more valences, X 2 may be absent. Furthermore, an anion corresponding to the surplus valence of the metal may be arranged with respect to the phosphorus compound.
- the metal Li, Na, K, Ca, Mg, and Al are preferable.
- the catalytic activity of the aluminum compound is improved, and the thermal stability of the resulting copolyester resin is also improved.
- the phosphorus compound preferably used as the polycondensation catalyst is at least one phosphorus compound selected from the compounds represented by the chemical formula (4) and the chemical formula (5).
- Irganox 1222 (manufactured by BASF) is commercially available.
- Irganox 1425 (manufactured by BASF) is commercially available and can be used.
- the copolymer polyester resin of the present invention preferably contains 10 to 100 ppm of phosphorus atoms derived from a phosphorus compound as a polymerization catalyst with respect to the total mass of the copolymer polyester resin.
- the amount of the phosphorus compound used in the polymerization catalyst according to the present invention is preferably such that the phosphorus atom remains (contains) 10 to 100 ppm relative to the total mass of the resulting copolymerized polyester resin, more preferably. It is 15 to 90 ppm, more preferably 20 to 80 ppm, particularly preferably 25 to 70 ppm, and most preferably 30 to 60 ppm. Residual amounts of phosphorus atoms exceeding the above upper and lower limits may cause a decrease in polymerization activity.
- the heat resistance and color tone of the resin can be improved by using a phosphorus compound.
- the cause is not certain, it is considered that the heat resistance of the copolyester resin is improved by the hindered phenol moiety in the phosphorus compound.
- the copolymerized polyester resin of the present invention is used for adhesion to a metal plate or the like, the thermal degradation of the copolymerized polyester resin in the adhesive layer is suppressed by adding a phosphorus compound, thereby adhering the copolymerized polyester resin in the adhesive layer. Can be improved. This is thought to be due to the suppression of the production of a medium molecular weight polyester resin due to thermal degradation.
- the polyester resin in the medium molecular weight region enters the irregularities on the surface of the metal plate, thereby inhibiting the adhesion of the copolyester resin for the adhesive layer and consequently reducing the adhesiveness of the copolyester resin for the adhesive layer.
- the residual amount (content) of the phosphorus compound is less than 10 ppm, the effect of improving the heat resistance is reduced, and as a result, the heat resistance and coloring improvement effect of the copolymerized polyester resin of the present invention may not be seen. .
- a metal-containing polycondensation catalyst such as an antimony compound, a titanium compound, a tin compound, or a germanium compound may be used in combination in order to further improve the catalytic activity.
- the antimony compound is preferably 30 ppm or less as an antimony atom with respect to the mass of the obtained copolyester resin
- the germanium compound is preferably 10 ppm or less as a germanium atom with respect to the mass of the copolyester resin to be obtained.
- the titanium compound is preferably 3 ppm or less as a titanium atom with respect to the mass of the obtained copolymerized polyester resin, and the tin compound has 3 ppm or less as a tin atom with respect to the mass of the obtained copolymerized polyester resin.
- metal-containing polycondensation catalysts such as antimony compounds, titanium compounds, tin compounds and germanium compounds as much as possible.
- a small amount of alkali metal, alkaline earth metal and at least one selected from the compound may coexist as the second metal-containing component.
- the coexistence of such a second metal-containing component in the catalyst system is effective in improving productivity by obtaining a catalyst component having an increased reaction rate in addition to an effect of suppressing the formation of diethylene glycol, and thus a higher reaction rate.
- the addition amount (mol%) is preferably 1 ⁇ 10 4 with respect to the number of moles of the dicarboxylic acid component constituting the copolymer polyester resin. -5 to 0.01 mol%.
- Alkaline metal, alkaline earth metal, or a compound thereof may be considered to be a residual amount because almost 100% of the usage amount remains even when placed in a reduced pressure environment during polyester polymerization.
- the polymerization catalyst according to the present invention has catalytic activity not only in the polycondensation reaction but also in the esterification reaction and transesterification reaction.
- the transesterification reaction between an alkyl ester of a dicarboxylic acid such as dimethyl terephthalate and a glycol such as ethylene glycol is usually carried out in the presence of a transesterification catalyst such as zinc.
- the catalyst of the present invention is used in place of these catalysts. You can also.
- the catalyst according to the present invention has catalytic activity not only in melt polymerization but also in solid phase polymerization or solution polymerization.
- the polymerization catalyst used in the present invention can be added to the reaction system at any stage of the polymerization reaction.
- it can be added to the reaction system at any stage before and during the esterification reaction or transesterification reaction, immediately before the start of the polycondensation reaction, or at any stage during the polycondensation reaction.
- the aluminum compound and the phosphorus compound of the present invention are preferably added immediately before the start of the polycondensation reaction.
- the carboxylic acid group terminal concentration (AVo) is 500 eq / ton or more and 1500 eq / ton or less. is important.
- the activity of the catalyst composed of the aluminum compound and the phosphorus compound is most effectively expressed from the relationship with the composition of the copolymerized polyester resin in the present invention.
- AVo is preferably 700 eq / ton or more and 1300 eq / ton or less.
- the reduction in reduced viscosity after heat-treating the copolyester resin of the present invention at 275 ° C. for 2 hours in a nitrogen atmosphere is 0.20 dl / g or less, and the color b value is 5 or less. For this purpose, this AVo is very important.
- the hydroxyl group terminal concentration (OHVo) is 1500 eq / ton or more and 3000 eq / ton or less. This is very important. When it is within the above range, the activity of the catalyst comprising the aluminum compound and the phosphorus compound is most effectively expressed from the relationship with the composition of the copolyester resin of the present invention. When OHVo is less than 1500 eq / ton, the polymerization reaction does not proceed. Conversely, even when OHVo exceeds 3000 eq / ton, the polymerization reaction is undesirably slowed.
- OHVo is more preferably 1600 eq / ton or more and 2800 eq / ton or less.
- the reduction in reduced viscosity after heat-treating the copolyester resin of the present invention at 275 ° C. for 2 hours in a nitrogen atmosphere is 0.20 dl / g or less, and the color b value is 5 or less.
- this OHVo is very important.
- the ratio of hydroxyl group terminals (OHV%) to the total terminal functional group concentration of the reaction intermediate product (oligomer) after completion of the esterification reaction is 55% or more and 75%. The following is preferable. When it is within the above range, the polymerization activity of the catalyst comprising the aluminum compound and the phosphorus compound is most effectively expressed.
- the copolymerized polyester resin of the present invention can be obtained by the above method, but the reduction in the reduced viscosity after heat-treating the copolymerized polyester resin at 275 ° C. for 2 hours in a nitrogen atmosphere is 0.20 dl / g. And the color b value is 5 or less.
- the reduction in the reduced viscosity and the color b value can be measured by the method described in the Examples section described later.
- the reduction in the reduced viscosity is preferably 0.18 dl / g or less, and the color b value is preferably 4.5 or less.
- the adhesive When the copolymerized polyester resin of the present invention is used as an adhesive, the adhesive has excellent color tone and durability.
- Antimony atom 1 g of a sample was wet-decomposed with a mixed solution of sulfuric acid / hydrogen peroxide solution. Next, sodium nitrite was added to make Sb atoms Sb 5+, and brilliant green was added to form a blue complex with Sb. After this complex was extracted with toluene, the absorbance at a wavelength of 625 nm was measured using an absorptiometer (manufactured by Shimadzu Corporation, UV-150-02), and the amount of Sb atoms in the sample was compared with a calibration curve prepared in advance. The color was determined.
- a mixed solution of sulfuric acid / nitric acid / perchloric acid or a mixed solution of sulfuric acid / hydrogen peroxide. was defined as orthophosphoric acid.
- molybdate was reacted in a 1 mol / L sulfuric acid solution to form phosphomolybdic acid, which was reduced with hydrazine sulfate to produce heteropoly blue.
- Absorbance at a wavelength of 830 nm was measured with an absorptiometer (manufactured by Shimadzu Corporation, UV-150-02). The amount of phosphorus atoms in the sample was quantified from a calibration curve prepared in advance
- the solution was filtered through a glass filter (3G) to obtain an aqueous solution of an aluminum compound.
- 3G glass filter
- 2.0 liters of an aqueous solution of the aluminum compound and 2.0 liters of ethylene glycol were charged into a flask equipped with a distillation apparatus at room temperature and normal pressure. After stirring for 30 minutes, a uniform water / ethylene glycol mixed solution was obtained. . Subsequently, the temperature was raised to 110 ° C., and water was distilled off from the solution. When the amount of distilled water reached 2.0 liters, the heating was stopped and the mixture was allowed to cool to room temperature to obtain an ethylene glycol solution of an aluminum compound.
- Example 1 A reaction vessel equipped with a stirrer, a thermometer, and a distillation cooler was charged with 50 parts by mass of terephthalic acid, 50 parts by mass of isophthalic acid, 20.5 parts by mass of ethylene glycol, and 34.5 parts by mass of neopentyl glycol, 0.35 MPa Under pressure, the temperature was gradually raised to 250 ° C., and the esterification reaction was carried out while removing the water distilled off over 4 hours. The oligomer obtained by esterification was sampled, the AVo (acid value) and OHVo (OH value) of the oligomer were measured, and OHV% (ratio of hydroxyl end groups) was calculated.
- an ethylene glycol solution of a phosphorus compound and an ethylene glycol mixed solution of an aluminum compound prepared by the above-described method for preparing a polycondensation catalyst solution were used in an amount of 0.04 mol% as phosphorus atoms with respect to the acid component in the polyester.
- initial polymerization under reduced pressure is performed to 1.3 kPa over 1 hour, the temperature is increased to 275 ° C., and later polymerization is performed at 0.13 kPa or less to obtain a copolyester resin.
- the time required for the late polymerization was 100 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.90 dl / g.
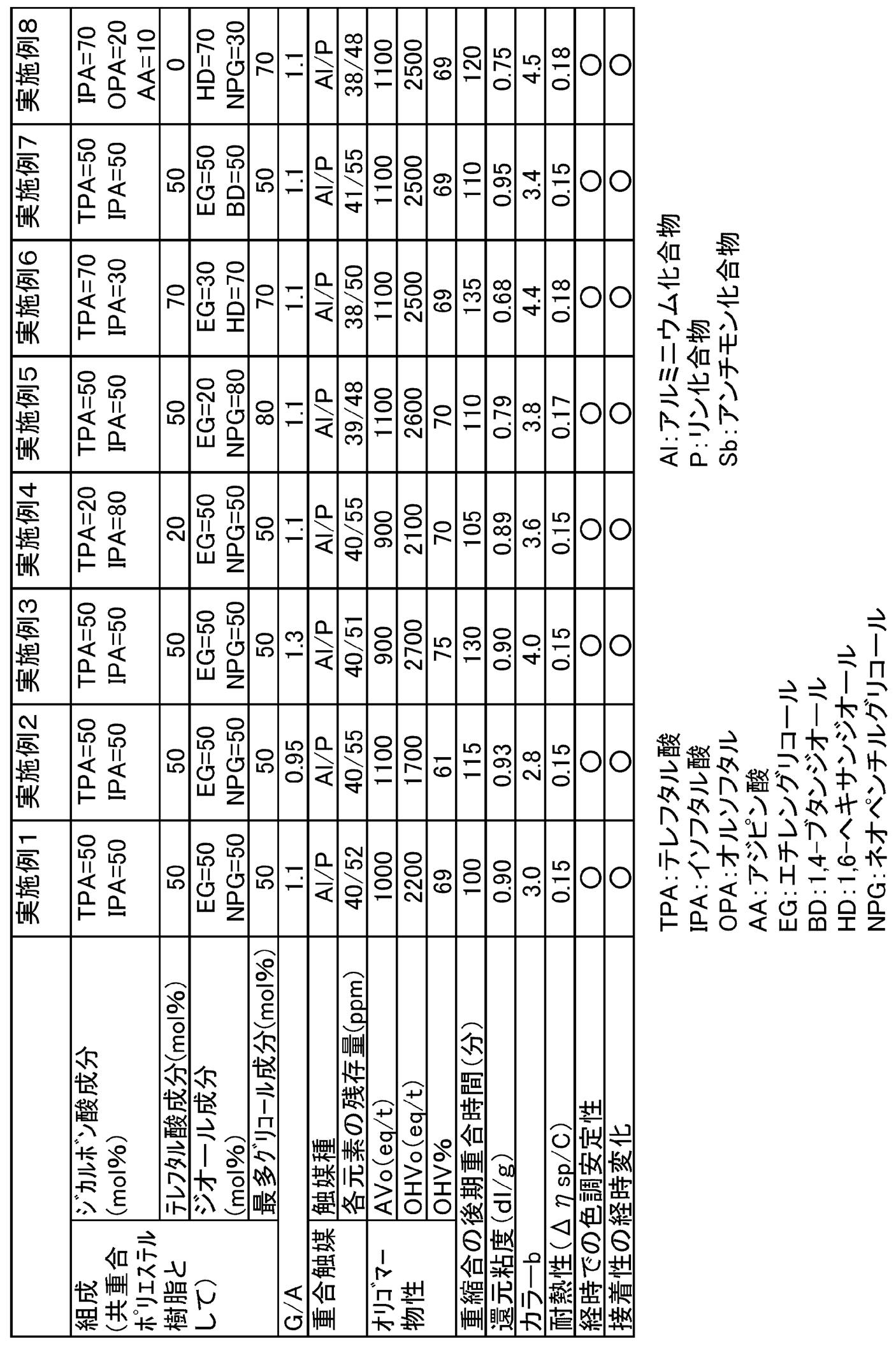
- Table 1 shows the physical properties of the obtained copolyester resin.
- 10 parts by mass of the copolymerized polyester resin thus obtained, 36 parts by mass of toluene, and 54 parts by mass of methyl ethyl ketone were charged into a reaction can equipped with a stirring blade, a thermometer, and a reflux condenser. The temperature was raised to 0 ° C. and dissolved completely over 3 hours. In this way, a solution of a copolyester resin was obtained.
- Table 1 shows the evaluation results of the properties of the resin solution thus obtained.
- Example 2 A reactor equipped with a stirrer, thermometer, and distillation cooler was charged with 50 parts by mass of terephthalic acid, 50 parts by mass of isophthalic acid, 17.7 parts by mass of ethylene glycol, and 29.8 parts by mass of neopentyl glycol.
- esterification reaction and catalyst addition initial polymerization, and late polymerization reaction were performed to obtain a copolyester resin.
- the time required for the late polymerization was 115 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.93 dl / g.
- Table 1 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 3 A reactor equipped with a stirrer, thermometer, and distillation cooler was charged with 50 parts by mass of terephthalic acid, 50 parts by mass of isophthalic acid, 26.1 parts by mass of ethylene glycol, and 37.6 parts by mass of neopentyl glycol.
- esterification reaction and catalyst addition initial polymerization, and late polymerization reaction were performed to obtain a copolyester resin.
- the time required for the late polymerization was 130 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.90 dl / g.
- Table 1 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 4 A reactor equipped with a stirrer, thermometer, and distillation cooler was charged with 20 parts by mass of terephthalic acid, 80 parts by mass of isophthalic acid, 20.5 parts by mass of ethylene glycol, and 34.5 parts by mass of neopentyl glycol.
- esterification reaction and catalyst addition initial polymerization, and late polymerization reaction were performed to obtain a copolyester resin.
- the time required for the late polymerization was 105 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.89 dl / g.
- Table 1 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 1 A reactor equipped with a stirrer, thermometer, and distillation cooler was charged with 50 parts by mass of terephthalic acid, 50 parts by mass of isophthalic acid, 8.2 parts by mass of ethylene glycol, and 55.1 parts by mass of neopentyl glycol.
- esterification reaction and catalyst addition initial polymerization, and late polymerization reaction were performed to obtain a copolyester resin.
- the time required for the late polymerization was 110 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.79 dl / g.
- Table 1 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 6 A reactor equipped with a stirrer, thermometer, and distillation cooler was charged with 70 parts by mass of terephthalic acid, 30 parts by mass of isophthalic acid, 12.3 parts by mass of ethylene glycol, and 54.7 parts by mass of 1,6-hexanediol.
- esterification reaction and catalyst addition initial polymerization, and late polymerization reaction were carried out to obtain a copolymerized polyester resin.
- the time required for the late polymerization was 135 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.68 dl / g.
- Table 1 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 7 A reaction can equipped with a stirrer, a thermometer, and a distillation cooler was charged with 50 parts by mass of terephthalic acid, 50 parts by mass of isophthalic acid, 20.5 parts by mass of ethylene glycol, and 29.8 parts by mass of 1,4-butanediol.
- esterification reaction and catalyst addition initial polymerization, and late polymerization reaction were performed to obtain a copolymerized polyester resin.
- the time required for the late polymerization was 110 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.95 dl / g.
- Table 1 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 8> In a reaction vessel equipped with a stirrer, thermometer, and distillation cooler, 20 parts by mass of orthophthalic acid, 70 parts by mass of isophthalic acid, 8.8 parts by mass of adipic acid, 54.7 parts by mass of 1,6-hexanediol, neopentyl 20.7 parts by mass of glycol was charged, and esterification reaction, catalyst addition, initial polymerization, and late polymerization reaction were performed in the same manner as in Example 1 to obtain a copolymerized polyester resin. The time required for the late polymerization was 120 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.75 dl / g. Table 1 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 1 A reactor equipped with a stirrer, thermometer, and distillation cooler was charged with 50 parts by mass of terephthalic acid, 50 parts by mass of isophthalic acid, 31.7 parts by mass of ethylene glycol, and 40.7 parts by mass of neopentyl glycol.
- esterification reaction and catalyst addition initial polymerization, and late polymerization reaction were performed to obtain a copolyester resin.
- the time required for the late polymerization was 180 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.88 dl / g.
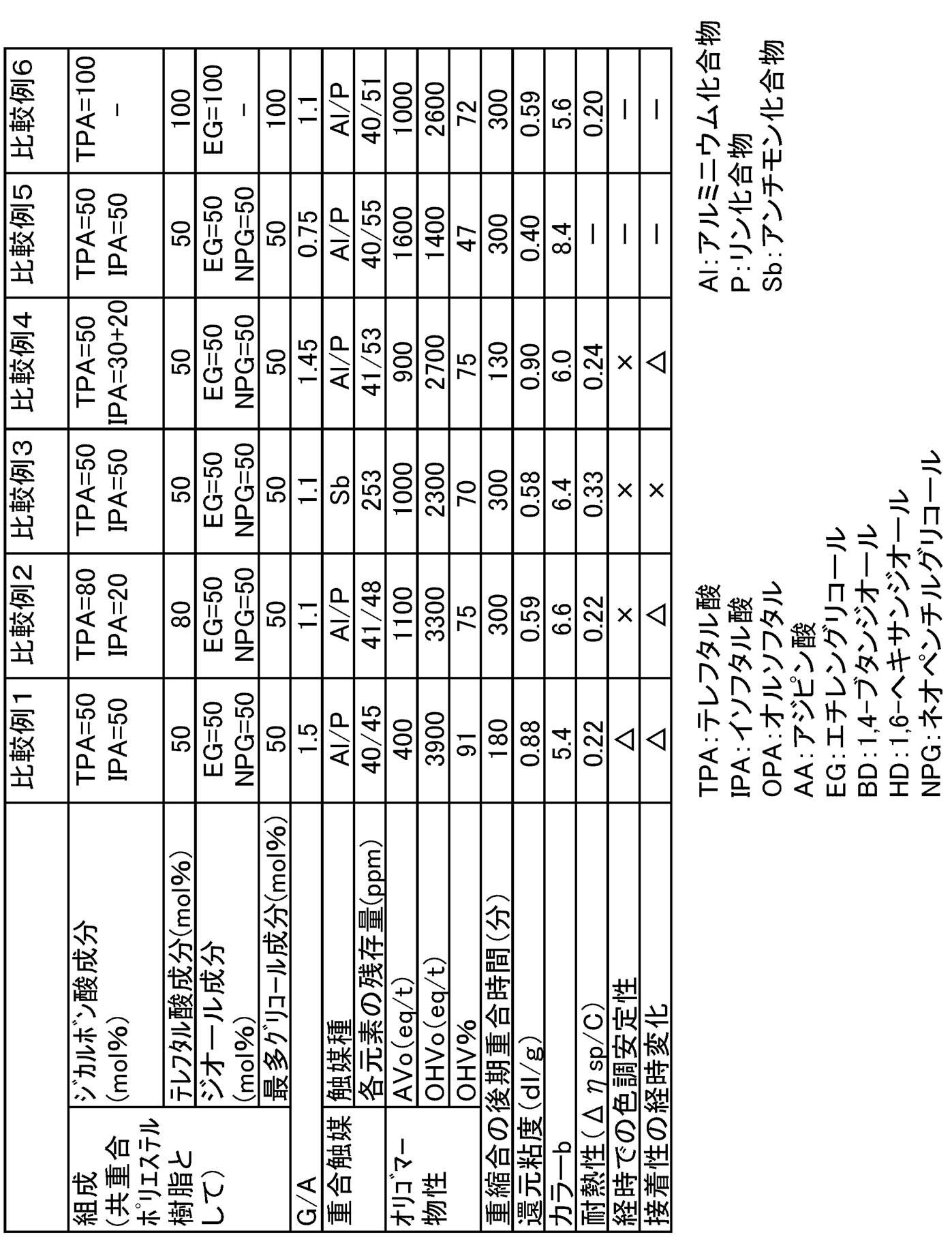
- Table 2 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- Example 1 A reactor equipped with a stirrer, a thermometer, and a distillation cooler was charged with 80 parts by mass of terephthalic acid, 20 parts by mass of isophthalic acid, 20.5 parts by mass of ethylene glycol, and 34.5 parts by mass of neopentyl glycol. In the same manner as above, esterification reaction and catalyst addition, initial polymerization, and late polymerization reaction were performed to obtain a copolyester resin. Even if the time required for the late polymerization exceeded 300 minutes, the reaction was terminated because no increase in melt viscosity was observed due to the polycondensation reaction.
- the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.59 dl / g.
- Table 2 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- the polycondensation catalyst solution was added to an ethylene glycol solution of antimony trioxide so as to be 0.04 mol% as antimony atoms with respect to the acid component in the polyester, and then 1.3 kPa over 1 hour.
- the initial polymerization was carried out under reduced pressure, the temperature was raised to 275 ° C., and the latter polymerization was carried out at 0.13 kPa or less to obtain a copolyester resin.
- the time required for the late polymerization was 300 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.58 dl / g.
- Table 2 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- the oligomer obtained by the second esterification was sampled, and the AVo (acid value) and OHVo (OH value) of the oligomer were measured to calculate OHV%.
- an ethylene glycol solution of a phosphorus compound and an ethylene glycol mixed solution of an aluminum compound prepared according to the above-described method for preparing a polycondensation catalyst solution 0.04 mol% as a phosphorus atom with respect to the acid component in the polyester, After adding it as 0.03 mol% as an aluminum atom, the pressure reduction initial polymerization is performed to 1.3 kPa over 1 hour, it rises to 275 degreeC, and also the latter polymerization is performed at 0.13 kPa or less, and copolyester A resin was obtained.
- the time required for the late polymerization was 130 minutes, and the reduced viscosity ( ⁇ sp / C) of the obtained copolyester resin was 0.90 dl / g.
- Table 2 shows the physical properties of the obtained copolyester resin. In the same manner as in Example 1, the characteristics of the resin solution were evaluated.
- the copolymer polyester resins of Examples 1 to 8 are excellent in color tone stability over time when processed into a varnish. Further, when used as an adhesive, it is excellent in long-term stability of adhesiveness with a substrate. Furthermore, the methods for producing the copolyester resins of Examples 1 to 8 have a high polycondensation rate and high productivity. On the other hand, Comparative Examples 1 to 4 outside the present invention are inferior in color tone stability and adhesive stability over time. In addition, Comparative Examples 1 to 3, 5, and 6 have a slow polycondensation rate and are inferior in productivity. In Comparative Example 5, the polycondensation rate was slow, and a copolymer polyester resin having a sufficient reduced viscosity could not be obtained. Comparative Example 4 was polymerized by a method according to Patent Document 2 and Patent Document 5, but could not obtain a copolyester resin satisfying the color tone and heat resistance.
- the copolyester resin of the present invention is superior in heat resistance and less colored compared to the prior art, so that when used as an adhesive, an adhesive composition excellent in adhesiveness and color tone stability can be obtained.
- the method for producing a copolyester resin of the present invention can advance the esterification reaction at a low temperature and in a short time compared to the prior art, it is easy to control the terminal group composition of the product after the esterification reaction. .
- the polycondensation rate is high, and a high-quality copolyester resin is obtained as a whole, and the productivity is dramatically increased.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Polyesters Or Polycarbonates (AREA)
- Adhesives Or Adhesive Processes (AREA)
Abstract
Description
本発明は、アンチモン、ゲルマニウムおよびチタン系以外の金属成分を触媒の主たる金属成分とする重縮合触媒を用いて製造される共重合ポリエステル樹脂であり、各種用途へ適応される接着剤に用いた場合に、優れた色調や耐久性を有する接着剤樹脂を得ることが出来る、共重合ポリエステル樹脂を提供するものである。さらに本発明は、重縮合速度が速く、品質と生産性を両立させた、該共重合ポリエステル樹脂の製造方法を提供するものである。
すなわち、本発明は、以下の構成からなる。
(1)共重合ポリエステル樹脂を窒素雰囲気下、275℃で2時間加熱処理した後の還元粘度の低下が、0.20dl/g以下である
(2)カラーb値が5以下である
(3)エステル化反応を開始する際の全ジカルボン酸成分のモル量(A)と全ジオール成分のモル量(G)の比(G/A)が0.8~1.4である
(4)エステル化反応終了後の反応中間生成物のカルボン酸末端基濃度が500~1500eq/ton、ヒドロキシル末端基濃度が1500~3000eq/tonである
本発明において、「共重合ポリエステル樹脂」とは、後記する重合触媒化合物を含むものである。「共重合ポリエステル」と言う化学物質以外のものを含む点では、一種の「組成物」とも言えるが、重合触媒化合物の量は微量であることから、本発明においては、「共重合ポリエステル樹脂」と表す。なお、簡略化して「共重合ポリエステル」と称する場合もある。
本発明に言う共重合ポリエステルとは、ジカルボン酸成分とジオール成分を主成分とするポリエステルであり、全ジカルボン酸成分の75モル%以下の割合でテレフタル酸を含むジカルボン酸成分と、2種類以上のジオール成分とから形成されるものをいう。
テレフタル酸を75モル%以下の割合とするのは、後述する理由による。テレフタル酸の割合の下限は特に無く、0モル%であっても構わない。
本発明にかかる重合触媒に用いられるアルミニウム化合物の使用量は、アルミニウム原子として、得られる共重合ポリエステル樹脂の全質量に対して1~80ppm残留(含有)するようにすることが好ましく、より好ましくは2~60ppmであり、更に好ましくは3~50ppmであり、特に好ましくは5~40ppmであり、最も好ましくは10~30ppmである。
上記を下回ると触媒活性不良となる可能性があり、上記を超えるとアルミニウム系異物生成を引き起こす可能性がある。
アルミニウム化合物は、ポリエステル重合時に減圧環境下に置かれても、使用量のほぼ100%が残留するので、使用量が残留量になると考えてよい。


また、X1は、金属が2価以上であって、X2が存在しなくても良い。さらには、リン化合物に対して金属の余剰の価数に相当するアニオンが配置されていても良い。
金属としては、Li、Na、K、Ca、Mg、Alが好ましい。


本発明にかかる重合触媒に用いられるリン化合物の使用量は、リン原子として、得られる共重合ポリエステル樹脂の全質量に対して10~100ppm残留(含有)するようにすることが好ましく、より好ましくは15~90ppmであり、更に好ましくは20~80ppmであり、特に好ましくは25~70ppmであり、最も好ましくは30~60ppmである。上記の上下限の範囲を超える量のリン原子が残存することで、重合活性を低下させる可能性がある。
リン化合物は、ポリエステル重合時に減圧環境下に置かれる際、その条件により、使用量の約10~30%が系外に除去される。そこで、実際は、数回の試行実験を行い、リン化合物のポリエステル中への残留率を見極めた上で、使用量を決める必要がある。
<評価方法>
(1)反応中間生成物(オリゴマー)のカルボン酸基末端濃度(AVo)の測定
オリゴマーを0.2g精秤し、20mlのクロロホルムに溶解し、0.1N-KOHエタノール溶液で、フェノールフタレインを指示薬として滴定し、樹脂106g当たりの当量(単位;eq/ton)を求めた。
オリゴマー0.5gを精秤し、アセチル化剤(無水酢酸ピリジン溶液0.5モル/L)10mlを加え、95℃以上の水槽に90分間浸漬した。水槽から取り出した直後、純水10mlを添加し室温まで放冷した。フェノールフタレインを指示薬として、0.2N-NaOH-CH3OH溶液で滴定した。試料を入れずに、ブランクも同じ作業を行った。常法に従い、上記AVoの値を使いOHVoを算出した。
上記方法で求めたOHVoとAVoとより、下記式に従って算出した。
OHV%={OHVo/(OHVo+AVo)}×100
共重合ポリエステル樹脂0.10gを、フェノール:テトラクロロエタン=60:40(質量比)の混合溶媒25cm3に溶かし、ウベローデ粘度管を用いて、30℃で測定した。
クロロホルム-d溶媒中、ヴァリアン社製核磁気共鳴分析計(NMR)ジェミニ-200を用いて、1H-NMR分析を行って、その積分比より決定した。
以下に示す方法で定量した。
試料1gを硫酸/過酸化水素水の混合液で湿式分解させた。次いで、亜硝酸ナトリウムを加えてSb原子をSb5+とし、ブリリアングリーンを添加してSbとの青色錯体を生成させた。この錯体をトルエンで抽出後、吸光光度計(島津製作所製、UV-150-02)を用いて、波長625nmにおける吸光度を測定し、予め作成した検量線から、試料中のSb原子の量を比色定量した。
試料1gを、炭酸ナトリウム共存下で乾式灰化分解させる方法、あるいは硫酸/硝酸/過塩素酸の混合液または硫酸/過酸化水素水の混合液で湿式分解させる方法によってリン化合物を正リン酸とした。次いで、1モル/Lの硫酸溶液中においてモリブデン酸塩を反応させてリンモリブデン酸とし、これを硫酸ヒドラジンで還元してヘテロポリ青を生成させた。吸光光度計(島津製作所製、UV-150-02)により波長830nmにおける吸光度を測定した。予め作成した検量線から、試料中のリン原子の量を定量した。
試料0.1gを6M塩酸溶液に溶解させ一日放置した後、純水で希釈し1.2M塩酸測定用溶液とした。調製した溶液試料を高周波プラズマ発光分析により求めた。
色差計(日本電色工業(株)製、ZE-2000)を用いて、共重合ポリエステル樹脂のチップの色差(L、a、b)を測定した。
共重合ポリエステル樹脂を窒素雰囲気下、常圧で、275℃、2時間加熱処理した。加熱処理前後の共重合ポリエステル樹脂の還元粘度(ηsp/C)を測定し、加熱処理後の還元粘度の低下(Δηsp/C)を求めた。
実施例、比較例で得られた共重合ポリエステル樹脂を固形分濃度が10質量%となるようにトルエン/メチルエチルケトン=40/60(質量比)の混合溶媒に溶解し、接着剤溶液を得た。この接着剤溶液を以下に示す方法で評価した。
色差計(日本電色工業(株)製、ZE-2000)を用いて、石英ガラス製10mm液体セル中の接着剤溶液の色差(L0,a0,b0)を測定した。その後、接着剤溶液をガラス製密閉容器に封入して60℃の恒温槽中で100時間保持した後に、同様に溶液の色差(L1,a1,b1)を測定した。熱処理前後の色調の変化量Δb(Δb=b1-b0)を求め、以下のように判定した。
○:Δb≦2
△:2<Δb≦5
×:Δb>5
接着剤溶液を25μmの二軸延伸PETフィルム上に乾燥後の厚みが3g/m2となる様に塗布し、120℃で3分間乾燥しコートフィルムを作製した。このコートフィルムを用いて接着層とティンフリースチール鋼鈑を、テスター産業社製ロールラミネータを用いて接着した。なお、ラミネートは温度180℃、圧力0.3MPa、速度1m/minで行った。このフィルムラミネート鋼鈑の接着強度は東洋ボールドウイン社製RTM100を用いて、25℃雰囲気下で引っ張り試験を行い、50mm/minの引っ張り速度で、90°剥離接着力を測定した。さらに、フィルムラミネート鋼鈑を80℃の恒温槽で72時間保持した後の接着強度を同様に測定した。熱処理前後のラミネート強度の保持率R(R=熱処理後の接着強度/熱処理前の接着強度×100(%))を求め、以下のように判断した。
○:R≧80%
△:60%≦R<80%
×:R<60%
(リン化合物のエチレングリコール溶液)
窒素導入管、冷却管を備えたフラスコに、常温常圧下、エチレングリコール2.0リットルを加えた後、窒素雰囲気下で攪拌しながら、リン化合物として化学式(4)で表されるIrganox1222(ビーエーエスエフ社製)200gを加えた。さらに2.0リットルのエチレングリコールを追加した後、設定温度を196℃として昇温し、内温が185℃以上になった時点から60分間還流下で攪拌した。その後加熱を止め、窒素雰囲気下を保ったまま、30分以内に120℃以下まで冷却し、リン化合物のエチレングリコール溶液を得た。
冷却管を備えたフラスコに、常温常圧下、純水5.0リットルを加えた後、攪拌しながら塩基性酢酸アルミニウム(ヒドロキシアルミニウムジアセテート)の200gを純水とのスラリーとして加えた。さらに全体として10.0リットルとなるよう純水を追加して常温常圧で12時間攪拌した。その後、設定温度100℃にて昇温し、内温が95℃以上になった時点から3時間還流下で攪拌した。攪拌を止め、室温まで放冷した。その際、未溶解粒子が見られた場合は、溶液をガラスフィルター(3G)にてろ過してアルミニウム化合物の水溶液を得た。
続いて、蒸留装置を備えたフラスコに、常温常圧下、前記アルミニウム化合物の水溶液2.0リットルとエチレングリコール2.0リットルを仕込み、30分間攪拌後、均一な水/エチレングリコール混合溶液を得た。次いで、設定温度を110℃として昇温し、該溶液から水を留去した。留出した水の量が2.0リットルになった時点で加熱を止め、室温まで放冷することでアルミニウム化合物のエチレングリコール溶液を得た。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール20.5質量部、ネオペンチルグリコール34.5質量部を仕込み、0.35MPaの加圧下、250℃まで徐々に昇温し、4時間かけて溜出する水を系外に除きつつエステル化反応を行った。エステル化で得られたオリゴマーをサンプリングし、オリゴマーのAVo(酸価)、OHVo(OH価)を測定し、OHV%(ヒドロキシル末端基の割合)を算出した。続いて、前記の重縮合触媒溶液の調製方法により作製したリン化合物のエチレングリコール溶液およびアルミニウム化合物のエチレングリコール混合溶液を、ポリエステル中の酸成分に対して、リン原子として0.04モル%、アルミニウム原子として0.03モル%となるように添加した後、1時間かけて1.3kPaまで減圧初期重合を行うとともに275℃まで上昇し、さらに0.13kPa以下で後期重合を行い、共重合ポリエステル樹脂を得た。後期重合にかかった時間は100分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.90dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。
次いで、撹拌翼、温度計、還流用冷却管を装備した反応缶内に、このようにして得られた共重合ポリエステル樹脂を10質量部、トルエンを36質量部、メチルエチルケトンを54質量部仕込み、50℃まで昇温を行い、3時間かけて完全に溶解した。このようにして共重合ポリエステル樹脂の溶液を得た。このようにして得られた樹脂溶液の特性評価結果を表1に示す。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール17.7質量部、ネオペンチルグリコール29.8質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は115分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.93dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール26.1質量部、ネオペンチルグリコール37.6質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は130分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.90dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸20質量部、イソフタル酸80質量部、エチレングリコール20.5質量部、ネオペンチルグリコール34.5質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は105分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.89dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール8.2質量部、ネオペンチルグリコール55.1質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は110分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.79dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸70質量部、イソフタル酸30質量部、エチレングリコール12.3質量部、1,6-ヘキサンジオールール54.7質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は135分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.68dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール20.5質量部、1,4-ブタンジオール29.8質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は110分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.95dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にオルソフタル酸20質量部、イソフタル酸70質量部、アジピン酸8.8質量部、1,6-ヘキサンジオール54.7質量部、ネオペンチルグリコール20.7質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は120分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.75dl/gであった。得られた共重合ポリエステル樹脂の物性を表1に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール31.7質量部、ネオペンチルグリコール40.7質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は180分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.88dl/gであった。得られた共重合ポリエステル樹脂の物性を表2に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸80質量部、イソフタル酸20質量部、エチレングリコール20.5質量部、ネオペンチルグリコール34.5質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、共重合ポリエステル樹脂を得た。後期重合にかかった時間は300分を超えても重縮合反応による溶融粘度の上昇が見られないため反応を終了した。得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.59dl/gであった。得られた共重合ポリエステル樹脂の物性を表2に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール20.5質量部、ネオペンチルグリコール34.5質量部を仕込み、0.35MPaの加圧下、250℃まで徐々に昇温し、4時間かけて溜出する水を系外に除きつつエステル化反応を行った。エステル化で得られたオリゴマーをサンプリングし、オリゴマーのAVo(酸価)、OHVo(OH価)を測定し、OHV%(ヒドロキシル末端基の割合)を算出した。続いて、前記の重縮合触媒溶液を、三酸化アンチモンのエチレングリコール溶液をポリエステル中の酸成分に対してアンチモン原子として0.04モル%となるように添加した後、1時間かけて1.3kPaまで減圧初期重合を行うとともに275℃まで上昇し、さらに0.13kPa以下で後期重合を行い、共重合ポリエステル樹脂を得た。後期重合にかかった時間は300分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.58dl/gであった。得られた共重合ポリエステル樹脂の物性を表2に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸30質量部、エチレングリコール23.2質量部、ネオペンチルグリコール33.8質量部を仕込み、0.35MPaの加圧下、250℃まで徐々に昇温し、4時間かけて溜出する水を系外に除きつつエステル化反応を行った。その後、オリゴマーを180℃以下に冷却し、イソフタル酸を20質量部仕込み、30分かけて190℃まで徐々に昇温し、第2エステル化反応を行った。第2エステル化で得られたオリゴマーをサンプリングし、オリゴマーのAVo(酸価)、OHVo(OH価)を測定し、OHV%を算出した。続いて、前記の重縮合触媒溶液の調製方法に従って作製したリン化合物のエチレングリコール溶液およびアルミニウム化合物のエチレングリコール混合溶液を、ポリエステル中の酸成分に対して、リン原子として0.04モル%を、アルミニウム原子として0.03モル%となるように添加した後、1時間かけて1.3kPaまで減圧初期重合を行うとともに275℃まで上昇し、さらに0.13kPa以下で後期重合を行い、共重合ポリエステル樹脂を得た。後期重合にかかった時間は130分、得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.90dl/gであった。得られた共重合ポリエステル樹脂の物性を表2に示す。実施例1と同様に、樹脂溶液の特性評価を行った。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸50質量部、イソフタル酸50質量部、エチレングリコール14質量部、ネオペンチルグリコール23.5質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行なった。後期重合にかかった時間は300分を超えても重縮合反応による溶融粘度の上昇が見られないため反応を終了した。得られた共重合ポリエステル樹脂の還元粘度(ηsp/C)は0.40dl/gと低いため、共重合ポリエステル樹脂の物性評価を実施しなかった。カラーb値のみ測定した。結果を表2に示す。
攪拌機、温度計、溜出用冷却機を装備した反応缶にテレフタル酸100質量部、および、エチレングリコール41.1質量部を仕込み、実施例1と同様な方法でエステル化反応および触媒添加、初期重合、後期重合反応を行ない、ポリエステル樹脂を得た。後期重合にかかった時間は300分を超えても重縮合反応による溶融粘度の上昇が見られないため反応を終了した。得られたポリエステル樹脂の還元粘度(ηsp/C)は0.59dl/gであった。得られたポリエステル樹脂の物性を表2に示す。得られたポリエステル樹脂は、トルエン/メチルエチルケトン混合溶媒に不溶のため、樹脂溶液の特性評価は行わなかった。
Claims (7)
- テレフタル酸を75モル%以下の割合で含むジカルボン酸成分と、2種類以上のジオール成分とを構成成分とする共重合ポリエステル樹脂において、下記(1)及び(2)を満足することを特徴とする共重合ポリエステル樹脂。
(1)共重合ポリエステル樹脂を窒素雰囲気下、275℃で2時間加熱処理した後の還元粘度の低下が、0.20dl/g以下である
(2)カラーb値が5以下である - 重合触媒であるアルミニウム化合物由来のアルミニウム原子、及び重合触媒であるリン化合物由来のリン原子をそれぞれ、共重合ポリエステル樹脂の全質量に対して、1~80ppm、10~100ppm含有する請求項1記載の共重合ポリエステル樹脂。
- 前記ジカルボン酸成分が、テレフタル酸を75モル%以下の割合で含み、テレフタル酸と、オルソフタル酸、イソフタル酸、アジピン酸、セバシン酸およびアゼライン酸の内、少なくとも1種以上を合わせた合計で80モル%以上含み、前記ジオール成分が、エチレングリコール、ネオペンチルグリコール、1,3-プロピレングリコール、1,4-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、および1,4-シクロヘキサンジメタノールの内、少なくとも2種以上の合計で80モル%以上含む請求項1または2に記載の共重合ポリエステル樹脂。
- アルミニウム化合物から選択される少なくとも1種、及びリン化合物から選択される少なくとも1種を含む重合触媒を用い、全ジカルボン酸成分の75モル%以下の割合でテレフタル酸を含むジカルボン酸成分と、2種類以上のジオール成分とを構成成分とする共重合ポリエステル樹脂の製造方法であって、下記(3)及び(4)を満足することを特徴とする共重合ポリエステル樹脂の製造方法。
(3)エステル化反応を開始する際の全ジカルボン酸成分のモル量(A)と全ジオール成分のモル量(G)の比(G/A)が0.8~1.4である
(4)エステル化反応終了後の反応中間生成物のカルボン酸末端基濃度が500~1500eq/ton、ヒドロキシル末端基濃度が1500~3000eq/tonである - エステル化反応終了後の反応中間生成物の全末端基に対するヒドロキシル末端基の割合が、55~75モル%であることを特徴とする請求項4記載の共重合ポリエステル樹脂の製造方法。
- 前記ジカルボン酸成分が、テレフタル酸を75モル%以下の割合で含み、テレフタル酸と、オルソフタル酸、イソフタル酸、アジピン酸、セバシン酸およびアゼライン酸の内、少なくとも1種以上を合わせた合計で80モル%以上含み、前記ジオール成分が、エチレングリコール、ネオペンチルグリコール、1,3-プロピレングリコール、1,4-ブタンジオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、および1,4-シクロヘキサンジメタノールの内、少なくとも2種以上の合計で80モル%以上含む請求項4または5に記載の共重合ポリエステル樹脂の製造方法。
- 請求項1~3のいずれかに記載の共重合ポリエステル樹脂を含む接着剤。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16746660.6A EP3255081B1 (en) | 2015-02-06 | 2016-02-03 | Copolymerized polyester resin and method for producing the same |
| CN201680008989.8A CN107207716B (zh) | 2015-02-06 | 2016-02-03 | 共聚聚酯树脂及其制造方法 |
| ES16746660T ES2865498T3 (es) | 2015-02-06 | 2016-02-03 | Resina de poliéster copolimerizada y procedimiento para producir la misma |
| JP2016512127A JP6011742B1 (ja) | 2015-02-06 | 2016-02-03 | 共重合ポリエステル樹脂、及びその製造方法 |
| US15/544,365 US10266645B2 (en) | 2015-02-06 | 2016-02-03 | Copolymerized polyester resin and method for producing the same |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015022032 | 2015-02-06 | ||
| JP2015-022033 | 2015-02-06 | ||
| JP2015-022032 | 2015-02-06 | ||
| JP2015022033 | 2015-02-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016125829A1 true WO2016125829A1 (ja) | 2016-08-11 |
Family
ID=56564169
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/053225 Ceased WO2016125829A1 (ja) | 2015-02-06 | 2016-02-03 | 共重合ポリエステル樹脂、及びその製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10266645B2 (ja) |
| EP (1) | EP3255081B1 (ja) |
| JP (1) | JP6011742B1 (ja) |
| CN (1) | CN107207716B (ja) |
| ES (1) | ES2865498T3 (ja) |
| TW (1) | TWI675861B (ja) |
| WO (1) | WO2016125829A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20220049049A1 (en) * | 2018-10-16 | 2022-02-17 | Toyobo Co., Ltd. | Polyester resin for heat-shrinkable film, heat-shrinkable film, heat-shrinkable label, and packaged product |
| WO2024070038A1 (ja) * | 2022-09-30 | 2024-04-04 | 東洋紡エムシー株式会社 | 共重合ポリエステル樹脂 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7711587B2 (ja) * | 2019-12-18 | 2025-07-23 | 東洋紡株式会社 | ポリエステル樹脂およびポリエステル樹脂の製造方法 |
| NL2025038B1 (en) * | 2020-03-03 | 2021-10-14 | Lamcoatings B V | Coil coating process |
| EP4130095B1 (en) * | 2020-03-26 | 2024-07-03 | Toyobo Co., Ltd. | Polyester resin and method for producing blow molded body formed of polyester resin |
| JP7697369B2 (ja) * | 2020-04-15 | 2025-06-24 | 東洋紡株式会社 | 共重合ポリエステル樹脂、成形品、熱収縮性フィルム、及び繊維 |
| TWI803790B (zh) * | 2020-11-24 | 2023-06-01 | 遠東新世紀股份有限公司 | 鞘芯型熱黏合纖維及不織布 |
| CN116355187B (zh) * | 2023-04-03 | 2025-06-27 | 北京化工大学 | 一种聚酯材料及其制备方法 |
| CN116512682B (zh) * | 2023-05-09 | 2024-05-24 | 深圳德昌裕新材料科技有限公司 | 一种化妆品用塑料管材及其制备工艺 |
| CN118725271A (zh) * | 2024-07-12 | 2024-10-01 | 四川大学 | 一种生物可降解共聚酯pbcat及其制备方法和应用 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02158620A (ja) * | 1988-12-10 | 1990-06-19 | Mitsubishi Rayon Co Ltd | 共重合ポリエステルの製造方法 |
| JP2004083620A (ja) * | 2002-08-22 | 2004-03-18 | Toyobo Co Ltd | 共重合ポリエステルの製造方法 |
| JP2004123984A (ja) * | 2002-10-04 | 2004-04-22 | Toyobo Co Ltd | 共重合ポリエステル |
| JP2005281381A (ja) * | 2004-03-29 | 2005-10-13 | Toyobo Co Ltd | ポリエステルならびにポリエステルの製造方法 |
| JP2005314515A (ja) * | 2004-04-28 | 2005-11-10 | Toyobo Co Ltd | ポリエステルの製造方法 |
| JP2007009155A (ja) * | 2005-07-04 | 2007-01-18 | Toyobo Co Ltd | ポリエステルの製造方法 |
| JP2007138139A (ja) * | 2005-07-12 | 2007-06-07 | Mitsubishi Chemicals Corp | 脂環式ポリエステル及びその製造方法ならびに樹脂組成物 |
| JP2007211089A (ja) * | 2006-02-08 | 2007-08-23 | Unitika Ltd | 耐光性が良好な可溶性共重合ポリエステル樹脂 |
| JP2008081576A (ja) * | 2006-09-27 | 2008-04-10 | Toyobo Co Ltd | ポリエステル樹脂の製造方法及びそれにより得られたポリエステル樹脂 |
| JP2010212272A (ja) * | 2009-03-06 | 2010-09-24 | Toyobo Co Ltd | 太陽電池用ポリエステルフィルムおよびその製造方法 |
| JP2011046829A (ja) * | 2009-08-27 | 2011-03-10 | Toyobo Co Ltd | 芳香族共重合ポリエステルの製造方法 |
| JP2011068880A (ja) * | 2009-08-28 | 2011-04-07 | Toyobo Co Ltd | 共重合ポリエステル製成形体 |
| JP2011089090A (ja) * | 2009-10-26 | 2011-05-06 | Toray Ind Inc | ポリエステル樹脂組成物、その製造方法、およびフィルム |
| WO2015060335A1 (ja) * | 2013-10-24 | 2015-04-30 | 東洋紡株式会社 | 共重合ポリエステル樹脂 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000234018A (ja) | 1999-02-16 | 2000-08-29 | Nippon Ester Co Ltd | 共重合ポリエステル |
| JP4078582B2 (ja) | 2001-02-22 | 2008-04-23 | 東洋紡績株式会社 | ポリエステルの製造方法 |
| JP2002249602A (ja) * | 2001-02-23 | 2002-09-06 | Toyobo Co Ltd | 配向ポリエステルフィルム |
| JP2002322250A (ja) | 2001-04-26 | 2002-11-08 | Toyobo Co Ltd | ポリエステルならびにポリエステルの製造方法 |
| JP2002327052A (ja) | 2001-04-27 | 2002-11-15 | Toyobo Co Ltd | ポリエステルならびにポリエステルの製造方法 |
| US6765070B2 (en) * | 2001-05-18 | 2004-07-20 | Mitsubishi Chemical Corporation | Copolymerized polyester resin composition and stretched film |
| JP4552107B2 (ja) | 2003-10-02 | 2010-09-29 | 東洋紡績株式会社 | ポリエステルならびにポリエステルの製造方法 |
| US20060094858A1 (en) * | 2004-10-28 | 2006-05-04 | Turner Sam R | Novel copolyester compositions with improved impact strength at low temperatures |
| JP4779412B2 (ja) | 2005-04-11 | 2011-09-28 | 東洋紡績株式会社 | 共重合ポリエステルの製造方法 |
| US20090215933A1 (en) | 2005-07-12 | 2009-08-27 | Mitsubishi Chemical Corporation | Alicyclic Polyester and Process for Producing the Same, and Resin Composition Using the Same |
| CN101218279A (zh) * | 2005-07-12 | 2008-07-09 | 三菱化学株式会社 | 脂环式聚酯、其制造方法以及树脂组合物 |
-
2016
- 2016-02-03 CN CN201680008989.8A patent/CN107207716B/zh active Active
- 2016-02-03 WO PCT/JP2016/053225 patent/WO2016125829A1/ja not_active Ceased
- 2016-02-03 JP JP2016512127A patent/JP6011742B1/ja active Active
- 2016-02-03 ES ES16746660T patent/ES2865498T3/es active Active
- 2016-02-03 EP EP16746660.6A patent/EP3255081B1/en active Active
- 2016-02-03 US US15/544,365 patent/US10266645B2/en active Active
- 2016-02-04 TW TW105103668A patent/TWI675861B/zh active
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02158620A (ja) * | 1988-12-10 | 1990-06-19 | Mitsubishi Rayon Co Ltd | 共重合ポリエステルの製造方法 |
| JP2004083620A (ja) * | 2002-08-22 | 2004-03-18 | Toyobo Co Ltd | 共重合ポリエステルの製造方法 |
| JP2004123984A (ja) * | 2002-10-04 | 2004-04-22 | Toyobo Co Ltd | 共重合ポリエステル |
| JP2005281381A (ja) * | 2004-03-29 | 2005-10-13 | Toyobo Co Ltd | ポリエステルならびにポリエステルの製造方法 |
| JP2005314515A (ja) * | 2004-04-28 | 2005-11-10 | Toyobo Co Ltd | ポリエステルの製造方法 |
| JP2007009155A (ja) * | 2005-07-04 | 2007-01-18 | Toyobo Co Ltd | ポリエステルの製造方法 |
| JP2007138139A (ja) * | 2005-07-12 | 2007-06-07 | Mitsubishi Chemicals Corp | 脂環式ポリエステル及びその製造方法ならびに樹脂組成物 |
| JP2007211089A (ja) * | 2006-02-08 | 2007-08-23 | Unitika Ltd | 耐光性が良好な可溶性共重合ポリエステル樹脂 |
| JP2008081576A (ja) * | 2006-09-27 | 2008-04-10 | Toyobo Co Ltd | ポリエステル樹脂の製造方法及びそれにより得られたポリエステル樹脂 |
| JP2010212272A (ja) * | 2009-03-06 | 2010-09-24 | Toyobo Co Ltd | 太陽電池用ポリエステルフィルムおよびその製造方法 |
| JP2011046829A (ja) * | 2009-08-27 | 2011-03-10 | Toyobo Co Ltd | 芳香族共重合ポリエステルの製造方法 |
| JP2011068880A (ja) * | 2009-08-28 | 2011-04-07 | Toyobo Co Ltd | 共重合ポリエステル製成形体 |
| JP2011089090A (ja) * | 2009-10-26 | 2011-05-06 | Toray Ind Inc | ポリエステル樹脂組成物、その製造方法、およびフィルム |
| WO2015060335A1 (ja) * | 2013-10-24 | 2015-04-30 | 東洋紡株式会社 | 共重合ポリエステル樹脂 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20220049049A1 (en) * | 2018-10-16 | 2022-02-17 | Toyobo Co., Ltd. | Polyester resin for heat-shrinkable film, heat-shrinkable film, heat-shrinkable label, and packaged product |
| US12134679B2 (en) * | 2018-10-16 | 2024-11-05 | Toyobo Co., Ltd. | Polyester resin for heat-shrinkable film, heat-shrinkable film, heat-shrinkable label, and packaged product |
| WO2024070038A1 (ja) * | 2022-09-30 | 2024-04-04 | 東洋紡エムシー株式会社 | 共重合ポリエステル樹脂 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6011742B1 (ja) | 2016-10-19 |
| ES2865498T3 (es) | 2021-10-15 |
| TWI675861B (zh) | 2019-11-01 |
| EP3255081A1 (en) | 2017-12-13 |
| EP3255081A4 (en) | 2018-10-10 |
| TW201638144A (zh) | 2016-11-01 |
| JPWO2016125829A1 (ja) | 2017-04-27 |
| US20180265627A1 (en) | 2018-09-20 |
| CN107207716A (zh) | 2017-09-26 |
| EP3255081B1 (en) | 2021-03-24 |
| US10266645B2 (en) | 2019-04-23 |
| CN107207716B (zh) | 2020-09-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6011742B1 (ja) | 共重合ポリエステル樹脂、及びその製造方法 | |
| TWI648305B (zh) | 共聚合聚酯樹脂、熱收縮性薄膜、成型品、片材及接著劑 | |
| JP6844748B2 (ja) | 共重合ポリエステル樹脂、成形品、及び熱収縮性フィルム | |
| JP6657760B2 (ja) | ポリエステルの製造方法 | |
| WO2013180215A1 (ja) | ポリエステル樹脂の製造方法 | |
| JP7697369B2 (ja) | 共重合ポリエステル樹脂、成形品、熱収縮性フィルム、及び繊維 | |
| JP7579527B1 (ja) | 共重合ポリエステル樹脂の製造方法 | |
| JP2008081576A (ja) | ポリエステル樹脂の製造方法及びそれにより得られたポリエステル樹脂 | |
| JP6236946B2 (ja) | ポリエステルの製造方法 | |
| JP5267795B2 (ja) | 共重合ポリエステル、それからなる粉体塗料用共重合ポリエステル組成物並びに粉体塗料 | |
| EP0119731B1 (en) | Copolyester of polyethylene terephthalate, process for making it and its use in producing molded articles | |
| JP5911734B2 (ja) | ポリエステル樹脂組成物及びその製造方法 | |
| JP2008001855A (ja) | ポリエステルおよびその用途 | |
| JPH04309521A (ja) | ボトル成形用ポリエチレンテレフタレート | |
| WO2023063218A1 (ja) | 共重合ポリエステル樹脂、成形品、熱収縮性フィルム、及び繊維 | |
| CN119137184A (zh) | 共聚聚酯树脂 | |
| JP2015110742A (ja) | ポリエステル樹脂組成物 | |
| US20150045530A1 (en) | Catalyst solution for use in production of polyester, and method for producing polyester resin using same | |
| JP2020050765A (ja) | ポリエステルの製造方法 | |
| JP2004256683A (ja) | 重縮合反応用触媒およびそれを用いるポリエステルの製造方法 | |
| JPH02245020A (ja) | ポリエステルの製造方法 | |
| JP2015147831A (ja) | ポリエステル組成物 | |
| JP2003231756A (ja) | ポリオルガノシロキサン化合物のスラリー化法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016512127 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16746660 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15544365 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2016746660 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |