WO2016136453A1 - マスターバッチの製造方法、該製造方法により得られるマスターバッチ、タイヤ用ゴム組成物及び空気入りタイヤ - Google Patents
マスターバッチの製造方法、該製造方法により得られるマスターバッチ、タイヤ用ゴム組成物及び空気入りタイヤ Download PDFInfo
- Publication number
- WO2016136453A1 WO2016136453A1 PCT/JP2016/053767 JP2016053767W WO2016136453A1 WO 2016136453 A1 WO2016136453 A1 WO 2016136453A1 JP 2016053767 W JP2016053767 W JP 2016053767W WO 2016136453 A1 WO2016136453 A1 WO 2016136453A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- rubber
- microfibrillated plant
- plant fiber
- mass
- fiber
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
- C08J3/22—Compounding polymers with additives, e.g. colouring using masterbatch techniques
- C08J3/226—Compounding polymers with additives, e.g. colouring using masterbatch techniques using a polymer as a carrier
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B60—VEHICLES IN GENERAL
- B60C—VEHICLE TYRES; TYRE INFLATION; TYRE CHANGING; CONNECTING VALVES TO INFLATABLE ELASTIC BODIES IN GENERAL; DEVICES OR ARRANGEMENTS RELATED TO TYRES
- B60C1/00—Tyres characterised by the chemical composition or the physical arrangement or mixture of the composition
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
- C08J3/22—Compounding polymers with additives, e.g. colouring using masterbatch techniques
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L15/00—Compositions of rubber derivatives
- C08L15/02—Rubber derivatives containing halogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L7/00—Compositions of natural rubber
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L7/00—Compositions of natural rubber
- C08L7/02—Latex
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2307/00—Characterised by the use of natural rubber
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2307/00—Characterised by the use of natural rubber
- C08J2307/02—Latex
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2309/00—Characterised by the use of homopolymers or copolymers of conjugated diene hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2401/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2401/02—Cellulose; Modified cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/02—Fibres or whiskers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/03—Polymer mixtures characterised by other features containing three or more polymers in a blend
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/06—Polymer mixtures characterised by other features having improved processability or containing aids for moulding methods
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/14—Polymer mixtures characterised by other features containing polymeric additives characterised by shape
- C08L2205/16—Fibres; Fibrils
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2310/00—Masterbatches
Definitions
- the present invention relates to a masterbatch production method, a masterbatch obtained by the production method, a tire rubber composition, and a pneumatic tire.
- the rubber composition For the purpose of reinforcing the rubber composition and improving the modulus (complex elastic modulus), there is a method of blending the rubber composition with microfibrillated plant fibers such as cellulose fibers as a filler.
- microfibrillated plant fiber has a strong self-aggregating power and poor compatibility with the rubber component, the dispersibility during rubber kneading is low. Therefore, even if it mix
- Patent Documents 1 to 4 disclose that cellulose fibers and a rubber latex are mixed in advance to improve the dispersibility of the cellulose fibers and improve the physical properties of the rubber. The point which improves the dispersibility of a cellulose fiber and improves rubber physical properties etc. still leaves the room for improvement.
- the present invention solves the above problems, further improves the dispersibility of the microfibrillated plant fiber, has excellent processability, low fuel consumption, tensile strength, elongation at break, air permeability, rigidity, fracture characteristics, adhesion performance,
- An object of the present invention is to provide a method for producing a masterbatch having improved rubber properties such as permanent strain resistance.
- the present invention comprises a step (I) of mixing rubber latex and microfibrillated plant fiber oxidized with an N-oxyl compound, and adjusting the pH of the resulting mixture to 2 to 6 for coagulation. It is related with the manufacturing method of a masterbatch containing.
- the content of the microfibrillated plant fiber with respect to 100 parts by mass of rubber in the master batch is preferably 10 to 50 parts by mass.
- the present invention also relates to a master batch obtained by the production method.
- the present invention relates to a rubber composition for tires produced using the master batch.
- the present invention relates to a pneumatic tire produced using the rubber composition.
- the rubber latex and the microfibrillated plant fiber oxidized with an N-oxyl compound are mixed, and the pH of the resulting mixture is adjusted to 2 to 6 to solidify (I )
- An improved tire rubber composition and pneumatic tire are obtained.
- the fiber can be highly dispersed in rubber and the physical properties of rubber can be improved by adopting the production method including the above step (I). it can.
- the microfibrillated plant fiber oxidized with the N-oxyl compound has good dispersion efficiency in the rubber latex, and the fiber is uniformly incorporated into the latex. It is assumed that the fibers are uniformly dispersed in the rubber. In particular, when natural rubber latex is used as the rubber latex, proteins and phospholipids in the rubber are removed, and at the same time, the fibers are uniformly dispersed, and the rubber physical properties are remarkably improved.
- the rubber latex is not particularly limited.
- modified natural rubber latex such as natural rubber latex and saponified natural rubber latex
- isoprene-based rubber latex such as epoxidized natural rubber latex and isoprene rubber latex
- epoxidized natural rubber latex and isoprene rubber latex
- modified natural rubber latexes such as natural rubber latex and saponified natural rubber latex can be preferably used.
- a normal natural rubber latex containing non-rubber components such as phosphorus and nitrogen
- the non-rubber component can be sufficiently removed without using a rubber latex from which a non-rubber component such as a special saponified natural rubber latex has been removed.
- Natural rubber latex is collected as the sap of natural rubber trees such as Hevea trees, and contains rubber, water, proteins, lipids, inorganic salts, etc., and the gel content in rubber is based on the complex presence of various impurities. It is considered a thing.
- raw latex field latex
- concentrated latex concentrated by centrifugation or creaming method purified latex, high ammonia latex added with ammonia by a conventional method
- Zinc oxide, TMTD and ammonia stabilized LATZ latex, etc. can be used as a natural rubber latex.
- Rubber latex such as natural rubber latex may be mixed with the microfibrillated plant fiber as it is.
- a saponified one may be used in advance.
- the saponification treatment can be performed by adding an alkali such as NaOH and a surfactant as necessary to natural rubber latex and allowing to stand at a predetermined temperature for a certain time. In addition, you may perform stirring etc. as needed.
- an alkali such as NaOH
- a surfactant as necessary to natural rubber latex and allowing to stand at a predetermined temperature for a certain time. In addition, you may perform stirring etc. as needed.
- the saponification treatment By performing the saponification treatment in the latex state, each particle of the natural rubber is uniformly treated, and the saponification treatment can be performed efficiently.
- the saponification treatment is performed, the phosphorus compound separated by the saponification is removed, so that the phosphorus content in the natural rubber contained in the prepared master batch can be suppressed.
- the protein in the natural rubber is de
- the alkali used for the saponification treatment sodium hydroxide, potassium hydroxide and the like are preferable.
- the surfactant is not particularly limited, and examples thereof include known nonionic surfactants such as polyoxyethylene alkyl ether sulfates, anionic surfactants, and amphoteric surfactants.
- a polyoxyethylene alkyl ether sulfate ester salt is preferable because it can be saponified.
- the addition amount of alkali and surfactant, the temperature and time of the saponification treatment may be appropriately set.
- the microfibrillated plant fiber oxidized with the N-oxyl compound has a primary hydroxyl group at the 6-position of carbon in the pyranose ring of cellulose oxidized to a carboxyl group or an aldehyde group, and has a cellulose I-type crystal structure. What has it can be used conveniently.
- Such specific microfibrillated plant fibers are disclosed in JP-A-2008-001728 and the like.
- the microfibrillated plant fiber is a fiber that is refined by surface oxidation of the primary hydroxyl group at the 6-position of carbon in the pyranose ring of cellulose to a carboxyl group or an aldehyde group and a salt thereof.
- a pyranose ring is a six-membered ring carbohydrate consisting of five carbons and one oxygen.
- this primary hydroxyl group is selectively oxidized. That is, natural celluloses are nanofibers at the time of biosynthesis, but many of these are converged by hydrogen bonds to form fiber bundles.
- the primary hydroxyl group at the 6-position of the carbon of the pyranose ring is selectively oxidized, and this oxidation reaction remains on the surface of the microfibril, so that the density is high only on the surface of the microfibril.
- a carboxyl group is introduced. Since the carboxyl groups are negatively charged and repel each other and dispersed in water, the aggregation of the microfibrils is hindered. As a result, the fiber bundle is unraveled in units of microfibrils and becomes cellulose nanofibers. From the viewpoint that the effects of the present invention can be obtained satisfactorily, those in which the primary hydroxyl group at the 6-position of carbon in the pyranose ring of cellulose is surface oxidized to a carboxyl group are preferred.
- the total amount of carboxyl groups and aldehyde groups present in the microfibrillated plant fiber is preferably 0.1 mmol / g or more, more preferably 0.2 mmol / g or more, based on the weight (absolutely dry) of the cellulose fiber. In addition, it is preferably 2.5 mmol / g or less, more preferably 2.2 mmol / g or less. Within the above range, the nanofibers can be uniformly dispersed. In the present invention, the above sum is expressed as the amount of charge in the microfibrillated plant fiber.
- Absolutely dry means that cellulose fiber accounts for 100% of the total weight.
- the amount of the carboxyl group is preferably 0.1 mmol / g or more, more preferably 0.2 mmol / g or more, and preferably 2.4 mmol / g, based on the weight (absolute dryness) of the cellulose fiber.
- it is preferably 2.1 mmol / g or less.
- the average fiber length of the microfibrillated plant fiber is preferably 50 nm or more, more preferably 150 nm or more, and the average fiber length is preferably 5000 nm or less, more preferably 2000 nm or less. If the thickness is less than 50 nm or exceeds 5000 nm, the effects of the present invention may not be obtained satisfactorily.
- the maximum fiber diameter of the microfibrillated plant fiber is preferably 1000 nm or less, more preferably 500 nm or less, still more preferably 30 nm or less, and the lower limit is not particularly limited. If it exceeds 1000 nm, the properties of the nanofiber are hardly exhibited and the dispersibility is also poor, so that the effects of the present invention may not be sufficiently exhibited.
- the number average fiber diameter of the microfibrillated plant fiber is preferably 2 to 150 nm, more preferably 2 to 100 nm, still more preferably 2 to 10 nm, and particularly preferably 2 to 5 nm. Within the above range, the nanofibers can be uniformly dispersed.
- the identification that the microfibrillated plant fiber has a type I crystal structure, the amount of aldehyde group and carboxyl group (mmol / g), the average fiber length maximum fiber diameter, and the number average fiber diameter are known methods. For example, it can be analyzed by the method described in JP-A-2008-001728. The number average fiber diameter and the average fiber length were measured with a scanning probe microscope (manufactured by Hitachi High-Tech Science Co., Ltd.) with a cellulose nanofiber fixed on a mica section (3000 nm ⁇ 3000 nm), and the fiber width for 50 fibers was measured. The number average fiber diameter was calculated. The average fiber length was calculated from the obtained observation image using image analysis software WinROOF (Mitani Corporation).
- Microfibrillated plant fibers oxidized using an N-oxyl compound for example, oxidize natural cellulose by using natural cellulose as a raw material, using an N-oxyl compound as an oxidation catalyst in water, and acting a co-oxidant.
- a dispersion in which natural cellulose is dispersed in water is prepared.
- the natural cellulose include purified cellulose isolated from cellulose biosynthetic systems such as plants, animals, and bacteria-producing gels. Natural cellulose can be subjected to a treatment for increasing the surface area such as beating. It is also possible to use natural cellulose that has been stored in Never Dry after isolation and purification.
- the dispersion medium of natural cellulose in the reaction is water, and the natural cellulose concentration in the aqueous reaction solution is usually about 5% or less.
- N-oxyl compound that can be used as an oxidation catalyst for cellulose refers to a compound capable of generating a nitroxy radical.
- 2,2,6,6-tetraalkylpiperidine-1-oxyl and derivatives thereof such as 4-hydroxy-2,2,6, 6-tetraalkylpiperidine-1-oxyl, 4-alkoxy-2,2,6,6-tetraalkylpiperidine-1-oxyl, 4-benzoyloxy-2,2,6,6-tetraalkylpiperidine-1-oxyl 4-amino-2,2,6,6-tetraalkylpiperidine-1-oxyl and the like, among which 2,2,6,6-tetramethylpiperidine-1-oxyl (hereinafter also referred to as TEMPO) and its Derivatives such as 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (hereinafter also referred to as 4-hydroxy TEMPO) 4-alkoxy-2,2,6,6-tetramethylpiperidine-1-oxyl (hereinafter also referred to as 4-alkoxy TEMPO), 4-benzo
- 4-hydroxy TEMPO derivatives include ethers of 4-hydroxy TEMPO having a linear or branched carbon chain in which the hydroxyl group of 4-hydroxy TEMPO is a compound having the following formulas (2) to (4). And derivatives obtained by esterification with carboxylic acid or sulfonic acid.
- R 5 represents a linear or branched carbon chain having 4 or less carbon atoms.
- R 6 represents a linear or branched carbon chain having 4 or less carbon atoms.
- R 7 represents a linear or branched carbon chain having 4 or less carbon atoms.
- 4-acetamido TEMPO represented by the following formula (5) in which the amino group of 4-amino TEMPO is acetylated and imparted appropriate hydrophobicity is inexpensive and uniformly This is preferable in that an oxidized cellulose can be obtained.
- a radical of an N-oxyl compound represented by the following formula (6), that is, an azaadamantane type nitroxy radical is also preferable in that cellulose can be efficiently oxidized in a short time.
- R 8 and R 9 are the same or different and each represents a hydrogen atom or a linear or branched alkyl group having 1 to 6 carbon atoms.
- the amount of the N-oxyl compound added is not particularly limited as long as the oxidized cellulose obtained can be sufficiently converted into a nanofiber, but is preferably 0.01 to 1 g of cellulose fiber (absolutely dry). It is ⁇ 10 mmol / g, more preferably 0.01 to 1 mmol / g, still more preferably 0.025 to 0.5 mmol / g.
- hypohalous acid, halous acid, perhalogen acid or salts thereof; hydrogen peroxide, perorganic acid, etc. can be used, and alkali metal hypohalite is preferable.
- alkali metal hypohalite is preferable.
- the addition amount of the alkali metal bromide is preferably 0.1 to 100 mmol / g, more preferably 0.1 to 10 mmol / g, still more preferably 0.5 to 5 mmol / g with respect to 1 g of cellulose fiber (absolutely dry).
- the amount of sodium hypochlorite added is preferably 0.5 to 500 mmol / g, more preferably 0.5 to 50 mmol / g, still more preferably 2.5 to 25 mmol / g.
- the pH of the aqueous reaction solution is preferably maintained in the range of about 8-11.
- the temperature of the aqueous solution can be about 4 to 40 ° C., for example, room temperature, and it is not particularly necessary to control the temperature.
- the amount of the co-oxidant added is preferably in the range of about 3.0 to 8.2 mmol / g with respect to 1 g of cellulose fiber (absolutely dry).
- the purification step compounds other than the reaction fiber and water contained in the reaction slurry such as unreacted hypochlorous acid and various by-products are removed from the system.
- a normal purification method can be employed. For example, a dispersion of high-purity (99% by mass or more) reactant fiber and water is prepared by repeating washing with water and filtration.
- the dispersion of the microfibrillated plant fiber is performed by subjecting the reaction product fiber (water dispersion) impregnated with water obtained in the step to dispersion in a solvent (dispersion step).
- the solvent as the dispersion medium is usually preferably water.
- alcohols, ethers, ketones and the like that are soluble in water may be used.
- a disperser used in the dispersion step a general-purpose disperser, a powerful and beating ability apparatus such as a homomixer under high speed rotation, a high-pressure homogenizer, or the like can be used.
- the dispersion of the microfibrillated plant fiber is used as a microfibrillated plant fiber oxidized by using an N-oxyl compound. Further, by drying the dispersion of the microfibrillated plant fiber, it can also be used as a microfibrillated plant fiber oxidized by using an N-oxyl compound. For drying, freeze-drying or the like can be employed.
- a binder in the dispersion of the microfibrillated plant fiber a compound having a very high boiling point such as a water-soluble polymer or saccharide and having an affinity for cellulose can be mixed.
- the microfibrillated plant fiber that can be dispersed again as a nanofiber in a solvent can be obtained.
- the amount of the binder added to the dispersion is desirably in the range of 10 to 80% by mass with respect to the reactant fiber.
- the microfibrillated plant fiber obtained by the drying can be used again as a microfibrillated plant fiber oxidized by using an N-oxyl compound by mixing in a solvent and applying an appropriate dispersion force. it can.
- the mixing of the rubber latex and the microfibrillated plant fiber oxidized with the N-oxyl compound may be performed by mixing the microfibrillated plant fiber as it is with the rubber latex, but the microfibrillated plant fiber aqueous solution ( It is preferable to mix with the rubber latex in the state of dispersion of the microfibrillated plant fiber, such as an aqueous solution in which the microfibrillated plant fiber is dispersed in water.
- the content (solid content) of the microfibrillated plant fiber in the aqueous solution is preferably 0.2 to 20% by mass, more preferably 0.5 to 10% by mass, and still more preferably 0.5 to 3% by mass. It is.
- the step of mixing rubber latex and the microfibrillated plant fiber to produce a mixture thereof can be prepared by sequentially dropping them, injecting them, etc., and then mixing them by a known method.
- the pH is preferably 2-5, more preferably 3-4.
- pH adjustment can be performed by adding an acid, a base, or the like.
- the rubber component in the coagulated product can be highly purified. Specifically, non-rubber components such as proteins and phospholipids in natural rubber are removed and purified, and fuel efficiency and processability are improved.
- the deterioration of the rubber is likely to proceed by removing the non-rubber component, etc., but since the decrease in the molecular weight of the rubber during storage is suppressed by adjusting the pH during solidification to a predetermined range, it is favorable. Heat aging resistance is obtained. Furthermore, a composite in which the microfibrillated plant fiber is uniformly dispersed in rubber is obtained. Therefore, the dispersibility of the microfibrillated plant fiber is remarkably enhanced, and the rubber physical properties are improved.
- the purification of natural rubber means removing impurities such as phospholipids and proteins other than natural polyisoprenoid components.
- Natural rubber has a structure in which the isoprenoid component is covered with the impurity component. By removing the component, the structure of the isoprenoid component changes, and the interaction with the compounding agent changes, resulting in energy. It is assumed that loss can be reduced, durability can be improved, and a better masterbatch can be obtained.
- Methods for coagulating the mixture include acid coagulation, salt coagulation, methanol coagulation and the like, but in order to coagulate the microfibrillated plant fiber in a uniformly dispersed state in the masterbatch, acid coagulation, salt coagulation or These combinations are preferred, and acid coagulation is more preferred.
- acid for coagulation include formic acid, sulfuric acid, hydrochloric acid, acetic acid and the like, and sulfuric acid is preferable from the viewpoint of cost.
- the salt include monovalent to trivalent metal salts (calcium salts such as sodium chloride, magnesium chloride, calcium nitrate, and calcium chloride).
- the coagulation of the mixture is preferably carried out under conditions such that the microfibrillated plant fiber is gently taken into the rubber latex.
- the temperature of the mixture is preferably 40 ° C. or less, and more preferably 35 ° C. or less.
- the coagulated product obtained in step (I) may be washed as necessary. Thereby, for example, when natural rubber latex is used as the rubber latex, the obtained coagulated product can be further washed to adjust the amount of phosphorus and the amount of nitrogen to more desired amounts.
- washing method examples include a method of centrifuging after diluting the rubber component with water, and a method of allowing the rubber component to stand still after diluting with the water to float or precipitate the rubber and discharge only the aqueous phase.
- the latex when centrifuging, the latex is first diluted with water so that the rubber content of the latex is 5 to 40% by mass, preferably 10 to 30% by mass, and then 1 to 60 at 5000 to 10000 rpm. Centrifugation may be performed for a minute, and washing may be repeated until a desired phosphorus content is obtained.
- washing may be repeated until the desired phosphorus content is obtained by repeatedly adding water and stirring.
- the washing method is not limited to these, and it may be washed by removing the liquid phase after neutralization with weak alkaline water such as sodium carbonate so that the pH is in the range of 6-7.
- the rubber After washing as necessary, it is usually dried by a known method (oven, reduced pressure, etc.).
- a known method for drying the rubber is kneaded with a biaxial roll, a Banbury mixer or the like after drying, a crumb-like masterbatch containing rubber and the microfibrillated plant fiber is obtained.
- the masterbatch is preferably formed into a sheet having a thickness of several centimeters by a rolling roll in order to improve unity and handling properties.
- the said masterbatch may contain another component in the range which does not inhibit the effect of this invention.
- the phosphorus content contained in the rubber content (solid content) in the master batch is preferably 200 ppm or less, and more preferably 120 ppm or less.
- the phosphorus content can be measured by a conventional method such as ICP emission analysis.
- Phosphorus is derived from phospholipids (phosphorus compounds).
- the said masterbatch 0.3 mass% or less is preferable and, as for the nitrogen content contained in the rubber
- the nitrogen content can be measured by a conventional method such as Kjeldahl method. Nitrogen is derived from protein.
- the content of the microfibrillated plant fiber with respect to 100 parts by mass of the rubber component is preferably 1 part by mass or more, more preferably 5 parts by mass or more, and further preferably 10 parts by mass or more. is there. If the amount is less than 1 part by mass, in the rubber composition containing the masterbatch, when trying to secure the necessary microfibrillated plant fiber, the amount of the rubber is excessively increased, the crosslinking density is lowered, and the fuel consumption is reduced. Sexuality may worsen. Moreover, this content becomes like this. Preferably it is 50 mass parts or less, More preferably, it is 30 mass parts or less, More preferably, it is 20 mass parts or less. When it exceeds 50 parts by mass, the dispersibility of the microfibrillated plant fiber may be lowered, and the fuel economy and elongation at break may be deteriorated.
- the rubber composition for tires of this invention contains the said masterbatch.
- a rubber composition in which the microfibrillated plant fibers are uniformly dispersed in the rubber can be obtained.
- the content of the rubber component derived from the master batch in 100% by mass of the rubber component is preferably 5% by mass or more, more preferably 10% by mass or more, and further preferably 15% by mass or more. is there. If it is less than 5% by mass, the content of the microfibrillated plant fiber contained in the rubber composition is too small, and the effects of the present invention may not be sufficiently obtained.
- the content of the isoprene-based rubber in 100% by mass of the rubber component is preferably 5% by mass or more, more preferably 10% by mass or more, and further preferably 15% by mass. If it is less than 5% by mass, excellent fuel efficiency may not be obtained.
- Rubber components that can be used in addition to isoprene rubber include butadiene rubber (BR), styrene butadiene rubber (SBR), ethylene propylene diene rubber (EPDM), chloroprene rubber (CR), acrylonitrile butadiene rubber (NBR), and butyl rubber (IIR).
- BR butadiene rubber
- SBR styrene butadiene rubber
- EPDM ethylene propylene diene rubber
- CR chloroprene rubber
- NBR acrylonitrile butadiene rubber
- IIR butyl rubber
- the content of the microfibrillated plant fiber is preferably 1 part by mass or more, more preferably 2 parts by mass or more, and further preferably 3 parts by mass or more with respect to 100 parts by mass of the rubber component. It is. Moreover, this content becomes like this. Preferably it is 50 mass parts or less, More preferably, it is 30 mass parts or less, More preferably, it is 20 mass parts or less. There exists a possibility that the effect by mix
- the rubber composition of the present invention includes reinforcing fillers such as carbon black and silica, zinc oxide, stearic acid, various anti-aging agents, softeners (oil, wax, etc.), and vulcanizing agents.
- reinforcing fillers such as carbon black and silica, zinc oxide, stearic acid, various anti-aging agents, softeners (oil, wax, etc.), and vulcanizing agents.
- Various materials generally used in the tire industry such as (sulfur, organic peroxide, etc.), vulcanization accelerators (sulfenamide-based, guanidine-based vulcanization accelerators, etc.) may be appropriately blended. .
- the above components are kneaded using a rubber kneader such as an open roll or a Banbury mixer, and then vulcanized. Can be manufactured.
- a rubber kneader such as an open roll or a Banbury mixer
- the rubber composition of the present invention includes a cap tread, a base tread, an under tread, a clinch apex, a bead apex, a sidewall, a breaker, an edge band, a full band, a breaker cushion rubber, a rubber for covering a carcass cord, and a run flat reinforcing layer. It can be used for tire members such as insulation, chafers, inner liners, belts, rolls, etc., but it can be suitably used for sidewalls and inner liners.
- the tire of the present invention is produced by a usual method using the rubber composition. That is, a rubber composition containing various materials as necessary is extruded in accordance with the shape of each member such as a tire sidewall and an inner liner at an unvulcanized stage, and is usually used on a tire molding machine. After forming an unvulcanized tire by molding by the method, it can be produced by heating and pressing in a vulcanizer.
- Examples of the tire of the present invention include a pneumatic tire and an airless (solid) tire. Among them, a pneumatic tire is preferable.
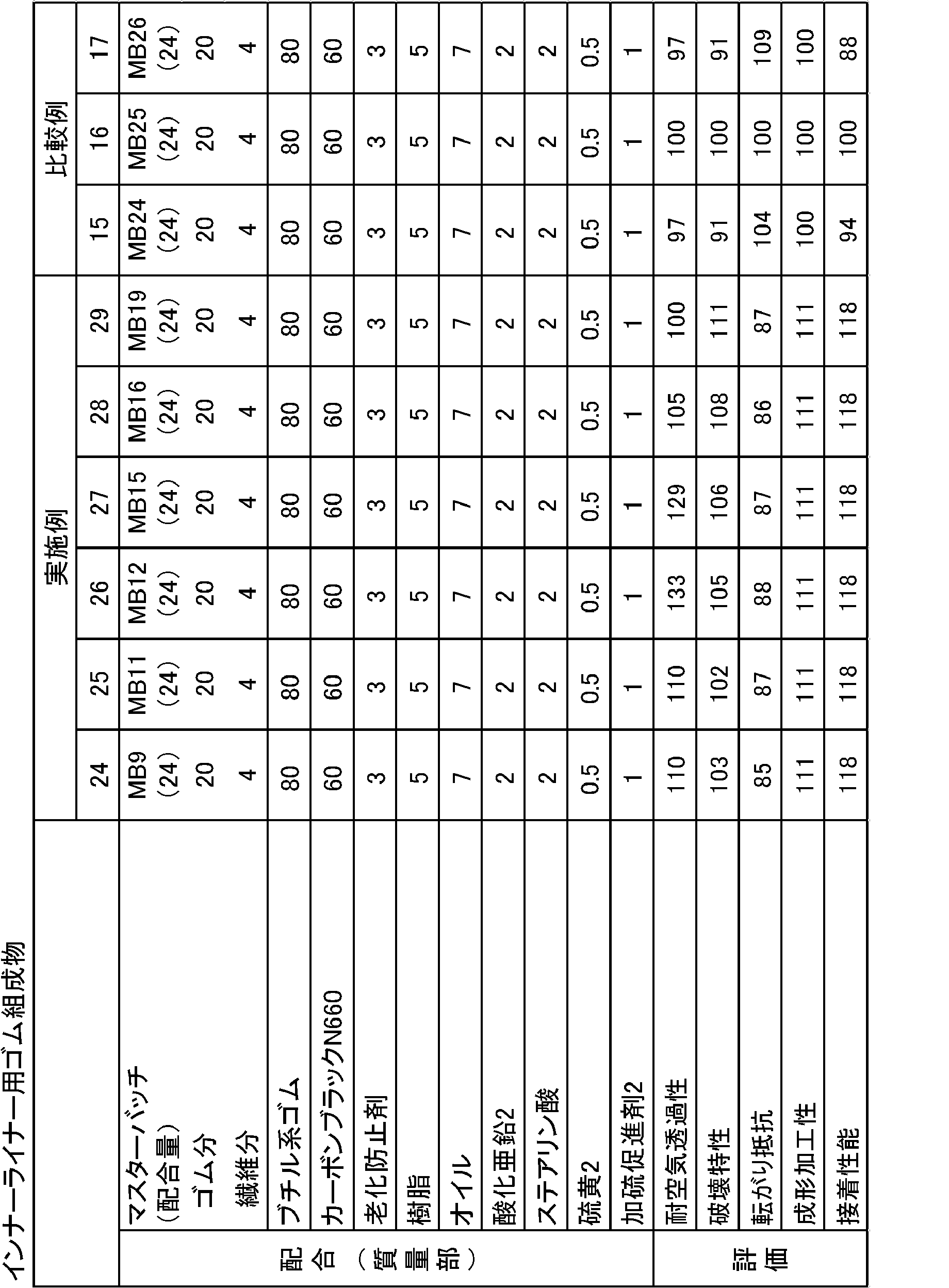
- Microfibrillated plant fiber 15 BiNFi-s (manufactured by Sugino Machine Co., Ltd.) (cellulose fine fiber processed by ultra-high pressure water jet technology, number average fiber diameter: 20-50 nm, average fiber length: 500-1000 nm)
- Microfibrillated plant fiber 16 Neofiber manufactured by Oji Bag Co., Ltd. (number average fiber diameter: 0.1 to 1.2 mm, average fiber length: 0.2 to 1.2 mm)
- Coagulant 1% sulfuric acid NR: TSR20 manufactured by Wako Pure Chemical Industries, Ltd.
- Purified NR prepared in the following production example Butyl rubber: Chlorobutyl HT1066 manufactured by ExxonMobil BR: BR150B manufactured by Ube Industries, Ltd. (cis content: 95% by mass) Carbon black N660: Dia Black N660 manufactured by Mitsubishi Chemical Corporation (N 2 SA: 28 m 2 / g, DBP absorption: 84 ml / 100 g) Carbon black N550: Dia Black N550 manufactured by Mitsubishi Chemical Corporation (N 2 SA: 41 m 2 / g, DBP absorption: 115 ml / 100 g) Anti-aging agent: NOCRACK 6C (N-phenyl-N ′-(1,3-dimethylbutyl) -p-phenylenediamine) (6PPD) manufactured by Ouchi Shinsei Chemical Co., Ltd.
- the reaction product is filtered through a glass filter, washed with a sufficient amount of water and filtered five times to impregnate 15% by mass of water with a solid content of 15% by mass.
- Reactant fibers were obtained.
- water was added to the reactant fiber to form a slurry having a solid content of 1% by mass.
- oxidized cellulose absolute dry
- 1.5 ml of 1M NaOH and 0.5 ml of 30% hydrogen peroxide water were added, and ultrapure water was added to adjust to 5% (w / v). Heated at 80 ° C. for 2 hours.
- Unwashed alkaline hydrolyzed oxidized cellulose was treated three times with an ultra-high pressure homogenizer (treatment pressure 140 MPa) to obtain a transparent gel dispersion (microfibrillated plant fiber 1).
- the total amount of carboxyl groups and aldehyde groups present in the microfibrillated plant fiber and the amount of carboxyl groups are 1.6 mmol / g and 1.5 mmol / g based on the weight of the cellulose fiber, and the maximum fiber diameter and The number average fiber diameter was 8.2 nm and 4.0 nm, and the average fiber length was 470 nm.
- the diameter of the aggregate (aggregated rubber) thus obtained was about 0.5 to 5 mm.
- the obtained agglomerates were taken out and immersed in 1000 ml of a 2% by weight aqueous sodium carbonate solution at room temperature for 4 hours, and then the rubber was taken out. To this, 2000 ml of water was added and stirred for 2 minutes to remove water as much as possible 7 times. Thereafter, 500 ml of water was added, 2% by mass formic acid was added until pH 4 was reached, and the mixture was allowed to stand for 15 minutes. Further, remove the water as much as possible, repeat the work of adding water again and stirring for 2 minutes three times, then squeezing the water with a water squeeze roll to form a sheet, and then drying at 90 ° C.
- NR was obtained.
- the phosphorus content contained in the highly purified NR was 92 ppm, and the nitrogen content was 0.07% by mass.
- the anti-aging dispersion was prepared by mixing 462.5 g of water with 12.5 g of Emulvin W, 12.5 g of Tamol NN9104, 12.5 g of Van gel B, and 500 g of Wingstay L (total 1000 g) for 16 hours using a ball mill. did.
- Example 1 (Masterbatch 1)
- the dispersion of the microfibrillated plant fiber 1 is diluted with water so that the solid concentration is 0.5% (w / v), and is treated for about 10 minutes with a high-speed homogenizer (rotation speed: 8000 rpm, T25 manufactured by IKA Japan).
- a uniform dispersion was prepared (viscosity was 7-8 mPas).
- the dispersion of the microfibrillated plant fiber 1 is 20 dry weight (solid content) of the microfibrillated plant fiber.
- Example 2 Masterbatch 2 (MB2) in the same manner as in Example 1 except that the dispersion of microfibrillated plant fiber 1 was added so that the dry weight (solid content) of the microfibrillated plant fiber was 10 parts by mass. Got.
- Example 3 (Comparative Example 1 (Masterbatch 3) A master batch 3 (MB3) was obtained in the same manner as in Example 1 except that the dispersion of the microfibrillated plant fiber 2 was used.
- Example 3 (master batch 5)
- the dispersion of the microfibrillated plant fiber 1 is diluted with water so that the solid concentration is 0.5% (w / v), and is treated with a high-speed homogenizer (rotation speed: 8000 rpm) for about 10 minutes to obtain a uniform dispersion.
- Produced (viscosity 7-8 mPas).
- DRC solid content concentration
- 25 g of 10% Emar E-27C aqueous solution and 60 g of 25% NaOH aqueous solution were added to 1000 g of the latex and saponified for 24 hours at room temperature. Reaction was performed to obtain a saponified natural rubber latex.
- the dispersion of the microfibrillated plant fiber 1 is used as the dry weight of the microfibrillated plant fiber.
- Solid content was added to 20 parts by mass and stirred for about 5 minutes with a homogenizer (rotation speed: 8000 rpm) to prepare a rubber latex dispersion.
- a coagulant was added with slow stirring to adjust the pH to 3-4, coagulate in a crumb shape, dehydrated, and dried at 40 ° C. for 12 hours to obtain a master batch 5 (MB5).
- Example 4 Masterbatch 6 (MB6) in the same manner as in Example 3 except that the dispersion of the microfibrillated plant fiber 1 was added so that the dry weight (solid content) of the microfibrillated plant fiber was 10 parts by mass. Got.
- the nitrogen content was quantified with a gas chromatograph after pyrolysis.
- the phosphorus content was determined using an ICP emission spectrometer (P-4010, manufactured by Hitachi, Ltd.).
- the dispersibility of the microfibrillated plant fiber is better than when the microfibrillated plant fiber 2 is used, and the nitrogen content of the rubber It can be seen that the content and phosphorus content are reduced. Further, when mixed with saponified natural rubber latex, the dispersibility was better.
- each of the number average fiber diameter, the average fiber length, and the weight of the cellulose fiber shown in Table 4 is the total amount of the carboxyl group and the aldehyde group present in the microfibrillated plant fiber, and the microfibrillated plant fiber 3-16 Was prepared.
- the obtained dispersion of each microfibrillated plant fiber was diluted with water so that the solid content concentration would be 1% (w / v), and treated with a high-speed homogenizer (rotation speed: 8000 rpm) for about 10 minutes to be uniform.
- a dispersion was prepared. With respect to 100 parts by mass of field latex (solid content concentration (DRC) 30% (w / v)), the obtained dispersion of each microfibrillated plant fiber is the dry weight (solid content) of the microfibrillated plant fiber.
- DRC solid content concentration
- a coagulant was added with slow stirring to adjust the pH to 3 to 4 to solidify in a crumb shape, dehydrated, dried in an oven at 60 ° C. for 12 hours, and master batches 9 to 23 shown in Table 4 ( MB9 to 23) were obtained.
- master batches 24 to 29 (MB24 to 29) using microfibrillated plant fibers 15, 2 and 16 were prepared and are shown in Table 4 together.
- Air permeability resistance> A rubber test piece having a diameter of 90 mm and a thickness of 1 mm made of the vulcanized rubber composition was prepared, and the air permeability coefficient (cc ⁇ cm / cm 2 ⁇ sec / cmHg) was determined in accordance with ASTM D-1434-75M. Calculated. Then, the air permeability coefficient of the reference comparative example was expressed as an index (air permeability resistance index) using the following formula as the reference (100). It shows that it is hard to permeate
- Air permeability resistance index) (Air permeability coefficient of reference comparative example) / (Each air permeability coefficient) ⁇ 100
- ⁇ Adhesion performance> The obtained vulcanized rubber composition was subjected to a tack test (adhesive strength measurement) using a tack test device manufactured according to JIS-T 9233.
- the measured value of the reference comparative example was set to 100, and the index was displayed by the following calculation formula. The larger the index, the better the adhesion performance.
- (Adhesive performance index) (Measured tack test value of each formulation) / (Measured tack test value of reference comparative example) ⁇ 100
- a test piece was prepared from a vulcanized rubber composition obtained according to JIS K6273, and the elongation remaining when it was freely contracted after being stretched to a specified elongation in an atmosphere at 25 ° C. was measured. Permanent strain was determined.
- the index was displayed using the reference comparative example as 100. The larger the index, the better the permanent set resistance.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
Abstract
Description
本発明のマスターバッチの製造方法は、ゴムラテックスと、N-オキシル化合物を用いて酸化処理されたミクロフィブリル化植物繊維とを混合し、得られた混合物のpHを2~6に調整して凝固させる工程(I)を含む。
本発明では、先ず、ゴムラテックスと、N-オキシル化合物を用いて酸化処理されたミクロフィブリル化植物繊維とを混合し、得られた混合物のpHを2~6に調整して凝固させる工程(I)が行われる。
なお、本発明において、上記総和をミクロフィブリル化植物繊維における荷電量として表す。絶乾とは、全重量中セルロース繊維が100%を占めるものをいう。
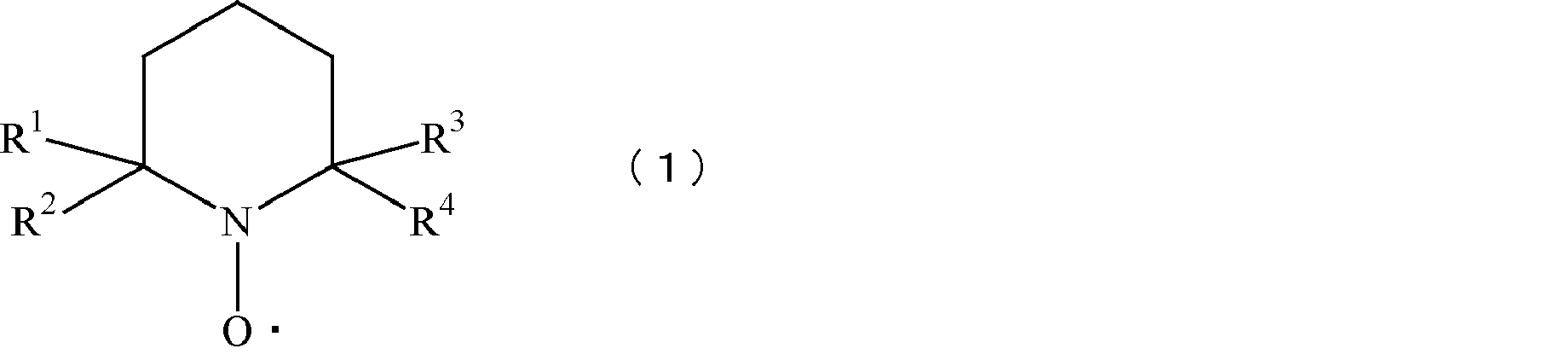





工程(I)で得られた凝固物(凝集ゴム及び前記ミクロフィブリル化植物繊維を含む凝集物)を、必要に応じて洗浄してもよい。これにより、例えば、ゴムラテックスとして天然ゴムラテックスを用いた場合、得られた凝固物に対して、更に洗浄処理を施してリン量や窒素量をより所望の量に調整できる。
なお、洗浄方法はこれらに限定されず、pHが6~7の範囲となるように炭酸ナトリウム等の弱アルカリ水で中和後、液相分を除去することで洗浄してもよい。
本発明の製造方法で調製されたマスターバッチは、該マスターバッチ中のゴム分(固形分)に含まれるリン含有量が、200ppm以下が好ましく、120ppm以下がより好ましい。ここで、リン含有量は、例えばICP発光分析等、従来の方法で測定することができる。リンは、リン脂質(リン化合物)に由来するものである。
本発明のタイヤ用ゴム組成物は、上記マスターバッチを含む。前記マスターバッチを用いることで、ゴム中に前記ミクロフィブリル化植物繊維が均一に分散したゴム組成物が得られる。その結果、混練工程でのゴム物性の低下防止、充填剤などの分散向上が実現し、優れた低燃費性、引張強度及び破断伸びを改善できる。
以下に、実施例で用いた各種薬品について説明する。
針葉樹漂白クラフトパルプ:日本製紙(株)岩国工場製
TEMPO:エボニック社製の2,2,6,6-テトラメチル-1-ピペリジン-N-オキシラジカル(上記式(1)中、R1~R4がメチル基で表される化合物)
臭化ナトリウム:マナック社製
次亜塩素酸ナトリウム:埼玉薬品(株)製のサイクロン
エマールE-27C(界面活性剤):花王(株)製のエマールE-27C(ポリオキシエチレンラウリルエーテル硫酸ナトリウム、有効成分27質量%)
NaOH:和光純薬工業(株)製のNaOH
Wingstay L(老化防止剤):ELIOKEM社製のWingstay L(ρ-クレゾールとジシクロペンタジエンとの縮合物をブチル化した化合物)
エマルビンW(界面活性剤):LANXESS社製のエマルビンW(芳香族ポリグリコールエーテル)
タモールNN9104(界面活性剤):BASF社製のタモールNN9104(ナフタレンスルホン酸/ホルムアルデヒドのナトリウム塩)
Van gel B(界面活性剤):Vanderbilt社製のVan gel B(マグネシウムアルミニウムシリケートの水和物)
フィールドラテックス:ムヒバラテックス社から入手したフィールドラテックス(天然ゴムラテックス)
ミクロフィブリル化植物繊維1、3~14:下記製造例で調製したミクロフィブリル化植物繊維(TEMPOを用いて酸化処理されたミクロフィブリル化植物繊維)
ミクロフィブリル化植物繊維2:(株)スギノマシン製のBiNFi-s Tシリーズ(カルボキシメチル化したセルロースナノファイバー、数平均繊維径:20~50nm、平均繊維長:500~1000nm)
ミクロフィブリル化植物繊維15:(株)スギノマシン製のBiNFi-s(超高圧ウォータージェット技術で加工したセルロース微細繊維、数平均繊維径:20~50nm、平均繊維長:500~1000nm)
ミクロフィブリル化植物繊維16:王子製袋(株)製のネオファイバー(数平均繊維径:0.1~1.2mm、平均繊維長:0.2~1.2mm)
凝固剤:和光純薬工業(株)製の1%硫酸
NR:TSR20
高純度化NR:下記製造例で調製
ブチル系ゴム:エクソンモービル社製のクロロブチルHT1066
BR:宇部興産(株)製のBR150B(シス含量:95質量%)
カーボンブラックN660:三菱化学(株)製のダイアブラックN660(N2SA:28m2/g、DBP吸収量:84ml/100g)
カーボンブラックN550:三菱化学(株)製のダイアブラックN550(N2SA:41m2/g、DBP吸収量:115ml/100g)
老化防止剤:大内新興化学工業(株)製のノクラック6C(N-フェニル-N’-(1,3-ジメチルブチル)-p-フェニレンジアミン)(6PPD)
樹脂:丸善石油化学(株)製のマルカレッツT-100AS(C5系樹脂)
オイル:出光興産(株)製のダイアナプロセスPA32
酸化亜鉛1:三井金属鉱業(株)製の酸化亜鉛2種
酸化亜鉛2:三井金属鉱業(株)製の亜鉛華1号
ステアリン酸:日油(株)製のビーズステアリン酸つばき
硫黄1:日本乾溜工業(株)製のセイミ硫黄(オイル分:10%)
硫黄2:軽井沢硫黄(株)製の粉末硫黄
加硫促進剤1:大内新興化学工業(株)製のノクセラーNS(N-tert-ブチル-2-ベンゾチアゾリルスルフェンアミド)
加硫促進剤2:大内新興化学工業(株)製のノクセラーMBTS(ジベンゾチアジルジスルフィド)
<マスターバッチ1~8の作製>
乾燥重量で5.00gの未乾燥の針葉樹漂白クラフトパルプ(主に1000nmを超える繊維径の繊維から成る)、39mgのTEMPO及び514mgの臭化ナトリウムを水500mlに分散させた後、15質量%次亜塩素酸ナトリウム水溶液を、1gのパルプ(絶乾)に対して次亜塩素酸ナトリウムの量が5.5mmolとなるように次亜塩素酸ナトリウムを加えて反応を開始した。反応中は3MのNaOH水溶液を滴下してpHを10.0に保った。pHに変化が見られなくなった時点で反応終了と見なし、反応物をガラスフィルターにてろ過した後、十分な量の水による水洗、ろ過を5回繰り返し、固形分量15質量%の水を含浸させた反応物繊維を得た。
次に、該反応物繊維に水を加え、固形分量1質量%スラリーとした。
酸化されたセルロース4g(絶乾)に1MのNaOH1.5ml、30%過酸化水素水0.5mlを添加し、超純水を加えて、5%(w/v)に調整した後、オートクレーブで80℃で2時間加熱した。
未洗浄のアルカリ加水分解処理後の酸化されたセルロースを超高圧ホモジナイザー(処理圧140MPa)で3回処理し、透明なゲル状分散液(ミクロフィブリル化植物繊維1)を得た。
なお、ミクロフィブリル化植物繊維に存在するカルボキシル基とアルデヒド基の量の総和及びカルボキシル基の量は、セルロース繊維の重量に対し、1.6mmol/g及び1.5mmol/gで、最大繊維径及び数平均繊維径は、8.2nm及び4.0nm、平均繊維長は470nmであった。
フィールドラテックスの固形分濃度(DRC)を30%(w/v)に調整した後、該ラテックス1000gに、10%エマールE-27C水溶液25gと25%NaOH水溶液60gを加え、室温で24時間ケン化反応を行い、ケン化天然ゴムラテックスを得た。次いで、下記老化防止剤分散体6gを添加し、2時間撹拌した後、更に水を添加してゴム濃度15%(w/v)となるまで希釈した。次いで、ゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~5mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出した。これに、水2000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、15分間放置した。更に、水を極力取り除き、再度水を添加して2分間撹拌する作業を3回繰返した後、水しぼりロールで水を絞ってシート状にした後、90℃で4時間乾燥して高純度化NRを得た。高純度化NR中に含まれるリン含有量は92ppm、窒素含有量は0.07質量%であった。
なお、上記老化防止剤分散体は、水 462.5gにエマルビンW 12.5g、タモールNN9104 12.5g、Van gel B 12.5g、Wingstay L 500g(合計1000g)をボールミルで16時間混合し、調製した。
ミクロフィブリル化植物繊維1の分散液を固形分濃度が0.5%(w/v)となるように水で希釈し、高速ホモジナイザー(回転数8000rpm、IKAジャパン製のT25)で約10分処理して均一分散液を作製した(粘度は7-8mPas)。
フィールドラテックス(固形分濃度(DRC)を30%(w/v))100質量部に対して、上記ミクロフィブリル化植物繊維1の分散液をミクロフィブリル化植物繊維の乾燥重量(固形分)が20質量部となるように添加し、ホモジナイザー(回転数8000rpm)で約5分撹拌してゴムラテックス分散液を作製した。次いで、ゆっくり撹拌(IKAジャパン製のEurostar)しながら凝固剤を添加してpH((株)堀場製作所製のpHメーターD51T)を3~4に調整してクラム状に凝固させ、脱水し、40℃12時間乾燥し、マスターバッチ1(MB1)を得た。
上記ミクロフィブリル化植物繊維1の分散液をミクロフィブリル化植物繊維の乾燥重量(固形分)が10質量部となるように添加した以外は、実施例1と同様の方法でマスターバッチ2(MB2)を得た。
上記ミクロフィブリル化植物繊維2の分散液を用いた以外は、実施例1と同様の方法でマスターバッチ3(MB3)を得た。
上記ミクロフィブリル化植物繊維2の分散液をミクロフィブリル化植物繊維の乾燥重量(固形分)が10質量部となるように添加した以外は、比較例1と同様の方法でマスターバッチ4(MB4)を得た。
ミクロフィブリル化植物繊維1の分散液を固形分濃度が0.5%(w/v)となるように水で希釈し、高速ホモジナイザー(回転数8000rpm)で約10分処理して均一分散液を作製した(粘度は7-8mPas)。
フィールドラテックスの固形分濃度(DRC)を30%(w/v)に調整した後、該ラテックス1000gに、10%エマールE-27C水溶液25gと25%NaOH水溶液60gを加え、室温で24時間ケン化反応を行い、ケン化天然ゴムラテックスを得た。得られたケン化天然ゴムラテックス(固形分濃度(DRC)を30%(w/v))100質量部に対して、上記ミクロフィブリル化植物繊維1の分散液をミクロフィブリル化植物繊維の乾燥重量(固形分)が20質量部となるように添加し、ホモジナイザー(回転数8000rpm)で約5分撹拌してゴムラテックス分散液を作製した。次いで、ゆっくり撹拌しながら凝固剤を添加してpHを3~4に調整してクラム状に凝固させ、脱水し、40℃12時間乾燥し、マスターバッチ5(MB5)を得た。
上記ミクロフィブリル化植物繊維1の分散液をミクロフィブリル化植物繊維の乾燥重量(固形分)が10質量部となるように添加した以外は、実施例3と同様の方法でマスターバッチ6(MB6)を得た。
上記ミクロフィブリル化植物繊維2の分散液を用いた以外は、実施例3と同様の方法でマスターバッチ7(MB7)を得た。
上記ミクロフィブリル化植物繊維2の分散液をミクロフィブリル化植物繊維の乾燥重量(固形分)が10質量部となるように添加した以外は、比較例3と同様の方法でマスターバッチ8(MB8)を得た。
B型粘度(60rpm、20℃)をVISCOMETER TV-10粘度計(東機産業(株)製)を用いて測定した。
窒素含有量は、熱分解後ガスクロマトグラフで定量した。
ICP発光分析装置(P-4010、(株)日立製作所製)を使用してリン含有量を求めた。
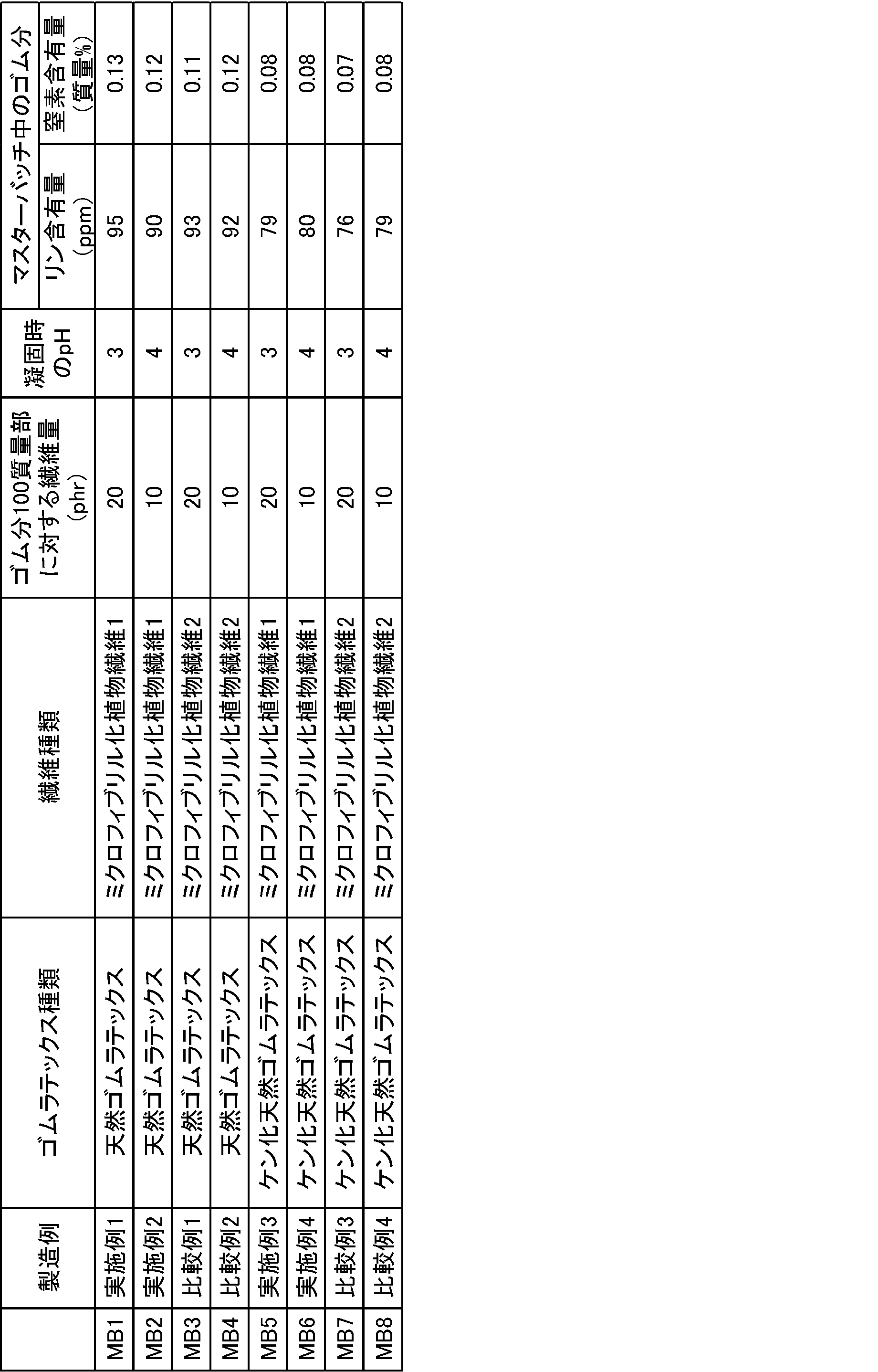
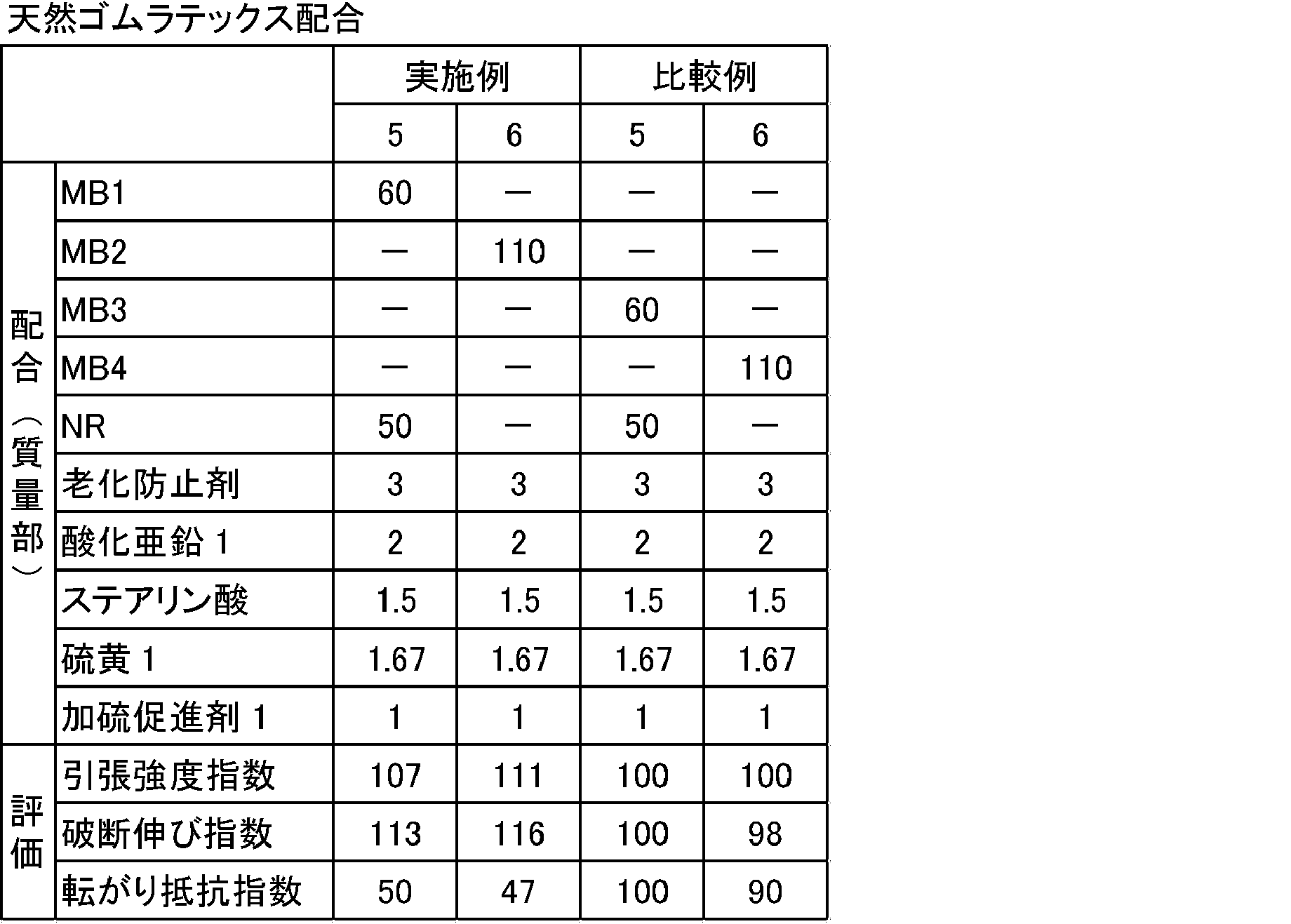
表2、3に示す配合処方にしたがい、(株)神戸製鋼製1.7Lバンバリーミキサーを用いて、硫黄及び加硫促進剤以外の薬品を混練りした。次に、オープンロールを用いて、得られた混練り物に硫黄及び加硫促進剤を添加して練り込み、未加硫ゴム組成物を得た。次に、得られた未加硫ゴム組成物を、150℃で30分間、2mm厚の金型でプレスし、加硫ゴム組成物を得た。得られた未加硫ゴム組成物及び加硫ゴム組成物を下記により評価した。結果を表2、3に示す。なお、基準比較例は比較例5及び7とした。
粘弾性スペクトロメーターVES((株)岩本製作所製)を用いて、温度70℃、初期歪み10%、動歪み1%、周波数10Hzの条件下で、各配合(加硫物)の損失正接(tanδ)を測定し、基準比較例の転がり抵抗指数を100として、下記計算式により算出した。転がり抵抗指数が小さいほど、転がり抵抗が低減され、好ましいことを示す。
(転がり抵抗指数)=(各配合のtanδ)/(基準比較例のtanδ)×100
JIS K6251「加硫ゴム及び熱可塑性ゴム-引張特性の求め方」に従い、引張強度及び破断伸びを測定した。下記の計算式、
引張強度指数=(各配合の破断応力)/(基準比較例の破断応力)×100
破断伸び指数=(各配合の破断伸び)/(基準比較例の破断伸び)×100
により引張強度指数、破断伸び指数を算出した。指数が大きい程、加硫ゴム組成物が良好に補強されており、ゴムの機械強度が大きく、ゴム物性に優れることを示す。

上述のミクロフィブリル化植物繊維1の製造方法をもとに、酸化剤の添加量を適宜調整し、酸化されたセルロースに対して添加するNaOHの濃度や添加量、オートクレーブによる反応時間を適宜調整することにより、表4に示す各数平均繊維径、平均繊維長、セルロース繊維の重量に対するミクロフィブリル化植物繊維に存在するカルボキシル基とアルデヒド基の量の総和量となるミクロフィブリル化植物繊維3~16を調製した。
更に、得られた各ミクロフィブリル化植物繊維の分散液を固形分濃度が1%(w/v)となるように水で希釈し、高速ホモジナイザー(回転数8000rpm)で約10分処理して均一分散液を作製した。
フィールドラテックス(固形分濃度(DRC)を30%(w/v))100質量部に対して、得られた各ミクロフィブリル化植物繊維の分散液をミクロフィブリル化植物繊維の乾燥重量(固形分)が20又は10質量部となるように添加し、ホモジナイザー(回転数15000rpm)で約5分撹拌してゴムラテックス分散液を作製した。次いで、ゆっくり撹拌しながら凝固剤を添加してpHを3~4に調整してクラム状に凝固させ、脱水し、オーブンにて60℃12時間乾燥し、表4に示すマスターバッチ9~23(MB9~23)を得た。
なお、比較例として、ミクロフィブリル化植物繊維15、2、16を用いたマスターバッチ24~29(MB24~29)を作製し、併せて表4に示す。
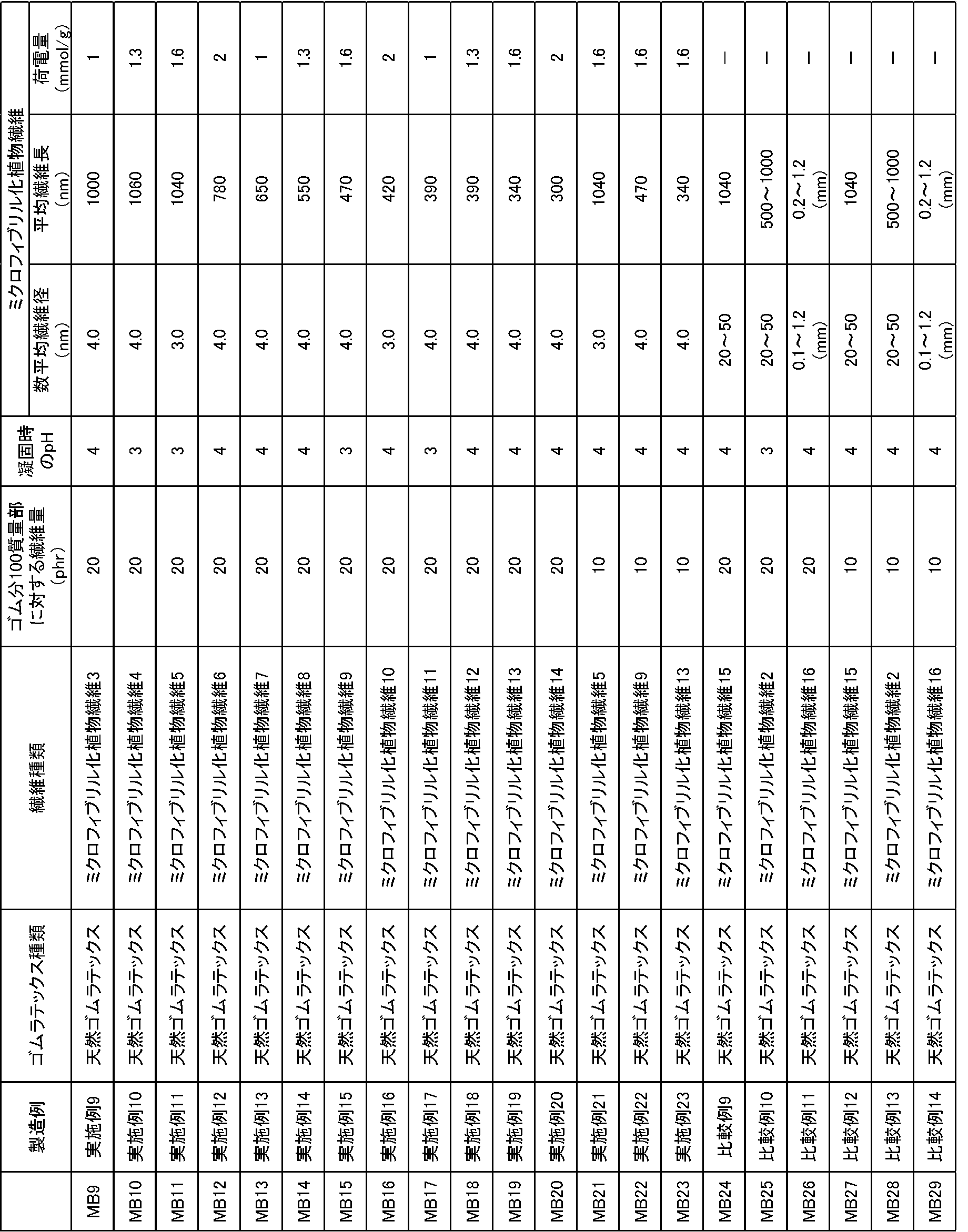
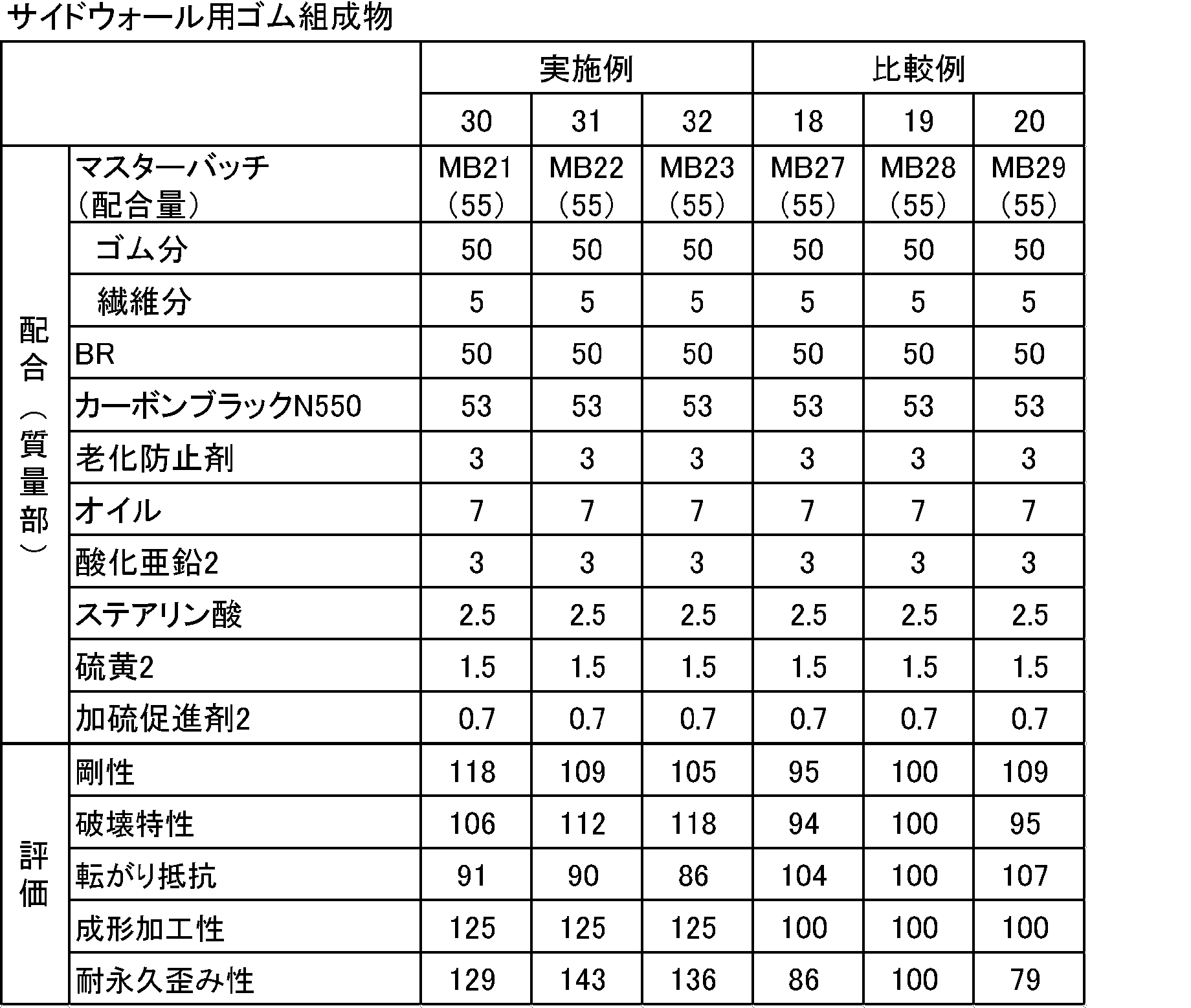
表5、6に示す配合処方にしたがい、(株)神戸製鋼製1.7Lバンバリーミキサーを用いて、硫黄及び加硫促進剤以外の薬品を混練りした。次に、オープンロールを用いて、得られた混練り物に硫黄及び加硫促進剤を添加して練り込み、未加硫ゴム組成物を得た。次に、得られた未加硫ゴム組成物を、150℃で30分間、2mm厚の金型でプレスし、加硫ゴム組成物を得た。得られた未加硫ゴム組成物及び加硫ゴム組成物を下記により評価した。結果を表5、6に示す。なお、基準比較例は比較例16、19とした。
得られた加硫ゴム組成物からなるゴム試験片直径90mmおよび厚さ1mm)を作製し、ASTM D-1434-75Mにしたがって、空気透過係数(cc・cm/cm2・sec/cmHg)をそれぞれ算出した。そして、下記計算式により基準比較例の空気透過係数を基準(100)としてそれぞれ指数表示した(耐空気透過性指数)。指数が大きいほど、空気を透過しにくく、耐空気透過性に優れることを示す。
(耐空気透過性指数)=(基準比較例の空気透過係数)/(各空気透過係数)×100
JIS K6251:2010に基づいて、得られた加硫ゴム組成物からダンベル状6号形試験片を作製し、該試験片を用いて25℃雰囲気下において、引張試験を実施して、破断強度TB(MPa)、破断時伸びEB(%)を測定した。そして、破壊エネルギーとして、TB×EB(MPa・%)/2を算出した。各配合の破壊エネルギーを算出し、基準比較例の破壊エネルギーを100として指数表示した。指数が大きいほど、破壊特性に優れることを示す。
粘弾性スペクトロメーターVES((株)岩本製作所製)を用いて、温度70℃、初期歪み10%、動歪み1%、周波数10Hzの条件下で、各配合(加硫物)の損失正接(tanδ)を測定し、基準比較例の転がり抵抗指数を100として、下記計算式により算出した。転がり抵抗指数が小さいほど、転がり抵抗が低減され、好ましいことを示す。
(転がり抵抗指数)=(各配合のtanδ)/(基準比較例のtanδ)×100
JIS K6300に準じて、130℃における未加硫ゴム組成物のムーニー粘度ML(1+4)を測定し、シート作製時の焼け性及び平坦性をシート加工性として目視観察した。これらの加工性を総合的に評価し、基準比較例を100として指数表示した。指数が100より小さいと、成形時のゴムの厚みが不安定となり、成形加工性に劣ることを示す。
得られた加硫ゴム組成物について、JIS-T 9233にもとづいて製作されたタックテスト装置を用いて、タックテスト(粘着力測定)を行った。基準比較例の測定値を100として、下記計算式により指数表示した。指数が大きいほど接着性能に優れる。
(接着性能指数)=(各配合のタックテスト測定値)/(基準比較例のタックテスト測定値)×100
(株)岩本製作所製の粘弾性スペクトロメーターVESを用いて、温度70℃、周波数10Hz、初期伸縮歪10%及び動歪2%の条件下で、加硫ゴム組成物の複素弾性率E*(MPa)を測定した。基準比較例のE*を100として、下記計算式により指数表示した。指数が大きいほど剛性が高いことを示す。
(剛性指数)=(各配合のE*)/(基準比較例のE*)×100
JIS K6273に準じて得られた加硫ゴム組成物から試験片を作製し、25℃雰囲気下において規定の伸びに伸長したあと自由に収縮させたときに残留する伸びを測定し、定伸長における引張永久ひずみを求めた。基準比較例を100として、指数表示した。指数が大きいほど耐永久歪み性が優れていることを示す。
Claims (5)
- ゴムラテックスと、N-オキシル化合物を用いて酸化処理されたミクロフィブリル化植物繊維とを混合し、得られた混合物のpHを2~6に調整して凝固させる工程(I)を含むマスターバッチの製造方法。
- 前記マスターバッチ中のゴム分100質量部に対する前記ミクロフィブリル化植物繊維の含有量が10~50質量部である請求項1記載のマスターバッチの製造方法。
- 請求項1又は2記載の製造方法により得られたマスターバッチ。
- 請求項3記載のマスターバッチを用いて作製したタイヤ用ゴム組成物。
- 請求項4記載のゴム組成物を用いて作製した空気入りタイヤ。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/547,215 US10428188B2 (en) | 2015-02-26 | 2016-02-09 | Method for producing master batch, master batch obtained by said production method, rubber composition for tire, and pneumatic tire |
| CN201680010009.8A CN107250222B (zh) | 2015-02-26 | 2016-02-09 | 母料的制造方法、用该制造方法制备的母料、轮胎用橡胶组合物和充气轮胎 |
| JP2016545368A JP6473161B2 (ja) | 2015-02-26 | 2016-02-09 | マスターバッチの製造方法、該製造方法により得られるマスターバッチ、タイヤ用ゴム組成物及び空気入りタイヤ |
| EP16755200.9A EP3255082B1 (en) | 2015-02-26 | 2016-02-09 | Method for producing master batch, master batch obtained by said production method, rubber composition for tire, and pneumatic tire |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-036883 | 2015-02-26 | ||
| JP2015036883 | 2015-02-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016136453A1 true WO2016136453A1 (ja) | 2016-09-01 |
Family
ID=56789295
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/053767 Ceased WO2016136453A1 (ja) | 2015-02-26 | 2016-02-09 | マスターバッチの製造方法、該製造方法により得られるマスターバッチ、タイヤ用ゴム組成物及び空気入りタイヤ |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10428188B2 (ja) |
| EP (1) | EP3255082B1 (ja) |
| JP (1) | JP6473161B2 (ja) |
| CN (1) | CN107250222B (ja) |
| WO (1) | WO2016136453A1 (ja) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018070387A1 (ja) * | 2016-10-13 | 2018-04-19 | 日本製紙株式会社 | ゴム組成物の製造方法 |
| WO2018101266A1 (ja) * | 2016-11-30 | 2018-06-07 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及びタイヤ |
| WO2018116661A1 (ja) * | 2016-12-21 | 2018-06-28 | 日本製紙株式会社 | 酸型カルボキシル化セルロースナノファイバー |
| JP2018119041A (ja) * | 2017-01-24 | 2018-08-02 | 住友ゴム工業株式会社 | マスターバッチの製造方法 |
| JP2018119072A (ja) * | 2017-01-26 | 2018-08-02 | 日本製紙株式会社 | マスターバッチ、ゴム組成物、及びそれらの製造方法 |
| CN108623856A (zh) * | 2017-03-16 | 2018-10-09 | 住友橡胶工业株式会社 | 充气轮胎 |
| JP2018162436A (ja) * | 2017-02-08 | 2018-10-18 | 日本製紙株式会社 | H型カルボキシル化セルロースナノファイバー |
| WO2018199191A1 (ja) * | 2017-04-27 | 2018-11-01 | 日本製紙株式会社 | マスターバッチ、ゴム組成物及びそれらの製造方法 |
| JP2018184515A (ja) * | 2017-04-25 | 2018-11-22 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物、タイヤ用ゴム組成物の製造方法及びタイヤ |
| JP2019172858A (ja) * | 2018-03-29 | 2019-10-10 | 日信工業株式会社 | ゴム組成物及びゴム組成物の製造方法 |
| JP2020007457A (ja) * | 2018-07-09 | 2020-01-16 | 日本製紙株式会社 | セルロースナノファイバーおよびセルロースナノファイバーを含有するゴム組成物の製造方法 |
| JP2020055962A (ja) * | 2018-10-03 | 2020-04-09 | Toyo Tire株式会社 | ゴムウエットマスターバッチの製造方法 |
| JP2020083913A (ja) * | 2018-11-15 | 2020-06-04 | 旭化成株式会社 | セルロースナノファイバー組成物 |
| JP2020147664A (ja) * | 2019-03-13 | 2020-09-17 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| KR20210081400A (ko) * | 2018-10-22 | 2021-07-01 | 비를라 카본 유.에스.에이., 인코포레이티드 | 엘라스토머 화합물들에서 나노셀룰로스 분산을 개선하기 위한 방법들, 및 엘라스토머 화합물들에 분산된 나노셀룰로오스를 함유하는 조성물들 |
| JP2022011982A (ja) * | 2020-06-30 | 2022-01-17 | 日本製紙株式会社 | 化学変性ミクロフィブリルセルロース繊維の製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180134079A1 (en) * | 2015-06-08 | 2018-05-17 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire and motorcycle tire |
| US11578142B2 (en) | 2016-12-21 | 2023-02-14 | Nippon Paper Industries Co., Ltd. | Acid type carboxylated cellulose nanofiber |
| JP6650476B2 (ja) * | 2018-02-27 | 2020-02-19 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| CN113677756A (zh) * | 2019-04-25 | 2021-11-19 | 日本制纸株式会社 | 胶乳浸渍液、橡胶组合物以及它们的制备方法 |
| EP4334364A1 (en) * | 2021-05-04 | 2024-03-13 | Nutrition & Biosciences USA 4, Inc. | Compositions comprising oxidized insoluble alpha-glucan |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009197122A (ja) * | 2008-02-21 | 2009-09-03 | Kao Corp | 樹脂組成物 |
| JP2013018918A (ja) * | 2011-07-13 | 2013-01-31 | Kao Corp | ゴム組成物及びその製造方法 |
| JP2013129767A (ja) * | 2011-12-22 | 2013-07-04 | Mitsubishi Chemicals Corp | ゴム組成物の製造方法、ゴム組成物、加硫ゴムおよびタイヤ |
| JP2013241586A (ja) * | 2012-04-25 | 2013-12-05 | Mitsubishi Chemicals Corp | ゴム改質材、ゴムラテックス分散液及びゴム組成物 |
| JP2014125607A (ja) * | 2012-12-27 | 2014-07-07 | Kao Corp | ゴム組成物 |
| JP2014139303A (ja) * | 2012-12-18 | 2014-07-31 | Mitsubishi Chemicals Corp | ゴム改質剤、ゴムラテックス分散液及びゴム組成物 |
| JP2014141637A (ja) * | 2012-12-25 | 2014-08-07 | Mitsubishi Chemicals Corp | セルロースナノファイバー含有ゴムマスターバッチ |
| JP2014141658A (ja) * | 2012-12-25 | 2014-08-07 | Mitsubishi Chemicals Corp | ゴム改質剤、ゴムラテックス分散液及びゴム組成物 |
| JP2015093882A (ja) * | 2013-11-08 | 2015-05-18 | 三菱化学株式会社 | 複合材の製造方法 |
| JP2015098576A (ja) * | 2013-10-17 | 2015-05-28 | 日信工業株式会社 | ゴム組成物の製造方法及びゴム組成物 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA74371B (en) * | 1973-02-15 | 1974-11-27 | Goodyear Tire & Rubber | Method of coagulating latices |
| JP3294901B2 (ja) * | 1993-05-20 | 2002-06-24 | 花王株式会社 | ゴム組成物 |
| JP3714773B2 (ja) | 1997-06-18 | 2005-11-09 | 住友ゴム工業株式会社 | 固形脱蛋白天然ゴムの製造方法 |
| JP2005036046A (ja) * | 2003-07-16 | 2005-02-10 | Sumitomo Rubber Ind Ltd | ゲル分の多い脱蛋白天然ゴムラテックスとその製造方法およびそれを用いた脱蛋白天然ゴム |
| JP5159190B2 (ja) * | 2007-06-29 | 2013-03-06 | 住友ゴム工業株式会社 | インナーライナー用ゴム組成物およびそれを用いたインナーライナーを有するタイヤ |
| JP4581116B2 (ja) | 2007-09-10 | 2010-11-17 | 住友ゴム工業株式会社 | 加硫ゴム組成物、空気入りタイヤおよびこれらの製造方法 |
| JP5524810B2 (ja) * | 2010-11-16 | 2014-06-18 | 住友ゴム工業株式会社 | 複合体、その製造方法、ゴム組成物及び空気入りタイヤ |
| JP2013043956A (ja) | 2011-08-25 | 2013-03-04 | Sumitomo Rubber Ind Ltd | ビードエーペックス用ゴム組成物及び空気入りタイヤ |
| JP5706863B2 (ja) * | 2012-01-16 | 2015-04-22 | 住友ゴム工業株式会社 | マスターバッチ、ゴム組成物及び空気入りタイヤ |
| JP2013177540A (ja) | 2012-02-08 | 2013-09-09 | Mitsubishi Chemicals Corp | ゴム改質材、ゴムラテックス分散液及びゴム組成物 |
| JP5966859B2 (ja) * | 2012-10-30 | 2016-08-10 | 王子ホールディングス株式会社 | 微細セルロース繊維分散液の製造方法 |
| JP6416749B2 (ja) * | 2013-03-14 | 2018-10-31 | 株式会社ブリヂストン | ゴム組成物の製造方法、ゴム組成物、加硫ゴムおよびタイヤ |
| JP6093639B2 (ja) * | 2013-04-30 | 2017-03-08 | 株式会社ブリヂストン | 接着剤組成物、繊維、ゴム部材、空気入りタイヤ及びランフラットタイヤ |
| JP6181426B2 (ja) * | 2013-05-23 | 2017-08-16 | 住友ゴム工業株式会社 | マスターバッチ、製造方法、ゴム組成物及び空気入りタイヤ |
| KR200478039Y1 (ko) * | 2015-01-20 | 2015-08-20 | 최인 | 등압착식 허리받침대 |
-
2016
- 2016-02-09 WO PCT/JP2016/053767 patent/WO2016136453A1/ja not_active Ceased
- 2016-02-09 CN CN201680010009.8A patent/CN107250222B/zh active Active
- 2016-02-09 EP EP16755200.9A patent/EP3255082B1/en active Active
- 2016-02-09 JP JP2016545368A patent/JP6473161B2/ja active Active
- 2016-02-09 US US15/547,215 patent/US10428188B2/en active Active
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009197122A (ja) * | 2008-02-21 | 2009-09-03 | Kao Corp | 樹脂組成物 |
| JP2013018918A (ja) * | 2011-07-13 | 2013-01-31 | Kao Corp | ゴム組成物及びその製造方法 |
| JP2013129767A (ja) * | 2011-12-22 | 2013-07-04 | Mitsubishi Chemicals Corp | ゴム組成物の製造方法、ゴム組成物、加硫ゴムおよびタイヤ |
| JP2013241586A (ja) * | 2012-04-25 | 2013-12-05 | Mitsubishi Chemicals Corp | ゴム改質材、ゴムラテックス分散液及びゴム組成物 |
| JP2014139303A (ja) * | 2012-12-18 | 2014-07-31 | Mitsubishi Chemicals Corp | ゴム改質剤、ゴムラテックス分散液及びゴム組成物 |
| JP2014141637A (ja) * | 2012-12-25 | 2014-08-07 | Mitsubishi Chemicals Corp | セルロースナノファイバー含有ゴムマスターバッチ |
| JP2014141658A (ja) * | 2012-12-25 | 2014-08-07 | Mitsubishi Chemicals Corp | ゴム改質剤、ゴムラテックス分散液及びゴム組成物 |
| JP2014125607A (ja) * | 2012-12-27 | 2014-07-07 | Kao Corp | ゴム組成物 |
| JP2015098576A (ja) * | 2013-10-17 | 2015-05-28 | 日信工業株式会社 | ゴム組成物の製造方法及びゴム組成物 |
| JP2015093882A (ja) * | 2013-11-08 | 2015-05-18 | 三菱化学株式会社 | 複合材の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3255082A4 * |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018070387A1 (ja) * | 2016-10-13 | 2018-04-19 | 日本製紙株式会社 | ゴム組成物の製造方法 |
| JP6990190B2 (ja) | 2016-10-13 | 2022-01-12 | 日本製紙株式会社 | ゴム組成物の製造方法 |
| JPWO2018070387A1 (ja) * | 2016-10-13 | 2019-08-08 | 日本製紙株式会社 | ゴム組成物の製造方法 |
| WO2018101266A1 (ja) * | 2016-11-30 | 2018-06-07 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及びタイヤ |
| JPWO2018101266A1 (ja) * | 2016-11-30 | 2019-10-24 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及びタイヤ |
| US11242448B2 (en) | 2016-11-30 | 2022-02-08 | Sumitomo Rubber Industries, Ltd. | Rubber composition for tire and tire |
| JP7070399B2 (ja) | 2016-11-30 | 2022-05-18 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及びタイヤ |
| WO2018116661A1 (ja) * | 2016-12-21 | 2018-06-28 | 日本製紙株式会社 | 酸型カルボキシル化セルロースナノファイバー |
| JP2018119041A (ja) * | 2017-01-24 | 2018-08-02 | 住友ゴム工業株式会社 | マスターバッチの製造方法 |
| WO2018139369A1 (ja) * | 2017-01-24 | 2018-08-02 | 住友ゴム工業株式会社 | マスターバッチの製造方法 |
| JP2018119072A (ja) * | 2017-01-26 | 2018-08-02 | 日本製紙株式会社 | マスターバッチ、ゴム組成物、及びそれらの製造方法 |
| JP7162422B2 (ja) | 2017-02-08 | 2022-10-28 | 日本製紙株式会社 | H型カルボキシル化セルロースナノファイバー |
| JP2018162436A (ja) * | 2017-02-08 | 2018-10-18 | 日本製紙株式会社 | H型カルボキシル化セルロースナノファイバー |
| CN108623856A (zh) * | 2017-03-16 | 2018-10-09 | 住友橡胶工业株式会社 | 充气轮胎 |
| US11014405B2 (en) | 2017-03-16 | 2021-05-25 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire |
| JP2018184515A (ja) * | 2017-04-25 | 2018-11-22 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物、タイヤ用ゴム組成物の製造方法及びタイヤ |
| US11225532B2 (en) | 2017-04-25 | 2022-01-18 | Sumitomo Rubber Industries, Ltd. | Rubber composition for tires, method for preparing rubber composition for tires, and tire |
| JP7069564B2 (ja) | 2017-04-25 | 2022-05-18 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物、タイヤ用ゴム組成物の製造方法及びタイヤ |
| WO2018199191A1 (ja) * | 2017-04-27 | 2018-11-01 | 日本製紙株式会社 | マスターバッチ、ゴム組成物及びそれらの製造方法 |
| JP6473550B1 (ja) * | 2017-04-27 | 2019-02-20 | 日本製紙株式会社 | マスターバッチ、ゴム組成物及びそれらの製造方法 |
| JP7044300B2 (ja) | 2018-03-29 | 2022-03-30 | 日立Astemo株式会社 | ゴム組成物及びゴム組成物の製造方法 |
| JP2019172858A (ja) * | 2018-03-29 | 2019-10-10 | 日信工業株式会社 | ゴム組成物及びゴム組成物の製造方法 |
| JP2020007457A (ja) * | 2018-07-09 | 2020-01-16 | 日本製紙株式会社 | セルロースナノファイバーおよびセルロースナノファイバーを含有するゴム組成物の製造方法 |
| JP2020055962A (ja) * | 2018-10-03 | 2020-04-09 | Toyo Tire株式会社 | ゴムウエットマスターバッチの製造方法 |
| JP7125883B2 (ja) | 2018-10-03 | 2022-08-25 | Toyo Tire株式会社 | ゴムウエットマスターバッチの製造方法 |
| JP2022508946A (ja) * | 2018-10-22 | 2022-01-19 | ビルラ カーボン ユー.エス.エー.,インコーポレイティド | エラストマー化合物中でのナノセルロース分散を改善するための方法、及びエラストマー化合物中に分散されたナノセルロースを含む組成物 |
| JP7739497B2 (ja) | 2018-10-22 | 2025-09-16 | ビルラ カーボン ユー.エス.エー.,インコーポレイティド | エラストマー化合物中でのナノセルロース分散を改善するための方法、及びエラストマー化合物中に分散されたナノセルロースを含む組成物 |
| JP2024045582A (ja) * | 2018-10-22 | 2024-04-02 | ビルラ カーボン ユー.エス.エー.,インコーポレイティド | エラストマー化合物中でのナノセルロース分散を改善するための方法、及びエラストマー化合物中に分散されたナノセルロースを含む組成物 |
| KR20210081400A (ko) * | 2018-10-22 | 2021-07-01 | 비를라 카본 유.에스.에이., 인코포레이티드 | 엘라스토머 화합물들에서 나노셀룰로스 분산을 개선하기 위한 방법들, 및 엘라스토머 화합물들에 분산된 나노셀룰로오스를 함유하는 조성물들 |
| KR102900229B1 (ko) * | 2018-10-22 | 2025-12-15 | 비를라 카본 유.에스.에이., 인코포레이티드 | 엘라스토머 화합물들에서 나노셀룰로스 분산을 개선하기 위한 방법들, 및 엘라스토머 화합물들에 분산된 나노셀룰로오스를 함유하는 조성물들 |
| US12378390B2 (en) | 2018-10-22 | 2025-08-05 | Birla Carbon U.S.A., Inc. | Methods for improving nanocellulose dispersion in elastomeric compounds, and compositions containing dispersed nanocellulose in elastomer compounds |
| JP7564813B2 (ja) | 2018-10-22 | 2024-10-09 | ビルラ カーボン ユー.エス.エー.,インコーポレイティド | エラストマー化合物中でのナノセルロース分散を改善するための方法、及びエラストマー化合物中に分散されたナノセルロースを含む組成物 |
| JP7266995B2 (ja) | 2018-11-15 | 2023-05-01 | 旭化成株式会社 | セルロースナノファイバー組成物 |
| JP2020083913A (ja) * | 2018-11-15 | 2020-06-04 | 旭化成株式会社 | セルロースナノファイバー組成物 |
| JP7261420B2 (ja) | 2019-03-13 | 2023-04-20 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| JP2020147664A (ja) * | 2019-03-13 | 2020-09-17 | 横浜ゴム株式会社 | タイヤ用ゴム組成物 |
| JP7574553B2 (ja) | 2020-06-30 | 2024-10-29 | 日本製紙株式会社 | 化学変性ミクロフィブリルセルロース繊維の製造方法 |
| JP2022011982A (ja) * | 2020-06-30 | 2022-01-17 | 日本製紙株式会社 | 化学変性ミクロフィブリルセルロース繊維の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107250222B (zh) | 2020-12-18 |
| JPWO2016136453A1 (ja) | 2017-11-30 |
| JP6473161B2 (ja) | 2019-02-20 |
| EP3255082B1 (en) | 2020-07-08 |
| EP3255082A1 (en) | 2017-12-13 |
| US20180016402A1 (en) | 2018-01-18 |
| EP3255082A4 (en) | 2018-10-24 |
| CN107250222A (zh) | 2017-10-13 |
| US10428188B2 (en) | 2019-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6473161B2 (ja) | マスターバッチの製造方法、該製造方法により得られるマスターバッチ、タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP5706863B2 (ja) | マスターバッチ、ゴム組成物及び空気入りタイヤ | |
| JP5763995B2 (ja) | ゴム組成物及びその製造方法 | |
| JP6082591B2 (ja) | ゴム組成物 | |
| KR101898303B1 (ko) | 개선된 탄성 중합체 배합물 | |
| JP5691463B2 (ja) | タイヤ用ゴム組成物 | |
| CN114341250B (zh) | 表面处理纳米纤维素母炼胶 | |
| JP6353169B2 (ja) | ゴム組成物及び空気入りタイヤ | |
| JP2016147996A (ja) | ミクロフィブリル化植物繊維・ゴム複合体及びその製造方法、並びに、ゴム組成物及び空気入りタイヤ | |
| US10414882B2 (en) | Method for producing masterbatch | |
| JP6386598B2 (ja) | マスターバッチの製造方法 | |
| JP5933302B2 (ja) | タイヤ用ゴム組成物、その製造方法及び空気入りタイヤ | |
| JP2020066699A (ja) | タイヤ用ゴム組成物 | |
| JP6181426B2 (ja) | マスターバッチ、製造方法、ゴム組成物及び空気入りタイヤ | |
| JP6880657B2 (ja) | マスターバッチの製造方法 | |
| JP2015010136A (ja) | タイヤ用ゴム組成物およびそれを用いた空気入りタイヤ | |
| JP2024089229A (ja) | エラストマー組成物及びタイヤ | |
| JP2025138328A (ja) | セルロースナノファイバー添加ウェットマスターバッチの製造方法及びゴム組成物の製造方法 | |
| JP7283667B2 (ja) | ナノセルロースマスターバッチ |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016545368 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16755200 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15547215 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2016755200 Country of ref document: EP |