WO2016171036A1 - ガス発生剤、及びそれを用いた発泡体の製造方法 - Google Patents
ガス発生剤、及びそれを用いた発泡体の製造方法 Download PDFInfo
- Publication number
- WO2016171036A1 WO2016171036A1 PCT/JP2016/061802 JP2016061802W WO2016171036A1 WO 2016171036 A1 WO2016171036 A1 WO 2016171036A1 JP 2016061802 W JP2016061802 W JP 2016061802W WO 2016171036 A1 WO2016171036 A1 WO 2016171036A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- generating agent
- gas generating
- aminoguanidine
- gas
- mass
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/06—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent
- C08J9/10—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent developing nitrogen, the blowing agent being a compound containing a nitrogen-to-nitrogen bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C281/00—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group
- C07C281/16—Compounds containing any of the groups, e.g. aminoguanidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C55/00—Saturated compounds having more than one carboxyl group bound to acyclic carbon atoms
- C07C55/02—Dicarboxylic acids
- C07C55/06—Oxalic acid
- C07C55/07—Salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/06—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent
- C08J9/10—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a chemical blowing agent developing nitrogen, the blowing agent being a compound containing a nitrogen-to-nitrogen bond
- C08J9/104—Hydrazines; Hydrazides; Semicarbazides; Semicarbazones; Hydrazones; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/29—Compounds containing one or more carbon-to-nitrogen double bonds
- C08K5/31—Guanidine; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K9/00—Use of pretreated ingredients
- C08K9/10—Encapsulated ingredients
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K3/00—Materials not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2201/00—Foams characterised by the foaming process
- C08J2201/02—Foams characterised by the foaming process characterised by mechanical pre- or post-treatments
- C08J2201/026—Crosslinking before of after foaming
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/22—Thermoplastic resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/26—Elastomers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/04—Homopolymers or copolymers of ethene
- C08J2323/06—Polyethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/16—Ethene-propene or ethene-propene-diene copolymers
Definitions
- the present invention relates to a gas generating agent and a method for producing a foamed body such as a thermoplastic resin or rubber using the same.
- ADCA Azodicarbonamide
- the advantages of ADCA are that the amount of generated gas is 200 mL / g or more compared to other commercially available chemical foaming agents, and the combination with foaming aids that have a relatively high decomposition start temperature of 200 to 210 ° C.
- the decomposition start temperature can be lowered to around 140 ° C, it can be applied to general-purpose plastics (for example, thermoplastic resins) and rubber, and the generated gas is mainly composed of nitrogen and self-extinguishing, so it is safe to handle. It is expensive.
- ADCA contains a small amount of ammonia gas in the gas generated at the time of foaming.
- This ammonia gas shows corrosiveness
- cyanic acid which is a decomposition product of ADCA, has sublimation properties and polymerizes after sublimation.
- corrosive cyanuric acid which tends to cause mold contamination (see, for example, Patent Document 1 and Non-Patent Document 1).
- OBSH 4,4′-oxybis (benzenesulfonylhydrazide)
- OBSH has a low decomposition initiation temperature of about 170 ° C., and is mainly used for ethylene-propylene-diene rubber (EPDM) for weather strips and chloroprene rubber (CR) for wet suits.
- EPDM ethylene-propylene-diene rubber
- CR chloroprene rubber
- OBSH has a small amount of generated gas of 120 mL / g, and it is necessary to increase the amount of addition in order to obtain a desired expansion ratio, which has problems in terms of economy and resource saving.
- guanidine derivatives such as guanidine salts, aminoguanidine salts, diaminoguanidine salts, and triaminoguanidine salts can also be used as gas generating agents.
- guanidine derivatives are also used for explosives, pharmaceuticals, antioxidants for fiber processing, soap resin stabilizers, and other various synthetic raw materials (see, for example, Non-Patent Document 2).
- gas generating agents containing carbonates, nitrates, and perchlorates of guanidine derivatives are disclosed for airbag systems (see, for example, Patent Documents 2 to 4).
- nitrates of the above guanidine derivatives are simple and versatile compounds that are synthesized, but alone generate less gas.
- the carbonate of the guanidine derivative has a decomposition start temperature of less than 100 ° C., which is greatly different from the molding temperatures of general-purpose plastics (for example, thermoplastic resins) and rubber. Therefore, the conventional nitrates and carbonates of the guanidine derivatives are difficult to use as gas generating agents for general-purpose plastics (for example, thermoplastic resins) and rubber chemical foaming agents such as ADCA and OBSH.
- an object of the present invention is to use a guanidine derivative that is easy to synthesize and has a decomposition start temperature that can be used as a chemical foaming agent for general-purpose plastics (for example, thermoplastic resins) and rubbers.
- the object is to provide a gas generating agent that has a large amount of gas and contains almost no corrosive gas such as ammonia in the generated gas.
- the present invention is as follows.
- X and Y are each independently a hydrogen atom or an amino group.
- Agent. [3] The gas generating agent according to [1] or [2], wherein the aminoguanidine compound is aminoguanidine.
- a foaming composition comprising the gas generating agent according to any one of [1] to [4] and a foamed material.
- the manufacturing method of a foam including the process of heating the foaming composition as described in [5] or [6].
- a decomposition starting temperature that can be used as a chemical foaming agent for, for example, general-purpose plastics (for example, thermoplastic resins) and rubber, using guanidine derivatives and oxalic acid, which are simple and versatile chemical raw materials. It is possible to provide a gas generating agent having a large amount of generated gas and containing almost no corrosive gas such as ammonia in the generated gas.
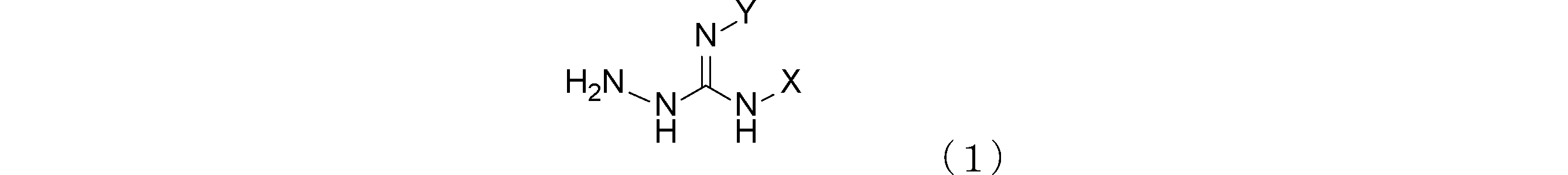
- the gas generating agent of this embodiment contains an oxalate salt of an aminoguanidine compound represented by the following general formula (1) (hereinafter also simply referred to as “aminoguanidine compound”).
- aminoguanidine compound represented by the following general formula (1)
- X and Y are each independently a hydrogen atom or an amino group.
- oxalic acid is used as the acid that forms a salt with the aminoguanidine compound.
- oxalates of aminoguanidine compounds such as aminoguanidine, it has a decomposition start temperature that can be used as a chemical foaming agent for general-purpose plastics (for example, thermoplastic resins) and rubbers. It is possible to obtain a gas generating agent that hardly contains corrosive gas such as ammonia.
- the amount of gas generated when the gas generating agent of the present embodiment is foamed is preferably 120 mL / g or more.
- the amount of ammonia generated when the gas generating agent of the present embodiment is foamed is preferably 10 mg / g or less, more preferably 5 mg / g or less, and even more preferably 1 mg / g.
- the decomposition start temperature of the gas generating agent of the present embodiment is preferably a decomposition start temperature that can be used as, for example, a chemical foaming agent for general-purpose plastics (for example, thermoplastic resins) and rubbers.
- the temperature is preferably 380 ° C., more preferably 110 to 250 ° C.
- the amount of gas generated when the gas generating agent is foamed, the amount of ammonia generated, and the decomposition start temperature can be measured by the methods described in the examples below.
- the oxalate salt of an aminoguanidine compound used in the present embodiment can be obtained by reacting an aminoguanidine compound and oxalic acid.
- the aminoguanidine compound is not particularly limited, and examples thereof include aminoguanidine, diaminoguanidine, triaminoguanidine and the like. Of these, aminoguanidine is preferable.
- Oxalic acid used in the above reaction is a known compound and can be obtained as a commercial product, and either oxalic anhydride or oxalic acid dihydrate may be used.
- an aminoguanidine compound that does not form a salt can also be used.
- an aminoguanidine compound forms a salt with an acid, stability is improved and it is easy to obtain.
- a salt of an aminoguanidine compound is not particularly limited.
- these acids it is preferable to use as a raw material an aminoguanidine compound that forms a salt with an acid having a lower acidity than oxalic acid.
- Examples of such salts of aminoguanidine compounds include aminoguanidine carbonate.
- reaction conditions for obtaining the oxalate of the aminoguanidine compound used for the gas generating agent of this embodiment from the carbonate of the aminoguanidine compound and oxalic acid will be described.
- 1 mol to excess of oxalic acid is used with respect to 1 mol of carbonate of aminoguanidine compound, and the mixture is stirred in a polar solvent such as water or alcohol at 0 to 100 ° C. for 10 minutes to 24 hours. It progresses by making it react.
- the target product is purified by a known method. For example, a method of obtaining a crude crystal by precipitating and isolating crystals by cooling with ice water or the like can be mentioned.
- the ratio of the aminoguanidine compound carbonate and oxalic acid used is preferably 1: 1 to 1: 100, more preferably 1: 1 to 1:10 in terms of molar ratio (carbonate: oxalic acid).
- the reaction may be performed at room temperature, or may be performed under heating as necessary.
- a polar solvent such as water or alcohol, it is preferably 0 to 100 ° C., more preferably 20 to 80 ° C. Do.
- the polar solvent is not particularly limited, and examples thereof include water, methanol, ethanol, propanol, isopropyl alcohol, butanol, and isobutyl alcohol. Water that is easy to purify the product and excellent in economic efficiency is preferable.
- the molar ratio of aminoguanidine compound to oxalic acid is preferably in the range of 1: 1 to 2: 1.
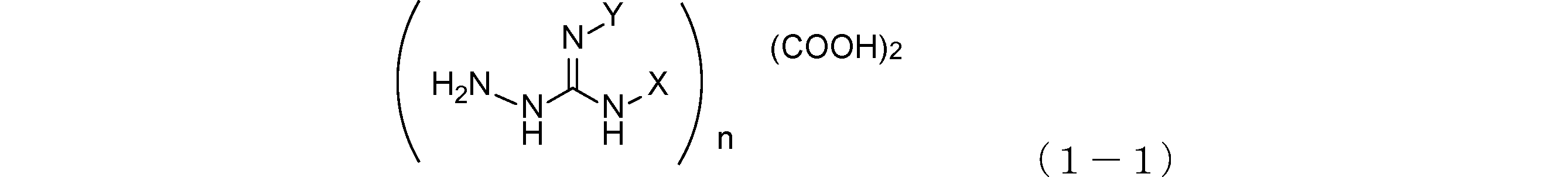
- the range is more preferably in the range of 1: 1 to 1.5: 1, and still more preferably in the range of 1: 1 to 1.2: 1. Therefore, the oxalate salt of the aminoguanidine compound used in this embodiment can also be represented by the following formula (1-1), for example.
- X and Y are each independently a hydrogen atom or an amino group, and n is a number in the range of 1.0 to 2.0.
- the oxalate of the aminoguanidine compound obtained as described above can be used as a gas generating agent.
- the gas generating agent of this embodiment can be suitably used as a chemical foaming agent for general-purpose plastics (for example, thermoplastic resins) and rubber, for example.
- the oxalate of the aminoguanidine compound used for the gas generating agent of the present embodiment is a corrosive gas such as ammonia at a foam molding temperature of general-purpose plastics (for example, thermoplastic resin) and rubber (for example, about 130 to 250 ° C.).
- the foam of high magnification can be obtained independently.
- the gas generating agent of the present embodiment can be suitably used as a chemical foaming agent for general-purpose plastics (for example, thermoplastic resins) and rubber, even when only the oxalate of an aminoguanidine compound is used.
- An auxiliary agent may be further contained in addition to the oxalate salt of the compound.
- the said adjuvant is not specifically limited, For example, an oxidizing agent, a crosslinking agent (C), nitrite, a hydrotalcite, etc. are mentioned.
- the gas generating agent of the present embodiment further contains such an auxiliary agent, generation of corrosive gas such as ammonia can be further suppressed, and the temperature at the time of producing the foam described later is also in a suitable range. It tends to be.
- the content of the oxalate salt of the aminoguanidine compound is the sum of the oxalate salt of the aminoguanidine compound and the auxiliary agent. It is preferably 0.5 to 95% by mass relative to the mass.
- said oxidizing agent For example, sodium percarbonate is mentioned.
- the said oxidizing agent may be used individually by 1 type, and may use 2 or more types together.
- the content of the oxidizing agent is preferably 0.5 to 95% by mass with respect to the total mass of the oxalate salt of the aminoguanidine compound and the auxiliary agent, and 5 to 50% by mass. % Is more preferable.
- the crosslinking agent (C) is not particularly limited, and examples thereof include dicumyl peroxide, di-t-butyl peroxide, 2,5-dimethyl-2,5-di- (t-butylperoxy) hexane, 2,5-dimethyl-2,5-di- (t-butylperoxy) hexyne, 1,3-bis (t-butylperoxyisopropyl) benzene, 1,1-bis (t-butylperoxy) -3 , 3,5-trimethylcyclohexane, 1,1-bis [t-butylperoxy] cyclohexane, n-butyl-4,4-bis (t-butylperoxy) valerate, benzoyl peroxide, p-chlorobenzoyl peroxide 2,4-dichlorobenzoyl peroxide, t-butyl peroxybenzoate, t-butyl perbenzoate, t-butyl
- the said crosslinking agent (C) may be used individually by 1 type, and may use 2 or more types together.
- the content of the crosslinking agent (C) is preferably 0.1 to 10% by mass with respect to the total mass of the oxalate salt of the aminoguanidine compound and the auxiliary agent. More preferably, the content is 5 to 1.5% by mass.
- the nitrite is not particularly limited, and examples thereof include sodium nitrite, potassium nitrite, calcium nitrite and the like. One selected from these can be used alone or in combination of two or more. . These nitrites are preferably finely divided powder.
- the content of nitrite is preferably 0.5 to 60% by mass, and preferably 15 to 55% by mass with respect to the total mass of the oxalate salt of the aminoguanidine compound and the auxiliary agent. % Is more preferable.
- the hydrotalcite is a crystalline composite metal hydroxide, and a hydrotalcite represented by the following general formula (H) is preferable.
- M 2+ is a divalent metal ion of a metal selected from the group consisting of Mg, Mn, Fe, and Zn
- M 3+ is a metal selected from the group consisting of Al, Fe, and Cr.
- Trivalent metal ion is an n-valent anion of a group selected from the group consisting of OH, F, Cl, Br, NO 3 , CO 3 and SO 4 , x is in the range of 0 ⁇ x ⁇ 0.33
- n is an integer and m is 0 or greater.
- m is preferably 0, but it varies depending on the dry state and storage state of the hydrotalcite, and m is not particularly limited as long as the effects of the present invention are not impaired.
- n is the valence of the anion, preferably 1 or 2, and more preferably 2.
- hydrotalcite in which M 2+ is Mg 2+ and M 3+ is Al 3+ is preferable, and the molar ratio of Al: Mg is preferably 2: 5 to 2:10 from the viewpoint of availability.
- the molar ratio of Al: Mg is 2: 5
- the molar ratio of Al to Mg is 2:10.
- the molar fraction x of Al is 0.17.
- the hydrotalcite has the effect of improving the reactivity of nitrite.
- hydrotalcite By blending hydrotalcite, it promotes the decomposition of ammonia gas that can be generated when the gas generant is heated, improves the reactivity of nitrite, suppresses the generation of nitrite gas, and generates nitrogen gas.
- the foaming property of the gas generating agent can be improved.
- the particle size of the hydrotalcite is not particularly limited, but in order to effectively act on the reaction between the oxalate and nitrite of the aminoguanidine compound, the dispersibility in the gas generating agent is increased. And finely divided hydrotalcite having a maximum particle size of 80 ⁇ m or less is more preferable.
- the content of hydrotalcite is preferably 1 to 40% by mass with respect to the total mass of the oxalate salt of the aminoguanidine compound and the auxiliary agent, and 5 to 25% by mass. It is more preferable that
- the stabilizer is not particularly limited.
- the pigment / filler is not particularly limited, and examples thereof include chrome yellow, carbon black, titanium dioxide, and calcium carbonate.
- a foam regulator For example, a maleic acid etc. are mentioned.
- the manufacturing method of the gas generating agent of this embodiment is not specifically limited, A general mixing method can be used.
- oxalate, nitrite and hydrotalcite of aminoguanidine compound are mixed using a high-speed mixer, ribbon blender, cone blender, etc., so that they are uniformly dispersed under conditions of a temperature of 60 ° C. or less and a time of about 5 minutes. do it.
- the gas generating agent of this embodiment may be a microcapsule type gas generating agent.
- the microcapsule type gas generating agent refers to a gas generating agent having a core-shell structure.
- a microcapsule-type gas generating agent for example, an aminoguanidine compound oxalate or a composition containing the above-mentioned auxiliary agent in addition to the aminoguanidine compound oxalate is used as a core component.
- a microcapsule type gas generating agent may be mentioned.
- the main component of the shell of the microcapsule type gas generating agent is not particularly limited, but is preferably, for example, polymethyl methacrylate, and includes a fatty acid salt or a surfactant described later as a dispersant.
- the polymethyl methacrylate is preferably a homopolymer having a methyl methacrylate monomer as a structural unit, but other monomers may be copolymerized within a range not impairing the effects of the present invention.
- Examples of other monomers include, but are not limited to, acrylic esters such as methyl acrylate, ethyl acrylate, butyl acrylate, and dicyclopentenyl acrylate, ethyl methacrylate, butyl methacrylate, and isobornyl methacrylate.
- examples include methacrylic acid esters, acrylonitrile, methacrylonitrile, vinylidene chloride, vinyl chloride, styrene, vinyl acetate, ⁇ -methylstyrene, chloroprene, neoprene, and butadiene.
- the cross-linking agent (D) used for producing the microcapsule-type gas generating agent is not particularly limited.
- ethylene glycol dimethacrylate, (poly) ethylene glycol, and trimethylolpropane tri (meth) acrylate are preferable.
- the addition ratio of the crosslinking agent (D) is preferably 1 to 5% by mass with respect to the mass of the microcapsule shell monomer (the total mass of methyl methacrylate monomer and other monomers).
- the ratio of the crosslinking agent (D) is less than 1% by mass, the capsule shell is not sufficiently crosslinked, and the decomposition residue generated when the core component is decomposed cannot be adsorbed efficiently. Therefore, the decomposition residue easily passes through the capsule shell. End up.
- the ratio of the crosslinking agent (D) exceeds 5% by mass, the capsule shell becomes hard and brittle, the capsule shell is broken when the core component is decomposed, and the effect of adsorbing the decomposition residue is lowered.
- the polymerization initiator used for producing the microcapsule type gas generating agent is not particularly limited, and those generally used in this field can be used.
- Examples of the polymerization initiator that can be used include dialkyl peroxides, diacyl peroxides, peroxyesters, peroxydicarbonates, and azo compounds.
- dialkyl peroxides such as methyl ethyl peroxide, di-t-butyl peroxide, dicumyl peroxide, isobutyl peroxide, benzoyl peroxide, 2,4-dichlorobenzoyl peroxide, 3, 5, Diacyl peroxides such as 5-trimethylhexanoyl peroxide, t-butylperoxypivalate, t-hexylperoxypivalate, t-butylperoxyneodecanoate, t-hexylperoxyneodecanoate, 1-cyclohexyl-1-methylethylperoxyneodecanoate, 1,1,3,3-tetramethylbutylperoxyneodecanoate, cumylperoxyneodecanoate, ( ⁇ , ⁇ -bis- Neodecanoyl peroxy) peroxyesthetics such as diisopropylbenzene Bis (4-tert-
- the microcapsule type gas generating agent can be produced by an emulsion polymerization method such as a suspension polymerization method.
- a microcapsule shell monomer for example, methyl methacrylate monomer
- a crosslinking agent D
- an oil phase O phase
- a solid phase S phase
- S phase core component
- S / O emulsion is prepared by adding a mixture of the above-mentioned adjuvants and the like in addition to the oxalate of aminoguanidine compound or the oxalate of aminoguanidine compound and stirring and mixing.
- the S / O / W suspension is prepared by adding this S / O emulsion to the aqueous phase (W phase) and mixing with stirring.
- This S / O / W suspension is charged into a pressure polymerization reactor such as an autoclave and subjected to pressure polymerization to obtain a cake-like substance.
- This cake-like substance is filtered off by a general method such as filtration or centrifugation, and the obtained residue is washed about 3 to 5 times with distilled water and then dried at a temperature of 40 to 60 ° C. for 24 hours with a dryer. By doing so, the microcapsule-type gas generating agent can be obtained.
- pressure polymerization conditions general conditions can be used. For example, the polymerization temperature is 40 to 70 ° C., the polymerization time is 10 to 24 hours, and the polymerization pressure is 0.2 to 0.3 MPa.
- the suspension polymerization is preferably carried out in the presence of a dispersion stabilizer or a dispersion stabilizing aid.
- the dispersion stabilizer is not particularly limited.
- silica, calcium phosphate, magnesium hydroxide, aluminum hydroxide, ferric hydroxide, barium sulfate, calcium sulfate, sodium sulfate, sodium chloride, calcium oxalate, calcium carbonate, Barium carbonate, magnesium carbonate or the like can be used.
- silica and calcium phosphate are preferable.
- the dispersion stabilizing aid is not particularly limited.
- a condensation product of diethanolamine and an aliphatic dicarboxylic acid a condensation product of urea and formaldehyde, polyvinylpyrrolidone, polyethylene oxide, polyethyleneimine, tetramethylammonium hydroxide, gelatin, Methyl cellulose, polyvinyl alcohol, dioctyl sulfosuccinate, polyglycerin fatty acid ester, sorbitan ester and the like can be used.
- a combination of colloidal silica and a condensation product can be mentioned.
- the condensation product is preferably a condensation product of diethanolamine and an aliphatic dicarboxylic acid, particularly preferably a condensation product of diethanolamine and adipic acid or a condensation product of diethanolamine and itaconic acid.
- a condensate is prescribed
- addition of an inorganic salt, particularly sodium chloride, sodium sulfate or the like, is suitable for obtaining a microcapsule type gas generating agent having a fine and uniform particle size.
- the microcapsule-type gas generating agent preferably contains a fatty acid salt or a surfactant as a dispersant. These fatty acid salts or surfactants are added to an oil phase (O phase) having a microcapsule shell monomer, a crosslinking agent (D), and a polymerization initiator when a microcapsule-type gas generating agent is produced. .
- the content of the fatty acid salt or the surfactant in the microcapsule-type gas generating agent is not limited to the core component (the compound represented by the general formula (1) or the compound represented by the general formula (1)). The content is preferably 0.01 to 3% by mass with respect to the composition containing the auxiliary agent and the like.
- the stability of the core component is sufficient, so that solid phase / oil phase droplets are prevented from coalescing and tend not to become aggregates during polymerization.
- the content is 3% by mass or less, the viscosity of the solid phase / oil phase droplets does not become excessively high, and the droplets hardly adhere to each other, and the particle size distribution of the microcapsule particles tends to be suppressed. is there.
- the fatty acid salt is not particularly limited.
- fatty acid amides such as stearic acid amide and arachidic acid amide, zinc stearate, calcium stearate, potassium stearate, aluminum stearate, lithium stearate, sodium stearate, stearic acid
- examples thereof include magnesium, zinc palmitate, zinc myristate, calcium palmitate, sodium palmitate and the like.
- these fatty acid salts it is preferably at least one selected from higher fatty acid salts having 15 to 22 carbon atoms, more preferably one or more higher fatty acids selected from the group consisting of potassium, calcium, lithium and magnesium. It is a metal salt.
- the surfactant is not particularly limited and includes known cationic surfactants, anionic surfactants, amphoteric surfactants, and nonionic surfactants. Although it does not specifically limit as a cationic surfactant, For example, alkyl trimethyl ammonium salt, dialkyl dimethyl ammonium salt, alkyl dimethyl benzyl ammonium salt etc. are mentioned.
- the anionic surfactant is not particularly limited.
- sulfonate type anionic surfactants such as alkylbenzene sulfonate, alkyl sulfosuccinate and allyl sulfonate, and sulfate type anionic interfaces such as alkyl sulfate and polyoxyethylene alkyl sulfate.
- sulfonate type anionic surfactants such as alkylbenzene sulfonate, alkyl sulfosuccinate and allyl sulfonate
- sulfate type anionic interfaces such as alkyl sulfate and polyoxyethylene alkyl sulfate.
- activators and lignin sulfites include activators and lignin sulfites.
- the nonionic surfactant is not particularly limited.
- sugar ester type nonionic surfactants such as sorbitan fatty acid ester and polyoxyethylene sorbitan fatty acid ester
- fatty acid ester type nonionic surfactant such as polyoxyethylene fatty acid ester.
- vegetable oil type nonionic surfactants such as polyoxyethylene castor oil, alcohol type nonionic surfactants such as polyoxyethylene alkyl ether, alkylphenols such as polyoxyethylene alkyl (C8-12) phenyl ether formalin condensate Type nonionic surfactant, polyoxyethylene / polyoxypropylene block polymer type nonionic surfactant such as polyoxyethylene / polyoxypropylene block polymer, polyaromatic ring type nonionic interface such as phenylphenyl ether Gender, and the like.
- nonionic surfactants are most preferred.
- an anionic surfactant, a cationic surfactant, and various additives can be used in combination as long as the effects of the present invention are not impaired.
- the HLB value (Hydrophile-Lipophile Balance) of the surfactant is preferably in the range of 0.01 to 16, more preferably 0.01 to 9, and still more preferably 0.01 to 3.
- the HLB value is larger than 16, the hydrophilicity of the surfactant is strong, and the solid phase in the solid phase / oil phase droplet becomes closer to the water phase. If the solid phase of the solid phase / oil phase droplet is close to the aqueous phase, the core component is collected near the surface of the microcapsule particles after polymerization, and the effect of reducing the decomposition residue when the core component is decomposed is small.
- the HLB value is 16 or less
- the surfactant has high lipophilicity
- the solid phase in the solid phase / oil phase droplets gathers at the center of the oil phase
- the core components gather at the center of the particles after polymerization.
- a microcapsule type gas generating agent is obtained, and the effect of reducing decomposition residue during heating is also high.
- the foaming composition of this embodiment contains the above-mentioned gas generating agent and foamed material.
- the material to be foamed contained together with the gas generating agent is not particularly limited, and examples thereof include a thermoplastic resin and / or rubber.
- the material to be foamed may be used alone or in combination of two or more.
- the thermoplastic resin include a vinyl chloride resin, a vinyl chloride copolymer resin, polyethylene, polypropylene, a polyolefin copolymer resin exemplified by an ethylene-propylene copolymer, a polystyrene resin, and an acrylonitrile-butadiene-styrene copolymer.
- the foaming composition of this embodiment may further contain a crosslinking agent (E).
- the crosslinking agent (E) contained in the foaming composition of the present embodiment may be the same as or different from the crosslinking agent (C) contained in the gas generating agent.
- the foam of this embodiment is a foam obtained by foaming the above-described foaming composition.
- the method for producing the foam of the present embodiment is not particularly limited as long as it includes a step of heating the foaming composition containing the gas generating agent and the material to be foamed. Manufacturing methods can be used.
- the foaming composition (unfoamed resin composition) can be prepared by kneading the thermoplastic resin, the crosslinking agent (E), and the gas generating agent described above with a heated kneading roll.
- the kneading temperature is preferably 90 to 130 ° C.
- the crosslinking agent (E) contained in the foaming composition is not particularly limited.
- the said crosslinking agent (E) may be used individually by 1 type, and may be used together 2 or more types. Further, the content of the crosslinking agent (E) in the foaming composition is preferably 0.1 to 10 parts by mass, and 0.5 to 1.5 parts by mass with respect to 100 parts by mass of the thermoplastic resin. It is more preferable that
- a foam of a resin composition (for example, a thermoplastic resin) is obtained by filling the unfoamed resin composition thus obtained in a mold and pressurizing with a press.
- a resin composition for example, a thermoplastic resin
- 100% of the unfoamed resin composition is filled in a mold having a thickness of 1 to 30 mm, and pressed with a press at 100 to 170 ° C. and 150 kg / cm 2 for 3 to 60 minutes, and 3 to 60 minutes.
- a sheet-like composition is obtained by water cooling. The obtained sheet-like composition is allowed to stand for 1 day, and then heated in an oven at 40 to 80 ° C. for 1 hour, and then heated at 130 to 250 ° C. for 150 to 600 seconds to obtain a foam of the resin composition.
- the usage-amount of the above-mentioned gas generating agent can be suitably selected according to the target expansion ratio, and there is no restriction
- it is a synthetic resin. 1 to 30 parts by mass is blended with 100 parts by mass of the material.
- Examples of the rubber used in this embodiment include natural rubber (NR), polyisoprene rubber, styrene-butadiene rubber (SBR), acrylonitrile-butadiene rubber, chloroprene rubber, ethylene-propylene rubber, ethylene-propylene-diene rubber, and butadiene rubber. However, it is not limited to these.
- General foam production conditions can be used as a method of preparing the foaming composition by blending the above gas generating agent with rubber and thereby producing the foam.
- a rubber material, a vulcanizing agent, a filler, a vulcanization accelerator, and the gas generating agent described above are uniformly dispersed with a kneading roll to obtain a foaming composition.
- the obtained foaming composition is put into an extruder heated to 70 to 90 ° C. to prepare an unvulcanized molded body.
- the resulting unvulcanized molded body is heated in an oven heated to 60 to 220 ° C. for 5 to 15 minutes to vulcanize and foam to obtain a foam of rubber material.
- the blending ratio of each component in the foaming composition is not particularly limited, but the vulcanizing agent is preferably blended in an amount of 0.1 to 10 parts by mass with respect to 100 parts by mass of the rubber material.
- the filler is preferably blended in an amount of 10 to 150 parts by mass with respect to 100 parts by mass of the rubber material.
- the vulcanization accelerator is preferably blended in an amount of 0.1 to 20 parts by mass with respect to 100 parts by mass of the rubber material.
- the amount of the gas generating agent used can be appropriately selected according to the target foaming ratio and is not particularly limited, but preferably 100 mass of rubber. 1 to 20 parts by mass with respect to parts.
- vulcanizing agent used by this embodiment, For example, sulfur is mentioned.
- specific examples of the filler used in the present embodiment are not particularly limited, and examples thereof include heavy, light calcium carbonate, and carbon black.
- Specific examples of the vulcanization accelerator used in the present embodiment are not particularly limited. Examples thereof include dithiocarbamate vulcanization accelerators and dithiocarbamic acid vulcanization accelerators. Specifically, for example, DM (dibenzylthiazol disulfide), dimethyldithiocarbamine zinc, and dibutyldithiocarbamine zinc.
- the foaming composition of this embodiment may contain other additives.
- additives include, but are not limited to, for example, Diana process oil, zinc oxide, stearic acid, calcium oxide, urea-based auxiliary, silicon dioxide (silica), talc, magnesium oxide, zinc stearate, calcium hydroxide , Barium stearate, dibasic lead phosphite, lead oxide and the like.
- the present invention will be described more specifically with reference to examples and comparative examples, but the present invention is not limited to these examples.
- the structure of the compound used for the gas generating agent is identified by CHN elemental analysis and melting point, and the decomposition start temperature, generated gas amount, and ammonia generation amount of the gas generating agent are identified.
- the evaluation was performed by the following methods.
- the decomposition start temperature of the gas generating agent is increased at 10 ° C./min using a differential thermal-thermogravimetric simultaneous measurement apparatus (EXSTAR6000 manufactured by SII Nanotechnology Co., Ltd.) at 25 ° C. as the start temperature. It measured on condition of.
- the decomposition start temperature was a temperature at which the weight decreased by 5% with respect to the weight at the start of temperature increase.
- Ammonia gas generated during the heating was collected in 0.1N hydrochloric acid, and ammonium ions in hydrochloric acid were quantified by ion chromatography (DX-320J, manufactured by Nippon Dionics Co., Ltd.). The amount of ammonia gas generated (mg) was determined.
- the obtained solid was subjected to elemental analysis using a simultaneous carbon, hydrogen, and nitrogen quantitative determination apparatus CHN coder MT-6 (Yanako Analytical Industrial Co., Ltd.).
- the calculated value was C, 21.96; H, 4.91; N, 34.14, measured values C, 21.78; H, 4.75; N, 34.22, confirming aminoguanidine oxalate.
- the molar yield was 71%.
- the melting point of the obtained solid was measured using a trace melting point measuring device BY-1 (manufactured by Yazawa Kagaku Co., Ltd.), and it was 211 to 212 ° C.
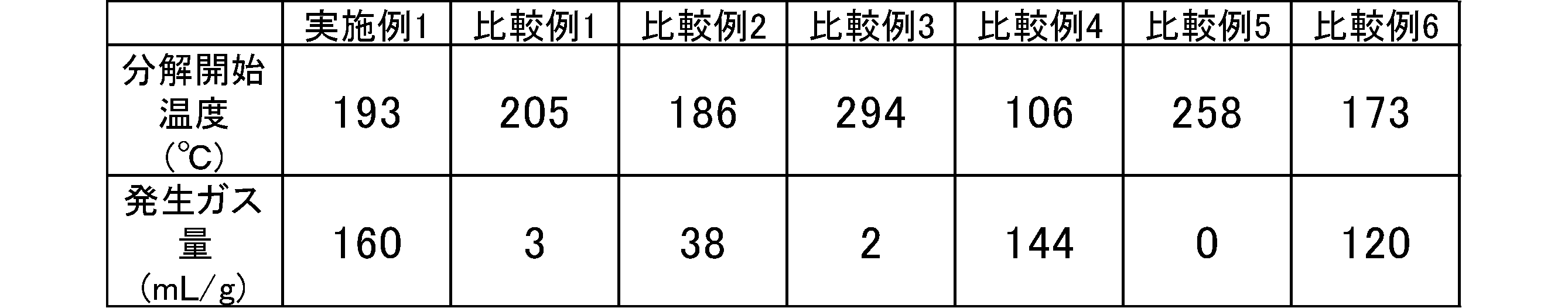
- Example 1 The decomposition start temperature and the amount of generated gas of aminoguanidine oxalate (2) obtained in Synthesis Example 1 were measured. Table 1 shows the decomposition start temperature and the amount of generated gas.
- Comparative Example 1 The decomposition start temperature and the amount of generated gas of guanidine oxalate (3) obtained in Comparative Synthesis Example 1 were measured under the same conditions as in Example 1. Table 1 shows the decomposition start temperature and the amount of generated gas.
- Example 2 The decomposition start temperature and the amount of generated gas of guanidine carbonate (manufactured by Tokyo Chemical Industry Co., Ltd.) were measured under the same conditions as in Example 1. Table 1 shows the decomposition start temperature and the amount of generated gas.
- Example 3 The decomposition start temperature and the amount of generated gas of guanidine nitrate (manufactured by Tokyo Chemical Industry Co., Ltd.) were measured under the same conditions as in Example 1. Table 1 shows the decomposition start temperature and the amount of generated gas.
- Example 4 The decomposition start temperature and the amount of generated gas of aminoguanidine carbonate (manufactured by Tokyo Chemical Industry Co., Ltd.) were measured under the same conditions as in Example 1. Table 1 shows the decomposition start temperature and the amount of generated gas.
- Example 5 The decomposition start temperature and the amount of generated gas of aminoguanidine nitrate (manufactured by SIGMA-ALDRICH) were measured under the same conditions as in Example 1. Table 1 shows the decomposition start temperature and the amount of generated gas.
- aminoguanidine, etc. can form a salt with oxalic acid to increase the amount of gas generated, or the decomposition start temperature is 120 ° C or higher, and can be used for general-purpose plastics (for example, thermoplastic resins) and rubber molding temperatures.
- Example 2 The method as described above, except that the amount of ammonia gas generated in aminoguanidine oxalate (2) (compound represented by formula (2)) obtained in Synthesis Example 1 was changed to 185 ° C. at the heating temperature in the block heater. Measured with The results are shown in Table 2.
- the gas generating agent of the present embodiment has a small ammonia gas generation amount, and is excellent in safety as a chemical foaming agent for general-purpose plastics (for example, thermoplastic resins) and rubber without contaminating the mold during molding. I understood it.
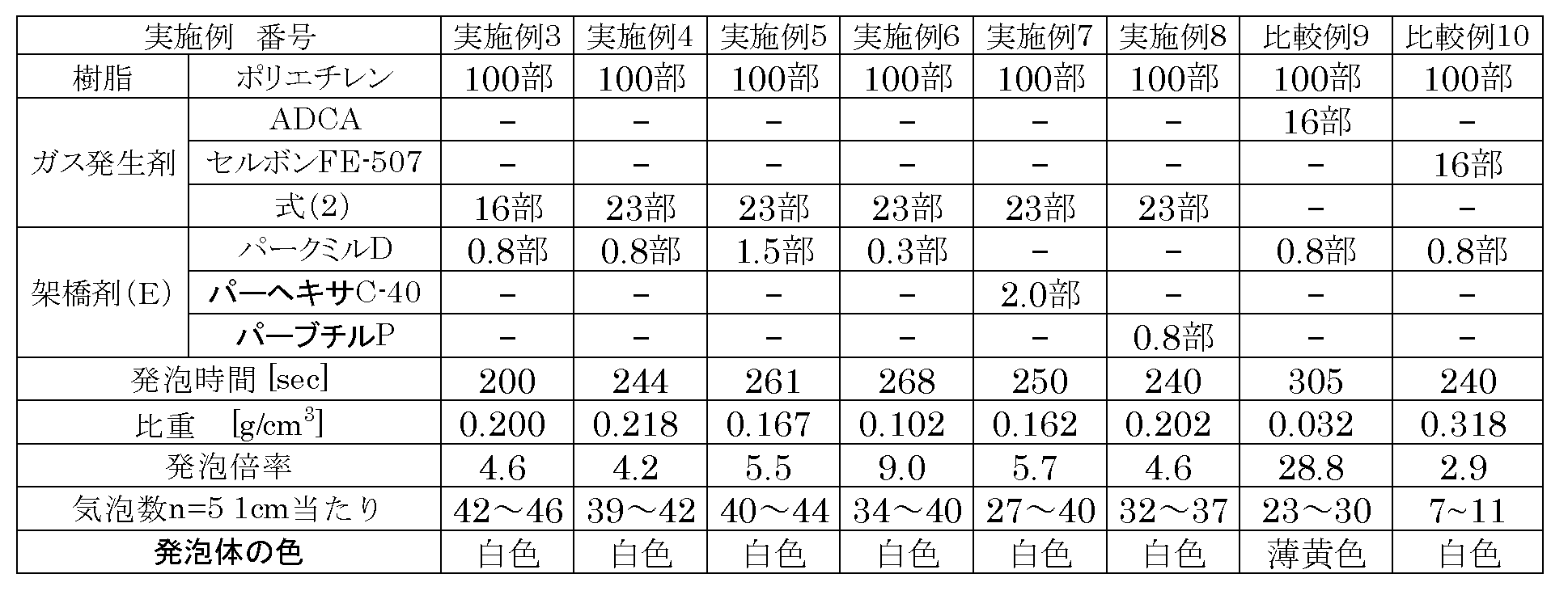
- Example 3 100 parts by mass of polyethylene (trade name “LE200M” manufactured by Nippon Polyethylene Co., Ltd.) was kneaded with an open roll heated to 120 ° C., and then aminoguanidine oxalate (2) (formula ( Compound represented by 2) 16 parts by mass was added and kneaded for 4 minutes and 30 seconds, followed by dicumyl peroxide (DCP) (trade name “Park Mill D”, manufactured by NOF Corporation) 0.8 mass The kneaded product was taken out from the open roll by kneading for 1 minute and 30 seconds. The kneaded material is charged so that the inner volume of the die (2 mm) of the press apparatus heated to 165 ° C.
- DCP dicumyl peroxide
- Example 4 Implemented except that the amount of aminoguanidine oxalate (2) (compound represented by formula (2)) obtained in Synthesis Example 1 was changed to 23 parts by mass and the heating time at 220 ° C. was changed to 244 seconds. A foam was obtained under the same conditions as in Example 3. The evaluation results of the obtained foam are shown in Table 3.
- Example 5 The addition amount of aminoguanidine oxalate (2) (compound represented by formula (2)) obtained in Synthesis Example 1 was changed to 23 parts by mass, the addition amount of DCP was changed to 1.5 parts by mass, and 220 A foam was obtained under the same conditions as in Example 3 except that the heating time at °C was 261 seconds. The evaluation results of the obtained foam are shown in Table 3.
- Example 6 The addition amount of aminoguanidine oxalate (2) (compound represented by formula (2)) obtained in Synthesis Example 1 was changed to 23 parts by mass, the addition amount of DCP was changed to 0.3 parts by mass, and 220 A foam was obtained under the same conditions as in Example 3 except that the heating time at °C was 268 seconds. The evaluation results of the obtained foam are shown in Table 3.
- Example 7 The amount of aminoguanidine oxalate (2) (compound represented by formula (2)) obtained in Synthesis Example 1 was changed to 23 parts by mass, and DCP was changed to 1,1-bis [t-butylperoxy].
- the foam was obtained under the same conditions as in Example 3 except that the weight was changed to 2.0 parts by mass of cyclohexane (trade name “Perhexa C-40”, manufactured by NOF Corporation) and the heating time at 220 ° C. was changed to 250 seconds. Obtained.
- the evaluation results of the obtained foam are shown in Table 3.
- Example 8 The amount of aminoguanidine oxalate (2) (compound represented by formula (2)) obtained in Synthesis Example 1 was changed to 23 parts by mass, and DCP was converted to di- (2-t-butylperoxyisopropyl). A foam was obtained under the same conditions as in Example 3 except that benzene (trade name “Perbutyl P”, manufactured by NOF Corporation) was changed to 0.8 parts by mass, and the heating time at 220 ° C. was changed to 240 seconds. . The evaluation results of the obtained foam are shown in Table 3.
- Example 9 A foam was obtained under the same conditions as in Example 3 except that the gas generating agent was changed to azodicarbonamide (ADCA) (manufactured by Eiwa Kasei Kogyo Co., Ltd.) and the heating time at 220 ° C. was changed to 305 seconds.
- ADCA azodicarbonamide
- Example 10 Foaming was performed under the same conditions as in Example 3 except that the gas generating agent was changed to sodium hydrogen carbonate (trade name “SELBON FE-507”, manufactured by Eiwa Kasei Kogyo Co., Ltd.) and the heating time at 220 ° C. was changed to 240 seconds. Got the body.
- the evaluation results of the obtained foam are shown in Table 3.
- the specific gravity, expansion ratio, and number of bubbles of the foam were measured as follows. Specific gravity: Measured with an electronic hydrometer MD-200S. Expansion ratio: Calculated by specific gravity of polyethylene (0.92 g / cm 3 ) / specific gravity of foam. Number of bubbles: measured with a microscope HIROX KH7700 2D measurement. From Table 3, it was found that a white foam was obtained by using the gas generating agent of the present invention.
- Example 9 100 parts by mass of saturated hydrocarbon rubber (trade name “EPT 4021” manufactured by Mitsui Chemicals, Inc.) is kneaded in a cooled (water-passed) roll machine, followed by carbon black (trade name “Asahi # 50UG” Asahi Carbon ( Co., Ltd.) 70 parts by mass, light calcium carbonate (Omi Chemical Co., Ltd.) 40 parts by mass, Diana Process Oil (trade name “PW-90” manufactured by Idemitsu Kosan Co., Ltd.) 45 parts by mass, zinc oxide ( 5 parts by mass of ZnO (Zinc Hua) Mitsui Metals Mining Co., Ltd.) and 1 part by mass of stearic acid (CH 3 (CH 2 ) 16 COOH, trade name “Tsubaki” manufactured by NOF Corporation) are added and kneaded.
- carbon black trade name “Asahi # 50UG” Asahi Carbon ( Co., Ltd.) 70 parts by mass
- light calcium carbonate (
- a kneaded material A was obtained.
- the kneaded material A was aged by storing at room temperature for 1 day or longer.
- the aged kneaded product A is kneaded with a cooled (water-passed) roll machine, and then a dithiocarbamate vulcanization accelerator (dimethyldithiocarbamine zinc, trade name “Noxeller PZ”, Ouchi Shinsei Chemical Co., Ltd.) 1.1 parts by mass, 1.7 parts by mass of a dithiocarbamic acid-based vulcanization accelerator (dibutyldithiocarbamine zinc, trade name “Noxeller BZ” manufactured by Ouchi Shinsei Chemical Co., Ltd.) are added and kneaded.
- a dithiocarbamate vulcanization accelerator dimethyldithiocarbamine zinc, trade name “Noxeller PZ”, Ouchi Shinsei Chemical Co., Ltd.
- the obtained sheet-like molded body was aged at room temperature for 1 day, and then heated in an oven at 180 ° C. for 10 minutes to obtain a foam.
- the foaming ratio of the obtained foaming agent was 1.280.
- a kneaded product C was produced in the same manner as the kneaded product B except that aminoguanidine oxalate (2) was not blended as a blank for calculating the expansion ratio.
- Specific gravity was measured.
- the expansion ratio was calculated from the specific gravity of the kneaded product C (0.860 g / cm 3 ) / the specific gravity of the foam (0.672 g / cm 3 ).
- the specific gravity was measured with an electronic hydrometer MD-200S.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
ADCAの優れた点としては、他の市販化学発泡剤と比較して発生ガス量が200mL/g以上と多いこと、分解開始温度が200~210℃と比較的高いものの発泡助剤との併用により分解開始温度を140℃付近まで下げることができ、汎用プラスチック(例えば、熱可塑性樹脂)及びゴムに展開可能であること、発生ガスが窒素を主成分とし、自己消火性のため取り扱い上安全性が高いこと、等が挙げられる。
一方、ADCAは、発泡時に発生ガス中に少量のアンモニアガスが含まれており、このアンモニアガスが腐食性を示すこと、ADCAの分解生成物であるシアン酸は昇華性があり、昇華後に重合して腐食性のシアヌル酸となり、金型汚染の原因になりやすいこと等の課題を有する(例えば、特許文献1及び非特許文献1参照)。
このようにADCAの欠点であるアンモニアや汚染性を嫌う分野には、発生ガスが窒素及び水だけで分解生成物も非汚染性である4,4'-オキシビス(ベンゼンスルホニルヒドラジド)(以下「OBSH」とも記す)がガス発生剤として使用される。OBSHは分解開始温度が約170℃と低く、主にウェザーストリップ用のエチレン-プロピレン-ジエンゴム(EPDM)やウェットスーツ用のクロロプレンゴム(CR)に用いられている。
しかしながら、OBSHは発生ガス量が120mL/gと少なく、所望の発泡倍率を得るには添加量を多くする必要があり、経済面、省資源の観点で課題を抱えている。
ADCA、OBSHの他に、グアニジン塩、アミノグアニジン塩、ジアミノグアニジン塩、トリアミノグアニジン塩等のグアニジン誘導体も、ガス発生剤として用いることができる。これらのグアニジン誘導体は、火薬用,医薬用,繊維処理加工用酸化防止剤,石けん樹脂安定剤、その他各種合成原料用にも用いられる(例えば、非特許文献2参照)。具体的には、例えば、エアバックシステム向けに、グアニジン誘導体の炭酸塩、硝酸塩、過塩素酸塩を含むガス発生剤が開示されている(例えば、特許文献2~4参照)。
そこで、本発明の課題は、合成が簡便で汎用的なグアニジン誘導体を用いて、例えば、汎用プラスチック(例えば、熱可塑性樹脂)及びゴムの化学発泡剤として使用可能な分解開始温度を有し、発生ガス量が多く、発生ガス中にアンモニア等の腐食性ガスをほとんど含まないガス発生剤を提供することである。
[1]
下記一般式(1)で表されるアミノグアニジン化合物のシュウ酸塩を含有する、ガス発生剤。

[2]
前記アミノグアニジン化合物のシュウ酸塩において、アミノグアニジン化合物とシュウ酸とのモル比(アミノグアニジン化合物:シュウ酸)が、1:1~2:1の範囲である、[1]に記載のガス発生剤。
[3]
前記アミノグアニジン化合物がアミノグアニジンである、[1]又は[2]に記載のガス発生剤。
[4]
マイクロカプセル型である、[1]~[3]のいずれかに記載のガス発生剤。
[5]
[1]~[4]のいずれかに記載のガス発生剤と被発泡材料とを含有する、発泡用組成物。
[6]
被発泡材料が、熱可塑性樹脂及び/又はゴムである、[5]に記載の発泡用組成物。
[7]
[5]又は[6]に記載の発泡用組成物を加熱する工程を含む、発泡体の製造方法。
[8]
[5]又は[6]に記載の発泡用組成物を発泡させた発泡体。
本実施形態のガス発生剤において、アミノグアニジン化合物と塩を形成する酸にシュウ酸を用いる。アミノグアニジン等のアミノグアニジン化合物のシュウ酸塩を用いることにより、汎用プラスチック(例えば、熱可塑性樹脂)及びゴムの化学発泡剤として使用可能な分解開始温度を有し、発生ガス量が多く、発生ガスにアンモニア等の腐食性ガスをほとんど含まないガス発生剤を得ることが可能となる。
本実施形態のガス発生剤の発泡時の発生ガス量は、120mL/g以上であることが好ましい。
本実施形態のガス発生剤の発泡時のアンモニア発生量は、10mg/g以下であることが好ましく、5mg/g以下であることがより好ましく、1mg/gであることがさらに好ましい。
本実施形態のガス発生剤の分解開始温度は、例えば、汎用プラスチック(例えば、熱可塑性樹脂)及びゴムの化学発泡剤として使用可能な分解開始温度であることが好ましく、具体的には、90~380℃であることが好ましく、110~250℃であることがより好ましい。
なお、本実施形態において、ガス発生剤の発泡時の発生ガス量、アンモニア発生量及び分解開始温度は、後述の実施例に記載の方法により測定することができる。
本実施形態に用いるアミノグアニジン化合物のシュウ酸塩は、通常、アミノグアニジン化合物:シュウ酸=1:1の割合で塩を形成しているが、アミノグアニジン化合物:シュウ酸=2:1の割合で塩を形成している場合もあり、両者の混合物の場合もある。本実施形態に用いるアミノグアニジン化合物のシュウ酸塩において、アミノグアニジン化合物とシュウ酸とのモル比(アミノグアニジン化合物:シュウ酸)は、1:1~2:1の範囲であることが好ましく、1:1~1.5:1の範囲であることがより好ましく、1:1~1.2:1の範囲であることがさらに好ましい。
したがって、本実施形態に用いるアミノグアニジン化合物のシュウ酸塩は、例えば、下記式(1-1)で表すこともできる。
本実施形態のガス発生剤は、アミノグアニジン化合物のシュウ酸塩だけからなる場合であっても汎用プラスチック(例えば、熱可塑性樹脂)及びゴムの化学発泡剤として好適に用いることができるが、アミノグアニジン化合物のシュウ酸塩以外に補助剤をさらに含有させてもよい。当該補助剤は、特に限定されないが、例えば、酸化剤、架橋剤(C)、亜硝酸塩、ハイドロタルサイト等が挙げられる。本実施形態のガス発生剤は、このような補助剤をさらに含有すると、アンモニア等の腐食性ガスの発生をより一層抑制することができ、後述する発泡体を製造する際の温度も好適な範囲となる傾向にある。
本実施形態のガス発生剤において、アミノグアニジン化合物のシュウ酸塩以外に補助剤を含有させる場合、アミノグアニジン化合物のシュウ酸塩の含有量は、アミノグアニジン化合物のシュウ酸塩と補助剤との合計質量に対して0.5~95質量%であること好ましい。
前記酸化剤としては、特に限定されないが、例えば、過炭酸ナトリウムが挙げられる。前記酸化剤は1種を単独で用いてもよく2種以上を併用してもよい。
本実施形態のガス発生剤において、酸化剤の含有量は、アミノグアニジン化合物のシュウ酸塩と補助剤との合計質量に対して0.5~95質量%であることが好ましく、5~50質量%であることがより好ましい。
前記架橋剤(C)としては、特に限定されないが、例えば、ジクミルパーオキシド、ジ-t-ブチルパーオキシド、2,5-ジメチル-2,5-ジ-(t-ブチルパーオキシ)ヘキサン、2,5-ジメチル-2,5-ジ-(t-ブチルパーオキシ)ヘキシン、1,3-ビス(t-ブチルパーオキシイソプロピル)ベンゼン、1,1-ビス(t-ブチルパーオキシ)-3,3,5-トリメチルシクロヘキサン、1,1-ビス[t-ブチルパーオキシ]シクロヘキサン、n-ブチル-4,4-ビス(t-ブチルパーオキシ)バレレート、ベンゾイルパーオキシド、p-クロロベンゾイルパーオキシド、2,4-ジクロロベンゾイルパーオキシド、t-ブチルパーオキシベンゾエート、t-ブチルペルベンゾエート、t-ブチルパーオキシソプロピルカーボネート、ジアセチルパーオキシド、ラウロイルパーオキシド、t-ブチルクミルパーオキシドが挙げられる。前記架橋剤(C)は1種を単独で用いてもよく2種以上を併用してもよい。
本実施形態のガス発生剤において、架橋剤(C)の含有量は、アミノグアニジン化合物のシュウ酸塩と補助剤との合計質量に対して0.1~10質量%であることが好ましく、0.5~1.5質量%であることがより好ましい。 前記亜硝酸塩としては、特に限定されないが、例えば、亜硝酸ナトリウム、亜硝酸カリウム、亜硝酸カルシウム等が挙げられ、これらの中からから選ばれる一種を単独でまたは二種以上を組み合わせて用いることができる。また、これらの亜硝酸塩は粉砕された微粉であることが好ましい。
本実施形態のガス発生剤において、亜硝酸塩の含有量は、アミノグアニジン化合物のシュウ酸塩と補助剤との合計質量に対して0.5~60質量%であることが好ましく、15~55質量%であることがより好ましい。
前記ハイドロタルサイトは、結晶性複合金属水酸化物であり、下記一般式(H)で表されるハイドロタルサイトが好ましい。
ここで、mは好ましくは0であるが、ハイドロタルサイトの乾燥状態や保管状態により変動するものであり、本発明の効果を損なわない範囲であれば特にmを限定するものではない。nはアニオンの価数であり、好ましくは1又は2、より好ましくは2である。
これらの中でも、M2+がMg2+、M3+がAl3+であるハイドロタルサイトが好ましく、Al:Mgのモル比は、入手のし易さから2:5~2:10が好ましい。例えば、Al:Mgのモル比が2:5である場合には、Alのモル分率x(x=Al/(Mg+Al))は0.29であり、AlとMgのモル比が2:10の場合には、Alのモル分率xは0.17である。
前記ハイドロタルサイトは、亜硝酸塩の反応性を向上させる作用を有する。ハイドロタルサイトを配合することにより、ガス発生剤の加熱時に発生し得るアンモニアガスの分解を促進し、亜硝酸塩の反応性を向上させることで亜硝酸ガスの発生を抑制し、窒素ガスの生成を促進しガス発生剤の発泡性を向上することができる。前記ハイドロタルサイトの粒子径は特に限定されるものではないが、アミノグアニジン化合物のシュウ酸塩と亜硝酸塩との反応に効果的に作用させるために、ガス発生剤中での分散性を高めることが好ましく、最大粒子径が80μm以下の微粉ハイドロタルサイトがより好ましい。
本実施形態のガス発生剤において、ハイドロタルサイトの含有量は、アミノグアニジン化合物のシュウ酸塩と補助剤との合計質量に対して1~40質量%であることが好ましく、5~25質量%であることがより好ましい。
本実施形態のガス発生剤の製造方法は特に限定されるものではなく、一般的な混合方法を用いることができる。例えば、アミノグアニジン化合物のシュウ酸塩、亜硝酸塩及びハイドロタルサイトを、高速ミキサー、リボンブレンダー、コーンブレンダー等を用いて、温度60℃以下、時間5分程度の条件で均一に分散するように混合すればよい。
本実施形態において、マイクロカプセル型のガス発生剤とは、コアシェル構造を有するガス発生剤のことをいう。
マイクロカプセル型のガス発生剤として、具体的には、例えば、アミノグアニジン化合物のシュウ酸塩、又はアミノグアニジン化合物のシュウ酸塩以外にさらに前記補助剤等を含有させた組成物をコア成分として有するマイクロカプセル型のガス発生剤が挙げられる。当該マイクロカプセル型のガス発生剤のシェルの主成分は、特に限定されないが、例えば、ポリメタクリル酸メチルであり、かつ分散剤として後述する脂肪酸塩または界面活性剤を含むことが好ましい。このようなマイクロカプセル型のガス発生剤を用いてゴムや熱可塑性樹脂に気泡を導入することで、加硫や架橋を阻害することなく、さらに分解残渣やVOC成分を発生することなくポリマー中に均一な気泡を導入できるため、架橋密度の低下が抑制され、かつ気泡状態が良好で発泡性に優れた架橋発泡体を得ることができる。
前記ポリメタクリル酸メチルは、メタクリル酸メチルモノマーを構成単位とするホモポリマーであることが好ましいが、本発明の効果を損なわない範囲で他のモノマーを共重合してもよい。他のモノマーとしては、特に限定されないが、例えば、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、ジシクロペンテニルアクリレート等のアクリル酸エステル類、メタクリル酸エチル、メタクリル酸ブチル、イソボルニルメタクリレート等のメタクリル酸エステル類、アクリロニトリル、メタクリロニトリル、塩化ビニリデン、塩化ビニル、スチレン、酢酸ビニル、α-メチルスチレン、クロロプレン、ネオプレン、ブタジエン等が挙げられる。
前記の界面活性剤のうち、非イオン界面活性剤が最も好適である。また、本発明の効果を阻害しない範囲において、陰イオン界面活性剤、陽イオン界面活性剤、及び各種添加物を併用することも可能である。
本実施形態の発泡用組成物において、上述のガス発生剤と共に含有させる被発泡材料としては、特に限定されないが、例えば、熱可塑性樹脂及び/又はゴムが挙げられる。被発泡材料は1種を単独で用いてもよく、2種以上を併用してもよい。上記の熱可塑性樹脂としては、例えば、塩化ビニル樹脂、塩化ビニル共重合樹脂、ポリエチレン、ポリプロピレン、エチレン-プロピレン共重合体で例示されるポリオレフィン共重合樹脂、ポリスチレン樹脂、アクリロニトリル-ブタジエン-スチレン共重合体(ABS樹脂)等が挙げられるが、これらに限定されるものではない。
また、本実施形態の発泡用組成物は、架橋剤(E)をさらに含有していてもよい。本実施形態の発泡用組成物に含有させる架橋剤(E)は、上述のガス発生剤に含有させる架橋剤(C)と同一であってもよく、異なっていてもよい。
本実施形態の発泡体を製造する方法としては、上述のガス発生剤と被発泡材料とを含有する発泡用組成物を加熱する工程を含む方法であれば特に限定されず一般的な発泡体の製造方法を用いることができる。例えば、熱可塑性樹脂、架橋剤(E)、及び上述のガス発生剤を、加熱した混練りロールで混練りし、発泡用組成物(未発泡樹脂組成物)を調製することができる。混練温度は好ましくは90~130℃である。ここで発泡用組成物中に含有させる架橋剤(E)としては、特に限定されないが、例えば、ジクミルパーオキシド、ジ-t-ブチルパーオキシド、2,5-ジメチル-2,5-ジ-(t-ブチルパーオキシ)ヘキサン、2,5-ジメチル-2,5-ジ-(t-ブチルパーオキシ)ヘキシン、1,3-ビス(t-ブチルパーオキシイソプロピル)ベンゼン、1,1-ビス(t-ブチルパーオキシ)-3,3,5-トリメチルシクロヘキサン、1,1-ビス[t-ブチルパーオキシ]シクロヘキサン、n-ブチル-4,4-ビス(t-ブチルパーオキシ)バレレート、ベンゾイルパーオキシド、p-クロロベンゾイルパーオキシド、2,4-ジクロロベンゾイルパーオキシド、t-ブチルパーオキシベンゾエート、t-ブチルペルベンゾエート、t-ブチルパーオキシソプロピルカーボネート、ジアセチルパーオキシド、ラウロイルパーオキシド、t-ブチルクミルパーオキシド等が挙げられる。前記架橋剤(E)は1種単独で用いてもよく2種以上併用してもよい。また、発泡用組成物中の架橋剤(E)の含有量は、熱可塑性樹脂100質量部に対して、0.1~10質量部であることが好ましく、0.5~1.5質量部であることがより好ましい。
本実施形態で用いられる充填剤の具体例としては、特に限定されないが、例えば、重質、軽質炭酸カルシウム、カーボンブラックが挙げられる。
本実施形態で用いられる加硫促進剤の具体例としては、特に限定されないが、例えば、ジチオカルバミン酸塩系加硫促進剤、ジチオカルバミン酸系加硫促進剤が挙げられ、具体的には、例えば、DM(ジベンジルチアゾル・ジスルフィド)、ジメチルジチオカルバミン亜鉛、ジブチルジチオカルバミン亜鉛が挙げられる。
本実施形態の発泡用組成物には、その他の添加剤を含んでいてもよい。その他の添加剤としては、特に限定されないが、例えば、ダイアナプロセスオイル、酸化亜鉛、ステアリン酸、酸化カルシウム、尿素系助剤、二酸化ケイ素(シリカ)、タルク、酸化マグネシウム、ステアリン酸亜鉛、水酸化カルシウム、ステアリン酸バリウム、二塩基性亜リン酸鉛、酸化鉛等が挙げられる。
ガス発生剤の分解開始温度は、示差熱-熱重量同時測定装置(エスアイアイ・ナノテクノロジー(株)製EXSTAR6000)を用いて、大気下、25℃を開始温度として10℃/minで昇温するという条件で測定した。分解開始温度は、昇温開始時の重量に対して、5%重量減少した温度とした。
ガス発生剤0.5gを試験管に取り、熱媒体として流動パラフィン10mLを添加した後、試験管とガスビュレットとをゴム管でつなぎ、試験管を60℃のオイルバスに浸した。その後、オイルバスを2℃/minの昇温速度で、240℃まで加熱した。当該加熱中に発生したガスをガスビュレットですべて捕集し、ガス発生剤1gあたりの発生ガス量(mL)を求めた。
ガス発生剤0.5gを試験管に取り、熱媒体として流動パラフィン1mLを添加した後、この試験管と、ねじ口試験管と、0.1N塩酸を100mL入れたねじ口瓶とを、この順にゴム管でつないだ。窒素ガスを流速0.4L/minで流しながら、ガス発生剤を入れた試験管をブロックヒーターにて、228℃になるまで加熱した。当該加熱中に発生したアンモニアガスを0.1N塩酸に捕集し、塩酸中のアンモニウムイオンをイオンクロマトグラフ(日本ダイオニクス(株)製DX-320J)にて定量して、ガス発生剤1gあたりの発生したアンモニアガス量(mg)を求めた。

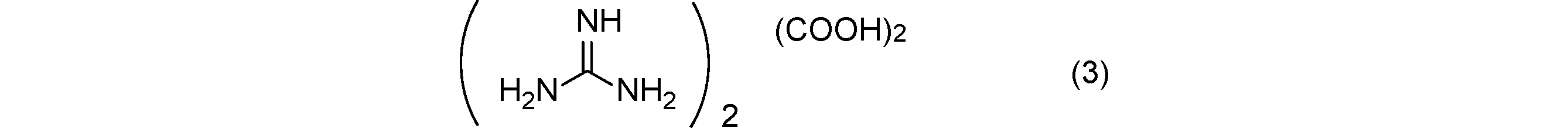
合成例1で得られたアミノグアニジンシュウ酸塩(2)の分解開始温度及び発生ガス量を測定した。分解開始温度及び発生ガス量を表1に示す。
比較合成例1で得られたグアニジンシュウ酸塩(3)の分解開始温度及び発生ガス量を実施例1と同様の条件で測定した。分解開始温度及び発生ガス量を表1に示す。
グアニジン炭酸塩(東京化成工業(株)製)の分解開始温度及び発生ガス量を実施例1と同様の条件で測定した。分解開始温度及び発生ガス量を表1に示す。
グアニジン硝酸塩(東京化成工業(株)製)の分解開始温度及び発生ガス量を実施例1と同様の条件で測定した。分解開始温度及び発生ガス量を表1に示す。
アミノグアニジン炭酸塩(東京化成工業(株)製)の分解開始温度及び発生ガス量を実施例1と同様の条件で測定した。分解開始温度及び発生ガス量を表1に示す。
アミノグアニジン硝酸塩(SIGMA-ALDRICH社製)の分解開始温度及び発生ガス量を実施例1と同様の条件で測定した。分解開始温度及び発生ガス量を表1に示す。
4,4'-オキシビス(ベンゼンスルホニルヒドラジド)(OBSH)(永和化成工業(株)製)の分解開始温度及び発生ガス量を実施例1と同様の条件で測定した。分解開始温度及び発生ガス量を表1に示す。
合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)のアンモニアガス発生量をブロックヒーターでの加熱温度を185℃に変えた以外は上記記載の方法で測定した。結果を表2に示す。
比較合成例1で得られたグアニジンシュウ酸塩(3)(式(3)で表される化合物)のアンモニアガス発生量を、上記記載と同様の条件で測定した。結果を表2に示す。
アゾジカルボンアミド(ADCA)(永和化成工業(株)製)のアンモニアガス発生量を、ブロックヒーターでの加熱温度を210℃に変えた以外は、上記記載と同様の条件で測定した。結果を表2に示す。

120℃に加熱されたオープンロールでポリエチレン(商品名「LE200M」日本ポリエチレン(株)製)100質量部を混練し、続いて合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)16質量部を添加して4分30秒混練し、続いてジクミルパーオキサイド(DCP)(商品名「パークミルD」、日油(株)製)0.8質量部を添加して1分30秒混練してオープンロールから混練物を取り出した。165℃に加熱されたプレス装置の金型(2mm)の内容積100%充填となるように混練物を投入し、プレス圧力50kg/cm2で3分間加圧し、続いてプレス圧力150kg/cm2で3分間加圧し、5分間水冷しシート状組成物を得た。得られたシート状組成物を1日静置した後、60℃のオーブンで1時間加熱し、220℃で244秒間加熱し、発泡体を得た。得られた発泡体の評価結果を表3に示す。
合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)の添加量を23質量部に変え、220℃で加熱する時間を244秒にした以外は実施例3と同様の条件で発泡体を得た。得られた発泡体の評価結果を表3に示す。
合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)の添加量を23質量部に変え、DCPの添加量を1.5質量部に変え、220℃で加熱する時間を261秒にした以外は実施例3と同様の条件で発泡体を得た。得られた発泡体の評価結果を表3に示す。
合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)の添加量を23質量部に変え、DCPの添加量を0.3質量部に変え、220℃で加熱する時間を268秒にした以外は実施例3と同様の条件で発泡体を得た。得られた発泡体の評価結果を表3に示す。
合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)の添加量を23質量部に変え、DCPを1,1-ビス[t-ブチルパーオキシ]シクロヘキサン(商品名「パーヘキサC-40」、日油(株)製)2.0質量部に変え、220℃で加熱する時間を250秒にした以外は実施例3と同様の条件で発泡体を得た。得られた発泡体の評価結果を表3に示す。
合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)の添加量を23質量部に変え、DCPをジ-(2-t-ブチルパーオキシイソプロピル)ベンゼン(商品名「パーブチルP」、日油(株)製)0.8質量部に変え、220℃で加熱する時間を240秒にした以外は実施例3と同様の条件で発泡体を得た。得られた発泡体の評価結果を表3に示す。
ガス発生剤をアゾジカルボンアミド(ADCA)(永和化成工業(株)製)に変え、220℃で加熱する時間を305秒にした以外は実施例3と同様の条件で発泡体を得た。得られた発泡体の評価結果を表3に示す。
ガス発生剤を炭酸水素ナトリウム(商品名「セルボンFE-507」、永和化成工業(株)製)に変え、220℃で加熱する時間を240秒にした以外は実施例3と同様の条件で発泡体を得た。得られた発泡体の評価結果を表3に示す。
比重:電子比重計 MD-200Sにより測定した。
発泡倍率:ポリエチレンの比重(0.92g/cm3)/発泡体の比重により算出した。
気泡数:マイクロスコープ HIROX KH7700 2D計測により測定した。
表3より本発明のガス発生剤を用いることで、白色の発泡体が得られることがわかった。
冷却(通水)されたロール機で飽和炭化水素ゴム(商品名「EPT 4021」三井化学(株)製)100質量部を混練し、続いてカーボンブラック(商品名「旭#50UG」旭カーボン(株)製)70質量部、軽微性炭酸カルシウム(近江化学工業(株)製)40質量部、ダイアナプロセスオイル(商品名「PW-90」出光興産(株)製)45質量部、酸化亜鉛(ZnO(亜鉛華)三井金属鉱業(株)製)5質量部、及びステアリン酸(CH3(CH2)16COOH、商品名「つばき」日油(株)製)1質量部を添加して混練して混練物Aを得た。当該混練物Aを1日以上、常温で保管して熟成させた。冷却(通水)されたロール機で前記熟成させた混練物Aを混練し、続いてジチオカルバミン酸塩系加硫促進剤(ジメチルジチオカルバミン亜鉛、商品名「ノクセラーPZ」大内新興化学工業(株)製)1.1質量部、ジチオカルバミン酸系加硫促進剤(ジブチルジチオカルバミン亜鉛、商品名「ノクセラーBZ」大内新興化学工業(株)製)1.7質量部を添加して混練し、続いて硫黄(商品名「微粉硫黄S」細井化学工業株式会社製)1.7質量部、酸化カルシウム(商品名「Fライム1300D」株式会社カルファイン製)5質量部、合成例1で得られたアミノグアニジンシュウ酸塩(2)(式(2)で表される化合物)5.25質量部、及び尿素系助剤(商品名「セルペーストK5」永和化成工業(株)製)6質量部を添加して混練してロール機から混練物Bを取り出した。当該混練物Bを押出機に投入し、ダイス温度50℃で押出成形し、シート状の成形体を得た。得られたシート状の成形体を1日常温で熟成(エージング)した後、180℃のオーブンで10分間加熱して発泡体を得た。得られた発泡剤の発泡倍率は1.280であった。なお、当該発泡倍率を算出するためのブランクとしてアミノグアニジンシュウ酸塩(2)を配合しなかった以外は上記混練物Bの製造方法と同様にして混練物Cを製造し、当該混練物Cの比重を測定した。当該発泡倍率は、混練物Cの比重(0.860g/cm3)/発泡体の比重(0.672g/cm3)により算出した。また、比重は、電子比重計 MD-200Sにより測定した。
Claims (8)
- 下記一般式(1)で表されるアミノグアニジン化合物のシュウ酸塩を含有する、ガス発生剤。

- 前記アミノグアニジン化合物のシュウ酸塩において、アミノグアニジン化合物とシュウ酸とのモル比(アミノグアニジン化合物:シュウ酸)が、1:1~2:1の範囲である、請求項1に記載のガス発生剤。
- 前記アミノグアニジン化合物がアミノグアニジンである、請求項1又は2に記載のガス発生剤。
- マイクロカプセル型である、請求項1~3のいずれか一項に記載のガス発生剤。
- 請求項1~4のいずれか一項に記載のガス発生剤と被発泡材料とを含有する、発泡用組成物。
- 被発泡材料が、熱可塑性樹脂及び/又はゴムである、請求項5に記載の発泡用組成物。
- 請求項5又は6に記載の発泡用組成物を加熱する工程を含む、発泡体の製造方法。
- 請求項5又は6に記載の発泡用組成物を発泡させた発泡体。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016563144A JP6118002B2 (ja) | 2015-04-23 | 2016-04-12 | ガス発生剤、及びそれを用いた発泡体の製造方法 |
| CN201680001766.9A CN106661423B (zh) | 2015-04-23 | 2016-04-12 | 气体发生剂以及使用该气体发生剂的发泡体的制造方法 |
| US15/567,910 US20180155277A1 (en) | 2015-04-23 | 2016-04-12 | Gas generating agent, and method for producing foam using the same |
| KR1020177031027A KR102477927B1 (ko) | 2015-04-23 | 2016-04-12 | 가스발생제, 및 이것을 이용한 발포체의 제조방법 |
| EP16783057.9A EP3196270B1 (en) | 2015-04-23 | 2016-04-12 | Gas-generating agent, and process for producing foamed object using same |
| US16/722,044 US11084782B2 (en) | 2015-04-23 | 2019-12-20 | Gas generating agent, and method for producing foam using the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015088657 | 2015-04-23 | ||
| JP2015-088657 | 2015-04-23 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/567,910 A-371-Of-International US20180155277A1 (en) | 2015-04-23 | 2016-04-12 | Gas generating agent, and method for producing foam using the same |
| US16/722,044 Division US11084782B2 (en) | 2015-04-23 | 2019-12-20 | Gas generating agent, and method for producing foam using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016171036A1 true WO2016171036A1 (ja) | 2016-10-27 |
Family
ID=57143066
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/061802 Ceased WO2016171036A1 (ja) | 2015-04-23 | 2016-04-12 | ガス発生剤、及びそれを用いた発泡体の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US20180155277A1 (ja) |
| EP (1) | EP3196270B1 (ja) |
| JP (1) | JP6118002B2 (ja) |
| KR (1) | KR102477927B1 (ja) |
| CN (1) | CN106661423B (ja) |
| WO (1) | WO2016171036A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018110842A1 (ko) * | 2016-12-12 | 2018-06-21 | 주식회사 동진쎄미켐 | 트리아미노 구아니딘 염 화합물을 포함하는 발포제 조성물 |
| WO2020188931A1 (ja) * | 2019-03-15 | 2020-09-24 | 永和化成工業株式会社 | ガス発生剤、発泡用組成物、発泡体、及び発泡体の製造方法 |
| JP2022535654A (ja) * | 2019-06-14 | 2022-08-10 | シーカ テクノロジー アクチェンゲゼルシャフト | 化学発泡剤を含む熱膨張性組成物 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102355601B1 (ko) | 2020-10-07 | 2022-02-07 | 주식회사 금양 | 친환경 발포제 조성물 및 이를 이용하여 제조된 친환경 발포체 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07309194A (ja) * | 1994-05-20 | 1995-11-28 | Sensor Technol Kk | エアバッグ用ガス発生剤 |
| JPH11314992A (ja) * | 1998-05-08 | 1999-11-16 | Daicel Chem Ind Ltd | ガス発生剤組成物 |
| JP2001139728A (ja) * | 1999-11-17 | 2001-05-22 | Bridgestone Corp | ゴム組成物 |
| WO2014061396A1 (ja) * | 2012-10-18 | 2014-04-24 | 株式会社ダイセル | ガス発生剤組成物 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1290418A (ja) * | 1969-12-26 | 1972-09-27 | ||
| JPS4830673B1 (ja) * | 1970-10-19 | 1973-09-21 | ||
| JPS5535359B2 (ja) | 1972-09-09 | 1980-09-12 | ||
| CN1132501A (zh) * | 1993-10-06 | 1996-10-02 | 尼古化学股份公司 | 气体发生器的推进剂 |
| US5756929A (en) | 1996-02-14 | 1998-05-26 | Automotive Systems Laboratory Inc. | Nonazide gas generating compositions |
| JPH1121364A (ja) | 1997-07-04 | 1999-01-26 | Pooren Kagaku Sangyo Kk | 発泡剤組成物及び発泡性熱可塑性重合体組成物 |
| JPH11292678A (ja) | 1998-04-15 | 1999-10-26 | Daicel Chem Ind Ltd | エアバッグ用ガス発生剤組成物 |
| JP2000086375A (ja) * | 1998-09-09 | 2000-03-28 | Daicel Chem Ind Ltd | ガス発生剤組成物 |
| US8012277B2 (en) * | 2007-04-13 | 2011-09-06 | Alliant Techsystems Inc. | Ionic liquid and a method of synthesizing an ionic liquid |
| JP5647606B2 (ja) * | 2009-06-19 | 2015-01-07 | 永和化成工業株式会社 | ガス発生剤 |
| EP2426160B1 (de) * | 2010-09-03 | 2014-01-15 | Sika Technology AG | Hitzehärtende Epoxidharzzusammensetzung mit Wasser als Treibmittel |
| DE102012222424A1 (de) * | 2012-12-03 | 2014-06-18 | Ludwig-Maximilians-Universität München | 3,3'-Dinitro-5,5'-Bistriazol-1,1'-diol |
| JP5924562B1 (ja) * | 2014-06-10 | 2016-05-25 | 三菱瓦斯化学株式会社 | 変性ゴム、それを用いたタイヤ用ゴム組成物及びタイヤ |
| US10173973B2 (en) * | 2014-06-10 | 2019-01-08 | Mitsubishi Gas Chemical Company, Inc. | Alkylidene aminoguanidine and salt thereof, modifying composition, modified rubber for tire, rubber composition for tire, and tire |
-
2016
- 2016-04-12 JP JP2016563144A patent/JP6118002B2/ja active Active
- 2016-04-12 US US15/567,910 patent/US20180155277A1/en not_active Abandoned
- 2016-04-12 EP EP16783057.9A patent/EP3196270B1/en active Active
- 2016-04-12 KR KR1020177031027A patent/KR102477927B1/ko active Active
- 2016-04-12 WO PCT/JP2016/061802 patent/WO2016171036A1/ja not_active Ceased
- 2016-04-12 CN CN201680001766.9A patent/CN106661423B/zh active Active
-
2019
- 2019-12-20 US US16/722,044 patent/US11084782B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07309194A (ja) * | 1994-05-20 | 1995-11-28 | Sensor Technol Kk | エアバッグ用ガス発生剤 |
| JPH11314992A (ja) * | 1998-05-08 | 1999-11-16 | Daicel Chem Ind Ltd | ガス発生剤組成物 |
| JP2001139728A (ja) * | 1999-11-17 | 2001-05-22 | Bridgestone Corp | ゴム組成物 |
| WO2014061396A1 (ja) * | 2012-10-18 | 2014-04-24 | 株式会社ダイセル | ガス発生剤組成物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3196270A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018110842A1 (ko) * | 2016-12-12 | 2018-06-21 | 주식회사 동진쎄미켐 | 트리아미노 구아니딘 염 화합물을 포함하는 발포제 조성물 |
| WO2020188931A1 (ja) * | 2019-03-15 | 2020-09-24 | 永和化成工業株式会社 | ガス発生剤、発泡用組成物、発泡体、及び発泡体の製造方法 |
| JPWO2020188931A1 (ja) * | 2019-03-15 | 2020-09-24 | ||
| US20220169815A1 (en) * | 2019-03-15 | 2022-06-02 | Eiwa Chemical Ind. Co., Ltd. | Gas generating agent, foamable composition, foam, and method of producing foam |
| JP2022535654A (ja) * | 2019-06-14 | 2022-08-10 | シーカ テクノロジー アクチェンゲゼルシャフト | 化学発泡剤を含む熱膨張性組成物 |
| JP7633942B2 (ja) | 2019-06-14 | 2025-02-20 | シーカ テクノロジー アクチェンゲゼルシャフト | 化学発泡剤を含む熱膨張性組成物 |
| US12534587B2 (en) | 2019-06-14 | 2026-01-27 | Sika Technology Ag | Thermally expandable compositions comprising a chemical blowing agent |
Also Published As
| Publication number | Publication date |
|---|---|
| CN106661423A (zh) | 2017-05-10 |
| US20200123101A1 (en) | 2020-04-23 |
| EP3196270B1 (en) | 2019-06-19 |
| EP3196270A1 (en) | 2017-07-26 |
| EP3196270A4 (en) | 2018-02-14 |
| US20180155277A1 (en) | 2018-06-07 |
| US11084782B2 (en) | 2021-08-10 |
| JPWO2016171036A1 (ja) | 2017-05-18 |
| CN106661423B (zh) | 2018-06-15 |
| KR102477927B1 (ko) | 2022-12-15 |
| JP6118002B2 (ja) | 2017-04-19 |
| KR20170140236A (ko) | 2017-12-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11084782B2 (en) | Gas generating agent, and method for producing foam using the same | |
| JP5392251B2 (ja) | ポリテトラフルオロエチレンファインパウダーの製造方法 | |
| JP4890415B2 (ja) | 金属シロキサン系化合物で表面処理された改質発泡剤、及びこれを含む高分子樹脂組成物 | |
| CN109414672A (zh) | 可热膨胀热塑性微球及其制备方法 | |
| CN107922519A (zh) | 改性聚四氟乙烯的水性乳化液、细粉及拉伸多孔体的制造方法 | |
| WO2015029916A1 (ja) | 熱膨張性微小球の製造方法 | |
| JP2010265421A (ja) | マイクロカプセル | |
| JP5647606B2 (ja) | ガス発生剤 | |
| JP6121638B2 (ja) | ガス発生剤、及びそれを用いた発泡体の製造方法 | |
| KR101518698B1 (ko) | 슬포니 하이드라이즈(Sulfonyl Hydrazide)를 포함하는 고분자 발포제의 제조방법 | |
| WO2006083041A1 (ja) | 熱発泡性マイクロスフェアー及びその製造方法並びに組成物 | |
| TW201333071A (zh) | 中分子量聚氧化伸烷之製造方法 | |
| WO2020188931A1 (ja) | ガス発生剤、発泡用組成物、発泡体、及び発泡体の製造方法 | |
| EP4454746A1 (en) | Expanded microspheres with excellent barrier properties | |
| JP5903225B2 (ja) | 発泡体製造用のガス発生剤 | |
| JPH08143697A (ja) | ポリプロピレン発泡体組成物 | |
| WO2006095567A1 (ja) | 内包物質の回収方法とその方法で得られうる新規な微小球 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016563144 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16783057 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2016783057 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016783057 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15567910 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20177031027 Country of ref document: KR Kind code of ref document: A |