WO2016194813A1 - エンザルタミド結晶形の製造方法 - Google Patents
エンザルタミド結晶形の製造方法 Download PDFInfo
- Publication number
- WO2016194813A1 WO2016194813A1 PCT/JP2016/065729 JP2016065729W WO2016194813A1 WO 2016194813 A1 WO2016194813 A1 WO 2016194813A1 JP 2016065729 W JP2016065729 W JP 2016065729W WO 2016194813 A1 WO2016194813 A1 WO 2016194813A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- enzalutamide
- solvent
- crystal
- type
- crystals
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/86—Oxygen and sulfur atoms, e.g. thiohydantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4166—1,3-Diazoles having oxo groups directly attached to the heterocyclic ring, e.g. phenytoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C331/00—Derivatives of thiocyanic acid or of isothiocyanic acid
- C07C331/16—Isothiocyanates
- C07C331/28—Isothiocyanates having isothiocyanate groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B33/00—After-treatment of single crystals or homogeneous polycrystalline material with defined structure
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B7/00—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the present invention relates to a novel method for producing enzalutamide crystal forms. It also relates to a novel method for producing the intermediate product.
- Enzalutamide (MDV3100) is a very useful oral androgen receptor inhibitor that can prevent castration-resistant prostate cancer from being promoted by androgen. Development of the enzalutamide drug substance in a solvent-free crystal form (hereinafter sometimes referred to as “A-type crystal”) is underway, but enzalutamide is a solvate that is a solvent addition form. The possibility that it is often formed in the process of conversion is suggested, and details thereof are not known (Patent Documents 1 and 2).
- 2-propanol and isopropyl acetate are mainly used as the final crystallization solvent, and when the solvent is used, in addition to A-form crystals of enzalutamide
- IPAc isopropyl acetate
- B-type crystal a crystal which is a 1 ⁇ 2 sum of 2-propanol
- it is necessary to go through a long drying process.
- it turned out that it may transfer from an A-type crystal to a B-type crystal by a drying process depending on drying conditions.
- a new enzalutamide in which 2-propanol itself and B-type crystals solvated with enzalutamide are reduced from the wet crystal. It is an object of the present invention to provide a method for producing a crystal form. Further, another object of the present invention is to verify other solvents that solvate with enzalutamide and their crystal forms, and to provide a method for producing a new enzalutamide crystal form in which these other crystal forms are also reduced.
- the inventors of the present invention newly included a washing step using a specific solvent after the crystallization step, thereby reducing the solvate in the form of a solvent addition form A-type crystals of enzalutamide Has been found to be able to be produced, and the present invention has been completed.
- the present invention relates to the following ⁇ 1> to ⁇ 7>.
- a method for producing an enzalutamide crystal form represented by the following formula including a crystallization step for obtaining a wet crystal of enzalutamide and a drying step for the wet crystal, wherein a good solvent and a poor solvent are provided after the crystallization step.
- the manufacturing method of the enzalutamide crystal form characterized by including the washing
- ⁇ 2> The method for producing an enzalutamide crystal form according to ⁇ 1>, wherein the washing step is performed before the drying step.
- ⁇ 3> The enzalutamide crystal form according to ⁇ 1> or ⁇ 2>, wherein the ratio of the good solvent to the poor solvent in the mixed solvent is from 1:99 to 99: 1 in a volume ratio.
- ⁇ 4> Any one of the above ⁇ 1> to ⁇ 3>, wherein the good solvent is at least one solvent selected from the group consisting of an acetic ester organic solvent, acetone, methyl ethyl ketone, tetrahydrofuran, and acetonitrile.
- ⁇ 5> Any one of the above ⁇ 1> to ⁇ 4>, wherein the poor solvent is at least one solvent selected from the group consisting of a hydrocarbon-based organic solvent, water, and methyl-tert-butyl ether.
- ⁇ 6> The method for producing an enzalutamide crystal form according to any one of ⁇ 1> to ⁇ 5>, wherein the good solvent is isopropyl acetate and the poor solvent is n-heptane.
- Thiophosgene is dissolved in a mixed solvent of a hydrocarbon-based organic solvent or a chlorine-based organic solvent and water, and 4-cyano-3-trifluoromethylaniline is converted into a hydrocarbon-based organic solvent or a chlorine-based organic solvent.
- a method for producing 4-cyano-3-trifluoromethylphenyl isothiocyanate comprising the step of dropping a solution dissolved in
- crystals (B-type crystals) that are 1 ⁇ 2 sum of enzalutamide 2-propanol are reduced without going through a high temperature and long drying step.
- a solvent-free enzalutamide crystal form (A-type crystal) can be obtained.
- enzalutamide A-type crystals in which the crystal form of enzalutamide, which is a solvate with a solvent other than 2-propanol, is also reduced can be obtained.
- FIG. 1 is a powder X-ray diffraction spectrum of an enzalutamide type A crystal.
- FIG. 2 is a powder X-ray diffraction spectrum of B-type crystals of enzalutamide.
- FIG. 3 is a powder X-ray diffraction spectrum of a C-type crystal of enzalutamide.
- FIG. 4 is a powder X-ray diffraction spectrum of D-form crystal of enzalutamide.
- FIG. 5 is a powder X-ray diffraction spectrum of an enzalutamide E-type crystal.
- FIG. 6 is a powder X-ray diffraction spectrum of F-type crystal of enzalutamide.
- FIG. 7 is scanning electron micrographs of enzalutamide A-type and B-type crystals.
- the present invention is a method for producing an enzalutamide crystal form represented by the following formula, including a crystallization step for obtaining a wet crystal of enzalutamide and a drying step for the wet crystal, wherein a good solvent and a poor solvent are provided after the crystallization step.
- a crystallization step for obtaining a wet crystal of enzalutamide
- a drying step for the wet crystal, wherein a good solvent and a poor solvent are provided after the crystallization step.
- Enzalutamide can be produced, for example, by the following reaction. That is, 2- (3-fluoro-4-methylcarbamoyl-phenylamino) -2-methyl-propionic acid methyl ester (hereinafter sometimes referred to as “compound (A)”) and 4-cyano-3-tri It can be obtained by heating and reacting a mixture of fluoromethylphenyl isothiocyanate (hereinafter sometimes referred to as “compound (B)”) and dimethyl sulfoxide (DMSO).
- compound (A) 2- (3-fluoro-4-methylcarbamoyl-phenylamino) -2-methyl-propionic acid methyl ester
- compound (B) 4-cyano-3-tri It can be obtained by heating and reacting a mixture of fluoromethylphenyl isothiocyanate (hereinafter sometimes referred to as “compound (B)”) and dimethyl sulfoxide (DMSO).
- Both compound (A) and compound (B) can be synthesized by a known method described in, for example, Patent Document 2, but it is difficult to control impurities, particularly dimer impurities, in the synthesis of compound (B). . Therefore, instead of the conventional method of dropping thiophosgene into a mixed solution of 4-cyano-3-trifluoromethylaniline in heptane-water, the following dropping method is used to control compound (B) with good impurity control. Since it can obtain, it is more preferable.
- Solvents for dissolving thiophosgene are hydrocarbon organic solvents such as hexane, ether organic solvents such as ether, acetate organic solvents such as ethyl acetate and isopropyl acetate, chlorine organic solvents such as methylene chloride and water. And a mixed solvent of an acetate solvent such as ethyl acetate and isopropyl acetate and water is more preferable.
- the mixing ratio of the acetate solvent and water is preferably 0.1: 1 to 20: 1 by volume.
- the solvent of 4-cyano-3-trifluoromethylaniline is a common hydrocarbon-based organic solvent such as hexane, an ether-based organic solvent such as ether, an acetate-based organic solvent such as ethyl acetate or isopropyl acetate, Chlorinated organic solvents such as methylene chloride are preferred, and acetate solvents such as ethyl acetate and isopropyl acetate are more preferred.
- the concentration of 4-cyano-3-trifluoromethylaniline in an acetic acid ester solvent solution is preferably 1 g / mL or less.
- the dropping speed of the acetate solvent solution of 4-cyano-3-trifluoromethylaniline is preferably 10 L or less per minute.
- the reaction temperature after the dropwise addition is preferably ⁇ 10 to 50 ° C., more preferably 0 to 30 ° C.
- the reaction time is preferably 0.1 to 24 hours, more preferably 1 hour or longer.
- the aqueous solution added to the organic layer may be water or a basic aqueous solution containing a salt such as sodium / potassium, and an aqueous potassium hydrogen carbonate solution is more preferable. Water may be added instead of the hydrocarbon solvent added after removing the water layer and concentrating.
- enzalutamide can be produced by sequentially performing the following steps. a. Dissolving compound (A) and compound (B) in a mixed solvent of DMSO and IPAc and stirring; b. Subsequently, 2-propanol (IPA) is dropped, c. Separating the organic layer, d. Adding a seed crystal of enzalutamide (A-type crystal) to the sorted organic layer; e. Obtaining wet crystals of enzalutamide, and f. A step of drying the wet crystal.
- step e. B type crystals are reduced by performing a washing step using a mixed solvent of a good solvent and a poor solvent after a step of obtaining wet crystals of enzalutamide (hereinafter sometimes referred to as “crystallization step”).
- the obtained solvent-free enzalutamide crystal form (A-type crystal) can be obtained.
- the washing step is performed before the step of drying the wet crystal (hereinafter sometimes referred to as “drying step”), the B-type crystal is easily transferred to the A-type crystal by a drying step under mild conditions. It is preferable because it can be made.
- the A-type crystals are easily transitioned to B-type crystals even at room temperature by suspending them in the presence of IPA. For this reason, there is a risk of transition from the A-type crystal to the B-type crystal in any state, such as between the crystallization process or between the crystallization process and the drying process, during the drying process, or after the drying process. Therefore, in order to stably obtain an A-type crystal of enzalutamide with a reduced B-type crystal, it is possible to transfer from the B-type crystal to the A-type crystal by long-time heating and drying under reduced pressure. It was necessary to remove the IPA.
- the B-type crystal can be easily transferred to the A-type crystal by washing the wet crystal with a mixed solvent of a good solvent and a poor solvent. Furthermore, residual IPA in the wet crystal that causes the transition to the B-type crystal can also be removed by the cleaning. Therefore, the transition from the A-type crystal to the B-type crystal can be prevented, and the A-form of enzalutamide in which the B-type crystal is completely reduced through the drying process under a mild condition without going through a drying process at a high temperature for a long time. A type crystal can be obtained.
- the B-type crystal in the system is transferred to the A-type crystal by solvent-mediated transfer.
- IPA remaining in the wet crystal is removed to the filtrate by solvent replacement with the mixed solvent.
- the good solvent in the mixed solvent is a solvent having a crystal solubility of 10 g / L or more at 25 ° C., and the solubility is preferably 30 g / L or more.
- Specific examples include acetate organic solvents such as ethyl acetate and isopropyl acetate, acetone, methyl ethyl ketone, tetrahydrofuran, and acetonitrile. Of these, isopropyl acetate is preferred from the viewpoint of solubility.
- the good solvent may be one type or two or more types.
- the poor solvent in the mixed solvent is a solvent having a crystal solubility of less than 5 g / L at 25 ° C., and the solubility is preferably less than 1 g / L.
- Specific examples include hydrocarbon organic solvents such as n-heptane and cyclohexane, water, and methyl-tert-butyl ether. Of these, n-heptane is preferred from the viewpoint of solubility.
- the poor solvent may be one type or two or more types.
- the volume ratio of the good solvent and the poor solvent is preferably 1:99 to 99: 1 in terms of solubility, more preferably 5:95 to 40:60, and further preferably 25:75 to 35:65. preferable.
- the combination of the good solvent and the poor solvent is most preferably a mixed solvent of isopropyl acetate and n-heptane in a volume ratio of 30:70.
- the wet crystals are suspended in the mixed solvent and stirred.
- the stirring time is preferably 5 minutes or more, more preferably 10 minutes or more, and further preferably 15 minutes or more. Further, stirring for about 1 hour as the upper limit is sufficient because the B-type crystal is transferred to the A-type crystal.
- the washing step may be performed once, but after washing, the liquid may be removed and the washing may be repeated twice or more. By repeating washing and liquid removal, the transition from the B-type crystal to the A-type crystal and the removal of the remaining IPA can be made more complete. It is not necessary to heat at the time of stirring, preferably 0 to 40 ° C, more preferably 5 to 30 ° C.
- drying step drying under reduced pressure is preferably performed at an external temperature of 25 to 65 ° C., and an external temperature of 45 to 55 ° C. is more preferable.
- the drying time is preferably 1 to 68 hours.
- the solvent used for a crystallization process is made into the mixed solvent of a good solvent and a poor solvent instead of IPA, The desired A-form crystal
- the enzalutamide crystal form includes a solvent-free A-type crystal, a B-type crystal that is a 1 ⁇ 2 sum of 2-propanol, and a C-type crystal that is a 1 ⁇ 2 sum of methanol depending on the solvent used.
- a D-type crystal that is a monohydrate of dioxane
- an E-type crystal that is a 1 ⁇ 2 sum of dioxane
- an F-type crystal that is a monohydrate of dimethyl sulfoxide.
- suspension purification using a mixed solvent of a good solvent and a poor solvent allows the transition from C-type crystals to E-type crystals to A-type crystals. Can be made.
- the solvents constituting the C-type crystal to the E-type crystal that is, methanol and dioxane can be removed from the wet crystal.
- the mixed solvent used in the washing step for obtaining the enzalutamide A-type crystals with reduced C-type crystals to E-type crystals should be the same as the mixed solvent used to reduce the B-type crystals. it can.
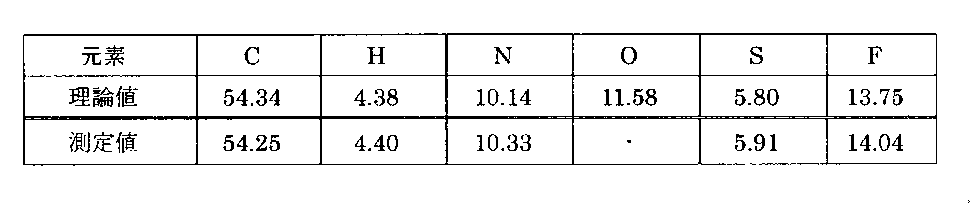
- Identification of enzalutamide A-type, B-type and C-type to F-type crystals can be performed by 1 H-NMR, elemental analysis, powder X-ray scattering (XRD), and differential scanning calorimetry (DSC). ) To confirm the thermophysical properties.
- the present invention will be specifically described below with reference to examples, but the present invention is not limited to these.
- ⁇ Evaluation method> the obtained crystal form is 1 H-NMR measurement with a nuclear magnetic resonance apparatus (JEOL, JNM-ECS400, 400 MHz), XRD measurement with a powder X-ray diffractometer (Rigaku, Miniflex), elemental analysis apparatus. (Measurement by Elemental Co., Micro cube and Thermo Fisher Scientific Co., Ltd., ion chromatogram ICS-3000), DSC measurement by differential scanning calorimeter (Ta Instruments, Q2000 V24.4 Build 116) went.
- DMSO-d 6 was used for A-type crystals to E-type crystals, and CDCl 3 -d 6 was used for F-type crystals.
- the conditions for DSC measurement were as follows. Temperature range: 20-230 ° C, sweep rate: 10 ° C / min, measurement atmosphere: N 2 gas (40 mL / min), stainless steel sample pan, completely sealed
- Example 1 Synthesis of 4-cyano-3-trifluoromethylphenyl isothiocyanate Isopropyl acetate (IPAc) (20 mL) / aqueous solution (56 mL) of thiophosgene (14.9 g) was prepared, and 4-cyano-3 A solution of trifluoromethylaniline (20 g) in IPAc solution (90 mL) was added dropwise over 30 minutes. The internal temperature was 4 ° C. The mixture was stirred as it was at an internal temperature of 4 ° C. for 5 minutes, and then allowed to stand for 30 minutes or more to separate the aqueous layer.
- IPAc aqueous solution
- IPAc solution 90 mL
- Example 2 Synthesis of enzalutamide type A crystals Under a nitrogen atmosphere, 2- (3-fluoro-4-methylcarbamoyl-phenylamino) -2-methyl-propionic acid methyl ester (33.0 g) and obtained in Example 1
- the obtained 4-cyano-3-trifluoromethylphenyl isothiocyanate (56.1 g) was dissolved in a mixed solvent of dimethyl sulfoxide (DMSO) (33 mL) / IPAc (66 mL), and the internal temperature was raised to 75 to 85 ° C. The mixture was stirred for 12 hours or more at the same temperature.
- DMSO dimethyl sulfoxide
- methanol (4.95 mL) was added dropwise at 55 to 80 ° C., and the mixture was stirred at the same temperature for 60 to 90 minutes. Thereafter, the mixture was cooled to 15 to 25 ° C., diluted with IPAc (198 mL) and purified water (99 mL), stirred at the same temperature for 10 to 30 minutes, and then allowed to stand for 30 to 45 minutes.
- 2-Propanol (IPA) (49.5 mL) was slowly added dropwise at an internal temperature of 15 to 25 ° C. to break the emulsion. The organic layer was separated and the line was washed with IPAc (15 mL).
- the separated organic layer was concentrated under reduced pressure until the liquid volume was around 165 mL.
- the concentrated liquid was heated to 80 to 85 ° C. and stirred at the same temperature for 30 to 60 minutes to completely dissolve the suspension.
- IPA 330 mL
- Atmospheric concentration was carried out until the liquid volume was around 660 mL.
- IPA 165 mL
- Atmospheric concentration was carried out until the liquid volume was around 264 mL.
- the internal temperature was adjusted to 75 to 85 ° C., and seed crystals were added.
- the mixture was cooled to an internal temperature of 55 to 65 ° C. at 10 to 20 ° C./hour, and stirred at the same temperature for 30 to 60 minutes. Subsequently, the internal temperature was cooled to 0 to 10 ° C. at 10 to 20 ° C./hour. After confirming that the internal temperature was 0-10 ° C., the slurry was transferred to a filter and filtered. After filtration, washing with IPA (138 mL) was performed twice.
- Example 3 Transition from enzalutamide B-type crystals to A-type crystals Under a nitrogen atmosphere, isopropyl acetate (3.0 mL) as a good solvent for enzalutamide B-type crystals (2.0 g) and n-heptane as a poor solvent ( 7.0 mL) was stirred at an internal temperature of 20 to 30 ° C. for 1 hour or longer. After stirring, the precipitated crystals were collected by filtration and washed with n-heptane (4.0 mL). The obtained crystals were dried under reduced pressure at 55 ° C. for 3 hours to obtain enzalutamide A-type crystals.
- the IPA content corresponds to 1 ⁇ 2 molar equivalent of the B-type crystal content.
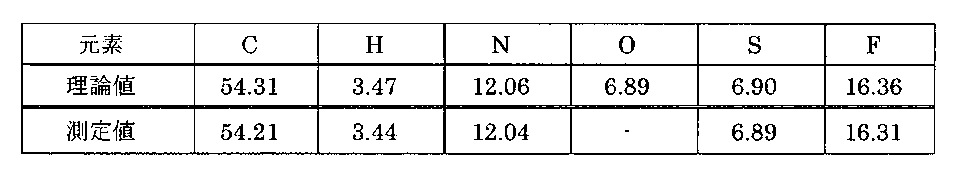
- Table 3 From this, it was found that even if the washing step using a mixed solvent of a good solvent and a poor solvent was performed for 1 minute, the enzalutamide B type crystal was transferred to the A type crystal. In addition, if the cleaning process was 5 minutes or more, 95% or more of the B-type crystals were transferred to the A-type crystals, and if 15 minutes or more, the B-type crystals were completely transferred to the A-type crystals. In Table 3, “ND” indicates that it was not detected.
- Example 4 Transition from enzalutamide C-type crystals to A-type crystals Under a nitrogen atmosphere, isopropyl acetate (0.45 mL) as a good solvent for enzalutamide C-type crystals (0.3 g) and n-heptane as a poor solvent ( 1.05 mL) was stirred at an internal temperature of 50 to 60 ° C. for 1 hour or longer. After stirring, the mixture was cooled to an internal temperature of 20 to 30 ° C. and stirred at the same temperature for 30 minutes or more. The precipitated crystals were collected by filtration and washed with n-heptane (1.0 mL). The obtained crystals were dried under reduced pressure at 25 ° C. for 4 hours to obtain enzalutamide A-type crystals.
- Example 5 Transition from enzalutamide D-type crystals to A-type crystals Under a nitrogen atmosphere, isopropyl acetate (0.45 mL) as a good solvent for enzalutamide D-type crystals (0.3 g) and n-heptane as a poor solvent ( 1.05 mL) was stirred at an internal temperature of 70 to 80 ° C. for 1 hour or longer. After stirring, the mixture was cooled to an internal temperature of 20 to 30 ° C. and stirred at the same temperature for 30 minutes or more. The precipitated crystals were collected by filtration and washed with n-heptane (1.0 mL). The obtained crystals were dried under reduced pressure at 25 ° C. for 2 hours to obtain enzalutamide A-type crystals.
- Example 6 Transition from enzalutamide E-type crystals to A-type crystals Under a nitrogen atmosphere, a mixed solution of enzalutamide E-type crystals (0.2 g), which is a good solvent, isopropyl acetate and a poor solvent, n-heptane, was heated at an internal temperature. The mixture was stirred at 70 to 80 ° C. for 1 hour or longer. After stirring, the mixture was cooled to an internal temperature of 20 to 30 ° C. and stirred at the same temperature for 30 minutes or more. The precipitated crystals were collected by filtration and washed with n-heptane (1.0 mL). The obtained crystals were dried under reduced pressure at 25 ° C. for 4 hours to obtain enzalutamide A-type crystals.
- solvent-free crystals of enzalutamide with reduced solvated crystals can be obtained under mild conditions.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Crystallography & Structural Chemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Glass Compositions (AREA)
- Crystals, And After-Treatments Of Crystals (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
Abstract
Description
エンザルタミド原薬の形態として、溶媒フリーである結晶形(以下、「A型結晶」と称することもある。)で開発が進められているが、エンザルタミドは、溶媒付加形態である溶媒和物が結晶化の過程でしばしば形成される可能性が示唆されており、その詳細については分かっていない(特許文献1及び2)。
そこで本発明では、エンザルタミド結晶形の製造過程における晶析工程でエンザルタミドのウェット結晶を得た後、当該ウェット結晶から、エンザルタミドと溶媒和する2-プロパノールそのもの及びB型結晶が低減された新たなエンザルタミド結晶形の製造方法を提供することを課題とする。さらには、エンザルタミドと溶媒和する他の溶媒及びその結晶形についても検証し、それら他の結晶形も低減された、新たなエンザルタミド結晶形の製造方法を提供することを課題とする。
<1>エンザルタミドのウェット結晶を得る晶析工程及び前記ウェット結晶の乾燥工程を含む、下記式で表されるエンザルタミド結晶形の製造方法であって、前記晶析工程の後に、良溶媒と貧溶媒との混合溶媒を用いた洗浄工程を含むことを特徴とするエンザルタミド結晶形の製造方法。

<3>前記混合溶媒における前記良溶媒と前記貧溶媒の割合が体積比で、1:99~99:1であることを特徴とする前記<1>又は<2>に記載のエンザルタミド結晶形の製造方法。
<4>前記良溶媒が酢酸エステル系有機溶媒、アセトン、メチルエチルケトン、テトラヒドロフラン及びアセトニトリルからなる群より選ばれる少なくとも1の溶媒であることを特徴とする前記<1>~<3>のいずれか1に記載のエンザルタミド結晶形の製造方法。
<5>前記貧溶媒が炭化水素系有機溶媒、水及びメチル-tert-ブチルエーテルからなる群より選ばれる少なくとも1の溶媒であることを特徴とする前記<1>~<4>のいずれか1に記載のエンザルタミド結晶形の製造方法。
<6>前記良溶媒が酢酸イソプロピルであり、前記貧溶媒がn-ヘプタンである、前記<1>~<5>のいずれか1に記載のエンザルタミド結晶形の製造方法。
<7>チオホスゲンを炭化水素系の有機溶媒又は塩素系有機溶媒と、水との混合溶媒に溶解して、4-シアノ-3-トリフルオロメチルアニリンを炭化水素系の有機溶媒又は塩素系有機溶媒で溶解した溶液を滴下する工程を含むことを特徴とする、4-シアノ-3-トリフルオロメチルフェニルイソチオシアネートの製造方法。
さらには、2-プロパノール以外の溶媒との溶媒和物であるエンザルタミドの結晶形も低減された、エンザルタミドのA型結晶を得ることができる。
また、本明細書において、“重量%”と“質量%”とは同じ意味を表す。


当該反応は終始、液-液の二層系で進み、過剰反応成績体を含め不純物を良好にコントロールすることができることから、化合物(B)を高収率で得ることができる。
酢酸エステル系溶媒と水の混合比は体積比で0.1:1~20:1が好ましい。
4-シアノ-3-トリフルオロメチルアニリンの酢酸エステル系溶媒溶液の滴下速度は毎分10L以下が好ましい。
有機層に加える水溶液は水もしくは、ナトリウム・カリウム等の塩を含む塩基性水溶液であればよく、炭酸水素カリウム水溶液がより好ましい。
水層を除去して濃縮した後に加える炭化水素系溶媒に代えて、水を加えてもよい。
a.化合物(A)及び化合物(B)をDMSOとIPAcの混合溶媒に溶解させ、攪拌する工程、
b.次いで2-プロパノール(IPA)を滴下する工程、
c.有機層を分取する工程、
d.前記分取した有機層にエンザルタミド(A型結晶)の種晶を添加する工程、
e.エンザルタミドのウェット結晶を得る工程、及び
f.ウェット結晶を乾燥する工程。
しかしながら、晶析工程後に、ウェット結晶を良溶媒と貧溶媒との混合溶媒を用いて洗浄することにより、B型結晶をA型結晶に容易に転移させることができる。さらには、B型結晶への転移の要因となるウェット結晶中の残留IPAも当該洗浄により除去することができる。そのため、A型結晶からB型結晶への転移を防ぐことができ、高温かつ長時間の乾燥工程を経ることなく、温和な条件の乾燥工程を経て完全にB型結晶が低減されたエンザルタミドのA型結晶を得ることができる。
具体的には、酢酸エチルや酢酸イソプロピルを始めとする酢酸エステル系有機溶媒、アセトン、メチルエチルケトン、テトラヒドロフラン、アセトニトリルなどが挙げられる。中でも酢酸イソプロピルが溶解度の点から好ましい。良溶媒は1種でも2種以上であってもよい。
具体的には、n-ヘプタンやシクロヘキサンを始めとする炭化水素系有機溶媒、水、メチル-tert-ブチルエーテルなどが挙げられる。中でもn-ヘプタンが溶解度の点から好ましい。貧溶媒は1種でも2種以上であってもよい。
洗浄工程は一度行えばよいが、洗浄した後に脱液をして、二度以上繰り返して洗浄を行ってもよい。洗浄と脱液を繰り返すことにより、B型結晶からA型結晶への転移と、残留しているIPAの除去を、より完全にすることができる。
攪拌時は加熱する必要はなく、0~40℃が好ましく、5~30℃がより好ましい。
乾燥工程は外温25~65℃にて減圧乾燥をすることが好ましく、外温45~55℃がより好ましい。乾燥時間は1~68時間が好ましい。
これらC型結晶~E型結晶についてもB型結晶と同様に、良溶媒と貧溶媒との混合溶媒を用いた懸濁精製を行うことにより、C型結晶~E型結晶からA型結晶へ転移させることができる。また、C型結晶~E型結晶を構成する溶媒、すなわち、メタノール、ジオキサンをウェット結晶から除去することができる。
<評価方法>
本実施例において、得られた結晶形は核磁気共鳴装置(JEOL製、JNM-ECS400、400MHz)による1H-NMR測定、粉末X線回折装置(リガク製、Miniflex)によるXRD測定、元素分析装置(エレメンタール社製、Micro cube及びサーモフィッシャーサイエンティフィック社製、イオンクロマトグラムICS-3000)による測定、示差走査熱量分析装置(Taインスツルメント製、Q2000 V24.4 Build 116)によるDSC測定を行った。
1H-NMRの溶媒はA型結晶~E型結晶についてはDMSO-d6を、F型結晶についてはCDCl3-d6を用いた。
XRD測定の条件は以下のとおりとした。
X線:CuKα、電圧-電流:30kV-15mA、測定範囲:2θ=0~35°、スキャンスピード:2°/分、発散スリット幅:可変、散乱スリット幅:4.2°、受光スリット幅:0.3mm、測定誤差:±0.5°
DSC測定の条件は以下のとおりとした。
温度範囲:20~230℃、掃引速度:10℃/分、測定雰囲気:N2ガス(40mL/分)、ステンレス製サンプルパン、完全密封
チオホスゲン(14.9g)の酢酸イソプロピル(IPAc)(20mL)/水溶液(56mL)を調製し、そこに4-シアノ-3-トリフルオロメチルアニリン(20g)をIPAc溶液(90mL)に溶解させたものを、30分かけて滴下した。内温は4℃だった。そのまま内温4℃にて5分間攪拌した後、30分以上静置し、水層を分離した。得られた有機層を減圧下濃縮し、濃縮残渣にn-ヘプタンを加えさらに減圧下80mL以下になるまで濃縮した。得られた濃縮残渣にIPAc(1mL)を加え内温40℃にて5分間撹拌した後、25℃にて種晶(10mg)を加え1時間撹拌し、内温4℃にて撹拌した後、ろ過することにより、4-シアノ-3-トリフルオロメチルフェニルイソチオシアネート(22.1g)を得た。
窒素雰囲気下、2-(3-フルオロ-4-メチルカルバモイル-フェニルアミノ)-2-メチル-プロピオン酸メチルエステル(33.0g)及び実施例1で得られた4-シアノ-3-トリフルオロメチルフェニルイソチオシアネート(56.1g)をジメチルスルホキシド(DMSO)(33mL)/IPAc(66mL)の混合溶媒に溶解し、内温75~85℃まで昇温させ、同温にて12時間以上攪拌した。反応終了後、55~80℃にてメタノール(4.95mL)を滴下し、同温にて60~90分攪拌した。その後、15~25℃に冷却し、IPAc(198mL)、精製水(99mL)にて希釈し、同温にて10~30分攪拌した後、30~45分静置した。内温15~25℃にて2-プロパノール(IPA)(49.5mL)をゆっくりと滴下しエマルジョンを破壊した。有機層を分取し、IPAc(15mL)にてラインの洗浄を実施した。
濃縮終了液に、60~70℃にて予め清澄濾過を実施したIPA(330mL)を70℃以上に保ちながら添加した。液量が660mL付近になるまで常圧濃縮を実施した。濃縮終了液に、60~70℃にて予め清澄濾過を実施したIPA(165mL)を70℃以上に保ちながら添加した。液量が264mL付近になるまで常圧濃縮を実施した。
内温を75~85℃に調整し、種晶を添加した。10~20℃/時間にて内温55~65℃へと冷却し、同温度にて30~60分攪拌した。引き続き、10~20℃/時間にて内温0~10℃へと冷却した。内温が0~10℃になったことを確認した後、スラリーを濾過機に移送し濾過を実施した。濾過後、IPA(138mL)による洗浄操作を2度実施した。
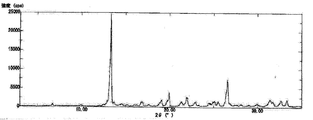
1H-NMR(DMSO-d6,400MHz):δ(ppm)=1.55(6H,s),2.81(3H,d,J=4.8Hz),7.34(1H),7.43(1H),7.79(1H),8.09(1H),8.30(1H),8.41(1H),8.46(1H)
XRD:2θ(°)=13.2,16.7,18.9,19.8,21.2,21.8,25.4,26.4
窒素雰囲気下、エンザルタミドA型結晶の精結晶(10.0g)のIPA(80mL)溶液を室温にて撹拌した。20~30℃にて、エンザルタミドB型結晶の種晶(10.2mg=0.1質量%)を添加し、同温度にて4日間撹拌した。撹拌後、析出した結晶をろ取した。その後、IPA(10mL)にて洗浄し、55℃にて4時間程度減圧乾燥を実施してIPAの1/2和物であるエンザルタミドB型結晶を10.3g得た。収率は96.2%だった。
1H-NMR(DMSO-d6,400MHz):δ(ppm)=1.04(3H,d,J=6.0Hz),1.55(6H,s),2.81(3H,d,J=4.8Hz),3.77(0.5H,m),4.36(0.5H,d,J=4.4Hz),7.34(1H),7.43(1H),7.79(1H),8.09(1H),8.30(1H),8.41(1H),8.46(1H)
XRD:2θ(°)=4.6,7.4,9.1,10.8,13.6,14.8,16.2,20.9,23.4,25.6

窒素雰囲気下、エンザルタミドB型結晶(2.0g)の良溶媒である酢酸イソプロピル(3.0mL)及び貧溶媒であるn-ヘプタン(7.0mL)の混合溶液を内温20~30℃にて1時間以上撹拌した。撹拌後、析出している結晶をろ取し、n-ヘプタン(4.0mL)にて洗浄した。得られた結晶を55℃にて3時間減圧乾燥を行い、エンザルタミドのA型結晶を得た。
良溶媒である酢酸イソプロピル(3.0mL)及び貧溶媒であるn-ヘプタン(7.0mL)の混合溶液を用いた攪拌時間を1分、5分、15分、30分又は1時間に変更した以外は、上記(実施例3)と同様にして、エンザルタミドのA型結晶を得た。
得られた結晶のXRDスペクトルのうち、A型結晶に由来するピークとIPAに由来するピークの各々の積分値から、IPAの含有量と、A型結晶とB型結晶の存在比率を求めた。なお、B型結晶はIPAの1/2和物であることから、IPAの含有量はB型結晶の含有量の1/2モル当量に相当する。結果を表3に示す。これより、良溶媒と貧溶媒との混合溶媒を用いた洗浄工程が1分であっても、エンザルタミドのB型結晶からA型結晶に転移することが分かった。また、洗浄工程が5分以上であればB型結晶のうち95%以上がA型結晶に転移し、15分以上であればB型結晶から完全にA型結晶に転移する結果となった。なお、表3中「N.D.」は未検出であったことを示す。
窒素雰囲気下、エンザルタミドA型結晶の精結晶(10.0g)のメタノール(45mL)溶液を内温50~60℃に昇温し、同温度にて15分間撹拌した。撹拌後、内温50℃付近にてn-ヘプタン(105mL)を45分間かけて滴下し、同温度にて15分以上撹拌した。撹拌後,内温20~30℃まで冷却し、同温度にて終夜撹拌した。析出した沈殿物を取得するため、デカンテーションを実施し更に、40℃以下にて減圧濃縮を行い、溶媒を完全に除去した。
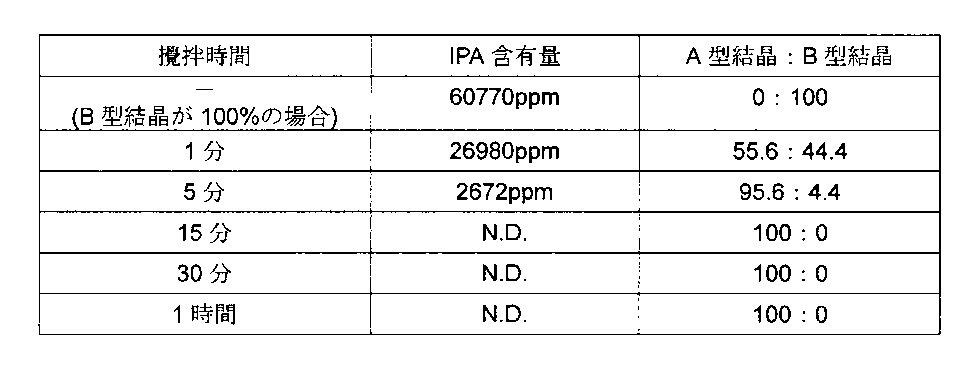
得られた結晶を、室温下減圧乾燥を実施して、メタノールの1/2和物であるエンザルタミドC型結晶を8.86g得た。収率は86.0%だった。
1H-NMR(DMSO-d6,400MHz):δ(ppm)=1.55(6H,s),2.81(3H,d,J=4.8Hz),3.17(1.5H,d,J=5.2Hz),4.11(0.5H,q,J=5.2Hz),7.34(1H),7.43(1H),7.79(1H),8.09(1H),8.30(1H),8.41(1H),8.46(1H)
XRD:2θ(°)=4.8,9.6,11.2,13.8,15.8,16.7,18.1,22.6,24.2,25.4
窒素雰囲気下、エンザルタミドC型結晶(0.3g)の良溶媒である酢酸イソプロピル(0.45mL)及び貧溶媒であるn-ヘプタン(1.05mL)の混合溶液を内温50~60℃にて1時間以上撹拌した。撹拌後、内温20~30℃へと冷却し、同温度にて30分以上攪拌した。析出している結晶をろ取し、n-ヘプタン(1.0mL)にて洗浄した。得られた結晶を25℃にて4時間減圧乾燥を行い、エンザルタミドのA型結晶を得た。
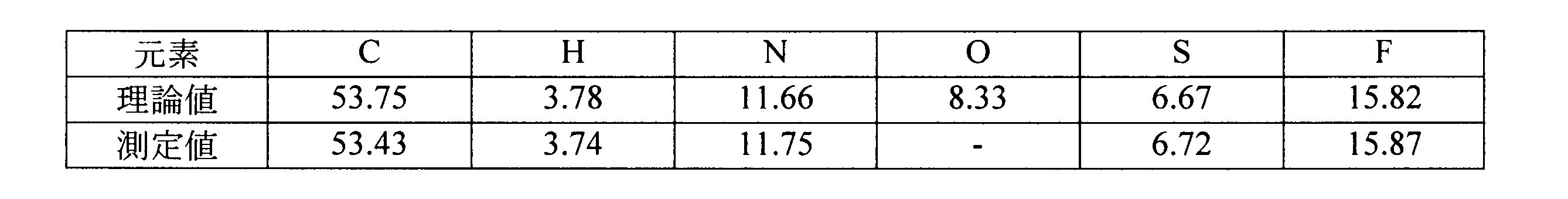
窒素雰囲気下、エンザルタミドA型結晶の精結晶(6.0g)のジオキサン(30mL)溶液を内温70℃付近に昇温し、溶解を確認した。溶解を確認後、15℃付近までゆっくり冷却した。このとき、20℃付近で結晶の析出が見られた。内温15℃にて終夜撹拌して析出した結晶をろ取した。その後、ジオキサン(6mL)にて洗浄し、室温下1時間、減圧乾燥を実施してジオキサンの1和物であるエンザルタミドD型結晶を4.72g得た。収率は66.1%だった。
1H-NMR(DMSO-d6,400MHz):δ(ppm)=1.55(6H,s),2.81(3H,d,J=4.8Hz),3.57(8H,s),7.34(1H),7.43(1H),7.79(1H),8.09(1H),8.30(1H),8.41(1H),8.46(1H)
XRD:2θ(°)=10.7,14.1,14.8,15.4,18.2,21.4,24.1
窒素雰囲気下、エンザルタミドD型結晶(0.3g)の良溶媒である酢酸イソプロピル(0.45mL)及び貧溶媒であるn-ヘプタン(1.05mL)の混合溶液を内温70~80℃にて1時間以上撹拌した。撹拌後、内温20~30℃へと冷却し、同温度にて30分以上攪拌した。析出している結晶をろ取し、n-ヘプタン(1.0mL)にて洗浄した。得られた結晶を25℃にて2時間減圧乾燥を行い、エンザルタミドのA型結晶を得た。
窒素雰囲気下、エンザルタミドA型結晶の精結晶(10.0g)のジオキサン(30mL)溶液を内温55℃に昇温し、同温下にて撹拌を起動した。内温50~60℃にてn-ヘプタン(70mL)を45分間以上かけて滴下した。滴下終了後、内温20~30℃に冷却し同温度にて終夜撹拌した。撹拌後、析出した結晶をろ取した。n-ヘプタンにて洗浄を実施した後、室温下2時間減圧乾燥を実施し、ジオキサンの1/2和物であるエンザルタミドE型結晶を10.71g得た。収率は97.8%だった。
1H-NMR(DMSO-d6,400MHz):δ(ppm)=1.55(6H,s),2.80(3H,d,J=4.8Hz),3.57(4H,s),7.34(1H),7.43(1H),7.79(1H),8.09(1H),8.30(1H),8.41(1H),8.46(1H)
XRD:2θ(°)=11.7,13.3,17.5,20.9,23.6,29.0

窒素雰囲気下、エンザルタミドE型結晶(0.2g)の良溶媒である酢酸イソプロピル及び貧溶媒であるn-ヘプタンの混合溶液を内温70~80℃にて1時間以上撹拌した。撹拌後、内温20~30℃へと冷却し、同温度にて30分以上攪拌した。析出している結晶をろ取し、n-ヘプタン(1.0mL)にて洗浄した。得られた結晶を25℃にて4時間減圧乾燥を行い、エンザルタミドのA型結晶を得た。
窒素雰囲気下、エンザルタミドA型結晶の精結晶(30.0g)のDMSO(30mL)溶液を内温100℃付近に昇温し、結晶の溶解を確認した。溶解確認後、内温40℃付近に冷却し、結晶の析出を確認した。引き続き、内温25℃付近に冷却し、同温下にて1時間撹拌した。同温度にて析出した結晶をろ取し、DMSO(40mL)にて洗浄を実施した。得られたウェット結晶を55℃にて終夜、減圧乾燥を実施し、DMSOの1和物であるエンザルタミドF型結晶を7.2g得た。収率は20.5%だった。
1H-NMR(CDCl3-d6,400MHz):δ(ppm)=1.62(6H,s),2.62(6H,s),3.07(3H,d,J=4.4Hz),6.74(1H,m),7.15(1H),7.25(1H),7.83(1H),7.95(1H),7.99(1H),8.28(1H)
XRD:2θ(°)=17.1,20.2,24.6

Claims (7)
- エンザルタミドのウェット結晶を得る晶析工程及び前記ウェット結晶の乾燥工程を含む、下記式で表されるエンザルタミド結晶形の製造方法であって、
前記晶析工程の後に、良溶媒と貧溶媒との混合溶媒を用いた洗浄工程を含むことを特徴とするエンザルタミド結晶形の製造方法。

- 前記洗浄工程が、前記乾燥工程の前に行われることを特徴とする請求項1に記載のエンザルタミド結晶形の製造方法。
- 前記混合溶媒における前記良溶媒と前記貧溶媒の割合が体積比で、1:99~99:1であることを特徴とする請求項1又は2に記載のエンザルタミド結晶形の製造方法。
- 前記良溶媒が酢酸エステル系有機溶媒、アセトン、メチルエチルケトン、テトラヒドロフラン及びアセトニトリルからなる群より選ばれる少なくとも1の溶媒であることを特徴とする請求項1~3のいずれか1項に記載のエンザルタミド結晶形の製造方法。
- 前記貧溶媒が炭化水素系有機溶媒、水及びメチル-tert-ブチルエーテルからなる群より選ばれる少なくとも1の溶媒であることを特徴とする請求項1~4のいずれか1項に記載のエンザルタミド結晶形の製造方法。
- 前記良溶媒が酢酸イソプロピルであり、前記貧溶媒がn-ヘプタンである、請求項1~5のいずれか1項に記載のエンザルタミド結晶形の製造方法。
- チオホスゲンを炭化水素系の有機溶媒又は塩素系有機溶媒と、水との混合溶媒に溶解して、4-シアノ-3-トリフルオロメチルアニリンを炭化水素系有機溶媒又は塩素系有機溶媒で溶解した溶液を滴下する工程を含むことを特徴とする、4-シアノ-3-トリフルオロメチルフェニルイソチオシアネートの製造方法。
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201680029674.1A CN107635969B (zh) | 2015-05-29 | 2016-05-27 | 恩杂鲁胺结晶形式的制造方法 |
| US15/577,007 US10118899B2 (en) | 2015-05-29 | 2016-05-27 | Production method of enzalutamide crystal form |
| EP16803248.0A EP3305770B1 (en) | 2015-05-29 | 2016-05-27 | Process for producing enzalutamide crystal form |
| HK18109971.4A HK1251218B (en) | 2015-05-29 | 2016-05-27 | Process for producing enzalutamide crystal form |
| ES16803248T ES2942618T3 (es) | 2015-05-29 | 2016-05-27 | Procedimiento para la fabricación de una forma cristalina de enzalutamida |
| KR1020177037241A KR102666017B1 (ko) | 2015-05-29 | 2016-05-27 | 엔잘루타미드 결정형의 제조 방법 |
| MX2017014958A MX385084B (es) | 2015-05-29 | 2016-05-27 | Método de producción de forma cristalina de enzalutamida. |
| PL16803248.0T PL3305770T3 (pl) | 2015-05-29 | 2016-05-27 | Sposób wytwarzania postaci krystalicznej enzalutamidu |
| CA2987563A CA2987563C (en) | 2015-05-29 | 2016-05-27 | Production method of enzalutamide crystal form |
| JP2017521901A JP6753396B2 (ja) | 2015-05-29 | 2016-05-27 | エンザルタミド結晶形の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015109805 | 2015-05-29 | ||
| JP2015-109805 | 2015-05-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016194813A1 true WO2016194813A1 (ja) | 2016-12-08 |
Family
ID=57442345
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/065729 Ceased WO2016194813A1 (ja) | 2015-05-29 | 2016-05-27 | エンザルタミド結晶形の製造方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US10118899B2 (ja) |
| EP (1) | EP3305770B1 (ja) |
| JP (1) | JP6753396B2 (ja) |
| KR (1) | KR102666017B1 (ja) |
| CN (1) | CN107635969B (ja) |
| CA (1) | CA2987563C (ja) |
| ES (1) | ES2942618T3 (ja) |
| MX (1) | MX385084B (ja) |
| PL (1) | PL3305770T3 (ja) |
| PT (1) | PT3305770T (ja) |
| WO (1) | WO2016194813A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021064123A1 (en) | 2019-10-03 | 2021-04-08 | Synthon B.V. | Pharmaceutical composition comprising enzalutamide |
| WO2025125565A1 (en) | 2023-12-13 | 2025-06-19 | Krka, D.D., Novo Mesto | Tablet comprising enzalutamide |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3990435A1 (en) * | 2019-06-27 | 2022-05-04 | Synthon B.V. | Process for preparation of enzalutamide |
| CN111217757B (zh) * | 2020-01-06 | 2021-03-19 | 武汉大学 | 一种恩杂鲁胺化合物及其药物组合物制剂 |
| CN111303042A (zh) * | 2020-03-25 | 2020-06-19 | 北京赛思源生物医药技术有限公司 | 一种恩杂鲁胺的新晶型 |
| CN114591246A (zh) * | 2022-03-25 | 2022-06-07 | 重庆华邦制药有限公司 | 一种恩扎卢胺的纯化方法 |
| CN115536591B (zh) * | 2022-09-27 | 2024-06-25 | 爱斯特(成都)生物制药股份有限公司 | 一种连续流制备恩扎卢胺的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014041487A2 (en) * | 2012-09-11 | 2014-03-20 | Dr. Reddy's Laboratories Limited | Enzalutamide polymorphic forms and its preparation |
| WO2016005875A1 (en) * | 2014-07-11 | 2016-01-14 | Shilpa Medicare Limited | An improved process for the preparation of enzalutamide |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4644737B2 (ja) * | 2005-05-13 | 2011-03-02 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | ジアリールヒダントイン化合物 |
| PL2538785T3 (pl) | 2010-02-24 | 2018-08-31 | Medivation Prostate Therapeutics Llc | Sposoby syntezy zwiazków diarylotiohydantoinowych i diarylohydantoinowych |
| US20130090417A1 (en) * | 2010-06-14 | 2013-04-11 | Solvay Specialty Polymers Italy S.P.A. | PVDF coating compositions |
| CN105188699B (zh) | 2013-10-14 | 2017-04-26 | 杭州普晒医药科技有限公司 | 恩杂鲁胺的固态形式及其制备方法和用途 |
| US9611225B2 (en) | 2014-01-27 | 2017-04-04 | Cadila Healthcare Limited | Process for preparation of androgen receptor antagonist |
| CN104610158A (zh) | 2015-02-03 | 2015-05-13 | 江苏慧博生物科技有限公司 | 一种恩杂鲁胺的精制方法 |
| ITUB20151204A1 (it) * | 2015-05-28 | 2016-11-28 | Olon Spa | Procedimento per la preparazione di enzalutamide |
-
2016
- 2016-05-27 MX MX2017014958A patent/MX385084B/es unknown
- 2016-05-27 CN CN201680029674.1A patent/CN107635969B/zh active Active
- 2016-05-27 PT PT168032480T patent/PT3305770T/pt unknown
- 2016-05-27 KR KR1020177037241A patent/KR102666017B1/ko active Active
- 2016-05-27 US US15/577,007 patent/US10118899B2/en active Active
- 2016-05-27 CA CA2987563A patent/CA2987563C/en active Active
- 2016-05-27 PL PL16803248.0T patent/PL3305770T3/pl unknown
- 2016-05-27 EP EP16803248.0A patent/EP3305770B1/en active Active
- 2016-05-27 JP JP2017521901A patent/JP6753396B2/ja active Active
- 2016-05-27 ES ES16803248T patent/ES2942618T3/es active Active
- 2016-05-27 WO PCT/JP2016/065729 patent/WO2016194813A1/ja not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014041487A2 (en) * | 2012-09-11 | 2014-03-20 | Dr. Reddy's Laboratories Limited | Enzalutamide polymorphic forms and its preparation |
| WO2016005875A1 (en) * | 2014-07-11 | 2016-01-14 | Shilpa Medicare Limited | An improved process for the preparation of enzalutamide |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3305770A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021064123A1 (en) | 2019-10-03 | 2021-04-08 | Synthon B.V. | Pharmaceutical composition comprising enzalutamide |
| WO2025125565A1 (en) | 2023-12-13 | 2025-06-19 | Krka, D.D., Novo Mesto | Tablet comprising enzalutamide |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2017014958A (es) | 2018-07-06 |
| PT3305770T (pt) | 2023-04-26 |
| CA2987563C (en) | 2024-04-09 |
| CN107635969A (zh) | 2018-01-26 |
| US10118899B2 (en) | 2018-11-06 |
| JP6753396B2 (ja) | 2020-09-09 |
| KR102666017B1 (ko) | 2024-05-16 |
| HK1251218A1 (en) | 2019-01-25 |
| EP3305770A4 (en) | 2018-12-12 |
| EP3305770B1 (en) | 2023-03-29 |
| US20180148417A1 (en) | 2018-05-31 |
| JPWO2016194813A1 (ja) | 2018-06-07 |
| CA2987563A1 (en) | 2016-12-08 |
| PL3305770T3 (pl) | 2023-06-26 |
| KR20180014015A (ko) | 2018-02-07 |
| EP3305770A1 (en) | 2018-04-11 |
| CN107635969B (zh) | 2021-08-17 |
| MX385084B (es) | 2025-03-14 |
| ES2942618T3 (es) | 2023-06-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6753396B2 (ja) | エンザルタミド結晶形の製造方法 | |
| JP5305248B2 (ja) | 新規ナテグリニド結晶 | |
| JP6378844B2 (ja) | 第6結晶形ソホスブビルの調製方法 | |
| KR20170023417A (ko) | L-카르노신 아연 착물의 제조 방법 | |
| US12528771B2 (en) | Method for producing centanafadine | |
| US1982675A (en) | Pbeparation of acetoacetanilid | |
| CN101910124B (zh) | 光学活性3-氨基吡咯烷盐、其制造方法以及3-氨基吡咯烷的光学拆分方法 | |
| CN105111188A (zh) | 一种埃索美拉唑镁三水合物晶型的制备方法 | |
| JP2008255065A (ja) | 塩酸サルポグレラートの工業的製造方法 | |
| CN104703967B (zh) | 氟伏沙明游离碱的精制方法及利用其的高纯度马来酸氟伏沙明的制备方法 | |
| WO2020110831A1 (ja) | チオラクトン誘導体の製造方法 | |
| JP2010229098A (ja) | イソインドリン誘導体のa型の結晶形の製造方法及びイソインドリン誘導体のa型の結晶形 | |
| JP2018080125A (ja) | インドリン化合物のβ型結晶の製造方法 | |
| JP4849374B2 (ja) | (±)2−(ジメチルアミノ)−1−{〔O−(m−メトキシフェネチル)フェノキシ〕メチル}エチル水素サクシナート塩酸塩のI形結晶とII形結晶の混晶の製造法 | |
| HK1251218B (en) | Process for producing enzalutamide crystal form | |
| JP3884063B2 (ja) | セフカペンピボキシルのメタンスルホン酸塩 | |
| JP6499948B2 (ja) | セレコキシブii型結晶の製造方法 | |
| WO2019146508A1 (ja) | 2-クロロアセト酢酸アミドの製造方法 | |
| JP7340534B2 (ja) | トレプロスチニルジエタノールアミン塩の多形形態bを製造するための方法 | |
| JP2019059688A (ja) | 結晶性l−カルノシン亜鉛錯体の製造方法 | |
| CN106432197B (zh) | 一种雷迪帕韦中间体单对甲苯磺酸盐、其晶型及其制备方法 | |
| JP6663232B2 (ja) | 新規結晶構造を有するアジルサルタン及びその製造方法 | |
| JP2022530334A (ja) | スガマデクスを調製するための新しいプロセス | |
| JP2011225491A (ja) | 4−アミノ−5−クロロ−2−エトキシ−n−〔〔4−(4−フルオロベンジル)−2−モルホリニル〕メチル〕ベンズアミドまたはその溶媒和物の新規な結晶およびその製造方法 | |
| JP2008115171A (ja) | 4´−{2−[(1s,2r)−2−ヒドロキシ−2−(4−ヒドロキシフェニル)−1−メチルエチルアミノ]エトキシ}−3−イソプロピル−3´,5´−ジメチルビフェニル−4−カルボン酸塩酸塩の結晶多形 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16803248 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017521901 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2017/014958 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15577007 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2987563 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20177037241 Country of ref document: KR Kind code of ref document: A |